extração em meio aquoso e concentração por processos de ... · Á embrapa agroindústria de...
TRANSCRIPT

Extração em meio aquoso e concentração por processos
de membranas de fibras solúveis a partir do bagaço de uva
branca
Carla Patrícia da Silva Ferreira
Dissertação para obtenção do Grau de Mestre em
Engenharia Química
Júri
Presidente: Professor Doutor Francisco Manuel da Silva Lemos (UTL-IST)
Orientadores: Professora Doutora Maria Norberta Neves Correia de Pinho
(UTL-IST)
Doutora Lourdes Maria Correa Cabral (Embrapa Agroindústria de
Alimentos, Rio de Janeiro)
Vogais: Professor Doutor Luís Miguel Minhalma (IPL - ISEL)
Professora Doutora Suely Pereira Freitas (UFRJ – EQ)
Julho, 2013

ii

iii
Agradecimentos
Esta dissertação veio encerrar um trabalho desenvolvido ao longo de vários meses, divididos
entre as cidades do Rio de Janeiro e de Lisboa. Antes de adiantar mais a respeito desta dissertação,
não posso deixar de expressar o meu mais sincero agradecimento a todos aqueles que direta ou
indiretamente me ajudaram e apoiaram a ultrapassar diversos obstáculos com os quais me deparei,
bem como no desenvolvimento de todo este trabalho.
Às orientadoras Professora Doutora Maria Norberta Pinho e Doutora Lourdes Cabral pela orientação,
apoio, ajuda e conhecimentos transmitidos durante o desenvolvimento de todo o trabalho. Um
especial agradecimento à Doutora Lourdes Cabral pelo carinho com que me acolheu na cidade
do Rio de Janeiro e como sua orientada.
À Professora Doutora Suely Pereira Freitas, responsável pelo Laboratório de Processamento de
Matérias Primas Vegetais da Escola de Química da Universidade Federal do Rio de Janeiro
(UFRJ), por todo o apoio e acompanhamento no desenvolvimento da parte experimental do
trabalho, bem como na dedicação que prestou a este trabalho, com ideias, rigor e exigência,
mostrando-se sempre disponível em todos as fases da dissertação.
Á Doutora Caroline Mellinger Silva, investigadora do Laboratório de Bioquímica da Embrapa
Agroindústria de Alimentos, pela incansável dedicação e apoio, pelas ideias e opiniões
construtivas, pelo conhecimento transmitido, pela amizade, disponibilidade e incentivo que
sempre ofereceu.
Á Carol Viêgas, Regina Cássia Mattos e Leilson Oliveira pela ajuda, amizade e por proporcionarem
bons momentos de laboratório.
Á Embrapa Agroindústria de Alimentos, Rio de Janeiro, por receber “A portuguesa” na sua tão
calorosa e familiar empresa. Em especial aos amigos e funcionários: Renata Tonon, Ana Paula
Gil Cruz, Natalia Eitel, Mônica Pagani, Luiz Fernando (Chorão), Sérgio Macedo Pontes (Filé),
William, Flávia Gomes dos Santos, Tatiana Candea (Tati), Rozana Moreira, Diego Renan Paim,
Rodrigo Gouvêa, Marcelly Santos e Joice Barbosa.
Ao Departamento de Bioquímica e Biologia Molecular da Universidade Federal do Paraná (UFPR),
em especial aos Professores Doutor Marcello Iacomini e Doutora Fernanda Simas Tosin pelas
análises de composição monossacarídica e experimentos de HPSEC e RMN.
Á Vinícola Aurora por gentilmente ceder a matéria-prima necessária à realização do trabalho
experimental.

iv
Á Beu por toda a amizade. Pelo admirável apoio, carinho e companheirismo.
Á Tia Sónia, por me ter recebido na sua casa de forma tão calorosa e me ter abraçado como família.
Aos “Césares” Mafalda Rodrigues, Joana Cabrita, Catarina da Cunha, Tiago Leiria de Brito e Marcelo
Oliveira; ao João Pedro Duarte e Inês Sarmento de Matos pela ajuda, amizade e apoio nos
meses de trabalho no Rio de Janeiro.
A todos os meus amigos e colegas, em especial à Leonor, João, André, Andreia, Carolina, Pipo,
Francisco, Noki, Caba, Jorge Neto, Lita, Pedro, Pedro da Cruz Gonçalves, Serrão, Raquel e
Sérgio Rodrigues (Máquina), pela amizade e apoio, e por estarem presentes nos bons e nos
maus momentos, contribuindo para o meu crescimento enquanto profissional e enquanto
pessoa.
A toda a minha família pelo apoio, carinho e preocupação. Em especial aos tios Zé e Dalva e aos
primos Adriana e Volker, que nesta fase mostraram um apoio e dedicação incondicionais.
Por último, um especial agradecimento aos meus pais, Fernanda e Domingos e ao meu irmão
Ricardo, por terem acreditado sempre em mim, pelo apoio incondicional, pela confiança, pela
compreensão e pelas oportunidades que me proporcionaram, incluindo a minha estadia no Rio
de Janeiro. Sem eles não seria a pessoa que sou.

v
Resumo
Os objectivos deste trabalho foram: avaliar as condições de extração de fibras solúveis a partir
do bagaço da uva branca Chardonnay, concentrar o extrato obtido com água (H) por ultrafiltração
(UF) e microfiltração (MF) e caraterizar os extratos e as frações quanto à sua estrutura polisacarídica,
atividade antioxidante in vitro e compostos fenólicos. Os rendimentos máximos, foram 2,16±0,012
g/100g bagaço (2% KOH, a 90ºC, relação substrato/solvente (R) de 1:4 e bagaço in natura),
0,904±0,0453 g/100g bagaço (a 90ºC, R de 1:4 e bagaço in natura), e 0,724±0,0100 g/100g bagaço
(a 50ºC, R de 1:4, concentração de Celulase de 350µL/18g de bagaço seco e bagaço in natura)
respetivamente, para solução alcalina, água e solução aquosa de celulase. Os processos de UF e MF
foram conduzidos num sistema semi-piloto em escala laboratorial, de configuração tubular contendo 4
módulos de membranas cerâmicas de 20 kDa e 0,1 µm e pressão aplicada à membrana de 15 bar e
2,5 bar, respetivamente. Ambos os processos concentraram as fibras. Os extratos H, enzimático (E) e
alcalinos (K) são constituídos por ramnose, arabinose, xilose, manose, galatose e glucose nas
seguintes proporções molar, respetivamente: H (4,6:29,8:3,5:8,5:14,5:39,1); E
(6,3:22,1:4,0:7,6:8,6:51,4); K (8,7:30,9:7,6:9,8:19,7:20,2). As frações obtidas por UF, concentrado (C)
e permeado (P) têm uma composição monossacarídica em ramnose, arabinose, xilose, manose,
galactose e glucose com a seguinte proporção molar, respetivamente: C
(12,8:29,1:3,4:14,9:24,3:15,5) e P (2,5:9,5:2,2:7,4:8,0:70,4). O extrato K e a fração C apresentaram
uma atividade antioxidante in vitro de 8423,9±221,73 mol Trolox/100 g bagaço e de 312,43±6,078
mol Trolox/100 g bagaço e um teor de compostos fenólicos de 1616,5±53,01 mg ácido gálico/100g
bagaço e 135,05±5,970 mg ácido gálico/100g bagaço, respetivamente.
Palavras-chave: Bagaço de uva, Extração alcalina, Extração enzimática, Extração aquosa, Ultrafiltração, Fibras
solúveis, Polissacarídeos.

vi

vii
Abstract
The aim of this work was to study the conditions of extraction of soluble fibres from white grape
pomace Chardonnay, concentrate the extract obtained with water (H) by ultrafiltration (UF) and
microfiltration (MF) and characterize the extracts and fractions for their polysaccharide structure, in
vitro antioxidant activity and content of phenolics. It was obtained a maximum mass of sugar in 100 g
of grape pomace of 0,904±0,0453 (90°C, ratio substrate/solvent (R) of 1:4 and the grape pomace was
in its natural size), 0,724±0,0100 (50°C, R of 1:4, concentration of cellulase 350μL/18g of dry grape
pomace and grape pomace in natura) and 2,16±0,012 (90°C, R of 1:4 and grape pomace in natura) by
the aqueous, enzymatic and alkali 2% extractions, respectively. The processes of UF and MF were
conducted in a semi-pilot scale laboratory setup of 4 tubular ceramic membranes modules of 20 kDa
and 0,1 µm, and pressure applied to the membrane 15 bar and 2,5 bar, respectively. Both processes
concentrated the soluble fibers. The aqueous (H), enzymatic (E) and alkali (K) extracts are composed
of rhamnose, arabinose, xylose, mannose, galactose and glucose in the following molar proportions,
respectively: H (4,6:29,8:3,5:8,5:14,5:39,1), E (6,3:22,1:4,0:7,6:8,6:51,4), K
(8,7:30,9:7,6:9,8:19,7:20,2). The fractions obtained by UF, concentrate (C) and permeate (P),
presented a monosaccharide composition of rhamnose, arabinose, xylose, mannose, galactose and
glucose in the following molar proportions, respectively: C (12,8:29,1:3,4:14,9:24,3:15,5) and P
(2,5:9,5:2,2:7,4:8,0:70,4). The extract K and fraction C showed an in vitro antioxidant activity of
8423,9±221,73 mol Trolox/100 g of grape pomace and 312,43±6,078 mol Trolox/100 g of grape
pomace and a content of phenolic compounds of 1616,5± 53,01 mg galic acid/100g of grape pomace
and 135,05±5,970 mg galic acid/100g of grape pomace, respectively.
Keywords: Wine grape pomace, Alkali Extraction, Enzymatic Extraction, Aqueous Extraction,
Ultrafiltration, Soluble Fibers, Polysaccharides

viii

ix
Nomenclatura
A –Área da membrana
CA – Concentração de açúcar em solução
Cb – Concentração no seio da alimentação
CE – Concentração de Celulase
CK – Concentração de KOH
Cm – Concentração máxima na superfície da membrana
D – Coeficiente efetivo de difusão ou difusividade efetiva
f – Fator de atrito
L – Espessura da camada de transferência
J, Jp – Fluxo; Fluxo de permeação
k – Coeficiente de transferência de massa
kB – Constante de Boltzmann
m0 – Massa de extrato da alimentação
mp – Massa do permeado medida no tempo de processo
R – Relação substrato/solvente
t – Tempo de processo
T – Temperatura
tt – Tempo de trituração do bagaço
δ – Espessura do filme junto à membrana
µ – Potencial químico

x
Lista de Abreviaturas
13C – Carbono 13
1H – Protão
AA – Atividade Antioxidante
ABTS•+
– 2,29-azinobis-(3-ethylbenzothiazoline-6-sulfonicacid)
AOAC – Association of Official Analytical Chemists
CuSO4 – Sulfato de Cobre
DGF - Deutsche Gesellschaft fur Fettewissenschaft (Sociedade Alemã de Ciência de Gorduras)
DAT – Dosagem de açúcares totais
FCV – Fator de concentração volumétrico
FT – Fenólicos Totais
GC-MC – Cromatografia líquido-gasosa acoplada à espectrometria de massa
HPSEC – High pressure size exclusion chromatography (Cromatografia de exclusão estérica)
HSQC – Heteronuclear Single Quantum Coherence (Ressonância Magnética Nuclear
multidimensional)
IR – Índice de Refração
KOH – Hidróxido de Potássio
MWCO – Molecular Weight Cut-Off (Limite de exclusão molecular da membrana)
NaBH4 – Boroidreto de Sódio
NaN3 – Azida de sódio
NaNO2 – Nitrito de sódio
p.c. – Ponto central do planeamento fatorial composto central
RMN – Ressonância Magnética Nuclear
TFA – Ácido trifluoroacético
UFRJ – Universidade Federal do Rio de Janeiro
UFPR – Universidade Federal do Paraná

xi
Índice
1. Introdução ......................................................................................................................... 1
1.1. Resíduos industriais e Bagaço da uva ............................................................................. 1
1.2. Processos de extração com solvente e separação por membranas ............................... 2
1.3. Estrutura da Dissertação .................................................................................................. 2
2. Revisão da Literatura e Enquadramento ......................................................................... 5
2.1. Uva e Vinho ...................................................................................................................... 5
2.2. Composição do Bagaço da Uva ....................................................................................... 7
2.2.1. Compostos Fenólicos ....................................................................................................... 7
2.2.2. Fibras Alimentares ........................................................................................................... 9
2.3. Transferência de massa ................................................................................................. 11
2.3.1. Processos de Extração Sólido-Líquido.................................................................. 12
2.3.2. Processos de Separação por Membranas ............................................................ 14
2.4. Objetivo da Dissertação ................................................................................................. 19
3. Parte Experimental ......................................................................................................... 21
3.1. Matéria-Prima ................................................................................................................. 21
3.2. Equipamentos................................................................................................................. 22
3.3. Procedimento Experimental ........................................................................................... 24
3.3.1. Extrações sólido-líquido de fibras solúveis ........................................................... 24
3.3.2. Microfiltração e Ultrafiltração ................................................................................. 25
3.3.3. Métodos Químicos ................................................................................................. 26
3.3.4. Métodos Espectrofotométricos .............................................................................. 28
3.3.5. Métodos Cromatográficos ..................................................................................... 29
3.3.6. Análises Espectroscópicas .................................................................................... 30
3.3.7. Métodos Estatísticos.............................................................................................. 31
4. Resultados e Discussão ................................................................................................. 33
4.1. Extração sólido-líquido ................................................................................................... 33
4.1.1. Determinação da condição ótima de extração ...................................................... 34
4.1.2. Comparação do processo para os três solventes estudados ............................... 43
4.2. Processos de Membranas .............................................................................................. 47
4.2.1. Microfiltração e Ultrafiltração ................................................................................. 47

xii
4.3. Análises de identificação químico-molecular e atividade antioxidante .......................... 51
4.3.1. Análise estrutural de Polissacarídeos ................................................................... 52
4.3.2. Atividade Antioxidante ........................................................................................... 59
4.3.3. Fenólicos Totais ..................................................................................................... 60
5. Conclusões ..................................................................................................................... 63
6. Perspetivas de trabalho futuro ....................................................................................... 65
7. Referências Bibliográficas .............................................................................................. 67
8. Anexos ............................................................................................................................ 75
8.A – Bagaço de uva branca sujeito aos 3 tempos de trituração estudados .......................... 75
8.B – Resultados da determinação da espessura do bagaço de uva branca ......................... 75
8.C – Curvas padrão e absorvâncias lidas para os ensaios de extração com e sem efeitos
cinéticos .......................................................................................................................... 75
8.D - Gráficos de superfície de resposta para determinação da melhor condição de extração
........................................................................................................................................ 83
8.E – Ajustes à 2ª Lei de Fick .................................................................................................. 91
8.F – Curvas padrão e absorvâncias lidas para os ensaios de Microfiltração e Ultrafiltração 93
8.G – Determinação da Atividade Antioxidante (AA) .............................................................. 95
8.H – Determinação dos Fenólicos Totais (FT) ....................................................................... 96

xiii
Lista de Figuras
Figura 1 – Representação esquemática de um bago de uva (Fonte: Petichateau) ..................... 5
Figura 2 – Esquema simplificado do processamento de vinho (Fonte: Adaptado de
Petitchateau) ............................................................................................................... 6
Figura 3 – Exemplo de compostos fenólicos flavonóides: Catequinas (A) e antocianinas (B)
(Fonte: Silva et al., 2010) ............................................................................................ 8
Figura 4 – Exemplo de compostos fenólicos não flavonóides: Moléculas dos ácidos
hidrocinâmico (A) e do hidroxibenzóico (B) (Fonte: Silva et al., 2010) ....................... 8
Figura 5 – Estruturas básicas de xilanas e xiloglucanas: Glucuronoarabinoxilana (A) e
Xiloglucana (B) (Fonte: Caffal e Mohnen., 2009) ...................................................... 10
Figura 6 – Representação esquemática do Extrator Rotocel (Fonte: Schweitzer, 1982) ........... 13
Figura 7 – Esquema geral de um processo de separação com membranas ............................. 14
Figura 8 – Representação esquemática dos fluxos de filtração: Filtração frontal (A); Filtração
tangencial (B) (Fonte: Adaptado de Alves, 2010) ..................................................... 15
Figura 9 – Esquema representativo do fenómeno de polarização da concentração (Fonte:
Adaptado de Pinho e Prazeres, 2008) ...................................................................... 15
Figura 10 – Representação esquemática dos intervalos de tamanhos de partículas removidas
por processos de filtração em unidades de micrómetros, Angstrom e de massa
molecular aproximada, Da (de cima para baixo na imagem) (Fonte: Adaptado do
portal de Engenharia Química) ................................................................................. 16
Figura 11 - Representação esquemática do módulo de membranas do tipo planar (Fonte:
Resende, 2011) ......................................................................................................... 17
Figura 12 – Representação esquemática do módulo de membranas do tipo tubular (Fonte:
Resende, 2011) ......................................................................................................... 17
Figura 13 – Representação esquemática do módulo de membranas do tipo fibras ocas (Fonte:
Resende, 2011) ......................................................................................................... 18
Figura 14 – Representação esquemática do módulo de membranas do tipo espiral (Fonte:
Resende, 2011) ......................................................................................................... 18
Figura 15 – Componentes constituintes do bagaço da uva branca em estudo (m/m) ............... 22
Figura 16 – Fotografia do Banho Termostatizado ...................................................................... 22
Figura 17 – Representação esquemática de módulo de membranas ........................................ 23
Figura 18 – Membrana de 20kDa ............................................................................................... 23
Figura 19 – Fotografia do módulo de membranas (ultra e microfiltração) .................................. 24
Figura 20 – Gráfico de pareto obtido através dos resultados dos extratos obtidos com água
pura ........................................................................................................................... 35
Figura 21 – Gráfico de pareto obtido através dos resultados dos extratos enzimáticos ............ 37
Figura 22 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 2% ....... 39
Figura 23 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 6% ....... 39
Figura 24 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 10% ..... 39

xiv
Figura 25 – Cinética em meio Aquoso ........................................................................................ 41
Figura 26 – Cinética em meio aquoso enzimático ...................................................................... 42
Figura 27 – Cinética em meio alcalino a 2% ............................................................................... 42
Figura 28 – Fotografia comparativa das soluções aquosa (A) e alcalina (B) antes da extração 43
Figura 29 – Figura comparativa dos extratos aquoso (A) e alcalino (B) ..................................... 43
Figura 30 – Figura comparativa dos resíduos das extrações aquosa (A) e alcalina (B) ............ 44
Figura 31 – Comparação dos valores máximos obtidos para a massa de açúcar extraída por
cada solvente estudado ............................................................................................ 45
Figura 32 – Representação gráfica comparativa da massa de açúcar extraída sem efeitos
cinéticos pelos solventes água e solução aquosa de Celulase ................................ 45
Figura 33 – Representação gráfica comparativa das cinéticas dos extratos para os 3 solventes
em estudo .................................................................................................................. 46
Figura 34 – Fluxo permeado de água em função da diferença de pressão aplicada a membrana
de microfiltração ........................................................................................................ 48
Figura 35 – Fluxo permeado de água em função da diferença de pressão aplicada à membrana
de ultrafiltração .......................................................................................................... 48
Figura 36 – Variação do fluxo de permeação do extrato obtido com água (Jp) em função do
factor de concentração volumétrico (FCV)................................................................ 49
Figura 37 – Fotografia comparativa das soluções de Alimentação (A), Concentrado (C) e
Permeado (P) do processo de Ultrafiltração ............................................................. 50
Figura 38 – Início da precipitação etanólica do permeado (P) e concentrado (C) obtidos por
ultrafiltração do extrato aquoso ................................................................................. 51
Figura 39 – Esquema sumário dos processos de extração e de tecnologia de membranas
realizados e amostras obtidas .................................................................................. 52
Figura 40 – HPSEC das amostras de extrato aquoso (A) e extrato enzimático (B) ................... 54
Figura 41 – Espectro de 13
C-RMN da amostra de extrato aquoso obtida nas melhores
condições estudadas (Realizado em D2O a 50ºC. Valores expressos em δ (ppm)) 55
Figura 42 – HPSEC da amostra de extrato alcalino (K) (Inserido: HPSEC de H) ...................... 56
Figura 43 – HPSEC da amostra do extrato bruto H (A) e da fração de concentrado, C, por
ultrafiltração do extrato aquoso (B) ........................................................................... 57
Figura 44 – Espectro de HSQC da fração de concentrado, C de ultrafiltração (Realizado em
D2O, 50ºC. Valores expressos em δ (ppm)) ............................................................. 58
Figura 45 - Figura comparativa do bagaço triturado durante os 3 tempos estudados ............... 75
Figura 46 – Curva Padrão de D-Glucose 1 ................................................................................. 81
Figura 47 – Curva Padrão de D-Glucose 2 ................................................................................. 81
Figura 48 – Curva Padrão de D-Glucose 3 ................................................................................. 81
Figura 49 – Curva Padrão de D-Glucose 4 ................................................................................. 82
Figura 50 – Curva Padrão de D-Glucose 5 ................................................................................. 82
Figura 51 – Curva Padrão de D-Glucose 6 ................................................................................. 82
Figura 52 – Curva Padrão de D-Glucose 7 ................................................................................. 83

xv
Figura 53 – Curva Padrão de D-Glucose 8 ................................................................................. 83
Figura 54 – Curva Padrão de D-Glucose 9 ................................................................................. 83
Figura 55 – Superfície de resposta para solvente água (Massa de açúcar f(R/T)) .................... 84
Figura 56 - Superfície de resposta para solvente água (Massa de açúcar f(tt/R)) ..................... 84
Figura 57 - Superfície de resposta para solvente água (Massa de açúcar f(T/tt)) ..................... 85
Figura 58 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(R/CE))
................................................................................................................................... 85
Figura 59 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(tt/R))
................................................................................................................................... 86
Figura 60 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(CE/tt))
................................................................................................................................... 86
Figura 61 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(R/T))
................................................................................................................................... 87
Figura 62 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(tt/R))
................................................................................................................................... 87
Figura 63 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(T/tt))
................................................................................................................................... 88
Figura 64 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(R/T))
................................................................................................................................... 88
Figura 65 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(tt/R))
................................................................................................................................... 89
Figura 66 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(T/R))
................................................................................................................................... 89
Figura 67 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(R/T))
................................................................................................................................... 90
Figura 68 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(tt/R))
................................................................................................................................... 90
Figura 69 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(T/tt))
................................................................................................................................... 91
Figura 70 – Ajuste à 2ª Lei de Fick para a cinética da extração aquosa (R = 0,972) ................. 92
Figura 71 – Ajuste à 2ª Lei de Fick para a cinética da extração enzimática (R = 0,968) ........... 92
Figura 72 – Ajuste à 2ª Lei de Fick para a cinética da extração alcalina a 2% (R = 0,922) ....... 93
Figura 73 – Curva Padrão de D-Glucose 7 ................................................................................. 94
Figura 74 – Curva Padrão de D-Glucose 10 ............................................................................... 95
Figura 75 – Curva padrão de Trolox ........................................................................................... 95
Figura 76 – Curva padrão de Ácido Gálico ................................................................................. 96

xvi

xvii
Lista de Tabelas
Tabela 1 – Comparação de diferentes configurações modulares (Fonte: Alves, 2010) ............. 18
Tabela 2 – Composição físico-química do bagaço da uva branca Vitis Vinifera Chardonnay ... 21
Tabela 3 – Variáveis independentes e seus níveis ..................................................................... 31
Tabela 4 – Planeamento fatorial composto central ..................................................................... 31
Tabela 5 – Densidades e massas específicas das amostras ..................................................... 34
Tabela 6 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Extrato aquoso .... 34
Tabela 7 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Extrato enzimático 36
Tabela 8 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Solução aquosa de
KOH ............................................................................................................................. 38
Tabela 9 – Coeficiente de difusão para cada solvente ............................................................... 43
Tabela 10 – Concentração de açúcar nas soluções de alimentação, concentrado e permeado,
dos processos de membranas de Microfiltração e Ultrafiltração ................................. 50
Tabela 11 – Composição monossacarídica dos extratos alcalino, enzimático e aquoso ........... 53
Tabela 12 – Composição monossacarídica dos permeados, concentrados e alimentações do
sistema de membranas de ultrafiltração (20 kDa) ....................................................... 56
Tabela 13 – Sinais referentes à correlação C1/H1 do espectro de HSQC de C ........................ 58
Tabela 14 – Atividade Antioxidante das amostras de extrato obtidas para os três solventes e
nas melhores condições selecionadas e das frações de concentrado e permeado do
processo de ultrafiltração ............................................................................................. 60
Tabela 15 – Composição de fenólicos totais (FT) na amostra de extrato alcalino obtido na
melhor condição selecionada e das frações de concentrado e permeado do processo
de ultrafiltração ............................................................................................................ 60
Tabela 16 – Espessuras medidas do bagaço da uva branca ..................................................... 75
Tabela 17 – Absorvâncias medidas nos extratos aquosos obtidos sem efeitos cinéticos ......... 76
Tabela 18 – Absorvâncias medidas nos extratos aquosos obtidos com efeitos cinéticos ......... 76
Tabela 19 – Absorvâncias medidas nos extratos enzimáticos obtidos sem efeitos cinéticos .... 77
Tabela 20 – Absorvâncias medidas nos extratos enzimáticos obtidos com efeitos cinéticos .... 77
Tabela 21 – Absorvâncias medidas nos extratos alcalinos a 2% obtidos sem efeitos cinéticos 78
Tabela 22 – Absorvâncias medidas nos extratos alcalinos a 2% obtidos com efeitos cinéticos 79
Tabela 23 – Absorvâncias medidas nos extratos alcalinos a 6% obtidos sem efeitos cinéticos 79
Tabela 24 – Absorvâncias medidas nos extratos alcalinos a 10% obtidos sem efeitos cinéticos
..................................................................................................................................... 80
Tabela 25 – Concentração de açúcar adimensionalizada dos extratos aquosos para estudo da
cinética ......................................................................................................................... 91
Tabela 26 – Concentração de açúcar adimensionalizada dos extratos enzimáticos para estudo
da cinética .................................................................................................................... 92
Tabela 27 – Concentração de açúcar adimensionalizada dos extratos alcalinos a 2% para
estudo da cinética ........................................................................................................ 93

xviii
Tabela 28 – Absorvâncias medidas nas frações do processo de Microfiltração ........................ 94
Tabela 29 – Absorvâncias medidas nas frações do processo de Ultrafiltração ......................... 94
Tabela 30 – Absorvâncias medidas para determinação da Atividade Antioxidante ................... 96
Tabela 31 – Absorvâncias medidas para determinação dos Fenólicos Totais .......................... 97

