pró- reitoria de graduação grazielle peres … · grazielle peres guimarÃes ... aos meus pais,...
TRANSCRIPT

GRAZIELLE PERES GUIMARÃES
AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS PORTADORES DE ANEMIA
FALCIFORME NO HOSPITAL DAS FORÇAS ARMADAS DE BRASÍLIA – DF
Projeto apresentado ao curso de graduação em
Biomedicina da Universidade Católica de
Brasília como parte do trabalho de conclusão
de curso da disciplina TCC II.
Orientadora: Profª. Msc. Cintia do Couto
Mascarenhas.
Co-orientador: Prof. Msc. Paulo Roberto
Sabino.
Co-orientador: Prof. Esp. Fábio de França
Martins.
Brasília
2010
Pró- Reitoria de Graduação
Biomedicina
Trabalho de Conclusão de Curso
AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS
PORTADORES DE ANEMIA FALCIFORME NO HOSPITAL DAS
FORÇAS ARMADAS DE BRASÍLIA – DF
Grazielle Peres Guimarães
Orientadora: Profa. Msc.Cintia do Couto Mascarenhas
Co-orientador: Prof. Msc. Paulo Roberto Sabino Jr
Co-orientador: Prof. Esp. Fábio de França Martins
Brasília - DF
2010

2
Projeto de TCC II, de autoria de Grazielle Peres Guimarães, intitulado “Avaliação da
prevalência de indivíduos portadores de anemia falciforme no Hospital das Forças Armadas
de Brasília – DF”, apresentado como requisito parcial para a obtenção do grau de Bacharel em
Biomedicina da Universidade Católica de Brasília no segundo semestre de 2010, defendida e
aprovada pela banca examinadora abaixo assinada:
____________________________________________________
Profª MSc. Cintia do Couto Mascarenhas
Orientadora
Doutoranda pela Faculdade de Ciências Médicas da UNICAMP
Pesquisadora voluntária – UCB
_____________________________________________________
Prof. Dr. Anderson Ferreira da Cunha
Examinador
Professor Adjunto – UFSCAR
____________________________________________________
Profª. Drª. Rosangela Vieira de Andrade
Examinador
Ciência Genômicas e Biotecnologia – UCB
Brasília
2010

3
Aos meus pais, Roberto e Lúcia.
Ao meu irmão, Higor.

4
AGRADECIMENTOS
A Deus, por ser o meu amparo, minha fortaleza e por me capacitar a alcançar meus
objetivos.
Aos meus pais, Roberto e Lúcia, pelo amor incondicional e por estarem sempre ao
meu lado.
A professora Cintia Mascarenhas, pela orientação, apoio e confiança.
Ao professor Paulo Sabino, pela disponibilidade e paciência em me ensinar a lidar
com as dificuldades da pesquisa científica.
Ao professor Fábio de França, pelo incentivo e disponibilidade em compartilhar
conhecimentos.
As minhas grandes amigas e companheiras de graduação Camilla e Karoline que
sempre estiveram ao meu lado, tornando meus dias de universidade mais compensadores.
Aos meus grandes amigos Artur, Edmércia e Ana Vera, pelos anos de amizade,
paciência e por transformarem meu cansaço em momentos de muita alegria.
A todos os meus professores pelo conhecimento transmitido e dedicação para a minha
formação profissional e moral.

5
Resumo
Referência: GUIMARÃES, Grazielle Peres. Título: Avaliação da prevalência de indivíduos
portadores de anemia falciforme no Hospital das Forças Armadas de Brasília – DF. 55 folhas.
Trabalho de Conclusão de Curso (Biomedicina) – Universidade Católica de Brasília, Águas
Claras, 2010.
A anemia falciforme é caracterizada pela presença da hemoglobina S em homozigose
(HbSS) e está entre as hemoglobinopatias mais prevalentes no Brasil e no mundo, sendo
observada principalmente na população afro descendente. A polimerização da HbS
desoxigenada ocasiona deformação, maior rigidez e fragilidade do eritrócito, além de anemia
hemolítica e episódios vasoclusivos. A adesividade aumentada das células falcizadas e
leucócitos ao endotélio causa diminuição do fluxo sanguíneo na microcirculação, resultando
no principal evento da anemia falciforme, que é a vasoclusão.
O trabalho objetivou realizar a avaliação da prevalência de portadores da HbS no
Hospital das Forças Armadas de Brasília e correlacionar esses dados com outros estudos de
prevalência no Distrito Federal.
Foram avaliados 1181 pacientes no período de setembro a outubro de 2010, após a
aprovação do Comitê de Ética em pesquisa do HFA, e assinatura do termo de consentimento
livre esclarecido pelos pacientes. Dentre estes, 61 fazem parte da triagem realizada a partir do
hemograma, teste de falcização, eletroforese manual e confirmação pela eletroforese por
capilaridade, 1057 a partir dos resultados de exames de hemoglobina glicada do equipamento
D-10 (Bio-Rad®
), o qual inclui resultados para os demais tipos de hemoglobina, e 64
pacientes com exames cadastrados para realização de eletroforese por capilaridade no
equipamento Minicap (SEBIA®
).
Das amostras avaliadas, nenhuma apresentou homozigose para hemoglobina S
(HbSS), enquanto trinta e seis pacientes apresentaram traço falciforme (HbAS) e três
apresentaram hemoglobinopatia SC (HbSC), com prevalência de 3,05% para traço falciforme
e 0,25% para hemoglobinopatia SC.
Foi possível observar que a média de idade entre os pacientes que apresentaram
hemoglobina HbS foi de 44,36 anos. Já a distribuição quanto ao sexo mostrou maior
incidência de HbS em pessoas do sexo feminino, representando 61,54% (24), enquanto para o
sexo masculino foi de 38,46% (15). Houve o predomínio de alterações de frações
hemoglobínicas em indivíduos declarados de cor parda.
A prevalência de portadores de traço falciforme obtida foi compatível com a de outros
estudos realizados no Brasil e no mundo, apresentando semelhança especial com os dados
disponíveis do Distrito Federal. Observou-se que o número de casos de traço falciforme foi
maior em relação aos outros fenótipos anômalos encontrados, enquanto a hemoglobinopatia
SC foi encontrada em menor número de indivíduos.
Nossos resultados indicam que existe relação entre a ampliação da triagem
laboratorial, para hemoglobinopatias, e o aumento do número de casos de HbS. Apesar da
concordância dos resultados obtidos com os da literatura, sugere-se a realização de estudos
maiores que possam contribuir para o melhor conhecimento do perfil epidemiológico das
hemoglobinopatias na região do Distrito Federal.
Palavras-chave: Anemia falciforme. Traço falciforme. Hemoglobina S.

6
ABSTRACT
Referência: GUIMARÃES, Grazielle Peres. Título: Evaluation of the prevalence of
individuals with sickle cell disease in the Armed Forces Hospital of Brasilia - DF. 55 folhas.
Trabalho de Conclusão de Curso (Biomedicina) – Universidade Católica de Brasília, Águas
Claras, 2010.
Sickle cell anaemia is characterized by the presence of hemoglobin S homozygous (HbSS)
and is among the most prevalent hemoglobinopathies in Brazil and worldwide, and mainly
observed in the population african descent. The polymerization of deoxygenated HbS causes
deformation, rigidity and fragility of the erythrocytes, and hemolytic anemia and episodes
vasocclusives. The increased adhesiveness of sickled cells and leukocytes to the endothelium
causes decreased blood flow in the microcirculation, resulting in the main event of sickle cell
disease, which is the vasocclusion.
The work has aimed at assessing the prevalence of carriers of HbS in the Armed Forces
Hospital of Brasilia and correlate these data with other prevalence studies in the Distrito
Federal.
We evaluated 1181 patients during the period September to October 2010 following the
approval of the Committee of Ethics of HFA, and signing the consent form for patients.
Among these, 61 are part of the screening conducted from the blood count, sickling test,
manual electrophoresis and confirmation by capillary electrophoresis, 1057 from the results of
tests for glycated hemoglobin Equipment D-10 (Bio-Rad ®
), the which includes results for the
other types of hemoglobin, and 64 patients enrolled to carry out examinations by capillary
electrophoresis equipment Minicap (Sebia ®
).
Of the samples tested, none had homozygous hemoglobin S (HbSS), while thirty-six patients
had sickle cell trait (HbS) and three had hemoglobinopathy SC (HbSC), with a prevalence of
3.05% for sickle cell trait and 0.25% for SC hemoglobinopathy.
It was observed that the average age among patients with hemoglobin HbS was 44.36 years.
The distribution by sex showed a higher incidence of HbS in feminine, accounting for 61.54%
(24), while for masculine was 38.46% (15). There was a predominance of changes in
hemoglobin fractions of individuals declared brown.
The prevalence of patients with sickle cell trait result was compatible with other studies in
Brazil and worldwide, with particular similarity to the available data from the Distrito
Federal. It was observed that the number of cases of sickle cell trait was higher in relation to
other abnormal phenotypes found, while the SC hemoglobinopathy was found in fewer
individuals.
Our results indicate that there is a relationship between the expansion of laboratory screening
for hemoglobinopathies, and the increased number of cases of HbS. Despite the concordance
of results with the literature, it is suggested to carry out larger studies that could contribute to
a better understanding of the epidemiology of hemoglobinopathies in the region of the
Distrito Federal.
Keywords: Sickle cell disease. Sickle cell trait. Hemoglobin S.

7
LISTA DE FIGURAS
Figura 1- Esquema de representação geográfica do gene da βS globina.
Identificação de áreas na África e Árabe-Índia (Linha pontilhada) de regiões que
apresentam índices expressivos de pessoas com anemia falciforme (linhas
vermelhas), situadas em maior parte da costa e parte central das regiões: Senegal,
Benin e Bantu................................................................................................................
Figura 2- Distribuição da Hb S em diferentes regiões brasileiras................................
Figura 3- Fisiopatologia da vasoclusão na anemia falciforme. (A) Substituição do
nucleotídeo (GTG GAG). (B) Polimerização da HbS, com ocorrência de
vasoclusão e hemólise intravascular . (C) Formação do eritrócito falcizado. (D)
Processo de vasoclusão mediado por eritrócitos falcizados, neutrófilos e
reticulócitos. EC= endotélio. R= reticulócito. ISC= célula irreversivelmente
falcizada. N= neutrófilo. RBC= célula vermelha..........................................................
Figura 4- Processo de adesão celular entre eritrócitos falcizados e endotélio
vascular. PS= fosfatidilsserina. TSP= trombospondina. FN= fibronectina. LM=
laminina. VWF= fator de von Willebrand. Ocorre aumento de moléculas de adesão
(α4β1, CD36, B-CAM/LU) em eritrócitos e reticulócitos na anemia falciforme
quando comparado com eritrócitos normais.................................................................
Figura 5- Representação de teste de falcização positivo em paciente com traço
falciforme......................................................................................................................
Figura 6- Diagrama eletroforético de paciente com traço falciforme pela técnica de
eletroforese por capilaridade. Concentração das frações de hemoglobina: HbA= 55,5
%, HbA2= 3,1 % e HbS= 41,4 %............................................................................
Figura 7- (A) Análise do gel de eletroforese manual de hemoglobina utilizando
tampão Tris-EDTA-Borato - pH 8,5. 1 a 3 e 5 a 9. Amostras com hemoglobina A
(banda mais escura) e hemoglobina A2 (banda mais clara). 10. Amostra controle -
paciente apresentando hemoglobinas A, A2 e F em níveis normais. (B) Em
evidência poço número 4 com amostra apresentando bandas de hemoglobina A
(banda mais escura), S e banda que sugere Hb A2 e HbC de acordo com o padrão de
bandas de hemoglobinas utilizado no estudo................................................................
Figura 8- (A) Análise do gel de eletroforese manual de hemoglobina utilizando
tampão Citrato pH 6,3. 1 a 3. e 5 a 9. Amostras com resultado de HbF e HbA. 10.
14
16
19
20
29
31
37

8
Amostra controle - paciente apresentando hemoglobinas A, A2 e F em níveis
normais. (B) Em evidência poço número 4 com amostra apresentando bandas de
hemoglobina F, A, S e banda desconhecida em relação ao padrão de bandas de
hemoglobinas utilizado no estudo..............................................................................
Figura 9- Diagrama eletroforético de paciente com traço falciforme que apresentou
fração de hemoglobina variante pela avaliação de eletroforese por capilaridade. A
zona nove (Z9) mostra a concentração de hemoglobina A (66,9 %), a zona sete (Z7)
apresenta a concentração de hemoglobina F (0,2 %), a zona cinco (Z5) apresenta a
concentração de hemoglobina S (29,6 %) e o início da zona três (Z3) apresenta a
concentração de hemoglobina A2 (1,4 %) e o final desta zona (Z3) evidencia uma
fração de hemoglobina variante cuja a concentração foi de1,9%.................................
38
38

9
LISTA DE GRÁFICOS
Gráfico 1- Distribuição quanto à cor da pele de pacientes com HbS...........................
Gráfico 2- Distribuição quanto à cor da pele entre pacientes do sexo masculino e
feminino que apresentaram HbS...................................................................................
Gráfico 3- Valores de concentração de hemoglobinas entre os 39 pacientes com
traço falciforme e hemoglobinopatia SC. Pacientes com traço falciforme
apresentaram índices maiores de hemoglobina A. Pacientes com hemoglobinopatia
SC apresentaram índices discretamente maiores de hemoglobina S em relação a
hemoglobina C..............................................................................................................
34
34
37

10
LISTA DE TABELAS
Tabela 1- Quantidade de pacientes analisados no estudo segundo as técnicas
utilizadas................................................................................................................................
Tabela 2- Número absoluto de casos de portadores de traço falciforme e
hemoglobinopatia SC avaliados no laboratório de análises clínicas do Hospital das
Forças Armadas, de maio a agosto de 2010..........................................................................
Tabela 3- Média dos índices hematimétricos de pacientes que apresentaram HbS.............
Tabela 4- Dados quanto ao sexo, idade, cor da pele, concentração das frações de
hemoglobina e índices hematimétricos dos 39 pacientes que apresentaram traço
falciforme e hemoglobinopatia SC........................................................................................
28
33
35
36

11
SUMÁRIO
1. INTRODUÇÃO .................................................................................................................. 13
1.1 DADOS EPIDEMIOLÓGICOS ...................................................................................... 15
2. REFERENCIAL TEÓRICO ............................................................................................. 17
2.1 MANIFESTAÇÕES CLÍNICAS ..................................................................................... 21 2.1.1 Crises vasoclusivas ....................................................................................................... 22
2.1.2 Crises aplásticas ........................................................................................................... 22
2.1.3 Crise de sequestro esplênico ........................................................................................ 23
2.1.4 Crises hemolíticas ........................................................................................................ 23
2.2 DIAGNÓSTICO ............................................................................................................... 24
2.3 TRATAMENTO ............................................................................................................... 25
3. JUSTIFICATIVA ............................................................................................................... 26
4. OBJETIVOS ....................................................................................................................... 27
4.1 OBJETIVO GERAL ........................................................................................................ 27
4.2 Objetivos Específicos ........................................................................................................ 27
5. MATERIAIS E MÉTODOS .............................................................................................. 28
5.1 CONTAGEM AUTOMATIZADA DE CÉLULAS E ANÁLISE MORFOLÓGICA
POR MICROSCOPIA ........................................................................................................... 28
5.2 TESTE DE FALCIZAÇÃO ............................................................................................. 29
5.3 ELETROFORESE DE HEMOGLOBINA EM PH ALCALINO E ÁCIDO .............. 30
5.4 ELETROFORESE DE HEMOGLOBINA POR CAPILARIDADE ........................... 30
5.5 CROMATOGRAFIA LÍQUIDA DE ALTA PERFORMANCE – HPLC .................. 31
5.6 ANÁLISE DOS DADOS .................................................................................................. 32
6. RESULTADOS ................................................................................................................... 33

