dispersÃo sÓlida como tecnologia para melhoria …
TRANSCRIPT

UNIVERSIDADE FEDERAL DE MATO GROSSO
CAMPUS UNIVERSITÁRIO DO ARAGUAIA
INSTITUTO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE
CURSO DE GRADUAÇÃO EM FARMÁCIA
JOÃO AUGUSTO GARCIA AGUIAR DO NASCIMENTO
DISPERSÃO SÓLIDA COMO TECNOLOGIA PARA
MELHORIA DAS PROPRIEDADES BIOFARMACÊUTICAS:
ÊNFASE EM ANTI-INFLAMATÓRIOS NÃO ESTEROIDAIS
Barra do Garças –MT
2018

UNIVERSIDADE FEDERAL DE MATO GROSSO
CAMPUS UNIVERSITÁRIO DO ARAGUAIA
INSTITUTO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE
CURSO DE GRADUAÇÃO EM FARMÁCIA
JOÃO AUGUSTO GARCIA AGUIAR DO NASCIMENTO
DISPERSÃO SÓLIDA COMO TECNOLOGIA PARA
MELHORIA DAS PROPRIEDADES BIOFARMACÊUTICAS:
ÊNFASE EM ANTI-INFLAMATÓRIOS NÃO ESTEROIDAIS
Monografia apresentada à banca examinadora do
Curso de Graduação de Bacharelado em
Farmácia do Campus Universitário do
Araguaia/UFMT, como requisito parcial para
obtenção do Título de Bacharel em Farmácia.
ORIENTADOR: Prof. Dr. Fernando Boldrini
Barra do Garças –MT
2018

JOÃO AUGUSTO GARCIA AGUIAR DO NASCIMENTO
DISPERSÃO SÓLIDA COMO TECNOLOGIA PARA
MELHORIA DAS PROPRIEDADES BIOFARMACÊUTICAS:
ÊNFASE EM ANTI-INFLAMATÓRIOS NÃO ESTEROIDAIS
Monografia apresentada à banca examinadora do
curso de Farmácia do Campus Universitário do
Araguaia/UFMT, como exigência para a
obtenção do Título de Bacharel em Farmácia.
BANCA EXAMINADORA
___________________________________________________
Prof. Dr. Fernando Boldrini
ORIENTADOR
___________________________________________________
Profª. Dra. Eliane Aparecida Suchara
EXAMINADORA
___________________________________________________
Prof. Dr. Claudemir Batalini
EXAMINADOR
Nota final:_____
Barra do Garças – MT
2018

RESUMO
A dispersão sólida é uma tecnologia muito empregada com a finalidade de melhorar a
biodisponibilidade oral de fármacos pouco solúveis como os anti-inflamatórios não esteroidas
(AINE's). Este trabalho teve por objetivo revisar a literatura sobre a aplicação da tecnologia
de dispersão sólida na melhoria das propriedades biofarmacêuticas de fármacos anti-
inflamatórios não esteroidais presentes em formas farmacêuticas sólidas orais. A plataforma
digital Web of Science foi a principal base de dados utilizada no período de 1961-2018.
Segundo o Sistema de Classificação Biofarmacêutica (SCB) os AINES’s podem ser da classe
II ou IV, em virtude de baixa solubilidade, e com isso devem ser empregadas novas
tecnologias, como as dispersões sólidas, para que seja possível a otimização do medicamento
em relação a sua biodisponibilidade. Isso decorre de propriedades típicas das dispersões
sólidas, tais como, redução total ou parcial de cristalinidade do fármaco e matriz, redução do
tamanho de partícula do fármaco, podendo alcançar o nível molecular e aumento de afinidade
do fármaco com o meio aquoso decorrente do uso de matrizes hidrofílicas. Os estudos
indicam que as dispersões sólidas são uma tecnologia eficiente para o aumento da
solubilidade dos AINE'S com resultados muito acima das formulações convencionais
disponibilizadas no mercado.
Palavras chave: Dissolução, Biodisponibilidade, AINE's.

ABSTRACT
Solid dispersion is a widely used technology for the purpose of improving the oral
bioavailability of poorly soluble drugs such as non-steroidal anti-inflammatory drugs
(NSAIDs). The objective of this work was to review the literature on the application of solid
dispersion technology in improving the biopharmaceutical properties of non - steroidal anti -
inflammatory drugs present in solid oral dosage forms. The Web of Science digital platform
was the main database used in the period 1961-2018. According to the Biopharmaceutical
Classification System (BCS), NSAIDs may be class II or IV, due to low solubility, and with
this new technologies, such as solid dispersions, must be employed, in order to optimize the
drug in relation to their bioavailability. This is due to the typical properties of the solid
dispersions, such as total or partial reduction of drug and crystalline matrix, reduction of drug
particle size, being able to reach the molecular level and increase of drug affinity with the
aqueous medium resulting from the use of hydrophilic matrices. The studies indicate that
solid dispersions are an efficient technology for increasing the solubility of NSAIDs with
results well above the conventional formulations available on the market.
Keywords: Dissolution, Bioavailability, NSAIDs.

LISTA DE FIGURAS
FIGURA 1 - Diferentes classes biofarmacêuticas de fármacos segundo o SCB ............. 18
FIGURA 2 - Processo do comprimido até a absorção ..................................................... 23
FIGURA 3 - Influências que o a matriz e o fármaco tem na dispersão ........................... 24
FIGURA 4 - Gerações das dispersões sólidas e suas características ............................... 26
FIGURA 5 - Perfil de dissolução por gerações de dispersão sólida ............................... 26
FIGURA 6 - Formas de dispersões sólidas ...................................................................... 27
FIGURA 7 - Exemplificação da extrutora por fusão a quente ......................................... 33
FIGURA 8 - Esquema de preparação da dispersão sólida por aspersão .......................... 36
FIGURA 9 - Cascata de inflamação ................................................................................. 38
FIGURA 10 - Estrutura molecular do Ibuprofeno ........................................................... 39
FIGURA 11 - Perfil de dissolução comparado com a MM do polímero ......................... 40
FIGURA 12 - Perfil de dissolução comparado com solvente usado na obtenção da DS . 40
FIGURA 13 - Comparação do perfil de dissolução da dispersão sólida com diferentes
tensoativos .........................................................................................................................
41
FIGURA 14 - Comparação da diferença e proporção de solventes (a) acetona; (b)
etanol, utilizados para a preparação de dispersão sólida de ibuprofeno e seu perfil de
dissolução ..........................................................................................................................
42
FIGURA 15 - Imagem de microscopia eletrônica de varredura comparando a dispersão
sólida 1:4 com acetona com a dispersão sólida 1:4 com etanol ........................................
43
FIGURA 16 - Estrutura molecular do Naproxeno ........................................................... 43
FIGURA 17 - Estrutura molecular do Cetoprofeno ......................................................... 44
FIGURA 18 - Imagem da Difração de Raio-X, (A) Cetoprofeno; (B) PM 30%
Cetoprofeno K30; (C) MF 30% Cetoprofeno PVPVA 6: 4; (D) MF 30% Cetoprofeno
PVA; (E) SD 30% Cetoprofeno PVP K30; (F) SD 30% Cetoprofeno PVPVA 6: 4; (G)
SD 30% Cetorpofeno PVA e (H) PVA puro .....................................................................
45
FIGURA 19 - (A) Perfil de dissolução cetoprofeno e PVP K30; (B) Perfil de
dissolução cetoprofeno PVP/VA; (C) Perfil de dissolução cetoprofeno e PVA; (D)
Comparação do perfil de dissolução de todas as dispersões sólidas .................................
46
FIGURA 20 - Possível mecanismo de liberação para a dispersão sólida com matriz
PVP-K30 ...........................................................................................................................
46
FIGURA 21 - Estrutura molecular do Piroxicam ............................................................. 47

FIGURA 22 - Imagem da difração de Raio-X do piroxicam puro, mistura fisica e
dispersão sólida para PVP K90 e PVP K17 ......................................................................
48
FIGURA 23 - Imagem de IVTF caracterização da amorfização da dispersão sólida ...... 48
FIGURA 24 - Comparação do perfil de dissolução das Dispersões sólidas com as
matrizes PVP K-90 e K17 .................................................................................................
49
FIGURA 25 - Estrutura molecular da Nimesulida ........................................................... 50

LISTA DE SIGLAS E ABREVIATURAS
AA - Ácido araquidônico
AAS - Ácido acetilsalicílico
AINE's - Anti-inflamatórios não esteroidais
Compritol 888 ATO® - Behenato de glicerilo
COX-1 - Ciclooxigenase-1
DS - Dispersão sólida
FDA - Food and Drug Administration
IVTF - Infra Vermelho com Transformador de Fourier
Gelucire® - Stearoil polioxil-32 glicerides
HPMC - Hidroxipropilmetilcelulose
Kollicoat IR ® - polivinil álcool/polietileno glicol
Lutrol ® F 68 - Poloxamer 188
MF - Mistura Fisíca
PEG - Polietilenoglicóis
PVA - Acetato de polivinila
PVP - Polivinilpirrolidona
SCB - Sistema de Classificação Biofarmacêutica
Soluplus® - Polivinil caprolactam-polivinil acetato-poliethileno glicol
TGI - Trato gastrointestinal
Tween 80® - Polissorbato 80

LISTA DE TABELAS
TABELA 1 - Fatores que interferem na biodisponibilidade .................................................. 16

Sumário 1. Objetivo ............................................................................................................... 12
1.1.Objetivo geral ................................................................................................ 12
1.2.Objetivo específico ........................................................................................ 12
2. Metodologia ......................................................................................................... 13
3. Revisão de literatura ............................................................................................ 14
3.1.Introdução ...................................................................................................... 14
3.2.Biofarmácia ................................................................................................... 15
3.2.1.Fatores que afetam a biodisponibilidade ..............................................15
3.2.2.Sistema de Classificação Biofarmacêutica ........................................... 18
3.3.Mecanismo de liberação ................................................................................ 22
3.3.1.Desintegração e dissolução ................................................................... 22
3.3.2.Mecanismo de liberação das dispersões sólidas ................................... 24
3.4.Dispersão Sólida ............................................................................................ 25
3.4.1.Dispersão sólida cristalina (1ª geração) ................................................ 27
3.4.2.Dispersão sólida amorfa (2ª geração) ................................................... 28
3.4.3.Dispersão sólida amorfa + adjuvante (3ª geração) ............................... 30
3.4.4.Dispersão sólida amorfa de liberação controlada (4ª geração) ............. 31
3.5.Método de preparação ................................................................................... 32
3.5.1.Fusão ..................................................................................................... 32
3.5.2.Solvente ................................................................................................ 34
3.5.3.Solvente-fusão ...................................................................................... 37
3.6.Anti-inflamatórios não esteroidais ................................................................ 37
3.6.1.Ibuprofeno ............................................................................................ 39
3.6.2.Naproxeno ............................................................................................ 43
3.6.3.Cetoprofeno .......................................................................................... 44
3.6.4.Piroxicam .............................................................................................. 47
3.6.5.Nimesulida ............................................................................................ 50

4. Conclusão ............................................................................................................ 51
5. Referências .......................................................................................................... 52

12
1. Objetivo
1.1. Objetivo geral
O presente trabalho tem como finalidade desenvolver uma revisão da literatura sobre a
tecnologia da dispersão sólida aplicada a melhoria das propriedades biofarmacêuticas de
fármacos anti-inflamatórios não esteroidais presentes em formas farmacêuticas sólidas
destinadas a administração oral.
1.2. Objetivo específico
• Descrever as diferentes técnicas de preparação de dispersões sólidas;
• Descrever as possíveis composições para as dispersões sólidas;
• Relacionar a composição das dispersões sólidas e seus mecanismos de
liberação;
• Analisar a viabilidade das dispersões sólidas para correção de problemas de
baixa solubilidade dos anti-inflamatórios não esteroidais;
• Desenvolvimento de revisão das principais anti-inflamatórios não esteroidais
desenvolvidos como dispersões sólidas.

13
2. Metodologia
Nesta revisão foi utilizada as base de dados Web of Science, Science Direct e
PubMed, nos períodos de 1961-2018. O inglês foi a língua empregada na busca dos artigos.
Foram utilzadas as palavras-chave Solid dispersion, Solid dispersion amorphous, first solid
dispersion, non-steroidal anti-inflammatory and solid dispersion, non-steroidal anti-
inflammatory e solid dispersion method.

14
3. Revisão de literatura
3.1. Introdução
Com o passar das décadas a indústria farmacêutica percebeu que não era possível a
utilização apenas de fármacos com os requisitos de boa dissolução e permeação, pois a
maioria dos novos fármacos desenvolvidos já não apresentavam mais essas características
físico-químicas (KU & DULIN, 2012; VAN DEN MOOTER et al., 2006).
Para racionalizar o desenvolvimento de fármacos com problemas de dissolução e
permeação, em 1995 Amidon et al. criaram o sistema de classificação biofarmacêutica (SCB),
no qual os fármacos foram classificados conforme as duas propriedades de solubilidade e
permeabilidade. O SCB auxilia na melhor compreensão das necessidades típicas de cada
fármaco. Logo, segundo o SCB, resolvendo as limitações de cada classe, a biodisponibilidade
de cada fármaco aumenta, viabilizando o uso de muitos na terapêutica. Surgiram então
variadas tecnologias para elucidar tais limitações, tanto na modificação estrutural do fármaco,
como suas características físico-químicas, forma farmacêutica, entre outros (JERMAIN;
BROUGH; WILLIAMS III, 2018; KORN & BALBACH, 2014).
Das classes do SCB, a classe II. é a que mais tem estudos para solucionar suas
limitações. A solubilidade em tese é mais fácil de resolver, pois tem menos fatores externos
ao fármaco envolvidos na problemática. Dentre todas as tecnologias que são utilizadas para
aumentar a solubilidade do fármaco, o método de dispersão sólida tem sido a mais
promissora. Dentro da classe II os anti-inflamatórios não esteroidais (AINE's) é uma das
classes do SCB que mais possuem fármacos (ADEBISI et al., 2016).
As dispersões sólidas consiste em uma metodologia de trabalho antiga que vem sendo
aprimorada durante anos. Hoje ela é definida como uma dispersão de um fármaco
cristalino/amorfo em uma ou mais matrizes, através de métodos de fusão, solvente ou
solvente-fusão. A metodologia será escolhida de acordo com a necessidade de cada fármaco
ou matriz trabalhada (THIRY et al., 2016; SOSNIK & SEREMETA, 2015).
As características das dispersões sólidas para melhorar a solubilidade e dissolução está
na diminuição drástica das partículas do fármaco, a mudança da partícula do estado cristalino
para o amorfo quando dispersa na matriz, a utilização de polímeros hidrofílicos na formulação
e mais recentemente, a adição de adjuvantes que diminuem a força de coesão das partículas
(KOJO et al., 2017).

