um sistema computacional para diagnosticar viroses de ... · um sistema computacional para...
TRANSCRIPT

Universidade Federal do Rio Grande do Norte
Centro de Tecnologia
Programa de Pós-Graduação em Engenharia Elétrica
Dissertação de Mestrado
Um Sistema Computacional para Diagnosticar
Viroses de Plantas Usando a Técnica de PCR com
Construção de “Primers Espécie-Específicos”
AUTOR: Kliger Kissinger Fernandes Rocha
ORIENTADOR: Prof. Dr. Luiz Marcos Garcia Gonçalves
CO-ORIENTADOR: Prof. Dr. Paulo Sérgio Marinho Lúcio
Natal/RN – Brasil
Abril de 2005

Um Sistema Computacional para Diagnosticar Viroses de
Plantas Usando a Técnica de PCR com Construção de
“Primers Espécie-Específicos”
Kliger Kissinger Fernandes Rocha
Aprovada, em 04 de abril de 2005, pela Comissão Examinadora formada pelos seguintes
membros:
_____________________________________________________________
Profa. Dra. Eliana Silva de Almeida – TCI-UFAL
_____________________________________________________________
Prof. Dr. José Alfredo Ferreira da Costa – UFRN
______________________________________________________________
Prof. Dr. Paulo Sérgio Marinho Lúcio – UFRN (Co-Orientador)
______________________________________________________________
Prof. Dr. Luiz Marcos Garcia Gonçalves – UFRN (Orientador)
NATAL, RN – BRASILAbril de 2005.

Universidade Federal do Rio Grande do Norte
Centro de Tecnologia
Programa de Pós-Graduação em Engenharia Elétrica
Kliger Kissinger Fernandes Rocha
Um Sistema Computacional para Diagnosticar
Viroses de Plantas Usando a Técnica de PCR com
Construção de “Primers Espécie-Específicos”
ORIENTADOR: Prof. D.Sc. Luiz Marcos Garcia Gonçalves
CO-ORIENTADOR: Prof. D.Sc. Paulo Sérgio Marinho Lúcio
Natal/RN – BrasilAbril de 2005
Dissertação submetida ao Programa de Pós-
Graduação em Engenharia Elétrica da
Universidade Federal do Rio Grande do Norte,
como parte dos requisitos necessários para
obtenção do grau de Mestre em Ciências
(M.Sc.).

� � � � � � � � � �
A Deus onde sempre encontro forças para
superar as dificuldades.
Aos meus pais; Consuelo Fernandes Rocha e
Manoel Ferreira da Rocha (in memorian),
exemplos de vida, mentores da minha evolução
profissional e moral.
As minhas irmãs, Kelly Cristina Fernandes
Rocha e Janine Fernandes Rocha, facilitadoras
de meu aprendizado, minha paz e felicidade.

ii
UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
Date: abril de 2005
Author: Kliger Kissinger Fernandes Rocha
Title: Um Sistema Computacional para Diagnosticar Viroses de Plantas Usando aTécnica de PCR com Construção de “Primers Espécie-Específicos”
Department: Programa de Pós-Graduação em Engenharia Elétrica
Degree: M.Sc. Convocation: May Year: 2005
Permission is herewith granted to Federal University of Rio Grande do Norte tocirculate and to have copied for non-commercial purposes, at its discretion, the abovetitle upon the request of individuals or institutions.
THE AUTHOR RESERVES OTHER PUBLICATION RIGHTS, AND NEITHERTHE THESIS NOR EXTENSIVE EXTRACTS FROM IT MAY BE PRINTED OROTHERWISE REPRODUCED WITHOUT THE AUTHOR’S WRITTEN PERMISSION.
THE AUTHOR ATTESTS THAT PERMISSION HAS BEEN OBTAINED FORTHE USE OF ANY COPYRIGHTED MATERIAL APPEARING IN THIS THESIS(OTHER THAN BRIEF EXCERPTS REQUIRING ONLY PROPERACKNOWLEDGEMENT IN SCHOLARLY WRITING) AND THAT ALL SUCH USE ISCLEARLY ACKNOWLEDGED.

iii
Agradecimentos
Agradeço principalmente à Deus, inteligência suprema, causa primária de todas as coisas, por
sempre me doar todas as condições necessárias para evoluir como consciência e tantos motivos
para agradecer e ser feliz.
Aos meus amorosos pais, por vários motivos: pela educação fundamentada na ética e no
amor responsável, pelo apoio financeiro, pelos exemplos de seres humanos louváveis na conduta
moral, por terem me concebido com muito amor, pelas renuncias, pela paciência, pelas crenças e
valores Cristãos... Enfim, por dedicarem-se tanto pela felicidade dos filhos.
Às minhas irmãs, pelo convívio amoroso que sempre facilita o meu sucesso e felicidade.
Aos prezados orientadores (Prof. Paulo Marinho e Prof. Luiz Marcos), modelos de
profissionais, por me aceitarem como orientando, pelas valiosas informações para minha
formação profissional, e principalmente por me incentivarem e investirem tanto na minha
qualificação profissional.
À banca examinadora, pelas importantes contribuições para melhorar este trabalho.
À CAPES, pelo apoio financeiro recebido.
Aos amigos: Hani e família, Sheila Mara e Susy; por todos os momentos que passamos
juntos compartilhando sentimentos e idéias que contribuíram para o meu sucesso neste trabalho.
E agradeço também aos colegas: Francisco Cardoso, César e Maristela Holanda.

iv
A todos os professores do meu histórico como estudante, pelos ensinamentos que
contribuíram para uma melhor compreensão deste trabalho, e ampliação da minha visão de
mundo. E, nesta fase de Pós-Graduação, agradeço em especial aos seguintes Professores: Dr.
Luiz Affonso Henderson Guedes de Oliveira pela orientação no Estágio Docente; e Dra. Patrícia
Sommer pelo incentivo e apoio.
Aos funcionários da UFRN que colaboraram, principalmente a Santana, por sempre ser
prestativo.
A todos que, de bom grado, contribuíram direta e indiretamente até em anonimato para a
minha formação profissional e não foram citados nestes agradecimentos.

v
Conteúdo
Resumo
Abstract
1 Introdução 11.1 Motivação para o Trabalho. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Organização da Dissertação. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2 Os Primers e a Técnica PCR 52.1 A técnica PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
3 O Problema do Projeto e Escolha de Primers 123.1 O Método Tradicional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
3.2 O Problema de Alinhamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153.2.1 O Algoritmo de Força Bruta. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
3.2.1.1 Algoritmo. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.3 O Problema da Construção de Primers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183.2.1 Temperatura de desnaturação (Tm) e tempo . . . . . . . . . . . . . . . . . . . . . . . . 183.3.2 Temperatura de Anelamento (TA) e construção de primer . . . . . . . . . . . . . 193.3.3 Comprimento do primer. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.3.4 Primers degenerados. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213.3.5 Temperatura de Extensão . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.3.6 Número de Ciclos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4 Uma Solução Computacional 244.1 O Sistema . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
4.1.1 O Módulo de Alinhamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294.1.2 O Módulo de Construção de Primers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.2 Programação, Plataforma e Interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5 Experimentos e Resultados 365.1 Experimentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
5.1.1 Cenário 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 385.1.2 Cenário 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
6 Conclusão e Perspectivas 56
Referências Bibliográficas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58

vi
Lista de Figuras
1.1 Etapas do ciclo de PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
2.1 Interfaces dos locais na Internet usados para alinhamento de seqüências de genomas 7
2.2 Primer na fita-molde com hidroxila livre e nucleotídeo com fosfato livre . . . . . . . . . 9
2.3 Os primeiros quatro ciclos de uma PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.4 Etapas de PCR (amplificação exponencial em 30 ciclos). . . . . . . . . . . . . . . . . . . . . . . 10
2.5 Termociclador. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.6 Ingredientes da reação in vitro da PCR. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
3.1 Exemplos de complementaridade inter- e intra-primers que resulta em problemas. . . 14
3.2 Gel de Eletroforese com vários tamanhos de fragmentos amplificados por PCR . . . . 14
3.3 Funcionamento do Algoritmo de Força Bruta . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
3.4 Seqüências de primers foram derivadas dos alinhamentos múltiplos de seqüência . . 21
4.1 Formato Fasta. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
4.2 Formato do arquivo de primers do programa. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
4.3 Tela inicial do programa. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
4.4 Tela mostra em que sequencia e posição da sequencia se encontra o primer. . . . . . . . 33
4.5 Tela de resultados que mostra as regiões especificas. . . . . . . . . . . . . . . . . . . . . . . . . . 34
4.6 Tela de resultados que mostra as regiões universais. . . . . . . . . . . . . . . . . . . . . . . . . . . 34
4.7 Tela mostrando os primers da região específica com seus respectivos parâmetros. . . 13
5.1 Tela de resultados do Entrez no NCBI mostrando 7 genomas de vírus da batata . . . . 39
5.2 Tela de Resultados do Blast-N para Potato Vírus V. . . . . . . . . . . . . . . . . . . . . . . . . . . 41
5.3 Tela de Resultados do Blast-N para Cherry rasp leaf virus RNA2. . . . . . . . . . . . . . . . 42
5.4 Tela de Resultados do Blast-N para Cherry rasp leaf virus . . . . . . . . . . . . . . . . . . . . 44

vii
5.5 Tela de Resultados do Blast-N para Potato Vírus Y . . . . . . . . . . . . . . . . . . . . . . . . . . 45
5.6 Tela de Resultados do Blast-N para Narcissus mosaic virus . . . . . . . . . . . . . . . . . . . . 47
5.7 Tela de Resultados do Blast-N para Potato Vírus M . . . . . . . . . . . . . . . . . . . . . . . . . . 48
5.8 Tela de Resultados do Blast-N para Potato Vírus A. . . . . . . . . . . . . . . . . . . . . . . . . . . 49
5.9 Tela de resultados do Entrez no NCBI com 4 genomas de vírus de meloeiro . . . . . . . 51
5.10 Tela de Resultados do Blast-N para Cucurbit yellow stunting disorder virus RNA2. . 52
5.11 Tela de Resultados do Blast-N para Cucurbit yellow stunting disorder virus RNA1. . 53
5.12 Tela de Resultados do Blast-N para Melon chlorotic leaf curl virus . . . . . . . . . . . . . . 54
5.13 Tela de Resultados do Blast-N para Melon necrotic spot virus . . . . . . . . . . . . . . . . . . 55

viii
Lista de Gráficos, Diagramas e Tabelas
3.1 Gráfico de Tempo de Execução de Força Bruta. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
5.1 Gráfico de performance da última versão do algoritmo de Força Bruta . . . . . . . . . . . . 37
4.1 Diagrama de Acessibilidade ao banco de dados. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.2 Diagrama de Arquitetura do Sistema. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.3 Diagrama do Módulo de Alinhamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.4 Diagrama do Módulo de Construção de Primers. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.1 Soluções do Sistema Dadas aos Diversos Problemas Abordados . . . . . . . . . . . . . . . . . 45
5.1 Tabela de Primers de Espécies de Viroses de Batata Obtida Pelo Programa Proposto . 39
5.2 Tabela de Primers de Espécies de Vírus do Meloeiro Obtida Pelo Programa Proposto. 50

ix
Resumo
Propõe-se uma solução computacional baseada no desenvolvimento de um software para
construir primers espécie-específicos, usados para melhorar o diagnóstico de viroses de planta
por PCR. Primers são indispensáveis à reação PCR, além de proporcionar a especificidade do
diagnóstico. Um primer é um fragmento de DNA sintético, curto e de fita simples, utilizado
como um iniciador na técnica PCR que flanqueia a seqüência que se deseja amplificar. Primers
espécie-específicos são primers que só indicam a região bem conhecida de início e término onde
a enzima polimerase vai amplificar, de uma determinada espécie, ou seja, é específica para
somente uma espécie. Assim, o objetivo principal deste trabalho é automatizar o processo de
escolha de primers, otimizando a especificidade dos primers escolhidos pelo método tradicional.
Palavras chaves: Design de Primer, PCR, Bioinformática

x
Abstract
It proposes a established computational solution in the development of a software to construct
species-specific primers, used to improve the diagnosis of virus of plant for PCR. Primers are
indispensable to PCR reaction, besides providing the specificity of the diagnosis. Primer is a
synthetic, short, single stranded piece of DNA, used as a starter in PCR technique. It flanks the
sequence desired to amplify. Species-specific primers indicate the well known region of
beginning and ending where the polymerase enzyme is going to amplify on a certain species, i.e.
it is specific for only a species. Thus, the main objective of this work is to automatize the
process of choice of primers, optimizing the specificity of chosen primers by the traditional
method.
Keywords: Primer Design, PCR, Bioinformatics

