anexo i resumo das caracterÍsticas do...
TRANSCRIPT

1
ANEXO I
RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO

1. DENOMINAÇÃO DO MEDICAMENTO PREZISTA 300 mg comprimidos revestidos por película. 2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA Cada comprimido revestido por película contém 300 mg de darunavir (sob a forma de etanolato). Excipiente: cada comprimido contém 1,375 mg do corante amarelo sunset FCF (E110). Lista completa de excipientes, ver secção 6.1. 3. FORMA FARMACÊUTICA Comprimido revestido por película. Comprimido de forma oval, cor de laranja, com a inscrição “300 MG” numa das faces e “TMC114” na face oposta. 4. INFORMAÇÕES CLÍNICAS 4.1 Indicações Terapêuticas PREZISTA, administrado em associação com 100 mg de ritonavir está indicado, em associação com outros medicamentos anti-retrovirais, no tratamento da infecção pelo vírus da imunodeficiência humana (VIH-1) em doentes adultos previamente muito tratados que não tenham respondido a mais do que um regime contendo um inibidor da protease (IP). Esta indicação baseia-se nas análises da resposta virológica e imunológica, às 24 semanas, em 2 ensaios controlados de fase II para determinação de dose e em informação adicional de estudos não controlados (ver secção 5.1). Quando se opta por iniciar o tratamento com PREZISTA administrado em associação com 100 mg de ritonavir deve prestar-se particular atenção à história terapêutica de cada doente e aos padrões de mutações associados aos diferentes fármacos. Os testes genotípicos ou fenotípicos (quando disponíveis) e a história terapêutica devem orientar a eleição do tratamento com PREZISTA. 4.2 Posologia e modo de administração A terapêutica deve ser iniciada por um médico com experiência no tratamento da infecção pelo VIH. PREZISTA deve ser sempre administrado oralmente com 100 mg de ritonavir, como potenciador farmacocinético, e em associação com outros medicamentos anti-retrovirais. Dever-se-á, consequentemente, consultar o Resumo das Características do Medicamento de ritonavir antes de instituir uma terapêutica com PREZISTA. Adultos: A dose recomendada de PREZISTA é de 600 mg duas vezes por dia administrados com ritonavir 100 mg, duas vezes por dia, e com alimentos. O tipo de alimentos não afecta a exposição ao darunavir (ver secções 4.4, 4.5 e 5.2). Crianças e adolescentes: PREZISTA não é recomendado em crianças e adolescentes devido à ausência de dados de segurança, eficácia e farmacocinética. Idosos: A informação disponível nesta população é limitada (ver secções 4.4 e 5.2). Compromisso hepático: O darunavir é metabolizado pelo sistema hepático. Portanto, o compromisso hepático grave pode provocar um aumento da exposição ao darunavir e um agravamento do seu perfil de segurança. Assim, PREZISTA deve ser utilizado com precaução em doentes com compromisso hepático ligeiro (Classe A de Child-Pugh) ou moderado (Classe B de Child-Pugh) e não deve ser
2

utilizado em doentes com compromisso hepático grave (Classe C de Child-Pugh), (ver secções 4.3, 4.4 e 5.2). Compromisso renal: Não é necessário efectuar ajustes posológicos em doentes com compromisso renal (ver secções 4.4 e 5.2). No caso de uma dose de PREZISTA e /ou ritonavir ter sido omitida durante 6 horas, deve recomendar-se ao doente que tome a dose prescrita de PREZISTA e ritonavir, com alimentos, assim que for possível. Se foi verificado que o doente não tomou a dose recomendada passadas mais do que 6 horas sobre a hora recomendada da toma, o doente não deve tomar a dose omissa, mas deve prosseguir com o esquema posológico inicial. Esta recomendação baseia-se na semivida de 15 h de darunavir na presença de ritonavir e no intervalo de administração recomendado de aproximadamente 12 horas. 4.3 Contra-indicações Hipersensibilidade à substância activa ou a qualquer um dos excipientes. Doentes com compromisso hepático grave (Classe C de Child-Pugh). A rifampicina não deve ser administrada com PREZISTA porque a co-administração pode causar uma elevada diminuição nas concentrações de darunavir, o que pode por seu lado diminuir significativamente o efeito terapêutico de darunavir (ver secção 4.5.). Produtos à base de plantas, contendo hipericão (Hypericum perforatum) não devem ser utilizados em simultâneo com PREZISTA devido ao risco de diminuição das concentrações plasmáticas e redução do efeito clínico do darunavir (ver secção 4.5). Está contra-indicada a administração simultânea de PREZISTA com 100 mg de ritonavir com substâncias activas cuja depuração está altamente dependente do CYP3A4 e para as quais as elevadas concentrações plasmáticas estão associadas com acontecimentos graves e/ou potencialmente fatais. Estas substâncias activas incluem por exemplo: anti-arrítmicos (amiodarona, bepridilo, quinidina, lidocaína sistémica), anti-histamínicos (astemizol, terfenadina), alcalóides da ergotamina (nomeadamente, di-hidroergotamina, ergonovina, ergotamina, metilergonovina), agentes da motilidade gástrica (cisaprida), neurolépticos (pimozida, sertindol), sedativos/hipnóticos (triazolam, midazolam administrado por via oral) e inibidores da reductase HMG-CoA (sinvastatina e lovastatina) (ver secção 4.5). 4.4 Advertências e precauções especiais de utilização Os doentes devem ser informados de que a actual terapêutica anti-retroviral não cura a infecção pelo VIH, não estando provado que previna a transmissão do VIH, por contacto sexual ou através do sangue. Devem continuar a adoptar-se medidas profilácticas adequadas. PREZISTA só deve ser utilizado em associação com 100 mg de ritonavir como potenciador da farmacocinética (ver secção 5.2). O aumento da dose de ritonavir em relação ao que é recomendado na secção 4.2, não afectou significativamente as concentrações de darunavir e não é recomendado. Idosos: Dada a informação limitada disponível sobre a utilização de PREZISTA em doentes com idade igual ou superior a 65 anos, recomenda-se precaução na administração de PREZISTA em doentes idosos, considerando a maior frequência de casos de redução da função hepática e de doenças e outras terapêuticas concomitantes (ver secções 4.2 e 5.2). Darunavir liga-se predominantemente à glicoproteína alfa-1-ácido. Esta ligação à proteína depende da concentração e indica saturação da ligação. Portanto, não se pode excluir o deslocamento da proteína de medicamentos que se ligam fortemente à glicoproteína alfa-1-ácido.
3

O darunavir contém um componente sulfonamida. PREZISTA deve ser utilizado com precaução em doentes com alergia à sulfonamida. Durante o programa de desenvolvimento clínico foram notificadas erupções cutâneas graves, incluindo eritema multiforme e síndrome de Stevens-Johnson. O tratamento com PREZISTA deve ser suspenso em caso de desenvolvimento de exantema grave. Doentes com condições clínicas co-existentes Doença hepática A segurança e eficácia de PREZISTA não foram estabelecidas em doentes com alterações hepáticas graves, pelo que PREZISTA está contra-indicado em doentes com compromisso hepático grave. PREZISTA deve ser utilizado com precaução em doentes com compromisso hepático ligeiro a moderado (ver secções 4.2, 4.3 e 5.2). Doentes com hepatite crónica B e C e tratados com combinações de terapêuticas anti-retrovirais têm um risco aumentado de sofrer acontecimentos adversos hepáticos graves e potencialmente fatais. Em caso de tratamento antiviral concomitante para a hepatite B e C devem ter-se em conta as informações constantes do resumo das características do medicamento daqueles fármacos. Doentes com disfunção hepática pré-existente incluindo hepatite crónica têm com maior frequência anomalias da função hepática durante o tratamento com combinações de anti-retrovirais, devendo ser monitorizados de acordo com a prática clínica. Se existir evidência de agravamento de doença hepática neste doentes, deve ser considerada a interrupção ou suspensão do tratamento. Doença renal Não são necessárias precauções especiais nem ajustes de dose em doentes com insuficiência renal. Uma vez que o darunavir e o ritonavir apresentam uma elevada ligação às proteínas plasmáticas, é improvável que sejam significativamente eliminados por hemodiálise ou por diálise peritoneal. Portanto, não são necessárias, nestes doentes, precauções especiais nem ajustes de dose (ver secções 4.2 e 5.2). Doentes hemofílicos Tem sido referido aumento dos casos de hemorragia, incluindo hematomas cutâneos espontâneos e hemartrose, em doentes com hemofilia tipo A e B tratados com IPs. Em alguns doentes foi administrado adicionalmente factor VIII. Em mais de metade dos casos notificados, o tratamento com IPs foi mantido ou reinstituído quando houve suspensão da terapêutica. Tem sido sugerida a existência de uma relação causal, mas o mecanismo de acção não se encontra esclarecido. Os doentes hemofílicos deverão, portanto, estar informados sobre a possibilidade de se verificar aumento dos casos de hemorragia. Diabetes Mellitus/Hiperglicemia Têm sido notificados casos de diabetes mellitus de novo, hiperglicemia ou exacerbação de diabetes mellitus preexistente em doentes submetidos a uma terapêutica anti-retroviral, incluindo IPs. Em alguns destes doentes a crise de hiperglicemia foi grave, associando-se, também, em alguns casos, a cetoacidose. Muitos doentes apresentavam condições médicas concomitantes, algumas das quais requeriam terapêutica com fármacos que têm sido associados ao desenvolvimento de diabetes mellitus ou hiperglicemia. Redistribuição de tecido adiposo e alterações metabólicas A terapêutica anti-retroviral combinada tem sido associada a redistribuição da gordura corporal (lipodistrofia) em doentes com infecção por VIH. Desconhecem-se presentemente quais as consequências a longo prazo destes efeitos. O conhecimento sobre o respectivo mecanismo é incompleto. Foi formulada a hipótese da existência de uma relação entre lipomatose visceral e IPs e lipoatrofia e NRTIs. Um risco maior de lipodistrofia tem sido associado a factores individuais, nomeadamente idade avançada e factores relacionados com os fármacos, tais como duração mais prolongada do tratamento anti-retroviral e perturbações metabólicas associadas. O exame clínico deverá incluir avaliação dos sinais físicos de redistribuição da gordura. Dever-se-á considerar a
4

determinação dos níveis de lípidos séricos e de glicemia em jejum. As dislipidemias devem ser tratadas de acordo com a prática clínica adequada (ver secção 4.8). Osteonecrose Embora a etiologia seja considerada multifactorial (incluindo utilização de corticosteróides, consumo de álcool, imunodepressão grave, índice de massa corporal elevado), foram descritos casos de osteonecrose, particularmente em doentes com doença avançada pelo VIH e/ou longa exposição à terapêutica anti-retroviral combinada . Os doentes devem ser aconselhados a procurar conselho médico se tiverem dores nas articulações, rigidez nas articulações ou dificuldade nos movimentos. Síndrome de reconstituição imunitária Em doentes com infecção pelo VIH com imunodepressão grave aquando da instituição da terapêutica anti-retroviral combinada, poder-se-á desenvolver uma reacção inflamatória a agentes patogénicos oportunistas em fase assintomática ou residual provocando situações clínicas graves ou agravamento dos sintomas. Tipicamente, estas reacções têm sido observadas nas primeiras semanas ou meses após a instituição da terapêutica anti-retroviral combinada. São exemplos relevantes a retinite a citomegalovírus, as infecções sistémicas ou localizadas a micobactérias e a pneumonia causada pelo Pneumocystis jiroveci (anteriormente conhecido como Pneumocystis carinii). Deve proceder-se à avaliação de quaisquer sintomas inflamatórios e à instituição de terapêutica, quando necessário. Adicionalmente, foi observada a reactivação de herpes simplex e herpes zoster, em ensaios clínicos com PREZISTA administrado em associação com 100 mg de ritonavir. Interacções medicamentosas Vários estudos de interacção foram realizados com darunavir em doses inferiores às recomendadas. Os efeitos da co-administração de medicamentos podem estar subestimados e recomendando-se monitorização clínica da segurança. Consultar a secção 4.5, para informação completa sobre interacções com outros medicamentos. PREZISTA comprimidos contém o corante amarelo sunset FCF (E110) que pode causar reacções alérgicas. 4.5 Interacções medicamentosas e outras formas de interacção Tanto o darunavir como o ritonavir são inibidores da isoforma CYP3A4. A co-administração de darunavir e ritonavir com fármacos que são essencialmente metabolizados pelo CYP3A4 poderá induzir aumentos das concentrações plasmáticas dos referidos fármacos, o que poderá potenciar ou prolongar os respectivos efeitos terapêuticos e reacções adversas. PREZISTA, administrado em associação com 100 mg de ritonavir não deve ser co-administrado com fármacos cuja depuração seja altamente dependente do CYP3A4 e para os quais a elevação das concentrações plasmáticas está associada a acontecimentos graves e/ou potencialmente fatais (índice terapêutico estreito). Nestes medicamentos incluem-se a amiodarona, o bepridil, a quinidina, a lidocaína sistémica, o astemizol, a terfenadina, o midazolam, o triazolam, a cisaprida, a pimozida, o sertindol, sinvastatina, lovastatina e os alcalóides da ergotamina (por ex., ergotamina, dihidroergotamina, ergonovina e metilergonovina) (ver secção 4.3). O efeito global no aumento da farmacocinética pelo ritonavir foi de aproximadamente 14 vezes na exposição sistémica de danuravir quando foi administrado, por via oral, uma dose única de 600 mg de darunavir em associação com 100 mg de ritonavir duas vezes por dia. Portanto, PREZISTA só pode ser administrado em associação com 100 mg de ritonavir como potenciador farmacocinético (ver secções 4.4 e 5.2). Vários estudos de interacção (ver quadros abaixo) foram realizados com doses mais baixas de darunavir do que as recomendadas. Os efeitos nos medicamentos associados podem encontrar-se subestimados, recomendando-se monitorização clínica de segurança. Medicamentos que afectam a exposição darunavir/ritonavir
5