15
O aumento da solubilidade é de suma importância e reduzir a quantidade de fármaco
ingerido para uma ação terapêutica eficaz reduz a possibilidade da ocorrência de efeitos
adversos. Por exemplo, a inibição da ciclooxigenase-1 (COX-1), que tem ação citoprotetora
da mucosa gastrointestinal, pode causar úlceras ou até insuficiência renal (SMITH et al.,
2012;BATLOUNI, 2010).
3.2. Biofarmácia
Biofarmácia é definida como as características físico-químicas dos fármacos, forma de
dosagem e via de administração que possam afetar o perfil e o tempo de absorção do
medicamento. A relação medicamento, dosagem e via de administração determina a sua
biodisponibilidade efetiva. Para que um medicamento tenha o efeito desejado é necessário que
o fármaco alcance o seu sítio ativo e permaneça nele por tempo suficiente; e isso irá depender
da rota de administração, da forma em que é administrada e a taxa de dissolução (AULTON,
2005).
3.2.1. Fatores que afetam a biodisponibilidade
A biodisponibilidade de um fármaco administrado pela via oral está ligada a vários
fatores intrínsecos e extrínsecos. Os fatores intrínsecos ao medicamento, isto é, dependentes
dele são: características físico-químicas do fármaco, forma farmacêutica, excipientes
utilizados na sua formulação, processo de fabricação, via de administração e farmacocinética
(AULTON, 2005).
Os fatores extrínsecos, ou seja, aqueles exógenos ao medicamento, e, por conseguinte
ao fármaco são: tempo de transito gastrointestinal, metabolização de primeira passagem,
condições patológicas e idade (AULTON, 2005).
Os fatores intrínsecos ao fármaco são os mais relevantes, visto que são exatamente
nesses fatores que as dispersões sólidas irão atuar, deixando o fármaco com partículas
menores, na sua forma amorfa, com excipientes que irão ajudar na dispersão e dissolução e
utilizando um processo de fabricação que seja ideal. (AULTON, 2005).
Como a tecnologia que envolve a preparação de dispersões sólidas visa maximizar a
redução do tamanho de partículas do fármaco, na sua forma amorfa, com o uso de excipientes
que irão ajudar na dispersão e dissolução, os fatores que estão ligados ao fármaco se tornam

16
os mais relevantes. Portanto, serão esses os fatores mais abordados. De modo geral, os
principais fatores que afetam a biodisponibilidade de fármacos pela via oral estão
apresentados na Tabela 1 (ALLEN & ANSEL, 2013).
Tabela 1 - Fatores que interferem na biodisponibilidade de fármacos (fonte: modificado de ALLEN &
ANSEL, 2013)
Principais fatores que afetam a biodisponibilidade de fármacos pela via oral
Fatores relacionados ao fármaco
• Tamanho de partícula
• Cristalinidade e forma amorfa
• Forma salina
• Hidratação
• Solubilidade lipídica e aquosa
• pH e pka
• Complexos
Características próprias da forma farmacêutica
• Taxa de desintegração de comprimidos
• Tempo de dissolução
• Idade do produto e condições de estocagem
• Processo de fabricação
Excipientes farmacêuticos
• Diluentes
• Aglutinantes
• Desintegrantes
• Lubrificantes
• Agentes suspensores
• Agentes molhantes
• Agentes reológicos
• Estabilizantes
Fisiologia e características do paciente
• Tempo de esvaziamento gástrico
• Tempo de trânsito intestinal
• Anormalidade gastrintestinal ou condição
patológica
• Conteúdo gástrico
• Presença de outros fármacos
• Alimentos
• Condição dos fluidos
• pH gastrintestinal
• Metabolismo do fármaco
A redução do tamanho de partícula do fármaco pouco solúvel afeta a
biodisponibilidade porque aumenta a área de superfície, consequentemente, aumenta a taxa de
dissolução. A micronização é uma estratégia muito empregada para essa finalidade, porém
pode não ser capaz de promover substancial aumento da biodisponibilidade. A preparação de
dispersões sólidas consiste justamente em reduzir o tamanho da partícula do fármaco. Por
exemplo, na técnica em que se emprega a fusão do componente da matriz hidrofílica, como o
polietilenoglicol (PEG), com o fármaco, se alcança alto nível de redução de tamanho,
podendo chegar a dispersar molecularmente. Uma vez em um fluido gastrintestinal, a matriz

17
se dissolve rapidamente, tornando mais fácil a dissolução do fármaco (ALLEN & ANSEL,
2013).
Muitos fármacos possuem não apenas uma forma cristalina, com forma, estrutura e
propriedades físicas definidas, mas várias. Polimorfismo é o fenômeno no qual um fármaco
possui dois ou mais diferentes cristais, chamados de polimorfos. Esses podem ter não apenas
diferenças na solubilidade, mas também atividade biológica. Quando não há estrutura
cristalina definida é dito que o fármaco se apresenta amorfo. A dispersão sólida pode interferir
na estrutura de arranjo molecular do fármaco podendo obter preferencialmente seu amorfo, ou
a estabilização de algum polimorfo mais adequado. Em geral a técnica visa obter o fármaco
amorfo porque nessa forma se torna mais solúvel. Por exemplo, a insulina aplicada via
subcutânea pode estar amorfa, solúvel, ou cristalina, precipitada. Na forma amorfa a insulina é
usada para ação rápida, já na cristalina pode ser usada para ação prolongada até ação
ultralenta (ALLEN & ANSEL, 2013).
Outros importantes fatores que podem ser destacados são a forma salina ou hidratada
em que fármaco pode se apresentar. Como sal um fármaco se torna mais solúvel, logo, mais
biodisponível. A teofilina é usada como broncodilatador e é uma base pouco solúvel, podendo
ser preparada, na forma de solução apenas em misturas hidroalcoólica, mas o seu sal
etilenodiamina é bem solúvel em água, portanto preferível a ser usado (ALLEN & ANSEL,
2013).
Um fármaco na forma hidratada, isto é, possui molécula de água e seu retículo
cristalino, possui menor solubilidade que sua forma anidra. A ampicilina anidra é muito mais
solúvel que a trihidratada, por exemplo (ALLEN & ANSEL, 2013).
Os excipientes farmacêuticos desempenham fundamental importância na constituição
da forma farmacêutica na qual o fármaco estará presente, cada categoria atuando de modo
particular. Porém, se não houver um trabalho rigoroso do desenvolvimento da formulação é
possível que possam interferir negativamente na biodisponibilidade do fármaco. Os diluentes,
por exemplo, para otimizar a dissolução do fármaco pouco solúvel, devem ser insolúveis,
como o amido, pois não competindo com a água favorecem a dissolução do fármaco, ao
contrário dos solúveis, como lactose. Os aglutinantes naturalmente dificultam a desintegração,
por promoverem a adesão interpartícula, mas em quantidades adequadas o prejuízo na
biodisponibilidade é minimizado. Mas não apenas a quantidade aumentada afeta, o tipo de
substância também pode interferir. Foi demonstrado que a hidróxi-metil-etilcelulose retardou
a dissolução da clorpropamida em comprimidos porque formou um revestimento ao redor das

18
partículas de fármaco. Os lubrificantes geralmente são hidrofóbicos, como o estearato de
magnésio, portanto, sua concentração deve ser controlada para não dificultar a dissolução do
fármaco (ALLEN & ANSEL, 2013; ROWE; SHESKEY; QUINN, 2009 ).
A própria técnica usada na preparação da forma farmacêutica pode interferir
negativamente na biodisponibilidade. No caso de formas sólidas obtidas por compressão se a
pressão exercida pela máquina de compressão for aumentada pode dificultar a dissolução,
uma vez que aumenta a coesividade interpartícula, gerando comprimidos muito duros. Outros
fatores importantes que o formulador farmacêutico deve se atentar é a velocidade de
compressão para comprimidos, o tipo e tamanho de cápsulas gelatinosas e o tamanho de
partícula comparativa. Nesse último caso, por exemplo, a taxa de dissolução de fármaco a
partir de uma suspensão será maior do que uma cápsula, e essa de um comprimido
(AULTON, 2005).
3.2.2. Sistema de Classificação Biofarmacêutica
Amidon et al., (1995) desenvolveram o Sistema de Classificação Biofarmacêutica
(SCB), que tem como objetivo a inclusão dos fármacos em 4 classes distintas, de acordo com
a suas características de permeabilidade e solubilidade, classe I: alta solubilidade e alta
permeabilidade, classe II: baixa solubilidade e alta permeabilidade, classe III: alta
solubilidade e baixa permeabilidade e por ultimo, classe IV: baixa solubilidade e baixa
permeabilidade (Figura 1).
Figura 1 - Diferentes classes biofarmacêuticas de fármacos segundo o SCB (fonte: modificado de Amidon et al.,
1995).

19
Segundo o SCB e seguido pelo Food and Drug Administration (FDA) (2017) um
fármaco terá uma alta solubilidade quando a maior dose oral do princípio ativo for
solubilizado em um volume menor ou igual a 250 mL, em uma faixa de pH de 1 - 6,8, em
uma temperatura 37 ± 1°C. O valor de 250 mL é referência, pois os protocolos de estudos de
bioequivalência preconizam que os pacientes precisam estar em jejum e ingerir a medicação
com um copo de 250 mL de água.
A alta permeabilidade será dada aos fármacos que tiverem sua biodisponibilidade
sistêmica maior ou igual a 85% do princípio ativo ingerido, baseando na verificação do
balanço de massas (FDA, 2017).
Fármacos e critérios de escolha de excipientes
A classe I é constituída pelos fármacos que apresentam alta solubilidade e alta
permeabilidade (AMIDON et al., 1995). Isso significa que o fármaco não precisa sofrer
nenhuma alteração em sua forma para que ele apresente boa biodisponibilidade. Podem ser
citados o metoprolol, propranolol e teofilina (WU & BENET, 2005).
A classe II, de baixa solubilidade e alta permeabilidade (AMIDON et al. 1995), é a
que mais tem crescido nos últimos anos, pois cerca de 70% de potenciais novos fármacos se
encaixam nesta classe (KU & DULIN, 2012) e 40% dos fármacos já em comercialização
tem pouca solubilidade (VAN DEN MOOTER et al., 2006; TAKAGI et al., 2006). Dentre
esses, incluem uma grande parte da classe AINE's como cetoprofeno, ibuprofeno, naproxeno
e piroxicam (WU & BENET, 2005; TSUME, 2014).
Fármacos da classe III têm como fator limitante a baixa permeabilidade, mas sua
solubilidade é alta. Porém, o fármaco não consegue permear a membrana do trato
gastrointestinal (TGI) de forma adequada, prejudicando a absorção, afetando a sua
biodisponibilidade (AMIDON et al., 1995). O captropil, furosemida, amoxicilina e
ciprofloxacin são exemplos de fármacos de classe III (WU & BENET, 2005).
Como as mucosas absortivas possuem composição lipofílica, logo é necessário
empregar meios para tornar o fármaco mais lipofílico quando este estiver na interface
(KAWABATA et al., 2010). Podem ser feitas alterações na estrutura química do fármaco,
com risco de mudar a sua ação farmacológica. Podem ser usados promotores de permeação
como tensoativos, polissacarídeos, ácidos graxos, sais biliares, entre outros (THANOU;
VERHOEF; JUNGINGER, 2001; FASANO, 1998), ou o aumento do tempo de permanência

20
do fármaco no local de absorção, através de novas tecnologias como sistema de vacina oral
(mucojet), sistema para retenção gástrica, e cápsulas telemétricas (CHAVDA; PATEL;
ANAND, 2010; LENNERNÄS & ABRAHAMSSON, 2005).
Na classe IV os fármacos apresentam baixa solubilidade e baixa permeabilidade.
Diante disso, esses fármacos apresentam grandes desafios para a correção da
biodisponibilidade. Fatores fisiológicos influenciam fortemente a absorção desses fármacos,
como tempo de esvaziamento gástrico e tempo de trânsito gastrointestinal, já que, por
consequência, é baixa nesse grupo (HÖRTER & DRESSMAN, 2001). São exemplos de
fármacos da classe IV a amoxicilina triidratada, eritromicina, doxiciclina, sulfametoxazol,
meloxicam, ritonavir, saquinavir e paclitaxel (WU & BENET, 2005).
Excipientes em formas farmacêuticas sólidas orais
Como os medicamentos orais são formulados contendo fármaco e excipientes, torna
necessário conhecer sua importância e critérios para seleção para fármacos específicos
conforme a SCB.
Na preparação de medicamentos orais existirão excipientes mais apropriados para
determinadas classes do SCB, na tentativa de melhorar a biodisponibilidade do fármaco no
TGI. O termo excipiente farmacêutico, ou adjuvante, como definição, é a substância inerte
que não interfere na ação farmacológica e que está presente em uma formulação para otimizar
suas características tanto de fabricação, como a melhoria do fluxo e compactação, quanto a
biodisponibilidade do fármaco, melhorando a estabilidade, desintegração e dissolução
(VILLANOVA, 2010; ROWE; SHESKEY; QUINN, 2009).
Em cada formulação farmacêutica é empregada uma variedade de excipientes que são
classificados em uma série de classes como diluentes, absorventes, veículos, agentes
suspensores, revestimentos, agentes estabilizantes, flavorizantes, corantes, edulcorantes,
agentes molhantes, desagregantes, deslizantes, dentre outros. Porém, as que apresentam maior
importância para as formas farmacêuticas sólidas de uso oral são secantes, aglutinantes,
desintegrantes, diluentes, lubrificantes e molhantes (ALLEN & ANSEL, 2013). Os agentes
secantes, são substâncias que por serem higroscópicas isto é absorve água, protegem a
formulação contra umidade (PAPAGEORGIOU et al., 2017; FERREIRA, 2008). Os
desintegrantes ou desagregantes são utilizados para ajudar na desintegração das formas
farmacêuticas sólidas no TGI. Essa classe é importante, pois a desintegração tem como

21
finalidade o aumento da área superficial e promovendo a dissolução do fármaco (ZHAO et
al., 2017; AKIN-AJANI; ITIOLA; ODEKU, 2016).
Os diluentes têm como objetivo adequar o volume do comprimido para uma devida
compressão, ou para o enchimento de cápsulas (SALVADOR et al., 2012). Além disso,
podem também contribuir na dissolução. A lactose, por exemplo, é um diluente hidrofílico,
podendo aumentar a molhabilidade das partículas e aumentando assim o potencial de
liberação do fármaco (MIRANDA; CARDOSO; MORAIS, 2013). Os lubrificantes atuam no
processo de preparação de comprimidos promovendo a fluidez do pó, isto é, seu fluxo,
evitando a subdosagem de fármaco, porém, interferem dificultando a desintegração
(MERLIM, 2011). Os agentes molhantes têm como finalidade a redução da tensão superficial
entre o fluido gastrintestinal e o sólido, aumentando assim a interação entre as parte e
melhorando a taxa de dissolução (FERREIRA, 2008).
Os excipientes usados para a classe I são aqueles que tendem a apenas manter a
estabilidade e aumentar as propriedades de fluxo, como a sílica coloidal, que tem
características higroscópicas, mantendo o pó seco. A utilização do talco para a lubrificação e
aumento do fluxo, e o amido como diluente para a formulação também são muito usados
(RIBEIRO & RAU, 2012; PRISTA et al., 1995).
Na classe II, por sua baixa solubilidade, é necessário utilizar excipientes que possam
auxiliar no aprimoramento desta limitação, isto é, a incorporação de um tensoativo, como o
lauril sulfato de sódio e o polisorbato 80, que terão como função reduzir a tensão superficial.
Consequentemente melhoram a molhabilidade dos fármacos em água, promovendo aumento
da solubilidade e da taxa de dissolução (CHEN et al., 2016; ASHFORD, 2005).
O amido glicolato de sódio interfere na formulação aumentando a solubilidade dos
fármacos e facilitando a desintegração dos mesmos, sendo então um excelente excipiente para
a melhora da dissolução. Manitol, para essa classe, aparenta ser melhor opção, por ter alta
solubilidade, ajudar na desintegração e por ter baixo risco a incompatibilidade entre outros
componentes da formulação (ROWE; SHESKEY; QUINN, 2009).
O lauril sulfato de sódio é empregado como agente molhante e lubrificante. Ele é
utilizado nessa classe para elevar a dissolução e a biodisponibilidade, pois é um eficiente
agente molhante. Com isso, favorece a uma maior interação entre o fármaco e a água, causada
pelo seu caráter aniônico e sua cadeia apolar de tamanho significativo, e contribui no aumento
da permeabilidade do fármaco em contato com as membranas absortivas (CHORILLI et al.,
2007; ASHFORD, 2005).

