simulação de propriedades estruturais e eletrônicas de...
TRANSCRIPT

Universidade de São Paulo Instituto de Física
Simulação de Propriedades Estruturais e Eletrônicas de Agregados, Líquidos Regulares e
Supercríticos.
Rafael Carvalho Barreto
Orientador: Prof. Dr. Sylvio R A Canuto
Tese de doutorado apresentada ao Instituto de Física para a obtenção do título de doutor em ciências.
São Paulo
2010

(contra capa)

(espaço reservado para o registro da biblioteca)

(vazio)

A ciência é o que nós compreendemos suficientemente bem para explicar a um
computador. A arte é tudo mais.
Donald Knuth
Ciência da computação tem tanto a ver com o computador como a Astronomia com o
telescópio, a Biologia com o microscópio, ou a Química com os tubos de ensaio. A
Ciência não estuda ferramentas, mas o que fazemos e o que descobrimos com elas.
Edsger Dijkstra
Não existe física computacional sem computador.
Rafael Barreto

(vazio)

Sumário
Agradecimentos ................................................................................................................ 8
Resumo ............................................................................................................................. 9
Abstract........................................................................................................................... 10
Capítulo 1 : Introdução ................................................................................................... 11
Capítulo 2 : Métodos de modelagem molecular em líquidos ......................................... 14
Capítulo 3 : Solvatocromismo do fenol .......................................................................... 16
3.1 Agregados de fenol e água.............................................................................. 19
3.2 Agregados de fenol e etanol ........................................................................... 24
3.3 Fase líquida: Simulação do fenol em água ..................................................... 41
3.4 Fase líquida: Simulação do fenol em etanol ................................................... 50
Capítulo 4 : Água supercrítica ........................................................................................ 57
4.1 Determinação do Ponto Crítico da Água usando o Modelo AMOEBA......... 62
Capítulo 5 : Retrospectiva, Conclusões e Perspectivas .................................................. 70
Apêndice A: Realizações no Doutorado......................................................................... 73
Apêndice B: Mecânica Molecular - Líquidos................................................................. 75
Apêndice C: Mecânica Quântica - Métodos Aproximativos.......................................... 83
Apêndice D: Campos de Força ....................................................................................... 89
Apêndice E: Aspectos Técnicos da Física Computacional............................................. 95
Referências ................................................................................................................... 101

8
Agradecimentos
Agradeço a todos, sem exceção. Agradeço especialmente a FAPESP pelo
apoio financeiro.

9
Resumo
Esta tese de doutorado investiga as propriedades espectroscópicas e
termodinâmicas de líquidos moleculares. Para este fim utilizam-se técnicas de
química quântica e modelagem na simulação computacional. Para simular as
propriedades termodinâmicas e flexibilidade molecular foi utilizada Simulação
Clássica (Monte Carlo e Dinâmica Molecular). Já para reproduzir as propriedades
eletrônicas, valeu-se de métodos de Química Quântica (post-HF e DFT). O efeito de
polarização foi introduzido pelo Método Seqüencial Monte Carlo/Mecânica Quântica
(S-MC/QM) e também via campo de força polarizáveis. Apresenta-se uma visão geral
destas técnicas no apêndice.
Este trabalho se divide em três partes. Na primeira parte são obtidas as
propriedades estruturais e espectroscópicas de agregados de fenol e água (1+1 e 1+2)
e fenol e etanol (1+1). Os modos vibracionais (inter e intramoleculares) e a energia de
ligação são calculados. Os resultados estruturais e o desvio solvatocrômico são
coerentes com dados experimentais e expectativas teóricas (para os diferentes
agregados). Na segunda parte é realizada a simulação do fenol em água utilizando o
protocolo S-MC/QM, com o respectivo cálculo do espectro eletrônico. É apresentado
o estudo das pontes de hidrogênio e as diferentes contribuições que determinam o
desvio solvatocrômico em solução. Em seguida é feita a simulação do fenol em etanol
utilizando dinâmica molecular e o campo de força flexível e polarizável AMOEBA.
As propriedades termodinâmicas da simulação estão em excelente concordância com
as expectativas teóricas e experimentais. Isso é surpreendente dado o fato que o
modelo não é parametrizado empiricamente. Por fim, é detalhado o trabalho realizado
com a água supercrítica. Foram executadas centenas de simulações a fim de se obter o
ponto crítico do modelo AMOEBA. São apresentadas as isotermas e os parâmetros da
equação de estado que determinam o comportamento do modelo nessa região.
Este documento amostra o conhecimento adquirido pelo aluno em diferentes
aspectos da física computacional, desde a parte técnica, os diversos modelos e
aproximações teóricas utilizadas em simulação e a capacidade de aplicação deste
conhecimento em problemas de interesse físico, os quais resultaram em dois artigos
publicados [12,22] e mais dois trabalhos em preparação.

10
Abstract
This thesis investigates the spectroscopic and thermodynamic properties of
molecular liquids. To this end, we use techniques of quantum chemistry and modeling
to do computational simulation. To simulate the thermodynamic properties and
molecular flexibility it was used Classical Simulation (Monte Carlo and Molecular
Dynamics). To reproduce the electronic properties it was used Quantum Chemistry
methods (post-HF and DFT). The polarization effect was introduced through the
Sequential Monte Carlo / Quantum Mechanics Method and polarizable force fields. It
is presented an overview of these techniques in the appendix.
This work is divided into three parts. In the first part is obtained spectroscopic
and structural properties of phenol and water (1+1 and 1+2) and phenol and ethanol
(1+1) clusters. The vibrational modes (intra and intermolecular) and binding energies
are calculated. The structural results and the solvatochromic deviations are consistent
with experimental data and theoretical expectations (for different clusters). In the
second part is performed the simulation of phenol in water using the S-MC/QM
protocol, followed by the calculation of the electronic spectrum. It is presented the
study of hydrogen bonding and the different contributions which determine the
solvatochromic deviation in solution. So, it is made the simulation of phenol in
ethanol using molecular dynamics and the polarizable and flexible force field
AMOEBA. The thermodynamic properties of the simulation are in excellent
agreement with theoretical expectations and experimental data. This is surprising
given the fact that the model is not parameterized empirically. Finally, it is detailed
the work on supercritical water. It has been performed hundreds of simulations in
order to obtain the critical point of the model AMOEBA. Isotherms are presented as
well as the parameters of the equation of state which determine the behavior of the
model in that region.
This document samples the acquired knowledge of the student in different
aspects of computational physics, from the technical side, the various models and
theoretical approaches used in simulation and the ability to apply this knowledge to
problems of physical interest, which resulted in two published articles [12,22] and
other two are in preparation.

11
Capítulo 1: Introdução
O estudo teórico das propriedades moleculares em líquidos não é simples. O
líquido tem densidade similar ao sólido, mas suas partículas fluem sem ter posição
definida. As forças de atração e repulsão impõem restrições de como suas partículas
podem se movimentar. Dado o grande número de possíveis conformações é necessária
cautela quando se tenta construir modelos teóricos. O problema é ainda mais
complexo já que não existe uma equação simples para descrever as forças
intermoleculares.
O surgimento da mecânica quântica estabeleceu um novo paradigma para o
estudo teórico das propriedades microscópicas dos líquidos [1]. Expressões analíticas
para as forças intermoleculares puderam ser obtidas para sistemas simples, como
gases nobres. Mas foi apenas com o surgimento do computador que se pode avançar
sobre sistemas mais complexos. Técnicas numéricas foram aplicadas a problemas cuja
abordagem analítica era impossível devido à grande quantidade de vínculos e
restrições. Mesmo com o avanço contínuo e acelerado guiado pelo desenvolvimento
computacional, a maioria dos problemas em meio solvente ainda é complicada o
suficiente para desafiar as soluções numéricas do tipo "força bruta". Inevitavelmente,
foram desenvolvidos modelos simplificados para descrever as forças intermoleculares
(chamados de campos de força), já que os cálculos quânticos (ainda) são inaplicáveis
para a maioria dos sistemas de interesse devido ao seu alto custo computacional.
Quando se trata de líquidos polares [2], uma boa descrição dos momentos de
multipolo é crucial. Tais interações são fundamentais para a conformação de
complexos moleculares, membranas e micelas, determinação do número médio de
pontes de hidrogênio, cálculo da constante dielétrica e típicas propriedades anômalas.
Moléculas apolares, como sistemas aromáticos e solventes utilizados em processos
químicos, ou moléculas complexas com regiões apolares, tais como lipídios e
proteínas, necessitam de tratamento diferenciado. Em tais moléculas é necessário
utilizar campos de força que descrevam bem os efeitos de indução, dispersão e termos
superiores da expansão multipolar. Para líquidos homogêneos, quando se utiliza
modelos simplificados para descrever as interações moleculares, a qualidade do
campo de força pode facilmente ser verificada pelas propriedades termodinâmicas
calculadas. Mas não existe a mesma facilidade quando se deseja estudar moléculas em
meio heterogêneo. Compreender o efeito do solvente sobre o soluto é a chave para

12
explicar resultados experimentais, desvendar interações específicas, entender
mudanças conformacionais e solvatocromismo. Potenciais que descrevem bem as
propriedades termodinâmicas do sistema podem não descrever corretamente a
interação específica soluto-solvete, e assim são necessários métodos mais avançados e
maior cuidado para avançar nesta área.
Ao fim do capítulo 2, são apresentadas algumas das técnicas utilizadas para
melhorar os campos de força tradicionais e incluir parte dos efeitos de polarização do
soluto causado pelo solvente.
Este trabalho dá atenção especial ao fenol. Fenóis são compostos orgânicos em
que um grupo álcool está diretamente ligado a um grupo aromático de
hidrocarbonetos. É um importante grupo funcional, encontrado em diversas reações
químicas e biológicas. Sua forma mais simples é o C6H5OH (Figura 1), conhecido
como fenol, benzenol, ácido carbólico, hidroxibenzeno, entre outros nomes. É mais
ácido que os alcoóis, podendo ser convertido quando em solução aquosa em fenolato
ou fenóxido. Estudar a interação dos fenóis com o meio solvente possui imensa
importância, tal como a compreensão de mecanismos de reação química e
interpretação do espectro de compostos complexos, interação de moléculas de
interesse biológico com o solvente, dentre outras aplicações tanto na indústria quanto
em laboratório. No capítulo 3 é apresentado parte do estudo feito com o fenol. Nesta
parte, destacam-se a influência das diferentes contribuições (bulk e pontes de
hidrogênio) para o desvio solvatocrômico do fenol em meio.
Figura 1: Molécula de fenol.

13
Na segunda parte deste trabalho, é apresentado o estudo da água em condições
supercríticas. O objetivo desta parte foi desenvolver o conhecimento e as ferramentas
necessárias para futuros estudos de solvatação molecular nessas condições. Fluidos
supercríticos (SCF), por definição, são todas as substâncias cuja temperatura e pressão
estão além do ponto crítico. Ao contrário do que se pode pensar, simular em
condições supercríticas não é tão simples quanto se simular em condições normais.
No ponto crítico, a descontinuidade nas propriedades físicas, a qual diferencia a fase
líquida da gasosa, desaparece. Na região supercrítica, o fluido exibe propriedades
completamente novas, as quais vêm despertando crescente interesse na última década
[3]. Dentre as mais intrigantes, se encontra a mudança drástica nas propriedades de
solvatação de fluidos comuns [4]. Nessa nova condição, algumas dessas substâncias
são chamadas de solventes verdes, pois são capazes de substituir solventes orgânicos
pesados prejudiciais ao ambiente. A água, em especial, com pressão crítica de 217
atm e temperatura crítica de 647 K, na região supercrítica apresenta baixa polaridade e
alta acidez [5]. A correta descrição e compreensão das propriedades físicas de
moléculas solvatadas a condições normais é um desafio por si só. Repetir esse estudo
em altas temperaturas e pressões, caso da água supercrítica, é um desafio ainda maior.
No capítulo 4 apresenta-se o estudo feito com simulação de água em condições
supercríticas. O resultado mais importante deste trabalho é a determinação do ponto
crítico da água utilizando o modelo polarizável AMOEBA.
*************************
Todas as variáveis (escalares, vetores ou tensores) utilizadas nas equações e
tabelas deste trabalho foram colocadas em itálico (q0, x…). Os vetores foram escritos
em negrito (µ, r…). Os tensores de segunda ordem, mesmo sendo variáveis, estão
apenas em negrito (Θ). Os operadores têm circunflexos (Â, Ĥ…). Em alguns
momentos do texto, alguns termos-chave foram destacados em negrito a primeira vez
que apareceram. Alguns termos utilizados na literatura, nomes de métodos, ou de
constantes foram colocados em itálico. Foi utilizado ponto (.) como separador
decimal, contrariando o padrão nacional. Todas as equações serão chamadas no meio
do texto pelo seu número entre parênteses, ou seja, (53) deve ser lido como "equação
53". Os usos do hífen e do trema estão de acordo com a gramática antiga, antes da
reforma ortográfica que uniu a gramática dos países de língua portuguesa.

14
Capítulo 2: Métodos de modelagem molecular em líquidos
Estudar as propriedades físicas de sistemas líquidos requer conhecimento
abrangente. Um líquido é um condensado de moléculas, as quais se guiam pela força
de interação de cada uma com todas as outras. Condensado implica que a força é em
média atrativa, e que a velocidade de seus constituintes não é em média suficiente
para que estes se dispersem. Porém, esta força não é grande o bastante para confinar
os movimentos das moléculas em torno de alguma posição de equilíbrio. E por mais
relevante que seja sua descrição microscópica, o estudo dos líquidos inicia-se
realmente por suas propriedades macroscópicas (termodinâmica). Seu formalismo
torna possível reconhecer as equações de estado de um sistema em equilíbrio,
independente de sua constituição microscópica. A partir de um pequeno conjunto de
princípios, como a conservação da energia e a maximização da entropia, é possível
relacionar quantidades extensivas (entropia e volume) com intensivas (temperatura e
pressão), e encontrar características inerentes da matéria tais como o calor específico
e a compressibilidade. A conexão entre as propriedades termodinâmicas e sua origem
microscópica é feita através da mecânica estatística. Como não há interesse em saber
a localização exata de cada molécula no espaço a cada instante, procura-se obter o
efeito médio desta multidão em cada propriedade observada. O efeito conjunto dessa
multidão de possíveis configurações é chamado de ensemble1.
Encontrar uma equação de estado de um líquido em equilíbrio através da
mecânica estatística enfrenta dois problemas fundamentais. Como calcular a energia
de uma determinada configuração e como ponderar sobre um número aparentemente
infinito de configurações. Isto poderia ser feito por meios analíticos, integrando-se
sobre o "espaço de configurações" acessíveis ao sistema. Infelizmente, mesmo
conhecendo a forma analítica da energia, o número de variáveis e vínculos dos
sistemas físicos de interesse torna tal procedimento quase sempre inviável. Portanto
são utilizados métodos numéricos (simulação computacional). E para se obter a
energia de uma configuração é necessário compreender a natureza das forças
moleculares. Estas possuem suas origens no eletromagnetismo. Classicamente, caso
fosse possível saber a exata distribuição de cargas, posições e velocidades de cada um
dos constituintes do sistema, saber-se-ia também a energia total de cada configuração.
1 Ensemble: Unidade ou grupo de partes complementares que contribuem para um efeito único. [Francês, do Francês Antigo, do Latim Tardio īnsimul, "ao mesmo tempo".]

15
Entretanto a distribuição de cargas e a própria conformação molecular só pode ser
conhecida através da mecânica quântica, elevando o problema a um novo grau de
complexidade. O estado do sistema pode ser representado por uma função de onda, e
os observáveis físicos por operadores que atuam nesta função. Entende-se por
observável aquilo que é passível de medição, como energia, posição, e momento, por
exemplo. Problemas simples como o oscilador harmônico e o átomo de hidrogênio
são resolvidos analiticamente com o uso da álgebra destes operadores. Problemas
mais elaborados, como o problema de três corpos, requerem métodos aproximativos,
pois recaem em equações diferenciais das quais não se conhece a solução. Assim o
cálculo da energia demanda uma série de aproximações e o extensivo uso de cálculo
numérico.
Os métodos de simulação computacional de líquidos, cálculo da energia de
cada configuração, os métodos quânticos utilizados e seus respectivos graus de
aproximação, assim como os campos de força derivados deste conhecimento estão nos
apêndices. No apêndice B são apresentados os métodos de Monte Carlo e o de
Dinâmica Molecular, utilizados neste trabalho para gerar configurações de líquidos.
No apêndice C são apresentados alguns métodos quânticos utilizados para obter as
propriedades eletrônicas dessas configurações. No apêndice D são apresentadas
algumas características dos campos de força utilizados nas simulações
computacionais. No apêndice E são apresentados alguns aspectos importantes da
tecnicalidade necessária para a realização da simulação computacional.

16
Capítulo 3: Solvatocromismo do fenol
Neste capítulo é apresentado o estudo do deslocamento espectral do fenol em
solvente. O solvente de principal interesse neste trabalho é a água. Entretanto,
resultados experimentais [6] para o deslocamento espectral do fenol em etanol
mostram que seu valor é contrário àquele obtido para água. É interessante notar que
ambos os solventes são polares, e ambos são capazes de fazer pontes de hidrogênio.
Este resultado levanta a questão sobre a origem do solvatocromismo do fenol em
função das pontes de hidrogênio formadas em solução. Desta maneira, foi realizada
simulação tanto de fenol em água quanto de fenol em etanol.
Inicialmente realizou-se a otimização de geometria da molécula de fenol
isolada, com o método perturbativo MP2 [96] e a base aug-cc-pVDZ [7]. A Tabela 1
mostra os resultados desta otimização. A posição de mínimo é garantida pelo fato de
todas as freqüências vibracionais serem reais. Nota-se que as distâncias CO e HO são
um pouco superestimadas (1%) em relação aos valores obtidos com espectroscopia de
microondas [53]. O valor para o momento de dipolo, que é função da distribuição de
cargas e da geometria, também é superestimado (4.5%). Os valores obtidos para as
constantes rotacionais, que são função inversa da geometria, são levemente
subestimados (2%).
Tabela 1: Resultados do cálculo da otimização da molécula de fenol isolada. A energia foi obtida com o método MP2/aug-cc-pVDZ. A energia de ponto zero (ZPE) é calculada dentro da
aproximação harmônica. As freqüências vibracionais são típicas para torção e estiramento OH.
Teórico Experimental [53] RCO (Å) 1.382 1.364 RHO (Å) 0.968 0.956 ΘHOC (º) 108.6 109.0 ΘHOCC (º) 0.0 0.0 µ (Debye) 1.28 1.224 ± 0.008
A 5.560 5.650 [8] B 2.577 2.619 I (GHz) C 1.761 1.789
E(hartree) -306.61069 — ZPE(kcal/mol) 65.09 — τHOCC (cm-1) 330 310 [18] νHO (cm-1) 3808 3656

17
A energia total é dada pela energia eletrônica E (incluindo a repulsão nuclear)
mais a energia de ponto zero (ZPE) associada com o movimento vibracional dos
núcleos (que por conveniência foi escrita separadamente). Na tabela se destacam os
dois modos vibracionais que são mais afetados pelas pontes de hidrogênio que o fenol
é capaz de realizar. O movimento de torção (HOCC) e o movimento de estiramento
(OH) da hidroxila desviam respectivamente 7% e 5% do valor obtido
experimentalmente por Bist et al.[18]. Nota-se que estas freqüências foram calculadas
na aproximação harmônica, e incluir correções anarmônicas fariam com que estas
freqüências diminuíssem em direção ao resultado experimental.
Obtida a geometria, calculou-se o espectro de absorção em fase gasosa. Os
métodos utilizados foram TDDFT [9] com o funcional B3LYP (TDB3LYP) [10],
TDHF [9], e CIS(D) [97]. Como o CISD não é um método extensivo, ele não deve ser
usado para estudar efeitos de solvente onde o número de moléculas explicitamente
consideradas muda [95]. Assim optou-se pelo método CIS (que é extensivo) e as
excitações duplas são incluídas de forma perturbativa com o cuidado de se preservar a
extensividade (veja apêndice C). Os cálculos para o espectro eletrônico foram
realizados utilizando-se a base aug-cc-pVDZ, a mesma das otimizações de geometria.
Os resultados são apresentados na Tabela 2. Os resultados obtidos são típicos. O
método CIS produz valores muito altos para a transição, o mesmo ocorrendo para
TDHF. Os métodos TDB3LYP e CIS(D) fornecem resultados melhores, entretanto
ainda superiores ao resultado experimental obtido por Baba e Suzuki [11].
Tabela 2: Resultados do cálculo da transição eletrônica para a molécula de fenol isolada.
π → π* 1º exc (cm-1) 2º exc (cm-1) CIS 46350 48770 CIS(D) 39810 51030 TDHF 44530 46290 TDDFT 39800 — Experimental [11] 36070 45400
Os métodos CIS, CIS(D) e TDHF resultaram em transição de natureza π → π*
para a primeira e segunda excitação (Figura 2). O método TDDFT possui resultado
equivalente ao método CIS(D) para o valor da primeira transição, entretanto nas
quatro excitações subseqüentes não houve mais nenhuma transição π → π*.

18
Figura 2: Caráter dos orbitais moleculares dominantes da primeira transição π → π* do fenol. O orbital da esquerda é associado ao estado fundamental, e o da direita ao estado excitado.
Adicionalmente estudou-se a variação do momento de dipolo no nível
CIS/aug-cc-pVDZ. Isto foi feito para inferir qual estado eletrônico do fenol
(fundamental ou excitado) interage mais com o solvente polar. Nota-se aqui que o
primeiro termo não nulo da expansão multipolar para moléculas polares neutras é o
momento de dipolo. Observou-se um decréscimo de 0.1 D do estado fundamental para
o estado excitado, o que sugeriria um sutil desvio para o azul em solvente polar. Este
raciocínio desconsidera completamente o efeito das pontes de hidrogênio, situação a
qual a proximidade entre os centros de carga impossibilita que se trunque a expansão
multipolar em seus primeiros termos.

19
3.1 Agregados de fenol e água
Antes de simular o fenol em água para obter seu solvatocromismo, faz-se
necessário estudar os possíveis agregados em fase gasosa [12]. De antemão, é crucial
compreender como esse desvio se comporta em relação à formação de pontes de
hidrogênio entre fenol e água. Portanto, estudou-se o complexo de fenol com uma
molécula de água em duas possíveis conformações para ponte de hidrogênio: água
doadora (W:Ph) e água aceitadora (Ph:W), como pode ser visto na Figura 3. As
geometrias foram otimizadas no nível de cálculo MP2/aug-cc-pVDZ, o mesmo nível
utilizado para o monômero.
Figura 3: Complexos otimizados de fenol com uma água em nível MP2/aug-cc-pVDZ.
Tabela 3: Resultados para a energia total eletrônica mais repulsão nuclear (hartrees), e as energias de ligação (kcal/mol), para os complexos com uma água em nível MP2/aug-cc-pVDZ.
Energia de Ligação (kcal/mol) Energia (hartrees) ∆E ∆E+ZPEa ∆E+ZPE
+BSSEb Água -76.26091 — — — Fenol -306.61069 — — — W:Ph -382.88005 5.30 3.82 2.63 Ph:W -382.88385 7.68 5.91 4.50 a ZPE: Inclusão da energia de ponto zero devido ao movimento nuclear. b BSSE: Correção do erro de superposição de base [13].
Os resultados obtidos para a energia total e de ligação são mostrados na
Tabela 3, em nível MP2/aug-cc-pVDZ. Nota-se que a energia de ligação é dada por

20
∆E = EA + EB - EAB. As três últimas colunas apresentam esta energia, considerando-se
primeiro apenas ∆E, depois ∆E corrigido em relação à energia de ponto zero (ZPE), e
finalmente corrigindo também o erro de superposição de base (BSSE). Esta última
coluna é a melhor estimativa da energia de ligação. Em concordância com a literatura,
a configuração de água aceitadora é mais estável e mais abundante [14,15]. Nota-se
que os experimentos de jet cooling reportados na literatura para obtenção de espectro
e geometria são realizados em temperaturas abaixo de 50K (kT ~ 0.1kcal/mol).
Considerando-se que o fator de Boltzmann, exp(-U/kT), determina as populações de
cada uma das possíveis conformações (onde U = EAB - (EA + EB) = -∆E). Estima-se
que os complexos de água aceitadora são oito ordens de grandeza2 mais prováveis que
os complexos de água doadora de ponte de hidrogênio, nas condições em que o
experimento foi realizado (~50K).
Todos os resultados obtidos para as geometrias foram submetidos ao cálculo
de freqüências para conferir se eram mesmo conformações de mínima energia (Tabela
4). Os valores de 161 para Ph:W e 137 para W:Ph correspondem ao modo de
estiramento intermolecular na ponte de hidrogênio, cujo resultado experimental
reportado para Ph:W é de 156 cm-1 [16]. Nota-se que há uma fortuita concordância, já
que este cálculo é feito dentro da aproximação harmônica, e portanto esperava-se um
valor superestimado.
Tabela 4: Resultado do cálculo das freqüências intermoleculares dos complexos de fenol e água, no nível MP2/aug-cc-pVDZ. Entre parênteses, o valor experimental obtidos da ref. [16].
νINTER (cm-1) Ph:W W:Ph
38 18 62 46 108 100
161 (156) 137 217 222 236 271
Os resultados para os modos de vibração intramoleculares do grupo OH do
fenol são apresentados na Tabela 5. Tais resultados para a fase gasosa são
equivalentes aos obtidos em nível CASSCF por Schumm et al.[17]. Ambos os
resultados para os complexos estão de acordo com o esperado. A freqüência do modo
2 p(Ph:W)/p(W:Ph) = exp((4.50-2.63)/0.1) ≈ exp(19) ≈ 108

21
de torção do grupo OH aumenta e o modo de estiramento diminui para o fenol doando
a ponte (Ph:W).
Tabela 5: Resultado do cálculo das freqüências intramoleculares do fenol (estiramento e torção do OH). Primeira linha para fase gasosa e depois para os complexos de fenol e água, no nível
MP2/aug-cc-pVDZ. Entre parênteses, os valores experimentais obtidos da ref [18].
Torção (cm-1)
Estiramento (cm-1)
Fenol 328 (310) 3808 (3656) W:Ph 290 3808 Ph:W 761 3628
Figura 4: Complexos do fenol com duas águas. Ambas apresentam energias equivalentes (ver Tabela 6).
Em seguida, motivados pelo trabalho experimental de Fuke e Kaya [21],
adicionou-se mais uma água a essas geometrias. Encontraram-se duas conformações
mostradas na Figura 4. Ambas as conformações estão de acordo com a literatura
[19,20]. Dado os erros dos métodos e nível de cálculo utilizados, as conformações
Ph:W:W1 e Ph:W:W2 apresentam a mesma energia de ligação (com diferença de
0.2kcal/mol em favor de Ph:W:W2), como visto na Tabela 6. Fato interessante é que a
segunda água na conformação Ph:W:W2 aparenta fazer um ponte do tipo H-pi, com
uma distância de 2.29Å do C1 (veja Figura 1 no Capítulo 1). Adicionalmente
procurou-se orientar esta ponte de hidrogênio (desta segunda água) sobre o C2 (o qual
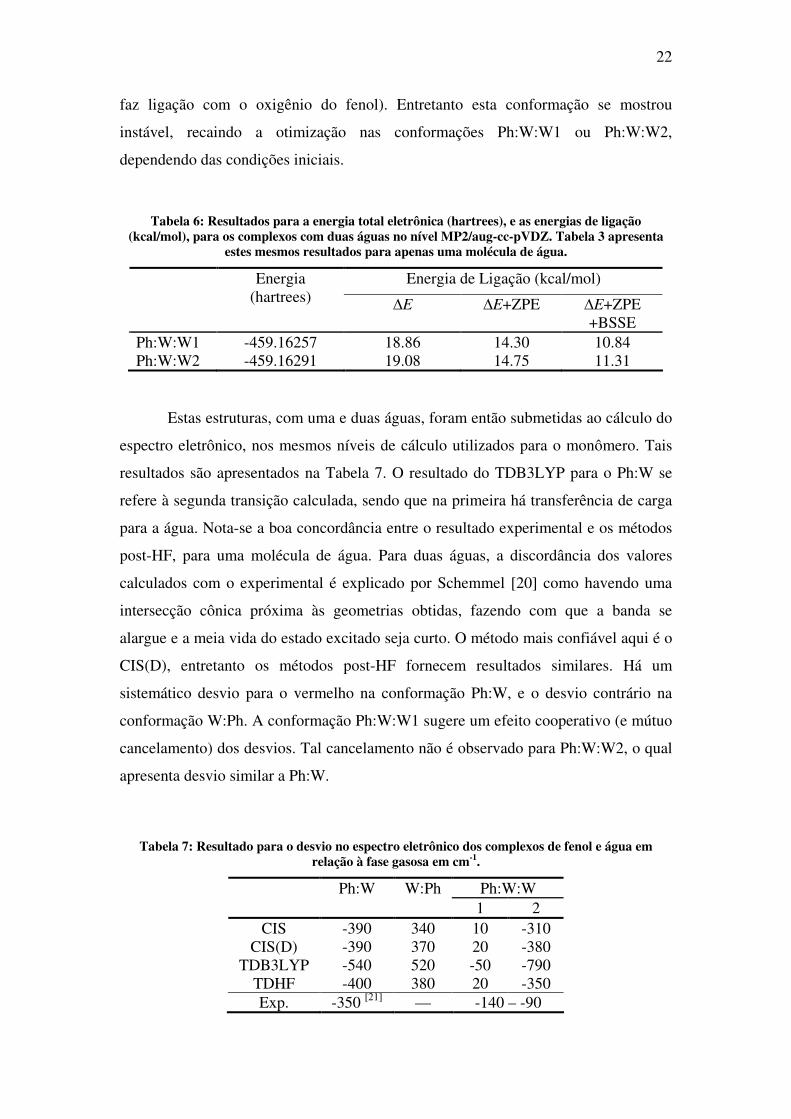
22
faz ligação com o oxigênio do fenol). Entretanto esta conformação se mostrou
instável, recaindo a otimização nas conformações Ph:W:W1 ou Ph:W:W2,
dependendo das condições iniciais.
Tabela 6: Resultados para a energia total eletrônica (hartrees), e as energias de ligação (kcal/mol), para os complexos com duas águas no nível MP2/aug-cc-pVDZ. Tabela 3 apresenta
estes mesmos resultados para apenas uma molécula de água.
Energia de Ligação (kcal/mol) Energia (hartrees) ∆E ∆E+ZPE ∆E+ZPE
+BSSE Ph:W:W1 -459.16257 18.86 14.30 10.84 Ph:W:W2 -459.16291 19.08 14.75 11.31
Estas estruturas, com uma e duas águas, foram então submetidas ao cálculo do
espectro eletrônico, nos mesmos níveis de cálculo utilizados para o monômero. Tais
resultados são apresentados na Tabela 7. O resultado do TDB3LYP para o Ph:W se
refere à segunda transição calculada, sendo que na primeira há transferência de carga
para a água. Nota-se a boa concordância entre o resultado experimental e os métodos
post-HF, para uma molécula de água. Para duas águas, a discordância dos valores
calculados com o experimental é explicado por Schemmel [20] como havendo uma
intersecção cônica próxima às geometrias obtidas, fazendo com que a banda se
alargue e a meia vida do estado excitado seja curto. O método mais confiável aqui é o
CIS(D), entretanto os métodos post-HF fornecem resultados similares. Há um
sistemático desvio para o vermelho na conformação Ph:W, e o desvio contrário na
conformação W:Ph. A conformação Ph:W:W1 sugere um efeito cooperativo (e mútuo
cancelamento) dos desvios. Tal cancelamento não é observado para Ph:W:W2, o qual
apresenta desvio similar a Ph:W.
Tabela 7: Resultado para o desvio no espectro eletrônico dos complexos de fenol e água em relação à fase gasosa em cm-1.
Ph:W:W Ph:W W:Ph 1 2
CIS -390 340 10 -310 CIS(D) -390 370 20 -380
TDB3LYP -540 520 -50 -790 TDHF -400 380 20 -350 Exp. -350 [21] — -140 – -90

23
Conclusões parciais
Foram analisadas as conformações de fenol e água (1+1 e 1+2) que realizavam
pontes de hidrogênio, a fim de compreender suas características estruturais,
energéticas e espectroscópicas. A conformação em que o fenol doa a ponte de
hidrogênio (Ph:W) mostrou-se ser a mais estável em 2.0 kcal/mol que a conformação
em que a água doa a ponte (W:Ph). Os modos vibracionais na aproximação harmônica
apresentam-se em excelente concordância com os resultados experimentais (diferença
da ordem de 5%). A primeira excitação eletrônica destas configurações (caráter
π→π*) apresenta desvio para o azul de 370m-1 quando a água é doadora de ponte
(W:Ph) e desvio para o vermelho de 390cm-1 quando o fenol é doador (Ph:W). Este
último está em excelente concordância com o resultado experimental de 350cm-1 para
o vermelho. Adicionalmente obtiveram-se conformações com duas águas, em que ao
mesmo tempo o fenol é doador e aceitador de ponte de hidrogênio. Observou-se que
neste caso existe um cancelamento parcial das duas contribuições, fornecendo um
desvio total de 20cm-1 para o azul.

