registro sanitÁrio de medicamentos novos as … · ceme – central de medicamentos cpmp -...
TRANSCRIPT

FUNDAÇÃO OSWALDO CRUZ ESCOLA NACIONAL DE SAÚDE PÚBLICA SÉRGIO AROUCA
MESTRADO EM SAÚDE PÚBLICA POLÍTICAS PÚBLICAS E SAÚDE
REGISTRO SANITÁRIO DE MEDICAMENTOS NOVOS AS NORMAS LEGAIS E UMA ANÁLISE DO MERCADO
BRASILEIRO
CÍNTIA MARIA GAVA
ORIENTADORES: JORGE ANTONIO ZEPEDA BERMUDEZ VERA LUCIA EDAIS PEPE
RIO DE JANEIRO, 2005

ii
CÍNTIA MARIA GAVA
REGISTRO SANITÁRIO DE MEDICAMENTOS NOVOS: AS NORMAS LEGAIS E UMA ANÁLISE DO MERCADO BRASILEIRO
Dissertação de Mestrado apresentada à Escola Nacional de Saúde Pública Sérgio Arouca, Fundação Oswaldo Cruz, como parte dos requisitos necessários à obtenção do título de Mestre em Saúde Pública.
ORIENTADORES: JORGE ANTONIO ZEPEDA BERMUDEZ
VERA LÚCIA EDAIS PEPE
BANCA EXAMINADORA: SUELY ROZENFELD
GERALDO LUCCHESE
Rio de Janeiro, 2005

iii
G279r Gava, Cíntia Maria
Registro sanitário de medicamentos novos: as normas legais e uma análise do mercado brasileiro. / Cíntia Maria Gava. Rio de Janeiro : s.n., 2005.
113 p., tab., graf.
Orientador: Zepeda Bermudez, Jorge Antonio Pepe, Vera Lúcia Edais
Dissertação de Mestrado apresentada à Escola Nacional de Saúde Pública Sérgio Arouca.
1.Uso de medicamentos. 2.Registros. 3.Indústria
farmacêutica. 3.Vigilância sanitária. 4.Inovação
organizacional. I.Título.

iv
“A saúde pública não é o objetivo da Indústria Farmacêutica que tende a
comporta-se como qualquer outro tipo de ramo industrial. Esta situação só pode
ser solucionada se o funcionamento e o papel da Indústria Farmacêutica forem
enquadrados em uma política farmacêutica definida pelos Estados como parte
fundamental da política sanitária dos países”.
OPAS/OMS, 1992. Documento de la Reunion de Expertos de la Subregion Andina.

v
A Antonio Gava, meu Pai
À Maria José Volpato, minha Mãe

vi
AGRADECIMENTOS
Agradeço a Deus por ter guiado meus passos para atuar como profissional de saúde pública.
Aos meus pais, pela vida e exemplo.
Aos professores que foram estímulo à minha caminhada.
Aos amigos que souberam ouvir e compartilhar.
Ao meu orientador, Jorge Antonio Zepeda Bermudez, pelo apoio, incentivo e exemplo como
profissional de saúde pública.
À minha orientadora, Vera Lúcia Edais Pepe, pelo incentivo, amizade e carinho dedicados.
A Sérgio de Andrade Nishioka, pela entrevista concedida.
A Agência Nacional de Vigilância Sanitária e a Escola Nacional de Saúde Pública Sérgio Arouca,
pelo apoio financeiro concedido para elaboração desta pesquisa por meio do convênio
ENSP/ANVISA – Pesquisa e Desenvolvimento Institucional em vigilância sanitária.

vii
RESUMO A entrada crescente de novos medicamentos nos mercados, acompanhada de intenso
investimento em propaganda e marketing destes produtos, realizado pelas empresas
farmacêuticas, é uma realidade em vários países do mundo na atualidade.
O controle sobre a qualidade, segurança e eficácia dos medicamentos cresceu nas últimas
décadas, impulsionado por trágicos episódios ocorridos no último século decorrentes dos efeitos
adversos atribuídos ao uso dos medicamentos.
Como os medicamentos apresentam riscos à saúde é necessária sua contínua regulação,
buscando-se a atualização dos meios usados para o controle da produção, comercialização e uso.
As autoridades regulatórias dos países controlam os medicamentos de diferentes formas e a
exigência do registro sanitário prévio à comercialização é uma das medidas fundamentais para a
proteção da saúde da população.
Considerando a importância do lançamento de novos medicamentos no mercado e do
registro sanitário, o presente trabalho apresenta inicialmente uma breve discussão sobre a
inovação no setor farmacêutico e em seguida descreve o processo de registro de medicamentos
novos em quatro autoridades sanitárias: FDA, EMEA, ANMAT e ANVISA. É feita uma
descrição sucinta sobre a evolução da regulação sanitária de medicamentos nos Estados Unidos e
no Brasil, são apresentadas algumas características estruturais importantes das autoridades
sanitárias selecionadas, as exigências legais para o registro de medicamentos novos e as
definições de medicamentos novos adotadas.
Por último, os quarenta e nove medicamentos novos registrados na ANVISA, nos anos de
2000, 2001 e 2002, são analisados quanto ao registro nas demais agências regulatórias, quanto à
classificação na FDA e ao tempo decorrido entre a solicitação e a concessão dos registros na
ANVISA e na FDA.
Foram encontradas diferenças na quantidade e qualidade nas informações oficiais
disponíveis, bem como em relação ao que é considerado novo medicamento pelas quatro
autoridades sanitárias estudadas. As exigências quanto ao tipo e volume de informações que
devem ser fornecidas pelos solicitantes de registro às autoridades sanitárias e os prazos legais
para análise da solicitação de registro também são distintas.

viii
A análise da relação de medicamentos novos registrados na ANVISA revelou que 75,5%
dos mesmos estão registrados na FDA, 73,5% na ANMAT e 55,1% na EMEA. No que se refere à
classificação da ANVISA para os medicamentos novos, o estudo apresentado por Reis (2004)
revelou que trinta e um medicamentos são compostos por novas entidades moleculares, mas
quando analisados quanto ao potencial terapêutico, apenas treze medicamentos foram
classificados como aplicações prioritárias pela FDA. Quanto aos prazos necessários para
concessão dos registros, houve diferenças relevantes, considerando os valores medianos, entre a
ANVISA e a FDA.
Palavras-chave: registro de medicamentos; vigilância sanitária; medicamentos novos;
autoridades sanitárias; inovação; Agência Nacional de Vigilância Sanitária; Agência Européia de
Medicamentos; Administração de Alimentos e Medicamentos; Administração Nacional de
Medicamentos, Alimentos e Tecnologias Médicas.

ix
ABSTRACT
There is a growing offer of new drugs in the market of many countries, by pharmaceutical
companies, accompanied of intense investment in propaganda and marketing of these products.
In the last century, the control of efficacy, safety and quality, have been stimulated by the tragic
episodes of adverse drug reactions attributed to the use of medicines.
Continuous drug regulation, from the production to commercialization and use as well as
searching for continuous update in regulatory actions are necessary since these drugs present
health risks. The regulatory authorities of the countries controls the life cycle of drugs in different
ways. The demand of previous drug registration to the commercialization constitutes one of these
regulatory measures, fundamental to the public health.
Considering the importance of the introduction of new drugs in the market and of the
sanitary register, this research presents initially a brief discussion about innovation in the
pharmaceutical sector. It also describes the process of new drug register in four sanitary
authorities: FDA, EMEA, ANMAT and ANVISA. A summary description of the evolution of
sanitary drug regulation in the United States and Brazil as well as some important structural
characteristics of the selected sanitary authorities, the legal requirements for the new drug register
and the adopted definitions of new drug by each one are presented.
Finally, the forty nine new drugs registered in ANVISA, during the years of 2000, 2001
and 2002, are analyzed regarding their registration in FDA, EMEA and ANMAT, their
classification in the FDA and the time elapsed between the submission and the approval by
ANVISA and FDA.
Differences were found in the amount and quality of available official information, as
well as in relation to the definition of new medicine, for all studied sanitary authorities.
Requirements about the type and volume of information that should be supplied by the applicant
pharmaceutical company to the sanitary authorities, as well as the legal periods of time spent for
registration analysis, legally specified, are also distinct.
The analysis of the new drug registered in ANVISA revealed that 75.5% of them are
registered in the FDA, 73,5% in ANMAT and 55,1% in EMEA. In relation to ANVISA’s
classification of these new drugs, the study presented by Reis (2004) revealed that thirty one are
new molecular entities. However, when analyzed for their therapeutic potential, only thirteen

x
drugs were classified as a priority application by FDA. Considering the median time between the
submission and the approval of the register, there were relevant differences between ANVISA
and FDA.
Key-words: drug registration; sanitary surveillance; new drug; sanitary authorities; innovation;
National Health Surveillance Agency (ANVISA); European Medicines Agency (EMEA); Food
and Drug Administration (FDA); National Administration of Medicine, Food and Medical
Technologies (ANMAT).

xi
SUMÁRIO
1. Introdução ...............................................................................................................................1
2. Inovação no setor farmacêutico e o desenvolvimento de medicamentos ...............................5
3. Regulação Sanitária de Medicamentos ..................................................................................14
4. Justificativas ...........................................................................................................................23
5. Objetivos ................................................................................................................................25
6. Metodologia ...........................................................................................................................26
7. Resultados e Discussões ........................................................................................................29
7.1 FDA ....................................................................................................................................29
7.2 EMEA ................................................................................................................................40
7.3 ANMAT .............................................................................................................................49
7.4 ANVISA ............................................................................................................................54
8. Registro de medicamentos novos nas quatro agências: algumas considerações ...................67
9. Medicamentos novos registrados na ANVISA 2000-2002 ...................................................78
10. Conclusões ...........................................................................................................................89
Referências Bibliográficas .........................................................................................................92
Anexo 1. Entrevista ...................................................................................................................97
Anexo 2. Quadro 8 - Relação de Medicamentos Novos Registrados na ANVISA nos anos de 2000, 2001 e 2002 ...................................................................................................................100 Anexo 3. Quadro 9 - Medicamentos novos registrados na ANVISA e na EMEA, FDA e ANMAT ..................................................................................................................................108 Anexo 4. Quadro 10 - Prazo em dias para o registro dos medicamentos na ANVISA e na FDA .................................................................................................................................................110

xii
LISTA DE SIGLAS
ANDA – ABBREVIATED NEW DRUG APPLICATION
ANMAT - ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS, ALIMENTOS Y
TECNOLOGIA MEDICA.
ANVISA – AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA
CATEME – CÂMARA TÉCNICA DE MEDICAMENTOS
CBER – CENTER FOR BIOLOGICS EVALUATION AND RESEARCH
CDER – CENTER FOR DRUG EVALUATION AND RESEARCH
CEE – COMUNIDADE ECONÔMICA EUROPÉIA
CEME – CENTRAL DE MEDICAMENTOS
CPMP - COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE
CTD – COMMON TECHNICAL DOCUMENT
CVMP - COMMITTEE FOR MEDICINAL PRODUCTS FOR VETERINARY USE
DGSP – DIRETORIA GERAL DE SAÚDE PÚBLICA
DIMED – DIVISÃO DE MEDICAMENTOS
DNSP – DEPARTAMENTO NACIONAL DE SAÚDE PÚBLICA
EMEA – EUROPEAN MEDICINES AGENCY d
EPAR – RELATÓRIO EUROPEU DE AVALIAÇÃO PÚBLICO
EU – EUROPEAN UNION
FDA - FOOD AND DRUG ADMINISTRATION
FIFARMA - FEDERAÇÃO LATINO-AMERICANA DA INDÚSTRIA FARMACÊUTICA
GATB - ALIANÇA GLOBAL PARA DESENVOLVIMENTO DE DROGAS PARA
A TUBERCULOSE
GEPEC – GERÊNCIA DE MEDICAMENTOS NOVOS, PESQUISA E ENSAIOS CLÍNICOS
GGMED – GERÊNCIA GERAL DE MEDICAMENTOS
ICDRA - INTERNATIONAL CONFERENCE OF DRUG REGULATORY
AUTHORITIES
ICH – INTERNATIONAL CONFERENCE ON HARMONIZATION
IND – INVESTIGATIONAL NEW DRUG
ISDB – INTERNATIONAL SOCIETY OF DRUG BULLETINS

xiii
MERCOSUL – MERCADO COMUM DO SUL
MSF – MÉDICOS SEM FRONTEIRAS
MSH – MANEGEMENT SCIENCIES FOR HEALTH
NDA – NEW DRUG APPLICATION
NME – NEW MOLECULAR ENTITY
OMS - ORGANIZAÇÃO MUNDIAL DA SAÚDE
OPAS – ORGANIZAÇÃO PANAMERICANA DA SAÚDE
OTC – OVER - THE - COUNTER
PNM – POLÍTICA NACIONAL DE MEDICAMENTOS
RDC – RESOLUÇÃO DA DIRETORIA COLEGIADA
SNFM – SERVIÇO NACIONAL DE FISCALIZAÇÃO DA MEDICINA
SNFMF - SERVIÇO NACIONAL DE FISCALIZAÇÃO DA MEDICINA E FARMÁCIA
SNVS – SECRETARIA NACIONAL DE VIGILÂNCIA SANITÁRIA
SPC – SUMMARY OF PRODUCT CHARACTERISTICS
VISA – VIGILÂNCIA SANITÁRIA
WHO – WORLD HEALTH ORGANIZATION

xiv
LISTA DE TABELAS
Tabela 1 - Diferenças entre o ensaio clínico controlado na Fase III e o uso de um fármaco na prática
clínica habitual ...................................................................................................................................9
Tabela 2 – Número de medicamentos registrados e classificados por potencial terapêutico segundo
país sede da empresa fabricante........................................................................................................81

xv
LISTA DE QUADROS
Quando 1 - Ranking das Aplicações de Novos Medicamentos (NDA) segundo critérios de classificação da FDA .......................................................................................................................................................37 Quadro 2 - Documento Técnico Geral (CTD) para Registro de Medicamentos na EMEA ......................44 Quadro 3 - Relatório Técnico para Registro de Medicamento Novo segundo RDC 136 de 23/05/2003...61 Quadro 4 - Exigências da RDC 136 segundo características dos Medicamentos Novos ..........................62 Quadro 5 - Estrutura e funções das autoridades sanitárias: FDA, EMEA, ANMAT e ANVISA .............68 Quadro 6 - Definições para medicamento novo adotadas pela FDA, EMEA, ANMAT e ANVISA .......72 Quadro 7 - Algumas características do processo de registro de medicamentos novos na FDA, EMEA, ANMAT e ANVISA .................................................................................................................................75

1
1. INTRODUÇÃO
No século XIX os terapeutas adeptos da medicina “ortodoxa”1 tentavam usar as novas
ciências biológicas ou médicas segundo princípios hipocráticos reconstruídos no Iluminismo e
disputavam espaço com outras doutrinas médicas como a homeopatia. No Brasil, na França e nos
Estados Unidos a homeopatia ameaçava a anátomo-clínica como uma concorrente de peso.
Mas a criação de técnicas de prevenção e tratamento etiológico, destacando-se as
contribuições de Pasteur com bases científicas nítidas, foi realmente impressionante para a
sociedade do século XIX e contribuíram decisivamente para a mudança do paradigma de controle
de doenças: a ciência médica assumiu a hegemonia na virada do século XIX. Segundo Sayd
(1998:133):
“(...).este prestígio significou a hegemonia do médico ortodoxo sobre os
demais, adeptos de outros sistemas terapêuticos como a homeopatia. A
linha entre o charlatanismo e uma medicina oficial se tornou nítida e
modificou inteiramente a situação de competição anterior entre os
terapeutas (...)”
O final do século XIX foi marcado pelo crescimento dos centros urbanos e pelo processo
de industrialização em várias partes do mundo. Somam-se a estas mudanças a proliferação de
doenças contagiosas e a necessidade de que a classe trabalhadora fosse mantida saudável, já que
dela dependia o sistema de produção (Costa, 2004).
No Brasil, a industrialização e o desenvolvimento da ciência e da tecnologia tiveram papel
decisivo no processo de produção de normas sanitárias pelo Estado neste período. Havia uma
pressão para que as atividades comerciais fossem padronizadas e que os produtos obedecessem
algumas normas de qualidade. A sociedade, preocupada com as denúncias freqüentes de
adulteração de alimentos e medicamentos, pressionava os órgãos públicos para que tomassem
medidas de controle sanitário (Costa & Rozenfeld, 2000).
1 Coulter (1973, apud Sayd,1998) chama a medicina anátomo-clínica de ortodoxa ao narrar a disputa entre
a American Medical Association e a homeopatia norte-americana.

2
A influência do setor industrial neste processo de regulamentação pode ser explicada pelo
interesse em expandir o mercado para seus produtos já que grande parte do comércio era
dominada pela produção artesanal, como no caso das preparações farmacêuticas de origem
vegetal. Nas décadas de 30 e 40 os medicamentos industrializados2 representavam menos de 25%
dos produtos vendidos nas farmácias (Laporte et al, 1993).
A importância assumida pela medicina científica e o crescimento da produção industrial
de medicamentos são fatores que se somam, no inicio do século XX, contribuindo um para a
legitimação e valorização do outro: os medicamentos eram fabricados segundo os princípios da
racionalidade científica e somente os terapeutas adeptos deste modelo teriam capacidade de
prescrevê-los (Sayd, 1998).
O desenvolvimento da química, da fisiologia e da farmacologia básica, durante a segunda
metade do século XX, associado ao aumento da demanda por serviços de saúde, garantiram a
introdução de vários produtos farmacêuticos no mercado. O aumento da utilização de fármacos
acentuou-se após a Segunda Guerra Mundial e o período entre as décadas de 1940 e 1960 ficou
conhecido como Idade de Ouro da Indústria Farmacêutica (Barros, 1995). Os efeitos benéficos
dos medicamentos foram profundamente percebidos neste período pela introdução de antibióticos
como penicilina e estreptomicina que permitiram salvar muitas vidas em condições anteriormente
impossíveis (Laporte et al, 1993).
O consumo cada vez maior de medicamentos, induzido maciçamente por meio de
propagandas e marketing dos produtos, teve como impacto cultural a substituição de formas
tradicionais de cuidado da saúde, gerando a dependência da população pela alopatia, e
conseqüentemente, pela classe médica e farmacêutica (Sayd, 1998). Para os dois principais
agentes envolvidos na cadeia terapêutica – o médico e o paciente – o medicamento passou a ter
uma função simbólica: para o médico, um medicamento adequadamente prescrito lhe outorgava
prestígio e reforçava seu poder sobre o paciente, e para o paciente, o sinal de uma boa consulta
médica tende a ser uma prescrição.
Os avanços tecnológicos permitiram a fabricação de uma variedade de medicamentos que
hoje estão presentes no mercado - obtidos de fontes naturais, sintetizados nos laboratórios e
2 Entende-se por medicamento o produto farmacêutico tecnicamente obtido ou elaborado, com finalidade profilática, curativa, paliativa ou para fins de diagnóstico (Brasil, 1976).

3
obtidos por processos de biotecnologia - comercializados em diferentes formas farmacêuticas
como comprimidos, cápsulas, xaropes, injetáveis, sprays, adesivos transdérmicos e cremes. A
grande variedade de apresentações comerciais dos medicamentos permite que o paciente tenha à
sua disposição formas farmacêuticas de uso mais conveniente ao tratamento, contribuindo para
adesão ao mesmo.
Entretanto, passados os anos de euforia da Idade de Ouro, houve um grande período de
letargia no que se refere à descoberta de fármacos que representassem avanços terapêuticos
efetivos e um incremento de “novidades” que não passavam de uma nova roupagem dada a
produtos já existentes no mercado (Barros, 1995). Esta situação apresenta-se como um problema
para os sistemas de saúde, pois estes medicamentos, registrados pelas autoridades sanitárias são
anunciados por seus fabricantes como medicamentos novos e acabam “conquistando” muitos
profissionais de saúde. Neste sentido, Dupuy & Karsenty (apud Barros, 1995:29) afirmam que:
“(...) prescrever a última novidade lançada no mercado significa
“competência”, o que favorece a boa imagem do médico pois assim
ele estará buscando uma eficácia maior para o tratamento; e mesmo a
indicação de produtos mais antigos pode provocar no paciente a idéia
de que seu caso não está merecendo o interesse que esperava.”
Mas uma questão importante não pode ser desconsiderada: os medicamentos também
podem representar riscos à saúde, causar sérios danos ao organismo dos indivíduos e, em casos
extremos, até a morte. A vulnerabilidade do consumidor no mercado de consumo é reforçada
pelo que se define como assimetria de informação (Piovesan, 2002: 60):
“(...) impossibilidade das pessoas disporem de todas as informações
necessárias para orientar suas escolhas de consumo, os produtores e
prestadores dispõem de informações sobre produtos e serviços que a
população não possui.”
Neste cenário, o registro sanitário de medicamentos torna-se um importante instrumento
de regulação sanitária por meio do qual o Estado deve atuar como mediador entre os interesses

4
das empresas produtoras de medicamentos, que desejam registrar seus produtos, e os interesses
da saúde pública, zelando por sua defesa e proteção. A regulação exercida pelo Estado é definida
por Camargo (apud Piveosan,2002:56) como:
“Uma função típica da materialização de políticas públicas, pois por
meio delas procura-se estabelecer uma ponderação de interesses
aparentemente conflitantes em situações que comprometem o
equilíbrio das relações entre agentes econômicos e usuários ou
consumidores.”
Uma das principais ações de proteção à saúde dos futuros consumidores é, portanto, a
avaliação da solicitação de registro de medicamentos, devendo esta ser conduzida com a
responsabilidade e o rigor necessários, uma vez que alguns dos efeitos dos medicamentos
somente serão identificados após o uso prolongado e por um número significativo de pessoas,
cada qual com sua singularidade.
Tendo em vista a importância da entrada de novos medicamentos nos mercados
considerou-se oportuno fazer inicialmente uma breve abordagem sobre a inovação no setor
farmacêutico, para em seguida analisar, o processo de registro de medicamentos novos nos
Estados Unidos, na Comunidade Européia, na Argentina e no Brasil, assim como os registros de
medicamentos novos, concedidos no Brasil, entre os anos de 2000 e 2002.

5
2. INOVAÇÃO NO SETOR FARMACÊUTICO E O DESENVOLVIMENTO DE MEDICAMENTOS
A inovação tecnológica é um componente fundamental para as empresas que desejam
competir no mercado atual e é obtida por meio de investimentos em atividades de pesquisa e
desenvolvimento.
Existem dois tipos de inovação: inovação não-linear e inovação linear (Schmid, 2002).
Um exemplo perfeito de inovação não-linear foi a descoberta da penicilina. Em 1928,
trabalhando com o vírus Influenza o pesquisador Alexander Fleming observou que não havia
crescimento da cultura de Staphilococcus aureus ao redor de certas regiões da placa de cultura
contaminadas por fungo. Fleming não pretendia descobrir um agente bactericida, mas por suas
observações e deduções acabou descobrindo a penicilina e abrindo caminho para o tratamento de
uma série de infecções.
A inovação linear ou lógica, por sua vez, é aquela inovação baseada em um conhecimento
prévio, a partir do qual se somam avanços incrementais. Segundo Schmid, a inovação linear é
uma forma de inovação orientada, com objetivos bem definidos. Um exemplo deste tipo de
inovação na Indústria Farmacêutica é o avanço em terapias existentes pela descoberta de
medicamentos mais seguros e eficazes a partir de uma mesma classe terapêutica.
No setor farmacêutico a inovação é um processo bastante variado e contínuo, podendo
ocorrer de duas formas: produção de medicamentos cujos fármacos são novas entidades
moleculares ainda não comercializadas e o desenvolvimento de medicamentos que já estão no
mercado por meio de pequenas alterações nos mesmos (NIHCM Foundation, 2002). Estas
alterações podem melhorar o perfil farmacocinético do medicamento, aumentando sua absorção,
diminuindo o número de doses diárias do medicamento, dentre outros benefícios. Em alguns
casos, as pesquisas com medicamentos reconhecidamente eficazes levam à descoberta de uma
nova indicação terapêutica para o mesmo, alterando-se apenas a concentração do fármaco no
medicamento. A aspirina (ácido acetilsalicílico) é um exemplo de medicamento antigo, usado
inicialmente como analgésico, antitérmico e antiinflamatório, que teve seu uso expandido para a
prevenção de doenças cardiovasculares, como angina pectoris, trombose venosa e episódio
isquêmico transitório devido a sua capacidade de inibição da atividade plaquetária (NAF/ENSP,
2002).

6
Segundo Schmid (2002) dificilmente o primeiro medicamento para o tratamento de uma
nova doença apresentará todas as propriedades que o tornem bem aceito pelos médicos e
pacientes e, até atingir o status ideal, são necessários de 10 à 15 anos de comercialização. Ainda
segundo o autor esta “otimização” é realizada pela Indústria Farmacêutica por meio de inovações
incrementais nos medicamentos já comercializados.
Esta discussão pode ser importante quando se percebe o uso constante da palavra
inovação por todos os tipos de empresas na publicidade de seus produtos. O termo é usado para o
lançamento de produtos genuinamente novos e para produtos originados de alterações em
produtos já comercializados.
A Sociedade Internacional de Boletins sobre Medicamentos (ISDB), em sua Declaração
sobre o Avanço Terapêutico no Uso de Medicamentos (ISDB, 2001,) definiu a inovação de
medicamentos por meio de três conceitos:
a) Conceito comercial: qualquer produto "me-too"3 recém-comercializado, novas
substâncias, novas indicações, novas formulações, e novos métodos de tratamento.
b) Conceito tecnológico: qualquer inovação industrial, uso de biotecnologia, introdução de
um novo sistema de liberação da substância (adesivo, aerossol, etc) ou a seleção de um
isômero ou um metabólito.
c) Conceito de avanço terapêutico: um novo tratamento que beneficia o paciente quando
comparado a opções previamente existentes.
Arnau & Laporte (1989) apresentam uma classificação qualitativa dos medicamentos
segundo seu valor terapêutico potencial: valor “elevado”, valor “relativo”, valor “duvidoso/nulo”
e valor “inaceitável”. Medicamentos de valor elevado são definidos pelos autores como aqueles
cuja eficácia foi demonstrada em ensaios clínicos controlados e também por aqueles que não
foram avaliados em ensaios clínicos controlados, mas cujo uso está justificado por apresentarem
efeitos imediatos e óbvios. Medicamentos de valor relativo são aqueles que, apesar de possuírem
um princípio ativo de valor potencial elevado, possuem também uma ou mais entidades químicas
com eficácia terapêutica duvidosa; os medicamentos de valor relativo são aqueles cuja eficácia
3 Me-too drug (medicamento similar; "eu também"): medicamento cuja estrutura química é similar ao medicamento de referência e cujo perfil farmacológico e terapêutico não difere significativamente deste. (ISDB,2001)

7
ainda não foi demonstrada de maneira convincente em ensaios clínicos controlados, para os quais
não foram associados efeitos indesejáveis graves ou freqüentes e os medicamentos classificados
como de valor inaceitável são aqueles que, devido sua composição, apresentam uma relação
risco/benefício claramente desfavorável em todas as circunstâncias.
Sob o ponto de vista da saúde pública, definir a inovação de um medicamento a partir do
conceito de avanço terapêutico é, sem dúvida, a melhor forma para se avaliar um novo
medicamento. A “novidade” usada pelos fabricantes na propaganda de seus medicamentos exerce
forte influência sobre os profissionais de saúde e sobre os pacientes que, na maioria das vezes,
passam a indicá-los e/ou utilizá-los sem ter acesso às informações sobre as verdadeiras vantagens
(ou desvantagens) terapêuticas trazidas pelos novos medicamentos.
Mas para que um novo medicamento esteja disponível no mercado atualmente são
necessárias várias etapas que incluem a pesquisa básica, os ensaios pré-clínicos (em culturas de
células e com animais), os ensaios clínicos em seres humanos e a aprovação para
comercialização.
Os Ensaios Clínicos Controlados
A Lei de Alimentos, Medicamentos e Cosméticos dos Estados Unidos, aprovada em 1938,
norteia as atividades de regulação da FDA (FDA, 1938), tendo sido aprovadas algumas emendas
importantes a esta lei como a Emenda Kafauver-Harris, em 1962. Dentre as exigências
estabelecidas pela referida Lei está a garantia de segurança dos medicamentos antes da
comercialização. A Lei conferiu maiores poderes e responsabilidades a FDA e definiu que não
mais o governo e sim os fabricantes dos medicamentos deveriam comprovar a segurança dos
mesmos antes da comercialização naquele país.
Mas para que a agência americana pudesse proceder à avaliação clínica da segurança dos
novos medicamentos foi necessário um grande esforço inicial, pois não havia naquele período
parâmetros pré-estabelecidos para o tipo de pesquisa que se pretendia e eram marcantes a
relatividade e a ambigüidade incorporadas à definição de segurança dos medicamentos naquele
período. A segurança era um atributo relativo, sua avaliação realizada por meio de julgamento
valorativo – incorporando subjetividade – e não apenas por medidas objetivas contidas nos
padrões de toxicidade vigentes. Neste cenário de “incertezas metodológicas” os ensaios clínicos

8
foram definidos pelos técnicos da FDA como a única forma para determinar a segurança dos
novos medicamentos (Oliveira, 2001).
Os padrões de referência para a realização de ensaios clínicos controlados foram
estabelecidos pela FDA em 1970, sendo definidas exigências metodológicas como (Marks, 1997
apud Oliveira, 2001): alocação aleatória (randomização) dos indivíduos nos grupos do estudo;
necessidade de grupo controle e elaboração do protocolo de pesquisa permitindo proceder à
análise quantitativa usando métodos estatísticos adequados. Os ensaios clínicos controlados são
definidos como estudos experimentais analíticos que partem da causa (exposição ao
medicamento) para o efeito por meio da observação e acompanhamento dos grupos experimental
e controle. A intervenção que se deseja avaliar é realizada em apenas um dos grupos
(experimental) – o outro grupo (controle) é usado como padrão para a comparação dos resultados
(Oliveira, 2001).
Ainda que a realização dos ensaios clínicos tenha sido um passo importante no processo
de regulação farmacêutica é importante salientar que os mesmos são realizados com uma amostra
da população, com características biológicas e história clínica particulares. Logo, os achados
clínicos correspondem à realidade da amostra escolhida e não à totalidade de indivíduos que
estarão expostos ao medicamento. Crianças, idosos, pacientes apresentando determinadas
patologias e gestantes são excluídos dos ensaios clínicos, mas também poderão utilizar o
medicamento quando o mesmo for liberado para comercialização. Desta forma, pode-se afirmar
que os ensaios clínicos possuem capacidade limitada na medida em que são incapazes de
determinar todas as possíveis reações adversas causadas por um medicamento e porque algumas
reações só aparecem com o uso prolongado.
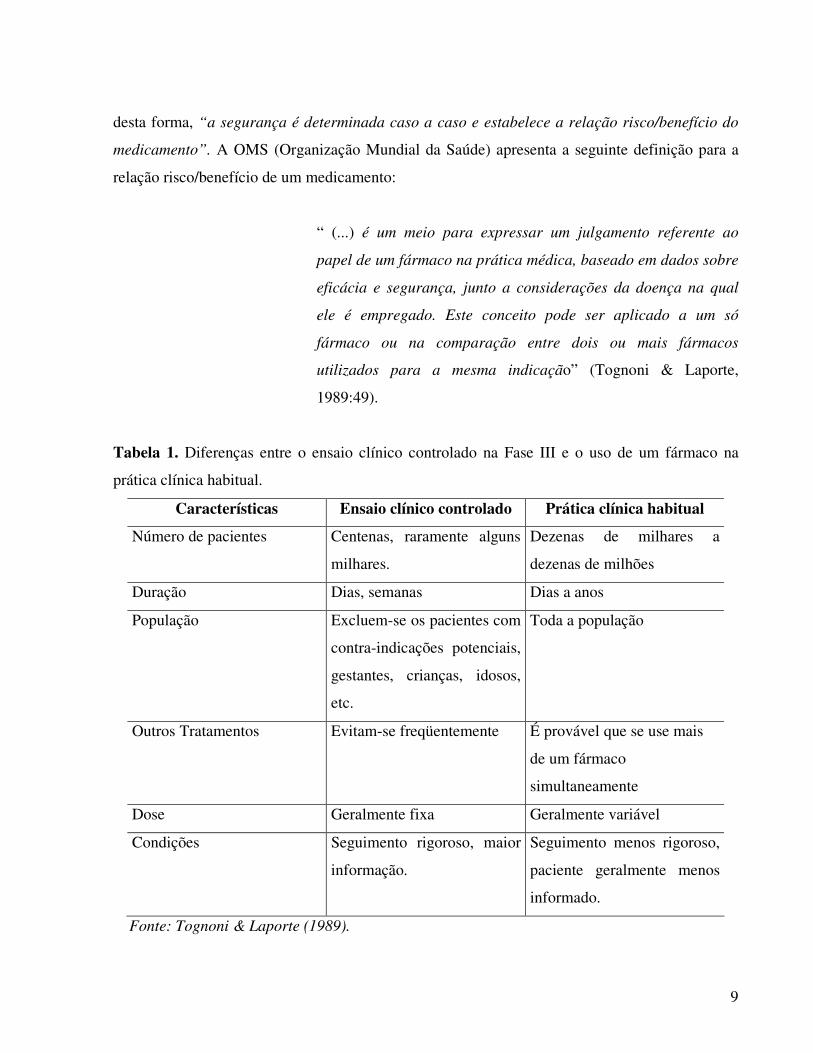
Tognoni & Laporte (1989) destacam algumas diferenças entre o ensaio clínico controlado
e o uso de fármacos na prática clínica habitual, apresentadas na Tabela 1. Segundo os autores,
“(...) os ensaios clínicos prévios a comercialização (...) são só uma primeira impressão parcial
dos seus efeitos potenciais.”
Os ensaios clínicos controlados com medicamentos são geralmente realizados para a
verificação da eficácia e segurança dos mesmos. Segundo o Drug Review Glossary, disponível na
página eletrônica da FDA (FDA, 2005d), nenhum medicamento é completamente seguro ou
incapaz de provocar efeitos adversos e os testes realizados com o medicamento antes de sua
comercialização devem demonstrar que o mesmo é seguro para a indicação clínica pretendida e,
9
desta forma, “a segurança é determinada caso a caso e estabelece a relação risco/benefício do
medicamento”. A OMS (Organização Mundial da Saúde) apresenta a seguinte definição para a
relação risco/benefício de um medicamento:
“ (...) é um meio para expressar um julgamento referente ao
papel de um fármaco na prática médica, baseado em dados sobre
eficácia e segurança, junto a considerações da doença na qual
ele é empregado. Este conceito pode ser aplicado a um só
fármaco ou na comparação entre dois ou mais fármacos
utilizados para a mesma indicação” (Tognoni & Laporte,
1989:49).
Tabela 1. Diferenças entre o ensaio clínico controlado na Fase III e o uso de um fármaco na
prática clínica habitual.
Características Ensaio clínico controlado Prática clínica habitual
Número de pacientes Centenas, raramente alguns
milhares.
Dezenas de milhares a
dezenas de milhões
Duração Dias, semanas Dias a anos
População Excluem-se os pacientes com
contra-indicações potenciais,
gestantes, crianças, idosos,
etc.
Toda a população
Outros Tratamentos Evitam-se freqüentemente É provável que se use mais
de um fármaco
simultaneamente
Dose Geralmente fixa Geralmente variável
Condições Seguimento rigoroso, maior
informação.
Seguimento menos rigoroso,
paciente geralmente menos
informado.
Fonte: Tognoni & Laporte (1989).