22
Na classe III o excipiente mais utilizado é a celulose microcristalina. Isto se deve ao
fato de ter ampla variedade de funções, como ser um excelente diluente, ter propriedades
lubrificantes, desagregantes e adsorventes. Apesar de ser insolúvel na água, auxilia na
desagregação da forma farmacêutica sem interferir na solubilidade do fármaco no meio
dissolvente (SILVA et al., 2015; DOELKER, 1993).
O diluente mais utilizado é o amido, pelas suas características inertes e por não ser
incompatível com os fármacos e outros excipientes (RIBEIRO & RAU, 2012). Por ter
propriedades de fluxo ruim é importante que tenha uma mistura com outro diluente de melhor
fluxo, como a celulose microcristalina, que tem como características de fluxo muito melhor
que o amido, melhorando até a desagregação do fármaco (DIAS et al., 2018; PRISTA et al.,
1995).
Para a classe IV é importante que os excipientes tenham múltiplas funções, pois é
necessário uma série de melhorias nas propriedades do fármaco para aumentar a sua
biodisponibilidade. Devem corrigir ao mesmo tempo a baixa solubilidade e permeabilidade do
fárrmaco. Os excipientes incluem molhantes, desintegrantes, e componentes que promovam
aumento da lipofilia do fármaco, como ésteres graxos, e seu particionamento, como os
polissorbatos. Diante do uso de componentes com ações muitas vezes antagônicas, aliado às
fracas características biofarmacêuticas do fármaco, torna-se um desafio desenvolver eficientes
medicamentos orais para fármacos desta classe do SCB (AULTON, 2005).
3.3. Mecanismo de liberação
3.3.1. Desintegração e dissolução
Para compreensão do mecanismo de liberação das dispersões sólidas, é necessário
conhecer as definições dos processos pelos quais o fármaco passa até sua liberação no meio
gastrointestinal (Figura 2)(AULTON, 2005).

23
Figura 2 - Processo do comprimido até a absorção (fonte: modificado de Aulton et al., 2005).
De acordo com a figura 2, o comprimido passa por três processos determinantes para
que a sua biodisponibilidade seja eficaz: desintegração, desagregação e dissolução
(AULTON, 2005).
A desintegração é o fracionamento do comprimido oral compacto em partículas. Já a
desagregação é o fracionamento dessas partículas em partículas ainda menores. A dissolução,
por sua vez, consiste na passagem do fármaco para sua forma solúvel para então ser
absorvido. É importante ressaltar que quanto maior a superfície de contato, isto é, quanto
menor o tamanho da partícula, maior será o seu potencial para a dissolução (AULTON, 2005).
Podem ser solucionados problemas com a desintegração e dissolução com ajustes na
formulação, como adição de desintegrantes, tensoativos ou a diminuição de aglutinantes; na
regulagem das máquinas de compressão dos pós; e na produção de comprimidos de
desintegração rápida (ALLEN & ANSEL, 2013; KHAN, 2011).
Para melhor abordar estratégias para solucionar a fraca solubilidade aquosa dos
fármacos da classe II do SCB torna-se importante conhecer melhor em que consiste o
fenômeno da dissolução. Dessa forma, abaixo a equação de Noyes-Whitney explica a
dissolução do fármaco na (equação 1):
Equação 1: 𝑑𝐶
𝑑𝑡=𝐷. 𝑆
ℎ(𝐶𝑠 − 𝐶)

24
em que:
• dC/dt, é a taxa de dissolução,
• D, o coeficiente de difusão,
• S, é a superfície de contato do sólido,
• h, será a espessura da camada de difusão,
• Cs, máximo da fármaco dissolvida até sua saturação,
• C, a concentração da fármaco em massa,
De acordo com a equação de Noyes-Whitney os fatores que afetam a taxa de
dissolução do fármaco são definidos como a área de superfície efetiva, o coeficiente de
difusão, a espessura da camada de difusão, a solubilidade da saturação, a quantidade de
fármaco dissolvido e o volume de dissolução (ALLEN e ANSEL, 2013).
3.3.2. Mecanismo de liberação das dispersões sólidas
Existem dois mecanismos de liberação de fármacos a partir de dispersão sólida: i-
quando o agente limitante é o fármaco, ii- quando o agente limitante é o carreador, isto é, a
própria dispersão sólida como sistema de liberação.
Figura 3 - Influências que a matriz e o fármaco tem na dispersão. (Fonte: modificado de Hallouard et al., 2016).
O que determina um mecanismo ou outro é a viscosidade do carreador quando entra
em contato com a água. O carreador pode se dissolver ou reter a água, formando uma camada
concentrada de carreador ou um gel (camada de saturação). Se o fármaco for totalmente

25
solúvel nessa camada de carreador, porém esse carreador ter a viscosidade suficiente para que
o fármaco dissolvido não consiga ser liberado para o TGI então o limitante será o carreador.
Mas se o fármaco tiver baixa solubilidade quando o carreador tiver na fase da camada de
saturação, o fármaco será o limitante (SUN, 2015; CRAIG, 2002).
Estes dois mecanismos ocorrem frequentemente em simultâneo, porque o fármaco
pode ser parcialmente solúvel ou aprisionado na camada de saturação. A melhora do perfil de
dissolução do fármaco quando a proporção de carreador em dispersões sólidas foi aumentada
depende de que o fármaco esteja melhor disperso e que a cristalinidade do fármaco se reduza
(VO; PARK; LEE, 2013).
3.4. Dispersão Sólida
A dispersão sólida foi inicialmente definida como uma mistura eutética de um fármaco
hidrofóbico, isto é, insolúvel em água, em uma matriz altamente solúvel e cristalina como a
ureia (SEKIGUCHI & OBI, 1961).
A dispersão sólida é definida atualmente como uma dispersão de um fármaco em uma
ou mais matrizes inertes no estado sólido. Estes sistemas de liberação podem ser produzidos
por variados métodos, dependendo apenas das características das matrizes ou do fármaco,
como os métodos de fusão, solvente e solvente-fusão. Alguns autores classificaram as
dispersões sólidas em gerações (Figura 4), seguindo a evolução das proposições tecnológicas
para a melhoria da solubilidade, assim como de outros problemas associados
(VASCONCELOS; SARMENTO; COSTA, 2007; VO; PARK; LEE, 2013).
O fator que faz com que a dispersão sólida aumente a biodisponibilidade de um
fármaco é pela melhor molhabilidade, porosidade e características polimórficas dentre outros
pontos positivos em comparação a outras técnicas para melhorar a solubilidade do fármaco.
Além disso, há outras vantagens, como a não aglomeração das partículas tanto no
armazenamento ou na dissolução, que acontece em formulações convencionais (LELEUX &
WILLIAMS, 2014).
O que impede a aglomeração das partículas na dispersão sólida é a interação entre a
matriz e o fármaco, permitindo uma redução de tamanho a níveis moleculares. A falta de
aglomeração em dispersões sólidas permite evitar o uso de novos processos nanotecnológicos,
que são trabalhosos e caros, e requerem estabilizadores, assim como equipamentos específicos
(GAO et al., 2013; LELEUX & WILLIAMS, 2014).

26
Figura 4 - Gerações das dispersões sólidas e suas características (fonte: modificado de Vo et al., 2013).
Dispersões sólidas podem ser classificadas com base em diferentes aspectos como seu
perfil de liberação, seus processos de preparação ou suas composições. Atualmente a
indicação de gerações indica classificação das dispersões sólidas de acordo com suas
composições..
Figura 5 - Perfil de dissolução por gerações de dispersão sólida fármaco puro (0), 1ª geração (1), 2ª geração (2),
3ª geração (3) e 4ª geração (4). (Fonte: modificado de Van Den Mooter, 2012).
Exemplificando na figura 5, a evolução das dispersões sólidas em comparação com as
matrizes, mostrando que independente da matriz, ela sempre será melhor que o fármaco puro
(0), entretanto a melhor dissolução está na geração 3 (3).

27
As dispersões sólidas, quando produzidas segue 3 modelos de dispersão (Figura 6), a
forma ideal para existir um bom resultado de dissolução é a forma (A), quando o fármaco
(representado pela parte vermelha) e a matriz (representada pela parte azul) são uniformes e
sem aglomeração, diferente da forma (B), onde caracteriza uma dispersão com fármaco
cristalino, pouca interação polímero fármaco, onde as moléculas do fármaco estão bem juntas,
dificultando a sua dissolução e a forma (C) onde apesar de existir um fármaco amorfo, ele não
tem uma boa interação com o polímero, indicando que pode acontecer uma separação de fase,
exemplificada na figura 6 (HUANG & DAI, 2014).
Figura 6 - Visão micromolecular das possíveis dispersões sólidas (Huang & Dai, 2014).
3.4.1. Dispersão sólida cristalina (1ª geração)
A primeira geração é a pioneira no processo de dispersão sólida, sendo elas cristalinas.
Foram desenvolvidas em 1961 por Sekiguchi & Obi utilizando uma mistura eutética de ureia
e sulfatiazol, método que consiste na mistura dos dois componentes, um que tenha alta
afinidade pela água que será o carreador e outro que será o fármaco geralmente com caráter
hidrofóbico alto. Obrigatoriamente o ponto de fusão é menor quando os componentes estão
juntos do que eles separados, caracterizando assim o ponto eutético. Quando resfriados eles
cristalizam no mesmo tempo formando uma dispersão microcristalina em água, o que tende a
deixar o composto mais solúvel, pois aumenta sua superfície de contato e sua molhabilidade.
No estudo notou-se uma melhora na dissolução do fármaco em meio biológico, no entanto, se
a mistura do fármaco e matriz não for exatamente na composição eutética, haverá uma
separação parcial dos dois componentes, levando à dispersão superfina do fármaco no
carreador (CHIOU & RIEGELMAN, 1971).

28
Logo depois, foram usados açúcares carreadores, como o manitol em solução sólida
(KANIG, 1964). Neste estudo constatou uma estabilidade em altas temperaturas e por ser um
açúcar a sua dissolução era melhor que a mistura eutética de ureia. A cristalinidade de tais
dispersões sólidas é no entanto também uma desvantagem significativa. Os compostos
cristalinos possuem maior energia livre de Gibbs que os amorfos (VAN DEN MOOTER,
2012). essa energia precisa ser superada para que moléculas do fármaco possa ser dissolvidas
no meio do solvente, portanto explica o por que um fármaco em qualquer forma cristalina,
será menos solúvel que a forma amorfa, pois não apresenta reticulo cristalino definido
(SINKO, 2008).
3.4.2. Dispersão sólida amorfa (2ª geração)
A segunda geração é caracterizada justamente pela utilização de carreadores amorfos,
inicialmente desenvolvidos no final da década de 60. Sua melhor eficiência na liberação do
fármaco em comparação com os cristalinos é explicada pela menor estabilidade
termodinâmica das dispersões sólidas amorfas (VIPPAGUNTA et al., 2007). As principais
matrizes utilizadas são os polímeros, que podem ser de origem natural, derivados da celulose,
ou sintética, como a polivinilpirrolidona (PVP) (XU et al., 2018; MOTALLAE; TAHERI;
HOMAYOUNI, 2018).
As matrizes amorfas também podem aumentar a molhabilidade e a dispersibilidade do
pó, bem como inibir o processo de precipitação do fármaco quando a dispersão sólida amorfa
entra em contato com a água (CHAUHAN; HUI-GU; ATEF, 2013). Estas propriedades,
juntamente com a taxa de dissolução rápida do carreador amorfo, devido à baixa estabilidade
termodinâmica das matrizes no estado amorfo, aumentam a solubilidade do fármaco e a taxa
de liberação (ZHANG et al., 2012).
Essas dispersões sólidas podem ser classificadas em três subtipos: i - as soluções
sólidas amorfas, quando o fármaco e o veículo são miscíveis e naturalmente homogêneos; ii -
suspensões sólidas amorfas quando acontece a falta de solubilidade do fármaco no pó
formando duas fases podendo ser ocasionada pelo ponto de fusão do fármaco ser muito alto;
iii - não ocorrer miscibilidade completa entre a matriz e fármaco, gerando um sistema misto
de ambos os sistemas descritos, que é a solução e dispersão sólida do fármaco na matriz
(VAN DEN MOOTER, 2012).

29
Os polímeros sintéticos mais utilizados nessa geração são os polietilenoglicóis (PEG)
(BLEY et al., 2010), a polivinilpirrolidona (PVP) (WLODARSKI, 2015) e os polímeros
naturais como os que tem como base principal a celulose, por exemplo a etilcelulose
(OHARA,2005), hidroxipropilmetilcelulose (HPMC) (FAN, 2018; DROOGE et al.,2006) e a
não derivada ciclodextrina (YUVARAJA & KHANAM, 2014).
Para a preparação de um adequado sistema formado entre o fármaco e a matriz é de
suma importância a seleção de um polímero adequado que tenha alta temperatura de transição
vítrea, forte interação com o fármaco e baixa higroscopicidade. Com isso, tende-se a
estabilizar o estado físico dos fármacos em dispersões sólidas, evitando-se acontecer
interações, como o surgimento de efeito plastificante entre alguns fármacos e determinados
polímeros (VASANTHAVADA et al.,2004; YOSHIOKA; HANCOCK; ZOGRAFI, 1994).
A ligação de hidrogênio entre o fármaco e o polímero é a principal força para
aumentar a molhabilidade da dispersão sólida, pois quanto mais fortes forem as ligações de
hidrogênio, menor será seu tamanho de partícula, aumentando sua superfície de contato, e
ainda impedindo a separação de fases por aumentar a estabilidade evitando a recristalização
(KARAVAS et al., 2007; VASANTHAVADA et al., 2005).
O tamanho da cadeia polimérica e/ou o massa molecular (MM) do polímero pode
influenciar diretamente na performance de dissolução da dispersão sólida. Isto é explicado
pela viscosidade adquirida pelo polímero a medida que sua cadeia polimérica aumenta, e
quanto maior a viscosidade menor será a taxa de dissolução. No entanto, uma alta viscosidade
do polímero também apresenta algumas vantagens, como a prevenção da recristalização do
fármaco durante o processo de preparação, armazenamento e no processo de dissolução
(LEUNER & DRESSMAN, 2000).
Geralmente, quando o fármaco está supersaturado na matriz ele perde sua estrutura
cristalina, se convertendo ao estado amorfo. Essa matriz geralmente também está amorfa, o
que caracteriza esse tipo de dispersão sólida. As características amorfas melhoram a
dissolução por apresentar menor energia necessária nesse processo (CHAUHAN; HUI-GU;
ATEF, 2013).
A dispersão sólida de segunda geração consistiu em um avanço significativo em
relação a primeira, melhorando sua dissolução e sua biodisponibilidade. Entretanto, ainda
existem alguns fatores que precisavam ser aprimorados, como a estabilidade físico-química da
dispersão. Devido a sua supersaturação, a separação de fases ocorre com certa facilidade,
podendo ocorrer recristalização quando entra em contato com o estômago ou motivada por

30
interferências externas como umidade, calor e idade do produto. Estudos sobre o
envelhecimento das dispersões sólidas são realizados por análise térmica, por espectroscopia
de infravermelho e cristalografia de raios X, porque podem indicar se com o passar do tempo
a dispersão perde sua solubilidade (BAIRD & TAYLOR, 2012; VASANTHAVADA, 2005).
3.4.3. Dispersão sólida amorfa + adjuvante (3ª geração)
Diante dos problemas apresentados pelas dispersões sólidas de segunda geração,
foram propostas novas alternativas como o uso de matrizes contendo misturas de tensoativos,
auto-emulsionantes ou agentes de inclusão/complexação. Estas previnem desvantagens da
segunda geração, tais como cristalização da dispersão durante o armazenamento ou
precipitação de fármaco in vivo por supersaturação em meio de dissolução (CHAUDHARI et
al., 2013).
O uso dos tensoativos pode previnir a nucleação e a aglomeração do fármaco na
matriz, melhorando a estabilidade física e química durante a preparação. Além disso, os
tensoativos reduzem a taxa de recristalização do fármaco no meio de dissolução e reduzem a
tensão superficial da dispersão sólida com o TGI, fazendo que o tempo de dissolução diminua
ainda mais, comparado com a 2ª geração (KIM et al., 2011).
Os tensoativos mais utilizados na geração III são: Inutec SP1® (XU et al., 2016;
SRINARONG et al., 2011), polissorbato (Tween 80) (KOJO et al.,2017; LI; ORMES;
TAYLOR, 2016; RASHID et al., 2015), macrogol 32 (Gelucire 44/14®) (FARAH et al.,
2017; VERMA; PATEL; PATEL, 2017), lauril sulfato de sódio (JUNG et al., 2016; MAGGI
et al., 2015), behenato de glicerilo (Compritol 888 ATO®) (ROBERTS et al.,2015;
JAGDALE et al., 2011), poloxâmer (MEDAREVIĆ et al., 2016; SONG; YOON; KIM, 2016)
e polietilenoglicol (Soluplus®) (ALBADARIN et al., 2017; ALSHAHRANI et al., 2015).
Os agentes de inclusão/complexação são utilizados como forma de evitar a
aglomeração do fármaco na dispersão sólida, pois quanto mais dispersas estiverem as
partículas maior a superfície de contato, o que auxilia a evitar que o fármaco precipite no
meio de dissolução. Isto ocorre porque o adjuvante evita a supersaturação no resfriamento
forçado durante a preparação da dispersão sólida. Esses fatores fazem com que a dispersão
sólida tenha um rendimento melhor (FATMI et al., 2015; JOUDIEH et al., 2009).
A utilização de adjuvantes na formulação foi benéfica para as dispersões sólidas, por
aumentar significativamente seu perfil de dissolução, pela melhor molhabilidade, dando mais

