polimorfismos do gene egfr e associaÇÃo com...
TRANSCRIPT

POLIMORFISMOS DO GENE EGFR E
ASSOCIAÇÃO COM FATORES PROGNÓSTICOS
DO CÂNCER DE MAMA
MARCELO SOBRAL LEITE
RIO DE JANEIRO
2012
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
INSTITUTO DE CIÊNCIAS BIOMÉDICAS
DEPARTAMENTO DE FARMACOLOGIA E QUÍMICA
MEDICINAL

Marcelo Sobral Leite
POLIMORFISMOS DO GENE EGFR E
ASSOCIAÇÃO COM FATORES PROGNÓSTICOS
DO CÂNCER DE MAMA
Orientadora:
Prof.ª Dr.ª Rosane Vianna Jorge
Rio de Janeiro
2012
Dissertação apresentada à
Pós-Graduação em Farmacologia e
Química Medicinal do Instituto de
Ciâncias Biomédicas da
Universidade Ferderal do Rio de
Janeiro, como requisito à obtenção
do título de mestre em
Farmacologia.

Leite, Marcelo Sobral.
Polimorfismos do gene EGFR e associação com fatores prognósticos
do câncer de mama Rio de Janeiro: UFRJ/ICB, 2012.141f.
Orientadora: Profª. Drª Rosane Vianna-Jorge.
Dissertação (Mmstrado)–UFRJ/ICB/Pós-graduação de Farmacologia
e Química Medicinal, 2012.
Referências Bibliográficas: f. 110-132.
1. Câncer de mama. 2.Polimorfismo. I. Título.
CDD:

...à minha mãe.

AGRADECIMENTOS
Gostaria de agradecer primeiramente a todas as mulheres,
pacientes de câncer de mama do Hospital do Câncer III do INCA, pelo carinho,
sorrisos, forças, emoções e aprendizados. Sinceros agradecimentos às
pacientes que aceitaram participar deste estudo e se esforçaram para
responder sinceramente ao questionário aplicado.
Este trabalho não teria sido realizado sem o apoio dos diversos
setores do Hospital do Câncer III do INCA. Muito obrigado a todos os colegas
dos Serviços de Psicologia, Serviço Social, Fisioterapia, em especial ao
Fisioterapeuta Ricardo Dias, Érica, Maria Gisele, Nayana, Elisângela, e a
Zuleica, a Penha e tantos outros. Obrigado a todos do Laboratório de Análises
Clínicas, a Tereza, Rita, Rosane; da Oncologia Clínica, ao Dr. Luis Guilherme,
a todos da Farmácia, do Centro de Estudos, do Serviço de Quimioterapia e
todas as enfermeiras.
Agradeço imensamente a colaboração do prof. Rodrigo Moura-Neto e a
todos de seu laboratório, Priscila, Tatiana; a colaboração da Dra Gisele
Vigdinal do Serviço de Patologia do INCA; a colaboração do Departamento de
Genética do CPq/INCA, em especial a Kelly, Leila, Priscila, Hanna, João; a
colaboração do prof. Marcelo Alex, e de todos os colegas da farmacologia do
INCA, Renata, João, Giu, Cínthias, Mateus, Fê, Vera, Vanessa, Thales; a todos
da Pesquisa Clínica e BNT, Maurício, Fernandinha, Vivi, Haynna, Andrea e
Valadão; e aos amigos do Grupo de Farmacologia Clínica e Assistência
Farmacêutica: Ju, Carol, Diogo, Vanessa, Sheyla, João, Tales, Taiana, Padula,
Hayra, Laura, Camila, Márcia... e em especial a Letícia Giacomin pela parceria
e a Rosane pela orientação.
Agradeço ao Departamento de Farmacologia e Química Medicinal da
UFRJ, as professoras Tereza Sollero, Cláudia Benjamin, Cláudia Martins e
Eline, os professores Paulo Melo e Newton, e a todos os alunos da pós pelo
apoio incondicional.
Agradeço a American Association for Cancer Research e a Avon
Fundation pelo apoio, oportunidade e incentivo. Aos colegas da Perinatal,
Chico, Simones, Adriano, Anna, Ana, Susana, Michael, Odália, Dr. Jofre e Dr.
Fernando, Izaura, Marta, Fábia, etc...
Agradeço a todos os meus amigos que me apoiaram e incentivaram, a
minha irmã Gê, a Paulinha, Jô, Carol, Om, Clara, Gracinha, Chamusca, Torvi,
Ariel, Blenda, Billy, Pryia, amigo Felipe, Enoque e todos da Mansão. Gio,
obrigado, sua força chegou aqui.
Por fim, minha eterna gratidão ao meu Mestre da Vida, Dr. Daisaku
Ikeda, Presidente da Soka Gakkai Internacional; e a todos os amigos místicos

da Associação Brasil SGI, Simas, Monique, Selma, Joanas, Cida, Wal,
Giovana, Ari, Naza, Earl Taylor, Márcia Nicolau e tantos maravilhosos
companheiros! Forte agradecimento ao Paulo, Vitor, Tom e todos os membros
do Tayio Ongakutai!
A minha afilhada Letícia, meus cumpadres Lázaro e Yara, pela ausência
necessária. A toda minha família, primo Augusto, Vanessa, prima Nina, Andrea
tia Celita, Tania, minha Vó, Laís e Alexandre. Meus sinceros agradecimentos
eternos ao meu pai, e a minha mãe.

"Se você quer descobrir alguma
coisa, tem que acordar e dormir com
essa coisa na cabeça. Não pode
dispersar. “
Octavio Augusto Ceva Antunes

Lista de Simbolos
º grau(s)
® marca registrada
% porcentagem
∑ somatória
≤ menor ou igual
≥ maior ou igual
< menor que
> maior
μ micro (10-6)
Lista de Abreviaturas
ác. ácido
ajus ajustado
Arg arginina
cr crude
IC95% Intervalo de confiança de 95%
Lys lisina
mg miligramas
min minuto(s)
N número de amostras
p/v peso por volume
pb pares de base
pN status linfonodal
pT tamanho tumoral
pTMN estadiamento TNM
p/ para
s/ sem

Lista de Siglas
ASCO American Society of Clinical Oncology
CAP College of American Pathologists
DNA ác. desoxirribonucleico
EGF epidermal growth factor.
EGFR receptor de EGF
ERR Estimativa de Risco de Recorrência da Doença
GSK-3ß glicogênio sintase cinase 3ß
HBEGF heparin‑binding EGF‑like growth factor
INCA Intituto Nacional do Câncer
IP3 trifosfato de inositol-1,4,5
MAPK cinases ativadas por mitógeno
NPI Nottingham Prognostic Index
NRG neuregulin
OR odds ratio
PCR polymerase chain reaction
PI3K fosfatidilinositol trifosfato cinase
PIP3 fosfatidilinositol trifosfato
RE Receptor de Estrogênio
RFLP restriction fragment length polymorphism
RNA ác. ribonucleico
RP Receptor de Progesterona
SNP single nucleotide polymorphism -
TCLE Termo de Consentimento Livre e Esclarecido
TGFα transforming growth factor‑α
UICC International Union Against Cancer
WHO World Health Organization

SUMÁRIO
RESUMO
ABSTRACT
1. INTRODUÇÃO......................................................................... 13
1.1. CÂNCER........................................................................... 13
1.2. CÂNCER DE MAMA........................................................ 20
1.3. POLIMORFISMOS GENÉTICOS...................................... 35
2. OBJETIVOS........................................................................... 49
2.1. OBJETIVO GERAL........................................................... 49
2.2. OBJETIVOS ESPECÍFICOS............................................. 49
3. MATERIAL E MÉTODOS....................................................... 50
3.1. DESENHO DO ESTUDO.................................................. 50
3.2. PACIENTES..................................................................... 50
3.3. FORMULÁRIOS............................................................... 51
3.4. VARIÁVEIS CLINICO-PATOLÓGICAS............................ 51
3.5. ANÁLISE DOS POLIMORFISMOS.................................. 60
3.6. ANÁLISE ESTATÍSTICA.................................................. 71
4. RESULTADOS....................................................................... 72
4.1. POPULAÇÃO................................................................... 72
4.2. ASSOCIAÇÕES GENÓTIPO X FENÓTIPO.................... 78
5. DISCUSSÂO.......................................................................... 101
6. CONCLUSÂO........................................................................ 110
7. REFERÊNCIAS..................................................................... 111
ANEXOS

RESUMO
Introdução: O receptor do fator de crescimento epidermal (EGFR) é um
receptor tirosina-cinase que desempenha papel crucial no crescimento celular,
regulando a diferenciação das células e tecidos. O EGFR encontra-se
frequentemente super expresso em muitos tumores, inclusive no câncer de
mama e contribui para uma proliferação anômala. Um polimorfismo do gene
EGFR é uma sequência de repetição CA no intron 1 (rs72554021). Sequências
mais curtas, i. e (CA)n ≤ 16 são associadas a maior atividade transcricional em
tumores de mama. Outro polimorfismo, uma troca de um único nucleotídeo,
R497K (rs11543848), localizado no exon 13, está associado a um prognóstico
favorável em carcinoma colorretal e em câncer de pulmão.O objetivo deste
estudo foi investigar a associação entre estes polimorfismos do gene EGFR e
variáveis histopatológicas com valor prognóstico em câncer de mama.
Materiais e métodos: A população de estudo foi formada por mulheres
brasileiras (≥ 18 anos de idade), com diagnóstico confirmado de câncer de
mama unilateral não metastático. O DNA genômico foi extraído de amostras de
sangue e a análise do tamanho de fragmento do intron 1 foi determinado por
eletroforese de capilar em 426 pacientes, enquanto o polimorfismo R497K foi
identificado em 472 pacientes por reações de PCR seguidas de digestão com
enzima de restrição. O perfil histopatológico foi determinado após a ressecção
do tumor. A associação entre os genótipos do EGFR e as características
histopatológicas foram analisadas pelo teste do chi-quadrado, com cálculo das
razões de chance (odds ratio, OR) simples e ajustadas, com seus intervalos de
confiança (IC95%). Resultados e discussão: Onze alelos diferentes foram
encontrados para o polimorfismo (CA)n, variando de 14 a 24 repetições,
formando 37 genótipos diferentes. O alelo mais frequente foi o de 16 repetições
CA (0,425; IC95%, 0,398-0,460). A frequência do alelo variante do R497K
(Lys), foi de 0,218 (IC95%, 0,192–0,245). Pacientes com os dois alelos longos,
i.e (CA)n > 16 repetições, mostraram menor chance de apresentar status de
receptor de progesterona negativo (ORajus = 0,324; IC 95%, 0,124–0,845).
Pacientes com pelo menos um alelo Lys mostraram menor chance de
apresentar status linfonodal pN2 ou pN3 (ORajus = 0,332; IC95%, 0.164–0.671),
o que contribui para melhor estadiamento TNM (ORajus = 0,389; IC95%, 0,202–
0,749) e melhor estimativa de risco de recorrência (OR = 0.470; IC95%, 0,278–
0,794) quando comparados aos pacientes com genótipo R497K predominante
(Arg/Arg). Pacientes com pelo menos um alelo Lys e com os dois alelos de
repetição CA longos mostraram menor estimativa de risco de recorrência (OR =
0,210; IC95%, 0,063–0,695). Conclusão: A combinação dos dados dos dois
polimorfismos do gene EGFR sugere que a presença das formas variantes
contribui para melhor prognóstico no câncer de mama.
Palavras-chave: EGFR, polimorfismo, câncer de mama, prognóstico.

ABSTRACT
Introduction: The epidermal growth factor receptor (EGFR) is a
transmembrane tyrosine kinase receptor, which plays an essential role in
growth by regulating the differentiation of cells and tissues. EGFR is frequently
overexpressed in many tumors, including breast cancer, and contributes to
unrestricted proliferation. One EGFR gene polymorphism is a (CA)n
dinucleotide repeat sequence (rs72554021) in intron 1. Shorter sequences, i.e
(CA)n ≤ 16 dinucleotide repeats are associated with increased EGFR
transcriptional activity in breast tumors. Other, single nucleotide polymorphism,
R497K (rs11543848), located in exon 13, is associated with favorable prognosis
in colorectal carcinoma and lung cancer. The aim of the present study was to
investigate the association between EGFR polymorphisms and
histopathological variables with prognostic value in breast cancer. Material and
method: The study population consisted of Brazilian women (≥ 18 years old)
with a confirmed diagnosis of unilateral non-metastatic breast cancer. Genomic
DNA was extracted from blood samples and the fragment length of the intron 1
was determined by capillary electrophoresis in 426 patients, whereas R497K
was identified in 472 patients by PCR-RFLP. The histopathological profile was
determined after tumor resection. The association between EGFR genotypes
and histopathological features was evaluated by the Chi-square test, with
calculation of the adjusted odds ratios (ORajus) and their 95% confidence
intervals (95%CI). Results and discussion: Eleven different (CA)n alleles were
found, ranging from 14 to 24 repeats, forming thirty seven different genotypes.
The major frequent allele was (CA)16 (0.425; 95%CI, 0.398–0.460).The
frequency of the R497K variant (Lysi) allele was 0.218 (95%CI; 0.192–0.245).
Patients with two long alleles, i.e. (CA)n > 16 repeats, showed a lower chance
of being negative for progesterone receptor (ORajus = 0.324; 95%CI, 0.124–
0.845). Patients with at least one Lys allele showed a lower chance of
presenting lymph node status pN2 or pN3 (ORajus = 0.332; 95%CI, 0.164–0.671),
which contributed for lower stages in TNM status, (ORajus = 0.389; 95%CI, 0.202–
0.749) and lower estimated risk of recurrence (OR = 0.470; 95%CI, 0.278–
0.794), than those who were Arg/Arg. Patients with at least one Lys allele and
with two long (CA)n alleles showed a lower estimated risk of recurrence (OR =
0.210; 95%CI, 0.063–0.695). Conclusion: The combined data of R497K and
(CA)n repeat polymorphism suggests that the presence of the variant forms of
EGFR contribute for better prognosis in breast cancer.
Key-words: EGFR, polymorphism, breast cancer, prognosis.

13
1 INTRODUÇÃO
1.1 CÂNCER
Câncer é o nome genérico dado a um conjunto de mais de 100 doenças
que têm em comum o crescimento desordenado de células que invadem
tecidos e órgãos. Dividindo-se rapidamente, estas células tendem a ter um
perfil metabólico anômalo e arranjo celular desordenado, determinando a
formação de tumores malignos, que podem espalhar-se para outras regiões do
corpo (Moore 1966).
O câncer pode ser considerado uma doença genética, uma vez que é
desencadeado por alterações no DNA da célula. No entanto, o câncer não é
necessariamente uma doença hereditária. Os cânceres humanos são, na sua
maioria, de origem somática, resultantes da interação de fatores genéticos e
ambientais (Perera et al. 1997).
A incidência de câncer no mundo vem aumentando rapidamente nos
últimos anos em decorrência do envelhecimento da população mundial e da
crescente exposição a fatores de risco ambientais. A Organização Mundial da
Saúde estimou que, no ano 2030, podem-se esperar 27 milhões de casos
incidentes de câncer, 17 milhões de mortes por câncer e 75 milhões de
pessoas vivas, anualmente, com câncer (WHO 2010). Assim, terapias cada vez
mais eficazes para o combate a esta enfermidade e a otimização de terapias
utilizadas atualmente se fazem necessárias.
No Brasil, as estimativas do Instituto Nacional do Câncer para o ano de
2012 (válidas também para 2013) apontam a ocorrência de 518.510 casos
novos de câncer, incluindo os casos de pele não melanoma, reforçando a
magnitude do problema do câncer no país (INCA 2011).
O quadro 1 mostra os 10 tumores com maiores estimativas de incidência
em 2012, excetuando-se o câncer de pele não melanoma, e suas respectivas
distribuições percentuais de acordo com o sexo (INCA 2011).

14
1.1.1 Biologia do câncer
A formação de um tumor é um processo complexo e frequentemente se
estende por décadas. A maioria dos tumores humanos se desenvolve a partir
de tecidos epiteliais. Os tumores oriundos destes tecidos são denominados
carcinomas (Peto 2001).
O aumento no grau de anormalidade tecidual de células em crescimento
e proliferação exacerbada é um processo comum no caminho da progressão
tumoral, no qual células consideradas normais se desenvolvem
progressivamente em fenótipos malignos, passando por etapas que mantém o
seguinte sentido: normal → hiperplásico → displásico → neoplásico →
metastático. A ciência busca entender a relação entre causa e efeito destes
eventos (Preston-Martins et al 1990).
Hanahan et al. (2000) enumeraram as seis alterações celulares básicas
que contribuem para a carcinogênese. Podem ser citadas: autossuficiência em
relação aos fatores de crescimento; insensibilidade aos inibidores de
crescimento; evasão à morte celular programada por apoptose; potencial
ilimitado de replicação; angiogênese sustentada; invasão tecidual;
disseminação à distância (figura 1).
Quadro 1: Distribuição proporcional dos dez tipos de câncer mais incidentes estimados para
2012 por sexo, exceto pele não melanoma (números arredondados para 10 ou múltiplos de 10)
(INCA 2011).

15
Assim, diversos genes associados a estes mecanismos e ao reparo aos
danos do DNA são atualmente estudados buscando a compreensão do
processo de carcinogênese (Hoeijmakers 2001; Wilson et al. 2002).
1.1.1.1 Fatores de crescimento
As células normais necessitam de sinais mitogênicos para desencadear
seus processos de transformação do estado quiescente para os estados ativo
ou proliferativo (Fedi et al. 1997). São sinais estimulatórios, mediados por
fatores de crescimento, que se ligam a receptores transmembranares, que
integram tal sinal de ligação a circuitos complexos dentro da célula (Alberts et
al. 2004). Em sequência ao evento inicial de ligação entre o fator de
crescimento e seu respectivo receptor, diversas moléculas mensageiras
realizam a transdução de sinal via uma rede de proteínas transdutoras que irão
desencadear atividades celulares, tais como a transcrição gênica no núcleo e o
repasse destas informações para células vizinhas (Alvarado et al. 2010). Esta
Figura 1: Capacidades adquiridas na carcinogênese.
Ocorrem de maneira semelhante nos processos de
desenvolvimento dos tumores, através de funções
originadas por diversos mecanismos e estratégias
celulares. Adaptado de Hanahan et al. (2000).

16
cadeia de eventos se inicia com a fosforilação de resíduos de tirosina na
porção intracelular do receptor e, para tanto, os domínios com atividade
tirosina-cinase cumprem papel crucial no mecanismo de transdução de sinal
proliferativo (Aaronson 1991; Avraham & Yarden 2011).
A família ErbB de receptores com atividade tirosina-cinase consiste de
quatro membros: receptor para fator de crescimento epidermal (EGFR; ErbB-
1), HER2 (ErbB-2 ou Her2-neu), HER3 (ErbB-3) e HER4 (ErbB-4) (figura 2);
que desempenham funções envolvidas nos processos de migração,
crescimento, adesão e diferenciação celular, eventos chaves na progressão
tumoral (Shepard et al. 2008; Flynn et al. 2009; Da Silva et al. 2010).
1.1.1.1.1 EGFR
O receptor do fator de crescimento epidermal (epidermal growth factor
receptor – EGFR), é uma estrutura transmembrana sensível a sinais
proliferativos representados pelo fator de crescimento epidermal (epidermal
growth factor – EGF) e pelo fator de transformação e crescimento α
(transforming growth factor-α – TGF-α). O receptor do fator de crescimento
Epidermal (EGFR) é uma glicoproteína transmembrana de 170 kDa, com
atividade tirosina-cinase, que tem um papel crucial na proliferação,
Figura 2: Representação esquemática dos ligantes da família ErbB de Homo sapiens. A semelhança entre as cadeia de aminoácidos dos os receptores varia de 53% entre EGFR e ErbB3 a 64% entre EGFR e ErbB2 (Jorissen 2003). Pelo ponto de vista tridimensional, as regiões extracelulares da região C-terminal se diferenciam mais entre os homólogos da família. Quando ligantes específicos interagem com o domínio cinase, ocorre a formação de dímeros e ativação dos domínios tirosina-cinase e promoção da atividade intracelular. A formação de homodímeros e heterodímeros é definida de acordo com os ligantes e a
interação entre os homólogos (Olayioye et al. 2000). HBEGF: heparin‑binding EGF‑like
growth factor; NRG: neuregulin; TGFα, transforming growth factor‑α; EGFR: epidermal
growth factor. Adaptado de Avraham & Yarden (2011).

17
diferenciação e motilidade de células normais e tumorais (Jimeno & Hidalgo
2006). O EGFR é uma proteína codificada pelo gene EGFR, que está situado
no braço longo do cromossomo 7, sendo codificado a 25 exons. É sintetizado a
partir de um precursor polipeptídico de 1210 resíduos e, após clivagem de
sequência da porção N-terminal, a proteína de 1186 resíduos é inserida na
membrana celular (Jorissen 2003).
A ligação de fatores específicos promove a formação de homodímeros
ou heterodímeros com outros membros da família erbB, desencadeando a
fosforilação de vários resíduos de tirosina do domínio intracelular (Olayioye et
al. 2000; Modjtahedi & Essapen 2009) (figura 3). Aqui, são descritos alguns
mecanismos que resultam na fosforilação de vários substratos intracelulares
que irão ativar diversas cascatas intracelulares (Shepard et al. 2008; Spano et
al. 2008).
A cascata de fosforilação desencadeada pela ativação do EGFR vai
atuar através da atividade de uma complexa rede de proteínas que atuam
como mensageiras da transdução de sinais proliferativos (Mendelsohn &
Baselga 2003; Jimeno & Hidalgo 2006; Flynn et al. 2009; Jorissen 2003) (figura
4). Um dos principais mecanismos do receptor envolve a ativação da proteína
Ras via um fator de troca de nucleotídeo de guanina, levando à atividade de
uma série de cinases ativadas por mitógeno (MAPK), como Raf, MEK, Erk1 ou
2, que irão regular a transcrição de genes e fatores de transcrição (c-myc, c-
Figura 3: Um receptor de fator de crescimento epidermal funcionando normalmente emite sinais (em vernelho) em resposta à interação com o ligante
Adaptado de Weinberg (2008).

