resposta inflamatória e disfunção mitocondrial em...
TRANSCRIPT

INSTITUTO OSWALDO CRUZ
Doutorado em Biologia Celular e Molecular
Resposta Inflamatória e Disfunção Mitocondrial em
Pacientes com Choque Séptico
André Miguel Japiassú
Rio de Janeiro
Novembro de 2009

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

Instituto Oswaldo CruzCurso de Pós-graduação em Biologia Celular e Molecular
André Miguel Japiassú
Resposta Inflamatória e Disfunção Mitocondrial em Pacientes
com Choque Séptico
Orientadores: Prof. Dr. Hugo Caire de Castro Faria Neto
Dr. Fernando Augusto Bozza
Rio de Janeiro2009
ii
Tese apresentada ao Instituto Oswaldo Cruz,
como parte dos requisitos para obtenção do
título de Doutor em Ciências.

iii

Instituto Oswaldo CruzCurso de Pós-graduação em Biologia Celular e Molecular
André Miguel Japiassú
Resposta Inflamatória e Disfunção Mitocondrial em Pacientes
com Choque Séptico
Orientadores: Prof. Dr. Hugo Caire de Castro Faria Neto
Dr. Fernando Augusto Bozza
EXAMINADORES:
Prof. Dr. Dumith Chequer Bou-Habib - Presidente
Prof. Dr. Gilberto Friedman
Dr. Aurélio Vicente Graça de Souza
Rio de Janeiro, 8 de Dezembro de 2009
iv

ÍNDICE
PáginaLista de abreviaturas viLista de figuras e tabelas xResumo xvAbstract xvi1 - Introdução 1
1.1 - Definições de sepse 11.2 - Epidemiologia da sepse 41.3 - Escores prognósticos 91.4 - Resposta endócrino–metabólica: Papel do cortisol 101.5 - Resposta inflamatória sistêmica e mediadores solúveis 131.6 - Disfunção mitocondrial na sepse 23
2 - Objetivos 293 - Painel de biomarcadores em pacientes com choque séptico 30
3.1 – Introdução 303.2 – Métodos 313.3 – Resultados 36 3.3.1 – Perfil de citocinas como marcadores de gravidade na sepse: análise multiplex 36 3.3.2 – Cinética de biomarcadores em pacientes com choque séptico 383.4 – Conclusões Parciais 63
4 - Disfunção Mitocondrial em Células Mononucleares do Sangue Periférico 644.1 – Introdução 644.2 – Métodos 654.3 - Resultados 684.4 – Conclusões Parciais 77
5 - Discussão 786 – Conclusões Finais 907 - Referências Bibliográficas 91Anexo 1: Protocolo de coleta de dados clínicos e escore SOFA 107Anexo 2: Termo de consentimento livre e esclarecido 110Anexo 3: Artigo Original “Cytokine profiles as markers of disease severity in sepsis: a multiplex
analysis”, publicado na revista Critical Care em abril de 2007
112
Anexo 4: Artigo de revisão “Revisiting steroid treatment for septic shock: molecular actions and
clinical effects - A review”, publicado em Memórias do Instituto Oswaldo Cruz, em agosto de
2009
120
Anexo 5 – Artigo original “Sepsis is a major determinant of outcome in HIV/AIDS critically ill
patients”, a ser submetido em novembro de 2009
138
v

Lista de abreviaturas:
ACCP/SCCM – Colégio Americano de Médicos do Tórax/Sociedade Americana de Medicina
Intensiva (American College of Chest Physicians/Society of Critical Care Medicine)
ACTH – Hormônio adreno-corticotrófico (Adrenocorticotropic Hormone)
ADP – adenosina 5´-difosfato
Akt – família de proteínas de sinalização intracelular
AP1 - proteína de ativação 1 (activating protein-1)
APACHE II – Avaliação de Fisiologia Aguda e Saúde Crônica (Acute Physiology and
Chronic Health Evaluation)
APC - Proteína C Ativada (activated protein C)
ANOVA – analysis of variance
ANT – trocador de nucleotídeos adenínicos
ATP – adenosine 5´-trifosfato
AUROC – Area under the receiver-operating-characteristic curve
BASES - Brazilian Sepsis Epidemiological Study
CARS - Compensatory Antiinflammatory Response Syndrome
CD – grupo de diferenciação
CDC – Centers for Diseases Control and Prevention
cDNA – DNA complementar
CIRCI - critical illness-related corticosteroid insufficiency
CLP - ligadura e punção cecal (cecal ligation and puncture)
CURB 65 – escore de avaliação de gravidade para pneumonia comunitária (Confusão, Uréia,
Respiração, Blood pressure e idade acima de 65 anos)
Cyt c - citocromo c
DNA – ácido desoxirribonucléico
EGTA – ácido tetraacético etileno glicol (ethylene glycol tetraacetic acid)
ELISA - Enzyme Linked Immuno Sorbent Assay
EPCR - proteína C de células endoteliais (Endothelial Cell Protein C Receptor)
vi

EUA – Estados Unidos da América
FCCP - carbonil cianeto p-(trifluorometoxi)fenilhidrazona
FiO2 – fração inspirada de oxigênio
G-CSF/GM-CSF - fator estimulador de colônias de granulócitos/granulócitos-monócitos
(granulocyte/ granulocyte-macrophage colony-stimulating factor)
GR – receptor de glicocorticóide (glucocorticoid receptor)
GRE – elementos responsivos a glicocorticóides (glucocorticoid responsive elements)
gp130 – glicoproteína 130
HBSS – solução salina balanceada de Hank
HCl – ácido clorídrico
HLA – antígeno leucocitário humano (human leukocyte antigen)
HMGB-1 - high mobility group box 1
ICAM-1 – molécula de adesão intercelular 1 (intercellular adhesion molecule-1)
ICD-9 CM - The International Classification of Diseases/WHO, 9th Revision, Clinical
Modification
IL – interleucina (interleukin)
IL-1ra – antagonista do receptor de IL-1 (Interleukin 1 receptor antagonist)
IL-6R – receptor de IL-6 (Interleukin 6 receptor)
IFN – interferon (interferon)
JAK – cinase da família Janus (Janus kinase)
JNK - c-Jun N-terminal kinase
LPS – lipopolissacarídeos (lipopolysaccharide)
KD – constante de dissociação
kDa – quilodaltons
KCl – cloreto de potássio
MCP-1 – proteína quimiotática para monócito-1 (monocyte chemoattractant protein-1)
MgCl2 – cloreto de magnésio
MIF - fator inibidor da migração de macrófagos (macrophage migration inhibitory factor)
MIP-1 – proteína inflamatória de macrófago-1(macrophage inflammatory protein-1)
vii

mmHg – milímetros de mercúrio
Mn-SOD – superóxido dismutase associada a Manganês
mRNA – RNA mensageiro
NFkB - fator nuclear-kB (nuclear factor-κB)
NK – natural killer (célula)
NO – óxido nítrico (nitric oxide)
O2 - oxigênio
OMS - Organização Mundial da Saúde
ONOO - peroxinitrito
p38 – família de proteínas de sinalização intracelular
PAI-1 - inibidor do ativador do plasminogênio (plasminogen-activator-inhibitor-1)
PaO2 – pressão parcial arterial de oxigênio
PAR-1 - receptor ativado por protease-1 (protease-activated receptor-1)
PBMC – peripheral blood mononuclear cell
PCRt - proteína C reativa titulada
PCT – procalcitonina (procalcitonin)
PROGRESS - Promoting Global Research Excellence in Severe Sepsis
PROWESS - Recombinant Human Protein C Worldwide Evaluation in Severe Sepsis
PTP – poro de transição de permeabilidade
ROC - Receiver Operating Characteristic
RNA - ácido ribonucléico
SAPS II - Simplified Acute Physiologic Score II
SARA – Síndrome de Angústia Respiratória Aguda (Acute Respiratory Distress Syndrome-
ARDS)
SDOM - Síndrome de Disfunção Orgânica Múltipla
SIDA – Síndrome de Imunodeficiência Adquirida
sIL-6R - receptor solúvel de IL-6 (soluble interleukin 6 receptor)
viii

SIRS – Síndrome de Resposta Inflamatória Sistêmica (Systemic Inflammatory Response
Syndrome)
SOFA – Avaliação de Disfunção Orgânica relacionada à Sepse (Sepsis-related Organ Failure
Assessment)
STAT – proteínas sinalizadoras e ativadoras de transcrição (signal transducers and activators
of transcription)
sTNF-R – receptor solúvel de TNF (soluble TNF receptor)
sTREM-1 - TREM-1 solúvel (soluble triggering receptor expressed on myeloid cells)
TF - fator tecidual (Tissue Factor)
TGFβ - transforming growth factor
Th-1/Th-2 – type 1/type 2 helper T cells
TLR – receptor tipo Toll (Toll-like receptor)
TNF - fator de necrose tumoral – (tumor necrosis factor)
TNFR – receptor de TNF (TNF receptor)
TYK2 – tirosina cinase (tyrosine kinase)
UQ - ubiquinona
UTI – Unidade de Tratamento Intensivo
ix

Lista de Figuras e Tabelas:
Tabela e Figuras PáginaTabela 1.1: Mortalidade hospitalar comparativa entre diferentes estudos
epidemiológicos referentes à sepse, sepse grave e choque séptico.
8
Figura 1.1: Esquema da cadeira transportadora de elétrons na membrana interna
mitocondrial.
27
Quadro 3.1 – Critérios de SIRS/sepse. 31Tabela 3.1 – Concentrações de citocinas em pacientes com sepse grave e choque
séptico.
37
Tabela 3.2 – Desempenho das citocinas para predição de mortalidade precoce. 37Tabela 3.3 – Desempenho das citocinas para predição de mortalidade em 28 dias. 37Tabela 3.4 – Características demográficas e clínicas de pacientes com choque
séptico de acordo com o desfecho em 28 dias.
39
Figura 3.1 – Comparação dos níveis de escore SOFA e citocinas entre pacientes
controles, sépticos sobreviventes e sépticos não-sobreviventes em 28 dias.
41
Tabela 3.5 – Níveis plasmáticos de citocinas e cortisol nos dias 1, 3, 5 e 7 após início
de choque séptico.
44
Figura 3.2 – Padrões de cinética de citocinas nos dias 1, 3, 5 e 7 após início de
choque séptico.
45
Tabela 3.6 – Comparação dos níveis de citocinas, proteína C reativa e cortisol no 1º
dia de choque séptico entre sobreviventes e não-sobreviventes.
46
Tabela 3.7 – Comparação da média dos níveis plasmáticos de citocinas, cortisol e
proteína C reativa nos dias 1, 3, 5 e 7 após início de choque séptico.
48
Tabela 3.8 - Comparação dos níveis máximos dos níveis plasmáticos de citocinas,
cortisol e proteína C reativa nos dias 1, 3, 5 e 7 após início de choque séptico.
49
Figura 3.3 – Evolução do escore SOFA e cinética de leucócitos totais, proteína C
reativa, cortisol e citocinas ao longo dos primeiros 7 dias de choque séptico, de
acordo com a mortalidade em 28 dias.
50
Figura 3.4 – Curvas Receiver Operating Characteristic (ROC) com análise de
sensibilidade e especificidade de citocinas e escores SAPS II e SOFA medidos no 1º
dia de choque séptico para predição de mortalidade até 28 dias.
54
Figura 3.5 – Curvas Receiver Operating Characteristic (ROC) com análise de
sensibilidade e especificidade da média dos níveis de citocinas e escores SAPS II e
SOFA medidos durante a 1ª semana de choque séptico para predição de mortalidade
até 28 dias.
55
Figura 3.6 – Curvas Receiver Operating Characteristic (ROC) com análise de
sensibilidade e especificidade dos níveis máximos de citocinas e escores SAPS II e
SOFA medidos durante a 1ª semana de choque séptico para predição de mortalidade
até 28 dias.
56
Tabela 3.9 - Comparação dos níveis de citocinas, proteína C reativa e cortisol no 1º
dia de choque séptico de acordo com mortalidade precoce (menos de 3 dias).
58
x

Figura 3.7 – Curvas Receiver Operating Characteristic (ROC) com análise de
sensibilidade e especificidade para predição de mortalidade precoce (menos de 3
dias) de pacientes com choque séptico.
59
Figura 3.8 – Níveis plasmáticos de cortisol se correlacionaram com os níveis de MIF
apenas em pacientes com choque séptico que não receberam esteróides venosos.
61
Figura 3.9 – Correlação entre o escore Sequential Organ Failure Assessment
(SOFA) e citocinas e cortisol plasmático em pacientes com choque séptico.
62
Tabela 4.1 – Características demográficas, gravidade de doença e desfecho
hospitalar de pacientes com choque séptico e controles.
69
Figura 4.1 – Consumo de oxigênio mitocondrial associado à síntese de ATP em
células mononucleares de sangue periférico (PBMC) em pacientes com choque
séptico e controles.
71
Figura 4.2 – Consumo de oxigênio mitocondrial em PBMC humanos e mortalidade
hospitalar.
73
Figura 4.3 – Correlação entre consumo de oxigênio mitocondrial em PBMCs
humanos e escore Sequential Organ Failure Assessment (SOFA).
74
Figura 4.4 – Conteúdo funcional de F1Fo-ATP sintase e trocador de nucleotídeo
adenínico (ANT) em PBMCs humanos.
76
Figura 5.1 – Representação esquemática do mecanismo proposto pelo qual o choque
séptico causa disfunção mitocondrial em PBMCs.
87
xi

xii
Para minha família, meu pilar.
Ao meu pai, saudades.
Ao meu filho, amor e esperança.

Agradecimentos
Este trabalho é fruto de esforço conjunto de uma grande equipe, entre profissionais que atuam
em laboratórios de pesquisa e em hospitais. A aproximação entre as equipes profissionais que
estão à beira do leito e os pesquisadores de laboratórios de pesquisa faz com que o
conhecimento de fenômenos presentes na prática clínica sejam entendidos de forma mais
completa. Servi de elo entre estes diversos profissionais e pretendo seguir estreitando a
relação entre bancada e beira do leito.
Agradeço a Fernando Bozza, amigo e orientador, que serve de exemplo para os intensivistas
que querem trabalhar com pacientes e têm curiosidade de entender a sepse amplamente.
Hugo Caire foi grande incentivador e facilitador do meu trabalho, e fiz grande amigo, além da
orientação da tese.
Patrícia Bozza tem espírito empreendedor e incrível capacidade de compreensão e
esclarecimento de processos e pessoas, fazendo com que coisas complicadas se tornem
simples; agradeço a grande ajuda em momentos difíceis do trajeto.
A Vera Koatz, pelo início da vida em pesquisa e seus conselhos, in memorian.
À equipe do laboratório de Imunofarmacologia, que me ajudou em todos os momentos da
tese; especialmente, Rachel Gomes, que participou técnica e intelectualmente em dosagens e
artigos publicados; Edson Assis, que foi companheiro e incentivador, nos momentos mais
tensos e complicados; Rodrigo Amâncio, que é parte do nosso time e grande amigo; Adriana
Broxado, a quem admiro pelas realizações e presteza para ajudar a quem quer que seja; e
Rose, que não deixa faltar absolutamente nada para que tenhamos rendimento máximo.
O pessoal do Instituto de Bioquímica Médica da UFRJ me acolheu muito bem e foi
fundamental para realizasse grande parte do trabalho da tese. Marcus Oliveira foi meu terceiro
orientador e ensinou muita Bioquímica. Joana D´Ávila realizou grande parte dos
experimentos que deram início à linha de pesquisa. Ana Paula Santiago desenvolveu
experimentos, me ensinou técnicas de bancada e se tornou amiga durante este ano. E Antonio
Galina, que atua como um guru e resolve problemas complexos quando nossa capacidade se
esgota. E aos pesquisadores Juliana e Wagner do Laboratório de Bioenergética Aplicada, que
nos auxiliaram de forma tão prestativa na parte técnica dos experimentos.
Bruna Fonseca, do Laboratório de Tecnologia Diagnóstica de Biomanguinhos-FIOCRUZ,
trabalhou conosco na dosagem multiplex de citocinas e graças a ela estes resultados foram
possíveis.
Ingebourg Georg, do Laboratório de Imunologia do IPEC-FIOCRUZ, possibilitou as
dosagens de proteína C reativa e cortisol que foram fundamentais para nossos resultados.
xiii

Ângela Santos, do mesmo laboratório, realizou todas estas dosagens comigo, com muita
paciência para agüentar um médico se “intrometendo” na rotina daquele laboratório.
Meus amigos formados nos hospitais por onde passei e coletei as amostras clínicas me deram
suporte para que realizasse meu doutorado. No hospital do Fundão (UFRJ), tenho
praticamente uma família que sempre me apoiou e agradeço a Ricardo Amorim, Rosa Vianna,
Analucia Mattera, Rosane Goldwasser, Cid Marcus David, toda equipe de enfermagem, e
residentes (Renata Carnevale e Fred Mohilla especialmente) que participaram na coleta de
dados e amostras clínicas. No Hospital Quinta D´Or, Roberto Costa, Odilon Neto, Alex
Coscia, Alessandra Longo, Renata Breves, Haroldo Falcão, Vera Veríssimo, Andre
Albuquerque e Jose Valério, que me deram apoio para trabalhar e continuar na pesquisa. No
IPEC-FIOCRUZ, me sinto bem acolhido na companhia de meus colegas infectologistas, a
quem agradeço através de Maria Isabel Fragoso e Lourdes Benamor como representantes
deste grupo, além de toda a equipe multiprofissional que me faz sentir em casa. Paula Luz
(IPEC) foi uma professora paciente na análise estatística de meandros do trabalho e nos
ajudou muito. Emersom Mesquita é companheiro e se inseriu durante este tempo na nossa
equipe. E na Casa de Saúde São José, Gustavo Nobre e Marcelo Kalichsztein, que deram a
oportunidade para fazer pesquisa profissionalmente na vida privada, e Gustavo Almeida,
Pedro Kurtz, Michel Cukier, Bruno Oliveira, Carlos Roberto Gondim e Ronaldo Vegni que
me ajudam na pesquisa além de suas atividades assistenciais.
Pacientes e familiares sempre foram compreensíveis em relação à pesquisa e agradeço a
todos, pois através deles é possível entender melhor os fenômenos da fisiopatologia da sepse.
Finalmente à minha família, que sempre teve paciência e me deu o apoio necessário para me
manter são neste longo período. Roberta minha companheira e incentivadora. Minha mãe
Gloria, exemplo e conselheira. Henrique, Ricardo, Maria Augusta, João Ricardo, Akeo, Gesy,
Claudia, Maria Luiza, Deco, Flavia, Ana Beatriz e Maria – amo vocês.
xiv

Resumo
A sepse representa um problema clínico de alta relevância, principalmente devido a sua
grande incidência em pacientes hospitalizados e aos seus altos índices de mortalidade. A
determinação precisa e a compreensão dos mecanismos fisiopatológicos envolvidos nesse
processo será de grande valia para o desenvolvimento de novas abordagens terapêuticas
visando a prevenção ou resolução da Síndrome de Disfunção Orgânica Múltipla (SDOM) e
redução da morbi-mortalidade. A inflamação sistêmica associada à sepse envolve a ativação
do sistema imune e neuroendócrino. A sepse leva tanto à produção excessiva de mediadores
pró-inflamatórios, incluindo-se citocinas, radicais do oxigênio e mediadores lipídicos, quanto
à produção de hormônios de estresse oriundos do eixo hipotálamo-hipófise-adrenal. Por outro
lado, há evidência de que ocorre inibição da atividade mitocondrial e consequentemente
redução da fosforilação oxidativa em diferentes tipos celulares de diversos tecidos.
Neste trabalho, demonstramos a cinética de um painel de citocinas durante a primeira semana
de choque séptico, analisando o desenvolvimento de disfunções orgânicas e a mortalidade.
Níveis significativamente maiores de citocinas permanecem após o primeiro dia de choque
séptico. Estabelecemos quatro padrões de cinética de biomarcadores nos primeiros sete dias
de choque séptico: queda exponencial (ex. IL-6), curva Gaussiana (ex. IL-10), níveis
crescentes (ex. IL-8) e níveis estáveis (ex. MIF). Maiores níveis de IL-6, IL-8, IL-10 e MCP-1
se associam com mortalidade precoce. Níveis elevados de MIF no 1º dia, média elevada de
IL-6 e níveis máximos de cortisol na primeira semana são bons preditores de mortalidade em
28 dias em pacientes com choque séptico. Há boa correlação entre a gravidade de disfunções
orgânicas com os níveis de MIF, IL-8, cortisol e MIP-1β.
Verificamos que células imunes circulantes apresentam disfunção mitocondrial, com redução
do consumo de oxigênio associado à síntese de ATP. O mecanismo desta disfunção se associa
à redução do conteúdo funcional da F1Fo-ATP sintase. A disfunção mitocondrial de células
mononucleares de sangue periférico se correlacionou com o grau de disfunções orgânicas e
com a mortalidade hospitalar de pacientes com choque séptico.
Foram identificados marcadores imunológicos e bioquímicos associados com a evolução de
pacientes com choque séptico para Síndrome de Disfunção Orgânica Múltipla.
xv

Abstract
Sepsis is a highly relevant clinical syndrome, due to its high incidence on hospitalized
population and high rate of mortality. The precise knowledge and understanding of
pathophysiological mechanisms of sepsis will be useful to the development of new diagnosis
and treatment strategies, leading to prevention or resolution of Multiple Organ Dysfunction
Syndrome (MODS) and reduction of morbi-mortality. Systemic inflammation associated with
sepsis involves the activation of the immune and neuroendocrine system. Sepsis triggers
increased production of proinflammatory mediators, including cytokines, oxygen free
radicals, lipid mediators and the production of stress hormones from the hypothalamus-
hypophysis-adrenal axis. Despite of excessive activation of inflammation, there is evidence of
mitochondrial dysfunction, with consequent oxidative phosphorylation inhibition in different
cell types from several tissues.
In our work, we performed the kinetics of a cytokine panel during the first week of septic
shock, analyzing also the evolution of organ dysfunctions and mortality. Highly significant
levels of cytokines persist beyond the first day of septic shock. Four patterns of kinetics were
established during the first seven days of septic shock: exponential decay (ex. IL-6), Gaussian
curve (ex. IL-10), increasing levels (ex. IL-8) and stable levels (ex. MIF). Higher levels of IL-
6, IL-8, IL-10 and MCP-1 were associated with early mortality. High MIF levels on the first
day, high mean levels of IL-6 and maximal levels of cortisol in the first week are good
predictors of 28th day mortality for septic shock patients. There is good correlation of organ
dysfunction severity and MIF, IL-8, cortisol and MIP-1β levels.
Mitochondrial dysfunction is present on circulating immune cells, with lower oxygen
consumption related to ATP synthesis. The mechanism of such dysfunction is associated with
reduction of F1Fo-ATP synthase functional content. Mitochondrial dysfunction of peripheral
blood mononuclear cells was correlated with severity of organ dysfunctions and hospital
mortality of septic shock patients.
There was identification of immunological and biochemical markers related to septic shock
patients evolution to Multiple Organ Dysfunction Syndrome.
xvi

1
1. Introdução
1.1 - Definições de sepse
A sepse representa um problema clínico de alta relevância, principalmente devido a
sua grande incidência em pacientes hospitalizados e aos seus altos índices de mortalidade
(Angus e cols., 2001). Sua incidência anual tem aumentado de maneira preocupante nas
últimas décadas, sendo estimada, nos Estados Unidos da América, em 2000, em 751.000
casos que causaram 215.000 mortes. Estima-se que o Brasil tenha cerca de 600.000 casos de
sepse anualmente (Sales Júnior e cols., 2006). Apesar dos constantes avanços obtidos na
terapêutica de suporte, assim como na antibioticoterapia, sua mortalidade continua sendo
extremamente elevada, variando de 40 a 70 % (Christaki & Opal, 2008). Por este motivo, a
determinação precisa e a compreensão dos mecanismos fisiopatológicos envolvidos nesse
processo será de grande valia para o desenvolvimento de novas abordagens terapêuticas
visando a redução da morbidade e da mortalidade na sepse.
A inflamação sistêmica associada à sepse envolve a ativação do sistema imune e
neuroendócrino. A sepse desencadeia uma produção excessiva de mediadores pró-
inflamatórios, incluindo citocinas, radicais do oxigênio e mediadores lipídicos, quanto a
produção de hormônios de estresse oriundos do eixo hipotálamo/hipófise/supra-renal
(Vanhorebeek & van den Berghe, 2006). Em estudos com animais de experimentação,
utilizando-se anticorpos neutralizantes ou proteínas recombinantes, ficou demonstrado que
algumas citocinas têm um papel central na sepse, podendo-se ressaltar fator de necrose
tumoral (TNF-α), interleucina (IL)1β, IL-6, IL-8, IL-10 e fator de inibição de migração de
macrófagos (MIF). Existe ainda uma importante interação entre moléculas de reconhecimento
de patógenos, conhecidas como receptores toll-like (TLR), e a produção de citocinas na sepse
(Bochud & Calandra, 2003).
Em 1991, realizou-se a Conferência de Consenso da ACCP/SCCM para o
estabelecimento de definições sobre sepse (Bone e cols., 1992). O termo Síndrome de
Resposta Inflamatória Sistêmica (SIRS) foi criado, com o objetivo de indicar uma série de
manifestações clínicas secundárias a insultos diversos, sendo que o mais freqüente é a
infecção. Sepse passou a ser definida como SIRS, acompanhada de um processo infeccioso
conhecido. Novas terminologias como sepse severa, choque séptico, hipotensão relacionada à
sepse e Síndrome de Disfunção Orgânica Múltipla (SDOM) passaram a ser usadas, e outros
estudos surgiram afirmando que estas terminologias representariam a evolução de um mesmo

2
processo (Rangel-Frausto e cols., 1995; Brun-Buisson e cols., 1995).
Após o Consenso de 1991, surgiram críticas à excessiva sensibilidade e à baixa
especificidade da nova terminologia. As variáveis utilizadas para definir sepse e SIRS como
temperatura corporal, frequência cardíaca e respiratória e leucometria, não possuem poder
preditivo para avaliar mortalidade em pacientes hospitalizados com febre. Uma reunião para a
discussão de novos critérios para sepse foi realizada no final de 2001 com estudiosos da área.
Foi sugerida a criação de um sistema de critérios mais específico, que incluiria fatores
predisponentes para o desenvolvimento de sepse, extensão da infecção (localizada ou
sistêmica), presença e magnitude de disfunções orgânicas e parâmetros de avaliação da
resposta imune (proteína C reativa titulada - PCRt, procalcitonina, IL-6). Deste modo, não se
limitaria apenas ao diagnóstico da sepse, mas também se atribuiria importância à gravidade da
infecção (Levy e cols., 2003). O novo sistema é chamado PIRO:
P – Predisposição: os pacientes com patologias associadas importantes têm maior chance de
apresentar quadros graves de infecção; além disto, parece existir um espectro de
apresentações de uma mesma infecção de acordo com a predisposição genética do indivíduo
(por exemplo, portadores de determinados polimorfismos para TNF-α, IL-10 ou CD14);
I – tipo de Infecção: demonstração de bactérias ou outros microorganismos em sítios
possíveis de infecção, em meios de culturas ou sorologias;
R – Resposta à infecção: presença de marcadores da resposta inflamatória, fisiológicos (ex.
choque) e laboratoriais (ex. PCRt);
O – Órgãos com disfunção: evidência clínica (hipotensão, oligúria, torpor) ou laboratorial
(elevação da creatinina, piora da troca gasosa, elevação de bilirrubinas ou trombocitopenia)
de disfunção de 1 ou mais órgãos.
Os pacientes com sepse grave e choque séptico estão predispostos a desenvolver
múltiplas disfunções orgânicas. Existe um efeito cumulativo do número de disfunções de
órgãos e sistemas em relação à sobrevida (Padkin e cols., 2003; Flaaten e cols., 2004;
Tanriover e cols., 2005; Vincent e cols., 2006; Engel e cols., 2007; Cheng e cols., 2007;
Rezende e cols., 2008). Pacientes com mais de 3 ou 4 disfunções apresentam mortalidade
acima de 60% (Vincent e cols., 2006). Assim sendo, estes pacientes constituem um subgrupo
de pacientes com sepse, que desafiam médicos e pesquisadores com sua alta letalidade. Por
isso criou-se o termo SDOM há mais de 20 anos, com prevalência de aproximadamente 15%
nos pacientes internados em unidades de tratamento intensivo (UTI) (Tilney e cols., 1973). A

3
aplicação vigorosa de suporte orgânico tem prevenido a letalidade precoce, mas por outro
lado estende a internação no UTI e cria oportunidade para o aparecimento de novos insultos e
progressão lenta da SDOM (Mongardon e cols., 2009). O manuseio clínico é baseado no
suporte de disfunções orgânicas, como hemodiálise para disfunção renal e ventilação
mecânica para dano pulmonar agudo, até que o paciente se recupere espontaneamente.
Sistemas de avaliação de disfunções orgânicas, como o escore Sequential Organ Failure
Assessment (SOFA), carecem de biomarcadores que se correlacionem com a evolução para
SDOM. A estratificação através do sistema PIRO pode ser interessante se conseguir
relacionar todos seus aspectos, mas os critérios de resposta do hospedeiro e de disfunções
orgânicas não foram aprofundados com o melhor conhecimento de fisiopatologia da sepse.
Por exemplo, critérios de resposta à infecção têm sido limitados à presença de taquicardia ou
hipotensão (Lisboa e cols., 2008; Rubulotta e cols., 2009), e não conseguiram identificar
biomarcadores para ajudar na predição do diagnóstico ou gravidade da sepse (Bozza e cols.,
2005).
Existem algumas teorias para o desenvolvimento da SDOM. Há dúvida se a SDOM é
um processo patológico irrecuperável ou adaptativo a um estresse inflamatório grave e
prolongado. Não se sabe se SDOM representa causa de morte ou ocorre interrupção ou não
prescrição de terapêuticas após tempo prolongado de internação na UTI (Sprung e cols.,
2003; Soares e cols., 2007). Os escores de avaliação para disfunções orgânicas, como SOFA
(Vincent e cols., 1996) e MODS (Marshall e cols., 1995), são incapazes de predizer o
prognóstico de cada paciente. Cortes histológicos de tecidos de pacientes que morrem de
SDOM apresentam pouco infiltrado inflamatório ao se comparar com tecidos de pacientes
politraumatizados que faleceram (Hotchkiss & Karl, 2003). Falências respiratória (Síndrome
de Angústia Respiratória Aguda - SARA) e renal (necrose tubular aguda) são disfunções
principalmente funcionais, já que a capacidade de recuperação destes tecidos é quase total
após meses. Pacientes mais graves permanecem maior tempo hospitalizados e recebem maior
número de intervenções invasivas, predispondo ao desenvolvimento de novas infecções. Uma
das teorias para este fenômeno é o desligamento bioenergético causado por lesão ou inibição
da atividade mitocondrial e consequentemente redução da respiração fosforilativa (Singer e
cols., 2004; Belikova e cols., 2007). Este desligamento bioenergético pode ser parte de um
processo patológico, no qual se desenvolve a SDOM, ou uma resposta adaptativa, na qual o
metabolismo corporal é reduzido enquanto a infecção é combatida e o organismo se torna
apto à recuperação e reversão de disfunções orgânicas/teciduais (Rudiger e cols., 2008).
Apesar das diferentes teorias relacionadas ao desenvolvimento da SDOM, a evolução de

4
disfunções orgânicas, biomarcadores, bioenergética e mortalidade não foram claramente
evidenciados no âmbito clínico.
1.2 - Epidemiologia da Sepse
Diferentes autores procuraram avaliar as definições de 1992, especialmente os
aspectos da epidemiologia clínica e microbiológica, suas inter-relações e impacto na
sobrevida dos pacientes.
Podemos dividir os estudos sobre epidemiologia da sepse em dois grandes grupos, os
estudos retrospectivos e os prospectivos. Entre os retrospectivos estão incluídos os de Angus
e Martin. Estes são grandes estudos populacionais baseados em bancos de dados de
internações hospitalares e utilizam códigos de diagnóstico pós-alta (ex. ICD-9CM - The
International Classification of Diseases/WHO, 9th Revision, Clinical Modification). Como
estes códigos são utilizados para pagamentos de internações e procedimentos hospitalares, e
não incluem características fisiológicas necessárias ao diagnostico de sepse, talvez sofram de
imprecisões e vieses difíceis de serem quantificados. Angus e colaboradores analisaram
6.621.559 internações hospitalares no ano de 1995 em 847 hospitais e identificaram 192.980
casos de sepse grave, estimando em 751.000 casos/ano nos EUA (300 casos por 100.000
habitantes ou 2,26 casos por 100 internações hospitalares) sendo que em torno de 383.000
(51,1%) receberam cuidados intensivos. A mortalidade hospitalar foi de em 28,6% e a
mortalidade na terapia intensiva de 34,1% (Angus e cols., 2001). Outro estudo analisou dados
de 750 milhões de internações hospitalares nos EUA entre 1979 e 2000, identificando
10.319.418 casos de sepse, com um importante incremento na incidência de sepse nestes 22
anos, em 1979 a incidência era de 82,7 casos/100.000 habitantes, contra 240,4/100.000 em
2000 (Martin e cols., 2003), mas apresentou um declínio significativo, de 27,8% entre 1979-
1984 para 17,9% entre 1995-2000. Embora sejam retrospectivos, são úteis para dimensionar o
problema da sepse dentro do contexto da saúde pública, especialmente devido aos seus
grandes números.
Quanto aos estudos prospectivos, embora tenham um número menor de pacientes
incluídos, estes estudos provavelmente são mais detalhados na descrição da gravidade dos
casos e precisos na análise de desfecho. O primeiro grande estudo prospectivo observacional
acompanhou 3708 pacientes admitidos em um hospital universitário durante 9 meses (Rangel-
Frausto e cols., 1995). Sessenta e oito porcento preenchiam mais de dois critérios para SIRS e
foram acompanhados por 28 dias. Durante o acompanhamento, 17% desenvolveram sepse,

5
13% sepse grave e 13% choque séptico. A mortalidade apresentou um aumento progressivo
entre SIRS, sepse, sepse grave e choque séptico: 7%, 16%, 20%, e 46%, respectivamente. Os
estágios de SIRS, sepse e choque séptico representam um contínuo hierárquico de intensidade
da resposta inflamatória sistêmica. Recente estudo alemão estratificou o risco de desenvolver
sepse grave a partir de sinais de SIRS: havia aumento cumulativo de 2 vezes de chance de
sepse por critério de SIRS (Engel e cols., 2007). Um estudo francês prospectivo e
multicêntrico identificou incidência, fatores de risco e mortalidade de 1052 pacientes com
sepse grave durante 2 meses (Brun-Buisson e cols., 1995). A incidência para sepse grave foi
de 90/1000 admissões na terapia intensiva, e 69/1000 para choque séptico. A mortalidade em
28 dias foi de 56% para pacientes com sepse grave e de 71% para pacientes com choque
séptico.
Estudos regionais sobre a epidemiologia da sepse grave foram publicados nos últimos
15 anos, com resultados heterogêneos. No EPISEPSIS, estudo realizado em 205 unidades de
terapia intensiva na França, durante um período de duas semanas em 2001, foram avaliadas
3738 admissões, sendo que 546 pacientes (14,6%) apresentavam critérios de sepse grave ou
choque séptico. A mortalidade foi de 35% em 30 dias e 41,9% em dois meses (Brun-Buisson
e cols., 2004). No Reino Unido a incidência de sepse grave foi 51/100.000 habitantes, com
prevalência de 27% de todas as internações hospitalares de 1995 a 2000 (Padkin e cols.,
2003). Os autores britânicos estimaram que 46% das diárias de UTI são utilizadas pelos
pacientes sépticos. Na Alemanha, a incidência nacional de sepse grave foi de 76 a 110 casos
por 100.000 habitantes, com mortalidade de 55% (Engel e cols., 2007). Na Espanha, a região
de Valencia apresentou incidência crescente da sepse grave em 4,7 a 5,4 casos por 100.000
habitantes entre 1995 e 2004 (Ballester e cols., 2008), enquanto é de 25/100.000 habitantes
em outras 14 regiões do mesmo país (Blanco e cols., 2008). A mortalidade hospitalar foi 42 e
54%, respectivamente nestes dois estudos espanhóis. Na Noruega a incidência de sepse grave
foi de 1,49 casos por 1000 habitantes, com 9,5 casos por 1000 internações hospitalares, e
idosos e aqueles com mais de 2 disfunções orgânicas apresentaram pior prognóstico (Flaatten,
2004). No estudo conduzido pelo Anzics Clinical Trials Group, da Oceania foram avaliados
5878 pacientes, dos quais quase 12% apresentaram sepse grave, com mortalidade hospitalar
de 37,5% (Finfer e cols., 2004).
Estudos em países em desenvolvimento mostraram alto grau de disfunções orgânicas e
alta mortalidade de pacientes com sepse grave: na Turquia, a mortalidade hospitalar foi 87%
(Tanriover e cols., 2005); mortalidade hospitalar de 51,2% na República Eslovaca (Záhorec e
cols., 2005); mortalidade de 50% de pacientes em hospital terciário na Tailândia (Khwannimit

6
e cols., 2009); 48,7% de pacientes cirúrgicos com sepse grave morreram em estudo
multicêntrico chinês (Cheng e cols., 2007).
Os dados referentes à epidemiologia da sepse no Brasil são recentes. O Brazilian
Sepsis Epidemiological Study (BASES) avaliou 1383 pacientes internados em cinco unidades
de terapia intensiva (três em São Paulo e duas em Santa Catarina) durante um período de
cinco meses (Silva e cols., 2004). Do total de 1383 pacientes incluídos, 415 pacientes (30,5%)
desenvolveram sepse, 241 (17,4%) sepse grave e 203 (14,7%) choque séptico. A taxa de
mortalidade encontrada foi de 33,9%, 46,9% e 52,2%, para sepse, sepse grave e choque
séptico, respectivamente. O estudo Sepse Brasil foi realizado em 75 UTIs de todas as regiões
brasileiras (Sales Júnior e cols., 2006). A incidência de sepse foi 16,7% (521 de 3128
pacientes admitidos em UTIs). Ocorreu sepse em 19,6% dos pacientes, sepse grave em 29,6%
e choque séptico em 50,8%. A idade média da população foi 61 anos, com leve predomínio
do sexo masculino e com tempo de internação na UTI 15 dias. Houve diferenças regionais:
pacientes da região Sudeste eram mais idosos e tiveram menor mortalidade que outros das
regiões Sul, Nordeste, Centro-Oeste e Norte. Grande parte dos pacientes tinha comorbidades
significativas (60%) e foi admitida por doenças não-cirúrgicas e pneumonia (65%). A
mortalidade global foi 46,6%, mas aumentou gradativamente de 16,7% com sepse, para
34,4% na sepse grave e 65,3% no choque séptico. Finalmente o estudo multicêntrico global
PROGRESS (Promoting Global Research Excellence in Severe Sepsis) também pôde
fornecer dados sobre características e prognóstico da sepse grave do Brasil, já que 982
brasileiros de 12750 pacientes foram incluídos. Os pacientes brasileiros apresentavam
características demográficas e gravidade de doença aguda (expressa por escores prognósticos)
semelhantes a de outros países (Argentina, Canadá, Índia, Alemanha e Austrália), mas
ficaram internados no hospital por mais tempo (média 33 dias contra 28 dias no grupo global)
e apresentaram maior mortalidade hospitalar (67,4% no Brasil, versus média geral de 49%).
Tomadas em conjunto destes 4 estudos, a taxa de mortalidade por sepse grave no Brasil é
57,9% (1008 óbitos do total de 1731 pacientes). Finalmente, Rezende e colaboradores
avaliaram a apresentação de sepse grave em 342 pacientes no Serviço de Emergência de um
hospital terciário de São Paulo. A incidência foi 6,4%, com elevação nos meses de inverno, e
presença de 2 ou mais disfunções orgânicas em 28% dos pacientes sépticos. A mortalidade
hospitalar foi 64%, e esteve associada a fatores como idade acima de 70 anos, sexo
masculino, pneumonia, presença de disfunções orgânicas e acidose metabólica.
As diferenças de mortalidade por sepse grave de estudos ao redor do mundo estão
representadas na Tabela 1.1. A mortalidade de 18,9% foi calculada a partir da relação entre

7
óbitos e casos de sepse grave de todos os estudos desde 1991 até o primeiro semestre de
2009; nesta análise está incluído o estudo de Martin e colaboradores (2003), que representa a
maioria absoluta em relação aos outros estudos. Se excluirmos os estudos anteriores ao
consenso de 1992 e o estudo de Martin (que possui coorte que foi acompanhada desde 1979),
a taxa de mortalidade hospitalar foi 32,5%. A análise de estudos exclusivamente brasileiros
demonstra mortalidade hospitalar de 59,1%.