12
7. DISCUSSÃO ....................................................................................................................... 39
8. REFERÊNCIAS ................................................................................................................. 43
ANEXOS ................................................................................................................................. 49
ANEXO A ................................................................................................................................ 49
ANEXO B ................................................................................................................................ 50
ANEXO C ................................................................................................................................ 51
ANEXO D ................................................................................................................................ 53
ANEXO E ................................................................................................................................ 55

13
1. INTRODUÇÃO
As hemoglobinopatias são doenças hereditárias caracterizadas pelo comprometimento
de grupamentos de genes, localizados no braço curto do cromossomo 16 e cromossomo 11,
responsáveis, respectivamente, pela síntese de cadeias globínicas do tipo alfa e beta das
moléculas de hemoglobina. Essas alterações podem causar modificações estruturais,
deficiência parcial ou total na síntese de cadeias globínicas, determinando o surgimento de
hemoglobinas variantes e das talassemias (SILVA, 2001; GALIZA NETO; PITOMBEIRA,
2003).
Assim sendo, as hemoglobinopatias, que correspondem a um amplo grupo de
alterações, são consideradas as doenças hematológicas de maior prevalência no mundo, sendo
apontadas como um problema de saúde pública mundial, devido ao aumento da freqüência e
das severidades apresentadas (FATTOUM, 2006).
No Brasil estas doenças também são um problema de saúde pública, devido ao alto
grau de miscigenação da população brasileira (RAMALHO, 2003). Segundo Bonini-
Domingos (2009), a incidência de determinadas hemoglobinopatias pode variar de acordo
com a fixação dos grupos raciais colonizadores de cada região.
Entre as hemoglobinopatias situa-se a anemia falciforme, que é caracterizada pela
presença em homozigose da hemoglobina S (HbSS) e é considerada uma doença muito
comum, de modo a apresentar expressiva distribuição em diferentes países, incluindo o Brasil
(GALIZA NETO; PITOMBEIRA, 2003).
A anemia falciforme é ocasionada pela substituição de um nucleotídeo adenina por
timina (A T), o que leva à troca de aminoácidos (ácido glutâmico valina) na posição 6
da cadeia globínica β. Esta doença possui considerável importância clínica, pois suas
peculiares manifestações, como úlcera de membros inferiores, priaprismo, complicações
cardíacas, pulmonares e neurológicas, são decorrentes tanto da anemia hemolítica crônica
quanto de fenômenos vasoclusivos, conduzindo a crises dolorosas agudas e ao dano tecidual
(SCHNOG et al., 2004; ZAGO, 2004; KATO et al., 2009).
A literatura destaca que esta doença possui diversos haplótipos, que são definidos
como distintas composições de sítios polimórficos de DNA presentes em uma região gênica
específica. Desta forma, os diferentes haplótipos associados ao segmento cromossômico da
beta-globina permitem a caracterização da presença da HbS em determinados grupos étnicos.

14
A classificação dos cinco haplótipos (Senegal, Benin, Banto, Asiático e Camarões)
característicos da anemia falciforme, foi estabelecida segundo a origem geográfica de cada
um (Figura 1) (BEZERRA, 2009).
Estudos apontam que os haplótipos: Senegal, Benin e Banto ocorrem com maior
frequência nas Américas, fato que pode ser explicado pelo grande tráfico de escravos. Já o
haplótipo Asiático apresenta maior incidência nas regiões da Arábia Saudita e Oriente Médio.
O haplótipo Camarões está limitado a regiões específicas da África (STUART; NAGEL,
2004). Assim, a origem multicêntrica desses haplótipos também pode estar relacionada à
grande variabilidade clínica da doença falciforme, que é correlacionada com a concentração
de hemoglobina fetal (HbF) presente em cada haplótipo ( SCHNOG et al., 2004).
Figura 1- Esquema de representação geográfica do gene da βS globina. Identificação de áreas na África e Árabe-
Índia (Linha pontilhada) de regiões que apresentam índices expressivos de pessoas com anemia falciforme
(linhas vermelhas), situadas em maior parte da costa e parte central das regiões: Senegal, Benin e Bantu. Fonte:
Adaptado de Stuart e Nagel (2004)
Os indivíduos que portam os haplótipos Senegal e Asiático possuem maior
concentração de HbF, o que determina a redução da gravidade dos sintomas do paciente. Os
portadores dos haplótipos Benin e Banto, por sua vez, apresentam níveis diminuídos de HbF,
ocasionando alterações clínicas mais severas. No Brasil existe o predomínio de indivíduos
com haplótipo Banto, compreendendo aproximadamente 66% dos casos de anemia
registrados. O restante dos casos está relacionado aos haplótipos Benin (32%) e Senegal (2%)
(ANVISA, 2002).

15
1.1 DADOS EPIDEMIOLÓGICOS
A expressiva heterogeneidade genética da população brasileira, resultante da
composição étnica diversificada de seus grupos formadores e do cruzamento entre eles,
possibilita tanto a interação de genes, como os responsáveis pela síntese de variantes
estruturais de hemoglobina, quanto a propagação destes genes para outras regiões e
populações do país, devido ao fluxo migratório vigente (BONINI-DOMINGOS, 2009;
SONATI; COSTA, 2008).
Desta forma, a grande freqüência e a importante repercussão clínica da doença
falciforme demonstram sua importância entre as hemoglobinopatias no Brasil e no mundo. O
que é enfatizado pela inclusão desta patologia em programas de saúde pública brasileiros,
como o PNTN (Programa Nacional de Triagem Neonatal), criado em 2001, e o Programa
Nacional de Atenção Integral as Pessoas com Doença Falciforme e outras
Hemoglobinopatias, criado em 2005, ambos do Ministério da Saúde (RAMALHO, et al.,
2003; BRASIL, 2005).
O reconhecimento das doenças falciformes como um problema de saúde pública no
Brasil foi determinante para a instituição tanto de programas de saúde publica quanto de leis
como, a Lei 14.482, de 16 de julho de 2007, que “torna possível a proposta de integralidade
das ações e da assistência aos portadores das doenças falciformes e outras
hemoglobinopatias”, o que torna possível, um melhor acesso aos serviços, a garantia da
informação e da qualificação dos profissionais que atendem essa população (BRASIL, 2008).
É importante ressaltar ainda, que este reconhecimento e conseqüente elaboração de
políticas públicas, como também da realização de medidas que visam o tratamento dos
portadores de doença falciforme, têm elevado de maneira significativa os custos financeiros
do governo federal (BRASIL, 2008).
A introdução da HbS no Brasil, determinada pela entrada maciça de escravos africanos
entre 1550 e 1850, configura a composição populacional, segundo o processo de
miscigenação das regiões brasileiras (RUIZ, 2007). De acordo com Cançado e Jesus (2007),
os estados que apresentam as maiores prevalências do traço falciforme são: Bahia (5,3%),
Pernambuco (4%), Rio de Janeiro (4%) e Minas Gerais (3%), enquanto os estados de São
Paulo (2,6%) e Rio Grande do Sul (2,0%) apresentam prevalência menor, o que confirma os
dados disponíveis sobre a distribuição histórica de povoamento geográfico brasileiro
(CANÇADO; JESUS, 2007) (Figura 2).

16
Na região Centro-Oeste, segundo Melo-Reis et al. (2006), o estado de Goiás possui
prevalência de 2,2% do traço falciforme, apresentando semelhança com os resultados dos
demais estados dessa região, como Mato Grosso do Sul (1,64%) (HOLSBACH, D. R. et al.,
2008) e Distrito Federal (3,23%) (DINIZ et al., 2009).
Figura 2- Distribuição da Hb S em diferentes regiões brasileiras. Fonte: Cançado e Jesus (2007)
Esses dados demonstram que existe freqüência similar de heterozigotos AS na
população brasileira (2% a 8%) (MURÃO; FERRAZ, 2007), quando comparada a algumas
regiões do continente africano como, a região Norte que apresenta prevalência entre 1% a 2%
e a região Sul com menos de 1%. Entretanto, é observada freqüência significativa na região
equatorial, variando de 10% a 40% de indivíduos com traço falciforme. De acordo com dados da
Organização Mundial da Saúde (OMS), aproximadamente cinco milhões de pessoas em todo
o mundo possuem anemia falciforme, sendo que 70% delas se encontram na África (OMS,
2006).

17
2. REFERENCIAL TEÓRICO
A anemia falciforme é a hemoglobinopatia de maior prevalência mundial, afetando,
sobretudo, pessoas afro descendentes (LOUREIRO; ROZENFELD; PORTUGAL, 2008).
Esta desordem é determinada pela presença de uma mutação pontual no gene da beta-globina,
que ocorre na posição 6 da sequência de aminoácidos, na qual uma base adenina (A) é trocada
por uma base timina (T) (GAG GTG), resultando na substituição de um ácido glutâmico
por uma valina (β6 Glu Val). O efeito dessa mudança de aminoácido é a formação de uma
hemoglobina variante, denominada hemoglobina S (HbS) (BALLAS; MOHANDAS, 1996).
Um dos resultados da troca do ácido glutâmico pela valina é a redução da solubilidade
da molécula de hemoglobina S (HbS) quando desoxigenada (BARBARINO; PLATT; KAUL,
2010). Tal fato, promove a união das moléculas de hemoglobina formando longas cadeias de
polímeros, que são estruturas tubulares formadas por filamentos de HbS dispostos em torno
de um eixo longitudinal do eritrócito (BOOKCHIN; LEW, 1996; VEKILOV, 2007).
O processo de polimerização da HbS promove a alteração tanto na forma do eritrócito,
surgindo o eritrócito falcizado, quanto na sua flexibilidade (ZAGO, 2004) (Figura 3). Assim,
a polimerização da HbS, quando está desoxigenada, constitui o evento fundamental da
patogênese da anemia falciforme, conferindo uma mudança na conformação do eritrócito que
perde sua forma de disco bicôncavo (BOOKCHIN; LEW, 1996).
Somente uma valina de cada tetrâmero de HbS polimerizada participa dos contatos
intermoleculares (ZAGO, 2004; BALLAS, 2005). Contudo, alguns fatores como a
concentração intra-eritrocitária da HbS e Fetal (HbF) e/ou associação com outras variáveis,
como doença falciforme associada a beta-talassemia, grau de desoxigenação das
hemoglobinas, pH, temperatura e formação de íons podem colaborar para a estimulação do
processo da polimerização (BALLAS, et al., 2010).
Os polímeros podem sofrer despolimerização quando são novamente reoxigenados,
revertendo a falcização da célula. Porém, repetidos episódios de falcização e reoxigenação do
eritrócito induzem a um estado irreversível da membrana celular alterada do eritrócito
(BALLAS, et al. 2010; ZAGO, 2004).
Além dos eventos intra-eritrocitários algumas variáveis extra-eritrocitárias também são
importantes para o desenvolvimento das manifestações clínicas, dentre as quais estão: o
número e atividade de leucócitos e plaquetas, quantidade de mediadores inflamatórios,

