universidade federal do rio de janeiro – ufrj instituto de...
TRANSCRIPT

Universidade Federal do Rio de Janeiro – UFRJ
Instituto de Ciências Biomédicas
Departamento de Histologia e Embriologia
Pós-Graduação em Ciências Morfológicas
Dissertação de Mestrado
“As características moleculares das leucemias agudas de
lactentes em coorte brasileira”
Autora: Mariana Emerenciano
2006

ii
Universidade Federal do Rio de Janeiro – UFRJ
Instituto de Ciências Biomédicas
Departamento de Histologia e Embriologia
Pós-Graduação em Ciências Morfológicas
Dissertação de Mestrado
“As características moleculares das leucemias agudas de
lactentes em coorte brasileira”
Autora: Mariana Emerenciano
Sob a orientação da Dra. Maria do Socorro Pombo-de-Oliveira
Tese submetida como requisito parcial
para obtenção do grau de Mestre em
Ciências Morfológicas.
2006

iii
AS CARACTERÍSTICAS MOLECULARES DAS LEUCEMIAS
AGUDAS DE LACTENTES EM COORTE BRASILEIRA
MARIANA EMERENCIANO
ORIENTADORA: DRA. MARIA DO SOCORRO POMBO-DE-OLIVEIRA
APROVADA EM: 14/03/2006
BANCA EXAMINADORA:
ISIS QUEZADO MAGALHÃES
MARIA ISABEL DORIA ROSSI
RAQUEL MAIA
IVONE OTAZU
JOSÉ CLÁUDIO CASALLI

iv
A estas crianças e a seus pais,
ofereço o meu empenho e demonstro
gratidão e respeito.

v
AGRADECIMENTOS
À Dra. MARIA DO SOCORRO POMBO DE OLIVEIRA, pela orientação,
incentivo e amizade durante a realização deste e de outros trabalhos, e por ter
acreditado em mim desde a iniciação científica, disponibilizando recursos e
transmitindo conhecimentos.
A todos os MÉDICOS, ENTREVISTADORES e demais profissionais
envolvidos no Grupo Colaborativo Brasileiro do Estudo de Leucemia Aguda dos
Lactentes, pelo empenho e dedicação. Contando com a colaboração de vocês,
este trabalho pode ser concretizado.
A todos os AMIGOS DO LABORATÓRIO, agradeço pela ajuda
desinteressada, pelos conhecimentos compartilhados e pelo companheirismo de
todo e cada dia. A ALESSANDRA DE JESUS FARO e CRISIANE WAIS
ZANROSSO meu carinho especial.
À MINHA FAMÍLIA dedico mais esta conquista. Vocês foram essenciais
para que esta tese fosse concluída, mesmo que talvez nem saibam como.
A LAURO CAVALCANTI DE SÁ, por todo carinho, incentivo, ternura e
compreensão em todos os momentos da realização deste trabalho e de nossas
vidas. Obrigada, meu amor.
A VOCÊ, que ao ler minhas palavras me incentiva a prosseguir.

vi
RESUMO As anormalidades cromossômicas freqüentemente ocorrem no período pré-
natal nas leucemias da infância, caracterizando um evento precoce na
leucemogênese. A maioria das anormalidades que ocorrem nos lactentes envolve
o gene MLL na região cromossômica 11q23. Nós descrevemos os achados
genético-moleculares de 207 casos de leucemia aguda do lactente (LAL),
pertencentes ao Grupo Brasileiro de Estudo Cooperativo da Leucemia Aguda do
Lactente. O diagnóstico de leucemia linfoblástica aguda (LLA) ou leucemia
mielóide aguda (LMA) foi feito seguindo a classificação morfológica e
imunofenotípica, seguida por citogenética convencional. As amostras foram então
testadas usando o RT-PCR para a detecção de fusões gênicas específicas. A
análise por FISH para os rearranjos do MLL foi realizada somente nos casos com
resultados negativos ou inconclusivos. As características das crianças com LAL
eram: 115 meninos e 92 meninas, idade entre 0 e 23 meses, média de 12 meses
de idade, 145 LLA e 62 LMA. Uma associação estatisticamente significativa foi
observada entre os casos LLA pró-B e aqueles MLL+vos (p = 0.0001), e entre o
grupo etário de 0-3 meses com os casos MLL+vos (p = 0.008). Dois casos raros de
LLA pró-T com MLL+vo foram encontrados. Além dos rearranjos do MLL, outras
aberrações moleculares foram detectadas, como TEL/AML1 (n=9), E2A/PBX1
(n=4), PML/RARA (n=4) e AML1/ETO (n=2). As análises de citogenética
revelaram hiperdiploidia (n=6), del(7) em dois casos e del(11)(q23) em 7 casos.
Nossos resultados corroboram que não somente os rearranjos do MLL, como
também outras anormalidades moleculares ocorrem antes do nascimento e
podem contribuir para a leucemogênese.

vii
ABSTRACT
Background. Chromosome abnormalities often occur prenatally in childhood
leukemia, characterizing an early event in leukemogenesis. The majority of the
abnormalities occurring in infants involve the MLL gene on chromosome band
11q23. We describe the molecular cytogenetics findings of 207 infant acute
leukemia (IAL) cases included in the Brazilian Collaborative Study Group of Infant
acute leukemia. Procedure. The diagnosis of acute lymphoblastic leukemia (ALL)
or acute myeloblastic leukemia (AML) was made according to morphology and
immunophenotyping classification, followed by conventional karyotyping. Samples
were then screened using RT-PCR for the presence of specific fusion genes. FISH
assay for MLL rearrangements was performed only in cases with negative or
inconclusive cytogenetic or PCR results. Results. The characteristics of children
with IAL were as follow: 115 boys and 92 girls, age range 0 to 23 months, mean
age 12 months, 145 ALL and 62 AML. A statistically significant association was
observed between pro-B ALL cases and MLL+ve (p = 0.0001) cases and the age
group 0-3 months with MLL+ve (p = 0.008) cases. Two rare cases of pro-T ALL with
MLL+ve were found. Other than MLL rearrangements, other molecular aberrations
were detected including TEL/AML1+ve (n=9), E2A/PBX1+ve (n=4), PML/RARA+ve
(n=4) and AML1/ETO+ve (n=2). Cytogenetic analysis revealed hyperdiploidy (n=6),
del(7) in two cases and del(11)(q23) in 7 cases. Conclusions. Our results endorse
that not only MLL rearrangements, but also other molecular abnormalities occur
before birth and may contribute to leukemogenesis.

viii
RESUMO .........................................................................................................................VI
ABSTRACT ........................................... .........................................................................VII
ÍNDICE DE FIGURAS ......................................................................................................IX
ÍNDICE DE TABELAS .................................. ....................................................................X
LISTA DE ABREVIATURAS.............................. ..............................................................XI
1. INTRODUÇÃO.............................................................................................................. 1
1.1 HEMATOPOESE NORMAL..................................................................................... 1 1.2 LEUCEMIAS AGUDAS............................................................................................ 4
1.2.1 Leucemia linfoblástica aguda............................................................................ 7 1.2.2 Leucemia mielóide aguda ............................................................................... 11
1.3 IMUNOFENÓTIPO X GENÓTIPO NAS LEUCEMIAS AGUDAS............................ 12 1.3.1 Os rearranjos do gene MLL ............................................................................ 15 1.3.2 A fusão gênica TEL/AML1 .............................................................................. 21 1.3.3 Hiperdiploidia cromossomal............................................................................ 24 1.3.4 LLA com a fusão gênica E2A/PBX1................................................................ 25 1.3.5 A fusão gênica AML1/ETO ............................................................................. 26
1.4 UMA DOENÇA DIFERENTE A CADA IDADE ....................................................... 27 1.5 FATORES DE RISCO............................................................................................ 30
1.5.1 Os fatores ambientais ..................................................................................... 31 1.5.2 Infecção viral .................................................................................................. 34
2. JUSTIFICATIVA DO ESTUDO......................... ........................................................... 34
3. OBJETIVOS....................................... ......................................................................... 36
3.1 GERAL .................................................................................................................. 36 3.2 ESPECÍFICOS ...................................................................................................... 36
4. MATERIAL E MÉTODOS .............................. ............................................................. 37
4.1 AMOSTRAS .......................................................................................................... 37 4.2 ETAPAS DIAGNÓSTICAS .................................................................................... 38
4.2.1 Análise morfológica ........................................................................................ 38 4.2.2 Separação de células mononucleares ............................................................ 38 4.2.3 Imunofenotipagem.......................................................................................... 39 4.2.4 Citogenética convencional e FISH .................................................................. 42 4.2.5 Genética-molecular......................................................................................... 42
4.3 ANÁLISE ESTATÍSTICA........................................................................................ 50
5. RESULTADOS...................................... ...................................................................... 51
6. DISCUSSÃO............................................................................................................... 60
7. CONCLUSÕES........................................................................................................... 67
8. REFERÊNCIAS........................................................................................................... 68
ANEXO 1 – Grupo Brasileiro de Estudo Colaborativo da Leucemia Aguda de Lactente….........82 ANEXO 2 – Banco de Dados ..……………………..…………………………………….…....83 ANEXO 3 – Produção bibliográfica durante o mestrado………….......……………….…....84

ix
ÍNDICE DE FIGURAS
Figura 1 – Hematopoese normal.............................................................................2
Figura 2 – Evolução clonal......................................................................................5
Figura 3 – Diferenciação da linhagem linfóide B.....................................................8
Figura 4 – Diferenciação da linhagem linfóide T...................................................10
Figura 5 – Diferenciação da linhagem mielóide....................................................12
Figura 6 – Parceiros do gene MLL........................................................................18
Figura 7 – Estrutura éxon/íntron do gene MLL......................................................19
Figura 8 – Modelo para a história natural das leucemias agudas da infância......24
Figura 9 – Subtipos moleculares da LLA em lactentes, crianças e adultos..........28
Figura 10 – Fatores de risco envolvidos no desenvolvimento da leucemia..........30
Figura 11 – Modelo esquemático das funções executadas pela topo-II...............33
Figura 12 – Corrida de eletroforese em gel de agarose dos produtos da
amplificação do gene GAPDH...............................................................................46
Figura 13 – Pontos de quebra e posições dos oligonucleotídeos iniciadores dos
genes incluídos na reação de RT-PCR multiplex..................................................49
Figura 14 – Exemplo de corrida de eletroforese em gel de agarose (2%) corado
com brometo de etídio dos produtos da amplificação do RT-PCR multiplex.........50
Figura 15 – Distribuição do número de casos por faixa etária de acordo com os
subtipos imunofenotípicos de leucemia…..............................................................52
Figura 16 – Resultados dos testes aplicados para verificar o status do gene MLL
no paciente com LLA pró-T....................................................................................58
Figura 17 – Distribuição do número de casos X faixa etária de acordo com os
rearranjos do MLL..................................................................................................63

x
ÍNDICE DE TABELAS
Tabela 1 – Subtipos celulares e alterações moleculares nas leucemias infantis..14
Tabela 2 – Associações dos AcMo utilizados no painel das leucemias agudas...40
Tabela 3 – Seqüência de oligonucleotídeos para a amplificação gênica
GAPDH..................................................................................................................45
Tabela 4 – Seqüência de oligonucleotídeos para a amplificação gênica das
principais translocações............................................. ..........................................47
Tabela 5 – Seqüência de oligonucleotídeos para a amplificação gênica rearranjos
do MLL...................................................................................................................48
Tabela 6 – Distribuição dos casos de LLA e LMA.................................................51
Tabela 7 – Características clínico-laboratoriais dos casos de LALs.....................53
Tabela 8 – Associação entre o gene MLL e características relacionadas.............54
Tabela 9 – Comparação de métodos usados na detecção do rearranjo do MLL. 55
Tabela 10 – Sumário das translocações cromossômicas X faixa etária...............56
Tabela 11 – Características dos casos raros de LAL............................................57

xi
LISTA DE ABREVIATURAS
AcMo – anticorpo monoclonal
ADN – ácido desoxirribonucléico
ADNc – ácido desoxirribonucléico complementar
ARN – ácido ribonucléico
CD – cluster of differentiation (grupamentos de diferenciação)
CSF – fator estimulador de colônia
DEPC – dietilpirocarbonato
dNTP – desoxinucleotídeos
DRM – doença residual mínima
DTT – ditiotreitol
EDTA – ácido etilenodiaminotetraacético
EGIL – European Group for Immunophenotyping Leukaemia
FAB – French-American-British group
FISH – fluorescent in situ hybridization hibridização in situ por fluorescência
FITC – Isotiocianato de fluoresceína
GAPDH – gliceraldeído - 3 - fosfato desidrogenase
HLA-DR – human leucocitary antigen-DR (antígeno leucocitário humano-DR)
LA – leucemia aguda
LAL – leucemia aguda de lactente
LLA – leucemia linfoblástica aguda
LLA-c – leucemia linfoblástica aguda comum

xii
LLA-pB – leucemia linfoblástica aguda de células precursoras B
LLA-T – leucemia linfoblástica aguda de linhagem T
LMA – leucemia mielóide aguda
MO – medula óssea
MPO – mieloperoxidase
NS – estastisticamente não significativo
pb – pares de bases
PBS – phosphate buffered saline (solução salina tamponada com fosfato)
PCR – polymerase chain reaction (reação em cadeia da polimerase)
PE – ficoeritrina
PerC5 – ficoeritrina – cianina 5.1
rpm – rotações por minuto
RT-PCR - reverse transcriptase polymerase chain reaction (reação em cadeia da
polimerase por transcriptase reversa)
SP – sangue periférico
SV – saco vitelínico
Taq – enzima Thermus aquaticus
TCR – T-cell receptor (receptor de célula-T)
Topo-II – topoisomerase II

xiii
EMERENCIANO, Mariana
As características moleculares das leucemias agudas de lactentes
em coorte brasileira / Mariana Emerenciano. -- Rio de Janeiro,
Universidade Federal do Rio de Janeiro, UFRJ, 2006.
Orientadora: Maria do Socorro Pombo-de-Oliveira
Dissertação de Mestrado em Ciências Morfológicas
Palavras-chave: 1.Leucemia aguda de lactente. 2.MLL 3. TEL/AML1.
4. AML1/ETO. 5. E2A/PBX1. 6. PML/RARA.

1
1. INTRODUÇÃO
As leucemias agudas (LAs) são doenças malignas decorrentes da
expansão clonal de um precursor hematopoético. Durante a evolução de uma
célula progenitora, é possível que ocorra uma anormalidade genômica e, outros
fatores celulares ou moleculares que favoreçam a expansão deste clone de
células defeituosas, caracterizadas por um fenótipo definido por moléculas de
superfície celular e pela etapa de diferenciação do precursor hematopoético (1).
Sendo assim, o entendimento da hematopoese normal é necessário para que sua
contrapartida patogênica possa ser discutida.
1.1 HEMATOPOESE NORMAL
A hematopoese se inicia no embrião e se prolonga por toda a vida do
indivíduo. Por volta do sétimo dia embrionário nas ilhotas sangüíneas do saco
vitelínico (SV), as células são especificadas a partir de precursores mesodérmicos
e são produzidas por meio de uma “onda” de hematopoese primitiva, consistindo
principalmente de eritrócitos nucleados (1). Esta onda transitória de hematopoese
primitiva é progressivamente substituída por uma segunda onda de hematopoese
definitiva, que garante a produção células especializadas linfóides, mielóides e
eritróides. À medida que a hematopoese primitiva vai se reduzindo, as células
produzidas no SV e num local hematogênico chamado de região aorta-gônada-
mesonéfrica colonizam o fígado fetal por volta do décimo primeiro dia
embrionário, este se torna o principal local da produção sangüínea durante a
gestação do embrião (1). Durante toda a vida uterina, o feto sofre as
transformações necessárias para adaptar o aparelho circulatório à sua futura

2
existência fora do útero. Próximo ao final da gestação, outra mudança ocorre e a
medula óssea (MO) passa a ser o local predominante da hematopoese definitiva e
assim permanece durante a vida pós-natal (2).
As células primitivas hematopoéticas possuem a propriedade de auto-
renovação e também a de gerar, através de divisão e diferenciação celular,
populações de células progenitoras encarregadas das principais linhagens de
células da medula óssea: eritróides, granulocíticas e monocíticas,
megacariocíticas e linfóides (Figura 1). Estes precursores multipotentes são
células altamente proliferativas que expressam receptores para fatores de
crescimento e de sobrevivência específicos, os fatores estimuladores de colônias
(CSFs), e passam por várias etapas sucessivas de diferenciação até que sejam
uma progênie matura e que não se divide, capaz de gerar os tipos celulares
específicos do sangue (3-5).
Figura 1 - Hematopoese normal*.
*Modificado de Hitzler JK e colaboradores (6).

3
O controle da proliferação, diferenciação e maturação desses diversos
tipos celulares é feito através de uma complexa interação molecular das células
com o microambiente da MO e depende de mecanismos que estabilizem e
mantenham programas específicos de expressão gênica (5). A função
desempenhada pelos fatores de transcrição é central neste processo, pois eles
atuam de modo combinatório para ativar ou conter conjuntos de genes-alvo no
ambiente celular adequado (7,8).
Muitas informações importantes sobre o papel dos fatores de transcrição
no desenvolvimento da linhagem hematopoética têm sido obtidas a partir de
estudos envolvendo bloqueio de ação ou superexpressão destes fatores (2,4,8).
Muitos genes foram identificados por estarem rompidos em conseqüência das
translocações cromossômicas encontradas nas leucemias. Pode-se citar como
exemplo os fatores de transcrição quiméricos, originados de translocações
cromossômicas, que ativam diversas cascatas transcricionais que, pelo menos
em parte, convergem para modificar o padrão de expressão normal de membros
da importante família dos genes HOX. Os fatores de transcrição HOX se ligam ao
ADN e regulam genes envolvidos tanto na diferenciação do embrião como da
célula–tronco hematopoética; eles também são importantes na auto-renovação e
na proliferação das células hematopoéticas (9). Os progressos no conhecimento
destes mecanismos da dinâmica hematopoética vêm sendo muito importantes
para o sucesso do tratamento das leucemias.

4
1.2 LEUCEMIAS AGUDAS
Assim como ocorre para outros tipos de câncer, o entendimento da
natureza das LAs ainda é um grande desafio. A leucemia é uma doença clonal, ou
seja, se origina de uma única célula e evolui a partir da aquisição de alterações
moleculares neste clone. A Figura 2 exemplifica didaticamente como ocorre uma
evolução do tipo clonal, ou seja, um “tumor” desenvolve-se a partir de sucessivos
ciclos de mutação e proliferação, podendo eventualmente gerar um clone
completo de células malignas. Em cada etapa, uma única célula sofre uma
mutação que potencializa a proliferação celular, de forma que sua progênie torne-
se o clone dominante. A proliferação deste clone então acelera a ocorrência da
próxima etapa da progressão do câncer, devido ao aumento do tamanho da
população celular que está sob o risco de sofrer uma mutação adicional. O
resultado é uma diversificação genética progressiva seguida por uma “seleção
natural” dos subclones mutantes dominantes.
As LAs foram inicialmente descritas como um grupo de doenças no qual
ocorre um descontrole dos mecanismos fisiológicos de proliferação e maturação
de células do sistema hematopoético. O organismo é muitas vezes incapaz de
reparar uma mutação e a célula alterada, chamada de blasto, começa a se
multiplicar descontroladamente.
Entre os sintomas, são freqüentes as infecções, pois o paciente fica
imunodeprimido, a anemia, pois a produção de glóbulos vermelhos decai à
medida que as células leucêmicas predominam na MO e, o sangramento
excessivo, pois a capacidade de coagulação diminui à medida que diminui o
número de plaquetas, conseqüente da diminuição dos megacariócitos (1).

5
Figura 2 - Evolução clonal*
Sob o ponto de vista morfológico e citoquímico, as LAs compreendem dois
grupos principais: mielóides e linfóides. Em adultos, aproximadamente 85% são
mielóides. Em crianças, ocorre o inverso, com 80% sendo linfóides. A leucemia
linfoblástica aguda (LLA) é a neoplasia mais comum na infância. Desde o início de
1990, com o aprimoramento de técnicas imunomoleculares que refinaram a
identificação das leucemias, o Prof. Mel Greaves e colaboradores defendem uma
hipótese de que mutações espontâneas são responsáveis pelo desenvolvimento
das leucemias na infância. O ambiente fetal é rico em células em diversos estágios
maturativos sob estímulos constantes para proliferação e diferenciação celular. As
mutações sucessivas neste período embrionário podem desempenhar um papel
importante nas codificações gênicas e predisporem a um desequilíbrio celular que
resulte em alterações malignas (10). De acordo com esta hipótese, uma ou mais
mutações seqüenciais ocorrendo no desenvolvimento de uma célula progenitora
*Modificado de Alberts, Molecular Biology of the cell, 2002

6
poderiam desviar o mecanismo normal de proliferação em curso, em um
determinado momento da infância.
O padrão alterado de auto-renovação e diferenciação das células-tronco
hematopoéticas que causam a leucemia pode resultar dos fatores de transcrição
quiméricos que, por sua vez, são gerados a partir das translocações
cromossômicas que unem porções de dois genes diferentes. Estas alterações
genéticas, portanto, contribuem para a transformação leucêmica das células-
tronco hematopoéticas ou de seus progenitores comprometidos através da
alteração de funções celulares. Elas alteram processos regulatórios essenciais
por manterem ou por aumentarem uma capacidade ilimitada de auto-renovação,
subvertendo aos controles de proliferação normal, bloqueando a diferenciação, e
promovendo resistência aos sinais de morte celular (apoptose) (11).
Muitos avanços foram conseguidos no entendimento da fisiopatologia
molecular da LA através da clonagem e da caracterização de fusões gênicas
recorrentes presentes em vários subtipos de LAs, que permitiram a identificação
de genes críticos para o processo leucemogênico (12). Além disso, a presença de
determinadas alterações pode estratificar os pacientes em distintos subgrupos
leucêmicos. O uso das análises de microarranjo para caracterizar as diferenças
na expressão gênica entre as leucemias com diferentes aberrações moleculares
solidificou a noção de que anormalidades cromossômicas específicas determinam
subgrupos leucêmicos únicos (13,14).
A necessidade de uma classificação mais abrangente das leucemias,
portanto se faz necessária pelo fato de que os protocolos quimioterápicos se
adaptam a cada tipo de leucemia e para pesquisas referentes aos fatores que

7
determinam sua etiologia.
No princípio, as classificações das leucemias baseavam-se nos aspectos
clínicos, morfológicos e citoquímicos. Em seguida, o emprego da citometria de
fluxo e anticorpos monoclonais, que permite o reconhecimento dos diferentes
tipos de células e de suas linhagens específicas, e os estudos moleculares
consolidaram as buscas dos fatores que contribuem para o processo
leucemogênico e as características do prognóstico.
1.2.1 Leucemia linfoblástica aguda
As LLAs podem ser causadas por um dano genético adquirido no ADN de
uma única célula na MO. Os efeitos são o crescimento incontrolável e exagerado
e o acúmulo de células chamadas linfoblastos e, o bloqueio da produção normal
de células da medula, levando a uma deficiência de células vermelhas (anemia),
plaquetas (trombocitopenia) e de células brancas no sangue.
Os principais tipos das LLAs são definidos por exames de
imunofenotipagem, subdivididos em dois grandes grupos oriundos de precursores
de células B e de linfócitos T (15). A LLA representa a maioria dos casos de
leucemia infantil e o pico de incidência ocorre aos 2-5 anos. Para explicar o pico
de ocorrência das LLAs da infância, foram elaboradas teorias envolvendo
mecanismos de uma resposta imune às infecções adquiridas na infância, ou
associações a infecções virais (16).