Darunavir e ritonavir são metabolizados pelo CYP3A. É de esperar que os medicamentos que induzem a actividade do CYP3A aumentem a depuração do darunavir e ritonavir, resultando na diminuição nas concentrações plasmáticas de darunavir e ritonavir (ex: rifampicina, rifabutina, hipericão, lopinavir). A associação de darunavir e ritonavir com outros medicamentos que inibem o CYP3A, pode diminuir a depuração de darunavir e ritonavir, o que pode resultar no aumento nas concentrações plasmáticas de darunavir e ritonavir (ex: indinavir, azóis sistémicos, tais como o cetonazol e clotrimazol). Estas interacções estão descritas nos quadros de interacção abaixo. Quadro de interacção Interacções entre darunavir e ritonavir e inibidores da protease, agentes anti-retrovirais para além dos inibidores da protease e outros medicamentos não-anti-retrovirais estão descritos nos quadros abaixo (aumento é indicado como “↑”, diminuição como “↓”, sem alteração com “↔”, duas vezes ao dia como “b.i.d.” e uma vez ao dia como “q.d”).
Interacções – Darunavir/ritonavir com inibidores da protease
A eficácia e segurança na utilização de PREZISTA com 100 mg de ritonavir e com outros inibidores da protease (ex: (fos)amprenavir, nelfinavir e tipranavir) não foi avaliada em doentes infectados pelo VIH. De um modo geral, a terapêutica dupla com inibidores da protease não é recomendada. Medicamento
associado Dose do
medicamento associado
(mg)
Dose de darunavir/ritonavir
(mg)
Medicamento avaliado
AUC Cmín
Lopinavir/ ritonavir
400/100 b.i.d. 300/100 b.i.d. Lopinavir Darunavir
↑37% ↓53%
↑72% ↓65%
Não se recomenda a co-administração de PREZISTA associado com 100 mg de ritonavir com lopinavir/ritonavir.
Saquinavir 1000 b.i.d. 400/100 b.i.d. Darunavir ↓26% ↓42% O estudo com saquinavir não mostrou nenhum efeito significativo do darunavir no
saquinavir. Não se recomenda a co-administração de PREZISTA associado com 100 mg de ritonavir com saquinavir.
Indinavir 800 b.i.d. 400/100 b.i.d. Indinavir ↑23% ↑125% Darunavir ↑24% ↑44% Quando utilizado em co-administração com PREZISTA associado com 100 mg de
ritonavir, em casos de intolerância, pode ser necessário um ajuste da dose de indinavir de 800 mg b.i.d. para 600 mg b.i.d..
Atazanavir 300 q.d. 400/100 b.i.d. Atazanavir ↔ ↔ Darunavir ↔ ↔ Darunavir/ritonavir não afectou significativamente a exposição ao atazanavir, no
entanto o IC de 90% para Cmín foi de 99-234%. Atazanavir pode ser utilizado com PREZISTA, administrado em associação com 100 mg de ritonavir.
Interacções – Darunavir/ritonavir com fármacos anti-retrovirais que não sejam inibidores da protease
Medicamento
associado Dose do
medicamento associado
(mg)
Dose de darunavir/ritonavir
(mg)
Medicamento avaliado
AUC Cmín
Efavirenz 600 q.d. 300/100 b.i.d. Efavirenz ↑21% ↑17% Darunavir ↓13% ↓31%
6

Efavirenz diminuiu as concentrações plasmáticas de darunavir como resultado da indução de CYP3A4. Darunavir/ritonavir aumenta as concentrações plasmáticas de efavirenz como resultado da inibição de CYP3A4. Está indicada a monitorização clínica para a toxicidade do sistema nervoso central, associado com o aumento na exposição ao efavirenz quando PREZISTA administrado em associação com 100 mg de ritonavir é co-adimistrado com efavirenz.
Nevirapina 200 b.i.d. 400/100 b.i.d. Nevirapina ↑27% ↑47% Darunavir ↔ ↔ Darunavir/ritonavir aumenta as concentrações plasmáticas de nevirapina como
consequência da inibição de CYP3A4. Uma vez que esta diferença não é considerada clinicamente relevante, a associação de PREZISTA, administrado em associação com 100 mg de ritonavir, com nevirapina, não requer ajustes posológicos.
Tenofovir 300 q.d. 300/100 b.i.d. Tenofovir ↑22% ↑37% Darunavir ↔ ↔ Como mecanismo de acção para o aumento nas concentrações plasmáticas de
tenofovir, propôs-se o efeito de ritonavir no transporte MDR-1 nos túbulos renais. A monitorização da função renal pode estar indicada quando PREZISTA administrado em associação com 100 mg de ritonavir, é co-administrado com tenofovir, particularmente em doentes com doença renal ou sistémica, ou em doentes medicados com agentes nefrotóxicos.
Zidovudina Didanosina Zalcitabina Entricitabina Estavudina Lamivudina Abacavir
Com base nas diferentes vias de eliminação dos outros NRTIs zidovudina, zalcitabina, entricitabina, stavudina, lamivudina, que são essencialmente excretados por via renal, e didanosina e abacavir para os quais o metabolismo não é mediado pelo CYP450, não é previsível a ocorrência de interacções medicamentosas entre estes fármacos e PREZISTA administrado em associação com 100 mg de ritonavir.
Interacções – Darunavir/ritonavir com medicamentos não anti-retrovirais co-administrados
Medicamento associado
Dose do medicamento
associado (mg)
Dose de darunavir/ritonavir
(mg)
Medicamento avaliado
AUC Cmín
Antiarrítmicos Digoxina 0,4 mg dose
única 600/100 b.i.d. Digoxina ↑60% ND
Darunavir/ritonavir aumenta as concentrações plasmáticas de digoxina. A inibição da Pgp pode ser uma explicação. Como a digoxina tem uma margem terapêutica estreita, recomenda-se prescrever inicialmente a dose mais baixa no caso da digoxina ser administrada a doentes sob terapêutica de darunavir/ritonavir. A dose de digoxina deve ser cuidadosamente titulada a fim de se obter o efeito clínico desejado enquanto se avalia o estado clínico global do doente.
Antibióticos Claritromicina 500 b.i.d. 400/100 b.i.d. Claritromicina ↑57% ↑174% Darunavir ↔ ↔ Darunavir/ritonavir aumenta as concentrações plasmáticas da claritromicina como
resultado da inibição do CYP3A4 e possível inibição da Pgp. Não se detectaram concentrações do metabolito 14-OH-claritromicina. Recomenda-se monitorização clínica e precaução. Em doentes com insuficiência renal deve ponderar-se a redução de dose de claritromicina.
Anticoagulantes
7

