informe final laboratorio biotecnologia_1
TRANSCRIPT

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 1/49
LABORATORIO BIOTECNOLOGIA
AMALIA MARTINEZ MORENO
CODIGO: 35220542
JOSE LUIS SABOGAL PRIETO
CODIGO: 80756972
TUTOR
GLAEHTER YHON FLOREZ
UNIVERSIDAD NACIONAL ABIERTA Y A DISTANCIA “UNAD”
ESCUELA DE CIENCIAS BASICAS E INGENIERIA
PROGRAMA DE INGENIERIA DE ALIMENTOS
CEAD JAG BOGOTA
ABRIL 2011

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 2/49
PRACTICA 1
ELECTROFORESIS DE PROTEÍNAS EN CONDICIONES DESNATURALIZANTES(SDS-PAGE)
INTRODUCCIÓN
Esta práctica se realizó con el fin de separar las proteínas de E-coli para identificar sutamaño molecular y propiedades de carga, se utilizo el método SDS- PAGE aunque comono es posible estimar el peso molecular de una proteína que tenga más de una cadenapolipectídica, pero si se pueden separar y purificar pequeñas muestras de proteína ydeterminar la secuencia parcial de aminoácidos.
Con el método de SDS- PAGE solo se pueden estimar los pesos moleculares de unaproteína comparándolos con pesos moleculares de otras ya conocidas llamadosmarcadores de pesos molecular.
OBJETIVOS
Conocer la técnica de electroforesis de proteínas en geles SDS-PAGE Separar diversas proteínas mediante electroforesis desnaturalizante en
geles de poliacrilamida. Determinar el peso molecular de las proteínas en estudio Analizar los resultados obtenidos.
MARCO TEÓRICO
La electroforesis es una técnica de separación de moléculas cargadas por migración en
un campo eléctrico. Las moléculas se separan en función de su carga eléctrica,desplazándose al electrodo de carga contraria y a mayor velocidad cuanto mayor es lacarga de la molécula.
Cada molécula se desplaza en el campo eléctrico alcanzando una velocidad constante.En el estado estacionario, la fuerza impulsora (fuerza del campo eléctrico) se equilibra conla resistencia al avance (fuerza de fricción hidrodinámica) en el medio en que se desplaza.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 3/49
Se define la movilidad electroforética como la velocidad de desplazamiento por unidad decampo eléctrico. En unas condiciones determinadas de electroforesis, la diferentemovilidad de cada molécula define su comportamiento y separación en el espacio.Diferentes moléculas tendrán diferente movilidad electroforética en un medio determinado.
Hay tres tipos de electroforesis:
De frente móvil.
Zonal.
Continua.
Procedimiento de separación de proteínas basada en la carga eléctrica
La separación de proteínas basada en su carga eléctrica depende de suspropiedades acido-básicas, las cuales se hallan determinadas por el número y lostipos de los grupos R ionizables de sus cadenas polipeptídicas. Debido a que lasproteínas difieren en su composición de aminoácidos y secuencia, cada proteínaposee propiedades ácido-básicas características. Los principios implicados en laseparación electroforética de las proteínas son mejor comprendidos si seconsidera la curva de valoración ácido-básica de una proteína globular decontenido aminoácido conocido.
La mayor parte de las proteínas globulares poseen puntos isoeléctricos (no haydesplazamiento de la proteína) situados entre un pH de 4,5 y 6,5. Si el pH de unaproteína se halla por encima del punto isoeléctrico la proteína posee una carganegativa neta y se desplazará hacia el ánodo (+). Su carga negativa aumenta enmagnitud a medida que el pH aumenta; contrariamente a un pH por debajo delpunto isoeléctrico la proteína poseerá carga positiva neta y se desplazará hacia elcátodo (-). En conclusión el conocimiento de las propiedades ácido-básicas de unaproteína determinada hace posible predecir su comportamiento en un campoeléctrico.
Electroforesis de zona
En la electroforesis de zona los componentes proteicos se separan en diferenteszonas, en las cuales la composición y cantidad de proteínas existentes en cadauna de las zonas separadas se determina por la aplicación de un colorante quetiñe a las proteínas. La electroforesis de zona puede separar una mezcla deproteínas basándose en la carga eléctrica y el tamaño molecular.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 4/49
La electroforesis en gel de poliacrilamida puede aislar grandes cantidades deproteínas purificadas. El tamaño del poro del gel de acrilamida, se puede ajustarpara optimizar la separación de la muestra de interés, geles con un porcentaje altode acrilamida (10 a 15 %T), son óptimos para separar proteínas de pequeñotamaño (menores de 50 KDa), geles de porcentajes menores (< 10%T), son los
indicados para separar proteínas mayores
Con la electroforesis en gel de poliacrilamida SDS, que es una variación de laelectroforesis de zona, se puede disociar en proteína oligomérica en sussubunidades y determinar sus pesos moleculares, este método sirve también paradeterminar el peso molecular de proteínas de una sola cadena.
La proteína se trata con el detergente dodecil sulfato de sodio (SDS), que disociala proteína en subunidades y despliega por completo cada cadena polipeptídicaformando un complejo polipéptido-SDS cilíndrico y largo. En este complejo la
cadena polipeptídica se halla cubierta por una capa de moléculas de SDS, de talmanera que sus cadenas hidrocarbonadas se hallan íntimamente asociadas porfuerzas hidrofóbicas a la cadena polipeptídica, y los grupos sulfato cargados queposee el detergente se hallan expuestos al medio acuoso. Tales complejoscontienen una relación constante de SDS a proteína y difieren solo en masa.
Cuando una proteína de cadena única tratada con SDS es sometida aelectroforesis en gel de tamiz molecular (poliacrilamida), su velocidad deemigración la determina principalmente la masa de la partícula SDS-polipéptidosegún el principio de exclusión molecular, lo que determina el peso molecular de
las proteínas. El campo eléctrico sólo suministra la fuerza impulsora para eltamizado molecular. Para calibrar un sistema de gel se hacen circular proteínas depeso molecular conocido que actúan como marcadores con el objeto de efectuarla comparación.
El tratamiento de muestras con agentes desnaturalizantes (SDS), provoca ladesnaturalización de las proteínas, perdida de la estructura secundaria y ladisociación de las subunidades. Las proteínas quedan cargadas negativamente ymigran del polo negativo (cátodo) al positivo (ánodo) durante la electroforesis.
Las proteínas separadas mediante SDS-PAGE, pueden ser visualizadas en el gelmediante diversos métodos de tinción: fluorescencia, plata y azul comassie,siendo el último el más utilizado. (Lehninger, 2ª Edición).

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 5/49
Metodología
Adicionar el persulfato de amonio y TEMED, mezclar suavemente el vial, ya
que una excesiva aireación pude interferir con la polimerización
Introducir la solución en el interior del set de vidrios usando una micropipeta
permitiendo que esta descienda a lo largo del espaciador sin formar burbujas
Adicionar las soluciones A, B, SDS y agua en un vial de 10 ml para el gel
separador.
Cuando una apropiada cantidad de la solución del gel separador ha sido
adicionada (alrededor de 1.5 cm por debajo de la parte superior del vidrio ó 0.5
cm por debajo del nivel del peine) suavemente adicionar 1.5 ml de agua para
mantener la superficie del gel plana.
Permita que el gel polimerice. Cuando el gel ha polimerizado, aparece una
interfase entre el gel separador y el agua
Adicionar las soluciones A, C, SDS y agua en un vial de 5 ml para el gel
concentrador.
Adicionar el persulfato de amonio y TEMED, mezclar suavemente el vial, ya que
una excesiva aireación pude interferir con la polimerización.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 6/49
Adicionar 400 ml de buffer de electroforesis en el interior y exterior de la cámara,
permitiendo humedecer los pozos, los cuales deben ser purgados con una micro
pipeta de 1000 l para evitar excesos o residuos de la polimerización.
Preparar las muestras en tubos eppendorf. Mezclar cada muestras de proteínas
con el respectivo buffer de carga 5X (i.e 20 l de muestra + 5 l de buffer carga)
para alcanzar una dilución (1:5) del buffer. El volumen de muestra puede ser de 5
Retirar el gel del sistema de fijación y colocarlo en el interior de la cámara para
ensamblarlo, asegurando las puertas en el compartimiento y colocar el interior
de la cámara en su respectivo tanque.
En la electroforesis SDS-PAGE las muestras deben colocadas en baño a ebullición
(92 ºC) por 3 min., para lograr una completa desnaturalización de las proteínas
por la acción del agente reductor 2-mercaptoetanol. Y centrifugar por 30 seg
para homogenizar.
En la electroforesis nativa-PAGE las muestras no se someten a un
calentamiento, ya que las proteínas de interés pueden perder su actividad
enzimática.
Introducidas cada una de las muestras en los respectivos pozos, teniendo
cuidado de no introducir burbujas, ya que pueden contaminar los pozo vecinos.
Introducir la solución sobre el gel separador hasta llenar el espacio libre y colocar
el respectivo peine de 10 pozos, asegurando de no atrapar burbujas en los
dientes de peine y permita que el gel polimerice y remueva el peine con cuidado.
En la electroforesis nativa-PAGE las muestras no se someten a un
calentamiento, ya que las proteínas de interés pueden perder su actividad
enzimática y introducidas cada una de las muestras en los respectivos pozos,
teniendo cuidado de no introducir burbujas, ya que pueden contaminar los
pozo vecinos.
Verificar que el frente de corrida (azul de Bromofenol) comience a descender
hacia el ánodo y Detener la electroforesis cuando el frente de corrido alance la
parte inferior del gel separador.
Apagar la fuente de poder y desconectar los electrodos para retirar el gel y
continuar con el proceso de teñido

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 7/49
TEÑIDO CON AZUL DE COOMASSIE R-250 DIAGRAMA DE FLUJO
Utilizar guantes durante todo el procedimiento para evitar manchar el gel con
los dedos.
Adicionar una pequeña cantidad de la solución de teñido (20 ml es suficiente).
Agitar suavemente el gel por 10 –15 min en un shaker. Cubrir el contenedor con
vinipel durante el teñido y desteñido Y enjuagar el gel con agua destilada, 3
veces
Recoger el gel y colocarlo en un contenedor plástico o de vidrio.Enjuagar el gel
con agua destilada, 3 veces.
Adicionar la solución de desteñido (cerca de 50 ml) hasta que las bandas
aparezcan Y Para desteñir completamente el gel, cambiar la solución de
desteñido y agitar toda la noche.
Sumergir el gel en la solución de conservación por 30-60 min.
Sumergir dos hojas (15x15 cm) de papel celofán en agua para hidratarlo y
colocar el gel entre las dos hojas. Se puede utilizar un vidrio (14x12 cm) para fijar
el celofán en sus extremos, los cuales deben ser sujetados con dos pinzas.
Finalmente se deja secar el gel a temperatura ambiente ó en una incubadora (37
ºC) por 2-4 horas. Antes de retirar el gel verificar que esté completamente seca
la superficie del gel. Luego cortarlo y almacenarlo.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 8/49
RESULTADOS
1. Determine el peso molecular en KDa, de la enzima mostrada en la figura 1.
Figura 1. Electroforesis correspondiente a la purificación de la enzimainulinfructotransferasa, de Arthrobacterureafaciens . Los marcadores de pesomolecular están dados en kilo daltons (Kda). M: marcadores de pesosmoleculares, 1: muestra patron, 2: muestra de precipitación con sulfato de amonio.Tomado de: Kelleyt R. L. 1986. Journal Of Bacteriology , Apr. P. 269-274 166, No.1
Inulin fructotransferasa

