e zinco a níouel, sobre enxofre cobalto, de...
TRANSCRIPT

EFEITOS
DIOXIDOREDUÇAO
AUG USTO CAMARA NEIVA
Engenheiro Metaturgista, Escota politécnica
da Universidade de São pauto, 1976
DE COBALTO, NíOUEL, ZINCO E
DE ENXOFRE SOBRE A ELETRO-DE MANGANÊS
Dissertação apresentada à Escola politécnicada Universidade de São Paulo para a obtençãodo Título de Mestre em Engenharia
Orientador: Prof. Dr. Eduardo Barchese
Professor Assistente Doutor do Departamentode Engenharia Metalúrgica da .Escota politéc-nica da Universidade de São Paulo
São Paulo, 1984
..%

AGRADECIMENTOS
Ao Prof. Eduardo Barehese, orientador, pelo interesse, entu-
siasmo e paciência sempre presentes durante todo este trabalho.
Ao Prof. Tharcisio Damy de Souza Santos, ao Prof. Renato Rocha
Vieira e ao Prof. Carlos Dias Brosch, pelo incentivo constante e pelas faci-
lidades oferecidas no desenvolvimento do trabalho experimental no Departa-
mento de Engenharia Metalirgica da Escola Politécnica da Universidade de São
Paulo.
Ao Prof. José Deodoro Trani Capocchi e ao Prof. Cyro Takano pelas
discussões e pela solicítude.
Ao Prof. Stephan Wolynec e ao EngQ D. K. Tanaka, do Instituto de
Pesquisas Tecnol6gicas, pelas facilidades oferecidas em seu laboratório.
Ao Prof. tt6tio Chagas, do Instituto de QuÍmica da Universidade
de São Pau1o, pelas facilidades oferecidas em seu laborat6rio.
Ao Prof. José Carlos DtAbreu e ao Prof. Maurício Leonardo Torem,
do Departamento de Ciôncia dos MaËeriaís e Metalurgia da Pontifícia Univer-
sidade Catõlicado Rio de Janeiror.péla gentileza d.emonstrada em minha vísita a seu laboratério.
Ao Prof. Andrã Paulo Tschiptschin e ao Prof. nétio Goldenstein,
pelo apoio constante e pelo auxí1io na utilização do microsc6pio eletrônico
de varredura.

I]
Aos colegas EngQ Ramiro da Conceição Nascímento, EngQ João Vi-
cente Carchedi Roxo, EngQ Nei Freitas de Quadros, EngQ Celso PonteseEngg
Antônio Augusto Marialva Neto pela amizade, pelo apoio e pelo auxí1ío,
sob diferenÈes formas, oferecidos na execuçã<, e conclusão deste trabalho.
Às colegas BeI. QuÍmica Ana Lúcia Exner Godoy, Ana Lúcia Rodri
gues de Paula e Ana Maria Coutinho de Souza pela amizade e pelo auxílio
nas etapas de impressão e apresentação deste trabalho.
Ao Prof, Francisco Ambrozío Filho e ao Prof" C6tio Xavier, do
InsËítuto de Pesquisas Energãticas e Nucleares, pelo incentivo e pelas
facilidades oferecidas ã conclusão do trabalho"
Aos T6cnicos MeËalurgisËas Ayr:ton Mazzvcato Leal e Regínaldo
Mariano, pelo freqtlente auxÍ1io durante a execução das experíências e
dos servíços fotográficos.
Ao T6cnico Mecânico Ricardo I'latanabe, pelo auxílío na execu-
ção de desenhos.
A todos os colegas e colaboradores do Departamento de Engenha
ria Metalúrgíca da Escola Polit6cníca da Universidade de São Paulo e do
DeparLamento de Materiais Metãlicos do InstiËuto de Pesquisas Energ6ti-
cas e Nucleares que de alguma forma contribuíram para que este trabalho
fosse concluído.
A meus pais, pelo estímulo e colaboração.
A Nena, pela colaboração e apoio em todas as etapas deste
trabalho, a Jûlia, Tânia e Sara pela compreensão diante do que thes im-
pôs sua execução"

III
RESI]MO
Diferentes adições de dióxido de enxofre foram combinadas com dife
rentes adições de cobalto, níquel e zinco para se estudar sua influência se
bre o processo de eletro-redução de manganês em soluções aquosas de sulfatos
de manganês e de amônio
Com os Ërinta eleÈrólitos assim obtidos, foram medidos o potencíal
de cat.odo e a eficiência de correnËe em diferentes densidades de corrente.
Avaliou-se o efeito das adições sobre a eficiência de corrente. Traçaram-se
curvas de polarização parciais das reações cat6dicas de deposíção do manga-
nês e desprendimento de-hidrogênio. Por meio del-as, interpretou-se o efeito
das adições sobre a eficiência de corr:ente e propôs-se um modelo de mecanis-
no de atuação das impurezas e do di6xido de enxofre, baseado na formação de
pares galvânicos impureza*metal, na variação da sobreLensão de hidrogênio,
na ocupação de locais ativos do dep6sito pelo enxofre, etc.
ComplemenËarmente, foram obtidas fotomicrografías do processo de de
posiçãor por microscopia óptica, e do dep6sito, por microscopia de varredura,
verificando-se que o dep6sito consiste de um substrato recoberËo por n6dulos
facetados (aglomerados ou não), que o desprendimento de hidrogênio 6 mais in
tenso sobre o substrato que sobre os nódulos, que as adições afetam a morfo
logía do dep6síto, etc.

IV
ABSTRACT
Different sulfur dioxide additions were cornbined with different co-
ba1t, nickel and zinc additions to study their influence on manganese el.ectro
winning in water solutions of manganese and ammonium sulphate.
I'üith the thirty electrolytes so obtained, the cathode potential and
the current efficiency with different current densities were measured. The
effect of the additions on the current efficiency r^ras evaluated. partial
polarization curves of the cathodic react.ions of manganese deposition and
hydrogen evolution were plotted" I^Iith their aid, the effect of the additions
on the current efficiency \,/as interpreted and a model of the acting mecha-
nism of the impurities and of the sulfur dioxide, based on the formation ofimpurity-rnetal galvanic pairs, on the variation of the hydrogen overpoten-
tial, on the occupation of actíve deposit sites by sulfur etc. \nras proposed"
As a complement, photomicrographs of the deposition process ürere
obtained by optical microscopy and of the deposit by scanning electron mi-
croscopy' whereby it r¿as found that the deposít consists in a substract co-
vered by faceted nodules (agglomerated or not), that the hydrogen evoluËion
lüas stronger on the substract than on the nodules, and that the additions
affect the morphology of the deposit.

V
suuÃnro
Agradecímentos
Resumo
Abs Ëract
Sumãrío
Notação
Lista de figuras
Lista de tabelas
1. OBJETIVO
2" CARACTERISTICAS E USOS DO MANGANÊS ¡LETNOLÍTTCO
3 " pRopu-çÃo rNpusTRTAJ, pE* MANGAJIÊS ELETROLÍTTCO
3"1 HTSTõRIA E PRODUçÃO ATUAL
3.2 PROCESSO CONSAGRADO DE PRODUçÃO IN MANGANES ELETROLÍTICO
3.3 CARACTERÍSTTCAS DA ETAPA ELETROLÍTTCA
3.3.1 Eficiência de corrente e consumo energético3 "3.2 Por que solução aquosa
3.3.3 Por que o ânion sulfato3,3.4 Papel do diafragma e do amônio
3.3.5 Por que anodo de liga chumbo-prata3.3.6 Por que catodo de aço inoxidãvel
3.4 VARIÃVEIS OPERACIONAIS DA ETAPA ELETROLÍICA
3.4"T Densidade de corrente catódica e tenõao apricada ã
3.4"2 Densidade de corrente anódica3"4"3 Concentração de sulfato de amônio3"4.t+ concentrção de Mn2*, pH e vazão do eletrólito3,4.5 Temperatura
c6lu1a
I
III
TV
V
VII ]
X
XIIT
l_
2
4
4
5
B
B
B
9
10
10
L2
L2
T2
13
13
13
L4

3.5 EFEITO DAS IMPUREZAS
3.6 PAPEL DO DIõXIDO DE ENXOFRE
4. ESTUDOS SOBRE MECAN.ISMOS
4.1 DESPRENDIMENTO CATõDICO DE HTDROGÉNIO
4.I.L Desprendímenro de hidrogênio a partir de HrO e Hr0+4.L"2 Desprendímento de hidrogênio a partír de Íons amônio4"L.3 Redução química de hidrogênio por íons Mn"o+ o.., ãto*o"
"rrd4.L"4 Efeíto de impurezas sobre o desprendimento de hidrogêniosobre manganês
4,L.5 Efeito de enxofre, selênio e telúrio sobre o desprendimentode hidrogênio sobí" *r"tr"ê; -------
4.2 DEPOSIçÃO CATõDICA DE MANGANÊS AcoMPANHADA DE DESPRENDIMENTO DEHIDROGÊNIO
4,2.r Estudos do rnstituto de Tecnología QuÍrnica de Dneprope-trowsk
Estudo de Barchese
Estudos do rnstituto de QuÍmica Geral e rnorgânica de KievEstudos do U. S. Bureau of MinesEstudos de Dhananjayan
Estudos de Fekete
Estudos de Agladze e Legran
Estudos de Tilak, Rajogopalan e Reddy
EsËudos de Radhakrishnamurty, Sathyanarayama e ReddyDOS MODELOS APRESENTADOS
Informaçoes e hipóteses mais consensuaisOutras hip6teses e informações importantesContradições entre diferentes modelos
5. s E METõDos
5. 1 PROGRAMA EXPERI}IENTAL
5.2 EQUIPAMENTOS
5.2.T Cãlula e complementos
5.2.2 Microscopia 6ptica para observação de deposição5.2-3 'Mícroscopia eretrônica para observação do depósito5.2.4 InsËrumentação e circuito e16trico
5.3 PREPARAçÃO DOS ELETRõlrros
5.4 DETERMINAçÃO DOS POTENCIAIS DE ELETRODO
5.5 DETERMTNAçÃO DAS EFrC]ÊUCrns DE CORRENTE
5"6 FOTOMICROGRAFTAS DA DEPOSIÇÃO
4.3
VI
15
L7
20
20
20
2L
23
25
2B
29
29
36
40
42
42
43
44
45
46
49
49
50
51
52
52
55
65
67
67
70
7T
73
79

VII
5.7 FOTOMICROGRAFIAS DO DEPÓSITO
6. RESULTADOS EXPERI}MNTAIS
6.1 EFrcrÊncras DE coRRENTE E porENcrArs DE cATODo
6.1.1 Reproduribilidade e precisão6.r.2 Resultados e cálculos completos para o eletrólito ng 30
6.1.3 Eficíência de corrente em função da densidade de correntee do potencial para Ëodos os eletrólitos
6.2 CURVAS DE POLARIZAçÃO
6.3 OBSERVAçÂO DA DEPOSTçÃO n DO DEPõSrTO
6.3.1 Seqtlência de deposição6,3.2 Observação do dep6sito
DISCUSSÃO
7.1 EFETTO DAS ADrçõES SOBRE A EFIC]ÊNCrA DE CORRINTE
7.I.1 Eficiêncía de corrente em função de densidade de corrente7.L"2 Efeito do cobalto sobre a eficiência de corrente7.r.3 Efeito do níquel sobre a eficiôncia de correnre7.I.4 Efeíto do zinco sobre a eficiência de corrente7.L,5 Efeito do di6xido de enxofre sobre a eficiência de
corrente7.2 MECANISMOS DE ATUAçÃO DAS ADIçõES
7.2.1 Resumo dos efeitos das adições7,2.2 Outras premissas para estabelecer o modelo7 .2.3 Mecanismo proposto
CONCLUSõES
9. REFERÊNCIAS åIBLIOGRÃFICAS
BO
B1
81
81
85
B.
90
93
105
105
108
TL4
LT4
LT4
LT6
L22
L25
130
138
138
L39
L42
147
rs4

VIII
NOTAç40
A - -area exposta do eletrodo de trabalho (A=1r03cm2¡
B - relação i""..""/id..de - eoncentração
e - elétronE - potencial de eletrodo reversível_oE- - potencial de eletrodo-padrao
EC - eficiência de correnteECM - eficiência de corrente mãxima (valor máximo da eficiência de correnËe
em função da densidade de corrente, para um dado eletr6lito)ECS - eletrodo de calomelano saturado
F - constante de Faraday (F=96 496 C.*o1-1 .r-I)H - hidroeânio adsorvido
act
I - corrente que atravessa a c6l-u1a
í-- * densidade de corrente de corrosão eletroquírnicacei- - densidade de corrente equivalenËe ä corrosão norrnalcqi¿ - densidade de corrente cat6dica totaliUC* * densídade de corrente em que ocorre a eficiôncia de corrente mãxima
i,, - densidade de corrente limite (da curva de polarização do mar,rganês)I].mi,, - densidade de corrente parcial do hidrogênio
LI
irr, - densidade de corrent.e parcial do manganêst -1 --1k - constante \r, "^/F .z (k=L,06g "-cm'.h '.A ')
1
\r" - massa atômica relativa do manganãs (çrr=Sa,939.mo1--r)
tt"lr, - massa do depõsito de manganôs
Mn, - manqanâs adsorvidoadP¿ - poËencial de catodo (durante a etapa de deposição)
PSCI,' - potencial de catodo em que ocorre a eficiência de corrente mãxima

ÏX
r - fator químico/eletroquímico (r=1 para etapas eletroquímicas)t- - duração da corrosão eletroquímicacet - duracão da corrosão normalcqrËr - duração da deposíção
z - número de oxidação do manganês na solução (z=2)
ã - coeficiente de transferência da reação anódica
È - coeficiente de rransferência da reação cat6dica
ß * fator de simetria
í * nümero de etapas anteriores ã etapa lenta
i - nümero de etapas posteriores ã etapa lentav - nimero de vezes que ocorre a etapa lenta ïìo processo total

X
LISTA DE FIGURAS
Fluxograma da Usina de Boulder City, do U. S. Bureau of Mines
Diagrama de equilíbrio potencial - pH do sistema manganês a 25oC
Curvas polarográficas de desprendimento de hidrogênio sobre manganês
Efeito dos íons amônio sobre as curvas polarográficas de desprendi-mento de hidrogênio sobre manganês
Representação esquemãtica das curvas de polari zação parciaís de de-posição de manganês e desprendímento de hidrogênio
Efeito de aditivos sobre a sobretensão de hidrogênio sobre manganês
Fotografia da cé1ula desmontada
C61u1a (vista parcíal e cortes longitudinais)
c61u1a (seções transversais)
c61u1a (legenda)
Fotografia da cé1u1a com seu sistema de circulação do eletróliroCirculação de elerrõlito e de ãgua de circulação
Fbtografia da cólu1a e do microsc6pio 6ptíco em posição de observaçao de uma deposição
Observação e regisËro fotográfico da deposição
CircuiËo el6trico
Fotografias do arranjo experimental
comparação enËre dois m6todos de medida da eficiêncía de corrente
Potencial de catodo em função do tempo, em deposição seguida decorrosão eletroquímica
Determinaçãg da^eficiência de corrente pela extrapolação de B at6oetxot'=0ce
6.4 Potencial de catodo em função do tempo, em deposição seguida decorrosão normal
3.1
3.2
4.r
4.2
4.3
4.4
5.1
5,2a
5.2b
5.2c
5.3
5.4
5.5
5.6
5.7
5.8
6.1
6.2
6.3
7
11
24
24
26
26
56
57
5B
59
63
64
65
66
6B
69
B4
B7
BB
B9

6.5 Eficiência de corrente em função da densidade de corrente
6.6 Curvas de polarização dos eletrólitos sem adição de imputezas
6.7 Curvas de polarizaçã.o dos eletrflitos com 0 r25 mg.dr-3 Co
6.8 Curvas de polarização dos eletrõlitos com 0r5 mg.dr-3 Co
6,g Curvas de polarízação dos eletr6litos com 1r0 mg.dr-3 Co
6.10 Curvas de'polarização dos eletrólitos com 0r5 mg.¿*-3 Ni
6.11 Curvas de poLarízação dos eletrólitos com 1,0 mg.¿r-3 tli
6.L2 Curvas de poLarização dos eletrólitos com 2r0 mg"dr-3 }{i
6.13 Curvas de polarízação dos eletrólitos com 10 mg.dr-3 zn
6.14 Curvas de poLarízação dos eletrólitos com 20 mg.d^-3 Zn
6.15 Curvas de polarízação dos eletrólitos com 40 mg.dr-3 Zo
6.16 Fotomicrografias da deposição com o eletrólito nQ 1 (microscópioóptico acopl-ado ã c61u1a, aumento 25x)
6.17 Fotomicrografias da deposição com o eletró1ito n9 19 (microsc6pioóptico acoplado ã cãlula, aumento 25x)
6.18 Fotomícrografías da camada inicial com eletrólitos sem adição deimpurezas
6.19 Fotomicrografias da camada inicial com eletr6litos com adiçoesde cobalto e de níque1
XI
9L
95
96
97
9B
99
100
101
LO2
103
104
106
t07
109
110
110
111
TL2
113
113
116
IL7
118
T2T
6.20
6.2L
6,22
6.23
6.24
7.r
7.2
Fotomicrografia de depósito sem diferenciação da camada inicial,com e1etr61íto com adição de zinco
Fotomicrografias de trinca na camada inicial
Fotomicrografias da camada superior
Fotomicrografias de glóbulos com faceËas lisas
Fotomicrograf.ía de dep6sito com facetas irregulares, com eletr6-1ítos corn adição de zinco
Efeito das adições de cobalto sobre ECM, i'a, " PgC¡.t
Efeitos das adições de cobalto sobre as cuïvas de polari zação parciais, em eletrãtito com 0,1 g.dm-3 de diõxido de enxofre
Efeito da adição de cobalto sobre as densid.ades de corrente parciais
Efeitos das adições de cobalto sobrg as curvas de polarização parciais, em e1eËré1ito" com 0 16 g dm-3 de dióxido de enxofre
7.3
7.4

7.5 Efeíto das adições de níquel sobre ECM, t'a* . pnCU
7.6 Efeitos das adições de nÍquel sobre aslcurvas de polarizaçãoparciais, em eletrólitos com 0rl g.dm-' de di6xido d.e enxofre
7.7 Efeitos das adições de nÍquel sobre asecurvas de polarizaçãoparciais, em eletr6litos com 016 E.dm'de dióxido de enxofre
7,8 Efeito das adições de zinco sobre ECM, inCU u pUCU
7.9 Efeito das adições de zínco sobre as curvas de polarização par-ciais, em eletr6litos com 0,1 g.drnj3 de dióxidã du un*ófre
7.10 Efeito das adições de zinco sobre at curvas de polari zação par-ciais, em eleti6titos com 0 ,6 g,dm-' de di6xido d.e enxof re
7.11 Efeito do aumenËo da adição de dióxido d.e enxofre sobre ECMiua" n P¡cpt
7.I2 Efeíto do aumento da adição de diõxido de enxofre sobre ECMiuat u PECu
7.13 Efeito do aumento da adição de díóxído de enxofre sobre ECMirCOO . PECM, em eletrólitos com e sem adição de zinco
7.14 EÍeítos das variaçõss de adição de dióxido de enxofre sobre ascurvas de polarizaçao parciais, em eletr6litos sem adição deimpurezas
7.15 Efeitos das variações de adição de dióxido de enxofre sobre ascuïvas de polarização parciais, em eletr6litos com Or5 mg.dm-3de cobalto
7 .16 Ef.eitos das variaçóescurvas de polarízaçaode níquel
7,L7 EfeíËos das variaçõescurvas de po1-ari zaçãode zinco
XII
L22
L23
124
125
L26
L27
131
131
132
de adição de di6xido de enxofre sobre asparciais, em.reletr6litos com 1r0 mg.dm-3
de adição de di6xido de enxofre sobre asparciaís, em eletr6litos com 20 mg.dm-3
133
135
L36
I37

XI]I
LISTA DE TABELAS
2.I Composição química tÍpica de manganês eletrolítico
3.1 Produção mundial de manganês eletrolítico
3.2 Limites máximos admissíveis e efeiÈos de impurezas meËãlicaspresentes no eletr6lito
4.L Densidade de corrente de Ëroca da reação de desprendimentocat6dico de hidrogênio sobre diferentes metais
5,2 Composição química dos eletr6litos
6.1 ReproduËibilidade na duração da corrosão eletroquímica (experiências preliminares para ãeterminação^da reprodutibilidade, cõmo e1etr61íro ng 1, iu = 5215 mA . cm-Z " td = 900s)
6.2 Reprodutibilidade na duração da corrosão química
6.3 Reprodutibilidade do potencial- de catodo (íU = 52,5 mA
6.4 Deposições seguidas de corrosão eletroquímica (eletrõlitoiO = 52,5 mA cm-2 " td = 900s)
6.5 Deposições seguidas de corrosão química (e1etró1ito nQ 30)
6.6 Eficiência de corrente e potencial de catodo em função dadade de corrente catódica (e1etr61íto nQ 30)
L6
2B
72
3
5
82
B3
B6
87
B9
90
92
9I
141
-)cm -)
nQ 30,
densi
6.7 Efíciências de correnËe mãximas e correspondentes densidades decorrente e potenciais
6.8
7"r
Potencial de catodo e densidades de correnËe parciais em funçãoda densidade de corrente catódica total (eletró1íto nQ 30)
Potenciais de eletrodo reversíveis

OBJETIVO
0 objetivo deste trabalho 6 estudar o efeito
três impu.rezas sobre a eËapa eletroquÍmica do processo
- -1 .nes eletrolr-f r-co.
de
de
O aditivo estudado -e o dióxido de enxofrer gue tem uso consagrad.o
na produção de manganês eletrolítico. Considera-se que sua adição ao eletrõ1i
to permite a obtenção de eficiências de corrente suficientemente elevadas pa-
ra garaîtir a economicidade do processo.
As impurezas estudadas são níquel, cobalto e zi"nco, gue gera.lmente
estão presentes nos eletr-olitos, originãrias dos min-erios de manganês . As
duas primeiras foram escolhidas por serem consideradas, dentre todas as impu-
rezas, as rnais prejudiciais ao processo, por reduzirem extremamente a eficiên
cía de corrente ou mesmo impedirem a deposição. A terceira, zínco, 6 por ve-
zes considerada prejudicial e por
tendo sido incluída neste estudo
gentes.
vezes considerada favorãve1
exatamente em função destes
um aditivo e de
obtenção de manga-
âo processo,
resulËados diver

z, cARACTERÍsTTcAS E usos DO MANcANÊs ¡l¡rRor.Írrco
O manganês eleËrolítico 6 a forma maís pura de manganês utiliza
da em quanËidades comercialmente significativas, atingindo purezas em tor-
no de ggrï7. Mn. Apesar dísto, seu custo ã competítivo com o de ferro*manga
nês de baixo carbono, o que possibilita o seu uso, por exemplo, em siderur
gia. Neste setore o manganês el-etrolítíco tem grande interesse como aditi*
vo pâra aços com al-tos reores de manganês e baixos teores de carbono, f6s-
- .-4foro e silício, como 6 o caso cle aços de baixa liga e alta resistência e
dos aços especiais.
OuËra aplicação importante do manganês eleËrolítico encontra-se
na produção de ligas não-ferrosas, em especial de alumínio e de cobre. Ele
tambóm pode ser utílizado na produção de eletrodos de solda, Ímãs cerâmi-
cos, compostos químicos de al-Ëa pureza, etc.
o manganês eletrolítico ã comerciaLizado sob a forma de pó, gra
nulos ou escamas. Sua composição química típica 6 apresentada na tabela
2,12

Tab. 2.7 - Composição química típica de manganês eletrolítico (ref.1)
elemento
Mn
S
P
Si
Fe
c
N
H
0
tipo baixo
nl drogen]-o
99,9
0,030
0 ,001
0,002
0,001
0,006
0 ,01
0,0006
0,3
teor (7.)
tipo baixo
oxi gênio
99,9
0 ,030
0,001
0 ,002
0,001
0 ,006
0,01
0,018
0r1
tiponitrogenado
94a960,030
0,001
0,002
0,001
0,006
4a6

3. PRODUçÃO INDUSTRIAL DE MANGANES ELETROI,ÍTICO
3.1. Hrsrõnra n pRotuÇÃo arual
Em 1918, Van Arsdal-e (ref . 2) realizou um dos primeiros trabal.hos
sobre a obtenção de manganês eletrolítico, em laborat6rio. Ern 1935, o United
States Bureau of Mines iniciou uma série de estudos em escala-piloto volta-
dos à definição de um processo comercial para sua produção; em 1939, como re
sultado destes estudos, formou-se a Electro-Manganese Corporation que, três
anos depois, ap6s terem sid.o solucionados inúmeros problemas referentes a pu-
rificação dos eletrõlitos, formação de compostos an6dicos, preparaçäo de ca-
todo, etc, iniciou a operação comercial de sua usina (ref. 1).
Destes três anos i-niciais e da operação de uma outïa usina-piloto
do U.S. Bureau of Mines saíram os principais parâmetros tecnológicos do pro-
cesso ainda hoje adotado. A produção norte-amer:icana cresceu muito nestes 40
anos, mas 6 suplantada atualmente pela da Ãfrica do Sulrque ini-ciou sua pro-
dução err 1955 e possui hoje as duas maiores produtoras de manganês eletrolí-
tico do mundo, a Emcor e a Delta-Manganese, voltadas principalmente para ex-
portação. A tabela 3.1 apresenËa uma relação dos principais paÍses produto*
res, sua produção anualea data de início de produção, com dados de suas usi
nas atualmente em operação:

Tab. 3.1. - Produção mund.ial de manganês elerrolítico em r97r (ref r)
parsln]-clo deprodução
usinas em operaçao
início de produçãoempresa operação tl ano
produçaoto ta1t/ano
Ãfrica do Sul 34000
Estados Unidos 27800
Japão 9600
Unlao Sovretrca e
'I che co-.b s Iovaq ur a
Chi na
3.2. PROCESSO CONSAGRADO DE PRODUçÃO DE MANGANES ELETROLÍTICO
0 processo consagrado de produção de manganês eletrolítico utiliza
solução aquosa de sulfatos de manganês e de amônio, com adição de di6xido de
enxofre ao eletr6lito, e exige purificação cuicladosa do eletr6lito. A figura
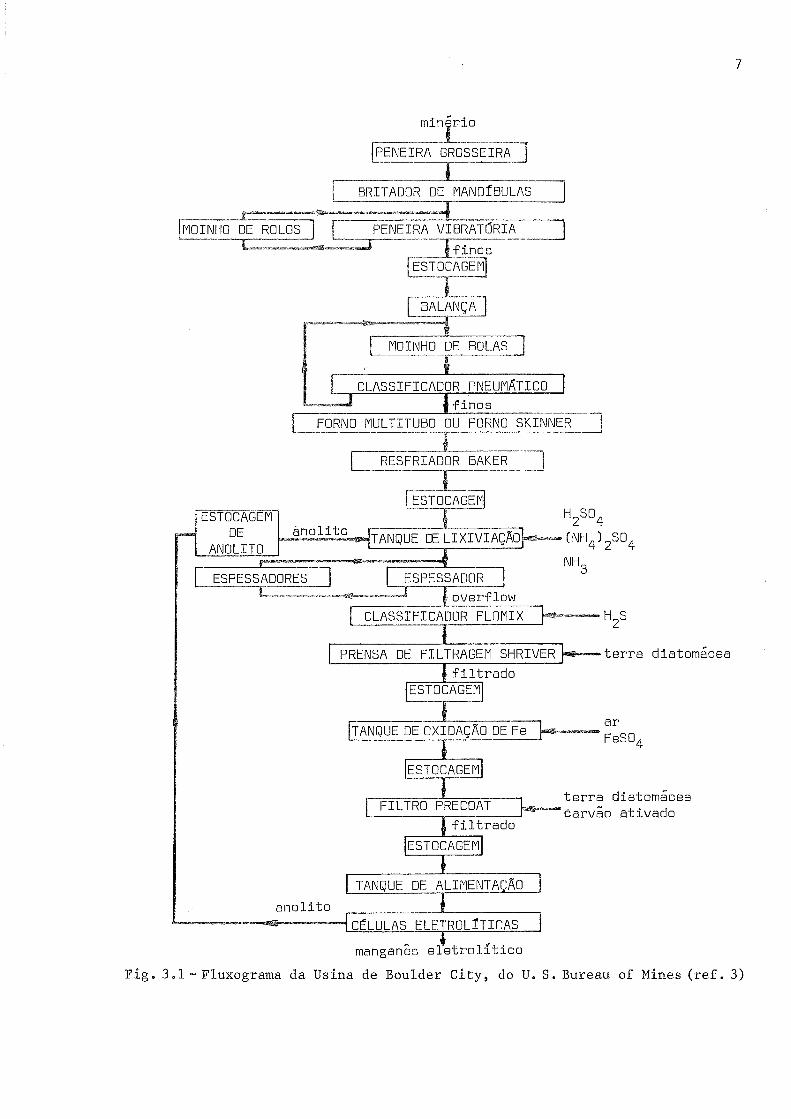
3.1 apresenta um exemplo de fluxograma do processo (ref. 3).
O processo inclui usualmente as seguintes etapas (ref. 4):
a) Preparação do min6rio para lixiviação, envolvendo usualmente
cominuição e redução pirometalúrgica do minério a MnO, Çuê é solúvel no ãci-
do sulfúrico diluído que serã urilizado na lixiviação.
L962 e
L965
L975
Delta Manganese
Emcor
Foote MineralUnion Carbide
American Potash
Toyo Soda
Chuo Denki
L97 4
1 955
1966
1954
1962
L97T
t94L
I 7500
16500
1 1000
9 100
7 700
6000
3600

b) Lixiviação, efetuada pelo anolito
se aËingir este pH, 6 necessãria alguma adição
Ëoe que conË6m usualmente de 25 a 40 g.¿oi3 ¿"
e 27 a 55 g.dr-3 du Mnsoo.
c) Borbulhamento com ar ou adição ¡þlgr6xido de hidrogênio (ref .
5)rpara reoxidar " F"3* os íons F.2+ pr""entes na solução, que seriam difí-
ceis de precipitar.
d) Neutralização da solução atã um pH em torno de 6,5, por meio de
hidr6xido de amônio, para que se precipitem, rra forma de hidr-oxidos, o ferro,
o alumÍnio, o molibdênio, o arsênio e o silício presenËes na solução.
e) Adição de 52- ã solução, na forma, por exemplo, de sulfeto <le
amônio, para remoção de cobre, zínco, níquel e cobalto¡ gue se precipitam na
forma de sulfetos e são retirad.os por filtração.
f) Adição_de di6xido de enxofre ã solução, promovendo a deposição
de manganês na sua forma alotr6pica q e aumentando a eficiência de corrente.
g) Eletrólise, realizada em c-elula provida de um diafragma poroso
que a divíde em um compartimento cat6dico e outro an6dico. No primeiro fica
um catodo de aço inoxidãvel e no segundo um anodo de liga Pb-Ag. A solução
obtida nas etapas anteriores ã continuamente introduzida no compartimento ca
t6dico, onde ocorrem as reações catódicas de deposição do manganês e despren
dimento do hidrogênio. Atravãs do diafragma poroso, a solução alcança o com-
partimento an6dico, onde ocorre a reação an6dica de desprendimento de oxigê-
nio, que the ímprime carãter ãcido. Esta solução, Çue ainda contem algum
manganês e praticamente todo o amônio inicial, sai do compartímento an6dico
eserã utíIizad.a como agente lixiviador na formação de novo eletrólito.
h) Precipitação de sais de magn6sio do eletrólito retirado, por
meio de uma torre de resfriamento (refs. 6, 7 e B), para evitar o contÍnuo
crescimento do teor de magnésio na solução recirculada. O magnãsio, originá-
6
recirculado, em pH 2r5. Para
de âcido sulfurico ao anoli-
Hzsoh, I4o g.dm-r de NH4SO4
PENE]RA GROSSETRA
BR]TADOR DË I"]ANDÍBUL-AS
PENEIRA VTBRATÕR]A
I]OÏNHO DE BOLAS
FORNO ITULTITUBO OU FORNO SK]NNER *l_t
TFTCADOR PNEUÍYATICO
L=-'ffl49ql-q¡[E¡,.-]ESTOCAGE
STOCAGEM
DE ano 1 ito
overj"1ow
filtrado
H,S0,LA
(NH4l 2So4
NH-J
terracarvao
Hzs
terra diatomácea
dietomáceaativado
filtrado
manganês el"etrolftico
TANQUE E LTX]VÏ
CLASSTFTCADOR FLOIY]X
ESPESSADORES
PRENSA DE F]LTRAGEH SHRIVER
ESTOCAGEIY
TANQUE DE OX]DAQAO DE FE
ESTOCAGE
FTLTRO PRECOAT
TANQUE DE ALTIYENTAÇÃO
AS ELETRÛLfT]CAS
Fíg. 3.1* F1-uxograma da Usina de Boulder City, do U. S. Bureau of Mines (ref. 3)

B
rio dos minãrios de manganês, apresenta um potencial de eletrodo-padrão mui-
to negativo (-2,363V, ref.9) e não se codeposita com o manganês como as de-
mais impurezasr p€ïrTrânêcendo na solução, que ,6 recirculada.
i) Tratamentos finais do produto, como moagem, desidrogenaçãoretc.
3.3. CARACTERÍSTICAS DA ETAPA ELETROLÍTICA
3.3.1. Eficiência de corrente e consumo energ-etico
O processo consagrado, descrito nos ítens anteriores, caractetíza-
se por um intenso desprendimento de hidrogênio associado à deposição do man-
ganês, em reação paralela que consome grande parcela da corrente catódica ts
tal. Assim, a eficiência de corrente do manganês, defínida pela relação en-
tre a corrente efetivamente utiTizada para redução do manganês e a corrente
total consumida, resulta mrril-o inferior a 1007", tendo valores usuais em tor-
no de 65%. Por este mot.ivor o consumo energético do processo 6 elevado, com
valores por volta de B a 10 k-t^lh.f.g¡lo (ref . 10).
3 '3 '2 ' -B-eË-quç- r.q-1-1¡ção-ag.uo.sa
A co-redução do hidrogênio, que reduz a eficiência de corrente, d!
ficilmente pode ser evitada. l'la série eletroquímica, o manganês ã o último
meËal que se consegue eletro-reduzír em meio aquoso; o potencialde eletrodo*
padrão para a redução de Mn2+ a Mn é -1,18v (ref.9).
Por que, então, não se evita a principal fonte de hidrogênio - a
ãgua -, rrti,1-izando-se outro tipo de eletrólito, como, por exemplo, os sais
fundidos? Ou, então, algum solvente orgânico? Estas hipõteses têm sido estu-

9
dadas e realmente conduzem a elevadas eficiências de corrente, mas ainda
apresentam inconvenientes. Em pri.rneiro lugar, estes eletrólitos são caros,
exigem equipamentos mais sofisticados e demandam maiores cuidados operacio-
nais que as soluções aquosas. No caso dos sais fundidos, há tambãm o proble-
ma da formação de dendritas do manganês depositado, que retãm o eletr6tito
quando retirado. Porisso, uma vez que é possível a obtenção de manganês ele-
trolítico por via aquosa - o q,r. não ocorre, por exemplo, com o alumínio
esta opção, embora difícil, 6 ainda a preferida.
3.3.3. Por que o ânion sulfato
Dos inúmeros ânions que, em princÍpío, poderiam ser usados para a
produção de manganês eletrolítico em solução aquosa, os que mereceram mais
estudo foram o sulfato e o cloreto.
As deposições realízad,as com cloreto apresentam (ref. 11) algumas
vantagens com relação ãs reaLizað,as com sulfato: permitem a aplicação de uma
tensão menor, admitem maiores concentrações de manganês no eletrólito e pos-
sibilitam maiores densidades de corrente.
Em contrapartida, com o uso de cloreto, o processo passa a incluir,
entr:e suas reações artódicas, o desprendimento de nitrogênio e de c1oro, com
a correspondente diminuição dos teores de NHr,+ e ,le Cl no eletró1ito a ser4
recirculado. Surgem assim dois problemas: a) o N[Ìr+ ã um insumo caro' e esta
reação exige que a sua reposição seja maior do que no processo com íon su1Ía
to; b) o cloro 6 corrosivo e Ë6xico, tornando-se necessãrio recolher cuidado
samente os gases desprendidos nos anodos. Alãm disto, a pr6pria solução de
cloretos 6 muito mais corrosiva que a solução de sulfatos, lrazend.o maiores
problemas para o equipamento. Por estes motivos, a opção usualmente adotada
são as soluções de sulfatos.

l0
3.3.4. Papel do diafragma e do amônio
Quanto maior a concentração de cátions Ht, maior será a taxa de
sua redução cat6¿ica a hidrogênio. Portanto, a diminuição do pH contribtri pa
ra a diminuição da eficiência de corrente do manganês. O diafragma contribui
para eviËar este abaixamento de pH, ao minimizar o refluxo do anolito - que
adquire carãLer ãcido devido ã reação anódica de desprendimento d.e oxigênio
- para a região do catodo,
Entretanto, não 6 apenas um pH baixo que deve ser evitado. Em pH
acima de 8,5 ocorre a precipitação de Mn(OH), (tig. 3.2), eue paralisa a de
posição. Assim, o ideal é que o pH se mantenha estãvel em torno de valores
pouco abaixo d.e B. Mas, em virtude do consumo de ânions H+ pela reação cat6
dica de desprendimento de hidrogênio, o pH do eletr-olito tende a aumentar
nas proximidades do catodo. O principal papel dos cãtions amônio é exatamen-
te o de manter este pH estável, pois eles tamponam a solução entre pH 7 e B
(ref. 12), A16m disso, o amônio ainda desloca para valores mais altos o pH
de precipitação do hidr6xido, e tambãm aumenta a condutividade el-etrica na
solução (ref. 4).
3.3.5. Por que anodo de liga chumbo-prata
Um dos problemas sãrios encontrados rro desenvolvimento inicial do
processo de produção de manganês eletrolítico pela Electro-Manganese Corpor-
ation e pelo U.S. Bureau of Mines residiu na formação de MnO, nos anodos. O
MnOrr além de consumir manganês, formava uma lama que prejudicava o sucesso¿-
da deposição. A solução foi o uso de anodos de ligas de chumbo, eue formavam,
apõs algum tempo de deposição, uma camada porosa e aderente de óxidos. Des-
t.as ligas, gue continham cobalto, arsênio, bismuto, telúrio, cãlcio, estanho,