Capítulo 1
Introdução
A Bioinformática visa compreender problemas em que questões biológicas delineiam questões
algorítmicas, bem como propor suas soluções. É uma área de pesquisa relativamente nova, com
um crescimento substancial de trabalhos.
Na primeira metade da década de 80, foi desenvolvido um método de amplificação de
seqüências de DNA que revolucionou a análise genética nestes últimos anos: a “reação em
cadeia da polimerase” (ou PCR, do inglês Polymerase Chain Reaction). Esta técnica possibilita
que múltiplas cópias de uma molécula de DNA sejam geradas por meio da amplificação
enzimática de uma seqüência de DNA escolhida. A técnica baseia-se na capacidade que a enzima
DNA polimerase tem de replicar seqüências de DNA, em certas condições laboratoriais, a partir
de um par de pequenos fragmentos iniciadores da fita molde, denominados de primers.
Figura 1.1 - Etapas do ciclo de PCR [41]

2
A Figura 1.1 ilustra a técnica. Através de variações alternadas e cíclicas de temperatura que
permitem a desnaturação (ex. 92ºC, abertura da fita dupla de DNA), anelamento (ex. 54ºC,
pareamento dos primers ou iniciadores) e extensão (ex. 72ºC, cópia da fita dupla original pela
incorporação de nucleotídeos nas fitas complementares). Assim, uma determinada seqüência de
DNA é replicada, ciclo após ciclo, em progressão geométrica (figura 2.3), o que torna possível
sua visualização em gel de eletroforese na forma de uma banda (figura 2.7).
O desenvolvimento dessa técnica e suas aplicações concederam ao americano Kary Mullis o
Prêmio Nobel em Química em 1993. As aplicações da técnica são inúmeras, conforme pode ser
verificado em revisão feita por Mullis, Rerré e Gibbs [23]. A PCR tem sido utilizada, por
exemplo, desde experimentos relacionados ao seqüenciamento de DNA até aplicações
comerciais na área de diagnose. Algumas variações da PCR levaram ao desenvolvimento de
outras técnicas poderosas na análise de diversidade genética, como: AFLP, RAPD, SAMPL e
SSR.
� � � � � � � � � � � � � � � � � � � " $ �
O trabalho desenvolvido foi inicialmente motivado pela necessidade de se estabelecer
condições técnicas de identificação por PCR de viroses em plantas no Estado do Rio Grande do
Norte. Especificamente, procurou-se desenvolver uma ferramenta em bioinformática que
aperfeiçoasse a escolha de primers ou oligonucleotídeos para a reação de PCR. Neste trabalho, o
aplicativo desenvolvido e otimizado evitaria a fabricação de oligonucleotídeos pouco eficientes
na identificação das viroses em plantas caso fossem escolhidos manualmente.
Para o desenvolvimento da ferramenta foi necessário, no entanto, especificar quais os
parâmetros que influenciariam na qualidade da amplificação por PCR. Estes parâmetros têm uma
influência direta na construção de primers. Por outro lado, há que considerar que a região do
genoma viral tem que garantir a especificidade do diagnóstico em nível de espécie do agente
causador da virose.
Uma das principais motivações que encontramos é a tentativa de diminuir ou eliminar o
prejuízo na fruticultura do Estado do Rio Grande do Norte, onde plantadores têm queimado
plantas em áreas agrícolas por causa de suspeita de apenas um ou alguns exemplares da planta
com virose. O problema é que muitas vezes não é virose, podendo ser outro agente patogênico
ou deficiência ou mesmo o excesso de nutrientes. Mas, para evitar o risco de uma epidemia,
infelizmente, quase sempre é preferido o modo drástico de resolver o problema; queimando toda
a área plantada. Isso gera prejuízo na agricultura de exportação do Estado, e do país.

3
A motivação biológica de ser um programa de primers para vírus é o fato de que, por serem
organismos altamente instáveis, compostos por genes mutantes e recombinantes, os vírus
pesquisados apresentam problemas quanto a sua erradicação.
Basicamente, o software contempla dois módulos: um módulo de alinhamento dos genomas de
vírus para separar as áreas polimórficas, e o segundo módulo é de construção de primers
específicos para diagnosticarem uma determinada espécie de vírus por PCR. Testes e
experimentos foram realizados e os resultados foram satisfatórios para genomas pequenos como
os de vírus. O presente trabalho já publicou resultados parciais em pôster no SIBGRAPI 2003
[44], e em artigo completo no 4th IEEE International Symposium on Bioinformatics and
Bioengineering (BIBE 2004) [45].
Mesmo usando o Algoritmo da Força Bruta [12], considerado um método simples, uma
intensa modificação realizada foi no sentido de evitar comparações desnecessárias e melhorou
em 75% a performance de tempo do programa.
A principal questão deste trabalho é propor uma estratégia de escolha de primers eficiente para
diagnosticar um patógeno (agente biológico capaz de causar doença) suspeito. A resposta a esta
questão avança a metodologia de diagnóstico molecular em nível de espécie, facilitando no
processo de tratamento.
Como contribuições deste trabalho podem-se citar a construção de um banco de dados,
contendo seqüências de vírus de plantas, e um sistema para alinhar e separar domínios de
seqüências. O banco de dados que este trabalho se refere é uma coleção ordenada de arquivos
semelhantes, em conformidade com um formato padrão de conteúdo. O banco de dados de
arquivos simples pode ser pesquisado devido à indexação. Contudo, à medida que a coleção de
arquivos simples fica cada vez maior, torna-se ineficaz trabalhar com ela.
Esta estratégia melhora a mineração de dados no banco de dados, encontrando seqüências e
gerando múltiplos alinhamentos. Essas seqüências podem compartilhar similaridades com
domínios e diferenciar entre domínios polimórficos. Trabalhou-se com domínios polimórficos
para construir primers com especificidade elevada.
Além disso, o programa desenvolvido possibilita também um estudo de polimorfismo por
possuir um módulo de alinhamento indicando regiões com polimorfismo e regiões similares
entre duas ou mais espécies de vírus. A construção de um banco de dados contendo arquivos de
vírus de plantas é também uma necessidade do sistema, além de otimizar o acesso e a mineração
de dados para tomada de decisões pelo pesquisador, pode ser fonte de pesquisa para o
pesquisador descobrir aspectos importantes da Filogenia de vírus. A Filogenia descreve a origem
e a evolução das espécies.

4
� � � � � � � � � � � � � � � � � � � � � � � � �
O Capítulo 2 trata dos trabalhos relacionados, discutindo a contribuição deste trabalho
comparado às publicações estudadas. Um apanhado do estado da arte é apresentado, incluindo
informações teóricas básicas, necessárias ao entendimento do problema tratado.
No Capítulo 3, apresentamos o histórico, especificação e detalhamento do problema abordado,
incluindo um estudo dos parâmetros necessários à técnica PCR.
O capítulo 4 aborda a solução teórica encontrada, apontando para possíveis formas de resolver
o problema. Neste Capítulo, apresentamos também a solução computacional adotada para
solucionar o problema e os módulos do programa implementados.
No capítulo 5 são mostrados os resultados de diversos experimentos e testes realizados para
validar o sistema computacional proposto.
Por fim, no Capítulo 6 é apresentada a conclusão sobre o trabalho, e perspectivas futuras
relacionadas ao projeto.

5
Capítulo 2
Os Primers e a Técnica PCR
Muitos trabalhos na área de Biologia Molecular estão relacionados com a construção e escolha
de primers para diagnóstico, sendo alguns deles estudados neste trabalho [01 a 11]. A maioria
desses usa programas somente para construção de primers sem prever por alinhamento que
região do genoma a ser amplificada seria ideal. A proposta deste trabalho é automatizar e
aperfeiçoar o processo com a estratégia do alinhamento antes da construção de primers.
Alguns métodos computacionais ou programas estão disponíveis atualmente para a construção
de primers [01,02,03], com uma finita probabilidade de produzir erros. Os trabalhos encontrados
na literatura descrevem os passos envolvidos no processo e os esforços dispensados para
automatizá-lo. A proposta geral é selecionar uma região para construir um primer onde a
probabilidade de erro de diagnóstico usando PCR seja baixa [01,03], considerando não somente
as regiões selecionadas visualmente, mas também regiões examinadas estatisticamente.
Convém ressaltar que, em relação aos programas disponíveis na Internet, públicos e privados,
o programa proposto neste trabalho tem várias vantagens, sendo a automação do processo de
diagnóstico para um usuário sem muitos conhecimentos técnicos de computação uma das
principais delas. O programa desenvolvido lista vários primers candidatos com os respectivos
atributos para a correta decisão do especialista. No modo como são listados os primers, pode-se
distinguir facilmente as vantagens dos candidatos.
No módulo de alinhamento do programa proposto, deseja-se comparar duas ou mais
seqüências genéticas, sendo esta, tradicionalmente, a operação básica de bioinformática. Através
da comparação de seqüências, podem-se obter várias informações, tais como: similaridade
(medida numérica que indica quão similares são duas seqüências); presença ou não de homologia
(indica se dois ou mais genes possuem uma história evolutiva comum); alinhamento entre
seqüências (forma de se colocar uma seqüência "em cima" da outra, de maneira a evidenciar a
correspondência entre caracteres ou subcadeias similares das seqüências); entre outras. Esta

6
metodologia não é aplicada pela maioria dos programas para projetar primers estudados neste
trabalho. O programa público Gene Fisher tem a mesma metodologia [40] de busca, mas usa o
CLUSTALW ou DCA como programa de alinhamento, dependendo da demora na Internet o
processo é moroso e não é considerado totalmente automatizado. Ainda, o programa proposto
neste trabalho não tem somente uma funcionalidade básica (construção de primers espécie-
específicos), mas também permite ao pesquisador estudar o polimorfismo de vírus em uma
mesma família, e entre taxonomias diferentes.
Existem alguns problemas no caso múltiplo que não existem no caso básico: a pontuação dos
alinhamentos; complexidade da abordagem que utiliza programação dinâmica pura (trata-se de
um problema NP - completo); criação de heurísticas que aumentem a velocidade da computação;
etc. Existem tanto estudos teóricos que atacam esses problemas quanto algoritmos que
implementam essas heurísticas (métodos de alinhamento em estrela, em árvore, e outros),
tornando possível, assim, viabilizar o alinhamento de múltiplas seqüências.
O estudo de algoritmos de Bioinformática teria sido incompleto se não fosse abordado um
tema cuja importância aumenta a cada dia: a comparação de seqüências genéticas em bancos de
dados. A busca de seqüências em bancos de dados permite determinar quais das centenas de
milhares de seqüências presentes no banco podem estar relacionadas a uma dada seqüência.
Nesse tipo de ambiente, a operação básica consiste em alinhar uma seqüência de consulta com as
seqüências do banco de dados.
Os atuais bancos de dados de seqüências já são gigantescos, e continuam a crescer numa taxa
exponencial, como por exemplo o “Genbank” em 2004 publicou 44.575.745.176 pares de bases e
40.604.319 seqüências [43]. Isso torna a aplicação de programação dinâmica pura inviável,
obrigando o uso de heurísticas, que aumentam bastante a velocidade dos alinhamentos (mas com
uma pequena probabilidade de perder alinhamentos verdadeiros).
Os dois programas de busca de seqüências mais usados na atualidade são o “FASTA” e o
“BLAST” (vide Figura 2.1). O FASTA foi o primeiro de todos os programas do tipo a ser
amplamente utilizado, enquanto o BLAST, posteriormente introduzido, trouxe uma série de
refinamentos. Quando de uma busca, ambos aplicam em primeiro lugar métodos heurísticos e,
após a obtenção de uma lista inicial de seqüências, métodos baseados em programação dinâmica
são usados para, finalmente, gerarem a lista final de hits. Esta palavra hits pode ser identificada
rapidamente pre-indexando todas as palavras da query e então consultando o índice na medida
que o BD é pecorrido. A Figura 2.1 mostra as telas de entrada de dados do FASTA e do BLAST,
respectivamente, acessíveis por seus web sites [42,43].

