síndrome adrenogenital. distúrbio presente no nascimento, caracterizado pela deficiência de...
TRANSCRIPT

Síndrome Adrenogenital

Síndrome Adrenogenital
Distúrbio presente no nascimento, caracterizado pela
deficiência de cortisol e uma superprodução de andrógeno
O que é:

INTERSEXO
• Sexo Genético ( determinado pela transmissão dos códigos genéticos paterno e materno )
• Sexo Gonadal ( Gônada que se formou determinada pelo sexo genético )
• Sexo Fenotípico (responsável pelo sexo Legal, Psico-social e de criação)

INTERSEXO • Intersexo: resulta de uma diferenciação imperfeita ou incompleta
dos órgãos genitais, ocasionando uma genitália ambígua.
• ADS é a situação em que não há acordo entre os vários sexos do indivíduo, ou seja, o sexo genético, retratado pela sua constituição cariotípica 46,XX ou 46,XY, o sexo gonadal/hormonal, e o sexo fenotípico. Desta forma, poderemos ter casos com e sem ambigüidade genital.
• Uma anomalia genital ocorre em 1 de cada 4.500 nascimentos.

Ambigüidade Genital• Pseudo-hermafroditismo: gônadas e genitália
externa discordantes entrte si.
- Masculino (genitália ambígua com testículo)
- Feminino (genitália ambígua com ovário)
• Hermafroditismo Verdadeiro: presença de tecido ovariano e testicular em um mesmo indivíduo.

Genitália Ambígua

Pseudo-Hermafroditismo Feminino

FISIOLOGIA DO CÓRTEX ADRENALDEFICIÊNCIAS ENZIMÁTICAS
ENVOLVIDAS


FEEDBACK NEGATIVO
• Cortisol circulante• Hormônio adrenocorticohipofisário(ACTH)• Hormônio Hipotalâmico liberador de
corticotrofina (CRH)
VIA HORMONAL DO CÓRTEX ADRENAL

CRHCRH
ACTHACTH
CortisolCortisol
AldosteronaAldosterona
Esteróides sexuaisEsteróides sexuais
(+)
(+)
Córtex adrenalCórtex adrenalDefeito enzimáticoDefeito enzimático
Hiperplasia Hiperplasia adrenaladrenal
FISI
OPAT
OLOG
IA
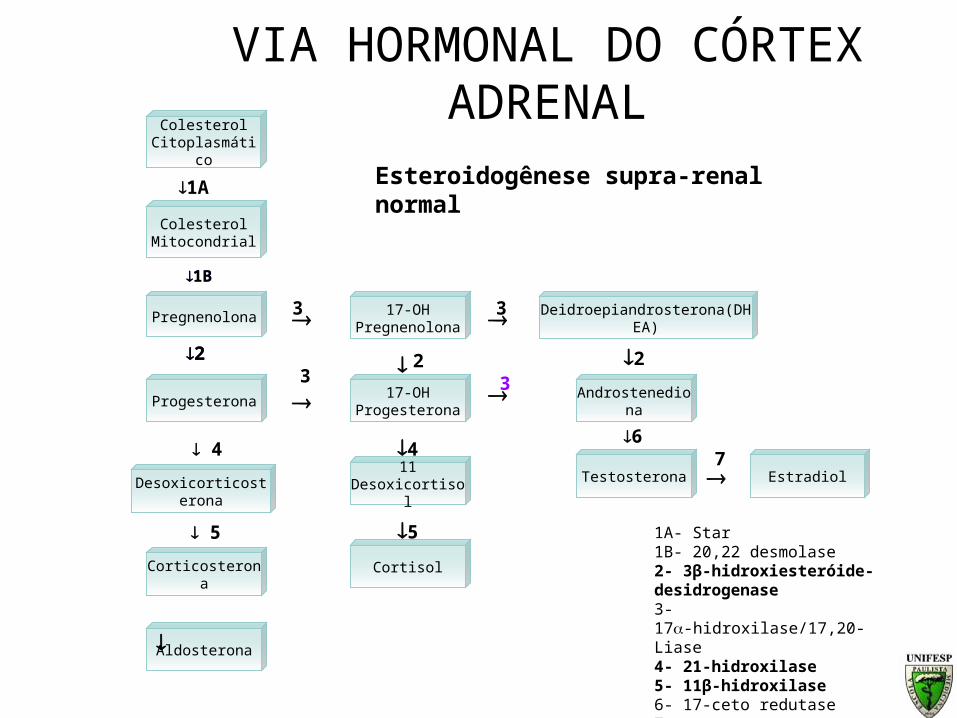
Esteroidogênese supra-renal normal
Colesterol Mitocondrial
Pregnenolona
Progesterona
Desoxicorticosterona
Corticosterona
Aldosterona
17-OHPregnenolona
17-OHProgesterona
11 Desoxicortisol
Cortisol
Deidroepiandrosterona(DHEA)
Androstenediona
5
4
1B
2
Colesterol Citoplasmático
1A
1B
2
1A- Star1B- 20,22 desmolase2- 3β-hidroxiesteróide-desidrogenase3- 17-hidroxilase/17,20-Liase4- 21-hidroxilase5- 11β-hidroxilase6- 17-ceto redutase7- aromatase
4
5
22
Testosterona
6
Estradiol7
3
3
3
3
VIA HORMONAL DO CÓRTEX ADRENAL

Definição• Excesso de androgênio
• Grupo de erros metabólicos inatos autossômicos recessivos
• Deficiência ou ausência total de uma determinada enzima envolvida na síntese dos esteroides adrenais (cortisol).
• Aumento na produção de androgênios responsáveis por efeitos virilizantes: essa deficiência de cortisol causa um aumento da secreção de ACTH, por feedback negativo, e portanto a hiperplasia supra renal e aumento na síntese de testosterona.
• Hiperplasia adrenal bilateral, com aumento de 10 a 15 vezes o seu peso normal devido ao aumento constante de ACTH. A maioria dos pacientes com HAC apresenta hiperplasia das células corticotróficas produtoras de ACTH.

Deficiência de 11β-HIDROXILASE

Epidemiologia• 1 em 100.000 a 200.000 nascimentos
mundo• < 5% casos de Hiperplasia Adrenal
Congênita• Alto grau de endogamia: judeus de origem
norte-africana, iranianos, marroquinos e negros

Genética• Uma das enzimas da familia do citocromo
P450 chamada CYP11• 2 genes – cromossomo 8• CYP11B1 (zona fasciculada)• Afeta síntese cortisol• CYP11B2 (zona glomerular)• Afeta síntese aldosterona-sintetase

GenéticaGenes CYP11B1 e CYP11B2
• Localizados braço longo cromossomo 8 (8q21)
• Ativos• Extremamente homólogos entre si• Apresentam 9 éxons, 8 íntrons e 7 kb de
extensão• Separados entre si por 40 kb• Homologia: 95% Éxons, 90% Íntrons, 93% aa

GenéticaGENE CYP11B1
- responsável pela hidroxilação c11 na zona fasciculada para
formação de cortisol- presença de duas cópias gênicas para 11 β -OH parece ter origem na duplicação do gene original
- conseqüências: especialização das funções da enzima especialização da função espacial de sua
síntese na glândula - 13/31 das mutaçõesmutações deletérias estão situadas entre os éxons 6 e
8 - OBS: constituindo um cluster (agrupamento) de mutações
sugerindo um hot-spot (região suscetível a mutações) no gene.

Gene CYP11B2
- expresso na zona glomerulosa das supra- renais - função: é capaz de sintetizar aldosterona hidroxilação de c18
oxidação de c18 hidroxilação de c11
GenéticaGenética

Fisiopatologia
Colesterol Mitocondrial
Pregnenolona
Progesterona
Desoxicorticosterona
Corticosterona
Aldosterona
17-OHPregnenolona
17-OHProgesterona
11 Desoxicortisol
Cortisol
Deidroepiandrosterona(DHEA)
5
4
1B
2
Colesterol Citoplasmático
1A
1B
2
4
5
22
Testosterona
Androstenediona
6
Estradiol7
1A- Star1B- 20,22 desmolase2- 3β-hidroxiesteróide-desidrogenase3- 17-hidroxilase/17,20-Liase4- 21-hidroxilase5- 11β-hidroxilase6- 17-ceto redutase7- Aromatase
X X
3
3
3
3

FisiopatologiaA falta da enzima:
• desoxicorticosterona e 11- desoxicortisol (composto S).
moderado androstenediona e testosterona( cortisol, ACTH)
atividade plasmática renina e aldosterona K+ sérico e alcalose metabólica(frequente)

Quadro Clínico
Fetos femininos: Virilização in úteroFetos masculinos: Sem ambiguidade genitalPeríodo pós-natal: Virilização em ambos os sexos
Hipertensão com ou sem alcalose hipocalêmica (Aprox. 2/3)Manifestação entre o 1º e 2º ano de vida

Aumento da velocidade de crescimento e da maturação esquelética
Aumento da massa muscular
Quadro Clínico

Pubarca Precoce (6 -- 10%)
Hirsutismo
Quadro Clínico

Acne
Quadro Clínico

Clitoromegalia
Quadro Clínico

Engrossamento da vozIrregularidade menstrual
InfertilidadeOvários Policísticos
Quadro Clínico

Deficiência de 3-β-hidróxiesteróide Desidrogenase

Epidemiologia
• Doença raríssima (1% casos HAC)• Forma clássica: descrita Bongiovanni
1961• Forma não-clássica: descrita Lobo &
Goebelsmann 1981

Genética
Genes HSD3B1 e HSD3B2• Mapeados por hibridização in situ no
cromossomo 13p.1• 4 éxons, 3 íntrons, 7,8 kb de extensão• Homologia na seqüência de aminoácidos
das isoenzimas de 93,5%

Gene HSD3B1- Codifica isoenzima do tipo 1- 372 aminoácidos- Se expressam na placenta e em tecidos periféricos (pele e
glândulas mamárias)
Genética

GenéticaGene HSD3B2
- Codifica isoenzima do tipo 2 - 371 aminoácidos - se expressam nas supra-renais e nas gônadas - mutaçõesmutações responsáveis pela forma clássica da doença
- com perda de salcom perda de sal - códon de terminação precoce na síntese protéica
- mutações pontuais- mutações do quadro de leitura
- sem perda de salsem perda de sal - mutações de troca de aminoácidos

Fisiopatologia
Colesterol Mitocondrial
Pregnenolona
Progesterona
Desoxicorticosterona
Corticosterona
Aldosterona
17-OHPregnenolona
17-OHProgesterona
11 Desoxicortisol
Cortisol
Deidroepiandrosterona(DHEA)
5
4
1B
2
Colesterol Citoplasmático
1A
1B
2
4
5
22
Testosterona
Androstenediona
6
Estradiol7
1A- Star1B- 20,22 desmolase2- 3β-hidroxiesteróide-desidrogenase3- 17-hidroxilase/17,20-Liase4- 21-hidroxilase5- 11β-hidroxilase6- 17-ceto redutase7- Aromatase
X X X
3
3
3
3

Fisiopatologia
A enzima :
• Requerida para a formação de todas as classes de hormônios esteroidogênicos.
• Também presente nas gônadas.• Deficiência: cortisol, aldosterona, androgênios e
estrogênios concentração sérica: DHEA, pregnenolona, 17-hidroxi-
pregnenolona

Quadro Clínico• PRÉ-NATAL • -Sexo feminino : Normal ou virilização leve a
moderada, perda de sal • -Sexo masculino: ambigüidade genital, perda
de sal • PÓS-NATAL • -Sinais de hiperandrogenismo leve a moderado• Acne, hirsutismo, pubarca, irregularidade
menstrual

Deficiência de 21-Hidroxilase

Deficiência de 21-Hidroxilase
• 1° caso relatado em 1965 – Luigi de Crecchio.
• B. Dupont e cols. – Associação Genética complexo HLA localizado no braço curto do cromossomo 6.

Deficiência de 21-hidroxilase- >90%
• Atua tanto nas vias produtoras de glicocorticóides quanto nas de mineralocorticóides
• Deficiência: androgênios, podendo ou não estar diminuída síntese de mineralocorticóides.
• Doença recessiva em que a consangüinidade dos pais não tem relevância significativa

Deficiência de 21-hidroxilase- >90%
Colesterol Mitocondrial
Pregnenolona
Progesterona
Desoxicorticosterona
Corticosterona
Aldosterona
17-OHPregnenolona
17-OHProgesterona
11 Desoxicortisol
Cortisol
Deidroepiandrosterona(DHEA)
5
4
1B
2
Colesterol Citoplasmáticp
1A
1B
2
4
5
22
Testosterona
Androstenediona
6
Estradiol7
1A- Star1B- 20,22 desmolase2- 3β-hidroxiesteróide-desidrogenase3- 17-hidroxilase/17,20-Liase4- 21-hidroxilase5- 11β-hidroxilase6- 17-ceto redutase7- Aromatase
X X
3
3
3
3

• Deficiência da enzima 21-hidroxilase (21-OH) - Localização do gene: 6p21.3 - Relacionado ao complexo de histocompatibilidade HLA - Gene: CYP21 ATIVO - Pseudogene: CYP21P INATIVO - Cópia quase idêntica (98% de homologia)
- Causa molecular da deficiência pode ser identificada em 75% a 85% dos alelos afetados
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

• CYP21 e CYP21P se intercalam com os genes das 2 formas do quarto componente do complemento (C4A e C4B)
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

• Gene CYP21 e gene CYP21P alta homologia: - Pode favorecer emparelhamento desigual das cromátides na
meiose: - deleções dos genes C4B e CYP21 com perda de 30kb - conversão em larga escala levando a inativação do gene - ambos os casos o produto final é um gene híbrido
- Mutações de pontos: substituição de adenina por guanina no
nucleotídeo 668 do intron 2 constitui a mutação mais freqüente CYP21P CYP21 ( microconversões)
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

• Avaliação do grau de comprometimento na atividade enzimática:
- clonagem do gene portador de uma mutação em particular - expressão da proteína in vitro
- ensaio da atividade enzimática de proteína resultante• Mutações “sem sentido” proteína truncada sem
atividade• Mutações com alteração do quadro de leitura falta
completa da atividade enzimática• Mutações de sentido trocado atividade enzimática em
graus variados
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

MUTAÇÕES ENVOLVIDAS• Formas clínicas da doença alteração
na atividade enzimática - Formas clássicas de deficiência: - Não perdedora de sal
- Perdedora de sal - Formas não clássicas de deficiência

● Forma perdedora de sal: - combinação de duas mutações que causam perda total
da atividade enzimática - mutações: - microconversões levando a vários genótipos,
sendo o principal o Sp2. - deleção e conversão gênica
- Populações européias e americanas: alta freqüência de deleção e conversões gênicas
- População brasileira: deleções são raras
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

● Forma não-perdedora de sal: - combinação de duas mutações que mantêm a atividade
enzimática de 1 a 4% - combinação de uma mutação que mantém a atividade
enzimática de 0% e uma de 1 a 4%. - mutações: - 60%: I172N (troca de isoleucina por asparagina), no
éxon 4 do gene CYP21 - deleção e conversão gênica em larga escala, sendo
que nesse caso o gene híbrido ainda apresenta CYP21
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

• Formas não clássicas de deficiência - perda moderada da atividade enzimática, entre 20 e 60% - mutações: - microconversões: P30L (substituição de prolina por
leucina ) e a V281L (substituição de prolina por leucina) - mutação P453S: substituição de prolina por serina no
éxon 10
MUTAÇÕES ENVOLVIDASMUTAÇÕES ENVOLVIDAS

QUADRO CLÍNICO (NPS)• Clássica: (PS)
tardia• Não Clássica: críptica

Clássica
• 90% SAG.
• 1: 14.500.
• sexo, mineralocortcóide.
• pré-natal.

Clássica (PS)
• 75% deficiência clássica 21-OH
• cortisol e aldosterona
• atividade enzimática

SINAIS
• hipercalemia, renina plasmática, ganho ponderal;
• vômitos, desidratação, colapso vascular, hipotensão;
CHOQUE

DIFERENÇAS
• masculino: sem hiperandrogenismo
• feminino: ambigüidade genital

CLÁSSICA (NPS)
• cortisol andrógenos
ambigüidade genital(mulher)

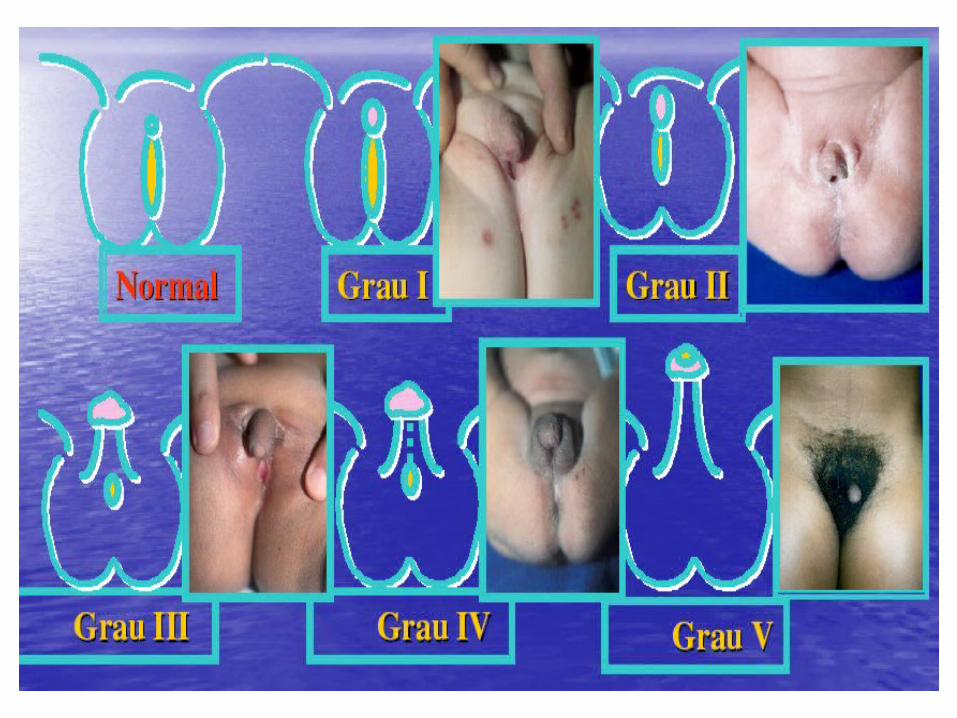

PSEUDO-HERMAFRODITISMO FEMININOPSEUDO-HERMAFRODITISMO FEMININO
Hipertrofia do clitóris, fusão labio-escrotal parcial, intróito vaginal e uretra tópicas, enrugamento da porção lábio escrotal (PraderII)

PSEUDO-HERMAFRODITISMO FEMININOPSEUDO-HERMAFRODITISMO FEMININO
Grau mais severo de virilização: formação peniana com uretra penoescrotal cordee e aspecto escrotal dos grandes lábios. .(Prader III)

PSEUDO-HERMAFRODITISMO FEMININOPSEUDO-HERMAFRODITISMO FEMININO
Clitóris muito virilizado, abertura do seio urogenital na extremidade do falo (uretra peniana) e fusão labio-escrotal completa

CLÁSSICA (NPS)
• Pós - natal:1. Virilização e clitoromegalia
2. Hirsutismo e pubarca
3. Acne e avanço de idade óssea;
4. Redução da altura final e infertilidade


CLÁSSICA (NPS)
• Pós – natal:1. Pubarca precoce e aumento de pênis;
2. Avanço da idade óssea e acne;
3. Hipertrofia muscular e baixa estatura final;
4. Engrossamento da voz;

NÃO CLÁSSICA
• tardia : hiperandrogenismo, puberdade precoce;
• críptica: assintomática;

DIAGNÓSTICODIAGNÓSTICO


AnamneseAnamnese
No caso da Hiperplasia Adrenal Congênita, verificar se No caso da Hiperplasia Adrenal Congênita, verificar se há casos semelhantes na família ou se houve mortes há casos semelhantes na família ou se houve mortes inexplicadas por desidratação, uma vez que trata-se de inexplicadas por desidratação, uma vez que trata-se de um tipo de ambigüidade genital que apresenta um tipo de ambigüidade genital que apresenta transmissão genética autossômica recessiva.transmissão genética autossômica recessiva.

Exame FísicoExame Físico
• Presença de mal-formaçõesPresença de mal-formações
• Estado de hidrataçãoEstado de hidratação
• Pilificação CorpóreaPilificação Corpórea
• Pressão ArterialPressão Arterial
• Exame dos genitaisExame dos genitais

Exame dos genitaisExame dos genitais GônadasGônadas – localização, tamanho e – localização, tamanho e
consistência.consistência.3 situações: 3 situações:
1. Não há gônadas palpáveis2. Ambas as gônadas são palpáveis
3. Apenas uma gônada palpável
Falo – caracterização do tamanho em relação às medidas consideradas normais.
Posicionamento do Meato uretral


Diagnóstico por Diagnóstico por ImagemImagem

“ “Para avaliação da Para avaliação da genitália interna (útero), genitália interna (útero), da associação com da associação com malformações do trato malformações do trato urinário, da presença de urinário, da presença de seio urogenital e, para seio urogenital e, para oferecer elementos ao oferecer elementos ao cirurgião para o cirurgião para o planejamento cirúrgico”planejamento cirúrgico”
Ultrassonografia + Genitografia

Diagnóstico Diagnóstico LaboratorialLaboratorial

Deficiência da 21-OHDeficiência da 21-OH [ ] sérica 17-OH-Progesterona [ ] sérica 17-OH-Progesterona Níveis Extremamente Elevados Diagnóstico Níveis Extremamente Elevados Diagnóstico das formas clássicasdas formas clássicas
Forma Perdedora de Sal
[aldosterona]
[11-desoxicorticosterona]
[renina plasmática]

Teste de Estímulo Rápido de Teste de Estímulo Rápido de ACTHACTH
• [ ] da 17-OH-P dosada após 60 min da adm de [ ] da 17-OH-P dosada após 60 min da adm de ACTHACTH
• Útil nos casos de genitália ambígua e forte Útil nos casos de genitália ambígua e forte suspeita de D21-OH, mas sem níveis suspeita de D21-OH, mas sem níveis diagnósticos de 17-OH-Pdiagnósticos de 17-OH-P
• Diag Dif da forma não clássica com o Diag Dif da forma não clássica com o hiperandrogenismo de causa não adrenalhiperandrogenismo de causa não adrenal
• Desnecessário nas formas clássicasDesnecessário nas formas clássicas• Realizado após 24h de vidaRealizado após 24h de vida

D11-D11-ββ-OH forma clássica-OH forma clássica
Diagnóstico Diagnóstico
Constatação de 3 hormônios principais:Constatação de 3 hormônios principais:
11-desoxicortisol sérico, DOC e da11-desoxicortisol sérico, DOC e dasubstância S-tetra-hidro-urinária(THS) substância S-tetra-hidro-urinária(THS)

D11-D11-ββ-OH forma não clássica-OH forma não clássica
• Etiologia incomumEtiologia incomum• Diagnóstico elevação do 11-Diagnóstico elevação do 11-
desoxicortisol em pelo menos 3 vezes os desoxicortisol em pelo menos 3 vezes os valores máximos obtidos pelo percentil 95 valores máximos obtidos pelo percentil 95 da população após estímulo com ACTH da população após estímulo com ACTH exógeno. exógeno.

Diag Dif de D21-OH com D11-Diag Dif de D21-OH com D11-ββ-OH-OH
• 17-OH-P 17-OH-P 11-11-ββ-OH-OH 21-desoxicortisol(21- 21-desoxicortisol(21-DF)DF)
Via ativada exclusivamente na D21-OHVia ativada exclusivamente na D21-OH [21-DF] importante no Diag Dif da D21-[21-DF] importante no Diag Dif da D21-
OH com formas não hipertensivas da D11-OH com formas não hipertensivas da D11-ββ-OH -OH

D3-D3-ββ-HSDII-HSDII
• Elevação dos esteróides Elevação dos esteróides ΔΔ5 (pregnenolona, 5 (pregnenolona, 17-OH-pregnenolona e DHEA) e diminuição 17-OH-pregnenolona e DHEA) e diminuição dos compostos dos compostos ΔΔ4 (progesterona, 4 (progesterona, 17OHP,androestenediona)17OHP,androestenediona)
• Critério baseado na relação de esteróide Critério baseado na relação de esteróide ΔΔ5 5 com o correspondente com o correspondente ΔΔ4 é controverso4 é controverso
• Melhor exame Melhor exame Busca de alteração Busca de alteração específica no gene da 3-específica no gene da 3-ββ-HSD tipo II pela -HSD tipo II pela análise do DNAanálise do DNA

ACONSELHAMENTO ACONSELHAMENTO GENÉTICO E GENÉTICO E
DIAGNÓSTICO PRÉ-DIAGNÓSTICO PRÉ-NATALNATAL

Aconselhamento e Aconselhamento e diagnóstico Pré-nataldiagnóstico Pré-natal
Objetivo:Objetivo:-evitar virilização dos genitais -evitar virilização dos genitais externos de crianças do sexo externos de crianças do sexo
femininofeminino-evitar desequilíbrios -evitar desequilíbrios
hidroeletrolíticos potencialmente hidroeletrolíticos potencialmente fataisfatais

Diagnóstico pré-natal da Diagnóstico pré-natal da deficiência da 21-hidroxilase:deficiência da 21-hidroxilase:
• Medida da concentração de 17-Medida da concentração de 17-hidroxiprogesterona no líquido amniótico – hidroxiprogesterona no líquido amniótico – “janela terapêutica” (abandonado) “janela terapêutica” (abandonado)
• Utilização da tipagem de HLA de células Utilização da tipagem de HLA de células obtidas a partir das punções amniótica e de obtidas a partir das punções amniótica e de vilosidade coriônica.vilosidade coriônica.

Punção de vilosidade Punção de vilosidade coriônicacoriônica
• precoce (11 a 13 semanas)precoce (11 a 13 semanas)• obtenção imediata de grande obtenção imediata de grande
número de células número de células • 1% de risco adicional de 1% de risco adicional de
abortamentoabortamento• Casos raros de anomalias de Casos raros de anomalias de
membros de fetosmembros de fetos
Punção de líquido amniótico Punção de líquido amniótico • tardia (15 a 16 semanas) tardia (15 a 16 semanas) • necessário o cultivo celular necessário o cultivo celular • 0,1a 0,2% de risco adicional 0,1a 0,2% de risco adicional
de abortamentode abortamento
Diagnóstico pré-natal da Diagnóstico pré-natal da deficiência da 21-hidroxilase:deficiência da 21-hidroxilase:

Necessidade de equipe Necessidade de equipe multidisciplinar:multidisciplinar:
• Endocrinologistas pediátricosEndocrinologistas pediátricos• GeneticistasGeneticistas• CitogeneticistasCitogeneticistas• Especialistas em biologia Especialistas em biologia
molecularmolecular• ObstetrasObstetras

Aconselhamento genético:Aconselhamento genético:
““Um processo que permite a indivíduos ou Um processo que permite a indivíduos ou famílias a tomada de decisões famílias a tomada de decisões conscientes e equilibradas a respeito da conscientes e equilibradas a respeito da procriação.”procriação.”
Ramalho (1986)Ramalho (1986)

Aconselhamento genético:Aconselhamento genético:
• Risco de 25% de haver uma nova criança Risco de 25% de haver uma nova criança afetada em cada gestação. (Autossômica afetada em cada gestação. (Autossômica Recessiva)Recessiva)
• Deixar bem claro e explicar isso aos pais, Deixar bem claro e explicar isso aos pais, considerando todas as esferas que são afetadas considerando todas as esferas que são afetadas pela doença (biopsicossocial).pela doença (biopsicossocial).

Teste do PezinhoTeste do Pezinho
plusplus (Fenilcetonúria e outras (Fenilcetonúria e outras Aminoacidopatias, Aminoacidopatias, hipotireoidismo congênito, hipotireoidismo congênito, hiperplasia adrenal congênita hiperplasia adrenal congênita e e fibrose cística)fibrose cística)
ampliado (Fenilcetonúria e outras Aminoacidopatias, hipotireoidismo congênito, hiperplasia adrenal congênita, fibrose cística, galactosemia, deficiência de biotinidase, toxoplasmose congênita)

Tratamento

Objetivos do Tratamento
• Reposição Hormonal,• Evitar a virilização,• Prevenir a desidratação
por perda de sal,• Normalização do
hiperandrogenismo sem afetar a velocidade de crescimento e
• Preservar a função gonadal, fertilidade e estatura final.

Períodos de Tratamento

Tratamento Pré-Natal

Tratamento Pré-Natal• Dexametasona 20µg/Kg/dia
dividido em 3 doses.• 9 semanas de Gestação: Biópsia
de vilo coriônico. Resultado + → Continua tratamento até parto.
• 16 a 18 semanas: Aminiocentese. Controle do tratamento: Dosagem sanguínea (mãe) de Cortisol e Estriol e dosagem de 17-OHP no líquido amniótico.

Tratamento Pós-Natal
• Tipos: Agudo e Crônico.• Agudo: • (Forma Perdedora de Sal), 20mL/Kg
solução salina (0,95%) em 1h.• Administrar Succinato de Sódio e
Hidrocortisona em bolus (50mg/m²) e 100mg/m² adicionada ao soro em 24h.
• Crise Controlada.

Tratamento Pós-Natal• Crônico: Suprimir níveis de ACTH e suprimir
função da Adrenal.• Até 2 anos: IM, 10 a 15mg/m² de Hidrocortisona
a cada 3 dias.• Após 2 anos: VO, 20 a 25mg/m² de
Hidrocortisona , 2 ou 3 X ao dia (dose maior à noite).
• Regressão dos sinais de hiperandrogenismo, normaliza PA e crescimento.

Tratamento Pós-Natal
• Deficiência de 21-Hidroxilase: • Forma perdedora de sal e• Forma não perdedora de sal.
• OBS: Dosagem sérica de 17-OHP e androstenediona é fonte de controle no tratamento dessa deficiência.

Tratamento Pós-Natal• Forma perdedora de sal:• Balanço de Na+ negativo. Causado por acumulo de
substratos (P e 17-OHP)→ ↑excreção de Na+ e ↑PRA. • Administração associada de Glicocorticóide
(prednisolona, dose de 2 a 4 mg/m², 2X ao dia) e Mineralocorticóide, (21-acetiloxi)-9α-fluorohidroxicortisona), (Florinef®), na dose única e oral de 0,05 a 0,15 mg/dia .

Tratamento Pós-Natal
• Forma não perdedora de sal:• VO, 20 a 25mg/m² de Hidrocortisona , 2
ou 3 X ao dia (dose maior à noite).• Prednisona e prednisolona podem ser
usadas na dose de 2 a 4 mg/m² dividido em 2X ao dia.
• A dose de dexametasona é de 0,25 mg/m², 1 vez ao dia.

Tratamento Pós-Natal
• Deficiência de 11β-Hidroxilase:• Reposição de Glicocorticóide: 10 a 20
mg/m² de Hidrocortisona oral dividido em 2 doses.
• Correção cirúrgica da genitália ambígua.• Uso de Espironolactona e Amiloride para
corrigir Hipocalemia e HAS leve.

Tratamento Pós-Natal
• Deficiência 3β-Hidroxiesteróide Desidrogenase:• Administração de 10 a 20 mg/m² de hidrocortisona
oral dividido em 2 doses (infância).• Administração de 2 a 4 mg/m² de Prednisona e
prednisolona dividido em 2X ao dia.• A dose de Dexametasona é de 0,25 mg/m², 1 vez
ao dia (adolescentes e adultos). • Correção da genitália ambígua.• No caso de perda de sal, trata-se com
administração de mineralocorticóide.

Tratamento Pós-Natal• Puberdade Precoce:• Ocorre na deficiência de
11β-Hidroxilase e 21-Hidroxilase.
• Trata-se com drogas progestacionais que suprimem a secreção de gonadotrofinas (Acetato de Ciproterona 75 a 100 mg/m² V.O. divididos em 2 doses ao dia).

Acompanhamento Pós-Natal

Controle do Tratamento
• Medidas de PRA são úteis no monitoramento da eficácia do tratamento de SAG por deficiência de 21-Hidroxilase e 3β-HSD.

Correção Cirúrgica da Genitália Externa
• Clitoroplastia,• Ampliação do intróito vaginal,• Abertura do seio urogenital e • Manutenção da glande clitoriana tópica.

Clitoroplastia

Novos Tratamentos
• Terapia Gênica: injeção intra adrenal de adenovírus não replicante contendo a seqüência genômica do CYP21 humano corrigida.
• Adrenalectomia: caso ocorra falha terapêutica.
• Uso de Análogo de GnRH e Hormônio de Crescimento.