24
3.2 Agregados de fenol e etanol
Nesta seção é apresentado o estudo dos agregados de fenol e etanol, sua
energia de ligação, e seu espectro vibracional e eletrônico [22]. A motivação principal
desta parte do trabalho é a comparação direta com o trabalho feito para os clusters de
fenol e água. Entretanto o trabalho com o etanol é muito menos simples que com a
água, e por isso se tornou muito mais extenso e detalhado. Há duas conformações
possíveis para o etanol: anticlinal e synclinal (Figura 5). Nota-se que a conformação
synclinal é quiral [23], e apresenta duas possibilidades equivalentes de posição do H1.
Figura 5: Conformações possíveis do etanol. À esquerda o synclinal (ou gauche) e à direita anticlinal (ou trans). A conformação synclinal é quiral.
Tabela 8: Resultados da otimização de geometria do etanol em nível MP2/aug-cc-pVDZ e comparação com resultados experimentais.
Synclinal Anticlinal Experimental [53] RCC (Å) 1.525 1.519 1.512 RCO (Å) 1.438 1.440 1.431 RHO (Å) 0.967 0.966 0.971 ΘOCC (º) 112.6 107.2 107.8 ΘHOC (º) 107.6 108.2 105 ΘHOCC (º) ±60.8 180.0 — µ (Debye) 1.76 1.62 1.68 / 1.44
A 33.920 34.413 34.194 / 34.891 [24] B 9.095 9.327 9.189 / 9.350
I (GHz)
C 8.032 8.096 8.099 / 8.135 E(hartree) -154.61341 -154.61381 — ZPE(kcal/mol) 50.39 50.42 — τHOCC (cm-1) 285 305 * νHO (cm-1) 3820 3830 3689 [25] * As conformações synclinal e anticlinal são acopladas nesse modo [26].

25
Inicialmente foram otimizadas as geometrias de ambos os monômeros de
etanol, com o mesmo nível de cálculo utilizado para o fenol (MP2/aug-cc-pVDZ). Na
Tabela 8, apresenta-se os resultados obtidos dessa otimização. Os resultados
experimentais para o momento de dipolo, reportados no Handbook of Chemistry and
Physics [53], são 1.68 ± 0.03 D (synclinal) e 1.44 ± 0.03 D (anticlinal). Estes valores
diferem respectivamente 5% e 12% do resultado calculado. Os desvios quadráticos
médios de cada geometria calculada em relação aos resultados advindos de
espectroscopia de microondas em fase gasosa [53] sugerem que a conformação mais
estável é a anticlinal. Este resultado é corroborado por outros trabalhos teóricos e
experimentais [23,27,28,24,26]. A diferença de energia calculada das duas
conformações em nível MP2/aug-cc-pVDZ (~0.25kcal/mol) é 100% maior que a
obtida via espectro rotacional de microondas (~0.12kcal/mol) [26,29]. Embora ambos
sejam muito pequenos, este valor deve ser usado com critério, notando-se que kbT
para 300K é aproximadamente 0.6kcal/mol. Dada esta pequena diferença de energia
entre as duas conformações (em relação à kbT), e do fato da conformação synclinal ser
duplamente degenerada, a abundância relativa em temperatura ambiente é de 6:4 em
favor da synclinal [23,28]. Ainda na Tabela 8, a distância calculada da ligação OH no
etanol é subestimada em relação ao resultado reportado experimentalmente, enquanto
que para o fenol esse valor é superestimado (veja Tabela 1). As geometrias de
equilíbrio reportadas neste trabalho são garantidas pelo cálculo das freqüências
vibracionais (em torno dos pontos de mínimo da superfície de energia potencial).
Verifica-se que o modo de torção τHOCC acopla as conformações synclinal e anticlinal,
e também a torção do metil em C2 [26], não sendo trivial sua análise. O modo de
estiramento νHO é aproximadamente 10cm-1 superior para a conformação anticlinal,
fato conhecido na literatura e relacionado com a interação dos lonepairs do oxigênio
com o hidrogênio do grupo metil [30].
Com o objetivo de se estudar o espectro eletrônico dos heterodímeros fenol-
etanol, procurou-se inicialmente quais conformações poderiam ser de mais baixa
energia. Para o caso do fenol doando a ponte de hidrogênio (Ph:Et), intuiu-se que os
lonepairs do oxigênio do etanol deveriam estar na direção do hidrogênio do fenol.
Dessa forma foram propostas seis conformações possíveis como esquematizado na
Figura 6. Nesse esquema, a bola vermelha representa o oxigênio, a cinza claro o
hidrogênio e a cinza escura os carbonos (esses representados apenas no fenol). A linha
saindo do oxigênio representa a cadeia carbônica do etanol. A nomenclatura de cada
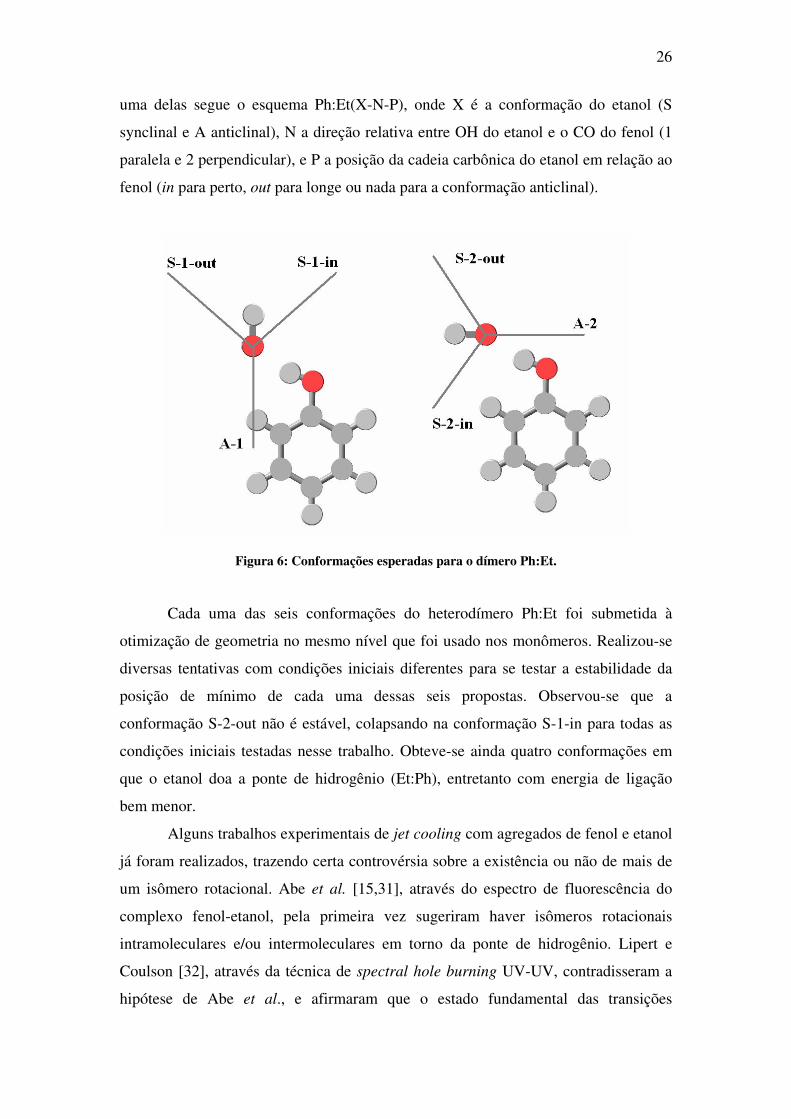
26
uma delas segue o esquema Ph:Et(X-N-P), onde X é a conformação do etanol (S
synclinal e A anticlinal), N a direção relativa entre OH do etanol e o CO do fenol (1
paralela e 2 perpendicular), e P a posição da cadeia carbônica do etanol em relação ao
fenol (in para perto, out para longe ou nada para a conformação anticlinal).
Figura 6: Conformações esperadas para o dímero Ph:Et.
Cada uma das seis conformações do heterodímero Ph:Et foi submetida à
otimização de geometria no mesmo nível que foi usado nos monômeros. Realizou-se
diversas tentativas com condições iniciais diferentes para se testar a estabilidade da
posição de mínimo de cada uma dessas seis propostas. Observou-se que a
conformação S-2-out não é estável, colapsando na conformação S-1-in para todas as
condições iniciais testadas nesse trabalho. Obteve-se ainda quatro conformações em
que o etanol doa a ponte de hidrogênio (Et:Ph), entretanto com energia de ligação
bem menor.
Alguns trabalhos experimentais de jet cooling com agregados de fenol e etanol
já foram realizados, trazendo certa controvérsia sobre a existência ou não de mais de
um isômero rotacional. Abe et al. [15,31], através do espectro de fluorescência do
complexo fenol-etanol, pela primeira vez sugeriram haver isômeros rotacionais
intramoleculares e/ou intermoleculares em torno da ponte de hidrogênio. Lipert e
Coulson [32], através da técnica de spectral hole burning UV-UV, contradisseram a
hipótese de Abe et al., e afirmaram que o estado fundamental das transições

27
observadas era devido a apenas um isômero. Cordes et al. [33], estudando o espectro
de ionização, apoiaram a interpretação de Lipert e Coulson. Eles procuraram explicar
o complexo espectro vibracional como uma combinação de modos intermoleculares
de um único confórmero. Spangenberg et al. [34], através de spectral hole burning
IR-UV e cálculos ab initio, contestaram as interpretações anteriores. Em apoio a Abe
et al., afirmaram que espectro era produzido por pelo menos dois isômeros rotacionais
com energias de excitação quase degeneradas. Spangenberg et al. apresenta 3
isômeros, os quais ele denomina anti, gauche(1) e gauche(2), que na notação deste
trabalho equivaleriam respectivamente à Ph:Et(A-1), Ph:Et(S-1-out) e Ph:Et(S-1-in).
As três geometrias apresentadas por Spangenberg et al., quando submetidas à
otimização em nível MP2/aug-cc-pVDZ, recaem nas mesmas geometrias apresentadas
neste trabalho. Tais otimizações de geometria podem ser encontradas na referência
42.
Figura 7: Conformações encontradas para Ph:Et(A-1) (esquerda) e Ph:Et(A-2) (direita).
A Figura 7 apresenta duas conformações encontradas para Ph:Et(A). O
heterodímero Ph:Et(A-1) possui energia de ligação (BE) de 6.36kcal/mol, enquanto
que Ph:Et(A-2) possui 6.14kcal/mol. Tais cálculos corrigem a energia de ponto zero
(ZPE), BSSE [13] e também o efeito de distorção na geometria dos monômeros. Em
cálculo similar, mas em nível HF/6-31G(d,p), Spangenberg et al. [34] reporta
4.49kcal/mol para o complexo Ph:Et(A-1). Está razoavelmente estabelecido na
literatura que MP2 fornece resultados equivalentes a CCSD(T) para a energia de
ligação em sistemas com pontes de hidrogênio [35], enquanto que HF fornece valores
subestimados [36]. Também é sabido que MP2 superestima a energia de ligação em
sistemas aromáticos onde os termos de dispersão são dominantes [37]. Por isso
espera-se que as energias e freqüências vibracionais calculadas neste trabalho estejam

28
levemente superestimadas em relação aos resultados experimentais, mas ainda
comparáveis a CCSD(T).
Tabela 9: Principais resultados da otimização para Ph:Et(A-1) e Ph:Et(A-2) em nível MP2/aug-cc-pVDZ. Subscrito, entre parênteses, a variação da geometria em relação ao monômero
calculado (veja Tabela 1 e Tabela 8).
Ph:Et(A-1) Ph:Et(A-2) RCC (Å) 1.519 (0.000) 1.519 (0.000) RCO (Å) 1.456 (0.016) 1.452 (0.012) RHO (Å) 0.968 (0.002) 0.967 (0.001) ΘOCC (º) 107.6 (0.4) 107.8 (0.6) ΘHOC (º) 108.2 (0.0) 108.8 (0.6)
Eta
nol
ΘHOCC (º) 173.5 (-6.5) 177.8 (-2.2) RCO (Å) 1.374 (-0.008) 1.371 (-0.011) RHO (Å) 0.982 (0.014) 0.980 (0.012) ΘHOC (º) 108.6 (0.0) 108.8 (0.2) Fe
nol
ΘHOCC (º) 7.8 (7.8) 13.7 (13.7) RH…O (Å) 1.803 1.821 ΘOH…O (º) 166.6 168.6 µ (Debye) 3.33 3.59 BE(kcal/mol) 6.36 6.14
Ainda no agregado Ph:Et(A-1), pela Tabela 9, nota-se que há maior mudança
no diedro ΘHOCC do etanol (-6.5º) em relação ao monômero. Já a conformação
Ph:Et(A-2) apresenta maior mudança no diedro ΘHOCC do fenol (13.7º). A distância da
ponte de hidrogênio RH…O no complexo Ph:Et(A-1) é 0.018Å mais próxima do que
em Ph:Et(A-2). Além da distância da ponte de hidrogênio, é relevante também notar a
distância do grupo metil do etanol ao oxigênio do fenol. O complexo Ph:Et(A-1), em
relação ao Ph:Et(A-2), apresenta o hidrogênio do metil mais próximo 0.016Å do
oxigênio fenólico. Isto reforça o resultado do complexo Ph:Et(A-1) ser mais estável
(maior energia de ligação) em relação ao Ph:Et(A-2). Entretanto a diferença de
energia é muito pequena, e deve-se ter cautela ao se utilizar esse resultado. O
complexo Ph:Et(A-2) possui momento de dipolo maior (em 8%) que Ph:Et(A-1), e
portanto espera-se que se estabilize mais em um ambiente polar.
A Figura 8 apresenta as conformações com etanol synclinal aceitando a ponte
de hidrogênio do fenol. Dentre elas, a conformação mais estável é a Ph:Et(S-1-in)
com 6.49kcal/mol de energia de ligação (Tabela 10). Ph:Et(S-1-in) é a conformação
com a maior energia de ligação de todos os complexos estudados (veja Tabela 9 e
Tabela 12), e é a que possui o metil do etanol mais próximo do oxigênio do fenol

29
(2.70Å). O complexo Ph:Et(S-2-in) possui a distância da ponte de hidrogênio menor
0.008Å em relação a Ph:Et(S-1-in). Entretanto seu metil é 0.179Å mais distante do
oxigênio fenólico e sua energia de ligação aproximadamente 0.5kcal/mol menor que a
conformação Ph:Et(S-1-in). As conformações Ph:Et(S-1-in) e Ph:Et(S-2-in)
deformam-se de maneira equivalente em relação aos monômeros, sendo maior a
deformação no fenol e menor no etanol.
Figura 8: Conformações encontradas para Ph:Et(S-1-in) (esquerda), Ph:Et(S-1-out) (direita) e Ph:Et(S-2-in) (acima). A conformação Ph:Et(S-2-out) não é estável e colapsa em Ph:Et(S-1-in).
A conformação Ph:Et(S-1-out) é a única com a cadeia carbônica do etanol
longe do anel fenólico. Em comparação com as outras duas conformações, ela
apresenta maior deformação no etanol e menor no fenol (veja Tabela 10). Esta
conformação também possui o maior momento de dipolo (3.62D). Spangenberg et al.
[34] encontra um isômero equivalente a este como o mais estável, com 4.85kcal/mol
de energia de ligação em nível HF/6-31G(d,p). Com o nível de cálculo MP2/aug-cc-
pVDZ, a conformação Ph:Et(S-1-out) tem energia de ligação intermediária dentre as
três synclinais aceitadoras. Esta conformação possui um hidrogênio do carbono C1
0.073Å mais próximo em comparação com a distância metil etanólico e oxigênio
fenólico do complexo Ph:Et(S-2-in) (veja Figura 5 e Figura 8). Note que Ph:Et(S-2-
in) tem energia de ligação menor que Ph:Et(S-1-out) (Tabela 10).
30
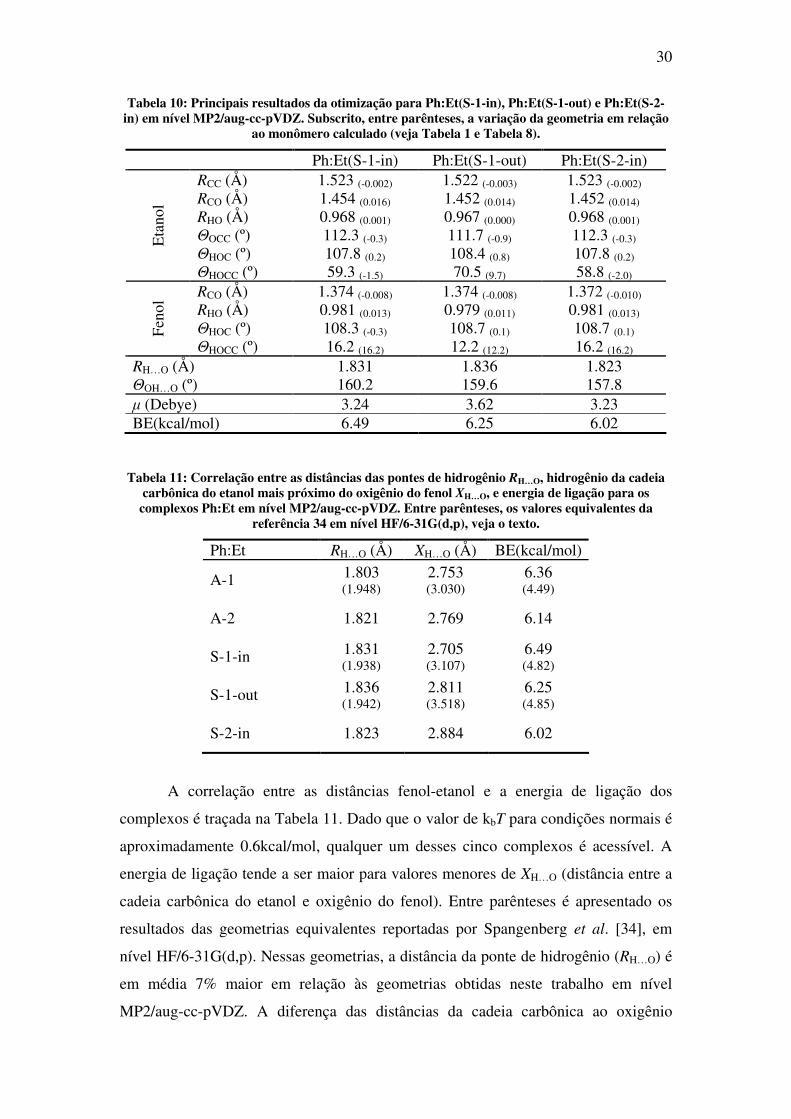
Tabela 10: Principais resultados da otimização para Ph:Et(S-1-in), Ph:Et(S-1-out) e Ph:Et(S-2-in) em nível MP2/aug-cc-pVDZ. Subscrito, entre parênteses, a variação da geometria em relação
ao monômero calculado (veja Tabela 1 e Tabela 8).
Ph:Et(S-1-in) Ph:Et(S-1-out) Ph:Et(S-2-in) RCC (Å) 1.523 (-0.002) 1.522 (-0.003) 1.523 (-0.002) RCO (Å) 1.454 (0.016) 1.452 (0.014) 1.452 (0.014) RHO (Å) 0.968 (0.001) 0.967 (0.000) 0.968 (0.001) ΘOCC (º) 112.3 (-0.3) 111.7 (-0.9) 112.3 (-0.3) ΘHOC (º) 107.8 (0.2) 108.4 (0.8) 107.8 (0.2)
Eta
nol
ΘHOCC (º) 59.3 (-1.5) 70.5 (9.7) 58.8 (-2.0) RCO (Å) 1.374 (-0.008) 1.374 (-0.008) 1.372 (-0.010) RHO (Å) 0.981 (0.013) 0.979 (0.011) 0.981 (0.013) ΘHOC (º) 108.3 (-0.3) 108.7 (0.1) 108.7 (0.1) Fe
nol
ΘHOCC (º) 16.2 (16.2) 12.2 (12.2) 16.2 (16.2) RH…O (Å) 1.831 1.836 1.823 ΘOH…O (º) 160.2 159.6 157.8 µ (Debye) 3.24 3.62 3.23 BE(kcal/mol) 6.49 6.25 6.02
Tabela 11: Correlação entre as distâncias das pontes de hidrogênio RH…O, hidrogênio da cadeia carbônica do etanol mais próximo do oxigênio do fenol XH…O, e energia de ligação para os
complexos Ph:Et em nível MP2/aug-cc-pVDZ. Entre parênteses, os valores equivalentes da referência 34 em nível HF/6-31G(d,p), veja o texto.
Ph:Et RH…O (Å) XH…O (Å) BE(kcal/mol)
A-1 1.803 (1.948)
2.753 (3.030)
6.36 (4.49)
A-2 1.821 2.769 6.14
S-1-in 1.831 (1.938)
2.705 (3.107)
6.49 (4.82)
S-1-out 1.836 (1.942)
2.811 (3.518)
6.25 (4.85)
S-2-in 1.823 2.884 6.02
A correlação entre as distâncias fenol-etanol e a energia de ligação dos
complexos é traçada na Tabela 11. Dado que o valor de kbT para condições normais é
aproximadamente 0.6kcal/mol, qualquer um desses cinco complexos é acessível. A
energia de ligação tende a ser maior para valores menores de XH…O (distância entre a
cadeia carbônica do etanol e oxigênio do fenol). Entre parênteses é apresentado os
resultados das geometrias equivalentes reportadas por Spangenberg et al. [34], em
nível HF/6-31G(d,p). Nessas geometrias, a distância da ponte de hidrogênio (RH…O) é
em média 7% maior em relação às geometrias obtidas neste trabalho em nível
MP2/aug-cc-pVDZ. A diferença das distâncias da cadeia carbônica ao oxigênio

31
fenólico (XH…O) é maior ainda: 10% para Ph:Et(A-1), 15% para Ph:Et(S-1-in) e 25%
para Ph:Et(S-1-out). Isto indica que a correlação eletrônica (origem da interação de
dispersão) é importante para descrever a interação entre a cadeia carbônica do etanol e
a anel fenólico, e conseqüentemente na determinação da geometria do complexo.
Para o caso do etanol doando a ponte de hidrogênio (Et:Ph) encontrou-se
apenas quatro conformações. Duas delas são pontes H-O, em que o etanol doa o
próton para o oxigênio do fenol. As outras duas são pontes do tipo H-π, ou seja, o
hidrogênio do etanol faz ponte com a nuvem π fenol. Estas conformações, com o
etanol doador (Figura 9), diferenciam-se pela maior distância da ponte de hidrogênio
em relação às configurações com etanol aceitador (diferença superior a 0.3Å). Ainda
em relação ao etanol aceitador, estas conformações com etanol doador apresentam
aproximadamente metade da energia de ligação (Tabela 12). Isto corrobora o caráter
ácido do fenol (pKa = 9.99) em relação ao etanol (pKa = 15.5) [53], i.e., o fenol doa o
próton mais facilmente (energeticamente favorável) do que o etanol.
Figura 9: Conformações encontradas para Et(S):Ph (superior-esquerda), Et(A):Ph (superior-direita), Et(S):π-Ph (inferior-esquerda) e Et(S):π-Ph (inferior-direita). As linhas tracejadas sobre
os átomos do anel foram colocadas apenas para facilitar a visualização.

32
Comparando a Tabela 12 com as anteriores, observa-se que o comprimento da
ligação RCO da molécula doadora se encurta em relação ao monômero. A mesma
ligação RCO da molécula aceitadora se alonga. Nota-se também que a ligação ROH se
alonga tanto na molécula doadora quanto na aceitadora. Esta regra é quebrada para as
configurações que formam ponte H-π. Como a interação da ponte não está sobre o
oxigênio do fenol, a ligação CO do fenol é pouco afetada. Os momentos dipolares das
conformações Et:Ph são em média menores que os das conformações Ph:Et. Isto
sugere que as estruturas Ph:Et irão se estabilizar melhor em solvente polar. A energia
de ligação das conformações Et:Ph também é expressivamente menor, sendo
aproximadamente metade daquela encontrada para Ph:Et. Tal resultado é coerente
com as energias de ligação obtidas para os cluster de fenol e água (seção anterior).
Tabela 12: Principais resultados da otimização para Et(S):Ph e Et(A):Ph em nível MP2/aug-cc-pVDZ. Subscrito, entre parênteses, a variação em relação ao monômero calculado (veja Tabela 1
e Tabela 8).
Et(S):Ph Et(A):Ph Et(S):π-Ph Et(A):π-Ph RCC (Å) 1.526 (0.001) 1.520 (0.001) 1.526 (0.001) 1.520 (0.001) RCO (Å) 1.436 (-0.002) 1.438 (-0.002) 1.435 (-0.003) 1.435 (-0.005) RHO (Å) 0.970 (0.003) 0.969 (0.003) 0.970 (0.003) 0.970 (0.004) ΘOCC (º) 113.0 (0.4) 107.4 (0.2) 112.4 (-0.2) 107.5 (0.3) ΘHOC (º) 108.5 (0.9) 108.1 (-0.1) 107.6 (0.0) 107.9 (-0.3)
Eta
nol
ΘHOCC (º) 70.1 (9.3) 179.6 (-0.4) 74.0 (10.2) 178.4 (-1.6) RCO (Å) 1.387 (0.005) 1.388 (0.006) 1.379 (-0.003) 1.379 (-0.003) RHO (Å) 0.969 (0.001) 0.969 (0.001) 0.969 (0.001) 0.968 (0.000) ΘHOC (º) 108.9 (0.3) 108.8 (0.2) 108.8 (0.2) 108.7 (0.1) Fe
nol
ΘHOCC (º) 2.4 (2.4) 6.9 (6.9) 0.3 (0.3) 1.2 (1.2) RH…O (Å) 2.117 2.182 — — ΘOH…O (º) 172.9 166.1 — — µ (Debye) 2.87 3.27 2.42 2.33 BE(kcal/mol) 3.66 3.01 3.64 3.36
A Tabela 13 apresenta as constantes rotacionais e a projeção do momento de
dipolo nos eixos principais dos complexos estudados. As constantes rotacionais dos
complexos em que o etanol doa a ponte são distintas daquelas em que o etanol aceita a
ponte. A constante rotacional do eixo principal (A) varia entre 2.2 a 2.5GHz com o
etanol aceitador (Ph:Et), enquanto que para o etanol doador (Et:Ph) não passa de
1.7GHz. Mesmo tendo constantes rotacionais próximas, os complexos em que o
etanol é doador (Et:Ph) apresentam diferentes componentes e valor total do momento
de dipolo. A mesma observação pode ser tomada sobre os complexos em que o etanol
33
recebe a ponte (Ph:Et). Destes complexos destacam-se Ph:Et(S-1-in) e Ph:Et(S-2-in),
os quais possuem constantes rotacionais e momento de dipolo total muito similares,
mas componentes µA, µB, e µC completamente diferentes. Entre parênteses, para as
conformações Ph:Et(A-1), Ph:Et(S-1-in) e Ph:Et(S-1-out) são apresentados os valores
das geometrias equivalentes obtidas em nível HF/6-31G(d,p) por Spangenberg et al.
[34]. O fato da constante rotacional no eixo principal (A) ser maior acusa que o etanol
está mais afastado do fenol, isto é, os monômeros estão menos empacotados (como
destacado pelo valor de XH…O na Tabela 11). Não é possível traçar uma comparação
direta dos momentos de dipolo entre esse trabalho e o de Spangenberg et al. pelo fato
de serem diferentes tanto as geometrias e quanto o nível de cálculo. Entretanto,
preserva-se a relação de que Ph:Et(S-1-out) tem o maior momento de dipolo, seguido
de Ph:Et(A-1) e depois Ph:Et(S-1-in).
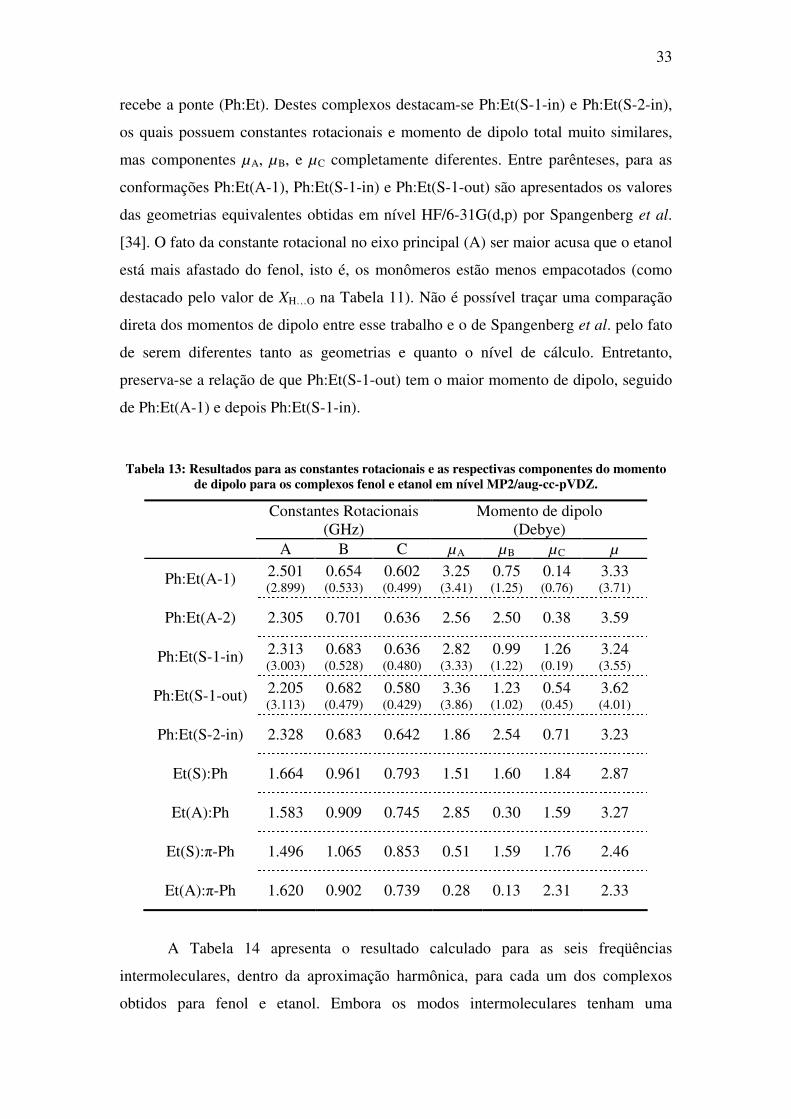
Tabela 13: Resultados para as constantes rotacionais e as respectivas componentes do momento de dipolo para os complexos fenol e etanol em nível MP2/aug-cc-pVDZ.
Constantes Rotacionais (GHz)
Momento de dipolo (Debye)
A B C µA µB µC µ
Ph:Et(A-1) 2.501 (2.899)
0.654 (0.533)
0.602 (0.499)
3.25 (3.41)
0.75 (1.25)
0.14 (0.76)
3.33 (3.71)
Ph:Et(A-2) 2.305 0.701 0.636 2.56 2.50 0.38 3.59
Ph:Et(S-1-in) 2.313 (3.003)
0.683 (0.528)
0.636 (0.480)
2.82 (3.33)
0.99 (1.22)
1.26 (0.19)
3.24 (3.55)
Ph:Et(S-1-out) 2.205 (3.113)
0.682 (0.479)
0.580 (0.429)
3.36 (3.86)
1.23 (1.02)
0.54 (0.45)
3.62 (4.01)
Ph:Et(S-2-in) 2.328 0.683 0.642 1.86 2.54 0.71 3.23
Et(S):Ph 1.664 0.961 0.793 1.51 1.60 1.84 2.87
Et(A):Ph 1.583 0.909 0.745 2.85 0.30 1.59 3.27
Et(S):π-Ph 1.496 1.065 0.853 0.51 1.59 1.76 2.46
Et(A):π-Ph 1.620 0.902 0.739 0.28 0.13 2.31 2.33
A Tabela 14 apresenta o resultado calculado para as seis freqüências
intermoleculares, dentro da aproximação harmônica, para cada um dos complexos
obtidos para fenol e etanol. Embora os modos intermoleculares tenham uma
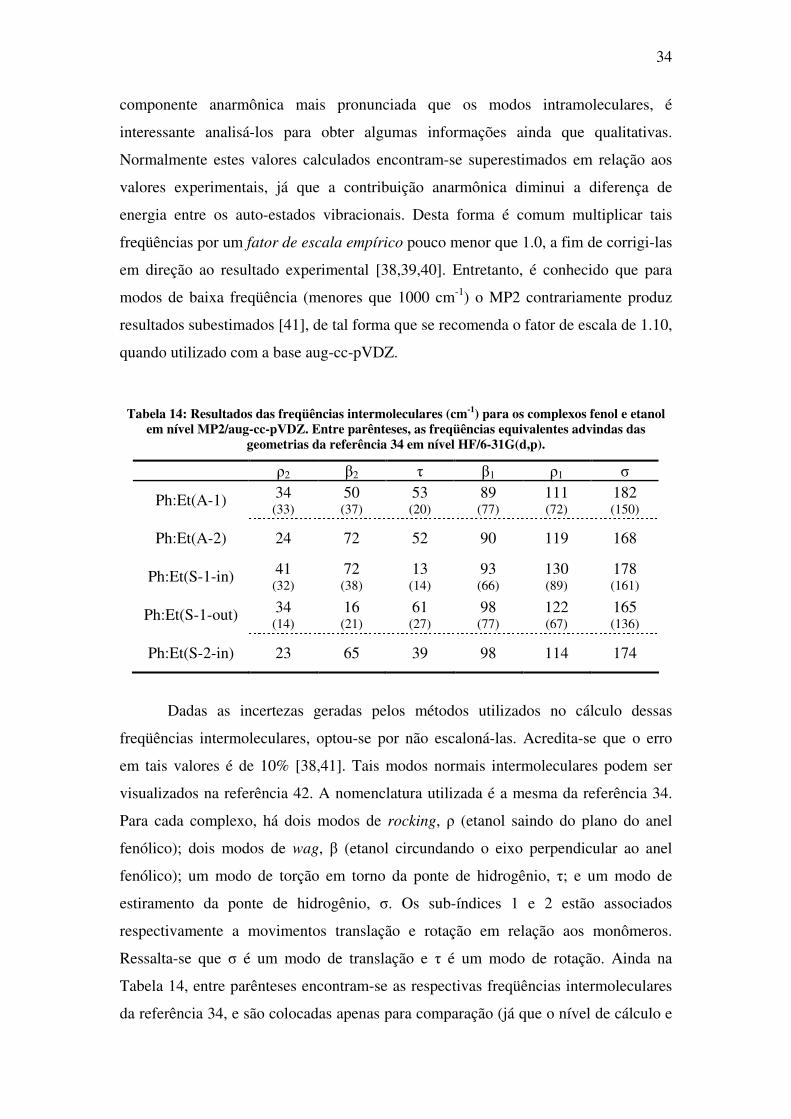
34
componente anarmônica mais pronunciada que os modos intramoleculares, é
interessante analisá-los para obter algumas informações ainda que qualitativas.
Normalmente estes valores calculados encontram-se superestimados em relação aos
valores experimentais, já que a contribuição anarmônica diminui a diferença de
energia entre os auto-estados vibracionais. Desta forma é comum multiplicar tais
freqüências por um fator de escala empírico pouco menor que 1.0, a fim de corrigi-las
em direção ao resultado experimental [38,39,40]. Entretanto, é conhecido que para
modos de baixa freqüência (menores que 1000 cm-1) o MP2 contrariamente produz
resultados subestimados [41], de tal forma que se recomenda o fator de escala de 1.10,
quando utilizado com a base aug-cc-pVDZ.
Tabela 14: Resultados das freqüências intermoleculares (cm-1) para os complexos fenol e etanol em nível MP2/aug-cc-pVDZ. Entre parênteses, as freqüências equivalentes advindas das
geometrias da referência 34 em nível HF/6-31G(d,p).
ρ2 β2 τ β1 ρ1 σ
Ph:Et(A-1) 34 (33)
50 (37)
53 (20)
89 (77)
111 (72)
182 (150)
Ph:Et(A-2) 24 72 52 90 119 168
Ph:Et(S-1-in) 41 (32)
72 (38)
13 (14)
93 (66)
130 (89)
178 (161)
Ph:Et(S-1-out) 34 (14)
16 (21)
61 (27)
98 (77)
122 (67)
165 (136)
Ph:Et(S-2-in) 23 65 39 98 114 174
Dadas as incertezas geradas pelos métodos utilizados no cálculo dessas
freqüências intermoleculares, optou-se por não escaloná-las. Acredita-se que o erro
em tais valores é de 10% [38,41]. Tais modos normais intermoleculares podem ser
visualizados na referência 42. A nomenclatura utilizada é a mesma da referência 34.
Para cada complexo, há dois modos de rocking, ρ (etanol saindo do plano do anel
fenólico); dois modos de wag, β (etanol circundando o eixo perpendicular ao anel
fenólico); um modo de torção em torno da ponte de hidrogênio, τ; e um modo de
estiramento da ponte de hidrogênio, σ. Os sub-índices 1 e 2 estão associados
respectivamente a movimentos translação e rotação em relação aos monômeros.
Ressalta-se que σ é um modo de translação e τ é um modo de rotação. Ainda na
Tabela 14, entre parênteses encontram-se as respectivas freqüências intermoleculares
da referência 34, e são colocadas apenas para comparação (já que o nível de cálculo e
35
as geometrias produzidas são diferentes). As caracterizações dos modos calculados τ,
ρ2 e β2 apresentados na tabela 3 da referência 34 são interpretadas de maneira distinta
neste trabalho, e podem ser conferidos na referência 43. Não foi possível também
caracterizar univocamente modo por modo com os resultados experimentais como
feito na referência 34. A princípio os confórmeros Ph:Et(A-1) e Ph:Et(A-2) poderiam
se intercambiar através dos modos de wag (β1 e β2), mas a barreira correspondente
não foi calculada. O mesmo vale para os confórmeros Ph:Et(S-1-out) e Ph:Et(S-2-in).
Os modos dos complexos em que o etanol doa a ponte de hidrogênio não foram
analisados. Estes se diferenciam substancialmente daquelas em que o etanol aceita,
dado o posicionamento do etanol sobre o anel fenólico. Nesses complexos (Et:Ph), o
etanol não interage apenas via ponte de hidrogênio, mas também com a nuvem π do
fenol e a cadeia carbônica do etanol. Uma complicação adicional é o fato da energia
de ligação ser menor, ou seja, o sistema é menos ligado. Isso implica que os erros nos
modos vibracionais são ainda maiores. Desta maneira, é muito difícil determinar e
caracterizar tais modos vibracionais.
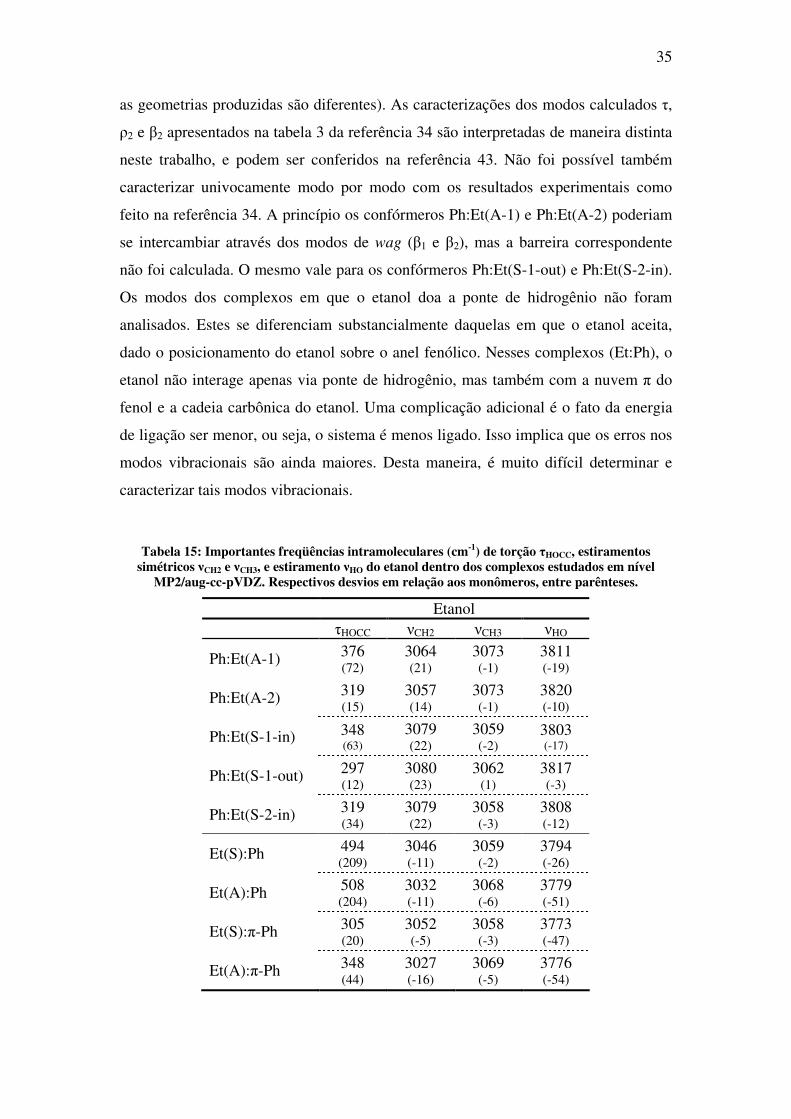
Tabela 15: Importantes freqüências intramoleculares (cm-1) de torção τHOCC, estiramentos simétricos νCH2 e νCH3, e estiramento νHO do etanol dentro dos complexos estudados em nível
MP2/aug-cc-pVDZ. Respectivos desvios em relação aos monômeros, entre parênteses.
Etanol τHOCC νCH2 νCH3 νHO
Ph:Et(A-1) 376 (72)
3064 (21)
3073 (-1)
3811 (-19)
Ph:Et(A-2) 319 (15)
3057 (14)
3073 (-1)
3820 (-10)
Ph:Et(S-1-in) 348 (63)
3079 (22)
3059 (-2)
3803 (-17)
Ph:Et(S-1-out) 297 (12)
3080 (23)
3062 (1)
3817 (-3)
Ph:Et(S-2-in) 319 (34)
3079 (22)
3058 (-3)
3808 (-12)
Et(S):Ph 494 (209)
3046 (-11)
3059 (-2)
3794 (-26)
Et(A):Ph 508 (204)
3032 (-11)
3068 (-6)
3779 (-51)
Et(S):π-Ph 305 (20)
3052 (-5)
3058 (-3)
3773 (-47)
Et(A):π-Ph 348 (44)
3027 (-16)
3069 (-5)
3776 (-54)