18
Jun, JNK e p38) associados ao início do ciclo de divisão e transformação
celular (Jimeno & Hidalgo 2006; Modjtahedi & Essapen 2009).
A ação da cascata de fosforilação do receptor EGFR desencadeia
também a formação de trifosfato de inositol-1,4,5 (IP3) e diacilglicerol, pela
ação catalítica da enzima fosfolipase C sobre o fosfatidilinositol exposto na
porção hidrofílica citoplasmática da bicamada lipídica. Estes eventos
desencadeiam a formação de fosfatidilinositol trifosfato (PIP3) pela ação da
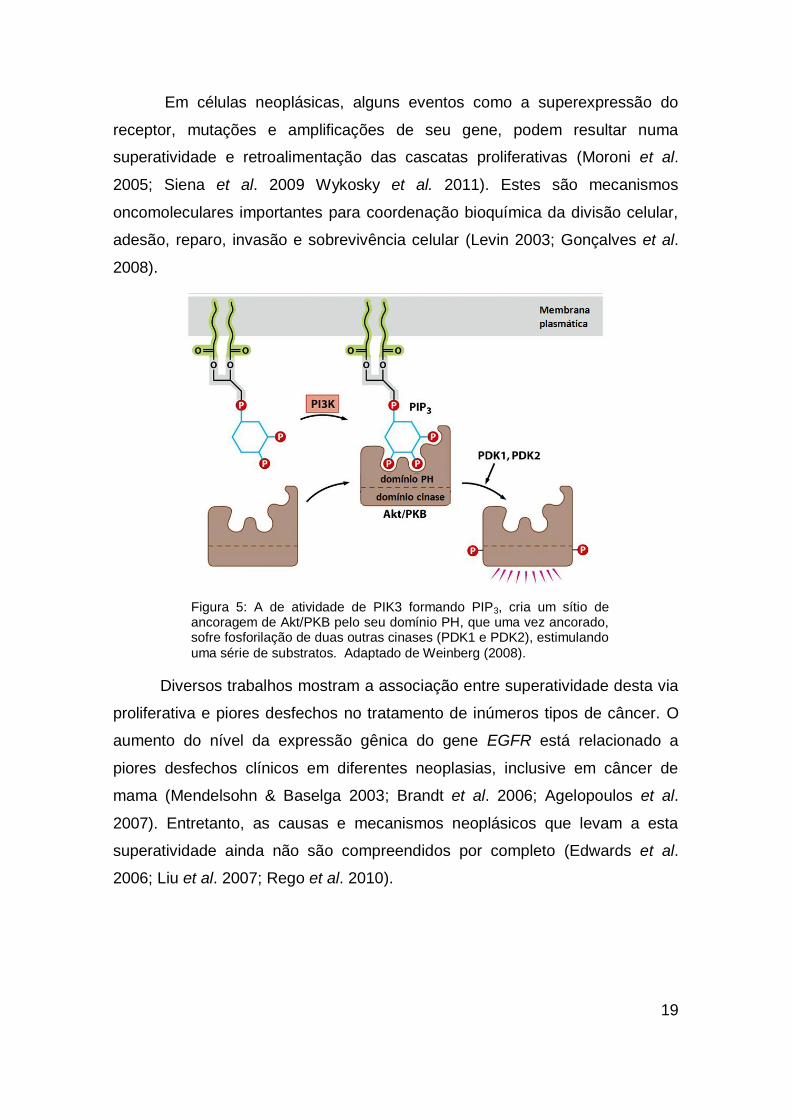
fosfatidilinositol trifosfato cinase (PI3K) (figura 5). PIP3 torna-se um sítio de
ancoragem de uma cinase treonina-serina chamada Akt/PKB que
desencadeará o trabalho de uma série de fatores de síntese proteica, como
mTOR (mammalian Target of Rapamycin) e de estimulação da proliferação
celular, como o glicogênio sintase cinase 3ß (GSK-3ß) (Kallergi et al. 2008;
Gori et al. 2009).
A ativação da via do PI3K via EGFR desempenha um papel central
numa série de rotas sinalizadoras associadas ao crescimento, mobilidade,
proliferação, diferenciação e a sobrevivência celular, além de influenciar a
redução da possibilidade de ativação do programa de apoptose (Zhang et al.
2006; Dong et al. 2009; Matsubara et al. 2010).
Figura 4: Exemplo de associação intermolecular para ativação de Ras via associação com Sos, um fator de troca de guanina. Uma proteína adaptadora Grb2 utiliza seus domínios SH3 para ligar Sos e seu domínio Sh2 para ligar-se a uma fosfotirosina numa proteína de sustentação Shc, que por sua vez, utiliza seu próprio domínio Sh2 para ancorar numa fosfotirosina do receptor
EGFR quando este é ativado. Adaptado de Weinberg (2008).
19
Em células neoplásicas, alguns eventos como a superexpressão do
receptor, mutações e amplificações de seu gene, podem resultar numa
superatividade e retroalimentação das cascatas proliferativas (Moroni et al.
2005; Siena et al. 2009 Wykosky et al. 2011). Estes são mecanismos
oncomoleculares importantes para coordenação bioquímica da divisão celular,
adesão, reparo, invasão e sobrevivência celular (Levin 2003; Gonçalves et al.
2008).
Diversos trabalhos mostram a associação entre superatividade desta via
proliferativa e piores desfechos no tratamento de inúmeros tipos de câncer. O
aumento do nível da expressão gênica do gene EGFR está relacionado a
piores desfechos clínicos em diferentes neoplasias, inclusive em câncer de
mama (Mendelsohn & Baselga 2003; Brandt et al. 2006; Agelopoulos et al.
2007). Entretanto, as causas e mecanismos neoplásicos que levam a esta
superatividade ainda não são compreendidos por completo (Edwards et al.
2006; Liu et al. 2007; Rego et al. 2010).
Figura 5: A de atividade de PIK3 formando PIP3, cria um sítio de ancoragem de Akt/PKB pelo seu domínio PH, que uma vez ancorado, sofre fosforilação de duas outras cinases (PDK1 e PDK2), estimulando
uma série de substratos. Adaptado de Weinberg (2008).

20
1.2 CÂNCER DE MAMA
1.2.1 Epidemiologia
O câncer da mama é o tipo de câncer que mais acomete as mulheres
em todo o mundo, tanto em países em desenvolvimento quanto em países
desenvolvidos. Cerca de 1,4 milhões de casos novos dessa neoplasia foram
esperados para o ano de 2008 em todo o mundo, o que representa 23% de
todos os tipos de câncer (WHO 2010).
Apesar de apresentar um bom prognóstico quando diagnosticado
precocemente e tratado imediatamente, o câncer de mama é responsável por
18% dos óbitos por câncer, em mulheres, por ano (INCA 2009). De acordo com
dados do INCA, esperam-se, para o Brasil, em 2012, 52.680 casos novos de
câncer da mama, com um risco estimado de 52 casos a cada 100 mil mulheres
(INCA 2011). Dessa forma, o câncer de mama é considerado um problema de
saúde pública no Brasil.
No estágio atual do conhecimento, não existem medidas específicas de
prevenção primária para o câncer de mama. A detecção precoce é o objetivo
principal, visando à diminuição da mortalidade e o aumento da sobrevida livre
de doença (Lee et al. 2012), com este objetivo, diversos países, inclusive o
Brasil, implantaram programas de rastreamento da doença com exame
mamográfico. (Boyle & Levin 2008; INCA 2011).
1.2.2 Fatores de risco
Embora não se conheça exatamente todo o mecanismo causal do
câncer de mama, não há dúvida de que a interação entre os fatores genéticos
e ambientais exerce papel fundamental na etiologia e na evolução dos casos
(Mill et al. 2010).
Dados clínicos, epidemiológicos e experimentais têm demonstrado que o
risco de desenvolvimento de câncer de mama esporádico está fortemente
relacionado à produção de esteroides sexuais. Condições endócrinas
moduladas pela função ovariana, como a menarca precoce, menopausa tardia

21
e gestação, assim como a utilização de estrógenos exógenos, são
componentes relevantes do risco de desenvolvimento do câncer de mama
(MacMahon et al. 1970; Lambe et al. 1996; Greenlee et al, 2000).
A utilização de contraceptivos hormonais orais e terapia hormonal pós-
menopausa podem causar pequenos aumentos no risco de câncer de mama
(Key et al. 2001). É controversa a hipótese de que o consumo de álcool (Kelsey
et al. 1996), o sedentarismo, a obesidade e a dieta lipídica aumentam o risco
de câncer de mama (Marchant, 1997; National Cancer Institute Bethesda 2008;
Mill et al. 2010).
Entre os fatores ambientais, a exposição à radiação ionizante é
sabidamente associada ao câncer de mama. Os campos eletromagnéticos
assim como os pesticidas também têm sido apontados como possíveis
responsáveis pelo aparecimento deste tipo de câncer (Kelsey et al. 1996; INCA
2011).
Estima-se que cerca de 5 a 10% dos casos de câncer de mama estejam
ligados a causas hereditárias de desenvolvimento do câncer. Nestes casos é
possível determinar claramente um padrão de herança familiar associada a
mutações em genes determinantes de susceptibilidade ao câncer (Easton
1993; Ford 1998). Os casos de câncer de mama hereditários têm como
importante característica o acometimento de mulheres jovens. Quando a
doença se apresenta antes dos 35 anos, em 25% dos casos existe uma
relação com fatores hereditários (Gomes et al. 2002; Anders et al. 2008).
BRCA1 e BRCA2 são genes humanos que estão na classe dos genes
conhecidos como supressores de tumor. Mutações nestes genes estão ligadas
ao aumento do risco de câncer de mama hereditário e câncer de ovário (Dapic
et al 2005). Mulheres com predisposição hereditária associada a mutações no
gene BRCA1 têm um risco estimado de 56-80% para o desenvolvimento do
câncer de mama (contra 11% na população geral) e de 16 a 60% para câncer
de ovário (contra 1,4 a 2,5% na população geral) (Miki et al 1994; Spurdle et al.
2011).

22
1.2.3 Diagnóstico
O advento da mamografia e sua modernização têm trazido benefício
para o diagnóstico do câncer de mama. Diante de leões suspeitas detectadas
pelos exames de imagem, a prática das biópsias percutâneas como punção
aspirativa com agulha fina, core-biopsy ou mamotomia possibilitou a
confirmação ou a invalidação de malignidade, evitando-se procedimentos mais
invasivos (Fumagalli & Sotiriou 2010; Leong & Zhuang 2011). O diagnóstico do
câncer de mama em estádio inicial tem sido associado a muitas variáveis, entre
elas, a posição sócio-econômica e o acesso à saúde oferecido à população
(Lanin et al. 1998; Richardson et al. 2001; Bradley et al. 2002; Lee et al. 2012).
O sistema de estadiamento usado com mais freqüência é o de
classificação TNM (T = tamanho; N = nódulos linfáticos comprometidos; M =
metástases a distância) da International Union Against Cancer (UICC, 2002).
Com relação ao Brasil, com base nos dados disponíveis de registros
hospitalares, 60% dos tumores de mama, em média, são diagnosticados em
estádio III ou IV (INCA 2005; Simon et al. 2009).
1.2.4 Patologia
As neoplasias de mama têm apresentação clínico-patológica e
comportamento biológico extremamente heterogêneo, em parte pela variedade
de tecidos envolvidos na gênese das lesões epiteliais, mesenquimais e
mioepiteliais, em parte pela composição genética de cada tipo histológico. O
conhecimento dos aspectos morfológicos, moleculares e bioquímicos tem sido
importante no sentido de auxiliar nas estratégias de tratamento local e
sistêmico (Peto 2001; Yenidunya et al. 2011). Dentre os fatores clínico-
patológicos, o tamanho do tumor, o grau tumoral, o estado do envolvimento dos
linfonodos regionais e a idade seguem sendo as variáveis de maior impacto no
prognóstico do câncer de mama (Arriagada et al. 2006; Soerjomataram et al.
2008; Anders et al. 2008). O tipo histopatológico, o grau nuclear, o nível de
expressão do receptor HER-2 e do receptor de estrogênio, entre outras

23
variáveis, também contribuem para refinar a classificação dos casos (Carvalho
et al. 2011; Danova et al. 2011).
1.2.4.1 Tipos histopatológicos
A classificação histopatológica baseia-se em características celulares e
padrão de crescimento de células sem considerar o local de origem da
neoplasia. Adjetivos como ductal ou lobular definem padrões de apresentação
morfológica com critérios definidos pela Organização Mundial de Saúde
(Travassoli & Devilee 2003) e pelo College of American Pathologist
(Fitzgibbons et al. 2000). Quanto ao aspecto morfológico, as neoplasias de
mama se apresentam na forma in situ ou invasiva.
1.2.4.1.1 Carcinomas in situ
Caracteriza-se por proliferação epitelial neoplásica, sem sinais de
ultrapassagem da membrana basal (Pinder & Ellis 2003). São comumente
considerados como uma etapa na sequência que antecede o carcinoma
invasivo, entretanto, é importante reconhecer que nem sempre o processo é
contínuo, e nem todos os carcinomas in situ evoluem para a forma invasiva
(Silverstein et al, 1996).
1.2.4.1.2 Carcinomas Invasivos
Apresentam-se geralmente constituídos por células epiteliais poligonais,
com atipia variável, distribuídas em agrupamentos coesos com a tendência de
formação de espaços em meio ao estroma com variáveis desmoplásicas e
infiltrados linfoides (Fitzgibbons et al. 1999). O tipo mais frequente é o ductal,
seguido de longe pelo carcinoma lobular. Entre os tipos especiais, alguns têm
comportamento biológico mais favorável em relação ao tipo ductal, como os
carcinomas tubular, mucinoso do tipo colóide, cribriforme infiltrativo, secretor e
adenocístico, enquanto outros têm comportamento mais agressivo, tais como o
metaplásico e o micropapilar invasivo (Di Saverio et al. 2008).
24
1.2.4.2 Grau Histológico
Os carcinomas invasivos são subdivididos de acordo com o grau de
diferenciação celular. Esta graduação busca estimar o quanto as células
neoplásicas se distanciam de um fenótipo normal (E. A. Rakha, Reis-filho, et al.
2010). Dados clínicos e patológicos vêm mostrando que o grau histológico é
um fator que, aliado ao status linfonodal, tem grande valor na predição de
desfecho da doença. Sua caracterização é relativamente simples, com custo
econômico baixo, requerendo apenas a avaliação de um patologista treinado
para análise microscópica de um corte tecido tumoral corado em hematoxilina-
eosina. (Galea et al. 1992; Walker 2003).
Dentre os sistemas mais usuais de graduação histológica, destacam-se
a classificação de Scarff-Bloom-Richardson, o sistema de graduação nuclear
de Fisher e o sistema de Nottingham. Em todos eles, o aumento do grau é
associado a piores desfechos clínicos (Harris et al. 2004).
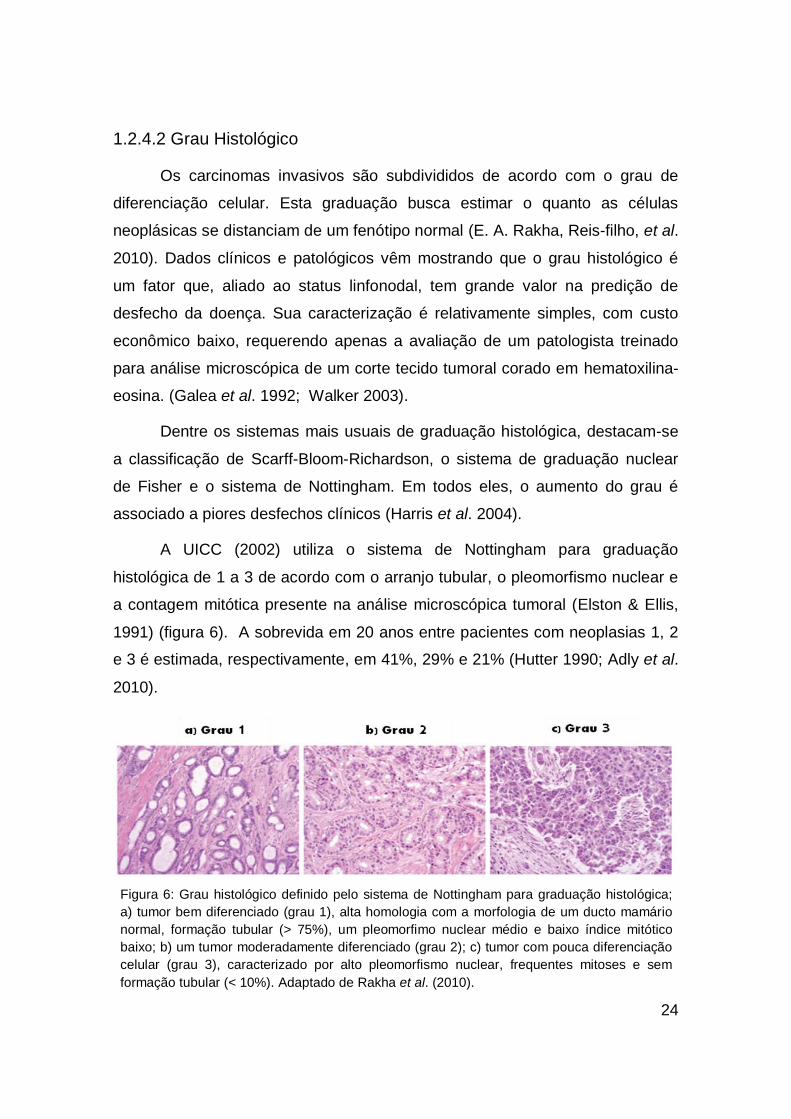
A UICC (2002) utiliza o sistema de Nottingham para graduação
histológica de 1 a 3 de acordo com o arranjo tubular, o pleomorfismo nuclear e
a contagem mitótica presente na análise microscópica tumoral (Elston & Ellis,
1991) (figura 6). A sobrevida em 20 anos entre pacientes com neoplasias 1, 2
e 3 é estimada, respectivamente, em 41%, 29% e 21% (Hutter 1990; Adly et al.
2010).
Figura 6: Grau histológico definido pelo sistema de Nottingham para graduação histológica;
a) tumor bem diferenciado (grau 1), alta homologia com a morfologia de um ducto mamário
normal, formação tubular (> 75%), um pleomorfimo nuclear médio e baixo índice mitótico
baixo; b) um tumor moderadamente diferenciado (grau 2); c) tumor com pouca diferenciação
celular (grau 3), caracterizado por alto pleomorfismo nuclear, frequentes mitoses e sem
formação tubular (< 10%). Adaptado de Rakha et al. (2010).

25
Nas últimas décadas, o grau histológico tem demonstrado imenso valor
preditivo da progressão da doença e na definição da terapia adjuvante
(Carvalho et al. 2011; Berruti et al. 2011).
1.2.4.3 Tamanho tumoral
O tamanho do tumor e a condição dos linfonodos axilares são os dois
mais importantes indicadores de prognóstico para o câncer de mama e,
portanto, constituem a base do estadiamento TNM, estabelecido e promulgado
pela União Internacional Contra o Câncer (Carter et al. 1989). Com o advento
da mamografia e a gradual difusão do método, associada a maior
conscientização da população sobre sua importância, vem-se observando em
alguns países desenvolvidos a redução significativa no tamanho dos tumores
quando do diagnóstico (Abreu & Koifman 2002).
É certo também que o tamanho do tumor está diretamente relacionado
ao risco de recidiva, sendo que, nos casos de pacientes com ausência de
comprometimento metastático dos linfonodos, o tamanho do tumor torna-se a
melhor característica preditora de recidiva (Arriagada et al. 2006; Fumagalli &
Sotiriou 2010). Os tumores de menor tamanho estão invariavelmente
relacionados a melhor prognóstico tanto para sobrevida global quanto para
sobrevida livre de doença, independentemente do tipo de tratamento aplicado
(Abreu & Koifman 2002; Soerjomataram et al. 2008).
1.2.4.4 Envolvimento linfonodal
A circulação linfática participa do processo de drenagem de substâncias
geradas pelo metabolismo celular, e, auxiliado pelo baço e pelo timo, compõe o
sistema linfático. Todo este sistema se encontra bastante hiperativo durante o
processo inflamatório, que é bem característico no câncer (Hirakawa 2009; Ji
2006). A detecção de células neoplásicas em linfonodos adjacentes a um tumor
primário caracteriza-se pela ocorrência de metástase linfonodal (Nakamura et

26
al. 2009). O sistema linfático é, portanto o sítio mais comum de metástases
(Wong & Hynes, 2007). Aparentemente, as células malignas chegam à linfa
muito mais facilmente do que à circulação ou a outros órgãos distantes. A
metástase linfonodal é, sobretudo, um marcador de prognóstico, pois existe
uma associação entre número de linfonodos acometidos e a ocorrência de
metástases à distância, com consequente redução da sobrevida em pacientes
oncológicos (Nakamura et al. 2009; Zwaans & Bielenberg 2007).
Os vasos linfáticos possuem menor densidade e pressão hidrostática do
que o sanguíneo. Possuem também mais invaginações, menor revestimento
muscular e lâmina basal descontínua, ao contrário do endotélio do sistema
circulatório sanguíneo (Wong & Hynes 2007; Hirakawa 2009). Estes fatores
contribuem para uma transferência de células neoplásicas do tecido primário
para a linfa de forma razoavelmente permissiva. Aliado a isso, o fluxo da
drenagem das células adjacentes ao tumor segue o fluxo em direção ao
linfonodos regionais. (Weinberg 2008). O linfonodo sentinela é a primeira
estrutura a receber as células cancerosas que se desprenderam do seu local
primário. O método de pesquisa de células tumorais no linfonodo sentinela
diminuiu substancialmente o número de esvaziamentos axilares
desnecessários na cirurgia oncológica (Qian et al. 2006).
Neste cenário, os nódulos linfáticos drenantes do tumor primário
poderiam funcionar como áreas de preparação, como um nicho onde células
tumorais ali permanecem em diferenciação, ganhando novas características
malignas até atingirem a circulação sistêmica com mais facilidade (Ochi et al.
2007). Ou então, a chegada de células neoplásicas nos linfonodos já
representaria um sinal de que as unidades celulares da massa tumoral primária
estariam num nível de diferenciação e malignidade tal, que seriam capazes de
invadir tecidos e nele se aderir (Nakamura et al. 2009; Hirakawa 2009).
O status linfonodal é um dos fatores mais importantes no prognóstico do
câncer de mama. O impacto negativo nas sobrevidas global e livre de doença é
modulado pelo tamanho da metástase, número de linfonodos acometidos e
localização das estruturas envolvidas (axilar, supra ou infraclavicular) (UICC
2002).

27
No câncer de mama, pacientes diagnosticados com invasão metastática
linfonodal possuem um risco 60% maior de mortalidade quando comparados
com pacientes sem invasão linfonodal (Schoppimann et al. 2004; Lee et al.
2006). A figura 7 mostra a correlação entre o número de linfonodos
comprometidos com metástases e a redução da sobrevida global e pacientes
com câncer de mama (Soerjomataram et al. 2008).
1.2.4.5 Receptores
Os receptores hormonais (RH) destacam-se dentre os marcadores
moleculares como definidores de perfil tumoral. De acordo com o nível de
amplificação, o status dos RH exibe uma associação com o perfil proliferativo
da neoplasia (Berry et al. 2006). Existem diferenças na probabilidade de
sobrevida e nas taxas e locais de recidiva entre o status positivo e negativo
para receptores hormonais (de estrogênio, e em menor extensão o de
progesterona). Estas diferenças aumentam de acordo com a fração de células
positivas e negativas e o valor prognóstico desaparece após dois anos (Hess at
Figura 7: Proporção da sobrevida cumulativa em 8162 pacientes
diagnosticados com câncer de mama na região sul da Holanda entre 1970
e 1994, acompanhadas até 2004, e agrupadas de acordo com o status
linfonodal (node): linfonodo negativo (-■-); 1 a 3 linfonodos positivos (-♦-);
de 4 a 9 linfonodos positivos (-▲-); 10 ou mais linfonodos positivos (-X-).
Adaptado de Soerjomataram et al. (2008).

