anexo i resumo das caracterÍsticas do...
TRANSCRIPT

1
ANEXO I
RESUMO DAS CARACTERÍSTICAS DO MEDICAMENTO

2
1. DENOMINAÇÃO DO MEDICAMENTO Zevalin 1,6 mg/ml, Kit para preparação radiofarmacêutica para perfusão 2. COMPOSIÇÃO QUALITATIVA E QUANTITATIVA Ibritumomab tiuxetan* 1,6 mg por ml Um frasco contém 3,2 mg de ibritumomab tiuxetan *produzido geneticamente por linhas celulares de Ovário de Hamster Chinês (OHC) e conjugado com o agente quelante MX-DTPA Zevalin é fornecido sob a forma de kit para a preparação de ibritumomab tiuxetan marcado radioactivamente com ítrio-90. A formulação final após marcação radioactiva contém 2,08 mg de ibritumomab tiuxetan num volume total de 10 ml. Excipientes, ver 6.1. 3. FORMA FARMACÊUTICA Kit para preparação radiofarmacêutica para perfusão 4. INFORMAÇÕES CLÍNICAS 4.1 Indicações terapêuticas Zevalin marcado radioactivamente com [90Y] está indicado no tratamento de doentes adultos com linfoma não Hodgkin (LNH) folicular de células B CD20+ em recidiva ou refractário ao rituximab. 4.2 Posologia e modo de administração Zevalin marcado radioactivamente com [90Y] só deve ser manipulado e administrado por pessoal qualificado com a devida autorização para utilização e manipulação de radionuclídeos num ambiente clínico apropriado. A sua preparação, utilização, transferência, conservação e eliminação estão sujeitas a regras e/ou autorização apropriadas. As perfusões devem ser administradas sob vigilância atenta de um médico experiente com a garantia de disponibilidade imediata de equipamento de reanimação (para as precauções relativas a produtos radiofarmacêuticos, ver também 4.4). Zevalin deve ser utilizado após o tratamento prévio com rituximab. Consulte as informações sobre o rituximab para uma orientação pormenorizada sobre a sua utilização. A solução para perfusão preparada deve ser administrada sob a forma de uma administração intravenosa lenta durante 10 minutos. Não utilizar sob a forma de injecção intravenosa em bólus. A solução de Zevalin marcado radioactivamente com [90Y] deverá ser preparada de acordo com a secção 6.6 “Instruções de utilização/manipulação”. Antes da administração ao doente, a percentagem de radioincorporação do Zevalin marcado radioactivamente com [90Y] preparado tem que ser verificada, de acordo com o procedimento descrito na secção 6.6. Se a pureza radioquímica média for inferior a 95%, a preparação não deve ser administrada. A dose recomendada é: - para doentes com 150.000 plaquetas ou mais por mm3: 15 MBq de Zevalin marcado
radioactivamente com [90Y] por kg de peso corporal até um máximo de 1200 MBq.

3
- para doentes com menos de 150.000 mas com mais de 100.000 plaquetas por mm3: 11 MBq de Zevalin marcado radioactivamente com [90Y] por kg de peso corporal até um máximo de 1200 MBq.
Zevalin marcado radioactivamente com [90Y] pode ser administrado directamente por perfusão desviando o fluxo de um saco de perfusão e administrando-o directamente no sistema. É necessário utilizar um filtro com baixo índice de ligação às proteínas de 0,2 ou 0,22 mícron no sistema entre o doente e o ponto de perfusão. Lavar o sistema com pelo menos 10 ml de solução (a 0,9 %) de 9 mg/ml de cloreto de sódio após a perfusão de Zevalin marcado radioactivamente com [90Y]. O tratamento consiste em duas administrações por via intravenosa de rituximab e uma administração de Zevalin marcado radioactivamente com [90Y] pela seguinte ordem: Dia 1: uma perfusão intravenosa de rituximab. Esquema posológico para a perfusão de rituximab: 250 mg/m2 de rituximab. Dia 8: uma perfusão por via intravenosa de rituximab pouco antes da administração de Zevalin marcado radioactivamente com [90Y]. Esquema posológico para a perfusão de rituximab: 250 mg/m2 de rituximab. Perfusão de Zevalin marcado radioactivamente com [90Y]: 10 minutos de perfusão por via intravenosa de Zevalin marcado radioactivamente com [90Y] administrada até uma dose máxima de 1200 MBq. Se a pureza radioquímica média for inferior a 95%, a preparação não deve ser administrada. Utilização repetida Não estão disponíveis informações sobre o tratamento repetido de doentes com Zevalin marcado radioactivamente com [90Y]. 4.3 Contra-indicações Hipersensibilidade a ibritumomab tiuxetan, a cloreto de ítrio, a outras proteínas de origem murina ou a qualquer dos excipientes. Gravidez e aleitamento. 4.4 Advertências e precauções especiais de utilização Os produtos radiofarmacêuticos devem ser utilizados apenas por pessoal qualificado com a devida autorização governamental para utilização e manipulação de radionuclídeos. Este produto radiofarmacêutico pode apenas ser recebido, utilizado e administrado por pessoas devidamente autorizadas em ambientes específicos para esse fim. A sua recepção, conservação, utilização, transferência e eliminação estão sujeitas às regulamentações e/ou licenças apropriadas das entidades oficiais locais competentes. Os produtos radiofarmacêuticos devem ser preparados pelo utilizador de forma a satisfazer os requisitos de segurança radioactiva e de qualidade farmacêutica. Devem tomar-se as precauções de assépsia adequadas, em conformidade com os requisitos das Boas Práticas de Fabrico de medicamentos. Zevalin marcado radioactivamente com [90Y] não deve ser administrado a doentes com probabilidade de desenvolverem sinais de toxicidade hematológica que constituam perigo de vida. Zevalin não deve ser administrado aos doentes referidos a seguir pois a sua segurança e eficácia não foi estabelecida: - doentes nos quais mais de 25% da medula óssea foi infiltrada por células de linfoma, - doentes que tenham recebido anteriormente radiação externa de feixe envolvendo mais de 25%
da medula óssea activa, - doentes com contagem de plaquetas <100.000/mm3 ou contagem de neutrófilos <1.500/mm3 e

4
- doentes que tenham recebido anteriormente transplante de medula óssea ou suporte de células estaminais. - crianças e adolescentes com menos de 18 anos de idade.
É preciso ter especial cuidado no que se refere à depleção da medula óssea. Doentes que tenham recebido proteínas derivadas de murino antes do tratamento com Zevalin devem ser testados para determinar a presença de anticorpos humanos anti-rato (AHAR). Doentes que tenham desenvolvido anticorpos humanos anti-rato podem ter reacções alérgicas ou de hipersensibilidade quando tratados com Zevalin ou com outras proteínas derivadas de murino. Foram referidas reacções anafilácticas e outras reacções de hipersensibilidade em menos de 1% dos doentes na sequência da administração intravenosa de proteínas. Deverão estar disponíveis os medicamentos para o tratamento de reacções de hipersensibilidade, por exemplo, adrenalina, anti-histamínicos e corticosteróides, para serem utilizados imediatamente em caso de uma reacção alérgica durante a administração de Zevalin. Depois da utilização de Zevalin, os doentes devem, de modo geral, ser testados para determinar a presença de anticorpos humanos anti-rato, antes de prosseguir o tratamento com proteínas derivadas de ratos. Não foram realizados estudos de longa duração em animais sobre o efeito na fertilidade e função reprodutiva. Devido à natureza do composto, tanto as mulheres em idade fértil como os homens, devem utilizar métodos contraceptivos eficazes durante o tratamento com Zevalin e nos 12 meses seguintes. Não foi estudada a segurança da imunização com qualquer vacina, em especial vacinas de vírus vivos, no seguimento da terapêutica com Zevalin. A capacidade de produzir uma resposta humoral primária ou anamnéstica a qualquer vacina também não foi estudada. 4.5 Interacções medicamentosas e outras formas de interacção Não são conhecidas quaisquer interacções com outros medicamentos. Não foram efectuados estudos formais sobre interacções do fármaco. 4.6 Gravidez e aleitamento Não foram efectuados estudos de reprodução em animais com ibritumomab tiuxetan. Sabendo-se que a IgG atravessa a barreira placentária e, devido à utilização concomitante de radiação, Zevalin não pode ser utilizado durante a gravidez. É necessário excluir todas as possibilidades de gravidez antes de iniciar o tratamento em mulheres. As mulheres em idade fértil, bem como os homens, deverão utilizar métodos contraceptivos eficazes durante o tratamento com Zevalin e nos 12 meses seguintes. Quando é necessário administrar Zevalin a mulheres em idade fértil, deve-se sempre recolher todas as informações sobre uma possível gravidez. Qualquer mulher a quem tenha faltado um período menstrual deve ser considerada grávida até prova em contrário e, nesse caso, deverão ser consideradas terapêuticas alternativas que não incluam radiação ionizante. Não se sabe se o ibritumomab tiuxetan é excretado no leite humano. Como a IgG humana é excretada no leite humano, e dado que não se conhece o potencial de absorção e de imunossupressão no bebé, as mulheres têm que deixar de amamentar. 4.7 Efeitos sobre a capacidade de conduzir e utilizar máquinas Dado que foram relatadas tonturas como efeito secundário muito frequente, o Zevalin pode afectar a capacidade de conduzir e utilizar máquinas. 4.8 Efeitos indesejáveis