7
Figura 2.1: Interfaces dos locais na Internet usados para alinhamento de seqüências de genomas.

8
O programa desenvolvido neste trabalho compara as seqüências obtidas de um banco de dados
com uma seqüência padrão, identificando e numerando as alterações encontradas. Além disso, é
possível no sistema concluir informações importantes cruzando estes resultados de alinhamento
com os dados dos respectivos vírus estudados.
O sistema desenvolvido forma uma parte do elo de ligação entre os dados biológicos de vírus,
de um lado, e as informações de seqüências genéticas, de outro, recebendo dados tanto de
arquivos Fasta de seqüenciadores quanto dos bancos de dados genéticos de vírus de plantas.
Esta ferramenta computacional é extremamente útil em comparação às outras de domínio
público, ao permitir um controle muito mais acurado no projeto de primers específicos, e uma
mineração de dados pelo relacionamento com o banco de dados de vírus.
� � � � � � � � � � � �
A técnica de PCR (do inglês Polymerase Chain Reaction, reação em cadeia da polimerase)
consiste numa reação em que uma região pequena e específica do genoma é amplificada por
síntese, pela polimerase de DNA. A reação em cadeia da polimerase possibilita a amplificação
de uma seqüência rara de DNA a partir de uma mistura complexa, sem a necessidade de
clonagem molecular. Esta técnica é amplamente utilizada em pesquisa básica, em medicina
forense e no diagnóstico de doenças genéticas e infecciosas.
Inicialmente, é necessária a construção por síntese química de dois oligonucleotídeos de DNA
ou primers (iniciadores) complementares, as extremidades de cada fita de DNA, flanqueando a
região de interesse. Estes oligonucleotídeos servem como iniciadores da síntese de DNA in vitro,
que é catalisada pela DNA polimerase, devido ao primer fornecer uma extremidade de hidroxila
livre onde a DNA polimerase catalisará a reação deste composto com o grupo fosfato de um
nucleotídeo correspondente à base nitrogenada da fita-molde (vide Figura 2.2).
Um ciclo de PCR começa com a desnaturação por calor (95°C), promovendo a separação da
fita dupla de DNA. A reação é resfriada na presença de um excesso dos dois oligonucleotídeos,
possibilitando a hibridização dos dois iniciadores com a seqüência complementar presente no
DNA alvo. Em seguida, a reação é incubada para atividade da DNA polimerase, produzindo
novas fitas de DNAs a partir dos iniciadores e utilizando quatro desoxirribonucleotídeos (dATP,
dCTP, dGTP e dTTP) (vide Figura 2.6) [24].
Cada novo ciclo da reação inicia-se com o aquecimento para desnaturação da dupla fita de
DNA, seguido de resfriamento para hibridação dos iniciadores e síntese de uma nova fita pela

9
DNA polimerase a partir dos iniciadores, sendo que as fitas de DNA recém sintetizadas servem
de molde no ciclo seguinte. Portanto, em cada ciclo é sintetizado o dobro do DNA produzido no
ciclo anterior. A Figura 2.3 mostra no terceiro ciclo, duas duplas fitas que apresentam o tamanho
correto sendo copiadas (as duas fitas com o mesmo tamanho). No quarto ciclo, 8 duplas fitas
que apresentam o mesmo tamanho são copiadas. Usualmente, são realizados entre 20 e 30 ciclos
para amplificação de um segmento de DNA específico dentro de um genoma (Figura 2.4) usando
uma máquina termocicladora (máquina que varia a temperatura da PCR em segundos, vide
Figura 2.5) e os ingredientes dentro de um tubo onde ocorre a reação mostrada na Figura 2.6.
Figura 2.2: Primer nafita-molde com hidroxila livre e nucleotídeo com fosfato livre
Figura 2.3: Os primeiros 4 ciclos de uma PCR (http:// allserv.rug.ac.be/ ~avierstr/index.html)
Grupo fosfato
Hidroxila
Fita-molde
Primer

10
Figura 2.4: Etapas de PCR (amplificação exponencial em 30 ciclos)
Figura 2.5: Termociclador
Figura 2.6: Ingredientes da reação in vitro da PCR

11
Nas primeiras iniciativas para amplificar fragmentos de DNA, utilizava-se a enzima DNA
polimerase da Escherichia coli, que possui atividade máxima a 37°C. Esta enzima deveria ser
adicionada a cada ciclo, pois o passo de desnaturação inativa a enzima. Um importante avanço
ocorreu com a descoberta de a enzima Taq DNA polimerase [25] oriunda da bactéria Thermus
aquaticus. A Taq DNA polimerase possui atividade ótima a 72°C e permanece razoavelmente
estável mesmo a 95°C e com isto, a enzima é adicionada somente no inicio do processo.

12
Capítulo 3
O Problema do Projeto e Escolha de Primers
Viroses de plantas constituem um sério problema que afeta a produção de plantas tropicais. No
caso de algumas plantas como o mamão, por exemplo, eles são responsáveis pelo abandono da
área contaminada pelo PRSV (Papaya Ring Spot Virus), PMV (Papaya Mosaic Virus) ou PMeV
(Papaya Meleira Virus) [27], como tem recentemente ocorrido no Brasil. Outros vírus de planta
poderiam ser citados aqui que apresentem as mesmas conseqüências. O controle de vírus de
planta empregado em produção de plantas é baseado na identificação dos sintomas da virose que
são visíveis quando a contaminação é potencialmente um risco para outras plantas. Estas plantas
são então eliminadas da plantação e normalmente queimadas.
A identificação prévia do vírus de planta pelas técnicas da biologia molecular representa uma
maneira eficiente para prevenir contaminações de vírus em grande escala e pode ser empregada
em muitas situações como a triagem de plantas importadas pelas instituições de segurança de
plantas. Estas técnicas moleculares estão baseadas na PCR que permite a amplificação de
seqüências de vírus específicos do DNA da planta afetada.
Reações de PCR específicas são mais eficientes usando oligonucleotídeos normalmente com
um tamanho entre 16 e 30 bases [05]. Estes primers específicos são escolhidos concordando
com seqüências conhecidas presentes no DNA amplificado do genoma, que neste caso são
regiões do genoma do vírus. Alguns critérios que permitem uma boa escolha de primers devem
ser considerados. É importante, por exemplo, evitar as seguintes falhas: formação de dímeros de
primers, auto-complementariedade (vide figura 3.1), temperatura de fusão muito baixa, e/ou
estabilidade interna incorreta.
Primers devem ser construídos com mecanismos de detecção de conteúdo de G+C baixo.
Estes critérios podem ser tratados por ferramentas de Bioinformática na escolha de primers sem
problemas da fonte de DNA e pode ser produzido pela escolha de qualquer primer para PCR. A

13
escolha de primers específicos para a identificação de vírus de plantas para serem usados no
diagnóstico de viroses deveria considerar, tendo em vista os critérios gerais acima, a
complexidade de famílias de vírus de plantas e diferentes espécies. Portanto, a construção do
primer é o principal fator a ser considerado e todos os outros parâmetros devem ser considerados
na sua construção. Por isso, esta abordagem computacional é eficiente para a identificação de
vírus de plantas.
� � � � � � � � � � � � � � � � � � � �
A escolha de primers pelo processo tradicional [07] é mais propícia a erros, pois, neste
processo, o pesquisador utiliza várias ferramentas diferentes para a escolha do par de primers,
conseqüentemente sofre a demora do processo por depender da Internet, e nem sempre os
primers escolhidos garantem a eficiência ou especificidade do diagnóstico. Isso acarreta prejuízo
com o gasto de primers ineficientes e demora no diagnóstico.
O processo tradicional de escolha de primers tem os seguintes passos:
1. O pesquisador deve procurar as seqüências do genoma do(s) vírus desejados em um banco de
dados. O banco mundial de dados genômicos é o Genbank [42]. O processo de copiar a
seqüência genômica de interesse do Genebank é demorado por ser bastante requisitado via
Internet.
2. Em seguida deve-se fazer um alinhamento múltiplo com as várias seqüências genômicas
suspeitas usando algum programa disponível na Internet. Isso é necessário para descobrir se
existe uma ou mais regiões espécie-específicas.
3. Tal região deve ter um tamanho de no mínimo 150 bases, pois será amplificada na técnica de
PCR pela escolha de dois primers. A visualização pela eletroforese torna-se mais eficiente
quanto maior for esta região (vide Figura 3.2). Além do tamanho da região a ser amplificada,
o pesquisador deve também se preocupar em encontrar de 14 a 20 bases iniciadoras nas
extremidades desta região, complementares a primers que tenham características similares.
4. Estas características similares devem ser calculadas cuidadosamente seguindo alguns
parâmetros. No entanto, os parâmetros usados para que os primers sejam específicos para
uma determinada espécie de vírus de planta não são controlados de modo eficiente pelos
programas de construção de primers também disponíveis na Internet.
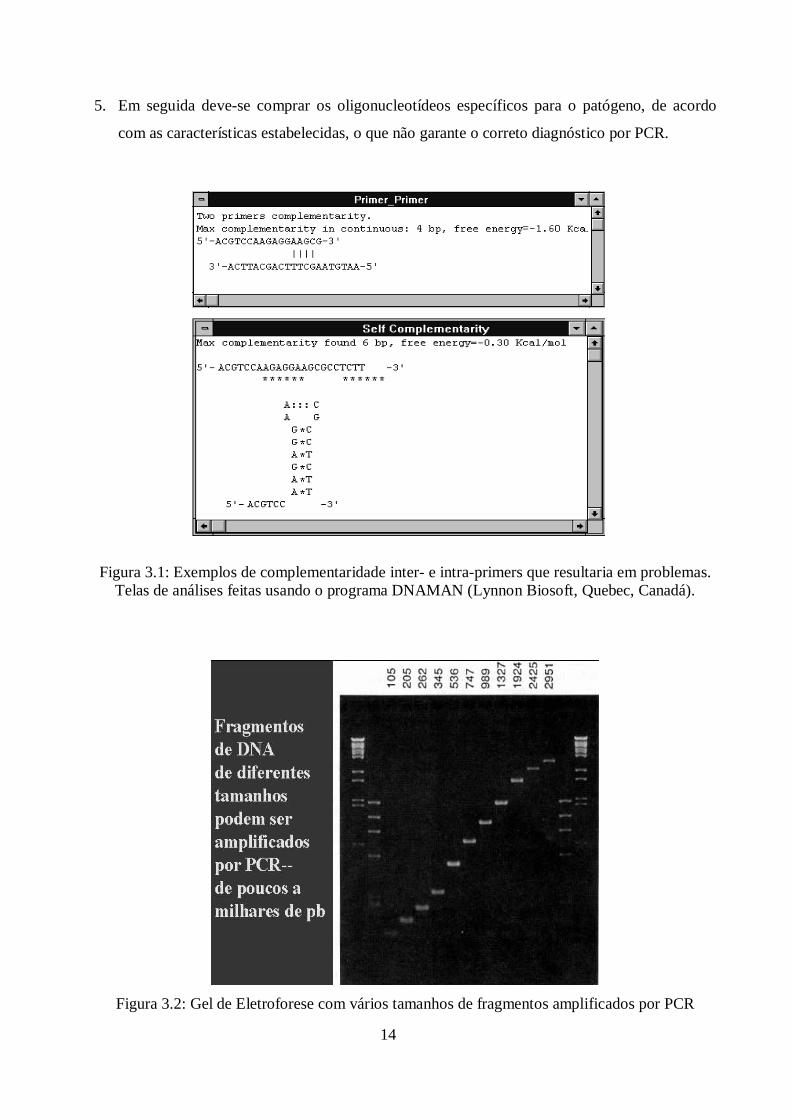
14
5. Em seguida deve-se comprar os oligonucleotídeos específicos para o patógeno, de acordo
com as características estabelecidas, o que não garante o correto diagnóstico por PCR.
Figura 3.1: Exemplos de complementaridade inter- e intra-primers que resultaria em problemas.Telas de análises feitas usando o programa DNAMAN (Lynnon Biosoft, Quebec, Canadá).
Figura 3.2: Gel de Eletroforese com vários tamanhos de fragmentos amplificados por PCR