11
-2 -l2,2r--,
o 3 4 5 6 l4t2ltto9B t3 t5 16
r'ld
'F{
Ê¡rQ)
{J;oP{
2rO
1,8
lr6MnOã
t,
t,
l,o0,8
or6
o,4
o12
o
- o,- o,4
- 0,6
- o,g
t,l12
1,4
1,6
-- lrg
Mn
-2 -l --L-,.--L__ ' l-_l_,--_ .l-_,._.-l_ -.-_l-.__ t*.-_ I____1 I r .,_L_-_I_--__L__
o I 2 3 4 5 6 7 I I tO ft t2 t3 t4 t5
pH
Fig. 3.2 * Diagrama de equilíbrio potencial-pH do sistema manganês-ãgua a
25ac (ref. 13)
2,?
2rO
1,8
1,6
l14
1,2
l,oo,B
o,6
o,4o,2o
-o,2-o,4-o,6- o,B
- l,o- 1,2
- 1,4
- 116
- l,B
Unro=

I2
antimônio, ferro, prata, manganês, etc, a que mel.hor atendia o compïomisso
entre bom comportamenËo como anodo, resistência mecânica e cusËo foi a de
chumbo com prata, cujos teores ficam em torno de 1 a 2157" (ref..4).
3.3.6. Por que catodo de aço inoxídãvel
Alumíní.o, cobre, titânio, aços comuns e aços inoxidãveis foram ex-
perimentados como catodos para a deposição de manganês. Diversos aspectos fg
ram analisados (ref. 4). E* primeiro lugar, o destacamento do depósito, que
se mostrou difÍciI no caso do alumínio e do cobre. Em segund.o 1ugar, a resis
tência ã corrosão provocada pelos respingos do eletr6lito acima do níve1 da
solução; sob este aspecto, os aços comuns mostraram-se inadequados. Em teï-
ceiro 1ugar, o custor guê descarLou o titânio, adequado sob os demais enfo-
ques. ResËou, assim, o aço inoxidãve1.
3.4. VARIÃ.VSTS OPERACIONAÏS DA ETAPA ELETROLfTICA
3.4.1. Densidade dè iorrente cat6dica e tensão aplicada ã c-e1u1a
A eficiêncía de corrente do manganês inicialmente cresce com o au-
mento de densidade de corrente cat6dica, passa por um mãximo e depois decres
ce; este máximo ocorre entre 40 e 50 r4.".-2¡ gue 6. a faíxa adotada no pro-
cesso industrial, por corresponder ao mínimo consumo energótico. Esta traría-
çao da eficiencia de corrente decorre do fato de que as densidades de cor-
rente parciais dos dois processos catódicos competitivos - a deposição de
manganês e o desprendimento de hidrogênio - crescem de maneira diferente com
a diminuição do potencial de catodo (Fíg. 4.3), A tensão aplicada aos ele-

13
Lrodos para a obtenção desËa corrente ã da ordem de 5 V (ref " 14)
3.4.2. Densidade de corrente an6dica
Como jã vímos , a liga Pb-Ag foi adotada porque e1a inibe a forma-
ção de MnO., devido ã formação de um óxido poroso e aderente. Para que isto
ocorra efícazmente, contudo, 6 necessária uma densidade de corrente anódica
em torno de 90 rA..*-2 (ref. 4), ou seja, cerca de duas vezes superior ã ae¡
sidade de corrente catódica adotada. Para se compatibilizarem estes valores,
a átea exposta do anodo ã cerca de metade da do catodo.
3.4.3. Concentra
Devído ao aumento d.a condutividade el6trica da solução e ao aumen-
to da ação tarnponante ao Unf, a eficiência de corrente do manganês cresce
com o aunento do teor de sulfaÈo de amônio (ref. 4). Contudo, segundo Louis
e Martin (que não Índicarn a origem de sua informação), a eficiência de cor-
rente volta a decrescer quando se usam teores acirna de 200 g.dr 3 de sulfato
de amôn.ío (ref. 15). Este fato pode ser atribuído ao aumento do desprendimen
to cle hidrogânio pela redução catódica do NHf a NH' descrita pela reação
4.8. (seção 4.L.2). Os teores usados industrialmente ficam na faixa de 120 a
_a150 g.dm ' de sulfato de amônio (ref. 4) .
1t3.4.4. Concentração de Mn'', pIl e vazão do eletrólito
Se o eletr6lito não fosse continuamente renovado, a concentração1t
Mn'' do catolito decresceria ã medida que a eletrodeposição de manganês
efetuasse. Da mesma forma, a concentração de H+ tanibém decresceria, em
de
SC

74
virtude da reação paralela de desprendimento de hidrogênio. Esta diminuição
da concentração de Mn2+, euê desfavorece cineticamente a reação de eleËrode-
posição do manganês, e o concomitante aumento do pH, que pode provocar a pre
cipitação do hidr6xido de manganês, são compensados pela contínua alimenta-
ção de eletr6lito novo. O pH, como já vimos, 6 controlado tambãm pela ação
tamponante dos Íons amônio e pela presença do diafragma.
Para uma dada taxa de formação de Mn e de.Hr, as concentrações de
,LI
Mn'- e de H' no caËolito dependerão da vazão e da composição do eletrólito
novo. Segundo Jacobs et a1. (ref. B), a f.aixa ideal de concentração de Mn2*
na região do catodo estã entre B e t6 g.dm-3, com um valor ótimo de L2g.dn-3.
Nas suas experiências, a solução de alimentação continha de 25 a 50 g.dt 3
de Mn2+ e tinha um pH em torno d.e 7. As vazões de alimentação do eletr-olito-1eram conËrol.adas de modo a se obter aquela concentração ideal, 12g.dm',
junto ao catodo. Com base nos dados de composição de eletró1ito novo, compo-
sição do eletrólito usado e eficiência de corrente, podemos calcular que es-
Las vazóes estejam entre 015 e 1,5 m3.*i-rr-1.A-1.
3.4.5. Temperatura
Temperaturas entre 30 e 40oC são consideradas as melhores para o
processo, havendo pouca diferença de consumo energ6tico dentro desta faíxa.
O uso de temperaËuras um pouco abaixo de 30oC pode levar a um pequeno aumento
na eficiência de corrente, mas, em contrapartída, concluz a uma diminuiçao da
condutividade elétrica da solução e subseqtlenËe aumento da tensão a ser apli-
cada ã célula; com isso, o consumo energ-etico aumenta. Com o aumento da tempe
ratura para acima de 40oC, o oposto ocorre; ou seja, a condutivi<lade cresce
mas a eficiência de corrente decresce (ref. 14).

15
3.5. EFEITO DAS IMPUREZAS
Um dos aspectos mais crÍticos do processo de eletro-redução de man
ganês ã o efeito extremamente prejudicial de impurezas metãlicas presentes no
eletr-olito, que reduzem muito a eficiência da correnËe e chegam, mesmo em teo
res baixos, a impedir a deposição. Como mostra a tabela 2.1, apenas 3 mg.dm-3
de cobalto, por exemplo, são suficientes para que a deposição não o.corra (ref.
16); o limite mãximo adnissível desta impureza no eletrólito 6 de O,S mg.dm-3,
no processo industrial (ref. B).
Uma outra indicação da importância do papel das impurezas 'e o fato
de que a simples diminuição de seus teores a valores realmente baixos é ca-
paz, por si só, de conduzix o processo a eficiências de corrente rnuito mais
elevadas que as usuais. Este fato foi demonstrado por Znamenskii, Gamali e
Stender (re.f. 17), que, atrav6s de uma cuidadosa purificação do eletrólito,ob
tiveram uma eficiência de corrente de 937", em laborat6rio. Louis e Martin
(ref. 15), contudo, contestam estes resultados: eles não obtiveram qualquer
aumento de eficiência de corrente com a purificação adicional de um eletr6li-
to de orígem industrial que permitia eficiências de corrente ð,e 657".
O prineiro estudo sistemãtico a respeito do efeito das impurezas
sobre a eficiência de corrente foi feito por Jacobs et a1. (ref.B), na usina-
piloto de Boulder City, do United States Bureau of Mines. Neste estudo, esta-
beleceram-se limiËes mãximos admissíveis de cada impureza no e1etr6lito. A ta
bela 3,2 apresenta estes valores, bern como dados e conclusões de outros auto
res sobre o efeit-o <lesfavorár¡el - ou, eventualmente, favorável - das ímpure-
zas.
Observanos, nesta tabela, Çue as impurezas mais prejudiciais 'são
cobalto, níquel, arsênio, antimônio, cobre e ferro, segundo o consenso de ro
dos os autores. Prata e zínco não contam com este consenso: em alguns estudos

T6
[ab, 3.2- Limites mãximos adnissíveis e efeitos de impurezas metãlicas presentes noe lerró lit<¡
lirnite mãxirno (ou faixaimpureza referência escudada) mg,dm-3 efeiÈos, condições, etc
Ni
B
18L9T6
20,27
8
7722
19l6
20,2r "24
8o
22
II8at
¿J
2516
20,2L¿o1.9
8
8
na
19
2829
22
30
I31191A
JI
0,5/r/50,3
(0,2 a 0,6)(0,5_a 2,0)
r /2ls0, 35
2,5 a 3,05
(0,5 a 4,0)(0,5_a 25)
La2/6a924124/24
515/t0 a 1515a20
2r5 a 3,O5
(5,0 a 40)(0,5_a 35)
( 100)(2,5 a 10)
5
15 a 20/20/30( 1,3 a 1,8)( 2,0 a 4,0)(>s)_
( 3ooo)
2
i> 25)(10 a 30)
(<25)
ro/20/30(5 a 1r0)(5 a l0)(0,3-a 15)
(5 a 110)(5,0 a 100)
( > 2ooo0)( 200)
(5 a 10)
( 200)
(2 a 20)
critãrios 1/II/IIIx; reduz EC
reduz EC
reduz sobretensão de hidrogênioreduz EC; forma par galvânicoreduz EC; forma par galvãnico
crírêrios Í/IT/IITi ) reduz ECreduz EC
reduz ECreduz EC
reduz sobretensão de hidrogênioreduz EC; forrna par galvânicoreduz EC; forma par galvânicoesll; critãrios IIlIIIr.; reduz ECAs'-; critérios r/IIIIII*; reduz ECreduz EC
cri!6rios I/IT/III'q; reduz EC
reduz ECredtz EC
reduz EC
reduz EC
reduz EC; forma par galvânicoreduz EC; forma par galvânicoreduz EC e qualidadereduz sobretensão de hidrogêniocrit6rio III,t; ïeduz EC
¡g2+; critãríos I/II/ITIrr; reduz EC
reduz ECmuda potencial de deposição de Mn
reduz EC e qualidadereduz EC
forma hidr6xido de alumínio
forma crosta no diafragma
criCério ffrt; reduz ECreduz EC e qualidadereduz sobretensão de hidrogênioreduz ECaumenta EC
crit6rios I/II/tttttl' reduz EC
reduz ECaumenEa EC
armenta sobretensão de hidrogênioaumenta EC
aumenta EC (com Se)
reduz E
nenhu¡n efeitoreduz ECnenhum efeitoaumenla EC; reduz qualidade
nenhum efeitonenhum efeiEo
aumenta EC
SbFe
Mo
Be
A1
Ca
Ag
Zn 8
/5I
l95¿33
2510
34L9
1(
I922
23
Mg
Bi
Na
Pb
* observação: os crit6rios adotados por Jacobs et al. (ref. B) Para os liuritesmaximos admissÍveis de impurezas no eletr6lito foram:Crit6rio 1 - Z_4 hotas^de,deposição sem decr6scímo significacivo
da efr-ciencla de correnteCrit6rio II - não afetar a mÍnirna densidade de corrente em que o-
corre deposiçãoCrit6rio III - duas horas dã deposição com apenas 22 de decréscimo
da eficiência de correnLe

L7
niostram-se prejudiciais ã eficiência de corrente, em outros mostram-se favg
ráveis à mesma. ttã tamb6rn impurezas metá1icas que mostraram apenas efeito
favorável, como o chumbo e o bismuto.
As impurezas prejudiciais são menos eletronegativas que o manganes
e com ele se codepositam. Alãm disso, todas e1as, com exceção do zi-nco, apïe
sentan uma baixa sobretensão de hidrogênio eur
como catodos, elas oferecem condições para um
drogênio, havendo a hipótese d.e que seja este
meio bãsico. Ou seja, agindo
intenso desprendimento de hi-
o principal mecanismo pe 1o
qual as ímpurezas reduzem a eficiência de corrente do manganês. Esta híp-ote-
se explicaria também o eventual efeito favorãvel do zinco, atribuído à eleva
da sobretensão de hidrogênio sobre este meta1. Estes aspectos são discutidos
com mais detalhes na seção 4.1.4.
Outra hip6tese aventada 6 a de que o efeito das impurezas se deva
à formação de pares galvânicos entre elas e o manganês depositado que, mais
eletronegativo, reËornaria ã solução. Esta proposição serã abordada na se-
Çao 4 . z.J.
3.6. PAPEL DO DIõXIDO DE ENXOFRE
Uma descoberta importante para o desenvolvímento do processo indus
trial de produção de manganês eletrolítico foi a de que somente se conseguia
obter dep-ositos espessos com boa eficiência de correnËe quando estes tinham
a forma de Mn-a , ê â de que este resultado estava relacionado com a presen-
ça de compostos de enxofre no el.etr61ito. Grube (ref . 36), por exenrplo, che-
gou perto de estabelecer um processo industrial, mas não obtinha eficiências
de corrente satisfatõrias. Seu depósito era de Mn-7
Diversos compostos redutíveis de enxofre mostraram aquele efeito

18
favorãvel quando adicionados ao eletr-olito (refs. 4, 25,37 e 38). Destes, o
mais utiTlzado foi o di-oxido de enxofre. Seu uso como aditivo ao eletrótito
do processo de eletro-redução de manganês foi patenteado por Shelton em 1938
(ref. 39). Desde então, o di6xido de enxofre vem sendo empregado na produção
industrial de manganês eletrolítico.
Segundo Dean (ref. 4), a quantidade de di6xido de enxofre utilíza-
da nas experiências da usina do United States Bureau of Mines em Boulder Ci-
ty (e provavelmente tamb6m na de Knoxville) era de 0,1 g.dt 3. Lewis, Scaife
e Swinkels (ref . 40) ciËam como típi ca a fai:ra de Orl a 0,2 g.¿* 3 ¿u di6xi-
do de enxofre, mas não indicam a origem desta informação.
Louis e Martin (ref. 15), omitindo tamb6m a origem de sua inforrna-
ção, afirmam que o di6xido de enxofre favorece linearmente a eficiência de
corrente atã um teor de 0r1 g.d* 3, e que incrementos subseqtlentes não mais
a modificam. DtAbreu e Torem (ref. 4I e 42), conËudo, encontraram uma. faixa
nruito mais ampla. Segundo eles, o efeito favorável do di6xido de enxofre 6
crescente at6 016 g.a* 3, e somente então se estabiliza.
Todos os autores que uËilizaram o dióxido de enxofre na eleËro-re-
dução do manganês verificaram a formação de Mn-a e o aumento da eficiência
de corrente. Nestes estudos, inúmeras hip6teses ou constatações sobre a ação
do dióxido de enxofre foram associados ãqueles efeitos:
a) evita a precipitação de MnO.OH (ref. 4);
b) evita a oxidação de Mn2+ a MnO,, no anodo (ref. 43);¿
c) reage com oxigãnio para formar ácido sulfúrico, sob ação catalí
tica de sulfato de manganês (refs . 44 e 45);
d) cria o meio redutor necessário ao processo (ref. 46);
e) 6. reð,uzíd,o a enxof re elementar, que f orma um colóide sobre o ca
todo (ref.. 4) t
f) suprime o efeito prejudicial das impurezas (refs . 77 e 48);

19
g) forma sulfetos com as impurezas (refs . 48 e 49);
h) facilita a transição de Mn-] para Mn-c (segundo o auLor em
questão, o manganês se depositaría inicialmente como Mn-7; ref.
4) - v. seção 4.2.4;
i) diminui o tamanho dos cristais do depósito, devido a aumento da
Èaxa de formação de núcleos de cristalizaçã.o e diminuição da ta
xa de crescimento (ref. S0);
j) aumenta a sobretensão de hidrogênio (refs. 23 e 5I) - v. seção
4 .r.5;
é reduzido a enxofre elementar, que envenena superfícies de ca*
talízação heterogênea da reação de desprendimento de hidrogênio
(ref s . 23 e 38) - v. seções 4.I.5 e 4.2.5;
aumenta a concentração de ãLomos de hidrogênio adsorvidos sobre
o dep6sito de manganês (ref. 38) - v. seção 4.2.5;
diminui a taxa de corrosão do manganês (refs. 46, 48, 49 e 52);
6 reduzido a enxofre elementar ou S2-, q,ru facilitam a redução
de Íons complexos de manganês e a eletrocristali zação do manga
nês, e reduzem a taxa de desprendimento de hidrogênio por meío
de reação química (ref. 53) - v. seções 4.7.3,4.I.5 e 4.2.I;
remove o efeito da dísparidade entre os parâmetros cristalinos
do substrato e do depõsito (ref. 54).
Em alguns dos ítens acima, índicamos outras seções nas quais hâ
mais detalhes sobre o assurito. Estas seções pertencem ao Capítulo 4, que
aborda os mecanismos de eletro-redução de manganês.
k)
r)
m)
n)
o)

20
4. ESTUDOS SOBRN MECANISMOS
O processo de deposiçäo catódica de manganês estã associado a des-
prendímento catódico de hídrogênio. Embora o desprendímenËo de hidrogênio
dê-se inicialmente sobre o catodo-base (de aço inoxidãvel, no processo indt,s
rrial), ele passa rapidamente a se dar sobre o depósito de manganês. Por es-
te motivo' r-Iossa atenção voltar-se-ã inicialmente aos mecanismos de desprendi
mento cat6dico de hÍdrogênio sobre catodo de manganê". ¡* seguida, abordare-
mos os mecanismos de deposição catódica de manganês, descrevendo resultados
e modelos existentes na literatura. ltro fina1, faremos um resumo comparativo
das conclusões mais importantes d.estes trabalhos.
4.1" DESpRENDTì4ENTO cATõDrco DE HTDROGÊNro
4"L.I" Desprendimento de. hidrogênio a partir .de H^!_S_ë30:
Mecanismos c1ãssicos
0 desprendimento catõdico de hiclrogênio a partir de HrO e
se segundo a seguinte s6rie de etapas consecutivas (ref. 9):
a) Transporte da esp6cie eletroativa para a superfície do
b) Formação e adsorção de átomos de hidrogênio:
":o* dã*
catodo.
H^0+e+H.+0Hzãd 4.L

e/ ou
c) Formaçáo da
mecanismo catalítico
e/ou mecanismo e 1e troquÍmi co
e /ou
e/ou mecanismo de
+-t30 +e + H"d*H20
mol-ecula de hidrogên.io :
H"d*H"d È Hz
2T
4.2
4.3
4.4
4.s
4.6
4.7
Hro +
+HgO +
H.+ead
H,+eao
Had
H+ H
+ H2+0H
+ HZ+HZ]
sH
l U,t2
emlss ao
d) Transporte da mo1-ecu1a de hidrogênio para a fase gasosa.
rAs reações 4.2. e 4.5, cuja esp-ecie eletroativa -e o HrO', são ca-
racteristicas de meio ãcido. As reações 4.1 e 4.4 sáo predominantes em meio
bãsico. Entretanto, elas pod.em ser tambãm importantes em meio ãcido, quando
a densidade de corrente cat6dica for suficientemente elevada para tornar o
+transporte de HrO uma etapa limitante.
Especie eletroativa principal e etapa limitante com caËodo de man-
ganês
Quando o catodo adsorve inten.samente o hidrogênioe torna-se merror a
energia de ativação da etapa de formação e adsorção de ãtomos de hidrogênio
(reações 4.I e/ou 4.2), e aumenta a energia de ativação da etapa de des-
sorção (reações 4.3 a 4.7). Assim, esta ú1tima etapa tende a ser a etapa len
ta do processo. Se, pelo contrârío, o catodo não adsorve inÈensarnente o hi-

22
drogênio, a etapa lenËa tende a ser a de formação e adsorção de ã.tomos de hi-
drogênío (reações 4.L elou 4.2),
A intensidade de adsorção pode ser avaliada pela entalpia de adsor-
ção ou pela fração recoberta pela espãcie adsorvida. Não conhecemos dados so-
bre est,es parâmetros entre manganês e hidrogênio em meio lÍquído. Em meio gaso
so¡ contudo, sabe-se que o manganês adsorve o hidrogênio com pequena intensida
de, uma vez que a entalpia de adsorção entre eles 6 menor que 52 kcal.rnol-] el
quanËo que as do cromo e do cobalto, por exemplo, são ð,e 74 e 64 irccaL.rof] r"u
pectivamente (ref. 55)"
B6langer e Vijh (ref. 56) baseiam-se neste faËo para sugerir qual se
ja a etapa lenta do processo de desprendimento de hídrogênio sobre manganês. Es
tes autores fizeram experíências de desprendimento de hidrogênio sobre manga-
nês em soluções H.SOO/NarSO4. A partir delas, eles puderam concluir que a eÈa-
pa lenEa 6 eletroquÍmica, umâ vez que suas curvas polarográficas (Fig. 4.I) a-
presentam um longo trecho com inclinação constante e igual a 140 mV.d6cada-| a
tendendo aproximadamente, segundo eles, ã f,ei de Tafel.
A16m disso, eles concluÍram que, mesmo em pH igual a 1r9, a espÉcie
eletroativa principal 6 HrO, uma vez q,re em densidades de corrente inferiores
ãs do Ërecho linear acíma cítado observa-se um trecho vertícal, correspondente
a uma densidade de corrente-limíte, atribuída ã etapa de transporte de UrOl As
sim, o processo seria constituido pelas reações 4.1 e 4.3 ou 4.4 (eles não dis
cutem as reações 4.6 e 4"7)" A etapa lenta, com base no baixo valor da ental*
pia de adsorção, seria a reação 4.1.
4.L,2 Desprendimento de hidr_ogênio a pa_rtir_de_íong_ arnijnio
Gamali e SËender (ref. 57) verificaram que o desprendimento de hi*
drogênio sobre catodo de manganês dá*se em potencíais menos negativos quando

se utilízam íons amônio em lugar de íons sódio no eletr-ol-ito suporte
4.2). Eles sugerem que isto se deva ã reação
23
(Fis.
4.8+NH. + e4
+ H"d * NH3
que ocorrería paralelamente à reação 4.1 ou 4.2. Entretanto, -e pouco provã-
ve1 que este mecanismo seja importanËe, uma vez que não se observa odoï de
amônia durante a eletrodeposição de manganês. Al-em disso, esta reação provo-
caría consumo dos ânions amônío, o que igualmente não se deteta experímental-
mente.
4.L.3 Redução química de hidrogênio por íons MnrU ou atomos Mn
-âd
Quando o desprendimento de hidrogênio se dá simultaneamente com a
deposição de manganâs, a curva de polarização do hidrogênio sofre uma modifi
cação qualítativa. Gamali, nybalrskaya, Trofimenko e Elina (ref. 53),e Losh-
karev, Stender e Galushko (ref. 58) verificaram que, na ausência de compos-
tos redutíveis de enxofre ou selênio, a curva de polarização d,o hidrogênio
passa a ter uma inclinação muito pequena na faixa intermed.iária de densida-
des de corrente (Ta.i-xa II, Fíg. 4.3). Como veremos na seção 4.2.I, estes au-
tores acreditam que, nesta faixa de densidades de corrente, haja grande dis-
ponibílidade de ãtomos de manganês apenas adsorvidos sobre o dep-osito ( rea-
ções 4.19 ou 4.20). Estes ãtomos, menos estãveis que os pertencentes ã estru
tura cristalina do dep6sito de manganês, poderiam ser reoxidados e retornar
à solução, ao inv-es de se integrarem ao depósito pela reação 4.2L. E o pro-
duto reduzido seria o hidrogênio, gue f.ícaría adsorvido sobre o dep6sito:
Mnro+ 2lro + Mrr2* * zlad+ zol 4.9

24
Ò),1 4f a' A_.-â
/áF*-
de corrente tA."r-2)-*-*J****
lidade):3 4) 5,2
vas polarográficas de
ntattgartês (ref . 57)
o-l!:t_
- /./ l-,H "lq'nI rt,6 -"r4- t'lE Þ' -,utI -o r: -,.a.--u'1.::,H#7i;'
* 0.51 3 ' densidade r
L---'*-r*"' -'-
-- I --'
-'-) - 1
l0 ' 10 -
Concentraçåo de (NH4l2504 [norma]
1) O,25 2) 1 3l
Efeito dos ions amônio sobre as curv
desprendimento de hidrogênio sobre ru
l-0 "_a
10 '
Fig. 4.1 - Curvas polarográficas de
sobre manganês (ref. 56)
Fig" 4 "2 *
-110
desprendimento de hídrogênio
densidade de
t¡lr-lcd
.r{oÊq¡.lJoÊl

e /ou
25
4.10')+rMn-+f
Mn + 2H^0'aoJ zlF'ad + 2H2O
Mnro + 2NHf Ê Mr,2* * 2uad + 2NH3e /ou
Caso a deposição de
sobre o depósito (reações 4.19
seja, haveria, nesta faixa de
dos íons Mn+ que, ao inv6s de
reação 4.24, seriam reoxidados
4.rr
manganês tenha uma etapa em que se forme Mrr+
ou 4.20), o mesmo raciocínio se aplicaria. Ou
densidades de corrente, grande disponibilidade
serem reduzidos e integrados ao dep6sito pela
2+aMn :
e/ou
* ')+Mn'+H^0 Mn''+H +OH¿-ad
++2+Mn +H30 =
Mn +H +H^0ao¿
4.L2
4.L3
ef ou 4.L4
Estâs reaçoes ocorreriam paralelamente às reações eletroquímicas
4.Lr 4.2 e 4.8. No caso de presença de compostos redutíveis de enxofre ou se
1ênio, elas deixariam de ocorrer. O motivo seria, segundo os autores, a me-
+^nor disponibilidade de MnuU ou Mn', em decorrência de modificaçoes em eta-
pês responsãveis pela for:mação destas esp-ecies no processo de deposição de
manganês (reações 4.20,4.2ir,4.23 e 4.24). Estes fatos serão abordados mais
adianËe, na seção 4.2.L
4.L.4 Efeito de impurezas sobre o desprend.imento de hidrogênio sobre
manganes
Itn* * Nuf * Mr,2* * H"d * NH3
Explicação baseada nas sobretensões de hidrogênio das impurezas

26
I
I
I
o1Jo
+_)ü(J
OJ-ü
'-lo'rJOC(Ð
+t)rl.
¡
regiao III
/'u//
t'4 -*'
,n¿¡rrÈ9\)il':
reg:låo T
jr,
I o¡1i,rri1-nrr., d¿r rJe n s :lt.i¿rc J e de cc:I'r'eil t. e r:¡¡ L cicl i c;:
Fíg. 4.3 - Represent,ação esquemãtica das curvas de polarização parciaisde deposição de manganês e desprendimento de hidrogênio(ref. 53)
n{) L_ ** _ r.__- . t*_._J
logaritmo da concentraçäo. r -1r. g.l_on " t]-tro )
regiåo ll
tltO-!>oltu Oúl .-lcco (o){JÞ00rof{ f{-o!O '-laÍ.
1) S0:J
,_Ð seoi
Efeito de aditivos sobre a sobreLensão de
sobre manganês (ref. 23)
,"-*-\
/ r--*{.. \/\
"/ \.
Fie. 4 ,4 - hidrogênio

2.7
Como expusemos na seção 3.5, a presença de impurezas metãlicas co-
mo cobalto, nÍquel, zinco, cobre, ferro ou arsênio no eletr-olito, mesmo em
pequenos teores, reduz drasËícamente a eficiência de corrente de manganês.
É interessante assinalar que todos esËes metais, com exceção do
zínco, apresentam uma baixa sobretensão de hidrogênio. Ou seja, em um mesmo
potencial, o desprendimento de hidrogênio sobre eles 6 muito mais intenso
que sobre o manganês, que apresenta uma sobretensão de hidrogênio razoave!-
mente elevada (v. tab. 4.1 ).
Este fato dã ensejo a uma explicação para a diminuição da eficiên-
cia de corrente quando estas impurezas estão presentes no eletrólito. Estan-
do todas elas situadas em potenciais menos negativos que o manganês na esca-
1a de potenciais-padrão de eletrodo, esËas impurezas com eIe se codepositam.
Depositadas sobre o catodo e expostas ã solução, elas se tornariam pequenos
catodos combaix4 sobretensão de hidrogênio. Sobre estas áreas, o desprendi-
mento de hidrogênio seria muito mais intenso que sobre o resLante d.o catod.o,
de manganês. Quanto maior o teor da impureza, maíor a'area exposta destes
catodos e maior o desprendimento de hidrogênio. E menor, portânto, a eficiên
cia de corrente do manganês.

Tab. 4,L - Densidade de corrente
co de hidrogênio sobre
metai s
2B
de troca da reação de desprendimento catõdi
diferente metais
densidade de
meio bãsi co
-q5.10'
-(3.10 '4. 10-6
3 . 10-6-13. 10
-13.10 t
-11.10_a
3.10'
-o1.10 "1.10-10
de- Ëroca (4."* 2¡
meio ácido
-L4.70 '-16.10 t
1. 10-10
7.10-15
6.10-10
2. 10-6_1a
2.ro "I . 10-6
1 " 10-B
1.10-11-o4.10 " (pH=6,5
4.LO-rz6.10-8
5. 10-11
referência
9
56
9
9
9
9
9
9
9
57
57
56
9
9
corrente
Co
'l
A1
Pb
Sn
Ni
Cd
Fe
Mn
tt
t9
il
Cu
Zn
4.L.5 Efeito de enxofre, selênio e telúrio sobre o desprend.imento
hidrogênio sobre manganês
Como citamos na seção 3.6, a introdução de compostos redutíveis de
enxofre eleva substancialmente a eficiência de corrente. Selênio e telúrioapresentam efeito semelhante ao do enxofre.
No modelo de mecanismo de deposição de manganês desenvolvido pelos
pesquisadores de Dnepropetrovsk (v. seção 4.2), os efeitos do enxofre e do
selênio são atribuídos ã sua atuação sobre uma das reações de eletro-redução
do manganês e sobre a reação química de desprendimento de hidrogênio descri-

29
ta na seção 4.L.3.
Anteriormente, contudo, Gamali e Stender (ref. 23) haviam verifica
do que os íons to3-, SeoS- e reol- elevam a sobretensão de desprendímen
to de hidrogênio sobre catodo de manganês, a partir de soluções aquosas de
sulfato de amônio (l'ig. 4.4). EsÉes autores observaram ainda que o manganês
retira estes íons da solução e fica recoberto por um filme branco, vermelho
ou preto, sobre o qual o desprendimento de hidrogênio deixa de ocorrer. Eles
sugerem que o enxofre elementar seja adsorvido sobre o catodo e envenene 1o-
cais ativos daquela reação.
Estes dados sugerem, porËanto, guê estes compostos tenham alguma
ação retard.adora sobre o pr6prio processo de desprendimento eletroquímico de
hidrogênio, uma vez que nestas e>çeriências não ocorria deposição de manga
nês.
4.2. ouposrÇÃo carõnrca nn Mar¡enrqÊs (¿coIæANHAIR nu onspnulÐrMnNro ln Hrrno-
cÊrlro )
4.2.I. Estudos do Ingtituto de Tecnologia Química de Dnepropetrovsk
Desde pelo menos 1950, um grupo de pesquisadores do Instituto de
Tecnologia Química F.E. Dzerzhinskii, em Dnepropetrovsk, na nepúb1ica Sovi6-
tica da Ucrânia, vem publicando trabalhos sobre os mecanismos de eletrodepo-
sição de manganÊs em soluções aq,rosas de sulfatos ou cloretos. Alguns destes
trabalhos jâ. f.orarn citados nas seções anteriores (refs.23, 43,50, 53, 54,
57 e 5B). Na presente seção, descreveremos algumas das conclusões por eles ob
tidas e o modelo de mecanismo que resulta deste conjunto de trabalhos.

30
Efeitos do íon amônio, segundo o modelo dos pesquisadores de
Dnepropetrovsk
Um dos efeitos da presença de íons amônio t'ro eletróliËo é a reaçãô
paralela de desprendimento de hidrogênio a partir destes íons, representada
pela reação 4.8 (ref . 57) e jã abordada na seção 4.I.2,
A reação 4.8 6 responsável pela presença de amônia ou hidr6xido de
amônio na camada de difusão junto ao catodo. Bondar, Gama1i e Stender (ref.
59) , analisando deposições reali zad,as com diferentes pHs e diferentes concen
-?++traçoes de Mn- , NH3 e NHO , concluem que os íons amônio e a amônia presen
tes na camada de difusão evitam a formação de hidr6xido de manganês e favore
cem a formação de complexos amoniacais de manganês. Estas reações seriam
representadas pelo seguinte equilÍbrio duplo (ref. 60):
Mn(OH), + 2unf + (n-2)Nrr4OHÈ Mr,2* * nNHOoH 4.I5a
1tMn'' + nNHoOH + nHrO 4.15b
A formação do complexo amoníacal teria lugar quando houvesse eleva
das concentrações de arnônia e sulfato de amôn.io. Se a concenÈração de sulfa
to de amônio for baixa, haverã formação de hidr6xido de manganês quando do
aumento do pH do eletrólito devído ao desprend.imento de hidrogênio.
0 n do equilíbrio 4.15b pode, segundo Trofimenko, Gamali e Vasi-
levskaya (ref" 61), assuruir valores de 1 a 4" Por meio de estu<los polarogrã-
ficos, estes autores conclulram que, das esp6cies contendo Mn2+, as predomi-
nantes em baixas concenrrações de NH, são Mr,2*, þCt"rl']2.
, þn(uH ì ù'
)+e MnNHr-' . Em altas conce-ntrações de NHr, a espãcie predominante -e
= þcxur>"]2.