5
A dose de radiação resultante da exposição terapêutica pode dar origem a malignidades secundárias e ao desenvolvimento de defeitos hereditários. É necessário assegurar que os riscos de exposição à radiação são inferiores aos da própria doença. Prevê-se que a maioria dos doentes manifeste reacções adversas. As frequências das reacções adversas a seguir referidas (muito frequentes ≥ 10%, frequentes ≥ 1 a <10%, pouco frequentes <1%) baseiam-se em dados de ensaios clínicos, independentemente da causalidade. Reacções anafilácticas e hipersensibilidade Foram referidas reacções anafilácticas e outras reacções de hipersensibilidade em menos de 1% dos doentes na sequência da administração intravenosa de proteínas. Deverão estar disponíveis os medicamentos para o tratamento de reacções de hipersensibilidade, por exemplo, adrenalina, anti-histamínicos e corticosteróides, para serem utilizados imediatamente em caso de uma reacção alérgica durante a administração de Zevalin. Reacções adversas hematológicas A toxicidade hematológica tem sido muito frequentemente observada em ensaios clínicos e é factor limitante da dose. O tempo mediano para atingir os níveis mais baixos (nadirs) de granulócitos e de plaquetas no sangue foi de cerca de 60 dias após o início do tratamento. Foi referida trombocitopenia de grau 3 ou 4, com tempos medianos de recuperação de 13 e 21 dias, e neutropenia de grau 3 ou 4, com tempos medianos de recuperação de 8 e 14 dias. Infecções Durante as primeiras 13 semanas após o tratamento com Zevalin, os doentes desenvolveram muito frequentemente infecções. Foram referidas frequentemente infecções de grau 3 e de grau 4. No período de seguimento dos doentes, as infecções ocorreram frequentemente. Destas infecções, as de grau 3 eram frequentes e as de grau 4 não frequentes. Malignidades secundárias Mielodisplasia ou leucemia mielóide aguda (LMA) foi referida em cinco de 211 doentes, associada ao tratamento com Zevalin. O risco de desenvolver mielodisplasia ou leucemia secundária após terapêutica com agentes alquilantes é bem conhecido. Como todos estes doentes foram previamente tratados com agentes alquilantes, os resultados disponíveis fornecem resultados insuficientes sobre a eventual contribuição de Zevalin para um risco aumentado de mielodisplasia, ou sobre o grau de risco. Incidência de reacções adversas por sistemas orgânicos O quadro abaixo indica os efeitos adversos por sistemas orgânicos: No total, foram muito frequentes as infecções, independentemente da causa; contudo, são referidas no quadro nos termos exactos em que foram relatadas.
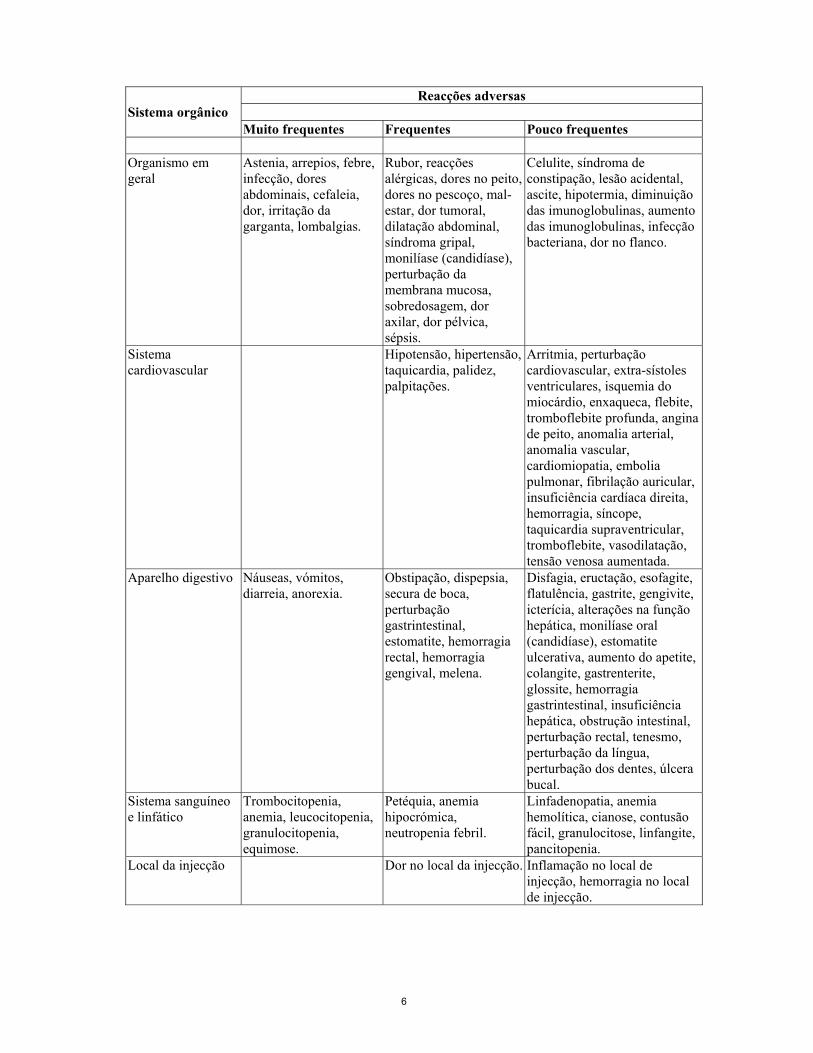
6
Reacções adversas
Sistema orgânico Muito frequentes Frequentes Pouco frequentes
Organismo em geral
Astenia, arrepios, febre, infecção, dores abdominais, cefaleia, dor, irritação da garganta, lombalgias.
Rubor, reacções alérgicas, dores no peito, dores no pescoço, mal-estar, dor tumoral, dilatação abdominal, síndroma gripal, monilíase (candidíase), perturbação da membrana mucosa, sobredosagem, dor axilar, dor pélvica, sépsis.
Celulite, síndroma de constipação, lesão acidental, ascite, hipotermia, diminuição das imunoglobulinas, aumento das imunoglobulinas, infecção bacteriana, dor no flanco.
Sistema cardiovascular
Hipotensão, hipertensão, taquicardia, palidez, palpitações.
Arritmia, perturbação cardiovascular, extra-sístoles ventriculares, isquemia do miocárdio, enxaqueca, flebite, tromboflebite profunda, angina de peito, anomalia arterial, anomalia vascular, cardiomiopatia, embolia pulmonar, fibrilação auricular, insuficiência cardíaca direita, hemorragia, síncope, taquicardia supraventricular, tromboflebite, vasodilatação, tensão venosa aumentada.
Aparelho digestivo Náuseas, vómitos, diarreia, anorexia.
Obstipação, dispepsia, secura de boca, perturbação gastrintestinal, estomatite, hemorragia rectal, hemorragia gengival, melena.
Disfagia, eructação, esofagite, flatulência, gastrite, gengivite, icterícia, alterações na função hepática, monilíase oral (candidíase), estomatite ulcerativa, aumento do apetite, colangite, gastrenterite, glossite, hemorragia gastrintestinal, insuficiência hepática, obstrução intestinal, perturbação rectal, tenesmo, perturbação da língua, perturbação dos dentes, úlcera bucal.
Sistema sanguíneo e linfático
Trombocitopenia, anemia, leucocitopenia, granulocitopenia, equimose.
Petéquia, anemia hipocrómica, neutropenia febril.
Linfadenopatia, anemia hemolítica, cianose, contusão fácil, granulocitose, linfangite, pancitopenia.
Local da injecção Dor no local da injecção. Inflamação no local de injecção, hemorragia no local de injecção.

7
Reacções adversas Sistema orgânico
Muito frequentes Frequentes Pouco frequentes Perturbações metabólicas e alimentares
Edema periférico, angio-edema, LDH aumentada, desidratação, hiperglicemia, aumento da fosfatase alcalina, edema, SGOT aumentada, perda de peso, aumento do azoto na ureia do sangue, hipocalcemia, SGPT aumentada.
Caquexia, aumento da creatinina, edema facial, hipercalcemia, hipoglicemia, hipocaliemia, bilirrubinemia, hiperlipemia, hipernatremia, hiperuricemia, hipomagnesemia, hipofosfatemia, sede, aumento de peso.
Sistema musculo-esquelético
Artralgia. Mialgia, dor óssea, cãibras nas pernas, miastenia.
Artrite, fractura óssea espontânea, disfunção das articulações, disfunção dos tendões.
Sistema nervoso Tonturas. Ansiedade, insónia, depressão, parestesia, hipestesia, sonolência, vasodilatação, alteração no andar.
Agitação, instabilidade emocional, tremores, ataxia, confusão, convulsão, alteração na coordenação, encefalopatia, hematoma subdural, hiperquinesia, nervosismo, neuralgia, neurite, aumento da salivação, perturbação da fala, pensamentos estranhos, contracção espasmódica, vertigens, retenção urinária.
Sistema respiratório Aumento da tosse, dispneia.
Rinite, infecção, broncoespasmo, sinusite, epistaxe, bronquite, alteração da voz, dor no peito, faringite, pneumonia.
Asma, efusão pleural, perturbação pulmonar, perturbação respiratória, expectoração aumentada, hipoventilação, hipoxia, perturbação pleural.
Pele e apêndices Prurido. Rash, urticária, sudação, sudação nocturna, perturbação cutânea, herpes simples.
Herpes zoster, rash maculopapuloso, secura da pele, acne, dermatite fúngica, eczema, furunculose, hipertrofia cutânea, drenagem de lesões, rash pustuloso.
Sentidos especiais Conjuntivite, ambliopia, visão anómala.
Diplopia, perturbação ocular, dor ocular, alteração do paladar, cataratas, secura ocular, hemorragia da retina, queratite, perturbação da lacrimação, otite externa, perda de paladar, zumbido.

