clonagem e expressÃo de membros da famÍlia...
TRANSCRIPT

CENTRO NACIONAL DE PESQUISA EM ENERGIA E MATERIAIS LABORATÓRIO NACIONAL DE BIOCIÊNCIAS
LABORATÓRIO DE MICROARRANJO- LMA 20º PROGRAMA BOLSAS DE VERÃO
CLONAGEM E EXPRESSÃO DE MEMBROS DA FAMÍLIA HUMANA DE NEKs CINASES
ENVOLVIDAS NA REGULAÇÃO DO CICLO CELULAR
CAROLLINE FERNANDA RODRIGUES ASCENÇÃO
CAMPINAS - SP 2011

ii
CENTRO NACIONAL DE PESQUISA EM ENERGIA E MATERIAIS LABORATÓRIO NACIONAL DE BIOCIÊNCIAS
LABORATÓRIO DE MICROARRANJO- LMA 20º PROGRAMA BOLSAS DE VERÃO
CLONAGEM E EXPRESSÃO DE MEMBROS DA FAMÍLIA HUMANA DE NEKs CINASES ENVOLVIDAS NA REGULAÇÃO
DO CICLO CELULAR
Relatório final das atividades realizadas durante o projeto desenvolvido no 20° Programa Bolsas de Verão de 05/01 a 25/02/11, elaborado por Carolline F. R. Ascenção, sob orientação do Dr. Jörg Kobarg e co-orientação do Dr. Diogo Ventura Lovato.
Dr. Jörg Kobarg - Orientador Dr. Diogo Ventura Lovato - Co-Orientador
CAMPINAS
2011

iii
“Que os vossos esforços desafiem as impossibilidades, lembrai-vos de que as grandes coisas do homem foram
conquistadas do que parecia impossível.” Charles Chaplin

iv
AGRADECIMENTOS
À Deus, por ter me proporcionado a oportunidade de cursar o
20° Programa Bolsas de Verão e por ter sido sempre meu guia amparo em todas as tempestades;
À minha mãe, Suely, minha grande incentivadora, por todo
cuidado, carinho e todas as sábias lições; Ao meu namorado Daniel por todo carinha, paciência e ajuda;
Ao 20° Programa Bolsas de Verão pela oportunidade de participação;
Ao Jörg pela escolha, confiança e orientação dada;
Ao Diogo pela orientação dada e pelos momentos científicos filosóficos que renderam sempre boas risadas;
A todo o grupo do LMA, especialmente ao Marcos, Eduardo, Daniel, Deivid, Priscila, Edmárcia, Gabi, Ângela, Ariane, Fernanda e Germana,
A técnica Maria Eugênia por toda disponibilidade de ajudar e boa vontade sempre;
Ao Laboratório Nacional de Biociências, pela estrutura cedida;
E a todos os colegas bolsistas, pelo companheirismo e pelos momentos de descontração.

v
Resumo
A divisão celular em mamíferos é impulsionada por um conjunto de proteínas
que constituem o sistema de controle, sendo este o responsável pela progressão correta
do ciclo celular. Essas proteínas são da classe de proteínas quinases (PKs), e agem
através de cascatas de fosforilação, orquestrando uma acurada sequência de eventos que
culminam com a regulação do tempo certo de entrada em mitose. Genes que codificam
para várias famílias de proteínas quinases são os mais comumente mutados em
situações que levam ao câncer em humanos. Proteínas cinases da família NIMA (Never
in Mitosis gene A) são um grupo da proteína cinases do tipo serina/treonina cinases
descritas pela primeira vez no fungo Aspergillus nidulans, no qual possui participação
no ciclo celular. Em humanos, os genes ortólogos a NIMA são chamados NEKs (NIMA
related Kinases) e compõem uma família gênica constituída por 11 membros que têm
domínios catalíticos altamente conservados, mas com regiões N e C-terminais variáveis.
NEKs têm sido descritas como tendo expressão aumentada em diferentes tipos de
câncer, entre eles podemos citar alguns tipos de câncer de mama e outras disfunções
orgânicas, como a doença policística do rim. Dentre os 11 membros da família de NEKs
cinase, NEK2 é melhor descrito estruturalmente até o momento. Contudo, muito pouco
se sabe sobre seus parceiros de interações proteína-proteína e seus substratos alvo em
estudos estruturais. Entre os outros membros da família de NEKs cinases, em particular,
NEK3 e NEK5 foram pouquíssimos estudados sendo que até o presente
momento,NEK5 não possui artigos publicados na literatura científica.Desta forma são
necessários estudos estruturais que elucidem sítios-chave de ligação a substrato e de
interações proteína-proteína que permitem o desenho racional de drogas.
Objetivando-se clonar os fragmentos gênicos codificantes para as NEKs cinases
2, 3 e 5 e os fragmentos gênicos codificantes para apenas os domínios de cinase das
NEKs 3 e 5, foi feita a amplificação via PCR de tais regiões gênicas. NEK2 foi
amplificada a partir de biblioteca de cDNA de HELA e NEK3 foi amplificada a partir
das bibliotecas de cDNAde leucócitos e cérebro fetal humano. Não foi obtido sucesso
nas tentativas de amplificação de NEK5 em nenhuma das bibliotecas de cDNA de
leucócitos humanos, cérebro fetal humano, medula óssea, mama e HELA. As regiões

vi
gênicas amplificadas de NEK2 e NEK3 foram clonadas em pGEM T easy e a
confirmação das clonagens foi feita por PCR de colônias e de sequenciamento. A partir
das sequências obtidas, foram feitas análises de similaridade em banco de dados
BLAST e encontrada similaridade com as NEKs 2 e 3 dispostas no banco de dados. A
construção pGEM T easy/NEK3 foi utilizada com DNA template para amplificar
apenas o domínio de cinase de NEK3, cuja amplificação foi obtida com êxito e tal
fragmento gênico foi clonado nos vetores de expressão pET 28a e pFastBacDual-eGFP.
Entretanto foram obtidas, após a transformação de E. coli DH 5α, colônias
transformantes e recombinantes apenas para a construção pET 28a/NEK3 kd e não para
a pFastBacDual-eGFP/NEK3 kd. Foi feita a transformação de E. coli BL 21 com a
construção pET 28a/NEK3 kd e a indução da expressão da proteína recombinante.
Todavia, foi observada, através de SDS-PAGE, uma banda protéica com o tamanho
esperado de 35 kDa tanto no lisado bacteriano transformado e induzido que continha a
construção pET 28a/NEK3 kd quanto no que continha apenas o vetor pET 28a vazio. O
DNA plasmidial da construção pGEM T easy/NEK2 foi utilizado, em várias
concentrações, para a transformação de uma quantidade variada de células da levedura
S. cerevisae L40, por choque térmico e por eletroporação. Entretanto, somente as
leveduras contendo os controles da transformação cresceram nas placas.

vii
ÍNDICE AGRADECIMENTOS ..................................................................................... iv
Resumo ............................................................................................................. v
1 - Introdução .................................................................................................... 1
2 - Objetivos .................................................................................................... 13
2.1- Objetivos Gerais ................................................................................... 13
2.2 - Objetivos específicos ........................................................................... 13
3 - Material e Métodos ..................................................................................... 15
3.1 Microrganismos utilizados ..................................................................... 15
3.2 - Plasmídeos utilizados ........................................................................... 15
3.2.1- pBTM 116 K (pLEXA) .................................................................. 15
3.2.2 - pET 28a ........................................................................................ 16
3.2.3 - pGEM T easy ................................................................................ 17
3.2.4 - pACT ............................................................................................ 17
3.2.5 – pFastBacDual ............................................................................... 18
3.3 - Desenho de oligonucleotídeos específicos para amplificar NEK3, NEK5,
NEK2, e para amplificar os domínios de cinases de NEK3 e NEK5......................... 19
NEK2 ....................................................................................................... 19
NEK3 ....................................................................................................... 20
Sequência do mRNA, disposta no GenBank (acesso: NM_2498.2). .......... 20
NEK5 ....................................................................................................... 22

viii
3.4 - Amplificação dos genes das NEKs 2, 3 e 5 e da região gênica
correspondente ao domínio de cinase das NEKs 3 e 5 via reação de PCR (Reação em
Cadeia de Polimerase) ............................................................................................. 23
3.4.1 - NEK2............................................................................................ 23
3.4.2 - NEK3............................................................................................ 23
3.4.3 - NEK5............................................................................................ 24
3.4.4 - Extração e purificação do DNA em gel de agarose ........................ 24
3.5 – Reações de clivagem ........................................................................... 24
3.5.1 - Clivagem de NEK2 e da região codificante para o domínio de cinase
de NEK3 ............................................................................................................. 24
3.5.2- Clivagem dos vetores pBTM 116 K e pET 28a e pFastBacDual ..... 25
3.6 – Reações de ligação .............................................................................. 25
3.6.1- Ligação dos fragmentos amplificados de NEK 2, 3 e 5 em pGEM T
easy vector .......................................................................................................... 25
3.6.2- Ligação dos fragmentos amplificados referente ao domínio de cinase
de NEK3 ao vetor pET 28a e ao vetor pFastBacDual e de NEK2 ao vetor pBTM
116 K .................................................................................................................. 25
3.7 – Transformação bacteriana ................................................................... 26
3.7.1 - Choque térmico ............................................................................. 26
3.7.2- Eletroporação ................................................................................. 26
3.8 - Seleção das colônias de E. coli transformantes e recombinantes ........... 27
3.9 - Sequenciamento de DNA e análise das sequências de nucleotídeos ...... 27
3.10 – Transformação da levedura Saccharyomyces cerevisae L40 .............. 27

ix
3.11 – Ensaio de mini-indução da expressão de pET 28a/NEK3 domínio de
cinase ...................................................................................................................... 29
4 – Resultados e Discussão .............................................................................. 31
4.1 - Amplificação dos genes das NEKs 2, 3 e 5 e da região gênica
correspondente ao domínio de cinase de NEK3 via reação de PCR (Reação em
Cadeia de Polimerase) ............................................................................................. 31
4.1.1- NEK2 ............................................................................................ 31
4.1.2 - NEK3............................................................................................ 33
4.1.3- NEK5 ............................................................................................ 36
4.2 – Clonagens, sequenciamentos e subclonagens ....................................... 39
4.3 – Ensaio de mini-indução da expressão de pET 28a/NEK3 domínio de
cinase ...................................................................................................................... 48
4.4 - Transformação da levedura Saccharyomyces cerevisae L40 ................. 50
5 – Conclusão .................................................................................................. 53
6 – Perspectivas ............................................................................................... 55
7 – Referências bibliográficas .......................................................................... 56

1
1 - Introdução
O câncer pode ser definido como um conjunto de centenas de doenças que têm
em comum o crescimento desordenado (maligno) de células do próprio organismo.
Essas células perdem a capacidade de limitar e controlar o seu próprio crescimento
passando, então, a multiplicarem-se muito rapidamente e sem nenhum controle. O
resultado desse processo desordenado de crescimento celular é uma produção em
excesso dos tecidos do corpo (que podem ser processos inflamatórios, infecciosos ou
mesmo os crescimentos celulares benignos), formando o que se conhece como tumor
(Alberts et al, 2002).
FIGURA 1 – Processo de metástase.
Em 2008, a Agência Internacional para Pesquisa sobre Câncer das Nações
Unidas (IARC) divulgou que o número de mortes por câncer chegou a 7,6 milhões em
todo o mundo. A expectativa da referida agência, divulgada em junho de 2010, é a de
que até 2030, 13,2 milhões de pessoas morram de câncer. A tabela 1 indica a
expectativa de novos casos de câncer, feita pela IARC, para o ano de 2011.

2
TABELA 1 – Estimativa de novos casos de câncer para 2011.
Fonte: IARC (Agência Internacional para Pesquisa sobre Câncer das Nações
Unidas).
Chama-se ciclo celular o conjunto de processos que se passam
numa célula viva entre duas divisões celulares. O ciclo celular consiste na intérfase e na
fase mitótica, que inclui a mitose e a divisão celular (citocinese) (Alberts et al, 2002).
A intérfase é todo o período de vida de uma célula situado entre duas fases de
mitose consecutivas. O primeiro período da interfase é o G1 (do inglês gap, intervalo).

3
Durante este período, a célula executa suas funções normais sem se ocupar com a
divisão celular. Ainda na intérfase, existe a fase S (do inglês synthesis), no qual ocorre a
replicação do DNA e duplicação do material genético. O último momento da intérfase é
dito G2. Nele, a célula certifica-se de que todo o DNA já foi duplicado e cresce um
pouco mais, se for necessário. Além disso, termina de duplicar os centríolos (ficando,
então, com dois pares), processo que iniciou no período S, e aparecem as proteínas que
formarão o fuso mitótico ou fuso acromático. Até o final da interfase, a célula tem pelo
menos dois pontos de checagem. O primeiro deles é no final do G1 no qual a célula
certifica-se se está grande o suficiente para se dividir, se o ambiente é propício a uma
divisão e se o DNA precisa ser reparado antes de iniciar o processo de replicação, típico
do período S. O outro ponto de checagem é no final do G2 no qual a célula volta a se
certificar se seu tamanho está apropriado e se todos os eventos que antecedem a mitose
foram realizados com precisão (De Robertis & De Robertis Jr., 1996). Em todos esses
pontos de checagem, observa-se em nível molecular, a atuação de proteínas, via de
regra, proteínas cinases, que por meio de cascatas de fosforilação, checam e sinalização
o momento correto de entrada em mitose. Tais proteínas, quando se apresentam
mutadas, podem ter comportamentos anormais de sinalização e checagem, culminando,
por exemplo, com a entrada precoce em mitose ou a incorreta sinalização de resposta ao
dano ao DNA antes que o material genético seja duplicado, levando a fenótipos que
podem ser caracterizados como câncer.
A mitose é a fase em que as células eucarióticas dividem seus cromossomos
entre duas células filhas. Este processo dura, em geral, de 90 a 120 minutos, e é dividido
em quatro etapas: prófase, metáfase, anáfase e telófase (Lodish et al, 2007) (Figura 2).

4
FIGURA 2 – Eventos do ciclo celular.
Assim como na intérfase, na mitose é necessária a presença de proteínas cinases
para controlar a correta passagem de uma de suas fases a outra, sendo imprescindível a
presença das referidas proteínas para que haja precisão na organização do fuso mitótico,
correta segregação dos cromossomos para as células filhas e inclusiva para que a célula
seja capaz de executar a citocinese. As PKs que participam da regulação do ciclo celular
são consideradas os mais promissores alvos terapêuticos contra diferentes tipos de
câncer. Os principais pontos de controle do ciclo celular (checkpoints) foram
identificados no final da fase G1 (ponto de restrição), na passagem de G2 para mitose e
dentro da própria mitose, na metáfase. As PKs que atuam nestes checkpoints incluem as
CDKs (cyclin dependent kinases), cinase Aurora, cinases tipo-Polo, ATR/ATM (para
revisão Malumbres & Barbacid, 2007 e Lapenna & Giordano, 2009) e as cinases do tipo
NEK. Perdas ou falhas nestes pontos de controle são associadas com o desenvolvimento
de células neoplásicas, uma vez que essas não obedecem mais ao ritmo de divisão
celular e acumulam mutações no DNA (para revisão Malumbres & Barbacid, 2007).
Entre as diversas famílias de PKs descritas, NEK-cinases são as menos estudadas e
menos caracterizadas funcionalmente. De acordo com Salem et al (2007), a proteína