Varfarina As concentrações de varfarina poderão ser afectadas quando co-administrada com darunavir/ritonavir. Recomenda-se monitorização do Quociente Internacional Normalizado (INR) quando a varfarina é associada ao PREZISTA, administrado em associação com 100 mg de ritonavir.
Anticonvulsivantes Fenobarbital Fenitoína Carbamazepina
O fenobarbital, a fenitoína e a carbamazepina são indutores das isoenzimas CYP450. PREZISTA, administrado em associação com 100 mg de ritonavir não deve ser utilizado em associação com estes medicamentos, uma vez que a sua co-administração poderá provocar reduções significativas das concentrações plasmáticas de darunavir.
Antifúngicos Voriconazol
A associação de voriconazol e darunavir, administrado em associação com 100 mg de ritonavir, não foi estudada. Voriconazol é metabolizado pelas isoenzimas do citocromo P450, CYP2C19, CYP2C9 E CYP3A4. O ritonavir que pode induzir algumas destas isoenzimas, pode diminuir as concentrações plasmáticas do voriconazol. Voriconazol não deve ser co-administrado com PREZISTA, administrado em associação com 100 mg de ritonavir, excepto se a avaliação da relação benefício/risco justificar a utilização de voriconazol.
Cetoconazol 200 b.i.d. 400/100 b.i.d. Cetoconazol Darunavir
↑212% ↑42%
↑868% ↑73%
Cetoconazol é um inibidor potente, bem como, substracto do CYP3A4. Recomenda-se monitorização clínica e precaução. Quando a co-administração de cetoconazol é necessária, a dose diária de cetoconazol não deve exceder 200 mg.
Itraconazol Itraconazol, à semelhança do cetoconazol, é um potente inibidor, bem como substracto do CYP3A4. A utilização sistémica concomitante de itraconazol e darunavir, administrado em associação com 100 mg de ritonavir, pode aumentar as concentrações plasmáticas de darunavir. Simultaneamente, as concentrações plasmáticas de itraconazol podem ser aumentadas pelo darunavir, administrado em associação com 100 mg de ritonavir. Recomenda-se monitorização clínica e precaução. Quando a co-administração de itraconazol é necessária, a dose diária de itraconazol não deve exceder 200 mg.
Clotrimazol A utilização sistémica concomitante de clotrimazol e darunavir, administrado em associação com 100 mg de ritonavir, pode aumentar as concentrações plasmáticas de darunavir. Isto foi confirmado usando um modelo farmacocinético de população. O aumento do valor da mediana da AUC24h de darunavir para os doentes medicados com clotrimazol relativamente à mediana global foi de 33%. Recomenda-se monitorização clínica e precaução quando a co-administração de clotrimazol é necessária.
Bloqueadores da entrada de cálcio Felodipina Nifedipina Nicardipina
Darunavir e ritonavir inibem o CYP3A4 e como resultado é de esperar um aumento nas concentrações plasmáticas dos antagonistas da entrada de cálcio, os quais são substratos do CYP3A4. Recomenda-se monitorização dos efeitos terapêuticos e adversos quando estes medicamentos são administrados em associação com PREZISTA e com 100 mg de ritonavir.
Inibidores da HMG-CoA redutase Lovastatina Sinvastatina
A lovastatina e a sinvastatina, são altamente dependentes do metabolismo da CYP3A4, sendo, portanto, de esperar que se registem aumentos marcados das concentrações plasmáticas quando co-administrados com darunavir, administrado em associação com 100 mg de ritonavir. Isto poderá causar miopatia, incluindo rabdomiólise. Como tal, é contra-indicada a utilização concomitante de PREZISTA, administrado em associação com 100 mg de ritonavir com lovastatina e sinvastatina (ver secção 4.3).
Atorvastatina 10 q.d. 300/100 b.i.d. Atorvastatina ↑ 3 a 4 ↑ 3 a 4
8
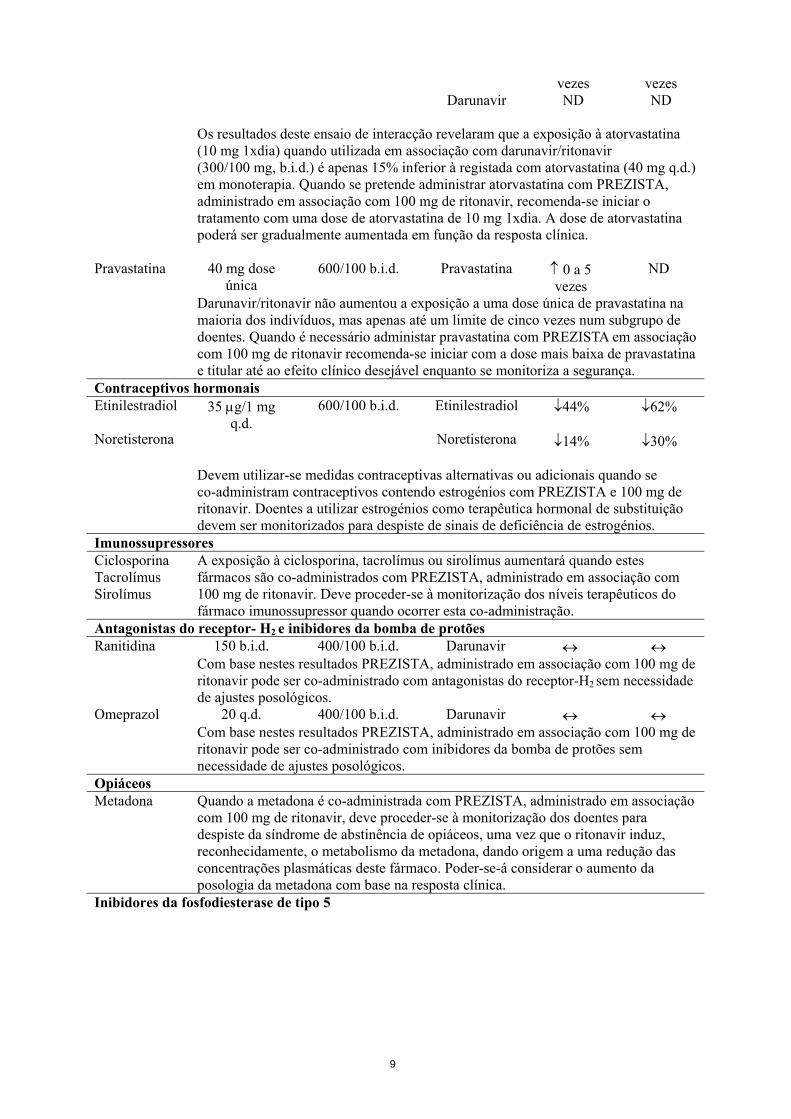
Darunavir
vezes ND
vezes ND
Os resultados deste ensaio de interacção revelaram que a exposição à atorvastatina
(10 mg 1xdia) quando utilizada em associação com darunavir/ritonavir (300/100 mg, b.i.d.) é apenas 15% inferior à registada com atorvastatina (40 mg q.d.) em monoterapia. Quando se pretende administrar atorvastatina com PREZISTA, administrado em associação com 100 mg de ritonavir, recomenda-se iniciar o tratamento com uma dose de atorvastatina de 10 mg 1xdia. A dose de atorvastatina poderá ser gradualmente aumentada em função da resposta clínica.
Pravastatina 40 mg dose única
600/100 b.i.d. Pravastatina ↑ 0 a 5 vezes
ND
Darunavir/ritonavir não aumentou a exposição a uma dose única de pravastatina na maioria dos indivíduos, mas apenas até um limite de cinco vezes num subgrupo de doentes. Quando é necessário administar pravastatina com PREZISTA em associação com 100 mg de ritonavir recomenda-se iniciar com a dose mais baixa de pravastatina e titular até ao efeito clínico desejável enquanto se monitoriza a segurança.
Contraceptivos hormonais Etinilestradiol Noretisterona
35 μg/1 mg q.d.
600/100 b.i.d. Etinilestradiol
Noretisterona
↓44%
↓14%
↓62%
↓30%
Devem utilizar-se medidas contraceptivas alternativas ou adicionais quando se co-administram contraceptivos contendo estrogénios com PREZISTA e 100 mg de ritonavir. Doentes a utilizar estrogénios como terapêutica hormonal de substituição devem ser monitorizados para despiste de sinais de deficiência de estrogénios.
Imunossupressores Ciclosporina Tacrolímus Sirolímus
A exposição à ciclosporina, tacrolímus ou sirolímus aumentará quando estes fármacos são co-administrados com PREZISTA, administrado em associação com 100 mg de ritonavir. Deve proceder-se à monitorização dos níveis terapêuticos do fármaco imunossupressor quando ocorrer esta co-administração.
Antagonistas do receptor- H2 e inibidores da bomba de protões Ranitidina 150 b.i.d. 400/100 b.i.d. Darunavir ↔ ↔ Com base nestes resultados PREZISTA, administrado em associação com 100 mg de
ritonavir pode ser co-administrado com antagonistas do receptor-H2 sem necessidade de ajustes posológicos.
Omeprazol 20 q.d. 400/100 b.i.d. Darunavir ↔ ↔ Com base nestes resultados PREZISTA, administrado em associação com 100 mg de
ritonavir pode ser co-administrado com inibidores da bomba de protões sem necessidade de ajustes posológicos.
Opiáceos Metadona Quando a metadona é co-administrada com PREZISTA, administrado em associação
com 100 mg de ritonavir, deve proceder-se à monitorização dos doentes para despiste da síndrome de abstinência de opiáceos, uma vez que o ritonavir induz, reconhecidamente, o metabolismo da metadona, dando origem a uma redução das concentrações plasmáticas deste fármaco. Poder-se-á considerar o aumento da posologia da metadona com base na resposta clínica.
Inibidores da fosfodiesterase de tipo 5
9

Sildenafil Vardenafil Tadalafil
Num ensaio de interacção, observou-se uma exposição sistémica comparável ao sildenafil após a administração de uma dose única de 100 mg de sildenafil isoladamente e de uma dose única de 25 mg de sildenafil co-administrada com darunavir/ritonavir (400/100 mg, b.i.d.). Recomenda-se precaução ao administrar inibidores da fosfodiesterase de tipo 5 concomitantemente com PREZISTA, administrado em associação com 100 mg de ritonavir. Caso esteja indicada a utilização concomitante de PREZISTA, administrado em associação com 100 mg de ritonavir com sildenafil, vardenafil ou tadalafil, recomenda-se a administração de sildenafil numa dose única máxima de 25 mg em 48 horas, vardenafil numa dose única máxima de 2,5 mg dose em 72 horas ou tadalafil numa dose única máxima de 10 mg em 72 horas.
Rifamicinas Rifabutina Rifabutina é um indutor e um substrato das isoenzimas CYP450. Espera-se que a
utilização concomitante de rifabutina, darunavir e ritonavir aumente a exposição à rifabutina e reduza a exposição ao darunavir; não se recomenda a administração concomitante de uma dose padronizada de rifabutina, pois pode estar associada a intolerância à rifabutina. Quando indicado, recomenda-se a administração da rifabutina numa posologia de 150 mg em dias alternados quando associada a PREZISTA, administrado em associação com 100 mg de ritonavir. Devem considerar-se as orientações oficiais sobre o tratamento adequado da tuberculose em doentes infectados pelo VIH.
Rifampicina A rifampicina é um potente indutor do metabolismo do CYP450. PREZISTA, administrado em associação com 100 mg de ritonavir não pode ser utilizado em associação com rifampicina, uma vez que a co-administração poderá provocar reduções significativas das concentrações plasmáticas de darunavir (ver secção 4.3).
Inibidores Selectivos da Recaptação da Serotonina (SSRIs) Paroxetina 20 q.d. 400/100 b.i.d. Paroxetina
Darunavir ↓39%
↔ ↓37%
↔ Sertralina 50 q.d. 400/100 b.i.d. Sertralina
Darunavir ↓49%
↔ ↓49%
↔ Quando se co-administram SSRIs com PREZISTA e ritonavir, a abordagem
recomendada consiste numa titulação da dose do SSRI com base na avaliação clínica da resposta antidepressora. Além disso, deve proceder-se à monitorização dos doentes que estão a ser tratados com uma dose estável de sertralina ou paroxetina e que iniciam um tratamento com PREZISTA, administrado em associação com 100 mg de ritonavir para avaliação da resposta antidepressora.
Esteróides Propionato de Fluticasona
Num ensaio clínico onde foi administrado a doentes saudáveis ritonavir 100 mg cápsulas, duas vezes ao dia, com 50 μg de propionato de fluticasona intranasal, 4 vezes ao dia e durante 7 dias, as concentrações plasmáticas de propionato de fluticasona aumentaram significativamente, enquanto os níveis intrínsecos de cortisol diminuíram aproximadamente 86% (intervalo de confiança de 90%, 82-89%). Quando o propionato de fluticasona é inalado, é esperado o aumento dos seus efeitos. Efeitos sistémicos dos corticosteróides, incluindo a síndrome de Cushing ou supressão supra-renal, foram notificados em doentes a receber ritonavir e propionato de fluticasona inalado ou administrado por via intranasal; tal podo ocorrer com outros corticosteróides metabolizados via CYP450 3A, como, por exemplo, o budesonido. Assim, a utilização concomitante de PREZISTA, administrado em associação com 100 mg de ritonavir, e este glucocorticóide não é recomendada, excepto se o potencial benefício do tratamento for superior ao risco dos efeitos sistémicos dos corticosteróides. Deve considerar-se a redução de dose deste glucocorticóide, com rigorosa vigilância dos seus efeitos locais e sistémicos ou mudança para um glucocorticóide que não seja substrato do CYP3A4 (por exemplo a beclometasona). Adicionalmente, em caso de suspensão da terapêutica com glucocorticóide, deve ser efectuada uma redução progressiva de dose, por um
10

período mais prolongado. Não são ainda conhecidos os efeitos do aumento da exposição sistémica a fluticasona, nos níveis plasmáticos do ritonavir.
Dexametasona A dexametasona sistémica induz CYP3A4, podendo, como tal, reduzir a exposição ao darunavir. Logo, recomenda-se precaução na utilização desta associação.
Outros Hipericão
PREZISTA, administrado em associação com 100 mg de ritonavir não deve ser utilizado concomitantemente com produtos que contenham hipericão (Hypericum perforatum) uma vez que a co-administração pode provocar reduções significativas das concentrações plasmáticas de darunavir e também de ritonavir. Isto é devido à indução de enzimas metabolizadoras pelo hipericão. Se um doente já estiver a tomar hipericão, interrompa a administração de hipericão e se possível, verifique as cargas virais. A exposição ao darunavir (e também a exposição a ritonavir) pode aumentar ao interromper a administração de hipericão. O efeito indutor pode persistir durante pelo menos 2 semanas após a interrupção do tratamento com hipericão (ver secção 4.3).
4.6 Gravidez e aleitamento Gravidez Não existem estudos adequados e bem controlados com darunavir na mulher grávida. Os estudos realizados em animais não mostraram efeitos nocivos directos no que diz respeito à gravidez, ao desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal (ver secção 5.3). PREZISTA, administrado em associação com 100 mg de ritonavir só deve ser utilizado durante a gravidez se o benefício potencial justificar o risco potencial. Aleitamento É recomendado que mulheres infectadas pelo VIH não amamentem os filhos, em nenhuma circunstância, por forma a evitar a transmissão do VIH. Desconhece-se se darunavir é excretado no leite humano. Os estudos realizados no rato demonstraram que darunavir é excretado no leite e doses elevadas (1000 mg/kg) causaram toxicidade. As mães deverão ser aconselhadas a não amamentar se estiverem a ser tratadas com PREZISTA. Fertilidade Não existem dados sobre o efeito de darunavir na fertilidade. Não foi demonstrado qualquer efeito de darunavir sobre o acasalamento ou a fertilidade no rato (ver secção 5.3). 4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas Não foram efectuados estudos sobre os efeitos de PREZISTA em associação com ritonavir sobre a capacidade de conduzir e utilizar máquinas. Foram, no entanto, referidos casos de tonturas em alguns doentes durante o tratamento com regimes contendo PREZISTA, administrado em associação com 100 mg de ritonavir, pelo que se deverá ter presente este facto ao avaliar a capacidade do doente conduzir e utilizar máquinas (ver secção 4.8). 4.8 Efeitos indesejáveis Os dados de segurança são baseados em estudos com formulações experimentais e comerciais de darunavir. A biodisponibilidade da formulação comercial de darunavir foi aproximadamente 20% superior relativamente às formulações experimentais. A segurança de darunavir foi avaliada num número limitado de doentes que tomaram medicamentos que poderiam interactuar durante os ensaios clínicos. A segurança de darunavir não foi avaliada em doentes que tomavam NNRTIs. Portanto, os dados obtidos não podem ser todos aplicáveis à utilização generalizada de darunavir. Os dados de segurança de PREZISTA 600 mg, administrado em associação com 100 mg de ritonavir, duas vezes ao dia, derivam de dados de 2 ensaios clínicos de fase IIb, ainda a decorrer,
11