8
Reacções adversas Sistema orgânico
Muito frequentes Frequentes Pouco frequentes Aparelho urogenital Infecção das vias
urinárias, disúria. Hemorragia vaginal, incontinência urinária, sensibilidade mamária, frequência urinária, albuminúria, hematúria, cálculo renal, insuficiência renal, noctúria, oligúria, perturbação urogenital, insuficiência na micção, fibromiomas uterinos degenerados.
4.9 Sobredosagem Em ensaios clínicos, verificaram-se casos de sobredosagem até 19,2 MBq/kg de Zevalin marcado radioactivamente com [90Y]. Foi observada toxicidade hematológica esperada, de grau 3 ou 4. Os doentes recuperaram destes sinais de toxicidade, e os casos de sobredosagem não estiveram associados a consequências graves ou fatais. Não há qualquer antídoto específico conhecido para a sobredosagem de Zevalin marcado radioactivamente com [90Y]. O tratamento consiste na interrupção da administração do Zevalin e em terapêutica de suporte que pode incluir factores de crescimento. Se disponível, deve ser administrado suporte autólogo das células estaminais para controlar a toxicidade hematológica. Em caso de administração acidental do radiofármaco precursor puro ítrio-90, consulte a informação sobre o ítrio-90. 5. PROPRIEDADES FARMACOLÓGICAS 5.1 Propriedades farmacodinâmicas Grupo farmacoterapêutico: anticorpos monoclonais, código ATC: L01X C O ibritumomab tiuxetan é um anticorpo monoclonal recombinante IgG1 kappa de murino, específico para o antigénio CD20 de células B. O ibritumomab tiuxetan tem como alvo o antigénio CD20 que se encontra à superfície dos linfócitos B malignos e normais. Durante a maturação das células B, o CD20 é inicialmente expresso na fase intermédia do linfoblasto B (pré-célula B), e perde-se durante a fase final da maturação das células B quando estas se transformam em células plasmáticas. Não se liberta da superfície da célula e não se internaliza aquando da ligação aos anticorpos. O anticorpo conjugado tem uma constante de afinidade aparente com o antigénio CD20 de aproximadamente 17 nM. O modo de ligação é muito restrito, sem qualquer reactividade cruzada com outros leucócitos ou outros tipos de tecido humano. Zevalin marcado radioactivamente com [90Y] liga-se especificamente a células B, incluindo células malignas de expressão CD20. O isótopo ítrio-90 é um emissor β puro e apresenta um comprimento médio de percurso de cerca de 5 mm. Este facto resulta na capacidade de destruir tanto as células alvo como as células circundantes. O tratamento prévio com rituximab é necessário para eliminar as células B em circulação, permitindo que o Zevalin forneça radiação de um modo mais específico aos linfomas. O rituximab é administrado numa dose reduzida em comparação com a monoterapia aprovada.

9
O tratamento com Zevalin marcado radioactivamente com [90Y] também conduz à depleção de células B CD20+ normais. A análise farmacodinâmica demonstrou que este foi um efeito temporário; a recuperação das células B normais iniciou-se num período de 6 meses e as contagens médias de células B situaram-se dentro do intervalo normal no período de 9 meses a seguir ao tratamento. A segurança e a eficácia do regime terapêutico com Zevalin foram avaliadas em dois ensaios multi-cêntricos incluindo um total de 197 indivíduos. O regime terapêutico com Zevalin foi administrado em duas fases (ver 4.2). A eficácia e a toxicidade de uma variação do regime terapêutico com Zevalin utilizando uma actividade reduzida de Zevalin [90Y], foi posteriormente avaliada num terceiro estudo que incluiu um total de 30 doentes com trombocitopenia ligeira (contagem de plaquetas entre 100.000 e 149.000 células/mm3). O Estudo 1 foi um estudo de um braço com 54 doentes com linfoma folicular em recidiva, refractário ao tratamento com rituximab. Os doentes eram considerados refractários se o seu último tratamento anterior com rituximab não tivesse resultado numa resposta completa ou parcial, ou se o tempo decorrido até à progressão da doença (TTP) tivesse sido < 6 meses. O endpoint primário de eficácia do estudo foi a taxa global de resposta (ORR) utilizando os Critérios de Resposta do Workshop Internacional (IWRC). Os endpoints secundários de eficácia incluíram o tempo decorrido até à progressão da doença (TTP) e a duração da resposta (DR). Numa análise secundária comparando a resposta objectiva ao regime terapêutico com Zevalin com a observada com o tratamento mais recente com rituximab, a duração mediana da resposta na sequência do regime terapêutico com Zevalin foi 6 meses versus 4 meses. O Quadro 1 resume os dados de eficácia resultantes deste estudo. O Estudo 2 foi um estudo multicêntrico, aleatorizado e controlado, comparando o regime terapêutico com Zevalin com o tratamento com rituximab. O estudo foi realizado em 143 doentes com linfoma não Hodgkin (LNH) de grau baixo ou folicular ou LNH de células B transformadas, em recidiva ou refractário. No total, 73 doentes foram submetidos ao regime terapêutico com Zevalin e 70 doentes receberam rituximab, administrado sob a forma de perfusão intravenosa a doses de 375 mg/m2, 4 vezes por semana. O endpoint primário de eficácia do estudo foi determinar a taxa global de resposta (ORR) utilizando os Critérios de Resposta do Workshop Internacional (IWRC) (ver Quadro 1). A taxa global de resposta (ORR) foi significativamente mais alta (80% vs. 56%, p = 0,002) para doentes tratados com o regime terapêutico de Zevalin. Os endpoints secundários, a duração da resposta e o tempo decorrido até à progressão não foram significativamente diferentes entre os dois grupos de tratamento.
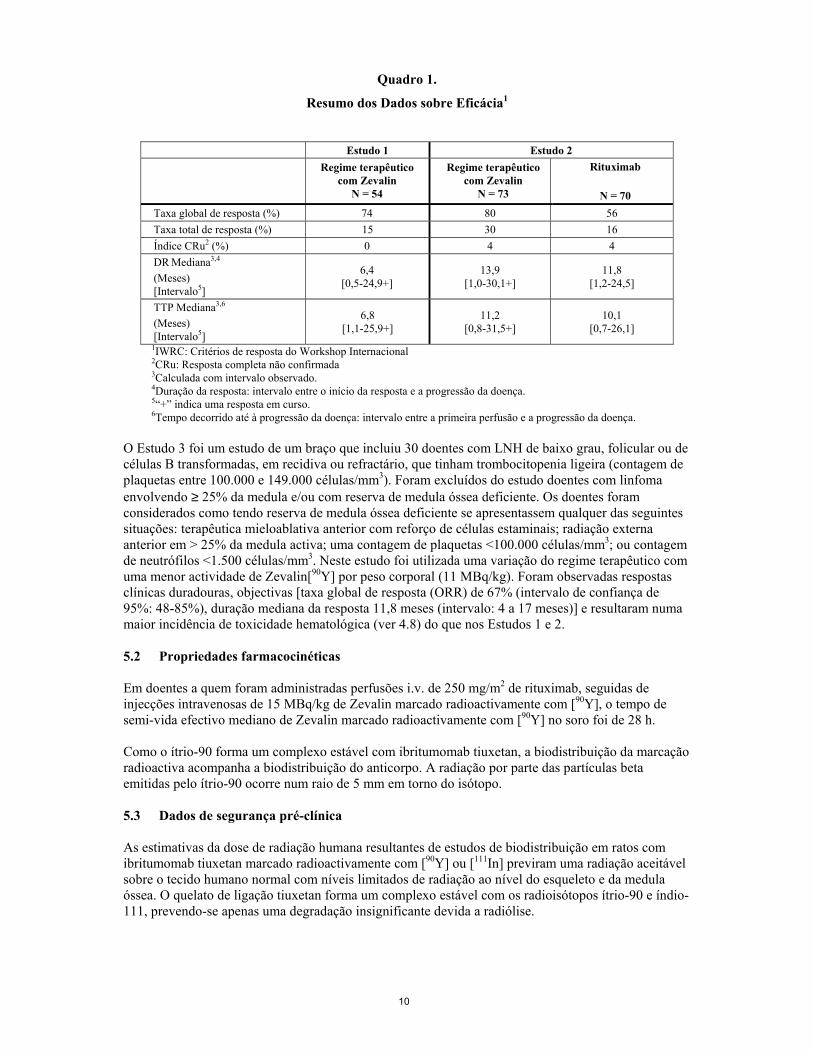
10
Quadro 1.
Resumo dos Dados sobre Eficácia1
Estudo 1 Estudo 2
Regime terapêutico
com Zevalin N = 54
Regime terapêutico com Zevalin
N = 73
Rituximab
N = 70 Taxa global de resposta (%) 74 80 56 Taxa total de resposta (%) 15 30 16 Índice CRu2 (%) 0 4 4 DR Mediana3,4 (Meses) [Intervalo5]
6,4 [0,5-24,9+]
13,9 [1,0-30,1+]
11,8 [1,2-24,5]
TTP Mediana3,6 (Meses) [Intervalo5]
6,8 [1,1-25,9+]
11,2 [0,8-31,5+]
10,1 [0,7-26,1]
1IWRC: Critérios de resposta do Workshop Internacional 2CRu: Resposta completa não confirmada 3Calculada com intervalo observado. 4Duração da resposta: intervalo entre o início da resposta e a progressão da doença. 5“+” indica uma resposta em curso. 6Tempo decorrido até à progressão da doença: intervalo entre a primeira perfusão e a progressão da doença.
O Estudo 3 foi um estudo de um braço que incluiu 30 doentes com LNH de baixo grau, folicular ou de células B transformadas, em recidiva ou refractário, que tinham trombocitopenia ligeira (contagem de plaquetas entre 100.000 e 149.000 células/mm3). Foram excluídos do estudo doentes com linfoma envolvendo ≥ 25% da medula e/ou com reserva de medula óssea deficiente. Os doentes foram considerados como tendo reserva de medula óssea deficiente se apresentassem qualquer das seguintes situações: terapêutica mieloablativa anterior com reforço de células estaminais; radiação externa anterior em > 25% da medula activa; uma contagem de plaquetas <100.000 células/mm3; ou contagem de neutrófilos <1.500 células/mm3. Neste estudo foi utilizada uma variação do regime terapêutico com uma menor actividade de Zevalin[90Y] por peso corporal (11 MBq/kg). Foram observadas respostas clínicas duradouras, objectivas [taxa global de resposta (ORR) de 67% (intervalo de confiança de 95%: 48-85%), duração mediana da resposta 11,8 meses (intervalo: 4 a 17 meses)] e resultaram numa maior incidência de toxicidade hematológica (ver 4.8) do que nos Estudos 1 e 2. 5.2 Propriedades farmacocinéticas Em doentes a quem foram administradas perfusões i.v. de 250 mg/m2 de rituximab, seguidas de injecções intravenosas de 15 MBq/kg de Zevalin marcado radioactivamente com [90Y], o tempo de semi-vida efectivo mediano de Zevalin marcado radioactivamente com [90Y] no soro foi de 28 h. Como o ítrio-90 forma um complexo estável com ibritumomab tiuxetan, a biodistribuição da marcação radioactiva acompanha a biodistribuição do anticorpo. A radiação por parte das partículas beta emitidas pelo ítrio-90 ocorre num raio de 5 mm em torno do isótopo. 5.3 Dados de segurança pré-clínica As estimativas da dose de radiação humana resultantes de estudos de biodistribuição em ratos com ibritumomab tiuxetan marcado radioactivamente com [90Y] ou [111In] previram uma radiação aceitável sobre o tecido humano normal com níveis limitados de radiação ao nível do esqueleto e da medula óssea. O quelato de ligação tiuxetan forma um complexo estável com os radioisótopos ítrio-90 e índio-111, prevendo-se apenas uma degradação insignificante devida a radiólise.