5
cinase NEK7 (NIMA-related kinases) está diretamente envolvida em eventos de
citocinese, pois camundongos mutantes knockdown para a referida proteína apresentam
fibroblastos multinucleados e com retardo na citocinese. Sabe-se também que na
ausência de outra cinase da família NEK, o membro NEK9, ocorre incorreta segregação
dos cromossomos durante a mitose (Lynne et al, 2005). Todas estas disfunções
causadas pela atividade incorreta de proteínas cinases, ou ausência das mesmas, são
capazes de evoluir para fenótipos de câncer.
Em humanos, existem quatro principais famílias de proteínas serino/treonina
cinases envolvidas na correta progressão das fases da mitose, no ciclo celular, são elas:
as cinases dependentes de ciclinas, CDKs; as Polo-like cinases, PLKs; as Aurora
cinases e as NIMA- related kinases.
Cinases dependentes de ciclinas são proteínas que devem se associar a uma
ciclina para se tornarem cinases ativas. São reguladores chave do ciclo celular. Existem
hoje aproximadamente 12 CDKs descritas até o momento, sendo a CDK1 (ou cdc2)
considerada como a cinase que, provavelmente, é a responsável por todas as transições
do ciclo celular (Fisher & Nurse, 1996). Isso é verdade em leveduras onde a atividade
cinase de CDK1 é necessária para as transições G1/S e G2/M (Durkacz; Carr & Nurse,
1986). Em células de mamíferos, no entanto, a atividade CDK1 é necessária apenas para
a transição G2/M de transição (Draetta & Beach, 1988).
As Polo-Like cinases formam uma família de quatro proteínas diferentes que
regulam muitos aspectos da progressão do ciclo celular. Elas compartilham pequenos
domínios conservados nomeados pólo-box que são requeridos para a localização das
referidas proteínas. De todas as Plks, Plk1 é a mais estudada e foi verificado estar
envolvida em eventos de progressões das fases da mitose sendo um homólogo da polo-
cinase de Drosophila melanogaster (Nigg, 1996). Plk2, Plk3 Plk4 são mais prováveis de
estarem envolvidas somente em intérfase. Sabe-se também que a atividade Plk4 é
necessária para a duplicação de centríolos, um evento que deve ser obtido antes de
entrar em mitose e necessários para a montagem do fuso mitótico bipolar (Habedanck et
al, 2005).
Aurora cinases foram primeiramente identificadas na Saccharyomyces
cerevisae e Drosophila melanogaster (Chan & Botstein, 1993). As células de levedura
possuem apenas uma Aurora cinase, Drosophila melanogaster e Caenorhabditis

6
elegans possuem duas (A e Tipo B) e em mamíferos foram descritas, até o momento,
três, chamadas Aurora A, B e C (Nigg, 2001). Sabe-se que os tipos A e
B evoluíram de um ancestral comum, enquanto o tipo C evoluiu a partir de do tipo B
(Brown et al, 2004). Por conseguinte, Aurora A tem funções distintas, enquanto as
Aurora B e C possuem funções relacionadas, apesar de todas as três cinases estarem
envolvidas no controle de muitos processos necessários para a mitose.
NIMA-related kinases pertencem a uma família muito grande de proteínas
cinases, com 13 diferentes NEKs em humanos, de NEK1 a NEK11 (NEK2A e NEK2B
e NEK11L e NEK11S) (O´Connell et al, 2004). Os membros da família NEK são
definidos pela homologia com a cinase NIMA (Nerver in mitosis gene A),
primeiramente identificada em Aspergillus nidulans como uma cinase envolvida na
progressão das fases da mitose do ciclo celular. Entre as diversas famílias de PKs
descritas, os membros da família relacionados à NIMA são os menos estudados e menos
caracterizados funcionalmente. Tem sido demonstrado que algumas cinases da família
NIMA possuem um papel como regulador mitótico essencial, pois sua ativação se faz
necessária para a fosforilação mitótico-específica na serina 10 da histona H3. Se a
fosforilação mitótica de H3 é bloqueada, a condensação cromossômica não ocorre e a
célula fica impedida de entrar em mitose. Da mesma forma, se não ocorre a
desfosforilação de H3, os cromossomos permanecem condensados durante a mitose.
NIMA é capaz de fosforilar H3 in vitro, o que sugere fortemente que é a responsável
por fosforilar H3 in vivo. Todavia, isso ainda não foi verificado experimentalmente.
Mutantes NIMA sensíveis à temperatura, de A. nidulans (figura 6), apresentam atraso
em G2, com cromatina não condensada de forma que não ocorre a transição G2/mitose.
São necessários altos níveis de NIMA para que ocorra a transição G2/mitose. Sabe-se
que NIMA exibe uma localização nuclear, mais precisamente de ligação à cromatina
durante a fosforilação de H3, na intérfase, e durante a mitose, NIMA está localizada no
fuso. Isso sugere que NIMA é necessária e suficiente para permitir a fosforilação de H3
e posterior entrada em mitose (De Souza et al, 2000).

7
FIGURA 3 – Mutantes NIMA de A. nidulans, sensíveis a temperatura.
Genes que codificam proteínas cinases da família NIMA foram inicialmente
descritos em humanos recebendo a nomenclatura de família das NEKs (NIMA related
kinases) (Quarmby & Mahjoub, 2005). Essa família é formada por domínios catalíticos
de cinases altamente conservados, mas com regiões N e C-terminais variáveis, que em
alguns casos são muito importantes para regulação e especificidade de substratos
(Surpili et al, 2003). A superexpressão de NEKs em células de mamíferos resultou no
desencadeamento precoce de eventos relacionados à mitose, incluindo condensação de
cromatina e despolimerização dos microtúbulos (Lu & Hunter, 1995). Com o advento
do genoma humano, foram anotados onze genes codificantes de NEKs sendo 13
produtos gênicos, uma vez que existem as isoformas de NEK2, NEK2A e NEK2B, e de
NEK11, NEK11L e NEK11S.
NEK1 é um dos membros mais estudados da família protéica das NEK cinases.
Sabe-se que camundongos mutantes para NEK1 apresentam o desenvolvimento da
doença policística do rim, kat 1 e kat 2, resultando num fenótipo pleiotrópico recessivo
que inclui a progressiva formação de cistos renais, esterelidade em machos, nanismo,
anormalidades olfativas, dimorfismos faciais, cistos de hidrocefalia, anemia e uremia.
Embora tenha sido sugerido que NEK1 tenha papéis no controle do ciclo celular,
meiose, gametogênese e no sistema nervoso (Arama et al, 1998), a função celular de
NEK1 até o momento permanece obscura. De todas as NEKs, NEK1 apresenta a maior

8
estrutura protéica primária prevista (figura 4), com 1.203 aminoácidos e 42% de
identidade com as NIMA de A. nidulans.
FIGURA 4 – Estrutura primária de NEK1.
Foi proposto por Mark et al (2008) que NEK1 apresenta uma localização
centrossomal e nuclear, devido ao motivo estrutural coiled-coil, durante o ciclo celular,
indicando um importante papel desta cinase na organizaçãodo centrossomo.
NEK2 apresenta 445 aminoácidos em sua estrutura primária e estrutura
tridimensional resolvida do domínio catalítico. Entretanto, muito pouco se sabe sobre
quais são todos os seus parceiros de interação proteína-proteína. Esta proteína é um
dímero capaz de sofrer trans-auto-fosforilação, possuindo um domínio de cinase N-
terminal e um domínio C-terminal que inclui um zíper de leucina. É uma proteína que
possui duas isoformas, NEK2A e NEK2B, e é sabido que NEK2A é requerida para a
correta separação dos centrossomos na transição G2/mitose, mas muito pouco se sabe
sobre NEK2B (Lynne et al, 2005). É a NEK que apresenta maior porcentagem de
identidade aminoacídica, 48%, com as NIMA de A. nidulans. Apesar da similaridade
com NIMAs, knockouts de NEK2 entram em mitose sugerindo que existem diferenças
significativas na regulação do ciclo celular entre fungos e humanos (Fletcher et al,
2005).
Atribuiu-se para NEK2, até o presente momento, as funções celulares na
coordenação da estrutura e função dos centrossomos para a correta progressão das fases
da mitose, regulação do núcleo protéico do cinetócoro através da ativação de Hec1
(High expressed in cancer) e papel de ativação de NEK11, quando translocada para o
nucléolo durante a transição G1/S. A deleção de NEK2 pode ocasionar o atraso na

9
mitose uma vez que não ocorre a correta separação dos centrossomos e aumento de
apoptose.
Até o momento, muito pouco se sabe sobre as funções e os parceiros de
interação proteína-proteína de NEK3. Foi relatado entre tumores de mama e NEK3 a
qual parece desempenhar papel importante na sinalização do receptor de prolactina,
especificamente em uma via que contribui para a progressão e mobilidade desses tipos
de cânceres (Miller et al, 2005). Outra função descrita é a regulação da acetilação de
microtúbulos em neurônios (Chang et al, 2009). Este membro da família das NEKs
cinases apresenta 489 aminoácidos e 42% de identidade com as NIMA de A. nidulans.
NEK4 é um dos membros da famíla das NEKs cinases recentemente identificado
e pobremente caracterizado. Sabe-se que NEK4 apresenta funções ciliares e flagelares e
sugere-se papel na regulação do ciclo celular (Takeno et al, 2007). Com relação à
estrutura primária, NEK4 apresenta 841 resíduos de aminoácidos e 39% de identidade
com as NIMA de A. nudulans.
Apesar de todos os estudos pós-genômicos, até o presente momento, nenhum
trabalho foi publicado caracterizando funcional e estruturalmente as NEKs 5 e 10 e
apenas um para NEK10 (Moniz & Stambolic, 2011). Sabe-se que NEK10 está presente
em tumores de mama, mas muito pouco se sabe sobre sua real atuação neste tipo de
câncer (Odefrey et al, 2010).
As NEKs 6 e 7 apresentam-se envolvidas em cascatas mitóticas de sinalização.
Possuem 87% de identidade entre seus próprios domínios de cinase. NEK6 apresenta
313 resíduos de aminoácidos em sua estrutura primária e NEK7 apresenta 302. Ao
contrário de NEK2, não dimerizam e nem sofrem trans-auto-fosforilação.Elas não
apresentam o domínio de cinase no N-terminal, mas sim no C-terminal (Takeno et al,
2007). Ambas são ativadas por NEK9 e já foi demonstrado que não apresentam
atividades e funções redundantes. A NEK6 humana parece ser requerida para a
progressão de eventos mitóticos, pois sua abundância e atividade de cinase são
aumentadas durante a mitose (Takeno et al, 2007). NEK6 não funcionais geram uma
parada na mitose. Se NEK7 possui um papel na mitose, isso é pouco claro. Sabe-se,
todavia, que NEK7 está diretamente envolvida em eventos de citocinese pois
camundongos mutantes knockdown, para a referida proteína, apresentam fibroblastos
multinucleados e com retardo na citocinese (Salem et al, 2010).

10
NEK9, além da função acima descrita de ativar NEK 6 e 7, parece estar
intimamente ligada com o correto alinhamento e a segregação dos cromossomos durante
mitose. Provavelmente, por afetar a organização do fuso. Sabe-se que esta cinase é
ativada pela auto-fosforilação da tirosina 210 no loop de ativação, e que apresenta-se
localizada nos centrossomos durante a mitose (Takeno et al, 2007). A deleção de NEK9
interfere com a formação do fuso bipolar, e células knockdown para NEK9 exibem
atrasada progressão G1/S (Takeno et al, 2007). Com relação à estrutura primária de
NEK9, sabe-se que esta apresenta 979 resíduos de aminoácidos, possuindo, ao contrário
das NEKs 6 e 7, o domínio catalítico na região N-terminal.
NEK8 é um membro da família das NEK cinases que, assim como NEK1, está
envolvida na doença policística do rim. Sabe-se que a superexpressão desta cinase, em
cultura de células, promove o aparecimento de células multinucleadas, sendo observado,
também, a redução do número de fibras de actina estressadas (Takeno et al, 2007). A
estrutura aminoacídica primária desta cinase mostra que a mesma possui 698
aminoácidos e domínio de cinase no N-terminal em concordância com a maioria dos
outros membros da família das NEKs cinases.
NEK11 apresenta-se em duas isoformas identificadas como NEK11L e NEK11S
(O’Connell et al, 2004). No entanto, muito pouco ainda se sabe sobre as modificações,
se pós-transcricionais ou pós-traducionais, que geram tais isoformas. Sua sequência
aminoacídica primária divide de 40% a 45% de identidade com as NIMA de A.
nudulans dentro de seu domínio catalítico N-terminal. Com relação às funções já
relatadas de NEK11, sabe-se que a mesma é ativada por NEK2 e translocada ao núcleo
onde está envolvida com a resposta ao dano ao DNA (O’Regan et al, 2007).
As quatro famílias de proteínas cinases descritas são intrinsecamente envolvidas
em acurados eventos de sinalização celular que culminam com uma organizada cascata
de ativação de proteínas e verificação de pontos–chave do ciclo celular (figura 6), para
que a célula possa iniciar eventos de divisão celular e saída de mitose somente se
realmente estiver “madura”, ou seja, se todos os eventos que antecedem cada uma das
etapas da intérfase ou da mitose estiverem completados com precisão.

11
FIGURA 6 – Cinases envolvidas na regulação da prófase, metáfase e citocinese.
Caso algum membro das quatro famílias protéicas descritas apresente-se mutado
ou ausente em determinados tipos de células, pode-se observar que os eventos celulares
que outrora ocorriam de maneira acurada podem apresentar-se falhos e ocorrer, por
exemplo, a entrada precoce em mitose e divisão celular além dos limites normais, atraso
ou ausência de citocinese com o consequente aparecimento de células multinucleadas e
poliplóides, incorreta segregação dos cromossomos, entre outras consequências. Esses
eventos anormais no ciclo celular podem levar ao aparecimento de inúmeros fenótipos
relacionados a doenças, dentre elas, o câncer, como está demonstrado na tabela 2.
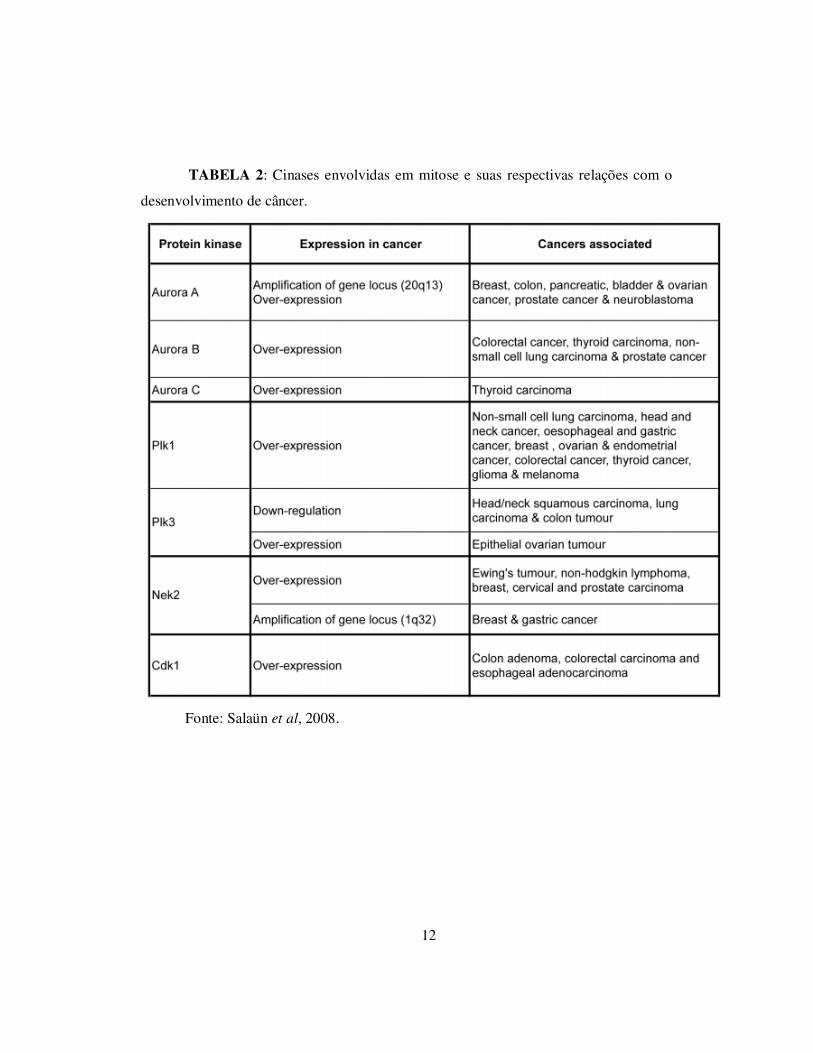
12
TABELA 2: Cinases envolvidas em mitose e suas respectivas relações com o
desenvolvimento de câncer.
Fonte: Salaün et al, 2008.

13
2 - Objetivos
2.1- Objetivos Gerais
O presente trabalho apresentou como objetivos gerais a clonagem e expressão do
domínio catalítico de NEK3 e NEK5 em Escherichia coli BL 21, clonagem do domínio
catalítico de NEK3 em pFastBacDual e a identificação de parceiros de interação
proteína-proteína com NEK2 por sistema de seleção de Duplo-Híbrido em leveduras.
2.2 - Objetivos específicos
• Amplificação via PCR dos fragmentos gênicos correspondentes à região
de domínio catalítico de NEK3 (a partir de uma biblioteca de cDNA de
tecido de leucócitos) e NEK5 (a partir de uma biblioteca de cDNA de
cérebro humano fetal).
• Clonagem dos fragmentos amplificados de NEK3 e NEK5 em pGEM T
para sequenciamento e confirmação das frames.
• Subclonagem dos fragmentos gênicos referentes aos domínios de cinase
de NEK3 e NEK5 em vetor de expressão pET 28a, transformação de
Escherichia coli BL 21 e indução da expressão das proteínas
recombinantes.
• Subclonagem do fragmento gênico referente aos domínios de cinase de
NEK3 em pFastBacDual.
• Amplificação via PCR de NEK2 (a partir de uma biblioteca de cDNA de
HELA) e clonagem em vetor pGEM T para sequenciamento e
confirmação da frame.
• Subclonagem em vetor pBTM 116 K para realização de sistema de
Duplo-Híbrido em leveduras.