1
1. Introdução
1.1. Resíduos industriais e Bagaço da uva
Nas últimas décadas, questões económicas, de saúde pública e ambientais têm vindo a
aumentar a preocupação com o volume elevado de resíduos gerados na indústria de alimentos
(Arvanitoyannis et al., 2006; González-Centeno et al., 2010). Um dos principais objetivos neste tema
atual é o de reclassificar esses resíduos, em subprodutos, com o intuito de os aproveitar e com vista
a diminuir os impactos ambientais e económicos a eles associados. Neste sentido, conceitos como
gestão de resíduos da cadeia produtiva são uma realidade dos dias de hoje aplicados em prole da
redução, eliminação ou aproveitamento dos resíduos industriais. Estes conceitos empregam uma
visão holística na qual toda a cadeia produtiva é avaliada com vista a uma diminuição da máxima
quantidade dos resíduos, de forma a garantir a rentabilidade e a sustentabilidade do processo e a
preservação ambiental. No que respeita a indústria alimentar, dada a elevada quantidade de resíduos
e subprodutos gerados o interesse é despertado tanto da parte da indústria como da área de
pesquisa científica para encontrar novas fontes de compostos bioativos que possam ser usados como
ingredientes funcionais no mesmo sector (González-Centeno et al., 2010).
Uma das frutas mais cultivadas no mundo é a uva. Segundo dados da FAOSTAT em 2011
foram produzidas cerca de 70 milhões de toneladas de uvas no mundo, sendo que 41% destas uvas
tiveram como destino a produção de vinho (cerca de 29 milhões de toneladas nesse ano). Ocupando
a 10ª posição na lista dos maiores produtores de vinho do mundo em 2008 (Ribeiro, 2011), Portugal é
um dos países onde a vitivinicultura atinge uma expressão económica considerável.
O bagaço da uva, constituído pelas peliculas, sementes e engaço, é um dos principais
subprodutos gerados na indústria de produção de vinho, cerca de 17,0 kg e 13,5 kg para 100 L de
vinho branco e vinho tinto produzidos, respetivamente (Silva, 2003). Este subproduto representa um
problema ambiental devido não só ao elevado volume produzido, bem como às características
poluentes que apresenta, como o baixo pH, elevados teores de compostos fenólicos, antibacterianos
e fitotóxicos, resistindo à degradação biológica (Bustamante et al., 2008). No entanto, a presença de
compostos bioativos naturais como fibras alimentares e compostos antioxidantes valorizam este
subproduto pela potencialidade de aplicações mais nobres, como na indústria farmacêutica,
cosmética, alimentar (Arvanitoyannis et al., 2006; González-Centeno et al., 2010) e de embalagens
(Siracusa et al., 2008; Deng e Zhao, 2011), bem como na utilização do bagaço como fertilizantes
biológicos (Arvanitoyannis et al., 2006; Bustamante et al., 2008). Dada a crescente tendência atual do
consumidor para a preocupação com as temáticas da saúde e do bem-estar, este subproduto torna-
se economicamente atrativo, sendo que a sua exploração e aproveitamento vai de encontro à
resolução do problema ambiental que o envolve.
Compondo cerca de 23,5±1,1 g de 100 g de bagaço fresco da uva Chardonnay proveniente da
vinificação (Gonzaléz-Centeno et al., 2010), as fibras alimentares, despertam interesse na
comunidade científica e industrial. Em virtude dos seus benefícios para a saúde e da sua estrutura

2
polissacarídica este material ganha importância junto do sector industrial (Arvanitoyannis et al., 2006;
Siracusa et al., 2008; Deng et al., 2011; Deng e Zhao, 2011).
A Embrapa Agroindústria de Alimentos, localizada na cidade do Rio de Janeiro, Brasil, nos
últimos anos tem conquistado uma sólida experiência no aproveitamento de resíduos e coprodutos
agroindustriais, tendo desenvolvido diversos projetos voltados para a extração de substâncias de
interesse comercial e produtos inovadores a partir de resíduos agroindustriais. Integrado num dos
projetos desta organização, e perante o que foi exposto acima, este trabalho debruça-se sobre o
estudo das fibras solúveis presentes no bagaço da uva proveniente da vinificação tinta, com o intuito
de as recuperar deste material com grande potencial tecnológico.
1.2. Processos de extração com solvente e separação por membranas
A extração sólido-liquido ou lixiviação é um processo com aplicação em variados processos
industriais, como na separação de metais do minério por extração ácida, na recuperação de açúcar
da beterraba através da água e na extração de óleos vegetais a partir de sementes oleaginosas, com
o auxílio de hexano (Farelo e Serrano). Este tipo de separação, muito utilizada na indústria alimentar,
é particularmente adequada para a separação de compostos de interesse do bagaço da uva, com a
adição de um solvente adequado à extração desse soluto. Variáveis como a afinidade soluto-
solvente, as condições de pressão e temperatura, tempo de extração, dimensões do substrato, e
quantidade de solvente adicionado, são determinantes para uma extração de qualidade. Sendo o
efluente deste processo uma solução de solvente contendo o soluto a recuperar, é importante e
necessário isolar este último, de forma obter-se um produto purificado.
A integração de tecnologia de membranas neste processo torna-se, portanto, vantajosa, dada a
sua fácil implementação, flexibilidade e baixo custo associado. Sendo considerados uma boa
alternativa aos processos de concentração clássicos (ex. evaporação a vácuo), estes processos são
adequados no fracionamento de misturas complexas, como o extrato do bagaço da uva, podendo
concentrar um determinado composto na corrente de concentrado e a obtenção de uma corrente de
permeado com o solvente. Dos inúmeros processos por tecnologia de membranas existentes, a
ultrafiltração apresenta-se como a operação indicada ao isolamento e/ou fracionamento de
macromoléculas como os polissacarídeos.
1.3. Estrutura da Dissertação
O bagaço da uva é um coproduto da produção de vinho com grande potencial no que respeita
a sua composição. É de extrema importância valorizá-lo de forma a reduzir o impacto que este tem no
ambiente, recuperando os compostos de interesse que dele fazem parte.
Neste trabalho pretende-se estudar a aplicação da extração sólido-liquido e do fracionamento
por processos de tecnologia de membranas ao bagaço da uva proveniente da vinificação, como
forma de recuperação de fibras solúveis.

3
Numa primeira fase do trabalho, estudam-se e selecionam-se as condições que originam
melhores rendimentos para a extração de fibras solúveis do bagaço da uva.
Na segunda parte, procede-se ao isolamento destas fibras solúveis do extrato obtido nas
condições selecionadas, a partir de processos de tecnologia de membranas de microfiltração e
ultrafiltração.
A última parte do trabalho consiste na identificação estrutural dos polissacarídeos dos extratos
e das frações obtidas por ultrafiltração, bem como na avaliação no que respeita a atividade
antioxidante in vitro e o teor de fenólicos.
Neste contexto, a presente dissertação encontra-se dividida em sete capítulos principais, com
os seguintes conteúdos:
Capítulo 1 – O presente, onde se pretendeu introduzir o tema em que se insere esta
dissertação.
Capítulo 2 – Revisão da literatura e enquadramento do tema da dissertação no estado atual
dos conhecimentos.
Capítulo 3 – Descrição das técnicas e metodologias aplicadas e dos equipamentos utilizados
para o cumprimento dos objetivos proposto.
Capítulo 4 – Resultados obtidos nas várias fases do trabalho, com respetiva discussão.
Capítulo 5 – Conclusões com base nos resultados obtidos e visando os objetivos inicialmente
propostos.
Capítulo 6 – Perspetivas para futuros trabalhos dentro da temática abordada.
Capítulo 7 – Referências bibliográficas utilizadas ao longo de todo o trabalho.

4

5
2. Revisão da Literatura e Enquadramento
2.1. Uva e Vinho
O berço da vitivinicultura no mundo localiza-se no sudeste de Anatólia, onde hoje é a Turquia
(Berkowitz,1996 e Adega, 2012) e remonta ao ano de 6000 a.c. (Berkowitz,1996). Em Portugal, a
longa tradição nessa área estende-se por todo o território nacional, fazendo parte da sua cultura e da
sua história. Apesar da sua pequena dimensão territorial, Portugal está entre os países que mais
produzem vinho no mundo, tendo em 2008 ocupado a décima posição a nível mundial e a 5ª posição
a nível europeu (Ribeiro, 2011). De acordo com a FAOSTAT, nesse mesmo ano, Portugal produziu
547962 toneladas de vinho, sendo que em 2011 foi verificado um aumento dessa produção,
registando-se um valor de 694612 toneladas.
Um cacho de uva é composto por uma parte herbácea, os engaços (3% a 6%) e pelo bago que
representa 94% a 97% do todo (Silva, 2006). A figura 1 mostra uma representação esquemática do
bago da uva que é constituído pela pelicula que representa 8 a 20% do peso do fruto, pela polpa (6%
do peso do bago) e pela semente que diz respeito a cerca de 75 a 85% (% peso fresco do bago)
(Silva, 2006).
Figura 1 – Representação esquemática de um bago de uva (Fonte: Petichateau)
Segundo Silva (2006) durante a maturação, a uva vai sofrendo transformações químicas e
físicas como acumulação dos açúcares, diminuição da acidez, aumento dos compostos fenólicos,
formação de aromas, evolução dos compostos azotados e migração das matérias minerais. Estas
características da uva são posteriormente transferidas para o vinho aquando do processamento das
mesmas.
A produção de vinho é influenciada por diversos fatores com grande impacto na qualidade final
do mesmo. Começando na vindima e terminando na garrafa, a vinificação é uma sequência de
processos físicos e químicos que pode culminar em diferentes tipos de vinho, dependendo do método
utilizado e dos fatores externos. A boa qualidade das uvas é o ponto de partida para a obtenção de
um vinho de qualidade, sendo o clima, o tipo de solo e o grau de maturação três fatores importantes
que atuam sobre esse parâmetro. Em linhas gerais, na elaboração do vinho branco, as uvas são
esmagadas, submetidas aos processos de desengace e prensagem e só depois da separação do
Engaço
Semente
Polpa
Película

6
bagaço do mosto, este último segue para o fermentador. Por outro lado, para produzir vinho tinto, o
bagaço segue com o mosto para o fermentador e só depois se dá a prensagem e o envelhecimento
em barris de carvalho ou garrafas (Infovini, 2009) (vide figura 2). A fase de fermentação do mosto
(com ou sem o bagaço) constitui o principal processo de vinificação. É nesta fase que se dá a
fermentação alcoólica, onde ocorre a transformação de açúcares em álcool, sob a ação de leveduras
com formação de dióxido de carbono (Campos, 2005).
Figura 2 – Esquema simplificado do processamento de vinho (Fonte: Adaptado de Petitchateau)
Tal como acontece noutras indústrias, a fabricação de vinho tem a ela associada uma elevada
produção de resíduos e subprodutos. Em paralelo com o aumento da quantidade de resíduos
industriais, tem crescido também a consciencialização dos impactos ambientais decorrentes das
atividades produtivas de vários sectores da sociedade. Sendo o sector vitivinícola, um sector com
grande importância na economia portuguesa, a gestão de resíduos provenientes desta indústria
torna-se não só importante devido às suas implicações ambientais, bem como na possibilidade
destes resíduos serem aproveitados para a produção de outros produtos de alto valor acrescentado.
O bagaço da uva proveniente da vinificação é produzido juntamente com resíduos, como o
folhelho, borras e sarro, sendo essencialmente constituído pelas sementes, pelicula e engaço (Silva,
2003; Campos, 2005). Segundo Silva (2003), em 100 litros de vinho branco e vinho tinto produzidos,
geram-se 32 kg e 25 kg de subprodutos, respetivamente. O bagaço representa cerca de 17,0 kg
desses subprodutos da vinificação branca e 13,5 kg dos subprodutos da vinificação tinta.

7
Estes subprodutos são atualmente utilizados como rações animais e adubos fertilizantes
(Campos, 2005; Ishimoto et al., 2007), sendo que, esta última prática já mostrou ter efeitos negativos
associados (Negro et al., 2003, Bustamante et al., 2008). Outra aplicação prevista pelo regulamento
do conselho europeu (CE) 1493/1999 sobre a organização comum do mercado vitivinícola é a de que
as vinícolas devem destinar o seu bagaço para as destilarias (produção de etanol) para que a prática
de fermentação do bagaço para outros fins seja abolida (Conselho da União Europeia, 1999;
Bustamante et al., 2008). No entanto, esta prática gera outros resíduos, como o bagaço exaurido e a
vinhaça (Bustamante et al., 2008). A nível industrial, o bagaço é já utilizado para a recuperação de
antocianinas (Arnous e Meyer, 2009; González-Centeno et al., 2010) e o óleo que é extraído das
sementes tem sido utilizado como suplemento nutricional, devido à sua atividade antioxidante
(Arvanitoyannis et al., 2006).
O bagaço da uva apresenta características poluentes que surgem da sua resposta biológica à
carência bioquímica de oxigénio, o seu baixo pH e elevado teor de matéria orgânica como ácidos,
carbohidratos, fenóis e compostos insaturados (Chatzilazarou et al., 2010). Por outro lado, para além
do baixo custo associado a estes subproduto (Deng et al., 2011), a composição rica em fibras e
compostos fenólicos torna-o interessante pela potencialidade de gerar produtos inovadores e
funcionais. Assim, o potencial destes compostos pode contribuir para um aproveitamento deste
subproduto de forma economicamente vantajosa e ambientalmente consciente.
2.2. Composição do Bagaço da Uva
Com o crescente interesse na exploração de resíduos e subprodutos gerados da produção de
vinho, os estudos realizados com o bagaço da uva têm, por consequência, seguido a mesma
tendência. As sementes e a película da uva são uma fonte rica em diversos compostos de alto valor
como etanol, ácidos tartáricos e málicos, ácido cítrico, óleo de sementes de uva, hidrocolóides,
proteínas, fibras alimentares e compostos fenólicos (Igartuburu et al., 1997; Arvanitoyannis et al,
2006). A composição química destes coprodutos é variável e depende do clima de crescimento das
videiras, da variedade da uva e das condições de processamento da mesma (Deng et al., 2011).
2.2.1. Compostos Fenólicos
Compostos fenólicos são metabólitos secundários das plantas (não exercem funções vitais na
mesma), normalmente sintetizados em resposta a reações de defesa da própria planta contra
agressões do meio ambiente (Silva et al., 2010). Estes compostos agem como antioxidantes devido à
sua capacidade de doar um hidrogénio e devido à estabilidade dos seus radicais intermédios, inibindo
assim a oxidação de outras moléculas dos alimentos, essencialmente, lípidos (Arvanitoyannis et al,
2006; Silva et al., 2010). Negro, Tommasi e Miceli (2003) nos seus estudos evidenciaram essa
relação, comprovando que a quantidade de compostos fenólicos conduz a um aumento da
capacidade antioxidante do extrato onde está presente.

8
Estas substâncias contêm na sua estrutura química anéis aromáticos com grupos hidroxílicos
ligados aos primeiros, e são divididos em duas subclasses: os flavonóides, polifenóis (catequina,
epicatequina, procianidinas e antocianinas), e os não flavonóides, fenóis simples ou ácidos (Silva et
al., 2010) (vide figuras 3 e 4). Os flavonóides são compostos presentes em grande quantidade no
bagaço da uva devida à fraca capacidade de extração dos mesmos aquando da etapa de
fermentação para produção de vinho (Arvanitoyannis et al., 2006).
Figura 3 – Exemplo de compostos fenólicos flavonóides: Catequinas (A) e antocianinas (B) (Fonte: Silva et al.,
2010)
Figura 4 – Exemplo de compostos fenólicos não flavonóides: Moléculas dos ácidos hidrocinâmico (A) e do hidroxibenzóico (B) (Fonte: Silva et al., 2010)
As antocianinas, grupo de fenólicos pertencentes à família dos flavonóides, são hidrossolúveis
e são as responsáveis pela cor dos frutos, flores e plantas (Harborne e Williams, 2000). A casca das
uvas escuras apresenta um valor elevado destas substâncias, tendo sido quantificado por Negro e
seus colaboradores (2003) em 0,64 g/L de extrato.
Muitos estudos foram conduzidos com o intuito de extrair compostos fenólicos do bagaço da
uva por meio de solventes orgânicos como o etanol (Negro et al., 2003) ou a acetona (Soares et al.,
2008) bem como da sua identificação e quantificação através de métodos como o de Folin-Ciocalteou
(Singleton e Rossi, 1965; Negro et al., 2003). Segundo a quantificação obtida por Negro e seus
colaboradores (2003), estes compostos encontram-se presentes no extrato obtido a partir do bagaço,
das sementes e das películas da uva numa concentração de 1,40 g/L, 2,86 g/L e 1,11 g/L,
respetivamente.
O interesse nestes compostos deve-se ao seu contributo benéfico para a saúde. Estes
mostram-se úteis no que respeitam doenças cancerígenas e do foro neurológico, e apresentam
efeitos positivos como antivirais, anti-inflamatórios, antialérgicos e na saúde gastrointestinal
(Harborne e William, 2000; Negro et al., 2003; Hervest-Hernández et al., 2009). O chamado
“Paradoxo Francês” mostrou que a incidência de doenças cardiovasculares no sul de França tem sido

9
baixa, apesar do consumo de gorduras saturadas. Os estudos revelaram que a presença de vinho
tinto na dieta desta região é o principal responsável para esses resultados (Giacomo e Taglieri, 2012;
Guilford e Pezzuto, 2011), devido à presença dos flavonóides com capacidade antioxidante, nessa
bebida. Outra questão que levou ao interesse pelo estudo de flavonóides é o facto destes, sendo
antioxidantes naturais considerados seguros, poderem vir a substituir ou associar-se a antioxidantes
sintéticos, com o intuito de diminuir a quantidade destes últimos nos alimentos (Soares, 2002).
2.2.2. Fibras Alimentares
Em geral, os resíduos derivados do processamento de frutas e vegetais contêm uma grande
quantidade de fibras alimentares (Ishimoto et al., 2007; Hervest-Hernández et al., 2009). Neste tipo
de resíduos, tal como o bagaço da uva proveniente do processamento de vinho, as fibras apresentam
uma melhor qualidade funcional quando comparadas com aquelas presentes em resíduos
provenientes do processamento de cereais (González-Centeno et al., 2010). As fibras são definidas
pelos Fisiologistas como “compostos alimentares resistentes à degradação por parte de enzimas
humanas” e pelos Químicos como o “conjunto de polissacarídeos não amiláceos, e lignina” (Graham
e Aman, 1991). De acordo com AACC (2001) fibras alimentares são partes de plantas ou
carbohidratos comestíveis resistentes à digestão e absorção no intestino grosso humano com
fermentação completa, ou parcial no intestino delgado. Nesta definição estão incluídas celulose,
hemicelulose, ligninas, gomas, celulose modificada, mucilagens, oligossacarideos, pectinas e amidos
resistentes e oligossacarideos não digeríveis pelo trato digestivo humano (Mellinger, 2006; Caffal e
Mohnen, 2009; Anderson et al., 2009). Existe também um novo conceito de fibras alimentares
antioxidantes (Tseng e Zhao, 2013), onde este material combina os benefícios das fibras alimentares
com os benefícios da atividade antioxidante dos compostos fenólicos, já descrita anteriormente neste
trabalho.
Segundo González-Centeno e colaboradores (2010) o bagaço de uva Chardonnay é composto
por 23,5 (±1,1 g) /100 g de bagaço fresco de fibras alimentares. Este material pode ser classificado
relativamente ao grau solubilidade em meio aquoso como fibras solúveis e fibras insolúveis, sendo
que o seu teor no bagaço de uva Carbenet Sauvignon é, segundo Ishimoto e seus colaboradores
(2007), de 3,4±0,5% (base seca) e de 60,7±2,2% (base seca), respetivamente, para um valor total de
fibras alimentares de 64,1±3,4% nesse bagaço.
As fibras obtidas a partir de subprodutos agroindustriais, essencialmente provenientes das
paredes celulares que os compõem, são constituídas por diferentes polissacarídeos, organizados
como cadeias de celulose ligadas a diferentes tipos de hemiceluloses e a menores quantidades de
lignina (Peng et al., 2009). A hemicelulose é considerada a maior fração de polissacarídeos das fibras
alimentares (Igartuburu et al., 1997; Peng et al., 2009). De entre as hemiceluloses destacam-se as
xilanas, mananas e glucanas com muitas variações estruturais entre elas, dependendo dos resíduos
agroindustriais dos quais provêm (Karkuráková et al,. 2000), sabendo-se que as mais abundantes nas
plantas são as moléculas da classe das xilanas (Peng et al., 2009) (vide figura 5).

10
Figura 5 – Estruturas básicas de xilanas e xiloglucanas: Glucuronoarabinoxilana (A) e Xiloglucana (B) (Fonte:
Caffal e Mohnen., 2009)
À ingestão de fibras alimentares, como xilo e manooligossacarídeos e os oligossacarideos
derivados de pectinas (Gullón et al., 2013), está associada a redução de doenças cardíacas, cancro
do colon, de diabetes, obesidade e certos distúrbios gastrointestinais, bem como a melhoria de
funções imunológicas (Graham e Aman, 1991; Anderson et al., 2009; González-Centeno et al., 2010;
Deng et al., 2011; Gullón et al., 2013). Relativamente à sua ação no organismo humano, mais
concretamente, no trato gastrointestinal, as fibras solúveis, viscosas ou fermentáveis (pectinas), são
fermentadas no colon do intestino grosso humano; já as insolúveis podem ser fermentadas numa
extensão limitada do colon, e têm ação espessante das excreções. (Graham e Aman, 1991; Anderson
et al., 2009). Na área dos biomateriais, as fibras mostram ser um material promissor no
desenvolvimento de plásticos biodegradáveis e formação de filmes, especialmente para o
acondicionamento de produtos industrializados e alimentos (Siracusa et al., 2008; Deng e Zhao,
2011). Como Deng e Zhao (2011) referem, os materiais biodegradáveis e comestíveis de
embalagens, mostram-se competitivos a nível mecânico e nas suas propriedades em processos de
transferência de massa quando comparados com os materiais sintéticos. Para além de poderem
transportar substâncias funcionais como antioxidantes e agentes antimicrobianos, este tipo de
embalagens têm a capacidade de aumentar o tempo de prateleira dos alimentos neles armazenados.
A extração de fibras alimentares tem sido tema de estudo já há várias décadas, tendo sido
iniciada com resíduos da indústria dos cereais (González-Centeno et al., 2010). Em 1988, Graham e
seus colaboradores conduziram vários ensaios de extração em vegetais e cereais, com o intuito de
estudar a influência das condições de extração na quantidade e tipo de fibras obtidas nos extratos.
Aoe et al. (1993) estudaram o melhor solvente para extrair fibras solúveis do farelo de arroz
desengordurado, chegando à conclusão que o solvente alcalino de Ca(OH)2 apresentou os melhores
resultados por ter produzido menos cor no extrato e uma composição e rendimento de fibras
desejáveis. Apesar destes e outros autores (Saulnier et al., 1995; Sun et al., 2001; Mellinger et al.,
2005a; Peng et al., 2009; Mellinger-Silva et al., 2011) privilegiarem os solventes alcalinos para a
extração de fibras solúveis como a hemicelulose e, segundo Peng e seus colaboradores (2009) este
ser um processo de extração promissor que extrai as lignoceluloses na sua totalidade sem impacto
ambiental, outros estudos conduzem a conclusões menos limitantes, constatando que a água é outro
bom solvente para este tipo de polissacarídeos, essencialmente hemiceluloses mais ramificadas
(Geng et al., 2006; Peng et al., 2009), sendo que o método de extração escolhido dependerá da

11
aplicação final destes materiais (Deng e Zhao, 2011). No que respeitam as extrações aquosas
enzimáticas, verificou-se que estas são utilizadas na recuperação de óleos vegetais (Santos e Ferrari,
2005; Mariano et al., 2009) e proteínas (Lopes Junior, et al., 2010) com bons resultados. No entanto,
uma vez que o tratamento enzimático em meio aquoso tem-se mostrado eficiente na degradação das
paredes dos tecidos vegetais (Santos e Ferrari, 2005), este desperta também interesse na extração
destas fibras presentes no bagaço da uva.
O fracionamento e concentração destes materiais são o passo subsequente às extrações dos
mesmos para posterior utilização industrial. Vários estudos têm também sido desenvolvidos nesta
área, com a aplicação de processos de ultrafiltração e nanofiltração na separação de polissacarídeos
(Gonçalves, 2002; Catarino, et al., 2008; Sun et al., 2011), bem como de precipitação etanólica (Peng
et al., 2009).
Destes processos de extração e fracionamento, obtêm-se assim produtos de valor
acrescentado, sendo que o resíduo final, essencialmente celulose, pode ser utilizado para derivados
de papel ou celulose (Peng et al., 2009) ou como matéria-prima para obtenção de bioetanol de
segunda geração (Silva, 2010), conseguindo-se assim o máximo aproveitamento do bagaço da uva
proveniente da vinificação.
2.3. Transferência de massa
O fenómeno de transferência de massa consiste no movimento de um componente num
sistema de várias componentes. Este fenómeno pode ocorrer por via de dois mecanismos: convectivo
e difusivo. O mecanismo convectivo de transferência de massa dá-se através de um fluido em
movimento, ao passo que a transferência de massa por difusão molecular é consequência de uma
diferença de concentrações (força motriz) onde um componente A, por razões termodinâmicas, é
difundido de uma zona mais concentrada para uma zona menos concentrada nesse mesmo
componente (Azevedo e Alves, 2009). Estes dois mecanismos podem coexistir e ter a mesma
importância no resultado da transferência de massa, ou um deles pode ter maior importância nesse
fenómeno.
A difusão molecular é matematicamente definida pelas Leis estabelecidas por Fick nos seus
estudos (Pinho e Prazeres, 2008). Para soluções binárias, ideais e diluídas e em estado estacionário,
a relação linear existente entre o fluxo difusivo do componente x, (Jx), e o gradiente de concentrações
desse componente, dCx/dz, é dada pela 1ª Lei de Fick, representada na equação 1.
(Equação 1)
Para sistemas em estado transiente, Fick propõe uma equação de conservação de massa,
onde relaciona a taxa de transferência de massa com o gradiente de concentração designada como a
2ª Lei de Fick (equação 2) (Pinho e Prazeres, 2008).
(Equação 2)

12
A constante de proporcionalidade existente nas duas leis descritas, D, é designada como a
difusividade ou coeficiente efetivo de difusão. Esta constante representa a facilidade com que o
componente A é difundido no sistema e a sua definição matemática varia de acordo com o estado
físico do sistema. Para soluções ideias, o coeficiente efetivo de difusão é dado pela equação 3.
(Equação 3)
onde f é o fator de atrito, kB a constante de Boltzmann e T a temperatura (Pinho e Prazeres,
2008).
A resolução matemática da 2ª Lei de Fick, tratando-se de uma situação de difusão
unidimensional e em estado transiente, pode ser escrita em coordenadas cartesianas, como
apresentado na equação 4. As condições consideradas foram a inicial onde para t=0 a concentração
Cx=0, e as condições fronteira onde se considerou simetria no processo difusivo unidirecional e que a
Cx na fronteira sólido/solução é igual à Cx para o tempo de +infinito.
(Equação 4)
onde é a concentração de soluto adimensionalizada, L é a espessura de
transferência de massa e t é o tempo de processo.
Considerando-se os três primeiros termos, obtém-se a equação 5.
(Equação 5)
Assim, através dos dados experimentais de concentração em função do tempo, é possível
estimar o coeficiente efetivo de difusão, D.
Industrialmente, estes fenómenos de transferência de massa dão-se em diversos tipos de
processos, como é o caso das extrações sólido-liquido e processos de membranas que serão
descritos nas próximas secções deste capítulo.
2.3.1. Processos de Extração Sólido-Líquido
A operação de Extração é uma operação de separação e/ou purificação muito comum a nível
industrial. Neste processo, a separação de um ou mais componentes (solutos) de uma mistura é
promovida com a adição de um outro composto, o solvente, sendo que o soluto é transferido de uma
fase para outra com a qual tem mais afinidade.
As extrações podem ser do tipo líquido-líquido ou sólido-líquido. No primeiro caso, ambos o
solvente e a alimentação encontram-se no estado líquido, ao passo que no tipo de extração sólido-
líquido o estado de agregação da alimentação é sólido. Na extração sólido-líquido, também
designada por lixiviação, o solvente entra em contacto com a matéria sólida (substrato) e o soluto de
interesse é transferido do último para o primeiro. No final do processo, obtém-se o resíduo (substrato
esgotado, refinado) e o extrato (solvente com soluto).