complementados com a informação de 2 ensaios abertos, num total de 458 doentes que iniciaram tratamento com a dose recomendada (doentes de novo). Quarenta por cento destes doentes apresentaram pelo menos uma reacção adversa relacionada com o fármaco. Nos doentes de novo, as reacções adversas notificadas com maior frequência (≥ 2%) com um grau de gravidade mínimo de 2, consideradas possivelmente relacionadas com PREZISTA, administrado em associação com 100 mg de ritonavir, consistiram em diarreia (2,6%), vómitos (2,2%) e hipertrigliceridemia (2,0%). A maioria das reacções adversas (RAs) notificadas durante o tratamento com PREZISTA, administrado em associação com 100 mg de ritonavir, duas vezes ao dia, tinha uma gravidade de grau 1 a 2. As reacções adversas de qualquer grau mais frequentemente descritas foram náusea (7,2%), diarreia (6,6%) e cefaleia (3,3%). Todas as outras reacções adversas foram notificadas em menos de 3% dos doentes. Um por cento dos doentes suspendeu o tratamento devido a reacções adversas. As reacções adversas são especificadas por classe de sistema de orgãos (CSO) e por frequência. Os efeitos indesejáveis são apresentados por ordem decrescente de gravidade dentro de cada classe de frequência. Em termos de frequência as reacções são definidas como frequentes (≥ 1/100 a < 1/10) e pouco frequentes (≥ 1/1000 a < 1/100). As reacções adversas referidas em doentes de novo (grau 1-4) estão resumidas a seguir. A frequência foi calculada com base em reacções adversas, consideradas pelos investigadores como atribuíveis (no mínimo, relação causal possível) ao PREZISTA, administrado em associação com 100 mg de ritonavir. CSO Reacção Adversa Frequência Exames complementares de diagnóstico
aumento da alanina aminotransferase, aumento da creatinina sérica, aumento da amilase sérica, aumento da aspartato aminotrasferase, aumento da gama-glutamiltransferase, aumento da lipase, aumento da glucose sérica, aumento da fosfatase alcalina sérica, aumento da ureia sérica,
pouco frequente
Cardiopatias enfarte do miocárdio, alterações no electrocardiograma, taquicardia, dilatação atrial
pouco frequente
Doenças do sangue e do sistema linfático
neutropenia, trombocitopenia pouco frequente
Doenças do sistema nervoso cefaleia, tonturas neuropatia periférica, parestesia, hipoestesia, perturbação da memória, sonolência, acidente isquémico transitório, alterações na atenção, coordenação anormal, síncope
Frequente pouco frequente
Afecções oculares queratoconjuntivite sicca pouco frequente Afecções do ouvido e do labirinto
Vertigens pouco frequente
Doenças respiratórias, torácicas e do mediastino
dispneia, tosse, soluços pouco frequente
12

Doenças gastrointestinais vómitos, diarreia, náuseas, dor abdominal, obstipação, flatulência, distensão abdominal, dispepsia boca seca, hipersecreção salivar, úlceração da língua
frequente pouco frequente
Doenças renais e urinárias Insuficiência renal aguda, insuficiência renal, nefrolitíase, disúria, noctúria, proteinúria
pouco frequente
Afecções dos tecidos cutâneos e subcutâneas
lipoatrofia, eritema multiforme*, dermatite medicamentosa, inflamação cutânea, sudorese nocturna, hiperhidrose, alopécia, erupção cutânea maculopapular, dermatite alérgica, eczema, erupção cutânea tóxica, pele seca, prurido, edema facial, urticária
pouco frequente
Afecções musculosqueléticas e dos tecidos conjuntivos
artralgia, dor nas extremidades, mialgia, osteopenia, osteoporose, cãibras musculares
pouco frequente
Doenças endócrinas hipotiroidismo pouco frequente Doenças do metabolismo e da nutrição
hipertrigliceridemia, anorexia diabetes mellitus, hipercolesterolemia, polidipsia, hiperlipidemia, redistribuição da gordura, redução do apetite, aumento de peso, hiponatremia, obesidade
frequente pouco frequente
Infecções e infestações Foliculite pouco frequente Vasculopatias Hipertensão, rubor pouco frequente Perturbações gerais e alterações no local de administração
astenia, fadiga pirexia, hipertermia, edema periférico, rigidez
frequente pouco frequente
Doenças dos órgãos genitais e da mama
ginecomastia, disfunção eréctil pouco frequente
Perturbações do foro psiquiátrico
insónia ansiedade, estado confusional, desorientação, irritabilidade, alteração do humor, pesadelos.
frequente pouco frequente
* notificada com outra dose Alterações laboratoriais As alterações laboratoriais clínicas decorrentes do tratamento (Grau 3 ou 4) observadas em doentes de novo e referidas numa percentagem igual ou superior a 2% dos indivíduos consistiram em aumento dos níveis de triglicéridos (8,6%), da amilase pancreática (6,6%), do colesterol total (4,9%), da gama-glutamiltransferase (Gama GT) (3,8%), do tempo tromboplastina parcial (PTT) (3,6%), lipase pancreática (3,5%), alaninaminotransferase (ALT) (2,4%), da aspartato aminotransferase (AST) (2,2%) e redução da contagem de leucócitos (6,4%), neutrófilos (4,7%), contagem total absoluta de neutrófilos (4,2%), linfócitos (3,8%). Todas as outras alterações dos parâmetros laboratoriais foram observadas em menos de 2% dos indivíduos.
13

Nos ensaios clínicos com PREZISTA, administrado em associação com 100 mg de ritonavir, têm sido relatados casos graves de erupção cutânea, incluindo Síndrome de Stevens-Johnson. A terapêutica anti-retroviral combinada tem sido associada a redistribuição da gordura corporal (lipodistrofia) em doentes infectados pelo VIH, incluindo perda de gordura subcutânea periférica e facial, aumento da gordura intra-abdominal e visceral, hipertrofia mamária e acumulação de gordura dorsocervical (pescoço de búfalo) (ver secção 4.4). A terapêutica anti-retroviral combinada tem sido igualmente associada a alterações metabólicas, nomeadamente hipertrigliceridemia, hipercolesterolemia, resistência à insulina, hiperglicemia e hiperlactacidemia (ver secção 4.4). Têm sido referidos casos de aumento dos níveis de Creatina-fosfoquinase, mialgia, miosite e, raramente, rabdomiólise durante a utilização de inibidores da protease, particularmente em associação com NRTIs. Foram descritos casos de osteonecrose, particularmente em doentes com factores de risco reconhecidos, SIDA em estadio avançado ou longa exposição à terapêutica anti-retroviral combinada. Desconhece-se a sua frequência (ver secção 4.4). Em doentes com infecção pelo VIH com défice imunitário grave na altura da instituição da terapêutica anti-retroviral combinada poder-se-á desenvolver uma reacção inflamatória a infecções residuais oportunistas ou assintomáticas (ver secção 4.4). Têm sido notificados casos de aumento espontâneo de hemorragias, em doentes hemofílicos, que tomam anti-retrovirais, inibidores da protease. Doentes co-infectados pelo vírus da hepatite B e/ou hepatite C De entre 458 doentes tratados com PREZISTA, administrado em associação com 100 mg de ritonavir, duas vezes ao dia, 59 estavam co-infectados com hepatite B ou C crónica. Doentes co-infectados têm maior probabilidade de registar elevação de transaminases hepáticas, basais e durante o tratamento do que os doentes sem hepatite viral crónica (ver secção 4.4). 4.9 Sobredosagem A experiência humana de sobredosagem aguda com PREZISTA, administrado em associação com 100 mg de ritonavir é limitada. Têm sido administradas a voluntários saudáveis doses únicas até 3200 mg da solução oral de darunavir isoladamente e até 1600 mg da formulação em comprimidos de darunavir em associação com ritonavir sem que se registassem efeitos sintomáticos adversos. Não existe antídoto específico para a sobredosagem com PREZISTA. O tratamento da sobredosagem com PREZISTA consiste em medidas gerais de suporte, incluindo monitorização dos sinais vitais e observação do estado clínico do doente. Quando indicado, deve procurar eliminar-se a substância activa não absorvida por emése ou lavagem gástrica. Pode também administrar-se carvão activado para ajudar à eliminação da substância activa não absorvida. Uma vez que o darunavir apresenta uma elevada ligação às proteínas, é improvável que a diálise seja benéfica na eliminação de uma quantidade considerável da substância activa. 5. PROPRIEDADES FARMACOLÓGICAS 5.1 Propriedades farmacodinâmicas Grupo farmacoterapêutico: Antiviral para administração sistémica, código ATC: J05AE10. Mecanismo de acção
14

O darunavir é um inibidor da protease do VIH-1 (KD de 4.5 x 10-12M). Inibe selectivamente a clivagem das poliproteínas Gag-Pol codificadas pelo VIH em células infectadas pelo vírus, prevenindo assim a formação de partículas víricas maturas infecciosas. Actividade antiviral in vitro O darunavir demonstra actividade contra estirpes laboratoriais e isolados clínicos de VIH-1 e estirpes laboratoriais de VIH-2 em linhas de linfócitos T com infecção aguda, células mononucleares de sangue periférico humano e monócitos/macrófagos humanos, apresentando valores medianos de CE50 entre 1,2 e 8,5 nM (0,7 e 5,0 ng/ml). O darunavir demonstra actividade antiviral in vitro contra um amplo painel de isolados primários de VIH-1 do grupo M (A, B, C, D, E, F, G) e do grupo O, apresentando valores de CE50 compreendidos entre < 0,1 e 4,3 nM. Estes valores de CE50 são muito inferiores a 50% dos limites da concentração de toxicidade celular de 87 µM a > 100 µM. O valor da CE50 de darunavir aumenta em um factor mediano de 5,4 em presença de soro humano. O darunavir revelou actividade antiviral sinérgica quando estudado em associação com os inibidores da protease ritonavir, nelfinavir ou amprenavir e actividade antiviral aditiva quando estudado em associação com os inibidores da protease indinavir, saquinavir, lopinavir, atazanavir ou tipranavir, com os NRTIs zidovudina, lamivudina, zalcitabina, didanosina, stavudina, abacavir, entricitabina ou tenofovir, com os NRTINs nevirapina, delavirdina ou efavirenz e com o inibidor da fusão enfuvirtide. Não foi observado antagonismo entre o darunavir e qualquer um destes fármacos anti-retrovirais. Resistência A selecção in vitro de vírus de estirpes do tipo selvagem do VIH-1, resistentes ao darunavir, demonstrou ser prolongada (até 2 anos). Os vírus seleccionados não conseguiram desenvolver-se em presença de concentrações de darunavir superiores a 220 nM. Os vírus seleccionados nestas condições, que apresentavam menor sensibilidade ao darunavir (limites: 6 – 21 vezes), continham 3 a 6 substituições de aminoácidos no gene da protease. Está em curso a identificação dos factores determinantes da redução da sensibilidade ao darunavir nestes vírus. A selecção in vitro de VIH-1 resistente ao darunavir (limites: alteração de 53 – 641 vezes os valores da CE50) a partir de 9 estirpes do VIH-1 contendo múltiplas mutações associadas a resistência a IPs mostraram que a protease do VIH-1 deverá apresentar, no mínimo, 8 mutações seleccionadas in vitro, do darunavir, para conferir resistência ao vírus (mutação [FC] > 10) relativamente ao darunavir. No POWER 1, 2 e 3 (ver subsecção Experiência clínica) as substituições de aminoácidos desenvolvidas durante o tratamento com PREZISTA administrado em associação com ritonavir (600 mg/100 mg, duas vezes ao dia) em mais de 20% dos isolados foram V32I e I54L. As outras substituições desenvolvidas em 10 a 20% dos isolados foram L33F, I47V e L89V. Resistência cruzada Darunavir apresenta uma redução < 10 vezes da sensibilidade contra 90% de 3309 isolados clínicos resistentes ao amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir e/ou tipranavir, demonstrando que os vírus resistentes à maioria dos IPs continuam a ser sensíveis ao darunavir. Experiência clínica A eficácia de PREZISTA administrado em associação com 100 mg de ritonavir em doentes sem terapêutica anti-retroviral prévia Não existem dados sobre a eficácia de darunavir em associação com 100 mg de ritonavir no tratamento de doentes infectados pelo VIH-1 sem terapêutica anti-retroviral prévia. A eficácia de PREZISTA administrado em associação com 100 mg de ritonavir em doentes previamente submetidos a terapêutica anti-retroviral A evidência da eficácia de PREZISTA, administrado em associação com 100 mg de ritonavir em doentes previamente submetidos a terapêutica fundamenta-se no seguinte: O POWER 1 e POWER 2 são ensaios aleatorizados, controlados, que envolvem uma fase inicial, para determinação da dose e uma segunda fase de longa duração em que todos os doentes aleatorizados para tratamento com PREZISTA, administrado em associação com 100 mg de ritonavir receberam a dose recomendada de 600/100 mg, 2 vezes ao dia.
15