14
• Screenen das colônias de leveduras recombinantes para a identificação
dos parceiros de interação proteína-proteína de NEK2 com proteínas
vindas de uma biblioteca de cDNA de HELA.

15
3 - Material e métodos
3.1 Microrganismos utilizados
Os microrganismos utilizados neste trabalho foram Escherichia coli,
especificamente as estirpes DH 5α, BL 21 e JM109, e Saccharyomyces cerevisae em
que foi utilizada a cepa L40. Os respectivos genótipos estão descritos na tabela 3.
TABELA 3 – Linhagem e genótipo dos microrganismos utilizados neste projeto
3.2 - Plasmídeos utilizados
3.2.1- pBTM 116 K (pLEXA)
pBTM 116 (figura 7) é um plasmídeo comumente utilizado nos procedimentos
de realização de sistema de Duplo-Híbrido em leveduras. Apresenta, próximo ao sítio
múltiplo de clonagem, uma sequência codificante para o domínio protéico de ligação ao
DNA de LexA que é transcrito fusionado à proteína de interesse e sob o controle do
promotor de ADHI (Alcohol deydrogenase I). O plasmídeo apresenta 5.693 pb e contém
o gene TRP que confere prototrofia à cepas de leveduras que são mutantes auxotróficos
para triptofano, funcionando como um marcador de seleção. Outro marcador de seleção
é a ampicilina que, para o vetor utilizado neste trabalho, foi substituído por canamicina
(K).
Linhagem Genótipo
DH 5α endA1 recA1 hsdR17 supE44 gyrA96 thi-1 relA1 ∆lacU169
(φ80lacZ∆M15) Tetr ∆(mcrA)183 ∆(mcrCB-hsdSMR-mrr)173 BL 21 – CodonPlus DE3 E. coli B F– ompT hsdS(rB– mB–) dcm+ Tetr gal λ(DE3) endA
Hte [argU proL Camr] [argU ileY leuW Strep/Specr] L40 MAT a; his3-200; trp1-901; leu3-3,112; ade2; LYS::(4lexAop-
HIS3); URA3::(8lexAop-lacZ). JM109 F’ traD36 proA+B+ lacIq ∆(lacZ)M15/ ∆(lacproAB) glnV44 e
14- gyrA96 recA1 relA1 endA1 thi hsdR17)

16
FIGURA 7 - Mapa físico do vetor pBTM116.
3.2.2 - pET 28a
pET 28a (figura 8) é um vetor comumente usado para expressão heteróloga em
bactéria. Este vetor possui 5.369 pb, codificando para o marcador de seleção
canamicina, que foi substituído por ampicilina no vetor utilizado neste trabalho; uma
cauda de histidina que, sob o controle do promotor T7, é expressa fusionada ao N-
terminal da proteína de interesse que será inserida no sítio múltiplo de clonagem; e uma
sequência substrato para a ação de trombina que, no vetor utilizado neste trabalho foi
substituída por uma sequência substrato para a ação da protease TEV.

17
FIGURA 8 - Mapa físico do vetor pET 28a.
3.2.3 - pGEM T easy
pGEM T easy é um vetor largamente utilizado em clonagem. O plasmídeo
possui uma timina ligada a cada uma das extremidades 3’, o que impede a
recircularização do plasmídeo e melhora a eficiência de ligação de produtos de PCR,
pois algumas polimerases termoestáveis, como a Taq, adicionam uma adenina nas
extremidades 3’ do fragmento amplificado (figura 9) (Clark et al, 1988). Esse vetor
possui 3,0 kb e apresenta o gene de resistência a ampicilina como marca de seleção,
origem de replicação do fago F1 (ori), parte do gene lacZ que codifica o fragmento
amino terminal da enzima β-galactosidase, sítio múltiplo de clonagem e os promotores
T7 e SP6 flanqueando a região de clonagem.
FIGURA 9 – Mapa físico do vetor pGEM T easy.
3.2.4 - pACT
pACT (figura 10) é um plasmídeo utilizado em metodologias de sistema de
Duplo-Híbrido em leveduras, uma vez que permite a transcrição do ativador
transcricional em leveduras, GAL 4, fusionado a sequências codificantes de proteínas de
uma biblioteca de cDNA de interesse. Neste trabalho, foi usado o pACT que continha
sequências de proteínas vindas de uma biblioteca de cDNA de HELA. É um vetor que
contém 5.566 pb, o marcador de seleção ampicilina, e contém o gene LEU, que confere
prototrofia às cepas de leveduras que são mutantes auxotróficos para leucina.

18
FIGURA 10 – Mapa físico do plasmídeo pACT.
3.2.5 – pFastBacDual
pFastBacDual é um vetor de expressão comumente usado em metodologias de
expressão de proteínas recombinates em células de insetos por Baculovírs.Este
plasmídeo é capaz de expressar duas proteínas simultaneamente usando o Bac-to-Bac
Baculovirus Expression System. A expressão em células eucarióticas é acionada pelo
promotor de poliedrina (PH) ou pelo promotor P10. Neste vetor, existe um códon ATG
de iniciação que elimina a necessidade de clonagem em frame. pFastBacDual (figura
11) possui 5.238 pb, codifica o gene para resistência a ampicilina (para a clonagem em
bactérias), o gene de resistência a gentamicina e sequência sinal de poliadenilação. Para
uso, neste trabalho, o pFastBacDual já estava com o gene repórter eGFP (enhanced
Green fluorescent protein) clonado no polilinker que está sob o controle do promotor
P10, e foi inserido a sequência de NEK3 domínio de cinase no polilinker que está sob o
controle do promotor de poliedrina.

19
FIGURA 11 – Mapa físico do vetor pFastBacDual.
3.3 - Desenho de oligonucleotídeos específicos para amplificar NEK3,
NEK5, NEK2, e para amplificar os domínios de cinases de NEK3 e NEK5
Com o objetivo de amplificar, via PCR (Polymerase chain reaction), foi feito
inicialmente uma busca, no banco de dados GenBank, das sequências gênicas para as
NEKs 2, 3 e 5 e desenhados, manualmente, primers específicos para a amplificação e
posterior clonagem de toda a região codificante das referidas NEKs. Para as NEKs 3 e
5, foram também desenhados primers internos que amplificassem os seus respectivos
domínios de cinase. Esses primers foram posteriormente analisados em programas
OligoCalc (http://www.basic.northwestern.edu/biotools/oligocalc.html) para verificar as
características destes e possíveis problemas de anelamento interno inespecífico
(hairpin), entre outros. Seguem as sequências gênicas das NEKs 2, 3 e 5 que foram
usadas para o desenho dos primers específicos.
NEK2
Sequência do mRNA, disposta no GenBank (acesso: NM_002497.2).
GGTTAAACGGGGCCCAAGGCAGGGGTGGCGGGTCAGTGCTGCTCGGGGGCTTCTCCATCCAGGTCCCTGGAGTTCCTGGTC
CCTGGAGCTCCGCACTTGGCGGCGCAACCTGCGTGAGGCAGCGCGACTCTGGCGACTGGCCGGCCATGCCTTCCCGGGCTGAGGACTA

20
TGAAGTGTTGTACACCATTGGCACAGGCTCCTACGGCCGCTGCCAGAAGATCCGGAGGAAGAGTGATGGCAAGATATTAGTTTGGAAA
GAACTTGACTATGGCTCCATGACAGAAGCTGAGAAACAGATGCTTGTTTCTGAAGTGAATTTGCTTCGTGAACTGAAACATCCAAACA
TCGTTCGTTACTATGATCGGATTATTGACCGGACCAATACAACACTGTACATTGTAATGGAATATTGTGAAGGAGGGGATCTGGCTAG
TGTAATTACAAAGGGAACCAAGGAAAGGCAATACTTAGATGAAGAGTTTGTTCTTCGAGTGATGACTCAGTTGACTCTGGCCCTGAAG
GAATGCCACAGACGAAGTGATGGTGGTCATACCGTATTGCATCGGGATCTGAAACCAGCCAATGTTTTCCTGGATGGCAAGCAAAACG
TCAAGCTTGGAGACTTTGGGCTAGCTAGAATATTAAACCATGACACGAGTTTTGCAAAAACATTTGTTGGCACACCTTATTACATGTC
TCCTGAACAAATGAATCGCATGTCCTACAATGAGAAATCAGATATCTGGTCATTGGGCTGCTTGCTGTATGAGTTATGTGCATTAATG
CCTCCATTTACAGCTTTTAGCCAGAAAGAACTCGCTGGGAAAATCAGAGAAGGCAAATTCAGGCGAATTCCATACCGTTACTCTGATG
AATTGAATGAAATTATTACGAGGATGTTAAACTTAAAGGATTACCATCGACCTTCTGTTGAAGAAATTCTTGAGAACCCTTTAATAGC
AGATTTGGTTGCAGACGAGCAAAGAAGAAATCTTGAGAGAAGAGGGCGACAATTAGGAGAGCCAGAAAAATCGCAGGATTCCAGCCCT
GTATTGAGTGAGCTGAAACTGAAGGAAATTCAGTTACAGGAGCGAGAGCGAGCTCTCAAAGCAAGAGAAGAAAGATTGGAGCAGAAAG
AACAGGAGCTTTGTGTTCGTGAGAGACTAGCAGAGGACAAACTGGCTAGAGCAGAAAATCTGTTGAAGAACTACAGCTTGCTAAAGGA
ACGGAAGTTCCTGTCTCTGGCAAGTAATCCAGAACTTCTTAATCTTCCATCCTCAGTAATTAAGAAGAAAGTTCATTTCAGTGGGGAA
AGTAAAGAGAACATCATGAGGAGTGAGAATTCTGAGAGTCAGCTCACATCTAAGTCCAAGTGCAAGGACCTGAAGAAAAGGCTTCACG
CTGCCCAGCTGCGGGCTCAAGCCCTGTCAGATATTGAGAAAAATTACCAACTGAAAAGCAGACAGATCCTGGGCATGCGCTAGCCAGG
TAGAGAGACACAGAGCTGTGTACAGGATGTAATATTACCAACCTTTAAAGACTGATATTCAAATGCTGTAGTGTTGAATACTTGGTTC
CATGAGCCATGCCTTTCTGTATAGTACACATGATATTTCGGAATTGGTTTTACTGTTCTTCAGCAACTATTGTACAAAATGTTCACAT
TTAATTTTTCTTTCTTCTTTTAAGAACATATTATAAAAAGAATACTTTCTTGGTTGGGCTTTTAATCCTGTGTGTGATTACTAGTAGG
AACATGAGATGTGACATTCTAAATCTTGGGAGAAAAAATAATGTTAGGAAAAAAATATTTATGCAGGAAGAGTAGCACTCACTGAATA
GTTTTAAATGACTGAGTGGTATGCTTACAATTGTCATGTCTAGATTTAAATTTTAAGTCTGAGATTTTAAATGTTTTTGAGCTTAGAA
AACCCAGTTAGATGCAATTTGGTCATTAATACCATGACATCTTGCTTATAAATATTCCATTGCTCTGTAGTTCAAATCTGTTAGCTTT
GTGAAAATTCATCACTGTGATGTTTGTATTCTTTTTTTTTTTCTGTTTAACAGAATATGAGCTGTCTGTCATTTACCTACTTCTTTCC
CACTAAATAAAAGAATTCTTCAGTT
Foram desenhados um par de primers para amplificar a região codificante para
NEK2, presente nesta sequência de mRNA e que apresenta um tamanho 1.335 pb.
Foward: NEK2pbtm116BamHI: 5’ GGATCCGTATGCCTTCCCGGGCTGAG 3’
Reverse: NEK2pbtm116PstI: 5’ CTGCAGCTAGCGCATGCCCAGGATCTG3’
Tais primers foram desenhados contendo os sítios para as enzimas de restrição
(sequência sublinhada) BamHI (GGATCC) e PstI (CTGCAG), para que pudessem gerar
a amplificação de um fragmento que seria, subsequentemente, clivado pelas já citadas
enzimas de restrição, e ligado ao vetor pBTM 116 K, para posteriores ensaios de Duplo-
Híbrido em leveduras.
NEK3
Sequência do mRNA, disposta no GenBank (acesso: NM_2498.2).
CACATAGCTAGTAAGTTCTAGCTAGCACTGAGTGCTGTGCCCGTGAAATTTATCTACATAGGCTTTCACTTAACCTGCAGACAGAACT
CAGTTAGTCGGGGACAATTTCCCTCAATGTTAACAGCACTGTTCCACCGCAACGTGGAACAACAGCTTTAAAACGTGCTCTTCGTAGG

21
CCCGGCTACTCCAAGAACAGTGCCTCCCGCCAGACCCAGGCGGCTTCCTTCACCCGCAACCCGAGAGACGACCCGCCGGGCCCGCCCC
GCGGAAGCCGCCGGTTGCCAGGCCAAGGAGTGGACTAGGGTCGCCGGGGAAGCGGTTTGGGAGAGCCCATGGTGACTGCGTGAGTGGA
GCCCAGCTGTGTGGATGCCCCAGCATGGATGACTACATGGTCCTGAGAATGATTGGGGAGGGCTCCTTCGGCAGAGCTCTTTTGGTTC
AGCATGAAAGCAGTAATCAGATGTTTGCCATGAAAGAAATAAGGCTTCCCAAGTCTTTCTCTAATACACAGAATTCTAGGAAGGAGGC
TGTTCTTTTAGCCAAAATGAAACACCCTAATATTGTTGCCTTCAAAGAATCATTTGAAGCTGAAGGACACTTGTATATTGTGATGGAA
TACTGTGATGGAGGGGATCTAATGCAAAAGATTAAACAGCAGAAAGGAAAGTTATTTCCTGAAGACATGATACTTAATTGGTTTACCC
AAATGTGCCTTGGAGTAAATCACATTCACAAGAAACGTGTGCTACACAGAGATATCAAGTCCAAGAATATCTTCCTCACTCAGAATGG
AAAAGTGAAATTGGGAGACTTTGGATCTGCCCGTCTTCTCTCCAATCCGATGGCATTTGCTTGTACCTATGTGGGAACTCCTTATTAT
GTGCCTCCAGAAATTTGGGAAAACCTGCCTTATAACAATAAAAGTGACATCTGGTCCTTGGGTTGCATCCTGTATGAACTCTGTACCC
TTAAGCATCCATTTCAGGCAAATAGTTGGAAAAATCTTATCCTCAAAGTATGTCAAGGGTGCATCAGTCCACTGCCGTCTCATTACTC
CTATGAACTTCAGTTCCTAGTCAAGCAGATGTTTAAAAGGAATCCCTCACATCGCCCCTCGGCTACAACGCTTCTCTCTCGAGGCATC
GTAGCTCGGCTTGTCCAGAAGTGCTTACCCCCCGAGATCATCATGGAATATGGTGAGGAAGTATTAGAAGAAATAAAAAATTCGAAGC
ATAACACACCAAGAAAAAAAACAAACCCCAGCAGAATCAGGATAGCTTTGGGAAATGAAGCAAGCACAGTGCAAGAGGAAGAACAAGA
TAGAAAGGGTAGCCATACTGATTTGGAAAGCATTAATGAAAATTTAGTTGAAAGTGCATTGAGAAGAGTAAACAGAGAAGAAAAAGGT
AATAAGTCAGTCCATCTGAGGAAAGCCAGTTCACCAAATCTTCATAGACGACAGTGGGAGAAAAATGTACCCAATACAGCTCTTACAG
CTTTGGAAAATGCATCCATACTCACCTCCAGTTTAACAGCAGAGGACGATAGAGGTGGTTCTGTAATAAAGTACAGCAAAAATACTAC
TCGTAAGCAGTGGCTCAAAGAGACCCCTGACACTTTGTTGAACATCCTTAAGAATGCTGATCTCAGCTTGGCTTTTCAAACATACACA
ATATATAGACCAGGTTCAGAAGGGTTCTTGAAAGGCCCCCTGTCTGAAGAAACAGAAGCATCGGACAGTGTTGATGGAGGTCACGATT
CTGTCATTTTGGATCCAGAGCGACTTGAGCCTGGGCTAGATGAGGAGGACACGGACTTTGAGGAGGAAGATGACAACCCCGACTGGGT
GTCAGAGCTGAAGAAGCGAGCTGGATGGCAAGGCCTGTGCGACAGATAATGCCTGAGGAAATGTTCCTGAGTCACGCTGAGGAGAGGC
TTCACTCAGGAGTTCATGCTGAGATGATCATGAGTTCATGCGACGTATATTTTCCTTTGGAAACAGAATGAAGCAGAGGAAACTCTTA
ATACTTAAAATCGTTCTTGATTAGTATCGTGAGTTTGAAAAGTCTAGAACTCCTGTAAGTTTTTGAACTCAAGGGAGAAGGTATAGTG
GAATGAGTGTGAGCATCGGGCTTTGCAGTCCCATAGAACAGAAATGGGATGCTAGCGTGCCACTACCTACTTGTGTGATTGTGGGAAA
TTACTTAACCTCTTCAAGCCCCAATTTCCTCAACCATAAAATGAAGATAATAATGCCTACCTCAGAGGGATGCTGACCACAGACCTTT
ATAGCAGCCCGTATGATATTATTCACATTATGATATGTGTTTATTATTATGTGACTCTTTTTACATTTCCTAAAGGTTTGAGAATTAA
ATATATTTAATTATGATTTA
Foi desenhado um par de primers para amplificar a região codificante para
NEK3, presente na sequência de mRNA apresentada acima, e que contém 1.521 pb.
Foward: 5’ ATGGATGACTACATGGTCCTGAGAATGATTG 3’
Reverse: 5’ TTATCTGTCGCACAGGCCTTGCCATCCAGCTC 3’
Como também era de interesse clonar a região codificante para o domínio de
cinase de NEK3 no vetor de expressão pET 28a, foram desenhados pares de primers
para tal região, que também continham uma sequência de reconhecimento das enzimas
de restrição SalI (GTCGAC) e BamHI (GGATCC). Tais sequências de reconhecimento
se encontram sublinhadas nas sequências dos primers.
Foward: 5’ GGATCCATGGATGACTACATGGTCCTGAGAATGA 3’
Reverse 5’ GTCGACTTATACGATGCCTCGAGAGAGAAGCGTTGT 3’