31
estabilidade a dispersão e pela diminuição do percentual de recristalização e precipitação das
mesmas. Entretanto, é necessário ter atenção e empregar o tensoativo correto pois o efeito
pode ser contrário e ocorrer o aumento da recristalização (CHAUDHARI & DUGAR, 2017).
3.4.4. Dispersão sólida amorfa de liberação controlada (4ª geração)
A quarta geração consiste em sistemas de liberação prolongada a partir de dispersões
sólidas, que tem como objetivo deixar a fármaco mais solúvel através de uma dispersão e que
ela tenha liberação controlada através de polímeros que tenham afinidade baixa com a água
(HUANG; WIGENT; SCHWARTZ, 2006).
Nesta geração ocorrem dois processos: primeiro é a promoção da melhora na
dissolução das dispersões sólidas; e segundo é a obtenção de liberação gradativa do fármaco.
Um dos problemas encontrados nas dispersões sólidas de classe III era a maior probabilidade
de perda da homogeneidade intrapartícula, pois quanto mais substâncias incorporadas no
preparo da dispersão maior a facilidade de que ocorra a separação de fases. Para evitar isso
são utilizado polímeros com características tensoativas ou de complexação junto com a
função principal de matriz, contribuindo para reduzir a probabilidade de separação na
preparação da dispersão (DEBOUZY et al., 2014; SKIBA et al., 2014).
A maior característica dessa geração é justamente os estudos envolvendo a liberação
controlada de fármacos pouco solúveis em água, com meia vida curta. Isso significa que o
fármaco hidrolisa muito rápido, até antes de alcançar ao sítio de melhor absorção, o intestino.
Foi proposta a utilização de polímeros que não tem ou tem afinidade baixa com a água, que
demoram se dissolver. Ao contrário das outras gerações, em que o objetivo era a liberação e
absorção rápida para que a biodisponibilidade fosse maior, nessa geração se deseja que o
fármaco demore para dissolver e que tenha sua liberação no intestino (HUANG; WIGENT;
SCHWARTZ, 2006).
Os principais mecanismos de liberação de fármacos nessa geração são, portanto, a
difusão e a erosão. Portanto, essa dispersão sólida é capaz de fornecer uma quantidade
adequada de fármaco por um período prolongado de tempo, melhorando a adesão do paciente
devido à administração menos frequente, menos efeitos colaterais e efeito mais constante ou
prolongado do fármaco (CUI et al., 2003; DESAI; ALEXANDER; RIGA, 2006).
As matrizes poliméricas mais utilizadas são aquelas com maior dificuldade de
dissolver, podendo ser citados os polímeros acrílicos (RATCLIFFE et al., 2015), polímeros

32
carboxivinílicos (BI; RAHMAN; LESTER, 2015; URAMATSU et al. 2015), acetato de
celulose (LIU et al., 2014), hidroxipropilcelulose (OSAWA et al., 2014; HUGHEY et al.,
2015) e polietilenos (TEJA et al., 2016).
3.5. Método de preparação
3.5.1. Fusão
O método de fusão caracteriza por fundir o fármaco e o carreador em um só utilizando
calor, depois usando o resfriamento para solidificar a solução obtida para, posteriormente,
pulverizar em partículas menores, assim primeiramente proposto por Sekiguchi et al. (1964).
Logo vieram modificações para tentar melhorar a técnica, com a suspensão do fármaco em
um carreador previamente fundido. Isso leva a menor interferência do calor no fármaco, tendo
assim menor possibilidade de uma modificação da estrutura ou degradação da molécula, o que
poderia interferir na sua biodisponibilidade (VIPPAGUNTA et al., 2007).
Quanto ao resfriamento, existem uma série de métodos para a realização, entre eles
temos a agitação em meio ao gelo (SHETH, 2011; SEKIGUCHI; OBI; UEDA, 1964),
imersão em nitrogênio líquido (SARODE et al., 2013; SAREEN; MATHEW; JOSEPH,
2012) e extrusão por fusão a quente (Hot-melt) (VASOYA, et al., 2018; ZHANG et al.,
2018; VERHOEVEN et al, 2009).
Extrusão por fusão a quente
A extrusão por fusão a quente (hot-melt) é a técnica mais empregada no método de
fusão (THIRY et al., 2016) devido a sua escalabilidade e por ser desenvolvida em um
processo único, misturando fármaco/matriz homogeneamente, simultaneamente com a fusão e
extrusão, para então ser resfriada, pulverizada, para finalmente formar a dispersão sólida
(VERHOEVEN et al., 2009).

33
Figura 7 - Exemplificação da extrutora por fusão a quente (Fonte: modificado de PATIL et al., 2016).
O parafuso rotativo induz uma mistura intensa que leva à desagregação das partículas
do fármaco na matriz. A extrusão por fusão a quente tem a vantagem de limitar o tempo de
residência do fármaco e carreador a temperaturas elevadas na extrusora, reduzindo também o
risco de alterações de componentes devido à temperatura. (PATIL; TIWARI; REPKA, 2016;
CROWLEY et al., 2007; REPKA, 2007).
Meltrex™ é um processo patenteado e modificado de extrusão por fusão a quente que
usa uma extrusora de parafuso duplo especial com dois funis independentes. Este
equipamento reduz o tempo de permanência do fármaco à alta temperatura para
aproximadamente 2 minutos e, portanto, limita o risco da sua alteração dos componentes
(BALASAHEB; BALAJI; AVINASH, 2014; PARVE et al., 2014).
A vantagem do processo de fusão está no custo mais barato e por não ser tóxico.
Porém o método sofre com dois problemas: a primeira está no fato de alguns fármacos serem
termolábeis, ou seja em altas temperaturas ele pode mudar sua estrutura química ou ser
degradado e a outra está na viscosidade das matrizes, principalmente as poliméricas,
dificultando a miscibilidade dos compostos, podendo ocorrer a separação das fases o que
impedirá que a dispersão sólida seja obtida (KOLAŠINAC et al., 2012).
Foram propostas melhorias para o método de fusão, pois ainda é a técnica mais prática
e escalonável existente para a preparação de dispersão sólida. Esses aperfeiçoamentos
consistem na redução da duração do aquecimento e a dispersão do fármaco na matriz já
fundida em vez de aquecer ambos os componentes ao mesmo tempo (MEHANNA;
MOTAWAA; SAMAHA, 2010).

34
Dois processos que empregam esta adaptação foram patenteados - MeltDose®
(Lifecycle Pharma A/S) e Lidose® (laboratório SMB). No processo MeltDose® um fármaco
incorporado no carreador fundido é pulverizado sobre partículas carreadores inertes,
utilizando equipamento de leito fluidizado. Com as partículas obtidas podem ser preparados
comprimidos ou cápsulas. (JOSHI et al., 2012). Com relação à tecnologia Lidose®, um
medicamento após a mistura com carreador fundido é colocado em cápsulas duras e resfriado
sob condições específicas e constantes (ALAM et al., 2012).
3.5.2. Solvente
Com a dificuldade encontrada no processo de fusão foi proposto primeiramente por
Chiou & Riegelman (1971) outro método que pudesse ser feito sem a utilização de calor. Isso
facilitaria a utilização de mais compostos farmacêuticos na preparação de dispersões sólidas,
pois incluiria os fármacos ou matrizes termolábeis. Diante disso, foi proposto o método de
solvente, que consiste em dissolver a matriz e fármaco em um solvente comum a eles e em
seguida evaporá-lo por variados métodos.
As dispersões sólidas produzidas pelo método de solventes tendem a ter uma estrutura
altamente porosa induzida pela remoção rápida do solvente e levando ao aumento da taxa de
dissolução do fármaco. A escolha do polímero como matriz hidrofílico também influencia a
porosidade de dispersão sólida: polímeros lineares frequentemente levam a maior porosidade
de dispersão sólida do que os ramificados (KALPANA et al., 2010).
Porém a técnica sofre de problema como a dificuldade de esgotamento total do
solvente utilizado, pois pode ter alta toxicidade, além da dificuldade em se encontrar um
solvente que dissolva tanto a matriz quanto ao fármaco (SAHOO et al., 2011). A mistura de
solventes pode ser utilizada, com intuito de se obter uma polaridade comum aos dois
componentes, assim como o emprego de tensoativo como o polisorbato 80 para aumentar a
solubilidade nos solventes (HU; LOU; HAGEMAN, 2018; RASHID et al., 2015). Deve-se ter
o cuidado nessa etapa, visto que a adição de uma quantidade grande dos tensoativos aumenta
a precipitação (CHAUDHARI & DUGAR, 2017).
Uma alternativa para evitar a utilização de solventes muito voláteis é a utilização do
método de coprecipitação e antissolvente. Nesse método, primeiramente se dissolve o fármaco
e a matriz em um solvente orgânico, e, posteriormente, acrescenta-se um antissolvente para
reduzir a solubilidade dessa solução até chegar ao ponto de precipitação de ambos

35
simultaneamente. A suspensão resultante é então filtrada e para a remoção dos resíduos de
solvente (SHAH et al., 2013).
A principal vantagem deste método em comparação com os outros métodos de
evaporação de solvente é a possibilidade de remoção do solvente utilizado para dissolução do
fármaco/matriz. Além disso, a filtração pode permitir a utilização de solvente menos volátil
durante a coprecipitação do fármaco/matriz. Porém, a mistura de solvente/antissolvente
durante a etapa de precipitação deste método pode ter um efeito plastificante, pela geração de
mobilidade molecular, gerando assim um rearranjo, podendo recristalizar o fármaco.
(SERTSOU et al., 2002).
Existem variados métodos para a remoção do solvente ou secagem, pois um dos
fatores mais importantes em uma dispersão sólida é a sua rápida solidificação para manter o
fármaco no estado amorfo. Quanto mais rápido esse solvente for eliminado, mais rapidamente
será realizada a solidificação e tende-se a obter mais facilmente o fármaco em estado amorfo.
Os métodos mais usados são: aquecimento em uma chapa quente (FINI et al., 2005), secagem
a vácuo (CHOI et al., 2018; SAMMOUR et al., 2006), evaporação rotativa (HU; LOU;
HAGEMAN, 2018; FRIZON et al., 2013), Secagem por pulverização/aspersão
(MOTALLAE; TAHERI; HOMAYOUNI, 2018; HERBRINK et al., 2017; RASHID et al.,
2015; GU; LINEHAN; TSENG, 2015), congelamento ultra rápido (GUPTA; MISHRA;
PATHAK, 2015), liofilização (ANSARI et al., 2015; WLODARSKI et al., 2015), secagem
por congelamento por aspersão (MEHTA, 2016; WANNING; SÜVERKRÜP;
LAMPRECHT, 2015) e fluidos supercríticos (OBAIDAT et al., 2017; YANG et al., 2015).
Dos métodos citados o mais utilizado nas dispersões sólidas de anti-inflamatórios é a secagem
por aspersão. A tecnologia de fluidos supercríticos é uma inovação recente e tem potencial
para um futuro breve ser utilizado como método de primeira escolha.
Secagem por aspersão
A secagem por aspersão (spray drying) é um processo rápido e contínuo para a
produção de pós secos, demonstrado na figura 8, a partir de um material disperso em meio
líquido através de um atomizador para uma câmara de secagem arrastada por um meio gasoso
aquecido. Devido à grande área de superfície específica oferecida pelas gotículas o solvente
evapora rapidamente e a dispersão sólida é formada em frações de segundos, o que pode ser
rápido o suficiente para evitar a separação de fases. Além disso, as dispersões sólidas

36
preparadas por secagem por aspersão consistem de partículas cujos tamanhos podem ser
planejados, através de vários fatores, entre eles o diâmetro do orifício do atomizador. A
secagem por aspersão geralmente produz fármaco no estado amorfo (SOSNIK &
SEREMETA, 2015).
Figura 8 - Esquema de preparação da dispersão sólida por aspersão (Fonte: modificado de Peighambardoust et
al. 2011).
A secagem dos pós passa por 4 etapas: (1) em que a matriz e o fármaco previamente
dissolvidos são bombeados para o atomizador, e sob pressão o líquido é pulverizado; (2)
posteriormente, as gotículas geradas são secas por gás/ar quente na câmara de secagem; (3) à
medida que o pó seco é arrastado pelo gás ele se sedimenta no ciclone, onde as partículas da
dispersão sólida se depositam no coletor; (4) o gás e umidade são eliminados pelo exaustor
(CHAN et al., 2015).
Fluídos supercríticos
Os fluidos supercríticos, como o dióxido de carbono (CO2), estão substituindo os
solventes convencionais. O método é importante porque apresenta as propriedades favoráveis
dos gases (como alta difusividade) e, ao mesmo tempo, baixa tensão superficial e baixa
viscosidade transmitida aos líquidos (NI et al., 2017).

37
A manipulação da pressão de fluidos supercríticos permite um controle preciso da
dissolução de muitos fármacos. Assim, a extração de fluidos supercríticos é particularmente
fácil, rápida e não requer temperatura elevada. A extração é realizada simplesmente reduzindo
a pressão (YANG et al., 2015).
A taxa de extração também pode controlar o tamanho da partícula e o nível de
dispersibilidade encontradas nas dispersões sólidas. Como exemplo, dois processos
patenteados (RightSize™ da XSpray Microparticles e Formuldisp™ da Pierre Fabre) foram
desenvolvidos (ALAM; AL-JENOOBI; AL-MOHIZE, 2013).
Pela não utilização de solvente faz com que a técnica tenha vantagens, pois um dos
problemas relacionados ao método de solvente é a utilização de grandes quantidades quando é
usado como antissolvente para o método de co-precipiação, o que causa resquícios de
solvente na dispersão sólida (DJERAFI et al., 2017).
3.5.3. Solvente-fusão
O método do solvente-fusão é a combinação do método de fusão e do método do
solvente. Neste método, os fármacos são dissolvidos num solvente adequado e misturados
com o veículo fundido, seguido de remoção do solvente e solidificação para formar dispersões
sólidas. A vantagem deste método é que a temperatura e o tempo de mistura são menores que
o método de fusão, protegendo assim o fármaco da degradação térmica. Além disso, o
carreador no estado fundido é mais facilmente disperso e dissolvido no solvente em
comparação com o método solvente (PATEL & PATEL, 2006).
De fato, o método e a técnica ideais para a preparação de dispersão sólida foram
escolhidos com base nas propriedades físico-químicas de medicamento e matriz. Em geral, a
secagem por aspersão e a extrusão a quente são mais comumente usadas devido à sua alta
escalabilidade e aplicabilidade.
3.6. Anti-inflamatórios não esteroidais
Para o estudo dos anti-inflamatórios não esteroidais (AINE's) é de fundamental
importância compreender o processo inflamatório para assim conhecer os diferentes
mecanismo de ação desses fármacos (GOLAN et al., 2009).