18
disponibilidade alterada de fatores vasomotores, grau de ativação endotelial e do sistema de
coagulação (BARBARINO; PLATT; KAUL, 2010; CAPPELLINI, 2007).
A polimerização além de ocasionar mudança estrutural do eritrócito também eleva a
expressão de marcadores de superfície na membrana do eritrócito falcizado, determinando um
aumento da aderência desses eritrócitos ao endotélio vascular, culminando no fenômeno
principal da anemia falciforme que é a vasoclusão (BALLAS; MOHANDAS, 1996; ROSSE
et al., 2000).
O processo da vasoclusão envolve diversas etapas, que se iniciam pela alteração da
adesão tanto de células falcizadas quanto de leucócitos ao endotélio vascular e também pela
interação direta entre os eritrócitos falciformes e os leucócitos aderentes, o que produz a
diminuição do fluxo sanguíneo tecidual (BARBARINO; PLATT; KAUL, 2010).
Nesse sentido, diversos estudos demonstram um aumento do número e das
propriedades adesivas dos leucócitos ao endotélio microvascular em indivíduos com anemia
falciforme. Os eosinófilos, por exemplo, aparecem em número elevado, evidenciando também
um aumento na expressão de vários tipos de moléculas de adesão, entre elas estão as
integrinas LFA-1, Mac-1 e VLA-4 que, promovem maior afinidade de adesão entre os
eosinófilos de indivíduos com anemia falciforme e a fibronectina. Outro fator relacionado à
elevação da adesividade desses à fibronectina é o aumento da produção de citocinas, em
especial a GM-CSF decorrente de processos inflamatórios (CANALLI et al., 2004).
Segundo Canalli et al. (2008), os neutrófilos de indivíduos com anemia falciforme
também apresentam maior capacidade em se aderirem a componentes da matriz extracelular
como, a fibronectina, e ainda a moléculas que são expressas por células endoteliais como, o
ICAM-1. Além disso, eles demonstram capacidade elevada de estimulação quimiotaxica, fator
importante para o desencadeamento do processo vasoclusivo (CANALLI et al., 2008).
Outro fator que contribui para o fenômeno de vasoclusão é a deficiência na produção
ou na ação do óxido nítrico (ON), aumentando o tônus vascular (TAYLOR et al., 2008).
Níveis alterados de ON estão diretamente relacionados com adesão celular, ativação
plaquetária e com os processos inflamatórios que ocasionam a vasoclusão. Esse distúrbio
vascular promove a diminuição do fluxo sanguíneo, contribuindo, concomitantemente, para a
desoxigenação dos eritrócitos e sua conseqüente falcização e obstrução do vaso (WEINER et
al., 2003). No entanto, estudos recentes indicam que a diminuição dos níveis de ON não
possuem um papel central no aumento das propriedades adesivas dos leucócitos, já a
diminuição dos níveis de GMPc pode aumentar significativamente a adesão endotelial
(CANALLI et al., 2008; ALMEIDA et al., 2008).
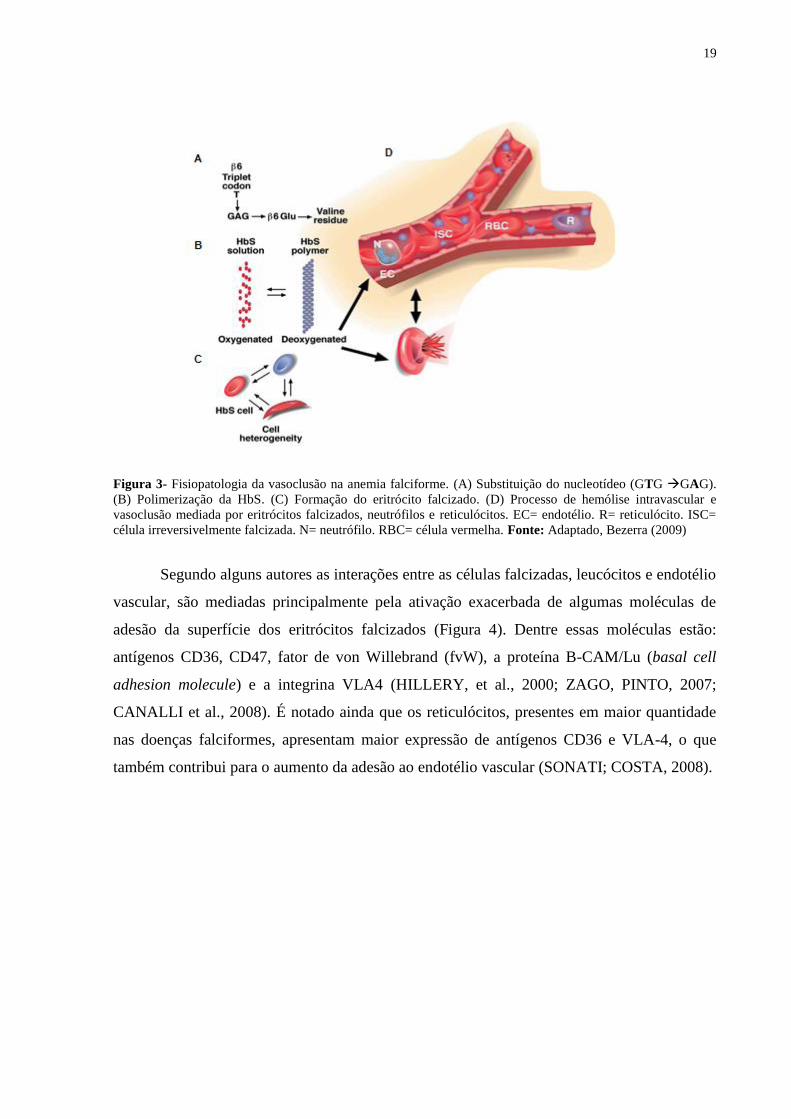
19
Figura 3- Fisiopatologia da vasoclusão na anemia falciforme. (A) Substituição do nucleotídeo (GTG GAG).
(B) Polimerização da HbS. (C) Formação do eritrócito falcizado. (D) Processo de hemólise intravascular e
vasoclusão mediada por eritrócitos falcizados, neutrófilos e reticulócitos. EC= endotélio. R= reticulócito. ISC=
célula irreversivelmente falcizada. N= neutrófilo. RBC= célula vermelha. Fonte: Adaptado, Bezerra (2009)
Segundo alguns autores as interações entre as células falcizadas, leucócitos e endotélio
vascular, são mediadas principalmente pela ativação exacerbada de algumas moléculas de
adesão da superfície dos eritrócitos falcizados (Figura 4). Dentre essas moléculas estão:
antígenos CD36, CD47, fator de von Willebrand (fvW), a proteína B-CAM/Lu (basal cell
adhesion molecule) e a integrina VLA4 (HILLERY, et al., 2000; ZAGO, PINTO, 2007;
CANALLI et al., 2008). É notado ainda que os reticulócitos, presentes em maior quantidade
nas doenças falciformes, apresentam maior expressão de antígenos CD36 e VLA-4, o que
também contribui para o aumento da adesão ao endotélio vascular (SONATI; COSTA, 2008).
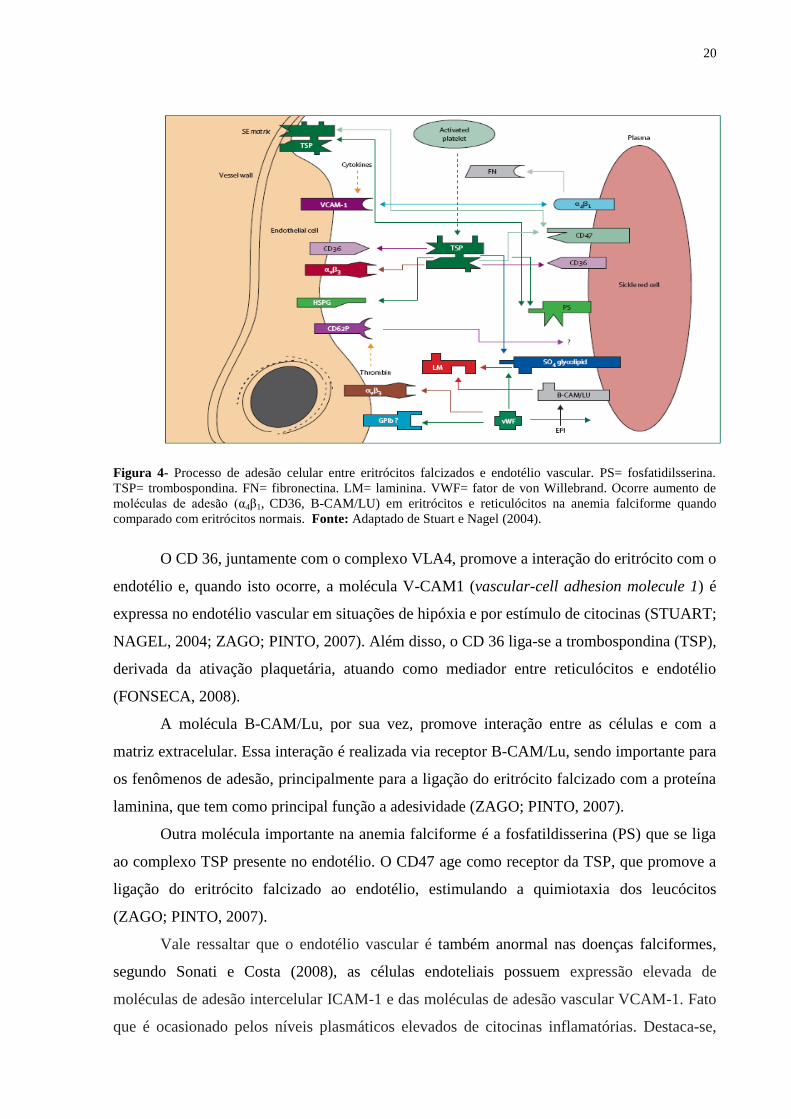
20
Figura 4- Processo de adesão celular entre eritrócitos falcizados e endotélio vascular. PS= fosfatidilsserina.
TSP= trombospondina. FN= fibronectina. LM= laminina. VWF= fator de von Willebrand. Ocorre aumento de
moléculas de adesão (α4β1, CD36, B-CAM/LU) em eritrócitos e reticulócitos na anemia falciforme quando
comparado com eritrócitos normais. Fonte: Adaptado de Stuart e Nagel (2004).
O CD 36, juntamente com o complexo VLA4, promove a interação do eritrócito com o
endotélio e, quando isto ocorre, a molécula V-CAM1 (vascular-cell adhesion molecule 1) é
expressa no endotélio vascular em situações de hipóxia e por estímulo de citocinas (STUART;
NAGEL, 2004; ZAGO; PINTO, 2007). Além disso, o CD 36 liga-se a trombospondina (TSP),
derivada da ativação plaquetária, atuando como mediador entre reticulócitos e endotélio
(FONSECA, 2008).
A molécula B-CAM/Lu, por sua vez, promove interação entre as células e com a
matriz extracelular. Essa interação é realizada via receptor B-CAM/Lu, sendo importante para
os fenômenos de adesão, principalmente para a ligação do eritrócito falcizado com a proteína
laminina, que tem como principal função a adesividade (ZAGO; PINTO, 2007).
Outra molécula importante na anemia falciforme é a fosfatildisserina (PS) que se liga
ao complexo TSP presente no endotélio. O CD47 age como receptor da TSP, que promove a
ligação do eritrócito falcizado ao endotélio, estimulando a quimiotaxia dos leucócitos
(ZAGO; PINTO, 2007).
Vale ressaltar que o endotélio vascular é também anormal nas doenças falciformes,
segundo Sonati e Costa (2008), as células endoteliais possuem expressão elevada de
moléculas de adesão intercelular ICAM-1 e das moléculas de adesão vascular VCAM-1. Fato
que é ocasionado pelos níveis plasmáticos elevados de citocinas inflamatórias. Destaca-se,

21
ainda, algumas proteínas de adesão como a P-selectina, a E-selectina, a fibronectina, a
laminina, e a integrina αVβ3, que são capazes de interagir com receptores de adesão expressos
pelos eritrócitos falcizados e pelos leucócitos, desempenhando um papel importante na
vasoclusão (SONATI;COSTA et al., 2008).
Concomitante à adesão celular e a polimerização da desoxi-HbS ocorre um aumento
da permeabilidade da membrana do eritrócito falcizado ao sódio, potássio, magnésio e cálcio,
resultando em uma alteração no gradiente de concentração normal da célula, o que causa
desidratação e aumenta a concentração de hemoglobina corpuscular média (CHCM)
(BOOKCHIN; LEW 1996; RINEHART, et al. 2010).
2.1 MANIFESTAÇÕES CLÍNICAS
Um evento característico da anemia falciforme é a anemia hemolítica resultante da
polimerização da hemoglobina, alteração de membrana e aumento de concentração de
hemoglobina corpuscular média (CHCM). Tais fatores propiciam o seqüestro e destruição
precoce das células falcizadas por meio do sistema monocítico fagocitário (ANVISA, 2002).
Esse sistema reconhece as imunoglobulinas, que estão presentes em níveis elevados na
membrana de células falcizadas, ocasionando a redução da vida média dos eritrócitos,
diminuição do hematócrito e hemoglobina além do aumento do número de reticulócitos
circulantes (ANVISA, 2002).
As etapas que fazem parte do processo de oclusão da microcirculação, com isquemia
de múltiplos órgãos, podem levar à cronicidade das lesões e ocorrência de significativas
manifestações clínicas típicas de pacientes com anemia falciforme. Dentre as manifestações
estão: crises dolorosas, aplásticas e de seqüestro esplênico, complicações cardíacas,
neurológicas, renais, pulmonares, oculares, ortopédicas e manifestações cutâneas (SCHNOG,
et al. 2004; ZAGO, 2004).

22
2.1.1 Crises vasoclusivas
A crise vasoclusiva, também denominada de crise dolorosa, é o evento mais comum
na anemia falciforme. A intensidade e o padrão da dor são bastante variados, indo desde
episódios agudos até episódios crônicos, muitas vezes, demandando atendimento hospitalar
emergencial (ANVISA, 2002).
De acordo com a Agência Nacional de Vigilância Sanitária (ANVISA), a crise
vasoclusiva consiste de “dor em extremidades, região lombar, abdome ou tórax, usualmente
associada a febre e urina escura ou vermelha”. As crises de dor frequentemente resultam de
necrose avascular da medula óssea, estando associadas ao aumento da pressão intramedular,
que é a causa mais freqüente de dor. Essa situação é evidenciada quando o paciente, após
análise laboratorial, apresenta necrose ou infiltrado neutrofílico em punção da região dolorosa
(ANVISA, 2002; STUART; NAGEL, 2004).
Stuart e Nagel (2004), relatam que indivíduos adultos com crises recorrentes de
vasoclusão, apresentando elevado grau de dor, tendem a possuir menor expectativa de vida,
comparado a indivíduos com menor nível de dor, indicando assim, que o aumento do estresse
oxidativo e inflamatório aceleram o processo de disfunção de determinados órgãos.
2.1.2 Crises aplásticas
Uma das manifestações agudas da anemia falciforme é a crise aplástica, que é oriunda
de uma insuficiência temporária da eritropoese, com diminuição da concentração de
hemoglobina e diminuição do número de reticulócitos, que ocorre geralmente nos primeiros
anos de vida (ZAGO, 2004). Porém, nos indivíduos com crise esta manifestação temporária é
suficiente para agravar o quadro de anemia, apesar da hiperplasia eritróide existente em
decorrência do mecanismo compensatório medular (STUART; NAGEL, 2004).

23
2.1.3 Crise de sequestro esplênico
A crise de sequestro esplênico representa uma das principais causas de morte na
primeira década de vida em pacientes com anemia falciforme (BALLAS et al., 2010). Ocorre
com maior freqüência após os seis meses de vida e tende a diminuir após os dois anos de
idade (ZAGO, 2004). No entanto, pode acontecer também em portadores de
hemoglobinopatia SC ou S/β-talassemia com incidência do primeiro evento mais tardiamente
do que na anemia falciforme (BALLAS et al., 2010).
Essa complicação leva a diminuição da concentração basal de hemoglobina que pode
chegar a 2g/dL, com reticulocitose (contagem de reticulócitos maior que 25% do valor
normal), trombocitopenia moderada ou severa, aumento rápido do baço, ocasionando choque
hipovolêmico, e hiperplasia da medula óssea. (DI NUZZO; FONSECA, 2004; BALLAS et
al., 2010).
A asplenia pode ser ocasionada em virtude de uma esplenomegalia decorrente da
congestão na polpa esplênica pelo bloqueio causado por grandes quantidades de eritrócitos
falcizados nos cordões esplênicos e sinusóides, e isso pode levar a ocorrência de infartos e
conseqüente atrofia e fibrose do órgão. Assim, devido às constantes agressões esplênicas tanto
a capacidade fagocítica mediada por proteínas que revestem microorganismos (opsoninas)
quanto a produção de anticorpos se tornam comprometidas, gerando a asplenia funcional (DI
NUZZO; FONSECA, 2004).
2.1.4 Crises hemolíticas
As crises hemolíticas, também definidas como crises hiper-hemolíticas, apresentam
redução acentuada de hemoglobina e elevada taxa de hemólise. Dessa forma, seu diagnóstico
é feito somente pela exclusão de outras causas de destruição de eritrócitos e pelo agravamento
da anemia. Frequentemente são associadas ao aumento de cerca de 25% na contagem de
reticulócitos e/ou presença de eritroblastos em sangue periférico (TAYLOR et al., 2008;
BALLAS et al., 2010).

