5910236 – física ii (química) – ffclrp – usp – prof. antônio...
TRANSCRIPT

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
1
A Energia Livre de Gibbs e o Potencial Químico
Nesta aula vamos apresentar uma grandeza termodinâmica
fundamental para a explicação de processos espontâneos na
natureza, como reações químicas e transições de fase. O uso dessa
grandeza termodinâmica é restrito ao caso em que o processo ocorre
a pressão e temperatura constantes. Em princípio, isso pode parecer
restritivo para a aplicabilidade de uma grandeza física, mas, se
pensarmos nos fenômenos biológicos, veremos que quase sempre
eles ocorrem à pressão constante (a pressão atmosférica) e à
temperatura constante (a temperatura do organismo), que variam
muito pouco durante as situações “normais” de funcionamento de
um organismo vivo.
Você pode se perguntar porque é necessário definir uma outra
grandeza termodinâmica para estudar processos espontâneos, pois a
entropia já desempenha este papel: os processos naturais, isto é,
irreversíveis, ocorrem sempre na direção do aumento da entropia do
universo:
ΔSuniverso = ΔSsistema + ΔSvizinhança ≥ 0.
Quando o sistema que passa pelo processo está isolado da sua
vizinhança, a variação na entropia do universo é a própria variação

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
2
na entropia do sistema (ΔSviz = 0). Neste caso, apenas o cálculo da
variação na entropia do sistema é suficiente para se determinar em
que sentido o processo deve ocorrer espontaneamente.
Porém, na maior parte dos casos de interesse, especialmente em
biologia, o sistema não está isolado do resto do universo. Em tais
casos, para se determinar a variação na entropia do universo deve-se
medir duas coisas: a variação na entropia do sistema e a variação na
entropia da sua vizinhança. Em geral, a variação na entropia do
sistema pode ser medida sem muita dificuldade, como no caso em
que ele está isolado, mas é muito difícil, para não dizer impossível,
medir a variação na entropia da vizinhança do sistema (isto
implicaria em ter que medir a variação na entropia do resto do
universo, com exceção do sistema).
Para processos a pressão e temperatura constantes, não é necessário
conhecer a variação na entropia do resto do universo para prever
qual a direção em que o processo ocorrerá espontaneamente. Para
mostrar isto, vamos considerar o sistema e a sua vizinhança em
equilíbrio térmico a uma temperatura T. A pressão P também será
constante durante o processo.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
3
Durante o processo, o sistema troca uma quantidade de calor qp com
a sua vizinhança. Como a pressão é constante, este calor é igual à
variação na entalpia do sistema:
sistemaHqp Δ= .
Do ponto de vista da vizinhança, a variação na sua entalpia é dada
por,
pqHH −=Δ−=Δ sistemavizinhança .
Portanto, podemos escrever para a vizinhança:
THH
S sistemavizinhançavizinhança T
Δ−=
Δ=Δ .
Sendo assim, podemos escrever a variação na entropia do universo
como,
( )sistemasistemasistema
sistemavizinhançasistemauniverso HSTTT
HSSSS Δ−Δ=
Δ−Δ=Δ+Δ=Δ
1
,
ou
( )sistemasistemauniverso STHT
S Δ−Δ−=Δ1
.
Note que agora conseguimos escrever a variação na entropia do
universo em termos apenas de variações de grandezas relativas ao
sistema.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
4
Vamos definir a energia livre de Gibbs do sistema, G, como
TSPVUTSHG −+=−= . (1)
Ela tem esse nome porque foi definida pela primeira vez pelo físico
norte-americano Josiah Willard Gibbs (1839-1903).
Note que G é uma função de estado do sistema, pois ela depende
apenas de variáveis de estado (P, V, T) e de outras funções de estado
(a entalpia e a entropia).
A variação de G é dada por
TSSTHG Δ−Δ−Δ=Δ .
Quando a temperatura T é constante, ΔT = 0, de maneira que
STHG Δ−Δ=Δ .
Observe que este é o mesmo termo que aparece na expressão obtida
anteriormente para a variação na entropia do universo em termos de
grandezas relativas apenas ao sistema.
Portanto, podemos escrever a variação da entropia do universo
durante um processo a temperatura e pressão constantes em termos
da variação da energia livre de Gibbs do sistema,
01≥Δ−=Δ sistemauniverso G
TS .