10
A eficácia de um tratamento (pelo uso de um medicamento) é a evidência de que ele
realmente funciona em pacientes com uma determinada doença e é avaliada sob condições
experimentais rigorosas, em geral, um ensaio clínico randomizado. A avaliação da eficácia de um
medicamento pode exigir uma metodologia tão rigorosa que o contexto da pesquisa torna-se
muito diferente do que é encontrado na prática. Por estes motivos as evidências sobre a
efetividade do medicamento, que informam se a intervenção funcionou quando realizada em
condições usuais da prática clínica, também devem ser consideradas (Duncan & Schmidt, 1996).
Neste sentido é importante que se reconheça o papel limitado dos ensaios clínicos
realizados com os medicamentos antes de sua comercialização e que os mesmos não conseguirão
abrangir todos os seus potenciais efeitos. A Farmacovigilância surge desta forma como um
valioso instrumento para proteção da saúde, sendo definida por Tognoni & Laporte (1989:49)
como “a identificação e a avaliação dos efeitos do uso, agudo e crônico, dos tratamentos
farmacológicos no conjunto da população ou em subgrupos de paciente expostos a tratamentos
específicos”. Originalmente a Farmacovigilância é voltada para o estudo dos efeitos desejáveis e
indesejáveis do medicamento, mas seus objetivos principais são identificar os efeitos indesejáveis
não descritos previamente, quantificar esses efeitos associados ao uso de determinados fármacos,
informar aos profissionais e tomar eventuais medidas administrativas (Carné & Laporte, 1989).
Queda na produção de novos medicamentos
Em um documento denominado Inovação ou Estagnação? Desafio e Oportunidade na
Etapa Crítica para Novas Tecnologias Médicas a FDA (2004a) declara que, apesar dos avanços
no campo da pesquisa básica, a produção de inovações significativas na área de saúde, incluindo
os medicamentos, tem deixado a desejar e aponta o ano de 2000 como o início da redução do
número de pedidos para registro de medicamentos contendo novas entidades moleculares em
agências de todo o mundo. A Indústria Farmacêutica tem se dedicado principalmente a encontrar
novas aplicações para medicamentos já comercializados e, no ano de 2000, somente 27% dos 98
medicamentos aprovados nos EUA eram novas entidades moleculares (Schmid & Smith, 2004).
É importante considerar ainda que o fato destes medicamentos possuírem novas entidades
moleculares não significa que representem avanços terapêuticos, conforme as definições
apresentadas anteriormente.

11
Segundo a FDA os equipamentos e técnicas de análise não passaram pelo processo de
inovação necessário para avaliar os novos produtos e os profissionais que trabalham com
pesquisa e desenvolvimento precisam usar ferramentas do século passado para avaliar os avanços
obtidos no presente século. O processo atual de descoberta de medicamentos baseado em técnicas
in vitro e em modelos animais é muitas vezes insuficiente para a compreensão da relevância
clínica do produto. A FDA destaca ainda que um medicamento que iniciava os ensaios clínicos
de Fase I, em 2000, não tinha maiores condições de chegar ao mercado que um medicamento que
tivesse iniciado a mesma fase em 1985, afirmando que esta “deficiência” em analisar novas
tecnologias pode ser uma das explicações na redução do lançamento de produtos com inovações
significativas no mercado.
Segundo os dados do Tufs Center for the Study of Drugs Development, baseados em
medicamentos aprovados nos Estados Unidos entre 1994 e 1998, são necessários, em média, 10 à
15 anos para um medicamento sair de um laboratório de pesquisa e estar disponível para um
paciente americano. Apenas 5 em 5000 compostos que entram em testes pré-clínicos (estudos em
laboratório e em animais) são liberados para testes clínicos (em seres humanos) e somente 1
destes 5 compostos é aprovado para comercialização (PhRMA, 2003).
Buscando reduzir esta deficiência tecnológica e as diferenças entre os resultados das
pesquisas básicas e a geração de novos produtos para a saúde, a agência americana declarou a
importância do desenvolvimento de novas ferramentas técnicas e científicas incluindo ensaios,
padrões, técnicas de modelagem computacional e biomarcadores que possam tornar o processo de
desenvolvimento de medicamentos mais eficiente e efetivo, resultando em produtos mais seguros
para os pacientes (FDAa, 2004).
Mas a explicação da redução da entrada de medicamentos que representam avanços
terapêuticos significativos no mercado, baseada apenas nas dificuldades técnicas e metodológicas
encontradas pela Indústria Farmacêutica, é limitada. Os interesses comerciais que norteiam os
investimentos em novos produtos ou na otimização de produtos já comercializados também
devem ser levados em conta. Um importante aspecto relacionado à inovação do mercado
farmacêutico é que ela ocorre preferencialmente envolvendo medicamentos que possuem um
mercado consumidor garantido para o tratamento de doenças crônicas ou oriundas do estilo de
vida. Segundo Trouiller (apud MSF, 2001) uma análise do desenvolvimento de medicamentos ao
longo das últimas duas décadas do século XX revelou que apenas 15 novos medicamentos foram

12
introduzidos no mercado para o tratamento das doenças tropicais, que afetam primordialmente as
populações dos países pobres e em desenvolvimento, enquanto no mesmo período foram
lançados 179 medicamentos para doenças cardiovasculares.
Uma doença mortal ou muito grave pode ser considerada negligenciada quando as opções
de tratamento são inadequadas ou não existem; quando seu mercado potencial de medicamentos é
insuficiente para provocar uma pronta resposta do setor privado em produzir tais medicamentos e
quando o interesse do governo em combater este tipo de doença é insuficiente. Malária,
tuberculose, doença de Chagas e tripanossomíase humana africana (doença do sono) são
exemplos de doenças negligenciadas.
Uma explicação muitas vezes apontada pela Indústria Farmacêutica para a queda do
número de novas entidades moleculares que estão entrando no mercado atualmente está ligada à
questão dos custos para desenvolvimento destes produtos. Entretanto, existe uma polêmica entre
aos custos divulgados pela Indústria Farmacêutica e aqueles divulgados por algumas pesquisas
que apresentam valores inferiores aos da Indústria. Em um trabalho publicado por J.A. DiMasi
et.al (1991), os autores declararam que o custo para desenvolvimento e lançamento de um novo
medicamento no mercado era de aproximadamente US$ 231 milhões. Este número foi usado em
trabalhos subseqüentes que alteraram alguns parâmetros usados pelo grupo de DiMasi, chegando
a valores como US$ 359 milhões, valor que ajustado para o dólar de 2000, correspondia a US$
473 milhões (MSF, 2001). Algumas estimativas recentes sobre os custos de desenvolvimento de
medicamentos revelaram resultados diferentes dos citados acima: utilizando o estudo original de
DiMasi, o grupo Public Citizen, uma organização sem fins lucrativos destinada à defesa dos
consumidores nos Estados Unidos, calculou o gasto com novos medicamentos em US$110
milhões, sem contar o custo de oportunidade (da receita potencial caso o capital fosse investido
em outra atividade) e a Aliança Global para Desenvolvimento de Drogas para a Tuberculose
(GATB) estimou o custo de um novo medicamento para a tuberculose em cerca de US$40
milhões, utilizando uma entidade química já identificada (MSF, 2001).
Verifica-se assim que o processo de inovação no setor farmacêutico apresenta aspectos
bastante polêmicos, como a definição do que é verdadeiramente uma inovação, os custos reais
associados ao processo de desenvolvimento de novos medicamentos e os interesses comerciais
das empresas farmacêuticas. Mas dentre as questões levantadas neste capítulo, uma pode ser
apontada como essencial sob o ponto de vista sanitário: a definição da inovação dos

13
medicamentos segundo o avanço terapêutico. Um medicamento só poderá ser considerado uma
verdadeira inovação, do ponto de vista terapêutico, se apresentar vantagens clínicas sobre os
medicamentos já comercializados, caso contrário, somente os fabricantes, e não os consumidores,
estarão sendo beneficiados com o lançamento de novos produtos no mercado.

14
3. REGULAÇÃO SANITÁRIA DE MEDICAMENTOS
O uso de preparações com propriedades curativas remonta ao início da história da
Humanidade. Achados arqueológicos demonstraram que a habilidade para elaborar um
medicamento já existia 16 séculos antes de Cristo (Costa & Rozenfeld, 2000). A civilização
grega utilizava o termo phármakon destacando que o medicamento possui ao mesmo tempo a
natureza de remédio (cura) e de veneno (dano à saúde) (Costa, 1999) e Galeno (131-210) já
chamava atenção de que potenciais efeitos venenosos existem em qualquer medicamento.
Entretanto, somente no final do século XVIII as preocupações de Galeno começaram a ser
efetivamente confirmadas e Withering, médico e botânico inglês, fez a descrição da intoxicação
por um fármaco – a intoxicação digitálica (Barros, 1982 apud Costa, 1999). No final do século
XIX surgiram os primeiros inquéritos formais quanto à segurança dos medicamentos como
resposta às reações adversas observadas em pacientes (Capellà et al.,1989 apud Costa, 1999).
Nas primeiras décadas do século XX os países mais desenvolvidos, preocupados com a
questão da incorporação descontrolada de novas tecnologias, começaram a estabelecer leis e criar
órgãos responsáveis para implementação destas leis, com o intuito de controlar a produção e a
comercialização de bens e serviços com potenciais riscos à saúde pública (Abraham,1995).
Segundo Tenner (1997 apud Lucchese, 2001), o progresso tecnológico impõe uma vigilância
cada vez maior, pois, geralmente, o uso de tecnologias mais avançadas pode produzir resultados
melhores (mais potentes), mas exige que sejam obtidos por meio de estreitos controles e critérios.
Neste cenário, os profissionais de saúde devem ser qualificados adequadamente para avaliar tais
tecnologias.
O Estado surge neste contexto como responsável pela proteção da saúde dos
consumidores, cabendo-lhe zelar pelos interesses coletivos, intervindo nas atividades de produção
e comercialização dos produtos e prestação de serviços. O poder de restringir direitos individuais
em favor dos direitos coletivos é inerente ao Poder Público, que intervém por meio de órgãos da
administração pública com poder de regulamentação e de polícia. Segundo Costa & Rozenfeld
(2000:16): “As ações de vigilância sanitária estão inseridas no âmbito das relações de produção
e consumo, onde se origina a maior parte dos problemas de saúde e sobre os quais é preciso
intervir”.

15
As atividades de regulação sanitária no Brasil foram influenciadas ao longo dos tempos
pelas mudanças no contexto político e econômico mundial. Do controle dos portos, dos
alimentos, do exercício profissional dos médicos e boticários e dos manufaturados importados a
atenção é dirigida para a produção interna do país, para as indústrias e produtos que podem
provocar riscos à saúde. No contexto atual, com a abertura econômica dos mercados em vários
países, a preocupação dos órgãos de vigilância sanitária está direcionada também à produção
externa e aos processos internacionais de regulamentação sanitária (Lucchese, 2001).
Na área dos produtos para saúde os medicamentos merecem atenção especial quando o
assunto é a avaliação do risco. Episódios como o de 1937, que levou à morte dezenas de pessoas
que utilizaram o Elixir de Sulfanilamida nos EUA, e o nascimento de crianças com focomelia no
final da década de 50, pelo uso da Talidomida durante a gravidez, chamaram a atenção das
autoridades regulatórias para a necessidade de se avaliar a segurança e a eficácia dos
medicamentos liberados para o consumo humano.
O número de produtos para a saúde que entram atualmente nos mercados é crescente e os
medicamentos acompanham esta tendência. Como conseqüência, a incorporação de
medicamentos recém lançados no mercado nas práticas de saúde é contínua, sendo facilitada pela
velocidade com que as informações chegam até os profissionais de saúde e consumidores.
Soares (1997, apud Costa, 1999:381) levanta uma questão importante sobre este fato ao
declarar que:
“(...) quanto maior a oferta de produtos farmacêuticos, mais difícil é
seu controle e uso correto: grande número de produtos registrados
piora a cadeia terapêutica e o nível sanitário de um país, pois no
mínimo aumenta a confusão no registro, dificulta o controle da
fabricação, distribuição, prescrição, dispensação e uso, torna
irrealizável o conhecimento sobre os produtos e dificulta o
estabelecimento de prioridades de trabalho”.
O Registro Sanitário de Medicamentos
O Processo de registro de medicamentos é uma das ações de vigilância sanitária cuja
responsabilidade compete aos órgãos de Estado e, atualmente, no Brasil, esta atividade compete à

16
Agência Nacional de Vigilância Sanitária (ANVISA) (Brasil, 1999). A Política Nacional de
Medicamentos (PNM), aprovada pela Portaria 3.916 de 30/10/1998, definiu o registro de
medicamentos como ato privativo do órgão competente do Ministério da Saúde, destinado a
conceder o direito de fabricação do produto no país (MS, 2001). A PNM tem como uma de suas
diretrizes a regulamentação sanitária de medicamentos e, neste aspecto, enfatiza o papel do gestor
federal no que se refere às questões como registro de medicamentos, autorização para
funcionamento de empresas e restrições e eliminações de produtos inadequados ao uso.
Seguindo o modelo de descentralização das ações de saúde, definido pelo Sistema Único
de Saúde, a PNM enfatiza a descentralização gradual das ações de vigilância sanitária do nível
federal para estados e municípios. Entretanto, atividades como registro de medicamentos e a
autorização para funcionamento de empresas são definidos como “papéis indelegáveis do gestor
federal” (MS, 2001).
Quando os atributos relacionados à qualidade, segurança e eficácia são considerados na
avaliação para concessão registro de medicamentos esta etapa pode se tornar decisiva para que
produtos de qualidade sejam liberados para o mercado. A avaliação destes critérios é feita por
meio da análise dos resultados dos testes conduzidos com o medicamento – controle de qualidade
química e microbiológica, ensaios toxicológicos e clínicos e dados sobre o processo de produção
do medicamento. A transparência e a confiabilidade no processo de registro de medicamentos são
fundamentais e esta atividade deve ser executada por profissionais capacitados que possam dispor
de condições adequadas de trabalho para avaliar criteriosamente a nova tecnologia e os impactos
de sua utilização na saúde pública.
O processo de registro de medicamentos e as exigências necessárias para a aprovação dos
mesmos têm sido considerados barreiras para a comercialização num mercado global em
crescimento. Aqueles que defendem a desregulação nesta área sugerem medidas como o registro
automático de medicamentos, o registro provisório e o limite de tempo para os processos de
avaliação dos dossiês para registro. As autoridades regulatórias têm sido pressionadas para que
revisem, simplifiquem e harmonizem métodos, técnicas e procedimentos administrativos
envolvidos no processo de registro (MSH,1997).
Em um documento elaborado em 1992 os especialistas da Subregião Andina ligados ao
Programa de Medicamentos Essenciais e Vacinas da OMS/OPAS apontaram justificativas
históricas para a existência do registro sanitário de medicamentos (OPAS/ OMS, 1992). A partir

17
da década de 50 a Indústria Farmacêutica teve um grande crescimento e as experiências com os
primeiros antibióticos foram muito bem sucedidas. Os medicamentos foram vistos pela sociedade
como um caminho promissor por meio do qual a ciência realizaria a cura para todo e qualquer
mal. Entretanto, no início da década de 60, a talidomida demonstrou que este caminho não estava
isento de riscos e sucessivamente outros medicamentos revelaram graves efeitos secundários.
Tornou-se necessário estabelecer normas mais complexas, sofisticadas, custosas e de larga
duração para a avaliação dos medicamentos antes da liberação dos mesmos para o mercado. No
referido documento os especialistas declararam que alguns fabricantes de medicamentos, com o
intuito de recuperar os investimentos em pesquisa e desenvolvimento e maximizar seus lucros,
lançaram medicamentos de eficácia questionável no mercado, omitiram efeitos adversos graves e
venderam medicamentos para os países em desenvolvimento e subdesenvolvidos que não
estavam autorizados à venda em países desenvolvidos.
Também são apontadas justificativas para a realização do registro sanitário de
medicamentos relacionadas às peculiaridades do mercado farmacêutico:
a) O paciente (consumidor) não é capaz de fazer o diagnóstico de seu estado de saúde e
definir o produto adequado para o seu tratamento; não é capaz de avaliar a utilidade e
eficácia do medicamento e o preço que será pago pelo mesmo;
b) O médico que prescreve o medicamento não é quem paga por ele e nem sempre está
capacitado suficientemente para avaliar a eficácia e segurança dos medicamentos;
c) As estratégias de promoção de venda dos medicamentos são destinadas
fundamentalmente aos prescritores e a transparência no mercado farmacêutico é objeto de
constante debate.
Verifica-se assim uma tensão neste campo entre os que defendem a importância do
registro sanitário de medicamentos para a proteção da saúde pública e aqueles que o consideram
mais uma barreira ao livre comércio. Os critérios para registro de medicamentos são apontados
pelos fabricantes como potenciais barreiras não alfandegárias ao comércio entre diferentes países,
já que as exigências sanitárias podem variar de um país para outro e, desta forma, impedir que um
medicamento registrado em determinado país seja registrado em outro. Neste sentido, dentre
outros objetivos, visando diminuir estas diferenças sanitárias legais e facilitar as trocas

18
comerciais entre os países foi iniciado um processo internacional denominado Harmonização dos
Regulamentos Técnicos Sanitários.
Harmonização dos Regulamentos Técnicos Sanitários
Conferência Internacional sobre a Harmonização(ICH)
Desde o final da década de 80, as maiores empresas farmacêuticas dos EUA, Europa e
Japão já pressionavam as autoridades regulatórias sanitárias daqueles países para que
padronizassem as exigências presentes em seus regulamentos sanitários. A principal justificativa
usada pelo setor farmacêutico era diminuir os gastos com o desenvolvimento de novos
medicamentos, principalmente com os ensaios clínicos e o tempo para liberação dos novos
produtos para o mercado (WHO, 2002a)
A Conferência Internacional sobre Harmonização (ICH) foi iniciada em abril de 1991,
formada por um Comitê Diretivo composto por seis representantes oficiais – dois da Comissão
Européia, dois da FDA e dois do Ministério da Saúde, Trabalho e Bem Estar do Japão.
Participam também do Comitê Diretivo seis representantes das respectivas federações da
Indústria Farmacêutica e autoridades do Canadá, Suíça e da OMS participam como observadores.
O objetivo principal da Conferência refere-se às exigências das autoridades regulatórias
relacionadas aos novos fármacos que, em sua maioria, são desenvolvidos na Europa Ocidental,
Estados Unidos ou Japão – estas três regiões contribuem com mais de 90% dos novos
medicamentos desenvolvidos no mundo (WHO, 2002a). A harmonização vale apenas para o
registro de medicamentos nestas três regiões, mas o objetivo é que no futuro a adoção dos
critérios harmonizados seja expandida para outros países, desenvolvidos ou em desenvolvimento.
A OMS tem estimulado a disseminação das diretrizes harmonizadas para os países que
não participam da Conferência por meio do desenvolvimento de normas e padrões de qualidade
internacionalmente reconhecidos. Os países, por sua vez, podem utilizar estes padrões
internacionais para elaboração de suas próprias normas, adaptando-os às suas realidades. A OMS
tem buscado cooperar na promoção da difusão de informações entre as autoridades regulatórias
de diversos países e um exemplo disto é a realização da Conferência Internacional das
Autoridades Regulatórias de Medicamentos (ICDRA), único fórum mundial onde os

19
representantes das entidades regulatórias regionais ou nacionais podem trocar informações e
debater sobre a questão da regulação de medicamentos. A Conferência é realizada a cada dois
anos desde 1980 e o encontro mais recente foi em Madri, na Espanha, em fevereiro de 2004. As
questões levantadas na Conferência influenciam a elaboração das resoluções da OMS que são
definidas na Assembléia Mundial de Saúde (WHO, 2002a).
O processo de harmonização dos regulamentos sanitários apresenta algumas questões que
precisam ser discutidas, tendo em vista as suas implicações para os países subdesenvolvidos e em
desenvolvimento. Ainda que a elevada qualidade científica dos manuais elaborados pela ICH
seja largamente reconhecida, o processo de discussão envolvendo 17 países industrializados
geralmente não leva em consideração as opiniões e necessidades dos demais países membros da
OMS, como os países em desenvolvimento. O objetivo de ter a OMS como instituição
observadora nos trabalhos da ICH é garantir que os interesses internacionais para proteção da
saúde pública sejam considerados. Porém, argumenta-se que ainda não foi criado nenhum
mecanismo operacional que garanta a participação adequada das autoridades regulatórias dos
países não membros da ICH nos processos de elaboração dos documentos técnicos, como dos
manuais (WHO, 2002b). A adoção de critérios de elevada complexidade técnica, estabelecidos
na ICH como padrão global, pode ter implicações sérias para a saúde pública dos países em
desenvolvimento. Empresas nacionais produtoras de medicamentos essenciais e genéricos,
necessários para a prevenção e tratamento de condições endêmicas locais e que não são
produzidos pelas empresas farmacêuticas multinacionais, podem ser afetadas (WHO, 2002b).
Segundo Lucchese (2001), para participar do processo de regulamentação sanitária
internacional e aproveitar os resultados de suas decisões para proteger sua população, os países
em desenvolvimento devem contar com uma estrutura que no presente não têm: de informação,
de documentação, de pessoal qualificado, de suporte científico-tecnológico e de uma estrutura
administrativa bem consolidada. Ainda segundo o autor, a agilização do processo de registro de
medicamentos exige um sistema de farmacovigilância ampliado e mais ágil, capaz de detectar
possíveis efeitos adversos e retirar do mercado os produtos inseguros. A harmonização dos
regulamentos técnicos cria a necessidade de que as agências tenham um alto grau de confiança
em suas decisões no menor tempo e com a menor exigência possível.

20
Conferência Pan-americana de Harmonização da Regulamentação Farmacêutica
Seguindo a tendência internacional para harmonização da regulamentação farmacêutica,
iniciada pelos países desenvolvidos no início da década de 90, a Organização Pan-americana da
Saúde (OPAS) iniciou os trabalhos de harmonização nas Américas, em 1997, com a realização da
I Conferência Pan-americana de Harmonização da Regulamentação Farmacêutica, em
Washington (EUA) (Lucchese, 2001).
A Conferência conta com a participação de representantes das autoridades regulatórias de
diferentes países e entidades representantes da Indústria Farmacêutica – A Federação Latino-
americana da Indústria Farmacêutica (FIFARMA), que representa majoritariamente as
multinacionais, e a Associação Latino-americana das Indústrias Farmacêuticas (ALIFAR), que
representa as indústrias de capital nacional da região. A segunda Conferência ocorreu em
novembro de 1999 e a terceira e última em abril de 2002, também em Washington (EUA). Dentre
as recomendações apresentadas na I Conferência podem ser destacadas (Lucchese, 2001):
• Adoção das boas práticas de fabricação de produtos farmacêuticos;
• Exigências de testes de bioequivalência e biodisponibilidade;
• Mudanças nas condições de venda dos produtos;
• Adoção das boas práticas clínicas;
• Fortalecimento das agências regulatórias;
• Processo de harmonização por blocos econômicos;
• Aproximação com a Conferência Internacional de Harmonização (ICH).
Na II Conferência foram discutidos alguns temas já abordados na I Conferência e temas
adicionais, como a falsificação de medicamentos e a classificação de medicamentos (com e sem
prescrição). Também foi validado o conceito de país de referência e os países latino-americanos
foram convocados a harmonizar a lista de países segundo critérios definidos.

21
Harmonização da Regulamentação Sanitária no Mercosul
A década de 1990 foi marcada por Reformas no aparelho de alguns Estados latino-
americanos, impulsionadas pelo baixo crescimento econômico, altas taxas de inflação,
desequilíbrio fiscal e dívida externa dos países. As reformas conduziram à abertura econômica
dos países ao comércio internacional por meio da redução das barreiras tarifárias e gerou um
ambiente favorável para a implementação de acordos bi ou multilaterais entre os países latino-
americanos (Lucchese, 2001).
Neste contexto, em 1991, quatro países da América do Sul – Brasil, Argentina, Paraguai e
Uruguai - formaram um bloco de integração econômica denominado Mercado Comum do Sul
(MERCOSUL), celebrado por meio do Tratado de Assunção. Dentre os pontos de negociação
previstos no Tratado está o compromisso de harmonização da legislação destes quatro países em
diversas áreas. O Tratado define a redução progressiva de barreiras tarifárias e a eliminação de
restrições não tarifárias (a regulamentação sanitária é um dos tipos mais importantes de barreira
não tarifária) (Senado Federal, 1996 apud Lucchese, 2001).
A harmonização dos regulamentos técnicos sobre alimentos, medicamentos, artigos de
higiene pessoal, cosméticos e perfumes, saneantes domiciliares e produtos de uso médico,
hospitalar, laboratorial e odontológico é realizada pelas comissões temáticas que formam cada
Subgrupo de Trabalho (SGT) do Mercosul, o órgão executivo mais importante.
Lucchese (2001) aponta alguns avanços no campo farmacêutico obtidos nas negociações
do Mercosul:
• O roteiro para inspeções: contêm todos os passos que um inspetor deve seguir para fazer
uma boa inspeção sanitária. É considerado o principal regulamento harmonizado do
Mercosul na área farmacêutica e farmoquímica e adotado como modelo em outros países
latino-americanos e como referência pela própria Indústria Farmacêutica;
• O Mercosul harmonizou um regime de inspeções conjuntas que permite que a autoridade
sanitária de um Estado Parte receptor do novo medicamento faça uma inspeção na
empresa do Estado Parte do produtor do medicamento de forma conjunta com a
autoridade sanitária do país sede da empresa produtora.

22
Considerando as questões abordadas neste capítulo, verifica-se que, embora o uso de
medicamentos pela humanidade remonte a muitos séculos atrás, a exigência de dados
relacionados à segurança e eficácia dos mesmos só foi estabelecida no século XX, principalmente
por meio da realização dos ensaios clínicos controlados. Efeitos adversos severos e mortes
marcaram algumas tragédias envolvendo o uso de medicamentos e chamaram a atenção das
autoridades sanitárias em vários países do mundo ao longo do século XX, reforçando a
importância do papel do Estado na regulação das relações de produção e consumo destes
produtos. O registro sanitário de medicamentos apresenta-se como um importante instrumento de
regulação, pois é por meio dele que o fabricante recebe autorização para comercializar seu
produto. Ainda que existam críticas à necessidade do registro sanitário de medicamentos, buscou-
se destacar neste capítulo a importância desta atividade para proteção da saúde coletiva.

23
4. JUSTIFICATIVAS
As discussões feitas até aqui sobre a inovação no setor farmacêutico, vigilância sanitária
de medicamentos, registro de medicamentos e harmonização de regulamentos técnicos sanitários
revelam que o medicamento adquiriu grande destaque no cenário social, econômico e político dos
países ao longo do século XX.
O registro sanitário de medicamentos, como descrito anteriormente, pode funcionar como
etapa importante para que a autoridade sanitária controle e avalie a qualidade, segurança e
eficácia dos produtos que estarão disponíveis no mercado. A presença de medicamentos seguros
e eficazes no mercado contribui para o uso racional dos mesmos, processo que vai desde a
prescrição até o consumo pelos pacientes. Barros (1995: 56) reforça este argumento ao afirmar
que:
“(...) é indubitável que a falta de medidas regulamentadoras estritas
facilita em grande medida a presença de uma oferta de qualidade
inferior (com sua conseqüente influência na forma como serão
prescritos e utilizados os medicamentos). No que se refere ao impacto
sobre a prescrição, são óbvias as dificuldades resultantes de uma
oferta de baixa qualidade (...)”.
Tratando-se de um medicamento novo, além dos aspectos de qualidade, segurança e
eficácia, considera-se importante também a avaliação de um outro aspecto: a inovação
terapêutica. Tendo em vista os impactos que a divulgação do lançamento de um novo
medicamento exerce sobre os consumidores, prescritores, profissionais de saúde em geral e
também sobre os setores dos governos responsáveis pelo controle e aquisição dos mesmos, o
tema traz para discussão algumas questões importantes: a inovação tem sido considerada um
atributo para a concessão do registro sanitário de medicamentos novos? Existem diferenças entre
o processo de registro de medicamentos novos no Brasil e em outros países? No que se refere à
inovação, como se apresentam os medicamentos registrados no Brasil?
Neste sentido, dada a importância das questões levantadas para o campo da saúde,
considerou-se oportuno a realização de um estudo do processo de registro de medicamentos

24
novos na ANVISA, na FDA, na EMEA e na ANMAT, bem como a análise de um conjunto de
medicamentos novos registrados na ANVISA nos anos de 2000, 2001 e 2002.

25
5. OBJETIVOS
5.1. OBJETIVOS GERAIS
Tendo em vista o cenário apresentado e as questões levantadas anteriormente, os objetivos
do presente trabalho são descritos a seguir:
1. Descrever o processo de registro de medicamentos novos em quatro autoridades sanitárias:
FDA, EMEA, ANMAT e ANVISA.
2. Analisar, de forma comparativa com as demais autoridades sanitárias, a relação de
medicamentos novos registrados no Brasil, no período de 2000 a 2002.
5.2. OBJETIVOS ESPECÍFICOS
1. Comparar as principais características estruturais e funcionais das autoridades sanitárias
selecionadas;
2. Descrever as principais exigências presentes nas normas atuais sobre o registro de
medicamentos novos das autoridades sanitárias selecionadas;
3. Descrever as definições adotadas para medicamentos novos;
4. Descrever as etapas do processo de registro de medicamentos novos nas autoridades
sanitárias selecionadas;
5. Analisar um conjunto de medicamentos novos registrados no Brasil, no período de 2000 a
2002, no que se refere ao registro destes medicamentos nas demais autoridades sanitárias
consideradas, à definição do país de origem do fabricante, à classificação dos medicamentos
segundo definições da RDC da ANVISA nº 136 de 29/05/03 para medicamentos novos e à
classificação dos medicamentos segundo potencial terapêutico definido pela FDA
6. Determinar os prazos para a concessão de registro dos medicamentos novos na ANVISA e na
FDA.