28
al, 2003). A presença de receptores hormonais com status positivo no tumor é
indicativo para tratamento antiestrogênico, porém, a positividade não garante
uma resposta favorável.
1.2.4.5.1 Receptor de estrogênio
O receptor de estrogênio (RE) é um regulador de crescimento celular,
proliferação e diferenciação. É o mais importante marcador biológico de
resposta terapêutica para o tratamento adjuvante do câncer de mama
(Partridge et al. 2003).
Na década de 1990, foram descritas isoformas deste receptor, e hoje
são descritos como receptor de estrogênio alfa (α) e beta (ß). São codificados
por genes distintos, mas apresentam analogia estrutural. Ambos os receptores
podem mediar transcrição gênica através do elemento de resposta ao
estrogênio ou por meio dos fatores de transcrição jus e fos. Pela primeira via,
ambos atuam estimulando atividade proliferativa, enquanto que pela via dos
fatores de transcrição, o RE-α estimula a proliferação celular enquanto o RE-ß
inibe (Paech 1997; Travis & Key 2003; Massarweh et al. 2008). Na última
década, identificou-se uma isoforma deste receptor situada na membrana
nuclear que pode estar ligada a uma rápida ação não genômica do estrogênio,
mediada por ativação de fatores de crescimento celular (Hanstein et al. 2004).
Os tumores com status RE- associam-se a maiores taxas de recidiva e
óbito nos 2 primeiros anos após cirurgia. O status de RE+ é um fator
determinante para indicação de terapia hormonal no câncer de mama (Hess at
al, 2003; Partridge et al. 2003; Geiger et al. 2004; Fisher et al. 2005; Stingl
2011).
1.2.4.5.2 Receptor de progesterona
O gene PR que codifica o receptor de progesterona (RP) é regulado pela
atividade de RE-α (Horwitz & McGuire 1975; Thakkar & Mehta 2011). Assim,
quando o RP está presente nas células neoplásicas, frequentemente significa

29
que a atividade do RE-α está funcional (Allred 2010). O RP é crucial no
desenvolvimento do lóbulo alveolar da mama, e é responsivo ao hormônio
progesterona, ativando algumas funções celulares, incluindo proliferação. É
representado por duas isoformas, RP-A e RP-B (Jacobsen et al. 2005). Maior
incidência de câncer de mama também é notada em mulheres que realizaram
terapia de reposição hormonal com estrogênio e progesterona, em comparação
com mulheres que realizaram esta terapia apenas com reposição de estrogênio
(Cui et al. 2005).
Com relação ao desfecho clínico, mulheres diagnosticadas com tumores
com status RP negativo (RP-) possuem pior sobrevida global do que quando o
RP é positivo (Hoefnagel et al. 2012; Bardou et al. 2003). Porém, o status de
RP sozinho tem um valor preditivo fraco (Allred 2010; Stingl 2011). Apesar de
existir uma associação entre a expressão de RE e RP, suas expressões podem
ter status diferentes nos tumores de mama (Rakha et al. 2010). O status do RP
também tem valor preditivo de resposta à terapia hormonal adjuvante no
câncer de mama: pacientes com status RP - apresentam pior tempo livre de
recorrência para este tratamento, independentemente do status do RE (Bardou
et al. 2003; Jacobsen et al. 2005; Jacobsen & Horwitz 2012)
1.2.4.5.3 HER2
Em células normais, o gene erbB2 codifica a proteína do receptor de
crescimento epidermal humano do tipo 2 (epidermal growth factor receptor type
2; HER2 ou ErbB2 ou ainda HER-2/neu), que irá desencadear respostas
celulares semelhantes aos receptores de sua família (Jorissen 2003; Murphy &
Modi 2009). Cerca de 15 a 30% dos tumores de mama superexpressam HER2
(Suo et al. 2002), o que implica em menor sobrevida global e livre de doença a
estes pacientes (Yonemori et al. 2010).
A superexpressão de HER2 é fator preditivo para indicação de terapia
com anticorpo monoclonal trastuzumab e outras terapias-alvo (Gori et al. 2009;
Yonemori et al. 2010;Sanpaolo et al. 2011). A associação deste anticorpo
monoclonal à quimioterapia com antraciclinas e taxanos demonstrou

30
significativa redução da mortalidade, recorrência da doença e ocorrência de
metástases (Viani et al. 2007). Porém, ainda é discutido qual tempo de uso do
trastuzumab adjuvante poderia trazer a melhor relação de benefícios
(Jahanzeb 2008).
1.2.4.5.4 EGFR
Em modelos de câncer de mama pré-clínicos, a superexpressão do
EGFR leva uma transformação maligna de células de camundongos (Di Fiori et
al. 1987). O EGFR encontra-se superexpresso em cerca de 20 a 40% dos
carcinomas de mama (Meche et al. 2009). Em neoplasias de mama, o EGFR
está associado à proliferação e à resistência a apoptose (Kersting et al. 2006;
Edwards et al. 2006).
Em 1987, já havia descrição da relação do status EGFR+ e pior
prognóstico do câncer de mama (menor sobrevida livre de doença e menor
sobrevida global) (Sainsbury et al 1987; Jones et al. 1996; Nieto et al. 2007).
Modelos experimentas de análise do perfil genético do câncer de mama
mostram que o gene EGFR está na lista dos principais genes associados à
ocorrência de metástase cerebral no câncer de mama (Bos et al. 2009).
Existem muitos estudos que fazem referência a pior desfecho no
tratamento do câncer de mama com superexpressão tumoral de EGFR (Xu et
al. 2005; Agelopoulos et al. 2007; Brand et al. 2011), sobretudo em pacientes
com tumores classificados como triplo negativo (RE-, RP- e HER2-). Quando
estas pacientes possuem status EGFR positivo, apresentam pior sobrevida e
menor tempo livre de recorrência (Tischkowitz et al. 2007; Bouchalova et al.
2009). Porém, ao contrário do que ocorre em outros carcinomas, nos tumores
de mama, a superexpressão do EGFR não está necessariamente associada à
amplificação de seu gene (Yu et al. 2008).

31
1.2.5 Estimativas de Prognóstico
O curso clínico do câncer de mama e a sobrevida variam de acordo com
as características próprias do tumor e da paciente. Esta variação é
determinada por uma série complexa de características que podem contribuir
para um desfecho favorável ou trazer complicações (Kelsey & Berkowitz 1988).
Fatores prognósticos em câncer de mama são parâmetros possíveis de serem
mensurados no momento do diagnóstico e que possuem um valor preditor da
sobrevida da paciente (Soerjomataram et al. 2008).
A aplicação e o conhecimento dos fatores prognósticos em câncer de
mama tem fundamental importância para a definição do tratamento. De uma
forma geral, quando estes fatores indicarem um desfecho desfavorável, o
tratamento seguirá para uma indicação mais agressiva. Por outro lado,
pacientes que possuem características indicativas de bom prognóstico,
receberão um tratamento mais conservador e de menor toxicidade (Chotteau-
Lelièvre et al. 2004; Fryback et al. 2006).
Sendo o câncer de mama uma doença bastante heterogênea, não é
surpreendente que as respostas aos tratamentos também possam variar. Neste
sentido, uma busca em construir modelos de predição de evolução clínica e
resposta terapêutica têm sido objeto de vários estudos nas últimas décadas
(Adly et al. 2010; Danova et al. 2011). Estes modelos contribuem para melhorar
a qualidade de vida do paciente e para definições de políticas em saúde
(Williams et al. 2006)
As características histopatológicas são fatores prognósticos clássicos e
continuam a ser a principal base de vários sistemas de agrupamento de casos,
como o estadiamento TNM e a categorização de St Gallen (UICC 2002;
Goldhirsch et al. 2005). Ao longo dos anos, os receptores hormonais passaram
a ter grande importância preditiva e orientadora da conduta terapêutica, sendo
incorporados aos modelos de classificação tumoral (Hind et al. 2007; Fisher et
al. 2005).
Com o desenvolvimento das técnicas de análise genética e moleculares,
a identificação de diferentes grupos de câncer de mama veio ganhando

32
diversidade, o que resultou no reconhecimento de subtipos como: basalóide,
HER2, luminais A e B (Bertucci et al. 2002; Voduc et al. 2010), que levam em
conta o status dos receptores hormonais, HER2 e outras proteínas como as
citoqueratinas. Os tipos luminais correspondem a 60% a 70% dos casos,
enquanto o tipo HER2, a 15% a 20% e o basalóide, a 10% a 15% (Blows et al.
2010; Wang et al. 2011).
Plataformas de testes genéticos que levam em conta o nível de
expressão de inúmeros genes no tumor têm diversificado ainda mais as
subclassificações biológicas, proporcionando modelos que verificam a
assinatura genética tumoral e assim, predições sobre as características
neoplásicas (Sørlie et al. 2001; Finak et al. 2006; Parisi et al. 2010).
Atualmente, existem alguns métodos de fácil classificação que têm
norteado os clínicos para decidir o tratamento. O Índice de Prognóstico de
Nottingham (Nottingham Prognostic Index - NPI), estabelecido desde os anos
1980, leva em conta o tamanho tumoral, o grau histológico e o status linfonodal
para inferir a provável sobrevida em 5 anos após cirurgia (Blamey et al. 1979;
Haybittle et al. 1982; Adly et al. 2010). No Reino Unido, o Índice de Prognóstico
de Oxford (Oxford Prognostic Index) foi elaborado para predição de sobrevida
num mais longo prazo (Campbell et al. 2010). A plataforma eletrônica Adjuvant!
(http://www.adjuvantonline.com) possibilita ao usuário a inclusão de
informações relevantes sobre o paciente e a doença e indica o melhor
esquema de tratamento adjuvante. Por fim, o consórcio Early Breast Cancer
Trialists’ Collaborative Group (2012) publica meta-análises periódicas,
avaliando estudos clínicos sobre respostas a esquemas de terapia
antineoplásica sistêmica, hormonioterapia e radioterapia.
1.2.6 Tratamento
Atualmente, o tratamento do câncer de mama é interdisciplinar. A correta
associação do tratamento cirúrgico, da radioterapia e dos tratamentos
sistêmicos é indispensável quando se pensa em maior sobrevida e controle
local da doença (Leite 1999; Mauri et al. 2008; Lee et al. 2012).

33
O tratamento cirúrgico tem como objetivo controlar a doença loco-
regional, estadiar cirurgicamente para estabelecer os grupos de alto risco para
recorrência local, orientar a terapia sistêmica, proporcionar maior sobrevida,
identificar grupos de maior risco de metástase à distância e sempre que
possível, evitar mutilação ou oferecer à paciente o benefício da reconstrução
mamária (Franco et al, 1997). Dentre os tipos de abordagem cirúrgica, os
principais são: cirurgias radicais (mastectomias), cirurgias conservadoras
(tumorectomia, quadrantectomia) (Veronesi et al 1990) e linfadenectomia axilar
(aliado ao estudo de linfonodo sentinela) (Piato 2002).
A irradiação da mama é considerada recomendada às mulheres
submetidas à cirurgia conservadora para o câncer de mama em estádios
iniciais. O tratamento baseia-se na irradiação de ondas de energia originadas
de material radioativo sobre o local definido pela equipe médica (INCA 2011).
Com o desenvolvimento das técnicas radiológicas, surgiu a radioterapia
tridimensional e a modulação de intensidade do feixe de radiação (IMRT), que
permitem proteger o coração e outros órgãos, diminuindo a chance de
toxicidade e proporcionando doses mais homogêneas (Vicini et al, 2002).
O tratamento farmacológico do carcinoma de mama envolve,
atualmente, a utilização de uma variedade de compostos, como
quimioterápicos antineoplásicos, incluindo a quimioterapia de compostos
citostáticos, compostos hormonais e modificadores de resposta biológica, em
particular representados pelos anticorpos monoclonais e os inibidores de
tirosina cinase (Goldhirsch et al. 2009; Bouchalova et al. 2010; Sanpaolo et al.
2011; Early Breast Cancer Trialists' Collaborative Group 2012).
As abordagens mais comuns de terapia sistêmica curativa se dividem
em neo-adjuvante e adjuvante.
1.2.6.1 Tratamento neoadjuvante
O tratamento neoadjuvante ou tratamento primário, consiste na
aplicação de procedimentos sistêmicos em pacientes portadores de carcinoma
localmente avançado da mama, contribuindo para cirurgias menores, com

34
possível benefício na sobrevida (Fisher et al, 1997, von Minckwitz et al. 2008).
Há, porém, discussões quanto ao risco de recorrência local em caso de cirurgia
conservadora (Barnadas 2010; Beasley & Olson 2010). Estudos randomizados
têm mostrado que a asssociação de docetaxel a esquemas com antraciclinas
oferece melhor resposta ao tratamento neoadjuvante (Valero, 2002; Smith et al,
2002) do que os esquemas com antraciclinas apenas, porém ainda há dúvidas
quanto aos riscos desta terapias, em decorrência da morbidade associada a
reações adversas (Huober et al. 2010).
1.2.6.2 Tratamento adjuvante:
O tratamento quimioterápico adjuvante contribui para aumento do tempo
livre de progressão da doença. Nos últimos anos, vários estudos clínicos e
meta-análises vêm abordando o favorecimento do uso de agentes como as
antraciclinas e os taxanos no tratamento antineoplásico adjuvante (Guimarães
2008; Jacquin et al. 2012). O uso da poliquimioterapia adjuvante por mais de
quatro meses reduz o risco de recorrência em 41% e o índice anual de morte
em 31% (Breast Cancer Study Group, 2002).
A hormonioterapia no tratamento adjuvante do câncer de mama tem tido
grande impacto positivo, beneficiando mulheres cujas neoplasias expressam
receptores hormonais positivos. Inúmeros estudos têm demonstrado que a
utilização de tamoxifeno, um antagonista do receptor de estrogênio, por 5 anos
após tratamento cirúrgico, reduz efetivamente o risco de recorrência, a
incidência de câncer de mama contralateral e a mortalidade em mulheres,
independentemente da faixa etária. (Hind et al. 2007). Os inibidores de
aromatase de terceira geração (anastrozol e letrozol) têm sido indicados para o
tratamento de pacientes resistentes ao tamoxifeno, por terem melhor
tolerabilidade do que os inibidores de progesterona, como o megestrol (Buzdar
et al 2001; Cuzick et al. 2010).
O advento da terapia anti-HER2 trouxe uma nova linha de tratamento
para inibir a progressão da doença (Gilmer et al. 2008). A partir daí, novos
candidatos a terapia-alvo vêm sendo estudados.

35
1.2.6.3 Terapia anti-EGFR
O EGFR foi proposto como um alvo molecular nos anos de 1980 (Sato et
al. 1983; Masui at al. 1984; Mendelson 2002). O Cetuximab é o anticorpo
monoclonal anti-EGFR que mais se destacou até agora e vem sendo utilizado
no tratamento do câncer colorretal e de cabeça e pescoço (Mendelsohn &
Baselga 2003; Flynn et al. 2009; Park et al. 2011).
Outros inibidores de tirosina-cinase com atividade anti-EGFR mostraram
alguma importância no tratamento do câncer de pulmão de células não
pequenas (Rosell et al. 2012), porém não obtiveram resultados promissores no
tratamento do câncer de mama (Ciardiello et al. 2006).
Na família dos inibidores de tirosina-cinase, o Lapatinib demonstrou
efeito anti-EGFR e anti-HER2 e tem alcançado resultados na terapia do câncer
de mama metastático quando associado a outros quimioterápicos (Bilancia et
al. 2007; Gilmer et al. 2008). Buscando melhorar os resultados com esta
terapia, estudos vêm sendo desenvolvidos para avaliar os valores preditivos do
EGFR e HER2 na resposta ao uso do Lapatinib em câncer de mama, bem
como suas prováveis vias de resistência (Polli et al. 2009; Andre et al. 2010;
Bouchalova et al. 2010).
1.3 POLIMORFISMOS GENÉTICOS
Polimorfismo genético é a ocorrência simultânea em uma mesma
população de duas ou mais formas distintas de um gene (alelos), que se
caracterizam por variações nas sequências de nucleotídeos, tais como
substituições, deleções, inserções e duplicação ou deleção de genes. Por
definição, um gene é considerado polimórfico quando o alelo menos frequente
ocorre na população com uma frequência de pelo menos 1% (Miller et al.,
2001). As variações alélicas podem resultar em alteração da expressão ou da
funcionalidade dos produtos gênicos.

36
É muito comum a ocorrência de duas possibilidades de nucleotídeos na
sequência do DNA do genoma humano, o que é chamado de substituição de
um único nucleotídeo (single nucleotide polymorphism - SNP). As combinações
possíveis geralmente formam 3 possibilidades de genótipos, que podem ou não
diferir em fenótipos (Voet et al. 2000).
Os polimorfismos de região microssatélites são trechos de DNA que
consistem em unidades repetidas de dois, três ou quatro nucleotídeos. O
número de unidades de nucleotídeos repetidos contidos dentro de qualquer
microssatélite pode diferir entre os dois cromossomos homólogos de uma
pessoa e entre pessoas na população (Easton et al. 2009). Uma determinada
região microssatélite é, portanto, um locus polimórfico, e os diferentes números
de unidades repetidas em um determinado microssatélite constituem os alelos
deste lócus (Lewin 2001).
A evolução das espécies é um fenômeno que possui relação com as
características adaptativas individuais dos indivíduos (Darwin 1859).
Polimorfismos são distribuídos por todo o DNA genômico dos organismos, e
compõem os fatores que proporcionam a diversidade de fenótipos e as
variabilidades adaptativas numa mesma espécie (Tan 2008).
Com o Projeto Genoma Humano e com o advento das técnicas de
mapeamento genético, um grande número de polimorfismos vêm sendo
estudado (Marzolini et al., 2004; Pirmohamed et al., 2011). Perguntas quanto a
frequência em determinadas populações, relação hereditária, possíves
associações fenotípicas e causas de patologias vêm tentando ser respondidas
(Thompson & Thompson 2002).
1.3.1 Polimorfismos e câncer
Vários estudos vêm sendo realizados buscando identificar os fatores de
risco associados ao desenvolvimento e à progressão do câncer. Variações
individuais na ativação ou detoxificação de carcinógenos exógenos, no
metabolismo de hormônios e no reparo de DNA contribuem para diferentes
perfis de susceptibilidade ao câncer (Irigaray et al., 2007). De forma

37
semelhante, variações na sensibilidade aos fármacos antineoplásicos podem
afetar a eficácia e a segurança do tratamento.
Por se tratar de uma doença complexa, com vários fatores causais,
diferentes perfis moleculares e de evolução clínica e cujo tratamento envolve
diversas abordagens terapêuticas, a avaliação de associação entre genótipos
individuais e desfechos clínicos no câncer de mama requer a realização de
estudos de epidemiologia molecular, com análise integrada dos fatores
genéticos e ambientais.
1.3.2 Polimorfismos do gene EGFR
Com relação às variáveis genéticas individuais, há especial interesse em
alterações que possam interferir na atividade de alvos moleculares
potencialmente envolvidos com o risco de progressão do câncer de mama,
como é o caso do gene EGFR, que possui inúmeros de pontos de variabilidade
genética.
Buscando melhor compreender os fatores que interferem nos níveis de
expressão de EGFR, sua estabilidade proteica, sua atividade e afinidade com
ligantes e como elas se relacionam com a sinalização celular, dois de seus
polimorfismos foram pesquisados. Uma vez que estes polimorfismos podem
estar associados a determinadas funções gênicas e proteicas, a classificação
genotípica poderia funcionar como um biomarcador de prognóstico da doença
ou como preditor de resposta a terapias antineoplásicas.
1.3.2.1 Polimorfismo de Repetição CA
Regiões microssatélites possuem valor ainda não completamente
elucidado sobre a replicação, transcrição e leitura do código genético.
Mecanismos envolvendo regiões microssatélites, regiões intrônicas e
processos de transcrição têm sido alvo de inúmeros estudos (Kashi et al. 1997;
Chorev & Carmel 2012). A função das regiões microssatélites não transcritas
tem sido relacionada à formação de nucleossomos, interferindo na ligação de