8
Tabela 1.1: Mortalidade hospitalar comparativa entre diferentes estudos epidemiológicos
referentes à sepse, sepse grave e choque séptico.
Mortalidade Autor Principal Ano N pacientes País
Sepse Sepse Grave Choque
Séptico
Greenman 1991 226 EUA 41%
Ziegler 1991 543 EUA 43%
Rangel-Frausto 1995 467 EUA 16% 20% 46%
Brun-Buisson 1995 1052 França - 56% 71%
Salvo 1995 67 Itália 36% 52% 81,8%
Sands 1997 1342 EUA 34%
Angus 2001 192980 EUA 28,6%
Alberti# 2002 3239 Europa 17-50% 25,5-56,3% 45,7-66,8%
Martin* 2003 4068819 EUA 17,9%
Annane 2003 8251 França - - 61,2%
Padkin 2003 15362 Reino Unido 47,3%
Brun-Buisson 2004 546 França - 41,9% -
Finfer 2004 691 Anzics1 - 37,5% -
Silva 2004 241 Brasil 33,9% 46,9% 52,2%
Flaatten 2004 6665 Noruega 13,5% 27% 29,3%
Sundararajan 2005 33741 Austrália 10,2% 31,1% -
Adrie 2005 713 França - 39% -
Tanriover 2005 63 Turquia - 87,3% -
Záhorec 2005 124 Rep. Eslovaca - 51,2% -
Sales Júnior 2006 521 Brasil 16,7% 34,4% 65,3%
Cheng 2007 318 China - 48,7% -
Engel 2007 415 Alemanha - 55,2% -
Moreno† 2008 2052 Global 35,4% 44,9% 52,5%
Ballester 2008 33767 Espanha - 42,5% -
Blanco 2008 311 Espanha - 54,3% -
Rezende 2008 342 Brasil - 64% -
PROGRESS 2009 12570 Global - 49,6% -

9
PROGRESS †† 2009 969 Brasil - 67,4% -
Kwannimit 2009 390 Tailândia - 21,8% 44,2%
Total 91-2009 4383114 - 18,9% -
Excluindo-se
Martin 2003
91-2009 305275 ‡
32,5% ‡
Estudos
brasileiros
2004-09 2073 ‡‡ 59,1% ‡‡
1Anzics - Australian and New Zealand Intensive Care Society; EUA – Estados Unidos da
América; PROGRESS - Promoting Global Research Excellence in Severe Sepsis; # -
mortalidade foi classificada de acordo com presença ou ausência de infecções adquiridas na
enfermaria e na UTI; * - correspondente ao ultimo período de observação do estudo (1995-
2000); † - parte do estudo multicêntrico SAPS 3; †† - parte do estudo multicêntrico
PROGRESS, com dados do desfecho de pacientes sépticos oriundos do Brasil; ‡ - Taxa
calculada excetuando o estudo de Martin e cols., 2003; ‡‡ - Taxa calculada apenas com
estudos brasileiros.
1.3 - Escores Prognósticos
Simplified Acute Physiology Score (SAPS) II
O escore prognóstico SAPS II foi criado no início da década de 90, com o intuito
de adaptar escores preexistentes (como o Acute Physiology and Chronic Health Evaluation –
APACHE II) à realidade de países europeus (Knaus e cols., 1985; Le Gall e cols., 1993). O
escore SAPS II usa um sistema de pontos baseado nos valores iniciais de 12 medidas
fisiológicas rotineiras (pressão arterial sistólica, frequência cardíaca, temperatura corporal,
oxigenação, débito urinário, ureia sérica, leucometria, sódio, potássio, bicarbonato e
bilirrubinas séricas), idade, admissão clínica ou cirúrgica e doenças preexistentes (neoplasias
metastáticas ou hematológicas e Síndrome de Imunodeficiência Adquirida - SIDA). Cada
parâmetro recebe uma pontuação específica, baseada em estudos de validação com milhares
de pacientes internados em UTI. Os valores anotados são os piores ao longo de 24 horas de
internação no CTI e a pontuação final varia entre 0 e 161 pontos. Uma equação logarítmica é
usada com a pontuação final para estimar a probabilidade de óbito. O escore tem sido usado
universalmente para estratificar e comparar vários grupos de pacientes gravemente doentes,
incluindo pacientes sépticos na entrada de protocolos de pesquisa. Porém este indicador não

10
tem validade para prever desfechos de morbidade (ex. uso de ventilação mecânica) ou
mortalidade que não seja a hospitalar.
Sequential Organ Failure Assessment (SOFA)
O escore SOFA foi criado em 1994 para descrever quantitativa e objetivamente o
grau de disfunção orgânica ao longo da internação de pacientes graves. Secundariamente, é
possível medir os efeitos de novas terapêuticas no curso da sepse e disfunções orgânicas com
a evolução deste escore (Vincent e cols., 1996). Os sistemas avaliados são: respiratório,
cardiovascular, hepático, coagulação, renal e neurológico. É dada uma pontuação de 0
(normal) a 4 (mais alterado) pontos dependendo do grau de alteração de cada órgão/sistema e
o pior valor é anotado diariamente. A pontuação varia entre 0 e 24 pontos. O sistema
cardiovascular é avaliado de acordo com a pressão arterial média e a necessidade e
intensidade de aminas vasopressoras. A função respiratória é avaliada pela relação da pressão
parcial arterial de oxigênio pela fração inspirada de oxigênio (PaO2/FiO2). A creatinina e a
diurese são utilizadas na avaliação da função renal. A queda na quantidade de plaquetas
discrimina a disfunção no sistema hematológico. A dosagem de bilirrubinas totais expressa a
função hepática. A disfunção neurológica é avaliada pela queda do escore de coma de
Glasgow.
Embora este indicador tenha sido criado para avaliar a gravidade de pacientes
com sepse, ele foi avaliado em relação ao prognóstico de pacientes com sepse grave.
Pacientes com pontuação acima de 11 pontos em qualquer momento da internação ou média
de pontuações acima de 5 pontos durante a permanência na UTI têm probabilidade de óbito
maior que 80% (Ferreira e cols., 2001).
Embora este escore seja eficaz no acompanhamento da gravidade de pacientes
sépticos, ele carece de acurácia para diagnóstico e prognóstico. Da mesma maneira, alguns
sistemas orgânicos não são contemplados neste escore, como o endócrino e o gastrointestinal.
São necessários mais estudos para o aperfeiçoamento deste escore, com o acréscimo de mais
parâmetros relacionados ao diagnóstico, gravidade e prognóstico de pacientes com sepse.
1.4 - Resposta endócrino–metabólica: Papel do cortisol
O cortisol é um hormônio glicocorticoide, secretado pela zona fasciculada das
glândulas adrenais a partir de estímulo do hormônio pituitário ACTH (adrenal corticotrophic

11
hormone). O cortisol estimula a gliconeogênese, lipólise e tem efeitos antiinflamatórios. Ele
faz parte da resposta fisiológica a estímulos inflamatórios por trauma e infecções, que são
acompanhados por alterações metabólicas em condições de jejum, com utilização de reservas
de energia (glicogêneo e ácidos graxos) principalmente para manutenção da função de órgãos
vitais como o cérebro e o sistema imune (Campbell, 2005). A resposta metabólica ao estresse
provoca alterações endócrinas, com efeito importante sobre o eixo hipotálamo-hipófise-
adrenal. Mudanças agudas no padrão de liberação de hormônios deste eixo incluem aumento
dos níveis plasmáticos de CRH (corticotropin releasing hormone), ACTH e cortisol, que
acompanham a maior liberação de citocinas também. No entanto, ao longo de dias após o
insulto primário ocorre uma redução progressiva e prolongada de atividade deste eixo e de
outros hormônios também, como os do eixo somatotrópico e tiroidiano (van den Berghe,
2000). Existe dúvida se isto significa uma exaustão do sistema endócrino, como uma
disfunção temporária, ou uma resposta adaptativa à necessidade de reduzir o metabolismo
corporal em face da invasão de microorganismos e inflamação sistêmica.
A relação entre o sistema imune e o eixo hipotálamo-hipófise-adrenal foi descrita
ainda no século XIX, quando Brown-Séquard notou maior chance de morte em pacientes
cirúrgicos que não apresentavam uma resposta adrenal normal (Brown-Séquard CE, 1856).
Estímulos adrenérgicos, citocinas e CRH são responsáveis pelo aumento dos níveis de
cortisol logo após cirurgias, traumas e sepse (Rivier & Vale, 1983; Harbuz e cols., 1992). Ao
longo de dias, os níveis de ACTH permanecem baixos, em contraste aos de cortisol ainda
elevados, podendo significar a regulação da produção e secreção de cortisol por outras
moléculas como endotelina, citocinas e receptores TLR2 e TLR4 (Vermes e cols., 1995;
Bornstein e cols., 2004; Emonts e cols., 2007).
O cortisol exerce uma série de efeitos moleculares que estão implicados na
resposta metabólica e inflamatória à sepse (Japiassú e cols., 2009). Os efeitos do cortisol são
mediados pelo receptor de glicocorticoide citossólico (GR), que se dissocia do complexo com
chaperonas e imunofilinas e é transportado ao núcleo, onde se liga a elementos responsivos a
glicocorticoides (GRE), que são regiões específicas em genes promotoras (Rhodes & Yamada
1995). O GR se liga a fatores de transcrição como proteína de ativação 1 (AP1), proteínas
sinalizadoras e ativadoras de transcrição (Stat) 3 e fator nuclear-kB (NF-kB), regulando a
produção de RNA mensageiro (mRNA) de citocinas e kinases (JNK e p38), regulando a
sinalização intracelular e os níveis de citocinas no plasma e nos tecidos (Meduri e cols.,
2005). Outros efeitos da ação do cortisol incluem efeitos anti-apoptóticos (por exemplo, em
timócitos) e inibição da vasodilatação generalizada da sepse (inibição de canais de potássio

12
adenosine 5´-trifosfato [ATP] sensíveis) (Bren e cols., 1999; d´Emmanuele di Villa Bianca e
cols., 2003; Delfino e cols., 2004).
A disfunção do eixo hipotálamo-hipófise-adrenal tem sido implicada no
prognóstico do pacientes com sepse grave e choque séptico, e serviu de base para o
tratamento com corticoides sintéticos nos casos mais graves. A insuficiência corticosteróide
associada a doença grave (critical illness-related corticosteroid insufficiency - CIRCI) é
definida como níveis paradoxalmente reduzidos de cortisol plasmático em doenças agudas
graves ou ausência de aumento de cortisol após estímulo com agonistas de ACTH (Marik e
cols., 2008). Níveis reduzidos de cortisol plasmático foram demonstrados na sepse,
pancreatite aguda grave, queimaduras extensas e insuficiência hepática (Murphy e cols.,
1993; Marik e cols., 2005; Widmer e cols., 2005; Annane e cols., 2006; Ho e cols., 2006). No
entanto, os casos mais graves e letais de sepse foram associados a níveis aumentados de
cortisol sérico (Melby & Spink, 1958; Salluh e cols., 2008b). Annane e cols (2000)
demonstrou que pacientes com níveis altos de cortisol plasmático e resposta deficiente ao
teste da cortrosina (com elevação menor que 9 µg/dl de cortisol após infusão de ACTH
sintético) têm pior prognóstico, comparativamente a pacientes com menores níveis de cortisol
e resposta adequada ao teste. Estes autores indicaram que a resposta à estimulação da
secreção de cortisol pela administração de ACTH sintético era mais relevante que os níveis
basais de cortisol em pacientes com sepse grave (Annane e cols., 2000).
A CIRCI também pode ser interpretada como um processo dinâmico, no qual
pode cursar com uma “exaustão” adrenal, com níveis aumentados precocemente após o início
da sepse e disfunção adrenal progressiva após alguns dias. Guzman & Guzman (2007)
observaram uma pequena coorte de pacientes que apresentaram níveis muito reduzidos de
cortisol plasmático após uma média de 6 dias da primeira dosagem ter sido normal ou alta
(inicialmente cortisol médio 42 µg/dl e repetição com nível médio 10 µg/dl); a terapia com
esteroides venosos foi eficaz na retirada de aminas vasopressoras nestes pacientes com
exaustão adrenal nesta pequena coorte.
Permanece desconhecida a relação entre diagnóstico de disfunção adrenal e o
benefício do tratamento com corticoides sintéticos (Japiassú e cols., 2009). Os estudos mais
recentes que avaliaram tanto a função adrenal quanto o tratamento não foram capazes de
identificar relação lógica entre função e resposta ao tratamento (Annane e cols., 2002; Elsouri
e cols., 2006; Morel e cols., 2006; de Jong e cols., 2007; Sprung e cols., 2008). Salluh e
colaboradores realizaram dois estudos sobre a função adrenal e pneumonia comunitária grave:
o cortisol plasmático foi o mais importante biomarcador preditor de mortalidade nesta

13
população; e o tratamento com esteroides sintéticos não parece ter mostrado benefício (Salluh
e cols 2008a; Salluh e cols., 2008b). Outro ponto aberto no estudo da função adrenal e sepse é
a relação entre as diversas citocinas e o cortisol plasmático. O cortisol age como contra-
regulador das ações do fator inibidor de migração de macrófagos (MIF), que age na
amplificação da resposta inflamatória induzindo a produção e liberação de citocinas como
TNF-α e IL-1 (Calandra e cols., 1995). MIF e cortisol apresentaram correlação positiva no
início do quadro de sepse, sendo maior nos pacientes que morrem (Beishuizen e cols., 2001).
Entretanto, outro estudo demonstrou correlação negativa em pacientes adultos e pediátricos
com sepse grave (Emonts e cols., 2007).
1.5 - Resposta inflamatória sistêmica e mediadores solúveis
Após a interação inicial patógeno-hospedeiro e o reconhecimento dos padrões
moleculares de patógenos, há ativação de genes relacionados à resposta inflamatória em
células da imunidade inata, na tentativa de coordenar os mecanismos de defesa tanto
humorais quanto celulares. As células mononucleares desempenham um papel chave neste
processo, tanto na produção de moléculas pró-inflamatórias quanto no desencadeamento de
mecanismos contra-reguladores. Alguns mediadores solúveis liberados nesta ocasião, efetores
da resposta imune, servem como marcadores gerais de atividade ou de funções específicas da
resposta imune.
Fator de Necrose Tumoral (TNF) e Interleucina-1 beta (IL-1β)
TNF-α e IL-1 são protótipos de citocinas pró-inflamatórias responsáveis por
muitos dos efeitos fisiopatológicos observados no choque endotóxico. Apesar de seus
receptores serem diferentes, TNF-α e IL-1β agem sinergicamente em várias situações. TNF-α
/IL-1β são liberados após 30-90 min do estímulo e ativam tanto uma segunda leva de
mediadores proteicos e lipídicos e radicais livres de oxigênio, bem como, são responsáveis
pelo aumento da expressão de moléculas de adesão, que vão resultar em migração celular
para os tecido (Thijs & Hack 1995). O TNF-α é produzido por várias células do sistema
imune, como linfócitos T, células NK ou mastócitos, mas tem como sua principal célula
produtora, o macrófago, especialmente após estímulos com LPS. É produzido como um
precursor de 26 kD e clivado em um peptídeo de 17 quilodaltons (kDa), sendo bioativo como
um homotrímero de 51 kDa. As respostas ao TNF-α são mediadas por dois receptores

14
(TNFR) trans-membranas distintos: o TNFR-I, de peso molecular de 55 kDa, também
chamado de p55 (CD 120a), e o TNFR-II, de peso molecular de 75 kDa conhecido como p75
(CD 120b).
Muitos dos efeitos biológicos do TNF-α parecem estar ligados a ativação do
receptor TNFR-I. Entre as principais ações do TNF-α estão: aumento da expressão de
moléculas de adesão na célula endotelial e indução da secreção de quimiocinas por
macrófagos e células endoteliais, levando a migração celular para o sítio de infecção. O
aumento da expressão de fator tecidual (TF) e do inibidor do ativador do plasminogênio (PAI-
1), assim como a inibição da trombomodulina, o que leva a um estado de pró-coagulante
(Hoffman & Cooper, 1995). As alterações hemodinâmicas observadas na sepse, como
diminuição da resistência vascular periférica, aumento da permeabilidade capilar e efeito
inotrópico negativo são observadas após administração de TNF-α. Em adição, TNF-α pode
levar à morte celular por necrose ou apoptose dependendo do tipo celular e do estado
metabólico.
Interleucina-6 (IL-6)
A interleucina 6 (IL-6) é uma glicoproteína de 21 a 28 kDa, originalmente
descrita como uma linfocina derivada de células T, que induzia a transformação de células B
ativadas em plasmócitos. Praticamente todas as células do corpo podem produzir IL-6 após
estímulos adequados, mas as principais produtoras são as células do sistema imune, como
linfócitos T, monócitos/macrófagos, endotélio e neutrófilos. Alguns estímulos importantes
para sua síntese e secreção são: endotoxina, TNF-α, IL-1 e o fator de crescimento de colônias
de granulócitos (G-CSF). Drogas que reduzem a atividade de TNF-α (por exemplo,
anticorpos anti-TNF) diminuem também os níveis séricos de IL-6. Glicocorticoides diminuem
a produção e secreção de IL-6 induzida por LPS, talvez em parte pela inibição mútua de IL-
1β e TNF-α. Em voluntários sadios, a administração de LPS na circulação sistêmica, leva a
um pico plasmático de IL-6 em 2 a 3 horas, retornando ao normal em 5 a 6 horas. Em
situações de injuria mais intensa esta cinética tende a ser mais prolongada (Barton 1997).
A IL-6 atua através da ligação com seu receptor transmembrana (IL-6R), ou com
o receptor solúvel (sIL-6R). O complexo receptor IL-6R, que é o responsável pela
intermediação das diferentes atividades biológicas da IL-6, é composto por duas
glicoproteínas distintas ligadas à membrana celular: uma é o receptor de reconhecimento IL-
6R, de 80 kDa; e a outra é a gp130, que funciona como elemento de transdução de sinais, de

15
130 kDa. O receptor solúvel (sIL-6R) liga-se à IL-6 e prolonga sua meia-vida plasmática.
Este complexo (IL-6/sIL-6R) é capaz de ativar células através da ligação direta com a
proteína gp130. A sinalização intracelular via IL-6R ou sIL-6R se dá pela formação de um
dímero de gp130, que ativa a fosforilação de JAKs (Janus kinase) e facilita o ancoramento
dos fatores STAT-1/STAT-3, com sua subsequente fosforilação. Subunidades monoméricas
de STAT formam homo e heterodímeros translocam-se para o núcleo, iniciando a expressão
gênica. (Jones e cols., 2001).
Entre as ações mais importantes da IL-6 está a indução de proteínas de fase aguda
no fígado, sendo capaz de aumentar, por exemplo, a síntese de proteína C reativa de 10 a 100
vezes. Outra ação importante no contexto da sepse é a ativação do gene para ICAM-1
(intercellular adhesion molecule-1) em células endoteliais, induzindo maior migração de
neutrófilos para o foco infeccioso. A IL-6 é capaz de modular a respostas de linfócitos,
levando à ativação de linfócitos T e diferenciação de linfócitos B. Existem algumas
evidências que a IL-6 inibe a apoptose de neutrófilos, aumentando sua sobrevida. Outra
propriedade da IL-6 é a ativação da fosfolipase A2, o que leva à maior disponibilidade de
ácido araquidônico e formação de eicosanoides.
A IL-6 pode induzir febre (em uma intensidade menor que IL-1 e TNF-α), no
entanto a administração de IL-6 é bem tolerada, não provocando uma resposta "sepsis-like"
tal como outras citocinas pró-inflamatórias. IL-6 induz a síntese de antagonista de receptor de
IL-1 (IL-1ra) e receptor solúvel de TNF-α (sTNFr), bem como inibe a síntese de IL-1 e TNF-
α induzida por LPS (Barton, 1997). Há indícios de ativação da coagulação por IL-6 após a
administração de endotoxina, sendo que, animais tratados com anticorpo neutralizantes anti-
IL-6 tiveram este efeito atenuado (van der Poll e cols., 1994), apesar que estes resultados não
foram confirmados em um estudo recente em humanos (Derhaschnig e cols., 2004).
IL-6 é bom preditor de desenvolvimento de SDOM e de mortalidade hospitalar
em pacientes com trauma grave e sepse (Pinsky e cols., 1993; Bernard e cols., 2001; Frink e
cols., 2009). A IL-6 apresenta cinética de aumento até 2-3 dias após o início de quadro de
SIRS/sepse (Oda e cols., 2005). Pacientes não-sobreviventes permanecem com níveis maiores
de IL-6 do que os sobreviventes, e ainda existe correlação desta citocina com o escore SOFA
(Oda e cols., 2005). Elevações persistentes de IL-6 até a alta hospitalar se associaram com
aumento da mortalidade após 1 ano em pacientes internados por pneumonia comunitária
grave (Yende e cols., 2008).
Interleucina 10 (IL-10)

16
A interleucina 10 é o protótipo da citocina antiinflamatória, e desempenha um
papel fundamental na regulação do processo inflamatório tanto local e quanto sistêmico. Ela é
produzida por linfócitos T CD4+ (Th2), linfócitos T CD8+ e células B,
monócitos/macrófagos, neutrófilos e células epiteliais. É produzida como um monômero de
18-20 kDa, que forma um homodímero em solução. Entre os principais estímulos para sua
produção estão LPS e citocinas pró-inflamatórias, como TNF-α e IL-1β.
A IL-10 se liga ao seu receptor celular, e ativa a fosforilação de JAK1 e tirosina
cinase (TYK) 2 (ambas são Janus family kinases) e a de STAT-3, culminando com inibição
da translocação nuclear de NF-kB. A síntese de IL-10 é inibida por IL-4, IL-1β e IFN-γ (Opal
e cols., 1998). Alguns hormônios exercem um efeito indutor da produção de IL-10, entre
outros, a adrenalina e glicocorticoides.
A IL-10 antagoniza a diferenciação de células T em T helper (Th) 1, em parte por
suprimir a síntese de interferon-γ, IL-12 e IL-18. Sua principal ação na sepse é a supressão da
expressão e síntese de citocinas pró-inflamatórias. IL-1, TNF-α, IL-6, IL-8, IL-12, IL-18, G-
CSF e GM-CSF, MIP-1β (macrophage inhibitor protein-1 beta) e óxido nítrico (NO) são
algumas substâncias que têm sua síntese inibida por IL-10. Animais geneticamente
deficientes para IL-10 exibem uma produção exagerada de TNF-α após estímulo com LPS
(Berg e cols., 1995). Entre os principais efeitos da IL-10 podemos destacar uma potente
indução de desativação de macrófagos e neutrófilos. Além da inibição da síntese de citocinas
pró-inflamatórias, a IL-10 diminui a síntese de radicais livres de oxigênio, NO e
prostaglandinas, bem como, reduz a degranulação e quimiotaxia de neutrófilos e se contrapõe
ao aumento de sobrevida de neutrófilos induzida por TNF. Em relação aos linfócitos, induz
crescimento e diferenciação de linfócitos B em plasmócitos, estimula a síntese de
imunoglobulinas, promovendo a resposta imune humoral.
Muitos dos componentes de uma fase de “imunoparalisia” podem ser atribuídos a
IL-10. Sua administração prévia ao estímulo inflamatório pode reduzir a resposta inflamatória
exagerada, mas se dada posteriormente ao início de uma infecção, pode piorar o prognóstico.
A administração exógena de IL-10 pode ter efeitos variáveis, dependendo do momento do
início do processo inflamatório, da bactéria causadora e do sítio primário de infecção (Opal &
Huber 2000, Oberholzer e cols. 2002).
Os níveis de IL-10 se elevam nos pacientes com SIRS/sepse, acompanhando
outras citocinas como TNF-α, IL-6 e IL-8 (Oberholzer e cols., 2005; Maier e cols., 2007).

17
Níveis elevados de IL-10 podem se manter elevados prolongadamente, chegando a predizer
pior prognóstico na alta de pacientes com pneumonia (Kellum e cols., 2007).
Quimiocinas (MCP-1/CCL2 e IL-8/CXCL8)
Existe um grupo de “citocinas quimiotáticas”, denominadas de quimiocinas, que
regulam o tráfego de vários leucócitos através de interações com seus receptores. As
quimiocinas foram classificadas de acordo com a posição dos resíduos de cisteínas, altamente
conservadas em suas estruturas. Até o momento existem quatro famílias, CXC (um resíduo de
aminoácido não conservado, entre as duas cisteínas na região N-terminal conservada), CC
(duas cisteínas encontram-se em justaposição), C (apenas um único resíduo de cisteína na
região conservada), CX3C (três resíduos não conservados, entre as duas cisteínas) (Baggiolini
e cols. 1997).
As quimiocinas atuam através de seus receptores conhecidos como receptores
com sete regiões transmembranas, estes são acoplados à proteína G e até o momento, foram
identificados 18 receptores para as quimiocinas (Murdoch & Finn 2000). Existe uma relação
promíscua entre esses receptores e as quimiocinas. Uma única quimiocina pode ligar-se a
vários receptores, bem como um único receptor pode promover a sinalização para diferentes
quimiocinas.
Algumas quimiocinas têm papel fisiopatológico na sepse, entre estas nos
deteremos em duas: MCP-1/CCL2 e IL-8/CXCL8.
Monocyte Chemoattractant Protein (MCP) 1/CCL2
A proteína quimiotática para monócitos-1 (MCP-1/CCL2) é uma CC quimiocina,
com 76 aminoácidos, com atividade sobre monócitos, células T, células NK, basófilos e
mastócitos. Foi originalmente identificada como um mediador do recrutamento e ativação
monocitária (Yoshimura e cols. 1989). Em adição à suas propriedades quimiotáticas, relatos
recentes sugeriram que esta quimiocina ativa células da linhagem monocítica, aumentando a
expressão da molécula de adesão CD11b/CD18. O MCP-1/CCL2 tem sido implicada em
doenças caracterizadas por um rico infiltrado monocítico como na aterosclerose, na artrite
reumatoide e na esclerose múltipla (Daly & Rollins 2003, Mahad & Ransohoff 2003,
Sheikine & Hansson 2004).

18
Diferentes autores demonstraram níveis elevados desta quimiocina no plasma de
pacientes sépticos (Bossink e cols., 1995). A administração de E.coli em babuínos resultou
em elevação dos níveis plasmáticos de MCP-1/CCL2 após 2h da administração, atingindo seu
pico entre 4-6 h, os níveis de MCP-1/CCL2 se correlacionaram aos de IL-8 (r=0.826 ) (Jansen
e cols., 1995). A administração exógena da MCP-1/CCL2 protegeu camundongos submetidos
a um modelo de infecção letal por Pseudomonas aeruginosa ou Salmonella typhimurium,
promovendo um controle da infecção e do crescimento bacteriano mais eficientes (Nakano e
cols. 1994).
O mecanismo pelo qual esta quimiocina exerce seus efeitos ainda não é
conhecido, mas a administração da proteína recombinante, em um modelo de endotoxemia,
protegeu estes animais, promovendo um decréscimo nos níveis de IL-12 e TNF,
acompanhado do aumento nos níveis da citocina antiinflamatória IL-10, sugerindo que a
MCP-1/CCL2 interfere no balanço entre citocinas pró e antiinflamatórias (Zisman e cols.,
1997). Já a neutralização da MCP-1/CCL2 resultou num impressionante aumento da
mortalidade em camundongos que receberam injeção de LPS. Entretanto, quando foi
administrada MCP-1/CCL2 recombinante, houve um acentuado aumento na sobrevida desses
animais (Zisman e cols., 1997). Matsukawa e colaboradores (Matsukawa e cols., 2000)
encontraram resultados semelhantes no modelo de peritonite polimicrobiana em
camundongos (CLP), onde a neutralização do MCP-1/CCL2 resultou em aumento de
mortalidade dos animais. Os autores justificam este achado por uma modificação do balanço
de citocinas pró e antiinflamatórias, onde o MCP-1 favorece uma resposta antiinflamatória e
imunomoduladora.
Nós recentemente observamos que animais geneticamente deficientes de MCP-
1/CCL2 são mais suscetíveis a uma dose sub-letal de LPS ou a peritonite polimicrobiana
induzida pela CLP (Gomes e cols., 2006). Esta maior suscetibilidade esteve associada a uma
redução na produção de IL-10 e um aumento na secreção do MIF.
O MCP-1 também parece participar dos efeitos antiinflamatórios da Proteína C
Ativada Recombinante Humana (APC) na sepse (Riewald e cols., 2002; Brueckmann e cols.,
2003; Riewald e cols., 2003). Os efeitos da APC se devem a ativação do receptor de proteína
C de células endoteliais (EPCR) e do receptor ativado por protease-1 (PAR-1). A ativação de
PAR-1 pela APC ocorre de maneira dependente de EPCR que funciona como um co-receptor
para a ativação de PAR-1. De fato, a sinalização pelo PAR-1 leva a indução de genes
protetores pela APC. Entre estes genes induzidos pela proteína C ativada se destaca o gene da

19
MCP-1/CCL2, que é seletivamente ativado por PAR-1 e parece ser um dos candidatos mais
importantes para o efeito da APC na sepse.
Interleucina-8 (IL-8)/CXCL8
A IL-8/CXCL8 foi inicialmente caracterizada como um fator quimiotático para
neutrófilos, a partir de sobrenadante de monócitos humanos estimulados com LPS.
Posteriormente foi purificada e clonada e desde então uma família de quimiocinas
estruturalmente relacionada foi identificada (CXC) (Walz e cols., 1987; Yoshimura e cols.,
1987; Schroder e cols., 1988; Schroder & Christophers, 1989).
Estudos de espectroscopia por ressonância nuclear magnética e cristalografia por
Raios-X sugerem que a IL-8/CXCL8 concentrada ou cristalizada ocorra como homodímero.
No entanto, em concentrações fisiológicas, diferentes autores sugerem que a forma funcional
é monomérica (Burrows e cols., 1994; Paolini e cols., 1994).
IL-8/CXCL8 pode ser produzida por leucócitos como monócitos, células T,
neutrófilos e células NK, bem como por células somáticas, como células endoteliais,
fibroblastos e células epiteliais. A IL-8/CXCL8 não é produzida constitutivamente, mas é
induzida por citocinas pró-inflamatórias como IL-1 e TNF (Strieter e cols., 1990). A IL-
8/CXCL8 também pode ser induzida por bactérias, vírus e seus produtos, o que pode resultar
em elevadas concentrações de IL-8/CXCL8 em fluidos dos sítios de infecção (Meduri e cols.,
1995).
A neutralização da IL-8/CXCL8 inibiu profundamente o recrutamento de
neutrófilos, induzido por LPS no modelo de pleurisia em coelhos (Broaddus e cols., 1994),
indicando que este fator é importante no modelo de inflamação aguda. Alguns trabalhos
demonstraram que durante a sepse há um aumento desta quimiocina no sangue (Marty e cols.,
1994). Estudos demonstraram que a proteína murina KC, foi capaz de induzir os mesmos
efeitos biológicos da IL-8 humana, tais como quimiotaxia de neutrófilos e aumento da
expressão da molécula de adesão CD11b/CD18 por essas células, sendo denominada então de
proteína murina homóloga funcional da IL-8 humana.
IL-8 está associada com o desenvolvimento de disfunção do tecido pulmonar em
pacientes com SIRS/sepse. Donnelly e colaboradores (1993) correlacionaram o aumento nos
níveis desta quimiocina no lavado bronco-alveolar de pacientes, com o desenvolvimento da
SARA. Além disto, demonstraram que os macrófagos alveolares são células importantes na

20
produção de IL-8, para subsequente influxo de neutrófilos. Estudos clínicos demonstraram
um aumento nos níveis da IL-8 no soro de pacientes sépticos (Oberholzer e cols., 2005;
Bozza e cols., 2007; Livaditi e cols., 2006) e nos pulmões de pacientes com a Síndrome da
Angústia Respiratória Aguda (SARA) (Chollet-Martin e cols., 1993; Meduri e cols., 1995;
Wiedermann e cols., 2004).
Interleucina 4 (IL-4)
A interleucina 4 (IL-4) é uma citocina produzida por linfócitos T, mastócitos e
basófilos e estimula a diferenciação e proliferação de células linfocitárias e a secreção de
imunoglobulina E por linfócitos B. Wu e cols (2008) verificaram níveis elevados de IL-4 em
pacientes com sepse grave. Embora os níveis de mRNA para IL-4 tenham sido elevados em
células monucleares de sangue periférico (PBMC) de pacientes que morreram por sepse
grave, os níveis séricos desta citocina no momento da admissão na UTI não se
correlacionaram com mortalidade.
Interleucina 5 (IL-5)
Interleucina 5 (IL-5) é uma citocina produzida por linfócitos T helper e
mastócitos, com função de estimular a diferenciação de células B e incrementar a secreção de
imunoglobulinas. A IL-5 também age na ativação de eosinófilos. O seu gene está localizado
próximo a outros de codificação de IL-3, IL-4 e GM-CSF, que são frequentemente co-
expressados em linfócitos T (van Leeuwen BH e cols., 1989). Esta citocina participa na
resposta inflamatória em vias aéreas de pacientes asmáticos (Bradding e cols., 1994). Seus
níveis em pacientes com sepse podem estar elevados, mas não parecem se associar com o
prognóstico dos pacientes (Bozza e cols., 2007).
Fator estimulante de colônias de granulócitos (G-CSF)
O G-CSF é uma glicoproteína que age como citocina, produzida em diferentes
tecidos para estimular hematopoiese e liberação de granulócitos na corrente sanguínea. O G-
CSF também estimula a diferenciação, função, proliferação e sobrevida de neutrófilos. Regula
estas funções através das vias de transdução/sinalização Janus kinase e Akt. O G-CSF é
produzido por células endoteliais, macrófagos e outras células do sistema imune, e está

21
envolvido em funções como quimiotaxia e fagocitose de neutrófilos. Este fator parece agir na
melhora da resposta microbicida de neutrófilos, incrementando a resposta imune inata
(Terashima e cols., 1995). Níveis elevados de G-CSF podem estar associados a maior
resposta inflamatória e aumento de citocinas em diversos tecidos.
G-CSF foi medido em pacientes com sepse grave e foi detectado em níveis
elevados nos primeiros dias após o início do quadro (Presneill e cols., CCM 2000). O G-CSF
se correlacionou com os níveis de IL-6, mas não foi preditor da evolução para SDOM nem da
mortalidade em 30 dias. Os seus níveis foram significativamente maiores nos pacientes com
choque séptico e apresentou cinética com queda exponencial a partir dos 2 primeiros dias
após o diagnóstico.
Fator inibidor da migração de macrófagos (MIF)
O fator inibidor da migração de macrófagos (MIF) foi originalmente descrito como
um produto de células T ativadas que inibe a migração de macrófagos peritoneais de cobaias
in vitro (David, J, 1966; Bloom & Bennett, 1966). Posteriormente, o MIF foi clonado e a sua
sequência do cDNA obtida (Weiser e cols., 1989). O MIF encontra-se pré-formado em
macrófagos, linfócitos e nas células corticotrópicas da glândula pituitária, sendo liberado em
grandes quantidades na circulação sanguínea após administração de LPS (Berhagen e cols.,
1993; Calandra e cols., 1994). Além disso, o MIF parece desenvolver um papel essencial na
sepse, uma vez que a sua inoculação simultânea com LPS causa um aumento na letalidade e a
sua neutralização com anticorpos, apresenta um papel protetor no choque endotóxico
(Berhagen e cols., 1993; Calandra e cols., 2000; Lehmann e cols., 2001). O MIF estimula a
produção de TNF-α por macrófagos e contrapõem os efeitos inibitórios dos glicocorticoides
na produção de citocinas como TNF-α, IL-6, IL-8 e na ativação celular (Calandra e cols.,
1995; Bacher e cols., 1996). A identificação do MIF como um hormônio de estresse capaz de
reverter os efeitos inibitórios de glicocorticoides, sugere um mecanismo utilizado pelo
sistema nervoso central na regulação da resposta inflamatória sistêmica.
Os estudos experimentais com modelos geneticamente deficientes para MIF
confirmaram o papel central desta citocina na sepse. Estes animais além de serem mais
resistentes que os controles a altas doses de LPS também fazem uma melhor eliminação de
bactérias Gram-negativas (Pseudomonas aeruginosa) instiladas no pulmão (Bozza e cols.,
1999). Apesar do seu papel proeminente como citocina pró-inflamatória, a deficiência do
gene de MIF aumenta a capacidade dos animais em eliminar Pseudomonas aeruginosa