11
Os estudos de toxicidade de dose única e de dose repetida do composto não radioactivo realizados em macacos cynomolgus não indicaram qualquer outro risco para além da depleção prevista das células B, decorrente da utilização de ibritumomab tiuxetan por si só, ou em combinação com rituximab. Não foram efectuados estudos sobre a toxicidade reprodutiva e de desenvolvimento, bem como sobre o potencial mutagénico e carcinogénico (ver secções 4.4 e 4.6). Não foram realizados estudos sobre o potencial mutagénico e carcinogénico de Zevalin. Devido à exposição a radiações ionizantes, resultante da marcação radioactiva, é necessário considerar um risco de efeitos mutagénicos e carcinogénicos. 5.4 Dosimetria de radiação O ítrio-90 decai por emissão de partículas beta de alta energia com uma semi-vida física de 64,1 horas (2,67 dias). O produto do decaimento radioactivo é o zircónio-90 estável. O comprimento do percurso das emissões beta (χ90) pelo ítrio-90 no tecido é de 5 mm. Foram efectuadas análises da dose de radiação absorvida esperada, utilizando produção de imagens quantitativas com o emissor de raios gama Zevalin marcado radioactivamente com [111In], análises sanguíneas e o programa informático MIRDOSE3. A dose de Zevalin marcado radioactivamente com [111In] para imagiologia foi sempre dada imediatamente após uma perfusão com rituximab a 250 mg/m², para destruir as células CD20+ periféricas e optimizar a biodistribuição. Após a administração de Zevalin marcado radioactivamente com [111In], foram obtidas imagens de todo o corpo em oito períodos de tempo, no máximo, com captação tanto de imagens anteriores como posteriores. Foram recolhidas amostras de sangue, em oito ocasiões diferentes, no máximo, utilizadas para calcular os tempos de residência para a medula vermelha. Com base em estudos de dosimetria com Zevalin marcado radioactivamente com [111In], realizados em 179 doentes tratados em quatro estudos clínicos, a dosimetria de radiação estimada para cada um dos órgãos individuais, após a administração de Zevalin marcado radioactivamente com [90Y] em actividades de 15 MBq/kg e 11 MBq/kg, foi calculada de acordo com a Dosimetria Clínica de Radiação Interna (MIRD) (Quadro 1). As doses estimadas de radiação absorvida por órgãos normais foram francamente inferiores aos limites superiores de segurança reconhecidos. Os resultados da dosimetria por doente individual não são indicadores de toxicidade com Zevalin marcado radioactivamente com [90Y] e, consequentemente, não se recomenda o procedimento geral da dosimetria.
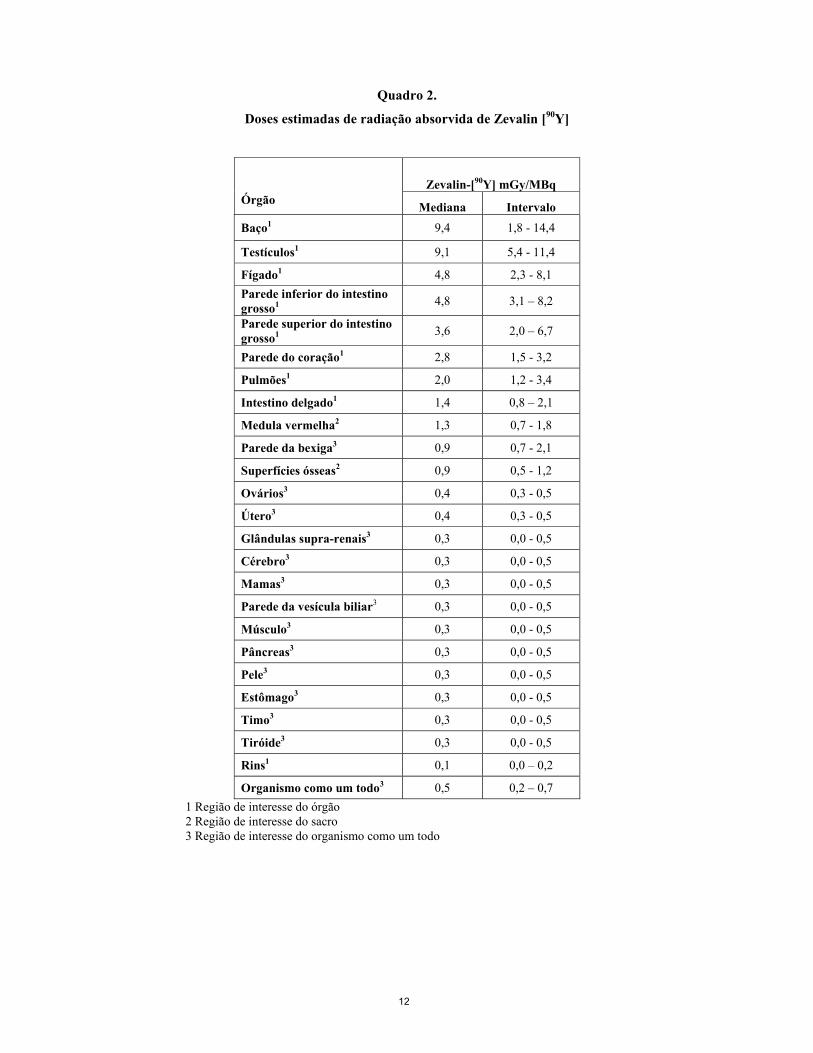
12
Quadro 2.
Doses estimadas de radiação absorvida de Zevalin [90Y]
Zevalin-[90Y] mGy/MBq Órgão Mediana Intervalo
Baço1 9,4 1,8 - 14,4
Testículos1 9,1 5,4 - 11,4
Fígado1 4,8 2,3 - 8,1 Parede inferior do intestino grosso1 4,8 3,1 – 8,2
Parede superior do intestino grosso1 3,6 2,0 – 6,7
Parede do coração1 2,8 1,5 - 3,2
Pulmões1 2,0 1,2 - 3,4
Intestino delgado1 1,4 0,8 – 2,1
Medula vermelha2 1,3 0,7 - 1,8
Parede da bexiga3 0,9 0,7 - 2,1
Superfícies ósseas2 0,9 0,5 - 1,2
Ovários3 0,4 0,3 - 0,5
Útero3 0,4 0,3 - 0,5
Glândulas supra-renais3 0,3 0,0 - 0,5
Cérebro3 0,3 0,0 - 0,5
Mamas3 0,3 0,0 - 0,5
Parede da vesícula biliar3 0,3 0,0 - 0,5
Músculo3 0,3 0,0 - 0,5
Pâncreas3 0,3 0,0 - 0,5
Pele3 0,3 0,0 - 0,5
Estômago3 0,3 0,0 - 0,5
Timo3 0,3 0,0 - 0,5
Tiróide3 0,3 0,0 - 0,5
Rins1 0,1 0,0 – 0,2
Organismo como um todo3 0,5 0,2 – 0,7 1 Região de interesse do órgão 2 Região de interesse do sacro
3 Região de interesse do organismo como um todo