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 9/49
CÁLCULO DE RESULTADOS
Marcadores de peso molecular (KDa) Log (KDa) Distancia de los marcadores (cm)
150 2,1761 1,4
100 2,0000 1,8
75 1,8751 2.150 1,6990 2,9
35 1,5441 3,6
25 1,3979 4,6
15 1,1761 6,2
Ecuación de la pendiente: Y = - 0,162 X + 2,343
Y = Logaritmo de KDa
PM = Antilogaritmo de Y
1 Muestra patrón(X₁)
2 Muestra de precipitación con(NH4)₂SO₄ (X₂) Y = - 0,162 X + 2,343
Peso Molecular(KDa)
3,4 1,7922 61,9726
5,0 1,5330 34,1129
1,8 2,0514 112,5641
2,4 1,9542 89,9912
3,4 1,7922 61,9726

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 10/49
ANÁLISIS DE RESULTADOS
Para los pesos molecular de la enzima inulinfructotransferasa, de 112,5641 y
89,9912 KDa, se puede observar que el pH de estas proteínas se encuentra pordebajo del punto isoeléctrico por lo cual estas poseen carga positiva neta ya quese desplazaron muy poco manteniéndose cerca del cátodo (-); por consiguienteposeen un alto peso molecular comparado con la proteína marcador, lo cual indicaque poseen cadenas largas de poli péptidos.
Las demás moléculas presentaron menor peso molecular y poseen cadenas mascortas de poli péptidos y se desplazaron un poco mas hacia el ánodo (+), por locual poseen un pH mas cercano al punto isoeléctrico.
CONCLUSIONES
Aunque no se pudo realizar con éxito la técnica de electroforesis deproteínas en geles SDS-PAGE, con el ejemplo enviado por el tutor se pudorealizar una lectura clara y comparativa de la migración de la enzimainulinfructotransferasa.
Mediante la técnica de electroforesis desnaturalizante en geles de
poliacrilamida se logran separar las muestras de proteínas. Se determinaron los pesos moleculares de la enzima
inulinfructotransferasa a partir de los pesos moleculares del marcador,como también su cercanía al punto isoeléctrico.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 11/49
PRACTICA 2
CINÉTICA DE CRECIMIENTO MICROBIANO
INTRODUCCION
Mediante la experimentación con diferentes sustratos, como medios de cultivo sedeterminarán el crecimiento de la E-coli, y se observará cuál de estos medios es elmás adecuado para su adaptación y crecimiento equilibrado.
Con un crecimiento equilibrado se puede medir la velocidad de crecimiento de un
cultivo, como también el de todos los componentes de la población, esta velocidadde crecimiento en un tiempo dado es proporcional al número o masa de la bacteriaE-coli, presente en este tiempo, finalmente la medida de esta masa se determinarapor medidas de absorbancia para cada uno de los cultivos realizados.
MARCO TEÓRICO
El crecimiento microbiano se puede considerar como un conjunto de reaccionesquímicas en cadena, que conducen en la síntesis de los constituyentes de labiomasa microbiana obtenida al final de la operación. Globalmente, el procesoobedece al principio de la conservación de la materia.
Cuando se introduce un microorganismo en un medio de cultivo que le conviene,desarrolla una actividad en relación a su composición y las condicionesambientales.
El microbio se reproduce, resulta un aumento de la concentración en biomasamicrobiana, la biosíntesis de los constituyentes celulares se hace a partir de los
compuestos del medio de cultivo.
La cinética describe las velocidades a la cual las reacciones químicas ybioquímicas se desarrollan en diferentes condiciones y constituye una de lasoperaciones más utilizadas por la ingeniería alimentaria y la biotecnología, por lotanto, es necesario conocer los diferentes mecanismos de crecimiento, así como,la forma de cuantificación de los mismos, sus formas de aplicación, las ventajas y

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 12/49
desventajas de los diferentes métodos, y sobre todo el monto económico de cadauno de ellos.
Para determinar la biomasa microbiana se utilizó el método de medición óptica.Esta interesante técnica se basa en el hecho de que la densidad óptica de
suspensión es proporcional a la masa en las partículas suspendidas. Así se midela absorción luminosa generalmente a 600nm o 700nm, de la suspensiónmicrobiana y por comparación con una escala de referencia se deduce laconcentración en biomasa. Las suspensiones de organismos filamentosos no sonapropiadas a esta técnica. Sin embargo se puede homogenizar la muestra encondiciones estandarizadas antes de hacer la medición.
Para medir la curva de crecimiento se dispone de una técnica de evaluacióncualitativa y cuantitativa de una población microbiana. Para ello se efectúa uncultivo de tipo clásico en el cual después de sembrar se observa el crecimiento
hasta el agotamiento del medio, manteniendo constantes las condicionesexteriores en particular el de la temperatura cuidando que sean favorables aldesarrollo.
El estudio se basa en seguir en función del tiempo la evolución de X concentracióncelular o la concentración de biomasa, según el tipo de microorganismo y el tipode método seleccionado para seguir el fenómeno. En todos los casos sedesenvuelve de la misma forma comportándose las diversas fases como son la delatencia que se presenta después de la siembra del microorganismo y es elperiodo de adaptación, en el curso en donde la célula sintetiza, en particular, las
enzimas que le son necesarias para metabolizar el sustrato que existe, en estafase no hay reproducción.
El crecimiento de poblaciones microbianas también puede ir acompañadas por laformación de productos finales tóxicos que causan la inhibición del crecimiento.Un ejemplo es la producción de etanol por levaduras y bacterias.
Los rendimientos de biomasa y de producto, son parámetros muy importantes, yaque representan la eficacia de la conversión del substrato en biomasa yproductos. Se definen como la biomasa o producto formado por la unidad de masade substrato consumido. (Alan, 1999)

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 13/49
METODOLOGIA
CINETICA Y CRECIMIENTO MICROBIANO
Tomamos una gradilla con 6 tubos de ensayo ylos marcamos desde el 0 hasta el 6.
Tomamos el inoculo de E-coli ya Preparado loagitamos y adicionamos 1ml al tubo 0 junto con el
extracto maltosa al 5% y agitamos.
Repetimos el procedimiento anterior con la tomade muestra en tubos de ensayo en los tiempos:9:30, 10:30, 11:30, 1:00, 2:00, 3:00, 4:00, min,cada tubo debidamente marcado con su respectivotiempo almacenarlo hasta el el crecimiento final.
Llevamos el tubo al refrigerador a 370C y tomamosel tiempo 0.
Luego sacamos todos los 6 tubos del refrigerador
y tomamos la absorbancia a cada uno de ellos
Agitamos y tomamos en un tubo de ensayo, unaalícuota de 2 ml en condiciones asépticas(utilizando un mechero) de cada medio de cultivo,el cual se utilizará como blanco (B).
Realización de la grafica

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 14/49
RESULTADOS
Ecuación para despejar la biomasa
Y= 0,19X + 0,005
Y= Abs (600nm) X= [biomasa]
GRAFICO DE E COLI EN VARIOS SUSTRATOS TIEMPO EN (h) VS BIOMASA EN (MG)
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 15/49
CINÉTICA Y CRECIMIENTO MICROBIANO
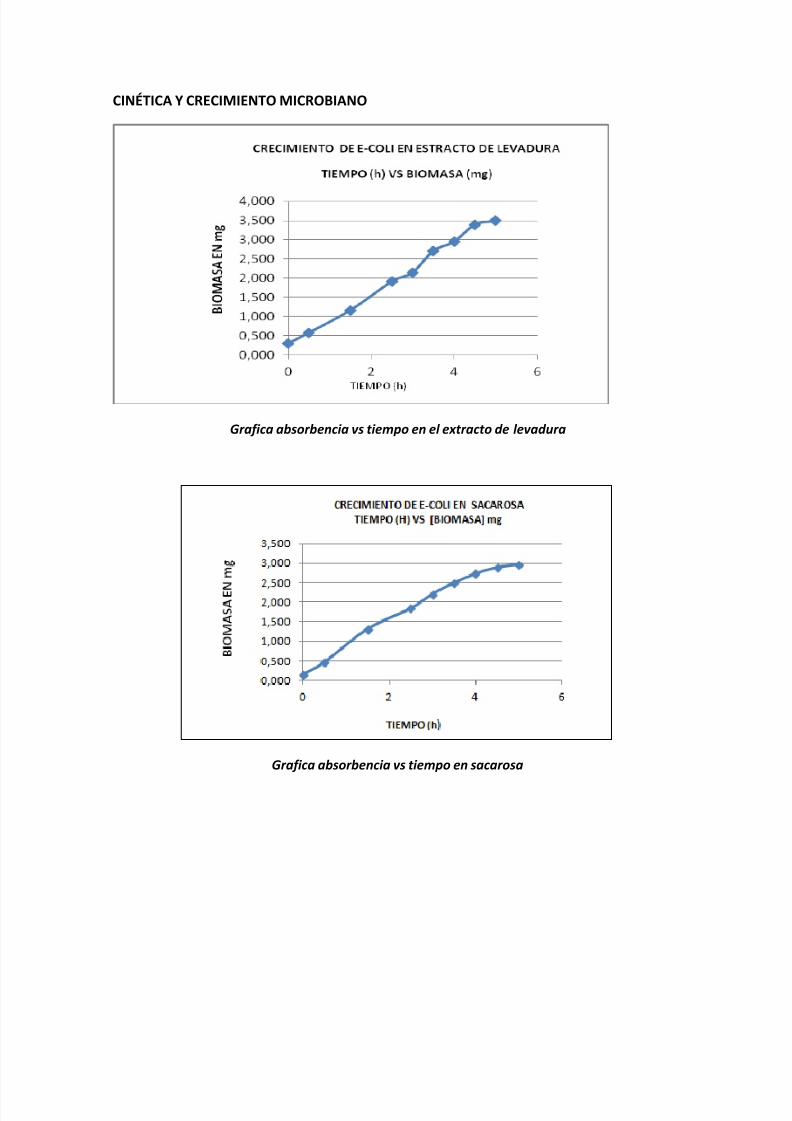
Grafica absorbencia vs tiempo en el extracto de levadura
Grafica absorbencia vs tiempo en sacarosa