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
5
Esta desigualdade pode ser reescrita como (lembre-se que a
temperatura T sempre é positiva),
0≤Δ sistemaG . (2)
Isto implica que, para T e P constantes, a desigualdade acima é
equivalente à segunda lei da termodinâmica:
Em qualquer processo que ocorre espontaneamente a pressão e
temperatura constantes, a energia livre de Gibbs do sistema nunca
aumenta.
Em outras palavras, a energia livre de Gibbs tende para um mínimo
a pressão e temperatura constantes. Esse mínimo é o estado de
equilíbrio do sistema.
Vamos ilustrar a aplicabilidade do conceito de energia livre de
Gibbs com um exemplo de transição de fase. Vamos considerar o
caso da transição água-gelo,
H2O(l) → H2O(s),
a três temperaturas diferentes: T = −10°C = 263,15 K, T = 0°C =
273,15 K e T = +10°C = 283,15 K.
5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
6
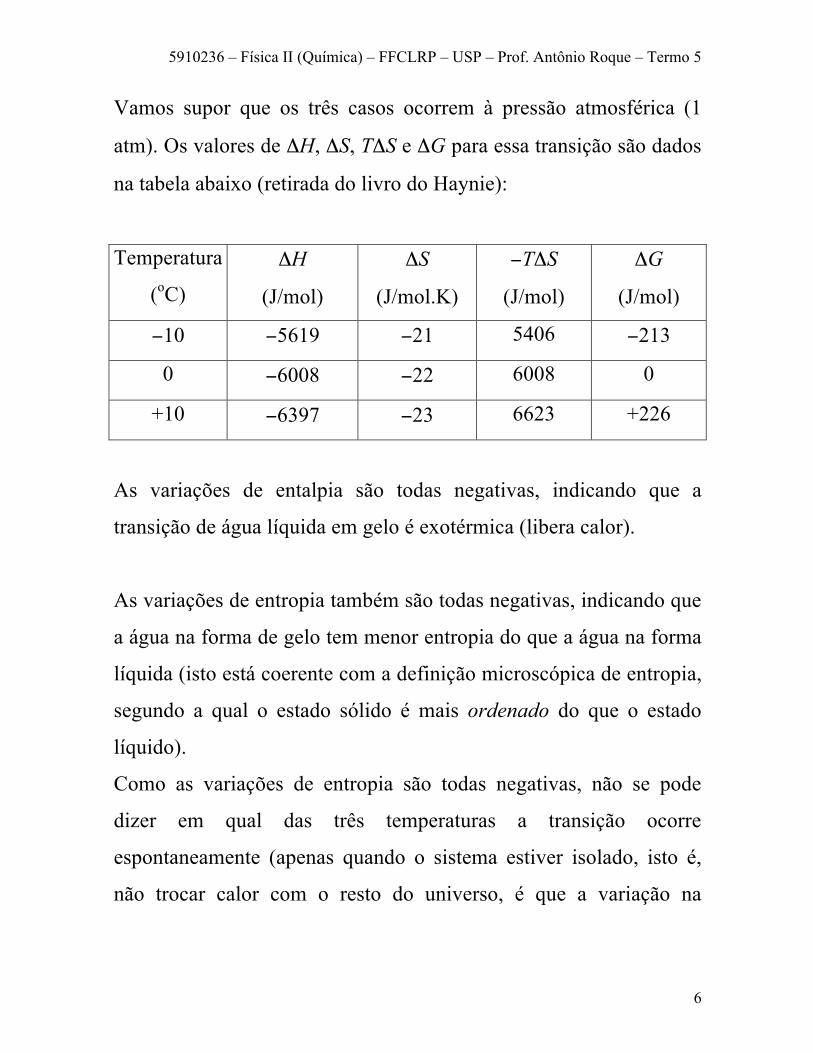
Vamos supor que os três casos ocorrem à pressão atmosférica (1
atm). Os valores de ΔH, ΔS, TΔS e ΔG para essa transição são dados
na tabela abaixo (retirada do livro do Haynie):
Temperatura
(oC)
ΔH
(J/mol)
ΔS
(J/mol.K)
−TΔS
(J/mol)
ΔG
(J/mol)
−10 −5619 −21 5406 −213
0 −6008 −22 6008 0
+10 −6397 −23 6623 +226
As variações de entalpia são todas negativas, indicando que a
transição de água líquida em gelo é exotérmica (libera calor).
As variações de entropia também são todas negativas, indicando que
a água na forma de gelo tem menor entropia do que a água na forma
líquida (isto está coerente com a definição microscópica de entropia,
segundo a qual o estado sólido é mais ordenado do que o estado
líquido).
Como as variações de entropia são todas negativas, não se pode
dizer em qual das três temperaturas a transição ocorre
espontaneamente (apenas quando o sistema estiver isolado, isto é,
não trocar calor com o resto do universo, é que a variação na