13
Industrialmente este processo dá-se através de contactos múltiplos entre ambas as fases
envolvidas no processo de separação. Com o intuito de aumentar a eficiência da separação, surgiu o
conceito de andares de equilíbrio (Azevedo e Alves, 2009). Estes permitem que as duas fases se
misturem em cada andar até se atingir o equilíbrio termodinâmico, sendo posteriormente separadas e
enviadas para os andares seguintes onde o processo é repetido com novas composições.
Esta transferência de massa é não só função do tipo de solvente utilizado, tendo em conta o
soluto a extrair, como também dependente de outras variáveis como: temperatura, relação
solvente/substrato, tempo de extração, pH, concentração do solvente, viscosidade, tamanho das
partículas, entre outras (Sun et al., 2001; Kemper, 2005; Mellinger et al., 2005a; Azevedo e Alves,
2009; Karacabey e Mazza, 2010).
A título de exemplo, na figura 6 apresenta-se o equipamento industrial de extração Rotocel.
Figura 6 – Representação esquemática do Extrator Rotocel (Fonte: Schweitzer, 1982)
O extrator Rotocel é constituído por um tambor rotativo com compartimentos separados onde
é alimentada a fase sólida, alimentação. Este tambor tem um fundo perfurado e movimento de
rotação em torno de um eixo vertical, encontrando-se sobre um tanque imóvel de diâmetro igual. O
substrato é introduzido num compartimento vazio do tambor. Durante uma rotação, o sólido é lavado
com o solvente alimentado por distribuidores de líquido, sendo que este último atravessa o substrato
e extrai o soluto de interesse. O líquido escorre através do fundo perfurado e é recolhido num
compartimento do tanque imóvel que também se encontra dividido. Daqui, o líquido é bombeado para
outro distribuidor de forma a lavar o sólido mais rico em soluto, repetindo-se várias vezes o mesmo

14
procedimento até o tambor fazer uma volta completa. Ao fim de uma volta, o refinado é descarregado
através de um parafuso. Esta sucessão de passagens assegura a realização de uma extração
contracorrente (Farelo e Serrano; Kemper, 2005) com vários andares de equilíbrio, de onde se obtém
o extrato e o resíduo da extração.
2.3.2. Processos de Separação por Membranas
A tecnologia de membranas tem vindo a crescer e a ganhar importância na indústria e na
sociedade. As suas principais aplicações incidem nas áreas de química, alimentação, medicina,
farmacêutica, têxtil, biotecnológicas, controlo ambiental e metalúrgica (Sun et al., 2011).
Em comparação com outros métodos tradicionais de separação, a separação de membranas
mostra-se uma opção vantajosa na medida em que opera à temperatura ambiente, em geral, sem
mudança de fase, requer um baixo investimento de capital, é energética e ambientalmente eficiente e
é um sistema compacto e de simplicidade operatória (Alves, 2010; Sun et al., 2011). Este tipo de
processos é utilizado com a finalidade de obter compostos com especificações previamente
determinadas ou impostas, separando, concentrando ou purificando os mesmos.
As membranas são incorporadas num sistema onde atuam como barreira semipermeável
fracionando uma corrente constituída pela solução a tratar, designada por alimentação, em permeado
(solução com os solutos com massa molecular inferior ao limite de exclusão molecular da membrana
(MWCO)) e concentrado (solução com solutos com massa molecular superior ao MWCO) por ação de
uma força motriz aplicada ao sistema. (vide figura 7).
Figura 7 – Esquema geral de um processo de separação com membranas
Neste processo, como no anteriormente descrito, o mecanismo de separação é a transferência
de massa, existindo, neste caso, uma barreira física. Esta transferência de massa é induzida por uma
força motriz, um gradiente, como a diferença de pressão, aplicada em operações de osmose inversa,
nanofiltração, ultrafiltração e microfiltração, a diferença de concentrações, aplicada em operações de
diálise e extração com membranas, e o potencial elétrico aplicado em operações de eletrodiálise
(Alves, 2010). Para além destes fatores, o sucesso e a qualidade da separação depende também da
natureza e o tipo de membrana utilizada.
Os processos de microfiltração e ultrafiltração são em muito semelhantes aos processos de
filtração tradicionais, na medida em que o mecanismo de separação é o de peneiração molecular,
existindo o limite de exclusão molecular (MWCO) que caracteriza as membranas. Estes processos
podem ser conduzidos de duas formas: filtração frontal (dead-end) e filtração tangencial (crossflow
Alimentação
Permeado
Concentrado
Membrana

15
filtration). A filtração frontal diz respeito ao processo onde o fluxo de alimentação é perpendicular à
membrana (vide figura 8A), enquanto que na filtração tangencial o fluxo dá-se de forma tangencial à
mesma (vide figura 8B). A vantagem da filtração tangencial face à filtração frontal é a existência de
um varrimento dos depósitos que se vão acumulando junto à membrana, evitando o seu entupimento.
Figura 8 – Representação esquemática dos fluxos de filtração: Filtração frontal (A); Filtração tangencial (B)
(Fonte: Adaptado de Alves, 2010)
No entanto, durante a operação em sistema de membranas, dada a seletividade das mesmas,
há solutos que, sendo rejeitados pela membrana, podem acumular-se na superfície da mesma,
provocando o designado fouling. Esta acumulação de uma camada concentrada na interface
membrana/solução, dá origem a um gradiente de concentrações e consequentemente um fluxo
difusivo em direção ao seio da alimentação, designando-se por polarização da concentração (vide
figura 9).
Figura 9 – Esquema representativo do fenómeno de polarização da concentração (Fonte: Adaptado de Pinho e
Prazeres, 2008)
Este fenómeno oferece uma resistência adicional à transferência de massa, provocando a
diminuição do fluxo de permeado ao longo do tempo. Assim, e com base nos conceitos já descritos
de transferência de massa, pode verificar-se que o transporte de um dado componente presente na
alimentação para e/ou através da membrana é feito por difusão e por convecção transversal
(JC), considerando o escoamento tangencial da alimentação completamente desenvolvido e em
estado estacionário (Pinho e Prazeres, 2008). Admite-se ainda que, junto à membrana, considerando
um escoamento unidirecional e laminar existe um filme de espessura δ (vide figura 9), onde a
A B

16
concentração de soluto aumenta e atinge o valor máximo na superfície da membrana, Cm. A jusante
dessa camada de filme, dá-se uma mistura completa e a concentração é constante e igual à
concentração no seio da alimentação, Cb (Pinho e Prazeres, 2008). Fazendo um balanço de massa
ao soluto, em estado estacionário, no filme de espessura δ, e pela teoria do filme, obtém-se uma
expressão matemática para o coeficiente de transferência de massa, k (equação 6), sendo que a
eficiência das operações de transferência de massa com membranas depende fortemente desse
coeficiente (Pinho e Prazeres, 2008).
(Equação 6)
Dado que a rejeição do soluto pela membrana é explicada por mecanismos de exclusão por
tamanho, assim como por interações físico-químicas entre os compostos e a camada seletiva da
membrana, cada um dos processos de separação com membranas foi desenhado de forma a permitir
que compostos sejam retidos dentro de uma escala específica de massa molar. Na figura 10 está
esquematizada a aplicação destes processos em função do tamanho de diferentes tipos de
partículas.
Figura 10 – Representação esquemática dos intervalos de tamanhos de partículas removidas por processos de
filtração em unidades de micrómetros, Angstrom e de massa molecular aproximada, Da (de cima para baixo na imagem) (Fonte: Adaptado do portal de Engenharia Química)
Membranas e Módulos:
Atualmente têm-se desenvolvido inúmeros tipos de membranas no que respeita o material e a
técnica de fabrico, por forma que este tipo de tecnologia vá de encontro a diversas aplicações. As
membranas que se encontram disponíveis no mercado hoje em dia são principalmente constituídas
por materiais orgânicos (vidro poroso, cerâmica, grafite, óxidos metálicos), materiais naturais
modificados (à base de celulose) ou são de origem sintética (poliamidas) (Alves, 2010).

17
Os módulos de membranas são também classificadas quanto ao tipo de configuração para o
qual são desenvolvidas, existindo membranas do tipo planar, tubular, fibras ocas e em espiral. Os
módulos do tipo planar, que suportam operações de osmose inversa, pervaporação, ultrafiltração,
eletrodiálise e microfiltração (Alves, 2010), são constituídos por membranas em forma de folha,
dispostas paralelamente, separadas por espaçadores e suportes porosos como representado
esquematicamente na figura 11.
Figura 11 - Representação esquemática do módulo de membranas do tipo planar (Fonte: Resende, 2011)
O módulo membranar do tipo tubular é constituído por uma membrana cilíndrica de material
polimérico ou cerâmico inserida num módulo com a mesma geometria e por um suporte poroso (vide
figura 12). Este tipo de módulos pode ser aplicado em operações de osmose inversa, ultrafiltração e
microfiltração (Alves, 2010).
Figura 12 – Representação esquemática do módulo de membranas do tipo tubular (Fonte: Resende, 2011)
Outro módulo de membranas com aplicação em operações de osmose inversa, pervaporação e
permeação gasosa é o do tipo de fibras ocas (Alves, 2010). Estas são usadas em forma de cartuchos
com centenas de fibras de pequeno diâmetro. Ao contrário do que acontece no módulo tubular, a
alimentação circula por fora das fibras e o permeado é recolhido no seu interior (vide figura 13)

18
Figura 13 – Representação esquemática do módulo de membranas do tipo fibras ocas (Fonte: Resende, 2011)
A configuração em espiral opera com processos de separação por membranas, principalmente
osmose inversa, pervaporação, permeação gasosa e ultrafiltração (Alves, 2010). Este módulo é
constituído por membranas planas, suportes e espaçadores que são fixados e enrolados em redor de
um tubo coletor central por onde flui o permeado (vide figura 14).
Figura 14 – Representação esquemática do módulo de membranas do tipo espiral (Fonte: Resende, 2011)
Existem vários fatores a considerar na escolha do melhor módulo não só económicos como
também de funcionalidade. Nesse sentido, e a título de comparação, apresenta-se na tabela 1, as
características de cada um destes tipos de módulos, bem como a compatibilidade destes a cada
operação.
Tabela 1 – Comparação de diferentes configurações modulares (Fonte: Alves, 2010)
Tubular Plano Espiral Fibras Ocas
Densidade de empacotamento Baixa Muito
Elevada
Muito
Elevada
Muito
Elevada
Custo de investimento/área Elevado Baixo Baixo Baixo
Tendência para “fouling” Baixa Elevada Elevada Elevada
Limpeza Fácil Difícil Difícil Difícil
Custos Operatórios Elevados Baixos Baixos Baixos
Possibilidade de substituição das
membranas Sim Sim Não Não

19
2.4. Objetivo da Dissertação
Integrada no projeto da Embrapa Agroindústria de Alimentos “Extração de substâncias de
interesse comercial e industrial a partir de coprodutos gerados na produção de vinhos e sucos de
uva” (Resuva), esta dissertação tem como principal objetivo o de extrair e fracionar os extratos de
fibras solúveis presentes no bagaço de uva proveniente do processo de vinificação tinta a partir da
uva branca Chardonnay.
Especificamente:
1) Determinar a melhor condição operacional de extração de fibras solúveis do bagaço da uva
branca proveniente da vinificação em tinto, tendo como parâmetro o teor de açúcares;
2) Fracionar os extratos por processos de tecnologia de membranas;
3) Identificar estruturalmente os polissacarídeos presentes nos extratos e nas frações obtidas
por processos de tecnologia de membranas;
4) Avaliar a capacidade antioxidante in vitro e o conteúdo de compostos fenólicos dos extratos
e das frações obtidas por processos de membranas.

20

21
3. Parte Experimental
No âmbito do presente trabalho foram realizados diversos ensaios experimentais, a fim de se
obter o conhecimento da melhor condição de extração de fibras solúveis do bagaço da uva branca e
do tipo de fibras obtidos por esse processo, bem como na separação das mesmas por processos de
membranas. Neste capítulo estão descritos os materiais e os procedimentos utilizados para esse fim,
bem como uma breve descrição dos métodos de análise realizados.
3.1. Matéria-Prima
O bagaço estudado neste trabalho foi o proveniente da uva branca Vitis Vinifera Chardonnay
submetida ao processo de vinificação em tinto. Todo o bagaço foi gentilmente cedido pela
Cooperativa Vinícola Aurora, localizada na cidade Bento Gonçalves, na região da Serra Gaúcha no
Estado do Rio Grande do Sul no Brasil, e proveniente da colheita de 2010/2011. Este bagaço foi
armazenado numa câmara de congelação a -16ºC até ao início da atividade experimental. A sua
composição físico-química está apresentada na tabela 2.
Tabela 2 – Composição físico-química do bagaço da uva branca Vitis Vinifera Chardonnay
Potássio (mg/kg) 4234,84
Magnésio (mg/kg) 180,75
Cálcio (mg/kg) 600,55
Fósforo (mg/kg) 572,09
Manganês (mg/kg) 5,33
Ferro (mg/kg) 20,38
Zinco (mg/kg) 4,06
Cobre (mg/kg) 39,66
Selénio (mg/kg) NQa
Humidade (g/100g) 82.47
Cinzas (g/100g) 1,02
Nitrogénio Total (g/100g) 0,41
Proteína (g/100g) 2,33
Extrato Étereo (g/100g) 1,55
Fibra Alimentar (g/100g) 11,81
Fitato (mg/g) 0,45
Carbohidratos (g/100g) 3,45
Valor Calórico (g/100g) 37,01 a NQ – não quantificável
A matéria-prima com que se realizou este trabalho não tem uma composição física
homogénea, sendo que, em média, é constituída pelos componentes apresentados na figura 15.

22
Figura 15 – Componentes constituintes do bagaço da uva branca em estudo (m/m)
3.2. Equipamentos
Para cumprir os objetivos propostos neste trabalho os ensaios experimentais foram agrupados
de acordo com o objetivo visado em, i) extração sólido-líquido e ii) microfiltração e ultrafiltração e iii)
identificação estrutural das fibras extraídas e concentradas por processos.
Para os ensaios de extração foram utilizados o triturador Walita e o banho termostatizado com
agitação (Dubnoff NT232; termostato: Comtemp, TCM 45) fornecido pela Novatecnica (vide figura
16), presente no Laboratório de Processamento de Matérias Primas Vegetais da Escola de Química
da Universidade Federal do Rio de Janeiro (UFRJ), Brasil, tendo todo o processo sido realizado neste
laboratório.
Figura 16 – Fotografia do Banho Termostatizado
Os processos de membranas efetuados neste trabalho foram, realizados na planta piloto de
operações unitárias da Embrapa Agroindústria de Alimentos, na cidade do Rio de Janeiro, Brasil.
Para a realização destes ensaios utilizou-se um sistema semi-piloto em escala laboratorial de
configurações do tipo: i) tubular de 3 módulos de membranas cerâmicas PALL de 0,1 m
(microfiltração) fornecidas pela Membralox, com área de membrana de 0,0165 m2
e ii) tubular de 4
módulos de membranas cerâmicas 20 kDa (ultrafiltração) com área de membrana de 0,022 m2.
O sistema é constituído por um tanque de alimentação, uma bomba, um permutador de calor,
um módulo de membranas dois manómetros e uma válvula de ajuste de pressão, como representado
na figura 17.
17%
21% 62%
Sementes
Caules/folhas
Películas

23
Figura 17 – Representação esquemática de módulo de membranas
Durante todo o processo foi necessário controlar a temperatura e a pressão do sistema. A
temperatura foi controlada através de um termopar colocado no tanque de alimentação e manipulada
através do caudal de água fria admitido ao permutador de calor. A variável pressão transmembrana
foi medida através da média das pressões à entrada e à saída do módulo de membranas, tendo sido
registada pelos manómetros instalados nessas posições e controlada por válvulas de pressão
presentes no sistema (vide figura 17).
As figuras 18 e 19 mostram, respetivamente, um corte da membrana cerâmica de 20 kDa e a
fotografia do módulo de membranas utilizado.
Figura 18 – Membrana de 20kDa
Tanque de
Alimentação Bomba Permutador de
calor
Módulo de
Membranas
Válvula de ajuste
de pressão

24
Figura 19 – Fotografia do módulo de membranas (ultra e microfiltração)
3.3. Procedimento Experimental
Este trabalho experimental começou com seleção da melhor condição de extração de fibras
solúveis do bagaço de uva branca em estudo, utilizando-se para esse efeito três solventes. Destes
três extratos provenientes dos três diferentes solventes estudados, só o extrato aquoso seguiu para
os processos de membranas subsequentes, questão que será discutida mais à frente neste trabalho
(vide Capitulo 4.2). Todas correntes do processo foram submetidas a diversas análises químicas para
que com esses resultados fosse possível dar resposta aos objetivos propostos. Estas análises foram
complementadas com o estudo das estruturas polisacarídicas de extratos selecionados.
3.3.1. Extrações sólido-líquido de fibras solúveis
Seleção das condições de extração
No âmbito do estudo da determinação da melhor condição de extração de fibras solúveis do
bagaço de uva branca, e como foi referido anteriormente, utilizaram-se três solventes: Água, solução
aquosa contendo Celulase (gentilmente cedida pelo Laboratório de Tecnologia Enzimática, no
Departamento Bioquímica do Instituto de Química da UFRJ, Rio de Janeiro Brasil. Enzima obtida do
Trichoderma reesei (RUT C30) com atividade de FPase de 2,65 UI/mL) e solução aquosa de
hidróxido de potássio (KOH) a 2, 6 e 10%.
Os ensaios relativos aos solventes água e solução aquosa de KOH nas três concentrações
estudadas tiveram como variáveis independentes a temperatura (70, 80 e 90ºC), o tempo de
trituração do bagaço (0, 3 e 6 minutos) e a relação substrato/solvente (1:2, 1:3 e 1:4). No que respeita
os ensaios realizados para o solvente aquoso contendo Celulase, a temperatura foi mantida
constante a 50ºC ao longo de todos os ensaios para maximizar a atividade de enzima, e considerou-
se a concentração de enzima em relação ao bagaço seco uma variável independente (350, 700 e

25
1050 µL de Celulase para 18 g de bagaço seco), mantendo-se as restantes variáveis estudadas para
os outros dois solventes (tempo de trituração do bagaço e relação substrato/solvente).
De forma a determinar-se a melhor condição com as variáveis independentes referidas, fixou-
se o tempo de extração em 5 horas, para garantir a extração máxima das fibras solúveis nas
condições em estudo. Os extratos obtidos foram analisados quanto á quantidade de açúcares totais
presentes (Dosagem de açúcar total) e com esses resultados, escolhida a melhor condição, dentro do
universo de variáveis estudadas.
Cinética de extração
Para a determinação da cinética da extração, realizaram-se ensaios nas melhores condições
de extração previamente estabelecidas para cada um dos solventes. As amostras foram mantidas no
banho por 30min, 1h, 1h30min, 2h, 2h30min, 3h, 3h30min, 4h, 4h30min e 5h. Após o período
estabelecido para cada amostra, realizou-se nova dosagem de açúcar total (DAT), para concluir a
seleção das variáveis de extração de fibras solúveis do bagaço da uva branca, no universo de
variáveis estudadas.
Todos os extratos alcalinos foram neutralizados com ácido acético concentrado glacial (99,7%
PA, M Tedia) para que se pudessem realizar as posteriores análises aos mesmos.
3.3.2. Microfiltração e Ultrafiltração
O extrato fibroso obtido por extração aquosa, nas melhores condições estudadas, foi
submetido a um processo de microfiltração e ultrafiltração com o objetivo de concentrar as fibras
solúveis presentes no mesmo.
Os ensaios foram realizados a 25±2°C, com uma pressão transmembrana de 2,5 bar para a
microfiltração e de 15 bar quando foram utilizadas as membranas de ultrafiltração. O caudal de
circulação registado para este sistema é de 900 L/h e cada membrana apresentou um diâmetro de
0,7 cm e comprimento de 25 cm. Estas condições levaram à obtenção de uma velocidade tangencial
de 6,50 m/s, para uma área de secção transversal de 0,38 cm2.
Para cada processo com membranas realizou-se uma extração do bagaço da uva na condição
previamente determinada como sendo a melhor, obtendo-se cerca de 2,70 kg de extrato que foi
posteriormente alimentado ao sistema de membranas. A cada 5 a 15 minutos de processo, foram
calculados os fluxos de permeado (Jp) e o fator de concentração volumétrico (FCV), através das
equações 7 e 8, respetivamente:
(Equação 7)
(Equação 8)
Onde m0 é a massa de extrato da alimentação, mp é a massa do permeado, medida no tempo
de processo t e A é a área da membrana.
Considerou-se o fim do ensaio sempre que se atingia o valor de 2 de FCV.