24
Sabe-se, entretanto, que a ocorrência de hemólise na anemia falciforme pode variar de
indivíduo para indivíduo segundo a expressão de determinados moduladores como a HbF, a
interação com a α-talassemia e ainda, pela influência de diferentes genes importantes na
fisiopatologia da falcização celular (TAYLOR et al., 2008; STEINBERG, 2005).
2.2 DIAGNÓSTICO
As doenças falciformes abrangem um grupo de genótipos distintos, compreendendo
desde a homozigose da HbS, que define a anemia falciforme, até as co-heranças com outras
hemoglobinas variantes ou com a β-talassemia (β0 e β
+) (STUART; NAGEL, 2004).
A anemia falciforme e a associação da HbS com a β0-talassemia constituem as formas
mais graves de manifestações clínicas, sendo seguidas pela hemoglobinopatia SC e a Sβ+-
talassemia (POWARS et al., 2002).
Em virtude da ampla variabilidade de doenças falciformes é necessário um diagnóstico
sensível e específico, para correta detecção das variantes hemoglobínicas, de acordo com a
peculiaridade de cada genótipo. Normalmente o diagnóstico inclui hemograma, técnicas de
eletroforese de hemoglobina e quantificação de hemoglobina fetal (HbF) e HbA2. Além disso,
pode-se realizar testes mais sensíveis como, a reação em cadeia da polimerase (PCR) e
cromatografia líquida de alta performance (HPLC) (ANVISA, 2002).
Nos casos de associação de HbS com a HbC o diagnóstico deve ser realizado mediante
associação de técnicas de eletroforese alcalina e ácida, que possibilitam a visualização de
bandas em diferentes posições, segundo suas respectivas cargas elétricas. Nos casos em que o
padrão eletroforético da anemia falciforme é semelhante, HbS/β-talassemia, HbS/ βδ-
talassemia e HbS/PHHF (Persistência Hereditária de Hemoglobina Fetal), é indispensável o
emprego de técnicas mais sensíveis e específicas como a reação em cadeia da polimerase
(PCR), podendo ser útil a análise quantitativa de HbA2 e de HbF. Pois normalmente a HbA2
encontra-se aumentada (acima de 3,5%) quando existe relação com β°-talassemia, e reduzida
em casos de HbS/ βδ talassemia (ANVISA, 2002; SCHNOG, et al., 2004).
No diagnóstico neonatal é de suma importância levar em consideração os tipos de
cadeias globínicas que estão sendo expressas neste período, pois a síntese predominante é de
hemoglobina Fetal (HbF). Dessa forma, a porcentagem de hemoglobinas presentes após o

25
sexto mês de vida compreende 95% de HbA, 2 a 3% de HbA2 e 0 a 2% de HbF (SILVA,
2001). Entretanto, se houver diagnóstico positivo para doenças falciformes em neonatos, eles
precisarão de nova avaliação laboratorial após o sexto mês de vida (LEES; DAVIES;
DEZATEUX, 2000).
Devido à importância do diagnóstico precoce das hemogloblobinopatias em neonatos
no Brasil, o Ministério da Saúde as incluiu no Programa Nacional de Triagem Neonatal
(PNTN), tornando obrigatória a realização de testes para detecção de hemoglobinopatias no
teste do pezinho (RAMALHO, 2003).
2.3 TRATAMENTO
O tratamento precoce é essencial para o aumento da sobrevida e do bem estar de
indivíduos portadores de hemoglobinas anormais. Todavia, os tratamentos não são curativos,
constituindo medidas que visam diminuir os efeitos da anemia crônica, crises de falcização e
susceptibilidade às infecções, que podem ser ocasionadas pela alteração imunológica e
asplenia funcional (DI NUZZO; FONSECA, 2004; ANVISA, 2002). Segundo a ANVISA
(2002), “deve ser realizada boa nutrição, profilaxia, diagnóstico e terapêutica precoce de
infecções”. Recomenda-se também evitar condições climáticas adversas e manter boa
hidratação, sendo necessário, ainda, o acompanhamento adequado em serviços especializados
e educação da família para melhor qualidade de vida do paciente (ANVISA, 2002).
Um dos medicamentos utilizados para o tratamento de doenças falciformes é a
hidroxiuréia (HU). Os efeitos benéficos para o seu uso devem-se à sua habilidade de reativar
genes γ globina e aumentar os níveis de HbF, o que reduz a polimerização da HbS nos
eritrócitos, proporcionando a diminuição dos eventos vasoclusivos e hemolíticos,
minimizando assim a freqüência das crises dolorosas (CANALLI et al., 2008; TAYLOR et
al., 2008).
Além da capacidade de aumentar a produção de HbF, a hidroxiuréia (HU) possui ação
antinflamatória e leucopênica, influenciando também, na hidratação do eritrócito, na adesão
endotelial e na produção de oxido nítrico (SONATI; COSTA, 2008).

26
3. JUSTIFICATIVA
A anemia falciforme é a hemoglobinopatia mais prevalente no Brasil e no mundo,
sendo observada principalmente na população afro descendente. Segundo o Programa
Nacional de Triagem Neonatal, estima-se que no Brasil, dos nascidos vivos, cerca de 3.500
possuam a doença falciforme e 200.000 possuam o traço falciforme (ARAUJO, 2007).
Devido à importância dessa doença como um problema de saúde pública no Brasil, ela
assume um lugar de destaque nos cuidados médicos, genéticos e psicossociais.
Portanto, a proposta de avaliação da prevalência de anemia falciforme na população de
Brasília-DF, contribuirá não somente para o melhor conhecimento do seu perfil
epidemiológico nessa região, mas também para que outras pesquisas sobre hemoglobinopatias
sejam desenvolvidas, no intuito de promover melhoria na qualidade de vida do paciente e
elaboração de medidas terapêuticas mais eficazes.

27
4. OBJETIVOS
4.1 Objetivo Geral
Avaliar a prevalência de indivíduos portadores de anemia falciforme no Hospital das
Forças Armadas de Brasília – DF.
4.2 Objetivos Específicos
- Diagnosticar indivíduos com anemia falciforme.
- Identificar a prevalência de anemia falciforme na população em estudo.
- Correlacionar os dados encontrados com os dados de outros estudos.
- Analisar o perfil das variáveis demográficas.
- Verificar se existe relação entre a ampliação da triagem laboratorial e o aumento da
freqüência de indivíduos portadores de anemia, traço e/ou associação falciforme.

28
5. MATERIAIS E MÉTODOS
O estudo foi realizado no período de maio a agosto de 2010, com pacientes atendidos
no Laboratório de análises clínicas do Hospital das Forças Armadas de Brasília-DF (HFA),
após a aprovação do Comitê de Ética em pesquisa do HFA, e assinatura do termo de
consentimento livre esclarecido pelos pacientes e controles (ANEXO A).
Foram selecionados 61 pacientes a partir da análise do eritrograma, teste de falcização,
eletroforese de hemoglobina e confirmação pela eletroforese por capilaridade. Além disso, foi
analisado o perfil hemoglobínico de 64 pacientes com exames cadastrados no laboratório para
realização de eletroforese por capilaridade no equipamento Minicap (SEBIA®
), e 1057
pacientes, cadastrados para realização do exame de hemoglobina glicada no equipamento D-
10 (Bio-Rad®
), o qual inclui resultados para os demais tipos de hemoglobina. Para estas
análises não foi possível o acesso aos dados dos hemogramas desses pacientes.
Tabela 1- Quantidade de pacientes analisados no estudo segundo as técnicas utilizadas.
Técnica N° de pacientes
*Hemograma 61
**Minicap (SEBIA®) 64
***D-10 (Bio-Rad®) 1057
TOTAL 1181
*Pacientes analisados a partir do hemograma, teste de falcização, eletroforese de hemoglobina manual e
confirmação pela eletroforese por capilaridade.
**Pacientes analisados a partir dos resultados de exames cadastrados para eletroforese por capilaridade no
equipamento Minicap (SEBIA®).
***Pacientes analisados a partir dos resultados de exames cadastrados para hemoglobina glicada no equipamento
D-10 (Bio-Rad®).
5.1 CONTAGEM AUTOMATIZADA DE CÉLULAS E ANÁLISE MORFOLÓGICA POR
MICROSCOPIA
As amostras de sangue periférico foram coletadas em tubos contendo anticoagulante
EDTA, seguindo para a realização do esfregaço sanguíneo utilizando 5µL de sangue e
coloração com corante Wright.

29
Para análise do eritrograma foram utilizados os valores de referência padronizados
pelo laboratório, de acordo com a idade dos pacientes (ANEXO B). A leitura dos índices
hematimétricos foi realizada por contador eletrônico de células (Beckman-Coulter®
), sendo
selecionados pacientes que apresentavam dosagem inferior a 12g/dL de hemoglobina e 35%
de hematócrito, valor aumentado de RDW e/ou presença elevada de eritroblastos e/ou
reticulócitos e, simultaneamente, foi feita análise morfológica das hemácias por microscopia
dos esfregaços sanguíneos com o propósito de identificar achados laboratoriais sugestivos da
presença de HbS em homozigose ou heterozigose. Foram selecionadas amostras de pacientes
que apresentavam anisocromia, anisocitose, policromasia e presença de drepanócitos. Um
grupo de indivíduos sabidamente saudáveis foi utilizado como controle para todas as técnicas
usadas nesse estudo.
5.2 TESTE DE FALCIZAÇÃO
Depois da triagem pela análise do hemograma foi realizado teste de falcização para a
avaliação qualitativa da presença ou ausência de HbS nos eritrócitos.
Em um tudo de hemólise, misturou-se 50 L de sangue e 100 L de solução de
metabissulfito de sódio a 2%. Foi colocada uma gota desta mistura em uma lâmina, que foi
coberta com uma lamínula e as bordas foram vedadas, para evitar a entrada de oxigênio.
Posteriormente, a lâmina foi conservada em câmara úmida e avaliada ao microscópio após 6,
24 e 48 horas para a verificação da existência de hemácias falcizadas.
O teste de falcização foi considerado positivo quando os eritrócitos dos pacientes
evidenciavam formas em foice.
Figura 5- Representação de teste de falcização positivo em paciente com traço falciforme.
Fonte: http://www.hemoglobinopatias.com.br/d-falciforme/diagnostico.htm.

30
5.3 ELETROFORESE DE HEMOGLOBINA EM PH ALCALINO E ÁCIDO
Para comprovar a existência de hemoglobinas anômalas, a eletroforese de
hemoglobinas foi realizada de acordo com os Protocolos de Metodologias Clássicas para o
Diagnóstico de Hemoglobinopatias da UNESP (Anexo C e D) com algumas adaptações.
A separação e indentificação da HbS e HbA foi feita inicialmente pela eletroforese em
pH alcalino utilizando tampão Tris-EDTA-Borato - pH 8,5. Após a análise da migração
eletroforética em pH alcalino, as amostras foram submetidas a eletroforese em pH ácido
utilizando tampão Citrato - pH 5,9 - no intuito de distinguir as HbS, HbD, HbC e HbE.
A partir das frações hemoglobínicas resultantes nos géis de agarose, realizou-se o
reconhecimento das hemoglobinas por meio da comparação dos padrões eletroforéticos
previamente conhecidos.
5.4 ELETROFORESE DE HEMOGLOBINA POR CAPILARIDADE
A análise de eletroforese por capilaridade foi utilizada em amostras que apresentaram
alterações significativas nas avaliações do hemograma, na eletroforese alcalina e ácida, e
também para aquelas já cadastradas no sistema do laboratório. Para esta análise utilizou-se o
equipamento Minicap (SEBIA®
), que possibilita a avaliação qualitativa e quantitativa dos
diferentes tipos de hemoglobinas através dos seus respectivos pontos isoelétricos.
Esta avaliação é realizada utilizando hemácias lavadas, com soro fisiológico, e
preparação automática do hemolisado, pelo equipamento. A seguir é feita uma diluição da
amostra em uma cúpula que contém uma cubeta ânodo. Na seqüência essa amostra diluída é
injetada no capilar por meio do contato da extremidade anódica desse capilar com a amostra
diluída. Logo após cerca de 10 a 20 µL desta amostra é aspirada para o interior do capilar. A
migração ocorre em capilares de silício sob temperatura constante que é controlada por um
sistema de efeito Peltier, no qual permite o aquecimento ou resfriamento do sistema de acordo
com a direção de passagem de uma corrente elétrica na junção de dois condutores metálicos
diferentes. Em seguida as hemoglobinas são separadas através de alta-voltagem e detectadas,
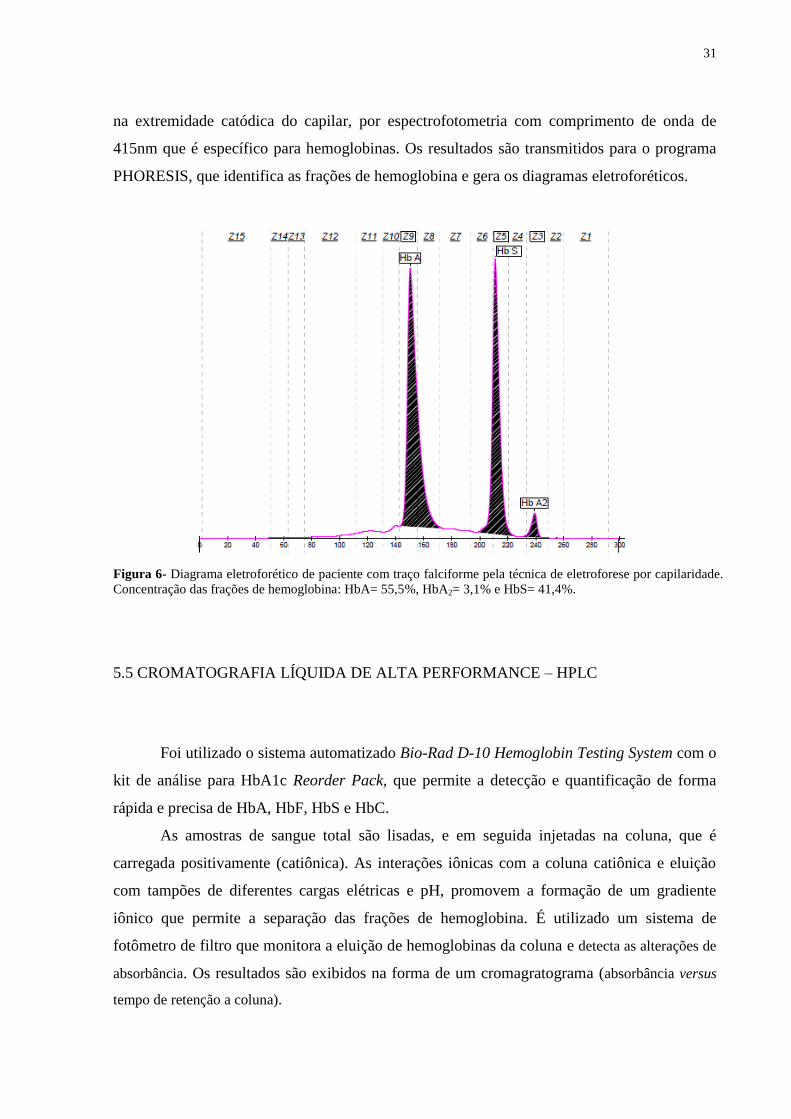
31
na extremidade catódica do capilar, por espectrofotometria com comprimento de onda de
415nm que é específico para hemoglobinas. Os resultados são transmitidos para o programa
PHORESIS, que identifica as frações de hemoglobina e gera os diagramas eletroforéticos.
Figura 6- Diagrama eletroforético de paciente com traço falciforme pela técnica de eletroforese por capilaridade.
Concentração das frações de hemoglobina: HbA= 55,5%, HbA2= 3,1% e HbS= 41,4%.
5.5 CROMATOGRAFIA LÍQUIDA DE ALTA PERFORMANCE – HPLC
Foi utilizado o sistema automatizado Bio-Rad D-10 Hemoglobin Testing System com o
kit de análise para HbA1c Reorder Pack, que permite a detecção e quantificação de forma
rápida e precisa de HbA, HbF, HbS e HbC.
As amostras de sangue total são lisadas, e em seguida injetadas na coluna, que é
carregada positivamente (catiônica). As interações iônicas com a coluna catiônica e eluição
com tampões de diferentes cargas elétricas e pH, promovem a formação de um gradiente
iônico que permite a separação das frações de hemoglobina. É utilizado um sistema de
fotômetro de filtro que monitora a eluição de hemoglobinas da coluna e detecta as alterações de
absorbância. Os resultados são exibidos na forma de um cromagratograma (absorbância versus
tempo de retenção a coluna).

32
Neste cromatograma está definida a porcentagem de hemoglobina e o tempo de retenção
característico de cada molécula de hemoglobina à coluna. O tempo de retenção é determinado
a partir do tempo em que a amostra se liga a coluna até o momento em que está ligação é
rompida (Manual de Instrução BIO-RAD, 2003). O tempo de retenção estimado pra Hb S é de
4.57 ± 0.028 minutos (ONDEI et al., 2007).
5.6 ANÁLISE DOS DADOS
Os dados obtidos foram analisados com o auxílio programa Excel (Office 2007), que
foi utilizado para o cálculo da prevalência e da média para os índices hematimétricos e de
concentração de hemoglobinas, de acordo com o sexo, idade e cor da pele.

