diedros conformacionais e sua aplicaÇÃo no estudo de estabilidade de...
TRANSCRIPT

PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 199
DIEDROS CONFORMACIONAIS E SUA APLICAÇÃO NO ESTUDO DE ESTABILIDADE DE BIOMOLÉCULAS
Anderson Hollerbach Klier 1
George Schayer Sabino 1
Sonaly Cristine Leal 1
Ana Flávia Arantes Pereira 2
Liege Aparecida Mapa 2
Luna Elisabeth Carvalho Ferreira 2
Nathália Martins Moreira 2
Paula Guimarães Chiesa 2
RESUMO: Os diedros, ângulos gerados entre os planos espaciais existentes entre quatro átomos consecutivos ligados por uma ligação simples ou ligação
sigma, podem ser utilizados para previsão da estabilidade conformacional. Tal estimativa é baseada na minimização de energia molecular das conformações
geradas após um giro de 360° graus no diedro requerido, que pode gerar até 360 conformações que auxiliam no entendimento sobre estabilidade de ligações
alternadas e eclipsadas. Ligações estas que estão intimamente relacionadas com a estabilidade de conformeros denominados anti ou antiperiplanar, anticlinal, sinclinal e gauche ou sinperiplanar. A análise dos diedros foi aplicada a glutationa, biomolécula essencial ao metabolismo de fase II e a seu
conjugado com a N-acetilimidoquinona.
PALAVRAS-CHAVE: diedro, conformação, biomolécula, estabilidade
INTRODUÇÃO
Diversas publicações recentes tem ressaltado a importân-
cia da análise conformacional na previsão da estabilidade mo-
lecular, e em última instância, a aplicabilidade de softwares na
química computacional a fim de facilitar o entendimento desta
estabilidade (FERREIRA, 2008; RAUPP, 2008; MARQUES, 2010,
KLIER 2012). As conformações em estruturas orgânicas básicas
geradas por carbonos tetraédricos ligados consecutivamente
por ligações covalentes simples podem ser analisadas através
do chamado ângulo diedro, conforme figura 1.

200 | PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785
Analisando a figura 1, pode-se visualizar dois planos ima-
ginários trigonais denominados ACH2CH2 e BCH2CH2, sendo os
carbonos metilênicos denominados de átomos 1 quando ligado
ao substituinte A , e 2 quando ligado ao substituinte B. Denomi-
na-se diedro, o ângulo formado entre os dois planos trigonais,
ou seja; se os planos ACH2CH2 e BCH2CH2 tem em comum a
ligação simples entre os átomos 1 e 2, podemos conservar a
posição do plano ACH2CH2 e girar o plano BCH2CH2 de 180° so-
bre a ligação entre os átomos 1 e 2. Assim, os substituintes A e
B que estavam espacialmente acima do eixo da ligação 1-2 e
abaixo do eixo da ligação 1-2, respectivamente, após o giro de
180° estarão ambos acima do eixo da ligação 1-2. Se aplicarmos
conceitos de representação estrutural em projeção, especifica-
mente a projeção de Newmann, à suposta estrutura elaborada
na figura 1, teríamos as projeções possíveis descritas a seguir
na figura 2.
Observa-se que dentre as conformações apresentadas,
três apresentam ligações alternadas (uma antiperiplanar e duas
sinclinais) e outras três apresentam ligações eclipsadas (uma
sinperiplanar e duas anticlinais. Considerando um giro limítrofe
de 60º para cada conformação, pode-se definir num esquema
gráfico a localização das geometrias sinperiplanar (SP), sincli-
nal (SC), anticlinal (AC) e antiperiplanar (AP) como esquemati-
zado abaixo na figura 3.
A simulação dos diedros pode ser feita com a utilização de softwares específicos que permitam sua análise, como o PcModel
(SILVA, 2006; ANDREI, 2003, BARREIRO, 1997). A simulação se inicia com a construção molecular escolhida, aqui repre-
sentada pelo 1,2-difeniletano, conforme figura 4.
PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 201
Estruturalmente o software difere os átomos por cores,
permitindo o reconhecimento estrutural da molécula com seus
respectivos grupos funcionais, quando for o caso. Construída
a estrutura molecular, a mesma é minimizada energeticamente
a fim de obtermos os dados de energia conformacional (MMx
Energy), entalpia de formação (Hf) e momento dipolar (Dip.