26
Limpeza e hidratação do sistema
Antes de serem utilizadas, ambas as membranas foram hidratadas fazendo-se passar pelo
módulo a seguinte sequência de soluções: água, uma solução de soda-caustica (1%) (em sistema
fechado) e novamente água. Cada uma destas fases de hidratação dura 20 minutos.
Depois de cada ensaio experimental, foi também necessário proceder à limpeza das
membranas para garantir que estas recuperam, pelo menos, 90% do fluxo inicial de permeação. Este
procedimento passou por fazer circular água em sistema aberto (sem recirculação) durante 20
minutos, seguindo-se uma lavagem em sistema fechado (com recirculação) com soda cáustica (25%)
durante outros 20 minutos. Até se atingir novamente um pH neutro, fez-se passar água pelo módulo
de membranas para posteriormente fazer-se uma limpeza cloro-alcalina com uma solução de 1% de
soda caustica e cloro 100 ppm, durante 20 minutos em sistema fechado. Por fim, lavaram-se
novamente as membranas com água até se atingir pH neutro. No final de cada lavagem mediu-se a
permeabilidade hidráulica para garantir que a membrana foi devidamente limpa.
3.3.3. Métodos Químicos
De forma a poder concluir-se a respeito dos procedimentos elaborados, foram também
realizadas análises químicas e físicas ao bagaço da uva e aos extratos obtidos. As análises químicas
realizadas a estes últimos apresentam parte das estruturas dos polissacarídeos obtidos nos
diferentes meios de extração, avaliando-se estas estruturas por métodos espectrofotométricos,
cromatográficos e espectrométricos. Estas análises foram realizadas nos laboratórios de Bioquímica
e de Físico-química da Embrapa Agroindústria de Alimentos, no Departamento de Bioquímica e
Biologia Molecular da Universidade Federal do Paraná e no Laboratório de Processamento de
Matérias Primas Vegetais da Escola de Química da UFRJ. Nos subcapítulos seguintes faz-se, uma
breve descrição das análises realizadas e dos métodos aplicados.
Caraterização físico-química do bagaço da uva branca
A caracterização físico-química do bagaço da uva branca submetida a vinificação tinta foi
determinada a partir das metodologias apresentadas abaixo, resultados estes que foram gentilmente
cedidos pelo laboratório de Físico-química da Embrapa Agroindústria de Alimentos:
Teor de Potássio, Magnésio, Cálcio, Fósforo, manganês, ferro, zinco e cobre por
mineralização pelo método 999.10, item 9.1.08, AOAC 2005 e a sua quantificação pelo
método 990.08, item 9.2.39, AOAC 2005,
Teor de cinzas pelo método 923.03, AOAC 2010,
Nitrogénio Total pelo método 2001.11 modificado, AOAC 2010 e fator 5,75 para
conversão do nitrogénio em proteínas totais,
Extrato Etéreo pelo método 945.38, AOAC 2010,
Teor de fibra alimentar pelo método 985.29, AOAC 2010,
Fitato pelo método 986.11 modificado, AOAC 2010,

27
Carbohidratos e valor calórico calculados segundo a Resolução – RDC n.º 360 de 23
de dezembro de 2003
Humidade total
A humidade total das amostras de bagaço fresco foi avaliada pela diferença entre a massa do
sólido húmido e a massa de sólidos totais, determinado pelo método gravimétrico nº 964.22 da AOAC
(2000), a uma temperatura de 105ºC. Esta metodologia foi realizada no Laboratório de
Processamento de Matérias Primas Vegetais da Escola de Química da UFRJ.
Obtenção dos polissacáridos totais (Precipitação Etanólica)
As amostras provenientes do estudo com os processos de membranas foram submetidas à
análise dos polissacáridos totais (Mellinger-Silva et al., 2011). Esta análise consiste na precipitação
etanólica dos polissacarídeos presentes em solução, adicionando 3 volumes de álcool etílico gelado
(95% P.A. da ACS) a cada uma das amostras. Depois de precipitadas todas as macromoléculas de
polissacáridos presentes, centrifugaram-se as soluções etanólica durante 15 minutos, a 9000 rpm e a
4ºC na ultracentrifuga da Thermo, gentilmente cedida pelo laboratório de Contaminantes e Resíduos
da Embrapa Agroindústria de Alimentos, para facilitar a posterior separação. Separado o precipitado
do sobrenadante, solubilizou-se o precipitado em água destilada, utilizando-se o menor volume
possível da mesma. A seguir, deixou-se a solução em Diálise over-night, utilizando-se membranas de
fibra de celulose da Spectra/Por® Dialysis Membrane e com limite de exclusão molecular (MWCO) de
3,500 kDa, de forma a remover todo o etanol ainda presente na solução. Por fim liofilizaram-se as
amostras no liofilizador L 101 da Liotop, ressolubilizou-se o pó liofilizado.
Hidrólise ácida total, redução e acetilação
Antes de se analisar a sua composição monossacarídica pelo método cromatográfico, as
amostras foram sujeitas a um pré-tratamento, onde sofreram hidrólise ácida, redução com NaBH4 e
posterior acetilação com uma mistura de anidrido acético e piridina.
A hidrólise ácida total dos polissacarídeos presentes nas amostras foi realizada utilizando 2 mg
da amostra e 1,0 mL de TFA 1 mol/L, a 100ºC durante 8h. Após hidrólise, a eliminação do ácido foi
realizada por evaporação em temperatura ambiente. O produto seco foi então ressuspendido em 1,0
mL de água destilada e reduzido com Boroidreto de Sódio (NaBH4), pH 10, em temperatura ambiente
durante 12h. De seguida, as frações foram neutralizadas com ácido acético glacial e a solução foi
novamente seca, desta vez sob pressão reduzida. O ácido bórico formado foi co-evaporado por meio
de sucessivas lavagens com metanol, na forma de borato de trimetila (Wolfrom e Thompson, 1963a).
Os alditos formados durante a redução efetuada foram então acetilados com uma mistura de anidrido
acético e piridina (1:1, v/v) durante 12h à temperatura ambiente (Wolfrom e Thompson, 1963b).
Interrompeu-se a acetilação adicionando gelo e os acetatos de alditóis foram extraídos com

28
clorofórmio. A piridina residual foi removida através de sucessivas lavagens com solução de CuSO4 a
5%. Estes acetatos foram preparados e analisados por Cromatografia liquido-gasosa acoplada à
espectrometria de massa (GC-MS), no Departamento de Bioquímica e Biologia Molecular da
Universidade Federal do Paraná (UFPR) localizada em Curitiba, Paraná, Brasil.
3.3.4. Métodos Espectrofotométricos
Dosagem de açúcar total (DAT)
Todas as amostras provenientes da extração sólido-líquido e dos processos de separação por
membranas foram submetidas a DAT, com o objetivo de quantificar os açúcares presentes e
consequentemente inferir quanto à quantidade de fibras solúveis extraídas e isoladas. Esta análise foi
realizada com base no Método do fenol-ácido sulfúrico, o qual apresenta uma sensibilidade de 8 a 70
µg de açúcar (Dubois et al., 1956), no laboratório de Bioquímica da Embrapa Agroindústria de
Alimentos, Rio de Janeiro, Brasil. De acordo com este método, adicionaram-se a 0,5 mL de amostra
solubilizada em água destilada, 0,5mL de Fenol 5% (p/v) (P.A., da Vertec - Química Fina) e 2,5 mL de
ácido sulfúrico concentrado (96% P.A. da Vertec - Química Fina). Com um comprimento de onda de
490nm, foram lidas as absorvâncias das soluções e dos brancos, recorrendo a um espectrofotómetro
(modelo SP-220 da Bioespectro, com células de quartzo) e utilizando como padrão a D-glucose (vide
Anexo 8.C) com concentração 200 µg/mL (97+% da Sigma Chemical Co.). Estas análises foram
realizadas em triplicado para garantir e demostrar a consistência dos resultados.
Este método de análise baseia-se na determinação de monossacáridos por hidrólise dos
carbohidratos totais presentes nas amostras através da reação com o ácido sulfúrico. Esta reação
forma o hidroximetilfurfural que, ao reagir com fenol, origina um complexo colorido. A intensidade da
cor deste complexo é diretamente proporcional à quantidade de açúcar presente em solução e é esta
que permite a leitura espectrofotométrica.
Uma vez que o bagaço em estudo foi submetido à vinificação, considerou-se que a presença
de açúcares livres nos extratos provenientes do mesmo fosse pequena. Assim, é correto afirmar que
os açúcares quantificados por este método são provenientes das fibras solúveis em estudo e não de
açúcares que poderiam encontrar-se livres em solução.
Dosagem de ácidos urónicos
Foi realizada a análise colorimétrica de ácidos urónicos de forma a detetar-se, caso existissem,
os monossacarídeos ácidos, principalmente os ácidos glucurónico e galacturónico. Este procedimento
segue o método do sulfamato-metahidroxibifenila e foi realizado apenas para a amostra de
concentrado do processo de ultrafiltração, pois tornou-se a fração com maior interesse químico do
estudo. Esta metodologia foi gentilmente efetuada no Departamento de Bioquímica e Biologia
Molecular da UFPR, tendo como base a descrita por Filisetti-Cozzi e Carpita, 1991, apresentando
uma sensibilidade de 0,97 a 38.8 µg de ácido urónico.

29
As amostras foram solubilizadas (0,4 mL) numa solução de ácido sulfâmico-sulfamato de
potássio (40 L, 4mol/L, pH 1,6), adicionando-se de seguida 2,4 mL de tetraborato de sódio (75 mol/L)
em ácido sulfúrico concentrado. Adicionou-se 80 L de m-hidroxibifenila às amostras, depois destas
terem sido aquecidas durante 20 minutos a 100ºC e posteriormente arrefecidas em banho de gelo. O
complexo colorido gerado, foi lido espectroscopicamente a 525nm, tendo como curva padrão a curva
de ácido galactorónico.
Análise da Atividade antioxidante (AA)
As amostras provenientes do processo de membranas de ultrafiltração e os extratos alcalino e
enzimático nas melhores condições de extração foram submetidas à análise da sua capacidade
antioxidante. A extração dos compostos com esta propriedade foi realizada segundo a metodologia
descrita por Rufino et al. (2007), onde se juntaram 10 mL de Metanol 10% (Impex, 99,8%) a 3 mL de
amostra seguida da extração com acetona a 70% (M,Tedia, 99,5%) do resíduo formado na extração
anterior. A quantificação dos compostos com capacidade antioxidante previamente extraídos realizou-
se segundo o método espectrofotométrico descrito por Re et al. (1999), utilizando o radical 2,29-
azinobis-(3-ethylbenzothiazoline-6-sulfonicacid) (ABTS•+), Etanol 95% e Trolox como padrão (vide
8.G). A leitura foi feita a 734nm, sendo que o radical se manteve na faixa de trabalho de 0,680 –
0,720 A, para garantir a estabilidade do mesmo e, consequentemente, a fiabilidade do método. Esta
análise foi realizada no Laboratório de Propriedade Físicas da planta piloto de operações unitárias da
Embrapa Agroindústria de Alimentos. Estas análises foram realizadas em triplicado para garantir e
demostrar a consistência dos resultados.
Fenólicos totais (FT)
Os fenólicos totais foram determinados nas amostras de permeado e concentrado da
ultrafiltração, bem como no extrato alcalino obtido nas melhores condições de extração determinadas,
no universo de variáveis estudado, no Laboratório de Propriedade físicas da planta piloto de
operações unitárias da Embrapa Agroindústria de Alimentos. Esta análise seguiu o método
espectrofotométrico proposto por Singleton e Rossi (1965) e modificado por Georgé et al. (2005),
onde se utiliza como padrão o ácido gálico e uma solução de Follin-Ciocalteau a 10% com carbonato
de sódio a 7,5% para a reação colorimétrica (vide Anexo 8.H). Estas análises foram realizadas em
triplicado para garantir e demostrar a consistência dos resultados.
Antes desta análise, as amostras foram submetidas a uma extração prévia com Acetona 70%,
dos compostos a analisar, sendo esta análise baseada na metodologia descrita por Parry et al.
(2005).
3.3.5. Métodos Cromatográficos
Todos os métodos cromatográficos apresentados neste trabalho foram realizados no
Departamento de Bioquímica e Biologia Molecular da UFPR pela Dra. Fernanda Simas Tosin, sob a
coordenação do Dr. Marcello Iacomini que gentilmente se predispuseram a fazê-los.

30
Cromatografia liquido-gasosa acoplada à espectrometria de massa (GC-MS)
Depois da hidrólise ácida total, redução e acetilação os monossacarídeos transformados a
alditóis de acetato foram quantificados por cromatografia liquido-gasosa (Cromatógrafo da Varian,
modelo 3300) acoplada à espectrometria de massa (espectrómetro de massa da Finnigan Mat,
modelo ITD 800). Utilizaram-se as colunas DB-225 de dimensões 30 m x 0,25 mm de diâmetro
interno e/ou DB-210. Utilizou-se Hélio ultra puro como gás de arraste, com um fluxo de 1,0 mol/min,
sendo que as amostras foram injetadas a uma temperatura de 50ºC, programada para aumentar
40ºC/min, até atingir a temperatura constante de 220ºC.
Cromatografia de exclusão estérica (HPSEC)
Com o objetivo de estudar o grau de homogeneidade das amostras provenientes do processo
de ultrafiltração e dos extratos alcalino e enzimático nas melhores condições determinadas,
realizaram-se análises de cromatografia de exclusão estérica de alta pressão (HPSEC).
As amostras foram solubilizadas em água Milli Q ou em uma solução de nitrito de sódio
(NaNO2) 0,1 mol/L com 200 ppm de Azida de Sódio (NaN3). As amostras foram filtradas em
membranas de acetato de celulose com porosidade de 0,22 m e posteriormente injetada no
cromatógrafo Waters equipado com detetor de índice de refração diferencial (modelo Waters 2410) e
de dispersão de luz em multiângulos (Wayatt Technology, modelo Dawn-DSP-F) e 4 colunas de gel
permeação em série, com limite de exclusão de 1x106,4x10
5 8x10
4 e 5x10
3 Da. Como eluente utilizou-
se o NaNO2 0,1 mol/L em NaN3, com fluxo de 0,6 mL/min.
3.3.6. Análises Espectroscópicas
As análises Espectroscópicas foram também realizadas no Departamento de Bioquímica e
Biologia Molecular da UFPR pela Dra. Fernanda Simas Tosin, sob a coordenação do Dr. Marcello
Iacomini.
13C - RMN
Foram obtidos espectros Ressonância Magnética Nuclear (RMN), compreendidos por análises
de carbono 13 (13
C) para as amostras de concentrado do processo de ultrafiltração e extrato alcalino
obtido na melhor condição de extração realizada, com o auxílio do espectrómetro Bruker, modelo
Avance-Drx-400. As análises foram realizadas em D2O, a 50-70ºC.
HSQC
A amostra de concentrado obtida após processos de separação com membrana de
ultrafiltração foi analisada por HSQC (Heteronuclear Single Quantum Correlation Spectroscopy), uma
técnica bidimensional de RMN, onde foram correlacionados os núcleos de 1H e
13C.

31
3.3.7. Métodos Estatísticos
Os dados obtidos a partir do planeamento fatorial composto central (vide tabelas 3 e 4),
adotado para seleção das variáveis de extração, foram avaliados pelo método de superfície de
resposta por meio do software Statistica 8.0.
Tabela 3 – Variáveis independentes e seus níveis
Níveis Ta (ºC) CE
a (µL/18g s.seco) R
a tt
a (min)
1 (-) 70 350 1:2 0
2 (p.c.) 80 700 1:3 3
3 (+) 90 1050 1:4 6 a T - Temperatura; CE - Concentração da solução aquosa de Celulase (atividade de FPase de 2,65 UI/mL); R - Relação substrato/solvente; tt - tempo de trituração do bagaço; p.c. – ponto central
Tabela 4 – Planeamento fatorial composto central1
Ensaios Ta,b
CEa,c
Ra,b,c
tta,b,c
1 T1 (-) C1 (-) R1 (-) tt1 (-)
2 T1 (-) C1 (-) R1 (-) tt3 (+)
3 T1 (-) C1 (-) R3 (+) tt1 (-)
4 T1 (-) C1 (-) R3 (+) tt3 (+)
5 T3 (+) C3 (+) R1 (-) tt1 (-)
6 T3 (+) C3 (+) R1 (-) tt3 (+)
7 T3 (+) C3 (+) R3 (+) tt1 (-)
8 T3 (+) C3 (+) R3 (+) tt3 (+)
9 T2 (p.c.) C2 (p.c.) R2 (p.c.) tt2 (p.c.)
10 T2 (p.c.) C2 (p.c.) R2 (p.c.) tt2 (p.c.)
11 T2 (p.c.) C2 (p.c.) R2 (p.c.) tt2 (p.c.) a T - Temperatura; CE - Concentração da solução aquosa de Celulase; R -
Relação substrato/solvente; tt - tempo de trituração do bagaço; p.c. – ponto central;
b Variável utilizada para os ensaios com os solventes água e solução
aquosa de KOH; c Variável utilizada para os ensaios conduzido com a solução
aquosa de Celulase.
Os ensaios foram realizados para todos os solventes (água, solução aquosa de KOH e solução
aquosa de Celulase) sendo que para o caso do processo conduzido com a solução aquosa de
Celulase, como já referido anteriormente, a temperatura foi mantida a 50ºC para garantir a atividade
máxima do enzima, sendo que a variável T foi substituída pela variável CE, nestes ensaios.
Através deste software foi também possível ajustar a 2ª Lei de Fick a uma função não linear,
para o estudo da cinética das extrações.
Como variável resposta deste planeamento fatorial, tanto para os dados cinéticos como para os
dados de extração a três variáveis, utilizaram-se os resultados da DAT realizadas para todas as
amostras, seguindo o procedimento já descrito neste capítulo. Os gráficos de pareto e de superfície
de resposta, com um nível de significância de 90%, foram usados para a discussão dos resultados.
1 Neste capítulo e nos capítulos subsequentes as amostras vão ser denominadas através da numeração
desta tabela fazendo-se anteceder de H, E ou K, quando se faz referência às amostras aquosas, enzimáticas ou alcalinas, respetivamente.

32

33
4. Resultados e Discussão
Neste capítulo faz-se referência aos resultados obtidos através dos procedimentos
experimentais já descritos no capítulo anterior. Discutem-se também esses resultados, tendo como
base uma análise crítica à viabilidade de uma possível aplicação industrial.
Para facilitar a leitura dos resultados e interpretação dos mesmos, o presente capítulo está,
organizado em 3 partes distintas. Em primeiro lugar são apresentados e discutidos os resultados
referentes aos ensaios de extração das fibras solúveis, com os respetivos resultados da DAT. A
segunda parte deste capítulo diz respeito aos ensaios de microfiltração e ultrafiltração do extrato
aquoso nas condições otimizadas de extração, com apresentação dos resultados da DAT realizada.
Por fim, apresentam-se e discutem-se características estruturais das fibras obtidas, bem como a
presença de grupos fenólicos. Com o intuito de se estabelecer uma relação estrutural destas
moléculas com uma possível atividade biológica, esta última parte também apresenta os resultados
da avaliação da atividade antioxidante in vitro do material em estudo.
4.1. Extração sólido-líquido
Como já referido no capítulo 3, foram realizadas extrações em triplicado em diferentes
condições de temperatura (T), tempo de trituração do bagaço (tt) (vide Anexo 8.A), relação
substrato/solvente (R), tipo de solvente utilizado e concentração das soluções aquosas de Celulase e
KOH (CE e CK respetivamente). Depois de realizados esses ensaios, procedeu-se ao estudo da
cinética da extração, nas melhores condições consideradas, ajustando-se os resultados à 2ª Lei de
Fick (equação 5 do capitulo 2.3). Neste subcapítulo serão apresentados os resultados obtidos com as
respetivas discussões referentes a estas duas temáticas.
Para a realização de todos estes ensaios de extração, utilizaram-se 200 g de solvente para as
amostras com R1 e R3 e 150 g de solvente quando se estudou a variável no ponto central, R2. A
massa de bagaço utilizada foi determinada com base na relação substrato/solvente que cada amostra
representa.
Para se chegar a um resultado passível de ser comparado com os demais, todos os valores de
massa de açúcar obtidos por DAT para cada amostra foram normalizados para uma base de 100g de
bagaço de uva.
É importante notar que a existência de possíveis variações nos valores de massa de açúcar
que não sejam esperadas podem ser justificadas pelo facto de as amostras de bagaço não
apresentarem uma composição homogénea (figura 15 do capítulo. 3.1).

34
4.1.1. Determinação da condição ótima de extração
Para os ensaios de extração realizados, foi seguro considerar a massa especifica dos extratos
enzimáticos e aquosos como igual à massa especifica da água a 25ºC2 e do extratos alcalinos como
a densidade da solução de KOH a 2%, 6% e 10%3. Isto foi possível dado que a diferença dos
referidos valores tabelados para os valores obtidos por medição das densidades num densímetro
PAAR DMA 46 a 22ºC4 não foi significativa que justificasse a medição da densidade de todos os
extratos. Na tabela 5, apresentam-se os valores de densidade medidos para uma solução de extrato
aquoso, uma solução de extrato alcalino e uma solução de extrato enzimático, bem como a massa
específica que será considerada doravante neste trabalho.
Tabela 5 – Densidades e massas específicas das amostras
Amostrasa Densidade
medida (22ºC)
Densidade
relativa
Massa específica considerada
(g/L) (Perry, 1999)
Celulase 0,9999 1,0017 997,045
Água 1,0008 1,0026 997,045
KOH (2%) 1,0304 1,0322 1017,500 a Mediram-se as densidades dos extratos mais fibrosos, os extratos cinéticos correspondentes ao tempo 5h.
No Anexo 8.C, apresentam-se as curvas padrão e as absorvâncias lidas para todos os extratos
obtidos através das extrações com e sem efeitos cinéticos, a partir das quais se pôde chegar aos
resultados que se apresentarão ao longo deste subcapítulo.
Extração sem efeitos cinéticos
Na tabela 6 são apresentados os resultados em massa de açúcar referentes aos ensaios de
extração, sem efeitos cinéticos, realizados com o solvente água.
Tabela 6 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Extrato aquoso
Amostras Massa de Açúcar (g/100 g bagaço)
H1 0,457±0,0142
H2 0,497±0,0047
H3 0,568±0,0314
H4 0,426±0,0398
H5 0,681±0,0193
H6 0,524±0,0238
H7 0,904±0,0453
H8 0,822±0,0284
H9 0,780±0,0187
H10 0,752±0,0105
H11 0,802±0,0145
2 Densidade da água: (25ºC) = 0,997045 (Perry, 1999)
3 Densidade da solução de KOH: 2%=1,0175; 6%=1,0544; 10%=1,0918 (Perry, 1999).
4 Densidade da água: (22ºC) = 0,99770 (Perry, 1999).

35
Analisando os resultados, pode-se afirmar que a amostra H7 (obtida a 90ºC, relação
substrato/solvente de 1:4 e bagaço in natura) é aquela que apresenta uma maior quantidade de
açúcares em solução, determinada pelo método fenol-ácido sulfúrico (Dubois et al., 1956) descrito no
Capitulo 3.3.4, e consequentemente uma maior quantidade de fibras solúveis.
Esta observação foi também suportada pelos resultados obtidos através do software Portable
Statistica 8. Com o auxílio do gráfico de pareto, (figura 20) e dos gráficos de superfície de resposta
em anexo (vide Anexo 8.D) é possível avaliar quais das variáveis estudadas são estatisticamente
significativas para o cumprimento do objetivo de extração das fibras solúveis.
Figura 20 – Gráfico de pareto obtido através dos resultados dos extratos obtidos com água pura
Verifica-se, por análise gráfica, que a eficiência de extração de fibras solúveis é mais sensível à
variação na temperatura, sendo que a massa de açúcar no extrato apresentou uma relação
diretamente proporcional a essa variável, como se pode observar nas figuras 55 e 57 do Anexo 8.D.
Nos níveis analisados, as variáveis relação substrato/solvente e tempo de trituração não mostraram
ser relevantes estatísticamente para o estudo.
No que respeita a variável temperatura, estes resultados já tinham sido observados em
trabalhos como o de Graham e colaboradores em 1988 e Geng et al. (2006), que verificou que
temperaturas mais altas favorecem o rendimento de extração das fibras com o solvente água.
Segundo Geng e seus colaboradores (2006) uma explicação para o aumento do rendimento com a
temperatura é o aumento da quebra das pontes de hidrogénio e níveis mais elevados de
hemiceluloses dissolvidas. A termodinâmica também justifica a influência do aumento de temperatura
no rendimento de extração de fibras solúveis. De acordo com Silbey, Alberty e Bawendi (2005) o
fenómeno de difusão descrito por Fick ocorre como resultado de um gradiente no potencial químico,
µ. Este potencial químico é a energia livre de um elemento numa solução estando portanto,
relacionado com a temperatura. Sabendo que um aumento de temperatura faz aumentar a energia de
um sistema e uma vez que este potencial químico é, para o fenómeno de difusão, a difusividade
pode-se então afirmar que um aumento de temperatura faz aumentar a difusão de um componente no
Pareto Chart of Standardized Effects; Variable: Massa de Açúcar (g/100g bagaço)
-,281376
-,363479
-,899204
1,275795
1,482764
2,59921
p=,1
Standardized Effect Estimate (Absolute Value)
2by3
1by3
(3)tt
1by2
(2)R
(1)T
-,281376
-,363479
-,899204

36
meio fluido (equação 3 do capitulo 2.3). Consequentemente, um aumento de temperatura faz com
que se obtenha uma maior quantidade de polissacarídeos em solução.
Relativamente à relação substrato/solvente espera-se que uma maior quantidade de água em
relação à quantidade de substrato favoreça a extração dos açúcares. Este resultado ocorre pelo facto
das moléculas de água, potencializadas pelo efeito da temperatura, serem responsáveis pelo
enfraquecimento da ultraestrutura molecular da parede celular, promovendo a libertação dos
polissacarídeos menos aderidos para o meio extrator. Assim, quanto maior a quantidade do solvente,
maior a interação entre este e o bagaço da uva e, consequentemente, maior a quantidade de fibras
(polissacarídeos representados pela quantidade de açúcar) solubilizadas. Entretanto, existe um limite
máximo de solvente que deve ser alimentado no extrator, e este valor está relacionado com a
solubilidade do soluto que se deseja extrair. O resultado obtido neste trabalho indicou que, na faixa
avaliada, a quantidade de solvente não foi o fator limitante.
Por último tem-se a variável tempo de trituração. Os resultados referentes a esta variável não
foram os esperados, na medida em que esta variável não apresentou significância estatística para a
extração das fibras solúveis. De acordo com a 2ª Lei de Fick (equação 2 do capitulo 2.3), a espessura
do material é uma das variáveis relevantes à transferência de massa, sendo que quanto menor a
espessura, menor será o tempo de difusão. No entanto, estes resultados podem ter duas possíveis
justificativas: o processo de trituração não foi eficiente para reduzir a espessura do bagaço ou o facto
desta variável ser, comparativamente, menos relevante que as demais. Este resultado mostra-se
vantajoso em contexto industrial, na medida em que menores custos estarão associados na etapa de
pré-tratamento do bagaço, tanto os custos de investimento como os custos de produção.
Desta forma, considerou-se que, para o solvente água e no universo das variáveis estudadas,
as condições operacionais selecionadas para a extração de fibras solúveis foram: 90ºC, relação
substrato/solvente de 1:4 e bagaço nas dimensões típicas do resíduo industrial.
No que respeita os ensaios efetuados com a solução aquosa contendo o enzima Celulase,
obtiveram-se os resultados apresentados na tabela 7, relativos à massa de açúcar determinada por
DAT nas soluções de extrato obtidas.
Tabela 7 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Extrato enzimático
Amostras Massa de Açúcar
(g/100 g bagaço)
E1 0,604±0,0165
E2 0,595±0,0168
E3 0,724±0,0100
E4 0,757±0,0142
E5 0,610±0,0128
E6 0,600±0,0149
E7 0,719±0,0137
E8 0,704±0,0331
E9 0,669±0,0076
E10 0,624±0,0283
E11 0,656±0,0383

