lucas ricardo fernandes figueiredo · 260°c e a adição de nwc aumentou a estabilidade térmica...
TRANSCRIPT

UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
CENTRO DE TECNOLOGIA
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIA E
ENGENHARIA DE MATERIAIS
PROPRIEDADES ADESIVAS DE POLÍMEROS BIODEGRADÁVEIS
DERIVADOS DO GLICEROL COM ADIÇÃO DE NANOWHISKERS DE
CELULOSE
LUCAS RICARDO FERNANDES FIGUEIREDO
NATAL- RN
2017

UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
CENTRO DE TECNOLOGIA
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIA E
ENGENHARIA DE MATERIAIS
PROPRIEDADES ADESIVAS DE POLÍMEROS BIODEGRADÁVEIS DERIVADOS DO
GLICEROL COM ADIÇÃO DE NANOWHISKERS DE CELULOSE
Lucas Ricardo Fernandes Figueiredo
Tese a ser apresentada ao Programa de Pós-
Graduação em Ciências e Engenharia de
Materiais como requisito parcial à obtenção
do título de doutor EM ENGENHARIA DE
MATERIAIS.
Orientador: Prof. Dr. José Daniel Diniz Melo
Co-orientador: Prof. Dr. Eliton Souto de Medeiros
Agência Financiadora: CAPES
NATAL – RN
2017

Ficha catalográfica

DEDICATÓRIA
Aos meus pais e meus irmãos.

1
1
MEMBROS DA BANCA EXAMINADORA DA TESE DE DOUTORADO
DE:
Lucas Ricardo Fernandes Figueiredo
BANCA EXAMINADORA:

2
2
“O problema são problemas demais
Se não correr atrás da maneira certa de
solucionar”
Chico Science

3
3
AGRADECIMENTOS
A todos que contribuíram direta e indiretamente para esse trabalho.
A capes pela bolsa concedida.
Ao LSR e LAMAB pelas análises realizadas.
Ao PPGCEM-UFRN pelas análises realizadas.
A Embrapa- São Carlos pelas análises de MET

4
4
RESUMO
“Adesivos verdes” têm despertado grande interesse científico e tecnológico como
alternativa aos produtos comerciais convencionais, que em geral, liberam substâncias tóxicas
e levam um longo período de tempo para degradar. Entretanto, as propriedades mecânicas
limitadas e a rápida degradação de alguns polímeros biodegradáveis limitam seu uso na
maioria das aplicações práticas. No presente trabalho, compósitos foram preparados usando
polímeros biodegradáveis e nanowhiskers de celulose (NWC) visando sua utilização como
adesivos para madeira. O objetivo da pesquisa foi estudar o efeito da adição dos
nanowhiskers de celulose (NWC) nas propriedades mecânicas e na degradação dos
polímeros. Os polímeros biodegradáveis sintetizados por policondensação à base de glicerol
e ácidos foram poli (adipato de glicerol) - PGA, poli (maleato de glicerol) - PGM, poli
(citrato de glicerol) - PGC, poli (ftalato de glicerol) - PGPh, poli (succinato de glicerol)
PGSu e poli (sebacato de glicerol) - PGS. Além desses, copolímeros poli (glicerol succinato-
co-maleato) - PGMSu, poli (glicerol succinato-co-adipato) - PGASu e poli (glicerol adipato-
co-maleato) - PGMA foram também sintetizados. Nanowhiskers de celulose (NWC) foram
adicionados aos polímeros para avaliação dos seus efeitos na melhoria das propriedades
mecânicas e no controle da taxa de degradação. Os materiais foram produzidos com a adição
de 0, 5, 10 e 20% em peso de NWC. Os polímeros puros e os compósitos foram analisados
por Calorimetria Diferencial de Varredura (DSC), Análise termogravimétrica (TGA),
difração de Raios X (DRX) e espectroscopia Infravermelho por transformada de Fourier
(FTIR). Juntas adesivas foram produzidas em corpos de prova de pinus (Pinus elliottii) e
angelim (Vatairea heteroptera Ducke) e submetidas a degradação por envelhecimento
acelerado e testes de cisalhamento sob tração. Juntas coladas produzidas com adesivos à
base de acetato de polivinila - PVAc e cianoacrilato foram também testadas, nas mesmas
condições, para fins de comparação. As análises térmicas indicaram que a maioria dos
polímeros biodegradáveis à base de glicerol testados são estáveis até aproximadamente
260°C e a adição de NWC aumentou a estabilidade térmica (Tonset) de alguns polímeros em
26°C até 48°C, enquanto outros não foram afetados. As análises de superfície de fratura por
microscopia eletrônica de varredura (MEV) após os testes de cisalhamento sugerem redução
de ductilidade com a incorporação de NWC ao adesivo. As medidas de adesão indicaram
que os adesivos verdes desenvolvidos a partir de polímeros de glicerol com 10 e 20% em
peso de NWC apresentaram resistência ao cisalhamento superior ao adesivo comercial à

5
5
base de PVAc. Os melhores resultados e resistência ao cisalhamento foram obtidos para
PGASu com 20% em peso de NWC (2,57 ± 0,36 MPa) e PGM com 20% em peso de NWC
(2,33 ± 0,43 MPa), enquanto que a resistência ao cisalhamento do acetato de polivinila -
PVAc foi de 1,58 ± 0,18 MPa. O envelhecimento resultou em maior resistência ao
cisalhamento de alguns adesivos. A melhoria mais significativa foi obtida para PGMA com
20% em peso de NWC, que atingiu (3,89 ± 0,74 MPa) após 250 h de envelhecimento,
portanto, maior que a resistência ao cisalhamento do cianoacrilato (3,12 ± 0,53 MPa). Em
última análise, os resultados apresentados neste trabalho sugerem que a adição de
nanowhiskers de celulose (NWC) é uma abordagem viável para ajustar as propriedades
mecânicas e degradação dos polímeros biodegradáveis.

6
6
ABSTRACT
“Green adhesives” are of great scientific and technological interest as an alternative to
conventional commercial products, which often release toxic substances and take long time
to degrade. However, the poor mechanical properties and fast degradation of some
biodegradable polymers limit their use in most practical applications. In the present work,
novel biodegradable composites were prepared using biodegradable polymers and cellulose
nanowhiskers (CNW) for their use as wood adhesives. The goal of the investigation was to
study the effect of the addition of CNW on the mechanical properties and degradation of the
polymers. The biodegradable polymers synthesized by polycondensation based on glycerol
and acids were poly(glycerol adipate) - PGA, poly(glycerol maleate) - PGM, poly(glycerol
citrate) - PGC, poly(glycerol phthalate) - PGPh, poly(glycerol succinate) - PGSu and
poly(glycerol sebacate) - PGS. In addition, the copolymers synthesized were poly(glycerol
succinate-co-maleate) - PGMSu, poly(glycerol succinate-co-adipate) – PGASu and
poly(glycerol adipate-co-maleate) – PGMA. Cellulose nanowhiskers (CNW) were added to
the polymers as an approach for improving the mechanical properties and controlling the
degradation rate. Composites were produced with the addition of 0, 5, 10 and 20 wt.% of
CNW. The neat polymers and composites were analyzed by Differential Scanning
Calorimetry (DSC), Thermogravimetric Analysis (TGA), X-ray diffraction (XRD), and
Fourier Transform Infrared Spectroscopy (FTIR). Adhesively bonded joints were produced
in test specimens of slash pine (Pinus elliottii) and angelim (Vatairea heteroptera Ducke)
and subjected to accelerated aging and tested under shear by tensile loading. Bonded joints
produced with polyvinyl acetate - PVAc and cyanoacrylate based adhesives were also tested
under the same conditions for comparison purposes. Thermal analyses indicated that most of
the biodegradable glycerol-based polymers tested are stable up to about 260°C and the
addition of CNW increased the thermal stability (Tonset) of some polymers by 26°C up to
48°C, while others were not affected. Analyses of the fracture surfaces after the shear tests
by Scanning Electron Microscopy (SEM) suggest less ductile fracture with the incorporation
of CNW to the adhesive. The shear strengths of the green adhesives developed from
glycerol with 10 and 20 wt.% CNW were greater than that of the commercial PVAc based
adhesive. The best results of shear strengths were obtained for PGASu 20 wt.% CNW (2.57
± 0.36 MPa) and PGM 20 wt.% CNW (2.33± 0.43 MPa), while the shear strength for
polyvinyl acetate - PVAc was 1.58 ± 0.18 MPa. Aging improved the shear strength of some
adhesives. The best result was obtained for PGMA 20 wt.% CNW, which reached (3.89 ±

7
7
0.74 MPa) after 250h of aging, thus greater than the shear strength of cyanoacrylate (3.12 ±
0.53 MPa). Ultimately, the results presented in this work suggest that the addition of
cellulose nanowhiskers (CNW) is a viable approach to tailor mechanical properties and
degradation of biodegradable polymers.

8
8
PUBLICAÇÕES
Patente depositada com título: Adesivos biodegradáveis de polímeros de
glicerol com adição de nanowhiskers de celulose (NWC). Número do
processo - BR 1020170219240

9
9
SUMÁRIO
1 INTRODUÇÃO ............................................................................................................. 18
2 FUNDAMENTAÇÃO TEÓRICA ............................................................................... 19
2.1 ADESIVOS ......................................................................................................... 19
2.1.1 Adesivos para madeira ..................................................................................... 21
2.2 POLÍMEROS DE GLICEROL .................................................................................. 25
2.2.1 Síntese e aplicações de polímeros de glicerol .................................................. 26
2.2.2 Uso de polímeros de glicerol como adesivos ................................................... 31
2.3 OBTENÇÕES E APLICAÇÕES DE NANOWHISKERS DE CELULOSE (NWC) ............. 32
3 OBJETIVOS .................................................................................................................. 37
4 MATERIAIS E PROCEDIMENTOS ......................................................................... 38
4.1 MATERIAIS ....................................................................................................... 39
4.1.1 Reagentes .......................................................................................................... 39
4.2 PROCEDIMENTO EXPERIMENTAL ....................................................................... 39
4.2.1 Extração dos NWC ........................................................................................... 39
4.2.2 Síntese dos polímeros e copolímeros ................................................................ 39
4.2.3 Preparação dos Compósitos ............................................................................. 42
4.2.4 Colagem dos adesivos de polímeros de glicerol .............................................. 44
4.2.5 Caracterizações dos polímeros e compósitos ................................................... 44
4.2.5.1 Calorimetria diferencial de varredura (DSC) ............................................ 44
4.2.5.2 Termogravimetria (TG) ............................................................................. 45
4.2.5.3 Espectroscopia na região do infravermelho por transformada de fourier
(FTIR). 45
4.2.5.4 Difratometria de raios X (DRX)................................................................ 45
4.2.5.5 Microscopia eletrônica de varredura (MEV) ............................................ 45
4.2.5.6 Microscopia de força atômica (AFM) ....................................................... 46
4.2.5.7 Microscopia eletrônica de transmissão (MET) ......................................... 46
4.2.5.8 Ensaio mecânico de cisalhamento sob tração ........................................... 46
4.2.5.9 Degradação por envelhecimento acelerado ............................................... 47
5 RESULTADOS E DISCUSSÃO .................................................................................. 48
5.1 SÍNTESE DOS POLÍMEROS PUROS. ...................................................................... 48
5.2 ESPECTROSCOPIA NA REGIÃO DO INFRAVERMELHO POR TRANSFORMADA DE FOURIER
(FTIR) DOS POLÍMEROS PUROS. ................................................................................... 49
5.3 DIFRATOMETRIA DE RAIOS X (DRX) ................................................................. 50
5.4 CALORIMETRIA EXPLORATÓRIA DIFERENCIAL (DSC) ....................................... 51
5.5 TERMOGRAVIMETRIA (TG) ............................................................................... 52
5.6 ENSAIO DE CISALHAMENTO SOB TRAÇÃO DOS POLÍMEROS PUROS EM MADEIRA DE
PINUS ELLIOTTII. ........................................................................................................... 56

10
10
5.7 ENSAIO DE CISALHAMENTO SOB TRAÇÃO DOS POLÍMEROS PUROS EM MADEIRA DE
ANGELIM ....................................................................................................................... 62
5.8 EXTRAÇÃO E CARACTERIZAÇÃO DOS NWC. ..................................................... 66
5.9 ESPECTROSCOPIA NA REGIÃO DO INFRAVERMELHO POR TRANSFORMADA DE FOURIER
(FTIR) DOS COMPÓSITOS. ............................................................................................ 68
5.10 CALORIMETRIA EXPLORATÓRIA DIFERENCIAL (DSC) DOS POLÍMEROS, COPOLÍMEROS
E COMPÓSITOS .............................................................................................................. 73
5.11 ESTUDO DA ESTABILIDADE TÉRMICA DOS NWC, POLÍMEROS E COMPÓSITOS POR TG
79
5.12 ENSAIO DE CISALHAMENTO SOB TRAÇÃO DOS POLÍMEROS PUROS E COMPÓSITOS EM
MADEIRA DE ANGELIM .................................................................................................. 87
5.13 ANÁLISE FRATOGRÁFICA DAS SUPERFÍCIES DE FRATURA POR MEV APÓS ENSAIO DE
CISALHAMENTO SOB TRAÇÃO ....................................................................................... 90
5.14 DEGRADAÇÃO POR ENVELHECIMENTO ACELERADO .......................................... 92
5.15 ENSAIO DE CISALHAMENTO SOB TRAÇÃO DOS POLÍMEROS PUROS E COMPÓSITOS EM
MADEIRA DE ANGELIM APÓS O ENVELHECIMENTO ACELERADO E MEV........................ 93
6 CONCLUSÕES ........................................................................................................... 105
7 REFERÊNCIAS .......................................................................................................... 106

11
11
ÍNDICE DE TABELAS
Tabela 1 - Classificação dos adesivos [30]. ............................................................................ 21
Tabela 2 - Propriedades físicas e químicas do glicerol adaptado da referência [3]................ 24
Tabela 3 - Síntese de polímeros a partir do glicerol [2, 3, 65] ............................................... 26
Tabela 4 - Estruturas químicas de vários ácidos/anidridos que podem ser usados para
produção de poliésteres através da reação com o glicerol. .............................................. 28
Tabela 5 – Condições de hidrólise e suas matérias prima - adaptado de, De Jesus Silva [106].
......................................................................................................................................... 34
Tabela 6 - Número ácido crítico para cada polímero. ............................................................ 41
Tabela 7 - Valores de Tg, Tm e ΔH dos polímeros de glicerol. ............................................. 52
Tabela 8 - Valores de Tonset, Tendset, DTGA (pico) e resíduo dos gráficos de termogravimetria
dos polímeros de glicerol. ................................................................................................ 55
Tabela 9 - valores de resistência ao cisalhamento dos polímeros de glicerol em madeira de
pinus. ................................................................................................................................ 56
Tabela 10 - Valores de resistência ao cisalhamento máximo dos polímeros de glicerol em
madeira de angelim. ......................................................................................................... 63
Tabela 11 - Valores da tg do PGA, PGM, PGSu e seus compósitos com 5,10 e 20% de
NWC. ............................................................................................................................... 76
Tabela 12 - Valores da tg do PGMSu, PGMA, PGASu e seus compósitos com 5,10 e 20%
de NWC. .......................................................................................................................... 78
Tabela 13 -Valores da degradação dos polímeros PGA, PGM e PGSu e compósitos 5, 10 e
20% de NWC ................................................................................................................... 83
Tabela 14 - Valores da degradação dos polímeros PGASu, PGMA e PGMSu e compósitos
com 5, 10 e 20% de NWC. .............................................................................................. 87
Tabela 15 – Resistência ao cisalhamento dos polímeros PGA, PGM e PGSu e compósitos
com 5, 10 e 20% de NWC. .............................................................................................. 88
Tabela 16 - Resistência ao cisalhamento dos copolímeros PGMSu, PGMA e PGASu e
compósitos com 5, 10 e 20% de NWC. ........................................................................... 89
Tabela 17 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos
da família do PGA e PGM. .............................................................................................. 94
Tabela 18 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos
da família do PGSu e PGMA. .......................................................................................... 95

12
12
Tabela 19 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos
da família do PGASu e PGMSu. ..................................................................................... 96
Tabela 20 - Valores de resistência ao cisalhamento x tempo de envelhecimento dos
compósitos com melhores resultados comparados com a cola branca comercial. .......... 98

13
13
LISTA DE FIGURAS
Figura 1 - Exemplos de falhas da união adesiva [22]. ............................................................ 19
Figura 2 – Esquemas dos fenômenos da adesão: a) por difusão b) ligação química c) forças
eletrostáticas e d) ancoragem mecânica [28]. .................................................................. 20
Figura 3 - Produção de biodiesel: reação de transesterificação [1]. ....................................... 23
Figura 4 - Reação do glicerol com triglicerídeos para obtenção de monoglicerídeos (MAG):
usados no tratamento de cabelos, em sobremesas e cremes [51]. .................................... 24
Figura 5 - Halogenação do glicerol obtendo epiclorohidrina, produto base para a resina epóxi
[62]. .................................................................................................................................. 25
Figura 6 - Reação de desidratação do glicerol, como produto a acroleína. ............................ 25
Figura 7 - Obtenção do poli(ftalato de glicerol), adaptado da referência [64]. ...................... 25
Figura 8 - Esquema da síntese de poli(éster amida) [75]. ...................................................... 27
Figura 9 - Esquema da Síntese de copolímeros de éter de diglicidil de glicerol [78, 80]. ..... 27
Figura 10 - Esquema de interação entre cadeias moleculares de celulose na região cristalina
e amorfas [45] .................................................................................................................. 33
Figura 11 - Fluxograma do procedimento experimental ........................................................ 38
Figura 12 - Esquema do procedimento experimental da síntese dos polímeros de glicerol. .. 40
Figura 13 – Produção dos corpos de prova dos polímeros: a) aplicação dos polímeros no
molde, b) polímeros no molde e c) polímeros curados. ................................................... 41
Figura 14 – Esquema das possíveis estruturas da reação do glicerol com diácidos, seus
respectivos pré-compósitos e os compósitos pós-cura. ................................................... 43
Figura 15 - Esquema das etapas de colagem: a) lixamento, b) aplicação dos adesivos, c)
sobreposição das placas de madeira e d) placas de madeira após prensagem. ................ 44
Figura 16 - Corpos de prova de acordo com a norma ASTM D906. .................................... 46
Figura 17 - Corpos de prova a serem ensaiados na câmara de envelhecimento acelerado. ... 47
Figura 18 - Sínteses do glicerol com diácidos, seus respectivos pré-polímeros e os polímeros
curados. ............................................................................................................................ 48
Figura 19 – Espectros de FTIR dos polímeros de glicerol. PGA - poli(adipato de glicerol),
PGM - poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh - poli(ftalato de
glicerol), PGSu - poli(succinato de glicerol), PGS - poli(sebacato de glicerol). ............. 49
Figura 20 - Difratometria de raios-x dos polímeros de glicerol. PGA - poli(adipato de
glicerol), PGM - poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh -

14
14
poli(ftalato de glicerol), PGSu - poli(succinato de glicerol), PGS - poli(sebacato de
glicerol). ........................................................................................................................... 50
Figura 21 - Gráficos de DSC dos polímeros: (a) aquecimento e (b) resfriamento. PGA -
poli(adipato de glicerol), PGM - poli(maleato de glicerol), PGC - poli(citrato de
glicerol), PGPh - poli(ftalato de glicerol), PGSu - poli(succinato de glicerol), PGS -
poli(sebacato de glicerol). ................................................................................................ 51
Figura 22 - Gráficos de TGA e DTGA dos polímeros de glicerol. PGA - poli(adipato de
glicerol), PGM - poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh -
poli(ftalato de glicerol), PGSu - poli(succinato de glicerol), PGS - poli(sebacato de
glicerol). ........................................................................................................................... 54
Figura 23 - Corpos de prova de cisalhamento sob tração em madeira de pinus ensaiados. ... 57
Figura 24 - MEV da madeira de Pinus, com aumentos de a)500x e b)1000x. ....................... 58
Figura 25 – MEV após falha sob tração do a) PGA e b) PGC. .............................................. 59
Figura 26 – MEV após falha sob tração do a) PGSu e b) PGM. ............................................ 60
Figura 27 - Imagens de MEV do a) PGPh e b) PGS após ensaio de cisalhamento sob tração.
......................................................................................................................................... 61
Figura 28 - Corpos de prova de cisalhamento sob tração na madeira de angelim após
ensaiados. ......................................................................................................................... 62
Figura 29 - Imagens de MEV do a) PGC, b) PGM, C) PGPh, PGSu após ensaio de
cisalhamento sob tração na madeira de angelim. ............................................................. 64
Figura 30 - Imagens de MEV do a) PGA, b) PGS e c) PGSu após ensaio de cisalhamento
sob tração na madeira de angelim. ................................................................................... 65
Figura 31 - Imagem do NWC após hidrólise ácida, diluído para liofilização e após a
liofilização. ...................................................................................................................... 66
Figura 32 - Imagem dos NWC por microscopia de força atômica (AFM)............................. 67
Figura 33 - Imagens dos NWC por microscopia eletrônica de transmissão (MET)............... 67
Figura 34 – Difratograma de raios-x dos Nanowhiskers de celulose. .................................... 68
Figura 35 – Espectro de FTIR do nanowhisker de celulose (NWC). ..................................... 69
Figura 36 - Espectros de FTIR dos polímeros (a) PGA, (b) PGM e seus compósitos com 5,
10 e 20% de NWC. .......................................................................................................... 70
Figura 37 - Espectros de FTIR do polímero PGSu e seus compósitos com 5, 10 e 20% de
NWC. ............................................................................................................................... 71
Figura 38 - Espectros de FTIR dos copolímeros a) PGMSu, b) PGASu, e seus compósitos
com 5, 10 e 20% de NWC. .............................................................................................. 72