38
proteínas relacionadas a transcrição nas regiões promotoras do gene (Buerger
et al. 2001).
No intron 1 do gene EGFR existe uma região microssatélite de repetição
de dinucleotídeos chamada de CA simple sequence repeat 1 (CA SSR I), ou,
polimorfismo de repetição CA (rs 72554020), onde ocorre uma variação de 14 a
25 repetições CA (figura 8). Um mecanismo de regulação transcricional
dependente do número de repetições CA tem sido descrito, com impacto sobre
o nível de síntese de pré-mRNA (Chi et al 1992). Gebhardt et al. (1999)
estudaram polimorfismos relacionados com a expressão aumentada de EGFR
nos tumores de mama, e demonstraram que o número de repetições CA no
intron 1 deste gene estaria inversamente relacionado à síntese de seu pré-
mRNA. Evidências deste mecanismo foram reforçadas por estudo do mesmo
grupo, que mostrou a associação entre alta expressão e ocorrência de alelos
da repetição CA mais curtos (Buerger et al. 2000).
A influência de uma proteína inibidora de transcrição do DNA nos alelos
com maior número de repetições CA é um modo de explicar esta associação.
Pode-se alternativamente especular que a região de repetição CA, por estar
situada entre a região promotora e a região enhancer, onde existem sítios de
ligação de elementos reguladores, a afinidade da formação de complexos
estaria comprometida nos alelos com tamanhos mais longos (Burkhard Brandt
et al. 2006).
Figura 8: Estrutura da região 5’ do gene EGFR. O início do gene como na maioria dos genes
estruturais, encontramos uma sequência tipo caixa GpC. O processo de transcrição do gene
EGFR inicia pela ligação de promotores em múltiplas regiões ricas em GC. Fator de transcrição
Sp1 entre outros contribuem para atividade do promotor. (Haley & Waterfield 1991). Dentre os
fatores que influenciam neste nível de transcrição, a existência de uma região de repetição de
dinucleotídeos (citosina-adenina) no intron 1 (CA SSR 1) logo antes de regiões enhancer 2
(+1,788 a +2,318) influenciam numa parada prematura de transcrição ou não. Adaptado de
Brandt et al. (2006).
39
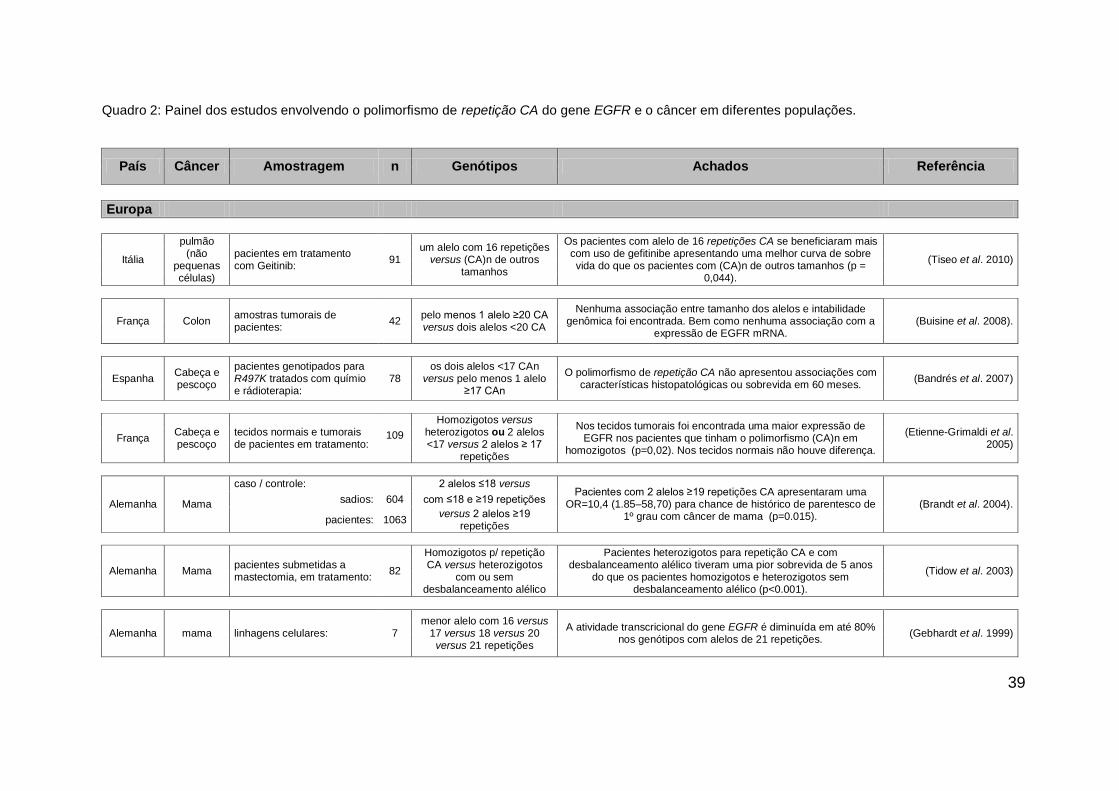
País Câncer Amostragem n Genótipos Achados Referência
Europa
Itália
pulmão (não
pequenas células)
pacientes em tratamento com Geitinib:
um alelo com 16 repetições
versus (CA)n de outros tamanhos
Os pacientes com alelo de 16 repetições CA se beneficiaram mais com uso de gefitinibe apresentando uma melhor curva de sobre vida do que os pacientes com (CA)n de outros tamanhos (p =
0,044).
(Tiseo et al. 2010) 91
França Colon amostras tumorais de pacientes:
42 pelo menos 1 alelo ≥20 CA versus dois alelos <20 CA
Nenhuma associação entre tamanho dos alelos e intabilidade genômica foi encontrada. Bem como nenhuma associação com a
expressão de EGFR mRNA. (Buisine et al. 2008).
Espanha Cabeça e pescoço
pacientes genotipados para R497K tratados com químio e rádioterapia:
78 os dois alelos <17 CAn
versus pelo menos 1 alelo ≥17 CAn
O polimorfismo de repetição CA não apresentou associações com características histopatológicas ou sobrevida em 60 meses.
(Bandrés et al. 2007)
França Cabeça e pescoço
tecidos normais e tumorais de pacientes em tratamento:
Homozigotos versus heterozigotos ou 2 alelos <17 versus 2 alelos ≥ 17
repetições
Nos tecidos tumorais foi encontrada uma maior expressão de EGFR nos pacientes que tinham o polimorfismo (CA)n em
homozigotos (p=0,02). Nos tecidos normais não houve diferença.
(Etienne-Grimaldi et al. 2005)
109
Alemanha Mama
caso / controle: 2 alelos ≤18 versus Pacientes com 2 alelos ≥19 repetições CA apresentaram uma
OR=10,4 (1.85–58,70) para chance de histórico de parentesco de 1º grau com câncer de mama (p=0.015).
(Brandt et al. 2004). sadios: 604 com ≤18 e ≥19 repetições
pacientes: 1063 versus 2 alelos ≥19
repetições
Alemanha Mama pacientes submetidas a mastectomia, em tratamento:
82
Homozigotos p/ repetição CA versus heterozigotos
com ou sem desbalanceamento alélico
Pacientes heterozigotos para repetição CA e com desbalanceamento alélico tiveram uma pior sobrevida de 5 anos
do que os pacientes homozigotos e heterozigotos sem desbalanceamento alélico (p<0.001).
(Tidow et al. 2003)
Alemanha mama linhagens celulares: 7 menor alelo com 16 versus
17 versus 18 versus 20 versus 21 repetições
A atividade transcricional do gene EGFR é diminuída em até 80% nos genótipos com alelos de 21 repetições.
(Gebhardt et al. 1999)
Quadro 2: Painel dos estudos envolvendo o polimorfismo de repetição CA do gene EGFR e o câncer em diferentes populações.
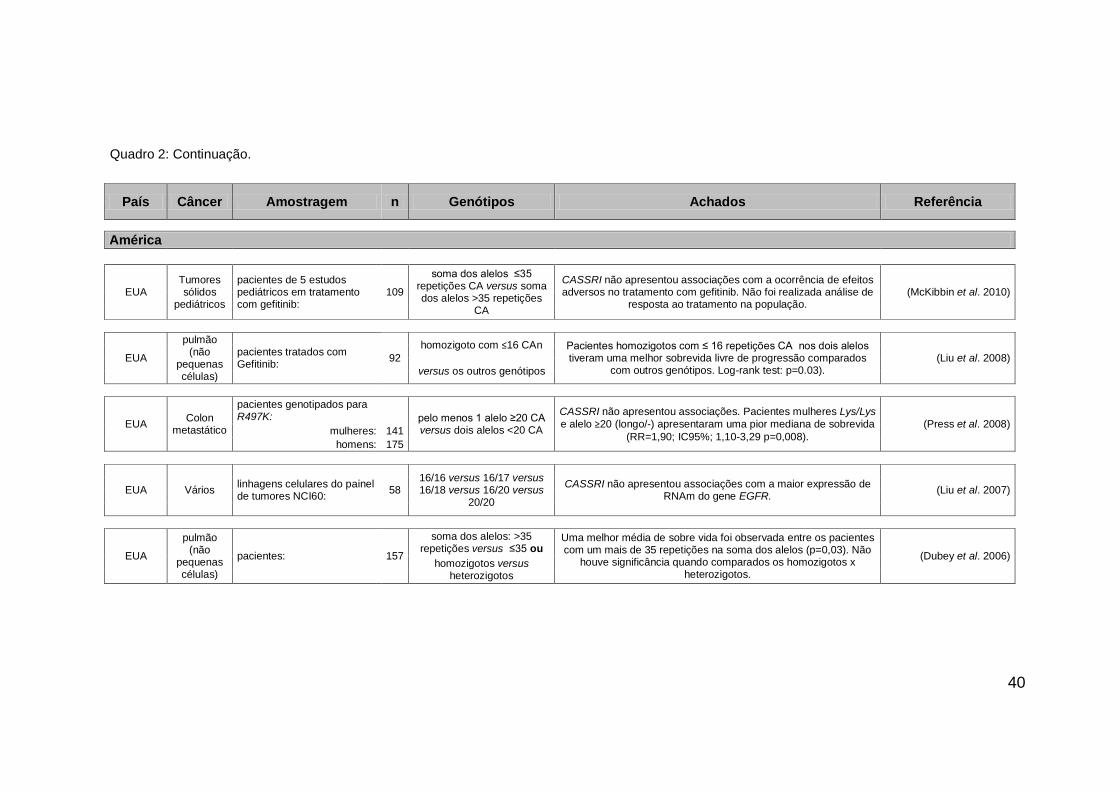
40
País Câncer Amostragem n Genótipos Achados Referência
América
EUA Tumores sólidos
pediátricos
pacientes de 5 estudos pediátricos em tratamento com gefitinib:
109
soma dos alelos ≤35 repetições CA versus soma dos alelos >35 repetições
CA
CASSRI não apresentou associações com a ocorrência de efeitos adversos no tratamento com gefitinib. Não foi realizada análise de
resposta ao tratamento na população. (McKibbin et al. 2010)
EUA
pulmão (não
pequenas células)
pacientes tratados com Gefitinib:
homozigoto com ≤16 CAn Pacientes homozigotos com ≤ 16 repetições CA nos dois alelos
tiveram uma melhor sobrevida livre de progressão comparados com outros genótipos. Log-rank test: p=0.03).
(Liu et al. 2008) 92
versus os outros genótipos
EUA Colon
metastático
pacientes genotipados para R497K: pelo menos 1 alelo ≥20 CA
versus dois alelos <20 CA
CASSRI não apresentou associações. Pacientes mulheres Lys/Lys
e alelo ≥20 (longo/-) apresentaram uma pior mediana de sobrevida
(RR=1,90; IC95%; 1,10-3,29 p=0,008).
(Press et al. 2008) mulheres: 141
homens: 175
EUA Vários linhagens celulares do painel de tumores NCI60:
58 16/16 versus 16/17 versus 16/18 versus 16/20 versus
20/20
CASSRI não apresentou associações com a maior expressão de RNAm do gene EGFR.
(Liu et al. 2007)
EUA
pulmão (não
pequenas células)
pacientes: 157
soma dos alelos: >35 repetições versus ≤35 ou
Uma melhor média de sobre vida foi observada entre os pacientes com um mais de 35 repetições na soma dos alelos (p=0,03). Não
houve significância quando comparados os homozigotos x heterozigotos.
(Dubey et al. 2006) homozigotos versus
heterozigotos
Quadro 2: Continuação.
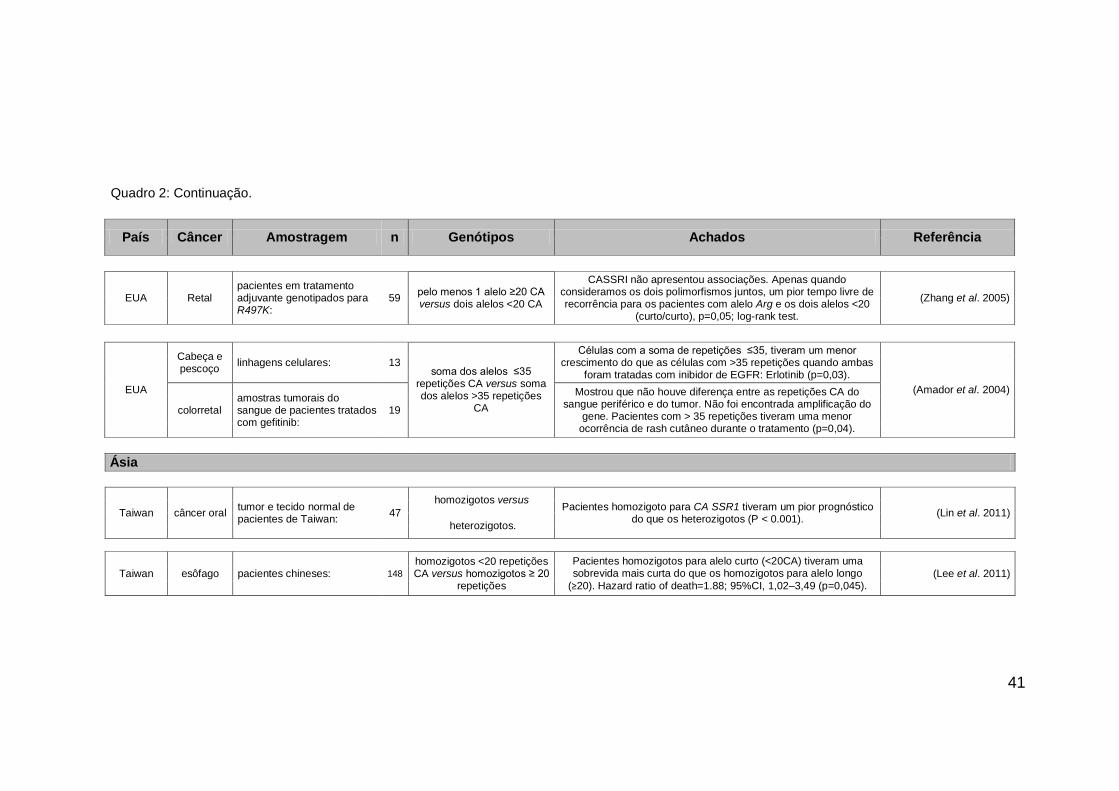
41
País Câncer Amostragem n Genótipos Achados Referência
EUA Retal pacientes em tratamento adjuvante genotipados para R497K:
59 pelo menos 1 alelo ≥20 CA versus dois alelos <20 CA
CASSRI não apresentou associações. Apenas quando consideramos os dois polimorfismos juntos, um pior tempo livre de recorrência para os pacientes com alelo Arg e os dois alelos <20
(curto/curto), p=0,05; log-rank test.
(Zhang et al. 2005)
EUA
Cabeça e pescoço
linhagens celulares: 13 soma dos alelos ≤35
repetições CA versus soma dos alelos >35 repetições
CA
Células com a soma de repetições ≤35, tiveram um menor crescimento do que as células com >35 repetições quando ambas
foram tratadas com inibidor de EGFR: Erlotinib (p=0,03).
(Amador et al. 2004)
colorretal amostras tumorais do sangue de pacientes tratados com gefitinib:
19
Mostrou que não houve diferença entre as repetições CA do sangue periférico e do tumor. Não foi encontrada amplificação do
gene. Pacientes com > 35 repetições tiveram uma menor ocorrência de rash cutâneo durante o tratamento (p=0,04).
Ásia
Taiwan câncer oral tumor e tecido normal de pacientes de Taiwan:
homozigotos versus
Pacientes homozigoto para CA SSR1 tiveram um pior prognóstico do que os heterozigotos (P < 0.001).
(Lin et al. 2011) 47
heterozigotos.
Taiwan esôfago pacientes chineses:
homozigotos <20 repetições CA versus homozigotos ≥ 20
repetições
Pacientes homozigotos para alelo curto (<20CA) tiveram uma sobrevida mais curta do que os homozigotos para alelo longo
(≥20). Hazard ratio of death=1.88; 95%CI, 1,02–3,49 (p=0,045). (Lee et al. 2011) 148
Quadro 2: Continuação.
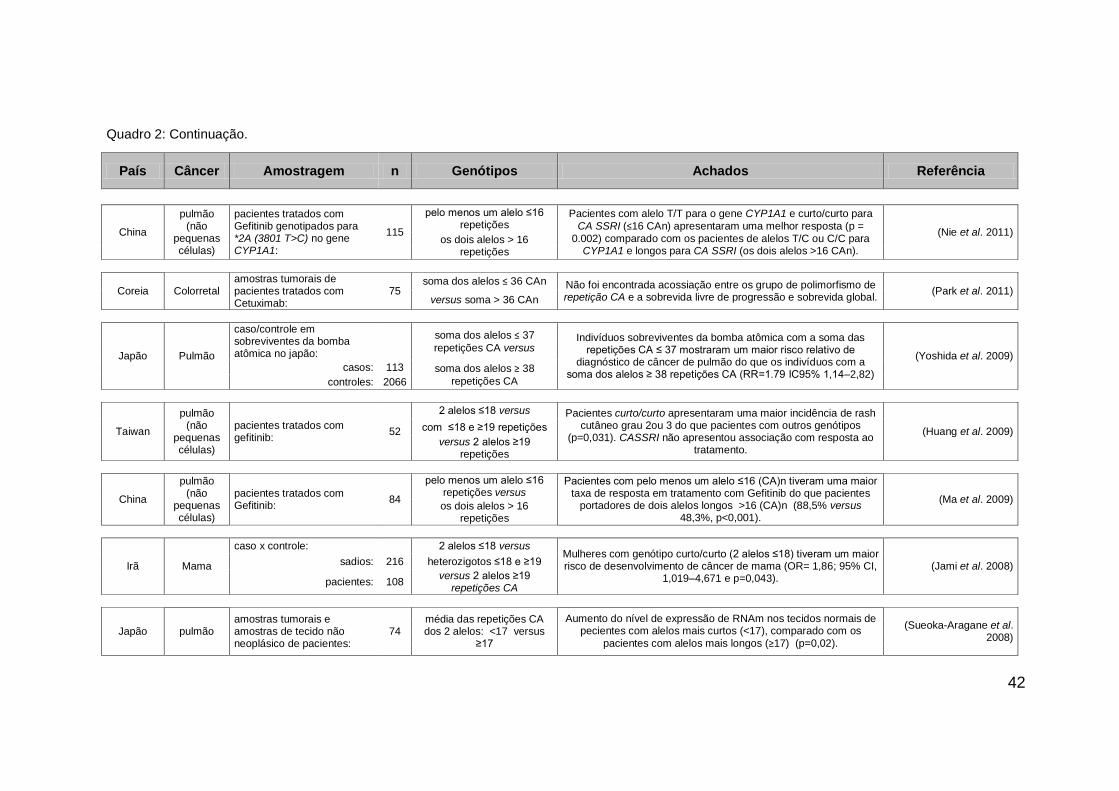
42
País Câncer Amostragem n Genótipos Achados Referência
China
pulmão (não
pequenas células)
pacientes tratados com Gefitinib genotipados para *2A (3801 T>C) no gene CYP1A1:
pelo menos um alelo ≤16 repetições
Pacientes com alelo T/T para o gene CYP1A1 e curto/curto para
CA SSRI (≤16 CAn) apresentaram uma melhor resposta (p =
0.002) comparado com os pacientes de alelos T/C ou C/C para CYP1A1 e longos para CA SSRI (os dois alelos >16 CAn).
(Nie et al. 2011) 115 os dois alelos > 16
repetições
Coreia Colorretal amostras tumorais de pacientes tratados com Cetuximab:
75 soma dos alelos ≤ 36 CAn Não foi encontrada acossiação entre os grupo de polimorfismo de
repetição CA e a sobrevida livre de progressão e sobrevida global. (Park et al. 2011)
versus soma > 36 CAn
Japão Pulmão
caso/controle em sobreviventes da bomba atômica no japão:
soma dos alelos ≤ 37
repetições CA versus Indivíduos sobreviventes da bomba atômica com a soma das
repetições CA ≤ 37 mostraram um maior risco relativo de diagnóstico de câncer de pulmão do que os indivíduos com a
soma dos alelos ≥ 38 repetições CA (RR=1.79 IC95% 1,14–2,82)
(Yoshida et al. 2009) casos: 113 soma dos alelos ≥ 38
repetições CA controles: 2066
Taiwan
pulmão (não
pequenas células)
pacientes tratados com gefitinib:
52
2 alelos ≤18 versus Pacientes curto/curto apresentaram uma maior incidência de rash cutâneo grau 2ou 3 do que pacientes com outros genótipos
(p=0,031). CASSRI não apresentou associação com resposta ao tratamento.
(Huang et al. 2009) com ≤18 e ≥19 repetições
versus 2 alelos ≥19 repetições
China
pulmão (não
pequenas células)
pacientes tratados com Gefitinib:
84
pelo menos um alelo ≤16 repetições versus
Pacientes com pelo menos um alelo ≤16 (CA)n tiveram uma maior taxa de resposta em tratamento com Gefitinib do que pacientes
portadores de dois alelos longos >16 (CA)n (88,5% versus 48,3%, p<0,001).
(Ma et al. 2009) os dois alelos > 16
repetições
Irã Mama
caso x controle: 2 alelos ≤18 versus Mulheres com genótipo curto/curto (2 alelos ≤18) tiveram um maior risco de desenvolvimento de câncer de mama (OR= 1,86; 95% CI,
1,019–4,671 e p=0,043). (Jami et al. 2008) sadios: 216 heterozigotos ≤18 e ≥19
pacientes: 108 versus 2 alelos ≥19
repetições CA
Japão pulmão amostras tumorais e amostras de tecido não neoplásico de pacientes:
74 média das repetições CA dos 2 alelos: <17 versus
≥17
Aumento do nível de expressão de RNAm nos tecidos normais de pecientes com alelos mais curtos (<17), comparado com os
pacientes com alelos mais longos (≥17) (p=0,02).
(Sueoka-Aragane et al. 2008)
Quadro 2: Continuação.
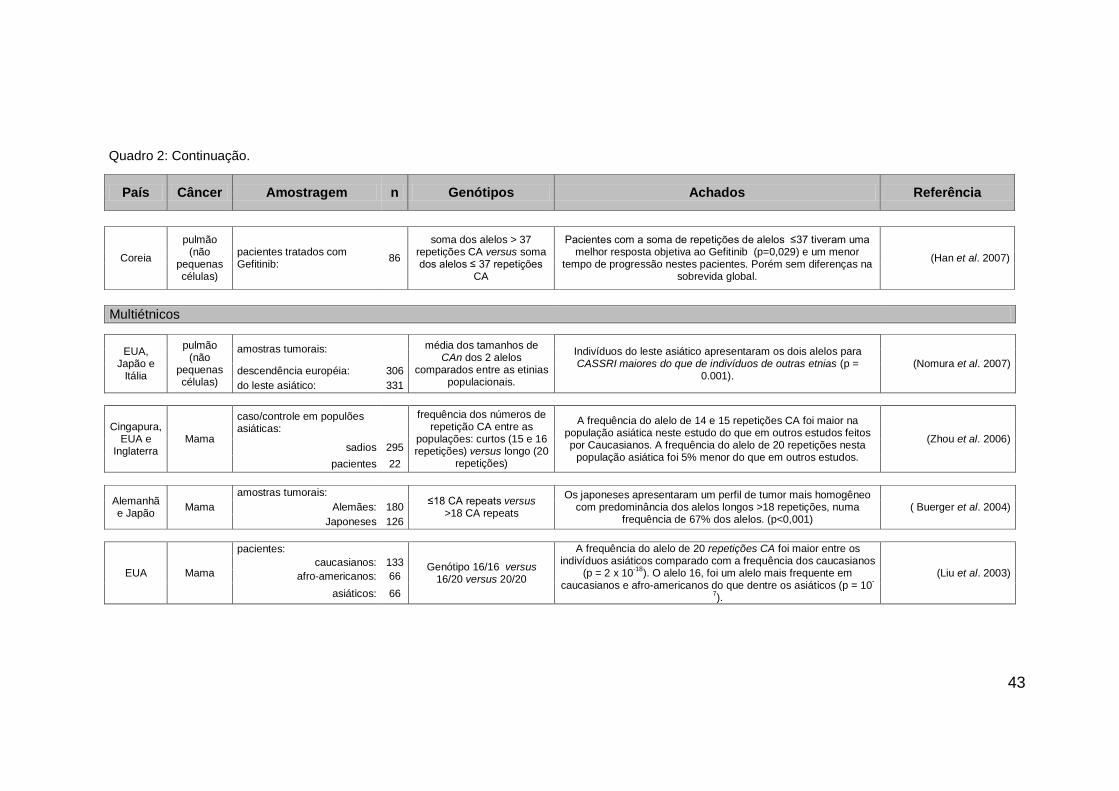
43
País Câncer Amostragem n Genótipos Achados Referência
Coreia
pulmão (não
pequenas células)
pacientes tratados com Gefitinib:
86
soma dos alelos > 37 repetições CA versus soma dos alelos ≤ 37 repetições
CA
Pacientes com a soma de repetições de alelos ≤37 tiveram uma melhor resposta objetiva ao Gefitinib (p=0,029) e um menor
tempo de progressão nestes pacientes. Porém sem diferenças na sobrevida global.
(Han et al. 2007)
Multiétnicos
EUA, Japão e
Itália
pulmão (não
pequenas células)
amostras tumorais: média dos tamanhos de
CAn dos 2 alelos comparados entre as etinias
populacionais.
Indivíduos do leste asiático apresentaram os dois alelos para CASSRI maiores do que de indivíduos de outras etnias (p =
0.001). (Nomura et al. 2007)
descendência européia: 306
do leste asiático: 331
Cingapura, EUA e
Inglaterra Mama
caso/controle em populões asiáticas:
frequência dos números de repetição CA entre as
populações: curtos (15 e 16 repetições) versus longo (20
repetições)
A frequência do alelo de 14 e 15 repetições CA foi maior na população asiática neste estudo do que em outros estudos feitos por Caucasianos. A frequência do alelo de 20 repetições nesta
população asiática foi 5% menor do que em outros estudos.
(Zhou et al. 2006)
sadios 295
pacientes 22
Alemanhã e Japão
Mama
amostras tumorais: ≤18 CA repeats versus
>18 CA repeats
Os japoneses apresentaram um perfil de tumor mais homogêneo com predominância dos alelos longos >18 repetições, numa
frequência de 67% dos alelos. (p<0,001) ( Buerger et al. 2004) Alemães: 180
Japoneses 126
EUA Mama
pacientes:
Genótipo 16/16 versus 16/20 versus 20/20
A frequência do alelo de 20 repetições CA foi maior entre os indivíduos asiáticos comparado com a frequência dos caucasianos
(p = 2 x 10-18
). O alelo 16, foi um alelo mais frequente em caucasianos e afro-americanos do que dentre os asiáticos (p = 10
-
7).
(Liu et al. 2003) caucasianos: 133
afro-americanos: 66
asiáticos: 66
Quadro 2: Continuação.