8
A LLA constitui diversos tipos de malignidade das linhagens de linfócitos
precursores B ou T, de acordo com a diferenciação interrompida e semelhante a
seus progenitores. As figuras 3 e 4 resumem as principais etapas da
diferenciação antigênica das células hematopoéticas e seus correspondentes
malignos (17-21).
Os estágios iniciais do desenvolvimento da célula B dependem de
interações entre moléculas da superfície celular e dos produtos secretados das
células estromais com seus receptores nos progenitores linfóides. Estes estágios
seqüenciais do desenvolvimento da célula B foram definidos por marcadores
DIFERENCIAÇÃO NORMAL CORRESPONDENTECORRESPONDENTE
MALÍGNOMALÍGNO
CÉLULATRONCOLINFÓIDE
LLA LLA ProPro-B-B
LLA-LLA-comumcomum
CÉLULAPRÉ-B LLA PRÉ-B
(µµµµ+)
CÉLULA BIMATURA LLA -B sIg M+
CD34HLADr
CD34CD19HLADrcCD79a
CD19HLADrCD10cCD79a
CD19HLADrCD10cCD22Cµ Ig
CD19HLADrCD22sIgM+κκκκ ou λ λ λ λ
µµµµ
TDT+
µ
µµ
µ
µ
TDT+
TDT+
TDT+
VLA-4
ÓR
GÃ
OS
LIN
FÓ
IDE
SS
EC
UN
DÁ
RIO
S
SAN
GU
E P
ER
IFÉ
RI C
O
sIgCD19
B
CD19sIgGsIgAsIgEsIgM
B
B
Plasmócito
Y
Y
Linfócito Bde Memória
Diagrama elaborado por Dr. Geraldo Cavalcanti Júnior
Figura 3 – Diferenciação da linhagem linfóide B

9
moleculares que podem ser usados para classificar as malignidades linfóides
como uma representação do impedimento da maturação e da imortalização em
etapas específicas da ontogenia da célula B. Vários dos fatores que controlam o
rearranjo ordenado e a expressão dos genes da imunoglobulina (Ig) foram
identificados. Alguns sinais gerados por intermediários durante o rearranjo do
gene da Ig, assim como pela molécula da Ig propriamente dita, foram
comprovados como sendo essenciais no processo de direcionamento do
desenvolvimento inicial da célula B. Os processos de ativação periférica da célula
B, de maturação da afinidade antigênica e de diferenciação terminal também são
altamente regulados (17,19,20).
Nos estágios mais precoces desde a célula pluripotente hematopoética e
os precursores totalmente comprometidos que sofrerão o rearranjo gênico do
receptor de célula T (TCR), existem diferenças significantes entre a linfopoese
fetal e a adulta. O grau de comprometimento dos precursores linfóides presentes
na MO ainda é parcialmente desconhecido, embora esteja claro que as células
pluripotentes hematopoéticas passam por alterações em seu desenvolvimento
antes de migrarem para o timo. Tanto os precursores multipotentes como aqueles
restritos à linhagem T foram isolados a partir de sangue fetal, sugerindo que
ambos podem povoar o timo. No timo, várias populações discretas de precursores
T antecedem o estágio do rearranjo gênico do TCR. Estas incluem precursores
que não estão exclusivamente comprometidos com a linhagem T, embora sejam
diferentes das células pluripotentes hematopoéticas. Estes precursores têm um
potencial para gerar células NK (natural killer), células B, células dendríticas e,
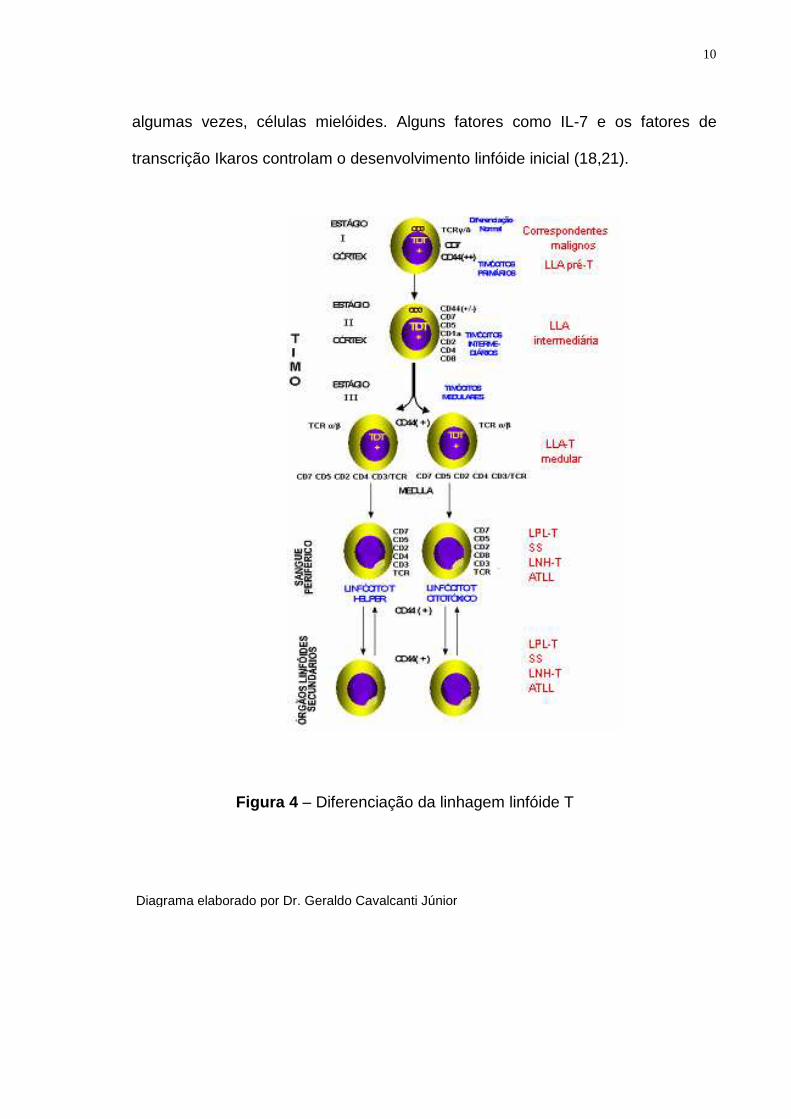
10
algumas vezes, células mielóides. Alguns fatores como IL-7 e os fatores de
transcrição Ikaros controlam o desenvolvimento linfóide inicial (18,21).
Figura 4 – Diferenciação da linhagem linfóide T
Diagrama elaborado por Dr. Geraldo Cavalcanti Júnior

11
Diversos antígenos intracitoplasmáticos e de superfície da membrana são
hoje reconhecidos através de um número progressivo de anticorpos monoclonais
(AcMo). Estas moléculas de leucócitos humanos se tornaram importantes no
estabelecimento de um diagnóstico preciso dos diversos subtipos de LLA (15).
1.2.2 Leucemia mielóide aguda
A leucemia mielóide aguda (LMA) é caracterizada pelo crescimento
incontrolável e exagerado e o acúmulo de células chamadas de mieloblastos. A
interrupção da produção normal de células da medula freqüentemente resulta em
insuficiência hematopoética (granulocitopenia, trombocitopenia, ou anemia), com
ou sem leucocitose (22) (Figura 5).
As LMAs correspondem a 85% das leucemias dos adultos. O risco
aumenta em dez vezes entre a idade de 30 anos (cerca de um caso por 100.000
pessoas) para a idade de 70 anos (cerca de um caso por 10.000 pessoas). Entre
os casos de leucemias infantis, apenas 15% são LMA (22). Entre os fatores de
risco associados ao desenvolvimento da LMA estão os distúrbios genéticos como
anemia de Fanconi e síndrome de Down, e exposição a doses muito altas de
irradiação, exposição a agentes químicos e à quimioterapia prolongada com
agentes alquilantes utilizados no tratamento de outros cânceres, têm sido
descritos como fatores de riscos associados ao desenvolvimento da LMA.
A idade do paciente, a contagem inicial dos glóbulos brancos no
hemograma e a presença de anormalidades cromossômicas influencia no
prognóstico. A evolução do sistema de classificação das LMAs que vai da
morfologia aos achados citogenético-moleculares, reflete o reconhecimento da

12
importância biológica de um subtipo específico (23). Apesar do refinamento no
diagnóstico dos subtipos de LMA e dos avanços nas estratégias terapêuticas,
ocorridos nas últimas décadas, a taxa de sobrevivência entre pacientes com idade
inferior a 65 anos é de apenas 40% (24).
1.3 IMUNOFENÓTIPO X GENÓTIPO NAS LEUCEMIAS AGUDAS
O padrão imunomolecular, as informações epidemiológicas e dados
etiopatológicos consistentes sugerem que as leucemias podem ser causadas por
mecanismos multifatoriais e que requerem um certo tempo para expansão do
clone leucêmico (25-28). Durante a evolução de uma célula progenitora, é
possível que ocorra uma anormalidade genômica e outros fatores celulares ou
moleculares, que favoreçam a expansão deste clone de células alteradas,
caracterizadas por um imunofenótipo definido por moléculas de superfície celular
e pela etapa de diferenciação do precursor hematopoético (29).
Assim, as LAs são doenças heterogêneas de progresso rápido que afetam
a maior parte das células ainda não diferenciadas. Atualmente, a classificação
CD34 CD33 CD13 CD65 CD15 / CD115 CD66
C
HLADR
CFU-GM Mieloblasto Metamielócito Pró-Mielócito Mielócito Bastão Segmentado
Figura 5 - Diferenciação da linhagem mielóide
a-MPO
Diagrama elaborado por Dr. Geraldo Cavalcanti Júnior

13
das LAs baseia-se em estudos de citometria de fluxo multiparamétrica, genética
molecular e citogenética.
A imunofenotipagem nos permite identificar a linhagem hematopoética da
qual as populações leucêmicas se originam. Um painel relativamente simples foi
proposto para a sub-classificação das leucemias pelo European Group for
Immunophenotyping Leukaemia (EGIL) (15). Através de técnicas de conjugação
dos anticorpos aos fluorocromos, e com técnicas de permeabilização, é possível o
acesso simultâneo aos marcadores de superfície e aos intracelulares.
Primeiramente, procede-se a marcação intracitoplasmática com os marcadores
mais específicos de linhagem expressos prematuramente e/ou somente no
citoplasma. Estes são: cCD79, cCD3 e mieloperoxidase (MPO), que são
altamente específicos para as linhagens B, T e mielóides, respectivamente (15).
Embora o cCD79 seja o marcador mais prematuro associado à linhagem B, o
cCD22 também foi proposto para caracterizar a linhagem B (30). Os blastos
leucêmicos na LMA e na LLA expressam antígenos normais de diferenciação
mielóide e linfóide, respectivamente. No entanto, mesmo nas leucemias com um
padrão de expressão de antígenos bem definido, são identificados casos em que
ocorre expressão aberrante de um marcador associado a uma outra linhagem
celular (31).
As técnicas moleculares estão auxiliando cada vez mais o diagnóstico e a
classificação das LAs (32). À medida que os marcadores moleculares vão sendo
identificados, as associações com idade, prognóstico, subtipo leucêmico e
expressão antigênica se solidificam (29,33-36). Os dados provenientes destes
estudos já nos permitem estabelecer associações entre as categorias genotípicas

14
das LAs e suas características imunofenotípicas (Tabela 1). A expressão de uma
única molécula raramente pode predizer um subtipo genético-molecular. Apenas
algumas exceções, como exemplo a fusão protéica CBFβ/SMMHC {produto da
inv(16)} (37) e a molécula aberrante de sulfato de condroitina NG2 (que se
correlaciona com as translocações envolvendo o gene MLL) (38,39). Apesar dos
contínuos esforços, alguns genótipos ainda apresentam o imunofenótipo bastante
variável, impedindo que se estabeleça a concordância completa entre as
categorias imunofenotípicas e genotípicas (29,32).
Tabela 1 – Subtipos celulares e alterações moleculares nas leucemias infantis
Subtipo leucêmico Alteração cromossômica Lesão molecular Freqüência (%)
LLA de células
progenitoras B
Translocações
na região 11q23
MLL/AF4, /ENL e outras
fusões gênicas
~85 (LLA de lactentes)
~5 (LLA em geral)
LLA de células
precursoras B (LLA-pB)
Hiperdiploidia Maior dosagem gênica ~35 (LLA-pB)
t(12;21)(p13;q22) TEL/AML1 ~20 (LLA-pB)
t(1;19)(q23;p13) E2A/PBX1 ~5 (LLA-pB)
t(9;22)(q34;q11) BCR/ABL ~5 (LLA-pB)
LLA de células
precursoras T
Deleção 1q-;
t(1;14)(p32;q11) SIL/SCL
~25 (LLA células
precursoras T)
LMA Translocações
na região 11q23
MLL/AF6, /AF9, /AF10 ou
outras fusões gênicas ~50 (LMA de lactentes)
t(8;21)(q22;q22) AML1/ETO ~15 (LMA em geral)
Adaptado de Greaves e Wiemels, 2003 (40)
O conhecimento destas categorias reflete em aplicações práticas que já
são realidade em muitos laboratórios. É possível, por exemplo, monitorar os

15
pacientes com LA durante e depois do tratamento, para detecção de doença
residual mínima (DRM), tanto pela identificação de marcadores aberrantes por
citometria de fluxo como por técnicas de reação em cadeia da polimerase (PCR) e
reação em cadeia da polimerase por transcriptase reversa (RT-PCR) (32,41). Este
tipo de associação ajuda também a explicar os mecanismos etiopatogênicos de
determinados subtipos de leucemias. A seguir estão descritos os principais
subgrupos leucêmicos, definidos por suas características genético-moleculares e
as expressões antigênicas freqüentemente encontradas nos blastos.
1.3.1 Os rearranjos do gene MLL
Os blastos de LLA com rearranjos do MLL são tipicamente CD19+, CD34+,
TdT+ e CD10- e, muitas vezes, expressam antígenos associados à linhagem
mielóide como o CD15 e/ou CD65 (42). Outros antígenos mielóides (CD13, CD33
e CD66c) podem ser positivos, já o CD20 é tipicamente negativo, correspondendo
aos estágios iniciais da diferenciação da célula B.
Um marcador importante das células leucêmicas que carregam rearranjos
do MLL é o homólogo de NG2, uma molécula de sulfato de condroitina que reage
com o anticorpo monoclonal 7.1, freqüentemente detectado nos casos de LLA
assim como nos de LMA. A expressão de NG2 é altamente sensível e específica
para rearranjos no MLL, chegando a alcançar quase 100% de especificidade nos
casos de LLA (38).

16
1.3.1.1 Estrutura e função do gene MLL e da proteín a MLL
O gene MLL é um homólogo humano do gene Trithorax da Drosophila (43),
que é um regulador crucial do desenvolvimento, incluindo a regulação da
expressão dos genes Hox, conforme mostrado em estudos de eliminação gênica
(44,45). O MLL ocupa uma extensão de aproximadamente 90 kb no cromossomo
11 na região 11q23 com quase 12 kb de seqüências codificadoras e codifica uma
proteína de 3.969 aminoácidos com um peso molecular de aproximadamente 430
kDa. A proteína MLL é clivada proteoliticamente pela Taspase1, uma protease
recentemente identificada (46), em um fragmento de 320-kd N-terminal e um
fragmento de 180 kd C-terminal, que interagem entre si para evitar a degradação
e conceder estabilidade à proteína (47).
Uma das principais funções da proteína MLL parece ser a manutenção da
expressão dos genes Hox como HoxA9 e HoxC8, que são genes essenciais na
regulação do desenvolvimento. A proteína MLL pode se ligar diretamente aos
promotores dos genes Hox e manter (mas não promover) seus níveis de
expressão através da modificação de histonas (48,49). O papel da proteína MLL
no desenvolvimento hematopoético normal foi identificada em vários estudos de
eliminação gênica. Em modelos experimentais foi possível estudar o papel do
MLL na hematopoese e também obter informações sobre a leucemogênese. Em
estudos realizados usando embriões de camundongos MLL-nulos, as células
hematopoéticas do saco vitelínico (50) e do fígado fetal prematuro (51) tinham
uma capacidade clonogênica reduzida, indicando um papel crucial da proteína
MLL no desenvolvimento das células sangüíneas. Estes resultados foram
importantes porque demonstraram que o MLL é necessário para a hematopoese

17
definitiva. Por outro lado, estes mesmos estudos de eliminação gênica deixaram
um perfil incompleto a respeito do papel do MLL na hematopoese porque os
camundongos MLL-nulos morriam precocemente.
1.3.1.2 Rearranjos do gene MLL nas leucemias
Os rearranjos do MLL são encontrados em 60-80% dos casos de
leucemias agudas de lactentes (LAL) geralmente encontrados em LLA pró-B, no
entanto, estes transcritos também podem ser vistos na LMA. Nas LLAs, o gene
AF4 é o parceiro mais comum entre os possíveis parceiros do MLL (Figura 6). É
(52).
Atualmente o MLL tem aproximadamente 40 parceiros de fusão descritos,
com diversas funções e domínios funcionais variáveis (51,53,54). As proteínas
quiméricas resultantes das fusões com o MLL, independentemente da localização
do parceiro de fusão, seja nuclear ou citoplasmático, formam consistentemente
padrões pontilhados no núcleo (55), indicando que a localização da fusão protéica
e, dos possíveis genes-alvo, é principalmente determinada pela atividade de
ligação ao ADN da porção correspondente ao MLL e não pelo parceiro de fusão.
Os principais genes-alvo desregulados transcricionalmente pelas proteínas
quiméricas resultantes das fusões do MLL no contexto da leucemogênese não
foram completamente identificados, mas vários estudos sugerem que os genes
Hox estão incluídos nesta lista. A expressão dos genes Hox foi identificada em
tecidos hematopoéticos normais, incluindo as células-tronco hematopoéticas, e foi
demonstrado que eles exercem um impacto significativo na proliferação e na
diferenciação (44,45). É possivelmente através da regulação dos genes Hox que

18
o MLL exerce um grande impacto no desenvolvimento hematopoético, conforme
mostrado em estudos in vitro e in vivo com células MLL-nulas (50,51). Acredita-se
que esta via de sinalização transcricional normal esteja alterada na
leucemogênese, como uma conseqüência da fusão gênica do MLL.
Figura 6 – Parceiros do gene MLL
Nas células leucêmicas com rearranjos do MLL, as regiões de ponto-de-
quebra genômicas estão em um fragmento que se estende do éxon 5 ao éxon 11
do gene MLL, esta região é chamada de região de concentração dos pontos-de-
quebra (breakpoint cluster region – bcr) (56) (Figura 7). A bcr do MLL tem vários
motivos que estão relacionados com a recombinação do ADN, como os sítios de
ligação à topoisomerase-II (topo-II) (57) e as seqüências Alu (56).

19
Figura 7 – Estrutura éxon/íntron do gene MLL*. A, o MLL ocupa 90 kb no cromossomo
11, consistindo de 36 éxons. B, mapa físico da bcr do gene MLL. As setas verticais indicam os
sítios consenso com a topoisomerase-II.
A presença destas seqüências de predisposição para a recombinação na
pequena região bcr indica que o rearranjo do MLL pode ser resultado de eventos
de quebra e recombinação do ADN. Os sítios de ligação da topo-II parecem estar
envolvidos em rearranjos do MLL relacionados a tratamentos com drogas que
atuam sobre a topo-II (quinolonas, etoposídios, teniposídios e antraciclinas) (58).
A mesma distribuição polarizada dos pontos-de-quebra na bcr do MLL vista em
leucemias secundárias ao tratamento, por exemplo, bcr telomérica, também foi
vista nas leucemias dos lactentes (59). Esta observação indicou que os
mecanismos de quebra do MLL nas LALs podem ser similares aos das LAs
secundárias a tratamentos quimioterápicos. Este achado levou à sugestão de que
a exposição fetal a alguns agentes com atividade inibidora da topo-II poderia ser o
evento inicial para o desenvolvimento das leucemias que afetam crianças com
* Modificado de Eguchi M e colaboradores, International Journal of Hematology, 2003.