15
� � � � � � � � � � � � � � � � � � � � � � #
Mesmo usando o Algoritmo da Força Bruta [12], considerado um método simples, uma
intensa modificação realizada foi no sentido de evitar comparações desnecessárias e melhorou
em 75% a performance de tempo do programa.
O exemplo canônico do algoritmo de Força Bruta é associado com o problema do caixeiro
viajante (TSP), um problema clássico NP-completo. Descreve um estilo de programação no qual
o programador confia no poder de processamento do computador em vez de usar sua própria
inteligência para simplificar o problema.
Para que a programação do algoritmo de Força Bruta seja considerada boa, dependeria do
contexto: se o problema não for terrivelmente grande, o tempo extra do processador gasto em
uma solução pelo Algoritmo de Força Bruta pode custar menos do que o tempo que o
programador faria para desenvolver um algoritmo mais inteligente. Adicionalmente, um
algoritmo mais inteligente pode implicar uma complexidade de longo prazo e custo de
manutenção que se justificaria pela melhoria da velocidade.
Convém ressaltar que foram feitas algumas modificações no algoritmo de Força Bruta. A
versão final atende perfeitamente a finalidade do programa, incluindo a especificação do
problema e o tempo da solução.
A interface executa um cruzamento de dados para obter a subseqüência, encontrando uma a
ser comparada com a seqüência do vírus. A lista de informações seguintes tem que ser
armazenada baseando-se em ocorrências de similaridade relativa: a identidade de seqüência
comparada, posição inicial e tamanho da subseqüência, e a posição da ocorrência. Veja
Diagrama 4.2 para mais detalhes sobre o sistema.
� � � � ' � � * � � # � � � 0 � 2 � 5 � 8 # �
O algoritmo de Forca Bruta é o algoritmo mais simples dentre os algoritmos de alinhamento.
Sua simplicidade resulta na sua baixa performance em velocidade, devido ao grande número de
comparações, às vezes sem necessidade.
O algoritmo funciona através de indexações na subcadeia B [26] (de tamanho m, indexada por
Índice B) e na cadeia-alvo A (de tamanho n, indexada por Índice A), efetuando sucessivas
comparações nos elementos de ambas. A partir do primeiro caracter da cadeia-alvo, compara-se

16
seqüencialmente cada caracter da cadeia-alvo com o caracter igualmente indexado da subcadeia,
enquanto forem iguais, ou até que se chegue ao fim da subcadeia (nesse caso, o casamento de
cadeias ocorreu na posição Índice A, e teve início na posição Índice A - m). Caso o fim da
subcadeia não tenha sido alcançado, significa que foi encontrado algum caracter diferente na
cadeia principal, e então deve se reiniciar todo o processo de comparação, posicionando Índice B
no início da subcadeia, e incrementando Índice A de uma posição, como segue no algoritmo
apresentado abaixo:
� � � � � � � � � � � � � � �
� � � � ! # $ & � ) � ! , . 0 2 ! 4 $ . 6 � 8 : 8 2 8 & = >? : $ ! . B . D � 6 G $ ! # : � B J $ . & . : M � : = J 2 6 Q $ ! # : � B J $ . & . : M � & =
Q . V B . D Q $ . ! $ 6 Y ! # & J # ! . � [ � ! ! ] : [ # . B J 2 J & � =a : # [ # �
# D d f >Q $ . ! $ D d i >J : j 4 . : $ � 6 # l d : = J 6 Q $ . ! $ d i = o . , .
p D d # >q D d f >
J : j 4 . : $ � 6 2 s q t d � s p t = J 6 q l d & = o . , .q D d q z f >
p D d p z f >) # & ~ J : j 4 . : $ � >
Q J q d & z f J : $ � �Q $ . ! $ D d p ~ & >
a D d # z f >) # & ~ J : j 4 . : $ � >
) # &
Um esquema do funcionamento do algoritmo de força bruta é apresentado na Figura 3.3,
assumindo que o texto é igual a “ALGORITMOS FAVORITOS” e o padrão P é igual a “RITO”.
Na tabela da página seguinte, a primeira coluna apresenta os valores do teste de mesa para o

17
algoritmo e a segunda traz o número de comparações efetuadas. Portanto, após 22 comparações,
o padrão P foi encontrado no texto T, iniciando-se no índice i = 15.
Essas duas comparações seqüenciais e alinhadas são responsáveis pela complexidade
quadrática O(m x n) para o pior caso (não encontrar B em A), já que o primeiro laço varre toda a
cadeia A (n interações para o pior caso), e para cada caracter de A, é feita uma varredura em B
(m interações para o pior caso).
Esse algoritmo pode ser facilmente mapeado para um algoritmo de busca numa matriz, onde
as colunas são os caracteres de A, e as linhas os caracteres de B, e a simples necessidade de
varrer toda a estrutura matricial para o pior caso dá-se a complexidade quadrática. Para o melhor
caso, a complexidade é O(1), e no caso médio O(n).
A garantia de que o algoritmo funciona está no fato de que todas as subcadeias possíveis na
cadeia-alvo são comparadas até que se encontrem caracteres diferentes nas mesmas.
Por outro lado, o ponto fraco do algoritmo reside nas indexações seqüenciais, e no excesso de
comparações desnecessárias realizadas no percurso ao longo da cadeia-alvo. Considerando-se
que essa cadeia alvo é um texto de grande porte, como uma revista, ou mesmo um livro, não se
poderia dar ao luxo de realizar tamanho número de operações desnecessárias. As modificações
do Algoritmo de Força Bruta foram ao sentido de evitar essas operações desnecessárias, e a
performance alcançada é satisfatória para o alinhamento de seqüências nucleotídicas de até
40000 bases de tamanho.
Figura 3.3: Funcionamento do Algoritmo de Força Bruta
Na figura 3.4, pode-se observar que o tempo de execução do algoritmo cresce
proporcionalmente ao tamanho do texto de entrada, tendo-se assim uma complexidade de O(n)

18
no caso médio. Ressaltamos que o algoritmo usando Força Bruta pode ser empregado nesta
ferramenta, pois dificilmente ela cairia no pior caso que é de O(nm).
Gráfico 3.1: Gráfico de Tempo de Execução de Força Bruta
� � � � � � � � � � � � � � � � � ! # % � � � � ) � � � �
Existem vários fatores que podem afetar a técnica PCR como: temperatura de desnaturação,
número de ciclos, tempo de extensão, uso de primers degenerados, comprimento do primer,
temperatura de anelamento. Dependendo destes fatores a PCR pode reagir melhor. Portanto, a
construção do primer é o principal fator a ser considerado e todos os outros parâmetros deveriam
ser considerados na sua construção.
Para resolver o problema vários fatores que podem afetar a técnica PCR foram tratados,
como: temperatura de desnaturação, número de ciclos, tempo de extensão, uso de primers
degenerados, comprimento do primer, temperatura de anelamento.
� � � � / 0 � � 3 � � � � ! � � � � � � � � � � ! � � # % > 0 � A � � � � 3
A associação da complementaridade especifica das bases devido às pontes de hidrogênio da
fita simples dos ácidos nucléicos é referida como “anelamento”: duas seqüências
complementares formarão pontes de hidrogênio entre suas bases complementares: (G) guanina
para (C) citosina, e (A) adenina para (T) timina ou (U) uracila e formam uma dupla fita estável,
molécula híbrida antiparalela. Uma pode fazer a fita simples de acido nucléico pela tendência de
anelamento, se ainda não é fita única, como a maioria dos vírus de RNA, por aquecê-lo a um
ponto acima da temperatura de fusão da forma dupla fita, e então bruscamente resfriá-lo: isto

19
assegura que as fitas separadas ou desnaturadas não reanelarão ou juntar-se-ão. Adicionalmente,
se o ácido nucléico é aquecido num tampão de força iônica menor do que 150mM de cloreto de
sódio, a temperatura de fusão é geralmente menor do que 100ºC – isso é o porquê que na PCR se
trabalha com temperaturas de desnaturação (Tm) entre 91 à 97ºC.
Sabe-se que a enzima Taq polimerase tem uma meia vida de 30 minutos à 95ºC, por isso, uma
enzima não deve fazer mais do que 30 ciclos de amplificação. Entretanto, é possível reduzir a
temperatura de desnaturação depois de aproximadamente 10 ciclos de amplificação. Como o
principal comprimento do DNA alvo é diminuído, por cópias de 300 pares de bases ou menos, a
temperatura de desnaturação pode ser reduzida a 88ºC por cópias com 50% (G+C) [10], ou seja,
uma pode desempenhar sua função um pouco mais de 40 ciclos sem muita diminuição da
eficiência da enzima.
O tempo em uma determinada temperatura é a razão principal para a desnaturação ou perda da
atividade da Taq polimerase. Desta forma, se uma reduz isto, o número de ciclos possíveis pode
ser aumentado se a temperatura é reduzida ou não. Normalmente o tempo de desnaturação é 1
minuto à 94ºC. Então é possível, para seqüências de copias curtas, reduzir isto para 30 segundos
ou menos. Aumentando a temperatura de desnaturação e diminuindo o tempo pode também
funcionar. Innis e Gelfand [5] recomendou 96ºC a 15 segundos.
� � � � � � � � � � � � � � � � � � � � � $ & � � * - $ � . � � � 2 3 $ � � 8 � �
O comprimento do primer e a seqüência são de importância crítica na configuração dos
parâmetros de uma amplificação bem sucedida: a temperatura de fusão ou temperatura de
anelamento de uma dupla hélice de DNA aumenta com o seu comprimento, e com o aumento do
conteúdo (G+C). Uma fórmula simples para calcular o Tm é dada por [9]:
Tm = 4(G + C) + 2(A + T)ºC.
Dessa forma, a temperatura de anelamento escolhida para a PCR depende diretamente do
comprimento e composição dos primers. Um dos métodos visa o uso de uma temperatura de
anelamento (Ta) de aproximadamente 5ºC abaixo do mínimo Tm do par de primers a ser usado
[5]. Um tratamento mais rigoroso de Ta é dado por Rychlik et al. [8]. Eles concluem que se a
Ta for aumentada por 1ºC em cada outro ciclo, a especificidade de amplificação e o rendimento
dos produtos menor do que 1kb de comprimento são aumentados. Uma conseqüência de ter a Ta
demasiado baixa é que um ou ambos os primers anelarão às outras seqüências, exceto o alvo

20
verdadeiro, então as más combinações internas de única-base ou anelamento parcial podem ser
tolerados. Isto é bom para amplificar alvos similares ou relacionados. Entretanto, pode conduzir
à amplificação "não-específica" e à redução conseqüente no rendimento do produto desejado se a
maioria das bases 3' for emparelhada com um alvo.
Uma conseqüência da Ta muito elevada é que uma quantidade muito pequena do produto será
produzida, assim como a probabilidade de um anelamento de primer é reduzida. Uma outra
consideração importante é que um par de primers com temperaturas muito diferentes de
anelamento pode nunca dar rendimentos apreciáveis de um produto único, e pode também
resultar em inadvertida amplificação "assimétrica" de fita simples da fita produto mais
eficientemente anelada ao primer.
O anelamento não demora muito tempo. A maioria de primers anelarão eficientemente em 30
segundo ou menos, a menos que Ta seja muito próxima da Tm, ou a menos que sejam
atipicamente longos.
� � � � � � � � � � � � � � � � � �
O comprimento melhor de um primer depende de seu conteúdo (A+T) e do Tm de seu
parceiro, se um tem o risco de ter problemas tais como descrito nos parâmetros anteriores. Fora
o Tm, uma consideração principal é que os primers devem ser complexos o bastante de modo
que a probabilidade do anelamento com as outras seqüências à exceção do alvo escolhido seja
muito baixa. Por exemplo, há uma possibilidade de ¼ (4-1) de encontrar um A, um G, um C ou
um T em toda a seqüência dada do DNA; há uma possibilidade 1/16 (16-1) de encontrar
qualquer seqüência dinucleotídica (por exemplo: AG); uma possibilidade de 1/256 de encontrar
uma dada seqüência de 4-bases. Assim, uma seqüência de dezesseis bases estará
estatisticamente presente somente uma vez em cada 416 bases (=4.294.967.296, ou 4 bilhões).
Isto é aproximadamente o tamanho do genoma do ser humano ou do milho, e 1000x maior do
que o tamanho do genoma de E. coli. Assim, a associação de um oligonucleotídeo de tamanho
maior do que 17 bases com sua seqüência alvo é um processo extremamente seqüência-
específico, igualmente à especificidade de anticorpos monoclonais em ligar às determinantes
antigênicas específicas. Conseqüentemente, 17 bases ou primers mais longos são usados
rotineiramente para a amplificação do DNA genômico de animais, plantas e microorganismos.
Um primer de comprimento muito longo pode significar que mesmo as altas temperaturas de
anelamento não são bastantes para impedir de se emparelhar em uma mau combinação e priming
não-específico.