22
instilada em pulmões. Este resultado foi confirmado e estendido com a descoberta que a
neutralização do MIF, com anticorpos, levou a uma melhor taxa de sobrevida em
camundongos com choque séptico induzido por CLP e infecção peritoneal com E coli.
Recentemente, foi demonstrado que o MIF regula a expressão de TLR4, molécula transdutora
de sinal do complexo do receptor de LPS (Roger e cols., 2001). Em modelos experimentais, a
expressão reduzida de TLR4 em macrófagos deficientes em MIF está relacionada a uma
produção diminuída de TNF-α por estas células quando estimuladas por LPS. No entanto,
ainda não foi demonstrado se o MIF regula a expressão de TLR-4 em humanos com sepse.
Em um estudo prospectivo, nós observamos uma correlação entre níveis séricos
maiores de MIF e pior prognóstico em pacientes sépticos (Bozza e cols., 2004). Níveis
plasmáticos de MIF e IL-6 diferiram significativamente entre pacientes que sobreviveram e
que faleceram. Do mesmo modo, uma diferença significativa dos níveis de MIF foi observada
quando se analisou apenas os pacientes do grupo com choque séptico. Beishuizen e cols
(2001), estudando 32 pacientes com choque séptico, observaram uma correlação significativa
entre níveis elevados de MIF na admissão e a letalidade. De fato, a análise com regressão
logística múltipla mostrou que o MIF foi um fator preditor de desfecho adverso em pacientes
com choque séptico. Gando e colegas estudaram 17 pacientes graves com SIRS e observaram
também uma correlação entre níveis maiores de MIF e letalidade (Gando e cols., 2001). No
estudo de Bozza e cols (2004), foram avaliados níveis de MIF e IL-6 prospectivamente, em
uma série de pacientes com sepse. Eles observaram que estas citocinas estão aumentadas em
pacientes com choque séptico e sepse, comparativamente a indivíduos saudáveis. Apesar de
diferenças importantes entre os níveis de MIF e IL-6 entre pacientes que sobreviveram e que
morreram, apenas os níveis de MIF mostraram um bom poder discriminativo para predizer a
letalidade relacionada a sepse. As concentrações elevadas de MIF parecem ser um marcador
precoce de mau prognóstico em pacientes sépticos internados no UTI.
MIF e glicocorticoides têm ações contrarregulatórias e ambos estão elevados
precocemente nos casos de sepse (Gando e cols., 2001; Emonts e cols., 2007). A relação entre
os níveis circulantes de MIF e de cortisol permanece controversa em estudos clínicos. Níveis
séricos de MIF foram correlacionados a maiores níveis de cortisol, demonstrando a integração
entre resposta inflamatória e sistema endócrino (Beishuizen e cols., 2001). Emonts e cols
verificaram, no entanto que os níveis de MIF parecem se correlacionar negativamente com os
de cortisol e positivamente com de ACTH. Maxime e cols (2005) demonstraram que o
tratamento com corticoides sintéticos (hidrocortisona ou metilprednisolona) reduziu os níveis

23
de MIF em sobrenadante de cultura de PBMCs de pacientes com choque séptico, o que pode
explicar o sucesso deste tratamento em parcela de pacientes com choque séptico.
Níveis de MIF permanecem elevados durante os primeiros dias de pacientes com sepse. Os
níveis permaneceram elevados por mais de 10 dias, comparativamente a pacientes internados
por outras razões (Beishuizen e cols., 2001). Níveis crescentes de MIF nas primeiras 48 horas
após o início de sepse grave foram fortemente associados com mortalidade precoce (Chuang e
cols., 2007).
O comportamento do MIF comparativamente a outros biomarcadores só foi melhor
estudado com IL-6, e teve desempenho melhor para predizer mortalidade em 28 dias. A
comparação entre o poder prognóstico do MIF em relação a um painel de citocinas não é
conhecida. Sua relação com cortisol plasmático ao longo do tempo e seus níveis após
tratamento com corticoides sintéticos também não foi explorada.
1.6 - Disfunção mitocondrial na sepse
A hipóxia tecidual é considerada uma das principais causas da supressão metabólica
associada à sepse e pode ser causada por hipoxemia, anemia ou perfusão inadequada.
Recentemente, um novo mecanismo foi proposto para explicar este quadro, chamado de
“hipóxia citopática” (Fink e cols., 2001). Este termo define a relação entre a baixa produção
de ATP e valores de pO2 normais ou até supranormais nos tecidos. Desta forma, mesmo na
presença de níveis adequados de O2 nas proximidades das mitocôndrias, estas não são capazes
de utilizá-lo para desempenhar suas funções normais. Sabe-se que após a reposição volêmica
e o equilíbrio do choque séptico, os níveis de pO2 teciduais são elevados, tanto quanto em
voluntários saudáveis (Boekstegers e cols., 1994). Disfunções de enzimas da cadeia
respiratória, como a citocromo c oxidase, são associadas a altos níveis teciduais de NO, que
também age na fisiopatologia do choque na sepse (Takehara e cols., 1995; Landry, 2001). A
partir da produção aumentada de NO, ocorre maior formação de peroxinitrito que, além de
inativar a citocromo c oxidase e induzir maior lesão de isquemia-reperfusão, inibe a F0F1-
ATPase e a aconitase (enzima do ciclo de Krebs que cataliza a reação de citrato em isocitrato)
(Castro e cols., 1994). A ativação da poliADP ribosil polimerase (PARP), enzima reparadora
de DNA e indutora de apoptose, age como NADase e depleta seus estoques, reduzindo a
fosforilação oxidativa (Zingarelli e cols., 1996).
Diversos mecanismos foram propostos para explicar este fenômeno, incluindo a
diminuição da disponibilidade de substratos para o ciclo de Krebs (como o piruvato), a

24
inibição de enzimas tanto do ciclo de Krebs quanto dos complexos da cadeia transportadora
de elétrons e o colapso do gradiente de prótons resultado do desacoplamento da respiração
mitocondrial (Crouser, 2002a). Assim, acredita-se que a hipóxia citopática aconteça tanto em
pacientes quanto em animais sépticos ou com endotoxemia e que os mecanismos citados
acima estejam envolvidos na fisiopatologia destas doenças (Crouser, 2004).
As primeiras associações estabelecidas entre a má distribuição de O2 tecidual e a
mortalidade de pacientes com sinais clínicos de falência circulatória, incluindo doentes com
falência cardíaca, sepse e hipovolemia, foram feitas há mais de 40 anos (Broder e Weil,
1964). Este estudo correlacionou os altos níveis de lactato sanguíneo com a mortalidade dos
pacientes, e concluiu que a falta de O2 seria a causa primária da lesão tecidual e disfunção
orgânica presentes no choque séptico. Questões relacionadas à validade da medida de lactato
plasmático como reflexo de hipóxia tecidual também surgiram nesta época (Harris e cols.,
1962), embora a associação entre a acidose sanguínea e a mortalidade tenha sido validada em
estudos posteriores (Hodgin & Sanford, 1965).
Outros estudos indicaram a possibilidade dos mecanismos responsáveis pela acidose e
pela oxigenação tecidual serem distintos durante a sepse (Mizock, 1984; van Deuren, 1995).
Estudos mais recentes sustentam a ideia de que a hipóxia tecidual desempenha um papel
importante na patogênese da falência orgânica associada à sepse (Sielenkamper e cols., 2000).
Entretanto, o aumento da oxigenação tecidual na tentativa de evitar o dano isquêmico, se
mostrou ineficaz podendo até mesmo ser prejudicial ao paciente com sepse (Hayes e cols.,
1994; Gattinoni e cols., 1995). Levando em consideração as discrepâncias observadas,
concluiu-se que os não-sobreviventes da sepse exibiam uma diminuição da capacidade de
aumentar o consumo de O2 em resposta ao aumento da disponibilidade tecidual de O2 (Hayes
e cols., 1997). Assim, pelo fato de a mitocôndria ser a principal organela responsável pelo
consumo de O2, postulou-se que a função desta organela estaria comprometida de alguma
forma durante a sepse. As informações disponíveis até o momento são aparentemente
conflitantes uma vez que em alguns casos observa-se uma inibição do consumo de O2,
indicando um efeito sobre os complexos da cadeia transportadora, enquanto que em outros
casos o desacoplamento é prevalente, sugerindo uma perda da eficiência da fosforilação
oxidativa, seja pela ativação de proteínas desacopladoras ou pela abertura de poro de
transição de permeabilidade ou ainda por alterações no metabolismo de adenosina 5´-
difosfato (ADP). A transportadora de elétrons na membrana interna mitocondrial está
representada na Figura 1.1.
Existem 5 estados metabólicos da respiração mitocondrial, a saber:

25
1 – sem consumo de oxigênio, antes da adição de substratos;
2 – adição de succinato (substrato para o complexo II) e início do consumo de oxigênio;
3 – adição de ADP e consumo de oxigênio máximo, associado à síntese de ATP;
4 – exaustão da fonte de ADP e desaceleração do consumo do oxigênio;
5 – cessação do consumo de oxigênio em condições de anóxia.
A disfunção mitocondrial na sepse pode se dar pelo desacoplamento e transição de
permeabilidade mitocondrial, com swelling e desacoplamento mitocondrial em resposta ao
LPS, tanto in vitro quanto in vivo. Estudos experimentais evidenciaram o desacoplamento em
modelos de injeção de endotoxina e choque hemorrágico (Mela e cols., 1971; Boczkowski e
cols., 1999). A respiração no estado 4 (induzido pela adição de inibidor da F1Fo-ATP sintase,
como oligomicina) foi relativamente aumentada em animais com indução da resposta
inflamatória, indicando desacoplamento. Esta alteração pode acontecer em tecidos musculares
(Boczkowski e cols., 1999), hepatócitos (Crouser e cols., 2002b) e no cérebro (D´Ávila e
cols., 2008). A alteração da estrutura morfológica de mitocôndrias hepáticas pode expressar
histologicamente a disfunção da organela, e ocorre por aumento da atividade de proteínas
desacopladoras ou pela abertura do poro de transição de permeabilidade (PTP) mitocondrial;
o tratamento com ciclosporina A, um inibidor da formação do PTP, é capaz de
restaurar/prevenir o dano a ultraestrutura de mitocôndrias (Crouser e cols., 2002b).
Clinicamente, foi demonstrado que estas alterações estruturais ocorrem em pacientes
que morrem com choque séptico, principalmente com desrregulação do controle da glicemia
(Vanhorebeek e cols., 2005).
Outro mecanismo importante é a inibição na atividade de enzimas mitocondriais.
Experimentalmente mostrou-se a redução do consumo de oxigênio em tecido hepático e
muscular em modelos murinos de sepse (Schumer e cols., 1970; Boveris e cols., 2002). A
respiração associada à síntese de ATP foi muito reduzida e a ultraestrutura mitocondrial se
alterou de maneira notável em vários tecidos após indução de sepse experimental, como
músculo esquelético, cardíaco e rim. Redução dos níveis de complexo I e IV e ATP foram
verificados clinicamente em tecidos muscular (Brealey e cols., 2002; Fredriksson e cols.,
2006) e hepático (Vanhorebeek e cols., 2005). Belikova e colaboradores demonstraram
redução do estado 3 (respiração induzida após adição de ADP) em PBMCs de pacientes com
sepse grave e menor expressão de human leukocyte antigen (HLA)-DR em monócitos
circulantes. Calvano e cols (2005) demonstraram que após injeção de endotoxina em

26
voluntários sadios, ocorre redução da expressão de genes codificadores de enzimas da cadeia
respiratória, como complexo I e F1F0-ATP sintase.
A menor quantidade e função de complexos da cadeia respiratória mitocondrial podem
ser efeitos da ação de óxido nítrico (NO) e espécies correlatas como o peroxinitrito (ONOO)
(Poderoso e cols., 1996; Cassina e cols., 1996). Além da hiporreatividade vasomotora
observada na sepse (Landry e cols., 1997), as espécies reativas de oxigênio atuam como
inibidores da atividade mitocondrial (Poderoso e cols., 1996). O estresse oxidativo ocasionou
o acúmulo precoce de produtos de peroxidação lipídica e de carbonilação de proteínas no
cérebro (Barrichello e cols., 2006), o que pode ajudar a explicar o desencadeamento da
encefalopatia ligada à sepse. Mecanismos de proteção anti-oxidativos estavam reduzidos
experimentalmente, como heat shock protein (HSP) 70 e a superóxido dismutase (Mn-SOD)
(Crouser e cols., 2006).

rotenonasuccinato + ADP oligomicin
FCC
ANT
AT
AD
atractilosídeo
potencial transmembrana
matriz
espaço intermembrana
ciclo de
Krebs
Figura 1.1 – Esquema da cadeia transportadora de elétrons na membrana interna mitocondrial.
Em verde, estão os complexos responsáveis pelo transporte de elétrons pela membrana
(complexo I – NADH desidrogenase; II – succinato desidrogenase; III – citocromo c redutase;
IV – citocromo c oxidase; V – F1FoATP sintase) e pelo efluxo de H+ para o espaço inter-
membranas. E amarelo, está representada a F1Fo-ATP sintase. Rotenona e oligomicina agem
como inibidores (seta tracejada) nos complexos I e IV, respectivamente. Succinato e ADP são
substratos para o complexo II. Carbonil cianeto p-(trifluorometoxi)fenilhidrazona (FCCP) é
um ionóforo de membrana que dissipa o gradiente de létrons e desacopla a respiração
mitocondrial (respiração sem síntese de ATP). UQ, ubiquinona; Cyt c, citocromo c; FAD –
dinucleotídeo flavina e adenina; NAD – dinucleotídeo nicotinamida e adenina; ADP –
adenosina difosfato; ATP – adenosina trifosfato.
27

28
Pacientes sépticos e politraumatizados frequentemente desenvolvem sinais de
disfunção orgânica múltipla ainda que os órgãos atingidos não tenham sido diretamente
afetados pelo insulto original. Esta falha orgânica é tradicionalmente atribuída ao efeito dos
mediadores inflamatórios circulantes que induzem modificações na circulação que resultam
em hipóxia e dano celular. Paradoxalmente, os tecidos oriundos de órgãos disfuncionantes
demonstram um perfil histológico frequentemente normal, com baixa incidência ou ausência
de células apoptóticas ou necróticas. Estes resultados sugerem que o comprometimento
orgânico é mais funcional do que estrutural e, portanto, potencialmente reversível (Brealey e
cols., 2004). A gravidade da sepse é acompanhada pela redução na utilização de oxigênio
embora os níveis teciduais deste gás não sejam alterados, sugerindo que o problema reside na
inibição do consumo de oxigênio e não na diminuição da perfusão tecidual. Entretanto a
natureza precisa do mecanismo pelo qual o consumo de oxigênio é inibido em pacientes
sépticos permanece obscura, já que a síntese de ATP parece estar reduzida na sepse,
colaborando para manutenção da SDOM.

29
2 - Objetivos
2.1 - Objetivo geral
- Estudar aspectos de inflamação e disfunções orgânicas (biomarcadores clínicos, citocinas,
cortisol e respiração mitocondrial) relacionados à gravidade e temporalidade da evolução da
Síndrome de Disfunção Orgânica Múltipla em pacientes com choque séptico.
2.2 - Objetivos específicos
- Analisar sequencialmente diferentes biomarcadores em pacientes com choque séptico;
- Analisar os níveis de diversos biomarcadores e a mortalidade precoce e mortalidade em 28
dias;
- Correlacionar os níveis de biomarcadores com o grau de disfunções orgânicas;
- Verificar se ocorre alteração da função mitocondrial de monócitos circulantes em pacientes
sépticos, e correlacionar com o prognóstico;
- Analisar possíveis mecanismos de disfunção mitocondrial em monócitos circulantes de
pacientes sépticos, comparativamente a pacientes controles.

30
3. Painel de biomarcadores em pacientes com choque séptico
3.1 – Introdução
Biomarcadores podem ser utilizados tanto para o diagnóstico quanto para o
acompanhamento do tratamento da sepse. Diferentes biomarcadores já foram estudados e se
correlacionam de modo distinto com o diagnóstico, com disfunções orgânicas específicas e
com o prognóstico da sepse. A interrelação entre vários dos biomarcadores clinicamente
utilizados e outros estudados experimentalmente não é bem conhecida. Um painel de
citocinas, quimiocinas e outros biomarcadores podem ajudar na detecção precoce da sepse,
auxiliando o diagnóstico e permitindo o início precoce do tratamento (Marshall & Reinhart,
2009). Este painel também pode ser útil para correlações com disfunções orgânicas
específicas e predizer o prognóstico a curto e médio prazo. É possível que um biomarcador
tenha bom desempenho para o diagnóstico da sepse, enquanto outro biomarcador seja melhor
para estratificação de gravidade ou predição de prognóstico. Dificilmente um biomarcador
isoladamente vai desempenhar boa acurácia para diagnóstico, análise de gravidade e
prognóstico simultaneamente. Finalmente, a visão mais ampla da resposta inflamatória
plasmática pode também fornecer conhecimento sobre a fisiopatologia da sepse grave, e
desvendar controvérsias tal como a do desequilíbrio entre resposta pró-inflamatória e
antiinflamatória, na figura dos conceitos de SIRS (Systemic Inflammatory Response
Syndrome) e CARS (Compensatory Antiinflammatory Response Syndrome).
Este estudo se destina a medir os níveis plasmáticos de 17 citocinas em pacientes com
sepse e choque séptico, simultaneamente através de uma plataforma multiplex, e correlacionar
os resultados com parâmetros clínicos avaliados no dia-a-dia do acompanhamento do paciente
séptico na UTI e melhorar a predição do prognóstico. Posteriormente, outros biomarcadores
são adicionados em população de maior gravidade (choque séptico) e faz-se o
acompanhamento através de medidas ao longo da primeira semana após o diagnóstico da
sepse. Através da cinética de parâmetros clínicos e citocinas, tenta-se encontrar padrões de
respostas inflamatórias associadas à mortalidade a curto e médio prazo.

31
3.2 – Métodos
3.2.1 – Pacientes
Foram incluídos pacientes com sepse grave ou choque séptico a partir das primeiras
24 horas do início do quadro, internados nas UTIs do Hospital Universitário Clementino
Fraga Filho (UFRJ), Hospitais Espanhol, Barra e Quinta D´Or e Casa de Saúde São José (Rio
de Janeiro), de acordo com os critérios de SIRS/sepse de Bone e cols., 1992 (Quadro 1), bem
como pacientes controle (internados em UTI com outros diagnósticos que não sepse ou
qualquer tipo de choque).
Quadro 3.1 – Critérios de SIRS/sepse, segundo Bone e cols., 1992.
1. Síndrome da Resposta Inflamatória Sistêmica (SIRS)
Dois ou mais dos seguintes critérios devem estar presentes :
Temperatura > 38o ou < 36o C
Frequência cardíaca > 90 batimentos por minuto
Frequência respiratória > 20 por minuto ou PaCO2 < 32 mmHg
Leucócitos > 12.000 ou < 4.000 mm3 ou > 10 % de bastões
2. Sepse
Critérios de SIRS , com a fonte de infecção presumida ou definida
3. Sepse grave
Sepse, disfunção orgânica, hipotensão ou hipoperfusão (incluindo, mas não se limitando, a
acidose láctica, oligúria ou alteração do estado mental)
4. Choque séptico
Hipotensão arterial – pressão arterial sistólica > 90 mmHg ou redução < 40 mmHg do usual
(apesar de reposição volêmica) – e alterações de perfusão tecidual e uso de vasopressores
Excluiu-se pacientes grávidas, menores de 18 anos ou que morreram com menos de 6
horas de permanência na UTI. Pacientes com neoplasias avançadas ou hematológicas e em
uso de quimioterapia também foram excluídos. Eles foram acompanhados clinica e
laboratorialmente através de coleta dos seguintes dados (pior valor dentro do mesmo dia):
pressão arterial, uso e dose de aminas vasopressoras, frequência cardíaca, frequência
respiratória, uso de ventilação mecânica, temperatura axilar, escala de coma de Glasgow,
leucograma, hematócrito, plaquetometria, dosagem de creatinina, bilirrubinas totais, gases

32
arteriais. Estes dados foram analisados também através de escores prognósticos: Simplified
Acute Physiology Score (SAPS) II ou Acute Physiology and Chronic Health Evaluation
(APACHE) II no 1o dia de entrada no estudo e Sequential Organ Failure Assessment (SOFA)
a cada 48 horas na primeira semana de choque séptico (Le Gall e cols., 1993; Vincent e cols.,
1996).
Materiais para culturas das fontes prováveis de infecção e hemoculturas (pelo menos 2
coletas de 10 ml de sangue de sítios de punção diferentes) foram coletados, conforme
recomendações do Centers for Disease Control and Prevention (CDC) (Horan e cols., 2008).
A coleta de culturas foi direcionada de acordo com os prováveis sítios de infecção,
determinados pelos exames clínicos e laboratoriais dos pacientes. O tratamento com
antibióticos foi decidido pelos médicos da UTI, conjuntamente com as orientações da
Comissão de Controle de Infecção Hospitalar.
Após a assinatura do consentimento pelo paciente ou por seu responsável, foram
coletadas amostras de 10 ml de sangue dos pacientes com sepse nos dias 1, 3, 5 e 7 após a
entrada no estudo. Os pacientes foram acompanhados até a alta hospitalar ou óbito.
Analisamos a letalidade precoce (até 3 dias após início do choque séptico) e em 28 dias. O
estudo foi aprovado pelo Comitê de Ética em Pesquisa do Hospital Universitário Clementino
Fraga Filho (UFRJ), sob o número 69/2004. O termo de consentimento pós-informado
encontra-se em anexo.
3.2.2 – Dosagem de macrophage migration inhibitory factor (MIF)
Os níveis de MIF no plasma foram determinados pelo método Enzyme Linked Immuno
Sorbent Assay (ELISA), utilizando-se pares de anticorpos comercializados pela R&D e/ou
Pharmingen.
3.2.3 - Ensaio de citocinas Multiplex (Luminex®)
O ensaio é realizado com a coleta de sangue, com separação de plasma por
centrifugação (2000 rpm, por 15 minutos). Com o kit de citocinas (interleucina [IL] 1 beta,
IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17, interferon [IFN] gama,
granulocyte colony-stimulating factor [G-CSF], granulocyte-macrophage colony-stimulating
factor [GM-CSF], monocyte chemoattractant protein [MCP] 1, macrophage inflammatory
protein-1 [MIP] beta e tumour necrosis factor [TNF] alfa) foi realizado o ensaio de acordo

33
com as instruções do fabricante (Bio-Rad, Hercules, CA, USA). De modo breve, amostras (50
µL) diluídos em tampão para plasma, foram colocados em placas de 96 poços com filtro. As
amostras foram incubadas com microesferas ligadas a anticorpo (50 µL em 2.000
esferas/poço) em temperatura ambiente por 30 minutos, em agitador de placa (300 rpm), em
ambiente escuro; seguido de 3 lavagens com solução contendo anticorpos secundários (25
µL/poço) adicionados aos poços e depois incubados em temperatura ambiente em agitador de
placas, por mais 30 minutos. Estreptavidina-PE (16 µg/ml em tampão do ensaio) foram
adicionados aos poços (50 µL), e incubada em temperatura ambiente por mais 10 minutos.
Proteínas não associadas às microesferas foram filtradas pelos poços, usando vácuo e nova
lavagem com tampão foi realizada. Após a última lavagem, 125 µL de tampão foi adicionado
em cada poço e nova incubação foi feita com agitador de placa por 1 minuto a 500 rpm e
depois por 3 minutos a 300 rpm. A análise dos dados foi realizada de acordo com o software
Bio-Plex Manager (Bio-Rad). A detecção de citocinas exibe altos graus de precisão intra e
inter-ensaios (10 a 20% de variação). A detecção de citocinas pelo Luminex xMAP é
comparável a ELISA (correlação r entre 0,75 a 0,99); a detecção de IL-6 em 31 pacientes
sépticos obteve correlação 0,815 e p<0,001 (Vignali e cols., 2003).
3.2.4 - Cortisol plasmático
O cortisol plasmático foi medido através de ensaio imunoenzimático (Vidas-cortisol,
Biomérieux, Lyon, France), com limite de detecção de 195 µg/dl.
3.2.5 - Proteína C Reativa Titulada (PCRt)
Os níveis de PCRt foram medidos através de método nefelométrico – kit comercial
Minineph, The Binding Site Group Ltd, Birmingham, UK, com limite de detecção 112 mg/l
na diluição 1/40.
3.2.6 - Análise estatística
As análises estatísticas foram realizadas com os programas SPSS para Windows,
versão 11.0 (SPSS Inc., Chicago, IL, USA) e GraphPad Prism para Windows, versão 4.0
(GraphPad Software, San Diego, CA, USA). As variáveis numéricas demográficas foram
expressas como mediana e intervalo interquartil (IQ 25-75%). Os valores das citocinas,

34
cortisol plasmático e PCRt foram mostradas como média e limites mínimo e máximo. Todas
as variáveis numéricas foram testadas para verificar a distribuição dos valores (teste de
normalidade). Se havia distribuição normal, usamos o teste t de Student para comparar as
médias, e com distribuição não-paramétrica aplicou-se o teste de Mann-Whitney. A
comparação de 3 ou mais grupos foi realizada com a análise de variância (ANOVA).
Variáveis categóricas foram analisadas com o teste qui-quadrado ou teste de Fisher.
Testamos as diferenças dos níveis de cada citocina e de cortisol e PCRt ao longo do 1º,
3º, 5º e 7º dias de choque séptico para avaliar a cinética (teste ANOVA ou Kruskall-Wallis), e
posteriormente comparamos as cinéticas entre sobreviventes e não-sobreviventes (desfecho
em 28 dias). Para efeito de comparação com dados clínicos, a evolução do escore SOFA
também foi demonstrada com as figuras de cinética dos biomarcadores.
O desfecho de interesse principal para a comparação dos níveis de biomarcadores foi a
mortalidade em 28 dias; secundariamente, os biomarcadores no subgrupo com mortalidade
precoce (definida com aquela ocorrida até 3 dias após o início do choque séptico) foram
analisados. Comparamos também os níveis de citocinas entre pacientes controles,
sobreviventes e não-sobreviventes do choque séptico (material suplementar). Fizemos
correlações entre os níveis dos biomarcadores no 1º dia de choque séptico e o grau de
disfunções orgânicas (escore SOFA), e entre os níveis de MIF e de cortisol plasmático (teste
de Pearson para variáveis de distribuição normal ou Spearman para variáveis não-
paramétricas). Para analisar os dados de cinética, testamos as comparações entre as médias e
os valores máximos de citocinas, cortisol e PCRt nos dias 1, 3, 5 e 7 de acordo com o
desfecho em 28 dias.
Curvas Receiver Operating Characteristic (ROC) foram construídas plotando valores
de sensibilidade e a diferença da unidade para a especificidade e calculando a maior área
abaixo da curva (AUROC). As áreas sob a curva ROC para mortalidade precoce e em 28 dias
foram calculados para biomarcadores e escores SAPS II e SOFA. As análises foram
realizadas com níveis no 1º dia de choque séptico, níveis médios e máximos de cada
parâmetro. O desempenho foi considerado bom se a AUROC fosse maior que 0,70. O cálculo
de ponto de corte foi calculado para os marcadores com AUROC acima de 0,65, através do
valor do parâmetro que apresentava o maior produto de sensibilidade vezes especificidade.
Realizamos regressão logística para identificar fatores independentemente associados
com a mortalidade em 28 dias, escolhendo na análise univariada os biomarcadores que foram
diferentes entre os grupos sobrevivente e não-sobrevivente em 28 dias (p valor inferior a 0,2)
e outros parâmetros que poderiam influenciar a mortalidade por choque séptico (idade e

35
escores SAPS II e SOFA). Razão de chances e intervalo de confiança de 95% foram
calculados para os fatores independentemente associados com a mortalidade em 28 dias.
Considerou-se que havia significância estatística se o p valor foi menor que 0,05.

36
3.3 – Resultados
3.3.1 – Perfil de citocinas como marcadores de gravidade na sepse: análise multiplex
(artigo publicado na revista Critical Care em 2007, volume 11, página R49)
3.3.1.1 - Características dos Pacientes
Sessenta pacientes foram incluídos; a mortalidade em 28 dias foi 48%. A idade foi de
64 anos (mediana, IQ 51-75), com predomínio do sexo masculino (60%). O escore SOFA no
1º dia foi 9 pontos (IQ 6-11), e 46 (76%) pacientes apresentavam choque séptico na inclusão
do estudo. O principal sítio de infecção foi pulmonar (60%); as culturas identificaram
microorganismos em 50 (83%) pacientes, com predomínio de bactérias Gram-negativas
(76%).
3.3.1.2 - Níveis plasmáticos de citocinas
O sistema multiplex verificou níveis detectáveis de citocinas em 69% das amostras.
Concentrações das interleucinas 6, 8 e 10 e MCP-1 foram detectadas em mais de 95% dos
ensaios. Os níveis de citocinas foram significativamente maiores nos pacientes com choque
séptico, quando comparadas aos pacientes com sepse grave (Tabela 3.1).
3.3.1.3 - Desempenho de citocinas para predição da mortalidade precoce e em 28 dias
A mortalidade precoce foi de 15%. Nestes pacientes, os níveis das citocinas IL-1β, IL-
4, IL-6, IL-8, MCP-1 e G-CSF apresentaram área sob a curva ROC acima de 0,70, com boa
discriminação para mortalidade precoce (Tabela 3.2).
Compararam-se os níveis de citocinas entre sobreviventes (n=31) e não-sobreviventes
(n=29). Pacientes sobreviventes apresentaram níveis significativamente menores das
seguintes citocinas: IL-1 beta (0,39 vs 1,30 pg/ml, p=0,05), IL-4 (0 vs 0,84 pg/ml, p=0,04),
IL-6 (1957 vs 6254 pg/ml, p=0,01), IL-8 (94,7 vs 281,4 pg/ml, p=0,001) e MCP-1 (268,4 vs
757,8 pg/ml, p=0,004).
Em relação à mortalidade até 28 dias, as citocinas que tiveram os melhores
desempenhos foram IL-8 e MCP-1, com áreas sob a curva ROC acima de 0,70 (Tabela 3.3).

37
Tabela 3.1 – Concentrações de citocinas em pacientes com sepse grave e choque séptico.
Valores são expressos em pg/ml e média (limites mínimo e máximo).
Citocina Sepse Grave
(n = 14)
Choque Séptico
(n = 46)
p valor
IL-1β 0,17 (0,00–0,79) 1,22 (0,01–7,33) 0,01
IL-6 1027(583–4854) 5632 (1889–12170) 0,007
IL-7 0,00 (0,00-0,00) 8,47 (0,60–13,56) <0,001
IL-8 52,6 (24.2–122,4) 145,3 (74,4–520,2) 0,01
IL-10 2,27 (0,95–11,72) 27,4 (6,8–116,3) <0,001
IL-13 0,27 (0,00–4,61) 7,21 (0,03–19,29) 0,008
IFN-γ 0,0 (0,0–22,8) 33,1 (0,0–116,7) 0,03
MCP-1 6,3 (0,0–372,2) 753,9 (324,6–1689,0) <0,001
Tabela 3.2 – Desempenho das citocinas para predição de mortalidade precoce. AUROC –
área sob a curva ROC.
Citocina AUROC (IC 95%) p valor
IL-8 0,78 (0,62–0,93) 0,01
IL-4 0,77 (0,59–0,94) 0,01
IL-6 0,76 (0,60–0,91) 0,01
MCP-1 0,74 (0,57–0,90) 0,02
G-CSF 0,73 (0,53–0,92) 0,04
IL-1β 0,71 (0,53-0,90) 0,04
Tabela 3.3 – Desempenho das citocinas para predição de mortalidade em 28 dias. AUROC –
área sob a curva ROC.
Citocina AUROC (IC 95%) p valor
IL-8 0,75 (0,63–0,88) 0,001
MCP-1 0,71 (0,59–0,84) 0,004
IL-6 0,68 (0,55–0,82) 0,01
IL-1β 0,65 (0,50–0,79) 0,05

38
Após análise multivariada através de regressão logística, o escore APACHE II e o logaritmo
dos níveis de MCP-1 foram independentemente associados com a mortalidade em 28 dias. A
razão de chances para mortalidade em 28 dias foi 1,41 (IC 95% 1,02-1,93) para o logaritmo
dos níveis de MCP-1 e 1,37 (IC 95% 1,12-1,66) para o escore APACHE II.
3.3.2 – Cinética de biomarcadores em pacientes com choque séptico
3.3.2.1 - Características dos Pacientes
Setenta e dois pacientes foram incluídos no estudo, porém 5 não foram incluídos na
análise final por conta de diagnóstico alternativo à sepse (2) e coleta de amostras clínicas
incompleta (3). As características gerais do grupo de 67 pacientes sépticos estão na Tabela
3.4. A idade foi de 69 anos (mediana, IQ 55-81), sendo 49% do gênero masculino. Patologias
cirúrgicas predominaram e foram responsáveis por 46 (68%) admissões desta população,
enquanto causas clínicas ocorreram em 21 (32%) dos pacientes. A maioria das internações
cirúrgicas foi por problemas gastrointestinais (84%) ou de vias biliares (7%), seguidas de
cirurgias torácicas (5%), ortopédicas (3%) e vasculares (1%). Em relação às causas clínicas,
insuficiência respiratória por pneumonias e doença pulmonar obstrutiva crônica foi mais
comum (47%); infecções de partes moles, leptospirose, coma e queimaduras extensas também
foram causas de internações.
O SAPS II mediano foi 53 pontos (IQ 45-60), com probabilidade óbito calculada em
53%. O escore SOFA no primeiro dia do choque séptico foi 9 pontos (IQ 6-11), com elevada
necessidade de suporte com ventilação mecânica invasiva (96%) e métodos dialíticos (36%).
O tempo mediano de internação na UTI foi 18,5 dias (IQ 10-31) e no hospital 33 dias (IQ 18-
64). A mortalidade neste grupo foi 10% nos primeiros 3 dias após o diagnóstico, 46% até 28
dias e 57% no hospital.
Infecções abdominais foram as responsáveis por sepse em 33 (49%) pacientes; outros
sítios de sepse foram o pulmonar (34%), bacteremia primária (9%), pele/tecidos moles (5%) e
trato urinário (3%). Houve isolamento de microorganismos em culturas coletadas em 48
(72%) pacientes. O predomínio de isolamento foi de bactérias Gram-negativas (78%),
seguido por Gram-positivas (29%) e isolamento de Candida sp em hemoculturas de 1 (2%)
paciente.

39
Tabela 3.4 – Características demográficas e clínicas de pacientes com choque séptico de
acordo com o desfecho em 28 dias. Valores são expressos como mediana (intervalo
interquartil). *-p<0.05, teste qui-quadrado ou teste t Student.
Todos pacientes
(n = 67)
Sobreviventes
(n = 36)
Não-sobreviventes
(n = 31)
Idade (anos) 69 (55-81) 70 (50-80) 69 (59-82)
Gênero masculino 33 (49%) 18 (50%) 15 (48%)
SAPS II (pontos) 53 (45-60) 51 (41-56) 58 (52-62)*
SOFA no 1º dia (pontos) 9 (6-11) 9 (6-10) 10 (8-13)*
Sítio de infecção
Abdominal
pulmonar
Sanguineo
Pele/tecidos moles
Trato urinário
37
21
5
2
2
20
11
3
1
1
17
10
2
1
1
Tipo de admissão
(clínica/cirúrgica)
18/49 10/26 8/23
Tempo de permanência
na UTI (dias)
18 (10-31) 31 (21-48)* 10 (4-15)
Tempo de permanência
no hospital (dias)
33 (18-64) 63 (41-93)* 18 (8-27)

40
3.3.2.2 - Níveis plasmáticos de citocinas e cortisol
Dez das dezessete citocinas apresentaram níveis de detecção no sistema multiplex
acima de 90% das 236 amostras de 67 pacientes sépticos e 11 pacientes controles: IL-5, IL-13
e IFN-γ (100%); IL-1β (99,5%); IL-8 (98,5%); IL-10 e MIP-1β (97,2%); IL-6 e G-CSF
(92,5%); e MCP-1 (91,6%).
Os níveis de citocinas e o escore SOFA foram maiores em pacientes com choque
séptico do que em pacientes controles (em período pós-operatório) (Figura 3.1). Observamos
que o comportamento dos biomarcadores varia quando se analisa a presença ou o prognóstico
da sepse. Algumas das citocinas demonstraram diferença significativa entre controles e
pacientes sépticos, embora não se faça diferenciação entre pacientes sépticos que
sobreviveram ou não (por exemplo, IL-10 e G-CSF). Escore SOFA e níveis de MIF, IL-6, IL-
8 e MCP-1 foram significativamente maiores em pacientes sépticos em relação aos controles,
além de aumentarem mais ainda naqueles que não sobreviveram.

controle sobrev não-sobrev0
10
20
SOFA
(pontos)
controle sobrev no-sobrev0
2500
5000
7500
10000
MIF
(pg/ml)
controle sobrev no-sobrev0
500
1000
1500
MCP-1
(pg/ml)
controle sobrev no-sobrev0
50
100
150
200
250
300
350
MIP-1b
(pg/ml)
controle sobrev no-sobrev
1
3
5
7
IL-1b
(pg/ml)
controle sobrev no-sobrev1.5
2.0
2.5
3.0
3.5
4.0
4.5
IL-5
(pg/ml)
p<0,001 p<0,001
p=0,003 p=0,21
p=0,20p=0,33
Figura 3.1 – Comparação dos níveis de escore SOFA e citocinas nas primeiras 24 horas de
inclusão no estudo entre pacientes controles pós-operatórios eletivos, sépticos sobreviventes e
sépticos não-sobreviventes em 28 dias. Legenda: sobrev – sobreviventes; não-sobrev – não-
sobreviventes.
41

controle sobrev no-sobrev0
5000
10000
15000
IL-6
(pg/ml)
controle sobrev no-sobrev0
400
800
1200
1600
IL-8
(pg/ml)
controle sobrev no-sobrev0
25
50
75
100
IL-10
(pg/ml)
controle sobrev no-sobrev0.0
0.5
1.0
1.5
2.0
2.5
IL-13
(pg/ml)
controle sobrev no-sobrev0
500
1000
1500
IFN-gamma
(pg/ml)
controle sobrev no-sobrev0
2500
5000
7500
10000
12500
G-CSF
(pg/ml)
p=0,14 p=0,005
p=0,37p=0,002
p=0,01p=0,03
Figura 3.1 – Comparação dos níveis de escore SOFA e citocinas nas primeiras 24 horas de
inclusão no estudo entre pacientes controles pós-operatórios eletivos, sépticos sobreviventes e
sépticos não-sobreviventes em 28 dias. Legenda: sobrev – sobreviventes; não-sobrev – não-
sobreviventes.
42

43
Em relação à cinética de biomarcadores em pacientes com choque séptico, as
interleucinas 5, 6, 8 e 10, MCP-1, MIP-1β e G-CSF demonstraram níveis significativamente
diferentes durante a cinética da primeira semana de choque séptico, quando se analisa os
níveis no grupo total de pacientes (p<0,05, teste ANOVA) (Tabela 3.5).
Todas as interleucinas (exceto IL-6), assim como MCP-1 e MIP-1β, apresentaram
níveis máximos da cinética no 3º dia de choque séptico. Uma queda exponencial foi vista na
análise de outras citocinas entre os dias 1 e 7 (exemplo, IL-6). No caso de outras citocinas,
valores estáveis permaneceram durante todo o período (exemplo, MIF). Assim, pudemos
identificar 4 padrões de comportamento de cinética no grupo: (A) queda exponencial a partir
do 1º dia (IL-6, IL-13, G-CSF e IFN-γ); (B) curva Gaussiana, com pico dos níveis de
citocinas em torno do 3º ou 5º dia (IL-1β, IL-5 e IL-10, MIP-1β, MCP-1 e cortisol); (C) níveis
crescentes durante a 1ª semana (IL-8); (D) níveis estáveis ao longo da 1ª semana (MIF)
(Figura 3.2).
3.3.2.3 - Níveis plasmáticos dos biomarcadores para mortalidade em 28 dias
A mortalidade em 28 dias foi 46%. Os biomarcadores se comportaram de modo
diferente quando se analisa apenas os níveis do 1º dia ou quando se vê a cinética como parte
da predição também. Na análise com valores plasmáticos de biomarcadores no 1º dia de
choque séptico, MIF e PCRt foram os biomarcadores que se diferenciaram entre
sobreviventes e não-sobreviventes (Tabela 3.6). Níveis de MIF em sobreviventes foram
menores que não-sobreviventes (2496 vs 3681 pg/ml, p=0,03). Os níveis de PCRt foram
menores em sobreviventes também (179,0 vs 316,8 mg/l, p=0,01).