31
EsËes autores veríficaram ainda que,
duzid.as eletroquimicamenËe no catodo: Mn2+ e
centrações de NHr, predomina a seguirrte reação:
1tMn'' +2e + Mn
Em altas concentrações de NHr,
do complexo amoniacal. Neste caso, a redu
da pela reação química de dissociação de
dominanËe nestas concentrações de NHr:
1tL' + 2NH,
2**zu + |4n + 2NH,
destas esp6cies,
[r"<*nrl ,]'.
predomina a redução
ção eletroquímica devef lc-
frnfxHr)oJ' , que -e
duas são re-
Em bai-xas con
4.16
e 1e troquími ca
ser precedi-
a espãcie pre
4.77
4. 18
2. * þ"rNH3) t[*,-"r,0]
F",rrr,f
Deposição em diferentes densidades de corrente
UÈilizando eletr-olitos isentos de compostos redutíveis de enxofre
ou selênio, Gamali, Trofimenko e Vorozhko (refs. 62 e 63) verificaram que se
definiam três faixas de densidade de corrente cat6dica (ou de potencial catõ
dico) bastante distintas (Fie. 4.3.):
Faixtt I: baixa,s d¿røidade's d¿ conzønfØ - Eormrse Mn-? de alta
perfeição, epitaxial com o catodo-base (de cobre). A curva polarogrãfica par
cial do manganês obedece ã tei de Tafel, o que indica controle eletroquími
co. Como as densidades de correnËe são baixas, a taxa de formação de NH.J
(reação 4.8) serã baixa, assim como serã baixa a Eaxa de consumo de Mn2+(rea

32
1t
çã,o A.16 ou 4,L9 ou 4.22). Assim, a esp-ecie Mn'' seria suficientemente abun-
dante nestas condições e a reação 4.16 (ou 4.19 ov 4.22) seria a predominan
Ëe.
FøLxct II: døuidctC¿ d¿ conn¿nt¿-n imitø do mctngctnù - co* maiores
densidades de corrente, o depósito começa a apresentar estrutura distorcida,
com traços de Mn-a. Além disso, observa-se a existência de uma densidade de
corrente-limite para a manganês. Isto 6 atríbuído a um limite na disponibir!1t
dade de Mn'' para a reação 4.16 (ou 4.L9 ou 4.22), seja pelo aumento da ta-
xa desta pr6pría reaçãorseja pelo deslocamento do ec1uilÍbrio 4.15 no sentido
da formação do complexo amoniacal de manganês, ãs custas do Mn2+. Este deslo
camento seria causado pelo aumento da concentração de NH, na camada de difu-
são do catodo, em virtude do aumento da taxa de desprendimento de hidrogênio
pela reação 4.8 (aumento correspondente ã elevação da densidade de corrente).
Como o desprendímento de hidrogênio não -e simultaneamente reËardado - pelo
contrário, ele ó acelerado, como visto na seção 4.1.3 - nestas densidades de
corrente, a eficiância de corrente do manganãs reduz-se muíto. A1ãm disso, o
aumento da taxa de desprendimento de hidrogânio em relação ã Èaxa de deposi-
ção de manganês 6 indícado como a causa da formação de urn depósiËo distorci-
do. Isto porque o hidrogênio - presente agora em grande quantidade - interfe
riria com o processo de cristaLízação do manganês. Os traços de Mn-a seríam
resultado tamb6m desta distorção do dep-osito. Com e1a, a energia necessäria
para cristalízaçã,o na forma ^f aumenËa, superando em alguns lugares a neces-
s'aría para cristalízaçãa na forma ø. Nestes locais, formar-se-ia Mn-a .
Faixa III: a.Ltfl^ d¿nsídctdøt d¿ conn¿ntØ - Caminhando-se para poten
ciais mais catõdicos, pode-se atingir os valores necessários para a redução
eletroquÍmica dos complexos amoniacais. Assim, riesta f.aLxa de densidades de
corrente ocorre esta nova reação,com controle eletroquímico, como indícado
pelo novo trecho linear da curva de polari zaçáo parcial do manganês. Nesta

33
f.aíxa, o manganês deposita-se na forma alotr6pica a .
Eletro-redução de manga€s - interdependência com o desprendimento
de hidrogênþ, segundo o modelo dos pesquisadores de Dneproge-
trovsk
Tanto a reação 4.16 quanto a 4.18 podem ser subdivididas em duas
etapas. A primeira etapa seria a redução eletroquímica do manganês, que fica
ria adsorvido sobre o dep-osito:
1tMn'' + 2e Mn
ad
+Mn,+2NH^aoJ[u"rNnr) 12*+2e
4.r9
4.20
4.27
l+.22
l, a1
0utra
manganês sobre o
A segunda etapa serÍa a cristalízaçã.o do manganês:
Mn Mnao
possibilidade
catodo:
seria uma etapa inicial de redução parcial do
2+Mn +e Mn
+e Mn + 2NHa
seguindo-se a redução final e
r' 1', L
fivn{llHr)rj''
Mn +e -> Mn
cristalizaçã.oz
4.24

34
As esp6cies inËermediãrias Mr"d or Mn+ são sugeridas por Gamali,
Rybalrskaya, Trofimenko e Elina (ref. 53). (Estes auËores não expliciÊam as
reações envolvidas; nós o fizemos aqui com o intuito de facilitar a exposi-
ção do modelo.) Segundo eles, a reação de cristalízaçã.o (reação 4.2I ou 4.24)
é retardada quando o depósito ao qual o manganês vai se integrar estã muito
di storcido .
Retardando-se a reação 4.2L ot 4.24, a concentração de seus reagen
+tes, Mnad ou Mn', aumenta. Com isso, acelera-se o desprendimento de hidrogê-
nio pelas reações 4.9 a 4.14 (seçáo 4.1.3) .
Como vimos no item anterior, o depósito aplîesenta-se distorcido
quando a deposição se reali za rLos potenciais correspondenËes à densidade de
corrente-limite do manganês, em eletr-olitos isentos de compostos redutíveis
de enxofre ou selênio.
Assim, segundo os autores, estaria explicada a coincidência entre
a aceleração do desprendimento de hidrogênio e o reËardamento de cleposição
do. manganês, que ocorrem nesta faixa de densidades de corrente (Faixa fI,
descrita. no item anterior e na figura 4.3.).
Comentã::io: Cremos haver uma contradição nesta explicação dos au
tores. Eles dizem que, em função da dificuldade de cristaLizaçáo, haveria
, ou Mn+ são exatamente os
proCutos das reações 4.19 ou 4.22, que esËão, segundo os autores, retarda-
das por falta do reagente Mn2+. Aliás, são os próprios autores clue dizem que
a causa do retardamento da cristalízação - o depósitc distorcido - ã resulta
do do retardamento da eËapa eletroquímica. Assim, como seria possível haver
disponibilidade de esp6cies que, alãm de serem o produto de uma reação retar
dada por falta ,Je reagente, são a causa do retardamento da etapa seguinte ?
Cremos ser razo'avel a interdependôncia sugerida entïe a distorção do dep-osi-
to e o aumento d.o desprendimento de hidrogênio, explicado pelas reações 4.9

35
a 4.I4. Entretanto, 6 necessãrio encontrar uma outra explicação para a dis-
torção do dep-osito - uma explicação independente da densidade de corrente-li
mite do manganôs -t pãTa que se possa acreditar na disponibilidade de *r"d+ou Mn nestas condições.
EfeÍtos do enxofre e do selênio, segundo o modelos dos pesquisado-
res de Dnepropetrovsk
A presença de compostos redutíveis de enxofre e de selênio propi-
cia a deposição de manganâs na forma alotr-opica o¿ (em lugar de na forma 7),
torna menos negativos os potenciais de deposição e aumenta a eficiôncia de
corrente de manganês.As experiências de Gamali, Rybaltskaya, Trofimenko e
Elina (ref. 53), interpreEadas à luz de seu modelo, permitem-lhes propor ex-
plicações para a modificação de potencial de deposição e da eficiência de
corrente. Segundo estes autores, aqueles compostos agem das seguintes manei
ras (rigs . 4.3 e 4.4):
a) Tazem desaparecer a den.sidade de corrente-limite do manganês:
Com estes compostos, a curva de polarização parcial do manganês
deixa de apresenta.r o trecho vertical que caracteriza urfla etapa quÍmica limi
tante na deposição a partir de Mn2+. Lsto ocorreria devido a uma mu<lança no
mecanismo de redução dos complexos amoniacais,(reação 4.20 ou 4.22), que pas
saria a oç.orrer em potenciais mais an6dicos. Esta mudança de mecanismo po-
deria ser atribuída, talvez, ã criação de pontes eletrônicas. O presenteefei
to ã mais pronunciado para o enxofre que para o selênio"
b) Facilitam a cristalízação do manganês:
Os compostosi redutíveis de enxofre e de selênÍo facilitariam a
cristalizaçáo do manganês (reação 4.2I ou 4.24). Com isto, diminuiria a con-
centração de Mn"U ou Mrr+ na superficie do catodo; conseqllentemente, diminui-

36
ria o desprendimento de manganês pelo mecanismo das reações 4.9 a 4.I4. Isto
explicaria o aumento da eficiência de corrente resultanËe destas adições. Ao
contrário do que ocorre com o efeiËo anterior, este efeito ã rnais acentuado
com selênio que com enxofre.
c) Diminuem a constante cin6tica das reações quÍmicas de desprendi-
mento de hidrogênio:
A velocidade de uma reação química 6 proporcional ã concentração
dos reagentes elevada a um dado expoente. 0s autores sugerem que os compos-
tos de enxofre e selênio diminuam o valor desta constante de proporcionalida
de - a constante cin6tica - nas reações quÍmicas de desprendimento de hidro-
gênio por meio de Mn"O ou Mn+ (reações 4.9 a 4.14).
4.2.2 Estudo de Barchese
Em estudos reaLizados na Universidade de Sao Pau1o, Barchese (ref.
5 ) utiLîzou tócnicas cronopotenciom-etricas para estudar a eletro-redução do
manganês. As variãveis analisadas foram a densidade de corrente do eletrodo
de trabalho e a concentração da espãcíe eletroativa, que são variãveis ine-
rentes ao m6todo cronopoËenciom-etrico, bem como a concentração de íons amô-
ni-o, o pH e a temperatura da solução"
MecanÍsmo proposËo
0 estudo evidenciou o seguinte mecanismo (refs. 5 e 64):
+NH,
+
2+Mn-
+oH =
NH3+H20
+ NH3 + OH Ë MnNH3OH
4.25
4.26

+-MnNH^OH + 2e + Mn +
J
1¿--+Mn'' +e + Mn
+-H +e + Had
ZEud + nz
+Mn *H"d -> Mn+H
NH3 + 0H
37
4.27
4.28
4.29
4.30
4.31
Discutiremos a seguir alguns aspecËos do mecanismo proposto.
O íon complexo e a parËicipação dos íons amônio, segundo o modelo
de Barchese
O manganês apresenta grande tendência a se hidrolisar. Jã citamos,
por exemplo, que o hidr-oxido de manganês, Un(Ott)r, forma-se a part.ir de pHs
relativamente baixos (fig.3'i2 ). Assim, -e razoâvel supor que o rnanganês te-
nha maior tendêncí a a formar Íons complexos conËendo o radical OH do que a
formar o aquo-Íon usualmente formado por outros metais.
Por outro lado, os importantes efeiËos da presença de íons amônio
sobre a eficiência de corrente e sobre o tamponamento da solução sugerem que
os íons complexos de manganês contenham, a1-em de OH , NH3.
Esses fatos e considerações levaram Barchese a ar.alísar a parËici
pação de complexos de forma Mn(NHr)*oH+ no processo de eletro-redução de man
ganês. Seus resultados experimentais levaram-no a concluir que este comple-
Xor com o coef icienLe x assr.rmindo valor igua1. a 1, realmente participa dos
mecanismos de tamponamento e de redução eletroquimica do manganês.

3B
Os equilíbrios representados pelas equações 4.25 e 4.26 estabele-
cem um sistema tampão que bloqueia a elevação do pH do catolito. Em pH bai-
xo, a concentração do íon complexo MnNHTOH+ (reação 4,26) -e muito baixa. O
aumento do pH favorece a formação deste íon, cuja redução (reação 4.27) pas-
sa a ocorrer. O potencial desta reação, segundo as conclusões de Barchese, é
mais anódico que o da redução de Mn2+.
Barchese discute a possível forrnação do íon complexo MnNH3+ ( pro-
posto, por exemplo, no modelo precedente; v. seção 4.2.L). Dois fatos levam-
no a concluir não ser esta a espécie participante no mecanismo de tamponamen
to e responsãvel pelo segundo mecanismo de eletro-redução de manganês ( con-
siderand.o-se como primeiro o mecanismo a partir de urr2*). Em primeiro lugar,
a constante de equilí¡rio deste complexo, segundo dados da literatura, ã se-
te ordens de grandeza superior à calculada por Barchese a partir de suas ex-
periências" Em segundo lugar, a eletro-redução de íons complexos amoniacais
semelhantes a este, como zn(NHr)O'*, a.r{r"r) o'*, cu(NHr) 2* " Aia(NH3)2+, dã-
se usualmente em potenciais mais negativos que a eletro-redução de aquo-íons
simples dos met.ais correspondentes. Segundo Barchese, como jã vimos, o manga
nês apresenta o comportamenËo oposto: a eletro-redução do Íon complexo dá-se
em potenciais menos negativos que a do Mn2+.
Eletro-redução de manganÊs - inlerdependência com o desprendimento
de hidrogÊnio, segundo o modelo de Barchese
Comr: vi'mos nos itens anteriores, a eletro*redução de manganês se
f.az, segundo Barchese, a partir de Mn2+ e de MnNH3OH*. Nas condições usuais
do processo, o autor verifica que a participação do íon complexo -e muito pe-
quena. A reduçã.o a partir de Mrr2+ seria, então, a mais importanËe. Com rela-
ção a ela, as experiências de Barchese indicaran claramente a existäncia

39
de duas etapas. A primeira, el.etroquímica, forma o íon Mn+ (reaçáo 4,28). A
segunda, química, consiste na redução deste íon por áËomos de hidrogênio ad-
sorvidos na superfície do catodo (reação 4,31). Esta etapa competiria com a
etapa final, também quÍmica, do mecanismo de desprendimenËo de hidrogênio
(reação 4.30), uma vez que ambas Ëêm como um de seus reagentes o hidrogênio
adsorvido.
4.2.3 Estudos do Tnstituto de Química Geral e Inorgânica de Kiev (ucrâ-
nia, URSS)
Complexos sulfatados de manganÊs
Kub l-anovskil , Be linskii e GorodskiÍ (,ref . 65) reali zaram deposi-
ções de manganês em soluções de sulfatos de manganês e de amônia, sem adição
de di6xido de enxofre ou de selênio. Nestas experiências, eles observaram a
ocorrência de uma densidade de corrente-limite para o manganês, semelhante ã
observada pelos pesquisadores de Dnepropetrovsk (seção 4.2.7).
Em trabalho anterior, Kublanovskii, Belinskií e Zozímovich ( ref.
66) havíam concluÍdo que o processo de eletro-redução de manganês era gover-
f .12-nado por compostos complexos sulfatados, como MnSO, e lUn(SO,,) Zlu L uJ
Porisso, neste novo trabalho (ref. 66), os autores analisaram o
efeito da variação das concentrações do Íon sulfato e do íon Mr,2* sobre o va
1or da densidade de corrente-limite. Eles concluíram que a reação químíca
lenta que provocå o surgímento da densidade de corrente*limíte seria a dis-
sociação d.aquele Íon complexo diretamente r Mn2*,
f'"c'o+r r]'-1t 1-
Mn'' + 2So1L+
4.32

40
Seguindo-se
1tMn-' + 2e Mn t n,a
4.JJ
0s autores sugerem que o retardamento do processo de deposição
ocorra at-e que se atinja um potencial suficientemente catódico para que se
dê a eletro-redução de alguma outra esp6cie eletroativa que não o Mrr2*. En-
trecanto, eles não estudaram esta faíxa de potenciais nem procuraram identi-
ficar esta outra esp6cie eletroaÈiva.
Efeito das impurezas - o nodelo dos pares galvânicos
As impurezas metã1icas que prejudicam a eletro-redução de manganês
estão situadas em potenciais mais anódícos que o deste na escala de po.ten-
ciais-padrão de eletrodo. Ou seja, qualquer uma destas impurezas formaria
com o manganês uma c-elula galvânica îa qual o manganês, agindo como anodo,
seria dissolvido pela solução e a impureza, agíndo como catodo, depositar-se
-ía.
A partir de suas experiências com cobalto, níquel e cobre, Zozí-
movich, Sirvab e Belinskii(refs. 16, 20 e 2I) afirmam que a formação de pares
galvânicos manganês/ímpureza seria o motivo da ação desfavorãve1 destas im-
púrezas.
Segundo estes autores, em virtud.e da distribuição não-uniforme da
densidade de corrente (e do potencial) sobre a superfície do catodo, a con-
centração das impurezas codepositadas resulta tamb6m não-uniforme sobre a
srrperfície. A região onde este fato se manifesta mais íntensamente -e a linha
do nÍve1 superíor do eletrõlito no caËodo, em contato com ar, onde se forma
um menisco. Nesta regÍão, a densidade de corrente catõdica 6 menor que a den

47
sidade de corrente catódica m-edia sobre o catodo. Esta redução de densid.a-
de de corrente (e- aumento do potencial) afeta mais íntensamente a deposição
de manganês qtte a das impurezas, eue são mais eletropositívas. Em decorrên-
cia, a concentração das impurezas nesta região do depósito se torna rnaior
que no restante do catodo.
Constitui-se, assim, um par galvânico, com esËa regiáo ríca em im-
púTezas atuando como caEodo e o restante do depósito atuando como anodo. For
mado o par galvânico, ele tende a se intensifícar, uma vez que estas impure-
zas apresentam uma baixa sobretensão de hidrogênio (seção 4"I.4). Assim, os
pares galvânicos provocariam a dísso1ução do manganês na região an6dica do
depósito e, ao mesmo tempo, reduziriam a deposição do manganês na região ca-
tódica do mesmo.
Segundo este raciocÍnio, o efeÍto das impurezas sería anulado se
houvesse uma d,istribuição mais uniforme da densidade de corrente caródica (e
de potencial cat6dÍco). Experiências realízadas com um catodo rotativo con-
firmaram este fato (ref. 16). Deposição realizadas com 25 mg.d*-3 d. nÍquel
apresentaram, com o eletrodo roËativo, eficiências de corrente do manganês
de 70,37.. Sem rotação, esta efíciência se reduziu a 3,27"" O único papel
da rotação, segundo a argumentação dos auLores, foi evitar o surgimento de
"centros de corrosão" do manganês, resultantes da não-uniformidade do poten-
cial e origem dos pares galvânicos.
Segundo Shvab (ref" 21), pode-se formar tambérn um par galvânico en
tre Mn-a e Mn-? . Ele afirma ainda que mesmo metais que não se codepositam
com o manganâs podem af.etar sua deposíção se formarem hj-dr6>lídos na região
alcalina próxima ao catodo. Estes hidróxidos reduziriam localmente a densida
de de correrrte, dando origem à codeposição heterogênea das demais impure-
zas, e favoreceria localmente a formação de Mn-a .

42
4.2.4 Estudos do U.S. Bureau of Mines
Dean (ref. 67) verifícou que as primeiras camadas depositadas do
manganês apï'esentam-se na forma alotr6picã 1 , seguindo-se uma camada de
Mn-a de granulação fina e depoi.s cristais macroscópicos de Mn-4. Schlain,
David e Prater (refs. 68) verificaram, posteriormenter gue soluções extrema-
mente puras levavam a deposições apenas de Mn-"f eonc.L;i.ndo, então, que a
passagem de Mn-7 para Mn-a assinal.ada por Dean devia-se à presença de cer-
tos compostos de enxofre no eletr61ito.
Como veremos a seguir, a deposição ínicial de Mn-y foi contestada
por Dhananjayan.
4.2.5 Estudos de Dhananjayan
Dhananjayan (ref. 38), do Laboratório Metalúrgico Nacional, em Jam
shedpur (Índia), analisou o teor de hidrogênio, a estrutrrra cristalina, o pe
tencial-padrão de eletrodo, o potencial de deposição e a sobretensão de hi-
drogênio de Mn-o e Mn-y obtidos em eletr-olitos conËendo ou não selênio,
telúrio e enxofre.
Segundo o autor, as primeiras camadas depositadas teriam uma estru
tura o extremamenËe fina ( e não Y , como supusera Dean; seção 4.2.4). Na
ausência d.e enxofre ou selênío, este depósito teria uma sobreËensão de hidro
gênio muito baixa, resultando um intenso desprendimento de hidrogênio, que
elevaria o pH do eletr-o1ito. O desprendimento de hidrogênio aumentaria a po-
Larízaçáo do catodo, atingindo-se o potencial de deposição de Mn-Y. Uma vez
iníciada, a deposição de Mn-Y teria prosseguimento em lugar da de Mn-o, em
vista de a sobretensão de hidrogênio de Mn-Y ser mais elevada que a de Mn-o.
Em eletr6litos contendo enxofre, selênio ou telúrio, ou com eleva

43
das densidades de corrente, o autor verificou que a deposição de Mn-cr não
se interrompe após as primeiras camadas. O depósito, constituído apenas de
Mn-o, , apresenta grande quantidade de hidrogênior gue the confere baixa dure
za e densidade. O autor sugere então que parte do hidrogênio eletro-reduzido
assume posições no reticulado do manganês ou forma complexos durante a depo-
sição de Mn-o, , e que tal fato esteja relacionado com a formação desta estru
tura em preferência ã estrutura Y.
Esta elevada concenËração de hidrogênio no dep-osito seria devida,
segundo sugere o autor, a uma atividade catalítica negativa do enxofre e do
selênío sobre a reação de combinação de ãtomos de hidrogênio (reaçã,o 4.7 e
4.30). Poderíamos sugerir uma explicação para o efeito semelhante resultante
do uso de elevadas densidades de corrente; nesta situação, aquela reação po-
deria tornar-se uma etapa lenta do processo, o que resulËaría em aumento de
concentração de ãtomos de hidrogênio sobre o dep6sito.
4.2.6 Estudos de Fekete
Fekete, do Instituto de Pesquisa dos Metais Não-Ferrosos, em Bu-
dapeste (.Hungria), realizou inúmeras experiências sobre o controle do pH na
eletro-redução do manganÊs (ref. 69). Suas principais conclusões foram a res
peito d-o papel do diafragma sobre este controle. Ao contrãrio do que se suge
re usualmente, Fekete afirma 'fue o ð.iafragna rrão impede o retorno de ãcido
sulfúrico do compartímento anõdico para o catódico. E, segundo o autor: sê-
ría exatamente este ãcido sulfúrico, formado nas reações an-odicas, o princí-
pal agente controlador do pll do catolito.
Fekete observou, também, que freqllentemente ocorria um súbito des-
controle do processo de eletro-redução de manganês. Uma deposição que se
viesse realízanð.o normalmente, sob controle, em pH em torno de 7 15, repenti-

44
namente podia se interromper', com elevação do pH e formação do hidróxido de
manganês. Segundo o autor, esËe fenômeno ocorre quando alguma parte do manga
nês depositado eventualmenLe se destaca do restante do caEoclo, perdend.o o
contato el6trico com o mesmo. Ocorre, então, a dissolução deste manganês na
solução. Fekete observou que a dissolução guÍmica do manganês nos e1etr61i-
tos em questão 6 acompanhada de desprendimento de hidrogênio, desprendimento
de amônia e pre:ipitação de hidr6xido de manganês. Com base nestes fatos,
ele sugeriu o seguinte mecanismo para este processo:
2HrO + MnSOo + 2NH40H + H2 4.34
Mn(ott), + (NH4) 2so4
4.3s
4.2.7 Estudos de Agladze e Legran
Agladze e Legran (refs.70 e 77), do Instituto Polit6cnico V.I.Lenin,
em Tbilisi (Georgia, URSS), verificaram que a ação tamponante das soluções
contend.o MnSOO e (NHO)rSOO é superior ã ação tamponante destes sulfatos
isolados, e que a adição do sulfato de arnônio eleva o pH de precipitação do
hidrõxido de manganês.
Para explicar este comportamento, eles propuseram o seguinte equi-
11 br]-o :
Mn + (NH,)^S0, +u/u
MnSO . + 2lrlH .0H +44
onde n assume os valores 4
Embora com o m6ri
* f *<*"r,"
]2* * ,,"*
e 6.
to de terem sido os primeiros autores
)++Mn- * n NH4 4.36
a sugerrr a

4s
participação de complexos amoniacais no processo, Agladze e Legran têm sido
contestados por terem proposto a reação dos íons Mrr2* ditutamente com os
-+ions NHO e nao com NHOOH (ou NHr), como proposto por Tilak, Rajogopalan e
Reddy (seção 4.2,8), pelos pesquisad.ores de Dnepropetrovsk (seção 4.2.I) e
por Barchese (seção 4.2.2) ,
Os Índices n propostos, com valores 4 ou 6, são tamb6m
considerados muito elevados por Tilak, Rajogopalan e Reddy (seção 4.2.8). Os
pesquisadores de Dnepropetrovsk, contudo, identificam a existência de comple
xos com n = 4 (seção 4.2,I) .
4.2.8 Estudos de Tilak, Rajogopalan e Reddy
Tilak, Rajogopalan e Reddy (ref.72), do Instituto Central de Pes-
quisa EletroquÍmica em Karaikudi (fndia), estudaram a ação tamponante das s9
luções de sulfato de manganês e sulfato de anônio, bem como a precipitação
de hidr6xido de manganês. Eles verificaram que soluções contendo apenas
Mnso, tamponam apenas em pHs de 2 a 3. A adição de (NH4) ,soO introduz uma
região de Ëamponamento em pIIs de 7 a B, ação esta muito mais intensa que a
do sulfato de amônio isoladamente. Eles confirmaram Ëambãm a elevação do pH
de precipitação de Mn(OH)2 com a introdução de sulfato de amônio.
Com estes fatos, eles conclufram haver uma interação específica en
tre os íons un] "
Mrr2*. Esta interação deveria ser a formação de um comple-4
xo: em equilíOrio envolvendo os aquo-Íons de manganês e a molãcula NHr.
A possibilidade de que este equilíbrio envolvesse o próprio íon
amônio, sugerida por Agladze e Legran (seção 4.2.7), foi descartada com o ar
gumento de que, se os equilíbrios envolvessem NHf,, não haveria motivo para a
inexistência de ação tarponån.te em pHs d.e 3 a 7, rlma vez que esses Íons es-
tão tamb6m presentes nestas faixas de pH. Jâ a amônia, sugerida pelos auto-

46
res, somente estã presente em quantidades signifícativas em pHs superíores
7, explicando-se assim a f.aixa de tamponamento observada.
Assim, o mecanismo proposto é o seguinËe:
NHr+H2oËllHf+oH
+ NH, + xHrO
4.1/
4. 38
A menos da explicitação dos aquo-ligantes HrO com os índices n e
D-x, o equilí¡rio 4.38 assemelha-se ao. exposto na seção 4.2.L (equilíbrio
4.75b). Entretanto, enquanto estes pesquisadores concluíram que o expoente x
pode assumir os valores 1, 2 e 4, Tilak, Rajogopalan e Reddy concluíram, a
partir de anãlise baseada em constantes de equilíbrio, que este valor seria
apenas um, e igual a 1.
Assim, o equilÍbrío 4.38 seria, simplesmente,
þ",*"r, (Hzo)"-*]'*
þ","ro,"]'.
lz,nJ 4.39
4.2.9 EsËudos de RadhakrishnamurËy, Sathyanaraygma e Reddy
Radhakrishnamurty e Reddy (ref. 73 e 74), do Departåmento de Quími
ca Inorgânica e Físico-Quírnica do Instituto de Ciãncia da Índia, em Bangalo-
re (Índia), estudar¿tm os mecanismos d.e eletro-redução de manganês sobre aço
inoxidãvel por meio de curvas polarogrãficas parciais. A eficiência de cor-
rente de manganês, cujo valor foi necessãrio para o traçado destas curvas,
foi obtida por um m6todo (ref.73) que viemos a adotar em nossas experiãncias
(segunda etapa do m-etodo descrito na seção 5.4.2),
l*" ,"r0, + NH3 Ë þ" *"r(H2o)"-r]'. + Hro

47
Mecanismo de eletro*redução de ganganãs
Estes auLores verificaram (ref. 74) que: a) a deposição catõdica
de manganês se dá por uma reação de primeira ordem em relação ao manganâs, ou
seja, dlogiMn/dlogcrrr=1 ; b) a densidade de corrente de troca e o coefíci-
ente de transferãncia desta reação não se alteram substancialmente com a va*
riação do pH entre 2 e 7; c) a declividade Tafel na deposição catõdica do man
ganês 'e tZO mV,d6cada-1; ¿) a declividade Tafel na dissolução anõdica do man-
ganôs ä 4O rnV,d6cada-1. Com estes dados eles sugeren o seguinte mecanismo:
')¿- * +Mn'' + e. -' Mn (etapa lenta) 4.40
+Mn+e+Mn 4.4L
EsËe mecanismo corresponde simplesmente ao desmembramento da reação
global (que envolve a tranferência toËal de dois el6trons) em duas reações su
cessivas, cada uma com Ëransferência de um único e16tron. Isto justifica-se
pelo faËo de que a probabilidade de transferËncia simultânea de dois el6trons
6 rnuito baixa (ref . 75) "
Os autores observaram comporLamento de prímeira ordem em relação ao
1tíon Mn'-e na reação cat6díca. Seu modelo 6 coerenLe com isto, poís a primeira
reação (4.40) ã lenta e apresenta índice estequiométrico unitário para a esp6*
1t-cie reagente Mn' , o que corresponderia, segundo a equação de Butler*Volmer
(ref. 75), ã seguinte expressão:
i = n " F ( Ê . "M'o . "ãnnó/nr- È . "Mrr2* "-ãrnÓ/nr,
Nos potenciaís suficienËemente cat6dicos para que se possa ignorar
a reação anódica, a ordem da reação seria porEanto definida pelo expoente
4.42

47a
de c*rr2+, ou seja, o valor 1.
O mecanismo proposto 6 tamb6m coerente com as declividades Tafel e4
contradas' uma vez que, de acordo com a equação de Butler*Volmer desenvolvida
para reações com mais de uma etapa (ref. 75), dada por:
i = io{ exp I filu+ r * rß ) Fn/Rr] - exp I Ci¡u+ rß) rn/nr] ] 4.43
as declividades Tafel são dadas por:
a¿ BT _ 2,3 Rr4.44
4 "4s_ _2r3RT _ 2r3RT"l -;-¡- - l-7v +'BTF
onde:
coeficiente de transferência da reação anódica
coeficíente de transferãncia da reação cat6dica
número de etapas anteriores ã eËapa lenta
número de etapas posteriores ã etapa lent.a
número de vezes que ocorre a etapa lenta no processo total
fator ígual a I para etapas eletroquÍmicas
faËor de simetria
No modelo proposto, tem*se f =1rf =0 , v*1 e rË1" Assim, Ëoman
do-se ß =0r5 , obt6m-se ä= 1r5 e ã= 0r5 , o que corresponde a declividades
b, = 40 mV.dãcada-l " bb = 720 mV.d6cada*1,
"oro observado experimentalmen-
te. É inEeressante observar que seriam obtidos resultados opostos, ou seja,
b^= L20 mV.däcada-l " bb = 40 mV.dãcada*l, se fosse proposta como lenta a
segunda reação (4,4I), pois terÍamos enËão f = O e T= f
-(-0*->0-<-Y--tY*
r-
ß*

47b
O mecanismo proposto não possui qualquer etapa que envolva espäcies
+H' ou OH . Este faro ä coerente com a independência observada entre o pH e
a densidade de corrente de troca e o coeficiente de transferência. Devido a
esta independência, os autores descartam outro modelo:
)t- -. +Mn''+OH+Mn'
Mn0H +e
+eMn
-)' MnOH (eLapa lenta)
+Mn+OH
4.43
4.44
4.4s
Este modelo, semelhante ao mecanísmo proposto por Bockris eLal. pa-
ra a deposíção e dissolução de ferro (refs. 75 e 76)r levaria äs mesmas declí
vidades anõdica e cat6díca observadas para o manganês, assim como a um compor
tamento de primeira ordem em relação ao Íon Mo2+ ,,.r reação catódica. Entretal
to, ele irnplicaria em um comportamento de primeira ordem em relação ao íon OH*,
o que, como jã cítado, não foi observado.
Efeito do ãcido se1ênico
Radhakrishnamurty e Reddy estudaram tamb6m a açã-o do ácido selênico
sobre o pïocesso (ref . 77). Eles verificaram que o selênio não a1Ëera a densi
dade de corrente de troca da reação de deposição do manganês, mas reduz a re*
ferente ã reação de desprendimento de hidrogãnio" Assím, seu efeito sobre a e
ficiêncía de corrente seria devido ao reËardamento do desprendimento de hidro
gênio.

4B
Desprendimento d_e hidrogênio - o filme de õxido
Radhakrishnamurty, Sathyamarayana e Reddy (ref. 7B) estudaram a cí-
nãtica de desprendimento cat6dico de hidrogênio sobre catodo de aço inoxidá-
vel, em soluções de sulfaËos de amônio e de manganês. Para que não houvesse
deposição cat6dica de manganês, foram utilizados potenciais mais anódicos que
os necessários para aquela reação. Os autores verificaram que suas curvas po*
laxogr'afícas, apõs apresenËarem um comportamento linear (obedecendo ã f,eí de
Tafel) nas baixas densidades de corrente, passam a se desviar deste comporta-
menËo em densídades de corrente acíma de 3 . tO-3 a. "*-2 " o valor da diferença
entre o potencial medido nesLa região e o potencial extrapolado do trecho 1i*
near moslrou*se proporcional ao valor da corrente, permitindo aos autores o
cãlcu1o de uma constante de proporcionalidade, por eles aËribuÍda a uma perda
ôhmica" Segundo os autores, ela seria devida ã presença de um filme de 6xido
semicondutor sobre o caËodo. A partir da consËante de proporcionalidade (con-
siderada como a resistência do fílme) e supondo que este filme tenha uma es-
pessura de 1008 , sua resistividade estaria em torno cle 1r5 . 707 0. cm , va
1-or que estã na faixa das resistividades dos semícondutores.

49
4.3 RESIIMO DOS MODELOS APRESENTADOS
De todas as informações e hip6teses apresenta<las neste capítu1o, re
lacionamos e resumimos adiante as que se nos afiguram mais importantes. No
primeiro item listaremos as mais fundarnentais e consensuais. No segundo, ou-
tras menos consensuais (mas não necessariamente menos fundamenËadas). No ter
ceiro, finalmente, expomos as contradições entre os modelos mais fundamenta-
dos.
4.3.I Informaeões e hipóteses mais consensuais
a) A eletro-redução de manganês faz-se a partir de duas ou maís esp6-
cies eleLroativas: o aquo-íon Mrr2+ e um ou mais íons complexos de
manganês . (V. seções 4.2.L e 4,2 .2)
Existqm equilíbrios que envolvem íons complexos de manganês (eletroa
tivos ou não), aquo-íorrc Mrr2*, NH3 e oH " Estes equilíbrios afetam
a eletro-redução do íon complexo eletroativo, elevam o pH de precipi
tação de hidrõxido de manganês e definem um mecanismo de tamponamen-
to da solução em pHs de 7 a 8. (.V. seção 4.2.2)
Os íons complexos de manganês contêm o ligante NHr. (V. seções 4,2.L
e 4 .2 .2.)
Existe interdependência entre mecanismc¡s de desprendimento de'hidro-
gônio e deposição de manganês, provavelmente através de reações quí-
micas entre espécies adsorvidas na superfície do catodo e íons pre*
sentes na solução. (V. seções 4.L.3,4.2.I e 4.2.2,)
O efeito das impurezas mais prejudiciais ao processo está de a1.gum
modo relacionado com a baixa sobretensão de hidrogênio por elas apre
sentada. (V. seções 4.L.4 e 4.2.3.)
b)
c)
d)
e)

50
f) Deposições reaLízadas em elevadas densidades de corrente catódica,ou
com eletrólitos contendo compostos redutíveis de enxofre ou selênio,
levam à forrnação de Mn-o, ; em caso contrário, forma-se Mn-y. (V. se-
ções 4.2.L e 4.2.5.)
g) Deposições realízad.as sem rcompostos redutíveis d.e enxofre ou se1ênio
apresentam uma densidade de corrente-limite na reação de eletro-redu
ção de manganês . (V. seções 4.2.L e 4.2.3,)
4.3.2 Oulras hipóteses e informações importantes
O complexo eletroativo de manganês cont6m, al-em do ligante NH,r o 1!
ganre oH . (v. seção 4 .2 .2 .)
0 complexo eleÈroativo de manganês conserva alguns dos ligantes HZO
do aquo-íon Mrr2* . (v. s eção 4 . z .2 ,)
A eletro-redução do aquo-íon Mrr2* dã-se em cluas etapas consecutivas.
A primeira ã eletroquÍmica e forma o íon Mr,+. A segunda -e química e
consiste na redução destes íons por ãtomos de hidrogênio adsorvido
a)
b)
c)
d)
e)
(v. seção 4.2.2.)
Existem diversos íons cornplexos
em equilíbrios entre si e com o
4.2.3.)
de manganês na solução, envolvidos1t
aquo-íon Mn''. (V. seções 4.2.L e
f)
O desprendimenËo cat6dico de hidrogênio sobre manganês pelos mecanisI
mos clãssicos dã-se a partir de HrO ( e não de HrO') e tem controle
eletroquímico, provavelmente devido ã eËapa de eletro-redução de HrO
a H-, e OH (V. seção 4.1.1.)âcl
Ocorre também desprendimento catódico de hidrogênio sobre manganês
a par:ír dos íons *t4*. (V. seção 4,7.2,)
Existe um mecanismo químico de redução de hidrogênio por ãtomos Mns)

51
ou íons Mn+ adsorvidos sobre o catodo. (V. seção 4,I.3.)
h) A formação de Mn-cr em deposições com elevada densidade de corrente
cat6dica ou en eletr6litos contendo compostos redutíveis de selênio
ou enxofre estã de algum modo relacionada com o aumento d.a concentra
ção de hidrogênio sobre o depósito, resultante do retardamento da
reação de combinação de átomos de hidrogênio. (V.seção 4.2.5.)
i) O efeito das impurezas mais prejudiciais ao processo, além de estar
relacionado com suas sobretensões de hidrogênio, pode estar relacio-
nado com seu potencial-padrão de eletrodo, mais positivo que o
manganês . (v. seção 4 .2.3.)
do
4.3.3 ContradiÇões entre diferentes modglos
0s modelos propostos pelos pesquisadores de Dnepropetrovsk (seção
4.2.I) e pelos pesquisadores de Kiev (seção 4.2.3) afirrnam que a esp6cie1t
eletroativa Mn'- é eletro-reduzida corn baixas sobretensões. Com maiores so-
bretensões, este mecanismo atinge uma corrente-limite d,e caráter químico.
Com sobretensões ainda maiores, surge um novo mecanismo constituído pela re-
dução de um Íon complexo de manganês.
Estes modelos pressupõem, portanËo, que: a) a eletro-redução de
Íons complexo se dê em potenciais mais negativos que o do aquo-íon Mrr2+; b)
exista uma etapa química precedente ã eletro-redução do Mn2+, e esta etapa
química se torna fimitante acima de dados potenciais.
Barchese verificou exatamenle o contrãrio (seção 4.2.2): a) a redu
ção do íon complexo dã-se em potenciais mais positivos que a do aquo-íonnt
Mtt''; b) não existe etapa química lenta precedente ã eletro-redução do aquo1t
-íon Mn''.