21
� � � � � � � � � � � � � � � � � � �
Para a amplificação de seqüências cognatas de organismos diferentes, ou para "PCR
evolucionário", um pode aumentar as possibilidades de obter produto construindo primers
"degenerados". Estes seriam, de fato, um grupo de primers que têm a possibilidade de anelar em
diversas posições na seqüência e amplificar uma variedade de seqüências relacionadas. Para o
exemplo, Compton [4] usa grupos de primers de 14 bases com 4 e 5 degenerâncias tanto primers
no sentido direto e reverso, respectivamente, para a amplificação da glicoproteína B (gB) de
vírus de herpes. A seqüência reversa do primer usada é como se segue:
TCGAATTCNCCYAA
Na seqüência acima, Y = T + C, e N = A + G + C + T. Degenerância reduz obviamente a
especificidade do(s) primer(s), significando que as oportunidades de não combinação são
maiores, e o ruído de fundo aumenta. Também, degenerância aumentada significa que a
concentração de primers individuais diminui. Assim degenerância maior do que 512-fold deve
ser evitada. Entretanto, primers com amplitude de degenerância de 256- e 1024-fold para a
amplificação bem sucedida e o seqüenciamento direto subseqüente de uma escala larga de
Mastreviruses de encontro a um fundo de DNA genômico de milho [6].

22
Figura 3.4: Seqüências de primers foram derivadas dos alinhamentos múltiplos de seqüência
A Figura 3.5 mostra as posições de má-combinação que foram usadas como degenerâncias de
4-bases para os primers (mostrados como asteriscos; 5 em F e 4 em R).
Apesar da degenerância, os primers podiam ser usados para amplificar uma seqüência de 250
bp de vírus, diferindo na seqüência tanto quanto acima de 50% for a seqüência alvo, e 60% total.
Poderiam também ser usados para detectar muito sensivelmente a presença do DNA do vírus da
estria do milho contra o DNA genômico do milho, nas diluições abaixo de 1/109 da seiva
infectada.
� � � � � � � � � � � � � � � � � � " $
Esta é normalmente 70 - 72ºC por 0,5 - 3 minutos. A enzima Taq polimerase tem
realmente uma atividade específica em 37ºC que é muito perto daquela do fragmento de Klenow
da polimerase I do DNA de E coli, que explica o aparente paradoxo que resulta quando um tenta
compreender como os primers que anelam em uma temperatura ótima podem então ser
alongados em uma temperatura consideravelmente mais alta. A resposta é que a elongação
ocorre do momento do anelamento, mesmo se este é passageiro, que resulta em uma estabilidade
consideravelmente maior. Em torno de 70ºC a atividade é ótima, e uma extensão do primer
ocorre em até 100 bases por segundo. Aproximadamente 1 minuto é suficiente para a
amplificação fidedigna das seqüências de 2 kb [5]. Produtos mais longos requerem tempos mais
longos: 3 minutos é uma boa medida para 3kb e produtos mais longos. Tempos mais longos
podem também ser úteis em ciclos mais atrasados quando a concentração do produto excede a
concentração da enzima (> 1nM), e quando dNTP e/ou esgotamento do primer pode se tornar
limitante.
Concentrações maiores do que 50mM de cloreto de potássio ou cloreto de sódio inibe a Taq,
mas algum é necessário para facilitar o anelamento do primer. A concentração de Mg2+ afeta o
anelamento do primer; Tm da fita molde, produto e associações da fita molde com o primer;
especificidade do produto; atividade e fidelidade da enzima. Taq requer Mg2+ livre, assim que

23
as atenuações forem feitas pelos dNTPs, primers e fita molde, todos os quais são quelados e
seqüestrados o cátion; destes, os dNTPs são os mais concentrado, assim que [ Mg2+ ] estivesse
entre 0,5 - 2.5mM maior do que a concentração de dNTP. Uma titulação deve ser executada
com variação da concentração de Mg2+ com todas as combinações novas da fita-molde-primer,
como estas podem diferir marcantemente em suas exigências, ainda sob as mesmas condições
das concentrações e ciclagem de tempos/temperaturas.
Algumas enzimas não necessitam adicionar proteína, outras são dependentes dela. Algumas
enzimas trabalham marcantemente melhor na presença de detergente, provavelmente porque
evita a tendência natural da enzima se agregar.
Concentrações de primer não deveriam ir acima de 1uM a menos que houvesse um grau
elevado de degenerância; 0.2uM é suficiente para primers homólogos. A concentração do
nucleotídeo não necessita estar acima de 50uM cada. Entretanto, os produtos longos podem
requerer mais.
� � � � � � � � � � � � � � � � �
O número dos ciclos de amplificação necessários para produzir uma banda visível no gel de
eletroforese depende em grande parte da concentração inicial do DNA alvo: Innis e Gelfand [5]
recomendam de 40 - 45 ciclos para amplificar 50 moléculas alvo, e de 25 - 30 para amplificar as
moléculas 3x105 à mesma concentração. Esta desproporcionalidade é devido a um efeito platô
bem conhecido, que é a atenuação na taxa exponencial da acumulação do produto em estágios
tardios de uma PCR, quando o produto alcança 0,3 - 1,0 nM (nanomolar). Isto pode ser causado
pela degradação dos reagentes (dNTPs, enzima); esgotamento de reagente (primers, dNTPs -
formam um problema com os produtos curtos, e tardios para produtos longos); inibição do
produto final (formação do pirofosfato); competição de reagentes por produtos não-específicos;
competição por ligação de primer pelo reanelamento do produto concentrado (de 10nM) [5]. O
produto desejado não sendo feito em 30 ciclos deve-se fazer exame de uma amostra pequena (1
microlitro) da mistura amplificada e reamplificá-la de 20 a 30 vezes em uma mistura de reação
nova, melhor do que estender o funcionamento para mais ciclos. Em alguns casos onde a
concentração da fita molde está limitando, isto pode dar um produto bom onde a extensão do
ciclo a 40 vezes não funciona.

24

25
Capítulo 4
Uma Solução Computacional
Pelo exposto nos Capítulos anteriores, o projeto de primers é fundamentalmente importante
em métodos de detecção baseados em PCR. Os critérios gerais para primers são muito simples.
Contudo, é difícil escolher primers bons para uma dada seqüência do molde. Não são somente
os cálculos. O mecanismo de alinhamento para escolher a região espécie-específica também é
muito sofisticado. Portanto, a ajuda computacional na construção de primer é uma tarefa crítica
na Bioinformática.
Há diversos serviços na Internet ou software autônomo fornecidos ao público para construção
de primers, tal como PRIDE (35), PRIMER MASTER (36), PRIMO (37), PrimeArray (38),
Primer3 (34), Prime (39) e Web Primer (http://genome-www2.stanford.edu/cgi-bin/SGD/web-
primer). Os usuários podem definir os parâmetros listados no menu destas ferramentas e então
obter diversos pares de primers para a seqüência do molde alvo. Entretanto, a maioria deles
somente analisa uma única seqüência de pesquisa. A proposta deste trabalho é definir, por
alinhamento de várias seqüências nucleotídicas, uma região ideal para construção de primers.
Assim, considerando as limitações descritas no item 3.3 desta dissertação, um grupo de regras
pode ser derivado para garantir a especificidade do diagnóstico:
1) Os primers devem ser 18 a 25 bases de comprimento;
2) A composição de base deve ser 50 a 60% (G+C);
3) Temperaturas de desnaturação entre 55 à 80ºC são preferidas;
4) Extremidades 3' de primers não deve ser complementar com outro primer, caso contrário
dímeros de primers serão formados preferencialmente a qualquer outro produto. A auto-
complementaridade de primer (habilidade de formar estruturas secundárias tais como
frisos) deve ser evitadas (veja Figura 3.1);
5) O produto amplificado por PCR indicado pelo par de primers não deve exceder 1200
pares de bases de comprimento;
6) Os parâmetros para o par de primers devem ser similares.

26
Quanto maior for o primer, maior será sua especificidade. Apesar disso, há problemas na PCR
com a construção de primers muito extensos. As variações de comprimento de primer de 18 a 25
nucleotídeos são aconselhadas e a configuração padrão do programa é 19. Em primers de
tamanho acima de 25 bases há maior probabilidade de formar estruturas secundárias (frisos pela
auto-complementaridade) ou dímeros entre o par de primers. Portanto, para diagnóstico,
experimentos apontam que os primers não devem exceder de 25 bases.
A composição de bases é importante devido às pontes de hidrogênio que conferem maior
estabilidade no anelamento entre citosina e guanina do que em adenina e timina. Por isso um
conteúdo de CG estável deve ser a partir de 50% e não pode exceder de 60% devido a elevada
reatividade destas bases nitrogenadas em parearem-se formando dímeros entre primers. No
programa proposto a configuração padrão do conteúdo de GC do par de primers varia de 50% a
60%. Primers com Tm menor do que 50ºC são excluídos.
A diferença aceitável de valores de Tm em um par de primers é de 5ºC. Critérios básicos para
a seleção de Tm de primer. A temperatura de anelamento (Ta) é determinada pela temperatura de
desnaturação (Tm). A temperatura de desnaturação é o parâmetro mais importante porque o
termociclador ao elevar e diminuir a temperatura muito rapidamente afeta também os primers
que devem ser capazes de anelarem-se a partir de 54 ºC e não deve passar de 80ºC para que
possibilite a desnaturação necessária na PCR.
Deve-se ter cautela com a distribuição de nucleotídeos formar um anagrama de bases bem
variado, principalmente nas extremidades. Ao usar um par de primers para diagnóstico deve-se
preocupar-se se ocorre inter-complementaridade entre os dois primers. Esse cuidado também é
nas extremidades porque em um primer também se pode formar estruturas secundárias como um
friso ou um círculo devido a complementaridade das extremidades.
O par de primers são os limites da região que vai ser amplificada ou copiada várias vezes. É
necessário estabelecer um tamanho mínimo de 100 bases, pois a partir deste tamanho o
fragmento pode ser melhor visualizado em um gel de eletroforese. O tamanho máximo deve ser
de 1200 bases porque a partir deste tamanho a polimerase terá diminuição de performance. Em
um minuto a polimerase é capaz de encadear até 2000 bases.
Há uma preocupação do par de primers possuírem parâmetros como Tm e conteúdo GC
similares, devido à variação de temperatura que são submetidos.
O software desenvolvido para diagnóstico de viroses em plantas, ajuda em selecionar primers
para uma região específica e bem conhecida do genoma viral para ser amplificada na reação da
PCR.