22
NEK5
Sequência do mRNA, disposta no GenBank (acesso: NM_199289).
CCCGGATCCTTCCGGGACGCTTCGTTGGCCCCGCGGAGCCGGCGGAGCAGTTATCTGTGGCCACAAGGAAAGTTATTTGTCTCTGTCT
TGGCAAGGCTGGGAGGAAAGTTTTAGCTAAGAACCTCAGCCCATTGGAGACCATGGATAAGTACGATGTGATTAAGGCCATCGGGCAA
GGTGCCTTCGGGAAAGCATACTTAGCTAAAGGGAAATCAGATAGCAAGCACTGTGTCATAAAAGAGATCAATTTTGAAAAGATGCCCA
TACAAGAAAAAGAAGCTTCAAAGAAAGAAGTGATTCTTCTGGAAAAGATGAAACATCCCAACATTGTAGCCTTCTTCAATTCATTTCA
AGAGAATGGCAGGCTGTTTATTGTAATGGAATATTGTGATGGAGGGGATCTCATGAAAAGGATCAATAGACAACGGGGTGTGTTATTT
AGTGAAGATCAGATCCTCGGTTGGTTTGTACAGATTTCTCTAGGACTAAAACATATTCATGACAGGAAGATATTACACAGGGACATAA
AAGCTCAGAACATTTTTCTTAGCAAGAACGGAATGGTGGCAAAGCTTGGGGACTTTGGTATAGCAAGAGTCCTGAATAATTCCATGGA
ACTTGCTCGAACTTGTATTGGAACACCTTACTACCTGTCCCCAGAGATCTGTCAGAATAAACCCTACAACAATAAAACGGATATTTGG
TCTCTTGGCTGTGTCTTATATGAGCTCTGCACACTTAAACATCCTTTTGAGGGTAACAACTTACAGCAGCTGGTTCTGAAGATTTGTC
AAGCACATTTTGCCCCAATATCTCCGGGGTTTTCTCGTGAGCTCCATTCCTTGATATCTCAGCTCTTTCAAGTATCTCCTCGAGACCG
ACCATCCATAAATTCCATTTTGAAAAGGCCCTTTTTAGAGAATCTTATTCCCAAATATTTGACTCCTGAGGTCATTCAGGAAGAATTC
AGTCACATGCTTATATGCAGAGCAGGAGCGCCAGCTTCTCGACATGCTGGGAAGGTGGTCCAGAAGTGTAAAATACAAAAAGTGAGAT
TCCAGGGAAAGTGCCCACCAAGATCAAGGATATCTGTGCCAATTAAAAGGAATGCTATATTGCATAGAAATGAATGGAGACCACCAGC
TGGAGCCCAGAAGGCCAGATCTATAAAAATGATAGAAAGACCCAAAATTGCTGCTGTCTGTGGACATTATGATTATTATTATGCTCAA
CTTGATATGCTGAGGAGGAGAGCCCACAAACCAAGTTATCACCCTATTCCTCAAGAAAATACTGGAGTTGAGGATTACGGTCAGGAAA
CGAGGCATGGTCCATCCCCAAGTCAATGGCCTGCTGAGTACCTTCAGAGAAAATTTGAAGCTCAACAATATAAGTTGAAAGTGGAGAA
GCAATTGGGTCTTCGTCCATCTTCTGCCGAGCCAAATTACAACCAGAGACAAGAGCTAAGAAGTAATGGAGAAGAGCCTAGATTCCAG
GAGCTGCCATTTAGGAAAAACGAAATGAAGGAACAGGAATATTGGAAGCAGTTAGAGGAAATACGCCAACAGTACCACAATGACATGA
AAGAAATTAGAAAGAAGATGGGGAGAGAACCAGAGGAGAACTCAAAAATAAGTCATAAAACCTATTTGGTGAAGAAGAGTAACCTGCC
TGTCCATCAAGATGCATCTGAGGGAGAAGCACCTGTGCAGATGGAATTTCGCTCTTGTTGCCCAGGCTGGAGTGCAATGGCACGATCT
TGGCTCACCGCAACCTCCGCCTCCCAGGACATTGAAAAAGACTTGAAACAAATGAGGCTTCAGAACACAAAGGAAAGTAAAAATCCAG
AACAGAAATATAAAGCTAAGAAGGGGGTAAAATTTGAAATTAATTTAGACAAATGTATTTCTGATGAAAACATCCTCCAAGAGGAAGA
GGCAATGGATATACCAAATGAAACTTTGACCTTTGAGGATGGCATGAAGTTTAAGGAATATGAATGTGTAAAGGAGCATGGAGATTAT
ACAGACAAAGCATTTGAAAAACTTCACTGCCCAGAAGCAGGGTTTTCCACGCAGACTGTAGCTGCTGTGGGAAACAGGAGGCAGTGGG
ATGGAGGAGCGCCTCAGACTCTGCTGCAGATGATGGCAGTGGCCGACATCACCTCCACCTGCCCCACGGGGCCTGACAGTGAGTCTGT
GCTTAGCGTCAGTCGTCAGGAAGGGAAGACCAAGGACCCGTACAGCCCAGTGCTCATCCTGATGTGATAGTCTACTTCTCACTATACA
CCCTATAGATCTTGTATCAGACACTTTCAAATATGTTGTTTTGATATCTCCCTATGCCTAAAGAGACTTGCCTGCTAATTATCCTGGA
CAAATGACCACTCTAGACATGCACATATAAAGGAGGAAAAATCCAGGATGCCACCTTCCTCAGAGTGAGGGAAAATGCGTGAGGAAGG
ACAGCTGACTTGATTTACCTTCCAAGGCCAGCTCAACAACTTTTTTTTCGATTTCATAGGGTCATTTTTTTTTTTTTAATTTTTATTT
TTTATTGATCATTCTTGGGTGTTTCTCGCAGAGGGGGATTTGGCAGGGTCATAGGACAATAGTGGAGGGAAGGTCAGCAGATAAACAA
GTGAACAAAGGTCTCTGGTTTTTCTAGGCAGAGGACCCAGTGGCCTTCCGCAGTGTTTGTGTCCCTGGGTACTTGAGATTAGGGAGTG
GTGATGATTCTTAACGAGCATGCTGCCTTCAAGCATCTGTTTAACAAAGCACATCTTGCACTGCCCTTAATCCATTCATCTCTGAGTG
GACACAGCACATGTTTCAGAGAGCACAGGGTTGGGGGTAAGGTCACCGATTAGCAGGATCCCAAGGCAGAAGAATTTTTCTTAGTACG
GAACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Foram desenhados um par de primers para amplificar a região codificante para
NEK5, presente nesta sequência de mRNA e que apresenta um tamanho de 2.124 pb.
Foward: 5’ ATGGATAAGTACGATGTGATTAAGGCCATC 3’

23
Reverse: 5’ TCACATCAGGATGAGCACTGGGCTGTACGG 3’
Como também era de interesse clonar a região codificante para o domínio de
cinase de NEK5 no vetor de expressão pET 28a, foram desenhados pares de primers
para tal região, que também continham uma sequência de reconhecimento das enzimas
de restrição Eco RI e SalI. Tais sequências de reconhecimento se encontram sublinhadas
nas sequências dos primers a seguir:
Foward: 5’ GAATTCATGGATAAGTACGATGTGATTAAGGCC 3’
Reverse: 5’ GTCGACTCATAAAAAGGGCCTTTTCAAAATGG 3’
3.4 - Amplificação dos genes das NEKs 2, 3 e 5 e da região gênica
correspondente ao domínio de cinase das NEKs 3 e 5 via reação de PCR (Reação
em Cadeia de Polimerase)
3.4.1 - NEK2
Para cada reação de PCR foi utilizado uma mistura contendo aproximadamente
100,0 ng de cDNA vindo de uma biblioteca de cDNA de HELA, 2,5 µL de tampão PCR
10X (Roche, 100 mM de Tris, 500 mM de MgCl2 e 1% de Triton X - 100), 1,0 µL de
cada primer (5 µM), 0,5 µL de solução 2,5 mM de dNTP (Invitrogen) e 0,5 µL da
enzima Taq polimerase (Roche, 1 U/µL), sendo o volume completado para 25,0 µL com
água deionizada autoclavada. Essa reação de PCR foi desenvolvida em termociclador
onde, inicialmente, foi incubada a 94ºC por 5 min, seguida pela realização de 35 ciclos
de amplificação. Cada ciclo consistia nas etapas de separação das fitas a 94°C por 30
segundos, anelamento dos primers (descritos no item 4.2), em que foi testado, em
reações de PCR gradiente, as temperaturas compreendidas entre 50°C-60°C por 30
segundos, e alongamento da fita a 72°C por 1,5 min. Ao final, foi realizado um ciclo a
72°C por 7 min. Os produtos do PCR foram visualizados em gel de agarose 1,0% com
brometo de etídeo (10 µg/mL).
3.4.2 - NEK3
Com o objetivo de amplificar a região codificante para a proteína NEK3 e a
região codificante para apenas seu domínio de cinase, foram realizadas reações de PCR

24
semelhantes às descritas para NEK2 no item anterior, variando-se os pares de primers
(descritos no item 4.2) e o tempo de alongamento da fita. O tempo de alongamento para
amplificar toda a região codificante de NEK3 foi de 2 minutos, e para o domínio de
cinase foi de 1 minuto.
3.4.3 - NEK5
De maneira semelhante às NEKs 2 e 3, foram feitas reações de PCR para tentar
amplificar as regiões codificantes, correspondentes a toda a proteína NEK5 e a
correspondente ao seu domínio de cinase, utilizando-se os primers específicos para tais
regiões já descritos no item 4.2. e um tempo de extensão da polimerização a 72°C de 2,5
min para toda a proteína e 1 min para o domínio de cinase.
3.4.4 - Extração e purificação do DNA em gel de agarose
As bandas correspondentes aos tamanhos esperados para os produtos
amplificados de NEK2, NEK3 e NEK5, proteínas inteiras e domínio de cinase, foram
extraídas do gel de agarose e tiveram seu DNA extraído e purificado do gel pelo
QIAquick Gel Extraction Kit, conforme procedimento indicado pelo fabricante. Ao
final, o DNA purificado foi eluído em 30 µL de água desmineralizada e autoclavada.
3.5 – Reações de clivagem
3.5.1 - Clivagem de NEK2 e da região codificante para o domínio de cinase
de NEK3
As reações foram preparadas utilizando-se 300,0 ng de DNA purificado de
NEK2 e de NEK3 (domínio de cinase). Como descrito no item 4.5.1, NEK2 foi clivada
com Bam HI e PstI para ser inserida no vetor pBTM 116 K usando-se 0,5 µL da enzima
Bam HI (Promega, 10 U/µL), 1,0 µL da enzima PstI (Promega, 10U/µL), 2,0 µL de
tampão H 10X, sendo o volume completado para 20,0 µL com água deionizada
autoclavada. A região amplificada via PCR, codificante para o domínio de cinase de
NEK3, foi clivada com as enzimas de restrição Bam HI e SalI para ser inserida no vetor
pET 28a usando-se 1,0 µL de enzima Bam HI (Promega, 10 U/µL), 1,0 µL da enzima
SalI (Promega, 10 U/µL), 2,0 µL de tampão D 10X, sendo o volume completado para

25
20,0 µL com água deionizada autoclavada. As reações foram incubadas por toda a noite
a 37ºC.
3.5.2- Clivagem dos vetores pBTM 116 K e pET 28a e pFastBacDual
As reações foram preparadas utilizando-se 1,0 µg de DNA plasmidial. pBTM
116 K foi clivado com as enzimas de restrição BamHI (Promega, 10 U/µL) e PstI
(Promega, 10 U/µL) utilizando-se 1,0 µL de cada enzima, 2,0 µL de tampão H 10X,
sendo o volume completado para 20,0 µL com água deionizada autoclavada. pET 28a e
pFast Bac Dual foram clivados com as enzimas Bam HI (Promega, 10 U/µL) e SalI
(Promega, 10 U/µL), sendo utilizado 1,0 µL de cada enzima, 2,0 µL de tampão D 10X,
sendo o volume completado para 20,0 µL com água deionizada autoclavada. As reações
foram incubadas por toda a noite a 37ºC.
3.6 – Reações de ligação
3.6.1- Ligação dos fragmentos amplificados de NEK 2, 3 e 5 em pGEM T
easy vector
Os fragmentos gênicos amplificados, correspondentes às regiões codificantes de
NEK 2, 3 e 5, foram eluídos do gel de agarose, como descrito no item 4.4, quantificados
em nanodrop e ligados ao vetor pGEM T easy (Promega) de acordo com as
recomendações do fabricante. A reação foi incubada a 16°C por 14 horas.
3.6.2- Ligação dos fragmentos amplificados referente ao domínio de cinase
de NEK3 ao vetor pET 28a e ao vetor pFastBacDual e de NEK2 ao vetor pBTM
116 K
Os fragmentos amplificados, digeridos e purificados, de NEK2 foi ligado ao
vetor pBTM 116 K e da região codificante para o domínio catalítico de NEK3, foi
ligado aos vetores e pET 28a e a pFastBacDual, utilizando-se, 100 ng de DNA
plasmidial e 300 ng do gene de interesse a ser inserido, 1,0 µL de tampão de reação 10X
(Fermentas - Tris-HCl pH 7,6, MgCl2 10 mM, ATP 1 mM, DTT 1 mM e 25 de
polietileno glicol-8000) e 1,0 µL de T4 DNA ligase (Fermentas - 1,0 U/mL). As reações
foram incubadas a temperatura de 16°C por 14 horas. Como a ligação de NEK3