44
Tabela 3.5 – Níveis plasmáticos de citocinas e cortisol nos dias 1, 3, 5 e 7 após início de
choque séptico. Os valores das citocinas estão expressos em média e intervalo de valores
mínimo e máximo; unidade = pg/ml.
Citocina Dia 1
(n = 67)
Dia 3
(n = 51)
Dia 5
(n = 38)
Dia 7
(n = 31)
p valor
IL-1β 3,69
(1,43-114,2)
6,01
(1,44-109,4)
1,71
(1,48-2,79)
1,70
(1,34-3,09)
0,61
IL-5 4,39
(1,59-165,6)
7,41
(1,56-280,6)
2,02
(1,63-4,07)
1,98
(1,63-3,01)
0,02
IL-6 4090
(0-187923)
965
(0-21901)
205
(0-2254)
59
(0-707)
<0,001
IL-8 292,6
(0-7268,0)
363,6
(0-7268,0)
55,3
(0-287,2)
50,5
(0-632,4)
0,01
IL-10 32,6
(0-1082,0)
161,6
(0-7279,0)
4,1
(0-38,0)
2,9
(0-46,5)
0,004
IL-13 7,72
(0-428,30)
8,39
(0,04-
315,70)
1,25
(0,07-6,07)
1,21
(0,11-6,47)
0,54
IFN-γ 258,6
(0-6761,0)
278,4
(0-8554,0)
94,2
(0-325,1)
91,6
(0-382,8)
0,42
MCP-1 228
(0-1347)
297
(0-7408)
97
(0-677)
186
(0-3709)
<0,001
MIP-1β 22,6
(0-162,1)
26,5
(0-518,7)
15,8
(0-117,3)
15,4
(0-214,1)
0,008
G-CSF 1020,0
(0-11377,0)
461,7
(0-6628,0)
12,2
(0-121,6)
13,2
(0-211,5)
0,002
MIF 3056
(218-9293)
3026
(470-7186)
3169
(143-12725)
2883
(884-8676)
0,94
Cortisol 48,5
(3,4-195,0)
56,0
(1,4-195,0)
- 39,5
(1,7-195,0)
0,09

0 2 4 6 82000
3000
4000
dias
MIF (pg/ml)
0 2 4 6 80
1000
2000
dias
G-CSF (pg/ml)
0 2 4 6 80.0
2.5
5.0
7.5
10.0
dias
IL-1b (pg/ml)
0 2 4 6 80
2000
4000
6000
8000
dias
IL-8 (pg/ml)
A
B
C
D
Figura 3.2 – Padrões de cinética de citocinas nos dias 1, 3, 5 e 7 após início de choque
séptico. (A) padrão de queda exponencial a partir do 1 dia (IL-6, IL-13, G-CSF e IFN-γ); (B)
curva Gaussiana, com pico dos níveis de citocinas em torno do 3º ou 5º dia (IL-1β, IL-5 e IL-
10, MIP-1β, MCP-1 e cortisol); (C) níveis crescentes durante a 1ª semana (IL-8); (D) níveis
estáveis ao longo da 1ª semana (MIF).
45

46
Tabela 3.6 – Comparação dos níveis de citocinas, proteína C reativa e cortisol no 1º dia de
choque séptico entre sobreviventes e não-sobreviventes. As unidades dos valores de citocinas
estão expressas em pg/ml, dos valores de cortisol em µg/dl e proteína C reativa em mg/l.
Citocina Sobreviventes
(n = 38)
Não-sobreviventes
(n = 31)
p valor
IL-1β 1,84 (1,44-3,67) 6,16 (1,43-114,20) 0,29
IL-5 1,81 (1,59-2,58) 1,95 (1,59-4,42) 0,28
IL-6 886 (0-8228) 1754 (0-14162) 0,22
IL-8 180 (0-2047) 457 (6-7268) 0,1
IL-10 8.6 (0-88) 26,7 (0-334,5) 0,17
IL-13 0,83 (0-6,48) 0,62 (0,05-2,45) 0,85
IFN-γ 140,7 (9,1-749,4) 186,7 (0-1368) 0,26
MCP-1 176,2 (0-1265) 314,6 (0-1347) 0,36
MIP-1β 24,9 (0-162.1) 19,9 (0-118,4) 0,35
G-CSF 989 (0-5370) 1171 (0-11377) 0,69
MIF 2496 (218-7651) 3681 (460-9293) 0,03
Cortisol 38,3 (3,4-132,0) 58,1 (3,8-195) 0,06
PCRt 179,0 (26,7-337,0) 316,8 (51-1250) 0,01

47
Uma vez que se analisam parâmetros da cinética dos biomarcadores, um painel
distinto de diferenças significativas em relação à mortalidade em 28 dias é visto. A média dos
níveis dos biomarcadores nos 1º, 3º, 5º e 7º dias é maior de modo geral naqueles que não
sobrevivem até o 28º dia, sendo que médias significativas maiores são encontradas para IL-6,
IL-8, IL-10, cortisol e PCRt (Tabela 3.7). Os valores médios em sobreviventes e não-
sobreviventes significativamente diferentes foram respectivamente: IL-6 351 vs 2282 pg/ml
(p=0,05); IL-8 83,8 vs 602,6 pg/ml (p=0,03); IL-10 61 vs 555 pg/ml (p=0,05); cortisol 34,6 vs
74,6 µg/dl (p<0,001); e PCRt 170,6 vs 293,2 mg/l (p=0,03).
Outra análise realizada foi do nível máximo dos biomarcadores durante a 1a semana
de choque séptico (Tabela 3.8). Novamente os níveis máximos são maiores no grupo de não-
sobreviventes, com exceção apenas de IL-5. Os biomarcadores com níveis plasmáticos
máximos significativamente maiores no grupo não-sobrevivente foram cortisol (100,5 vs 46,3
µg/dl, p<0,001) e PCRt (381,0 vs 250,5 mg/l, p=0,04).

48
Tabela 3.7 – Comparação da média dos níveis plasmáticos de citocinas, cortisol e proteína C
reativa nos dias 1, 3, 5 e 7 após início de choque séptico. As unidades dos valores de citocinas
estão expressas em pg/ml, dos valores de cortisol em µg/dl e proteína C reativa em mg/l.
Citocina Sobreviventes
(n = 35)
Não-sobreviventes
(n = 26)
p valor
IL-1β 1,73 (1,51-2,70) 2,02 (1,51-4,54) 0,18
IL-5 1,95 (1,65-2,52) 1,88 (1,60-2,72) 0,20
IL-6 351 (0-4114) 2282 (0-16726) 0,05
IL-8 83,8 (1,0-877,6) 602,6 (8,9-3826,0) 0,03
IL-10 61,0 (0,8-635,7) 555,1 (6,3-4855,0) 0,05
IL-13 0,97 (0,12-3,64) 0,81 (0,22-2,65) 0,76
IFN-γ 105,1 (13,6-410,9) 169,2 (13,7-597,5) 0,11
MCP-1 93,3 (0-678,2) 290,6 (0-1393,0) 0,10
MIP-1β 15,7 (0-62,7) 20,2 (0-71,9) 0,30
G-CSF 388,4 (0,3-2691,0) 858,4 (0-9002,1) 0,53
MIF 2649 (529-7174) 3341 (1006-6872) 0,12
Cortisol 34,6 (5,7-110,7) 74,6 (3,8-195,0) <0,001
PCRt 170,6 (35,3-465,4) 293,2 (58,0-1250,0) 0,03

49
Tabela 3.8 - Comparação dos níveis máximos dos níveis plasmáticos de citocinas, cortisol e
proteína C reativa nos dias 1, 3, 5 e 7 após início de choque séptico. As unidades dos valores
de citocinas estão expressas em pg/ml, dos valores de cortisol em µg/dl e proteína C reativa
em mg/l.
Citocina Sobreviventes
(n = 35)
Não-sobreviventes
(n = 26)
p valor
IL-1β 1,85 (0-3,67) 9,49 (1,0-114,2) 0,71
IL-5 2,13 (0-4,06) 1,93 (0-4,42) 0,21
IL-6 942 (0-8228) 4659 (0-58562) 0,08
IL-8 197,7 (0-2047,0) 971,5 (0-7268,0) 0,15
IL-10 41,1 (0-1082,0) 520,5 (0-7279,0) 0,17
IL-13 3,61 (0-47,66) 15,70 (1,0-428,3) 0,46
IFN-γ 167,0 (0-749,4) 507,8 (0-8554,0) 0,49
MCP-1 212,5 (0-1356) 759,9 (0-7408) 0,12
MIP-1β 42,3 (0-395,5) 64,8 (0-518,7) 0,64
G-CSF 1095 (0-5370) 1245 (0-11377) 0,91
MIF 3599 (0-12725) 4158 (0-9293) 0,38
cortisol 46,3 (11.6-132,0) 100,5 (3,8-195,0) <0,001
PCRt 250,5 (0-694,3) 381,0 (72,0-1250,0) 0,04
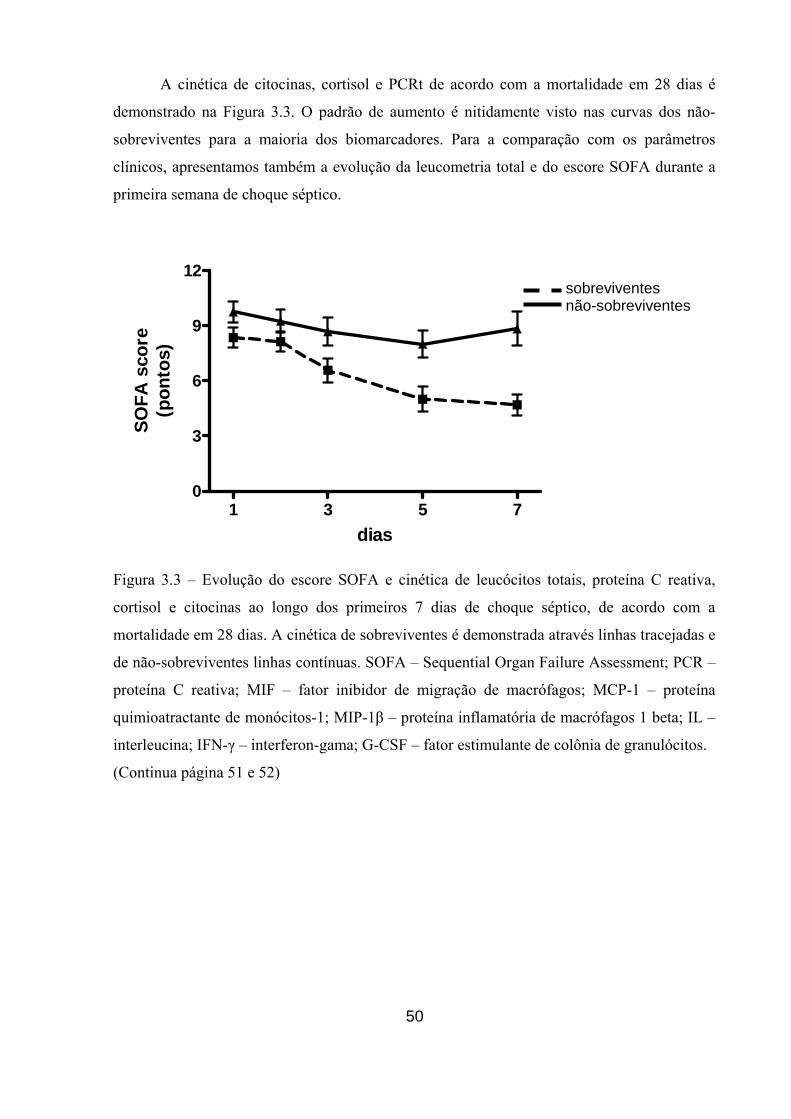
A cinética de citocinas, cortisol e PCRt de acordo com a mortalidade em 28 dias é
demonstrado na Figura 3.3. O padrão de aumento é nitidamente visto nas curvas dos não-
sobreviventes para a maioria dos biomarcadores. Para a comparação com os parâmetros
clínicos, apresentamos também a evolução da leucometria total e do escore SOFA durante a
primeira semana de choque séptico.
1 3 5 70
3
6
9
12survivorsnonsurvivors
dias
SOFA
sco
re(p
onto
s)
sobreviventes não-sobreviventes
Figura 3.3 – Evolução do escore SOFA e cinética de leucócitos totais, proteína C reativa,
cortisol e citocinas ao longo dos primeiros 7 dias de choque séptico, de acordo com a
mortalidade em 28 dias. A cinética de sobreviventes é demonstrada através linhas tracejadas e
de não-sobreviventes linhas contínuas. SOFA – Sequential Organ Failure Assessment; PCR –
proteína C reativa; MIF – fator inibidor de migração de macrófagos; MCP-1 – proteína
quimioatractante de monócitos-1; MIP-1β – proteína inflamatória de macrófagos 1 beta; IL –
interleucina; IFN-γ – interferon-gama; G-CSF – fator estimulante de colônia de granulócitos.
(Continua página 51 e 52)
50

1 3 5 710000
12500
15000
17500
20000
dias
leucócitos
(por m
m3)
1 3 5 70
90
180
270
360
450
dias
PCR
1 3 5 70
25
50
75
100
125
dias
cortisol
(mcg/dl)
1 3 5 7500
1500
2500
3500
4500
5500
dias
MIF
(pg/ml)
1 3 5 70
250
500
750
1000
1250
dias
MCP-1 (pg/ml)
1 3 5 70
20
40
60
dias
MIP-1b
(pg/ml)
51

1 3 5 70
5
10
15
20
25
dias
IL-1b
(pg/ml)
1 3 5 70
5
10
15
20
25
30
35
dias
IL-5
(pg/ml)
1 3 5 70
1000
2000
3000
4000
dias
IL-6
(pg/ml)
1 3 5 70
500
1000
1500
dias
IL-8
(pg/ml)
1 3 5 70
500
1000
1500
2000
2500
dias
IL-10
(pg/ml)
1 3 5 70
10
20
30
40
dias
IL-13
(pg/ml)
1 3 5 70
250
500
750
1000
dias
IFN-gamma
(pg/ml)
1 3 5 70
600
1200
1800
dias
G-CSF
(pg/ml)
52

53
O desempenho dos biomarcadores foi comparado com a de escores aplicados
clinicamente nas UTIs, como SAPS II e SOFA. Encontramos comportamento heterogêneo
dos biomarcadores em relação à mortalidade em 28 dias, dependendo da análise dos níveis no
1º dia ou da cinética. A análise com níveis dos biomarcadores no 1º dia de choque séptico foi
semelhante à realizada anteriormente pelo nosso grupo com MIF e IL-6 (Bozza e cols., 2004)
e no sistema multiplex (Bozza e cols., 2007). Demonstrou que MIF obteve o melhor
desempenho, com AUROC 0,73 (IC95% 0,57-0,89, p=0,01), que foi muito semelhante ao
escore SAPS II (AUROC 0,73, IC95% 0,57-0,89, p=0,01). A IL-10 apresentou AUROC 0,70
(IC95% 0,53-0,86, p=0,03). O escore SOFA do 1o dia teve desempenho apenas mediano
AUROC 0.68 (IC95% 0,57-0,85, p=0,07) (Figura 3.4).
Com a análise dos valores médios e máximos durante a 1ª semana de choque séptico,
o desempenho passa a ser bom para cortisol e IL-6. Os níveis médios de cortisol
demonstraram AUROC 0,77 (IC95% 0,63-0,92, p=0,001) e os de IL-6 AUROC 0,70 (IC95%
0,54-0,86, p=0,02). Os valores de média do escore SOFA durante a 1ª semana apresentou
AUROC 0,70 (IC95% 0,56-0,84, p=0,01) (Figura 3.5).
Os níveis máximos de cortisol e IL-6 demonstraram boa performance para
mortalidade em 28 dias: cortisol AUROC 0,81 (IC95% 0,69-0,93, p<0,001) e IL-6 0,70
(IC95% 0,55-0,84, p=0,02). Nesta análise, o escore SOFA máximo da 1ª semana obteve
desempenho inferior a estes dois biomarcadores (AUROC 0,63, IC95% 0,49-0,78, p=0,10)
(Figura 3.6).

1 - especificidade
sens
ibili
dade
1 - especificidade
sens
ibili
dade
Ponto de corte S / E AUROC IC 95% p valor
MIF 3201 pg/ml 70/82 0,73 0,58-0,89 0,009
IL-8 27,3 pg/ml 85/50 0,67 0,51-0,84 0,05
IL-10 2,7 pg/ml 80/59 0,69 0,53-0,85 0,04
PCRt 192,2 mg/l 70/64 0,63 0,46-0,81 0,14
SOFA 9,5 pontos 55/68 0,63 0,46-0,80 0,15
SAPS II 52,5 pontos 70/68 0,73 0,57-0,89 0,01
Figura 3.4 – Curvas Receiver Operating Characteristic (ROC) de citocinas e escores SAPS II
e SOFA medidos no 1º dia de choque séptico para predição de mortalidade até 28 dias. O
quadro abaixo expressa pontos de corte com melhor sensibilidade e especificidade, áreas sob
a curva ROC com intervalo de confiança de 95% (IC95%) e p valor. S / E –
sensibilidade/especificidade; AUROC – área sob a curva ROC.
54
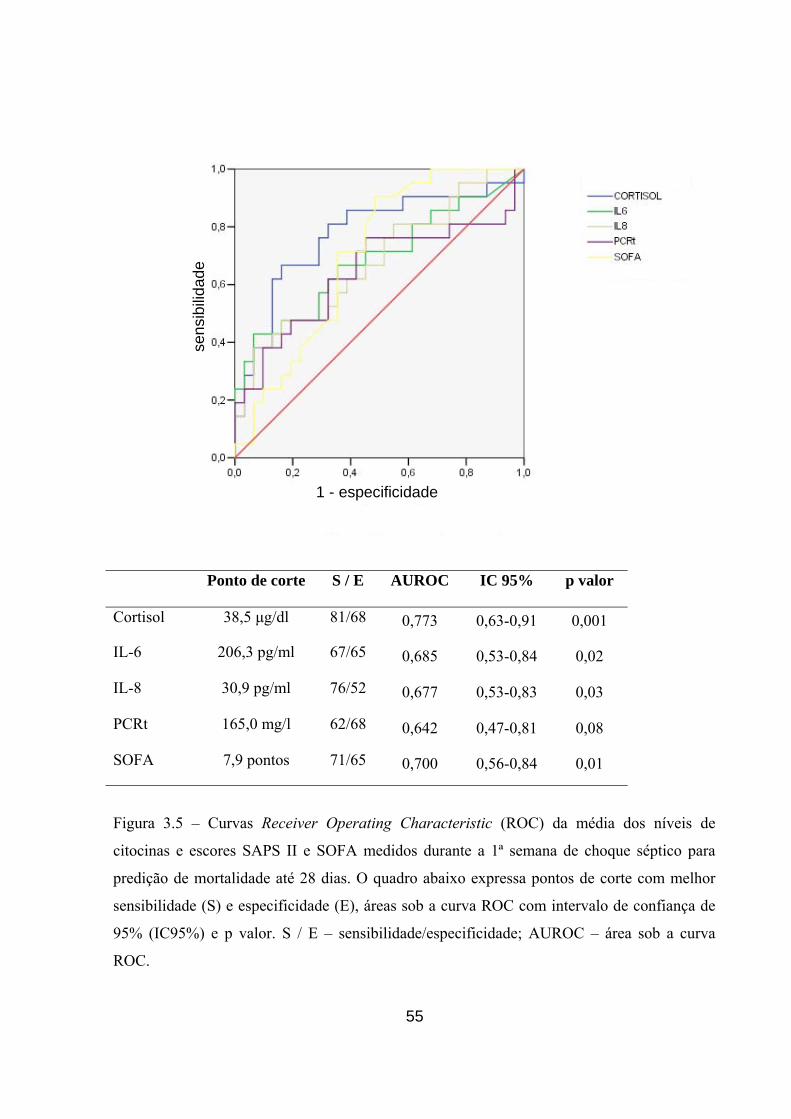
1 - especificidade
sens
ibilid
ade
1 - especificidade
sens
ibilid
ade
Ponto de corte S / E AUROC IC 95% p valor
Cortisol 38,5 µg/dl 81/68 0,773 0,63-0,91 0,001
IL-6 206,3 pg/ml 67/65 0,685 0,53-0,84 0,02
IL-8 30,9 pg/ml 76/52 0,677 0,53-0,83 0,03
PCRt 165,0 mg/l 62/68 0,642 0,47-0,81 0,08
SOFA 7,9 pontos 71/65 0,700 0,56-0,84 0,01
Figura 3.5 – Curvas Receiver Operating Characteristic (ROC) da média dos níveis de
citocinas e escores SAPS II e SOFA medidos durante a 1ª semana de choque séptico para
predição de mortalidade até 28 dias. O quadro abaixo expressa pontos de corte com melhor
sensibilidade (S) e especificidade (E), áreas sob a curva ROC com intervalo de confiança de
95% (IC95%) e p valor. S / E – sensibilidade/especificidade; AUROC – área sob a curva
ROC.
55

1 - especificidade
sens
ibilid
ade
1 - especificidade
sens
ibilid
ade
Ponto de corte S / E AUROC IC 95% p valor
Cortisol 59,3 µg/dl 76/75 0,81 0,69-0,93 <0,001
IL-6 235,9 pg/ml 81/56 0,70 0,55-0,84 0,02
PCRt 272,7 mg/l 57/69 0,61 0,45-0,78 0,17
SOFA 9,5 pontos 67/56 0,63 0,49-0,78 0,10
Figura 3.6 – Curvas Receiver Operating Characteristic (ROC) dos níveis máximos de
citocinas e escores SAPS II e SOFA medidos durante a 1ª semana de choque séptico para
predição de mortalidade até 28 dias. O quadro abaixo expressa pontos de corte com melhor
sensibilidade (S) e especificidade (E), áreas sob a curva ROC com intervalo de confiança de
95% (IC95%) e p valor. S / E – sensibilidade/especificidade; AUROC – área sob a curva
ROC.
56

57
Foi realizada análise multivariada para melhor discriminar quais os fatores
relacionados à mortalidade em 28 dias de maneira independente. Os parâmetros incluídos na
análise foram: idade; SAPS II; escore SOFA no 1º dia, médio e máximo; PCRt no 1º dia,
médio e máximo; cortisol no 1º dia, médio e máximo; MIF no 1º dia e médio; IL-6 no 1º dia,
médio e máximo; IL-8 no 1º dia, médio e máximo; IL-10 no 1º dia, médio e máximo; MCP-1
no 1º dia, médio e máximo. A fim de escolher o parâmetro que mais se diferenciou entre
sobreviventes e não-sobreviventes, foi escolhido o melhor momento de cada biomarcador (1º
dia, valor médio ou máximo). A regressão logística apontou que os níveis máximos de
cortisol (razão de chances 1,03 [IC95% 1,01-1,05]) e os níveis médios de IL-6 (razão de
chances 1,00 [IC95% 1,00-1,01]) na primeira semana foram independentemente associados
com a mortalidade em 28 dias.
3.3.2.4 - Níveis plasmáticos dos biomarcadores para mortalidade precoce (até 3dias)
A mortalidade nos primeiros 3 dias após início de choque séptico foi de 10%. Houve
aumento significativo dos seguintes biomarcadores nos pacientes não-sobreviventes no 1º dia
de choque séptico: IL-6 (1224 vs 2198 pg/ml, p=0,03); IL-8 (193 vs 1262 pg/ml, p<0,01); IL-
10 (30,7 vs 74,7 pg/ml, p=0,02); e MCP-1 (198,2 vs 538,1 pg/ml, p=0,02) (Tabela 3.9). Este
padrão visto para mortalidade precoce foi bem distinto do visto para mortalidade em 28 dias.
Para predição de mortalidade precoce o biomarcador que apresentou melhor
performance através da análise da área sob a curva ROC (AUROC) foi IL-8, com área de 0,81
(intervalo de confiança de 95% 0,64-0,98, p=0,02) (Figura 3.6). As interleucinas 6 e 10
apresentaram AUROC de 0,72 (IC95% 0,53-0,91, p=0,09) e 0,74 (IC95% 0,55-0,93, p=0,06),
respectivamente, com tendência à significância estatística. SOFA no 1º dia do choque séptico
teve desempenho ruim para mortalidade precoce AUROC de 0,58 (IC95% 0,31-0,86, p>0,2),
enquanto SAPS II apresentou AUROC 0,72 (IC95% 0,53-0,91, p=0,09), demonstrando
desempenho semelhante ou inferior às interleucinas 6, 8 e 10 (Figura 3.7).
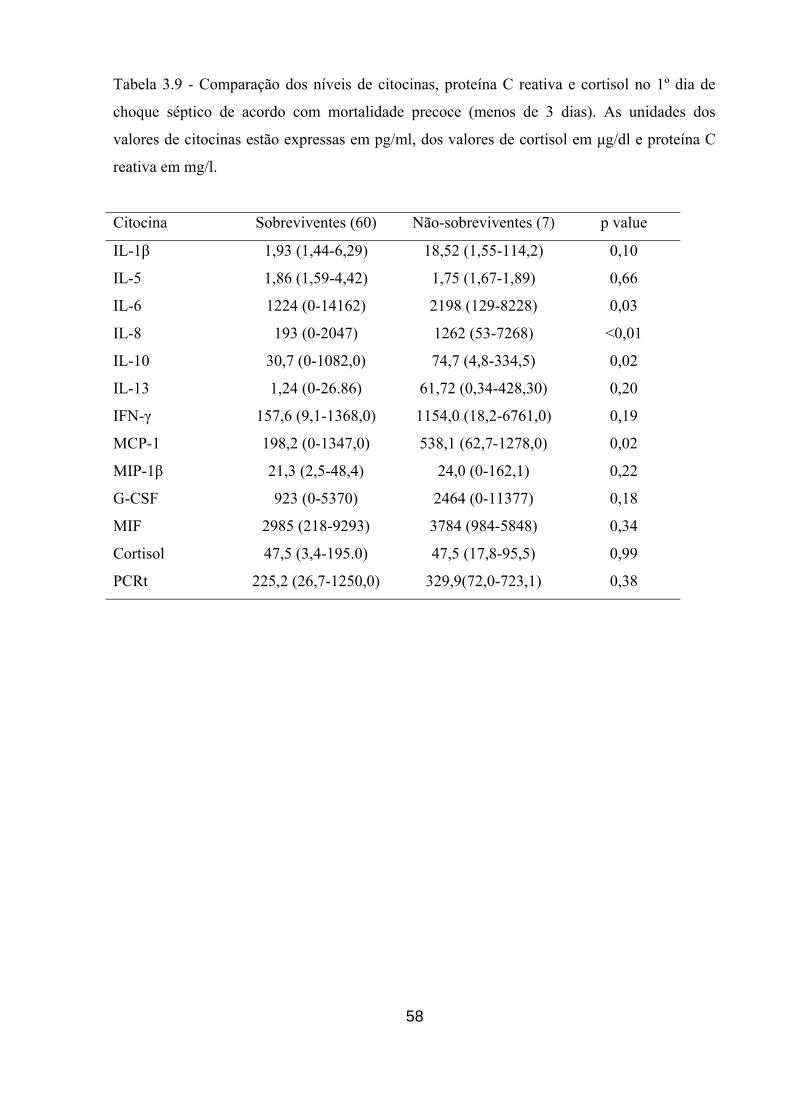
58
Tabela 3.9 - Comparação dos níveis de citocinas, proteína C reativa e cortisol no 1º dia de
choque séptico de acordo com mortalidade precoce (menos de 3 dias). As unidades dos
valores de citocinas estão expressas em pg/ml, dos valores de cortisol em µg/dl e proteína C
reativa em mg/l.
Citocina Sobreviventes (60) Não-sobreviventes (7) p value
IL-1β 1,93 (1,44-6,29) 18,52 (1,55-114,2) 0,10
IL-5 1,86 (1,59-4,42) 1,75 (1,67-1,89) 0,66
IL-6 1224 (0-14162) 2198 (129-8228) 0,03
IL-8 193 (0-2047) 1262 (53-7268) <0,01
IL-10 30,7 (0-1082,0) 74,7 (4,8-334,5) 0,02
IL-13 1,24 (0-26.86) 61,72 (0,34-428,30) 0,20
IFN-γ 157,6 (9,1-1368,0) 1154,0 (18,2-6761,0) 0,19
MCP-1 198,2 (0-1347,0) 538,1 (62,7-1278,0) 0,02
MIP-1β 21,3 (2,5-48,4) 24,0 (0-162,1) 0,22
G-CSF 923 (0-5370) 2464 (0-11377) 0,18
MIF 2985 (218-9293) 3784 (984-5848) 0,34
Cortisol 47,5 (3,4-195.0) 47,5 (17,8-95,5) 0,99
PCRt 225,2 (26,7-1250,0) 329,9(72,0-723,1) 0,38

1 - especificidade
sens
ibilid
ade
1 - especificidade
sens
ibilid
ade
Ponto de corte S / E AUROC IC 95% p valor
IL-8 105,6 pg/ml 86/72 0,84 0,71-0,97 0,004
IL-10 4,7 pg/ml 100/56 0,79 0,64-0,94 0,01
MCP-1 58,1 pg/ml 100/56 0,78 0,63-0,93 0,02
IL-6 359,1 pg/ml 86/64 0,76 0,62-0,89 0,03
SAPS II 59,5 pontos 71/74 0,74 0,55-0,92 0,04
SOFA 11,5 pontos 57/82 0,63 0,39-0,87 0,26
Figura 3.7 – Curvas Receiver Operating Characteristics (ROC) com análise de sensibilidade
e especificidade para predição de mortalidade precoce (menos de 3 dias) de pacientes com
choque séptico. O quadro abaixo expressa as áreas sob a curva ROC, com intervalo de
confiança de 95% (IC95%) e p valor. S / E – sensibilidade/especificidade; AUROC – área sob
a curva ROC.
59

60
3.3.2.5 - Correlação entre níveis plasmáticos de MIF e cortisol
A relação entre os níveis plasmáticos de MIF e cortisol no 1º dia de choque séptico foi
analisada. A princípio, não houve correlação significativa entre os níveis destes
biomarcadores no grupo total de pacientes. No entanto, como o estudo foi conduzido entre
2004 e 2009, as recomendações de tratamento de choque séptico que vigoraram durante a
maior parte deste período indicavam administração imediata com esteroides sintéticos no 1º
dia de tratamento. Trinta e nove (58%) pacientes receberam esteroides antes da primeira
coleta de plasma para o estudo. Pacientes que receberam esteroides venosos apresentaram
níveis plasmáticos ligeiramente maiores de MIF (3293 vs 2762 pg/ml, p=NS). No entanto, os
níveis de MIF plasmático só se correlacionaram positivamente nos pacientes que não
receberam esteroides venosos no 1º dia de tratamento do choque séptico (Figura 3.8).
3.3.2.6 - Correlação entre biomarcadores e escore SOFA
O escore SOFA pontua o grau de 6 disfunções orgânicas, como já explicado na
metodologia. Os níveis de biomarcadores no 1º dia de choque séptico foram correlacionados
com a pontuação deste escore também avaliado no mesmo dia. Os níveis de MIF, cortisol, IL-
8 e MP-1b foram os que obtiveram correlação significativa com o escore SOFA (Figura 3.9).
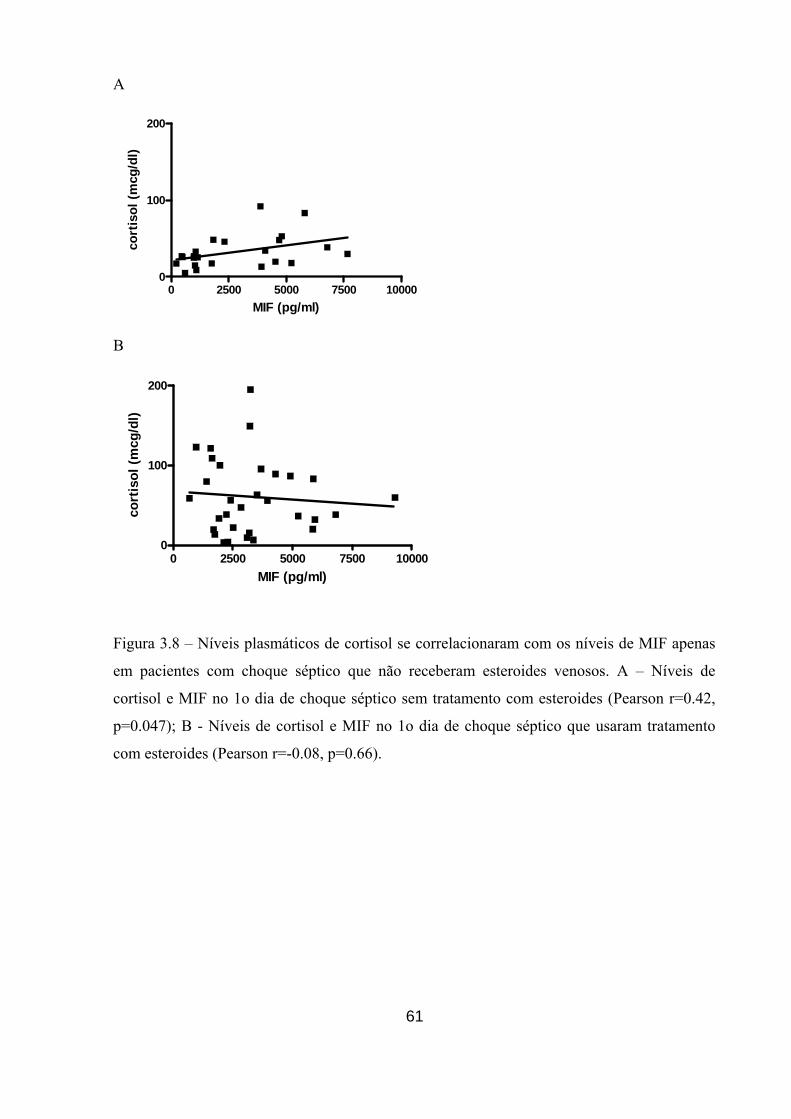
A
0 2500 5000 7500 100000
100
200
MIF (pg/ml)
cort
isol
(mcg
/dl)
B
0 2500 5000 7500 100000
100
200
MIF (pg/ml)
cort
isol
(mcg
/dl)
Figura 3.8 – Níveis plasmáticos de cortisol se correlacionaram com os níveis de MIF apenas
em pacientes com choque séptico que não receberam esteroides venosos. A – Níveis de
cortisol e MIF no 1o dia de choque séptico sem tratamento com esteroides (Pearson r=0.42,
p=0.047); B - Níveis de cortisol e MIF no 1o dia de choque séptico que usaram tratamento
com esteroides (Pearson r=-0.08, p=0.66).
61

0 5 10 15 200
2500
5000
7500
10000
SOFA
MIF
(pg/
ml)
0 5 10 15 200
50
100
150
200
250
SOFA
cort
isol
(mcg
/dl)
0 5 10 15 20
500
1500
2500
SOFA
IL-8
(pg/
ml)
0 5 10 15 200
100
200
SOFA
MIP
-1b
(pg/
ml
r = 0.41 p=0.002
r = 0.38 p=0.004
r = 0.37 p=0.005
r = 0.30 p=0.02
)
Figura 3.9 – Correlação entre o escore Sequential Organ Failure Assessment (SOFA) e
citocinas e cortisol plasmático em pacientes com choque séptico: MIF (A), IL-8 (B), MIP-1β
(C) e cortisol levels (D). MIF e cortisol apresentaram distribuição paramétrica e se aplicou o
teste de Pearson; IL-8 e MIP-1β foram analisados com o teste de correlação de Spearman
(variáveis não-paramétricas).
62

63
3.4 – Conclusões Parciais
a. Um perfil de citocinas pode identificar com maior acurácia a gravidade e o
prognóstico de pacientes com sepse grave ou choque séptico;
b. Pacientes com choque séptico exibem níveis significativamente maiores de várias
citocinas (IL-1β, IL-6, IL-7, IL-8, IL-10, IL-13, IFN-γ e MCP-1) do que pacientes controles e
com sepse ou sepse grave;
c. Interleucinas 6, 8 e 10 e MCP-1 apresentaram alta sensibilidade e especificidade para
identificar pacientes com mortalidade precoce (menos de 3 dias);
d. MCP-1 se associou independentemente com mortalidade em 28 dias em população
mista de pacientes com sepse grave e choque séptico;
e. Os níveis de citocinas são significativamente diferentes durante a primeira semana de
choque séptico e identificamos 4 padrões de cinética (decrescente exponencial; Gaussiano;
crescente; estável);
f. Os biomarcadores de melhor desempenho para predição de mortalidade em 28 dias
são: MIF e IL-8 no 1º dia de choque séptico; média e níveis máximos de cortisol e IL-6
durante a primeira semana de choque séptico;
g. Níveis máximos de cortisol e média dos níveis de IL-6 durante a primeira semana se
correlacionaram independentemente com a mortalidade em 28 dias;
h. Os níveis de MIF, cortisol, IL-8 e MP-1β se correlacionaram significativamente com o
escore SOFA.