27
O sistema computacional na sua primeira versão consiste de dois módulos: um módulo para
alinhamento das seqüências nucleotídicas e separação das regiões polimórficas, outro módulo
para construção de primers usando os melhores parâmetros de primers eficientes no diagnóstico
por padrão ou configurados pelo pesquisador.
A Tabela 4.1 pode resumir os tratamentos dados aos problemas solucionados neste trabalho.
PROBLEMAS ATUAIS SOLUÇÕES DO SISTEMA
Prejuizo no agronegócio do RN e Brasil;Análise prévia do diagnóstico por PCR emelhor tratamento;
Busca de genomas em banco de dados(demorado);
Busca mais rápida, devido banco de dadoslocalmente instalado;
Alinhamento múltiplo (demorado, falho);Alinhamento mais rápido do que na Internet,correto;
Escolher região a ser amplificada (manual);Escolha automática rápida detalhada(parâmetros);
Parâmetros controlados de modo ineficiente; Controle mais eficiente para diagnóstico;
Problema performance de tempo do algoritmo.Modificações evitando comparaçõesdesnecessárias.
Tabela 4.1: Soluções do Sistema Dadas aos Diversos Problemas Abordados
� � � � � � � � � �
O objetivo do sistema é automatizar o processo tradicional de escolhas de primers espécie-
específicos. O sistema tem dois módulos: um módulo para alinhamento de seqüências e outro
para projetar ou construir primers. Este sistema deve ser modelado orientado a objeto usando
UML [48]. No processo de desenvolvimento do software orientado a objetos é necessário ter
uma visão mais ampla do que a oferecida pelo modelo estático do sistema, pois este não reflete a
mudança dos objetos e seu comportamento através do tempo [47].
Os bancos de dados de arquivos simples são o tipo de banco de dados que os não-especialistas
entendem com mais facilidade. Um banco de dados de arquivos simples não é realmente um
banco de dados, é simplesmente uma coleção ordenada de arquivos semelhantes, geralmente em
conformidade com um formato padrão de conteúdo [46]. Os bancos de dados de arquivos
simples organizam os dados de forma sensata e podem ser pesquisados devido a indexação, e,
portanto, não são essencialmente ruins. Entretanto quando eles ficam cada vez maiores, como é
o caso dos bancos de dados biológicos em geral, faz-se necessário um sistema de gerenciamento
de banco de dados, como Postgree, ou SQL Server, ou Oracle.

28
A proposta inicial do sistema atendeu as expectativas com um índice unidimensional em um
banco de dados de arquivos simples indexado. A coleção de arquivos texto tem o formato Fasta
(vide figura 4.1). Muitos usuários de dados de seqüências biológicas armazenam e acessam as
seqüências localmente, utilizando o Sequence Retrieval System (SRS), um sistema de indexação
de arquivos simples projetado para utilização com dados biológicos.
Figura 4.1: Formato Fasta
O sistema não tem controle de acesso por senha ou preocupações maiores de segurança por
não estar acessível pela Internet, e sim localmente. No entanto, faz-se necessário criar um
sistema de segurança do banco de dados para desenvolvimento, manutenção, e prevenção de
problemas futuros. O sistema sempre estará em fase contínua de desenvolvimento e outros
processos podem ser implementados. O processo de assegurar-se de que a informação seja
alcançada somente em maneiras autorizadas é chamado controle de acesso. O controle de acesso
é um de diversos objetivos possíveis da segurança dentro de um sistema computacional; ilustra-
se no Esquema 4.1, que mostra a informação do banco de dados sendo transferida para e do
programa do usuário de acordo com um grupo de regras de acesso. O controle de fluxo da
informação visa prevenir o escape da segurança da informação. E todo o sistema desenvolvido
deverá possuir documentação.

29
Analisando o sistema tradicional de escolha de primers espécie específicos constatamos os
seguintes problemas:
• A busca em banco de dados públicos, o alinhamento de genomas e a construção de
primers são demorados por depender do tráfego via Internet.
• Utiliza-se vários programas diferentes para obter os primers espécie-específicos.
• Cada etapa do processo implica em copiar e colar resultados manualmente, isso pode
gerar algum erro.
• A visualização dos dados do processo é menos compreensível, visto que tem interfaces
diferentes sendo utilizadas.
• O alinhamento múltiplo usado em um programa público na Internet demora e pode dar
dados imprecisos.
O sistema proposto resolve todos esses problemas do método tradicional:
• O processo de construção de primers espécie-específicos é automatizado portanto não
é necessário utilizar vários programas, e o tempo de resposta em comparação ao
método tradicional é mais rápido.
• Utiliza-se apenas a interface de um programa para obter os resultados.
• Não é necessário copiar e colar manualmente os dados. O fluxo de dados é dirigido
para obter os resultados.
• O processo completo para obter os primers espécie-específicos é melhor
compreendido, por ter uma única interface, e por ser uma interface compreensível
facilitando a visualização dos dados sendo processados ou transformados. E, pode-se
voltar etapas do processo para entender o processamento.
• O alinhamento múltiplo de seqüências até 40000 bases de comprimento tem no pior
caso um tempo de resposta inferior a três minutos.
A arquitetura do sistema é mostrada no diagrama 4.2. A entrada do programa são arquivos
texto no formato Fasta e a saída são primers com os parâmetros otimizados por padrão ou
estabelecidos pelo usuário no sentido 3’-5’ou 5’-3’. Os arquivos texto estão armazenados
localmente na máquina no formato Fasta, onde se pode obter os genomas de várias espécies de
vírus de plantas que estão disponíveis nos bancos de dados públicos, ou que foram seqüenciados
no laboratório do usuário. O usuário escolhe as espécies suspeitas que deseja estabelecer uma
comparação e submete ao alinhamento múltiplo global. Em seguida o programa pode mostrar as
regiões comuns (regiões universais) e incomuns (regiões espécie-específicas) entre os genomas.

30
As regiões espécie-específicas são processadas em um grupo de regras ou parâmetros otimizados
para desenhar primers. No entanto, o usuário tem a liberdade de decidir o tamanho do fragmento
que quer amplificar, o tamanho do primers, a média do conteúdo de C+G, e a temperatura de
desnaturação antes do processamento das regiões espécie-específicas para desenhar os primers.
Nos resultados finais ou saída do programa, pode-se escolher em uma lista de primers fornecida
pelo programa, um par de primers limítrofes de parâmetros similares.
Diagrama 4.1: Acessibilidade do Banco de Dados
Diagrama 4.2: Arquitetura do Sistema
Processando…
Arquivos de vírus de planta
Seqüência de virus query
Detalhando…
Arquivo .txt Formato Fasta
BANCODE
DADOS
Infraestrutura de Bioinformática
GERENCIAMENTO DA INFORMAÇÃO
ENTRADA:Seqüência query
Cálculos do pipeline
SAÍDA:Lista de Primers de vírus / seqüência.
Ex.: 3’5’| Tm |C+G| 5’3’ |AGTC|54 ºC |52%|TCAG
Ambiente de mineração de dadosRegião espécie-específica da seqüência
query 3’5’ Ex.: (XXAGTCXX…)
Dados de vírus de planta
VírusPlanta
IrParaSeqüênciaVírus
�USUÁRIOBANCO
DEDADOS
Controle de Acesso
PROGRAMA
SERVIDOR

31
� � � � � � � � � � � � � � � � � � � � �
Inicialmente, desenvolveu-se um módulo de alinhamento (vide Diagrama 4.3), que aceita
como entrada arquivo texto com seqüências nucleotídicas em formato Fasta de vários
comprimentos e fornece como saída: domínios com similaridades e domínios espécie-
específicos. Um domínio espécie-específico é uma seqüência do genoma viral que está presente
somente em uma das espécies como resultado de uma comparação entre genomas.
O alinhamento é feito usando o algoritmo de Força Bruta modificado, usando uma string ou
tamanho de palavra configurado pelo usuário. A primeira seqüência escolhida no banco de
dados é a seqüência query, ou seja, é a seqüência que será fragmentada em várias strings e
comparada com as seqüências subseqüentes de modo global. O resultado do alinhamento será
mostrado na seqüência query. Depois, pode-se fazer um detalhamento do resultado indicando as
regiões universais e as regiões espécie-específicas.
Diagrama 4.3: Diagrama do Módulo de Alinhamento
� � � � � � � � � � � � � � � � � � � � � # � � � � �O módulo subseqüente “constrói” os primers usando por padrão parâmetros otimizados ou
parâmetros especificados pelo usuário (vide Diagrama 4.4). Uma lista de primers candidatos é

32
exibida com seus respectivos parâmetros para ajudar o usuário a minimizar primers ineficientes,
e estabelecer o tamanho do fragmento de DNA que deve ser amplificado na reação de PCR da
Biologia Molecular. Os parâmetros otimizados já foram listados no conjunto de regras derivados
para garantir a especificidade do diagnóstico no início deste capítulo.
A idéia da aplicação é usar regiões espécie-específicas de genomas virais para construir
primers de alta especificidade para diagnosticar vírus de plantas usando PCR. Primers devem
estar prontos para anelarem-se ao ácido nucléico alvo em uma localização prevista e ser
estendida pela enzima Taq Polimerase.
O resultado é uma lista de primers com seus respectivos parâmetros. Os parâmetros
mostrados são: tamanho, porcentagem do conteúdo de C+G, temperatura de desnaturação, e
primer complementar. O primer complementar é a versão 5’-3’ do primeiro primer listado. No
par de primers deve necessariamente ser um no sentido 3’-5’ e outro no sentido 5’-3’ de primers
diferentes e de distância conhecida.
Diagrama 4.4: Diagrama do Módulo de Construção de Primers

33
� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �
A ferramenta usada no desenvolvimento do sistema foi o Borland Delphi 5.0, para ambiente
Windows. A escolha dessa ferramenta decorreu do fato de a mesma ser visual e orientada a
objetos, bem como possuir boa integração com aplicações de bancos de dados. Ambos os fatos
contribuíram para a diminuição do tempo de desenvolvimento, ao evitar a preocupação com a
API do Windows e o acesso às tabelas Paradox.
A interface é compreensível, pois é intuitivo o modo de utilizar o programa ao observar as
telas. O programa permite que o usuário especifique o tamanho da string de busca ou
subseqüência no alinhamento entre seqüências, como mostra o item 1 na tela apresentada na
Figura 4.3. O tamanho de string padrão é 10.
Há na tela inicial do programa o botão de arquivos de seqüências (vide figura 4.3, item 2) para
acionar a tela de busca do arquivo texto do sistema operacional e assim o usuário pode buscar os
arquivos texto com seqüências de vírus em formato Fasta (vide figura 4.1). As seqüências são
mostradas no item 7 da figura 4.3 e a primeira seqüência é a seqüência query ou seqüência de
busca, onde todas as seqüências subsequentes serão comparadas, e onde os resultados serão
mostrados.
Há também o botão de arquivos de primers (vide figura 4.3, item 3). Este botão ao ser
acionado mostra a tela de busca do arquivo texto do sistema operacional, e o usuário pode
escolher um arquivo texto mostrando primers, cada um já escolhido para um respectivo vírus
(vide figura 4.2). Este arquivo de extensão .txt não tem seqüências no formato Fasta, mas lista
primers em texto padronizado para o programa proposto. E mostra os primers específicos de
vírus no item 6 da figura 4.3.
O botão Processar mostrado na figura 4.3 no item 4 serve para verificar se os primers são
realmente específicos para as seqüências de vírus adicionadas no programa. Ao acionar este
botão uma tela de resultados será exibida mostrando em que ordem de seqüência nucleotídica
viral se encontra o primer e sua respectiva posição de base inicial (vide figura 4.4).
O botão Localizar RE’s da figura 4.3 no item 5 serve para localizar as regiões específicas da
primeira seqüência de genoma de vírus do arquivo texto adicionado (vide figura 4.3, item 6).
Este botão acionará a tela de resultados (vide figura 4.4), que mostra uma região ou as regiões
específicas existentes na primeira seqüência do arquivo de seqüências. Quando não há região
específica a tela mostra todas as bases com a letra X. E quando há regiões específicas mostra na
primeira seqüência nucleotídica viral fragmentos em modo texto representadas pelas letras: G, C,
A e T. Pode-se também visualizar as regiões comuns ou universais entre os vírus escolhidos