37
O maior valor de massa de açúcar obtido nestes ensaios corresponde à amostra E4, que foi
obtida a partir do bagaço triturado por 6 minutos, concentração de 350 µL de Celulase /18 g de
bagaço e relação bagaço/solvente de 1:4.
Analisando o gráfico de pareto da figura 21 e os gráficos de superfície de resposta referentes a
estes ensaios (vide Anexo 8.D) observa-se que a massa de soluto no extrato depende fortemente da
relação substrato/solvente.
Figura 21 – Gráfico de pareto obtido através dos resultados dos extratos enzimáticos
Dos gráficos de superfície de resposta das figuras 58 e 59 do anexo 8.D, confirma-se que a
relação substrato/solvente apresentou um efeito significativo na quantidade de açúcar extraído. Tais
resultados já tinham sido observados por Mariano, et al. (2009) e Santos e Ferrari (2005). As
variáveis concentração de Celulase, CE em relação ao bagaço seco e tempo de trituração não
apresentaram ter um papel importante na extração de fibras solúveis, como se pode observar gráfico
de pareto da figura 21 e na curva de superfície de resposta da figura 60 do anexo 8.D. Relativamente
à variável tempo de trituração, observa-se, novamente, uma incoerência nos resultados
comparativamente com aquilo que era esperado, sendo que as possíveis justificações para este facto
já foram discutidas aquando da análise dos resultados da extração aquosa. Quanto à variável
concentração de Celulase, CE, os resultados indiciam que a adição deste enzima no meio aquoso não
favoreceu a extração de fibras. Isto confirma a importância da temperatura na extração destes
materiais. Como os ensaios com a solução enzimática foram conduzidos a 50ºC, a esta temperatura,
a força motriz não é tão elevada quanto quando se opera com temperaturas mais elevadas, como foi
constatado nos resultados para a extração com água.
Neste sentido, considerou-se que, para o universo de variáveis estudado, as melhores
condições de extração com a solução aquosa contendo Celulase foram: relação substrato/solvente de
1:4, menor concentração do enzima (350µL/18g de bagaço seco) e bagaço in natura. Estes dados
correspondem à amostra E3, que apresentou quantidade de açúcar próxima daquela observada para
E4.
Pareto Chart of Standardized Effects; Variable: Massa de Açúcar (g/100g bagaço)
-,006265
,6010979
-,791144
-,815242
-1,15195
8,26097
p=,1
Standardized Effect Estimate (Absolute Value)
(3)tt
2by3
(1)CE
1by3
1by2
(2)R
,6010979
-,791144
-,815242
-1,15195

38
Por último, estudou-se o solvente hidróxido de potássio (KOH). Nestes ensaios, além da
temperatura, tempo de trituração e relação substrato/solvente, foi também estudada a variável
concentração, CK. Na tabela 8 estão apresentados os resultados em massa de açúcar, referentes às
3 concentrações de KOH estudadas.
Tabela 8 – Massa de açúcar obtida para os ensaios sem efeitos cinéticos: Solução aquosa de KOH
Amostras
Massa de Açúcar
(g/100 g bagaço)
KOH 2% KOH 6% KOH 10%
K1 1,84±0,003 1,46±0,005 1,31±0,024
K2 1,72±0,047 1,45±0,038 1,32±0,021
K3 2,04±0,060 1,91±0,033 1,71±0,055
K4 1,82±0,011 2,39±0,010 1,79±0,059
K5 1,81±0,018 1,75±0,025 1,53±0,025
K6 1,65±0,030 1,99±0,021 1,75±0,004
K7 2,16±0,012 2,58±0,038 1,96±0,019
K8 2,01±0,028 2,41±0,052 2,12±0,042
K9 2,31±0,088a 2,25±0,066 1,69±0,022
K10 1,92±0,052 1,88±0,040 1,11±0,019
K11 1,79±0,030 1,79±0,024 1,92±0,035 a Este valor não foi contabilizado, uma vez que, da análise em triplicado que foi
realizada ao ponto central, apresenta um grande desvio à média.
Observando a tabela 8, pode-se inferir que os maiores valores de massa de açúcar foram
obtidos nos ensaios K7, a 2 e 6% de KOH, e K8 a 6% de KOH (p<0,05), com K8 a apresentar como
diferença nas variáveis utilizadas em K/, o tempo de trituração. Adicionalmente, o maior rendimento
foi alcançado quando se usou a solução aquosa de KOH a 6%.
Analisando e comparando os gráficos de pareto (vide figuras 22, 23, 24) e os gráficos de
superfície de resposta do Anexo 8.D. para os 3 tipos de ensaios realizados, constata-se que
estatisticamente, a sensibilidade dos resultados para as variáveis estudadas é mais complexa.

39
Figura 22 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 2%
Figura 23 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 6%
Figura 24 – Gráfico de pareto obtido através dos resultados dos extratos Alcalinos a 10%
Pareto Chart of Standardized Effects; Variable: Massa de Açúcar (g/100g bagaço)
,060592
-,126925
,3311355
,6940912
-1,06987
1,673196
p=,1
Standardized Effect Estimate (Absolute Value)
1by3
2by3
(1)T
1by2
(3)tt
(2)R
Pareto Chart of Standardized Effects; Variable: Massa de Açúcar (g/100g bagaço)
,1320203
-,228146
-,584526
,8164453
2,284124
3,984748
p=,1
Standardized Effect Estimate (Absolute Value)
2by3
1by2
1by3
(3)tt
(1)T
(2)R
-,584526
,8164453
Pareto Chart of Standardized Effects; Variable: Massa de Açúcar (g/100g bagaço)
,0187046
-,09746
,3254964
,532317
1,411472
1,931362
p=,1
Standardized Effect Estimate (Absolute Value)
2by3
1by2
1by3
(3)tt
(1)T
(2)R

40
Relativamente à variável com maior importância estatística, esta manteve-se concordante em
todos os ensaios nas 3 concentrações estudadas, sendo a massa de açúcar mais sensível à
quantidade de solvente adicionada na etapa de extração, R. A importância desta variável era
esperada, na medida em que uma maior relação solvente/substrato promove uma maior interação
entre a solução alcalina e o bagaço da uva, tal como já foi justificado para as extrações anteriores.
Neste contexto e relativamente à concentração de KOH em solução aquosa, seria de esperar
que houvesse um aumento da massa de açúcar em solução, com o aumento da concentração deste,
tal como foi apresentado por Sun et al. (2001). Contudo, e à semelhança do que se observa nos
resultados de Mellinger et al. (2005a), não existe uma relação direta entre o aumento da
concentração de açúcares no meio com o aumento da concentração de KOH. Neste caso, após ter
sido alcançada a condição de saturação (entre 6% e 10%) o aumento da concentração da solução
alcalina não favoreceu o rendimento da extração do soluto pretendido.
No caso dos ensaios para a concentração de KOH de 2% e 10%, a variável temperatura
demonstrou ser ter pouca importância estatística para a extração de açúcar do bagaço da uva em
estudo. Todavia, para os ensaios de KOH a 6%, e de forma análoga à extração aquosa e enzimática,
a massa de açúcar extraída foi sensível ao aumento da temperatura, bem como à relação solvente
substrato, como abordado acima nesta discussão. Neste caso, o aumento da temperatura e da
relação substrato/solvente promoveu uma maior interação entre a solução alcalina e o bagaço da
uva, principalmente, ao maior potencial de transporte do soluto para o meio aquoso.
A variável tempo de trituração do bagaço não foi significativa para as diferentes concentrações
de KOH estudadas (p<0,10), como pode ser observado nas figuras 22, 23 e 24. Assim, constata-se,
portanto, que, à semelhança dos resultados obtidos para os outros solventes, esta etapa de pré-
tratamento pode ser dispensada.
Os trabalhos reportados na literatura não se mostraram conclusivos no que respeita a
temperatura de extração. Alguns autores utilizam temperaturas mais amenas, 50ºC e 60ºC, para a
extração de fibras (Peng et al, 2009 e Aoe et al, 1993), respetivamente e outros utilizam temperaturas
elevadas, 100ºC e 120ºC, respetivamente (Mellinger et al, 2005 e Ju et al, 2011). Neste contexto, tal
como se considerou para os dois solventes já discutidos anteriormente e pela importância que esta
variável assumiu para a extração com KOH a 6%, para este trabalho selecionou-se a temperatura
mais elevada (90ºC), como sendo a melhor para a extração de fibras solúveis em solução aquosa de
KOH.
Entretanto, uma análise critica aos resultados acima, permite concluir que, em maior escala
pode ser mais seguro utilizar uma solução de KOH 2% em vez de KOH 6% para a extração de fibras
solúveis. Apesar da amostra K7 para a solução de KOH 6% apresentar uma massa cerca de 20%
superior à mesma amostra obtida com a solução de KOH 2%, o manuseamento de concentrações
mais elevadas desta base acarreta riscos operacionais, que teriam que ser salvaguardados e com
isto, mais custos operacionais estariam associados. Outra questão que favorece o uso da solução de
KOH 2% em detrimento da solução de KOH 6% é a necessidade de neutralização dos extratos após
extração. Em contexto industrial, isto implicaria um gasto acrescido não só com o ácido neutralizante

41
como novamente com as medidas de segurança necessárias à utilização do mesmo, dada a sua
natureza corrosiva e inflamável.
Assim, para prosseguir com o estudo, assumiu-se que as condições mais apropriadas para a
extração de fibras solúveis com a solução aquosa de KOH, e salvaguardando eventuais erros de
análise, seriam: concentração de 2%, temperatura de 90ºC, relação substrato/solvente de 1:4 e
bagaço sem pré-tratamento, o que remete à mostra K7 do planeamento apresentado (tabelas 3 e 4).
Cinética das Extrações
A partir das condições selecionadas foi possível estudar a cinética das extrações para cada um
dos solventes. Os resultados referentes à cinética da extração aquosa das fibras solúveis do bagaço
da uva branca estão apresentados no gráfico da figura 25.
Figura 25 – Cinética em meio Aquoso
Pode verificar-se na figura 25 um ligeiro aumento na massa de açúcar presente em solução de
extrato ao longo do tempo, observando-se que até aos 90 minutos de extração é onde se obtém o
maior aumento gradiente (aumento de 22% 13,5% da massa em relação ao tempo 30 e 60 minutos,
respetivamente).
A cinética da extração enzimática está ilustrada na figura 26.
0,0
0,2
0,4
0,6
0,8
1,0
0 30 60 90 120 150 180 210 240 270 300
Mas
sa A
çúca
r (g
/10
0g
bag
aço
)
Tempo (min)
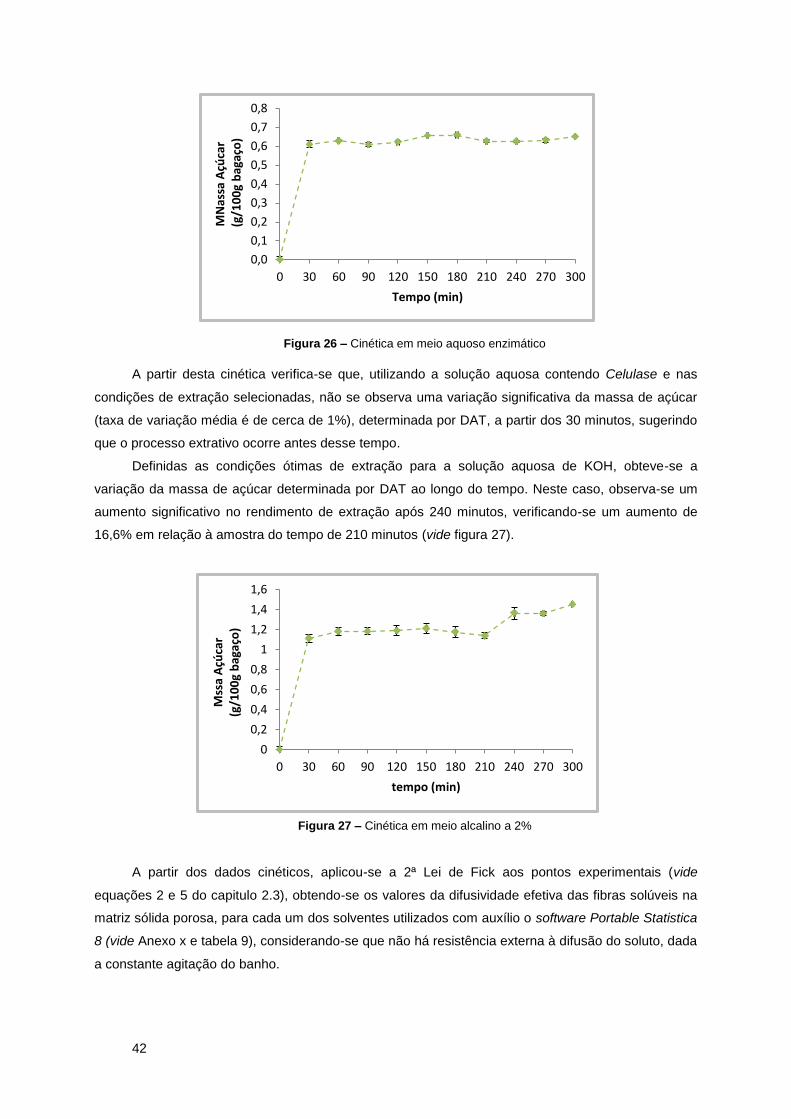
42
Figura 26 – Cinética em meio aquoso enzimático
A partir desta cinética verifica-se que, utilizando a solução aquosa contendo Celulase e nas
condições de extração selecionadas, não se observa uma variação significativa da massa de açúcar
(taxa de variação média é de cerca de 1%), determinada por DAT, a partir dos 30 minutos, sugerindo
que o processo extrativo ocorre antes desse tempo.
Definidas as condições ótimas de extração para a solução aquosa de KOH, obteve-se a
variação da massa de açúcar determinada por DAT ao longo do tempo. Neste caso, observa-se um
aumento significativo no rendimento de extração após 240 minutos, verificando-se um aumento de
16,6% em relação à amostra do tempo de 210 minutos (vide figura 27).
Figura 27 – Cinética em meio alcalino a 2%
A partir dos dados cinéticos, aplicou-se a 2ª Lei de Fick aos pontos experimentais (vide
equações 2 e 5 do capitulo 2.3), obtendo-se os valores da difusividade efetiva das fibras solúveis na
matriz sólida porosa, para cada um dos solventes utilizados com auxílio o software Portable Statistica
8 (vide Anexo x e tabela 9), considerando-se que não há resistência externa à difusão do soluto, dada
a constante agitação do banho.
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0 30 60 90 120 150 180 210 240 270 300
MN
assa
Açú
car
(g/
10
0g
bag
aço
)
Tempo (min)
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
0 30 60 90 120 150 180 210 240 270 300
Mss
a A
çúca
r
(g/1
00
g b
agaç
o)
tempo (min)

43
Tabela 9 – Coeficiente de difusão para cada solvente
Solventes D (m2/s)
Água (1,50±0,167) x10-15
Solução aquosa com Celulase (3,96±0,844) x10-15
Solução de KOH 2% (1,60±0,297) x10-15
Analisando os valores obtidos, verifica-se que os coeficientes de difusão para as extrações
com água e a solução aquosa de KOH a 2% não variam estatisticamente. Constata-se também, que
a extração com a solução aquosa de Celulase foi aquela que apresentou um valor de coeficiente de
difusão superior, cerca de 2,5 vezes maior que os demais.
Representando o coeficiente de difusão a taxa de transporte de massa observada em cada
processo, e confrontando estes resultados com os resultados das massas de açúcar extraídas por
cada solvente, verifica-se que, apesar de se obter melhores rendimentos para a água e para a
solução aquosa de KOH, é para a solução aquosa de Celulase que se observa uma taxa de
transporte de soluto relativa superior, alcançando o equilíbrio mais rapidamente, como se observou
na figura 26.
4.1.2. Comparação do processo para os três solventes estudados
Ao longo de todo o processo de extração realizado e da análise de resultados efetuada,
verificaram-se algumas similaridades e algumas diferenças entre os extratos obtidos para cada um
dos solventes estudados. Assim, considerou-se importante nesta fase do trabalho, comparar e
documentar aquilo que foi verificado.
As diferenças visuais foram aquelas que logo à partida foram verificadas, aquando da
realização dos ensaios experimentais (vide figuras 28, 29 e 30).
Figura 28 – Fotografia comparativa das soluções aquosa (A) e alcalina (B) antes da extração
Figura 29 – Figura comparativa dos extratos aquoso (A) e alcalino (B)
A B
A B

44
Figura 30 – Figura comparativa dos resíduos das extrações aquosa (A) e alcalina (B)
Como se pode observar pelas figuras, os extratos e resíduos alcalinos apresentaram uma cor e
uma consistência distintas daquilo que se observa para os extratos e resíduos aquosos5. Estas
características observadas para cada uma das soluções é evidente assim que se misturam o bagaço
com o solvente em análise como mostra a figura 28. No que respeita os extratos (vide figura 29), as
amostras provenientes da extração alcalina apresentaram uma cor avermelhada escura e uma maior
viscosidade em comparação com as soluções aquosas. Já os resíduos dos dois tipos de extração em
discussão apresentaram diferenças novamente a nível de cor bem como a nível de consistência do
material. Observou-se que o resíduo alcalino apresentou uma consistência mais pastosa, quando
comparado com o resíduo originado a partir da extração aquosa (vide figura 30).
A manifestação de cor por parte dos extratos alcalinos, já vinha sido observada por Aoe e seus
colaboradores (1993), que concluíram ser uma característica desvantajosa para aplicações
alimentares, devido à necessidade de clarificação (bleaching) dos extratos. Relativamente à
viscosidade e consistência dos extratos e resíduos, respetivamente, da extração alcalina, estas
podem ser justificadas pela capacidade alcalina de quebrar ligações de lignina e de lignina com
polissacarídeos (celulose e hemicelulose) (Mellinger et al., 2005a; Sun et al., 2001; Peng et al., 2009),
bem como de romper a parede celular por hidrólise de ésteres urónicos e acéticos, e por inchaço
(swelling) da celulose, com a consequente diminuição da cristalinidade da mesma (Peng, et al.,
2009). Sendo que estas macromoléculas conferem rigidez, impermeabilidade e resistência aos
tecidos vegetais, a quebra das ligações das mesmas como consequência das forças polímero-
solvente serem superiores, provoca então o enfraquecimento da estrutura vegetal da qual fazem
parte.
Outro tema com interesse para uma discussão comparativa, é a massa de açúcar extraída por
cada um dos solventes estudados, como apresentado na figura 31.
5 Não se apresentam as imagens referentes ao extrato enzimáticos porque são em tudo semelhantes às
do extrato aquoso.
A B

45
Figura 31 – Comparação dos valores máximos obtidos para a massa de açúcar extraída por cada solvente
estudado
Constata-se que a massa de açúcar extraída nas condições de extração selecionadas para o
solvente alcalino é 2,4 vezes superior à massa extraída quando a água pura é o solvente e quase 3
vezes superior quando se utiliza a solução aquosa de Celulase no processo de extração. Esta grande
diferença de rendimentos entre o solvente alcalino e os restantes, já verificada por outros autores
como Geng et al. (2006), é justificada pela intensidade das forças aquosa e alcalina. Sabe-se que a
libertação de hemicelulose da parede celular vegetal está relacionada com a ligação da lignina-
hemicelulose bem como com as fortes pontes de hidrogénio existentes entre polissacarídeos e outros
componentes, que dificultam a libertação deste material (Peng et al., 2009). Assim, e de acordo com
os resultados, contata-se que enquanto a força alcalina rompe com a estrutura da parede celular do
extrato vegetal, a força exercida pela água apenas enfraquece essa estrutura (Sun et al., 2001;
Mellinger et al., 2005a; Ju et al., 2011).
Outra questão que se considerou interessante discutir e comparar, foram os resultados dos
solventes água pura e solução aquosa de enzima (vide figura 32).
Solvente/Amostra 1 2 3 4 5 6 7 8 p.c.
Água 70ºC 90ºC 80ºC
Celulase 50ºC 50ºC 50ºC
Figura 32 – Representação gráfica comparativa da massa de açúcar extraída sem efeitos cinéticos pelos
solventes água e solução aquosa de Celulase
0,904
0,757
2,156
Água
Cellulase
KOH 2%
Solv
en
te
Massa Máxima de Açúcar (g/100g bagaço)
0,0
0,2
0,4
0,6
0,8
1,0
1 2 3 4 5 6 7 8 p.c.
Mas
sa d
e A
çuca
r (g
/10
0g
bag
aço
)
Amostra
Água
Cellulase

46
Uma vez que, como se verificou anteriormente neste capítulo, a concentração de enzima se
mostrou pouco significativa estatisticamente para a extração de fibras do bagaço da uva, é possível
comparar cada uma das amostras extraídas através desse solvente com as da extração aquosa, por
se tratar de extratos obtidos em condições semelhantes (vide figura 32). Dessa forma, verifica-se por
análise gráfica da figura 32 que para as temperaturas de 80 e 90ºC (amostras 5 a p.c.), a água é o
solvente que atinge um maior rendimento de extração de fibras, quando comparado com a solução
aquosa de enzima, verificando-se o inverso para temperaturas mais baixas (amostras 1 a 4). Estas
observações mostram que para temperaturas baixas a presença de Celulase na solução de solvente
pode ser vantajosa (amostras 1 a 4), contudo, com o aumento de temperatura, esta ultima variável
ganha maior importância face ao tipo de solvente utilizado. Isto vem reforçar a ideia apresentada
neste capítulo de que a temperatura é uma variável importante neste processo de extração. No
entanto, se for necessário operar com temperaturas brandas, o enzima pode auxiliar na remoção de
fibras do bagaço.
Para completar esta análise comparativa dos solventes água pura e solução enzimática, e
ainda no tema da variável temperatura, construiu-se um gráfico comparativo das cinéticas de cada
um dos solventes estudados (figura 33).
Figura 33 – Representação gráfica comparativa das cinéticas dos extratos para os 3 solventes em estudo
Por análise da figura 33 e como complemento às observações realizadas anteriormente neste
trabalho, considerou-se seguro afirmar que, a adição de Celulase ao solvente se mostrou pouco
significativa. Isto ocorreu, provavelmente, na medida em que a temperatura de atividade máxima do
enzima é baixa para uma extração eficiente de fibras solúveis. Comparando os resultados cinéticos
da água pura e da solução aquosa de Celulase a 50ºC, verifica-se que aos 30 minutos de extração,
ambos os solventes apresentam massa de açúcar equivalente em solução. No entanto, acima dos 30
minutos verificou-se um aumento da massa de açúcar extraída pela água pura a 90ºC, não se
observando o mesmo para a solução enzimática. Estes resultados vêm mostrar, mais uma vez, que
um aumento da temperatura promoveu num maior rendimento de extração dos materiais fibrosos do
bagaço da uva.
0,0
0,5
1,0
1,5
2,0
0 30 60 90 120 150 180 210 240 270 300
Mas
sa d
e A
çúca
r (g
/10
0g
bag
aço
)
tempo (min)
Cellulase Água KOH 2%

47
É, no entanto, importante ressalvar que estes resultados não mostram a ineficiência do enzima
Celulase enquanto extratora de materiais fibroso, mas sim a sua menor capacidade de ação em
substratos cujas ligações são fortes e requerem não só um solvente com elevada força extrativa
como também uma temperatura elevada de operação.
Ainda, com o auxílio da figura 33, pode mais uma vez constatar-se que a força alcalina é
aquela que apresenta um melhor rendimento de extração, quando comparada com as forças aquosas
e enzimáticas, nas condições de extração estudadas.
4.2. Processos de Membranas
Dos três solventes estudados no processo de extração previamente descrito e discutido,
apenas o extrato aquoso foi submetido aos processos de membranas subsequentes, dado que,
comparativamente com o extrato enzimático apresentou melhores resultados. Quando comparado
com o extrato alcalino apresentou resultados menos satisfatórios, no entanto, uma vez que trabalhar
com um solvente como o KOH acarreta riscos para a saúde, quando não são salvaguardadas as
medidas de segurança, optou-se por um solvente ao qual não estivessem associados riscos de
maior, dada a natureza académica deste trabalho. Outro motivo para, nesta fase do trabalho, não se
ter trabalho com o solvente de KOH é a necessidade de se neutralizar os extratos depois da extração
realizada a uma maior escala que as anteriormente discutidas (obtiveram-se cerca de 2,7 kg de
extrato nesta fase do trabalho), o que implicaria um gasto elevado de ácido acético e novamente o
manuseamento de uma grande quantidade de um composto corrosivo e inflamável.
Os ensaios realizados por processos de membranas foram efetuados em modo de
concentração, fazendo-se recircular o concentrado para o tanque de alimentação e recolhendo-se o
permeado. Assim, a concentração de solutos concentrados pela membrana vai aumentando ao longo
do processo.
Em cada ensaio, utilizaram-se cerca de 2,72 kg de extrato proveniente da extração aquosa
realizada a esta escala, com uma razão substrato/solvente de 1:4, a uma temperatura de ≈90ºC e
sem submeter o bagaço a qualquer tipo de processo de trituração. Realizaram-se estas extrações
para um tempo de 3 horas, tendo como base os resultados observados para a cinética de extração
aquosa. Concentraram-se as soluções até se atingir um FCV de 2.
4.2.1. Microfiltração e Ultrafiltração
A separação das fibras provenientes do processo de extração aquosa, foi estudado num
sistema de membranas de microfiltração e de ultrafiltração. Para o primeiro módulo de membranas
realizou-se apenas um ensaio, sendo que para o módulo de ultrafiltração, foram realizados dois
ensaios (apresentados como 20 kDaI e 20 kDaII), cujos resultados são apresentados ao longo deste
subcapítulo. A tecnologia de ultrafiltração (20 kDa) foi aplicada neste trabalho com o intuito de se
concentrarem as fibras solúveis em solução de extrato, já que estes polissacarídeos ficam, em geral,
retidos em membranas com este limite de exclusão. Já as membranas de microfiltração (0,1 µm, 300