15
15
Figura 39 - Espectros de FTIR do copolímero PGMA, e seus compósitos com 5, 10 e 20%
de NWC. .......................................................................................................................... 73
Figura 40 - Gráficos de DSC do PGA e seus compósitos com 5,10 e 20% de NWC. ........... 74
Figura 41 - Gráficos de DSC do, a) PGM, b) PGSu e seus compósitos com 5,10 e 20% de
NWC. ............................................................................................................................... 75
Figura 42 - Gráficos de DSC do a) PGASU, b) PGMA e seus compósitos com 5,10 e 20%
de NWC ........................................................................................................................... 77
Figura 43 - Gráficos de DSC do PGMSu e seus compósitos com 5,10 e 20% de NWC. ...... 78
Figura 44 - Gráfico de TGA do Nanowhisker de celulose e sua derivada. ............................ 79
Figura 45 - Gráfico de a) TGA e b) DTGA do polímero PGA e seus compósitos com 5,10 e
20% de NWC ................................................................................................................... 80
Figura 46 - Gráfico de a) TGA e b) DTGA o polímero PGM e seus compósitos com 5,10 e
20% de NWC ................................................................................................................... 81
Figura 47 - Gráfico de a) TGA e b) DTGA do polímero PGSu e seus compósitos com 5,10 e
20% de NWC ................................................................................................................... 82
Figura 48 - Gráfico de a) TGA e b) DTGA do copolímero PGMSu e seus compósitos com
5,10 e 20% de NWC. ....................................................................................................... 84
Figura 49 - Gráfico de a) TGA e b) DTGA do copolímero PGMA e seus compósitos com
5,10 e 20% de NWC. ....................................................................................................... 85
Figura 50 - Gráfico de a) TGA e b) DTGA do copolímero PGASU e seus compósitos com
5,10 e 20% de NWC. ....................................................................................................... 86
Figura 51 - Comparação dos compósitos com melhores resultados de tensão com os adesivos
comerciais. ....................................................................................................................... 89
Figura 52 - Micrografia da madeira de a) angelim e b) pinus após fratura criogênica. ......... 90
Figura 53 - Imagens de MEV do a) PGA, b) PGM, c) PGSu, d) PGA 10%, e) PGM 10% e f)
PGSu 10%, após ensaio de cisalhamento sob tração. ...................................................... 91
Figura 54 – Micrografias de MEV do, a) PGMSu, b) PGMA, c) PGASu, d) PGMSu 10%, e)
PGMA 10% e f) PGASu 10%, após ensaio de cisalhamento sob tração, aumentado
2000x. .............................................................................................................................. 92
Figura 55 - Corpos de prova do PGSu a) antes e b) após ciclo de 150h de envelhecimento. 92
Figura 56 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos
compósitos da família do PGA e PGM ............................................................................ 93
Figura 57 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos
compósitos da família do PGSu e PGMA. ...................................................................... 94

16
16
Figura 58 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos
compósitos da família do PGSu e PGMA. ...................................................................... 96
Figura 59 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento dos
compósitos com melhores resultados comparados com a cola branca comercial. .......... 97
Figura 60 - Micrografias do PGA e PGA 10% em diferentes períodos de envelhecimento
após o cisalhamento sob tração. a) PGA 150h, b) PGA 250h c) PGA 350h, d) PGA10%
150h, e) PGA10% 250h e f) PGA10% 350h ................................................................... 99
Figura 61 - Micrografias do PGM e PGM10% em diferentes períodos de envelhecimento
após o cisalhamento sob tração. a) PGM 150h, b) PGM 250h c) PGM 350h, d)
PGM10% 150h, e) PGM10% 250h e f) PGM10% 350h ............................................... 100
Figura 62 - micrografias do PGSu e PGSu 10% em diferentes períodos de envelhecimento
após o cisalhamento sob tração. a) PGSu 150h, b) PGSu 250h c) PGSu 350h, d) PGSu
10% 150h, e) PGSu 10% 250h e f) PGSu 10% 350h .................................................... 101
Figura 63 - Micrografias do PGMA e PGMA 10% em diferentes períodos de
envelhecimento após o cisalhamento sob tração. a) PGMA 150h, b) PGMA 250h c)
PGMA 350h, d) PGMA10% 150h, e) PGMA10% 250h e f) PGMA10% 350h ........... 102
Figura 64 - Micrografias do PGASu e PGASu10% em diferentes períodos de
envelhecimento após o cisalhamento sob tração. a) PGASu 150h, b) PGASu 250h c)
PGASu 350h, d) PGASu10% 150h, e) PGASu10% 250h e f) PGASu10% 350h ......... 103
Figura 65 - Micrografias do PGMSu e PGMSu10% em diferentes períodos de
envelhecimento após o cisalhamento sob tração. a) PGMSu 150h, b) PGMSu 250h c)
PGMSu 350h, d) PGMSu10% 150h, e) PGMSu10% 250h e f) PGMSu10% 350h ...... 104

17
17
SÍMBOLOS E ABREVIATURAS OU SIGLAS
ABREVIATURAS OU SIGLAS
DSC – Calorimetria diferencial de varredura
DTG – Derivada da curva de TG
DRX – Difratômetro de raios-X
FRX – Fluorescência de raios-X
MEV – Microscopia eletrônica de varredura
FTIR – Espectrometria na região do infravermelho por transformada de Fourier
IPDI – Diisocianato de isoforona
IDA – Diisocionato de hexametileno
SÍMBOLOS
∑ - Somatório
Ɵ – Teta

18
18
1 INTRODUÇÃO
O esgotamento das reservas mundiais de petróleo e o impacto da crescente poluição
ambiental têm levado pesquisadores à busca por materiais alternativos e renováveis, com
destaque para o biodiesel, produzido a partir de gorduras animais e óleos vegetais, e seu
subproduto, o glicerol, que corresponde a cerca de 10% da sua produção [1, 2]. O glicerol
apresenta combinações físicas e químicas que fazem dele uma substância muito versátil,
sendo utilizado nas indústrias alimentícias, farmacêutica, de cosméticos e química [3, 4]. A
síntese de materiais poliméricos a partir do glicerol tem um grande potencial, seja pela sua
modificação química ou por reação com poliácidos ou ácidos graxos. Novos materiais vêm
sendo estudados para uma grande variedade de aplicações que inclui encapsuladores de
drogas e de nutrientes agrícolas e enxertos para a engenharia de tecidos [5, 6].
Adesivos comerciais à base de acetatos de vinila ou ureia-formaldeído que em sua
maioria podem ser tóxicos e até liberam gases nocivos à saúde. Novas formas de
desenvolver ou aperfeiçoar adesivos biodegradáveis que substituam os comerciais têm sido
estudadas, como os adesivos selantes de polímeros de glicerol, adesivos à base de soja,
hidrogéis adesivos para recuperação de córneas e adesivos de soja reforçados com
nanowhiskers de celulose [5, 7–9]. A adição de reforços pode melhorar as propriedades
mecânicas, térmicas e de barreira dos polímeros de glicerol, tornando sua aplicação ainda
mais abrangente, com a possibilidade de novos usos, em larga escala.
Os whiskers ou nanowhiskers de celulose, obtidos por hidrólise ácida, apresentam
biodegradabilidade, não toxicidade e elevada resistência mecânica. Por essas características
são amplamente utilizados como reforços em compósitos e (bio)nanocompósitos [10–20].
Medeiros et.al [21] mostraram que a resistência mecânica e a biodegradação de polímeros
do glicerol podem ser controladas com a adição de NWC. O algodão, por ter porcentagem
elevada de celulose (implicando em maior cristalinidade) na sua composição, é o material de
maior abrangência na obtenção de NWC para formulações de compósitos. Além disso, o
algodão necessita apenas de hidrólise ácida para obtenção dos whiskers, dispensando
tratamentos prévios de branqueamento com bases fortes, já que seu teor de lignina é
relativamente pequeno.
O objetivo deste trabalho foi desenvolver polímeros de glicerol como adesivos para a
madeira e estudar a modificação de suas propriedades de adesão e resistência à degradação
pela incorporação de nanowhiskers de celulose.

19
19
2 FUNDAMENTAÇÃO TEÓRICA
2.1 Adesivos
Adesivos são materiais que quando aplicados entre duas superfícies são capazes de
conservá-las unidas. Os adesivos podem ser descritos pela sua forma física (adesivo líquido
e de fita) ou finalidades (adesivo para papel, metais, plásticos e borrachas). Esses materiais
funcionam principalmente por propriedades de adesão, que é o resultado de forças
intermoleculares (van der Waals) de duas substâncias diferentes; porém atuam também as
forças de coesão, onde atuam apenas em uma substância [22–24].
A maneira como se dá o processo de união entre as duas superfícies é diretamente
relacionada ao processo de falha obtido conforme mostra a Figura 1. Os principais tipos de
falha e/ou ruptura são a ruptura coesiva, que ocorre quando uma falha se propaga no seio do
polímero que constitui o adesivo ou no aderente, ruptura interfacial quando ocorre entre o
adesivo e os suportes aderentes; ruptura alternada, que combina o comportamento das
rupturas coesivas e adesivas; e ruptura do suporte que ocorre no suporte e não no adesivo,
sendo o adesivo mais resistente que o material do suporte [22, 24].
Figura 1 - Exemplos de falhas da união adesiva [22].

20
20
A adesão é o fenômeno interfacial ou a energia de separação de dois substratos,
enquanto adesivo é o material que promove a união entre os mesmos. As principais teorias
de adesão, que explicam a maioria de fenômenos observados, podem ser classificadas em:
teoria mecânica, adsorção, difusão e eletrostática.
Na teoria mecânica, a adesão se dá através da penetração do adesivo em substratos
porosos, levando à formação de ganchos fortemente presos ao substrato após a solidificação
do adesivo. Essa adesão melhora com tratamentos superficiais, que resultam em uma
microrrugosidade do substrato, aumentando a resistência e a durabilidade devido ao
encravamento (ancoramento) mecânico. A teoria da difusão, aplicada a polímeros, se dá
através da difusão de segmentos de cadeias, podendo ser visualizada como a mesma
produzida na adesão mecânica, só que agora em nível molecular. Este mecanismo não é
aplicável a materiais substancialmente diferentes, obtendo a melhor adesão efetiva quando
existe uma solubilidade mútua entre adesivo e material aderido [23, 25].
Os fenômenos de adesão são indicados na Figura 2. A teoria da adsorção dá-se
através das forças secundárias intermoleculares (van der Waals) ou ligações primárias. O
adesivo e o substrato ficam suficientemente próximos e permitem a adsorção física, como
também podem formar ligações primárias na interface, adsorção química. A adesão
eletrostática pode ocorrer em materiais com diferentes eletronegatividades, quando postos
em contato, e pode ocorrer uma transferência de elétrons do material com menor para o de
maior eletronegatividade [26, 27].
a) b) c) d)
Figura 2 – Esquemas dos fenômenos da adesão: a) por difusão b) ligação química c) forças eletrostáticas e d)
ancoragem mecânica [28].
Algumas superfícies podem estar fracamente ligadas, provocando um elo tênue que
reduz a resistência mecânica. Entretanto, alguns procedimentos podem ser tomados para
melhorar esta ligação, como a preparação da topografia da superfície a ser colada, o
processo de colagem (formulação/prensagem) e condições ambientais (temperatura/

21
21
umidade), que são fatores de grande importância que devem ser levados em consideração
para a colagem [29].
Os adesivos podem ser classificados pela origem do seu componente primário, como
mostrado na Tabela 1.
Tabela 1 - Classificação dos adesivos [30].
Tipo de adesivo Exemplos
Naturais Glutina (couro, pele, ossos), caseína (leite), albumina (sangue), soja,
batatas, trigo, borracha natural
Termoplásticos Poliacetato de vinila (PVAc), “hot-melt”, polietileno, borracha sintética.
Elastômeros Poliisoprenos, poliuretanos, poliolefinas, silicone.
Termorrígidos
Uréia-formaldeído (UF), Melamina formaldeído (MF), Fenol-
formaldeído (FF), Resorcina-formaldeído (RF), Melamina-uréia-
formaldeído (MUF), Fenol-melamina-uréia-formaldeído (FMUF),
Resorcina-fenol formaldeído (RFF), Tanino-formaldeído (TF),
Licor sulfito, Isocianato (MDI)
Diversos fatores como, deformação, viscosidade, reologia, difusibilidade,
solubilidade, penetração, rendimento, tensões térmicas, tempos de cura e armazenamento
devem ser analisados em um adesivo na avaliação de seu desempenho. A interface entre o
adesivo e o aderente é também um fator crítico nas propriedades de adesão [25].
2.1.1 Adesivos para madeira
Polímeros orgânicos de origem natural ou sintética são ingredientes químicos
fundamentais nas formulações de adesivos para madeira. O principal uso destes adesivos é
na fabricação de materiais de construção, aglomerados, portas, janelas e quadros, produtos
de madeira laminada de fábrica e na montagem de móveis e armários, fabricação de
produtos de madeira de engenharia.
As propriedades da madeira influenciam diretamente na mobilidade e penetrabilidade
do adesivo. Dentre estas propriedades, podem ser destacadas as propriedades anatômicas
(diferentes dimensões e porosidades), químicas (diferentes porcentagens de celulose (40-
47%), hemicelulose (20-30%) e lignina (6-30%), variação no pH (3-6), físicas (densidade e
umidade), além das mecânicas (tensões externas e internas) [31–33].

22
22
Alguns princípios básicos devem ser observados para o processo de colagem de
madeira, como ângulo de contato e umectação, elos de conexão e funções de mobilidade do
adesivo (fluidez, transferência, penetração, umedecimento e solidificação) [31].
Diferentes classes de polímeros são empregadas para colagem de madeira. Os
termofixos, com muitas reticulações, apresentam propriedades mecânicas superiores,
elevada resistência à umidade e a produtos químicos. Por estas razões, estes polímeros são
muito utilizados como adesivos estruturais. Os termoplásticos com suas longas cadeias,
apresentam em geral menores resistências ao calor, a esforços mecânicos e a umidade [34].
Dentre os adesivos utilizados para colagem da madeira, destaca-se o poli(acetato de
vinila) (PVAc) por apresentar baixo custo, facilidade e segurança no manuseio, além de ser
inodoro, não inflamável, de secagem rápida e alta estabilidade. No entanto, o PVAc
apresenta limitações em condições úmidas e temperaturas elevadas. Diferentes autores
estudam formas de melhorar essas limitações, como Kaboorani e Riedl [35, 36], que
adicionaram dois tipos de nanoargilas hidrofílicas e melanina ao PVAc, e conseguiram
melhorar sua estabilidade térmica e reduzir a hidrofilicidade. A combinação com ureia
formaldeído (UF) também foi estudada para melhorar as propriedades mecânicas deste
material [37].
Adesivos à base de formaldeído, dependendo da estrutura molecular, podem
apresentar excelentes resistência à umidade e baixo custo. Moya et al. [38] estudaram a
adição de montmorillonita a ureia-formaldeído e observaram um aumento na resistência da
união e na estabilidade térmica. A adição de pentaborato de amônio a ureia-formaldeído foi
estudada por Gao et al. [39] que demostraram melhorias na colagem e nas propriedades
mecânicas.
Alguns adesivos podem apresentar toxicidade elevada. Adesivos de ureia-
formaldeído podem liberar baixas concentrações de gás de formaldeído a partir de produtos
de madeira coladas, especialmente sob condições quentes e úmidas. O formaldeído pode
reagir com as proteínas do corpo humano e causar irritação e inflamação das membranas de
olhos, nariz e garganta, e ainda podem ser cancerígenos. Os diisocianatos são
sensibilizadores capazes de causar asma ocupacional. Os adesivos termoplásticos quando
utilizados em espaços pequenos, sem ventilação, o solvente pode acumular-se no ar e até
provocar uma explosão, se for inflamado [40, 41].
O grande número de resíduos gerados, toxicidade e a necessidade de reduzir o uso de
derivados do petróleo fazem com que a busca por adesivos renováveis e biodegradáveis seja
ampliada. Zhang et al. [42] desenvolveram adesivos à base de amido de milho

23
23
copolimerizados com silano e acetato de vinila, onde foi alcançado um aumento na
estabilidade térmica comparado com o adesivo a base de milho, o que melhora a resistência
de ligação e resistência à água. O estudo de Cheng et al. [43] investigou as propriedades de
adesivos de proteína de soja SPA, modificado pelo 2-octeno-1- anidrido succínico (AOS),
em diferentes concentrações, e obteve melhorias na adesão.
Polímeros produzidos a partir do glicerol estão também sendo estudados para uso
como adesivos, como os poliésteres de glicerol com diácidos, usados como adesivos
cirúrgicos [5]. Glicerol, que tem formula molecular, 1,2,3 propanotriol, e nome comercial
glicerina [4,5], é um material de grande interesse científico e tecnológico. Com o crescente
interesse da comunidade internacional por combustíveis de menores impactos ambientais e
produzidos a partir de fontes renováveis, a produção de biodiesel (Figura 3) a partir de
gorduras animais e óleos vegetais foi uma das alternativas que mais se desenvolveu. No
entanto, a produção do biodiesel também gera resíduos e co-produtos que devem ser
aproveitados. A cada 9 m3 de biodiesel produzido, 1m3 de glicerol é gerado, Figura 3.
Figura 3 - Produção de biodiesel: reação de transesterificação [1].
O glicerol apresenta combinações físicas e químicas que fazem dele uma substância
muito versátil. É uma molécula flexível que forma ligações intra e intermoleculares
possibilitando 126 conformações moleculares. As indústrias alimentícias, de
farmacêuticos/cosméticos e químicos, são os maiores consumidores de glicerol [45, 46]. A
Tabela 2 mostra as propriedades citadas.

24
24
Tabela 2 - Propriedades físicas e químicas do glicerol adaptado da referência [3].
Formula química C3H8O3
Massa molecular 92,09 g/mol
Densidade 1,261 g/cm3
Viscosidade 1,5 Pa.s
Ponto de fusão 18,2 °C
Ponto de ebulição 290 °C
Calorias 4,32 kcal/g
Ponto de fulgor 160 °C
Tensão superficial 64,00 mNm-1
Coeficiente de temperatura - 0,00598 mN (Mk)-1
A indústria farmacêutica apresenta-se como a principal consumidora mundial de
glicerol, na forma de cremes (Figura 4), loções, agentes hidratantes e suavizantes [47–49],
além de pomadas e xaropes [11].
Figura 4 - Reação do glicerol com triglicerídeos para obtenção de monoglicerídeos (MAG): usados no
tratamento de cabelos, em sobremesas e cremes [51].
Em alimentos, além do uso como espessantes [52, 53] e adoçantes [54], o glicerol
pode ser adicionado à dieta de animais, pois uma vez absorvido, converte-se em glicose
para a produção de energia [55–57].
Na indústria química, o glicerol é aproveitado como reagente na produção de
nitroglicerina [58]. Quando em solução aquosa, resulta na obtenção do gás hidrogênio (H2)
[59]. Na presença de catalisadores metálicos e de hidrogênio, o glicerol pode ser
hidrogenado a 1,2-propanodiol (propilenoglicol), 1,3-propanodiol, ou etilenoglicol [60, 61].
A halogenação do glicerol pode formar produtos intermediários para a obtenção de resinas
epóxi, Figura 5.

25
25
Figura 5 - Halogenação do glicerol obtendo epiclorohidrina, produto base para a resina epóxi [62].
Através da desidratação do glicerol, a acroleína (propenal), Figura 6, é obtido um
produto intermediário importante na área industrial, empregado como herbicida ou para
fabricar aminoácidos [63].
Figura 6 - Reação de desidratação do glicerol, como produto a acroleína.
A síntese de materiais poliméricos a partir do glicerol tem também um grande
potencial, seja pela sua modificação química, ou por reação com poliácidos ou ácidos
graxos. Um exemplo é o poli(ftalato de glicerol) ou glyptal (Figura 7), produto da reação
entre glicerol e anidrido ftálico, usado como adesivo e em tintas, secativas ou não.
Figura 7 - Obtenção do poli(ftalato de glicerol), adaptado da referência [64].
2.2 Polímeros de glicerol
Pesquisas científicas são realizadas para o aprimoramento dos polímeros de glicerol
existentes, bem como para o desenvolvimento de novas formas de utilização para esses
materiais.

26
26
2.2.1 Síntese e aplicações de polímeros de glicerol
Diferentes polímeros podem ser obtidos a partir do glicerol, como mostra a Tabela 3.
Tabela 3 - Síntese de polímeros a partir do glicerol [2, 3, 65]
Reação Monômero Polímero
Glicerol e
uréia/CO2/dicarboimidazol Carbonato de glicerol
Poliésteres, policarbonatos,
poliuretanas e poliamidas
Glicerol e ácidos carboxílicos Éster de glicerol Poligliceróis
Glicerol e H2 Propileno glicol Poliésteres, policarbonatos,
poliuretanas
Desidratação do glicerol,
seguida da oxidação da
acroleína
Ácido acrílico Copolímeros e poli(ácido acrílico)
Combinação com poliácidos e
óleos vegetais - Reação de
policondensação e oxidativas,
ácido sebácico e lático
Glicerol Poliésteres, polímeros termofixos e
oligômeros do glicerol
Policarbonatos de glicerol podem ser obtidos por diferentes rotas. Umas dessas rotas
é com monóxido de carbono e oxigênio com catalisadores metálicos (Cu ou PdCl2); outra é
com dióxido de carbono (CO2) tendo como catalisador óxido de magnésio/bases orgânicas,
dentre outros; outra rota é a reação com ureia na presença de ácidos de Lewis. Carbonatos de
dialquila também podem ser utilizados na presença de uma gama de catalisadores como, por
exemplo, hidróxido de potássio, de sódio, óxidos de cálcio e magnésio. Estes são bastante
utilizados em aplicações biomédicas e farmacêuticas [66–68]
Os poliuretanos de glicerol podem ser utilizados para transporte de medicamentos
para controlar sua liberação ou como estruturas preparadas na engenharia de tecidos. Uma
das formas de fabricação dos poliuretanos de glicerol é a partir de poliésteres de glicerol que
reagem com diisocianato de isoforona (IPDI), diisocianato de hexametileno(IDH) ou ácido
oleico [69–73].
Poliésteres de glicerol reagem com aminas (ex: etileno diamina, 1,3-diamino-2-
hydroxi-propano) para obterem poli(éster amida) (Figura 8), usados em produtos para
cuidados pessoais [74–77].