64
4 - Disfunção Mitocondrial em Células Mononucleares do Sangue Periférico
4.1 – Introdução
As evidências atuais apontam para a relevância da disfunção mitocondrial e
imunossupressão na patogênese da sepse. A função mitocondrial é afetada em monócitos de
pacientes com sepse (Adrie e cols., 2001; Belikova e cols., 2007), mas os mecanismos
responsáveis por esta disfunção e suas consequências clínicas ainda são desconhecidas.
Nós avaliamos a respiração mitocondrial em células mononucleares do sangue
periférico em 20 pacientes com choque séptico e 18 pacientes controles internados em 2
UTIs. As células mononucleares de pacientes com choque séptico exibiram significativa
redução da respiração induzida por adição de succinato e ADP (estado 3), que se associou
com o desfecho hospitalar e com o grau de disfunções orgânicas (escore SOFA). Apontamos
também para disfunção específica na F1Fo ATP-sintase, que foi inibida com menores doses
de oligomicina que as células de pacientes controles, enquanto as doses do inibidor do
trocador de nucleosídeos adenínicos (ANT) foram semelhantes em sépticos e controles. Este
mecanismo pode ser central na redução da capacidade bioenergética de células
mononucleares circulantes e pode contribuir para a supressão de resposta imune vista em
pacientes com Síndrome de Disfunção Orgânica Múltipla (SDOM) (Monnerat, 2005).

65
4.2 – Métodos
4.2.1 - Pacientes
Foram incluídos pacientes com diagnóstico de choque séptico nas primeiras 24 horas
de evolução, após obtenção de termo de consentimento livre e esclarecido de familiares e/ou
responsáveis, em 3 UTIs (Hospital Clementino Fraga Filho – UFRJ, Hospital Quinta D´Or e
Casa de Saúde São José, Rio de Janeiro, Brasil). Excluímos pacientes grávidas e menores de
18 anos.
Os critérios de SIRS/sepse estão detalhados anteriormente (Bone e cols., 1992).
Choque séptico foi definido como a presença de hipotensão persistente, apesar de reposição
volêmica adequada (40 ml/kg de soluções cristalóides ou 20 ml/kg de colóides, em 2 horas) e
necessidade de agente vasopressor para manter pressão arterial média acima de 70 mmHg. Os
dados demográficos, fisiológicos e laboratoriais foram coletados para cálculo dos escores
Simplified Acute Physiologic Score (SAPS) II no 1º dia de internação na UTI e Sequential
Organ Failure Assessment (SOFA) nas primeiras 24 horas de choque séptico (Le Gall e cols.,
1993; Vincent e cols., 1996). Dados de culturas de prováveis sítios de infecção e resultados de
microbiologia foram coletados. Os pacientes com choque séptico foram tratados de acordo
com o guideline Surviving Sepsis Campaign (primeira versão em 2004 e revisão em 2008),
incluindo reposição volêmica agressiva nas primeiras 6-12 horas de choque, antibioticoterapia
de largo espectro e início de esteroide venoso em hipotensões graves apesar de uso de
vasopressor(es). O desfecho principal foi a mortalidade hospitalar. O grupo controle se
constitui de pacientes internados nas mesmas UTIs, sem choque ou evidência de infecção;
majoritariamente foram internados para monitorização após cirurgias de médio e grande
porte.
4.2.2 – Células
Vinte mililitros de sangue total foram coletados de punção de veias periféricas. A
separação de células mononucleares de sangue periférico (PBMC) foi feita através de
gradiente de Ficoll. A seguir, o sangue foi diluído em solução de dextran 40 por 30 minutos e
o sobrenadante foi colocado por cima de solução de Ficoll-Hypaque a 100% e centrifugado
por 30 minutos, com rotação de 1050 rpm. As células mononucleares foram coletadas do
nível da coluna logo acima da solução de Ficoll, com diluição em sal balanceado de Hank

66
(HBSS) sem cálcio e magnésio, e nova centrifugação foi realizada por 8 minutos, a 1500
rotações por minuto. As células em forma de palheta eram diluídas para 2 ml de HBSS e
colocadas em recipiente com gelo (para temperatura a 0-4º C). A contaminação desta solução
com outros tipos celulares foi verificada através de citometria de fluxo com material de 1
paciente séptico e 2 controles, e neutrófilos e granulócitos representaram apenas 2,5% da
fração de células medidas. A avaliação da respiração mitocondrial era imediatamente
conduzida após isolamento de células mononucleares de sangue periférico (PBMCs).
4.2.3 – Medidas de consumo de oxigênio
A respiração mitocondrial em PBMCs foi realizada através de oxímetro com cubeta
com eletrodo de Clark (Yellow Springs Instrument, Model 5300, USA) nos experimentos de
2005 até 2008, e com oxígrafo de alta resolução (Oroboros Inc, Áustria) a partir de 2008. Para
o estudo do consumo de oxigênio, utilizamos tampão de respiração modificado (10 mM Tris-
HCl, pH 7,4, 0,32 M manitol, 5 mM fosfato inorgânico, 50 mM KCl, 4 mM MgCl2, 1 mM
ethylene glycol tetraacetic acid - EGTA), com quantidade de PBMC variável de 3,5 x 106
células a 1,0 x 107 células por cubeta e temperatura estável a 37º C. O experimento começava
com a adição através de seringa Hamilton de digitonina (0,005% w/v) para a permeabilização
de membranas celulares, rotenona (1µg/ml) para inibição do complexo I e adição de succinato
(2 mM) para dar início ao estado 2 de respiração mitocondrial. A seguir, foi adicionado ADP
(0,5 mM), e esperava-se a estabilização do fluxo de próton, atingindo o valor de consumo de
oxigênio no estado 3. Oligomicina (2 µg/ml), inibidora da atividade de F1Fo ATP sintase, foi
adicionada para induzir respiração mitocondrial tipo estado 4, que está relacionada ao escape
de prótons e redução do consumo de oxigênio. A respiração mitocondrial em PBMCs foi
essencialmente devido a atividade mitocondrial, já que era totalmente inibida após adição de
cianeto de potássio (1uM). A razão de controle respiratório foi obtida pela divisão dos
voalores de consumo de oxigênio no estado 3 pelo estado tipo 4, e indicava a integridade de
membrana interna mitocondrial (Chance e cols., 1955). Após estabilização da respiração
induzida por oligomicina, adicionamos carbonil cianeto p-(trifluorometoxi)fenilhidrazona
(FCCP) na concentração de 0,3 µM, para induzir a taxa de respiração máxima no estado
desacoplado.
Com o objetivo de acessar o conteúdo funcional de F1Fo-ATP sintase e do trocador de
nucleotídeos adenínicos (ANT), conduzimos uma titulação de oligomicina (inibidora da
F1Fo-ATP sintase) com doses de 0,5 ng/ml ou atractilosídeo (inibidor irreversível de ANT)

67
com doses de 0,25 nmol/ml após a respiração estado 3, até que houvesse estabilização da
respiração estado 4. Construímos gráficos com as taxas de respiração estado 3 contra as
mínimas doses de oligomicina ou atractilosídeo capazes de inibir totalmente a respiração
estado 4, calculando este valor com o encontro entre as curvas de respiração nos estados 3 e
4. Este cálculo estima o conteúdo funcional da F1Fo-ATP sintase e de ANT, e os valores
foram representados como ng oligomicina/ml/106 células e nmols atractilosídeo/ml/106
células (Trzcionka e cols., 2007).
4.2.4 – Análise estatística
A análise de dados foi realizada com o pacote GraphPad Prism para Windows, versão
4.0 (GraphPad Software, San Diego, USA). As variáveis clínicas foram expressas como
média e desvio padrão, o consumo de oxigênio foram expressas em barras com erro padrão de
cada condição. As comparações entre as variáveis numéricas foram realizadas através do teste
t de Student ou análise de variância (ANOVA), com pós-teste de Tukey para comparações
por pares. Correlações estatísticas entre o escore SOFA e a respiração mitocondrial nos
estados 3 e 4 foram feitas com o teste de Spearman. Significância estatística foi considerada
quando o p valor foi menor que 0,05.

68
4.3 – Resultados
4.3.1 – Características dos pacientes
Estudamos uma população de 18 pacientes em período pós-operatório (controles) e 20
pacientes com choque séptico, cujas características estão na Tabela 1. A idade foi similar nos
2 grupos, e como esperado, a gravidade de doença aguda foi maior no grupo com sepse
(SAPS II 51,5 ± 12,5 vs 27,1 ± 9,1 pontos, p<0,01). O grau geral de disfunção orgânica
também foi diferente estatisticamente, com escore SOFA no dia de estudo de 8,9 ± 3,4 pontos
nos pacientes sépticos e 1,9 ± 2,0 pontos nos pacientes controles (p<0,01). As fontes de
infecção foram abdominais em 10 pacientes sépticos, pulmonar em 7, bacteremia primária em
2 e urinária em 1 paciente. Todos os pacientes com sepse apresentavam choque no dia de
coleta de sangue, 95% usou ventilação mecânica e 46% necessitou suporte dialítico por
falência renal aguda durante a permanência na UTI. A mortalidade hospitalar foi de 50% no
grupo com choque séptico; todos pacientes do grupo controle tiveram alta hospitalar.

69
Tabela 4.1 – Características demográficas, gravidade de doença e desfecho hospitalar de
pacientes com choque séptico e controles. Variáveis contínuas foram expressas como media ±
desvio padrão.
Controle
(n = 18)
Choque Séptico
(n = 20)
Idade (anos) 70,3 ± 12,7 69,3 ± 14,0
Sexo masculino 12 11
Tipo de internação (cirúrgica/clínica) 16/2 15/5
Sítio de infecção
Abdominal
Pulmonar
Bacteremia primária
Urinário
-
-
-
-
10
7
2
1
SAPS II (pontos) 27,1 ± 9,1 51,5 ± 12,5
SOFA (pontos) 1,9 ± 2,0 8,9 ± 3,4
Ventilação mecânica 5 19
Terapia de suporte renal 0 9
Tempo de permanência na UTI (dias) 4,1 ± 3,8 22,4 ± 13,3
Mortalidade até 28 dias 0 9
Mortalidade hospitalar 0 10

70
4.3.2 – Respiração mitocondrial
Os exemplos de traçados de consume de oxigênio em paciente controle (linha cinza) e
com choque séptico (linha preta) estão na Figura 4.1A. Após permeabilização dos PBMCs, o
consumo de oxigênio acelera após a adição de ADP, porém a inclinação da reta durante
estado 3 no paciente séptico (linhas hachuradas) é menor que no controle. Entretanto, o
consumo de oxigênio durante o estado 4 e o desacoplamento são semelhantes nos 2 pacientes.
Na figura 4.1B, mostramos a quantificação da respiração mitocondrial nos PBMCs de
controles (barras brancas) e pacientes com choque séptico (barras pretas) em diferentes
estados metabólicos. Ocorre significativa redução do consumo de oxigênio induzida por
adição de ADP nos pacientes sépticos em relação aos controles (5,60 vs. 9,89 nmols
O2/min/107 células respectivamente, p<0,01). No entanto, não houve diferença entre os 2
grupos na respiração mitocondrial após adição de succinato (5,43 vs 4,09 nmols O2/min/107
células), oligomicina (1,58 vs 1,90 nmols O2/min/107 células) ou FCCP (9,78 vs 6,93 nmols
O2/min/107 células). A razão de controle respiratório foi significativamente menor no grupo
com choque séptico (controle = 7,10 vs choque séptico = 5,27, p=0,04) (Figura 4.1C).
A respiração mitocondrial de PBMCs foi significativamente diferente de acordo com o
desfecho hospitalar (Figura 4.2). Pacientes não-sobreviventes de choque séptico apresentaram
respiração mitocondrial estimulada por ADP (estado 3) sigficativamente menor que controles
e sobreviventes (não-sobreviventes=4,56 vs sobreviventes=6,64 vs controles=10,27 nmols
O2/min/107 células; p=0,004, ANOVA). Em relação à mesma análise da respiração
mitocondrial induzida por oligomicina (estado tipo 4), não houve diferença significativa entre
os grupos (controles=2,35 vs sobreviventes=1,88 vs não-sobreviventes=1,28 nmols
O2/min/107 células).
O consumo reduzido de oxigênio em PBMCs humanos se correlacionou
especificamente com o aumento do escore SOFA (Figura 4.3). O consumo de oxigênio
induzido por ADP se correlacionou negativamente com o escore SOFA do mesmo dia da
medida (Spearman r = - 0.46, p=0.005), com controles (triângulos brancos) demonstrando
maiores taxas respiratórias e menores pontuações do SOFA. Pacientes que não sobreviveram
ao choque séptico (quadrados pretos) demonstraram baixo consumo de oxigênio e alta
pontuação do escore SOFA, enquanto os sobreviventes (círculos cinzas) apresentaram
resultados híbridos. A respiração mitocondrial induzida por oligomicina não apresentou
correlação com o escore SOFA (Spearman r = - 0.12, p=0.49).

A
20 min.
50 n
mol
sO
2/mL
Sepse
Succ
ADP
oligo
FCCP
Controle
20 min.
50 n
mol
sO
2/mL
Sepse
Succ
ADP
oligo
FCCP
Controle
B C
71
0.0
2.5
5.0
7.5
10.0
12.5
cons
umo
O2
(nm
ol/m
in/1
07ce
ls)
*
succinate
C S
ADP
C S
oligo
C S
FCCP
C S 0
3
6
9
#Ta
xa d
e C
ontr
ole
Res
pira
tório
0
3
6
9
0.0
2.5
5.0
7.5
10.0
12.5
0.0
2.5
5.0
7.5
10.0
12.5
cons
umo
O2
(nm
ol/m
in/1
07ce
ls)
*
succinate
C S
ADP
C S
oligo
C S
FCCP
C S 0
3
6
9
#Ta
xa d
e C
ontr
ole
Res
pira
tório
0
3
6
9
C S

72
Figura 4.1 – Consumo de oxigênio mitocondrial associado à síntese de ATP em células
mononucleares de sangue periférico (PBMC) em pacientes com choque séptico e controles. A
– Traçados representativos de consumo de oxigênio de PBMC permeabilizados de pacientes
controle (linha cinza) e com choque séptico (linha preta), com adições de Succ (succinato) 10
mM; ADP (adenosina 5'-difosfato) 1 mM; oligo (oligomicina) 4 mg/mL; FCCP (carbonil
cianeto p-(trifluorometoxi)fenilhidrazona) 0,3 mM. As retas hachuradas indicam a taxa de
respiração estado 3 usada para comparações entre pacientes controles e com choque séptico.
B – Avaliação das taxas de consumo de oxigênio de PBMC permeabilizadas de controles (C)
(n=18, barras brancas) e pacientes com choque séptico (S) (n = 20, barras pretas) em
diferentes estados metabólicos. C – Taxa de controle respiratório (RCR) de PBMCs de
controles (n=18, barras brancas) e pacientes com choque séptico (n=20, barras pretas), obtida
pela divisão do consumo de oxigênio após adição de ADP (estado 3) pelo consumo após
oligomicina (estado tipo 4). Os dados foram expressos como barras com média ± erro padrão
para cada condição. * p<0,01 ANOVA e teste de Tukey.

3
6
9
12
73
Figura 4.2 – Consumo de oxigênio mitocondrial em células mononucleares de sangue
periférico e mortalidade hospitalar. Consumo de oxigênio de PBMCs de controles (barras
brancas), sobreviventes de choque séptico (barras cinzas) e não-sobreviventes (barras pretas)
foi verificada após estímulo com ADP (A) e oligomicina (B). As variáveis foram
representadas como barras com média ± erro padrão para cada condição. * p<0,05 ANOVA e
teste de Tukey.
cons
P(n
mol
/min
/107
cel
s)
cons
umo
O2
após
olig
o(n
mol
/min
/107
cel
s)
*
umo
O2
após
AD
3
6
9
12
3
6
9
12on
sP
(nm
ol/m
in/1
07 c
els)
cons
umo
O2
após
olig
o(n
mol
/min
/107
cel
s)
*
3
6
9
12
AD
apó
s um
o O
2c
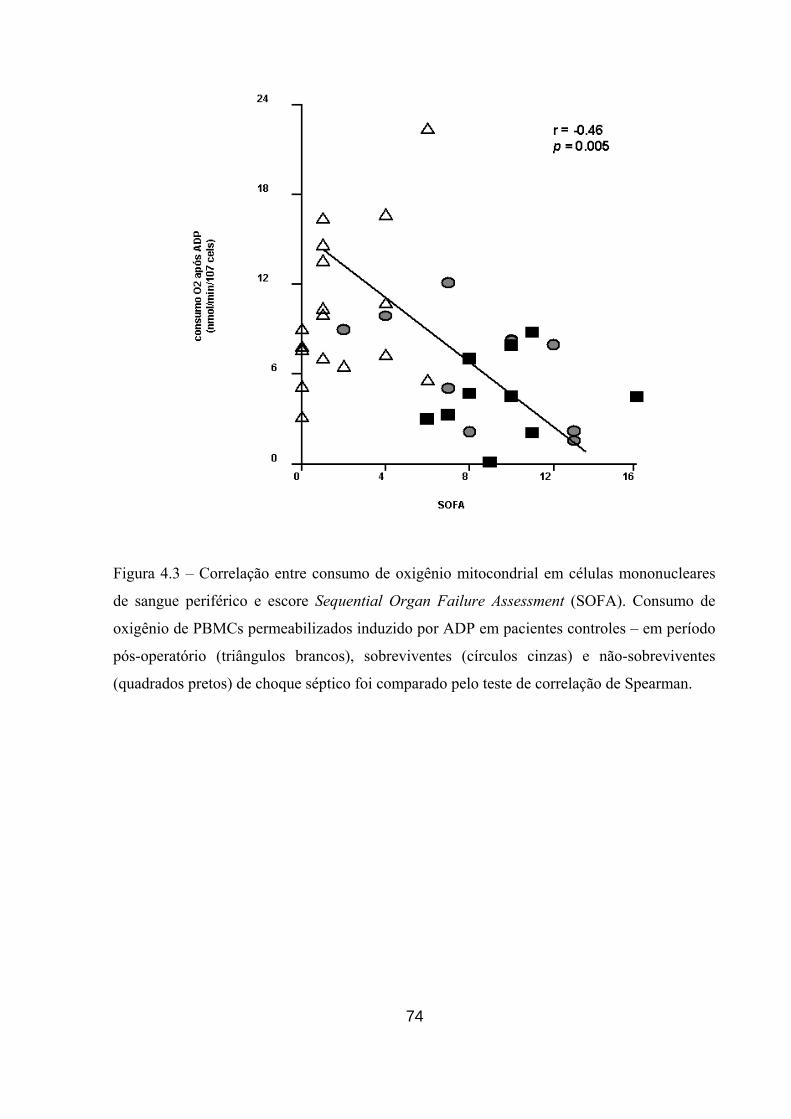
Figura 4.3 – Correlação entre consumo de oxigênio mitocondrial em células mononucleares
de sangue periférico e escore Sequential Organ Failure Assessment (SOFA). Consumo de
oxigênio de PBMCs permeabilizados induzido por ADP em pacientes controles – em período
pós-operatório (triângulos brancos), sobreviventes (círculos cinzas) e não-sobreviventes
(quadrados pretos) de choque séptico foi comparado pelo teste de correlação de Spearman.
74

75
Uma análise do seguimento de 5 sobreviventes de choque séptico mostrou um
aumento de 2,9 vezes na respiração mitocondrial induzida por ADP depois de 1 semana do
início do quadro (1º dia: 4,33; 7º dia: 12,46 nmols O2/min/107 células). Houve significativa
recuperação da taxa de controle respiratório nestes 5 pacientes após 7 dias (dia 1: 2,48; dia 7:
3,52; p=0,02, teste t pareado).
Finalmente, determinamos o conteúdo do complexo F1Fo-ATP sintase funcional,
assim como de ANT nos PMBCs de pacientes controles e sépticos, através de experimentos
com adições sucessivas de oligomicina e atractilosídeo para indução do estado tipo 4. A
inibição do consumo de oxigênio de PMBCs de pacientes com choque séptico ocorreu após
adição de doses menores de oligomicina que nos pacientes controles, resultando em menores
valores da constante de dissociação (KD) nos sépticos (sépticos 0,21 vs controles 1,29;
p=0,03, valor de melhor discriminação em regressão não-linear) (Figura 4.4A). O conteúdo de
complexo F1Fo-ATP sintase, determinado pela menor quantidade de oligomicina necessária
para inibir totalmente o consumo de oxigênio após adição de ADP, foi significativamente
menor nos pacientes sépticos (0,51 vs 1,00 ng oligo/mL/106 células; p=0,02, teste t de
Student) (Figura 4.4B). O mesmo procedimento foi realizado com a inibição de ANT pelo
atractilosídeo (Figura 4.4C), porém não observamos diferenças do consumo de oxigênio
mitocondrial, com valores semelhantes de KD entre PBMCs de pacientes controles e sépticos
(0,002 vs 0,015, respectivamente). O conteúdo funcional de ANT também foi semelhante
entre controles e sépticos (controles 0,50 vs sépticos 0,26 nmols atractilosídeo/mL/106
células) (Figura 4.4D).

A B
0 1 2 3 41
10
100
controlseptic
oligomicina (ng/mL/106 cels)
% in
ibiç
ão d
e co
nsum
o O
2ap
ós A
DP
(nm
ol/m
in/1
07 c
els)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
cont
eúdo
F1F
o-A
TP s
inth
ase
(ng
olig
o/m
L/10
6ce
ls)
C S
*
0.00
0.25
0.50
0.75
cont
eúdo
AN
T(n
mol
sat
ract
ilosi
deo
/mL/
106
cels
)
0.0 0.4 0.8 1.2 1.6 2.00
20
40
60
80
100
atractilosideo (nmols/mL/106 cels)
controlseptic
C D
C S
% in
ibiç
ão d
e co
nsum
o O
2ap
ós A
DP
(nm
ol/m
in/1
07 c
els)
0 1 2 3 41
10
100
controlseptic
oligomicina (ng/mL/106 cels)
% in
ibiç
ão d
e co
nsum
o O
2ap
ós A
DP
(nm
ol/m
in/1
07 c
els)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
cont
eúdo
F1F
o-A
TP s
inth
ase
(ng
olig
o/m
L/10
6ce
ls)
C S
*
0.00
0.25
0.50
0.75
cont
eúdo
AN
T(n
mol
sat
ract
ilosi
deo
/mL/
106
cels
)
0.0 0.4 0.8 1.2 1.6 2.00
20
40
60
80
100
atractilosideo (nmols/mL/106 cels)
controlseptic
C D
C S
% in
ibiç
ão d
e co
nsum
o O
2ap
ós A
DP
(nm
ol/m
in/1
07 c
els)
Figura 4.4 – Conteúdo funcional de F1Fo-ATP sintase e trocador de nucleotídeo adenínico
(ANT) em células mononucleares de sangue periférico. A – Titulação do consumo de
oxigênio induzido por ADP após sucessivas adições de oligomicina para alcançar estado tipo
4 em controles (círculos brancos, n=5) e pacientes com choque séptico (círculos pretos, n=9).
B – Conteúdo funcional de F1Fo-ATP sintase de PBMCs de controles (barra branca) e
pacientes sépticos (barra preta), demonstrado como quantidade de oligomicina necessária
para inibir totalmente a respiração mitocondrial no estado 3. C – Titulação do consumo de
oxigênio induzido por ADP após sucessivas adições de atractilosídeo para alcançar estado
tipo 4 em controles (círculos brancos, n=5) e pacientes com choque séptico (círculos pretos,
n=9). D – Conteúdo funcional de ANT de PBMCs de controles (barra branca) e pacientes
sépticos (barra preta), demonstrado como quantidade de atractilosídeo necessária para inibir
totalmente a respiração mitocondrial no estado 3. * p=0,02, teste Mann-Whitney.
76

77
4.4 – Conclusões Parciais
a. Ocorre redução significativa da respiração mitocondrial induzida após adição de ADP
em células mononucleares de sangue periférico de pacientes com choque séptico;
b. A respiração mitocondrial associada à síntese de ATP também se correlaciona
negativamente à gravidade de disfunções orgânicas;
c. Esta redução da respiração no estado metabólico associado à síntese de ATP é mais
acentuada em pacientes que não sobrevivem, e está associada à mortalidade hospitalar;
d. A disfunção da F1Fo-ATP sintase foi o mecanismo responsável pela redução da
respiração mitocondrial associada à síntese de ATP, demonstrada pela inibição por doses
menores de oligomicina nos pacientes com choque séptico do que em pacientes controles.

78
5 - Discussão
A sepse é a principal causa de morte nas unidades de Tratamento Intensivo ao redor
do mundo, estimando-se em 700.000 mortes anuais nos Estados Unidos e cerca de 400.000 no
Brasil (Angus e cols., 2001; Westphal 2009). O custo para cuidados de pacientes com sepse
grave no Brasil está estimado em 17 bilhões de reais por ano e cada paciente custa de 38 a 56
mil reais por internação hospitalar (Feijó e cols., 2009). Este gasto pode ser substancialmente
melhor empregado se a sepse for reconhecida adequadamente. Na fisiopatologia da sepse,
ocorre ativação sistêmica de células mediadoras de inflamação, como neutrófilos,
monócitos/macrófagos e linfócitos, com produção intensa de citocinas, mediadores lipídicos e
espécies reativas de oxigênio, associada com vasodilatação generalizada, aumento da
permeabilidade vascular e hipercoagulabilidade. As citocinas liberadas medeiam boa parte
destas alterações, seja no órgão alvo do sítio da infecção, ou em órgãos à distância. As
citocinas têm sido estudadas como biomarcadores do diagnóstico e do tratamento de
SIRS/sepse. Os biomarcadores podem auxiliar o raciocínio clínico para decisões de
manutenção ou modificação do tratamento do paciente séptico (Marshall & Reinhart, 2009).
Entre os biomarcadores mais utilizados estão o leucograma e a proteína C reativa (PCRt). O
leucograma está incluído nos critérios de SIRS propostos no início da década de 1990, e a
presença de leucopenia (abaixo de 4000/mm3) ou leucocitose (acima de 12000/mm3) ou a
presença de mais de 10% de bastões no exame provê 1 ponto do critério de SIRS. Povoa e
cols estudaram a PCRt em comparação ao leucograma para o diagnóstico de infecções na UTI
e verificaram que o diagnóstico é mais precoce cerca de 2 dias com o uso da PCRt. A PCRt
apresentou bom desempenho para o diagnóstico de infecção de pacientes internados em UTI.
As alterações do leucograma se mostraram semelhantes nos grupos com e sem infecção
(Póvoa e cols., 2005).
O sistema PIRO, proposto após reunião de consenso em 2001, inclui a avaliação de
critérios ampliados para o diagnóstico da sepse (Levy e cols., 2003). Este sistema foi criado
para ajudar na estratificação de diagnóstico e gravidade de pacientes com sepse, adaptando o
conceito do sistema TNM (tamanho do tumor, acometimento de linfonodos e presença de
metástases) para estratificação de neoplasias de órgãos sólidos. A presença de fatores
predisponentes, tipo de infecção, resposta do hospedeiro e disfunção orgânica depende
amplamente da população estudada, e existe uma lacuna de estudos que associam todos estes
parâmetros na literatura. Em relação à avaliação da resposta do hospedeiro, a inclusão de
biomarcadores como PCRt, procalcitonina e IL-6 pode tornar o diagnóstico mais específico,

79
já que os critérios de SIRS enfrentam críticas por estarem sujeitos a baixa especificidade
(Brun-Buisson e cols., 1995; Vincent, 1996). Os critérios de SIRS foram criados para tornar o
diagnóstico de sepse mais sensível, já que as definições até a década de 80 eram heterogêneas
ao redor do mundo e acarretava o reconhecimento tardio dos casos (Vincent, 1996). Porém,
ao longo dos anos, o diagnóstico de sepse passou a ser mais comum, pois houve uma
expansão do número de leitos de Tratamento Intensivo e aumento da prevalência de pacientes
em risco para sepse internados em hospitais. O contraponto mais discutido atualmente é a
baixa especificidade do diagnóstico da sepse, que pode acarretar erros de avaliação de casos
de febre ou choque de pacientes graves e também aumento do uso de antibióticos e resistência
bacteriana (Kumar, 2009). A melhora da identificação precisa de infecções no paciente grave
é um dos principais focos de pesquisa em sepse (Marshall & Reinhart 2009). Outro aspecto
avaliado no sistema PIRO é a avaliação de disfunções de órgãos e tecidos (Singh, 2006).
Disfunções cardiovascular e respiratória, expressas como choque e lesão pulmonar aguda,
respectivamente, ocorrem precocemente no quadro de sepse e aumentam o risco de
mortalidade precoce, enquanto disfunções renal e hepática aparecem após alguns dias e
predizem pior prognóstico no período de dias a semanas (Moreno e cols., 1999). A presença e
o grau de disfunções orgânicas não se correlacionam com o prognóstico com boa acurácia.
A avaliação sistematizada da aplicação do sistema PIRO em pacientes com sepse
ainda está em fase inicial. Somente após 8 anos, 3 estudos avaliaram o sistema PIRO,
validando seu uso para o prognóstico de sepse por pneumonia comunitária grave, pneumonia
associada a ventilação mecânica e em uma coorte de estudo epidemiológico da sepse grave
(Rello e cols., 2008; Lisboa e cols., 2008; Rubulotta e cols., 2009). Entretanto, em nenhum
destes estudos biomarcadores foram usados para o reconhecimento ou o prognóstico da sepse
grave. Este fato demonstra que ou não se avalia biomarcadores para diagnóstico de sepse
grave de modo sistemático nos estudos, ou ainda não se encontrou um biomarcador com bom
desempenho para este fim. Com este intuito, estudamos a avaliação de vários biomarcadores
simultaneamente para avaliar a gravidade da sepse e também seu prognóstico.
O sistema multiplex para dosagem de citocinas tem excelente eficiência para detecção
em amostras clínicas (Vignali, 2000). Em nosso estudo, a dosagem simultânea de 17 citocinas
em pacientes com diagnóstico recente de sepse grave e choque séptico identificou níveis
detectáveis em 69% das amostras, com melhor desempenho para IL-6, IL-8, IL-10 e MCP-1
(detectados em mais de 95% dos ensaios) (Bozza e cols., 2007). No estudo de cinética, IL-1β,
IL-5, IL-6, IL-8, IL-10, IL-13, IFN-γ, MIP-1β, G-CSF e MCP-1 foram detectados em mais de
90% das amostras. De modo geral, o aproveitamento de detecção de citocinas por este método

80
é igual ou superior a dosagem por técnicas tradicionais como ELISA. A eficácia do uso desta
técnica supera o uso de ELISA, pois permite a dosagem simultânea de um painel de 17
citocinas aliada à necessidade de pequenas alíquotas de amostras clínicas. Citocinas como
TNF-α, que estão associadas a alterações na sepse murina, são detectados em taxas menores
que 50% no plasma de pacientes sépticos (Marks e cols., 1990; Abraham e cols., 1998).
Verificamos também que as citocinas se comportaram de maneira heterogênea de
acordo com os desfechos analisados (correlação com disfunções orgânicas, mortalidade
precoce ou em 28 dias). A mortalidade precoce se associou com níveis elevados de IL-6, IL-
8, IL-10 e MCP-1, que apresentaram desempenho para predição deste desfecho melhor que os
escores de risco SAPS II e SOFA medido no 1º dia de choque séptico. Quando se analisa a
mortalidade em 28 dias, os melhores desempenhos são do MIF no 1º dia de choque séptico,
nível médio e máximo de cortisol e médio de IL-6 durante a primeira semana de choque
séptico. E os níveis de MIF, cortisol, IL-8 e MP-1b se correlacionaram significativamente
com o escore SOFA.
Estas citocinas também foram comparadas no nosso estudo com biomarcadores
aplicados na prática clínica como a PCRt. Os níveis de PCRt do 1º dia de choque séptico, a
média e o máximo da primeira semana foram maiores em pacientes que faleceram até 28 dias,
porém a sensibilidade e especificidade para mortalidade em 28 dias foram inferiores de outros
biomarcadores, como IL-6, IL-8 e cortisol plasmático. Outros estudos avaliaram isoladamente
PCRt ou procalcitonina (PCT), e verificaram que seus níveis estão aumentados em pacientes
com sepse grave, se dispondo para diagnóstico de infecção em pacientes graves (Luzzani e
cols., 2003; Povoa e cols., 2005). Mas o desempenho destes biomarcadores para o
prognóstico é ruim, principalmente porque sua especificidade é baixa (em torno de 45-50%)
(Clech e cols., 2004; Silvestre e cols., 2009). Algumas citocinas foram usadas como
marcadoras de diagnóstico e prognóstico de pacientes com sepse grave, como no estudo
Recombinant Human Protein C Worldwide Evaluation in Severe Sepsis (PROWESS), que
avaliou o tratamento de pacientes com sepse grave com APC recombinante. Os níveis séricos
de IL-6 se correlacionaram com o escore APACHE II e a mortalidade hospitalar (Bernard e
cols., 2001). Presneill e cols verificaram que os níveis de G-CSF estão muito elevados na
sepse e no choque séptico e são significativamente maiores que em pacientes sem sepse, mas
não prediz mortalidade (Presneill e cols., 2000). O GM-CSF não foi detectado na maioria dos
pacientes neste estudo. Isto aponta que talvez o G-CSF seja útil no diagnóstico, mas não na
predição de prognóstico. Níveis elevados de IL-4 também estavam presentes em pacientes
com sepse grave, porém seu desempenho de predição de prognóstico não foi satisfatório (Wu

81
e cols., 2008). Outros autores demonstraram que IL-8 medida no 1º dia de diagnóstico de
sepse se associou com a mortalidade em 28 dias, com melhor desempenho que o escore
APACHE II e que IL-6, IL-10 e IL-12 (Livaditi e cols., 2007). A nossa avaliação com várias
citocinas e PCRt permitiu a comparação com um biomarcador de uso amplo na prática diária
dos cuidados de pacientes graves.
O melhor desempenho com a análise dos níveis de biomarcadores no 1º dia de choque
séptico para marcar a mortalidade em 28 dias foi do MIF. Os níveis de MIF foram úteis na
identificação de pacientes com sepse, já que estava em níveis significativamente menores em
pacientes controles. MIF permitiu também a estratificação de gravidade, com níveis ainda
maiores em pacientes não-sobreviventes. Quando se comparou os níveis de MIF com o escore
SOFA (Figura 3.1), verificou-se grande semelhança das diferenças entre controles e pacientes
sépticos sobreviventes e não-sobreviventes. Um estudo anterior do nosso grupo já havia
demonstrado que os níveis de MIF são gradativamente maiores quanto maior a gravidade da
sepse (Bozza e cols., 2004). Seu desempenho foi melhor em relação ao prognóstico, com
AUROC de 0,79, enquanto a IL-6 apresentou AUROC 0,68 para mortalidade hospitalar. No
estudo de avaliação de cinética, a diferença dos níveis de MIF no 1º dia de choque séptico
marcaram a mortalidade em 28 dias, corroborando achados de estudos prévios. No entanto, a
cinética de MIF foi muito semelhante entre sobreviventes e não-sobreviventes, com curvas
estáveis durante a 1ª semana de choque séptico. Possíveis explicações para este fato são que
os estímulos para a liberação de MIF permaneçam aumentados durante um período
prolongado, como por exemplo níveis elevados de cortisol ou tratamento com esteroides
venosos (Maxime e cols., 2005; Emonts e cols., 2007). A quase totalidade dos pacientes na
nossa coorte recebeu esteroides sintéticos para o tratamento do choque séptico (58% no
primeiro dia do diagnóstico). Nossos resultados são antagônicos a outro estudo que
demonstrou uma cinética crescente de MIF nos 2 primeiros dias de sepse grave prediz
mortalidade em 3 e 7 dias (Chuang e cols., 2007). Verificamos que a cinética de cortisol
também foi marcada por níveis altos e estáveis, principalmente nos não-sobreviventes, e que
os níveis de MIF e cortisol no início do quadro estavam positivamente correlacionados (nos
pacientes sem tratamento com esteroides). O uso de esteroides sintéticos é uma explicação
para o aumento discreto dos níveis de MIF nos sobreviventes nos dias 5 e 7 após início do
choque séptico. Os níveis de MIF no 1º dia de choque séptico apresentaram sensibilidade de
70% e especificidade de 82% para o ponto de corte de 3201 pg/ml, e AUROC 0,73 para
mortalidade em 28 dias, e foi melhor que os escores SAPS II e SOFA, IL-6 e PCRt.