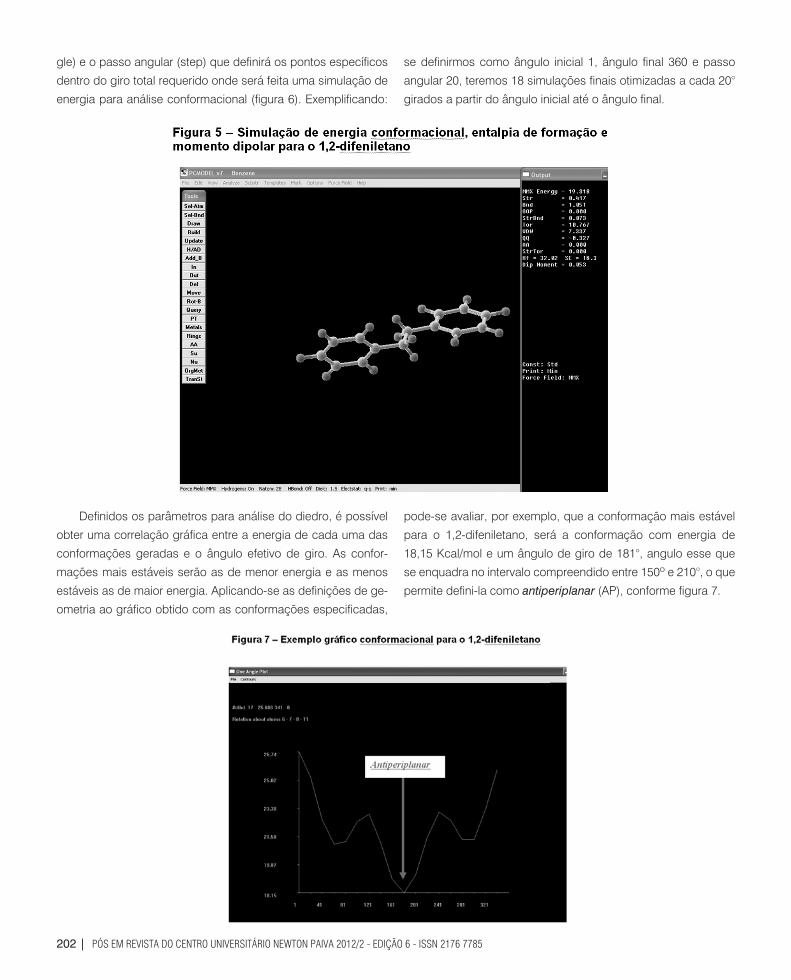
Moment), como pode ser observado na figura 5.
Observa-se valores simulados de 19,318 Kcal/mol para a
energia conformacional, 32,02 Kcal/mol para a entalpia de forma-
ção e um momento dipolar de 0,053 Debies. Otimizada a ener-
gia molecular, faz-se a marcação do diedro com a escolha dos
quatro átomos consecutivos a serem analisados, definindo-se o
ângulo inicial de giro (start angle), o ângulo final de giro (final an-
202 | PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785
gle) e o passo angular (step) que definirá os pontos específicos
dentro do giro total requerido onde será feita uma simulação de
energia para análise conformacional (figura 6). Exemplificando:
se definirmos como ângulo inicial 1, ângulo final 360 e passo
angular 20, teremos 18 simulações finais otimizadas a cada 20°
girados a partir do ângulo inicial até o ângulo final.
Definidos os parâmetros para análise do diedro, é possível
obter uma correlação gráfica entre a energia de cada uma das
conformações geradas e o ângulo efetivo de giro. As confor-
mações mais estáveis serão as de menor energia e as menos
estáveis as de maior energia. Aplicando-se as definições de ge-
ometria ao gráfico obtido com as conformações especificadas,
pode-se avaliar, por exemplo, que a conformação mais estável
para o 1,2-difeniletano, será a conformação com energia de
18,15 Kcal/mol e um ângulo de giro de 181°, angulo esse que
se enquadra no intervalo compreendido entre 150º e 210°, o que
permite defini-la como antiperiplanar (AP), conforme figura 7.

PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 203
A partir do gráfico obtido é possível analisar individualmente
cada uma das conformações geradas, acessando a denomina-
ção do arquivo salvo, que fornece a lista de estruturas com to-
dos os dados de cada uma das conformações, figura 8.
Acessando-se cada uma das conformações da lista de
estruturas pertinente, pode-se ainda calcular a distância entre
dois átomos quaisquer ou o ângulo gerado por três átomos
consecutivos, o que pode ser observado na figura 9, onde a
distância entre dois dos carbonos aromáticos de ciclos distin-
tos foi estimada em 6,786 Aº e o ângulo gerado por três dos
carbonos aromáticos benzênicos, estimada em 120°.