52
5. PROGBAMA E)GER-IqNTAL, EQUIPA:MENTOS E MÉTODOS
5.1 PROGRAMA EXPERIMENTAL
O presente trabalho voltou-se ao estudo da influência das impure-
zas cobalto, níque1 e zinco e do aditivo dióxido de enxofre sobre a eficiên-
cia de corrente do processo de eletrodeposição cat6dica de manganês acompa-
nhada de desprendimento catódico de hidrogênio, em soluções de sulfatos de
manganês e amônio.
Com este objetivo, foram estabelecidas trinta composições de e1e-
trólito combinando diferentes adições destas "ímpurezas" e do dióxido de en-
xofre, e medidos o poËencial de catodo e a eficiência de correnËe referentes
a deposições reali zadas com estes eletr6litos em diferentes densidades de
corrente.
Com estes resultados, traçaram-se curvas relacionando a efíciência
de corrente com a densidade de corrente e o potencial, bem como curvas de po
Larização das reações catõAicas.
Complementarmente, foram obtidas f<;tomicrografias do processo de
deposíção por meio de ní.croscópio 6ptico adaptado ã c-elula de deposição, e
fotomicrografías dos depósj-tos obtidos, por microscopia eletrônica de var-
redura.

53
Parâmetros fixos
A composição-base, o pH, a vazão e a temperaturâ dos eletrólitos
foram mantidos fixos, com valores dentro das faixas do processo industrial
(v. seção 3.4):
MnSO,+
(NH4) 2So4
pH
vazão
temperatura
_10,6 mo1.dm '
-11,0 mol.dm '
7rO
I -10,2 dm'.h '
3ooc
Adições de dióxido de enxofre e de cobalto, níque1 e zinco
As trinËa composições estudadas do eletrótito correspondem ao ele-
tr6lito-base fixo acima citado acrescido de diferentes adições de di6xido de
enxofre combinad.as com diferentes adições dos met.ais considerados como "impu
rezas", ou seja, cobalto, níquel e zinco.
Trâs valores de adição foram utilizados para o di-oxido de enxofre.
O primeiror 0rf g.am 3, correspondeu ao valor considerado adequado nas expe-
riências do U.S. Bureau og Mines (ref . 4) e amplamente utiiLlzad.o desd.e en-
tão.0s demais valores, 0r3 e 016 g.dr-3, visaram cobrir uma faixa de adi-
ções na qua1, segundo DrAbreu e Torem (refs . 4L e 42), o efeito favorãvel do
di"oxido de enxofre continua a crescer com o aumento de sua adição. (V. seção
3.6.)
Para cada um dos três valores de adição de dióxido de enxofre, fo-
ram estudados dez eletr6litos: um sem adição de impurezas, nove com adição
de impurezas. Estes nove eletról j tos correspondem a três valores de adiçãr: cle

54
cada uma das três impurezas. Um destes três valores corresporide ao limite
acima do qual, segundo Jacobs (ref. B), a deposição de manganês torna-se in-
viãve1 (v. seção 3.5). Os dois demais valores são respectivamente a metade
e o dobro daquele
A tabela 5.1 mostra os valores das adições utíLLzadas para cada um
dos e1etr61itos, numerados de 1 a 30:
Tab. 5"1 - AdÍções aos eletr:ólitos
adições de "impur ezas" (*g.a* 3)
Co
o, 2s | 0,5I
I
,r,lta adição
de
soz
(a . ar-:,0' 1
0'1 0'5
Ni
1'0 40
Zn
20
número de
i dentifi cação
do
eletró 1i to0r3
0'6
Densidade de corrente catódica
Foram utilizados oito valores de densidarle de corrente cat-odi ca
17 ,4 22,g 30,2 39,8 52,5 69,2 gL,z I20 *4. "r-2
EsËes valoïes formam uma progressao geoni-etrica, de modo a ficarem
equidistantes na escala logarítrica utíIizað.a nos gráficos das curvas polaro
idt
1
2
3
4
5
6
7
B
9
10
11
T2
13
L4
15
I6
I7
1B
L9
20
27
22
23
24
25
26
27
28
29
30

55
gráficas. AproximadamenËe no centro da faixa estão as densidades de corren-
te que conduzem a eficiências de corrente mais elevadas e que são utiLízað,as
industrialmente (entre 40 e 50 *4."*-2, como vimos na seção 3.4.1).
5" 2, EQUTPAMENTOS
5.2.I Cãlula e complementos
cé1u1 a
As figuras 5.1, 5.2 e 5.3 mostram a c61u1a utilízada nas expe-
riêncías. Ela 6 feita de acrílico, tem aproximadamente 15 cm3 e foi constr'l
da de modo a permitir observação visual do processo eletroquímico atrav6s de
um microscópio de pequeno aumento. Esta observação visual f.az-se atrav-es da
parede de acrílico que, para melhor imagem, possui espessura de apenas 2 mm
ã frente do eletrodo de trabalho. A cã1ula dispõe de orifícios para entrada
e saída de e1etr6lito, para saÍda do hidrogênio formado e para introdução de
um capilar de Luggin. Ela possui tamb6m um diafragma de vidro poroso, gue a
divide em dois compartimentos. Em cada um deles existe uma janela, contra as
quais são colocad.os o eletrodo de trabalho e o contra-eletrodo.
A áiea exposta d.o eletrodo de trabalho 6 delimitada por um anel de
PVC, e a do contra-eletrodo por um anel 0 ("O-ring") de neoprene apoiado em
um suporte de PVC.
A vedação dos anãis de PVC faz-se por meio de anóis
fler comprimindo-se os eletrodos e os an-eis contra a c-e1ula por
fusos de aperto fixados a barras de alumínio.
Para f acílidade de limpeza, a cã1ula -e desmontável e
de vidro poroso pode ser retirado. A vedação do conjunto f.az-se
0, de neopre-
meio de para-
o diafragma
por meio de

urna junta de neoprene. O aperto é
tro prisioneiros de latão que são
feito por meio de
fixados a uma das
56
porcas montadas em qua-
peças de acrÍ1ico.
HI#
Fig. 5.1 - Fotografia da célula desmontada
€ Ìt,
È,1
:_
offioo
--1 2
_t
CORTE A-A
COMPARTTMENTO DO
ELETRODO DE TRABALHO
Et
l*t'=frFul
Fig. 5"2 a - cãlula (vista frontal e cortes longitudinais)
I ,qlI k--k_ 59
ViSTA FRCruTAL
(representados apenas o corpo e. a tampa deacrílico, o diafragma e os prisioneiros)
It-CûNTF B-B
COMPARTIMENTO DO
CONTRA-ELETRODO
Escala;1:1
þgge!yþ-: ríg. 5 "2 c (tt.\¡

6
srçAûc-c .INTERIOR DA CÉLULA, COM INTERIOR DA CÉLULA E
ENTRADAS E SAÍDAS DE ELETRóLITO REBAIXO PARA OBSERVAçÃO VISUAL
observação: a _posição das seções aqui representadas estã índicada na Vista Frontal (Fig" 5"Z aj
Fig. 5"2b - Cãlula (seções transversais)
16o.-U!I
sEçp,0 Ð-D
--¡iI
I
I
I
r-Jc\
s[ÇA,t
S]STEMA DE FECHAMENTO DAE DE POSICIONAMENTO DASDE FTXAçÃO DOS ELETRODOS
4scala:1:1Legenda: Fig. 5.2 c
rrt__-L
CELULAPT¿,CAS
(.¡¡

1
2
3456
7
- Corpo principal (acrílico)- Tampa (acrílico)- Junta de vedação (neoprene)- Diafragma poroso (vidro sinËerizado)- Compartimento do eletrodo de trabalho- Eletrodo de trabalho (aço ABNT-316)- Anel de vedação, em PVC; delimitador da ã-
rea exposta do eletrodo de Ërabalho- An6is O ("O-rings") de neoprene para veda-
ção das janelas para os eletrodos- Janela para o e1eËrodo de trabalho- Entrada de eletrólito- orifício de saída de hidrogãnio- Tubo de saída de hidrogênio (PVC)- Capilar de Luggin (vidro)- Orifício para o capilar de Luggin- Rebaixo para observação visual do eletrodo
I
9
1011T213L415
Lesenda das f ieuras 5.2 a e 5.2b2
de trabalho16 - Contactos e16Ëricos (latão)17 - Fios de cobre ligados ao circuito elãtrico18 - Isoladores elétricos (madeira)19 - Parafusos de fixação dos eletrodos
20
2t
22232425262728
29303132
-Placa posterior, de alumínio, para f.íxação doeletrodo de trabalho (e tarnb6m da c61u1a)
- Prisioneiros de latão , para fixação da placaposterior e fechamento da tampa
- Porcas de aço para fixação da placa posterior- Molas para afastamento da placa posterior- Porcas de aço para fechamento da tampa- Orifícios para fixação da célula ã bancada- Compartimento do contra-eletrodo- Contra-eletrodo (liga chumbo-prata com 2Z Ag)-Anel O ("O-ring'o) de neoprene, delimitador da
ãrea exposta do conlra-eletrodo- SuporÈe do contra-eletrodo (PVC)
- Janela para o contra-eletrodo- Saída de eletr6lito-P1aca frontal, de alumínio, para fíxação do
contra-eletrodo- Prisioneiros de latão, para fíxação da placa
frontal e fechamento da !*p.- Porcas de aço para fixaçao da placa fronËal e
fechamento da tampa- nn6is de PVC para afaslamento da placa frontal
Fig" 5"2 c - Cãlula (legenda)
33
34
35
L¡\o

60
Eletrodo de trabalho e contra-eletrodos
Os eletrodos de trabalho são de chapa inoxidãvel ABNT- 316 com 2mm
de espessura, cortada em peças de aproximadamente 20 x 20 ,*,.2. ,ut^ uniformi-
zação da qualidade superficíal, eles eram lixados seqtlencialmente em lixas de
240, 320, 400 e 600 grãos por polegada e em seguida polidos com 6xido de cro-
mo. A superfÍcie exposËa é circular, com 1r03 cm2. Antes da deposição, o ele-
trodo era lavado com tricloroetileno e 'agua deioní zada. Um mesmo eletrodo de
trabalho era uËilízado para três deposições seguidas (o depõsito de manganês
era consumido por corrosão, entre as deposições), sem ser retirado do eleËr6-
lito. Caso fosse ultrapassado este número, não mais se obtinha reprodutibili-
dade de resultados, seja em termos de eficiêncía de corrente, seja em termos
cle potencial de eletrodo. Uma vez telírado, um eletrodo podia ser novamente
polido e então reutilizado.
O conËra-eletrodo é ae liga chumbo-prata com 27" de prata, obtido
da forma exposta a seguir. Fundiu-se 1174 kg de chumbo em um cadinho de carbo
neto de silício aquecido por maçarico de GLP, e adicionaram-se lentamente
L7,36 g de prata em g16bu1os. Quando bem homogeneízad.a, ap-os trinta minutos,
a Liga foí vazada em um molde cilíndrico bipartido de ferro fundido. Do taru-
go obtid.o, foram usinados os anodos utilizados nas experiências prelíminares,
com lr5Z de prat.a. Para as experiências definitivas, visando-se aumentar o
efeito passivador da prata (v. seção 3.3.5), preparou-se nova 1iga, com 2% de
prata. Para tal, fundiu-se o restante do tarugo anterior, adícíonou-se a pra-
ta necessãria e vazou-se um novo tarugo, em operação semelhante ã anterior.'
Da parte central deste novo tarugo, ußinou-se o anodo, na forma de um disco
com 12 nrn de diârnetro e 3 mm de espessura. A superfície exposta do anodo Ë
circular, com 0r5 cm2" Como as deposições tinham curta duração, a camada pas-
sivadora aderente - como descrita na seção 3.3.5 - não chegava a se consti-

tuir, formando-'se
que freqtlente-mente
pelo eletrólito.
sobre o anodo
se desËacava,
6r
um depósito marrom escuro de baixa aderência
acumulando-se na c-elula ou sendo al:rastada
Ponte salina
0 capilar de Luggin comunica-se com um reservatório de vidro onde
estã mon,tado o eletrodo de referência (seção 5.2.4). O reservatório, que con.
t6m o mesmo eletr6lito que a célula, estã dentro de um recipiente de acrÍli-
co ond.e circula termostatizað,a a aproximadamente 3loC. O sistema -e mostrado
nas figuras 5.3 e 5.4.
Circulação de elerrólito
As figuras 5.3 e 5.4 mostram o sistema de circulação de eletr-oli
to. EsËe 6 alirnentado de um reservatório de acrílico, que possuí uma serpen-
rina ínterna de vidro por onde circula ãgua termostati zada a aproximadamente
31oc. A altura de fixação do reservatório é regulãvel, de modo a se poder
manter constante o níve1 de eletrólito. Este cuidado 6 necessário para que
se mantenha o níve1 adequado do eletrõlito no reservatório da ponte salina,
uma vez que estes dois reservatórios se intercomunicam através da c61ula, e
o níve1 de eletr6lito nos mesmos resulta aproximadamente igual (o mesmo tam-
b6m ocorre, aliãs, com o tubo de saída do hidrogênio, que fica cheio de ele-
tr6lito at6 o nível em questão).
O eleËr61ito entra na cé1u1a pelo compartimento do eletrodo de tra
balho (catodo, na deposição) e sai pelo compartimento do contra-eletrodo(ano
do, na deposição), cle onde vai para um erlenmeyer. Para se obter a vazão d,e-
sejada, o eletr6tito ã aspirado por meio d.e uma trompa d'âgua ligada ao er-

lenmeyer
tubo de
62
ou, pelo contrário, ã contido por meio de uma válvula colocada no
s aída.
Ap6s filtragem do e1etr61ito, este 6 recolocado no reservat-orio de
são
de
alimentação.
(Observação: O eletrólito circulado tem composição quírnica muito
pr6xima da inicial, pois o manganês que dele 6 retirado, na etapa de deposi-
Ção, retorna em seguida, na etapa de corrosão do dep6sito - v. seção 5.5. O
único manganês que não retorna é o que compõe o dep6sito sobre o contra-ele-
trodo, mas esta quantidade é pequena em relação ã contida no eletrõlito.
Em vista dísto, o eletrótito pode ser reutili-zado).
Sistema de termostatizaÇão
A ponte salína e o reservatório de alímentação de eletrólito
ËermostaËizad.os por 'agua contínuamente fornecida por um reservatório.)
12 dm' dotado de uma resistência elétrica de 2000 I^I e de um termostato tipo
liga-desliga. Este sistema, esquematizado na flígura 5.4, fornece cerca de
- 3 -1 -+o --+olcm".s'dc'agua a 31-1"C, mantendo a temperatura do eletrõlito em 30:1"C
nos reservatórios (g temperatura do eletr-olito é acompanhada por meio de um
termômetro de mercúrio mantido no reservat-orio de alirnentação) . Como o tempo
de permanência do e1eËr61ito na c-elula 6 pequeno - cerca de 4 minutos - sua
temperatura se mantém dentro daquela faixa durante todo o processo eleËroquÍ
mico.

63
Fíg. 5.3 - Fotografia da
de eletró1íto
c61u1a com seu sistema de circulaçao

64
JLO
*,
I
I
I
I
I
I
I
I
¡
I
I
I
I
I
I
I
I
I
I
I
I
I
I
iI
I
I
I
II
I
CONVENCOES:-----_-L-
l=E --) elerrólito
t l + ãgua (para termostat ízagão)
Lesenda:
1 - cãlula (2ocm3, acrílico)2 - DiaÍragma poroso (vidro sinÈerizado)3 - Compartimento do eletrodo de trabalho4 - Compartimento do contra.-el-etrodo5 - Entrada de eletr6lito na cóLula6 - Capilar de Luggin (vidro)7 .- Saída de hidrogânio rla cãLrrl-aB * SaÍda de eLerr6Lito da c6'lui-a9 - Reservat6rio alimenfadc¡r de eLetr6Ii
to (200cm3, acrílíco)10 - TermômeÈro de mercúrio11 - Reservat6rio de eletr6lito da ponte
saLina (50cm3, viclro)12 '- Eletrodo de ref erâncíaL3 - Col.una de saÍda de hídrogênio (vídro)1l+'- Pinça rle ì4ohr.15 - Tromp a .,1'âgua (pVC)
Fig. 5.4 - Circulação de elerróliro e
T6L71B
'.19
2A
27222324
25
26
de
- Erlenmeyer (eletr6lito usado)- Fíltro (eIetró1ito recirculado)- Reservat6rio termostalízador pa
ra âgua (20dm3, plãstico)* Entrada de água corrente* Resistência e16trica (2k!ü)'- Termostato liga-desliga* Saída de água- Ladrao- Serpentina termostaËizadora do
alimentador (vidro)'- Recípiente termosta-t ízador da
pontã salina (150crn3, acrí1ico)- SaÍdas de água corrente
água de termostaËização

65
5.2.2 Microscopia 6ptica para observação de deposição
Um microsc6pio estereoscópico PZO MSt 130124L0 foí montado perante
o eletrodo de trabalho, com eixo óptico em posição hori zontaL, permitindc ob
servação visual da forrnação e corrosão do dep6sito de manganês e do despren-
dimento de hidrogênio, aËrav-es da parede de acrílico da c61u1a.
Para se obter registro fotográfico destas imagens, adaptou-se a um
dos tubos oculares do microsc6pio um conjunto Leítz para fotomicroscopia, dc
tado de cànara Leica MDa, ocular Leítz Periplan 10>rl"I e tubo com prisma para
focali zação e fotomettia. Para o acoplamento, utilizou-se uma cinta de bor-
racha enËre os dois tubos oculares e um suporte especial de PVC para apoio
da ocular. O aumento línear máximo obtido sobre o negaËivo foi de doze vezes.
Utilizou-se ainda um t'flash'r eletrônico Canon Speedlight 1554r euê ficava a
100 mm do eletrodo de trabalho. As figuras 5.5 e 5.6 mostram este conjunto:
Fig. 5.5 - Fotografia da c6lula e
do microscópio óptico
em posição de observa-
ção de uma deposição

66
VISUPER I OI
REBAT I MEN
SOBRE
PLANO A_
AN\/ 'b
ri
5;;
STA il)RElT0
lr¡'!1 1B
U I1 I
¡\t'o
/h-I
IC
LÈI
18
OCULAR FOTOGRAFICA E
DO TUBO PORTA.PRISMA(rscau 1:z)
- c6lu1a- Paredes da c61u1a (acrí1ico)- Eletrodo de trabalho- Rebaixo -para observação- Microsc6pio estereosðópico- Objetiva- Tubo ocular esquerdo, sem a
ocular orígínal- Tubo ocular direito- Conjunto fotomicrogrãfico- Tubo porta-prisma
Legende:
11 - Tubo de fíxação12 - Ocular fotogrãfica13 * Suporte de pVC para a ocular14 - Anel de adaptação (borracha)15 - Câmara forográfíca.l.6 * Visor17' "Flash" eletrônico18 - Fonte de iluminação permanente19 - Base do microsc6pío20 - Cantoneiras fixas da bancada21 - Braços articulados da bancada
Escala: 1:5
1
234567
B
9
10
VISTA LATËRAL
DIREITA
DETALHE
DA FIXAçAO DA
¡'ig" 5.6 * observaçãa e regí.sËr' fo'ogrãf ico cra deposição

67
5.2.3 Microscopia eletrônica para observação do depósito
Para observação dos dep6sitos obtidos, foi utilizado um microsco-
pio eletrônico de varredura marca AMR, modelo 900.
5.2.4 Instrumentação e circuito e16trico
Como veremos adiante (seção 5.5), utilizou-se um m6todo de determi
nação de eficiência de correnËe que exigía que, logo após a deposição do man
ganês no eletrodo de trabalho, este manganês fosse devolvido ã.solução por
meio de corrosão química ou eletroquímica. Para se poder reai-J-zar a corrosão
eletroquímica com uma dada corrente imediatamenËe ap6s uma depostçáo rea1.íza
da com ouËra corrente, foram utilizadas duas fontes retificadoras estabilíza
das. Uma delas foi uma Tectrol TC-10-05-S (que aËinge até 10V e/ou 5A) e a
outra foi uma Hewlett-Packard Harrison 6209 B (que atinge at6 320V
120 mA).
ef ou
Como se vê no círcuito mostrado na figura 5.7, para se f.igarem os
eletrodos da cé1ula a uma das fontes, ã outra ou a nenhuma delasr utilízou-
se uma chave comutadora bipolar de três posições " Uma outra chave bipolar ,
de d.uas posições, foi utilízada para que se pudesse empregar um multímetro
digital (Analog M-3525) t.anËo como amperímetro quanto como voltímetro. Como
amperímetrorele era utílizado para a pr-e-fixação da corrente nas duas fon-
tes . Como voltímetro, ele era utilizado para a medida do potencial do e1e-
trodo de trabalho, atrav6s do eletrodo de referência (de calomelano, não-sa*
turado, marca Tngold, modelo 303-Pa-Ns-K7). Para acompanhamento da estabili-
dade da corrente durante o processo eletroquímico foram utilizados ainda
dois multímetros anal6gicos, cada um ligado a uma das fontes. Um deles era
um Hartmann & Braun Multavi If e o outro um Normameter GI,rI .

6B
s trl6 tl l
a) rixação da correntede deposição
b) Fixação da corrente decorrosão eletroquímica
d) Corrosão eletroquímica
!çe9"4qt1 - Fonte retificadora estabílízada2 - FonËe retificadora estabiLízada3 - Multímetro anal6gico4 - Multímetro anal6gico5 - Chave (posições I, II ou Itr)6 - Chave (posições I ou tr )7 - MultÍmetro digitalB'- c6lu1.a9,- El-etrodo de trabalho
l-0 * Eletrodo de ref erêncial-1 - Contra-eLetrodo
A - amperímetroV - voltímetro
s tml6 ttrl
ì
e) Corrosão química ou intervalo entre deposições -
c) Deposição
Fig. 5.7 - Círcuito el6trico

69
b)
5.8
Jh-
-\h*.à.!gø!
vista lateral
- Fotografias do
a) vista superior
arranjo experimental

70
5.3 PREPARAçÃO DOS ELETRõLITOS
Os eletrólitos foram preparados a
seguintes reageïÌtes químicos "pro analisit':
part.ir de âgua deionizada e dos
Merck
Merck
Merck
B d A Allied Chemical Qeel
Fi sher
E Cl Dra
E cibra
MnSOO.Hr0
(NH4) 2SO4
CoSO, .7HZO
NiS04 .6H20
ZnSOO.THZO
NarS0,
NH40H
Inicialmente, foram
p.a.
p. a.
p.a.
p. a.
p. a.
p.a.
p. a.
preparadas as seguintes solrrções-estoque:
A-
B-
C_
D-
E_
MnSO,L+
(NH4) 2So4
CoS0O
NiS04
ZwSO O
NarS0,
1
0,67 mo1.dm '_1
1,1 mol.dm'
0,0017 mol.dm-3
0,0034 mol.dm 3
0,068 mo1.dm-3
01048 ino1.¿* 3
Com estas soluções-estoque, prepararam-se, por volumetria, 400 crn3
de cada um dos trinta eletr6litos desejados. Estes eletrólitos eram obtidos
com pH em torno de 3 a 4. Ap6s aquecídos atle a temperatura fixaða para as

7T
experiências, 30oC, e pouco antes de serem utilizados, os eletr-olitos tinham
seu pH elevado para 7 pela adição de hidr-oxido de amônio. Para o controle
desta adição, utíIízou-se um peagâmetro digital Phoenix modelo MS-2, com e1e
trodo Bradley-James (eletrólito:KC1 suspenso em ge1), calibrado por meio de
soluções- tarnpão Qee1.
Com base nos teores mãximos de impurezas (e nos teores mínimos dos
componentes principais) asse.gurados pelos fornecedores dos reagentes quími-
cos p.a. utílizados, e considerando-se que o teor de impurezas presentes lìa
ãgua deionízad.a 6 muito inferior ao provenienËe daqueles reagentes, calcula-
mos as cornposíções quÍmicas dos trinta eletrólitos preparados. Estas compo-
sições são apresentadas na tabela 5.2. Observamos que, a menos das adições
feitas intencionalmente, apenas uma impureza se aproximou dos limites mãxi-
mos de concentração estabelecidos por Jacobs para 24 horas de deposição (v.
seção 3.5). Esta impureza f.oí o níquel, gue atingiu um Èeor máximo de
-20r6 mg-dm'nos eletr-olitos em que não foi adicionado intencionalmente; esËe
teor representa 602 daquele limite mãximo admissível.
5.4. DETERMINAçÃO DOS POTENCIAIS DE ELETRODO
Como exposto em 5,2.4, o potencial de eletrodo foi nedido em relação
a um eletrodo de referência de calomelano não-saturado ligado a um multíme
tro digital. Na escala utilizada para esËa medida (escala de 2 V), a impedân
cia interna do multímetro era da ordem ð,e 25 a 50 kO , segundo determinação
indireta. EsËa determinação foi feita medindo-se com o multÍmetro a tensão
aplicada aos polos de uma resistência de precisão (leeds-Northrup) para se
passa.r uma corrente conhecida, pré-fixada, fornecida por uma fonte estabili-
zada (Hewlett-Packard Harrison 62098); a pr-e-fixação da corrente fora feita

Îab. 5.2 - Conposição quÍmica dos e1eÈr61itos
û¡mero
do ele-
r16tiro
I2J
456
7
I9
10111t13L415L6t71819202I2223¿42526na
282930
teores calcuLados com base nos valores nãxinos e mÍninos assegûrados peros fornecedores dcs reagentes utilizados,-3( g.dm )' I ( r,g. ar-3 ¡¡fn M4 so, I co Ni zn pb Fe cu As cd ca Mg Na K cl po, co-I
ué srg N¿r .4 _ - Z *03 CO3nlit t,l'
i:li : iüi 2',1 1,5 1,s 0,73 <10-3'0,13 7:3 6,0 7s u:.0 6,4 a,67 0,020 r,3 0,22rr t' 0160 oreo øit f, ,, r, rr ,, " n 2L2 r 'r ,, 'r,
:l " o,fo O;25 0,60 6rL !r rr , . ,, " ', 418 rr * , rr t' 0'66
l: " g'lq o;?5 0,60 6,r '¡ ,, ,r ,! ,, " '¡ 75 'Ìr ', ' rr " 1'3
:: " oreo orzs 0,60 6,L , ,r r! , ,, " " 2L2 !t t' * ,r 't 0'22
:l " O,iO Oi¡O 0,60 6,! , rr r¡ , ,, " '! 418 !r rr rr , " 0'66
l: " lrso orso lroo 6rr rr rr ' , ¡¡ rr r' 75 rr 'r rr 'r
" 1'3t¡ttO'OOO'SO0,606rL,¡r,¡,,,"t'2L2rr,,'t0'22
:l " OrfO trç 0160 6rI ri ,, , rr , rr rr 4LS . 'r ' ,r " 0'66
:: " !r:o 1;ô 0,60 6rr ¡, ,, ,, !r ,, " " 75 r' ', , ,' " l'3tr tr A160 1.;0 0160 611 r, rr rr ' rr
tt rr 2L2 !r rr rr , tt 0'22
¡t rr Or10 Ir1 6rL !r rr , , ,, " " 418 r rr r . " 0'66It rr Or:O - Ir1 611. ,, rr ,r , r,
tr tr 75 r rr . rr " 1'3trrtO,OO-1r1
6rL,r,,,,"'t2L2rrtr,tt0'22tr tr OrfO - 116 6rL ¡r , Ír * ,, " tt 418 tt rr , , tt 0'66
Ittr0r3o-1166rLrrr,,r¡,,""75rrr,tt1'3rr It 0160 - Lr6 611 rr rr rr r, ,, " 't 2I2 r rr rr ,
n 0'22rr tr O,fO - 216 6,! rr r¡ , rr ,, " " 418 tt r r r
t' 0'66ttrtor:o-2166rl ,rrr,,,,""75rrrrr,tt1'3rÌ tt O,OO - 216 611 ,r rr ,r , ,, " t' 2L2 t rr r r " 0'22rr tr orro - 0160 16rl rr ¡r r, ,r ,,
tt " 418 t rr ,, ,, " o'66rr rr or:o - 0160 16,11 ¡r rr rr rr ,, " " 75 rt rr r r
tt 1'3tt r¡ 0;60 - 0160 16rl ?r ,r * , ,, " tt 2r2 rr rr rr !r
tt o'22rf rt 0,1O - 0160 z6rL ,r ,, , , ,, " rt 418 rr , r rr " 0'66It rt 0r3o - 0160 26rL ,. ,r * , rr
rt tr 75 r r rr rr tt 1'3
rr rt Oreo - 0160 26rL l, ,, rr ,r ,, t' " 2L2 t rr , , 't o'22
tt ir oiro - 0,60 46rL r ,, r! ,r ,, " " 418 tr rr r rr " 0'66tt tr 0130 - 0160 46rL ,r , , r, ,, " " 75 tr r rr rr
tt 1'3!t rt o,oo - 0,60 46rL ,t , rr . ,, " t' 2I2 rr r rr r 't o'22
' ,' 4Lg , ,, rr , :: 9'9u
!t'J

73
com auxílio do próprio multímetro.
Considerou-se como potencial de deposíção PO o valor medido a par-
tir do quinto minuto de deposição, uma vez que naquele momento o potencíal,
já se mostrava sensivelmente estãvel. Esta estabilidade começava a se defi-
nír jâ a partir do segundo ou Ëerceiro minuto de deposição, como mostïam as
figuras 6.2 e 6.4.
Nossos potenciais de eletrodo foram expressos na escala do eletro-
do de calomelano saturado (ECS) " Este valor é ligeíramente di.fer:ente daquele
que medimos, uma vez que nosso eletrodo de referência era de calomelano não-
saturado. Esta diferença, correspondente ã diferença entre a concentração de
cloreto de potãssio em nosso eletrodo e o limite de saturação deste sal, foí
determinada inúmeras vezes anËes das deposições medindo-se a diferença de po
tencíal entre o nosso eletrodo e um de calomelano saturado, em uma solução
de cloreto de potássio.
5.5 DETERM]NAÇÃO DAS EFICIÊNCIAS DE CORRENTE
Primeira etapa - cã1cu1o de EC para um valor de td
A primeira etapa consiste no cãlcu1o da eficiência de corrente pa-
-9ra um dos oito valores de iU (escolhemos iU = 52,5 mA.crn "), atrav6s da t6c-
nica utiLízada por Louis e Martin (ref. 15) e exposta a seguir.
Efetuam-se deposições com a densidade de corrente cat-odica id e
com duração tU, formando-se um dep-osito de massa r5rr. Findo este tempo, in-
verËe-se a polaridade dos eletrodos, aplicando-se tensões que provoquem cor-
rosão eletroquímica do depósito com diferenËes densidades de corrente anódi-
.r i"u (adotamos sete valores de i"u: 1912, 26r0, 35,1, 47 15, 64,3, 87,0 e

_aII7,6 mA.cm '). E"t"
consumirã o dep6sito
Sendo i acq
corrente), a massa do
74
corrosão eletroquímica, aliada a uma corrosão normal,
em um tempo a". diferente para cada i"" (FiS. 6.2).
taxa de corrosão normal (expressa como densidade de
dep6sito consumida será dada pela 1ei de Faraday:
t"lr, = k(i.q * i".)'t"u
sendo
onde rn,, -e a massa atômica do manganês,!1n
a constanËe de Faraday e z 6. o número de
A eficiência de corrente EC 6
sa teórica equivalenËe ã carga e1ãtrica
5.1
5,2
A 6 a ãrea exposta do eletrodo, F é
oxidação do manganês na solução.
dacla pela relação entre rhn e a mas
consumida na deposição:
m.MnAFz
mMnEC= 5.3
k i.d
ou, pelas equações 5.1
td
5.3:
irEC= "q
tt +i- r-
dd
5.4td
Não podemos ainda calcular EC, pois não conhecemos i -,. Pela equa-cq
ção 5.1, contudo, vemos que quando i". tende a infinito, t." tende a zeto,
uma vez que rur-, , k u t"n são valores finitos e k 6 diferente de zeto.
Mas se t tende a zero, a primeira parceLa da equação 5.4, í^^.t^^/í".t^ 'cecqcedd
tenderã também a zero, pois o produto i".t" 6 diferente de zero e i 'e
ddcefinito. Assim, o valor da segunda parcela da equação 5.4 - parcela que <1e-
signaremos B - tenderá a EC.
irce ce
td

75
Em resumo, sendo
ir^ceceli =--
i- r-dd
ouando i'ce tende a infinito, B tende a EC.
5.5
paracada í , traçando' ce-
= 0 e tomando como ECce
Determinamos EC, portanto, calculando. B
a curva B x 1/i". , extrapolanclo-a at6 o eixo L/í
o valor de B nesta inËersecção (Fig. 6.3).
Segunda etapa - cãlculo de EC para os demais valores de
Poderíamos reutilizar a mesma t-ecnica para calcular as eficiências
de corrente associadas aos seis demais valores de id. Com o resultado da
primeira etapa, contudo, podemos adotar agora uma t6cnica mais simples, pro-
posta por Radhakrisnhamurty e Reddy (ref.73) e exposta a seguir.
Efetuam-se deposições com os oiËo valores de iO e. com duração tU;
finda a deposição, desliga-se o circuito e permite-se a corrosão normal do
depósito, com uma duraÇão t"n para cada iU (Fie. 6,4). A massa do dep6sito
será agora dada por:
nL- = k"i . tIvln cq cq5.6
Pelas equaçoes 5.3 e 5.6, teremos
iu
EC=
Como
primeira etapa,
i .rcq cq
i.. r-dd
jã conhecemos o valor de EC associado ao
podemos calcular t"n pela equação 5.7
5.7
valor iU adotado na
Em seguida, pela

76
mesma equação, calculamos EC para todos os demais valores de i..d
Comentãrios
Explicado o m-etodo utilizado, cabem aqui três comentãrios:
a) Aparentemente, poderíamos ter economizado sete das oito experi-
ências da primeira etapa, calculando t"O e EC para um dado iU com apenas
uma deposição sequida de corrosão eletroquímica com qualquer valor de i." "outra seguida de corrosão normal . Isto porque t"n e EC seriam as únicas
incõgnitas nas equações 5.4 e 5.7 e teríamos, assim, duas equações e duas in
cógnitas. Mas a taxa de corrosão normal é afetada pela corrosão eleEroquí-
mica. Assim, o valor de t"n 6 diferente nas duas equações citadas, inva-
lidando o m-etodo símplificad.o aventado.
b) Poderíamos ter pensando, tambãm, em calcultt í"0 jã no :final
da prirneíra etapae pela equação 5.4. TerÍamos economizado, assim, uma expe-
riôncia d.a segunda etapa. Mas, como visto no comentãrio anterioï, a taxa de
corrosão normal ã afeEada pela corrosão eletroquímica. Porisso, como deseja
mos o valor de i correspondente a i = 0, devemos calculã-lo com dadoscq^ce'da pr-opria segunda etapa.
c) Em lugar do m6todo adotado ou, ao menos, em lugar da prímeira
etapa, poderÍamos ter recorrido a m6todos gravim6tricos para calcular EC ou
í^^, Não o fízemos para evitar a possível imprecisão provocada por eventuaiscq
destacamentos do dep6sito nas etapas preceden.tes ã pesagem - retirada da c6-
lu1a, banho passivador, lavagem com ãgua e álcool, secagem -, bem como pela
corrosão normal que ocorre entre o tãrmíno da deposição e a imersão do dep6
siËo no banho passivador"

77
Determinaeão jlos tempos de corrosão
O momento de esgotamento do depósito de manganês pela corrosao
seja a normal, seja a normal maís a eletroquÍmica é indicado por
uma rãpída alteração no potencial de eletrodo. Tal como fizeram Radhakrishna
murty e Reddy (ref. 73) - no caso deles, apenas para corrosão normal -, "t!lizamos esta alteração para determinação dos tempos de corrosão. Para isso,
anotãvamos o valor do potencial de eletrodo a íntervalos curtos (5 em 5 s,
nos trechos maís críticos), traçãvamos a curva potencial x tempo e com ela
identificãvamos o momento da alteração do potencial de eletrodo. Obtínhamos
assim os tempos t"" ou ,"q, como exemplificado nas figuras 6.2 e 6.4.
O final de corrosão era tarnbém indicado pela cessação do desprendi
mento de hidrogênio que acompanha o processo. Entretanto, a sinrples observa-
ção visual seria inadequada para identif.icar com segurança este momentorpois
inúmeras bolhas de hiclrogênio permanecem sobre o eletrodo-base por algum tem
pcl após esgotado o manganês.
Tempos de deposição
Ao proporem o método de determinação da eficiência de corrente por
corrosão normal (v. seção anterior), Radhakrishnamurty e Reddy (ref. 73 )
alertam que a espessura do depósito deve ser pequena, para que não haja va-
ríação da taxa de corrosão durante o consumo de manganês . Para taL, o tempo
de deposição, segundo eles, deve ser curto. No exemplo mostrado por estes.â.Lr
tores, a deposição durava cerca de um minuto.
Considerações semelhantes caberiam, então, ao mãtodo de corrosão

/ó
eletloquímica uËilizado por Louis e Martin (ref. 15). Afinal, este m-etodo
tamb6m pressupõe uma taxa de corrosão normal constante durante a corrosão.
Entretanto, estes autores experimentaram tempos de deposição de 5 a 60 minu-
tos e verifícar¿rm que os tempos que levavam a resultados de maior coerência
eram os de quinze minutos.
Em nossas experiências preliminares, verificamos que o tempo de de
posição não afetava sensivelmente a relação a"O/tU ou 4."/ad. Assim,o tem
po de deposição foi definido de modo que grande parte de deposicão ocorresse
sob um potencial- de catodo já sensivelmente estabilizado (o que ocorria a
partir do segundo ou terceiro minuto, como já mencionãramos na seção 5.4), e
de modo que o tempo de corrosão resultanËe fosse suficientemente longo para
permitir uma medida adequadamente precisa. Com este enfoque, foram considera
dos suficientes, e adotados, os tempos de 600 s para os dois valores mais
elevados de id, 900 s para os quatro valores intermediãrios e 1200 s
os dois menores.
pata
RepetïÇão de experiências
Com algumas exceções, cada experíência foi realizada uma unica
vez, sem repetição. Exceções a isto foram as experiências preliminares, des-
tinadas a veríficar precisäo e reprodutibilidade do r4étodo (v. seção 6.1 )
Outra exceção foi a repetição de experíências que apresentaram alguma anoma-
lia em seu decorrer, como, por exemplo, a formação de unia grande bolha de h!
drogênio sobre o depósito - reduzíndo a superfÍcie exposta do catodo -, ou o
contato acidental do capilar de Luggin com o dep6sito, prejudicando-o local-
mente.
Mas a principal exceção foi
ções em todas as experiências com iO
exe.ução sistemática de três deposi-
5215 rnA.cm- - seguidas de corrosao
a

79
normal (seção 6.1). Adotou-se esta prãtica porque o valor de a.O obtido
nestas experiências, ao ser utilizado no cálculo d. i"O, constítui-se em elo
de ligação entre os dados das duas etapas de nosso mãtodo, afetando o cá1cu-
1o de EC para todos os valores de iU. As eventuais dispersões que incidam
nas demais experiâncias irão afetaï um único valor de EC e podem, portanto,
ser compensadas ao se traçarem as curvas de polarizaçã.o.
5.6 FOTOMICROGRAFIAS DA DEPOSIÇÃO
Todas as deposições, e subseqtlentes corrosões, foram acompanha-
das visualmente atrav-es do equipamento descrito na seção 5.2.2. A registro
fotogrãfico, contudo, restringiu-se a deposições realízadas na densidade de
correnËe ð,e 52r5 rA."*-2, exceto nas experiências preliminares, nas quais
diferentes situações foram regisËradas. Os registros foram feitos nos momen-
tos maís significativos da deposição, como o início, os primeiros sinais da
deposição do manganês, o começo da formação de g1óbu1os, etc.
Conf orme citado na seção 5.2 "2, uËi lizou-se r¡n ttf lash" eletrônico
como fonte de ilurninação. Como nosso sistema não dispunha de um diafragma
que pudesse ser re-gulado de acordo com as tabelas de dístância "flash"-obje-
to, a densidade correta do negativo foi obtida variando-se o tempo de revela
ção. Para o filme Kodak Plus-X f35 (ASA L25), o tempo de revelação finalmen-
te adotado foi d.e L2 mínutos, a lBoC, em tanque pequeno, com revelador Kodak
Microdol-X 1:1. Este tempo'e 507" superior ao recomendado pelo fabricante pa-
ra revelação normal nestas condições.