38
A inflamação passa por etapas, na cascata de inflamação (Figura 9). Ela se inicia pelos
eucosanoides, que são produtos principalmente do ácido araquidônico (AA), um ácido graxo
de cadeia poli-insaturada, ômega 6 (GOLAN et al., 2009).
Existem variadas vias para o AA e que irão levar a diferentes eucosanoides, entre eles
a via dos isoeicosanoides, lipoxigenase, epoxigenase P450 e a cicloxigenase (COX), que vai
ter como produto final sintetizado as prostaglandinas, prostaciclina e tromboxano que são os
prostanoides (KATZUNG & TREVOR, 2017).
Importante salientar que COX, é dividida em duas subclasses de enzimas, COX-1 e
COX-2, capazes de transformar o AA em prostanóides específicos. Essas enzimas têm
funções distintas, como por exemplo, a COX-1 age no processo de citoproteção gástrica, e a
COX-2 age nos processos inflamatórios. Apesar disso, em alguns casos elas podem atuar em
conjunto, como nos casos dos AINE's (BATLOUNI, 2010).
Processo estimulador da membrana
Ácido araquidônico
Cicloxigenase
Prostaglandinas Tromboxano Prostaciclina
Modulação dos leucócitos
Inflamação
Figura 9 - Cascata de inflamação (fonte: modificado de KATZUNG & TREVOR, 2017.)
Logo, o mecanismo de atuação dos AINE's será de modo geral justamente na COX,
podendo ser de modo não seletivo, inibindo as duas COX´s, como o ibuprofeno, ácido
acetilsalicílico (AAS), piroxicam, naproxeno e diclofenaco; ou inibindo seletivamente a
COX-2, como o valdecoxibe, parecoxibe e celecoxibe (KATZUNG & TREVOR, 2017).
De modo geral, sendo uma das classes mais antigas e com uma grande importância
comercial, o desafio encontrado pela indústria é inovar essa classe de fármacos. A estratégia
mais comum é a síntese de novos fármacos, porém, praticamente estão esgotadas as inovações
em torno de grupos farmacofóricos inovadores. Nesse sentido, atualmente, os lançamentos de

39
novos AINE'S se baseiam em pequenas modificações de grupos ligados ao sítio
farmacofórico, obtenção de novos fármacos COX-2 seletivos, ou a síntese de pró-fármacos
(DEQUEKER et al., 1998).
Diante das limitações das estratégias de síntese, as ferramentas tecnológicas
disponíveis para correção de características biofarmacêuticas típicas dos AINE'S tornaram-se
muito atrativas. Entre as razões destacam-se o número variado de opções tecnológicas, o
menor tempo de desenvolvimento do medicamento e o menor custo final (JERMAIN;
BROUGH; WILLIAMS III, 2018).
A baixa solubilidade aquosa dos AINE'S causa baixa desintegração nas formas
farmacêuticas sólidas, como comprimido, com consequente baixa taxa de dissolução, como já
comentado anteriormente. Os aspectos biofarmacêuticos relacionados devem ser superados
para elevar a fraca dissolução, seguida da melhora da biodisponibilidade (ADEBISI et al.,
2016).
Com esse objetivo, uma série de inovações têm sido utilizadas como complexos de
inclusão (PEREVA,2016), adição de sais (KORN & BALBACH, 2014), pró-fármacos
(AMIN, 2015) nanosuspensões (MÖSCHWITZER, 2013), co-cristais (THAKURIA, 2013),
micelas (LU & PARK, 2013), microemulsões (HE; HE; GAO, 2010), nanoemulsões (KOTTA
et al., 2012), nanopartículas lipídicas (NASERI, 2015) e a utilização de dispersão sólida
(JERMAIN; BROUGH; WILLIAMS III, 2018).
Com a finalidade de se melhorar a dissolução, obtendo consequentemente mais rápida
ação farmacológica, a tecnologia da dispersão sólida demonstra ser relevante alternativa às
demais existentes. São muitos os estudos envolvendo essa tecnologia contendo AINE's e
muitas são as estratégias utilizadas. Neste trabalho elencamos os AINE's mais usados na
terapêutica estudados como dispersão sólida e apresentamos os trabalhos mais relevantes para
cada fármaco individualmente.
3.6.1. Ibuprofeno
CH3
CH3
CH3
O
OH
Figura 10 - Estrutura molecular do ibuprofeno (Produzido em: ACD/CHEMSKETCH, 2008).

40
Existem muitos estudos sobre dispersões sólidas contendo ibuprofeno em diferentes
matrizes poliméricas. Um dos primeiros estudos foram Najib et al. (1986), que avaliaram as
propriedades do ibuprofeno na matriz polimérica de PVP, utilizando o método de evaporação
por solvente. Foi observado um aumento na dissolução do fármaco em comparação com
formulações orais convencionais. Foi demonstrado que a massa molar (MM) do polímero
interferiu na liberação, quanto maior a MM menor foi a extensão da liberação (Figura 11). O
mecanismo de liberação envolveu o pH, demonstrado pela absorção e dissolução do fármaco
em pH's extremos, 8,5 e 1,1, explicada pela ionização do grupo ácido carboxílico em meio
básico e pela protonação do grupo amida em meio ácido. A escolha do solvente (Figura 12) e
do tensoativo (Figura 13), demonstrou ser relevante para aumentar o perfil de dissolução da
dispersão sólida.
Figura 11 - Perfil de dissolução comparado com a MM do polímero (fonte: Najib et al., 1986).
Figura 12 - Perfil de dissolução comparado com solvente usado na obtenção da DS. (1) Dispersão sólida
ibuprofeno com cloroformio; (2) Dispersão sólida ibuprofeno com etanol; (3) mistura física; (4) fármaco puro
(fonte: Najib et al., 1986).

41
Figura 13 - Comparação do perfil de dissolução da dispersão sólida com diferentes tensoativos (fonte: Najib et
al., 1986).
A melhor dissolução de uma dispersão sólida em relação a escolha de um solvente,
pode ser explicada pela velocidade de evaporação que o solvente tem (Figura 12). Quanto
mais rápida for a evaporação, menor será o tempo de reorganização das cadeias cristalinas da
dispersão, deixando-o com maior característica amorfa.
Quando tensoativos aniônicos foram utilizados houve substancial melhora no perfil de
dissolução, quando comparado a tensoativos de cargas diferentes do fármaco, isto é,
catiônicos ou não-iônicos. No caso, o lauril sulfato de sódio tem carga negativa (aniônico),
portanto, semelhante a carga do ibuprofeno ionizado, já a cetrimida tem carga positiva
(catiônico) e o span 60 não-iônica (Figura 13).
Além do PVP (ROSSMANN et. al., 2014), outros polímeros também foram usados na
preparação de dispersões sólidas como: PEG, (NEWA et al, 2008; KHAN & JIABI, 1998;
SHAKHTSHNEIDER et al., 1996), poloxamer 188 (NEWA et al., 2007), mistura de PVP/VA
(MONEGHINI et al., 2008), polivinil álcool/polietileno glicol (Kollicoat IR®) (XU et al.,
2009). Em todos os trabalhos analisados fica evidente que técnica usada e o tamanho da
cadeia polimérica são os fatores mais importantes que afetam a dissolução do fármaco.
Quanto maior for a MM e quanto mais cristalino for o estado físico do fármaco na matriz
menor será taxa de dissolução.
AKTER et al., (2015) fizeram um estudo com a finalidade de identificar qual polímero
seria mais eficiente na liberação do ibuprofeno em dispersão sólida. Foram utilizados como
matrizes o PVP, Poloxamer 407, PEG 4000, PEG 6000. Os melhores resultados foram obtidos
com a utilização da mistura de ibuprofeno, polaxamer 407 e PEG 6000 na proporção 1:1:1
42
preparados pelo método de fusão, pois demonstrou a maior taxa de dissolução. A análise dos
espectros de Infravermelho com Transformador de Fourier (IVTF) indicou que o fármaco
passou da sua forma cristalina para amorfa na forma de dispersão sólida.
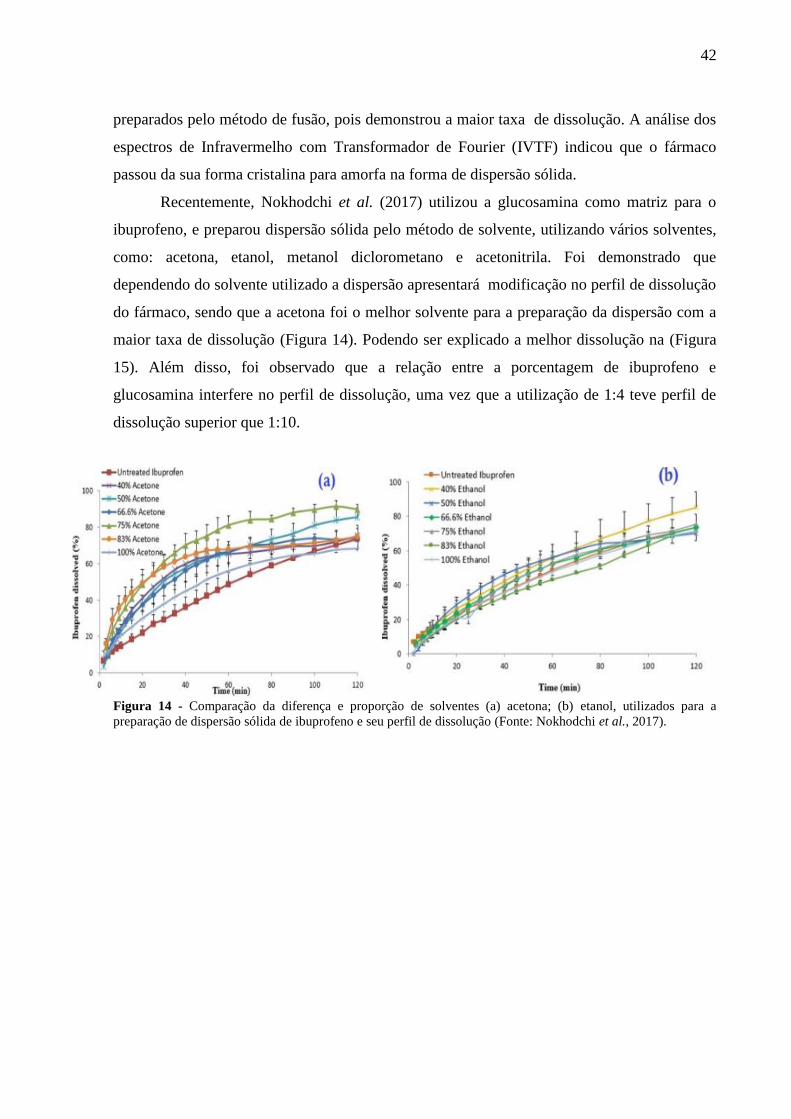
Recentemente, Nokhodchi et al. (2017) utilizou a glucosamina como matriz para o
ibuprofeno, e preparou dispersão sólida pelo método de solvente, utilizando vários solventes,
como: acetona, etanol, metanol diclorometano e acetonitrila. Foi demonstrado que
dependendo do solvente utilizado a dispersão apresentará modificação no perfil de dissolução
do fármaco, sendo que a acetona foi o melhor solvente para a preparação da dispersão com a
maior taxa de dissolução (Figura 14). Podendo ser explicado a melhor dissolução na (Figura
15). Além disso, foi observado que a relação entre a porcentagem de ibuprofeno e
glucosamina interfere no perfil de dissolução, uma vez que a utilização de 1:4 teve perfil de
dissolução superior que 1:10.
Figura 14 - Comparação da diferença e proporção de solventes (a) acetona; (b) etanol, utilizados para a
preparação de dispersão sólida de ibuprofeno e seu perfil de dissolução (Fonte: Nokhodchi et al., 2017).

43
Figura 15 - Imagem de microscopia eletrônica de varredura comparando a dispersão sólida 1:4 (terceiro
quadrante) com acetona com a dispersão sólida 1:4 com etanol (quarto quadrante) (Fonte: Nokhodchi et al.,
2017).
Quando analisado em microscopia eletrônica de varredura, fica evidente a diferença
morfológica das dispersões quando utilizado solventes diferentes, explicando o melhor perfil
de dissolução quando utilizado a acetona e não o etanol.
3.6.2. Naproxeno
Figura 16 - Estrutura molecular do Naproxeno (Produzido em: ACD/CHEMSKETCH, 2008).
Mura et al. (1996) desenvolveram dispersões sólidas com a matriz PEG, com
diferentes tamanhos da cadeia polimérica com massa molecular (MM) de 4000, 6000 e 20000
pelos métodos de fusão e de solvente. A caracterização no estado sólido não identificou
associação compatível com mudança de estado cristalino do fármaco. Em relação ao fármaco

44
puro foi obtida melhora considerável na dissolução do anti-inflamatório, previstas pela melhor
molhabilidade e menor tamanho de partículas observadas. Tanto a técnica empregada na
preparação das dispersões sólidas, assim como a MM da matriz não causaram diferenças
significativas na dissolução. De acordo com outros estudos era previsto que a utilização de
polímeros de cadeias maiores causassem redução da taxa de dissolução, o que não foi
observado, talvez decorrente da falta de amorfização do naproxeno.
Adibkia et al. (2013) fizeram dispersões sólidas envolvendo as matrizes crospovidona
e HPMC, utilizando o método de solvente seguido de secagem por aspersão, em diferentes
proporções fármaco:matriz (1:0,5; 1:1 e 1:2). Foi avaliada a taxa de dissolução, tanto no pH 3
(estômago) quanto pH 7,4 (intestino). Em geral a dissolução em meio alcalino é muito
superior do que em meio ácido, tanto com a crospovidona a HPMC. A formulação
fármaco:crospovidona 1:1 otimizada apresentou 90% do fármaco dissolvido em 60 minutos,
enquanto que o fármaco puro apenas 40% foi dissolvida.
3.6.3. Cetoprofeno
Figura 17 - Estrutura molecular do Cetoprofeno (Produzido em: ACD/CHEMSKETCH, 2008).
A primeira dispersão sólida publicada com esse AINE's foi feita por Rogers &
Anderson (1982), utilizando ureia como matriz, através de uma mistura eutética.
Margarit et al. (1994) utilizaram na matriz PEG 6000 para a preparação de dispersão
sólida, pelo método de solvente. O método envolveu a utilização de álcool e evaporação em
estufa. Foi observado que à medida que se aumentou a quantidade de fármaco em relação ao
polímero sua velocidade de dissolução abaixou, embora tendo sido observadas taxas de
dissolução mais elevadas em todas as proporções cetoprofeno:PEG em comparação ao
fármaco puro. Para as proporções fármaco:matriz 10:90, 20:80 e 50:50, foram determinados,
respectivamente, 1,9; 3,2 e 4 minutos para a dissolução. Já para o fármaco puro 80% se
dissolveram em 88,5 minutos.

45
Chan et al. (2015) preparam dispersões sólidas com matrizes poliméricas hidrofílicas,
PVP K30; PVP:VA (6:4) e PVA, através do método de solvente, seguido por secagem por
atomização. Foram obtidas dispersões sólidas amorfas com as matrizes PVP K30 e PVP:VA;
e dispersão sólida parcialmente cristalina com PVA (Figura 18). Neste trabalho foi observado
resultado inesperado, pois apesar do fato da DS estar no estado amorfo, isso não causou
melhora na dissolução, quando comparado a DS cristalina (Figura 19). Com isso, foi
demonstrado que a interação fármaco-polímero pode proporcionar melhor dissolução do
fármaco em DS cristalina. Se a dissolução do polímero em DS contendo fármaco amorfo for
muito rápida, poderá causar a precipitação ou recristalização do fármaco durante a sua
liberação, mudando as característica da DS e resultando em um perfil de liberação lenta
(Figura 20).
Figura 18 - Imagem da Difração de Raio-X, (A) Cetoprofeno; (B) PM 30% Cetoprofeno K30; (C) MF 30%
Cetoprofeno PVPVA 6: 4; (D) MF 30% Cetoprofeno PVA; (E) SD 30% Cetoprofeno PVP K30; (F) SD 30%
Cetoprofeno PVPVA 6: 4; (G) SD 30% Cetorpofeno PVA e (H) PVA puro (Fonte: Chan et al., 2015).
A difração de raio-x mostrou que as dispersões sólidas com as matrizes PVP-K30 e
PVP/VA não apresentaram picos, representando amorfização das amostras, o que não
aconteceu na dispersão sólida com PVA, obtendo picos de cristalinidade.