13
6. INFORMAÇÕES FARMACÊUTICAS 6.1 Lista dos excipientes Frasco de ibritumomab tiuxetan:
Cloreto de sódio Água para injectáveis
Frasco de acetato de sódio:
Acetato de sódio Água para injectáveis
Frasco de formulação tamponada:
Solução de albumina humana Fosfato dissódico dodecahidratado Hidróxido de sódio Dihidrogenofosfato de potássio Cloreto de potássio Ácido pentético Ácido clorídrico, diluído Água para injectáveis
6.2 Incompatibilidades Este medicamento não deve ser misturado com outros medicamentos 6.3 Prazo de validade 3 anos Recomenda-se a utilização imediata após a marcação radioactiva. A estabilidade química e física durante a utilização foi demonstrada durante 8 horas a uma temperatura entre 2°C e 8°C e protegida da luz. 6.4 Precauções especiais de conservação Conservar entre 2°C e 8°C (no frigorífico). Não congelar. Conservar na embalagem de origem a fim de proteger da luz. Após a marcação radioactiva: Conservar entre 2°C e 8°C (no frigorífico) e proteger da luz. A conservação deverá ser feita em conformidade com as regulamentações nacionais relativas a materiais radioactivos. 6.5 Natureza e conteúdo do recipiente 2 ml de solução de ibritumomab tiuxetan num frasco (vidro de tipo I) com fecho de borracha (bromobutilo revestido a teflon) 2ml de solução de acetato de sódio num frasco para injectáveis (vidro de tipo I) com fecho de borracha (bromobutilo revestido a teflon) 10 ml de formulação tamponada num frasco para injectáveis (vidro de tipo I) com fecho de borracha (bromobutilo revestido a teflon) frasco de reacção vazio de 10 ml (vidro de tipo I) com fecho de borracha (bromobutilo revestido a teflon)

14
Tamanho da embalagem de 1 kit 6.6 Instruções de utilização e manipulação e eliminação Leia atentamente as instruções completas antes de iniciar o procedimento de preparação. Devem ser utilizadas técnicas assépticas e as precauções adequadas à manipulação de materiais radioactivos. Devem utilizar-se luvas à prova de água na preparação e durante a determinação da pureza radioquímica de Zevalin marcado radioactivamente com [90Y]. Não foram observadas quaisquer incompatibilidades entre o Zevalin e os conjuntos de perfusão. A administração de produtos radiofarmacêuticos cria riscos para outras pessoas decorrentes da radiação externa ou contaminação de derrames de urina, vómitos, etc. Assim, devem tomar-se as precauções relativas a protecção contra radiação em conformidade com a regulamentação local. Quaisquer produtos não utilizados ou material de desperdício devem ser eliminados de acordo com os requisitos locais. Os materiais contaminados devem ser eliminados como resíduos radioactivos pela via autorizada. Características do ítrio-90 • Recomendam-se as seguintes características mínimas do ítrio-90: Concentração de radioactividade no momento da utilização
1,67 a 3,34 GBq/ml
Actividade extraível total a fornecer no momento da utilização
≥ 1,48 GBq correspondendo a 0,44 ml a 0,89 ml de solução de ítrio-90
Concentração de HCl 0,035-0,045 M Identificação do cloreto Positivo Identificação do ítrio Positivo Pureza radioquímica da solução de cloreto de ítrio-90
≥ 95% de ítrio 90 iónico livre
Endotoxinas bacterianas ≤ 150 EU/ml Esterilidade Nenhum crescimento Pureza radionuclídica do teor em estrôncio-90 ≤ 0,74 MBq de estrôncio-90 /
37 GBq de ítrio-90 Impurezas metálicas Metais totais* ≤ 50 ppm Metais individuais* ≤ 10 ppm de cada * Os metais a incluir têm que se basear no processo de fabrico específico. O controlo destes metais pode ser obtido quer através da validação do processo como do teste de libertação.

15
• Testes adicionais eventualmente necessários para a avaliação da conformidade: Impurezas específicas do processo Carbono orgânico total (ex.: quelantes orgânicos) Abaixo do limite de quantificação* Resíduos do processo (ex.: amónia, nitrato) Abaixo do limite de quantificação* Impurezas Alfa totais Abaixo do limite de quantificação* Outras impurezas Beta totais (não estrôncio-90) Abaixo do limite de quantificação* Impurezas Gama totais Abaixo do limite de quantificação* * Necessita de ser incluído como teste de libertação ou controlado através da validação do processo se se situar acima do limite de quantificação Instruções para marcação radioactiva do Zevalin com Ítrio-90: Para a preparação de Zevalin marcado radioactivamente com [90Y], tem que ser utilizado cloreto de ítrio-90 estéril, isento de pirogénios, da qualidade especificada acima. Antes da marcação radioactiva, trazer o kit de Zevalin refrigerado à temperatura ambiente (25°C). Limpar os fechos de borracha de todos os frascos do kit e do frasco de cloreto de ítrio-90 refrigerados com um algodão embebido em álcool e deixar secar ao ar. Colocar o frasco de reacção do kit numa protecção adequada para utilização (plástico revestido por chumbo). Passo 1: Transferir a solução de acetato de sódio para o frasco de reacção Utilizando uma seringa esterilizada de 1 ml, transferir a solução de acetato de sódio para o frasco de reacção. O volume da solução de acetato de sódio adicionada é equivalente a 1,2 vezes o volume do cloreto de ítrio-90 a ser transferido no passo 2. Passo 2: Transferir o cloreto de ítrio-90 para o frasco de reacção De forma asséptica, transferir 1500 MBq de cloreto de ítrio-90 com uma seringa esterilizada de 1 ml para o frasco de reacção que contém a solução de acetato de sódio transferida no passo 1. Misturar completamente revestindo toda a superfície interna do frasco de reacção. Misturar por inversão, com rotação do recipiente, evitando a formação de espuma ou agitar a solução. Passo 3: Transferir a solução de ibritumomab tiuxetan para o frasco de reacção Utilizando uma seringa esterilizada de 2 a 3 ml, transferir 1,3 ml de solução de ibritumomab tiuxetan para o frasco de reacção. Misturar completamente, revestindo toda a superfície interna do frasco de reacção. Misturar por inversão, com rotação do recipiente, evitando a formação de espuma ou agitar a solução. Incubar a solução de cloreto de ítrio-90 /acetato/ibritumomab tiuxetan à temperatura ambiente durante cinco minutos. Um tempo de marcação superior a seis minutos ou inferior a quatro minutos dará origem a uma radioincorporação inadequada. Passo 4: Adicionar a formulação tamponada ao frasco de reacção Utilizando uma seringa de 10 ml com uma agulha de maior calibre (18-20 G), recolher formulação tamponada, resultando num volume combinado total de 10 ml. Após o período de incubação de cinco minutos, adicionar a formulação tamponada ao frasco de reacção, concluindo a incubação. Imediatamente antes desta adição, retirar um volume igual de ar do frasco de reacção para normalizar a pressão. Adicionar cuidadosamente a formulação tamponada, pela parte lateral do frasco de reacção. Não produzir espuma, não abanar nem agitar a mistura. Passo 5: Proceder ao ensaio da solução de Zevalin marcado radioactivamente com [90Y] quanto à sua radioactividade específica. A pureza radioquímica do preparado marcado radioactivamente aplica-se enquanto mais de 95% de ítrio-90 estiver incorporado no anticorpo monoclonal. Antes da administração ao doente, a percentagem de radioincorporação do Zevalin marcado radioactivamente com [90Y] preparado tem que ser verificada, de acordo com o procedimento descrito a seguir. Atenção: A dose do doente não pode ultrapassar 1200 MBq.

16
Instruções para determinar a percentagem de radioincorporação O ensaio de radioincorporação para a pureza radioquímica é realizado por cromatografia instantânea em camada fina (ITLC) e deve ser realizado de acordo com o seguinte procedimento:
Materiais necessários não fornecidos no kit de Zevalin: - Câmara de revelação para cromatografia - Fase móvel: solução (a 0,9%) de 9 mg/ml de cloreto de sódio, isenta de bacteriostáticos - Tiras para cromatografia instantânea de camada fina (ITLC) (por ex., placas de gel de sílica
(SG) para ITLC, Art.º N.º 61885, Gelman Sciences, Ann Arbor, Michigan, EUA ou equivalente; dimensões: 0,5 cm x 6 cm, origem: 1,4 cm, linha de corte: 3,5 cm, frente do solvente: 5,4 cm)
- Frascos de cintilação - Cocktail de cintilação líquida (por exemplo, Ultima Gold, catálogo N.º 6013329, Packard
Instruments, EUA ou equivalente) Procedimento de ensaio: 1.) Adicionar aproximadamente 0,8 ml da solução de cloreto de sódio a 0,9% à câmara de revelação, assegurando-se de que o líquido não atingirá a marca de origem de 1,4 cm na tira para cromatografia instantânea de camada fina (ITCL). 2.) Utilizando uma seringa para insulina de 1 ml com uma agulha de 25 a 26 G, colocar uma gota em suspensão (7-10 µL) de Zevalin marcado radioactivamente com [90Y] sobre a tira para cromatografia instantânea de camada fina na sua origem . Colocar uma gota em cada tira de cada vez, pondo a revelar três tiras ITLC. Pode ser necessário efectuar uma diluição (1:100) antes da aplicação do Zevalin marcado radioactivamente com [90Y] nas tiras ITLC. 3.) Colocar a tira de ITLC na câmara de revelação e deixar a frente do solvente passar para além da marca dos 5,4 cm. 4.) Retirar a tira de ITLC e cortar ao meio na linha de corte de 3,5 cm. Colocar cada metade em frascos de cintilação separados aos quais se deve acrescentar 5 ml da mistura LSC (por exemplo, Ultima Gold nº de catálogo 6013329, Packard Instruments, EUA ou equivalente). Contar cada frasco num contador beta ou num contador apropriado durante um minuto (CPM), registar as contagens líquidas, corrigidas em relação às anteriores. 5.) Calcular a Pureza Radioquímica média (PRQ) da seguinte maneira: 6.) % média de PRQ = CPM líquidas da metade inferior x 100 CPM líquidas da metade superior + CPM líquidas da metade inferior 7.) Se a pureza radioquímica média for inferior a 95%, a preparação não deve ser administrada. 7. TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO Schering AG 13342 Berlim Alemanha 8. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO EU/1/03/264/001 9. DATA DA PRIMEIRA AUTORIZAÇÃO/RENOVAÇÃO DA AUTORIZAÇÃO DE
INTRODUÇÃO NO MERCADO