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
7
entropia pode ser usada para prever que o sistema vai para o estado
de água líquida).
Porém, a variação de G nos permite dizer que, abaixo de 0°C, a água
congela espontaneamente (ΔG < 0); acima de 0°C o estado de gelo é
instável e a transformação ocorre espontaneamente na direção
oposta, isto é, de gelo para água líquida; e para T = 0°C o processo é
reversível, indicando que água e gelo coexistem em equilíbrio.
Podemos sumarizar isto da seguinte forma:
• ΔGsistema < 0: processo ocorre espontaneamente na direção
da seta;
• ΔGsistema = 0: processo reversível, não há uma direção
preferencial e as fases estão em equilíbrio;
• ΔGsistema > 0: processo não ocorre espontaneamente na
direção da seta, mas sim na direção contrária.
Para entender a origem do nome “energia livre”, consideremos um
sistema que passa por uma transformação reversível a pressão e
temperatura constantes. Nessa transformação, o sistema troca uma
quantidade de calor qrev = TΔS e realiza um trabalho wrev = PΔV +
w’rev.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
8
Os termos PΔV e w’rev indicam, respectivamente, o trabalho de
expansão volumétrica e outros tipos de trabalho realizados durante o
processo reversível (por exemplo, o trabalho elétrico para fazer
circular uma corrente elétrica por um circuito ou o trabalho químico
responsável pela síntese de novas moléculas durante uma reação
química).
Aplicando a definição de energia livre de Gibbs a esta situação:
.revrev STVPwqSTVPUG Δ−Δ+−=Δ−Δ+Δ=Δ
Substituindo os valores qrev = TΔS e wrev = PΔV + w’rev e
simplificando:
.'revwG −=Δ
Vimos na aula 14 que o máximo trabalho que um sistema pode
realizar é aquele feito quando o processo é reversível. Portanto,
podemos escrever w’rev = w’
max.
A equação acima nos diz então que:
.'maxwG −=Δ
O sinal negativo indica que o valor de G diminui durante o processo
(algo que já sabemos). Mas o módulo da variação de G é igual ao
máximo trabalho diferente do de expansão (PΔV) que o sistema
pode realizar a temperatura e pressão constantes:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
9
.'maxwG =Δ
Daí vem o nome energia livre, pois ela representa a máxima energia
de que um sistema pode dispor para realizar um tipo de trabalho que
não seja do tipo expansão volumétrica, como um trabalho químico
ou elétrico, por exemplo.
Existe uma outra função de estado também denominada de “energia
livre” em termodinâmica, que é a energia livre de Helmholtz. Ela é
definida como:
.TSUA −=
Pode-se mostrar que a sua variação durante um processo é igual ao
máximo trabalho (incluindo o de expansão PΔV) que um sistema
pode realizar.
Pode-se também mostrar que, a temperatura e volume constante, um
processo ocorre espontaneamente se,
.0≤ΔA
Como, em geral, os processos químicos e biológicos ocorrem a
pressão constante, a energia livre de Gibbs é muito mais usada
nesses campos do que a energia livre de Helmholtz.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
10
Falamos acima de trabalho químico. Devido à importância desse
tipo de trabalho em reações bioquímicas, vamos agora nos
aprofundar um pouco no seu estudo.
O trabalho de expansão feito por um sistema é dado por:
.VPw Δ=
Há outros tipos de trabalho (mecânico, eletromagnético ou químico)
que podem ser feitos por ou sobre um sistema. Nesses casos,