46
Figura 19 - (A) Perfil de dissolução cetoprofeno e PVP K30; (B) Perfil de dissolução cetoprofeno PVP/VA; (C)
Perfil de dissolução cetoprofeno e PVA; (D) Comparação do perfil de dissolução de todas as dispersões sólidas
(Fonte: Chan et al., 2015).
O perfil de dissolução de dissolução da DS com matriz PVP/VA obteve resultados que
são explicados na literatura. Onde demonstra que a utilização de dispersão sólida melhora o
perfil de dissolução quando comparados a mistura física e fármaco puro, o que também
acontece com a matriz PVA, apesar de ter característica cristalina, o aumento da área
superficial e as características hidrofílicas do polímero, fazem com que seu perfil de
dissolução seja mais eficaz. Entretanto quando utilizado a matriz PVP-K30 obteve resultados
diferentes do que se espera de uma dispersão sólida de liberação rápida.
Figura 20 - Possível mecanismo de liberação para a dispersão sólida com matriz PVP-K30 (Fonte: Chan et al.,
2015)
A taxa de liberação lenta do fármaco pode ser explicada pelas características
extremamente hidrofílica da matriz, dissolvendo rapidamente, o fármaco por sua vez

47
hidrofóbico, ficou retido no meio de dissolução recristalizando e podendo ter ocorrido a
aglomeração reduzindo sua área superficial, levando a essa liberação lenta.
3.6.4. Piroxicam
Figura 21 - Estrutura molecular do Piroxicam (Produzido em: ACD/CHEMSKETCH, 2008).
A primeira dispersão sólida contendo piroxicam foi desenvolvida por Fernandez et al.
(1992), que utilizou PEG 4000 como matriz utilizando o método de fusão-solvente. No
estudo, diferentes proporções de fármaco:matriz foram usadas e foi observado através de
espectrometria de infravermelho que o fármaco se manteve disperso até o limite de 40 %.
Após isso o piroxicam se apresentou insolúvel na matriz polimérica.
Anos depois, Tantishaiyakul et al. (1999), utilizou outras matrizes, o PVP K17 e K90,
polímeros baseados na vinilpirrolidona com MM diferentes, para a preparação da dispersão
sólida pelo método de solvente, obtendo amorfização das dispersões, tanto para PVP-K17
quanto para K90 utilizando difração por Raio-X (Figura 22) e confirmada por IVTF (Figura
23) . Foi observado que a proporção de 1:5 e 1:6 de piroxicam: PVP K17 foi melhor que PVP
K90 (Figura 24).

48
Figura 22 - Imagem da difração de Raio-X do piroxicam puro, mistura fisica e dispersão sólida para PVP K90 e
PVP K17 respectivamentes (Fonte: Tantishaiyakul et al., 1999).
A utilização da difração de Raio-X determinou que não havia picos de cristalinidade
na DS 1:2 (Piroxicam:PVP K-90) e na DS 1:3 (Piroxicam: PVP K-17), entretanto a utilização
de um método mais sensível foi necessário para obter a proporção exata de amorfização total
da dispersão sólida.
Figura 23 - Imagem de IVTF caracterização da amorfização da dispersão sólida (Fonte: Tantishaiyakul et al.,
1999).
Utilizando o IVTF foi possível definir o desaparecimento da banda que caracteriza a
cristalinidade do piroxicam, e a formação da dispersão sólida amorfa nas proporções 1:3 e 1:4
com PVP K-90 e com PVP K-17 nas proporções 1:5 e 1:6.

49
Figura 24 - Comparação do perfil de dissolução das Dispersões sólidas com as matrizes PVP K-90 e K17
respectivamente (Fonte: Tantishaiyakul et al., 1999).
O fator que causa o melhor perfil de dissolução do PVP- K17, em comparação ao K-
90, é a sua MM. À medida que a MM polimérica aumenta a sua viscosidade é elevada,
dificultando a difusão do fármaco através da camada de saturação da partícula da DS gerando
uma taxa de dissolução menor. A dispersão sólida com PVP-K17 no estado amorfo 1:5 e 1:6
obteve resultados cerca de 40 vezes melhor que o fármaco puro, sendo assim a melhor opção.
Wu et al. (2009) prepararam dispersões sólidas empregando a técnica de precipitação
por antissolvente seguindo de secagem por aspersão (spray drying). A dispersão sólida obtida
pela precipitação por antissolvente obteve melhores resultados na dissolução. A dispersão se
dissolveu nos primeiros 5 minutos do teste e a sua dissolução foi pelo menos 20 vezes mais
rápida que o fármaco na sua forma pura.
Adebisi et al. (2016) trabalharam com a glucosamina como matriz utilizando o método
de solvente seguido por secagem por aspersão para a preparação da dispersão sólida. Na
proporção piroxicam:glucosamina 2:1 foi obtida a maior taxa de dissolução. Esse resultado
pode ser explicado pelo tamanho reduzido (2,5 µm) e esfericidade da partícula, pela
amorfização da glucosamina e piroxicam e pela carga de superfície quase nula. Como o
piroxicam passou por uma diminuição da área superficial utilizando a secagem por
atomização seria esperado melhor dissolução, mas isso não aconteceu. A otimização só foi
obtida após redução das forças coesivas das partículas, o que somente ocorreu após adição da
glucosamina. Com isso a taxa de dissolução aumentou, pois a carga eletrostática coesiva, ou
seja, a tensão superficial diminuiu, aumentando a molhabilidade da partícula.

50
3.6.5. Nimesulida
Figura 25 - Estrutura molecular da Nimesulida (Produzido em: ACD/CHEMSKETCH, 2008).
Gohel & Patel (2003) prepararam dispersão sólida de nimesulida com variados tipos
de promotores de dissolução como PEG 400, Propilenoglicol, PVP K30, os tensoativos lauril
sulfato de sódio, e polisorbato 80, e lactose e celulose microcristalina como matrizes pelo
método de evaporação de solvente. No estudo as formulações contendo PVP, misturado com
os tensoativos lauril sulfato de sódio e polisorbato 80 apresentaram melhor taxa de dissolução,
obtendo 70 % em 120 minutos, contra 15 % com o fármaco puro. Dessa forma, foi
demonstrado que a nimesulida pode ter sua biodisponibilidade aumentada quando presente em
dispersão sólida formada por PVP, tensoativos e lactose ou celulose microcristalina.
Moneghini et al. (2009) utilizou micro-ondas para a preparação de dispersão sólida.
Foram utilizados como matrizes o Stearoil polioxil-32 glicerides (Gelucire ® 50/13) e o
Poloxamer 188 (Lutrol ®F 68), ambos com função tensoativa. A irradiação por micro-ondas
reduziu a cristalinidade do fármaco. Para avaliar a estabilidade da estrutura física das
dispersões sólidas um estudo de estocagem por 6 meses foi realizado. Após isso não foi
obsevada recristalização ou mudança de fase do fármaco na dispersão sólida. No estudo de
liberação em 13 minutos 90% do fármaco dissolvido, enquanto o fármaco puro nesse mesmo
tempo teve 18%. Este estudo demonstrou um grande avanço, pois com o uso do micro-ondas
não foi necessária a utilização de solventes para a preparação de dispersão sólida.

51
4. Conclusão
Durante a revisão é notória a evolução histórica das dispersões sólidas, com um
evidenciado aumento da dissolução e por consequência elevada biodisponibilidade de
fármacos pouco solúveis, enfatizando os anti-inflamatórios não esteroidais (AINE's). A
melhoria dessa biodisponibilidade faz com que tenhamos uma menor quantidade de fármaco
ingerido, para atividade biológica eficaz, considerando que AINE´s podem causar efeitos
adversos, como problemas gástricos pela inibição da COX. Os estudos na área de dispersão
sólida para a correção de problemas biofarmacêuticos são promissores diante dos avanços
observados no aprimoramento dos métodos de obtenção, no aumento da estabilidade, dos
tipos de matrizes e adjuvantes a serem trabalhados e dos métodos de estudos da associação da
matriz/fármaco/adjuvante com maior potencial de dissolução.

52
5. Referências
ACD/CHEMSKETCH, ADVANCED CHEMISTRY DEVELOPMENT, INC. ACD/Labs
Release: 12.00 Product version: 12.00. Build, v. 29305, 2008.
ADEBISI, Adeola O. et al. Solid-state, triboelectrostatic and dissolution characteristics of
spray-dried piroxicam-glucosamine solid dispersions. Colloids and Surfaces B:
Biointerfaces, v. 146, p. 841-851, 2016.
ADIBKIA, Khosro et al. Physicochemical characterization of naproxen solid dispersions
prepared via spray drying technology. Powder technology, v. 246, p. 448-455, 2013.
AKIN‐AJANI, Olufunke D.; ITIOLA, Oludele A.; ODEKU, Oluwatoyin A. Evaluation of the
disintegrant properties of native and modified forms of fonio and sweet potato
starches. Starch‐Stärke, v. 68, n. 1-2, p. 169-174, 2016.
AKTER, Farhana et al. Study of dissolution characteristics of ibuprofen by different polymers
and solid dispersion techniques. International Journal of Pharmaceutical Sciences and
Research, v. 6, n. 4, p. 1528, 2015.
ALAM, Mohd Aftab et al. Solid dispersions: a strategy for poorly aqueous soluble drugs and
technology updates. Expert opinion on drug delivery, v. 9, n. 11, p. 1419-1440, 2012.
ALAM, Mohd Aftab; AL-JENOOBI, Fahad I.; AL-MOHIZEA, Abdullah M. Commercially
bioavailable proprietary technologies and their marketed products. Drug Discovery Today, v.
18, n. 19-20, p. 936-949, 2013.
ALBADARIN, Ahmad B. et al. Development of stability-enhanced ternary solid dispersions
via combinations of HPMCP and Soluplus® processed by hot melt extrusion. International
journal of pharmaceutics, v. 532, n. 1, p. 603-611, 2017.
ALLEN, Loyd; ANSEL, Howard C. Ansel's pharmaceutical dosage forms and drug
delivery systems. Lippincott Williams & Wilkins, 2013.

53
ALSHAHRANI, Saad M. et al. Stability-enhanced hot-melt extruded amorphous solid
dispersions via combinations of Soluplus® and HPMCAS-HF. AAPS PharmSciTech, v. 16,
n. 4, p. 824-834, 2015.
AMIDON, Gordon L. et al. A theoretical basis for a biopharmaceutic drug classification: the
correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical
research, v. 12, n. 3, p. 413-420, 1995.
AMIN, Muhammad et al. Cellulose ether derivatives: a new platform for prodrug formation
of fluoroquinolone antibiotics. Cellulose, v. 22, n. 3, p. 2011-2022, 2015.
ANSARI, Muhammad Tayyab et al. Preparation and characterization of solid dispersions of
artemether by freeze-dried method. BioMed research international, v. 2015, 2015.
ASHFORD, M. Biodisponibilidade – fatores físico-químicos e relacionados à forma
farmacêutica. In: AULTON, M. E. Delineamento de formas farmacêuticas. 2. ed. Porto
Alegre: Artmed, 2005. cap. 17, p. 261-262.
AULTON, Michael E. Taylor K. Aulton’s pharmaceutics: the design and manufacture of
medicines. 4th ed. New York: Churchill livingstone Elsevier, 2005.
BAIRD, Jared A.; TAYLOR, Lynne S. Evaluation of amorphous solid dispersion properties
using thermal analysis techniques. Advanced drug delivery reviews, v. 64, n. 5, p. 396-421,
2012.
BALASAHEB, PARVE; BALAJI, T. E. L. I.; AVINASH, BIRAJDAR. Solid dispersions: An
overview on solubility enhancement of poorly water soluble drugs. International Journal of
Pharma and Bio Sciences, v. 5, n. 3, p. 7-25, 2014.
BATLOUNI, Michel. Anti-inflamatórios não esteroides: efeitos cardiovasculares, cérebro-
vasculares e renais. Arq Bras Cardiol, v. 94, n. 4, p. 556-63, 2010.

54
BI, Yunxia; RAHMAN, Mohammed Abdul; LESTER, James David. Solid dispersion of
poorly soluble compounds comprising crospovidone and at least one water-soluble
polymer. U.S. Patent Application n. 14/344,705, 8 jan. 2015.
BLEY, Heike; FUSSNEGGER, Bernd; BODMEIER, Roland. Characterization and stability
of solid dispersions based on PEG/polymer blends. International Journal of
Pharmaceutics, v. 390, n. 2, p. 165-173, 2010.
CHAN, Siok-Yee et al. The characterization and dissolution performances of spray dried
solid dispersion of ketoprofen in hydrophilic carriers. asian journal of pharmaceutical
sciences, v. 10, n. 5, p. 372-385, 2015.
CHAUDHARI, S. P. et al. Nanosuspension: A novel approaches in drug delivery system. Int
J Pharm Res Rev, v. 2, p. 30-9, 2013.
CHAUDHARI, Smruti P.; DUGAR, Rohit P. Application of surfactants in solid dispersion
technology for improving solubility of poorly water soluble drugs. Journal of Drug Delivery
Science and Technology, v. 41, p. 68-77, 2017.
CHAUHAN, Harsh; HUI-GU, Chong; ATEF, Eman. Correlating the behavior of polymers in
solution as precipitation inhibitor to its amorphous stabilization ability in solid
dispersions. Journal of pharmaceutical sciences, v. 102, n. 6, p. 1924-1935, 2013.
CHAVDA, H.; PATEL, C.; ANAND, I. Biopharmaceutics classification system. Systematic
reviews in pharmacy, v. 1, n. 1, p. 62, 2010.
CHEN, Yuejie et al. Sodium lauryl sulfate competitively interacts with HPMC-as and
consequently reduces oral bioavailability of posaconazole/HPMC-as amorphous solid
dispersion. Molecular pharmaceutics, v. 13, n. 8, p. 2787-2795, 2016.
CHIOU, Win Loung; RIEGELMAN, Sidney. Pharmaceutical applications of solid dispersion
systems. Journal of pharmaceutical sciences, v. 60, n. 9, p. 1281-1302, 1971.

55
CHOI, Jin-Seok et al. Solid dispersion of dutasteride using the solvent evaporation method:
Approaches to improve dissolution rate and oral bioavailability in rats. Materials Science
and Engineering: C, v. 90, p. 387-396, 2018.
CHORILLI, Marlus et al. Toxicologia dos cosméticos. Latin American Journal of
Pharmacy, v. 26, n. 1, p. 144, 2007.
CRAIG, Duncan QM. The mechanisms of drug release from solid dispersions in water-
soluble polymers. International journal of pharmaceutics, v. 231, n. 2, p. 131-144, 2002.
CROWLEY, Michael M. et al. Pharmaceutical applications of hot-melt extrusion: part
I. Drug development and industrial pharmacy, v. 33, n. 9, p. 909-926, 2007.
CUI, Fude et al. Design of sustained-release nitrendipine microspheres having solid
dispersion structure by quasi-emulsion solvent diffusion method. Journal of controlled
release, v. 91, n. 3, p. 375-384, 2003.
DEBOUZY, Jean-Claude et al. Interaction study of an amorphous solid dispersion of
cyclosporin A in poly-alpha-cyclodextrin with model membranes by 1H-, 2H-, 31P-NMR and
electron spin resonance. Journal of drug delivery, v. 2014, 2014.
DEQUEKER, J. et al. Improvement in gastrointestinal tolerability of the selective
cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: results of the
Safety and Efficacy Large-scale Evaluation of COX-inhibiting Therapies (SELECT) trial in
osteoarthritis. British journal of rheumatology, v. 37, n. 9, p. 946-951, 1998.
DESAI, J.; ALEXANDER, K.; RIGA, A. Characterization of polymeric dispersions of
dimenhydrinate in ethyl cellulose for controlled release. International journal of
pharmaceutics, v. 308, n. 1-2, p. 115-123, 2006.
DIAS, Iara Lucia Tescarollo et al. Avaliação das propriedades tecnológicas de granulados de
amido e lactose e para produção de comprimidos por compressão direta. Ciência &
Inovação, v. 1, n. 1, 2018.