26
domínio de cinase ao vetor pEt 28a não funcionou na proporção de 1:3 (um de vetor
para três de inserto) ,foram tentadas várias outras proporções, como as de 1:2 e 1:5.
3.7 – Transformação bacteriana
3.7.1 - Choque térmico
A partir de cada reação de ligação, descritas no item 4.6, foi realizada a
transformação de células competentes de E. coli DH 5α e de BL 21. Após a
centrifugação da reação de ligação por um minuto a 14.000 × g em microcentrífuga
Eppendorff, foram adicionados 5 µL da solução em 100 µL de solução de células
competentes DH 5α. A mistura foi incubada em gelo por 20 minutos. Em seguida, foi
realizado um choque térmico nas células, a 42ºC por 90 segundos, retornando-as para o
gelo por mais 2 minutos, para que elas incorporassem o plasmídeo. Foram adicionados
800 µL de meio SOC às células, e feita a incubação por uma hora a 37ºC, sob agitação a
200 rpm. O material foi centrifugado e ressuspendido em 100 µL do mesmo meio. Ao
final, 100 µL da cultura de transformação foram plaqueados em placas contendo 20,0
g/L de LB (Luria-Bertaini) e 50 µg/mL de ampicilina. As placas foram incubadas a
37ºC, por um período de 12 horas. Para as transformações em que a reação de ligação
foi feita usando-se como vetor o pGEM T easy, foi adicionado às placas, X-gal 20
µg/mL. Este protocolo representa uma adaptação feita a partir de Sambrook et al
(1989).
3.7.2- Eletroporação
Uma vez que nem todas as transformações, via choque-térmico, funcionaram de
maneira eficiente, algumas reações de ligação foram adicionadas a alíquotas de células
eletrocompetentes das cepas JM109 e BL 21 de E. coli. Foram adicionados 5 µL da
reação de ligação a 100 µL de solução de células eletrocompetentes e a mistura foi
incubada no gelo por 15 minutos. Em seguida, a mistura reacional foi disposta em
cubeta para eletroporação, de 0,2 cm (Biorad) previamente resfriadas no gelo, e
submetidas á um pulso de 2,5 kV, 25 µF e 200 Ω em eletroporador (Biorad). Após o
pulso, foram adicionados 800 µL de meio SOC às células, e feita a incubação por uma
hora a 37ºC, sob agitação a 200 rpm. O material foi centrifugado e ressuspendido em

27
100 µL do mesmo meio. Ao final, 100 µL da cultura de transformação foram
plaqueados em placas contendo 20,0 g/L de LB (Luria-Bertaini) e 50 µg/mL de
ampicilina. As placas foram incubadas a 37ºC por um período de 12 horas. Para as
transformações em que a reação de ligação foi feita usando-se como vetor o pGEM T
easy, foi adicionado às placas X-gal 20 µg/mL (Sambrook et al, 1989).
3.8 - Seleção das colônias de E. coli transformantes e recombinantes
Após incubação das placas contendo as células transformadas, foi feita a seleção
de 25 colônias brancas por placa, das quais foi feita uma réplica de cada. A seleção das
colônias recombinantes e transformantes foi feita por meio de PCR de colônias usando
como molde, na PCR descrita no item 4.3, a colônia de interesse ao invés de DNA.
Cada colônia recombinante selecionada contendo o fragmento de interesse foi
crescida em um tubo com meio líquido, contendo 5,0 mL de LB (20 g/L) e 5,0 µL de
ampicilina (50 mg/mL) ou canamicina (10 mg/mL). Os tubos foram incubados a 37ºC
por 12 horas, sob agitação de 200 rpm.
A extração do DNA plasmidial contendo um dos fragmentos foi feita utilizando
o Spin Miniprep Kit (QIAGEN), conforme procedimento indicado pelo fabricante. Ao
final, o DNA foi eluído em 50 µL de água desmineralizada estéril.
3.9 - Sequenciamento de DNA e análise das sequências de nucleotídeos
Assim que foram obtidos todos os plasmídeos, estes foram submetidos a reações
de sequenciamento para a verificação da integridade das ORFs (Open Reading Frame),
permitindo prosseguir aos passos de subclonagem do domínio de cinase de NEK3 em
vetores de expressão pET 28a, para posterior transformação de E. coli BL 21, e de
NEK2 no vetor pBTM 116 K, para a realização da metodologia de Duplo-Híbrido em
leveduras.
3.10 – Transformação da levedura Saccharyomyces cerevisae L40
Com o objetivo de realizar o sistema de Duplo-Híbrido em leveduras para
encontrar possíveis parceiros de interação proteína-proteína de NEK2 e de transcritos de
uma biblioteca de cDNA de HELA, inicialmente foi feita a transformação de L40 com a
construção pBTM 116 K/NEK2.

28
Uma colônia grande e isolada de L40 foi inoculada em 50 µL de água
desmineralizada estéril, foi feito vórtex do inóculo por 20 segundos, e estes 50 µL foi
inoculado em 50 mL de meio YPD (1% de extrato de levedura, 2% de peptona e 2% de
glicose). O inóculo foi então submetido à rotação de 200 rpm, 30°C por 24 horas. Após
este período, foi feita uma alíquota de 1 mL deste inóculo para ser usada em cada
transformação, que foi centrifugada a 5.000 rpm por 3 minutos a temperatura ambiente.
O sobrenadante foi descartado e as células ressuspendidas em 200 µL de tampão de
transformação (1.340 µL de PEG 50%, 200 µL de acetato de lítio 2M, 200 µL de TE
10X e 200 µL de DTT 1M). As células foram submetidas ao vórtex e adicionado 5 µL
de DNA de esperma de salmão (DNA carreador) 10 mg/mL, previamente desnaturado a
95°C por 10 min. Foi acrescentado 1,0 µL da construção pBTM 116 K/NEK2 (1 µg/µL)
e como controles positivos da transformação, foi utilizado 1,0 µL do vetor pBTM 116 K
(1,3 1µg/µL) e 2,5 µL do vetor pACT (800 ng/µL), como controles positivos de auto-
ativação, foram usadas, numa mesma transformação, 2,0 µL das construções pBTM 116
K/FEZ (1-392) (1,0 µg/µL) e 2,5 do vetor pACT (800 ng/µL), e também foi feito um
controle negativo de transformação utilizando-se água ao invés de DNA e um controle
negativo de interação co-transformando as células com 1,0 µL do vetor pBTM 116 K
(1,3 1µg/µL) e 2,5 do vetor pACT (800 ng/ µL). Após a adição do DNA às células,
estas foram levadas ao banho-maria a 45°C por 40 min e submetidas a uma agitação
eventual. Após este período, as células foram centrifugadas a 3.000 rpm por 5 min a
temperatura ambiente, foram descartados 100 µL do sobrenadante e plaqueados em
meio seletivo SD-W (0,14% de YNB sem aminoácidos e sem sulfato de amônio, 0,5%
de (NH4)2SO4, 2% de glicose, 0,002% de adenina, 0,01% de leucina, 0,002% de
histidina e 1,8% de ágar bacteriológico) as que continham apenas o plasmídeo pBTM
116 K e em meio SD-WL (0,14% de YNB sem aminoácidos e com sulfato de amônio,
0,5% de (NH4)2SO4, 2% de glicose, 0,002% de adenina, 0,002% de histidina e 1,8% de
ágar bacteriológico) as que continham os plasmídeos pBTM 116 K e pACT. As placas
foram mantidas em estufa a 30°C por 3 a 4 dias.
Uma vez que o protocolo acima descrito não de mostrou eficiente para a
incorporação do plasmídeo pBTM116 K-NEK2, foi feita uma tentativa de produzir
células L40 eletrocompetentes e realizar a eletroporação das mesmas como descrito para
transformação de células de Pichia pastoris (Pichia Expression Kit- Invitrogen) .

29
Inicialmente uma colônia isolada de L40 foi inoculada em 200 mL de meio YPD em
erlenmeyer de 2L de capacidade e incubada a 30ºC por 24 h sob agitação de 200 rpm.
Após este período, foram feitas medidas espectrofotométricas, de D.O. 600, a fim de
verificar se as células estavam na fase estacionária (D.O.600 entre 1,3 e 1,5) para que se
pudesse iniciar o preparo das células eletrocompetentes. Posteriormente, as células
foram coletadas por centrifugação a 1.500 g por 5 min a 4ºC e ressuspensas em 250 mL
de água destilada estéril gelada. Seguidamente foram submetidas à centrifugação nas
mesmas condições anteriores e ressuspensas em 125 mL de água destilada estéril
gelada. As células foram centrifugadas e ressuspensas em 10 mL de sorbitol 1 M
gelado, posteriormente foram centrifugadas e ressuspensas em 1 mL de sorbitol 1 M
gelado para um volume final de aproximadamente 1,5 mL. As células competentes
foram utilizadas imediatamente na transformação, pois apesar das células poderem ser
congeladas, sua eficiência diminui significativamente. Para cada transformação, 80 µL
de células eletrocompetentes foram homogeneizadas com 1,0 µL da construção pBTM
116 K/NEK2 (1 µg/µL) e como controles positivos da transformação, foi utilizado 1,0
µL do vetor pBTM 116 K (1,3 1µg/µL) e 2,5 µL do vetor pACT (800 ng/µL), como
controles positivos de auto-ativação, foram usadas, numa mesma transformação, 2,0 µL
das construções pBTM 116 K/FEZ (1-392) (1,0 µg/µL) e 2,5 do vetor pACT (800
ng/µL), e também foi feito um controle negativo de transformação utilizando-se água ao
invés de DNA e um controle negativo de interação co-transformando as células com 1,0
µL do vetor pBTM 116 K (1,3 1µg/µL) e 2,5 do vetor pACT (800 ng/ µL). A mistura
foi transferida para uma cubeta de 0,2 cm (Biorad) e incubada em gelo por 5 min. As
células foram submetidas a eletroporação de acordo com os seguintes parâmetros: 1,5
kV, 25 µF e 400 Ω. Imediatamente foi adicionado 1 mL de sorbitol 1M gelado, e o
conteúdo da cubeta foi transferido para um tubo de microcentrífuga. Um volume de
200-600 µL foi semeado em placas de Petri contendo meio seletivo as placas foram
levadas a estufa a 30°C por 3-4 dias.
3.11 – Ensaio de mini-indução da expressão de pET 28a/NEK3 domínio de
cinase
Com o objetivo de verificar se havia expressão do domínio de cinase de NEK3
nos clones de E. coli BL 21 que continham a construção pET 28a/NEK3 domínio de

30
cinase, foi feito um teste de mini-indução de expressão. Inicialmente, foi inoculada uma
colônia de E. coli BL 21 em 5 mL de meio LB (Lauria-Bertani, 20g/L) e este inóculo foi
submetido à agitação de 200 rpm a 37°C até que atingisse a D.O. (densidade óptica)
A600nm entre 0,6 e 0,8. Atingida a D.O., o pré-inóculo foi dividido em dois para que uma
amostra pudesse ter a expressão da proteína recombinante induzida e a outra não, e
então foram adicionados ao pré-inóculo que seria induzido, 2,5 µL de IPTG 1M e
ambos os inóculos, com e sem IPTG, foram submetidos à agitação de 200 rpm por 37°C
por toda a noite. A separação das proteínas extraídas foi realizada por meio de SDS-
PAGE no tampão de corrida (Tris-HCl 25 mM; glicina 200 mM; EDTA 1 mM e SDS
3,5 mM), com voltagem constante de 50 V. A concentração de acrilamida e bis-
acrilamida utilizadas foram de 12,5% (p/v) (Laemmli et al, 1970). Após a eletroforese,
o gel foi revelado pela incubação em solução corante [metanol 45% (v/v), etanol 95%
(v/v) e Comassie Brilhant Blue R250 0,001% (p/v)], por 12 horas e, em seguida, com
uma solução descorante [metanol 25% (v/v) e ácido acético glacial 7,5% (v/v)].
31
4 – Resultados e discussão
4.1 - Amplificação dos genes das NEKs 2, 3 e 5 e da região gênica
correspondente ao domínio de cinase de NEK3 via reação de PCR (Reação em
Cadeia de Polimerase)
4.1.1- NEK2
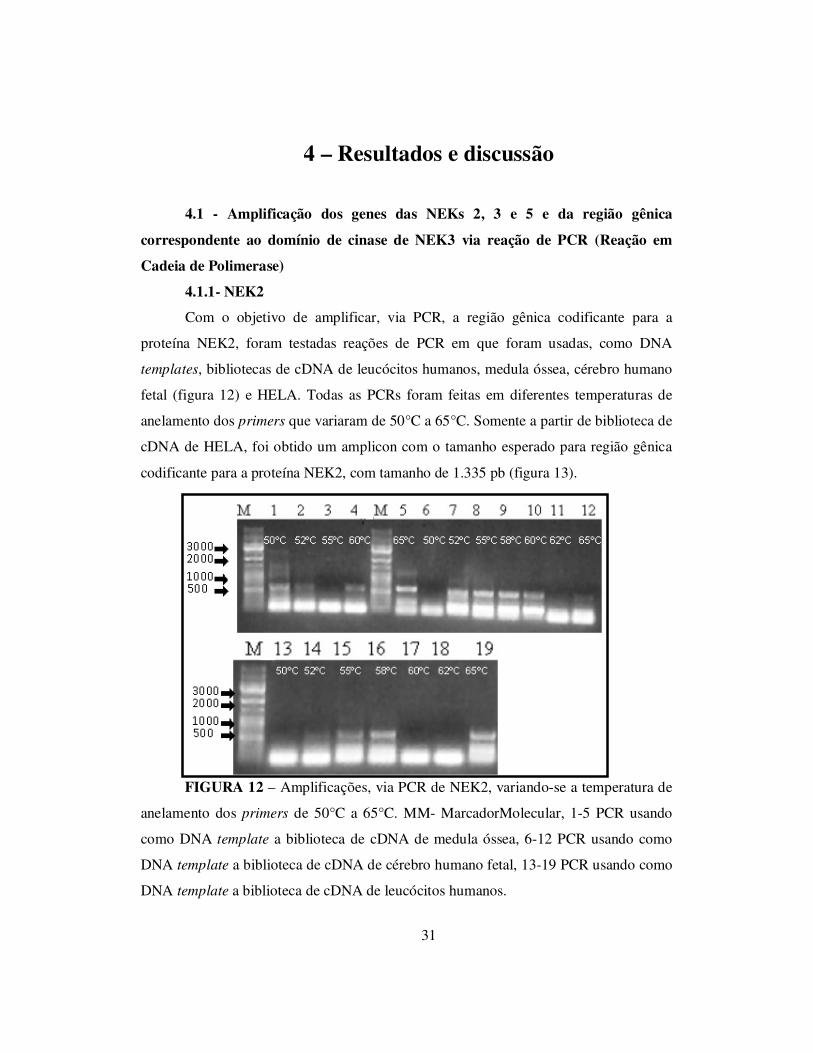
Com o objetivo de amplificar, via PCR, a região gênica codificante para a
proteína NEK2, foram testadas reações de PCR em que foram usadas, como DNA
templates, bibliotecas de cDNA de leucócitos humanos, medula óssea, cérebro humano
fetal (figura 12) e HELA. Todas as PCRs foram feitas em diferentes temperaturas de
anelamento dos primers que variaram de 50°C a 65°C. Somente a partir de biblioteca de
cDNA de HELA, foi obtido um amplicon com o tamanho esperado para região gênica
codificante para a proteína NEK2, com tamanho de 1.335 pb (figura 13).
FIGURA 12 – Amplificações, via PCR de NEK2, variando-se a temperatura de
anelamento dos primers de 50°C a 65°C. MM- MarcadorMolecular, 1-5 PCR usando
como DNA template a biblioteca de cDNA de medula óssea, 6-12 PCR usando como
DNA template a biblioteca de cDNA de cérebro humano fetal, 13-19 PCR usando como
DNA template a biblioteca de cDNA de leucócitos humanos.

32
FIGURA 13 – Amplificações, via PCR de NEK2, variando-se a temperatura de
anelamento dos primers de 50°C a 65°C. M- MarcadorMolecular, 1-6 PCR usando
como DNA template a biblioteca de cDNA de HELA.
Visando clonar em pGEM T easy (Promega) os fragmentos gênicos de NEK2,
para avaliar a integridade de tal sequência amplificada, via sequenciamento, foram
feitas reações de PCR, em grande quantidade, nas condições de melhor amplificação (a
partir de biblioteca de cDNA de HELA e usando a temperatura de anelamento dos
primers de 55°C), e que foram aplicadas (figura 14) e extraídas de gel de agarose, como
descrito no item 3.4.4.