17
10. DATA DA REVISÃO DO TEXTO

18
ANEXO II A. FABRICANTE DA SUBSTÂNCIA ACTIVA DE ORIGEM
BIOLÓGICA E TITULAR DA AUTORIZAÇÃO DE FABRICO RESPONSÁVEL PELA LIBERTAÇÃO DO LOTE
B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO
C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO

19
A. FABRICANTE DA SUBSTÂNCIA ACTIVA DE ORIGEM BIOLÓGICA E TITULAR DA AUTORIZAÇÃO DE FABRICO RESPONSÁVEL PELA LIBERTAÇÃO DO LOTE
Nome e endereço do fabricante da(s) substância(s) activa(s) de origem biológica IDEC Pharmaceuticals Corp. 3030 Callan Road San Diego, CA 92121 EUA Nome e endereço do fabricante responsável pela libertação do lote Schering AG Müllerstrasse 178 13342 Berlin ALEMANHA B. CONDIÇÕES DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO • CONDIÇÕES OU RESTRIÇÕES RELATIVAS AO FORNECIMENTO E À
UTILIZAÇÃO IMPOSTAS AO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO
Medicamento sujeito a prescrição médica restrita (ver anexo I: resumo das características do medicamento, 4.2.). • OUTRAS CONDIÇÕES C. OBRIGAÇÕES ESPECÍFICAS A SEREM CUMPRIDAS PELO TITULAR DA
AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO O titular da autorização de introdução no mercado deverá executar o estudo clínico abaixo referido dentro dos prazos indicados; os resultados desse estudo estarão na base da reavaliação anual da relação risco/benefício. Área Descrição Data de
cumprimento
ASPECTOS CLÍNICOS
Estudo Schering 304820 em curso
Este estudo destina-se a avaliar a eficácia e a segurança do tratamento com ibritumomab tiuxetan para utilização com 90ítrio em comparação a interrupção de tratamento em pacientes com linfoma folicular não-Hodgkin em estádio III ou IV que obtiveram uma remissão parcial ou total após a quimioterapia de primeira linha. Este estudo é um estudo de fase III prospectivo multicêntrico com distribuição aleatória. Os resultados da eficácia (sobrevida sem progressão) estarão disponíveis dois anos após a inclusão do último paciente, prevista para 2006. Por razões estatísticas, não poderão ser realizadas avaliações intercalares da eficácia. Serão fornecidas todos os anos avaliações médicas da segurança. O protocolo do estudo pode ser obtido mediante pedido.
O relatório final será apresentado até 31.12.07.

20
ANEXO III
ROTULAGEM E FOLHETO INFORMATIVO

21
A. ROTULAGEM

22
INDICAÇÕES A INCLUIR NA EMBALAGEM EXTERIOR OU, CASO ESTA NÃO EXISTA, NO ACONDICIONAMENTO PRIMÁRIO {NATUREZA/TIPO} 1. DENOMINAÇÃO DO MEDICAMENTO Zevalin 1,6 mg/ ml Kit para preparação radiofarmacêutica para perfusão Ibritumomab tiuxetan 2. DESCRIÇÃO DO(S) PRINCÍPIO(S) ACTIVO(S) Um ml contém ibritumomab* tiuxetan 1,6 mg por ml Um frasco contém 3,2 mg de ibritumomab tiuxetan *produzido geneticamente por linhas celulares de Ovário de Hamster Chinês (OHC) e conjugado com o agente quelante MX-DTPA 3. LISTA DOS EXCIPIENTES 4. FORMA FARMACÊUTICA E CONTEÚDO Kit para preparação radiofarmacêutica para perfusão (em frascos): 3,2 mg de ibritumomab tiuxetan numa solução de cloreto de sódio 2 ml de solução de acetato de sódio 10 ml de formulação tamponada: solução de albumina humana, fosfato dissódico dodecahidratado, hidróxido de sódio, dihidrogenofosfato de potássio, cloreto de potássio, ácido pentético, ácido clorídrico diluído, água para injectáveis Frasco de reacção vazio (10 ml) 5. MODO E VIA(S) DE ADMINISTRAÇÃO Tem que ser combinado com ítrio [90Y] antes da administração por via intravenosa. Consultar o Resumo das Características do Medicamento do ítrio-90 para instruções sobre a preparação e a utilização. 6. ADVERTÊNCIA ESPECIAL DE QUE O MEDICAMENTO DEVE SER MANTIDO
FORA DO ALCANCE E DA VISTA DAS CRIANÇAS Manter fora do alcance e da vista das crianças 7. OUTRAS ADVERTÊNCIAS ESPECIAIS, SE NECESSÁRIO Deve ser administrado apenas por pessoal autorizado

23
8. PRAZO DE VALIDADE VAL. (MM/AAAA) Recomenda-se a utilização imediata após a marcação radioactiva. A estabilidade química e física durante a utilização foi demonstrada durante 8 horas a uma temperatura entre 2°C e 8°C e protegida da luz. 9. CONDIÇÕES ESPECIAIS DE CONSERVAÇÃO Conservar a 2°C - 8°C (no frigorífico) Não congelar Conservar no recipiente de origem a fim de proteger da luz 10. CUIDADOS ESPECIAIS QUANTO À ELIMINAÇÃO DO MEDICAMENTO NÃO
UTILIZADO OU DOS RESÍDUOS PROVENIENTES DESSE MEDICAMENTO, SE FOR CASO DISSO
11. NOME E ENDEREÇO DO TITULAR DA AUTORIZAÇÃO DE INTRODUÇÃO NO
MERCADO Schering AG, 13342 Berlim, Alemanha 12. NÚMERO(S) DA AUTORIZAÇÃO DE INTRODUÇÃO NO MERCADO EU/1/03/264/001 13. NÚMERO DO LOTE DE FABRICO Lote {número} 14. CLASSIFICAÇÃO GERAL RELATIVA AO FORNECIMENTO Medicamento sujeito a receita médica 15. INSTRUÇÕES DE UTILIZAÇÃO

24
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {NATUREZA/TIPO} 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO Zevalin 1,6 mg/ml i.v. Solução de ibritumomab tiuxetan 2. MODO DE ADMINISTRAÇÃO 3. PRAZO DE VALIDADE VAL. (MM/AAAA) 4. NÚMERO DO LOTE Lote {número} 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 3,2 mg/2 ml

25
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {NATUREZA/TIPO} 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO Zevalin 1,6 mg/ml i.v. Solução de acetato de sódio 2. MODO DE ADMINISTRAÇÃO 3. PRAZO DE VALIDADE VAL. (MM/AAAA) 4. NÚMERO DO LOTE Lote {número} 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 2 ml

26
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {NATUREZA/TIPO} 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO Zevalin 1,6 mg/ml i.v. Solução de formulação tamponada 2. MODO DE ADMINISTRAÇÃO 3. PRAZO DE VALIDADE VAL. (MM/AAAA) 4. NÚMERO DO LOTE Lote {número} 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE 10 ml

27
INDICAÇÕES MÍNIMAS A INCLUIR EM PEQUENAS UNIDADES DE ACONDICIONAMENTO PRIMÁRIO {NATUREZA/TIPO} 1. DENOMINAÇÃO DO MEDICAMENTO E VIA(S) DE ADMINISTRAÇÃO Zevalin 1,6 mg/ml i.v. Frasco de reacção 2. MODO DE ADMINISTRAÇÃO 3. PRAZO DE VALIDADE VAL. (MM/AAAA) 4. NÚMERO DO LOTE Lote {número} 5. CONTEÚDO EM TERMOS DE PESO, VOLUME OU UNIDADE vazio

28
B. FOLHETO INFORMATIVO

29
FOLHETO INFORMATIVO
Leia atentamente este folheto antes de tomar o medicamento. - Conserve este folheto. Pode ter necessidade de o reler. - Se tiver questões ou dúvidas sobre alguma coisa, pergunte ao seu médico ou farmacêutico. - Este medicamento foi-lhe prescrito a si pessoalmente, não deverá dá-lo a outras pessoas. Poderá ser-lhes prejudicial, ainda que apresentem os mesmos sintomas. Neste folheto: 1. O que é Zevalin e para que é utilizado 2. Antes de utilizar Zevalin 3. Como utilizar Zevalin 4. Efeitos secundários possíveis 5. Conservação do Zevalin 6. Outras informações Zevalin 1,6 mg/ ml Kit para preparação radiofarmacêutica para perfusão Ibritumomab tiuxetan A substância activa é ibritumomab tiuxetan (3,2 mg/ 2 ml). Os outros ingredientes são cloreto de sódio; acetato de sódio; hidróxido de sódio; solução de albumina humana; fosfato dissódico dodecahidratado; dihidrogenofosfato de potássio; cloreto de potássio; ácido clorídrico diluido; ácido pentético; água para injectáveis. Titular da Autorização de Introdução no Mercado e Fabricante: Schering AG, 13342 Berlim, Alemanha 1. O que é Zevalin e para que é utilizado O Zevalin pertence a uma família de medicamentos (designados por anticorpos monoclonais selectivos) que atingem algumas células do corpo. É fornecido sob a forma de uma solução num kit de quatro frascos. É utilizado com uma substância radioactiva designada por ítrio, que dá ao Zevalin uma carga radioactiva muito pequena para utilizar quando atingir o seu alvo. Zevalin é utilizado para o tratamento do linfoma não Hodgkin. Trata-se de uma doença maligna de certos glóbulos brancos designados por linfócitos. Zevalin é utilizado no caso de um tratamento anterior não ter dado resultado ou ter deixado de dar resultado. Os glóbulos brancos designados por linfócitos participam normalmente no combate do organismo contra a doença. Em pessoas com linfoma não Hodgkin, o organismo produz demasiados linfócitos anormais, que não funcionam correctamente e que podem tomar o lugar das células sãs na medula óssea (onde se forma a maior parte das células sanguíneas novas) e no sangue. Tal pode conduzir a infecções, anemia, hematoma, hemorragia excessiva ou até mesmo insuficiência dos órgãos. A doença pode também causar um aumento do tamanho dos nódulos linfáticos e de outros órgãos como, por exemplo, o fígado e o baço.