33
6. RESULTADOS
Foram avaliados 1181 pacientes no período de maio a agosto de 2010. Dentre estes, 61
fazem parte da triagem realizada a partir do eritrograma, teste de falcização, eletroforese
manual e confirmação pela eletroforese por capilaridade, 64 a partir dos resultados de exames
já cadastrados no equipamento Minicap (SEBIA®) e 1057 a partir dos resultados do D-10
(Bio-Rad®).
Os casos positivos para HbS foram divididos em dois grupos de pacientes: os
portadores de traço falciforme (HbAS) e os portadores de HbS em associação com outras
variantes estruturais de hemoglobina (HbSC, SE, SD). Das amostras avaliadas, nenhuma
apresentou homozigose S (HbSS).
Trinta e seis pacientes apresentaram traço falciforme (HbAS) e três apresentaram
hemoglobinopatia SC (HbSC), com prevalência de 3,05% para traço falciforme e 0,25% para
hemoglobinopatia SC (Tabela 2).
Foi possível observar que a média de idade entre os pacientes que apresentaram
hemoglobina HbS foi de 44,36 anos anos (com média de 50,21 anos para o sexo feminino e
35 anos para o sexo masculino). Já a distribuição quanto ao sexo mostrou maior incidência de
HbS em pessoas do sexo feminino, representando 61,54% (24), enquanto para o sexo
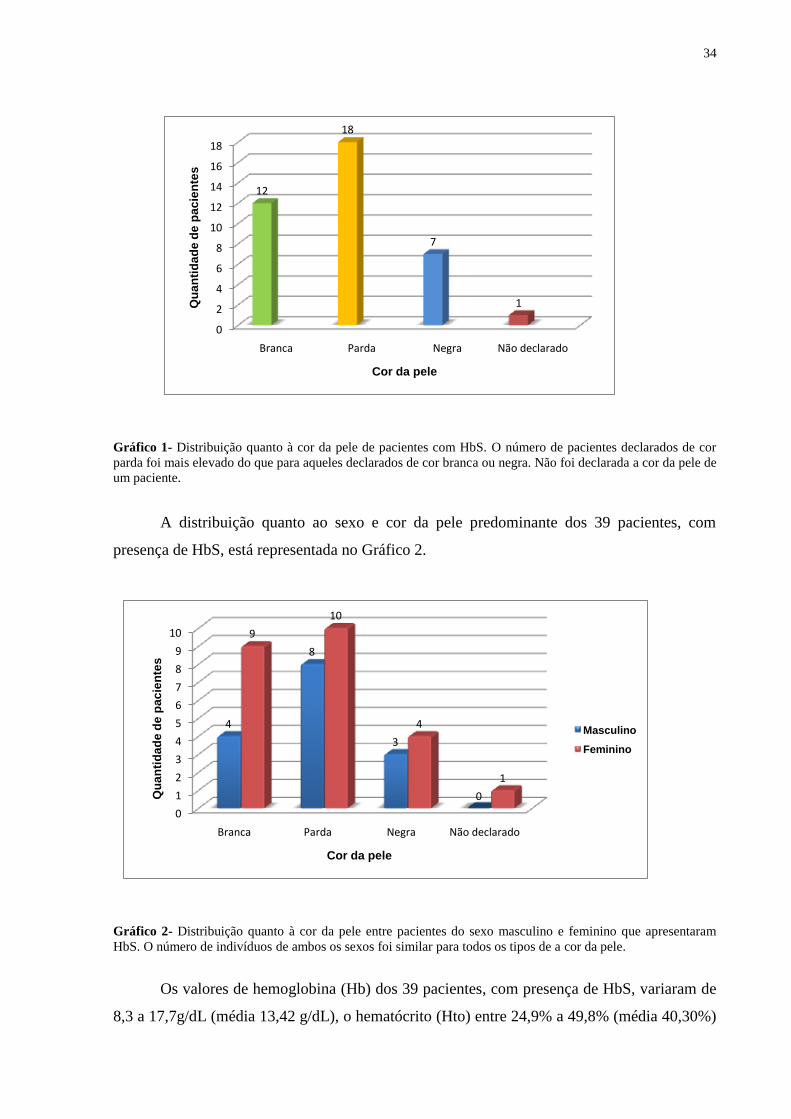
masculino foi de 38,46% (15) (Tabela 2). Houve o predomínio de alterações de frações
hemoglobínicas em indivíduos declarados de cor parda (Gráfico 1). A cor da pele de apenas
um paciente não foi informada.
Tabela 2- Número absoluto de casos de portadores de traço falciforme e hemoglobinopatia SC avaliados no
laboratório de análises clínicas do Hospital das Forças Armadas, de maio a agosto de 2010.
Tipo de Hemoglobina n Prevalência
% HbAS 36 3,05 HbSC 3 0,25
Total com HbS 39 3,30 *Outros 1145 96,95
HbS segundo o sexo n Frequência
% Feminino com HbS 24/39 61,54
Masculino com HbS 15/39 38,46
*Presença de 1110 pacientes com frações de hemoglobinas normais (HbAA), 18 pacientes com traço para
hemoglobina C (HbC), 16 pacientes com aumento de hemoglobina A2 e 01 paciente com Persistência Hereditária
de Hemoglobina Fetal (PHHF)
34
0
2
4
6
8
10
12
14
16
18
Branca Parda Negra Não declarado
12
18
7
1Qu
an
tid
ad
e d
e p
ac
ien
tes
Cor da pele
Gráfico 1- Distribuição quanto à cor da pele de pacientes com HbS. O número de pacientes declarados de cor
parda foi mais elevado do que para aqueles declarados de cor branca ou negra. Não foi declarada a cor da pele de
um paciente.
A distribuição quanto ao sexo e cor da pele predominante dos 39 pacientes, com
presença de HbS, está representada no Gráfico 2.
0
1
2
3
4
5
6
7
8
9
10
Branca Parda Negra Não declarado
4
8
3
0
9
10
4
1
Qu
an
tid
ad
e d
e p
ac
ien
tes
Cor da pele
Masculino
Feminino
Gráfico 2- Distribuição quanto à cor da pele entre pacientes do sexo masculino e feminino que apresentaram
HbS. O número de indivíduos de ambos os sexos foi similar para todos os tipos de a cor da pele.
Os valores de hemoglobina (Hb) dos 39 pacientes, com presença de HbS, variaram de
8,3 a 17,7g/dL (média 13,42 g/dL), o hematócrito (Hto) entre 24,9% a 49,8% (média 40,30%)
35
e o VCM de 67 a 102fL (média 87,47fL) para traço falciforme, enquanto para
hemoglobinopatia SC os valores de Hb, Hto e VCM permaneceram entre 10,3 a 11,4 g/dL
(média 10,87 g/dL), 30,4% a 34,8 (média 32,77%) e 87 a 93fL (média 88,33fL),
respectivamente (Tabela 3).
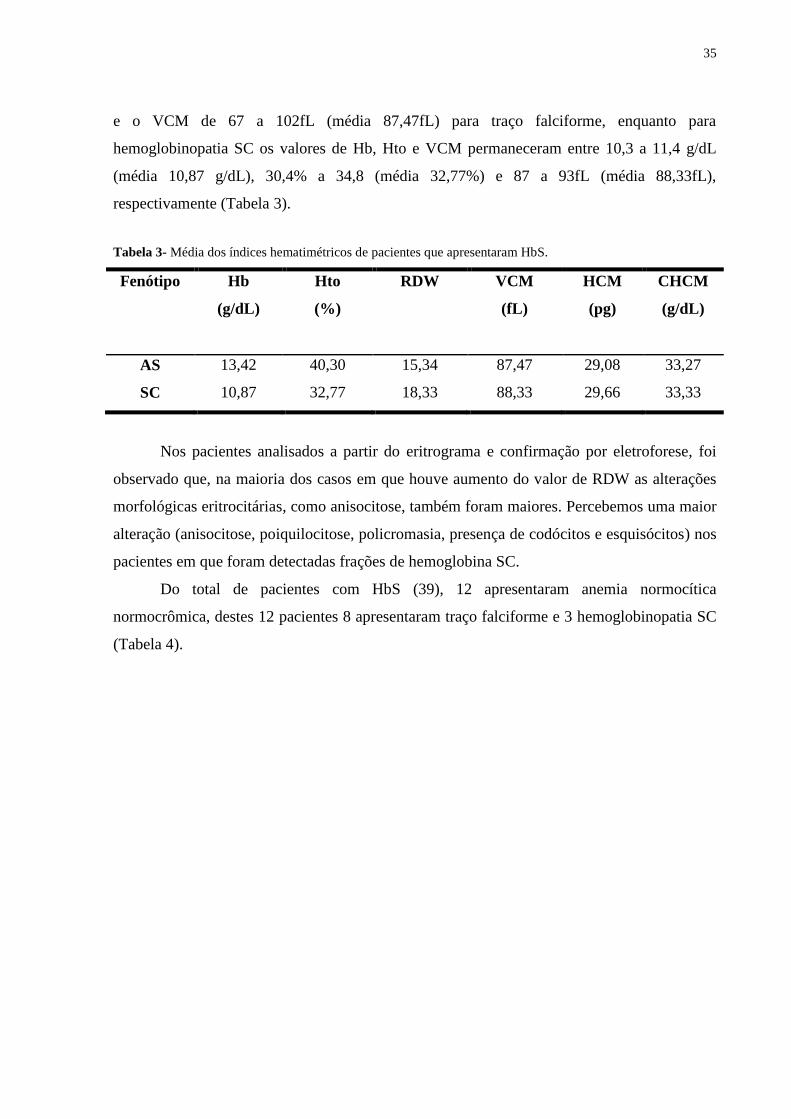
Tabela 3- Média dos índices hematimétricos de pacientes que apresentaram HbS.
Fenótipo
Hb
(g/dL)
Hto
(%)
RDW VCM
(fL)
HCM
(pg)
CHCM
(g/dL)
AS 13,42 40,30 15,34 87,47 29,08 33,27
SC 10,87 32,77 18,33 88,33 29,66 33,33
Nos pacientes analisados a partir do eritrograma e confirmação por eletroforese, foi
observado que, na maioria dos casos em que houve aumento do valor de RDW as alterações
morfológicas eritrocitárias, como anisocitose, também foram maiores. Percebemos uma maior
alteração (anisocitose, poiquilocitose, policromasia, presença de codócitos e esquisócitos) nos
pacientes em que foram detectadas frações de hemoglobina SC.
Do total de pacientes com HbS (39), 12 apresentaram anemia normocítica
normocrômica, destes 12 pacientes 8 apresentaram traço falciforme e 3 hemoglobinopatia SC
(Tabela 4).
36
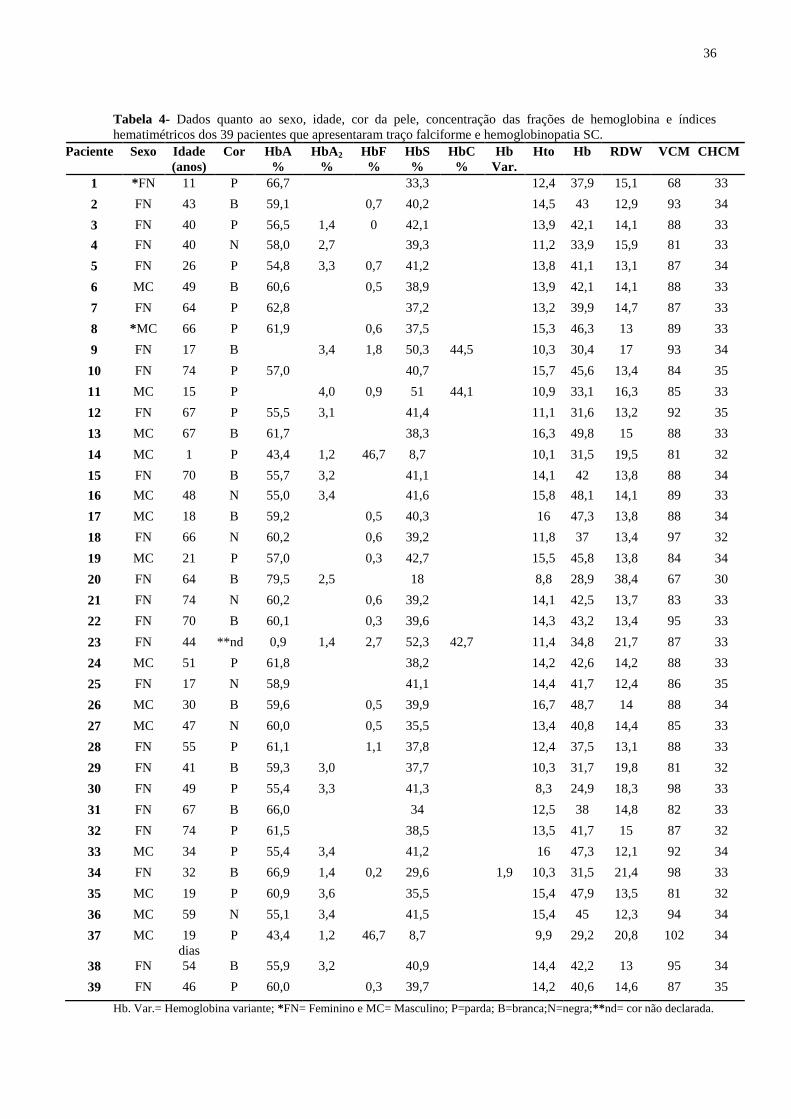
Tabela 4- Dados quanto ao sexo, idade, cor da pele, concentração das frações de hemoglobina e índices
hematimétricos dos 39 pacientes que apresentaram traço falciforme e hemoglobinopatia SC.
Paciente Sexo Idade
(anos)
Cor HbA
%
HbA2
%
HbF
%
HbS
%
HbC
%
Hb
Var.
Hto Hb RDW VCM CHCM
1 *FN 11 P 66,7 33,3 12,4 37,9 15,1 68 33
2 FN 43 B 59,1 0,7 40,2 14,5 43 12,9 93 34
3 FN 40 P 56,5 1,4 0 42,1 13,9 42,1 14,1 88 33
4 FN 40 N 58,0 2,7 39,3 11,2 33,9 15,9 81 33
5 FN 26 P 54,8 3,3 0,7 41,2 13,8 41,1 13,1 87 34
6 MC 49 B 60,6 0,5 38,9 13,9 42,1 14,1 88 33
7 FN 64 P 62,8 37,2 13,2 39,9 14,7 87 33
8 *MC 66 P 61,9 0,6 37,5 15,3 46,3 13 89 33
9 FN 17 B 3,4 1,8 50,3 44,5 10,3 30,4 17 93 34
10 FN 74 P 57,0 40,7 15,7 45,6 13,4 84 35
11 MC 15 P 4,0 0,9 51 44,1 10,9 33,1 16,3 85 33
12 FN 67 P 55,5 3,1 41,4 11,1 31,6 13,2 92 35
13 MC 67 B 61,7 38,3 16,3 49,8 15 88 33
14 MC 1 P 43,4 1,2 46,7 8,7 10,1 31,5 19,5 81 32
15 FN 70 B 55,7 3,2 41,1 14,1 42 13,8 88 34
16 MC 48 N 55,0 3,4 41,6 15,8 48,1 14,1 89 33
17 MC 18 B 59,2 0,5 40,3 16 47,3 13,8 88 34
18 FN 66 N 60,2 0,6 39,2 11,8 37 13,4 97 32
19 MC 21 P 57,0 0,3 42,7 15,5 45,8 13,8 84 34
20 FN 64 B 79,5 2,5 18 8,8 28,9 38,4 67 30
21 FN 74 N 60,2 0,6 39,2 14,1 42,5 13,7 83 33
22 FN 70 B 60,1 0,3 39,6 14,3 43,2 13,4 95 33
23 FN 44 **nd 0,9 1,4 2,7 52,3 42,7 11,4 34,8 21,7 87 33
24 MC 51 P 61,8 38,2 14,2 42,6 14,2 88 33
25 FN 17 N 58,9 41,1 14,4 41,7 12,4 86 35
26 MC 30 B 59,6 0,5 39,9 16,7 48,7 14 88 34
27 MC 47 N 60,0 0,5 35,5 13,4 40,8 14,4 85 33
28 FN 55 P 61,1 1,1 37,8 12,4 37,5 13,1 88 33
29 FN 41 B 59,3 3,0 37,7 10,3 31,7 19,8 81 32
30 FN 49 P 55,4 3,3 41,3 8,3 24,9 18,3 98 33
31 FN 67 B 66,0 34 12,5 38 14,8 82 33
32 FN 74 P 61,5 38,5 13,5 41,7 15 87 32
33 MC 34 P 55,4 3,4 41,2 16 47,3 12,1 92 34
34 FN 32 B 66,9 1,4 0,2 29,6 1,9 10,3 31,5 21,4 98 33
35 MC 19 P 60,9 3,6 35,5 15,4 47,9 13,5 81 32
36 MC 59 N 55,1 3,4 41,5 15,4 45 12,3 94 34
37 MC 19
dias
P 43,4 1,2 46,7 8,7 9,9 29,2 20,8 102 34
38 FN 54 B 55,9 3,2 40,9 14,4 42,2 13 95 34
39 FN 46 P 60,0 0,3 39,7 14,2 40,6 14,6 87 35
Hb. Var.= Hemoglobina variante; *FN= Feminino e MC= Masculino; P=parda; B=branca;N=negra;**nd= cor não declarada.
37
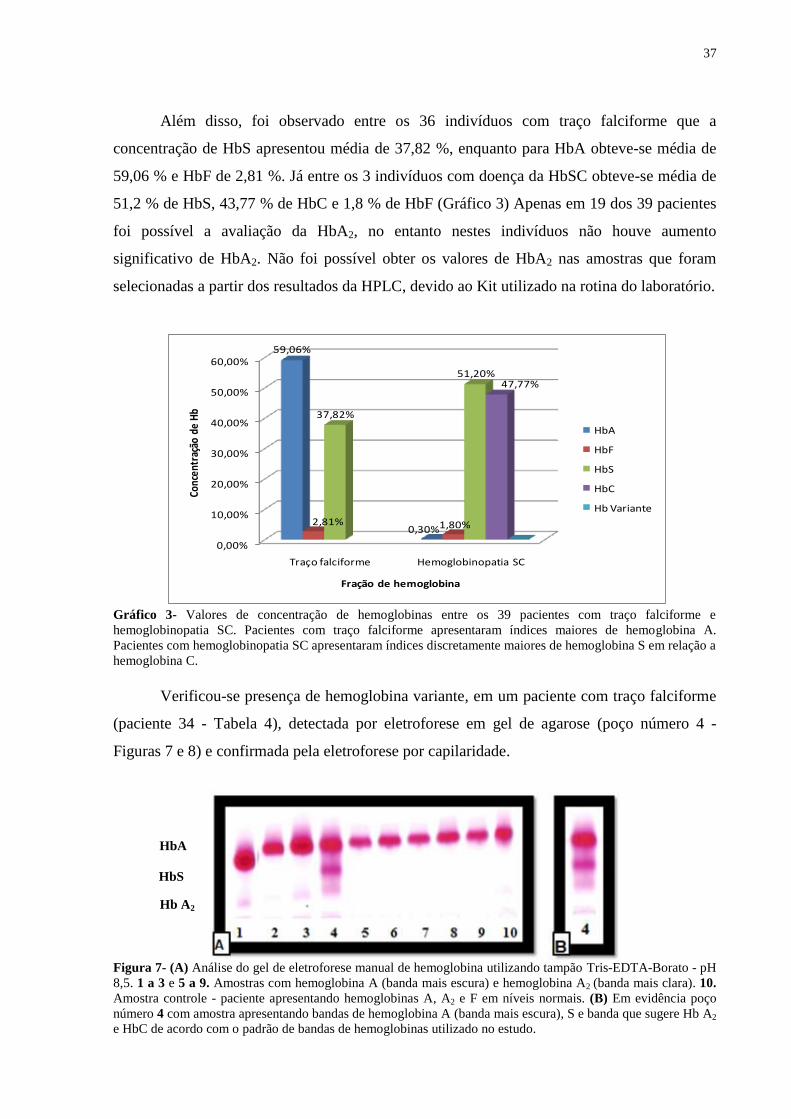
Além disso, foi observado entre os 36 indivíduos com traço falciforme que a
concentração de HbS apresentou média de 37,82 %, enquanto para HbA obteve-se média de
59,06 % e HbF de 2,81 %. Já entre os 3 indivíduos com doença da HbSC obteve-se média de
51,2 % de HbS, 43,77 % de HbC e 1,8 % de HbF (Gráfico 3) Apenas em 19 dos 39 pacientes
foi possível a avaliação da HbA2, no entanto nestes indivíduos não houve aumento
significativo de HbA2. Não foi possível obter os valores de HbA2 nas amostras que foram
selecionadas a partir dos resultados da HPLC, devido ao Kit utilizado na rotina do laboratório.
Gráfico 3- Valores de concentração de hemoglobinas entre os 39 pacientes com traço falciforme e
hemoglobinopatia SC. Pacientes com traço falciforme apresentaram índices maiores de hemoglobina A.
Pacientes com hemoglobinopatia SC apresentaram índices discretamente maiores de hemoglobina S em relação a
hemoglobina C.
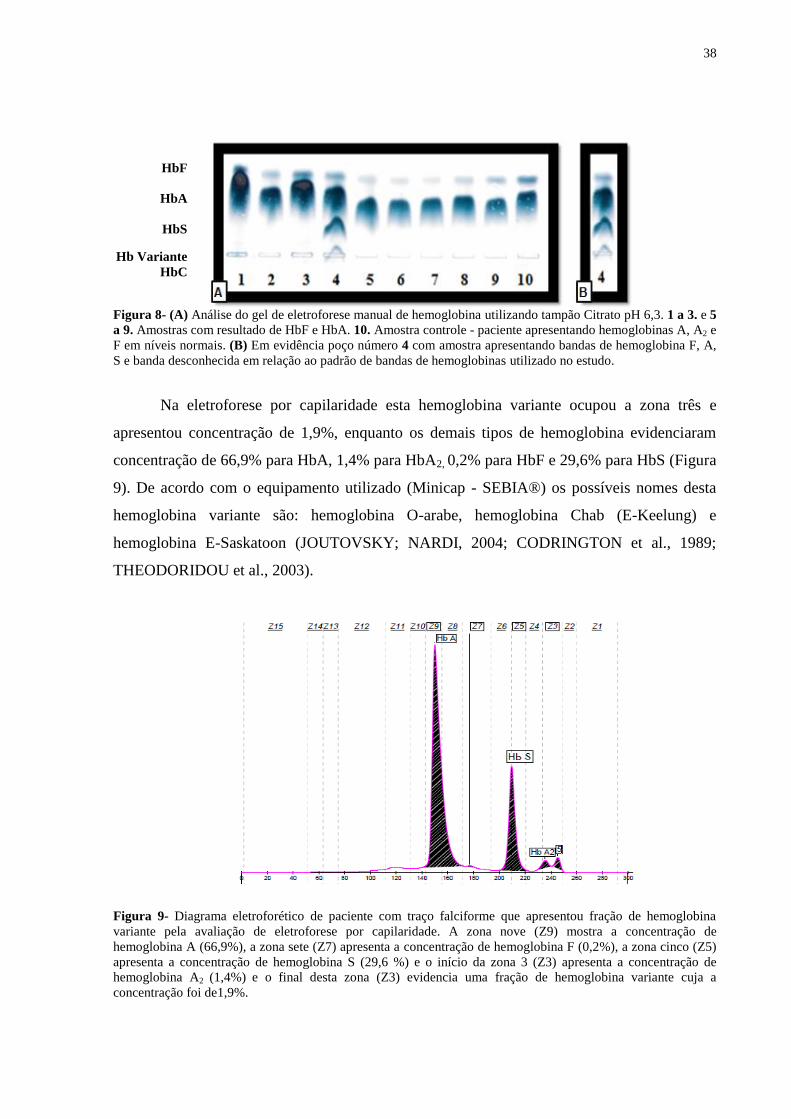
Verificou-se presença de hemoglobina variante, em um paciente com traço falciforme
(paciente 34 - Tabela 4), detectada por eletroforese em gel de agarose (poço número 4 -
Figuras 7 e 8) e confirmada pela eletroforese por capilaridade.
Figura 7- (A) Análise do gel de eletroforese manual de hemoglobina utilizando tampão Tris-EDTA-Borato - pH
8,5. 1 a 3 e 5 a 9. Amostras com hemoglobina A (banda mais escura) e hemoglobina A2 (banda mais clara). 10.
Amostra controle - paciente apresentando hemoglobinas A, A2 e F em níveis normais. (B) Em evidência poço
número 4 com amostra apresentando bandas de hemoglobina A (banda mais escura), S e banda que sugere Hb A2
e HbC de acordo com o padrão de bandas de hemoglobinas utilizado no estudo.
HbA
HbS
Hb A2
0,00%
10,00%
20,00%
30,00%
40,00%
50,00%
60,00%
Traço falciforme Hemoglobinopatia SC
59,06%
0,30%2,81% 1,80%
37,82%
51,20%47,77%
Conc
entr
ação
de
Hb
Fração de hemoglobina
HbA
HbF
HbS
HbC
Hb Variante
38
Figura 8- (A) Análise do gel de eletroforese manual de hemoglobina utilizando tampão Citrato pH 6,3. 1 a 3. e 5
a 9. Amostras com resultado de HbF e HbA. 10. Amostra controle - paciente apresentando hemoglobinas A, A2 e
F em níveis normais. (B) Em evidência poço número 4 com amostra apresentando bandas de hemoglobina F, A,
S e banda desconhecida em relação ao padrão de bandas de hemoglobinas utilizado no estudo.
Na eletroforese por capilaridade esta hemoglobina variante ocupou a zona três e
apresentou concentração de 1,9%, enquanto os demais tipos de hemoglobina evidenciaram
concentração de 66,9% para HbA, 1,4% para HbA2, 0,2% para HbF e 29,6% para HbS (Figura
9). De acordo com o equipamento utilizado (Minicap - SEBIA®) os possíveis nomes desta
hemoglobina variante são: hemoglobina O-arabe, hemoglobina Chab (E-Keelung) e
hemoglobina E-Saskatoon (JOUTOVSKY; NARDI, 2004; CODRINGTON et al., 1989;
THEODORIDOU et al., 2003).
Figura 9- Diagrama eletroforético de paciente com traço falciforme que apresentou fração de hemoglobina
variante pela avaliação de eletroforese por capilaridade. A zona nove (Z9) mostra a concentração de
hemoglobina A (66,9%), a zona sete (Z7) apresenta a concentração de hemoglobina F (0,2%), a zona cinco (Z5)
apresenta a concentração de hemoglobina S (29,6 %) e o início da zona 3 (Z3) apresenta a concentração de
hemoglobina A2 (1,4%) e o final desta zona (Z3) evidencia uma fração de hemoglobina variante cuja a
concentração foi de1,9%.
HbF
HbA
HbS
Hb Variante
HbC