36
Tabela 16: Importantes freqüências intramoleculares (cm-1) de torção τHOCC, estiramento simétrico ν(CH)5, e estiramento νHO do fenol dentro dos complexos estudados em nível MP2/aug-cc-
pVDZ. Respectivos desvios em relação aos monômeros, entre parênteses.
Fenol τHOCC ν(CH)5 νHO
Ph:Et(A-1) 785 (456)
3232 (-4)
3521 (-287)
Ph:Et(A-2) 732 (402)
3233 (-3)
3561 (-247)
Ph:Et(S-1-in) 710 (381)
3232 (-4)
3546 (-262)
Ph:Et(S-1-out) 704 (375)
3232 (-4)
3580 (-228)
Ph:Et(S-2-in) 733 (404)
3233 (-3)
3549 (-259)
Et(S):Ph 338 (8)
3237 (1)
3794 (-14)
Et(A):Ph 337 (7)
3237 (1)
3795 (-13)
Et(S):π-Ph 328 (-1)
3239 (3)
3800 (-8)
Et(A):π-Ph 335 (6)
3239 (3)
3802 (-6)
A Tabela 15 apresenta os resultados calculados para algumas freqüências
vibracionais intramoleculares do etanol para os complexos estudados, e sua variação
em relação aos monômeros isolados (veja Tabela 8). A Tabela 16 faz o mesmo para o
fenol (veja Tabela 1). Em relação à ponte de hidrogênio, como esperado, os modos de
torção (τHOCC) desviam para o azul (valores positivos), enquanto que os modos de
estiramento (νHO) desviam para o vermelho (valores negativos). Para os complexos
em que o fenol doa a ponte de hidrogênio, Ph:Et(A-1) fornece o estiramento e a torção
de maior desvio, tanto para o fenol quanto para o etanol. Os complexos em que o
etanol doa a ponte de hidrogênio possuem desvios menores em relação àquelas em
que o etanol aceita. O desvio para o vermelho dos modos de estiramento (νHO) pode
ser correlacionado com a variação do comprimento OH da ligação [44] (veja Tabela 9
e Tabela 10). O maior alargamento de ligação ocorre para o Ph:Et(A-1) (0.014Å) em
concordância com o maior desvio para a freqüência (-287cm-1). A boa concordância
desta correlação é apresentada na Tabela 17. A Figura 10 ilustra esta boa
concordância para todos os pontos.
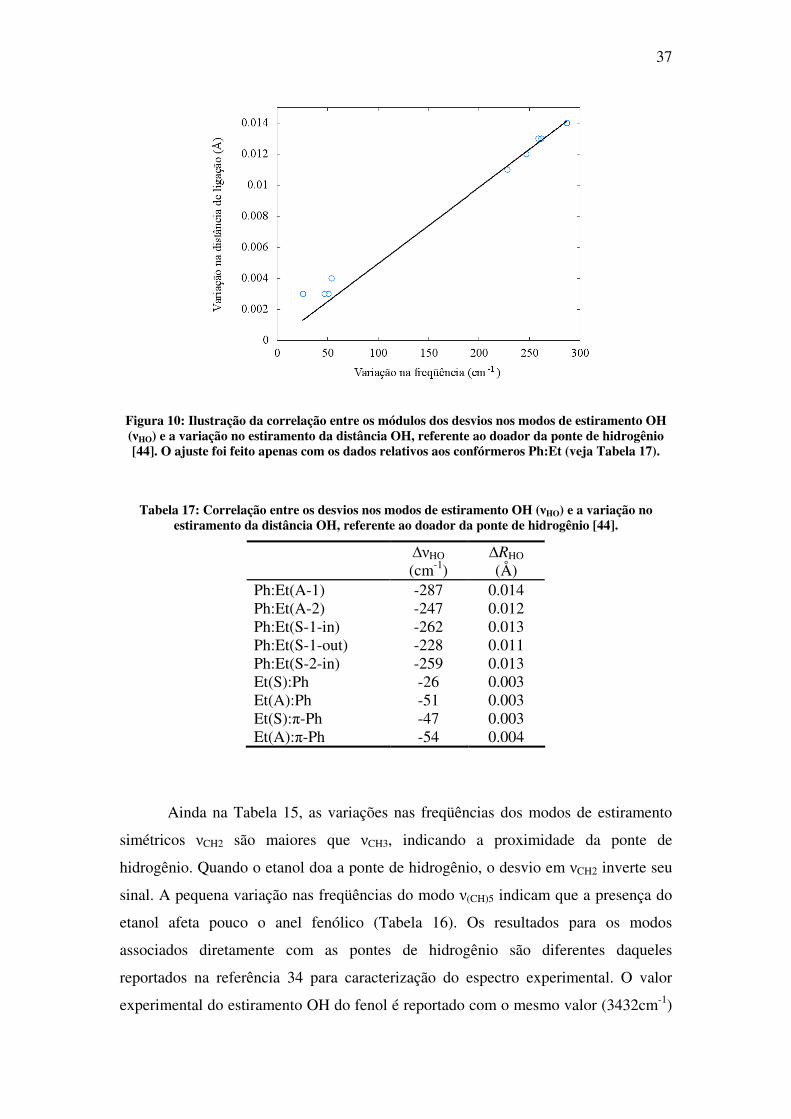
37
Figura 10: Ilustração da correlação entre os módulos dos desvios nos modos de estiramento OH (νHO) e a variação no estiramento da distância OH, referente ao doador da ponte de hidrogênio [44]. O ajuste foi feito apenas com os dados relativos aos confórmeros Ph:Et (veja Tabela 17).
Tabela 17: Correlação entre os desvios nos modos de estiramento OH (νHO) e a variação no estiramento da distância OH, referente ao doador da ponte de hidrogênio [44].
∆νHO (cm-1)
∆RHO (Å)
Ph:Et(A-1) -287 0.014 Ph:Et(A-2) -247 0.012 Ph:Et(S-1-in) -262 0.013 Ph:Et(S-1-out) -228 0.011 Ph:Et(S-2-in) -259 0.013 Et(S):Ph -26 0.003 Et(A):Ph -51 0.003 Et(S):π-Ph -47 0.003 Et(A):π-Ph -54 0.004
Ainda na Tabela 15, as variações nas freqüências dos modos de estiramento
simétricos νCH2 são maiores que νCH3, indicando a proximidade da ponte de
hidrogênio. Quando o etanol doa a ponte de hidrogênio, o desvio em νCH2 inverte seu
sinal. A pequena variação nas freqüências do modo ν(CH)5 indicam que a presença do
etanol afeta pouco o anel fenólico (Tabela 16). Os resultados para os modos
associados diretamente com as pontes de hidrogênio são diferentes daqueles
reportados na referência 34 para caracterização do espectro experimental. O valor
experimental do estiramento OH do fenol é reportado com o mesmo valor (3432cm-1)

38
para os complexos em que o etanol recebe a ponte (Ph:Et). Os valores calculados com
MP2/aug-cc-pVDZ variam de 3521cm-1 para o Ph:Et(A-1) até 3580cm-1 para Ph:Et(S-
1-out). Os respectivos valores calculados por Spangenberg et al. [34] diferem apenas
4cm-1. A freqüência de estiramento OH do etanol anticlinal é em média maior do que
a do etanol synclinal, nos complexos estudados (veja na Tabela 15). Tal resultado era
esperado por Spangenberg et al. [34], e junto com as freqüências νCH2 e νCH2 do
etanol, foi utilizado na interpretação do espectro experimental. Entretanto não é
possível associar de maneira única cada um dos complexos com os espectros
experimentais apresentados, como feitos na referência 34.
Tabela 18: Desvio padrão das freqüências vibracionais (cm-1) utilizando o fator de escala empírico de 0.95, em relação às freqüências experimentais associadas com o etanol anticlinal (A)
e synclinal (S) reportados por Spangenberg et al. [34].
Ph:Et A S A-1 34 31 A-2 23 20 S-1-in 32 26 S-1-out 26 18 S-2-in 31 25
Utilizando metodologia similar a de Sinha et al. [41], procurou-se um fator de
escala empírico para multiplicar as freqüências intermoleculares calculadas a fim de
fornecer o menor desvio padrão em relação às freqüências experimentais
intramoleculares apresentadas na referência 34. As freqüências calculadas podem ser
visualizadas na referência 42. Utilizando os modos de estiramento νOH, e os modos
simétricos νCH2 e νCH3 do etanol e ν(CH)5 do fenol, obteve-se um fator de 0.95 (com
apenas duas casas de precisão como sugerido por Irikura et al. [38]). Este resultado é
coerente com o valor de 0.96, recomendado por Sinha et al. [41] para freqüências
acima de 1000cm-1, com o nível de cálculo MP2/aug-cc-pVDZ. A Tabela 18
apresenta os desvios padrão obtidos com o fator de escala de 0.95 (ver referência
[41]).
A conformação Ph:Et(A-1), cuja conformação equivalente é utilizada no
trabalho de Spangenberg et al. [34], é a que menos combina (maior desvio padrão)
com as freqüências experimentais associadas ao complexo com etanol anticlinal. O
mesmo vale para a conformação Ph:Et(S-1-in). Com a informação apenas do desvio

39
padrão, poder-se-ia dizer que as conformações que melhor se ajustam às freqüências
experimentais são Ph:Et(A-2) e Ph:Et(S-1-out).
Finalmente, com o intuito de se comparar os resultados do espectro eletrônico
obtidos para os agregados de fenol e água, submeteram-se ao cálculo quântico os nove
complexos de fenol e etanol. Os resultados para o desvio em relação ao fenol isolado
são apresentados na Tabela 19. Como esperado [20,21,31,32,33,34] quando o fenol
doa a ponte de hidrogênio, a excitação eletrônica (de caráter π → π*) desvia para
menores freqüências, apresentando-se o contrário para o caso do fenol aceitando a
ponte de hidrogênio. Resultado similar, mas de menor desvio, é obtido para os
agregados que formam ponte H-π. Os resultados obtidos com TDHF, CIS e CIS(D)
apresentam valores similares quando o fenol doa a ponte de hidrogênio (Ph:Et).
TDB3LYP tem reciprocidade com os valores obtidos para fenol e água (Tabela 7). Ele
apresenta desvios maiores que os outros métodos, tanto para etanol doador quanto
aceitador. Para o caso das configurações H-π, TDB3LYP fornece um resultado
contrário ao esperado. É interessante notar que, para alguns agregados, a primeira
excitação com TDB3LYP possui caráter de transferência de carga intermolecular,
sendo que a transição π → π* aparece apenas na segunda excitação eletrônica. Os
valores experimentais reportados são -405cm-1 e -395cm-1 [31] e são associados
respectivamente com etanol anticlinal e synclinal por Spangenberg et al. [34]. Os
confórmeros que mais combinam com estes valores são respectivamente Ph:Et(A-1) e
Ph:Et(S-1-out). Não há resultado experimental para o etanol doador de ponte de
hidrogênio (Et:Ph). Mas para todos os métodos e clusters (com exceção do
TDB3LYP), as transições calculadas apresentam desvio para o azul.
Tabela 19: Resultados dos desvios do espectro eletrônico (cm-1) da transição π → π* do fenol para os complexos fenol e etanol em relação ao monômero. Diferentes métodos utilizando a mesma
base aug-cc-pVDZ. Os valores experimentais provêm das referências 31 e 34.
TDB3LYP TDHF CIS CIS(D) Exp. Ph:Et(A-1) -510 -377 -388 -377 Ph:Et(A-2) -708 -495 -497 -508
-405
Ph:Et(S-1-in) -456 -315 -320 -345 Ph:Et(S-1-out) -548 -360 -365 -359 Ph:Et(S-2-in) -761 -425 -423 -444
-395
Et(S):Ph 260 313 296 129 Et(A):Ph 230 337 310 172
—
Et(S):π-Ph -40 251 247 40 Et(A):π-Ph -16 203 202 25
—

40
Conclusões parciais
Foram obtidos nove diferentes agregados fenol-etanol (1+1), dos quais seis
nunca foram antes reportados. As geometrias foram otimizadas em nível MP2/aug-cc-
pVDZ, e sua estabilidade foi garantida através do cálculo dos modos vibracionais. As
energias de ligação foram calculadas considerando-se a energia de ponto zero,
correções de counterpoise da base e efeitos de deformação de geometria. A
configuração mais estável encontrada foi Ph:Et(S-1-in) (veja Figura 8) com energia de
ligação de 6.5 kcal/mol, condizente com as expectativas teóricas e experimentais (veja
o texto). Todas as configurações em que o fenol doa a ponte de hidrogênio possuem
energia de ligação calculada variando entre 6.0 e 6.5 kcal/mol. Por outro lado, quando
o fenol é aceitador, a energia de ligação é menor, variando entre 3.0 e 3.7 kcal/mol.
As freqüências inter e intramoleculares foram analisadas, e considerações sobre seus
valores, fatores de escala e correlação com as mudanças geométricas foram feitas. Em
especial foi caracterizada a transição π → π* do fenol. Observou-se que esta transição
apresenta um desvio sistemático para o vermelho quando o etanol é aceitador de ponte
de hidrogênio (Ph:Et), assim como um desvio para o azul quando este é doador
(Et:Ph). Este resultado está em plena concordância com as expectativas experimentais
e com os resultados obtidos para a água (veja a seção anterior).