27
27
Figura 8 - Esquema da síntese de poli(éster amida) [75].
Copolímeros são estudados na busca de melhores propriedades que aquelas
oferecidas pelos homopolímeros. Neste sentido, éter de diglicidila e propileno glicol são
usados como monômeros para produzir novos copolímeros (Figura 9) na presença de
diferentes catalisadores [78]. Poli(monometacrilato de glicerol) e o poli(metacrilato de
benzila) foram sintetizados para obtenção de nanopartículas esféricas do copolímero [79].
Figura 9 - Esquema da Síntese de copolímeros de éter de diglicidil de glicerol [78, 80].
Poliésteres de glicerol podem ser alcançados partindo da reação entre glicerol e
diácidos/anidridos de diferentes cadeias e massas molares tais como ácidos ftálico, sebácico,
fumárico e anidridos maleico, ftálico [81, 82]. Estes poliésteres podem ser sintetizados

28
28
através da reação do glicerol com diversos ácidos ou seus respectivos anidridos como, por
exemplo, ácidos dicarboxílicos como sebácico, adípico, maleico, etc., por policondensação,
que consiste na condensação sucessiva de grupos funcionais reativos existentes nos
materiais iniciais, aumentando o tamanho das moléculas até essas atingirem o tamanho de
uma cadeia polimérica [83]. A Tabela 4 exemplifica alguns ácidos comumente usados na
síntese de poliésteres.
Tabela 4 - Estruturas químicas de vários ácidos/anidridos que podem ser usados para produção de poliésteres
através da reação com o glicerol.
A degradação térmica e a cura antecipada durante a síntese são os grandes problemas
dessa reação, que podem ser evitados mantendo o balanceamento estequiométrico,
utilizando atmosfera inerte e mantendo a temperatura de reação correta, que normalmente
ocorre entre 120 e 250º C, assim como o controle do número ácido (NA), por titulação [21,
68, 84].
Nome Estrutura química Nome Estrutura química Nome Estrutura química
Ácido malônico
Ácido
tartárico
Ácido Málico
Ácido succínico
Ácido
Fumárico
Ácido
Ascórbico
Ácido
Glutárico
Ácido
Itacônico
Ácido
Aspártico
Ácido Adípico
Ácido Cítrico
Anidrido
Maléico
Ácido Pimélico
Ácido Ftálico
Anidrido Ftálico
Ácido Sebácico
Ácido
Cumárico
Anidrido
Succínico

29
29
A policondensação é um dos métodos mais utilizados na produção de polímeros de
alta massa molar. Diversas reações podem ocorrer simultaneamente durante uma reação de
policondensação, sendo que a principal reação implica na interação de dois grupos
funcionais, que pertencem a diferentes moléculas, resultando na formação de uma nova
molécula, com possibilidade de diferentes estruturas e propriedades físicas e químicas [85].
Agach et al. [86] sintetizaram poli(succinato de glicerol) e em seguida acrescentaram
ácido lauroil ou glicerol α-monolaurate, sem solvente nem catalisador, totalmente de origem
biológica, com tamanhos controláveis e topologia, como novos agentes tensoativos. O
poli(succinato de glicerol) (mistura de reagentes com razões molares diferentes) foram
introduzidos em um balão equipado com um aparelho de Dean-Stark e a mistura foi
aquecida a 190ºC, sob agitação mecânica constante e pressão atmosférica por 24 h. A
mesma síntese foi preparada para ácido lauriol e poli(succinato de glicerol), e o ácido lauriol
e glicerol α-monolaurate em poli(succinato de glicerol), só que com 12h e 24h,
respectivamente. Estes lauroil poliésteres formaram espumas abundantes e estáveis,
apresentaram uma grande capacidade de molhabilidade e exibiram excelentes propriedades
de solubilização micelar quando comparados com os padrões comerciais. Tais agentes
tensoativos de origem biológica e oligoméricas biodegradáveis são excelentes candidatos
como substitutos de ésteres de glicerol, como surfactantes para cosméticos.
Baharu et al. [87] conduziram pesquisas para sintetizar novos polímeros elásticos via
poliesterificação de glicerol (G) com ácidos succínico (AS) e azelaico (AZ), como um valor
agregado para um subproduto de óleo de palma. Os reagentes pré-misturados foram
colocados num balão de três bocas, equipado com um condensador de entrada, gás
nitrogênio e termômetro. A mistura de monômeros foi aquecida e misturada de 160-165 °C,
em um banho de óleo de silicone durante 2h em atmosfera de nitrogênio. O pré-polímero
resultante das composições (1:1:0; 1:0.5:0.5; 1:0.75:0.25; G:AZ:AS, respectivamente) foram
utilizados para preparação de película sem purificação adicional e lançados as placas de Petri
e deixados num forno a 125 °C durante 48 h. A adição de AS na formulação melhorou a
massa molar do produto final. A estabilidade térmica aumenta à medida que a concentração
de AZ aumenta. A eficiência com que a água é removida é crucial para as propriedades
desejadas do produto final. Estes materiais podem ser ainda mais explorados por
transmitirem biofuncionalidade para os polímeros, por exemplo, por biomoléculas
associadas, tais como proteínas.
Zhang et al. [88] sintetizaram e caracterizaram poliésteres com e sem ramificações
de glicerol e ácido adípico. Os reagentes foram misturados e aquecidos em um balão de três

30
30
bocas a 140 ºC com óxido de dibutil estanho como catalisador e conduzido até o termino da
reação, removendo a água liberada pela purga do vaso reacional com um fluxo contínuo de
N2. HBPEs de estequiometria que variaram de 1,52-2,16 foram sintetizados e modelados
utilizando o formalismo de Macosko e Miller (cálculo de massa molar) para validar o
modelo. A correspondência entre o peso molecular determinado e os previstos foi
considerada excelente.
Uma avaliação técnico-econômica da produção de poliésteres a partir de glicerol e
ácido adípico para ser integrado a instalações de produção de biodiesel foi realizada por
Bueno et al. [89]. O processo de reação foi realizado em dois reatores de tanque agitado,
simultaneamente, em 148ºC e 0,01 MPa, sendo o tempo de residência diferentes entre eles.
No reator, ácido adípico e glicerol (1:1; 1:1,5; 1:2, respetivamente) foram misturados em
balão de fundo redondo de 500 mL equipado com um agitador mecânico, um condensador
de Claisen, com adaptador de vácuo e N2. Os dados experimentais mostraram que a
proporção molar de 1:1 é a mais adequada e o processo não só é tecnicamente viável, mas
também economicamente promissor, resultando em custos para o polímero de 1,7 € / kg
quando valores de 5 € / kg de mercado podem ser encontrados.
Poli(sebacato de glicerol) (PGS) com proporções molares de 1:1, 2:3 e 3:2 de
glicerol e ácido sebácico foram sintetizados através de uma reação de policondensação, para
um substituto de estroma corneano por Salehi et al. [90]. Os reagentes foram misturados em
um frasco de vidro hermeticamente fechado e imerso em banho de silicone, aquecido a 120 °
C sob fluxo de gás N2 e agitado com um rotor a 50 rpm em vácuo (20 kPa) durante 24h. O
pré-polímero foi curado a 135 °C em uma placa de Petri, em forno de vácuo (a 20 kPa).
Estudos in vitro sugerem que o PGS com diferentes composições é satisfatoriamente
biocompatível em células da córnea. Para aplicações oftálmicas, a sobrevivência da célula
sobre os filmes foi bem sucedida.
Outros autores [91] produziram fibras elastoméricas por electrospinning (core/shell),
usando o núcleo/invólucro de poli(álcool vinílico) (PVA) como um escudo temporário e
PGS pré-polímero (misturados em proporção molar de 2:3, sob uma atmosfera de N2 a 130º
C durante 24 h). Em seguida o PGS foi reticulado por tratamento térmico e o PVA foi
parcialmente removido por dissolução em água. A citocompatibilidade em combinação com
as fibras macias e propriedades mecânicas elastoméricas fazem do PGS 2:3 um material
fibroso promissor para aplicações em engenharia de tecidos moles.

31
31
2.2.2 Uso de polímeros de glicerol como adesivos
Os adesivos de glicerol começaram o seu uso na década de 1930, com a produção da
resina glyptal, resultante da reação do glicerol com anidrido ftálico. No entanto, o
surgimento dos produtos petroquímicos como o polietileno, poliamidas e polipropileno, com
propriedades mecânicas, de processamento e flexibilidade melhores, tornara-os esquecidos.
No entanto, os “polímeros do petróleo” com longo tempo de degradação, acima de 100 anos,
e grande acúmulo de descartáveis, aumentaram o interesse de pesquisadores por fontes
biodegradáveis e renováveis como os polímeros de glicerol.
Os adesivos de polímeros de glicerol apresentam potencial para uso em diversas
áreas como química e biomedicina e os estudos em nanotecnologia abriram uma
oportunidade para desenvolvimento de uma nova geração desses adesivos. Krinstaff et al.
[92] sintetizaram polímeros de glicerol como adesivos oftalmológicos para a reparação de
lacerações de córnea, o poli(succinato-co-metacrilato de glicerol) (PGLSA-MA), com os
metacrilatos nas extremidades da cadeia, em diferentes gerações (G0, G1, G2 e G3). A
aplicação destes híbridos copolímeros lineares-dendríticos fotoligáveis a uma laceração 4,1
mm linear seguida por fotólise na presença de um foto-iniciador resulta em fechamento da
ferida. A formulação G1 teve o melhor desempenho, e resistiu a uma pressão de cerca de
170 mm Hg, bem acima do normal, a pressão intraocular (15 e 20 mm Hg).
O Copolímero PGS-co-LA de poli(sebacato de glicerol) (PGS) e ácido láctico (LA)
foram sintetizados por Chen et al. [5] em substituição a selantes fibrina e colágeno, pois há
sérias preocupações sobre a contaminação desses por vírus transmitidos pelo sangue, para
investigar a sua força adesiva e citotoxicidade. O PGS (razão molar 1:1, glicerol e ácido
succínico) e os copolímeros (razão molar 1:1:1 glicerol, ácido succínico e ácido láctico)
foram sintetizados em 3 temperaturas, 110, 115 e 120 ºC, por policondensação. A adição de
ácido láctico a PGS causou aumento do grau de polimerização antes da reticulação
significativa e a presença de maiores grupos álcool sobre as cadeias de polímeros, o que
aumenta o cisalhamento e a força de ligação de hidrogênio com os tecidos vedantes,
melhorando significativamente a citocompatibilidade em comparação com o PGS puro, além
de terem uma força de aderência mais elevada do que os selantes de fibrina, tornando-o um
candidato promissor a selante.
Mamiński et al. [93] sintetizaram dois polímeros hiperramificados partindo do
carbonato de glicerol e 1,1,1 – trihidroximetilpropano ou bisfenol A, como novos
componentes para adesivos em madeira. O carbonato de glicerol e 1,1,1 –

32
32
trihidroximetilpropano foram obtidos a 160º C em um balão de 3 bocas equipado com um
agitador magnético, um termômetro e funil de gotejamento durante 17h. O carbonato de
glicerol e bisfenol em proporção de 1:5 molar foi sintetizado da mesma forma do anterior,
porém em 20h a 170º C. O desenvolvimento de sistemas mostrou claramente que adesivos
baseados em poligliceróis hiper-ramificados foi possível, pois a resistência ao cisalhamento
das linhas de colagem foi maior do que os de madeira.
2.3 Obtenções e aplicações de nanowhiskers de celulose (NWC)
A crescente conscientização da população em relação aos problemas ambientais
somada a uma legislação ambiental cada vez mais rigorosa em muitos países tem aumentado
o interesse da comunidade científica e industrial por materiais biodegradáveis e renováveis.
Compósitos de matrizes poliméricas que utilizam materiais biodegradáveis com dimensões
nanométricas são de grande interesse por apresentarem melhorias nas suas propriedades
ópticas, térmicas e mecânicas, com pequenas frações de fase dispersa. Nanowhiskers de
celulose (NWC) ganharam destaque por serem um material de fonte renováveis com grande
disponibilidade na natureza (fibras vegetais) e custo relativamente baixo [94].
Uma grande variedade de fibras vegetais tem despertado interesse científico e
tecnológico pelas suas propriedades químicas, físicas e mecânicas, como algodão, sisal,
coco, juta, rami, curauá, fibra de bagaço de cana de açúcar, soja, resíduos de madeira, casca
de arroz e trigo [95–97]. As fontes vegetais de celulose possuem constituintes como
hemicelulose, ácidos graxos, pigmentos e lignina. Os materiais lignocelulósicos possuem
grupos hidroxila polares na superfície devido à predominância de celulose e lignina. No
processo de biopolimerização (glicoses formando cadeias poliméricas) regiões amorfas são
geradas, dando descontinuidade aos sítios cristalinos [98]. A interação entre cadeias
moleculares de celulose na região cristalina e amorfas está representado na Figura 10. No
intuito de melhorar a cristalinidade, e consequentemente suas propriedades, a obtenção de
nanowhiskers de celulose tem sido estudada.

33
33
Figura 10 - Esquema de interação entre cadeias moleculares de celulose na região cristalina e amorfas [45]
Os whiskers de celulose são regiões que crescem sob condições controladas, o que
permite a formação de regiões altamente cristalinas, conferindo fibras celulósicas de formas
cilíndricas, retangulares ou quadradas na base. Com pelo menos uma das dimensões igual ou
menor que 100 nm, são considerados nanowhiskers. Sua estrutura ordenada pode conferir
mudanças significativas em algumas propriedades tais como propriedades ópticas,
magnéticas, elétricas, ferromagnéticas, dielétricas, condutividade, biodegradabilidade,
capacidade de renovação, não toxicidade e elevada resistência mecânica [47–49, 99].
A hidrólise ácida é o método mais empregado para obtenção dos nanowhiskers de
celulose. O processo diminui as regiões amorfas na celulose, enquanto os segmentos
cristalinos continuam intactos, desde que tempos muito longos não sejam usados. Diferentes
morfologias e graus de cristalinidade podem ser obtidos de acordo com a variação da origem
da celulose e também das condições de hidrólise [97, 100]. Nanowhiskers podem ser
isolados a partir de diferentes fontes celulósicas, vegetais (algodão, casca de arroz e de coco,
carauá e bambu, mandacaru, etc.) [100–103] e animais (tunicados) (Tabela 5) [104, 105].

34
34
Tabela 5 – Condições de hidrólise e suas matérias prima - adaptado de, De Jesus Silva [106].
Na hidrólise ácida podem ser utilizados os ácidos sulfúrico e clorídrico. O ácido
clorídrico é menos comum em relação à hidrólise com ácido sulfúrico, que nas
concentrações de 64 a 65% p/p, constitui o ácido que mais vem sendo utilizado em estudos
para o isolamento dos NWC. O processo para isolá-los a partir de matérias primas
celulósicas consiste de várias etapas, tendo início no pré-tratamento da matéria prima,
passando pela hidrólise, seguida da neutralização, e podendo chegar à filtração da suspensão
de NWC [106, 107].
A adição dos nanowhiskers de celulose (NWC) pode melhorar as propriedades de
compósitos, devido à elevada área superficial específica (m2 / g de material). Além disso,
NWC são biodegradáveis, apresentam baixa densidade e são produzidos de fontes
renováveis. [45, 95, 103].
Santos et al. [108] avaliaram a influência de nanowhiskers de celulose de linter de
algodão nas propriedades físicas de filmes de gelatina de tilápia plastificado com glicerol,
produzidas pela técnica de fundição. A resistência à tração, módulo de elasticidade e a
barreira de vapor de água dos filmes foram melhoradas pela adição de NWC enquanto o
alongamento e a transparência do filme foram prejudicados.
Araki e Mishima [10] adicionaram pequenas quantidades de nanowhiskers de
algodão preparado por hidrólise com ácido clorídrico, como subsequente enxerto na
Matéria prima
Ácido
Concentração
(p/p)
Temperatura
(ºC)
Tempo
Relação
ácido/matéria
prima (mL/g)
Polpas kraft branqueadas de
conífera e folhosa H2SO4 64 45 Variável Variável
Conífera e papel de filtro H2SO4 64 45 45 10
Fibra de rami H2SO4 65 55 30 -
Línter de algodão H2SO4 64 50 45 17,54
Papel de filtro H2SO4 64 45 60 20
Fibra de algodão H2SO4 65 63 30 -
Polpa kraft branqueada de
conífera H2SO4 65 70 10 10
Polpa kraft branqueada de
conífera HCl 4N 80 225 35
Línter de algodão H2SO4 65 45,54,63 e 72 30 -
Celulose microcristalina H2SO4 65 72 30 -
Tunicados H2SO4 45 55 780 -

35
35
superfície de monometóxi poli(etileno glicol) (mPEG), e o compósito mostrou notável
birrefringência(formação de dupla refração) e uma estabilidade térmica aumentada.
Mauricio et al. [109] descrevem a preparação de uma composição de hidrogel de
nanowhiskers celulose (NWC) de bagaço de cana com amido em um ultrassom assistida por
emulsão, para liberação controlada de fármacos. A taxa de liberação de droga tornou-se 2,9
vezes mais lenta quando NWC foi adicionado.
NWCs extraídos com ácido sulfúrico (H2SO4) foram usados por Gao et al. [110]
para melhorar o desempenho de adesivo à base de farinha de soja. Os resultados mostraram
que a utilização do NWC na formulação adesiva melhorou a resistência à água em 20% e
foram observados na seção transversal do adesivo curado, menos falhas e fendas, bem como
uma superfície lisa, após a incorporação de NWC.
Os efeitos dos NWC em polímeros biodegradáveis como o PLA e amido estão bem
disseminados na literatura. Lee et al. [111] estudaram a incorporação de 1, 2 e 3% em peso
de NWC em matriz PLA usando um método de moldagem em solução, onde os NWC
mostraram efeitos intensificadores sobre as propriedades de tração, estabilidade térmica e
dimensional, além do aumento da cristalinidade.
Alves et al. [112] investigaram as propriedades de NWC e gelatinas, de diferentes
concentrações, em filmes de amido (3%) plastificado com glicerol (20%), onde as
propriedades mecânicas dos filmes estudados aumentaram significativamente com as
concentração de gelatina e NWC dentro dos filmes de amido de milho. Os filmes com baixo
teor de gelatina e de NWC apresentaram máximas temperaturas de degradação.
Poli(sebacato de glicerol) com NWC foram estudados por Zhou et.al [113]
sintetizado por policondensação de 1:1 razão molar de glicerol (grau de pureza 99%) e o
ácido sebácico (pureza 99%) a 130 ° C durante 24 h sob atmosfera de N2 com agitação
mecânica. O pré-polímero foi dissolvido em álcool etílico. Diferentes concentrações (0, 1, 2,
3, e 4% em peso) de NWC foram misturadas com uma solução a 50% em peso de PGS pré-
polímero em solução de álcool etílico e agitada magneticamente durante 30 min em
temperatura ambiente. Os resultados mostraram que a adição de NWC em PGS resultou uma
melhoria significativa da resistência à tração e módulo de elasticidade, assim como a
densidade de ligações cruzadas e a hidrofilicidade dos PGs. Os resultados de DSC indicam
que a adição de NWC melhorou tanto a capacidade de cristalização quanto a capacidade de
mobilidade da cadeia do PGS.
Estudos dos resultados dos NWC em polímeros de glicerol apresentam-se muito
escassos, principalmente quando se trata de novos polímeros. Medeiros et al. [21]

36
36
sintetizaram um novo polímero biodegradável baseado em glicerol, anidrido succínico e
anidrido maleico, poli(succinato-co-maleato de glicerol), poli(GlySAMA), através de
policondensação de fusão e testado como uma matriz para os compósitos com nanowhiskers
de celulose (NWC) (em concentrações de 1, 2 e 4%). A polimerização (razão molar de 2:2:1
de glicerol, anidrido succínico, anidrido maleico respectivamente) foi realizada em um balão
de 500 ml equipado com agitador e condensador, a 130º C sob atmosfera de N2 com 0,18%
em peso de octanoato de estanho como catalizador. O curso da reação foi seguido pelo
número ácido (NA) do polímero através de titulação. Os resultados mostram que este
polímero à base de glicerol é termicamente estável, uma consequência provável da sua
estrutura, que é reticulado e, além disso, o comportamento de biodegradação dos compósitos
é dependente e controlado pela quantidade de NWC adicionado, o que demonstra que a
degradação de poli(Gly SAMA) pode ser controlada ajustando o conteúdo NWC no
polímero.
Na literatura não há descrições do uso do NWC em polímeros de glicerol para uso
como adesivos.

37
37
3 OBJETIVOS
O objetivo deste trabalho foi estudar os efeitos da adição de nanowhiskers de celulose
nas propriedades adesivas de polímeros obtidos a partir de glicerol e ácidos graxos e
anidrido maleico.
As principais contribuições do trabalho são:
• Estudar a aderência de poliésteres de glicerol: poli(adipato de glicerol),
poli(maleato de glicerol) e poli(citrato de glicerol), poli(ftalato de glicerol),
poli(sebacato de glicerol), poli(succinato de glicerol) em superfícies de
madeira.
• Entender os efeitos da adição de NWC nas propriedades de polímeros
biodegradáveis obtidos a partir de glicerol como adesivos para madeira.
• Entender o processo de envelhecimento dos polímeros biodegradáveis obtidos
a partir de glicerol em aplicação como adesivos para madeira, como também o
efeito da adição de NWC no processo de envelhecimento.

38
38
4 MATERIAIS E PROCEDIMENTOS
O procedimento experimental para a confecção dos polímeros e compósitos, como
também para suas caracterizações está apresentado no fluxograma ilustrado na Figura 11. A
etapa experimental consistiu da síntese e caracterização dos polímeros de glicerol, colagem
em dois tipos de madeiras com diferentes graus de rigidez, sendo as madeira de Pinus (Pinus
elliottii) e Angelim (Vatairea heteroptera Ducke), e em seguida testes de resistência
mecânica em corpos de prova submetidos a cisalhamento sob tração e posterior análise por
microscopia eletrônica de varredura (MEV). Um estudo de degradação dos compósitos foi
também realizado seguido dos ensaios de cisalhamento sob tração e análise por MEV.
Figura 11 - Fluxograma do procedimento experimental

39
39
4.1 Materiais
4.1.1 Reagentes
Os reagentes utilizados na síntese dos polímeros e copolímeros foram glicerina
(95%, Chemco, indústria & comércio LTDA), anidrido maleico (99%, Vetec) e os ácidos
cítrico (99,5%), succínico (99%), adípico (99,8%), sebácico (99%) e ftálico (99,5%) (Vetec).
Além desses, foram utilizados fenolftaleína (0,5% Química moderna), álcool isopropílico
(IPA, 96,65%, Química moderna) Metil-etil-cetona (MEK, 99%, Vetec), tolueno (99,5%,
Vetec), hidróxido de potássio (KOH, 85%, Nuclear), catalisador octanoato de estanho
C4H9SnO (OH) (Sigma aldrich) e gás argônio (White Martins). Fibras de algodão foram
utilizadas para extração dos nanowhiskers de celulose. Madeiras do tipo Pinnus ellioti
(pinus) e Vatairea heteroptera Ducke (Angelim) adquiridas em madeireira local, foram
utilizadas para a confecção das juntas coladas.
4.2 Procedimento experimental
4.2.1 Extração dos NWC
Os NWC foram extraídos por hidrólise ácida com uma razão de fibras de algodão por
solução de 1g por 20 ml. Estas, por sua vez, foram tratadas em uma solução aquosa de ácido
sulfúrico (98%) (1:1) a 45 °C sob agitação constante por 60 minutos, promovendo a quebra
das cadeias da fase amorfa da celulose. As amostras foram centrifugadas a 3000 rpm em
centrífuga (Centerium Scientific K3 Series) por 15 min para retirada do ácido sobrenadante,
sendo o processo repetido por duas vezes. Em seguida, as amostras seguiram para diálise em
membrana por 24 h, sob fluxo contínuo de água, para eliminação de resíduos do processo de
extração até o material atingir pH neutro. Após cálculo da concentração, foram diluídos para
0,004% e liofilizadas por 2 dias em um liofilizador Enterprise II Terroni, adaptado de [21,
103]. Foram caracterizados por AFM e MET e medidos por um programa de análise de
dados (image j).
4.2.2 Síntese dos polímeros e copolímeros
Para a síntese dos polímeros de glicerol (Figura 12) manteve-se a razão estequiométrica
1:1 dos grupos [OH]:[COOH]. Os polímeros foram sintetizados em temperaturas de 120 °C

40
40
a 150 °C, sob agitação constante e atmosfera de argônio. Foi adicionado 0,01% em peso do
catalisador 2-etilexanoato de estanho). Os cálculos dos números ácidos foram feitos para
cada reação seguindo as equações de Carothers, onde o valor de extensão de polimerização
crítica para polímeros (Pc) e da funcionalidade média do polímero obtido (ʄav) são
destacados na Eq. (1) e na Eq. (2). A monitoração da reação foi feita por titulação em uma
bureta automática (Metrohm) com uma solução padronizada de hidróxido de potássio, com
concentração de 0,11 g/ml. Durante a síntese, amostras de polímero foram retiradas para que
a reação não parasse. Foram pesadas amostras de aproximadamente de 0,2-0,3g de polímero
para serem solubilizadas em 5 ml de solução de metil-etil-cetona/tolueno/álcool isopropílico
(25:50:25 v/v). Para calcular o número ácido (NA), foram utilizados a massa de polímero
solubilizada e o volume de KOH adicionado e sua concentração (Eq.3) [84].
(1)
(2)
(3)
Onde Pc é valor de extensão de polimerização crítica, ʄav é a funcionalidade média, NA o número ácido, n é
número de mols, C concentração, V volume, M massa molar.
Figura 12 - Esquema do procedimento experimental da síntese dos polímeros de glicerol.