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 16/49
Grafica absorbencia vs tiempo en glucosa
Grafica absorbencia vs tiempo en maltosa

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 17/49
Grafica absorbencia vs tiempo en fructosa
Grafica absorbencia vs tiempo en galactosa
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 18/49
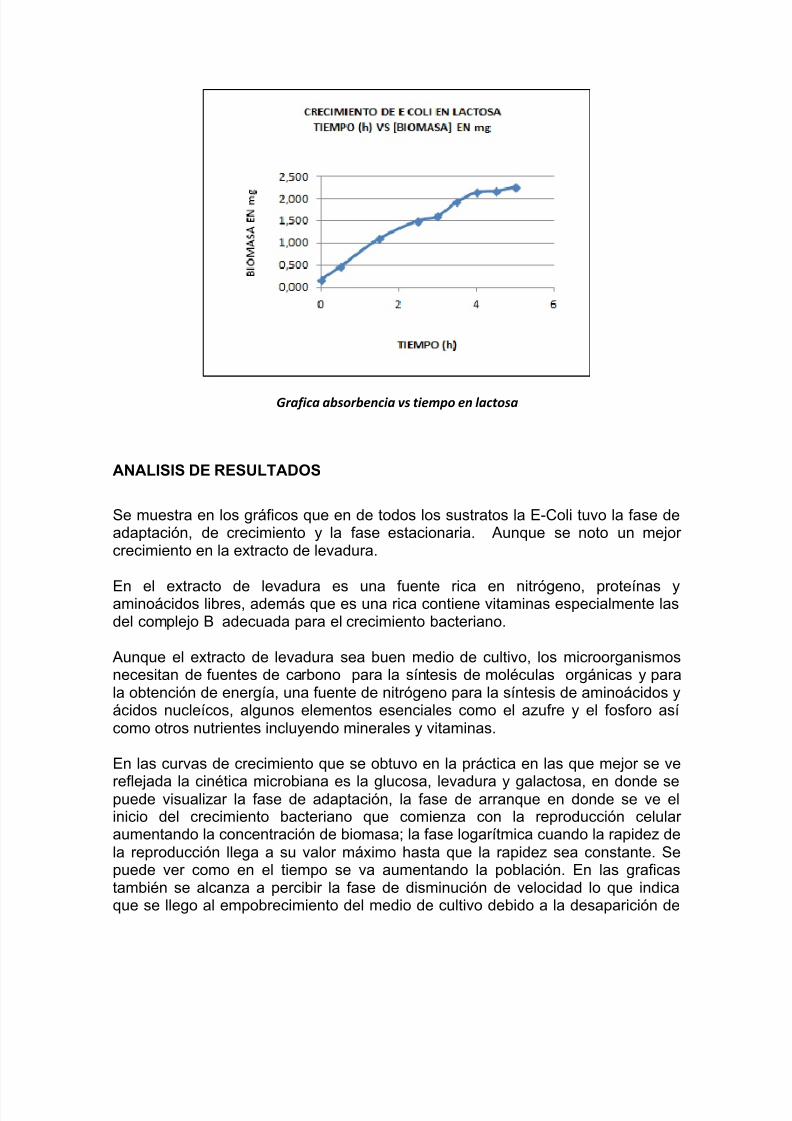
Grafica absorbencia vs tiempo en lactosa
ANALISIS DE RESULTADOS
Se muestra en los gráficos que en de todos los sustratos la E-Coli tuvo la fase deadaptación, de crecimiento y la fase estacionaria. Aunque se noto un mejorcrecimiento en la extracto de levadura.
En el extracto de levadura es una fuente rica en nitrógeno, proteínas yaminoácidos libres, además que es una rica contiene vitaminas especialmente lasdel complejo B adecuada para el crecimiento bacteriano.
Aunque el extracto de levadura sea buen medio de cultivo, los microorganismosnecesitan de fuentes de carbono para la síntesis de moléculas orgánicas y parala obtención de energía, una fuente de nitrógeno para la síntesis de aminoácidos yácidos nucleícos, algunos elementos esenciales como el azufre y el fosforo asícomo otros nutrientes incluyendo minerales y vitaminas.
En las curvas de crecimiento que se obtuvo en la práctica en las que mejor se ve
reflejada la cinética microbiana es la glucosa, levadura y galactosa, en donde sepuede visualizar la fase de adaptación, la fase de arranque en donde se ve elinicio del crecimiento bacteriano que comienza con la reproducción celularaumentando la concentración de biomasa; la fase logarítmica cuando la rapidez dela reproducción llega a su valor máximo hasta que la rapidez sea constante. Sepuede ver como en el tiempo se va aumentando la población. En las graficastambién se alcanza a percibir la fase de disminución de velocidad lo que indicaque se llego al empobrecimiento del medio de cultivo debido a la desaparición de

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 19/49
uno o varios compuestos necesarios para el crecimiento, aunque no se indica bienla fase de muerte. (Scriban, 1985).
CONCLUSIONES
La e- coli tuvo un buen comportamiento de crecimiento en la sacarosa al igual queen los sustratos de levadura, lactosa y glucosa, se pudo comparar los resultadosde crecimiento en los diferentes tipos de sustratos, en donde indica que en dondemenor hubo crecimiento fue en la galactosa.
Mediante las graficas de cinética de crecimiento se ve que la velocidad decrecimiento es proporcional a la concentración de células ya presentes. Durante elcrecimiento exponencial el peso seco celular aumenta. Se deduce que ladensidad celular después del tiempo se relaciona con la densidad presenteoriginalmente.
Se comprueba como los factores principalmente que afectan la rapidez decrecimiento son la concentración de substrato, la temperatura y el pH por elproducto. (Alan, 1999)
El ciclo de crecimiento debió ser mejor en la glucosa debido a ser gran fuente decarbono y energía pero se presento en la sacarosa el cual es un azúcardisacárido compuesto por fructosa y glucosa, el crecimiento mayor en este azúcarpudo haberse dado debido a que la sacarosa quizás estuviera hidrolizada yhubiera mayor cantidad de glucosa que azúcar compuesto por presencia de algúnacido.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 20/49
ACTIVIDAD ENZIMATICA E INVOLIZACION DE LA INVERTASA
INTRODUCCIÓN
Mediante esta práctica se puede determinar la actividad enzimática de la invertasaque es una enzima con capacidad de hidrolizar la sacarosa en fructosa y glucosa,esta enzima es proveniente de varios tipos de microorganismos. A nivel industrialel Aspergillus Niger es el microorganismo más utilizado para la producción deinvertasa.
El propósito de esta práctica es el estudio del funcionamiento de la enzimaglucosa oxidasa y la relación que existe entre la velocidad de la reacción
enzimática en diferentes concentraciones de sustrato utilizando 1ml de enzima,por lo que se debe tener en cuenta que la cantidad de enzima debe ser pequeñaen comparación a la concentración de sustrato para que la concentración deenzima sustrato no modifique la concentración del sustrato.
OBJETIVOS
Determinar la actividad enzimática de la invertasa Inmovilización de la enzima libre en alginato de sodio por atrapamiento
MARCO TEÓRICO
La palabra enzima se deriva del griego que significa (en las levaduras) y fue usadapor Kuhne en 1872. Sin embrago, el primer informe publicado acerca de la
capacidad de los extractos celulares (malta) para llevar a cabo una reaccióncaracterística de las células vivas (la hidrólisis de almidón de glucosa) fue escritoinicialmente en 1833 por Payen y Persoz. Más tarde, Buchner (1897) demostró lacapacidad de los extractos de levadura para catalizar la conversión del azúcar aalcohol, mientras que Emil Fischer (1894) demostró la especificidad de lasenzimas para su sustrato. Subsecuentemente, Summer (1926) cristalizo la primeraenzima la ureasa la hidroliza la urea a CO2 y NH3 y mostro que las enzimas eran

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 21/49
proteínas. Fue hasta la década de los años 70 que se conoció la composiciónquímica precisa de las enzimas mediante la secuencia de la ribonucleasa, juntocon los estudios de conformación tridimensional por cristalografía de rayos X.Estos trabajos permitieron que se postulara el mecanismo de acción enzimática,
junto con las proposiciones para la forma en que esta se regula.
Por último en 1969 se sintetizo químicamente la primera enzima (ribonucleasa), apartir de los aminoácidos precursores y, aunque su actividad y pureza eranescasas se demostró que las enzimas no son cualitativamente distintas a loscatalizadores no biológicos. (Alan, 1999)
Uno de los modelos más útiles en la investigación sistemática de las velocidadesenzimáticas, fue propuesto por Leonor Michaelis y Maud Menten en 1913. Elconcepto del complejo enzimas- sustrato, enunciado por primera vez por VictorHenri en 1903 es fundamental para la cinética de Michaelis – Mentel. Cuando se
une el sustrato S en el sitio activo de la enzima, se forma un complejointermediario (ES). Durante el estado de transición el sustrato se convierte enproducto. Tras un lapso breve, el producto se disocia de la enzima. (Mckee, 2009)
Hipótesis de Michaelis Menten: Al establecer la ecuación de la velocidad,Michaelis y Menten toman como principio de que la velocidad de transformacióndel complejo ES en E+P es muy lenta, con relación a la velocidad de la ruptura delcomplejo que vuelve a dar E+S. Esta hipótesis significa que K3 <k2 y que E y ESestán en equilibrio.
Los estudios de cinética enzimática corresponden siempre a esta parte lineal de lacurva lo que implica el trabajar a velocidades iníciales. En estas condiciones alprincipio de la reacción la concentración en producto es muy débil; puededespreciarse así como la reacción inversa de la transformación del producto ensustrato. (Scriban, 1985)