41
3.3 Fase líquida: Simulação do fenol em água
Concluído o estudo dos agregados, fez-se a simulação computacional do fenol
em água [12]. Com o mesmo nível de cálculo usado para a otimização de geometria
da molécula isolada (MP2/aug-cc-pVDZ), utilizou-se o método CHELPG [45] para
ajustar cargas clássicas sobre os sítios atômicos que melhor descrevessem os
momentos de multipolo quânticos (de fase gasosa). Com a geometria da molécula
isolada, as cargas CHELPG e parâmetros Lennard-Jones estabelecidos na literatura
para o cálculo de fenol em água [46], construiu-se um modelo não polarizado para se
realizar a simulação clássica. Este modelo foi submetido à simulação em condições
normais no ensemble NpT (298K, 1atm), com 500 moléculas de água. Os parâmetros
utilizados para a água são os do modelo SPC de Berendsen [47]. Este modelo de fenol
foi polarizado através do processo iterativo utilizado em trabalhos anteriores pelo
grupo. Nesta metodologia, realiza-se uma simulação clássica, da qual é retirado um
número reduzido de configurações estatisticamente descorrelacionadas. Tais
configurações são submetidas ao cálculo quântico, sendo o fenol colocado
explicitamente e as camadas de solvatação descritas por cargas pontuais. Os
resultados deste método carregam toda a informação estatística do ensemble, e é
capaz de polarizar eficientemente o soluto. A novidade deste procedimento
introduzida por Coutinho et al. [48] e adaptada dos trabalhos de Aguilar e
colaboradores [49], é que ao invés de se fazer N cálculos quânticos para se fazer a
média, realiza-se apenas um único cálculo quântico com a média das configurações.
Isto só é possível porque o solvente é descrito por cargas pontuais e o soluto não é
flexível (ou seja, rígido) durante a simulação. Após a primeira iteração, novamente as
cargas no fenol são re-obtidas via CHELPG, mas que agora contempla polarização.
Para que o efeito desta polarização do soluto afete a estrutura do solvente a sua volta,
o processo de simulação é repetido iterativamente até que a variação do momento de
dipolo da molécula de referência flutue em torno de uma média (dentro de um desvio
aceitável). Neste ponto, considera-se que o soluto e o solvente estão em equilíbrio
eletrostático.
42
0 1 2 3 41.2
1.4
1.6
1.8
2.0
2.2
2.4
2.6
Dip
ole
mom
ent (
D)
Iteration number
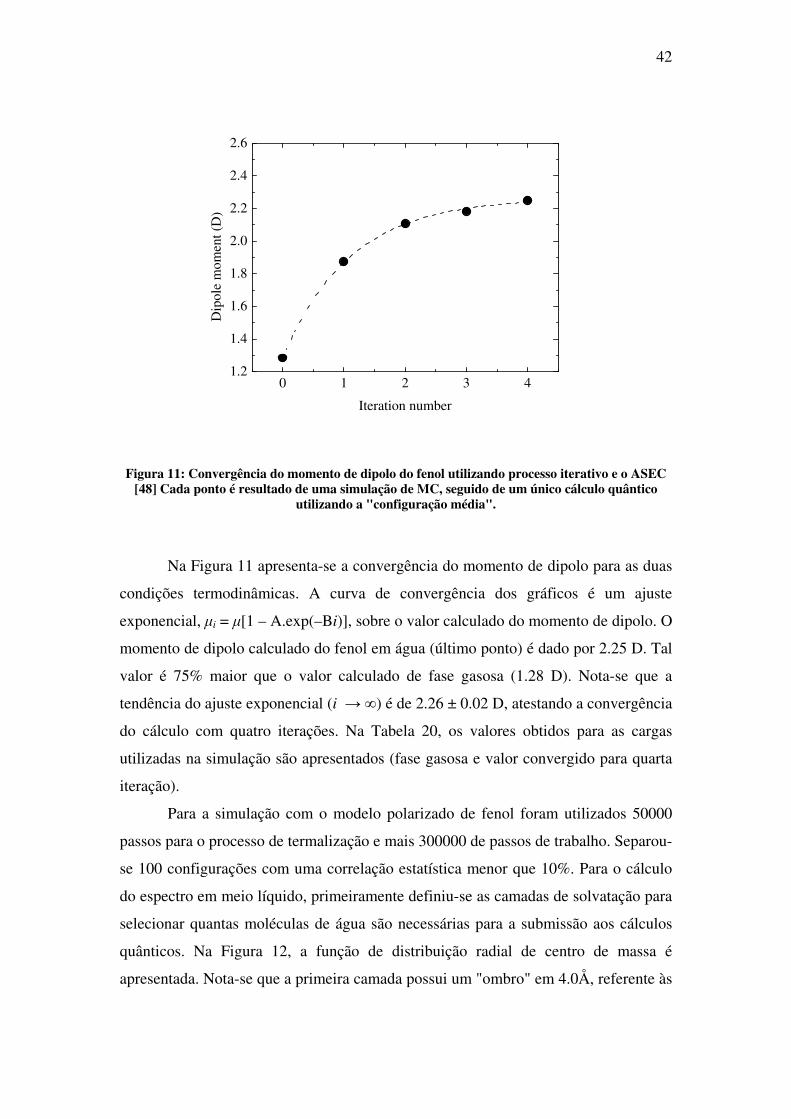
Figura 11: Convergência do momento de dipolo do fenol utilizando processo iterativo e o ASEC [48] Cada ponto é resultado de uma simulação de MC, seguido de um único cálculo quântico
utilizando a "configuração média".
Na Figura 11 apresenta-se a convergência do momento de dipolo para as duas
condições termodinâmicas. A curva de convergência dos gráficos é um ajuste
exponencial, µi = µ[1 – A.exp(–Bi)], sobre o valor calculado do momento de dipolo. O
momento de dipolo calculado do fenol em água (último ponto) é dado por 2.25 D. Tal
valor é 75% maior que o valor calculado de fase gasosa (1.28 D). Nota-se que a
tendência do ajuste exponencial (i → ∞) é de 2.26 ± 0.02 D, atestando a convergência
do cálculo com quatro iterações. Na Tabela 20, os valores obtidos para as cargas
utilizadas na simulação são apresentados (fase gasosa e valor convergido para quarta
iteração).
Para a simulação com o modelo polarizado de fenol foram utilizados 50000
passos para o processo de termalização e mais 300000 de passos de trabalho. Separou-
se 100 configurações com uma correlação estatística menor que 10%. Para o cálculo
do espectro em meio líquido, primeiramente definiu-se as camadas de solvatação para
selecionar quantas moléculas de água são necessárias para a submissão aos cálculos
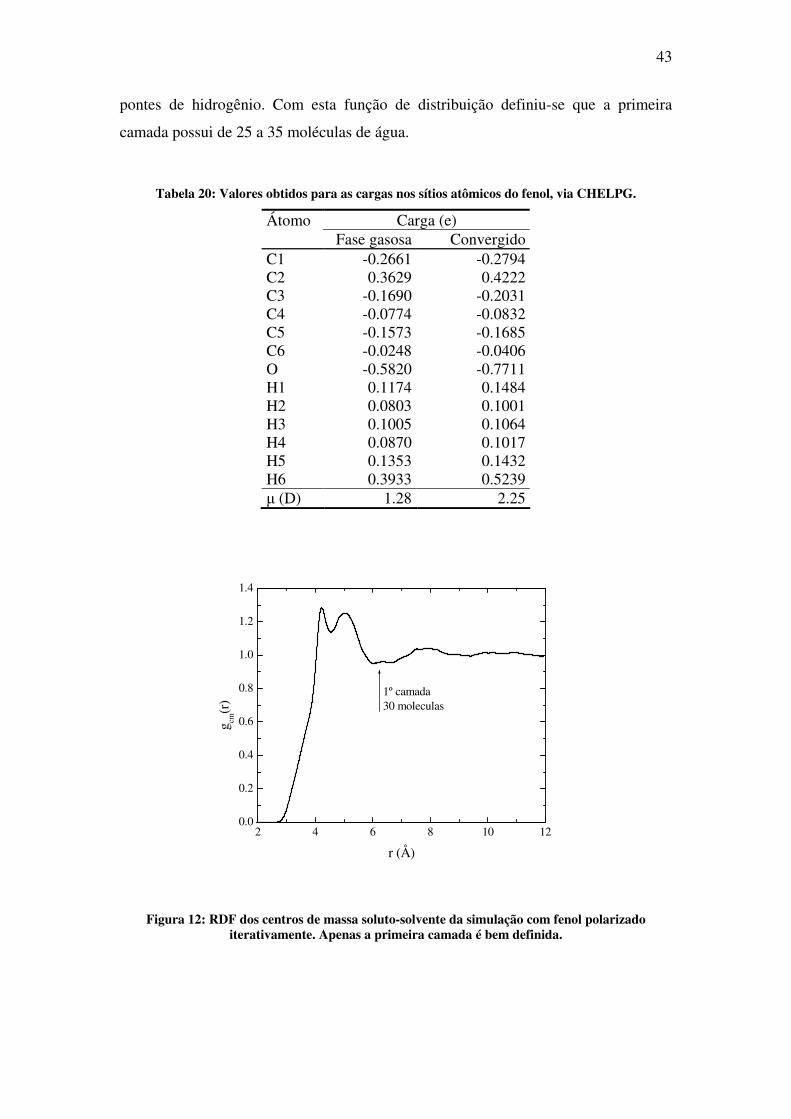
quânticos. Na Figura 12, a função de distribuição radial de centro de massa é
apresentada. Nota-se que a primeira camada possui um "ombro" em 4.0Å, referente às
43
pontes de hidrogênio. Com esta função de distribuição definiu-se que a primeira
camada possui de 25 a 35 moléculas de água.
Tabela 20: Valores obtidos para as cargas nos sítios atômicos do fenol, via CHELPG.
Átomo Carga (e) Fase gasosa Convergido C1 -0.2661 -0.2794 C2 0.3629 0.4222 C3 -0.1690 -0.2031 C4 -0.0774 -0.0832 C5 -0.1573 -0.1685 C6 -0.0248 -0.0406 O -0.5820 -0.7711 H1 0.1174 0.1484 H2 0.0803 0.1001 H3 0.1005 0.1064 H4 0.0870 0.1017 H5 0.1353 0.1432 H6 0.3933 0.5239 µ (D) 1.28 2.25
2 4 6 8 10 120.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1º camada30 moleculas
g cm(r
)
r (Å)
Figura 12: RDF dos centros de massa soluto-solvente da simulação com fenol polarizado iterativamente. Apenas a primeira camada é bem definida.
44
O cálculo quântico do espectro é bastante custoso devido à necessidade do uso
de moléculas explícitas de solvente (com o conseqüente aumento de elétrons e
funções base). Tendo em mente que este custo tem um fator maior que N4, optou-se
por se fazer o cálculo das 100 configurações descorrelacionadas com diferentes
quantidades de moléculas de água explícitas, iniciando-se em zero e aumentado de
cinco em cinco até se obter a convergência. Optou-se também por se utilizar uma base
menor (6-31G) do que a utilizada no soluto (aug-cc-pVDZ). Isto pode ser feito apenas
porque a excitação é localizada no fenol. O método quântico utilizado foi o TDHF,
que forneceu resultados para o desvio equivalentes ao CIS(D). Outro fator que pesou
pela escolha desse método foi que seu custo é quase igual ao CIS, mas com resultado
um pouco melhor para o valor absoluto (veja seções anteriores).
0 5 10 15 20 25300
400
500
600
700
800 Águas explícitas + cargas pontuais ASEC
Des
vio
solv
atoc
rôm
ico
(cm
-1)
Número de moléculas de água explícitas
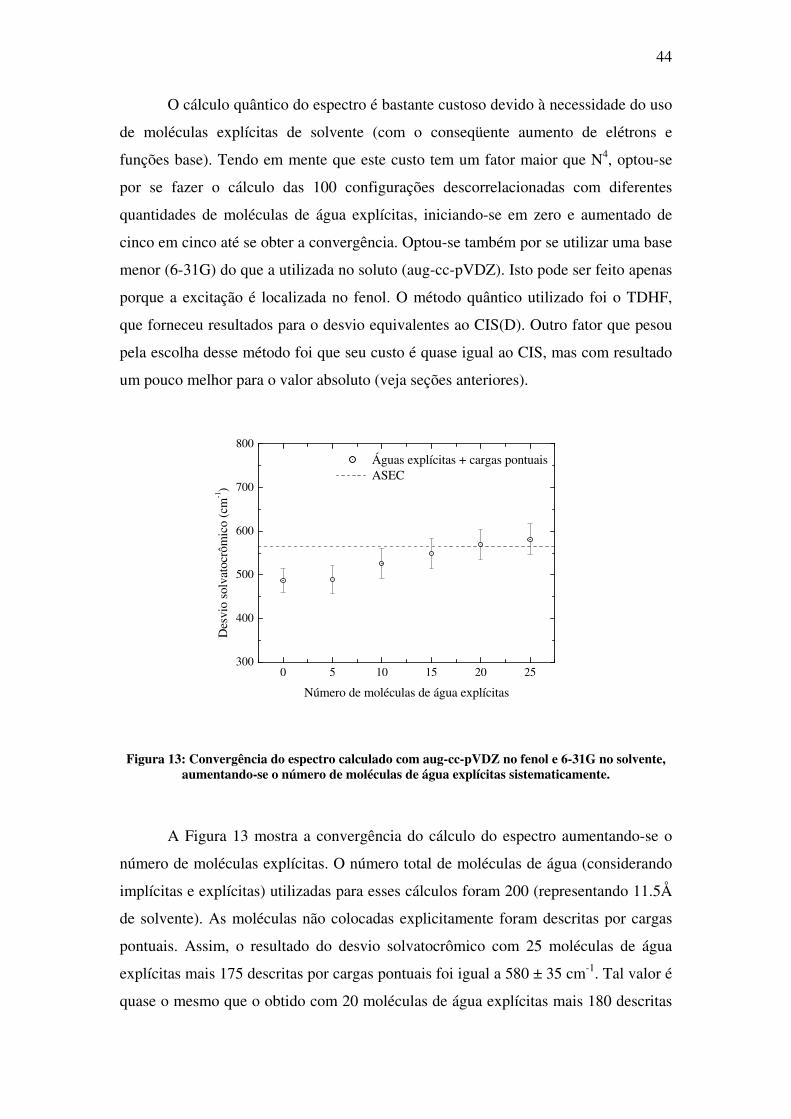
Figura 13: Convergência do espectro calculado com aug-cc-pVDZ no fenol e 6-31G no solvente, aumentando-se o número de moléculas de água explícitas sistematicamente.
A Figura 13 mostra a convergência do cálculo do espectro aumentando-se o
número de moléculas explícitas. O número total de moléculas de água (considerando
implícitas e explícitas) utilizadas para esses cálculos foram 200 (representando 11.5Å
de solvente). As moléculas não colocadas explicitamente foram descritas por cargas
pontuais. Assim, o resultado do desvio solvatocrômico com 25 moléculas de água
explícitas mais 175 descritas por cargas pontuais foi igual a 580 ± 35 cm-1. Tal valor é
quase o mesmo que o obtido com 20 moléculas de água explícitas mais 180 descritas
45
por cargas pontuais, 570 ± 34 cm-1, e que é coincidentemente similar ao resultado do
ASEC (565 cm-1). Destaca-se aqui que o ASEC deveria corresponder ao resultado da
média de cargas pontuais (485 ± 27 cm-1). Entretanto, Aguilar e colaboradores [49]
mostram que o método de carga média tende a fornecer maiores energias do que a
média dos cálculos de cada configuração descrita por cargas pontuais. Este argumento
é calcado na polarizabilidade (já que a configuração média suprime a polarização para
diferentes direções). Quanto maior a polarizabilidade, maior o erro na energia. É de se
esperar, portanto que o estado excitado resulte em energia maior ainda, aumentando-
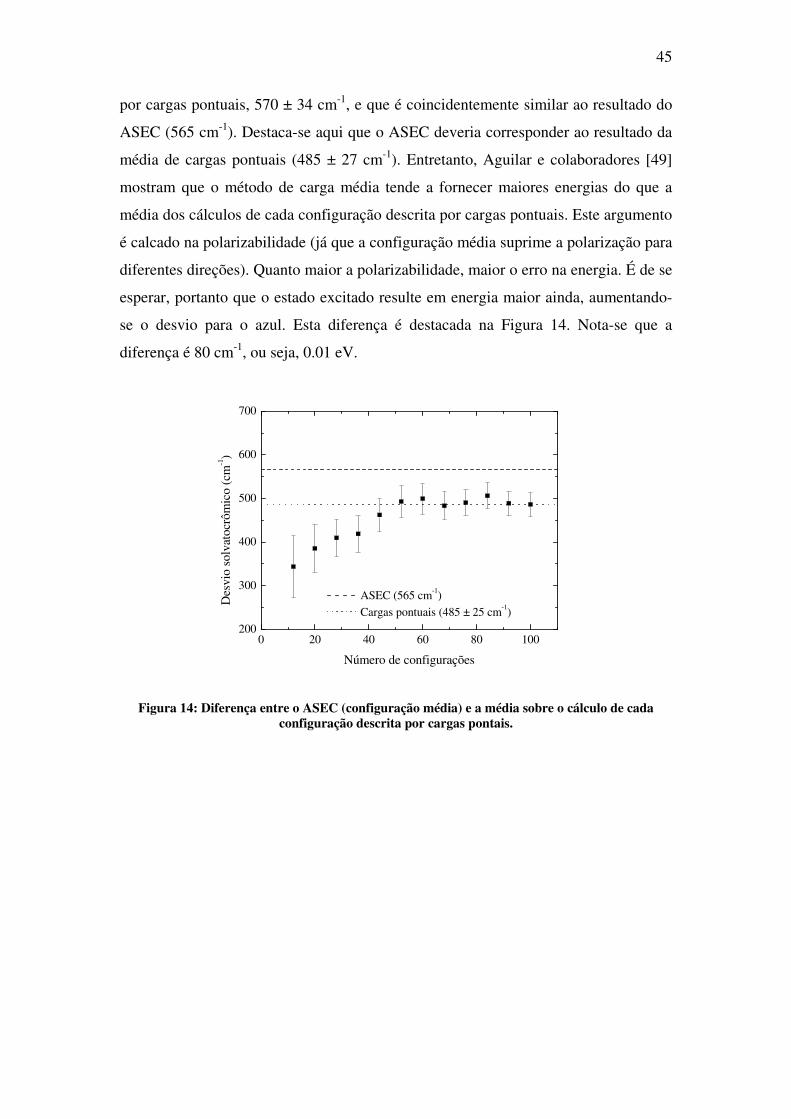
se o desvio para o azul. Esta diferença é destacada na Figura 14. Nota-se que a
diferença é 80 cm-1, ou seja, 0.01 eV.
0 20 40 60 80 100200
300
400
500
600
700
ASEC (565 cm-1) Cargas pontuais (485 ± 25 cm-1)
Des
vio
solv
atoc
rôm
ico
(cm
-1)
Número de configurações
Figura 14: Diferença entre o ASEC (configuração média) e a média sobre o cálculo de cada
configuração descrita por cargas pontais.
46
0 5 10 15 20 250
2
4
6
8
Hor
as d
e C
PU p
or c
onfi
gura
ção
Número de moléculas de água explícitas
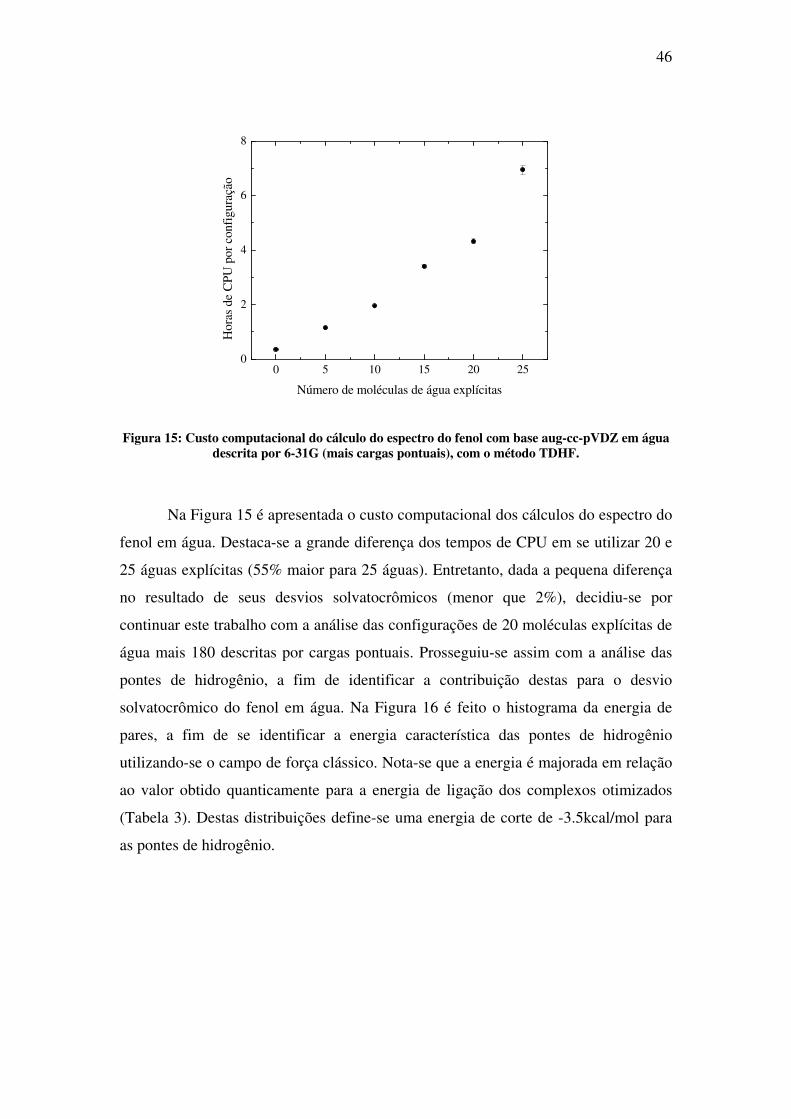
Figura 15: Custo computacional do cálculo do espectro do fenol com base aug-cc-pVDZ em água
descrita por 6-31G (mais cargas pontuais), com o método TDHF.
Na Figura 15 é apresentada o custo computacional dos cálculos do espectro do
fenol em água. Destaca-se a grande diferença dos tempos de CPU em se utilizar 20 e
25 águas explícitas (55% maior para 25 águas). Entretanto, dada a pequena diferença
no resultado de seus desvios solvatocrômicos (menor que 2%), decidiu-se por
continuar este trabalho com a análise das configurações de 20 moléculas explícitas de
água mais 180 descritas por cargas pontuais. Prosseguiu-se assim com a análise das
pontes de hidrogênio, a fim de identificar a contribuição destas para o desvio
solvatocrômico do fenol em água. Na Figura 16 é feito o histograma da energia de
pares, a fim de se identificar a energia característica das pontes de hidrogênio
utilizando-se o campo de força clássico. Nota-se que a energia é majorada em relação
ao valor obtido quanticamente para a energia de ligação dos complexos otimizados
(Tabela 3). Destas distribuições define-se uma energia de corte de -3.5kcal/mol para
as pontes de hidrogênio.
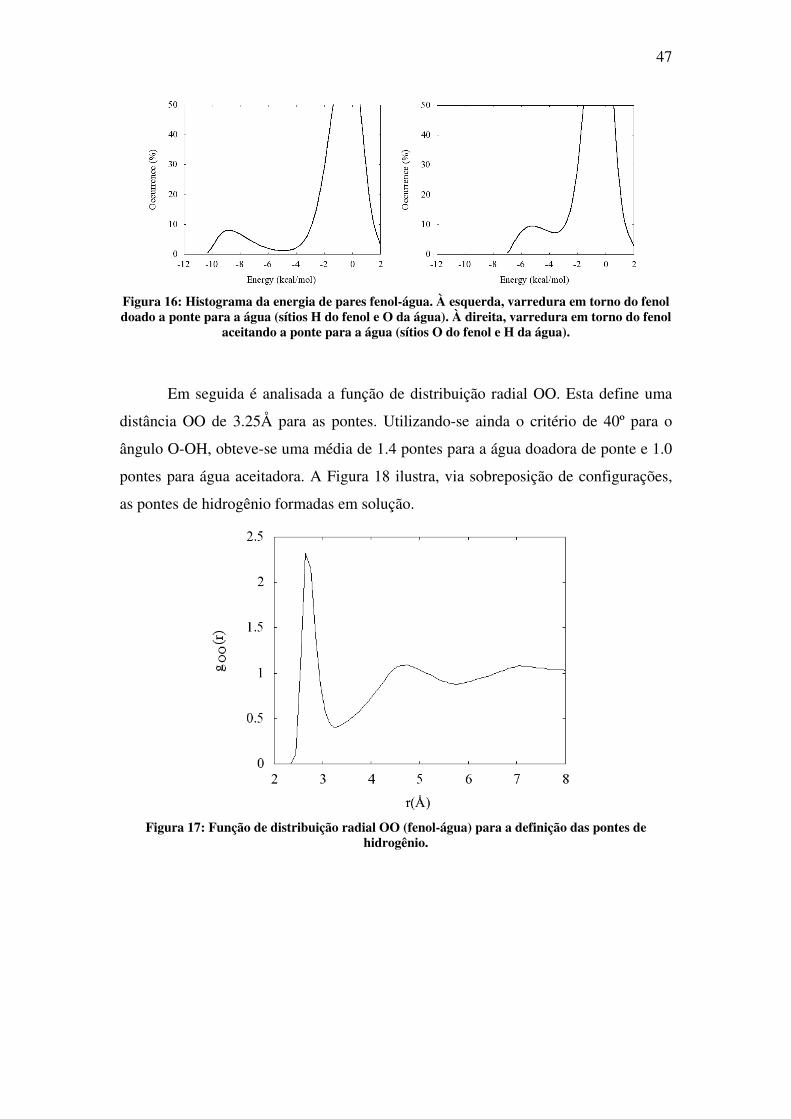
47
Figura 16: Histograma da energia de pares fenol-água. À esquerda, varredura em torno do fenol doado a ponte para a água (sítios H do fenol e O da água). À direita, varredura em torno do fenol
aceitando a ponte para a água (sítios O do fenol e H da água).
Em seguida é analisada a função de distribuição radial OO. Esta define uma
distância OO de 3.25Å para as pontes. Utilizando-se ainda o critério de 40º para o
ângulo O-OH, obteve-se uma média de 1.4 pontes para a água doadora de ponte e 1.0
pontes para água aceitadora. A Figura 18 ilustra, via sobreposição de configurações,
as pontes de hidrogênio formadas em solução.
Figura 17: Função de distribuição radial OO (fenol-água) para a definição das pontes de
hidrogênio.

48
Figura 18: Sobreposição das configurações de líquido contemplando apenas as pontes de
hidrogênio. À esquerda, pontes com água aceitadora (média de 1.0 pontes por configuração). À direita, pontes com água doadora (média de 1.4 pontes por configuração).
Assim, foi submetido ao cálculo quântico apenas as águas formadoras de
ponte de hidrogênio, excluindo-se das configurações do líquido todas as outras
moléculas de água. O desvio calculado apenas com as águas aceitadoras de pontes de
hidrogênio resultou em uma média de -435 ± 15 cm-1. O desvio com as águas
doadoras, 480 ± 20 cm-1. Assim como obtido para os complexos otimizados, seus
valores absolutos efetivamente se anulam. Dado o fato que o desvio do fenol em água
vai para o azul da mesma forma que a configuração otimizada W-Ph (Tabela 7), cria-
se a expectativa que em solução as água doadoras de ponte de hidrogênio dêem a
maior contribuição para o desvio total. Entretanto, ao se realizar o cálculo com ambos
os tipos de água (doadora e aceitadora), obtém-se somente 135 ± 25 cm-1 (25% do
valor obtido para o líquido, isto é com 20 moléculas de água explícitas e mais 180
descritas por cargas pontuais). De fato, ao se fazer o cálculo das configurações do
líquido, mas agora excluindo as moléculas de água que fazem as pontes, obtém-se 415
± 35 cm-1 (73% do valor obtido para o líquido). Conclui-se deste resultado que a
contribuição majoritária para o para o desvio do espectro do fenol em água não é dado
pelas pontes de hidrogênio, mas sim pelo efeito do bulk.
Tabela 21: Resultados para o espectro do fenol em água, analisando as diferentes contribuições.
Pontes Ph-W
Pontes W-Ph
Pontes Ph-W+ W-Ph
Líquido sem pontes
Líquido Exp. [6]
TD-HF -435 ± 15 480 ± 20 135 ± 25 415 ± 35 570 ± 35 460

49
Conclusões parciais
Realizou-se simulação de fenol em água (1+500) no ensemble NpT (1 atm,
298K) a fim de verificar a origem do desvio solvatocrômico do fenol. A água foi
descrita por um campo de força simples (SPC/E) e o fenol foi polarizado de acordo
com o protocolo Iterativo S-MC/MQ, utilizando o ASEC. Verificou-se que o ASEC é
um método eficiente e econômico para produzir a polarização no soluto, e que
também fornece uma boa estimativa para o desvio solvatocrômico. A função de
distribuição radial apontou que a primeira camada de solvatação do fenol é composta
por 30 moléculas de água. Verificou-se que utilizar todas as moléculas de água da
primeira camada para realização do cálculo de excitação eletrônica teria um alto custo
computacional. Foi então feita uma abordagem alternativa, onde se aumentou
sistematicamente o número de moléculas explícitas consideradas no cálculo, até se
obter a convergência. O valor calculado (570±35cm-1) está em bom acordo com o
resultado experimental (460cm-1). Adicionalmente foram identificadas as
contribuições para este desvio solvatocrômico. Verificou-se que o desvio do espectro
do fenol para o azul quando solvatado em água é causado basicamente pelo efeito do
bulk em torno do fenol, e não pelas pontes de hidrogênio como se pensava
inicialmente.
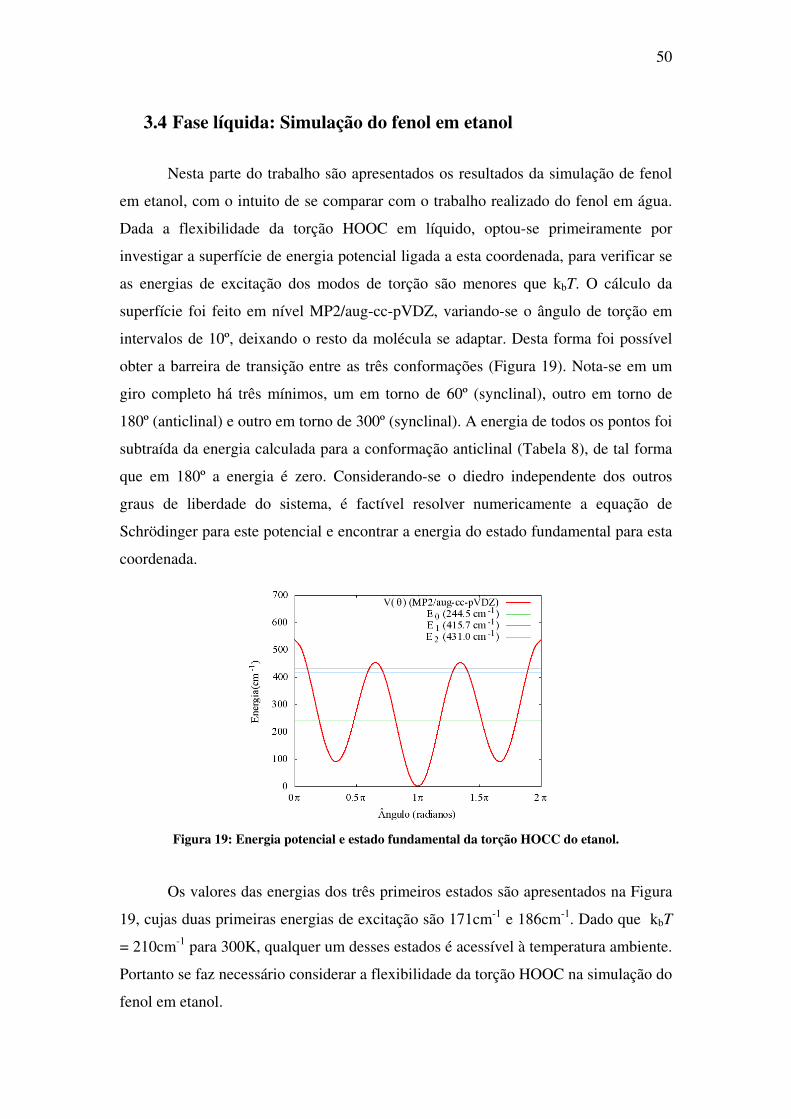
50
3.4 Fase líquida: Simulação do fenol em etanol
Nesta parte do trabalho são apresentados os resultados da simulação de fenol
em etanol, com o intuito de se comparar com o trabalho realizado do fenol em água.
Dada a flexibilidade da torção HOOC em líquido, optou-se primeiramente por
investigar a superfície de energia potencial ligada a esta coordenada, para verificar se
as energias de excitação dos modos de torção são menores que kbT. O cálculo da
superfície foi feito em nível MP2/aug-cc-pVDZ, variando-se o ângulo de torção em
intervalos de 10º, deixando o resto da molécula se adaptar. Desta forma foi possível
obter a barreira de transição entre as três conformações (Figura 19). Nota-se em um
giro completo há três mínimos, um em torno de 60º (synclinal), outro em torno de
180º (anticlinal) e outro em torno de 300º (synclinal). A energia de todos os pontos foi
subtraída da energia calculada para a conformação anticlinal (Tabela 8), de tal forma
que em 180º a energia é zero. Considerando-se o diedro independente dos outros
graus de liberdade do sistema, é factível resolver numericamente a equação de
Schrödinger para este potencial e encontrar a energia do estado fundamental para esta
coordenada.
Figura 19: Energia potencial e estado fundamental da torção HOCC do etanol.
Os valores das energias dos três primeiros estados são apresentados na Figura
19, cujas duas primeiras energias de excitação são 171cm-1 e 186cm-1. Dado que kbT
= 210cm-1 para 300K, qualquer um desses estados é acessível à temperatura ambiente.
Portanto se faz necessário considerar a flexibilidade da torção HOOC na simulação do
fenol em etanol.

51
Este trabalho foi realizado como o programa TINKER [50], e com o campo de
força flexível e polarizável AMOEBA [51]. Foi realizada dinâmica molecular no
ensemble NpT, em condições normais de pressão e temperatura (1 atm, 300K). O
algoritmo de integração utilizado foi o de Verlet, em conjunto com o termostato e
barostato de Berendsen. O passo de integração utilizado foi de 0.5ps, suficiente para
simulação com modelo flexível em condições normais. Foi utilizado o protocolo
padrão do AMOEBA, o qual não restringe os graus de liberdade internos das
moléculas. Também foi utilizado a correção de Ewald cujo efeito principal é corrigir a
interação coulombiana de longa distância. Foram utilizadas duas etapas de
termalização: uma de 100ns, e outra de 50ns. Na Figura 20 são apresentados os
valores da energia potencial intermolecular por molécula em cada passo. Espera-se
que tal valor seja coerente com a energia potencial do etanol puro. Vale lembrar que o
AMOEBA utiliza um potencial polarizável que depende da vizinhança, e não é
possível escrever esse potencial como uma soma de interação de pares. Portanto não
há como saber exatamente a contribuição do fenol para esta energia.
Figura 20: Energia potencial das configurações geradas durante o processo de termalização (últimos 50ns). A flutuação em torno de -9.6 kcal/mol indica que o sistema está próximo do
equilíbrio térmico.
52
Figura 21: Densidade durante o processo de termalização (últimos 50ns). A flutuação (sobe e desce) característica do uso do barostato indica que o sistema está próximo do equilíbrio térmico.
Na Figura 21 são apresentados os valores da densidade durante a segunda
parte do processo de termalização. Nota-se que a densidade inicialmente próxima a
0.785 g/cm3, diminui gradualmente até 0.775 g/cm3, para então subir novamente. Esta
flutuação (em torno de 0.78 g/cm3) serve de indicativo de que o sistema está
termalizado.
Garantida a termalização, efetuou-se a simulação. O passo de integração
utilizado foi o mesmo da termalização (0.5fs), entretanto efetuou-se 150ns de
simulação (300 mil passos). Na Figura 22 é apresentada a convergência do valor
médio da energia potencial por partícula. O erro apresentado é o erro estatístico
(desvio padrão dividido pela raiz do número de configurações descorrelacionadas).
Observa-se que o valor médio está muito bem convergido ao final da simulação. O
número total de configurações descorrelacionadas é obtido com o auxílio da função de
autocorrelação da energia (Figura 23). O número total de configurações
descorrelacionadas é 30 (tomando o 15% de correção entre as configurações, em um
intervalo de tempo de 5000ps). Desta forma, será necessário realizar pelo menos outra
simulação do mesmo tamanho, antes de realizar cálculos quânticos sobre essas
configurações. É conhecido que as propriedades quânticas calculadas convergem a
partir de 60 configurações descorrelacionadas. No trabalho realizado com a água,
procurou-se produzir 100 configurações descorrelacionadas.
53
Figura 22: Convergência do valor médio da energia potencial durante a simulação (150ns). O erro apresentado é o erro estatístico. Destaca-se o valor convergido (-9.548 kcal/mol).
Figura 23: Autocorrelação da energia potencial intermolecular. Com um intervalo de aproximadamente 5ns obtém-se menos de 15% de correlação.
A Figura 24 apresenta o histograma da energia potencial da simulação.
Observa-se que a distribuição é simétrica e se ajusta bem a curva teórica (gaussiana).
Este é um bom indicativo (mas não garante) que as configurações produzidas estão de
acordo com uma distribuição de Boltzmann (coerente com o ensemble NpT).
Ressalta-se que o termostato de Berendsen (utilizado na dinâmica molecular) não
garante a mesma distribuição do ensemble.
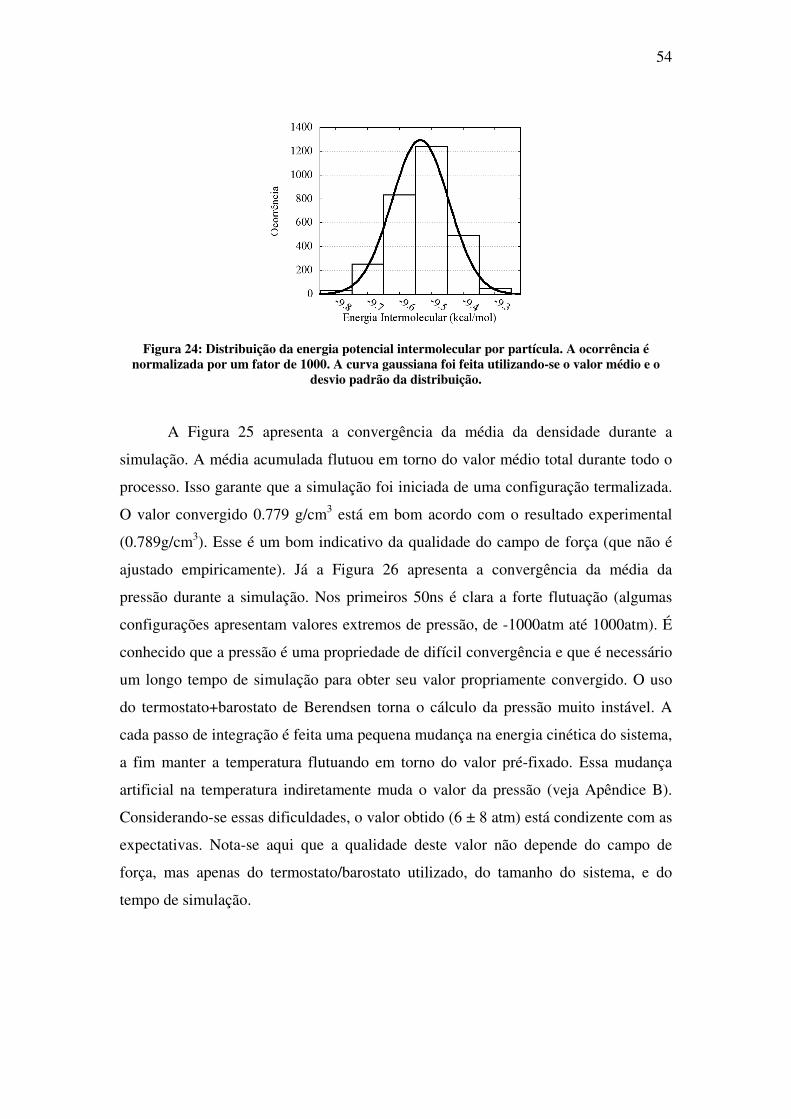
54
Figura 24: Distribuição da energia potencial intermolecular por partícula. A ocorrência é normalizada por um fator de 1000. A curva gaussiana foi feita utilizando-se o valor médio e o
desvio padrão da distribuição.
A Figura 25 apresenta a convergência da média da densidade durante a
simulação. A média acumulada flutuou em torno do valor médio total durante todo o
processo. Isso garante que a simulação foi iniciada de uma configuração termalizada.
O valor convergido 0.779 g/cm3 está em bom acordo com o resultado experimental
(0.789g/cm3). Esse é um bom indicativo da qualidade do campo de força (que não é
ajustado empiricamente). Já a Figura 26 apresenta a convergência da média da
pressão durante a simulação. Nos primeiros 50ns é clara a forte flutuação (algumas
configurações apresentam valores extremos de pressão, de -1000atm até 1000atm). É
conhecido que a pressão é uma propriedade de difícil convergência e que é necessário
um longo tempo de simulação para obter seu valor propriamente convergido. O uso
do termostato+barostato de Berendsen torna o cálculo da pressão muito instável. A
cada passo de integração é feita uma pequena mudança na energia cinética do sistema,
a fim manter a temperatura flutuando em torno do valor pré-fixado. Essa mudança
artificial na temperatura indiretamente muda o valor da pressão (veja Apêndice B).
Considerando-se essas dificuldades, o valor obtido (6 ± 8 atm) está condizente com as
expectativas. Nota-se aqui que a qualidade deste valor não depende do campo de
força, mas apenas do termostato/barostato utilizado, do tamanho do sistema, e do
tempo de simulação.

55
Figura 25: Convergência da média da densidade durante a simulação (150ns). O valor convergido é destacado pela linha verde (0.779 g/cm3). A convergência é obtida a partir de 100ns.
A escala do gráfico é a mesma da Figura 21. O erro estatístico é utilizado.
Figura 26: Convergência da pressão durante a simulação (150ns). Verifica-se que a convergência (dentro do erro estatístico) é obtida a partir de 60ns. Nota-se entretanto que as flutuações são
muito grandes.