41
41
A partir das quantidades estequiométricas de grupos COOH e OH dos monômeros,
um valor de extensão polimérica crítica (p) de 0,555 foi encontrado para o PGC e de 0,833
para os demais polímeros. O valor equivalente ao número de ácido crítico de KOH / g de
cada polímero foi calculado (Tabela 6).
Tabela 6 - Número ácido crítico para cada polímero.
Pré-polímeros Massa molar dos ácidos (g/mol) Nº ácido crítico
PGA - poli(adipato de glicerol) 146,14 270,31
PGM - poli(maleato de glicerol) 98,06 351,10
PGC - poli(citrato de glicerol) 192,12 221,28
PGPh - poli(ftalato de glicerol) 166,14 246,55
PGSu - poli(succinato de glicerol) 118,09 312,55
PGS - poli(sebacato de glicerol) 202,24 212,79
Após cerca de 2h de síntese (tendo variação dependendo do ácido utilizado), os pré-
polímeros foram formados e utilizados posteriormente na etapa de colagem (Figura 13).
Figura 13 – Produção dos corpos de prova dos polímeros: a) aplicação dos polímeros no molde, b) polímeros
no molde e c) polímeros curados.

42
42
4.2.3 Preparação dos Compósitos
As preparações dos compósitos seguiram o mesmo procedimento citado para os
polímeros. No entanto, os NWC foram adicionados ao pré-polímero, na forma de pó, em
percentuais de 5, 10 e 20% m/m, até a completa homogeneização, e em seguida utilizados na
etapa de colagem.
Para a fabricação dos compósitos foram utilizados os polímeros que apresentaram os
maiores valores de resistência nos ensaios de cisalhamento e sintetizados copolímeros entre
eles, com a finalidade de se obter polímeros flexíveis, rígidos e intermediários, visando
melhorar ainda mais a resistência dos adesivos.
O número ácido (NA) foi usado como parâmetro para as composições dos
compósitos. Os NWC em pó foram adicionados aos polímeros quando atingido o seu
número ácido crítico (Tabela 6). Com os compósitos homogeneizados, as madeiras foram
coladas, prensadas, e levadas à cura e caracterizadas. Amostras dos compósitos foram
também preparadas e curadas em molde de silicone para caracterizações. A Figura 14
representa algumas possíveis estruturas químicas da reação de formação dos compósitos de
glicerol.

43
43
OH
OH
OH
+
O
OH
O
OH
O
OH
O
OH
C1
0H
30 O
4S
n
OO
O
AN
IID
RID
O
MA
LÉ
ICO
GLIC
ER
OL
O
OH
O
O
O
O
O
O
R R
O
O
OH
O
O
O
O
O ORR
O
Es
tufa
12
0ºC
O OH
O
O
O
OR
OR
O O
NW
C
O
O
OR
O
OOR
O
OH
O
OH
OH
O
OH
OH
OH
OH
nP
GS
u / N
WC
nO
OR
OR
O
O
O
O
OH
O
OH
OH
O
OH
OH
OH
OH
PG
A / N
WC
OR
OR
O
O
O
O
O
OH
O
OH
OH
O
OH
OH
OH
OH
48
h
O
O
O
OOR
OO
O
O
R
O
OH
O
OH
OH
O
OH
OH
OH
OH
n
PG
AS
u / N
WC
OR
O
O
OH
O
O
O
O
OR
O
OH
O
OH
AC
ID0
S
UC
CIN
ICO
+
O
OH
O
OH
AC
IDO
A
DIP
IC O
O
OR
OO
OR
O
O
O
O
O
OH
O
OH
OH
O
OH
OH
OH
OH
n
PG
AM
/ N
WC
O
OR
O
O
OH
O
OO
R
O
O
OH
O
OH
+O
OO
n
OO
O+
O
OH
O
OH
OR
O
O
O
OH
O
OO
R
O
n
PG
MS
u / N
WC
OR
O
O
OOR
O
O
O
O
O
OH
O
OH
OH
O
OH
OH
OH
OH
PG
M / N
WC
AC
IDO
A
DIP
IC O
AC
ID0
S
UC
CIN
ICO
AC
ID0
S
UC
CIN
ICO
AC
ID0
S
UC
CIN
ICO
AN
IID
RID
O
MA
LÉ
ICO
AN
IID
RID
O
MA
LÉ
ICO
Figura 14 – Esquema das possíveis estruturas da reação do glicerol com diácidos, seus respectivos pré-
compósitos e os compósitos pós-cura.

44
44
4.2.4 Colagem dos adesivos de polímeros de glicerol
Antes da realização da colagem, placas de madeira de pinus e angelim com
dimensões de 20 cm de comprimento, 10 cm de largura e 0,5 cm de espessura e foram
lixadas com lixa grão 80, no sentido da fibra. Três gramas (3 g) do pré-polímero foram
aplicados na madeira com o auxílio de um pincel. Em seguida, as placas foram unidas e
prensadas em prensa hidráulica (Marconi) sob pressão de 3 toneladas-força, por 24 h, a 120
ºC. Para completar o processo de cura, os corpos de prova foram colocados em estufa na
temperatura de 120 ºC por 24 h, como mostra o esquema da Figura 15.
Figura 15 - Esquema das etapas de colagem: a) lixamento, b) aplicação dos adesivos, c) sobreposição das
placas de madeira e d) placas de madeira após prensagem.
4.2.5 Caracterizações dos polímeros e compósitos
4.2.5.1 Calorimetria diferencial de varredura (DSC)
As análises por calorimetria diferencial de varredura (DSC) foram realizadas em um
equipamento Shimadzu DSC-60 utilizando-se amostras de 8-10 mg em panela de alumínio,
numa faixa de temperatura de -50 a 300 ºC, sob taxa de aquecimento de 5 ºC/min e
atmosfera de nitrogênio (50 ml/min).

45
45
4.2.5.2 Termogravimetria (TG)
Análises termogravimétricas foram realizadas em um aparelho Shimadzu modelo
DTG 60H, usando 8-10 mg de amostra em cadinho de alumina sob atmosfera de nitrogênio
(50ml/min), e em uma faixa de temperatura de 25 a 600 ºC, com taxa de aquecimento de 5
ºC/min.
4.2.5.3 Espectroscopia na região do infravermelho por transformada de fourier (FTIR).
Todas as amostras dos polímeros de glicerol puros e com NWC, foram maceradas em
almofariz de ágata com auxílio de nitrogênio líquido e peneiradas em peneira ABNT 80#
(mesh - 32 µm) sendo em seguida pesadas e separadas para as análises. O equipamento
utilizado foi um espectrofotômetro Shimadzu Fourier Transform Infrared
Spectrophotometer, modelo IR Affinity-1 FTIR-8400S (Kyoto, Japan) modo ATR (reflexão
total atenuada), onde foram obtidos os espectros de absorção no infravermelho com
comprimento de onda de 4000 a 500 cm-1 e com resolução de 1 cm-1, 64 varreduras, para
identificação das ligações características dos poliésteres de glicerol.
4.2.5.4 Difratometria de raios X (DRX)
Na difração de raios X foi usado um equipamento DRX Shimadzu D6000, operando com
50kV, 100 mA e radiação de CuK (1,54 Å) e com ângulo 2θ entre 5 e 80º e um passo de 2º. O
DRX foi utilizado principalmente para caracterização dos NWC, pois os polímeros puros são
amorfos.
4.2.5.5 Microscopia eletrônica de varredura (MEV)
A interação entre polímero e reforço, assim como os corpos de prova após o
cisalhamento sob tração foram analisados por microscopia eletrônica de varredura (MEV)
em um LEO 1430 Zeiss, com voltagens de 10, 11 e 15 kV. Antes das análises, as amostras
foram fraturadas em nitrogênio líquido e recobertas com ouro em um EMITEC K550X.

46
46
4.2.5.6 Microscopia de força atômica (AFM)
A morfologia dos nanowhiskers de celulose (NWC) foi avaliada em um microscópio
de força atômica Shimadzu modelo SPM 9600. As imagens de AFM foram obtidas no modo
dinâmico, com uma taxa de varredura de 1Hz com ponta de Si com raio de curvatura inferior
a 10nm.
4.2.5.7 Microscopia eletrônica de transmissão (MET)
As amostras foram preparadas a partir da suspensão de nanofibras neutra, diluída e
submetida ao ultrasonificador (Branson 450) por 2 min. Uma gota desta suspensão foi
depositada sob uma grade de cobre com filme de Formvar (400 mesh – Ted Pella) e mantida
em dessecador por 24 h. Após este período, sob a grade com a amostra, foi depositada 1 gota
de uma solução 1,5% de acetato de uranila para corar e foi mantida no dessecador por 48 h.
Em seguida as amostras foram analisadas em um microscópio eletrônico de transmissão
Tecnai™ G2 F20, no modo STEM, em campo escuro (Dark Field).
4.2.5.8 Ensaio mecânico de cisalhamento sob tração
Os corpos de prova para os ensaios mecânicos foram confeccionados de acordo com
a norma ASTM D906 (Figura 16). Os ensaios de cisalhamento sob tração foram realizados
em máquina universal de ensaios mecânicos Shimadzu modelo AG-X 10 kN, a uma
velocidade de ensaio de 0,5 mm/min, temperatura de 21 ºC, sendo a área colada de 5,5 cm2.
A resistência ao cisalhamento foi determinada utilizando-se 7 corpos de prova para cada
uma das condições avaliadas.
Figura 16 - Corpos de prova de acordo com a norma ASTM D906.

47
47
4.2.5.9 Degradação por envelhecimento acelerado
Os ensaios de degradação foram realizados em câmara de envelhecimento Atlas UV-
test, com lâmpadas de ultravioleta (UV-A) seguindo a norma ASTM G 154 (Standard
Practice for Operating Fluorescent Ultraviolet (UV) Lamp Apparatus for Exposure of
Nonmetallic Materials). Todos os corpos de prova submetidos ao envelhecimento acelerado
(Figura 17) foram ensaiados em ciclos alternados, 20h de radiação a 60 °C e 4h de vapor
d’água, até atingir os tempos estimados (150, 250 e 350 h). Após cada tempo de ensaio, os
corpos de prova foram cisalhados sob tração.
Figura 17 - Corpos de prova a serem ensaiados na câmara de envelhecimento acelerado.

48
48
5 RESULTADOS E DISCUSSÃO
5.1 Síntese dos polímeros puros.
Amostras de polímeros puros foram preparadas para cura em molde de silicone por
48h em atmosfera de argônio. A cor, consistência e viscosidade dos polímeros de glicerol
variam de acordo com o número ácido (NA). Algumas possíveis estruturas para a síntese de
policondensação do glicerol com os ácidos é mostrada na Figura18.
OH
OH
OH
+
O
OHO
OHO
O
O
O
O
O
O
O
O
O
O
O R
R
R
O
OH
O
OH
O
O
O
O
O
O
O
O
O
O
O
O
R
R
R
O
OH
O
OH O
O
O
O
O
O
O
O
O
O
O
O
R
R
R
O
OH
O
OH
O OH
OH
O
O
O
O
O
OO
O
O O
OH
O
O
O O
OH
O
O
O O
OH
R
R
R
R
R
R
C10
H30
O4Sn
OOH
O
OH
OOO
O
O
O
O
O
O
O R
O R
O R
O
O
O
O
O
R
O
O
O
O
O
O
O
O
R
O
O
R
ÁCIDO SUCCINICO
ÁCIDO ADIPICO
ÁCIDO CÍTRICO
ÁCIDO FTÁLICO
ANIDRIDO MALEICO
ÁCIDO SEBACICO
GLICEROL
O
OH
O
O
O
O
O
O
R
R
O
O
O
O
O
O
O
O
R
ROH
O
OH
O
O
O
O
O
O R
R
O
Estufa 120ºC
O
O
O
OO
O
O O
OH
O
O
O O
OH
R
R
R
R
OH
O
O
R
O
OH
O
O
O
O
O
R
O
OH
O
O
O
O R
O R
O
O
PGSU
PGA
PGS
PGC
PGPh
PGM
Figura 18 - Sínteses do glicerol com diácidos, seus respectivos pré-polímeros e os polímeros curados.

49
49
5.2 Espectroscopia na região do infravermelho por transformada de
fourier (FTIR) dos polímeros puros.
Os espectros de FTIR dos diferentes tipos de poliésteres de glicerol são apresentados
na Figura 19. As bandas características de ligações ésteres são apresentadas entre 1670-1750
cm-1 (C=O) e 1020-1280 cm-1 (C–O) o que corrobora com o que foi visto no gráfico da
Figura 18 com bandas entre 1710-1724 cm-1 e 1060-1250 cm-1. Além de apresentar as
bandas características de ligações C-H, entre 2932-2946 cm-1. As bandas não intensas em
torno de 3460 cm-1 correspondem aos grupos das hidroxilas encontrados nos polímeros
curados, possivelmente da água da policondensação ou ainda do grupo OH da molécula de
glicerol, que mesmo em proporção molar estequiométrica, não reagiu [87, 91, 114, 115].
4000 3500 3000 2500 2000 1500 1000 500
3461
1126
1136
3461
2940
2946
2932
1710
1716
1710
1724
1720
PGC
PGS
PGSu
PGPh
PGA
Comprimento de onda (cm-1)
Tra
nsm
ita
ncia
(u
.a)
PGM
1720
Figura 19 – Espectros de FTIR dos polímeros de glicerol. PGA - poli(adipato de glicerol), PGM - poli(maleato
de glicerol), PGC - poli(citrato de glicerol), PGPh - poli(ftalato de glicerol), PGSu - poli(succinato de glicerol),
PGS - poli(sebacato de glicerol).
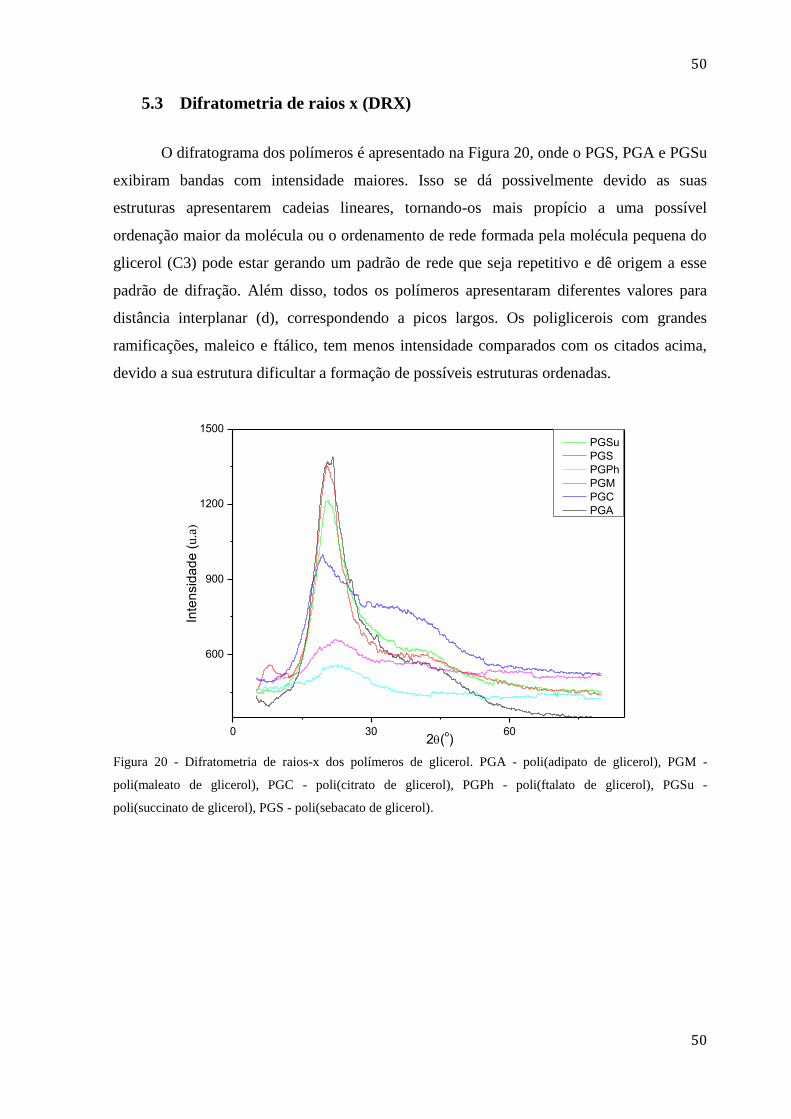
50
50
5.3 Difratometria de raios x (DRX)
O difratograma dos polímeros é apresentado na Figura 20, onde o PGS, PGA e PGSu
exibiram bandas com intensidade maiores. Isso se dá possivelmente devido as suas
estruturas apresentarem cadeias lineares, tornando-os mais propício a uma possível
ordenação maior da molécula ou o ordenamento de rede formada pela molécula pequena do
glicerol (C3) pode estar gerando um padrão de rede que seja repetitivo e dê origem a esse
padrão de difração. Além disso, todos os polímeros apresentaram diferentes valores para
distância interplanar (d), correspondendo a picos largos. Os poliglicerois com grandes
ramificações, maleico e ftálico, tem menos intensidade comparados com os citados acima,
devido a sua estrutura dificultar a formação de possíveis estruturas ordenadas.
0 30 60
600
900
1200
1500
Inte
nsid
ad
e (
u.a
2(o)
PGSu
PGS
PGPh
PGM
PGC
PGA
Figura 20 - Difratometria de raios-x dos polímeros de glicerol. PGA - poli(adipato de glicerol), PGM -
poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh - poli(ftalato de glicerol), PGSu -
poli(succinato de glicerol), PGS - poli(sebacato de glicerol).

51
51
5.4 Calorimetria exploratória diferencial (DSC)
Através do DSC estudou-se o comportamento térmico dos poliésteres de glicerol,
Figura 21. O primeiro ciclo de aquecimento mostra picos endotérmicos relativo a suas
transições térmicas, como mostrado na Tabela 7.
-50 0 50 100 150 200 250
-8
-4
0
Flu
xo d
e c
alo
r (
mW
)
Temperatura ( °C)
PGA
PGC
PGSu
PGS
PGPh
PGM
(a)
50 100 150 200 250
-1
0
1
Temperatura ( °C)
Flu
xo d
e c
alo
r (
mW
)
PGM
PGPh
PGC
PGSu
PGS
PGA
(b)
Figura 21 - Gráficos de DSC dos polímeros: (a) aquecimento e (b) resfriamento. PGA - poli(adipato de
glicerol), PGM - poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh - poli(ftalato de glicerol),
PGSu - poli(succinato de glicerol), PGS - poli(sebacato de glicerol).

52
52
O PGS por ter cadeia linear longa (10 carbonos), conseguem uma organização
molecular, como discutido no item 5.3 e mostrado na Figura 19, possivelmente por seus
carbonos reativos ficarem distantes, impossibilitando assim muitas ligações cruzadas, com
Tg em -31 ºC e Tm1 em -3 ºC e a Tm2 em 40 ºC, valores estes próximos aos relatados em
estudos anteriores [6, 115], ou ainda, o mais provável que essa Tm1 próximo de 0 ºC está
associado a água na amostra, os picos podem aparecer ligeiramente inferior a 0 °C devido a
impurezas dissolvidas pela umidade ou no gás de purga, e a Tm2 seja uma relaxação
molecular do polímero (fraca transição endotérmica), ou seja, a transição vítrea (Tg) aparece
como um pico de fusão endotérmico (Tm) [116]. Os polímeros PGA e PGSu apresentam Tg
em -30 e -12 ºC, respectivamente. O PGC, PGM e o PGPh com grupos muito rígidos tendem
a apresentar Tg’s mais elevadas, em 33, 49 e 59 ºC, respectivamente. Nas temperaturas entre
170 e 250 ºC observa-se outra transição térmica possivelmente causada por evaporação da
água da policondensação, corroborando com o que é observado na termogravimetria (Figura
21). O resfriamento, por não ser feito com arrefecimento de nitrogênio, não apresentou
temperaturas de cristalização no PGS. Uma observação importante a ser feita sobre esses
polímeros estudados é que o único que apresentou temperatura de fusão e que há relatos na
literatura foi o PGS, sendo assim classificado como elastômero. Os outros, PGA, PGSu,
PGC, PGPh e PGM não apresentam Tm, sendo, portanto, mais próximos de polímeros
termofixos.
Tabela 7 - Valores de Tg, Tm e ΔH dos polímeros de glicerol.
5.5 Termogravimetria (TG)
As Figuras 22 a e b, mostram respectivamente os gráficos de perda de massa e
derivadas das perdas de massa dos polímeros, e na Tabela 8 os valores da Tonset (°C) Tendset
Amostra Tg (ºC) Tm1 (ºC) Tm2 (ºC) ΔH (J/g)
PGS - poli(sebacato de glicerol) -31 -3 40 13,43
PGA - poli(adipato de glicerol) -30 - -
PGSu - poli(succinato de glicerol) -12 - -
PGC - poli(citrato de glicerol) 33 - - -
PGPh - poli(ftalato de glicerol) 59 - - -
PGM - poli(maleato de glicerol) 49 - - -

53
53
(°C), pico de DTGA (°C) e resíduos a 600°C (% massa). As curvas apresentam duas etapas
de degradação, exceto para o PGC com três etapas. A primeira alteração na curva de
derivada (DTGA) para o PGC ocorre, entre 170 e 260 ºC, corresponde à perda de moléculas
de água do processo ou a perda de voláteis, podendo estar relacionada à presença de
hidroxilas que não reagiram. Nesse mesmo intervalo, 170-260 ºC, ocorre a primeira derivada
dos outros polímeros estudados, PGS, PGA, PGSu, PGM e PGPh, pelo mesmo motivo
apresentado para o PGC. Entre 265 e 350 ºC, no PGC, por ser hidrofílico, ocorre a quebra
das ligações de grupos laterais contendo carbonos terciários, facilitando assim o início do
processo de formação de radicais livres que iniciam a cisão do polímero, como é observado
pelo valor de sua Tonset em 267 °C, a menor dentre os polímeros aqui mostrados. Acima de
350 até aproximadamente 500 ºC, as ligações das cadeias poliméricas são degradadas
através das rupturas [117].