Os doentes infectados pelo VIH-1, considerados elegíveis para participarem nestes ensaios falharam previamente mais do que um regime contendo IP. PREZISTA, administrado em associação com 100 mg de ritonavir combinado com um regime de base optimizado (RBO) foi comparado com um grupo de controlo tratado com um regime de IP(s), seleccionado pelo investigador, em associação com um RBO. O RBO consistia em, pelo menos, 2 NRTIs com ou sem enfuvirtide (ENF). POWER 3: obtiveram-se dados adicionais idênticos sobre a eficácia do PREZISTA, administrado em associação com 100 mg de ritonavir 600/100 mg, 2 vezes por dia juntamente com o RBO, nos doentes submetidos previamente a tratamento que participaram nos ensaios não aleatorizados, TMC114-C215 e TMC114-C208. Análise de resultados às 24 semanas O quadro a seguir apresenta os dados de eficácia das análises realizadas às 24 semanas relativos à dose recomendada de 600 mg de PREZISTA, administrado em associação com 100 mg de ritonavir, 2 vezes por dia dos ensaios agrupados POWER 1 e POWER 2 bem como da análise realizada às 24 semanas do POWER 3. Dados agrupados do POWER 1 e POWER 2 POWER 3 Características basais Mediana de níveis plasmáticos ARNdo VIH-1
4,52 log10 cópias/ml 4,60 log10 cópias/ml
Mediana de contagem de células CD4+
153x106 células/l 115x106 células/l
Resultados PREZISTA/ritonavir 600/100 mg 2 vezes por dia n=131
Controlo n=124
Diferença entre tratamentos
PREZISTA/ritonavir 600/100 mg 2 vezes por dia n=327
Alteração do ARN do VIH-1 log em relação aos valores basais (log10 cópias/ml)a
-1,89 (-2,11; -1,68)f
-0,48 (-0,65; -0,31)f
-1,41 (LSMb) (-1,67; -1,14)f
-1,73 (-1,87; -1,58)f
Alteração da contagem de células CD4+ em relação aos valores basais (x 106/l)c
92 (73; 112)f
17 (0; 35)f
75 (LSMb) (49; 101)f
80 (69; 90)f
ARN do VIH ≥ 1 log10 abaixo dos valores basaisd
92 (70%) (62%; 78%)f
26 (21%) (14%; 28%)f
49% (39%; 60%)e, f
217 (66%) (61%; 71%)f
ARN do VIH < 400 cópias/mld
82 (63%) (54%; 71%)f
23 (19%) (12%; 25%)f
44% (33%; 55%)e, f
193 (59%) (54%; 64%)f
ARN do VIH < 50 cópias/mld
59 (45%) (37%; 54%)f
15 (12%) (7%; 18%)f
33% (23%; 43%)e, f
141 (43%) (38%; 48%)f
a Aos doentes que não completaram o tratamento é atribuída falência: aos doentes que suspenderam prematuramente o tratamento é atribuída uma alteração igual a zero 0.
b As diferenças entre tratamentos baseiam-se nas Médias dos Quadrados Mínimos (MQM) de um modelo ANOVA incluindo os factores de estratificação. Valores de P < 0,001.
c Imputação a LOCF (última observação efectuada) d Imputações de acordo com o algoritmo TLOVR. e Intervalos de confiança acerca das diferenças observadas nas taxas de resposta; valores de P < 0,001; f Iintervalos de confiança de 95%. Genótipo ou fenotipo basais e resultados virológicos No POWER 1, 2 e 3 a presença inicial de 3 ou mais mutações V11I, V32I, L33F, I47V, I50V, I54L ou M, G73S, L76V, I84V ou L89V foi associada à diminuição da resposta virológica ao PREZISTA, administrado em associação com 100 mg de ritonavir. A presença de mutações individuais foi associada com uma mediana de 10 mutações associadas a resistência a IPs da lista de mutações IAS-USA.
16

Resposta a PREZISTA administrado em associação com ritonavir (600/100 mg, 2 vezes por dia) segundo genótipo inicial*: Segundo a análise POWER 1, 2 e 3. Número de mutações iniciais*
Alteração da carga viral log10, às 24 semanas
Proporção de doentes com redução ≥ 1log10, às 24 semanas
Proporção de doentes com < 50 cópias/ml, às 24 semanas
0-2 -2.1 78% 213/274
50% 138/274
3 -1,12 45% 26/58
22% 13/58
≥4 -0,46 27% 11/41
10% 4/41
* Número de mutações da lista de mutações associadas com uma diminuição da resposta a PREZISTA/ritonavir (V11I, V32I, L33F, I47V, I50V, I54L ou M, G73S, L76V, I84V ou L89V)
O fenótipo basal ao darunavir (alteração na susceptibilidade, relativamente à referência) demonstrou ser um factor preditivo dos resultados virológicos. Resposta a PREZISTA administrado em associação com ritonavir (600/100 mg, 2 vezes por dia), por fenótipo basal ao darunavir: Segundo a análise estudos POWER 1, 2 e 3. Fenótipo basal ao darunavir n=349
Proporção de doentes com redução ≥ 1 log10, às 24 semanas
Proporção de doentes com < 50 cópias/ml, às 24 semanas
< 10 82% 201/244
53% 129/244
10 – 40 44% 27/62
26% 16/62
> 40 40% 17/43
14% 6/43
Resultados a longo prazo Adicionalmente, existem dados que confirmam a eficácia a longo prazo de PREZISTA, administrado em associação com ritonavir 600/100 mg 2 vezes por dia, em doentes previamente submetidos a tratamento durante um período até 48 semanas, obtidos na análise agrupada do POWER 1 e POWER 2. No que se refere aos doentes que atingiram a semana 48 ou que interromperam o tratamento antes da análise da semana 48 indicam que a proporção de doentes com pelo menos uma queda de 1 log diminuiu com o tempo (de 69% para 61%); no entanto, a mesma percentagem de doentes apresentaram cargas virais indetectáveis (< 50 cópias de ARN do VIH /ml) na semana 24 e na semana 48 respectivamente (45%). Este medicamento foi sujeito a uma “Autorização de Introdução no Mercado Condicionada”. Isto significa que se aguardam evidências adicionais sobre este medicamento. A Agência Europeia do Medicamento (EMEA) irá rever anualmente nova informação sobre o medicamento e este RCM será actualizado se necessário. 5.2 Propriedades farmacocinéticas Avaliaram-se as propriedades farmacocinéticas do darunavir, co-administrado com ritonavir, em voluntários saudáveis adultos e em doentes infectados por VIH-1. A exposição ao darunavir foi maior nos doentes infectados por VIH-1 do que em indivíduos saudáveis. O aumento da exposição ao darunavir em doentes infectados por VIH-1 comparativamente com indivíduos saudáveis poderá ser explicado pela presença de concentrações mais elevadas da glicoproteína alfa-1-ácido (AAG) em doentes com infecção por VIH-1, resultando numa maior ligação do darunavir à AAG plasmática, e consequentemente, em maiores concentrações plasmáticas.
17

O darunavir é metabolizado principalmente pela CYP3A. O ritonavir inibe a CYP3A, aumentando, assim, consideravelmente as concentrações plasmáticas do darunavir. Absorção O darunavir foi rapidamente absorvido após administração oral. A concentração plasmática máxima de darunavir, na presença de uma dose baixa de ritonavir, é geralmente atingida no período de 2,5-4,0 horas. A biodisponibilidade oral absoluta de uma dose única de 600 mg de darunavir em monoterapia foi de aproximadamente 37%, aumentando para cerca de 82% na presença de 100 mg, 2 vezes por dia, de ritonavir. O efeito de potenciação global da farmacocinética de ritonavir traduziu-se por um aumento de cerca de 14 vezes da exposição sistémica ao darunavir quando se administrou por via oral uma dose única de 600 mg de darunavir em associação com ritonavir numa dose de 100 mg, 2 vezes por dia (ver secção 4.4). Quando administrado sem alimentos, a biodisponibilidade relativa do darunavir em presença de uma dose baixa de ritonavir é 30% inferior à registada quando o fármaco é administrado com alimentos. Consequentemente, PREZISTA comprimidos deve ser tomado com ritonavir e com alimentos. O tipo de alimentos não afecta a exposição ao darunavir. Distribuição A ligação do darunavir às proteínas plasmáticas é de aproximadamente 95%. O darunavir liga-se principalmente à glicoproteína alfa-1-ácido plasmática. Após administração intravenosa, o volume de distribuição de darunavir isolado foi de 88,1 ± 59,0 L (média ±DP) e aumentou para 131 ± 49,9 l (média ±DP), na presença de 100 mg de ritonavir, administrado duas vezes ao dia. Metabolismo As experiências in vitro com microssomas hepáticos humanos (HLMs) indicam que o darunavir sofre principalmente um metabolismo oxidativo. O darunavir é amplamente metabolizado pelo sistema CYP hepático e quase exclusivamente pela isoenzima CYP3A4. Um ensaio realizado com 14C-darunavir em voluntários saudáveis revelou que a maioria da radioactividade presente no plasma após uma dose única de 400/100 mg de darunavir com ritonavir foi devida ao fármaco original. Foram identificados pelo menos 3 metabolitos oxidativos de darunavir no homem; todos estes metabolitos revelaram uma actividade pelo menos 10 vezes inferior à actividade do darunavir contra o VIH de tipo selvagem. Eliminação Após a administração de uma dose de 400/100 mg de 14C-darunavir com ritonavir, foram recuperados nas fezes e urina, respectivamente, cerca de 79,5% e 13,9% da dose de 14C-darunavir administrada. O darunavir inalterado correspondeu a cerca de 41,2% e 7,7% da dose administrada, detectada nas fezes e na urina, respectivamente. A semivida de eliminação terminal do darunavir foi de aproximadamente 15 horas quando associado ao ritonavir. A depuração intravenosa do darunavir em monoterapia (150 mg) e em presença de uma dose baixa de ritonavir foi de 32,8 l/h e 5, l/h, respectivamente. Populações Especiais Pediátrica Não existem dados de farmacocinética disponíveis em crianças e adolescentes Idosos Uma análise farmacocinética da população em doentes infectados por VIH revelou que a farmacocinética de darunavir não apresenta diferenças consideráveis nos limites etários avaliados (18 a 75 anos) de doentes com infecção por VIH (n=12, idade ≥ 65) (ver secção 4.4). Contudo, apenas dados limitados estavam disponíveis em doentes com mais de 65 anos de idade. Sexo Uma análise farmacocinética da população revelou que a exposição ao darunavir é ligeiramente superior (16,8%) nas mulheres com infecção por VIH do que nos homens. Esta diferença não é clinicamente relevante.
18