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 22/49
Metodología
3. ACTIVIDAD ENZIMÁTICA
Cálculos y Resultados
[Glucosa]= ia xAbsorbancml mg
320,0
/1,0
G + H2O Acido gluconico + H2O2
1 mol de glucosa produce una mol de acido glucónico
Ecuación de biomasa
Y= 0,19x + 0,005 y= abs (600 mn) x= [biomasa mg/ml]
Medir las Observancias de cada tubo con sus
respectivos tiempos.
En una gradilla colocar 7 tubos de ensayo
En un vaso de precipitados de 50 ml, adicionar
10 ml de buffer acetatos al 20%, pH 4.6 y 5ml de
solución de glucosa
Atemperar en baño termostatado a 40 ˚C durante 5
min.
Adicionar 2 ml de la solución de la enzima invertasa, inmediatamente
cronometrar el tiempo tomando un alícuota de 10 ml con una micro pipeta y
depositarla en el tubo de ensayo marcado con 0, luego sumergir este tubo en
un vaso de precipitados en ebullición por 5 min para inactivar la enzima.
Repetir la toma de muestra en los tubos marcados con 6, 4, 2, 1, 0.1,
0.01 e inactivar, en los tiempos 0, 2, 5, 10, 20 y 60 minutos sin
interrupción.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 23/49
RESULTADOS
ACTIVIDAD ENZIMATICA
0,025
TIEMPO
(minutos)
ABSORVACIA[GLUCOSA]
mg/ml
MOLES DE
GLUCOSA
MOLES DE
ACIDOGLUCONICO
MASA DE
ACIDOGLUCONICO
mg /min
0 0 0 0 0 0
0,08 0,0002 0,0001 0 0 0,0001
0,15 0,005 0,0016 0,000009 0,000009 0,0017
0,22 0,007 0,0022 0,000012 0,000012 0,0024
0,26 0,01 0,0031 0,000017 0,000017 0,0034
0,38 0,015 0,0047 0,000026 0,000026 0,0051
0,47 0,02 0,0063 0,000035 0,000035 0,0068
1 0,025 0,0078 0,000043 0,000043 0,0085
1,13 0,03 0,0094 0,000052 0,000052 0,0102
1,31 0,036 0,0113 0,000062 0,000062 0,0123
1,4 0,04 0,0125 0,000069 0,000069 0,0136
1,55 0,045 0,0141 0,000078 0,000078 0,0153
2,15 0,05 0,0156 0,000087 0,000087 0,017
2,35 0,054 0,0169 0,000094 0,000094 0,0184
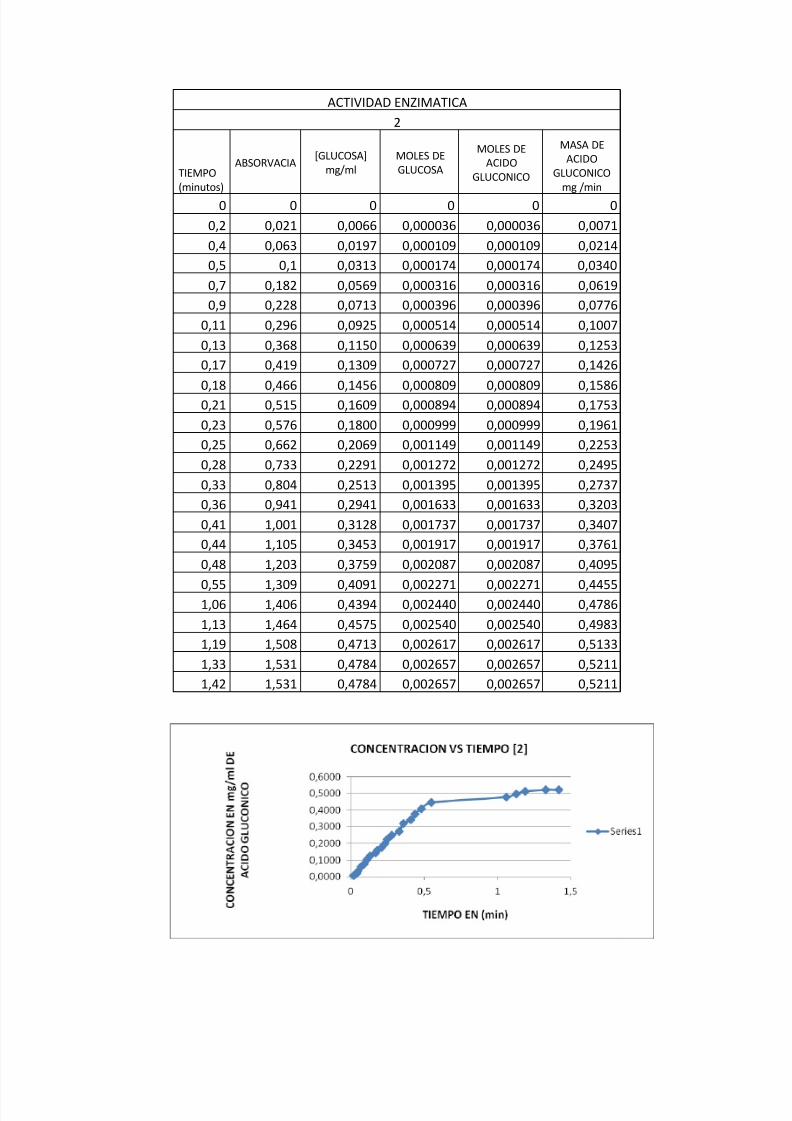
Grafica: Actividad Enzimática de la enzima glucosa oxidasa en sustrato al [0,25].
m= pendiente de velocidad enzimática en los primeros momentos
mnml mg m //01545.015.026.0
0017.00034.0

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 24/49
ACTIVIDAD ENZIMATICA
0,05
TIEMPO
(minutos)
ABSORVACIA[GLUCOSA]
mg/ml
MOLES DE
GLUCOSA
MOLES DE
ACIDO
GLUCONICO
MASA DE
ACIDO
GLUCONICO
mg /min
0 0 0 0 0 0
0,04 0,003 0,0009 0,000005 0,000005 0,001
0,11 0,008 0,0025 0,000014 0,000014 0,0027
0,18 0,015 0,0047 0,000026 0,000026 0,0051
0,27 0,023 0,0072 0,00004 0,00004 0,0078
0,38 0,035 0,0109 0,000061 0,000061 0,0119
0,53 0,047 0,0147 0,000082 0,000082 0,016
1,08 0,059 0,0184 0,000102 0,000102 0,0201
1,22 0,07 0,0219 0,000121 0,000121 0,0238
1,33 0,076 0,0238 0,000132 0,000132 0,0259
1,57 0,084 0,0263 0,000146 0,000146 0,0286
2,13 0,09 0,0281 0,000156 0,000156 0,0306
2,24 0,1 0,0313 0,000174 0,000174 0,034
2,32 0,106 0,0331 0,000184 0,000184 0,0361
2,53 0,11 0,0344 0,000191 0,000191 0,0374
3,09 0,12 0,0375 0,000208 0,000208 0,0408
3,2 0,13 0,0406 0,000226 0,000226 0,0442
Grafica: Actividad Enzimática de la enzima glucosa oxidasa en sustrato al [0,05].
m= pendiente de velocidad enzimática en los primeros momentos
mnml mg m //0340.011.038.0
0027.00119.0
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 25/49
ACTIVIDAD ENZIMATICA
2
TIEMPO
(minutos)
ABSORVACIA[GLUCOSA]
mg/ml
MOLES DE
GLUCOSA
MOLES DE
ACIDO
GLUCONICO
MASA DE
ACIDO
GLUCONICO
mg /min
0 0 0 0 0 0
0,2 0,021 0,0066 0,000036 0,000036 0,0071
0,4 0,063 0,0197 0,000109 0,000109 0,0214
0,5 0,1 0,0313 0,000174 0,000174 0,0340
0,7 0,182 0,0569 0,000316 0,000316 0,0619
0,9 0,228 0,0713 0,000396 0,000396 0,0776
0,11 0,296 0,0925 0,000514 0,000514 0,1007
0,13 0,368 0,1150 0,000639 0,000639 0,1253
0,17 0,419 0,1309 0,000727 0,000727 0,1426
0,18 0,466 0,1456 0,000809 0,000809 0,1586
0,21 0,515 0,1609 0,000894 0,000894 0,1753
0,23 0,576 0,1800 0,000999 0,000999 0,1961
0,25 0,662 0,2069 0,001149 0,001149 0,2253
0,28 0,733 0,2291 0,001272 0,001272 0,2495
0,33 0,804 0,2513 0,001395 0,001395 0,2737
0,36 0,941 0,2941 0,001633 0,001633 0,3203
0,41 1,001 0,3128 0,001737 0,001737 0,3407
0,44 1,105 0,3453 0,001917 0,001917 0,3761
0,48 1,203 0,3759 0,002087 0,002087 0,4095
0,55 1,309 0,4091 0,002271 0,002271 0,44551,06 1,406 0,4394 0,002440 0,002440 0,4786
1,13 1,464 0,4575 0,002540 0,002540 0,4983
1,19 1,508 0,4713 0,002617 0,002617 0,5133
1,33 1,531 0,4784 0,002657 0,002657 0,5211
1,42 1,531 0,4784 0,002657 0,002657 0,5211