54
54
0 50 100 150 200 250 300 350 400 450 500 550 600 650 700 750
0
25
50
75
100
PGM
PGC
PGSU
PGS
PHPh
PGA
% M
assa
Temperatura( ºC ) (a)
0 50 100 150 200 250 300 350 400 450 500 550 600 650 700
0,01
0,00
-0,01
-0,02
-0,03
-0,04
-0,05
% M
ass
a
Temperatura ( °C)
PGA
PGC
PGM
PGSU
PGS
PGPh
(b)
Figura 22 - Gráficos de TGA e DTGA dos polímeros de glicerol. PGA - poli(adipato de glicerol), PGM -
poli(maleato de glicerol), PGC - poli(citrato de glicerol), PGPh - poli(ftalato de glicerol), PGSu -
poli(succinato de glicerol), PGS - poli(sebacato de glicerol).

55
55
Os polímeros PGS, PGA e PGSu, por apresentarem cadeias com maior mobilidade
molecular, entre os polímeros estudados, tendem a degradar em temperaturas maiores. O
PGS com cadeia linear (10 carbonos), portanto com mobilidade molecular alta, exibe valor
de Tonset de 337 °C, o maior entre os polímeros analisados [6]. O PGA e o PGSu com cadeias
de seis e quatro carbonos, respectivamente, porém com mobilidade menor que o PGS,
apresentam valores de Tonset próximos, em aproximadamente 310 ºC [87, 118].
O PGM e o PGPh, por terem cadeias com grupos laterais grandes, e por conseguinte,
mobilidade dificultada, degrada em temperaturas mais baixas, quando comparadas com as
cadeias mais flexíveis. Os valores de Tonset de 275 °C para o PGPh e 290 °C para o PGM são
mostrados na Tabela 8.
Tabela 8 - Valores de Tonset, Tendset, DTGA (pico) e resíduo dos gráficos de termogravimetria dos polímeros de
glicerol.
AMOSTRAS PGA PGM PGSu PGS PGC PGPh
Tonset (°C) 309,1 290,8 310,4 337,1 267,5 275,8
Tendset (°C) 417,8 453,0 430,8 485,3 434,1 412,6
Pico de DTGA
(°C) 365,3 380,1 386,1 415,9 377,4 350,7
Resíduos a 600°C
(% massa) 2,5 19,2 2,4 3,7 12,1 0,3

56
56
5.6 Ensaio de cisalhamento sob tração dos polímeros puros em madeira de
Pinus elliottii.
Observa-se na Tabela 9 os valores de resistência ao cisalhamento dos polímeros de
glicerol em madeira de pinus. O polímero de glicerol com anidrido maleico, PGM,
apresentou entre os polímeros de glicerol utilizados a maior tensão, com valor 26% maior
que a cola comercial branca (PVAc). O PGA exibiu a menor tensão, sendo 36,3% menor que
a cola branca. O PGS, PGSu, PGC e PGPh apresentaram tensão de cisalhamento máximo
próximo da cola branca, cerca de 22% menor.
Tabela 9 - valores de resistência ao cisalhamento dos polímeros de glicerol em madeira de pinus.
Detalhes das rupturas das juntas coladas testadas em cisalhamento de acordo com a
norma ASTM D5573, com os polímeros aderidos à madeira são mostrados na Figura 23. O
poli(adipato de glicerol) (PGA), poli(maleato de glicerol) (PGM) e o poli(succinato de
glicerol) (PGSu) apresentaram ruptura coesiva na madeira, as colas comerciais poli(acetado
de vinila) (COLA BRANCA) e cianoacrilato (TEK BOND) além do poli(ftalato de glicerol)
(PGPh) e poli(sebacato de glicerol) (PGS) exibiram ruptura adesiva, enquanto que, para a
junta colada com poli(citrato de glicerol) (PGC) a ruptura foi mista (coesiva e adesiva).
Polímeros de glicerol
Resistência ao cisalhamento máximo
(MPa)
PGA - poli(adipato de glicerol) 0,67 ± 0,24
PGS - poli(sebacato de glicerol) 0,83 ± 0,48
PGPh - poli(ftalato de glicerol) 0,82 ± 0,40
PGM - poli(maleato de glicerol) 1,34 ± 0,67
PGSu - poli(succinato de glicerol) 0,96 ± 0,47
PGC - poli(citrato de glicerol) 0,80 ± 0,39
Cianoacrilato - TEK BOND 2,16 ± 1,06
PVAc - COLA BRANCA 1,06 ± 0,63

57
57
Figura 23 - Corpos de prova de cisalhamento sob tração em madeira de pinus ensaiados.
A madeira de pinus, apresentada na imagem microscópica da Figura 24, evidencia
sua porosidade, conferindo-lhe características de material com baixo módulo de elasticidade.

58
58
(a)
(b)
Figura 24 - MEV da madeira de Pinus, com aumentos de a)500x e b)1000x.

59
59
As Figuras 25 e 26 mostram as imagens do MEV dos polímeros com suas
respectivas rupturas , as quais foram mostradas na Figura 1.
(a)
(b) Figura 25 – MEV após falha sob tração do a) PGA e b) PGC.

60
60
(a)
(b)
Figura 26 – MEV após falha sob tração do a) PGSu e b) PGM.
Na Figura 27 ficam evidentes as camadas de adesivo que recobrem a madeira, o que
corrobora com que foi visto na Figura 22, a ruptura adesiva do PGPh e PGS.
Os valores de tensão de cisalhamento e as imagens do MEV mostram o potencial
dos polímeros de glicerol para esse tipo de madeira, os quais em sua maioria suportaram as
tensões até o rompimento da madeira de pinus.

61
61
(a)
(b)
Figura 27 - Imagens de MEV do a) PGPh e b) PGS após ensaio de cisalhamento sob tração.

62
62
5.7 Ensaio de cisalhamento sob tração dos polímeros puros em madeira de
angelim
Como vários corpos de prova com madeira de pinus apresentaram fratura coesiva na
madeira, testes foram realizados com corpos de prova de Angelim, que apresenta
propriedades mecânicas superiores. Corpos de prova de cisalhamento sob tração na madeira
de angelim após ensaiados são mostrados na Figura 28. Os resultados mostraram que os
polímeros aderidos na madeira apresentaram uma ruptura coesiva adesiva.
Figura 28 - Corpos de prova de cisalhamento sob tração na madeira de angelim após ensaiados.
Observa-se na Tabela10 os valores de tensão de cisalhamento máximo para cada
polímero de glicerol, assim como dos adesivos comerciais. O PGM e o PGA exibiram entre
os polímeros de glicerol utilizados as maiores tensões de cisalhamento máximo e o PGPh a
menor. A utilização do anidrido maleico como agente compatibilizante de fibras naturais já
é conhecida por melhorar a interação entre a madeira e o poli(glicerol maleato),
corroborando com o melhor valor de tensão de cisalhamento máximo encontrado para o
PGM [119, 120]. Os resultados de tensão do adesivo comercial cola branca são muito
próximo dos melhores resultados dos polímeros de glicerol, do PGM e PGA. O PGSu
apresentou valores intermediários entre os polímeros, 36,6% menor que o PGM, e quando
comparado com a cola branca essevalor foi 38,6% menor. O PGS e o PGC foram 47,5%

63
63
menor, o PGPh apresentou valor de tensão cerca de 50% menor que PGM, PGA e cola
branca, tendo sido o pior resultado nesse estudo. Quando comparado com o cianoacrilato
(tek bond) todos os polímeros apresentaram valores inferiores de resistência ao
cisalhamento.
Tabela 10 - Valores de resistência ao cisalhamento máximo dos polímeros de glicerol em madeira de angelim.
As Figura 29 e 30 exibem as imagens de MEV das superfícies de falha de corpos de
prova testados sob tração na madeira de angelim utilizando PGC, PGM, PGPh, PGA, PGS e
PGSu. Nas imagens 29a, b e c, observa-se a ruptura adesiva com fratura frágil, mostrada nos
círculos, deixada por alguns espaços vazios, particularidade da estrutura molecular rígida
dos polímeros, que em sua estrutura apresentam dupla ligação (PGM), anel aromático
(PGPh) ou molécula pequena com grupos laterais grandes (PGC) dificultando a mobilidade.
A ductilidade do PGA, PGS e PGSu fica explícita nas imagens de MEV da Figuras 30 a, b e
c, uma vez que suas estruturas com cadeias lineares na sua maioria formado por carbono (C)
e hidrogênio (H), lhes conferem uma mobilidade que os tornam flexíveis, suportando
deformações antes de romperem.
Polímeros de glicerol
Resistência ao cisalhamento máximo
(MPa)
PGA - poli(adipato de glicerol) 1,52 ± 0,321
PGS - poli(sebacato de glicerol) 0,83 ± 0,173
PGPh - poli(ftalato de glicerol) 0,77 ± 0,436
PGM - poli(maleato de glicerol) 1,53 ± 0,124
PGSu - poli(succinato de glicerol) 0,97 ± 0,208
PGC - poli(citrato de glicerol) 0,84 ± 0,178
Cianoacrilato - TEK BOND 3,12 ± 0,532
PVAc - COLA BRANCA 1,58 ± 0,180
a

64
64
(a)
(b)
(c)
Figura 29 - Imagens de MEV do a) PGC, b) PGM, C) PGPh, PGSu após ensaio de cisalhamento sob tração na
madeira de angelim.

65
65
(a)
(b)
(c)
Figura 30 - Imagens de MEV do a) PGA, b) PGS e c) PGSu após ensaio de cisalhamento sob tração na madeira
de angelim.

66
66
5.8 Extração e caracterização dos NWC.
O processo para obtenção dos NWC por extração em ácido sulfúrico é relatado em
trabalhos anteriores como sendo de elevada eficiência [47, 121, 122]. Após diluição, as
amostras de NWC foram submetidas à análise por microscopia de força atômica (AFM) e
microscopia eletrônica de transmissão (MET). Na Figura 31 são apresentadas imagens do
NWC após hidrolise ácida, diluído para liofilização e após a liofilização. A liofilização deixa
os NWC em forma de um pó fino, posteriormente utilizado para a fabricação dos
compósitos, como pode-se observar na Figura 31.
Figura 31 - Imagem do NWC após hidrólise ácida, diluído para liofilização e após a liofilização.
Para avaliação da morfologia, os nanowhiskers foram depositados sobre uma
superfície de mica recém-clivada numa concentração de 0.001% (m/v). A avaliação da
morfologia dos nanowhiskers por AFM e MET é importante, pois, as propriedades
apresentadas pelo adesivo irão depender da estrutura em que o material celulósico se
encontra (Figuras 32 e 33). Através das imagens obtidas, pode-se constatar que a morfologia
apresentada é característica de nanowhiskers de celulose, com estruturas alongadas na forma
de agulhas e diâmetro inferior ao comprimento longitudinal [123]. Pode-se concluir também
que a hidrólise ácida não provocou a formação de estruturas particuladas, que possuem
propriedades diferentes às apresentadas pelos nanowhiskers de celulose. Os nanowhiskers
mostrados nas Figuras 32 e 33 exibiram diâmetro médio de 12,7 nm e comprimento médio
de 291,3 nm, que resulta em uma razão de aspecto de 22,9.

67
67
Figura 32 - Imagem dos NWC por microscopia de força atômica (AFM).
Figura 33 - Imagens dos NWC por microscopia eletrônica de transmissão (MET)
O difratograma de raios-x apresentado na Figura 34 mostra os picos característicos
dos NWC, corroborando com a literatura para o polimorfismo cristalino da celulose em
aproximadamente 15, 17, 22, e 34,5º [21, 124].

68
68
10 20 30 40
0
50
100
150
200
250
300
350
400
450
Inte
nsity (
u.a
)
2
Figura 34 – Difratograma de raios-x dos Nanowhiskers de celulose.
5.9 Espectroscopia na Região do Infravermelho por Transformada de
Fourier (FTIR) dos compósitos.
O espectro contendo as bandas características do NWC pós-liofilização, na forma de
pó, são expostos na Figura 35. As bandas em 1028 e 1058 cm-1 são atribuídas as ligações
(C-O) localizados nas posições 3 e 6 da cadeia celulósica, 1104 cm-1 indica ligação C-H de
álcool, em 1159 cm-1 ocorre alongamento da ligação C-O-C encontrado na cadeia da xilose,
um dos componentes presentes na celulose. além disso, 1426 e 2917 cm-1 são bandas de
estiramento C-H e grupos -CH2- da celulose cristalina. Em 1646 cm1 é atribuído ao O-H
absorvido, normalmente de moléculas de água. Em 3292 e 3339 cm-1 tem-se bandas de
ligações –O-H livre. Valores correspondentes são encontrados na literatura [125, 126].

69
69
4000 3500 3000 2500 2000 1500 1000 500
Tra
nsm
itanc
ia (
%)
Comprimento de onda ( cm-1
)
1028
1058
1159
1426
1646
2917
3292
3339
1104
Figura 35 – Espectro de FTIR do nanowhisker de celulose (NWC).
As Figuras 36 e 37 apresentam os espectros dos polimeros PGA, PGM e PGSu e de
seus respectivos compósitos contendo 5, 10 e 20% em massa de NWC, pois como relatado
anteriormente, foram os polímeros escolhidos com os melhores resultados de resistência sob
tração. As bandas características de ligações ésteres são encontradas em torno 1715 cm-1
(C=O) 1115 cm-1 (C-O) 2930 cm-1 (C-H) e em torno de 3400cm-1 (O-H) [127] o que
corrobora com o que foi visto no gráfico com bandas entre 1710-1724 cm-1 e 1060-1250 cm-
1. As bandas características de ligações C-H, entre 2932-2946 cm-1, também podem ser
observadas, e em torno de 3460 cm-1 correspondem aos grupos das hidroxilas (valores
mostrados no item 5.1). As bandas em torno de 2930 cm-1 (C-H) e 3400 cm-1 (O-H) e outra
em 1036 cm-1 (C-O) são típicas das ligações celulósicas (Figura 30) [128].

70
70
4000 3000 2000 1000
PGA 20%
PGA 10%
PGA 5%
PGA
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
(a)
4000 3000 2000 1000
PGM 20%
PGM 10%
PGM 5%
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
PGM
(b)
Figura 36 - Espectros de FTIR dos polímeros (a) PGA, (b) PGM e seus compósitos com 5, 10 e 20% de NWC.

71
71
4000 3000 2000 1000
PGSu 20%
PGSu 10%
PGSu 5%
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
PGSu
Figura 37 - Espectros de FTIR do polímero PGSu e seus compósitos com 5, 10 e 20% de NWC.
Nas Figuras 38 e 39 são mostrados os espectros dos copolímeros PGMSu
(poli(maleato-co-succinato de glicerol)), PGASu (poli(adipato-co-succinato de glicerol)) ,
PGMA (poli(maleato-co-adipato de glicerol)) e seus respectivos compósitos com 5, 10 e
20% em massa de NWC. As bandas características para os poliésteres discutidas para os
polímeros PGA, PGM e PGSu (Figuras 36 e 37) também são encontradas nos copolímeros e
seus compósitos. Uma banda característica da estrutura molecular do anidrido maleico se
sobressai no PGM e nos copolímeros que contém o anidrido, C=C em aproximadamente
1640 cm-1 [129].

72
72
4000 3000 2000 1000
PGMSu 20%
PGMSu 10%
PGMSu 5%
PGMSu
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
(a)
4000 3000 2000 1000
PGASu 20%
PGASu 10%
PGASu 5%
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
PGASu
(b)
Figura 38 - Espectros de FTIR dos copolímeros a) PGMSu, b) PGASu, e seus compósitos com 5, 10 e 20% de
NWC.

73
73
4000 3000 2000 1000
PGMA 20%
PGMA 10%
PGMA 5%
Tra
nsm
itâ
ncia
(u
.a.)
Comprimento de onda (cm-1)
PGMA
Figura 39 - Espectros de FTIR do copolímero PGMA, e seus compósitos com 5, 10 e 20% de NWC.
5.10 Calorimetria Exploratória Diferencial (DSC) dos polímeros, copolímeros e
compósitos
As análises de DSC (Figuras 40 e 41) para os polímeros, copolímeros e os
compósitos exibem as transições vítreas (Tg) e outras duas transições a partir de 50 ºC. Os
valores das Tg´s obtidos dos gráficos, encontram-se na Tabela 11. A adição dos NWC
alterou de forma significativa as Tg´s dos compósitos de matriz de PGA, PGSu, PGASu e
PGMSu. A adição de 5% m/m de NWC reduziu em 16°C a Tg dos compósitos em relação
ao PGA, saindo de -21°C para -37°C. Nos compósitos PGA 10 e 20% os valores da Tg não
foram alterados de forma significativa em relação ao PGA, ficando em -36 e -35°C
respectivamente.
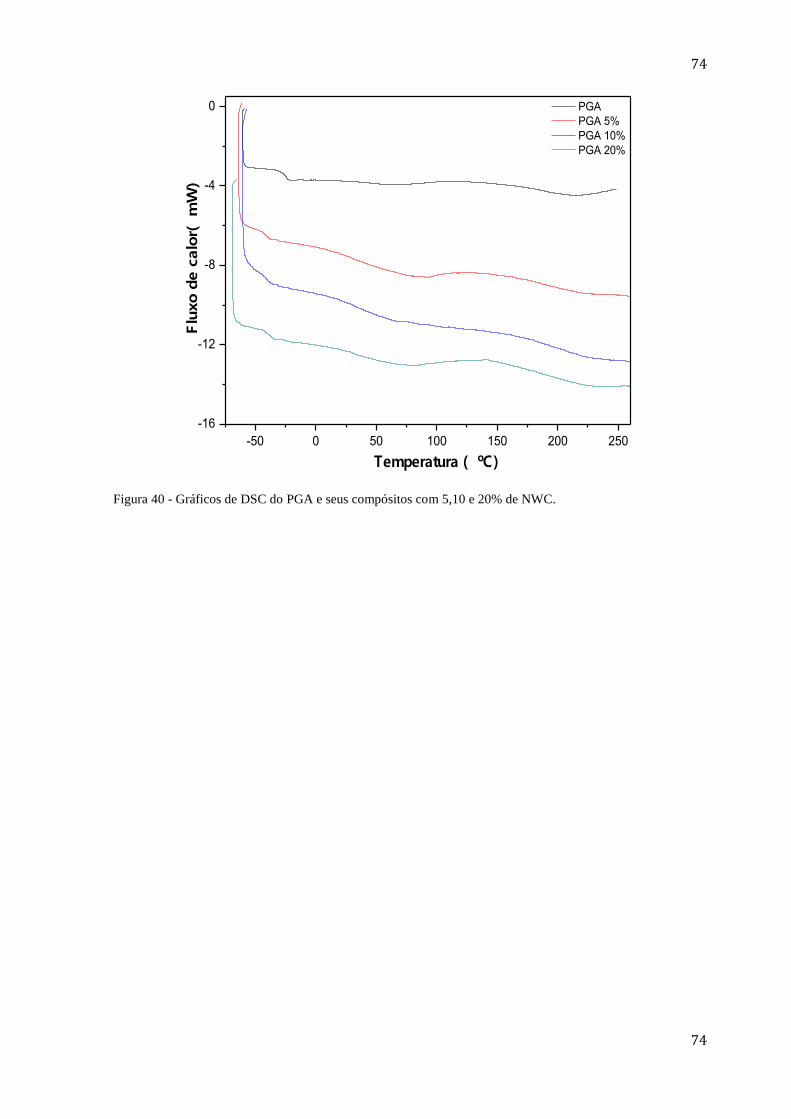
74
74
-50 0 50 100 150 200 250
-16
-12
-8
-4
0
Flu
xo d
e c
alo
r(m
W)
Temperatura ( ºC)
PGA
PGA 5%
PGA 10%
PGA 20%
Figura 40 - Gráficos de DSC do PGA e seus compósitos com 5,10 e 20% de NWC.