Insuficiência renal Os resultados de um estudo de equilíbrio de massas realizado com 14C-darunavir com ritonavir revelaram que cerca de 7,7% da dose administrada de darunavir são excretados na urina sob a forma inalterada. Embora o darunavir não tenha sido estudado em doentes com insuficiência renal, uma análise farmacocinética da população revelou que a farmacocinética do darunavir não foi significativamente afectada nos doentes com insuficiência renal moderada (CrCl entre 30-60 ml/min., n=20) infectados por VIH (ver secções 4.2 e 4.4). Compromisso hepático O darunavir é essencialmente metabolizado e eliminado pelo fígado. Num estudo de dose múltipla com PREZISTA administrado em associação com ritonavir (600/100 mg) duas vezes ao dia, foi demonstrado que os parâmetros farmacocinéticos no estado estacionário de darunavir, em doentes com compromisso hepático ligeiro (Classe A de Child-Pugh) a moderado (Classe B de Child-Pugh), foram comparáveis com os parâmetros de doentes saudáveis. O efeito do compromisso hepático grave na farmacocinética do darunavir não foi estudado (ver secções 4.2, 4.3 e 4.4). 5.3 Dados de segurança pré-clínica Foram efectuados estudos de toxicologia animal, com níveis de exposição superiores aos níveis de exposição dos ensaios utilizando darunavir em monoterapia, no ratinho, rato e cão, e em associação com ritonavir no rato e no cão. Nos estudos toxicológicos de dose repetida realizados em ratinhos, ratos e cães, o tratamento com darunavir exerceu apenas efeitos limitados. No rato, os órgãos alvo identificados foram o sistema hematopoiético, o sistema de coagulação sanguínea, o fígado e a tiróide. Foi observada uma redução variável, mas limitada, dos parâmetros relacionados com os eritrócitos, para além de aumento do tempo parcial da tromboplastina activada. Foram observadas alterações no fígado (hipertrofia dos hepatócitos, vacuolação, aumento das enzimas hepáticas) e na tiróide (hipertrofia folicular). No rato, a associação de darunavir com ritonavir conduziu a uma pequeno aumento do efeito nos parâmetros da série eritróide, fígado e tiróide e aumento a incidência de fibrose dos ilhéus pancreáticos (apenas em ratos machos), quando comparado com o tratamento com darunavir isolado. No cão, não foram identificados achados de toxicidade major ou órgãos alvo com exposições equivalentes à exposição clínica correspondente à dose recomendada. Num estudo efectuado no rato, o número de corpos luteínicos e implantações uterinas diminuíram, na presença de toxicidade materna. Contudo, não se observaram efeitos sobre o acasalamento ou a fertilidade durante o tratamento com darunavir em doses máximas de 1000 mg/kg/dia e com níveis de exposição inferiores (AUC – 0,5 vezes) aos registados no homem na dose clinicamente recomendada. Até aos mesmos níveis posológicos, não se detectou teratogenicidade no rato e no coelho tratados com darunavir em monoterapia nem no ratinho tratado em associação com ritonavir. Os níveis de exposição foram inferiores aos registados no homem com a dose clínica recomendada. Uma avaliação do desenvolvimento pré- e pós-natal no rato revelou que o darunavir, com e sem ritonavir, provocou uma redução transitória do aumento de peso corporal das crias pré-desmame e houve um pequeno atraso na abertura do olhos e ouvidos. O darunavir em associação com ritonavir provocou uma redução do número de cachorros que demonstraram resposta ao 15ºdia de aleitamento e reduziu a sobrevivência dos cachorros, no período de aleitamento. Estes efeitos podem ser secundários à exposição dos cachorros ao fármaco, via leite e/ou toxicidade materna. A administração do darunavir em monoterapia ou em associação com ritonavir não afectou as funções após desmame. Foi observado um aumento da mortalidade e, em alguns animais, convulsões, em ratos juvenis que receberam dosagens directas do 12ºao 25ºdia de vida. Não foram completados os estudos de carcinogenicidade a longo prazo realizados com darunavir em roedores. Obtiveram-se, no entanto, resultados negativos com darunavir no ensaio de mutação reversa de Ames in vitro e no ensaio de aberração cromossómica in vitro em linfócitos humanos, ambos realizados na ausência e em presença do sistema de activação metabólica. O darunavir não induziu
19

lesões cromossómicas no teste do micronúcleo in vivo no ratinho, até níveis de exposições muito semelhantes aos níveis de exposição terapêuticos, em humanos. 6. INFORMAÇÕES FARMACÊUTICAS 6.1 Lista de excipientes Núcleo do comprimido Celulose microcristalina Sílica coloidal anidra Crospovidona Estearato de magnésio Revestimento do comprimido Álcool polivinílico – parcialmente hidrolisado Macrogol 3350 Dióxido de titânio (E171) Talco Amarelo sunset FCF (E110) 6.2 Incompatibilidades Não aplicável. 6.3 Prazo de validade 2 anos 6.4 Precauções especiais de conservação O medicamento não necessita de quaisquer precauções especiais de conservação. 6.5 Natureza e conteúdo do recipiente Um frasco de plástico de polietileno de alta densidade (HDPE) contendo 120 comprimidos com fecho de polipropileno (PP) resistente à abertura por crianças. Um frasco 6.6 Precauções especiais de eliminação Não existem requisitos especiais. 7. TITULAR DA AUTORIZAÇÃO DA INTRODUÇÃO NO MERCADO Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Bélgica 8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
20

9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
10. DATA DA REVISÃO DO TEXTO Informação pormenorizada sobre este medicamento está disponível na Internet no site da Agência Europeia do Medicamento (EMEA) http://www.emea.europa.eu/.
21

ANEXO II A. TITULAR DE AUTORIZAÇÃO DE FABRICO RESPONSÁVEL
PELA LIBERTAÇÃO DO LOTE B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO
TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
22

A TITULAR DE AUTORIZAÇÃO DE FABRICO RESPONSÁVEL PELA LIBERTAÇÃO DO LOTE
Nome e endereço do fabricante responsável pela libertação do lote Janssen-Cilag SpA Via C. Janssen IT-04010 Borgo San Michele Latina Itália B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO • CONDIÇÕES OU RESTRIÇÕES RELATIVAS AO FORNECIMENTO E UTILIZAÇÃO
IMPOSTAS AO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Medicamento sujeito a receita médica restrita (ver anexo I: resumo das características do medicamento, secção 4.2). • CONDIÇÕES OU RESTRIÇÕES RELATIVAS À UTILIZAÇÃO SEGURA E EFICAZ
DO MEDICAMENTO Não aplicável. • OUTRAS CONDIÇÕES Sistema de farmacovigilância O titular da autorização de introdução no mercado deve assegurar que o sistema de farmacovigilância está implementado e em funcionamento, antes do início da comercialização e enquanto o medicamento for comercializado. Plano de gestão de risco O titular da autorização de introdução no mercado compromete-se a realizar as actividades de farmacovigilância adicionais descritas no Plano de Farmacovigilância. C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO TITULAR DA
AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO O titular da autorização de introdução no mercado deverá completar o programa de estudos abaixo referido dentro dos prazos indicados. Os resultados desses estudos estarão na base da reavaliação da relação risco/benefício durante a avaliação do pedido de renovação . Área Descrição Data limite
Clínica 1 Deve ser submetido o relatório final do estudo de interacção TMC114-C163 (Ensaio de fase I, aberto, aleatorizado, cruzado em voluntários saudáveis para estudar a interacção farmacocinética entre rifabutina e TMC114, co-administrado com baixas doses de ritonavir no estado estacionário).
31 Outubro 2007
23

Clínica 2 Deve ser submetido o relatório final do estudo de interacção TMC114-C123 (Ensaio de fase I, aberto, aleatorizado, cruzado em voluntários saudáveis para investigar a interacção farmacocinética entre didanosina e TMC114, co-administrado com baixas doses de ritonavir no estado estacionário).
30 Abril 2007
Clínica 3 Deve ser submetido o relatório final às 48 semanas (análise primária) do estudo TMC114-C214 (Ensaio aleatorizado, controlado, aberto, para comparar a eficácia, segurança e tolerabilidade de TMC114/RTV versus LPV/RTV no tratamento de doentes infectados pelo VIH-1 previamente submetidos a TARV) e deve incluir uma análise para avaliar o efeito da co-administração de nevirapina e efavirenz no darunavir; para além disto deve conter uma estimativa da variabilidade intra-individual.
31 Julho 2007
Deve ser fornecido o relatório final às 96 semanas do estudo TMC114-C214.
Q3 2008
Clínica 4 Deve ser submetido o relatório final às 96 semanas do estudo TMC114-C202 (Ensaio de fase II aleatorizado, controlado, com ocultação parcial para investigar a resposta da dose de TMC114/RTV em doentes infectados pelo VIH-1, classe 3, com terapêutica anti-retroviral prévia, seguidos por um período aberto com a dose recomendada de TMC114/RTV).
31 Julho 2007
Deve ser submetido o relatório final às 144 semanas do estudo TMC114-C202.
Q3 2008
Clínica 5 Deve ser submetido o relatório final às 96 semanas do estudo TMC114-C213 (Ensaio de fase II aleatorizado, controlado, com ocultação parcial para investigar a resposta da dose de TMC114/RTV em doentes infectados pelo VIH-1, classe 3, com terapêutica anti-retroviral prévia, seguidos por um período aberto com a dose recomendada de TMC114/RTV).
30 Abril 2007
Deve ser submetido o relatório final do estudo às 144 semanas do estudo TMC114-C213.
Q1 2008
Clínica 6 Deve ser submetido o relatório final às 96 semanas do estudo TMC114-C215 (Ensaio aberto de TMC114/RTV em doentes infectados pelo VIH-1 previamente submetidos a terapêutica.
31 Dezembro 2007
Deve ser submetido o relatório final do estudo às 144 semanas do estudo TMC114-C215.
Q3 2008
Clínica 7 Deve ser submetido o relatório do estudo TMC114-C208, no cut-off Q2 2007 (Ensaio aberto de TMC114/RTV em doentes infectados pelo VIH-1 aleatorizados nos ensaios TMC114-C201, TMC114-C207 ou em ensaios de fase I seleccionados pelo promotor)
31 Dezembro 2007
Clínica 8 Deve ser submetido o relatório do estudo TMC114-C209, no cut-off Q2 2007 (Ensaio aberto de segurança de TMC114 em associação com baixas doses de RTV e outros ARV em doentes infectados com VIH-1 previamente submetidos a várias terapêuticas sem opções de tratamentos ou com opções limitadas).
31 Dezembro 2007
24

Clínica 9 Devem ser submetidos os resultados do braço de tratamento de darunavir que não receberam o candidato NNRTI (TMC125) para os dois estudos seguintes: -relatório final do estudo TMC125-C206 na semana 24 (análise primária) (Estudo de fase II aleatorizado, com dupla ocultação, controlado com placebo para ainvestigar a eficácia, tolerabilidade e segurança de TMC125 como parte de um regime anti-retroviral incluindo TMC114/RTV e um RBO seleccionado pelo investigador em doentes infectados peloVIH-1 sem opções de tratamentos ou com opções limitadas). -deve ser submetido relatório do estudo final do estudo TMC125-C216 na semana 24 (análise primária) (Estudo de fase III aleatorizado, com dupla ocultação, controlado com placebo para investigar a eficácia, tolerabilidade e segurança de TMC125 como parte de um regime anti-retroviral, incluindo TMC114/RTV e um RBO seleccionado pelo investigador em doentes infectados pelo VIH-1 sem opções de tratamentos ou com opções limitadas).
31 Outubro 2007
25

ANEXO III
ROTULAGEM E FOLHETO INFORMATIVO
26

A. ROTULAGEM
27

INDICAÇÕES A INCLUIR NO ACONDICIONAMENTO SECUNDÁRIO E NO ACONDICIONAMENTO PRIMÁRIO EMBALAGEM EXTERIOR / ROTULAGEM DO FRASCO 1. DENOMINAÇÃO DO MEDICAMENTO PREZISTA 300 mg comprimidos revestidos por película darunavir 2. DESCRIÇÃO DO(S) PRINCÍPIO(S) ACTIVO(S) Cada comprimido revestido por película contém 300 mg de darunavir (sob a forma de etanolato). 3. LISTA DOS EXCIPIENTES Contém amarelo sunset FCF (E110). 4. FORMA FARMACÊUTICA E CONTEÚDO 120 comprimidos revestidos por película 5. MODO E VIA(S) DE ADMINISTRAÇÃO Via oral Consultar o folheto informativo. 6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DO ALCANCE E DA VISTA DAS CRIANÇAS Manter fora do alcance e da vista das crianças. 7. OUTRA(S) ADVERTÊNCIA(S) ESPECIAL(IS), SE NECESSÁRIO 8. PRAZO DE VALIDADE VAL: 9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO
28