20
idade inferior a 12 meses. Nestes casos, os rearranjos são os marcadores mais
freqüentemente encontrados (27,60).
1.3.1.3 A fusão do MLL é suficiente para causar a l eucemia?
As fusões gênicas encontradas nas LAs são consideradas como evento
iniciador da transformação leucêmica. Existe um debate bastante intenso no que
diz respeito à necessidade de mudanças genéticas adicionais nas LAs com
fusões do MLL. De acordo com a biologia do câncer e também com a história
natural das leucemias, sugere-se que pelo menos duas lesões genéticas
independentes em vias de sinalização complementares sejam necessárias para a
ocorrência de uma malignidade. Foi sugerido que as alterações genéticas que
afetam a diferenciação (p.ex., via fusões gênicas) requereriam um complemento
por uma alteração no controle do ciclo celular (p.ex., ativação de quinase) (61).
Uma característica relevante da história natural das LLAs de lactentes
assim como das leucemias secundárias com fusões gênicas do MLL é o
brevíssimo período pré-clínico ou latência da doença – média de 6 e 7 meses,
respectivamente (62,63). Estes dados sugerem que é possível que as fusões
gênicas do MLL sejam suficientes para a leucemogênese ou que as mutações
secundárias essenciais sejam rapidamente adquiridas. A concordância muito alta
de leucemia em gêmeos idênticos com LLA (quase 100% (64)) também sugere
que as fusões gênicas do MLL sejam suficientes ou que todas as alterações
genéticas necessárias sejam adquiridas no período pré-natal. Por outro lado, mas
não contrariamente, um estudo realizado com camundongos demonstrou que os
parceiros de fusão do MLL desempenham um papel importante na determinação

21
de algumas características biológicas das leucemias associadas ao MLL. Os
autores deste estudo verificaram que a transformação mielóide nos camundongos
com leucemias induzidas pelas fusões MLL/FKHRL1 e MLL/AFX tinham latências
e apresentações clínicas diferentes do que nas leucemias induzidas pelas fusões
MLL/AF10 ou MLL/ENL, que são notavelmente mais agressivas (65).
Algumas observações indicam um papel ativo de uma ou mais mutações
adicionais na leucemogênese com fusão do MLL. Em número substancial de
casos de lactentes diagnosticados com LLA - MLL positivo, estão também
presentes alterações cromossômicas ou mudanças genéticas sutis (66). Além
disso, aproximadamente 20% dos casos MLL-positivos de LAL apresentam
mutações no domínio de ativação do Flt3, resultando numa ativação aberrante da
quinase Flt3 (67). Nos casos em que o Flt3 está mutado, o inibidor específico de
Flt3 PCK412 pode inibir a proliferação celular aberrante in vitro e in vivo (67),
sugerindo que as células leucêmicas com fusão do MLL são dependentes da
ativação da quinase Flt3 para a sobrevivência. Esta hipótese está de acordo com
os resultados derivados de um modelo usando galinhas com leucemia MLL/ENL –
positivo, nas quais a fusão conseguia gerar a leucemia in vivo mas somente em
combinação com uma quinase ativada (68).
1.3.2 A fusão gênica TEL/AML1
A t(12;21)(p13;q22), não detectável por citogenética convencional, que une
os genes TEL e AML1, pode ser detectada por RT-PCR e/ou hibridização in situ
por fluorescência (FISH) em aproximadamente 25% dos casos de LLA da
infância, despontando como o rearranjo mais freqüente nas leucemias infantis . A

22
maioria dos pacientes positivos tem idade entre 1 e 12 anos ao diagnóstico, com
um pico entre 2 e 5 anos (69,70). Os casos de LLA que apresentam o transcrito
gênico TEL/AML1 apresentam exclusivamente fenótipo de célula precursora B,
sendo a maioria casos de LLA comum. Estes pacientes são caracterizados por
uma baixa contagem leucocitária ao diagnóstico (<50 000/l), e até o momento, a
t(12;21) não foi encontrada em LMA ou LLA-T e, sua freqüência em pacientes
adultos é baixa (71). Fenotipicamente, a maioria destes casos expressam CD19+,
CD34+, TdT+ e CD10+, ocorrendo em muitos casos a expressão aberrante do
CD33 e/ou CD13 (29).
A translocação t(12;21)(p13;q22) gera uma fusão gênica que inclui a
porção 5’ do TEL, um membro da família ETS de genes de fatores de transcrição,
e quase toda a região codificante de um outro fator de transcrição, o gene AML1,
que codifica a subunidade α do fator de ligação ao núcleo, um regulador
independente da formação das células-tronco hematopoéticas definitivas (72,73).
O fator de transcrição quimérico TEL/AML1 conserva um domínio de interação
essencial proteína-proteína do TEL e as seqüências de ligação ao ADN e de
regulação transcricional do AML1 [(também chamado CBF(α)] (72,73). O fator de
transcrição TEL é necessário para o retorno das células hematopoéticas
progenitoras para o seu local de origem na MO (74), enquanto o AML1 é o
componente de ligação ao ADN do fator de transcrição heterodimérico [CBF(α)
mais CBF(β)] chamado de fator de ligação ao núcleo, que exerce um papel central
na hematopoese (73,75). Os genes Hox provavelmente agem a jusante na
cascata de transcrição iniciada pelo fator de ligação ao núcleo (9).

23
Um efeito notável da fusão protéica TEL/AML1 é a inibição da atividade
transcricional que é normalmente iniciada quando AML1 se liga a uma região do
ADN chamada de seqüência “enhancer” do núcleo (76). A ligação do AML1 a esta
seqüência recruta outros fatores de transcrição e co-ativadores para esta região,
e o complexo protéico resultante regula a transcrição. Este complexo inclui
histona acetilases, que adicionam grupos acetil às histonas ligadas ao ADN,
causando assim mudanças na conformação da cromatina que intensificam a
transcrição dos genes-alvo. Assim como o AML1, a fusão protéica TEL/AML1
anormal é capaz de se ligar à seqüência “enhancer” do núcleo, mas ao invés de
ativar a transcrição, ela recruta histonas desacetilases, que induzem ao
fechamento da estrutura da cromatina e, conseqüentemente, à inibição da
transcrição . Estas mudanças na cascata normal de transcrição mediada pelo
AML1 alteram tanto a capacidade de auto-renovação como a de diferenciação
das células-tronco hematopoéticas (75,77).
A grande maioria (70-80%) dos casos com a fusão gênica TEL/AML1
apresenta deleção do alelo TEL normal (78). Esta deleção do alelo não-
rearranjado teria importante papel na patogênese da LLA da infância (77), pois, as
evidências sugerem que a fusão gênica TEL/AML1 normalmente acontece no
período pré-natal, como um evento precoce ou iniciador (64) e, que um evento
genético adicional ou secundário seria necessário para a transformação
leucêmica (28,79,80). Nestas leucemias, está cada vez mais evidente que,
independentemente das mudanças cromossômicas intra-uterinas, o evento pós-
natal ou as exposições que favorecem as mudanças genéticas secundárias,

24
inclusive a deleção do TEL, são cruciais para a maioria dos casos de leucemia na
infância (27) (Figura 8).
O processo leucemogênico constituído de mais de uma etapa parece ser
uma regra para as leucemias pediátricas com TEL/AML1 ou AML1/ETO (40).
Embora a fusão TEL/AML1 normalmente ocorra in utero (28,81), assim como a
fusão MLL/AF4 (26,82), o período de latência até o desenvolvimento da leucemia
é algumas vezes prolongado. Esta situação contrasta com os períodos de latência
muito curtos (em média, 6 meses após o nascimento) da maioria dos casos de
leucemia com a fusão MLL/AF4 (79).
Figura 8 – Modelo para a história natural das LAs da infância
1.3.3 Hiperdiploidia cromossomal
As alterações genéticas nas células leucêmicas incluem além das
alterações estruturais, alterações no número de cromossomos (ploidia). A ploidia
pode ser determinada diretamente pelo clássico método de contagem do número
de cromossomos num cariótipo metafásico, ou por um método indireto alternativo
que mede o conteúdo de ADN por citometria de fluxo.
Modificado de A Zelent e cols., 2004

25
A hiperdiploidia é a anormalidade cromossomal numérica mais comum na
LLA, caracterizada pela presença de 51 a 65 cromossomos por célula leucêmica,
correspondendo a um índice de ADN entre 1.16 e 1.6 (83). As conseqüências da
hiperdiploidia incluem uma propensão celular para entrar em apoptose e o
prognóstico é geralmente favorável. Este subtipo de LLA costuma ocorrer num
pico de idade pré-escolar. As trissomias (presença de 3 cópias de um
cromossomo) são as anormalidades mais vistas em casos de hiperdiploidia (84).
O imunofenótipo destes blastos corresponde à linhagem de célula
precursora B, em sua maioria CD19+, TdT+, CD10+ (muitas vezes
superexpresso) e CD45-. Os CD22 e CD24 também são tipicamente positivos e,
os antígenos mielóides, CD13, CD33 e CD 65 são usualmente negativos (85).
Normalmente os casos de hiperdiploidia são encontrados na ausência de
rearranjos do MLL e das fusões gênicas TEL/AML1 e BCR/ABL. Raros casos de
ocorrência mútua foram descritos (71,86).
1.3.4 LLA com a fusão gênica E2A/PBX1
A translocação recorrente t(1;19)(q23;p13) ocorre em aproximadamente 5-
6% das crianças e, em menos do que 5% dos adultos com LLA. Em 90-95%
destes casos, esta translocação leva à fusão gênica E2A/PBX1 (87). Existe uma
forte correlação entre esta fusão e os casos de LLA pré-B expressando IgM
citoplasmática, embora alguns casos raros em LLA pró-B, LLA comum, LLA-T e
LMA já tenham sido descritos (35,88). O imunofenótipo típico destes casos mostra
uma expressão homogênea de CD19, CD10 e imunoglobulina citoplasmática,
ausência completa de CD34 e, ausência parcial de CD20 (89).

26
A presença desta translocação confere um prognóstico desfavorável e está
freqüentemente correlacionada à presença de riscos clínicos como elevada
contagem celular, altos níveis séricos de desidrogenase láctea e envolvimento do
sistema nervoso central (35).
Após buscas pela fusão gênica E2A/PBX1 em amostras obtidas ao
nascimento e estudos moleculares, até o momento, as evidências sugerem que a
mesma deriva de uma origem pós-natal, ao contrário do que é encontrado para
outras translocações [t(12;21), t(8;21), e t(4;11)], que parecem ocorrer
predominantemente no período pré-natal (90).
1.3.5 A fusão gênica AML1/ETO
A translocação t(8;21)(q22;q22) é encontrada em aproximadamente 7%
das LMA e em aproximadamente 40% das LMA-M2 (22). A clonagem desta
translocação levou à identificação do AML1, que codifica uma unidade de ligação
do ADN ao fator de transcrição CBF(β), que tem um papel central na
hematopoese (73,75). Interessantemente, as translocações t(16;16) e t(12;21),
associadas respectivamente à LMA e à LLA, também envolvem o complexo
transcripcional AML1-CBF(β), fazendo deste o mais freqüente alvo de rearranjos
gênicos nas LAs (76).
A fusão AML1/ETO é encontrada tanto nas LMA de adultos como de
crianças, com ocorrência equilibrada entre os sexos, e com uma incidência
relativamente maior nos pacientes mais jovens (22). Assim como para outras
fusões (MLL/AF4 e TEL/AML1), os estudos sugerem que a fusão AML1/ETO se
origina in utero (26,91).

27
O imunofenótipo característico das LMA com t(8;21) é de blastos imaturos
mostrando expressão alta de CD34 e co-expressão de CD19. A expressão de
TdT na população de blastos imaturos é comum e, aparecem características de
assincronia de maturação com co-expressão de CD34 em alta intensidade,
concomitantemente com CD15 e mieloperoxidase (MPO) em alta intensidade. A
expressão do CD13 é usualmente elevada e a expressão do CD33 é
relativamente baixa. O CD56 é freqüentemente expresso e em alguns estudos foi
considerado um marcador de pior prognóstico (33,92).
1.4 UMA DOENÇA DIFERENTE A CADA IDADE
Os estudos clinico-epidemiológicos demonstraram que a leucemia infantil
não é uma doença homogênea. As diferenças, como visto anteriormente, estão
refletidas tanto na morfologia e nos marcadores imunofenotípicos de superfície
celular quanto em níveis bem mais sutis, como as translocações, deleções
gênicas ou trocas simples de bases nucleotídicas em determinados genes. Além
de toda esta diversidade imunomolecular, a LA infantil também está dividida em
subgrupos que variam de acordo com a idade (Figura 9). Este achado
clínico/biológico ajuda por exemplo a explicar as diferenças na sobrevida entre
lactentes (< 1 ano), crianças (entre 2 e 10 anos) e adultos (93).
Atualmente existem evidências importantes sugerindo que as fusões
gênicas normalmente encontradas nas leucemias pediátricas freqüentemente se
originam no período pré-natal durante a hematopoese fetal. Alguns estudos
investigativos exploram a hipótese de que os eventos aberrantes ocorram durante
a vida intra-uterina, usando como ferramenta molecular de investigação a

28
seqüência genômica da fusão gerada pela translocação cromossômica como um
marcador único e clonotípico. Um primeiro estudo envolve a investigação da
relação clonal entre leucemias concordantes em gêmeos monozigóticos (64).
Uma explicação plausível para este fenômeno clínico foi dada em 1975, quando
Boyse e Clarkson sugeriram que a leucemia concordante em gêmeos poderia ser
resultante do quimerismo sangüíneo, com a leucemia iniciando em um feto e
depois se propagando ao outro gêmeo através da comunicação vascular de uma
mesma placenta (94). Vários casos foram descritos ao longo dos anos, inclusive
um caso de leucemia em gêmeos siameses ocorrendo o fenótipo clínico após a
separação cirúrgica dos gêmeos (95).
Figura 9 – Subtipos moleculares da LLA em lactentes, crianças e adultos
A evidência convincente que fortalece a idéia da “metástase
intraplacentária” veio de um estudo internacional de casos de leucemia
concordante entre gêmeos. Este estudo utilizou as regiões de quebra genômicas
Modificado de M GREAVES, 2002

29
únicas nas fusões do MLL (82)e TEL/AML1 (28,81), e os rearranjos clonotípicos
de IGH e TCR (96,97). Em cada par de gêmeos investigado, foi demonstrado que
suas células leucêmicas compartilhavam a mesma seqüência de fusão gênica ou
seqüências de IGH/TCR. Como estes eventos clonais não-constitutivos e
adquiridos são compartilhados por cada par de gêmeos, a única explicação
sustentável é a de uma única origem celular seguida por ‘metástase’ (64).
Estes estudos nos gêmeos também foram informativos em outros
aspectos. Primeiramente, eles demonstraram que a latência pós-natal poderia ser
variável, mesmo entre gêmeos de um mesmo par, e poderia ser prolongada (em
até 14 anos) (28). Segundo, como a concordância entre os gêmeos variou de
50% ou mais para os casos de lactentes a até 5-10% em casos pediátricos com
maior idade, é provável que eventos genéticos adicionais e pós-natais sejam
necessários para complementar o evento da fusão gênica precoce (64,93).
Para reiterar a hipótese da origem pré-natal foi realizado um teste ainda
mais direto em pacientes não-gêmeos com leucemia. Foi realizado um
rastreamento da seqüência da fusão gênica no ADN arquivado (desde o período
neonatal) de pacientes com diagnóstico de leucemia (26,91). Estes estudos
confirmaram a origem pré-natal da fusão gênica (MLL/AF4) na LAL, e mostraram
que muitas e provavelmente a maioria das fusões TEL/AML1 (na LLA) e
AML1/ETO (na LMA) se originam in utero. Assim sendo, a idade ao diagnóstico
e/ou os distintos subgrupos biológicos são muito provavelmente os reflexos de
seus diferentes mecanismos etiológicos.

30
1.5 FATORES DE RISCO
Com base em tudo o que foi visto até aqui, é possível que raramente a
leucemia da criança envolva eventos genéticos herdados. Ao contrário, acredita-
se que as mutações nas células hematopoéticas ocorram mais freqüentemente in
utero ou, em alguns casos, durante a infância. Conforme mencionado
anteriormente, sugere-se que as mutações adicionais seriam necessárias para a
transformação leucêmica e que, na maioria das vezes, estes eventos ocorreriam
no período pós-natal, num clone pré-leucêmico (28,64,77,80,98). Saber como
estas mutações são adquiridas é a questão principal na etiologia da doença. A
princípio, são os estudos epidemiológicos que nos revelam as primeiras
informações nesta busca. Na figura 10 estão relacionados alguns dos fatores
associados com um risco aumentado para o desenvolvimento de leucemia.
Figura 10 – Fatores de risco envolvidos no desenvolvimento da leucemia (1,25).

31
As LALs representam o melhor modelo para os estudos epidemiológicos
moleculares: (i) A maioria dos casos de lactentes (aproximadamente 85% com
LLA e 65% com LMA) têm rearranjos na região 11q23 envolvendo o gene MLL
(36,64,99). (ii) Neste grupo, as evidências moleculares da ocorrência in utero dos
rearranjos do MLL são irrefutáveis (26,82). (iii) A janela crítica de exposições está
muito definida pelo tempo de exposição aos fatores de risco (59,100).
1.5.1 Os fatores ambientais
Algumas associações significativas já foram testadas e confirmadas,
apontando fatores de risco que influenciam na etiologia da leucemia. Estes
incluem um peso acima de 4.000 g ao nascimento (associado a um risco
aumentado 2 vezes maior de desenvolver LMA e LLA nas crianças menores de
dois anos), o consumo materno de álcool durante a gravidez (com um risco 1,5
vez maior associado com LMA infantil), e história materna de aborto ou parto de
natimorto (riscos aumentados em 2 a 5 vezes de desenvolver LLA ou LMA nos
lactentes) (101,102).
A exposição à radiação ionizante e a certos agentes químicos são
exemplos de fatores associados ao desenvolvimento da LA. Entre os
sobreviventes das explosões de bombas atômicas no Japão, o índice de casos de
leucemia é elevado. Em um estudo que analisou uma população de 73.330
sobreviventes em Hiroshima e Nagasaki, a curva de um efeito estimado de dose-
resposta entre a radiação ionizante e a mortalidade causada por leucemia foi
muito íngreme (P < 0.002), com um fator de 2.4, entre àqueles que relataram
queda dos pelos dentro do prazo de 60 dias após o bombardeamento, quando

32
comparados aos que não sofreram este sinal de exposição aguda à radiação
(103,104).
A exposição química (benzeno, por exemplo) também está associada ao
risco de desenvolver leucemia, no entanto, está diretamente relacionado à
atividade da enzima NAD(P)H que o metaboliza. Um estudo que determinou os
polimorfismos da enzima NQO1 que detoxifica o benzeno mostrou que as
leucemias com rearranjos do MLL apresentavam maior número de genótipos com
baixa atividade enzimática de NQO1 (heterozigotos CT ou homozigotos TT no
nucleotídeo 609) do que os controles (OR, 2.5; P = 0.015). Ao contrário, nenhuma
associação foi encontrada em outros grupos de leucemias pediátricas com as
fusões TEL-AML1 (n = 50) ou hiperdiploidia (n = 29). No subgrupo das leucemias
de lactentes que continham a fusão MLL-AF4 (n = 21), a associação era ainda
maior com os genótipos que determinam função reduzida ou nula de NQO1 (OR,
8.12; P = 0.00013) (105).
1.5.1.1 Agentes inibidores da topoisomerase II
A explicação mais plausível para as fusões gênicas do MLL no
cromossomo 11q23 dos lactentes recai sobre a exposição materna durante o
período gestacional a agentes químicos, como epidofilotoxinas e antracilcinas,
que causam quebras no ADN por meio da inibição da função da topoisomerase II
(topo-II) (60). A topo-II desempenha um papel crucial durante a replicação, sua
potencial ação na aquisição das translocações foi primeiramente notada
comparando-se os rearranjos do MLL em lactentes com aqueles observados em
leucemias secundárias ao uso de drogas que a têm como alvo (106,107).

33
Primeiramente a topo-II gera quebras na dupla-fita (Figura 11A) que será religada
depois do desenovelamento do ADN (Figura 11B). As drogas que atuam sobre a
enzima, interrompem o processo enquanto as fitas de ADN estão separadas,
deixando-as livres e mais propensas à ocorrência de fusões gênicas (108).
Considerando-se que o rearranjo do MLL acontece in utero na LAL e que
as células no feto em desenvolvimento têm um grau elevado de proliferação, a
exposição intra-uterina a drogas, alimentos, ou a fatores ambientais que inibem a
topo-II é considerada como um fator de risco bastante aceito para o início dos
rearranjos do MLL (27,109).
Figura 11 – Modelo esquemático das funções executadas pela topo-II
Uma grande variedade de componentes naturais e sintéticos, incluindo
agentes quimioterápicos, antibióticos a base de quinolonas, flavonóides
encontrados em comidas e bebidas, catequinas, resina, metabólitos do benzeno
(108,110,111), e até mesmo estrógenos podem inibir a topo-II . Em recente
A B
Topo -II
Dupla -fita ADN

34
estudo epidemiológico internacional, a exposição transplacentária a drogas que
danificam o ADN, como uma droga anti-inflamatória não-esteroidal (dipirona) e um
pesticida, foi associada ao desenvolvimento das LALs envolvendo fusões gênicas
do MLL (25). Embora os efeitos da dieta, medicação e exposições ambientais
sejam reduzidos em relação ao uso de quimioterápicos, a capacidade reduzida de
um feto e/ou sua mãe em desintoxicar tais agentes poderia potencializar a
susceptibilidade do feto em desenvolver leucemia (112,113).
1.5.2 Infecção viral
Desde a década de 80, com os primeiros trabalhos de Kinlen e Greaves,
existe a suspeita de que as infecções virais também poderiam desempenhar um
papel na patogênese da leucemia humana (10,114). Esta hipótese foi resultado
da observação de que a pouca idade em que se dá início a LLA corresponde ao
período em que o sistema imune está em desenvolvimento, e talvez, mais
vulnerável aos efeitos oncogênicos de certos vírus. Por enquanto, não há
associações diretas entre exposição viral pré-natal e o risco de leucemia. No
entanto, há um crescente suporte epidemiológico para a idéia de que o estresse
proliferativo associado a exposições virais deficientes durante a infância e
respostas imunológicas anormais devido ao retardo na ocorrência das infecções
comuns, desempenham um papel causal na LLA-comum (LLA-c) (27,93).
2. JUSTIFICATIVA DO ESTUDO
Todo projeto de pesquisa deriva de um processo dinâmico. Nosso grupo de
trabalho desenvolve projetos que buscam respostas de cunho etiopatogênico nas

35
LALs. Para tal, utilizamos uma metodologia multidisciplinar que inclui análise
morfológica, testes com citometria de fluxo (imunofenotipagem) e análises
moleculares. Com estas ferramentas foi iniciado um estudo multicêntrico e
multidisciplinar, com objetivo de investigar a etiologia das alterações moleculares
que comprometem a região 11q23 nas LALs, com atenção especial para a
exposição a fatores exógenos que danificam as estruturas de ADN. Além das
publicações científicas (25,31,115-118) e colaborações com pesquisadores
nacionais e estrangeiros, este estudo resultou num verdadeiro banco de
informações e associações clínico-biológicas e ambientais das LALs do Brasil.
Entre outras, as associações entre subtipos celulares, moleculares e a
idade dos pacientes são cada vez mais encontradas nas leucemias de forma
geral. As LLAs pró-B em neonatos com rearranjos do gene MLL e as LLAs-c em
crianças (2-10 anos) com hiperdiploidia cromossômica ou a fusão gênica
TEL/AML1 são exemplos clássicos. A hipótese da ocorrência intra-uterina
contempla algumas das translocações e/ou mutações encontradas nas leucemias.
No entanto, no grupo de casos analisados preliminarmente para o estudo
epidemiológico, foram encontrados casos raros que questionam algumas das
associações já estabelecidas.
Em vista dos relatos acima, propusemos um rastreamento nos casos de
lactentes, buscando pelas alterações genético-moleculares envolvidas na
leucemogênese. Estes dados certamente contribuirão nas discussões que ainda
polemizam os fóruns de reflexão sobre a etiopatogenia da doença.

36
3. OBJETIVOS
3.1 GERAL
• Identificar as alterações moleculares associadas às LAs infantis que
provavelmente se iniciam na vida intra-uterina;
3.2 ESPECÍFICOS
• Caracterizar através de testes imunomoleculares os marcadores mais
frequentes em LAs de crianças com idade inferior a 24 meses;
• Estabelecer um algoritmo de testes imunofenotípicos e citogenético-
moleculares capazes de caracterizar as alterações do gene MLL nas
LALs;
• Prover dados biológicos consistentes ao estudo epidemiológico
exploratório sobre LA infantil e fatores de risco ambientais.