56
Conclusões parciais
Realizou-se a simulação do fenol em etanol com o modelo flexível e
polarizável AMOEBA, utilizando o ensemble NpT. As propriedades termodinâmicas
calculadas são coerentes com os resultados esperados para esse sistema. A energia
potencial por molécula é -9.548 ± 0.016 kcal/mol, e está em excelente acordo com
simulações realizas com o potencial empírico OPLS (-9.69 kcal/mol) [52]. A
densidade calculada aqui 0.779 g/cm3 também se compara muito bem com o valor
experimental (0.789g/cm3) e com o resultado do OPLS (0.787 g/cm3) [52]. É
interessante notar que o AMOEBA não é parametrizado para reproduzir os resultados
experimentais, e portanto essa boa concordância é um forte indicativo de que este
modelo produz estruturas confiáveis.
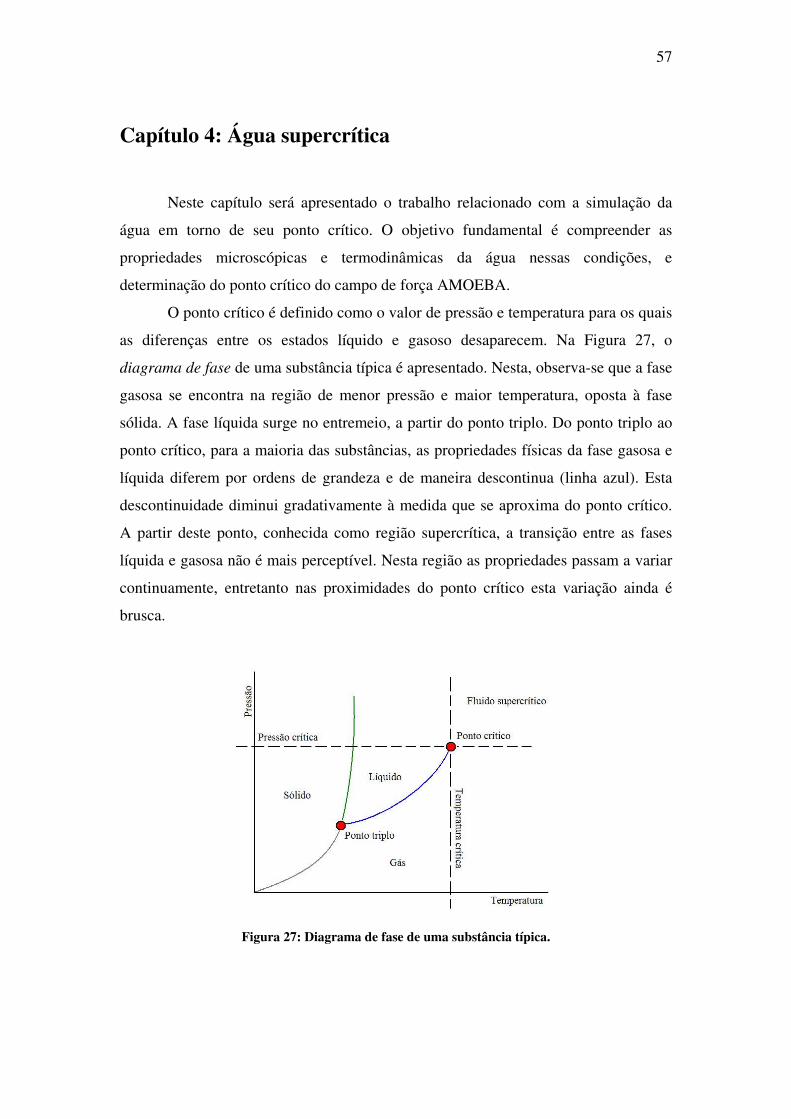
57
Capítulo 4: Água supercrítica
Neste capítulo será apresentado o trabalho relacionado com a simulação da
água em torno de seu ponto crítico. O objetivo fundamental é compreender as
propriedades microscópicas e termodinâmicas da água nessas condições, e
determinação do ponto crítico do campo de força AMOEBA.
O ponto crítico é definido como o valor de pressão e temperatura para os quais
as diferenças entre os estados líquido e gasoso desaparecem. Na Figura 27, o
diagrama de fase de uma substância típica é apresentado. Nesta, observa-se que a fase
gasosa se encontra na região de menor pressão e maior temperatura, oposta à fase
sólida. A fase líquida surge no entremeio, a partir do ponto triplo. Do ponto triplo ao
ponto crítico, para a maioria das substâncias, as propriedades físicas da fase gasosa e
líquida diferem por ordens de grandeza e de maneira descontinua (linha azul). Esta
descontinuidade diminui gradativamente à medida que se aproxima do ponto crítico.
A partir deste ponto, conhecida como região supercrítica, a transição entre as fases
líquida e gasosa não é mais perceptível. Nesta região as propriedades passam a variar
continuamente, entretanto nas proximidades do ponto crítico esta variação ainda é
brusca.
Figura 27: Diagrama de fase de uma substância típica.
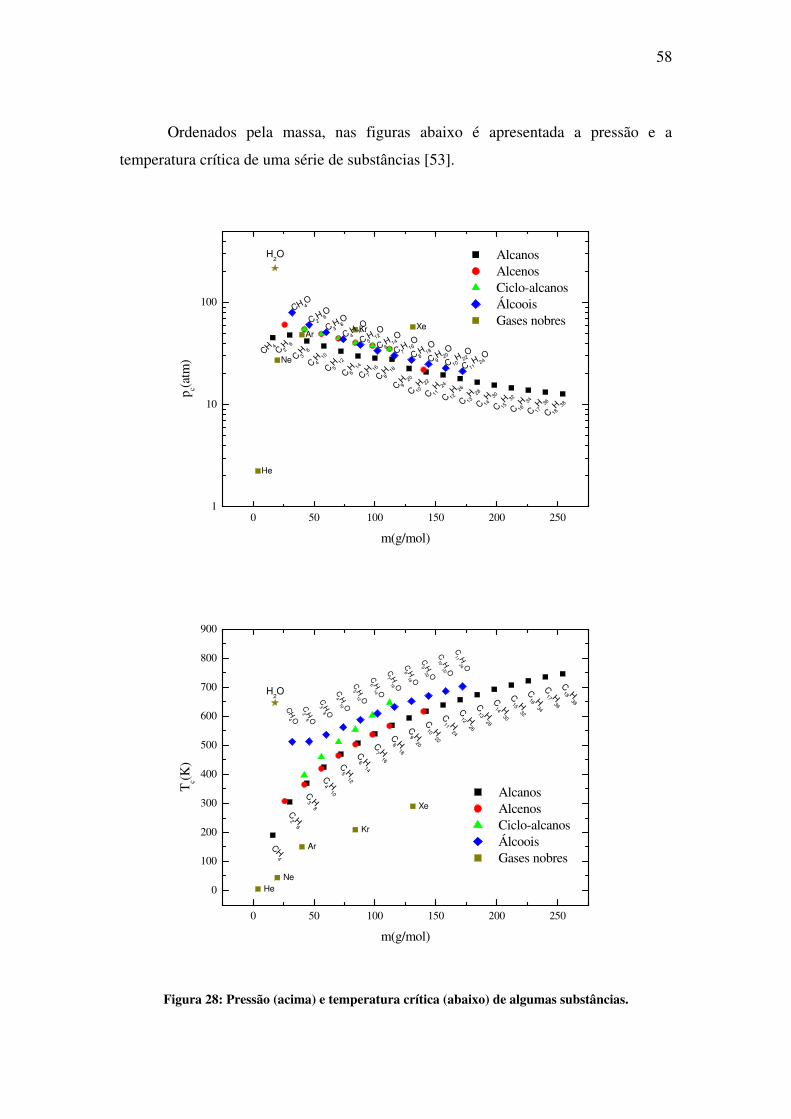
58
Ordenados pela massa, nas figuras abaixo é apresentada a pressão e a
temperatura crítica de uma série de substâncias [53].
0 50 100 150 200 2501
10
100
CH 4
C 2H 6
C 3H 8
C 4H 10
C 5H 12
C 6H 14
C 7H 16
C 8H 18
C 9H 20
C 10H 22
C 11H 24
C 12H 26
C 13H 28
C 14H 30
C 15H 32
C 16H 34
C 17H 36
C 18H 38
CH 4O
C 2H 6
O
C 3H 8
O
C 4H 10
O
C 5H 12
O
C 6H 14
O
C 7H 16
O
C 8H 18
O
C 9H 20
O
C 10H 22
O
C 11H 24
O
He
Ne
ArKr Xe
H2O
Alcanos Alcenos Ciclo-alcanos Álcoois Gases nobres
p c(atm
)
m(g/mol)
0 50 100 150 200 250
0
100
200
300
400
500
600
700
800
900
CH4
C2 H
6
C3 H
8
C4 H
10
C5 H
12
C6 H
14
C7 H
16
C8 H
18
C9 H
20
C10 H
22
C11 H
24
C12 H
26
C13 H
28
C14 H
30
C15 H
32
C16 H
34
C17 H
36
C18 H
38
CH
4 O
C2 H
6 O
C3 H
8 O
C4 H
10 O
C5 H
12 O
C6 H
14 O
C7 H
16 O
C8 H
18 O
C9 H
20 O
C10 H
22 OC
11 H24 O
He
Ne
Ar
Kr
Xe
H2O
Alcanos Alcenos Ciclo-alcanos Álcoois Gases nobres
Tc(K
)
m(g/mol)
Figura 28: Pressão (acima) e temperatura crítica (abaixo) de algumas substâncias.
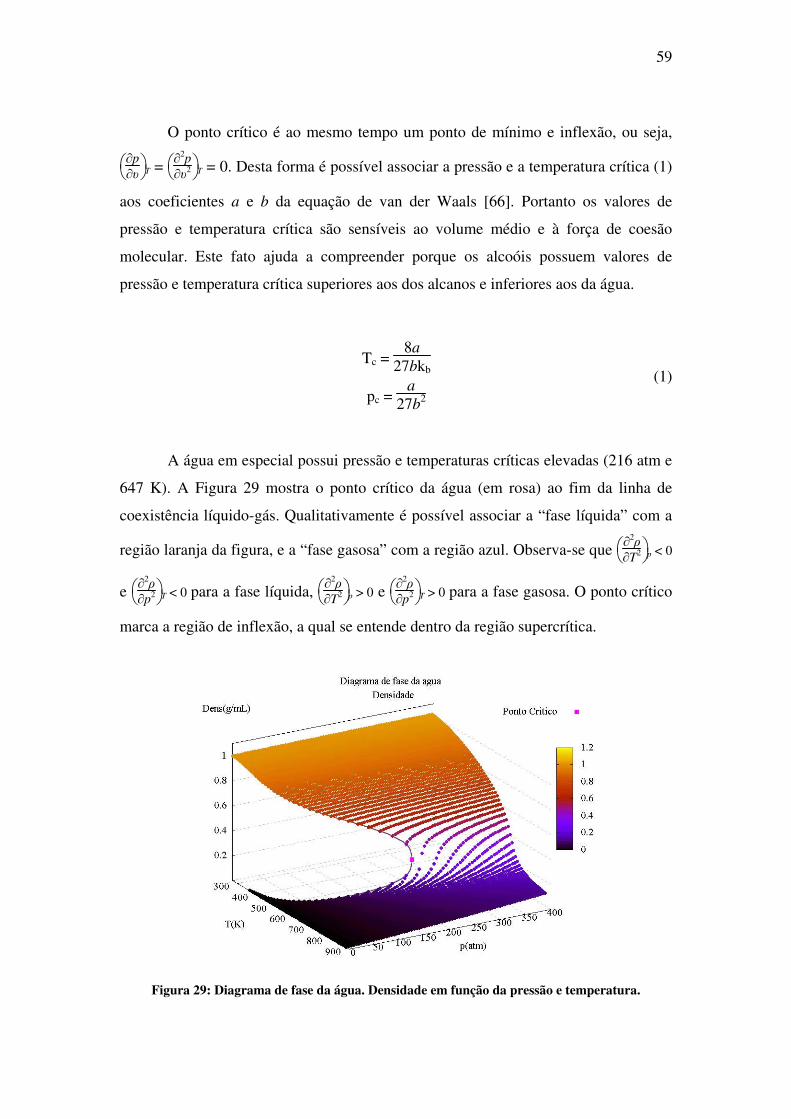
59
O ponto crítico é ao mesmo tempo um ponto de mínimo e inflexão, ou seja,
∂p
∂υ T =
∂2p
∂υ2 T = 0. Desta forma é possível associar a pressão e a temperatura crítica (1)
aos coeficientes a e b da equação de van der Waals [66]. Portanto os valores de
pressão e temperatura crítica são sensíveis ao volume médio e à força de coesão
molecular. Este fato ajuda a compreender porque os alcoóis possuem valores de
pressão e temperatura crítica superiores aos dos alcanos e inferiores aos da água.
Tc = 8a
27bkb
pc = a
27b2
(1)
A água em especial possui pressão e temperaturas críticas elevadas (216 atm e
647 K). A Figura 29 mostra o ponto crítico da água (em rosa) ao fim da linha de
coexistência líquido-gás. Qualitativamente é possível associar a “fase líquida” com a
região laranja da figura, e a “fase gasosa” com a região azul. Observa-se que
∂2ρ
∂T2 p < 0
e
∂2ρ
∂p2 T < 0 para a fase líquida,
∂2ρ
∂T2 p > 0 e
∂2ρ
∂p2 T > 0 para a fase gasosa. O ponto crítico
marca a região de inflexão, a qual se entende dentro da região supercrítica.
Figura 29: Diagrama de fase da água. Densidade em função da pressão e temperatura.

60
Ainda na Figura 29 observa-se que a densidade em torno do ponto crítico
dobra em uma ou duas unidades de pressão e temperatura. Da mesma forma, outras
propriedades físicas como a energia e o calor específico também sofrem imensas
flutuações nesta região. Realizar simulações computacionais em tais condições é um
grande desafio teórico que vem sendo realizado desde o final dos anos 80 [54].
Experimentalmente sabe-se que a rede de pontes de hidrogênio característica da água
em condições normais é parcialmente desfeita. Em seu lugar formam-se agregados de
diferentes tamanhos, formando regiões descontínuas [55]. Isto implica um maior
tempo de simulação (ou quantidade de partículas) para se obter as mesmas
propriedades termodinâmicas calculadas em condições normais. Os métodos de
cálculo de primeiros princípios ainda são demasiadamente caros e pouco eficientes
para serem utilizados nessas condições. É sabido que as propriedades termodinâmicas,
como densidade e pressão, não são bem descritas por cálculos DFT [56]. Isso se deve
ao fato de que tais propriedades são muito sensíveis a pequenas imprecisões na
energia de troca e correlação, mesmo em condições normais [56]. Desta forma tais
simulações, quando realizadas, são feitas com densidade constante, ignorando-se
completamente algumas propriedades como pressão e entalpia de vaporização [57].
Mesmo com o grande avanço computacional dado pelo processamento distribuído nos
últimos anos, as simulações ab initio ainda utilizam um número muito reduzido de
moléculas [58]. Por outro lado, os modelos clássicos rígidos podem não descrever
corretamente as grandes flutuações que surgem no ambiente supercrítico. É sabido
que os modelos polarizáveis costumam não propiciar resultados melhores para a
pressão e temperatura crítica do que os modelos não-polarizáveis [56]. A explicação
desta aparente falha reside no fato de que tais modelos costumam ser parametrizados
para descrever as propriedades termodinâmicas em condições normais. Outro aspecto
importante é que a polarizabilidade usada em tais modelos normalmente é a de fase
gasosa [56]. Desta maneira costuma-se fazer um balanço entre o custo computacional,
capacidade de reproduzir bem o ambiente complexo e descontínuo da região
supercrítica, e as propriedades termodinâmicas em torno de dessa região.
Surpreendentemente um dos modelos de água mais utilizado é o SPC/E [47, 59].
Estabelecido para este tipo de simulação em um importante trabalho de Guissani e
Guillot [60], o SPC/E subestima um pouco a pressão crítica (186atm calculado contra
217atm experimental) e concorda muito bem com a densidade crítica (0.326g/cm3

61
calculado contra 0.322g/cm3 experimental) e a temperatura crítica (651.7K calculado
contra 647.1K experimental). Não se deve esquecer, entretanto, que nesta região as
moléculas de água estão sujeitas a um ambiente extremamente inomogêneo, e que o
SPC/E é um modelo rígido e não polarizável. Isto implica que mesmo fornecendo boa
concordância para algumas propriedades termodinâmicas, este modelo deve perder
detalhes importantes da interação na vizinhança molecular. Após investigar a
literatura, decidiu-se investigar a estrutura microscópica e as propriedades
termodinâmicas da água em torno da região supercrítica utilizando um modelo
flexível e polarizável. O modelo escolhido foi o AMOEBA [51], proposto por Ren e
Ponder em 2002. Desde então, tal modelo se mostrou eficiente para reproduzir
propriedades termodinâmicas e estruturais de diversos sistemas, tanto para sistemas
moleculares [61, 62] quanto iônicos [63].
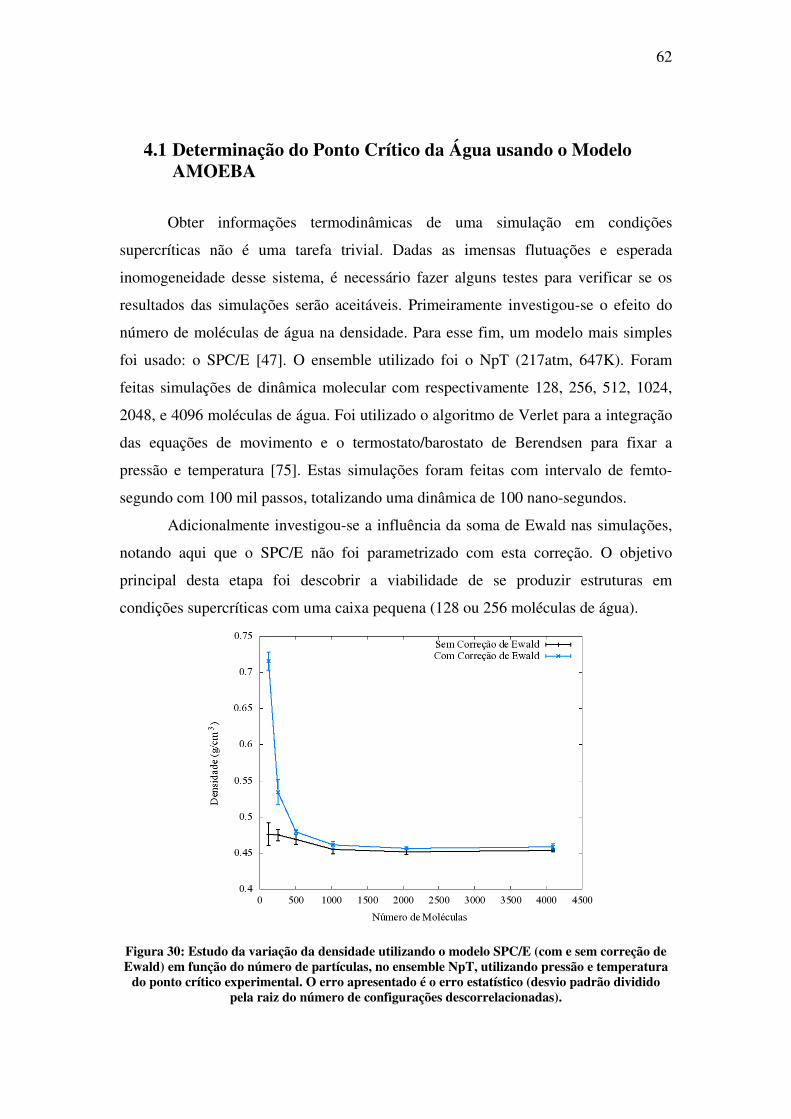
62
4.1 Determinação do Ponto Crítico da Água usando o Modelo AMOEBA
Obter informações termodinâmicas de uma simulação em condições
supercríticas não é uma tarefa trivial. Dadas as imensas flutuações e esperada
inomogeneidade desse sistema, é necessário fazer alguns testes para verificar se os
resultados das simulações serão aceitáveis. Primeiramente investigou-se o efeito do
número de moléculas de água na densidade. Para esse fim, um modelo mais simples
foi usado: o SPC/E [47]. O ensemble utilizado foi o NpT (217atm, 647K). Foram
feitas simulações de dinâmica molecular com respectivamente 128, 256, 512, 1024,
2048, e 4096 moléculas de água. Foi utilizado o algoritmo de Verlet para a integração
das equações de movimento e o termostato/barostato de Berendsen para fixar a
pressão e temperatura [75]. Estas simulações foram feitas com intervalo de femto-
segundo com 100 mil passos, totalizando uma dinâmica de 100 nano-segundos.
Adicionalmente investigou-se a influência da soma de Ewald nas simulações,
notando aqui que o SPC/E não foi parametrizado com esta correção. O objetivo
principal desta etapa foi descobrir a viabilidade de se produzir estruturas em
condições supercríticas com uma caixa pequena (128 ou 256 moléculas de água).
Figura 30: Estudo da variação da densidade utilizando o modelo SPC/E (com e sem correção de Ewald) em função do número de partículas, no ensemble NpT, utilizando pressão e temperatura
do ponto crítico experimental. O erro apresentado é o erro estatístico (desvio padrão dividido pela raiz do número de configurações descorrelacionadas).

63
Figura 31: À esquerda: Configuração aleatória com 256 moléculas de água (SPC/E, NpT, utilizando o ponto crítico experimental). As moléculas são representadas pelo método de
"superfície" (união das superfícies de van der Waals de cada molécula). É visível regiões sem nenhuma molécula de água, criando buracos na estrutura que atravessam a caixa de simulação.
À direita: Configuração aleatória com 500 moléculas de água (SPC/E, NpT, utilizando as condições normais de pressão e temperatura, proveniente da simulação de fenol em água). Não
há descontinuidades visíveis na estrutura.
A Figura 30 apresenta o valor da densidade para o SPC/E, com e sem a
correção de Ewald, para os diferentes tamanhos de caixa (número de moléculas).
Originalmente o TINKER (programa utilizado para fazer as simulações com o
AMOEBA) não possibilitava que se definisse a fronteira da caixa como raio de corte
(isso é necessário no ensemble NpT, já que o tamanho da caixa muda durante a
simulação). Desta forma o TINKER foi modificado para que o raio de corte das
interações fosse sempre igual à metade do tamanho da caixa. Com menos de 256
moléculas de água a utilização ou não da soma de Ewald muda consideravelmente o
resultado da simulação. Portanto o uso desta correção é relevante nestas condições se
o número de partículas é reduzido. A densidade obtida para valores superiores a 512
moléculas de água (aproximadamente 0.45g/cm3) é razoavelmente boa comparada
com a experimental (0.3g/cm3), e corrobora o fato do SPC/E ser amplamente utilizado
em simulações na região supercrítica. A Figura 31 apresenta uma das configurações
com 256 moléculas de água. Sua estrutura é formada de agregados e "vacâncias", de
tal forma que em cada direção é possível contar um ou dois agregados, cada um com
poucas moléculas de água. Desta forma, deve-se imaginar que a simulação não é
realizada com moléculas de água, mas sim com agregados formados por estas
moléculas. Tais agregados interagem e trocam partículas entre si. Isso implica que as

64
condições periódicas de contorno criam um vínculo artificial quando há poucas
moléculas de água no sistema. Este é um bom indicativo do porque há tanta variação
na densidade quando se simula com um número reduzido de partículas (Figura 30). A
única maneira de contornar este problema é utilizar o máximo número possível de
partículas nas simulações. Entretanto o custo computacional escalona no mínimo com
N2 (dado que o raio de corte utilizado aqui é sempre metade da caixa). Há também o
fato que o campo de força AMOEBA [51] utiliza um processo autoconsistente para
gerar os multipolos induzidos em cada sítio atômico, fazendo dele pelo menos 40
vezes mais dispendioso computacionalmente que o SPC/E. Uma simulação com 100
nano-segundos leva cinco dias com 256 moléculas de água3, e são necessárias
algumas centenas de simulações para se determinar o ponto crítico com alguma
precisão.
Após compreender o que seria enfrentado, foi realizada a busca pelo ponto
crítico da água utilizando o modelo AMOEBA (aparentemente não reportado na
literatura). Sabendo-se que 128 moléculas de água não descreveriam adequadamente a
estrutura da água supercrítica, e devido aos limitantes custos computacionais, optou-
se por utilizar 256 moléculas de água. Esta é a mesma quantidade de moléculas
utilizadas nas simulações de Guissani e Guillot para a determinação do ponto crítico
do SPC/E em 1993 [60]. Realizou-se uma primeira simulação em torno do ponto
crítico experimental, no ensemble NpT, e se percebeu que o resultado para a
densidade era surpreendentemente subestimado (em torno de 0.13 g/cm3). Foi
utilizada a correção de Ewald (dado o protocolo do AMOEBA), com ewald-fraction
igual a 0.25 (utilizando 7 vetores k, o que produziu uma diminuição do tempo de
cálculo sem afetar a precisão do potencial). Da mesma forma que nos testes iniciais
com o SPC/E, foi utilizado o algoritmo de integração de Verlet e o
termostato/barostato de Berendsen [75]. A geometria da água foi mantida fixa através
do algoritmo rattle [64].
Em seguida, foram feitas aproximadamente 200 simulações a partir deste
ponto, diminuindo-se a temperatura e aumentando-se a pressão, em busca da transição
de fase líquido/gás (Figura 32).
3 Valor aproximado considerando-se o cálculo serial em processador 64bits com 2.4GHz e 4MB de cache, sem a utilização de instruções específicas.
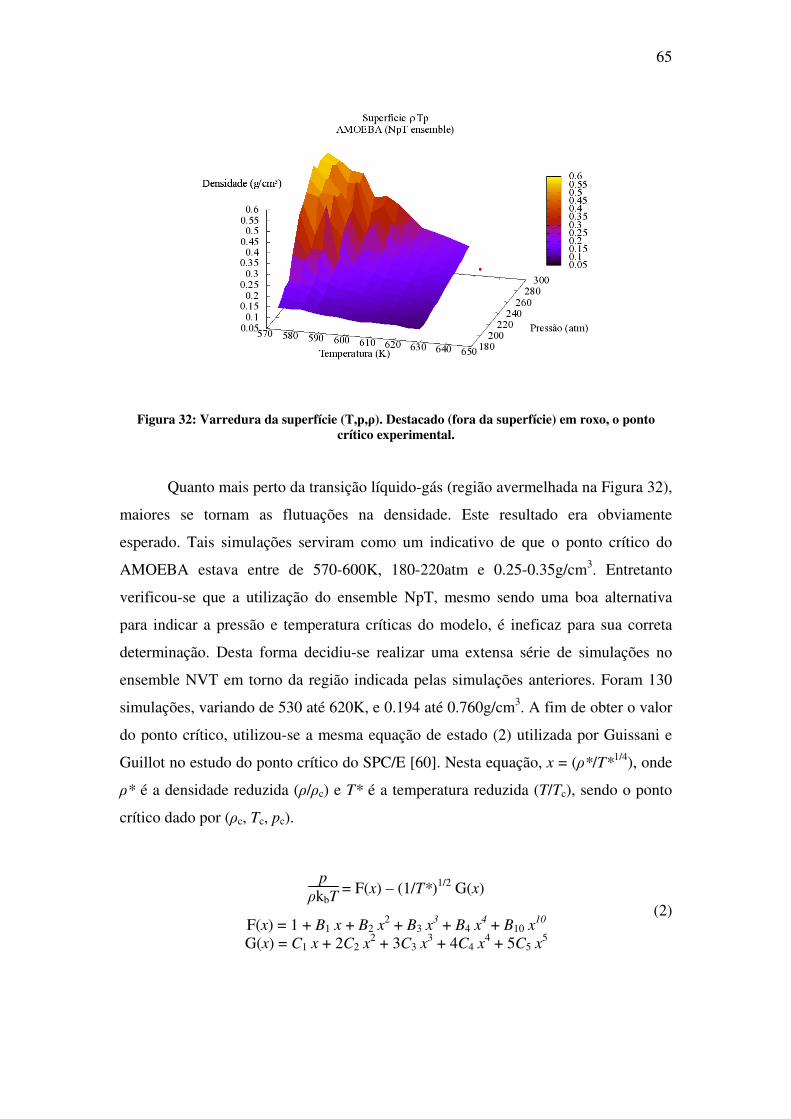
65
Figura 32: Varredura da superfície (T,p,ρ). Destacado (fora da superfície) em roxo, o ponto crítico experimental.
Quanto mais perto da transição líquido-gás (região avermelhada na Figura 32),
maiores se tornam as flutuações na densidade. Este resultado era obviamente
esperado. Tais simulações serviram como um indicativo de que o ponto crítico do
AMOEBA estava entre de 570-600K, 180-220atm e 0.25-0.35g/cm3. Entretanto
verificou-se que a utilização do ensemble NpT, mesmo sendo uma boa alternativa
para indicar a pressão e temperatura críticas do modelo, é ineficaz para sua correta
determinação. Desta forma decidiu-se realizar uma extensa série de simulações no
ensemble NVT em torno da região indicada pelas simulações anteriores. Foram 130
simulações, variando de 530 até 620K, e 0.194 até 0.760g/cm3. A fim de obter o valor
do ponto crítico, utilizou-se a mesma equação de estado (2) utilizada por Guissani e
Guillot no estudo do ponto crítico do SPC/E [60]. Nesta equação, x = (ρ*/T*1/4), onde
ρ* é a densidade reduzida (ρ/ρc) e T* é a temperatura reduzida (T/Tc), sendo o ponto
crítico dado por (ρc, Tc, pc).
p
ρkbT = F(x) – (1/T*)1/2 G(x)
F(x) = 1 + B1 x + B2 x2 + B3 x
3 + B4 x4 + B10 x
10 G(x) = C1 x + 2C2 x
2 + 3C3 x3 + 4C4 x
4 + 5C5 x5
(2)
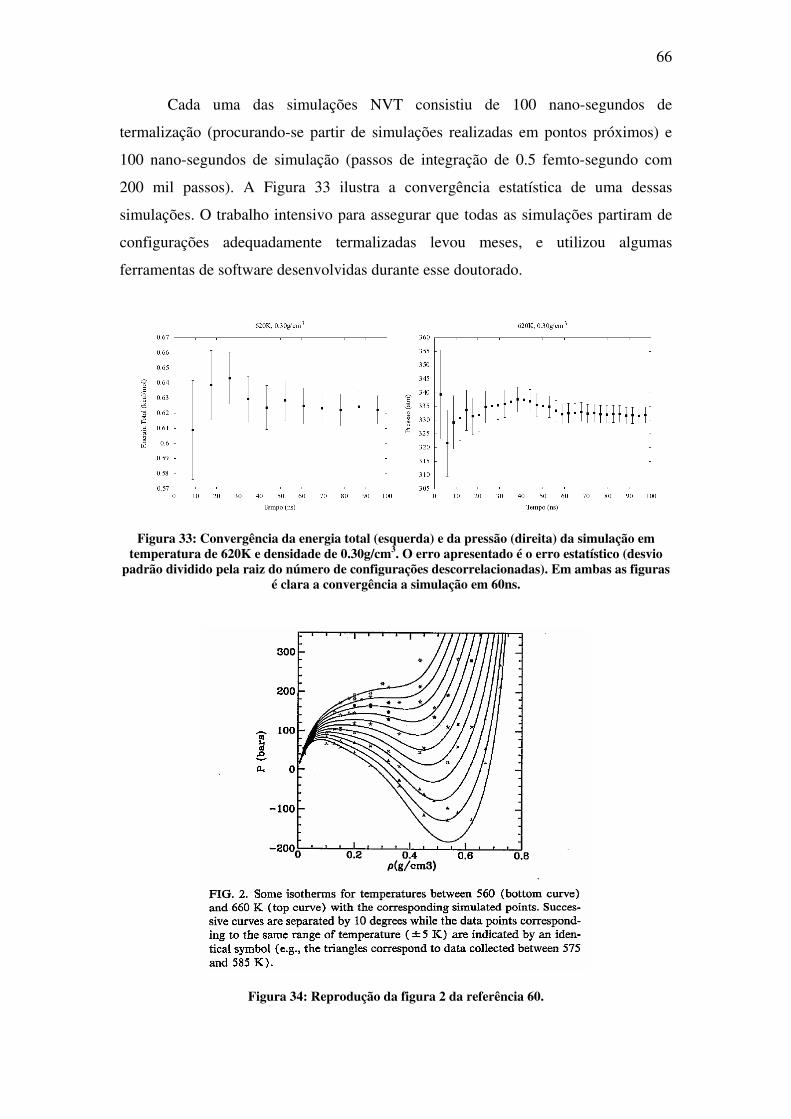
66
Cada uma das simulações NVT consistiu de 100 nano-segundos de
termalização (procurando-se partir de simulações realizadas em pontos próximos) e
100 nano-segundos de simulação (passos de integração de 0.5 femto-segundo com
200 mil passos). A Figura 33 ilustra a convergência estatística de uma dessas
simulações. O trabalho intensivo para assegurar que todas as simulações partiram de
configurações adequadamente termalizadas levou meses, e utilizou algumas
ferramentas de software desenvolvidas durante esse doutorado.
Figura 33: Convergência da energia total (esquerda) e da pressão (direita) da simulação em temperatura de 620K e densidade de 0.30g/cm3. O erro apresentado é o erro estatístico (desvio
padrão dividido pela raiz do número de configurações descorrelacionadas). Em ambas as figuras é clara a convergência a simulação em 60ns.
Figura 34: Reprodução da figura 2 da referência 60.