39
7. DISCUSSÃO
O estudo realizado buscou avaliar a prevalência de indivíduos portadores da HbS na
população atendida pelo Hospital das Forças Armadas de Brasília, com o intuito de verificar
se existe relação entre a freqüência de indivíduos portadores de anemia, traço e/ou associação
falciforme com a ampliação da triagem laboratorial para hemoglobinas anormais.
Os resultados, indicaram prevalência de 3,30% de indivíduos portadores da HbS na
população estudada, sendo que dentre estes observou-se prevalência de 3,05% de traço
falciforme. A prevalência observada é compatível com a relatada em estudos semelhantes
realizados na população do Distrito Federal, os quais evidenciaram prevalência de 2,06% de
traço falciforme em doadores de sangue e de 3,23% em recém-nascidos (VERAS et al., 1998;
DINIZ et al., 2009).
Acredita-se que a alta prevalência de portadores de traço falciforme no Distrito
Federal é decorrente de sua posição geográfica (DINIZ et al., 2009), pois está situado,
relativamente, próximo (em um raio de 1300 km) a estados que registram as maiores taxas de
traço falciforme, de acordo com outros estudos, e que demandam grande contingente
migratório. Entre estes estados incluem-se Minas Gerais, Rio de Janeiro e Bahia, o qual
possui elevados índices de indivíduos com anemia falciforme, cerca de 10% de sua
população, principalmente na cidade de Salvador.
Podemos dizer também que existe a possibilidade de que estes altos índices de traço
falciforme encontrados no Distrito Federal sejam resultantes do grande fluxo migratório de
pessoas da região Nordeste durante o período de construção de Brasília (DINIZ et al., 2009).
Assim, de acordo com Diniz et al. (2009), o Distrito Federal ocupa a quarta posição entre as
unidades federativas que apresentam os maiores índices de traço falciforme.
A análise das variáveis demográficas, como sexo e idade, indicou que existe maior
freqüência de casos de traço falciforme entre pessoas do sexo feminino, cuja média de idade
foi de 50,21 anos. O resultado aponta que a maior parte da população feminina estudada não
se encontra em idade reprodutiva, diminuindo assim, a chance de surgirem novos portadores
do traço falcêmico. Em caso de união de mulher portadora de traço falciforme com outro
portador heterozigoto AS ou portador que possua outras variantes estruturais da hemoglobina,
os descendentes poderão apresentar traço ou anemia falciforme, ou associação da HbS com
uma hemoglobina variante, como no caso da hemoglobinopatia SC.