44
Alguns estudos avaliando a associação deste polimorfismo com o risco
de desenvolvimento do câncer, com o processo de carcinogênese e com a
resposta à terapia antineoplásica foram desenvolvidos nos últimos anos. No
quadro 2, podemos visualizar um painel dos principais achados nestes estudos.
Nos trabalhos com populações ocidentais, o alelo de 16 repetições CA é
descrito como o mais frequente, enquanto que os trabalhos que envolvem
populações asiáticas descrevem a ocorrência do alelo de 20 repetições CA
como o alelo mais frequente. Estes dados sugerem que indivíduos asiáticos
possuem uma frequência de alelos maiores, ou com um maior número de
repetições CA do que as populações ocidentais (Liu et al. 2003; Buerger et al.
2004). Não são conhecidos trabalhos com a análise deste polimorfismo na
população brasileira.
Analisando todos os trabalhos de forma global, nota-se uma dificuldade
de classificação dos grupos genotípicos gerados pelos alelos de repetição CA.
Nos quadros aqui representados, verificamos diferentes formas de agrupar os
genótipos do polimorfismo de repetição CA. Não existe, portanto, um ponto de
corte ou um limite de tamanho usual na literatura que classifique exatamente os
alelos como curto ou longo.
1.3.2.2 Polimorfismo R497K
O outro polimorfismo escolhido para este estudo representa uma
mudança de um único nucleotídeo na região codificante (G > A), no exon 13,
gerando uma substituição de uma Arginina (Arg) por uma Lisina (Lys) no códon
497 (R497K) (Moriai et al. 1993) (figura 9). Moriai et al. (1994) demonstraram
que células expressando EGFR com Lys nesta posição apresentavam menor
resposta ao crescimento na presença dos ligantes TGF-α e EGF, além de
apresentarem indução reduzida dos efetores fos, jun, myc.
O EGFR, por ser um receptor a ativado pela formação de homodímeros
ou heterodímeros com outros homólogos de sua família, possui um mecanismo
de acoplamento, atividade e ciclização ainda não completamente estabelecido
(Endres et al. 2011). Estudos cristalográficos suspeitam que alguns

45
mecanismos refinados de acoplamento e dimerização entre os monômeros e
seus domínios possam interferir na atividade do receptor (Alvarado et al. 2010)
(figura 10). Suspeita-se que o polimorfismo R497K e outros polimorfismos em
regiões adjacentes possam interferir na formação dos dímeros e a estabilidade
destas ligações (Choura et al. 2010).
Alguns estudos avaliando a associação deste polimorfismo com o risco
de desenvolvimento câncer, com a carcinogênese e com a resposta à terapia
antineoplásica foram desenvolvidos nos últimos anos e estão compilados no
quadro 3. Entre eles, Wang et al. (2007) observaram uma associação entre a
forma variante R497K e uma diminuição no nível de fosforilação e de ativação
do EGFR em tumores de pacientes com carcinoma colorretal. Estes pacientes
portadores do alelo variante apresentaram menor envolvimento linfonodal,
menor ocorrência de metástases e, portanto, um melhor prognóstico.
Figura 9: Representação esquemática da
estrutura do receptor EGFR e suas unidades.
Os domínios extra celulares I, II, III e IV
encontram-se destacados. No domínio IV, na
posição 497, o sítio de troca de aminoácidos
arginina (Arg) por um lisina (Lys) gerada pela
ocorrência do polimorfismo R497K. Adaptado
de Flynn et al. (2009)

46
O único estudo encontrado envolvendo a pesquisa deste polimorfismo
em pacientes com câncer de mama foi publicado por um grupo da Tunísia,
(Kallel et al. 2009), que observou uma distribuição genotípica diferente entre as
pacientes com linfonodos negativos.
País Câncer Amostragem n Alelo Lys
Achados Referência
Europa
França colon pacientes metastáticos tratados com cetuximab
32 23,4%
Pacientes com a variante Lys e mostraram com melhor sobrevida do que os
pacientes selvagens quando tratados com cetuximab. (p = 0,041)
(Gonçalves et al. 2008)
Espanha cabeça e pescoço
pacientes tratados com quimio e radioterapia
76 29,6%
Quando combinado com polimorfismo de repetição CA, os pacientes homozigotos
selvagens e heterozigotos tiveram um pior resposta ao tratamento. (p = 0,034)
(Bandrés et al. 2007)
Quadro 3: Painel dos estudos envolvendo o polimorfismo R497K do gene EGFR e o câncer em
diferentes populações.
Figura 10: Representação esquemática dos domínios extra celulares do receptor EGFR (I, II,
III, IV). Modelo de mecanismo para uma cooperação negativa da atividade do receptor. (A)
Posicionamento de receptores EGFR em forma ligada em homodímeros ou na forma
monomérica. (B) Ligação de um único ligante (rosa) na molécula da esquerda; o domínio I
(azul) e o domínio III (amarelo) se deslocam para acoplar a molécula da direita pelo colapso
entre os domínios II (verde) de cada receptor. (C) Quando outra molécula ligante se envolve
com o receptor da direita, o espaço de acoplamento não é sensibilizado da mesma forma do
que no primeiro evento, deixando a estrutura dimérica assimétrica e por conseguinte, não
contribuindo para a ativação consistente da cascata de fosforilação. (D) Os dois ligantes
realizando o mesmo acoplamento, contribuindo para uma dimerização simétrica. Adaptado
de Alvarado et al. (2010).
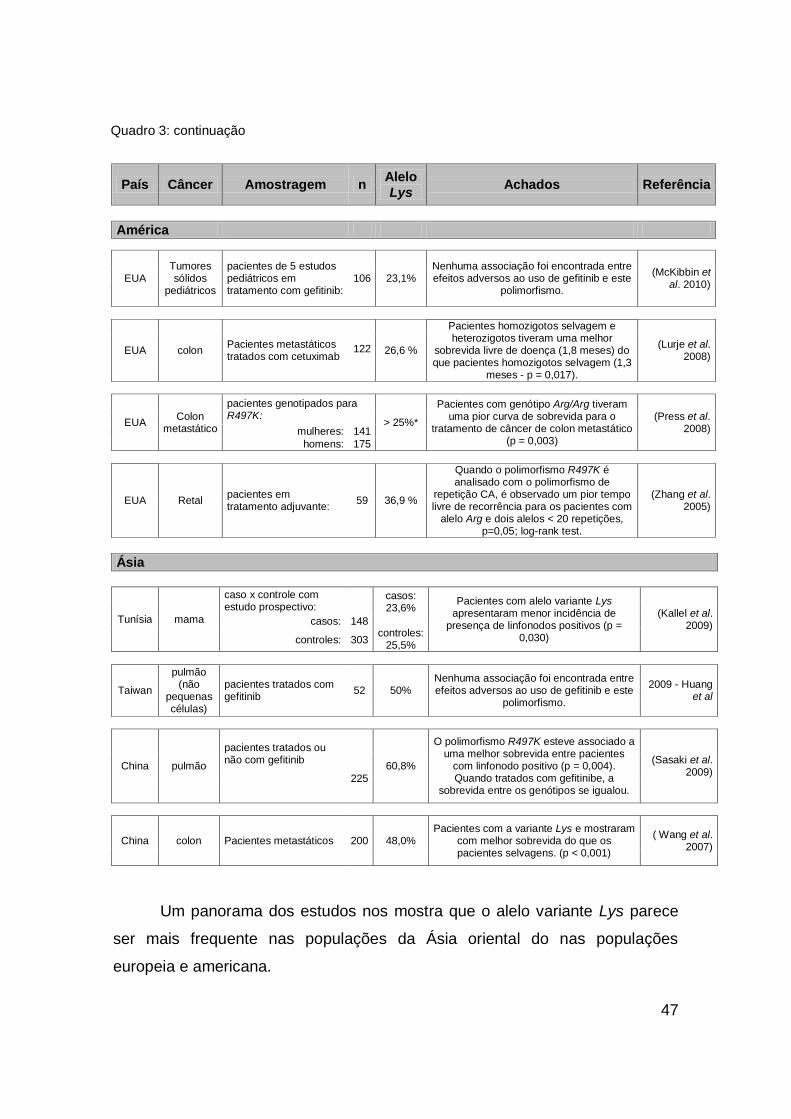
47
País Câncer Amostragem n Alelo
Lys Achados Referência
América
EUA Tumores sólidos
pediátricos
pacientes de 5 estudos pediátricos em tratamento com gefitinib:
106 23,1% Nenhuma associação foi encontrada entre efeitos adversos ao uso de gefitinib e este
polimorfismo.
(McKibbin et al. 2010)
EUA colon Pacientes metastáticos tratados com cetuximab
26,6 %
Pacientes homozigotos selvagem e heterozigotos tiveram uma melhor
sobrevida livre de doença (1,8 meses) do que pacientes homozigotos selvagem (1,3
meses - p = 0,017).
(Lurje et al. 2008)
122
EUA Colon
metastático
pacientes genotipados para R497K:
> 25%*
Pacientes com genótipo Arg/Arg tiveram uma pior curva de sobrevida para o
tratamento de câncer de colon metastático (p = 0,003)
(Press et al. 2008) mulheres: 141
homens: 175
EUA Retal pacientes em tratamento adjuvante:
59 36,9 %
Quando o polimorfismo R497K é analisado com o polimorfismo de
repetição CA, é observado um pior tempo livre de recorrência para os pacientes com
alelo Arg e dois alelos < 20 repetições, p=0,05; log-rank test.
(Zhang et al. 2005)
Ásia
Tunísia mama
caso x controle com estudo prospectivo:
casos: 23,6%
Pacientes com alelo variante Lys apresentaram menor incidência de
presença de linfonodos positivos (p = 0,030)
(Kallel et al. 2009) casos: 148
controles: 25,5%
controles: 303
Taiwan
pulmão (não
pequenas células)
pacientes tratados com gefitinib
52 50% Nenhuma associação foi encontrada entre efeitos adversos ao uso de gefitinib e este
polimorfismo.
2009 - Huang et al
China pulmão
pacientes tratados ou não com gefitinib
O polimorfismo R497K esteve associado a uma melhor sobrevida entre pacientes
com linfonodo positivo (p = 0,004). Quando tratados com gefitinibe, a
sobrevida entre os genótipos se igualou.
(Sasaki et al. 2009)
225
60,8%
China colon Pacientes metastáticos 200 48,0% Pacientes com a variante Lys e mostraram
com melhor sobrevida do que os pacientes selvagens. (p < 0,001)
( Wang et al. 2007)
Um panorama dos estudos nos mostra que o alelo variante Lys parece
ser mais frequente nas populações da Ásia oriental do nas populações
europeia e americana.
Quadro 3: continuação

48
Zhang et al (2005) demonstraram que o variante Lys em combinação
com o maior número de repetições CA do intron 1 em pacientes com câncer
colorretal está associado a menor recorrência de doença durante tratamento
com quimioterapia e radioterapia adjuvante.
Sugere-se também, que a forma variante do polimorfismo R497K (Lys)
pode estar associada a um prognóstico favorável em câncer avançado de
pulmão. Sasaki et al. (2009) demonstraram que pacientes Lys tiveram uma
melhor sobrevida entre os pacientes que fizeram quimioterapia à base de
platina.
Estes dados justificam a importância da análise da genotipagem do
EGFR e suas associações com os fatores prognósticos em estudo clínico, e
avaliação de sua contribuição como marcador de predição do desfecho do
tratamento adjuvante da doença.

49
2 OBJETIVOS
2.1 OBJETIVO GERAL
• Determinar a magnitude de associação entre polimorfismos genéticos do
gene EGFR e variáveis clínico-patológicas com valor prognóstico para o
câncer de mama.
2.2 OBJETIVOS ESPECÍFICOS
Determinar a prevalência dos polimorfismos de repetição CA e R497K
do gene EGFR pacientes com câncer de mama.
Determinar a magnitude da associação destes polimorfismos com o
prognóstico de pacientes diagnosticadas com câncer de mama.

50
3 MATERIAL E MÉTODOS
3.1 DESENHO DO ESTUDO
Trata-se de um estudo observacional exploratório em coorte hospitalar
de mulheres diagnosticadas com câncer de mama, no Hospital do Câncer
III/INCA. A coorte consistiu de uma série de casos de pacientes com indicação
de cirurgia curativa.
As participantes deste estudo fizeram parte do projeto “Polimorfismos
genéticos e evolução clínica, resposta terapêutica e reações adversas em
pacientes submetidas ao tratamento do câncer de mama”, aprovado pelo
Comitê de Ética em Pesquisa do INCA, aprovado em 12 de fevereiro de 2009,
sob registro nº 129/08 (Anexo 1). Esse projeto teve o intuito de documentar a
ocorrência das principais complicações associadas ao tratamento
antineoplásico em pacientes com câncer de mama, e avaliar a influência de
variáveis sócio-demográficas, terapêuticas, clínicas e genéticas, relacionadas
ao tumor e ao indivíduo.
As pacientes aqui analisadas foram convidadas a participar deste estudo
no período de 12 de fevereiro de 2009 a 1º de abril de 2011. Todas as
participantes assinaram o Termo de Consentimento Livre e Esclarecido (anexo
2).
3.2 PACIENTES
3.2.1 Critérios de inclusão
Foram consideradas elegíveis para o estudo mulheres com câncer de
mama primário, não-metastático, que receberam indicação inicial de tratamento
cirúrgico com intenção curativa, que aceitaram participar da investigação e
conseguiram responder às perguntas durante entrevista inicial. As mulheres
que, após a cirurgia, foram diagnosticadas com câncer de mama bilateral não
foram incluídas no estudo.

51
3.2.2 Critérios de exclusão
Foram excluídas do estudo as mulheres que solicitaram desligamento da
participação no estudo.
3.3 FORMULÁRIOS
Dados sócio-demográficos foram obtidos através de entrevista no
momento do recrutamento (Anexo 3). As pacientes incluídas foram
acompanhadas através de consultas ao prontuário para obtenção de
informações sobre a cirurgia, tratamento adjuvante e laudo histopatológico
(Anexo 4). Todos os dados foram registrados em formulário padronizado
específico e em banco de dados eletrônico utilizando o programa Microsoft
Office Excel (versão 2007).
3.4 VARIÁVEIS CLÍNICO-PATOLÓGICAS
3.4.1 Idade:
Foi considerada a idade na data do diagnóstico do câncer de mama. Na
análise, a idade foi estudada como variável contínua e no formato dicotômico.
Quando analisada no formato dicotômico, as categorias de idade foram
definidas no momento do diagnóstico como “superior a 36 anos”,
representando o grupo de melhor prognóstico versus “menor ou igual a 35 anos
de idade”, representando o grupo de pior prognóstico (Aebi et al, 2000).
3.4.2 Classificação histológica:
A classificação histológica foi definida por médico patologista do Serviço
de Patologia Clínica do INCA, com base na análise microscópica de cortes da
peça cirúrgica. De acordo com a apresentação morfológica, e utilizando os
critérios definidos pela Organização Mundial de Saúde (Tavassoli, 2003) e o
College of American Pathologists (Fitzgibbons et al, 1999), os tumores das
pacientes deste estudo receberam uma das seguintes classificações:

52
carcinoma ductal in situ;
carcinoma lobular in situ;
carcinoma ductal invasivo;
carcinoma lobular invasivo;
carcinoma misto.
Quando analisada no formato dicotômico, a classificação histológica foi
agrupada em pacientes diagnosticadas com carcinoma invasivo versus
pacientes diagnosticadas com carcinoma in situ, representando os grupos de
melhor e pior prognóstico respectivamente. Neste formato, os pacientes
diagnosticados com carcinoma misto foram retirados da análise.
3.4.3 Grau histológico:
Seguindo as recomendações da UICC (2002), foi utilizado o sistema de
Nottingham (Elston & Ellis, 1991) para graduação histológica, que prevê índices
de 1 a 3 de acordo com as seguintes características microscópicas tumorais:
arranjo tubular, pleomorfismo nuclear e contagem mitótica.
No arranjo tubular, os índices se referem à proporção de estruturas
tubulares na lesão. O pleomorfismo nuclear é avaliado levando-se em
consideração o tamanho do núcleo, contornos, uniformidade da cromatina e
variação de forma e tamanho, enquanto a atividade mitótica é avaliado
contando-se o número de figuras em 10 campos microscópicos contíguos de
grande aumento. O somatório dos índices pode variar de 3 a 9. A graduação
final foi obtida através de laudo histopatológico do Serviço de Patologia Clínica
do INCA que define como:
grau 1: somatório de 3 a 5;
grau 2: somatório de 6 a 7;
grau 3: somatório de 8 ou 9 pontos.
Quando analisado no formato dicotômico, o grau histológico foi agrupado
em pacientes classificadas como grau 1 ou 2 versus pacientes classificadas

53
como grau 3, representando os grupos de melhor e pior prognóstico
respectivamente.
3.4.4 Tamanho tumoral:
O tamanho tumoral foi anotado de acordo com o laudo histopatológico
de cada paciente, divulgado pelo Serviço de Patologia Clínica do INCA. As
medidas foram obtidas dos cortes histológicos da peça cirúrgica.
A categorização do tamanho tumoral (pT) foi baseada na Classificação
de Tumores Malignos TNM (UICC 2002). Para tal, foi levada em conta a
medida de maior diâmetro do tumor invasivo relatada no laudo histopatológico.
Nos casos onde foram encontrados tumores múltiplos, o valor de maior
diâmetro foi levado em conta para fins de estadiamento pT nas seguintes
categorias:
pTis: presença exclusiva de carcinoma ductal ou lobular in situ
independente da extensão;
pT1: Tumor com 2 cm ou menos em sua maior dimensão;
pT2: Tumor com mais de 2 cm, porém não mais de 5 cm em sua
maior dimensão;
pT3: Tumor com mais de 5 cm em sua maior dimensão;
pT4: Tumor de qualquer tamanho com extensão direta à parede
torácica ou à pele.
Quando analisado no formato dicotômico, o tamanho tumoral foi
agrupado independentemente da classificação histológica, em tumores com até
2 cm versus tumores maiores do que 2 cm, representando os grupos de melhor
e pior prognóstico respectivamente.

54
3.4.5 Grau de envolvimento linfonodal:
O número de linfonodos axilares retirados foi obtido pelo exame
histopatológico da peça cirúrgica oriunda da pesquisa do linfonodo sentinela
e/ou durante linfadenectomia axilar.
O número de linfonodos positivos para metástase regional foi anotado no
laudo histopatológico divulgado pelo Serviço de Patologia Clínica do INCA,
independentemente do nível do esvaziamento axilar (I, II ou III) secundário ou
não à biópsia do linfonodo sentinela. O grau de envolvimento linfonodal (pN) foi
categorizado de acordo com a Classificação de Tumores Malignos TNM (UICC
2002):
pN0: ausência de metástase em linfonodos regionais.
pN1: de 1 a 3 linfonodo(s) metastáticos;
pN2: de 4 a 9 linfonodos metastáticos;
pN3: metástase em 10 ou mais linfonodos.
Quando analisado no formato dicotômico, o grau de envolvimento
linfonodal foi agrupado como pN0 ou pN1 versus pN2 ou pN3, representando
os grupos de melhor e pior prognóstico respectivamente.
3.4.6 Status do Receptor de Estrogênio:
O status de amplificação do Receptor de Estrogênio (RE) nos tumores
pesquisados foi obtido através de reações de imuno-histoquímica em cortes
tumorais parafinados, pelo método de incubação em panela de pressão, em
solução de acido cítrico a pH 6,0, utilizando o anticorpo Clone SP1
(Neomarkers, Fremont, EUA). Foi anotada a porcentagem de marcação para
RE, de acordo com o laudo histopatológico divulgado pelo Serviço de Patologia
Clínica do INCA. Os tumores foram considerados RE-positivos quando o
percentual de células marcadas foi igual ou superior a 1% (NIH Consens,
2000).

55
Está variável foi analisada no formato dicotômico, sendo os grupos
categóricos definidos como RE-positivo versus RE-negativo, representando os
grupos de melhor e pior prognóstico, respectivamente.
3.4.7 Status do Receptor de Progesterona:
O status de amplificação do Receptor de Progesterona (RP) nos tumores
pesquisados foi obtido através de reações de imuno-histoquímica em cortes
tumorais parafinados, pelo método de incubacão em panela de pressão, em
solução de ácido cítrico a pH 6,0, utilizando o anticorpo Clone 16 (Novocastra
Laboratories, Newcastle, Reino Unido). Foi anotada a porcentagem de
marcação para RP, de acordo com o laudo histopatológico divulgado pelo
Serviço de Patologia Clínica do INCA. Os tumores foram considerados RP-
positivos quando o percentual de células marcadas foi igual ou superior a 1%
(NIH Consens, 2000).
Esta variável foi analisada no formato dicotômico, sendo os grupos
categóricos definidos como RP-positivo versus RP-negativo, representando os
grupos de melhor e pior prognóstico, respectivamente.
3.4.8 Status HER2:
O status de amplificação do Receptor do Fator de Crescimento
Epidermal do tipo 2 (HER2) nos tumores pesquisados foi obtido através de
reações de imunohistoquímica em cortes tumorais parafinados, pelo método de
Incubação em forno de micro-ondas, em solução de ácido cítrico a pH 6,0,
utilizando o anticorpo policlonal de coelho (Dako Denamark A/S, Glostrup,
Dinamarca). A gradação da reação imuno-histoquímica à positividade foi
definida em escores (0, 1+, 2+ e 3+) de acordo com as recomendações da
American Society of Clinical Oncology e do College of American Pathologists
(ASCO/CAP, 2007).
No laudo histopatológico divulgado pelo Serviço de Patologia Clínica do
INCA, o status HER2 foi definido da seguinte forma:

56
HER2 negativo: escore 0 ou 1+;
HER2 indeterminado: escore 2+;
HER2 positivo: escore 3+.
Algumas amostras tumorais em que o status HER2 foi considerado
indeterminado foram encaminhadas para pesquisa do gene HER2 por
hibridização in situ por fluorescência (FISH), efetuando a razão da média de
sinais do gene HER2 e da porção centromérica cr17, através do HER2 FISH
DAKO pharmDx Kit (Dako Denamark A/S, Glostrup, Dinamarca).
A presença de amplificação do gene HER2 foi considerada quando a
relação HER2/Cr17 foi superior a 2,2. Quando a relação foi superior a 1,8 e
inferior a 2,2, o teste foi determinado inconclusivo e quando igual ou inferior a
1,8, o gene HER2 foi considerado não amplificado (ASCO/CAP, 2007). O
resultado do teste inconclusivo manteve o status indeterminado para o HER2
daquela amostra. O status HER2 foi assumido como positivo ou negativo
quando os seguintes resultados foram encontrados; respectivamente: superior
a 2,2 ou igual ou inferior a 1,8.
Quando analisado no formato dicotômico, o status HER2 foi
categorizado como HER2-negativo versus HER2-positivo, representando os
grupos de melhor e pior prognóstico respectivamente. Neste formato, as
pacientes com status HER2 indeterminado foram retirados da análise.
3.4.9 Subclassificação biológica:
A identificação de diferentes perfis fenotípicos do câncer de mama pode
ser determinada segundo o nível de expressão de uma lista de genes,
caracterizando subgrupos denominados: luminal A, luminal B, basal-símile e
tipo HER2 (Sotiriou et al , 2003). Estes subgrupos fenotípicos possuem valor
prognóstico e podem eventualmente definir candidatos a alvos terapêuticos
(Sørlie et al, 2001).
Neste estudo, com os resultados da análise dos receptores de
estrogênio, progesterona e HER2 disponíveis no laudo histopatológico

57
divulgado pelo Serviço de Patologia Clínica do INCA, foi possível agrupar os
tumores na seguinte subclassificação biológica (Huober at al, 2010):
lumial A: tumores RE+, RP+, HER2 -;
lumial B: tumores RE+, RP-, HER2- ou RE+, RP-, HER2+ ou RE-,
RP+, HER2- ou RE+, RP+, HER2+;
tipo HER2: tumores RE-, RP+, HER2+ ou RE-, RP-, HER2+;
triplo negativo: RE-, RP-, HER2-.
3.4.10 Estadiamento:
A União Internacional Contra o Câncer, através da Classificação de
Tumores Malignos TNM, agrupa os estádios da doença de acordo com o
tamanho tumoral, o grau de envolvimento linfonodal e o número de metástases
à distância, no momento do diagnóstico. Uma vez que neste estudo não foram
incluídos pacientes com tumores metastáticos, o estadiamento foi feito de
acordo com o tamanho tumoral e o grau de envolvimento linfonodal obtidos nos
laudos histopatológicos, sendo caracterizados os seguintes níveis:
estádio 0: pTis, pN0;
estádio I: pT1, pN0;
estádio IIA: pT1, pN1 ou pT2, pN0;
estádio IIB: pT2, pN1 ou pT3, pN0;
estádio IIIA: pT1, pN2 ou pT2, pN2 ou pT3, pN1 ou pT3, pN2;
estádio IIIB: pT4, qualquer N (exceto pN3);
estádio IIIC: qualquer T, pN3.
Quando analisado no formato dicotômico, os níveis de estadiamento
foram agrupados em estádios 0, I, IIA ou IIB versus estádios IIIA, IIIB ou IIIC,
representando os grupos de melhor e pior prognóstico respectivamente.