quantidades equivalentes a P (variável intensiva) e V (variável
extensiva) são usadas para se escrever uma expressão para w.
Um caso importante de trabalho feito por ou sobre um sistema
ocorre quando se adicionam partículas a ele ou se retiram partículas
dele. Quando o número de moles ni de uma dada espécie química i é
aumentado em Δni, esperamos intuitivamente que a energia interna
do sistema aumente. Da mesma forma, quando o número de moles
de uma dada espécie química diminui, a energia interna do sistema
deve diminuir.
O número de moles é uma variável extensiva, como o volume.
Portanto, por similaridade com a expressão para o trabalho de
expansão, define-se a seguinte expressão para o trabalho associado a
uma variação no número de moles da espécie i:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
11
,iin nwi
Δ= µ (3)
onde a variável µi é denominada potencial químico da componente i.
Ainda em analogia com a expressão w = PΔV, podemos dizer que µi
deve ser uma variável intensiva.
No caso geral em que o sistema é composto por várias espécies
químicas (por exemplo, r), o trabalho associado às variações nos
números de moles de cada uma delas é escrito como:
∑=
Δ=r
iiiQ nw
1
.µ (4)
Este trabalho é chamado de trabalho químico.
Considerando apenas o trabalho de expansão volumétrica e o
trabalho químico, podemos escrever a variação da energia interna de
um sistema como:
∑=
Δ+Δ−=Δr
iii nVPqU
1.µ
A convenção de sinais para esta expressão no que diz respeito a q e
PΔV é a mesma que já vem sendo usada. No caso do trabalho
químico, o sinal positivo foi usado para indicar que uma adição de
partículas de uma dada dada espécie química i ao sistema implica
em um aumento da sua energia interna.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
12
Para um processo reversível,
.revrev STqTqS Δ=⇒=Δ
Substituindo qrev na expressão acima para ΔU:
∑=
Δ+Δ−Δ=Δr
iii nVPSTU
1.µ
Pela definição de energia livre de Gibbs:
⇒−+= TSPVUG
.TSSTPVVPUG Δ−Δ−Δ+Δ+Δ=Δ⇒
Substituindo a expressão para ΔU obtida acima nesta expressão:
.1
TSSTPVVPnVPSTGr
iii Δ−Δ−Δ+Δ+Δ+Δ−Δ=Δ ∑
=
µ
Simplificando:
.1∑=
Δ+Δ−Δ=Δr
iii nTSPVG µ
Para processos a temperatura e pressão consantes (ΔP=ΔT=0),
.1∑=
Δ=Δr
iii nG µ
Se impusermos que apenas moléculas da espécie química i podem
ser variadas:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
13
.ii nG Δ=Δ µ
Este resultado já era esperado, pois o máximo trabalho diferente do
de expansão feito sobre o sistema quando se altera a quantidade ni de
moléculas da espécie química i a pressão e temperatura constantes é
o trabalho feito de forma reversível (lembre-se que usamos a
igualdade ΔS = qrev/T no início de nossos cálculos), w’rev = µiΔni.
A expressão acima é usada para definir o potencial químico.
O potencial químico da i-ésima componente química de uma
solução contendo r componentes é definido como (mantendo-se a
pressão, a temperatura e as quantidades das outras espécies químicas
presentes constantes):
.i
i nG
ΔΔ
=µ
Em termos infinitesimais:
.,, ijnTPi
i dndG
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛=µ (5)
No estudo de um sistema qualquer (líquidos, gases, etc), é preciso
obter uma expressão para G em termos de parâmetros
termodinâmicos (P, T, V, ni) para, usando a definição acima,
encontrar uma expressão para o potencial químico. Antes de darmos