37
4. MATERIAL E MÉTODOS
4.1 AMOSTRAS
Foram incluídas nesta análise amostras de crianças com idade inferior a 24
meses (lactentes) portadores de LA, procedentes de diversos estados brasileiros,
sem restrição de idade, raça ou sexo e isentos de tratamento quimioterápico
prévio. As amostras destes pacientes foram obtidas após exames clínicos e
hemogramas sugestivos de leucemia e encaminhadas, para fins diagnósticos ao
nosso laboratório no Instituto Nacional de Câncer (INCA), Rio de Janeiro-RJ, no
período de janeiro de 2000 a fevereiro de 2005. Somente foram inseridas as
amostras de crianças cujos pais ou responsáveis legais concordaram com a
participação no estudo formalizado através da assinatura do “Termo de
consentimento livre e esclarecido”.
• Foram coletadas amostras de MO e/ou sangue periférico (SP) dos
pacientes em uma seringa esterilizada (5-10 ml) com anticoagulante
EDTA;
• 4 lâminas com esfregaços de MO e/ou SP foram enviadas para a
realização da análise morfológica;
As amostras de MO e/ou SP, tiveram seu imunofenótipo caracterizado pela
citometria de fluxo multiparamétrica conforme tabela 2.

38
4.2 ETAPAS DIAGNÓSTICAS
4.2.1 Análise morfológica
A análise morfológica inicial foi feita pela coloração convencional por May-
Grünwald-Giemsa (MGG) complementada pela técnica citoquímica pelo Negro de
Sudan. Esta análise preliminar permitiu a distinção entre LLA e LMA e a aplicação
dos critérios definidos pelo grupo French-American-British (FAB) (119).
4.2.2 Separação de células mononucleares
Técnica de separação por Ficoll-Hypaque-Hystopaque®
• As amostras foram inicialmente centrifugadas a 1500 rpm por 5 minutos,
para separação do plasma. Este foi retirado do tubo. A amostra restante
foi diluída com meio RPMI e homogeneizada;
• Nos casos com hiperleucocitose (>30 x 109 leucócitos/l) a etapa inicial
consistia na separação celular para obtenção das células mononucleares,
baseada em gradiente de centrifugação através de Ficoll-Hypaque-
Hystopaque® (densidade 1077);
• Todo o conteúdo das amostras diluídas foram cuidadosamente
depositados em tubos falcon de 15 ml, contendo Ficoll- Hystopaque®, na
proporção de 1:3 ml de amostra diluída e centrifugados a 1500 rpm por
25minutos;
• Após a centrifugação forma-se um anel na interfase do tubo, contendo as
células mononucleares. Este anel foi recolhido com pipeta Pasteur e

39
transferido para um novo tubo; o volume foi novamente completado com
RPMI para um volume de 10 ml;
• O tubo foi homogeneizado e centrifugado por 5 minutos a 1500 rpm; o
sobrenadante foi descartado e as células foram ressuspendidas em 2,0 ml
de RPMI;
• A viabilidade celular foi avaliada através da coloração pelo azul de tripano
e as células foram observadas e contadas em câmara de Newbauer;
• As células mononucleadas foram então distribuídas na concentração de
1,0 - 1,6 x 106/ml para realização da imunofenotipagem e citospins;
4.2.3 Imunofenotipagem
As células foram colocadas em tubos apropriados, para leitura no citômetro
de fluxo, em concentração de 106 células e incubadas durante 30 minutos com 10
µl do anticorpo primário. As incubações foram feitas com duplas ou triplas
associações de anticorpos monoclonais (AcMo) específicos marcados com
diferentes fluorocromos (fluoresceína ou ficoeritrina).
A aquisição e análise dos casos foram executadas em citômetro de fluxo,
seja no Epics-Coulter® ou no FacScan®, Becton Dickinson, existentes no setor,
onde foram contadas 104 células por cada tubo. Para as análises
citofluorimétricas foi utilizado um painel de AcMos capaz de reconhecer as
seguintes moléculas de superfície: CD45, CD14, CD15, CD34, CD117, CD19,
CD10, CD20, CD7, CD3, CD4, CD8, CD13, CD33, CD61, CD34, CD42, CD38,
HLA-DR, 7.1 e glicoforina-a. Os AcMos utilizados foram adquiridos com diversos
fabricantes como Coulter, Imunotech, Becton-Dickson, Pharmigen e Dako. As
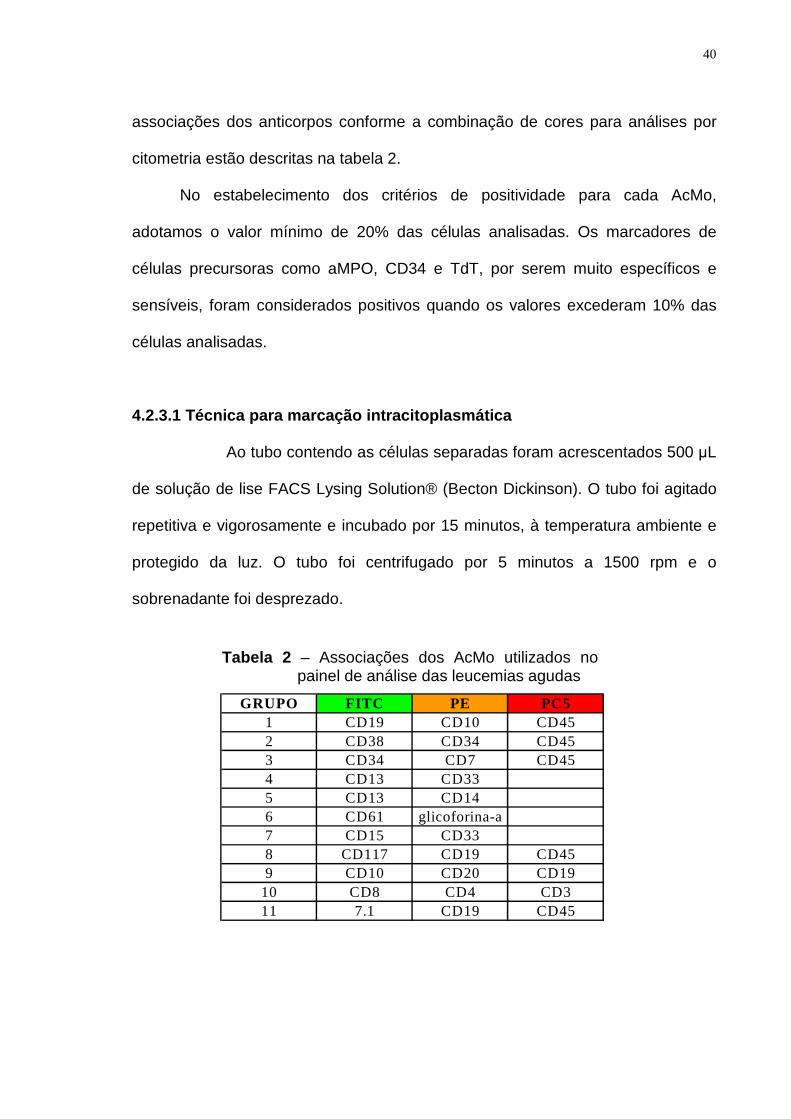
40
associações dos anticorpos conforme a combinação de cores para análises por
citometria estão descritas na tabela 2.
No estabelecimento dos critérios de positividade para cada AcMo,
adotamos o valor mínimo de 20% das células analisadas. Os marcadores de
células precursoras como aMPO, CD34 e TdT, por serem muito específicos e
sensíveis, foram considerados positivos quando os valores excederam 10% das
células analisadas.
4.2.3.1 Técnica para marcação intracitoplasmática
Ao tubo contendo as células separadas foram acrescentados 500 µL
de solução de lise FACS Lysing Solution® (Becton Dickinson). O tubo foi agitado
repetitiva e vigorosamente e incubado por 15 minutos, à temperatura ambiente e
protegido da luz. O tubo foi centrifugado por 5 minutos a 1500 rpm e o
sobrenadante foi desprezado.
GRUPO FITC PE PC51 CD19 CD10 CD452 CD38 CD34 CD453 CD34 CD7 CD454 CD13 CD335 CD13 CD146 CD61 glicoforina-a7 CD15 CD338 CD117 CD19 CD459 CD10 CD20 CD19
10 CD8 CD4 CD311 7.1 CD19 CD45
Tabela 2 – Associações dos AcMo utilizados no painel de análise das leucemias agudas

41
Foram adicionados 500 µl de solução detergente (TWEEN 20), para
permeabilizar a membrana celular, e a solução foi homogeneizada. Procedeu-se
nova centrifugação por 5 minutos a 1500 rpm, o sobrenadante foi desprezado e
então os AcMos (anti-CD22, CD3, IgM, aMPO, CD79a e CD13) foram
acrescentados em combinações de acordo com fluorocromo conjugado (PE ou
FITC).
4.2.3.2 Técnica para marcação de superfície celular
Adicionou-se ao tubo contendo as células separadas (5-15µl) de cada
AcMo, em simples, duplas ou triplas associações, conjugados com FITC, PE e
PerC5). O tubo foi homogeneizado e incubado por 20 minutos, a temperatura
ambiente e protegido da luz. Foram acrescentados 500 µl de PBS em cada tubo,
o tubo foi centrifugado por 5 minutos a 1500 rpm e o sobrenadante foi descartado.
Nos casos onde o AcMo não era conjugado a nenhum fluorocromo, foram
adicionados 2,5 µl de um segundo anticorpo conjugado a FITC ou PE,
constituindo a segunda camada e incubou-se por 20 minutos. As células foram
ressuspensas em 500 µl de PBS para serem analisadas no citômetro de fluxo.
4.2.3.3 Técnica para marcação por lise de hemáceas
Nos casos em que havia baixa leucometria ou pouco material para estudo,
a marcação foi feita por lise de hemáceas. Em um tubo de 5 ml foram colocados
50 µl de sangue e 5-10 µl de anticorpo. Após 20 minutos, adicionou-se 500 µl de
solução de lise de hemáceas, deixando o tubo em repouso por mais 10 minutos.
O tubo foi centrifugado por 5 minutos a 1500 rpm e o sobrenadante foi

42
descartado. Foram acrescentados 500 µl de PBS e o tubo foi novamente
centrifugado por 5 minutos a 1500 rpm. As células foram ressuspensas em 500 µl
de PBS para serem analisadas no citômetro de fluxo.
4.2.4 Citogenética convencional e FISH
A presença de anormalidades cromossômicas foi pesquisada através de
estudos do cariótipo, utilizando metodologia previamente descrita (120) e
realizados por Maria Luiza Macedo Silva, PhD e Gilena Dantas de Brito, Ms no
Laboratório de Citogenética/CEMO – Instituto Nacional de Câncer. As análises de
metáfases (quando disponíveis) e intérfases foram documentadas de acordo com
os critérios descritos por Hopman e colaboradores (121). Ao menos 200 células
foram analisadas por amostra usando o programa Cytovision, Applied Imaging. A
hibridização foi realizada usando MLL sonda bicolor que detecta a quebra
seguindo os protocolos da Vysis Inc. G.D.B. foi responsável por todos os
resultados de FISH aqui reportados.
4.2.5 Genética-molecular
As amostras de MO e/ou SP foram mantidas congeladas em freezer –80 ºC
ou em tanque de nitrogênio líquido, conforme rotina do laboratório, até seu
processamento inicial de acordo com os protocolos de estudo.
Para investigar o status do MLL, primeiramente foi realizada a citogenética
convencional, seguida pela RT-PCR nos casos inconclusivos ou negativos. O
rastreamento final foi por análise de FISH, realizada em 36 casos inconclusivos

43
e/ou negativos, ou em 7 casos com material biológico insuficiente para outros
testes. Para aqueles casos que foram negativos para o rearranjo do MLL, foi
realizada uma investigação adicional pela fusão gênica TEL/AML1 nas LLAs de
células precursoras B (LLA-pB) e, para a fusão gênica E2A/PBX1 nos casos LLA-
pB TEL/AML1-vos. Nos casos de LMA MLL-vos também foi testada a presença da
fusão gênica AML1/ETO.
4.2.5.1 Purificação de ácido ribonucléico (ARN)
O material que não continha células separadas passou por uma lise inicial
das hemáceas. Em tubo eppendorf contendo SP ou MO, adicionou-se 500 µl de
solução de lise de hemáceas e por inversão o tubo foi homogeneizado. Incubou-
se por 10 minutos a temperatura ambiente. Centrifugou-se por 10 minutos a
12000 rpm, desprezando-se o sobrenadante. Ao final desta etapa, acrescentou-se
TRIZOL LS Reagent (Invitrogen, EUA) em uma proporção de 3:1 (para cada 250
µl de suspensão celular adicionou-se 750 µl de TRIZOL), homogeneizando com
a pipeta até completa dissolução dos grumos. Após incubação em temperatura
ambiente por 5 minutos, foram acrescentados 200 µl de clorofórmio,
homogeneizando com pipeta e agitando-se vigorosamente por 15 segundos. A
solução resultante foi centrifugada a 4 ºC por 15 minutos a 12500 rpm e a fase
transparente (superior) de cada tubo foi transferida para novos tubos livres de
agentes deteriorantes de ácido ribonucléico (ARN). O ARN foi precipitado com
500 µl de álcool isopropílico 100% e gelado. As amostras foram incubadas a
temperatura ambiente por 10 minutos. A seguir, centrifugou-se a amostra a 4 ºC
por 15 minutos a 12500 rpm, desprezando-se o sobrenadante. Acrescentou-se

44
1000 µl de etanol 75%, seguido novamente de centrifugação refrigerada por 5
minutos a 7500 rpm. Retirou-se todo o álcool e o pellet preso ao tubo foi
dissolvido com água tratada com DEPC por pelo menos 15 minutos. O material foi
identificado e armazenado.
4.2.5.2 Síntese da fita de ADN complementar (ADNc)
Para cada 5 µg (3 a 5 µl) de ARN a ser transcrito, acrescentou-se 2 µl de
enzima que digere o ADN (10 U/µl), 2 µl de solução tampão 10X (500 mM Tris, pH
8,0, 100 mM MgCl2 e água tratada com DEPC para completar um volume final de
20 µl. Esta mistura foi incubada por 30 minutos a 37 ºC. Ao final, foram
adicionados 2 µl de EDTA 25 mM e incubou-se por 15 minutos a 65 ºC para
inibição da reação.
A seguir, cada amostra foi dividida em 2 tubos, contendo 10 µl cada. Um
dos tubos era identificado como RT e o outro como n-RT. A cada tubo,
acrescentou-se 1 µl de solução com mistura de dNTPs (deoxyNucleotide mix 10
mM; Invitrogen, EUA) e 1 µl de solução de iniciadores oligo (dT) 0,5 µg/µl
(Invitrogen, EUA). O conteúdo foi incubado por 5 minutos a 65 ºC. Ao final deste
processo, acrescentou-se ao tubo 4 µl do tampão de síntese 5X (First Strand
Buffer; Invitrogen, EUA), 2 µl de DTT 0,1 M (Invitrogen, EUA) e 1 µl do inibidor de
substâncias que digerem o ARN. As amostras foram levemente homogeneizadas.
Após esta etapa, ao tubo RT foi acrescentado 1 µl da enzima transcriptase
(Rnase H-Reverse Transcriptase 200 U/µl; Invitrogen, EUA) e todos os tubos
foram incubados em banho-maria a 37 ºC por uma hora. Ao final, as amostras
foram armazenadas a –20 ºC.

45
4.2.5.3 RT-PCR para amplificação do gene GAPDH
Para comprovar a integridade das amostras de ADNc, foi realizada a RT-
PCR para amplificação do gene GAPDH. Este gene de referência endógena ou
housekeeping codifica a proteína gliceraldeído-3-fosfato desidrogenase (GAPDH),
expressa em todas as células nucleadas. A seqüência de oligonucleotídeos
utilizada está descrita na tabela 3. O tamanho do produto de PCR amplificado é
de 419 pb.
Tabela 3 – Seqüência de oligonucleotídeos para a amplificação gênica GAPDH
PRIMER SEQÜÊNCIA (5’ – 3’)
GAPDH senso TGACCCCTTCATTGACCTCA
GAPDH anti-senso AGTCCTTCCACGATACCAAA
Para a reação de PCR foram utilizados 3 µl de ADNc. A solução adicionada
ao ADNc foi preparada contendo uma solução final de 50 µl com as seguintes
quantidades: iniciadores 0,4 µM, dNTPs 200 µM, solução tampão 1X (Invitrogen,
Brasil), cloreto de magnésio 2,5 mM (Invitrogen, Brasil), água de ampola para
completar o volume e 1U de Taq-polimerase 5U/µl (Taq DNA Polymerase;
Invitrogen, Brasil).
O termociclador utilizado foi o PCR-System 9700 (Gene Amp, Applied
Biosystems, EUA), utilizando-se as seguintes temperaturas e número de ciclos:
desnaturação inicial a 94 ºC por 3 minutos, 35 ciclos de desnaturação a 94 ºC por
30 segundos, anelamento a 60 ºC por 30 segundos e extensão a 72 ºC por 45
segundos e, extensão final a 72 ºC por 10 minutos. Os produtos de PCR foram

46
visualizados em gel de agarose corado com brometo de etídio através de luz
ultravioleta (Figura 12).
Figura 12 – Corrida de eletroforese em gel de agarose (1,5%) corado com
brometo de etídio dos produtos da amplificação do gene GAPDH
4.2.5.4 RT-PCR para amplificação dos genes MLL/AF4, TEL/AML1, E2A/PBX1
e AML1/ETO
As fusões gênicas resultantes das translocações t(4;11)(q21;q23),
t(12;21)(p13;q22), t(1;19)(q23;p13) e t(8;21)(q22;q22) foram pesquisadas através
de RT-PCR (simples, ninho ou semi-ninho) utilizando-se as condições e os
iniciadores divulgados pelo BIOMED-1 Concerted Action: Investigation of minimal
residual disease in acute leukemia (41). Na tabela 4 estão descritos os
oligonucleotídeos utilizados para este estudo.
O termociclador usado foi o PCR-System 9700 (Gene Amp, Applied
Biosystems, EUA), utilizando-se as seguintes temperaturas e número de ciclos:
desnaturação inicial a 94 ºC por 2 minutos, 35 ciclos de desnaturação a 94 ºC por
100 pb 1 2 3 4 5 6 7 8
419 pb

47
30 segundos, anelamento a 60 ºC por 60 segundos e extensão a 72 ºC por 60
segundos.
Tabela 4 – Seqüência de oligonucleotídeos para a amplificação gênica das
principais translocações
NOME SEQÜÊNCIA (5’ – 3’)
MLL-A CCG CCT CAG CCA CCT AC
MLL-C AGG ACC GCC AAG AAA AGA
AF4-B TGT CAC TGA GCT GAA GGT CG
TEL-A TGC ACC CTC TGA TCC TGA AC
AML1-B AAC GCC TCG CTC ATC TTG C
E2A-A CAC CAG CCT CAT GCA CAA C
PBX-B TCG CAG GAG ATT CAT CAC G
E2A-C CAC CCT CCC TGA CCT GTC T
PBX1-D GGC CTG CTC GTA TTT CTC C
AML1-A CTA CCG CAG CCA TGA AG AAC C
ETO-B AGA GGA AGG CCC ATT GCT GAA
Para a fusão gênica MLL/AF4 foi feito o RT-PCR ninho, com os iniciadores
MLL-A, AF4-B para a primeira reação e MLL-C e AF4-B para a segunda. O
transcrito TEL/AML1 foi procurado por RT-PCR simples, com os iniciadores TEL-
A e AML1-B. Para a análise da fusão gênica E2A/PBX1, também foi utilizado o
RT-PCR ninho, com os iniciadores E2A-A e PBX-B para a primeira reação e E2A-
C e PBX-D para a segunda. Por fim, o RT-PCR semi-ninho (utilizando os mesmos
oligonucleotídeos que foram usados na primeira reação) foi usado na amplificação
da fusão gênica AML1/ETO, com os iniciadores AML1-A e ETO-B.