30
Zevalin é um tratamento com um alvo específico que visa os glóbulos brancos anormais. Depois de ser combinado com a substância radioactiva ítrio, Zevalin transporta doses muito pequenas de radiação para os locais de crescimento de células anómalas e mata-as. 2. Antes de utilizar Zevalin Não tome Zevalin:
• Se tem uma alergia (se é hipersensível) ao componente activo (ibritumomab tiuxetan), ao cloreto de ítrio, a proteínas de rato ou a qualquer outro ingrediente do Zevalin. Ver a lista na área sombreada, em cima à direita. Se acha que este é o seu caso, avise o seu médico.
• Se estiver grávida ou com hipóteses de o estar. Não deve tomar Zevalin se estiver grávida; ver conselhos pormenorizados, mais à direita.
• Se estiver a amamentar. Não deve tomar Zevalin se estiver a amamentar; ver conselhos pormenorizados, mais à direita. Informe o seu médico.
Poderá necessitar de especial cuidado com Zevalin O seu médico terá que pensar cuidadosamente se deve ou não tomar Zevalin em alguns casos:
• Se um quarto ou mais da sua medula óssea contiver células anómalas malignas
• Se mais de um quarto da sua medula óssea tiver sido submetida a radiação externa (um tipo de radioterapia)
• Se o número das suas plaquetas sanguíneas for menor que 100.000/mm3 ou se o número de glóbulos brancos for inferior a 1.500/mm3.
• Se tiver feito anteriormente um transplante de medula óssea ou tiver recebido células sanguíneas indiferenciadas.
Se tiver recebido outro tipo de tratamento com anticorpos antes do tratamento com Zevalin, é possível que tenha uma reacção alérgica (de hipersensibilidade). Poderá ser necessário, portanto, fazer análises para detecção de anticorpos especiais. O seu médico dir-lhe-á se este é o seu caso. Após o tratamento com Zevalin, e se o seu médico tencionar tratá-lo com qualquer outro anticorpo, informe o médico sobre o seu tratamento com Zevalin. Isto pode ajudar a evitar uma possível reacção alérgica (de hipersensibilidade). A segurança da imunização com qualquer vacina, em especial vacinas de vírus vivos, após terapêutica com Zevalin não foi estudada. A capacidade do organismo de produzir uma resposta a qualquer vacina também não foi estudada. Zevalin não deve ser administrado a crianças e adolescentes A segurança e o efeito de Zevalin em crianças e adolescentes ainda não foram testados. Conduzir e utilizar máquinas É possível que o Zevalin afecte a sua capacidade de conduzir ou utilizar quaisquer ferramentas ou máquinas, uma vez que as tonturas são um efeito secundário muito frequente. Tome cuidado e não tente conduzir nem utilizar máquinas até ter a certeza de não estar afectado. Tomar quaisquer outros medicamentos Informe o seu médico se estiver a tomar ou tiver tomado recentemente outros medicamentos, incluindo medicamentos sem receita médica.

31
3. Como utilizar Zevalin O Zevalin ser-lhe-á administrado apenas por um profissional experiente e só será manuseado pelo pessoal do hospital devidamente qualificado. Antes de lhe ser administrado o Zevalin, ser-lhe-á administrado outro medicamento – denominado rituximab – para aumentar o efeito de Zevalin. As primeiras duas perfusões do seu tratamento são de rituximab. Irá ter 2 consultas no hospital, com uma semana de intervalo.
• Na primeira consulta (dia 1) ser-lhe-á administrada
o Uma perfusão de rituximab
• Na segunda consulta (dia 8) ser-lhe-á administrada
o Uma perfusão de rituximab
o Uma perfusão de Zevalin logo a seguir
No total ser-lhe-ão administradas 3 perfusões, o que concluirá o tratamento. Uma perfusão significa que o medicamento é administrado gota a gota na veia. As perfusões de Zevalin têm normalmente uma duração de cerca de 10 minutos. As perfusões de rituximab podem durar uma ou duas horas. Que quantidade de Zevalin é administrada O médico calculará a sua dose individual. Esta dependerá do seu peso corporal e do número de plaquetas sanguíneas. Depois de tomar Zevalin A quantidade de radiação à qual o seu organismo estará exposto durante um ciclo de Zevalin é inferior à da radioterapia. Com este tipo de radioactividade, não há efeito directo da radiação fora do organismo. Não estará a expor outras pessoas à radiação. Uma pequena parte dessa radioactividade sairá do seu organismo através da urina. O restante deteriora-se no organismo, não deixando quaisquer resíduos radioactivos. Existe apenas uma simples precaução a tomar devido à radioactividade:
• Lave as mãos com especial cuidado depois de urinar durante uma semana após a perfusão de Zevalin.
• O seu médico retirar-lhe-á pequenas amostras de sangue a intervalos regulares por forma a monitorizar os números de plaquetas sanguíneas e de glóbulos brancos. Estes tem tendência a revelar alguma escassez por volta do dia 60 após o início do tratamento. Consequentemente, o seu médico certificar-se-á de que são tomadas todas as medidas no caso de ocorrerem infecções.
Precauções relativamente à gravidez • Se tiver possibilidades de engravidar, use um contraceptivo fiável (por exemplo, a
pílula, implantes, diafragma, preservativos ou DIUs). A possibilidade de uma gravidez deve ser excluída antes do início do tratamento, durante o tratamento e durante um ano após o tratamento.
• Os homens aos quais foi administrado Zevalin e que podem vir a ser pais devem adoptar um contraceptivo fiável durante o tratamento e no ano seguinte ao fim do tratamento.
Aleitamento

32
• Consulte o seu médico antes de começar a amamentar após a conclusão do tratamento, pois os anticorpos são excretados no leite materno.
4. Efeitos secundários possíveis Zevalin pode ter efeitos secundários e é provável que a maioria das pessoas que o toma tenha alguns deles. Devido ao facto de muitos efeitos secundários se assemelharem aos efeitos da doença, nem sempre se sabe ao certo se a reacção observada nos nossos estudos se deve ou não ao Zevalin.
• Se estiver preocupado(a) com algum destes efeitos secundários, consulte o seu médico.
Segue-se uma lista dos efeitos secundários possíveis de acordo com as partes do organismo que afectam e a sua frequência. Muito frequentes significa que 10 ou mais pessoas em cada 100 têm probabilidades de os ter. Frequentes: entre 1 a 10 pessoas em cada 100 têm probabilidades de os ter. Não frequentes: menos de 1 pessoa em cada 100 têm probabilidades de ter estes efeitos. Organismo em geral e total Muito frequentes: Fraqueza, arrepios, febre, infecção bacteriana, cefaleia, dor, irritação na garganta, dores de estômago, dores nas costas. Frequentes: Rubor, sensação geral de mal-estar; dores no peito, no pescoço, nas axilas, na zona pélvica ou na área do tumor; distensão do estômago; sintomas semelhantes aos da gripe; infecções fúngicas (por exemplo, sapinhos); afecções dos tecidos húmidos que revestem diversas partes do corpo como, por exemplo, a boca e o nariz, reacções alérgicas. Não frequentes: Sintomas semelhantes aos de uma constipação, temperatura corporal baixa, dor lateral; ferimento acidental; pele quente, sensível e vermelha (por vezes febre). Sistema cardio-circulatório Frequentes: Hipotensão, hipertensão, batimento cardíaco acelerado ou irregular, pele pálida. Não frequentes: Alterações na frequência cardíaca, perturbações cardio-circulatórias, aumento do número de pulsações, dores no peito, doença do músculo cardíaco, perturbações nos vasos sanguíneos no coração e à volta deste, incluindo coágulos e fluxo sanguíneo reduzido; problemas cardíacos que provocam retenção de líquido nas pernas e nos pés, hemorragia, desmaio, enxaqueca; artéria pulmonar bloqueada, inchaço e vermelhidão ao longo de uma veia, dilatação dos vasos sanguíneos, aumento da pressão venosa. Sistema digestivo, fígado e vesícula biliar Muito frequentes: Sensação de enjoo (náuseas), vómitos, diarreia, perda de apetite. Frequentes: Boca seca, inflamação na boca, hemorragia das gengivas; obstipação, indigestão, hemorragia rectal, fezes muito escuras quase pretas, perturbações do sistema digestivo. Não frequentes: Infecção fúngica na boca, úlceras bucais, inchaço e hemorragia das gengivas, problemas dentários; língua inchada, vermelha ou inflamada e outros problemas na língua; dificuldade em engolir, inflamação do esófago (garganta), problemas de estômago, inchaço devido à acumulação de fluído à volta do estômago, eructação, excesso de gases, aumento do apetite; icterícia, função hepática anormal ou insuficiência hepática, inflamação no sistema digestivo ou na vesícula biliar; hemorragia ou obstrução no sistema digestivo, sensação de esvaziamento incompleto dos intestinos, outros problemas rectais. Sistema sanguíneo e linfático Muito frequentes: Nódoas negras, menor número de plaquetas, de glóbulos vermelhos ou brancos. Frequentes: Pontos vermelhos sob a pele, deficiência de células vermelhas sanguíneas, depleção das células brancas sanguíneas, originando febre. Não frequentes: Glândulas inchadas no pescoço, destruição de glóbulos vermelhos, lábios azulados ou pele azulada, aumento dos glóbulos brancos, inflamação dos vasos linfáticos, diminuição dos níveis