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 26/49
Grafica: Actividad Enzimática de la enzima glucosa oxidasa en sustrato al [2.00].
m= pendiente de velocidad enzimática en los primeros momentos
mnml mg m //81.004.009.0
0214.00619.0
ACTIVIDAD ENZIMATICA
4
TIEMPO
(minutos)
ABSORVACIA[GLUCOSA]
mg/ml
MOLES DE
GLUCOSA
MOLES DE
ACIDO
GLUCONICO
MASA DE
ACIDO
GLUCONICO
mg /min
0 0 0 0 0 0
0,02 0,026 0,0081 0,000045 0,000045 0,0088
0,04 0,063 0,0197 0,000109 0,000109 0,0214
0,05 0,1 0,0313 0,000174 0,000174 0,0340
0,07 0,14 0,0438 0,000243 0,000243 0,0477
0,09 0,182 0,0569 0,000316 0,000316 0,0619
0,11 0,228 0,0713 0,000396 0,000396 0,0776
0,13 0,296 0,0925 0,000514 0,000514 0,1007
0,17 0,368 0,1150 0,000639 0,000639 0,1253
0,18 0,419 0,1309 0,000727 0,000727 0,1426
0,21 0,46 0,1438 0,000798 0,000798 0,1566
0,23 0,515 0,1609 0,000894 0,000894 0,1753
0,25 0,574 0,1794 0,000996 0,000996 0,1954
0,28 0,662 0,2069 0,001149 0,001149 0,2253
0,33 0,733 0,2291 0,001272 0,001272 0,2495
0,36 0,804 0,2513 0,001395 0,001395 0,2737
0,41 0,914 0,2856 0,001586 0,001586 0,3111
0,44 1,001 0,3128 0,001737 0,001737 0,3407
0,48 1,105 0,3453 0,001917 0,001917 0,3761
0,55 1,203 0,3759 0,002087 0,002087 0,4095
1,06 1,309 0,4091 0,002271 0,002271 0,4455
1,13 1,406 0,4394 0,002440 0,002440 0,4786
1,19 1,464 0,4575 0,002540 0,002540 0,4983
1,33 1,568 0,4900 0,002721 0,002721 0,5337
1,42 1,531 0,4784 0,002657 0,002657 0,5211
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 27/49
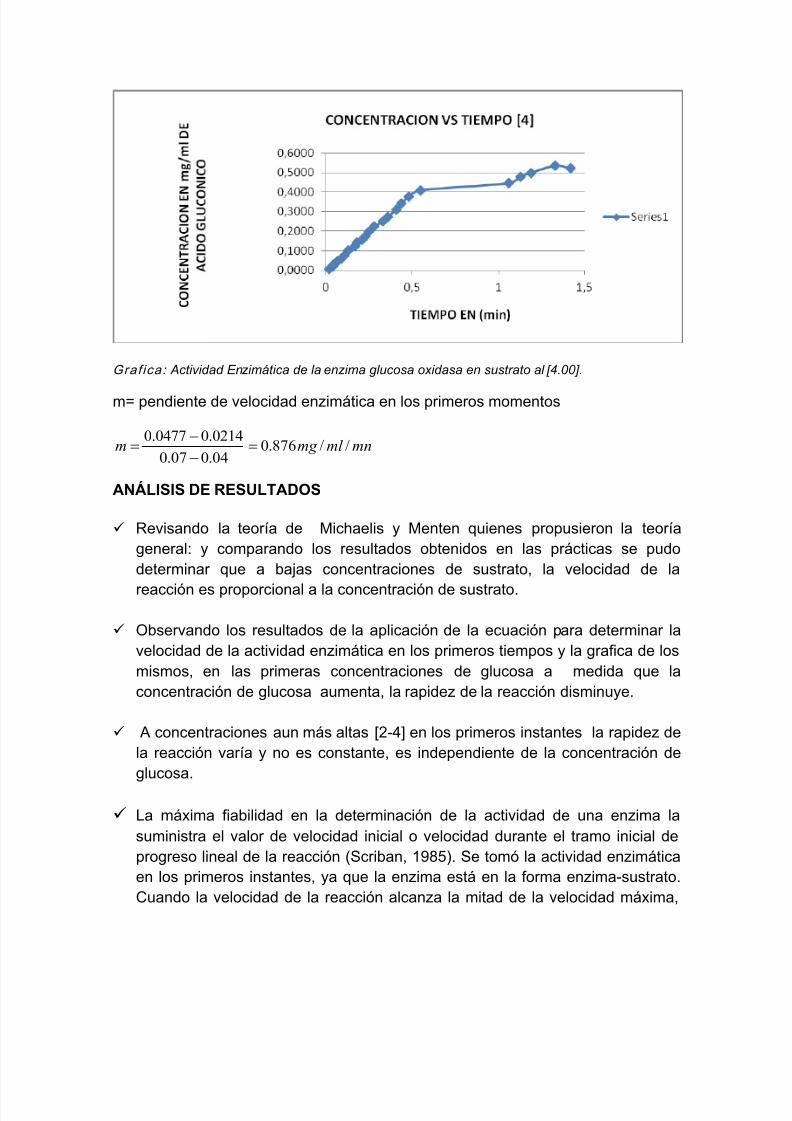
Grafica: Actividad Enzimática de la enzima glucosa oxidasa en sustrato al [4.00].
m= pendiente de velocidad enzimática en los primeros momentos
mnml mg m //876.004.007.0
0214.00477.0
ANÁLISIS DE RESULTADOS
Revisando la teoría de Michaelis y Menten quienes propusieron la teoríageneral: y comparando los resultados obtenidos en las prácticas se pudodeterminar que a bajas concentraciones de sustrato, la velocidad de la
reacción es proporcional a la concentración de sustrato.
Observando los resultados de la aplicación de la ecuación para determinar lavelocidad de la actividad enzimática en los primeros tiempos y la grafica de losmismos, en las primeras concentraciones de glucosa a medida que laconcentración de glucosa aumenta, la rapidez de la reacción disminuye.
A concentraciones aun más altas [2-4] en los primeros instantes la rapidez dela reacción varía y no es constante, es independiente de la concentración deglucosa.
La máxima fiabilidad en la determinación de la actividad de una enzima lasuministra el valor de velocidad inicial o velocidad durante el tramo inicial deprogreso lineal de la reacción (Scriban, 1985). Se tomó la actividad enzimáticaen los primeros instantes, ya que la enzima está en la forma enzima-sustrato.Cuando la velocidad de la reacción alcanza la mitad de la velocidad máxima,

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 28/49
indicando la cantidad de sustrato (Km), el cual puede ser asimilado a la inversade la relación de la glucosa oxidasa por la glucosa.
Se puede observar en las graficas la caída de la velocidad de reacción que sedebe a la disminución significativa de la concentración de sustrato, aunque
también pudo haber influido otros factores como el aumento de laconcentración de producto, cambios de pH, inactivación de la enzima.
La actividad catalítica de la glucosa oxidasa se determino midiendo lavelocidad inicial de reacción, que es la pendiente de la curva de progreso(curva de producto formado ó sustrato transformado frente al tiempo) en eltiempo cero como lo indican las graficas.
La disminución del sustrato indica la liberación de productos por la actividad de
la enzima y por ende una degradación progresiva del sustrato (Alan, 1999).
Inicialmente, las reacciones transcurren linealmente en las [0,01-2-4]pudiéndose tomar la pendiente de esta recta como velocidad inicial. A tiemposmás largos, el progreso de la reacción deja de ser lineal, debido al consumo deproducto por la enzima.
CONCLUSIONES
Mediante la práctica se determino que en las primeras concentraciones alaumentar la concentración la velocidad de la actividad enzimáticadisminuyo debido a que el tiempo de reacción aumentó.
Se puede observar que en las [0,025- 0,05] el tiempo de reacción delsustrato enzima, al aumentar la concentración de glucosa el tiempo dereacción disminuyo.
En [2-4] el tiempo de reacción fue directamente proporcional a laconcentración, ya que la caída de la velocidad de reacción se obtuvo enmayor tiempo a mayor concentración de sustrato.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 29/49
PRACTICA 3.EXTRACCIÓN Y ELECTROFORESIS DEL ADN
INTRODUCCIÓN
Con el desarrollo de esta práctica se extraerá el ADN de la bacteria E-coli, el cualse pretende separar, cuantificar y analizara por medio de la electroforesis encubeta horizontal en gel de agarosa.
El termino electroforesis se usa para describir la migración de una molécula opartícula cargada bajo la influencia de un campo eléctrico. Muchas moléculascomo son aminoácidos, péptidos, proteínas, nucleótidos, ácidos nucleídos, poseengrupos ionizantes y existen en solución como especies cargadas ya sea comocationes o como aniones. Estas moléculas inmersas en gel de agarosa y cargadasse van a separar en función de su carga aplicando un voltaje a través deelectrodos.
OBJETIVOS
Conocer la metodología para extraer ADN de la E-coli. Cuantificar el ADN extraído de la E-coli por medio de la técnica de la
Electroforesis el gel de agarosa. Determinar el peso molecular de las moléculas del ADN extraído. Analizar los resultados obtenidos.
MARCO TEORICO
Los ácidos nucleídos son macromoléculas o polímeros formados por la repeticiónde monómeros llamados nucleótidos, unidos por enlaces fosfodiéster: Formandoentre sí cadenas largas de polinucleótidos por lo cual estas molécula puede llegara tamaños gigantes, de millones de nucleótidos.
Los nucleótidos se clasifican en: ADN (ácido desoxirribonucléico) y ARN (ácido
ribonucléico), estos se diferencian por la pentosa (desoxirribosa y ribosa), lasbases nitrogenadas (purínica y pirimidínica), por la estructura o forma de suscadenas (cadena doble, monocatenaria, etc.) y por la masa molecular (la del ADNes mayor que la del ARN).
El ADN esta conformado por dos cadenas polinucleotídicas unidas entre si, estascadenas pueden ser de forma lineal como el ADN de células eucariotas o puede