BO
5.7 FOTOM]CROGRAFIAS DO DEPõSITO
Para observação do dep-osito de manganês em microscõpio eletrônico
de varredura (descrito na seção 5.2.3), foi necessárío realLzar deposições
especiais, pois as deposições destÍnadas ã obtenção das curvas parcíais de
polarizaçã.o eram seguidas de corrosão do depósito, como exposto na seção 5.5.
Nestas deposições especiais, o eletrodo de trabalho era retira<lo
da c61u1a logo após interromper-se a circulação de eletr-olito e desligar - se
o circuito elãtrico. Retirado da c61u1a, o eletrodo era imediatamente imer-
so em uma solução de dicromato de potássío a 57" em peso, destinada a pas-
sivar o dep6sito.
Toda esta seqüência era executada com a maior rapLdez possfvel, pa
ra que não ocorresse muíta corrosão do dep-osito pelo eletrólito, ou sua oxi-
dação pelo ar.
IJma vez passi'vada, a amostra e:--a lavada com água e com ãlcoo1 etí-
lico e depois era secada. Era então imediatamente colocada na câmara de ob-
servação do microsc6pio eletrônico, sob vãcuo.
Para obtenção de sinal eletrônico mais intenso, todas as fotomicro
grafias foram tíradas com a amostra vol.tada em direção ao sensor eletrônico
e, para ta1, inclinada de 45 a 600 em relação ao feixe de el-etrons.
Foram observados os dep-osítos obtidos com todos os trinta eletr6li
tos estudados. Nestas experíências, a densidade de corrente cat6dica foi de
_2J.52,5 rn|.cm " e o tempo de deposiçáo foi de dez minutos.

B1
6. RNSULTADOS E)GERIMENTATS
6,L EFICIÊNCIAS DE CORRENTE E POTENCIAIS DE CATODO
6.I.I Reprodutibílidade e precisão
Tempos de corrosão e1eËroquÍmica
Na seqüência experímental normal, não houve repetição de experiên-
cias de deposição seguida de corrosão eletroquírni ca. Pa:ra mostrar a reprodu-
tibilidade obtida nestes resultados, apresentamos na tabela 6.1 alguns dados
-adas experiências preliminares, realízað,as com o eletrólito nQ 1 ( 0r1 g.dm'
de dióxido de enxofre, nenhuma adição de impurezas):

B2
Tab. 6.1. - Reprodutibilidade na duração da corrosão eletroquÍmica (expe-
riências preliminares para determinação da reprodutibilidade,
com o eletrólito ng 1, i, = 52,5 mA..*.-2 " tO = 900 s)
densidade de
correntean6dica
-2.(mA. cm )
L9,2
26,0
35,1
47 ,5
64,3
87 ,0
LL7 ,6
duração da corrosão eletroquÍmica
expert_enc]- am-edía
,.) ^
(s)
desvio-padrão
erro
relativo(z)
560
522
448
322
326
2II
202
494
459
44r
372
293
237
L79
507
509
410
326
272
233
180
520
497
433
344
297
227
L87
35,2
33,4
20 12
27 ,7
27 12
74,3
L3,4
6'8
6r7
at I
8r1
a,"-6r3
712
Na etapa de
qllência experimental
como citado na seção
trinta eletr6litos:
Tempos de corrosão normal
deposição seguida de corrosão normal ,
previa a repetição de experiências com
5.5. Na tabeLa 6.2, apresentanos estes
a proprra se-_1
i.O=52,5 mA.cm',
dados para os

83
Tab. 6.2 - Reprodutibilidade na duração da corrosão normal (íð,=52,5 mA ." 2,
tO = 900 s)
e 1e rró 1i ro
n9
01
02
03
04
05
06
o7
OB
09
10
11
T2
13
L4
15
I6
T7
1B
L9
20
2T
22
23
24
25
26
27
28
29
30
duraçao da corrosao
experíência nQ
xxI2 xx13 xxl4
normal (s)
rnédia
843
1043
1 200
633
850
101 7
373
533
577
140
177
237
840
927
I203
653
793
r037
387
517
607
t293
L537
1927
947
L240
1620
537
770
10 70
desvio
padrão
/, ()
5B
46
40
46
60
15
35
35
10
L2
15
44
4s
42
32
46
55
23
25
25
75
B5
90
55
35
B9
25
50
53
errorelativo
(7.)
900
1 100
1 190
590
900
960
390
500
610
130
190
250
890
930
I250
690
820
1000
360
540
630
L220
1620
1 840
1000
L220
L720
560
720
109 0
820
1 100
L250
640
840
1080
370
530
540
150
170
220
810
880
11 70
630
740
I 100
400
490
580
r370
145 0
2020
890
12 B0
1550
s40
820
1110
810
101 0
I 160
670
810
10 10
360
570
580
140
L70
240
820
970
1 190
640
820
10 10
400
520
610
r290
15 40
1920
950
L220
1590
510
770
10 10
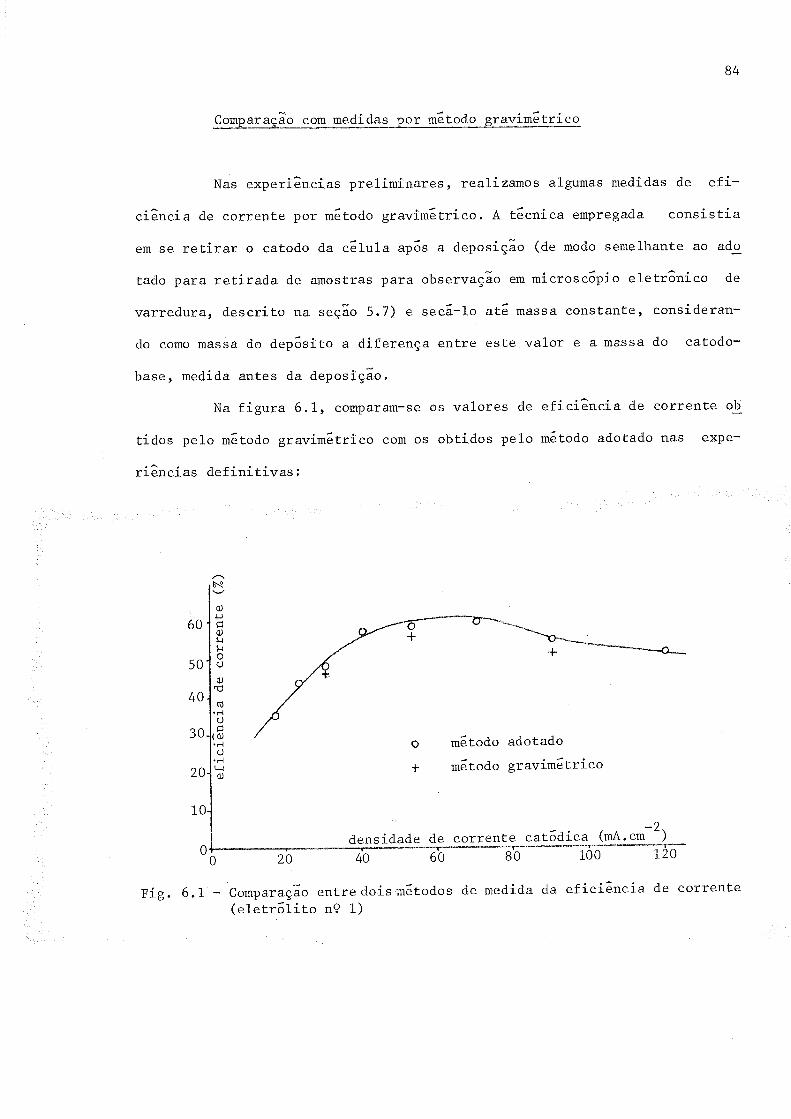
84
Comparação com medidas por m"etodo gravim-etrico
Nas experiências preliminares, realizamos algumas medidas de efi-
ciência de corrente por método gravim-etrico. A técnica empregada consistia
em se retirar o catodo da c-elula após a deposição (de modo semelhante ao ado
tado para retirada de amostras para observação em microscópio eletrônico de
varredura, descrito na seção 5.7) e secá-lo at6 massa constante' consideran-
do como massa do dep-osito a diferença entre este valor e a massa do catodo-
base, medi.da antes da deposição.
Na figura 6.1, comparam-se os valores de eficiência de corrente ob
tidos pelo método gravimãtrÍco com os obtídos pelo m6todo adotado nas expe-
rt-encras det]-nrt]-vas :
È..e
AJ
]Jda.)
ItoCJ
CJ.Ú
cd
'r'{(.)
É(a)'rlc)
q¡a)
60
50
40
30
20
método
mãtodo
adotado
gravim6trico
o
L_
densidade de
20 60
Comparação entre dois mãtodos(eletrólito nQ 1)
corrente cat6dica (tn."*-2)100
da eficiência
1
d
40 BO
de medida e correnteFig. 6,

B5
Potenciais de catodo
Com cada eletró1ito, realizaram-se dez deposições com a densidade
de corrente catódíca de 52,5 nA."*-2 (sete deposições na primeira etapa do
m6todo experimental, três na segunda), como explicado na seção 5.5. Para mos
trar a reproduLibílidade obtida na medição dos potenciais de catodo, apre-
sentamos estes valores na tabela 6.3,. Nas demais densidades de correnËe em-
pregad.as não houve repetição de experiências, cJe sorte que não podemos fazer
avaliação semelhante.
6.I.2 Resultados e cá1cu1os completos para o eJ-etrõlito nQ 30
O m6todo de determinação da eficiência de corrente e do potencial
de caËodo para oito valores de densidade de corrente catõdica, descrito n.a
seção 5.5, foi aplicado aos trinta eletr6litos sob estudo'. A partir destes
valores foram traç¿.das as curvas de polarizaçáo. A. guiza de exemplo, expore-
mos a seguir Èodos os resultados experimentais primários e etapas de cãlculo
para o eletrólíto n9 30 (Q,6 g.dr-3 de dióxido de enxofre, 40 rng.dt 3 de zin
co). Na seção seguinte, expomos os resultados finais correspondentes aos de-
mais eletr6litos "
Deposições seguidas de corrosão eletroquímica - cálculo da efici-
ência de corrente para íd =2_.)
-L5 mA.cm
Para este eletr6lito (como para os demaís) , realí-zaram-se sete de-
-nposições com densidade de cor:rente catódica id = 5215 d.cïì ' e duração
fU = 900 s, seguidas de corrosão eletroquímica com diferentes valores de den
sidade de corrente i A figura 6.2 mostra' como exemplo, o potencial de
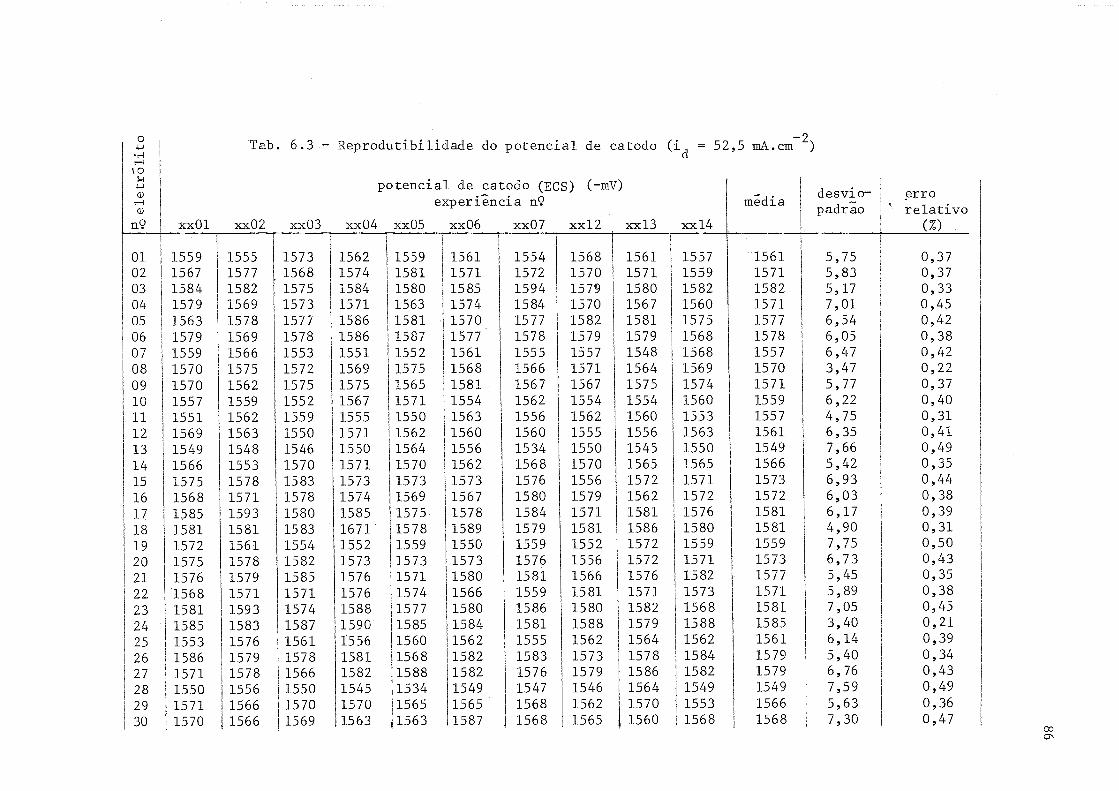
+J.rtF-l
10]J0.)
F-la)
n9
0l02030405060708f)q
1011T2137415T6L718L9202L2223¿42526272B2930
Tab. 6.3.- Reprodutibilidade do potencial de catodo (i¿
xx01 :c<02 xx03
155 9L567I5B4L5791563r57 9155915 7015 701557155 115691,5491566L57 5156815 851581L57 2r57 5t57 61s6B1581158515531586I57I1550L57 Tr570
1555r577T5821569ls7Bl-569l-566L57 5L5621559156215631548155315 78T57 L
159 315 81156 r-
r5781579L57 L159 31583r57 6
t579157 8L556L5661566
poËencial de catodo (ECS) (-mV)experiência nQ
xx04 xx05 xx06 xx07 xx12 xx13
L57 3r568L57 5r57 3L577t57 B
1553r572r57 51552155 915501546I57015 83r578158015 83r554l.58215857571157 475871561L57815661550r570L569
1562L57 4i584l-57I158615 86155 17569r57 5I5671555r57l1550I57 T
r573r57 415 857671r552r573L57 6157 615881 590ls s6158 1t5B2l-545L57 015 63
1559158 11580156315 81T5877s52r57 5L565r57 I155 0L562756415 70L573L569r57 5L57 815 59L573I57LL57 4L577158515601568r5 881534L5651563
1561L57L15 85L57 41570r5771561156 I158 1L554156315601556t562r57 3156lL57 815891550L57 31580156615801584L562L5821582rs4915651587
L554I572r59415 84r577r578155 5L566L56715621s5615607534156 BL57615 801584r579L559L57 615 B1155915 8615 8115551583L57 6L5471568156 8
156815 70rs79157015B2r579L557I57Il-567r55475621555155015 701556r579L57Lls 81L5521556156615 8115 8015 B8t562r57 3t579\546L5621 s65
= 52,5 inA. cm-2)
156 1757 I1580l-5671581t5791548t564r57 5
7554t_560155615451565157 27562r5811586r57 2
t572L57 6757L1582757 91564L57 B1s 86t564L570r560
xx14
L5571559t5821560L57 515681568r569757 41560155 3156315501565I57 T
L572L57615801559T5] L
7582Is7 3156 815 8875627584T5B2T5491553156 B
m-e¿iadesvio-padrão
1561r5771582I57 T
157715 78l-557L570L5771s591557156 t1549l-566r573t572158115 81L559r573r577T57I15 8115 85156 rr579r57915491566156 B
5,755,835,177 ,016,546,056,473,475,776,224,7 5
6,357 ,665,426,936r036,114,907,756,7 35,45
_errorelativo
0,370,370, 330,450,420, 3B0,420,220,310,400,310,4!0,490,350,440, 380,390,310,500,430, 350,380,450,2r0,390 r340,430,490, 360,47
5, 897 ,053,406,I45,406,767 ,595,637 ,30
Oo
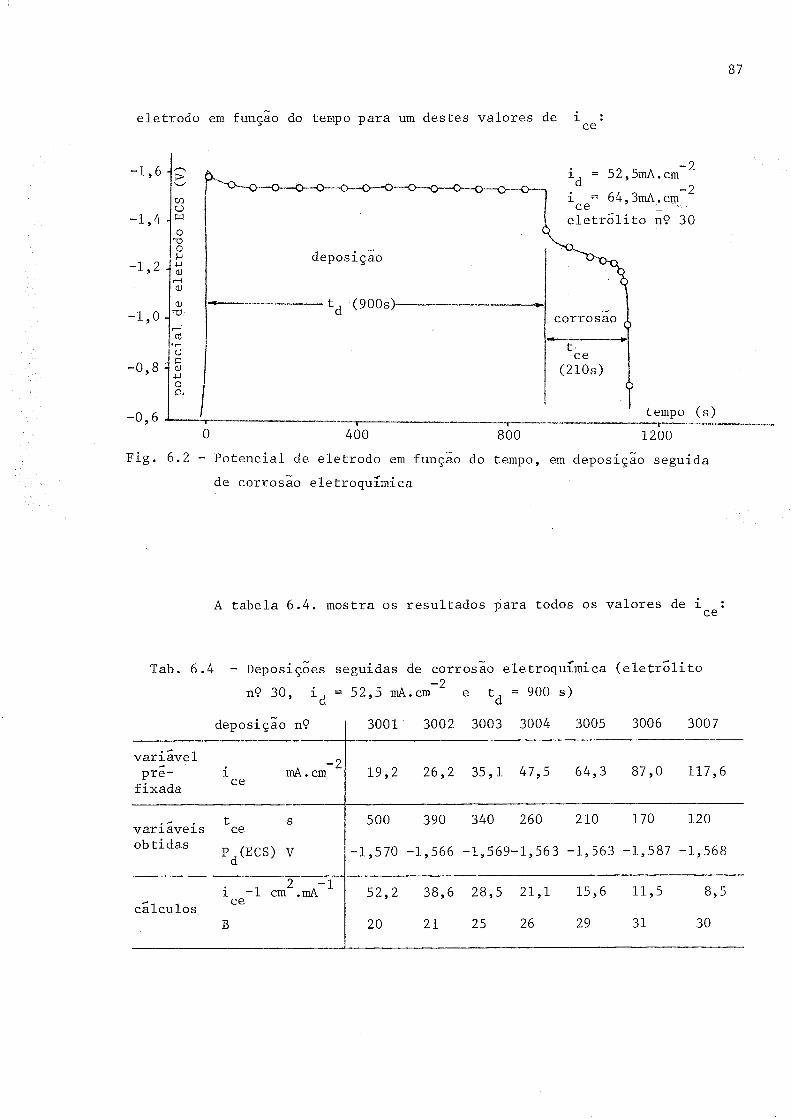
B7
eletrodo em funçao do tempo para um destes valores de ice
íU = 52r5mA.cm
í""= 6413mA,,.-cm
eletr6lito nq
corrosao
tce
(210s )
tempo (s)
800 1200
tempo, em deposição seguida
-L r6
-L r4
-L 12
-1,o
-0, 8
-0, 6
depos i ção
0 400
Fig. 6,2 - Potencial de eletrodo em função do
de corrosão eletroquímica
v)(Jt4oo¡r+JCJr-lqJ
c.)
$.Fl(.)
ka.)
]Joã
-L
-¿
30
A tabela 6.4. mostra os resultados para todos os valores de i
Tab. 6.4 - Deposições
nQ 30, íð.
deposição nQ
eletroquími ca ( eletrólito= 900 s)
3004 3005 3006 30
seguidas de
= 5215 rnA.cm
corrosão_.)
e t-d
3003
variáve1pre-
fixada
varlavelsob tidas
tce
P , (ECS)cl
i -1ce
B
3001 3002
500 390
-1,570 -7,566
mA. cm 19 12 26,2 35,1 47 ,5 64,3 87,0 7r7 ,6
r70 720
-1,587 -1,568
S
V
340 260 2r0
-L,569-7,563 -r,563., _1L,Icm .mA 52,2
20
L5 16
29
11,5
31
B'5
30
38,6 28,5 2r,L
21 25 26cãlculos

Para obtermos a eficiência de coïrente, traçamos o grãfico_ -1função de i^^' lfignrr 6.3). Extrapolando a curva obtida at6 a"ce
(i + *), obtemos a eficiência da corrente para í, = 5215 d."*-2,'ceq
eletrólito n9 30. Como vemos, este valor ê. l+Z:
88
deBem
origem
com o
B(%)
(etet16lito nelJ
(id=52,5m4. .^-2)
3020loo ¿lo. -t . ,lce (cm".
de corrente pe1-a
50
mA-l )
extrapolaçãoFig. 6,3 - Determinação da eficiênciade B at6 o eixo i;t = O
Deposições seguidas de corrosäo -nor4.e!
corrente para os demais valores de iO
- cãlculo da eficiência de
Nesta segunda etapa,
sidade de corrente catódica iU,
petições de experiências com iO
se, nesta etapa, dez deposições
exemplo, o potencial de catodo
idt
realízamos deposições com oito valores de den
seguidas de corrosão normal. Como houve re-
= 52,5 o'rA."t-2 ç,r. seção 5.4.4), realíza;raÍÍL-
por eletrótito. A f igur a 6 .4. mostra, como
em função do tempo para um destes valores de

B9
.-r r6
-l 14
-L 12
-1r 0
=0, B
-0, 6
_L
30cJ>r)f¡lo.úot{+JAJ
F-lCJ
oF.l
Fld
.ÈCJ
OJ
¡Jop'
deposição
t. (900s)(1
corrosao
t (870s)cq
id = 39,8mA.cm
eletrólito nQ
tempo (s)
0 400 800 1200 1600 2000 2400
Fig. 6,4 - Potencial de eletrodo em função do Ëempo, em deposição seguida
de corrosão normal
A Tabela 6.5 mostra estes resultados para todos os valores de iO:
Tab. 6.5 - Deposições seguidas de corrosão lorma1 (e1etr61ito ng 30)
deposição nQ 3008 3009 3010 30rl 3012 3013 3014 3015 3016 3017
varl avers
pré- f :'-xadas
-am.m
s
Id
td
L7,4 22,9 3O,2
1200 1200 900
39,8 52,5 52,5
900 900 900
52,5 69,2 gL,2 I2O,2
900 900 600 600
.LvarraveLs cq
obtidas P,(ECS)o
250 460 610
-L,532 -I,542 -1,.554
s
v
870 1090 1110 1010 1190 770 890
-L,562 -1,565 -1,560 -1,568 -L,573 -L,576 -1,583
Para i" = 5215 rnA." 2, a m-edia dos tempos de corrosão normal tdcqfoi 1070s (m6aia dos valores 1090, 1110 e 1010s referentes ãs deposições n9
30L2,3013 e 3014, respectivamente, mostrados na Ëabela 6.5). Com este valor
e com a eficiência de correnËe determinada no item anËerior, calculamos a
taxa de corrosão normal, do manganês neste eletrõlito, expressa em densidade
de corrente, por meio da equação 5.7:

90
.oI=
cq
EC i. r,clo 0 34 .52,5 900
otcq
10 70
-¿
Conhecido este
ra os demaís valores de
r .ioBç= cQ cq
io td
A tabela
sidade de corrente
lores do potencial
va1or, podemos
rd, pera equaçao
= 15 mA.cm
a eficiência de
exemplo, paîa
corrente pa
-)iU=I7 r4mA.dm -:calcular
5.7 . Por
6.6 apresenta
cat-odíca, com
de catodo (já
2s0 15 = LBZ17,4 . 1200
este resultado para
o eletr-olito n9 30,
mostrados na tabela
os oito valores de den-
OS VA-e
6
Ltaz rLovamenËe
.s):
Tab
variãve1pr-e-
fixada
17,4 22,9 30,2 39,8 52,5 69,2 9r,2 I20
variãve1
medidaL,532 1"542 r,554 L,562 r,564 7,57 3 r,576 1 ,583
variáve 1
calculada
6.1.3, Eficiência de corrente em função da densidade de corrente e do
potencial para todos os elet161iËos
A partir de tabelas semelhantes ã tabel.a 6.6, apresentamos a efi-
ciência de correnËe em função da densidade de corrente, para todos os trinËa
eletr-oliËos estudados, na figura 6.5. Com todos os eletr6litos as curvas da
figura 6.5 apresentam um mãximo da eficiência de corrente dentro da faixa es
tudada de densidades de corrente. Os valores destas "eficíências de corrente
máximas" (ECM) e das densidades de corrente (iuar) e potenciais (nua") em
que elas ocorrem estão apr:esentaclos na tabel a 6.7 .
6.6 - Eficiência de corrente
densidade de corrente
e potencíal de catodo em função da
catódica (e1etrólito nQ 30)
t_.o
Pd (ECS)

çn
60
L¡
2õ
0
z)0,lg.dm - S0,
r")
\0 ,F ,:-r{___.o--*_ìo/.
e,0 120_?,r.d (mA.cn -)
u1/"7\i."JIII
I^a
ì
i!
tj
Ec ([)I
h illI
II.-t+t¡II
I241
I
I
^t
ì'.is Ì-i\-?q)-:J {.¡---F,'lj /-'-..-i:-r-- -__ -? : ¿ù '-È_ -LJÊ
' /+ r--.+_
c '+r e-0 :i0,rd (nLå.cn -)
Flg, 6.5 - Eficiência <ie corretìte
I
Ib0 -T
ii
qo-ilI
2aj
i
o..l
i 2'J
/")
-1,/
2/
B0 L20
i- (ma.cm-2¡
z-;-=+?o *-. - &'-.\ 'e'-- B'---" È ---tl-
-?0,69.dm - S0,
?
;' €----Õ------o---_,* ; *-s --o-
frr " \-..lã*o'o-r- -=- ---o-: L.F--
f;*'-J?**=. - -Ê----c-
+_, +
, *-12u ar\..--,20 L-\-
Ec (z)II
6 0-lII
. ^J
I
I
iI
I
zc(7")I
60ii
uo-lI
I
¡o)I
è/ntÅ,s - -ú-2-6fJz- -+-;-*"ÞÊ
'+.\- -u--+-
É.
í7,"nr-n,--o- - -3- - * - ^i.J l9-'*- * LLr/' '--¡_._- -
:¡:---------:T-^uu l tu^io. (mA . cn-¿ )
t+0
/-
c
em runçao
adi.ção decobalt o
(ng. dn-3)_o zero__a 0,25
- ---o 0,5
29\'+.-.-_. -1-È_r_._. +_
::*_ _ _^_"}-...--:-+
80 li. (mA.cn
(l
3
40 80 izc-r.r rña ^rJ
a)r(:' \usr'e
da densidade de corrente
EC
6C
(
I¡I
Iiì
iI
jI
II
-270
7")
+ i,0
40
24 ¡-og"u¿5,/- .1È ^n'Ð- i'
H\ V¿ U\'Ê' r*.-+-9c/-) / \¡¡-
adição cenìquel
. . _1.(:ng, . d:n J.)
_o zcro
20
s0:
c
J
+
+
i20(nc. cm-2)
A 0,5o 1,0
+ 2,4
'ì'-.*-
adição deztnco
(rng.dn-3)_o zeto__^ i0
n ?fl
80 i20-1.1d (n;É . cII: 'J
+4A
\oH

92
Tab. 6.7 - Eficiências de corrente máximas e correspondentes densidades de
corrente e potenciais
adição deimpure zas
_2(me.dm')
adição desoz
(g . ¿*-3)
numelodo
e1etr6-li to
ECM
(7.)
iua*_a
(mA. cm')
Puctn
(7")
SEM IMPUREZAS
0r 1
0'30r6
1
2
J
6I646B
706060
-r ,57 3-1,580-1,585
COBALTO
0 r250'10r30r6
45
6
50545B
404s38
- 1,560-r ,57 5
-r,57 5
0'50'10r30'6
7o
9
363B3B
3B4035
-1,543-1,563-L,563
1r00r 1
0r30r6
1011T2
T41818
404032
-1'543-r,546-1,550
NÍQUEL
0 r5
0r10'30r6
13I415
5B5964
604544
-1,570-r,57 7
-L,57 7
1r00'10'30r6
16L718
525556
334042
-r,545*r,57 4
-7 ,57 5
2rO0'10'30r6
T92027
364040
353532
-1,540- 1, 555.-1,560
ZINCO
100' I0r30r6
222324
646666
52-
4548
*7,570-r,57 5
-1,585
200r10r30r6
252627
42474B
304043
-r,542.-r,567-I,57 5
400r 1
0'30r6
2B2930
303+36
353540
-1.,534-1,553-1,563

93
6.2. CURVAS DE BgLA¡I?{Ç^\g
As curvas de polarizaçáo catódíca representam a relação entre o
potencial de câtodo (ou a sobretensão cat6dica) e a densidade de corrente ca
t6dica, para um dado processo eletroquímico. No caso estudado' o processo ca
tódico global subdivide-se em dois - a deposição de manganês e o desprendi -
mento de hidrogênio -, associando-se a cada um dos mesmos uma densidade de
corrente parcial, iM' e i", respectivamente. Sua soma representa a densidade
de corrente catódica tota1, id.
A relação entre irr, . id corresponde ã eficiência de iorrente
da deposição de manganês, EC. Por exemplo, para iU= 17,4 mA."*2 (e1etr61i
to nQ 30):
i¿ . EC = I7,4 0, 18 = 3, 1 mA. cm-2
= id (1 - EC) = r7,4 A,82 = !4 ,n¿"."t 2
-Mn
tH
A tabela 6"8 apresenta estes resultados para
densidade de corrente cat6dica, com o eletr-olito nQ 30,
valores do potencial de catodo (já mostrados nas tabelas
os oito valores de
e traz novamente os
6.5 e 6.6):

94
Tab. 6.8 - Potencial de catodo
ção da densidade de
e densidades de corrente
corrente cat6dica totalparciais em fun-
(eletrólito ng30)
varíáve1pr6-fixada i¿ *¿. "r-2 Ll ,4 22,g 30,z 39, B 52,5 69,2 9I,2 l2O
yariãvqlme<lida Pd(ECS) -v r,532 I1542 1,554 1,562 1,564 L,573 L,576 1,583
varlavelscalculadas -)mA. cm
tM'iH
3r1
L4
517
T7
10 L4 18 20 L9 23
20 26 3s 49 72 97
A partir de tabelas semelhantes
vas cle polatízação totais e parciaís r para
nas figuras 6.6 a 6.L52
ã tabela 6.8, apresentamos as cur-
os Lrinta e1etr6litos estudados,

95
-1,60
-1 ,55
-1,60
-1 ,55
-1r500,001
-1 ,60
-l ,55
-1 ,500,001
Fig, 6,6 - Curvas
eletr6lito nq 1
-?SOZ - 0,1g.dm "
eletr6lito nQ 2I
SOZ - 0,3g " drn "
densidade decorrente catódica
(4. cm-2).61,
t')cn(Jt4oEo4JcüoOJ
FldO
()lJoÀ
_ b)(nUT4
oo+Jdo
a)€r{$
oHc).tJoÈ
dçl;/ri
H-- -Mn-"
total _*
-1 ,500 ,001
.t
eletrólito nq
S0Z - 0,6g.dm
0'1
H---Mn -'-
total **_
densidade decorrente catódica
(e. cm-2)
0r1
H***Mn *'-
¡6¡¿l ***
densidade decorrente catódica
(R. cn-2)
0,01
0,01
.nd'.N' ,ln{ ^/
c) 3
-3
,{.
c0¡oHc.)lJ
Ê
0,01
d9 ¡olarízação dos eletrólitos0r1
sem adição de impurezas
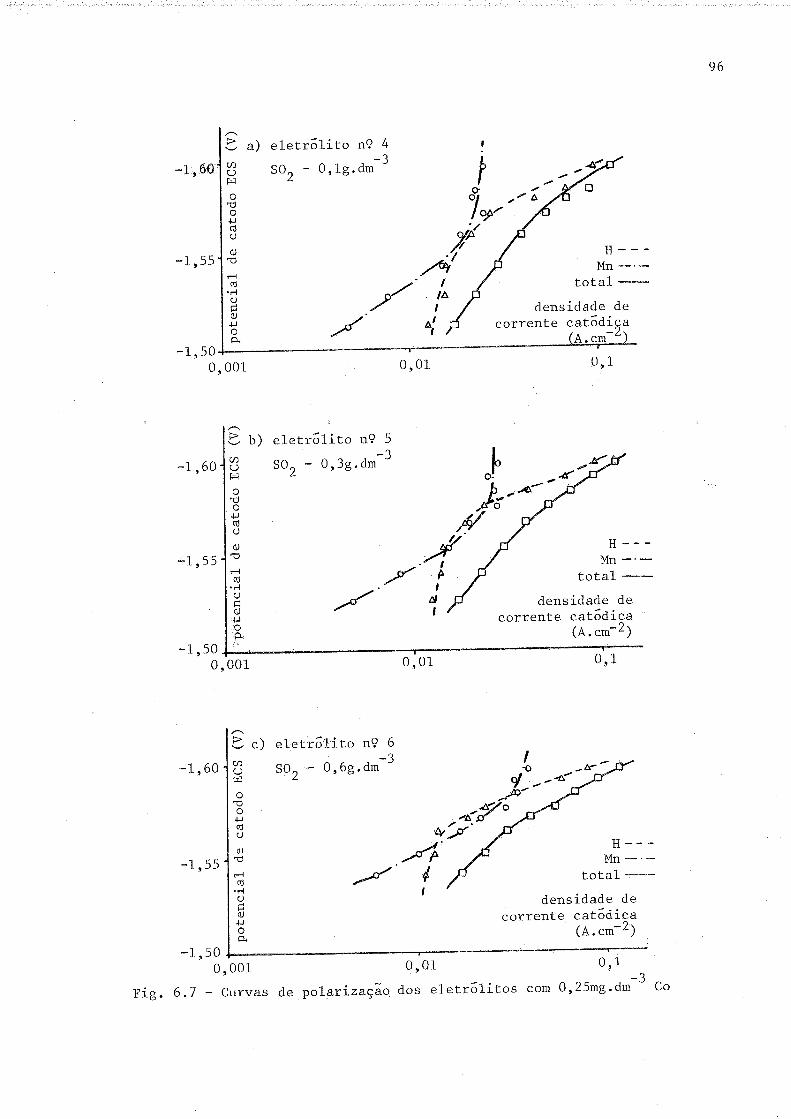
96
-1 , -55
'- A)(n(Jtrlo€oÐdoo€
Fl(ú
ooÐoÈ
t//ry
-{ tA,
dI
3¡)<nC)trloolJ(gO
o.d
r+cù.¡UÉAJ.uot+
i- ç)
U)(-)t4oEoÐdC)
0lÉr-l(Ù
.rlc)HoÐop.
eletr6lito nq 4^
SOZ - 0,1g.dm -
eletr6lito nq 5_?
SOZ - 0,3g.dm -
corrente catódiça(4. cm-')
H---Mn -'*¡s¡¿l **-
densidade de
0rl-1 ,500 ,001 0,01
0,01
"þÅ,
'*1 ,60
-1 ,55
-1 ,500,001
-:l-,60
-1 ,55
-1 r500,001
6.7 - Curvas
eletr6lito nQ
SOZ - 0,69.dm
-d
H---Mn -'-
total -_
densidade decorrente catódica
(A' cm-2¡
o'l
.F
H---Mn -'*total
-densidade decorrente catódica
(¿'. crn-2)
6
-3q
A.E/o.dd )t-r
"f
0,01 0, I
de polarização dos eletrólitos com 0,25mg'dm
þo
ol
o¡.
Fig.t-, co

97
v a) eletrõlito nqtnH so^ - o.le.dmt'o.oo+JCd
oAJ
.FJ
r-rd'rtL)
H---
total
H ---Mn *'-
total -densidade de
H---Mn *.-
total .-'*"_
densidade de
densidade decorrente catódica
(R. cm-2 ¡
7
-3-1 ,60
-1,55
-1 ,500
-1,60
-1r55
-1r500
H0)Ðoo.
-F
,d
"l.o
c
/"ëy
/l¿
{IIIlI
,F>i- b)(n(J
odo]JdL)
a)dF-l
Cü
.rlOHa)]Joê.
,001
,001
0ro1 0r1
eletrólito nQ B
SOZ - 0,3g.dm -¡
"f/'ol,{
N
0ro1
eletrólito nQ 91
SOZ - 0,6g.dm -
0,01
de polarizaçã.o dos eletrólitos
corrente catódica(A ' cm-2 ¡
0r1
-1 ,60
-1,55
-1 ,500,001
Fig. 6.8 - Curvas
corrente catódiÊa(4. cm-¿)
0r1
com o,5mg"dt-3 co
.p-ê î"-f -l
vc)U)CJt'l
€o]JdoU.d
F-lcd
OËa)
oè

9B
CNUf¡lo
id
+Jdoa).rl
r-lCd.¡oÊCJ.lJoo.
a) eletrólito nq
SOZ - 0,1g.dm
I
oÊþj.
,p
10-J
-1 ,60
-1 ,55
-1 ,50
H---Mn -'-total *"*
densidade de
0,001
i/ b) eletr6litoan:ì so^ - 0,3s.
¿o
'Úo+JcùO
AJ€
corrente catídicra(4. cm-- )
0r1
0r1
0r1
com 1,Omg.dt-3 co
0,01
-1 ,60
-1,55
11
t
þ
nQa
-Jm
o
I
dm
o
/o
c) eletró1ito n9 12
SOZ - 0,6e. dm-r,
dio
o/i
<nQtrlo.úo.lJdoCJ€
r-'l(d
'rlOÊ0.)
oor
r{doHAJ.¡JU'ÃÃ
-l ,500 ,001 0r01
*-6n
0,01
de polari zação dos eletró1itos
-1 ,60
-1 ,55
-1 ,500,001
6.9 - Curvas
densidade decorrente cat6dica
(4. cm-¿ )
densidade decorrente catõdíca
(4. cm-¿)
Fig.