67
Executar as simulações e conferir seus resultados é uma tarefa hercúlea.
Porém encontrar os coeficientes B e C (e também o ponto crítico) é uma tarefa ainda
mais complexa. A equação de estado utilizada para ajustar os pontos possui 12
parâmetros livres. Para encontrar estes parâmetros utilizou-se o método de mínimos
quadrados implementado no programa gnuplot [65]. Os valores iniciais para estes
coeficientes foram os mesmos reportados na referência 60 para os coeficientes da
água real. Aparentemente os coeficientes encontrados para a água com modelo
AMOEBA produzem isotermas que se ajustam melhor aos dados da simulação do que
às curvas e dados apresentados na referência 60 para a água com o modelo SPC/E
(reproduzido aqui na Figura 34). Na Tabela 22 é apresentado o resultado desse ajuste.
Com exceção dos coeficientes B10 e C5 (que determinam o comportamento da equação
nas condições subcríticas e normais) os parâmetros obtidos para o AMOEBA são
mais próximos daqueles ajustados para os dados experimentais do que os obtidos para
o SPC/E. Isso implica que em torno do ponto crítico, o AMOEBA apresenta uma
variação nas propriedades termodinâmicas que é mais próxima da água real do que o
modelo SPC/E. O ponto crítico para o AMOEBA (575K, 0.33g/cm3, 205atm)
apresenta a pressão crítica mais próxima do valor experimental (217.7atm). Por outro
lado, o ponto crítico do SPC/E (651.7K, 0.326g/cm3, 186.5atm) possui uma melhor
temperatura crítica quando comparada com o valor experimental (647.1K). Ambos os
campos de força fornecem uma excelente densidade crítica considerando-se a
imprecisão do ajuste.
Tabela 22: Parâmetros ajustados para a equação de estado (2). Os valores para SPC/E e Experimental foram retirados da referência 60. Nota-se que o ponto crítico experimental é
647.1K, 0.322g/cm3 e 217.7atm, e que os valores para "Experimental" apresentados na tabela mostram que a equação de estado não é exata.
SPC/E AMOEBA Experimental Tc(K) 651.7 575 652.3 ρ(g/cm3) 0.326 0.33 0.299 p(atm) 186.5 205 229.5
B1 8.480 6.414 6.630 B2 -13.45 -6.120 -5.849 B3 8.795 2.395 2.099 B4 -1.810 -0.2227 -0.1672 B10 2.297(10-4) 10-5 2.87(10-17) C1 11.32 8.004 8.151 C2 -9.127 -3.652 -3.492 C3 4.440 0.936 0.835 C4 -0.9888 -0.06623 -0.0514 C5 7.831(10-2) -2.2(10-3) 2.178(10-3)
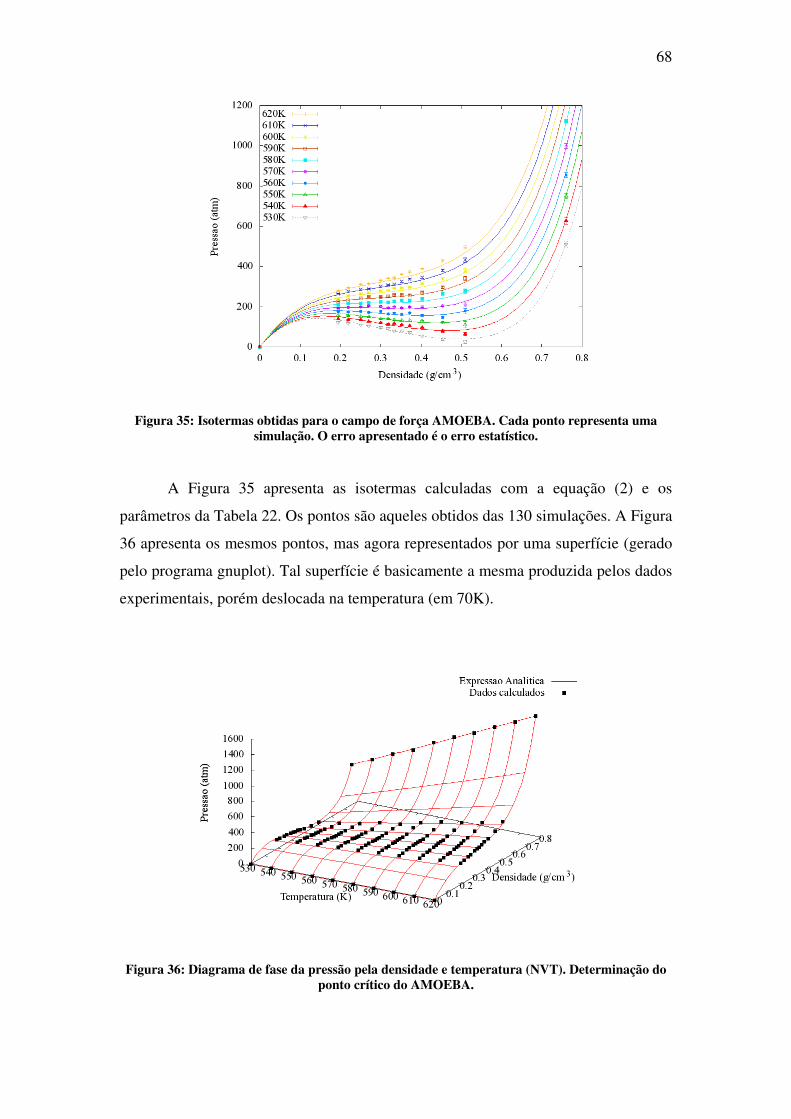
68
Figura 35: Isotermas obtidas para o campo de força AMOEBA. Cada ponto representa uma simulação. O erro apresentado é o erro estatístico.
A Figura 35 apresenta as isotermas calculadas com a equação (2) e os
parâmetros da Tabela 22. Os pontos são aqueles obtidos das 130 simulações. A Figura
36 apresenta os mesmos pontos, mas agora representados por uma superfície (gerado
pelo programa gnuplot). Tal superfície é basicamente a mesma produzida pelos dados
experimentais, porém deslocada na temperatura (em 70K).
Figura 36: Diagrama de fase da pressão pela densidade e temperatura (NVT). Determinação do ponto crítico do AMOEBA.

69
Conclusões parciais
Foram realizadas centenas de simulações da água em torno do ponto crítico
experimental, utilizando diferentes modelos e diferentes números de partículas.
Observou-se que as propriedades termodinâmicas (e por conseqüência as estruturais)
são sensíveis ao número de moléculas na simulação quando esta é menor que 256.
Portanto conclui-se que este número é o mínimo aceitável para reproduzir com
alguma fidelidade as propriedades da água em torno do ponto crítico e na região
supercrítica. Adicionalmente foi determinado o ponto crítico do campo de força
polarizável AMOEBA. Seu ponto crítico (575K, 0.33g/cm3, 205atm) se compara
surpreendentemente bem com o resultado experimental (647K, 0.322g/cm3, 217.7atm)
dado o fato que ele não foi parametrizado para isso. Dado que este campo de força
apresenta polarização e flexibilidade, ele se torna uma alternativa mais confiável para
realização de simulações nesta região do que os modelos não flexíveis e não
polarizáveis comumente utilizados.

70
Capítulo 5: Retrospectiva, Conclusões e Perspectivas
Este projeto de doutorado foi iniciado no início de 2007 com o objetivo de
estudar as propriedades espectroscópicas do fenol em agregados, soluções e em
condições supercríticas. A simulação do fenol em condições normais, utilizando
diferentes solventes, campos de força e métodos de química quântica, foi realizada
com sucesso. O aluno desenvolveu os conhecimentos necessários para simular e
analisar não apenas propriedades termodinâmicas e eletrônicas em solução, mas
também interpretar mudanças conformacionais e desvios no espectro vibracional,
estimativas de energia de ligação, efeitos de polarização e deconvoluir suas diferentes
contribuições. Devido a limitações computacionais, não foi possível completar o
estudo da solvatação do fenol em água supercrítica (e não há perspectiva realista para
a realização dessa parte). Entretanto, foi realizado um estudo sistemático das
propriedades termodinâmicas e estruturais em condições supercríticas, incluindo a
determinação do ponto crítico do modelo polarizável AMOEBA. Conclusões parciais
de cada parte do trabalho apresentado nesta tese estão presentes no final de cada
seção.
Durante o último ano e meio, foi investido um enorme esforço para adquirir
conhecimentos relativos à programação em C/C++ (em especial otimização de código
e programação eficiente orientada a objeto), programação em paralelo (com o uso de
threads de processamento e comunicação inter-processual via sockets), e manutenção
de grandes projetos colaborativos com o uso de software do tipo subversion. Este
estágio se iniciou com a necessidade de desenvolver um programa para separar as
configurações da simulação de dinâmica molecular (principalmente relativo às
simulações em condições supercríticas) e fazer uma análise adequada sobre eles.
Partindo de scripts básicos em shell para uma implementação amadora em C, o
programa começou a ser escrito na virada de 2008 para 2009 como uma ampla
ferramenta de separação de configurações de acordo com as mais diversas opções
possíveis (distância, energia, ângulo de ligação, vetor definido pelo usuário, multi-
seleção, lista reversa, negativa, produção de distribuição radial, angular, vetorial,
energética...). Batizado de magno, logo se tornou um enorme bloco de difícil
manutenção, e se percebeu a necessidade de melhorar a técnica de programação e a
abordagem sobre ele. Após adquirir o conhecimento necessário para executar esta

71
tarefa, iniciou-se o processo de reescrita, executando isso de maneira modular e
organizada. Utilizando programação orientada a objeto, foram criadas classes de uso
universal para operar de maneira simples e eficiente sobre arquivos, buffers de texto,
listas, e variáveis dinâmicas. Ao mesmo tempo, foram criadas classes específicas para
operar sobre átomos, moléculas, campos de força, vetores e matrizes. Tais
ferramentas (classes) são otimizadas e eficientes, e obviamente não são restritas
"separação de configurações". Toda a dificuldade da otimização de algoritmos
básicos, operações de input/output e programação em paralelo são incluídas nessas
classes, facilitando e aumentando a flexibilidade do programador. Tais ferramentas
podem ser utilizadas em qualquer programa que execute operações sobre átomos e
moléculas. Isto implica que elas não estão restritas ao magno, mas podem se
manifestar em um projeto maior. Desta maneira foi iniciado o projeto OpenQMMM,
onde estas ferramentas de software podem ser desenvolvidas em alto nível de código
e organização, em colaboração aberta. A maior vantagem da "colaboração aberta" é a
minimização do tempo de programação e detecção de falhas (bugs), já que diferentes
programadores têm acesso ao mesmo código e desfrutam do trabalho em conjunto. O
exemplo mais simples do sucesso desse tipo de abordagem é o sistema operacional
linux. Há outras dezenas de projetos nesse mesmo princípio, entretanto não há
nenhum projeto nacional nesta área (mecânica molecular e mecânica quântica). A
pretensão principal após o término do doutorado é investir em tempo integral neste
projeto, aprimorando os programas existentes e desenvolvendo novos programas.
Na parte condizente à solvatação do fenol em etanol, simulações com modelo
flexível e polarizável foram realizadas, e aprimoramentos no programa magno estão
sendo feitas para a separação das configurações e sucessiva submissão destas ao
cálculo quântico. Este trabalho será fundamental para explicar a origem do desvio
solvatocrômico para o vermelho do fenol em etanol.
A próxima etapa da simulação da água supercrítica é produzir a contagem das
pontes de hidrogênio e tamanho dos agregados. Este resultado é fundamental na
comparação com o modelo rígido SPC/E, largamente utilizado na literatura.
Adicionalmente pretende-se realizar o cálculo da constante dielétrica e do espectro
eletrônico para comparação de tais propriedades com resultados experimentais.

72
Pretende-se executar a simulação do fenol em água supercrítica com pelo
menos 256 moléculas de água. Inicialmente pretendia-se realizar simulação clássica
com modelo polarizável e flexível. Entretanto, dada a provável desprotonação do
fenol (levando-o a fenoxi) em tais condições, é necessário realizar dinâmica ab initio.
O programa escolhido para este fim é o CP2K, com DFT, funcional PBE e base m-
DZVP. Infelizmente, dado o alto custo computacional e a falta de máquinas, não há
perspectiva para o início dessas simulações. Para se ter uma comparação realista do
custo dos métodos utilizados neste trabalho, a simulação ab initio com 64 moléculas é
aproximadamente 30 vezes mais lenta do que a realizada com o AMOEBA com 256
moléculas, e 600 vezes mais lenta que com o SPC/E, também com 256 moléculas.

73
Apêndice A: Realizações no Doutorado
• Disciplinas obrigatórias
o Mecânica Quântica II;
o Teoria de Grupos e Simetrias em Física do Estado Sólido e Física
Molecular.
• Artigos publicados:
o R.C. Barreto, K. Coutinho, H.C. Georg, S. Canuto, "Combined Monte
Carlo and quantum mechanics study of the solvatochromism of phenol
in water. The origin of the blue shift of the lowest pi-pi* transition",
Phys. Chem. Chem. Phys. 11 (2009) 1388.
o R.C. Barreto, S. Canuto, "Characterization and spectroscopy analysis
of phenol-ethanol hydrogen bonded clusters", Chem. Phys. Lett. 496
(2010) 236
• Participação em eventos:
o XIV Simpósio Brasileiro de Química Teórica (Poços de Caldas/MG
2007) com apresentação de pôster: "Espectro do Fenol em Água em
Condições Normais e Supercríticas, incluindo polarização do soluto";
o II Escola de Computação de Alto Desempenho (São Carlos/SP 2007)
com apresentação de pôster: "Montando um Sistema Integrado de
Desktops";
o II Encontro Anual da Rede Theo-Nano (São Paulo/SP 2007) com
apresentação oral: "Espectro do Fenol em Água";
o II Simpósio de Estrutura Eletrônica e Dinâmica Molecular (Brasília/DF
2008) com apresentação do pôster: "Análise do espectro eletrônico do
fenol em água, separação de diferentes contribuições".
• Estágio:
o Estada de 4 meses (setembro a dezembro de 2008) dentro do programa
CAPES/GRICES-FCT de cooperação com o grupo do Prof. Benedito
José Costa Cabral na Universidade de Lisboa, Portugal, desenvolvendo
conhecimentos de dinâmica molecular.
• Conhecimentos acadêmicos extraordinários:

74
o Montagem, instalação, configuração e manutenção do cluster do Grupo
Física Molecular e Modelagem. Parte deste trabalho pode ser
encontrada em http://ac.if.usp.br/~barreto/cluster.htm
o Criação e manutenção do projeto de software livre OpenQMMM, cujo
objetivo primário é desenvolver e aprimorar ferramentas de software
utilizadas nos trabalhos relacionados com simulação molecular e
cálculo quântico. O objetivo secundário é criar um projeto sólido de
colaboração open source entre desenvolvedores no país (algo
inexistente até o momento). A página do projeto e informações podem
ser encontradas em https://sourceforge.net/projects/openqmm.

75
Apêndice B: Mecânica Molecular - Líquidos
Cada propriedade termodinâmica é obtida através da média desta sobre todas
as configurações acessíveis ao sistema. Como as coordenadas moleculares variam
continuamente, a soma torna-se uma integral, a qual deve ser efetuada sobre uma
determinada região desse espaço de configurações. Cada ponto deste espaço
representa uma diferente configuração de posição e velocidade de cada partícula. Isto
implica em 6 coordenadas por partícula (x, y, z, vx, vy, vz) e portanto qualquer função
relacionada a este espaço possui 6N variáveis (onde N é o número de partículas). O
método numérico mais simples de integração consiste em dividir certo intervalo x em
n porções de tamanho dx, e somar a função n vezes. Desta forma a integração
numérica de uma função com 6N variáveis necessita, a princípio, da ordem de n6N
operações para ser realizada. Considerando-se que um processador moderno realiza
da ordem de 1010 operações por segundo, a integração numérica de um sistema com
algumas dezenas de partículas é completamente impraticável.
Uma das soluções mais inteligentes para se evitar a integração numérica sobre
todo o espaço consiste em avaliar apenas uma ínfima, mas representativa, parte do
ensemble. Mais precisamente, isso significa fazer uma amostragem. Pressupondo-se
que se conheça a distribuição das configurações, poder-se-ia sorteá-las em quantidade
suficiente para se obter o valor convergido da propriedade física que se tem interesse
em calcular. Desta forma é utilizada a simulação computacional para gerar
configurações segundo as condições de contorno e os vínculos que determinam o
ensemble. Há duas maneiras de se realizar esta tarefa: determinista e estocástico. O
determinista consiste em gerar uma configuração a partir de outra de maneira
dinâmica; utilizando-se das forças de interação para atualizar as coordenadas do
sistema, através de incrementos da variável temporal. Este método é conhecido por
dinâmica molecular. O método estocástico consiste em produzir configurações por
meios aleatórios, sorteando-as segundo a distribuição característica do ensemble. Este
método de amostragem estocástica é conhecido como Monte Carlo.
Monte Carlo
O método de Monte Carlo (MC) consiste em sortear pontos do espaço das
configurações. Cada ponto é representado por Γk = Γ{R,P}, onde k numera a
configuração, R se refere ao conjunto de coordenadas de posição e P de momento, de

76
todas as partículas. O valor médio de uma função ƒ(R,P) é definido pela (3). Nesta,
w(Γk) é a probabilidade de o sistema ser encontrado na configuração Γk, e M é o
número total de pontos utilizados no cálculo da média [66].
⟨ƒ(R,P)⟩ = ∑
M
k=1 ƒ(R,P) w(Γk)
∑ M
k=1 w(Γk)
(3)
Se todas as configurações sorteadas forem equiprováveis, w(Γk) = 1. Esse é o
caso do ensemble microcanônico (NVE). Como a energia potencial é dependente das
distâncias intermoleculares e a cinética pode assumir apenas valores positivos, apenas
uma complicada e descontínua superfície do espaço das configurações é acessível
neste ensemble. A alternativa direta é o uso do ensemble canônico, cujas condições de
contorno são NVT. Desta forma o número de configurações acessíveis aumenta, já que
a energia flutua em torno de um valor médio. O ensemble canônico é caracterizado
pela distribuição de probabilidades w(Γk) = exp(–βEk), onde Ek é a energia da
configuração Γk e β = 1/kbT (onde kb é a constante de Boltzmann e T é a temperatura).
Com isto, utiliza-se o teorema da eqüipartição da energia, no qual cada coordenada
quadrática na equação da energia (x2, y2, z2, px2, py
2, pz2) produz uma contribuição de
½kbT para a energia interna do sistema. A energia cinética total é dada por (3/2)NkbT.
Portanto para obter a energia total do sistema utilizando o método de MC no ensemble
canônico basta sortear configurações de posição R e utilizá-las na (4), generalizando-
se este procedimento a qualquer propriedade estrutural.
⟨E⟩ = 32 NkbT +
∑ M
k=1 Uk(R) exp( )– β Uk(R)
∑ M
k=1 exp( )– β Uk(R)
(4)
Método de Metropolis
O uso do ensemble canônico reduz a dificuldade de se sortear as
configurações. Porém uma análise atenta da função energia potencial em um
condensado mostra que esta pode aumentar algumas ordens de grandeza com
pequenas diminuições das distâncias intermoleculares [67]. Tal constatação implica

77
que a maior parte do espaço das configurações possui peso estatístico quase nulo,
como se as partículas tendessem a se localizar centradas em pequenas células
individuais definidas pelo seu volume livre, υf* = ∫dυ exp(– β U(R)) [1]. Portanto
sortear com a mesma probabilidade qualquer configuração Γ{R} ainda é um método
muito ineficiente. Metropolis et al., em 1953, apresentou uma forma de contornar esta
ineficiência [68]. Ao invés de escolher configurações aleatoriamente e depois
executar uma média ponderada, faz-se um MC modificado, em que se escolhem as
configurações Γ{R} segundo seu peso estatístico para então realizar uma média
simples. Isto é feito com a ajuda de uma cadeia de Markov [69,70]. Nesta, uma
configuração Γj é obtida a partir de outra Γk fazendo-se {xj = xk + αξk}, onde x
contempla todas as 3N coordenadas de posição, α é o deslocamento máximo
(arbitrário) na direção x, e ξ é um número aleatório entre –1 e 1. Feito isto, calcula-se
a energia da nova configuração. Se ∆U = Uj – Uk ≤ 0, o movimento é aceito e a nova
configuração é registrada. Se ∆U > 0, o movimento é aceito com probabilidade w(Γk
→ Γj) = exp(– β ∆U); ou seja, sorteia-se um número ξ entre 0 e 1; se ξ ≤ w(Γk → Γj), o
movimento é aceito e a configuração Γj é registrada; se ξ > w(Γk → Γj), o movimento
é rejeitado, registrando-se novamente a configuração Γk. As configurações obtidas
com tal procedimento reproduzem a distribuição canônica, e qualquer propriedade
⟨ƒ(R)⟩ pode ser calculada através de uma média simples sobre o valor ƒk(R) referente
a cada ponto Γk selecionado (5).
⟨ƒ(R)⟩ = 1M
∑ M
k=1 ƒk(R) (5)
Para a utilização deste método no ensemble isotérmico-isobárico (NpT), deve-
se incluir o termo relacionado ao trabalho mecânico (pV) associado à variação do
volume. Como a soma sobre as coordenadas de posição R é vinculada ao volume V do
sistema, o número de pontos acessíveis à variável R é proporcional ao hiper-volume
VN do espaço das configurações. Para eliminar este vínculo, faz-se a transformação {xi
= L qi}; onde L é proporcional à raiz cúbica do volume, de forma que dR = VN dQ,
onde Q é a nova coordenada de posição adimensional, independente do volume do
sistema. Como conseqüência o teste de Metropolis se modifica: se [∆U + p∆V –
(N/β)ln(Vj/Vk)] ≤ 0, o movimento é aceito e a nova configuração é registrada; caso
contrário, ele é aceito com probabilidade w(Γk → Γj) = exp[– β(∆U + p∆V) + N ln(Vj/Vk)].

78
Dinâmica Molecular
Outra forma de se amostrar o espaço das configurações de um líquido é
através da evolução temporal do sistema. Isto é feito partindo-se de um conjunto
inicial de posições e velocidades (Γ(t) = Γ{R,P}) e tendo em mãos uma expressão
para a energia potencial do sistema (U(R)). Com isto é possível calcular as forças
entre as partículas (F(r) = -∇∇∇∇rU(R)), e finalmente utilizar as leis de Newton para
atualizar suas posições e velocidades, (Γ(t+δt)). Isso foi realizado pela primeira vez
em 1956 por Alder e Wainwright [71], para estudar a transição de fase em um sistema
de esferas rígidas. Em 1964, Rahman foi o primeiro a simular um líquido real
(argônio) [72]. Os métodos de integração mais utilizados para resolver as equações de
movimento e obter Γ(t+δt) a partir de Γ(t) são aqueles conhecidos por diferenças
finitas [75]. Há dezenas de possíveis algoritmos que utilizam diferentes aproximações.
A escolha do algoritmo depende de certos requisitos básicos. É desejável que o passo
de integração (δt) seja o maior possível, de tal forma que se economize tempo de
computação e que se produza o maior número possível de configurações
descorrelacionadas (como será explicado nas próximas seções). É também desejável
que o algoritmo seja o mais rápido (simples) possível ao mesmo tempo em que
reproduza fielmente a trajetória clássica e satisfaça as leis de conservação. Desta
forma, o algoritmo de integração pode variar com o sistema a ser simulado e o
ensemble (as propriedades que se deseje manter constante). Em especial, os graus de
liberdade moleculares afetam a escolha do método de integração. O passo de
integração utilizado para atualizar os movimentos intermoleculares pode não ser (e
comumente não é) adequado para os intramoleculares.
Algoritmo de Integração
Provavelmente o algoritmo de integração mais utilizado em dinâmica
molecular seja o de Verlet [73]. Isto se deve apenas a sua simplicidade, rapidez e
eficiência. Tal método parte da expansão de Taylor da posição em torno de t,
avançado e atrasado por δt, r(t+δt) e r(t–δt) (6).
r(t+δt) = r(t) + r'(t) δt + ½ r''(t) δt2 +… r(t–δt) = r(t) – r'(t) δt + ½ r''(t) δt2 –…
(6)

79
Somando as expressões (6) e desprezando os termos de quarta ordem e
superiores, é possível obter a posição r(t+δt) utilizando apenas a posição atual e a
anterior, e a aceleração atual (7). Ao se subtrair as expressões (6) e desprezando os
termos superiores a segunda ordem, obtém-se a velocidade r'(t) (8).
r(t+δt) = 2 r(t) – r(t–δt) + r''(t) δt2 (7)
r'(t) = r(t+δt) – r(t–δt)
2 δt (8)
Desta forma, a integração da trajetória utilizando o algoritmo de Verlet produz
um erro da ordem de δt4 para as posições e δt2 para as velocidades. Deve-se ressaltar
que apenas o erro nas posições é cumulativo durante o processo de integração, e
apenas esse erro afeta a qualidade da trajetória obtida em relação ao resultado exato.
Considerando-se aqui como "resultado exato" aquele que seria obtido utilizando-se
δt→0, e precisão computacional infinita. Outra característica notável do algoritmo de
Verlet é que ele é centrado, isto é, r(t+δt) e r(t–δt) participam de maneira igual nas
equações e podem ser invertidos. Isto faz do algoritmo reversível no tempo. Isto
implica que ao utilizar forças conservativas, é garantida a conservação do momento
linear [75].
Há dois algoritmos algebricamente equivalentes ao algoritmo de Verlet: leap-
frog e Verlet velocidade. O algoritmo conhecido por leap-frog utiliza passos
intermediários da velocidade, r'(t+δt/2) [75]. Ele pode ser compreendido a partir das
expansões da posição em torno de t e t+δt (9).
r(t+δt) = r(t) + r'(t) δt + ½ r''(t) δt2 +… r(t) = r(t+δt) – r'(t+δt) δt + ½ r''(t+δt) δt2 –…
(9)
Subtraindo as expressões (9), obtém-se a posição r(t+δt) (10). A velocidade no
passo intermediário é dada pela aproximação r'(t+½δt) = ½ [ r'(t+δt) + r'(t) ], e o
termo de segunda ordem é na verdade de terceira ordem, já que em primeira
aproximação r'''(t+½δt) δt = [ r''(t+δt) – r''(t) ].
r(t+δt) = r(t) + ½ [ r'(t+δt) + r'(t) ] δt + – ¼ [ r''(t+δt) – r''(t) ] δt2 +…
(10)
Desprezando os termos superiores à segunda ordem da equação (10), e
tomando sua derivada temporal (atrasado em meio passo de integração), obtêm-se as

80
expressões para o algoritmo leap-frog (11). O uso dessas expressões elimina parte do
erro de truncamento presente em (7).
r(t+δt) = r(t) + r'(t+½δt) δt r'(t+½δt) = r'(t–½δt) + r''(t) δt
(11)
O algoritmo mais moderno, chamado de Verlet velocidade, melhora a precisão
da velocidade em relação aos algoritmos anteriores [75]. Dado por (12), a velocidade
em t+δt é deduzida da mesma forma que a posição é obtida para o leap-frog, isto é, a
velocidade é obtida com a derivada da expressão (10).
r(t+δt) = r(t) + r'(t) δt + ½ r''(t) δt2 r'(t+δt) = r'(t) + ½ [ r''(t+δt) + r''(t) ] δt
(12)
Termostato e Barostato
Uma vez que a técnica de dinâmica molecular evolui no tempo as posições das
partículas em um dado volume, conservando assim o momento e a energia total do
sistema, o ensemble natural deste tipo de simulação é NVE. Desta forma, para se
simular em outros ensembles é necessária a utilização de termostatos e barostatos,
para fixar a temperatura e a pressão. Isto é feito imaginando-se que o sistema está
conectado a um reservatório. É possível também fixar o potencial químico, mas isso
foge do escopo deste trabalho. Há muitas propostas de como acoplar o reservatório à
caixa de simulação. Nenhuma delas é exata, e cada uma parte de conjuntos diferentes
de princípios para justificar o acoplamento. Antes de apresentar as utilizadas neste
trabalho, é necessário compreender como se calcula temperatura e pressão. Como
ilustração, para um sistema atômico, a temperatura e pressão instantâneas são dadas
respectivamente por (13) e (14), onde T = ⟨T(t)⟩, p = ⟨P(t)⟩ e V = ⟨V(t)⟩ [75]. O termo
de virial W(t) é dado por (15), onde rij(t) é a distância interatômica, e u(rij) o
respectivo potencial de interação.
T(t) = 1
3N ∑ N
i mi |r'i(t)|
2 (13)
P(t) = [ NkbT(t) + W(t) ] / V(t) (14)
W(t) = – ⅓ ∑ N
i ∑ N
j>i rij(t) · ∇∇∇∇u(rij) (15)

81
A idéia mais simples de termostato é multiplicar as velocidades por (T0/T(t))½
a cada passo da simulação, onde T0 é a temperatura do reservatório. A mesma idéia
poderia ser aplicada para a pressão, mas para isso seria necessário um processo
iterativo já que a variação do volume afeta o resultado do virial (15). De qualquer
forma, tal mudança abrupta nas coordenadas a cada passo de simulação para manter
fixa a temperatura e/ou pressão não é desejável. Isto perturbaria de forma artificial e
imprevisível a dinâmica do sistema. A proposta mais simples para evitar essa abrupta
descontinuidade é dada pelo termostato/barostato de Berendsen [74]. Para o
termostato, considera-se que a cada passo de simulação o sistema esteja sujeito a um
fluxo de temperatura instantânea dado por T'(t) = [ T0 - T(t) ] / τT, onde τT é uma
constante de acoplamento maior que o passo de integração (τT > δt). Desta forma, a
variação da temperatura pode ser realizada de maneira arbitrariamente suave. O efeito
deste fluxo é transformar as velocidades: r'(t) → λ(t) r'(t), onde λ(t) é dado por (16).
λ(t) = 2
1 + δt T'(t)T(t) (16)
Para o barostato de Berendsen, o fluxo de pressão instantânea tem a mesma
forma, P'(t) = [ p0 - P(t) ] / τp. Entretanto, como não se sabe a priori como o termo de
virial muda com o volume do sistema, utiliza-se uma aproximação relacionada com a
compressibilidade isotérmica (17). Nesta, βT é um parâmetro característico do campo
de força que está sendo utilizado. Com o auxílio de (17), o efeito da pressão externa
sobre o sistema se manifesta na transformação das posições: r(t) → µ(t) r(t), sendo
que µ(t) é dado por (18).
P'(t) = – V'(t)βT V(t)
(17)
µ(t) = [ 1 – δt βT P'(t) ]⅓ (18)
Não há garantia de que o termostato/barostato de Berendsen reproduza
corretamente o ensemble canônico ou o das pressões [75]. Os valores de τT e τp devem
ser os maiores possíveis para perturbar o mínimo possível a dinâmica do sistema. Em
especial, o barostato de Berendsen produz resultados equivocados à medida que o
valor de βT diferencia da verdadeira compressibilidade isotérmica do sistema em
estudo. Entretanto, tomando as devidas precauções, o termostato/barostato de

82
Berendsen é bastante eficiente, e dada a sua simplicidade, justifica-se o fato de ser
amplamente utilizado.
Correlação Estatística
O método de Monte Carlo Metropolis (MCM) e o de Dinâmica Molecular
possuem o inconveniente de produzir configurações correlacionadas. Configurações
correlacionadas fornecem pouca, quando nenhuma, informação sobre o ensemble. Isto
se deve ao fato que tais configurações estão muito próximas no espaço das
configurações acessíveis ao sistema, o qual se quer amostrar. A correlação entre duas
configurações separadas por um grande número de passos é pequena, e diminui cada
vez mais à medida que o número de passos aumenta. O intervalo de passos necessário
para obter a descorrelação estatística é conhecido através da função de autocorrelação
[75]. A função de correlação entre dois conjuntos ordenados, {xj} e {yj}, é definida
pela (19). Nesta, ∆x = xj – ⟨x⟩ é o desvio da média, enquanto que σx = ⟨∆x2⟩½ é o
desvio padrão. Se ∆x = ∆y, para todo j, então C(x,y) = 1; caso contrário -1 < C(x,y) <
1. Quanto mais próximo de zero, é dito que mais descorrelacionados são os conjuntos
x e y.
C(x,y) = ⟨∆x∆y⟩σxσy
(19)
A função de autocorrelação da energia é obtida se fazendo xj = Uj e yj = Uj+k,
de forma que C(x,y) = C(k); onde k é um determinado número de passos. Portanto
C(k) fornece a correlação da energia entre configurações separadas por k passos. Nos
sistemas utilizados neste trabalho, o intervalo típico de descorrelação (definido como
C(k) < 15%) para um líquido a pressão e temperatura normais é da ordem de 103
passos. Tomando K como o número de configurações descorrelacionadas obtidas, o
erro estatístico associado à média de uma propriedade ƒ(R) é igual a σƒ/K½, onde σƒ é
o desvio padrão da distribuição da propriedade. O erro estatístico, também chamado
de erro padrão, está associado com o tamanho da amostragem (distribuição) e fornece
a incerteza sobre uma medida.