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 30/49
ser circular como el ADN de las células procariotas, el de las mitocondrial ycloroplastos eucarioticos.
Las moléculas del ADN portan la información para el desarrollo de lascaracterísticas biológicas de un individuo, como también la información para que
las células realicen sus funciones.Según sea la composición del ADN, este se puede desnaturalizar o romper lospuentes de hidrógeno entre las bases, conformando ADN de cadena simple. El
ADN de algunos virus es monocatenario es decir esta formado por un solopolinucleótido, sin cadena complementaria.
El ADN contiene la información y el ARN expresa dicha información, pasando deuna secuencia lineal de nucleótidos a una secuencia lineal de aminoácidos en unaproteína. Para expresar la información del ADN deben haber varias etapas para locual existen varios tipos de ARN, como el ARN mensajero, el ARN detransferencia y el ARN ribosómico.
Para extraer el ADN se requieren de varias etapas, primero se debe romper lapared celular y la membrana plasmática para acceder al núcleo de la célula, luegose debe romper la membrana nuclear para que quede libre el ADN, finalmente sedebe proteger el ADN de enzimas que puedan degradarlo, para aislarlo de debehacer precipitar en alcohol.
La extracción de ADN de una muestra celular se basa en el hecho de que losiones de la solución salina son atraídos hacia las cargas negativas del ADN,permitiendo su disolución y posterior extracción de la célula. Se empieza por lisar(romper) las células mediante un detergente, en este caso SDS, centrifugar yagregar una disolución tampón (buffer TBE-5X) en la que se disuelve el ADN. Enese momento, el tampón contiene ADN y todo un surtido de restos moleculares:
ARN, carbohidratos, proteínas y otras sustancias en menor proporción. Lasproteínas asociadas al ADN, de gran longitud, se habrán fraccionado en cadenasmás pequeñas y separadas de él por acción del detergente, como también se handesnaturalizado por acción del calor. Sólo queda, por tanto, extraer el ADN de esamezcla de tampón y detergente, para lo cual se utiliza un alcohol (isopropanol).
La electroforesis en gel de agarosa (en cubetas horizontales), es la más usadapara separar, identificar, purificar, analizar y caracterizar ácidos nucleídos. Estegel se comporta como un tamiz molecular el cual permite separar moléculascargadas en función de su tamaño y forma, por lo general los ácidos nucleídosmigran desde el electrodo negativo hacia el positivo, ya que estas poseen carganegativa en el esqueleto azúcar-fosfáto; las moléculas de menor tamaño migraranmas rápido hacia el ánodo (polo positivo) que las de mayor tamaño.
Aplicando marcadores de peso molecular a la electroforesis, los cuales sonfragmentos de ADN de tamaño conocido, se puede calcular el tamaño aproximadodel ADN en estudio.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 31/49
Los ácidos nucleídos separados en gel de agarosa, se pueden visualizar pormedio de tinción con colorantes fluorescentes, los cuales se pueden observar bajoluz ultravioleta y con lo cual se puede evaluar su integridad y estimar suconcentración mediante análisis comparativo con patrones de concentración
conocida.Los colorantes fluorescentes actúan mediante la inserción entre los pares debases que conforman el ácido nucleído. El bromuro de etidio es ampliamenteutilizado para visualización del ADN y ARN, es altamente toxico y reactivo, conpropiedades mutagénicas, por lo cual se debe manipular con la debida proteccióny precaución.
Tomado de: Duina Posso Duque, Thaura Ghneim Herrera. 2008. Uso deMarcadores Microsatélites para la Estimación de Diversidad Genética en Plantas.Ediciones IVIC.
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 32/49
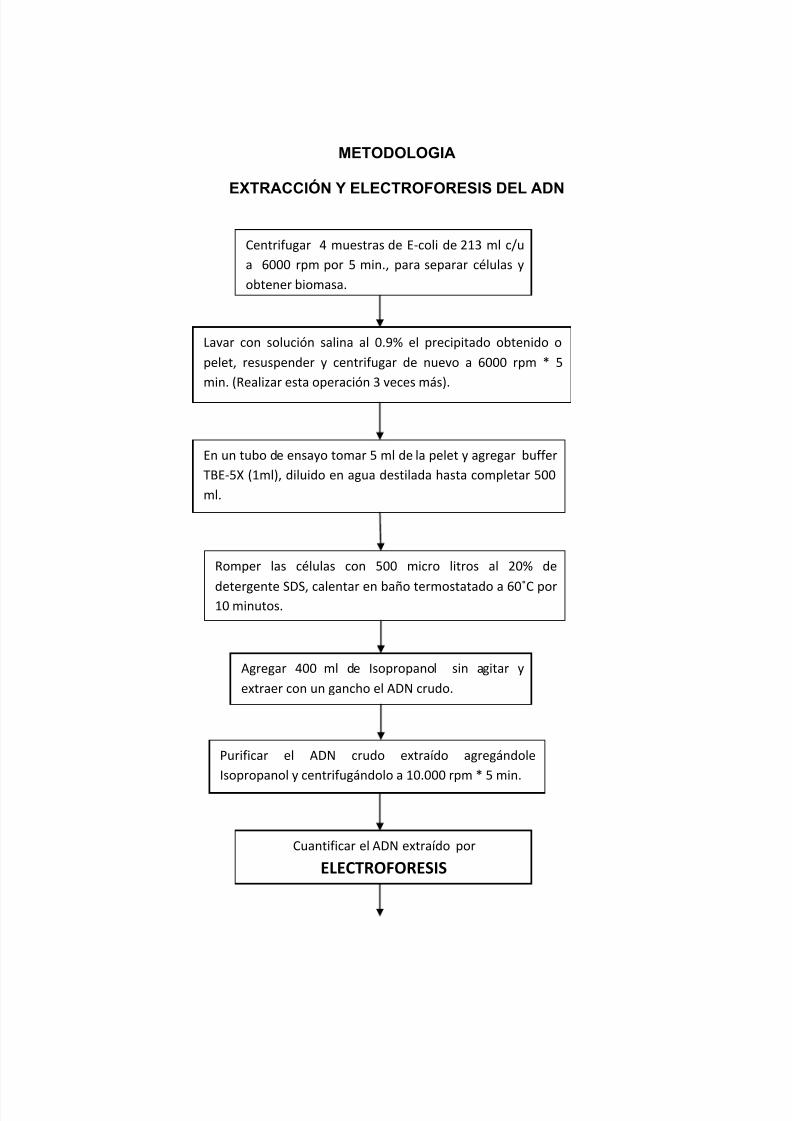
METODOLOGIA
EXTRACCIÓN Y ELECTROFORESIS DEL ADN
Centrifugar 4 muestras de E-coli de 213 ml c/u
a 6000 rpm por 5 min., para separar células y
obtener biomasa.
Lavar con solución salina al 0.9% el precipitado obtenido o
pelet, resuspender y centrifugar de nuevo a 6000 rpm * 5
min. (Realizar esta operación 3 veces más).
En un tubo de ensayo tomar 5 ml de la pelet y agregar buffer
TBE-5X (1ml), diluido en agua destilada hasta completar 500
ml.
Romper las células con 500 micro litros al 20% de
detergente SDS, calentar en baño termostatado a 60˚C por10 minutos.
Agregar 400 ml de Isopropanol sin agitar y
extraer con un gancho el ADN crudo.
Purificar el ADN crudo extraído agregándole
Isopropanol y centrifugándolo a 10.000 rpm * 5 min.
Cuantificar el ADN extraído por
ELECTROFORESIS

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 33/49
Preparar gel de agarosa al 0.8%, para 100ml: Tomar 0,8
ml de garosa y 100 ml de buffer TBE 0,5 X, calentar a
baño termostatado hasta dilución completa.
Dejar enfriar hasta 37 ˚C y agregar el bromuro de
etidio de alto metoxilo (con guantes), mezclar bien
evitando que se formen burbujas.
Agregar la solución lentamente por un extremo de la
bandeja de corrida, retirando las burbujas que se
formen. Dejar polimerizar la agarosa y retirar el peine.
Para la corrida, llenar el tanque de la cámara de
electroforesis con buffer TBE 0,5 X hasta cubrir el gel.
A la muestra de ADN extraída, agregarle buffer de tinción
Tris borato EDTA que debe estar diluido, luego con una
micro pipeta agregar 20 micro litros de muestra en cada
pozo.
Conectar los electrodos de la cámara a la fuente de
poder a 8 voltios por ½ hora para que corran las
muestras.
Para visualizar la separación de las muestras en el gel, se
introduce en un transiluminador de luz ultravioleta (UV),
luego tomarle una fotografía a la placa para realizar el análisis
del resultado.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 34/49
RESULTADOS (-) cátodo
(+) ánodo
ANÁLISIS DE RESULTADOS
En esta imagen tomada de la electroforesis del ADN, bajo un transiluminador deluz ultravioleta, se puede observar la migración de las moléculas de ADN demenor peso molecular hacia el ánodo y las de mayor peso molecular se mantienenmás cerca al cátodo. Las bandas mas gruesas equivalen a mayor cantidad deproteína y las más delgadas equivalen a menor cantidad de proteína.
CONCLUSIONES
En la práctica de electroforesis de ADN no se pudo realizar en el laboratorio yaque no se conto con los elementos suficientes para elaborarla.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 35/49
PRACTICA 4
INMOVILIZACION ENZIMATICA
Justificación
La utilización de la técnica de inmovilización enzimática es importante en elcampo de los alimentos porque proporcionan mejorar las condicionesorganolépticas de los mismos.
Objetivo
Describir las diferentes técnicas de inmovilización enzimática
Emplear la inmovilización enzimática en alginato de sodio poratrapamiento
Marco teórico
La inmovilización de enzimas es un proceso en el que se confina o localiza a laenzima en una región definida del espacio, para dar lugar a formas insolubles queretienen su actividad catalítica y que pueden ser reutilizadas repetidamente.Posteriormente esta definición se ha ampliado a aquel proceso por el cual serestringen, completa o parcialmente, los grados de libertad de movimiento deenzimas, orgánulos, células, etc. por su unión a un soporte.
En general, los métodos de inmovilización se suelen clasificar en dos grandescategorías:
1) Retención física
2) Unión química

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 36/49
Métodos de inmovilización de enzimas por retención física
Atrapamiento
El proceso de inmovilización se lleva a cabo mediante la suspensión de la enzimaen una solución del monómero. Seguidamente se inicia la polimerización por uncambio de temperatura o mediante la adición de un reactivo químico. Elatrapamiento, requiere poca cantidad de enzima para obtener derivados activos.Como ventaja adicional, la enzima no sufre ninguna alteración en su estructura.De todas formas, el atrapamiento requiere un control riguroso de las condicionesde polimerización, así como la comprobación de que la naturaleza química delproceso no altera los grupos reactivos de la proteína.
Unión químicaSe debe procurar que la inmovilización incremente la afinidad por el sustrato,disminuya la inhibición, amplié el intervalo de pH óptimo y reduzca las posiblescontaminaciones microbianas. Además el soporte debe tener resistencia mecánicaadecuada a las condiciones de operación del reactor y ser fácilmente separabledel medio líquido para que pueda ser reutilizado
Unión covalenteLa metodología de la unión covalente se basa en la activación de grupos químicosdel soporte para que reaccionen con nucleó filos de las proteínas De entre los 20aminoácidos diferentes que se encuentran en la estructura de las enzimas, losmás empleados para la formación de enlaces con el soporte son principalmente lalisina, la cisteína, la tirosina y la histidina, y en menor medida la metionina, eltriptófano, la arginina y el ácido aspártico y glutámico. El resto de aminoácidos,debido a su carácter hidrófobo, no se encuentran expuestos hacia el exterior de lasuperficie proteica, y no pueden intervenir en la unión covalente.
La inmovilización altera significativamente el comportamiento de las enzimas. Enprimer lugar se producen cambios en su estabilidad. En segundo lugar, la enzimainmovilizada es un sistema heterogéneo en el cual todos los componentes queintervienen en el proceso catalítico (pH, sustratos, productos, inhibidores,cofactores, activadores, etc.) se encuentran en interface: en el medio de reaccióny en la fase constituida por el soporte con la enzima.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 37/49
Efectos en la actividad enzimáticaTras una inmovilización, la actividad de la enzima puede disminuir e inclusoperderse por diversas razones. Si pierde totalmente la actividad enzimática puede
ser debido a que:1. la unión al soporte se produce de tal forma que el paso del sustrato al centroactivo está Impedido.2. los grupos reactivos del soporte reaccionan con algún aminoácido que formeparte del centro activo o que sea esencial para la actividad catalítica de la enzima.3. la inmovilización puede originar un cambio conformacional que da lugar a unaforma Inactiva.
Procedimiento
1. Preparar una solución de 50 Ml de alginato de sodio al 3% ( p/v ) enagua destilada, agitar lentamente hasta su disolución, la cualpresentara una viscosidad alta.
2. Disolver el alginato, adicionar y mezclar 3 ml de solución enzimáticainvertasa ( 0,025 % p/v en buffer de acetatos 0.1 M de pH 4,6
3. Preparar 250 ml de cloruro de calcio en agua destilada4. Repartir la solución de alginato- enzima en jeringas de 10 ml de
capacidad, gotear suavemente sobre la solución de cloruro de calcio.5. Con ayuda de un filtro de papel, separar y lavar las esferas formadas
con agua destilada.6. Pesar 2 gramos de esferas y colocarlas en 50 ml de una solución de
sacarosa al 10% p/v en agua destilada, incubar a 40°C por 30minutos. Antes de adicionar las esferas en la solución de sacarosa,tomar de esta, una alícuota de 2 ml como tiempo cero; al final de los30 minutos, tomar otra alícuota de 2 ml y marcar como tiempo 30.
7. A las muestras tomadas determinantes concentración de glucosa conayuda del kit enzimático glucosa GOD- PAD
8. Un cambio de color rosado establece la producción de glucosaproveniente de la sacarosa, lo cual implica actividad hidrolitica de lainvertasa inmovilizada.
7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 38/49
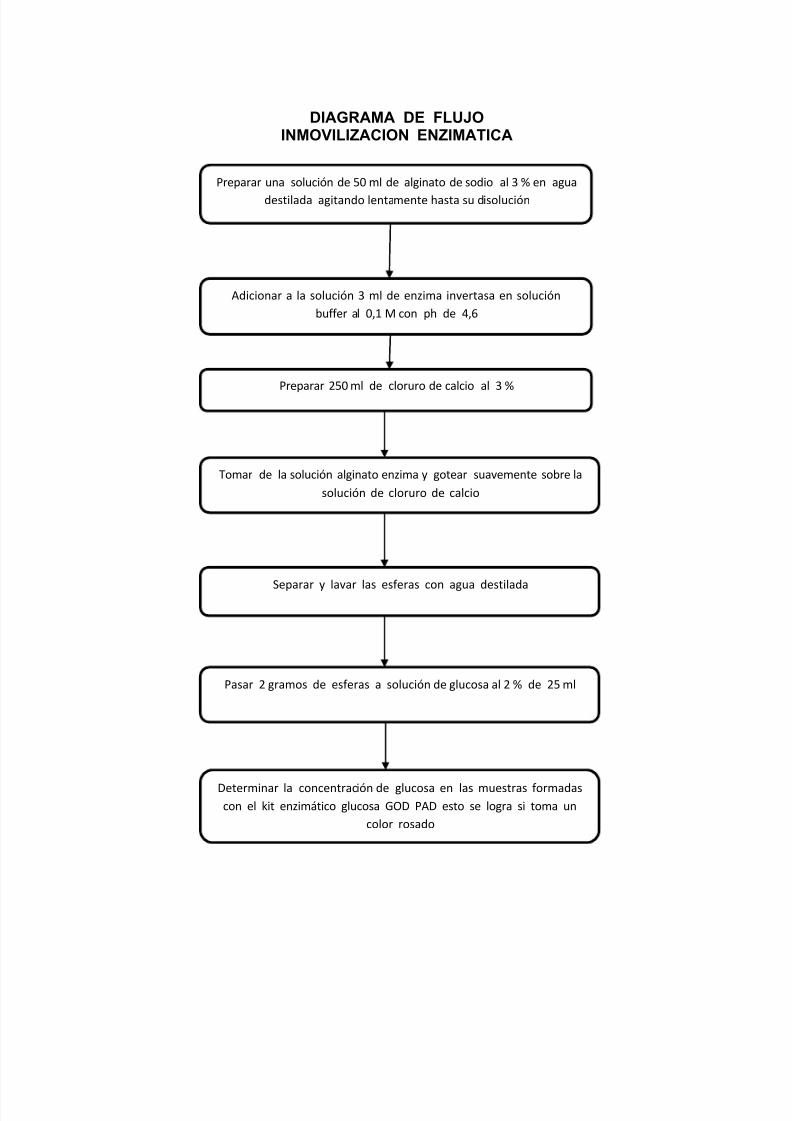
DIAGRAMA DE FLUJO
INMOVILIZACION ENZIMATICA
Preparar una solución de 50 ml de alginato de sodio al 3 % en agua
destilada agitando lentamente hasta su disolución
Adicionar a la solución 3 ml de enzima invertasa en solución
buffer al 0,1 M con ph de 4,6
Preparar 250 ml de cloruro de calcio al 3 %
Tomar de la solución alginato enzima y gotear suavemente sobre la
solución de cloruro de calcio
Separar y lavar las esferas con agua destilada
Pasar 2 gramos de esferas a solución de glucosa al 2 % de 25 ml
Determinar la concentración de glucosa en las muestras formadas
con el kit enzimático glucosa GOD PAD esto se logra si toma un
color rosado