99
-1, 6o
-1r55
*1 ,60
-1 ,55
3a)CNUf'lo'doÐcdo(J
Fl(d
.JoÊAJ
og
eletró1ito nq 13I
SOZ - 0,1g.dm '
¿
eletrólito nQ 14^
So^ - O.3s.dm -¿
É
{ff/densidade de
corïente catódica(4. cm-¿)
-r'r3
-1 ,500 ,001
ct)(Jt'lU.oo]JdCJ
oúr.ld.¡odC)]Jop.
c) eletró1ito nQ 15
SOZ - 0,6g.dm-3
-1,55
-1,500, 001
,/
+'-/
0,01
0,01
dos eletró1itos
densidade decorrente catódiça
(l' cm-2¡
corrente catódica(4. cm ¿)
0r1I
-tcom 0,5mg.dm -
H---Mn *'-
total -
H
Mn
total
H---Mn --'*
totalrlensidade de
-"1
vb)cnQTd
oFal
o.lJcd(-)
o
r-td
'r{C)Ha)ÐoÈ
Fig. 6.10 - Curvasde PorarlzaçaoNi

100
-1r55
-1 ,50
i/ a).J)(Jt4oo+Jdoo€r-l(d
'r{OÉCJ
]Joo.
¿b)U)
þlo€oÐCO
oCJ
r-4doc)ÐoÈ
vc)v)(Jtq
o.Úo]JdCJ
GJ.o
r-lCd
'rlOHCJ
+Jop.
eletr6lito nq 16I
SOZ - 0,1g.dm -
0,01
eletrólito nq 17â
SOZ * 0,3g.dm -
0, 01
eletrólíto nq 18ô
-1S0Z - 0,6g.dm -
0, 01
de polarização dos eletrõlitos
0,001
-1, 60
-1 ,55
-1, 60
-1 ,55
-1 ,500
Fie. 6. ll -,001Curvas
0, l'ô
-1com 1,Omg.dm'
densídade decorrente cat6dica
(4. cm ')
densidade decorrente catódica
(a.cm-2)
densidade decorrente catódica
(¡'. cm-2)
Ni

-1,55
-1,60
-1r55
-1,60
-1 ,55
*1 ,500,001
Fig. 6,L2 - Curvas de
eletr6lito n9 19_?
SOZ - 0,1g.dm -
eletró1ito nQ 21ô
SOZ - 0,6g.dm -
/ densidade de
,/
frb
/
ct)(Jt¡lo.úoJJcd
oa.)€
r{d
'rlokAJ.!og
,É
ar h\(n(J
oo4Jdocl
''ú
rlCÙ
'rlc)ÉCJ
{Jo
/a
dI
tc)(/)(_)
t'lU.ÚoçdoC)
r-l(d
.F{
oHa.).lJotr
//o.4; /t corrente catódita
(4. cm-')
0r01
0,01
0r10,001
eletrólito nQ 20a
SOZ - 0,3g.dm -
corrente catódica(4. cm-')
-1 ,500
0'L,001
iÊ
/o9- ,.N
./..Þ' ,Á /,//,r'dN,n
0, 01
poLarízação dos eletr6litos
corrente catódica(4. cm-2)
0rlcom 2,Omg, dm-3
H---Mn---
total __*
H---Mn -'-
total -densidade de
H---Mn -'-total
-densidade de
101
NJ
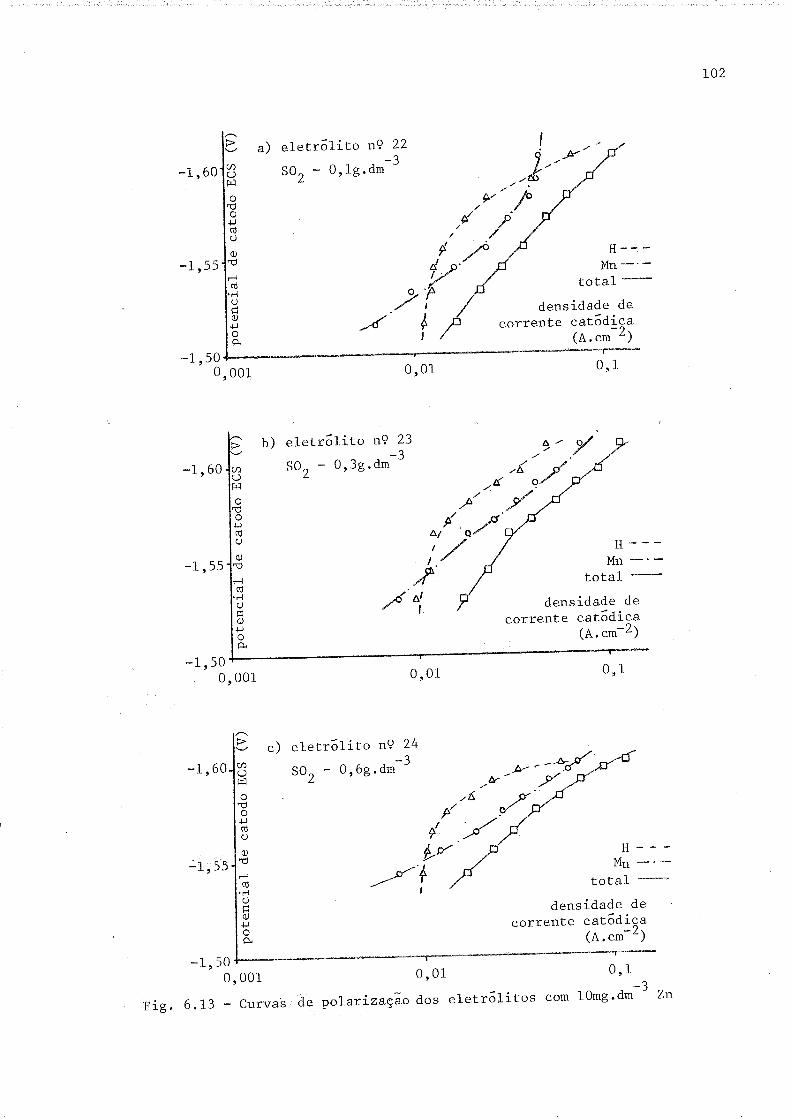
TO2
I
ç_l¿t/g
'{ al/l
---"t +
U)UtllorJoÐ($CJ
AJ€Ì--1
Cd.¡L9ll¡loJI ¡-¡
le
eletrólito nQ 22ô
-1SOZ * 0,1g.dm "-1r60
'-1 ,55
-1, 60
-1 ,55(drloHoÐoè
*1r500, 001
-1 ,60
-1; 55
-1,50o, ool
Fig" 6.13 - Curvas
-v
H---luln *' -
total -densidade de
0, l.
H---Mn -'-total "-..*
densidade de
o-
a)LN(J[¡]
oo.lJ(doCJ
r-lidl.nt9tblulolo
]J(doCJ1J
.-ld'rloÉa.)
]JcÊ
0-1 ,50
0 ,
^/"P
{tÕÍ'f;
,P
I
{,Ét/
corrente cat6dica(A.cm ¿)
01 0,01
0,01
oIJdO
0)
b) eletrólito n9 23
so^ - 0.3e.d*-3¿
c) eletrólito sQ 24.-3
S0^ - U'bg.dmI
corrente catódíca(4. cm-z)
MzÁr'-d
o
þv
0,01
de polari zação d.os eletr6litos
0"1
H---Mn -'"-
total --'
densidade decorrenLe catódica
(A ' cm-')
0r1
com 1omg"d*-3
^-"^{,
Zn

103
Þ^rva)
U)Ot¡lo€o]JdC)
q)
r-{d
'rloka)]Jo
-b)aî)Ut4o.do+J(Û
oq).o
F{d
.Flo,q¡+JoÈ
-o
./^{fz./*'
{r'-.r f
A
-c)at)(J[¡]
ooÐcdoqJ.ú
d'rloHolJo
eletrólito nq
S0Z - 0,1g.dm
25
-3
eletr6lito n9
SOZ - 0,6g.dm
H---Mn *'-
total -"-
H
Mn
total
densidade de
corrente catódica(4. cm-r)
0,01 0r10,001
-1 ,55
-1r55
0,001
Fig" 6.L4 - Curvas
eletrólito n9
SOZ - 0,3g.dm
26
-3
001s0l
10,
,P'
2l-3
0,01
0r01
dos
oX
densidade decorrente cat6dica
(4. cm-z)
' 0r1 .eletr6litos com 20mg.dm -
densidade decorrente catódica
(4. cm-2)
c1e polari zaçao Zn

104
-1,55
-1 ,60
-1 ,55
-1 ,60
-1 ,55
eletrólito nq 28-?
SOZ - 0,1g.dm -
eletrólito n9 29ô
SOZ - 0,3g.dm '
H---Mn -'-total
-
H ---Mn -'-
total -_densidade de
HMn
totaldensidade de
vdg,
-1r500
va)(nc)
o.d
.lJdoAJ.d
F-ld.¡Oda)+Joè
/"
vb)U)
trlo€o+Jcd(.)
GJ.o
r-l(d
CJ
HAJ
Ðoo.
vc)(n(JËd
.do+Jd(-)
c)n3
r-ld.¡(.)
Ho.uoÈ
-Ê--
ç
ATdensidade de
corrente catõdica(A' cm-2¡
0,01 0r1,001
oa,/v
corrente catódica(n. cm-z)
-1 ,50 0, 01 0'10, 001
eletr6lito n9 30^
SOZ - 0,6g.dm -
þF
"ß' g'
0l0,
-1 ,5001
Curvas
0, 01
de polari zação dos eletrólitos
corrente cat6dica(4. cm-/)
0r1a
com 40mg.dm "Fig" 6 "I5Zn

105
6.3 OBSERVAçÃO DA DEPOSIÇÃO E DO DEPõSITO
A observação do depósito e do processo de deposição por meio de m!
croscopia eletrônica d.e varredura, no primeiro caso, e de microscopia -opti-
ca, no segundo, evidenciou que, na maioria dos eletr-olitos estudados, o dep-o
sito de manganês consistiu de uma camada fina e lisa inicial sobreposËa por
uma cam4da mais espessa facetada, formada a partir de glóbulos isolados.
Nas deposições a parËir de eletró1itos contendo adições de zinco,
esta camada inicial tem espessura tão fina que se mostra indiferenciada da
camada superior quando observada no microsc6pio eletrônico de varredura.
6 . 3. 1 Seq{iência de deposicão
As figuras 6.16 e 6"77 apresentam seqtlências de deposição para os
eletr-olitos números 1 e 19, respectivamente. O primeiro, sem adição de impu-
rezas, apresenta um intenso desprendimento inicial de hidrogênio, que dimi-
nui rapidamente (a partir do d-ecimo segundo de deposição, aproximadamente )
ao se formar a camada inicial de manganâs; sobre esta camada, começam a se
formar, aproxímadamente aos 90 s de deposição, colônias de g16bu1os, sobre
as quais o desprendimento de hidrogênio é praticamente inexistente; este des
prendimento passa a ocorrer apenas sobre as regiões não-recobertas da camada
inicial. O segundo, com adição de 2r0 mg.d*-3 de nÍque1, apresenta a forma-
ção da camada inicial semelhante ã do caso anterior, mas a formação dos 91ó
bulos da segunda camada ã mais lenta e estes estão mais separados entre si;
o clesprendimento de hidrogênio tambãm diminui ap-os a formação da camada ini-
cia1, mas permanece constante durante o prosseguimento da deposição.

a) 2s
c) 360s
e) 1020s
E,iê..i,¡ì.6t6 - Fo tomicrograf i as
6ptico acoplado ã
da deposição com
cãlula, aumento
106
b) 90s
f) 1800s
o eletrr6ïit¡25x)
d) 600s
agrr.l (microsc6pio

a) 3s
c) 480s
e) 960s
!'].i:g,.,.....',:.'6'- *'7:, - Fotomicrograf ias
óptico acoplado ã
da deposição com
cãlu1a, aumento
L07
b) 1B0s
d) 780s
f) 2400s
o e1èti.6-ïåf:o
25x)
n9'.1r9 (microscópio

108
A observação da deposição não permitiu identificar diferenças en-
tre as deposições feitas a partir dos diversos eletr6litos estudados, exceto
com relação ã velocidade de formação da segunda camada, constituída de g16bg
los isolados ou aglomerados; esta velocidade foi menor e os glóbulos se mos-
traram mais isolados com os eletr-olitos que continham adição de ímpurezas ;
e1a foi maior' com glóbulos mais aglomerados, quando não havia adição de im-
purezas. Isto é mostrado nos exemplos das figuras 6.16 e 6.77, respecËiva-
mente.
O desprendimento de hidrogênio também se apresentou semelhante em
todas as seqtlências. E1e 6 extremamente ínLenso ao se ligar o circuito elé-
trico, quando o catodo-base de aço inoxidável não estã ainda recoberto por
manganês. Nestes ínstantes iniciaís, o catodo*base fica inteiramente recober
to de bolhas de hídrogÊnior guê crescem rapidamente at6 diâmetros entre 0,2
e 1r0 nrn e então se desprendem, subindo at6. a supe-rfÍcie d.o e1etr6lito. Com
o ínício da formação do depósito, este desprendimento inÈenso se restringe
às áreas que eventrtalmente permaneçam não-recobertas. Nas 'areas recobertas
pelo manganôs, o desprendimento Jmuito menos intenso e as bolhas que se
desprendem têm diâmetros muito menores, da ordem de 0,05 a 0 12 rm. A16m dis-
so, ao invés de ser homogêneo como sobre o aço inoxidãvel, o desprendimen-
to passa a ocorrer preferencialmente nas irregularidades do dep6sito e nas
arestas entre os gl6bulos e o dep6sito liso inicial. Quando estas arestas
desaparecem (ou seja, quando a camada inicial fica totalmente recoberta pela
segunda camada) , o desprendimento aparentemente reduz-se ainda mais.
6,3.2 Observação do depósito
0 depósito apresentou duas camadas nitidamente diferenciadas com
todos os eletrólitos que não continham adição de zinco. Mesmo quando a cama-

109
da superior se apresentava muito compacta, era possíve1 observar a camada
inicial ao se procurar a borda do depósito. Nos eletr-olítos com adição de
zinco, não se observou a camada inicial sequer nesta região.
Ao observarmos a camada inicial junto ã borda do dep6sito ( local
delimitado pelo anel de PVC, como descrito na seção 5.2.I), verificamos ser
ela constituÍda de pequenos g16bu1os, com cerca de 0,5 a 4,0 Um de diâmetro,
cuja morfologia nã-o conseguimos discernír por falta de definição.
O diâmetro destes glóbu1os, que podemos, grosseiramente, consíde-
rar como da mesma ordem de grandeza da espessura da camada inicial , 'e af.eta-
do pela adição de cobalto e nÍquel. E1e passa de 3rA - 4,0 U*, nos eletróli-
Ëos sem impurezas, para 1r0 - 1r5 pm, com adição de cobalto, ou 0r5 - 1r0 Um,
com adição de niquel.
A figura 6.18 mostra exemplos de fotomicrograf.ias da camada iniÉ
a1 com eletrólito sem impurezas:
a) MEV -
Fig. 6 ,LB
25Ox - eletr6lito nQ 1 b) MEV - 45x - eletrólito nQ 3
- Fotomicrografías da camada ínícial com el-e-tró1itos sem adição de
impurezas

110
A figura 6"19 mosËra exemplos de fotomicrografias da camada inicíal
obtida em deposições com eletr6litos contendo cobalto e níquel:
a) MEV - 700x - elerr6liro nQ 12 b) MEV - 950x - elerróliro ng 16
Fig. 6,L9 - Fotomícrografías da camada inicial com eletrólitos com adições de
cobalto e de níque1
Nos depósiEos resultantes dos eletr6litos corn adíção de zinco, não
se conseguiu diferenciar a camada inicial e o resËante do depõsito. A figura
6.20 apresenta exemplo desÈes tipos de dep6sito:
MEV -
Fotomicrografia de
com eletr6lito com
170x - eletrólito nQ 22
depósito sem diferenciaçãoadição de zinco
Fig. 6.20 - da camada inicial,

111
A figura 6.21 mostra mais um exemplo da camada inicial, cuja obser-
vação foi favorecida pela presença de uma trinca. Este tipo de trinca foi um
faËo fortuíto, não observado em qualquer outro local do dep6siËo ou em qual-
quer outro depósito:
a)
Fig.
MEV - 70x - eletr6lito n9 4
6.2L - Fotomicrografias de
b) MEV -
Erinca na camada
1400x - eletr6lito n9 4
inicial
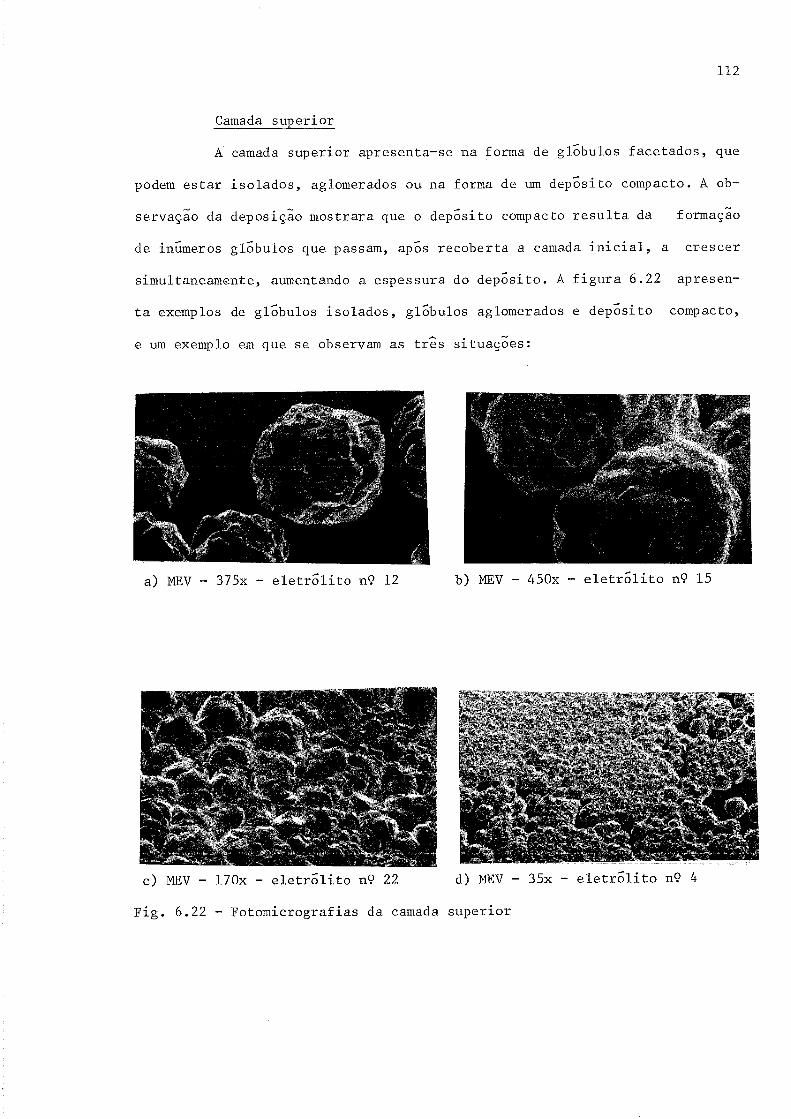
1L2
Camada superior
A camada superior apïesenta-se na forma de g1-obulos facetados, que
pod.em estar isolados, aglomerados ou na forrna de um depósito compacto. A ob-
servação da deposição mostïara que o dep6siËo compacto resulta da formação
de inúmeros gl6bulos que passam, ap-os recoberta a camada inícial, a cr,escer
simultaneamente, aumentando a espessura do dep6sito. A figura 6.22 apresen-
ta exemplos de gl-obulos isolados, g16bu1os aglomerados e dep-osito compacËo,
e um exemplo em que se observam as três situações:
a) wv - 375x - eletrólito nQ 12
c) }ßV - 170x - eletr6lito nQ 22 d) MEV - 35x - eletrólíto nQ 4
Fig. 6.22 - Fotomicrografias da camada superior
b) MEV - 450x - eletr6lito n9 15

Nos dep6sitos obtidos a partír de alguns ele
pequenas de diõxido de enxofre, encontïam-se gl6bulos
tam-se lisas e perfeitas, como mostrarn os exemplos da
trótitos com
cuj as facetas
figura 6.23t
113
adições
apresen-
a) mv - 90Ox - elerr6lito nQ 7 b) Ißv - 800x - eletrólito ng 13
Fig. 6,23 - Fotomicrografias de g16bulos com facetas lisas
Quando o eletr6l.ito tem adição de zinco, as faceEas do dep6sito a-
presentam-se sempre irregulares e com defeitos, como mostra a figura 6.242
Fig. 6.24 -
MEV
Fotomicrografialitos com adição
- B5Ox - eletról íto nQ 22
de dep6sito com facetas irregulares, com eletr6-de zinco

L7.4
7. DTSCUSSAO
7.1. EFEITO DAS ADIçõES SOBRE A EFICIÊNCIA DE CORRENTE
7,I.L - Eficiência de corrente em função da densidade de corrente
Pelas curvas da figura 6.5 (eficiência de corrente em função da
densidade de corrente catõdica total), observamos que, com qualquer eletró1i
to, a eficiência de corrente 6 mais alta na faixa central das densidades de
corrente utilizadas do que abaixo ou acima desta faixa. Ou seja, todas as
curvas apresentam um mãximo - que denominamos "eficiência de corrente mãxi-
ma", ou ECM - dentro desËa faixa. Este mãximo ocorre em uma clada densiclade
de corrente, inCM, e em dado potencial de eletrodo, PECM.
A forma d.as curvas de polarização explica a existência deste mãxi-
mo.
As curvas de po1-arização parciais de manganês apresentam a.umento
da inclinação com o aumento do logaritmo da densidade de corrente tota1, po-
dendo-se distinguir uma região inicial, nos potenciais mais an-odicos, com in
clinação pequena e aproximadamente constante, uma região de transição e uma
região final, nos potenciais mais cat6dicos, de grande inclinação (aproxima-
damente vertical, em alguns eletr6litos com grandes adições de impurezas e
pequenas adições de di6xido de enxofre).
As curvas de polari"açã.o parcíaís de hidrogänio apresentam diminui
ção da inclinação com o aumento do logaritmo da densidade de corrente total,
podendo-se observar uma região inicial (nos potenciais mais anódicos) de grair

115
de inclinação e .uma região f inal de inclinação muito pequena. EsLa
região ocorre na mesma faixa de potencial do trecho de grande inclinação das
curvas parciais do manganês.
Este afasËamento enËre as curvas do hidrogênio e as do manganês
deslocando-se para a direita as primeiras e mantendo-se verËicais as segun-
das - corresponde ã grande diminuíção da eficiência cle corrente veríficada
nas densidad.es de corrente mais elevadas. O rnáximo de eficiência de corrente,
por sua vez, ocorre em densidades de correrite um pouco inferíores ãs do iní-
cio do afastamento enLre as curvas. Abaixo destas densidades de corrente, a
eficiência de corïente diminui porque as inclinações dos trechos iniciais
das curvas do manganês são nenores que as das curvas de hidrogênio.
Os valores das eficiêncías de corrente máxímas e das densidades de
corrente e potenciais correspondentes, ouseja, ECM, iSCl,, . PECM, variaram
dentro de uma ampla faixa. 0 menor valor de ECM f.oí I47", gue ocorreu em uma
iuC* d" 4OmA.cm-2 . rr PECM de -1 ,543 vrcom o eletr-olito nQ 10, que tem acli
ções de 0r1 g.dr 3 de di6xido cle enxofre e 1,0 mg.d* 3 de cobalto. O maior
valor de ECM f.oí 687" e ocorreu com o eletr-olito nQ 3, que tem adi-
ção de A,6 g.d*-3 de di6xido de enxofre. Os valores máximo e mínimo de iua"
foran 32 e 70 r¿.."t 3, respecËivamente, e os d" P'CU foram -11585 e -1,534V,
respecËivamenËe.
Analisaremos a seguir o efeito do di6xido de enxofre, do cobalto,
do níque1 e do zinco sobre estes valores. Estas adições afetam todos os parâ
meËros das curvas de:polarízação ou seja, a posição das duas curvas par-
ciais, a definição mais nítida ou menos nítida de regiões distintas na cur*
va d.o manganês, a densidade de correnËe em que ocorre o trecho de grande in-
clinação da curva do manganês, eËc.

116
7.L.2 - Ef eito do cobalto sobre a ef.íciência de corrente
As adiçoes do cobalto sempre dimínuem a
xima, ECM, e o potencial em que e1a ocorre, PECM,
de dióxído de enxofre. A densidade de corrente em
bãm diminui com estas adições, exceto na passagem_1
cobalto, com 0r1 g.dm ' de dióxido de enxofre, na_a
ra 40 mA.cm '. A figura 7.1 mostra estes efeitos:
eficiência de corrente má-
qualquer que seja a adição
que ocorre ECM, i'a*, Ëam-
de 0, 5 para 1r0 mg.¿* 3 ¿.
qual e1a aumenta de 38 pa-
3
N
80I
=()l¡J
fL
o o,25 q5 l,o o
' Cobolto ( mg. d m-3 I
o o,js..dm-3 so^-zFig. 7.I * Efeito das adições de
0,26 q5 tpCobolto ( mg. d nts )
o o,25 0,5 t,o
Cobolto (mg.dm-3l,-3-3x 0,3g.dm - S0, A. 0,6g.dm - S0,
cobalto sobre ECM, i¡ClU " PACU
A figura 7.2 mosLra os efeitos das adições de cobalto sobre as cur
vas polarogr'af.ícas parciais de manganês e hidrogênio, em eletrólitos com
a
0,1 g.dm ' de diõxido de enxofre.
Por que ocorre a dirninuição da eficiência de corrente máxima? E1a
ocorre porque, como vemos , a að,íção de cobalto diminuí inËensamente a depo-
sição de manganês, ou seja, desloca a curva do manganês para a esquerda (den
'E9
e
Et¡J
t=()lr¡
60
il

sidades de corrente menores), ao mesmo tempo em gue aumenta
de hidrogênio, ou seja, desloca a curva do hidrogênio para
qualquer potencial. Assim, a eficiência de corrente diminui
tencial e, porËanto, a eficiência de corrente mãxima Ëambãrn
tenciais mais caËódicos (parte superior das curvas, uma vez
adoËada 6 o negativo do potencial), ambos esËes efeitos são
Esta diferença é mais pronunciada para a curva d.o manganês,
balto altera sua forma, definindo um trecho aproximadamente
-1, ,
/
Ì
-1 ,55
TI7
o desprendimento
a direita, em
em qualquer po-
diminui. Nos po-
que a abcissa
mais intensos,
uma vez que o co
vertícal na sua
U)Utrlo€o{JdCJ
c).o
r-lCd
.FloHc)!oÀ
I /r',ø -"&?,1 .f /
'¿ n -.o' -/-¿ r?r -f
e1.
e1.
e1 .
e1.
oPncu
0, 01
(sem impurezas)
(0,25mg . dr-3co)(0,5 mg.dr-3co)(1,0 mg.dr-3co)
densidade de
corrente parcialde manganês (a.c*-2)
0r1
/ ,r'-
-1, 5040,
*001
1
4
7
10-1 ,60 <))(_)trl
ioo]J(Ú
oCJ
F-ld
'rloHo4JoÈ
-1,55
-1 ,500,001 0,01
Fig. 7.2' - Efeitos das adições de cobalto sobreparciais, em eletrõljtos com 0rlg.dm
0r1
as curvas de polarizaçãoa-' d" di6xido de enxofre
ødens idade
corrente parcialhidrogênio (4. cm
de
de-)-)
'/ d

118
parte final. Quanto maior a adição de cobalto, maís clara a existência desËe
trecho de grande inclinação e menor o valor da densidade de corrente em que
o mesmo ocorre.
Comparemos numericamente o efeito do cobalËo em duas faixas de po-
tencial, novamente com eletrólitos contendo 0,f g.a*-3 de dióxido de enxo-
fre. Em -11580 V, que está dentro da faixa de potenciais em que ocorre o tre
cho de grande inclinação da curva do manganês, a adição de lr0mg.dn 3 U" "o-
balto dininui oito vezes a densidade de corrente do manganês e aumenta qua-
Ëro vezes a do hidrogênio, em relação ao eletr6lito sem adição de impurezas.
Em -1,520V, gu€ estã dentro d.a faixa de potenciais correspondentes ãs densi-
dades de corrente que antecedem ao trecho de grande inclinação das curvas de
manganês, a adição de 1,0 mg.dm-3 de cobalto diminui apenas três vezes a den
sidade de corrente do manganês e aumenta duas vezes a do hidrogênio. A figu-
ra 7.3 apresenta estes valores de densidade de corrente:
t20
100
80
60
40
20
00102040adição-¿" "oU.fto {*g.ar-3¡
Fie. 7.3 - Efeito da adição de cobalro
01020401adição de cobalto (rg.dr-')
sobre as densidades de corrente parciais
*1,520V

119
Por que o potenci"l PECM, correspondente à eficiência de corrente
mãxima¡ âurrr€rrtâ com adições de cobalto? l{a figura 7.2, assinalamos em cada
curva o correspondente potencial PUC''. Como vemos, este valor 'e. razoavelmen-
te cat6dico, -1,573 V, para o eletrólito sem adição de impurezas; nesta fai-
xa de potencial, a diminuição da densidade de correnLe do manganês quando se
adiciona cobalto ã muito intensa, devido ã jã comentada formação do trecho
de grande inclinação. Em potenciais um pouco mais anódicos, 6 menor a dimi-
nuição da densidade de corre-nte do manganês. Como a diferença entre os efei-
tos sobre a curva do hidrogãnio não 6 tão grande, resulta uma menor dininui-
ção da eficiência de corrente nestes potenciaís mais anódicos, onde se esta-
belece, portanto, o novo máximo de eficiência de correnËe. Com Or25r 0r5 e
a,J1r0 fng.dm " de cobalto, os potenciais Pua* são, respectivamente, -1,560,
-1,543 e -1,543 V. Na ú1tima adição não ocorre modificação do potencial,por-
euer com 0,5 mg.dr 3 de cobalto, a eficiência de correnËe mãxima 6 atingida
fora da f.aíxa correspondente ao trecho de grande inclinação da curva do man-
ganês. Isto não ocorre nas iiltimas adições de cobal.to dos eletrólitos que
cont6m 0r3 ou 016 g.dr 3 de diõxido de enxofre, pois nestes casos, as efi-
ciências de corrente máximas ocorrem em potenciais mais catõdicos, ainda den
tro d.o Èrecho verLical das curvas do manganês (v. Tab. 6.7, Fig. 7.L e Fig.
7.4) .
Por que decrescer'mas não sempre, a densidade de corrente iECMr.oI
respondente ã eficiência de corrente mãxima? Esta densidade de corrente cor-
responde, evidentemente, ã soma das densidades de corrente parciais de man-
ganês e de hidrogênio neste potencía1. Como vimos, as adições de cobalto di-
minuem mais a deposição de manganês do que aumentam o desprendimento de hi-
drogânio. Assim, sua soma diminuiria mesmo "" PECM se mantivesse fixo. Como
PECM ". torna mais an6dico, esta soma diminui com mais razão ainda, pois em
qualquer curva polarogrãfica cat6dica o aumento do potencial corresponde a

r20
uma diminuição da densidade de corrente. Verificamos, contudo, llue no último-?
aumento da adição de cobalto (de 0 15 para 1,0 mg.dm -) o valor d. igCl,t se
mant6m constante, com 0r3 g.d*-3 de di6xido de enxofre, ou mesmo chega a
crescer, com 0rl g.d;3 de di6xido de enxofre. Isto ocorre porque nos eletró
litos com estas adições a diminuição da densidade de corrente do manganês
passa a ser aproximadamente igual ou mesmo menor que o aumento da de hidro-
gênio. Além disso, no caso do eletr6lito com 0,1 g.d;3 de cli6xido de 'enxo-
fre, o potencirl PSCM se mantém fixo, como exposto no parâgraÍo anterior, e,
portanto, deixa de afeËar iUa*.
O efeito do cobalto não ã afetado qualitativaaente pelo aumento da
adição de dióxido de enxofre, que apenas diminui a distância entre as curvas
(ou seja, diminui a intensidade do efeito do cobalto) e as desloca para po-
tenciais mais caËódicos. Como exenplo, a figura 7.4 apresenta o efeito do cg
balto sobre as curvas de polarização parciais dos eletrólitos com 016 g.dm 3
de dióxido de enxofre:

t2L
*1r500
-1r60
-l- ,55
-l ,60
-1, 55
e1.
e1.
e1.
e1.
,001
(nQ[']o
.lJdoo
r-id.rlCJ
Éc.)+JoÈ
v
<n
td
+J(dCJ
CJ
r-{ñ.¡ora)+JoÊ.
0,01(sem ímpurezas)
(0,25mg. dr-3co)(0,5 mg.dr-3co)(1,0 mg.d*-3co)
0,01
densidade de
corrente Parcial_t
de manganês (A.cm ')
0r1
de
de-L.
)
0r1
as curvas de polari zaçã"o
-3 d. diõxido de enxofre
dens idade
corrente parcialhidrosênio (A.cm
J
6
9
L2
-1 ,500 ,001
- Efeitos das adições de cobalto sobre
parciais, em eletrólitos com 0r69.dm
Fig. 7.4

L22
7.1.3 Efeito do nÍquel sobre a eficiência de corrente
Como mostra a figura 7.5, os efeitos da adição do níque1 são quali
tativamente semelhanËes aos da adição de cobalto, embora menos intensos. En-
quanto, por exemplo, 1r0 mg.dr-3 de cobalto abaixa a eficiência de corrente
mãxima de 61 para L47", 2,0 rg.a* 3 de níquel abaixam-se apenas a 367" ( exem-
plos com os eletrõlitos n9s L,10 e 19, com 0,1 g.dt-3 de dióxido de enxo-
fre, a partir de dados da tabela 6.7 ou das figuras 7.1 e 7.5)"
I
=()o.t¡J
ü=()ul
1.O 2p O
Ni¡uel ( mg.dm-3 I
-3o D,1g.dm " S0,
* Efeíto das adições
..Él¡9$zr
o,5 l,o 2,o
Níquel ( mg.am-s)
X o,3g.om-3 so,
de níquel sobre ECM,
\o.'\,o
o q5 r,o 29Nrhuel ( mg.dnis
ô
.^ 0,6g"dm " S0,
incu " Pnc¡.r L
20
q5
Fíe. 7 .5
(\¡Eq
Ê,
=ol¡¡
60