56
DJERAFI, Rania et al. Supercritical antisolvent co-precipitation of rifampicin and ethyl
cellulose. European Journal of Pharmaceutical Sciences, v. 102, p. 161-171, 2017.
DOELKER, E. Comparative compaction properties of various microcrystalline cellulose types
and generic products. Drug Development and Industrial Pharmacy, v. 19, p. 2399-2471,
1993.
FAN, Na et al. Impact of HPMC on inhibiting crystallization and improving permeability of
curcumin amorphous solid dispersions. Carbohydrate polymers, v. 181, p. 543-550, 2018.
FARAH, Nabil et al. Pharmaceutical Composition Comprising Solid Dispersion of BCS
Class II Drugs with Gelucires. U.S. Patent Application n. 15/400,539, 4 maio 2017.
FASANO, Alessio. Innovative strategies for the oral delivery of drugs and peptides. Trends
in biotechnology, v. 16, n. 4, p. 152-157, 1998.
FATMI, Sofiane et al. Amorphous solid dispersion studies of camptothecin-cyclodextrin
inclusion complexes in PEG 6000. Acta Pol Pharm, v. 72, n. 1, p. 179-192, 2015.
FERNANDEZ, Mercedes et al. Characterization of solid dispersions of
piroxicam/polyethylene glycol 4000. International journal of pharmaceutics, v. 84, n. 2, p.
197-202, 1992.
FERREIRA, AO. Guia prático de farmácia magistral. 3. ed. v. l. 1. São Paulo:
Pharmabooks, v.1, p.736, 2008.
FINI, Adamo et al. Diclofenac salts, II. Solid dispersions in PEG6000 and Gelucire
50/13. European journal of pharmaceutics and biopharmaceutics, v. 60, n. 1, p. 99-111,
2005.

57
FOOD AND DRUG ADMINISTRATION et al. Guidance for industry: waiver of in vivo
bioavailability and bioequivalence studies for immediate-release solid oral dosage forms
based on a biopharmaceutics classification system. Food and Drug Administration, Silver
Spring, MD, 2017.
FRIZON, Fernando et al. Melhoria da taxa de dissolução da loratadina em dispersões sólidas
de polivinilpirrolidona K-30 por métodos de solventes. Powder technology , v. 235, p. 532-
539, 2013.
GAO, Kezheng et al. Cellulose nanofibers/multi-walled carbon nanotube nanohybrid aerogel
for all-solid-state flexible supercapacitors. RSC Advances, v. 3, n. 35, p. 15058-15064, 2013.
GOHEL, M. C.; PATEL, L. D. Processing of nimesulide-PEG 400-PG-PVP solid dispersions:
preparation, characterization, and in vitro dissolution. Drug development and industrial
pharmacy, v. 29, n. 3, p. 299-310, 2003.
GOLAN, David et al. Princípios de farmacologia: a base fisiopatológica da farmacoterapia.
In: Princípios de farmacologia: a base fisiopatológica da farmacoterapia. 2009..
GU, Bing; LINEHAN, Brian; TSENG, Yin-Chao. Optimization of the Büchi B-90 spray
drying process using central composite design for preparation of solid
dispersions. International journal of pharmaceutics, v. 491, n. 1-2, p. 208-217, 2015.
GUPTA, Rahul; MISHRA, A. K.; PATHAK, A. K. A Critical Review on Different
Pharmaceutical Aspects of Solid Dispersion Technique for Solubility Enhancement. Int J
Pharm Biol Sci, v. 5, p. 119-28, 2015.
HALLOUARD, Francois et al. Solid dispersions for oral administration: an overview of the
methods for their preparation. Current pharmaceutical design, v. 22, n. 32, p. 4942-4958,
2016.
HE, Cai-Xia; HE, Zhong-Gui; GAO, Jian-Qing. Microemulsions as drug delivery systems to
improve the solubility and the bioavailability of poorly water-soluble drugs. Expert opinion
on drug delivery, v. 7, n. 4, p. 445-460, 2010.

58
HERBRINK, Maikel et al. Improving the solubility of nilotinib through novel spray-dried
solid dispersions. International journal of pharmaceutics, v. 529, n. 1-2, p. 294-302, 2017.
HÖRTER, D.; DRESSMAN, J. B. Influence of physicochemical properties on dissolution of
drugs in the gastrointestinal tract1. Advanced drug delivery reviews, v. 46, n. 1-3, p. 75-87,
2001.
HU, Xian-Yue; LOU, Hao; HAGEMAN, Michael J. Preparation of Lapatinib Ditosylate Solid
Dispersions Using Solvent Rotary Evaporation and Hot Melt Extrusion for Solubility and
Dissolution Enhancement. International Journal of Pharmaceutics, 2018.
HUANG, Jingjun; WIGENT, Rodney J.; SCHWARTZ, Joseph B. Nifedipine molecular
dispersion in microparticles of ammonio methacrylate copolymer and ethylcellulose binary
blends for controlled drug delivery: effect of matrix composition. Drug development and
industrial pharmacy, v. 32, n. 10, p. 1185-1197, 2006.
HUANG, Yanbin; DAI, Wei-Guo. Fundamental aspects of solid dispersion technology for
poorly soluble drugs. Acta Pharmaceutica Sinica B, v. 4, n. 1, p. 18-25, 2014.
HUGHEY, Justin R. et al. The incorporation of low-substituted hydroxypropyl cellulose into
solid dispersion systems. Drug development and industrial pharmacy, v. 41, n. 8, p. 1294-
1301, 2015.
JAGDALE, S. C. et al. Preparation and Characterization of Metformin Hydrochloride—
Compritol 888 ATO Solid Dispersion. Journal of young pharmacists: JYP, v. 3, n. 3, p.
197, 2011.
JERMAIN, Scott V.; BROUGH, Chris; WILLIAMS III, Robert O. Amorphous solid
dispersions and nanocrystal technologies for poorly water-soluble drug delivery–An
update. International journal of pharmaceutics, v. 535, n. 1-2, p. 379-392, 2018.
JOSHI, Meenakshi et al. Solid Dispersion: A Technology for the Improvement of Oral
Bioavailability of Poorly Soluble Drugs. Current Pharma Research, v. 2, n. 4, p. 668, 2012.

59
JOUDIEH, Samer et al. Cyclodextrin polymers as efficient solubilizers of albendazole:
complexation and physico-chemical characterization. Journal of nanoscience and
nanotechnology, v. 9, n. 1, p. 132-140, 2009.
JUNG, Hyuck Jun et al. Improved oral absorption of tacrolimus by a solid dispersion with
hypromellose and sodium lauryl sulfate. International journal of biological
macromolecules, v. 83, p. 282-287, 2016.
KALPANA, Patidar et al. Solid Dispersion: Approaches, Technology involved, Unmet need
& Challenges. Drug Invention Today, v. 2, n. 7, 2010.
KANIG, Joseph L. Properties of fused mannitol in compressed tablets. Journal of
pharmaceutical sciences, v. 53, n. 2, p. 188-192, 1964.
KARAVAS, Evangelos et al. Investigation of the release mechanism of a sparingly water-
soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug,
particle size distribution and drug–polymer interactions. European journal of
pharmaceutics and biopharmaceutics, v. 66, n. 3, p. 334-347, 2007.
KATZUNG, Bertram G.; TREVOR, Anthony J. Farmacologia Básica e Clínica-13. McGraw
Hill Brasil, 2017.
KAWABATA, Yohei et al. Novel crystalline solid dispersion of tranilast with high
photostability and improved oral bioavailability. European Journal of Pharmaceutical
Sciences, v. 39, n. 4, p. 256-262, 2010.
KHAN, Gul Majid; JIABI, Zhu. Preparation, characterization, and dissolution studies of
ibuprofen solid dispersions using polyethylene glycol (PEG), talc, and PEG-talc as dispersion
carriers. Drug development and industrial pharmacy, v. 24, n. 5, p. 455-462, 1998.
KHAN, Tarique et al. An approach for rapid disintegrating tablet: A Review. International
Journal of Pharmaceutical Research & Development, v. 3, n. 3, p. 170-183, 2011.

60
KIM, Min-Soo et al. Enhanced bioavailability of sirolimus via preparation of solid dispersion
nanoparticles using a supercritical antisolvent process. International journal of
nanomedicine, v. 6, p. 2997, 2011.
KOJO, Yoshiki et al. Improved oral absorption profile of itraconazole in hypochlorhydria by
self-micellizing solid dispersion approach. European Journal of Pharmaceutical Sciences,
v. 97, p. 55-61, 2017.
KOLAŠINAC, Nemanja et al. Solubility enhancement of desloratadine by solid dispersion in
poloxamers. International journal of pharmaceutics, v. 436, n. 1-2, p. 161-170, 2012.
KORN, Christian; BALBACH, Stefan. Compound selection for development–Is salt
formation the ultimate answer? Experiences with an extended concept of the “100 mg
approach”. European Journal of Pharmaceutical Sciences, v. 57, p. 257-263, 2014.
KOTTA, Sabna et al. Exploring oral nanoemulsions for bioavailability enhancement of poorly
water-soluble drugs. Expert opinion on drug delivery, v. 9, n. 5, p. 585-598, 2012.
KU, M. Sherry; DULIN, Wendy. A biopharmaceutical classification-based Right-First-Time
formulation approach to reduce human pharmacokinetic variability and project cycle time
from First-In-Human to clinical Proof-Of-Concept. Pharmaceutical development and
technology, v. 17, n. 3, p. 285-302, 2012.
LELEUX, Jardin; WILLIAMS, Robert O. Recent advancements in mechanical reduction
methods: particulate systems. Drug development and industrial pharmacy, v. 40, n. 3, p.
289-300, 2014.
LENNERNÄS, Hans; ABRAHAMSSON, Bertil. The use of biopharmaceutic classification of
drugs in drug discovery and development: current status and future extension. Journal of
pharmacy and pharmacology, v. 57, n. 3, p. 273-285, 2005.

61
LEUNER, Christian; DRESSMAN, Jennifer. Improving drug solubility for oral delivery
using solid dispersions. European journal of Pharmaceutics and Biopharmaceutics, v. 50,
n. 1, p. 47-60, 2000.
LI, Na; ORMES, James D.; TAYLOR, Lynne S. Leaching of lopinavir amorphous solid
dispersions in acidic media. Pharmaceutical research, v. 33, n. 7, p. 1723-1735, 2016.
LIU, Haoyu et al. Synthesis and structure–property evaluation of cellulose ω-carboxyesters
for amorphous solid dispersions. Carbohydrate polymers, v. 100, p. 116-125, 2014.
LU, Ying; PARK, Kinam. Polymeric micelles and alternative nanonized delivery vehicles for
poorly soluble drugs. International journal of pharmaceutics, v. 453, n. 1, p. 198-214,
2013.
MAGGI, Lauretta et al. Improvement of the dissolution behavior of gliclazide, a slightly
soluble drug, using solid dispersions. Journal of Drug Delivery Science and Technology, v.
26, p. 17-23, 2015.
MARGARIT, Maria Victoria; RODRÍGUEZ, Inés Carmen; CEREZO, Antonio. Physical
characteristics and dissolution kinetics of solid dispersions of ketoprofen and polyethylene
glycol 6000. International journal of pharmaceutics, v. 108, n. 2, p. 101-107, 1994.
MEDAREVIĆ, Djordje P. et al. Dissolution rate enhancement and physicochemical
characterization of carbamazepine-poloxamer solid dispersions. Pharmaceutical
development and technology, v. 21, n. 3, p. 268-276, 2016.
MEHANNA, Mohammed M.; MOTAWAA, Adel M.; SAMAHA, Magda W. In sight into
tadalafil–block copolymer binary solid dispersion: mechanistic investigation of dissolution
enhancement. International journal of pharmaceutics, v. 402, n. 1-2, p. 78-88, 2010.
MEHTA, Piyush. Dry powder inhalers: a focus on advancements in novel drug delivery
systems. Journal of drug delivery, v. 2016, 2016.

62
MERLIM, Érika de Azevedo. Diminuição do tempo de desintegração de uma formulação
dispersível a partir de alterações no tempo de mistura com lubrificante. 2011.
MIRANDA, Letícia Paula; CARDOSO, Mayara Gois; DE MORAES, Anamaria Junqueira.
PROPOSTA DE FORMULAÇÕES PARA EXCIPIENTES-PADRÃO DE FÁRMACOS
CLASSIFICADOS PELO SISTEMA DE CLASSIFICAÇÃO BIOFARMACÊUTICA. e-
RAC, v. 3, n. 1, 2013.
MONEGHINI, Mariarosa et al. Microwave generated solid dispersions containing
ibuprofen. International journal of pharmaceutics, v. 361, n. 1-2, p. 125-130, 2008.
MONEGHINI, Mariarosa; ZINGONE, Guglielmo; DE ZORDI, Nicola. Influence of the
microwave technology on the physical–chemical properties of solid dispersion with
Nimesulide. Powder technology, v. 195, n. 3, p. 259-263, 2009.
MÖSCHWITZER, Jan P. Drug nanocrystals in the commercial pharmaceutical development
process. International journal of pharmaceutics, v. 453, n. 1, p. 142-156, 2013.
MOTALLAE, Saeed; TAHERI, Azade; HOMAYOUNI, Alireza. Preparation and
characterization of solid dispersions of celecoxib obtained by spray-drying ethanolic
suspensions containing PVP-K30 or isomalt. Journal of Drug Delivery Science and
Technology, v. 46, p. 188-196, 2018.
MURA, P. et al. Properties of solid dispersions of naproxen in various polyethylene
glycols. Drug development and industrial pharmacy, v. 22, n. 9-10, p. 909-916, 1996.
NAJIB, Naji M.; SULEIMAN, M.; MALAKH, A. Characteristics of the in vitro release of
ibuprofen from polyvinylpyrrolidone solid dispersions. International journal of
pharmaceutics, v. 32, n. 2-3, p. 229-236, 1986.
NASERI, Neda; VALIZADEH, Hadi; ZAKERI-MILANI, Parvin. Solid lipid nanoparticles
and nanostructured lipid carriers: structure, preparation and application. Advanced
pharmaceutical bulletin, v. 5, n. 3, p. 305, 2015.

63
NEWA, Madhuri et al. Preparation and evaluation of fast dissolving ibuprofen-polyethylene
glycol 6000 solid dispersions. Drug delivery, v. 15, n. 6, p. 355-364, 2008.
NEWA, Madhuri et al. Preparation, characterization and in vivo evaluation of ibuprofen
binary solid dispersions with poloxamer 188. International journal of pharmaceutics, v.
343, n. 1-2, p. 228-237, 2007.
NI, Huaiwei et al. Supercritical fluids at subduction zones: Evidence, formation condition,
and physicochemical properties. Earth-science reviews, v. 167, p. 62-71, 2017.
NOKHODCHI, Ali et al. The use of various organic solvents to tailor the properties of
ibuprofen–glucosamine HCl solid dispersions. Chemical Engineering Research and Design,
v. 117, p. 509-519, 2017.
OBAIDAT, Rana M. et al. Using supercritical fluid technology (SFT) in preparation of
tacrolimus solid dispersions. AAPS PharmSciTech, v. 18, n. 2, p. 481-493, 2017.
OHARA, Toshio et al. Dissolution mechanism of poorly water-soluble drug from extended
release solid dispersion system with ethylcellulose and
hydroxypropylmethylcellulose. International journal of pharmaceutics, v. 302, n. 1-2, p.
95-102, 2005.
OSAWA, Mizuho et al. Characterization of Sulindac Solid Dispersion with Hydroxypropyl
Cellulose Prepared by Hot Melt Extrusion. Journal of Pharmaceutical Science and
Technology, Japan, v. 74, n. 2, p. 160-169, 2014.
PAPAGEORGIOU, Myrsini et al. Novel Isocyanate-Modified Carrageenan Polymer
Materials: Preparation, Characterization and Application Adsorbent Materials of
Pharmaceuticals. Polymers, v. 9, n. 11, p. 595, 2017.
PARVE, B. et al. Solubility enhancement techniques: a review. World J. Pharm. Pharm.
Sci, v. 3, p. 400-422, 2014.