40
Embora a presença de HbS não apresente relação com o sexo, observou-se no estudo
que a prevalência de pacientes com HbS do sexo feminino foi superior a encontrada em
pacientes do sexo masculino. Os resultados corroboram os obtidos por Loureiro e Rozenfeld
(2005) e contradizem os resultados do trabalho realizado em recém-nascidos de Fortaleza, no
qual foram analisados 389 pacientes e foi relatada maior prevalência de HbS em pacientes do
sexo masculino, compreendendo 6,3% dos pacientes avaliados (PINHEIRO et al., 2006).
Entretanto cabe notar que o presente estudo contou com maior número de pessoas do sexo
feminino, o que pode ter influenciado a maior prevalência em indivíduos do sexo feminino.
De acordo com dados do Censo do Instituto Brasileiro de Geografia e Estatística
(IBGE) do ano de 2000, a distribuição da população residente na região Centro-Oeste segundo
a cor da pele mostra que 46,2% das pessoas se declararam brancas, 49,42% pardas e 3,53%
negras. Já a comparação entre as variáveis cor e sexo mostrou que entre pessoas do sexo
masculino, 44,98% são de cor branca, 50,35% de cor parda e 3,83% de cor negra e para
pessoas do sexo feminino, observou-se as seguintes porcentagens 47,41%; 48,50% e 3,23%,
respectivamente (IBGE, 2000). Os resultados obtidos permitem notar que a presença da HbS
na população estudada obedeceu às características relativas à cor da pele para esta região
segundo o IBGE. Concluímos então, que a triagem de hemoglobinopatias não deve se basear
apenas em relação à etnia do indivíduo, no que se refere à região Centro-Oeste. Pois dentre os
39 pacientes que apresentaram traço falciforme ou hemoglobinopatia SC, 13 se declararam
com cor de pele branca.
Quanto aos achados hematológicos para os portadores de traço falciforme, percebeu-se
que grande parte dos casos (27 de um total de 39) não apresentaram valores alterados de Hb,
Hto, VCM, RDW e HCM, sendo similares, portanto, a outros resultados descritos na literatura
(MURÃO et al., 2007; TRIPETTE et al., 2009). Do mesmo modo, observou-se a discreta
alteração morfológica eritrocitária e também, concentração de HbA maior do que a de HbS,
revelando que os resultados são compatíveis com as características da heterozigose AS.
Entre os casos de hemoglobinopatia SC, no entanto, os achados hematológicos
revelaram índices diminuídos de hemoglobina e hematócrito, fato que está relacionado ao
grau leve de anemia presente nos pacientes. Entretanto, a presença de anemia em pacientes
com hemoglobinopatia SC ocorre em menor intensidade do que nos portadores de anemia
falciforme (NAGEL; FABRY; STEINBERG, 2003; TRIPETTE et al., 2009). Nesse sentido,
os resultados desses dois parâmetros estão de acordo com dados de Tripette et al. (2009), pois
os valores de Hb e Hto apresentaram médias menores para a hemoglobinopatia SC (Hb
10,87g/dL; Hto=32,77%) e também para o traço falciforme (Hb 13,4g/dL; Hto=40,30%),

41
obedecendo à forma de classificação clínica de cada fenótipo de acordo com os valores
diminuídos de hemoglobina e hematócrito (SS<SC<AS) (TRIPETTE et al., 2009).
É importante lembrar que a apresentação clínica da hemoglobinopatia SC é
considerada mais branda do que a anemia falciforme (NAGEL; FABRY; STEINBERG,
2003).
Outro achado relevante foi o aumento do RDW proporcional a expressividade das
alterações morfológicas do eritrócito. As alterações morfológicas encontradas podem sugerir
que as células SC apresentaram certo grau de densidade, proporcionando, assim, aumento da
incidência de células com formas alteradas, fato que foi constatado por Nagel e colaboradores
(2003) em estudo anterior. Ao contrário do que foi indicado por Ballas et al. (1987) não foi
observada microcitose ou hipercromia nas células SC, o que indica que não houve
desidratação intensa do eritrócito, sendo a microcitose e a hipercromia evidenciadas como
efeito secundário da desidratação (BALLAS et al., 1987).
O valor médio do CHCM dos três pacientes com hemoglobinopatia SC, foi menor
(33,33pg) do que a média de 37pg indicada em outros estudos (ROMERO et al., 2004),
sugerindo que no período de avaliação do eritrograma dos pacientes, não houve relação direta
com o aumento da densidade e desidratação do eritrócito, indicando menor probabilidade de
ocorrer formação de polímeros devido ao efeito da concentração de HbS na célula SC.
O valor do VCM entre estes pacientes, com hemoglobinopatia SC, permaneceu
normal, corroborando os estudos que estimam as alterações mais frequentes entre os
indivíduos que carregam o gene da βS globina, pois se sabe que este índice pode estar normal
ou diminuído nesta doença (HASHMI et al., 2008).
A concentração média da HbS e HbC nos indivíduos que apresentaram dupla
heterozigose foi de 51,2% para Hb S e de 43,77% para HbC, evidenciando maior
concentração de HbS, o que reforça os dados de estudos prévios, os quais relatam que
pacientes com hemoglobinopatia SC apresentam níveis de HbS superiores ao de HbC
(ONDEI et al., 2007).
Notou-se também que a média de concentração de HbS, que foi de 36,71% nos
pacientes com traço falciforme e de 51,2% nos pacientes com a hemoglobinopatia SC,
coincidiu com as médias descritas pela literatura: 40% e 50%, respectivamente. Dessa forma,
os dados obtidos corroboram a observação de Nagel et al. (2003) de que existe semelhança
entre o traço falciforme e hemoglobinopatia SC, pois pacientes com essas alterações genéticas
possuem níveis de HbS menores do que aqueles que são portadores de anemia falciforme,
cujo índice de HbS varia de 90% a 95% (HASHMI et al., 2008). No entanto, o conhecimento

42
de que ocorre maior desidratação nas células SC, comparada às células AS, demonstra uma
das diferenças entre o traço falciforme e a hemoglobinopatia SC, o que pode ser evidenciado
de acordo com suas respectivas características clínicas, sendo a hemoglobinopatia SC
considerada moderadamente severa, enquanto a heterozigose AS é considerada assintomática
(NAGEL; FABRY; STEINBERG, 2003).
Apesar da heterozigose AS raramente apresentar complicações, um estudo anterior
sugere que a ocorrência da viscosidade aumentada do sangue, ocasionada por determinadas
situações de estresse, contribui para a ativação de maior número de selectinas que, são
capazes de desencadear perturbações significativas da microcirculação (TRIPETTE et al.,
2009).
Este fato chama a atenção no presente estudo em virtude da população analisada, que é
composta por indivíduos que estão constantemente submetidos a fatores ambientais e físicos,
considerados de risco para aqueles que portam o gene da β S globina, por se tratar de militares.
Os resultados sugerem que a prevalência encontrada foi compatível com a de
resultados de outras investigações realizadas no Distrito Federal e no Brasil. Assim, é
importante considerar a necessidade de ampliação do rastreamento das hemoglobinopatias na
rotina laboratorial, e do aumento da realização de novos estudos com amostras mais
significativas.
Nesse sentido, com a realização de estudos futuros, almeja-se contribuir para o melhor
conhecimento do perfil epidemiológico das hemoglobinopatias na região do Distrito Federal.
Além disso, visa-se auxiliar o diagnóstico e o esclarecimento sobre medidas profiláticas e
terapêuticas, com o intuito de promover a melhoria na qualidade de vida do paciente e, ainda,
despertar o interesse de segmentos governamentais para a elaboração de medidas explicativas
e de conscientização da sociedade em geral.

43
8. REFERÊNCIAS
AGENCIA NACIONAL DE VIGILANCIA SANITÁRIA (ANVISA). Manual de
diagnóstico e tratamento de doenças falciformes. Brasília: ANVISA, 2002.
ALMEIDA, C. B. et al. High expression of the cGMP-specific phosphodiesterase, PDE9A, in
sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD
neutrophils. British Journal of Haematology. V. 142, p 836–844, 2008.
ARAUJO, Paulo Ivo C.. O autocuidado na doenca falciforme. Revista Brasileira de
Hematologia e Hemoterapia .V.29, N.3, p. 239-246, 2007.
BALLAS, S. K. Pain management of sickle cell disease. Hematology/Oncology Clinics of
North America. V. 19, N°.5, p. 785-802, 2005.
BALLAS, S. K.et al.The Xerocytosis of Hb SC Disease. Blood. V. 69. N° 1, p. 124-130,
1987.
BALLAS, S. K.et al. Definitions of the phenotypic manifestations of sickle cell disease.
American Journal of Hematology. V 1, N° 85 p. 6-13, 2010.
BALLAS, S. K.; MOHANDAS, N. Pathophysiology of vaso-occlusion.
Hematology/Oncology Clinics of North America. V. 10, N°. 6, p. 1221-39, 1996.
BARBARINO, G. A., PLATT, M. O., KAUL, D. Sickle Cell Biomechanics. Annual Review
of Biomedical Engineering. V. 12, 2010.
BEZERRA, M. A. C. Determinação das propriedades adesivas e funcionais em glóbulos
vermelhos, neutrófilos e plaquetas de pacientes com hemoglobinopatia SC, S/β
Talassemia e Talassemia intermediária. 2009. 102f. Dissertação (Doutorado em -
Fiopatologia Médica) - Universidade Estadual de Campinas, Campinas.
BONINI-DOMINGOS, C. R. As hemoglobinopatias e a diversidade genética da população
brasileira. Revista Brasileira de Hematologia e Hemoterapia, V.31, N°.6, p. 401-401, 2009.
BOOKCHIN, R. M.; LEW, V. L. Pathophysiology of sickle cell anemia.
Hematology/Oncology Clinics of North America. V. 10, N°. 6, p. 1241-53, 1996.

44
BRASIL. Ministério da saúde. Portaria nº 1.018, de 1º de julho de 2005. Institui no âmbito
do Sistema Único de Saúde, o Programa Nacional de Atenção Integral as Pessoas com
Doença Falciforme e outras Hemoglobinopatias. Diário Oficial da republica Federativa do
Brasil. Brasília, DF, 4 ago. 2005. Disponível em:
<ftp://ftp.saude.sp.gov.br/ftpsessp/bibliote/informe_eletronico/iels.jul.05/Iels124/U_PT-MS-
GM-1018_010705.pdf>. Acesso em: 05 set. 2010.
BRASIL. Ministério da saúde. Programa de atenção integral às pessoas com doenças
falciformes e outras hemoglobinopatias da cidade de São Paulo. São Paulo, Abril de 2008.
Disponível em:
<http://ww2.prefeitura.sp.gov.br/arquivos/secretarias/saude/popnegra/DoencaFalciforme_Pro
gramaAtencaoIntegral.pdf > Acesso em: 06 out. 2010.
CANALLI, A. A. et al. Increased adhesive properties of neutrophils in sickle cell disease may
be reversed by pharmacological nitric oxide donation. Haematologica. V. 93, N° 4, p 605-
609, 2008.
CANALLI, A.A et al. Increased adhesive properties of eosinophils in sickle cell disease
Experimental Hematology. V. 32, p. 728–734, 2004.
CANÇADO, R. D.; JESUS ,J. A. A doença falciforme no Brasil. Revista Brasileira de
Hematologia e Hemoterapia. V. 29, N°. 3, p. 203-206, 2007.
CAPPELLINI, M. D. Coagulation in the pathophysiology of hemolytic anemias. American
Society of Hematology. p. 74-78, 2007.
CHANG, J. et al. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle
cell mice through rapid inhibition of neutrophil adhesion. Blood. V. 111, N°. 2, p. 915-23,
2008.
CODRIGTON, J.F. Hb Chad or alpha 223(B4)Glu-Lys beta 2 observed in members of a
Surinam family in association with alpha-thalassemia-2 and with Hb S. Hemoglobin. V. 13,
N°. 6, p. 543-556, 1989.
DI NUZZO, D. V. P.; FONSECA, S. F. Anemia falciforme e infecção. Jornal de Pediatria.
V. 80, N°. 5, p. 347-54, 2004.
DINIZ, D. et al. Prevalência do traço e da anemia falciforme em recém-nascidos do Distrito
Federal, Brasil, 2004 a 2006. Caderno de Saúde Pública.V. 25, N°. 1, p. 188-194, 2009.

45
FATTOUM, S. Hemoglobinopathies in Tunisia. An updated review of the epidemiologic and
molecular data. La Tunisie Medicale. V. 84, N°. 11, p 687-96, 2006.
FONSECA, G. H. H. Diagnóstico de hipertensão pulmonar em indivíduos adultos com
doença falciforme. 2008. f 153. Dissertação (Mestrado em Ciências) - Universidade de São
Paulo, São Paulo.
GALIZA NETO, G. C.; PITOMBEIRA, M. S. Aspectos moleculares da anemia falciforme.
Jornal Brasileiro de Patologia e Medicina Laboratorial.V. 39, N°. 1, p. 51-56, 2003.
HASHMI, N. K. et al. Chromatographic analysis of HbS for the diagnosis of various sickle
cell disorders in Pakistan. Annals of hematology. V. 87,p. 639–645, 2008.
HILLERY, C. A. et al. Hydroxyurea therapy decreases the in vitro adhesion of sickle
erythrocytes to thrombospondin and laminin. British Journal of Haematology. V. 109, p.
322-7, 2000.
HOLSBACH, D. R. et al. Ocorrência de hemoglobina S no estado de Mato Grosso do Sul,
Brasil •Jornal Brasileiro de Patologia e Medicina Laboratorial. V. 44, N°. 4, p. 277-282,
2008.
INSTITUTO BRASILEIRO DE GEOGRAFIA E ESTATÍSTICA (IBGE). Pesquisa
nacional por amostra de domicílios 1999: Brasil, grandes regiões, unidades da
federação e regiões metropolitanas. Síntese de indicadores 1999. Rio de Janeiro,
2000. Disponível em:
<http://www.ibge.gov.br/seculoxx/arquivos_xls/palavra_chave/populacao/cor.shtm> Acesso
em: 12 set. 2010.
JOUTOVSKY, A. NARDI, M. Hemoglobin C and Hemoglobin O-Arab Variants Can Be
Diagnosed Using the Bio-Rad Variant II High-Performance Liquid Chromatography System
Without Further Confirmatory Tests. Archives of Pathology & Laboratory Medicine
V. 128, No. 4, pp. 435-439, 2004.
KATO, G. J. et al. Endogenous nitric oxide synthase inhibitors in sickle cell disease:
abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis,
organ dysfunction and death. British Journal of Haematology. V. 145, N°. 4, p. 506-13,
2009.
LEES, C. M., DAVIES, S. DEZATEUX, C. Neonatal screening for sickle cell disease.
Cochrane Database of Systematic Reviews. V. 2, 2000.

46
LOUREIRO, M. M.; ROZENFELD, S.; PORTUGAL, R. D. Acute clinical events in patients
with sickle cell disease: epidemiology and treatment. Revista Brasileira de Hematologia e
Hemoterapia. V. 30, N°. 2, p. 95-100, 2008.
LOUREIRO, M. M.; ROZENFELD, S. Epidemiology of sickle cell disease hospital
admissions in Brazil. Revista de Saúde Pública. V. 39, N°. 6, 2005.
MELO-REIS, P. R. et al. Prevalência de talassemias e hemoglobinas variantes no estado de
Goiás, Brasil • Jornal Brasileiro de Patologia e Medicina Laboratorial. V. 42, N. 6, p. 425-
430, 2006.
MURÃO, M.; FERRAZ, M. H. Traço falciforme: heterozigose para hemoglobina S. Revista
Brasileira de Hematologia e Hemoterapia. V. 29, N° 3, p. 223-225, 2007.
NAOUM, P. C.; NAOUM, F. A.; NAOUM, P. F. Doença falciforme. Disponível em: <
http://www.hemoglobinopatias.com.br/d-falciforme/diagnostico.htm>. Acesso em: 17 out. 2010.
NAOUM, P. C. Interferentes eritrocitários e ambientais na anemia falciforme. Revista
Brasileira de Hematologia e Hemoterapia. V. 22, N° 1, p. 05-22, 2000.
NAGEL, R. L., FABRY, M. E. STEINBERG, M. H. The paradox of hemoglobin SC disease.
Blood Reviews. V. 17, p. 167–178, 2003.
ONDEI , L. S. et al. HPLC determination of hemoglobins to establish reference values with
the aid of statistics and informatics. Genetics and Molecular Research. V. 6, N° 2, p 453-
460, 2007.
ORGANIZAÇÃO MUNDIAL DA SAÚDE (OMS). Sickle cell anemia. In: fifty-ninth world
health assembly. Report by the secretariat - provisional agenda - item 11.4, 24 de abril 2006.
PINHEIRO, L. S. et al. Prevalência de hemoglobina S em recém-nascidos de Fortaleza:
importância da investigação neonatal. Revista Brasileira de Ginecologia e Obstetrícia. V.
28, N°. 2, p. 122-125, 2006.
POWARS, D. R. et al. Outcome in hemoglobin SC disease: a four-decade observational study
of clinical, hematologic, and genetic factors. American Journal of Hematology. V. 70, N°.
3, p. 206-215, 2002.