83
Apêndice C: Mecânica Quântica - Métodos Aproximativos
Em 1926, Erwin Schrödinger demonstrou que a quantização poderia ser
compreendida como um problema de autovalores, e forneceu a solução analítica (não
relativística) para o espectro do átomo de hidrogênio utilizando o formalismo
hamiltoniano [76], reconhecendo-o como um problema de autovalores Hψ(r) = Eψ(r).
A equação de Schrödinger para uma partícula é dada por (20); onde ψ(r) é a função de
onda associada a uma partícula de massa m sujeita a um potencial U(r), com energia
E. Esta é convenientemente escrita como.
– ħ2
2m ∇2ψ(r) + U(r) ψ(r) = E ψ(r) (20)
⌡⌠dr ψ*(r)ψ(r) = 1 (21)
No mesmo ano, Max Born postulou que a probabilidade de encontrar a
partícula entre r e r + dr descrita pela função de onda ψ(r), é w(r) = ψ*(r)ψ(r) [77],
onde ψ*(r) é o conjugado complexo de ψ(r), em analogia à chance de encontrar o
fóton em uma onda eletromagnética. Portanto, o valor médio de qualquer observável
A representado pelo operador  pode ser obtida segundo a (22), notando que ^r = r e ^p
= – iħ∇∇∇∇. A integral da (22) convenientemente escrita como ⟨A⟩ = ⟨ψ|Â|ψ⟩.
⟨A⟩ = ⌡⌠dr ψ*(r)Âψ(r) (22)
"As leis fundamentais necessárias para o tratamento matemático de grande
parte da física e de toda a química estão plenamente conhecidas, e a dificuldade
reside apenas no fato de que a aplicação dessas leis leva a equações que são muito
complexas para serem resolvidas" [78]. Os problemas envolvendo muitos corpos
costumam recair em um conjunto de equações acopladas cuja solução analítica não se
conhece. A solução do problema de vários corpos interagentes, tanto em mecânica
clássica quanto em mecânica quântica, pode ser obtida com métodos de integração
numérica [79]. Dado que o custo destes métodos cresce muito rapidamente com o
número de partículas, tal procedimento é impraticável. Uma alternativa viável para a
solução do problema de vários corpos se encontra nos métodos aproximativos. Isto é
feito alterando-se a forma da interação, U(r) → U'(r), ou equivalentemente, o tipo de
solução Ψ(r) → Ψ'(r). Sendo que estas alterações podem ter origem empírica

84
(utilizando ajustes experimentais), teórica (desprezando termos pouco influentes) ou
híbrida. As aproximações são aplicadas de acordo com o sistema, as propriedades que
se queira estudar, e o erro que elas acarretam.
Aproximação de Born-Oppenheimer
Uma aproximação comumente utilizada é a de Born-Oppenheimer [80], em
que é feita a separação das funções de onda nuclear e eletrônica. Parte-se do princípio
de que a energia cinética nuclear pode ser tratada como uma pequena perturbação
sobre o movimento eletrônico. Toma-se a partição: H = H0(Re,Rn,Pe) + V(Pn), onde n
e e se referem respectivamente aos núcleos e elétrons, R e P aos operadores de
posição e momento. O sistema não perturbado H0 ψσ(Re;Rn) = εσ(Rn) ψσ(Re;Rn) é a
solução da parte eletrônica e depende parametricamente das posições nucleares (Rn).
Expande-se então a solução exata em termos dessas autofunções (23).
Ψ(Re,Rn) = ∑ σ
χσ(Rn) ψσ(Re;Rn) (23)
Integrando-se H Ψ(Re,Rn) = E Ψ(Re,Rn) apenas nas coordenadas eletrônicas,
obtém-se um conjunto de equações acopladas que pode ser escrito na forma matricial:
[ Hn(Rn) + He(Rn) ] χ(Rn) = E χ(Rn). Sendo (Hn(Rn))νσ = ⟨ψν|V(Pn)|ψσ⟩ e (He)νσ =
εσ(Rn)δνσ. A aproximação toma apenas os termos diagonais de Hn(Rn), e portanto
também implica na aproximação adiabática, ou seja, o movimento nuclear não altera o
estado eletrônico. Os termos não diagonais de Hn(Rn) são chamados de acoplamento
vibrônico e estão além da aproximação de Born-Oppenheimer.
Hartree e Hartree-Fock
Uma das primeiras abordagens para encontrar a função de onda eletrônica foi
desenvolvida em 1928 por Douglas Hartree [81,82]. Nesta aproximação imagina-se
que cada elétron se move no campo médio de todos os outros. Isto implica que o
potencial inter-eletrônico pode ser escrito como uma soma de potenciais de um
elétron, U'e(Re) = ∑ u(i). A função multi-eletrônica ψσ(Re;Rn) é então escrita como um
único produto de funções de um elétron (produto de Hartree), ψ'σ(Re;Rn) = ∏ φi(i). O
índice subscrito i caracteriza o orbital de cada elétron i (na posição ri).
Em 1929, John Clark Slater reconheceu a necessidade de escrever o produto
de Hartree ψ'σ(Re;Rn) como um determinante |[φn(i)]|. Ele propôs também que a função

85
de spin deveria ser incluída no orbital φn(i), obtendo notável sucesso em prever o
espectro de diversos átomos [83]. A fundamentação teórica das equações foi obtida de
maneira independente por Vladimir Fock, em 1930 [84], e historicamente o método
passou a ser chamado de Hartree-Fock (HF). O operador de Fock F(i) é a soma da
energia cinética e do potencial médio sobre o elétron i. Ao atuar sobre o orbital φn(i)
fornece o autovalor εn: F(i) φn(i) = εn φn(i).
Combinação Linear de Orbitais Atômicos (LCAO) e as funções base
A maneira comumente usada para escrever os orbitais moleculares φn(i) em é
através de uma combinação de orbitais localizados nos átomos {αk}. A idéia de
combinar orbitais atômicos para descrever orbitais moleculares é anterior ao seu uso
em HF [85,86,87]. De um modo geral, o método LCAO escreve φ'n(i) = ∑ cknαk com
uma base fixa de orbitais atômicos αk. Em 1932, Slater propôs uma forma analítica
simples para descrever o spin-orbital de sistemas atômicos, φ'n(i) = ∑ crbexp(-ar),
onde c, b e a variam em cada termo [88]. Em 1950, Samuel Boys demonstrou que se
fosse escolhida uma base de gaussianas, as integrais poderiam ser analiticamente
resolvidas [89].
Em 1951, independentemente, Clemens Roothaan [90] e George Hall [91]
formalizaram o método HF via LCAO para obtenção de orbitais moleculares. Estas
equações são escritas em forma matricial (F – ε'nS)cn = 0; nesta, F é a matriz de Fock
(Fmn = ⟨φm(i)|F(i)|φn(i)⟩), S é a matriz de sobreposição (Smn = ⟨φm(i)|φn(i)⟩), cn é o vetor
dos coeficientes das funções base, e ε'n são as raízes, cujo número é igual ao número
de funções base utilizadas. O uso de funções base para descrever os orbitais
moleculares introduz o erro de base finita, já que é impossível tratar um conjunto
infinito de funções base computacionalmente. Portanto, de acordo com o princípio
variacional, a energia do orbital LCAO é maior ou igual à energia exata do orbital de
Fock, εn ≤ ε'n.
Teoria do Funcional da Densidade (DFT)
Em 1964, Pierre Hohenberg e Walter Kohn [92] propuseram dois teoremas
que estabeleceram a idéia de que a energia do sistema quântico está conectada
biunivocamente com a densidade eletrônica (24). O primeiro teorema: "O potencial
externo V(Re) é um funcional único de ρ(r)" estabelece que a energia do estado

86
fundamental também seja um funcional único da densidade. O segundo teorema é
uma extensão do método variacional para a densidade. Portanto, basta conhecer a
forma funcional exata da energia E[ρ] para encontrar a energia do estado fundamental
de um sistema quântico. Desta forma: E[ρ] = T[ρ] + U[ρ] + V[ρ], onde T[ρ] é a energia
cinética eletrônica, U[ρ] é o potencial inter-eletrônico e V[ρ] é o potencial dos elétrons
com os núcleos. Apenas este último termo possui expressão analítica: V[ρ] = ∫dr V(r)
ρ(r). A principal vantagem deste modelo é que substitui uma complicada função de
3N coordenadas ψ(Re;Rn), pela densidade eletrônica que depende apenas de 3.
ρ(r1) = N
∏ N
k=2 ⌡⌠
V
d3rk |ψ(Re;Rn)|2 (24)
Em 1965, Walter Kohn e Lu Jeu Sham [93] propuseram uma maneira
autoconsistente de calcular E[ρ]. É utilizado um sistema fictício de "elétrons não
interagentes" sujeitos a um potencial externo Vs[ρ], cuja densidade eletrônica é por
definição a mesma densidade do sistema real. A energia do sistema de Kohn-Sham
(KS) é obtida via uma função de onda auxiliar ψs(Re) e recai em equações de uma
partícula análogas ao método de HF. Desta forma: Es[ρ] = Ts[ρ] + Vs[ρ], onde Ts[ρ] é a
energia cinética do sistema fictício. O potencial Vs[ρ] é escrito como a soma de 3
termos: Vs[ρ] = V[ρ] + VH[ρ] + VXC[ρ], onde VH[ρ] é o potencial de Hartree (auto-
interação coulombiana da densidade eletrônica) e VXC[ρ] é o potencial de troca e
correlação, e carrega todos os efeitos quânticos do sistema de muitas partículas.
O método mais utilizado para VXC[ρ] é conhecido por "Aproximação de
Densidade Local" (LDA). Nesta, VXC[ρ] depende apenas da densidade no ponto em
que é calculada: VXC-LDA[ρ] = ∫dr εXC(ρ) ρ(r). Outra abordagem utilizada, que também
é local, é a "Aproximação do Gradiente Generalizado" (GGA). Nesta considera-se o
gradiente da densidade: VXC-GGA[ρ] = ∫dr εXC(ρ,∇∇∇∇ρ) ρ(r). Por fim, é muito utilizado em
sistemas moleculares potenciais híbridos que incluem parte da energia de troca exata
obtida da teoria de HF.
Post-HF e a correlação eletrônica
A aproximação de HF transforma um problema de N elétrons em N problemas
de um elétron, ao custo de perder a correlação do movimento eletrônico; cada elétron
está sujeito apenas ao campo médio produzido por todos os outros. Por esse motivo, a

87
diferença entre a energia eletrônica exata e a energia obtida com o método de Hartree-
Fock é chamada de correlação eletrônica [94]. O método DFT-KS inclui parte da
energia de correlação de maneira ad hoc. Há dezenas de propostas para o funcional de
troca e correlação. Entretanto nenhum deles é exato ou pode ser sistematicamente
melhorado em direção ao resultado exato. Felizmente é possível utilizar a função de
onda HF como base para métodos que fornecem a solução exata. Tais métodos são
conhecidos por post-HF.
Método de interação de configurações (CI)
O CI possui a mesma metodologia do método variacional utilizado para
resolver as equações de Roothaan. Escreve-se a função de N elétrons como uma
expansão de produtos anti-simétricos (25). O primeiro termo é a solução de HF, com
os orbitais de mais baixa energia ocupados; o segundo termo conta sobre todas as
excitações simples (isto é, um orbital ocupado n é trocado por um orbital virtual ν); o
terceiro termo conta sobre todas as excitações duplas (dois orbitais n e m são trocados
por orbitais virtuais ν e µ); e assim sucessivamente. Quando apenas o primeiro termo
da série é computado, o método é chamado de CIS; quando o primeiro e o segundo
são contados, CISD; e assim sucessivamente; quando todos os termos possíveis são
incluídos, o método é chamado de Full-CI. O método CI é comumente utilizado para
obter os estados excitados da função eletrônica molecular ψσ(Re;Rn).
ψσ(Re;Rn) = A0,σ|[φ]|0 + ∑ Aνn,σ|[φ]|
νn + ∑ A
νµnm,σ|[φ]|
νµnm … (25)
Truncar (25) no segundo termo (CIS) não melhora o resultado para o estado
fundamental. Este resultado é conhecido por teorema de Brillouin, ⟨|[φ]|νn|Ĥ||[φ]|0⟩ = 0
[95].
Møller-Plesset de ordem 2 (MP2)
Em 1934, Christian Møller e Milton Plesset aplicaram a teoria de perturbação
de Rayleigh-Schrödinger utilizando uma soma de operadores de Fock como
hamiltoniano não-perturbado [96], Ĥ(0) = ∑ F(i). A perturbação é a diferença entre o
potencial exato e o potencial de uma partícula, V = Ue – ∑ û(i). As funções de onda
do problema não perturbado são os determinantes construídos com os orbitais de
Fock, ψ(0)σ (Re;Rn) = |[φ]|
ν…n…, cuja energia é a soma dos autovalores, E
(0)σ = ∑ εn. A

88
correção de primeira ordem para a energia (E(1)σ = ⟨ψ
(0)σ |V|ψ
(0)σ ⟩) do estado fundamental
recupera a energia de HF, ou seja, EHF = E(0)σ + E
(1)σ (σ = 0). Conseqüentemente a
correlação eletrônica é obtida a partir da correção de segunda ordem chamada de
MP2: E(2)0 = ⟨ψ
(0)0 |V|ψ
(1)0 ⟩. Nestas equações ψ
(0)0 é a função de onda de HF, ou seja,
|[φ]|0. A correção de primeira ordem para a função de onda (26) é então escrita
utilizando-se configurações ψ(0)τ , que nada mais são do que substituições simples,
duplas, triplas, etc., em relação à solução de HF: ψ(0)τ = |[φ]|
ν…n…. Desta forma, apenas as
substituições duplas sobrevivem em MP2, de tal forma que a correção de segunda
ordem é dada em termos de orbitais de uma partícula por (27).
|ψ(1)0 ⟩ = ∑
∞ τ≠0
|ψ(0)τ ⟩ ⟨ψ
(0)τ |V|ψ
(0)0 ⟩
(E(0)0 – E
(0)τ )
(26)
E(2)0 = ∑ ∞
n<m,ν<µ |⟨nm|V|νµ⟩ – ⟨nm|V|µν⟩|2
(εn + εm – εν – εµ) (27)
CIS(D)
MP2 é um método eficiente para incluir correlação eletrônica sobre o estado
fundamental. CIS por usa vez não altera o estado fundamental, mas é o método mais
simples de se obter os estados excitados. Existem técnicas híbridas capazes de incluir
as correções perturbativas de MP2 sobre os estados de referência CIS [97,98]. Neste
trabalho foi utilizado o CIS(D) por ser um método extensivo [95] e por ter um custo
computacional igual a N5 (similar a MP2). CIS(D) é a segunda ordem da expansão
perturbativa do método de coupled-cluster para estados excitados (não apresentado
aqui), restrito a substituições simples e duplas, onde o termo de ordem zero é a função
CIS e o termo de primeira ordem se anula [98]. Formalmente a correção CIS(D) é
escrita como: ECIS(D) = ⟨ψ
(0)σ |V|U2ψ
(0)σ ⟩ + ⟨ψ
(0)σ |V|T2U1ψ
(0)σ ⟩, onde U1 e U2 produzem
substituições simples e duplas de acordo com CI (isto é, coeficientes determinados
pelo método variacional), enquanto que T2 produz substituições duplas de acordo com
MP2 (isto é, coeficientes determinados pelo método perturbativo). Sendo que nesta
representação, a correção MP2 é E(2)0 = ⟨ψ
(0)0 |V|T2ψ
(0)0 ⟩ enquanto que a função de onda
CIS é dada por |ψCIS⟩ = |U1ψ(0)0 ⟩.

89
Apêndice D: Campos de Força
Mesmo conhecendo a natureza quântica da matéria e os principais métodos de
solução do problema de muitos corpos, não é um processo trivial descrever um
sistema com centenas de moléculas (milhares de elétrons). O custo para se calcular a
energia de um sistema com muitas moléculas através de métodos quânticos é
excessivamente elevado. É necessário simplificar as interações moleculares o máximo
possível sem entretanto obscurecer sua conexão com a formulação quântica original.
Expansão multipolar
A princípio, uma molécula pode ser descrita como uma distribuição de cargas.
O potencial de interação de duas distribuições (a e b) de cargas pontuais fixas é dado
pela (28); onde qi é o valor de cada carga em a e qj em b; rij = |(ri + ra) – (rj + rb)| é a
distância entre as cargas i e j; ra é a posição do centro das cargas i, e rb das cargas j.
Escrevendo rab = rb – ra, tomando rab > ri e rab > rj é possível utilizar (29) e expandir
(28) na forma (30). Tal expansão é conhecida como expansão multipolar [99].
U(r) = ∑ i ∑ j qiqj
rij (28)
1rij
= ∑ ∞
n = 0 ∑ ∞
m = 0
∇∇∇∇
n
i ∇∇∇∇m
j 1rij
ri = rj = 0
(· ri)n(· rj)
m
n!m! (29)
U(r) = qaqb
rab +
µbqa – µaqb
r3ab
· rab + µb · µa
r3ab
+
+
Θbqa – 3µaµb + Θaqb
r5ab
(· rab)2 + …
(30)
qa = ∑ i qi
µa = ∑ i qiri
Θa = ∑ i qi
32 riri –
12 1
(31)
Termo de Coulomb
Um método aproximado de representar a distribuição de cargas em uma
molécula é utilizar um termo na forma (32), em que se escolhe um número reduzido
de sítios de carga. Esse potencial é definido como a soma de todas as interações
coulombianas entre os sítios de carga qi de uma molécula A e os sítios de carga qj de

90
uma molécula B. Os valores das cargas e a posição desses sítios são ajustados a fim de
reproduzir o efeito médio da expansão multipolar (30).
(UC)ij = qiqj
rij (32)
Os efeitos de polarização instantânea devido às mudanças dinâmicas da
vizinhança molecular podem ser incluídos de forma média ou como cargas flutuantes.
O termo de Coulomb não contempla os efeitos quânticos de troca e correlação, de tal
forma que estes são incluídos através de termos ligados às interações de van der
Waals.
Equação de van der Waals
Em 1873, Johannes van der Waals deduziu uma equação de estado que
descreve de maneira continua o estado líquido e o gasoso [100]. A equação de van der
Waals (33) é uma modificação da equação de estado do gás ideal (pV = NkbT),
incluindo-se o volume das moléculas (υ → υ – b) e a força de coesão (p → p + a/υ2).
Os parâmetros a e b são conhecidos como constantes de van der Waals e seus valores
são obtidos experimentalmente para cada substância, dentro de determinados
intervalos de temperatura e pressão.
p +
a
υ2 (υ – b) = kbT (33)
Em 1902, Kamerlingh Onnes [101] expressou os resultados de suas
observações sobre diversos gases na forma da equação de estado empírica (34). Os
coeficientes A … F foram chamados por Onnes de coeficientes de virial, sendo função
da temperatura, e podendo ser relacionados às constantes de van der Waals: A = kbT e
B = bkbT – a.
pυ = A + B
υ + C
υ2 + D
υ4 + E
υ6 + F
υ8 (34)
É possível calcular o segundo virial (B) teoricamente, em sistemas cujas
moléculas possam ser descritas por um potencial de pares esfericamente simétrico do
tipo u(rij) [101]. Nesta expressão, (35), β = 1/kbT.

91
B = 2πβ ⌡⌠
0
∞
dr r2 {1 – exp[-β u(r)]} (35)
Termo de Lennard-Jones
Em 1924, Lennard-Jones procurou ajustar o potencial u(r) segundo o potencial
empírico intermolecular (36), partindo de resultados experimentais para B. Ele
estudou quais expoentes (n e m) poderiam ser utilizados para descrever sistemas reais,
ao mesmo tempo procurando os melhores parâmetros que se ajustassem ao argônio
[101]. Nesta equação, o termo com λn é repulsivo e o termo com λm é atrativo.
Lennard-Jones não obteve resultados conclusivos, a não ser a constatação teórica que
n > m ≥ 4.
u(r) = λn
rn – λm
rm (36)
Em 1927, S. C. Wang utilizando teoria de perturbação de segunda ordem
mostrou que a interação entre dois átomos de hidrogênio contém um termo atrativo
proporcional a 1/r6 [102,103]. No mesmo ano, Heitler e London utilizando teoria de
perturbação de primeira ordem mostraram que a força repulsiva entre átomos com
camadas fechadas aumenta rapidamente na medida em que a distância diminui [85].
Em 1928, Slater interpretou esta força repulsiva como um efeito de interferência,
tornando-se apreciável com a interpolação das funções de onda atômicas; e portanto,
caindo exponencialmente com a distância [102]. Partindo do método de Heitler e
London, e dos resultados de Wang para o hidrogênio, Slater obteve uma forma
analítica (em primeira aproximação) para a interação entre dois átomos de hélio (37).
Nesta equação, r é dado em unidades de raio de Bohr (a0) e a energia em 10-15 J. O
segundo coeficiente virial (35) calculado com o potencial (37) está em excelente
acordo com a experiência [104].
u(r) = 7.7 exp(-2.43 r) – 0.67
r6 (37)
Em 1930, London obteve a correlação entre as polarizabilidades e o potencial
atrativo entre duas moléculas [105,106], confirmando a forma obtida por Wang para
dois átomos de hidrogênio.
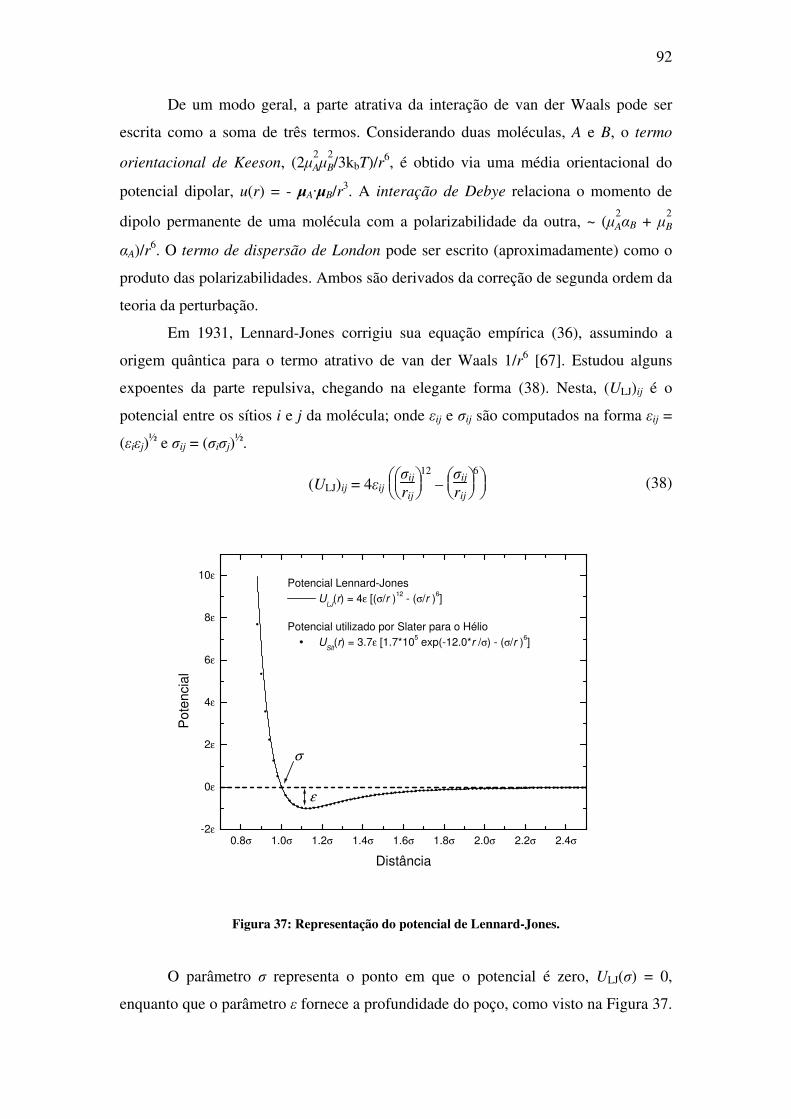
92
De um modo geral, a parte atrativa da interação de van der Waals pode ser
escrita como a soma de três termos. Considerando duas moléculas, A e B, o termo
orientacional de Keeson, (2µ2Aµ
2B/3kbT)/r6, é obtido via uma média orientacional do
potencial dipolar, u(r) = - µA·µB/r3. A interação de Debye relaciona o momento de
dipolo permanente de uma molécula com a polarizabilidade da outra, ~ (µ2AαB + µ
2B
αA)/r6. O termo de dispersão de London pode ser escrito (aproximadamente) como o
produto das polarizabilidades. Ambos são derivados da correção de segunda ordem da
teoria da perturbação.
Em 1931, Lennard-Jones corrigiu sua equação empírica (36), assumindo a
origem quântica para o termo atrativo de van der Waals 1/r6 [67]. Estudou alguns
expoentes da parte repulsiva, chegando na elegante forma (38). Nesta, (ULJ)ij é o
potencial entre os sítios i e j da molécula; onde εij e σij são computados na forma εij =
(εiεj)½ e σij = (σiσj)
½.
(ULJ)ij = 4εij
σij
rij
12
–
σij
rij
6
(38)
0.8σ 1.0σ 1.2σ 1.4σ 1.6σ 1.8σ 2.0σ 2.2σ 2.4σ
-2ε
0ε
2ε
4ε
6ε
8ε
10ε
σ
ε
Po
ten
cia
l
Distância
Potencial Lennard-Jones
ULJ
(r) = 4ε [(σ/r )12
- (σ/r )6]
Potencial utilizado por Slater para o Hélio
USlt
(r) = 3.7ε [1.7*105 exp(-12.0*r /σ) - (σ/r )
6]
Figura 37: Representação do potencial de Lennard-Jones.
O parâmetro σ representa o ponto em que o potencial é zero, ULJ(σ) = 0,
enquanto que o parâmetro ε fornece a profundidade do poço, como visto na Figura 37.

93
O potencial utilizado por Slater para o Hélio (37) obtido via teoria de perturbação,
possui εi = 1.77 (kcal/mol) e σi = 2.62 (Å), e se ajusta muito bem à curva fornecida
por ULJ na região do mínimo.
Para átomos, o potencial de Lennard-Jones é o termo mais importante do
potencial. Para moléculas que a núvem eletrônica não possui simetria esférica, os
termos eletrostáticos devem ser considerados. Isto é feito através do potencial de
Coulomb (32). Os termos da expansão multipolar (30) podem ser atrativos ou
repulsivos, dependendo da orientação relativa do par de moléculas, e portanto
definem a orientação relativa das moléculas em um líquido. Enquanto que o potencial
Lennard-Jones é sempre repulsivo para rij < 21/6σij e atrativo rij > 21/6σij, e definem
como e o quanto estas moléculas se empacotam.
Método Seqüencial Monte Carlo/Mecânica Quântica (S-MC/MQ)
A idéia do método S-MC/MQ é bastante simples [107]. Utiliza-se potenciais
do tipo Coulomb e Lennard-Jones para realizar a simulação do líquido e gerar as
estruturas. Usualmente são realizadas simulações da ordem de 100 mil passos de
MCM ou Dinâmica Molecular (com intervalo de tempo de 1fs), para temperatura
ambiente. Através da função de autocorrelação da energia, separa-se um número
reduzido de configurações estatisticamente descorrelacionadas. Através da função de
distribuição radial [75], determina-se a quantidade de moléculas em cada uma das
camadas de solvatação em torno da molécula referência. Sobre essas configurações
selecionadas, são realizados cálculos quânticos. As propriedades calculadas levam em
consideração a convergência tanto com o número de partículas quanto com o número
de configurações. Este protocolo tem sido usado com sucesso nos últimos anos para o
estudo de diversos sistemas [108].
O potencial de pares utilizado nas simulações clássicas é normalmente
ajustado para reproduzir as propriedades experimentais de fluido. Como não se
conhece previamente o momento de dipolo da molécula em fase líquida, os efeitos de
polarização induzida normalmente são incluídos implicitamente. Assim o momento de
dipolo é majorado de 10% a 20% em relação ao seu valor em fase gasosa [109], e em
seguida são ajustados os parâmetros Lennard-Jones. Porém moléculas em diferentes
ambientes sofrem polarizações distintas. Estudar moléculas solvatadas em diferentes
solventes, utilizando-se o mesmo conjunto de cargas pode resultar em resultados
errôneos. O método S-MC/MQ pode ser utilizado iterativamente a fim de se evitar

94
esse problema e polarizar adequadamente a molécula de interesse de acordo com o
solvente utilizado na simulação [110]. A polarização do soluto é feita partindo-se de
cargas iniciais não polarizadas (fase gasosa) ou pré-polarizadas, e executando-se
normalmente a simulação. As estruturas selecionadas (com solvente descrito por
cargas pontuais) são utilizadas para recalcular as cargas nos sítios atômicos do soluto.
Então este soluto é novamente levado a simulação e o processo é repetido até que as
propriedades ligadas à polarização (como o momento de dipolo) convirjam. Este
método foi utilizado no estudo de fenol em água apresentado neste trabalho [12].
Modelos polarizáveis
Em 1981, Thole propôs uma maneira eficiente de se calcular a polarização
molecular na vizinhança de cargas [111]. O método consiste em tratar cada átomo
como um dipolo difuso (i.e., não pontual). Uma vez conhecida a polarizabilidade em
cada átomo, seria possível calcular a polarização em um desses centros sujeito à
distribuição de cargas externas. Tal procedimento é autoconsistente, e leva em
consideração o efeito mutuo que as moléculas polarizadas causam umas sobre as
outras. Em seguida, Stone propôs um protocolo para obter a distribuição as cargas e
polarizabilidades sobre os sítios atômicos, a partir de cálculos de primeiros princípios
[112]. Uma vez conhecidas as distribuições de carga e polarizabilidades as quais
reproduzissem com precisão os momentos de multipólo quânticos, restaria apenas
incluir as interações de van der Waals no cálculo da energia total. Existem alguns
modelos que se utilizam dessa idéia de polarização de Thole [113,114]. O modelo
utilizado neste trabalho é o AMOEBA [51], através do programa TINKER [50].

95
Apêndice E: Aspectos Técnicos da Física Computacional
O computador é uma máquina idealizada por matemáticos e realizada por
físicos em meados da década de 1940 [115,116]. Desde então, se tornou uma
ferramenta essencial para aplicação e teste de teorias cuja abordagem analítica é
inviável. No princípio os físicos dominavam-na completamente, e eram capazes de
criar programas específicos escritos diretamente em linguagem de máquina.
Entretanto o computador evoluiu, tornou-se um produto e se popularizou. Ele deixou
de ser uma ferramenta exclusiva de uso científico, para se tornar um equipamento
eletrônico de múltiplas funcionalidades (tais como multimídia e internet). E à medida
que a complexidade dos computadores aumentava, os físicos deixavam de lado os
detalhes de seu funcionamento. O conhecimento do hardware foi delegado a
engenheiros da computação, de software a cientistas da computação, e a operação em
si foi delegada aos técnicos de informática e analistas de sistemas. Essa partição de
funções era inevitável dada a abrangência de conhecimentos e técnicas que surgiram à
medida que o computador se desenvolveu. Entretanto ignorar completamente o
funcionamento desta ferramenta implica em seu uso ineficiente. Saber como
aperfeiçoar um programa, e operar eficientemente e saber os limites da máquina
implica em mais resultados, mais hipóteses, mais verificações, e conseqüentemente
maior produção científica.
Este apêndice propõe uma mudança de paradigma. O objetivo deste apêndice
não é transformar a ferramenta em objeto de estudo, mas mostrar que o conhecimento
desta é vital para o físico que a utiliza. E também chamar a atenção que a ignorância
sobre ela pode trazer prejuízos e atrasos.
Física computacional e o Computador
Formalmente física computacional é uma área de pesquisa multidisciplinar
que investiga problemas que fazem parte, ao mesmo tempo, da física e da ciência da
computação. A ciência da computação estuda os algoritmos (seqüência de instruções),
suas aplicações, formalizações matemáticas e suas implementações. Os fundamentos
da ciência da computação e as "ferramentas de computação" surgiram muito antes do
computador moderno, e pode-se dizer que nasceram juntamente com a matemática.
Entretanto a definição formal de algoritmo e computador foi estabelecida apenas no
começo do século XX [115]. Mas foi apenas com a segunda guerra mundial e a

96
necessidade de quebrar códigos de guerra que o desenvolvimento do computador se
acelerou [116]. John Von Neumann e colaboradores desenvolveram as bases do
computador moderno. Publicado em um trabalho de 1945 [117], tal design passou a
ser conhecido por "arquitetura de von Neumann". Ele se caracteriza primariamente
por guardar o programa a ser executado junto com os dados na memória física do
computador. Pode-se afirmar que o uso da simulação computacional para resolver
problemas físicos também se estabeleceu neste período. A solução do problema da
fissão nuclear por amostragem estatística via simulação computacional foi sugerida
em 1946 pelo matemático Stanislaw Ulam, que era do grupo de pesquisa de von
Neumann [118]. (Nascia assim o método de Monte Carlo.)
Hardware
Por mais de uma década a computação científica ficou restrita a grandes
instituições de pesquisa capazes de comprar ou produzir, e manter seus computadores.
Eles eram caros, gigantes e consumiam muita energia. O surgimento dos
semicondutores na década de 1950 possibilitou a substituição das válvulas pelos
transistores. Na década de 1960, os computadores já eram menores, mais rápidos,
mais confiáveis, com menor necessidade de consumo e mais baratos de se produzir.
Em 1965, Gordon Moore (co-fundador da Intel) publicou um trabalho mostrando que
o número de transistores dobrara a cada ano desde o surgimento dos circuitos
integrados em 1958 [119]. Posteriormente a projeção foi corrigida para dois anos, e se
manteve desde então. Na década de 1970, eram comuns circuitos integrados e
microprocessadores, diminuindo ainda mais os custos de produção de computadores.
Na década de 1980, surgiram os computadores pessoais, popularizando seu uso em
casa (desktops). Os computadores pessoais se desenvolveram rapidamente, mas fazer
computação científica ainda era restrito aos mainframes dos grandes centros de
pesquisa até a década de 1990. Em 1994, surgiu o Beowulf, o primeiro cluster
computacional formado de desktops configurado para fazer computação científica
através de processamento distribuído [120]. A partir de 2000, com a popularização
dos processadores de múltiplos núcleos, o conceito de processamento distribuído
passou a ser dominante. Em diversos centros de pesquisa, clusters de baixo custo
rapidamente substituíram caríssimos mainframes que se tornavam obsoletos.
Entretanto, desde 2007 um novo paradigma vem se estabelecendo com o uso das
unidades de processamento gráfico (GPU) para execução massiva de cálculos em

97
paralelo. As GPUs não se comportam como a arquitetura de von Neumann, sendo
capazes de fazer centenas de operações simultâneas (enquanto que a CPU faz apenas
uma operação por vez).
Hoje é comum encontrar clusters híbridos, que utilizam ao mesmo tempo
servidores de alta capacidade e desktops de baixo custo, ambos equipados com placas
gráficas prontas para serem utilizadas por programas de investigação científica.
Entretanto montar tais clusters não é uma tarefa trivial, já que isto demanda
conhecimentos técnicos (de hardware e software) e noções mercadológicas (isto é,
saber o valor real do hardware que se está negociando). Considerando a lei de Moore
como válida, a cada dois anos os computadores dobram de capacidade. Isso implica
que um computador comprado há dois anos vale agora metade do que foi pago. Com
um ano de idade o computador cai a 70% do seu valor. Desta forma é vital que ao
efetuar uma compra, o grupo de pesquisa coloque a máquina em funcionamento o
mais rápido possível. É comum ver grupos de pesquisa gastando mais dinheiro que o
necessário, e por fim obtendo menos recursos computacionais. O atraso causado pela
burocracia ou pela ineficiência das pessoas encarregadas de realizar a instalação e
configuração normalmente são as causas desse prejuízo. Outra consideração crucial
relaciona qualidade/quantidade/preço. Existem diferentes configurações de hardware
no mercado em um largo espectro de capacidades e custos para diferentes aplicações.
É comum encontrar vendedores tentando fornecer configurações inadequadas, ou por
má fé, ou por não compreender como a máquina será utilizada. Este problema só pode
ser contornado através do conhecimento das técnicas numéricas utilizadas nos
programas de simulação e das capacidades dos hardwares oferecidos.
Para ilustrar estes aspectos, é apresentado abaixo o resultado obtido para o
cálculo dos modos vibracionais (absorbância) da vitamina B12, utilizando 2 diferentes
métodos (HF e B3LYP), onde B3LYP foi executado com pseudo potencial
(LANL2DZ para o cobre) e sem pseudo potencial (6-31G* para o cobre). A base dos
outros átomos foi 6-31G, com exceção do nitrogênio que teve 6-31G*.
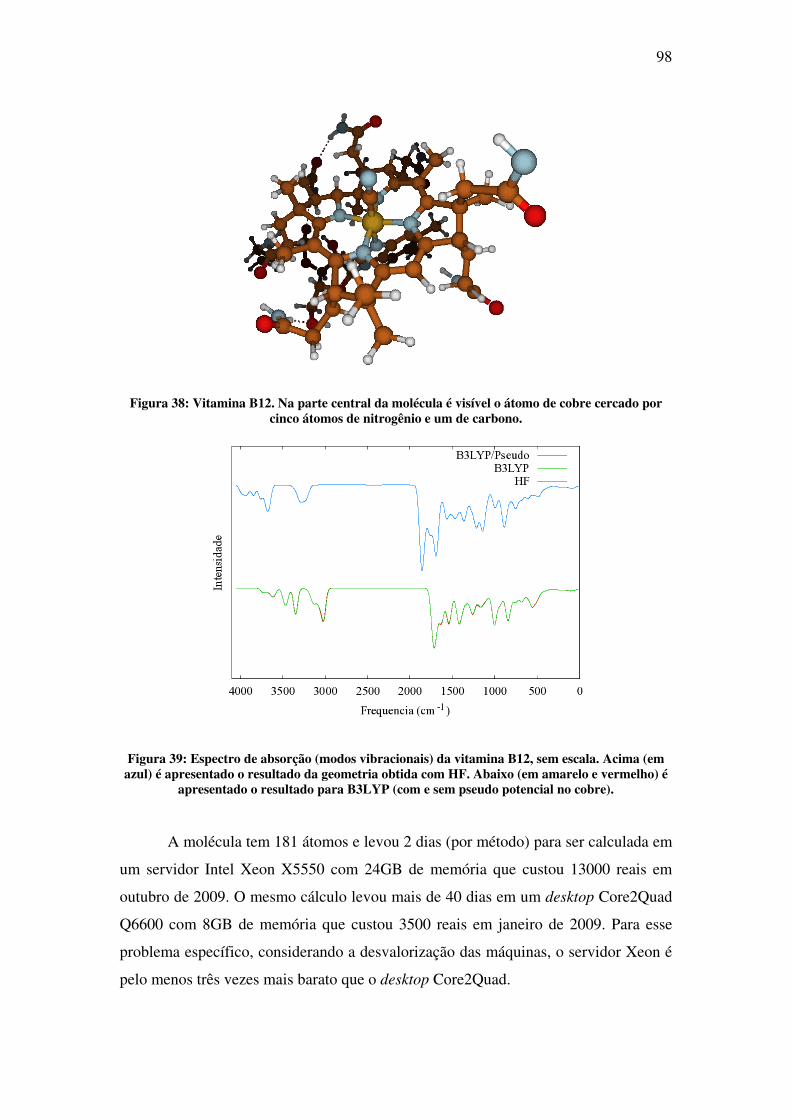
98
Figura 38: Vitamina B12. Na parte central da molécula é visível o átomo de cobre cercado por cinco átomos de nitrogênio e um de carbono.
Figura 39: Espectro de absorção (modos vibracionais) da vitamina B12, sem escala. Acima (em azul) é apresentado o resultado da geometria obtida com HF. Abaixo (em amarelo e vermelho) é
apresentado o resultado para B3LYP (com e sem pseudo potencial no cobre).
A molécula tem 181 átomos e levou 2 dias (por método) para ser calculada em
um servidor Intel Xeon X5550 com 24GB de memória que custou 13000 reais em
outubro de 2009. O mesmo cálculo levou mais de 40 dias em um desktop Core2Quad
Q6600 com 8GB de memória que custou 3500 reais em janeiro de 2009. Para esse
problema específico, considerando a desvalorização das máquinas, o servidor Xeon é
pelo menos três vezes mais barato que o desktop Core2Quad.