123
Todos os efeitos da adição do nÍquel sobre a eficiência de corïen-
te mãxima, sobre P'CM . sobre irc* .*plicam-se pelos mesmos motivos expostos
a respeito do cobalto. Como mostra a figura 7.6, os efeitos do níquel sobre
as curvas de po1-arização parciais são semelhantes aos do cobalto, stas menos
intensos. Assim, estes efeitos, ou seja, a definição de um trecho aproximada
mente vertical no final da curva do manganês, a diurinuição da deposição de
manganês especialmente nos poËenciais mais cat6dícos, correspondentes ao
trecho aproximadamente verticali e o atunento do desprendimento do hidrogê-
nió, levam aos mesmos comportamentos finais que o cobalto.
-1 ,60 (n(Jtrl
o]JdU
CJ.o
r-1d
.FlCJ
q)ÐoÈ
-1r55
-1 ,50
densidade de' corrente parcial
_,)de manganês (4. cm -)
0r10,001 0,01(sem impurezas)
(o,5mg.¿m-3 ni)(1,Omg.¿m-3 ui)(2,Omg.¿m-3 ni)(/j
Qþloño]JcúO
c).o
È-'l(Ú
.Joo4Jog
1
13
I6
L9
-1 ,55
-1,500,001 0,01
Fig. 7.6 - Efeitos das adições de níquel sobre as
parciais, em eletrõlitos com O,19.dm-3
el.e1.
el.e1
dens idade
corrente parcialhidrogênio (A.cm
de
de_)')
0r1
curvas de polarizaçã.o
de di6xido de enxofre

124
0 efeito do níquel tamb6m não é qualitaËivamente afetado pelo au-
mento da adição de dióxido de enxofre, que, tal como observado no caso do cg
balto, apenas diminui a distância enËre as curvas e as d.esloca para poten-
ciais mais catódicos. A figur a 7.7 apresenËa o efeito do níquel sobre as cur
vas de polarízação parciais dos eletr-olitos contendo 0r6 g.dm-3 de di6xido
de enxofre:
-1r60
-1r55
densidade de
corrente parcialde manganês (a.cr-2)
0,01(sern ímpurezas)
(o,5mg.¿m-3 ui)(l,omg.¿rn-3 ui)(2,}mg. ¿rn-3 Ni)
-1r55
densidade de
corrente parcial de
hidrogênio (4. c^ 2)
0,01
adições de níquel sobre as
eletrõ1itos com 0,69.dm-3
-1 ,500 ,001
CN(Jtrlo
.Ú
.tJCd
U
o.Ú
r-{d
.F{
oHG)+Joor
t;t:l.dloI .¡¡
lilËL:001
itorcia.
I
OS
ai
0r1
-1r60
e1.
el.e1.
e1.
3
L5
18
2L
50L0rc
lfe i)arc
Ef
pa
-1,
s das
i", em
0r1
curvas de polarizaçã.o
de di6xido de enxofre
Fie. 7 ,7
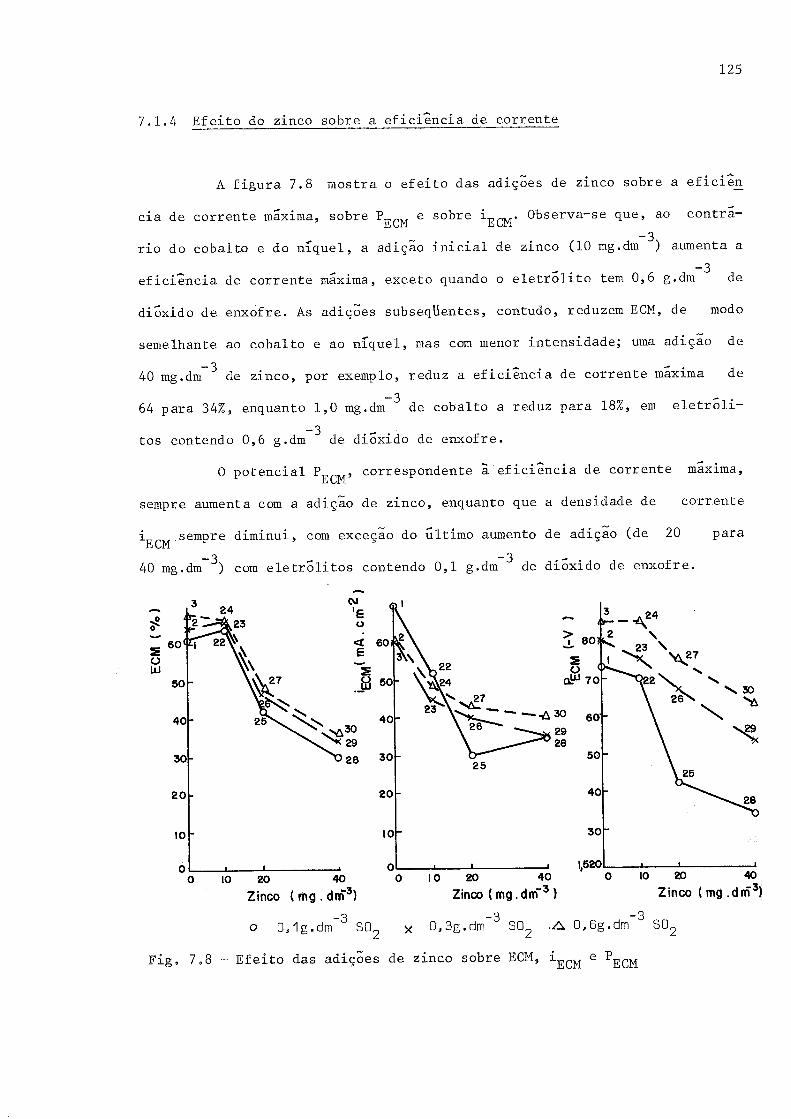
L25
7.L4 Ef eito do zinco sobre a ef iciência de corrente
A figura 7.8 mosËra o efeito das adições de zinco sobre a eficiên
cia de corrente mãxima, sobre PECn u sobre ira*. Observa-se que' ao contrã-
rio do cobalto e do níquel, a adíção inicial de zinco (10 rng.¿*-3) aumenta a
eficiêncía de corrente mãxima, exceto quando o eletr6lito tem 0,6 g.d*-3 de
di-oxido de enxofre. As adições subseqtlentes, contudo, reduzem ECM, de modo
semelhante ao cobalto e ao nÍque1, mas com menor intensidade; uma adição de
_e40 mg.¿nr-'de zincor por exempi'o, reduz a eficiência de corrente mãxima de
64 para 34%, enquanto 1r0 mg.dt-3 de cobalto a reduz para I87., em eletr-o1i-
tos conËendo 0,6 g.d*-3 de di-oxido de enxofre.
O potencirl PECM, correspondente ã eficiência de corrente mãxima,
sempre aumenta com a adição de zinco, enquanËo que a densidade de corrente
iUC, sempre dirninui, com exceção do ú1timo aumento de adição (de 20 para
-a -?40 mg.dr-') com eletr6litos contendo 0,1 g.dm'de di-oxido de enxofre.
N'E()
e
-=.r3
J=o
o,l¡J \lo\
-q,30
2928
3029
2ø
lo 20 ¿to
Zinco ( mg. dnîs)
o o,1g.dm-3 so,
* Efeito das adições
Zinco ( mg. am-3 )
x o,3g.dm-3 so, .a. o,sg.dm-3
de zinco sobre ECM, ígClt " PnCl,t
æZinco
Soz
¿lo
( mg.oni'3!
24
Fig. 7 .B

r26
A f.í-gura 7.9 mostra o efeito das adições de zínco sobre as curvas
de polari zação parciais de manganês e de hidrogênio, com eletr6litos com
_20r1 g.dm'de di-oxido de enxofre. Observa-se que, tal qual o cobalto e o ní-
quel, o zinco desloca as curvas do manganês para densidades de corrente mais
baixas, especialmente nos potenciais mais catódicos, e Ëende a definir um
trecho aproximadamente vertical no final das curvas. Sobre as eurvas de hi-
drogênio, contudo, há uma diferença: a primeira adição de zinco, 10 mg.d*-3,
diminui a densidade de corrente de hiclrogênio, ao contrário das adições de
-1 ,60
-1,55
-1r50
U)Q[¡]
oÈo.lJdoAJ
rid
'rlOÉ(.).lJ
È
0, 001
¿|-¿ de
el.e1.
el.e1.
1
22
25
28
0,01(sem impurezas)
( lOmg . dr-3 zr,)
( 2omg . dr-3 zr,)
(4omg. ¿*-3 zrr)
densidade de
corrente parcial-2
manganês (A.cm -)
0r1
-1 ,60 (n(Jf¡loEo.u(üCJ
q)ÎJrlcd
" r''lUÉq),l-.)
oo.
-1r55
-1 ,500,001
- Efeitos das
parciais, em
,/
lt,p
0,01 0r1
adições de zinco sobre as curvas de polarização
eletrólitos com 0r19.dt-3 d. dióxido de enxofreFig. 7,9

L27
cobalt.o ou nÍquel. As adições subseqtlentes seguem o efeito observado com co*
balto e níquel, aumentando o desprendimento de hidrogênio.
Com 0r3 ou 016 g.d**3 de dióxido de enxofre, os efeitos de zínco
observados nos eletrõlitos com 0rl g.d**3 de di6xido de enxofre repetem-se,
qualitativamente. A figura 7.10r Que apresenta o efeito do zinco sobre as
curvas de polarização parciais de elerrólitos contendo O16 g"dr-3 de di6xido
de enxofre, ilustra este fato:
-1, 60
*1r55
densidade de
corrente parcialde manganês (a.cr-2)
U)(Jt{oo+Jdo(.)
F-l
0r01
impurezas )
.dm - zn)
,dm' Zn)
.dm ' zn)
-1r55
<nOtrloo+JdC)
o
r-l(d
.rlU
CJ]Joo.
-1, 500 ,001
-1, 60
d.JoHqiTJoÊ.
-1, 500, 001 0,01
Fig. 7.L0 - Efeitos das adições de zíncoparciais, em eletrólitos com
0r1
dens idade
corrente parcialhidrogênio (4" cm
0r1
as curvas de polari zaçãoJ, de dioxido de enxofre
e1
e1
e1
e1
3
24
27
30
(sem
(1Omg
(2)ne
(40me
de
de-)
sobre
0,69.dm
,'i

128
Analisemos, então, a variação da eficiência de corrente mãxima com
as adições de zinco.
Com a adíção de 10 rg.d* 3 de zinco, a eficiência de corrente au-
menËa nos potenciais nos quais a diminuição da densidade de corrente de man-
ganês ã menor que a do hidrogênio. Com 0,1 g.dm-3 de di6xido de enxofre, ís-
Èo ocorre em qualquer potencial mais an6dico que -11580 V. Assim, como a efí
ciência de corrente máxima do eletr6lito sem adição de impurezas oco:rre em
-I,573 V, ECM aumenta. Por exemplo, do eletrõliÈo n9 1 para o nQ 2-2, ECM ¿s-_,)
menËa de 61 para 64% (v. Fig. 7.8 ou Tab. 6.7). Com 0,3 B.dm ' de dióxido de
enxofre, a adição de 10 rg.ar 3 de zinco provoca uma diminuição levemente
maior do desprendimento de hidrogênio do que da deposição de manganês, em
qualquer potencial (e, portanto, tambãm .* PnCu); assim, a eficiência de cor
rente mãxima aumenta um pouco. Com 016 g.d* 3 de di6xido de enxofre, contu-
do, a eficiência de corrente s6 aumenta em potenciais mais an6dicos que
-L,575 V, e diminui em pobenciais mais catódicos que este; como a eficiâncía
de corrente mãxima deste e1etr6lito, quando Sem adição deimpurezas (e1etr61i
to nQ 3) ocorre em -11585 V, a eficiência de correnËe mãxima diminui com a
adição de 10 ng.dm 3 de zinco.

t29
_1As adições de 20 e de 40 mg.dm ' de zinco provocam a diminuição da
eficiência de corrente' em qualquer potencial, coln qualquer das adíções estu-
dadas de di-oxido de enxofre, uma vez que estas adições de zinco diminuem a
deposição de manganês e a,rmentam o desprendimento de hidrogênio em qualquer
potencial. Porisso, ECM diminui com estas adições
O aumento do potencirl PSCI", com as adições de zínco deve-se aos
mesmos fatos expostos na anãliså do cobalto e do nÍquel: a eficiência de cor
rente mãxima dos eletrólitos sem impurezas ocorre em potenciais :razoavelmen-
te catfidicos, nos quais a adíção de í-mpurezas provoca o surgimento do trecho
aproximadamente vertical na curva cle polarização p"r"ial do manganês; nestes
potenciais, como jã vimos, a diminuição da densidade de corrente do manga-
nês, causada por estas adições, ã muiËo intensa; em potenciais um pouco mais
an6dicos, esta diminuição -e menos intensa e, portanto, a eficiência de cor-
rente cresce mais ou decresce menos; assim, PECM d""loca-se para estes poten
ciais. As alterações de comporËamento das curvas de hídrogênio com a varia-
ção do potencial são menores que as das curvas de manganês, e porisso são as
de manganês que deter:minam o sentido da vari.ação de P'CM.
A densÍdade de corrente catódica total correspondenËe a ECM decres
-?ce com a adição de 10 mg.dm ' de zinco porque ambas as densídades de corren-
te parciais decrescem com esta adição, Ëanto pelo efeito da pr-opria adição
em dado potencia.l fixo como pelo efeito do aumento d" P'CU. Com as adições
subseqtlentes de zi-nco, iECM decresce menos porque a densidade de corrente do
manganês continua decrescendo por efeito das adições, mas a de hidrogênio
passa a crescer. Há uma situação em que incu.h"Ba a crescer: trata*se da
passagem do eletr-olito com 20 para o com 40 ng.d*-3 de zinco, com 0r1 g.d*-3
d.e dt-oxldo de enxofre. Neste caso, o aumento da densidade de corrente do hi-
drogênio sobrepuja a dirninuição da de mattganês, ê â densidade de corrente to
tal aumenta; isso ocorre polque, nestes eletr-o1itos, PECM sofre variação pe-

130
quena com a adição de zinco, pot estar pr6ximo aos potenciai,s mais anfidicos,
nos quais ocorre grande diminuição da eficiência de corrente.
7 .r.5 Efeito do dióxido de enxofre sobre a eficiência de corrente
As figuras 7.11 a 7.I3 apresentam os efeitos das adições de di6xi-do de enxofre sobre a efíciência de corrente m-axima e sobre o potencial e a
densidade de correnËe em que e1a ocorre.
Como vemos, o aumento_a
0r3 g.dm ' sempre eleva o valor
jam as adíções de impurezas.
o aumento subseq'rlente, de 0r3 para 0r6 g.dr-3, aumenta a ef iciên-
cia de corrente mãxima apenas quando o eletr6lito não tern adição de impure-
zas ov quando esta adição 6 pequena; quando as adições de impurezas são gran
des, ECM não se altera significativamente.
Esta diferença de comportamento deve explicar as divergentes con-
clusões sobre o limite de atuação do di-oxido de enxofre ¡ eue podem ser devi-
das a diferenças nos teores de impurezas dos eletrõlitos utilizados pelos
diversos autores.
da adição de dióxido d.e enxofre de 0,1 para
de ECM e diminui o d" PECM, quaisquer que se
iECu r" altera pe
que não guardam
iECl4 r."rltantes
resultantes das
Pelas figuras 7.IL a 1.I3, observamos também que
1as adições de dióxido de enxofre, mas em sentidos diversos
qualquer correlação símp1es com as adições de impurezas.
Observamos ainda que as alterações em ECM, pECM u
das adições de dióxido d.e enxofre são muito menores que as
aclições de cobalto, níquel ou zinco

131
Ë
=ol¡¡
60
NIFIÉ
=ou¡
¡
õolP 70
àe
=()t,J Þ
o o,l o,3 q6 O O/ 93 016 o ql o,5 0,6soe(g.drnsI soz(g.drñ-3) soz(g.drlis)
o sem impurezas À 0,25mg"¿m3co fl 0,5mg.¿m3co + l,Omg.¿m3co
fig" 7.LL - Efeito do aumento da adíçãó ¿e dí6xido de enxofre sobre ECM,
incu " PECM , em eletr6litos com e sem adição de cobalËo
--El8
v_À'.'-'--Îa';F'--'
2t
oqt 93 q6 oo/ 93 016 oqt q3 q6
soz (g . cm-3) so a ( g. om-s) so2 ( g.dm'sl
o sem impurezas A 0,5mg'arn3tli tr l,Omg.om3tli + 2,omg.¿m3ltli
Fig. 7 "L2 - Efeito do aumento da adíção de dióxido de enxofre sobre ECM,
ircu " PECM, em eletrõlítos com e sem adição de níquel
Þt3út6
ft9
-vl7
ã
I
J
=()lrJ
fL
N,Êo<Ê
=,ol¡J
-+2l
IlF'''v
t9
3
4

t32
.9o
-C)lrJ 50
NrEo<c
=()Lr¡
I
\_
.22\r zs -ÅoY/26 -Ã27"c,8.lso
29trl25
;t=()
a!¡J V22
or3 016
soz (g. om-31
qg 016
So2 ( g. om-sl
{* 4o
q3 96So2(g. dm3l
orl
I
i'ñ¿' /'T28
-? -1n 2omg. dm " zn + 4omg. dni" zrt
de di6xído de enxofre sobre ECM,
com e sem adíção de zinco
o sem
Fie. 7 .I3
impurezas A 1omg.¿ni3 zn
- Efeito do aumento da adição
iuCM "
PECM , em eletr61itos

133
A f igura 7.14 mostra que, com eletr-olitos sern adição de impurezas,
o aumento da adição de di6xido de enxofre diminui tanto a densidade de cor-
rente parcial do manga4ês como a de hidrogênio, em qualquer potencial. A efí
ciência de corrente aumeriËa porque a diminuição do desprendim-ento de hidro-
gênio ã mais pronunciada que a da deposição de manganês, em toda a faixa de
poËenciais estudados.
-1r60 d'¿/
-1 ,55
-1r50
,:,, densidade de
corrente parcial)
de manganês (A.cm--)
U)(Jf¡lo.do]J(dO
c)
r-'{d
' r'lU
AJ+JoÈ
0, 001
1
2
3
-1,60
-1r 5
-1r500,001
Fig. 7,I4 - Efeitos das var,iações
curvas de polarizaçäoimpurezas
0r1
dióxido de enxofre sobre
eletr6litos sem adições
el.e1.
el.
0rol-1(0,1g.dm -
^(0,3g.dm -_i
(O,6g.dm -
0r1
dens idade
corrente parcialhidrogênio (A.cm
so2)
so2)
s02)c/)Q
o.o
.tJ($O
0)'ur-ld
'riOHqJ
+J
a
de
de_.,
')
0,01
de adição
parcíais,AS
de
de
em

134
Em potenciais pouco mais cat-odicos que -L,573 (eU* do eletr6lito_1
com 0,1 g.dm'de dióxido de enxofre), a diminuição da densidade de corrente
de manganês torna-se menor com o aumento do potencial, enquanto que a dimi-
nuição da de hidrogênio mantãm-se aproximadamente constante. Porisso, PECM
desloca-se para estes potenciais mais catódicos quando se aumenta a adição
de dióxido de enxof re.
O valor d" iEclt, neste casoe dirninui com o aumento da adição de d!
6xido de enxofre porque este aumento provoca a diminuição das duas densida-
des de corrente parciais (em um dado potencial fixo), e porque esta diminui
ção não chega a ser contrabalançada pela diminuição, acima comenËada,
PECM, que provoca o aumento das duas densidades de corrente.
de
As figuras 7.15,7"L6 e 7.Il mostram que a adição de impurezas mo-
difica o comportamento do diõxido de enxofre. O aumento de sua adição conti-
nua diminuindo a densidade de corrente de hidrogênio. A densidade de cor-
rente d.o manganês, contudo, s6 diminui nos potenciais mais anódícos. Itros po-
tenciais mais catõdicos, correspondentes ao trecho de grande inclinação das
curvas d,o manganês, o aumento da adição de di6xido de enxofre aumenta a den-
sidade de corrente do manganês, anulando em parte a formação do trecho apro-
ximadamente vertical pelas impurezas.
Nestes potenciais, portanto, a eficiência de corrente cresce muito
com o aumento da adição de dióxido de enxofre, uma vez que a deposição do
manganês diminui. Este efeito, contudo, ocorre em potenciais nos quais as
eficiências de corrente, mesmo com o efeito do dióxido de enxofre, são muito
baixas. A eficiência de corrente mãxíma continua ocorrendo fora da faixa do
trecho aproximadamenËê vertical da curva do manganes e, portanto, e pouco
afetada pela anulação deste efeito pelo di-oxido de enxofre
Com a presença de impurezas, o aumento da adição
xofre diminui o potenci.l P'CU mais intensamente do que nos
de di6xido de en-
eletrólitos sem

135
adição de impurezas. Isto ocorre por dois motivos. Por um lado, o aumento da
adição de di6xido de enxofre (principalmente a primeira, de 0,1 para-e0,3 g.dm -) provoca uma intensa diminuição da densidade de corrente do hidro
gênio em potenciais um pouco mais catõdicos gr" PECtui dos eletrólitos com_')
0,1 g.dm'de dióxido d.e enxofre. Por outro lado,como o dióxido de enxofr:e
tende a anular a formação do trecho aproximadanente vertical das curvas de po
Larização de manganês, características dos eletr6litos com adições de impu-
rezas, a densidade de corrente do manganês diminui pouco ou mesmo aumenta
-1, 60
-1 ,55
U)Qt¡lo.Ú
.¡Jdoo
.Ú
r-l(0
.F1
oHc.)ÐoÈ
(nUtrloo]J(úoo.Ú
F-{cd
oÉ0)]J
o.
,llt{t'll/t
.,4t,'4.1/ t/,/'.t /
.' ./ //-"-'¿ t
t -/./ .'-./ -//¿'
-çl "
densidade de
corrente parcialde manganês (4..r-2)
-1,500 ,001
--sl.--- --el.el.
0,01a
(0,1g.dm -a
(0,3g.dm -ô
(0,6g.dm'
7
B
9
0rl
s02)
so2)
so2)
.d{u.oá';-
.lt ,r'
-1r60
-1r55
-1,500,001
Fig. 7.15 - Efeitos das variaçõescurvas de pol-ariração
de cobalto
dens idade
corrente parcialhidrogênio (A'. cm
0r1
di6xido de enxofre sobre as
eletrólitos com 0,5mg.d*-3
,/), ,¿
irI
de
de_,)')
0, 01
de adição
parciais,de
em
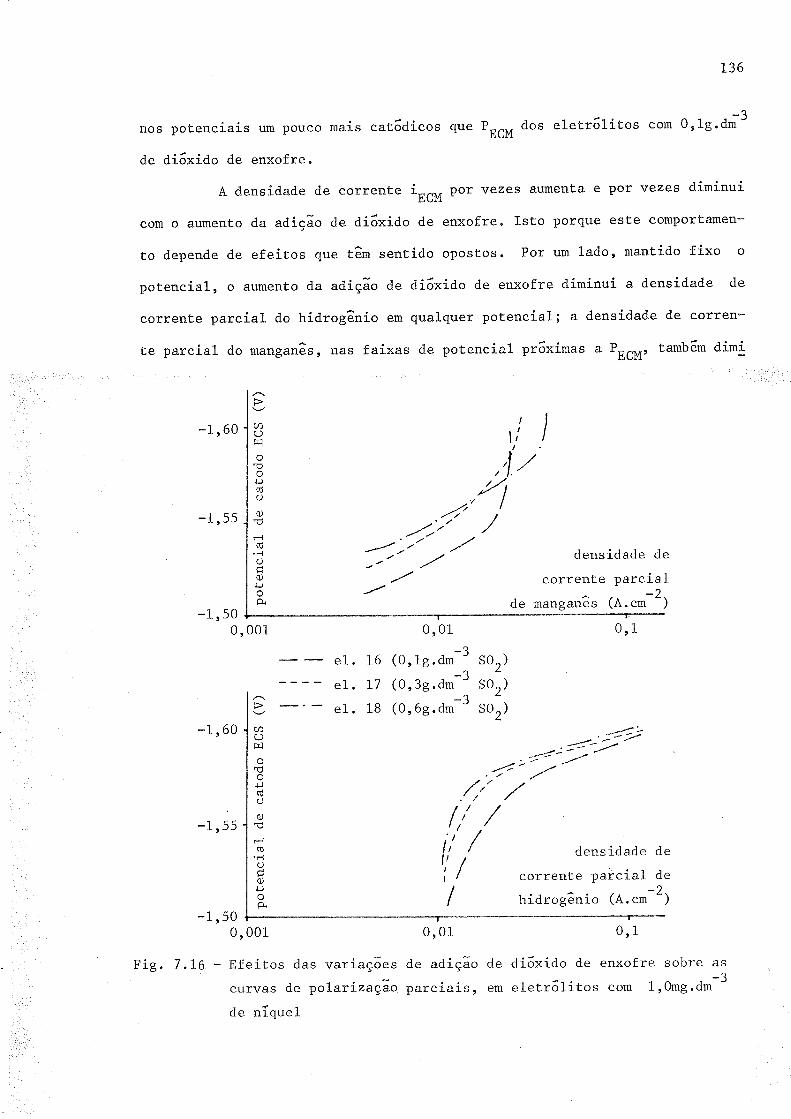
L36
nos potenciais um pouco mais cat6dicos er" PECM dos eletr6liros com 0rtg.a*-3
de di6xido de enxofre.
A densídade de correna" iEC* por vezes aumenta e por vezes dimínui
com o aumento da adição de di6xido de enxofre. Isto porque este comportamen*
to depende de efeiËos que têm sentido opostos" Por um lado, mantido fixo o
potencial, o aumento da adição de di6xido de enxofre díminui a densídade de
correnËe parcial do hidrogênio em qualquer potencial; a densidade de corren-
Ee parcial do manganês, nas faixas de potencial pr6ximas t PECM, tamb6n dirni
-1 ,60
r¡
s02)
so2)
so2)
v)C)rd
o.Úo.lJadoqJ
EFld.JoCJ
+Jo
t,l I,'l 'l
-1 ,55
./
-r/¡/'/o/-'t' -/-¿-//,/' ,"'
-" -/
0,01
(0, 1g. dm
(0,39.dm
(0, 69. dm
0r0l
de adição
parciais,
densidade de
corrente parcialde manganês (A.cm-')
0r1-1r50
0, 001
e1.
e1.
e1.
L6
L7
1B
-3
-3
-J
-T,60 c.l)(Jtdo€o.lJdCJ
(J.o
Flcd
.FiOÉo]Joa.
-1 ,55
-1 ,500,001
Fig. 7,16 - Efeitos das variaçõescurvas de polari zaçã.o
de níquel
0r1
dióxido de enxofre sobre as
eletrólitos com 1,Omg.dt-3
dens idade
corrente parcialhidrogênio (A.cm
de
de_,-)
de
em

1_3 7
nui, ou então aumenta pouco. Por outro lado, ambas as densidades de corrente
parciais aumentam com a diminuição do potencial e, como vimos' o aumento da
adíção de di6xido de enxofre sempre diminui PUa*. As figuras 7.L4 e 7.L5
mostraram exemplos nos quais prevalece o efeito da diminuição das densidades
de corrente parciai" u iua" decresce com o aumento da adição de di6xido de
enxofre. As figuras 7.16 e 7.17 mostrarâm exempl-os nos quaís prevalece o e-
feito do potencial e iECM "t.uce com este aumento de adição.
-I 160 CAUt'loodo0).o
r-l(ü
.FiOÉG)lJ
*
,l/
-1,55
-1,
-1,60
-1,55
_1r,
Fie, 7,L7 -
/."'r' /¿//t//--/
densidade de
corrente parcial-,)de manganês (A.cm ')
0r1
ۋ?''
-.t'
500 ,001
ele1
el
25
26
27
0ro1
(0, 1g. dm
(0,39.dm
(0, 69. dm
-J
-3
-3
de
de,)I
so2 )
so2)
so2)
cn(J14
o€oÐad(-)
clËr-{d
'rlCJ
Éo.p
o,
de
em
4t' ,/¿'¿/ '/-/t ./
./ t ,/'
dióxido de
eletró1 itos
/: t500,001
Efeitos das variaçoescurvas de polarizaçäode zinco
densidade
corrente parcialhidrogênio (A.cm
0r1
enxofre
com
0,01
de adição
parcíais,sobre as
1
20mg.dm '

t3B
7.2 I.{ECANTSMOS pE ATUAÇAO pAS AprÇOES
7.2.L Resumo dos efeitos das adições
Para estabelecermos um mecanismo de atuação das adições de cobal-
to, níquel, zínco e dióxido de enxofre, partimos dos seus efeitos sobre as
curvas de polarizaçã.o quee resumidamente, são:
a) Quaísquer adições ou aumentos de adição de cobalto ou níque1 d!
minuem a densidade de corrente parcial de deposição de manganês e aumentâm a
de desprendimento de hidrogênio, em qualquer pot.encial, mas principalmenLe
nos poËenciais mais catódicos. O mesmo ocorre com. as duas adições maíores de
zinco.
b) O efeito do zinco 6 menos intenso que o do níquel, e este ê me-
nos intenso que o do cobalto.
c) Uma adição pequena de zinco diminui Ëanto a deposição de manga-
nês como o desprendimento de hidrogênio.
d) Nos potenciais mais an6dicos, o aumento de adição de di6-
xido de enxofre diminui tanto a deposição de manganês como o de-s-
prendimento de hidrogênio.
e) Nos potenciais mais cat6dicos, o aumento da adição de di-oxido
de enxofre diminui tanto a deposição de manganês como o desprendimento de h!
drogênio, quando o eletr6lito não recebe adição de cotialto, níque1 ou zinco;
quando e1e tem tais adições, o aumento da adição de di6xido de enxofre tam-
b6m dirninui o desprendimento de hidrogênio, mas aumenta a deposição cle man-
ganês.

139
7.2.2 0utras premissas para estabelecer o modelo
Al-em dos diversos efeitos das adições, resumidos na seção ante-
rior, várias premissas ou línhas de raciocínio foram utíIízadas na definição
de um modelo de mecanismo. Resumidamente, elas são as seguintes:
a) Curvas de polarização parciaís traçadas com base no val.or das
eficiências de correnËe representam na realidade o processo g1oba1 e não ne-
cessariamente as reações eletroquÍmicas parciais reais.
Isto ocorre porque o m6todo utilizado não mede diretamente as cor-
rentes de deposição de manga.nês e de despr:end.imento de hidrogênio. Ele mede
a massa de manganês resultante do processo g1obal. Se, por exemplo, o manga-
nês eletroquimicamente depositado for parcialmente consumido por alguma rea-
ção quírnica, a massa de manganês resultante sobre o catodo será menor que a
correspondente ao processo eletroquÍmico. Neste caso, a curva obtida não re-
presentarâ a real corrente parcial de deposição.
Da mesma forma, se houver alguma reação química que resulte em de-
posição de manganês paralelamente ã reação eletroquímica, novamente a curva
obtida representarâ. o processo globa1 e não apenas o eletroquímico.
F, claro, contudo, guê, em regime estacionãrio, o resultado global
das reações de redução (ou seja, deposição de manganês e desprendimento de
hidrogênio) será efeito apenas de processo eletroquímico. Em outras pala-
vras, como os únicos produtos significativos do processo catódico são manga-
nês e hidrogênio, e como não existe alimen|ação de agentes redutores ou oxi-
dantes, qualquer consumo químico do manganês deposítaclo serã realizado ne-
cessariamente ãs custas de desprendimento químico de hidrogênio, e vice-ver-
sâr de modo que, globalmente, as Leis de Faraday sejam obedecidas.
0 mesmo ocorre se as reações adicionais forem tamb6m eletroquí-
micas, como ocorre na formação de pares galvânicos (v. seção 4.2..3, ref.16),

pois neste caso o desprendirnento de hidrogênio
respondente, em termos el-etricos, ã dissolução
ca.
140
região catódica seria cor-
*anganês na região an6di-
na
de
b) Apesar <le estarem presenres em teores muiËo pequenos, o cobal-
to, o níque1 e o zinco apresentam potenciais de eletrodos reversíveis muito
mais an6dicos que o do manganês nas soluções estudadas e, portanto' muito
mais distantes dos potenciais utilizados em nosso estudo. 0s poËenciais de
eletrodo-padrão das reações de deposição de cobalto, níque1 e zinco' na esca
1a de hidrogênio, são -0,277, -0,250 e -0,7628 v' respectivamente (ref" 9).
Como suas concentrações são baixas, seus poËenciais de eletrodo reversíveis
serão muito mais cat6dicos. De acordo com a expressão de Nernst (ref. 9), e
tomand.o as atividades como concentrações (uma vez que as concentrações são
baixas) , temos:
E=Eo R T.lnc 7.7
A tabela 7.1 apresenta esËes potenciais para as concentrações uti-
Lízadas em nosso trabal.ho.
Estes valores são mais an6¿icos que o potencial de eletrodo rever-
sível do manganês, cujo potencial de eletro-padrão é -1,180 V (ref. 9), e cu
jo potencial de eletrodo reversível, portanto, 6 ainda mais catódico. Os pe
tenciais utíIízados em nosso esËudo estiverarn entre -Lr77 e -1r82 V, na esca
la de hidrogênio. Assim, estas impurezas certamenËe se codepositam com o man
ganês na faixa de potenciais estudada.

L4I
Tab. 7,I - Potenciais de eletrodo(escala de hidrogênio,
4..revel:s 1ve1s
T=298K)
concentração_1
(mg dm ')_â(mol d* ')
EO(v)
E(v)
cobal to 0,25
0r5
1'0
0r5
1r0
2r0
10
20
40
-610"10-6
-510"
1 0-6_(
10'10-5
1O-4
10- 4
_L10
-0,277il
il
-0,250tt
lr
-0 ,7 63
lt
il
nÍque1
zlnco
c) O enxofre do diõxido de enxofre adicionado ao eletr-olíto 6 co-
reduzido no catodo com o manganêsr urnê vez que esta adição eleva o teor de
enxofre no depósito (ref. 38).
d) O enxofre ã tensoativo e deve ocupar locais ativos na superfí-
cie do depósito (ref. 23 e 38) , podendo concorrer com o manganês, com o hi-
drogênio e com as impurezas em seus processos de deposição ou desprendimento.
e) A presença de dióxido de enxofre no eletrólito eleva a sobreten
são de hidrogênio d.o manganês (Fig. A.4, ref . 23); como mostrou Dha-nanjayan,
a adição de di-oxido de enxofre possibilita a deposição de manganês em sua
forma alotrópica a, ao aumentar a sobretensão desta fase, tornando-a maior
que a de Mn-J (seção 4.2.5, ref. 38).
f) Os resultados experimentaís de Zosimovich, Shvab e Belinskii
(ref. 16, seção 4.2.3), em suas deposições com eletrodo rotativo, são coeren
tes com seu modelo de pares galvânicos impurezafmanganès.
4tL
Br4
l17
8r5
I'7314
1'5
3'1
6rr
2
2
2
2
2
2
2.
2
2
-o,435
-0,427
-0,41 8
-0,400
-0, 39 1
-0,382
-0,876
-o,867
-0,858

142
7.2.3 Mecanismo proposto
Com base nos efeitos de cobalto, níquel, zinco e dióxido de enxo-
fre observados em nosso trabalho (resumidos na seção 7.2.I) e nas premissas
e raciocínios resumidos na seção 7.2.2, estabelecemos o seguinte mecanismo
de atuação destas adições:
AtuaÇão do cobalto, níquel e zinco; os pares galvãnicos
Devido ã ¿istri¡uição não-uniforme da corrente (e do potencía1) so
bre a superfície do catodo, a codeposição clas impurezas ou a corrosão do man
ganês se dão de maneira não-uniforme sobre esta superfície. Como o potencial
de eletrodo reversível destas impurezas 6 mais an6dico que o do manganês, a
deposição ou corrosão das mesmas devem ser menos afeLadas que as de manganês
pelas variações de potencial dentro da nossa faíxa de estudo. Assim, nas
ãreas em que o potencial se torna mais an6dico, a deposição de manganês dimi
nui mais (ou sua corrosão aumenta maís) do que a de cobalto, níquel ou zín-
co; nesLas áreas, então, o depósito se torna maís rieo, em termos relativos,
destas impurezas. Como esËas impurezas apresentam potencial de eletrodo re-
versível muito mais an6dico que o cle manganês, forma-se um par galvânico,
constituído, por um lado, pelas regiões ricas em impurezas, Í.ormando um cato
do, e, por outro, pelas regiões mais puras, formando um anodo. Nesta região
anódica do depósito ocorre a reação de corrosão do manganês. Na região cató-
dica, a reação de desprendimento de hidrogênio.
Estas duas reações, embora eletroquímicas, não estão vínculadas ã
corrente total que atravessa a cãlula, uma vez que ambos os seus polos se si
tuam no eletrodo de trabalho. O efeíto destas duas reações, portanto, soma-
se ao efeito das reações cat6dicas correspondentes à corrente que atravessa

t43
a cé1ula e, no resultado g1obal, aumenËam o desprendimen.to do hidrogênio e
diminuem a deposição de manganês, o que se reflete nas curvas polarogrãfi-
cas parciais, uma vez que elas são obtidas a partir da massa de manganês re-
sultantes ao final da deposição.
DiferenÇas entre cobalto, níquel e zinco;_a sobretensão de hidro-:
gênio
A intensidade ou mesmo o sentido de atuação das impurezas nos pa-
res galvânicos estão vinculados a seu potencial de eletrodo reversíve1, que
afeta a deposição da impureza e a diferença de potencial. toËa1 do par galvâ-
nico, e ã sua sobretensão de hidrogênío, que afeta a intensidade do despren-
dimento de hidrogênio (e tamb6m, portanto, da corrosão do manganês). A sobre
tensão de hidrogênio tamb6m afeta, evidentemente, o desprendimento de hidrogê
nio vinculado à corrente que aLravessa a célu1a"
A formação de um par galvânico cria condíções termodinâmicas para
as duas reações eletroquímicas citadas, adicionais ãquelas vinculadas ã pas-
sagem de corrente pela cã1ula. A sobretensão de hídrogênio constitui um fa-
tor cin-etico que af.e:'a não apenas as reações deste par galvânico como tanb6m
as vinculadas à correnËe da cãlula.
No caso de cobalto e níquel, estes dois fatores (o "termodinãnico'i
e o cinético) se aliam para aumentar o desprendimento de hidrogênio e dimi-
nuir a deposição de manganês, uma vez que, além de formarem os pares galvâni
cos, estas impurezas apresentam uma baixa sobretensão de hidrogênío" A pr6-
pria intensidade dos efeitos pode ser vinculada ã sobretensão de hidrogênio,
uma vez que o cobalto, Quê apresenta uma sobretensão de hidrogênio mais bai-
xa que o níquel , afeta mais que este a intensidade do desprendimento de hi-
drogênio e da deposição de manganês.