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 39/49
RESULTADOS
ANALISIS DE RESULTADOS
- La oxidasa se inmovilizo aplicando la técnica de atrapamiento, usando unpolímero natural como lo es el alginato el cual se puede polimerizar atemperaturas bajas (25ºC). Debido a que estas condiciones son muysuaves el alginato es uno de los polímeros más usados.
Se usa el alginato porque atrapa la enzima en partículas moldeadas de unpolímero en un modelo. Usándose el alginato en medio como el cloruro decalcio.
Se realizo una emulsión de una solución acuosa de la enzima en buffer deacetatos 0.1 M de pH 4.6, el cual actuó como un disolvente orgánicoinmiscible en agua para formar microgotas acuosas. Al agregar la soluciónde alginato a la emulsión agitada, se forma una membrana sobre lasuperficie de las microgotas, las cuales se dispersaron en una fase acuosa.
CONCLUCION
La inmovilización enzimática esta destinada a la perdida de actividad , opara retener ciertas características de la enzima utilizada.Donde la invertasa entra en un sistema heterogéneo en el que suscomponentes intervienen en un proceso catalítico, es decir su ph, sustratos,
productos, inhibidores se encuentran en una interfase.Por lo que su actividad enzimática se afecta por efectos de tipo difusional esdecir su difusión de los sustratos hacia el centro activo de la enzima seimpide por resistencias de tipo externo e interno.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 40/49
CUESTIONARIO BIOTECNOLOGIA
1. Explique la metodología para la determinación de la actividad enzimática de lassiguientes proteínas:
1.1. Glucosa Isomerasa.
Se realiza una curva de calibración con diluciones de 1 * 10-4 a 1 * 10 _ 6M obtenidas de una solución madre de p- nitrofenol ( NO2C6H4OH ) a 0,1M, se mide la absorbancia a 400 nm en el espectrofotómetro. Para la extraerla enzima se cortan las vainas finamente del producto y se agrega fosfato desodio 0,1 M de ph 7 en relación peso volumen.
1.2. Proteasa.
Método d e Balls y Hoov er conocido como el método de coagulación de la leche,es, sin duda, uno de los métodos mas expeditos para determinar actividadenzimática de una proteinasa. Se toma como índice de actividad proteolítica, eltiempo necesario para que una cantidad conocida de enzima coagule undeterminado volumen de solución de leche. La actividad se expresa en términosde unidades de leche coagulada por g de preparado enzimático. La unidad deleche coagulada se la define coma la cantidad en peso de un preparadoenzimático necesario para coagular 5 ml de solución de leche estándar por minuto,
cuando la temperatura es de 40°C y el pH 6. La principal desventaja de estemétodo radica en el hecho de no medir la proteólisis total, es decir, la hidrólisis deun substrato de proteína, pero, por el contrario, mide la actividad de la coagulaciónde la leche. Afortunadamente, sin embargo, la coagulación de la leche parece seruna propiedad característica de los componentes proteolíticos de este grupo deenzimas y puede ser usada con seguridad en la medición proteolítica.
Según algunos autores, las preparaciones es necesario activarlas antes con obien con exceso de cianuro.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 41/49
DETERMINACIÓN DE PAPAÍNA
Fundamento :
El método se basa en medir el aminoácido tirosina que se libera por la acción de la
enzima sobre las proteínas, usando el reactivo de Folin.Reactivos:
Solución de cianuro de sodio 2,0 activador de la enzima Substrato hemoglobina: 20 mg/ml de hemoglobina se disuelven en
solución tampón de borato 0,1 M, de ph 7,5 Acido tricloroacetico 0,3 M Solución de NaOH 0,4 N Reactivo del fenol según Folin- ciocalteu
Prepar ación d e extrac to en zimático :
El producto pesado y molido se extrae con solución tampón de borato 0,1 M de pH7,5. Luego se centrifuga separándose el sobrenadante que se usará en ladeterminación.
Procedimiento :
En primer lugar, debe activarse la enzima, para lo cual se toma una alícuota delextracto enzimático problema y se le agrega 0,25 ml de cianuro sódico 2,0 M,manteniéndose la mezcla a 25°C por 3 minutos.
A 5 ml de solución de hemoglobina se le agrega 1 ml del extracto activado y semantiene a 35,5°C por 10 minutos; para agregar luego 10,0 ml de ácidotricloroacético 0,3 1\1 y centrifugar. Del sobrenadante se toman 5 ml, se agregan10,0 ml de NaOH 0,5 N y 3 ml del reactivo de Folin (dilución 1 + 2); se lee laabsorbancia a 750 nm.
1.3. Amilasa.
Método de la Ma lto sa : Se basa en suspender 10 g de harina en 46 ml de
solución tampón de pH 4,5-4,8 e incubar por 1 hora a 30°C. Esto permite que laenzima presente en la harina actúe sobre el almidón y lo convierte en maltosa, laque se determina por el método del ferricianuro. El valor de maltosa se definecomo los mg de maltosa producidos por 10 g de harina bajo las condiciones yaseñaladas.
Fundamento :

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 42/49
Los grupos reductores liberados del almidón se miden por su propiedad de reducirel ácido 3,5-dinitrosalicilico.
React ivos :
Enzima : Diluir una suspensión cristalina 1: 500 (o el extracto problema),determinar la concentración proteicamg/ml = x 0,81. Para trabajar, diluir 1 microgramo/ml.
Substrato : Solución al 1% de almidón en fosfato de sodio 0,02 M de pH 6,9 conNaCl 0,006 M (para alfa-amilasa) y solución al 1 % de almidón en acetato de sodio0,016 M, de pH 4,8 (para beta-amilasa).
React ivo d e color : 1 g de ácido dinitrosalicílico se disuelve en 20 ml de NaOH2Ny 50 ml de agua, se agregan 30 g de sal de Seignette (tartrato de Na y K) y sediluye a 100 ml, protegiéndola del .
Procedimiento :
0,5 ml de solución de la enzima o del extracto problema se mezclan con 0,5 ml desubstrato. Se incluye un blanco con 0,5 ml de agua en remplazó de la enzima oextracto problema. Se incuba a 25°C por 3 minutos, sé agrega 1 ml de reactivode color, se calienta en agua hirviendo por 5 minutos y luego se enfría. Seagregan 10 ml de agua y se lee a 540 nm (filtro verde) contra el blanco.
Se establece una curva con maltosa (0,3-3,0 m moles) para convertir las lecturascolorimétricas a unidades de actividad enzimática, o sea, que libera un micromol
de azúcar reductor, calculado como maltosa por minuto y a 25°C bajo lascondiciones del ensayo.
La maltosa puede también determinarse por el método de Munson y Walker,usando la reacción de Fehling I, 11, o bien por titulación yodométrica de Cu no,reducido a
2. Una de los métodos más utilizados para el rompimiento celular es la Zonicación,describa y explique el fundamento físico de esta técnica.
Consiste en la aplicación de ultrasonidos a una suspensión celular. La intensaagitación producida destruye las membranas celulares. Dependiendo de lafrecuencia, intensidad y energía aplicada se pueden destruir asimismo lasestructuras subcelulares e incluso solubilizar complejos proteicos. Se suele aplicaren frio para evitar el sobrecalentamiento de las muestras que podría provocar ladesnaturalización de las proteínas. Se transmite una corriente eléctrica a unsistema mecánico que la convertirá en vibraciones de alta intensidad generándose