64
PATEL, M. M.; PATEL, D. M. Fast dissolving valdecoxib tablets containing solid dispersion
of valdecoxib. Indian journal of pharmaceutical sciences, v. 68, n. 2, 2006.
PATIL, Hemlata; TIWARI, Roshan V.; REPKA, Michael A. Hot-melt extrusion: from theory
to application in pharmaceutical formulation. AAPS PharmSciTech, v. 17, n. 1, p. 20-42,
2016.
PEREVA, Stiliyana et al. Efficiency of “cyclodextrin-ibuprofen” inclusion complex
formation. Journal of Drug Delivery Science and Technology, v. 35, p. 34-39, 2016.
PEIGHAMBARDOUST, S. H.; TAFTI, A. Golshan; HESARI, J. Application of spray drying
for preservation of lactic acid starter cultures: a review. Trends in Food Science &
Technology, v. 22, n. 5, p. 215-224, 2011.
PRISTA L. N., Alves A. C. e Morgado R., 1995. Tecnologia Farmacêutica, I Volume, 5ª
edição. Fundação Calouste Gulbenkian. Lisboa.
RASHID, Rehmana et al. Effect of hydroxypropylcellulose and Tween 80 on
physicochemical properties and bioavailability of ezetimibe-loaded solid
dispersion. Carbohydrate polymers, v. 130, p. 26-31, 2015.
RATCLIFFE, Liam PD et al. Polymerization-induced self-assembly of all-acrylic diblock
copolymers via RAFT dispersion polymerization in alkanes. Macromolecules, v. 48, n. 23, p.
8594-8607, 2015.
REPKA, Michael A. et al. Pharmaceutical applications of hot-melt extrusion: Part II. Drug
development and industrial pharmacy, v. 33, n. 10, p. 1043-1057, 2007.
RIBEIRO, Virginia Strapassoni; RAU, Carina. QUALITY OF RAW MATERIALS USED
AT HANDLING PHARMACIES IN CURITIBA AND METROPOLITAN AREA. Visão
Acadêmica, v. 13, n. 3, 2012.

65
ROBERTS, Matthew et al. Preparation and characterization of Compritol 888 ATO matrix
tablets for the sustained release of diclofenac sodium. Pharmaceutical development and
technology, v. 20, n. 4, p. 507-512, 2015.
ROGERS, J. A.; ANDERSON, A. J. Physical characteristics and dissolution profiles of
ketoprofen-urea solid dispersions. Pharmaceutica Acta Helvetiae, v. 57, n. 10-1, p. 276-281,
1982.
ROSSMANN, Matthias; BRAEUER, Andreas; SCHLUECKER, Eberhard. Supercritical
antisolvent micronization of PVP and ibuprofen sodium towards tailored solid
dispersions. The Journal of Supercritical Fluids, v. 89, p. 16-27, 2014.
ROWE, R. C.; SHESKEY, P. J.; QUINN, M. E. Handbook of Pharmaceutical Excipients 6th
edition Pharmaceutical Press. London, England, p. 637, 2009.
SAHOO, Nanda Gopal et al. Dissolution enhancement of a poorly water-soluble antimalarial
drug by means of a modified multi-fluid nozzle pilot spray drier. Materials Science and
Engineering: C, v. 31, n. 2, p. 391-399, 2011.
SALVADOR, Milena; CESCONETTO Juliana S.; SOARES, Érica C. S.; FRANÇA, Patrícia
O. Avaliação da influência de diferentes diluentes sobre os parâmetros de resistência física e
desintegração de comprimidos de ibuprofeno obtidos por compressão direta. Infarma-
Ciências Farmacêuticas, v. 23, n. 7/8, p. 44-51, 2012.
SAMMOUR, Omaima A. et al. Formulation and optimization of mouth dissolve tablets
containing rofecoxib solid dispersion. AAPS PharmSciTech, v. 7, n. 2, p. E167-E175, 2006
.
SAREEN, Swati; MATHEW, George; JOSEPH, Lincy. Melhoria na solubilidade de fármacos
solúveis em água pobres por dispersão sólida. Revista Internacional de Investigação
Farmacêutica , v. 2, n. 1, p. 12, 2012.
SARODE, Ashish L. et al. Extrusão por fusão a quente (HME) para dispersões sólidas
amorfas: ferramentas preditivas para processamento e impacto de interações droga-polímero

66
na supersaturação. Revista Européia de Ciências Farmacêuticas , v. 48, n. 3, p. 371-384,
2013.
SEKIGUCHI, Keiji; OBI, Noboru. Studies on Absorption of Eutectic Mixture. I. A
Comparison of the Behavior of Eutectic Mixture of Sulfathiazole and that of Ordinary
Sulfathiazole in Man. Chemical and Pharmaceutical Bulletin, v. 9, n. 11, p. 866-872, 1961.
SEKIGUCHI, Keiji; OBI, Noboru; UEDA, Yoshio. Studies on Absorption of Eutectic
Mixture. II. Absorption of fused Conglomerates of Chloramphenicol and Urea in
Rabbits. Chemical and Pharmaceutical Bulletin, v. 12, n. 2, p. 134-144, 1964.
SERTSOU, Gabriel et al. Factors affecting incorporation of drug into solid solution with
HPMCP during solvent change co-precipitation. International journal of pharmaceutics, v.
245, n. 1-2, p. 99-108, 2002.
SHAH, Navnit et al. Improved human bioavailability of vemurafenib, a practically insoluble
drug, using an amorphous polymer‐stabilized solid dispersion prepared by a
solvent‐controlled coprecipitation process. Journal of pharmaceutical sciences, v. 102, n. 3,
p. 967-981, 2013.
SHAKHTSHNEIDER, T. P. et al. The mechanochemical preparation of solid disperse
systems of ibuprofen-polyethylene glycol. International journal of pharmaceutics, v. 130,
n. 1, p. 25-32, 1996.
SHETH, N. S. Formulation and evaluation of solid dispersion of olanzepine. International
Journal of Pharmaceutical Sciences and Research, v. 2, n. 3, p. 691, 2011.
SILVA, Joel Rocha et al. Otimização do processo de compressão de comprimidos de
hidroclorotiazida. Ensaios e Ciência: C. Biológicas, Agrárias e da Saúde, v. 17, n. 5, 2015.
SINKO, Patrick J. Martin: físico-farmácia e ciências farmacêuticas. Arthmed, Porto Alegre,
p. 528-578, 2008.

67
SKIBA, M. et al. Preparation and characterization of amorphous solid dispersions of
nimesulide in cyclodextrin copolymers. Journal of nanoscience and nanotechnology, v. 14,
n. 4, p. 2772-2779, 2014.
SMITH, Francine G. et al. Cyclooxygenase (COX) inhibitors and the newborn
kidney. Pharmaceuticals, v. 5, n. 11, p. 1160-1176, 2012.
SONG, Chung Kil; YOON, In-Soo; KIM, Dae-Duk. Poloxamer-based solid dispersions for
oral delivery of docetaxel: Differential effects of F68 and P85 on oral docetaxel
bioavailability. International journal of pharmaceutics, v. 507, n. 1-2, p. 102-108, 2016.
SRINARONG, Parinda et al. Surface‐active derivative of inulin (Inutec® SP1) is a superior
carrier for solid dispersions with a high drug load. Journal of pharmaceutical sciences, v.
100, n. 6, p. 2333-2342, 2011.
SUN, Dajun D.; LEE, Ping I. Probing the mechanisms of drug release from amorphous solid
dispersions in medium-soluble and medium-insoluble carriers. Journal of Controlled
Release, v. 211, p. 85-93, 2015.
TAKAGI, Toshihide et al. A provisional biopharmaceutical classification of the top 200 oral
drug products in the United States, Great Britain, Spain, and Japan. Molecular
pharmaceutics, v. 3, n. 6, p. 631-643, 2006.
TANTISHAIYAKUL, V.; KAEWNOPPARAT, N.; INGKATAWORNWONG, S. Properties
of solid dispersions of piroxicam in polyvinylpyrrolidone. International Journal of
Pharmaceutics, v. 181, n. 2, p. 143-151, 1999.
TEJA, Surikutchi Bhanu et al. Drug-excipient behavior in polymeric amorphous solid
dispersions. Journal of Excipients and Food Chemicals, v. 4, n. 3, p. 1048, 2016.
THAKURIA, Ranjit et al. Pharmaceutical cocrystals and poorly soluble drugs. International
journal of pharmaceutics, v. 453, n. 1, p. 101-125, 2013.
THANOU, M.; VERHOEF, J. C.; JUNGINGER, H. E. Chitosan and its derivatives as
intestinal absorption enhancers. Advanced drug delivery reviews, v. 50, p. S91-S101, 2001.

68
THIRY et al. Continuous production of itraconazole-based solid dispersions by hot melt
extrusion: preformulation, optimization and design space determination. International
journal of pharmaceutics, v. 515, n. 1-2, p. 114-124, 2016.
TSUME, Yasuhiro et al. The Biopharmaceutics Classification System: subclasses for in vivo
predictive dissolution (IPD) methodology and IVIVC. European Journal of Pharmaceutical
Sciences, v. 57, p. 152-163, 2014.
URAMATSU, Shunji et al. Base for dry solid dispersion, solid dispersion containing the
base, and composition containing the dispersion. U.S. Patent n. 9,101,617, 11 ago. 2015.
VAN DEN MOOTER, Guy et al. Evaluation of Inutec SP1 as a new carrier in the formulation
of solid dispersions for poorly soluble drugs. International journal of pharmaceutics, v.
316, n. 1-2, p. 1-6, 2006.
VAN DEN MOOTER, Guy. The use of amorphous solid dispersions: A formulation strategy
to overcome poor solubility and dissolution rate. Drug Discovery Today: Technologies, v. 9,
n. 2, p. e79-e85, 2012.
VAN DROOGE, D. J. et al. Characterization of the molecular distribution of drugs in glassy
solid dispersions at the nano-meter scale, using differential scanning calorimetry and
gravimetric water vapour sorption techniques. International journal of pharmaceutics, v.
310, n. 1-2, p. 220-229, 2006.
VASANTHAVADA, Madhav et al. Phase behavior of amorphous molecular dispersions II:
Role of hydrogen bonding in solid solubility and phase separation kinetics. Pharmaceutical
research, v. 22, n. 3, p. 440-448, 2005.
VASANTHAVADA, Madhav et al. Phase behavior of amorphous molecular dispersions I:
Determination of the degree and mechanism of solid solubility. Pharmaceutical research, v.
21, n. 9, p. 1598-1606, 2004.

69
VASCONCELOS, Teofilo; SARMENTO, Bruno; COSTA, Paulo. Solid dispersions as
strategy to improve oral bioavailability of poor water soluble drugs. Drug discovery today, v.
12, n. 23-24, p. 1068-1075, 2007.
VASOYA, Jaydip et al. Development of Solid Dispersion by Hot Melt Extrusion Using
Mixtures of Polyoxylglycerides with Polymers as Carriers for Increasing Dissolution Rate of
a Model Poorly Soluble Drug. Journal of pharmaceutical sciences, 2018.
VERHOEVEN, Ellen et al. Influence of polyethylene glycol/polyethylene oxide on the
release characteristics of sustained-release ethylcellulose mini-matrices produced by hot-melt
extrusion: in vitro and in vivo evaluations. European Journal of Pharmaceutics and
Biopharmaceutics, v. 72, n. 2, p. 463-470, 2009.
VERMA, Somya; PATEL, Urmi; PATEL, Rakesh P. Formulation and evaluation of
ivermectin solid dispersion. Journal of Drug Delivery and Therapeutics, v. 7, n. 7, p. 15-
17, 2017.
VILLANOVA, Janaina CO; ORÉFICE, Rodrigo L.; CUNHA, Armando S. Aplicações
farmacêuticas de polímeros. Polímeros: Ciência e Tecnologia, v. 20, n. 1, 2010.
VIPPAGUNTA, Sudha R. et al. Factors affecting the formation of eutectic solid dispersions
and their dissolution behavior. Journal of pharmaceutical sciences, v. 96, n. 2, p. 294-304,
2007.
VO, Chau Le-Ngoc; PARK, Chulhun; LEE, Beom-Jin. Current trends and future perspectives
of solid dispersions containing poorly water-soluble drugs. European journal of
pharmaceutics and biopharmaceutics, v. 85, n. 3, p. 799-813, 2013.
WANNING, Stefan; SÜVERKRÜP, Richard; LAMPRECHT, Alf. Pharmaceutical spray
freeze drying. International journal of pharmaceutics, v. 488, n. 1-2, p. 136-153, 2015.

70
WLODARSKI, K. et al. Physicochemical properties of tadalafil solid dispersions–Impact of
polymer on the apparent solubility and dissolution rate of tadalafil. European Journal of
Pharmaceutics and Biopharmaceutics, v. 94, p. 106-115, 2015.
WU, Chi-Yuan; BENET, Leslie Z. Predicting drug disposition via application of BCS:
transport/absorption/elimination interplay and development of a biopharmaceutics drug
disposition classification system. Pharmaceutical research, v. 22, n. 1, p. 11-23, 2005.
WU, K. E. et al. Formation and characterization of solid dispersions of piroxicam and
polyvinylpyrrolidone using spray drying and precipitation with compressed
antisolvent. Journal of pharmaceutical sciences, v. 98, n. 7, p. 2422-2431, 2009.
XU, Hui et al. Extended Tacrolimus Release via the Combination of Lipid-based Solid
Dispersion and HPMC Hydrogel Matrix Tablets. Asian Journal of Pharmaceutical
Sciences, 2018
XU, L. et al. Improvement of dissolution rate of ibuprofen by solid dispersion systems with
Kollicoat IR using a pulse combustion dryer system. Journal of Drug Delivery Science and
Technology, v. 19, n. 2, p. 113-118, 2009.
XU, Wei-Juan et al. Enhanced dissolution and oral bioavailability of valsartan solid
dispersions prepared by a freeze-drying technique using hydrophilic polymers. Drug
delivery, v. 23, n. 1, p. 41-48, 2016.
YANG, Gang et al. Improved dissolution and bioavailability of silymarin delivered by a solid
dispersion prepared using supercritical fluids. asian journal of pharmaceutical sciences, v.
10, n. 3, p. 194-202, 2015.
YOSHIOKA, Minoru; HANCOCK, Bruno C.; ZOGRAFI, George. Crystallization of
indomethacin from the amorphous state below and above its glass transition
temperature. Journal of pharmaceutical sciences, v. 83, n. 12, p. 1700-1705, 1994.

71
YUVARAJA, K.; KHANAM, J. Enhancement of carvedilol solubility by solid dispersion
technique using cyclodextrins, water soluble polymers and hydroxyl acid. Journal of
pharmaceutical and biomedical analysis, v. 96, p. 10-20, 2014.
ZHANG, Meimei et al. Formulation and delivery of improved amorphous fenofibrate solid
dispersions prepared by thin film freezing. European Journal of Pharmaceutics and
Biopharmaceutics, v. 82, n. 3, p. 534-544, 2012.
ZHANG, Qinxiu et al. Effect of HPMCAS on recrystallization inhibition of nimodipine solid
dispersions prepared by hot-melt extrusion and dissolution enhancement of nimodipine
tablets. Colloids and Surfaces B: Biointerfaces, v. 172, p. 118-126, 2018.
ZHAO, Junshu et al. The Impact of Disintegrant Type, Surfactant, and API Properties on the
Processability and Performance of Roller Compacted Formulations of Acetaminophen and
Aspirin. The AAPS journal, v. 19, n. 5, p. 1387-1395, 2017.