33
de todas as células sanguíneas, dando origem possivelmente a cansaço, uma sucessão de infecções frequentes, hemorragias e febre; alterações nas proteínas do sangue que combatem a doença. À volta do local da injecção Frequentes: Dor no local da injecção. Não frequentes: Inchaço, vermelhidão, calor, dor e hemorragia no local da injecção. Sistema metabólico e de eliminação de resíduos Frequentes: Inchaço causado pela retenção de líquidos nos braços e nas pernas e noutros tecidos; inchaço da cara, lábios, boca, língua ou garganta; perda de fluidos corporais, níveis elevados de açúcar no sangue, níveis elevados de enzimas hepáticas, alteração de certos valores do sangue, perda de peso. Não frequentes: Perda de peso acentuada, aumento de peso, níveis baixos de açúcar no sangue, aumento de gorduras no sangue, sede. Ossos, articulações e músculos Muito frequentes: Dor nas articulações. Frequentes: Músculos doridos, dores nos ossos, cãibras nas pernas, os músculos enfraquecem e cansam-se facilmente. Não frequentes: ossos facilmente quebradiços, problemas nas articulações ou nos tendões, artrite. Sistema nervoso e sentidos Muito frequentes: Tonturas. Frequentes: Sensação de ansiedade, dificuldades em dormir, depressão, formigueiro, diminuição da sensibilidade de toque, sonolência, dilatação dos vasos sanguíneos, conjuntivite, problemas de visão, andar anormal ou instável. Não frequentes: Agitação, perturbação emocional, confusão, hiperactividade não habitual, nervosismo; tremor, convulsões, contracção espasmódica, coordenação deficiente; doença da função cerebral, pensamentos incoerentes, hematoma entre o crânio e o cérebro; dor aguda ou inflamação ao longo dos nervos, perturbação da fala, tonturas (vertigens); visão dupla, dores nos olhos, cataratas, secura ocular, hemorragia na parte posterior do olho, doença da córnea, alterações na secreção lacrimal; infecção do ouvido externo, ruído persistente nos ouvidos (tinido); perda ou alterações do paladar, aumento da salivação, perturbações oculares, retenção urinária. Sistema respiratório e pulmonar Muito frequentes: Aumento da tosse, dificuldade em respirar. Frequentes: Pieira, nariz a pingar, infecção; sensação de tensão no nariz, bochechas e por trás dos olhos; hemorragia nasal, bronquite, alteração da voz, pneumonia, inflamação da garganta e desconforto ao engolir. Não frequentes: Asma, perturbações respiratórias e pulmonares, aumento da expectoração, redução da função pulmonar, perturbação do diafragma. Pele Muito frequentes: Comichão. Frequentes: Rash, erupções cutâneas, sudação (incluindo sudorese nocturna), herpes (herpes simplex), problemas de pele. Não frequentes: Zona (herpes zóster), pele seca ou inflamada, acne, doença fúngica da pele, bolhas múltiplas, espessamento da pele, feridas em supuração, rash com borbulhas. Sistema urinário e reprodutor Frequentes: Infecção das vias urinárias, dor ao urinar. Não frequentes: Perda de controlo da bexiga, frequência urinária, sangue e proteínas na urina, pedra no rim, insuficiência renal, micção nocturna, micção deficiente, micção em menor quantidade que o habitual, perturbação do sistema urinário e reprodutor, tecido fibroso degenerativo no útero, hemorragia vaginal, sensibilidade mamária. Tumores e doenças malignas

34
Numa pequena percentagem (inferior a 3 em 100) de doentes oncológicos que participaram em alguns estudos com Zevalin, foram referidos outros tipos de cancro relacionados com o sangue: mielodisplasia ou leucemia mielóide aguda (LMA). O risco destes cancros secundários ocorrerem após a terapêutica com agentes alquilantes é já bem conhecido pelos médicos. Um tratamento preliminar com agentes alquilantes é, no entanto, um procedimento padrão antes do tratamento com Zevalin. Por isso, é difícil saber se o Zevalin contribui para o risco de desenvolver uma doença cancerígena secundária. • Se tiver problemas com algum destes efeitos, ou detectar outros efeitos não mencionados neste
folheto, informe o seu médico ou farmacêutico.
5. Conservar Zevalin Conservar a 2°C - 8°C (no frigorífico). Não congelar. Conservar na embalagem original para proteger da luz. Após a marcação radioactiva: Conservar entre 2°C e 8°C (no frigorífico) e proteger da luz. A conservação deverá ser efectuada de acordo com as regulamentações nacionais relativamente a materiais radioactivos.

35
6. Outras informações Para quaisquer informações sobre este medicamento, queira contactar o representante local do titular da autorização de introdução no mercado. België/Belgique/Belgien Genesis Pharma S.A. J.E. Mommaertslaan 14 B-1831 Diegem Tel: 02-712 85 00
Italia Italfarmaco S.p.A. Viale F. Testi, 330 I-20126 Milano Tel: 02-64431
Česká republika Schering s.r.o. – člen koncernu Šafaříkova 17 120 00 Praha 2 Tel: 271 730 661
Ísland Thorarensen Lyf ehf Lynghálsi 13 IS-110 Reykjavík Sími/Tel: + 354 530 7100
Danmark Schering AS Herstedřstervej 27-29 DK-2620 Albertslund Tel: 43 29 09 99
Cyprus A. POTAMITIS MEDICARE LTD 62, Arch. Kyprianou Avenue 2059 Strovolos Nicosia CYPRUS Tel: +357 22-313611
Deutschland Schering Deutschland GmbH Max-Dohrn- Strasse 10 D-10589 Berlin Tel: 0130-11 23 22
Latvija Schering AG pārstāvniecība Latvijā Ģertrūdes iela 3 Rīga, LV 1010 Tel: +371-784 55 63
Ελλάδα SCHERING ΕΛΛΑΣ Α.Ε. Κύπρου 12-14 & Λεωφ. Ηρακλείου 466 141 22 ΗΡΑΚΛΕΙΟ τηλ. 210-2723192
Lietuva UAB “Schering” Vytenio g. 4 LT-2009 Vilnius, Lietuvos Respublika Tel: +370 686 51 037
Espańa Schering Espańa S.A. C. Méndez Alvaro, 55 E-28045 Madrid Tel: 902 24 62 46
Luxembourg/Luxemburg N.V. Schering S.A. J.E. Mommaertslaan 14 B-1831 Diegem, Belgique/Belgien Tel: +32 2-712 85 00
Eesti SCHERING AG EESTI FILIAAL Pärnu mnt. 139 E 11317 Tallinn Tel: 06-55 85 65
Magyarország Schering KFT Szépvölgyi út 35-37 1037 Budapest Tel: 01-453 80 10
France Schering S.A. Rue de Toufflers F-59390 Lys-Lez-Lannoy Tel: 03 20 20 80 80
Malta Alfred Gera & Sons Ltd. New Street in Triq II – Milied QORMI QRM 09 Tel: 02-44 61 13
Ireland Nederland

36
HE Clissmann 44 Dartmouth Square IRL-Dublin 6 Tel: 01-6 68 85 66
Schering Nederland BV Postbus 116 NL-1380 AC Weesp Tel: 0294-46 24 24
Norge Schering AG Postboks 180 N-1321 Stabekk Tel: + 47 67 59 20 00
Slovenija Schering AG Berlin, Podružnica za Slovenijo Dunajska cesta 22 1511 Ljubljana Tel: 01-300 10 50
Österreich Schering Wien Ges.m.b.H. Postfach 50 A-1147 Wien Tel: (01) 9 70 37
Suomi/Finland Schering Oy Eerikinkatu 24 FIN -00100 Helsinki Puh. 09-6 85 04 40
Polska Schering AG Spółka Akcyjna Oddział w Polsce Ul. Migdałowa 4 02-796 Warszawa Tel: 022-645 13 00
Sverige Schering Nordiska AB Box 912 S-175 29 Järfälla Tel: 08-7 28 42 00
Portugal Schering Lusitana Lda. Estrada Nacional 249, km 15 Apartado 16 P-2726-901 Mem Martins Tel: 021-9 26 81 10
United Kingdom Schering Health Care Ltd. The Brow GB-Burgess Hill, West Sussex RH15 9NE Tel: 01444-23 23 23
Slovenská republika Schering Slovakia s.r.o. Obchodná ul. 2 811 06 Bratislava Tel: 02-54 41 0317
Este folheto foi aprovado pela última vez em {data}.