82
A cinética de citocinas durante a evolução da sepse grave demonstrou 4 padrões
distintos. Interleucinas 1, 5 e 10, MIP-1β e MCP-1 se comportaram com níveis elevados até o
3º dia, com posterior queda significativa. Um padrão de queda exponencial a partir do 1º dia
foi visto com IL-6, IL-13, IFN-γ e G-CSF. Níveis estáveis de MIF e crescentes de IL-8
completaram os diferentes perfis durante a 1ª semana de choque séptico. Estudos recentes
demonstraram cinéticas de algumas citocinas durante o início do acompanhamento de
pacientes com sepse. Oda e colaboradores demonstraram cinética de queda exponencial de
IL-6 durante a primeira semana de sepse, com queda mais rápida em pacientes que
sobrevivem (Oda e cols., 2005). Eles também correlacionaram os níveis de IL-6 com o escore
SOFA e os níveis séricos de plaquetas. Van Zoelen e cols demonstraram que os níveis da
citocina High Mobility Group Box-1 (HMGB1) estão elevados em pacientes sépticos com
pneumonia e peritonite nos dias 1 e 4 de acompanhamento, e esta citocina se correlacionou
com IL-6, IL-8 e IL-10 nas medidas de cinética (Van Zoelen e cols., 2007), que
permaneceram em níveis elevados durante as primeiras 72 horas da sepse. Oberholzer e cols
(2005) mediram os níveis de¨TNF-α, receptor solúvel de TNF (TNFR), e interleucinas 6, 8 e
10 por 4 dias em coorte de 124 pacientes após o início de sepse grave, e somente IL-6 foi
preditora independente de mortalidade. Porém neste estudo, as variações temporais de
cinética das citocinas não se alteraram significativamente neste período, talvez pelo curto
tempo de observação, já que algumas citocinas aumentaram até o 3º dia na nossa coorte. A
cinética de IL-6 no nosso estudo, por exemplo, apresentou queda exponencial em
sobreviventes, enquanto permaneceu elevada até o 3º dia em não-sobreviventes (Figura 3.3).
Os estudos com avaliação de cinética mais prolongada são escassos e corroboram
parcialmente nossos resultados. Maier e cols (2007) mediram semanalmente TNF-α, IL-6, IL-
8, IL-10 e TNFR no plasma de 352 pacientes com politrauma grave em vários hospitais da
Alemanha. Os níveis destas citocinas se associaram com a evolução da SDOM tardiamente, e
IL-6 e TNFR se associaram com mortalidade hospitalar. Não houve diferenças nas cinéticas
de IL-6, IL-8 e IL-10 entre sobreviventes e não-sobreviventes na 1ª semana após o trauma
grave, embora IL-6 tenha aumentado em torno do 4º dia nos não-sobreviventes. A dosagem
de citocinas foi realizada duas vezes na primeira semana e apenas uma vez por semana
posteriormente, não caracterizando a visualização de uma cinética detalhada. Os autores
recomendaram o uso de IL-6 e TNFR para prever o aparecimento de SDOM tardiamente. Wu
e cols (2009) estudaram a cinética de IL-6, IL-10, IL-12, transforming growth factor beta 1
(TGFβ-1) e IFN-γ durante a 1ª semana em 76 pacientes com sepse grave, dos quais menos
que 50% tinha choque séptico. A interleucina 6 foi associada com a presença de choque e IL-

83
10 foi associada independentemente com mortalidade. Medidas iniciais e cinéticas das outras
citocinas neste estudo não se diferenciaram entre sobreviventes e não-sobreviventes. Existem
diferenças entre estes 2 estudos e os nossos resultados. Nossa coorte incluiu pacientes mais
graves, o que torna os resultados mais significativos em população mais homogênea (todos
tinham choque séptico). O painel de biomarcadores no nosso trabalho é mais amplo, e inclui
biomarcadores aplicados no dia-a-dia na UTI, como PCRt, e também o cortisol, que está
envolvido no eixo imunoendócrino ativado na sepse. Outra diferença é na forma de analisar
os resultados. Traçamos um paralelo entre análises bem conhecidas de evolução do escore
SOFA para disfunções orgânicas e o prognóstico de pacientes com sepse. Ferreira e cols
(2001) estudaram valores do escore SOFA durante a internação de pacientes em UTIs gerais e
verificaram que a média de SOFA de todos os dias de internação e o valor máximo do escore
apresentaram melhor predição de mortalidade que o SOFA do 1º dia de acompanhamento.
Fizemos estas análises para todos os biomarcadores, assim como para o escore SOFA.
Encontramos resultados significativamente heterogêneos se a análise é realizada apenas com
dados do 1º dia de choque séptico (como na maioria dos estudos com biomarcadores na
sepse) ou com os valores médios e máximos durante a evolução na 1ª semana. É possível que
interações entre os diversos biomarcadores sejam mais elucidativas que a análise isolada de
cada um para com o desfecho. Para isso, seria necessário fazer análise combinatória entre os
vários parâmetros, para explorar amplamente todas as possibilidades. Uma opção é calcular a
relação entre duas ou mais citocinas (Mainer e cols., 2007). Fizemos a análise da relação entre
os valores de IL-6 e IL-10, partindo do princípio que pacientes com maior relação teria
padrão predominantemente pró-inflamatório e aqueles com menor relação teriam padrão anti-
inflamatório, porém não obtivemos resultados significativos. Isto é demonstrado pelos
gráficos de cinética, nos quais citocinas pró-inflamatórias apresentaram padrão semelhante de
cinética que as anti-inflamatórias.
Finalmente, os melhores desempenhos para os valores de cinética para predição de
mortalidade em 28 dias foram de cortisol plasmático. Nossos resultados são semelhantes aos
vistos em outros estudos. Melby e Spink (1958) já haviam demonstrado que o cortisol sérico é
significativamente maior em pacientes que morrem de choque séptico há 5 décadas. Annane e
cols (2000) demonstraram que pacientes sépticos com cortisol sérico maior que 34 µg/dl
apresentavam maior mortalidade, principalmente se não havia aumento do cortisol após
administração de análogo de ACTH, indicando disfunção adrenal. O cortisol é marcador de
gravidade e mortalidade em pacientes com pneumonia comunitária grave (Christ-Crain e
cols., 2007; Salluh e cols., 2008b). No estudo de Salluh e cols., o cortisol sérico no início da

84
internação para tratamento de pneumonia apresentou melhor desempenho (AUROC 0,77)
para mortalidade do que os escores APACHE II e o CURB-65 (escore que avalia confusão
mental, ureia plasmática, taquipneia, hipotensão arterial e idade superior a 65 anos) e
biomarcadores como PCRt e D-dímero. No nosso trabalho, o cortisol plasmático foi o
biomarcador com melhor desempenho com a análise dos valores médios e máximos durante a
1ª semana de choque séptico e foi independentemente associado à mortalidade em 28 dias.
O painel analisado no nosso estudo é a oportunidade de associar desfechos clínicos,
como a gravidade de disfunções orgânicas e a mortalidade, com diferentes padrões de
biomarcadores. Citocinas como IL-6 e IL-8 são boas preditoras da evolução para SDOM e
morte até 3 dias do diagnóstico de choque séptico. Outros marcadores são preditores de
prognóstico, tanto no primeiro dia de sepse (MIF), quanto durante a primeira semana (cortisol
e IL-6). A associação de alguns destes biomarcadores aos escores prognósticos já utilizados
no dia-a-dia nas UTIs pode elevar o desempenho de risco para SDOM e mortalidade.
A sepse está associada com alterações metabólicas e existem evidências que apontam
um papel crítico da atividade mitocondrial na patogênese da SDOM (Crouser e cols., 2006;
Belikova e cols., 2007; D´Ávila e cols., 2008). A disfunção mitocondrial pode ocorrer por
alterações da atividade de complexos da cadeia fosforilativa, dissipação do gradiente de
elétrons e desacoplamento da fosforilação oxidativa. Estas alterações parecem acontecer em
diferentes graus em diferentes tecidos. Por exemplo, no tecido cerebral as alterações de fluxo
de elétrons e de desacoplamento da respiração já foram demonstradas experimentalmente
(D`Ávila e cols., 2008). No tecido muscular, complexos I e IV estão em concentração
reduzida em pacientes com sepse grave (Brealey e cols., 2002; Fredriksson e cols., 2006). O
controle estrito da glicemia teve influência na arquitetura e na concentração de complexo IV
de hepatócitos de pacientes com sepse grave (Vanhorebeek e cols., 2005).
Estudos recentes associaram critérios de disfunção celular ou de tecidos com a
redução da atividade bioenergética vista na sepse grave. A redução da respiração mitocondrial
em estado 3 (induzida após adição de ADP) em PBMCs de voluntários saudáveis ocorreu
quando se incubou estas células com o plasma de pacientes com sepse grave, ao passo que a
expressão de antígeno HLA-DR também foi significativamente reduzida em monócitos de
pacientes sépticos (Belikova e cols., 2007). A fraqueza muscular generalizada decorrente de
polineuromiopatia também foi associada a redução de complexos mitocondriais I e IV e de
ATP em músculos intercostais e de perna (Fredriksson e cols., 2006). No entanto, a
correlação funcional de alterações mitocondriais com parâmetros clínicos e mortalidade não
foi analisada. Isto se deve talvez pela dificuldade de conseguir amostras de tecidos de

85
pacientes sépticos sem métodos invasivos, como biópsia de tecidos. A nossa escolha pelo
estudo de células mononucleares circulantes se impôs pela facilidade em acessá-las através de
punção venosa periférica, além de saber que elas podem participar da patogênese da
“imunoparalisia”, que deixa os pacientes com sepse grave mais predispostos a infecções
secundárias durante o decorrer de dias a semanas (Monnerat, 2005). Nossos resultados
mostraram que PBMCs de pacientes com choque séptico apresentaram redução significativa
da respiração mitocondrial no estado 3, que se inicia quando se adiciona ADP e expressa o
estado metabólico para síntese de ATP. Este resultado foi obtido com a comparação de
PBMCs de pacientes igualmente internados em UTI, em período recente de pós-operatórios
(24-48 horas). Curiosamente, a respiração mitocondrial nos outros estados metabólicos
(estado 2 após adição de succinato e estado 4 após oligomicina) não foi diferente entre células
de controles e sépticos. Isto indica que a principal alteração na respiração mitocondrial em
PBMCs está no estado associado à síntese de ATP. E esta alteração ocorre nas primeiras 24
horas após início do choque séptico, e pode ser um dos estímulos mais precoces para a gênese
da SDOM. Já havia evidência de redução do estado 3 em PBMCs, com incubação de células
de voluntários sadios com plasma de pacientes com sepse grave (Belikova e cols., 2007). Este
mesmo trabalho mostrou a presença de desacoplamento da respiração mitocondrial, após
adição de FCCP, entretanto as evidências são indiretas, já que, além de células de voluntários
saudáveis, o resultado estatístico foi obtido através de alteração da taxa de respiração
mitocondrial com porcentagem entre antes e depois da adição de FCCP. Nossos resultados
demonstram que não há alteração de desacoplamento energético, já que o nível de consumo
de oxigênio volta ao nível da respiração estado 3 tanto em pacientes sépticos quanto em
controles (Figura 4.1). A “saúde” mitocondrial, expressa pela relação entre estado 3 e 4,
também se mostrou significativamente reduzida em PBMCs sépticos.
A respiração mitocondrial no estado metabólico para síntese de ATP foi associada ao
grau de disfunções orgânicas e à mortalidade hospitalar. Quanto menor o consumo de
oxigênio após adição de ADP, maior o escore SOFA na população estudada (Figura 4.3).
Vemos de maneira clara que pacientes controles estão concentrados na parte esquerda do
gráfico, com maior consumo de oxigênio induzido por ADP e menores escores SOFA,
enquanto os pontos de pacientes sépticos que morreram estão na parte direita do gráfico, com
maior SOFA e menor consumo de oxigênio. Existe heterogeneidade de resultados entre os
pacientes com choque séptico que sobreviveram. Esta correlação entre disfunções orgânicas e
medidas de estruturas ou atividade mitocondrial foram feitas anteriormente com focos
diferentes. Brealey encontrou correlação entre as doses de noradrenalina usada no tratamento

86
do choque séptico e os níveis de complexo I mitocondrial em células musculares (Brealey e
cols., 2002). Níveis de complexos I e IV reduzidos se associaram à menor força muscular
respiratória em pacientes com ventilação mecânica (Fredriksson e cols., 2006).
O consumo de oxigênio induzido por ADP foi decrescente na análise da Figura 4.2,
quando se analisa pacientes controles, sobreviventes e não-sobreviventes, com diferença
significativa entre os 3 subgrupos (p<0,05, ANOVA). Em apenas 5 pacientes que
sobreviveram à sepse, a taxa de controle respiratório aumentou significativamente após 1
semana, indicando melhora na eficiência para síntese de ATP, porém sem comparação ainda
em pacientes que sobreviveram por 1 semana, mas faleceram no seguimento do estudo. A
biogênese mitocondrial é reconhecida como um mecanismo de reparo de processos
patológicos, porém sem evidências na sepse e na SDOM até o momento (Nisoli e cols., 2003;
Protti e cols., 2006).
A redução no consumo de oxigênio induzido por ADP em células mononucleares de
sangue periférico de pacientes sépticos foi causada pela disfunção da F1Fo-ATP sintase. O
estado metabólico 3, no qual ocorre síntese de ATP, é modulado pela função da enzima F1Fo-
ATP sintase e pela quantidade de substratos ADP e fosfato transportados para o citossol
mitocondrial pelo ANT e carreador de fosfato, respectivamente. Conduzimos experimento
com titulação de inibidores destas vias, com exceção do carreador de fosfato que não tem
inibidor específico atualmente, para apontar o provável mecanismo responsável pela redução
do estado 3 no choque séptico (Figura 4.4). Adições repetidas de pequenas doses de
oligomicina (inibidor da F1Fo-ATP sintase) e atractilosídeo (inibidor do ANT) foram
realizadas para encontrar a dose mínima destes para suprimir o consumo de oxigênio induzido
após ADP. O conteúdo funcional de ANT foi semelhante nos 2 grupos, enquanto o conteúdo
de F1Fo-ATP sintase foi significativamente menor em células mononucleares de sangue
periférico de pacientes com choque séptico. A representação esquemática dos mecanismos de
disfunção mitocondrial em células mononucleares de sangue periférico de pacientes com
choque séptico está na Figura 5.1.
Este resultado corrobora achado de proteômica de voluntários após injeção de LPS,
nos quais os genes envolvidos na fosforilação oxidativa, como da F1Fo-ATP sintase, têm
expressão reduzida (Calvano e cols., 2005). Alguns componentes da fosforilação podem ser
inibidos em condições de estresse oxidativo (Kantrow e cols., 2000). Doenças inflamatórias
podem influenciar na respiração mitocondrial através de mediadores, como as citocinas. A
adição de citocinas pode promover a inibição da F1Fo-ATP sintase em modelos

experimentais in vitro com adição de TNF-α (Shchepina e cols., 2002; Cortés-Hernández e
cols., 2005; Samavati e cols., 2008) e IL-6 (Berthiaume e cols., 2003).
ciclo de Krebs
espaço intermembrana
matriz
potencial transmembrana
ANT
ADP?
ATP
UCP
perfusãotecidual
hidrólise ATP ?
?
NO
Figura 5.1: Representação esquemática do mecanismo proposto pelo qual o choque séptico
causa disfunção mitocondrial em PBMCs. O retângulo amarelo representa a F1Fo-ATP
sintase, que apresenta conteúdo funcional reduzido. As setas vermelhas indicam possíveis
mecanismos que ainda permanecem desconhecidos para a respiração mitocondrial de PBMCs
na sepse. A participação de proteínas desacopladoras (UCP) e do conteúdo funcional do
trocador de nucleotídeos adenínicos (ANT) permanece inconclusiva. Os complexos da cadeia
transportadora de elétrons geram gradiente de prótons (H+) através da membrana interna
usando oxigênio como ponte de elétrons. Os números em romano indicam a classificação dos
complexos da cadeia fosforilativa. UQ, ubiquinona; Cyt c, citocromo c; F1Fo, F1Fo ATP
sintase; FAD – dinucleotídeo flavina e adenina; NAD – dinucleotídeo nicotinamida e adenina;
ADP – adenosina difosfato; ATP – adenosina trifosfato.
87

88
A perda do potencial de membrana mitocondrial já foi mostrada em casos de sepse
grave na fase mais tardia, que é correlacionada com o aparecimento de marcadores de
apoptose (Adrie e cols., 2001). O aumento da apoptose de PBMCs, especificamente
linfócitos, foi demonstrada em grupo de pacientes com sepse grave (Hotchkiss e cols., 2005).
A correlação entre a funcionalidade da maquinaria de fosforilação oxidativa mitocondrial e a
sinalização intracelular a partir de estímulo inflamatório ainda não foi explorada. É sabido
que a IL-6 tem ação indireta na inibição da F1Fo-ATP sintase através da sinalização a partir
de receptores celulares com tirosina quinase (Berthiaume e cols., 2003), mas as ações de
outros citocinas, como MIF e MCP-1 que estão associadas a maior mortalidade na sepse
grave, ainda não foram estudadas. A análise conjunta entre citocinas e atividade mitocondrial
dos nossos pacientes proporcionaram apenas correlação com tendência a significância
estatística de MIF com respiração mitocondrial no estado 3 (p=0,056; r=0,30). Interpretamos
que o tamanho da população para esta análise de correlação ainda é pequena, mas pode ser
promissora. Estudos experimentais podem ser conduzidos no sentido de entender o quanto a
sinalização intracelular após estímulos inflamatórios e infecciosos pode influenciar a
atividade mitocondrial. Atualmente a sinalização mais conhecida é de ativação de caspases
por citocromo c liberado por mitocôndrias, com ativação de apoptose (Hotchkiss e cols,
2005).
Nossos resultados são pioneiros na demonstração funcional da fosforilação de ADP
mitocondrial em células imunes de pacientes sépticos. No entanto, devemos aprofundar o
conhecimento sobre a disfunção da enzima F1Fo-ATP sintase em PBMCs com estudos
quantitativos da enzima nas células mononucleares circulantes (Western blot). Sabemos que o
conteúdo funcional está reduzido, mas resta saber se ocorre deficiência quantitativa ou
qualitativa, e verificar o peso destas alterações. Os experimentos com atractilosídeo, inibidor
do ANT, foram inconclusivos em relação à inibição da respiração mitocondrial, mesmo
usando doses muito pequenas sem diferenças entre pacientes sépticos e controles. Não se sabe
se o conteúdo deste trocador está reduzido em estados inflamatórios ou outro fator pode inibir
seu funcionamento in vitro.
A medida de concentrações de complexos da cadeia fosforilativa ficou limitada neste
estudo. A quantidade de células, e consequentemente o isolamento de mitocôndrias, é
limitada pela coleta de sangue, que se exagerada pode depauperar a condição clínica de
pacientes com choque. Trabalhos que utilizam amostras de tecidos têm a vantagem de isolar
mitocôndrias ou adquirir grande quantidade de células e dosar complexos e produtos de
mitocôndrias (Brealey e cols, 2002; Vanhorebeek e cols., 2005; Fredrikssen e cols., 2006).

89
Com o uso de oxígrafo de alta resolução a partir da segunda metade do nosso trabalho, passou
a ser possível armazenar parte das células extraídas de sangue total dos pacientes e
possibilitará a complementação destas medidas.
O cálculo de consumo de oxigênio em PBMCs intactos (sem permeabilização) pode
apontar para diferenças no uso de oxigênio entre pacientes sépticos e controles; as figuras que
mostram consumo de oxigênio sugerem haver alguma redução no consumo de oxigênio,
mesmo no estado 2 ou 4, embora seja muito mais visível no estado 3. A utilização de
oxigênio por mitocôndrias de PBMCs de pacientes sépticos pode ser deficiente por
mecanismos ainda desconhecidos.
A evolução da respiração mitocondrial ao longo do tempo pode identificar correlações
clínicas com determinadas disfunções orgânicas específicas e desfecho; temos dados de
recuperação clínica com aumento da respiração e na taxa de controle respiratório, mas não
sabemos se a ausência da recuperação nesta taxa também prediz ou acompanha prognóstico
ruim em não-sobreviventes. Não afastamos a participação de radicais livres de oxigênio na
inibição da F1Fo-ATP sintase, embora a respiração após adição de FCCP não aponte para
diferenças de desacoplamento energético entre controles e pacientes sépticos.
O tamanho amostral de 70 pacientes é comum a outros estudos (Oda e cols., 2005;
Bozza e cols., 2007; Wu e cols., 2009) e limita de certa forma alguns resultados. Este
tamanho amostral é relativamente pequeno para aprofundar certas análises de correlação com
disfunções orgânicas específicas, porque a variância dos resultados de alguns biomarcadores,
como IL-6 e MCP-1, é alta, diferentemente de estudos experimentais com animais isogênicos.
Nós transformamos alguns valores de citocinas para unidade logarítmica para poder analisar e
homogenizar os resultados (Bozza e cols., 2007).
Finalmente, espera-se que nossos resultados contribuam para o melhor conhecimento
de fisiopatologia da sepse grave, melhorem a acurácia do diagnóstico e da avaliação da
gravidade do quadro de sepse e possibilitem o entendimento mais amplo da evolução da
Síndrome de Disfunção Orgânica Múltipla associada à sepse.

90
6 - Conclusões finais:
- Um perfil temporal de citocinas ampliou o conhecimento de fisiopatologia da sepse em
relação a aspectos clínicos como disfunções orgânicas e mortalidade;
- Citocinas e outros biomarcadores apresentam quatro padrões de cinética durante a primeira
semana após início de choque séptico, o que corresponde a diferentes aplicações clínicas para
diagnóstico, gravidade e prognóstico de pacientes com choque séptico;
- Encontramos biomarcadores preditores de mortalidade precoce (IL-6, IL-8, IL-10 e MCP-1)
e mortalidade em 28 dias (MIF, cortisol e IL-6);
- Células mononucleares circulantes de pacientes com choque séptico apresentam disfunção
mitocondrial, que se correlaciona com a gravidade das disfunções orgânicas e mortalidade
hospitalar;
- A redução do conteúdo funcional da F1Fo-ATP sintase é o mecanismo responsável pela
disfunção mitocondrial em células mononucleares circulantes de pacientes com choque
séptico.

91
Referências bibliográficas
Abraham E, Anzueto A, Gutierrez G, Tessler S, San Pedro G, Wunderink R, Nogare A, Nasraway S, Berman S, Cooney R, Levy H 1988. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock – NORASEPT II. Lancet 351: 929-933.
Adrie C, Bachelet M, Vayssier-Taussat M, Russo-Marie F, Bouchaert I, Adib-Conquy M, Cavaillon JM, Pinsky MR, Dhainaut JF, Polla BS 2001. Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am J Respir Crit Care Med 164: 389-395.
Adrie C, Alberti C, Chaix-Couturier C, Azoulay E, De Lassence A, Cohen Y, Meshaka P, Cheval C, Thuong M, Troché G, Garrouste-Orgeas M, Timsit JF 2005. Epidemiology and economic evaluation of severe sepsis in France: age, severity, infection site, and place of acquisition (community, hospital, or intensive care unit) as determinants of workload and cost. J Crit Care 20:46-58.
Alberti C, Brun-Buisson C, Burchardi H, Martin C, Goodman S, Artigas A, Sicignano A, Palazzo M, Moreno R, Boulmé R, Lepage E, Le Gall R 2002. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med 28:108-121.
Alberti C, Brun-Buisson C, Goodman SV, Guidici D, Granton J, Moreno R, Smithies M, Thomas O, Artigas A, Le Gall J 2003. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med 168:77-84.
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR 2001. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303-1310.
Annane D, Sébille V, Troché G, Raphaël JC, Gajdos P, Bellissant E 2000. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA 283:1038-1045.
Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troché G, Chaumet-Riffaud P, Bellissant E 2002. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 288:862-871.
Annane D, Aegerter P, Jars-Guincestre MC, Guidet B 2003. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med 168:165-172.
Annane D, Maxime V, Ibrahim F, Alvarez JC, Abe E, Boudou P 2006. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am J Respir Crit Care Med 174:1319-1326.
Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R 1997. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol 150:235-246.
Baggiolini M, Dewald B, Moser B 1997. Human chemokines: an update. Annu Rev Immunol 15:675-705.

92
Ballester JCA, Ballester F, González Sánchez A, Almela Quilis A, Colomer Rubio E, Peñarroja Otero C 2008. Epidemiology of sepsis in the Valencian Community (Spain), 1995-2004. Infect Control Hosp Epidemiol 29:630-634.
Barichello T, Fortunato JJ, Vitali AM, Feier G, Reinke A, Moreira JC, Quevedo J, Dal-Pizzol F 2006. Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Crit Care Med 34:886-889.
Barton BE 1997. IL-6: insights into novel biological activities. Clin Immunol Immunopathol 85:16-20.
Beale R, Reinhart K, Brunkhorst FM, Dobb G, Levy M, Martin G, Martin C, Ramsey G, Silva E, Vallet B, Vincent JL, Janes JM, Sarwat S, Williams MD 2009. Promoting Global Research Excellence in Severe Sepsis (PROGRESS): lessons from an international sepsis registry. Infection 37:222-232.
Belikova I, Lukaszewicz AC, Faivre V, Damoisel C, Singer M, Payen D 2007. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit Care Med 35: 2702-2708.
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr 2001. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344:699-709.
Bernard N, Matecki S, Py G, Lopez S, Mercier J, Capdevila X 2003. Effects of prolonged mechanical ventilation on respiratory muscle ultrastructure and mitochondrial respiration in rabbits. Intensive Care Med 29:111-118.
Beishuizen A, Thijs LG, Haanen C, Vermes I 2001. Macrophage Migration Inhibitory Factor and Hypothalamo-Pituitary-Adrenal Function during Critical Illness. J Clin Endocrinol Metabol 86:2811-2816.
Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N, Grunig G, Rennick D 1995. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest 96:2339-2347.
Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R 1993. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365:756-759.
Berthiaume F, MacDonald AD, Kang YH, Yarmush ML 2003. Control analysis of mitochondrial metabolism in intact hepatocytes: effect of interleukin-1beta and interleukin-6. Metab Eng 5:108-123.
Boczkowski J, Lisdero CL, Lanone S, Samb A, Carreras MC, Boveris A, Aubier M, Poderoso JJ 1999. Endogenous peroxynitrite mediates mitochondrial dysfunction in rat diaphragm during endotoxemia. FASEB J 13:1637-1646.
Boekstegers P, Weidenhöfer S, Kapsner T, Werdan K 1994. Skeletal muscle partial pressure of oxygen in patients with sepsis. Crit Care Med 22:640-650.
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ 1992. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine Chest 101:1644–1655.

93
Bornstein SR, Schumann RR, Rettori V, McCann SM, Zacharowski K 2004. Toll-like receptor 2 and Toll-like receptor 4 expression in human adrenals. Horm Metab Res 36:470-473.
Bossink AW, Paemen L, Jansen PM, Hack CE, Thijs LG, Van Damme J 1995. Plasma levels of the chemokines monocyte chemotactic proteins-1 and -2 are elevated in human sepsis. Blood 86:3841-3847.
Boveris A, Alvarez S, Navarro A 2002. The role of mitochondrial nitric oxide synthase in inflammation and septic shock. Free Radic Biol Med 33:1186-1193.
Bouchud PY, Calandra T 2003. Pathogenesis of sepsis: new concepts and implications for future treatment. BJM 326:262-266.
Blanco J, Muriel-Bombín A, Sagredo V, Taboada F, Gandía F, Tamayo L, Collado J, García-Labattut A, Carriedo D, Valledor M, De Frutos M, López MJ, Caballero A, Guerra J, Alvarez B, Mayo A, Villar J 2008. Incidence, organ dysfunction and mortality in severe sepsis: a Spanish multicentre study. Crit Care 12:R158.
Bloom BR, Bennett B 1966. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 153:80-82.
Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR 1999. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 189:341-346.
Bozza FA, Gomes RN, Japiassu AM, Soares M, Castro-Faria-Neto HC, Bozza PT, Bozza MT 2004. Macrophage Migration Inhibitory Factor Levels Correlate with Fatal Outcome in Sepsis. Shock 22:309-313.
Bozza FA, Bozza PT, Castro Faria Neto HC 2005. Beyond sepsis pathophysiology with cytokines: what is their value as biomarkers for disease severity? Mem Inst Oswaldo Cruz 100 Suppl 1:217-221.
Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, Bozza MT, Castro-Faria-Neto HC, Bozza PT 2007. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care 11:R49.
Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R, Heusser CH, Howarth PH, Holgate ST 1994. Interleukin-4, -5, and -6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol 10:471-480.
Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M 2002. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360: 219-223.
Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M 2004. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 286:R491-497.
Brem AS, Bina RB, Mehta S, Marshall J 1999. Glucocorticoids inhibit the expression of calcium-dependent potassium channels in vascular smooth muscle. Mol Genet Metab 67: 53-57.
Broaddus VC, Boylan AM, Hoeffel JM, Kim KJ, Sadick M, Chuntharapai A, Hebert CA 1994. Neutralization of IL-8 inhibits neutrophil influx in a rabbit model of endotoxin-induced pleurisy. J Immunol 152:2960-2967.

94
Broder G, Weil MH 1964. Excess lactate: an index of reversibility of shock in human patients. Science 143:1457-1459.
Brown-Séquard CE 1856. Recherches expérimentales sur la physiologie et la pathologie des capsules surrénales. C R Acad Sci [D] Paris 43:422– 425.
Brueckmann M, Marx A, Weiler HM, Liebe V, Lang S, Kaden JJ, Zieger W, Borggrefe M, Huhle G, Konstantin Haase K 2003. Stabilization of monocyte chemoattractant protein-1-mRNA by activated protein C. Thromb Haemost 89:149-160.
Brun-Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, Mercier JC, Offenstadt G, Régnier B 1995. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA 274:968-74.
Brun-Buisson C, Meshaka P, Pinton P, Vallet B 2004. EPISEPSIS: a reappraisal of the epidemiology and outcome of severe sepsis in French intensive care units. Intensive Care Med 30:580-588.
Burrows SD, Doyle ML, Murphy KP, Franklin SG, White JR, Brooks I, McNulty DE, Scott MO, Knutson JR, Porter D 1994. Determination of the monomer-dimer equilibrium of interleukin-8 reveals it is a monomer at physiological concentrations. Biochemistry 33:12741-12745.
Calandra T, Bernhagen J, Mitchell RA, Bucala R 1994. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med 179:1895-1902.
Calandra T, Bucala R 1995. Macrophage migration inhibitory factor: a counter-regulator of glucocorticoid action and critical mediator of septic shock. J Inflamm 47:39-51.
Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP 2000. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med 6:164-170.
Calandra T, Gerain J, Heumann D, Baumgartner JD, Glauser MP 1991. High circulating levels of interleukin-6 in patients with septic shock: evolution during sepsis, prognostic value, and interplay with other cytokines. The Swiss-Dutch J5 Immunoglobulin Study Group. Am J Med 91:23-29.
Calandra T, Roger T 2003. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3:791-800.
Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF 2005. A network-based analysis of systemic inflammation in humans. Nature 437:1032-1037.
Campbell I 2005. Adrenocortical hormones. Anaesth Intensive Care Med 6:326-329. Cassina A, Radi R 1996. Differential inhibitory action of nitric oxide and peroxynitrite on
mitochondrial electron transport. Arch Biochem Biophys 328:309-316. Castro L, Rodriguez M, Radi R 1994. Aconitase is readily inactivated by peroxynitrite, but
not by its precursor, nitric oxide. J Biol Chem 269:29409-29415. Chance B, Williams GR 1955. Respiratory enzymes in oxidative phosphorylation. I. Kinetics
of oxygen utilization. J Biol Chem 217: 383-393.

95
Cheng B, Xie G, Yao S, Wu X, Guo Q, Gu M, Fang Q, Xu Q, Wang D, Jin Y, Yuan S, Wang J, Du Z, Sun Y, Fang X 2007. Epidemiology of severe sepsis in critically ill surgical patients in ten university hospitals in China. Crit Care Med 35:2538-2546.
Chollet-Martin S, Montravers P, Gibert C, Elbim C, Desmonts JM, Fagon JY, Gougerot-Pocidalo MA 1993. High levels of interleukin-8 in the blood and alveolar spaces of patients with pneumonia and adult respiratory distress syndrome. Infect Immun 61:4553-4559.
Christ-Crain M, Stolz D, Jutla S, Couppis O, Müller C, Bingisser R, Schuetz P, Tamm M, Edwards R, Müller B, Grossman AB 2007. Free and total cortisol levels as predictors of severity and outcome in community-acquired pneumonia. Am J Respir Crit Care Med 176:913-920.
Christaki E, Opal SM 2008. Is the mortality rate for septic shock really decreasing? Curr Opin Crit Care 14:580-586.
Chuang CC, Wang ST, Chen WC, Chen CC, Hor LI, Chuang AY 2007. Increases in serum macrophage migration inhibitory factor in patients with severe sepsis predict early mortality. Shock 27:503-506.
Clec'h C, Ferriere F, Karoubi P, Fosse JP, Cupa M, Hoang P, Cohen Y 2004. Diagnostic and prognostic value of procalcitonin in patients with septic shock. Crit Care Med 32:1166-1169.
Cortés-Hernández P, Domínguez-Ramírez L, Estrada-Bernal A, Montes-Sánchez DG, Zentella-Dehesa A, de Gómez-Puyou MT, Gómez-Puyou A, García JJ 2005. The inhibitor protein of the F1F0-ATP synthase is associated to the external surface of endothelial cells. Biochem Biophys Res Commun 330:844-849.
Crouser ED, Julian MW, Blaho DV, Pfeiffer DR 2002a. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med 30: 276-284.
Crouser ED, Julian MW, Joshi MS, Bauer JA, Wewers MD, Hart JM, Pfeiffer DR 2002b. Cyclosporin A ameliorates mitochondrial ultrastructural injury in the ileum during acute endotoxemia. Crit Care Med 30: 2722-2728.
Crouser ED 2004. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 4:729-741.
Crouser ED, Julian MW, Huff JE, Mandich DV, Green-Church KB 2006. A proteomic analysis of liver mitochondria during acute endotoxemia. Intensive Care Med 32:1252-1262.
Daly C, Rollins BJ 2003. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: therapeutic opportunities and controversies. Microcirculation 10:247-257.
Damas P, Ledoux D, Nys M, Vrindts Y, De Groote D, Franchimont P, Lamy M 1992. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg 215:356-362.
David JR 1966. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci U S A 56:72-77.
D’Avila JCP, Santiago APSA, Amâncio RT, Galina A, Oliveira MF, Bozza FA 2008. Sepsis induces brain mitochondrial dysfunction. Crit Care Med 36:1925-1932.

96
de Jong MF, Beishuizen A, Spijkstra JJ, Groeneveld AB 2007. Relative adrenal insufficiency as a predictor of disease severity, mortality, and beneficial effects of corticosteroid treatment in septic shock. Crit Care Med 35:1896-1903.
Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C 2004. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood 104:4134-4141.
d'Emmanuele di Villa Bianca R, Lippolis L, Autore G, Popolo A, Marzocco S, Sorrentino L, Pinto A, Sorrentino R 2003. Dexamethasone improves vascular hyporeactivity induced by LPS in vivo by modulating ATP-sensitive potassium channels activity. Br J Pharmacol 140:91-96.
Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, Gea-Banacloche J, Keh D, Marshall JC, Parker MM, Ramsay G, Zimmerman JL, Vincent JL, Levy MM 2004. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Intensive Care Med 30:536-555.
Derhaschnig U, Bergmair D, Marsik C, Schlifke I, Wijdenes J, Jilma B 2004. Effect of interleukin-6 blockade on tissue factor-induced coagulation in human endotoxemia. Crit Care Med 32:1136-1140.
Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, Grant IS, Pollok AJ, Haslett C 1993. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 341:643-647.
Elsouri N, Bander J, Guzman JA 2006. Relative adrenal insufficiency in patients with septic shock; a close look to practice patterns. J Crit Care 21:73-77.
Emonts E, Sweep FCGJ, Grebenchtchikov N, Geurts-Moespot A, Knaup M, Chanson AL, Erard V, Renner P, Hermans PWM, Hazelzet JA, Calandra T 2007. Association between High Levels of Blood Macrophage Migration Inhibitory Factor, Inappropriate Adrenal Response, and Early Death in Patients with Severe Sepsis. Clin Infect Dis 44:1321-1328.
Engel C, Brunkhorst FM, Bone HG, Brunkhorst R, Gerlach H, Grond S, Gruendling M, Huhle G, Jaschinski U, John S, Mayer K, Oppert M, Olthoff D, Quintel M, Ragaller M, Rossaint R, Stuber F, Weiler N, Welte T, Bogatsch H, Hartog C, Loeffler M, Reinhart K 2007. Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med 33:606-618.
Feijó J, Westphal GA, Filho MC, Martins SF, Silva FN, Bagnati F, Araújo JP, Kaepfer KM, Bork JC. Avaliação institucional de uma nova metodologia para identificação precoce da sepse em pacientes hospitalizados em um hospital público. Disponível em http://www.saudebrasilnet.com.br/votasepse/trabalhos/005_PR.pdf, em 03/11/2009.
Ferreira FL, Bota DP, Bross A, Mélot C, Vincent JL 2001. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 286:1754-1758.
Finfer S, Bellomo R, Lipman J, et al. Adult-population incidence of severe sepsis in Australian and New Zealand intensive care units. Intensive Care Med, 2004;30:589-596.
Fink MP 2001. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin 17:219-37.
Flaatten H 2004. Epidemiology of sepsis in Norway in 1999. Crit Care 8:R180-184.

97
Fredriksson K, Hammarqvist F, Strigård K, Hultenby K, Ljungqvist O, Wernerman J, Rooyackers O 2006. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am J Physiol Endocrinol Metab 291:E1044-E1050.
Frink M, van Griensven M, Kobbe P, Brin T, Zeckey C, Vaske B, Krettek C, Hildebrand F 2009. IL-6 predicts organ dysfunction and mortality in patients with multiple injuries. Scand J Trauma Resusc Emerg Med 17:49.
Gando S, Nishihira J, Kobayashi S, Morimoto Y, Nanzaki S, Kemmotsu O 2001. Macrophage migration inhibitory factor is a critical mediator of systemic inflammatory response syndrome. Intensive Care Med 27:1187-1193.
Gattinoni L, Brazzi L, Pelosi P, Latini R, Tognoni G, Pesenti A, Fumagalli R 1995. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med 333:1025-1032.
Gomes RN, Bozza FA, Amâncio RT, Japiassú AM, Vianna RC, Larangeira AP, Gouvêa JM, Bastos MS, Zimmerman GA, Stafforini DM, Prescott SM, Bozza PT, Castro-Faria-Neto HC 2006. Exogenous platelet-activating factor acetylhydrolase reduces mortality in mice with systemic inflammatory response syndrome and sepsis. Shock 26:41-49.
Greenman RL, Schein RM, Martin MA, Wenzel RP, MacIntyre NR, Emmanuel G, Chmel H, Kohler RB, McCarthy M, Plouffe J 1991. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA Sepsis Study Group. JAMA 266:1097-1102.
Guzman JA, Guzman CB 2007. Adrenal exhaustion in septic patients with vasopressor dependency. J Crit Care 22:319-323.
Harbuz MS, Rees RG, Eckland D, Jessop DS, Brewerton D, Lightman SL 1992. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology 130:1394-1400.
Harris P, Bateman M, Gloster J 1962. Relations between the cardio-respiratory effects of exercise and the arterial concentration of lactate and pyruvate in patients with rheumatic heart disease. Clin Sci 23:531-543.
Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D 1994. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med 330:1717-1722.
Hayes MA, Timmins AC, Yau EH, Palazzo M, Watson D, Hinds CJ 1997. Oxygen transport patterns in patients with sepsis syndrome or septic shock: influence of treatment and relationship to outcome. Crit Care Med 25:926-936.
Ho JT, Al-Musalhi H, Chapman MJ, Quach T, Thomas PD, Bagley CJ, Lewis JG, Torpy DJ 2006. Septic shock and sepsis: a comparison of total and free plasma cortisol levels. J Clin Endocrinol Metab 91:105-114.
Hodgin UG, Sanford JP 1965. Gram-negative rod bacteremia. An analysis of 100 patients. Am J Med 39:952-60.
Hoffman M, Cooper ST 1995. Thrombin enhances monocyte secretion of tumor necrosis factor and interleukin-1 beta by two distinct mechanisms. Blood Cells Mol Dis 21:156-167.