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
14
um exemplo disso, vejamos outra propriedade importante do
potencial químico.
Consideremos a seguinte situação (veja a figura a seguir), que serve
como modelo para muitos processos em que ocorre transporte de
matéria através de membranas semi-permeáveis.
Seja um sistema fechado composto por dois sistemas simples
conectados por uma parede rígida e diatérmica, permeável apenas a
uma das espécies químicas (por exemplo, ni) que constituem o
sistema.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
15
Para este caso, os números de moles das outras espécies químicas
são fixos e constantes, assim como os volumes dos dois subsistemas,
mas as energias internas e os números de moles da espécie i dentro
dos volumes 1 e 2 podem variar, sujeitos às restrições:
. constante )1()2()2()1( UUUU Δ−=Δ⇒=+
. constante )1()2()2()1(iiii nnnn Δ−=Δ⇒=+
Para determinar a direção do fluxo de matéria, vamos fazer o
seguinte raciocínio. Vamos supor que os dois sistemas simples estão
à mesma temperatura T, mas que exista uma pequena diferença entre
os seus potenciais químicos da espécie i (por exemplo, sem perda de
generalidade, µ(1)i > µ(2)
i).
Quando o sistema é deixado livre para evoluir nas condições
estabelecidas acima, começa a haver um fluxo da componente i
através da membrana semipermeável até que o equilíbrio seja
atingido. A variação total (isto é, dos sistemas 1 e 2) da entropia
entre o instante inicial e o estado fnal de equilíbrio é positiva,
( ) ( ) .021total >Δ+Δ=Δ SSS
Como fazer para determinar as variações nas entropias dos dois
sistemas? Volte para a página 10 e tome a seguinte equação:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
16
∑=
Δ+Δ−Δ=Δr
iii nVPSTU
1.µ
Isolando ΔS:
∑=
Δ−Δ
+Δ
=Δr
iii nTT
VPTUS
1.1
µ
No caso em questão, os volumes V(1) e V(2) e os números de moles de
todas as espécies químicas com exceção da i-ésima não variam.
Portanto, as variações nas entropias dos sistemas 1 e 2 são,
respectivamente:
( ) )1()1()1(
1 1ii n
TTUS Δ−Δ
=Δ µ
e
( ) )1()2()1(
)2()2()2(
2 11iiii n
TTUn
TTUS Δ+
Δ−=Δ−
Δ=Δ µµ ,
onde se usou as duas condições de conservação da página 13 na
segunda equação. Substituindo estas duas expressões na
desigualdade para ΔStotal:
⇒>Δ+Δ− 011 )1()2()1()1(iiii n
Tn
Tµµ
( ) 0)1()1()2( >Δ−⇒ iii nµµ .