58
3.4.11 Prognóstico de Nottingham
O Índice de Prognóstico de Nottingham (NPI - Nottingham Prognostic
Index) é usado para determinar o prognóstico/probabilidade da sobrevida
global durante os 5 anos após a abordagem cirúrgica do câncer de mama
(Haybittle et al, 1982; Galea et al. 1992). Seu valor ou índice é calculado
usando três critérios patológicos, o tamanho do tumor ressecado, o status
linfonodal e o grau histológico do tulmor, aplicados na seguinte formula:
NPI = [0.2 x S] + N + G
Onde:
(S) = tamanho do tumor em centímetros;
(N) = o número de linfonodos metastáticos;
o pN0 = 2;
o pN1 = 2;
o pN2 ou pN3 = 3.
(G) = o grau histológico:
o grau 1 = 1;
o grau 2 = 2;
o grau 3 = 3;
Dependendo do valor do score NPI encontrado com a aplicação da
fórmula, cada paciente foi categorizada num grupo de prognóstico com base na
estimativa de sobrevida em 5 anos de pós operatório (quadro 4).

59
Quando analisado no formato dicotômico, o NPI foi agrupado em
moderado a excelente versus ruim, representando os grupos de melhor e pior
prognóstico respectivamente.
3.4.12 Estimativa de risco de recorrência:
No momento atual, a escolha do tratamento adjuvante do câncer de
mama envolve a observação do tipo e do grau histológico, tamanho do tumor,
invasão linfonodal, expressão de receptores hormonais e HER2, idade e outros
marcadores moleculares.
A fim de nortear as condutas terapêuticas para o tratamento do câncer
de mama, o Serviço de Oncologia do INCA (2011) utiliza consensos de dados
da literatura (Early Breast Cancer Trialists' Collaborative Group, 2005) para
definir uma Estimativa de Risco de Recorrência da Doença (ERR) que
categoriza a paciente com base nos fatores prognósticos conhecidos (quadro
5).
Quadro 4: Definição dos níveis de Risco de Estimativa de Recorrência.

60
Quando analisada no formato dicotômico, a ERR foi agrupada em risco
baixo ou intermediário versus risco elevado.
3.5 ANÁLISE DOS POLIMORFISMOS
3.5.1 Colheita de sangue
Uma amostra de sangue periférico de cada paciente (5 ml) foi colhida
por profissional treinado, aproveitando a rotina hospitalar de exames
laboratoriais já existente. Uma agulha acoplada a uma seringa foi introduzida
na veia de mais fácil acesso e uma amostra do sangue colhido foi logo
repassada para tubo apropriado. Todos os cuidados foram tomados, de forma
a tornar mínimos os riscos de infecção, contaminação, sangramento e dor.
3.5.2 Extração do DNA genômico
A extração do DNA genômico foi realizada a partir de células
mononucleares de amostras de sangue periférico colhido de cada paciente,
utilizando-se o sistema “blood genomic prep mini spin kit” (GE Heathcare,
Buckinghamshire, UK), de acordo com os procedimentos recomendados pelo
fabricante. O DNA foi armazenado a -20°C.
Quadro 5: Definição dos níveis de Risco de Estimativa de Recorrência.

61
3.5.3 Identificação do polimorfismo
O método utilizado para amplificação das regiões de DNA estudadas
neste trabalho foi a técnica de Reação em Cadeia da Polimerase (Polymerase
chain reaction – PCR). Esta técnica permite-nos obter uma grande quantidade
de DNA da região pretendida partindo de uma amostra relativamente pequena.
As sequências nucleotídicas foram identificadas pelo programa BLAST
disponível no portal do National Center for Biotechnology Information (NCBI,
2009). Os iniciadores senso e reverso utilizados para amplificação das regiões
de interesse foram desenhados a partir da observação das sequências
identificadas no portal do NCBI e confirmadas por análises de sequenciamento.
3.5.4 Genotipagem do polimorfismo de repetição CA
3.5.4.1 Amplificação da região de interesse
Para genotipar este polimorfismo situado no intron 1 do gene EGFR, tal
região foi amplificada por PCR a partir de uma mistura de reação de 10 μL
contendo: 0,4 μl de DNA genômico do paciente, 200 μM de dNTPs, 2,5 nM de
MgCl2, 50 mM de KCl, 10 mM de tris-HCl (pH 9.0), 0,25 U de Taq polimerase e
0,4 μM de cada oligonucleotídeo iniciador, sendo o senso: 5'-
CTCCACCTGCAGCCCTTC-3', marcado com fluorescência 6-FAMTM (Life
Thechnology, Carlsbad, EUA) e o reverso: 5'-CAAGGTCTGGGAACCACTGT-
3'. A mistura de reação foi processada em termociclador automático VeritiTM
(Applied Biosystems, Foster City, USA) nas seguintes condições: desnaturação
inicial a 95ºC por 5 min, seguido de 35 ciclos com 30 segundos de
desnaturação a 95ºC, 30 segundos de hibridização a 60 ºC e 30 segundos de
extensão a 72ºC; com uma etapa final de extensão a 72ºC por 5 minutos.
Os produtos de reação foram analisados por eletroforese em gel de
agarose a 2% (p/v) na presença de brometo de etídeo e visualizados sob luz
UV. O resultado foi a geração de fragmento(s) amplificado(s) de
aproximadamente 200 pb (figura 11).

62
3.5.4.2 Análise de tamanho de fragmento
Os produtos amplificados foram analisados por eletroforese de capilar
para obtenção dos tamanhos exatos referentes a cada alelo de cada paciente.
Inicialmente, 1μL do produto amplificado foi diluído em 3μL de
formamida high dye (pH 7,0). Utilizando placa de 96 poços, foram adicionados
em cada um deles: 1 μL do produto diluído, 0,5μL de padrão de peso molecular
GeneScan-400HD (Applied Biosystems, Foster City, EUA) e 8,5 μL de
formamida high dye (pH 7,0). A placa foi incubada a 95ºC por 3 minutos e logo
em seguida, mantida a 4ºC por 2 minutos.
A placa foi inserida em aparelho sequenciador automático modelo ABI
3130 (Applied Biosystems, Foster City, EUA) onde as misturas foram
submetidas à eletroforese de capilar de 30 cm em polímero POP7.
Após aproximadamente uma hora e meia, os fragmentos de DNA
marcados com a fluorescência FAMTM foram separados de acordo com seus
Figura 11: Observação sob luz UV do produto da
amplificação de sequencia do intron 1 do gene
EGFR de 3 pacientes (1 a 3) após eletroforese em
gel de agarose 2% (p/v). Obtenção de produto
esperado de aproximadamente 200 pb (p: padrão
de peso molecular de 50pb).
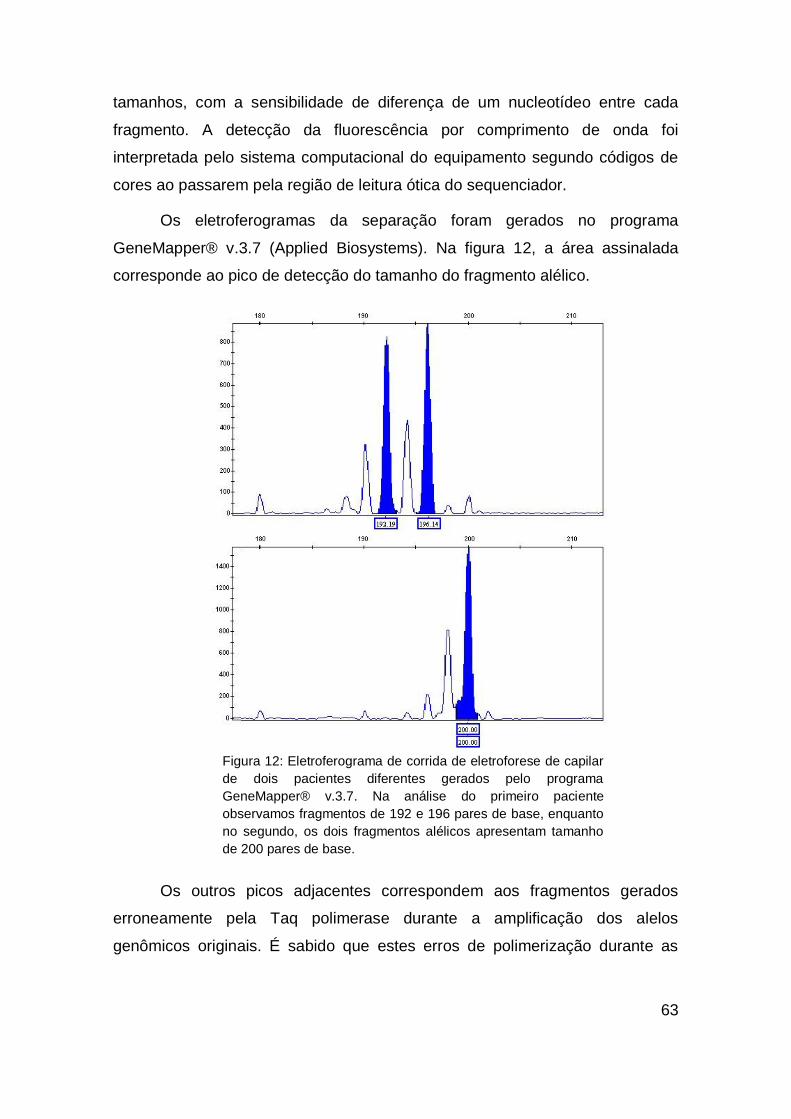
63
tamanhos, com a sensibilidade de diferença de um nucleotídeo entre cada
fragmento. A detecção da fluorescência por comprimento de onda foi
interpretada pelo sistema computacional do equipamento segundo códigos de
cores ao passarem pela região de leitura ótica do sequenciador.
Os eletroferogramas da separação foram gerados no programa
GeneMapper® v.3.7 (Applied Biosystems). Na figura 12, a área assinalada
corresponde ao pico de detecção do tamanho do fragmento alélico.
Os outros picos adjacentes correspondem aos fragmentos gerados
erroneamente pela Taq polimerase durante a amplificação dos alelos
genômicos originais. É sabido que estes erros de polimerização durante as
Figura 12: Eletroferograma de corrida de eletroforese de capilar
de dois pacientes diferentes gerados pelo programa
GeneMapper® v.3.7. Na análise do primeiro paciente
observamos fragmentos de 192 e 196 pares de base, enquanto
no segundo, os dois fragmentos alélicos apresentam tamanho
de 200 pares de base.

64
reações de amplificação em cadeia geram fragmentos de tamanhos menores
do que os originais genômicos (Ellegren 2004).
As pacientes que apresentaram dois alelos de tamanhos diferentes na
análise foram consideradas heterozigotas para o polimorfismo de repetição CA,
ou seja, apresentaram número de repetições diferente em cada alelo. As
pacientes que não apresentaram tamanhos de alelos diferentes na análise
foram consideradas homozigotas para este polimorfismo.
3.5.4.3 Confirmação por sequenciamento direto
A fim de estimar a correspondência entre o tamanho do fragmento
amplificado e o número exato de repetições CA neste fragmento, sete amostras
de pacientes homozigotas (figura 13) foram selecionadas para realização de
sequenciamento direto do DNA genômico empregando o método de
terminação em cadeia (Sanger et al., 1977).
A reação de sequenciamento foi composta pela mistura de DNA
genômico (1-3 ng), 1,5 μl de tampão de reação contendo os
dideoxinucleotídeos fluorescentes ddATP, ddCTP, ddGTP, ddTTP disponíveis
no kit ABI PRISM Dye Terminator v.3.1 (Applied Biosystems, Foster City, USA),
3,2 pmols dos iniciadores específicos senso ou reverso anteriormente descritos
para amplificação de região de interesse do exon 13 do gene EGFR e água
deionizada para volume final de 10 μl. As misturas de reação foram
processadas em termociclador automático VeritiTM (Applied Biosystems, Foster
City, USA) programado para uma desnaturação inicial por um minuto a 96°C,
seguida de 25 ciclos de 15 segundos de desnaturação a 96ºC, 15 segundos de
hibridização a 50 ºC e 4 minutos de extensão a 60ºC.
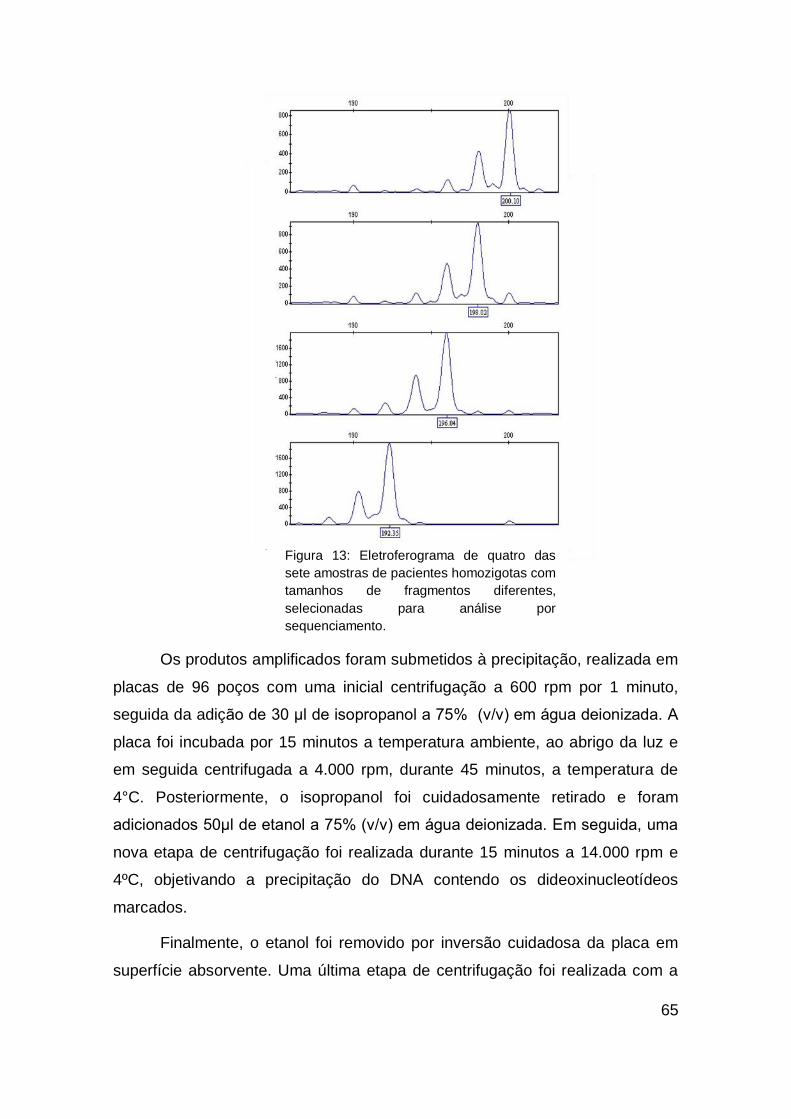
65
Os produtos amplificados foram submetidos à precipitação, realizada em
placas de 96 poços com uma inicial centrifugação a 600 rpm por 1 minuto,
seguida da adição de 30 μl de isopropanol a 75% (v/v) em água deionizada. A
placa foi incubada por 15 minutos a temperatura ambiente, ao abrigo da luz e
em seguida centrifugada a 4.000 rpm, durante 45 minutos, a temperatura de
4°C. Posteriormente, o isopropanol foi cuidadosamente retirado e foram
adicionados 50μl de etanol a 75% (v/v) em água deionizada. Em seguida, uma
nova etapa de centrifugação foi realizada durante 15 minutos a 14.000 rpm e
4ºC, objetivando a precipitação do DNA contendo os dideoxinucleotídeos
marcados.
Finalmente, o etanol foi removido por inversão cuidadosa da placa em
superfície absorvente. Uma última etapa de centrifugação foi realizada com a
Figura 13: Eletroferograma de quatro das
sete amostras de pacientes homozigotas com
tamanhos de fragmentos diferentes,
selecionadas para análise por
sequenciamento.

66
placa invertida até alcançar 600 rpm. A placa foi incubada a 60°C durante 10
minutos sob abrigo da luz para completa evaporação do etanol. Após esta
etapa, a placa foi mantida a -20ºC até o momento de ser encaminhada a
eletroforese em sequenciador automático.
Os poços da placa contendo as amostras de produto amplificado e
precipitado foram reconstituídos em 10 uL de formamida high dye (pH 7,0). A
placa foi submetida à etapa de choque térmico a 95°C por dois minutos,
objetivando a quebra de estruturas secundárias que possam impedir a correta
separação dos fragmentos de DNA marcados e em seguida, foi submetida à
eletroforese em capilar de 30 cm em sequenciador automático modelo ABI
3130 (Applied Biosystems, Foster City, EUA).
Após aproximadamente uma hora e meia, os fragmentos de DNA
marcados foram separados de acordo com seus tamanhos (diferença de um
nucleotídeo entre cada fragmento), e sequenciados por emissão de
fluorescência em diferentes comprimentos de onda para cada nucleotídeo, ao
passarem pela região de leitura ótica do sequenciador. Os comprimentos de
onda eram interpretados pelo sistema computacional do equipamento segundo
códigos de cores (azul, vermelho, verde e amarelo para cada nucleotídeo).
As sequências obtidas foram analisadas pelo programa Geneious 5.5
(Biomatters Ltd, Auckland, Nova Zelândia). Após o emparelhamento das fitas
geradas por cada um dos dois iniciadores, senso e reverso, foi possível estimar
o número de repetições CA existentes nos fragmentos de tamanho conhecido
analisados (figura 14).
Figura 14: Eletroferograma de sequenciamento dos produtos amplificados do intron 1 do gene
EGFR pelo programa Geneious 5.5. Emparelhamento da sequencia senso e reverso de um
paciente com tamanho de fragmentos de 192 pares de base. Na análise é possível observar
uma sequencia com 16 repetições CA.
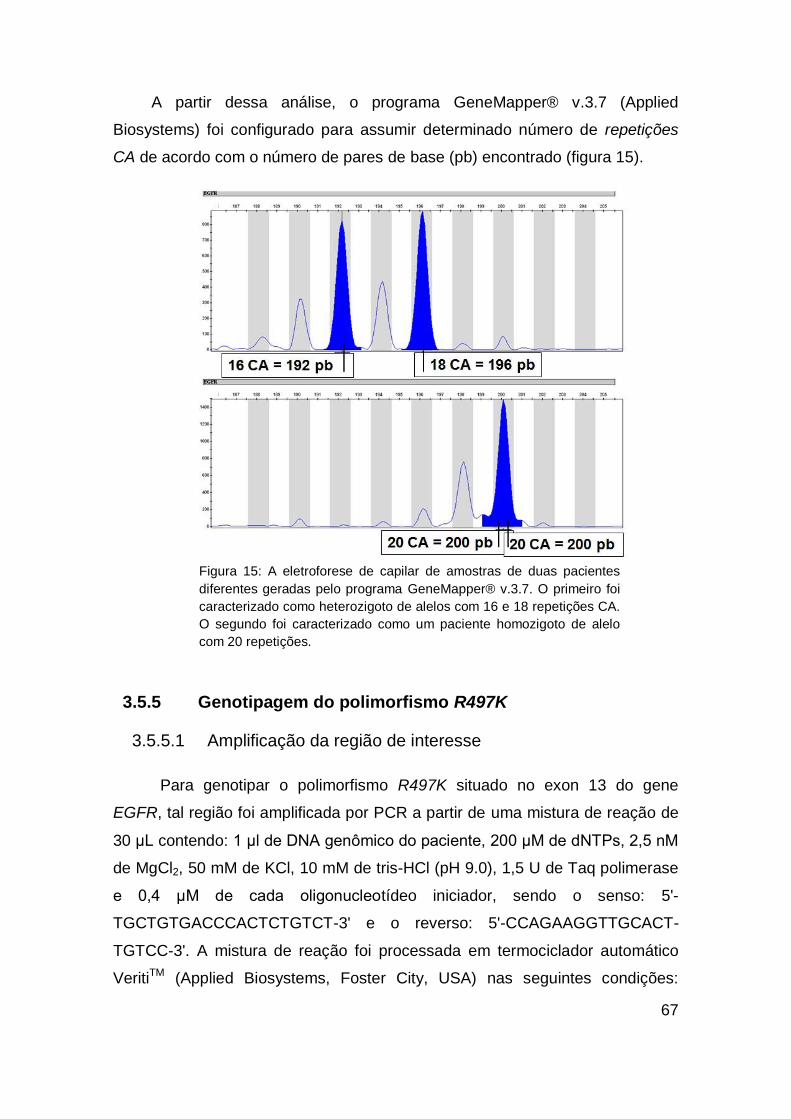
67
A partir dessa análise, o programa GeneMapper® v.3.7 (Applied
Biosystems) foi configurado para assumir determinado número de repetições
CA de acordo com o número de pares de base (pb) encontrado (figura 15).
3.5.5 Genotipagem do polimorfismo R497K
3.5.5.1 Amplificação da região de interesse
Para genotipar o polimorfismo R497K situado no exon 13 do gene
EGFR, tal região foi amplificada por PCR a partir de uma mistura de reação de
30 μL contendo: 1 μl de DNA genômico do paciente, 200 μM de dNTPs, 2,5 nM
de MgCl2, 50 mM de KCl, 10 mM de tris-HCl (pH 9.0), 1,5 U de Taq polimerase
e 0,4 μM de cada oligonucleotídeo iniciador, sendo o senso: 5'-
TGCTGTGACCCACTCTGTCT-3' e o reverso: 5'-CCAGAAGGTTGCACT-
TGTCC-3'. A mistura de reação foi processada em termociclador automático
VeritiTM (Applied Biosystems, Foster City, USA) nas seguintes condições:
Figura 15: A eletroforese de capilar de amostras de duas pacientes
diferentes geradas pelo programa GeneMapper® v.3.7. O primeiro foi
caracterizado como heterozigoto de alelos com 16 e 18 repetições CA.
O segundo foi caracterizado como um paciente homozigoto de alelo
com 20 repetições.