33
FIGURA 14 – 1- Amplificação, via PCR de NEK2, em grande quantidade, a
partir de biblioteca de cDNA de HELA e com temperatura de anelamento dos primers
de 55°C, MM- MarcadorMolecular.
4.1.2 - NEK3
Com o objetivo de amplificar, via PCR, a região gênica codificante para toda a
proteína NEK3, foram testadas reações de PCR em que foram usadas, como DNA
templates, bibliotecas de cDNA de leucócitos humanos (figura 15) e de cérebro humano
fetal (figura 16). Todas as PCRs foram feitas em diferentes temperaturas de anelamento
dos primers que variaram de 50°C a 65°C. Em todas as reações de PCR foi observado a
amplificação de um fragmento com o tamanho esperado de 1.521 pb.
FIGURA 15 – MM- Marcador Molecular, 1-4 Amplificações, via PCR, da
região gênica codificante para toda a proteína NEK3 a partir de bibliotecas de cDNA de
cérebro humano fetal, variando-se a temperatura de anelamento dos primers de 50°C a
65°C. Sendo obtido, em todas as temperaturas,o amplicon no tamanho esperado de
1.521pb.
34
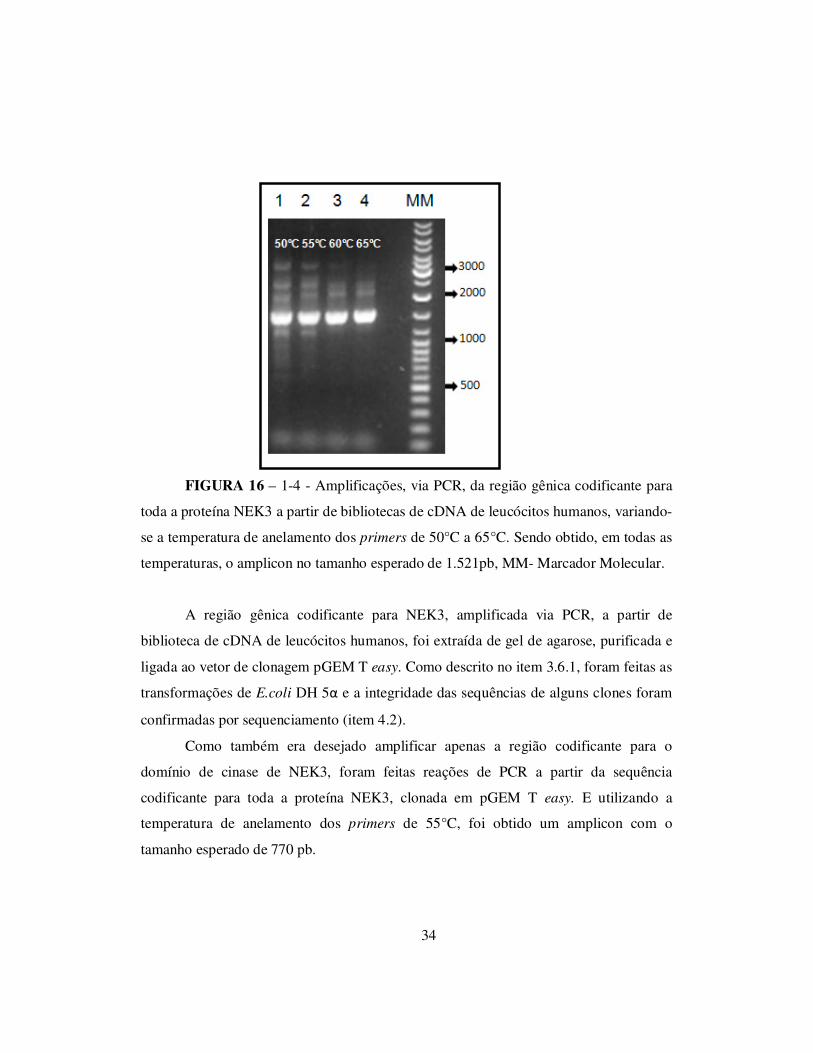
FIGURA 16 – 1-4 - Amplificações, via PCR, da região gênica codificante para
toda a proteína NEK3 a partir de bibliotecas de cDNA de leucócitos humanos, variando-
se a temperatura de anelamento dos primers de 50°C a 65°C. Sendo obtido, em todas as
temperaturas, o amplicon no tamanho esperado de 1.521pb, MM- Marcador Molecular.
A região gênica codificante para NEK3, amplificada via PCR, a partir de
biblioteca de cDNA de leucócitos humanos, foi extraída de gel de agarose, purificada e
ligada ao vetor de clonagem pGEM T easy. Como descrito no item 3.6.1, foram feitas as
transformações de E.coli DH 5α e a integridade das sequências de alguns clones foram
confirmadas por sequenciamento (item 4.2).
Como também era desejado amplificar apenas a região codificante para o
domínio de cinase de NEK3, foram feitas reações de PCR a partir da sequência
codificante para toda a proteína NEK3, clonada em pGEM T easy. E utilizando a
temperatura de anelamento dos primers de 55°C, foi obtido um amplicon com o
tamanho esperado de 770 pb.
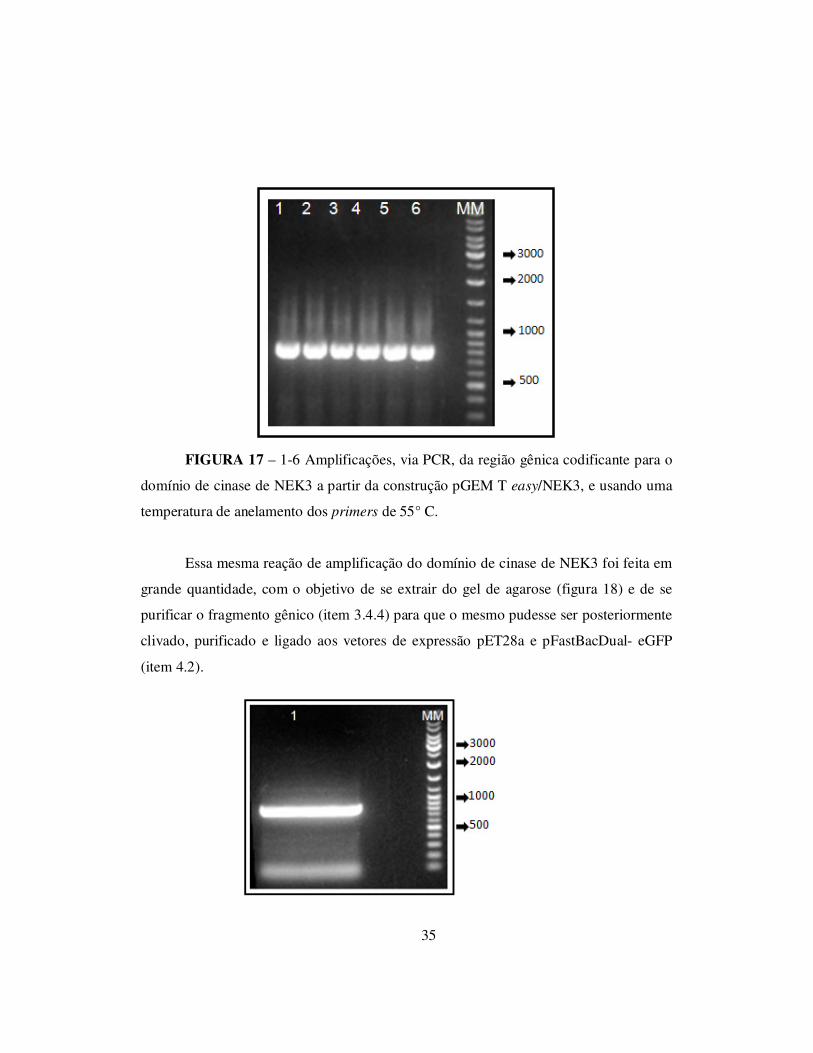
35
FIGURA 17 – 1-6 Amplificações, via PCR, da região gênica codificante para o
domínio de cinase de NEK3 a partir da construção pGEM T easy/NEK3, e usando uma
temperatura de anelamento dos primers de 55° C.
Essa mesma reação de amplificação do domínio de cinase de NEK3 foi feita em
grande quantidade, com o objetivo de se extrair do gel de agarose (figura 18) e de se
purificar o fragmento gênico (item 3.4.4) para que o mesmo pudesse ser posteriormente
clivado, purificado e ligado aos vetores de expressão pET28a e pFastBacDual- eGFP
(item 4.2).

36
FIGURA 18 – Amplificação, via PCR, em grande quantidade, da região gênica
codificante para o domínio de cinase de NEK3, a partir da construção pGEM T
easy/NEK3, e usando uma temperatura de anelamento dos primers de 55°C.
4.1.3- NEK5
Com o objetivo de amplificar, via PCR, a região gênica codificante para toda a
proteína NEK5, foram testadas reações de PCR em que foram usadas, como DNA
templates, bibliotecas de cDNA de leucócitos humanos, cérebro humano fetal, medula
óssea, HELA e mama. As reações de PCR foram feitas em diferentes temperaturas de
anelamento dos primers, que variaram de 50°C a 65°C.
FIGURA 19 – Amplificação, via PCR da região gênica codificante para toda a
proteína NEK5 a partir de biblioteca de cDNA de 1- leucócitos humanos, 2- cérebro
fetal humano, 3- medula óssea, MM- Marcador Molecular. Todas as reações foram
feitas com temperatura de anelamento dos primers de 55°C.
As bandas que apresentavam tamanhos próximos ao esperado para a região
gênica que codifica toda a proteína NEK5, 2.124 pb, foram extraídas do gel de agarose,
purificadas (item 3.4.4) e clonadas em pGEM T easy. Como nos clones sequenciados
que continham os segmentos gênicos com tamanho próximo a 2.124 pb não foi
confirmada a presença de NEK5, foram feitas outras tentativas de clonar tal gene.
Inicialmente, foram feitas reações de PCR utilizando a biblioteca de cDNA de
leucócitos humanos, variando-se a temperatura de anelamento dos primers de 50°C a
65°C (figura 20).
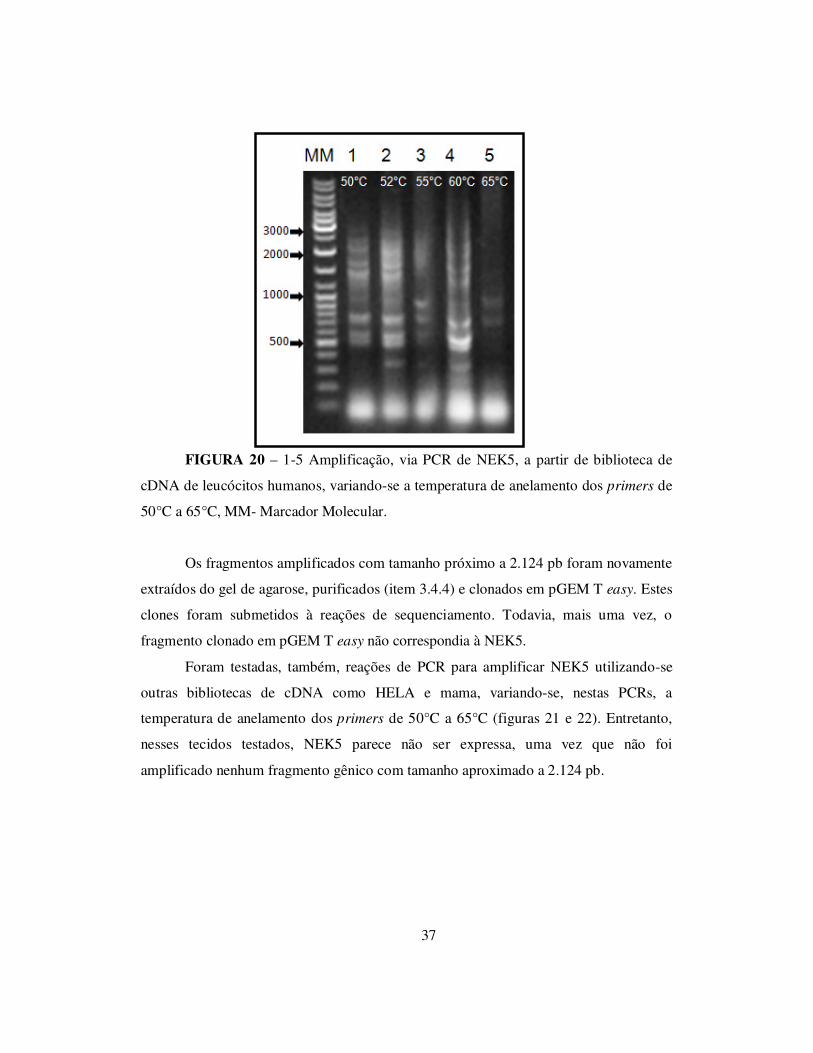
37
FIGURA 20 – 1-5 Amplificação, via PCR de NEK5, a partir de biblioteca de
cDNA de leucócitos humanos, variando-se a temperatura de anelamento dos primers de
50°C a 65°C, MM- Marcador Molecular.
Os fragmentos amplificados com tamanho próximo a 2.124 pb foram novamente
extraídos do gel de agarose, purificados (item 3.4.4) e clonados em pGEM T easy. Estes
clones foram submetidos à reações de sequenciamento. Todavia, mais uma vez, o
fragmento clonado em pGEM T easy não correspondia à NEK5.
Foram testadas, também, reações de PCR para amplificar NEK5 utilizando-se
outras bibliotecas de cDNA como HELA e mama, variando-se, nestas PCRs, a
temperatura de anelamento dos primers de 50°C a 65°C (figuras 21 e 22). Entretanto,
nesses tecidos testados, NEK5 parece não ser expressa, uma vez que não foi
amplificado nenhum fragmento gênico com tamanho aproximado a 2.124 pb.
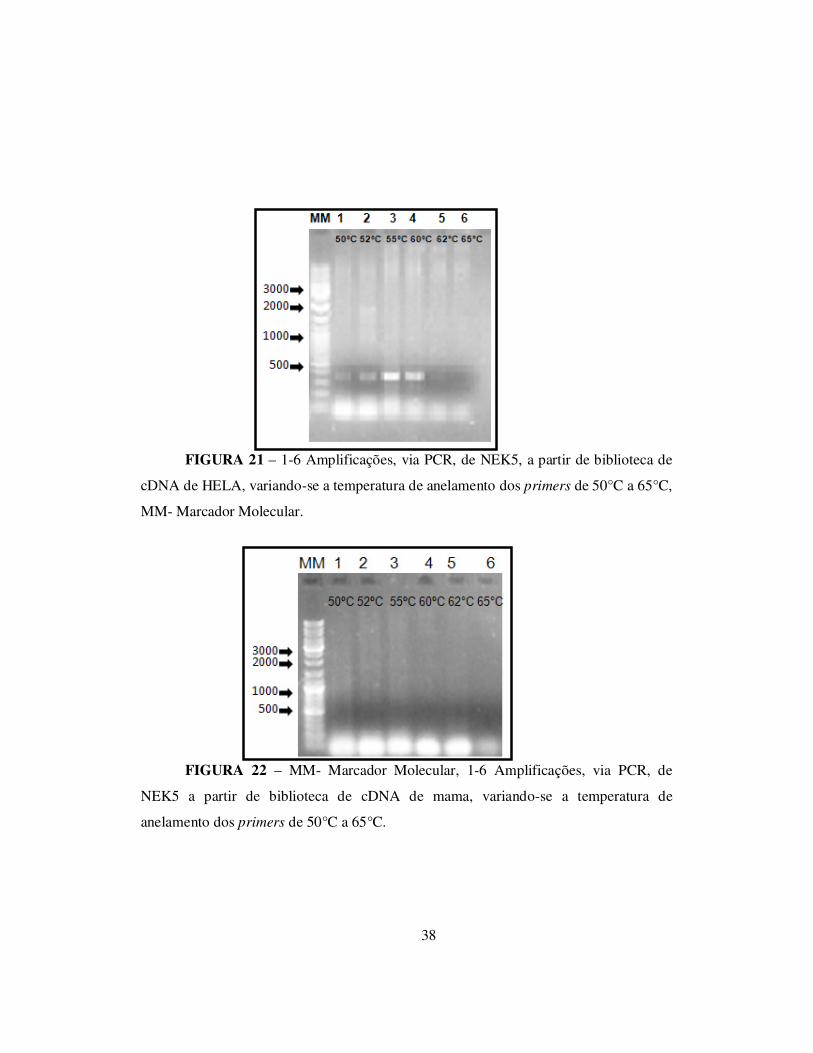
38
FIGURA 21 – 1-6 Amplificações, via PCR, de NEK5, a partir de biblioteca de
cDNA de HELA, variando-se a temperatura de anelamento dos primers de 50°C a 65°C,
MM- Marcador Molecular.
FIGURA 22 – MM- Marcador Molecular, 1-6 Amplificações, via PCR, de
NEK5 a partir de biblioteca de cDNA de mama, variando-se a temperatura de
anelamento dos primers de 50°C a 65°C.