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
17
Se µ(1)i > µ(2)
i, a desigualdade acima só é satisfeita se Δn(1)i < 0. Ou
seja, a quantidade de partículas da espécie i é reduzida no
compartimento onde o potencial químico desta espécie é menor.
Portanto, a matéria tende a fluir de regiões de maior potencial
químico para regiões de menor potencial químico.
O potencial químico pode ser visto como uma espécie de “potencial”
para o fluxo de matéria. Uma diferença no potencial químico entre
os dois sistemas fornece uma “força generalizada” para o fluxo de
matéria.
O potencial químico fornece uma espécie de “força generalizada”
não apenas para o fluxo de matéria de um ponto a outro, mas
também para mudanças de fase e reações químicas. Portanto, o
potencial químico desempenha um papel muito importante na
modelagem e descrição de processos químicos e biológicos.
Vamos agora dar um exemplo do uso da energia livre de Gibbs e do
potencial químico para o estudo de um processo de importância em
biofísica. Algumas técnicas matemáticas a serem usadas nesta parte
da aula talvez sejam desconhecidas por vocês; caso isto aconteça,
deixem de lado a tentativa de entender as passagens matemáticas e

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
18
prestem atenção ao texto para tentar entender o seu conteúdo.
Depois que vocês entenderem bem o conteúdo físico do exemplo,
podem voltar a ler para tentar refazer as passagens matemáticas. No
fundo, é assim que a maioria dos físicos e biofísicos fazem ao ler um
texto com muitas deduções matemáticas.
Seja um sistema fluido formado por n1 moles de um único
componente 1 (água, por exemplo) para o qual o potencial químico é
µ10 (o superíndice 0 indica que o componente 1 está sozinho). Então,
vamos supor que seja adicionado a 1 um segundo componente 2 (o
soluto) em uma quantidade muito pequena formada por n2 moles
onde
12 nn << .
A condição acima caracteriza a solução como diluída. Pode-se
mostrar (isto não será feito aqui) que a energia livre de Gibbs para
uma solução diluída como esta pode ser escrita como:
,lnln),(),(),,,(21
22
21
112
01121 nn
nRTnnn
nRTnTPnTPnnnPTG+
++
++≅ ψµ
onde ψ é uma função qualquer de P e T.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
19
Como estamos na aproximação de solução diluída, podemos
expandir o logaritmo do terceiro termo na expressão acima até
primeira ordem em n2/n1:
1
2
1
221
1
1
1lnlnnn
nnnn
n−≈
+=
+ .
Podemos também desprezar n2 em relação a n1 no logaritmo do
quarto termo da expressão:
.lnln1
2
21
2
nn
nnn
≈+
Desta forma, a expressão anterior pode ser escrita como:
.ln),(),(),,,(1
2222
01121 n
nRTnRTnTPnTPnnnPTG +−+≅ ψµ
Usando agora a equação (5),
ijnTPii dn
dG
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛=
,,
µ ,
para calcular os potenciais químicos do solvente e do soluto
obtemos:
,),(),,( 20121 RTxTPxTP −= µµ (6)
e
,ln),(),,( 222 xRTTPxTP +=ψµ (7)
onde se usou a definição da fração molar do soluto:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
20
.1
2
21
22 n
nnn
nx ≈+
=
Vamos fazer uma análise do que estes resultados significam.
A equação para µ1 nos dá o potencial químico do solvente (água).
Vemos que ele é igual ao potencial químico que o solvente teria se
fosse puro (µ10) menos o termo x2RT. Isto quer dizer que o potencial
químico do solvente quando há soluto misturado nele é menor do
que quando o solvente está sozinho. E quanto maior a quantidade de
soluto diluído no solvente, menor o potencial químico do solvente.
Como já vimos que o movimento de matéria se dá no sentido de
uma região de maior potencial químico para uma de menor potencial
químico, isto explica termodinamicamente o fenômeno da osmose,
que corresponde à passagem de solvente (água) de um
compartimento em que há menos soluto dissolvido nele para um
recipiente em que há mais soluto diluído nele (veja a figura abaixo).

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
21
A equação para µ1 implica também que a redução no potencial
químico do solvente depende apenas da fração molar do soluto
(multiplicada por duas constantes, R e T). Isto é usado em físico-
química e biofísica experimental para a determinação do número de
partículas (e também da natureza) do soluto em solução (a partir de
medidas do desvio do potencial químico do solvente em relação ao
caso puro).
Vamos considerar uma situação experimental como a do osmômetro
de Pfeffer1 em que um balão com uma membrana permeável a água
e com um tubo vertical é cheio com uma solução contendo água e
1 Wilhelm Pfeffer (1845-1920) foi um botânico alemão que desenvolveu o seu osmômetro em 1877.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
22
um soluto (açúcar, por exemplo) e é imerso em um recipiente
contendo água pura (veja a figura abaixo).
A membrana permite apenas a passagem de água, impedindo a
passagem das moléculas de soluto. Após a imersão, água do
recipiente entra no balão aumentando a pressão hidráulica no seu
interior e fazendo com que solução suba pela coluna vertical.
Após algum tempo, chega-se a um equilíbrio em que a solução no
interior do tubo vertical para de subir quando atinge uma certa altura
heq (medida a partir da superfície da água no recipiente, como na
figura acima).
Vamos assumir que a pressão hidráulica no recipiente contendo água
pura é mantida constante e igual a P0, enquanto que a pressão
hidráulica no balão pode ser alterada devido à altura da solução no
tubo. Como há uma diferença de potencial químico da água entre o