48
Os controles positivos foram as linhagens celulares RS4;11, REH,
KAZUMI-1 e 697, respectivamente, para as translocações t(4;11), t(12;21), t(8;21)
e t(1;19). Em algumas reações, pacientes positivos pela citogenética convencional
foram usados como controles positivos.
4.2.5.5 Reação RT-PCR multiplex para a amplificação dos principais
rearranjos do gene MLL
Para a detecção das fusões gênicas resultantes das translocações
t(4;11)(q21;q23), t(11;19)(q23;p13.3), t(9;11)(p21-22;q23), e t(11;19)(q23;q13.1),
utilizou-se a técnica de RT-PCR multiplex previamente descrita(122). A tabela 5
contém as seqüências dos nucleotídeos utilizadas.
Tabela 5 – Seqüência de oligonucleotídeos para os rearranjos do MLL
NOME SEQÜÊNCIA (5’ – 3’)
MLL 3759 CCC AAA GAA AAG CAG TAG TGA
AF4 1607 TTT GTA GGG AAA GGA AAC TTG
AF9 1825 GAT TTG CTT TGC TTT ATT GG
ENL 216 CGT ACC CCG ACT CCT CTA CT
ENL 1399 GCC TGA CAC CTT GCT GTT

49
Os parceiros mais freqüentes do MLL são rastreados por esta técnica. A
realização de reações simples após a detecção do rearranjo permite a
identificação do parceiro exato em cada caso. A figura 13 é um esquema
mostrando os pontos de quebra e as posições dos genes usados no multiplex.
Figura 13 – Pontos de quebra (setas verticais) e posições dos oligonucleotídeos
iniciadores (setas horizontais) dos genes incluídos na reação de RT-PCR
multiplex.
As fusões gênicas MLL/AF4, MLL/AF9 e MLL/ENL foram amplificadas
numa mesma reação. A reação multiplex de RT-PCR foi realizada em 50 µl de 1,1
X PCR Buffer, 2,0 mM de MgCl2, 0,8 mM de dNTPs, 1 U de Taq polimerase, e os
ML AF4
AF6
AF9
AF1
ELL
ENL

50
oligonucleotídeos iniciadores numa concentração final de 0,5 µM. Realizou-se 35
ciclos de amplificação numa máquina termocicladora contendo o seguinte
programa: 95 ºC por 5 minutos, 35 ciclos de 95 ºC por 30 segundos, 60 ºC por 30
segundos, e 72 ºC por 1 minuto, 72 ºC por 5 minutos.
Figura 14 – Exemplo de corrida de eletroforese em gel de agarose (2%) corado
com brometo de etídio dos produtos da amplificação do RT-PCR multiplex para
detecção dos rearranjos do MLL. Coluna 1 mostra o controle positivo. Colunas 2, 4 e 13
mostram pacientes negativos para as fusões procuradas. Colunas 3 e 5-12 mostram pacientes
positivos para alguma das fusões procuradas. Coluna 14 mostra o controle negativo.
4.3 ANÁLISE ESTATÍSTICA
A análise estatística foi realizada através do programa SPSS 13.0 (SPSS
Inc., 2004). Para a avaliação estatística das freqüências e das associações entre
os dados clínicos e a presença ou ausência das anormalidades cromossômicas, o
teste Pearson Chi-Square foi utilizado. Um valor de p menor ou igual a 0.05 foi
considerado significativo.
100 pb 1 2 3 4 5 6 7 8 9 10 11 12 13 14 50 pb
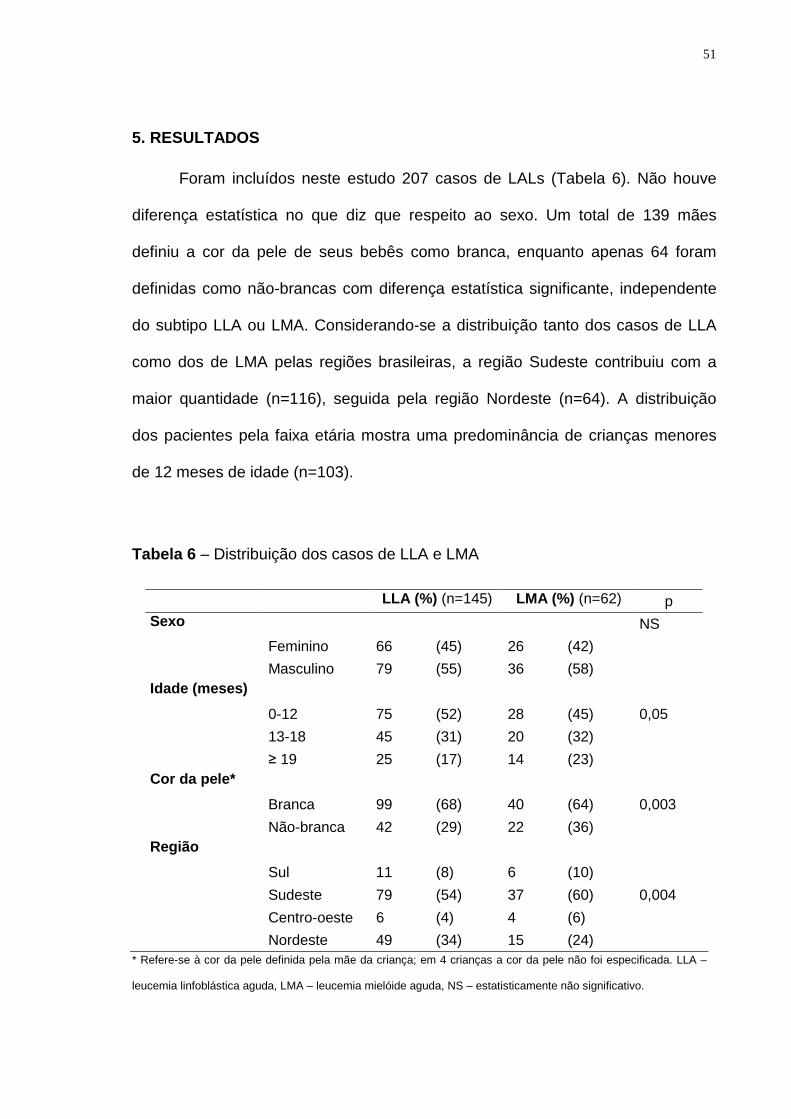
51
5. RESULTADOS
Foram incluídos neste estudo 207 casos de LALs (Tabela 6). Não houve
diferença estatística no que diz que respeito ao sexo. Um total de 139 mães
definiu a cor da pele de seus bebês como branca, enquanto apenas 64 foram
definidas como não-brancas com diferença estatística significante, independente
do subtipo LLA ou LMA. Considerando-se a distribuição tanto dos casos de LLA
como dos de LMA pelas regiões brasileiras, a região Sudeste contribuiu com a
maior quantidade (n=116), seguida pela região Nordeste (n=64). A distribuição
dos pacientes pela faixa etária mostra uma predominância de crianças menores
de 12 meses de idade (n=103).
Tabela 6 – Distribuição dos casos de LLA e LMA
LLA (%) (n=145) LMA (%) (n=62) p Sexo NS Feminino 66 (45) 26 (42) Masculino 79 (55) 36 (58) Idade (meses) 0-12 75 (52) 28 (45) 0,05 13-18 45 (31) 20 (32) ≥ 19 25 (17) 14 (23) Cor da pele*
Branca 99 (68) 40 (64) 0,003
Não-branca 42 (29) 22 (36)
Região
Sul 11 (8) 6 (10)
Sudeste 79 (54) 37 (60) 0,004 Centro-oeste 6 (4) 4 (6) Nordeste 49 (34) 15 (24)
* Refere-se à cor da pele definida pela mãe da criança; em 4 crianças a cor da pele não foi especificada. LLA –
leucemia linfoblástica aguda, LMA – leucemia mielóide aguda, NS – estatisticamente não significativo.
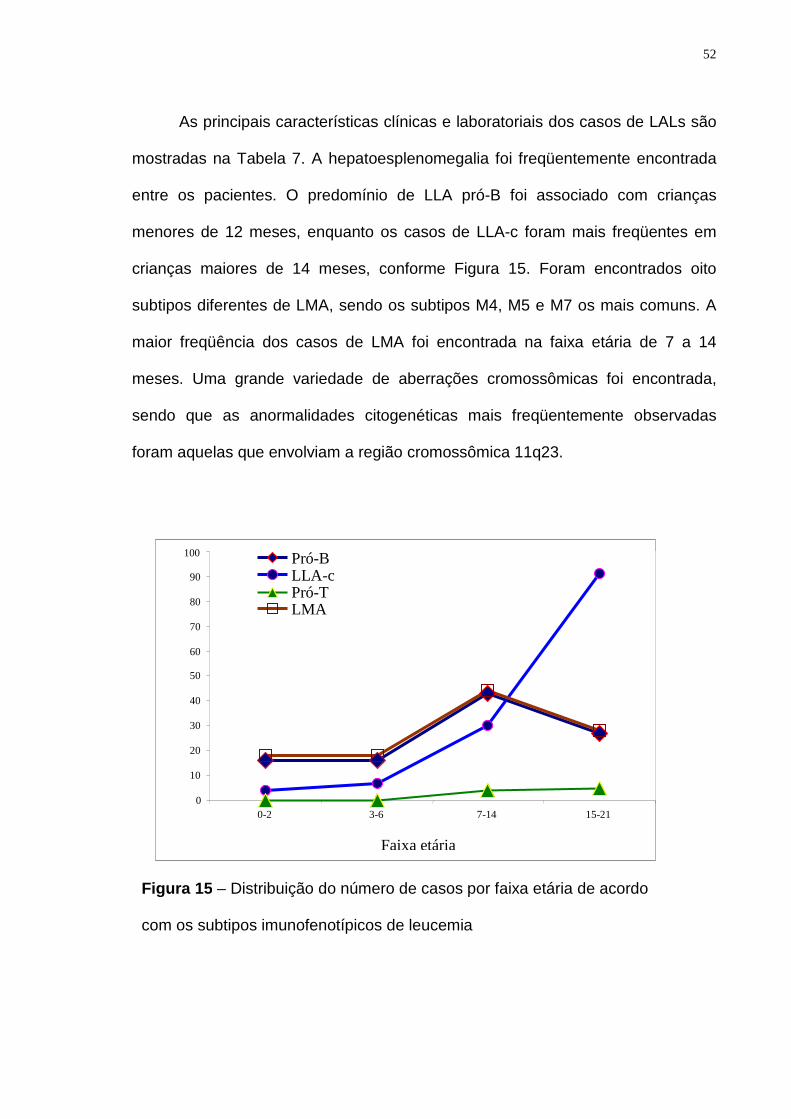
52
As principais características clínicas e laboratoriais dos casos de LALs são
mostradas na Tabela 7. A hepatoesplenomegalia foi freqüentemente encontrada
entre os pacientes. O predomínio de LLA pró-B foi associado com crianças
menores de 12 meses, enquanto os casos de LLA-c foram mais freqüentes em
crianças maiores de 14 meses, conforme Figura 15. Foram encontrados oito
subtipos diferentes de LMA, sendo os subtipos M4, M5 e M7 os mais comuns. A
maior freqüência dos casos de LMA foi encontrada na faixa etária de 7 a 14
meses. Uma grande variedade de aberrações cromossômicas foi encontrada,
sendo que as anormalidades citogenéticas mais freqüentemente observadas
foram aquelas que envolviam a região cromossômica 11q23.
0
10
20
30
40
50
60
70
80
90
100
0-2 3-6 7-14 15-21
Pró-B LLA-c Pró-T LMA
Figura 15 – Distribuição do número de casos por faixa etária de acordo
com os subtipos imunofenotípicos de leucemia
Faixa etária
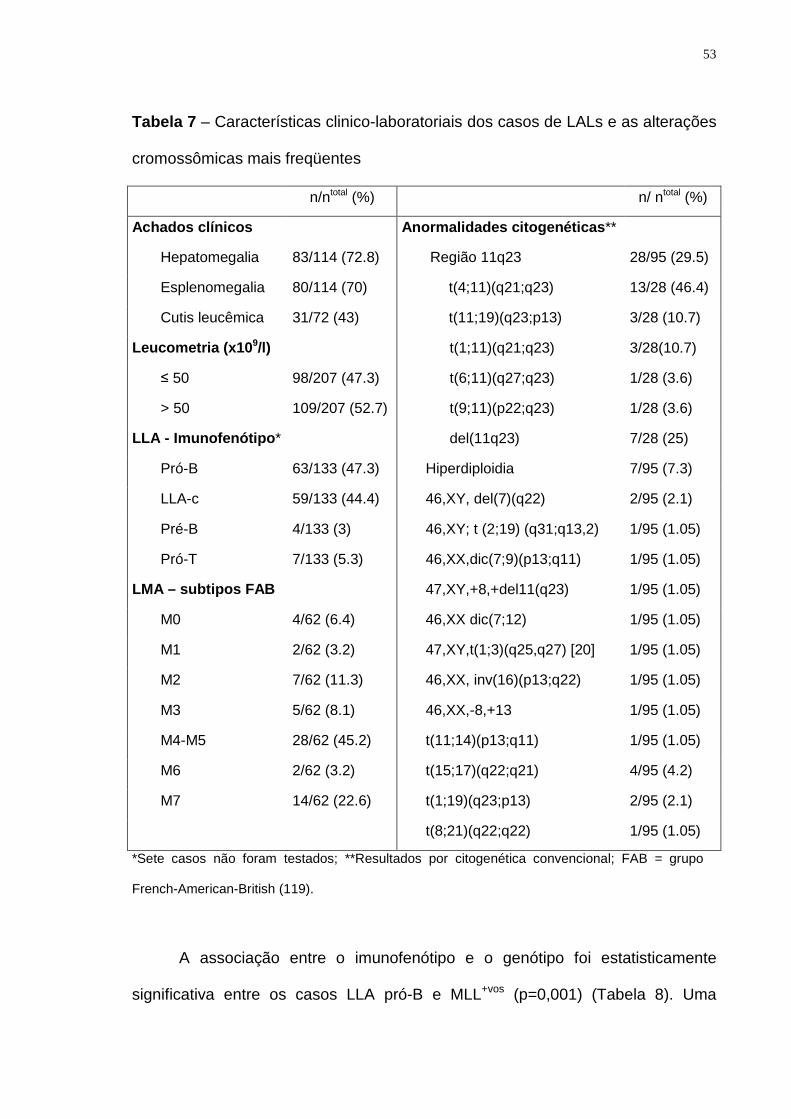
53
Tabela 7 – Características clinico-laboratoriais dos casos de LALs e as alterações
cromossômicas mais freqüentes
n/ntotal (%) n/ ntotal (%)
Achados clínicos Anormalidades citogenéticas **
Hepatomegalia 83/114 (72.8) Região 11q23 28/95 (29.5)
Esplenomegalia 80/114 (70) t(4;11)(q21;q23) 13/28 (46.4)
Cutis leucêmica 31/72 (43) t(11;19)(q23;p13) 3/28 (10.7)
Leucometria (x10 9/l) t(1;11)(q21;q23) 3/28(10.7)
≤ 50 98/207 (47.3) t(6;11)(q27;q23) 1/28 (3.6)
> 50 109/207 (52.7) t(9;11)(p22;q23) 1/28 (3.6)
LLA - Imunofenótipo * del(11q23) 7/28 (25)
Pró-B 63/133 (47.3) Hiperdiploidia 7/95 (7.3)
LLA-c 59/133 (44.4) 46,XY, del(7)(q22) 2/95 (2.1)
Pré-B 4/133 (3) 46,XY; t (2;19) (q31;q13,2) 1/95 (1.05)
Pró-T 7/133 (5.3) 46,XX,dic(7;9)(p13;q11) 1/95 (1.05)
LMA – subtipos FAB 47,XY,+8,+del11(q23) 1/95 (1.05)
M0 4/62 (6.4) 46,XX dic(7;12) 1/95 (1.05)
M1 2/62 (3.2) 47,XY,t(1;3)(q25,q27) [20] 1/95 (1.05)
M2 7/62 (11.3) 46,XX, inv(16)(p13;q22) 1/95 (1.05)
M3 5/62 (8.1) 46,XX,-8,+13 1/95 (1.05)
M4-M5 28/62 (45.2) t(11;14)(p13;q11) 1/95 (1.05)
M6 2/62 (3.2) t(15;17)(q22;q21) 4/95 (4.2)
M7 14/62 (22.6) t(1;19)(q23;p13) 2/95 (2.1)
t(8;21)(q22;q22) 1/95 (1.05)
*Sete casos não foram testados; **Resultados por citogenética convencional; FAB = grupo
French-American-British (119).
A associação entre o imunofenótipo e o genótipo foi estatisticamente
significativa entre os casos LLA pró-B e MLL+vos (p=0,001) (Tabela 8). Uma
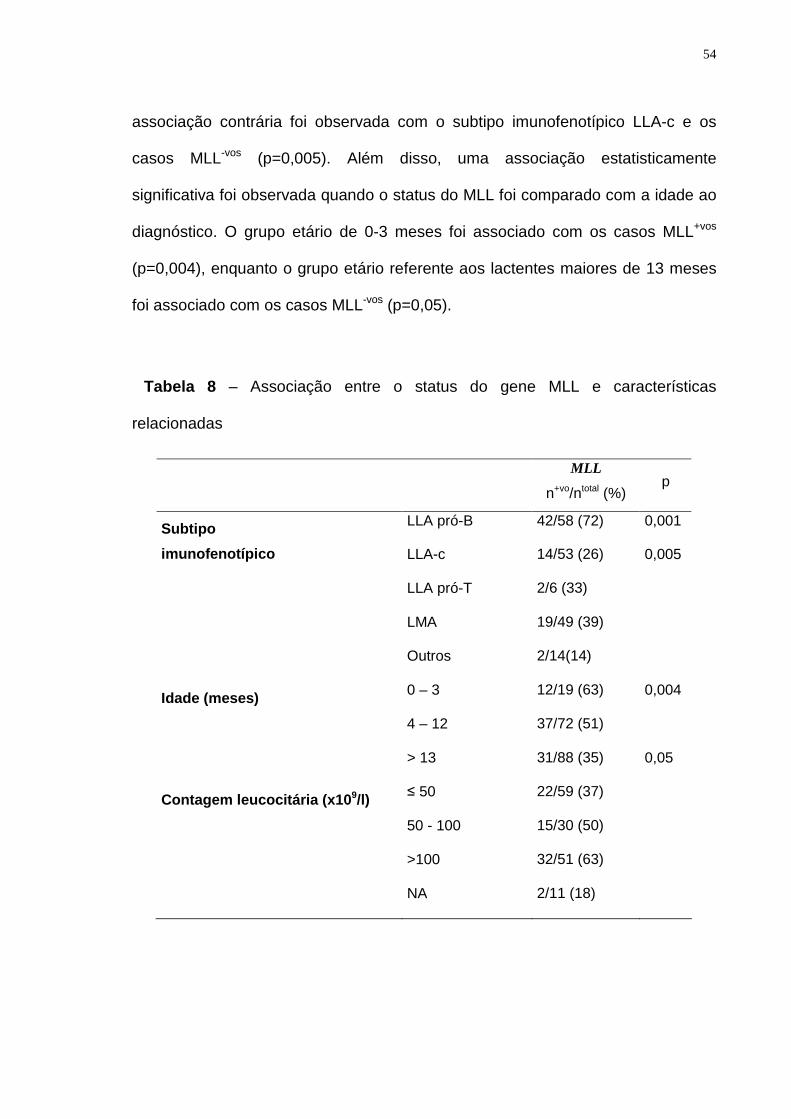
54
associação contrária foi observada com o subtipo imunofenotípico LLA-c e os
casos MLL-vos (p=0,005). Além disso, uma associação estatisticamente
significativa foi observada quando o status do MLL foi comparado com a idade ao
diagnóstico. O grupo etário de 0-3 meses foi associado com os casos MLL+vos
(p=0,004), enquanto o grupo etário referente aos lactentes maiores de 13 meses
foi associado com os casos MLL-vos (p=0,05).
Tabela 8 – Associação entre o status do gene MLL e características
relacionadas
MLL
n+vo/ntotal (%) p
Subtipo LLA pró-B 42/58 (72) 0,001
imunofenotípico LLA-c 14/53 (26) 0,005
LLA pró-T 2/6 (33)
LMA 19/49 (39)
Outros 2/14(14)
Idade (meses) 0 – 3 12/19 (63) 0,004
4 – 12 37/72 (51)
> 13 31/88 (35) 0,05
Contagem leucocitária (x10 9/l) ≤ 50 22/59 (37)
50 - 100 15/30 (50)
>100 32/51 (63)
NA 2/11 (18)
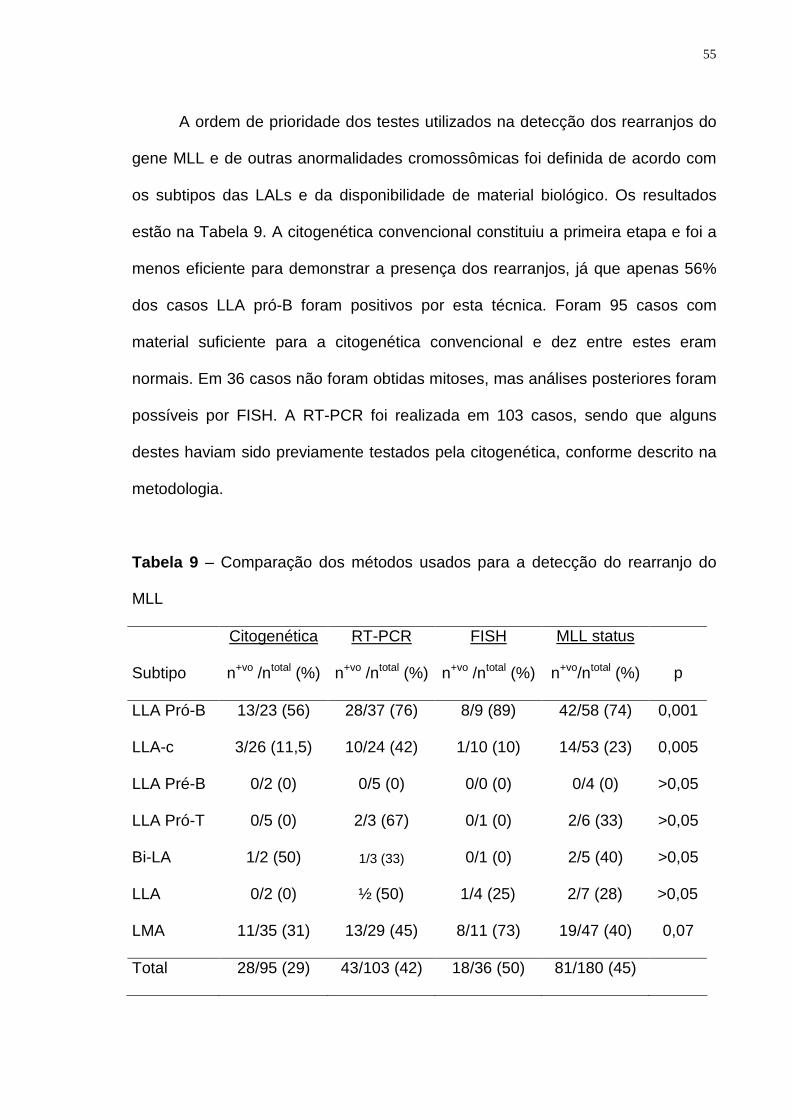
55
A ordem de prioridade dos testes utilizados na detecção dos rearranjos do
gene MLL e de outras anormalidades cromossômicas foi definida de acordo com
os subtipos das LALs e da disponibilidade de material biológico. Os resultados
estão na Tabela 9. A citogenética convencional constituiu a primeira etapa e foi a
menos eficiente para demonstrar a presença dos rearranjos, já que apenas 56%
dos casos LLA pró-B foram positivos por esta técnica. Foram 95 casos com
material suficiente para a citogenética convencional e dez entre estes eram
normais. Em 36 casos não foram obtidas mitoses, mas análises posteriores foram
possíveis por FISH. A RT-PCR foi realizada em 103 casos, sendo que alguns
destes haviam sido previamente testados pela citogenética, conforme descrito na
metodologia.
Tabela 9 – Comparação dos métodos usados para a detecção do rearranjo do
MLL
Citogenética RT-PCR FISH MLL status
Subtipo n+vo /ntotal (%) n+vo /ntotal (%) n+vo /ntotal (%) n+vo/ntotal (%) p
LLA Pró-B 13/23 (56) 28/37 (76) 8/9 (89) 42/58 (74) 0,001
LLA-c 3/26 (11,5) 10/24 (42) 1/10 (10) 14/53 (23) 0,005
LLA Pré-B 0/2 (0) 0/5 (0) 0/0 (0) 0/4 (0) >0,05
LLA Pró-T 0/5 (0) 2/3 (67) 0/1 (0) 2/6 (33) >0,05
Bi-LA 1/2 (50) 1/3 (33) 0/1 (0) 2/5 (40) >0,05
LLA 0/2 (0) ½ (50) 1/4 (25) 2/7 (28) >0,05
LMA 11/35 (31) 13/29 (45) 8/11 (73) 19/47 (40) 0,07
Total 28/95 (29) 43/103 (42) 18/36 (50) 81/180 (45)