48
kDa) foram usadas em carater meramente investigativo, para que fosse possível o fracionamento
destes polímeros através da massa molecular dos mesmos.
Antes de cada ensaio experimental, determinou-se a Permeabilidade Hidráulica (Lp) de cada
uma das membranas, visto ser um parâmetro importante na caracterização das mesmas. Este
parâmetro representa a capacidade de permeação da água pura da membrana e é definido como a
quantidade de fluido permeado por unidade de tempo, área de superfície e pressão aplicada. A sua
determinação experimental passou por medir o fluxo de água que foi permeada pelo módulo de
membranas em estudo (Jp), em função da pressão transmembrana (ΔP). Através da representação
linear destas variáveis com ordenada na origem nula (vide figuras 34 e 35) obteve-se a Lp a partir do
declive da mesma (equação 9).
(Equação 9)
Figura 34 – Fluxo permeado de água em função da diferença de pressão aplicada a membrana de microfiltração
Assim, pode dizer-se que a membrana de 0,1 m apresenta uma permeabilidade hidráulica de
212,14 kg/(h.m
2.bar).
Figura 35 – Fluxo permeado de água em função da diferença de pressão aplicada à membrana de
ultrafiltração
No caso da membrana de 20 kDa, correspondente à Ultrafiltração, a permeabilidade hidráulica
registada foi de 184,02 kg/(h.m2.bar) (média dos dois ensaios).
Jp = 212,14 P R² = 0,9108
0
500
1000
1500
0 1 2 3 4
Jp (
kg/h
.m2)
ΔP (bar)
Jp = 184,29 P R² = 0,9993
Jp = 183,74 P R² = 0,9995
0
1000
2000
3000
0 5 10 15
Jp (
kg/h
.m2 )
P (bar)
20 kDA I 20 kDA II

49
Antes de se passar à análise da composição das soluções de permeado e concentrado,
estudou-se a evolução do fluxo de permeação (Jp) com o aumento do fator de concentração
volumétrico (FCV), para as condições operatórias referidas, nos dois módulos de membranas com
que se trabalhou (vide figura 36).
Figura 36 – Variação do fluxo de permeação do extrato obtido com água (Jp) em função do factor de
concentração volumétrico (FCV)
Verifica-se que quando foi utilizado o módulo de membranas de 0,1 m, que requer uma
pressão mais baixa, obtém-se um maior fluxo de permeação, quando comparado com os dois
ensaios realizados com o módulo de membranas de 20 kDa. No entanto, esta tendência verificou-se
apenas e com clareza a partir de 30 minutos de processo, aquando da estabilização do fluxo de
permeação. Na fase inicial do processo, verifica-se em ambas as membranas utilizadas, um
decréscimo de fluxo ao longo do tempo, sendo que este declínio é mais acentuado para módulo de
membranas de 20 kDa (ultrafiltração). Mais concretamente observa-se um declínio do fluxo de cerca
de 46% e de 51% (média aritmética dos dois ensaios realizados) para os módulos de membranas de
0,1 m e 20 kDa, respetivamente.
Realizadas as filtrações através das membranas de microfiltração e ultrafiltração, verificou-se
que o concentrado apresentou uma coloração nitidamente mais escura que a do permeado e que a
da solução de alimentação, o extrato aquoso (vide figura 37).
0
20
40
60
80
100
120
140
1 1,2 1,4 1,6 1,8 2
J p (
kg/h
.m2 )
FCV
0,1 mm
20 kDA I
20 kDA II
T≈ 25ºC
v = 6,50 m/s
ΔP0.1µm = 2,5 bar
ΔP20kDA = 15 bar

50
Figura 37 – Fotografia comparativa das soluções de Alimentação (A), Concentrado (C) e Permeado (P) do
processo de Ultrafiltração
Esta coloração indiciou a presença de uma maior quantidade de polissacarídeos na solução
concentrada que na solução de permeado, como seria de esperar. Comparando a solução de
alimentação com a de concentrado, pode também constatar-se que, sendo mais escura, a solução de
concentrado está mais concentrada em fibras do que a solução de alimentação.
Para sustentar e complementar estes resultados visuais, realizou-se a dosagem de açúcar total
(DAT) (vide Anexo8.F) para as três soluções de ambos os sistemas de membranas utilizados
(Microfiltração e Ultrafiltração). Na tabela 10, estão apresentados os resultados dessa dosagem.
Tabela 10 – Concentração de açúcar nas soluções de alimentação, concentrado e permeado, dos processos de
membranas de Microfiltração e Ultrafiltração
Microfiltração Ultrafiltração
a
Concentração de açúcar
em solução (g/L)
Concentração de açúcar
em solução (g/L)
Alimentação 1,59±0,052 2,20±0,041
Concentrado 1,96±0,037 2,31±0,045
Permeado 1,16±0,003 1,10±0,053 a Estes resultados foram obtidos fazendo-se a média aritmética dos dois ensaios de ultrafiltração realizados;
b Estas massas foram obtidas tendo em conta a densidade da água a 25ºC [Perry, 1999].
Verifica-se então que em qualquer uma das membranas, conseguiu concentrar-se as fibras
solúveis. É possível que o permeado apresente também polissacarídeos de baixa massa molar, no
entanto, outras análises químicas são necessárias para se investigar tal conteúdo. Este resultado é
coerente já que os polissacarídeos são moléculas grandes que, em geral, apresentam massas
moleculares maiores que 15 kDa, podendo chegar a mais de 1x106 Da. Ainda, segundo Sun e seus
colaboradores (2011), este resultado era espectável na medida em que estes afirmam que na última
década a ultrafiltração é o processo de separação de membranas que tem sido utilizada para separar
polissacarídeos.
Por forma a confirmar esta concentração de polissacarídeos na fração de concentrado e
observar a presença de polímeros de alta massa molecular, procedeu-se à análise dos
C P A

51
polissacarídeos totais por precipitação etanólica das frações obtidas por filtração com a membrana de
20 kDa (ultrafiltração). Com esta análise verificou-se, a formação de um precipitado etanólico na
amostra de concentrado, não se verificando qualquer tipo de precipitado na amostra de permeado,
como se observa na figura 38.
Figura 38 – Início da precipitação etanólica do permeado (P) e concentrado (C) obtidos por ultrafiltração do
extrato aquoso
Nesta figura observa-se a formação da agregação molecular que, após algumas horas e sob
arrefecimento, forma um precipitado no fundo da proveta. Isto vem mostrar que, a extração com água
nas condições que se consideraram ótimas foi eficiente na recuperação de polímeros de alta massa
molar, não sendo estes extratos apenas constituídos de metabólitos secundários. Por outro lado, e
comparando as figuras das soluções etanólica do permeado e do concentrado, pode constatar-se que
houve concentração dessas macromoléculas no concentrado, dado que não se observa a existência
de polímeros de alta massa molecular na solução de permeado.
4.3. Análises de identificação químico-molecular e atividade antioxidante
Para complementar os estudos elaborados até esta fase do trabalho, decidiu-se estudar mais
aprofundadamente os polissacarídeos fibrosos que foram extraídos e posteriormente separados por
processos de membranas. Neste subcapítulo, estão então apresentados os resultados relativos à
análise dos polissacarídeos nos extratos obtidos e nas frações de ultrafiltração, no que respeita a sua
identificação estrutural. Considerou-se também interessante, no decorrer do trabalho experimental,
determinar os fenólicos totais e analisar a atividade antioxidante in vitro de amostras selecionais,
sendo que esses resultados serão também apresentados neste item do trabalho.
C P
Precipitado etanólico
(macromoléculas)

52
4.3.1. Análise estrutural de Polissacarídeos
Os extratos obtidos através dos três solventes nas condições selecionadas anteriormente como
sendo as melhores (extratos brutos), bem como as frações obtidas através do processo de
Ultrafiltração foram analisados quanto às características estruturais dos polissacarídeos que os
compõem. Por forma a facilitar a compreensão da origem de cada amostra referida neste capítulo,
apresenta-se um esquema resumo na figura 39 que sumariza as amostras obtidas por cada processo
anteriormente realizado.
Figura 39 – Esquema sumário dos processos de extração e de tecnologia de membranas realizados e
amostras obtidas
As análises desta etapa do estudo compreendem a composição monossacarídica dos pools de
polissacarídeos de cada amostra, a análise cromatográfica de exclusão estérica (HPSEC) que
consiste numa técnica analítica que tem como objetivo a separação de populações de moléculas com
base no seu tamanho e por meio das conformações que estas assumem no solvente em que estão
inseridas e as análises espectroscópicas de ressonância magnética nuclear de carbono-13 (13
C-
RMN) e a técnica bidimensional de HSQC, que correlaciona os espectros de ressonância de 1H e
13C.
Para as análises de HPSEC utilizaram-se dois métodos de deteção dos polissacarídeos: um
detetor por dispersão de luz cujos resultados estão representados pelas curvas a vermelho das
figuras que se apresentam neste capitulo e um detetor por índice de refração (IR) representado pelas
curvas a azul nessas mesmas figuras. Em condições ideias as duas curvas sobrepor-se-iam, no

53
entanto, como tal não foi verificado devido à diferença de sensibilidade, vão apenas ser analisados os
resultados referentes ao IR, que apresentou resultados mais fidedignos.
Extratos Brutos
As análises das composições monossacarídicas dos diferentes extratos obtidos através dos
solventes água, e soluções de KOH 2% e Celulase estão apresentadas na tabela 11.
Tabela 11 – Composição monossacarídica dos extratos alcalino, enzimático e aquoso
Amostras Composição monossacarídica (mol%)
a
Rhab
Fucb Ara
b Xyl
b Man
b Gal
b Glc
b AU
b
K 8,7 3,1 30,9 7,6 9,8 19,7 20,2 nd
E 6,3 - 22,1 4 7,6 8,6 51,4 nd
H 4,6 - 29,8 3,5 8,5 14,5 39,1 nd a analisados em GC-MS, na forma de alditóis acetato, coluna OV-225, temperatura 40-220ºC.
b Rha – ramnose; Fuc – fucose; Ara – arabinose; Xyl – xilose; Man – manose; Gal – galactose; Glc – glucose; AU –
ácidos urónicos totais; nd – não determinado.
Pode observar-se que as amostras E e H apresentam percentuais monossacarídicos
condizentes entre si. O extrato enzimático apresenta, no entanto, uma maior quantidade de glucose,
ao passo que no extrato aquoso se observa, para além desse monossacarídeo, um teor elevado em
arabinose e galactose. O elevado percentual de glucose presente no extrato enzimático pode ser
justificado pela atividade do enzima utilizada, Celulase, fragmentando a celulose em estruturas mais
simples, e mais suscetíveis à hidrólise ácida nas condições adotadas neste estudo.
No entanto, ao observar-se os perfis cromatográficos por cromatografia de exclusão estérica
(HPSEC) (vide figura 40), verifica-se uma grande similaridade de perfis entre H e E.

54
Figura 40 – HPSEC das amostras de extrato aquoso (A) e extrato enzimático (B)
Estas figuras mostram a presença de três populações de polissacarídeos, todo de baixa massa
molecular, dado o seu tempo de eluição ser após os 60 minutos de corrida. Nestas três populações
observa-se um pico maior e mais homogéneo nos dois extratos, e outros dois menores que coeluíram
próximos da saída do solvente. Assim, pode supor-se que a capacidade extrativa dos dois solventes
é muito similar no que respeita os tipos de polissacarídeos extraídos, mesmo tendo usado um enzima
capaz de romper as ligações celulósicas da parede celular. Estes resultados, e em paralelo com os
rendimentos de extração obtidos anteriormente neste trabalho, vêm mostrar que, apesar desta
capacidade de rutura deste enzima, a branda temperatura de 50ºC não foi suficiente para transferir os
polissacarídeos da parede celular para o líquido extrator, de maneira tão eficiente quanto a água pura
a 90ºC.
Dada a similaridade do material e ao maior interesse em aprofundar os estudos na
identificação molecular dos polissacarídeos extraídos em água, a amostra H foi analisada por
ressonância magnética nuclear do carbono-13 (13
C RMN) (figura 41).

55
Figura 41 – Espectro de 13
C-RMN da amostra de extrato aquoso obtida nas melhores condições
estudadas (Realizado em D2O a 50ºC. Valores expressos em δ (ppm))
A figura apresenta ampla variedade de sinais, com a presença de 3 sinais na região anomérica
em δ 109,1; 97,7 e 93,1. O sinal em δ 109,1 é referente às unidades de α-Araf (Mellinger et al.,
2005b; Cipriani et al., 2006; Tischer et al., 2002), enquanto o sinal δ 97,7 pode ser proveniente e
unidades de α-GalpA, ou de terminais redutores de α-Ara ou β-Gal (Gorin e Mazurek, 1975; Tischer et
al., 2002). O sinal de δ 93,1 é referente a terminais redutores de α-Ara ou β-Gal (Gorin e Mazurek,
1975), podendo advir de monossacarídeos ou dissacarídeos livres em solução (Tischer et al., 2002).
Relativamente ao extrato alcalino (K), constata-se a presença intensa de arabinose e galactose
(vide tabela 11), provavelmente provenientes de arabinogalactanas e/ou ramnogalacturonanas
(Mellinger et al., 2005b; Arnous e Meyer, 2009). A presença de uma maior quantidade de xilose no
extrato alcalino comparativamente com os restantes mostra a possibilidade do início da extração de
xilanas, que por serem mais fortemente ligadas à parede celular, requerem o uso de soluções com
força alcalina para serem extraídas (Mellinger et al, 2005a; Peng et al., 2010). A análise desta
composição monossacarídica vem, novamente, complementar a premissa de que a força alcalina
rompe com as ligações intermoleculares da parede celular enquanto a força aquosa apenas as
enfraquece (Sun et al, 2011), conseguindo extrair mais abundantemente os polissacarídeos aderidos
a esta parede (Geng et al., 2006; Peng et al., 2009; Mellinger et al., 2005a).
O cromatograma de exclusão estérica (HPSEC) da amostra K vem mostrar e confirmar a
eficiência da extração alcalina do bagaço da uva. Na figura 42 observa-se que este solvente foi capaz
de extrair diferentes populações de moléculas do bagaço da uva, de forma mais eficiente quando
comparada à extração aquosa, apresentando, no mínimo, 5 picos identificados de 1 a 5 no
cromatograma.

56
Figura 42 – HPSEC da amostra de extrato alcalino (K) (Inserido: HPSEC de H)
Extratos Aquosos fracionados por Membranas
No que respeita os resultados referentes às frações obtidas por ultrafiltração, os resultados são
apresentados na tabela 12.
Tabela 12 – Composição monossacarídica dos permeados, concentrados e alimentações do sistema de
membranas de ultrafiltração (20 kDa)
Amostras Composição monossacarídica (mol%)
a
Rhab
Fucb Ara
b Xyl
b Man
b Gal
b Glc
b AU
b
AI 12 - 24,2 2,7 16,8 28 16,3 nd
AII 13 - 24,4 5 14,7 25 17,9 nd
A 12,5 - 24,3 3,8 15,7 26,6 17,1 nd
PI 2 - 10,1 2,4 6,8 9,1 69,6 nd
PII 3,1 - 8,9 2 8 7 71 nd
P 2,5 - 9,5 2,2 7,4 8,0 70,4 nd
CI 11 - 29,9 3,2 16 24,8 15,1 dd
CII 14,4 1,6 28 3,7 13,6 23 15,7 dd
C 12,8 trc 29,1 3,4 14,9 24,3 15,5 d
d
a analisados em GC-MS, na forma de alditóis acetato, coluna OV-225, temperatura 40-220ºC.
b Rha – ramnose; Fuc –
fucose; Ara – arabinose; Xyl – xilose; Man – manose; Gal – galactose; Glc – glucose; AU – ácidos urónicos totais; nd – não determinado.
c tr – traços.
d – determinado, mas não foi levado em consideração para fins comparativos entre as
outras frações (valor de 25,4mol%)
As amostras realizadas em duplicado mostraram uma composição monossacarídica coerente
entre si, pelo que foi possível realizar a média aritmética das amostras e com ela realizar uma análise
comparativa entre as três amostras.
Nas amostras de alimentação (A) e concentrado (C), a elevada presença de concentrações de
ramnose, arabinose, galactose e manose mostra a possível diversidade molecular, onde podem ser
encontradas pectinas, arabinogalactanas, xiloglucanas e/ou mananas, formadas a partir destas
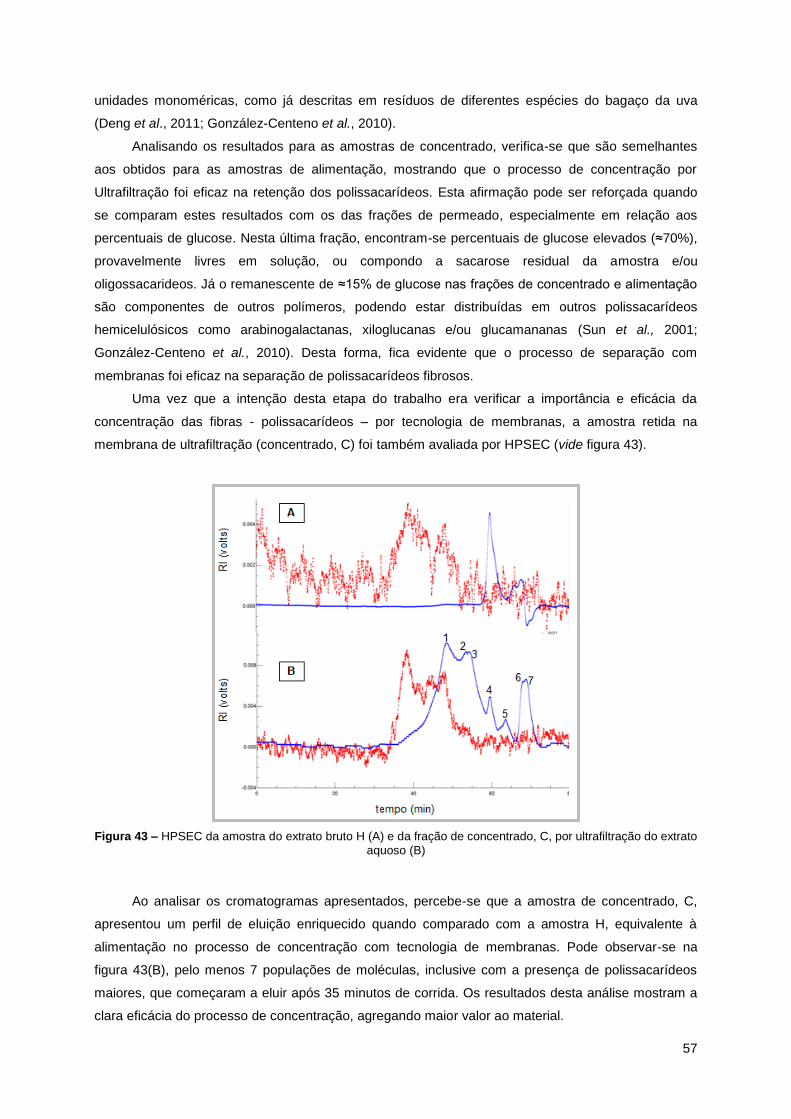
57
unidades monoméricas, como já descritas em resíduos de diferentes espécies do bagaço da uva
(Deng et al., 2011; González-Centeno et al., 2010).
Analisando os resultados para as amostras de concentrado, verifica-se que são semelhantes
aos obtidos para as amostras de alimentação, mostrando que o processo de concentração por
Ultrafiltração foi eficaz na retenção dos polissacarídeos. Esta afirmação pode ser reforçada quando
se comparam estes resultados com os das frações de permeado, especialmente em relação aos
percentuais de glucose. Nesta última fração, encontram-se percentuais de glucose elevados (≈70%),
provavelmente livres em solução, ou compondo a sacarose residual da amostra e/ou
oligossacarideos. Já o remanescente de ≈15% de glucose nas frações de concentrado e alimentação
são componentes de outros polímeros, podendo estar distribuídas em outros polissacarídeos
hemicelulósicos como arabinogalactanas, xiloglucanas e/ou glucamananas (Sun et al., 2001;
González-Centeno et al., 2010). Desta forma, fica evidente que o processo de separação com
membranas foi eficaz na separação de polissacarídeos fibrosos.
Uma vez que a intenção desta etapa do trabalho era verificar a importância e eficácia da
concentração das fibras - polissacarídeos – por tecnologia de membranas, a amostra retida na
membrana de ultrafiltração (concentrado, C) foi também avaliada por HPSEC (vide figura 43).
Figura 43 – HPSEC da amostra do extrato bruto H (A) e da fração de concentrado, C, por ultrafiltração do extrato
aquoso (B)
Ao analisar os cromatogramas apresentados, percebe-se que a amostra de concentrado, C,
apresentou um perfil de eluição enriquecido quando comparado com a amostra H, equivalente à
alimentação no processo de concentração com tecnologia de membranas. Pode observar-se na
figura 43(B), pelo menos 7 populações de moléculas, inclusive com a presença de polissacarídeos
maiores, que começaram a eluir após 35 minutos de corrida. Os resultados desta análise mostram a
clara eficácia do processo de concentração, agregando maior valor ao material.

58
Ainda com o intuito de conhecer melhor a estrutura química dos polissacarídeos que foram
extraídos e concentrados por membranas, a fração C foi submetida a análise HSQC. O espectro
apresentado na figura 44 apresenta uma alta complexidade, contendo 14 sinais na região anomérica
para a correlação C1/H1, representada entre δ 109,6/5,74 e 98,4/5,56.
Figura 44 – Espectro de HSQC da fração de concentrado, C de ultrafiltração (Realizado em D2O, 50ºC.
Valores expressos em δ (ppm))
Para uma melhor compreensão dos dados do espectro de HSQC da fração C, a tabela 13
apresenta os sinais referentes à correlação C1/H1, confirmando a presença da ampla diversidade
estrutural do material.
Tabela 13 – Sinais referentes à correlação C1/H1 do espectro de HSQC de C
Sinais
C1/H1 (ppm) Unidade
a
109,6/5,74
α-Araf 107,9/5,59
107,9/5,56
107,6/5,64
104,3/4,94 β-Galp
103,4/4,96
102,9/4,99 α-Manp / β-Xylp /
β-GlcpA
100,3/5,45 O-CH3-α-GalpA
99,8/5,43 α-GalpA / α-Manp /
α-Glcp 99,6/5,67
(C1/H1)

59
99,3/5,40
99,1/5,63
98,4/5,78 α-Rhap
98,4/5,56
a As unidades sugeridas, de acordo com a literatura, são: α-Araf: alfa-arabinofuranose; β-
Galp: beta-galactopiranose; α-Manp: alfa-manopiranose; β-Xylp: beta-xilopiranose; β-GlcpA: ácido-beta-glucurônico; O-CH3-α-GalpA: ácido-alfa-galacturônico metil-esterificado;α-GalpA: ácido-alfa-galacturônico; α-Glcp: alfa-glucopiranose; α-Rhap: alfa-ramnopiranose.
Ao analisar conjuntamente a composição monossacarídica, os cromatogramas de HPSEC e o
espectro de HSQC pode-se supor que a amostra C apresenta rica variedade molecular, com a
provável presença de arabinogalactana(s) (Kacuráková et al., 2010; Geng et al., 2006; González-
Centeno et al., 2010) e/ou polissacarídeos pécticos, sugerido a partir do alto conteúdo de ácido
urónico da amostra (25,4% mol) (Deng et al., 2011; Kacuráková et al., 2010; González-Centeno et al.,
2010), além da presença dos outros monossacarídeos que compõem a amostra (mol%): 9,5% Rha;
22,1% Ara; 2,5% Xyl; 11% Man; 18% Gal; 11,5% Glc. A presença de xiloglucanas e/ou glucomananas
não são descartadas, já que são polissacarídeos frequentemente encontrados na película da uva
(Igartuburu et al., 1997; Arnous e Meyer, 2009; Deng et al., 2011).
A literatura apresenta dados para os assinalamentos encontrados no espectro baseados na
caracterização química de polissacarídeos previamente purificados de um organismo (Tischer et al.,
2002; Simas et al., 2008; Steinhorn et al., 2011). Na tabela 14, são apresentadas algumas dessas
possibilidades, de acordo com a composição monossacarídica da fração C. No entanto, para que se
possa realizar uma caracterização química fina deste material, é preciso fracionar esta amostra,
isolando cada polissacarídeo em uma nova fração purificada. Depois, por meio das análises aqui
descritas, juntamente com a análise de metilação, pode-se esclarecer a caracterização química de
cada um destes polímeros presentes na amostra C. Os dados aqui apresentados cumprem com a
proposta inicial deste estudo e estudos futuros serão necessários para dar continuidade na análise
química dos polissacarídeos do bagaço da uva.
4.3.2. Atividade Antioxidante
Analisadas as estruturas polissacarídicas presentes nos extratos e fração de concentrado,
estudou-se outra propriedade destas amostras, a capacidade antioxidante (vide Anexo 8.G). A tabela
14 apresenta os resultados obtidos.

60
Tabela 14 – Atividade Antioxidante das amostras de extrato obtidas para os três solventes e nas melhores
condições selecionadas e das frações de concentrado e permeado do processo de ultrafiltração
Amostras AA
a
( mol Trolox/100 g bagaço)
K
8423,9±221,73 H
366,14±10,45
C
312,43±6,078 P
200,52±12,956
E 83,312±7,9044 a
AA – atiividade antioxidante; K – extrato alcalino 2% obtido nas condições de extração selecionadas; H - extrato aquoso obtido nas condições de extração selecionadas; C - fração de concentrado por ultrafiltração; P - fração de permeado por ultrafiltração; E - extrato enzimático obtido nas condições de extração selecionadas.
A partir dos resultados obtidos verifica-se que o concentrado obtido por ultrafiltração do extrato
aquoso apresenta maior atividade que o permeado e uma atividade antioxidante similar à
apresentada para a alimentação desse processo de membranas, o que vem garantir que esta
propriedade não é perdida no processo de concentração. Inclusive, pode sugerir que a interação
molecular entre as fibras e os compostos fenólicos podem estar agindo de forma cinética para a
atividade observada. Verifica-se também que o extrato alcalino é aquele que apresenta uma maior
atividade antioxidante, sendo cerca de 23 vezes superior ao maior valor de AA obtido para as
restantes amostras.
4.3.3. Fenólicos Totais
Muitos autores já relataram que a atividade antioxidante está relacionada primeiramente com a
quantidade de compostos fenólicos presentes (Karacabey e Mazza, 2010). Nesse sentido,
quantificaram-se os fenólicos presentes na solução que apresentou maior atividade antioxidante
(extrato alcalino) e nas soluções de concentrado e permeado da ultrafiltração. Os resultados foram
agrupados na tabela 15 e tiveram como base os dados presentes em Anexo (vide anexo 8.H).
Tabela 15 – Composição de fenólicos totais (FT) na amostra de extrato alcalino obtido na melhor condição
selecionada e das frações de concentrado e permeado do processo de ultrafiltração
Amostras FT
(mg ácido gálico/100g bagaço)
K 1616,5±53,01
P 73,018±0,8133
C 135,05±5,970
Do que se observa dos resultados obtidos, a solução de concentrado apresenta uma maior
quantidade de fenólicos que a fração permeada pelo módulo de membranas de ultrafiltração, cerca
de 1,85 vezes mais. A solução alcalina é aquela que apresenta uma grande quantidade de fenólicos,
sendo uma ordem de grandeza superior à quantidade de fenólicos observada na solução de
concentrado. Pode então dizer-se que, dado que o principal objetivo era o de extrair e concentrar as
fibras, estas vêm ligadas a compostos com atividade antioxidante, interagindo com as mesmas,
enriquecendo o material final. Segundo Sun e seus colaboradores (2001) radicais ácidos como

61
grupos carboxílicos ou fenólicos, ionizados na solução alcalina, podem também promover a
solubilização das hemiceluloses e resíduos de lignina, o que pode justificar a diferença significativa
dos resultados do extrato alcalino para os restantes.
Através destes resultados podemos sugerir que a atividade antioxidante pode ser resultado do
conjunto de compostos fenólicos e fibras solúveis, podendo existir sinergismo entre elas, indo-se de
encontro ao conceito de fibras antioxidantes (Tseng and Zhao, 2013).