75
75
-50 0 50 100 150 200 250
-8
-4Flu
xo d
e c
alo
r (
mW
)
Temperatura( ºC)
PGM
PGM 5%
PGM 10%
PGM 20%
(a)
-50 0 50 100 150 200 250
-12
-8
-4
0
Temperatura ( ºC)
Flu
xo d
e c
alo
r (
mW
)
PGSu
PGSu 5%
PGSu 10%
PGSu 20%
(b)
Figura 41 - Gráficos de DSC do, a) PGM, b) PGSu e seus compósitos com 5,10 e 20% de NWC.
Nos compósitos do PGSu a redução mais significativa da temperatura de transição
vítrea foi no PGSu 20%, que apresentou redução de 4,2 °C no polímero e 20 °C no

76
76
compósito. Os poliésteres de glicerol com cadeias com carbonos lineares tendem a ser mais
maleáveis, como o PGA (Tg = -21°C) e PGSu (Tg = 4,2°C), e suas Tg´s ocorrem em valores
de temperatura próximos ou menores que 0°C. A incorporação dos NWC nesses polímeros
deslocou a transições vítreas para temperaturas mais baixas, o que ocorreu possivelmente
devido à estrutura da celulose apresentar muitos O-H (hidrofilicidade) e formar H2O,
evidenciado no evento térmico entre 50 e 100 °C, apenas nos compósitos, atuando como
uma espécie de ‘plastificante’, como relatado na literatura [17, 130]. Outra observação é a
redução do tempo de cura desses polímeros, que ocorreu em 48 h no polímero puro e passou
a ser de 24-30 h nos compósitos. Outros autores [131] relataram a forte interação entre carga
e matriz alterando os valores de Tg. Nos compósitos com PGM, sua estrutura molecular
dificultou a mobilidade, tornando-os mais rígidos e, consequentemente, sua Tg ocorre em
temperaturas altas. Mesmo os NWC formando H2O ente 50 e 100 ºC, diferentemente do
PGA e PGSu, sua transição vítrea não foi alterada de forma significativa, como mostrado na
Tabela 11.
Tabela 11 - Valores da tg do PGA, PGM, PGSu e seus compósitos com 5,10 e 20% de NWC.
Tg (°C) - Tg (°C) - Tg (°C)
PGA -21 PGM 50 PGSu 4,2
PGA 5% -37 PGM 5% 51 PGSu 5% 5,3
PGA 10% -35 PGM 10% 53 PGSu 10% -1,5
PGA 20% -35 PGM 20% 54 PGSu 20% -20
Nas Figuras 42 e 43 são mostrados os gráficos dos copolímeros PGASu, PGMA,
PGMSu e seus respectivos compósitos com 5,10 e 20% de NWC. No PGMSU a Tg ocorre
em um valor intermediário (20°C) entre o PGSu e PGM, isso porque a estrutura do PGSu
apresenta 4 carbonos apenas (baixa maleabilidade) e PGM tem mais rigidez, deslocando-a
para temperatura maiores (Tabela12). Com os compósitos ocorre o mesmo efeito do PGSu,
ocorrendo uma redução da Tg (efeito da H2O).

77
77
-50 0 50 100 150 200 250
-12
-8
-4
Temperatura( ºC)
Flu
xo d
e c
alo
r (
mW
)
PGASu
PGASu 5%
PGASu 10%
PGASu 20%
(a)
-50 0 50 100 150 200 250
-8
-4
0
Temperatura ( °C)
Flu
xo
de
ca
lor
(mW
)
PGMA
PGMA 5%
PGMA 10%
PGMA 20%
(b)
Figura 42 - Gráficos de DSC do a) PGASU, b) PGMA e seus compósitos com 5,10 e 20% de NWC

78
78
-50 0 50 100 150 200 250
Flu
xo d
e c
alo
r (
mW
)
PGMSu
PGMSu 5% NWC
PGMSu 10% NWC
PGMSu 20% NWC
Temperatura ( ºC) Figura 43 - Gráficos de DSC do PGMSu e seus compósitos com 5,10 e 20% de NWC.
Assim como no PGMSu, o PGMA apresentou Tg intermediária (Tabela 12), entre
PGA e PGM, porém nos seus compósitos não houve alteração significativa. Nos compósitos
do PGASu, a Tg reduziu consideravelmente, de 1,6°C para -23ºC (PGASu 20%) ratificando
o efeito ‘plastificante’ da H2O, já discutido anteriormente.
Tabela 12 - Valores da tg do PGMSu, PGMA, PGASu e seus compósitos com 5,10 e 20% de NWC.
Tg (°C) - Tg (°C) - Tg (°C)
PGMSU 20 PGMA -18 PGASu 1,6
PGMSU 5% -4 PGMA 5% -17 PGASu 5% -18
PGMSU 10% -3,5 PGMA 10% -16 PGASu 10% -20
PGMSU 20% -7 PGMA 20% -16,9 PGASu 20% -23

79
79
5.11 Estudo da estabilidade térmica dos NWC, polímeros e compósitos por TG
Na Figura 44 é apresentada a análise de TGA e DTGA dos nanowhiskers. A celulose
tem naturalmente caráter hidrofílico, como observado pela DTGA no primeiro evento de 25
°C até 105 °C, indicado pela perda de H2O por evaporação. No entanto, esta perda de massa
pode ser também atribuída a compostos de baixa massa molar presentes na superfície dos
NWC. O evento térmico ocorrido entre 200 °C a 400 °C é associado a decomposição da
celulose (processo de pirólise). É sabido que dependendo do tempo de extração por hidrólise
ácida, os grupos hidroxilas da celulose (OH) são substituídos por grupos sulfatos (SO42-),
onde os mesmos diminuem a energia de ativação, causando o fracionamento das cadeias e
como consequência a redução da temperatura na qual ocorre a degradação. Acima de 450 °C
cinzas e compostos com cadeias degradadas são formados [17, 131–136].
0 100 200 300 400 500 600 700
0
20
40
60
80
100
NWC
Derivada
Temperatura (°C)
ma
ssa
(%
)
-0,9
-0,8
-0,7
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
0,0
0,1
dM
/dT
(%/°C
)
Figura 44 - Gráfico de TGA do Nanowhisker de celulose e sua derivada.
Nas Figuras 45, 46 e 47 são mostrados os gráficos de TGA dos polímeros,
copolímeros e compósitos, como também as curvas de DTGA. Os dados de degradação
encontram-se resumidos na Tabela 13.

80
80
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura ( ºC )
PGA
PGA 5%
PGA 10%
PGA 20%
(a)
100 200 300 400 500 600
Temperatura ( ºC )
dm
/dt (
%/°C
)
PGA
PGA 5%
PGA 10%
PGA 20%
(b)
Figura 45 - Gráfico de a) TGA e b) DTGA do polímero PGA e seus compósitos com 5,10 e 20% de NWC

81
81
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura (°C)
PGM
PGM 5%
PGM 10%
PGM20%
(a)
(b)
Figura 46 - Gráfico de a) TGA e b) DTGA o polímero PGM e seus compósitos com 5,10 e 20% de NWC
100 200 300 400 500 600
Temperatura ( ºC )
dm
/dt (
%/°C
)
PGM
PGM 5%
PGM 10%
PGM 20%

82
82
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura (°C)
PGSu
PGSu 5%
PGSu 10%
PGSu 20%
(a)
100 200 300 400 500 600
Temperatura (ºC )
dm
/dt
(%/°
C)
PGSu
PGSu 5%
PGSu 10%
PGSu 20%
(b)
Figura 47 - Gráfico de a) TGA e b) DTGA do polímero PGSu e seus compósitos com 5,10 e 20% de NWC
O pico de degradação dos polímeros e compósitos é registrado acima dos 300°C,
onde inicia-se a ruptura das cadeias poliméricas, assim como ocorre a decomposição da
celulose. Nota-se que a adição dos NWC desloca para valores maiores a temperatura de
degradação quando comparado com os polímeros puros. Alguns autores como, Planes et.al
[137] atribuem esse aumento da estabilidade térmica, à compatibilidade dos NWC com a
matriz, resultado da forte interação das ligações de hidrogênio, como também da boa
dispersão da carga [21, 138].

83
83
Com base nos dados de degradação (Tonset), os compósitos PGA 20% e PGM 20%
aumentaram em 33°C e 27°C, respectivamente, a estabilidade térmica em relação a sua
matriz, enquanto que o PGSu 20% não apresentou qualquer aumento de estabilidade térmica
em relação ao polímero puro.
Tabela 13 -Valores da degradação dos polímeros PGA, PGM e PGSu e compósitos 5, 10 e 20% de NWC
Nas Figuras 48, 49 e 50 são mostrados os gráficos de TGA dos copolímeros e
compósitos, como também suas curvas de DTGA. Os dados de degradação encontram-se
resumidos na Tabela 14.
AMOSTRAS PGA PGA
5%
PGA
10%
PGA
20% PGM
PGM
5%
PGM
10%
PGM
20% PGSu
PGSu
5%
PGSu
10%
PGSu
20%
Tonset (°C) 292,54 321,9 365,97 325,44 196,27 253,35 206,33 223,19 302,9 303,54 294,67 299,87
Tendset (°C) 377,94 397,95 375,16 443,44 461,33 477,47 472,25 468,12 401,79 382,24 417,19 412,12
Pico de
DTGA (°C) 348,95 365,65 372,24 377,81 350,21 353,62 378,78 383,47 365,13 361,71 381,13 370,83
Resíduos a
600°C
(% massa)
7,84 7,82 6,27 4,50 25,31 25,56 20,89 14,27 5,26 1,34 4,24 6,69

84
84
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura (°C)
PGMSu
PGMSu 5%
PGMSu 10%
PGMSu 20%
(a)
100 200 300 400 500 600
Temperatura ( ºC )
dm
/dt (
%/°C
) PGMSu
PGMSu 5%
PGMSu 10%
PGMSu 20%
(b)
Figura 48 - Gráfico de a) TGA e b) DTGA do copolímero PGMSu e seus compósitos com 5,10 e 20% de
NWC.

85
85
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura (°C)
PGMA
PGMA 5%
PGMA 10%
PGMA20%
(a)
100 200 300 400 500 600
Temperatura ( ºC )
dm
/dt (
%/°C
)
PGMA
PGMA 5%
PGMA 10%
PGMA 20%
(b)
Figura 49 - Gráfico de a) TGA e b) DTGA do copolímero PGMA e seus compósitos com 5,10 e 20% de NWC.

86
86
0 100 200 300 400 500 600
0
20
40
60
80
100
% M
assa
Temperatura (°C)
PGASu
PGASu 5%
PGASu 10%
PGASu 20%
(a)
100 200 300 400 500 600
Temperatura ( ºC )
dm
/dt (
%/°C
)
PGASu
PGASu 5%
PGASu 10%
PGASu 20%
(b)
Figura 50 - Gráfico de a) TGA e b) DTGA do copolímero PGASU e seus compósitos com 5,10 e 20% de
NWC.
Com base nos dados de degradação (Tonset), os compósitos dos copolímeros PGMA
(poli(maleato-co-adipato de glicerol)) e PGMSu (poli(maleato-co-succinato de glicerol))
apresentaram aumento da estabilidade térmica de 48°C e 29°C, respectivamente, com a

87
87
incorporação da carga, em comparação com os polímeros, enquanto que para o PGASu
(poli(adipato-co-succinato de glicerol)) a estabilidade térmica permaneceu inalterada.
Tabela 14 - Valores da degradação dos polímeros PGASu, PGMA e PGMSu e compósitos com 5, 10 e 20% de
NWC.
5.12 Ensaio de cisalhamento sob tração dos polímeros puros e compósitos em
madeira de Angelim
Os valores de tensão máxima de cisalhamento dos polímeros poli(adipato de
glicerol) (PGA), poli(maleato de glicerol) (PGM) e poli(succinato de glicerol) (PGSu) e seus
compósitos com 5,10 e 20% de NWC são exibidos na Tabela 15, onde a influência da carga
fica evidente nos valores de tensão quando comparados com os polímeros puros.
Para os compósitos do PGA, observa-se um aumento da resistência ao cisalhamento
com a adição de NWC. O PGA 20% exibe os melhores resultados de resistência do grupo,
sendo 23% maior que o polímero puro. No caso do PGA 10%, observou-se uma redução da
resistência ao cisalhamento em comparação com o PGA 5%, provavelmente devido a
formação de aglomerados. O PGA apresenta a solidificação rápida (tempo em aberto) da
região da superfície, contudo a adição dos nanowhiskers em grande quantidade, 20% por
exemplo, e a colagem não sendo feita rapidamente, pode ocasionar aglomerados, que
influencia diretamente nas propriedades dos mesmos.
No grupo do PGSu e PGM, a incorporação da carga melhorou os valores de tensão,
como relatado por Medeiros et al. e outros [16, 21, 139]. Para os compósitos do grupo PGM,
observou-se o mesmo comportamento dos compósitos de matriz PGA, com uma redução da
resistência ao cisalhamento dos compósitos de PGM 10% em comparação com o PGM 5%,
provavelmente devido à formação de aglomerados. Os valores de resistência voltam a
AMOSTRAS PGASU PGASU
5%
PGASU
10%
PGASU
20% PGMA
PGMA
5%
PGMA
10%
PGMA
20% PGMSU
PGMSU
5%
PGMSU
10%
PGMSU
20%
TONSET (°C) 307,57 297,34 315,42 307,47 242,80 272,36 240,15 291,08 301,67 294,04 334,00 330,68
TENDSET (°C) 420,45 380,80 391,15 440,19 435,56 458,30 456,98 436,76 349,94 383,67 408,38 377,43
PICO DE
DTGA (°C) 357,67 351,27 371,96 398,40 347,86 380,06 391,36 368,55 335,28 358,31 378,78 355,97
RESÍDUOS
A 600°C
(% MASSA)
2,31 2,29 0,36 10,58 13,58 24,92 14,16 6,20 2,076 1,92 2,053 1,66

88
88
aumentar para o compósito PGM 20%. Já no caso dos compósitos de matriz PGSu, os
valores de resistência ao cisalhamento mais elevados foram observados para os materiais
com 10% de NWC. Os melhores resultados de tensão máxima de cisalhamento foram o
PGSu 10% e PGM 20%. Em todos os casos, os resultados indicam que a adição de NWC
elevou a resistência ao cisalhamento, entretanto, os resultados sugerem a influência de
fatores, como a processabilidade da reação, colagem, cura, e formação de aglomerados.
Tabela 15 – Resistência ao cisalhamento dos polímeros PGA, PGM e PGSu e compósitos com 5, 10 e 20% de
NWC.
Resistência ao
cisalhamento
(MPa)
- Resistência ao
cisalhamento
(MPa)
- Resistência ao
cisalhamento
(MPa)
PGA 1,54 0,11 PGM 1,54 0,14 PGSu 0,98 0,17
PGA 5% 1,61 0,04 PGM 5% 2,24 0,46 PGSu 5% 1,57 0,09
PGA 10% 1,45 0,05 PGM 10% 1,95 0,23 PGSu 10% 2,20 0,23
PGA 20% 1,90 0,04 PGM 20% 2,33 0,43 PGSu 20% 1,52 0,07
Para os compósitos dos copolímeros apresentados na Tabela 16, no grupo do PGMSu
a incorporação da carga aumentou os valores de tensão para todos os compósitos quando
comparados com o copolímero, chegando a um valor 28,6% maior no caso do PGMSu 20%.
O PGMSu 5 e 10% aumentaram 19 e 7%, respectivamente, em relação ao copolímero sem
NWC.
Para os compósitos do grupo do PGMA e PGASu a adição da carga melhorou as
propriedades de tensão, com exceção do PGASu 5 e 10%, com redução de 8 e 15%,
respectivamente, quando comparados com o PGASu sem NWC. O PGMA 5 e 10% tiveram
aumentos de 9 e 69%, respectivamente, em comparação com o PGMA sem NWC. O PGMA
20% com valores de tensão maiores, 29% e o PGASu 20% com valores 36 % a mais do que
os copolímeros.

89
89
Tabela 16 - Resistência ao cisalhamento dos copolímeros PGMSu, PGMA e PGASu e compósitos com 5, 10 e
20% de NWC.
Os compósitos com melhores resultados de resistência ao cisalhamento das juntas
coladas estão apresentados nas Tabelas 15 e 16, comparados com a cola branca e
cianoacrilato (Figura 51). O PGSu 10% e o PGM 10%, mostraram-se com valores de
resistência superiores a cola branca (com valor de tensão de 1,59 MPa) 27,2 e 18,3%
respectivamente. Os compósitos PGASu 20% e o PGM 20% se destacam com relação a
tensão em comparação com a cola branca, com valores de 38 e 31% a mais,
respectivamente. O compósito que mais se aproxima dos valores de tensão do adesivo
cianoacrilato (Tek Bond) é o PGASu 20%, com valor médio 17% menor. Os dados
apresentados na Figura 51 indicam que, embora o adesivo cianoacrilato tenha apresentado
resistência ao cisalhamento maior, a dispersão nos resultados foi também maior.
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
Ten
sao
Maxim
a (
N/m
m2)
cola branca
tek bond
PGA 10%
PGA 20 %
PGM 10%
PGM 20 %
PGSu 10%
PGSu 20 %
PGASu 10%
PGASu 20 %
PGMA 10%
PGMA 20 %
PGMSu 10%
PGMSu 20 %
Figura 51 - Comparação dos compósitos com melhores resultados de tensão com os adesivos comerciais.
Resistência ao
cisalhamento
(MPa)
- Resistência ao
cisalhamento
(MPa)
- Resistência ao
cisalhamento
(MPa)
PGMSU 1,57 0,17 PGMA 1,08 0,06 PGASu 1,89 0,16
PGMSU 5% 1,88 0,20 PGMA 5% 1,18 0,14 PGASu 5% 1,74 0,04
PGMSU 10% 1,68 0,23 PGMA 10% 1,83 0,23 PGASu 10% 1,59 0,03
PGMSU 20% 2,20 0,35 PGMA 20% 1,40 0,17 PGASu 20% 2,57 0,36

90
90
5.13 Análise fratográfica das superfícies de fratura por MEV após ensaio de
cisalhamento sob tração
Na Figura 52 são apresentas imagens das superfícies das amostras de angelim e de
pinus, na qual identificam-se para as duas madeiras, suas linhas vasculares. Na madeira de
pinus a quantidade de poros é visivelmente maior, o que facilita uma maior adesão, e a
porosidade baixa é um dos principais fatores que confere a madeira de angelim maior
resistência, consequentemente, uma adesão mais dificultada.
a) b)
Figura 52 - Micrografia da madeira de a) angelim e b) pinus após fratura criogênica.
Nas imagens da Figura 53 são apresentadas as superfícies dos polímeros PGA, PGM e
PGSu com a incorporação dos nanowhiskers de celulose após o ensaio de cisalhamento sob
tração. As micrografias mostram as características estruturais de cada polímero. O PGA
(Figura 53a) e o PGSu (Figura 53c) apresentam estruturas que sugerem mobilidade
molecular maior, comparados com o PGM (Figura 53b). Contudo, nas micrografias com
10% de NWC (Figura 53 d, e, f), observa-se além da deformação, a formação de vazios,
característicos de estruturas rígidas (NWC). O PGM (Figura 53 b) tem característica de
menor ductilidade, ocasionado pela mobilidade restrita do polímero, que é intensificada pela
adição de carga, Figura 53 e.

91
91
Figura 53 - Imagens de MEV do a) PGA, b) PGM, c) PGSu, d) PGA 10%, e) PGM 10% e f) PGSu 10%, após
ensaio de cisalhamento sob tração.
Na Figura 54 são mostradas as micrografias das superfícies de fratura dos
copolímeros PGMSu, (poli(maleato-co-succinato de glicerol)), PGMA (poli(maleato-co-
adipato de glicerol)) e PGASu (poli(adipato-co-succinato de glicerol)), e dos compósitos
com 10% de NWC. Nas micrografias da Figura 54 a e b, observa-se características de
ductilidade do PGSu e PGA, nas bordas das fendas deformadas, bem como a rigidez do
PGM nos vazios das próprias fendas. Na imagem do PGASu (Figura 54 c), a ductilidade é
também predominante, mostrando apenas as rugosidades, sem fendas. Nas micrografias 54
d, e, f, a rigidez dos NWC influencia diretamente na formação de fendas com característica
de arrancamento rápido. Apenas na Figura 54 f esse efeito é diminuído, pois o copolímero
tem maleabilidade alta, oriunda de dois polímeros lineares, PGSu e PGA. As setas indicam
as fendas.
a
d
b c
e f

92
92
Figura 54 – Micrografias de MEV do, a) PGMSu, b) PGMA, c) PGASu, d) PGMSu 10%, e) PGMA 10% e f)
PGASu 10%, após ensaio de cisalhamento sob tração, aumentado 2000x.
5.14 Degradação por envelhecimento acelerado
As amostras ensaiadas após envelhecimento acelerado em diferentes tempos, 150, 250
e 350 h apresentaram comportamentos distintos de resistência à degradação. Alguns
adesivos não resistiram ao envelhecimento, desprendendo totalmente da madeira no
primeiro ciclo (Figura 55). Alguns fatores como a colagem manual, os aglomerados de
NWC e a própria estrutura química do polímero, podem ter acarretado esse desprendimento.
Figura 55 - Corpos de prova do PGSu a) antes e b) após ciclo de 150h de envelhecimento.
a b c
d e f
a b

93
93
5.15 Ensaio de cisalhamento sob tração dos polímeros puros e compósitos em
madeira de Angelim após o envelhecimento acelerado e MEV
Os valores de resistência ao cisalhamento em função do tempo de degradação dos
compósitos da família do PGA e do PGM são exibidos na Figura 56.
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Te
nsa
o m
axim
a (
MP
a)
Tempo (h)
PGA
PGA 5%
PGA 10%
PGA 20%
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGM
PGM 5%
PGM 10%
PGM 20%
Figura 56 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos compósitos da família
do PGA e PGM
A Tabela 17, assim como as Tabelas 18 e 19, expõem para alguns compósitos,
variações nos valores de resistência ao cisalhamento que, como já exposto anteriormente, se
devem ao processo de colagem manual, o tempo em aberto da colagem e a presença de
bolhas de ar resultantes do processo de síntese.
Como mostrado no item 5.3, adição de NWC eleva a resistência ao cisalhamento sob
tração dos compósitos. Autores como Medeiros et al. [21] e Goetz et al. [140] relatam a
interação química que pode ocorrer entre os NWC e os polímeros, além de alterar a
degradação. O compósito de PGA com até 10% de NWC não exibe degradação de
propriedades nos tempos testados, como mostra os valores apresentados na Tabela 17. O
PGA 20%, assim como no item 5.3, desprendeu durante o ensaio de envelhecimento,
provavelmente porque os 20% de NWC incorporados nos polímeros favoreceram a
formação de aglomerados, agindo como concentrador de tensão, e assim fragilizando o
adesivo, como consequência facilitando o desprendimento da madeira, quando submetidos a
umidade e temperatura.
A família do PGM não apresentou ruptura durante o ensaio de envelhecimento. O
PGM 5 e 10%, apresentaram aumento de resistência ao cisalhamento de 53 e 48% em 350h