144
No caso do zinco, o fator t'termodinâmicott, ou seja, a formação do
par galvânico,6 semelhante ao caso do cobalto e do níquel. O fator cin-eti-
cor contudo' tem sentido oposto, uma vez que sua sobretensão de hidrogênio 6
muito elevada. Com pequenas adições (10 mg.d* 3 de zinco), prevalece este fa
tor e o desprendimento de hidrogênio total dirninui. Com as adições maiores,
prevalece o efeito de formação do par galvânico, e ocorre aumento do despren
dimento de hidrogênio e diminuição da deposição de manganês. Ainda assirn, es
te efeíto é menos inËenso do que com o níque1 e o cobalto.
Efeito do aumento de adição de cobalto, níquel òu zinco
O aumento de adíção de cobalto, níque1 ou zinco aumenta o efeito
de formação de pares galvânicos porque a quantidade destas impurezas no depó
sito aumenta, seja pela mudança de seu potencial de eletrodo reversível para
valores mais anõdicos (Tab. 7 .I) , se sua deposição estiver sob controle ele-
troquímico, seja pela mudança da velocidade das reações químicas ou de trans
porte, se sua deposição estiver em corrente-lirnite.
Efeito do diõxido de enxofre
A intensidade da reação de deposição do manganês 6 diminuída pela
presença de enxofre, proveniente do dióxido de enxofre, nos locais ativos da
superficie do manganês. A intensidade da reação de desprendimento de hidrogô
nio tamb6m 6 diminuída, seja por esEa ocupação dos locais ativos, seja pela
alteração de sobretensão de hidrogênio com a modificação estrutural do manga
nês.
Quando a solução não cont6m teor elevado de impurezas, ocorrem ape
nas estes efeitos t è ã intensidade de ambas as reações diminui em qualquer

145
potencial.
Quando, por6m, a solução cont6m impurezas, os pares galvânicos en-
tão formados são afetados pelo diõxido de enxofre. Pode-se atribuir seu efei
to, por um lado, a um aumento de sobretensão de hidrogênio ou a ocupação de
locais ativos na superfície das regiões ricas em impurezas, diminuind.o assim
o desprendimento de hidrogênio. Ou poile-se, por outro lado, atribuí-1o à ocu
pação dos locais ativos na região pobre em impurez,as, d.íminuíndo assim a die
solução de manganês. Em qualquer caso, o efeito final 6. red.uzír o efeito do
par galvânico, dimínuindo igualmente a dissolução de manganês e o desprendi-
mento de hidrogênio a e1a vinculado.
Assim, quando a solução conËém impurezas, os efeitos do di6xido de
enxofre sobre o desprendimento de hidrogênío correspondente ã corrente que
aËravessa a c61u1a se somam aos efeitos sobre o par galvãnico, e ocorre uma
grande diminuição do desprendimento total de hidrogênio. Com relação ãs rea-
ções do manganês, entretanto, os efeitos do dióxído de enxofre têm sentido
oposto entre si, diminuindo tanto a deposição como a dissolução do mesmo"Nos
potenciais mais anódicos, prevalece o efeíto sobre a deposição, e sua in-
tensidade total diminui. Nos potenciais mais cat6dicos, prevalece o
sobre a dissolução, e a íntensidade t.oËal da deposição aumenta.
ef ei Ëo
Efeito da mudança de potencial
Nos potenciais mais anódicos, a densidade de corrente de manganês
-e muito mais baixa que nos potenciais mais catõdicos. Assim, a relação entre
as quantidades de enxofre e manganês no dep6sito , para um dado valor de adi-
ção de diõxido de enxofre, deve ser maior nos potenciais mais an6dicos.
Nestes potenciais mais an6dicos, porisso, mesmo uma pequena adição
de di6xido de enxofre -e capaz de se contrapor ao efeito dos pares gah.âni-

r46
cos, e o aumento da adição pouco acrescenta a esta ação. Este aumento de adi
Ção, contudo, tem efeito sobre a reação de deposição do manganês, dimínuind.o
seu va1or, e porisso este efeito prevalece sobre a diminuição da reação de
dissolução do manganês.
Nos potenciais mais catódicos, por outro lado, a adição pequena de
di6xido de enxofre resulta em uma relação enxofre/manganês menor d.o que nos
potenciais mais an6dicos, e porisso se mosËra íncapaz de se contrapor ao
efeito dos pares galvânicos. Assim, o efeito dos pares galvânicos resulta
muito mais intenso nesta faixa de potenciais, e, a16m disso, muito mais sen-
sível ao aumento da adição de di6xido de enxofre, que passa, com estes maio-
res teores, a se contrapor ãquele efeito. Isto explica, por um 1ado, porque
as impurezas têm efeitos mais intensos nos potenciais mais cat-odicos e, por
outro lado, porque, nestes potenciais, o efeito do dióxido'de enxofre sobre
os pares galvânicos prevalece sobre seu efeito sobre a deposição de manga-
nês, resultando um aumento na intensidade total de deposição de mangattês.
Evidentemente, este comportamento de dióxido de enxofre nos poten-
ciais maís cat6dicos ocorre apenas quando existem os pares galvânicos, ou se
ja, quando o eletr6tito cont6m cobalto, níque1 ou zinco. Em caso contrário,
seu comportamenËo serã semelhante ao que ocorre nos potenciais mais anódi-
cos, nos quais o aumento de sua adição climinui tanto o desprendimento de hi-
drogênio como a deposição de manganês.

147
8. CONCLUSõES
As conclusões expostas a seguir têm validade apenas dentro das fai
xas experimentais deste trabalho._a
1. a) Aumentanclo-se a densidade de corrente catódica desde \714 A.c^' atã
-)120 A.cm', a eficiência de corrente cresce, passa por um mãximo e em
seguida decresce, com qualquer dos eletrólitos estudados.
b) Isto ocorre porque nos potenciais mais an6dicos (densidades de corren-
te totais menores) a densidade de corrente parcial do manganês á muito
baixa, mas ela aumenta mais rapidamente que a do hidrogênio com a di-
minuição do potencial (aurnento da densidade de corrente total); ou se-
ja, nesta região a curva polarográfica do *anganês tem inclinação me-
nor que a de hidrogênio e a eficiência de cortente aumenta; a partir
de um dado potencíal, contudo, a situação se inver:te, € â curva do man
ganês passa a ter inclinação muito grande e a do hidrogênío inclinação
muito pequena, e a eficiência de corrente passa a diminuir.
2. Os valores das eficiências de corrente mãximas variaram entre L4 e 687".
a) Qualquer adição ou aumenËo de adição de cobalto ou nÍque1 diminui a
etlclenc]-a de corrente maxrma.
b) Isto ocorre porque estas adições diminuem a densidade de corrente par-
cial do manganês e aumentam a de hidrogênio, em qualquer potencial.
_aa) A adíção de 10 mg.dm-'de zinco aumenta a eficiência de corrente rnãxi-
3.
4.

148
ma quando o eletrólito tem 0r1 ou 0,3 g.dr-3 de dióxido de enxofre e
diminui a eficiência de corrente mãxima quando esLe tem 0,6 g.dr-3 de
dióxido de enxofre.
b) Isto ocorre porque esta adição de zínco diminui tanto a densidade de
corrente de manganês como a de hidrogênio, em qualquer potencial; com1
0,1 g.dm'de dióxido de enxofre, a diminuição da densidade de corren
te do hidrogênio 6 maior que a da de mangattâs, na faixa de potenciais
em que ocorre a eficiência de corrente máxima; com 0,3 g.¿r 3 a" di6xi
do de enxofre, isto ocorre em qualquer potencial; com 016 g.dm-3 de
di6xido de enxofre, isto só ocorre fora da faíxa de potencíais em que
ocorre a eficiência de corrente mãxima, e dentro da faixa ocorre o
oposto.
c) Adições de 20 e de 40 rg.dr 3 de zinco diminuem a eficiência de
rente máxima.
cor-
d) IsËo ocorre porque estas adições aumentam a densidade de corrente par-
cial de hidrogênio e diminuem a de manganês, ern qualquer potencial.
e) I divergência enËre resultados de diferentes estudos sobre o efeito do
zinco pode ser atribuída aos comportamentos acima descritos, ou seja,
ao fato de a adição de zinco aumentar ou diminuir a eficiência de cor-
rente conforme o valor desta própria adição, conforme o potencial (ou
densidade de corrente) e conforme o valor da adição de di6xido de enxo
fre ao e1etr6lito.
5. a) Qualquer aumento de adição de di6xido de enxofre aumenta a eficiência
de corrente máxima quando o eletrólito não tem adições de impurezas ou
quando estas adições são pequenas. Quando o eletrólito conË6m grandes
adições de impurezas, o aumento de 0,1 para 0,3 g.dn-3 de dióxido de
enxofre aumenta a eficiência de corrente mãxima, mas o de 0r3 paraI
0,6 g.dm ' não tem efeíto significativo sobre ela.

I4.9
b) Isto ocorre porque o aumento da adição de dióxido de enxofre diminui
ambas as densidades de corrente parcíais, na faixa de potenciais em
que ocorre a eficiência de corrente mãxima, mas dimínui mais a de hi-
drogênio. Nos el.etr61íËos sem impurezas ou com pequenas adíções das
mesmas, esta diferença 6 grande nos dois aumentos da adição de di-oxido
de enxofre. Nos eletr6litos com maiores adíções de impurezas, o aumen-_a
to da adição de di6xido de enxofre de 0r3 para 0,6 g.dm'diminui mui-
to pouco as duas densidades de corrente e, portanto, a. díferença entre
estas diminuições 6 pequena e a eficiência de correnLe pouco se alte-
ra, rLa faixa de potenciais em gue ocorrem as eficiências de correnËe
mãximas.
c) A divergência entre diferentes estudos sobre a faíxa de atuação do dió
xido de enxofre pode ser atribuída a diferenças nos teores de irnpure-
zas em seus eletrólitos ou na faixa de potenciais de caËodo utí!ízað,a.
6. O potencial em que ocorre a eficiência de
-1,585 e -1,534 V, na escala de eletrodo
corrente mãxima variou entre
de calornelano saturado.
a) O potencial em que ocorre a eficiência de corrente máxima torna-se ma-
is anõdico com qualquer adição ou aumento de adição de cobalro, níqueI
ou zinco (com uma exceção em que e1e não se altera), e torna-se mais
catódico com qualquer aumento de adição de di-oxido de enxofre.
IsËo se deve ao fato de que a eficiência de corrente mãxima ocorre em
potenciais urn pouco mais an6dicos que aqueles correspondentes ao Ëre-
cho de grande inclinação na curva polarogrâf.íca do manganãs. Como este
trecho de grande inclinação passa a ocorrer em potenciais mais anõdi-
cos com a adição ou o aumento da adíção de cobalto, níque1 ou zinco,
e passa a ocorrer em potenciais mais catódicos com o aumento da adição
de diõxido de enxofre, a eficiência de corrente mãxima acompanha este
b)

150
comportamento.
8. A densidade de corrente catódica
maxima variou entre 32 a 70 mA.cm
em
-z
que ocorre a eficiência de corrente
9, a) O valor da densidade de corrente cat6dica em que ocorre a eficiência
de corrente mãxima diminui, com algumas exceções, com a adição ou o au
mento de adição de cobalto, níque1 ou zinco; com o aumento das adições
de di-oxido de enxofre, este valor diminui ou aumenta, sem regra clara,
conforme as diferentes adições de impurezas utilizadas.
b) Este comportãnento sem regras claras ou sujeito a e*ceções explica-se
pelo fato de que a alteração desta densidade de corrente é regida por
fenômenos que têm sentidos opostos. Em qualquer poÈencíal fixo, as adi
ções ou aumentos de adição de cobalto, níquel ou zinco díminuem a den-
sidade de corrente parcial do manganês, que 6 uma das parcelas de den-
sidade de corrente total, mas em compensação aumentam (com exceção da
adição de 10 *g.d* 3 de zinco) a densidade de corrente parcialdehidro
gêníor llue 6 a outra parcela da densidade de corrente total; a1ém dis-
so, o potencial em que ocorre a eficiência de corrente máxima não é fí
xo; ele se desloca para valores mais anódicos com estas adições, o que
desloca ambas as densidades de corrente parciais para valores menores.
No caso do dióxido de enxofre, o aumento de sua adição diminui ambas
as densidades de corrente parcíais, em um potencial fixo (dentro da
faixa correspondente ãs eficiências de corrente mãximas), mas em com-
pensação o potencial se desloca para valores mais catõdicos, o que des
loca ambas as densidades de corrente para valores maiores.
10. As curvas de polarização parciais de manganês apresentam aumento de incli
nação com o aumento da densidade de corrente total, podendo-se distinguir
uma região inicial (nos potenciais mais an6dicos) com inclinação pequena

a)11
b)
e aproximadamente constante,
de grande inclinação.
151
uma região de transição e uma região final
diminuem
mals ano
tem gran-
manganes,
corrente
As adições ou aumentos de adição de cobalto, níquel ou zinco
as densidades de corïente parciais de manganês nos potenciais
dicos (região inícial das curvas).
As adições ou aumentos de adição de cobalto, níquel ou zi.nco
de efeito sobre a região de grande inclinação das curvas do
definindo-a mais claramente e deslocando-a para densidades de
menores e potenciais mais an6dicos.
\2. a) O aumento da adição de di6xido de enxofre diminui as densidades de cor
rente parciaís de manganês na região inicial e na região de transição
das curvas polarográficas parciais de manganês.
b) O aumento da adição de dióxido de enxofre tende a se contrapor à ação
das impurezas rLa região final das curvas polarogrãficas parciais de
manganês, dirninuindo sua inclinação e deslocando-as para d.ensidades de
corrente maíores. Nos eletrólitos sem adição de impurezas, nos quais
não se define esta região final de grande inclínação, o efeito do dió-
xido de enxofre nesta faixa de potenciais se assemelha aos verificados
nos potenciais mais an6dicos.
13. As curvas de polarizaçã.o parciais de hidrogênio apresentam diminuição de
inclinação com o aumento da densidade de correnËe tota1, podendo-se obser
var uma região inicial (nos potenciais mais anódicos) de grande inclina-
ção e uma região final de inclinação extremamente pequena.
14. a) Todas as adições ou aumentos de adição de cobalto ou níque1 aumentam a
densidade de corrente parcial de hidrogênio em qualquer pote-ncia1 e
deslocam para potenciais mais an6dicos a região final de pequena incli
nação. As adições maiores de zínco (20 e 40 mg.¿r 3) apresentam estes

752
mesmos efeitos._,)
b) As adições de 10 mg.dm ' de zinco diminuem a densidade de corrente
parcial de hidrogênio, principalmente nos potenciais mais anódicos.
15. Os aumenLos de adição de dióxido de enxofre diminuem a densidade de cor-
rente parcial de hidrogênio em qualquer potencial e desloca para poten-
ciais mais catódicos a região final de pequena inclinação das curvas pola
rogrâficas parcíais de hidrogênio.
I6 O depósito de manganês consiste de um substrato fino sobreposto por
camada faceËada formada a partir de g16bulos isolados ou agrupados.
uma
17. Durante a deposição, o desprendimento de hidrogênio 6 muito intenso nos
instantes iniciais, sobre o catodo-base de aço inoxidável. Em poucos se-
gundos, o desprendimento dirninui, com a formação do substrato de manga-
nês. O desprendimento de hidrogênio sobre a camada superior facetada -e ms
nor do que sobre o substrato inicial, de modo que durante a formação da
camada superior o desprendimento se restringe ãs áreas do substrato ainda
não-recoberras. Após o recobrimento tota1, o clesprend.imento diminui.
18. a) Com a adição de zinco, o substrato, observado durante a deposição, se
mostra indiferenciado da camada superior, quando observado no micro.sc6
pio eletrônico de varredura.
b) O substrato é formado por g16bulos de 0,5 a 1rO pm de diâmetro nos
eletr6litos com adição de níque1, 1,0 a 1,5 ¡lm nos com adição de co-
balto e 0,5 a 4,0 pm nos sem adíção de impurezas.
c) ua camada superior, os eletrólitos que contôm adição <le zinco apresen*
tam dep6sitos com facetas irregulares e com defeitos.
a) Os resultados experimentais evídenciam um mecanismo de atuação de co-
balto, níquel, zínco e diõxido de enxofre.
b) Cobalto, níquel e zinco, codepositados, formam pares galvânicos com o
t9

c)
manganês, aumentando o desprendimento
do a deposição total de manganês.
1.53
total de hidrogênio e reduzin-
No caso de cobalto e níquel, a atuação dos pares galvânicos 6 reforça-
da pela baixa sobretensão de hidrogênio apresentada por esËas impure-
zas. No caso de zlnco, sua elevada sobretensão de hidrogênio cond.uz,
com pequena adição, a uma diminuição do desprendimento de hidrogênio;
com maiores adições, prevalece o efeito dos pares galvânicos por ele
formados "
O dióxido de enxofre atua tanto sobre os processos eleËroquímicos cor-
respondentes ã corrente que atïavessa a c61u1a como sobre os cor-
respondentes aos pares galvânicos, ao ocupar locais ativos na superfí-
cie do depósito e elevar a sobretensão de hidrogênio, reduzindo assim
a intensidade de Ëodas as reações. Nos potenciais mais anódicos, o
efeito sobre os pares galvânicos 6 intenso desde as menores adições, e
porisso o aumento da adição tem mais efeito sobïe a própria reação de
deposição de manganês, recluzindo-a, do que sobre os pares galvãnicos.
Nos potenciais mais catódicos, o efeito de suas pequenas adições -e me-
nor e, porisso, a atuação dos pares galvânicos 6 mais intensa que nos
potenciais anódícos; com adições maíores, o dióxido de enxofre passa a
ter efeito sobre os pares galvânicos e porisso esta 6 a atuação predo-
minante do aumento da adição de dí6xido de enxofre nestes potenciais
mais cat6dicos.
d)

L54
1.
9. RTFERINCIAS BIBL]OGRÃFICAS
IIARRIS, M. - The history, production and
Ãfrica do Sul, L977 (texto envíado pe
uses of electrolytic manganese
1o autor).
vAN AR.SDALE, c.D. & MATER, C.c.in sulphate solutions. Trans
- The electrolytic behavior of manganese
. An. E1ect. Soc., 33, 109-29, 1918.
3. JACOBS, J.H
ation of.; HUNTER, J.lnl.; CHURCHT^IARD, P IO{TCKERBOCKER,
lant at Boulder
.E. ;
lotR.G. -
Ci ty,0per-
4.
5.
6.
Ëhe electrolyti c manganese pl Neva-
4". U.S. Bureau of Mines Bulletin 463" L946 apud Ref. 4.
DEAN, R.J. - Elgctrol--ytic _mgnganeçe end its e_lloys. Nova lorque, N.Y.,Ronald, 1952.
BARCHESE' E. - Aplicação da croqgpotencíometria ao estudo de obtenção de
manganês eletrolítico. São Pau1o, \977 (Tese de Doutoramento, EscolaPolitãcnica, Universidade de São Paulo).
MANTELL, C.L. -E lectrochem.
Commerci a1 production of elecËrolytic manganese
232-43, L94B..apud Ref . 7.
Trans.
Soc., 94,
7. MANTELL, C.L.
164, 92-5,
- Two decades
L963.
of electrolytic manganese production, E & ldl,
8. JACOBS,
R.G.
J.H. ; HUNTER, J
- First 2 years
.lri.; YARROL, I^I.H.;
operation of the
CHURCHI^IARD, P.E. & KNICKERBOCKER,
Bureau of Mines electrolytic

155
manganese pilot plant. Trans. Am. Inst. Min. & Met. Engrs., I59r 408 -28, 1944 apud Ref. 4. Ref. 15.
9. ANTROPOV, L. - Theoretical electrochemistry. Moscou, MIR, 7972.
10. AGLADZE, R.I. - Electrolytic manganese and electrolysis of chlorides. In:P."ports of fift , 31-40.
Nova lorque, NY, V.V. Stender (ed.), Consultants Bureau, 7965.
11. LEI4IIS, J.E.; SCAIFE, P.H. & SI,IINKELS, D.A.J. - Electrolytic manganese
metal from chloride electrolytes. I. Study of deposítion conditions.J. App1. Electrochem. ,6 (3), 199-209, L976.
12. TTLAK, B.V.K.S.R.A.; RAJAGOPALAN. J.R. & REDDY, A.K.N. - Buffer character-istics of manganous sulphate + amnonium sulphate electrodepositionbaths. Trans. Faraday Soc.,5B, 795-804, 7962.
13. PCIURBAIX, M. - Atlas of electrochemi,cal egui-lilrie in aqueous solutions.Londres, Pergamon, 1966 apud Ref. 15.
14. JACOBS, J.H.; CHURCHI^IARD, P.E. & KNICKERBOCKER, R.c. - The effect ofcertain variables on the electrodeposition of manganese. Trans.
Electroch. Soc., 86, 383-93, 1944.
15. LOUIS, P. & MARTIN, J.P. - Increased current efficiency during the
eleqlfo¿epqliligt q-I_-T9ry?t?99 II_og splp!?!g 9199!to1y!9s. NationalInstitute for Metallur:gy , 26Eh November, 1976, Report lI9 1859 (ISBN 0
86999 353 4) , 'Ãftica do Sul.
16. ZOSIMOVICH, D.P.; SHVAB, N.A. & BELINSKII, V.N. - Etrectrodepositíon of
manganese on a rotatíng electrode in the presence of impurities.Elektrokhimiya, 3(12), 1481-5 , 1967.
L7. ZNAMENSKI, G.N.; GAMALI, f .V. & STENDER, V.V. - Characteristics of the
elecËrolytic deposition of metals from solutions of "extraordinary"purity. 4E"d. Nauk SSR, I37, 335-7, L961.

156
18. AGLADZE, R.I. & GOFMAN, N.T. - The influence of certain additives on the
electrolysis of manganese ín the presence of impurities. Elektrokhim.
Margaqlsa, Akad. Nauk Gruzin SSR, 1, 53-68,1.957 apud Ref.12 e Ref.39.
19. MANTELL, C.L.
impuritiesFERMENT, G.
manganese
- Current-potential of trace
236, 7rB-25, 7966.
u
IN
eff ec ts
. A]ME,
20. SHVAB, N.A. - Mechanism of
tion of manganese from
ll-jz), 140-3, 1975 aPud
electrowinning, lfglg
the action of meË41 ions on the electrodeposi-aqueous solutions. Ukr. Khím, Zh. (Russ. Ed.),
Chem. Abstr., 33, 87130c, 7975
2L.
)'l
SHVAB, N"A. - Mechanism of the effecË
on manganese electrodeposiËion from
Nauk Ukr. RSR, Ser.B, 33(1), 58-60,
I50224L, L97L.
AGLADZE, R.I. & PACHUASHVILI, E.M. - The influencearsenic, antimony and sodium in the process of
mangarrese. Elekt.rokhim. MarganËsa, Akad. Nauk
1957 aPud nef. 15.
23. cAl'fALT, I.V. & STENDER, V.V. - Ef f ect of some
the overvoltage for Ëhe hydrogen evolutionKhim., 35(11), 2436-9, 1962.:
of cobalt, nickel, and copper íons
aqueous solutions. Dopov, Akad.
1971 apud Chem. Abstr., 742
of iron, aluminium,
obtaining electrolyticGrtzin. SSR, 1, 169-83,
impurities and additives on
on manganese. Zr. Prík7.
24. SULIAKAS, A.; JUSEVICIENE, G.; JANICKS, J. - Cathodie polarízaËion during
mangaÍrese elec_trodeposition lrom an electrolyte with a nícke1 impurity
and selenite addition. Chmr. Chern,_!ecþr191_r , lech. 4c4qLu Isvy-tyrng Resp.
¿ , 73-6 aPud Chem' Abstr"86:9945 v, 1977.
25. SHVAB, N.A.; BELINSKII¡ V.N.; ZOSIMOVICH, D.P. - Combíned díscharge of
zínc, cadmium, and copper íons wiËh manganese and its effect on
electrolysis. Dopov, Akad. Nauk SSR, Ser. B, 33(B), 722-5, 1971. apud
!!"rr- n¡glf., 752 I36478L, I97I.

r57
26. KUODYTE, M.; SIILIAKAS, A.; JANICKIS, J. - Effect of selenite and selenate
ions on the electrodeposiËion of manganese in the presence of copper
impurities in the electrolyte. Che*. Chem, Technol..rTech.
Isvytymo Resp. J Result. Panaudojimo Konf. Medziaga 1975, 5-6 apud
Chem. Abstr., 88:160603u, 1978.
he
1,
27 cOcrsHvrl-r, N. & AGLADZE, R.r. - The
electrolysis of manganese sulfate.169-83 , 1957 apud ^Ref . 15 "
influence of molybdenum ions on tSoolschch. Akad. Nauk Brunz. SSR'
- Effect of treryllium ions on28. KOUDTTE , M. ; YANITSKIf ,
electrodeposition of
I.V.; SULIAKAS, A.
manganese. Tezísy I(onf " llrolodykh
89:119598d, 1978.
Dokl . - Resp.
Uch. - Khim. , 2nd, 7, 103*4, 1977 apud Chem. Abstr.'
29. JANICKIS, J.; SULIAKAS, A.;beryllir:rn impurities on
TSR Mokslu Akad. Darb.,
KUODYTE, M. - Effect of
the electrodeposiËion of
copper, silver, and
y-manganese. Liet.
Ser, B, (5)' 49-57, L978 apud Chem. Abstr.,
922223036e, I978.
30. ESTADOS UNIDOS. U.S. Patent 2.810.684. Diaphragm for electror¿inning of
manganese. CROMI,IELL, R.H. & PARSONS, I^I.4. apud Ref . 15.
31. KUODYTE, If.; YANITSKII, I.V.; SULIAKAS, A. - Effect of a silver impurity
on manganese electrodeposition. Issled. Ob1. Elektoosazhdeniya Met.,
Mater. Resp . Konf . E lekËrokhim. Li _t. SSR, 15 th , 57-6L ' L97 7 apud
Chem. Abstr., BB:I7926Ic, 1978.
JL SIiVAB, N.A. - Mechanism of the action of
deposition of manganese from aqueous
4L(2),140-3, L975 apud Ref. 15"
AGLADZE, R-.I. & DZHINCHARADZE, M.D. - Ef fect of
plating of manganese. Elektrokhim. Margantsa,
Chem. Abstr., 84:23637m, I976.
metal ions on the electro-solutíons. Ukr. Khim. Zh.,
magnesium on electro-
å 773-7, 1975 apud
33. ESTADOS UNIDOS. U.S. Patent 3.821.096. Electrodepositing Mn metal. LAI,
SAN-CHENG, 28 jun. 74 apud Chem. Abstr., B?zP66I29y' 7975.
34

35. SHUL'GrNA, N.P. - Ef f ect
Uch. Zap. Perm. Univ.,of bismuth on electrodeposition of manganese.
Abstr., 77:(229), 127-30, I97O apud Chem.
108693g, 1972.
36. GRUBE, G. - Process for making elecËrolytic manganese apud FOERSTER, F.
Handbuch der Angewandten Physikalischen Chemie, p. 560. Bredig, 1923
apud Ref. 4.
158
tn37 . I4ANTELL, C.L
manganese
38. DHANANJAYAN,
and gamma
. & SHAH, B.G. -electrowinning.
l{. - Mechanism
modifications.electrodeposi tionElectrochem. Soc.
of manganese in alpha
, Lrl, 1006-11, 1970.
CurrenÈ-potential effects of additives
Tferiq. 4IlG , U, 591-2, 1967.
of
J.
39. ESTADOS UNIDOS. U.S. Patent 2.LIg.560. Electrolytic manganese. SHELTON,
S.M., j.ro. 7,1938 apud Ref . 4.
40. LEI^IIS, J.E . ; SCA
metal from ch
Electrochem.,
IfE, P.H.; SIüINKELS, D.A.J. - El,ectrolytic manganese
loride electrolyËes. II. Effect of additives. J. Appl.g(5), 453-62, 1976.
41. D|ABREU, J.C. & TOREM, M.L. - Efeito de alguns aditivos na eletrólise de
manganês em meio sulfúrico. In: XXXV Congresso Anual da Associação Bra
sileira de Metais, São Paulo,6-L1 de julho de 1980. São Paulo, ABM,
1980.
42. TOREM, M.L. - Efeito de alguns aditivos na eletrocristalizaçao do manga-
nês em meio sulfúrico. Rio, 1980 (Dissertação de Mestrado, Departamen
to de Ciância dos Materiais e Metalurgia, Pontifícia Universida<le Cat-o
lica do Rio de Janeiro)
43. STENDER, V.V. &
of manganese
1963.
44. II^TASAKI, I" & CARLSON. JR., W.J.
for electrowinning manganese.
LOSHKAREV, Yu.M. - Experiments on the electrodeposition
from chloride solutions. Zh. Prikl. Khim.' 36(5), 1033-40,
- A continuous-weíghing laboratory ce11
Trans. AIME, 24L, 308-10, 1968.

159
45. COIIGHANOI{R, D.R. & KRAUSE, F.E. - Ind. Eng. 9!:t., 4, 6L- , 1965
apud Ref . 44,
46. I.Z.D. AN CRUZ SSR (editora) Electrochemistry of manganese (em russo).
Tbilisi, 1957 apud Ref. 44 e Ref. 53.
47. AGLADZE, R.I. - Metallurg., 9, 15, 1939 apud Ref. 23.:
48. PEREC, M. * Met. Giesser.eilech, 3, 388- , 1953 apud. Ref . 40.
49. YANTTSKTT, IV.; SHULYAKAS, A.K.; STUL'PINAS, B.B. - Tr. Akad. Nauk. LitSSR, Ser. !., 2, 25, I07- , 1961 apud Ref. 53.
50. VOROZHKO, A.V.; POLESYA, A.F.; GAMALI, I.V.; PIKAT,OV, G.V. - Structure of
electrolyËically deposited manganese. Elektrokhimiy:1, 11(1)' L46-9,
1975.
51. yANrTSKrr, r.V. & SUL'P]NAS, B.B. - Zr. Prikl. Khim., 19, 7776- , 7957
apud Ref. 53.
52. SHVAB, N.A. & ZOSIMOVICH, D.P. Ukr. Khirn. Zt., 3!, 569- 'L969 apud
Ref. 53.
53. GAMALT, r.V.; RYBAT,'SKAYA, G.E.; TROÏTMENKO, V.V.; ELTNA, V.V. - Kinetic
features of the joint evolution of manganese and hydrogen during
electrolysis of pure elecËrolytes and electrolytes with added sulfurand compounds Elektrokh.i*i,y1, l:1(3) , 454-7, 1975.
54. GAIVIALI, I.V.; DANILOV, F.I.; STENDER, V.V. - Dimensional correspondence
ín the electrodeposition of manganese. Zh. Prikl. i<him,, 37(2),
337-42, 1964.
55. EHRLICH, G. - fn: Third congress on catalysis, vo1. 1' 't'J.M.H. Sachtler,
G.C.A. Schuit, P. ZwieËering (editores), North Hol1and, Amsterdã, 1965
apud Ref . 56.

160
56. BELANGER, A. & VIJH, A.K. - Hydrogen evolution reaction on vanadium,
chromium, manganese, cobalt. J . E lectrochem. Soc. , L2):(2.) , 225-30 ,1974 .
57. GAMALI, I.V. & STENDER, V.V. - Overvoltage of hydrogen evolution on
manganese. ah. Prik1. Khim., 35(1), 127-32, 1962.
58. LOSHKAREV, Yu. M.; STENDER, V.V.; GALUSHKO, V.P. - Cathodic process in
the electrolysis of solutions of manganese chloride. In: ReporËs of
Fifth All-Union Seminar on Applied Electrochemistry, 4l-7. Nova Iorque,
N.Y., V.V; Stender (ed.), Consultants Bureau, L965.
59. BONDAR, R.U.; GAMALI, I.V.; STENDER, V.V. - Electrodeposition of manganese
from amoniacal solutions . Zh. Prikl. Khim., 40(5), 1025-30, 1967.----
60. BONDAR, R.U. & GAMALI, I.V. - Influence of anrnonium ion and ammonia
contents in the el.ectrolyte on el.ectrodeposition of manganese. Zh.
Prikl. Khim., 43(6), L244-7, L970.
61. TROFIMENKO, V.V.; GAMALI, I.V.; VASILEVSKAYA, A.V. - Reduction of manga-
nese on a mercury dropping electrode from ammoníum=armnonia solutíons,
Elek-rrokhimiyj, 7(B), 1230-5, I97I.
62. GAMALI, I.V.; VOROZHKO, A.V.; TROFIMENKO, V.V. - Correlation of micro-
structure and conditions for electrodeposition of Y*manganese.
Elektrokhimiya, B(B) , LI29-34, 1972.
63. GAMALI, I.V.; TROFII4ENKO, V.V.; V0R0ZHK0, A.V. - Characteristics of
electrodeposition of manganese from pure electrolytes. Zþ. Prik1.
Khim, 47 (9) , 2035-9, L974.

64, BARCHESE, E. - Comunicação pessoal.
65. KUBLANOVSKII, V.S.; BELINSKII, V.N
electrodeposition of manganese
khimiya, !!_(O) , 586-9, L975.
66. KUBLANOVSKII ' V.S.;Ukranian
BELINSKII, V
of First Repub 1í can
67 . DEAN, R.S. ; ANDERSON, C.T.;its al1oys. U.S. Bureau
apud Ref. 4.
68. SCHLAIN, D. & PRATER,
Electfochem. Soc.,
161
. ; GORODSKII, A.V. - Mechanism of
from a sulphate electrolyte. Elektro-
.N.; ZOSIMOVICH, D.P. - In: Transactions
Conference on Electrochemistry (em russo)ParË. 1, Nankova Ilmka, Kiev, p. L52, 1973 apud Ref. 65.
MOSS; CRISAP; AMBROSE, P.M. - Manganese and
of Mines. Report of Investigations 3477 , L939
J.D. - Electrodeposition of ganrna manganese. Trans,
94, 58-73, 1948.
69. FEKETE,
the
L. - The control of pH in manganese electrolysis. Proceedings ofResearch InsËitute for Non-Ferrous Metals, Budapeste, 281-5, I97I
70. AGLADZE, R.I. - The complex ion of mixed solutions of manganese and
anrnonium sa1ts. J. Gen. Chem. (U.S.S.R.), 10, 340-6, 1940 apud Chem.
Abstr. 34:72021L, 1940.
71. AGLADZE, R.r.so lut i ons .
4582h.
& LEGRAN, A.B. -Zhur. Fiz. Khim.
Electrode poten¿ia1 of
, ZLr, II22-7, 1950 apud
manganese ln aqueous
Chem. Abstr. 45:
72. TTLAK, B.V.K.S.R.A.; RAJAGOPALAN, S.R., REDDY, A.K.N. _ Buffer characLerislics
of manganous sulphate+aûmoníum sulphate electrodeposition baths. Iægt-,
Faraday Soc., 59, 795-804, L962,
73. RADHAKRISHNAMURTHY, P. & REDDY, A.K.N. - A novel method
derermination. Journal of Appliecl ElecËrochemístry,
of
5:
current efficiency
39*4Lu 1975"

L62
74. RADHAI(RISHNAì{URTY, P. & REDDY, A.K"N. - The mechanism of manganese
electrodeposition. Journal of Applied Electrochemistry, 4, 3I7-2I"
L97 4.
75. BOCKRTS, J. O'M. & R-EDDY, A.K.N. * Modern ElgqtreehgrTri$Ly., v. 2,
Nova lorque, Plunum, L973.
76. BOCKRIS, J. O'M.; DRAZIC, D.; DESPIC, A.R. * Elektrochim. Acta, 4,
325, L96I, apud ref. 74,
77. RADHAKRISHNAI"TURTY, P. & R-EDDY, A.K.N. * Mechanism of actíon of selenius
acid in Ëhe electrodeposition of manganese" ¡-ggl"?!_gålpp.!l"d
EleqtrochemisËry, 7, LL3-7, L977.
78. RADHÄKRISIINAMURTY, P.; SATHYANARAYANA, S.; REDDY' A.K"N. - Kinectics of
hydrogen evolution reaction on a stainless steel electrode. Journal
of Appl:!ecl E_legËrochelnlstry, 7, 51-5, L977 "
Este trabalho foi realizado com apoio de bolsa de mestrado
e verbas do CNPq, no Programa PRONUCLEAR, de L979 a L982.