34
(vide figura 4.6). Na tela de resultados (Figura 4.5) são mostradas também as configurações dos
primers que serão desenhados. As configurações que podem ser modificadas pelo usuário são:
número mínimo de bases, número máximo de bases, intervalo de temperatura de desnaturação
(Tm) em graus Celcius, e intervalo de porcentagem de conteúdo G+C. Há tambem na leta de
resultados dois botões; um para detalhar as regiões específicas, e outro para detalhar as regiões
universais. Ao ser acionado um dos botões ele mostrará uma outra tela (vide figura 4.6) com os
primers desenhados de acordo com as configurações estabelecidas na tela de resultados.
A tela de primers específicos mostrada na figura 4.7 mostra as regiões específicas detalhadas
com um número em ordem crescente no sentido 5’-3’, a posição da base inicial e a posição da
base final na primeira seqüência viral, e o comprimento da região espécie-específica em número
de bases. Ao selecionar um tamanho de região específica, mostrará nesta mesma tela os
respectivos primers que podem ser desenhados nesta região de acordo com as configurações
estabelecidas. Cada primer de uma região espécie-específica é listado com sua respectiva:
seqüência de bases representadas em letras, temperatura de desnaturação em graus Celcius (Tm),
porcentagem do conteúdo G+C, tamanho em número de bases, posição na região espécie
específica, e o primer complementar. Ao selecionar a seqüência de bases do primer nesta seção,
será mostrada na seção Seqüência, as bases pintadas na cor verde e toda a região especie
específica pintada na cor vermelha. O usuário pode copiar a seqüência verde. Para saber o
tamanho de fragmento de DNA que será copiado várias vezes na técnica e PCR para diagnosticar
a espécie de virus, é preciso saber o resultado da subtração da posição inicial do primeiro primer
e a posição final do segundo primer da primeira seqüência viral ou seqüência query.
Figura 4.2: Formato do Arquivo de Primers do Programa

35
Figura 4.3: Tela inicial do programa
Figura 4.4: Tela mostra em que seqüência e posicão da seqüência se encontra o primer

36
Figura 4.5: Tela de resultados que mostra as regiões especificas
Figura 4.6: Tela de resultados que mostra as regiões universais

37
Figura 4.7: Tela mostrando os primers da região específica com seus respectivos parâmetros

38
Capítulo 5
Experimentos e Resultados
No sistema desenvolvido, a adaptação do algoritmo de força bruta prolongada. A busca
executa uma verificação em todas as seqüências em ordem para obter a subseqüência, usando um
tamanho de seqüência de nucleotídeos configurada pelo usuário. Uma lista de subseqüências é
armazenada baseada na ocorrência de similaridade relativa: ID da seqüência comparada, posição
inicial e tamanho das subseqüências. A figura 4.1 ilustra o sistema final projetado com base em
modificações propostas. Este sistema também pode ser usado para análise de polimorfismo.
Realizou-se testes com o Algoritmo de Força Bruta modificado para algumas seqüências de
vírus selecionados e os resultados mostram que os primers não são ambíguos para cada
seqüência. O tempo gasto para casar uma seqüência única de comprimento 40.000 bases (pior
caso de teste) com outra seqüência de 1000 bases, usando uma string de busca de tamanho 10,
foi de 2 minutos e 43 segundos, usando uma máquina com processador AMD Athlon 2,4 GHz,
256 MB de memória RAM. Executaram-se vários experimentos com o algoritmo, principalmente
testando o tempo de execução com seqüências de tamanhos variados, em relação à versão
original. A figura 5.1 mostra um resultado mais extensivo demonstrando a performance do
algoritmo. No último caso, o banco de dados de seqüências cresceu, denotando dados mais
precisos.
Foi realizado um experimento para algumas seqüências de vírus de planta selecionadas em
banco de dados público e os resultados mostram que os primers também não são ambíguos para
cada seqüência. Na versão atual, executou-se uma comparação algorítmica do tempo com
tamanhos variáveis da seqüência. O gráfico na figura 5.1 mostra que o programa proposto no
pior caso (40.000 pares de bases de comprimento) tem uma performance tempo aproximado de 3
minutos.

39
Performance de Tempo do Programa
720
14037
293
163157
1201059474604330140
300
600
900
1200
0 4 8 12 16 20 24 28 32 36 40
Número de bases da seqüência nucleotídica em Kb (1000 bases)
Tempo(segundos)
Algoritmo de Força Bruta Algoritmo de Força Bruta Modificado
Gráfico 5.1: Gráfico de performance da última versão do algoritmo de Força Bruta
� � � � � � � � � � � �
Dois cenários de experimentos com viroses de planta foram executados para verificar se o
programa implementado atende ao objetivo proposto. A situação é descobrir uma virose de
planta de um determinado cultivar, através da técnica de PCR, usando os primers projetados pelo
programa proposto, sem ter conhecimento dos sintomas da planta doente. A limitação é que as
opções de primers para diagnóstico é de acordo com os genomas disponíveis nos bancos de
dados de genomas de vírus de planta. Tanto na Bioinformática quanto na Biologia Molecular, os
experimentos validaram o programa proposto.

40
� � � � � � � � � � � �
A simulação é para diagnosticar viroses de batata (Solanum tuberosum). Os genomas
completos de viroses de Solanum tuberosum disponíveis no banco de dados de domínio público
NCBI são sete, até 16 de setembro de 2004, como mostra na figura 5.3. Em seguida, obteve-se o
formato Fasta de cada genoma de vírus da batata. Todos os sete genomas foram processados pelo
programa proposto com o cuidado de manter as configurações padronizadas para todos. O
alinhamento foi feito com uma string de tamanho 10, e em 25 segundos obteve-se o resultado
usando o processador AMD Athlon 2,4 GHz, 256 MB de memória RAM. O programa usa o
primeiro genoma da lista como alvo para alinhar os subseqüentes.
Para cada genoma, obteve-se um par de primers com características iguais (tamanho, Tm e
conteúdo G+C) configuradas na tabela 5.1. Estas características estão otimizadas segundo Innis
e Gelfand [05]. O tamanho do primer foi de 21 bases por ser mais específico e menos provável
de formar problemas de complementaridade (vide figura 3.1). Quanto maior o primer mais
específico ao alvo, porém também se considera que primers muito longos podem formar frisos
com a intra-complementaridade. Tendo em vista isso, as extremidades dos primers foram
escolhidas procurando não formar dímeros entre o par de primers nem frisos intra-primer. A
temperatura de desnaturação (Tm em ºC) escolhida foi 54,36 por ser um valor
experimentalmente eficiente na reação. A porcentagem de guanina e citosina (% G+C) foi de
52.38% também estável experimentalmente no anelamento da reação de PCR. O tamanho do
fragmento do genoma do vírus amplificado por PCR foi entre 150 à 1200 pares de bases por ser
um tamanho de extensão viável para a enzima polimerase expandir na reação. Este tamanho é
limitado pelo par de primers escolhido.
Tabela 5.1: Primers de espécies de viroses de batata obtida pelo programa proposto

41
Figura 5.1: Tela de resultados do Entrez no NCBI mostrando 7 genomas de vírus da batata.
Os pares de primers específicos obtidos de cada espécie de vírus também foram testados
usando o Blast-n do NCBI e comprova-se que são específicos em nível de espécie, em 16 de
dezembro de 2004. E em todos os primers 5`-3` de viroses de batata, constata-se a especificidade
com o vírus respectivo, porque o Blast-N alinhou no banco de dados com várias seqüências de
genomas parciais e com o genoma completo de cada vírus analisado. Além disso, as outras
seqüências alinhadas que não são de vírus têm um E-value alto, indicando que foram alinhadas
ao acaso (vide figuras 5.4 a 5.10). E-value (Expectation value) é um número, resultado de
cálculos estatísticos, que indica o grau de "validade" de um alinhamento. Quanto menor o E-
value mais significativo é o alinhamento. O BLAST mostra os E-values do alinhamento
colocando-os em ordem decrescente de significância.

42
Figura 5.2: Tela de Resultados do Blast-N para Potato Vírus V

43
Figura 5.3: Tela de Resultados do Blast-N para Cherry rasp leaf virus RNA2

44
� �
Figura 5.4: Tela de Resultados do Blast-N para Cherry rasp leaf virus

45
Figura 5.5: Tela de Resultados do Blast-N para Potato virus Y

46
Figura 5.6: Tela de Resultados do Blast-N para Narcissus mosaic virus

47
Figura 5.7: Tela de Resultados do Blast-N para Potato virus M

48
Figura 5.8: Tela de Resultados do Blast-N para Potato virus A

49
� � � � � � � � � � � �
O Rio Grande do Norte é o maior exportador de melão do Brasil. Neste cenário simula-se um
experimento para diagnosticar viroses do meloeiro (Cucumis melo L). Os genomas completos de
viroses de Cucumis melo L disponíveis no banco de dados de domínio público NCBI são quatro,
até 16 de setembro de 2004, como mostra na figura 5.11. Em seguida, obtive-se o formato Fasta
de cada genoma de vírus do meloeiro. Todos os quatro genomas foram processados pelo
programa proposto com o cuidado de manter as configurações padronizadas para todos. O
alinhamento foi feito com uma string de tamanho 10, e em 13 segundos obteve-se o resultado
usando um processador AMD Athlon 2,4 GHz, 256 MB de memória RAM. O programa usa o
primeiro genoma da lista como alvo para alinhar os subseqüentes.
Para cada genoma obteve-se um par de primers com características iguais (tamanho, Tm e
conteúdo G+C) configuradas na tabela 5.2. Estas características estão otimizadas segundo Innis
e Gelfand [05]. O tamanho do primer foi de 21 bases por ser mais específico e menos provável
de formar problemas de complementaridade (vide figura 3.1). Quanto maior o primer mais
específico ao alvo, porém também se considera que primers muito longos podem formar frisos
com a intra-complementaridade. Tendo em vista isso, as extremidades dos primers foram
escolhidas procurando não formar dímeros entre o par de primers nem friso intra-primer. A
temperatura de desnaturação (Tm em ºC) escolhida foi 54,36 por ser um valor
experimentalmente eficiente na reação. A porcentagem de guanina e citosina (% G+C) foi de
52.38% também estável experimentalmente no anelamento da reação de PCR. O tamanho do
fragmento do genoma do vírus amplificado por PCR foi entre 150 à 1200 pares de bases por ser
um tamanho de extensão viável para a enzima polimerase expandir na reação. Este tamanho é
limitado pelo par de primers escolhido.
Tabela 5.2: Tabela de Primers de Espécies de Vírus do Meloeiro Obtida Pelo Programa Proposto

50
Figura 5.9: Tela de resultados do Entrez no NCBI com 4 genomas de vírus de meloeiro.
Os pares de primers específicos obtidos de cada espécie de vírus também foram testados
usando o Blast-n do NCBI e comprova-se que são específicos em nível de espécie, em 16 de
dezembro de 2004. E em todos os primers 5`-3` de viroses do meloeiro, constata-se a
especificidade com o vírus respectivo, porque o Blast-N alinhou no banco de dados com várias
seqüências de genomas parciais e com o genoma completo de cada vírus analisado. Além disso,
as outras seqüências alinhadas que não são de vírus têm um E-value alto, indicando que foram
alinhadas ao acaso (vide figuras 5.12 a 5.15).