94
94
de ensaio, respectivamente, em relação ao polímero puro sem envelhecimento. Os resultados
apresentados pelo compósito de PGM/NWC podem ser atribuídos ao anidrido maleico, que
teoricamente reage estequiometricamente de forma igualitária, porém partes do
ácido/anidrido não reagem por completo, sendo que essas partes contribuem para a
compatibilização entre os materiais, pois sua estrutura molecular permite uma maior
interação entre o polímero e carga.
Tabela 17 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos da família do PGA
e PGM.
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
PGA 1,54 ± 0,11 2,99 ± 0,33 2,36 ± 0,45 2,91 ± 0,60 PGM 1,54 ±0,14 3,08 ± 0,1 2,76 ± 0,27 1,64 ± 0,15
PGA
5%
1,61 ± 0,04 2,32 ± 0,12 1,71 ± 0,24 0,37 ± 0,03 PGM
5%
2,24 ± 0,46 4,18 ± 0,19 2,12 ± 0,12 2,52 ± 0,21
PGA
10%
1,45 ± 0,05 3,6 ± 0,67 2,24 ± 0,22 2,67 ± 0,35 PGM
10%
1,95 ± 023 2,8 ± 0,42 1,98 ± 0,1 2,44 ± 0,24
PGA
20%
1,90 ± 0,04 2,12 ± 0,32 0 0 PGM
20%
2,23 ± 0,4 2,07 ± 0,08 2,64 ± 0,41 1,94 ± 0,33
A família do PGSu foi a que apresentou as piores respostas ao envelhecimento
acelerado, como mostrado na Figura 57 e na Tabela 18. Após 250h de envelhecimento todos
os corpos de prova romperam sem aplicação de esforço mecânico externo.
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGSu
PGSu 5%
PGSu 10%
PGSu 20%
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGMA
PGMA 5%
PGMA 10%
PGMA 20%
Figura 57 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos compósitos da família
do PGSu e PGMA.
A adesão dos compósitos de PGSu na madeira se mostrou ineficiente para esse teste.
Esse polímero apresenta uma cadeia linear menor, com ligações cruzadas, logo tendo uma

95
95
restrição no seu movimento macromolecular. Mesmo com a adição de um material rígido
(NWC), a exposição à umidade e temperatura em tempos elevados faz com que as reações
por hidrólise ocorram de forma rápida.
A família do PGMA exibiu excelentes resultados ao envelhecimento acelerado nas
condições estabelecidas. A combinação de um monômero com maior mobilidade,
comparado ao PGSu, e outro com função reconhecida de compatibilizante, além da rigidez
dos NWC, tornaram os melhores adesivos estudados nesse trabalho, como mostrado na
Tabela 18 e gráfico 57. Os resultados de resistência ao cisalhamento do PGMA 10 e 20%
após o ensaio de envelhecimento de 350h, foram de 33 e 71%, respectivamente, maiores que
o PGMA na mesma condição, possivelmente pela compatibilização do anidrido maleico com
o polímero e a carga, além dos NWC nessas proporções não terem aglomerados para o
PGMA, indicando que podem ter retardado a degradação nesse compósito, oferecendo assim
excelentes resultados.
Tabela 18 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos da família do
PGSu e PGMA.
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
PGSu 0,98 ± 0,17 1,31 ± 0,24
0 0 PGMA 1,08 ±0,10 3,89 ± 0,67 1,75 ± 0,21 2,21 ± 0,17
PGSu
5%
1,57 ± 0,1 2,04 ± 0,07 0 0 PGMA
5%
1,18 ± 0,10 2,99 ± 0,18 1,87 ± 0,03 1,77 ± 0,21
PGSu
10%
2.2 ± 0,23 2,12 ± 0,12 0 0 PGMA
10%
1,83 ± 0,23 3,81 ± 0,32 3,35 ± 0,1 2,96 ± 0,25
PGSu
20%
1,52 ± 0,21 2,77 ± 0,34 0 0 PGMA
20%
1,40 ± 0,17 3,56 ± 0,26 3,89 ± 0,34 3,78 ± 0,20
A família do PGASu mostrou resultados negativos quando submetidos ao ensaio de maior
tempo de envelhecimento, Tabela 19. Copolímeros com linearidade tendem a um número maior de
carbonos terciários, que apresenta força de ligação é pequena, implicando em energias menores para
quebra-las. A incorporação dos NWC surtiu o efeito esperado até 250h de ensaio, Figura 58. Com o
maior tempo de exposição à umidade e luz UV, possivelmente, a quebra das ligações nos carbonos
terciários ocorreu, como também os aglomerados, acarretando na ruptura dos corpos de prova.

96
96
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGASu
PGASu 5%
PGASu 10%
PGASu 20%
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGMSu
PGMSu 5%
PGMSu 10%
PGMSu 20%
Figura 58 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento nos compósitos da família
do PGSu e PGMA.
A família do PGMSu apresentou bons resultados de tensão máxima de cisalhamento,
apenas o PGMSu 5% rompeu após o envelhecimento de 350h. Esse resultado para o PGMSu
5%, pode ser atribuído a problemas no processo, como a presença de bolhas, já que na
incorporação de maiores quantidades de carga, 10 e 20%, os corpos de prova não romperam.
Diferente do copolímero PGMA, que também contém um monômero de anidrido maleico e
apresentou melhores resultados com a incorporação da carga, os compósitos de PGMSu
apresentaram valores de resistência ao cisalhamento menores que o puro para tempos de
exposição maiores que 250h. O PGMSu 10 e 20% ficaram com valores de 38 e 10,1%,
respectivamente, menores que o PGMSu.
Tabela 19 - Valores de resistência ao cisalhamento x tempo envelhecimento nos compósitos da família do
PGASu e PGMSu.
Resistência
ao
cisalhamento
(MPa )
0h
Resistência
ao
cisalhamento
(MPa )
150h
Resistência
ao
cisalhamento
(MPa )
250 h
Resistência
ao
cisalhamento
(MPa )
350h
Resistência
ao
cisalhamento
(MPa )
0h
Resistência
ao
cisalhamento
(MPa )
150h
Resistência
ao
cisalhamento
(MPa )
250 h
Resistência
ao
cisalhamento
(MPa )
350h
PGASu
1,89 ± 0,16 1,92 ± 0,10 2,33 ± 0,23 1,29 ± 0,11 PGMSu
1,57 ±0,17 2,48 ± 0,07 2,99 ± 0,33 2,87 ± 0,45
PGASu
5% 1,74 ± 0,04 2,50 ± 0,4 2,01 ± 0,13 0 PGMSu
5% 1,88 ± 0,20 3,05 ± 0,31 1,66 ± 0,22 0
PGASu
10% 1,59 ± 0,1 2,67 ± 0,17 1,49 ± 0,13 0 PGMSu
10% 1,68 ± 023 2,78 ± 0,14 1,78 ± 0,06 1,77 ± 0,1
PGASu
20% 2,57 ± 0,36 3,32 ± 0,22 0 0 PGMSu
20% 2,20 ± 0,35 2,31 ± 0,17 1,38 ± 0,21 2,58 ± 0,28

97
97
O gráfico da Figura 59 e a Tabela 20, apresentam os valores comparativos dos
melhores compósitos estudados com a cola branca comercial (PVAc).
0 150 300
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
Tempo (h)
Te
nsa
o m
axim
a (
MP
a)
PGA
PGM 10%
PGMSu 10%
PGMSu 20%
PGMA 10%
PGMA 20%
COLA BRANCA
Figura 59 - Gráficos de tensão máxima de cisalhamento x tempo de envelhecimento dos compósitos com
melhores resultados comparados com a cola branca comercial.
Os valores da Tabela 20 mostram os compósitos com melhores resultados no teste de
cisalhamento sob tração comparados com à cola branca, mesmo após 350h de
envelhecimento. Apenas o PGMSu 10% apresentou valores menores que a cola branca, com
8% a menos. O PGMA 20% exibiu 98% de aumento em relação a cola branca, sendo o
melhor resultado dentre os compósitos. Os outros compósitos, PGA 10%, PGM 10%,
PGMSu 20% e PGMA 10%, também apresentaram valores maiores, 39%, 30%, 35% e 55%,
respectivamente.

98
98
Tabela 20 - Valores de resistência ao cisalhamento x tempo de envelhecimento dos compósitos com melhores
resultados comparados com a cola branca comercial.
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
Resistência
ao
cisalhamento
(MPa)
0h
Resistência
ao
cisalhamento
(MPa)
150h
Resistência
ao
cisalhamento
(MPa)
250 h
Resistência
ao
cisalhamento
(MPa)
350h
PGA
10% 1,45 ± 0,05 3,60 ± 0,07 2,24 ± 0,22 2,67 ± 0,35 PGMA
10% 1,83 ±0,23 3,81 ± 0,32 3,35 ± 0,33 2,96 ± 0,25
PGM
10% 2,24 ± 0,46 2,80 ± 0,42 1,98 ± 0,10 2,49 ± 0,24
PGMA
20% 1,40 ± 0,17 3,56 ± 0,26 3,89 ± 0,34 3,78 ± 0,20
PGMSu
10% 1,88 ± 0,20 2,78 ± 0,14 1,78 ± 0,06 1,77 ± 0,10
COLA
BRANCA 1,53 ± 0,32 2,87 ± 0,21 2,75 ± 0,31 1,91 ± 0,14
PGMSu
20% 2,20 ± 0,35 2,31 ± 0,17
1,38 ± 0,21 2,58 ± 0,28 - - - - -
As imagens da Figura 60 exibem as micrografias do PGA e PGA 10% com
diferentes períodos de envelhecimento, após os ensaios de cisalhamento sob tração. Os
compósitos de PGA com até 10% de NWC apresentaram bons resultados de tensão de
cisalhamento, como mostra a Tabela 17, e suas micrografias corroboram com isso. Nas
imagens da Figura 60, pode-se observar deformações dos polímeros com mobilidade
molecular facilitada, característica do PGA, como também a presença de vazios. Nas
micrografias da Figura 60 observa-se que para o envelhecimento de até 350h nos compósitos
com 10% de NWC, não há alterações das características morfológicas da superfície de
fratura. No entanto como discutido no item 5.15, os compósitos com 20% de carga
romperam durante o envelhecimento.

99
99
Figura 60 - Micrografias do PGA e PGA 10% em diferentes períodos de envelhecimento após o cisalhamento sob
tração. a) PGA 150h, b) PGA 250h c) PGA 350h, d) PGA10% 150h, e) PGA10% 250h e f) PGA10% 350h
a d
c
b e
f

100
100
A Figura 61 exibe as micrografias do PGM e PGM10% com diferentes períodos de
envelhecimento, após os ensaios de cisalhamento. As imagens 61 a, b e c, sugerem
característica de fratura com menor ductilidade do PGM, polímero com mobilidade restrita.
As imagens 61 d, e, f, indicam a possível interação dos NWC com o polímero, onde mesmo
tendo os espaços vazios, mostra que não ouve degradação a ponto de romper o compósito,
mesmo com 350h de envelhecimento ou com 20% de NWC incorporado (Tabela 17).
Figura 61 - Micrografias do PGM e PGM10% em diferentes períodos de envelhecimento após o
cisalhamento sob tração. a) PGM 150h, b) PGM 250h c) PGM 350h, d) PGM10% 150h, e)
PGM10% 250h e f) PGM10% 350h
a
b
c
d
e
f

101
101
Na Figura 62 são apresentadas as micrografias do PGSu e PGSu 10% após o
cisalhamento sob tração, após terem sido expostos a diferentes períodos de envelhecimento.
Nas micrografias observam-se superfícies de fratura indicando ruptura com mobilidade
molecular reduzida. No entanto com as imagens não são observadas as alterações que
ocorreram após as 150 h de envelhecimento (ver Tabela 18 e Figura 57).
Figura 62 - micrografias do PGSu e PGSu 10% em diferentes períodos de envelhecimento após o
cisalhamento sob tração. a) PGSu 150h, b) PGSu 250h c) PGSu 350h, d) PGSu 10% 150h, e) PGSu 10%
250h e f) PGSu 10% 350h
a d
c
b e
f

102
102
Na Figura 63 são mostradas micrografias das superfícies de fratura do PGMA e
PGMA 10% como diferentes períodos de envelhecimento e após o ensaio de cisalhamento.
Compósitos e polímeros puros não apresentaram variações de aspecto da superfície de
fratura. Observa-se a interação entre os componentes, devido a adesão dos NWC ao
copolímero, acarretando nos excelentes resultados do mesmo no cisalhamento sob tração,
assim como no não rompimento com a incorporação de alto valor de carga (20%).
Figura 63 - Micrografias do PGMA e PGMA 10% em diferentes períodos de envelhecimento após o
cisalhamento sob tração. a) PGMA 150h, b) PGMA 250h c) PGMA 350h, d) PGMA10% 150h, e)
PGMA10% 250h e f) PGMA10% 350h
a
b
c
d
e
f

103
103
Na Figura 64 são apresentadas as micrografias do PGASu e PGASu 10% expostos a
diferentes períodos de envelhecimento, após o cisalhamento sob tração. Assim como no
PGSu não foram observadas rupturas no envelhecimento ocorridas após 250h.
Figura 64 - Micrografias do PGASu e PGASu10% em diferentes períodos de envelhecimento após o
cisalhamento sob tração. a) PGASu 150h, b) PGASu 250h c) PGASu 350h, d) PGASu10% 150h, e)
PGASu10% 250h e f) PGASu10% 350h
a
b
c
d
e
f

104
104
Na Figura 65 são apresentadas as micrografias do PGMSu e PGMSu 10% expostos a
diferentes períodos de envelhecimento, após o cisalhamento sob tração. Assim como o
PGMA, o PGMSu apresentou possíveis interações (discutidas no item 5.15) e assim sendo,
bons resultados de cisalhamento sob tração. Essas interações ficam evidentes na Figura 65,
assim como as propriedades de resistência da carga (NWC) e dos monômeros (ácido
succínico e anidrido maleico), já comentadas anteriormente.
Figura 65 - Micrografias do PGMSu e PGMSu10% em diferentes períodos de envelhecimento após o
cisalhamento sob tração. a) PGMSu 150h, b) PGMSu 250h c) PGMSu 350h, d) PGMSu10% 150h, e)
PGMSu10% 250h e f) PGMSu10% 350h
a
b
c
d
e
f

105
105
6 CONCLUSÕES
Os resultados apresentados mostraram que a adição dos NWC melhorou as propriedades
adesivas dos polímeros de glicerol e foi possível obter os compósitos (adesivos) com tensão
ao cisalhamento maior que a cola comercial PVAc. As tensões ao cisalhamento foram
diretamente dependentes da concentração do NWC. Os compósitos PGM, PGMSu e PGMA
com 10 e 20% de NWC mostraram os melhores resultados dentre os polímeros/compósitos
após os tempos de envelhecimento utilizados, tendo como o mais promissor o PGMA 10 e
20% de NWC.
Os compósitos estudados mostraram-se promissores para o uso como adesivos verdes
em madeiras de Pinus e Angelim.

106
106
7 REFERÊNCIAS
1. Leoneti AB, Aragão-Leoneti V, de Oliveira SVWB (2012) Glycerol as a by-product of
biodiesel production in Brazil: Alternatives for the use of unrefined glycerol. Renew Energy
45:138–145. doi: 10.1016/j.renene.2012.02.032
2. Umpierre AP, Machado F (2013) Gliceroquímica e valorização do glicerol. Rev Virtual Quim
5:106–116.
3. Ayoub M, Abdullah AZ (2012) Critical review on the current scenario and significance of
crude glycerol resulting from biodiesel industry towards more sustainable renewable energy
industry. Renew Sustain Energy Rev 16:2671–2686. doi: 10.1016/j.rser.2012.01.054
4. Shah EM (2014) Base material for pharmaceutical and/or cosmetic cream (herbal
composition for itchy or infected skin).
5. Chen Q, Liang S, Thouas G a. (2011) Synthesis and characterisation of poly(glycerol
sebacate)-co-lactic acid as surgical sealants. Soft Matter 7:6484. doi: 10.1039/c1sm05350g
6. Gaharwar AK, Patel A, Dolatshahi-Pirouz A, et al (2015) Elastomeric nanocomposite
scaffolds made from poly(glycerol sebacate) chemically crosslinked with carbon nanotubes.
Biomater Sci 3:46–58. doi: 10.1039/C4BM00222A
7. Gao Q, Li J, Shi SQ, et al (2012) Soybean meal-based adhesive reinforced with cellulose
nano-whiskers. BioResources 7:5622–5633. doi: 10.15376/biores.7.4.5622-5633
8. Grinstaff MW (2007) Designing hydrogel adhesives for corneal wound repair. Biomaterials
28:5205–14. doi: 10.1016/j.biomaterials.2007.08.041
9. Czech Z, Wilpiszewska K, Tyliszczak B, et al (2013) Biodegradable self-adhesive tapes with
starch carrier. Int J Adhes Adhes 44:195–199. doi: 10.1016/j.ijadhadh.2013.03.002
10. Araki J, Mishima S (2015) Steric stabilization of “charge-free” cellulose nanowhiskers by
grafting of poly(ethylene glycol). Molecules 20:169–84. doi: 10.3390/molecules20010169
11. Bondeson D, Oksman K (2007) Polylactic acid/cellulose whisker nanocomposites modified
by polyvinyl alcohol. Compos Part A Appl Sci Manuf 38:2486–2492. doi:
10.1016/j.compositesa.2007.08.001
12. Çetin NS, Tingaut P, Özmen N, et al (2009) Acetylation of cellulose nanowhiskers with vinyl
acetate under moderate conditions. Macromol Biosci 9:997–1003. doi:
10.1002/mabi.200900073
13. Domingues RMA, Gomes ME, Reis RL (2014) The potential of cellulose nanocrystals in
tissue engineering strategies. Biomacromolecules 15:2327–2346. doi: 10.1021/bm500524s
14. Dufresne A (2010) Processing of polymer nanocomposites reinforced with polysaccharide
nanocrystals. Molecules 15:4111–4128. doi: 10.3390/molecules15064111
15. Luiz De Paula E, Mano V, Pereira FV (2011) Influence of cellulose nanowhiskers on the
hydrolytic degradation behavior of poly(d,l-lactide). Polym Degrad Stab 96:1631–1638. doi:
10.1016/j.polymdegradstab.2011.06.006
16. Miao C, Hamad WY (2013) Cellulose reinforced polymer composites and nanocomposites: A
critical review. Cellulose 20:2221–2262. doi: 10.1007/s10570-013-0007-3
17. Pereira ALS, Nascimento DM Do, Souza Filho MDSM, et al (2014) Improvement of
polyvinyl alcohol properties by adding nanocrystalline cellulose isolated from banana
pseudostems. Carbohydr Polym 112:165–172. doi: 10.1016/j.carbpol.2014.05.090
18. Petersson L, Kvien I, Oksman K (2007) Structure and thermal properties of poly(lactic
acid)/cellulose whiskers nanocomposite materials. Compos Sci Technol 67:2535–2544. doi:
10.1016/j.compscitech.2006.12.012
19. Takagi H, Nakagaito a. N, Bistamam MS a. (2013) Extraction of cellulose nanofiber from

107
107
waste papers and application to reinforcement in biodegradable composites. J Reinf Plast
Compos 32:1542–1546. doi: 10.1177/0731684413494109
20. Wang Y, Cao X, Zhang L (2006) Effects of cellulose whiskers on properties of soy protein
thermoplastics. Macromol Biosci 6:524–531. doi: 10.1002/mabi.200600034
21. Medeiros ES, Offeman RD, Klamczynski AP, et al (2014) Synthesis, Characterization and
Nanocomposite Formation of Poly(glycerol succinate-co-maleate) with Nanocrystalline
Cellulose. J Polym Environ 1–8. doi: 10.1007/s10924-014-0643-1
22. DA SILVA, Lucas FM; MAGALHÃES, António G.; MOURA MF (2007) Juntas adesivas
estruturais. Publindústria, Porto
23. Petrie EM, Edward M (2000) Handbook of Adhesives and Sealants. McGraw - Hill, New
York
24. Antonio Pizzi KLM (2011) Wood Adhesives. Taylor & Francis group, Washington
25. POCIUS A V. (2012) Adhesion and adhesives technology: an introduction. Carl Hanser
Verlag GmbH Co KG
26. Makkonen L (2012) Ice Adhesion —Theory, Measurements and Countermeasures. Volume
26,:
27. Gardner DJ, Blumentritt M, Wang L, Yildirim N (2014) Adhesion Theories in Wood
Adhesive Bonding. Rev Adhes Adhes 2:127–172. doi: 10.7569/RAA.2014.097304
28. Gorb SN, Koch K (2014) From sticky to slippery: Biological and biologically-inspired
adhesion and friction. Beilstein J Nanotechnol 5:1450–1. doi: 10.3762/bjnano.5.157
29. Sina Ebnesajjad (2011) Handbook of Adhesives and Surface Preparation. Handb Adhes Surf
Prep. doi: 10.1016/B978-1-4377-4461-3.10004-5
30. shields J (2013) adhesives Handbook, 3a. batler & tanner LTDA
31. KRETSCHMANN DE et al. (2012) Mechanical properties of wood, v. 5, p. 3. Wisconsin-
Madison,
32. Ou R, Xie Y, Wolcott MP, et al (2014) Effect of wood cell wall composition on the
rheological properties of wood particle/high density polyethylene composites. Compos Sci
Technol 93:68–75. doi: 10.1016/j.compscitech.2014.01.001
33. Menezes WM de, Santini EJ, Souza JT de, et al (2014) Modificação térmica nas propriedades
físicas da madeira. Ciência Rural 44:1019–1024. doi: 10.1590/S0103-84782014000600011
34. Taurus B (2009) Adhesive Bonding of Wood Materials. doi: 10.1016/B978-0-12-373580-
5.50043-0
35. Kaboorani A, Riedl B (2011) Effects of adding nano-clay on performance of polyvinyl
acetate (PVA) as a wood adhesive. Compos Part A Appl Sci Manuf 42:1031–1039. doi:
10.1016/j.compositesa.2011.04.007
36. Kaboorani A, Riedl B (2011) Improving performance of polyvinyl acetate (PVA) as a binder
for wood by combination with melamine based adhesives. Int J Adhes Adhes 31:605–611.
doi: 10.1016/j.ijadhadh.2011.06.007
37. Costa NA, Ferra J, Martins J, et al (2014) Impact of thermal treatment on bonding
performance of UF/PVAc formulations. Int Wood Prod J 5:212–216. doi:
10.1179/2042645314Y.0000000075
38. Moya R, Rodríguez-Zúñiga A, Vega-Baudrit J, Álvarez V (2015) Effects of adding nano-clay
(montmorillonite) on performance of polyvinyl acetate (PVAc) and urea-formaldehyde (UF)
adhesives in Carapa guianensis, a tropical species. Int J Adhes Adhes 59:62–70. doi:
10.1016/j.ijadhadh.2015.02.004
39. Gao W, Du G, Ma H, Li J (2015) Dynamic mechanical analysis (DMA) of urea formaldehyde
(UF) resin modified by ammonium pentaborate (APB) as wood adhesive. Polym Compos n/a-