98
Horan TC, Andrus M, Dudeck MA 2008. CDC/NHSN surveillance definition of health care–associated infection and criteria for specific types of infections in the acute care setting. Am J Infect Control 36:309-332.
Hotchkiss RS, Karl IE 2003. The pathophysiology and treatment of sepsis. N Engl J Med 348:138-150.
Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE 2005. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Imunnol 174: 5110-5118.
Hubbard WJ, Bland KI, Chaudry IH 2004. The role of the mitochondrion in trauma and shock. Shock 22: 395-402.
Jansen PM, van Damme J, Put W, de Jong IW, Taylor FB, Jr., Hack CE 1995. Monocyte chemotactic protein 1 is released during lethal and sublethal bacteremia in baboons. J Infect Dis 171:1640-1642.
Japiassú AM, Salluh JI, Bozza PT, Bozza FA, Castro-Faria-Neto HC 2009. Revisiting steroid treatment for septic shock: molecular actions and clinical effects--a review. Mem Inst Oswaldo Cruz 104:531-548.
Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM 2001. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. Faseb J 15:43-58.
Kantrow SP, Tatro LG, Piantadosi CA 2000. Oxidative stress and adenine nucleotide control of mitochondrial permeability transition. Free Radic Biol Med. 28: 251-260.
Karlsson S, Varpula M, Ruokonen E Pettilä V, Parviainen I, Ala-Kokko TI, Kolho E, Rintala EM 2007. Incidence, treatment, and outcome of severe sepsis in ICU-treated adults in Finland: the Finnsepsis study. Intensive Care Med 33:435-443.
Kellum JA, Kong L, Fink MP, Weissfeld LA, Yealy DM, Pinsky MR, Fine J, Krichevsky A, Delude RL, Angus DC 2007. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med 167:1655-1663.
Knaus WA, Draper EA, Wagner DP, Zimmerman JE 1985. APACHE II: a severity of disease classification system. Crit Care Med 13:818-829.
Khwannimit B, Bhurayanontachai R 2009. The epidemiology of, and risk factors for, mortality from severe sepsis and septic shock in a tertiary-care university hospital setting. Epidemiol Infect 137:1333-1334.
Kumar A 2009. Optimizing Antimicrobial Therapy in Sepsis and Septic Shock. Crit Care Clin 25:733-751.
Landry DW, Levin HR, Gallant EM, Ashton RC Jr, Seo S, D'Alessandro D, Oz MC, Oliver JA 1997. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 95:1122-1125.
Landry DW, Oliver JA 2001. The pathogenesis of vasodilatory shock. N Engl J Med 345:588-595.
Le Gall JR, Lemeshow S, Saulnier F 1993. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA 270:2957-2963.
Lehmann LE, Novender U, Schroeder S, Pietsch T, von Spiegel T, Putensen C, Hoeft A, Stuber F 2001. Plasma levels of macrophage migration inhibitory factor are elevated in patients with severe sepsis. Intensive Care Med 27:1412-1415.

99
Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med 29:530-538.
Levy RJ 2007. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock 28:24-28.
Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS 2004. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock 21:110-114.
Lisboa T, Diaz E, Sa-Borges M, Socias A, Sole-Violan J, Rodríguez A, Rello J 2008. The ventilator-associated pneumonia PIRO score: a tool for predicting ICU mortality and health-care resources use in ventilator-associated pneumonia. Chest 134:1208-1216.
Livaditi O, Kotanidou A, Psarra A, Dimopoulou I, Sotiropoulou C, Augustatou K, Papasteriades C, Armaganidis A, Roussos C, Orfanos SE, Douzinas EE 2006. Neutrophil CD64 expression and serum IL-8: sensitive early markers of severity and outcome in sepsis. Cytokine 36:283-290.
Luzzani A, Polati E, Dorizzi R, Rungatscher A, Pavan R, Merlini A 2003. Comparison of procalcitonin and C-reactive protein as markers of sepsis. Crit Care Med 31:1737-1741.
Mahad DJ, Ransohoff RM 2003. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 15:23-32.
Maier B, Lefering R, Lehnert M, Laurer HL, Steudel WI, Neugebauer EA, Marzi I 2007. Early versus late onset of multiple organ failure is associated with differing patterns of plasma cytokine biomarker expression and outcome after severe trauma. Shock 28:668-674.
Marks JD, Marks CB, Luce JM, Montgomery AB, Turner J, Metz CA, Murray JF 1990. Plasma tumor necrosis factor in patients with septic shock. Mortality rate, incidence of adult respiratory distress syndrome, and effects of methylprednisolone administration. Am Rev Respir Dis 141:94-97.
Marik PE, Gayowski T, Starzl TE 2005. The hepatoadrenal syndrome: a common yet unrecognized clinical condition. Crit Care Med 33:1254-1259.
Marik PE, Pastores SM, Annane D, Meduri GU, Sprung CL, Arlt W, Keh D, Briegel J, Beishuizen A, Dimopoulou I, Tsagarakis S, Singer M, Chrousos GP, Zaloga G, Bokhari F, Vogeser M 2008. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 36:1937-1949.
Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ 1995. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 23:1638-1652.
Marshall JC, Reinhart K 2009. Biomarkers of sepsis. Crit Care Med 37:2290-2298. Martin GS, Mannino DM, Eaton S, Moss M 2003. The epidemiology of sepsis in the United
States from 1979 through 2000. N Engl J Med 348:1546-1554. Marty C, Misset B, Tamion F, Fitting C, Carlet J, Cavaillon JM 1994. Circulating interleukin-
8 concentrations in patients with multiple organ failure of septic and nonseptic origin. Crit Care Med 22:673-679.

100
Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Strieter RM, Kunkel SL 1999. Endogenous monocyte chemoattractant protein-1 (MCP-1) protects mice in a model of acute septic peritonitis: cross-talk between MCP-1 and leukotriene B4. J Immunol 163:6148-6154.
Maxime V, Fitting C, Annane D, Cavaillon JM 2005. Corticoids Normalize Leukocyte Production of Macrophage Migration Inhibitory Factor in Septic Shock. J Infect Dis 191:138–144.
Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A 1995. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest 108:1303-1314.
Meduri GU, Muthiah MP, Carratu P, Eltorky M, Chrousos GP 2005. Nuclear factor-kappaB- and glucocorticoid receptor alpha- mediated mechanisms in the regulation of systemic and pulmonary inflammation during sepsis and acute respiratory distress syndrome. Evidence for inflammation-induced target tissue resistance to glucocorticoids. Neuroimmunomodulation 12:321-38.
Mela L, Bacalzo LV Jr, Miller LD 1971. Defective oxidative metabolism of rat liver mitochondria in hemorrhagic and endotoxin shock. Am J Physiol 220:571-577.
Melby JC, Spink WW 1958. Comparative studies on adrenal cortical function and cortisol metabolism in healthy adults and in patients with shock due to infection. J Clin Invest 37:1791-1798.
Mizock B 1984. Septic shock. A metabolic perspective. Arch Intern Med 144:579-585. Mongardon N, Dyson A, Singer M 2009. Is MOF an outcome parameter or a transient,
adaptive state in critical illness? Curr Opin Crit Care 15:431-436. Monneret G 2005. How to identify systemic sepsis-induced immunoparalysis. Advances in
Sepsis 4:42-49. Morel J, Venet C, Donati Y, Charier D, Liotier J, Frere-Meunier D, Guyomarc'h S, Diconne
E, Bertrand JC, Souweine B, Papazian L, Zeni F 2006. Adrenal axis function does not appear to be associated with hemodynamic improvement in septic shock patients systematically receiving glucocorticoid therapy. Intensive Care Med 32:1184-1190.
Moreno R, Vincent JL, Matos R, Mendonça A, Cantraine F, Thijs L, Takala J, Sprung C, Antonelli M, Bruining H, Willatts S 1999. The use of maximum SOFA score to quantify organ dysfunction/failure in intensive care. Results of a prospective, multicentre study. Intensive Care Med 25:686-696.
Moreno RP, Metnitz B, Adler L, Hoechtl A, Bauer P, Metnitz PG 2008. Sepsis mortality prediction based on predisposition, infection and response. Intensive Care Med 34:496-504.
Murdoch C, Finn A 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood 95:3032-3043.
Murphy JF, Purdue GF, Hunt JL 1993. Acute adrenal insufficiency in the patient with burns. J Burn Care Rehabil 14:155-157.
Nakano Y, Kasahara T, Mukaida N, Ko YC, Nakano M, Matsushima K 1994. Protection against lethal bacterial infection in mice by monocyte-chemotactic and -activating factor. Infect Immun 62:377-383.
Nicholls DG, Ferguson SJ: Bioenergetics 3, 3 ed. London, Academic Press, 2002.

101
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO 2003. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299:896-899.
Oberholzer A, Oberholzer C, Moldawer LL 2002. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med 30 Supl 1:S58-S63.
Oberholzer A, Souza SM, Tschoeke SK, Oberholzer C, Abouhamze A, Pribble JP, Moldawer LL 2005. Plasma cytokine measurements augment prognostic scores as indicators of outcome in patients with severe sepsis. Shock 23:488-493.
Oda S, Hirasawa H, Shiga H, Nakanishi K, Matsuda K, Nakamua M 2005. Sequential measurement of IL-6 blood levels in patients with systemic inflammatory response syndrome (SIRS)/sepsis. Cytokine 29:169-175.
Opal SM, Wherry JC, Grint P 1998. Interleukin-10: potential benefits and possible risks in clinical infectious diseases. Clin Infect Dis 27:1497-1507.
Opal SM, Huber CE 2000. The role of interleukin-10 in critical illness. Curr Opin Infect Dis 13:221-226.
Padkin A, Goldfrad C, Brady AR, Young D, Black N, Rowan K 2003. Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med 31:2332-2338.
Paolini JF, Willard D, Consler T, Luther M, Krangel MS 1994. The chemokines IL-8, monocyte chemoattractant protein-1, and I-309 are monomers at physiologically relevant concentrations. J Immunol 153:2704-2717.
Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E 1993. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 103:565-575.
Poderoso JJ, Carreras MC, Lisdero C, Riobó N, Schöpfer F, Boveris A 1996. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85-92.
Póvoa P, Coelho L, Almeida E, Fernandes A, Mealha R, Moreira P, Sabino H 2005. C-reactive protein as a marker of infection in critically ill patients. Clin Microbiol Infect 11: 101–108.
Presneill JJ, Waring PM, Layton JE, Maher DW, Cebon J, Harley NS, Wilson JW, Cade JF 2000. Plasma granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor levels in critical illness including sepsis and septic shock: relation to disease severity, multiple organ dysfunction, and mortality. Crit Care Med 28:2344-2354.
Protti A, Carré J, Frost MT, Taylor V, Stidwill R, Rudiger A, Singer M 2007. Succinate recovers mitochondrial oxygen consumption in septic rat skeletal muscle. Crit Care Med 35:2150-2155.
Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP 1995. The natural history of the systemic inflammatory response syndroms (SIRS). JAMA 273:117-123.
Rello J, Rodriguez A, Lisboa T, Gallego M, Lujan M, Wunderink R 2009. PIRO score for community-acquired pneumonia: a new prediction rule for assessment of severity in

102
intensive care unit patients with community-acquired pneumonia. Crit Care Med 37:456-462.
Rezende E, Silva JM Jr, Isola AM, Campos EV, Amendola CP, Almeida SL 2008. Epidemiology of severe sepsis in the emergency department and difficulties in the initial assistance. Clinics (São Paulo) 63:457-464.
Rhodes C, Yamada Y 1995. Characterization of a glucocorticoid responsive element and identification of an AT-rich element that regulate the link protein gene. Nucleic Acids Res 23:2305-2313.
Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W 2002. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296:1880-1882.
Riewald M, Petrovan RJ, Donner A, Ruf W 2003. Activated protein C signals through the thrombin receptor PAR1 in endothelial cells. J Endotoxin Res 9:317-321.
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M 2001. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 345:1368-1377.
Rivier C, Vale W 1983. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature 305:325-327.
Roger T, David J, Glauser MP, Calandra T 2001. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414:920-924.
Rubulotta F, Marshall JC, Ramsay G, Nelson D, Levy M, Williams M 2009. Predisposition, insult/infection, response, and organ dysfunction: A new model for staging severe sepsis. Crit Care Med 37:1329-1335.
Rudiger A, Stotz M, Singer M 2008. Cellular processes in sepsis. Swiss Med Wkly 138:629-634.
Sales Junior JA, David CM, Hatum R, Souza PCSP, Japiassú A, Pinheiro CTS, Friedman G, Silva OB, Dias MD, Koterba E, Dias FS, Piras C, Luiz RR 2006. Sepse Brasil: estudo epidemiológico da sepse em unidades de terapia intensiva brasileiras. Rev Bras Ter Intens 18:9-17.
Salluh JI, Povoa P, Soares M, Castro-Faria-Neto HC, Bozza FA, Bozza PT 2008a. The role of corticosteroids in severe community-acquired pneumonia: a systematic review. Crit Care 12:R76.
Salluh JI, Bozza FA, Soares M, Verdeal JCR, Castro-Faria-Neto HC, Silva JRL, Bozza PT 2008b. Adrenal response in severe community-acquired pneumonia: Impact on outcomes and disease severity. Chest 134:947-954.
Salvo I, de Cian W, Musicco M, Langer M, Piadena R, Wolfler A, Montani C, Magni E 1995. The Italian SEPSIS study: preliminary results on the incidence and evolution of SRIS, sepsis, severe sepsis and septic shock. Intensive Care Med 21 Supl 2:S244-S249.
Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M 2008. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem 283:21134-21144.
Sands KE, Bates DW, Lanken PN, Graman PS, Hibberd PL, Kahn KL, Parsonnet J, Panzer R, Orav EJ, Snydman DR, Black E, Schwartz JS, Moore R, Johnson BL Jr, Platt R 1997. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA 278:234-240.

103
Schroder JM, Christophers E 1989. Secretion of novel and homologous neutrophil-activating peptides by LPS-stimulated human endothelial cells. J Immunol 142:244-251.
Schroder JM, Mrowietz U, Christophers E 1988. Purification and partial biologic characterization of a human lymphocyte-derived peptide with potent neutrophil-stimulating activity. J Immunol 140:3534-3540.
Schumer W, Erve P, Kapica SK, Moss GS 1970. Endotoxin effect on respiration of rat liver mitochondria. J Surg Res 10:609-612.
Shchepina LA, Pletjushkina OY, Avetisyan AV, Bakeeva LE, Fetisova EK, Izyumov DS, Saprunova VB, Vyssokikh MY, Chernyak BV, Skulachev VP 2002. Oligomycin, inhibitor of the F0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis. Oncogene 21:8149-8157.
Sheikine Y, Hansson GK 2004. Chemokines and atherosclerosis. Ann Med 36:98-118. Sielenkämper AW, Yu P, Eichelbrönner O, MacDonald T, Martin CM, Chin-Yee IH, Sibbald
WJ 2000. Diaspirin cross-linked Hb and norepinephrine prevent the sepsis-induced increase in critical O2 delivery. Am J Physiol Heart Circ Physiol 279:H1922-H1930.
Silva E, Pedro Mde A, Sogayar AC, Mohovic T, Silva CL, Janiszewski M, Cal RG, de Sousa EF, Abe TP, de Andrade J, de Matos JD, Rezende E, Assunção M, Avezum A, Rocha PC, de Matos GF, Bento AM, Corrêa AD, Vieira PC, Knobel E 2004. Brazilian Sepsis Epidemiological Study (BASES study). Crit Care 8:R251-260.
Silvestre J, Póvoa P, Coelho L, Almeida E, Moreira P, Fernandes A, Mealha R, Sabino H 2009. Is C-reactive protein a good prognostic marker in septic patients? Intensive Care Med 35:909-913.
Sims NR, Blass JP 1986. Expression of classical mitochondrial respiratory responses in homogenates of rat forebrain. J Neurochem 47:496-505.
Singer M 2007. Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med 35 Supl 9:S441-S448.
Singh S, Evans TW. Organ dysfunction during sepsis. Intensive Care Med 2006; 32:349-360. Soares M, Terzi RG, Piva JP 2007. End-of-life care in Brazil. Intensive Care Med 33:1014-
1017. Sprung CL, Cohen SL, Sjokvist P, Baras M, Bulow HH, Hovilehto S, Ledoux D, Lippert A,
Maia P, Phelan D, Schobersberger W, Wennberg E, Woodcock T 2003. End-of-life practices in European intensive care units: the Ethicus Study. JAMA 290:790-797.
Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, Weiss YG, Benbenishty J, Kalenka A, Forst H, Laterre PF, Reinhart K, Cuthbertson BH, Payen D, Briegel J 2008. Hydrocortisone therapy for patients with septic shock. N Engl J Med 358:111-124.
Strieter RM, Chensue SW, Basha MA, Standiford TJ, Lynch JP, Baggiolini M, Kunkel SL 1990. Human alveolar macrophage gene expression of interleukin-8 by tumor necrosis factor-alpha, lipopolysaccharide, and interleukin-1 beta. Am J Respir Cell Mol Biol 2:321-326.
Sundararajan V, Macisaac CM, Presneill JJ, Cade JF, Visvanathan K 2005. Epidemiology of sepsis in Victoria, Australia. Crit Care Med 33:71-80.
Takehara Y, Kanno T, Yoshioka T, Inoue M, Utsumi K 1995. Oxygen-dependent regulation of mitochondrial energy metabolism by nitric oxide. Arch Biochem Biophys 323:27-32.

104
Tanriover MD, Guven GS, Sen D, Unal S, Uzun O 2006. Epidemiology and outcome of sepsis in a tertiary-care hospital in a developing country. Epidemiol Infect 134:315-322.
Terashima T, Soejima K, Waki Y, Nakamura H, Fujishima S, Suzuki Y, Ishizaka A, Kanazawa M 1995. Neutrophils activated by granulocyte colony-stimulating factor suppress tumor necrosis factor-alpha release from monocytes stimulated by endotoxin. Am J Respir Cell Mol Biol 13:69-73.
Thijs LG, Hack CE 1995. Time course of cytokine levels in sepsis. Intensive Care Med 21 Supl 2:S258-S263.
Tilney NL, Bailey GL, Morgan AP 1973. Sequential system failure after rupture of abdominal aortic aneurysms: an unsolved problem in postoperative care. Ann Surg 178:117-122.
Trzcionka M, Withers KW, Klingenspor M, Jastroch M 2008. The effects of fasting and cold exposure on metabolic rate and mitochondrial proton leak in liver and skeletal muscle of an amphibian, the cane toad Bufo marinus. J Exp Biol 211:1911-1918.
Van den Berghe G 2000. Novel insights into the neuroendocrinology of critical illness. Eur J Endocrinol 143:1-13.
Van der Poll T, Levi M, Hack CE, ten Cate H, van Deventer SJ, Eerenberg AJ, de Groot ER, Jansen J, Gallati H, Buller HR 1994. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med 179:1253-1259.
Van Deuren M, van der Ven-Jongekrijg J, Bartelink AK, van Dalen R, Sauerwein RW, van der Meer JW 1995. Correlation between proinflammatory cytokines and antiinflammatory mediators and the severity of disease in meningococcal infections. J Infect Dis 172:433-439.
Van Leeuwen BH, Martinson ME, Webb GC, Young IG 1989. Molecular organization of the cytokine gene cluster, involving the human IL-3, IL-4, IL-5, and GM-CSF genes, on human chromosome 5. Blood 73:1142-1148.
Van Zoelen M, LAterre PF, Van Veen S, Van Till JW, Wittebole X, Bresser P, Tanck MW, Dugernier T, Ishizaka A, Boermeester MA, Van der Poll T 2007. Systemic and local high mobility group box 1 concentrations during severe infection. Crit Care Med 35:2799-2804.
Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G 2005. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 365:53-59.
Vanhorebeek I, Van den Berghe G 2006. The neuroendocrine response to critical illness is a dynamic process. Crit Care Clin 22:1-15.
Vermes I, Beishuizen A, Hampsink RM, Haanen C 1995. Dissociation of plasma adrenocorticotropin and cortisol levels in critically ill patients: possible role of endothelin and atrial natriuretic hormone. J Clin Endocrinol Metab 80:1238-1242.
Vignali DA 2000. Multiplexed particle-based flow cytometric assays. J Immunol Methods 243:243-255.
Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, Reinhart CK, Suter PM, Thijs LG 1996. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. Intensive Care Med 22:707-710.

105
Vincent JL 1997. Dear SIRS, I'm sorry to say that I don't like you... Crit Care Med 25:372-374.
Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, Moreno R, Carlet J, Le Gall JR, Payen D 2006. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med 34:344-353.
Walz A, Peveri P, Aschauer H, Baggiolini M 1987. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem Biophys Res Commun 149:755-761.
Yende S, D'Angelo G, Kellum JA, Weissfeld L, Fine J, Welch RD, Kong L, Carter M, Angus DC 2008. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med 177:1242-1247.
Yoshimura T, Matsushima K, Oppenheim JJ, Leonard EJ 1987. Neutrophil chemotactic factor produced by lipopolysaccharide (LPS)-stimulated human blood mononuclear leukocytes: partial characterization and separation from interleukin 1 (IL 1). J Immunol 139:788-793.
Yoshimura T, Yuhki N, Moore SK, Appella E, Lerman MI, Leonard EJ 1989. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett 244:487-493.
Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG, Clark SC, David JR 1989. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A 86:7522-7526.
Westphal GA, Feijó J, Andrade PS, Trindade L, Suchard C, Monteiro MAG, Martins SF, Nunes F, Filho MC 2009. Estratégia de detecção precoce e redução de mortalidade na sepse grave. Rev Bras Ter Intens 21:113-123.
Wiedermann FJ, Mayr AJ, Kaneider NC, Fuchs D, Mutz NJ, Schobersberger W 2004. Alveolar granulocyte colony-stimulating factor and alpha-chemokines in relation to serum levels, pulmonary neutrophilia, and severity of lung injury in ARDS. Chest 125:212-219.
Widmer IE, Puder JJ, Konig C, Pargger H, Zerkowski HR, Girard J, Muller B 2005. Cortisol response in relation to the severity of stress and illness. J Clin Endocrinol Metab 90:4579-4586.
Wu HP, Wu CL, Chen CK, Chung K, Tseng JC, Liu YC, Chuang DY 2008. The interleukin-4 expression in patients with severe sepsis. J Crit Care 23:519-524.
Wu HP, Chen CK, Chung K, Tseng JC, Hua CC, Liu YC, Chuang DY, Yang CH 2009. Serial cytokine levels in patients with severe sepsis. Inflamm Res 58:385-393.
Záhorec R, Firment J, Straková J, Mikula J, Malík P, Novák I, Zeman J, Chlebo P 2005. Epidemiology of severe sepsis in intensive care units in the Slovak Republic. Infection 33:122-128.
Ziegler EJ, Fisher CJ Jr, Sprung CL, Straube RC, Sadoff JC, Foulke GE, Wortel CH, Fink MP, Dellinger RP, Teng NN 1991. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. N Engl J Med 324:429-436.

106
Zingarelli B, O'Connor M, Wong H, Salzman AL, Szabó C 1996. Peroxynitrite-mediated DNA strand breakage activates poly-adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol 156:350-8
Zisman DA, Kunkel SL, Strieter RM, Tsai WC, Bucknell K, Wilkowski J, Standiford TJ 1997. MCP-1 protects mice in lethal endotoxemia. J Clin Invest 99:2832-2836.

107
Anexo 1 – Protocolo de coleta de dados e pontuação do escore Sequential Organ Failure
Assessment (SOFA).
PROTOCOLO CHOQUE SÉPTICO – SDOM
No: ___ NOME: ____________________ IDADE: __ anos SEXO: __ PRONT: ______
internação na UTI: __/__/____ inclusão no estudo em: __/__/____ origem: __________
DIAGNÓSTICO: ________________________________________________________
choque séptico __ / sepse grave __; sítio de infecção: ___________
culturas: _______________________________________________________________
germe: ________________________________________________________________
antibioticoterapia: _______________________________________________________
__ __ __ __/___; SAPS II: ___ prob óbito: ___ %
SOFA D1= D3= D5= D7=
PaO2/FiO2
PAM
aminas
plaquetas
creatinina
bilirrubinas
Glasgow
leucócitos
PCRt
vent mec
corticoide
Swan-Ganz
hemodiálise
insulina
lactato
DESFECHO: ____________ data: __/__/____

108
Escore Sequential Organ Failure Assessment (SOFA).
Sistema 0 ponto 1 ponto 2 pontos 3 pontos 4 pontos
Respiratório
PaO2/FiO2
> 400 301-400 201-300 101-200 < 100
Cardiovascular PAM > 70
mmHg
PAM < 70
mmHg
Uso de
dobutamina
Dopamina <
15 µg/kg/min
OU
Noradrenalin
a < 0,1
µg/kg/min
Dopamina >
15 µg/kg/min
OU
Noradrenalin
a > 0,1
µg/kg/min
Renal Creatinina
< 1,2 mg/dl
Creatinina
1,2-1,9
mg/dl
Creatinina
2,0-3,4
mg/dl
Creatinina
3,5-4,9 mg/dl
OU diurese <
500 ml/dia
Creatinina >
5,0 mg/dl OU
diurese < 200
ml/dia
Hematológico
(plaquetometria
, 1000/mm3)
> 150 100-150 50-100 20-50 < 20
Hepático
(bilirrubinas
totais séricas,
mg/dl)
< 1,2 1,2-1,9 2,0-5,9 6,0-11,9 > 12,0
Neurológico
(escala de
Glasgow,
pontos)
15 13-14 10-12 6-9 < 6

Referência: os escores prognósticos SAPS II e SOFA estão descritos no site
http://www.sfar.org/t/spip.php?article60 , onde se pode calcular estes e outros escores
aplicados na Medicina Intensiva.
109

110
Anexo 2: Termo de Consentimento LIVRE E ESCLARECIDO
TÍTULO
Inflamação na Síndrome de Disfunção Múltipla Orgânica – envolvimento de citocinas, proteínas da
coagulação e disfunção mitocondrial.
JUSTIFICATIVA E OBJETIVOS
Durante as infecções graves, nosso organismo produz várias substâncias envolvidas no combate aos
microorganismos, estas substâncias podem por outro lado ter efeitos prejudiciais, quando em excesso.
Este estudo visa entender melhor os fenômenos ligados às infecções graves e futuramente contribuir
para o desenvolvimento de novas terapias para estas infecções.
PROPOSTA DO ESTUDO
O Sr(a) _________________________________ está sendo convidado a participar deste estudo, para
estudar a presença de substâncias produzidas pelo nosso organismo em resposta a infecções graves.
EXPLICAÇÃO DOS PROCEDIMENTOS
Será realizada a coleta de uma amostra de sangue de 10 (dez) ml, através de uma punção de veia
periférica, utilizando-se material estéril e descartável. Este procedimento é semelhante à coleta de
sangue para exames laboratoriais de rotina. Será coletado nos dias 1, 3, 5, 7 e 14 após início do estudo.
BENEFÍCIOS
No seu caso não há benefícios diretos, mas este estudo poderá ajudar a entender melhor os fenômenos
ligados às infecções graves e futuramente contribuir para o desenvolvimento de novas terapias para
estas infecções.
DESCONFORTOS E RISCOS
Os desconfortos que podem ocorrer são aqueles relacionados a uma retirada normal de sangue para
exame, como dor no local da punção venosa e formação de um hematoma local. Este estudo não
implica em riscos, nem em qualquer modificação do tratamento empregado ou administração de
medicamentos experimentais.
PARTICIPAÇÃO VOLUNTÁRIA NO ESTUDO
A participação neste estudo é voluntária. Você pode se recusar a participar, bem como cancelar sua
participação a qualquer momento do estudo. Esta decisão não afetará de nenhuma maneira os cuidados
médicos que lhe serão oferecidos.

111
CONFIDENCIALIDADE
O seu nome não será mencionado em publicações ou relatórios produzidos para este estudo.
Entretanto seu prontuário médico poderá ser consultado pelos profissionais envolvidos no estudo.
SE VOCÊ TEM DÚVIDAS
Se você tiver qualquer dúvida sobre o estudo, por favor telefone para o Dr. André Japiassú no telefone
2562-1311 ou Dr. Fernando Bozza no telefone 3865-9620.
CONSENTIMENTO PARA A PARTICIPAÇÃO NO ESTUDO
A sua assinatura significa que você leu este formulário ou que ele foi lido para você, que lhe foram
dadas todas as explicações sobre o estudo, que você recebeu respostas para as suas dúvidas, está
satisfeito com as informações que lhe foram dadas e concordou com a participação no estudo.
____________________________ ______________________
Assinatura (Paciente) Data
Se o paciente não é capaz de consentir:
A sua assinatura, como representante legal do paciente, significa que você leu este formulário ou que
ele foi lido para você, que lhe foram dadas todas as explicações sobre o estudo, que você recebeu
respostas para as suas dúvidas, está satisfeito com as informações que lhe foram dadas e concordou
com a participação do paciente no estudo.
________________________ não é capaz de dar o seu consentimento.
Nome do Paciente (em letra de forma)
_______________________________ __________________________
Nome do Representante Legal Grau de parentesco com o paciente
(em letra de forma)
____________________________ ______________________
Assinatura (Representante legal) Data

Anexo 3 – Artigo Original “Cytokine profiles as markers of disease severity in sepsis: a
multiplex analysis”, publicado na revista Critical Care, em 2007, volume 11, página R49
(participação como 3º autor).
112

113

114
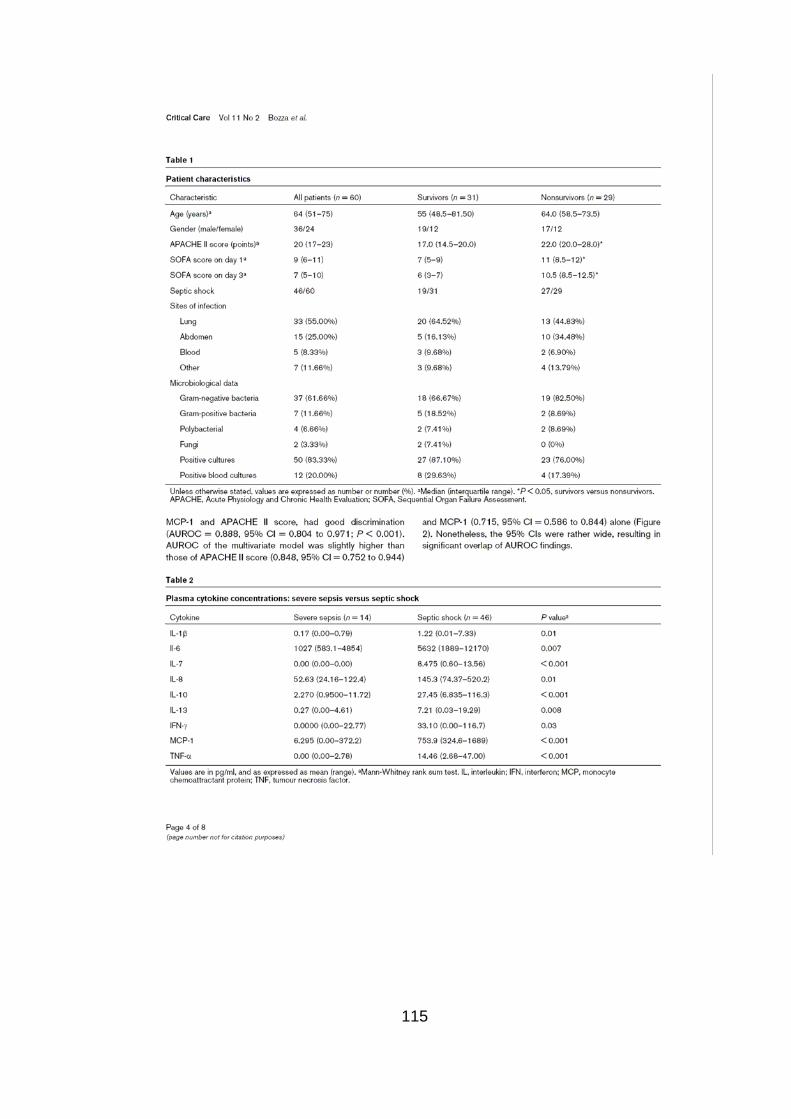
115

116

117

118

119

Anexo 4 – Artigo de revisão “Revisiting steroid treatment for septic shock: molecular actions
and clinical effects - A review”, publicado na revista Memórias do Instituto Oswaldo Cruz,
em 2009, volume 104, páginas 531-548.
120

121

122

123

124

125

126

127

128

129

130

131

132

133

134

135

136

137

138
Anexo 5 – Artigo submetido à publicação na revista Critical Care, em 2 de fevereiro de 2010.
“Sepsis is a major determinant of outcome in HIV/AIDS critically ill patients”
Japiassú AM, Amâncio RT, Mesquita EC, Medeiros DM, Bernal HB, Nunes EP, Luz PM,
Grinsztejn B, Bozza FA
Instituto de Pesquisa Clínica Evandro Chagas, Fundação Oswaldo Cruz, Rio de Janeiro, RJ,
Brazil.
Laboratório de Imunofarmacologia, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de
Janeiro, RJ, Brazil.
Address requests for reprints to: Fernando A. Bozza MD, PhD. ICU, Instituto de Pesquisa
Clínica Evandro Chagas, Fundação Oswaldo Cruz, Av. Brasil, 4365, Manguinhos, Rio de
Janeiro, RJ, Brazil, 21040-900, Phone: (+55-21) 3865-9595; Fax: (+55-21) 2290-4532, E-
mail: [email protected] or [email protected]
Keywords: HIV, AIDS, Sepsis, prognosis, intensive care.
Text word count: 3405 words

139
Abstract (338 words)
Background: Natural history of HIV/AIDS has been changing over the last years, with a
significant improvement in survival and quality of life of patients. However, new challenges
have arisen to the management of the critically ill HIV/AIDS patients. Severe sepsis has
emerged as a common cause of ICU admission for those living with HIV/AIDS.
Contrastingly, HIV/AIDS patients have been systematically excluded from sepsis studies,
limiting the understanding of the impact of sepsis in this population. We prospectively
followed HIV/AIDS critically ill patients to evaluate the consequences of severe sepsis on the
short- and long-term survival.
Methods: We collected demographic data and HIV/AIDS characteristics at ICU admission.
Severity of illness, incidence of severe sepsis, sites of infection and microbiologic results
were registered. Twenty-eighth day, hospital and 6-month outcomes were obtained for all
patients. We calculated the Kaplan-Meier survival function stratified by sepsis, and Cox
proportional hazards regression analysis measured the effect of sepsis, as well as potential
factors, on 28-day and 6-month mortalities.
Results: During the 2-year study period, 88 HIV/AIDS critically ill patients were admitted to
the ICU. The three main causes of ICU admission were acute respiratory failure (29%), severe
neurological dysfunction (23%) and severe sepsis (20%). Seventy percent of patients had
opportunist infections, median CD4 count was 75 cells/mm3 and 45% were on antiretroviral
therapy. Severe sepsis occurred on 44 (50%) patients, mainly due to lower respiratory tract
infections. Length of stay (11 vs 7 days, p=0.03) and hospital mortality (66% vs 34%,
p=0.002) were significantly higher for septic HIV/AIDS patients, and severe sepsis
determined the highest hazard ratio for 28-day (adjusted HR 6.83, 95%CI 1.87-24.8) and 6-
month (adjusted HR 5.20 95%CI 1.38-9.97) mortalities. The survival of septic and non-septic
patients was significantly different at 28-days and 6-months follow-up times (log-rank and
Peto test p<0.001).
Conclusions: Severe sepsis has emerged as a major cause of admission and mortality for
hospitalized HIV/AIDS patients. We found that severe sepsis is the main risk factor for
hospital mortality, significantly impacting short- and longer-term survival of HIV/AIDS
critically ill patients.