204 | PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785
OBJETIVOS
Dessa forma, os objetivos do presente trabalho foram a
obtenção de dados de energia conformacionais, momentos
dipolo e entalpia de formação, simulados no software PcMo-
del através da rotação de diedros desejados, a fim de avaliar
a aplicabilidade destes dados no entendimento de estabilida-
de conformacional de fármacos e biomoléculas estruturais.
Como moléculas protótipo foram estudados a glutationa, im-
portante biomolécula de conjugação no metabolismo de Fase
II, e o conjugado desta com a N-acetilimidoquinona, principal
metabólito de Fase I do paracetamol.
MATERIAL E MÉTODOS
As simulações de minimização de energia, distâncias inte-
ratômicas e ângulos de giro de diedro foram obtidas no software
PcModel 7.2 Serena software, utilizando-se o template do próprio
software para simulação peptídica. Os diedros estipulados tanto
na glutationa como no conjugado foram marcados nos carbonos
consecutivos da estrutura do aminoácido central cisteína e os ami-
noácidos glicina e ácido glutâmico foram otimizados na conforma-
ção antiperiplanar entre seus grupamentos mais polares antes da
formação das ligações peptídicas com a cisteína.
RESULTADOS E DISCUSSÃO
Após as minimizações iniciais obtidas para a molécula da
glutationa; que é um tripeptídeo de conjugação formado pelos
aminoácidos ácido glutâmico, cisteína e glicina, é possível ob-
servar que na conformação minimizada energeticamente, a mo-
lécula se apresenta fortemente estabilizada por uma interação
do tipo ligação de hidrogênio intramolecular. Essa interação é
gerada entre as carboxilas terminais dos aminoácidos glicina e
ácido glutâmico, interação esta que se apresenta delimitada na
coloração amarela, com uma energia molecular total de - 8,745
Kcal/mol, conforme figura 10.
A partir desta conformação obtida o diedro de giro foi deli-
mitado como demonstrado abaixo entre os átomos de 1, 2, 3 e
4 e as conformações obtidas utilizando-se um passo angular ou
step de 5 º, identificados na figura 11.

PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 205
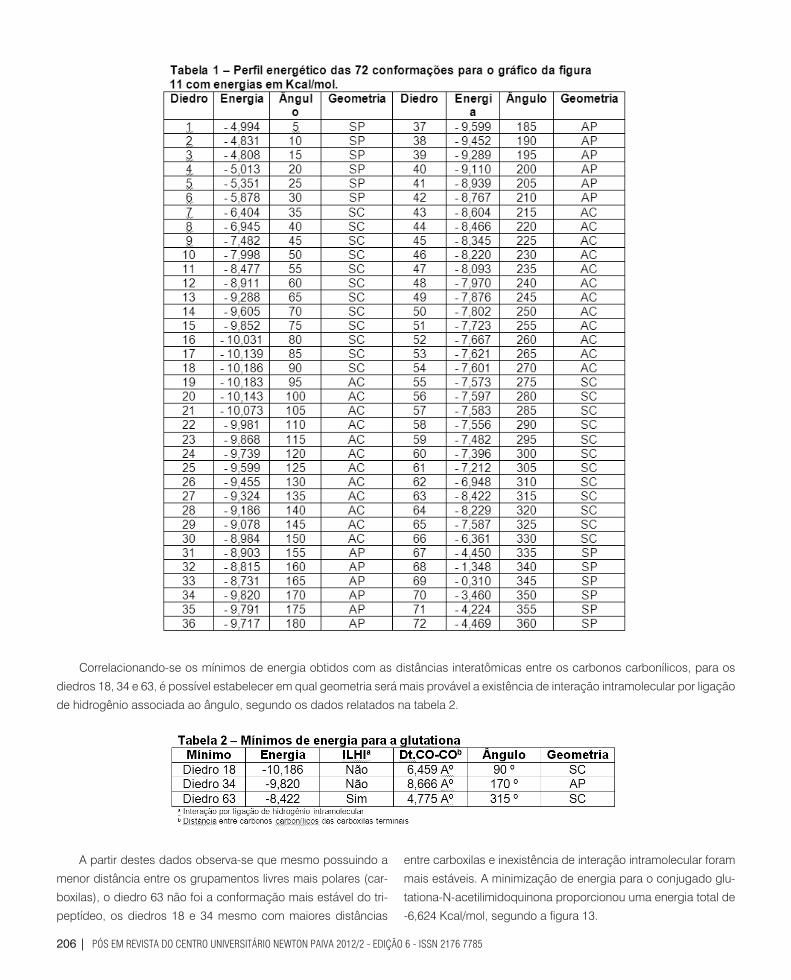
O gráfico que correlaciona a energia e o ângulo de giro para a glutationa é apresentado na figura 12.