51
Figura 5.10: Tela de Resultados do Blast-N para Cucurbit yellow stunting disorder virus RNA2

52
Figura 5.11: Tela de Resultados do Blast-N para Cucurbit yellow stunting disorder virus RNA1

53
Figura 5.12: Tela de Resultados do Blast-N para Melon chlorotic leaf curl virus

54
Figura 5.13: Tela de Resultados do Blast-N para Melon necrotic spot virus

55
Capítulo 6
Conclusão e Perspectivas
Eu obtive um diferencial em relação aos softwares existentes em domínio público e em
domínio privado. O diferencial do sistema proposto está na estratégia para realizar um
diagnóstico em nível de espécie de modo eficiente. A estratégia é procurar similaridades e não-
similaridades nas regiões do genoma do vírus. Regiões onde ocorrem não-similaridades são
regiões espécie-específicas, ou seja, os primers projetados a partir desta região serão específicos
para apenas uma espécie suspeita. O sistema também possibilita ao usuário configurar a
estringência do alinhamento, bem como os parâmetros de primers ótimos para o diagnóstico.
Estringência é o número de bases da string ou palavra de busca (tamanho da subseqüência). A
priori, o sistema tem uma configuração padrão baseada em medidas otimizadas da literatura
pesquisada.
A estratégia de escolher regiões específicas de uma espécie antes da construção de primers é
eficiente para diagnosticar uma virose suspeita. A resposta avança então a metodologia de
diagnóstico molecular em nível de espécie de vírus e de outros patógenos facilitando no processo
de tratamento. Além de beneficiar o controle de pragas na agricultura de importação e
exportação, pode-se fazer um estudo de polimorfismo entre espécie e estudar a filogenia entre
espécies de microorganismos.
O programa proposto para diagnóstico de vírus de planta pode ser aplicado para outros
patógenos com uma simples adaptação de sua interface e algoritmo. A base principal é o módulo
de alinhamento que pode ser tão simples quanto usar o algoritmo de força bruta. O alinhamento
de seqüências de vírus também pode ser útil para identificar marcadores moleculares. As
modificações implementadas no Algoritmo de Bruta melhoraram consideravelmente sua
performance de tempo. Justifica-se o uso deste algoritmo porque em geral o tamanho de uma
seqüência de um vírus de planta não é maior do que 40.000 bases, e o processamento foi
realizado em 2 minutos e 43 segundos, sendo satisfatório em comparação ao tempo de demora
usando outros programas de alinhamento e construção de primers disponíveis na Internet.

56
Ao longo dos últimos anos, a pesquisa na área de genômica e de bioinformática vem obtendo
crescente importância no país. Projetos bem sucedidos, como os da Xylella fastidiosa, da cana-
de-açúcar, do câncer e outros apenas comprovam essa afirmação. Nesse contexto, a criação de
um programa para identificar viroses em plantas torna-se mais um passo na aquisição de
excelência por pesquisadores brasileiros nessa área.
O estudo desenvolvido é importante também devido à escassez de pesquisas realizadas no
Brasil que proponham técnicas qualitativas de diagnóstico da Biologia Molecular. De um modo
geral, o trabalho alcançou o objetivo proposto, entretanto resta resolver a escolha do par de
primers limitando a seqüência a ser amplificada. Alem disso, pretende-se criar o banco de dados
de genoma de vírus de planta e o banco de primers.
Para versões posteriores do programa sugere-se o uso do Algoritmo de Needleman-Wunsch
[48] para alinhamento global de seqüências nucleotídicas, indicado para alinhamento de
genomas de outros patógenos com genoma superior a 40000 pares de bases [49]. Tendo em vista
que a metodologia adotada pode ser utilizada e adaptada para outros trabalhos com diferentes
patógenos.

57
Referências Bibliográficas
[01] Thomson K. at al., Identification of Zucchini yellow mosaic potyvirus by RT-PCR andanalysis of sequence variability, Journal of Virological Methods, 55, p. 83-96. ed.Addison-Wesley, 1995.
[02] Gitton F. at al., A two-step multiplex RT-PCR method for simultaneous detection of soil-borne wheat mosaic virus and wheat spindle streak mosaic virus from France, PlantPathology, 48, p. 635-641, 1999.
[03] Antoniw, J., A new method for designing PCR primers specific for groups of sequencesand its application to plant viruses, Molecular Biotechnology, 4, 111- 119, 1995.
[04] Compton T., Degenerate primers for DNA amplification. pp. 39-45 in: PCR Protocols (Innis, Gelfand, Sninsky and White, eds.); Academic Press, New York, 1990.
[05] Innis M. A. and Gelfand D. H., Optimization of PCRs. pp. 3-12 in: PCR Protocols(Innis, Gelfand, Sninsky and White, eds.); Academic Press, New York, 1990.
[06] Rybicki E.P. and Hughes F.L., Detection and typing of maize streak virus and otherdistantly related geminiviruses of grasses by polymerase chain reaction amplificationof a conserved viral sequence. Journal of General Virology 71:2519-2526, 1990.
[07] Rybicki E. P., PCR primer design and reaction optimization. Department of Molecularand Cell Biology, University of Cape Town, 2001.
[08] Rychlik W., Spencer W.J. and Rhoads R.E., Optimization of the annealing tempera-ture for DNA amplification in vitro. Nucleic Acids Research 18 (21):6409-6412, 1990.
[09] Thweatt R., Goldstein S. and Reis R.J.S., A universal primer mixture for sequencedetermination at the 3' ends of cDNAs. Analytical Biochemistry 190:314-316, 1990.
[10] Yap E.P.H. and McGee JO'D, Short PCR product yields improved by lower denaturationtemperatures. Nucleic Acids Research 19 (7):1713, 1991.
[11] Exposito at al. PCR Strategy for Identification and Differentiation of Smallpox andOther Orthopoxviruses. Journal of Clinic Microbiology, 95, p. 2069-2076. AmericanSociety for Microbiology, 1995.
[12] Cormen, T. H., Leiserson, C. E., Rivest, R.L., Introduction to Algorithms. MIT Press,1990.
[13] Elmasri, R., Navathe, S. B., Fundamentals of Database Systems. Addison-Wesley. 3rd ed.2000.

58
[14] Hammond, J., Calef, C., Larder, B., Schinazi, R., Mellors, J. W., Mutations in RetroviralGenes Associated with Drug Resistance, 1999.
[15] National Center for Biotechnology Information. URL http://www.ncbi.nlm.nih.gov.Acessado em junho de 2004.
[16] Setubal, J., Meidanis, J., Introduction to Computational Molecular Biology. Boston:PWS, p. 1-103, 1997.
[17] Silberschatz, A., Korth, H. F., Sudarshan, S., Sistema de Banco de Dados. MakronBooks, São Paulo, 3ª ed. 1999.
[18] Stanford HIV RT and Protease Sequence Database. URL http://hivdb.stanford.edu/hiv.Acessado em junho de 2004.
[19] Baxevanis, A. D., Ouellette, B. F. F., Bioinformatics: A Practical Guide to the Analysisof Genes and Proteins. John Wiley & Sons, 1998.
[20] Hillier L. and Green P., OSP: a Computer Program for Choosing PCR and DNASequencing Primers, PCR Methods and Applications, 1, pp. 124-128, 1991.
[21] Lowe T., Sharefkin J., Yang S. Q., and Dieffenbach, A Computer Program for selectionof oligonucleotide primers for polymerase chain reactions, Nuc. Acids Res., 18, pp.1757-1761, 1990.
[22] Lucas K., Busch M., Mossinger S., and Thompson J. A., An Improved MicrocomputerProgram for finding Gene- or Gene Family-Specific Oligonucleotides Suitable asPrimers for Polymerase Chain Reactions or as Probes, Comp. Appl. Biosci., 7,pp. 525-9, 1991.
[23] Mullis, K.B., Ferré, F., and Gibbs, R.A. The Polymerase Chain Reaction. Birkhäuser,1994.
[24] Nascimento A., Espreafico E.M., Larson M.L., Monesi N., Rossi N..M., Rodrigues V.Tecnologia do DNA Recombinante. USP, 2003.
[25] Saiki R.K., Gelfand D.H., Stoffel S., Scharf S.J., Higuchi R., Horn G.T., Mullis K.B.,Erlich H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA-polymerase. Science 239: 487-491, 1988.
[26] Pádua A., Santana A.J., Bonfim P.S., Algoritmos de Busca em texto.URL http://www.buscaemtexto.hpg.com.br. Acessado em junho de 2004.
[27] Lima, R.C.A., Lima, J.A.A., Souza Jr., M.T., Pio-Ribeiro, G. & Andrade, G.P., Etiologiae estratégias de controle de viroses do mamoeiro no Brasil. Fitopatologia Brasileira.26:689-702, 2001.
[28] Cuticchia A.J., Arnold J., Timberlake W.E.; RT "PCAP: probe choice and analysispackage - set of programs to RT aid in choosing synthetic oligomers for contigmapping."; RL Comput. Appl. Biosci. 9:201-203, 1993.

59
[29] Engels W.R.; RT "Contributing software to the Internet: the Amplify program."; RLTrends Biochem. Sci. 18:448-450, 1993.
[30] Proutski V.Y., Sokur O.V.; RT "New original software for optimized design of RT PCRprimers."; RL (In) Abstracts of the IX International Conference RL "Progress in ClinicalVirology",(August 1994, Stockholm).
[31] Usdin K.; RT "Hypercard-based data management tools for molecular biologists."; RLComput. Appl. Biosci. 8:107-111, 1992.
[32] Napiwotzki J., Becker A., Damian M.; RT "Primer Design - a new program to choosePCR primers RT and oligonucleotide probes."; RL Medizinische Genetic, Programmeand Abstracts, Vol. 2, 1995.
[33] RA Resnick, R.; RT "Primers! for the WWW"; URL http://www.williamstone.com.Acessado em julho 2004.
[34] Rozen,S. and Skaletsky,H. Primer3 on the WWW for general users and for biologistprogrammers. Methods Mol. Biol., 132, 365–386, 2000.
[35] Haas,S., Vingron,M., Poustka,A. and Wiemann,S. Primer design for large scalesequencing. Nucleic Acids Res., 26, 3006–3012, 1998.
[36] Proutski,V. and Holmes,E.C. Primer Master: a new program for the design and analysisof PCR primers. Comput. Appl. Biosci., 12, 253–255, 1996
[37] Li,P., Kupfer,K.C., Davies,C.J., Burbee,D., Evans,G.A. and Garner,H.R. PRIMO: aprimer design program that applies base quality statistics for automated large-scale DNAsequencing. Genomics, 40, 476–485, 1997.
[38] Raddatz,G., Dehio,M., Meyer,T.F. and Dehio,C. PrimeArray: genome-scale primerdesign for DNA-microarray construction. Bioinformatics, 17, 98–99, 2001.
[39] Eberhardt,N.L. A shell program for the design of PCR primers using genetics computergroup (GCG) software (7.1) on VAX/VMS systems. Biotechniques, 13, 914–917, 1992.
[40] Meyer,F., Schleiermacher,C. and Giegerich,R. Gene-Fisher software support for thedetection of postulated genes, 1995.
[41] Andy Vierstraete Homepage. URL http://allserv.rug.ac.be/~avierstr/index.html. Acessadoem maio de 2004.
[42] BLAST. URL http://www.genbank.org/blast. Acessado em maio de 2004.
[43] NCBI GenBank Statistics. http://www.ncbi.nlm.nih.gov/Genbank/genbankstats.html.Acessado em fevereiro de 2005.
[44] Rocha K. K. F., Medeiros C., Monteiro M., Gonçalves L. M., Marinho P. Primer Designfor Automated Virus Diagnosis in Plants. SIBGRAPI 2003, p036. URLhttp://w3.impa.br/~lhf/sib2003/p036.pdf Acessado em maio de 2004.

60
[45] Rocha K. K. F., Medeiros C., Monteiro M., Gonçalves L. M., Marinho P. Design ofSpecie-Specific Primers for Virus Diagnosis in Plants with PCR. BIBE 2004: 149-158.URL http://csdl.computer.org/comp/proceedings/bibe/2004/2173/00/21730149abs.htm.Acessado em fevereiro de 2005.
[46] Gibas, C., Jambeck, P. Trad. Cristina de Amorim Machado. Desenvolvendobioinformática. Rio de Janeiro: Campus, 2001. 440p.
[47] Furlan, J. D. Modelagem de Objetos Através da UML - The Unified Modeling Language,Makron Books,São Paulo, 1998.
[48] Rumbaugh, J.; Blaha, M.; Premerlani, W.; Eddy, F.; Lorensen, W.; Object-OrientedModeling and Design, Prentice-Hall International, New Jersey, 1991.
[49] Needleman, S. B. and Wunsch, C. D. (1970) J. Mol. Biol. 48, 443-453.
[50] Ye, Liang, Huang, Xiaoqiu. MAP2: multiple alignment of syntenic genomic sequences;Nucl. Acids Res. 2005 33: 162-170