108
108
n/a. doi: 10.1002/pc.23422
40. (2010) Adhesives with wood materials : bond formation and performance. 190:10.1-10.24.
41. (2012) Handbook of Wood Chemistry and Wood Composites, Second Edition. CRC Press
42. Zhang Y, Ding L, Gu J, et al (2015) Preparation and properties of a starch-based wood
adhesive with high bonding strength and water resistance. Carbohydr Polym 115:32–7. doi:
10.1016/j.carbpol.2014.08.063
43. Cheng HN, Dowd MK, He Z (2013) Investigation of modified cottonseed protein adhesives
for wood composites. Ind Crops Prod 46:399–403. doi: 10.1016/j.indcrop.2013.02.021
44. Beatriz A, Araújo YJK, Lima DP (2011) Glycerol: a brief history and their application in
stereoselective syntheses. Quim Nova 34:306–319. doi: 10.1590/S0100-40422011000200025
45. Zhou C, Wu Q (2012) Recent development in applications of cellulose nanocrystals for
advanced polymer-based nanocomposites by novel fabrication strategies.
46. Le Goff KJ, Jouanneau D, Garnier C, Aubry T (2014) Gelling of cellulose nanowhiskers in
aqueous suspension. J Appl Polym Sci 131:8476–8481. doi: 10.1002/app.40676
47. Eichhorn SJ (2011) Cellulose nanowhiskers: promising materials for advanced applications.
Soft Matter 7:303. doi: 10.1039/c0sm00142b
48. Suh YJ, Kil DS, Chung KS, et al (2011) Natural Nanocontainer for the Controlled Delivery of
Glycerol as a Moisturizing Agent. J Nanosci Nanotechnol 11:661–665. doi:
10.1166/jnn.2011.3194
49. steve cochram and Michael anthonavage (2015) Lipids and Skin Health. doi: 10.1007/978-3-
319-09943-9
50. Pando D, Caddeo C, Manconi M, et al (2013) Nanodesign of olein vesicles for the topical
delivery of the antioxidant resveratrol. J Pharm Pharmacol 65:1158–67. doi:
10.1111/jphp.12093
51. Rezende MJC, Dos Santos NBL (2012) Produção de Monoacilgliceróis: Rotas e
Catalisadores. Rev Virtual Química 4:118–129. doi: 10.5935/rvq.v4i2.217
52. Sanz T, Falomir M, Salvador A (2015) Reversible thermal behaviour of vegetable oil
cellulose ether emulsions as fat replacers. Influence of glycerol. Food Hydrocoll 46:19–27.
doi: 10.1016/j.foodhyd.2014.11.030
53. Denis Ruhlmann J-MS (2014) Associative acrylic thickening agent containing polyglycerols
and its use to increase the open time of thin or thick films.
54. Tulbure DA, Ognean M, Ognean C-F, Danciu I Effects of Some Sweeteners on Gingerbread
Propertie s – Water Sorption. In: 2014, 20(1), 21-25.
55. Hales KE, Bondurant RG, Luebbe MK, et al (2013) Effects of crude glycerin in steam-flaked
corn-based diets fed to growing feedlot cattle. J Anim Sci 91:3875–80. doi: 10.2527/jas.2012-
5944
56. E Hanczakowska, K Weglarzy BS Effect of adding crude or refined glycerol to pig diets on
fattening performance, nutrient digestibility and carcass evaluation. Ann Anim Sci,Vol 10,
No 1 67–73.
57. Madrid J, Villodre C, Valera L, et al (2013) Effect of crude glycerin on feed manufacturing,
growth performance, plasma metabolites, and nutrient digestibility of growing-finishing pigs.
J Anim Sci 91:3788–95. doi: 10.2527/jas.2013-5684
58. Lal Nand, Srivastava Neerja (2014) Bioremediation of Glycerol Trinitrate (GTN) Explosive.
J Funct Environ Bot 4:55–70.
59. Manfro RL, Ribeiro NFP, Souza MMVM (2013) Production of hydrogen from steam
reforming of glycerol using nickel catalysts supported on Al2O3, CeO2 and ZrO2. Catal
Sustain Energy 1:60–70. doi: 10.2478/cse-2013-0001

109
109
60. Srinivasa Godavarthy, Wei-Yang Su, Ralph M. Diguilio, Stan Harville MWF (2014) Process
for the production and purification of propylene glycol.
61. Salazar JB, Falcone DD, Pham HN, et al (2014) Selective production of 1,2-propanediol by
hydrogenolysis of glycerol over bimetallic Ru–Cu nanoparticles supported on TiO2. Appl
Catal A Gen 482:137–144. doi: 10.1016/j.apcata.2014.06.002
62. Bell BM, Briggs JR, Campbell RM, et al (2008) Glycerin as a renewable feedstock for
epichlorohydrin production. The GTE process. Clean - Soil, Air, Water 36:657–661. doi:
10.1002/clen.200800067
63. Pagliaro M, Rossi M (2010) The Future of Glycerol.
64. Gooch JW (2002) Emulsification and Polymerization of Alkyd Resins.
65. De Araújo Medeiros M, Lago RM (2011) Polimerização do glicerol: uma reação simples e
versátil para produzir diferentes materiais a partir do coproduto do biodiesel. Quim Nova
34:1079–1084. doi: 10.1590/S0100-40422011000600028
66. Geschwind J, Frey H (2013) Poly(1,2-glycerol carbonate): A Fundamental Polymer Structure
Synthesized from CO 2 and Glycidyl Ethers. Macromolecules 46:3280–3287. doi:
10.1021/ma400090m
67. Sonnati MO, Amigoni S, Taffin de Givenchy EP, et al (2013) Glycerol carbonate as a
versatile building block for tomorrow: synthesis, reactivity, properties and applications.
Green Chem 15:283–306. doi: 10.1039/C2GC36525A
68. Zhang H, Grinstaff MW (2014) Recent advances in glycerol polymers: chemistry and
biomedical applications. Macromol Rapid Commun 1906–1924. doi:
10.1002/marc.201400389
69. Zhang J, Tu W, Dai Z (2013) Transparent polyester polyol-based polyurethane coatings: the
effect of alcohols. J Coatings Technol Res 10:887–895. doi: 10.1007/s11998-013-9527-x
70. Velayutham TS, Majid WHA, Ahmad AB, et al (2009) Synthesis and characterization of
polyurethane coatings derived from polyols synthesized with glycerol, phthalic anhydride and
oleic acid. Prog Org Coatings 66:367–371. doi: 10.1016/j.porgcoat.2009.08.013
71. Sivak WN, Pollack IF, Petoud S, et al (2008) LDI-glycerol polyurethane implants exhibit
controlled release of DB-67 and anti-tumor activity in vitro against malignant gliomas. Acta
Biomater 4:852–62. doi: 10.1016/j.actbio.2007.10.012
72. Mequanint, Kibret, Patel A, Bezuidenhout and D Synthesis, Swelling Behavior, and
Biocompatibility of Novel Physically Cross-Linked Polyurethane- block -Poly(glycerol
methacrylate) Hydrogels. In: Biomacromolecules 2006, 7, 883 - 891.
73. Hu S, Wan C, Li Y (2012) Production and characterization of biopolyols and polyurethane
foams from crude glycerol based liquefaction of soybean straw. Bioresour Technol 103:227–
33. doi: 10.1016/j.biortech.2011.09.125
74. Bettinger CJ, Bruggeman JP, Borenstein JT, Langer RS (2008) Amino alcohol-based
degradable poly(ester amide) elastomers. Biomaterials 29:2315–25. doi:
10.1016/j.biomaterials.2008.01.029
75. Cheng H, Hill PS, Siegwart DJ, et al (2011) A novel family of biodegradable poly(ester
amide) elastomers. Adv Mater 23:H95-100. doi: 10.1002/adma.201003482
76. Kathryn S. Hayes, Charles R. Frihart RJW (1986) Novel poly(ester-amide) hot-melt
adhesives.
77. Richard C Macqueen MSP (1998) Ester-terminated polyamides of polymerized fatty acids
useful in formulating transparent gels in low polarity liquids.
78. Kasetaite S, Ostrauskaite J, Grazuleviciene V, et al (2013) Copolymers of glycerol and
propylene glycol diglycidyl ethers with aromatic dithiols. J Appl Polym Sci n/a-n/a. doi:
10.1002/app.39697

110
110
79. Cunningham VJ, Alswieleh AM, Thompson KL, et al (2014) Poly(glycerol
monomethacrylate)–Poly(benzyl methacrylate) Diblock Copolymer Nanoparticles via RAFT
Emulsion Polymerization: Synthesis, Characterization, and Interfacial Activity.
Macromolecules 47:5613–5623. doi: 10.1021/ma501140h
80. Pagliaro M, Ciriminna R, Kimura H, et al (2009) Recent advances in the conversion of
bioglycerol into value-added products. Eur J Lipid Sci Technol 111:788–799. doi:
10.1002/ejlt.200800210
81. Gandini A, Lacerda TM (2015) From monomers to polymers from renewable resources:
recent advances. Prog Polym Sci. doi: 10.1016/j.progpolymsci.2014.11.002
82. Stanley S, Karo W (1998) Sourcebook of Advanced Polymer Laboratory Preparations | 978-
0-12-618605-5 | Elsevier. https://www.elsevier.com/books/sourcebook-of-advanced-polymer-
laboratory-preparations/sandler/978-0-12-618605-5. Accessed 3 Apr 2015
83. JUNIOR SVC (2010) Ciência dos polímeros: um texto básico para tecnólogos e engenheiros.,
2a. Artliber,
84. ODIAN, George G.; ODIAN G (2004) Principles of polymerization. Wiley-Interscience, New
York
85. Kricheldorf H (2014) Polycondensation. doi: 10.1007/978-3-642-39429-4
86. Agach M, Delbaere S, Marinkovic S, et al (2013) Synthesis, characterization,
biodegradability and surfactant properties of bio-sourced lauroyl poly(glycerol-succinate)
oligoesters. Colloids Surfaces A Physicochem Eng Asp 419:263–273. doi:
10.1016/j.colsurfa.2012.12.006
87. Baharu MN, Kadhum AAH, Al-Amiery AA, Mohamad AB (2014) Synthesis and
characterization of polyesters derived from glycerol, azelaic acid, and succinic acid. Green
Chem Lett Rev 8:31–38. doi: 10.1080/17518253.2014.991810
88. Zhang T, Howell BA, Dumitrascu A, et al (2014) Synthesis and characterization of glycerol-
adipic acid hyperbranched polyesters. Polymer (Guildf) 55:5065–5072. doi:
10.1016/j.polymer.2014.08.036
89. Bueno L, Toro C, Martín M (2015) Techno-economic evaluation of the production of
polyesters from glycerol and adipic acid. Chem Eng Res Des 93:432–440. doi:
10.1016/j.cherd.2014.05.010
90. Salehi S, Fathi M, Javanmard SH, et al (2015) Fabrication and characterization of
biodegradable polymeric films as a corneal stroma substitute. Adv Biomed Res 4:9. doi:
10.4103/2277-9175.148291
91. Xu B, Li Y, Zhu C, et al (2015) Fabrication, mechanical properties and cytocompatibility of
elastomeric nanofibrous mats of poly(glycerol sebacate). Eur Polym J 64:79–92. doi:
10.1016/j.eurpolymj.2014.12.008
92. Degoricija, L ; Johnson, CS; Wathier, M ; Kim, T ; Grinstaff M (2007) hoto cross-linkable
biodendrimers as ophthalmic adhesives for central lacerations and penetrating keratoplasties.
Invest Ophthalmol Vis Sci Vol.48(5):pp.2037-2042. doi: 10.1167/iovs.06-0957
93. Mamiński M, Witek S, Szymona K, Parzuchowski P (2013) Novel adhesive system based on
1,3-dimethylol-4,5- Dihydroxyethyleneurea (DMDHEU) and hyperbranched polyglycerols.
Eur J Wood Wood Prod 71:267–275. doi: 10.1007/s00107-013-0680-9
94. CHIRAYIL, Cintil Jose; MATHEW, Lovely; THOMAS S Review of recent research in nano
cellulose preparation from different lignocellulosic fibers. Rev. Adv. Mater. Sci, v. 37, p. 20-
28, 2014.
95. ZHANG Hao,SHE Ying,ZHENG Xue,CHEN He-yu PJ (2013) The Preparation and
Modification and Applications of Nanocrystalline Cellulose. Heilongjiang Pulp Pap. 2013–2:
96. Cao X, Wang X, Ding B, et al (2013) Novel spider-web-like nanoporous networks based on

111
111
jute cellulose nanowhiskers. Carbohydr Polym 92:2041–2047. doi:
10.1016/j.carbpol.2012.11.085
97. Ludueña LN, Vecchio A, Stefani PM, Alvarez V a. (2013) Extraction of cellulose
nanowhiskers from natural fibers and agricultural byproducts. Fibers Polym 14:1118–1127.
doi: 10.1007/s12221-013-1118-z
98. Faruk O, Bledzki AK, Fink H-P, Sain M (2012) Biocomposites reinforced with natural fibers:
2000–2010. Prog Polym Sci 37:1552–1596. doi: 10.1016/j.progpolymsci.2012.04.003
99. Le Goff KJ, Jouanneau D, Garnier C, Aubry T (2014) Gelling of cellulose nanowhiskers in
aqueous suspension. J Appl Polym Sci 131:n/a-n/a. doi: 10.1002/app.40676
100. Morais JPS, Rosa M de F, de Souza Filho M de sá M, et al (2013) Extraction and
characterization of nanocellulose structures from raw cotton linter. Carbohydr Polym 91:229–
35. doi: 10.1016/j.carbpol.2012.08.010
101. Hu W, Wang L, Wu Q, Wu H (2014) Preparation, characterization and microwave absorption
properties of bamboo-like β-SiC nanowhiskers by molten-salt synthesis. J Mater Sci Mater
Electron 25:5302–5308. doi: 10.1007/s10854-014-2305-4
102. Johar N, Ahmad I, Dufresne A (2012) Extraction, preparation and characterization of
cellulose fibres and nanocrystals from rice husk. Ind Crops Prod 37:93–99. doi:
10.1016/j.indcrop.2011.12.016
103. Rosa MF, Medeiros ES, Malmonge J a., et al (2010) Cellulose nanowhiskers from coconut
husk fibers: Effect of preparation conditions on their thermal and morphological behavior.
Carbohydr Polym 81:83–92. doi: 10.1016/j.carbpol.2010.01.059
104. Lavoine N, Desloges I, Dufresne A, Bras J (2012) Microfibrillated cellulose - its barrier
properties and applications in cellulosic materials: a review. Carbohydr Polym 90:735–64.
doi: 10.1016/j.carbpol.2012.05.026
105. Bondeson D, Oksman K (2007) Dispersion and characteristics of surfactant modified
cellulose whiskers nanocomposites. Compos Interfaces 14:617–630. doi:
10.1163/156855407782106519
106. De Jesus Silva D, D'Almeida MLO (2009) Nanocristais de celulose. O Pap 70:34–52.
107. De Figueirêdo MCB, De Freitas Rosa M, Lie Ugaya CM, et al (2012) Life cycle assessment
of cellulose nanowhiskers. J Clean Prod 35:130–139. doi: 10.1016/j.jclepro.2012.05.033
108. Santos TM, Souza Filho MDSM, Caceres CA, et al (2014) Fish gelatin films as affected by
cellulose whiskers and sonication. Food Hydrocoll 41:113–118. doi:
10.1016/j.foodhyd.2014.04.001
109. Mauricio MR, da Costa PG, Haraguchi SK, et al (2015) Synthesis of a microhydrogel
composite from cellulose nanowhiskers and starch for drug delivery. Carbohydr Polym
115:715–22. doi: 10.1016/j.carbpol.2014.07.063
110. Gao Q, Li J, Shi SQ, et al (2012) SOYBEAN MEAL-BASED ADHESIVE REINFORCED
WITH CELLULOSE NANO-WHISKERS. BioResources 7:5622–5633. doi:
10.15376/biores.7.4.5622-5633
111. Lee JH, Park SH, Kim SH (2014) Surface modification of cellulose nanowhiskers and their
reinforcing effect in polylactide. Macromol Res 22:424–430. doi: 10.1007/s13233-014-2064-
3
112. Alves JS, dos Reis KC, Menezes EGT, et al (2015) Effect of cellulose nanocrystals and
gelatin in corn starch plasticized films. Carbohydr Polym 115:215–22. doi:
10.1016/j.carbpol.2014.08.057
113. Zhou L, He H, Jiang C, He S (2015) Preparation and characterization of poly ( glycerol
sebacate )/ cellulose nanocrystals elastomeric composites. Appl Polym Sci. doi:
10.1002/app.42196

112
112
114. Domiguez-Sánchez JAM-T; AL-M; MÁ, Dyna 2015 (2015) Synthesis and characterization of
polymers based on citric acid and glycerol: Its application in non-biodegradable polymers.
Dyna 82 (190).
115. Cai W, Liu L (2008) Shape-memory effect of poly (glycerol-sebacate) elastomer. Mater Lett
62:2175–2177. doi: 10.1016/j.matlet.2007.11.042
116. Thomas LC (2010) Interpreting Unexpected Events and Transitions in DSC Results. Gov Inf
Q 27:423–430.
117. Halpern JM, Urbanski R, Weinstock AK, et al (2014) A biodegradable thermoset polymer
made by esterification of citric acid and glycerol. J Biomed Mater Res - Part A 102:1467–
1477. doi: 10.1002/jbm.a.34821
118. Moura CVR de, Nunes ASL, Moita Neto JM, et al (2012) Synthesis and characterization of
polyesters from glycerol by-product of biodiesel production. J Braz Chem Soc 23:1226–1231.
doi: 10.1590/S0103-50532012000700005
119. You YC, Jiao JL, Li Z, Zhu CY (2003) Synthesis and swelling behavior of crosslinked
copolymers of neutralized maleic anhydride with other monomers. J Appl Polym Sci
88:2725–2731. doi: 10.1002/app.11804
120. McCarron PA, Woolfson AD, Donnelly RF, et al (2004) Influence of plasticizer type and
storage conditions on properties of poly(methyl vinyl ether-co-maleic anhydride) bioadhesive
films. J Appl Polym Sci 91:1576–1589. doi: 10.1002/app.13228
121. Abdul Khalil HPS, Bhat AH, Ireana Yusra AF (2012) Green composites from sustainable
cellulose nanofibrils: A review. Carbohydr Polym 87:963–979. doi:
10.1016/j.carbpol.2011.08.078
122. Eichhorn SJ, Dufresne a., Aranguren M, et al (2010) Review: Current international research
into cellulose nanofibres and nanocomposites. J Mater Sci. doi: 10.1007/s10853-009-3874-0
123. de Morais Teixeira E, Corrêa AC, Manzoli A, et al (2010) Cellulose nanofibers from white
and naturally colored cotton fibers. Cellulose 17:595–606. doi: 10.1007/s10570-010-9403-0
124. Flauzino Neto WP, Silvério HA, Dantas NO, Pasquini D (2013) Extraction and
characterization of cellulose nanocrystals from agro-industrial residue - Soy hulls. Ind Crops
Prod 42:480–488. doi: 10.1016/j.indcrop.2012.06.041
125. Hossain KMZ, Ahmed I, Parsons AJ, et al (2012) Physico-chemical and mechanical
properties of nanocomposites prepared using cellulose nanowhiskers and poly(lactic acid). J
Mater Sci 47:2675–2686. doi: 10.1007/s10853-011-6093-4
126. Kavkler K, Gunde-Cimerman N, Zalar P, Demšar A (2011) FTIR spectroscopy of
biodegraded historical textiles. Polym Degrad Stab 96:574–580. doi:
10.1016/j.polymdegradstab.2010.12.016
127. Pavia DL (2008) Introduction to spectroscopy. ., 4th ed. Cengage Learning
128. Tammer M (2004) G. Sokrates: Infrared and Raman characteristic group frequencies: tables
and charts. Colloid Polym Sci 283:235–235. doi: 10.1007/s00396-004-1164-6
129. Xiaofei Zhao, Lixin Liu, Hongxia Dai, Chunxi Ma, Xiaohong Tan RY (2009) Synthesis and
Application of Water-Soluble Hyperbranched Poly(ester)s from Maleic Anhydride and
Glycerol. JournalofAppliedPolymerScience 113:3376–3381. doi: DOI 10.1002/app.29965
130. Siqueira G, Bras J, Dufresne A (2010) Cellulosic bionanocomposites: A review of
preparation, properties and applications. Polymers (Basel) 2:728–765. doi:
10.3390/polym2040728
131. Habibi Y, Lucia LA, Rojas OJ (2010) Cellulose nanocrystals: Chemistry, self-assembly, and
applications. Chem Rev 110:3479–3500. doi: 10.1021/cr900339w
132. Chen Y, Liu C, Chang PR, et al (2009) Bionanocomposites based on pea starch and cellulose
nanowhiskers hydrolyzed from pea hull fibre: Effect of hydrolysis time. Carbohydr Polym

113
113
76:607–615. doi: 10.1016/j.carbpol.2008.11.030
133. Fahma F, Iwamoto S, Hori N, et al (2010) Isolation, preparation, and characterization of
nanofibers from oil palm empty-fruit-bunch (OPEFB). Cellulose 17:977–985. doi:
10.1007/s10570-010-9436-4
134. Guo C, Zhou L, Lv J (2013) Effects of expandable graphite and modified ammonium
polyphosphate on the flame-retardant and mechanical properties of wood flour-polypropylene
composites. Polym Polym Compos 21:449–456. doi: 10.1002/app
135. Yu HY, Zhang DZ, Lu FF, Yao J (2016) New Approach for Single-Step Extraction of
Carboxylated Cellulose Nanocrystals for Their Use As Adsorbents and Flocculants. ACS
Sustain Chem Eng 4:2632–2643. doi: 10.1021/acssuschemeng.6b00126
136. Morais JPS, Rosa MDF, De Souza Filho MDSM, et al (2013) Extraction and characterization
of nanocellulose structures from raw cotton linter. Carbohydr Polym 91:229–235. doi:
10.1016/j.carbpol.2012.08.010
137. Planes M, Brand J, Lewandowski S, et al (2016) Improvement of the thermal and optical
performances of protective polydimethylsiloxane space coatings with cellulose nanocrystal
additives. ACS Appl Mater Interfaces. doi: 10.1021/acsami.6b09043
138. Kaboorani A, Riedl B, Blanchet P, et al (2012) Nanocrystalline cellulose (NCC): A
renewable nano-material for polyvinyl acetate (PVA) adhesive. Eur Polym J 48:1829–1837.
doi: 10.1016/j.eurpolymj.2012.08.008
139. Oksman K, Mathew a. P, Bondeson D, Kvien I (2006) Manufacturing process of cellulose
whiskers/polylactic acid nanocomposites. Compos Sci Technol 66:2776–2784. doi:
10.1016/j.compscitech.2006.03.002
140. Goetz L, Mathew A, Oksman K, et al (2009) A novel nanocomposite film prepared from
crosslinked cellulosic whiskers. Carbohydr Polym 75:85–89. doi:
10.1016/j.carbpol.2008.06.017