26
6. METODOLOGIA
O presente trabalho é um estudo descritivo do processo de registro de medicamentos
novos realizado por autoridades sanitárias de diferentes países. As autoridades sanitárias
selecionadas para o estudo foram: Administração de Alimentos e Medicamentos (FDA) dos
Estados Unidos da América, Agência Européia de Medicamentos (EMEA), Administração
Nacional de Medicamentos, Alimentos e Tecnologia Médica (ANMAT), da Argentina e Agência
Nacional de Vigilância Sanitária (ANVISA), do Brasil.
A escolha destas instituições pode ser justificada por alguns motivos. A agência
americana é, em muitas situações, tomada como modelo de órgão regulador, foi criada no início
do século XX e já acumula uma experiência importante no campo regulatório. Desta forma,
decidiu-se que seria importante estudar este órgão considerado “modelo”. A EMEA, por sua vez,
apesar de representar interesses sanitários de países europeus que possuem órgãos sanitários mais
antigos, é uma agência nova, criada no início da década de 90 para representar diversos países
europeus que possuem papel de destaque no mercado farmacêutico mundial. Por meio da escolha
destes dois órgãos regulatórios buscou-se incluir no estudo países que têm grande importância no
contexto que foi abordado.
A inclusão da ANVISA no estudo teve como razão principal o interesse pelo estudo do
processo de registro de medicamentos novos no Brasil. A escolha da ANMAT pode ser explicada
pelo interesse em pesquisar este processo em outro país latino-americano e em desenvolvimento,
como a Argentina.
O estudo teve como instrumentos para sua realização as seguintes fontes de dados:
pesquisa documental e entrevista aberta. Foram realizadas consultas às páginas eletrônicas das
autoridades regulatórias: http://www.fda.gov/cder/index.html (FDA), http://www.emea.eu.int/
(EMEA), http://www.anmat.gov.ar/principal.htm (ANMAT) e http://www.anvisa.gov.br
(ANVISA) visando à obtenção de informações oficiais das agências referentes ao processo de
registro de medicamentos novos. A pesquisa foi feita em livros, relatórios, revistas especializadas
e em outros tipos de publicação que continham informações relevantes para a discussão do tema.
A entrevista com o gerente da unidade da ANVISA responsável pelo registro de
medicamentos novos foi realizada após aprovação do projeto de pesquisa pelo Comitê de Ética
em Pesquisa da Escola Nacional de Saúde Pública Sérgio Arouca (CEP/ENSP), estando a

27
presente pesquisa registrada no Conselho Nacional de Ética em Pesquisa (CONEP). Foi obtida
autorização do profissional responsável pela Gerência Geral de Medicamentos (GGMED) da
ANVISA para que o entrevistado pudesse conceder a entrevista e este último assinou Termo de
Consentimento Livre e Esclarecido. A entrevista foi aberta e gravada e as perguntas feitas ao
entrevistado estão apresentadas no Anexo 1. O texto correspondente às respostas fornecidas pelo
entrevistado foi submetido à análise e aprovação do mesmo que também autorizou a divulgação
de seu nome no trabalho.
A relação de medicamentos novos registrados na ANVISA nos anos de 2000, 2001 e
2002, analisada no presente trabalho (Quadro 8, Anexo 2), foi extraída do estudo apresentado por
Reis (2004). A referida relação é composta pelos medicamentos novos registrados e que estavam
sendo comercializados no primeiro semestre de 2003. Desta forma, convém destacar que o
número total de medicamentos novos registrados na ANVISA, no período de 2000 a 2002, é
maior que o número de medicamentos analisados no estudo.
As datas de entrada do pedido de registro e de concessão dos mesmos pela FDA e pela
ANVISA foram obtidas nas páginas eletrônicas das referidas agências. Na página eletrônica da
FDA, digitando-se o nome do medicamento em campo específico, obtém-se o número da NDA
(New Drug Apllication) referente ao medicamento e o histórico de análise e aprovação do
mesmo, que por sua vez fornece a carta de aprovação da NDA enviada pelo Center for Drug
Evaluation and Research (CDER) ao solicitante de registro. Nesta carta consta a data de entrada
do pedido de registro ou de “aplicação” de um novo medicamento na FDA. Vale ressaltar que
houve dificuldade em definir qual a data seria considerada como a data de início da avaliação do
pedido de registro pelos técnicos da FDA, pois a referida carta de aprovação relaciona uma séria
de datas nas quais houve recebimento de documentos enviados pelo solicitante ao CDER,
compondo um histórico do contato oficial entre a agência e o solicitante. Como na maior parte
das cartas de aprovação (28) não havia detalhamento dos documentos entregues pelo solicitante
foi considerada como data de início da análise do dossiê a primeira data apresentada na carta de
aprovação, a data de protocolo dos dossiês no CDER.
Para a coleta das datas referentes ao início e fim da análise do processo de registro de
medicamentos novos na ANVISA foi acessado o banco de dados de medicamentos registrados na
ANVISA4. Na página que fornece os dados sobre o registro do produto, incluindo a data de
4 http://www7.anvisa.gov.br/datavisa/Consulta_Produto/consulta_medicamento.asp

28
aprovação de registro, está disponível também o número do “processo” aberto na agência para o
referido produto. Este número foi inserido em outro campo da página eletrônica5 onde foi
possível verificar o trâmite de documentos na agência, como a data de entrada do pedido de
registro do medicamento. Assim como ocorreu com a maioria dos medicamentos registrados na
FDA, na página eletrônica da ANVISA não há informação sobre a data de início da análise do
pedido de registro, apenas as datas referentes à tramitação dos documentos. Foi considerada
como data de início do processo de análise a data de protocolo do pedido de registro.
A preocupação com a definição destas datas está em não obter prazos diferentes daqueles
que foram necessários aos técnicos das duas agências para avaliar os medicamentos e decidir
sobre a concessão do registro. Porém, como descrito acima, estas datas não estavam claramente
definidas nas páginas eletrônicas da ANVISA e da FDA e, desta forma, é importante destacar que
no presente estudo os prazos obtidos para autorização dos registros de medicamentos foram
considerados como o tempo (dias) decorrido entre as datas de protocolo dos pedidos de registro
nas referidas agências e as datas de deferimento (aprovação) dos mesmos. Não foram
descontados os períodos nos quais a análise técnica do registro pode ter sido suspensa, como por
exemplo, para cumprimento de exigência feita pela autoridade sanitária ao solicitante do registro.
5 http://www7.anvisa.gov.br/datavisa/Consulta_Processos/Consulta_Processo.asp

29
7. RESULTADOS E DISCUSSÕES
7.1 ADMINISTRAÇÃO DE ALIMENTOS E MEDICAMENTOS (FDA)
A origem da FDA remonta ao século XIX, à criação da Divisão de Química que era ligada
ao Departamento de Agricultura dos Estados Unidos, em 1862. Em 1901 esta Divisão foi
transformada em Departamento de Química e com a publicação da Lei de Alimentos e
Medicamentos, em 1906, foi iniciada a era moderna da agência. O nome atual – Administração
de Alimentos e Medicamentos - foi adotado em julho de 1930.
A FDA está situada atualmente dentro do Departamento de Saúde e Serviços Humanos
(HHS), principal instituição do governo americano voltada para a proteção da saúde da população
e que é responsável pela execução de mais de trezentos programas voltados para as mais diversas
atividades no campo da saúde (FDA, 2005b).
A FDA é formada por oito Centros: Centro de Avaliação e Pesquisa de Produtos
Biológicos (CBER), Centro de Avaliação e Pesquisa de Medicamentos (CDER), Centro de
Equipamentos e Produtos Radiológicos para a Saúde (CDRH), Centro de Segurança Alimentar e
Nutrição (CFSAN), Centro de Medicamentos Veterinários (CVM), Centro Nacional de Pesquisa
Toxicológica (NCTR), Departamento do Diretor (OC) e Departamento de Assuntos Regulatórios
(ORA) (FDA, 2005c). A FDA conta atualmente com um corpo de 10.446 funcionários
compreendendo químicos, farmacologistas, médicos, microbiologistas, veterinários,
farmacêuticos, advogados, dentre outros profissionais (FDA, 2005b).
Regulação Sanitária de Medicamentos nos Estados Unidos
No início do Século XX existiam muitos medicamentos adulterados, falsificados,
denominados medicamentos de patente no mercado americano. Mas apesar desta denominação
estes medicamentos não eram patenteados, seus fabricantes usavam fórmulas secretas para
garantir seus mercados. Os medicamentos produzidos pelas empresas farmacêuticas disputavam o
mercado com estes medicamentos de patente e eram vendidos principalmente para os médicos.
Eram chamados de medicamentos éticos – pois eram produtos de “elevada qualidade, produzidos

30
a partir de preparações padronizadas, ao contrário dos medicamentos de patente” (Abraham,
1995).
O controle estatal americano sobre os medicamentos adulterados foi iniciado em 1906
quando a Lei dos Alimentos e Medicamentos Puros, a primeira lei para o controle da adulteração
de medicamentos nos Estados Unidos, foi aprovada. As atividades de regulação do setor
farmacêutico também eram desenvolvidas na Europa e neste período foi aprovada uma lei muito
similar à americana na Inglaterra. As leis protegiam os consumidores dos falsos anúncios sobre
os benefícios dos medicamentos, mas permitia que os fabricantes dos medicamentos de patente
mantivessem as fórmulas dos mesmos sob segredo. Mas apesar desta proteção o volume de
vendas dos medicamentos industrializados aumentou significativamente nas primeiras décadas do
século XX e pouca importância era dada à possibilidade de ocorrência de efeitos adversos
severos e à necessidade de garantia de segurança e eficácia dos medicamentos (Abraham, 1995).
Entretanto, em 1938, um episódio foi capaz de chamar a atenção das autoridades
americanas: a morte de mais de 100 pessoas (maioria crianças) que utilizaram um xarope de
Sulfanilamida. Este fármaco era largamente utilizado naquele período e nenhum problema quanto
à sua segurança havia ocorrido até então. Em outubro de 1937, uma empresa obteve autorização
para comercializar a substância sob a forma líquida, usando, porém uma substância tóxica como
solvente, o dietilenoglicol. Os testes de toxicidade (em animais) com os compostos resultantes do
dietilenoglicol foram negligenciados e o nome do solvente tóxico não estava presente no rótulo
do produto (Abraham, 1995). Como conseqüência da tragédia, o governo americano aprovou, em
junho de 1938, a Lei de Alimentos, Medicamentos e Cosméticos. Por meio desta nova lei as
empresas farmacêuticas ficaram obrigadas a realizar estudos de segurança com os medicamentos
e passou a ser exigido também o registro dos medicamentos na FDA para a comercialização. Até
então as empresas farmacêuticas não eram obrigadas a solicitar autorização governamental para
comercializar seus medicamentos nos Estados Unidos e a FDA apenas tinha poderes para retirar
do mercado aqueles medicamentos que apresentassem riscos à saúde pública (Abraham, 1995).
O desenvolvimento da química, da fisiologia e da farmacologia básica durante a segunda
metade do século XX, associado ao aumento na demanda por serviços de saúde, garantiram a
introdução de vários produtos farmacêuticos no mercado. O aumento da utilização de
medicamentos acentuou-se após a Segunda Guerra Mundial e os efeitos benéficos dos mesmos
foram profundamente percebidos durante as décadas de 40 e 50 pela introdução de antibióticos

31
como penicilina e estreptomicina que permitiram salvar muitas vidas em condições até então
improváveis (Laporte et al., 1993).
O consumo cada vez maior de medicamentos, promovido maciçamente pelos laboratórios
fabricantes por meio de propagandas e marketing dos produtos, teve, segundo Sterky (1985 apud
Laporte et al.,1993), um impacto cultural óbvio: aumento da dependência da alopatia (e
consequentemente da classe médica e farmacêutica) pela população e a substituição de formas
tradicionais de cuidado da saúde. O controle das autoridades sanitárias sobre o uso indevido dos
medicamentos, sobre a propaganda dos medicamentos que em sua maioria só destacavam os
efeitos benéficos (quase milagrosos) dos mesmos e sobre a eficácia destes produtos era ainda
muito incipiente. Neste cenário surge então um novo medicamento: a Talidomida.
Lançada no mercado alemão, em outubro de 1957, pelo laboratório Chemie Grunenthal, a
Talidomida foi indicada inicialmente como sedativo e o fabricante anunciava que o produto não
apresentava toxicidade. O pesado investimento em marketing fez do produto um dos líderes de
venda na Alemanha naquele período. O uso da Talidomida não ficou restrito à Alemanha e no
início da década de 60 vinte países tinham licença para produzí-la e /ou distribuí-la. (Oliveira et
al, 1999). O crescimento das vendas do produto foi acompanhado dos primeiros relatos médicos
sobre reações adversas, como constipação intestinal, tonteiras, perda de memória e polineurites.
O fabricante, por sua vez, buscava minimizar estes relatos atribuindo-os ao uso de altas doses do
medicamento e por tempo prolongado (Mokhiber,1995 apud Oliveira et al.,1999).
Os relatos médicos sobre o nascimento de crianças com má-formação congênita,
caracterizada pelo desenvolvimento defeituoso dos ossos longos dos braços e das pernas e mãos e
pés variando entre aspecto normal e o rudimentar (síndrome denominada focomelia pela
semelhança dos recém - nascidos com a forma externa das focas), começaram a surgir na
Alemanha a partir de 1959. Em 1961, durante o North Rhein-Westphalia Pediatric Meeting,
realizado em Dusseldorf, na Alemanha, Lenz apresentou os resultados de uma pesquisa onde
foram analisados 34 casos de recém–nascidos com graves deformidades nas extremidades. O
pediatra levantou publicamente a possibilidade das anomalias congênitas terem sido provocadas
pelo consumo da Talidomida durante a gestação (Mellin & Katzenstein,1962 apud Oliveira et al.,
1999).
A FDA não havia autorizado o registro da Talidomida nos Estados Unidos, pois segundo
a agencia americana, o fabricante não apresentou resultados satisfatórios sobre a segurança do

32
medicamento (Abraham, 1995). No contexto da tragédia a FDA saiu fortalecida por ter impedido
um desastre maior com a população americana e, estimulado pela repercussão dos fatos, em
outubro de 1962, o Congresso Americano aprovou a Emenda Kefauver-Harris. Esta emenda
fortaleceu o controle da FDA sobre os medicamentos e os fabricantes foram obrigados a fornecer
não somente provas de segurança, mas também de eficácia de seus produtos (FDA, 2004b). Foi
estabelecida a exigência das solicitações de Investigação de Novo Medicamento (IND) e
Aplicação de Novo Medicamento (NDA) que deveriam ser feitas pelo solicitante de registro do
medicamento a FDA (Abraham, 1995).
Outras normas importantes sobre os medicamentos foram aprovadas nos EUA nos anos
seguintes, dentre as quais podem ser destacadas (FDA, 2004b):
1- Ato de Competição de Preço de Medicamentos e Prazo de Restauração de Patente
(1984): esta Lei aumentou o número de medicamentos que poderiam ser submetidos a uma NDA
abreviada (ANDA). Este processo de avaliação para registro tem como objetivo a redução de
custos e de tempo para registro de medicamentos genéricos. O Prazo de Restauração de Patente
referia-se aos 17 anos de proteção legal que era concedida para a patente de um medicamento.
2- Ato das taxas sobre medicamentos de prescrição (1992): por meio desta lei os
fabricantes de medicamentos vendidos sob prescrição médica passaram a pagar taxas para a
aplicação dos novos medicamentos e suplementos – uma taxa anual por fabricante e uma taxa
anual por produto. Estes recursos foram utilizados pela FDA na contratação de mais de 700
profissionais no final do ano de 1997 com o objetivo de acelerar o processo de revisão das NDA.
3- Lei de Modernização da FDA (1997): esta lei causou uma das mais amplas mudanças
na lei de Alimentos, Medicamentos e Cosméticos em 35 anos (depois da Emenda Kefauver-
Harris, em 1962) e teve impacto importante no registro de medicamentos, pois foram
estabelecidas normas para o processo acelerado de registro e orientações para os revisores das
NDA. Por meio deste processo é dada autorização para a comercialização de novos
medicamentos sem que se tenha obtido resultados clínicos conclusivos (requeridos no processo
de registro normal) sobre a eficácia dos mesmos. A FDA argumenta que a aprovação de registro
acelerada é concedida para alguns medicamentos novos indicados para patologias graves e que
carecem de tratamento satisfatório.
Nesta modalidade de registro, medidas menos tradicionais, denominadas desfechos
substitutos (surrogate endpoints) são usadas para avaliar a eficácia dos medicamentos. Os

33
desfechos substitutos são resultados laboratoriais que não permitem uma “medida” direta de
como o paciente se sente, como seu organismo funciona ou sobrevive, mas que permitem
“prever” o benefício. Um exemplo de desfecho substituto pode ser a diminuição da carga viral
por curtos períodos de tempo em um paciente que esteja usando um antiretroviral. Muitos
medicamentos antiretrovirais estão sendo aprovados para comercialização nos EUA desta forma
(FDA, 2004c). O medicamento Mesilato de Imatinib (Glivec), indicado para tratamento oral de
pacientes com Leucemia Mielóide Crônica, foi submetido ao registro acelerado baseado em
resultados de três estudos de Fase II que revelaram redução substancial dos níveis de células
cancerígenas na medula óssea e no sangue. O fabricante do medicamento (Novartis
Pharmaceuticals) recebeu autorização para Investigação de Novo Medicamento (IND) em abril
de 1998 e a FDA recebeu a solicitação para Aplicação de Novo Medicamento (NDA) em
fevereiro de 2001. O medicamento foi aprovado dois meses e meio depois, em maio de 2001
(Meadows, 2002).
Registro de medicamentos novos na FDA
O processo de registro de medicamentos na FDA é regulamentado pela Lei dos
Alimentos, Medicamentos e Cosméticos de 1938, citada anteriormente, e tem sido baseado na
Aplicação de Novo Medicamento (NDA) desde 1962, quando foi aprovada a Emenda Kefauver-
Harris (emenda à Lei de 1938).
Empresas, instituições de pesquisa e outras instituições que desejam testar seus
medicamentos em seres humanos nos Estado Unidos devem inicialmente solicitar uma
autorização para Investigação de Novo Medicamento (IND) a FDA. Este documento deve
conter informações como a fórmula estrutural do princípio ativo, os resultados dos ensaios pré-
clínicos (em culturas de células e animais) realizados em laboratório com a substância,
informações sobre o processo de produção do medicamento e o planejamento que será adotado
para os testes em seres humanos. Os ensaios com o medicamento em seres humanos,
denominados ensaios clínicos, só podem ser realizados após revisão dos dados da IND pela
FDA e por um grupo formado por cientistas e outros profissionais que supervisionarão a
pesquisa clínica. Estes supervisores são responsáveis pela aprovação dos protocolos clínicos
onde estão definidos os indivíduos que participarão dos ensaios clínicos, a relação de testes e

34
procedimentos que serão realizados, o medicamento e as doses que serão estudadas, a duração
dos testes e os objetivos dos estudos. É necessário também que o grupo de supervisores se
certifique que os indivíduos selecionados para o estudo estejam informados sobre os riscos dos
testes para sua saúde e que tenham dado consentimento expresso sobre sua participação na
pesquisa clínica. Observadas as exigências éticas e legais para realização da pesquisa
envolvendo seres humanos, os ensaios clínicos são realizados em quatro fases consecutivas
(Meadows, 2002):
Fase I: os testes são normalmente conduzidos com voluntários sadios e os objetivos são
determinar os efeitos adversos mais freqüentes do medicamento e como o mesmo é
metabolizado e eliminado do organismo humano. Visam avaliar principalmente a segurança
do medicamento. O número de indivíduos envolvidos nesta fase pode variar de 20 a 80.
Fase II: se os estudos da Fase I não demonstrarem níveis de toxicidade inaceitáveis do
medicamento, os estudos da Fase II poderão ser iniciados. Nesta etapa busca-se avaliar
principalmente a eficácia do medicamento em pacientes doentes, mas também são avaliados
os aspectos relacionados à segurança. O número de indivíduos envolvidos nesta fase pode
variar de 100 a 300.
Fase III: se os ensaios realizados na Fase II revelarem que o medicamento apresenta eficácia
clínica para a indicação proposta, os ensaios da Fase III poderão ser iniciados. Estes ensaios
têm por objetivo dar continuidade à avaliação de segurança e eficácia do medicamento. O
número de indivíduos envolvidos nesta fase pode variar de algumas centenas a 3000.
Fase IV: é realizada quando o medicamento já se encontra no mercado, podendo ser usado por
um grande número de indivíduos. Nesta fase são avaliados os novos usos do medicamento,
novas populações de pacientes e podem ser monitorados os efeitos adversos que surgem com o
uso prolongado.
O contato entre os profissionais da FDA com os solicitantes de registro pode variar
conforme a necessidade de informações para subsidiar o processo de desenvolvimento e
avaliação do produto. Ao término dos ensaios clínicos de Fase II, normalmente acontece uma
reunião onde os profissionais da FDA e o fabricante discutem sobre como os ensaios clínicos
da Fase III deverão ser conduzidos.

35
Aplicação de Novo Medicamento (NDA)
As etapas descritas acima devem ser observadas pelos fabricantes que pesquisam e
desenvolvem seus medicamentos nos Estados Unidos. Estas empresas e aquelas que
desenvolveram seus produtos fora dos EUA e que desejam registrar e comercializar seus
medicamentos nos EUA devem solicitar a FDA a autorização para Aplicação de Novo
Medicamento (NDA). Esta solicitação deve ser feita ao Centro de Avaliação e Pesquisa de
Medicamentos (CDER) da FDA, que será responsável pela avaliação da NDA. Tratando-se de
medicamentos biológicos esta solicitação deverá ser feita ao CBER.
O volume de informação que deve estar presente na NDA pode variar de um produto
para o outro, dependendo da natureza deste, mas de forma geral, uma NDA deve ser composta
de seções relacionadas aos seguintes tópicos (FDA, 2005d):
1- Estrutura química do medicamento, processos de produção e controle;
2- Dados sobre amostra, métodos de validação da embalagem e rotulagem;
3- Dados farmacológicos e toxicológicos dos ensaios pré-clínicos;
4- Dados clínicos (farmacocinética e biodisponibilidade);
5- Relatório atualizado sobre dados de segurança do medicamento (geralmente 120
dias após a entrada do pedido de NDA);
6- Informações sobre a patente do produto.
Para o início do processo de revisão, o CDER utiliza um sistema de classificação das
NDA que permite diferenciar os níveis de inovação dos medicamentos. Esta classificação é
feita a partir da caracterização do tipo químico e do potencial terapêutico dos medicamentos
(NIHCM, 2002):
Tipo químico do medicamento: classificação referente ao fármaco do medicamento. Este
fármaco pode ser novo, pode estar contido em um medicamento já aprovado ou ser derivado de
um fármaco presente em um medicamento já aprovado.
Potencial terapêutico do medicamento: referente à eficácia clínica do medicamento quando
comparada à eficácia de medicamentos já disponíveis no mercado, destinados ao diagnóstico,

36
tratamento ou prevenção da mesma patologia, tendo por base as evidências disponíveis no
período da revisão para aprovação.
A partir da análise do tipo químico, o CDER classifica os medicamentos que são
apresentados para o registro da seguinte forma (FDA, 2005d):
1. Novas Entidades Moleculares (NME): medicamentos contendo fármacos ainda não
aprovados para o mercado americano;
2. Novo sal de um medicamento já aprovado;
3. Nova formulação de medicamento já aprovado;
4. Nova associação de dois ou mais medicamentos;
5. Medicamentos já comercializados (novo fabricante);
6. Nova indicação para medicamento já comercializado.
Os medicamentos que se enquadram nos itens 2, 3 e 4 são denominados pelo CDER como
IMD (Incrementally Modified Drugs).A FDA permite que um novo fabricante produza um
medicamento que já está sendo produzido e comercializado por outro fabricante em duas
situações diferentes: permite que um novo fabricante produza um medicamento que já está sendo
produzido por outra empresa e permite o registro de medicamentos que estavam no mercado
antes da promulgação da Emenda Kefauver-Harris, em 1962. Como visto anteriormente, esta
Emenda estabeleceu os requerimentos de eficácia para o registro de novos medicamentos e foi
aplicada de forma retroativa para os medicamentos aprovados após a Lei dos Alimentos,
Medicamentos e Cosméticos de 1938 que, por sua vez, exigia a apresentação de dados de
segurança. Os medicamentos que entraram no mercado antes desta data (1938) são assumidos
como seguros e eficazes pela FDA, a menos que surjam evidências contrárias (NIHCM, 2002).
A partir da análise do potencial terapêutico, a revisão da NDA pode ser classificada
como padrão ou prioritária. Se o medicamento não apresentar nenhuma vantagem clínica sobre
os medicamentos já comercializados, o CDER denomina a revisão desta NDA como padrão. Para
atingir o status prioritário, o novo medicamento deve demonstrar avanço terapêutico de uma das
quatro formas (NIHCM, 2002):
1. Evidências de aumento da eficácia no tratamento, diagnóstico ou prevenção de uma doença;
2. Eliminação ou substancial redução de reações adversas que limitam o tratamento;

37
3. Aumento da adesão do paciente ao tratamento;
4. Evidências de segurança e eficácia para uma nova sub-população de pacientes.
Segundo as características químicas do medicamento e do seu perfil terapêutico, as NDA
podem ser organizadas segundo o “grau de inovação” apresentado pelo produto (NICHM, 2002):
Quadro 1- Ranking das Aplicações de Novos Medicamentos
(NDA) segundo critérios de classificação da FDA
Tipo de NDA Nível de inovação
NME prioritário Maior inovação
NME padrão
IMD prioritário
IMD padrão
Outros medicamentos Menor inovação
A definição de prioridade para a revisão da NDA foi estabelecida pela FDA em 1976 e até
1992 a agência usava três categorias para classificar as NDA segundo o avanço terapêutico: A, B
e C. Os medicamentos da classe A apresentavam ganho terapêutico significativo sobre os
medicamentos comercializados, os medicamentos da classe B apresentavam ganho terapêutico
moderado e os medicamentos da classe C apresentavam uma pequena vantagem ou nenhuma
vantagem sobre os medicamentos presentes no mercado. Este sistema foi aplicado de forma
retroativa para os medicamentos registrados a partir de 1962. Em 1992 a agência converteu este
sistema em duas categorias a partir das quais os medicamentos anteriormente classificados como
“A” ou “B” passaram a ter sua NDA classificada como “prioritária” e os medicamentos
classificados como “C” passaram a ter sua NDA classificada como “padrão” (DiMasi, 2000).
DiMasi (2000) destaca que, embora a significância clínica definitiva dos medicamentos
seja desconhecida durante o processo de revisão da NDA, já que outros possíveis usos e efeitos
do medicamento são desconhecidos até que sejam usados na prática clínica diária, há uma forte
correlação positiva entre a classificação da FDA e a significância clínica dos medicamentos.
A partir da solicitação de revisão da NDA a FDA possui um prazo de sessenta dias para
decidir se o dossiê apresentado pelo fabricante contém as informações necessárias para sua

38
revisão, podendo ser enviado para avaliação técnica (FDA, 2005d). O CDER faz a revisão das
NDAs classificadas como “padrão” em, no mínimo, dez meses ou aproximadamente 300 dias, já
para os medicamentos considerados “prioritários”, o processo de avaliação leva em média seis
meses (Meadows, 2002).
A equipe de revisores do CDER que faz a revisão da NDA varia conforme o tipo de
medicamento apresentado para registro, se for um medicamento indicado para tratamento de
câncer, a equipe responsável pertence à Divisão de Oncologia, a NDA de um medicamento
contraceptivo será encaminhada para revisão na Divisão de Medicamentos do Sistema
Reprodutor e Urológico, e assim por diante. A revisão da NDA é feita inicialmente por revisores
primários e em seguida por um grupo de supervisores. Os Diretores dos Departamentos
geralmente é que tomam a decisão final quando o medicamento que está sendo avaliado é uma
Nova Entidade Molecular e decidem também sobre a mudança de status de um medicamento de
venda sob prescrição médica para medicamento de venda livre (OTC). As outras situações são
decididas ao nível das Divisões do próprio CDER.
Para se certificar que o processo de revisão e aprovação da NDA será baseado em dados
atualizados sobre a segurança do medicamento, o CDER exige que o solicitante do registro
forneça dados sobre os estudos de segurança do produto em períodos determinados: quatro meses
após a entrada do pedido de revisão da NDA na FDA, após o envio da carta de aprovação da
NDA ao solicitante e em outros períodos, se necessário. O relatório enviado pelo solicitante deve
informar a ocorrência de novas reações adversas e alterações importantes na freqüência e na
gravidade de efeitos adversos já conhecidos.
Quanto às inspeções nos locais de fabricação do medicamento, quando uma NDA é
submetida para revisão no CDER, a mesma é cadastrada em um banco de dados que é
monitorado pela Divisão de Investigações Científicas da FDA que designa revisores de campo
para realização de inspeções e verificação da validade dos dados citados na NDA (FDA, 2004c).

39
Processo de Revisão da NDA
Aplicante da NDA (solicitante do registro)
NDA
NDA completa?
Enviada para solicitante
Revisão pelo CDER (Divisões)
Médica
Farmacologia
Química
Biofarmacêutica
Estatística
Microbiologia
Reuniões com Comitê Consultivo Reuniões com Aplicante
Revisão completa e aceitável?
Informações adicionais
Revisão pelo solicitante
Revisão da bula adequada?
Inspeção local adequada?
Aguardando resultado
satisfatório
NDA Action
Não
Não
Não Não
Sim
Sim Sim
Sim
Fonte: http://www.fda.gov/cder/handbook/irb.htm

40
7.2 AGÊNCIA EUROPÉIA DE MEDICAMENTOS (EMEA)
A Agência Européia de Medicamentos (EMEA) foi criada pelo Regulamento nº 2309/93
da Comunidade Econômica Européia (CEE), mas suas atividades só foram iniciadas em fevereiro
de 1995. A sede da agência está localizada em Londres, na Inglaterra.
A EMEA é uma das dezesseis agências especializadas (agências comunitárias) existentes
atualmente no âmbito da Comunidade Européia, organismos públicos criados por meio da
legislação européia que possuem personalidade jurídica própria e têm por objetivo a realização de
tarefas de natureza técnica, científica ou gerencial (Garattini & Bertelle, 2004):
O "triângulo institucional" formado pelo Conselho da União Européia, Parlamento
Europeu e Comissão Européia é responsável pela elaboração da legislação (diretivas,
regulamentos e decisões) que se aplica em toda a União Européia e, de forma geral, a Comissão
Européia propõe a legislação que é então adotada pelo Conselho e pelo Parlamento. No âmbito da
União Européia, a EMEA está subordinada à Diretoria Geral de Empresas da Comissão Européia
e não à Diretoria de Saúde Pública e as suas atividades são regulamentadas pela legislação
européia (Garattini & Bertelle, 2004).
A EMEA é composta por um conselho de administração e por um grupo técnico. O
conselho de administração é formado por dois representantes de cada Estado-Membro, dois
representantes da Comissão Européia e dois representantes indicados pelo Parlamento Europeu.
O grupo técnico é composto por dois comitês científicos, responsáveis pela elaboração dos
pareceres sobre questões relacionadas à avaliação dos produtos farmacêuticos para uso humano, o
Committee for Proprietary Medicinal Products (CPMP), e para uso veterinário, o Committee for
Veterinary Medicinal Products (CVMP) (Garattini & Bertelle, 2004).
O CPMP é formado por dois representantes de cada país que compõe a União Européia
(EU), indicados por seus governantes. A União Européia é atualmente formada por vinte e cinco
países: Alemanha, Bélgica, França, Itália, Luxemburgo, Países Baixos, Dinamarca, Irlanda,
Reino Unido, Grécia, Espanha, Portugal, Áustria, Finlândia, Suécia, Chipre, República Tcheca,
Estônia, Hungria, Letônia, Lituânia, Malta, Polônia, Eslováquia e Eslovênia (Europa, 2005)
Dentre as principais funções do CPMP estão a avaliação dos pedidos de registro de
medicamentos feitos pela Indústria Farmacêutica e a emissão do parecer, favorável ou não ao
registro do medicamento para a Diretoria Geral de Empresas da Comissão Européia. O CPMP

41
pode solicitar o apoio de consultores externos para a resolução de questões mais complexas
relativas aos medicamentos.
Registro de medicamentos na União Européia
Os procedimentos para registro de medicamentos na União Européia são denominados
procedimento centralizado e procedimento descentralizado. Por meio destes mecanismos os
fabricantes de medicamentos obtêm autorização para que seus produtos sejam comercializados
em mais de um país na União Européia. Quando o fabricante deseja comercializar seu produto
em apenas um país da União Européia, o procedimento escolhido será a solicitação de registro
nacional (European Commission, 2002).
O procedimento descentralizado é usado para a solicitação de registro da maior parte dos
medicamentos. Por meio deste procedimento uma empresa farmacêutica solicita a autorização
para registro de seu medicamento à autoridade sanitária oficial de determinado país da União
Européia e, obtendo o registro, a empresa pode solicitar a aprovação para a comercialização do
medicamento em outros países da Comunidade, baseando-se no princípio do reconhecimento
mútuo entre os países que compõe a União Européia. Quando um Estado-Membro não reconhece
o registro para comercialização de um medicamento registrado em outro país da Comunidade, a
EMEA assume o papel na arbitragem deste processo, cabendo à Comissão Européia tomar a
decisão final a partir do parecer emitido pela EMEA (European Commission, 2002).
O Procedimento Centralizado é realizado quando o fabricante solicita o registro do
produto diretamente à EMEA. Um registro aprovado por meio do procedimento centralizado
garante que o medicamento poderá ser comercializado em todos os países da União Européia.
Existe um grupo de medicamentos que só pode ser comercializado na União Européia se
registrado por meio do procedimento centralizado (Parlamento Europeu, 2004).
1. Medicamentos para uso humano que contenham uma substância ativa nova ainda não
autorizada na Comunidade Européia, e cuja indicação terapêutica seja o tratamento de uma das
seguintes patologias:
— síndrome de imunodeficiência adquirida,

42
— neoplasias,
— doenças neurodegenerativas,
— diabetes
2. Medicamentos desenvolvidos por meio de um dos seguintes processos biotecnológicos:
— tecnologia do DNA recombinante;
— expressão controlada da codificação de genes para proteínas biologicamente ativas em
procariontes e eucariontes, incluindo células mamíferas transformadas;
— métodos de hibridoma e de anticorpos monoclonais.
3. Medicamentos designados como medicamentos órfãos, conforme o Regulamento (CE) n.o
141/2000
4. Medicamentos veterinários destinados como potenciadores de rendimento a fim de promover o
crescimento dos animais tratados ou aumentar a sua produtividade.
Os medicamentos que não se enquadram nas categorias listadas acima podem obter
autorização de introdução no mercado concedida pela EMEA se o solicitante do registro
demonstrar que o medicamento constitui uma inovação significativa no plano terapêutico,
científico ou técnico, capaz de justificar uma concessão de registro comunitário (Parlamento
Europeu, 2004)
Quando o medicamento representa um grande interesse do ponto de vista da saúde
pública, principalmente quando se refere a uma inovação terapêutica, o requerente pode solicitar
um procedimento de avaliação acelerado para o seu produto, devendo o pedido ser devidamente
fundamentado. Se o CPMP aceitar o pedido, o prazo para emissão do parecer para a Comissão
Européia é reduzido de 210 dias para 150 dias (Parlamento Europeu, 2004).

43
Os fabricantes de medicamentos que desejam solicitar o registro de seus produtos na
EMEA devem entregar o dossiê do produto contendo informações referentes à qualidade,
segurança e eficácia do medicamento e estas informações devem estar organizadas em módulos
(referentes à qualidade, segurança e eficácia) que compõem o Documento Técnico Geral (CTD)
(Quadro 2) para o Registro de Medicamentos de Uso Humano (Comissão Européia, 2002).

44
Quadro 2-Documento Técnico Geral (CTD) para Registro de Medicamentos na EMEA
Qualidade Segurança Eficácia
1-Princípio Ativo: Informações sobre o fabricante, processo de obtenção da substância, critérios de aceitação de resultados, descrição do processo de validação; Caracterização da substância ativa: estrutura e isomerismo; Controle de qualidade: processos analíticos e estudos de estabilidade. 2- Dados sobre o Medicamento: Descrição e composição Desenvolvimento Farmacêutico: composição das formulações usadas nos ensaios clínicos e descrição dos perfis de dissolução destas formulações; Processo de produção: controle em processo, processo de validação, controle de qualidade dos excipientes e estudos de estabilidade do medicamento.
1-Sumário Não-Clínico: deve fornecer um resumo compreensivo dos resultados dos testes não-clínicos com os seguintes tópicos: Farmacologia: resultados farmacodinâmicos primários e secundários, segurança farmacológica, interações farmacodinâmicas do medicamento; Farmacocinética: métodos analíticos e dados sobre absorção, distribuição, metabolismo, excreção e interações farmacocinéticas do medicamento; Toxicologia: estudo de toxicidade de dose única, toxicidade de dose múltipla, carcinogenicidade, toxicidade reprodutiva e tolerância local. 2-Panorama dos Estudos Não-Clínicos Deve apresentar uma avaliação integrada e crítica dos dados farmacológicos, farmacocinéticos e toxicológicos obtidos a partir dos ensaios não-clínicos com o medicamento. Interpretação dos dados, relevância clínica dos resultados, relação entre os dados obtidos nos ensaios de segurança e os aspectos de qualidade do medicamento e implicações dos resultados não-clínicos para o uso seguro do medicamento em seres humanos.
1-Sumário Clínico: resumo detalhado da informação clínica obtida dos estudos clínicos: resultados dos estudos individuais, comparação dos resultados entre os estudos e persistência de eficácia e/ou efeitos de tolerância ao medicamento; 2-Panorama Clínico: documento que fornece as análises críticas, as conclusões e implicações dos dados apresentados no Sumário Clínico, as limitações e sucessos do processo de desenvolvimento do medicamento e os resultados dos estudos clínicos. Breve sumário do conhecimento científico existente que dá suporte à investigação do uso do medicamento para a indicação clínica proposta; metodologia adotada para o ensaio clínico; declaração de conformidade com as Boas Práticas Clínicas; Avaliação dos benefícios e riscos a partir dos estudos clínicos, informando como os dados de segurança e eficácia subsidiam a definição da dose e indicação terapêutica proposta; questões não resolvidas durante o processo de desenvolvimento do medicamento; fundamentos das informações contidas na bula;

45
O solicitante de registro de medicamento na EMEA é obrigado a fornecer também os
dados sobre a comercialização do medicamento em outros países, qualquer resultado novo ou
diferente sobre a segurança do medicamento e sobre as ações regulatórias relacionadas à
segurança do mesmo. Além destas informações, o solicitante deverá também apresentar no CTD
as conclusões sobre os riscos e benefícios do medicamento, quando o mesmo é comparado com
alternativas farmacológicas disponíveis para tratamento da doença ou com nenhum tratamento
farmacológico, quando esta for uma opção clinicamente aceitável.
No caso de medicamentos que contenham substâncias ativas presentes na composição de
medicamentos já comercializados, mas que ainda não tenham sido associadas para fins
terapêuticos, devem ser fornecidos os resultados dos novos ensaios pré-clínicos ou clínicos
relativos à associação (Parlamento Europeu, 2004).
As autoridades sanitárias responsáveis pela fiscalização das empresas produtoras dos
medicamentos registrados na EMEA são as autoridades dos Estados-Membro que concederam a
autorização de fabricação do medicamento. A inspeção nestas empresas pode ser realizada
durante o processo de avaliação do pedido de registro do medicamento.
Os critérios adotados pela agência européia para desenvolvimento e registro de
medicamentos na União Européia são influenciados atualmente pelo processo de harmonização
dos regulamentos técnicos sanitários, descrito anteriormente. Desta forma, os manuais elaborados
pela EMEA para orientação das empresas farmacêuticas contêm regras para a fabricação e
registro de medicamentos que já foram harmonizadas com outros países.
Etapas do processo de registro de medicamentos na EMEA:
O procedimento para registro de medicamentos na EMEA pode ser resumido nas
seguintes etapas (Garattini & Bertelle, 2004):
1- Etapa de Validação: até o 15º dia após o recebimento do pedido de registro o
secretariado da EMEA verifica se o dossiê apresentado pelo solicitante contém todas as
informações necessárias à avaliação técnica.
2- Avaliação do CPMP: dois membros do CPMP são selecionados como relatores
(indicados geralmente pelo fabricante e pelo próprio CPMP). Estes relatores são

46
responsáveis pela coordenação das atividades de avaliação do dossiê, pelo contato com
o fabricante, pela solicitação de informações necessárias durante o processo de
avaliação e pela apresentação do relatório de avaliação à equipe do CPMP. O CPMP
emite o parecer para a Comissão Européia geralmente 7 meses após a entrada do pedido
de registro na EMEA.
3- Decisão da Comissão Européia: a Comissão Européia tem três meses após o
recebimento do parecer do CPMP para decidir se concorda ou não com o Comitê. A
decisão é obtida por consenso entre os integrantes ou pelo voto da maioria
(aproximadamente 300 dias após a entrada do pedido de registro na EMEA).
4- Nos casos em que o pedido de registro é negado, a empresa pode entrar com recurso que
será avaliado pelo CPMP depois de terem sido indicados dois relatores, reiniciando o
processo.

47
Etapas do processo de avaliação para registro de medicamentos na EMEA
Após a autorização para registro, os resultados da avaliação do dossiê são resumidos em
um documento denominado Sumário de Características do Produto (SPC). Também é elaborado
um Relatório Europeu de Avaliação Pública (EPAR) para o medicamento registrado que fica
disponível na página eletrônica da EMEA.
Quando o fabricante é incapaz de fornecer dados suficientes sobre o medicamento que
pretende registrar (indicação clínica do novo medicamento é pouco conhecida e rara, os
conhecimentos científicos sobre a doença e o medicamento são insuficientes, etc) a EMEA pode
Validação de Dossiê Secretariado da EMEA
Dossiê incompleto devolvido
para solicitante
Dossiê completo encaminhado para
2 relatores
Apresentação do Relatório de
Avaliação para CPMP
Emissão do Parecer para Comissão Européia
Decisão da Comissão Européia
3 meses
10 m
eses

48
conceder uma aprovação temporária. Neste caso o fabricante assume o compromisso de fornecer,
durante o período de comercialização do produto, dados suficientes para a obtenção do registro
definitivo (Parlamento Europeu, 2004).
O registro sanitário de medicamentos na União Européia deve ser renovado cinco anos
após a concessão do registro e, a partir de então, possui validade ilimitada. Além disso, qualquer
autorização não utilizada durante três anos consecutivos, isto é, que não tenha dado origem à
introdução de um medicamento no mercado dos Estados-Membros durante este período, é
considerada caducada (Parlamento Europeu, 2004).

49
7.3 ADMINISTRAÇÃO NACIONAL DE MEDICAMENTOS, ALIMENTOS E
TECNOLOGIA MÉDICA (ANMAT)
A Administração Nacional de Medicamentos, Alimentos e Tecnologia Médica (ANMAT)
foi criada na Argentina por meio do Decreto Presidencial nº 1490, de 20/08/1992. Naquele
período foi realizada na Argentina uma série de atividades com vistas à revisão e adequação das
funções básicas do Estado e a criação da agência ocorreu em observância ao Decreto Presidencial
nº 1269/92, que estabeleceu as Políticas de Saúde na Argentina, denominadas Políticas
Substantivas (Argentina, 1992a). A quarta e última Política Substantiva do referido Decreto tem
como um dos seus objetivos:
“(...) adecuar la estructura de la SECRETARIA DE SALUD a fin de
posibilitar a dicho organismo el desarrollo de las líneas estratégicas
de transformación propuestas, favoreciéndose así la conformación de
un ente técnico con capacidad para liderar la salud en el desarrollo
con equidad, implementar la planificación estratégica en
normatización, en fiscalización y conducción superior, y garantizar
una efectiva acción sanitaria en todo el ámbito nacional, poniéndose
énfasis en el desarrollo de las acciones de promoción y protección
(ítem 4.1.1.)”.
A ANMAT é um órgão descentralizado, uma autarquia com autonomia financeira e suas
atividades possuem abrangência nacional. Está ligada administrativamente à Secretaria de
Políticas de Saúde e Fiscalização Sanitária do Ministério da Saúde e Ação Social da Argentina,
responsável pela elaboração das normas técnicas e científicas que norteiam as atividades da
agência.

50
ANMAT na hierarquia do Estado Argentino (ANMAT, 2005a)
As ações de controle e fiscalização da ANMAT são executadas por diferentes unidades e
compreendem drogas, produtos químicos, medicamentos, materiais para diagnóstico e
tecnologias médicas, alimentos industrializados e aditivos de alimentos, produtos de higiene e
cosméticos (Organograma 1) (ANMAT, 2005b)
O Instituto Nacional de Medicamentos é responsável pelo registro e controle de drogas,
medicamentos, reativos e elementos de diagnóstico, cosméticos e outros produtos usados em
medicina humana. Este controle é realizado por meio de estudos farmacotécnicos, biológicos,
farmacológicos e toxicológicos e compreendem as etapas de fabricação, fracionamento,
importação ou exportação, depósito e comercialização dos produtos. A Direção de Avaliação de
Medicamentos é responsável pela avaliação clínica dos medicamentos que se encontram em fase
de registro e daqueles que já estão sendo comercializados (Argentina, 2003).
ANMAT
Instituto Nacional de Medicamentos
Instituto Nacional de Alimentos
Direção de Avaliação de Medicamentos
Direção de Tecnologia Médica
Direção de Planificação e Relações Institucionais
Direção de Coordenação e Administração
Direção de Assuntos Jurídicos
Nação Argentina
� Poder Executivo
� Ministério da Saúde e Ação Social
� Secretaria de Políticas de Saúde e Fiscalização Sanitária
� ANMAT
Organograma 1- Estrutura da ANMAT

51
Registro de Medicamentos
O registro de medicamentos na Argentina é regulamentado atualmente pelo Decreto
150/92, que segue as definições estabelecidas na Lei 16.463, de 1964, conhecida como Lei dos
Medicamentos (Argentina, 1992b). A ANMAT adota duas listas de países de referência a partir
das quais são definidos os critérios para o processo de registro de medicamentos. As listas são
compostas pelos seguintes países:
Lista 1
Estados Unidos, Suíça, Japão, Suécia, Israel, Canadá,
Áustria, Alemanha, França, Reino Unido, Países Baixos,
Bélgica, Dinamarca, Espanha e Itália.
A empresa que deseja registrar um medicamento na ANMAT deverá entregar a
solicitação de registro de medicamento incluindo as seguintes informações (Argentina, 1992b):
a- Sobre o produto: nome proposto, fórmula do produto, forma farmacêutica, classificação
farmacológica (classificação internacional de medicamentos da OMS) e condições de
dispensação;
b- Informação técnica: método de controle, validade, método de produção (em conformidade
com as Boas Práticas de Fabricação vigentes) e dados sobre a biodisponibilidade do produto;
c- Informações que deverão estar contidas na embalagem.
d- Projetos de bulas: ações farmacológicas e terapêuticas atribuídas ao produto com indicações
clínicas precisas, advertências e precauções, interações medicamentosas e efeitos adversos,
posologia, doses máximas e mínimas, forma de administração e apresentações;
Lista 2
Austrália, México, Brasil, Cuba, Chile, Finlândia, Hungria,
Irlanda, China, Luxemburgo, Noruega e Nova Zelândia.

52
Os medicamentos registrados em pelo menos um dos países que compõem a Lista 1
poderão ser importados e registrados na Argentina e este registro será automático. O solicitante
do registro deve fornecer apenas os seguintes dados:
1- Autorização de registro em pelo menos um país da Lista 1;
2- Projetos de rótulos e etiquetas do produto;
3- Projetos de bula;
4- Dados de biodisponibilidade do produto.
Para as três situações descritas a seguir, o pedido de registro na ANMAT deverá observar
as exigências dos tópicos a, b, c e d, descritos anteriormente, e fornecer também a documentação
que comprove a eficácia e segurança do medicamento para o uso proposto:
1- Medicamentos a serem produzidos na Argentina e que são novidade no país;
2- Medicamentos importados de algum país da Lista 2, autorizado e comercializado no país
de origem e sem similares registrados na Argentina;
3- Medicamentos fabricados em países não incluídos na Lista 1 e 2 e não autorizados para
comercialização em nenhum país da Lista 1.
Os medicamentos importados a serem registrados na Argentina devem ser registrados e
comercializados nos países de origem. Quanto ao prazo para decisão sobre autorização de
registro, a partir da entrega de solicitação de registro do medicamento, o Ministério da Saúde e
Ação Social possui 120 dias para decidir sobre o pedido de registro. Sendo necessária a
verificação da planta onde o medicamento é produzido o prazo para avaliação do produto será
considerado a partir desta verificação. A renovação do registro de medicamento deve ser feita a
cada cinco anos (Argentina, 1992b). É considerado medicamento novo na ANMAT o
medicamento que satisfaz uma das condições apresentadas abaixo:
1- Para o qual não exista medicamento similar registrado e comercializado na Argentina;
2- Para o qual não exista similar registrado no mercado de pelo menos um dos países que
compõe a Lista 1 dos países de referência.

53
Desta forma, conforme estabelecido pelo Decreto 150/92, verifica-se que na Argentina
um medicamento é registrado como medicamento novo se não tiver sido registrado em nenhum
país que compõem a Lista 1. Se o medicamento já estiver registrado e for importado de um dos
países que compõem a Lista 1 será registrado na Argentina como medicamento similar.
Segundo Lucchese (2001) esta legislação foi conquistada pelas empresas nacionais, visto
que o país não reconhecia patentes na área farmacêutica até 1997. A legislação permitia agilidade
e competitividade às empresas domésticas, que podiam copiar rapidamente as inovações lançadas
em qualquer dos países mais desenvolvidos e colocá-las no mercado argentino. Atualmente a
Argentina reconhece patentes de medicamentos e os medicamentos registrados como similares na
ANMAT e que estão sob proteção patentária não podem ser fabricados por outras empresas até
que o prazo da patente seja expirado (Argentina, 1996).

54
7.4 AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA (ANVISA)
Regulação sanitária de medicamentos no Brasil - antecedentes
As primeiras regulamentações na área de medicamentos surgiram no Brasil no início do
século XX, período em que se faziam muitas denúncias sobre a qualidade dos medicamentos e
dos alimentos presentes no mercado nacional, como descrito por Monteiro Lobato em 1918:
“Em matéria de drogas nem é bom falar. Iodofórmio adulterado com
enxofre. Emetina fabricada com sais de quina. Quinino e aspirina
feitos com lactose. Óleos minerais e medicinais clarificados com ácido
sulfúrico impuríssimo, contendo arsênico. E, cúmulo, 914 em ampolas
que não passa de finíssimo fubá de milho amarelo. São Paulo virou o
paraíso da fraude bromatológica. Indefesa como está a cidade,
confiada a uns fiscais que fiscalizam para si, os desalmados
envenenam-se por todas as vias e amontoam fortunas colossais à custa
da saúde alheia. São duas coisas que, arre! Valem a pena: falsificar e
fiscalizar” (Lobato, 1964 apud Rozenfeld, 1998).
Este período é caracterizado pelo crescimento econômico do país, impulsionado pela
produção de café e o aquecimento do comércio internacional. As cidades portuárias receberam
atenção especial: era necessário que suas estruturas sanitárias estivessem em condições
adequadas, pois eram rotas de exportação de produtos para várias partes do mundo e porque
assim favoreciam a vinda de imigrantes para o aumento da força de trabalho. Mas os problemas
sanitários não foram resolvidos de imediato e epidemias, como a febre amarela, tornaram-se
sérias ameaças à população e à economia do país (Costa, 2004).
Oswaldo Cruz assume o comando da Diretoria Geral de Saúde Pública (DGSP) e em 1904
é aprovado o Decreto nº 5.156 que estabelecia o novo Regulamento dos Serviços Sanitários da
União, contendo normas para o controle sanitário no país. Na área de medicamentos foi
elaborada, em 1918, uma política de medicamentos oficinais e no Instituto Oswaldo Cruz
passaram a ser produzidos medicamentos para o controle de doenças endêmicas e epidêmicas,
como a quinina para o tratamento da malária (Costa, 2004). Em 1920 é criado o Departamento

55
Nacional de Saúde Pública (DNSP) e em 1923 foi editado um importante documento sanitário no
país, o Decreto nº 16.300, que permaneceu por muitos anos como o Regulamento Sanitário
Federal. Em relação ao controle sanitário sobre os medicamentos, o DNSP, por meio do referido
Decreto, tinha como competência (Costa, 2004:126).
“(...) a fiscalização de produtos farmacêuticos (...) expostos à venda e
são estabelecidas multas e penas de prisão inafiançáveis para os
falsificadores de alimentos e outros produtos sob o controle do DNSP,
como medicamentos e produtos biológicos.”
Ainda segundo a autora, a venda de preparados farmacêuticos, anti-sépticos e
desinfetantes, nacionais ou importados, só poderia ocorrer a partir da licença concedida pelo
DNSP. O Decreto não continha exigências sobre a eficácia e segurança dos produtos – o
fabricante detinha a credibilidade sobre estes atributos.
No que se refere ao controle da produção de medicamentos, o Decreto Federal nº
19.606/1931 estabeleceu as normas para o controle sanitário e para a atuação da Indústria
Farmacêutica no Brasil (Zubioli, 1992 apud Rozenfeld, 1998). Neste Decreto reafirma-se a
necessidade de licenciamento prévio à produção e comercialização de produtos farmacêuticos e
outros produtos de interesse da saúde quando fabricados por particulares (Costa, 2004). Em 1941
foi criado o Serviço Nacional de Fiscalização da Medicina (SNFM) por meio do Decreto 3.171 e
em 1942 são criadas as Comissões de Biofarmácia e a Comissão de Revisão da Farmacopéia.
Segundo Costa & Rozenfeld (2000) a Comissão de Biofarmácia foi responsável pela elaboração
de várias normas que restringiram a produção e a comercialização de produtos neste período.
Nas décadas de 30 e 40 houve um expressivo desenvolvimento do ramo químico-
farmacêutico no Brasil, que foi acompanhado da criação de um importante regulamento para a
Indústria Farmacêutica – o Decreto nº 20.397/46. Este Decreto apresentava normas para as
especialidades farmacêuticas, produtos oficinais e biológicos, produtos psicotrópicos e
entorpecentes, produtos químico-farmacêuticos, anti-sépticos e desinfetantes e normas para o
funcionamento dos estabelecimentos fabricantes destes produtos. Segundo Costa (2004), o
referido Decreto não apresentava exigências que dificultassem o licenciamento das
especialidades farmacêuticas, este era concedido desde que houvesse a legalização do

56
estabelecimento produtor e do responsável técnico. A licença para o medicamento tinha validade
de dez anos, fim dos quais deveria ser renovada. O processo de licenciamento era baseado no
Relatório Técnico do medicamento e não eram exigidas informações referentes às contra-
indicações, eficácia e segurança do produto.
Nas décadas de 60 e 70 o Serviço Nacional de Fiscalização da Medicina e Farmácia
(antigo SNFM) e a Comissão de Biofarmácia do Ministério da Saúde proscreveram ou
restringiram vários produtos presentes no mercado brasileiro.
Em 1971 foi criada a Central de Medicamentos (CEME) que tinha como funções a
regulação da produção e da distribuição de medicamentos em todo o país. A década de 70 foi
muito importante historicamente para ao campo da vigilância sanitária brasileira, pois neste
período foram criadas importantes normas, como as Leis 5.991/73 e 6.360/76. A Lei 5.991/73
(Brasil, 1973) revogou o Decreto Federal nº 19.606, de 1931, e estabeleceu o controle sanitário
do comércio de drogas, medicamentos, insumos farmacêuticos e correlatos. Esta Lei incorporou o
termo medicamento no lugar de especialidade farmacêutica (Costa, 2004).
Na reforma administrativa promovida pelo governo do presidente Ernesto Geisel, em
1976, o controle de produtos foi agregado ao controle dos portos, aeroportos e fronteiras e estas
atividades foram identificadas como pertencentes à área de vigilância sanitária. Neste período a
fiscalização do exercício profissional, um dos primeiros objetos de regulamentação e controle
sanitário estatal, passou a ser realizada pelos conselhos das respectivas profissões e um grande
número de estabelecimentos de prestação de serviços era objeto de vigilância sanitária estadual e
municipal (Lucchese, 2001).
A Lei 6.360, de 1976, estabeleceu normas sanitárias para medicamentos, drogas, insumos
farmacêuticos, cosméticos, produtos de higiene e perfumes, saneantes domissanitários e seus
elementos (embalagens e rotulagem), estabelecimentos produtores, meios de transporte e
propaganda. Introduziu também o termo vigilância sanitária em seu sentido amplo, não restrito
ao conceito de fiscalização, incluiu o elemento qualidade como componente da vigilância
sanitária de produtos e os fabricantes de medicamentos ficaram obrigados a informar à autoridade
sanitária as reações nocivas provocadas pelo uso de seus produtos (Brasil, 1976).
Os primeiros anos da década de 80 foram marcados pelo Movimento Sanitário que
apresentava propostas reformistas para o setor saúde no Brasil. Neste período, alguns
profissionais envolvidos no referido “Movimento” foram convocados para atuar em importantes

57
setores ligados à saúde e houve também uma aproximação entre os profissionais de vigilância
sanitária, os demais profissionais da saúde e as organizações de consumidores. No ano de 1986
ocorreram dois importantes eventos: a 8ª Conferência Nacional de Saúde e a Conferência
Nacional de Saúde do Consumidor, esta última tendo sido definida por Costa (2004) como o
“mais significativo e amplo debate sobres as questões do campo sanitário”. Na área dos
medicamentos foram publicadas duas importantes normas direcionadas para as substâncias e
produtos cujo uso deveria ser controlado: as Portarias DIMED nº 27 e nº 28, de 1986, que
estiveram em vigência até final década de 90.
No início da década de 90 o Governo do presidente Fernando Collor de Melo promoveu a
primeira etapa da Reforma do Estado brasileiro, caracterizada, dentre outros fatores, pela redução
do tamanho do Estado, desmonte das instituições de protecionismo e estatismo, liberalização do
comércio internacional e do investimento estrangeiro, desregulamentação do setor privado e
privatizações de várias empresas estatais. A Secretaria Nacional de Vigilância Sanitária (SNVS),
então vigente, foi também atingida pela onda de reformas – a prioridade era a agilidade
administrativa e o atendimento dos pedidos das empresas. O número de funcionários da SNVS
foi reduzido, ainda que insuficiente para o atendimento da demanda existente (Lucchese, 2001).
Em agosto de 1990 o Governo Federal lançou o Projeto Inovar e, segundo Costa & Rozenfeld
(2000) “(...) neste período foram liberados irregularmente registros para uma grande quantidade
de produtos, principalmente medicamentos, desconsiderando-se a importância das análises
técnico-científicas.”
Os primeiros anos da década de 90 foram bastante conturbados para SNVS. A mudança
freqüente dos Secretários não permitia a continuidade das ações que se pretendiam implementar
na área, denúncias de corrupção e o desgaste dos profissionais que trabalhavam na SNVS
revelavam a necessidade de uma discussão mais séria sobre quais seriam os rumos da vigilância
sanitária no Brasil. Em meio ao estado de crise, em setembro de 1990 foi publicada a Lei 8.080 –
Lei Orgânica da Saúde – que definiu a Vigilância Sanitária como:
“(...) um conjunto de ações capaz de eliminar, diminuir ou prevenir
riscos à saúde e de intervir nos problemas sanitários decorrentes do
meio ambiente, da produção e circulação de bens e da prestação de
serviços de interesse da saúde.”

58
Publicada em 26 de agosto de 1994, a Portaria GM nº 1.565 estabeleceu o Sistema
Nacional de Vigilância Sanitária, definindo a abrangência do sistema, a distribuição das
competências entre a União, estados e municípios, os procedimentos para a articulação política e
administrativa destas três esferas de governo no Sistema Único de Saúde e as bases para a
descentralização das ações de vigilância sanitária.
Em janeiro de 1995, no início do primeiro Governo Fernando Henrique Cardoso, a nova
direção da SNVS tinha como objetivo dar continuidade ao projeto de reestruturação completa da
vigilância sanitária federal e transformar a SNVS em uma autarquia de caráter especial. Diante
das pressões do seguimento produtivo e enfrentando sérios problemas, como a falsificação de
medicamentos, o projeto de criação de uma agência regulatória forte ganhou destaque naquele
período.
O processo de reformulação da SNVS foi concluído em janeiro de 1999 quando então foi
aprovada no Congresso Nacional a Medida Provisória nº 1.791 que resultou na Lei nº 9.782, de
26/01/1999. Esta Lei definiu o Sistema Nacional de Vigilância Sanitária e criou a Agência
Nacional de Vigilância Sanitária - ANVISA. A partir deste fato o Sistema Nacional de Vigilância
Sanitária ficou formado pelos seguintes órgãos: ANVISA, Conselho Nacional de Secretários
Estaduais de Saúde (CONASS), Conselho Nacional de Secretários Municipais de Saúde
(CONASEMS), Centros de Vigilância Sanitária Estaduais, do Distrito Federal e Municipais
(VISAS), Laboratórios Centrais de Saúde Pública (LACENS), Instituto Nacional de Controle de
Qualidade em Saúde (INCQS), Fundação Oswaldo Cruz (FIOCRUZ) e Conselhos Estaduais,
Distrital e Municipais de Saúde.
Momento atual - A Agência Nacional de Vigilância Sanitária (ANVISA)
A ANVISA, definida pela Lei 9.782 como uma agência regulatória – uma autarquia sob
regime especial, vinculada ao Ministério da Saúde e caracterizada pela independência
administrativa, estabilidade de seus dirigentes e autonomia financeira, é responsável pela
coordenação do Sistema Nacional de Vigilância Sanitária. Os órgãos de vigilância sanitária
estaduais ligados às secretarias estaduais de saúde são responsáveis pela coordenação das ações
de vigilância sanitária no âmbito estadual e executam as principais ações de fiscalização do
sistema nacional.Os órgãos de vigilância sanitária municipais são responsáveis pela execução das

59
ações sanitárias locais, como o licenciamento e o monitoramento de estabelecimentos comerciais,
de criadouros de animais em áreas urbanas e de prestadores de serviços de interesse da saúde.
No que se refere às competências da ANVISA, além da coordenação do Sistema, a Lei nº
9.782 (Brasil, 1999), em seu Artigo 7º, definiu uma série de outras atividades: estabelecer
normas, propor, acompanhar e executar as políticas, as diretrizes e as ações de vigilância
sanitária; autorizar o funcionamento de empresas de fabricação, distribuição, importação e
comercialização dos produtos relacionados na referida lei; conceder registros de produtos;
coordenar as ações de vigilância sanitária realizadas por todos os laboratórios que compõem a
rede oficial de laboratórios de controle de qualidade em saúde e fomentar o desenvolvimento de
recursos humanos para o sistema e a cooperação técnico-científica nacional e internacional.
O registro de medicamentos como atividade competente ao órgão federal de vigilância
sanitária já havia sido enfatizada pela Política Nacional de Medicamentos (PNM), aprovada pela
Portaria 3.916, de 30/10/1998, alguns meses antes da aprovação da Lei nº 9782. A PNM definiu o
registro de medicamentos como “ato privativo do órgão competente do Ministério da Saúde
destinado a conceder o direito de fabricação do produto no país” (MS, 2001).
Estrutura da ANVISA
A ANVISA é formada por uma Diretoria Colegiada, responsável por sua direção, uma
Procuradoria, uma Corregedoria, uma Ouvidoria, um Conselho Consultivo e unidades
especializadas responsáveis por diferentes atividades.
A unidade da ANVISA atualmente responsável pelo processo de avaliação dos pedidos de
registro de medicamentos novos é a Gerência de Medicamentos Novos, Pesquisa e Ensaios
Clínicos (GEPEC), ligada à Gerência Geral de Medicamentos (GGMED) que por sua vez está
subordinada à Direção da agência. A GGMED é composta ainda pela Gerência de Medicamentos
Genéricos, Gerência de Isentos, Fitoterápicos e Homeopáticos, Gerência de Medicamentos
Similares, Unidade de Produtos Biológicos e Hemoterápicos, Unidade de Farmacovigilância e
Unidade de Produtos Controlados.
A GEPEC possui uma série de competências dentro da agência (ANVISA, 2000):
planejar, coordenar, orientar e fomentar as atividades técnicas e operacionais relativas a produtos

60
sujeitos a vigilância sanitária em pesquisas envolvendo seres humanos; planejar, coordenar e
orientar as atividades técnicas e normativas relativas ao registro de medicamentos novos e
analisar e emitir parecer circunstanciado e conclusivo nos processos referentes a registro de
medicamentos novos, tendo em vista a identidade, qualidade, finalidade, atividade, eficácia,
segurança, risco, preservação e estabilidade dos produtos sob o regime de vigilância.
Registro de medicamentos novos na Anvisa
O registro de medicamentos novos na ANVISA é atualmente regulamentado pela
Resolução da Diretoria Colegiada (RDC) da ANVISA nº 136, de 29 de maio de 2003. O
Regulamento Técnico estabelecido por meio desta RDC, definido como “Regulamento Técnico
para Medicamentos Novos ou Inovadores com Princípios Ativos Sintéticos ou Semi-Sintéticos”,
deve ser aplicado para (ANVISA, 2003a):
1. Registro de medicamentos novos com princípios ativos sintéticos ou semi-sintéticos
associados ou não;
2. Registro de novas formas farmacêuticas, novas concentrações, nova via de administração
e indicações no País com princípios ativos sintéticos ou semi-sintéticos por parte de
empresas não detentoras de registro inicial daquele(s) principio(s) ativo(s);
3. Registro de produto resultante de:
3.1. Alteração de propriedades farmacocinéticas;
3.2. Retirada de componente ativo de produto já registrado;
3.3.Sais novos, isômeros, embora a entidade molecular correspondente já tenha sido
autorizada.
Na primeira parte do Regulamento são apresentadas as exigências anteriores à etapa de
registro, referentes à procedência do medicamento e aos ensaios clínicos com o mesmo. Se o
produto novo a ser registrado for de origem nacional o fabricante deve apresentar os protocolos
de pesquisa clínica e os resultados do andamento destas pesquisas. Para os medicamentos novos
fabricados fora do país (importados), cujos fabricantes desejam fazer os estudos clínicos de Fase
III no Brasil, é necessário a apresentação do protocolo de pesquisa e os resultados de seu
andamento. A terceira situação refere-se à realização de estudos de Fase III com medicamento de

61
origem estrangeira que terminará de ser fabricado no Brasil, neste caso o fabricante deverá
notificar a agência para a produção de lotes piloto.
No ato do protocolo de pedido de registro a empresa deverá apresentar, dentre outros
documentos, a cópia do protocolo da notificação da produção de lotes-piloto e o Certificado de
Boas Práticas de Fabricação e Controle (BPFC) emitidos pela ANVISA para a linha de produção
na qual o produto classificado como medicamento novo será fabricado, ou ainda, cópia do
protocolo de solicitação de inspeção para fins de emissão do referido certificado.
O Quadro 3, apresentado a seguir, contem um resumo das informações técnicas que
devem estar presentes no Relatório Técnico que o fabricante precisa fornecer para protocolar o
pedido de registro de medicamento novo junto à ANVISA.
Quadro 3 - Relatório Técnico para Registro de Medicamento Novo segundo RDC 136 de 23/05/2003
Informações técnicas do(s) princípio(s) ativo(s): fórmula estrutural; fórmula molecular; peso
molecular; sinonímia e referência completa; forma física do sal; ponto de fusão; solubilidade; rotação óptica
específica; propriedades organolépticas; possíveis isômeros; polimorfismo (descriminando as características do
polimorfo utilizado e de outros relacionados ao princípio ativo); relação sal/base e os excessos utilizados; espectro de
infravermelho da molécula; outras análises necessárias à correta identificação e quantificação da(s) molécula(s); rota e
descrição da síntese do fármaco (listar solventes utilizados, solventes residuais e respectiva concentração); estudos de
estabilidade do fármaco; farmacodinâmica (mecanismo de ação(s), posologia); dados de farmacocinética de cada
princípio ativo na formulação; relatório de produção; controle de qualidade de todas as matérias-primas utilizadas e do
produto acabado; material de embalagem primária.
Relatório de ensaios pré-clínicos: toxicidade aguda, sub-aguda e crônica, toxicidade reprodutiva, atividade
mutagênica, potencial oncogênico.
Relatório de ensaios clínicos (comprovação da eficácia terapêutica) acompanhado de referências bibliográficas
quando disponíveis. A apresentação destas informações deve seguir a ordem: estudos clínicos fase I, II, III. A Anvisa
poderá rever os dados dos estudos clínicos de Fase III para averiguar se as amostras e as diferenças de resultados entre
os grupos que receberam diferentes intervenções foram suficientes para a obtenção de significância estatística e
clínico-epidemiológica.
Prazo de validade: resultados do estudo de estabilidade acelerada de três lotes piloto utilizados nos testes e estudos
de estabilidade de longa duração em andamento;
Dados gerais: texto de bula e esboço de "layout" de rótulo e embalagem;
Relatório contendo o preço atualizado no varejo do medicamento em países onde ele já esteja sendo comercializado.
Se o produto novo ainda não for comercializado em outro país, encaminhar proposta de preço do produto no varejo (a
falta deste documento não impede a submissão, mas impede a aprovação final do produto).

62
Para os medicamentos que se enquadram em alguma das características apontadas no
Quadro 4, apresentado a seguir, o Regulamento estabelece outras exigências que deverão ser
observadas pelo solicitante de registro.
Quadro 4 – Exigências da RDC 136 segundo características dos Medicamentos Novos
Características dos medicamentos Exigências
Associações medicamentosas ou duas ou mais
apresentações em uma mesma embalagem para uso
concomitante ou seqüencial
Estudos de biodisponibilidade relativa dos princípios
ativos associados e cada princípio ativo isolado;
Ensaios clínicos controlados para cada indicação
terapêutica, provando que associações com as
mesmas doses tenham um efeito aditivo ou sinérgico
sem aumento dos riscos quando comparados com
cada princípio ativo isoladamente, ou que a
associação com dose menor de pelo menos um dos
princípios ativos obtenha o mesmo benefício com
riscos iguais ou menores quando comparados com
uma associação com doses conhecidas.
Estudos que demonstrem que a associação previne o
advento de resistência microbiana quando se tratar de
antibióticos. São aceitas até no máximo 3 princípios
ativos na mesma formulação por apresentação oral ou
injetável.
Novas formas farmacêuticas, concentrações, nova via de administração e indicações no país
com princípios ativos sintéticos ou semi-sintéticos por parte de empresas não detentoras
de registro inicial daquele(s) principio(s) ativo(s).
Resultados dos estudos de Fase III - empresas que
descobrirem uma nova indicação terapêutica no país
para um fármaco registrado por uma outra empresa,
na mesma concentração e mesma forma
farmacêutica;
Resultados dos estudos de Fase II e III - empresas que
descobrirem uma nova concentração, e/ou forma
farmacêutica, e/ou via de administração no País para a
mesma indicação terapêutica para um fármaco
registrado por uma outra empresa (estes estudos estão
dispensados, sendo substituídos pela prova de
biodisponibilidade relativa quando estiverem dentro
da faixa terapêutica já aprovada);

63
Os fabricantes ou seus representantes que pretendem importar medicamentos novos, além
dos dispositivos anteriores, terão que apresentar outros documentos e informações dentre os quais
podem ser destacados:
a) Especificar a fase em que o medicamento será importado: produto terminado, produto a granel
ou na embalagem primária;
b) Apresentar autorização da empresa fabricante do medicamento para o registro, representação
comercial e uso da marca no Brasil, quando cabível;
c) Cópia do Certificado de Boas Práticas de Fabricação e Consumo (BPFC), emitido pela
ANVISA para a empresa fabricante e atualizado por linha de produção. No caso da ANVISA
ainda não ter realizado inspeção na empresa fabricante é aceito o comprovante do pedido de
inspeção sanitária, acompanhado do certificado de boas práticas de fabricação de produtos
farmacêuticos por linha de produção, emitido pelo órgão responsável pela Vigilância Sanitária do
país fabricante. A ANVISA pode efetuar a inspeção da empresa fabricante no país ou bloco de
origem, conforme legislação específica;
d) Comprovante de registro emitido pela autoridade sanitária de um país onde esteja localizada a
empresa e o respectivo texto de bula;
e) Metodologia de controle de qualidade físico-química, química, microbiológica e biológica a
ser realizado pelo importador, de acordo com a forma farmacêutica, do produto terminado, granel
ou na embalagem primária.
No que se refere ao item “d”, citado acima, convém destacar que a Lei 6360, de 1976, em
seu Artigo 18, estabelece que o registro de medicamentos e insumos farmacêuticos de
procedência estrangeira depende também da comprovação de registro no país de origem, onde foi
fabricado o medicamento. Na impossibilidade do cumprimento da referida exigência, deve ser
apresentada comprovação de registro em vigor, emitida pela autoridade sanitária do país onde é
comercializado o medicamento ou autoridade sanitária internacional, e aprovado em ato próprio
da Agência Nacional de Vigilância Sanitária do Ministério da Saúde.

64
Etapas para o Registro de Medicamentos Novos
Convém destacar que a relação de medicamentos novos analisada a seguir foi registrada
entre os anos de 2000 e 2002 quando ainda estava em vigência a Instrução Normativa nº 01 da
SNVS, de 30/09/1994. Entretanto, o processo de registro de medicamentos novos na ANVISA,
descrito neste trabalho, teve como base a norma atual, a RDC nº 136 da ANVISA, de 29/05/2003,
O processo para registro de medicamento novo na ANVISA inicia-se quando o solicitante
protocola o pedido de registro na ANVISA. O § 3º do Artigo 12, da Lei 6.360, de 23 de setembro
de 1976, estabelece que o registro será concedido no prazo máximo de 90 (noventa) dias, a contar
da data de entrega do requerimento, salvo nos casos de inobservância da referida Lei ou de seus
regulamentos.
As etapas referentes a este processo não estavam disponíveis na página eletrônica da
agência no período em que a pesquisa foi realizada e, desta forma, para melhor compreensão e
descrição do modelo de registro de medicamentos novos adotado atualmente no Brasil foi
realizada uma entrevista com um importante ator neste processo – o atual Gerente da unidade da
ANVISA responsável pelo registro de medicamentos novos, a Gerência de Medicamentos Novos,
Pesquisa e Ensaios Clínicos (GEPEC), Sérgio de Andrade Nishioka. As perguntas feitas ao
entrevistado e as informações fornecidas pelo mesmo estão apresentadas no Anexo 1.
A partir das informações obtidas na entrevista foi elaborada uma representação
esquemática do processo de registro de medicamentos novos na ANVISA, apresentada a seguir:

65
Entrada do pedido de registro
(Protocolo da ANVISA)
GEPEC
O registro de medicamentos tem validade de cinco anos no Brasil. A renovação do
registro deve ser requerida no primeiro semestre do último ano do qüinqüênio de validade,
considerando-se automaticamente revalidado, independentemente de decisão, se esta não tiver
sido proferida até a data do término da validade do registro (Brasil,1976). O §8º do Artigo 12, da
Análise Farmacotécnica
Realizada por Técnicos da própria
GEPEC
Análise de Segurança e Eficácia
Realizada por Consultores externos
Pareceres avaliados pela
GEPEC
Parecer da GEPEC
GGMED e Diretoria da Agência Decisão sobre o registro do
medicamento

66
Lei 6360, de 1976, estabelece que o registro do produto que não for industrializado no primeiro
período de validade (5 anos) não será revalidado.
A RDC 136, de 2003, estabelece ainda que para a renovação do registro todas as
empresas deverão apresentar documento comprobatório de venda do produto no período de
vigência do registro, os números das notas fiscais e a relação de estabelecimentos compradores
em um máximo de 3 (três) notas por forma farmacêutica.

67
8. REGISTRO DE MEDICAMENTOS NOVOS NAS QUATRO AGÊNCIAS:
ALGUMAS CONSIDERAÇÕES
A descrição dos processos para registro de medicamentos novos nas quatro autoridades
sanitárias, apresentada até aqui, permite que sejam feitas algumas comparações relacionadas à
organização estrutural das autoridades, à definição de medicamento novo adotada e ao processo
de registro. Os dados obtidos estão apresentadas nos Quadros 5, 6 e 7 a seguir.
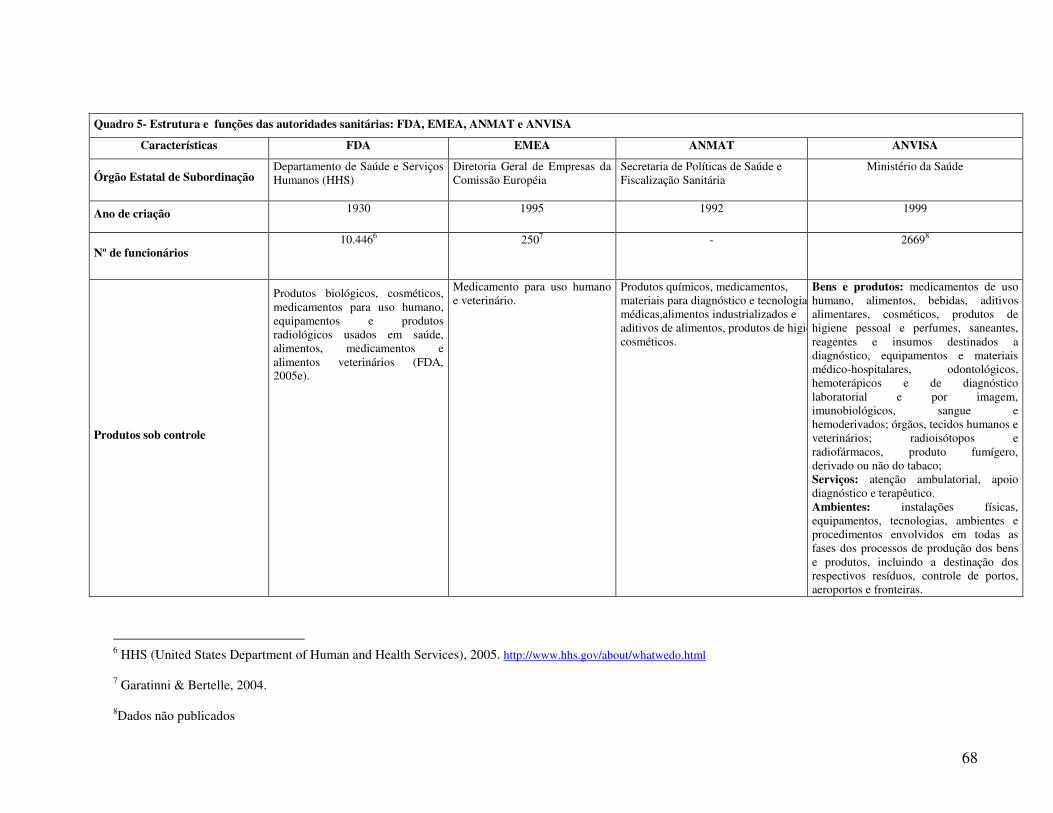
68
Quadro 5- Estrutura e funções das autoridades sanitárias: FDA, EMEA, ANMAT e ANVISA
Características FDA EMEA ANMAT ANVISA
Órgão Estatal de Subordinação Departamento de Saúde e Serviços Humanos (HHS)
Diretoria Geral de Empresas da Comissão Européia
Secretaria de Políticas de Saúde e Fiscalização Sanitária
Ministério da Saúde
Ano de criação 1930 1995 1992 1999
Nº de funcionários 10.4466
2507 - 26698
Produtos sob controle
Produtos biológicos, cosméticos, medicamentos para uso humano, equipamentos e produtos radiológicos usados em saúde, alimentos, medicamentos e alimentos veterinários (FDA, 2005e).
Medicamento para uso humano e veterinário.
Produtos químicos, medicamentos, materiais para diagnóstico e tecnologias médicas,alimentos industrializados e aditivos de alimentos, produtos de higiene e cosméticos.
Bens e produtos: medicamentos de uso humano, alimentos, bebidas, aditivos alimentares, cosméticos, produtos de higiene pessoal e perfumes, saneantes, reagentes e insumos destinados a diagnóstico, equipamentos e materiais médico-hospitalares, odontológicos, hemoterápicos e de diagnóstico laboratorial e por imagem, imunobiológicos, sangue e hemoderivados; órgãos, tecidos humanos e veterinários; radioisótopos e radiofármacos, produto fumígero, derivado ou não do tabaco; Serviços: atenção ambulatorial, apoio diagnóstico e terapêutico. Ambientes: instalações físicas, equipamentos, tecnologias, ambientes e procedimentos envolvidos em todas as fases dos processos de produção dos bens e produtos, incluindo a destinação dos respectivos resíduos, controle de portos, aeroportos e fronteiras.
6 HHS (United States Department of Human and Health Services), 2005. http://www.hhs.gov/about/whatwedo.html 7 Garatinni & Bertelle, 2004. 8Dados não publicados
69
Unidade responsável pela avaliação do dossiê para registro de medicamento novo
Centro de Avaliação e Pesquisa de Medicamentos (CDER)
Centro de Avaliação e Pesquisa de Produtos Biológicos (CBER)
Comitê de Produtos Farmacêuticos (CPMP)
Instituto Nacional de Medicamentos e
Direção de Avaliação de Medicamentos
Gerência de Medicamentos Novos, Pesquisa e Ensaios Clínicos (GEPEC)
Profissionais responsáveis pela avaliação do dossiê
Avaliadores do CDER Consultores externos
Técnicos dos Comitês Consultores Externos
_ Profissionais da GEPEC Consultores externos

70
Em relação às características dos órgãos sanitários apresentadas no Quadro 5 algumas
considerações importantes podem ser feitas. As atividades da EMEA são reguladas pelo órgão
responsável pela direção das empresas no âmbito da União Européia, a Diretoria Geral de
Empresas, e não pelo órgão responsável pela área de saúde, a Diretoria Geral de Saúde e Proteção
do Consumidor. Como a agência européia tem por função o controle sanitário dos medicamentos
que entram na Comunidade Européia seria mais apropriado, do ponto de vista da saúde pública,
que a EMEA estivesse subordinada aos interesses da Diretoria Geral de Saúde e Proteção do
Consumidor ao invés de estar ligada à Diretoria Geral de Empresas. Garattini (2005) refere-se a
este fato como uma anomalia que levanta a suspeita de que os produtos farmacêuticos têm maior
importância para o mercado do que para a saúde de quem os consome. Este autor refere-se ainda
ao fato da menor transparência desta agência em relação à FDA no que diz respeito à
disponibilização das informações sobre o pedido e análise dos registros. Outra crítica é quanto à
não exigência de estudos comparativos para a determinação do lugar que o medicamento ocupa
no arsenal terapêutico.
A FDA é o órgão sanitário mais antigo dentre os que foram analisados, tendo sido criada
em 1930, enquanto os demais foram criados na década de 90. Os países da Comunidade Européia
contam com órgãos sanitários nacionais mais antigos, e no Brasil, apesar de ter existido órgãos
responsáveis pelo controle sanitário anteriores à ANVISA, como a Secretaria Nacional de
Vigilância Sanitária (SNVS), a agência atual foi criada há menos de uma década.
No que se refere ao campo de atuação dos órgãos, a ANVISA apresenta maior número de
atividades sob sua responsabilidade, regulando produtos e bens, serviços e ambientes
relacionados à saúde. Entretanto vale salientar que a ANVISA, na área de medicamentos, é
responsável pela regulação das atividades relacionadas ao ciclo dos medicamentos (desde a
pesquisa até a comercialização), incluindo a concessão de registro, a autorização de
funcionamento das empresas e o seguimento pós-comercialização. Mas, com exceção da inspeção
internacional das empresas produtoras de medicamentos que é de responsabilidade da ANVISA,
as atividades de inspeção de ambientes e fiscalização sanitária das empresas situadas no Brasil
estão sob a responsabilidade dos órgãos estaduais e municipais de vigilância sanitária (Brasil,
1999).
As atividades de inspeção sanitária são realizadas no âmbito da Comunidade Européia
pelas autoridades sanitárias dos países que fornecem as informações necessárias ao

71
desenvolvimento das atividades na EMEA. A FDA é responsável pela inspeção dos
estabelecimentos envolvidos nas etapas de pesquisa, desenvolvimento e produção de
medicamentos, contando ela própria com profissionais específicos para o exercício destas
atividades. O Instituto Nacional de Medicamentos da ANMAT é o órgão responsável pela
fiscalização da produção, distribuição e venda dos medicamentos na Argentina.

72
Quadro 6- Definições para medicamento novo adotadas pela FDA, EMEA, ANMAT e ANVISA.
FDA EMEA ANMAT ANVISA
NDA para medicamentos como:
Novas Entidades Moleculares (NMEs): Novo sal de um medicamento já aprovado Nova formulação de medicamento já aprovado Nova associação de dois ou mais medicamentos; Medicamentos já comercializados (novo fabricante) Nova indicação para medicamento já comercializado;
Substância química, biológica ou radiofarmacêutica ainda não autorizada como um produto medicinal na União Européia; Isômero, mistura de isômeros, um complexo, um derivado ou um sal de uma substância já autorizada como um produto medicinal na União Européia, mas que apresente diferenças relacionadas à segurança e eficácia. Substância biológica previamente autorizada como um produto medicinal na União Européia, mas que apresente diferença na estrutura molecular, na matéria prima ou no processo de fabricação; Substância radiofarmacêutica (radionucleotídeo ou ligante) ainda não autorizada como um produto medicinal na União Européia (European Commission Enterprise Directorate-General, 2004).
Medicamento não registrado em nenhum país que compõem a Lista 1 dos países de referência adotada pela ANMAT.
Medicamentos com novo princípio ativo sintético ou semi-sintético associado ou não; Novas formas farmacêuticas, concentrações, via de administração e indicações no país com princípios ativos sintéticos ou semi-sintéticos por parte de empresas não detentoras de registro inicial daquele(s) principio(s) ativo(s).
Produto resultante de alteração de propriedade farmacocinética, retirada de componente ativo de produto já registrado, sais novos, isômeros, embora a entidade molecular correspondente já tenha sido autorizada.
As informações apresentadas no Quadro 6 revelam que as definições para medicamentos
novos adotadas pelas agências sanitárias são abrangentes, incluindo desde medicamentos com
novas entidades moleculares até medicamentos com fármacos já comercializados.
A definição adotada pela FDA merece alguns comentários: para serem registrados na
FDA os medicamentos precisam ter suas NDAs submetidas para a avaliação pelo CDER e,
conforme descrito anteriormente, estas NDAs são classificadas segundo o tipo químico e o
potencial terapêutico dos medicamentos, ou seja, os medicamentos são classificados segundo a
inovação que apresentam. A informação sobre esta classificação é fornecida pela FDA em sua
página eletrônica, juntamente com as informações sobre o registro.
A classificação de medicamento novo adotada pela EMEA e pela ANVISA é também
abrangente, incluindo os medicamentos denominados me too, versões modificadas de produtos já

73
comercializados. Entretanto, diferentemente da FDA, estas agências não fornecem informações
em suas páginas eletrônicas sobre a classificação dos medicamentos novos registrados.
A ANMAT, por sua vez, não possui uma classificação em termos químicos para os
medicamentos novos. O conceito de medicamento novo adotado não é justificável em termos
sanitários, pois o fato do medicamento estar registrado em um país considerado “rigoroso” pela
ANMAT não representa nenhuma garantia de que o medicamento é seguro e eficaz e mais, que
deva realmente ser registrado e consumido pela população argentina.
No que se refere à ANVISA, o registro de medicamento me too como medicamento novo
tem gerado alguns questionamentos e, neste sentido, a ANVISA disponibilizou em sua página
eletrônica, em junho de 2004, seu posicionamento quanto ao registro de medicamentos novos
considerados me too (ANVISA, 2004). Neste documento é declarada a dificuldade e mesmo a
impossibilidade, em certas situações, de se classificar um medicamento como me too no
momento de seu registro:
“(...) alguns atributos que permitiriam que se fizesse essa qualificação
só podem ser verificados depois da comercialização e utilização em
grande escala do produto. Entre esses atributos podem ser citadas a
ocorrência ou não de eventos adversos raros, a identificação de
subgrupos de indivíduos que potencialmente se beneficiariam do novo
medicamento de forma diferenciada e a descoberta de novas
indicações terapêuticas para esse medicamento em particular (...) a
dificuldade de se classificar um produto como me too no momento de
seu registro impossibilita o estabelecimento de regras que regulem o
registro de medicamentos sob tal ótica.”
Por meio do referido documento a agência enfatiza ainda que a legislação brasileira
vigente não subsidia o indeferimento de pedido de registro de medicamentos novos pela
possibilidade de serem classificados como me too.
É importante considerar os argumentos apontados pela agência no sentido de que alguns
efeitos dos medicamentos apenas serão percebidos com o uso prolongado e por um grande

74
número de indivíduos. Entretanto, quando se considera o número de medicamentos e
apresentações presentes no mercado brasileiro e se verifica que a oferta é bastante significativa, a
restrição do registro de medicamentos me too deveria ser considerada ou minimamente estes
produtos deveriam ser classificados como tais durante o registro. Garattini (2005) considera
importante que os critérios de aprovação do registro de medicamentos levem em consideração
não apenas a qualidade, a segurança e a eficácia dos medicamentos, mas também que apresentem
maior vantagem terapêutica, seja em termos de maior atividade, menor toxicidade, possibilidade
de maior adesão ou menor preço.

75
Quadro 7 – Algumas características do processo de registro de medicamentos novos na FDA, EMEA, ANMAT e ANVISA
Características FDA EMEA ANMAT ANVISA
Normas Legais vigentes atualmente
Lei Federal de Alimentos, Medicamentos e Cosméticos (1938)
Emenda Kefauver-Harris(1962)
Regulação do Conselho Europeu nº 2309/936, Diretiva 93/41/EEC e normas complementares
Lei 16.463 Decreto 150 de 1992
Lei 6.360 de 23/09/1976 RDC da ANVISA nº 136 29/05/03
Verificação do dossiê antes da avaliação técnica
60 dias para verificar se o dossiê está completo
Etapa de validação do dossiê
Não consta informação Não ocorre
Inspeção nos locais de fabricação durante a avaliação do pedido de registro
Divisão de Investigações Científicas (revisores de campo) – validade dos dados contidos na NDA
Inspetores das autoridades sanitárias dos Estados-Membro
Quando necessário ocorre a verificação da planta onde o medicamento é produzido
Não consta informação nas fontes pesquisadas
Prazo legal para decisão sobre o registro
6 meses -NDA Prioritária
10 meses – NDA Padrão
210 dias para emissão do parecer para a Comissão
300 dias para decisão da Comissão
120 dias 90 dias
Registro acelerado de medicamentos
Aprovação de NDA sem resultados clínicos conclusivos (estudos em fase clínica II)
Medicamentos novos para patologias graves
Inovações terapêuticas -revisão do dossiê em 150 dias
Registro automático para medicamentos registrados em pelo menos 1 país da Lista 1 dos países de referência.
Não é previsto

76
Renovação do registro de medicamento novo
O registro não precisa ser renovado.
Cinco anos após o registro. A partir de então tem validade ilimitada.
A cada 5 anos A cada 5 anos
Exigências de dados de segurança do produto no período pós- registro
Dados atualizados sobre segurança: 120 dias após submissão da NDA, após envio da carta de aprovação e em outros períodos.
Dados sobre comercialização em outros países e ações regulatórias sobre a segurança do produto
Riscos e benefícios comparados a outros medicamentos ou a nenhum tratamento farmacológico
Não constam nas fontes pesquisadas
Na renovação do registro a empresa deve apresentar os estudos de Fase IV (se houver) e também os dados de farmacovigilância. Estes últimos podem ser requisitados pela ANVISA antes do prazo para renovação.

77
As informações apresentadas no Quadro 7 revelam algumas diferenças importantes no
processo de registro de medicamentos novos nas quatro agências estudadas. Quanto à inspeção
dos locais de produção dos medicamentos, durante o período de avaliação do dossiê para registro,
verificou-se que esta etapa não consta nas normas brasileiras analisadas. Conforme visto
anteriormente, ao protocolar o pedido de registro de medicamento novo, a empresa deve
apresentar o Certificado de Boas Práticas de Fabricação e Controle (BPFC), emitido pela
ANVISA, para a linha de produção na qual o produto será fabricado ou a cópia do protocolo de
solicitação de inspeção para emissão do Certificado. Mas não está prevista, como nas normas da
FDA e da EMEA, a realização de inspeção durante a análise do dossiê.
O prazo legal para concessão do registro também merece destaque. Dentre as quatro
agências a ANVISA possui legalmente o menor prazo para decidir sobre a aprovação ou não do
registro, 90 dias, seguida da ANMAT com 120 dias.
O processo acelerado de registro de medicamentos não é previsto legalmente no Brasil.
Em fevereiro de 2004 a GEPEC disponibilizou na página eletrônica da ANVISA seu
posicionamento quanto ao tema. Segundo a GEPEC, tal mecanismo de aprovação exige
acompanhamento rigoroso, uma vez que é condicionado à apresentação de evidências adicionais
de eficácia e segurança e, atualmente, a ANVISA não está estruturada e organizada para realizar
este tipo de monitoramento. Considerando os aspectos relacionados à segurança dos
medicamentos e os argumentos apresentados pela GEPEC, é realmente mais adequado à
realidade brasileira que este tipo de procedimento não esteja sendo realizado na ANVISA.

78
9. MEDICAMENTOS NOVOS REGISTRADOS NA ANVISA 2000-2002
Os dados apresentados no Quadro 8 (Anexo 2) referentes aos 49 medicamentos, marcas,
ano de registro na ANVISA, ano de comercialização no Brasil, fabricante, classificação de
medicamento novo segundo ANVISA e potencial terapêutico segundo FDA foram obtidos do
estudo realizado por Reis (2004). As indicações terapêuticas aprovadas para comercialização
foram obtidas nas bulas dos medicamentos por meio de consulta às páginas eletrônicas das
empresas fabricantes, onde também foram obtidos os nomes dos países sede destas empresas.
Todos os medicamentos foram comercializados até o segundo ano após o registro. Os gráficos
apresentados a seguir foram construídos com base nos dados contidos no Quadro 8.
O Gráfico 1 revela que do total de 49 medicamentos novos registrados na ANVISA a
maior parte (21) foi produzida por empresas farmacêuticas com sede nos Estados Unidos. O
número de empresas com sede no Brasil (8) se iguala ao número de empresas com sede na Suíça
e o restante dos medicamentos (12) é fabricado por empresas com sede em outros países
europeus. Estes números confirmam o destaque americano e europeu no setor farmacêutico e a
importância que estas empresas multinacionais possuem no mercado farmacêutico brasileiro.
Gráfico 1 - Distribuição de medicamentos segundo país sede
do fabricante
0
5
10
15
20
25
Dinam
arca
Holan
da
Franç
a
Suécia
Alem
anha
Suíça
Brasil
EUA
Países sede do fabricante
Núm
ero
de m
edic
am
ento
s
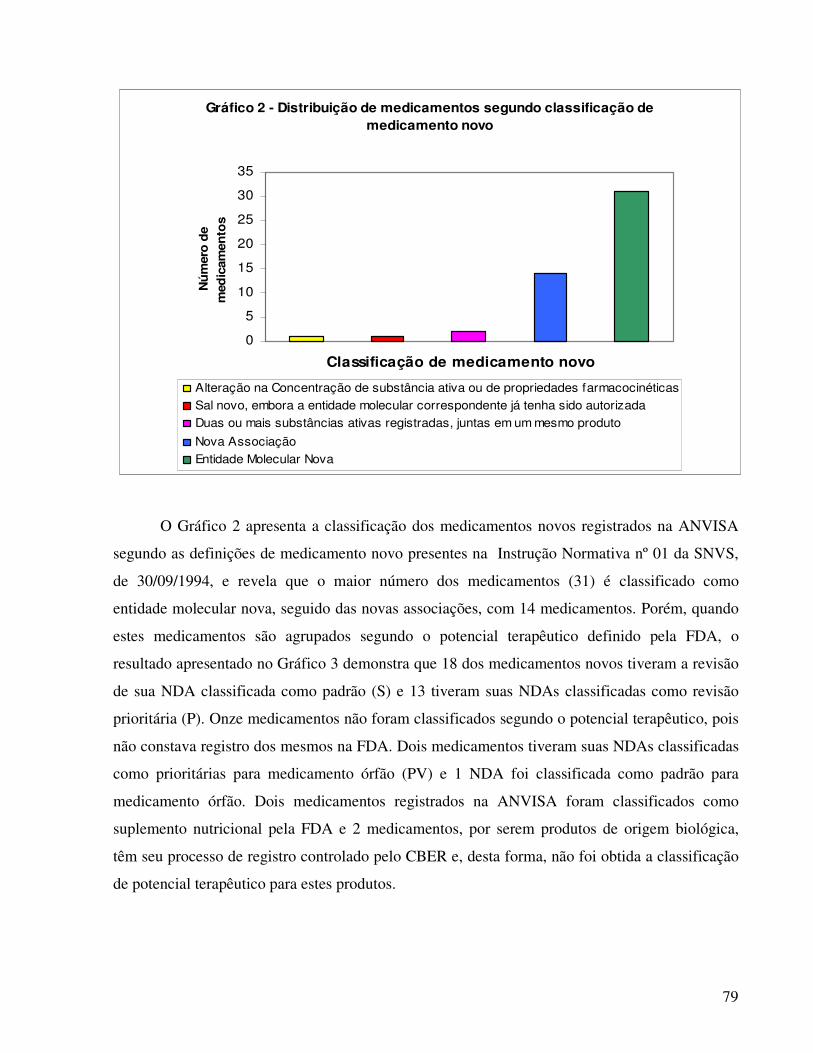
79
Gráfico 2 - Distribuição de medicamentos segundo classificação de
medicamento novo
0
5
10
15
20
25
30
35
Classificação de medicamento novo
Nú
mero
de
med
icam
en
tos
Alteração na Concentração de substância ativa ou de propriedades farmacocinéticas
Sal novo, embora a entidade molecular correspondente já tenha sido autorizada
Duas ou mais substâncias ativas registradas, juntas em um mesmo produto
Nova Associação
Entidade Molecular Nova
O Gráfico 2 apresenta a classificação dos medicamentos novos registrados na ANVISA
segundo as definições de medicamento novo presentes na Instrução Normativa nº 01 da SNVS,
de 30/09/1994, e revela que o maior número dos medicamentos (31) é classificado como
entidade molecular nova, seguido das novas associações, com 14 medicamentos. Porém, quando
estes medicamentos são agrupados segundo o potencial terapêutico definido pela FDA, o
resultado apresentado no Gráfico 3 demonstra que 18 dos medicamentos novos tiveram a revisão
de sua NDA classificada como padrão (S) e 13 tiveram suas NDAs classificadas como revisão
prioritária (P). Onze medicamentos não foram classificados segundo o potencial terapêutico, pois
não constava registro dos mesmos na FDA. Dois medicamentos tiveram suas NDAs classificadas
como prioritárias para medicamento órfão (PV) e 1 NDA foi classificada como padrão para
medicamento órfão. Dois medicamentos registrados na ANVISA foram classificados como
suplemento nutricional pela FDA e 2 medicamentos, por serem produtos de origem biológica,
têm seu processo de registro controlado pelo CBER e, desta forma, não foi obtida a classificação
de potencial terapêutico para estes produtos.
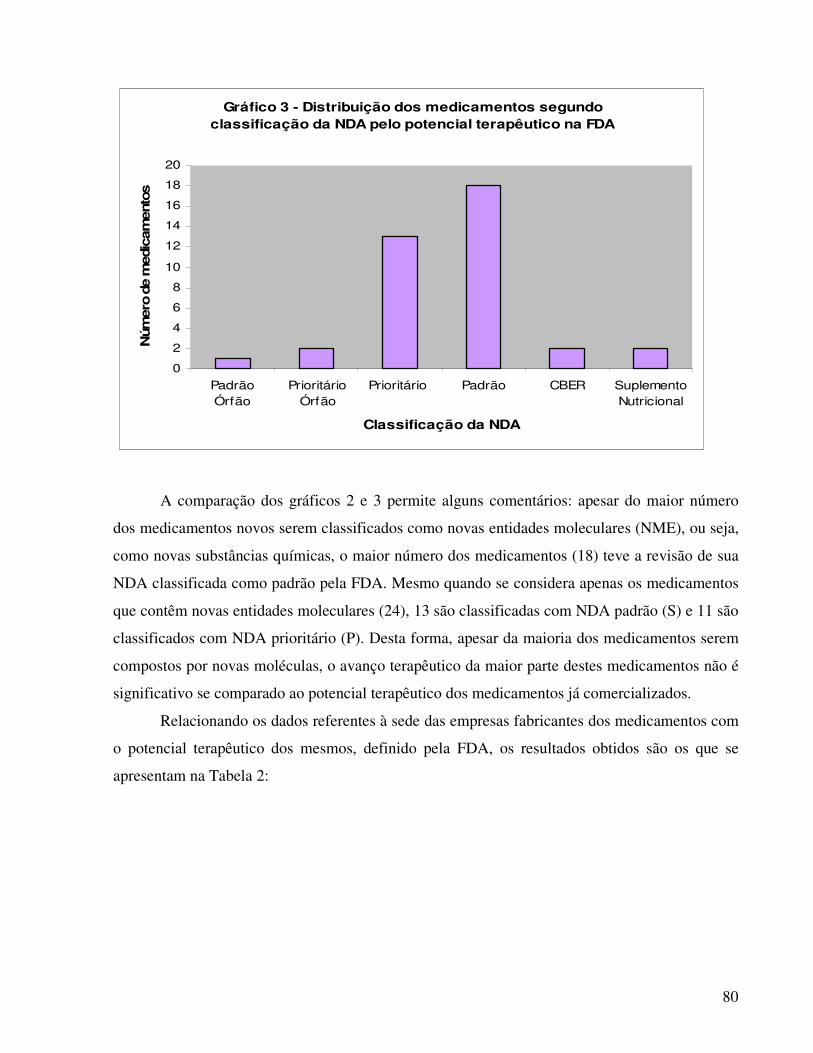
80
Gráfico 3 - Distribuição dos medicamentos segundo
classificação da NDA pelo potencial terapêutico na FDA
0
2
4
6
8
10
12
14
16
18
20
PadrãoÓrfão
PrioritárioÓrfão
Prioritário Padrão CBER SuplementoNutricional
Classificação da NDA
Núm
ero
de m
edic
am
ento
s
A comparação dos gráficos 2 e 3 permite alguns comentários: apesar do maior número
dos medicamentos novos serem classificados como novas entidades moleculares (NME), ou seja,
como novas substâncias químicas, o maior número dos medicamentos (18) teve a revisão de sua
NDA classificada como padrão pela FDA. Mesmo quando se considera apenas os medicamentos
que contêm novas entidades moleculares (24), 13 são classificadas com NDA padrão (S) e 11 são
classificados com NDA prioritário (P). Desta forma, apesar da maioria dos medicamentos serem
compostos por novas moléculas, o avanço terapêutico da maior parte destes medicamentos não é
significativo se comparado ao potencial terapêutico dos medicamentos já comercializados.
Relacionando os dados referentes à sede das empresas fabricantes dos medicamentos com
o potencial terapêutico dos mesmos, definido pela FDA, os resultados obtidos são os que se
apresentam na Tabela 2:
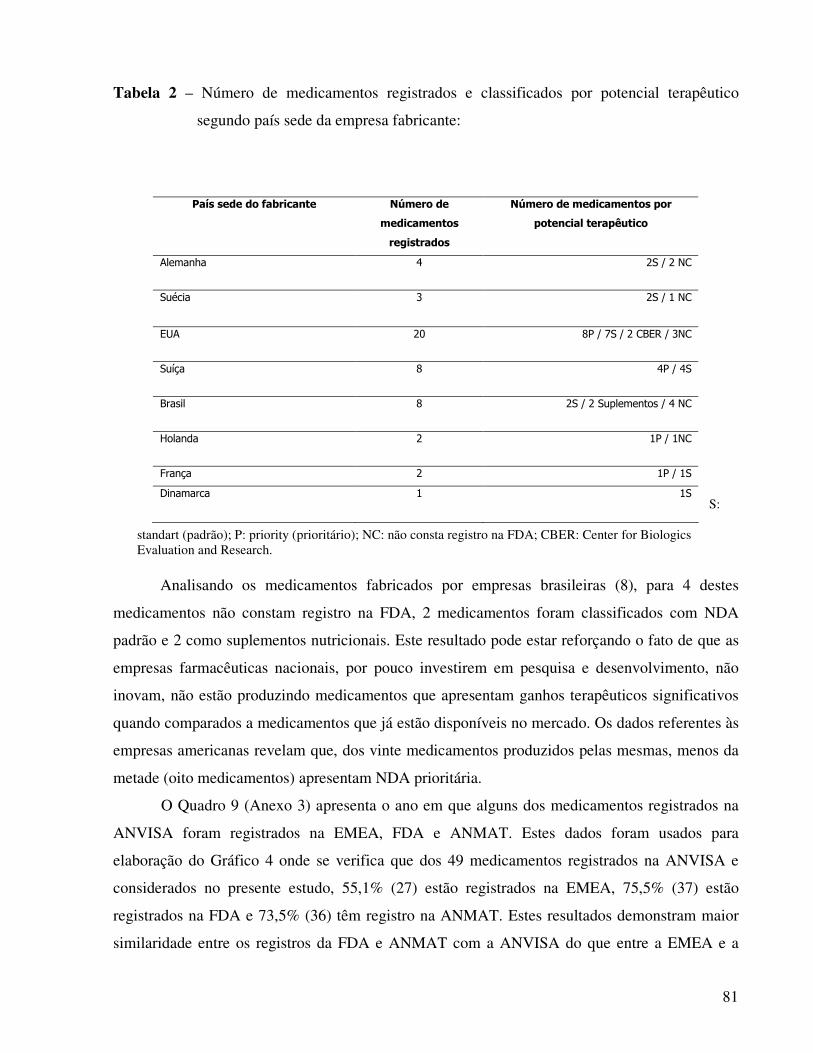
81
Tabela 2 – Número de medicamentos registrados e classificados por potencial terapêutico
segundo país sede da empresa fabricante:
S:
standart (padrão); P: priority (prioritário); NC: não consta registro na FDA; CBER: Center for Biologics Evaluation and Research.
Analisando os medicamentos fabricados por empresas brasileiras (8), para 4 destes
medicamentos não constam registro na FDA, 2 medicamentos foram classificados com NDA
padrão e 2 como suplementos nutricionais. Este resultado pode estar reforçando o fato de que as
empresas farmacêuticas nacionais, por pouco investirem em pesquisa e desenvolvimento, não
inovam, não estão produzindo medicamentos que apresentam ganhos terapêuticos significativos
quando comparados a medicamentos que já estão disponíveis no mercado. Os dados referentes às
empresas americanas revelam que, dos vinte medicamentos produzidos pelas mesmas, menos da
metade (oito medicamentos) apresentam NDA prioritária.
O Quadro 9 (Anexo 3) apresenta o ano em que alguns dos medicamentos registrados na
ANVISA foram registrados na EMEA, FDA e ANMAT. Estes dados foram usados para
elaboração do Gráfico 4 onde se verifica que dos 49 medicamentos registrados na ANVISA e
considerados no presente estudo, 55,1% (27) estão registrados na EMEA, 75,5% (37) estão
registrados na FDA e 73,5% (36) têm registro na ANMAT. Estes resultados demonstram maior
similaridade entre os registros da FDA e ANMAT com a ANVISA do que entre a EMEA e a
País sede do fabricante Número de
medicamentos
registrados
Número de medicamentos por
potencial terapêutico
Alemanha 4 2S / 2 NC
Suécia 3 2S / 1 NC
EUA 20 8P / 7S / 2 CBER / 3NC
Suíça 8 4P / 4S
Brasil 8 2S / 2 Suplementos / 4 NC
Holanda 2 1P / 1NC
França 2 1P / 1S
Dinamarca 1 1S
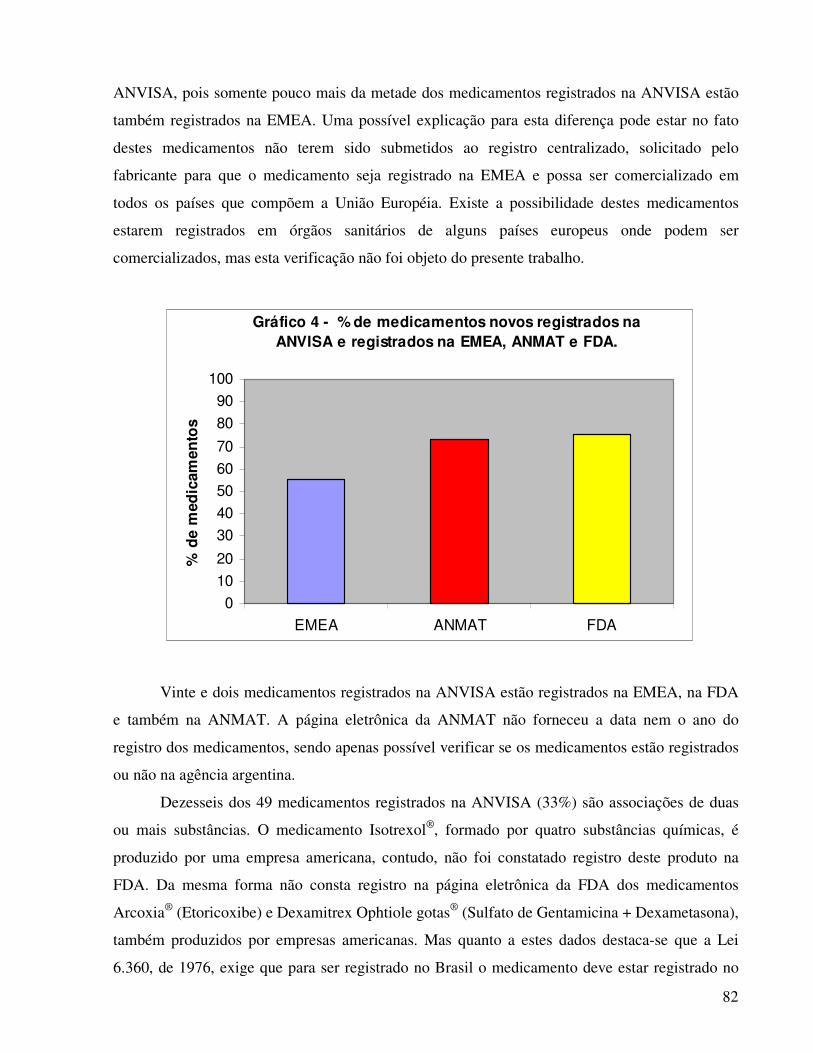
82
ANVISA, pois somente pouco mais da metade dos medicamentos registrados na ANVISA estão
também registrados na EMEA. Uma possível explicação para esta diferença pode estar no fato
destes medicamentos não terem sido submetidos ao registro centralizado, solicitado pelo
fabricante para que o medicamento seja registrado na EMEA e possa ser comercializado em
todos os países que compõem a União Européia. Existe a possibilidade destes medicamentos
estarem registrados em órgãos sanitários de alguns países europeus onde podem ser
comercializados, mas esta verificação não foi objeto do presente trabalho.
Gráfico 4 - % de medicamentos novos registrados na
ANVISA e registrados na EMEA, ANMAT e FDA.
0
10
20
30
40
50
60
70
80
90
100
EMEA ANMAT FDA
% d
e m
ed
icam
en
tos
Vinte e dois medicamentos registrados na ANVISA estão registrados na EMEA, na FDA
e também na ANMAT. A página eletrônica da ANMAT não forneceu a data nem o ano do
registro dos medicamentos, sendo apenas possível verificar se os medicamentos estão registrados
ou não na agência argentina.
Dezesseis dos 49 medicamentos registrados na ANVISA (33%) são associações de duas
ou mais substâncias. O medicamento Isotrexol®, formado por quatro substâncias químicas, é
produzido por uma empresa americana, contudo, não foi constatado registro deste produto na
FDA. Da mesma forma não consta registro na página eletrônica da FDA dos medicamentos
Arcoxia® (Etoricoxibe) e Dexamitrex Ophtiole gotas® (Sulfato de Gentamicina + Dexametasona),
também produzidos por empresas americanas. Mas quanto a estes dados destaca-se que a Lei
6.360, de 1976, exige que para ser registrado no Brasil o medicamento deve estar registrado no
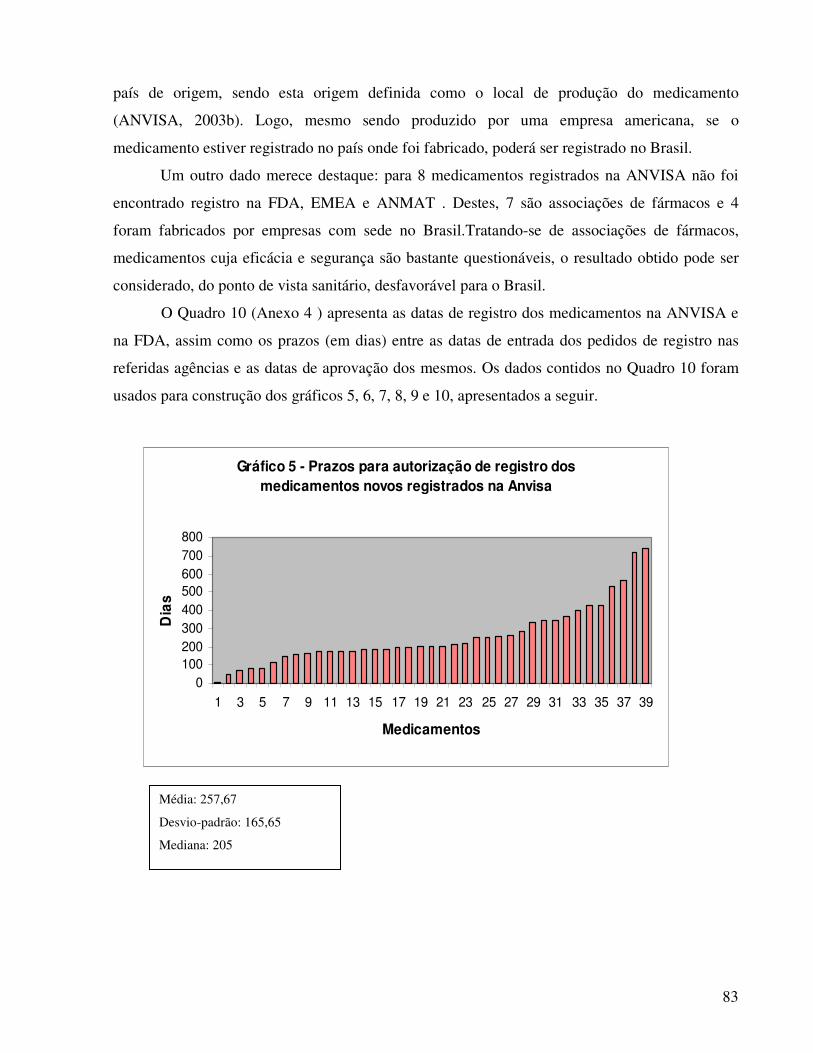
83
país de origem, sendo esta origem definida como o local de produção do medicamento
(ANVISA, 2003b). Logo, mesmo sendo produzido por uma empresa americana, se o
medicamento estiver registrado no país onde foi fabricado, poderá ser registrado no Brasil.
Um outro dado merece destaque: para 8 medicamentos registrados na ANVISA não foi
encontrado registro na FDA, EMEA e ANMAT . Destes, 7 são associações de fármacos e 4
foram fabricados por empresas com sede no Brasil.Tratando-se de associações de fármacos,
medicamentos cuja eficácia e segurança são bastante questionáveis, o resultado obtido pode ser
considerado, do ponto de vista sanitário, desfavorável para o Brasil.
O Quadro 10 (Anexo 4 ) apresenta as datas de registro dos medicamentos na ANVISA e
na FDA, assim como os prazos (em dias) entre as datas de entrada dos pedidos de registro nas
referidas agências e as datas de aprovação dos mesmos. Os dados contidos no Quadro 10 foram
usados para construção dos gráficos 5, 6, 7, 8, 9 e 10, apresentados a seguir.
Gráfico 5 - Prazos para autorização de registro dos
medicamentos novos registrados na Anvisa
0100200300400500600700800
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39
Medicamentos
Dia
s
Média: 257,67
Desvio-padrão: 165,65
Mediana: 205
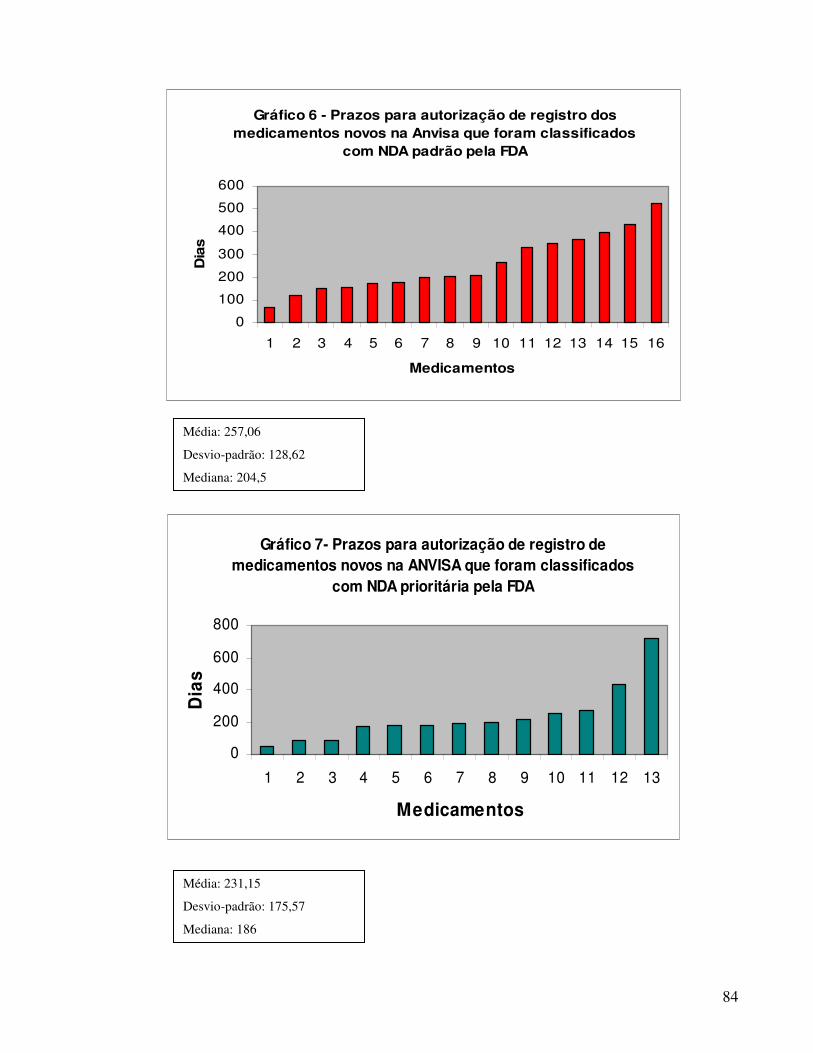
84
Gráfico 6 - Prazos para autorização de registro dos
medicamentos novos na Anvisa que foram classificados
com NDA padrão pela FDA
0
100
200
300
400
500
600
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Medicamentos
Dia
s
Gráfico 7- Prazos para autorização de registro de
medicamentos novos na ANVISA que foram classificados
com NDA prioritária pela FDA
0
200
400
600
800
1 2 3 4 5 6 7 8 9 10 11 12 13
Medicamentos
Dia
s
Média: 257,06
Desvio-padrão: 128,62
Mediana: 204,5
Média: 231,15
Desvio-padrão: 175,57
Mediana: 186

85
O Gráfico 5 foi obtido a partir dos prazos para registro de 39 medicamentos na ANVISA.
Para sete medicamentos não constavam dados para o cálculo do prazo de registro e para outros
três medicamentos a página eletrônica da ANVISA, referente ao produto, não estava disponível
no momento da pesquisa. Considerando que a série de dados (prazos em dias) apresenta uma
distribuição assimétrica em todos os gráficos obtidos, a medida estatística que foi utilizada na
análise dos resultados apresentados a seguir foi a mediana, tendo em vista que esta medida de
tendência central sofre menos influência de valores extremos. Os valores encontrados para média
e desvio-padrão estão também apresentados.
A mediana obtida para os prazos em dias necessários para a aprovação de registro dos
medicamentos novos na ANVISA, apresentados no Gráfico 5, foi de 205 dias. Este dado revela
que os prazos para registro de 50% dos medicamentos apresentados foram superiores a 205 dias e
que 50% dos prazos foram inferiores a 205 dias. Considerando que o prazo limite para decisão
sobre de registro de medicamentos na ANVISA é de 90 dias (Brasil, 1976), o valor encontrado
para a mediana ultrapassa em 115 dias o prazo máximo (90 dias) estabelecido na Lei. A mediana
dos prazos para registro dos medicamentos novos com NDA classificada como padrão pela FDA
(16), na Anvisa, foi de 204,5 dias (Gráfico 6) e para o registro dos medicamentos novos
classificados com NDA prioritária foi de 186 dias, conforme dados do Gráfico 7.
Esta mesma análise foi feita com os medicamentos novos registrados na ANVISA e que
também estão registrados na FDA, verificando-se o prazo para aprovação das NDA na FDA. O
Gráfico 8 foi elaborado a partir dos prazos para aprovação das NDAs de 32 medicamentos
registrados na FDA e a mediana obtida foi de 314,5 dias. Para cinco medicamentos registrados na
FDA não foi possível obter os prazos para análise e aprovação das NDAs. Dois medicamentos
foram registrados pelo CBER, dois foram registrados como suplemento alimentar e para um
medicamento a carta de aprovação da NDA não estava disponível na página eletrônica da FDA
no momento da consulta. O Gráfico 9 apresenta os prazos para aprovação das NDA classificadas
como padrão na FDA e a mediana encontrada foi de 350 dias. O Gráfico 10, elaborado a partir
dos prazos para aprovação das NDAs classificadas como prioritária, apresenta mediana de 231
dias. Verifica-se, desta forma, que as medianas encontradas para os dados dos Gráficos 9 e 10
ultrapassam os prazos definidos pela FDA para aprovação das NDAs padrão e prioritária, 300 e
180 dias, respectivamente.
Comparando os resultados obtidos para o registro dos 39 medicamentos novos na
ANVISA com os resultados obtidos para a aprovação das NDAs de 32 destes medicamentos na

86
FDA, algumas considerações podem ser feitas. Os valores encontrados para as medianas diferem
significativamente entre as duas agências, a FDA apresenta valores medianos maiores que a
ANVISA. A mediana do Gráfico 5 (205 dias) para os 39 medicamentos registrados na ANVISA é
menor que a mediana do Gráfico 8 (314,5 dias) para os 32 medicamentos registrados na FDA. A
maior diferença foi encontrada entre as medianas referentes aos prazos para registro de
medicamentos com NDA padrão, 204,5 dias para a ANVISA e 350 dias para a FDA (Gráficos 6 e
9).Vale destacar novamente que estes prazos foram obtidos entre as datas de protocolo dos
pedidos de registro de medicamentos e as datas de aprovação dos mesmos, nas duas agências, ou
seja, não foram excluídos os períodos nos quais, possivelmente, por motivos diversos, a análise
do pleito tenha sido suspensa. Entretanto, ainda que razões desconhecidas para o presente
trabalho possam ter influenciado no tempo para aprovação dos registros, os resultados obtidos
indicam que, de forma geral, o processo de análise e aprovação das solicitações de registro dos
medicamentos foi mais demorada na FDA do que na ANVISA.
Este resultado chama a atenção, pois comparada a FDA, a ANVISA possui um número
menor de técnicos e também menos experiência acumulada para análise dos aspectos
relacionados à qualidade, segurança e eficácia dos novos medicamentos. No entanto, o processo
de revisão dos dossiês foi mais rápido na ANVISA. Uma das possíveis explicações para este fato
pode estar na data de aprovação dos registros na ANVISA e na FDA, apresentadas no Quadro 10
(Anexo 4). Para 18 dos 49 medicamentos não foi possível obter a data de aprovação do registro
em pelo menos uma das duas agências. Considerando os 31 registros restantes, 24 foram
aprovados primeiro na FDA e, posteriormente, na ANVISA. Sete medicamentos foram
registrados primeiro na ANVISA. A existência de registro do medicamento na FDA pode ter
influenciado a tomada de decisões na ANVISA e, de alguma forma, acelerado o processo de
aprovação do registro na agência brasileira.
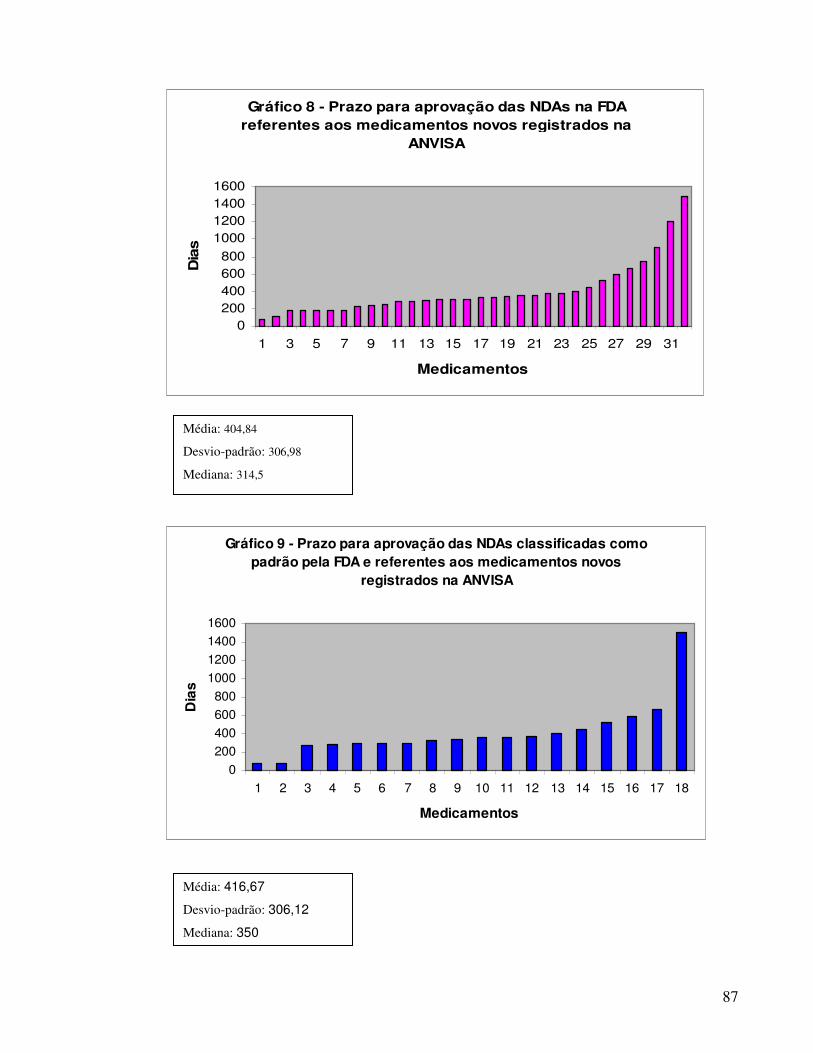
87
Gráfico 8 - Prazo para aprovação das NDAs na FDA
referentes aos medicamentos novos registrados na
ANVISA
0200400600800
1000120014001600
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31
Medicamentos
Dia
s
Gráfico 9 - Prazo para aprovação das NDAs classificadas como
padrão pela FDA e referentes aos medicamentos novos
registrados na ANVISA
0
200
400
600
800
1000
1200
1400
1600
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Medicamentos
Dia
s
Média: 404,84
Desvio-padrão: 306,98
Mediana: 314,5
Média: 416,67
Desvio-padrão: 306,12
Mediana: 350

88
Gráfico 10- Prazo para autorização das NDAs classificdas
como prioritárias na FDA e referentes aos medicamentos
novos registrados na ANVISA
0
200
400
600
800
1000
1 2 3 4 5 6 7 8 9 10 11 12 13 14
Medicamentos
Dia
s
Média: 309,57
Desvio-padrão: 228,22
Mediana: 231

89
10. CONCLUSÕES
Os resultados e considerações apresentadas nos capítulos anteriores permitem que sejam
feitas algumas conclusões sobre o processo de registro de medicamentos novos nas quatro
autoridades sanitárias selecionadas: FDA, EMEA, ANMAT e ANVISA. Quanto à ANVISA
também são apresentadas algumas recomendações referentes aos pontos abordados. Dentre os
resultados obtidos no presente estudo alguns foram destacados e estão comentados nos parágrafos
seguintes.
A pesquisa de informações oficiais nas páginas eletrônicas das autoridades sanitárias
selecionadas permitiu a obtenção de dados importantes, mas a qualidade e a quantidade de
informações fornecidas variaram significativamente entre as fontes acessadas. A página
eletrônica da FDA apresentou um número maior de informações, maior facilidade de acesso aos
dados e estes estavam apresentados de forma mais organizada. A página eletrônica da EMEA
apresentou um volume muito grande de informações, mas estas não estavam organizadas de
forma a facilitar a compreensão dos dados e a coleta das informações necessárias. A página
eletrônica da ANMAT apresentou um volume muito reduzido de informações e, quando
disponíveis, estavam incompletas (como visto, não foi possível a obtenção do ano de registro dos
medicamentos na ANMAT). A página eletrônica da ANVISA forneceu um volume insuficiente
de informações, estando ausentes alguns dados importantes como as etapas para o registro dos
medicamentos novos, a classificação dos medicamentos segundo as definições da RDC 136, de
2003, e as datas do período de avaliação das solicitações de registro não estavam disponíveis de
forma clara.
Considerando que a ANVISA possui uma página eletrônica e que alguns dados estão
disponíveis para consulta do público em geral, é fundamental que esta informação seja de boa
qualidade. Dada a importância do registro sanitário de medicamentos e a necessidade de
transparência neste processo, tanto para os profissionais de saúde e consumidores quanto para o
setor regulado, a ANVISA deveria dar um pouco mais de atenção a estes aspectos,
disponibilizando um número maior de informações e melhorando a qualidade do que já está
disponível em sua página eletrônica.
As normas legais que orientam o processo de registro de medicamentos novos
estabelecem que os atributos como qualidade, segurança e eficácia são fundamentais para que um
medicamento seja registrado nas quatro autoridades sanitárias. Porém, nenhuma das normas

90
analisadas apresentou exigências quanto à inovação, ao avanço terapêutico para que o
medicamento seja registrado como medicamento novo. A classificação segundo potencial
terapêutico utilizada pela FDA determina o prazo no qual a NDA será avaliada, mas não restringe
o registro quando o medicamento não apresenta inovação terapêutica. Somado a este dado, as
definições para medicamentos novos são abrangentes em todas as quatro autoridades estudadas e
também não são uniformes. A definição de medicamento novo adotada pela ANMAT é a que
mais se diferencia, uma vez que a definição da “novidade” é feita a partir da existência do
registro em outros países selecionados e não baseada em critérios de inovação do produto.
Ao contrário da FDA, as normas da EMEA e da ANVISA não classificam os
medicamentos registrados e não estabelecem prioridade para a análise da solicitação de registro.
A ANMAT, por sua vez, tem seu processo de registro baseado em listas de países de referência.
A priorização adotada pela FDA pode servir como modelo para as agências regulatórias de outros
países. Estabelecer prazos distintos para a análise dos pedidos de registro é adequado, pois os
medicamentos diferem no nível de inovação e essencialidade sanitária. Desta forma, seria
adequado que as demais agências, ao definirem os critérios de priorização nas análises dos
dossiês, considerassem as inovações terapêuticas trazidas pelos produtos e as necessidades de
saúde das populações.
As autoridades sanitárias dos países onde as atividades de pesquisa e desenvolvimento são
escassas, como do Brasil e da Argentina, adotam prazos legais menores para avaliação e
concessão de registro que países como EUA e países europeus, onde se realiza pesquisa e
desenvolvimento e os avaliadores detêm mais informações sobre os produtos que vão analisar. O
prazo para concessão do registro sanitário de medicamentos no Brasil é ainda norteado por uma
Lei de 1976 que estabeleceu um prazo de 90 dias. O desenvolvimento científico-tecnológico
ocorrido nas últimas décadas trouxe também a necessidade introduzir uma série de exigências
para a comprovação da qualidade, segurança e eficácia dos novos medicamentos, tornando mais
lento o processo de concessão de registro sanitário. Considerando estes aspectos, há necessidade
de se atualizar a legislação brasileira em vigor, estabelecendo, inclusive, critérios de priorização
nas análises dos pleitos e de revisão e cancelamento de registros de medicamentos.
Com relação ao conjunto de medicamentos novos registrados na ANVISA, nos anos de
2000, 2001 e 2002, a maior parte destes medicamentos (31) são quimicamente novos. Contudo,
verificou-se que quando os medicamentos foram classificados segundo o potencial terapêutico

91
definido pela FDA, o maior número, 18 medicamentos, não apresentaram avanço terapêutico
sobre medicamentos já comercializados que justificasse prioridade na revisão de suas NDAs.
Os resultados dos prazos em dias para registro dos 49 medicamentos novos na ANVISA
entre os anos de 2000 e 2002 e dos prazos para o registro de 32 destes medicamentos na FDA
revelaram que a ANVISA concedeu o registro para os medicamentos em prazos menores que a
FDA. Ainda que este resultado seja baseado em valores medianos e considerando os aspectos
apresentados no capítulo anterior, o dado obtido contraria de certa forma, uma tendência geral em
afirmar que agência brasileira é morosa em suas atividades, principalmente no que se refere à
concessão de registro sanitário.
A verificação do registro dos 49 medicamentos novos registrados na ANVISA na FDA,
EMEA e ANMAT revelou que uma quantidade significativa destes medicamentos está registrada
na FDA e na ANMAT, 37 medicamentos e 36 medicamentos, respectivamente. Já o número de
medicamentos registrados na EMEA foi menor, 27 medicamentos. Logo, ainda que os dados
relacionados ao processo de registro de medicamentos novos, como as exigências legais, as
definições de medicamento novo e os prazos para análise dos dossiês tenham apresentado
diferenças entre as autoridades regulatórias estudadas, a constatação de um número elevado de
registro dos medicamentos na FDA e na ANMAT pode representar que aquelas diferenças não
foram suficientes para produzir um quadro distinto na oferta destes medicamentos em países
como os EUA, Argentina e Brasil. A situação da EMEA é particular, pois, como visto
anteriormente, para ser registrado na EMEA o medicamento deve pertencer a um dos grupos
apresentados anteriormente ou o solicitante do registro deve demonstrar que o medicamento
constitui uma inovação significativa, capaz de justificar uma concessão de registro comunitário.
Este fato, porém, não descarta a possibilidade de que alguns destes medicamentos estejam
registrados em alguns países da União Européia.

92
REFERÊNCIAS BIBLIOGRÁFICAS ABRAHAM, J.,1995. Partial Progress? The Development of American and British Drug
Regulation.Science, Politics and the Pharmaceutical Industry,pp.36-86, New York: St. Martin”s Press.
ANMAT,2005a. <http://www.anmat.gov.ar/principal.html> ANMAT, 2005b. 23/01/05 <http://www.anmat.gov.ar/Institucional/nueva%20estructura.pdf ANVISA (Agência Nacional de Vigilância Sanitária), 2003a. Resolução RDC Nº 136 de
29/05/2003.Aprova o Regulamento Técnico para Medicamentos Novos Com Princípios Ativos Sintéticos ou Semi Sintéticos. http://www.anvisa.gov.br/medicamentos/registro/doc_novo.htm
ANVISA, 2003b. Resolução RDC nº 35, de 25 de fevereiro de 2003. Determina todos os estabelecimentos Distribuidores e Fracionadores de Insumos Farmacêuticos o cumprimento das diretrizes estabelecidas no Regulamento Técnico de Boas Práticas de Distribuição e Fracionamento de Insumos Farmacêuticos
http://e-legis.bvs.br/leisref/public/showAct.php?id=9570&mode=PRINT_VERSION ANVISA,2000. Portaria nº 593 de 25/08/2000. Aprova o Regimento Interno e o Quadro Demonstrativo
de Cargos em Comissão da Agência Nacional de Vigilância Sanitária. Republicada no DOU de 22/12/00.
ANVISA, 2004. Posicionamento da ANVISA sobre o registro de medicamentos novos considerados
como me-too. 9/6/2004. GEPEC/ANVISA <http://www.anvisa.gov.br/medicamentos/registro/metoos.htm>
ARGENTINA, 1992a.Decreto 1269 de 20/07/1992. Políticas Nacionales de Salud Ministerio de Salud
y Acción Social. <http://www.msal.gov.ar/htm/site/Digesto_Rec_Hum_Salud/decretos/decreto_1269-92.html>
ARGENTINA, 1992b. Decreto nº 150 de 23/01/92 (Texto ordenado de acuerdo com las modificacionaes de los Decretos nº 1.890/92 y 177/93). Normas para el Registro, Elaboracion, Fraccionamiento, Prescripcion, Expendio de Medicamentos.05/08/05. http://www.anmat.gov.ar/principal.htm
ARGENTINA, 1996. Ley nº 24.766 de 20/12/1996. Ley de Confidencialidad sobre información y productos que estén legitimamente bajo control de uma persona y se divulge indebidamente de manera contraria a los usos comerciales honestos. <http://www.ftaa-alca.org/intprop/natleg/Argentina/L 24766.asp
ARGENTINA, 2003.Decision Admnistrativa 22 de 14/03/2003. Apruébase la estructura organizativa del primer nível operativo del citado organismo descentralizado dependiente de la

93
Secretaría de Políticas, Regulación y Relaciones Sanitarias del Ministerio de Salud.23/01/05 <http://www.anmat.gov.ar/Institucional/nueva%20estructura.pdf
ARNAU, J.M. & LAPORTE, J-R.,1989.Promoção do Uso Racional dos Medicamentos e Preparação
de Guias Farmacológicos. In: Epidemiologia do Medicamento. Princípios Gerais (J-R. Laporte, G. Tognoni & S. Rozenfeld), pp. (57-74), São Paulo – Rio de Janeiro: Hucitec-Abrasco.
BARROS, J.A.C.,1995. Propaganda de Medicamentos - Atentado à Saúde?São Paulo: Editora
Hucitec/Sobravime BRASIL, 1973. Lei nº 5.991 de 17 de dezembro de 1973. Dispõe sobre o controle sanitário do
comércio de drogas, medicamentos, insumos farmacêuticos e correlatos, e dá outras providências. D.O.U. de 19/12/1973
BRASIL, 1976. Lei nº 6.360, de 23/09/1976. Dispõe sobre a vigilância sanitária a que ficam sujeitos
os medicamentos, as drogas, os insumos farmacêuticos e correlatos, cosméticos, saneantes e outros produtos, e dá outras providências. D.O.U. 24/09/1976.
BRASI, 1999. Lei nº 9.782 de 26/01/1999. Define o Sistema Nacional de Vigilância Sanitária, cria a Agência Nacional de Vigilância Sanitária, e dá outras providências. D.O.U. de 27.01.1999, Seção 1, pág. 1.
CARNÉ, X. & LAPORTE, J.R.,1989. Metodologia Epidemiológica Básica em Farmacologia. In: Epidemiologia do Medicamento. Princípios Gerais (J-R. Laporte, G. Tognoni & S. Rozenfeld), pp. (125-138), São Paulo – Rio de Janeiro: Hucitec-Abrasco.
COSTA,E.A.,1999.Vigilância Sanitária: Proteção e Defesa da Saúde. São Paulo: Hucitec/Sobravime. COSTA, E.A., 2004. Vigilância Sanitária. Proteção e Defesa da Saúde.Segunda Edição Aumentada -
São Paulo: Sociedade Brasileira de Vigilância de Medicamentos.
COSTA,E.A.& Rosenfeld,S.,2000.Constituição da Vigilância Sanitária no Brasil. Fundamentos da Vigilância Sanitária (S.Rosenfeld, org.), pp.15-40, Rio de Janeiro: Editora FIOCRUZ.
DiMASI, J.A., HANSEN, R.W., GRABOWSKI, H.G. & LASAGNA, L., 1991. Cost of innovation in
the pharmaceutical industry. Journal of Health Economics (10/02/1992). pp.107-142. DiMASI, J.A., 2000.New Drug Innovation and Pharmaceutical Industry Structure: trends in the output
of pharmaceutical firms. Drug Information Journal,34:1109-1194.
DUNCAN, B.B. & SCHMIDT, M.I., 1996. Epidemiologia Clínica e a Medicina Baseada em Evidências. In: Medicina Ambulatorial: Condutas Clínicas em Atenção Primária. (Duncan B., Schmidt, M.I. & Giugliani E.R.J.).2.ed. Porto Alegre: Artes Médicas.
Comissão Européia, 2002.Notice to Applicants. Medicinal Products for Human Use, Volume 2B. User Guide for the Apllication Form, August 2002. <http://pharmacos.eudra.org./f2/eudralex/vol-2/b/ugfinalrev0augu02.pdf

94
EUROPA, 2005. O portal da União Européia. 20/01/05. http://europa.eu.int/abc/index_pt.htm
EUROPEAN COMISSION, 2002. Pharmaceuticals in the European Union. http://europa.eu.int EUROPEAN COMISSION ENTERPRISE DIRECTORATE-GENERAL,2004. Notice to Applicants.
Volume 2A Procedures for Marketing Authorization, Chapter 1, Marketing Authorization.Brussels<http://pharmacos.eudra.org/F2/eudralex/vol2/A/v2a_chap1%20_r2_2004-02.pdf
FDA, 1938. Federal Food, Drug and Cosmetic Act.13/03/05
<http://www.fda.gov/opacom/laws/fdcact/fdctoc.htm>
FDA, 2004a.Innovation or Stagnation? Challenge and Opportunity on the Critical Path to New Medical Technologies. U.S.Department of Health and Human Services. March 2004. >http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html
FDA, 2004b.The Evolution of U.S. Drug Law.15/12/04 http://www.fda.gov/fdac/special/newdrug/benlaw.html FDA,2004c. Benefit vs. Risk: How CDER Approves New Drugs.(05/12/04) <http://www.fda.gov/cder/about/whatwedo/testtube-5.pdf> FDA,2005a. Drug Review Glossary.15/02/05 <http://www.fda.gov/fdac/special/newdrug/bengloss.html
FDA,2005b. Department of Health Human Service: What We Do
25/02/05<http://www.hhs.gov/about/whatwedo.html/>
FDA,2005c. FDA Organization. (13/03/05) <http://www.fda.gov/opacom/7org.html> FDA, 2005d. The CDER Handbook. 14/02/05 <http://www.fda.gov/cder/handbook/ndabox.htm FDA,2005e.What FDA Regulates.(13/03/05) http://www.fda.gov/comments/regs.html GARATTINI,S.& BERTELLE, V., 2004. The role of the EMEA in regulating pharmaceutical
products.In: Regulating Pharmaceuticals in Europe: striving for efficiency, equity e quality (E. Mossialos, M. Mrazek & T. Walley., ed.), pp. 80-96, England: Open University Press.
GARATTINI, S., 2005. EMEA: For patients or for industry?. Pharmacoeconomics, 23(3): 207-8.

95
ISDB (International Society of Drug Bulletins), 2001.Declaração da ISDB sobre o Avanço Terapêutico no Uso de Medicamentos. Paris, 15-16 de novembro de 2001.05/09/04 >http://www.isdbweb.dspace.it/pag/>
LAPORTE, J.R.,BAKSAAS, I. & LUNDE, P.K.M, 1993. General background. In: Drug utilization
studies: methods and uses (M.N.G., Dukes,). pp. 5-22: WHO Regional Publications. European series; nº 45.
LUCCHESE, G.,2001.Globalização e Regulação Sanitária: Os Rumos da Vigilância Sanitária no Brasil.Tese de Doutorado, Rio de Janeiro: Escola Nacional de Saúde Pública, Fundação Oswaldo Cruz.
MEADOWS, M.,2002. The FDA's Drug Review Process: Ensuring Drugs are Safe and Effective. FDA Consumer Magazine. July-August 2002., Volume 36, Number 4. (05/12/04) > http://www.fda.gov/fdac/features/2002/402_drug.html
MS (Ministério da Saúde), 2001.Política Nacional de Medicamentos. Departamento de Atenção Básica, Secretaria de Políticas de Saúde, Ministério da Saúde.
MSF (Médicos sem Fronteiras), 2001. Desequilíbrio Fatal – A Crise em Pesquisa e Desenvolvimento de Drogas para Doenças Negligenciadas.Campanha de Acesso a Medicamentos Essenciais e Grupo de Trabalho de Drogas para Doenças Negligenciadas.Genebra: MSF.
MSH (Management Sciences for Health), 1997. Managing Drug Supply: The Selection, Procurement, Distribution, and Use of Pharmaceuticals. Second edition, revised and expanded. Published by Kumarian Press, Inc. USA.
NAF/ENSP (Núcleo de Assistência Farmacêutica / Escola Nacional de Saúde Pública), 2002. Fundamentos Farmacológicos- Clínicos dos Medicamentos de Uso Corrente 2002. Rio de Janeiro:ENSP,2002. <http://www.anvisa.gov.br/divulga/public/livro_eletronico/INDEX.HTM>
NIHCM Foundation (National Institute for Health Care Management),2002. Changing Patterns of Pharmaceutical Innovation.12/12/03.<http://www.nihcm.org/innovations.pdf>
OLIVEIRA, M.A.,2001.Tecnociência, Ativismo e a Política do Tratamento da AIDS.Tese de Doutorado.,Rio de Janeiro:COPPE/Universidade Federal do Rio de Janeiro.
OLIVEIRA, M.A.; BERMUDEZ, J.A.Z.& SOUZA, A.C.M.,1999. Talidomida no Brasil: vigilância com responsabilidade compartilhada?. Cadernos de Saúde Pública, 15 (1):99-112.
OPAS/OMS (Organização Panamericana da Saúde / Organização Mundial da Saúde),1992. Políticas
de Autorizaniòn de Productos Farmacêuticos. Documento de la Reunion de Expertos de la Subregion Andina. Quito, Ecuador.WHO/DAP/93.2
PARLAMENTO EUROPEU, 2004. Regulamento (CE) nº 726/2004, de 31 de março de 2004.
Estabelece Procedimentos Comunitários de Autorização e de Fiscalização de Medicamentos para

96
uso humano e veterinário e institui uma Agência Européia de Medicamentos. Estrasburgo: Jornal Oficial da União Européia,L 136/ 1-33.
PhRMA (Pharmaceutical Research and Manufacturers of America),2003. New Drugs Approval in 2002.Washington.p.1-19. 12/01/03. <http://www.rtk.org/perspectives/2003-02-11.91.pdf>
PIOVESAN, M.F., 2002. A Construção Política da Agência Nacional de Vigilância Sanitária. Dissertação de Mestrado, Rio de Janeiro: Escola Nacional de Saúde Pública, Fundação Oswaldo Cruz.
REIS, A.L.A.,2004. Novos Produtos no Mercado Farmacêutico: padrão de difusão e preços. Tese de Doutorado, Rio de Janeiro: Escola Nacional de Saúde Pública, Fundação Oswaldo Cruz.
ROZENFELD,S.,1998. Farmacovigilância: elementos para discussão e perspectivas.Cadernos de Saúde Pública, 14 (2):237-250.
SAYD, J.D.,1998. Higiene, o Desperdício de Panacéia. In: Mediar, Medicar, Remediar: aspectos da terapêutica na medicina ocidental, pp.127-155, Rio de Janeiro: EdUERJ(Saúde & Sociedade).
SCHMID,E.F.& SMITH, D.A.,2004. Is pharmaceutical R&D just a game of chance or can strategy make a difference? Drug Discovery Today, 9 (1):18-26
SCHMID,E.F.,2002.Should scientific innovation be managed? Drug Discovery Today,7(18):941-945.
TOGNONI G. & LAPORTE J-R., 1989. Estudos de Utilização de Medicamentos e de Farmacovigilância.In:Epidemiologia do Medicamento. Princípios Gerais (J-R. Laporte, G. Tognoni & S. Rozenfeld), pp. 43-56, São Paulo – Rio de Janeiro: Hucitec-Abrasco.
WHO (World Health Organization), 2002a 10th International Conference of Drug Regulatory Authorities (ICDRA).Geneva: WHO.
WHO, 2002b. The Impact of Implementation of ICH Guidelines in Non-ICH Countries. Regulatory Support Series, nº 9. Geneve:WHO.
http://www.emea.eu.int/index/indexh1.htm http://www7.anvisa.gov.br/datavisa/Consulta_Produto/consulta_medicamento.asp http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ http://www.anmat.gov.ar/principal.htm http://www.emea.eu.int/pdfs/human/ich/288799enq.pdf

97
ANEXO 1
Entrevista
Entrevista realizada em 07/11/2004
Entrevistado: Sérgio de Andrade Nishioka – Gerente GEPEC/GGMED/ANVISA
Questão 1- Quais as etapas necessárias para o registro de medicamentos novos na ANVISA?
Nishioka destacou que a análise segue as exigências da RDC atual, a RDC 136, cujos pontos
mais importantes foram relacionados no Quadro 3. A análise do dossiê para registro entregue pelo
solicitante do registro é realizada em duas etapas: análise farmacotécnica e análise de segurança e
eficácia. Na análise farmacotécnica são avaliados os dados referentes à qualidade do produto (onde é
produzido, como é produzido, testes de estabilidade, área de produção,etc.). Esta etapa é realizada por
farmacêuticos da GEPEC e, segundo Nishioka, raramente é solicitado um consultor ad hoc.Na análise
de segurança e eficácia são avaliados os resultados dos ensaios pré-clínicos e clínicos com o produto.
Esta análise não é realizada por profissionais da ANVISA e sim por consultores externos. Nishioka
destacou que a utilização de consultores externos nesta etapa ocorre pela dificuldade em recrutar
pessoal qualificado para realizar este tipo de atividade na agência
Questão 2- Como são escolhidos os consultores para avaliação de segurança e eficácia?
Sobre a escolha dos consultores externos, o entrevistado informou que esta escolha é feita a
partir de um banco de consultores composto por professores universitários ou médicos conceituados
nas suas especialidades (que já participaram deste processo na agência) ou por meio de consulta às
Sociedades Médicas que indicam, dentre seus associados, aquele mais capacitado para avaliar a
segurança e a eficácia do medicamento em questão. Informou também que a agência verifica
inicialmente se os consultores escolhidos para participar do processo de análise possuem algum
conflito de interesse potencial, como a participação na investigação clínica do produto no país e se o
mesmo é acionista de alguma empresa farmacêutica.
O número de consultores convidados para análise de segurança e eficácia varia conforme o tipo
de medicamento novo: quando se trata de medicamento genuinamente novo no país os dados são

98
enviados para dois consultores externos, quando o medicamento é uma nova indicação terapêutica ou
nova forma farmacêutica, por exemplo, os dados são enviados para um consultor apenas.
Questão 3- Quais os profissionais responsáveis pela decisão sobre o registro do medicamento?
Após a análise dos dados de segurança e eficácia os consultores emitem seus pareceres para a
GEPEC (solicita-se aos consultores que os pareceres sejam enviados após trinta dias de análise).
Nishioka enfatizou que a GEPEC possui autonomia para concordar ou não com o parecer emitido,
podendo autorizar ou não o registro do medicamento novo. Enfatizou, porém, que para a maioria dos
medicamentos há concordância entre o parecer dos consultores e a avaliação da GEPEC, mas já houve
casos de discordância.
Questão 4- Como você avalia o processo de registro de medicamentos novos atualmente na
ANVISA?
Nishioka destacou que houve uma evolução no processo de avaliação da segurança e eficácia
dos medicamentos novos submetidos ao registro ao longo do tempo. A criação das Câmaras Técnicas e,
mais recentemente, da CATEME, representaram um avanço, mas segundo o entrevistado, durante a
vigência da CATEME também existiram problemas relacionados à eficiência e transparência no
processo de registro. Informou que em média eram analisados em cada reunião da CATEME dez
dossiês e, como conseqüência disto, o processo de registro era muito mal documentado e não
apresentava clareza no que se referia à tomada de decisão, favorável ou não ao registro. Acrescentou
que esta situação ocasionava muitos problemas com os solicitantes de registro quando a decisão da
CATEME era de não autorizar o registro do medicamento, pois as decisões não havia sido
documentadas de forma correta e a agência não tinha subsídios técnicos para argumentar e defender
suas decisões.
Sobre o momento atual, com a GEPEC, Nishioka julgou que a análise do processo de registro
está muito boa no que se refere à transparência das decisões, mas em relação à qualidade da avaliação
dos medicamentos a serem registrados, afirmou não dispor de instrumentos para medir o nível desta
qualidade.

99
Questão 5 – Qual a definição de medicamento novo adotada pela ANVISA para o registro?
Sobre o conceito de medicamento novo adotado pela agência, o entrevistado chamou atenção
para a RDC 136 que considera o medicamento novo no país e que esta definição permite considerar um
medicamento já registrado e usado em outro país há anos como uma inovação no Brasil. Para Nishioka
o conceito adotado atualmente é muito mais amplo do que se deseja – destacando o fato da legislação
brasileira permitir o registro de pró-fármacos como medicamentos novos.
A empresa que deseja registrar um medicamento entra com pedido de registro de medicamento
novo na agência. Se o medicamento não corresponder a nenhuma das definições para medicamento
novo estabelecidas pela RDC nº 136 a GEPEC indefere o registro de medicamento novo, orientando a
empresa a entrar com pedido de registro apropriado.
Segundo Nishioka, o registro de medicamento denominado me too como medicamento novo
contribui para a regulação do mercado, tornando-o mais competitivo. A outra vantagem apontada pelo
entrevistado é que alguns grupos populacionais podem adaptar-se melhor a um medicamento me too de
uma determinada empresa do que ao primeiro medicamento inovador do qual o me too difere em
alguma característica. A definição de me too é mais fácil de ser dada quando o medicamento já está no
mercado, sendo usado por um número grande de indivíduos, só assim podem ser comparados os
produtos quanto ao efeito que produzem. No momento do registro é impossível fazer este tipo de
avaliação.
Acrescentou também que se o fabricante de um medicamento similar faz um estudo clínico para
uma nova indicação (para qual o produto de referência ainda não foi testado), o fabricante do similar
pode pedir registro da nova indicação terapêutica no país, passar a ser o medicamento de referência
para aquela indicação, pode receber um novo nome fantasia e necessariamente recebe um novo
registro. Isto também acontece para uma nova concentração de princípio ativo.
Nihioka enfatizou também que durante o processo de avaliação para registro os profissionais da
GEPEC procuram ter muita atenção no que se refere a indicação terapêutica para qual o medicamento
foi testado, pois esta indicação é que será aprovada. Finaliza afirmando que o “médico até pode indicar
o medicamento novo para outras patologias, mas a indicação oficial é aquela que foi aprovada pela
agência”.
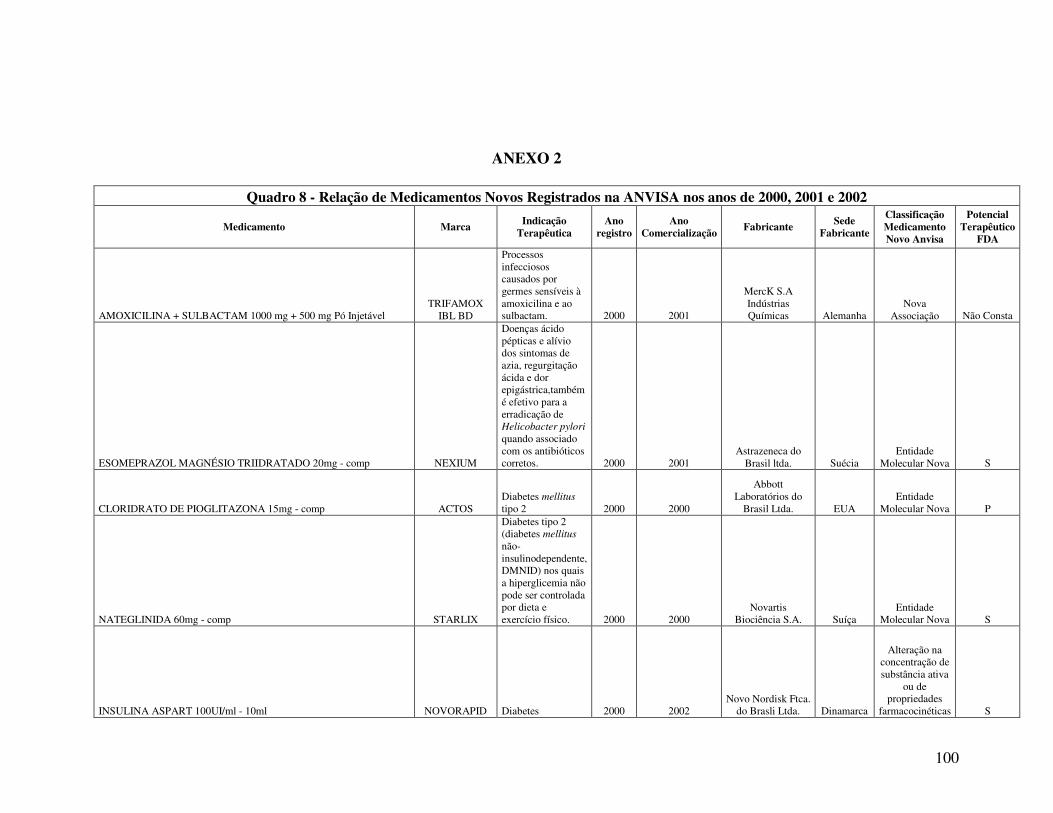
100
ANEXO 2
Quadro 8 - Relação de Medicamentos Novos Registrados na ANVISA nos anos de 2000, 2001 e 2002
Medicamento Marca Indicação Terapêutica
Ano registro
Ano Comercialização
Fabricante Sede Fabricante
Classificação Medicamento Novo Anvisa
Potencial Terapêutico
FDA
AMOXICILINA + SULBACTAM 1000 mg + 500 mg Pó Injetável TRIFAMOX
IBL BD
Processos infecciosos causados por germes sensíveis à amoxicilina e ao sulbactam. 2000 2001
MercK S.A Indústrias Químicas Alemanha
Nova Associação Não Consta
ESOMEPRAZOL MAGNÉSIO TRIIDRATADO 20mg - comp NEXIUM
Doenças ácido pépticas e alívio dos sintomas de azia, regurgitação ácida e dor epigástrica,também é efetivo para a erradicação de Helicobacter pylori quando associado com os antibióticos corretos. 2000 2001
Astrazeneca do Brasil ltda. Suécia
Entidade Molecular Nova S
CLORIDRATO DE PIOGLITAZONA 15mg - comp ACTOS Diabetes mellitus tipo 2 2000 2000
Abbott Laboratórios do
Brasil Ltda. EUA Entidade
Molecular Nova P
NATEGLINIDA 60mg - comp STARLIX
Diabetes tipo 2 (diabetes mellitus não-insulinodependente, DMNID) nos quais a hiperglicemia não pode ser controlada por dieta e exercício físico. 2000 2000
Novartis Biociência S.A. Suíça
Entidade Molecular Nova S
INSULINA ASPART 100UI/ml - 10ml NOVORAPID Diabetes 2000 2002 Novo Nordisk Ftca.
do Brasli Ltda. Dinamarca
Alteração na concentração de substância ativa
ou de propriedades
farmacocinéticas S

101
MONONITRATO DE ISOSSORBIDA + ACIDO ACETILSALICILICO 60mg + 75mg - cap gel VASCLIN
Profilaxia e tratamento da angina do peito e da isquemia miocárdica com ação antiagregante plaquetária. 2000 2001
Libbs farmacêutica Ltda Brasil
Nova Associação Não consta
CILOSTAZOL 50mg- comp CEBRALAT
Doença vascular periférica, para redução do sintoma da claudicação intermitente e na prevenção da recorrência de acidente vascular cerebral. 2002 2002
LDZ Comércio, Importação e
Exportação Ltda (Libbs) Brasil
Entidade Molecular Nova S
FLUTRIMAZOL 0,01g/ml solução tópica spray MICETAL
Tratamento tópico de micoses cutâneas superficiais, produzidas por fungos sensíveis : candidíase cutânea; pitiríase versicolor 2000 2002
Laboratórios Biossintética Ltda Brasil
Entidade Molecular Nova Não consta
PIMECROLIMUS 10mg/g creme - 15g ELIDEL
Tratamento em curto prazo (agudo) e em longo prazo dos sinais e sintomas da dermatite atópica (eczema) . 2002 2002
Novartis Biociência S.A. Suíça
Entidade Molecular Nova S
BUTIRATO DE HIDROCORTISONA 1mg/g creme - 15 g LOCOID Dermatites 2000 2000 Eurofarma
Laboratórios Ltda. Brasil
Sal novo, embora a entidade
molecular corespondente já
tenha sido autorizada S
ETONOGESTREL 68mg solução dermatológica - implante IMPLANON Contracepção 2000 2001
Organon do Brasil Indústria e
Comércio Ltda. Holanda Entidade
Molecular Nova Não consta
LUTROPINA ALFA (R-LH) 75UI pó liófilo inj LUVERIS
Em mulheres com insuficiência de LH e FSH, em associação com o FSH. 2001 2002
Serono Produtos Farmacêuticos
Ltda. Suíça Entidade
Molecular Nova S
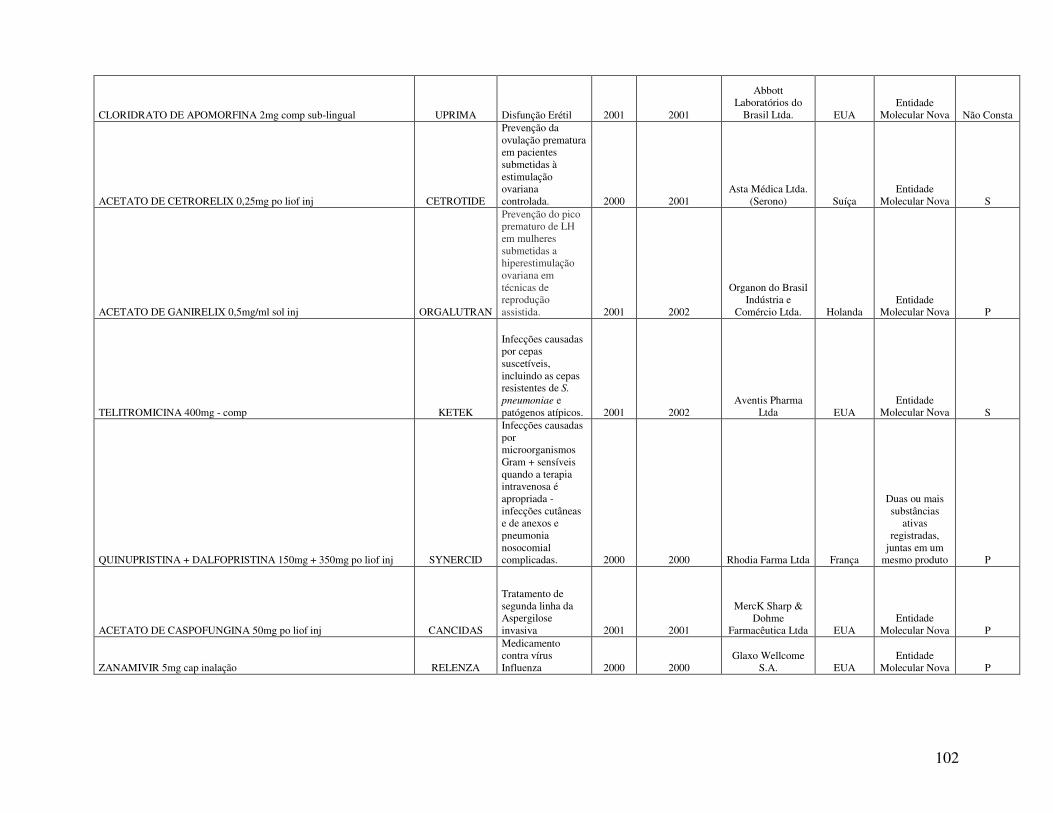
102
CLORIDRATO DE APOMORFINA 2mg comp sub-lingual UPRIMA Disfunção Erétil 2001 2001
Abbott Laboratórios do
Brasil Ltda. EUA Entidade
Molecular Nova Não Consta
ACETATO DE CETRORELIX 0,25mg po liof inj CETROTIDE
Prevenção da ovulação prematura em pacientes submetidas à estimulação ovariana controlada. 2000 2001
Asta Médica Ltda. (Serono) Suíça
Entidade Molecular Nova S
ACETATO DE GANIRELIX 0,5mg/ml sol inj ORGALUTRAN
Prevenção do pico prematuro de LH em mulheres submetidas a hiperestimulação ovariana em técnicas de reprodução assistida. 2001 2002
Organon do Brasil Indústria e
Comércio Ltda. Holanda Entidade
Molecular Nova P
TELITROMICINA 400mg - comp KETEK
Infecções causadas por cepas suscetíveis, incluindo as cepas resistentes de S. pneumoniae e patógenos atípicos. 2001 2002
Aventis Pharma Ltda EUA
Entidade Molecular Nova S
QUINUPRISTINA + DALFOPRISTINA 150mg + 350mg po liof inj SYNERCID
Infecções causadas por microorganismos Gram + sensíveis quando a terapia intravenosa é apropriada - infecções cutâneas e de anexos e pneumonia nosocomial complicadas. 2000 2000 Rhodia Farma Ltda França
Duas ou mais substâncias
ativas registradas,
juntas em um mesmo produto P
ACETATO DE CASPOFUNGINA 50mg po liof inj CANCIDAS
Tratamento de segunda linha da Aspergilose invasiva 2001 2001
MercK Sharp & Dohme
Farmacêutica Ltda EUA Entidade
Molecular Nova P
ZANAMIVIR 5mg cap inalação RELENZA
Medicamento contra vírus Influenza 2000 2000
Glaxo Wellcome S.A. EUA
Entidade Molecular Nova P

103
OSELTAMIVIR 75mg - cap gel dura TAMIFLU
Tratamento da gripe e profilaxia da gripe em adultos e adolescentes com mais de 13 anos. 2000 2000
Produtos Roche Químicos e
Farmacêuticos S.A. Suíça Entidade
Molecular Nova P
LOPINAVIR + RITONAVIR 133,3mg + 33,3mg cap gel mole KALETRA AIDS 2000 2001
Abbott Laboratórios do
Brasil Ltda. EUA
Duas ou mais substâncias
ativas registradas,
juntas em um mesmo produto P
TEMOZOLOMIDA 5mg cap gel dura TEMODAL Antineoplásico 2000 2001
Indústria Química e Farmacêutica
Schering Plough S.A. EUA
Entidade Molecular Nova PV
MESILATO DE IMATINIB 50mg - cap gel dura GLIVEC
Leucemia mielóide crônica (LMC) em crise blástica, fase acelerada ou em fase crônica, em caso de falta de controle hematológico aos 3 meses ou falta de resposta citogenética. 2001 2001
Novartis Biociência S.A. Suíça
Entidade Molecular Nova PV
EXEMESTANO 25mg drágeas AROMASIN Antineoplásico 2000 2000 Pharmacia Brasil
Ltda. Suécia Entidade
Molecular Nova SV
PEGINTERFERON ALFA 2 B 50mcg/0,5ml - po liof inj PEGINTRON Agentes Imunoestimulantes 2001 2001
Indústria Química e Farmacêutica
Schering Plough S.A. EUA
Entidade Molecular Nova CBER
SIROLIMUS 1mg/ml solução oral RAPAMUNE
Profilaxia da rejeição de órgãos em pacientes transplantados renais. 2000 2001
Laboratórios Wyeth-Whitehall
Ltda. EUA Entidade
Molecular Nova S
INFLIXIMAB 10mg/ml po liof REMICADE
Artrite reumatóide, espondilite anquilosante e doença de Crohn: 2000 2001
Indústria Química e Farmacêutica
Schering Plough S.A. EUA
Entidade Molecular Nova CBER