39
4.2 – Clonagens, sequenciamentos e subclonagens
Como descrito dos itens 3.4 ao 3.9, as reações de PCR das NEKs 2, 3 e 5 foram
extraídas e purificadas, a partir do gel de agarose; ligadas ao vetor de clonagem pGEM
T easy; e as reações de ligação foram usadas para transformar cepas de clonagem de E.
coli (JM109 e DH 5α) por choque térmico e por eletroporação (em situações em que a
transformação, via choque térmico, não se mostrou eficiente). Foram feitas reações de
PCR de colônias para saber quais colônias eram transformantes e recombinantes para as
construções pGEM T easy/NEK2, pGEM T easy/NEK3 e pGEM T easy/NEK5 (figuras
23, 24, 25 e 26).
FIGURA 23 – 1-19 Reações de PCR de colônias de 19 clones de E. coli DH 5α
contendo a construção pGEM T easy/NEK2, MM – Marcador Molecular.
FIGURA 24 – 1-9 Reações de PCR de colônias de 9 clones de E. coli DH 5α
contendo a construção pGEM T easy/NEK3, MM – Marcador Molecular.

40
FIGURA 25 – 1-9 Reações de PCR de colônias de 9 clones de E. coli DH 5α
contendo a construção pGEM T easy/NEK5. Essa construção foi feita a partir de um
inserto cujo tamanho era 2.124 pb e que fora amplificado a partir de biblioteca de cDNA
de cérebro humano fetal; MM – Marcador Molecular.
FIGURA 26 - 1-9 Reações de PCR de colônias de 9 clones de E. coli DH 5α
contendo a construção pGEM T easy/NEK5. Essa construção foi feita a partir de um
inserto cujo tamanho era 2.124 pb e que fora amplificado a partir de biblioteca de cDNA
de leucócitos humanos; MM – Marcador Molecular.

41
Os clones 1-6 confirmados pela PCR de colônias, contendo a construção pGEM
T easy/NEK2, foram sequenciados utilizando-se o primer T7 promoter que possui
anelamento no vetor de clonagem pGEM T easy. A sequência obtida (figura 27) foi
alinhada contra o banco de dados do NCBI (National Center for Biotechnology
Information) utilizando programa BLAST (Basic Local Alignment Search Tool) para
que fosse verificada e confirmada sua integridade.
FIGURA 27 – Sequência obtida para o clone 1 contendo a construção pGEM T
easy/NEK2. Foi feito o sequenciamento apenas com o primer T7 promoter, sendo
obtida uma sequência cujo tamanho é 1.015 pb.
FIGURA 28 – Análise de similaridade da sequência obtida para NEK2 contra o
banco de dados BLAST.

42
FIGURA 29 - Alinhamento da sequência obtida para NEK2 com a sequência de
NEK2 disposta no banco de dados do NCBI utilizadondo BLAST. Existem algumas
regiões nas quais é possível observar GAPs; e outras regiões, nas quais é possível

43
observar que a base presente em uma sequência é diferente da outra, mas somente em
regiões de fim das reações de sequenciamento.
Os clones 1-6 confirmados pela PCR de colônias, contendo a construção pGEM
T easy/NEK3, foram sequenciados utilizando-se, da mesma forma que para a
construção pGEM T easy/NEK2, o primer T7 promoter que possui anelamento no vetor
de clonagem pGEM T easy. A sequência obtida (figura 30) foi alniada por BLAST
(Basic Local Alignment Search Tool) para que fosse verificada e confirmada sua
integridade.
FIGURA 30 – Sequência obtida para o clone 1 contendo a construção pGEM T
easy/NEK3. Foi feito o sequenciamento apenas com o primer T7 promoter, sendo
obtida uma sequência cujo tamanho é 1.097 pb.
FIGURA 31 – Análise de similaridade da sequência obtida para NEK3 contra o
banco de dados NCBI utilizando o programa BLAST.

44
FIGURA 32 - Alinhamento da sequência obtida para NEK3 com a sequência de
NEK3 disposta no banco de dados do NCBI (National Center for Biotechnology
Information) utilizando programa BLAST. Como evidenciado pela figura, a sequência
obtida é idêntica à do banco de dados BLAST.
Para NEK5, todas as sequências obtidas, dos sequenciamentos dos clones
contendo a contrução pGEM T easy/NEK5, não correspondiam à NEK5 humana
presente em banco de dados.
Uma vez que as sequências de NEK2 e NEK3 estavam com correta orientação
das frames, foram escolhidos os clones 1 das construções pGEM T easy/NEK2 e pGEM
T easy/NEK3 para se prosseguir com as subclonagens.
O DNA plasmidial contendo a construção pGEM T easy/NEK2 foi clivado com
as enzimas de restrição PstI (Promega) e BamHI (Promega), como descrito no item 3.5;

45
e o inserto liberado, correspondente a NEK2, foi extraído do gel de agarose, purificado,
quantificado e ligado ao vetor pBTM 116 K previamente digerido e purificado com PstI
e BamHI (itens 3.5 e 3.6). Foram feitas as transformações por eletroporação da cepa
JM109 de E. coli, e 6 colônias transformantes foram escolhidas para extrair o DNA
plasmidial e proceder aos testes de confirmação da subclonagem que, neste caso, foram
a digestão da construção pBTM 116 K/NEK2 com as enzimas de restrição BamHI
(Promega) e Hind III (Promega), já que estas possuíam tampões compatíveis (o tampão
de BamHI permite atividade de 100% de Hind III) de modo que fosse possível realizar a
digestão dupla, o que não era o caso de PstI e BamHI. Analisando o mapa físico do
vetor pBTM 116 K, é possível notar que se o vetor vazio for digerido com BamHI e
Hind III, liberará um fragmento de 859 pb e, caso NEK2 esteja clonada, o fragmento
liberado apresentará 2.159 pb. Foi verificado que, em todos os seis clones, era liberado,
após a digestão, um fragmento de DNA com tamanho aproximado de 2.159 pb, o que
confirma a clonagem de NEK2 em pBTM 116 K (figura 33).
FIGURA 33 – 1-6 Reações de clivagens das construções pBTM 116 K/NEK2
com BamHI e Hind III. MM- Marcador Molecular. As reações foram incubadas a 37°C
por 4 horas.
Outro teste foi feito para a confirmação da clonagem de NEK2 em pBTM 116 K,
em que o DNA plasmidial de 3 dos 6 clones foram submetidos à reações de PCR,
utilizando os primers específicos para amplificar NEK2. Como controle negativo das
reações de PCR, foi utilizado, em triplicata, o DNA plasmidial do vetor pBTM 116 K
vazio (figura 34).

46
FIGURA 34 – MM- Marcador Molecular, 1-3 PCR do DNA plasmidial de
pBTM 116 K/NEK2 dos clones 1, 2 e 3, respectivamente, 4-6 PCR, em triplicata, do
DNA plasmidial de pBTM 116 K vazio.
Foram extraídos os DNAs plasmidiais dos clones 1, 2 e 3, e estes foram
submetidos à reações de sequenciamento para a confirmação da clonagem de NEK2 em
pBTM 116 K. Todavia, tais reações de sequenciamento, utilizando-se os primers T7
promoter e os primers específicos para NEK2, não funcionaram.
O DNA plasmidial do clone 1 foi usado para as transformações, por choque
térmico e eletroporação da cepa L40 de S. cerevisae (item 3.10), para a realização do
Duplo-Híbrido.
O DNA plasmidial contendo a construção pGEM T easy/NEK3 foi utilizado
como DNA template para a amplificação, via PCR, do domínio de cinase de NEK3.
Após a amplificação, o fragmento gênico correspondente ao domínio de cinase de
NEK3 (770 pb) foi extraído do gel de agarose, purificado (item 4.1.2) e clivado com as
enzimas de restrição Bam HI (Promega) e SalI (Promega), e depois, ligado aos vetores
de expressão pET 28a e pFastBacDual-eGFP, previamente digeridos com as mesmas
enzimas e purificados. Foram feitas as transformações por choque térmico da cepa DH
5α de E. coli com as construções pET 28a/NEK3 kd e pFastBacDual-eGFP/NEK3 kd.

47
Entretanto, só foram conseguidas colônias transformantes e recombinantes, confirmadas
via reações de PCR de colônias, para a construção pET28a/NEK3 kd (figura 35).
FIGURA 35 – MM- Marcador Molecular, 1-11 Reações de PCR de colônias das
colônias transformantes para confirmar quais clones continham a construção pET
28a/NEk3 kd.
Foram extraídos os DNAs plasmidiais dos clones 1, 2 e 5, e estes foram
submetidos à reações de sequenciamento para a confirmação da clonagem do domínio
de cinase de NEK3 no vetor de expressão pET 28a. Todavia, tais reações de
sequenciamento, utilizando-se os primers T7 promoter e os primers específicos para o
domínio de cinase de NEK3, não funcionaram. Para tentar confirmar tal clonagem, foi
feito, também, a digestão dos DNAs plasmidiais dos clones 1, 2 e 5 com as enzimas de
restrição Bam HI e XbaI (Promega), pois estas possuíam tampões compatíveis (o
tampão de BamHI permite atividade de 100% de XbaI) de modo que fosse possível
realizar a digestão dupla, o que não era o caso de SalI e BamHI. Analisando o mapa
físico do vetor pET 28a, é possível notar que se o vetor vazio for digerido com BamHI e
XbaI, o vetor será apenas linearizado, pois ambas as enzimas estão no sítio polilinker; e
caso o domínio de cinase de NEK3 esteja clonado, o fragmento liberado apresentará em
torno de 770 pb. Foi verificado que nos clones 1 e 2 era liberado, após a digestão, um
fragmento de DNA com tamanho aproximado de 770 pb (figura 36).

48
FIGURA 36 – 1, 2 e 5 Reações de clivagens das construções pET 28a/NEK3 kd
com BamHI e XbaI. MM- Marcador Molecular. As reações foram incubadas a 37°C por
4 horas.
Foram feitas transformações de E. coli BL 21 DE 3 por choque térmico e
eletroporação com os DNAs plasmidiais do vetor pET 28a vazio e dos clones 1 e 2, para
que fossem feitos ensaios de mini-indução da expressão (item 3.11).
4.3 – Ensaio de mini-indução da expressão de pET 28a/NEK3 domínio de
cinase
As colônias transformantes de E. coli BL 21 DE 3, contendo as construções pET
28a/NEK3 e contendo apenas o pET 28a vazio, foram inoculadas em LB até D.O de 0,6
e, logo após as frações, foram divididas em dois tubos cada, em que, em um deles, a
expressão da proteína recombinante foi induzida pela adição de IPTG e no outro não.
Ambos os tubos foram incubados a 37°C por 200 rpm por toda a noite. No dia seguinte,
a separação das proteínas do lisado bacteriano foi feita por SDS-PAGE (item 3.11)
(figura 37).
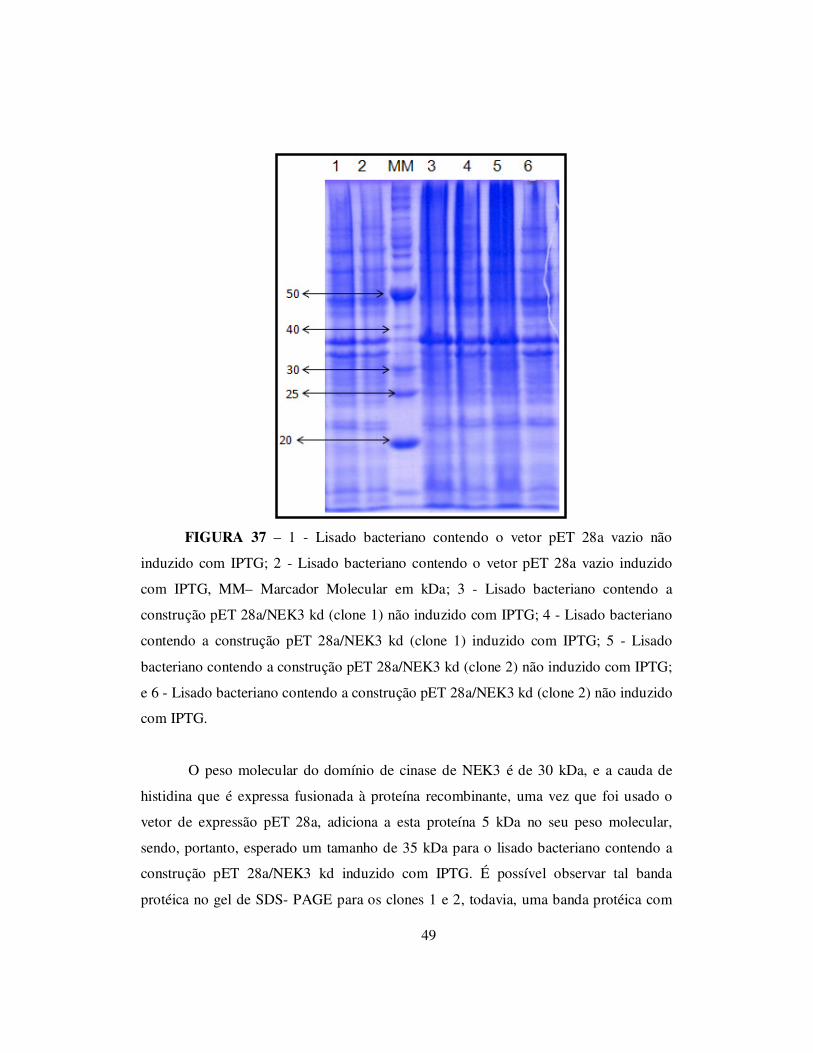
49
FIGURA 37 – 1 - Lisado bacteriano contendo o vetor pET 28a vazio não
induzido com IPTG; 2 - Lisado bacteriano contendo o vetor pET 28a vazio induzido
com IPTG, MM– Marcador Molecular em kDa; 3 - Lisado bacteriano contendo a
construção pET 28a/NEK3 kd (clone 1) não induzido com IPTG; 4 - Lisado bacteriano
contendo a construção pET 28a/NEK3 kd (clone 1) induzido com IPTG; 5 - Lisado
bacteriano contendo a construção pET 28a/NEK3 kd (clone 2) não induzido com IPTG;
e 6 - Lisado bacteriano contendo a construção pET 28a/NEK3 kd (clone 2) não induzido
com IPTG.
O peso molecular do domínio de cinase de NEK3 é de 30 kDa, e a cauda de
histidina que é expressa fusionada à proteína recombinante, uma vez que foi usado o
vetor de expressão pET 28a, adiciona a esta proteína 5 kDa no seu peso molecular,
sendo, portanto, esperado um tamanho de 35 kDa para o lisado bacteriano contendo a
construção pET 28a/NEK3 kd induzido com IPTG. É possível observar tal banda
protéica no gel de SDS- PAGE para os clones 1 e 2, todavia, uma banda protéica com