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
23
recipiente em que ela está em estado puro e o interior do balão, água
irá fluir através da membrana semipermeável para dentro do balão
até que se atinja o equilíbrio.
No equilíbrio, os potenciais químicos da água dos dois lados devem
ser iguais:
),,,()0,,( 2101 xTPTP µµ =
onde P é a pressão hidráulica exercida pela coluna de solução no
tubo.
Usando a equação (6) para µ1:
RTxTPTP 2010
01 ),(),( −= µµ .
Expandindo µ10(P,T) em torno de P0,
( )..),(),(),( 00
01
001
01 PP
PTPTPTP −
∂
∂+=
µµµ
Vamos agora pegar a equação para ΔG da página 11, só que escrita
em termos de variações infinitesimais:
.1∑=
+−=r
iiidnSdTVdPdG µ

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
24
Como G é uma variável de estado, dG tem que ser uma diferencial
exata. Diferenciais exatas devem satisfazer às chamadas condições
de Cauchy2. Uma delas é a seguinte:
in
i
Pi PnV
⎟⎠
⎞⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛
∂∂ µ
.
Fazendo i = 1 nessa equação e escrevendo o volume da solução
como V = n1v, onde v é o volume ocupado por um mol de solvente,
temos que:
.1 vP
in
=⎟⎠
⎞⎜⎝
⎛∂∂µ
Substituído isto na equação para µ10(P,T) expandida em torno de P0:
( ).),(),( 0001
01 PPvTPTP −+= µµ
Comparando esta equação com a equação para µ10(P,T) não
expandida, temos que:
( ) ,20 RTxPPv =−
ou,
( ) .20 RT
VnPP =−
Definindo a concentração do soluto como:
2Augustin Louis Cauchy (1789-1857) foi um matemático francês.

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
25
,2sol Vnc =
Podemos escrever finalmente:
( ) .sol0 RTcPP =− (8)
A pressão osmótica é definida como sendo igual à pressão
hidrostática que a solução no interior da coluna vertical do
osmômetro de Pfeffer exerce sobre a solução. Isto é, a pressão
osmótica de uma solução é a pressão hidráulica que precisa ser
aplicada à solução para evitar que haja fluxo osmótico de solvente
através de uma membrana semi-permável.
Baseado em medidas experimentais feitas com o seu osmômetro,
Pfeffer mostrou que a pressão osmótica é proporcional à
concentração de soluto e cresce continuamente à medida que a
temperatura da solução aumenta.
Em 1886, o físico-químico holandês Jacobus Henricus van’t Hoff
(1852-1911) definiu matematicamente a pressão osmótica como
sendo proporcional ao produto da concentração de soluto pela
temperatura absoluta. A constante de proporcionalidade é a
constante universal dos gases R:

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
26
RTCΣ=π ,
onde π indica a pressão osmótica (para diferenciar da pressão
hidráulica) e CΣ é a soma das concentrações de todas as espécies de
soluto:
.1∑=
Σ =r
iicC
No caso em que só há uma espécie de soluto:
.solcC =Σ
Como na condição de equilíbrio em um osmômetro de Pfeffer a
diferença de pressão hidrostática entre a coluna que contém o soluto
e o recipiente que contém o solvente tem que ser igual à pressão
osmótica, van’t Hoff usou sua definição para deduzir a chamada lei
de van’t Hoff para soluções diluídas,
( ) .0 RTCPP Σ=−
Note que esta é exatamente a equação (8) deduzida acima. Portanto,
a lei de van’t Hoff para soluções diluídas pode ser deduzida a partir
das leis da termodinâmica.
Porém, como acontece com todas as deduções baseadas na
termodinâmica, apenas as relações macroscópicas corretas foram
obtidas. Os mecanismos microscópicos responsáveis pela pressão

5910236 – Física II (Química) – FFCLRP – USP – Prof. Antônio Roque – Termo 5
27
osmótica não são revelados pela análise termodinâmica e é
necessária a aplicação de uma teoria cinética para modelar o
fenômeno de osmose a partir de interações moleculares.