99
Linguagens de programação e Compiladores
Durante a década de 1940, os programas de computador eram escritos em
linguagem de máquina, que são executados diretamente pela CPU. No início dos anos
50, surgiu a linguagem assembly. Esta utiliza uma representação simbólica
(expressões mnemônicas) para representar endereços de memória e operações. Um
utilitário chamado assembler traduz as expressões da linguagem assembly para a
linguagem de máquina. Em 1954, surgiu na IBM o FORTRAN (FORmula
TRANslator), a primeira linguagem abstrata e portável, e por isso chamada de "alto
nível" [121]. Para transformar a linguagem de alto nível em linguagem de máquina, é
necessário utilizar um compilador. O compilador diferencia-se do assembler pela
capacidade de interpretar as instruções em sua forma abstrata (de alto nível),
aperfeiçoá-la, e então traduzir para linguagem de máquina. O FORTRAN e os
compiladores se desenvolveram junto com o computador. Ao mesmo tempo surgiram
dezenas de outras linguagens de programação. Entre as décadas de 1960 e 1970
surgiram paradigmas que são utilizados até hoje (programação matricial, funcional,
orientada a objeto, de sistema, lógica, estruturada...) [122]. Em especial, em 1968,
Edsger Dijkstra publicou um artigo condenando o uso da operação GOTO em
qualquer linguagem de alto nível [123]. Durante os anos 80, a programação orientada
a objeto, de sistema e o uso extensivo de módulos consolidaram C (e depois C++)
como a linguagem mais utilizada de todos os tempos. A linguagem C surgiu em 1972
para ser usada em conjunto com o sistema operacional Unix [124]. Ela é utilizada no
kernel do Linux e é a base para linguagens de "altíssimo nível" como Java e
linguagens interpretadas como Perl e Python. O C++ é "C com classes" (orientação a
objeto), e possui também outros aprimoramentos. O C++ é projetado para produzir
códigos mais curtos e organizados que C. O custo disso é que se a classe que
comporta um objeto for muito complexa, o programa em C++ tende a ser menos
eficiente. Evidentemente isso depende da habilidade do programador, já que um
código em C é compilado normalmente pelo compilador de C++. O C ainda oferece
recursos de baixo nível, como operações em bits e uso de código assembly. Isso faz
do C uma linguagem extremamente eficiente e versátil. Entretanto, durante muitos
anos, a linguagem C mostrou-se inferior ao FORTRAN para uso em computação
científica. Isso se devia primariamente à escassez de boas bibliotecas matemáticas
para C. Essa deficiência foi rapidamente eliminada com popularização do Linux e o
uso de softwares livres baseados em C. Hoje, não há diferença no desempenho se um

100
programa é escrito em FORTRAN ou C. E isso é natural, já que ambos serão
compilados (aperfeiçoados e transformados em linguagem de máquina). Existem
inclusive programas que convertem um código em FORTRAN para C (mas não o
contrário, já que FORTRAN não possui recursos de baixo nível e de sistema).
Aparentemente, o compilador atual mais eficiente para códigos de computação
científica na arquitetura x86(-64) é o da Intel. Há outros conhecidos no mercado como
o da Portland (PGI) e o da GNU. Mas o compilador Intel se destaca mesmo em
processadores AMD (a concorrente em fabricação de processadores). Obviamente os
desenvolvedores do compilador conhecem muito bem a estrutura e as instruções da
arquitetura, e são capazes de produzir códigos de máquina altamente eficientes. Em
alguns casos, o compilador Intel produz programas duas vezes mais rápidos que os
outros compiladores.

101
Referências
[1] J.E. Lennard-Jones, "Liquid State", Proc.Phys.Soc.52(1940)729
[2] G.N. Lewis, "The Atom and the Molecule", J.Chem.Am.Soc.38(1916)762
[3] R. Noyori, "Supercritical Fluids: Introduction", Chem.Rev.99(1999)353
[4] J.M. De Simone, "Practical Approaches to Green Solvents", Science297(2002)799
[5] P.E. Savage, "Organic Chemical Reactions in Supercritical Water",
Chem.Rev.99(1999)603
[6] Y.P. Morozova, O.N. Chaikovskaya and O.K. Bazyl, "Influence of Solvents on the
Spectral and Luminescent Properties of Phenol", RussianPhys.J.46(2003)62
[7] T.H. Dunning Jr., "Gaussian basis sets for use in correlated molecular calculations.
I. The atoms boron through neon and hydrogen", J.Chem.Phys.90(1989)1007
[8] N.W. Larsen, "Microwave spectra of the six mono-13C-substituted phenols and of
some monodeuterated species of phenol. Complete substitution structure and absolute
dipole moment", J.Mol.Struct.51(1979)175
[9] S. Hirata, M. Head-Gordon, R.J. Bartlett, "Configuration interaction singles, time-
dependent Hartree-Fock, and time-dependent density functional theory for the
electronic excited states of extended systems", J.Chem.Phys.111(1999)10774; A.
Dreuw, M. Head-Gordon, "Failure of time-dependent density functional theory for
long-range charge-transfer excited states: The zincbacteriochlorin-bacterlochlorin and
bacteriochlorophyll-spheroidene complexes", J.Am.Chem.Soc.126(2004)3007
[10] A.D. Becke, "Correlation energy of an inhomogeneous electron gas: A
coordinate-space model", J.Chem.Phys.88(1988)1053; C. Lee, W. Yang, R.G. Parr,
"Development of the Colle-Salvetti correlation-energy formula into a functional of the
electron density", Phys.Rev.B37(1988)785; B. Miehlich, A. Savin, H. Stoll, H. Preuss,
"Results obtained with the correlation energy density functionals of becke and Lee,
Yang and Parr", Chem.Phys.Lett.157(1989)200
[11] H.Baba e S.Suzuki, "Electronic Spectra and Hydrogen Bonding. I. Phenol and
Naphthols", J.Chem.Phys.35(1961)1118

102
[12] R.C. Barreto, K. Coutinho, H.C. Georg, S. Canuto, "Combined Monte Carlo and
quantum mechanics study of the solvatochromism of phenol in water. The origin of
the blue shift of the lowest pi-pi* transition", Phys.Chem.Chem.Phys.11(2009)1388
[13] F.B. van Duijneveldt, J.G.C.M. van Duijneveldt-van de Rijdt, J.H. van Lenthe,
"State-of-the-art in Counterpoise Theory", Chem.Rev.94(1994)1873.
[14] M. Ito, "Supersonic jet spectroscopy for the study of hydrogen bonding",
J.Mol.Struct.177(1988)173
[15] H. Abe, N. Mikami, M. Ito, "Fluorescence excitation spectra of hydrogen-bonded
phenols in a supersonic free jet", J.Phys.Chem.86(1982)1768
[16] K. Mueller-Dethlefs, O. Dopfer, "ZEKE Spectroscopy of Complexes and
Clusters", Chem.Rev.94(1994)1845
[17] S. Schumm, M. Gerhards, W. Roth, H. Gier, K. Kleinermanns, "A CASSCF
study of the S0 and S1 states of phenol", Chem.Phys.Lett.263(1996)126
[18] H.D. Bist, J.C.D. Brand, D.R. Willians, "The vibrational spectrum and torsion of
phenol", J.Mol.Spect.24(1967)402
[19] E. S. Kryachko, H. Nakatsuji, "Ab Initio Study of Lower Energy Phenol−Water
1≤n≤4 Complexes: Interpretation of Two Distinct Infrared Patterns in Spectra of
Phenol−Water Tetramer", J.Phys.Chem.A106(2002)731
[20] D. Schemmel and M. Schültz, "Phenol-water 1<=n<=3 revisited: An ab initio study
on the photophysics of these clusters at the level of coupled cluster response theory",
J.Chem.Phys.127(2007)174304
[21] K. Fuke, K. Kaya, "Electronic absorption spectra of phenol---(H2O)n and
(phenol)n as studied by the MS MPI method", Chem.Phys.Lett.94(1983)97
[22] R.C. Barreto, S. Canuto, "Characterization and spectroscopy analysis of phenol-
ethanol hydrogen bonded clusters", Chem.Phys.Lett.496(2010)236
[23] R.A. Shaw, H. Wieser, R. Dutler, A. Rauk, "Vibrational Optical Activity of (S)-l-
d-Ethanol", J.Am.Chem.Soc.112(1990)5401
[24] J.C. Pearson, K.V.L.N. Sastry, E. Herbst, F.C. De Lucia, "The Millimeter- and
Submillimeter-Wave Spectrum of Gauche-Ethyl Alcohol",
J.Mol.Spect.175(1996)246; J.C. Pearson, K.V.L.N. Sastry, M. Winnewisser, E.

103
Herbst, F.C. De Lucia, J.Phys.Chem.Ref.Data24(1995)1; A. Maris, W. Caminati, B.
Velino, C.M. Andrews, B.J. Howard, "Free and pulsed jet rotational spectra and van
der Waals motions of ethanol ... argon", Chem.Phys.Lett.399(2004)39
[25] J.H.S. Green, "Thermodynamic Properties of Organic Oxygen Compounds. Part
5 - Ethyl Alcohol", Trans.FaradaySoc.57(1961)2132; G.M. Barrow, "Heat Capacity,
Gas Imperfection, Infrared Spectra and Internal Rotation Barriers of Ethyl Alcohol",
J.Chem.Phys.20(1952)1739; E.K. Plyler, "Infrared Spectra Of Methanol, Ethanol,
And Normal-Propanol", J.Res.Nat.Bur.Stand.48(1952)281
[26] J.C. Pearson, C.S. Brauer, B.J. Drouin, "The asymmetric top–asymmetric frame
internal rotation spectrum of ethyl alcohol", J.Mol.Spect.251(2008)394
[27] E.E. Fileti, P. Chaudhuri, S. Canuto, "Relative strength of hydrogen bond
interaction in alcohol-water complexe", Chem.Phys.Lett.400(2004)494
[28] M. Ehrbrecht, F.J. Huisken, "Vibrational Spectroscopy of Ethanol Molecules and
Complexes Selectively Prepared in the Gas Phase and Adsorbed on Large Argon
Clusters", J.Phys.Chem.A101(1997)7768
[29] R.K. Kakar, C.R. Quade, "Microwave rotational spectrum and internal rotation in
gauche ethyl alcohol", J.Chem.Phys.72(1980)4300
[30] P.J. Krueger, J. Jan, H.J. Wieser, "Infrared spectra: Intramolecular trans lone
pair inter-action with α-CH bonds and the stability of conformers in alcohols and
thiols", J.Mol.Struct.5(1970)375; M. Oki, H. Iwamura,
Bull.Chem.Soc.Japan.32(1959)950.
[31] H. Abe, N. Mikami, M. Ito, "Dispersed fluorescence spectra of hydrogen-bonded
phenols in a supersonic free jet", J.Phys.Chem.86(1982)2567
[32] R.J. Lipert, S.D. Colson, "Persistent Spectral Hole Burning of Molecular Clusters
In a Supersonic Jet", J.Phys.Chem.93(1989)3894
[33] E. Cordes, O. Dopfer, T.G. Wright, K. Muller-Dethlefs, "Vibrational
Spectroscopy of the Phenol-Ethanol Cation", J.Phys.Chem.97(1993)7471.
[34] D. Spangenberg, P. Imhof, W. Roth, Ch. Janzen, K. Kleinermanns, "Phenol-
(Ethanol)1 Isomers Studied by Double-Resonance Spectroscopy and ab Initio
Calculations", J.Phys.Chem.A103(1999)5918

104
[35] R. Rivelino, S. Canuto, "Theoretical Study of Mixed Hydrogen-Bonded
Complexes: H2O···HCN···H2O and H2O···HCN···HCN···H2O",
J.Phys.Chem.A105(2001)11260
[36] J. Sponer, P. Hobza, "MP2 and CCSD(T) study on hydrogen bonding, aromatic
stacking and nonaromatic stacking", Chem.Phys.Lett.267(1997)263
[37] S. Tsuzuki, T. Uchimaru, K. Matsumura, M. Mikami, K. Tanabe, "Effects of the
higher electron correlation correction on the calculated intermolecular interaction
energies of benzene and naphthalene dimers: comparison between MP2 and CCSD(T)
calculations", Chem.Phys.Lett.319(2000)547
[38] K.K. Irikura, R.D. Johnson III, R.N. Kac, "Uncertainties in Scaling Factors for ab
Initio Vibrational Frequencies", J.Phys.Chem.A109(2005)8430
[39] M.E. Dunn, T.M. Evans, K.N. Kirschner, G.C. Shields, " Prediction of Accurate
Anharmonic Experimental Vibrational Frequencies for Water Clusters, (H2O)n, n = 2 -
5", J.Phys.Chem.A.110(2006)303
[40] B. Mennucci, C.O. da Silva, "A quantum mechanical strategy to investigate the
structure of liquids: The cases of acetonitrile, formamide, and their mixture",
J.Phys.Chem.B112(2008)6803
[41] P. Sinha, S.E. Boesch, C. Gu, R.A. Wheeler, A.K. Wilson, "Harmonic
Vibrational Frequencies: Scaling Factors for HF, B3LYP, and MP2 Methods in
Combination with Correlation Consistent Basis Sets", J.Phys.Chem.A108(2004)9213
[42] http://fig.if.usp.br/~barreto/phenol
[43] http://www-public.rz.uni-duesseldorf.de/~pc1/abinitio/
[44] A. Novak, "Hydrogen bonding in solids correlation of spectroscopic and
crystallographic data", Struct.Bond.18(1974)177
[45] C.M. Breneman, K.B. Wiberg, "Determining atom-centered monopoles from
molecular electrostatic potentials. The need for high sampling density in formamide
conformational analysis", J.Comput.Chem.11(1990)361
[46] R.C. Guedes, K. Coutinho, B.J. Costa Cabral, S. Canuto, C.F. Correia, R.M.
Borges dos Santos, and J.A. Martinho Simões, "Solvent Effects on the Energetics of
the Phenol O−H Bond: Differential Solvation of Phenol and Phenoxy Radical in

105
Benzene and Acetonitrile", J.Phys.Chem.A107(2003)9197; W.L. Jorgensen, T.B.
Nguyen, "Monte Carlo simulations of the hydration of substituted benzenes with
OPLS potential functions ", J.Comput.Chem.14(1993)195
[47] H.J.C. Berendsen, J.R. Grigera, T.P. Straatsma, "The missing term in effective
pair potentials", J.Phys.Chem.91(1987)6269
[48] K. Coutinho, H.C. Georg, T. Fonseca, V. Ludwig, S. Canuto, "An efficient
statistically converged average configuration for solvent effects",
Chem.Phys.Lett.437(2007)148
[49] M.L. Sanchez, M.A. Aguilar, F.J. Olivares Del Valle, "Study of solvent effects
by means of averaged solvent electrostatic potentials obtained from molecular
dynamics data", J.Comput.Chem.18(1997)313; M.L. Sanchez Mendoza, M.A.
Aguilar, F.J. Olivares Del Valle, "A mean field approach that combines quantum
mechanics and molecular dynamics simulation: the water molecule in liquid water",
J.Mol.Struct.:Theochem426(1998)181; M.E. Martin, M.L. Sanchez, F.J. Olivares Del
Valle, M.A. Aguilar, "A multiconfiguration self-consistent field/molecular dynamics
study of the (n→π*)1 transition of carbonyl compounds in liquid water",
J.Chem.Phys.113(2000)6308
[50] TINKER 4.2 (2004), "Software Tools for Molecular Design",
http://dasher.wustl.edu/tinker/
[51] P. Ren, J.W. Ponder, "A Consistent Treatment of Inter- and Intramolecular
Polarization in Molecular Mechanics Calculations", J.Comput.Chem.23(2002)1497;
P. Ren, J. W. Ponder, "Polarizable Atomic Multipole Water Model for Molecular
Mechanics Simulation", J.Phys.Chem.B107(2003)5933
[52] L. Saiz, J.A. Padró, E. Guàrdia, "Structure and Dynamics of Liquid Ethanol",
J.Phys.Chem.B101(1997)78
[53] D.R. Lide, Handbook of Chemistry and Physics, CRC Press, 1993
[54] Y. Kataoka, "Studies of liquid water by computer simulations. V. Equation of
state of fluid water with Carravetta–Clementi potential", J.Chem.Phys.87(1987)589;
R.D. Mountain, "Molecular dynamics investigation of expanded water at elevated
temperatures", J.Chem.Phys.90(1989)1866

106
[55] P. Postorino, R.H. Tromp, M-A. Ricci, A. K. Soper, G. W. Neilson, "The
interatomic structure of water at supercritical temperatures", Nature366(1993)668;
A.K. Soper, F. Bruni, M.A. Ricci, "Site-site pair correlation functions of water from
25 to 400 °C: Revised analysis of new and old diffraction data",
J.Chem.Phys.106(1997)247; T. Tassaing, M-C. Bellissent-Funel, B. Guillot, Y.
Guissani, "The partial pair correlation functions of dense supercritical water",
Europhys.Lett.42(1998)265
[56] B. Guillot, "A reappraisal of what we have learnt during three decades of
computer simulations on water", J.Mol.Liq.101(2002)219
[57] R. Scipioni, D.A. Schmidt, M. Boero, "A first principles investigation of water
dipole moment in a defective continuous hydrogen bond network",
J.Chem.Phys.130(2009)024502; M. Guidon, F. Schiffmann, J. Hutter, J.
VandeVondele, "Ab initio molecular dynamics using hybrid density functionals",
J.Chem.Phys.128(2008)214104, P.L. Silvestrelli, M. Parrinello, "Structural,
electronic, and bonding properties of liquid water from first principles",
J.Chem.Phys.111(1999)3572
[58] N. L. Doltsinis, M. Burchard, W. V. Maresch, A. D. Boese, T. Fockenberg, "Ab
initio molecular dynamics study of dissolved Sio2 in supercritical water",
J.Theo.Comput.Chem.6(2007)49; B.S. Mallik, A.Chandra, "Vibrational Spectral
Diffusion in Supercritical D2O from First Principles: An Interplay between the
Dynamics of Hydrogen Bonds, Dangling OD Groups, and Inertial Rotation",
J.Phys.Chem.A112(2008)13518; R. Scipioni, D.A. Schmidt, M. Boero, "A first
principles investigation of water dipole moment in a defective continuous hydrogen
bond network", J.Chem.Phys.130(2009)024502;
[59] T.L. Fonseca, K. Coutinho, S. Canuto, "Probing supercritical water with the n-
pi(*) transition of acetone: A Monte Carlo/quantum mechanics study",
J.Chem.Phys.126(2007)034508 ; M.S. Skaf, D. Laria, "Dielectric relaxation of
supercritical water: Computer simulations", J.Chem.Phys.113(2000)3499; A.A.
Chialvo, P.T. Cummings, "Hydrogen bonding in supercritical water",
J.Chem.Phys.101(1994)4466

107
[60] Y. Guissani, B. Guillot, "A computer simulation study of the liquid-vapor
coexistence curve of water", J.Chem.Phys.98(1993)8221
[61] P. Ren, J.W. Ponder, "Temperature and Pressure Dependence of the AMOEBA
Water Model", J.Phys.Chem.B108(2004)13427
[62] T.D. Rasmussen, P. Ren, J.W. Ponder, F. Jensen, "Force Field Modeling of
Conformational Energies: Importance of Multipole Moments and Intramolecular
Polarization", Int.J.QuantumChem.107(2007)1390
[63] A. Grossfield, P. Ren, J.W. Ponder, "Ion solvation thermodynamics from
simulation with a polarizable force field", J.Am.Chem.Soc.125(2003)15671; J.P.
Piquemal, L. Perera, G.A. Cisneros, P. Ren, L.G. Pedersen and T.A. Darden,
"Towards accurate solvation dynamics of divalent cations in water using the
polarizable amoeba force field: from energetics to structure",
J.Chem.Phys.125(2006)054511
[64] H.C. Andersen, "RATTLE: A "Velocity" Version of the SHAKE Algorithm for
Molecular Dynamics Calculations", J.Comp.Phys.52(1983)24
[65] T. Williams, C. Kelley, GNUPLOT: Famous scientific plotting package, 2004,
http://sourceforge.net/projects/gnuplot
[66] S.R.A. Salinas, Introdução à Física Estatística, Edusp, 1999
[67] J.E. Lennard-Jones, "Cohesion", Proc.Phys.Soc.43(1931)461
[68] N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, E. Teller,
"Equations of State Calculations by Fast Computing Machines",
J.Chem.Phys.21(1953)1087
[69] C.M. Grinstead, J.L. Snell, Introduction to Probability, American Mathematical
Society, 2006; http://www.dartmouth.edu/~chance/
[70] W.K. Hastings, "Monte Carlo Sampling Methods Using Markov Chains and
Their Applications", Biometrika57(1970)97
[71] B.J. Alder, T.E. Wainwright, "Phase Transition for a Hard Sphere System",
J.Chem.Phys.27(1957)1208

108
[72] A. Rahman, "Correlation in the Motion of Atoms in Liquid Argon",
Phys.Rev.136(1964)A405
[73] L. Verlet, "Computer experiments on classical fluids. I. Thermodynamical
properties of Lennard-Jones fluids", Phys.Rev.159(1967)98
[74] H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. DiNola, J.R. Haak,
"Molecular Dynamics with Coupling to an External Bath",
J.Chem.Phys.81(1984)3684
[75] M.P. Allen, D.J. Tildesley, Computer Simulation of Liquids, Oxford University
Press, Oxford, 1987; D.C. Rapaport, The Art of Molecular Dynamics Simulation,
Cambridge University Press, Cambridge, 1995; D. Frenkel, B. Smit, Understanding
Molecular Simulation, Academic Press, 2002
[76] E. Schrödinger, "An Undulatory Theory of the Mechanics of Atoms and
Molecules", Phys.Rev.28(1926)1049; E. Schrödinger, "Quantisierung als
Eigenwertproblem", Ann.Phys.79(1926)361
[77] M. Born, "On Quantum Mechanics II", Z.Phys.35(1926)557
[78] P.A.M. Dirac, "Quantum Mechanics of Many-Electron Systems",
Proc.Roy.Soc.A123(1929)714
[79] W.H. Press, S.A. Teukolsky, W.T. Vetterling, B.P. Flannery, Numerical Recipes
in C: The art of scientific computing, Cambridge, 1992
[80] M. Born, R. Oppenheimer, "Quantum theory of the molecules",
Ann.Phys.389(1927)457
[81] D.R. Hartree, "The Wave Mechanics of an Atom with a Non-Coulomb Central
Field", Proc.Camb.Phil.Soc.24(1928)89.
[82] J.C. Slater, "The Self Consistent Field and the Structure of Atoms",
Phys.Rev.32(1928)339
[83] J.C. Slater, "The Theory of Complex Spectra", Phys.Rev.34(1929)1293
[84] V. Fock, "Näherungsmethoden zur Lösung des Quantenmechanischen
Mehrkörperproblems", Z.Phys.61(1930)126

109
[85] W. Heitler and F. London, "Wechselwirkung neutraler Atome und homöopolare
Bindung nach der Quantenmechanik", Zeits.Phys.44(1927)455
[86] J.E. Lennard-Jones, "The Electronic Structure of Some Diatomic Molecules",
Trans.FaradaySoc.25(1929)668
[87] G.G. Hall, "The Lennard-Jones paper of 1929 and the foundations of molecular
orbital theory", Adv.Quant.Chem.22(1991)1
[88] J.C. Slater, "Analytic Atomic Wave Functions", Phys. Rev. 42, 33-43 (1932)
[89] S.F. Boys, "Electronic wavefunctions. I. A general method of calculation for
stationary states of any molecular system", Proc.Roy.Soc.A200(1950)542
[90] C.C.J. Roothaan, "New Developments in Molecular Orbital Theory",
Rev.Mod.Phys.23(1951)69
[91] G.G. Hall, "The molecular orbital theory of chemical valency VIII. A method of
calculating ionization potentials", Proc.Roy.Soc.A205(1951)541
[92] P. Hohenberg, W. Kohn, "Inhomogeneous Electron Gas",
Phys.Rev.136(1964)B864
[93] W. Kohn, L.J. Sham, "Self-Consistent Equations Including Exchange and
Correlation Effects", Phys.Rev.140(1965)A1133
[94] P.O. Lowdin, "Correlation Problem In Many-Electron Quantum Mechanics .1.
Review Of Different Approaches And Discussion Of Some Current Ideas",
Adv.Chem.Phys.2(1959)207
[95] J.D.M. Vianna, A. Fazzio, S. Canuto, Teoria Quântica de Moléculas e Sólidos,
Livraria da Física, 2004
[96] Chr. Møller, M.S. Plesset, "Note on an Approximation Treatment for Many-
Electron Systems", Phys.Rev.46(1934)618
[97] M. Head-Gordon, R.J. Rico, M. Oumi, T.J. Lee, "A doubles correction to
electronic excited states from configuration interaction in the space of single
substitutions", Chem.Phys.Lett.219(1994)21
[98] J.B. Foreman, M. Head-Gordon, J.A. Pople, M.J. Frisch, "Toward a Systematic
Molecular Orbital Theory for Excited States", J.Phys.Chem.96(1992)135

110
[99] J.D. Jackson, Classical Electrodynamics, Wiley, 1999
[100] J.D. van der Waals, Over de Continuïteit van den Gasen Vloeistoftoestand,
Thesis, Leiden, 1873
[101] J.E. Lennard-Jones, "On the Determination of Molecular Fields. II From the
Equation of State of a Gas", Proc.Roy.Soc.A106(1924)463
[102] J.C. Slater, "The Normal State of Helium", Phys.Rev.32(1928)349
[103] S.C. Wang, Phys.Zeits.28(1927)663
[104] J.C. Slater, J.G. Kirkwood, "The van der Waals Forces in Gases",
Phys.Rev.37(1931)682
[105] R. Eisenschitz, F. London, "The relation between the van der Waals forces and
the homopolar valence forces", Zeits.Phys.60(1930)491
[106] F. London, "Theory and systematics of molecular forces",
Zeits.Phys.63(1930)245
[107] S. Canuto, K. Coutinho, "Solvent effects from a sequential Monte Carlo -
Quantum mechanical approach" Adv.QuantumChem.28(1997)89
[108] K. Coutinho, R.C. Guedes, B.J. Costa Cabral, S. Canuto, "Electronic
polarization of liquid water: converged Monte Carlo-quantum mechanics results for
the multipole moments", Chem.Phys.Let.369(2003)345; R.C. Guedes, K. Coutinho,
B.J. Costa Cabral, S. Canuto, "Differential hydration of phenol and phenoxy radical
and the energetics of the phenol O-H bond in solution",
J.Phys.Chem.B107(2003)4304; K. Coutinho, S. Canuto, "The sequential Monte Carlo-
quantum Mechanics Methodology. Application to the Solvent Effects in the Stokes
Shift of Acetone in Water", J.Mol.Struct.(Theochem)632(2003)235
[109] J. Gao, X. Xia, "A priori evaluation of aqueous polarization effects through
Monte Carlo QM-MM simulations", Science258(1992)631
[110] H.C. Georg, K. Coutinho, S. Canuto, "Converged electronic polarization of
acetone in liquid water and the role in the n-pi* transition",
Chem.Phys.Lett.429(2006)119; H.C. Georg, K. Coutinho, S. Canuto, "Solvent effects
on the UV-visible absorption spectrum of benzophenone in water: A combined Monte

111
Carlo quantum mechanics study including solute polarization",
J.Chem.Phys.126(2007)034507
[111] B.T. Thole, "Molecular polarizabilities calculated with a modified dipole
interaction", Chem.Phys.59(1981)341
[112] A.J. Stone, "Distributed Multipole Analysis, or How to Describe a Molecular
Charge-Distribution", Chem.Phys.Lett.83(1981)233; A.J. Stone, "Distributed
Multipole Analysis - Methods And Applications", Mol.Phys.56(1985)1047; A.J.
Stone, "Distributed Polarizabilities", Mol.Phys.56(1985)1065;
[113] L.X. Dang, T.M. Chang, J.Chem.Phys.106(1997)8149
[114] C.J. Burnham, S.S. Xantheas, J.Chem.Phys.116(2002)1500
[115] K. Gödel, "On formally undecidable propositions of Principia Mathematica and
related systems I", Mon.Math.Phys.38(1931)173; A.M. Turing, "On Computable
Numbers, with an Application to the Entscheidungsproblem",
Proc.Lond.Math.Soc.42(1936)230;
[116] N. Metropolis, J. Howlett, G. Rota, "A history of computing in the twentieth
century", Academic Press, 1980
[117] J. von Neumann, "First Draft of a Report on the EDVAC", University of
Pennsylvania, 1945
[118] N. Metropolis, "The Beginning Of The Monte Carlo Method", Los Alamos
Science, Special Issue, 1987
[119] G.E. Moore, "Cramming more components onto integrated circuits",
Electronics38(1965)
[120] http://www.ibiblio.org/pub/linux/docs/HOWTO/archive/Beowulf-
HOWTO.html
[121] J.W. Backus, H. Stern, I. Ziller, R.A. Hughes, R. Nutt, R.J. Beeber, S. Best, R.
Goldberg, L.M. Haibt, H.L. Herrick, R.A. Nelson, D. Sayre, P.B. Sheridan, "The
FORTRAN Automatic Coding System". Western joint computer conference:
Techniques for reliability (Los Angeles, California: Institute of Radio Engineers,
American Institute of Electrical Engineers, ACM), 1957, pg. 188

112
[122] R.L. Wexelblat, "History of Programming Languages", Academic Press, 1981
[123] E.W. Dijkstra, "Go To Statement Considered Harmful",
Comm.ACM11(1968)147
[124] B.W. Kernighan, D.M. Ritchie, "The C Programming Language", Prentice Hall,
1978