50
tamanho igual também aparece no lisado bacteriano contendo o vetor pET 28a vazio,
induzido e não induzido com IPTG. Tais resultados, somados ao fato de que não foi
possível confirmar a integridade da sequência do domínio de cinase de NEK3 clonada
em pET 28a, sugerem fortemente que tal banda protéica poderia não ser o domínio de
cinase de NEK3.
Em virtude disso, foi tentada, novamente, a clonagem do domínio de cinase de
NEK3 em um outro vetor pET 28a, em diferentes proporções inserto: vetor nas reação
de ligação e tentando-se realizar a transformação de estirpes de E. coli DH 5α e JM109
por choque térmico e eletroporação, respectivamente. Entretanto, por falta de tempo,
não foi possível fazer as confirmações dessas clonagens.
4.4 - Transformação da levedura Saccharyomyces cerevisae L40
Com o objetivo de realizar o sistema de Duplo-Híbrido em leveduras para
encontrar possíveis parceiros de interação proteína-proteína de NEK2 e de transcritos de
uma biblioteca de cDNA de HELA, inicialmente foi feita a transformação de L40 com a
construção pBTM 116 K/NEK2 vinda do clone 1.
Foram testadas diferentes condições de transformação em três tentativas de
transformação, nas quais variou-se a concentração de DNA plasmidial (construção
pBTM 116 K/NEK2) de 0,1 µg a 3,0 µg, a quantidade de células de L40 e o método de
transformação (choque térmico e eletroporação). Em todas as transformações, houve
crescimento satisfatório dos controles positivos utilizados (descritos no item 3.10) e não
houve crescimento do controle negativo da transformação.
Na primeira tentativa de transformação, foram utilizadas células de L40 com
D.O de 0,8 e quantidades do DNA plasmidial pBTM 116 K/NEK2 que variaram de 100
ng a 1 µg; as concentrações dos DNAs plasmidiais das construções que foram usadas
como controle foram sempre de 1 µg. Como só cresceram leveduras nas placas dos
controles positivos, foi feita uma segunda tentativa em que foi usada uma quantidade
maior de células de L40 com D.O de 1,6, e a quantidade de DNA plasmidial pBTM 116
K/NEK2 variou de 0,5 a 3 µg; os controles foram usados nas mesmas condições que na
primeira transformação. Novamente, só os controles positivos cresceram e foi testado
um protocolo alternativo de transformação em que foram preparadas células
eletrocompetentes de L40, e feita a transformação por eletroporação como descrito no
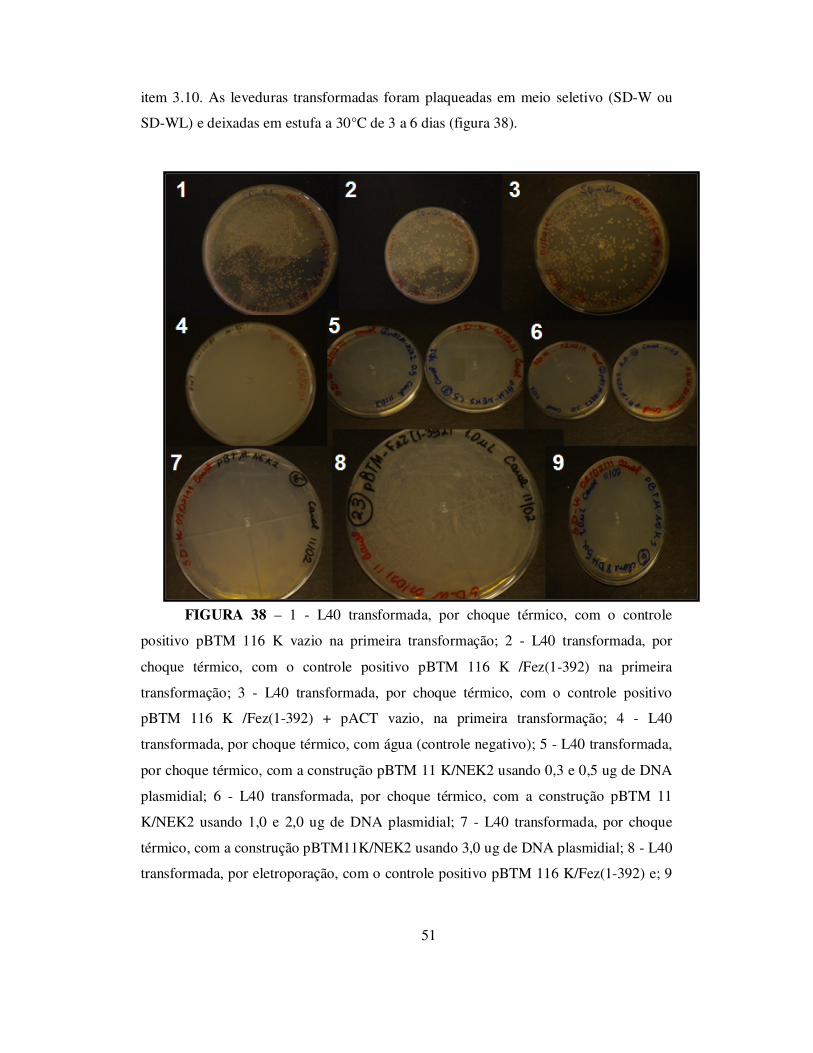
51
item 3.10. As leveduras transformadas foram plaqueadas em meio seletivo (SD-W ou
SD-WL) e deixadas em estufa a 30°C de 3 a 6 dias (figura 38).
FIGURA 38 – 1 - L40 transformada, por choque térmico, com o controle
positivo pBTM 116 K vazio na primeira transformação; 2 - L40 transformada, por
choque térmico, com o controle positivo pBTM 116 K /Fez(1-392) na primeira
transformação; 3 - L40 transformada, por choque térmico, com o controle positivo
pBTM 116 K /Fez(1-392) + pACT vazio, na primeira transformação; 4 - L40
transformada, por choque térmico, com água (controle negativo); 5 - L40 transformada,
por choque térmico, com a construção pBTM 11 K/NEK2 usando 0,3 e 0,5 ug de DNA
plasmidial; 6 - L40 transformada, por choque térmico, com a construção pBTM 11
K/NEK2 usando 1,0 e 2,0 ug de DNA plasmidial; 7 - L40 transformada, por choque
térmico, com a construção pBTM11K/NEK2 usando 3,0 ug de DNA plasmidial; 8 - L40
transformada, por eletroporação, com o controle positivo pBTM 116 K/Fez(1-392) e; 9

52
- L40 transformada, por eletroporação, usando 1,0 ug de DNA plasmidial da construção
pBTM 116 K/NEK2.
Em nenhuma das tentativas de transformação de L40 foi conseguido obter
leveduras transformadas com a construção pBTM 116 K /NEK2. Houveram alguns
trabalhos publicados em que foi feito o sistema de Duplo-Híbrido em leveduras usando
a NEK2 como isca, dentre os quais foi observado que nenhum deles havia utilizado
cepa L40 de S. cerevisae. Fry et al (1999) utilizou a cepa Y190 de S. cerevisae para
transformar a levedura com a construção que continha a NEK2; nesta cepa, utiliza-se,
ao invés do vetor pBTM116 K, o vetor pAS-2. Chen et al (2002) utilizou uma série de
cepas diferentes de S. cerevisae, dentre elas, pode-se citar YPH 499, WHL 101 e
WHL6502. Em nenhum artigo foi encontrado o uso de sistema de Duplo-Híbrido em
leveduras usando a NEK2 como isca e transformação da cepa L40. Como não foi
alcançado fazer a transformação da cepa L40, pode-se supor que, talvez, a proteína
NEK2, quando expressa em L40, torna-se tóxica às células, ou impeça, de alguma
forma, a transcrição da enzima necessária à biossíntese de triptofano. Como L40 é um
mutante auxotrófico para triptofano, sem a biossíntese de tal enzima, as células são
incapazes de sobreviver em meio SD-W. Uma última suposição seria a de que a
construção pBTM 116 K/NEK2 simplesmente não foi capaz de ser introduzida em L40
por nenhuma das concentrações do DNA plasmidial utilizado, e em nenhum dos dois
métodos de transformação que foram testados.

53
5 – Conclusão
Com o objetivo de clonar os fragmentos gênicos codificantes para as NEKs
cinases 2, 3 e 5, e os fragmentos gênicos codificantes para apenas os domínios de cinase
das NEKs 3 e 5, foi feita a amplificação via PCR de tais regiões gênicas. NEK2 foi
amplificada a partir de biblioteca de cDNA de HELA e NEK3 foi amplificada a partir
das bibliotecas de cDNA de leucócitos e cérebro fetal humano. Não foi obtido sucesso
nas tentativas de amplificação de NEK5 em nenhuma das bibliotecas de cDNA de
leucócitos humanos, cérebro fetal humano, medula óssea, mama e HELA. As regiões
gênicas amplificadas de NEK2 e NEK3 foram clonadas em pGEM T easy e a
confirmação das clonagens foi feita por PCR de colônias e de sequenciamento. A partir
das sequências obtidas, foram feitas análises de similaridade em banco de dados
BLAST e encontrada similaridade com as NEKs 2 e 3 dispostas no banco de dados. Foi
feita, também, uma análise da integridade das sequências e verificado que não havia
mutações ou deleções nas sequências obtidas, e que as mesmas estavam na fase de
leitura correta.
A construção pGEM T easy/NEK3 foi utilizada com DNA template em reações
de PCR em que se objetivava amplificar apenas o domínio de cinase de NEK3. A
amplificação do domínio de cinase de NEK3 foi obtida com êxito e tal fragmento
gênico foi clonado nos vetores de expressão pET 28a e pFastBacDual-eGFP. Entretanto,
foram obtidas, após a transformação de E. coli DH 5α, colônias transformantes e
recombinantes apenas para a construção pET 28a/NEK3 kd e não para a pFastBacDual-
eGFP/NEK3 kd. Foi feita a transformação de E. coli BL 21 com a construção pET
28a/NEK3 kd e a indução da expressão da proteína recombinante. Todavia, foi
observada, através de SDS-PAGE, uma banda protéica com o tamanho esperado de 35
kDa tanto no lisado bacteriano transformado e induzido que continha a construção pET
28a/NEK3 kd, quanto no que continha apenas o vetor pET 28a vazio. Novas reações de
ligação e transformações foram feitas, porém, em virtude do limite de tempo, não foi
possível obter as confirmações dos clones.

54
O DNA plasmidial da construção pGEM T easy/NEK2 foi utilizado, em várias
concentrações, para a transformação de uma quantidade variada de células da levedura
S. cerevisae L40, por choque térmico e por eletroporação. Entretanto, somente as
leveduras contendo os controles da transformação cresceram nas placas.

55
6 – Perspectivas
Os resultados do presente trabalho abrem como perspectivas futuras:
• Otimização da transformação de S. cerevisae L40 com a construção
pBTM 116 K/NEK2.
• Clonagem de NEK2 em vetor pAS- 2 para a transformação da cepa Y190
de S. cerevisae.
• Nova clonagem do domínio de cinase de NEK3 em pET 28a e em
pFastBacDual-eGFP e posterior expressão da proteína recombinante.
• Amplificação de NEK5 e de seu domínio de cinase a partir de bibliotecas
de cDNA de outros órgãos e tecidos como útero, linfonodo, fígado, rim
entre outros.

56
7 – Referências bibliográficas
Alberts, B. Molecular Biology of the Cell, 4th ed., New York: Garland, 2002. Arama, E.; Yanai, A.; Kilfin, G.; Bernstein, A.; Motro, B. Murine NIMArelated kinases
are expressed in patterns suggesting distinct functions in gametogenesis and a
role in the nervous system. Oncogene, Apr 9; 16 (41),1813-23, 1998. Brown, J.R.; Koretke, K.K.; Birkeland, M.L.; Sanseau, P.; Patrick D.R. Evolutionary
relationships of Aurora kinases: implications for model organism studies and the
development of anti-cancer drugs. BMC Evol. Biol., Oct 12; 4(1):39, 2004. Chan, C.S.; Botstein, D. Isolation and characterization of chromosome-gain and
increase-in-ploidymutants in yeast. Genetics, Nov 135(3): 677-91, 1993.
Chang, J.; Baloh, R.; Milbrandt, J. The NIMA-family kinase Nek3 regulates
microtubuleacetylation in neurons. Washington University, Journal of Cell Science, 122, p.2274-2282, 2009.
Chen, Y.; Riley, D.J.; Zheng, L.; Chen, P.L.; Lee, W.H. Phosphorylation of the Mitotic
Regulator Protein Hec1 by Nek2 Kinase Is Essential for Faithful Chromosome
Segregation. University of Texas. JBC Papers in Press,Oct 16 (51), 49408-49416, 2002.
De Robertis, E. & De Robertis Jr., E. M. Biologia Celular e Molecular, Fundação
Calouste Gulbenkian, Lisboa, 1996. De Souza, C.P.C.; Osmani, A.H.; Wu, L.; Spotts, J.L.; Osmani, S.A. Mitoic histone h3
phosphorylation by the nima kinase in Aspergillus nidulans. Cell, Aug 4,102 (3): 293-302, 2000.
Draetta, G.; Beach, D. Activation of cdc2 protein kinase during mitosis in human cells:
cell cycledependent phosphorylation and subunit rearrangement. Cell, Jul1; 54 (1): 17-26, 1988.
Draetta, G.; Luca, F.; Westendorf, J.; Brizuela, L.; Ruderman, J.; Beach, D. Cdc2
protein kinase is complexed with both cyclin A and B: evidence for proteolytic
inactivation of MPF. Cell, Mar 10; 56 (5):829-38, 1989. Durkacz, B.; Carr, A.; Nurse, P. Transcription of the cdc2 cell cycle control gene of the
fission yeast Schizosaccharomyces pombe. EMBO J, Feb; 5(2): 369–3731986. Fisher, D.L.; Nurse, P. A single fission yeast mitotic cyclin B p34cdc2 kinase promotes
both S-phase andmitosis in the absence of G1 cyclins. EMBO J, Feb 15; 1996.

57
Fletcher, L.; Cerniglia, G.J.; Yen, T.J.; Muschel, R.J. Live cell imaging reveals distinct
roles in cell cycle regulation for Nek2A and Nek2B. Biochim Biophys Acta, Jun 30;1744(2):89-92 2005.
Fry, A.M.; Arnaud, L.; Nigg, E.A. Activity of the Human Centrosomal Kinase, Nek2,
Depends on an Unusual Leucine Zipper Dimerization Motif. J. Biol. Chem. Jun 4;274(23):16304-10, 1999.
Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4
functions in centriole duplication. Nat Cell Biol, Nov;7(11):1140-6, 2005. Hanks, S.K. Genomic analysis of the eukaryotic protein kinase superfamily: a
perspective. Genome Biol., Apr 29; 4(5):11 2003. Knighton, D.R.; Bell, S.M.; Zheng, J. 2.0 A refined crystal structure of the catalytic
subunit of cAMP-dependent protein kinase complexed with a peptide inhibitor and
detergent. Acta Crystallogr. D Biol. Crystallogr. May 1;49(Pt 3):357-6, 1993. Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor,
S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine
monophosphate-dependent protein kinase. Science. ed. 253 October; 2(10): 1559–1573. 1991.
Laemmli, U. K. Cleavage of strutucturalprotein during the assembly of the head of
bacteriophage T4. Nature, Aug 15, 227, 680 - 685, 1970. Lapenna, S & Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nature
Rev Drug Discov , Jul 8, 547-566, 2009.
Lu, K.P. & Hunter, T. Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell, May 5;81(3):413-24, 1995.
Malumbres, M & Barbacid, M. Cell cycle kinases in cancer. Curr Opin Genet,
Mar;11(3):291-302 2007. Manning, G.; Whyte, D.B. The protein kinase complement of the human
genome. Science, Dec 6,Vol 298 no. 5600,p. 1912-1934 , 2002. Mark, C.W. & Lynne, M.Q. The NIMA-family kinase, Nek1 affects the stability of
centrosomes and ciliogenesis. Biochemical and Biophysical Research Communications, Nov 6, Vol 389; p52-56; 2010.
Nigg, E.A. Polo-like kinases: positive regulators of cell division from start to finish.
Curr Opinion in Cell Biol, Dec;Vol. 10,p. 776-783, 1998. Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev
Mol Cell Biol, Jan;2(1):21-32, 2001.

58
O’Connell, M.J.; Krien, M.J.; Hunter, T. The NIMA-related protein kinases in mitotic
control. Trends Cell Biol. May;13(5):221-8, 2003.
O'Regan, L.; Blot, J.; Fry, A. Mitotic regulation by NIMA-related kinases. Cell Division, Aug 29; 2, 25, 2007.
Odefrey, F.; Stone, J.; Gurrin, L.C.; Byrnes, G.B.; Apicella, C.; Dite, G.S.; Cawson,
J.N.; Giles, G.G.; Treloar, S.A.; English, D.R.; Hopper, J.L.; Southey, M.C. Common genetic variants associated with breast cancer and mammographic
density measures that predict disease. Cancer Res. 2010 Feb 15;70(4):1449-58. Quarmby, L.M. & Mahjoub, M.R. Caught Nek-ing: cilia and centrioles. J. Cell. Sci.
118, 5161-5169 , 2005. Salaün, P.; Rannou,Y.; Prigent, C. Cdk1, Plks, Auroras, and Neks: the mitotic
bodyguards. Adv Exp Med Biol; 617: 41–56, 2008 . Salem, H.; Rachmin, I.; Yissachar, N.; Cohen, S.; Amiel, A.; Haffner, R.; Lavi,
L.; Motro, B. Nek7 kinase targeting leads to early mortality, cytokinesis
disturbance and polyploidy. Oncogene, Jul 15; 29 (28):4046-57, 2010.
Sambrook, J.; Fritsch, J.F.; Maniatis, T.Molecular cloning: a laboratory manual, Second edition, 1989.
Stout, T.J.; Foster, P.G.; Matthews, D.J. High-throughput structural biology in drug
discovery: protein kinases. Curr. Pharm., 2004.
Surpili, M.J.; Delben, T.M.; Kobarg, J. Identification of proteins that interact with the
central coiled coil region of the human protein kinase NEK7 involved in the cell
cycle regulation. Biochemistry,42(51):15369-15376, 2003. Vlahopoulos, S. & Zoumpourlis, V.C. JNK: a key modulator of intracellular
signaling. Biochemistry (Mosc), Aug;69(8):844-54, 2004.