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 43/49
ondas de ultrasonido que generan millones de burbujas microscópicas, las cualesse expanden y colapsan contra las células causando la ruptura de su membrana.El fundamento físico de esta técnica se llama CAVITACION es el fenómenopor el cual la presión total lugar de la más baja presión en todo el sistemaalcanza la presión de vapor del líquido bombeado. El agua hervirá y se
formarán burbujas de vapor. Luego el agua y las burbujas son impulsadas haciaafuera por el impulsor a la parte de mayor presión de la bomba donde lasburbujas colapsan e implotan originando desprendimiento de materiales.
3. Determine la sensibilidad de dos métodos de determinación de proteínas, juntocon las tinciones de comassie y nitrato de plata.
Azul comassie sensibilidad hasta 50 ng
Fluorescencia sensibilidad hasta 50 ng
Coloracion de plata sensibilidad hasta 1 o 2 ng
Tinción argentica de geles de poliacrilamida SDS
La tinción con plata fue introducida por Kerényi y Gallyas como un procedimientoextremadamente sensible para detectar pequeñas cantidades de proteínas engeles La técnica se ha ampliado para el estudio de otras macromoléculas
biológicas que han sido separadas en una variedad de soportes. La tinción clásicacon coomassie brilliant blue, por lo usual puede detectar una banda de 50 ng deproteína, la tinción de plata aumenta la sensibilidad típicamente unas 50 veces.Muchas variables pueden influir en la intensidad del color y cada proteína tienesus propias características de tinción, las claves para el éxito en una tinciónargéntica son, material de vidrio limpio, reactivos puros y agua de alta pureza.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 44/49
4. Defina y explique y el fundamento de las siguientes cromatografías:4.1. Cromatografía preparativa
se usa para purificar suficiente cantidad de sustancia para un uso posterior, más
que para análisis.
La cromatografía preparativa comprende un amplio campo de aplicaciones, desdeel aislamiento de 1 µg. de muestra para identificación espectroscópica hasta elaislamiento de un compuesto puro de una mezcla de 100 g. La cromatografíapreparativa se diferencia de la técnica básica en muchos aspectos, desde lapreparación de la papilla. Ésta debe hacerse con menos agua que de ordinario.Una papilla más espesa ayuda a estabilizar el grosor de las placas que se precisapara una carga de mayor magnitud. El espesor óptimo oscila desde un mínimo de1 mm. hasta un máximo de 2 mm.
Las placas utilizadas en cromatografía preparativa son placas grandes, deaproximadamente 20x20 cm. que permiten separar aproximadamente 1 g. demezcla utilizando unos 35 g. de adsorbente.
4.2. Cromatografía exclusión por tamañoLa cromatografía de exclusión por tamaños ( SEC ), a la que a menudo nosreferimos como cromatografía de permeacion de gel ( GPC ), es una técnicade caracterización de polímeros que proporciona la distribución completa depesos moleculares de una muestra y sus distintos promedios: es una técnicarelativa, no suministra valores absolutos, si no que requiere un calibrado con
fracciones mono dispersas del mismo polímero. También es posible utilizaren el calibrado un polímero distinto del caracterizado, pero en este caso, elcalibrado se basa en la comparación de volúmenes hidrodinámicos y se llamacalibrado universal.
4.3. Cromatografía de intercambio iónicoLa cromatografía de intercambio iónico (o cromatografía iónica) es unproceso que permite la separación de iones y moléculas polares basado en laspropiedades de carga de las moléculas. Puede ser usada en casi cualquier tipo demolécula cargada, incluyendo grandes proteínas,
pequeños nucleótidos y aminoácidos.La cromatografía de intercambio iónico conserva los analitos basándose en lasinteracciones de Coulomb. La fase estacionaria muestra en la superficie gruposfuncionales iónicos que interactúan con iones de carga opuesta del analito. Estetipo de cromatografía se subdivide a su vez en la cromatografía de intercambiocatiónico y cromatografía de intercambio aniónico:

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 45/49
La cromatografía de intercambio catiónico retiene cationes cargadospositivamente debido a que la fase estacionaria muestra un grupo funcionalcargado negativamente, como un acido fosfórico
La cromatografía de intercambio de aniones retiene aniones usando gruposfuncionales cargados positivamente, como un catión de amonio cuaternario.
R-A-H+ + M+ + B- <--> R-A-M+ + H+ + B- La cromatografía de intercambio iónico separa las proteínas de acuerdo a sucarga neta, la cual depende la composición de la fase móvil. Ajustando el ph o laconcentración de iones de la fase móvil, varias moléculas de proteína pueden serseparadas
4.4. Cromatografía de intercambio catiónico
La cromatografía de intercambio catiónico retiene cationes cargados positivamentedebido a que la fase estacionaria muestra un grupo funcional cargado
negativamente, como un acido fosfórico
4.5. Cromatografía de fase reversa
La cromatografía en fase reversa (RPC) permite separar moléculas en base a supolaridad. El principio de la cromatografía en fase reversa es semejante al de lacromatografía en capa fina. Sin embargo, aquí la fase estacionaria es departículas de sílice químicamente modificadas con hidrocarburos saturados,insaturados o aromáticos de diferentes tipos. Esto convierte a la fase estacionariaen una matriz apolar. Por lo tanto, para este tipo de cromatografías se empleanmezclas de solventes polares, tales como agua, acetonitrilo, acetato de etilo,acetona y alcoholes alifáticos.
5. Como se calcula el factor de purificación de una enzima y el rendimiento deesta, cual es su significado practico.

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 46/49
El factor de purificación de una enzima se calcula de la siguiente forma: se cuenta con la actividad especifica de la enzima inicial que se mide
por mg proteína total siendo igual a U= mg. Así mismo se debe contar con la actividad especifica final de la misma
enzima.
Para obtener el factor de purificación debemos dividir la actividadespecifica final entre la inicial el valor que nos da es el factor.
6. Cuales son los métodos de concentración de proteínas escala laboratorio masutilizados y cual es su fundamento físico.
No existe un sistema totalmente satisfactorio para determinar la concentraciónproteica de una muestra. La elección de un método u otro se basa en la naturalezade la proteína y del resto de componentes de la muestra y en la sensibilidad yprecisión requeridos.
Existen diversos métodos que permiten la cuantificación de proteínas, entrelos métodos mas usados tenemos:
METODO DESCRIPCION
Folin fenol o de Lowry Basado en la formación de un complejocoloreado entre el Cu ++ y los nitrógenos

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 47/49
de los enlaces peptídicos reacción de Biuret,Para resaltar el color formado en esta reacción yaumentar la sensibilidad, se hace reaccionar
posteriormente con el reactivo de Folin que al ser
reducido por los residuos de aminoácidosaromáticos tirosina y triptofano da un colorazulado.
Ventajas
Color estable Alta precisión y exactitud
Aplicaciones
Es el método mas usado Sirve de método estándar Excelente para proteínas de membrana
Coomassie o de Bradford Basado en la unión directa del colorante azul deCoomasie a la estructura terciaria de lasproteínas y aminoácidos específicos. El colorantecambia de color de marrón a azul al unirse aproteínas.
Ventajas
Bajo limite de detección Compatible con agentes reductores Rapidez
Aplicaciones
Se emplea cuando se necesita rapidez Para muestras con agentes reductores
Método espectrofotométrico Se basa en la propiedad de las proteínas detener un máximo de absorción a 280 nm debidoal contenido en los aminoácidos tirosina y

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 48/49
triptófano. La variabilidad en estos aminoácidosen diferentes proteínas obliga a añadir un factorde corrección aportado por la absorbancia delenlace peptídico a 205 nm. La concentración se
puede obtener aplicando la siguiente expresión:
Ventajas
Alta sensibilidad no hay perdida de la muestra
Aplicaciones
Para muestras muy escasas y valiosas
BIBLIOGRAFIA
Electroforesis de ácidos nucleídos en geles de agarosa. Aislamiento ycaracterización electroforética de DNA plasmídico.
Carmen Alicia Padilla Peña, Jesús Diez Dapena, Emilia Martínez Galisteo, José Antonio Bárcena Ruiz, Concepción García Alfonso
Departamento de Bioquímica y Biología Molecular, Campus Universitario deRabanales, Edificio Severo Ochoa, 14071-Córdoba.
Alan, S. (1999). BIOTECNOLOGIA PARA INGENIEROS. Mexico D.F.: LIMUSA.
Lehninger. (1985). BIOQUIMICA (Segunda ed.).

7/24/2019 Informe Final Laboratorio Biotecnologia_1
http://slidepdf.com/reader/full/informe-final-laboratorio-biotecnologia1 49/49
Mainero, F. X. (2000). LA INGENIERIA GENETICA Y LA NUEVABIOTECNOLOGIA (segunda ed.). Mexico, 2000: LA CIENDIA PARA TODOS.
Mckee, T. M. (2009). BIOQUIMICA LAS BASE MOLECULARES DE LA VIDA(Cuarta ed.). Mexico: Mc Graw Hill.
Perez, H. M. (2000). ELECTROFORESIS EN GELES DE POLOACRILAMINA;Fundamentos Actualidad e Importancia. Mexico D.F.: UNIV.
Renneberg, R. (2009). BIOTECNOLOGIA. España: REVERTÉ.
Scriban, R. (1985). BIOTECNOLOGIA. Paris: EL MANUAL MODERNO.
Duina Posso Duque, Thaura Ghneim Herrera. 2008. Uso de MarcadoresMicrosatélites para la Estimación de Diversidad Genética en Plantas. EdicionesIVIC. Protocolos de laboratorio UEG 2009.
Nelson, DL., Cox, MM. (2000) Lehninger Principles of Biochemistry, 3rd Ed., WorthPublishers, USA.
http://www.joseacortes.com/practicas/extraccionADN.htm
http://iie.fing.edu.uy/investigacion/grupos/gti/timag/trabajos/2006/electroforesis/
By Carolina Etchart & Marcelo Lavagna
http://www.ivic.ve/ecologia/ueg/formatos/Electroforesis%20de%20ADN%20en%20geles%20de%20agarosa.pdf
http://laguna.fmedic.unam.mx/~evazquez/0403/ecuacion%20de%20michaelis4.html.
http://www.mitecnologico.com/lb/Main/EcuacionDeLineweaverBurk
http://es.wikipedia.org/wiki/Cin%C3%A9tica_enzim%C3%A1tica
http://es.wikipedia.org/wiki/Cin%C3%A9tica_de_Michaelis-Menten