140
Introduction
The long-term survival of patients with human immunodeficiency virus (HIV) has markedly
improved since the introduction of highly active anti-retroviral therapy (HAART). Estimates
for 2007 indicate that 33 million individuals were living with HIV; and it is expected that this
number will continue to grow, in particular in third world urban centers [1]. Recent studies
have analyzed HIV/AIDS critically ill patients’ characteristics, especially comparing pre and
post-HAART eras, with an emphasis on causes of admission and risk factors for mortality [2-
7]. Sepsis has emerged as an important cause of ICU admission for HIV/AIDS patients, and
its incidence has increased since the 1980s, contrary to the decreasing trend observed for
acute respiratory insufficiency and P jiroveci pneumonia [8]. Other studies have shown that
bacterial infections are common causes of mortality in patients with HIV, irrespective of
HAART use [9,10,11].
Epidemiological studies have shown that a fraction of 1-10% of the septic patients are
composed of individuals with HIV/AIDS; with the variation being mostly due to regional
differences in the prevalence of HIV and ICU admission practices [12-19]. In general,
HIV/AIDS septic patients are younger, have similar number of organ failures, but have lower
rates of ICU admission when compared to other patients with severe sepsis, including those
with worse prognosis, such as metastatic cancer or severe liver disease [20].
Despite the significant increases in survival and quality of life, HIV/AIDS patients have been
systematically excluded from sepsis studies, limiting the understanding of the impact of sepsis
in this population. To date, few studies have evaluated the effect of severe sepsis on survival
of HIV/AIDS critically ill patients. In this study, we prospectively followed HIV/AIDS
critically ill patients to evaluate the impact of severe sepsis on the short- and long-term
survival.
Patients and Methods
Design and Setting
This prospective cohort study was conducted at the ICU of the Instituto de Pesquisa Clínica
Evandro Chagas (IPEC), Fundação Oswaldo Cruz, Rio de Janeiro, Brazil. IPEC has provided
care to HIV/AIDS patients in Rio de Janeiro since 1986 and, currently, more than two
thousand adult patients are actively followed at the HIV/AIDS clinic.
At our institution, generally only patients for whom potentially lifespan-extending treatment
is available are considered for ICU admission. Critically ill unstable patients, with indication

141
for intensive care and monitoring and/or need for immediate procedures or interventions are
eligible for ICU admission. Treatment-limitation decisions are made in the ICU when patients
do not recover from acute illness despite intensive care, or control of baseline condition has
not been achieved. These decisions are shared by ICU team, HIV/AIDS specialist and family
members.
This study was supported by institutional funds. The institutional review board approved the
study and waived the need for informed consent. The study conduct did not interfere with
patient management decisions.
Participants, Data Collection and Definitions
All consecutive HIV-infected patients admitted to the ICU from June 2006 to May 2008 were
included in this study. AIDS cases and the occurrence of AIDS-defining diseases were
defined by the Centers for Disease Control and Prevention (CDC) definitions, and HAART
was initiated when CD4 cell count was below 200-350 cells/mm3 [21]. Time since AIDS
diagnosis was calculated from AIDS onset until ICU admission. Recent AIDS onset was
defined as those occurring 2 months prior to admission. The CD4 cell count was considered
when obtained within 3 months of ICU admission or measured during the first week of ICU
entry. HAART was defined as the regular use of at least 2 nucleoside reverse transcriptase
inhibitor (NRTI) plus a protease inhibitor (PI) or a nonnucleoside reverse transcriptase
inhibitor (NNRTI) or a PI and a NNRTI in combination [20].
In case of multiple ICU admissions, only the first was considered. ICU causes of admission
were divided into acute respiratory insufficiency, sepsis, severe neurological disturbances,
heart disease, complications of solid organ or hematological neoplasms, metabolic
disturbance, and gastrointestinal complications. Readmissions were defined for patients
returning to ICU during the same hospitalization.
A standardized data entry form was created for the collection of demographic data, main
admission diagnosis, opportunistic infections, comorbidities, location before ICU admission
(emergency room, ward or other hospital), laboratory results based on computerized
laboratory records review, ICU and hospital length of stay. The following variables were
collected during the first day of ICU stay: expanded Simplified Acute Physiology Score
(SAPS) II [22,23], Sequential Organ Failure Assessment (SOFA) score [24], and use of
vasopressor agents. Performance Status (PS) was calculated based on patient or surrogate
information [25]. During ICU stay, the need for mechanical ventilation for more than 24
hours, use of renal support devices, development of shock (use of vasopressor agent to

142
maintain mean arterial pressure higher than 70 mmHg) and Acute Lung Injury (defined as
PaO2/FiO2 ratio less than 300) were also recorded.
Sepsis definitions were based on ACCP/SCCM Consensus Conference [26]; sepsis was
present if there was a presumed or confirmed infection, associated with at least two of the
following: tachycardia > 90 beats per minute; tachypnea > 30 cycles per minute (or
hypocapnia less than 32 mmHg); fever (> 38º C) or hypothermia (< 36º C); and leucocytosis
(> 12000/mm3) or leucopenia (< 4000/mm3) or presence of more than 10% immature forms.
Severe sepsis was defined if there was any organ dysfunction, sepsis-induced hypotension or
elevated serum lactate levels. Septic shock was defined by sepsis with hypotension that
persisted after adequate infusion of fluids and need of vasopressor agents.
Infections were classified as community-acquired or nosocomial, according to the cutoff time
point 48 hours of hospital admission. Definitions of lower pulmonary tract infection,
bloodstream infection, and urinary tract infection were made following the CDC definitions
[27]. Appropriate biological material was sampled from suspected sites of infection.
Microbiological data were collected and analyzed according to the local Infection Control
Committee.
Statistical Analysis
Continuous variables were summarized as medians and interquartile ranges. We compared the
distribution of continuous variables using the t-test, and of categorical variables using the χ2
test. We calculated the Kaplan-Meier survival function stratified by sepsis. The log-rank and
Peto tests were used to evaluate if the estimated survival functions are significantly different
by strata. Two outcomes were of interest: mortality within 28 days and 6 months from entry
to the ICU. We used Cox proportional hazards regression analysis to measure the effect of
sepsis, as well as other factors, on 28-days and 6-months mortalities. We also estimated the
effect of sepsis while controlling for potential confounders such as age, CD4 cell count, time
since AIDS diagnosis, HAART, location before ICU, and cardiovascular and respiratory
dysfunctions, in a multivariate model. The proportionality assumption in Cox’s regression
was evaluated by the estimating the correlation between survival time and Shoenfeld's
standardized residuals. We used the statistical software R, version 2.9 (www.r-project.org) for
all statistical analysis.
Results

143
Patient characteristics
During the 2-year study period, there were 107 ICU admissions of HIV/AIDS critically ill
patients; 19 of which were readmissions. We analyzed the first ICU admission of 88 patients;
their characteristics are shown in Table 1. Briefly, median age was 40 years, and there was a
male predominance (76%). Median CD4 cell count was 75 (interquartile range: 32-227)
cells/mm3. The median time since AIDS diagnosis was 40 months. Twenty-five patients
(28%) had recently been diagnosed with AIDS; forty patients (45%) were using HAART. The
percentage of patients using the three drug classes was: NRTI (97%), PI (67%) and NNRTI
(35%). HAART use was not found to impact hospital survival (50% mortality for HAART
and non-HAART subjects).Patients not using HAART were younger (37 vs 43 years, p=0.03),
had more opportunistic infections (83% vs 55%, p<0.01), and less time since AIDS diagnosis
(3.5 vs 68.5 months, p<0.01).
Acute respiratory failure was the cause of ICU admission for 29% of the patients, which was
caused by bacterial pneumonia (n=10), Pneumocystis jiroveci pneumonia (n=8), tuberculosis
(n=6) and cardiogenic pulmonary edema (n=2). Severe neurological complications were the
cause of ICU admission in 20 patients (23%). Other causes of ICU admission were: severe
sepsis (n=18), decompensate heart disease (n=9), gastrointestinal complications (n=6),
metabolic dysfunction secondary to renal dysfunction or lactic acidosis related to drug
adverse effects (n=5), and complications of high-grade non-Hodgkin´s lymphoma (n=4)
(Table 1). Sixty-two patients (70%) had at least one opportunistic infection; P jiroveci
pneumonia and neurotoxoplasmosis were the most frequent opportunistic infections.
In-hospital mortality
We observed 49% in-hospital mortality. Patients with non-Hodgkin´s lymphoma and
gastrointestinal complications presented high mortality (100% and 83%, respectively), while
patients presenting with acute respiratory failure, neurological disorders and sepsis had
moderate mortality (46%, 45% and 50%, respectively). SAPS II (54 vs 44 points, p < 0.001)
and SOFA score on day 1 (7 vs 4 points, p < 0.001) were significantly higher for nonsurvivors
(Table 2). Early shock (use of vasopressor agents in the first 24 hours since admission) was
more common in nonsurvivors (30 vs 13%, p < 0.05). Nonsurvivors used more life-support
devices, such as mechanical ventilation (84 vs 38%, p < 0.01) and renal support therapy (30
vs 7%, p < 0.01). Severe sepsis or septic shock was observed with higher frequency for the
nonsurvivor group (67 vs 33%, p < 0.01). Other demographic characteristics such as age,
gender, admission from emergency or ward, and Performance Status were similar in the two

144
groups. There was no statistically significant difference between the two groups regarding
CD4 cell counts, time since AIDS diagnosis and HAART use.
Severe sepsis in HIV/AIDS patients
Forty-four patients (50%) presented with severe sepsis at ICU admission or during ICU stay.
In comparison with non-septic patients, septic patients were more likely to come from wards
(75% vs 45%, p<0.01), spent more time in ICU (11 vs 7 days, p=0.03), were more severely ill
(SAPS II, 56 vs 44 points, p<0.001; and SOFA on day 1, 7 vs 3 points, p<0.001), and needed
mechanical ventilation more often (86 vs 34%, p<0.001). We did not observe differences on
CD4 cell count or time since AIDS diagnosis between septic and non-septic groups. Hospital
mortality was significantly higher in septic when compared to non-septic patients (66% vs
34%, p=0.002). Lung was the most common site of infection (52%), followed by primary
bloodstream infections (38%), venous catheter-related bacteremia (7%) and urinary tract
infections (3%) (Table 3). In 90% of the cases, nosocomial infections were the source of
sepsis in our cohort. Microbiology of infections was mostly composed of Gram-negative rods,
such as Pseudomonas aeruginosa (10), Klebsiella pneumoniae (6), Enterobacter sp (5),
Escherichia coli (3), Acinetobacter sp (3), Serratia marcenses (3), and Staphylococcus sp (9).
Mycobacterium tuberculosis was the main ethiologic agent of severe sepsis in 6 (14%)
patients. There were also single cases of S maltophilia, C difficile, C freundi, B cepacia, and
Candida sp. Bacteremia was detected in 43% (19) of the septic patients, irrespectively of the
site of infection.
28th day and 6-month survival
The survival expectation of septic and non-septic patients was significantly different both at
28-days and 6-months follow-up timepoints (Figure 1, p-values for log-rank and Peto tests <
0.001). Using Cox proportional hazards regression, we estimated that the hazard of death at
28-days for septic patients was 4-times higher than for non-septic patients (HR 4.17 95%CI
1.96-8.90). Other factors associated with an increased hazard of death at 28-days were being
in a hospital ward before entry to the ICU, and cardiovascular and respiratory dysfunctions
(Table 4). The effect of sepsis on 28-days mortality was even stronger in the multivariate
model, that is, when controlling for confounders. The adjusted hazard of death at 28-days was
almost 7-fold higher (HR 6.83 95%CI 1.87-24.8) for septic patients compared to non-septic
when controlling for age, CD4 cell count, time since AIDS diagnosis, HAART, location
before ICU, and cardiovascular and respiratory dysfunctions. When considering 6-months of

145
follow-up, sepsis also determined the highest hazard of death both in the crude (HR 3.25
95%CI 1.75-6.16) and adjusted (HR 5.20 95%CI 1.38-9.97) analyses (Table 4). No significant
difference of 28-day or 6-month mortality was retrieved when on-admission or ICU-acquired
severe sepsis was analyzed separately. When comparing the two follow-up periods, 28-days
and 6-months, we noticed that, although the effect of sepsis was stronger early during follow-
up, it remained significant at 6 months.
Discussion
The improvement of HIV/AIDS management has led to changes in the observed clinical
manifestations and outcomes of these patients in critical care units, with a decreasing trend in
admissions due to opportunistic infections while an opposing trend has been observed for
other infectious and metabolic diseases [28]. The current phase of the HIV/AIDS epidemic
poses new challenges to the management of the HIV/AIDS critically ill. Severe sepsis has
emerged as a common cause of hospital admission for those living with HIV/AIDS [2,8,29].
In this prospective study, we demonstrated that severe sepsis is the main risk factor for
hospital mortality, significantly impacting short- and long-term survival of HIV/AIDS
critically ill patients.
Half of our patients presented with severe sepsis, either as the admission cause or by
developing this condition during ICU stay. An increasing incidence of severe sepsis as the
cause of ICU admission in the HIV/AIDS population has been observed in the last 2 decades.
Casalino et al reported that severe sepsis at ICU admission increased from 16% in the pre-
HAART era to 22% in the post-HAART era [29]. Huang et al showed that severe sepsis was
the only cause of ICU admission for HIV/AIDS patients that increased from the period 1981-
1985 to 2000-2003, while respiratory failure significantly decreased [8]. Overall, severe septic
patients constitute a varying fraction of HIV/AIDS ICU population, ranging from 12% to 31%
of these patients [2-7, 29-32]. Although our observed fraction of severe septic patients at ICU
admission is within the range reported in the literature, we found that an additional significant
proportion of patients developed severe sepsis during ICU stay. This finding emphasizes the
importance of severe sepsis development in the ICU, and points out to a subgroup of patients
that is being overlooked in studies that only considering those who presented with severe
sepsis at ICU admission.
Sepsis related mortality was increased in both short and longer-term follow-up. Indeed, septic
patients had significantly higher in-hospital mortality than non-septic patients. Thyrault et al

146
studied a cohort of septic AIDS patients and reported 46% 28-days mortality and only 4%
survival after 1 year [33]. Although the results of the latter study are relevant, it is important
to note that it was conducted during the pre-HAART era, when the long-term prognosis of
AIDS patients after hospitalizations was very poor. A 4-year retrospective cohort study
conducted in the early HAART period (1995-1999), reported 68% in hospital mortality for
AIDS critically ill patients with bacterial sepsis [2]. The latter study identified bacterial cause
of sepsis and APACHE III score greater than 80 points as risk factors for hospital mortality.
We also found that cardiovascular and respiratory dysfunctions increased the hazards of
death, especially in the short-term. Respiratory dysfunction is closely associated to
mechanical ventilation use and the presence of severe sepsis. Cardiovascular dysfunction
could also be related to the presence of shock and use of vasoactive agents, which are more
frequent in septic patients. Use of mechanical ventilation [3,5,29,32,34], higher APACHE II
or SAPS II scores [2,29,32,34], use of HAART during ICU stay [32], and opportunistic
infections [3,5,34] have been found to impact hospital mortality in a heterogeneous group of
studies. Low CD4 cell counts, time since AIDS diagnosis, HAART treatment or AIDS stage
of disease, on the other hand, were not identified as risk factors for mortality in the post-
HAART era [4,6,34]. In our cohort, almost 30% of patients had a recent AIDS diagnosis and
were, therefore, HAART naïve, and an additional 16% of the patients were non-compliant
HAART users. We found that these groups, who correspond to almost 50% of the patients in
our study, were not subject to higher hospital mortality. Recent data has shown a greater
impact of ICU care rather than HIV/AIDS management as major risk factors for death.
Ventilatory management with lower tidal volumes was shown to be protective for mortality
due to acute lung injury in critically ill patients, and this strategy was associated with lower
mortality among HIV/AIDS patients with respiratory insufficiency [35].
Severe sepsis was strongly associated with worse outcomes, and was caused by bacterial
infections, mainly of nosocomial origin. Nosocomial infections appear to be more common in
patients with acquired immunodeficiency syndrome (AIDS) compared to those not HIV-
infected [36]. Nosocomial infections have been associated with immunosuppression level,
prior antibiotic use and greater exposure to invasive devices such as intravenous catheters
[37]. The CD4 cell count in our cohort was very low, and this could be associated with greater
use of antibiotic for opportunistic infections prophylaxis or for treatment of bacterial
infections contributing to a high rate of antibiotic resistance and consequently, the
development of nosocomial infection. Pneumonia and bloodstream infections were the main
sites of infections for almost all septic patients, and hospital-acquired bacteria composed the

147
major part of the microbiology of severe infections. Lower tract respiratory infection was also
the main site of infection on two other studies. Thyrault et al reported on the characteristics of
28 septic shock AIDS patients admitted to ICU, mostly due to nosocomial pneumonia caused
by Gram-negative and Gram-positive bacteria [33]. Rosenberg et al verified that pneumonia
was the cause of 65% of bacterial sepsis in HIV/AIDS critically ill patients, mainly caused by
P aeruginosa, S aureus and K pneumoniae [38]. A high incidence of nosocomial bacteremia
was also common in HIV/AIDS patients with lower CD4 cell count [39,40]. In our
population, bacteremia was diagnosed in 43% of the patients. Bloodstream infections can
negatively impact on HIV/AIDS patients´ outcomes, with higher mortality rates occurring in
this group than among those without bacteremia [37, 40-43]. Microbiology of the septic
patients in our study was very similar to the reported non-HIV infected patients. We observed
a predominance of Gram-negative and Gram-positive bacteria, but also Mycobacterium
tuberculosis, which was the main pathogen associated with severe sepsis in 5 out of 44
patients in our cohort. Tuberculosis, which is highly prevalent in developing countries, has
been found to cause sepsis with a high rate of bacteremia in AIDS patients [40, 44-48].
The main contribution of our study relies on the fact that sepsis is the most important risk
factor for mortality in patients with HIV/AIDS admitted to ICU, irrespective of HAART use.
Other studies have not evaluated the diagnosis of severe sepsis or septic shock as independent
variable on survival analysis, whereas they remained giving greater worth to the presence of
opportunistic infections or immunosuppression surrogates (CD4 cell count or viral load).
Otherwise, our study has limitations. The study was conducted in a single center specialized
in HIV/AIDS patients care, and a multicenter study is needed to confirm our findings
regarding severe sepsis on HIV/AIDS critically ill patients. Although there was no
prospective comparison with a cohort without HIV, there are similarities in the microbiology
of infections and incidence of organ dysfunctions with non HIV/AIDS population reported in
the literature [20,49,50]. The high frequency of nosocomial infection does not allow us to
extrapolate our results to community-acquired septic patients. The long-term effects of sepsis
were not evaluated with quality of life questionnaires or with functional capacity examination.
The incidence of sepsis is increasing worldwide, with a projected increase of more than 15%
for the next decade [12,36]. Although international efforts sought to reduce sepsis mortality,
increased survival has not been observed in low-and middle-income countries (LMIC) [18,51-
53]. New strategies to reduce the impact of sepsis, especially in LMIC are timely. Those
strategies should be able to deal with specific subgroups of patients, including the HIV/AIDS
population. In this setting, the prevention of both nosocomial and community-acquired

148
infections should be incorporated into sepsis guidelines as a cost-effective measure for
LMICs. Finally, biomarkers and functional immunoassays must be studied in order to
understand the difference on sepsis pathophysiology between general septic patients and the
HIV/AIDS septic patients.
In conclusion, we demonstrated that severe sepsis was the main risk factor for hospital
mortality in our cohort, significantly impacting short- and longer-term survival of HIV/AIDS
critically ill patients. Mortality was shown to be more dependent on critically illness factors as
the presence of sepsis and the severity of organ dysfunction than the HIV/AIDS related
characteristics, such as the level of immunodeficiency, use of HAART or time since AIDS
diagnosis. A high level of suspicion related to sepsis diagnosis coupled with initiatives related
to nosocomial infection and sepsis prevention could contribute to decrease mortality in
critically ill HIV/AIDS patients.
List of abbreviations:
HIV/AIDS; Human Immunodeficiency Virus/ Acquired Immunodeficiency Syndrome;
HAART: Highly Active Anti-retroviral Therapy; NRTI: nucleoside reverse transcriptase
inhibitor; PI protease inhibitor, NNRTI: nonnucleoside reverse transcriptase inhibitor
APACHE: Acute Physiology and Chronic Health Evaluation Score; SAPS: simplified acute
physiology score; ICU: intensive care unit; SOFA: Sequential Organ Failure Assessment.
ACCP/SCCM: American College of Chest Physicians/Society of Critical Care Medicine;
CD4: cluster of differentiation 4; PS: Performance Status
Competing interests:
The authors declare that they have no competing interests.
Authors' contributions
All authors made substantial contribution to the study design and methods. AMJ, DMM and
FAB conceived of the study. AMJ, RTA, ECM and HBB collected clinical and
microbiological data. PML performed the data analysis. AMJ, PML and FAB drafted the

149
manuscript and DMM, EPN and BG critically revised it for important intellectual content. All
authors read and approved the final version of the manuscript.
References
1. Karim SSA, Karim QA, Gouws E, Baxter C: Global Epidemiology of HIV-AIDS.
Infect Dis Clin N Am 2007, 21:1-17.
2. Afessa B, Green B: Clinical Course, Prognostic Factors, and Outcome Prediction
for HIV Patients in the ICU. The PIP (Pulmonary Complications, ICU Support, and
Prognostic Factors in Hospitalized Patients with HIV) Study. Chest 2000, 118:138-145.
3. Morris A, Creasman J, Turner J, Luce JM, Watcher RM, Huang L: Intensive Care of
Human Immunodeficiency Virus–infected Patients during the Era of Highly Active
Antiretroviral Therapy. Am J Resp Crit Care Med 2002, 166:262-267.
4. Narasihman M, Posner AJ, DePalo VA, Mayo PH, Rosen MJ: Intensive Care in
Patients With HIV Infection in the Era of Highly Active Antiretroviral Therapy. Chest
2004, 125:1800–1804.
5. Khouli H, Afrasiabi A, Shibli M, Hajal R, Barrett CR, Homel P: Outcome of
Critically Ill Human Immunodeficiency Virus–Infected Patients in the Era of Highly
Active Antiretroviral Therapy. J Intensive Care Med 2005, 20:279-285.
6. Palacios R, Hidalgo A, Reina C, de la Torre MV, Márquez M, Santos J: Effect of
antiretroviral therapy on admissions of HIV-infected patients to an intensive care unit.
HIV Med 2006, 7:193-196.
7. Powell K, Davis JL, Morris AM, Chi A, Bensley MR, Huang L: Survival for
Patients With HIV Admitted to the ICU Continues to Improve in the Current Era of
Combination Antiretroviral Therapy. Chest 2009, 135:11-17.
8. Huang L, Quartin A, Jones D, Havlir DV: Intensive Care of Patients with HIV
Infection. N Engl J Med 2006, 355:173-181.
9. Rosen MJ, Narasimhan M: Critical care of immunocompromised patients: Human
immunodeficiency virus. Crit Care Med 2006, 34:S245-S250.
10. Davaro RE, Thirumalai A: Life-Threatening Complications of HIV Infection. J
Intensive Care Med 2007, 22:73-81.
11. Grinsztejn B, Veloso VG, Friedman RK, Moreira RI, Luz PM, Campos DP, Pilotto
JH, Cardoso SW, Keruly JC, Moore RD: Early mortality and cause of deaths in patients
using HAART in Brazil and the United States. AIDS 2009; 23:2107-14.)

150
12. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR:
Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med 2001, 29:1303-1310.
13. Martin GS, Mannino DM, Eaton S, Moss M: The Epidemiology of Sepsis in the
United States from 1979 through 2000. N Engl J Med 2003, 348:1546-1554.
14. Annane D, Aegerter P, Jars-Guincestre MC, Guidet B: Current Epidemiology of
Septic Shock. Am J Resp Crit Care Med 2003, 168:165-172.
15. The EPISEPSIS Study Group: EPISEPSIS: a reappraisal of the epidemiology and
outcome of severe sepsis in French intensive care units. Intensive Care Med 2004, 30:580-
588.
16. Ballester JCA, Ballester F, Sánchez AG, Quilis AA, Rubio EC, Otero CP:
Epidemiology of Sepsis in the Valencian Community (Spain), 1995–2004. Infect Control
Hosp Epidemiol 2008, 29:630–634.
17. Blanco J, Muriel-Bombín A, Sagredo V, Taboada F, Gandía F, Tamayo L, Collado J,
García-Labattut A, Carriedo D, Valledor M, et al.: Incidence, organ dysfunction and
mortality in severe sepsis: a Spanish multicentre study. Crit Care 2008, 12:R158.
18. Khwannimit B, Bhurayanontachai R: The epidemiology of, and risk factors for,
mortality from severe sepsis and septic shock in a tertiary-care university hospital
setting. Epidemiol Infect 2009, 137;1333–1341.
19. Beale R, Reinhart K, Brunkhorst FM, Dobb G, Levy M, Martin G, Martin C, Ramsey
G, Silva E, Vallet B, et al.: Promoting Global Research Excellence in Severe Sepsis
(PROGRESS): lessons from an international sepsis registry. Infection 2009, 37:222-232.
20. Mrus JM, Braun LA, Yi MS, Linde-Zwirble WT, Johnston JA: Impact of HIV/AIDS
on care and outcomes of severe sepsis. Crit Care 2005, 9:R623-R630.
21. US Department of Health Human Services, Centers for Disease Control and
Prevention (CDC): Impact of expanded AIDS surveillance case definition on AIDS case
reporting. Morb Mort Wkly Rep 1993, 16:308–311.
22. Le Gall JR, Lemeshow S, Saulnier F: A new Simplified Acute Physiologic Score
(SAPS II) based on an European/North American multicenter study. JAMA 1993,
270:2957-2963.
23. Le Gall JR, Neumann A, Hemery F, Bleriot JP, Fulgencio JP, Garrigues B, Gouzes C,
Lepage E, Moine P, Villers D: Mortality prediction using SAPS II: an update for French
intensive care units. Crit Care 2005, 9:R645-R652.

151
24. Vincent JL, Moreno R, Takala J, Willatts S, de Mendonça A, Bruining H, Reinhart
CK, Suter PM, Thijs LG: The SOFA (Sepsis-related Organ Failure Assessment) score to
describe organ dysfunction/failure. Intensive Care Med 1996, 22:707-710.
25. Zubrod CG, Schneiderman M, Frei E III: Appraisal of methods for the study of
chemotherapy of cancer in man: Comparative therapeutic trial of nitrogen mustard and
triethylene thiophosphoramide. J Chron Dis 1960, 11:7–33.
26. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM,
Sibbald WJ: Definitions for sepsis and organ failure and guidelines for the use of
innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee.
American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992,
101:1644-1655.
27. Horan TC, Andrus M, Dudeck MA: CDC/NHSN surveillance definition of health
care–associated infection and criteria for specific types of infections in the acute care
setting. Am J Infect Control 2008, 36:309-332.
28. Pacheco AG, Tuboi SH, May SB, Moreira LFS, Ramadas L, Nunes EP, Merçon M,
Faulhaber JC, Harrison LH, Schechter M: Temporal Changes in Causes of Death Among
HIV-Infected Patients in the HAART Era in Rio de Janeiro, Brazil. J Acquir Immune
Defic Syndr 2009, 51:624-630.
29. Casalino E, Wolff M, Ravaud P, Choquet C, Bruneel F, Regnier B: Impact of
HAART advent on admission patterns and survival in HIV-infected patients admitted to
an intensive care unit. AIDS 2004, 18:1429-1433.
30. Casalino E, Mendoza-Sassi G, Wolff M, Bédos JP, Gaudebout C, Regnier B, Vachon
F: Predictors of Short- and Long-term Survival in HIV-Infected Patients Admitted to
the ICU. Chest 1998, 113:421-429.
31. Dickson SJ, Batson S, Copas AJ, Edwards SG, Singer M, Miller RF: Survival of
HIV-infected patients in the intensive care unit in the era of highly active antiretroviral
therapy. Thorax 2007, 62:964-968.
32. Croda J, Croda MG, Neves A, Santos SS: Benefit of antiretroviral therapy on
survival of human immunodeficiency virus-infected patients admitted to an intensive
care unit. Crit Care Med 2009, 37:1605-1611.
33. Thyrault M, Gachot B, Chastang C, Souweine B, Timsit JF, Bédos JP, Regnier B,
Wolff M: Septic shock in patients with the acquired immunodeficiency syndrome.
Intensive Care Med 1997, 23:1018-1023.

152
34. Vincent B, Timsit JF, Auburtin M, Schortgen F, Bouadma L, Wolff M, Regnier B:
Characteristics and outcomes of HIV-infected patients in the ICU: impact of the highly
active antiretroviral treatment era. Intensive Care Med 2004, 30:859-866.
35. Davis JL, Morris A, Kallet RH, Powell K, Chi AS, Bensley M, Luce JM, Huang L:
Reduced Mortality in HIV-infected Patients with Low Tidal Volume Ventilation Is
Associated with Acute Ling Injury. Thorax 2008, 63:988-993.
36. Angus DC, Wax RS: Epidemiology of sepsis: an update. Crit Care Med 2001; Suppl
7:109-116.
37. Petrosillo N, Pagani L, Ippolito G; Gruppo HIV e Infezioni Ospedaliere: Nosocomial
infections in HIV-positive patients: an overview. Infection 2003, Suppl 2:28-34.
38. Rosenberg AL, Seneff MG, Atiyeh L, Wagner R, Bojanowski L, Zimmerman JE: The
importance of bacterial sepsis in intensive care unit patients with acquired
immunodeficiency syndrome: Implications for future care in the age of increasing
antiretroviral resistance. Crit Care Med 2001, 29:548-556.
39. Meynard JL, Guiguet M, Fonquernie L, Lefebvre B, Lalande V, Honore I, Meyohas
MC, Girard PM: Impact of highly active antiretroviral therapy on the occurrence of
bacteremia in HIV-infected patients and their epidemiologic characteristics. HIV Med
2003, 4:127-132.
40. Arthur G, Nduba VN, Kariuki SM, Kimari J, Bhatt SM, Gilks CF: Trends in
bloodstream infections among human immunodeficiency virus-infected adults admitted
to a hospital in Nairobi, Kenya, during the last decade. Clin Infect Dis 2001, 33:248-256.
41. Hung CC, Hsueh PR, Hsieh SM, Liu CJ, Chen MY, Luh KT: Bacteremia and
fungemia in patients with advanced human immunodeficiency vírus (HIV) infection in
Taiwan. J Formos Med Assoc 1998, 97:690-697.
42. Tumbarello M, Tacconelli E, Donati KG, Citton R, Leone F, Spanu T, Cauda R: HIV-
Associated Bacteremia: How It Has Changed in the Highly Active Antiretroviral
Therapy (HAART) Era. J Acquir Immune Defic Syndr 2000, 23:145-151.
43. Vidal F, Mensa J, Martínez JA, Almela M, Marco F, Gatell JM, Richart C, Soriano E,
Jiménez de Anta MT: Pseudomonas aeruginosa Bacteremia in Patients Infected with
Human Immunodeficiency Virus Type 1. Eur J Clin Microbiol Infect Dis 1999, 18:473–
477.
44. Grinsztejn B, Veloso VG, Pilotto JH, Campos DP, Keruly JC, Moore RD:
Comparison of clinical response to initial highly active antiretroviral therapy in the

153
patients in clinical care in the United States and Brazil. J Acquir Immune Defic Syndr
2007, 45:515-520.
45. Schmaltz CA, Santanna FM, Neves SC, Velasque LD, Lourenço MC, Morgado MG,
Rolla VC, Lopes GS: Influence of HIV Infection on Mortality in a Cohort of Patients
Treated for Tuberculosis in the Context of Wide Access to HAART, in Rio de Janeiro,
Brazil. J Acquir Immune Defic Syndr, in press.
46. Pacheco AG, Durovni B, Cavalcante SC, Lauria LM, Moore RD, Moulton LH,
Chaisson RE, Golub JE: AIDS-Related Tuberculosis in Rio de Janeiro, Brazil. PLoS One
2008, 3:e3132.
47. Waddell RD, Lishimpia K, von Reyn CF, Chintua C, Babooa KS, Kreiswirthb B,
Talbotc EA, Karagas MR: Study Group Bacteremia due to Mycobacterium tuberculosis
or M. bovis, Bacille Calmette-Guerin (BCG) among HIV positive children and adults in
Zambia. AIDS 2001, 15:55-60.
48. Grinsztejn B, Fandinho FC, Veloso VG, João EC, Lourenço MC, Nogueira SA,
Fonseca LS, Werneck-Barroso E: Mycobacteremia in patients with the acquired
immunodeficiency syndrome. Arch Intern Med 1997; 157:2359-63.)
49. Silva E, Pedro MA, Sogayar ACB, Mohovic T, Silva CLO, Janiszewski M, Cal RGR,
Sousa EF, Abe TP, Andrade J et al.: Brazilian Sepsis Epidemiological Study (BASES
study). Crit Care 2004, 8:R260.
50. Sales-Júnior JA, David CM, Hatum R, Souza PCSP, Japiassú A, Pinheiro CTS,
Friedman G, Silva OB, Dias MD, Koterba E et al.: An Epidemiological Study of Sepsis in
Intensive Care Units. Sepsis Brazil Study. Rev Bras Ter Intens 2006, 18:9-17.
51. Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K,
Angus DC, Brun-Buisson C, Beale R et al.: Surviving Sepsis Campaign: international
guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 2008,
36:296-327.
52. Becker JU, Theodosis C, Jacob ST, Wira CR, Groce NE: Surviving sepsis in low-
income and middle-income countries: new directions for care and research. Lancet Infect
Dis 2009, 9:577-582.
53. Cheng AC, West TE, Limmathurotsakul D, Peacock SJ: Strategies to Reduce
Mortality from Bacterial Sepsis in Adults in Developing Countries. PLoS Med 2008,
5:1173-1179.

154
Table 1 – Demographics, HIV/AIDS characteristics and causes of admission to ICU of all
patients.
Patients (n = 88)
Age (years) 40 (31-47)
Male gender 67 (76%)
Time since AIDS diagnosis (months) 40 (2-90)
Recent AIDS diagnosis 25 (28%)
CD4 cell count (per mm3) 75 (32-227)
HAART 40 (45%)
Cause of admission
Acute respiratory failure
Coma/torpor
Sepsis
Decompensate heart disease
Gastrointestinal diseases
Metabolic disturbance
High-grade lymphoma
26 (29%)
20 (23%)
18 (20%)
9 (10%)
6 (7%)
5 (6%)
4 (5%) Continuous values are shown as median and interquartile interval. ICU – intensive care unit;
AIDS – Acquired Immunodeficiency Syndrome; HAART – highly active antiretroviral
therapy.

155
Table 2 – Comparison of patients according to hospital survival.
Survivors
(n = 45)
Non-survivors
(n = 43)
Age (mean ± SD) 42 (35-50) 38 (29-46)
Male gender (%) 36 (80%) 31 (72%)
Location before ICU (ward, %) 24 (53%) 29 (67%)
Performance status 3 or 4 1 (0-2) 2 (1-3)
CD4 count < 50 mm3 (%) 20 (44%) 21 (49%)
Length of time from AIDS diagnosis 39 (2-92) 50 (2-85)
Recent (< 3 months) AIDS diagnosis 13 (29%) 13 (30%)
HAART prescription 20 (44%) 20 (46%)
SAPS II expanded (points) 44 (37-54) 54 (46-67)†
SOFA D1 (points) 4 (1-7) 7 (4-10)†
Mechanical ventilation 17 (38%) 36 (84%)†
Use of vasopressors on day 1 6 (13%) 15 (30%)*
Renal support 3 (7%) 13 (30%)†
Severe sepsis / septic shock (%) 15 (33%) 29 (67%)†
ICU length of stay (days) 9 (5-17) 10 (4-14)
Hospital length of stay (days) 24 (12-58) 15 (10-24)
ICU – Intensive Care Unit; HAART – Highly Active Anti-Retroviral Therapy; AIDS –
Acquired Imunodeficiency Syndrome; SAPS – Simplified Acute Physiology Score; SOFA –
Sequencial Organ Failure Assessment. Significant differences are shown as * p<0.05; †
p<0.01.

156
Table 3 – Site of infection and microbiological data of septic shock AIDS patients.
Site of infection N
Pulmonary 23 (52%)
Primary bacteremia 17 (38%)
Venous catheter-related infection 3 (7%)
Urinary tract infections 1 (3%)
Microbiology of infection N
P aeruginosa 10 (23%)
K pneumoniae 6 (13%)
M tuberculosis 6 (13%)
Enterobacter sp 5 (11%)
S aureus 5 (11%)
Staphylococcus negative-coagulase 4 (9%)
E coli 3 (7%)
A calcoaceticus 3 (7%)
S marcenses 3 (7%)
Other 5 (11%)

157
Table 4 – Crude and adjusted hazard ratios (HR) and 95% confidence intervals for factors
associated with 28-days and 6-months mortality as estimated using Cox proportional hazards
regression.
28-days mortality 6-months mortality
Crude HR
(95% CI)
Adjusted HR
(95% CI)
Crude HR
(95% CI)
Adjusted HR
(95% CI)
Age (< 40 years) 1.96
(0.99-3.89)
1.52
(0.84-2.77)
Location before
ICU (ward)
2.28*
(1.10-4.73)
1.93*
(1.02-3.64)
Cardiovascular
dysfunction
2.97*
(1.23-7.13)
2.12*
(1.05-4.30)
Respiratory
dysfunction
2.21*
(1.01-4.86)
1.58
(0.82-3.02)
Severe
sepsis/Septic shock
4.17†
(1.96-8.90)
6.83†
(1.87-24.8)
3.25†
(1.75-6.16)
5.20†
(1.38-9.97)
Time since AIDS
diagnosis (< 60
days)
0.96
(0.46-2.00)
0.95
(0.49-1.84)
CD4 count (< 50
cells/mm3)
1.16
(0.51-2.65)
1.55
(0.76-3.18)
HAART 1.31
(0.68-2.52)
1.21
(0.67-2.18)
ICU – Intensive Care Unit; HAART – Highly Active Anti-Retroviral Therapy; AIDS –
Acquired Imunodeficiency Syndrome. Significant differences are shown as * p<0.05; †
p<0.01. Adjusted HR include adjustments for age, CD4 count, time since AIDS diagnosis,
HAART, location before ICU, and cardiovascular and respiratory dysfunctions.
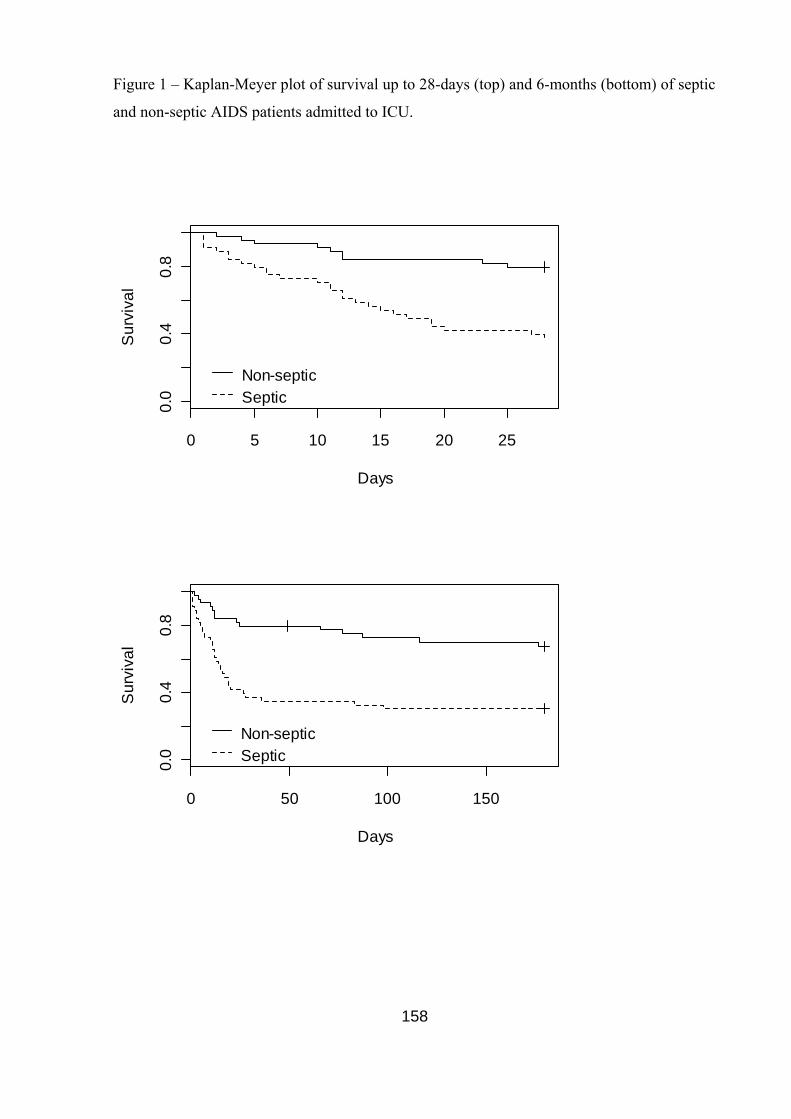
Figure 1 – Kaplan-Meyer plot of survival up to 28-days (top) and 6-months (bottom) of septic
and non-septic AIDS patients admitted to ICU.
0 5 10 15 20 25
0.0
0.4
0.8
Days
Sur
viva
l
Non-septicSeptic
0 50 100 150
0.0
0.4
0.8
Days
Sur
viva
l
Non-septicSeptic
158

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo