síntese, caracterização e avaliação de compostos de nióbio como
TRANSCRIPT

sid.inpe.br/mtc-m19@80/2010/05.27.16.36-TDI
SINTESE, CARACTERIZACAO E AVALIACAO DE
COMPOSTOS DE NIOBIO COMO CATALISADOR
ACIDO EM REACAO MODELO
Janiciara Botelho da Silva
Tese de Doutorado do Curso de Pos-Graduacao em Engenharia e Tecnologia
Espaciais/Ciencia e Tecnologia de Materiais e Sensores, orientada pelo Dr. Jose
Augusto Jorge Rodrigues, aprovada em 27 de maio de 2010.
URL do documento original:
<http://urlib.net/8JMKD3MGP7W/37J5PMB>
INPE
Sao Jose dos Campos
2010

PUBLICADO POR:
Instituto Nacional de Pesquisas Espaciais - INPE
Gabinete do Diretor (GB)
Servico de Informacao e Documentacao (SID)
Caixa Postal 515 - CEP 12.245-970
Sao Jose dos Campos - SP - Brasil
Tel.:(012) 3208-6923/6921
Fax: (012) 3208-6919
E-mail: [email protected]
CONSELHO DE EDITORACAO E PRESERVACAO DA PRODUCAO
INTELECTUAL DO INPE (RE/DIR-204):
Presidente:
Dr. Gerald Jean Francis Banon - Coordenacao Observacao da Terra (OBT)
Membros:
Dra Inez Staciarini Batista - Coordenacao Ciencias Espaciais e Atmosfericas (CEA)
Dra Maria do Carmo de Andrade Nono - Conselho de Pos-Graduacao
Dra Regina Celia dos Santos Alvala - Centro de Ciencia do Sistema Terrestre (CST)
Marciana Leite Ribeiro - Servico de Informacao e Documentacao (SID)
Dr. Ralf Gielow - Centro de Previsao de Tempo e Estudos Climaticos (CPT)
Dr. Wilson Yamaguti - Coordenacao Engenharia e Tecnologia Espacial (ETE)
Dr. Horacio Hideki Yanasse - Centro de Tecnologias Especiais (CTE)
BIBLIOTECA DIGITAL:
Dr. Gerald Jean Francis Banon - Coordenacao de Observacao da Terra (OBT)
Marciana Leite Ribeiro - Servico de Informacao e Documentacao (SID)
Deicy Farabello - Centro de Previsao de Tempo e Estudos Climaticos (CPT)
REVISAO E NORMALIZACAO DOCUMENTRIA:
Marciana Leite Ribeiro - Servico de Informacao e Documentacao (SID)
Yolanda Ribeiro da Silva Souza - Servico de Informacao e Documentacao (SID)
EDITORACAO ELETRONICA:
Viveca Sant´Ana Lemos - Servico de Informacao e Documentacao (SID)

sid.inpe.br/mtc-m19@80/2010/05.27.16.36-TDI
SINTESE, CARACTERIZACAO E AVALIACAO DE
COMPOSTOS DE NIOBIO COMO CATALISADOR
ACIDO EM REACAO MODELO
Janiciara Botelho da Silva
Tese de Doutorado do Curso de Pos-Graduacao em Engenharia e Tecnologia
Espaciais/Ciencia e Tecnologia de Materiais e Sensores, orientada pelo Dr. Jose
Augusto Jorge Rodrigues, aprovada em 27 de maio de 2010.
URL do documento original:
<http://urlib.net/8JMKD3MGP7W/37J5PMB>
INPE
Sao Jose dos Campos
2010

Dados Internacionais de Catalogacao na Publicacao (CIP)
Silva, Janiciara Botelho da.Si38s Sıntese, caracterizacao e avaliacao de compostos de niobio
como catalisador acido em reacao modelo / Janiciara Botelho daSilva. – Sao Jose dos Campos : INPE, 2010.
200 p. ; (sid.inpe.br/mtc-m19@80/2010/05.27.16.36-TDI)
Tese (Doutorado em Engenharia e Tecnologia Espaci-ais/Ciencia e Tecnologia de Materiais e Sensores) – Instituto Na-cional de Pesquisas Espaciais, Sao Jose dos Campos, 2010.
Orientador : Dr. Jose Augusto Jorge Rodrigues.
1. Hidratacao de etileno 2. Producao do etanol. 3.Catalisadoracido solido. 4. Catalisador de niobia. 5. Catalisador misto niobia-alumina I.Tıtulo.
CDU 544.476.2
Copyright c© 2010 do MCT/INPE. Nenhuma parte desta publicacao pode ser reproduzida, arma-zenada em um sistema de recuperacao, ou transmitida sob qualquer forma ou por qualquer meio,eletronico, mecanico, fotografico, reprografico, de microfilmagem ou outros, sem a permissao es-crita do INPE, com excecao de qualquer material fornecido especificamente com o proposito de serentrado e executado num sistema computacional, para o uso exclusivo do leitor da obra.
Copyright c© 2010 by MCT/INPE. No part of this publication may be reproduced, stored in aretrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying,recording, microfilming, or otherwise, without written permission from INPE, with the exceptionof any material supplied specifically for the purpose of being entered and executed on a computersystem, for exclusive use of the reader of the work.
ii



v
“Deus está aqui nesse momento. Sua presença é real em meu viver!
Entregue sua vida e seus problemas. Fale com Deus, ele vai ajudar você.
Oh! Oh! Deus ti trouxe aqui, para aliviar os teus sofrimentos.
È ele é o autor da fé do princípio ao fim, em todos os teus tormentos.
E ainda se vier noites traiçoeiras, se a cruz pesada for,
CRISTO ESTARÁ CONTIGO!
O mundo pode até fazer você chorar,
MAS DEUS TI QUER SORRINDO!
Seja qual for o seu problema.
FALE COM DEUS,
Ele vai ajudar você!
Após a dor vem a ALEGRIA!
Pois DEUS É AMOR e não ti deixará sofrer.
Oh! Oh! DEUS TI TROUXE AQUI, para aliviar os teus sofrimentos.
É ELE O AUTOR DA FÉ
Do princípio ao fim, em todos os teus tormentos.
E ainda se vier noites traiçoeiras, sua a cruz pesada for,
CRISTO ESTARÁ CONTIGO!
O mundo pode até fazer você chorar,
MAS DEUS TI QUER SORRINDO!
ELE TI QUER SORRINDO!”
(Padre Marcelo Rossi)

vi

vii
À minha família: meu esposo Alexandre, minha mãe Maria,
minhas irmãs Eudivania e Jaciara, meu sobrinho Murilo e
meu pai Ieudes (in memoriam).

viii

ix
AGRADECIMENTOSAGRADECIMENTOSAGRADECIMENTOSAGRADECIMENTOS
Ao Instituto Nacional de Pesquisas Espaciais (INPE) de maneira geral por sua estrutura
organizacional, que possibilitou um aprendizado com qualidade. Meus sinceros agradecimentos ao
Dr. Demétrio Bastos pela oportunidade e pelo incentivo.
Ao Prof. Dr. José Augusto Rodrigues pela disponibilidade em me orientar e por todo o
suporte dispensado a mim ao longo desses anos que permitiram a concretização deste trabalho.
À pós-graduação em Engenharia e Tecnologia Espacial na Área de Concentração em Ciência
de Materiais e Sensores pela oportunidade de realização deste curso de doutorado.
À Companhia Brasileira de Metalurgia e Mineração pelo fornecimento das amostras de
nióbio, o HY-340, que foi a base principal para realização do projeto de doutorado.
Ao Prof. Dr. Claude Potvin da Université Pierre et Marie Curie na França e à Profª. Dra.
Maria Felipa Gomes Ribeiro do Instituto Superior Técnico em Lisboa, por toda presteza em realizar
as análises de Raios X e Adsorção de piridina monitorada por infravermelho, respectivamente, que
foram fundamentais para as discussões deste trabalho.
A todos do Laboratório Associado de Sensores e Materiais do Centro de Tecnologias
Especiais do Instituto Nacional de Pesquisas Espaciais (LAS/CTE/INPE) por terem disponibilizado
a infraestrutura para realização das análises de Raio X e MEV, em especial: à Maria Lúcia pelas
análises de MEV, “Seu” Chico pela ajuda com o equipamento de Raio X, e ao João Paulo pelas dicas
e ajuda nos dois equipamentos, enfim a todos pela boa vontade e simpatia com que sempre me
atenderam em São José dos Campos.
A todos os professores que contribuíram para o meu aprendizado durante este curso e pela
convivência privilegiada, em especial Dr. Chen, Dra. Maria do Carmo, Dr. Paulo Rappl, Dr. Gilberto
Marques. Aos colegas de curso pelo apoio durante as disciplinas e pela amizade: Henrique, Felipe,
Gustavo, Willian, Carina, Emílio e Beatriz.
A todos do Laboratório Associado de Combustão e Propulsão pela colaboração, pelo
incentivo e pelos momentos de descontração no cafezinho, em especial: Dr. David, Dr. Turíbio,
Cristiane, Tertulino, Jorge Jofre e Jalusa. Ao Dr. Ricardo Vieira pelas dicas e conversas que muito me

x
ajudaram. Meus agradecimentos às bibliotecárias Inês e Cida, pela simpatia e presteza com que
sempre me atenderam.
Aos amigos do laboratório de catálise do Laboratório Associado de Combustão e Propulsão,
pelo apoio, pelos ensinamentos compartilhados, por toda a ajuda e, principalmente, pelo carinho,
meus sinceros agradecimentos ao Jorge Damião, Sayuri, Maria Aparecida, Frederico, Paula Caroline,
Ricardo e Juninho. Meus sinceros agradecimentos à Dra. Mariza Zacarias e ao Dr. Luiz Claudio
Bastos sempre tão solícitos às minhas necessidades.
Aos amigos da cúpula, pelos esclarecimentos nos momentos de estudo, pelas brincadeiras nos
momentos de descontração, pelo apoio e amizade, obrigada: Maura, Anton, Sandro, Leonardo e
César. Aos amigos do alojamento que tornaram os dois anos de moradia inesquecíveis, em especial:
Marina, Renauld, Manoel, Vicent. Alexandra, Mara, Olimpya, German, Flugel, Izabelly, Rafael
Castelo, Ana e Judith.
Aos amigos que perto ou longe sempre estiveram ao meu lado, incentivando, torcendo,
apoiando ou, simplesmente, me ouvindo, meus agradecimentos à: Jomar, Aline, Luiz Eduardo,
Sherlan, André Leopoldo, José de Ribamar, Eliane, Jethânia, Lorena, Jemmla, Silvio, Yone, Janyeid
e Lucy. Em especial meus agradecimentos aos professores e amigos, Dr. Edmar Marques e Dra.
Aldálea Marques, pelo apoio e principalmente pela amizade.
Às pessoas responsáveis por eu ter chegado até aqui, meus agradecimentos especiais à minha
família: minhas irmãs Eudivania e Jaciara pelas palavras de incentivo; meu sobrinho e afilhado,
Murilo, por ser tão carinhoso e compreensivo; minha mãe, Maria do Socorro, por todo seu amor, suas
palavras de apoio incondicional e pelas preces nos momentos que precisei. E meu amor, Alexandre, por
adiar seus projetos em favor desse meu sonho, por estar ao meu lado em todos os momentos. Só quero
que saibam que vocês são a razão de tudo o que faço. Graças a DEUS eu tenho uma família
maravilhosa! Que ele nos proteja e nos una cada vez mais em seu infinito amor!
Enfim, o mais importante, meus agradecimentos a Deus, por ter me concedido a capacidade,
a oportunidade e a serenidade necessária para desenvolver este trabalho. E a Virgem Maria por sua
infinita bondade em guiar meus passos sempre que pedi sua intervenção.

xi
RESUMO
O presente trabalho tem como objetivo a síntese, a caracterização e avaliação catalítica de catalisadores contendo nióbio em sua formulação, na reação de hidratação de etileno. Os desempenhos destes foram comparados com os catalisadores comerciais alumina e ZEÓLITA Y, ambos calcinados em diferentes temperaturas, sendo a alumina proveniente da desidratação de uma pseuboemita (CATAPAL A). Os catalisadores à base de nióbio foram obtidos a partir do precursor óxido de nióbio hidratado comercial, material fornecido pela Companhia Brasileira de Metalurgia e Mineração (CBMM) com a denominação HY-340, empregando um processo de autoclavagem (hidrotratamento) que resultou em um catalisador com propriedades texturais e reológicas superiores àquelas apresentadas pelo HY-340 calcinado diretamente. Também foram preparados catalisadores mistos (Al-Nb) a partir de diferentes teores de HY-340 e de CATAPAL A, e estes foram submetidos ao mesmo processo de autoclavagem empregado para o HY-340. Os catalisadores contendo nióbio e alumina-nióbio foram submetidos às mesmas temperaturas nas quais foram calcinados os catalisadores comerciais HY-340, CATAPAL A e ZEÓLITA Y. Todos os materiais foram caracterizados através das seguintes técnicas: difratrometria de raios X, termogravimetria, volumetria de nitrogênio, quimissorção com amônia, quimissorção com piridina (FTIR), microscopia eletrônica de varredura. A avaliação catalítica dos materiais foi realizada através da reação modelo de hidratação de etileno, conduzida a 220oC e sob pressão parcial constante de água. Os produtos gerados foram analisados por cromatografia gasosa, empregando para esta finalidade detectores de condutividade térmica e ionização de chama. De acordo com os resultados o melhor desempenho catalítico foi obtido com o produto HY-340 autoclavado (AHY-340) e calcinado a 300oC, superando a ZEOLITA Y, um catalisador normalmente indicado em reações que necessitam de sítios ácidos. Este catalisador AHY-340 além de apresentar a maior atividade foi muito seletivo na produção de etanol, fato este que não ocorre com a ZEÓLITA Y, uma vez que este produziu o éter etílico além do etanol. O processo de hidrotratamento empregado neste trabalho, além de duplicar a área específica, aumentou significativamente a acidez do composto de nióbio comercial (HY-340), principalmente a acidez de Bronsted. A presença destes sítios e a elevada atividade tornam este material promissor para outras aplicações catalíticas que envolvam sítios ácidos, entre elas, o craqueamento catalítico fluido, processo presente na maioria das refinarias de petróleo. Os materiais mistos contendo teores diferentes de nióbio e alumina não apresentaram bom desempenho catalítico na reação de hidratação de etileno, provavelmente porque a metodologia de preparação empregada não propiciou a formação sítios de Bronsted, conforme ficou evidenciado nos resultados das análises de acidez, empregando a piridina como molécula sonda.

xii

xiii
SYNTHESIS, CHARACTERIZATION AND EVALUATION OF NIOBI UM COMPOUNDS AS ACID CATALYSTS IN MODEL REACTION
ABSTRACT
The objective of this work was to synthesize, characterize and evaluate catalytic behavior of niobium-based catalysts in ethylene hydration reaction. Their performance was compared to those of commercially available alumina and ZEOLITE Y catalysts, both calcined at different temperatures; the alumina was derived from pseudoboehmite dehydration (CATAPAL A). Niobium-based catalysts were obtained from a commercial hydrated niobium oxide precursor (HY-340) which was provided by CBMM (Companhia Brasileira de Metalurgia e Mineração), employing an autoclaving process (hydrothermic treatment) that resulted in a catalyst with textural and rheological properties superior to those presented by calcined HY-340. Al-Nb mixed catalysts were also prepared from different grades of HY-340 and CATAPAL A, which underwent the same autoclaving procedure employed for HY-340. The catalysts containing niobium and alumina-niobium were submitted to the same temperatures as commercial catalysts HY-340, CATAPAL A, and zeolite Y. All materials were characterized through the following techniques: X-ray diffraction, thermogravimetry, nitrogen volumetry, ammonia chemisorption, pyridine chemisorption (FTIR), and scanning electron microscopy. Catalytic evaluation of the materials was performed through a ethylene hydration model reaction, run at 220°C under constant water vapor pressure. Products generated were analyzed by gas chromatography using thermal conductivity and flame ionization detectors. Results showed that the best catalytic performance was obtained with the autoclaved HY-340 product (AHY-340) calcined at 300°C, which performed better than zeolite Y, a catalyst normally recommended in reactions that require acid sites. This AHY-340 catalyst, in addition to having higher activity, was very selective in ethanol production, something that didn’t occur with zeolite Y, since the latter produced ethylic ether in addition to ethanol. The hydrotreatment employed in this work, in addition to doubling specific area, significantly increased the acidity of the commercial niobium compound (HY-340), mainly Brönsted acidity. Presence of these sites and elevated activity make this material promising for other catalytic applications involving acid sites, among them catalytic fluid cracking, a process which is present in most petroleum refineries. Mixed materials containing different grades of niobium and alumina didn’t have good catalytic performance in the ethylene hydration reaction, probably because the preparation methodology employed didn’t allow formation of Brönsted sites, as was shown in the acidity analyses employing pyridine as the probe molecule.

xiv

xv
LISTA DE FIGURAS
Figura 3.1 - Posição central dos alcoóis na química orgânica . ......................... 7
Figura 3.2 - Produção industrial de etanol por fermentação . .......................... 10
Figura 3.3 – Derivados petroquímicos do etileno (eteno) . .............................. 13
Figura 3.4 - Produção industrial do etanol por hidratação indireta do etileno . 15
Figura 3.5 - Produção industrial de etanol por hidratação direta do etileno .... 17
Figura 3.6 - Mecanismo geral - regra de Markovinikov ................................... 20
Figura 3.7 – Mecanismo simplificado da reação de hidratação de alcenos em
fase homogênea .............................................................................................. 22
Figura 3.8 - (a) Estrutura do H-Nb2O5. (losangos) NbO6 na forma octaedrica,
(�) Nb em sítio tetraédrico. (b) Projeção da estrutura paralela do T-Nb2O5 no
plano [001]; () oxigênio, (○,●) Nb no sítio tetraédrico . .................................. 28
Figura 3.9 - Estrutura do nióbio isopoliácido (H8Nb6O19) ................................ 30
Figura 3.10 - Superfície do óxido de nióbio mostrando a vacância do oxigênio..
......................................................................................................................... 31
Figura 3.11 - Natureza química e espécies nióbio na catálise heterogênea . . 32
Figura 3.12 - Processamento da bauxita e produção de aluminas . ................ 38
Figura 3.13 – Estruturas cristalinas da alumina . ............................................. 40
Figura 3.14 - Classificação dos oxi-hidróxidos de alumínio . ........................... 41
Figura 3.15 - Superfície das aluminas antes (a) e após (b) a ativação segundo
o Modelo de Peri, sendo que, (+) denota uma subcamada de Al3+ . ................ 43
Figura 3.16 - Configurações do grupo OH na superfície da alumina com suas
respectivas cargas residuais (σσσσOH), de acordo com o modelo de Knözinger-
Ratnasamy . ..................................................................................................... 44
Figura 3.17 – Configurações das hidroxilas na superfície de uma γγγγ-alumina no
modelo de Busca–Lorenzelli (B-L), com base nas freqüências dos estiramentos
ννννOH . .................................................................................................................. 45
Figura 3.18 – (a) Interação dipolo-dipolo entre as hidroxilas na γ-alumina no
modelo de Peri; (b) uma representação da interação dipolo-dipolo na
configuração geminal das hidroxilas negligenciada no modelo T-M . .............. 46

xvi
Figura 3.19 – Os três tipos de estado da superfície de uma gibsita . .............. 47
Figura 3.20 - Tipos de seletividade com peneiras moleculares . ..................... 49
Figura 3.21 - Estruturas FAU e LTA a partir de tetraedros de TO4 . ................ 53
Figura 3.22 - Esquema representativo para formulação da lei de Bragg . ....... 59
Figura 4.1 – Programação da temperatura de calcinação. .............................. 69
Figura 4.2 – Fluxograma da autoclavagem do óxido de nióbio (HY). .............. 71
Figura 4.3 – Esquema do reator de quartzo utilizado no teste catalítico. ........ 76
Figura 4.4 – Unidade de avaliação catalítica do LCP/INPE. ............................ 78
Figura 5.1 - Termograma do hidróxido de alumínio (catapal A)....................... 82
Figura 5.2 - Termograma da zeólita Y. ............................................................ 83
Figura 5.3 – Termograma da nióbia (HY). ....................................................... 84
Figura 5.4 – Termograma da nióbia autoclavada (AHY). ................................ 85
Figura 5.5 - Fluxo de calor comparando as duas nióbias (HY e AHY). ........... 85
Figura 5.6 - Difratograma do hidróxido de alumínio (CAT) calcinada a 300 °C/ 6
horas. ............................................................................................................... 86
Figura 5.7 - Difratograma do hidróxido de alumínio (CAT) calcinado a 400 °C/ 6
horas. ............................................................................................................... 87
Figura 5.8 - Difratograma da zeólita Y (ZEO) calcinada a 300 °C/ 6 horas. .... 88
Figura 5.9 - Difratograma do óxido de nióbio (HY) calcinado a 300 °C/6 horas.
......................................................................................................................... 89
Figura 5.10 – Difratograma do óxido de nióbio calcinado a 500 °C / 6 horas. .. 90
Figura 5.11 – Difratograma do óxido de nióbio autoclavado calcinado a 500 °C/
6 horas. ............................................................................................................ 91
Figura 5.12 – Microscopia eletrônica de transmissão do AHY calcinado a 300
°C por 6 horas. ................................... .............................................................. 92
Figura 5.13 – Difratograma do óxido de nióbio autoclavado (AHY) calcinada a
600 °C/6 horas. ................................... ............................................................. 93
Figura 5.14 - Imagens obtidas por MEV dos materiais: HY, AHY, CAT e ZEO
calcinados a 600 °C por 6 horas. .................. ................................................... 94
Figura 5.15 - Isotermas de adsorção-dessorção da CATAPAL A. ................... 96

xvii
Figura 5.16 – Distribuições de poros da CATAPAL A (CAT), obtidas por
volumetria de nitrogênio, em função da temperatura de calcinação. ............... 97
Figura 5.17 – Isotermas de adsorção-dessorção da ZEÓLITA Y. ................... 98
Figura 5.18 – Distribuições de poros da ZEÓLITA Y, obtidas por volumetria de
nitrogênio, em função da temperatura de calcinação. ...................................... 99
Figura 5.19 – Isotermas de adsorção-dessorção da HY. .............................. 101
Figura 5.20 - Distribuição de poros da HY por adsorção de nitrogênio. ........ 101
Figura 5.21– Isotermas de adsorção-dessorção da HY. ............................... 103
Figura 5.22 – Distribuição de poros da AHY por adsorção de nitrogênio. ..... 103
Figura 5.23 – Comparação das isotermas e da distribuição de poros da HY e
AHY calcinadas a 500 °C por 6 horas. .............. ............................................. 104
Figura 5.24 - Gráfico da produção de etanol a partir do etileno, utilizando como
catalisador o AHY calcinado a 300 °C. ............. .............................................. 105
Figura 5.25 – Ilustração da linha catalítica que foi mantida aquecida durante
todo o experimento. ........................................................................................ 107
Figura 5.26 - Limpeza da superfície das CATAPAL A em função do tempo. 108
Figura 5.27 – Produção de etanol a partir do etileno utilizando como catalisador
as CATAPAL A calcinadas em diferentes temperaturas. ............................... 109
Figura 5.28 – Limpeza da superfície das ZEÓLITA Y em função do tempo. . 110
Figura 5.29 – Produção de etanol a partir do etileno utilizando como catalisador
as ZEÓLITA Y calcinadas em diferentes temperaturas. ................................. 111
Figura 5.30 - Produção de éter etílico a partir do etileno utilizando como
catalisador as ZEÓLITAS Y calcinadas a diferentes temperaturas. ............... 112
Figura 5.31 - Mecanismo simplificado de geração do éter a partir do etileno.113
Figura 5.32 – Limpeza da superfície dos HY em função do tempo. .............. 114
Figura 5.33 - Produção de etanol a partir do etileno utilizando como catalisador
os HY calcinados em diferentes temperaturas. .............................................. 115
Figura 5.34 – Limpeza da superfície dos AHY em função do tempo. ............ 116
Figura 5.35 - Produção de etanol a partir do etileno utilizando como catalisador
as AHY calcinadas em diferentes temperaturas. ............................................ 117

xviii
Figura 5.36 – Acidez total, corrigidos pelas áreas específicas, dos materiais
catalíticos em suas respectivas temperaturas de calcinação. ........................ 119
Figura 5.37 – Evolução da acidez total, ao longo de seis semanas
consecutivas, dos materiais catalíticos calcinados a 300 °C. ......................... 120
Figura 5.38 - Isotermas de adsorção-dessorção e distribuições porosas da
AHY por volumetria de nitrogênio. .................................................................. 121
Figura 5.39 - Difratogramas de raios X das AlX%Nb calcinada a 400 °C/6
horas. ............................................................................................................. 123
Figura 5.40 - Isotermas de adsorção-dessorção das Al%Nb preparadas com
diferentes teores de nióbia (AHY). ................................................................. 124
Figura 5.41 – Produção de etanol, obtida a partir do etileno, utilizando, como
catalisadores, misturas Al%Nb calcinadas a 400 °C. ..................................... 126
Figura 5.42 – Comparação da produção de etanol, obtida a partir do etileno,
utilizando como catalisadores as misturas Al%Nb, AHY400 e a CAT400. ..... 127
Figura 5.43 – Comparação da produção de etanol, obtida a partir do etileno,
utilizando como catalisadores as misturas Al30%Nb calcinada a 400 °C,
AHY300, HY300, ZEO30 e a CAT300. ........................................................... 128
Figura 5.44 - Espectros de FTIR relativos à adsorção da piridina para as
amostras AHY300, AHY400 e CAT400 (Tdessorção = 150 ºC; P = 10-6 mbar).
....................................................................................................................... 130
Figura 5.45 - Espectros de FTIR relativos à adsorção da piridina para as
amostras AHY400, Al30Nb400, Al20Nb400, Al5Nb400 e CATA400 (Tdesorção
= 150 ºC; P = 10-6 mbar). ............................................................................... 131
Figura 5.46 – Gráfico dos resultados para a reação de etanol a partir do
etileno, utilizando como catalisador os materiais calcinadas a 300 °C. .......... 133

xix
LISTA DE TABELAS
Tabela 3.1 – Espécies de nióbia aquoso na faixa de pH de 14,5 a 0,55 ........ 27
Tabela 3.2 - Breve histórico da catálise com as peneiras moleculares . .......... 50
Tabela 3.3 - Características dos poros de algumas peneiras moleculares . .... 52
Tabela 3.4 - Tipos de cromatografia. ............................................................... 61
Tabela 4.1 - Bandas associadas aos centros ácidos de Lewis e Brönsted. .... 80
Tabela 5.1 - Área específica da catapal A, diâmetro médio e volume de poros
da Catapal A, obtidos por volumetria de N2. ..................................................... 97
Tabela 5.2 – Área específica da zeólita Y, diâmetro médio e volume de poros
obtidos por volumetria de N2. ......................................................................... 100
Tabela 5.3 - Área específica da HY, diâmetro médio e volume dos poros
obtidos por volumetria de N2. ......................................................................... 102
Tabela 5.4 - Área específica, diâmetro médio e volume de poros da AHY,
obtidos por volumetria de N2. ......................................................................... 104
Tabela 5.5 – Acidez total, corrigidos pelas áreas específicas, dos materiais
catalíticos em suas respectivas temperaturas de calcinação. ........................ 118
Tabela 5.6 - Evolução da acidez total, ao longo de seis semanas consecutivas,
dos materiais catalíticos calcinados a 300 °C. .... ........................................... 120
Tabela 5.7 – Volumetria de nitrogênio e quimissorção de amônia para as
misturas Al%Nb. ............................................................................................. 125
Tabela 5.8 – Bandas no infra-vermelho (1400-1700 cm-1) região de adsorção
da piridina em sólidos ácidos ........................................................................ 129
Tabela 5.9 – Bandas associadas aos centros ácidos de Lewis e Brönsted. .. 129
Tabela 5.10 - Absorvâncias integradas e concentrações dos centros ácidos de
Lewis e Brönsted obtidas para as diferentes amostras. ................................. 132

xx

xxi
LISTA DE ABREVIAÇÕES
INPE – Instituto Nacional de Pesquisas Espaciais
CBMM – Companhia Brasileira de Mineração e Metalurgia
LCP – Laboratório Associado de Combustão e Propulsão
MEV – Microscopia Eletrônica de Varredura
FTIR – Infravermelho com transformada de Fourier
CAT – Alumina comercial ( Catapal A)
HY – Nióbio precursor (HY-340 da CBMM)
AHY – Nióbio HY-340 após procedimento hidrotérmico
ZEO Y – Zeólita Y comercial

xxii

xxiii
SUMÁRIO
Pág.
CAPÍTULO 1 – INTRODUÇÃO ........................... ............................................ 1 1.1 A importância do etanol ....................... ................................................... 1
CAPÍTULO 2 – OBJETIVO ............................. .................................................5 2.1 Objetivo Geral ................................ .......................................................... 5 2.2 Objetivo Específico............................ ...................................................... 5 CAPÍTULO 3 – REVISÃO BIBLIOGRÁFICA ................ .................................. 7 3.1 O Etanol ...................................... .............................................................. 8 3.1.1 Redução do Acetaldeído ..........................................................................8 3.1.2 Fermentação ........................................................................................... 8 3.1.3 Hidratação do Etileno ..............................................................................11 3.2 As Olefinas ................................... ............................................................ 11 3.2.1 Etileno ..................................................................................................... 12 3.3 Hidratação .................................... ............................................................ 14 3.3.1 Hidratação Indireta do Etileno ................................................................. 14 3.3.2 Hidratação Direta do Etileno ................................................................... 16 3.3.3 O mecanismo Geral de Hidratação ......................................................... 19 3.4 Catalisador Ácido Sólido à Base de Nióbio ..... ..................................... 24 3.4.1 Conceitos Fundamentais Referentes ao Nióbio ..................................... 24 3.4.2 As reservas de Nióbio e suas Aplicações................................................ 25 3.4.3 Estrutura da Nióbia ................................................................................. 26 3.4.4 Propriedades Ácidas da Nióbia ............................................................... 29 3.4.5 Aplicações Catalíticas da Nióbia ............................................................. 32 3.5 Considerações sobre a Alumina ................. ............................................37 3.5.1 Informações Gerais ................................................................................. 37 3.5.2 Morfologia da Alumina ............................................................................ 39 3.5.3 Aplicações Catalíticas ............................................................................. 41 3.6 Considerações sobre as Zeólitas ............... ............................................ 48 3.6.1 Histórico e Conceito ................................................................................ 48 3.6.2 Estrutura Geral e a Zeólita Y .................................................................. 52 3.6.3 Aplicações das Zeólitas .......................................................................... 54 3.7 Métodos de Caracterização ..................... ............................................... 56 3.7.1 Termogravimetria .................................................................................... 56 3.7.2 Microscopia Eletrônica de Varredura (MEV) ........................................... 57 3.7.3 Difratometria de Raios X ......................................................................... 57 3.7.4 Volumetria de Nitrogênio ........................................................................ 60 3.7.5 Cromatografia Gasosa ............................................................................ 61 3.7.6 Quimissorção de Amônia ........................................................................ 62

xxiv
3.7.7 Espectroscopia na Região do Infravermelho com Transformada de Fourier ........................................................................................................ 63 CAPÍTULO 4 – MATERIAIS E METODOLOGA ............... .............................. 65 4.1 Materiais e Equipamentos ...................... ................................................ 66 4.1.1 Materiais ................................................................................................. 66 4.1.2 Equipamentos.......................................................................................... 67 4.2. Preparação dos Catalisadores ................. ............................................. 67 4.2.1 Catalisadores de Alumina, Zeólita e Nióbia ............................................67 4.2.2 Procedimento da Calcinação ................................................................. 68 4.2.3 Processo de Autoclavagem do Catalisador de Nióbio ............................69 4.2.4 Catalisador Misto Nióbia-Alumina ...........................................................71 4.3 Caracterização dos Catalisadores .............. ........................................... 72 4.3.1 Termogravimetria .................................................................................... 72 4.3.2 Microscopia Eletrônica de Varredura (MEV) ........................................... 72 4.3.3 Difratometria de Raios X (DRX) .............................................................. 73 4.3.4 Volumetria de Nitrogênio ........................................................................ 73 4.3.5 Volumetria de Amônia ............................................................................ 74 4.3.6 Teste Catalítico Acompanhado por Cromatografia a Gás .................... .75 4.3.7 Volumetria de Piridina .............................................................................79 CAPÍTULO 5 – RESULTADOS E DISCUSSÃO ............... ............................. 81 5.1 Análise Térmica ............................... ....................................................... 81 5.1.1 Termograma da Catapal A..................................................................... 81 5.1.2 Termograma da Zeólita Y ..................................................................... 82 5.1.3 Termograma da Nióbia e da Nióbia Autoclavada ................................. 83 5.2 Difratometria de Raios X ...................... ................................................. 86 5.2.1 Difratometria da Catapal A .................................................................... 86 5.2.2 Difratometria da Zeólita Y ..................................................................... 88 5.2.3 Difratometria do óxido de Nióbio – HY .................................................. 89 5.2.4 Difratometria do óxido de Nióbio autoclavado - AHY ............................ 90 5.3 Microscopia Eletronica de Varredura (MEV) ..... ....................................93 5.4 Caracterização por Volumetria de Nitrogênio ... ................................... 94 5.4.1 Volumetria de Nitrogênio da Catapal A .................................................. 95 5.4.2 Volumetria de Nitrogênio da Zeólita Y ................................................... 98 5.4.3 Volumetria de Nitrogênio da HY ........................................................... 100 5.4.4 Volumetria de Nitrogênio da AHY ......................................................... 102 5.5 Testes dos Materiais na Reação Modelo – Hidrata ção do Etileno .... 105 5.5.1 Teste Catalítico com as Catapal A ........................................................ 108 5.5.2 Teste Catalítico com a Zeólita Y ........................................................... 110 5.5.3 Teste Catalítico com os HY não Autoclavadas ..................................... 113 5.5.4 Teste Catalítico Utilizando os HY Autoclavadas ................................... 115 5.6 Determinação da Acidez Total .................. ............................................ 117 5.7 Catalisadores Mistos – Al%Nb .................. ........................................... 122 5.7.1 Caracterização dos Catalisadores Mistos ............................................. 122 5.8. Caracterização por FTIR ...................... ................................................. 128

xxv
6 CONCLUSÕES ........................................................................................... 135 SUGESTÕES PARA TRABALHOS FUTUROS .................. ......................... 137 REFERÊNCIAS BIBLIOGRÁFICAS ........................ ..................................... 139 APÊNDICE A ........................................ ......................................................... 149 APÊNDICE B ........................................ ......................................................... 151 APÊNDICE C ................................................................................................. 153 APÊNDICE D ................................................................................................. 155 APÊNDICE E ................................................................................................. 157 APÊNDICE F ................................................................................................. 159 APÊNDICE G ................................................................................................. 161 APÊNDICE H ................................................................................................. 163 APÊNDICE I ...................................................................................................165 APÊNDICE J ........................................ ......................................................... 167 APÊNDICE L ........................................ ......................................................... 169 APÊNDICE M ................................................................................................ 171 APÊNDICE N ................................................................................................. 173

xxvi

1
CCAAPPÍÍTTUULLOO 11
11 IINNTTRROODDUUÇÇÃÃOO
11..11 AA iimmppoorr ttâânncciiaa ddoo eettaannooll
O etanol possui inúmeras aplicações, que serão abordadas ao longo
deste trabalho, porém, entre todas, existe uma que cada vez mais vem se
tornando importante: a utilização como combustível. Em 1908 o etanol foi o
combustível da eleição de Henry Ford, no emblemático carro Ford T (1). Na
década de 70, a crise internacional de energia provocada pelos países
produtores de petróleo, devido ao embargo da Organização dos Países
Exportadores de Petróleo (OPEP), levou todos os importadores a mobilizar a
comunidade cientifica em busca de fontes alternativas de energia e acelerar
projetos de prospecção e perfuração de novos poços de petróleo (1, 2).
Os Estados Unidos, visando diminuir a dependência do petróleo
estrangeiro e fomentar a economia agrícola, voltou-se para o etanol, gerado a
partir do milho. No Brasil, a alternativa encontrada foi a criação do Plano
Nacional do Álcool (Proálcool), em 1975, através do processo de fermentação
da cana de açúcar (1, 2).
No início do século XX, a descoberta de poços de petróleo nos Estados
Unidos condicionou o desenvolvimento dos primeiros motores automotivos ao
uso da gasolina e mais tarde do diesel. Porém, hoje são conhecidos os
grandes problemas ambientais causados pela emissão de gases poluentes,
que, infelizmente, vêem aumentando no passar dos anos. A vantagem do
etanol é que sua molécula tem apenas um átomo de oxigênio, assim sua
combustão é completa, além de ser naturalmente biodegradável (1-3), e as
emissões de monóxido de carbono (CO) são menores que em outros

2
combustíveis, como a gasolina, atribuindo-lhe maior qualidade e menor caráter
poluente.
O aumento do consumo de energia e a busca por fontes alternativas
para o futuro são preocupações do mundo atual e refletem a imensa
dependência do ser humano por energia. Vários países, tanto desenvolvidos
como em desenvolvimento, estão adotando programas para a utilização de
uma matriz enérgica mais limpa.
A busca por fontes alternativas de energia advem de motivos diversos -
políticos, econômicos, energéticos e ambientais – para cada país.
Dependentes ou não da importação de petróleo, os países desenvolvidos ou
em desenvolvimento têm investido cada vez mais recursos em pesquisas na
área de combustíveis alternativos. Desde os anos 80 até os dias atuais vem se
difundindo o uso da mistura etanol/gasolina por todo o mundo. No final dos
anos 90 e início do século XXI, motivados por políticas ambientais que o
mundo vem tentando adotar, com o intuito de reduzir a emissão de gases que
causam o efeito estufa, mais países passaram a analisar a possibilidade de
implementar programas de bicombustíveis.
O Japão, por exemplo, adotou a política ambiental de reduzir a emissão
de gases poluentes através da adição de etanol na gasolina, da ordem de 10 %
desde 2008. Anteriormente, a legislação japonesa permitia, porém, não
obrigava o emprego da mistura de 3 % de álcool na gasolina (2). Para os
Estados Unidos, no ano de 2000, já que a produção fora de aproximadamente
2 milhões de galões de etanol, o presidente em exercício, George W. Bush,
recomendou ao Congresso levar adiante o aumento da cota energética que
elevaria a produção para 5 bilhões de galões até 2012 (1).
O etanol, obtido a partir do processo de fermentação da cana-de-açúcar,
também pode ser produzido a partir de subprodutos do petróleo, atualmente,
lançados na atmosfera, como por exemplo, o etileno. Com recursos e
tecnologia adequados, e a adição de água ao etileno - na presença de um

3
catalisador eficiente, pode-se obter etanol desse subproduto perdido, o que só
traria benefícios (4).
Geralmente, o processo catalítico da produção de etanol envolve
catalisadores ácidos, os quais podem atuar em processo homogêneo ou
heterogêneo. No caso particular da catálise heterogênea, o interesse se
restringe aos catalisadores sólidos. Mais especificamente, o desenvolvimento
de novos catalisadores ácidos que sejam insolúveis, estáveis e facilmente
regeneráveis, tem sido aguardado ansiosamente pelos ambientalistas e pelas
indústrias químicas (5), principalmente as do refino de petróleo.
Visando ao desenvolvimento de catalisadores empregados no refino de
petróleo e explorando as características ácidas dos compostos a base de
nióbio, cujas maiores reservas encontram-se no Brasil, decidiu-se investigar
neste trabalho, a atividade catalítica em uma reação que exija a presença de
sítios ácidos, no caso, a reação de hidratação de uma olefina, o etileno.

4

5
CCAAPPÍÍTTUULLOO 22
22 OOBBJJEETTIIVVOOSS
22..11 OObbjjeett iivvoo ggeerraall
O objetivo geral deste trabalho consiste em sintetizar, caracterizar e
avaliar novos catalisadores visando à possível aplicação na indústria do refino
de petróleo. O material de partida é fornecido pela Companhia Brasileira de
Mineração e Metalurgia (CBMM) e os catalisadores produzidos terão seus
desempenhos catalíticos avaliados na reação de hidratação do etileno. Os
trabalhos experimentais foram executados nos laboratórios do grupo de
catálise do Laboratório Associado de Combustão e Propulsão (LCP) do
Instituto Nacional de Pesquisas Espaciais (INPE). Para cumprir o presente
trabalho, alguns objetivos específicos foram propostos.
22..22 OObbjjeett iivvooss eessppeeccííff iiccooss
• Sintetizar catalisadores de nióbia com elevada área específica;
• Caracterizar a morfologia e a textura dos catalisadores de nióbia;
• Avaliar o comportamento catalítico dos catalisadores na reação de
hidratação do etileno, conduzida em fase gasosa e na pressão
ambiente;
• Sintetizar catalisadores alumina-nióbia, com diferentes razões atômicas
Al/Nb;
• Caracterizar a morfologia e a textura dos catalisadores alumina-nióbia;
• Avaliar os catalisadores mistos alumina-nióbia na reação de hidratação de
etileno, conduzida também em fase gasosa e na pressão ambiente e

6
• Comparar o desempenho catalítico dos catalisadores de nióbia e de
alumina-nióbia, desenvolvidos neste trabalho, com os obtidos com
catalisadores ácidos, tais como uma zeólita e uma alumina, ambas
comerciais, na mesma reação de hidratação de etileno, sob as mesmas
condições reacionais.

7
oo CCAAPPÍÍTTUULLOO 33
33 RREEVVIISSÃÃOO BBIIBBLLIIOOGGRRÁÁFFIICCAA
Na química orgânica os alcoóis ocupam uma posição central. Eles
podem ser preparados a partir de muitas classes de compostos (alquenos,
halogenetos de alquila, cetonas, ésteres, aldeídos, entre outras) e também
podem ser transformados em um grande número de outros compostos,
conforme pode-se observar na Figura 1.
Figura 3.1 - Posição central dos alcoóis na química orgânica (1).
O etanol ou álcool etílico (CH3CH2OH) é conhecido há tempos pela
humanidade na forma de bebidas, como: cervejas, vinhos, licores e destilados.
Inicialmente, utilizado para definir um fino pó de antimônio, o nome origina-se
do árabe al-kuhul, termo reelaborado e, mais tarde, ampliado pelos alquimistas
medievais englobando todos os produtos da destilação (6).

8
33..11 OO eettaannooll
O etanol é uma molécula muito simples que apresenta baixo peso
molecular, líquido incolor, ponto de ebulição igual a 78 ºC, odor característico,
sendo volátil, inflamável e solúvel em água. Por ser miscível assim como a
grande maioria dos líquidos de baixo peso molecular, o etanol é utilizado em:
produtos farmacêuticos; na fabricação de tintas, vernizes, perfumes; e como
combustível (7). Como todo álcool, o etanol não existe em estado livre na
natureza, no entanto, pode ser obtido por três processos ao nível industrial:
redução do acetaldeído, fermentação e hidratação do etileno (1, 4).
3.1.1 Redução do Acetaldeído
O processo de obtenção do etanol através da redução do acetaldeído
desperta interesse apenas acadêmico, já que esta rota de obtenção é
economicamente inviável (1).
3.1.2 Fermentação
A fermentação é um processo conhecido desde a antiguidade e até hoje
o etanol é preparado da mesma forma (6).
Esse é o processo dominante, correspondendo a 90 % da produção
mundial do etanol, podendo ser obtido a partir de uma série de matérias-primas
como: açúcares (melaço, cana-de-açúcar, beterraba), amidos (milho, trigo,
aveia, mandioca, arroz, batata doce) e celulose (madeira, resíduos industriais
ou agrícolas). O Saccharomyces cerevisae e Zymonas mobilis são alguns
microorganismos utilizados, e as reações podem ser representadas como (1):

9
No Brasil, por exemplo, o etanol é obtido através do processo de
fermentação da cana-de-açúcar. Os demais países utilizam a beterraba e o
milho como matérias-primas e, por isso, o etanol é também chamado de “álcool
de cereais” (6).
O processo de fermentação do açúcar é ilustrado na Figura 2. A
produção se inicia numa bateria de três fermentadores que operam em um
processo continuo e, após passar pelo 3º fermentador, o conteúdo vai para o
decantador, onde recebe uma mistura de água, etanol, e óleo (o decantador
libera biomassa e proteínas). Em seguida, ocorre uma recirculação dessa
mistura, sendo que os 25 % retornam para o primeiro fermentador e os 75 %
restantes são enviados para um filtro prensa, que produz um composto rico em
proteínas vendido como ração animal (1).
C12H22O11 + H2O → 2 C6H12O6
Açúcares
C6H12O6 → 2 C2H5OH + 2 CO2
Celulose C6H12O6 → 2 C2H5OH + 2 CO2
[C6H10O5]x + x H2O → x C6H12O6
Amido
C6H12O6 → 2 C2H5OH + 2 CO2

10
Figura 3.2 - Produção industrial de etanol por fermentação (1).
Fora do Brasil a hidratação do etileno é o principal processo de
fabricação do etanol, a qual pode ser direta ou indireta. Atualmente, estima-se
que cerca de 80 % do etanol produzido nos Estados Unidos seja conduzida
através da hidratação do etileno (6). O etileno, também denominado olefina, é
um subproduto do processo de beneficiamento do petróleo, cujo processo de
hidratação é mais complexo que os processos de redução do acetaldeído ou
fermentação, já citados.

11
3.1.3 Hidratação do etileno
Para compreender como ocorre esse processo, primeiramente é
necessária uma abordagem química das olefinas: o que são olefinas, sua
importância e o que vem a ser processo de hidratação propriamente dito.
33..22 AAss oolleeff iinnaass
As olefinas são hidrocarbonetos cujas ligações entre carbonos são
realizadas através de ligações duplas em cadeias abertas, as quais podem ser
normais ou ramificadas. Elas não são encontradas no petróleo bruto; sua
origem vem de processos físico-químicos realizados durante o refino, como o
craqueamento, e possuem características e propriedades diferentes dos
hidrocarbonetos saturados (7).
A palavra olefina é um sinônimo usado para alcenos, que são
hidrocarbonetos insaturados, ou seja, de cadeia aberta tendo CnH2n como
fórmula molecular. As propriedades físicas desses compostos dependem de
sua cadeia carbônica, por exemplo: compostos de cadeias com menos de
cinco átomos de carbono são gases incolores, e são os de maior interesse para
aplicação industrial. Já os que contêm cinco ou mais carbonos são líquidos
incolores, exceto os que possuem uma cadeia carbônica muito grande (o que
não é comum), os quais podem ser sólidos (7, 8).
Não há formação de olefinas no óleo cru, caso haja a quantidade
formada é muito pequena, o que significa que a razão custo-benefício não é
compensadora. Porém, durante o refino, que envolve processos físico-químicos
de beneficiamento do óleo cru, a quantidade e a diversidade das olefinas
geradas são consideráveis (9).
Como as olefinas são muito reativas, sua utilização em misturas
químicas é limitada, pois elas podem sofrer reação de polimerização e/ou
oxidação quando estocadas. No entanto, de maneira geral, elas são muito
importantes para a indústria, porque além da possibilidade de sua aplicação

12
direta, elas podem ser utilizadas na síntese de diversos outros produtos, como
por exemplo: etanol, acetaldeído, ácido acético, óxido de etileno, polietileno,
polipropileno etc. Quando se fala em reações de conversões químicas ou
outros processos do setor petroquímico, as olefinas são compostos
intermediários importantíssimos, como por exemplo: propeno, buteno e eteno,
podendo até serem consideradas “subprodutos” do refino do petróleo,
enquanto que em outras reações são importantes precursores de produtos
comercializados (9). Neste trabalho a olefina em estudo foi o eteno, mais
conhecida por etileno - nome comercial.
3.2.1. Etileno
O etileno, alceno mais simples, cuja fórmula molecular é C2H4, é uma
molécula plana e com ângulos de ligação C-C-H de 121º e H-C-H de 118º,
valores muito próximos a 120º. A ligação dupla carbono-carbono é composta
de uma ligação forte, denominada ligação sigma (σ) com cerca de 100
kcal/mol, e uma ligação mais fraca, denominada ligação pi (π) com cerca de 45
cal/mol (7).
O etileno é o derivado químico gerado em maior quantidade durante o
processo de refino, podendo ser obtido a partir de outros compostos, como: o
etano, o gasóleo pesado ou do próprio óleo cru, dependendo somente da
necessidade econômica. As misturas fornecidas pelas refinarias de petróleo, ou
pelas companhias de gás natural, são separadas e seus compostos
intermediários, ou seja, os “subprodutos” são convertidos em precursores
reativos, como é o caso do etileno (9).
Durante o processo de refino ocorrem conversões químicas, muito
complexas na maioria das vezes, que formam vários produtos. No caso do
etileno, especificamente, a Figura 3 apresenta um fluxograma dos seus
possíveis derivados químicos.
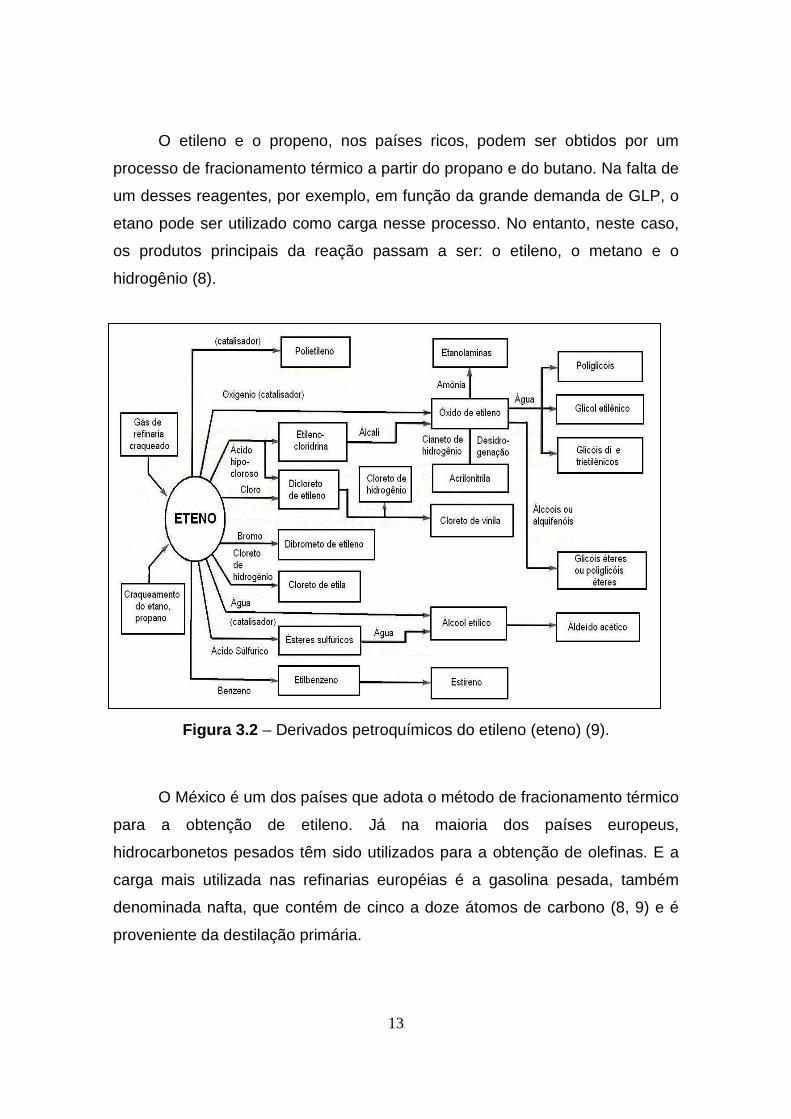
13
O etileno e o propeno, nos países ricos, podem ser obtidos por um
processo de fracionamento térmico a partir do propano e do butano. Na falta de
um desses reagentes, por exemplo, em função da grande demanda de GLP, o
etano pode ser utilizado como carga nesse processo. No entanto, neste caso,
os produtos principais da reação passam a ser: o etileno, o metano e o
hidrogênio (8).
Figura 3.2 – Derivados petroquímicos do etileno (eteno) (9).
O México é um dos países que adota o método de fracionamento térmico
para a obtenção de etileno. Já na maioria dos países europeus,
hidrocarbonetos pesados têm sido utilizados para a obtenção de olefinas. E a
carga mais utilizada nas refinarias européias é a gasolina pesada, também
denominada nafta, que contém de cinco a doze átomos de carbono (8, 9) e é
proveniente da destilação primária.

14
Como importantes conversões químicas na indústria do petróleo podem
ser citadas: craqueamento, polimerização, alquilação, hidrogenação,
isomerização, hidrocraqueamento e hidratação (9). A conversão química de
maior interesse para o desenvolvimento deste trabalho é a hidratação.
33..33 HHiiddrraattaaççããoo
A hidratação é uma reação que pode envolver inúmeras transformações
químicas, como por exemplo: transformar alcinos em compostos carbonílicos;
epóxidos em glicóis; nitrilas em amidas; e alcenos em alcoóis ou éteres (5).
Dentre os produtos químicos obtidos por hidratação catalítica os mais
importantes são: etanol, 2-propanol, propanona e 2-butanol. Os dois processos
sintéticos importantes de hidratação, para a produção do etanol, ocorrem ou na
forma de hidratação direta do etileno, ou hidratação indireta do etileno (9).
3.3.1. Hidratação Indireta do Etileno
Esse processo ocorre em duas etapas: na esterificação e na hidrólise.
Na etapa de hidrólise dos ésteres sulfúricos é originado, além do etanol, um
subproduto, que é o éter dietílico. Sabe-se que a principal impureza do etileno
é o etano, sendo que no caso especifico dessa reação, o rendimento é de 90 %
de etanol e 5 a 10 % de éter, o que já é uma grande desvantagem (1, 5, 9).
Esterificação
CH2=CH2 + H2SO4 → C2H5O-SO3H
ou
2CH2=CH2 + H2SO4 → (C2H5O)2SO2
Hidrólise dos ésteres sulfúricos
C2H5O-SO3H → C2H5OH + H2SO4
ou
(C2H5O)2SO2 → (C2H5)2O + H2SO4

15
A Figura 3.4 mostra como ocorre industrialmente esse processo: o
etileno diluído é colocado em contato com o ácido sulfúrico (98 %) em
contracorrente em uma série de colunas de absorção a 80 ºC e 1,3-1,5 bar. Por
ser uma reação exotérmica, a temperatura e o volume têm que ser controlados,
no caso, o controle térmico é feito com um sistema exterior de arrefecimento, e
o de volume, é realizado por uma medição rigorosa desses reagentes (1).
Figura 3.3 - Produção industrial do etanol por hidratação indireta do etileno (1).
Os gases que não são absorvidos saem no topo como gases para
combustão. Os ésteres sulfúricos e o éter dietílico absorvidos são hidrolisados
(a quantidade de água que é adicionada diminui a concentração do ácido para
50 %). Os produtos da hidrólise entram em seguida em uma série de colunas
de separação (stripping), as quais separam os produtos das impurezas, no
caso, do ácido sulfúrico. Este último é direcionado para um ciclo de
recuperação (sua concentração é feita em evaporadores sob vácuo). Já os

16
componentes voláteis, que saem do topo das colunas de separação, sofrem
um processo de lavagem (scrubbing) com água ou soda cáustica diluída e,
finalmente, o etanol é separado do éter dietílico por destilação fracionada (1, 9).
3.3.2. Hidratação direta do etileno
A reação em si não poderia ser mais simples, tanto que chega a
contrastar com o nível de complexidade do processo (1, 9).
É uma reação muito exotérmica (∆H2980= - 45 kJ/mol), e o fato de
acarretar uma diminuição do número de mols, indica que as condições ótimas
de reação são a baixas temperaturas - acima de 100 °C, a fim de impedir a
condensação de vapores de água, e a elevadas pressões. Em relação à
pressão, esta não pode elevar-se muito, acima de 70 bar pode ocorrer a
polimerização do etileno. Há uma aceleração dessa reação com o auxílio
catalítico, geralmente, ácido fosfórico (H3PO4) depositado na superfície de um
suporte de sílica (1).
A Figura 5 apresenta, esquematicamente, o processo industrial. Os
gases liberados no reator são condensados parcialmente, e sofrem dois
processos, lavagem e scrubbing: no primeiro, os vestígios de ácido fosfórico
são neutralizados, e ocorre a condensação de uma fração de água/álcool; no
segundo, depois da lavagem com água e uma purga, os gases restantes são
comprimidos e reciclados.
Hidratação Direta CH2=CH2 + H2O → C2H5OH

17
Figura 3.4 - Produção industrial de etanol por hidratação direta do etileno (1).
Na etapa seguinte, a mistura água/etanol é enviada para a primeira
coluna de destilação, para que componentes leves, como éteres, sejam
removidos; e depois, para a segunda, na qual é separada a água do azeótropo
de etanol. Ressalte-se que caso haja a formação de acetaldeído, este é
convertido em álcool por hidrogenação catalítica com níquel. O rendimento em
etanol em relação ao etileno é de 98,5 %. Infelizmente, sob condições
reacionais de 230-300 ºC e 60-80 bar o ácido fosfórico é perdido com o
transporte reacional e, conseqüentemente, ocorre uma queda na atividade
catalítica. Porém, o nível de atividade pode ser mantido pela adição contínua
de ácido fosfórico para alimentar a reimpregnação do catalisador no reator de
hidratação (1).
No processo utilizado pela companhia Shell, por exemplo, etileno e vapor
de água são introduzidos em um reator cíclico, mantido a 300 ºC e 70 atm de

18
pressão, empregando como catalisador o ácido sílico-fosfórico. A conversão
em cada ciclo é de 4 a 25 %, mas a conversão global alcança o valor de 92 %.
Tanto a hidratação indireta como a direta possuem pontos negativos. O
processo indireto, além da presença de subprodutos, como éter dietílico, requer
um agente ácido, por exemplo, ácido sulfúrico altamente concentrado (95-98
%). Esse agente é responsável pela forte corrosão dos reatores, por acidentes
de intoxicação, por queimaduras e por alto custo de recuperação do
catalisador, portanto, é um processo que tem sido abandonado. O emprego de
catalisadores ácidos no processo de hidratação direta, como o ácido fosfórico
suportado, também ocasiona problemas de corrosão no equipamento devido à
volatilização de produtos corrosivos, e ainda sua instabilidade térmica provoca
declínio na atividade catalítica (1, 5, 9).
Fougret, Atkins e Holderich (10) testaram a atividade catalítica do ácido
fosfórico em vários suportes: titânia pura, óxido de zinco/titânia, misto de
sílica/titânia, zircônia e ácido nióbico na reação de hidratação de etileno
(realizada em um reator de fluxo continuo a 50 bar e 300 ºC). Os resultados
mostraram que a natureza do ácido fosfórico não muda, quando impregnado
em sílica. E o mais importante, o fato da sílica ter poros pequenos, conservou
por mais tempo o ácido fosfórico na forma líquida, ou seja, ácido fosfórico livre,
que é pré-requisito para a atividade catalítica na reação de hidratação do
etileno. Dos materiais testados, foram observados desempenhos catalíticos
apenas nos catalisadores de sílica, de titânia e do misto sílica/titânia. A falta de
atividade nos demais catalisadores, como óxido de zinco/titânia, pode ser
explicada pela interação do ácido fosfórico impregnado nesses suportes.
Em outro trabalho apresentado por Fougret e Holderich (11) os
resultados mostraram que os catalisadores impregnados em alumina, zircônia
e fosfato de cério, empregados na hidratação de etileno, apresentaram
conversão maior que o catalisador de ácido fosfórico em sílica. Apesar disso, a
quantidade de ácido fosfórico arrastado durante a reação era muito grande, o
que não ocorreu no caso da sílica. No caso do ácido fosfórico, associado ao

19
fosfato de ferro, a interação foi muito pequena, provavelmente não existia ácido
fosfórico líquido livre, o que explica a quase inatividade desse material na
reação de hidratação. Realizar uma correlação entre a acidez e a atividade
ainda não era possível, porém, soube-se através deste trabalho que a atividade
era mais dependente do ácido fosfórico líquido na superfície do suporte.
3.3.3. O mecanismo geral de hidratação
O mecanismo químico das reações dos alcenos resume-se praticamente
à ligação dupla carbono-carbono, formada por uma ligação forte (σ) e uma
ligação mais fraca (π). Segundo a termodinâmica, as reações ocorrem em
função da quebra da ligação π (mais fraca) e, posteriormente, a substituição
desta por duas novas ligações σ (mais fortes). A própria geometria dos alcenos
já ajuda nesse mecanismo, pois os elétrons π ficam mais expostos tornando o
acesso aos mesmos, na maioria das vezes, cineticamente mais fácil. A ligação
π tem a capacidade de doar elétrons (base de Lewis), portanto, os melhores
reagentes para a reação de adição são aqueles que procuram elétrons, neste
caso, ácidos de Lewis ou reagentes eletrofílicos (9).
Geralmente, todas as reações que envolvem alcenos, tanto as de
eliminação quanto as de adição (reação reversa), possuem alguns pontos em
comum, por exemplo, é regra que nesse mecanismo os elétrons só sejam
transferidos aos pares, e que os catalisadores possuam centros ácidos e
básicos para auxiliar nessa etapa. Esse par ácido-base pode estar presente
tanto na solução como na superfície do sólido, e as duas reações (eliminação e
adição) podem ser efetuadas com catalisadores homogêneos e heterogêneos.
No caso da reação homogênea, catalisada por ácido forte ou base em solução,
o estudo é mais fácil, pois o mecanismo de eliminação e adição, catalisado por
ácido forte ou base em solução, já foi totalmente elucidado pelo esforço em
conjunto de pesquisadores na área de química orgânica (12).

20
O conhecimento adquirido com as reações homogêneas vem sendo
utilizado para a compreensão dos mecanismos heterogêneos. A química
orgânica desenvolveu toda uma nomenclatura para as reações homogêneas,
que esta também está sendo adotada para as reações catalíticas
heterogêneas, por exemplo, a letra E significa reação de eliminação e os
números 1 e 2 designam a molecularidade, respectivamente, reações mono ou
bimoleculares. Na catálise heterogênea o passo mais importante é a quebra da
ligação π. Por exemplo, em uma reação de eliminação bimolecular (E2), isso só
ocorre havendo um par de elétrons de centros ácidos ou básicos na superfície.
Nesse caso, a etapa determinante da velocidade é a eliminação
monomolecular (E1), na qual ocorre a ionização do reagente e a formação do
carbocátion, o qual se decompõe rapidamente para formar o alceno (12). A
regra de Markovinikov (Figura 3.6), do mecanismo geral das adições
eletrofílicas ou nucleofílicas, pode ser apresentada de forma geral como um
processo polar em duas etapas, mostradas a seguir.
Figura 3.5 - Mecanismo geral - regra de Markovinikov (7).
Ataque eletrofilico determinando a velocidade
Ataque nucleofilico rápido no carbocátion
ou
1 ) Etapaa
2 ) Etapaa
+ +lenta+H
-Z C C +
H
C C -Z
rápidoC C
H Z + -
Z C C +
H
+C C +
HZH
rápidoC C
H Z H +
rápido
C C
H Z+ +
H

21
O início do mecanismo reacional ocorre com a adição do próton na
ligação dupla, com a formação de um carbocátion convencional (complexo π)
onde o próton está situado entre os dois átomos de carbono. Em seguida, o
próton é transferido para um dos átomos de carbono e, obedecendo à regra de
Markovinikov (7), para o carbono que está ligado ao maior número de átomos
de hidrogênio (ligação σ). A última etapa é a formação da ligação entre a
espécie Z- e o segundo átomo de carbono; no caso especifico do etanol, Z é
uma hidroxila (OH) (12).
Quando a reação em estudo é uma adição (reação reversa), o
mecanismo envolve primeiramente a adição de um HZ (Z pode ser um –Cl, -Br,
-OH, etc) na ligação dupla C=C. A primeira etapa é a quebra da ligação dupla,
formando um carbocátion por um curto tempo de vida, antes da ligação π ser
quebrada, ressaltando-se que todo o processo ocorre com a molécula
adsorvida na superfície do sólido (12).
No processo de eliminação, o mecanismo corresponde a uma reação de
eliminação E1, a qual é catalisada por um ácido protônico forte. O mecanismo
da reação de adição heterogênea, por exemplo, é acionado pela água na
presença de um catalisador de Bronsted. Resumidamente, pode-se dizer que
os catalisadores de hidratação funcionam com trocas de íons ácidos e, nas
reações de eliminação ou adição, eles são de natureza ácido-básica e podem
ser encontrados nos óxidos ou óxidos mistos, como por exemplo: alumina,
sílica, zircônia, magnesita, sílica-alumina, sílica-magnesita (12).
A superfície de um catalisador sólido ácido-base não é, particularmente,
fácil de compreender, dependendo de seu volume e sua composição
cristalográfica. Todo óxido catalítico tem uma estrutura cristalográfica imperfeita
e nesta encontra-se uma apreciável quantidade de água adsorvida. São
justamente essas imperfeições cristalográficas que fazem toda a diferença para
a atividade catalítica, pois são nelas que se formam os grupos hidroxilas. Estes
grupos são, normalmente, anfóteros, dependendo da acidez do reagente,
porém, na maior parte do tempo eles atuam como centros ácidos, fornecendo

22
prótons para o reagente. A força desses ácidos de Bronsted depende da
estrutura do centro ativo, por exemplo: o grupo hidroxila (-OH) em sílica pura
não apresenta quase nenhuma acidez, enquanto que na superfície da alumina
os átomos exibem uma forte acidez, a qual chega a se igualar à do ácido
sulfúrico concentrado (12).
No caso especifico dos alcenos, que é o escopo deste trabalho, a reação
de hidratação é catalisada por ácido, que na presença de uma molécula de
água, forma álcool. O mecanismo reacional homogêneo para hidratação de
alcenos em meio aquoso ácido é apresentado, na Figura 3.7, de forma
simplificada. A protonação do alceno produz um carbocátion intermediário, que
determina a velocidade da reação global (5, 13).
Figura 3.6 – Mecanismo simplificado da reação de hidratação de alcenos em
fase homogênea (5).
De acordo com Kazansky (13), o mecanismo da transformação de
hidrocarbonetos catalisada por ácido foi originalmente sugerido por Bronsted.
Segundo essa teoria, os intermediários ativos das reações catalisadas por
ácido seriam formas protonadas de substratos, estes sendo mais reativos que
suas moléculas iniciais. Entretanto, a principal desvantagem dessa
aproximação clássica é a descrição muito formal das espécies protonadas, na
teoria não se menciona a estrutura real das moléculas ou suas interações com
o meio reacional. O mecanismo tinha que ser mais realista e, para isso, seria
necessária uma descrição molecular mais detalhada das etapas intermediárias
das reações catalisadas por ácidos, mas isso na época em que Bronsted
formulou sua teoria era impossível.
+ lentarápidoH O2 + +
H C C+
H C C +
H C C
OH

23
Agora, com o progresso técnico – cientifico, ficou mais fácil fazer uma
descrição molecular e explicar que se houver diferença na solvatação das
espécies protonadas, normalmente, haverá alterações no processo reacional, o
que causa discrepâncias nos produtos intermediários e, conseqüentemente,
nos produtos finais. Tudo depende da natureza e da concentração dos ácidos,
ou do solvente; por exemplo, nos super-ácidos existem íons carbônio e alquil
livres, enquanto que em solução aquosa os íons carbônio estão ligados aos
alquil formando espécies intermediárias (13).
Na catálise heterogênea, a superfície de um óxido tem sítios de
diferentes forças ácidas e básicas, capaz de catalisar diferentes reações.
Sendo possível determinar a distribuição dos sítios em termos de acidez e
basicidade podendo correlacioná-los frente à sua atividade.
Hoje é de grande importância o desenvolvimento de um novo catalisador
comercial mais eficiente, porque o utilizado atualmente (ácido fosfórico
suportado) ainda exibe alguns problemas de vaporização de ácido fosfórico
durante o processo, o que provoca, além da corrosão do equipamento, uma
diminuição na atividade catalítica.
Nesta busca surgiram vários catalisadores, um destes foi o trióxido de
tungstênio (WO3) suportado em zircônia (ZrO2). Este sistema foi estudado na
hidratação de propeno e na hidrólise de éster, tendo sido avaliado também na
hidratação de etileno. Os experimentos foram realizados em um reator contínuo
de leito fixo, a 473 K (198 ºC) e a pressão atmosférica. Os resultados
mostraram que a atividade máxima para a hidratação do etileno foi obtida a
1073 K (798 ºC), com uma razão W/Zr=0.4, maior que nos catalisadores
típicos. Uma das razões para esse resultado é a alta hidrofobicidade desse
catalisador, o que torna necessário salientar que tão importante quanto as
propriedades ácidas é a hidrofobicidade da superfície catalítica, pois o
desempenho da reação catalisada por ácido depende da presença de água
(14).

24
Existem na literatura artigos mencionando sólidos ácidos para a hidratação
de etileno, mas esses catalisadores não se tornaram comerciais devido à
inviabilidade econômica, à dificuldade de reprodução e/ou à atividade catalítica.
É necessário o desenvolvimento de um novo catalisador que seja de fácil
regeneração, não libere vapor corrosivo, bem como ativo e seletivo para a
reação de hidratação de etileno. Levando em consideração todos os pontos já
apresentados, a presente tese teve como objetivo principal o desenvolvimento
de um catalisador sólido à base de nióbio. Mas, porque utilizar o nióbio?
33..44 CCaattaall iissaaddoorr áácciiddoo ssóóll iiddoo àà bbaassee ddee nniióóbbiioo
3.4.1. Conceitos fundamentais referentes ao nióbio
O nióbio (Nb), elemento químico de número atômico 41, foi descoberto
em 1801, pelo químico inglês Charles Hatchett, que na época o nomeou
Colúmbio em homenagem ao descobridor da América, de onde veio o mineral
columbita, do qual o metal foi separado. Como inicialmente esse metal não
apresentou potencial de aplicação, foi esquecido. Todavia, cinqüenta anos
depois, o químico alemão Henrich Rose o redescobriu e o chamou de nióbio,
uma homenagem à filha do mitológico rei Tântalo, Níobe (deusa das lágrimas).
A literatura (15) começou a descrever a aplicação do nióbio em 1925, quando
foi utilizado com o intuito de substituir o tungstênio na produção de ferramentas
de aço. Até 1930, o nióbio não tinha importância industrial, mas apesar do alto
custo e da dificuldade de obtenção, passou a ser utilizado industrialmente por
sua eficácia na prevenção de corrosão intergranular em aços inoxidáveis (15-
18).
Como metal puro, o nióbio é mole e dúctil, sua estrutura cúbica de corpo
centrada permite um fácil deslizamento das suas camadas. Suas propriedades
químicas são semelhantes às do tântalo, tais como (15-18):
• Alta resistência à corrosão por ácidos minerais, com exceção do ácido
fluorídrico;

25
• Alta resistência ao ataque pela maior parte das substâncias orgânicas e
• Reage com oxigênio e nitrogênio em temperaturas acima de 300 ºC.
3.4.2. As reservas de nióbio e suas aplicações
A importância do nióbio (Nb) começou com as descobertas de depósitos
de pirocloro no Canadá (Oka) e no Brasil (Araxá), que ocorreram quase
simultâneamente, na década de 1950. A abundância do nióbio na crosta
terrestre é de aproximadamente 20 ppm, sendo encontrado, usualmente com o
tântalo em minerais (Fe,Mn)M2O6 em que (M=Nb, Ta). Os principais produtores
de nióbia são o Brasil, com cerca de 60 % da produção total, o Canadá, a
Nigéria e o Zaire (19).
O óxido de nióbio pode ser obtido a partir de dois processos distintos
(15, 16):
1) Do processamento da columbita-tantalita – a Columbita é uma mistura
isomórfica entre a niobita - (Fe, Mn) (Nb,Ta)2O6 e a tantalita (FeMn)(TaNb)2O6 .
É o processo mais difundido, em que o óxido de nióbio é obtido como
subproduto do tântalo.
2) Do pirocloro, cuja fórmula química é (Ca,Na)2(Nb,Ti,Ta)2O6(OH,F,O) –
Este processo é utilizado exclusivamente pela Companhia Brasileira de
Metalurgia e Mineração (CBMM), sendo o mais empregado atualmente e
responsável por mais de 90 % da produção mundial.
Com o início da exploração, na década de 1950, o nióbio tornou-se
abundante e ganhou importância no desenvolvimento de novos materiais.
Assim, ligas de nióbio foram desenvolvidas para utilização nas indústrias tanto
espacial quanto nuclear e, também, para fins relacionados à
supercondutividade. Citando algumas importantes utilizações do Nb2O5, temos:
superligas de níquel empregadas como componentes em turbinas de aviões;
fios de liga nióbio-titânio supercondutores, utilizados na fabricação de

26
equipamentos de ressonância magnética para diagnósticos médicos; microliga
na fabricação de automóveis, podendo ser utilizada para a exploração de óleo
e gás; liga leve na fabricação de jóias, por seu brilho levemente azulado
quando polido; nanomateriais, dispositivos optoeletrônicos e catalisadores (15-
18).
O óxido de nióbio (V) é um sólido insolúvel, de cor branca, sendo estável
ao ar e podendo ser muitas vezes descrito como anfótero; no entanto, é mais
caracterizado como inerte. Sua estrutura é extremamente complicada e
apresenta um amplo polimorfismo (19).
3.4.3. Estrutura da nióbia
O pentóxido de nióbio (Nb2O5) apresenta uma estrutura que envolve um
octaedro NbO6 ligado pelas bordas e cantos. A estrutura NbO2 só existe
quando a razão do oxigênio é mantida próxima a dois, por exemplo, um óxido
de composição NbO2.09 apresenta linhas de difração de raios X que são
características do pentóxido, mesmo que contenha somente um pequeno
excesso de oxigênio. Reduzindo o Nb2O5 (1300 – 1700 ºC) se produz o
monóxido de nióbio (NbO), de cor cinza, uma estrutura cúbica que apresenta
condutividade metálica, as linhas de difração de raios X começam aparecer no
NbO1.04, enquanto que os óxidos NbO0.94 e NbO0.87 mostram linhas de raios X
características do metal (19).
O pentóxido hidratado, mais conhecido como ácido nióbio, é obtido a
partir de um precipitado branco com indeterminada quantidade de água, isso
acontece quando os complexos solúveis do metal são hidrolisados ou quando a
solução de nióbia é acidificada. Em solução aquosa existem diferentes tipos de
espécies iônicas do óxido de nióbio como: [NbO2-(OH)4-3, Nb6O19
-8, HxNb6O19–(8-
x), sendo x=1,2 ou 3, e Nb12O36-12]. Estas espécies presentes são determinadas
em função do pH da solução e da concentração do óxido de nióbio, como
mostra a Tabela 1 (18).

27
Tabela 3.1 – Espécies de nióbia aquoso na faixa de pH de 14,5 a 0,55 (19).
pH da solução Espécies
>14,5 NbO2-(OH)4 3-
14,5 Nb6O19 -8
11,5 HxNb6O19 (8-x)-
6,5 Nb12O36 -12, Nb2O5.nH2O
3,65 Nb12O36 -12, Nb2O5.nH2O
0,55 Nb2O5.nH2O
O óxido de nióbio amorfo aumenta o grau de cristalinidade e forma fases
mais estáveis de Nb2O5 entre 300 e 1000 ºC. Os resultados (20) mostraram
uma diminuição muito grande da área específica do óxido de nióbio em função
do aumento da temperatura, devido à formação de grandes cristalitos de
Nb2O5.
Como unidade estrutural o óxido de nióbio amorfo Nb2O5.nH2O possui o
octaedro distorcido (NbO6), o pentaedro (NbO7) e o hexaedro (NbO8), esse
começa a cristalizar em baixa temperatura, a chamada forma T, do alemão tief
para baixo, ou γ, a aproximadamente 500 ºC, mas, a cristalização ocorre mais
rapidamente em altas temperaturas, até aproximadamente 830 ºC, quando
ocorre à transição M (“temperatura-média”) ou β, e a forma começa a tomar
aparência (19, 20).
Existe também uma fase intermediária, a fase TT-Nb2O5 (300 - 500 ºC)
que possui uma célula unitária pseudohexagonal com um defeito constitucional
de um átomo de oxigênio por célula unitária, e a forma pentagonal (tetragonal e
bipirâmide) com seis ou sete átomos de oxigênio coordenados ao átomo de Nb.
Essa transição continua mais rapidamente a altas temperaturas (1000 ºC) e
aquecida por 4 horas quando ocorre a completa conversão. Até que acima
1000 ºC ocorre a terceira transformação, a forma H ou α, que é a forma mais

28
estável termodinamicamente. Essas transições polimórficas acontecem
lentamente, são irreversíveis, e em temperaturas que ainda não estão bem
definidas (19, 20).
A forma H da nióbia (Figura 3.8-a) apresenta uma estrutura que consiste
na formação de blocos de octaedro NbO6 (3x4 e 3x5) que dividem o canto com
o octaedro do seu próprio bloco e a borda com o octaedro em outro bloco. Um
dos 28 átomos em cada célula unitária está presente em um sítio tetraédrico,
onde ocorre a junção do bloco. A forma T, visualizada na Figura 3.8-b,
apresenta uma estrutura totalmente diferente, a célula unitária contém 42
átomos de oxigênio posicionados (grandes círculos abertos). Oito íons da
nióbia estão presentes como um octaedro distorcido e outros oito íons como
uma bipirâmide pentagonal (19, 20).
Figura 3.7 - (a) Estrutura do H-Nb2O5. (losangos) NbO6 na forma octaedrica,
(�) Nb em sítio tetraédrico (19). (b) Projeção da estrutura paralela
do T-Nb2O5 no plano [001]; () oxigênio, (○,●) Nb no sítio
tetraédrico (19).

29
Muitas estruturas do pentóxido de nióbio podem ser agrupadas em baixas
e em altas temperaturas, porém o comportamento da cristalização depende do
material de partida utilizado, de impurezas que podem estar presentes e
alguma interação com outros componentes. A forma como ocorrem essas
interações, afeta as propriedades físicas (mobilidade) e químicas (redutibilidade
e acidez) do sistema catalítico contendo nióbia (19).
3.4.4 Propriedades ácidas da nióbia
Diferentes pesquisadores (21-23) descobriram que a interação do óxido
de nióbio com suportes de superfícies básicas resulta na formação de uma
estrutura altamente distorcida, enquanto que a interação com superfícies
ácidas resulta na formação de grupos NbO6, NbO7 e NbO8. A atividade
catalítica da superfície do óxido de nióbio depende do processo de preparação
e também está relacionada à ligação Nb=O. Numa reação de adição, por
exemplo, o número de coordenação dos átomos de nióbio influencia os sítios
ácidos e, portanto, a acidez, porém, a relação entre a estrutura e a reatividade
ainda não está totalmente esclarecida.
O óxido de nióbio hidratado (ácido nióbico - Nb2O5.nH2O) tem uma alta
força ácida (Ho= -5,6 ~ -8,2) e apresenta sobre sua superfície sítios ácidos de
Lewis, cujo número aumenta com o aumento da temperatura de pré-tratamento
acima de 500ºC, e sítios ácidos de Bronsted, que são mais abundantes a
100ºC e diminuem em alta temperatura.
A Figura 3.9 mostra a estrutura do óxido de nióbio como um possível
isopoliácido de composição H8Nb6O19, apresentando oito prótons sobre oito
faces triangulares de um octaedro formado por seis átomos de nióbio. Esses
prótons são estáveis e é essa característica que aumenta a força acida do
óxido de nióbio (24).

30
Figura 3.8 - Estrutura do nióbio isopoliácido (H8Nb6O19) (24).
Segundo diferentes trabalhos realizados (21, 22 e 24) o processo de
desidratação altera imediatamente a estrutura octaédrica distorcida NbO6
devido à remoção da água coordenada, mas não perturba, apenas distorce um
pouco a estrutura octaédrica NbO6. Se a distorção fosse forte, ou seja, alta
distorção da estrutura octaédrica NbO6, a ligação estabelecida seria Nb=O a
qual está associada com sítios ácidos de Lewis. Já uma distorção leve da
estrutura octaédrica, com grupos NbO7 e NbO8, só apresenta ligações do tipo
Nb-O e estas estão associadas aos sítios ácidos de Bronsted.
Existem, de acordo com a literatura (24), comportamentos diferentes para
a adsorção de água, metanol e etileno sobre uma superfície ordenada do óxido
de nióbio (Nb2O5), ou sobre uma superfície com defeitos. Os defeitos de
formação produzidos pelas vacâncias de oxigênio representam uma importante
função na adsorção de moléculas na superfície do óxido, por exemplo: a água
e o metanol são adsorvidos associativamente, e o etileno é fracamente
adsorvido na superfície do óxido ordenado à temperatura ambiente, porém, na
superfície do óxido com defeito a adsorção de água e metanol acontece de
forma dissociativa e torna-se forte a adsorção de etileno. Portanto a vacância
do oxigênio, representada na Figura 3.10, está relacionada com a origem da
acidez do óxido de nióbio.

31
Figura 3.9 - Superfície do óxido de nióbio mostrando a vacância do oxigênio
(24).
Outro ponto importante para a avaliação das propriedades ácidas é a
escolha da base, pois, segundo o conceito fundamental da teoria de Bronsted,
a transferência do próton é do ácido para a base, e dependendo do tipo de
base e da reação, muitos sítios ácidos deixam de ficar acessíveis na superfície.
A água pode ser utilizada como base, pois ela satisfaz muitos requisitos, dentre
estes, é uma molécula pequena, capaz de acessar facilmente todos os sítios
ácidos. As reações com água são quantitativas e equilibradas, e as espécies
formadas podem ser mais facilmente identificadas e sua concentração
determinada (25).
De acordo com a literatura (25) existem íons H3O+ e espécies H2O...OH,
e ocorre um aumento contínuo na concentração de íons H3O+ com o número
de moléculas de água. Por exemplo, numa hidratação completa 50 % dos sítios
ácidos do óxido de nióbio ficam ionizados, com isso apenas um de cada dois
sítios ácidos é forte. Tal fato é atribuído ao completo efeito de diluição e ao
sinergismo entre sítios ácidos de Bronsted e de Lewis, presentes no ácido
nióbico.
Enfim, o pentóxido de nióbio hidratado, Nb2O5.nH2O (ácido nióbico),
exibe, consideravelmente, uma alta força na presença de vapor de água, e por
todas as propriedades já apresentadas, ele vem sendo estudado e utilizado
como catalisador ácido em reações de desidratação, alquilação, condensação
e hidratação (26).

32
É importante ressaltar que o Brasil detém a maior reserva de nióbio do
mundo, e o desenvolvimento de tecnologias que utilizem esse elemento é
crucial para um melhor aproveitamento e valorização do mesmo.
3.4.5 Aplicações catalíticas da nióbia
O interesse em aplicações da nióbia na área catalítica vem crescendo
consideravelmente em vários grupos de pesquisas (19, 27-35). Isso se deve ao
avanço tecnológico, como as técnicas espectroscópicas, bem como físicas e
químicas.
Desde a última década, vem crescendo o interesse em óxido de nióbio
com estrutura mesoporosa, preparada só com nióbio ou sob a forma de um
óxido misto, contendo, por exemplo: nióbio e molibdênio; nióbio e alumínio;
nióbio e tungstênio, assim como outros catalisadores como: sulfetos, nitretos e
carbetos à base de nióbia (Figura 3.11) (29-38).
Figura 3.10 - Natureza química e espécies nióbio na catálise heterogênea (29).
Os compostos de nióbio exibem propriedades especiais, as quais
nenhum de seus vizinhos na tabela periódica possui, como: estabilidade e forte
interação do suporte com o metal, que são características importantes para um

33
catalisador (27-30). Mas, também exibem pontos desfavoráveis que são as
baixas mobilidades de oxigênio e redutibilidade, além do ponto de fusão muito
elevado (1512ºC). No entanto, para a catálise o mais importante é a
temperatura Tamman, na qual os átomos começam a difundir para a superfície.
Para a nióbia a temperatura Tamman é 620 ºC, valor não muito elevado, se
comparado às temperaturas de reações catalíticas típicas entre 200 e 600 ºC
(29). Outra característica da nióbia importante para a catálise é sua acidez, que
é muito dependente da temperatura de calcinação. Depois de calcinado a
400ºC, na maioria das vezes, o óxido de nióbio forma sítios ácidos de
Bronsted, porém, sob elevadas temperaturas de calcinação aumenta
relativamente o número de sítios ácidos de Lewis. Segundo a literatura (29), os
sítios de Lewis estão presentes em todos os sistemas de óxido de nióbio
suportados, mas os sítios de Bronsted são limitados aos sistemas Nb2O5/Al2O3
e Nb2O5/SiO2.
O óxido de nióbio e os óxidos mistos de nióbio, incluindo Nb2O5-SiO2,
Nb2O5-Al2O3 e Nb2O5-zeólita, têm apresentado resultados interessantes, como
a atividade catalítica de Mo-Ni/Nb2O5-Al2O3 para remoção de enxofre e
nitrogênio do gás-óleo mostrou-se muito mais efetiva que o Mo-Ni/Al2O3. Isto
em função da presença do nióbio no suporte que teria aumentado a acidez da
superfície (33). Na reação de hidrocraqueamento do cumeno o catalisador
Mo/Nb2O5 mostrou-se 60 vezes mais efetivo que o Mo/Al2O3 (37). Há trabalhos
(38) que relatam que sulfetos de nióbio exibem alta atividade catalítica em
reações de craqueamento e isomerização, pois a atividade intrínseca do NbS3
é maior do que a de MoS2.
Quando óxidos diferentes são misturados, materiais com diferentes
propriedades ácidas podem ser formados. De acordo com o ponto de vista de
Védrine e colaboradores (39), que resumiram trabalhos já publicados sobre
misturas de óxidos, há três diferentes propostas para explicar a acidez dos
óxidos mistos: a primeira é que óxidos mistos podem existir como uma solução
sólida, neste caso, o óxido em maior quantidade, porcentagem mássica, impõe

34
seu próprio ambiente de cátions ao óxido em menor quantidade, e se a carga
do cátion em menor quantidade for menor que a do cátion em maior quantidade
aparece a acidez, especificamente, a acidez de Bronsted, formada em função
do balanço dos cátions. A segunda proposta diz que sítios ácidos de Lewis e
de Bronsted podem ser formados se no óxido misto, o óxido em menor
quantidade preservar o seu número de coordenação ao redor dos íons do óxido
em maior quantidade. E, finalmente, a terceira proposta sugere que a acidez
aparece no limite onde dois óxidos estão em contato, uma espécie de fronteira.
Após a realização desse trabalho eles mostraram que um óxido misto Nb2O5-
Al2O3, com uma razão molar de 1:1, apresentou propriedades ácidas maiores
que a nióbia e a alumina separadamente.
Wachs e colaboradores (40) descreveram a estrutura e reatividade dos
óxidos metálicos do grupo V, inclusive comparando as propriedades físico-
químicas do Nb2O5 e V2O5. A redutibilidade do Nb2O5 é mais difícil que a do
V2O5, sendo que a área específica do Nb2O5 utilizado foi menor (1,9 m2/g) que
a do V2O5 (3,5 m2/g). Através de experimentos de quimissorção de metanol a
100ºC, foi constatado que o Nb2O5 mesmo com uma área específica menor,
tem cinco vezes mais sítios ativos em sua superfície que o V2O5, isto devido à
diferença de morfologia na superfície de ambos os óxidos. Segundo esses
autores o método de preparação (oxalato, alcoóxidos ou álcalis) não afeta a
estrutura molecular das espécies nióbia na superfície, mas pode afetar sua
dispersão, ou seja, sua densidade superficial. Por exemplo, catalisadores
suportados de nióbia possuem em sua superfície sítios das espécies Nb (+5),
os quais podem estar presentes de forma isolada (NbO4) ou na forma
polimerizada mono-oxo (NbO6).
No entanto, Schmal (41) et al., preparando catalisadores suportados
Nb2O5/Al2O3, verificaram que a natureza do precursor de nióbio influencia
significativamente a distribuição da nióbia sobre a alumina. Os resultados
mostraram que quando o precursor utilizado é o complexo de oxalato de
amônia e nióbia, há a formação de multicamadas de nióbia sobre a alumina, de

35
forma homogênea, porém, quando o precursor utilizado é apenas oxalato de
nióbio ocorre a formação de ilhas de nióbia sobre a superfície da alumina.
Portanto, a natureza do precursor afeta significativamente a redução das
espécies de nióbia. Com base nos resultados os autores concluíram que a
adição de óxido de nióbio diminuiu a fração dos sítios ácidos de Lewis (LAS-do
inglês Lewis Acid Sites) e aumentou a fração dos sítios ácidos de Bronsted
(BAS-do inglês Bronsted Acid Sites), isso ocorreu independentemente do tipo
de precursor utilizado.
Yangcheng et al. (42) prepararam um catalisador suportado Nb2O5/α-
Al2O3 para emprego na hidratação de óxido de etileno, produzindo monoetileno
glicol. Os resultados apresentados foram melhores que os obtidos com a
zeólita HZSM-5 e outros catalisadores sólidos. Este catalisador suportado
exibiu uma excelente estabilidade, não havendo desativação após 1000 horas
de teste, característica muito importante quando comparada à de outros
catalisadores já conhecidos, como o ácido sulfúrico e outros convencionais.
Em trabalho posterior (43), esse mesmo grupo estudou o efeito do
MgAl2O4 na acidez do catalisador suportado Nb2O5/α-Al2O3 e também na
reação de hidratação de óxido de etileno, e verificou que a modificação da α-
Al2O3 com MgAl2O4 leva a um aumento na basicidade e na resistência
mecânica do suporte. Esse material mostrou-se menos ácido que a α-Al2O3
pura e a acidez do catalisador triplo Nb2O5/ MgAl2O4/α-Al2O3 diminui com o
crescente aumento de MgAl2O4, sendo muito mais fraco que a de Nb2O5 puro.
Apesar dessas alterações, o desempenho do catalisador ainda foi muito bom,
havendo apenas uma redução, na seletividade para o produto monoetileno
glicol (de 100 % para 90,6 %), porém, a estabilidade permaneceu excelente
durante teste de 1000 horas.
Rehim e colaboradores (44) estudaram reações ácido-base,
desidratação de isopropanol a 180 °C, isomerização do 1-buteno a 75 °C e
desalquilação do cumeno a 450 °C, utilizando como c atalisador uma alumina
suportada em nióbia. Os resultados de adsorção de piridina, analisada por

36
infravermelho, mostraram que fracos sítios ácidos de Lewis foram criados pela
adição de nióbia na superfície da alumina, ao mesmo tempo em que fortes
sítios de Lewis foram cobertos. Os diferentes comportamentos observados nas
reações ácido-base ocorreram em função do tipo de reação frente ao nióbio,
por exemplo: a diminuição da atividade catalítica da reação de desidratação do
isopropanol foi causada pela diminuição da concentração de sítios básicos,
determinada por quimissorção de CO. A atividade na isomerização do 1-buteno
também diminuiu, porém, neste caso, o principal fator foi a diminuição da
concentração dos sítios ácidos de Lewis associados à alumina, pois a adição
de nióbia altera as vizinhanças dos sítios da alumina. Por outro lado, a criação
de sítios de Bronsted pela adição de nióbia aumentou a atividade da reação de
desalquilação do cumeno, sendo que sítios associados à espécie
tridimensional da nióbia parecem mais efetivos para essa reação.
Barros e colaboradores (45) impregnaram a nióbia com uma zeolita
ZSM-5, a fim de atribuir novas propriedades a esse material. De acordo com os
resultados obtidos, os autores mostraram que a adição do pentóxido de nióbio
diminuiu o volume de poros e a área específica da zeólita, porque o pentóxido
de nióbio poderia ter bloqueado os canais e cavidades da ZSM-5. Os
resultados de adsorção de piridina, monitorados por infravermelho, indicaram a
presença de sítios ácidos de Bronsted e de hidrogênio. Os resultados
indicaram a presença de uma monocamada de nióbio na superfície da ZSM-5.
Após toda a abordagem apresentada sobre aplicações catalíticas da
nióbia, fica evidente o grande potencial que esse material possui sozinho ou
com outros óxidos, e como catalisador mássico ou suporte. No entanto, a
compreensão do mecanismo das reações ainda contém muitas lacunas a
serem preenchidas, em função da dificuldade da própria caracterização da
nióbia.
Já que o presente trabalho visa o estudo da nióbia mássica e da mistura
Nb2O5-Al2O3 como catalisadores na reação de hidratação de etileno, é
necessário abordar as aluminas, suas características e particularidades.

37
33..55 CCoonnssiiddeerraaççõõeess ssoobbrree aa aalluummiinnaa
3.5.1. Informações gerais
A alumina é o nome freqüentemente utilizado em mineralogia, cerâmica
e ciências dos materiais para designar o óxido de alumínio - Al2O3 (46). Mesmo
antes de Cristo, os persas já fabricavam potes e recipientes de argila que
continham o óxido de alumínio, que os egípcios já utilizavam na fabricação de
cosméticos, medicamentos, corantes e tecidos (47).
Em 1821, o francês Berthier descobriu perto da aldeia de Les Baux, no
sul da França, um minério avermelhado que continha 52 % de óxido de
alumínio, tratava-se da bauxita. No ano de 1855, Henry Saint-Claire Deville
apresentou, em uma exposição em Paris, o primeiro lingote de um metal mais
leve que o ferro, era o alumínio, obtido pela primeira vez por via química. Em
1886, tornou-se público o processo de obtenção do alumínio, por meio da
redução eletrolítica da alumina dissolvida em banho fundido de criolita
conhecido como processo Hall-Héroult, o que permitiu o estabelecimento da
indústria global do alumínio (47). No entanto, foi Karl Joseph Bayer, entre os
anos de 1887 e 1888, quem desenvolveu o processo de refino da bauxita
mundialmente conhecido (48).
A principal função do Processo Bayer era a produção de alumina, com
fins metalúrgicos, para a indústria do alumínio. Porém, o processo tornou-se
uma fonte de hidróxido de alumínio puro de baixo custo para a indústria
química, utilizado até os dias de hoje. As maiores minas de extração de bauxita
estão na Austrália, Iugoslávia, Jamaica, Grécia e Brasil, e elas fornecem
matéria-prima para diversas áreas (49).

38
Figura 3.11 - Processamento da bauxita e produção de aluminas (49).
A alumina é hoje um dos produtos inorgânicos puros fabricados em
maior escala, e embora sua produção ainda esteja mais voltada para a
produção do alumínio na forma metálica, sua aplicação em outras áreas vem
crescendo consideravelmente. O desenvolvimento de pesquisas em materiais
cerâmicos à base de óxido de alumínio (alumina) tem se intensificado nas
últimas décadas, principalmente, pelo baixo custo e por suas características
físicas e químicas. O objetivo das pesquisas atuais é explorar as aplicações
potenciais da alumina, que ainda não são aproveitadas integralmente. (49).
A primeira aplicação prática da alumina ocorreu no início do século XX,
como isolante para velas de ignição e equipamentos de laboratório, depois
vieram as aplicações na eletrônica e engenharia mecânica, isto levando em
consideração apenas sua alta resistência mecânica (50). Nos últimos anos, as

39
aplicações que mais têm despertado o interesse são: material para
revestimento (blindagem), instrumentos cirúrgicos, azulejos resistentes à
abrasão, pigmentos, e o uso como catalisadores e suportes, este em função de
suas diferentes estruturas cristalinas (51-58).
3.5.2. Morfologia da alumina
Apesar da aparente simplicidade da fórmula Al2O3, as características da
alumina dependem de uma série de fatores, como; forma cristalina, impurezas
e microestrutura. Os estudos já realizados indicam a existência de sete fases
cristalográficas principais, que são: alfa, gama, delta, eta, theta, kappa e chi
(Figura 3.13), dependendo do precursor e da temperatura na qual o tratamento
térmico é realizado.
A α-Al2O3 é um óxido de alumínio completamente anidro, preparada a
partir dos hidróxidos de alumínio, ou oxi-hidróxidos, acima de 1200 °C, se a
temperatura aplicada for menor, então se tem as chamadas aluminas de
transição, cada uma com suas respectivas propriedades. O aquecimento de
uma gibbsita, por exemplo, a 150 °C gera boemita mi crocristalina, e a 400 °C
resulta na série de aluminas gama, que inclui os tipos chi (χ), eta (η) e gama
(γ). Em temperaturas mais altas, cerca de 1000 °C, é formada a série de
aluminas delta (δ); esta contém muito poucos grupos OH e inclui as variedades
kapa (κ), theta (θ) e delta (δ), são muito mais cristalinas que as aluminas da
variedade gama (γ). As γ e η-aluminas raramente são encontradas em fases
puras, são conhecidas como aluminas ativadas e são as mais importantes
cataliticamente, devido às suas propriedades de quimissorção distintas (54,
58).
As aluminas de transição são estabilizadas pelas baixas energias de
superfície, e a mais abordada pela literatura é a γ- Al2O3, uma forma
policristalina com alta área específica, apresentando propriedades estruturais e
aplicações muito diversificadas, principalmente, na área da catálise (54-59).

40
Figura 3.12 – Estruturas cristalinas da alumina (54).
De modo geral o termo alumina é, normalmente, utilizado para designar
o conjunto de sólidos iônicos obtidos pelo aquecimento das formas amorfas e
cristalinas de Al(OH)3 e AlO(OH). A existência de um grande número de oxi-
hidróxidos de alumínio, diferentes entre si química e fisicamente, foi um fator
determinante no desenvolvimento dos vários tipos de alumina (Figura 3.14),
que estão atualmente no mercado, pois, a estrutura de uma determinada
alumina depende do seu grau de hidroxilação (54, 58).

41
Figura 3.13 - Classificação dos oxi-hidróxidos de alumínio (54).
As aluminas totalmente hidroxiladas correspondem aos trihidróxidos
(Al(OH)3) e têm como formas cristalinas a bayerita, a gibbsita e a nordstrandita.
Em determinadas condições que impedem a incorporação de hidroxilas, ou sob
tratamento térmico, são formados os oxi-hidróxidos, a boemita e a diáspora. A
pseudo-boemita, um oxi-hidróxido pouco cristalino com água em excesso, é
formada em substratos planares. Outras formas de alumina são discutidas,
porém, não se sabe ao certo se realmente são fases diferentes, ou seja, novas
fases; ou apenas distorções do retículo cristalino pela presença de impureza,
ou água adsorvida (54).
3.5.3. Aplicações catalíticas
Nas últimas décadas, houve um aumento do interesse no uso de
hidróxidos e oxi-hidróxidos, como precursores das aluminas de transição
empregadas como suportes ou catalisadores. Enquanto suporte catalítico, a
alumina é muito utilizada, em função de ser um material de baixo custo e

42
estruturalmente estável, podendo ser preparada com uma grande variedade de
volume de poros e distribuição dos diâmetros de poros (54, 57,58).
Na área acadêmica, as aluminas puras são amplamente utilizadas há
muito tempo, como catalisadores para reações que envolvem a ativação de
ligações, por exemplo, hidrogênio-hidrogênio, carbono-hidrogênio e carbono-
carbono. Isso evidencia as propriedades químicas da superfície da alumina,
que permitem desse material utilização como catalisador em uma série de
reações ácido-base, como: isomerização e epoxidação de olefinas,
halogenação de aromáticos, desidratação de alcoóis, entre outras (5, 54-59).
Um fato importante a ser discutido sobre as aluminas é em relação às
questões energéticas dos grupos iônicos presentes em sua superfície, onde a
terminação do cristalito é realizada pelos grupos OH.
Há alguns modelos discutindo a existência de diferentes freqüências de
estiramento OH, observados pela espectroscopia na região do infravermelho,
quando aluminas de transição são expostas à água (60-63). O modelo de Peri
(62), por exemplo, propôs que a γ-alumina tem um plano (100) completamente
hidroxilado e os íons Al3+ estão localizados em uma camada logo abaixo, em
sítios octaédricos. A desidroxilação com a remoção aleatória de pares OH foi
investigada através de simulações matemáticas, no início, sem a criação de
sítios defeituosos, mas com uma subseqüente formação de defeitos por íons
Al3+ expostos e íons O2-. Com base nas discussões matemáticas, Peri (62)
identificou espécies na superfície, cujas concentrações eram interdependentes
e controladas pela temperatura de ativação da alumina (Al+3), e cinco tipos de
grupos OH- cercados por O2- observados por espectroscopia na região do
infravermelho.
A Figura 3.15 apresenta a proposta de Peri para os cinco sítios que
apareceram após a desidroxilação da γ-alumina a temperaturas entre 600 e
700° C. O grupo OH no sítio A, por exemplo, tem quatro óxidos adjacentes e,
por isso foi considerado o mais básico, devido ao efeito indutivo dos óxidos,

43
sendo aquele que apresentou o maior número de onda. Seguindo o mesmo
raciocínio, o sítio E é o mais ácido.
Figura 3.14 - Superfície das aluminas antes (a) e após (b) a ativação segundo
o Modelo de Peri (62), sendo que, (+) denota uma subcamada de Al3+ (62).
O outro modelo descrito na literatura foi o proposto por Knözinger e
Ratnasamy (60), que além de explicar os dados de infravermelho, foi coerente
com outros dados químicos e espectroscópicos da época. Os resultados
obtidos por Knözinger-Ratnasamy indicaram cinco configurações de grupos OH
diferentes na superfície das aluminas (Figura 3.16) e, como estes grupos
teriam cargas diferentes, deveriam ter propriedades diferenciadas. A freqüência
de vibração foi relacionada à carga residual do grupo OH (σOH) correspondendo
à soma da carga do ânion com a carga do cátion, dividida pelo número de
coordenação do cátion.
A banda de mais alto número de onda, de acordo com as leituras no
infravermelho foi a 3800 cm-1, que corresponde à configuração Ib, de carga
residual mais negativa, e a banda de 3700 cm-1 corresponde à configuração III.
De acordo com isso, as intensidades relativas das bandas de OH variam de
a) b)

44
acordo com o tipo de alumina estudada, pois, dependem consideravelmente da
distribuição na superfície do material (54).
Figura 3.15 - Configurações do grupo OH na superfície da alumina com suas
respectivas cargas residuais (σOH), de acordo com o modelo de Knözinger-
Ratnasamy (54).
Os modelos de Knözinger-Ratnasamy (K-R), Tsyganenko–Mardilovich (T-
M) e Busca–Lorenzelli (B-L) ajudaram a elucidar algumas lacunas sobre as
propriedades reativas da alumina. Esses modelos demonstraram a
sensibilidade da utilização do infravermelho, ao realizar as medidas dos
primeiros vizinhos da hidroxila (OH-Al), e do segundo vizinho, em que o
número de anions em volta dos íons Al3+ (OH-Al-X) independe se X é um íon
óxido ou outro OH.
No modelo B-L (Figura 3.17) três vizinhos foram incluídos na
representação, porque, para algumas configurações era importante determinar
se o íon Al3+ estava adjacente ou não a uma vacância de sítio catiônico. Estes
sítios poderiam estar presentes de qualquer forma, de acordo com a
quantidade de Al2O3, ou seja, uma disposição OH-Al-X-Al era completamente
diferente de OH-Al-X-� (60). Esta distinção não foi considerada para os grupos
II e III no modelo K-R, talvez porque na época não havia muitas possibilidades
para fundamentar as discussões, que pudessem explicar que devido à alta
diversidade estrutural dessas espécies suas bandas eram mais largas.

45
Figura 3.16 – Configurações das hidroxilas na superfície de uma γ-alumina no
modelo de Busca–Lorenzelli (B-L), com base nas freqüências dos estiramentos
νOH (60).
Outro fato, que não foi considerado por esses modelos, fora as
interações dipolo-dipolo (Figura 3.18), sendo que estas se mostraram
importantíssimas no modelo de Peri (62). O modelo T-M, na realidade,
enfatizou que pares OH geminais (OH-Al-OH) poderiam ser encontrados em
alguma face, mas, determinava que as interações dipolo-dipolo fossem
ignoradas, sem nenhuma justificativa (60).

46
Figura 3.17 – (a) Interação dipolo-dipolo entre as hidroxilas na γ-alumina no
modelo de Peri; (b) uma representação da interação dipolo-dipolo na
configuração geminal das hidroxilas negligenciada no modelo T-M (60).
Na realidade, ainda há muito a ser discutido, principalmente, a nível
nanométrico, mas apesar das discordâncias e questionamentos, há um
consenso entre os pesquisadores, de que a adsortividade e as propriedades
reativas da alumina são governadas pelas hidroxilas da superfície, ou seja, as
espécies OH.
Estudos realizados por calorimetria da dissolução em alta temperatura
da alumina (64, 65) mostraram que a diferença de entalpia entre as fases alfa e
gama, com áreas semelhantes, diminui com o aumento das áreas, o que
implica numa menor energia livre de superfície para a gama alumina. Os
resultados experimentais não apresentaram uma diferença significativa na
energia de superfície entre os polimorfos hidratados. Os pesquisadores
justificaram tal fato em função da adsorção das moléculas de água, que
segundo eles, cobriam os defeitos da superfície da alumina ao se ligarem aos
íons de coordenação incompleta. Ainda segundo esses trabalhos a molécula
de água poderia se adsorver de duas maneiras na superfície da alumina: a
primeira, em um processo de quimissorção, existindo como íons hidroxila
ligados a Al3+ em várias configurações e também poderiam existir moléculas de
água quimissorvidas através de pontes de hidrogênio; a segunda seria através
a) b)

47
de um processo de fisissorção, dependendo da temperatura e da pressão
parcial da água.
Um artigo recente (66) corroborou com as conclusões anteriores.
Segundo este, a diferença de acidez na superfície de uma alumina reflete a
diferença de sua composição química e que os íons alumínio afetam,
consideravelmente, a acidez, enquanto que os íons óxidos afetam a
alcalinidade da superfície. A Figura 3.19 apresenta os três estados da
superfície de uma gibsita: o primeiro obtido através da adsorção física da água,
o segundo por adsorção química e o terceiro com as pontes de oxigênio (66).
Figura 3.18 – Os três tipos de estado da superfície de uma gibsita (66).
Comparando os três tipos de oxi-hidróxido de alumínio, pôde-se
compreender que a diferença fundamental da morfologia da superfície poderia
ser atribuída aos tipos e à combinação de uma ou duas hidroxilas ligadas aos
íons alumínio. Quanto mais hidroxilas ligadas ao alumínio, maior a acidez
dessa superfície, e quanto mais oxigênios maior a basicidade (66).
Peri (63) já havia divulgado um estudo de adsorção de amônia na γ-
alumina por infravermelho, no qual ele fez questionamentos e toda uma
discussão dos prováveis tipos de sítios ácidos (Bronsted e Lewis) existentes na
alumina. No entanto, a literatura diverge muito quanto ao mecanismo de ação
das aluminas como catalisador, alguns pesquisadores defendendo a idéia de
que somente sítios de Bronsted participam do processo reacional, enquanto

48
outros acham que os sítios de Lewis é que são os ativos, e há ainda quem
acredite que a atividade é dada em função da acidez total.
Compreender as reações na superfície das aluminas é complexo, porque
envolve uma série de fatores como: cinética de hidratação, transformação de
fase, inclusão de impurezas, etc. No entanto, é muito importante estabelecer
mecanismos reacionais para aperfeiçoar as reações industriais de muitos
processos.
Na indústria, são inúmeras as reações químicas que utilizam algum tipo
de catalisador e, de acordo com o objetivo deste trabalho, considerou-se
pertinente comparar os resultados obtidos, utilizando os materiais em estudo à
base de óxido de nióbio, com os já conhecidos pelos meios acadêmicos e
industriais, no caso escolheu-se uma zeólita. As zeólitas são sólidos
microporosos utilizados como catalisadores ácidos, na maior parte dos
processos industriais envolvendo catálise ácida.
33..66 CCoonnssiiddeerraaççõõeess ssoobbrree aass zzeeóóll ii ttaass
3.6.1. Histórico e conceito
O conhecimento sobre as zeólitas iniciou-se com a descoberta da stilbita
em 1756, descrita primeiramente pelo mineralogista sueco Baron Axel
Cronstedt, ele não chegou a concluir que as pedras “ferviam” sob aquecimento,
devido à água armazenada nos seus microporos, característica esta que deu
origem a seu nome (do grego: zeo=ferver e lithos=pedra) (59, 67-69).

49
Weigel e Steinholf, em 1925, foram os primeiros a constatar que a
chabazita absorvia seletivamente moléculas orgânicas menores e rejeitava as
maiores. Contudo, as zeólitas só apresentaram grande interesse após a
década de cinqüenta, quando foi possível sua síntese em escala industrial.
Após muitas pesquisas, depois de duzentos anos aproximadamente, McBain
criou em 1932, o conceito de peneira molecular, que se aplica a sólidos
porosos capazes de adsorver seletivamente (Figura 3.20) moléculas de
diferentes tamanhos (67-69).
Figura 3.19 - Tipos de seletividade com peneiras moleculares (59, 68).

50
Há aproximadamente quatro décadas que as zeólitas, como a estilbita
de Cronstedt, começaram a ser empregadas como catalisadores para as mais
variadas reações químicas. Um breve histórico dos principais
desenvolvimentos da ciência relacionados à catálise com peneiras moleculares
é mostrado na Tabela 3.2.
Tabela 3.2 - Breve histórico da catálise com as peneiras moleculares (59, 68).
Séc. VIII Jabir Ibn Haiyan (Geber) descreve a desidratação de álcool para produzir
éter utilizando ácido sulfúrico como catalisador
1756 Cronstedt descobre as zeólitas (em grego, zeo=que ferve, lithos=pedra)
1836 Berzelius cria o termo “catálise” (em grego, katalusis significa dissolução)
1845 Síntese hidrotérmica de quartzo a partir de gel de sílica por Schafhautle
1862 St. Claire Deville faz a 1ª síntese (hidrotérmica) de zeólita: a levinita
1895 Ostwald estabelece a natureza cinética da catálise
1911 Sabatier sugere existirem intermediários na superfície de catalisadores
1925 Weigel e Steinhoff observam o efeito de peneira molecular na chabazita
1930 Primeira determinação de estrutura de zeólita é feita por Taylor e Pauling
1932 McBain introduz o conceito de peneira molecular
1936 Craqueio catalítico: Processo Houdry com SiO2-Al2O3
1938 Brunauer, Emmet e Teller descrevem método para medir área específica
1945 Richard Barrer classifica zeólitas baseado em propriedades de peneira
molecular 1948 Milton sintetiza zeólitas de estrutura desconhecida na natureza
1962 Utilização de zeólitas como catalisadores de craqueamento
1968 A seletividade de forma nas zeólitas é descoberta
1982 Wilson sintetiza uma série de peneiras moleculares de aluminofosfatos
1983 A Enichem, na Itália, introduz a TS-1, a primeira peneira molecular redox
1985 Conversão do metanol em gasolina: Processo Mobil com ZSM-5
1986 N. Herron (Du Pont) cria a peneira barco-na-garrafa (Co-salen+zeólita Y)
1988 Davis sintetiza a VPI-5, uma peneira molecular com poros de 12 Å

51
A partir da aplicação pioneira em processos de craqueamento de
petróleo, em 1962, as zeólitas assumiram a posição de catalisador de suma
importância na indústria química, chegando a ser considerada a pedra filosofal
do químico contemporâneo, um catalisador ideal. O mecanismo desse
catalisador onírico funcionaria como uma pinça molecular, imobilizando cada
molécula de substrato na posição apropriada para romper somente a ligação
química necessária, a fim de formar o produto esperado com altíssima
atividade e seletividade absoluta.
De acordo com a definição clássica, o termo zeólita abrange somente
aluminossilicatos cristalinos hidratados de estrutura aberta, constituída por
tetraedros de SiO4 e AlO4 ligados entre si pelos átomos de oxigênio. No
entanto, o uso desse termo já foi estendido para designar estruturas análogas
contendo também tetraedros de outros elementos (PO4, GaO4, etc.).
Atualmente, as zeólitas abrangem, quimicamente, os aluminossilicatos
cristalinos hidratados, de estrutura aberta, constituída por tetraedros TO4 (T =
Si, Al, B, Ge, Fe, P, Co...) ligados entre si através de átomos de oxigênio, e a
fórmula química por célula unitária é Mx/n [(AlO 2)x (SiO2)y]. m H2O, em que: M
é o cátion de valência n, m é o número de moléculas de água e (x + y) é o
número de tetraedros por célula unitária (67-69).
A União Internacional de Química Pura e Aplicada (IUPAC) classifica as
zeólitas utilizando um código de três letras baseado somente na estrutura,
independente da composição química, exemplificado na Tabela 3.3. Os
microporos das zeólitas são classificados de acordo com o tamanho: poros
pequenos (< 4 Å), médios (4 – 6 Å), grandes (6 – 8 Å), ou supergrandes (> 8
Å). Peneiras moleculares com poros maiores que 20 Å, com paredes amorfas,
são classificadas como mesoporosas (68).

52
Tabela 33 - Características dos poros de algumas peneiras moleculares (68).
3.6.2. Estrutura geral e a zeólita y
As estruturas das peneiras moleculares são baseadas em uma rede
tridimensional, na qual tetraedros TO4, que são unidades primárias de
construção, estão interconectados através dos átomos de oxigênio. A união de
tais tetraedros leva à formação de um octaedro truncado, o qual consiste de 24
tetraedros TO4, dando origem a uma estrutura básica chamada unidade
sodalita, ou cavidade β, encontrada nas zeólitas A, X e Y. Se as unidades
sodalitas ligarem-se pelos prismas hexagonais surge então a estrutura faujasita
(FAU), com uma “supercavidade” (φ =1,20 nm) interconectada por “janelas”
contendo 12 átomos T. Uma faujasita (Zeólita X) apresenta como célula unitária
a fórmula Na56[(AlO2)56(SiO2)136].250H2O (70).
Tamanho do
microporo
Diâmetro
do poro
(Å)
Nome
comum
Símbolo
estrutural
Dimensio-
nalidade*
Maior molécula
adsorvível
Pequeno 4,1 Zeólita A LTA 3 n-hexano
médio 5,3x5,6
3,9x6,3
5,5x6,2
TS-1, ZSM-5
AIPO-11
ZSM-12
MFI
AEL
MTW
3
1
1
ciclohexano
ciclohexano
-
grande 7,3
7,4
~6x~7
AIPO-5
Zeóltia X, Y
Zeólita β
AFI
FAU
BEA
1
3
3
neopentano
tributilamina
-
supergrande 7,9x8,7
12,1
13,2x4,0
AIPO-8
VPI-5
Cloverita
AET
VFI
CLO
1
1
3
-
Triisopropilbenzeno
-
Mesoporoso 15-100 MCM-41 1 -
* Dimensionalidade 1=canais unidirecionais. 2=canais cruzados. 3=canais nas três direções x, y e z.

53
De acordo com a sua Unidade de Construção Secundária (UCS), esse
material pode ser classificado, de acordo com a Internacional Zeolite
Association (IZA), como DR6 (Double Six Ring), o qual possui 12 tetraedros na
abertura da cavidade principal (supercavidade) e cerca de 7,5 Å de abertura de
canal. A zeólita Y possui cela unitária cúbica, com dimensões de,
aproximadamente, 25 Å, e com 192 tetraedros TO4 (T = Si, Al), e a união
destes tetraédros forma octaedros truncados, cuja associação origina a célula
unitária, representada na Figura 3.21 (70).
Figura 3.20 - Estruturas FAU e LTA a partir de tetraedros de TO4 (68).
Como os canais e os poros são os responsáveis pela difusão e
acessibilidade dos substratos a sítios específicos existentes nas zeólitas, sua
natureza e distribuição são fundamentais para determinar as propriedades
físico-químicas das peneiras moleculares. Segundo a literatura (67), a estrutura
e distribuição desses canais variam consideravelmente, sendo que, há três
classes principais identificadas:

54
1) unidimensional – não ocorre a interconexão entre os canais, como por
exemplo, ocorre na zeólita L;
2) bidimensional – a interconexão ocorre, mas, é restrita a alguns planos,
como acontece na mordenita;
3) tridimensional – subdivide-se em dois tipos: canais que possuem
diâmetros diferentes em função da direção cristalográfica, por exemplo
na gmelinita; e com os diâmetros de todos os canais idênticos,
independente da direção, como, por exemplo, a zeólita Y.
As principais aplicações das zeólitas são em adsorção e catálise. A zeólita
tipo Y é mundialmente usada como catalisador de craqueamento de petróleo
desde os anos 60. No Brasil, é produzida pela Fábrica Carioca de
Catalisadores (FCC), para uso pela PETROBRAS em suas refinarias. A
produção mundial de zeólita sintética é estimada em 1,5 milhões t/ano, sendo
que grande parte se destina à manufatura de detergentes, e cerca de 1/3 aos
processos catalíticos (69).
3.6.3. Aplicações das zeólitas
A eficiência das zeólitas em catálise se deve a algumas características
peculiares, que esses materiais possuem (69):
1) Alta área superficial e capacidade de adsorção;
2) Propriedades de adsorção que variam num amplo espectro desde
altamente hidrofóbicas a altamente hidrofílicas;
3) Uma estrutura que permite a criação de sítios ativos, tais como sítios
ácidos, cuja força e concentração podem ser controladas de acordo com
a aplicação desejada;
4) Tamanho de canais e cavidades compatíveis com a maioria das
moléculas das matérias-primas usadas na indústria e

55
5) Uma complexa rede de canais que lhes confere diferentes tipos de
seletividade de forma, de reagente, de produto e de estado de transição.
As zeólitas têm o potencial de promover uma série de reações químicas
sobre a sua superfície ácida, sendo que a maioria desses processos está na
área de petroquímica e de refino de petróleo. Dentre algumas destas
aplicações tem-se (59, 71): hidroisomerização um processo de obtenção da
gasolina com índice de octanagem elevada; isomerização de hidrocarbonetos
aromáticos para se obter orto e para-xileno, a partir de hidrocarbonetos
aromáticos em C8; alquilação do tolueno para formar matérias primas de alto
valor agregado, como os xilenos; processos MTG (Metanol To Gasoline) e
MTO (Metanol To Olefins); reações de craqueamento e hidrocraqueamento em
que frações pesadas de óleo são convertidas em frações leves empregadas
como combustíveis.
Segundo a literatura (71), a reação secundária de formação de gases
leves a partir de gasolina é inibida devido à rápida formação de compostos
aromáticos e isoparafinas sobre os sítios ácidos das zeólitas, e o catalisador
usado normalmente para o craqueamento é a zeólita Y. A razão das zeólitas
terem se tornado uma tecnologia promissora em catálise heterogênea foi
devida às vantagens que elas apresentaram em relação aos catalisadores
homogêneos (59).
Os sítios ácidos da zeólita encontram-se no interior de seus poros,
portanto, pode ser manuseada muito mais facilmente do que, por exemplo, o
ácido sulfúrico. Além disso, alguns tipos possuem acidez cerca de 10 milhões
de vezes mais fortes do que o ácido sulfúrico concentrado (59, 69). Por isso,
inicialmente, as zeólitas foram consideradas um grande bônus para o meio
ambiente, chegando até a serem classificadas como resíduo não perigoso pela
EPA (Agência Norte-Americana de Proteção ao Meio Ambiente). Entretanto,
esse conceito foi revisto, em 1996, pelo fato de que, após serem usados, nas
unidades petroquímicas em especial, tais catalisadores tornaram-se poluentes
em potencial, devido a três fatores básicos: o teor, normalmente, considerável

56
de metais pesados; a presença de compostos altamente cancerígenos,
presentes nos inevitáveis depósitos de coque, que se alojam sobre sua
superfície no decorrer de seu uso, que é a principal causa de desativação das
zeólitas, e a elevada ácido-basicidade desses materiais, muito superior à dos
solos.
33..77 MMééttooddooss ddee ccaarraacctteerr iizzaaççããoo
3.7.1. Termogravimetria
O campo da análise térmica compreende vários métodos dos quais os
principais são os seguintes: ebuliometria, calorimetria, titulações entálpicas ou
termométricas, termogravimetria (TG), termogravimetria derivada (TGD),
análise térmica diferencial (ATD), análise de evolução térmica gasosa,
calorimetria exploratória diferencial (CED). No caso específico da
caracterização de catalisadores, através de análise térmica, os métodos mais
empregados são TG, TGD e ATD, para avaliar a estabilidade térmica dos
catalisadores (72-73).
A TG é usada para se estudar o caminho detalhado das alterações que o
aquecimento pode provocar nas substâncias, objetivando estabelecer a faixa
de temperatura, nas quais o material adquire composição química definida ou
temperatura, em que se inicia algum processo de decomposição, sinterização,
mudança de fase, etc. Assim as curvas de variação de massa em função da
temperatura, obtidas a partir de uma termobalança, permitem chegar a
algumas conclusões sobre a composição e estabilidade dos compostos
intermediários e sobre a composição do composto formado após aquecimento.
O conjunto das técnicas TG/DTG também pode ser utilizado para estimar a
acidez de catalisadores heterogêneos a partir do método de termodessorção
de uma base quimiossorvida na superfície do material (72-73).

57
3.7.2. Microscopia Eletrônica de Varredura (MEV)
Uma imagem de MEV consiste em uma análise da topografia da
superfície da amostra. Esta é obtida por reflexão de feixe de elétrons pela
superfície da amostra e, para isso, é necessário que essa superfície seja
condutora. As amostras de materiais não condutores necessitam de
recobrimento com uma fina camada de um metal condutor (0, a 5 nm) e pouco
suscetível à oxidação, geralmente, utiliza-se o ouro (74-75).
O uso de microscópicos eletrônicos modernos, com poder de resolução
da ordem de nanômetros permite, por exemplo: determinar um diâmetro médio,
no caso de partículas esféricas; medir o tamanho de determinadas partículas;
visualizar partículas metálicas nos suportes; etc. No caso específico da
caracterização de catalisadores, para se estimar o tamanho das partículas, é
necessário que sejam preparadas várias amostras do mesmo catalisador, e
obter um número suficiente de fotos que representem a distribuição das
partículas na amostra. As ampliações devem permitir um aumento final entre 5
e 105 vezes, mas só devem ser computadas as partículas que estejam na
distância focal correta e isentas de astigmatismo (74-75).
3.7.3. Difratometria de raios X
Os raios X, descobertos em 1895 por William Röntgen, definidos como
radiações eletromagnéticas, cujo comprimento de onda varia de 0,1 a 100 Å,
fundamentam a base da técnica de difração de raios X. Esta técnica usa essas
radiações, de maneira bem controlada em um equipamento específico, para se
determinar propriedades de um determinado material, e possui muitas
aplicações: identificação da estrutura cristalina e de suas fases; identificação
de fases; análise quantitativa de fases; textura e análise de tensão (75-76).
A análise pode ser realizada com o material na forma de sólidos e pós -
monocristais, matrizes, folhas e fibras. As amostras consistem em monocristais
de 0,1 a 0,5 mm de lado e pós (da ordem de gramas). Apesar de ser muito

58
empregada em catálise, principalmente para determinação da estrutura
cristalina dos materiais sintetizados, a técnica apresenta também as suas
limitações, por exemplo:
• É usada apenas em materiais cristalinos, pois em materiais
amorfos, geralmente, não reproduzem difração;
• Os picos sobrepostos podem dificultar a identificação na análise
quantitativa;
• Os efeitos de matriz, materiais fortemente difratados, podem
encobrir os fracamente difratados e
• As amostras fluorescentes podem elevar a linha de difração ou
podem causar saturação em certos tipos de detectores.
Se um feixe de raios-X com uma dada freqüência incidir sobre um átomo
isolado, elétrons desse átomo serão excitados e vibrarão com a freqüência do
feixe incidente. Estes elétrons vibrando, emitirão raios-X em todas as direções
com a mesma freqüência do feixe incidente, ou seja, o átomo isolado espalha o
feixe incidente de raios-X em todas as direções. A equação básica da difração,
chamada Lei de Bragg, é nλλλλ = 2.d.sen θθθθ, em que: n é a ordem de reflexão
(n={1,2,3,...}), λλλλ é o comprimento de onda, d é a distância interplanar e θθθθ é o
ângulo de incidência entre os planos reticulados. Essa equação pode ser
melhor compreendida pela análise matemática da Figura 3.22 que representa
um plano cristalino (76-77).

59
Figura 3.21 - Esquema representativo para formulação da lei de Bragg (76).
O princípio de obtenção dos raios-x consiste em se excitar átomos ou
íons no interior de uma fonte selada - mantida sob alto vácuo - a qual é um
tubo que consiste basicamente de um filamento aquecido (cátodo), geralmente
de tungstênio, funcionando como fonte de elétrons, e um alvo (ânodo) que
pode ser formado por diversos metais (cobre, molibdênio, cobalto, etc). A
aplicação de uma diferença de potencial entre o cátodo e o ânodo, faz com que
os elétrons emitidos pelo filamento incandescente sejam acelerados em
direção do ânodo e, quando estes colidem com o metal do ânodo ocorre a
transformação da energia cinética adquirida pelos elétrons em calor e, em
menor extensão em raios-X. Através de uma pequena abertura, essa radiação
primária deixa o tubo e segue em direção ao material a ser analisado.
Um método bastante empregado para a análise de raios-x é o método
do pó, o qual é aplicado em materiais difíceis de preparar na forma de
monocristais. O método consiste basicamente em uniformizar a amostra de
modo a torná-la um pó fino e homogêneo. Quando esse pó é colocado no porta
amostra do equipamento, um grande número de pequenos cristalitos é
orientado em todas as direções possíveis. Dessa forma, quando um feixe de
raios-X atravessa o material, um número significante de partículas está

60
orientado de tal forma que a condição de Bragg, para a reflexão de cada
possível distância interplanar, seja obedecida (76-77).
3.7.4. Volumetria de nitrogênio
Em 1938, Brunauer, Emmett e Teller desenvolveram uma equação para a
adsorção de gases em multicamadas na superfície de sólidos (72, 78-79):
em que:
V= volume de N2 adsorvido à pressão relativa P/P0;
Vm = volume de N2 para cobrir o adsorvato com uma monocamada;
P0 = pressão de saturação do N2 líquido;
Esta equação, denominada BET, se baseia na hipótese de que as forças
responsáveis pela condensação do gás são também responsáveis pela atração
de várias moléculas para a formação de multicamadas. Um dos métodos mais
comuns de determinação da área específica de um sólido se baseia na
determinação da quantidade necessária de um adsorvato para formar uma
monocamada sobre a superfície a ser medida.
O estudo do fenômeno de adsorção é feito com o objetivo de se obter
informações sobre a área específica e a estrutura porosa de um sólido, através
da construção de uma isoterma de adsorção. A área específica, ou seja, a área
de superfície total do sólido por unidade de massa é o parâmetro crucial a ser
determinado, pois é na superfície que toda reação catalítica se processa. O
estudo da estrutura porosa de um catalisador, as determinações do diâmetro e
do volume poroso são importantes, porque estão relacionados à área total do
sólido (72, 78-79).

61
3.7.5. Cromatografia gasosa
A cromatografia pode ser definida como sendo um método físico-químico
de separação na qual os constituintes da amostra a serem separados são
fracionados entre duas fases, uma fase estacionária de grande área, e a outra
um fluído insolúvel que percola através da primeira (80). A cromatografia se
baseia então na partição entre uma fase móvel líquida ou gasosa e uma fase
estacionária líquida ou sólida. A Tabela 4 mostra os diversos tipos de
cromatografia, dependendo da fase móvel e da fase estacionária.
Tabela 3.4 - Tipos de cromatografia.
Nas análises realizadas por cromatografia gasosa (CGS), existe uma
série de variáveis que influenciam na boa separação, dentre eles podemos citar
como principais:
• Tipo de gás de arraste: o gás de arraste consiste num gás inerte que
atravessa a coluna transportando a mistura a ser separada.
• Vazão do gás de arraste: a vazão do gás de arraste influencia
diretamente a maior ou menor difusividade dos constituintes da mistura
pela coluna.
• Temperatura na coluna: parâmetro de grande importância para se
aperfeiçoar a separação dos componentes que percolam uma coluna.
Fase Móvel Fase Estacionária Cromatografia Abreviação
Gás Sólido Gás-Sólido CGS
Gás Líquido Gás-Líquido CGL
Líquido Sólido Líquido-Sólido CLS
Líquido Líquido Líquido-Líquido CLL

62
• Tipo da coluna: as colunas podem se apresentar de 2 formas: colunas
empacotadas - são fabricadas depositando um filme da fase estacionária
escolhida sobre um material inerte chamado de suporte; e colunas
capilares - são fabricadas depositando-se um filme finíssimo de 0,1
mícron da fase estacionária nas paredes de um tubo capilar.
• Tipo de fase estacionária: a escolha de uma fase sólida adequada é de
fundamental importância na separação, visto que deve apresentar
afinidade eletrostática suficiente para se promover a perfeita separação.
Portanto de acordo com cada grupo de substâncias usa-se um
determinado tipo de fase estacionária. São exemplos de fases
estacionárias: porapak, cromossorb, etc (80).
3.7.6. Quimissorção de amônia
A técnica de dessorção termoprogramada de amônia (NH3-TPD) é
provavelmente a técnica mais utilizada para a caracterização de acidez em
sólidos ácidos. Existem muitas variações de métodos, mas a técnica
tipicamente envolve a saturação dos sítios ácidos na superfície com amônia,
seguida de uma rampa de temperatura sob fluxo de gás inerte.
Alternativamente, a rampa de temperatura pode seguir até um valor fixo, após
o qual a dessorção é realizada isotermicamente (72, 79, 81).
Nos últimos anos, tem-se determinado a acidez de catalisadores ácidos
também por quimissorção de amônia por pulso. Nesta análise, a superfície do
material, previamente limpo, é submetida à injeção do gás amônia, puro,
estando a uma temperatura controlada, aproximadamente 120 °C. Essas
injeções são repetidas até que seja observada a saturação do material,
monitorado pela altura ou pela área do pico, fornecidas por um TCD acoplado a
um sistema de detecção (79, 81).

63
3.7.7. Espectroscopia na região do infravermelho co m transformada de
Fourier
Espectroscopia na região do infravermelho é uma das técnicas mais
comuns e utilizadas em diversas áreas. Baseia-se fundamentalmente na
medida de absorção em freqüências de infravermelho por uma amostra
posicionada no caminho do feixe de radiação infravermelha. As radiações
infravermelhas apresentam comprimentos de onda típicos que variam de 0,78 a
1000 µm e números de onda variando de 13.000 a 10 cm-1. Os espectros de
infravermelho, geralmente, são gráficos apresentados na forma de número de
onda ou comprimento de onda (eixo X) versus absorbância ou transmitância
(eixo Y) (82).
A adsorção de moléculas-sonda acompanhada por infravermelho com
transformada de Fourier (FTIR) é particularmente interessante para a obtenção
de informações sobre a força e distribuição dos sítios. De maneira geral,
sólidos ácidos podem exibir tanto sítios de Bronsted como de Lewis, e
utilizando diferentes moléculas pode-se quantificar a acidez de alguns
materiais. Diferentes moléculas podem sondar diferentes tipos de sítios ácidos,
e as moléculas que interagem fracamente são mais específicas que aquelas
que interagem fortemente e podem, portanto, prover informações mais
detalhadas sobre os sítios ácidos. As moléculas como piridina, aminas e NH3
formam uma ligação química com os prótons dos grupos hidroxila, dando
informações sobre a concentração dos sítios ácidos (82, 83).

64

65
CCAAPPÍÍTTUULLOO 44
44 MMAATTEERRIIAAIISS EE MMEETTOODDOOLLOOGGIIAA
Neste Capítulo são apresentados e detalhados os materiais,
equipamentos e procedimentos experimentais utilizados na preparação,
caracterização e no teste reacional dos catalisadores em estudo.
Grande parte de todo o desenvolvimento experimental deste trabalho -
envolvendo bancada de preparação dos catalisadores, metodologia de
calcinação, processo de caracterização por análise térmica, adsorção de
nitrogênio, quimissorção de amônia e os testes catalíticos -, foi realizada no
LCP, Laboratório Associado de Combustão e Propulsão, do Instituto Nacional
de Pesquisas Espaciais (INPE). Ainda no INPE, no Laboratório Associado de
Materiais e Sensores (LAS), foram realizadas as medidas de caracterização
utilizando o microscópico eletrônico de varredura (MEV) e o difratrômetro de
raios X (DRX).
No quadro de intercâmbio INPE – Université Paris VI, e graças a toda
presteza do prof. Dr. Claude Potvin desta universidade, foram obtidas as
medidas de infravermelho com transformada de Fourier de alguns materiais
selecionados. Foram obtidas ainda algumas medidas de DRX, por cortesia do
grupo, que foram colocadas como apêndice e permitiram uma boa comparação
com as medidas realizadas no LAS.

66
44..11 MMaatteerr iiaaiiss ee eeqquuiippaammeennttooss
4.1.1. Materiais
� Reagentes para o preparo dos catalisadores: Faujazita comercial,
zeólita Y, denominada neste trabalho ZEO; hidróxido de alumínio
comercial (bohemita - Catapal A), denominada por CAT; óxido de
nióbio HY-340, simplesmente HY. Tanto a ZEO como a CAT foram
doadas pelo CENPES (Petrobrás), e o HY pela CBMM (Companhia
Brasileira de Mineração e Metalurgia). Além destes três principais
reagentes, ainda utilizou-se o ácido oxálico comercial (VETEC) e
água destilada e deionizada.
� Gases para pré-tratamento e caracterização: oxigênio, hélio,
nitrogênio, argônio, amônia e piridina, todos com grau de pureza
99,99 % da White Martins.
� Gases para o teste catalítico: hidrogênio, ar sintético, etileno e hélio,
todos com grau de pureza 99,99 % da White Martins.
� Vidrarias, utilitários e outros: reator de quartzo, célula volumétrica de
vidro (específico para BET), célula de quartzo formato em U e funil
de teflon (específico para Chembet), unidade catalítica de vidro,
microsseringa de vidro 5000 µL, bolhômetro de vidro, erlenmeyer,
béquer de vidro, bastão de vidro, funil de vidro, vidro de relógio,
balão volumétrico, funil de buchner, cadinhos de porcelana,
nitrogênio líquido (White Martins), coluna de aço inox (Porapak Q),
autoclave de aço inox, peneira inox 105 µm, porta-amostra de aço
inox para o MEV, pinça metálica, fita adesiva de carbono, álcool
isopropílico, válvulas de posto, manômetros, cápsula de teflon,

67
cronômetro, mangueiras de silicone, luva de amianto, lã de vidro,
pincel, papel de filtro e espátulas.
4.1.2. Equipamentos
� Para o preparo dos catalisadores e pré-tratamentos: Balança
analítica, estufas, bomba de vácuo, capela com exaustor, chapa
aquecedora, forno com fluxo de gás, termopares e controladores de
temperaturas.
� Para a caracterização dos catalisadores: Termobalança modelo 92-
16.18 (Setaran), difratômetro de raio X modelo PW 1830 (Philips),
microscópio eletrônico de varredura modelo JEOL (JSM 5310),
NOVA 1000 e Chembet 3000 (ambos Quantachrome), Infravermelho
com transformada de Fourier, controladores de vazões gasosas e
balança analítica.
� Para o teste catalítico: Balança analítica, termopares, controladores
de temperatura, forno, controladores mássicos de vazões,
cromatógrafo a gás com detectores de condutividade térmica (TCD)
e ionização de chama (FID), modelo 3800 (Varian), acoplado a um
computador com software para aquisição dos dados.
44..22 PPrreeppaarraaççããoo ddooss ccaattaall iissaaddoorreess
4.2.1. Catalisadores de alumina, zeólita e nióbia
O procedimento adotado para estes três materiais, inicialmente, foi
submeter, individualmente, todos eles (ZEO, CAT e HY) a uma peneira de
abertura 105 µm, com o auxílio de um pincel. Depois de adquirir a

68
granulometria desejada, os materiais foram pesados e armazenados em
recipientes fechados na temperatura ambiente. Desse estoque foram retiradas
alíquotas de aproximadamente 12 g. Essa massa foi transferida para um
cadinho de porcelana, e levada ao forno, sob fluxo de oxigênio (1200 mL/min),
para realizar o processo de calcinação, de acordo com a temperatura desejada.
4.2.2. Procedimento da calcinação
Neste trabalho, optou-se por calcinar os materiais em quatro
temperaturas diferentes (300, 400, 500 e 600 °C ), buscando assim uma melhor
comparação e discussão, do comportamento de um material X em função da
variação da temperatura, e também comparar o comportamento desse material
X com os demais, quando calcinados à mesma temperatura.
Todos os catalisadores foram submetidos à mesma programação de
temperatura durante a calcinação, dividida em duas etapas: a primeira seguiu
uma taxa de aquecimento de 0,37 °C/min, desde a temperatura ambiente até
130 °C, permanecendo nesta por uma hora; na segunda etapa, partindo de 130
°C e obedecendo à taxa de aquecimento de 0,75 °C/min, ao atingir a
temperatura desejada, permaneceu nessa temperatura por 6 horas (Figura
4.1).

69
Figura 4.1 – Programação da temperatura de calcinação.
Ao atingir a duração total do processo de calcinação o forno foi
desligado e, depois de vinte minutos, a vazão de oxigênio também foi
interrompida. A etapa seguinte foi aguardar o resfriamento do forno, até
aproximadamente 40 °C, para abri-lo. Esse tempo de espera variou de acordo
com a temperatura final do forno, por exemplo, numa calcinação conduzida a
300 °C esperou-se, aproximadamente, 7 horas até a temperatura diminuir para
abrir o forno; já numa calcinação a 600 °C esse tempo de resfriamento foi,
aproximadamente, de 30 horas.
4.2.3. Processo de autoclavagem do catalisador de n ióbio
O HY foi utilizado de duas formas, a primeira apenas peneirando a
massa e calcinando a temperatura desejada (itens 4.2.1 e 4.2.2.), e a segunda
foi submetendo-o a um tratamento hidrotérmico, denominado autoclavagem,
Vazão de Oxigênio a 1200 mL/min
1ª taxa: 0,37 °C/min
130°C por 1 h
Mantido por 6 h
2ª taxa: 0,75 °C/min
Temperatura Final
Temperatura Ambiente

70
gerando assim um HY com características um pouco diferentes das iniciais,
este novo material sendo chamado AHY (óxido de nióbio autoclavado).
Para preparar o AHY selecionou-se granulometricamente o HY, em
peneira de abertura 105 µm com o auxílio de um pincel. Pesou-se 25 g desse
material e essa massa foi transferida para uma cápsula de teflon, onde foram
adicionados 100 mL de uma solução recém preparada de ácido oxálico 10 %.
Houve a formação de uma suspensão dentro da cápsula, que foi
homogeneizada com o auxilio de um bastão de vidro. Logo em seguida, a
cápsula de teflon foi fechada e o conjunto inserido em uma autoclave de aço
inox, a qual foi colocada em uma estufa pré-aquecida a 170 °C.
Inicialmente, houve a necessidade do monitoramento da temperatura
interna da estufa, pois, com a abertura da estufa o equilíbrio térmico a 170 oC
teve que ser restabelecido. O tempo de monitoramento levou,
aproximadamente, quarenta minutos, e já com a estufa a 170 °C iniciou-se o
processo de autoclavagem realizado durante 8 horas a 170 °C e mais 9 horas
de envelhecimento. Este último procedimento, o envelhecimento propriamente
dito, foi realizado desligando a estufa e deixando a cápsula inox, contendo a
amostra, fechada dentro da estufa por mais 9 horas.
Após o tempo de envelhecimento, a estufa foi aberta para resfriar a
autoclave, cuja temperatura era de aproximadamente 90 °C, mais rapidamente,
até atingir a temperatura ambiente. Em seguida, a autoclave foi levada a uma
capela e sob exaustão, a cápsula foi despressurizada, sendo que durante essa
etapa foram observadas pequenas liberações de gás carbônico (CO2),
detectadas por um sensor específico. Constatado que todo o CO2 foi liberado, a
cápsula foi aberta, e em seguida homogeneizada, com o auxilio de um bastão
de vidro, para enfim levar o material à filtração a vácuo, utilizando um funil de
buchner com papel de filtro comum. O material sólido obtido foi transferido para
um vidro de relógio, pesado, e deixado na estufa a 40 °C, sob circulação
forçada, por 24 horas.

71
Depois de secado, o material obtido foi novamente pesado, para fins de
cálculos de rendimento do processo, e iniciou-se novamente o processo de
peneiramento (peneira de abertura 105 µm) para finalmente obter o AHY
pronto para ser calcinado. O fluxograma apresentado na Figura 24 descreve
todo o procedimento de autoclavagem do HY até transformar-se em AHY.
Figura 4.2 – Fluxograma da autoclavagem do óxido de nióbio (HY).
4.2.4. Catalisador misto nióbia-alumina
Os catalisadores mistos alumina-nióbia (denominados AlX%Nb ), em que
X representa a porcentagem em massa de nióbia (Nb) em relação à massa de
alumina (Al), foram preparados com cinco composições: Al5%Nb, Al10%Nb,
Al20%Nb, Al30%Nb e Al40%Nb , isso com o objetivo de obter catalisadores
mais ativos que aluminas e nióbias puras, buscando um efeito sinérgico entre
os dois óxidos.

72
O preparo dos catalisadores Al%Nb iniciou-se com o processo de
peneiração do CAT e do HY (peneira de abertura 105µm), seguido das
pesagens e mistura dos precursores nas respectivas proporções, e depois a
autoclavagem, como descrito no item 4.2.3, encerrando com o peneiramento e
a calcinação. Esta foi fixada a 400 °C, porém, obedecendo às taxas e ao tempo
de aquecimento descritos no item 4.2.2. A temperatura escolhida foi de 400 oC
em função do bom desempenho obtido em testes preliminares realizados com
os catalisadores preparados na reação de hidratação do etileno.
44..33 CCaarraacctteerr iizzaaççããoo ddooss ccaattaall iissaaddoorreess
4.3.1. Termogravimetria
A análise termogravimétrica (TG) é uma técnica na qual a variação de
massa de uma amostra, ou seja, perda ou ganho, de uma amostra é medida
em função da temperatura e/ou do tempo. É uma técnica importante para
caracterizar materiais, porque permite obter informações sobre a formação e
estabilidade dos compostos químicos.
As curvas de TG deste trabalho foram obtidas em uma termobalança,
modelo 92-16.18 (Setaram) no laboratório do LCP/INPE. As análises de TG da
ZEO, CAT, HY e AHY foram efetuadas sob fluxo de argônio com 20 % de
oxigênio, e a faixa de temperatura estudada foi da temperatura ambiente (25
°C) até 700 °C, com uma taxa de aquecimento de 5 °C /min.
4.3.2. Microscopia Eletrônica de Varredura (MEV)
A microscopia eletrônica de varredura (MEV) é uma técnica importante
para realizar a análise morfológica dos materiais. Pequenas quantidades dos
catalisadores em pó foram colocadas, cuidadosamente, sobre uma fita de
carbono adesiva, esta fixada na superfície de um porta-amostra de aço inox,
própria do equipamento; a seguir, este porta-amostra foi levado até a câmara,

73
onde por sputtering, houve a deposição direta de um filme de ouro sobre os
pós. Finalmente, o porta-amostras foi levado ao MEV para proceder as análises
de cada amostra.
O equipamento utilizado foi um microscópio eletrônico de varredura da
marca JEOL, modelo JMS 5310, pertencente ao LAS/INPE.
4.3.3. Difratometria de Raios X (DRX)
A técnica de difração de raios X, baseada na Lei de Bragg, foi
empregada para a identificação das fases cristalinas presentes nos pós dos
materiais em estudo. Isso foi feito por comparação dos dados obtidos com os
armazenados nas fichas Joint Committee on Powder Diffraction Standards
(JCPDS) (84). Ao encontrar a que identificava o material, foi gerado, com o
auxilio de um programa computacional desenvolvido pelo laboratório de
catálise do LCP/INPE, o difratograma coletado no equipamento juntamente
com o da ficha escolhida.
O equipamento utilizado foi um difratômetro de raios X da marca
PHILIPS, modelo PW 1830, instalado no LAS/INPE. As condições
estabelecidas para a obtenção dos difratogramas foram: radiação CuKα obtida
em 40 kV (com corrente de filamento em 25 A), varredura com passo angular
de 0,05o e intervalos de medição de acordo com o pó em análise: para os de
HY e AHY, o intervalo adotado foi 10o < 2θ < 70o; para CAT e as misturas
AlNb, o intervalo foi 10o < 2θ < 80o; e para ZEO, o intervalo adotado foi 5o < 2θ
< 80o.
4.3.4. Volumetria de nitrogênio
A adsorção superficial de N2 gera informações sobre a área superficial
do material em estudo e sua porosidade. Essa técnica está fundamentada na

74
adsorção física das moléculas do gás, variando a pressão do gás N2 injetado
sobre a amostra.
Através dos dados da pressão relativa e do volume de N2 adsorvido
foram obtidas as isotermas de adsorção e de dessorção do gás. As curvas
foram obtidas em um equipamento da marca QUANTACHROME, modelo Nova
1000, pertencente ao LCP/INPE. Inicialmente, foi pesada em uma célula de
vidro, uma massa do pó em estudo (aproximadamente 0,2 g), em seguida essa
célula, contendo o material, foi submetida a um tratamento térmico a 200 °C,
por 2 horas sob vácuo, para remoção de impurezas adsorvidas na superfície do
material.
O valor da área específica foi calculado pela equação de BET (72), cujo
modelo é o mais aceito para interpretar as isotermas de adsorção e de
dessorção, a partir da formação de uma monocamada do gás adsorvido na
superfície externa e nos poros das partículas. Tal cálculo foi efetuado pelo
próprio software do equipamento NOVA1000. Para a determinação da
distribuição de tamanhos de poros foi utilizado o método proposto por Barret,
Joyner e Halenda (BJH), também utilizado pelo próprio software do
equipamento, cujos cálculos envolvidos baseiam-se na equação de Kelvin e
são válidos para diferentes formatos de poros (72, 85).
4.3.5. Volumetria de amônia
A adsorção de amônia por pulsos é uma das técnicas mais conhecidas
para se determinar a acidez total de um material. Neste trabalho, a acidez total
foi calculada utilizando tal técnica, ou seja, injetando volumes conhecidos de
gás amônia até a completa saturação do material.
As análises de acidez total foram realizadas em um aparelho da marca
CHEMBET, modelo 3000, disponível no LCP/INPE. Em uma célula de quartzo,
própria do equipamento, foi inserida uma pequena quantidade de lã de vidro,
apenas para segurar o pó, depois foi pesada uma quantidade do pó em estudo

75
(aproximadamente 0,15 g). Essa célula foi acoplada ao equipamento e
submetida a um tratamento térmico a 200 °C por 1 hora, passando hélio (130
mL/min) para ajudar a arrastar as impurezas.
Após o tratamento térmico, monitorou-se a temperatura da célula,
através de um termopar interno, até que atingisse 60 °C, isto mantendo a vazão
de hélio. A seguir, foram iniciadas as injeções de amônia passando pelo
material, efetuadas com o uso de uma micro-seringa de vidro (5000 µL), e
registradas através de um detector de condutividade térmica (TCD). A liberação
da quantidade de amônia de cada injeção, não adsorvida pelo material, foi
registrada como uma curva pelo próprio software do equipamento.
As injeções foram repetidas até não ser mais registrada adsorção de
amônia pelo material, o que foi feito através das comparações das áreas ou
das alturas dos picos registrados. Ainda foi possível comparar as medidas de
saturação com uma medida, em branco, realizada logo no início do
experimento, ou seja, uma injeção sem passar por dentro da célula (passando
somente pelo by-pass), o que tornou possível conhecer a área ou a altura do
pico de gás amônia correspondente aos 5000 µL.
4.3.6. Teste catalítico acompanhado por cromatograf ia a gás
O experimento catalítico proporciona a continuidade ou não de todo um
projeto. Antes de realizar qualquer tipo de caracterização, primeiramente, é
necessário saber se o material em estudo é ativo. No caso específico deste
trabalho, buscou-se o desenvolvimento de catalisadores para as reações de
hidratação de olefinas, com base na reação modelo de hidratação de etileno.
Todas as medidas foram realizadas em uma unidade catalítica acoplada
a um cromatógrafo a gás, sistema disponível no LCP/INPE. Para efetuar a
separação dos reagentes e dos produtos formados, utilizou-se uma coluna
empacotada PORAPAK Q.

76
� Pesagem do material:
O reator de quartzo foi instalado na linha catalítica e mantido sob vazão
de hélio (20 mL/min), durante 10 min. A seguir, o reator foi completamente
fechado e pesado. Em seguida, pesou-se cerca de 0,5 g do catalisador, e este
na forma de pó foi colocado, através de um funil, sobre a placa porosa interna
do reator. O reator contendo a amostra foi levado à linha catalítica novamente
para a passagem de hélio por 10 min, e depois de fechado, foi novamente
pesado. No final do teste catalítico esse procedimento foi repetido.
Figura 4.3 – Esquema do reator de quartzo utilizado no teste catalítico.

77
� Pré-tratamento dos catalisadores antes dos Testes Catalíticos:
Todos os catalisadores foram submetidos a um pré-tratamento para
retirar impurezas. O pré-tratamento foi realizado em reator de quartzo inserido
em um forno. O forno foi programado a uma taxa de aquecimento de 5 °C/min,
partindo da temperatura ambiente (26 °C) até 200 °C, sobre vazão de hélio
constante (20 mL/min), mantendo-se esta temperatura por, aproximadamente,
120 min. A seguir, foram efetuadas as injeções de 6 em 6 minutos, que foram
monitoradas pelo TCD do cromatógrafo.
� Estabilização do sistema e acendimento do FID:
Ao finalizar o tempo de tratamento térmico, o TCD foi desligado e iniciou-
se o processo de resfriamento do sistema, que demorou uma hora e trinta
minutos, mantendo a vazão de hélio passando pelo reator. Após atingir a
temperatura de estabilização para ligar o FID (detector por chama), o reator foi
fechado para os ajustes de vazões da etapa seguinte.
Para acender o FID ajustaram-se as vazões da mistura gasosa de
arraste, hélio a 11,4 mL/min; hidrogênio, essencial para manter a chama
acessa a 6,1 mL/min; e ar sintético a 16,5 mL/min.
� Reação de Hidratação do etileno:
Para a reação de hidratação de etileno a vazão de hélio foi ajustada em
10,24 mL/min e a vazão de etileno em 10,43 mL/min. A temperatura ideal para
a reação de hidratação é de 220 °C, segundo a literatura (5).
Após o tratamento térmico, ainda com o reator fechado, foi realizado o
monitoramento da estabilização da vazão da mistura reacional (etileno/hélio):
primeiramente a vazão foi passada por um saturador, contendo água
deionizada mantida a 50 °C, e depois pelo condensad or, também contendo
água deionizada, mantido a 40 °C, ou seja, a reação de hidratação foi realizada
com a pressão de vapor da água, aproximadamente, em 55 mmHg. A vazão de

78
etileno/hélio, arrastando o vapor d´água, foi estabilizada e monitorada pelas
coletas automáticas a cada cinco minutos, passando a mistura apenas pelo
by-pass da linha, ou seja, por fora do reator. O sinal observado foi um pico,
logo após meio minuto da coleta realizada. Esse pico foi identificado como o
etileno, através do padrão. A Figura 4.4 ilustra a unidade catalítica de teste
existente no LCP/INPE.
Figura 4.4 – Unidade de avaliação catalítica do LCP/INPE.
Com todos os parâmetros otimizados e estabilizados, o reator, contendo
o material catalítico em estudo, foi aberto, e a passagem pelo by-pass foi
fechada, a fim de direcionar o fluxo para o interior do reator. Assim, a mistura
gasosa contendo hélio/etileno entrou em contato com o catalisador já
arrastando o vapor d´água, proveniente do sistema condensador-saturador.
Foram adotados procedimentos padrões para permitir a repetibilidade das

79
medidas, tendo-se a preocupação de seguir todo um procedimento, com
etapas e tempos otimizados. A vazão total dos gases foi controlada através de
medidas freqüentes, utilizando-se para esse fim bolhômetro. As temperaturas
do saturador e do condensador permaneceram estabilizadas com o emprego
de banhos térmicos.
4.3.7. Volumetria de piridina
Medidas de adsorção de amônia são importantes, porém, só fornecem
informação geral da acidez total do material. Já as medidas realizadas por
adsorção de piridina, monitorada por FTIR, são importantes porque identificam
os tipos de sítios existentes no material analisado. Isso devido ao fato da
molécula de piridina, sendo específica, poder adsorver-se fortemente, ou não, à
superfície do material em estudo.
As amostras escolhidas para adsorver a piridina, monitoradas por FTIR,
foram selecionadas após a reação de hidratação do etileno, ou seja, apenas os
materiais que produziram bons resultados no teste catalítico. Isso devido à
dificuldade de disponibilidade de tal técnica, sendo que os resultados
apresentados, neste trabalho, foram obtidos em função da presteza e do
conhecimento da prof. Dra. Maria Filipa Ribeiro do Instituto Superior Técnico
em Portugal. O estudo da acidez das amostras CAT400, AHY400 , AHY300,
Al30%Nb, Al20%Nb e Al5%Nb foi realizado seguindo a dessorção de piridina,
por FTIR, in situ, usando pastilhas auto-suportadas com 5-10 mg/cm2.
As amostras foram submetidas a um pré-tratamento a 200 ºC, por duas
horas, sob pressão de 10-6 mbar. Após esta etapa, diminuiu-se a temperatura
para 150 ºC e colocou-se a pastilha em contacto com 1 mbar de piridina
durante 10 min. Depois, o excesso de piridina foi removido, fazendo-se uma
evacuação no sistema a 150 ºC durante 30 min com vácuo secundário (P =10-6
mbar).

80
Logo a seguir, foi registrado o primeiro espectro (a 150 ºC), depois a
temperatura foi elevada para 200 ºC (mantendo-se a pastilha nesta
temperatura durante 30 min), e registrou-se o segundo espectro, procedimento
esse que foi repetido a 250 ºC.
Na Tabela 4.1 estão apresentados os comprimentos de onda associados
a cada banda, de acordo com a bibliografia (86), as bandas detectadas por
FTIR podem ser associadas à piridina coordenada em centros ácidos de Lewis
e Bronsted.
Tabela 4.1 - Bandas associadas aos centros ácidos de Lewis e Brönsted.
A partir da absorvância integrada das bandas localizadas nos intervalos
de comprimentos de onda 1445-1455 e 1540-1545 cm-1, para cada
temperatura, foi possível determinar a concentração dos centros ácidos de
Lewis e Bronsted, respectivamente, de acordo com a seguinte equação (87):
m
SAIC
××=
ε
Em que:
C – Concentração (mmol/g)
S – Área Superficial da pastilha (cm2)
AI – Absorvância Integrada (cm-1)
m – Massa da pastilha (mg)
ε - Coeficiente de extinção molar (cm/µmol)
Lewis Lewis + Bronsted Bronsted
comp. de onda (cm -1) 1455-1450 1620-1600 1575 1490 1545 1635

81
CCAAPPÍÍTTUULLOO 55
55 RREESSUULLTTAADDOOSS EE DDIISSCCUUSSSSÃÃOO
55..11 AAnnááll iissee ttéérrmmiiccaa
5.1.1 Termograma da catapal A
Os primeiros experimentos realizados foram os termogravimétricos, pois
através deles ter-se-ia uma avaliação do comportamento de cada material,
quando estes fossem submetidos ao processo de tratamento térmico.
Conforme se pode observar na Figura 5.1, que apresenta o resultado obtido
com a catapal A, a faixa de temperatura para estudo variou da temperatura
ambiente (25 °C) até 700 °C.
Observam-se, na Figura 5.1, duas curvas que correspondem às perdas
de massa e o fluxo de calor, ambas em função do aumento da temperatura. A
curva referente ao fluxo de calor apresenta duas modas, a primeira em
aproximadamente 100 °C, atribuída à perda de água d o hidróxido de alumínio;
e a segunda, registrada em aproximadamente 450 °C s e deve à formação da γ-
alumina, segundo a literatura (54).

82
Figura 5.1 - Termograma do hidróxido de alumínio (catapal A).
5.1.2 Termograma da zeólita Y
O termograma apresentado na Figura 5.2 foi obtido com a análise da
zeólita Y, podendo-se observar, na curva do fluxo de calor, uma única moda
bem acentuada, localizada a 130 °C, o que corresponde a perdas de água e
outras impurezas voláteis.
0 100 200 300 400 500 600 700
16
17
18
19
20
21
22
23
24
-15
-12
-9
-6
-3
0
3
Termograma
Flu
xo d
e C
alor
(µµ µµV
)
Mas
sa (
mg)
Temperatura (ºC)
Fluxo: Argônio/20%O2
Taxa: 5ºC/min
450o C
104o C
Catapal A
Fluxo de Calor

83
Figura 5.2 - Termograma da zeólita Y.
5.1.3 Termograma da nióbia e da nióbia autoclavada
A análise realizada com a nióbia pura, o óxido de nióbio (HY) (Figura
5.3), mostrou além da perda de massa, através da curva TG (cor amarela),
uma grande liberação de calor a 562 °C, comportamen to este característico de
uma transição de fase do material, quando submetido ao tratamento térmico.
0 100 200 300 400 500 600 700
20,0
20,5
21,0
21,5
22,0
22,5
23,0
23,5
-12
-10
-8
-6
-4
-2
0
2
4
Fluxo: Argônio/20%O2
Taxa: 5ºC/min
Termograma
Flu
xo d
e C
alor
(µµ µµV
)
Mas
sa (
mg)
Temperatura (ºC)
130 oC
Zeólita Y
Fluxo de Calor

84
Figura 5.3 – Termograma da nióbia (HY).
Na Figura 5.4, pode-se visualizar o termograma da nióbia autoclavada
(AHY), quando se observou que seu comportamento é bem parecido ao da HY
pura. Conforme se pode evidenciar, há a liberação significativa de calor, só que
neste caso, há uma temperatura superior, a 600 °C, registrada pelo
monitoramento do fluxo de calor, a qual é característica da formação de uma
nova fase cristalina. A Figura 5.5 mostra uma comparação entre os fluxos de
calor obtidos com HY e AHY, visualizando uma melhor compreensão das
análises.
0 100 200 300 400 500 600 700
8,8
9,2
9,6
10,0
10,4
10,8
-6
-4
-2
0
2
4 Termograma
Flu
xo d
e C
alor
(µµ µµV
)
Mas
sa (
mg)
Temperatura (ºC)
120oC
Fluxo: Argônio/20%O2
Taxa: 5ºC/min
562oC
HY
Fluxo de Calor

85
Figura 5.4 – Termograma da nióbia autoclavada (AHY).
Figura 5.5 - Fluxo de calor comparando as duas nióbias (HY e AHY).
0 100 200 300 400 500 600 70013,5
14,0
14,5
15,0
15,5
16,0
16,5
17,0
-6
-4
-2
0
2
4
6
Termogramagra
Flu
xo d
e C
alor
(µµ µµV
)
Mas
sa (
mg)
Temperatura (ºC)
100o C
Fluxo: Argônio/20%O2
Taxa: 5ºC/min
600o C
AHY
Fluxo de Calor
0 100 200 300 400 500 600 700
-6
-4
-2
0
2
4
6
600oC
562oC
Comparação do Fluxo de Calor do HY e AHY - Taxa: 5º C/min
Flu
xo d
e C
alor
do
AH
Y
(( (( µµ µµV
)
AHY
Flu
xo d
e C
alor
do
HY
(µµ µµV
)
Temperatura (ºC)
-4
-2
0
2
4
HY

86
A fim de identificar as transições de fases ocorridas nas nióbias (HY e
AHY) e caracterizar melhor os demais materiais em estudo, foram realizadas
as análises com o auxílio da técnica de difratometria de raios X.
55..22 DDii ffrraattoommeettrr iiaa ddee rraaiiooss XX
5.2.1 Difratograma do hidróxido de alumínio (cata pal A)
O difratograma apresentado na Figura 5.6 caracteriza o hidróxido de
alumínio (CAT - catapal A) calcinado a 300 °C por 6 horas e, segundo a
literatura, trata-se de um oxi-hidróxido de alumina (84), ou seja, uma pseudo-
boemita. Na Figura 5.7, referente ao hidróxido de alumínio calcinado a 400 °C
por 6 horas, os picos determinados de difração indicam a formação de uma
alumina de transição, também chamada de γ-alumina.
Figura 5.6- Difratograma do hidróxido de alumínio (CAT) calcinada a 300 °C/ 6
horas.

87
Figura 5.7 - Difratograma do hidróxido de alumínio (CAT) calcinado a 400 °C/ 6
horas.
Os mesmos picos característicos da γ-alumina foram observados na
catapal A (CAT) após calcinações a 500 e 600 °C, am bas por 6 horas.
Ressalta-se que esses mesmos materiais foram também analisados na
Universidade de Paris VI, conforme pode ser constatado nos difratogramas do
Apêndice A, e os resultados obtidos foram semelhantes aos mostrados neste
capítulo, obtidos no LAS/INPE. Estes resultados também são coerentes com as
transformações observadas no termograma descrito no item 5.1.1

88
5.2.2 Difratograma da zeólita Y (ZEO)
O difratograma de raios X da zeólita Y (ZEO) calcinada a 300 °C por 6
horas (Figura 5.8) apresenta picos característicos de uma faujazita, segundo a
comparação com a literatura (84). Um resultado já esperado, pois se conhecia
a origem do material doado pelo CENPES ao LCP/INPE.
Figura 5.8 - Difratograma da zeólita Y (ZEO) calcinada a 300 °C/ 6 horas.
O aumento da temperatura de calcinação desta zeólita não provocou
alteração significativa no difratograma. Registrou-se apenas uma diminuição da
intensidade dos picos de difração, conforme pode ser verificado no Apêndice B,
o que também é coerente com a análise térmica (Figura 5.2), na qual não se
evidenciou grandes perdas, ou transformação com o aumento da temperatura.

89
5.2.3 Difratograma do óxido de nióbio (HY)
Os difratogramas de raios X obtidos com o óxido de nióbio (HY)
calcinado a 300 e 400 °C, sempre por 6 horas, mostraram a presença de uma
nanoestrutura organizada, conforme é possível observar na Figura 5.9. Não há
evidências da formação de uma rede cristalina definida, porque tais materiais
são amorfos aos raios X.
Figura 5.9 - Difratograma do óxido de nióbio (HY) calcinado a 300 °C/6 horas.
Quando o HY foi calcinado a 500 (Figura 36) e 600 °C (Apêndice C),
durante 6 horas, a difração de raios X mostrou uma microestrutura cristalina
defina, que segundo a literatura (19) é a fase T da nióbia, também denominada
fase γ. Este fato condiz com a transição de fase, observada na análise térmica
(Figura 5.3).

90
Figura 5.10 – Difratograma do óxido de nióbio calcinado a 500 °C/ 6 horas.
5.2.4 Difratograma do óxido de nóbio autoclavado (A HY)
Com o óxido de nióbio (AHY) calcinado a 500 °C/ 6 h oras (Figura 5.11) o
resultado de DRX indicou a existência de uma mistura de óxido de nióbio e oxi-
hidróxido de nióbio segundo a literatura (84), ambos com baixo grau de
cristalinidade. Esse mesmo resultado foi observado nos difratogramas do AHY
calcinado a 300 e 400 °C por 6 horas, mostrados no Apêndice D.

91
Figura 5.11 – Difratograma do óxido de nióbio autoclavado calcinado a 500 °C/
6 horas.
O fato da AHY calcinado a 500 °C não apresentar fas e cristalina bem
definida foi um diferencial nestes resultados, pois, o HY nesta temperatura já
possui uma única fase cristalina, conforme se pode comparar com as Figuras
apresentadas no Apêndice E. Mas este fato é coerente com os obtidos por
termogravimetria (Figura 5.5), onde ficou evidente através da comparação do
fluxo de calor das nióbias, que o tratamento de autoclavagem, ao qual a nióbia
foi submetida, retardou a transição de fase que ocorreria a 562 para 600 °C.
De fato a AHY permaneceu uma mistura de óxido e hidróxido de nióbio,
quando calcinado a 300 até 500 °C, porém, o AHY quando calcinado a 300 °C
já apresenta uma nanoestrutura organizada, conforme foi verificado através do
microscópio de transmissão (Figura 5.12), obtidos gentilmente pelo prof. Dr
Claude Potvin da Universidade de Paris VI. Sendo que a fase cristalina foi
evidenciada quando este material foi calcinado a 600 °C por 6 horas, conforme
é possível verificar através da Figura 5.13. Os resultados obtidos na

92
Universidade de Paris VI mais uma vez confirmaram os obtidos no INPE
(Apêndice F).
Figura 5.12 – Microscopia eletrônica de transmissão do AHY calcinado a 300
°C por 6 horas.

93
Figura 5.13 – Difratograma do óxido de nióbio autoclavado (AHY) calcinada a
600 °C/6 horas.
55..33 MMiiccrroossccooppiiaa EElleettrroonniiccaa ddee VVaarrrreedduurraa ((MMeevv))
As micrografias obtidas por MEV foram efetuadas visando ampliar as
informações de caracterização, uma alternativa para avaliar a textura dos
materiais. Conforme se observa na Figura 5.13 e no Apêndice G, as imagens
mostram que os estados de aglomeração do HY e do AHY predominaram até
mesmo nas altas temperaturas (500 e 600 °C), o mesmo pode ser observado
nas micrografias das catapal A e da zeólita Y.

94
HY com ampliação 5000X AHY com ampliação 5000X
HY 600°C
AHY 600°C
CAT com ampliação 5000X ZEO com ampliação 5000X
CAT 600°C
ZEO 600°C
Figura 5.14 - Imagens obtidas por MEV dos materiais: HY, AHY, CAT e ZEO
calcinados a 600 °C por 6 horas.
55..44 CCaarraacctteerr iizzaaççããoo ppoorr vvoolluummeettrr iiaa ddee nnii tt rrooggêênniioo
A área específica e a porosidade são duas propriedades importantes na
catálise heterogênea, pois, enquanto a área específica influencia na quantidade
dos sítios ativos em um catalisador sólido, a geometria e o volume de poros

95
controlam os fenômenos de transporte, podendo determinar a seletividade nas
reações catalíticas.
5.4.1 Volumetria de Nitrogênio da catapal A
De acordo com a literatura (88), as aluminas, como é o caso da
CATAPAL A, apresentam normalmente poros com diâmetro entre 30 e 1 000 Å,
dependendo, entre outros fatores, do tamanho dos cristais do hidróxido
precursor, no caso, a pseudoboehmita. O tratamento térmico, a que foi
submetida, para originar a γ-alumina, não alterou significativamente a estrutura
porosa, ou seja: o formato externo dos cristais da pseudoboehmita é mantido
durante a conversão a cristais de γ-alumina.
A Figura 40 mostra as análises de volumetria de nitrogênio da CATAPAL
A em função da variação de temperatura de calcinação, e no apêndice H pode-
se visualizar separadamente o formato de cada uma. Comparando os
resultados obtidos com os formatos apresentados na literatura (72), pode-se
indicar que as isotermas da CATAPAL A são do tipo IV, ou seja, um sólido
contendo uma mistura de microporos e macroporos, e suas histereses são do
tipo H1, que caracteriza poros regulares com formato cilíndrico e/ou poliédrico
com extremidades abertas.

96
Figura 5.15 - Isotermas de adsorção-dessorção da CATAPAL A.
No gráfico da distribuição de poros (Figura 5.15) observa-se que os
tratamentos térmicos a que a CATAPAL A foi submetida causaram algumas
modificações em suas características texturais. Por exemplo: enquanto na
CAT300 os poros estão mais concentrados na região entre 35 e 50 Å, com o
aumento da temperatura, como na CAT600, essa região está mais concentrada
em poros entre 60 e 70 Å.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
40
80
120
160
200
240
280V
olum
e A
dsor
vido
(cm
2 .g-1)
P/P0
CAT 300 CAT 400 CAT 500 CAT 600

97
Figura 5.16 – Distribuições de poros da CATAPAL A (CAT), obtidas por
volumetria de nitrogênio, em função da temperatura de
calcinação.
No entanto essas alterações nas modas não causaram mudanças muito
significativas nos valores de área específica e do volume médio dos poros,
conforme se verifica na Tabela 6.
Tabela 5.1 - Área específica da catapal A, diâmetro médio e volume de poros
da Catapal A, obtidos por volumetria de N2.
CAT Área (m 2/g) Dp (Å) Vp (cm 3/g)
300 238 52 0,31 400 286 51 0,37 500 252 63 0,40 600 208 76 0,40
0 20 40 60 80 100 120 140 160
0,0
0,4
0,8
1,2
1,6
2,0V
olum
e de
por
os (
cm3 .g
-1)
Diâmetro de poros (Å)
CAT 300 CAT 400 CAT 500 CAT 600

98
5.4.2 Volumetria de Nitrogênio da ZEÓLITA Y
A Figura 42 apresenta as isotermas de adsorção-dessorção das
amostras de ZEÓLITA Y. Conforme pode ser observado, não há alterações
relevantes no formato das isotermas em função do aumento da temperatura de
calcinação a que esse material foi submetido.
Figura 5.17 – Isotermas de adsorção-dessorção da ZEÓLITA Y.
De acordo com a literatura (72), essas isotermas podem ser
classificadas como do tipo IV, e considerando o formato das histereses como
sendo do tipo H3, elas indicam a presença de microporos associados a
macroporos na forma de fendas ou placas paralelas. Na Figura 5.17, está
apresentada a distribuição de tamanho dos poros desse material em função da
temperatura de calcinação.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0170
180
190
200
210
Vol
ume
Ads
orvi
do (
cm.g
-1)
P/P0
ZEO 300 ZEO 400 ZEO 500 ZEO 600

99
Figura 5.18 – Distribuições de poros da ZEÓLITA Y, obtidas por volumetria de
nitrogênio, em função da temperatura de calcinação.
Comparando os resultados dos quatro tratamentos térmicos, não são
observadas alterações relevantes, exceto o aumento de poros na região de 60
a 100 Å na zeólita calcinada a 600 °C. A Tabela 5.1 8 apresenta outras
informações relevantes obtidas com auxílio dessa técnica, tais como: a área
específica (determinada a partir dos dados obtidos por BET), o diâmetro médio
dos poros e o volume de poros da zeólita (determinado pelo método BJH).
0 20 40 60 80 100 120 140 160
0,00
0,04
0,08
0,12
0,16
0,20
Diâmetro de poros (Å)
Vol
ume
de p
oros
(cm
3.g
-1) ZEO 400
ZEO 500
0 20 40 60 80 100 120 140 160
Diâmetro de poros (Å)
ZEO 600
0,00
0,04
0,08
0,12
0,16
0,20
Vol
ume
de p
oros
(cm
3.g
-1)
ZEO 300

100
Tabela 5.2 – Área específica da zeólita Y, diâmetro médio e volume de poros
obtidos por volumetria de N2.
ZEO Área (m 2/g) Dp (Å) Vp (cm 3/g)
300°C 573 22 0,31 400°C 520 23 0,30 500°C 524 23 0,30 600°C 564 22 0,31
5.4.3 Volumetria de Nitrogênio da HY
A HY se mostrou sensível às alterações de temperaturas, conforme
pode ser visualizado na Figura 5.18, na qual se podem observar
comportamentos distintos para o formato das isotermas. Por exemplo: a HY
calcinada a 300 °C possui isotermas que se assemelham as do tipo IV, ou seja,
um material contendo microporos, porém a HY, quando calcinada a 600 °C,
apresentou um formato tipo II, que caracteriza um material não poroso.
Os gráficos das distribuições porosas, apresentados na Figura 5.19 são
coerentes com esta discussão e verifica-se que a HY calcinada a 600 °C
(HY600), por exemplo, não apresenta poros. Por outro lado, a HY500
apresenta uma boa distribuição de poros (de 30 a 80 Å), provavelmente devido
à transição apresentada pela HY nessa temperatura, em que o material antes
amorfo passa a apresentar uma fase cristalina, conforme foi visualizado nos
difratogramas.

101
Figura 5.19 – Isotermas de adsorção-dessorção da HY.
Figura 5.20 - Distribuição de poros da HY por adsorção de nitrogênio.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
10
20
30
40
50
60
Vol
ume
Ads
orvi
do (
cm3 .g
-1)
P/P0
HY 300 HY 400 HY 500 HY 600
20 40 60 80 100 120 140 160
0,0
0,1
0,2
0,3
0,4
0,5
0,6
Vol
ume
de p
oros
(cm
3 .g-1)
Diâmetro de poros (Å)
HY 300 HY 400 HY 500 HY 600

102
Analisando os dados obtidos (Tabela 5.3) observa-se uma diminuição
acentuada nos valores da área específica e do volume de poros, em função da
elevação das temperaturas de calcinação (Tabela 5.3).
Tabela 5.3 - Área específica da HY, diâmetro médio e volume dos poros
obtidos por volumetria de N2.
HY Área (m 2/g) Dp (Å) Vp (cm 3/g)
300 102 35 0,08 400 73 38 0,07 500 44 66 0,07 600 17 45 0,02
5.4.4 Volumetria de nitrogênio da AHY
O óxido de nióbio autoclavado (AHY) apresentou um comportamento
mais simétrico, em relação ao material sem tratamento hidrotérmico (HY),
quando submetido às mesmas temperaturas de calcinação (Figura 5.20).
As isotermas da AHY calcinada a 300 e 400 °C aprese ntaram formatos
semelhantes, do tipo IV, que caracterizam materiais contendo microporos
associados a macroporos, e com histereses do tipo H3, ou seja, com poros
com formatos de cunhas, cones e/ou placas paralelas. A isoterma da AHY
calcinada a 500 °C pode ser atribuída também como s endo do tipo IV, porém
os poros já poderiam ser classificados como do tipo H2, que são cilíndricos,
abertos e fechados com estrangulações. Por outro lado, o material calcinado a
600 oC (AHY600) apresentou-se com isoterma do tipo II, ou seja, um material
sem poros. As distribuições de poros mostraram-se semelhantes às da HY,
conforme se pode observar na Figura 5.21.

103
Figura 5.21– Isotermas de adsorção-dessorção da HY.
Figura 5.22 – Distribuição de poros da AHY por adsorção de nitrogênio.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
20
40
60
80
100
120
Vol
ume
Ads
orvi
do (
cm2 .g
-1)
P/P0
AHY300 AHY400 AHY500 AHY600
0 20 40 60 80 100 120 140 1600,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Vol
ume
de p
oros
(cm
3 .g-1)
Diâmetro de poros (Å)
AHY300 AHY400 AHY500 AHY600

104
Na Tabela 5.4 estão os resultados da análise da AHY, em que se
observa que o tratamento hidrotérmico aumentou os volumes dos poros e a
área específica, em relação à HY, quando submetida às mesmas temperaturas
de calcinação.
Tabela 5.4 - Área específica, diâmetro médio e volume de poros da AHY,
obtidos por volumetria de N2.
AHY Área (m 2/g) Dp (Å) Vp (cm 3/g)
300 205 38 0,19 400 167 44 0,18 500 98 63 0,15 600 22 49 0,02
Na Figura 5.22 pode-se diferenciar melhor a HY da AHY, ambas
calcinadas a 500 °C/6 h, já que nesta temperatura a HY já tem uma fase
cristalina bem definida e a AHY ainda não, segundo a análise dos
difratogramas de ambas.
Figura 5.23 – Comparação das isotermas e da distribuição de poros da HY e
AHY calcinadas a 500 °C por 6 horas.
0 20 40 60 80 100 120 140 160
0,0
0,1
0,2
0,3
0,4
Vol
ume
de p
oros
(cm
3 .g-1)
Diâmetro de poros (Å)
AHY 500 HY 500
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
20
40
60
80
100 AHY 500 HY 500
Vol
ume
Ads
orvi
do (
cm2 .g
-1)
P/P0
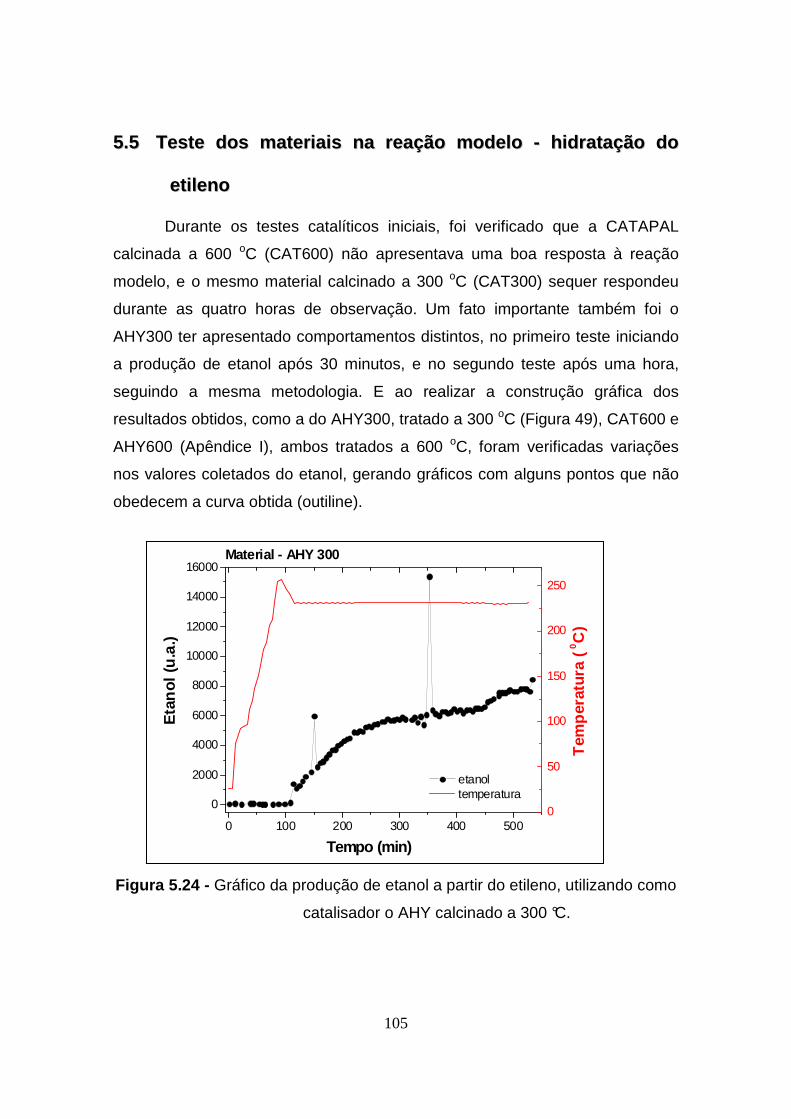
105
55..55 TTeessttee ddooss mmaatteerr iiaaiiss nnaa rreeaaççããoo mmooddeelloo -- hhiiddrraattaaççããoo ddoo
eett ii lleennoo
Durante os testes catalíticos iniciais, foi verificado que a CATAPAL
calcinada a 600 oC (CAT600) não apresentava uma boa resposta à reação
modelo, e o mesmo material calcinado a 300 oC (CAT300) sequer respondeu
durante as quatro horas de observação. Um fato importante também foi o
AHY300 ter apresentado comportamentos distintos, no primeiro teste iniciando
a produção de etanol após 30 minutos, e no segundo teste após uma hora,
seguindo a mesma metodologia. E ao realizar a construção gráfica dos
resultados obtidos, como a do AHY300, tratado a 300 oC (Figura 49), CAT600 e
AHY600 (Apêndice I), ambos tratados a 600 oC, foram verificadas variações
nos valores coletados do etanol, gerando gráficos com alguns pontos que não
obedecem a curva obtida (outiline).
Figura 5.24 - Gráfico da produção de etanol a partir do etileno, utilizando como
catalisador o AHY calcinado a 300 °C.
0 100 200 300 400 500
0
2000
4000
6000
8000
10000
12000
14000
16000T
empe
ratu
ra (
0C
)
Eta
nol
(u.a
.)
Tempo (min)
etanol
0
50
100
150
200
250
Material - AHY 300
temperatura

106
Então, após alguns testes realizados, variando a temperatura do banho
térmico, aprimorando as medições de vazões, e consultando a literatura foi
constatado: primeiro, que o etanol estava ficando condensado em alguns
pontos da linha catalítica e, provavelmente, também nos poros do catalisador, o
que provocava certo acúmulo desse produto, e de tempos em tempos registros
numéricos irreais de etanol gerado, que foram os outlines observados nos
gráficos; segundo, que a oscilação da temperatura do banho também
influenciava os resultados coletados, ou seja, pequenas elevações de
temperatura também alteravam a geração do etanol e, em terceiro lugar, que
todo o material catalítico, mesmo depois de calcinado, necessitaria ser
submetido a um tratamento térmico sob fluxo de hélio, para que toda impureza
adsorvida fosse retirada, ou seja, um procedimento de limpeza da superfície.
A fim de solucionar tais problemas, a linha catalítica (Figura 5.24) foi
recoberta com uma resistência térmica, para mantê-la aquecida durante a
reação, evitando assim os pontos de condensação do etanol. Essa resistência
térmica foi monitorada e mantida, através de um controlador de temperatura, a
150 °C. Quanto ao problema relacionado ao banho térmico, foram empregados
dois banhos, e não somente um, a fim de obter pressões gasosas mais
precisas / exatas durante o processo. O primeiro banho foi mantido a 50 °C, e o
segundo a 40 °C, sendo que 40 °C é a temperatura adequada à reação em
estudo.

107
Figura 5.25 – Ilustração da linha catalítica que foi mantida aquecida durante
todo o experimento.
Por fim, foi adotado o procedimento de limpeza das superfícies dos
materiais em estudo: todos foram aquecidos da temperatura ambiente até 200
°C (taxa de 5 °C/min), esta escolhida por ser menor à da reação em si (220 °C)
e também por ser inferior à menor das temperaturas de calcinação (300 °C); os
materiais permaneceram por 120 minutos a 200 °C, so b vazão constante de
hélio (20 mL/min), para garantir limpeza completa da superfície. A partir da
adoção desses novos procedimentos, todos os materiais catalíticos foram
novamente testados, individualmente, e os resultados estão apresentados nos
próximos tópicos.

108
5.5.1 Teste Catalítico com as CATAPAL A
A Figura 5.25 mostra os gráficos construídos a partir do tratamento
térmico das CATAPAL A, construídos com a saída de água, monitorada pelo
TCD do cromatógrafo, em função do tempo, sendo que todos os dados já estão
corrigidos pelas respectivas áreas específicas de cada CAT. Conforme se
observa, toda a água adsorvida, considerada impureza para esta reação, é
completamente eliminada nesse pré-tratamento.
Figura 5.26 - Limpeza da superfície das CATAPAL A em função do tempo.
A constatação da retirada de toda a água da superfície dos materiais é
importante, porque garante que a água que participará da reação virá do vapor
de água arrastado pelo fluxo da mistura de etileno e hélio, quando este passar
pelo conjunto condensador - saturador da linha catalítica, mantido a 40 °C (55
mmHg).
0 10 20 30 40 50 60 70 80 90 100 110
0
25
50
75
100
125
Fluxo de He: 22 ml/min
Tem
pera
tura
(ºC
)
Tempo (min)
CAT 300 CAT 400 CAT 500 CAT 600
50
100
150
200
Águ
a (u
.a.)
.m-2

109
Após encerrar o tratamento térmico, o cromatógrafo foi ajustado para o
monitoramento com o FID, e todas as vazões foram devidamente reajustadas.
Foi então iniciada a reação de hidratação do etileno para produzir etanol,
utilizando como catalisador as CATAPAL A calcinadas a diferentes
temperaturas. A Figura 5.26 apresenta os resultados obtidos com as CATAPAL
A, devidamente corrigidos por suas respectivas áreas específicas.
Figura 5.27 – Produção de etanol a partir do etileno utilizando como catalisador
as CATAPAL A calcinadas em diferentes temperaturas.
Os resultados mostram uma tendência de maior atividade para os
catalisadores que apresentam uma maior acidez total, ou seja, a catapal
calcinada a 400 °C e a catapal calcinada a 600 °C, embora todos tenham
apresentado um baixo desempenho. Este baixo desempenho implica em
trabalhar no limite de detecção do cromatógrafo, o que justificaria a incoerência
0 20 40 60 80 100 120 140 160 180 200 220 240
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
2,0
CAT300 CAT400 CAT500 CAT600
Eta
nol (
u.a.
).m
-2
Tempo (min)

110
dos resultados obtidos com a catapal calcinada a 600 °C ser menor que a
catapal calcinada a 400 °C para a geração de etanol .
5.5.2 Teste Catalítico Utilizando a ZEÓLITA Y
Os experimentos com as ZEÓLITA Y trouxeram informações muito
interessantes para o estudo realizado. A Figura 53 mostra os gráficos do
procedimento de limpeza das superfícies das ZEÓLITA Y, monitorado pela
saída de água em função do tempo, ressaltando-se que todos os dados foram
corrigidos pelas respectivas áreas específicas de cada material.
Figura 5.28 – Limpeza da superfície das ZEÓLITA Y em função do tempo.
Após encerrar o tratamento térmico, ajustar as vazões dos gases e o
cromatógrafo, para monitorar a reação pelo FID, foi iniciada a reação de
hidratação do etileno para produzir etanol, utilizando como catalisador a
ZEÓLITA Y. A Figura 5.28 mostra os resultados obtidos com as zeólitas,
0 15 30 45 60 75 90 105
0
5
10
15
20
25
30
35
40
45
ZEO 300 ZEO 400 ZEO 500 ZEO 600
Fluxo de He: 22 ml/minLimpeza de superfície - ZEO
Tem
pera
tura
(ºC
)
Tempo (min)
50
100
150
200
Águ
a (u
.a.)
.m-2

111
calcinadas em diferentes temperaturas, devidamente corrigidas por suas áreas
específicas.
Figura 5.29 – Produção de etanol a partir do etileno utilizando como catalisador
as ZEÓLITA Y calcinadas em diferentes temperaturas.
Verificou-se que independentemente da temperatura na qual a ZEÓLITA
Y foi calcinada inicialmente, a quantidade de etanol gerada foi
aproximadamente a mesma para todos os materiais. Um fato interessante
verificado foi que as zeólitas, além da geração do etanol, também produziram
outra substância, conforme apresentado no Apêndice J. Esse segundo produto
gerado foi devidamente identificado, após injeções de padrões, como sendo o
éter etílico.
A diferença observada nos resultados das zeólitas, além da geração de
etanol e éter etílico, foi que a seletividade em éter etílico sofreu uma grande
0 20 40 60 80 100 120 140 160 180 200 220
0
2
4
6
8
10
Eta
nol (
u.a.
).m
-2
Fluxo de He+etileno: 20 ml/min
ZEO300 ZEO400 ZEO500 ZEO600
Tempo (min)

112
influencia das temperaturas nas quais as zeólitas foram calcinadas. Conforme
se observa na Figura 5.29 evidencia-se que, exceto a ZEO300 (que produziu
mais etanol que éter), as demais ZEÓLITAS produziram muito mais éter etílico
que etanol.
Figura 5.30 - Produção de éter etílico a partir do etileno utilizando como
catalisador as ZEÓLITAS Y calcinadas a diferentes
temperaturas.
Uma possível explicação para isso pode estar relacionada a uma
alteração na relação entre os sítios de Bronsted e de Lewis, com o aumento da
temperatura. E a geração de éter etílico a partir da hidratação do etileno é
também uma reação relativamente simples, de acordo com a literatura (7) e
conforme o mecanismo apresentado na Figura 5.30.
0 20 40 60 80 100 120 140 160 180 200 220 240
0
50
100
150
200
250
Éte
r et
ílico
(u.
a.).
m-2
Fluxo de He+etileno: 20 ml/min
ZEO300 ZEO400 ZEO500 ZEO600
Tempo (min)

113
Figura 5.31 - Mecanismo simplificado de geração do éter a partir do etileno.
5.5.3 Testes Catalíticos com os HY não Autoclavados
A Figura 5.31 apresenta as curvas construídas a partir do
monitoramento, pelo TCD do cromatógrafo, da saída de água em função do
tempo das HY, e todas já corrigidas pelas respectivas áreas específicas.
Conforme se observa a HY 600 possuía mais água adsorvida em sua
superfície que as demais.
1 ) Etapaa
2 ) Etapaa
ou
+H C C
H H
H
H
H
OH.... C C
H H
H
H
H
O..H
H+ 2H Oráp ido
C C+
H H
H H
H
C C +
H H
H H
H + C C
H H
H
H
H
H ..O..
C C
H H
H
H
H
O..H
+ C C
H H
H H
H
3 ) Etapaa
C C
H H
H
H
H
O..H
+ C C
H H
H H
H+
H C C
H H
H H
HC C
H H
HH
H
O....
+ +H C C lenta C C +
H

114
Figura 5.38 – Limpeza da superfície dos HY em função do tempo.
Depois de finalizado os procedimentos de limpeza da superfície de cada
HY, estes foram testados na reação modelo, sob as mesmas condições já
discutidas para os demais materiais. Conforme se verificou (Figura 58), os
resultados obtidos com os HY300 e HY400 foram melhores em relação aos
obtidos com os HY500 e HY600, provavelmente, por apresentarem áreas
maiores que os demais. E mais importante, os HY geram apenas etanol a partir
da hidratação do etileno, em quantidade maior que as CATAPAL A e as
ZEÓLITAS Y, mesmo possuindo uma área um pouco menor que essas.
0 10 20 30 40 50 60 70 80
0
100
200
300
400
500
600
700
HY300 HY400 HY500 HY600
Fluxo de He: 22 ml/minLimpeza de superfície - HY
Tem
pera
tura
(ºC
)
Tempo (min)
0
50
100
150
200
Águ
a (u
.a.)
.m-2

115
Figura 5.33 - Produção de etanol a partir do etileno utilizando como catalisador
os HY calcinados em diferentes temperaturas.
5.5.4 Teste catalítico utilizando os HY autoclavado s
Os experimentos realizados com a nióbia após o tratamento
hidrotérmico, processo de autoclavagem que resultou na AHY, seguiram os
mesmos procedimentos já adotados para os demais materiais, ou seja,
primeiramente foi realizada a limpeza da superfície e posteriormente o teste
com a reação modelo, lembrando que cada catalisador foi analisado
separadamente. A Figura 5.33 mostra os resultados obtidos com a limpeza da
área de cada AHY calcinada, sendo que esses resultados já foram corrigidos,
por suas respectivas áreas específicas.
0 25 50 75 100 125 150 175 200 225 250
0
5
10
15
20
25
30Fluxo de He+etileno: 20 ml/min
HY300 HY400 HY500 HY600
Eta
nol (
u.a.
).m
-2
Tempo (min)

116
Figura 5.34 – Limpeza da superfície dos AHY em função do tempo.
Os resultados obtidos com a reação de hidratação do etileno foram
muito bons, conforme pode ser visualizado na Figura 5.34. Primeiro, porque
AHY foram seletivas em etanol durante a reação e, segundo, porque a
quantidade de etanol gerada na AHY calcinada a 300 °C/6horas foi maior que
com todos os demais materiais já testados até aqui, até mesmo maior que a
obtida com a Zeólita Y, um material bem conhecido na literatura por suas
propriedades ácidas. Este resultado foi muito interessante para o presente
trabalho, uma vez que sua proposta principal foi a de agregar um maior valor
comercial ao óxido de nióbio.
0 10 20 30 40 50 60 70 80 90 100
0
200
400
600
800
1000
0
50
100
150
200
Tem
pera
tura
(ºC
)
Águ
a (u
.a.)
.m-2
Tempo (min)
Fluxo de He: 22 ml/minLimpeza de superfície - AHY
AHY300 AHY400 AHY500 AHY600

117
Figura 5.35 - Produção de etanol a partir do etileno utilizando como catalisador
as AHY calcinadas em diferentes temperaturas.
Com base nos resultados promissores obtidos com as AHY, decidiu-se
realizar uma melhor caracterização da natureza das superfícies de alguns dos
materiais avaliados na reação catalítica, a fim de identificar em que o material
autoclavado (AHY) diferenciava em relação aos demais. Uma das principais
características de um bom catalisador para a reação de hidratação de etileno
está relacionada à quantidade e a natureza dos sítios ácidos presentes em sua
superfície.
55..66 DDeetteerrmmiinnaaççããoo ddaa aacciiddeezz ttoottaall
Seguindo o procedimento descrito no item 4.3.5 foram realizadas
análises de quimissorção de amônia em todos os materiais. Conforme se
0 20 40 60 80 100 120 140 160 180 200 220 240
0
10
20
30
40
50Fluxo de He+etileno: 20 ml/min
AHY300 AHY400 AHY500 AHY600
Eta
nol (
u.a.
).m
-2
Tempo (min)

118
observa na Tabela 5.5 e na Figura 5.35, o maior valor de acidez total registrado
a 300 °C foi o da AHY, sendo coerente com os dados obtidos no teste
catalítico. No entanto, nenhuma discrepância foi observada entre a AHY e a
HY, ambas calcinadas a 600°C, pois, nesta temperatu ra, os dois materiais
apresentaram valores de acidez total expressivos e semelhantes (esses já
corrigidos por suas respectivas áreas específicas), resultado esse discrepante
em relação ao desempenho no teste catalítico.
Tabela 5.5 – Acidez total, corrigidos pelas áreas específicas, dos materiais
catalíticos em suas respectivas temperaturas de calcinação.
Materiais 300°C
(µµµµmol/m 2) 400°C
(µµµµmol/m 2) 500°C
(µµµµmol/m 2) 600°C
(µµµµmol/m 2)
CAT 0,69 1,69 1,27 2,15
ZEO 4,51 4,07 3,01 2,81
AHY 7,97 4,88 5,26 16,45
HY 6,68 5,91 6,41 15,33

119
Figura 5.36 – Acidez total, corrigidos pelas áreas específicas, dos materiais
catalíticos em suas respectivas temperaturas de calcinação.
No apêndice L estão apresentadas tabelas comparativas dos resultados
obtidos por volumetria de nitrogênio e quimissorção de amônia. Em função das
divergências encontradas e outras observações experimentais, como a
reprodução de alguns resultados, foram realizados monitoramentos da acidez
total dos quatro materiais calcinados a 300 °C, ao longo de seis semanas, a fim
de verificar possíveis modificações. Dos quatro materiais analisados, a
CATAPAL A, calcinada a 300 oC (CAT300) não sofreu nenhuma modificação
significativa no valor de sua acidez total. No entanto, o valor da acidez total da
ZEÓLITA Y (ZEO300) e das nióbias (AHY300 e HY300), todas tratadas a
300oC, diminuiu ao longo desse tempo, em relação à primeira medida realizada
com o material recém preparado. Na Tabela 5.6 e na Figura 5.36 são
apresentados esses resultados, corrigidos pelas respectivas áreas específicas.
300
400
500
600
0
3
6
9
12
15
18
C A T A H Y H Y Z E O
M ateriais
C A T A H Y H Y Z E O
Aci
dez
Tot
al (
µµ µµmol
.m-2)
Temperatura ( oC
)
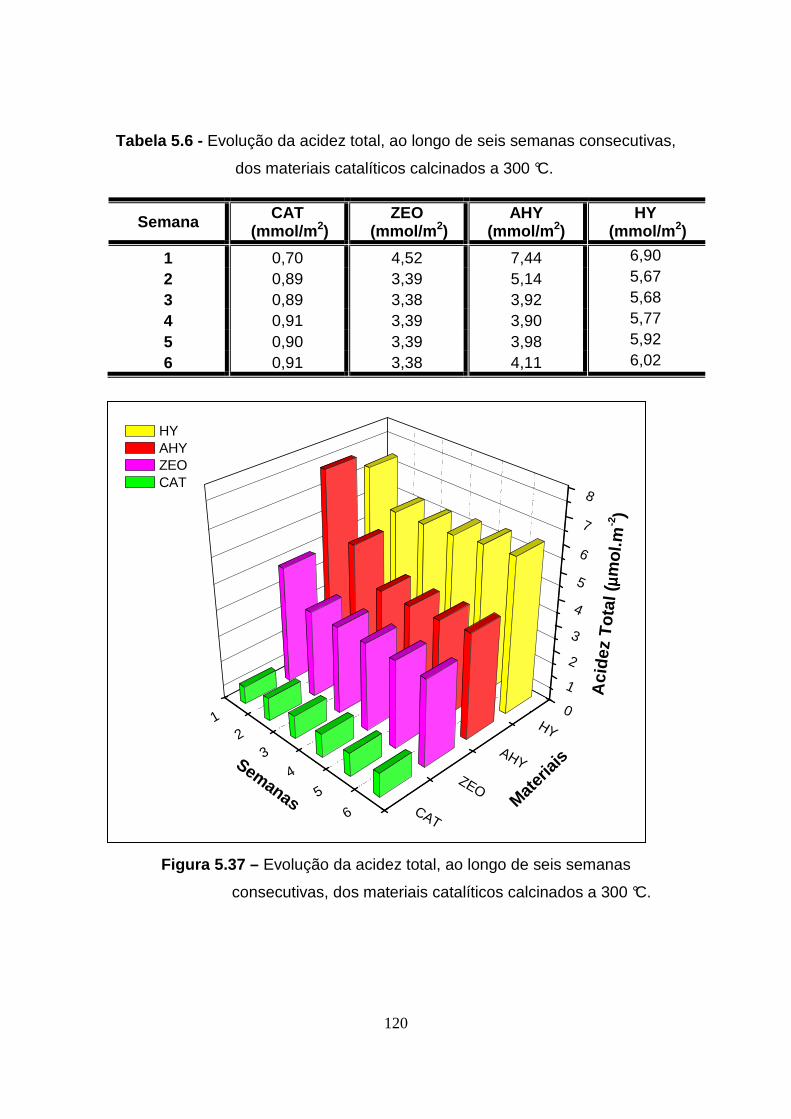
120
Tabela 5.6 - Evolução da acidez total, ao longo de seis semanas consecutivas,
dos materiais catalíticos calcinados a 300 °C.
Figura 5.37 – Evolução da acidez total, ao longo de seis semanas
consecutivas, dos materiais catalíticos calcinados a 300 °C.
Semana CAT (mmol/m 2)
ZEO (mmol/m 2)
AHY (mmol/m 2)
HY (mmol/m 2)
1 0,70 4,52 7,44 6,90
2 0,89 3,39 5,14 5,67
3 0,89 3,38 3,92 5,68
4 0,91 3,39 3,90 5,77
5 0,90 3,39 3,98 5,92
6 0,91 3,38 4,11 6,02
12
34
5
6
0
1
2
3
4
5
6
7
8
HY
AHYZEO
CAT M
ater
iais
Aci
dez
To
tal (
µµµµmo
l.m-2)
Semanas
HY AHY ZEO CAT

121
Paralelamente, foram realizadas também análises de volumetria de N2
para verificar se ocorrem modificações das áreas específicas. Os resultados da
ZEÓLITA Y e da CATAPAL A não mostraram qualquer alteração, ou seja, o
formato das isotermas, os valores das áreas específicas e as distribuições de
poros permaneceram inalterados. No entanto, as nióbias (HY e AHY)
apresentaram alterações, tanto o formato da isoterma mudou como os valores
de suas áreas diminuíram.
O monitoramento do óxido de nióbio autoclavado e calcinado a 300 oC
(AHY300), após 3 meses, mostrou uma diminuição da área específica de 205
m2/g para 102 m2/g, o mesmo aconteceu com as demais temperaturas estudas
e, também com HY. A Figura 5.37 mostra os resultados obtidos por volumetria
de nitrogênio da AHY 500, quando recém preparada, e esta após 3 meses
armazenada no laboratório, a temperatura ambiente, em recipiente
devidamente tampado e ao abrigo da luz.
Figura 5.38 - Isotermas de adsorção-dessorção e distribuições porosas da
AHY por volumetria de nitrogênio.
20 40 60 80 100 120 140
0,0
0,1
0,2
0,3
0,4
0,5
0,6
Vol
ume
Ads
orvi
do (
cm3 .g
-1)
AHY 500 (após 3 meses) AHY 500 (recém preparado)
Diâmetro de poros (Å)
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
20
40
60
80
100
120
Vol
ume
Ads
orvi
do (
cm3.g
-1)
P/P0
AHY 500 (após 3 meses) AHY 500 (recém preparado)

122
Baseando-se nos resultados encontrados para os materiais puros,
decidiu-se preparar compostos mistos contendo alumínio e nióbio, a partir da
CATAPAL A (alumina) e da Nióbia, em cinco proporções diferentes,
submetendo essas misturas ao tratamento hidrotérmico, gerando as Al%Nb,
que também foram caracterizadas e posteriormente testadas na reação de
hidratação do etileno.
55..77 CCaattaall iissaaddoorreess mmiissttooss –– AAll%%NNbb
Todas as cinco misturas (5, 10, 20, 30 e 40% em massa de nióbia em
alumina) foram calcinadas a 400 °C por 6 horas, seg uindo a mesma
programação de temperatura das amostras puras. Essa temperatura foi
escolhida após analisar os resultados dos materiais separadamente, pois, a
400 °C a CATAPAL A apresentou uma boa resposta na r eação modelo, e a
AHY manteve suas características principais para uma boa produção de etanol
a partir do etileno. A seguir, procederam-se as etapas de caracterização, tais
quais foram realizadas para os materiais anteriores.
5.7.1 Caracterização dos catalisadores mistos
A caracterização por MEV, assim como as dos outros materiais já
discutidos, não apresentaram resultados significativos, pois todas as amostras
mostraram-se amorfas, conforme podem ser visualizadas no apêndice M. E as
análises de difratometria de raios X forneceram resultados interessantes. A
Figura 5.39 mostra os difratogramas das cinco misturas após serem
autoclavadas, secas e peneiradas, respectivamente.

123
10 30 50 70 90
2Theta
Al5Nb
Al20Nb
Al30Nb
Al40Nb
Al10Nb
Inte
nsi
dad
e d
a a
mo
stra
(u.a
.)
Figura 5.39 - Difratogramas de raios X das AlX%Nb calcinada a 400 °C/6
horas.
Analisando a Figura 5.38 observa-se que o difratograma referente à mistura
Al5%Nb possui características semelhantes as da catapal A pura, ou seja,
picos característicos de uma γ-alumina. E à medida que se aumentou a
quantidade de HY os difratogramas foram registrando a perda dos picos
característicos da catapal A. Conforme pode-se observar a mistura Al40%Nb
possui difratograma característico somente da HY pura.
Os resultados obtidos por volumetria de nitrogênio dos catalisadores
contendo diferentes teores de Nb (Figura 5.39) mostraram isotermas parecidas
às da CATAPAL A pura.

124
Figura 5.40 - Isotermas de adsorção-dessorção das Al%Nb preparadas com
diferentes teores de nióbia (AHY).
A Tabela 5.7 apresenta os valores obtidos de área específica, volume e
diâmetro dos poros, dessas misturas, e acidez total, podendo-se observar que
entre elas os resultados da Al10%Nb se destacam das demais. As distribuições
de poros das misturas podem ser visualizadas no apêndice N.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
40
80
120
160
200
240V
olum
e A
dsor
vido
(cm
.g-1)
P/P0
Al5% Nb Al10% Nb Al20% Nb Al30% Nb Al40% Nb

125
Tabela 5.7 – Volumetria de nitrogênio e quimissorção de amônia para as
misturas Al%Nb.
Dando continuidade aos experimentos, as misturas foram submetidas à
avaliação catalítica na reação de hidratação de etileno, seguindo a mesma
metodologia dos demais catalisadores apresentados anteriormente, ou seja,
limpeza da superfície e posteriormente a reação em si. Na Figura 66, estão
apresentadas as curvas construídas com a geração de etanol, corrigidas por
suas respectivas áreas específicas. De acordo com esses dados, observa-se
que todas as misturas apresentaram desempenhos catalíticos muito baixos,
apesar de serem levemente superiores ao da CATAPAL A. Essa baixa
atividade conduziu a resultados aparentemente incoerentes, mas que estão
relacionados ao limite de detecção do cromatógrafo.
Al com %Nb Vp (cm 3/g) Dp (Å) Área (m 2/g) Acidez
(µµµµmol/g) Acidez/Área
(µ(µ(µ(µmol/m 2)
5% 0,25 52 192 508 2,65 10% 0,37 41 360 598 1,66 20% 0,36 66 217 362 1,67 30% 0,26 61 169 357 2,11 40% 0,35 48 285 396 1,39

126
Figura 5.41 – Produção de etanol, obtida a partir do etileno, utilizando, como
catalisadores, misturas Al%Nb calcinadas a 400 °C.
Fazendo uma comparação dos catalisadores mistos que obtiveram os
melhores resultados (Al5%Nb, Al20% e Al30%) com os seus correspondentes
puros calcinados a 400 °C, observou-se que a resposta do AHY400 foi muito
boa em relação às misturas, mas, estas por sua vez também apresentaram
geração de etanol um pouco maior que a CAT400, como pode ser verificado na
Figura 5.41.
0 20 40 60 80 100 120 140 160 180 200 220 240
0
1
2
3
4
5
6
7 Al5% Nb Al10% Nb Al20% Nb Al30% Nb Al40% Nb
Eta
nol (
u.a.
).m
-2
Tempo (min)
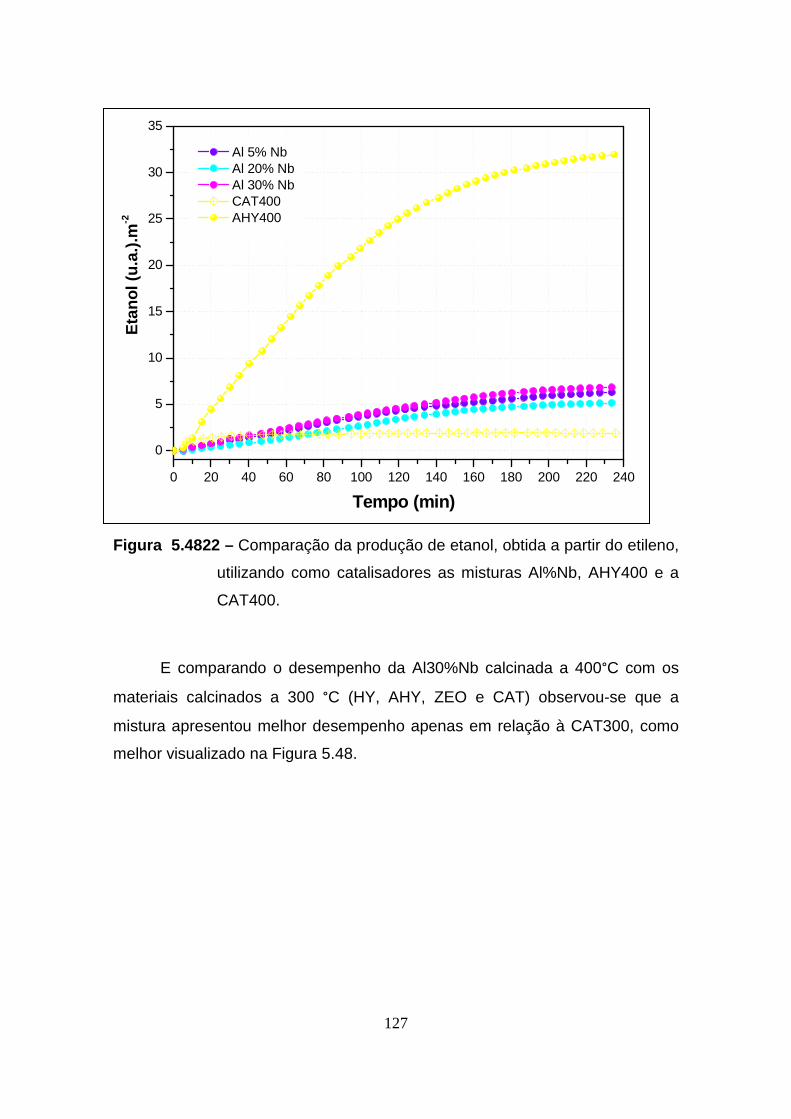
127
Figura 5.4822 – Comparação da produção de etanol, obtida a partir do etileno,
utilizando como catalisadores as misturas Al%Nb, AHY400 e a
CAT400.
E comparando o desempenho da Al30%Nb calcinada a 400°C com os
materiais calcinados a 300 °C (HY, AHY, ZEO e CAT) observou-se que a
mistura apresentou melhor desempenho apenas em relação à CAT300, como
melhor visualizado na Figura 5.48.
0 20 40 60 80 100 120 140 160 180 200 220 240
0
5
10
15
20
25
30
35
Al 5% Nb Al 20% Nb Al 30% Nb CAT400 AHY400
Eta
nol (
u.a.
).m
-2
Tempo (min)

128
Figura 5.233 – Comparação da produção de etanol, obtida a partir do etileno,
utilizando como catalisadores as misturas Al30%Nb calcinada a
400 °C, AHY300, HY300, ZEO30 e a CAT300.
A fim de compreender melhor esses resultados foram realizados os
experimentos de acidez específica, utilizando como sonda a piridina, pois esta
molécula é capaz de determinar simultaneamente os sítios de Lewis e de
Bronsted existentes em uma superfície.
55..88 CCaarraacctteerr iizzaaççããoo ppoorr FFTTIIRR
Segundo a literatura (86), a piridina pode se ligar à superfície de
materiais sólidos ácidos de forma coordenada, o que caracteriza a existência
de um sítio ácido de Lewis, ou podendo se ligar na forma de um íon piridinion,
0 50 100 150 200 250
0
10
20
30
40
50
Eta
nol (
u.a.
).m
-2
Tempo (min)
Al30b AHY300 HY300 ZEO300 CAT300

129
o que caracteriza a existência de um sítio ácido de Bronsted, conforme
apresentado na Tabela 5.8.
Tabela 5.8 – Bandas no infravermelho (1400-1700 cm-1) região de adsorção da
piridina em sólidos ácidos (86).
Então de acordo com a bibliografia (86), as bandas detectadas por FTIR
nas amostras podem ser associadas à piridina coordenada em centros ácidos
de Lewis e de Brönsted. Na Tabela 5.9 estão apresentados, de forma
resumida, os comprimentos de onda típicos associados a cada banda.
Tabela 5.9 – Bandas associadas aos centros ácidos de Lewis e Brönsted.
Comp. onda (cm -1)
Lewis Lewis + Brönsted Brönsted 1455-1450 1620-1600 1575 1490 1545 1635
Os espectros apresentados na Figura 5.43 correspondem às medidas
realizadas à temperatura de dessorção de 150 ºC e representam a diferença
entre o espectro obtido após contato com piridina e o espectro da pastilha após
o pré-tratamento, isto para cada amostra. Para o cálculo das concentrações
considerou-se os coeficientes de extinção molar associados a cada tipo de
centro ácido, apresentados na bibliografia (87) cujos parâmetros são 2,22
cm/µmol e 1,67 cm/µmol para os centros ácidos de Lewis e Brönsted,
respectivamente.
Piridina liga da ao hidrogênio
Piridina ligada de forma coordenada Íon piridinion
1400-1477 1447-1460 1485-1490 1488-1503 1485-1500
1540 1580-1600 ~1580
1600-1633 ~1640

130
Figura 5.44 - Espectros de FTIR relativos à adsorção da piridina para as
amostras AHY300, AHY400 e CAT400 (Tdessorção = 150 ºC; P = 10-6 mbar).
Os resultados obtidos com a CAT400 indicaram a presença apenas de
sítios ácidos de Lewis. Enquanto que os resultados obtidos com AHY300 e a
AHY400 apresentaram características únicas, correspondentes à adsorção em
sítios ácidos de Lewis e de Bronsted, sendo que a AHY300 apresentou um
número de centros ácidos, tanto de Lewis quanto de Bronsted, relativamente
mais elevados que a AHY400, conforme se visualiza na Tabela 5.10.
1700 1650 1600 1550 1500 1450 14000,0
0,1
0,2
0,3
0,4
1446
1609
Abs
orvâ
ncia
(u.
a.)
No de ondas (cm -1)
AHY300 AHY400 CAT400
1619
1450

131
As amostras das misturas Al30%Nb, Al20%Nb e Al5%Nb apresentaram
um comportamento intermediário entre a CAT400 e a AHY400, não
apresentando centros ácidos de Bronsted (Figura 5.44).
Figura 5.45 - Espectros de FTIR relativos à adsorção da piridina para as
amostras AHY400, Al30Nb400, Al20Nb400, Al5Nb400 e CATA400 (Tdesorção
= 150 ºC; P = 10-6 mbar).
1400 1450 1500 1550 1600 1650 1700
0,0
0,1
0,2
0,3
0,4 AHY400 CAT400 Al5Nb Al20Nb Al30Nb
Abs
orvâ
ncia
(u.
a.)
No de ondas (cm -1)

132
Tabela 5.10 - Absorvâncias integradas e concentrações dos centros ácidos de
Lewis e Brönsted obtidas para as diferentes amostras.
AHY300 (m = 14,50 mg) Temp. Absorvância integrada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 2,92 0,47 182 39 200 1,94 0,26 121 21 250 1,14 0,11 71 10
AHY400 (m = 15,87 mg)
Temp. Absorvância integada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 2,59 0,30 147 22 200 1,57 0,11 90 8 250 0,69 0 39 0
CAT400 (m = 16,51 mg) Temp. Absorvância integrada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 2,64 0 145 0 200 1,10 0 60 0 250 0,46 0 25 0
Al30%Nb (m = 16,86 mg) Temp. Absorvância integrada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 2,60 0 139 0 200 1,23 0 66 0 250 0,61 0 39 0
Al20%Nb (m = 13,84 mg) Temp. Absorvância integrada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 1,90 0 124 0 200 1,03 0 68 0 250 0,44 0 29 0
Al5%Nb (m = 17,90 mg) Temp. Absorvância integrada (cm-1) Concentração (µmol/g) (ºC) Centros de Lewis Centros de Brönsted Centros de Lewis Centros de Brönsted 150 2,57 0 130 0 200 1,16 0 59 0 250 0,34 0 17 0

133
É importante ressaltar que os testes catalíticos com a reação de
hidratação do etileno foram efetuados a 220 oC e, consequentemente,
participam da reação os sítios ácidos suficientemente fortes para
permanecerem ativos a essa temperatura. De acordo com os resultados de
determinação dos sítios ácidos, a principal característica a ser destacada é o
fato da AHY300 possuir tanto sítios ácidos de Bronsted quanto de Lewis em
maiores quantidades que os demais catalisadores, justificando seu melhor
desempenho catalítico na reação de hidratação de etileno (Figura 5.45).
Figura 5.46 – Gráfico dos resultados para a reação de etanol a partir do
etileno, utilizando como catalisador os materiais calcinadas a 300 °C.
Sob o ponto de vista da acidez de Bronsted, um sólido ácido é capaz de
doar, ou no mínimo transferir parcialmente, um próton, associando-se aos
anions de uma superfície. Do ponto de vista de Lewis, um sólido ácido é aquele
0 40 80 120 160 200 240
0
10
20
30
40
50
Tempo (min)
Eta
nol (
u.a.
).m
-2
AHY300 CAT300 HY300 ZEO300

134
capaz de aceitar um par de elétrons. Portanto, quando a superfície ácida reage
com uma base de Lewis, uma nova ligação é formada, e é claramente diferente
de uma reação de oxi-redução, na qual a transferência de um ou dois elétrons
é completa.
Conceitos como esses são muito importantes para fundamentar e
desenvolver novos materiais catalíticos ácidos, pois, é grande o número de
reações que os utilizam. Mas, sua importância e a necessidade industrial têm
ultrapassado a velocidade das descobertas da química fundamental e muitos
acabam sendo utilizados lançando mão de métodos experimentais.
A literatura [29] relata que a nióbia depois de calcinada a 400 °C forma
sítios ácidos de Bronsted, e que uma vez aumentando a temperatura de
calcinação aumenta-se os sítios de Lewis. Neste trabalho foi verificado que a
HY calcinada a 300 °C já possui sítios de Bronsted, e quando se aumenta a
temperatura para 400 °C há uma diminuição da quantidade de sítios ácidos,
tanto os de Lewis quanto os de Bronsted, neste caso, corrobora com a
divulgação de Ushikubo [24].
A alumina estudada, no caso a catapal A, uma psedoboemita, quando
calcinada a 400 °C apresentou somente sítios ácidos de Lewis, e não uma
soma dos sítios ácidos (Bronsted + Lewis). Neste caso, em particular, essa
propriedade justificou seu menor desempenho na reação catalítica de
hidratação do etileno em relação à nióbia, esta apresentou tanto sítios de Lewis
quanto de Bronsted.
De acordo com relatos anteriores [39] esperava-se que os resultados
dos experimentos de geração de etanol utilizando as misturas fossem ter
melhor desempenho que os materiais puros. No entanto, ocorreu o contrário, e
verificou-se que o melhor material para ser utilizado na reação de hidratação de
etileno foi a nióbia autoclavada, já que as misturas apresentaram resultados
melhores apenas em relação à alumina pura, a catapal A.

135
CCAAPPÍÍTTUULLOO 66
66 CCOONNCCLLUUSSÕÕEESS
Diante dos resultados apresentados e das discussões realizadas nos
capítulos anteriores, as principais conclusões deste trabalho são:
1) Os materiais HY e AHY apresentaram resultados na hidratação do etileno a
etanol muito superiores ao do catalisador normalmente utilizado nessa reação,
a zeólita Y, devendo-se ressaltar, além disso, que diferentemente deste
catalisador mais utilizado, o emprego dos óxidos de nióbio HY e AHY não
conduziram à formação de éter etílico mas, exclusivamente, à formação de
etanol sendo, portanto, tais óxidos mais ativos e seletivos para a produção de
etanol.
2) A principal diferença, e que teve grande relevância neste trabalho, foi o
tratamento hidrotérmico dado a HY, produzindo a AHY, pois, este melhorou as
propriedades da HY sem alterar suas características intrínsecas, como pôde
ser constatado, ao logo de toda a discussão realizada nos processos de
caracterização, e na reação catalítica. As respostas da HY e da AHY foram
semelhantes, no entanto, o diferencial que duplicou a geração de etanol na
reação estudada foi em função do procedimento de autoclavagem.
3) O processo de autoclavagem (hidrotratamento) do ácido nióbico hidratado,
comercialmente disponibilizado pela Companhia Brasileira de Metalurgia e
Mineração (CBMM), como HY-340, conferiu ao produto propriedades texturais
e reológicas que o torna muito interessante a reações catalíticas heterogêneas.
O aumento significativo do valor da área específica tem relação direta com o
número de sítios catalíticos, e a melhora nas propriedades reológicas viabiliza
sua obtenção na forma de moldados (extrudados), característica essencial nas
aplicações industriais.

136
4) O processo de autoclavagem (hidrotratamento) do produto comercial HY-340
também aumentou significativamente a acidez total, assim como os sítios de
Lewis e de Bronsted, característica muito importante em diversas reações
catalíticas que envolvem sítios ácidos, entre elas, o craqueamento catalítico
fluido, presente nas reinarias de petróleo.
5) O óxido nióbico hidratado (HY-340) autoclavado, além de ser o mais ativo, é
o mais seletivo a etanol, não conduzindo à formação de éter, produto
observado quando se emprega a zeólita Y.
6) O material autoclavado (AHY) mais ativo foi o calcinado a 300 oC,
provavelmente, porque apresenta a maior quantidade de sítios ácidos de
Bronsted e de Lewis na sua superfície.
7) Atividade catalítica na reação de hidratação de etileno somente foi
observada nos catalisadores que apresentam sítios de Bronsted em sua
superfície, caso dos óxidos de nióbio HY e AHY. O baixo desempenho obtido
com a Catapal A indica que somente a presença de sítios de Lewis não garante
uma boa atividade na reação de hidratação de etileno.
8) A adição de óxido de nióbio ao hidróxido de alumínio (Catapal), antes da
etapa de hidrotratamento (autoclavagem), não conduziu a catalisadores
significativamente mais ativos na reação de hidratação de etileno, embora
tenha se observado um pequeno aumento na produção de etanol em relação à
Catapal A pura. Esta constatação pode ser atribuida ao fato de que a
metodologia empregada na preparação dos óxidos mistos não aumentou a
quantidade de sítios ácidos, principalmente os de Bronsted.

137
CCAAPPÍÍTTUULLOO 77
SSUUGGEESSTTÕÕEESS PPAARRAA TTRRAABBAALLHHOOSS FFUUTTUURROOSS
De acordo com os resultados obtidos e as discussões apresentadas
neste trabalho, pode-se sugerir como trabalhos futuros:
1) Com relação à síntese do catalisador AHY, material comercial autoclavado,
investigar mudanças no processo que possam maximizar as propriedades
texturais, principalmente a área específica, de forma a aumentar a quantidade
e a concentração de sítios ácidos Bronsted, sítios estes que influenciam
diretamente a atividade catalítica na reação de hidratação de etileno;
2) Avaliar o catalisador à base de nióbio autoclavado em outras reações que
exigem a presença de sítios ácidos, entre elas, o craqueamento catalítico
fluido, processo presente nas refinarias de petróleo, desta forma, agregando
valor ao nióbio, cujas reservas deste minério estão concentradas no Brasil;
3) Otimizar o processo de autoclavagem, modificando alguns parâmetros de
processo, tais como temperatura e tempo de hidrotratamento, e/ou adicionando
ao material um composto que estabilize as suas propriedades texturais e
ácidas.
4) Buscar um novo processo de síntese de óxidos mistos ou mistura de óxidos,
ambos contendo Nb e Al, que resulte em catalisadores ácidos, sobretudo em
superfícies que contenham sítios de Bronsted.

138

139
RREEFFEERREENNCCIIAASS BBIIBBLLIIOOGGRRÁÁFFIICCAASS
(1) SARAIVA, I. J. Case study: produção de etanol. In: ______. Custos e impactes ambientaisno projecto de processos químico s. Coimbra, Portugal: DEQFCTUC, 2003. Cap. 6. Disponível em: <http://www.eq.uc.pt/~ines/seminario/ciappq08.html>. Acesso em: 24 set. 2007.
(2) FIGUEIRA, S.R. Os programas de álcool como combustíveis nos EUA, no Japão e na União Européia e as possibilidad es de exportação do Brasil. 2005. 246 p. Tese (Doutorado em Economia Aplicada) - USP/ESALQ, Piracicaba, 2005. Disponível em: <http://www.teses.usp.br/teses>. Acesso em: 01 mar. 2007.
(3) MARQUES, F. FAPESP lança programa para impulsionar pesquisa em bioenergia. Pesquisa FAPESP , n. 149, p. 20-25, jul. 2008.
(4) SANTOS, C. A. P. Álcool gel: a revolução. Household & Cosméticos , v. 8, n. 45, 2007. Disponível em: <http://www.profpc.hpg.ig.com.br/alcool.html>. Acesso em: 24 set. 2007.
(5) IZUMI, Y. Hydration/hydrolysis by solid acids. Catalysis Today , v. 33, n. 4, p. 371-409, 1997. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 maio 2006.
(6) SANTOS, C. A. P. Álcool gel: a revolução. Household & Cosméticos , v. 8, n. 45, 2007. Disponível em: <http://www.freedom.inf.br/artigos_tecnicos/20020409/89_90.asp>. Acesso em: 24 set. 2007).
(7) ALLINGER,N.; CAVA, M. P.; JONG, D. C. de; JOHNSON, C. R.; LEBEL, N. A.; STEVENS, C. L. Química orgânica . Rio de Janeiro: Livros Técnicos e Científicos, 1976. 961p. v. 2.
(8) Wikipedia olefina. Disponível em: < http://omega.ilce.edu.mx:3000/sites/ciencia/volumen1/ciencia2/39/html/sec_13.html> Acesso em: 25 de setembro de 2007).
(9) SHREVE, R. N.; BRINK Jr., J. A. Indústria de processos químicos. Rio de Janeiro: Guanabara Dois, 1980. 717p.

140
(10) FOUGRET, C.M.; ATKINS, M. P.; HOLDERICH, W. F. Influence of the carrier on the catalytic performance of impregnated phosphoric acid in the hydration of ethylene. Applied Catalysis A : General, v. 181, n. 1, p. 145-156, 1999. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 maio 2006.
(11) FOUGRET, C.M.; HOLDERICH, W. F. Ethylene hydration over metal phosphates impregnated with phosphoric acid. Applied Catalysis A : General, v. 207, n. 1, p. 295-301, 2001. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 maio 2006.
(12) CORMA, A.; GARCÍA, H. Organic reactions catalyzed over solid acids. Catalysis Today , v. 38, n. 3, p. 257-308, 1997.
(13) KAZANSKY, V. B. Solvation as a main factor that determines the strength os liquid superacids and selectivity of the acid-catalyzed reactions of olefins. Catalysis Today , v. 73, n. 1-2, p. 127-137, 2002. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 maio 2006.
(14) CHU, W.; ECHIZEN, T.; KAMIYA Y.; OKUHARA, T. Gas-phase hydration of ethene over tungstena-zirconia. Applied Catalysis A: General, v. 259, n. 1, p. 99-205, 2004. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 maio 2006.
(15) COMPANHIA BRASILEIRO DE METALURGIA E MINERAÇÃO. Nióbio. Disponível em: <http://www.cbmm.com.br> Acesso em: 16 de maio de 2006.
(16) Tabela periódica online – nióbio. Disponível em: <http://www.moderna.com.br/química/química_am/tperiodica/0004>. Acesso em: 17 de maio de 2006.
(17) BANCO NACIONAL DO DESENVOLVIMENTO ECONÔMICO E SOCIAL. Nióbio. Disponível em: <http://www.bndes.gov.br/conhecimento> Acesso em: 17 de maio de 2006.
(18) TAVARES, E. Nióbio- a riqueza que o Brasil precisa. Revista Decifra-me. Disponível em: <http:// www.mamore.net/port/niobio.htm> Acesso em: 17 de maio de 2006.
(19) NOWAK, I.; ZIOLEK, M. Niobium compounds: preparation, characterization, and application in heterogeneous catalysis. Chemical Reviews , v. 99, n. 12, p. 3603-3624, Dec. 1999. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.

141
(20) JEHNG, J.M.; WATCHS, I. E. Structural chemistry and Raman spectra of Niobium oxides. Chemical Materials , v.3, n. 1, p.100-107, 1991. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(21) JEHNG, L. M.; WACHS, L. E. The molecular structures and reactivity of supported niobium oxide catalysts. Catalysis Today , v.8, n. 1, p. 37-55, 1990. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(22) DENG, H. T.; KERNS, K. P.; CASTLEMAN Jr., A. W. Formation, structures, and reactivities of niobium oxide cluster ions. Journal Physical Chemistry , v. 100, n. 32, p. 13386-13392, Aug. 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(23) MAURER, S. M.; KO, E. I. Structural and acidic characterization of niobia aerogels. Journal of Catalysis , v. 135, n. 1, p. 125-134, May 1992. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(24) USHIKUBO, T.; KOIKE, Y.; WADA, K.; XIE, L.; WANG, D.; GUO, X. Study of the structure of niobium oxide by X-ray absorption fine structure and surface science techniques. Catalysis Today , v. 28, n. 1-2, p. 59-69, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(25) BATAMACK, P.; VINCENT, R.; FRAISSARD, J. The acidity of niobic studied by 1H broad-line NMR at 4K and 1H MAS NMR at room temperature: comparison with other solid acids. Catalysis Today , v.28, n. 1-2, p. 31-39, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(26) OKAZAKI, S.; KUROSAKI, A. Acidic properties and catalytic activities of niobic acid treated with phosphoric acid. Catalysis Today , v.8, n. 1, p. 113-122, 1990. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(27) TANABE, K. Catalytic application of niobium compounds. Catalysis Today , v.78, n. 1-4, p. 65-77, 2003. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.
(28) TANABE, K.; OKASAKI, S. Various reactions catalyzed by niobium compounds and materials. Applied Catalysis A: General, v.133, n. 2, p. 191-218, 1995. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 set. 2006.
(29) ZIOLEK, M. Niobium – containing catalysts – the state of the art. Catalysis Today , v.78, n. 1-4, p. 47-64, 2003. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 setembro 2006.

142
(30) SUN, Q.; FU, Y.; YANG, H.; AUROUX, A.; SHEN, J. Dehydration of methanol t imethyl ether over Nb2O5 and NbOPO4 catalysts: microcalorimetric ant FT-IR studies. ;Journal of Molecular Catalysis A: Chemical, v. 275, n. 1-2, p. 183-193, Sept. 2007. Disponível em: <http://www.sciencedirect.com> . Acesso em: 20 set. 2007.
(31) SILVA, C. L. T. da; CAMORIM, V. L. L.; ZOTIN, J. L.; PEREIRA, M. L. R. D.; FARO Jr., A. da C. Surface acidic properties of alumina-supported niobia prepared by chemical vapour deposition and hydrolysis of niobium pentachloride. Catalysis Today , v.57, n. 3-4, p. 209-217, 2000. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(32) USHIKUBO, T. Recent topics of research and developpment of catalysis by niobium and tantalum oxides. Catalysis Today , v.57, n. 3-4, p. 331-338, 2000. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 set. 2006.
(33) WEISSMAN, J. G. Niobia-alumina supported hydroprocessing catalysts: relationship between activity and support surface acidity. Catalysis Today , v.28, n. 1-2, p. 159-166, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(34) ICHIKUNI, N.; SHIRAI, M.; IWASAWA, Y. Surface structures and catalytic properties of supported niobium oxides. Catalysis Today , v.28, n. 1-2, p. 49-58, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(35) BRAGA, V. S.; DIAS, J. A.; DIAS, S. C. L.; MACEDO, J. L. Catalyst materials based on Nb2O5 supported on SiO2-Al2O3: preparation and structural characterization. Chemical Materials , v.17, n. 3, p.690-695, Feb. 2005. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 set. 2006.
(36) BRAYNER, R.; RODRIGUES, J.A.J.; CRUZ, G. M. ;Synthesis and molding of niobium oxynitrides with macropores generation: reactivity and stability in cyclohexane dehydrogenation. ;Catalysis Today , v.57, n. 3-4, p. 219-223, 2000. Disponível em: <http://www.sciencedirect.com>. Acesso em: 11 set. 2006.
(37) SANTOS, A.C.B. dos.; GRANGE, P.; FARO Jr., A.C. Effect of support sulphidation on the hydrocracking activity of niobia-supported nickel and molybdenum catalysts. Applied Catalysis A: General, v.178, n. 1, p. 29-38, 1999. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(38) GEANTET, C.; AFONSO, J.; BREYSSE, M.; ALLALI, N.; DANOT, M. Niobium sulfides as catalysts for hydrotreating reactions. Catalysis Today ,

143
v.28, n. 1-2, p. 23-30, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(39) VÉDRINE, J.C.; COUDURIER, G.; OUQOUR, A.; PRIES DE OLIVEIRA, P.G.; VOLTA, J.C. Niobium oxide based materials as catalysts for acidic and partial oxidation type reactions. Catalysis Today , v. 28, n. 1-2, p. 3-15, 1996. Disponível em: <http://www.sciencedirect.com>. Acesso em: 22 out. 2006.
(40) WACHS, I. E.; BRIAND, L. E.; JEHNG, J.M.; BURCHAM, L.; GAO, X. Molecular structure and reactivity of the group V metal oxides. Catalysis Today , v.57, n. 3-4, p. 323-330, 2000. Disponível em: <http://www.sciencedirect.com>. Acesso em: 14 mar. 2007.
(41) SCHMAL, M.; MENDES, F.M.T.; PEREZ, C.A.; SOARES, R.R.; NORONHA, F.B. Ammonium complex of niobium as a precursor for the preparation of Nb2O5/Al2O3. Catalysis Today , v.78, n. 1-4, p.449-458, 2003. Disponível em: <http://www.sciencedirect.com>. Acesso em: 14 mar. 2007.
(42) LI, Y.; YAN, S.; YUE, B.; YANG, W.; XIE, Z.; CHEN, Q.; HE, H. Selective catalytic hydration of ethylene oxide over niobium oxide supported on alumina. Applied Catalysis A: General, v. 272, n. 1-2, p. 305-310, 2004. Disponível em: <http://www.sciencedirect.com>. Acesso em: 14 mar. 2007.
(43) LI, Y.; YAN, S.; YUE, B.; YANG, W.; XIE, Z.; CHEN, Q.; HE, H. Selective catalytic hydration of ethylene oxide over niobium oxide supported on alumina. Journal of Molecular Catalysis A: Chemical, v. 226, n. 2, p. 285-290, Feb. 2005. Disponível em: <http://www.sciencedirect.com>. Acesso em: 14 mar. 2007.
(44) REHIM-ABDEL, M. A.; SANTOS, A. C. dos; CAMORIM, V. L. L.; FARO Jr., A. da C. Acid-base reactions on alumina-supported niobia. Applied Catalysis A: General, v. 305, n. 2, p. 211-218, 2006. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 nov. 2007.
(45) BARROS, I. C. L.; BRAGA, V. S.; PINTO, D. S.; MACEDO, J. L. de; FILHO, G. N. R.; DIAS, J. A.; DIAS, S. C. L. Effects of niobium addition on ZSM-5 studied by thermal and spectroscopy methods. Microporous and Mesoporous Materials , v. 109, p. 485-493, 2008. Disponível em: <http://www.sciencedirect.com>. Acesso em: 16 nov. 2008.
(46) WIKIPEDIA. Óxido de alumínio . Disponível em: <http://pt.wikipedia.org/wiki%C3%93xido_de_alum%C3%ADnio>. Acesso em: 01 mar. 2007.

144
(47) ASSOCIAÇÃO BRASILEIRA DO ALUMÍNIO (ABAL). O alumínio : história. São Paulo: ABAL, s.d. Disponível em:< http://www.abal.org.br/aluminio/historia.asp>. Acesso em: 01 mar. 2007.
(48) ASSOCIAÇÃO BRASILEIRA DO ALUMÍNIO (ABAL). Alumínio para futuras gerações : a indústria brasileira do alumínio e o desenvolvimento sustentável. São Paulo: ABAL, 2000. Disponível em: <http://www.abal.org.br/manuais/html/pdf/futuras_geracoes_parte2.pdf>. Acesso em: 01 mar. 2007.
(49) MISRA, C. Industrial alumina chemicals . Washinton, D. C.: America Chemical Society, 1986. 165p.
(50) CONSTANTINO, V. R. L.; ARAKI, K.; SILVA, D. DE O.; OLIVEIRA, W. de. Preparação de compostos de alumínio a partir da bauxita: considerações sobre alguns aspectos envolvidos em um experimento didático. Química Nova , v. 25, n. 3, p. 490-498, 2002.
(51) ANSELL, S.; KRISHNAN, S.; WEBER, J. K. R.; FELTEN, J.J.; NORDINE, P.C.; BEO, M.A.; PRICE, D.L.; SABOUNGI, M. L. Structure of liquid aluminum oxide. Physical Review Letters , v. 78, n. 3, p. 464-466, 1997.
(52) AUROUX, A., GERVASINI, A. Infrared spectroscopic study of the acidic character of modified alumina surfaces. Adsorption Science & Technology , v. 21, n. 8, p. 721-737, 2003.
(53) BAUMANN, T.F.; GASH, A.E.; CHINN, S.C.; SAWVEL, A.M.; MAXWELL, R.S.; SATCHER, J. H. Synthesis of high-surface-area alumina aerogels without the use of alkoxide precursors. Chemistry of Materials , v. 17, n.2, p. 395-401, 2005.
(54) CASTEL, B. Les alumines et leurs applications . Paris: Nathan, 1990. 74p.
(55) CASTRO, R. H. R.; GOUVÊA, D. Efeito do íon Mn como aditivo na transição de fase γ→ α da alumina. Cerâmica , v. 49, p. 55-60. 2003.
(56) OIKAWA, T.; OOKOSHI, T.; TSUNEHIRO, T. A new heterogeneous olefin metathesis catalyst composed of rhenium oxide and mesoporous alumina. Microporous and Mesoporous Materiais , v. 74, n. 1-3, p. 93-103, 2004.
(57) TETTENHORST, R.; HOFMANN, D. A. Crystal chemistry of boehmite. Clays and Clay Minerals , v. 28, n. 5, p. 373-380, 1980.

145
(58) SANTOS, P. S.; SANTOS, H.S.; TOLEDO, S. P. Standard transition aluminas: electron microscopy studies. Materials Research , v. 3, n. 4, p. 104-114, 2000. Disponível em: http://www.scielo.br. Acesso em: 06 mar. 2007.
(59) CIOLA, R. Fundamentos da catálise . São Paulo: Editora Moderna e EDUSP, 1981. 297p.
(60) LAMBERT, J. F.; CHE, M. The molecular approach to supported catalysts synthesis: state of the art and future challenges. Journal of Molecular Catalysis A: Chemical, v. 162, n. 1-2, p. 5-18, 2000. Disponível em: <http://www.sciencedirect.com>. Acesso em: 07 mar. 2007.
(61) PERI, J. B. Infrared and gravimetric study of the surface of γ-alumina . The Journal of Physical Chemistry , v. 69, n. 1, p. 211-219, 1965.
(62) ______. A model for the surface of γ-alumina. The Journal of Physical Chemistry , v. 69, n. 1, p. 220-230, 1965.
(63) ______. Infrared study of adsorption of ammonia on dry γ-alumina. The Journal of Physical Chemistry , v. 69, n. 1, p. 231-239, 1965.
(64) BAGWELL, R. B.; MESSING, G. L. Effect of seeding and water vapor on the nucleation and growth of alpha-Al2O3 from gamma- Al2O3. Journal of the American Ceramic Society, v. 82, n. 4, p. 825-832, Apr. 1999. . Disponível em: <http://www.sciencedirect.com>. Acesso em: 07 mar. 2007.
(65) BAGWELL, R. B.; MESSING, G. L.; HOWELL, P. R. The formation of alpha-Al2O3 from theta-Al2O3: The relevance of a “critical size” and diffusional nucleation or “synchro-shear? Journal of Materials Science , v. 36, n. 7, p. 1833-1841, Apr. 2001. Disponível em: <http://www.sciencedirect.com>. Acesso em: 07 mar. 2007.
(66) YANG, X.; SUN, Z.; WANG, D.; FORSLING, W. Surface acid-base properties and hydration/dehydration mechanisms of aluminum (hydr)oxides. Journal of Colloid and Interface Science , v. 308, n. 2, p. 395–404, Apr. 2007. Disponível em: <http://www.sciencedirect.com>. Acesso em: 04 fev. 2009.
(67) PIRES, E. L. Oxidação seletiva de cicloexano catalisada por zeól ita Y contendo terras raras . Tese (Doutorado em Ciências) - Universidade Estadual de Campinas (UNICAMP), São Paulo, 1999.
(68) LUNA, F. J.; SCHUCHARDT, U. Modificação de zeólitas para uso em catálise. Química Nova , v. 24, n. 6, p. 885-892, 2001. Disponível em: <http://www.scielo.br>. Acesso em: 10 mar. 2007.

146
(69) AFONSO, J. C.; PONTES, A. B.; SANTOS, E. S.; MENEZES, M. S.; AGUIAR, R. M. Reciclagem química de zeólitas comerciais desativadas. Química Nova , v. 27, n. 2, p. 315-319, mar. 2004. Disponível em: <http://www.scielo.br>. Acesso em: 10 mar. 2007.
(70) BROUSSARD, L.; SHOEMAKER, D. P. The structures of synthetic molecular sieves. Journal of American Chemical Society , v. 82, n. 5, p. 1041-1051, 1960.
(71) JEWUR, S. S. Química de zeólitas e catálise. Química Nova , v. 8, n. 2, p. 100-104, abr. 1985.
(72) SILVA, J. B.; RODRIGUES, J. A. J.; NONO, M. C. A. Caracterização de materiais catalíticos . São José dos Campos: INPE, 2006. 66p. (INPE-15252-PUD/198).
(73) BROWN, M. E. Introduction to thermal analysis: techniques and applications. New York: Springer, 1988. 280p.
(74) MANNHEIMER, W. A. Microscopia dos materiais : uma introdução. São Paulo: Sociedade Brasileira de Microscopia e Microanálise, 2002. 226p.
(75) MALISKA, A. M. Microscopia eletrônica de varredura . Apostila (Usuários de MEV): Florianópolis: Universidade Federal de Santa Catarina, 2006. 97p. Apostila (Usuários de MEV).
(76) SHACKELFORD, J. F. Introduction to materials science for engineers . New York: MacMillan, 1992. 600p.
(77) CULLITY, B. D. Elementes of x-ray diffraction . New York: Addison-Wesley, 1959. 514p.
(78) LOWELL, S.; SHIELDS, J. Powder surface area and porosity . New York: John Wiley, 1979. 462p.
(79) BARRICHELLO, N. J.; FARO Jr., A. da C. Caracterização de catalisadores . Rio de Janeiro: Instituto Brasileiro de Petróleo, 1995. 111p.
(80) CIOLA, R. Fundamentos as cromatografia a gás . São Paulo: Edgard Blücher, 1985.
(81) SHARMA, P.; PATEL, A. Tungstophosphoric acid supported onto hydrous zirconia: physicochemical characterization and esterification of 1° and

147
2° alcohol. Bulletin of Materials Science , v. 29, n. 5, p. 439–447, Oct. 2006. Disponível em: <http://www.sciencedirect.com>. Acesso em: 07 mar. 2007.
(82) SETTLE, F. Handbook of instrumental techniques for analytical chemistry. New York: Prentice Hall, 1997.
(83) BRUNNER, E. Characterization of solid acids by spectroscopy. Catalysis Today . v. 38, n. 3, p. 361–376, Nov. 1997. Disponível em: <http://www.sciencedirect.com>. Acesso em: 07 mar. 2007.
(84) INTERNATIONAL CENTRE FOR DIFFRACTION DATA. Joint Committee on Powder Diffraction Standards. Selected powder diffraction data for metals and alloys . Newton Square, PA: JCPDS, 1978. 2v. (PUBL.DBMA-1-27).
(85) ROUQUEROL, J.; ROUQUEROL, F.; SING, K. Adsorption by powders and porous solids . London: Academic Press, 1998.
(86) CORMA, A. Inorganic solid acids and their use in acid-catalyzed hydrocarbon reactions. Chemical Reviews , v. 95, n. 3, p. 559-614, May 1995. Disponível em: <http://www.sciencedirect.com>. Acesso em: 2 dez. 2009.
(87) EMEIS, C. A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. Journal of Catalysis , v. 141, n. 2, p. 347-354, Jun. 1993. Disponível em: <http://www.sciencedirect.com>. Acesso em: 2 dez. 2009.
(88) MOUREI, G. T.; MORGADO JR., E. E.; FIGUEIREDO, C. M. C. Controle de porosidade em aluminas para fins catalíticos: uma revisão. Boletim Técnico da Petrobrás. Rio de Janeiro, v. 42, n. 1/4, p. 36-44, jan./dez. 1999.

148

149
APÊNDICE A
Difratograma de raios X da CATAPAL A calcinada a 600 °C/6 horas obtido no
LAS/INPE.
Difratograma de raios X, obtido na Universidade de Paris VI, da CATAPAL A
calcinada a 300 °C/6 horas (linha vermelha) e a 600 °C/6 horas (linha preta).

150

151
APÊNDICE B
Difratograma de raios X da ZEÓLITA Y calcinada a 600 °C/6 horas obtido no
LAS/INPE.
Difratograma de raios X da ZEÓLITA Y calcinada a 300 °C/6 horas obtido na
Universidade de Paris VI.

152

153
AAPPEENNDDIICCEE CC
Difratogramas de raios X do HY calcinado a 300 e 400 °C/6 horas, obtidos na Universidade de Paris. VI.
Difratograma de raios X do HY calcinado a 600 °C/6 horas obtido no LAS/INPE

154

155
APÊNDICE D Difratograma de raios X do AHY calcinado a 300 °C/6 horas obtido no LAS/INPE.
Difratograma de raios X do AHY calcinado a 400 °C/6 horas obtido no
LAS/INPE.

156

157
APÊÊNNDDIICCEE EE
Difratograma de raios X do HY calcinado a 500 °C/6 horas obtido no LAS/INPE.
Difratograma de raios X do AHY calcinado a 500 °C/6 horas obtido no
LAS/INPE.

158

159
AAPPÊÊNNDDIICCEE FF
Difratogramas de raios X do AHY calcinado a 300 e 400 °C por 6 horas, obtido na Universidade de Paris VI. . Difratograma de raios X do AHY calcinado a 600 °C/6 horas obtido na Universidade de Paris VI.

160

161
AAPPÊÊNNDDIICCEE GG
Imagens obtidas por MEV com aumento de 5000x AHY calcinado a 300 ºC AHY calcinado a 400 ºC AHY calcinado a 500 ºC
HY calcinado a 300 ºC HY calcinado a 400 ºC HY calcinado a 500 ºC
CAT calcinado a 300 ºC CAT calcinado a 400 ºC CAT calcinado a 500 ºC
ZEO calcinado a 300 ºC ZEO calcinado a 400 ºC ZEO calcinado a 500 ºC

162

163
AAPPÊÊNNDDIICCEE HH
Isotermas de adsorção-dessorção da CATAPAL A em função da temperatura
de calcinação.
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
P/P0
CAT600
CAT 500
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00
40
80
120
160
200
240
280
Volu
me
Adso
rvid
o (c
m2 .g
-1)
P/P0
CAT 400
0
40
80
120
160
200
240
280
Volu
me
Adso
rvid
o (c
m2 .g
-1)
CAT 300

164

165
AAPPÊÊNNDDIICCEE II
Gráfico da reação catalítica de formação do etanol, a partir da hidratação do
etileno, utilizando como catalisador o AHY calcinado a 600 °C.
0 50 100 150 200 250 300 350 400
0
150
300
450
600
750
900
Tem
para
tura
(
0 C)
Eta
nol (
u.a.
)
Tempo (min)
Etanol
0
50
100
150
200
250
Material - AHY 600
temperatura
Gráfico da reação catalítica de formação do etanol, a partir da hidratação do
etileno, utilizando, como catalisador, CATAPAL A calcinada a 600 °C.
0 50 100 150 200 250 300 350
0
50
100
150
200
250
300
Tem
pera
tura
(
0 C)
Eta
nol (
u.a.
)
Tempo (min)
Etanol
0
50
100
150
200
250
Material - CAT 600
Temperatura

166

167
AAPPÊÊNNDDIICCEE JJ
Gráfico da reação catalítica de formação do etanol, a partir da hidratação do
etileno, utilizando como catalisador a ZEÓLITA Y calcinada a 300 °C.
.
0 50 100 150 200 2500
10000
20000
30000
40000
50000
60000
70000Fluxo de He+etileno: 20 ml/min
Eta
nol (
u. a
.) e
Éte
r (u
. a.)
Etil
eno
(u. a
.)
Tempo (min)
Etileno
0
1000
2000
3000
4000
5000
6000
Etanol)Éter Etílico

168

169
AAPPÊÊNNDDIICCEE LL
Dados comparativos das análises de volumetria de nitrogênio e quimissorção
de amônia dos catalisadores calcinados em diferentes temperaturas.
HY Vp (cm 3/g) Dp (Å) Área (m 2/g) Acidez ( µµµµmol/g) Acidez/Área (µ (µ (µ (µmol/m 2)
300 0,08 35 102 655 6,42 400 0,07 38 73 478 6,55 500 0,07 66 44 307 6,98 600 0,02 46 17 276 16,24
AHY Vp (cm 3/g) Dp (Å) Área (m 2/g) Acidez ( µµµµmol/g) Acidez/Área (µ (µ (µ (µmol/m 2)
300 0,18 40 181 1443 7,97 400 0,17 45 152 742 4,88 500 0,16 63 100 526 5,26 600 0,04 52 29 477 16,45
CAT Vp (cm 3/g) Dp (Å) Área (m 2/g) Acidez ( µµµµmol/g) Acidez/Área (µ (µ (µ (µmol/m 2)
300 0,31 52 238 185 0,78 400 0,37 51 286 482 1,69 500 0,40 64 252 305 1,21 600 0,40 77 208 475 2,28
ZEO Vp (cm 3/g) Dp (Å) Área (m 2/g) Acidez ( µµµµmol/g) Acidez/Área (µ (µ (µ (µmol/m 2)
300 0,31 22 573 2486 4,34 400 0,30 23 520 2339 4,50 500 0,30 23 524 1715 3,27 600 0,31 23 564 1568 2,78

170

171
AAPPÊÊNNDDIICCEE MM
Imagens obtidas por MEV dos catalisadores Al5%Nb, Al10%Nb, Al20%Nb,
Al30%Nb e Al40%Nb, todos calcinados a 400 °C durante 6 horas.
Material – Misturas (aumento de 5000x) Al5 %Nb Al10 %Nb
Al20 %Nb Al30 %Nb
Al40 %Nb

172
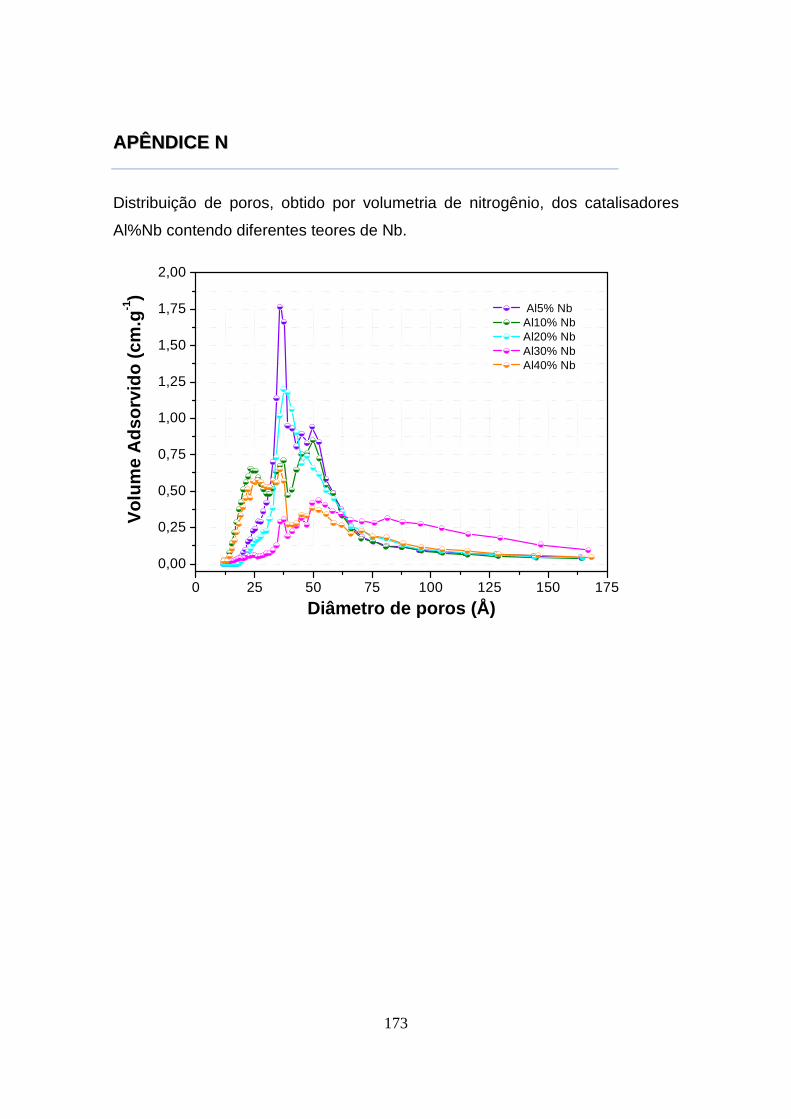
173
AAPPÊÊNNDDIICCEE NN
Distribuição de poros, obtido por volumetria de nitrogênio, dos catalisadores
Al%Nb contendo diferentes teores de Nb.
0 25 50 75 100 125 150 175
0,00
0,25
0,50
0,75
1,00
1,25
1,50
1,75
2,00
Vol
ume
Ads
orvi
do (
cm.g
-1)
Diâmetro de poros (Å)
Al5% Nb Al10% Nb Al20% Nb Al30% Nb Al40% Nb

174