10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE FOR CASO DISSO
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Bélgica 12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO EU /0/00/000/000 13. NÚMERO DO LOTE Lote: 14. CLASSIFICAÇÃO QUANTO À DISPENSA AO PÚBLICO Medicamento sujeito a receita médica. 15. INSTRUÇÕES DE UTILIZAÇÃO 16. INFORMAÇÃO EM BRAILLE (apenas aplicável à embalagem exterior) prezista 300 mg
29

B. FOLHETO INFORMATIVO
30

FOLHETO INFORMATIVO: INFORMAÇÃO PARA O UTILIZADOR
PREZISTA – 300 mg – comprimidos revestidos por película darunavir
Leia atentamente este folheto antes de tomar o medicamento. - Conserve este folheto. Pode ter necessidade de o reler. - Caso ainda tenha dúvidas, fale com o seu médico ou farmacêutico. - Este medicamento foi receitado para si. Não deve dá-lo a outros; o medicamento pode ser-lhes
prejudicial mesmo que apresentem os mesmos sintomas. - Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não
mencionados neste folheto, informe o seu médico ou farmacêutico. Neste folheto: 1. O que é PREZISTA e para que é utilizado 2. Antes de tomar PREZISTA 3. Como tomar PREZISTA 4. Efeitos secundários possíveis 5. Como conservar PREZISTA 6. Outras informações 1. O QUE É PREZISTA PARA QUE É UTILIZADO O que é PREZISTA? PREZISTA é um medicamento anti-retroviral, utilizado no tratamento da infecção pelo Vírus da Imunodeficiência Humana (VIH). Pertence a um grupo de fármacos denominado inibidores da protease. PREZISTA actua reduzindo a quantidade de VIH presente no seu corpo. Desta forma, irá melhorar o seu sistema imunitário e reduzir o risco de desenvolvimento de doenças associadas à infecção pelo VIH. Para que é utilizado? PREZISTA é utilizado no tratamento de adultos infectados pelo VIH e que não responderam suficientemente a outros medicamentos anti-retrovirais. PREZISTA deve ser administrado em associação com uma dose baixa de ritonavir e com outros medicamentos anti-VIH. O seu médico falará consigo sobre qual será a associação de medicamentos melhor para o seu caso. 2. ANTES DE TOMAR PREZISTA PREZISTA é para ser tomado em associação com doses baixas de ritonavir e outros medicamentos anti-retrovirais. Portanto, é importante que leia cuidadosamente o folheto informativo que é fornecido com esses medicamentos. Se tiver quaisquer outras questões sobre os medicamentos prescritos, pergunte ao seu médico ou farmacêutico. Não tome PREZISTA - se tem alergia (hipersensibilidade) ao darunavir, a outros componentes de PREZISTA ou a
ritonavir. - Se tem problemas de fígado graves. Pergunte ao seu médico se não tem a certeza sobre a
gravidade dos seus problemas de fígado. Alguns testes adicionais poderão ser necessários. Não combine PREZISTA com qualquer um dos seguintes medicamentos - astemizol ou terfenadina (para tratamento dos sintomas de alergia) - midazolam ou triazolam (para tratamento de problemas de insónias e/ou ansiedade) - cisaprida (para tratamento de perturbações gástricas)
31

- pimozida ou sertindol (para tratamento de perturbações psiquiátricas) - alcalóides da ergotamina (como ergotamina, di-hidroergotamina, metilergonovina, utilizados
para tratamento de enxaquecas e dores-de-cabeça) - amiodarona, bepridil, quinidina, lidocaína sistémica (para tratamento de certas doenças do
coração, ex: batimento cardíaco anormal) - lovastataina e sinvastatina (medicamentos para baixarem os níveis de colesterol) - rifampicina (medicamentos para o tratamento de algumas infecções como a tuberculose) - produtos que contenham hipericão (Hypericum perforatum) Se estiver a utilizar qualquer um destes medicamentos interrogue o seu médico sobre a possibilidade de mudar para outro medicamento. Tome especial cuidado com PREZISTA PREZISTA não é uma cura para a infecção pelo VIH. PREZISTA não reduz o risco de transmissão do VIH para terceiros por contacto sexual ou contaminação sanguínea. Deverá, portanto, continuar a utilizar medidas de precaução apropriadas. As pessoas que tomam PREZISTA podem continuar a desenvolver infecções ou outras doenças associadas à infecção por VIH. Deve manter um contacto regular com o seu médico. PREZISTA foi administrado a um número limitado de doentes com idade igual ou superior a 65 anos. Se pertence a este grupo etário, fale com o seu médico para saber se pode utilizar este medicamento. Informe o seu médico sobre a sua situação Assegure-se que revê os sete pontos a seguir referidos e informe o seu médico se algum é aplicável à sua situação. - Informe o seu médico se já teve problemas de fígado, incluindo hepatite B ou C. O seu médico
poderá avaliar o grau de gravidade da sua doença hepática antes de decidir se pode tomar PREZISTA.
- Informe o seu médico se tem diabetes. PREZISTA, pode aumentar os níveis de açúcar no sangue.
- Informe imediatamente o seu médico se apresentar quaisquer sintomas de infecção. Alguns doentes com infecção pelo VIH avançada e antecedentes de uma infecção oportunista, podem desenvolver sinais e sintomas de inflamação resultantes de uma infecção anterior logo após iniciar-se um tratamento anti-VIH. Pensa-se que estes sintomas são devidos a uma melhoria da resposta imunitária do organismo, que permite ao organismo combater as infecções que se encontrem eventualmente presentes sem sintomas óbvios.
- Informe o seu médico se detectar alterações na gordura corporal. Pode ocorrer redistribuição, acumulação ou perda de gordura corporal nos doentes que estão a ser tratados com uma associação de medicamentos anti-retrovirais.
- Informe o seu médico se tem hemofilia. PREZISTA, podem aumentar o risco de hemorragia. - Informe o seu médico se é alérgico a sulfonamidas (ex: utilizados para tratar certas infecções). - Informe o seu médico se notar alguns problemas musculo-esqueléticos. Alguns doentes
tomando associações de terapêutica anti-retroviral podem desenvolver uma doença nos ossos denominada osteonecrose (morte do tecido ósseo causada por uma perda de fornecimento de sangue aos ossos). Alguns dos muitos factores de risco para o desenvolvimento desta doença são a duração da terapêutica anti-retroviral de associação, utilização de corticosteróides, consumo de álcool, imunodepressão grave, índice de massa corporal elevado. Sinais da osteonecrose são rigidez nas articulações, dores nas articulações (especialmente na anca, joelho e ombro) ou dificuldade nos movimentos. Se notar qualquer destes sintomas, por favor informe o seu médico.
Tomar outros medicamentos PREZISTA pode interagir com outros medicamentos. Informe o seu médico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos obtidos sem receita médica. Existem alguns medicamentos que não poderá combinar com PREZISTA. Estes são mencionados no título “Não combine PREZISTA com qualquer um dos seguintes medicamentos:”
32

Na maioria dos casos PREZISTA pode ser combinado com outros medicamentos anti-VIH, pertencentes a outras classes de medicamentos (ex: NRTI (análogos nucleosídeos inibidores da transcriptase inversa (reversa)), NNRTI (análogos não nucleosídeos inibidores da transcriptase inversa (reversa)) e inibidores de entrada). A combinação de PREZISTA com ritonavir não foi testada com todos os IPs (inibidores da protease). Assim, informe sempre o seu médico se está a tomar outro medicamento anti-VIH e siga sempre cuidadosamente as suas instruções sobre que medicamentos devem ser combinados. Os efeitos de PREZISTA podem ser reduzidos se utilizar qualquer um dos seguintes produtos. Informe o seu médico se está a utilizar: - fenobarbital, fenitoína, carbamazepina (medicamentos para prevenir a ocorrência de crises
convulsivas) - dexametasona (esteróides). Os efeitos de outros medicamentos podem ser afectados pelo tratamento com PREZISTA. Informe o seu médico se está a tomar: - felodipina, nifedipina, nicardipina (medicamentos para doenças do coração), uma vez que o
efeito terapêutico ou os efeitos secundários indesejáveis deste medicamentos podem ser aumentados.
- varfarina (medicamentos utilizados para reduzir a coagulação do sangue) uma vez que o efeito terapêutico ou os efeitos secundários indesejáveis deste medicamentos podem estar alterados; o seu médico pode ter necessidade de analisar o seu sangue.
- contraceptivos orais e terapêutica hormonal de substituição. PREZISTA pode reduzir a sua eficácia. Quando utilizado para controlo da natalidade deverá associar os contraceptivos hormonais com outros métodos de controlo, tais como preservativos.
- pravastatina, atorvastatina (medicamentos para baixar os níveis de colesterol). Pode observar-se um aumento do risco de perturbações do tecido muscular. O seu médico avaliará qual o regime mais adequado para baixar os níveis de colesterol é melhor para a sua situação específica.
- ciclosporina, tacrolímus, sirolímus (medicamentos para o seu sistema imunitário) uma vez que o efeito terapêutico ou os efeitos secundários indesejáveis destes medicamentos por ser aumentado. O seu médico poderá pretender submetê-lo a alguns testes adicionais.
- propionato de fluticasona (medicamentos para controlo da asma). A sua utilização deve apenas efectuar-se após avaliação clínica e sob supervisão pelo seu médico quanto aos efeitos secundários dos corticosteróides.
Poderá ser necessário alterar a dose de outros medicamentos uma vez que os efeitos terapêuticos ou os efeitos secundários indesejáveis destes medicamentos ou de PREZISTA podem ser influenciados quando associados. Informe o seu médico se está a utilizar: - digoxina (medicamento para tratar certas doenças do coração) - cetoconazol, itraconazol, clotrimazol (medicamentes contra infecções fúngicas). O voriconazol
deve apenas ser tomado após avaliação clínica - rifabutina (medicamento contra infecções bacterianas) - sildenafil, vardenafil, tadalafil (medicamentos para a disfunção eréctil) - claritromicina (antibióticos) - paroxetina, sertralina (medicamentos para tratamento da depressão e ansiedade) - metadona Tomar PREZISTA com alimentos e bebidas Ver a secção 3 ‘Como tomar PREZISTA com alimentos e bebidas’. Gravidez e aleitamento Informe imediatamente o seu médico se está grávida ou a amamentar. Grávidas ou mães a amamentar não devem tomar PREZISTA, a não ser se especificamente recomendado pelo médico. É
33

recomendado que as mães infectadas pelo VIH não amamentem os filhos devido à possibilidade do seu bebé ser infectado pelo VIH, através do leite materno. Condução de veículos e utilização de máquinas Não conduza nem utilize quaisquer máquinas caso sinta tonturas durante o tratamento com PREZISTA. Informações importantes sobre alguns componentes de PREZISTA PREZISTA comprimidos contém o corante amarelo sunset FCF (E110) que pode provocar reacções alérgicas. 3. COMO TOMAR PREZISTA Tome PREZISTA sempre de acordo com as indicações do médico. Fale com o seu médico se tiver dúvidas. Mesmo que se sinta melhor, não deixe de tomar PREZISTA sem falar com o seu médico. PREZISTA não se destina a crianças e adolescentes, pois não foi estudado em doentes com menos de 18 anos de idade. Instruções - A dose habitual de PREZISTA é de dois comprimidos duas vezes por dia. - Tome sempre PREZISTA em conjunto com ritonavir. PREZISTA não actua adequadamente
sem o ritonavir. - De manhã tome dois comprimidos de 300 miligramas de PREZISTA com 100 mg de ritonavir. - À noite tome dois comprimidos de 300 miligramas de PREZISTA com 100 mg de ritonavir. - Tome PREZISTA com alimentos. PREZISTA não actua adequadamente sem alimentos. O tipo
de alimentos não é importante. - Engula os comprimidos com uma bebida, por exemplo, água ou leite. Remover a tampa de segurança infantil
O frasco de plástico possui um fecho de segurança infantil devendo ser aberto da seguinte forma: - Pressione a tampa roscada de plástico para baixo rodando-a no sentido
contrário ao dos ponteiros do relógio. - Retire a tampa desenroscada.
Se tomar mais PREZISTA do que deveria Contacte imediatamente o seu médico ou farmacêutico. Caso se tenha esquecido de tomar PREZISTA Caso se recorde no período de 6 horas deverá tomar os comprimidos imediatamente. Tome sempre o medicamento com ritonavir e com alimentos. Caso se recorde após 6 horas, não tome a dose que se esqueceu e tome as doses seguintes conforme habitual. Não tome uma dose a dobrar para compensar a dose que se esqueceu de tomar. Não pare de tomar PREZISTA sem falar previamente com o seu médico A terapêutica para o VIH poderá aumentar a sua sensação de bem estar. Não deixe de tomar PREZISTA mesmo que se sinta melhor. Fale primeiro com o seu médico. Caso ainda tenha dúvidas sobre a utilização deste medicamento, fale com o seu médico.
34