68
desnaturação inicial, a 95ºC por 5 minutos, seguido de 35 ciclos de 30
segundos de desnaturação a 95ºC, 30 segundos de hibridização a 56 ºC e 30
segundos de extensão a 72ºC; com uma etapa final de extensão a 72ºC por 5
minutos. Os produtos de reação foram analisados por eletroforese em gel de
agarose a 2% (p/v) na presença de brometo de etídeo e visualizados sob luz
UV. O resultado foi a geração de um fragmento amplificado de 156 pb (figura
16).
3.5.5.2 Digestão com enzima de restrição
A digestão foi realizada em uma mistura de reação de 15μl contendo 5μl
do produto de DNA amplificado do paciente, 5U da enzima de restrição BstNI
(Bio Labs, New England, UK) e tampão de reação correspondente. A mistura
foi incubada por uma hora a 60ºC. Logo após, receberam a adição de mais 2 U
da enzima BstNI, seguido de mais 2 horas de incubação a 60ºC.
Os produtos da digestão foram analisados por eletroforese em gel de
agarose 2% (p/v) corados com brometo de etídeo e visualizados sob luz UV.
Figura 16: Observação sob luz UV do produto
da amplificação de sequencia do exon 13 do
gene EGFR de 3 pacientes (1 a 3) após
eletroforese em gel de agarose 2% (p/v).
Obtenção de produto esperado de
aproximadamente 150 pb (p: padrão de peso
molecular de 50pb).
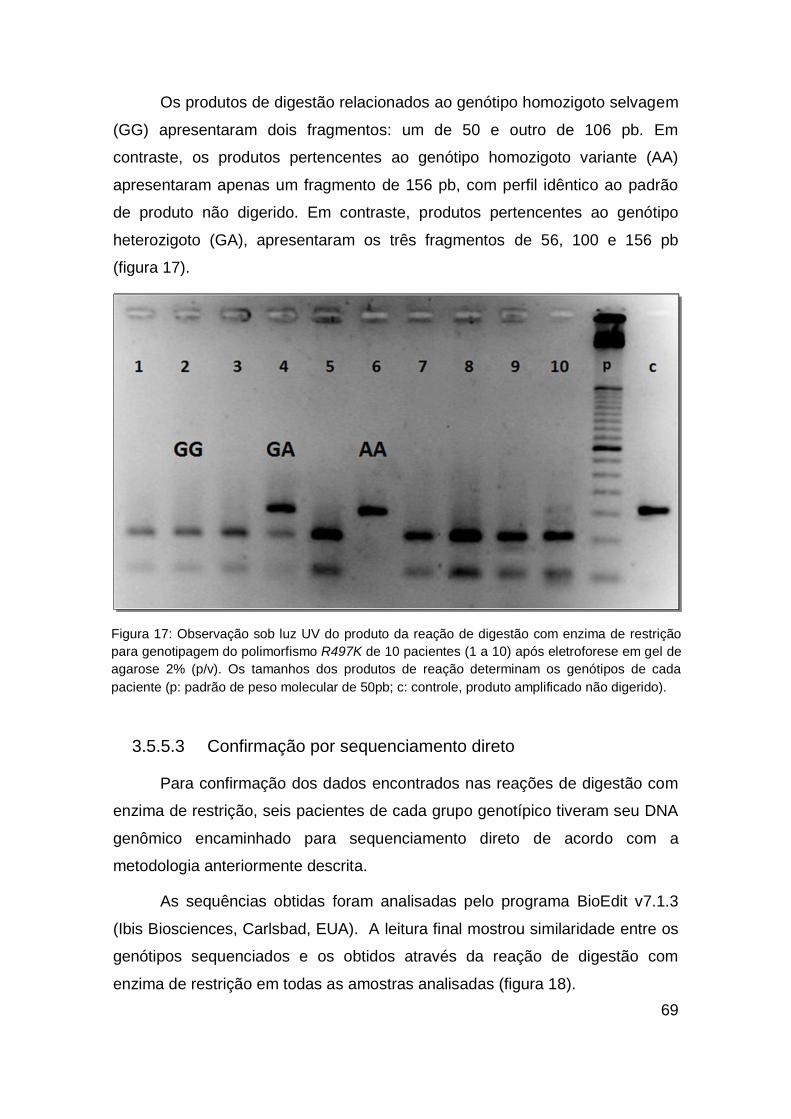
69
Os produtos de digestão relacionados ao genótipo homozigoto selvagem
(GG) apresentaram dois fragmentos: um de 50 e outro de 106 pb. Em
contraste, os produtos pertencentes ao genótipo homozigoto variante (AA)
apresentaram apenas um fragmento de 156 pb, com perfil idêntico ao padrão
de produto não digerido. Em contraste, produtos pertencentes ao genótipo
heterozigoto (GA), apresentaram os três fragmentos de 56, 100 e 156 pb
(figura 17).
3.5.5.3 Confirmação por sequenciamento direto
Para confirmação dos dados encontrados nas reações de digestão com
enzima de restrição, seis pacientes de cada grupo genotípico tiveram seu DNA
genômico encaminhado para sequenciamento direto de acordo com a
metodologia anteriormente descrita.
As sequências obtidas foram analisadas pelo programa BioEdit v7.1.3
(Ibis Biosciences, Carlsbad, EUA). A leitura final mostrou similaridade entre os
genótipos sequenciados e os obtidos através da reação de digestão com
enzima de restrição em todas as amostras analisadas (figura 18).
Figura 17: Observação sob luz UV do produto da reação de digestão com enzima de restrição
para genotipagem do polimorfismo R497K de 10 pacientes (1 a 10) após eletroforese em gel de
agarose 2% (p/v). Os tamanhos dos produtos de reação determinam os genótipos de cada
paciente (p: padrão de peso molecular de 50pb; c: controle, produto amplificado não digerido).
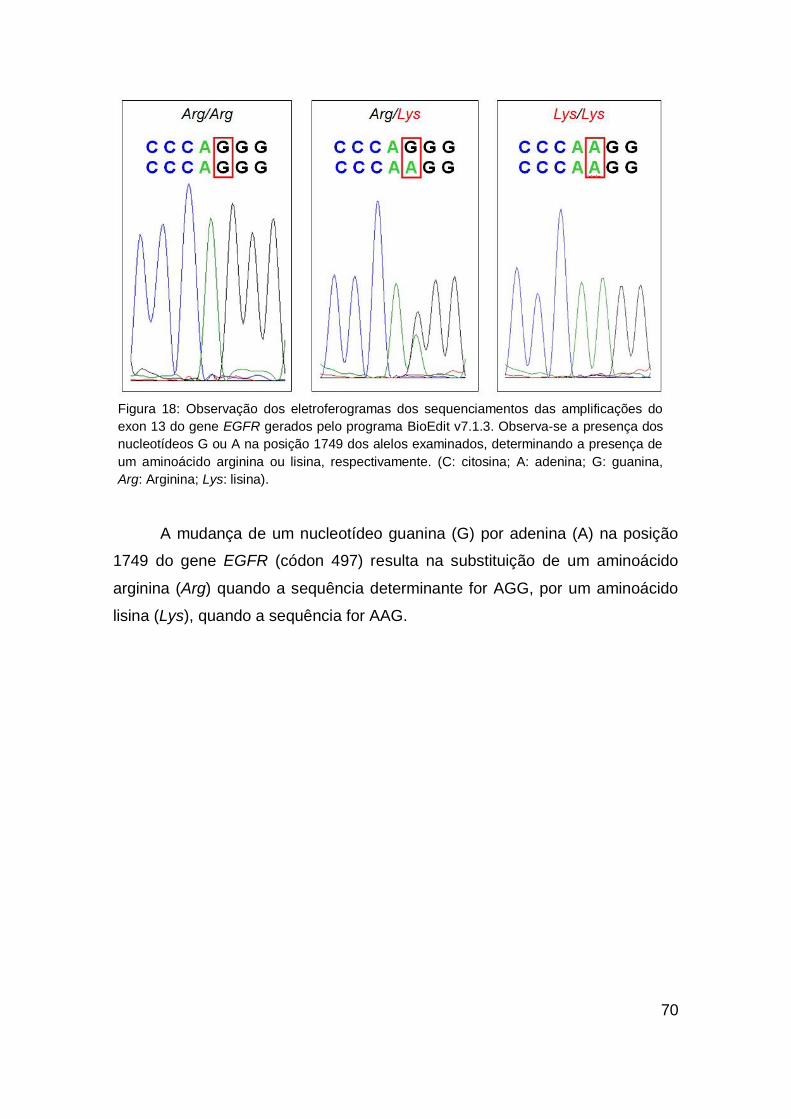
70
A mudança de um nucleotídeo guanina (G) por adenina (A) na posição
1749 do gene EGFR (códon 497) resulta na substituição de um aminoácido
arginina (Arg) quando a sequência determinante for AGG, por um aminoácido
lisina (Lys), quando a sequência for AAG.
Figura 18: Observação dos eletroferogramas dos sequenciamentos das amplificações do
exon 13 do gene EGFR gerados pelo programa BioEdit v7.1.3. Observa-se a presença dos
nucleotídeos G ou A na posição 1749 dos alelos examinados, determinando a presença de
um aminoácido arginina ou lisina, respectivamente. (C: citosina; A: adenina; G: guanina,
Arg: Arginina; Lys: lisina).

71
3.6 ANÁLISE ESTATÍSTICA
Para gerenciamento das análises estatísticas, foi utilizado o programa
SPSS (Statistical Package for the Social Sciences, versão 15.0).
Foi realizado um estudo descritivo da população em estudo, através da
análise das medidas de tendência central e de dispersão e distribuição de
freqüência para as variáveis independentes.
Os resultados encontrados na identificação dos polimorfismos foram
utilizados para estimar a frequência alélica. A aderência ao princípio de Hardy-
Weinberg foi avaliada pelo teste do qui-quadrado.
O teste do qui-quadrado foi usado para comparação de dados de
frequência, com exceção das comparações com frequencia menor que 5, em
que usou-se o teste de Fisher.
O cálculo de tendência da incidência dos fenótipos de acordo com a
presença da variante alélica polimórfica foi realizado por associação linear.
A avaliação de associações entre as variáveis clínico-patológicas e a
presença de polimorfismos foi realizada através do cálculo de razão de
chances (OR - odds ratio), considerando um intervalo de confiança de 95%.
Os ajustes nos cálculos de razão de chance pelas variáveis de
interferência foram realizados por regressão binária.
Neste estudo, o nível de significância para rejeição da hipótese de
nulidade foi de 5% (p ≤ 0,05). Os resultados com valor de p menor do que 0,05
foram destacados em negrito.

72
4 RESULTADOS
4.1 POPULAÇÃO
4.1.1.1 Recrutamento
Durante o período de recrutamento, foi possível selecionar 540
pacientes com diagnóstico de câncer de mama, das quais 492 foram incluídas.
Os genótipos para o polimorfismo de repetição CA e para o polimorfismo
R497K foram obtidos em 426 e 472 pacientes, respectivamente (quadro 6).
Portanto, dentre as 426 pacientes genotipadas para o polimorfismo de
repetição CA, todas foram também genotipadas para o polimorfismo R497K.
4.1.1.1 Descrição da população
As características clinico-patológicas das 472 pacientes que tiveram seu
DNA analisado para pelo menos um polimorfismo são descritas na tabela 1.
Não parece haver diferença no perfil descritivo da população quando as 48
Quadro 6: Fluxograma de inclusão e análise da população do estudo.
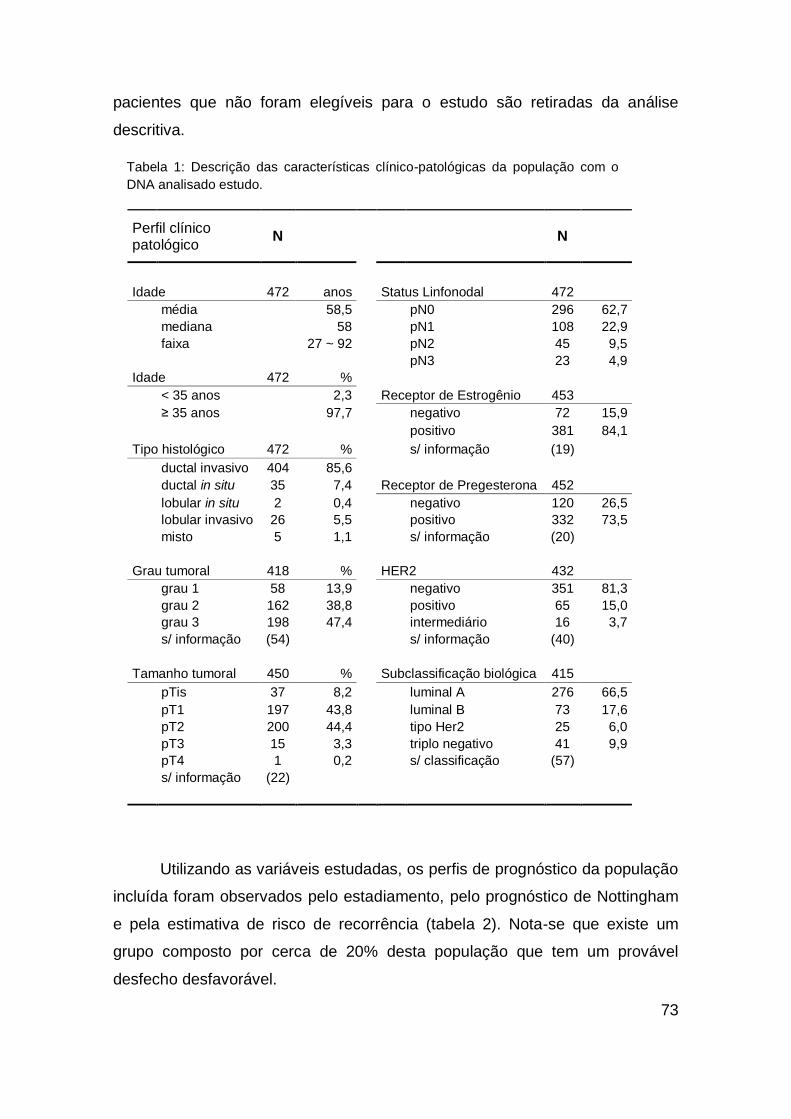
73
pacientes que não foram elegíveis para o estudo são retiradas da análise
descritiva.
Perfil clínico patológico
N N
Idade 472 anos Status Linfonodal 472
média 58,5 pN0 296 62,7
mediana 58 pN1 108 22,9
faixa 27 ~ 92 pN2 45 9,5
pN3 23 4,9
Idade 472 %
< 35 anos 2,3 Receptor de Estrogênio 453
≥ 35 anos 97,7 negativo 72 15,9
positivo 381 84,1
Tipo histológico 472 % s/ informação (19)
ductal invasivo 404 85,6
ductal in situ 35 7,4 Receptor de Pregesterona 452
lobular in situ 2 0,4 negativo 120 26,5
lobular invasivo 26 5,5 positivo 332 73,5
misto 5 1,1 s/ informação (20)
Grau tumoral 418 % HER2 432
grau 1 58 13,9 negativo 351 81,3
grau 2 162 38,8 positivo 65 15,0
grau 3 198 47,4 intermediário 16 3,7
s/ informação (54) s/ informação (40)
Tamanho tumoral 450 % Subclassificação biológica 415
pTis 37 8,2 luminal A 276 66,5
pT1 197 43,8 luminal B 73 17,6
pT2 200 44,4 tipo Her2 25 6,0
pT3 15 3,3 triplo negativo 41 9,9
pT4 1 0,2 s/ classificação (57)
s/ informação (22)
Utilizando as variáveis estudadas, os perfis de prognóstico da população
incluída foram observados pelo estadiamento, pelo prognóstico de Nottingham
e pela estimativa de risco de recorrência (tabela 2). Nota-se que existe um
grupo composto por cerca de 20% desta população que tem um provável
desfecho desfavorável.
Tabela 1: Descrição das características clínico-patológicas da população com o
DNA analisado estudo.

74
Estadiamento e prognóstico N
Estadiamento (pTNM) 451 %
0 37 8,2
I 141 31,3
II A 138 30,6
II B 66 14,6
III A 45 10,0
III B 1 0,2
III C 23 5,1
s/ classificação (21)
Prognóstico de Nottingham 374
excelente 22 5,9
bom 72 19,3
moderado 200 53,5
ruim 80 21,4
s/ classificação (98)
Risco de Recorrência 427
baixo 25 5,9
intermediário 316 74,0
alto 86 20,1
s/ classificação (45)
4.1.1.2 . Polimorfismo de repetição CA
Observando os resultados dos ensaios de genotipagem de 426
pacientes para o polimorfismo de repetição CA, foi detectada a ocorrência de
alelos contendo entre 14 e 24 repetições CA (gráfico 1). Os alelos de 16 e de
20 repetições foram os alelos mais frequentes na população e suas frequências
alélicas podem ser observadas na tabela 4. A análise de todos os tamanhos
alélicos encontrados na população indica 37 genótipos diferentes.
Tabela 2: Descrição das características patológicas
pelas estimativas de prognóstico.
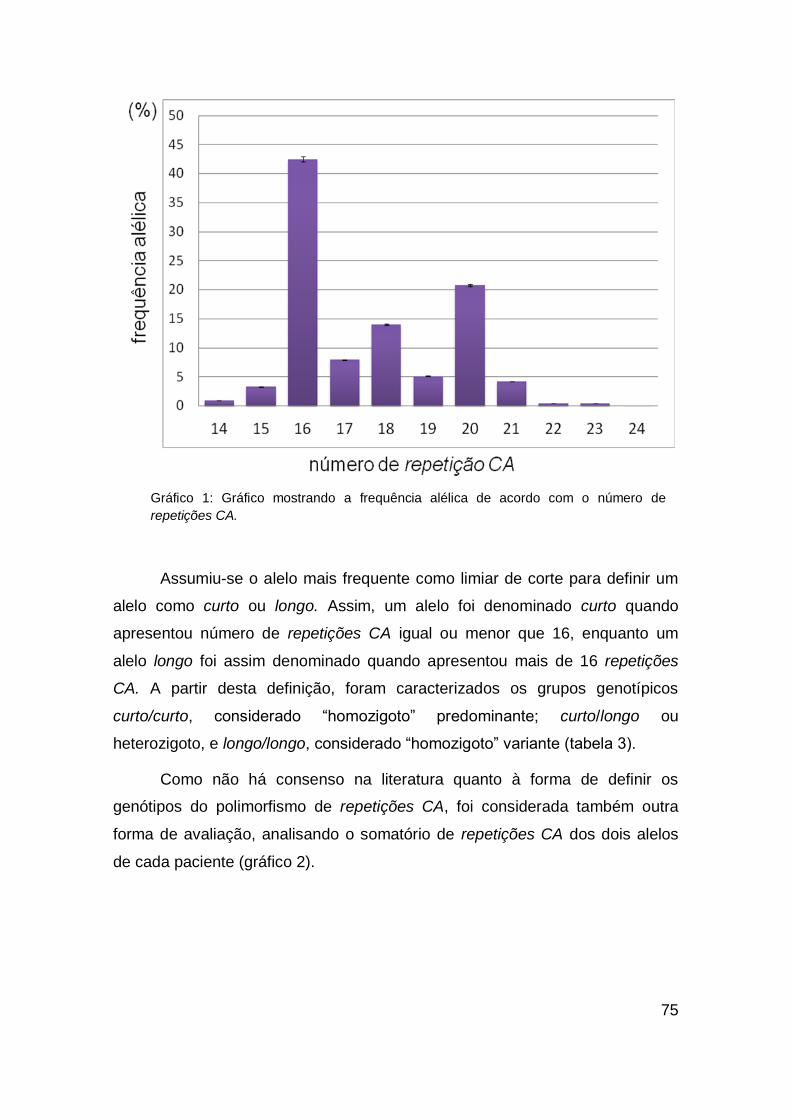
75
Assumiu-se o alelo mais frequente como limiar de corte para definir um
alelo como curto ou longo. Assim, um alelo foi denominado curto quando
apresentou número de repetições CA igual ou menor que 16, enquanto um
alelo longo foi assim denominado quando apresentou mais de 16 repetições
CA. A partir desta definição, foram caracterizados os grupos genotípicos
curto/curto, considerado “homozigoto” predominante; curto/longo ou
heterozigoto, e longo/longo, considerado “homozigoto” variante (tabela 3).
Como não há consenso na literatura quanto à forma de definir os
genótipos do polimorfismo de repetições CA, foi considerada também outra
forma de avaliação, analisando o somatório de repetições CA dos dois alelos
de cada paciente (gráfico 2).
Gráfico 1: Gráfico mostrando a frequência alélica de acordo com o número de
repetições CA.
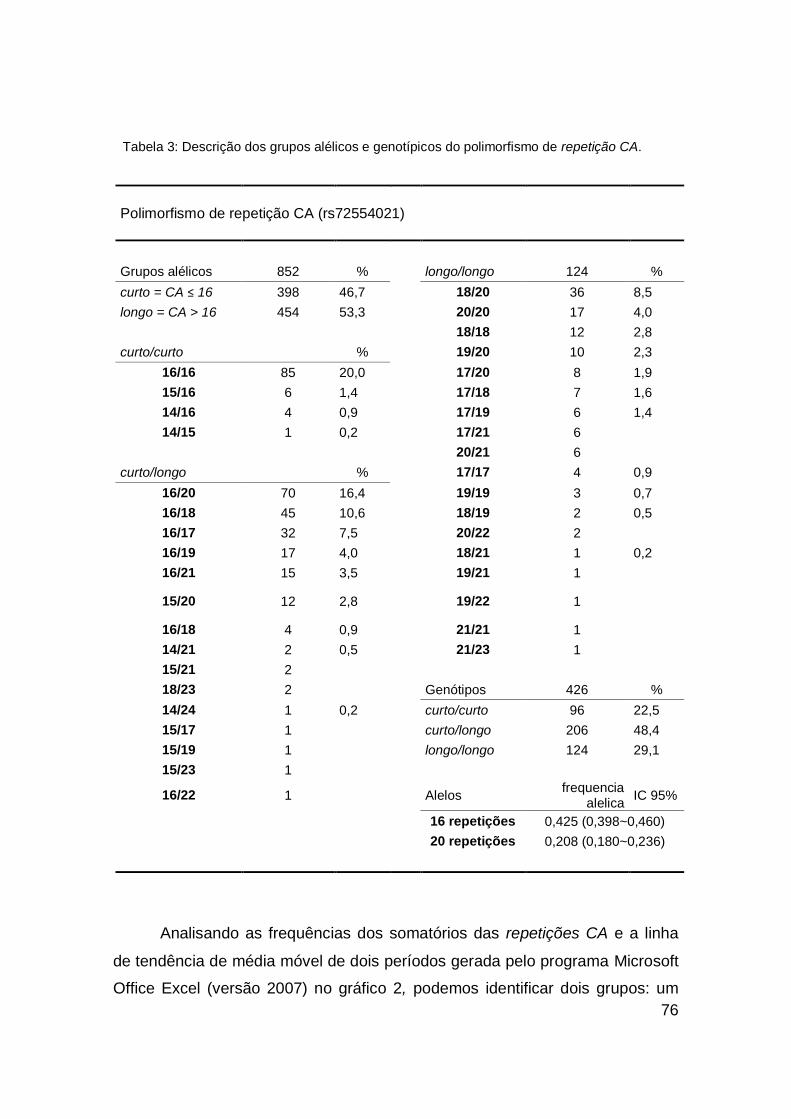
76
Polimorfismo de repetição CA (rs72554021)
Grupos alélicos 852 % longo/longo 124 %
curto = CA ≤ 16 398 46,7 18/20 36 8,5
longo = CA > 16 454 53,3 20/20 17 4,0
18/18 12 2,8
curto/curto % 19/20 10 2,3
16/16 85 20,0 17/20 8 1,9
15/16 6 1,4 17/18 7 1,6
14/16 4 0,9 17/19 6 1,4
14/15 1 0,2 17/21 6
20/21 6
curto/longo % 17/17 4 0,9
16/20 70 16,4 19/19 3 0,7
16/18 45 10,6 18/19 2 0,5
16/17 32 7,5 20/22 2
16/19 17 4,0 18/21 1 0,2
16/21 15 3,5 19/21 1
15/20 12 2,8 19/22 1
16/18 4 0,9 21/21 1
14/21 2 0,5 21/23 1
15/21 2
18/23 2 Genótipos 426 %
14/24 1 0,2 curto/curto 96 22,5
15/17 1 curto/longo 206 48,4
15/19 1 longo/longo 124 29,1
15/23 1
16/22 1 Alelos frequencia
alelica IC 95%
16 repetições 0,425 (0,398~0,460)
20 repetições 0,208 (0,180~0,236)
Analisando as frequências dos somatórios das repetições CA e a linha
de tendência de média móvel de dois períodos gerada pelo programa Microsoft
Office Excel (versão 2007) no gráfico 2, podemos identificar dois grupos: um
Tabela 3: Descrição dos grupos alélicos e genotípicos do polimorfismo de repetição CA.
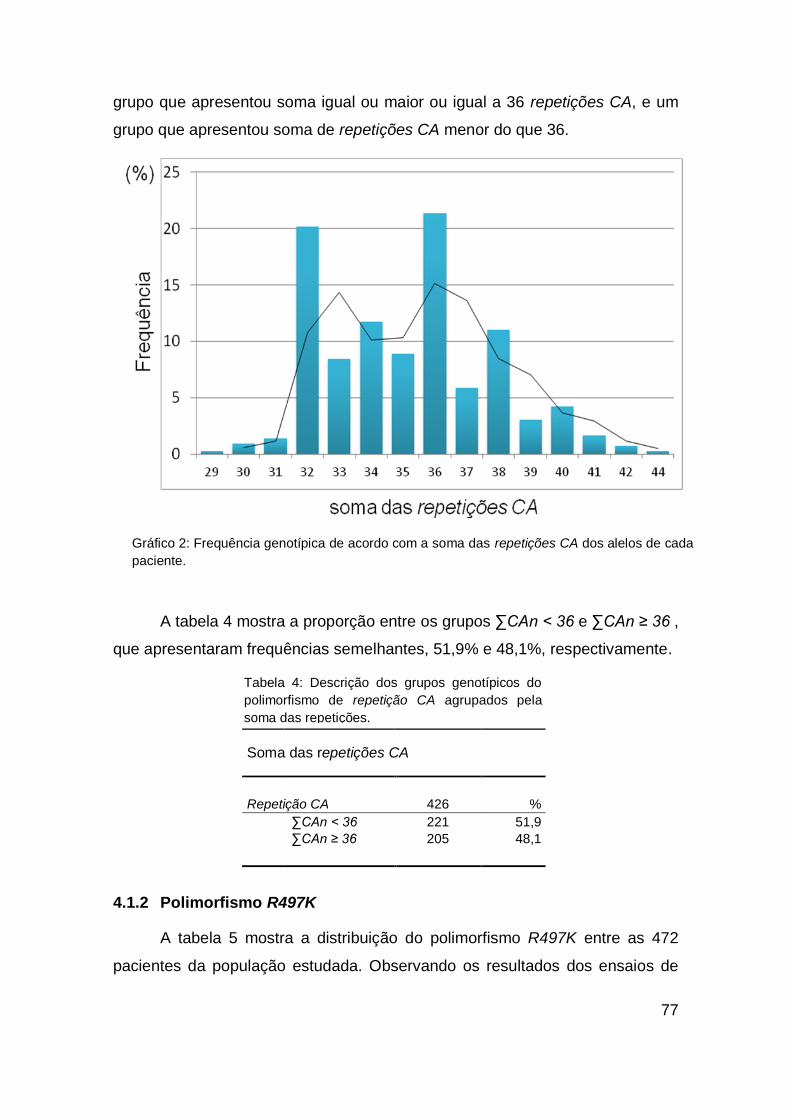
77
grupo que apresentou soma igual ou maior ou igual a 36 repetições CA, e um
grupo que apresentou soma de repetições CA menor do que 36.
A tabela 4 mostra a proporção entre os grupos ∑CAn < 36 e ∑CAn ≥ 36 ,
que apresentaram frequências semelhantes, 51,9% e 48,1%, respectivamente.
Soma das repetições CA
Repetição CA 426 %
∑CAn < 36 221 51,9
∑CAn ≥ 36 205 48,1
4.1.2 Polimorfismo R497K
A tabela 5 mostra a distribuição do polimorfismo R497K entre as 472
pacientes da população estudada. Observando os resultados dos ensaios de
Gráfico 2: Frequência genotípica de acordo com a soma das repetições CA dos alelos de cada
paciente.
Tabela 4: Descrição dos grupos genotípicos do
polimorfismo de repetição CA agrupados pela
soma das repetições.