104
SULFATO DE GLICOSAMINA 250mg - cap gel dura DINAFLEX
Artrose primária e secundária, osteocondrose, espondilose, condromalacia patelar e periartrite escapuloumeral. 2000 2001
Zodiac produtos Farmacêuticos
Ltda. Brasil Entidade
Molecular Nova Suplemento Nutricional
ETORICOXIB 60mg - comp ARCOXIA
Osteoartrite e artrite reumatóide; tratamento agudo da gota; alívio da dor aguda e crônica; tratamento da dismenorréia primária. 2002 2002
MercK Sharp & Dohme
Farmacêutica Ltda EUA Entidade
Molecular Nova Não consta
RISEDRONATO SODICO 5mg - comp ACTONEL
Tratamento e prevenção da osteoporose em mulheres no período pós-menopausa; 2000 2000
Aventis Pharma Ltda. EUA
Entidade Molecular Nova S
ZALEPLOM MICRONIZADO 5mg cap gel dura SONATA Tratamento da insônia 2000 2000
Laboratórios Wyeth-Whitehall
Ltda. EUA Entidade
Molecular Nova S
DESLORATADINA 5mg comp DESALEX
Rinite alérgica, alívio dos sintomas associados à urticária idiopática crônica, como prurido, e redução do tamanho e número de erupções cutâneas. 2001 2002
Indústria Química e Farmacêutica
Schering Plough S.A. EUA
Entidade Molecular Nova S
ETABONATO DE LOTEPREDNOL 5mg/ml sus oft -5ml LOTEPROL Antiinflamatório oftálmico 2000 2002
Bausch Lomb Indústria òtica
Ltda. EUA Entidade
Molecular Nova S
BIMATOPROST 0,3mg/ml sol oft LUMIGAN
Medicamentos Mióticos e antiglaucomatosos 2001 2001
Allergan Produtos FarmacêuticosLtda. EUA
Entidade Molecular Nova P
TRAVOPOST 0,004% sol oftálmica TRAVATAN
Medicamentos Mióticos e antiglaucomatosos 2001 2001
Alcon Laboratórios do Brasil Ltda EUA
Entidade Molecular Nova P
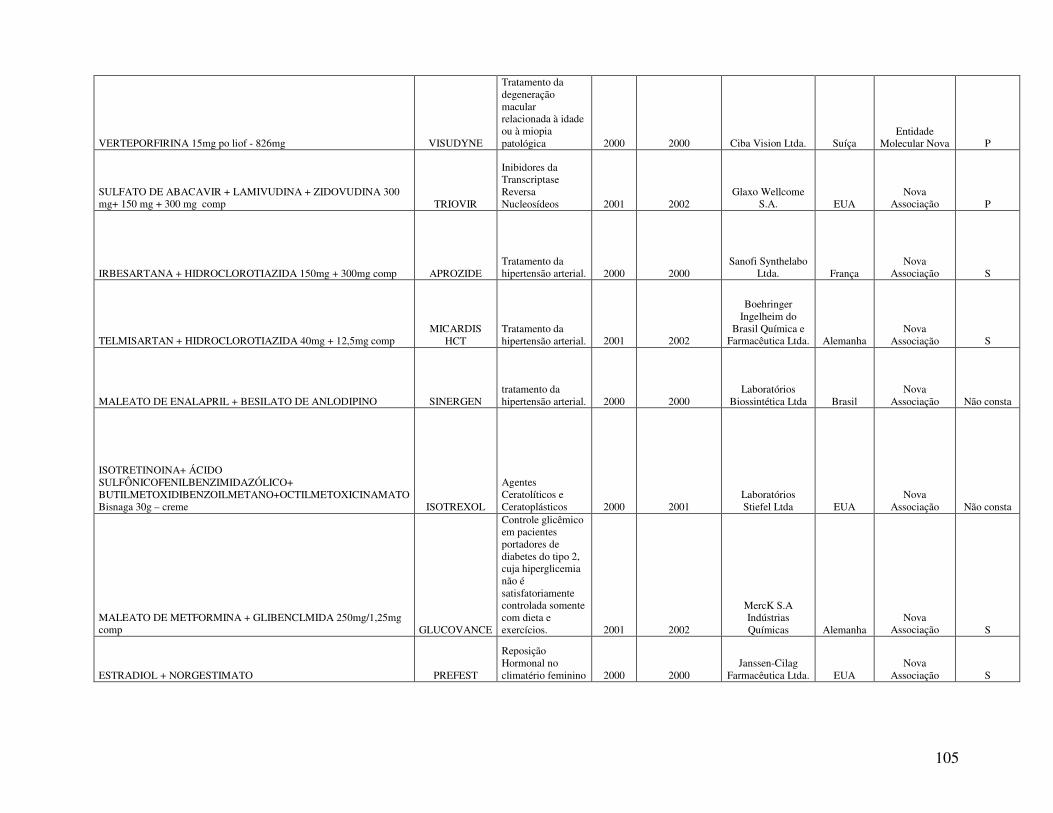
105
VERTEPORFIRINA 15mg po liof - 826mg VISUDYNE
Tratamento da degeneração macular relacionada à idade ou à miopia patológica 2000 2000 Ciba Vision Ltda. Suíça
Entidade Molecular Nova P
SULFATO DE ABACAVIR + LAMIVUDINA + ZIDOVUDINA 300 mg+ 150 mg + 300 mg comp TRIOVIR
Inibidores da Transcriptase Reversa Nucleosídeos 2001 2002
Glaxo Wellcome S.A. EUA
Nova Associação P
IRBESARTANA + HIDROCLOROTIAZIDA 150mg + 300mg comp APROZIDE Tratamento da hipertensão arterial. 2000 2000
Sanofi Synthelabo Ltda. França
Nova Associação S
TELMISARTAN + HIDROCLOROTIAZIDA 40mg + 12,5mg comp MICARDIS
HCT Tratamento da hipertensão arterial. 2001 2002
Boehringer Ingelheim do
Brasil Química e Farmacêutica Ltda. Alemanha
Nova Associação S
MALEATO DE ENALAPRIL + BESILATO DE ANLODIPINO SINERGEN tratamento da hipertensão arterial. 2000 2000
Laboratórios Biossintética Ltda Brasil
Nova Associação Não consta
ISOTRETINOINA+ ÁCIDO SULFÔNICOFENILBENZIMIDAZÓLICO+ BUTILMETOXIDIBENZOILMETANO+OCTILMETOXICINAMATO Bisnaga 30g – creme ISOTREXOL
Agentes Ceratolíticos e Ceratoplásticos 2000 2001
Laboratórios Stiefel Ltda EUA
Nova Associação Não consta
MALEATO DE METFORMINA + GLIBENCLMIDA 250mg/1,25mg comp GLUCOVANCE
Controle glicêmico em pacientes portadores de diabetes do tipo 2, cuja hiperglicemia não é satisfatoriamente controlada somente com dieta e exercícios. 2001 2002
MercK S.A Indústrias Químicas Alemanha
Nova Associação S
ESTRADIOL + NORGESTIMATO PREFEST
Reposição Hormonal no climatério feminino 2000 2000
Janssen-Cilag Farmacêutica Ltda. EUA
Nova Associação S
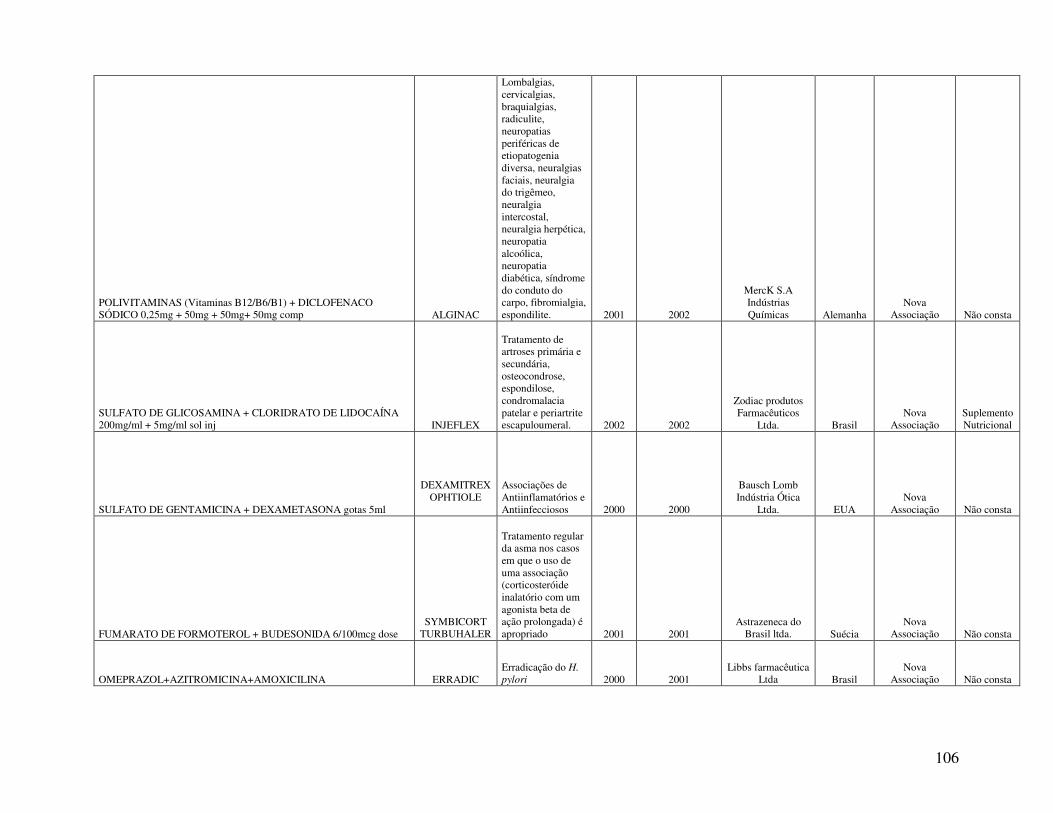
106
POLIVITAMINAS (Vitaminas B12/B6/B1) + DICLOFENACO SÓDICO 0,25mg + 50mg + 50mg+ 50mg comp ALGINAC
Lombalgias, cervicalgias, braquialgias, radiculite, neuropatias periféricas de etiopatogenia diversa, neuralgias faciais, neuralgia do trigêmeo, neuralgia intercostal, neuralgia herpética, neuropatia alcoólica, neuropatia diabética, síndrome do conduto do carpo, fibromialgia, espondilite. 2001 2002
MercK S.A Indústrias Químicas Alemanha
Nova Associação Não consta
SULFATO DE GLICOSAMINA + CLORIDRATO DE LIDOCAÍNA 200mg/ml + 5mg/ml sol inj INJEFLEX
Tratamento de artroses primária e secundária, osteocondrose, espondilose, condromalacia patelar e periartrite escapuloumeral. 2002 2002
Zodiac produtos Farmacêuticos
Ltda. Brasil Nova
Associação Suplemento Nutricional
SULFATO DE GENTAMICINA + DEXAMETASONA gotas 5ml
DEXAMITREX OPHTIOLE
Associações de Antiinflamatórios e Antiinfecciosos 2000 2000
Bausch Lomb Indústria Ótica
Ltda. EUA Nova
Associação Não consta
FUMARATO DE FORMOTEROL + BUDESONIDA 6/100mcg dose SYMBICORT
TURBUHALER
Tratamento regular da asma nos casos em que o uso de uma associação (corticosteróide inalatório com um agonista beta de ação prolongada) é apropriado 2001 2001
Astrazeneca do Brasil ltda. Suécia
Nova Associação Não consta
OMEPRAZOL+AZITROMICINA+AMOXICILINA ERRADIC Erradicação do H. pylori 2000 2001
Libbs farmacêutica Ltda Brasil
Nova Associação Não consta

107
TEGASERODE 2mg -comp ZELMAC
Tratamento da dor, do desconforto e da distensão abdominais e da alteração do funcionamento intestinal em pacientes com a síndrome do cólon irritável (SCI). 2002 2002
Novartis Biociência S.A. Suíça
Entidade Molecular Nova P
Fonte: Reis, 2004 (modificado) S = standart (padrão) P = priority (prioritário) V= medicamento órfão CBER = Center for Biologic Evaluation and Research CDER = Center for Drug Evaluation and Research

108
ANEXO 3
Quadro 9 - Medicamentos novos registrados na ANVISA e na EMEA, FDA e ANMAT
Medicamento Sede Fabricante
Registro ANVISA
Registro EMEA
Registro FDA
Registro ANMAT
AMOXICILINA + SULBACTAM 1000 mg + 500 mg Pó Injetável Alemanha 2000 Não Consta Não Consta Não Consta
ESOMEPRAZOL MAGNÉSIO TRIIDRATADO 20mg - comp Suécia 2000 Não Consta 2001 Sim
CLORIDRATO DE PIOGLITAZONA 15mg - comp EUA 2000 2000 1999 Sim
NATEGLINIDA 60mg - comp Suíça 2000 2001 2000 Sim
INSULINA ASPART 100UI/ml - 10ml Dinamarca 2000 1999 2000 Sim
MONONITRATO DE ISOSSORBIDA + ACIDO ACETILSALICILICO 60mg + 75mg - cap gel Brasil 2002 Não Consta Não Consta Não Consta
CILOSTAZOL 50mg- comp Brasil 2002 Não Consta 2004 Sim
FLUTRIMAZOL 0,01g/ml solução tópica spray Brasil 2000 Não Consta Não Consta Não Consta
PIMECROLIMUS 10mg/g creme - 15g Suíça 2002 Não Consta 2001 Sim
BUTIRATO DE HIDROCORTISONA 1mg/g creme - 15 g Brasil 2000 Não Consta 2002 Sim
ETONOGESTREL 68mg solução dermatológica - implante Holanda 2000 Não Consta Não Consta Sim
LUTROPINA ALFA (R-LH) 75UI pó liófilo inj Suíça 2001 2000 2004 Sim
CLORIDRATO DE APOMORFINA 2mg comp sub-lingual EUA 2001 2001 Não Consta Sim
ACETATO DE CETRORELIX 0,25mg po liof inj Suíça 2000 1999 2000 Sim
ACETATO DE GANIRELIX 0,5mg/ml sol inj Holanda 2001 2000 1999 Sim
TELITROMICINA 400mg - comp EUA 2001 2001 2004 Sim
QUINUPRISTINA + DALFOPRISTINA 150mg + 350mg po liof inj França 2000 Não Consta 1999 Sim
ACETATO DE CASPOFUNGINA 50mg po liof inj EUA 2001 2001 2001 Sim
ZANAMIVIR 5mg cap inalação EUA 2000 Não Consta 1999 Sim
OSELTAMIVIR 75mg - cap gel dura Suíça 2000 2002 1999 Não Consta
LOPINAVIR + RITONAVIR 133,3mg + 33,3mg cap gel mole EUA 2000 2001 2000 Sim
TEMOZOLOMIDA 5mg cap gel dura EUA 2000 1999 1999 Sim
MESILATO DE IMATINIB 50mg - cap gel dura Suíça 2001 2001 2003 Sim
EXEMESTANO 25mg drágeas Suécia 2000 2000 1999 Sim
PEGINTERFERON ALFA 2 B 50mcg/0,5ml - po liof inj EUA 2001 2000 2001 Sim
SIROLIMUS 1mg/ml solução oral EUA 2000 2001 1999 Sim
INFLIXIMAB 10mg/ml po liof EUA 2000 1999 1998 Sim
SULFATO DE GLICOSAMINA 250mg - cap gel dura Brasil 2000 Não Consta Suplemento Não Consta
ETORICOXIB 60mg - comp EUA 2002 2004 Não Consta Sim
RISEDRONATO SODICO 5mg - comp EUA 2000 Não Consta 1998 Sim
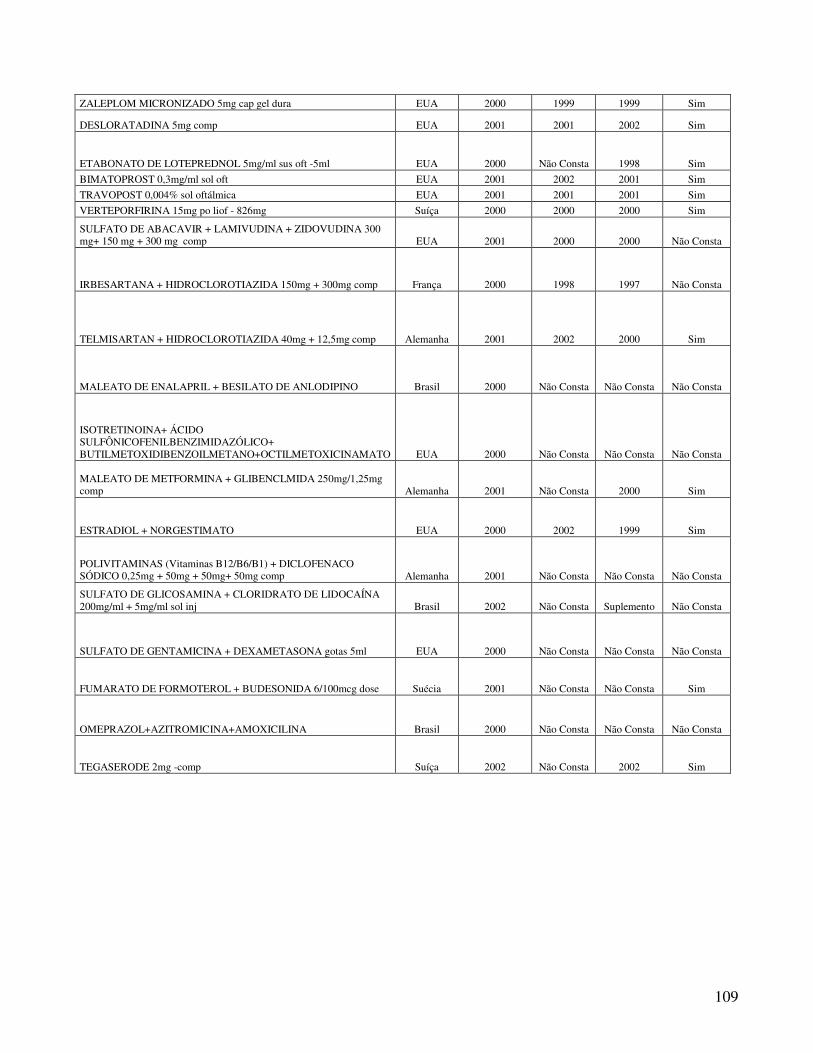
109
ZALEPLOM MICRONIZADO 5mg cap gel dura EUA 2000 1999 1999 Sim
DESLORATADINA 5mg comp EUA 2001 2001 2002 Sim
ETABONATO DE LOTEPREDNOL 5mg/ml sus oft -5ml EUA 2000 Não Consta 1998 Sim
BIMATOPROST 0,3mg/ml sol oft EUA 2001 2002 2001 Sim
TRAVOPOST 0,004% sol oftálmica EUA 2001 2001 2001 Sim
VERTEPORFIRINA 15mg po liof - 826mg Suíça 2000 2000 2000 Sim
SULFATO DE ABACAVIR + LAMIVUDINA + ZIDOVUDINA 300 mg+ 150 mg + 300 mg comp EUA 2001 2000 2000 Não Consta
IRBESARTANA + HIDROCLOROTIAZIDA 150mg + 300mg comp França 2000 1998 1997 Não Consta
TELMISARTAN + HIDROCLOROTIAZIDA 40mg + 12,5mg comp Alemanha 2001 2002 2000 Sim
MALEATO DE ENALAPRIL + BESILATO DE ANLODIPINO Brasil 2000 Não Consta Não Consta Não Consta
ISOTRETINOINA+ ÁCIDO SULFÔNICOFENILBENZIMIDAZÓLICO+ BUTILMETOXIDIBENZOILMETANO+OCTILMETOXICINAMATO EUA 2000 Não Consta Não Consta Não Consta
MALEATO DE METFORMINA + GLIBENCLMIDA 250mg/1,25mg comp Alemanha 2001 Não Consta 2000 Sim
ESTRADIOL + NORGESTIMATO EUA 2000 2002 1999 Sim
POLIVITAMINAS (Vitaminas B12/B6/B1) + DICLOFENACO SÓDICO 0,25mg + 50mg + 50mg+ 50mg comp Alemanha 2001 Não Consta Não Consta Não Consta
SULFATO DE GLICOSAMINA + CLORIDRATO DE LIDOCAÍNA 200mg/ml + 5mg/ml sol inj Brasil 2002 Não Consta Suplemento Não Consta
SULFATO DE GENTAMICINA + DEXAMETASONA gotas 5ml EUA 2000 Não Consta Não Consta Não Consta
FUMARATO DE FORMOTEROL + BUDESONIDA 6/100mcg dose Suécia 2001 Não Consta Não Consta Sim
OMEPRAZOL+AZITROMICINA+AMOXICILINA Brasil 2000 Não Consta Não Consta Não Consta
TEGASERODE 2mg -comp Suíça 2002 Não Consta 2002 Sim

110
ANEXO 4
Quadro 10 - Prazo em dias para o registro dos medicamentos na ANVISA e na FDA
Medicamento Data Registro Anvisa
Prazo Registro Anvisa (dias)
Data Registro FDA
Prazo Registro FDA (dias)
Classificação Medicamento Novo Anvisa
Potencial Terapêutico
FDA
AMOXICILINA + SULBACTAM 1000 mg + 500 mg Pó Injetável 24/5/2000 6 Não consta Não Nova Associação Não Consta
ESOMEPRAZOL MAGNÉSIO TRIIDRATADO 20mg - comp 29/12/2000 198 02/20/2001 445 Entidade Molecular Nova S
CLORIDRATO DE PIOGLITAZONA 15mg - comp 2/5/2000 186 07/15/1999 181 Entidade Molecular Nova P
NATEGLINIDA 60mg - comp 6/6/2000 117 22/12/2000 371 Entidade Molecular Nova S
INSULINA ASPART 100UI/ml - 10ml 28/12/2000 206 6/7/2000 213
Alteração na concentração de substância ativa ou de propriedades farmacocinéticas S
MONONITRATO DE ISOSSORBIDA + ACIDO ACETILSALICILICO 60mg + 75mg - cap gel Não Consta Não Consta Não Consta Não Consta Nova Associação Não consta
CILOSTAZOL 50mg- comp 5/6/2002 177 01/15/1999 340 Entidade Molecular Nova S
FLUTRIMAZOL 0,01mg/ml 6/4/2000 162 Não Consta Não Consta Entidade Molecular Nova Não consta
PIMECROLIMUS 10mg/g creme - 15g Não Consta Não Consta 12/13/2001 363 Entidade Molecular Nova S
BUTIRATO DE HIDROCORTISONA 1mg/g creme - 15 g 28/1/2000 261 22/5/2002 76
Sal novo, embora a entidade molecular corespondente já tenha sido autorizada S
ETONOGESTREL 68mg sol dermatológica implante 20/4/2000 343 Não Consta Não Consta Entidade Molecular Nova Não consta
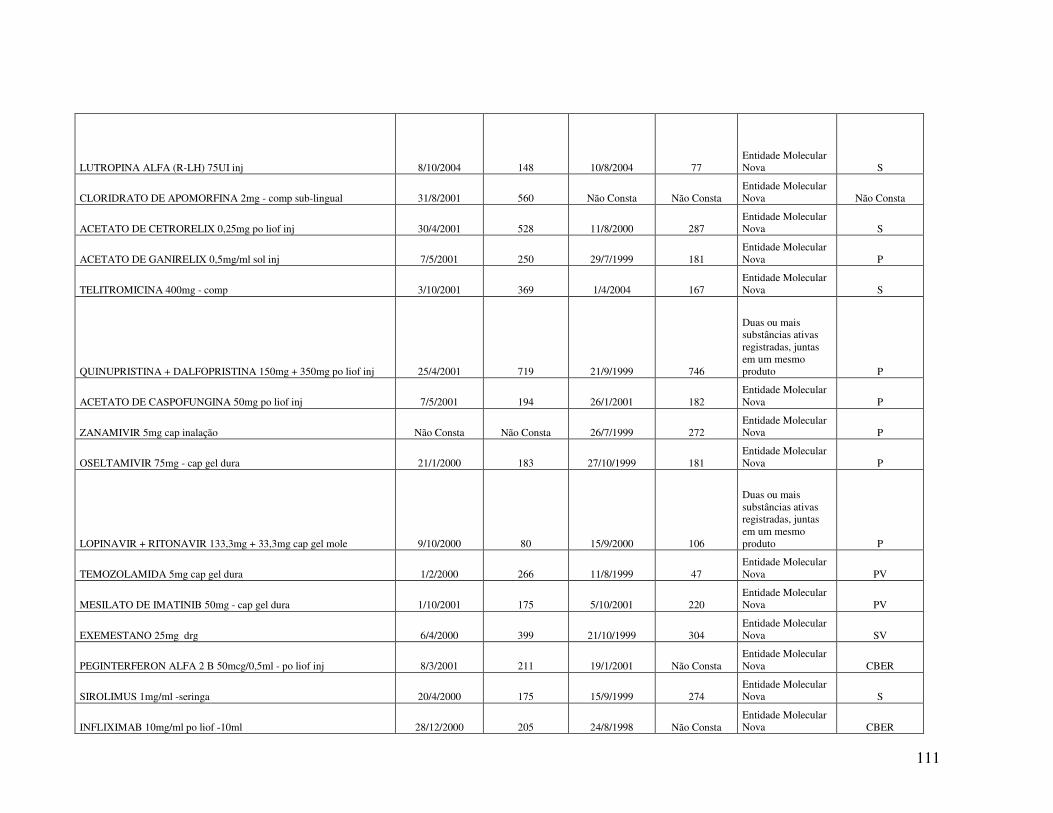
111
LUTROPINA ALFA (R-LH) 75UI inj 8/10/2004 148 10/8/2004 77 Entidade Molecular Nova S
CLORIDRATO DE APOMORFINA 2mg - comp sub-lingual 31/8/2001 560 Não Consta Não Consta Entidade Molecular Nova Não Consta
ACETATO DE CETRORELIX 0,25mg po liof inj 30/4/2001 528 11/8/2000 287 Entidade Molecular Nova S
ACETATO DE GANIRELIX 0,5mg/ml sol inj 7/5/2001 250 29/7/1999 181 Entidade Molecular Nova P
TELITROMICINA 400mg - comp 3/10/2001 369 1/4/2004 167 Entidade Molecular Nova S
QUINUPRISTINA + DALFOPRISTINA 150mg + 350mg po liof inj 25/4/2001 719 21/9/1999 746
Duas ou mais substâncias ativas registradas, juntas em um mesmo produto P
ACETATO DE CASPOFUNGINA 50mg po liof inj 7/5/2001 194 26/1/2001 182 Entidade Molecular Nova P
ZANAMIVIR 5mg cap inalação Não Consta Não Consta 26/7/1999 272 Entidade Molecular Nova P
OSELTAMIVIR 75mg - cap gel dura 21/1/2000 183 27/10/1999 181 Entidade Molecular Nova P
LOPINAVIR + RITONAVIR 133,3mg + 33,3mg cap gel mole 9/10/2000 80 15/9/2000 106
Duas ou mais substâncias ativas registradas, juntas em um mesmo produto P
TEMOZOLAMIDA 5mg cap gel dura 1/2/2000 266 11/8/1999 47 Entidade Molecular Nova PV
MESILATO DE IMATINIB 50mg - cap gel dura 1/10/2001 175 5/10/2001 220 Entidade Molecular Nova PV
EXEMESTANO 25mg drg 6/4/2000 399 21/10/1999 304 Entidade Molecular Nova SV
PEGINTERFERON ALFA 2 B 50mcg/0,5ml - po liof inj 8/3/2001 211 19/1/2001 Não Consta Entidade Molecular Nova CBER
SIROLIMUS 1mg/ml -seringa 20/4/2000 175 15/9/1999 274 Entidade Molecular Nova S
INFLIXIMAB 10mg/ml po liof -10ml 28/12/2000 205 24/8/1998 Não Consta Entidade Molecular Nova CBER
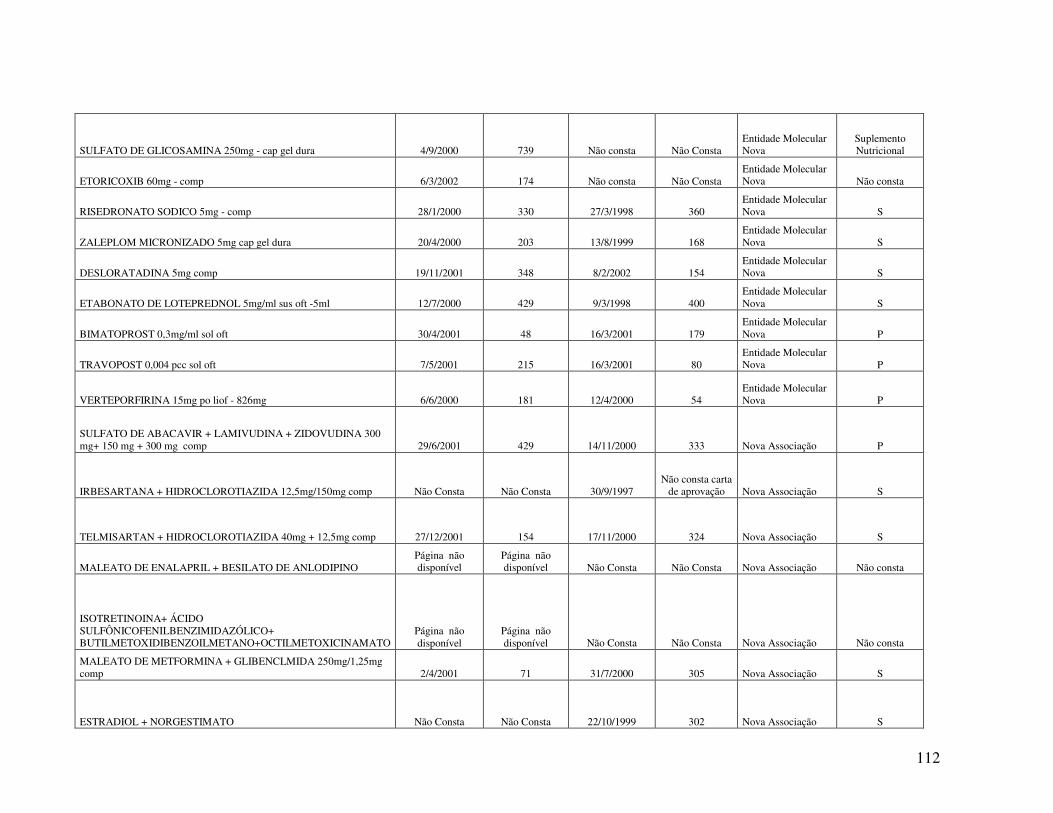
112
SULFATO DE GLICOSAMINA 250mg - cap gel dura 4/9/2000 739 Não consta Não Consta Entidade Molecular Nova
Suplemento Nutricional
ETORICOXIB 60mg - comp 6/3/2002 174 Não consta Não Consta Entidade Molecular Nova Não consta
RISEDRONATO SODICO 5mg - comp 28/1/2000 330 27/3/1998 360 Entidade Molecular Nova S
ZALEPLOM MICRONIZADO 5mg cap gel dura 20/4/2000 203 13/8/1999 168 Entidade Molecular Nova S
DESLORATADINA 5mg comp 19/11/2001 348 8/2/2002 154 Entidade Molecular Nova S
ETABONATO DE LOTEPREDNOL 5mg/ml sus oft -5ml 12/7/2000 429 9/3/1998 400 Entidade Molecular Nova S
BIMATOPROST 0,3mg/ml sol oft 30/4/2001 48 16/3/2001 179 Entidade Molecular Nova P
TRAVOPOST 0,004 pcc sol oft 7/5/2001 215 16/3/2001 80 Entidade Molecular Nova P
VERTEPORFIRINA 15mg po liof - 826mg 6/6/2000 181 12/4/2000 54 Entidade Molecular Nova P
SULFATO DE ABACAVIR + LAMIVUDINA + ZIDOVUDINA 300 mg+ 150 mg + 300 mg comp 29/6/2001 429 14/11/2000 333 Nova Associação P
IRBESARTANA + HIDROCLOROTIAZIDA 12,5mg/150mg comp Não Consta Não Consta 30/9/1997 Não consta carta
de aprovação Nova Associação S
TELMISARTAN + HIDROCLOROTIAZIDA 40mg + 12,5mg comp 27/12/2001 154 17/11/2000 324 Nova Associação S
MALEATO DE ENALAPRIL + BESILATO DE ANLODIPINO Página não disponível
Página não disponível Não Consta Não Consta Nova Associação Não consta
ISOTRETINOINA+ ÁCIDO SULFÔNICOFENILBENZIMIDAZÓLICO+ BUTILMETOXIDIBENZOILMETANO+OCTILMETOXICINAMATO
Página não disponível
Página não disponível Não Consta Não Consta Nova Associação Não consta
MALEATO DE METFORMINA + GLIBENCLMIDA 250mg/1,25mg comp 2/4/2001 71 31/7/2000 305 Nova Associação S
ESTRADIOL + NORGESTIMATO Não Consta Não Consta 22/10/1999 302 Nova Associação S

113
POLIVITAMINAS (Vitaminas B12/B6/B1) + DICLOFENACO SÓDICO 0,25mg + 50mg + 50mg+ 50mg comp 13/7/2001 248 Não consta Não Consta Nova Associação Não consta
SULFATO DE GLICOSAMINA + CLORIDRATO DE LIDOCAÍNA 200mg/ml + 5mg/ml sol inj 22/3/2002 283 Não consta Não Consta Nova Associação
Suplemento Nutricional
SULFATO DE GENTAMICINA + DEXAMETASONA gotas 5ml Não Consta Não Consta Não consta Não Consta Nova Associação Não consta
FUMARATO DE FORMOTEROL + BUDESONIDA 6/100mcg dose Página não disponível
Página não disponível Não consta Não Consta Nova Associação Não consta
OMEPRAZOL+AZITROMICINA+AMOXICILINA Não Consta Não Consta Não consta Não Consta Nova Associação Não consta
TEGASERODE 2mg -comp 5/6/2002 79 24/7/2002 146 Entidade Molecular Nova P