62

63
5. Conclusões
No presente trabalho estudou-se o processo de extração de fibras solúveis do bagaço da uva
branca Vitis Vinifera Chardonnay proveniente do processo de produção de vinho tinto e
concentraram-se os extratos obtidos com água pura por processos de tecnologia de membranas de
ultrafiltração e microfiltração. Os extratos do processo de extração e as frações obtidas por
concentração por ultrafiltração foram também analisados quanto à sua estrutura polisacarídica,
atividade antioxidante in vitro e teor de polifenóis totais.
Assim, é possível afirmar que os objetivos delineados inicialmente para esta dissertação foram
cumpridos, tendo-se chegado às seguintes conclusões:
Dos três solventes estudados (água e soluções aquosas de Cellulase e de hidróxido de
potássio) o solvente alcalino foi aquele que apresentou melhores rendimentos no que respeita a
massa de açúcar presente em solução de extrato. Concluiu-se que, de entre as três concentrações
de KOH estudadas, o solvente aquoso de concentração de 2% de KOH foi aquele que, agregando
questões económicas, de segurança e de rendimento de extração, apresentou os melhores
resultados, tendo-se obtido uma massa máxima de açúcar de 2,16±0,012 g/100 g de bagaço de uva
nas condições de 90ºC, relação substrato/solvente de 1:4 e bagaço in natura. No entanto, crê-se que
a necessidade de neutralização do extrato alcalino e a coloração escura do mesmo possam trazer
desvantagens ao uso do solvente KOH no panorama industrial, na medida em que estão associados
custos de operação acrescidos, quando comparados com os restantes solventes estudados, como o
uso de ácido neutralizante e clarificação (bleaching) dos extratos na indústria alimentar. No que
respeitam os restantes dois solventes, para a extração com água obteve-se um rendimento maximo
de extração de 0,904±0,0453 g/100 g de bagaço de uva (a 90ºC, relação substrato/solvente de 1:4 e
bagaço da uva in natura), e para a extração com solução enzimática obteve-se uma massa máxima
de açúcar de 0,724±0,0100 g/100 g de bagaço de uva (a 50ºC, relação substrato/solvente de 1:4 e
concentração de Celulase de 350µL/18g de bagaço seco).
Com os resultados das cinéticas das extrações com os três, pode concluir-se que os dados de
coeficiente de difusão, para os processos de extração das fibras solúveis, em meio aquoso e em meio
alcalino apresentaram taxas de transporte de massa similares porém inferior ao observado no
processo enzimático (2,6 vezes maior).
Ambos os processos de tecnologia de membranas, microfiltração (MF) e ultrafiltração (UF),
concentraram os açúcares provenientes da solução de extracto obtido nas condições seleccionadas
com água pura. A eficiência da concentração no processo de UF foi também confirmada por
precipitação etanólica, através da qual se concluiu que não existem polissacarídeos de massa
molecular elevada na fração de permeado do processo de ultrafiltração.
No que respeita a identificação estrutural polisacarídica concluiu-se que as amostras de extrato
enzimático e aquoso têm capacidade extrativa muito similar por apresentarem composição
monossacarídica e HPSEC semelhantes. Com este resultado, e em paralelo com os rendimentos de

64
extração, conclui-se que, apesar da capacidade de rutura de ligações celulósicas da parede celular
por parte do enzima, a branda temperatura de 50ºC não foi suficiente para transferir os
polissacarídeos para o meio extrator. Ainda no que respeitam resultados do extrato aquoso, conclui-
se por análise do espectro 13
C-RMN que este é constituído por unidades como α-Araf e α-GalpA ou
de terminais redutores α-Ara ou β-Gal.
Relativamente ao extrato alcalino, quando comparado com os anteriores, conclui-se que com
este solvente se conseguiu extrair uma maior diversidade molecular, como por exemplo, xilanas, por
análise do HPSEC e da composição monossacarídica. Estes resultados permitiram também concluir
que a força alcalina quebra ligações de moléculas da parede celular, ao passo que a força aquosa
apenas as enfraquece, conseguindo apenas extrair moléculas menos aderidas à parede.
A composição monossacarídica das frações obtidas por ultrafiltração levaram a concluir que o
processo de concentração através desta membrana foi eficiente e que o concentrado e a alimentação
podem ser constituídos por moléculas de pectinas, arabinogalactanas, xiloglucanas e/ou mananas. A
análise HPSEC do concentrado quando comparada com a do extrato aquoso levou à conclusão e
confirmação de que o processo de ultrafiltração concentrou as fibras solúveis do bagaço da uva,
tendo-se encontrado, pelo menos, 7 populações de moléculas na fração de concentrado. Através da
análise HSQC a que a fração de concentrado foi submetida, e em conjunto com as análises da
composição monossacarídica e HPSEC dessa amostra, concluiu-se que esta pode ser constituída por
arabinogalactana(s) e/ou polissacarídeos pécticos, sugerido a partir do alto conteúdo de ácido urónico
da amostra (25,4% mol). A presença de xiloglucanas e/ou glucomananas não são descartadas, já que
são polissacarídeos frequentemente encontrados na película da uva (Igartuburu et al., 1997; Arnous e
Meyer, 2009; Deng et al., 2011).
Por fim, no que respeita a capacidade antioxidante das amostras analisadas, concluiu-se que a
amostra relativa ao extrato alcalino a 2%, obtido a 90ºC, durante 5 horas com uma relação substrato
solvente de 1:4, é aquela que apresenta uma maior atividade antioxidante de 8423,9±221,73 mol
Trolox/100 g bagaço, cerca de 23 vezes superior ao valor obtido para o extrato aquoso obtido nas
mesmas condições, durante 3 horas. Concluiu-se também que a concentração das fibras por
processos de tecnologia de membranas de ultrafiltração não altera a capacidade antioxidante do
extrato, garantindo-se que se mantém esta propriedade.
A presenta de compostos fenólicos no extrato alcalino (1616,5±53,01 mg ácido gálico/100g
bagaço) e de menor quantidade na fração de concentrado (135,05±5,970 mg ácido gálico/100g
bagaço) veio também sugerir a aplicação sinérgica das fibras solúveis e destes composto fenólicos,
no que respeita a atividade antioxidante medida.
Este trabalho veio mostrar que a extração de compostos de interesse do bagaço da uva, como
as fibras solúveis, pode ajudar a garantir uma cadeia sustentável na indústria vinificadora. Os
resultados obtidos no estudo realizado sugerem a possibilidade de utilização do bagaço da uva
branca submetida à vinificação tinta como fonte de fibras solúveis com atividade antioxidante,
utilizando tecnologias de extração sólido-líquido e de membranas de ultrafiltração.

65
6. Perspetivas de trabalho futuro
Triturar o bagaço para obtenção de partículas em pó e reavaliar o rendimento do processo.
Estudar variáveis como o pH e a pressão na fase de extração das fibras solúveis.
Estudar a cinética da extração para tempos inferiores a 30 minutos e analisar a tendência.
Estudar a influência da pressão nos processos de Ultrafiltração e Microfiltração.
Investigar os polissacarídeos da fração alcalina e compará-los àqueles observados para a
extração aquosa, fazendo uma comparação mais aprofundada entre elas;
Depois de extraídos os compostos de interesse estudar a aplicabilidade do bagaço na indústria
de celulose ou no desenvolvimento de ração animal, uma vez que este material é rico em
algumas hemiceluloses e celulose;
Separar os compostos fenólicos e as fibras e avaliar a identidade molecular delas, bem como a
atividade antioxidante separadamente.
Avaliar uma possível reciclagem do permeado como solvente na etapa de extração.
Avaliar a composição química das frações do permeado e investigar diferentes aplicações,
como por exemplo, atividade probiótica.

66

67
7. Referências Bibliográficas
AACC report; The definition of dietary fibres; American Association of Cereal Chemists; 46, 112-126;
2001.
Adega; Noticia: Pesquisadores descobrem lugar de origem da viticultura, Revista “Adega”, Dezembro
de 2012, consultado em Maio de 2013; disponível em:
http://revistaadega.uol.com.br/Edicoes/0/Artigo275067-1.asp.
Alves, A.M.; Modulo de Processos de Separação com Membranas, Parte 1: Introdução aos processos
de Separação com Membranas; Ultrafiltração e Osmose Inversa; Disciplina de Processos de
Separação II; Instituto Superior Técnico; Secção de Folhas; 2010/2011.
AOAC (Association of Official Analytical Chemists); Official Methods of Analysis; W. Horwitz (ed),
Washington D. C 2005.
AOAC (Association of Official Analytical Chemists); Official Methods of Analysis; W. Horwitz (ed),
Washington D. C 2010.
Anderson, J.M.; Baird, P.; Davis, R.H.; Ferreri, S.; Knudtson, M.; Koraym, A.; Waters V.; Williams, C.L;
Health benefits of dietary fiber; Nutrition Reviews, Vol. 67(4), p.188–205; 2009.
Aoe S.; Oda, T.; Tatsumi, K.; Yamauchi, M.; Ayano Y.; Extration of Soluble Fibers from Defatted Rice
Bran; American Association of Cereal Chemists; vol.70, nº4; p.423-425; 1993.
Arnous, A; Meyer, A.S.; Quantitative Prediction of Cell Wall Polysaccharides composition in Grape
(Vitis Vinifera L.) and Apple (Malus domestica) skins from Acid Hidrolysis Monosaccharide Profiles;
Journal of Agricultural and Food Chemistry; v.57, p. 3611-3619; 2009.
Arvanitoyannis, I.S.; Ladas, D.; Mavromatis, A.; Potential uses and applications of treated wine waste:
a review; International Journal of Food Science and Technology, v.41, p.475-485, 2006.
Azevedo, E.G.; Alves, A.M.; Engenharia de Processos de Separação; Coleção Ensino da Ciência e
da Tecnologia, IST Press; Instituto Superior Técnico, Julho de 2009.
Berkowitz, M.; World's Earliest Wine. Archeology Archive, vol. 49, num.5, 1996, consultado em Maio
de 2013; disponível em: http://archive.archaeology.org/9609/newsbriefs/wine.html.

68
Bustamante, M.A.; Moral, R., Paredes, C.; Pérez-Espinosa, A; Moreno-Caselles J. and Pérez-;Murcia,
M.D.; Agrichemical characterization of the solid by-products and residues from the winery and distillery
industry; Waste Management, c.28, p.372-380, 2008.
Caffal, K.H.; Mohnen, D.; The structure, function and biosynthesis of plant cell wall pectic
polysaccharides; Carbohydrate Research; v.334, p.1879-1900, 2009.
Campos, L.M.A.; Obtenção de extratos de bagaço de uva Cabernet sauvignon (Vitis Vinifera L.);
Universidade Federal de Santa Catarina - Engenharia de Alimentos – Centro Tecnológico, 2005.
Catarino, I; Minhalma, M.; Beal, L.L.; Mateus, M.; Pinho, M.N.; Assessment of saccharide fractionation
by ultrafiltration and nanofiltration; Journal of Membrane Science; v.312; p.34-40; 2008.
Cippriani, T.R.; Mellinger, C.G.; Souza, L.M.; Baggio, C.H.; Freitas, C.S.; Marques, M.C.A.; Gorin,
P.A.J.; Sassaki, G.L.; Iacomini, M.; A Polysaccharide from a Tea (Infusion) of Maytenus ilicifolia
Leaves with Anti-ulcer Protective Effects; Journal of Natural Products; v. 69, 1018-1021; 2006.
Conselho da União Europeia; Regulamento (CE) n° 1493/1999 do Conselho de 17 de Maio de 1999
que estabelece a organização comum do mercado vitivinícola; Jornal Oficial das Comunidades
Europeias; L179; 1999.
Chatzilazarou C.; Katsoyannos E.; Gortzi O.; Lalas S.; Paraskevopoulos Y.; Dourtoglou E.; Tsaknis J.;
Removal of Polyphenols from Wine Sludge Using Cloud Point Extraction, Journal of the Air & Waste
Management Association, 60:4, 454-459, 2010.
Deng, Q.; Zhao, Y.; Physicochemical, Nutritional, and Antimicrobial Properties of Wine Grape
(cv.Merlot) Pomace Extract-Based Film; Journal of Food Science; V.76, no.3; p.E309-E317; 2011.
Deng, Q; Penner, M.H.; Zhao, Y; Chemical composition of dietary fiber and polyphenols of five
different varieties of wine pomace skins; Food Research International; v44, p. 2712-2720; 2011.
Dubois, M.; Gilles, K. A.; Hamilton, J. K.; Rebers, P. A.; Smith, F. Colorimetric method for
determination of sugars and related substances.; Analytical Chemistry, v. 28, p. 350-356, 1956.
Filisetti-Cozzi, T.M.C.C.; Carpita, N.C. Measurement of uronic acids without interference from neutral
sugars. Analytical. Biochemistry, v, 197, p. 157-162, 1991.
FAOSTAT - Food and Agriculture Organization; Production: Crops processed; consultado em Maio de
2013; disponível em: http://faostat3.fao.org/home/index.html#VISUALIZE.

69
Farelo, M.F.C; Serrano, M.L.; Processos Químicos I, Teoria e Problemas; Instituto Superior Técnico;
Secção de Folhas; Lisboa.
Geng, Z. C.; Sun, J. X.; Liang, S. F.; Zhang, F. Y.; Zhang, Y. Y.; Xu, F.; Sun, R. C.; Characterization of
Water- and Alkali-Soluble Hemicellulosic Polymers from Sugarcane Bagasse; International Journal of
Polymer Analysis and Characterization; V. 11, Issue 3, p.209-226; 2006.
Georgé, S.; Brat, P.; Alter, P. and Amiot, M.J. Rapid determination of polyphenols and vitamin C in
plant-derived products.; Journal of Agricultural and Food Chemistry, v. 53, p. 1370-1373, 2005.
Giacomo, G. D.; Taglieri L.; Production of Red Wine Polyphenols as Ingredient for Food and
Pharmaceutical Industry”; International Journal of Food Science and Nutrition Engineering; v.2, p.12-
15; 2012.
Gonçalves, F.M.S.; Optimização da Clarificação e Estabilização Tartárica de Vinhos por Processos de
Membranas. Influência das Macromoléculas e da Composição Iónica; Dissertação para obtenção do
grau de Doutor em Engenharia Química; Instituto Superior Técnico; 2002.
González-Centeno, M.R.; Rosseló, C.; Simal, S.; Garau, M.C.; López, F.; Femenia, A.; Physico-
chemical properties of cell wall material obtained from tem grape varieties and their byproducts: grape
pomaces and stems; Food Science and Technology; v.43, p.1580-1586; 2010.
Gorin, P.A.J.; Mazurek, M.; Further Studies on the Assignment of Signals in 13C Magnetic Resonance
Spectra of Aldoses and Derived Methyl Glycosides; Canadian Journal of Chemistry, v.53(8), p. 1212-
1223; 1975.
Graham, H.; Rydberg, M.B.G.; Aman, P.; Extraction of soluble Dietary Fiber; Journal of Agricultural
and Food Chemistry; v.36; p.494-497; 1988.
Graham H.; Aman, P.; Nutritional aspects of dietary fibres; Animal Feed Science and Tehcnology;
v.32, p.143-158; 1991.
Guilford J.M.; Pezzuto, J.M.; Wine and Health: A Review; American Journal of Enology and Viticulture;
62:4; 471-486; 2011.
Gullón B.; Gómez, B.; Martínez-Sabajanes, M.; Yáñes, R.; Parajó, J.C.; Alonso, J.L; Pectic
oligosaccharides: Manufacture and functional properties; Food Science and Technology; v.30, p.153-
161, 2013.

70
Harborne, J.B.; Williams,C.A; Advances in flavonoid research since 1992. Phytochemistry, Oxford, v.
52, p. 481- 504,2000.
Hervest-Hernández, D.; Pintado, C.; Rotger, R.; Goñi, I.; Stimulatory role of grape pomace
polyphenols on Lactobacillus acidophilus growth; International Journal of Food Microbiology; v.136,
p.119-122; 2009.
Igartuburu, J.M.; Pando, E.; Luis, F.R.; Gil-Serrano, A; An Acid xyloglucan from grape skins;
Phytochemistry; vol.48, nº8, p.1037-1312; 1997.
Infovini; Como se faz um vinho, 2009, consultada em Maio de 2013; disponível em:
http://www.infovini.com/pagina.php?codNode=18016.
Ishimoto, E.Y.; Capriles, V.D.; Matias, A.G.; Baccarin, A.; Torres, E.A.F.S; Formulating Functional
Popsicle with Wine and Grape Juice By-Products; Acta Horticulturae (ISHS); v.84, p.1603-1606; 2007.
Ju, Y.H.; Huynh, L.H.; Kasim, N.S.; Guo, T.J.; Wang, JH.; Analysis of soluble and insoluble fractions of
alkali and subcritical water treated sugarcane bagasse; Carbohydrate Polymers; v.83, p.591-599,
2011.
Lopes Junior, C.O.; Amorim, A.C.P.; Souza, M.W.S.; Silva, V.D.M.; Silva, M.R.; Pinto, M.C.S.; Otimização da extração enzimática da proteína do feijão; Maringá, v. 32, n. 3, p. 319-325, 2010.
Karacabey, E.; Mazza, G.; Optimization of antioxidant activity of grape cane using response surface
methodology; Food Chemistry, v.119, p.343-348; 2010.
Karkuráková, M.; Capek, P.; Wellner, N.; Ebringerová, A; FT-IR study of plant cell model compounds:
pectic polysaccharides and hemicelluloses; Carbohydrate Polymers; v. 43, p. 195-203, 2000.
Kemper, T.G.; Bailey’s Industrial Oil and Fat Products; Sixth Edition; Six Volume Set.; Cap. 2: Oil
Extraction; John Wiley & Sons, Inc.; 2005.
Mariano, R.G.B.; Couri, S.; Freitas, S.P.; Enzymatic Technology to improve oil extraction from
Caryocar brasiliense Camb.(Pequi) pulp; Revista Brasileira de Fruticultura, v.31, n.3, p.637-643; 2009.
Mellinger, C.G.; Carbonero, E.R.; Cipriani, T.R.; Gorin, P.A.J.; Iacomini, M.; Xylans from the Medical
Herb Phyllanthus niruti; Journal of Natural Products; v.68, p.129-132; 2005a.

71
Mellinger, C.G.; Carbonero, E.R.; Noleto, G.R.; Cipriani, T.R.; Oliveira, M.B.M.; Gorin, P.A.J.; Iacomini,
M.; Chemical and Biological Properties of an Arabinogalactan from Phyllanthus Niruri; Journal of
Natural Products, v.68, p.1479-1483; 2005b.
Mellinger, C.G.; Caracterização Estrutural e Atividade Biológica de Carboidratos de Phyllanthus niruri
(quebra-pedra); Tese de Doutoramento; Curitiba, 2006.
Mellinger-Silva, C.; Simas-Tosin, F.; Schiavinni, D.N.; Werner, M.F.; Baggio, C.H.; Pereira, I.T.; Silva,
L.M.; Gorin, P.A.J.; Iacomini, M.; Isolation of a gastroprotective arabinoxulan from sugarcane bagasse;
Bioresource Technology; v.102, p.10524-10528; 2011.
Negro C.; Tommasi L.; Miceli A.; Phenolic compounds and antioxidant activity from red grape marc
extracts; Bioresource Technology; v.87 p. 41–44; 2003.
Parry, J.; Su, L., Luther, M., Zhou K., Yurawecz, M. P., Whittaker, P.; Fatty acid composition and
antioxidant properties of cold-pressed marionberry boysenberry red raspberry and blue-berry seed
oils.; Journal of Agricultural and Food Chemistry, v. 53(3), p. 566–573, 2005.
Peng, F.; Ren, J.L.; Xu, F.; Bian, J.; Peng, P.; Sun, R.C.; Comparative study of Hemicelluloses
obtained ny Graded Ethanol Precipitation from Sugarcane Bagasse; Journal of Agricultural and Food
Chemistry; v.57, p.6305-6317; 2009.
Peng, F.; Ren, J.L.; Xu, F.; Bian, J.; Peng, P.; Sun, R.C.; Fractional study of alkali-soluble
hemicelluloses obtained by graded ethanol precipitation from sugarcane bagasse. Journal of
Agricultural and Food Chemistry; v.58, p.1768-1776; 2010.
Perry, R.H; Green, D.W; Chemical Engineers' Handbook; McGraw Hill Handbooks, cap.2, 1999.
Petitchateau; Vinificações, consultada em Maio de 2013; disponível em:
http://www.petitchateau.com.br/tudo-sobre-vinho/da-uva-ao-vinho/vinificacoes/.
Pinho, M.N; Prazeres, D.M.; Fundamentos de Transferência de Massa; Coleção Ensino da Ciência e
da Tecnologia; IST Press; Instituto Superior Técnico; Abril de 2008.
Portal de Engenharia Química; Processos de Separação: Membranas; consultado em Maio de 2013,
disponível em:
http://labvirtual.eq.uc.pt/siteJoomla/index.php?Itemid=206&id=57&option=com_content&task=view

72
Re, R.; Pellegrini, N.; Proteggente A.; Pannala, A.; Yang, M. and Rice-Evans, C.; Antioxidant activity
applying an improved ABTS radical cation decolorization assay; Free Radical Biology & Medicine, Vol.
26, nos
9/10, p. 1231–1237, 1999.
Ribeiro, J.C; O sector dos vinhos em Portugal: alguns dados e notas de política, Abril 2011
consultado em Novembro de 2012; disponível em: http://regioes.blogspot.com.br/2011/04/o-sector-
dos-vinhos-em-portugal-alguns.html.
Resende, A.M.F.R.N.; Separação de polissacáridos em vinho branco; Dissertação para obtenção de
grau de Mestre em Engenharia Química; Instituto Superior Técnico, Universidade Técnica de Lisboa,
2011.
Rufino, M. S.M et al.; Metodologia Científica: Determinação da atividade antioxidante total em frutas
pela captura do radical ABTS.+
.; Comunicado Técnico (Embrapa Agroindústria Tropical), 2007.
Saulnier, L; Marot, C; Chanliaud, E.; Thibault, J.F.; Cell wall polysaccharide interactions in maize bran;
Cabohydrate Polymers; v.26, p.279-287; 1995.
Santos, R.D.; Ferrari, R.A.; Extração aquosa enzimática de óleo de Soja; Ciências e Tecnologia de
Alimentos; Campinas; v.25(1), p.132-138; 2005.
Schweitzer, P. A.; Handbook of Separation Techiques for Chemical Engineers; McGraw-Hill ; Nova
Iorque, 1982.
Silbey, R.J.; Alberty, R.A.; Bawendi, M.G.; Physical Chemistry; 4th
edition; John Wiley & Sons, Inc.;
2005.
Silva, L.M.L.R.; Caracterização de subprodutos da vinificação, Millenium: Revista do ISPV, Portugal,
nº28, p.123-133, Outubro 2003.
Silva, J.M.R; Composição da uva, do mosto e do vinho; 100º Curso Intensivo de Vinificação;
Laboratório Ferreira Lapa Sector de Enologia; Universidade Técnica de Lisboa, Setembro de 2006.
Silva M.L.C.; Costa R.S.; Santana A.S; Koblitz, M.G.B.; Compostos fenólicos, carotenóides e
atividade antioxidante em produtos vegetais, Ciências Agrárias, Londrina, v.31, p.669-682,
Julho/Setembro 2010.
Silva, N.L.C.; Produçao de Bioetanol de segunda geração a partir de biomassa residual da industria
de celulose; Dissertação para obtenção do grau de Mestre em Ciências (M.Sc.) Escola de Química,
Universidade Federal do Rio de Janeiro, 2010.