4. EFEITOS SECUNDÁRIOS POSSIVEIS Como os demais medicamentos, PREZISTA pode causar efeitos secundários, no entanto estes não se manifestam em todas as pessoas. Ao tratar uma infecção pelo VIH nem sempre é fácil identificar quais são os efeitos secundários causados por PREZISTA e quais os causados por outros medicamentos que está a tomar ou quais são os causados pela própria infecção pelo VIH. Efeitos secundários frequentes (isto significa menos do que 1 a 10 doentes, mas mais do que 1 a 100 doentes afectados por estes efeitos). - vómitos, diarreia, náuseas, dor abdominal, obstipação, flatulência (gazes), distensão do
abdómen (barriga), indigestão, perda de apetite. - cefaleias, tonturas, perda ou falta de força, fadiga, insónia. - aumento de um tipo de gordura no sangue. - erupções cutâneas. A erupção cutânea é geralmente ligeiro a moderado. Uma erupção cutânea
pode ser um sintoma de uma situação rara grave. Portanto, é importante contactar o seu médico se desenvolver exantema. O seu médico aconselhá-lo-á sobre como lidar com os seus sintomas ou se deve interromper o tratamento com PREZISTA.
Efeitos secundários pouco frequentes (isto significa menos do que 1 a 100 doentes, mas mais do que 1 a 1000 doentes afectados por estes efeitos). - Ataque cardíaco, electrocardiograma anormal, coração com batimentos acelerados, dilatação do
coração. - Entorpecimento, formigueiro ou dor nas mãos ou pés, perda de sensibilidade cutânea, perda de
memória, dormência, sonolência, acidente vascular cerebral menor (AVC), perturbação na atenção, desmaio.
- Problemas com o seu equilíbrio. - Dificuldade em respirar, tosse, soluços. - Boca seca, excesso de saliva, úlceras na língua. - Insuficiência renal, passagem da urina dolorosa, frequente ou excessiva, por vezes com excesso
de proteínas, pedras no rim, diminuição da função renal. - Perda de gordura por debaixo da pele, inflamação da pele, suores nocturnos, perda de cabelo,
eczema, transpiração excessiva, inflamação de folículos capilares, pele seca, comichão, olhos secos, face inchada, urticária.
- Dor nas articulações, dor nas extremidades, dor muscular ou cãibras, osteoporose. - Diabetes, diminuição do apetite, sede anormal, alterações no corpo associadas com
redistribuição na gordura, aumento de peso, obesidade, diminuição na actividade da tiróide. - Pressão arterial elevada, rubor. - Febre, inchaço dos membros inferiores devido a retenção de fluidos, arrepios. - Nos homens desenvolvimento excessivo das glândulas mamárias, disfunção eréctil - Ansiedade, sensação de confusão ou desorientação, irritabilidade, alterações do humor,
pesadelos. - Alterações em alguns valores químicos ou de células sanguíneas. Estes podem observar-se nos
resultados de análises ao sangue. O seu médico explicar-lhe-á isto. Exemplos são: diminuição na contagem de glóbulos brancos, diminuição na contagem de plaquetas, níveis elevados de colesterol, níveis elevados de gordura no sangue, níveis baixos de sódio.
Alguns efeitos secundários são típicos de medicamentos anti-VIH da mesma família que o PREZISTA. Estes são: - aumento dos níveis sanguíneos de açúcar e agravamento da diabetes. - dor, sensibilidade ou fraqueza musculares. Em casos raros, estas perturbações musculares foram
graves. - alterações do perfil corporal devido a redistribuição da gordura. Estas alterações podem incluir
perda de gordura nas pernas, braços e face, aumento da gordura no abdómen (barriga) e noutros órgãos internos, aumento do volume mamário e acumulação de gordura na nuca (bossa de búfalo). Desconhecem-se presentemente a causa e os efeitos a longo prazo sobre a saúde destas condições.
35

Se algum dos efeitos secundários se agravar ou se detectar quaisquer efeitos secundários não mencionados neste folheto, informe o seu médico. 5. COMO CONSERVAR PREZISTA Manter fora do alcance e da vista das crianças. Não utilize PREZISTA após o prazo de validade impresso na embalagem exterior e no frasco, a seguir às letras VAL. O prazo de validade corresponde ao último dia do mês indicado. PREZISTA não necessita de quaisquer precauções especiais de conservação. Os medicamentos não devem ser eliminados na canalização ou no lixo doméstico. Pergunte ao seu farmacêutico como eliminar os medicamentos de que já não necessita. Estas medidas irão ajudar a proteger o ambiente. 6. OUTRAS INFORMAÇÕES Qual a composição de PREZISTA - A substância activa é o darunavir. Cada comprimido contém 300 mg de darunavir sob a forma
de etanolato. - Os outros componentes são a celulose microcristalina, sílica coloidal anidra, crospovidona,
estearato de magnésio. O revestimento contém álcool poli(vinílico) – parcialmente hidrolizado, macrogol 3350, dióxido de titânio (E171), talco, amarelo sunset FCF (E110).
Qual o aspecto de PREZISTA e conteúdo da embalagem Comprimido revestido por película, de forma oval, cor-de-laranja, mencionando TMC114 numa das faces e 300MG na face oposta. Frasco de plástico contendo 120 comprimidos. Titular da Autorização de Introdução no Mercado Janssen-Cilag International NV, Turnhoutseweg 30, 2340 Beerse, Bélgica Fabricante Janssen-Cilag SpA, Via C. Janssen, 04010 Borgo San Michele, Latina, Itália Para quaisquer informações sobre este medicamento, queira contactar o representante local do Titular da Autorização de Introdução no Mercado: België/Belgique/Belgien TIBOTEC, een divisie van, une division de, eine Division der JANSSEN-CILAG NV/SA Roderveldlaan 1 B-2600 Berchem Tél/Tel: +32 3 280 54 11
Luxembourg/Luxemburg TIBOTEC, une division de, eine Division der JANSSEN-CILAG NV/SA Roderveldlaan 1 B-2600 Berchem Belgique/Belgien Tél: +32 3 280 54 11
36

България Представителство на TIBOTEC, дивизия на Johnson & Johnson, d.o.o. ж.к. Младост 4 Бизнес Парк София, сграда 4 София 1715 Тел.: +359 2 976 94 00
Magyarország TIBOTEC, a JANSSEN-CILAG Kft. divíziója H-2045 Törökbálint, Tó Park Tel: +36 23 513 800
Česká republika TIBOTEC, divize JANSSEN-CILAG s.r.o. Karla Engliše 3201/06 CZ-150 00 Praha 5 - Smíchov Tel: +420 227 012 222
Malta AM MANGION LTD. Mangion Building, New Street off Valetta Road MT-Luqa LQA 06 Tel: +356 25402600 - 10
Danmark TIBOTEC, en division af JANSSEN-CILAG A/S Hammerbakken 19 DK-3460 Birkerød Tlf: +45 45 94 82 82
Nederland TIBOTEC, een divisie van JANSSEN-CILAG B.V. Postbus 90240 NL-5000 LT Tilburg Tel: +31 13 583 73 73
Deutschland JANSSEN-CILAG GmbH Raiffeisenstr. 8 D-41470 Neuss Tel: +49 2137 955-0
Norge TIBOTEC, en divisjon av JANSSEN-CILAG AS Hoffsveien 1D N-0275 Oslo Tlf: +47 24 12 65 00
Eesti TIBOTEC, JANSSEN-CILAG Polska Sp z o.o. Eesti filiaal Weizenbergi 20b EE-10150 Tallinn Tel: +372 626 6500
Österreich TIBOTEC, eine Division von JANSSEN-CILAG Pharma GmbH Pfarrgasse 75 A-1232 Wien Tel: +43 1 610 300
Ελλάδα TIBOTEC, τμήμα της JANSSEN-CILAG Φαρμακευτική Α.Ε.Β.Ε. Λεωφόρος Ειρήνης 56 GR-151 21 Πεύκη, Αθήνα Tηλ: +30 210 61 40 061
Polska TIBOTEC, oddział JANSSEN-CILAG Polska Sp. z o.o. ul. Szyszkowa 20 PL-02-285 Warszawa Tel: +48 22 668 01 50
España JANSSEN-CILAG, S.A. división TIBOTEC Paseo de las Doce Estrellas, 5-7 Campo de las Naciones E-28042 Madrid Tel: +34 91 722 81 00
Portugal TIBOTEC, uma divisão da JANSSEN-CILAG FARMACÊUTICA, LDA. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835
France TIBOTEC, une division de JANSSEN-CILAG 1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 44 44
România TIBOTEC, subsidiară a Janssen-Cilag, Johnson & Johnson d.o.o. Sipotul Fantanilor no. 8, Sect. 1 010 157 Bucureşti Tel: +40 21 312 1169
37

Ireland TIBOTEC, a division of JANSSEN-CILAG Ltd. Saunderton High Wycombe Buckinghamshire HP14 4HJ - UK Tel: +44 1494 567 444
Slovenija TIBOTEC za Janssen-Cilag, del Johnson&Johnson d.o.o. Šmartinska cesta 53 SI-1000 Ljubljana Tel: +386 1 401 18 30
Ísland TIBOTEC, deild hjá JANSSEN-CILAG c/o Vistor hf. Hörgatún 2 IS-210 Garðabær Sími: +354 535 7000
Slovenská republika TIBOTEC, divízia Johnson & Johnson s.r.o. Plynárenská 7/B SK-824 78 Bratislava Tel: +421 233 552 600
Italia TIBOTEC, una divisione di JANSSEN-CILAG SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1
Suomi/Finland TIBOTEC JANSSEN-CILAG OY Metsänneidonkuja/Skogsjungfrugränden 8 FI-02130 Espoo/Esbo Puh/Tel: +358 9 4155 5300
Κύπρος Βαρνάβας Χατζηπαναγής Λτδ, 7 Ανδροκλέους CY-1060 Λευκωσία Τηλ: +357 22 755 214
Sverige TIBOTEC, en division inom JANSSEN-CILAG AB Box 7073 S-192 07 Sollentuna Tel: +46 8 626 50 00
Latvija TIBOTEC, JANSSEN-CILAG Polska Sp. z o.o. filiāle Latvijā Baznīcas iela 20/22 Rīga, LV-1010 Tel: +371 7039805
United Kingdom TIBOTEC, a division of JANSSEN-CILAG Ltd. Saunderton High Wycombe Buckinghamshire HP14 4HJ - UK Tel: +44 1494 567 444
Lietuva UAB “Johnson & Johnson” Šeimyniškių g. 1A LT-09312 Vilnius Tel: +370 5 278 68 88
Este folheto foi aprovado pela última vez em xxx 2006 A este medicamento foi dada uma “autorização condicional”. Isto significa que se espera mais informação sobre este medicamento. A Agência Europeia do Medicamento (EMEA) irá rever anualmente nova informação sobre o medicamento e este folheto será actualizado se necessário. Informação pormenorizada sobre este medicamento está disponível na Internet no site da Agência Europeia do Medicamento (EMEA) http://www.emea.europa.eu/.
38