56
No total, 61 pacientes com LLA-pB foram testados por RT-PCR para
verificar a presença da fusão gênica TEL/AML1. Dentre estes, nove (15%) foram
positivos e tinham uma alta concordância com o imunofenótipo LLA-c (sete
casos). Em 23 casos LLA-pB foi realizada a RT-PCR para a detecção da fusão
gênica E2A/PBX1. Foram quatro casos positivos e todos apresentavam o
imunofenótipo típico, caracterizando o estágio pré-B da diferenciação, que é
definido pela expressão citoplasmática mas não de superfície da IgM. Dois foram
diagnosticados com a translocação cromossômica t(1;19)(q23;p13) tanto pela
citogenética convencional quanto por RT-PCR. Onze casos de LMA foram
investigados para a presença da fusão gênica AML1/ETO e, dois casos positivos
foram encontrados. Um deles também teve a translocação detectada por
citogenética convencional. A Tabela 10 sumariza os resultados finais destas
fusões gênicas de acordo com a faixa etária.
Tabela 10 – Sumário das translocações cromossômicas de acordo com a faixa
etária
0-12 months 13-23 months
Fusão gênica n+vo /ntotal (%) n+vo /ntotal (%) p
MLL 50/86 (58%) 31/78 (40%) 0,011
TEL/AML1 4/29 (14%) 5/32 (16%) 1,000
E2A/PBX1 1/10 (10%) 3/13 (23%) 0,604
AML1/ETO 1/6 (17%) 2/5 (40%) 0,545

57
Esta série de casos de LALs apresentou alguns resultados incomuns
quanto aos achados da citogenética convencional e molecular (Tabela 11). No
total, foram cinco casos MLL+vos com imunofenótipos atípicos. Dois entre estes
foram diagnosticados com LLA pró-T. Um menino de 14 meses de idade
apresentava infiltração da MO com linfoblastos caracterizados por células
CD7+/cCD3+, enquanto os CD19, CD10 e os marcadores mielóides eram
negativos.
Tabela 11 – Características dos casos raros de LAL
Sexo Idade
(meses)
Leuco
(x109/l) Subtipo Citogenética convencional e molecular
1 M 14 57.1 LLA Pró-T del(11)(q23); MLL/AF4+vo
2 F 20 330 LLA Pró-T t(4;11)(q21;q23); MLL/AF4+ vo
3 M 12 26.1 LMA-M7 del(11)(q23); MLL+ vo
4 F 21 2.7 LMA-M7 MLL/AF4+ vo
5 F 21 107.1 LMA-M7 t(11;19)(q23;p13); MLL/ENL+ vo
6 M 1 7.6 LLA-c TEL/AML1+ vo; MLL- vo
7 F 3 132.5 LLA-c TEL/AML1+ vo; MLL/AF4- vo
8 M 7 152.3 LLA-c TEL/AML1+ vo; MLL/AF4- vo
9 F 5 140.1 ND* TEL/AML1+ vo; MLL/AF4- vo
10 F 14 150.1 LLA Pré-B t(1;19)(q23;p13); E2A/PBX1+ vo
11 M 19 82.1 LLA Pré-B E2A/PBX1+ vo
12 F 22 55.7 LLA Pré-B E2A/PBX1+ vo
13 F 10 20.8 LLA Pré-B t(1;19)(q23;p13); E2A/PBX1+ vo
14 M 14 3.9 LLA-c 46,XY,del(7)(q22), t(9;11)(p22;q23)
15 F 20 5.32 LMA-M2 t(8;21)(q22;q22)
16 M 18 36.5 LMA -M2 AML1/ETO+ vo
17 F 14 9.5 LMA -M4 46,XX, inv(16)(p13;q22)
18 M 16 10.9 LMA -M3 t(15;17)(q22;q21); PML/RARA+ vo
19 M 14 78.9 LMA -M3 t (15,17)(q22;q21)
20 M 17 5.1 LMA -M3 t(15;17)(q22;q21)
*ND= subtipo imunofenotípico não definido, porque o CD10 não foi usado;
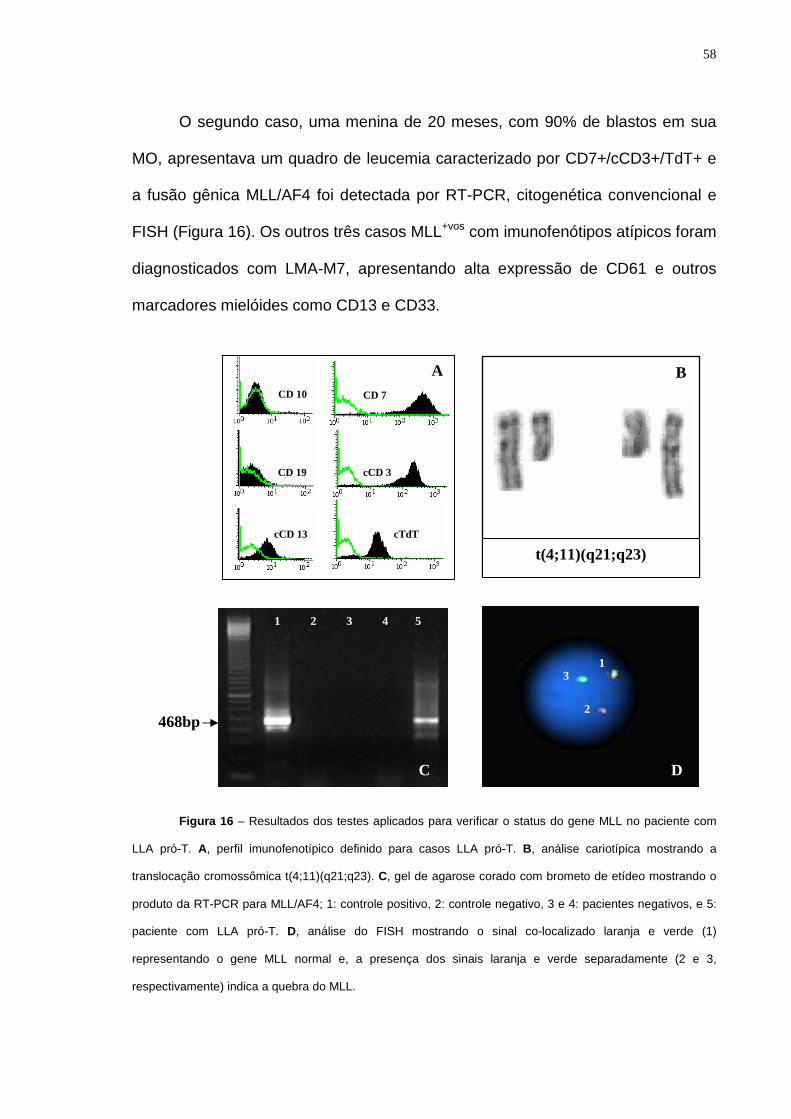
58
O segundo caso, uma menina de 20 meses, com 90% de blastos em sua
MO, apresentava um quadro de leucemia caracterizado por CD7+/cCD3+/TdT+ e
a fusão gênica MLL/AF4 foi detectada por RT-PCR, citogenética convencional e
FISH (Figura 16). Os outros três casos MLL+vos com imunofenótipos atípicos foram
diagnosticados com LMA-M7, apresentando alta expressão de CD61 e outros
marcadores mielóides como CD13 e CD33.
Figura 16 – Resultados dos testes aplicados para verificar o status do gene MLL no paciente com
LLA pró-T. A, perfil imunofenotípico definido para casos LLA pró-T. B, análise cariotípica mostrando a
translocação cromossômica t(4;11)(q21;q23). C, gel de agarose corado com brometo de etídeo mostrando o
produto da RT-PCR para MLL/AF4; 1: controle positivo, 2: controle negativo, 3 e 4: pacientes negativos, e 5:
paciente com LLA pró-T. D, análise do FISH mostrando o sinal co-localizado laranja e verde (1)
representando o gene MLL normal e, a presença dos sinais laranja e verde separadamente (2 e 3,
respectivamente) indica a quebra do MLL.
468bp
1 2 3 4 5
C
CD 10
CD 19
cCD 13
CD 7
cCD 3
cTdT
A B
t(4;11)(q21;q23)
D
3
2
1

59
No grupo das LLAs, duas crianças com hiperdiploidia eram menores do
que 16 meses de idade. Entre os lactentes com LMA, dois casos apresentavam
trissomia do cromossomo 8 e uma deleção na região 11q23. Deleções na região
cromossômica 11q23 foram encontradas em outros sete casos. Entre estes, cinco
eram MLL+vo e dois eram MLL-vo. Os casos positivos foram confirmados somente
por RT-PCR em dois pacientes, somente por FISH em outros dois pacientes e,
tanto por RT-PCR como por FISH em um dos pacientes. Um paciente MLL+vo
[t(9;11)(p22;q23)] também continha uma anormalidade cariotípica adicional,
caracterizada por del(7)(q22).
Uma menina e um menino, 18 e 20 meses respectivamente, com LMA-M2
de acordo com a classificação FAB (119) e, com o imunofenótipo definido por a-
MPO/CD33/CD19+ tiveram a translocação t(8;21)(q22;q22) detectada por
citogenética convencional e RT-PCR. A citogenética convencional ainda revelou a
presença da translocação cromossômica t(15;17)(q22;q21), resultando na fusão
gênica PML/RARα em quatro amostras de LAL. As idades ao diagnóstico eram
14, 16, 17 e 21 meses e todos apresentavam o subtipo LMA-M3 (123). Por fim,
uma garota de 14 meses com LMA-M4, apresentava o cariótipo
46,XX,inv(16)(p13;q22).

60
6. DISCUSSÃO
O risco da leucemia infantil reflete uma combinação de fatores que incluem
a predisposição hereditária, as exposições exógenas a agentes com potencial
leucemogênico e, eventos ao acaso (98). A ocorrência de alterações
cromossômicas e moleculares está diretamente associada a estes fatores. Estas
alterações genéticas tornaram-se marcadores de diagnóstico e também são
importantes para o conhecimento da patogênese da leucemia. O grupo de LAL
tem sido especialmente importante na compreensão da etiologia das leucemias.
Neste contexto, as leucemias de novo restritas aos lactentes com fusões do gene
MLL merecem uma atenção especial (34,82,98).
Em estudo colaborativo internacional anterior, foi realizada uma
investigação do tipo caso-controle, a fim de testar a hipótese de que certas
exposições ambientais poderiam aumentar o risco de LAL com rearranjos do MLL.
Vários fatores de exposição durante a gravidez foram identificados como
possíveis causadores das fusões do MLL e, conseqüentemente das LALs. As
taxas de risco aumentado foram encontradas somente nas leucemias com MLL+vo:
OR= 2.31 para drogas que danificam o ADN (p=0.03), e OR= 9.68 para o uso de
pesticidas (p=0.003). Neste estudo, no entanto, o desconhecimento do status do
gene MLL em alguns casos reduziu significativamente a força estatística dos
resultados (25). Este fato levou a realização de um amplo estudo em uma coorte
brasileira de LAL com os seguintes objetivos: (i) conduzir um estudo
imunofenotípico e molecular em crianças menores de dois anos de idade com LA,
para determinar as associações com fatores de risco selecionados em LALs de
diferentes regiões do Brasil; (ii) caracterizar as anormalidades moleculares

61
freqüentemente encontradas em LALs no Brasil. Os resultados referentes aos
estudos moleculares estão aqui descritos. Os resultados preliminares a respeito
dos fatores de risco associados com marcadores moleculares e com as
exposições maternas transplacentárias destes casos apontaram a exposição a
hormônios como um fator de risco proeminente (124).
Entre outras muitas técnicas disponíveis para a detecção de anormalidades
cromossômicas, a análise cariotípica clássica, RT-PCR e FISH são especialmente
responsáveis pelos avanços nas abordagens moleculares para a avaliação
biológica das LAs. Como cada técnica tem suas vantagens e desvantagens,
existe uma necessidade de combinar métodos para realizar uma análise confiável
do status do gene MLL em um estudo exploratório complexo. Por exemplo, a
citogenética que é plausível por proporcionar uma visão geral dos esfregaços
cromossômicos não é tão sensível como os métodos de PCR. A análise
cariotípica requer cromossomos em metáfase e, as amostras de MO de lactentes
são às vezes insuficientes. A análise por RT-PCR simples de genes quiméricos
específicos é rápida, mas detecta somente uma translocação por reação. A
análise por FISH pode ser usada para revelar se o MLL está rearranjado, mas é
um método limitado para a detecção de eventos raros, e não fornece nenhuma
informação sobre a fusão gênica específica gerada. Esta desvantagem pode ter
implicações clínicas já que diferenças no prognóstico têm sido descritas
dependendo do gene parceiro do MLL. Além disso, é uma técnica cara para um
país com outras prioridades como o Brasil, tornando a combinação de métodos
uma alternativa bastante plausível.

62
A LAL freqüentemente se apresenta com alta contagem leucocitária e um
imunofenótipo com o CD10 negativo. Na maioria dos casos (85-95%) de LALs os
rearranjos do gene MLL na região 11q23 estão presentes nos casos de LLA e
decresce nos casos de LMA (34,36). Estas características biológicas esperadas
também foram encontradas neste estudo, que mostrou os rearranjos do MLL em
ambos os subgrupos de LA, respeitando as diferentes proporções entre LLA e
LMA. Os casos MLL+vos foram estatisticamente associados ao grupo etário 0-3
meses (p = 0,004), ao subtipo LLA pró-B (p = 0,001) e, a altas contagens
leucocitárias (p = 0,009). É importante mencionar que uma porcentagem relevante
dos casos MLL+vos foi encontrada no grupo etário de 12-23 meses (Tabela 8),
sustentando nossa escolha ao incluir estas crianças neste estudo. No outro
extremo, os casos MLL-vos foram estatisticamente associados ao grupo etário >14
meses (p = 0,05) e ao imunofenótipo LLA-c (p = 0,005).
Em relação à positividade dos rearranjos do MLL, este estudo encontrou
uma freqüência menor do que aquela freqüentemente descrita na literatura (85-
95%). Foram 81 casos positivos entre 180 casos analisados. No entanto,
devemos considerar que este estudo incluiu casos de diversos subtipos
imunofenotípicos, havendo praticamente o mesmo número de casos de LLA pró-B
e LLA-comum. Além disso, a faixa etária analisada foi de 0 a 23 meses e,
conforme visto na figura 17, a partir dos 12 meses de idade, a positividade do
MLL decai significativamente.
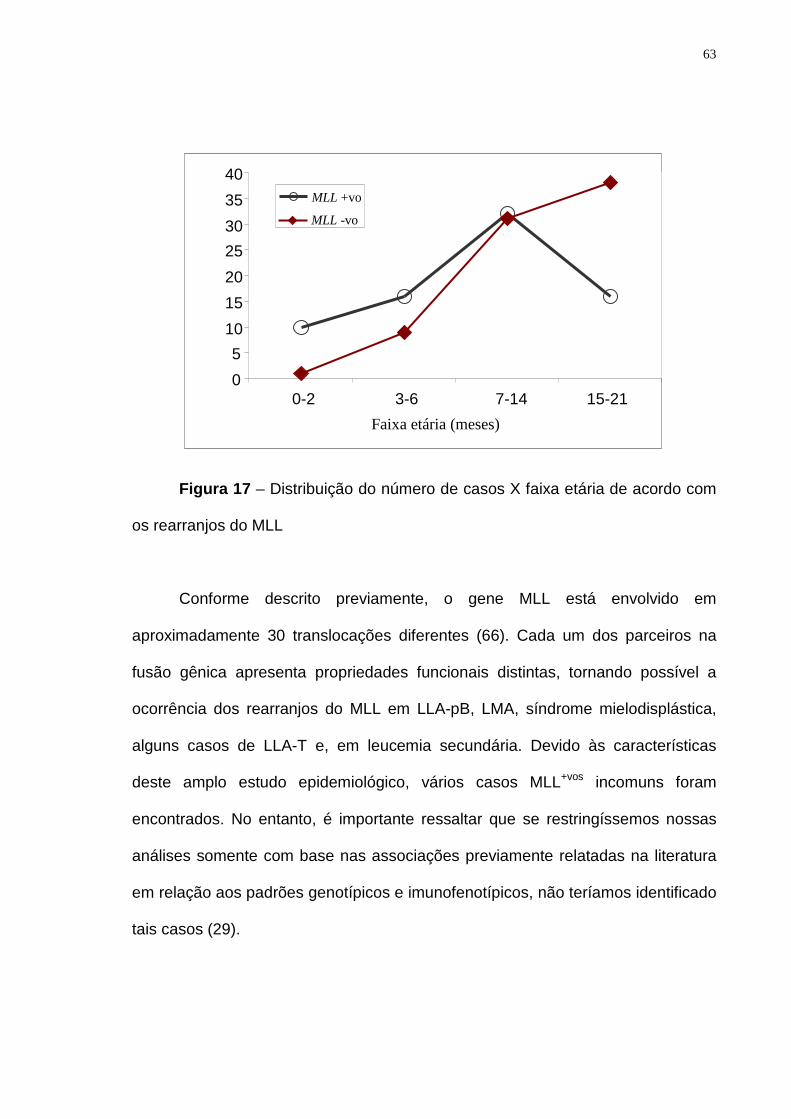
63
Figura 17 – Distribuição do número de casos X faixa etária de acordo com
os rearranjos do MLL
Conforme descrito previamente, o gene MLL está envolvido em
aproximadamente 30 translocações diferentes (66). Cada um dos parceiros na
fusão gênica apresenta propriedades funcionais distintas, tornando possível a
ocorrência dos rearranjos do MLL em LLA-pB, LMA, síndrome mielodisplástica,
alguns casos de LLA-T e, em leucemia secundária. Devido às características
deste amplo estudo epidemiológico, vários casos MLL+vos incomuns foram
encontrados. No entanto, é importante ressaltar que se restringíssemos nossas
análises somente com base nas associações previamente relatadas na literatura
em relação aos padrões genotípicos e imunofenotípicos, não teríamos identificado
tais casos (29).
0
5
10
15
20
25
30
35
40
0-2 3-6 7-14 15-21
Faixa etária (meses)
MLL +vo
MLL -vo

64
A fusão gênica TEL/AML1 foi descrita como sendo exclusivamente
associada ao subtipo LLA-c e isto foi confirmado neste estudo. Sua freqüência de
ocorrência está em torno de 25% das leucemias pediátricas de células
precursoras B. O pico de ocorrência de LLA-c com a fusão gênica TEL/AML1 está
em 4 anos de idade (variando entre 2 e 12 anos de idade) (70). O rastreamento
realizado neste estudo revelou quatro casos positivos para TEL/AML1 em
pacientes menores do que um ano de idade. Os estudos retrospectivos de
marcadores de LA indicam que a maioria das leucemias pediátricas
provavelmente têm origem intra-uterina através de alterações cromossômicas
(26,91). Estas incluem as fusões gênicas do MLL, TEL/AML1 e AML1/ETO. Ao
contrário das fusões que envolvem o MLL, que poderiam ser suficientes para
causar a leucemia por si só (53,64), os estudos em gêmeos com LA indicam que
a fusão gênica TEL/AML1, normalmente vista em LLA-c, requer alterações
genéticas pós-natais adicionais para gerar a leucemia (28,79). Nossa suposição é
que nestes casos de LAL com TEL/AML1+vo, os fatores de risco aos quais as
crianças foram expostas durante a gravidez foram suficientes para gerar estas
alterações genéticas adicionais.
Estudos dos marcadores de clonalidade IGH mostraram 100% de
positividade para a lesão genética nos esfregaços sangüíneos neonatais
(125,126). Nós relatamos sete casos de LAL, todos LLA-c, apresentando
hiperdiploidia em seus cariótipos. Uma possível exceção no que diz respeito à
origem intra-uterina é a fusão E2A/PBX1, considerando-se que a maioria dos
esfregaços sangüíneos neonatais é negativa para esta translocação. Mesmo
Embora rara, esta lesão foi descrita nos esfregaços sangüíneos neonatais com

65
uma freqüência em torno de 10% (90). Provavelmente duas meninas da nossa
coorte de estudo figuram nesta baixa porcentagem, já que elas tinham 10 e 14
meses ao diagnóstico. A presença e a descrição de eventos secundários em tais
casos têm sido investigadas buscando-se por associações que poderiam estar
envolvidas na leucemogênese.
Outros achados da citogenética convencional incluem deleções na região
cromossômica 11q23 (n=5) e deleções no cromossomo 7 (n=2). Embora as
translocações sejam as anormalidades mais comuns, as deleções também podem
ocorrer. A LAL está freqüentemente associada com anormalidades envolvendo
esta região, outros casos perinatais de LA mostrando estas aberrações já foram
descritos (127).
Uma outra fusão gênica que provavelmente também se origina in utero
deriva da translocação t(8;21)(q22;q22) e está associada com a LMA-M2 de novo,
com uma incidência relativamente maior em pacientes jovens (33). Em LMAs, há
também o relato de um caso com esfregaço neonatal positivo para PML/RARA e,
um outro positivo para CBFβ/MYH11 (128). Dois entre os 11 casos LMA-M2
(18%) pesquisados neste grupo para a fusão AML1/ETO foram positivos. Além
disso, três casos interessantes com o transcrito PML/RARA formado em
conseqüência da translocação t(15;17)(q22;q21) foram encontrados. Todos
apresentavam uma morfologia tipicamente do subtipo LMA-M3. Estes casos
foram discutidos com relação a uma maior incidência de LMA-M3 previamente
vista em estudos na população latina da Europa e dos Estados Unidos (123). Os
estudos epidemiológicos demonstraram que a LMA-M3 raramente ocorre em
crianças menores de dois anos de idade. Entretanto, uma alta freqüência de LMA-

66
M3 em crianças latinas levou à hipótese de que a leucemia promielocítica poderia
estar relacionada com razões étnicas e/ou com exposições a fatores ambientais
específicos.
Este trabalho contribuiu ainda com os resultados do estudo epidemiológico
que investigou o comportamento materno (exposições durante a gravidez) através
do Grupo Brasileiro de Estudo Colaborativo da Leucemia Aguda de Lactente,
fornecendo a caracterização molecular de um número maior de casos de
lactentes, cujas mães responderam ao questionário sobre os fatores de risco
ambientais (129).
Os dados apresentados acima também contribuem com casos novos e
interessantes sobre a LAL. Até o momento, os estudos têm contemplado
associações de exposição somente relacionadas às anormalidades do gene MLL.
O presente estudo sustenta a idéia de que o tempo de exposição necessário para
gerar as lesões genéticas e a leucemia difere entre os casos e, reforça a noção
de que as LALs com fusões do gene MLL têm uma etiologia única que
possivelmente envolve a exposição transplacentária a compostos químicos que
danificam o ADN. Além disso, nossos resultados sugerem que não somente os
rearranjos do MLL como também outras anormalidades gênicas ocorrem antes do
nascimento e podem contribuir para a leucemogênese. Assim, os hábitos
maternos devem ser cuidadosamente investigados para que os possíveis fatores
que causam o surgimento precoce da LA sejam compreendidos e evitados.

67
7. CONCLUSÕES
� A freqüência de positividade do gene MLL nesta coorte foi de 45%,
ressaltando-se que a faixa etária foi de 0 – 23 meses e que diferentes
subtipos de LLA e LMA foram incluídos;
� Uma alta prevalência de casos com MLL rearranjado foi encontrada em
crianças com idade inferior a 3 meses (63%). Por outro lado, nas crianças
com idade igual ou superior a 13 meses a prevalência dos rearranjos do
MLL foi de 35%, superior aos 5% encontrados nas LAs em geral;
� Embora pouco freqüente nos lactentes, entre as LLAs de células
precursoras B foi encontrada a presença da fusão TEL/AML1 restrita ao
subtipo LLA-comum (CD10+);
� O período e os fatores de exposição que levaram à leucemia dos casos
TEL/AML1 positivos devem ser melhor estudados, pois a latência foi
bastante curta, levando-se em consideração a história natural de no
mínimo dois eventos para a ocorrência deste subtipo de leucemia;
� Os casos atípicos aqui descritos sugerem que a grande relação entre
imunofenótipo e genótipo não é absoluta, pois LLA-T e LMA-M7 podem ter
rearranjos envolvendo o MLL;
� Para um estudo com base epidemiológica, a combinação de métodos
diagnósticos para a verificação do status do gene MLL é uma alternativa
bastante plausível.

68
8. REFERÊNCIAS 1. ZAGO MA, FALCAO RP, PASQUINI R. HEMATOLOGIA FUNDAMENTOS
E PRÁTICA. SÃO PAULO: ATHENEU, 2001; PP. 15-73.
2. DZIERZAK E, MEDVINSKY A& DE BM (1998). QUALITATIVE AND QUANTITATIVE ASPECTS OF HAEMATOPOIETIC CELL DEVELOPMENT IN THE MAMMALIAN EMBRYO. IMMUNOL TODAY, 19: 228-236.
3. OGAWA M (1993). DIFFERENTIATION AND PROLIFERATION OF HEMATOPOIETIC STEM CELLS. BLOOD, 81: 2844-2853.
4. SPANGRUDE GJ, HEIMFELD S& WEISSMAN IL (1988). PURIFICATION AND CHARACTERIZATION OF MOUSE HEMATOPOIETIC STEM CELLS. SCIENCE, 241: 58-62.
5. TAICHMAN RS (2005). BLOOD AND BONE: TWO TISSUES WHOSE FATES ARE INTERTWINED TO CREATE THE HEMATOPOIETIC STEM-CELL NICHE. BLOOD, 105: 2631-2639.
6. HITZLER JK, ZIPURSKY A (2005). ORIGINS OF LEUKAEMIA IN CHILDREN WITH DOWN SYNDROME. NAT REV CANCER, 5: 11-20.
7. ORKIN SH (2000). DIVERSIFICATION OF HAEMATOPOIETIC STEM CELLS TO SPECIFIC LINEAGES. NAT REV GENET, 1: 57-64.
8. SHIVDASANI RA, ORKIN SH (1996). THE TRANSCRIPTIONAL CONTROL OF HEMATOPOIESIS. BLOOD, 87: 4025-4039.
9. BUSKE C, HUMPHRIES RK (2000). HOMEOBOX GENES IN LEUKEMOGENESIS. INT J HEMATOL, 71: 301-308.
10. GREAVES MF (1988). SPECULATIONS ON THE CAUSE OF CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA. LEUKEMIA, 2: 120-125.
11. HANAHAN D, WEINBERG RA (2000). THE HALLMARKS OF CANCER. CELL, 100: 57-70.
12. ROWLEY JD (1998). THE CRITICAL ROLE OF CHROMOSOME TRANSLOCATIONS IN HUMAN LEUKEMIAS. ANNU REV GENET, 32: 495-519.
13. ARMSTRONG SA, STAUNTON JE, SILVERMAN LB, PIETERS R, DEN BOER ML, MINDEN MD, SALLAN SE, LANDER ES, GOLUB TR& KORSMEYER SJ (2002). MLL TRANSLOCATIONS SPECIFY A DISTINCT GENE EXPRESSION PROFILE THAT DISTINGUISHES A UNIQUE LEUKEMIA. NAT GENET, 30: 41-47.

69
14. YEOH EJ, ROSS ME, SHURTLEFF SA, WILLIAMS WK, PATEL D, MAHFOUZ R, BEHM FG, RAIMONDI SC, RELLING MV, PATEL A, CHENG C, CAMPANA D, WILKINS D, ZHOU X, LI J, LIU H, PUI CH, EVANS WE, NAEVE C, WONG L& DOWNING JR (2002). CLASSIFICATION, SUBTYPE DISCOVERY, AND PREDICTION OF OUTCOME IN PEDIATRIC ACUTE LYMPHOBLASTIC LEUKEMIA BY GENE EXPRESSION PROFILING. CANCER CELL, 1: 133-143.
15. BENE MC, CASTOLDI G, KNAPP W, LUDWIG WD, MATUTES E, ORFAO A& VAN'T VEER MB (1995). PROPOSALS FOR THE IMMUNOLOGICAL CLASSIFICATION OF ACUTE LEUKEMIAS. EUROPEAN GROUP FOR THE IMMUNOLOGICAL CHARACTERIZATION OF LEUKEMIAS (EGIL). LEUKEMIA, 9: 1783-1786.
16. SANDLER DP, ROSS JA (1997). EPIDEMIOLOGY OF ACUTE LEUKEMIA IN CHILDREN AND ADULTS. SEMIN ONCOL, 24: 3-16.
17. BURROWS PD, COOPER MD (1997). B CELL DEVELOPMENT AND DIFFERENTIATION. CURR OPIN IMMUNOL, 9: 239-244.
18. LECLERCQ G, PLUM J (1996). THYMIC AND EXTRATHYMIC T CELL DEVELOPMENT. LEUKEMIA, 10: 1853-1859.
19. LETARTE M, VERA S, TRAN R, ADDIS JB, ONIZUKA RJ, QUACKENBUSH EJ, JONGENEEL CV& MCINNES RR (1988). COMMON ACUTE LYMPHOCYTIC LEUKEMIA ANTIGEN IS IDENTICAL TO NEUTRAL ENDOPEPTIDASE. J EXP MED, 168: 1247-1253.
20. RUDIN CM, THOMPSON CB (1998). B-CELL DEVELOPMENT AND MATURATION. SEMIN ONCOL, 25: 435-446.
21. SHORTMAN K, WU L (1996). EARLY T LYMPHOCYTE PROGENITORS. ANNU REV IMMUNOL, 14: 29-47.
22. LOWENBERG B, DOWNING JR& BURNETT A (1999). ACUTE MYELOID LEUKEMIA. N ENGL J MED, 341: 1051-1062.
23. VARDIMAN JW, HARRIS NL& BRUNNING RD (2002). THE WORLD HEALTH ORGANIZATION (WHO) CLASSIFICATION OF THE MYELOID NEOPLASMS. BLOOD, 100: 2292-2302.
24. STONE RM, O'DONNELL MR& SEKERES MA (2004). ACUTE MYELOID LEUKEMIA. HEMATOLOGY (AM SOC HEMATOL EDUC PROGRAM ), 98-117.
25. ALEXANDER FE, PATHEAL SL, BIONDI A, BRANDALISE S, CABRERA ME, CHAN LC, CHEN Z, CIMINO G, CORDOBA JC, GU LJ, HUSSEIN H, ISHII E, KAMEL AM, LABRA S, MAGALHAES IQ, MIZUTANI S, PETRIDOU E, DE OLIVEIRA MP, YUEN P, WIEMELS JL& GREAVES MF (2001). TRANSPLACENTAL CHEMICAL EXPOSURE AND RISK OF

70
INFANT LEUKEMIA WITH MLL GENE FUSION. CANCER RES, 61: 2542-2546.
26. GALE KB, FORD AM, REPP R, BORKHARDT A, KELLER C, EDEN OB& GREAVES MF (1997). BACKTRACKING LEUKEMIA TO BIRTH: IDENTIFICATION OF CLONOTYPIC GENE FUSION SEQUENCES IN NEONATAL BLOOD SPOTS. PROC NATL ACAD SCI U S A, 94: 13950-13954.
27. GREAVES MF (1997). AETIOLOGY OF ACUTE LEUKAEMIA. LANCET, 349: 344-349.
28. WIEMELS JL, FORD AM, VAN WERING ER, POSTMA A& GREAVES M (1999). PROTRACTED AND VARIABLE LATENCY OF ACUTE LYMPHOBLASTIC LEUKEMIA AFTER TEL-AML1 GENE FUSION IN UTERO. BLOOD, 94: 1057-1062.
29. HRUSAK O, PORWIT-MACDONALD A (2002). ANTIGEN EXPRESSION PATTERNS REFLECTING GENOTYPE OF ACUTE LEUKEMIAS. LEUKEMIA, 16: 1233-1258.
30. STASI R, DEL PG, VENDITTI A, BRUNO A, SUPPO G, ARONICA G, DI CG& PAPA G (1995). LINEAGE IDENTIFICATION OF ACUTE LEUKEMIAS: RELEVANCE OF IMMUNOLOGIC AND ULTRASTRUCTURAL TECHNIQUES. HEMATOL PATHOL, 9: 79-94.
31. EMERENCIANO M, BOSSA Y, ZANROSSO CW, ALENCAR D, CAMPOS M, DOBBIN J, CARRIÇO K& POMBO-DE-OLIVEIRA MS (2004). FREQÜÊNCIA DE IMUNOFENÓTIPOS ABERRANTES EM LEUCEMIAS AGUDAS. REVISTA BRASILEIRA DE CANCEROLOGIA, 50: 183-189.
32. MACINTYRE EA, DELABESSE E (1999). MOLECULAR APPROACHES TO THE DIAGNOSIS AND EVALUATION OF LYMPHOID MALIGNANCIES. SEMIN HEMATOL, 36: 373-389.
33. ANDRIEU V, RADFORD-WEISS I, TROUSSARD X, CHANE C, VALENSI F, GUESNU M, HADDAD E, VIGUIER F, DREYFUS F, VARET B, FLANDRIN G& MACINTYRE E (1996). MOLECULAR DETECTION OF T(8;21)/AML1-ETO IN AML M1/M2: CORRELATION WITH CYTOGENETICS, MORPHOLOGY AND IMMUNOPHENOTYPE. BR J HAEMATOL, 92: 855-865.
34. CHEN CS, SORENSEN PH, DOMER PH, REAMAN GH, KORSMEYER SJ, HEEREMA NA, HAMMOND GD& KERSEY JH (1993). MOLECULAR REARRANGEMENTS ON CHROMOSOME 11Q23 PREDOMINATE IN INFANT ACUTE LYMPHOBLASTIC LEUKEMIA AND ARE ASSOCIATED WITH SPECIFIC BIOLOGIC VARIABLES AND POOR OUTCOME. BLOOD, 81: 2386-2393.

71
35. CRIST WM, CARROLL AJ, SHUSTER JJ, BEHM FG, WHITEHEAD M, VIETTI TJ, LOOK AT, MAHONEY D, RAGAB A, PULLEN DJ& . (1990). POOR PROGNOSIS OF CHILDREN WITH PRE-B ACUTE LYMPHOBLASTIC LEUKEMIA IS ASSOCIATED WITH THE T(1;19)(Q23;P13): A PEDIATRIC ONCOLOGY GROUP STUDY. BLOOD, 76: 117-122.
36. SORENSEN PH, CHEN CS, SMITH FO, ARTHUR DC, DOMER PH, BERNSTEIN ID, KORSMEYER SJ, HAMMOND GD& KERSEY JH (1994). MOLECULAR REARRANGEMENTS OF THE MLL GENE ARE PRESENT IN MOST CASES OF INFANT ACUTE MYELOID LEUKEMIA AND ARE STRONGLY CORRELATED WITH MONOCYTIC OR MYELOMONOCYTIC PHENOTYPES. J CLIN INVEST, 93: 429-437.
37. VISWANATHA DS, CHEN I, LIU PP, SLOVAK ML, RANKIN C, HEAD DR& WILLMAN CL (1998). CHARACTERIZATION AND USE OF AN ANTIBODY DETECTING THE CBFBETA-SMMHC FUSION PROTEIN IN INV(16)/T(16;16)-ASSOCIATED ACUTE MYELOID LEUKEMIAS. BLOOD, 91: 1882-1890.
38. BEHM FG, SMITH FO, RAIMONDI SC, PUI CH& BERNSTEIN ID (1996). HUMAN HOMOLOGUE OF THE RAT CHONDROITIN SULFATE PROTEOGLYCAN, NG2, DETECTED BY MONOCLONAL ANTIBODY 7.1, IDENTIFIES CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIAS WITH T(4;11)(Q21;Q23) OR T(11;19)(Q23;P13) AND MLL GENE REARRANGEMENTS. BLOOD, 87: 1134-1139.
39. WUCHTER C, HARBOTT J, SCHOCH C, SCHNITTGER S, BORKHARDT A, KARAWAJEW L, RATEI R, RUPPERT V, HAFERLACH T, CREUTZIG U, DORKEN B& LUDWIG WD (2000). DETECTION OF ACUTE LEUKEMIA CELLS WITH MIXED LINEAGE LEUKEMIA (MLL) GENE REARRANGEMENTS BY FLOW CYTOMETRY USING MONOCLONAL ANTIBODY 7.1. LEUKEMIA, 14: 1232-1238.
40. GREAVES MF, WIEMELS J (2003). ORIGINS OF CHROMOSOME TRANSLOCATIONS IN CHILDHOOD LEUKAEMIA. NAT REV CANCER, 3: 639-649.
41. VAN DONGEN JJ, MACINTYRE EA, GABERT JA, DELABESSE E, ROSSI V, SAGLIO G, GOTTARDI E, RAMBALDI A, DOTTI G, GRIESINGER F, PARREIRA A, GAMEIRO P, DIAZ MG, MALEC M, LANGERAK AW, SAN MIGUEL JF& BIONDI A (1999). STANDARDIZED RT-PCR ANALYSIS OF FUSION GENE TRANSCRIPTS FROM CHROMOSOME ABERRATIONS IN ACUTE LEUKEMIA FOR DETECTION OF MINIMAL RESIDUAL DISEASE. REPORT OF THE BIOMED-1 CONCERTED ACTION: INVESTIGATION OF MINIMAL RESIDUAL DISEASE IN ACUTE LEUKEMIA. LEUKEMIA, 13: 1901-1928.

72
42. LUDWIG WD, RIEDER H, BARTRAM CR, HEINZE B, SCHWARTZ S, GASSMANN W, LOFFLER H, HOSSFELD D, HEIL G, HANDT S, HEYLL A, DIEDRICH H, FISCHER K, WEISS A, VOLKERS B, AYDEMIR U, FONATSCH C, GOKBUGET N, THIEL E& HOELZER D (1998). IMMUNOPHENOTYPIC AND GENOTYPIC FEATURES, CLINICAL CHARACTERISTICS, AND TREATMENT OUTCOME OF ADULT PRO-B ACUTE LYMPHOBLASTIC LEUKEMIA: RESULTS OF THE GERMAN MULTICENTER TRIALS GMALL 03/87 AND 04/89. BLOOD, 92: 1898-1909.
43. TKACHUK DC, KOHLER S& CLEARY ML (1992). INVOLVEMENT OF A HOMOLOG OF DROSOPHILA TRITHORAX BY 11Q23 CHROMOSOMAL TRANSLOCATIONS IN ACUTE LEUKEMIAS. CELL, 71: 691-700.
44. YU BD, HESS JL, HORNING SE, BROWN GA& KORSMEYER SJ (1995). ALTERED HOX EXPRESSION AND SEGMENTAL IDENTITY IN MLL-MUTANT MICE. NATURE, 378: 505-508.
45. YU BD, HANSON RD, HESS JL, HORNING SE& KORSMEYER SJ (1998). MLL, A MAMMALIAN TRITHORAX-GROUP GENE, FUNCTIONS AS A TRANSCRIPTIONAL MAINTENANCE FACTOR IN MORPHOGENESIS. PROC NATL ACAD SCI U S A, 95: 10632-10636.
46. HSIEH JJ, CHENG EH& KORSMEYER SJ (2003). TASPASE1: A THREONINE ASPARTASE REQUIRED FOR CLEAVAGE OF MLL AND PROPER HOX GENE EXPRESSION. CELL, 115: 293-303.
47. HSIEH JJ, ERNST P, ERDJUMENT-BROMAGE H, TEMPST P& KORSMEYER SJ (2003). PROTEOLYTIC CLEAVAGE OF MLL GENERATES A COMPLEX OF N- AND C-TERMINAL FRAGMENTS THAT CONFERS PROTEIN STABILITY AND SUBNUCLEAR LOCALIZATION. MOL CELL BIOL, 23: 186-194.
48. MILNE TA, BRIGGS SD, BROCK HW, MARTIN ME, GIBBS D, ALLIS CD& HESS JL (2002). MLL TARGETS SET DOMAIN METHYLTRANSFERASE ACTIVITY TO HOX GENE PROMOTERS. MOL CELL, 10: 1107-1117.
49. YOKOYAMA A, WANG Z, WYSOCKA J, SANYAL M, AUFIERO DJ, KITABAYASHI I, HERR W& CLEARY ML (2004). LEUKEMIA PROTO-ONCOPROTEIN MLL FORMS A SET1-LIKE HISTONE METHYLTRANSFERASE COMPLEX WITH MENIN TO REGULATE HOX GENE EXPRESSION. MOL CELL BIOL, 24: 5639-5649.
50. HESS JL, YU BD, LI B, HANSON R& KORSMEYER SJ (1997). DEFECTS IN YOLK SAC HEMATOPOIESIS IN MLL-NULL EMBRYOS. BLOOD, 90: 1799-1806.

73
51. YAGI H, DEGUCHI K, AONO A, TANI Y, KISHIMOTO T& KOMORI T (1998). GROWTH DISTURBANCE IN FETAL LIVER HEMATOPOIESIS OF MLL-MUTANT MICE. BLOOD, 92: 108-117.
52. PUI CH, EVANS WE (1998). ACUTE LYMPHOBLASTIC LEUKEMIA. N ENGL J MED, 339: 605-615.
53. AYTON PM, CLEARY ML (2001). MOLECULAR MECHANISMS OF LEUKEMOGENESIS MEDIATED BY MLL FUSION PROTEINS. ONCOGENE, 20: 5695-5707.
54. COLLINS EC, RABBITTS TH (2002). THE PROMISCUOUS MLL GENE LINKS CHROMOSOMAL TRANSLOCATIONS TO CELLULAR DIFFERENTIATION AND TUMOUR TROPISM. TRENDS MOL MED, 8: 436-442.
55. YANO T, NAKAMURA T, BLECHMAN J, SORIO C, DANG CV, GEIGER B& CANAANI E (1997). NUCLEAR PUNCTATE DISTRIBUTION OF ALL-1 IS CONFERRED BY DISTINCT ELEMENTS AT THE N TERMINUS OF THE PROTEIN. PROC NATL ACAD SCI U S A, 94: 7286-7291.
56. GU Y, ALDER H, NAKAMURA T, SCHICHMAN SA, PRASAD R, CANAANI O, SAITO H, CROCE CM& CANAANI E (1994). SEQUENCE ANALYSIS OF THE BREAKPOINT CLUSTER REGION IN THE ALL-1 GENE INVOLVED IN ACUTE LEUKEMIA. CANCER RES, 54: 2326-2330.
57. BROEKER PL, SUPER HG, THIRMAN MJ, POMYKALA H, YONEBAYASHI Y, TANABE S, ZELEZNIK-LE N& ROWLEY JD (1996). DISTRIBUTION OF 11Q23 BREAKPOINTS WITHIN THE MLL BREAKPOINT CLUSTER REGION IN DE NOVO ACUTE LEUKEMIA AND IN TREATMENT-RELATED ACUTE MYELOID LEUKEMIA: CORRELATION WITH SCAFFOLD ATTACHMENT REGIONS AND TOPOISOMERASE II CONSENSUS BINDING SITES. BLOOD, 87: 1912-1922.
58. FELIX CA (2001). LEUKEMIAS RELATED TO TREATMENT WITH DNA TOPOISOMERASE II INHIBITORS. MED PEDIATR ONCOL, 36: 525-535.
59. CIMINO G, RAPANOTTI MC, BIONDI A, ELIA L, LO CF, PRICE C, ROSSI V, RIVOLTA A, CANAANI E, CROCE CM, MANDELLI F& GREAVES M (1997). INFANT ACUTE LEUKEMIAS SHOW THE SAME BIASED DISTRIBUTION OF ALL1 GENE BREAKS AS TOPOISOMERASE II RELATED SECONDARY ACUTE LEUKEMIAS. CANCER RES, 57: 2879-2883.
60. ROSS JA, POTTER JD& ROBISON LL (1994). INFANT LEUKEMIA, TOPOISOMERASE II INHIBITORS, AND THE MLL GENE. J NATL CANCER INST, 86: 1678-1680.

74
61. GILLILAND DG (2002). MOLECULAR GENETICS OF HUMAN LEUKEMIAS: NEW INSIGHTS INTO THERAPY. SEMIN HEMATOL, 39: 6-11.
62. GREAVES MF (1996). INFANT LEUKAEMIA BIOLOGY, AETIOLOGY AND TREATMENT. LEUKEMIA, 10: 372-377.
63. MEGONIGAL MD, CHEUNG NK, RAPPAPORT EF, NOWELL PC, WILSON RB, JONES DH, ADDYA K, LEONARD DG, KUSHNER BH, WILLIAMS TM, LANGE BJ& FELIX CA (2000). DETECTION OF LEUKEMIA-ASSOCIATED MLL-GAS7 TRANSLOCATION EARLY DURING CHEMOTHERAPY WITH DNA TOPOISOMERASE II INHIBITORS. PROC NATL ACAD SCI U S A, 97: 2814-2819.
64. GREAVES MF, MAIA AT, WIEMELS JL& FORD AM (2003). LEUKEMIA IN TWINS: LESSONS IN NATURAL HISTORY. BLOOD, 102: 2321-2333.
65. SO CW, CLEARY ML (2003). COMMON MECHANISM FOR ONCOGENIC ACTIVATION OF MLL BY FORKHEAD FAMILY PROTEINS. BLOOD, 101: 633-639.
66. HARRISON CJ, CUNEO A, CLARK R, JOHANSSON B, LAFAGE-POCHITALOFF M, MUGNERET F, MOORMAN AV& SECKER-WALKER LM (1998). TEN NOVEL 11Q23 CHROMOSOMAL PARTNER SITES. EUROPEAN 11Q23 WORKSHOP PARTICIPANTS. LEUKEMIA, 12: 811-822.
67. ARMSTRONG SA, KUNG AL, MABON ME, SILVERMAN LB, STAM RW, DEN BOER ML, PIETERS R, KERSEY JH, SALLAN SE, FLETCHER JA, GOLUB TR, GRIFFIN JD& KORSMEYER SJ (2003). INHIBITION OF FLT3 IN MLL. VALIDATION OF A THERAPEUTIC TARGET IDENTIFIED BY GENE EXPRESSION BASED CLASSIFICATION. CANCER CELL, 3: 173-183.
68. SCHULTE CE, VON LM, STEINLEIN P, BEUG H& WIEDEMANN LM (2002). MLL-ENL COOPERATES WITH SCF TO TRANSFORM PRIMARY AVIAN MULTIPOTENT CELLS. EMBO J, 21: 4297-4306.
69. SHURTLEFF SA, BUIJS A, BEHM FG, RUBNITZ JE, RAIMONDI SC, HANCOCK ML, CHAN GC, PUI CH, GROSVELD G& DOWNING JR (1995). TEL/AML1 FUSION RESULTING FROM A CRYPTIC T(12;21) IS THE MOST COMMON GENETIC LESION IN PEDIATRIC ALL AND DEFINES A SUBGROUP OF PATIENTS WITH AN EXCELLENT PROGNOSIS. LEUKEMIA, 9: 1985-1989.
70. ROMANA SP, POIREL H, LECONIAT M, FLEXOR MA, MAUCHAUFFE M, JONVEAUX P, MACINTYRE EA, BERGER R& BERNARD OA (1995). HIGH FREQUENCY OF T(12;21) IN CHILDHOOD B-LINEAGE ACUTE LYMPHOBLASTIC LEUKEMIA. BLOOD, 86: 4263-4269.

75
71. BORKHARDT A, CAZZANIGA G, VIEHMANN S, VALSECCHI MG, LUDWIG WD, BURCI L, MANGIONI S, SCHRAPPE M, RIEHM H, LAMPERT F, BASSO G, MASERA G, HARBOTT J& BIONDI A (1997). INCIDENCE AND CLINICAL RELEVANCE OF TEL/AML1 FUSION GENES IN CHILDREN WITH ACUTE LYMPHOBLASTIC LEUKEMIA ENROLLED IN THE GERMAN AND ITALIAN MULTICENTER THERAPY TRIALS. ASSOCIAZIONE ITALIANA EMATOLOGIA ONCOLOGIA PEDIATRICA AND THE BERLIN-FRANKFURT-MUNSTER STUDY GROUP. BLOOD, 90: 571-577.
72. GOLUB TR, BARKER GF, STEGMAIER K& GILLILAND DG (1996). INVOLVEMENT OF THE TEL GENE IN HEMATOLOGIC MALIGNANCY BY DIVERSE MOLECULAR GENETIC MECHANISMS. CURR TOP MICROBIOL IMMUNOL, 211: 279-288.
73. SPECK NA, GILLILAND DG (2002). CORE-BINDING FACTORS IN HAEMATOPOIESIS AND LEUKAEMIA. NAT REV CANCER, 2: 502-513.
74. WANG LC, SWAT W, FUJIWARA Y, DAVIDSON L, VISVADER J, KUO F, ALT FW, GILLILAND DG, GOLUB TR& ORKIN SH (1998). THE TEL/ETV6 GENE IS REQUIRED SPECIFICALLY FOR HEMATOPOIESIS IN THE BONE MARROW. GENES DEV, 12: 2392-2402.
75. LORSBACH RB, DOWNING JR (2001). THE ROLE OF THE AML1 TRANSCRIPTION FACTOR IN LEUKEMOGENESIS. INT J HEMATOL, 74: 258-265.
76. HIEBERT SW, LUTTERBACH B& AMANN J (2001). ROLE OF CO-REPRESSORS IN TRANSCRIPTIONAL REPRESSION MEDIATED BY THE T(8;21), T(16;21), T(12;21), AND INV(16) FUSION PROTEINS. CURR OPIN HEMATOL, 8: 197-200.
77. ZELENT A, GREAVES M& ENVER T (2004). ROLE OF THE TEL-AML1 FUSION GENE IN THE MOLECULAR PATHOGENESIS OF CHILDHOOD ACUTE LYMPHOBLASTIC LEUKAEMIA. ONCOGENE, 23: 4275-4283.
78. RAYNAUD S, CAVE H, BAENS M, BASTARD C, CACHEUX V, GROSGEORGE J, GUIDAL-GIROUX C, GUO C, VILMER E, MARYNEN P& GRANDCHAMP B (1996). THE 12;21 TRANSLOCATION INVOLVING TEL AND DELETION OF THE OTHER TEL ALLELE: TWO FREQUENTLY ASSOCIATED ALTERATIONS FOUND IN CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA. BLOOD, 87: 2891-2899.
79. MAIA AT, KOECHLING J, CORBETT R, METZLER M, WIEMELS JL& GREAVES M (2004). PROTRACTED POSTNATAL NATURAL HISTORIES IN CHILDHOOD LEUKEMIA. GENES CHROMOSOMES CANCER, 39: 335-340.

76
80. MORI H, COLMAN SM, XIAO Z, FORD AM, HEALY LE, DONALDSON C, HOWS JM, NAVARRETE C& GREAVES M (2002). CHROMOSOME TRANSLOCATIONS AND COVERT LEUKEMIC CLONES ARE GENERATED DURING NORMAL FETAL DEVELOPMENT. PROC NATL ACAD SCI U S A, 99: 8242-8247.
81. FORD AM, BENNETT CA, PRICE CM, BRUIN MC, VAN WERING ER& GREAVES M (1998). FETAL ORIGINS OF THE TEL-AML1 FUSION GENE IN IDENTICAL TWINS WITH LEUKEMIA. PROC NATL ACAD SCI U S A, 95: 4584-4588.
82. FORD AM, RIDGE SA, CABRERA ME, MAHMOUD H, STEEL CM, CHAN LC& GREAVES M (1993). IN UTERO REARRANGEMENTS IN THE TRITHORAX-RELATED ONCOGENE IN INFANT LEUKAEMIAS. NATURE, 363: 358-360.
83. LOOK AT, ROBERSON PK, WILLIAMS DL, RIVERA G, BOWMAN WP, PUI CH, OCHS J, ABROMOWITCH M, KALWINSKY D, DAHL GV& . (1985). PROGNOSTIC IMPORTANCE OF BLAST CELL DNA CONTENT IN CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA. BLOOD, 65: 1079-1086.
84. HEEREMA NA, SATHER HN, SENSEL MG, ZHANG T, HUTCHINSON RJ, NACHMAN JB, LANGE BJ, STEINHERZ PG, BOSTROM BC, REAMAN GH, GAYNON PS& UCKUN FM (2000). PROGNOSTIC IMPACT OF TRISOMIES OF CHROMOSOMES 10, 17, AND 5 AMONG CHILDREN WITH ACUTE LYMPHOBLASTIC LEUKEMIA AND HIGH HYPERDIPLOIDY (> 50 CHROMOSOMES). J CLIN ONCOL, 18: 1876-1887.
85. LAVABRE-BERTRAND T, JANOSSY G, IVORY K, PETERS R, SECKER-WALKER L& PORWIT-MACDONALD A (1994). LEUKEMIA-ASSOCIATED CHANGES IDENTIFIED BY QUANTITATIVE FLOW CYTOMETRY: I. CD10 EXPRESSION. CYTOMETRY, 18: 209-217.
86. TABERNERO MD, BORTOLUCI AM, ALAEJOS I, LOPEZ-BERGES MC, RASILLO A, GARCIA-SANZ R, GARCIA M, SAYAGUES JM, GONZALEZ M, MATEO G, SAN MIGUEL JF& ORFAO A (2001). ADULT PRECURSOR B-ALL WITH BCR/ABL GENE REARRANGEMENTS DISPLAYS A UNIQUE IMMUNOPHENOTYPE BASED ON THE PATTERN OF CD10, CD34, CD13 AND CD38 EXPRESSSION. LEUKEMIA, 15: 406-414.
87. HUNGER SP (1996). CHROMOSOMAL TRANSLOCATIONS INVOLVING THE E2A GENE IN ACUTE LYMPHOBLASTIC LEUKEMIA: CLINICAL FEATURES AND MOLECULAR PATHOGENESIS. BLOOD, 87: 1211-1224.
88. RAIMONDI SC, BEHM FG, ROBERSON PK, WILLIAMS DL, PUI CH, CRIST WM, LOOK AT& RIVERA GK (1990). CYTOGENETICS OF PRE-B-

77
CELL ACUTE LYMPHOBLASTIC LEUKEMIA WITH EMPHASIS ON PROGNOSTIC IMPLICATIONS OF THE T(1;19). J CLIN ONCOL, 8: 1380-1388.
89. TROUSSARD X, RIMOKH R, VALENSI F, LEBOEUF D, FENNETEAU O, GUITARD AM, MANEL AM, SCHILLINGER F, LEGLISE C, BRIZARD A& . (1995). HETEROGENEITY OF T(1;19)(Q23;P13) ACUTE LEUKAEMIAS. FRENCH HAEMATOLOGICAL CYTOLOGY GROUP. BR J HAEMATOL, 89: 516-526.
90. WIEMELS JL, LEONARD BC, WANG Y, SEGAL MR, HUNGER SP, SMITH MT, CROUSE V, MA X, BUFFLER PA& PINE SR (2002). SITE-SPECIFIC TRANSLOCATION AND EVIDENCE OF POSTNATAL ORIGIN OF THE T(1;19) E2A-PBX1 FUSION IN CHILDHOOD ACUTE LYMPHOBLASTIC LEUKEMIA. PROC NATL ACAD SCI U S A, 99: 15101-15106.
91. WIEMELS JL, CAZZANIGA G, DANIOTTI M, EDEN OB, ADDISON GM, MASERA G, SAHA V, BIONDI A& GREAVES MF (1999). PRENATAL ORIGIN OF ACUTE LYMPHOBLASTIC LEUKAEMIA IN CHILDREN. LANCET, 354: 1499-1503.
92. FERRARA F, DI NR, ANNUNZIATA M, COPIA C, LO PC, BOCCUNI P, SEBASTIO L& DEL VL (1998). IMMUNOPHENOTYPIC ANALYSIS ENABLES THE CORRECT PREDICTION OF T(8;21) IN ACUTE MYELOID LEUKAEMIA. BR J HAEMATOL, 102: 444-448.
93. GREAVES M (1999). MOLECULAR GENETICS, NATURAL HISTORY AND THE DEMISE OF CHILDHOOD LEUKAEMIA. EUR J CANCER, 35: 1941-1953.
94. CLARKSON BD, BOYSE EA (1971). POSSIBLE EXPLANATION OF THE HIGH CONCODDANCE FOR ACUTE LEUKAEMIA IN MONOZYGOTIC TWINS. LANCET, 1: 699-701.
95. POMBO DE OLIVEIRA MS, WAD EL SEED FE, FORONI L, MATUTES E, MORILLA R, LUZZATTO L& CATOVSKY D (1986). LYMPHOBLASTIC LEUKAEMIA IN SIAMESE TWINS: EVIDENCE FOR IDENTITY. LANCET, 2: 969-970.
96. FORD AM, POMBO-DE-OLIVEIRA MS, MCCARTHY KP, MACLEAN JM, CARRICO KC, VINCENT RF& GREAVES M (1997). MONOCLONAL ORIGIN OF CONCORDANT T-CELL MALIGNANCY IN IDENTICAL TWINS. BLOOD, 89: 281-285.
97. MAIA AT, VAN DV, V, HARRISON CJ, SZCZEPANSKI T, WILLIAMS MD, GRIFFITHS MJ, VAN DONGEN JJ& GREAVES MF (2003). PRENATAL ORIGIN OF HYPERDIPLOID ACUTE LYMPHOBLASTIC LEUKEMIA IN IDENTICAL TWINS. LEUKEMIA, 17: 2202-2206.

78
98. GREAVES M (2002). CHILDHOOD LEUKAEMIA. BMJ, 324: 283-287.
99. CIMINO G, LO CF, BIONDI A, ELIA L, LUCIANO A, CROCE CM, MASERA G, MANDELLI F& CANAANI E (1993). ALL-1 GENE AT CHROMOSOME 11Q23 IS CONSISTENTLY ALTERED IN ACUTE LEUKEMIA OF EARLY INFANCY. BLOOD, 82: 544-546.
100. FELIX CA, HOSLER MR, WINICK NJ, MASTERSON M, WILSON AE& LANGE BJ (1995). ALL-1 GENE REARRANGEMENTS IN DNA TOPOISOMERASE II INHIBITOR-RELATED LEUKEMIA IN CHILDREN. BLOOD, 85: 3250-3256.
101. ROSS JA, DAVIES SM, POTTER JD& ROBISON LL (1994). EPIDEMIOLOGY OF CHILDHOOD LEUKEMIA, WITH A FOCUS ON INFANTS. EPIDEMIOL REV, 16: 243-272.
102. ROSS JA, PERENTESIS JP, ROBISON LL& DAVIES SM (1996). BIG BABIES AND INFANT LEUKEMIA: A ROLE FOR INSULIN-LIKE GROWTH FACTOR-1? CANCER CAUSES CONTROL, 7: 553-559.
103. DAINIAK N (2002). HEMATOLOGIC CONSEQUENCES OF EXPOSURE TO IONIZING RADIATION. EXP HEMATOL, 30: 513-528.
104. NERIISHI K, STRAM DO, VAETH M, MIZUNO S& AKIBA S (1991). THE OBSERVED RELATIONSHIP BETWEEN THE OCCURRENCE OF ACUTE RADIATION EFFECTS AND LEUKEMIA MORTALITY AMONG A-BOMB SURVIVORS. RADIAT RES, 125: 206-213.
105. WIEMELS JL, PAGNAMENTA A, TAYLOR GM, EDEN OB, ALEXANDER FE& GREAVES MF (1999). A LACK OF A FUNCTIONAL NAD(P)H:QUINONE OXIDOREDUCTASE ALLELE IS SELECTIVELY ASSOCIATED WITH PEDIATRIC LEUKEMIAS THAT HAVE MLL FUSIONS. UNITED KINGDOM CHILDHOOD CANCER STUDY INVESTIGATORS. CANCER RES, 59: 4095-4099.
106. FELIX CA, LANGE BJ, HOSLER MR, FERTALA J& BJORNSTI MA (1995). CHROMOSOME BAND 11Q23 TRANSLOCATION BREAKPOINTS ARE DNA TOPOISOMERASE II CLEAVAGE SITES. CANCER RES, 55: 4287-4292.
107. LOVETT BD, LO NL, RAPPAPORT EF, BLAIR IA, OSHEROFF N, ZHENG N, MEGONIGAL MD, WILLIAMS WR, NOWELL PC& FELIX CA (2001). NEAR-PRECISE INTERCHROMOSOMAL RECOMBINATION AND FUNCTIONAL DNA TOPOISOMERASE II CLEAVAGE SITES AT MLL AND AF-4 GENOMIC BREAKPOINTS IN TREATMENT-RELATED ACUTE LYMPHOBLASTIC LEUKEMIA WITH T(4;11) TRANSLOCATION. PROC NATL ACAD SCI U S A, 98: 9802-9807.

79
108. HANDE KR (1998). ETOPOSIDE: FOUR DECADES OF DEVELOPMENT OF A TOPOISOMERASE II INHIBITOR. EUR J CANCER, 34: 1514-1521.
109. ROSS JA, POTTER JD, REAMAN GH, PENDERGRASS TW& ROBISON LL (1996). MATERNAL EXPOSURE TO POTENTIAL INHIBITORS OF DNA TOPOISOMERASE II AND INFANT LEUKEMIA (UNITED STATES): A REPORT FROM THE CHILDREN'S CANCER GROUP. CANCER CAUSES CONTROL, 7: 581-590.
110. FRANTZ CE, CHEN H& EASTMOND DA (1996). INHIBITION OF HUMAN TOPOISOMERASE II IN VITRO BY BIOACTIVE BENZENE METABOLITES. ENVIRON HEALTH PERSPECT, 104 SUPPL 6: 1319-1323.
111. KULLING SE, ROSENBERG B, JACOBS E& METZLER M (1999). THE PHYTOESTROGENS COUMOESTROL AND GENISTEIN INDUCE STRUCTURAL CHROMOSOMAL ABERRATIONS IN CULTURED HUMAN PERIPHERAL BLOOD LYMPHOCYTES. ARCH TOXICOL, 73: 50-54.
112. BIONDI A, CIMINO G, PIETERS R& PUI CH (2000). BIOLOGICAL AND THERAPEUTIC ASPECTS OF INFANT LEUKEMIA. BLOOD, 96: 24-33.
113. PUI CH, CAMPANA D& EVANS WE (2001). CHILDHOOD ACUTE LYMPHOBLASTIC LEUKAEMIA--CURRENT STATUS AND FUTURE PERSPECTIVES. LANCET ONCOL, 2: 597-607.
114. KINLEN LJ (1989). INFECTIVE CAUSE OF CHILDHOOD LEUKAEMIA. LANCET, 1: 378-379.
115. EMERENCIANO M, GUDELO ARIAS DP, COSER VM, DE BRITO GD, EDO SILVA ML& POMBO-DE-OLIVEIRA MS (2005). MOLECULAR CYTOGENETIC FINDINGS OF ACUTE LEUKEMIA INCLUDED IN THE BRAZILIAN COLLABORATIVE STUDY GROUP OF INFANT ACUTE LEUKEMIA. PEDIATR BLOOD CANCER.
116. MAGALHAES IQ, POMBO-DE-OLIVEIRA MS, BENNETT CA, CORDOBA JC, DOBBIN J, FORD AM& GREAVES MF (2000). TEL-AML1 FUSION GENE FREQUENCY IN PAEDIATRIC ACUTE LYMPHOBLASTIC LEUKAEMIA IN BRAZIL. BR J HAEMATOL, 111: 204-207.
117. MAGALHAES IQ, SPLENDORE A, EMERENCIANO M, CORDOBA MS, CORDOBA JC, ALLEMAND PA, FERRARI I& POMBO-DE-OLIVEIRA MS (2005). TRANSIENT NEONATAL MYELOPROLIFERATIVE DISORDER WITHOUT DOWN SYNDROME AND DETECTION OF GATA1 MUTATION. J PEDIATR HEMATOL ONCOL, 27: 50-52.
118. ZANROSSO CW, HATAGIMA A, EMERENCIANO M, RAMOS F, FIGUEIREDO A, FELIX TM, SEGAL SL, GUIGLIANI R, MUNIZ MT& POMBO-DE-OLIVEIRA MS (2005). THE ROLE OF

80
METHYLENETETRAHYDROFOLATE REDUCTASE IN ACUTE LYMPHOBLASTIC LEUKEMIA IN A BRAZILIAN MIXED POPULATION. LEUK RES.
119. CATOVSKY D, MATUTES E, BUCCHERI V, SHETTY V, HANSLIP J, YOSHIDA N& MORILLA R (1991). A CLASSIFICATION OF ACUTE LEUKAEMIA FOR THE 1990S. ANN HEMATOL, 62: 16-21.
120. SILVA ML, LAND MG, MARADEI S, OTERO L, VEITH M, BRITO G, KLUMB C, FERNANDEZ T& POMBO-DE-OLIVEIRA MS (2002). TRANSLOCATION (11;11)(P13- P15;Q23) IN A CHILD WITH THERAPY-RELATED ACUTE MYELOID LEUKEMIA FOLLOWING CHEMOTHERAPY WITH DNA-TOPOISOMERASE II INHIBITORS FOR LANGERHANS CELL HISTIOCYTOSIS. CANCER GENET CYTOGENET, 135: 101-102.
121. HOPMAN AH, VOORTER CE& RAMAEKERS FC (1994). DETECTION OF GENOMIC CHANGES IN CANCER BY IN SITU HYBRIDIZATION. MOL BIOL REP, 19: 31-44.
122. ANDERSSON A, HOGLUND M, JOHANSSON B, LASSEN C, BILLSTROM R, GARWICZ S, NILSSON PG, MITELMAN F& FIORETOS T (2001). PAIRED MULTIPLEX REVERSE-TRANSCRIPTASE POLYMERASE CHAIN REACTION (PMRT-PCR) ANALYSIS AS A RAPID AND ACCURATE DIAGNOSTIC TOOL FOR THE DETECTION OF MLL FUSION GENES IN HEMATOLOGIC MALIGNANCIES. LEUKEMIA, 15: 1293-1300.
123. MENDES WL, COSER VM, RAMOS G, PEREIRA W, LOPES LF& DE OLIVEIRA MS (2004). THE APPARENT EXCESS OF ACUTE PROMYELOCYTIC LEUKEMIA IN INFANT ACUTE LEUKEMIAS IN BRAZIL. HAEMATOLOGICA, 89: ELT16.
124. POMBO-DE-OLIVEIRA, M. S., EMERENCIANO, M., ARAÚJO, P. I. C., MAGALHAES, I. Q., PINHEIRO, V. R. P., AND KOIFMAN.F. MATERNAL EXPOSURES AND RISK OF INFANT ACUTE LEUKEMIA IN BRAZIL. BLOOD (ASH ANNUAL MEETING ABSTRACTS) 104(11), 1077. 2004. REF TYPE: ABSTRACT
125. PANZER-GRUMAYER ER, FASCHING K, PANZER S, HETTINGER K, SCHMITT K, STOCKLER-IPSIROGLU S& HAAS OA (2002). NONDISJUNCTION OF CHROMOSOMES LEADING TO HYPERDIPLOID CHILDHOOD B-CELL PRECURSOR ACUTE LYMPHOBLASTIC LEUKEMIA IS AN EARLY EVENT DURING LEUKEMOGENESIS. BLOOD, 100: 347-349.
126. TAUB JW, KONRAD MA, GE Y, NABER JM, SCOTT JS, MATHERLY LH& RAVINDRANATH Y (2002). HIGH FREQUENCY OF LEUKEMIC CLONES IN NEWBORN SCREENING BLOOD SAMPLES OF CHILDREN WITH B-

81
PRECURSOR ACUTE LYMPHOBLASTIC LEUKEMIA. BLOOD, 99: 2992-2996.
127. ISAACS H, JR. (2003). FETAL AND NEONATAL LEUKEMIA. J PEDIATR HEMATOL ONCOL, 25: 348-361.
128. MCHALE CM, WIEMELS JL, ZHANG L, MA X, BUFFLER PA, FEUSNER J, MATTHAY K, DAHL G& SMITH MT (2003). PRENATAL ORIGIN OF CHILDHOOD ACUTE MYELOID LEUKEMIAS HARBORING CHROMOSOMAL REARRANGEMENTS T(15;17) AND INV(16). BLOOD, 101: 4640-4641.
129. POMBO-DE-OLIVEIRA, M. S., KOIFMAN.S., AND BRAZILIAN COLLABORATIVE STUDY GROUP OF INFANT ACUTE LEUKEMIA. INFANT ACUTE LEUKEMIA AND MATERNAL EXPOSURES DURING PREGNANCY. CANCER EPIDEMIOLOGY, BIOMARKERS AND PREVENTION . 2006. REF TYPE: ABSTRACT

82
ANEXO 1

83
ANEXO 2

84
ANEXO 3