73
Simas, F.F.; Gorin, P.A.J.; Wagner, R.;Sassaki, G.L.; Bonkerner, A.; Iacomini, M.; Comparison of
structure of gum exudate polysaccharides from the trunk and fruit of the peach tree (Prunus persica);
Carbohydrate Polymers; v. 71, I.2, p.218-228; 2008.
Singleton, V. L. and Rossi, J. A.; Colorimetry of total phenolics with phosphomolybdic-phosphotungstic
acid reagents; American Journal of Enology and Viticulture, v.16, p.144-168, 1965.
Siracusa, V.; Rocculib, P.; Romanib, S.; Rosab, M.D.; Biodegradable polymers for food packaging: a
review; Trends in Food Science & Technology; v.19, p.634-643; 2008.
Soares, S.E.; Ácidos fenólicos como antioxidantes; Revista de Nutrição, Campinas; 15(1):71-81;
Janeiro/Abril 2002.
Soares, M.; Welter, L.; Kuskoski, E.M.; Gonzaga, L.; Fett, R.; Compostos fenólicos e atividade
antioxidante da casca de uvas Niágara e Isabel; Revista Brasileira de Fruticultura, Jaboticabal, São
Paulo; v.30, n.1, p.59-64; Março 2008.
Statistica 8.0 High Performance Analytic Software Solutions, StatSoft, 2007.
Steinhorn, G.; Sims, I.M.; Carnachan, S.M.; Carr, A.J.; Schlothauer, R.; Isolation and characterisation
of arabinogalactan-proteins from New Zealand kanuka honey; Food Chemistry; V.128, I.4, , p.949–
956; 2011.
Sun, R.C.; Fang, J.M.; Tomkinson, J.; Geng, Z.C.; Liu, J.C.; Fractional isolation, physico-chemical
characterization and homogeneous esterification of hemicelluloses from fast-growing poplar wood;
Carbohydrate Polymers; v.44, p.29-39; 2001.
Sun, H.; Qi, D.; Xu, J.; Juan, S.; Zhe, C.; Fractionation of polysaccharides from rapeseed by
ultrafiltration: Effect of molecular pore size and operation conditions on the membrane performance;
Separation and Purification Technology; v.80, p.670–676; 2011.
Tischer, C.A; Gorin, P.A.J.; Iacomini, M.; The free reducing oligosaccharides of gun Arabic: aids for
structural assignments in the polysaccharide; Carbohydrate Polymers; v.47, p.151-158; 2002.
Tseng, A.; Zhao, Y.; Wine grape pomace as antioxidant dietary fibre for enhancing nutritional value
and improving storability of yogurt and salad dressing; Food Chemistry; v.138, p. 356-365; 2013.
Wolfrom,M. L.; Thompson, A. Reduction with sodium borohydride. Methods Carbohydrates Chemistry,
v. 2, p. 65-68, 1963a.

74
Wolfrom,M. L.; Thompson, A.; Acetylation. Methods Carbohydrates Chemistry, v. 2, p. 211-215,
1963b.

75
8. Anexos
8.A – Bagaço de uva branca sujeito aos 3 tempos de trituração estudados
Figura 45 - Figura comparativa do bagaço triturado durante os 3 tempos estudados
8.B – Resultados da determinação da espessura do bagaço de uva branca
A espessura do bagaço foi medida por um micrómetro digital da Fowler, IP54 de precisão
0,000005 pol.
Tabela 16 – Espessuras medidas do bagaço da uva branca
Medida (pol) Medida (mm)
1,11E-01 4,44E-03
1,16E-01 4,64E-03
6,80E-02 2,72E-03
9,70E-02 3,88E-03
6,50E-02 2,60E-03
9,00E-02 3,60E-03
1,03E-01 4,12E-03
9,00E-02 3,60E-03
1,16E-01 4,64E-03
Média 9,51E-02 3,80E-03
Desvio Padrão 1,90E-02 7,60E-04
Coeficiente de Variação 19,97%
8.C – Curvas padrão e absorvâncias lidas para os ensaios de extração com e
sem efeitos cinéticos
Nas tabelas que se seguem, apresentam-se as absorvâncias lidas e a referência às curvas
padrão às quais estas estão associadas.
Nas tabelas 17 e 18 estão agrupadas as absorvâncias lidas para os extratos aquosos sem e
com efeitos cinéticos, respetivamente, e os respetivas referências às curvas padrões às quais as
6 min 3 min 0 min

76
primeiras estão associadas. Para estas análises, diluíram-se 10µL de amostra de extrato em 490µL
água destilada.
Tabela 17 – Absorvâncias medidas nos extratos aquosos obtidos sem efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de Variação
2 H1
0,533
3,11% 0,557
-
2 H2
-
0,95% 0,520
0,527
2 H3
0,307
5,53% 0,332
-
2 H4
0,242
9,35% 0,212
-
2 H5
-
2,84% 0,733
0,763
3 H6
0,693
4,55% 0,759
0,725
2 H7
-
5,01% 0,449
0,482
3 H8
-
3,45% 0,525
0,500
2 H9
0,583
2,40% 0,557
0,563
2 H10
-
1,39% 0,565
0,554
2 H11
0,586
1,81% 0,585
0,604
Tabela 18 – Absorvâncias medidas nos extratos aquosos obtidos com efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de Variação
6 30 min
0,372
1,03% 0,366
0,365
6 1horas
0,415
2,30% 0,423
0,404
6 1h30
0,472
2,80% 0,499
0,483
6 2horas
0,508
3,89% 0,488
0,470
7 2h30
0,528
2,30% 0,506
0,525
6 3horas
0,528
4,77% 0,484
0,490
6 3h30
0,546
2,07% 0,553
0,531
6 4horas 0,589
2,13% 0,565

77
0,582
6 4h30
0,558
2,31% 0,560
0,537
6 5horas
0,564
2,45% 0,538
0,545
As tabelas 19 e 20 que se seguem apresentam as absorvâncias lidas para os extratos
enzimáticos sem e com efeitos cinéticos, respetivamente, e os respetivas referências às curvas
padrões às quais as primeiras estão associadas. Para estas análises, diluíram-se 10µL de amostra de
extrato em 490µL água destilada.
Tabela 19 – Absorvâncias medidas nos extratos enzimáticos obtidos sem efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de
Variação
5 E1
0,576
2,73% 0,570
0,600
5 E2
0,672
2,82% 0,638
0,643
5 E3
0,359
1,39% 0,350
0,358
5 E4
0,367
1,88% 0,381
0,373
5 E5
0,586
2,10% 0,566
0,588
5 E6
0,620
2,48% 0,590
0,605
5 E7
0,350
1,91% 0,342
0,337
5 E8
0,342
4,70% 0,347
0,373
5 E9
0,450
1,13% 0,440
0,444
1 E10
0,419
4,53% 0,393
-
5 E11
0,420
5,84% 0,415
0,461
Tabela 20 – Absorvâncias medidas nos extratos enzimáticos obtidos com efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de
Variação
7 30 min
0,349
3,11% 0,371
0,364
7 1horas
0,354
2,94% 0,374
0,371
7 1h30
-
0,77% 0,363
0,367
7 2horas 0,379
1,59% 0,389

78
0,378
7 2h30
0,396
0,51% 0,394
0,398
7 3horas
0,403
0,00% 0,403
-
9 3h30
0,312
1,47% 0,308
0,303
9 4horas
0,304
1,31% 0,311
0,311
9 4h30
0,314
0,98% 0,316
0,310
9 5horas
0,312
1,58% 0,322
0,317
As tabelas 21 e 22 dizem respeito às absorvâncias lidas para os extratos alcalinos de
concentração 2% sem e com efeitos cinéticos, respetivamente, e os respetivas referências às curvas
padrões às quais as primeiras estão associadas. Para estas análises, diluíram-se 10µL das amostras
3 e 4 de extrato em 490µL água destilada, sendo que para as restantes amostras fez-se uma diluição
de 5 µL das mesmas em 495µL água destilada.
Tabela 21 – Absorvâncias medidas nos extratos alcalinos a 2% obtidos sem efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de Variação
4 1
0,987
0,18% 0,987
0,990
4 2
0,867
2,75% 0,893
0,916
4 3
0,991
2,92% 1,039
1,046
4 4
0,952
0,58% 0,948
0,959
4 5
0,867
0,98% 0,880
0,864
4 6
0,856
1,83% 0,862
0,886
4 7
0,491
0,57% 0,495
-
5 8
0,485
1,38% 0,474
0,486
5 9
0,741
3,81% 0,782
-
5 10
0,608
2,71% 0,638
0,610
5 11
0,553
1,69% 0,572
0,562

79
Tabela 22 – Absorvâncias medidas nos extratos alcalinos a 2% obtidos com efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de
Variação
8 30 min
0,393
2,53% 0,379
0,398
8 1horas
0,415
3,65% 0,394
0,423
8 1h30
0,414
3,05% 0,390
0,398
8 2horas
0,402
3,02% 0,427
0,415
8 2h30
0,395
4,13% 0,425
0,397
8 3horas
0,377
3,98% 0,402
0,406
8 3h30
0,395
4,52% 0,361
0,376
8 4horas
0,507
2,27% 0,486
0,474
8 4h30
0,459
4,43% 0,465
0,498
8 5horas
0,490
1,64% 0,505
0,492
Por fim, as tabelas 23 e 24 apresentam os valores de absorvâncias lidas para os extratos
alcalinos de concentração 6% e 10% sem efeitos cinéticos e os respetivas referências às curvas
padrões às quais as primeiras estão associadas. Para estas análises, diluíram-se 5µL de cada
amostra em 490µL água destilada.
Tabela 23 – Absorvâncias medidas nos extratos alcalinos a 6% obtidos sem efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de Variação
5 1
0,699
0,36% 0,697
0,702
5 2
0,713
2,61% 0,751
0,729
5 3
0,463
1,75% 0,476
0,461
5 4
0,561
4,17% 0,555
0,599
5 5
0,775
1,44% 0,797
0,791
5 6
0,922
1,04% 0,928
0,941
5 7
0,654
1,47% 0,635
0,645
5 8 0,564
2,16% 0,587

80
0,584
5 9
0,682
2,94% 0,707
0,723
5 10
0,600
2,13% 0,588
0,575
5 11
0,567
1,33% 0,579
0,565
Tabela 24 – Absorvâncias medidas nos extratos alcalinos a 10% obtidos sem efeitos cinéticos
Padrão Amostras Absorvância Coeficiente de
Variação
6 1
0,709
1,80% 0,699
0,684
6 2
0,803
1,62% 0,826
0,826
6 3
0,514
3,21% 0,499
0,482
6 4
0,520
3,26% 0,536
0,555
6 5
0,763
1,67% 0,755
0,780
6 6
0,999
0,21% 1,000
1,003
6 7
0,571
0,98% 0,582
0,579
6 8
0,590
1,99% 0,602
0,614
6 9
0,642
1,33% 0,645
0,629
6 10
0,376
1,74% 0,389
0,380
6 11
0,731
1,85% 0,745
0,718
Sempre que se efetuou um ensaio de Dosagem de açúcar total (DAT) e o espectrofotómetro
não se encontrava ainda em operação, montava-se uma nova curva padrão de D-Glucose. Esta curva
é traçada com base na Lei de Lambert-Beer (equação 10) que descreve uma relação de linearidade
entre a absorvância (Ab) lida com a concentração de glucose (Cglu).
(Equação 10)
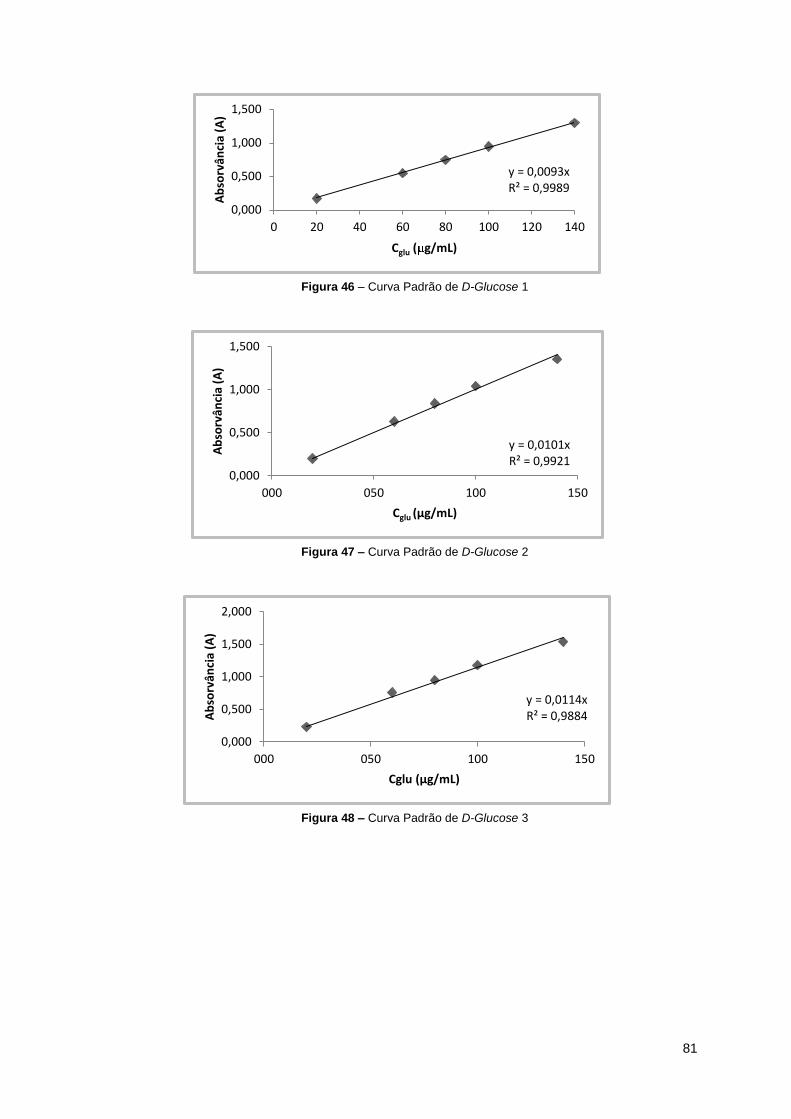
81
Figura 46 – Curva Padrão de D-Glucose 1
Figura 47 – Curva Padrão de D-Glucose 2
Figura 48 – Curva Padrão de D-Glucose 3
y = 0,0093x R² = 0,9989
0,000
0,500
1,000
1,500
0 20 40 60 80 100 120 140A
bso
rvân
cia
(A)
Cglu ( g/mL)
y = 0,0101x R² = 0,9921
0,000
0,500
1,000
1,500
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)
y = 0,0114x R² = 0,9884
0,000
0,500
1,000
1,500
2,000
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)

82
Figura 49 – Curva Padrão de D-Glucose 4
Figura 50 – Curva Padrão de D-Glucose 5
Figura 51 – Curva Padrão de D-Glucose 6
y = 0,0094x R² = 0,9982
0,000
0,200
0,400
0,600
0,800
1,000
1,200
1,400
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)
y = 0,0094x R² = 0,9988
0,000
0,500
1,000
1,500
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)
y = 0,0109x R² = 0,9881
0,000
0,500
1,000
1,500
2,000
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)

83
Figura 52 – Curva Padrão de D-Glucose 7
Figura 53 – Curva Padrão de D-Glucose 8
Figura 54 – Curva Padrão de D-Glucose 9
8.D - Gráficos de superfície de resposta para determinação da melhor
condição de extração
Através do software Statistica 8.0 foi possível obter os gráficos de superfície de resposta para
os três solventes em estudo. As figuras 55 a 69 apresentam então essas representações gráficas
para os solventes água, solução aquosa de Celulase, KOH 2%, 6% e 10%.
y = 0,0117x R² = 0,9966
0,000
0,500
1,000
1,500
2,000
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)
y = 0,0134x R² = 0,9865
0,000
0,500
1,000
1,500
2,000
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)
y = 0,0087x R² = 0,9981
0,000
0,200
0,400
0,600
0,800
1,000
1,200
1,400
000 050 100 150
Ab
sorv
ânci
a (A
)
Cglu (µg/mL)

84
Figura 55 – Superfície de resposta para solvente água (Massa de açúcar f(R/T))
Figura 56 - Superfície de resposta para solvente água (Massa de açúcar f(tt/R))
Solvente: Água
> 0,9
< 0,9
< 0,8
< 0,7
< 0,6
Solvente: Água
> 0,8
< 0,8
< 0,7
< 0,6

85
Figura 57 - Superfície de resposta para solvente água (Massa de açúcar f(T/tt))
Figura 58 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(R/CE))
Solvente: Água
> 0,8
< 0,8
< 0,7
< 0,6
< 0,5
Solvente: Solução aquosa de Celulase
> 0,74
< 0,74
< 0,72
< 0,7
< 0,68
< 0,66
< 0,64
< 0,62
< 0,6

86
Figura 59 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(tt/R))
Figura 60 - Superfície de resposta para solvente aquoso de Celulase (Massa de açúcar f(CE/tt))
Solvente: Solução aquosa de Celulase
> 0,74
< 0,74
< 0,72
< 0,7
< 0,68
< 0,66
< 0,64
< 0,62
< 0,6
< 0,58
Solvente: Solução aquosa de Celulase
> 0,67
< 0,67
< 0,66
< 0,65

87
Figura 61 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(R/T))
Figura 62 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(tt/R))
Solvente: Solução aquosa KOH (2%)
> 2,1
< 2,1
< 2
< 1,9
< 1,8
Solvente: Solução aquosa KOH (2%)
> 2,1
< 2,1
< 2
< 1,9
< 1,8
< 1,7

88
Figura 63 - Superfície de resposta para solvente aquoso de KOH 2% (Massa de açúcar f(T/tt))
Figura 64 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(R/T))
Solvente: Solução aquosa KOH (2%)
> 2
< 2
< 1,9
< 1,8
Solvente: Solução aquosa KOH (6%)
> 2,4
< 2,4
< 2,2
< 2
< 1,8
< 1,6
< 1,4

89
Figura 65 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(tt/R))
Figura 66 - Superfície de resposta para solvente aquoso de KOH 6% (Massa de açúcar f(T/R))
Solvente: Solução aquosa KOH (6%)
> 2,4
< 2,4
< 2,2
< 2
< 1,8
< 1,6
Solvente: Solução aquosa KOH (6%)
> 2,2
< 2,2
< 2,1
< 2
< 1,9
< 1,8
< 1,7

90
Figura 67 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(R/T))
Figura 68 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(tt/R))
Solvente: Solução aquosa KOH (10%)
> 2
< 2
< 1,8
< 1,6
< 1,4
Solvente: Solução aquosa KOH (10%)
> 1,9
< 1,9
< 1,8
< 1,7
< 1,6
< 1,5
< 1,4

91
Figura 69 - Superfície de resposta para solvente aquoso de KOH 10% (Massa de açúcar f(T/tt))
8.E – Ajustes à 2ª Lei de Fick
Através do software Statistica 8.0, foi também possível fazer o ajuste não linear à 2ª Lei de
Fick, através dos resultados cinéticos obtidos. Para tal foi necessário adimensionalizar a
concentração de açúcar obtida por DAT. Neste anexo estão apresentadas as tabelas com as
concentrações de açúcar adimensionalizadas (C*) para cada solvente estudado e o respetivo ajusto
obtido através do programa mencionado.
Tabela 25 – Concentração de açúcar adimensionalizada dos extratos aquosos para estudo da cinética
Tempo (h) Concentração açúcar (µg/mL)
C*
0 0 1
0,5 1686,54 0,384
1 1899,08 0,300
1,5 2223,24 0,171
2 2241,59 0,164
2,5 2220,80 0,172
3 2296,64 0,142
3,5 2492,35 0,064
4 2654,43 0
Solvente: Solução aquosa KOH (10%)
> 1,9
< 1,9
< 1,8
< 1,7
< 1,6
< 1,5

92
Figura 70 – Ajuste à 2ª Lei de Fick para a cinética da extração aquosa (R = 0,972)
Tabela 26 – Concentração de açúcar adimensionalizada dos extratos enzimáticos para estudo da cinética
Tempo (h) Concentração açúcar (µg/mL)
C*
0 0 1
0,5 1544,16 0,152
1 1565,53 0,141
1,5 1559,83 0,144
2 1632,48 0,104
2,5 1692,31 0,071
3 1722,22 0,055
3,5 1768,20 0,029
4 1773,95 0,026
4,5 1800,77 0,012
5 1821,84 0
Figura 71 – Ajuste à 2ª Lei de Fick para a cinética da extração enzimática (R = 0,968)

93
Tabela 27 – Concentração de açúcar adimensionalizada dos extratos alcalinos a 2% para estudo da
cinética
Tempo (h)
Concentração açúcar (µg/mL)
C*
0 0 1
0,5 2910,45 0,213
1 3064,68 0,171
1,5 2990,05 0,192
2 3094,53 0,163
2,5 3027,36 0,182
3 2947,76 0,203
3,5 2815,92 0,239
4 3606,97 0,025
4,5 3537,31 0,044
5 3699,00 0
Figura 72 – Ajuste à 2ª Lei de Fick para a cinética da extração alcalina a 2% (R = 0,922)
8.F – Curvas padrão e absorvâncias lidas para os ensaios de Microfiltração e
Ultrafiltração
Nas tabelas 28 e 290estão agrupadas as absorvâncias lidas para as frações de Microfiltração e
Ultrafiltração, respetivamente, e os respetivas referências às curvas padrões às quais as primeiras
estão associadas. Para estas análises, diluíram-se 10µL de amostra de extrato em 490µL água
destilada.

94
Tabela 28 – Absorvâncias medidas nas frações do processo de Microfiltração
Padrão Amostras Absorvância Coeficiente de
Variação
7 Alimentação
0,365
3,26% 0,367
0,387
7 Concentrado
0,453
1,89% 0,453
0,468
7 Permeado
-
0,26% 0,272
0,273
Tabela 29 – Absorvâncias medidas nas frações do processo de Ultrafiltração
Padrão Amostras Absorvância Coeficiente de
Variação
10 AI
0,415
2,94% 0,424
0,400
10 RI
0,437
2,93% 0,455
0,430
10 PI
0,201
2,31% 0,203
0,210
10 AII
0,412
1,73% 0,406
0,398
10 RII
0,407
0,88% 0,414
0,412
10 PII
0,199
4,02% 0,188
0,217
Sempre que se efetuou um ensaio de Dosagem de açúcar total (DAT) e o espectrofotómetro
não se encontrava ainda em operação, montava-se uma nova curva padrão de D-Glucose. Esta curva
é traçada com base na Lei de Lambert-Beer (equação 10) que descreve uma relação de linearidade
entre a absorvância (Ab) lida com a concentração de glucose (Cglu).
Figura 73 – Curva Padrão de D-Glucose 7
y = 0,0117x R² = 0,9966
0,000
0,500
1,000
1,500
2,000
000 050 100 150
Ab
sorv
ânci
a (A
)
Cgluc (µg/mL)

95
Figura 74 – Curva Padrão de D-Glucose 10
8.G – Determinação da Atividade Antioxidante (AA)
Os valores de atividade antioxidante foram medidos espectrofotometricamente e calculados
com base na curva padrão de Trolox apresentada na figura 76.
Figura 75 – Curva padrão de Trolox
O cálculo desta propriedade das amostras analisadas foi feito através das equações seguintes.
Onde mextrato e mbagaço são, respetivamente as massas de extrato e de bagaço da extração e o
declive é o declive obtido a partir da regressão linear dos pontos da curva padrão de ácido gálico.
As leituras realizadas em triplicado apresentam-se agrupadas na tabela 30.
y = 0,0092x R² = 0,9995
0,000
0,500
1,000
1,500
000 050 100 150
Ab
sorv
ânci
a (A
)
Cgluc (µg/mL)
y = 0,0008x R² = 0,9989
,000
,100
,200
,300
,400
,500
,600
,000 200,000 400,000 600,000 800,000
Ab
sorv
ânci
a
Concentração Trolox ( M)

96
Tabela 30 – Absorvâncias medidas para determinação da Atividade Antioxidante
Amostra mamostra
(g) Vextrato (mL)
Camostra (g/L)
Abs.controlo Abs. Trolox ( M) eq
PI
2,9365 25,00 117,4600 0,691 0,561 162,50
2,9377 25,00 117,5080 0,691 0,576 143,75
2,9316 25,00 117,2640 0,691 0,618 -
H
2,9245 25,00 116,9800 0,690 0,621 -
2,9452 25,00 117,8080 0,690 0,595 118,75
2,9051 25,00 116,2040 0,690 0,600 112,50
PII
2,9175 25,00 116,7000 0,687 0,585 127,50
2,9281 25,00 117,1240 0,687 0,580 133,75
2,9377 25,00 117,5080 0,687 0,587 125,00
RI
2,9125 25,00 116,5000 0,690 0,485 256,25
2,9233 25,00 116,9320 0,690 0,489 251,25
2,8614 25,00 114,4560 0,690 0,493 246,25
RII
2,9119 25,00 116,4760 0,691 0,535 195,00
2,9237 25,00 116,9480 0,691 0,528 203,75
2,9406 25,00 117,6240 0,691 0,507 -
K
3,1182 25,00 124,7280 0,688 0,493 243,75
3,0871 25,00 123,4840 0,688 0,502 232,50
3,1027 25,00 124,1080 0,688 - -
E
2,9186 25,00 116,7440 0,687 0,668 23,75
2,9325 25,00 117,3000 0,687 0,672 -
2,9545 25,00 118,1800 0,687 0,665 27,50
Onde mamostra é a massa de amostra que foi analisada, Vextrato é o volume de extrato utilizado para extrair os compostos fenólicos, Abs. é a absorvância lida e Abs.controle é a absorvância lida para o radical.
8.H – Determinação dos Fenólicos Totais (FT)
A quantidade de Fenólicos totais foi medida espectrofotometricamente e calculada com base
na curva padrão de ácido gálico apresentada na figura 77.
Figura 76 – Curva padrão de Ácido Gálico
y = 0,0105x R² = 0,9958
0,000
0,200
0,400
0,600
0,800
1,000
1,200
0,000 20,000 40,000 60,000 80,000 100,000 120,000
Ab
sorv
ânci
a
Concentração ác. gálico (mg/L)

97
O cálculo da quantidade de fenólicos totais (FT) das amostras analisadas foi feito através das
equações seguintes.
Onde mextrato e mbagaço são, respetivamente as massas de extrato e de bagaço da extração e o
declive é o declive obtido a partir da regressão linear dos pontos da curva padrão de ácido gálico.
As leituras realizadas em triplicado apresentam-se agrupadas na tabela 31.
Tabela 31 – Absorvâncias medidas para determinação dos Fenólicos Totais
Amostra mamostra
(g) Abs. Abs. – Abs.B
Cácido gálico
(mg/L) CFenólicos
(mg/g)
K
4,3614 0,777 0,764 78,76 4,51
4,0244 0,701 0,688 70,93 4,41
4,0275 0,674 0,661 68,14 4,23
P
6,1312 0,093 0,080 8,25 0,34
6,4872 0,127 0,114 11,75 0,45
6,1057 0,122 0,109 11,24 0,46
R
6,8282 0,233 0,220 22,68 0,83
6,7034 0,240 0,227 23,40 0,87
6,6195 0,246 0,233 24,02 0,91
Onde mamostra é a massa de amostra que foi analisada, Abs. é a absorvância lida e Abs.branco é a absorvância lida para o branco.