47
RAMALHO, A. S. A Portaria no 822/01 do Ministério da Saúde e as peculiaridades das
hemoglobinopatias em saúde pública no Brasil. Caderno de Saúde Pública. V. 19, N°. 4, p.
1195-1199, 2003.
ROMERO, J. R. et al. Expression of HbC and HbS, but not HbA, results in activation of K-Cl
cotransport activity in transgenic mouse red cells. Blood. V. 103, N. 6, 2004.
ROSSE, W. F. et al. New Views of Sickle Cell Disease Pathophysiology and Treatment. Hematology. p. 2-17, 2000.
RINEHART, et al. Determinants of erythrocyte hydration. Current Opinion in Hematology.
V. 17, N° 3, p. 191-7, 2010.
RUIZ, M. A. Anemia falciforme: Objetivos e resultados no tratamento de uma doença de
saúde pública no Brasil. Revista Brasileira de Hematologia e Hemoterapia. V. 29, N°. 3, p.
203-206, 2007.
SCHNOG, J. B. et al. Sickle cell disease; a general overview. Netherlands Journal of
Medicine. V. 62, N°. 10, p. 364-74, 2004.
SILVA, N. M. Influência da talassemia α+ nos índices hematimétricos VCM e HCM e nos
percentuais de Hb anômala em portadores da HbC. 2001. 71f. Dissertação (Mestrado em -
Ciências Médicas) - Universidade Estadual de Campinas, Campinas.
SONATI M. F.; COSTA F. F. The genetics of blood disorders: hereditary
hemoglobinopathies. Jornal de Pediatria. V. 84, N° 4, 2008.
STEINBERG, M. H. Predicting clinical severity in sickle cell anaemia. British Journal of
Haematology. V. 129, p. 495-481, 2005.
STUART, M. J.; NAGEL, R. L. Sickle-cell disease. The Lancet. V. 364, N°. 9442, p. 1343-
60, 2004.
TAYLOR, J. G. et al. Chronic hyper-hemolysis in sickle cell anemia: association of vascular
complications and mortality with less frequent vasoocclusive pain. PLoS One. V. 3, N°. 5,
2008.

48
THEODORIDOU, S. et al. The first case of a compound heterozygosity for Hb E-Saskatoon
and Hb S. Haematologica. V. 88, N°. 3, p. 33, 2003.
TRIPETTE, J. Red blood cell aggregation, aggregate strength and oxygen transport potential
of blood are abnormal in both homozygous sickle cell anemia and sickle-hemoglobin C
disease. Haematologica. V. 94, N° 8, p. 1060-1065, 2009.
VEKILOV, P. G. Sickle-cell haemoglobin polymerization: is it the primary pathogenic event
of sickle-cell anaemia?. British Journal of Haematology, V. 139, p. 173–184, 2007.
VERAS, M. S. et al. Prevalência do traço falciforme em doadores de sangue do Distrito
Federal / Prevalence of the falciform trace in blood donors from Distrito Federal. Revista de
saúde do Distrito Federal. V. 9, N°. 1, p. 9-12, 1998.
WEINER, D. L. et al. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive
crisis in pediatric patients with sickle cell disease. Journal Of the American Medical
Association. V. 289, N° 9, p. 1136-42, 2003.
ZAGO, M. A.; PINTO, A. C. S. Fisiopatologia das doenças falciformes: da mutação genética
à insuficiência de múltiplos órgãos. Revista Brasileira de Hematologia e Hemoterapia.
V.29, N°. 3, p. 207-214, 2007.
ZAGO, M. A. Hematologia: Fundamentos e Prática. Rio de Janeiro, Atheneu. 2004.

49
ANEXOS
ANEXO A
TERMO DE CONSENTIMENTO LIVRE ESCLARECIDO
Eu, __________________________________________________________________
( )(Concordo) ( )(Discordo) que o material coletado para realização dos exames
constantes nesta guia, assim como os resultados destas análises, os quais estarão
armazenados nas bases de dados do LAC, sejam utilizados na composição de pesquisas
e outros trabalhos executados pelo laboratório com finalidades acadêmicas e científicas,
inclusive para publicação, desde que seja preservado o sigilo da minha identidade.
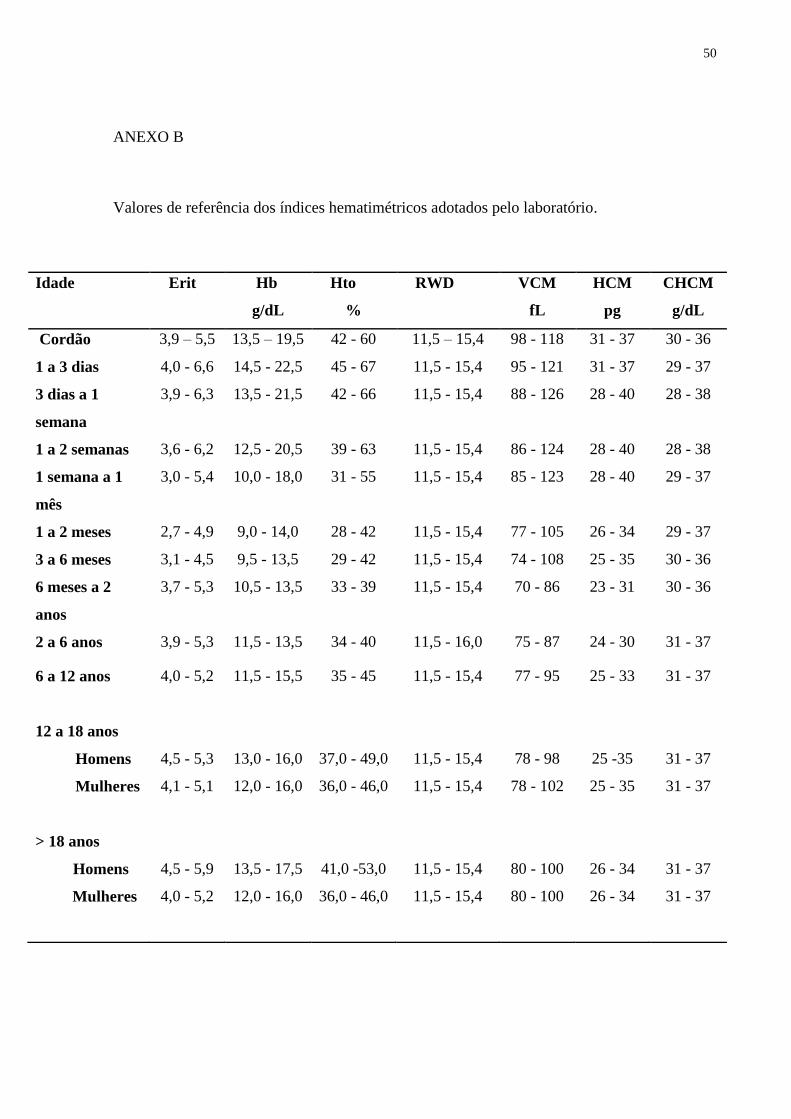
50
ANEXO B
Valores de referência dos índices hematimétricos adotados pelo laboratório.
Idade Erit
Hb
g/dL
Hto
%
RWD
VCM
fL
HCM
pg
CHCM
g/dL
Cordão 3,9 – 5,5 13,5 – 19,5 42 - 60 11,5 – 15,4 98 - 118 31 - 37 30 - 36
1 a 3 dias 4,0 - 6,6 14,5 - 22,5 45 - 67 11,5 - 15,4 95 - 121 31 - 37 29 - 37
3 dias a 1
semana
3,9 - 6,3 13,5 - 21,5 42 - 66 11,5 - 15,4 88 - 126 28 - 40 28 - 38
1 a 2 semanas 3,6 - 6,2 12,5 - 20,5 39 - 63 11,5 - 15,4 86 - 124 28 - 40 28 - 38
1 semana a 1
mês
3,0 - 5,4 10,0 - 18,0 31 - 55 11,5 - 15,4 85 - 123 28 - 40 29 - 37
1 a 2 meses 2,7 - 4,9 9,0 - 14,0 28 - 42 11,5 - 15,4 77 - 105 26 - 34 29 - 37
3 a 6 meses 3,1 - 4,5 9,5 - 13,5 29 - 42 11,5 - 15,4 74 - 108 25 - 35 30 - 36
6 meses a 2
anos
3,7 - 5,3 10,5 - 13,5 33 - 39 11,5 - 15,4 70 - 86 23 - 31 30 - 36
2 a 6 anos 3,9 - 5,3 11,5 - 13,5 34 - 40 11,5 - 16,0 75 - 87 24 - 30 31 - 37
6 a 12 anos 4,0 - 5,2 11,5 - 15,5 35 - 45 11,5 - 15,4 77 - 95 25 - 33 31 - 37
12 a 18 anos
Homens 4,5 - 5,3 13,0 - 16,0 37,0 - 49,0 11,5 - 15,4 78 - 98 25 -35 31 - 37
Mulheres
4,1 - 5,1 12,0 - 16,0 36,0 - 46,0 11,5 - 15,4 78 - 102 25 - 35 31 - 37
> 18 anos
Homens 4,5 - 5,9 13,5 - 17,5 41,0 -53,0 11,5 - 15,4 80 - 100 26 - 34 31 - 37
Mulheres
4,0 - 5,2 12,0 - 16,0 36,0 - 46,0 11,5 - 15,4 80 - 100 26 - 34 31 - 37

51
ANEXO C
ELETROFORESE DE Hb EM pH ALCALINO (Protocolos de Metodologias Clássicas
para o Diagnóstico de Hemoglobinopatias- UNESP, 2003)
Amostra: sangue total com EDTA
Material necessário:
Filme de agarose para separação eletroforética das Hb em pH alcalino (Hb alcalina)
Seringa micro-volume
Tampão alcalino TRIS pH 9,5 ±0,2 CELMGEL
Hemolisante (do Kit)
Corante Ponceau S (5,0 g CELM + 50,0 g de ácido tricloroacético)
Descorante para eletroforese de Hb (100mL ácido acético glacial + 50 mL metanol
P.A, completando com água até 1000mL)
Placas de Petri
Secador
Aparelho CELM FEA® – 250
Procedimento
Preparação do hemolisado
Centrifugar parte do sangue total com EDTA ou heparina e desprezar o plasma.
Lavar hemácias com solução salina 0,9% ( para cada 100µL de hemácias acrescentar
200µL de solução fisiológica) até obter sobrenadante límpido.
Misturar partes iguais das hemácias e hemolisante (para 100µL de hemácias 100µL de
hemolisante), em um tubo de ensaio.
Agitar vigorosamente em vortex por aproximadamente 30 segundos.
Determinar a concentração de hemoglobina no aparelho de hemograma e ajustar a
concentração de Hb em aproximadamente 1g/dl, diluindo com água destilada ou deionizada
até atingir concentração desejada.
O hemolisado obtido é estável por 24 horas quando armazenado entre 2 e 8º C.
Eletroforese
Com seringa de micro-volume, pipetar 1,0µL de cada hemolisado em uma cavidade
numerada no gel alcalino.

52
Ligar o aparelho CELM FEA® – 250 e ajustar para 30 minutos e 150 volts.
Colocar o gel alcalino no porta filme, coincidindo os pólos positivos e negativos do
filme e da cuba.
Colocar 80 mL de tampão alcalino gelado (2 a 8ºC), na cuba para cobrir o eletroldo
metálico.
Colocar o porta filme com o gel alcalino dentro da cuba ligá-la.
Após os 30 minutos deve-se desligar e retirar o porta-filme da cuba e deixar escorrer o
excesso de tampão em papel toalha.
Retirar o gel alcalino e colocá-lo a 60ºC (secador) até secar completamente, por
aproximadamente 15 minutos.
Mergulhar o gel em placa Petri, contendo o corante Ponceau S, por 10 minuto, sem
agitação.
Retira-lo do corante Ponceau e colocá-lo em outra placa contendo o descorante (ácido
acético / metanol), por 10 minutos.
Repetir o procedimento acima com descorante limpo por 10 minutos.
Completar a descoloração deixando o gel alcalino mergulhado por 10 minutos ou até
sua total descoloração em ácido acético 5,0%.
Secar completamente o gel alcalino à 60ºC (secador).

53
ANEXO D
ELETROFORESE DE Hb EM pH ÁCIDO (Protocolos de Metodologias Clássicas
para o Diagnóstico de Hemoglobinopatias- UNESP, 2003)
Amostra: sangue total com EDTA
Material necessário:
Filme de agarose para diferenciação da Hb S, C e F(Hb ácida)
Seringa micro-volume
Tampão citrato pH 6,3 ±0,2 CELMGEL
Hemolisante (do Kit)
Corante Amido-Black (Negro amido 0,1% em ácido acético a 5%)
Descorante para eletroforese de Hb (ácido acético a 5%)
Placas de Petri
Secador
Aparelho CELM FEA® – 250
Procedimento
Preparação do hemolisado
Centrifugar parte do sangue total com EDTA ou heparina e desprezar o plasma.
Lavar hemácias com solução salina 0,9% ( para cada 100µL de hemácias acrescentar
200µL de solução fisiológica) até obter sobrenadante límpido.
Misturar partes iguais das hemácias e hemolisante (para 100µL de hemácias 100µL de
hemolisante), em um tubo de ensaio.
Agitar vigorosamente em um vórtex por aproximadamente 30 segundos.
Determinar a concentração de hemoglobina no aparelho de hemograma e ajustar a
concentração de Hb em aproximadamente 1g/dl, diluindo com água destilada ou deionizada
até atingir concentração desejada.
O hemolisado obtido é estável por 24 horas quando armazenado entre 2 e 8º C.
Eletroforese
Com seringa de micro-volume, pipetar 0,5µL de cada hemolisado em uma cavidade
numerada no gel ácido. Aguardar alguns segundos para que o hemolisado penetre no gel e
aplicar mais 0,5µL do hemolisado.
Ligar o aparelho CELM FEA® – 250 e ajustar para 55 minutos e 50 volts.

54
Colocar o gel ácido no porta-filme, coincidindo o pólo positivo da cuba com o pólo
negativo do gel ácido.
Colocar 90 mL de tampão citrato pH 6,3 ±0,2 gelado (2 a 8ºC), na cuba para cobrir o
eletroldo metálico.
Colocar o porta filme com o gel alcalino dentro da cuba ligá-la.
Após os 55 minutos deve-se desligar e retirar o porta-filme da cuba e deixar escorrer o
excesso de tampão em papel toalha.
Retirar o gel ácido e colocá-lo em uma placa de Petri com o corante Amido-Black por
5 minutos, sem agitação.
Retira-lo do corante Amido – Black e colocá-lo em outra placa contendo o descorante
(ácido acético 5%), por 5 minutos.
Retirar o excesso de descorante do gel e colocá-lo à 60º (secador) até secá-lo
completamente, por aproximadamente 15 minutos.
Completar descoloração deixando o gel ácido mergulhado em ácido acético 5% até
que o fundo fique transparente, se necessário trocar o descorante.
Colocar o gel ácido em uma placa de Petri com água destilada por 2 minutos.
Secar completamente o gel ácido a 60ºC (secador).

55
ANEXO E