Considerando o perfil gráfico da figura 12 associado à ta-
bela 1 (abaixo), observa-se a alternância entre máximos e mí-
nimos de energia caracterizando nitidamente a existência con-
formacional de quatro máximos de energia a 15 º (-4,808 Kcal/
mol), a 165 º (8,731 Kcal/mol), a 310 º (-6,948 Kcal/mol) e a 345º
(-0,310 Kcal/mol), respectivamente nas geometrias sinperipla-
nar, antiperiplanar, sinclinal e sinperiplanar. Em contrapartida
observam-se três mínimos de energia a 90 º com -0,186 Kcal/
mol, a 170 º com -9,820 Kcal/mol e a 315 º com -8,422 Kcal/mol,
sendo as geometrias sinclinal, antiperiplanar e sinclinal.
206 | PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785
Correlacionando-se os mínimos de energia obtidos com as distâncias interatômicas entre os carbonos carbonílicos, para os
diedros 18, 34 e 63, é possível estabelecer em qual geometria será mais provável a existência de interação intramolecular por ligação
de hidrogênio associada ao ângulo, segundo os dados relatados na tabela 2.
A partir destes dados observa-se que mesmo possuindo a
menor distância entre os grupamentos livres mais polares (car-
boxilas), o diedro 63 não foi a conformação mais estável do tri-
peptídeo, os diedros 18 e 34 mesmo com maiores distâncias
entre carboxilas e inexistência de interação intramolecular foram
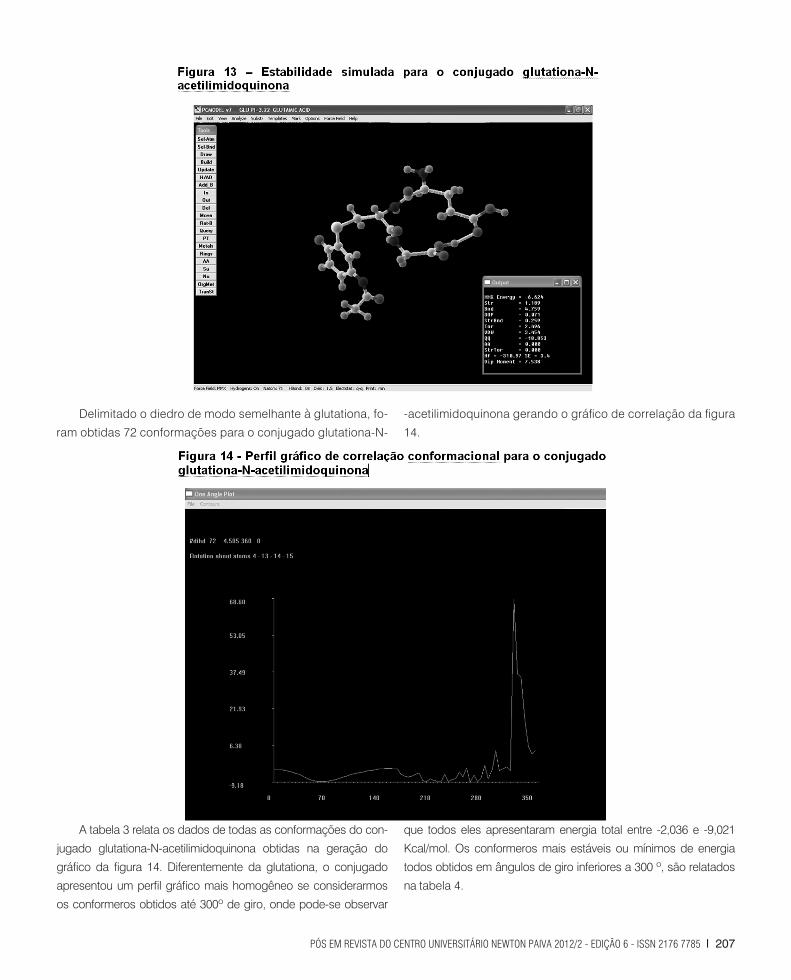
mais estáveis. A minimização de energia para o conjugado glu-
tationa-N-acetilimidoquinona proporcionou uma energia total de
-6,624 Kcal/mol, segundo a figura 13.
PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 207
Delimitado o diedro de modo semelhante à glutationa, fo-
ram obtidas 72 conformações para o conjugado glutationa-N-
-acetilimidoquinona gerando o gráfico de correlação da figura
14.
A tabela 3 relata os dados de todas as conformações do con-
jugado glutationa-N-acetilimidoquinona obtidas na geração do
gráfico da figura 14. Diferentemente da glutationa, o conjugado
apresentou um perfil gráfico mais homogêneo se considerarmos
os conformeros obtidos até 300º de giro, onde pode-se observar
que todos eles apresentaram energia total entre -2,036 e -9,021
Kcal/mol. Os conformeros mais estáveis ou mínimos de energia
todos obtidos em ângulos de giro inferiores a 300 º, são relatados
na tabela 4.

208 | PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785
Entre os mínimos do conjugado glutationa-N-acetilimidoqui-
nona houve menor variação de energia total entre -9,021 e -9,181
Kcal/mol e todos os conformeros apresentaram interação intramo-
lecular por ligação de hidrogênio. Entretanto, a interação intramole-
cular do diedro 13 ocorre entre o grupamento amino da glicina e a
carbonila da
amida entre o ácido glutâmico e cisteina, o que explica a maior
distância interatômica entre os carbonos carbonílicos das carboxi-
las terminais. Os diedros 54 e 56 apresentaram o mesmo padrão
de interação intramolecular dos mínimos da glutationa, entre os car-
bonos carbonílicos das carboxilas terminais, o que diminui conside-
ravelmente a distância interatômica entre as carbonilas.

PÓS EM REVISTA DO CENTRO UNIVERSITÁRIO NEWTON PAIVA 2012/2 - EDIÇÃO 6 - ISSN 2176 7785 l 209
A partir de 330º de giro observa-se um aumento substan-
cial na energia molecular total, gerando um máximo de energia
com 68,803 Kcal/mol no diedro 66. Os diedros entre 330° e 360º
apresentaram elevação da energia molecular total, compatível
com a compressão estérica gerada pela proximidade entre o
ciclo aromático da N-acetilimidoquinona e o ciclo da glutationa
momentaneamente estabilizado pela interação intramolecular,
conforme figura 15.
CONCLUSÃO
Considerando as limitações do próprio software bem como
a rigidez conformacional do método de simulação, uma vez que
simulamos somente os diedros do aminoácido central cisteina,
as simulações se mostraram totalmente aplicáveis as biomolé-
culas escolhidas proporcionando uma visão mais próxima da
realidade no aspecto tridimensional. Além disso, as simulações
permitiram prever as possíveis interações pertinentes à estabili-
zação da forma cíclica momentânea de peptídios em sua forma
isolada ou associada a metabólitos exógenos.
REFERÊNCIASFERREIRA, P.F.M., JUSTI, R.S. Modelagem e o “Fazer Ciência”. Quim. Nova na escola, n.28, 2008.
RAUPP, D., SERRANO, A., MARTINS, T.L.C. A evolução da química computacional e sua contribuição para a educação em química. Revis-ta Liberato, v.9, n.12, 2008.
KLIER, A.H. Conformações do cicloexano: um modelo de estudo no PcModel.Pós em Revista, n. 5, 2012.
SILVA, T.H.A. Practica III.3 Modelagem molecular com o auxílio do com-putador, 2006. Disponível em http://old.iupac.org/publications/cd/medi-cinal chemistry/Practica-III-3.pdf
ANDREI, C.C., FERREIRA, D.T., FACCIONE, M., FARIA, T.J. Da Química Medicinal à Química Combinatória e Modelagem Molecular: um curso prático. Barueri, SP: Manole, 2003. 154p.
MARQUES, M.V., RUSSOWSKY, D., FONTOURA, L.A.M. Análise Con-formacional de Compostos de Biginelli com Atividade Antineoplásica. Eclet. Química, v.35, n.4, 2010.
BARREIRO, E.J., RODRIGUES, C.R., ALBUQUERQUE, M.G., RABELLO DE SANT’ANNA, C.M., ALENCASTRO, R.B. Modelagem Molecular: Uma Ferramenta para o Planejamento Racional de Fármacos em Química
Medicinal. Quim. Nova, v.20, n.1, 1997.
NOTAS DE RODAPÉ1 Docentes do Centro Universitário Newton Paiva
2 Discentes do Curso de Farmácia do Centro Universitário Newton Paiva