78
genotipagem, foi encontrada uma frequência de 21,8% para o alelo variante A
(adenina), representado pelo fenótipo Lys. Analisando a distribuição genotípica
descrita na tabela 5, observamos que a população estudada encontra-se
dentro do equilíbrio de Hardy-Weinberg.
Grupos genotípicos R497K (rs11543848)
Alelos 944 frequência alélica (IC 95%)
Arg 739 0,782 (0,755~0,808)
Lys 205 0,218 (0,192~0,245)
Genótipos 472 %
Arg/Arg 285 60,4
Arg/Lys 169 35,8
Lys/Lys 18 3,8
Equilíbrio de Hardy-Weinberg
X2 = 1,329736298
p = 0,248852312
4.2 ASSOCIAÇÕES FENÓTIPO X GENÓTIPO
4.2.1 Polimorfismo de repetição CA
A seguir analisaremos a distribuição dos grupos genotípicos de repetição
CA em função das características clínico-patológicas. Na tabela 6, são
apresentadas as variáveis de idade e de descrição morfológica dos tumores,
não havendo diferenças significativas para a distribuição dos grupos
genotípicos de repetição CA.
Tabela 5: Descrição dos grupos alélicos e genotípicos do polimorfismo de
repetição CA agrupados pela soma das repetições.
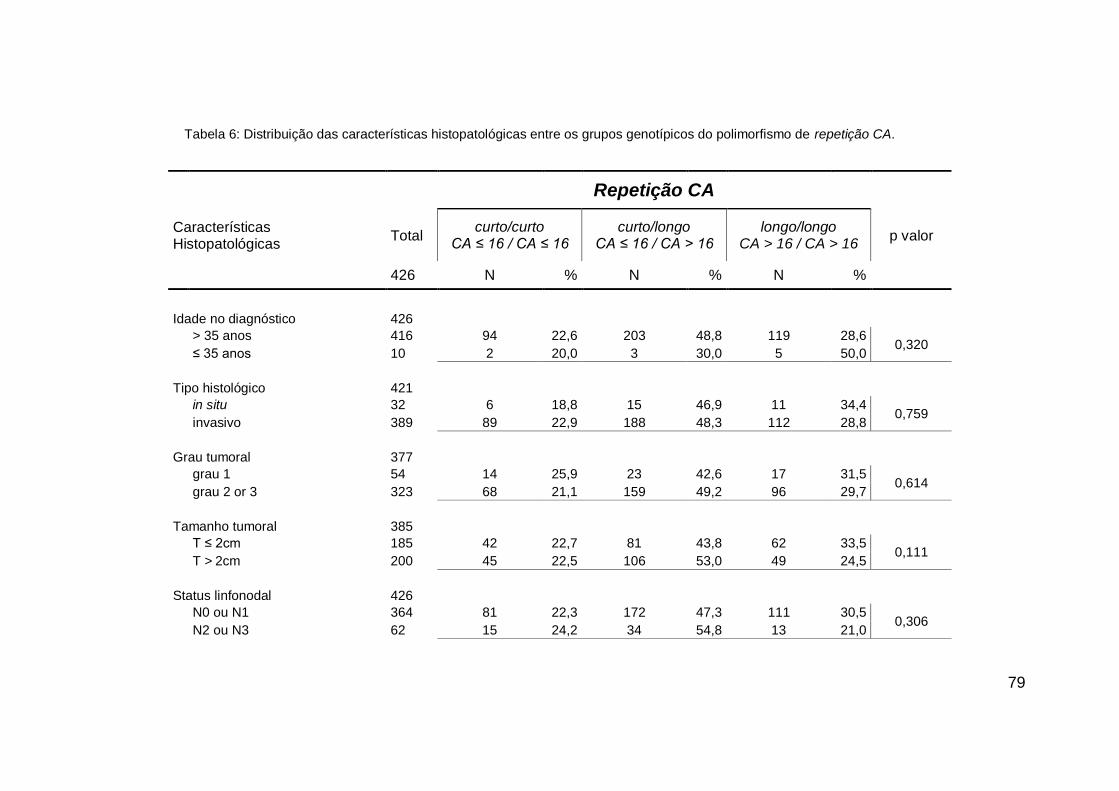
79
Repetição CA
Características Histopatológicas
Total curto/curto
CA ≤ 16 / CA ≤ 16 curto/longo
CA ≤ 16 / CA > 16 longo/longo
CA > 16 / CA > 16 p valor
426 N % N % N %
Idade no diagnóstico 426
> 35 anos 416 94 22,6 203 48,8 119 28,6 0,320
≤ 35 anos 10 2 20,0 3 30,0 5 50,0
Tipo histológico 421
in situ 32 6 18,8 15 46,9 11 34,4 0,759
invasivo 389 89 22,9 188 48,3 112 28,8
Grau tumoral 377
grau 1 54 14 25,9 23 42,6 17 31,5 0,614
grau 2 or 3 323 68 21,1 159 49,2 96 29,7
Tamanho tumoral 385
T ≤ 2cm 185 42 22,7 81 43,8 62 33,5 0,111
T > 2cm 200 45 22,5 106 53,0 49 24,5
Status linfonodal 426
N0 ou N1 364 81 22,3 172 47,3 111 30,5 0,306
N2 ou N3 62 15 24,2 34 54,8 13 21,0
Tabela 6: Distribuição das características histopatológicas entre os grupos genotípicos do polimorfismo de repetição CA.
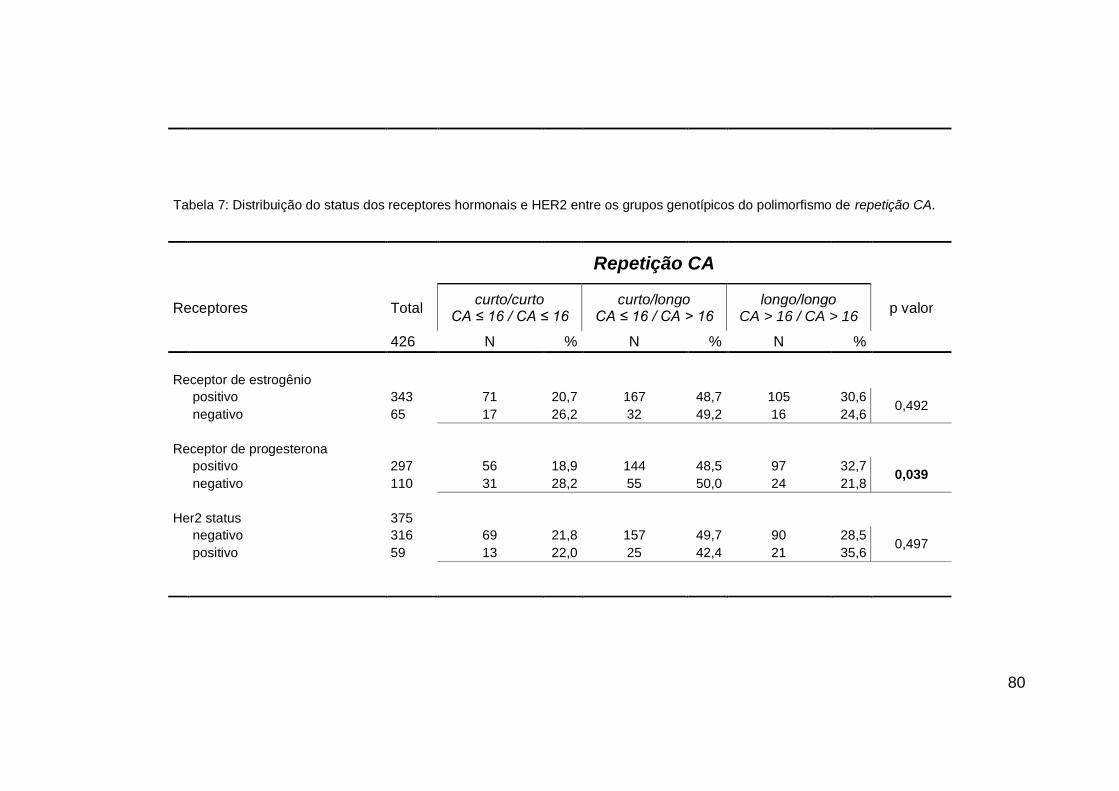
80
Repetição CA
Receptores Total curto/curto
CA ≤ 16 / CA ≤ 16 curto/longo
CA ≤ 16 / CA > 16 longo/longo
CA > 16 / CA > 16 p valor
426 N % N % N %
Receptor de estrogênio
positivo 343 71 20,7 167 48,7 105 30,6 0,492
negativo 65 17 26,2 32 49,2 16 24,6
Receptor de progesterona
positivo 297 56 18,9 144 48,5 97 32,7 0,039
negativo 110 31 28,2 55 50,0 24 21,8
Her2 status 375
negativo 316 69 21,8 157 49,7 90 28,5 0,497
positivo 59 13 22,0 25 42,4 21 35,6
Tabela 7: Distribuição do status dos receptores hormonais e HER2 entre os grupos genotípicos do polimorfismo de repetição CA.
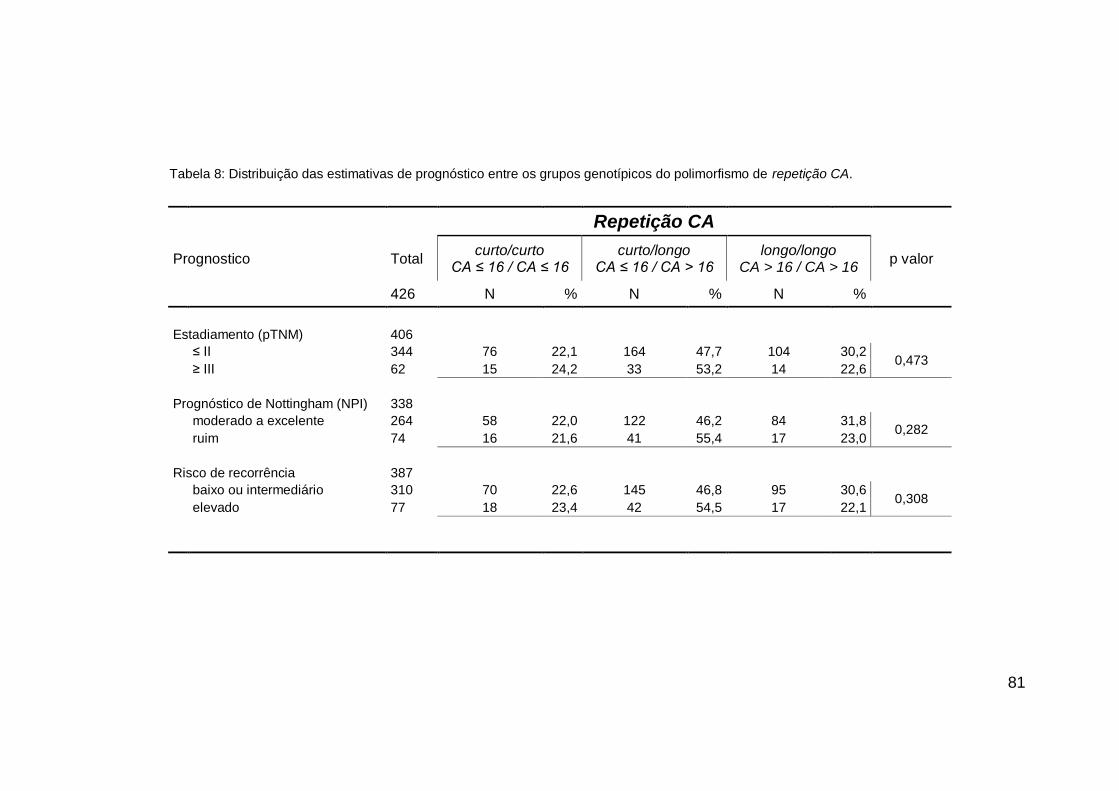
81
Repetição CA
Prognostico Total curto/curto
CA ≤ 16 / CA ≤ 16 curto/longo
CA ≤ 16 / CA > 16 longo/longo
CA > 16 / CA > 16 p valor
426 N % N % N %
Estadiamento (pTNM) 406
≤ II 344 76 22,1 164 47,7 104 30,2 0,473
≥ III 62 15 24,2 33 53,2 14 22,6
Prognóstico de Nottingham (NPI) 338
moderado a excelente 264 58 22,0 122 46,2 84 31,8 0,282
ruim 74 16 21,6 41 55,4 17 23,0
Risco de recorrência 387
baixo ou intermediário 310 70 22,6 145 46,8 95 30,6 0,308
elevado 77 18 23,4 42 54,5 17 22,1
Tabela 8: Distribuição das estimativas de prognóstico entre os grupos genotípicos do polimorfismo de repetição CA.

82
Na tabela 7, estão apresentados os perfis de expressão de receptores e
nota-se que a distribuição dos genótipos de repetição CA é diferente em função
do status do receptor de progesterona.
Na tabela 8, são avaliadas as variáveis de estimativa de prognóstico não
havendo diferenças significativas para a distribuição dos grupos genotípicos de
repetição CA.
Dentre as características clinico-patológicas estudadas, o status de
progesterona foi a única variável que demonstrou ter um p valor menor do que
0,05 para a diferença entre as distribuições genotípicas de repetição CA. No
gráfico 3, observamos a tendência de diminuição da ocorrência do status RP-
entre os grupos genotípicos de repetição CA (p = 0,011).
Embora as razões de chance entre os grupos curto/longo e curto/curto
ou entre os grupos longo/longo e curto/longo ultrapassem o valor de referência
(OR = 1,0), as pacientes com genótipo longo/longo têm por volta de 50%
menos chance de ter um status RP- do que pacientes com genótipo curto/curto
ou curto/longo (tabela 9). Quando ajustamos o cálculo de OR para outras
Gráfico 3: Tendência de diminuição da proporção de pacientes com
status RP negativo entre os grupos genotípicos de repetição CA.

83
variáveis que também poderiam estar associadas ao status de RP-, a
associação entre genótipo longo/longo e o status RP- manteve-se
estatisticamente significativa.
Repetição CA
Características curto/curto + curto/longo
longo/longo
OR = 1,000 OR cr (IC95%)
OR ajus (IC95%)
Receptor de progesterona 0,575 (0,344~0,961)
negativo 0,324 (0,124~0,845)
OR: odds ratio; ORcr: OR crude; OR ajus: OR ajustado:
IC 95%: intervalo de confiança de 95%
(RP) OR ajus p/ idade, tipo histológico, grau, pT, pN, RE e HER2.
A definição de pontos de corte para classificação dos alelos com
repetição CA em curtos ou longos para este polimorfismo revela desafios
devido à variabilidade dos tamanhos entre os alelos na população e a
indefinição sobre a real associação entre determinado tamanho ou grupo
específico de repetições CA e o perfil transcricional diferenciado.
Assim, também foi analisada a influência das características clínico-
patológicas sobre a distribuição os grupos genotípicos de repetição CA,
divididos de acordo com a soma alélica de repetições CA de cada paciente:
∑CAn < 36 versus ∑CAn ≥ 36. Novamente, apenas o status de RP- afetou a
proporção entre os grupos ∑CAn < 36 e ∑CAn ≥ 36 (tabela 10), isto é, as
pacientes com genótipo ∑CAn ≥ 36 têm por volta de 50% menos chance de ter
um status RP- do que as pacientes com ∑CAn < 36. Quando ajustamos este
cálculo para outras variáveis que também poderiam estar associadas ao status
Tabela 9: Cálculo de razão de chances para o status RP negativo
entre os grupos genotípicos de Repetição CA.de repetição CA.

84
de RP negativo, a associação entre genótipo ∑CAn ≥ 36 e o status RP negativo
continua estatisticamente significativa.
Repetição CA
Características ∑CAn < 36 ∑CAn ≥ 36
OR = 1,000 OR cr (IC95%)
OR ajus (IC95%)
Receptor de progesterona 0,536 (0,343~0,838)
negativo 0,277 (0,125 ~ 0,613)
OR: odds ratio; ORcr: OR crude; OR ajus: OR ajustado:
IC 95%: intervalo de confiança de 95%
(RP) OR ajus p/ idade, tipo histológico, grau, pT, pN, RE e HER2.
Visto que o diagnóstico de tumor com status RP- reflete pior prognóstico
para o tratamento do câncer de mama, deste resultado infere-se que a
ocorrência do genótipo variante do polimorfismo de repetição CA, representado
pela presença de dois alelos longos (CAn > 16) ou pela soma dos alelos maior
ou igual a 36 (∑CAn ≥ 36), poderia estar associado a um perfil de melhor
prognóstico da doença.
4.2.2 Polimorfismo R497K
A seguir, analisaremos a distribuição dos grupos fenotípicos gerados
pelo genótipo do polimorfismo R497K em função das características clínico-
patológicas, estadiamento e estimativas de prognóstico. Os resultados de
distribuição estão apresentados na tabela 11, 12 e 13.
Tabela 10: Cálculo de razão de chances para o status RP negativo
entre os grupos genotípicos de Repetição CA, agrupados pela soma
alélica de cada paciente.
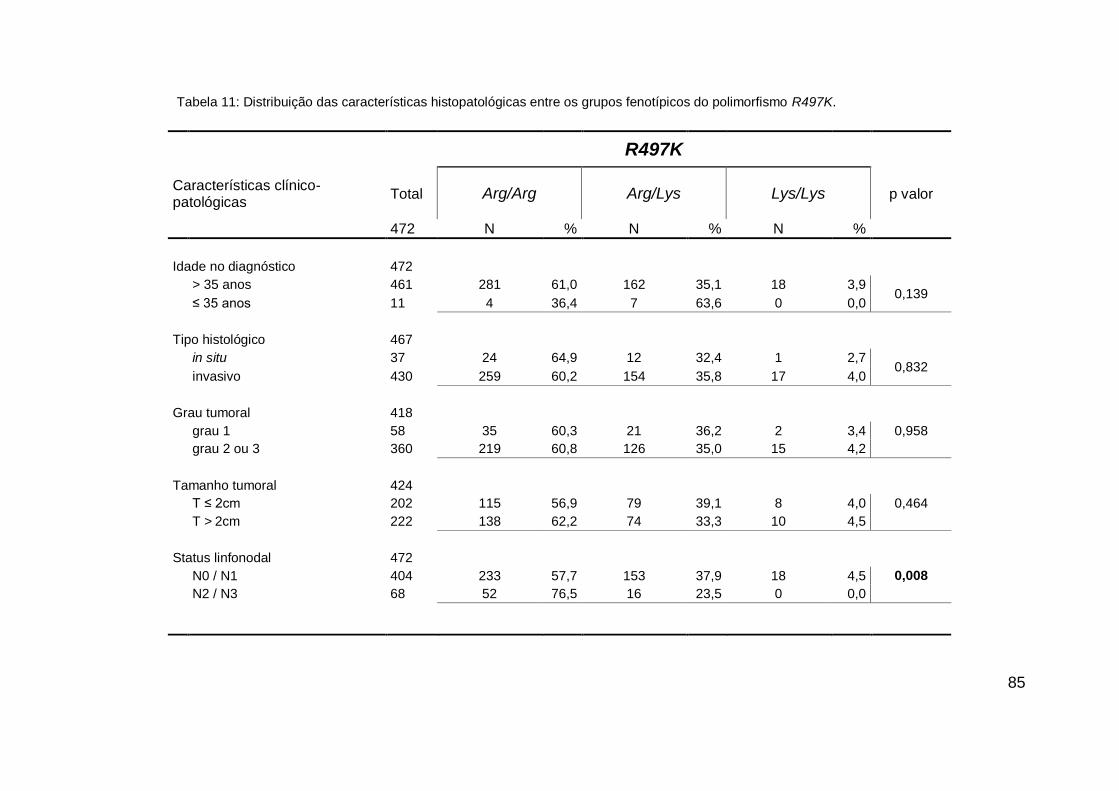
85
R497K
Características clínico-patológicas
Total Arg/Arg Arg/Lys Lys/Lys p valor
472 N % N % N %
Idade no diagnóstico 472
> 35 anos 461 281 61,0 162 35,1 18 3,9 0,139
≤ 35 anos 11 4 36,4 7 63,6 0 0,0
Tipo histológico 467
in situ 37 24 64,9 12 32,4 1 2,7 0,832
invasivo 430 259 60,2 154 35,8 17 4,0
Grau tumoral 418
grau 1 58 35 60,3 21 36,2 2 3,4 0,958
grau 2 ou 3 360 219 60,8 126 35,0 15 4,2
Tamanho tumoral 424
T ≤ 2cm 202 115 56,9 79 39,1 8 4,0 0,464
T > 2cm 222 138 62,2 74 33,3 10 4,5
Status linfonodal 472
N0 / N1 404 233 57,7 153 37,9 18 4,5 0,008
N2 / N3 68 52 76,5 16 23,5 0 0,0
Tabela 11: Distribuição das características histopatológicas entre os grupos fenotípicos do polimorfismo R497K.
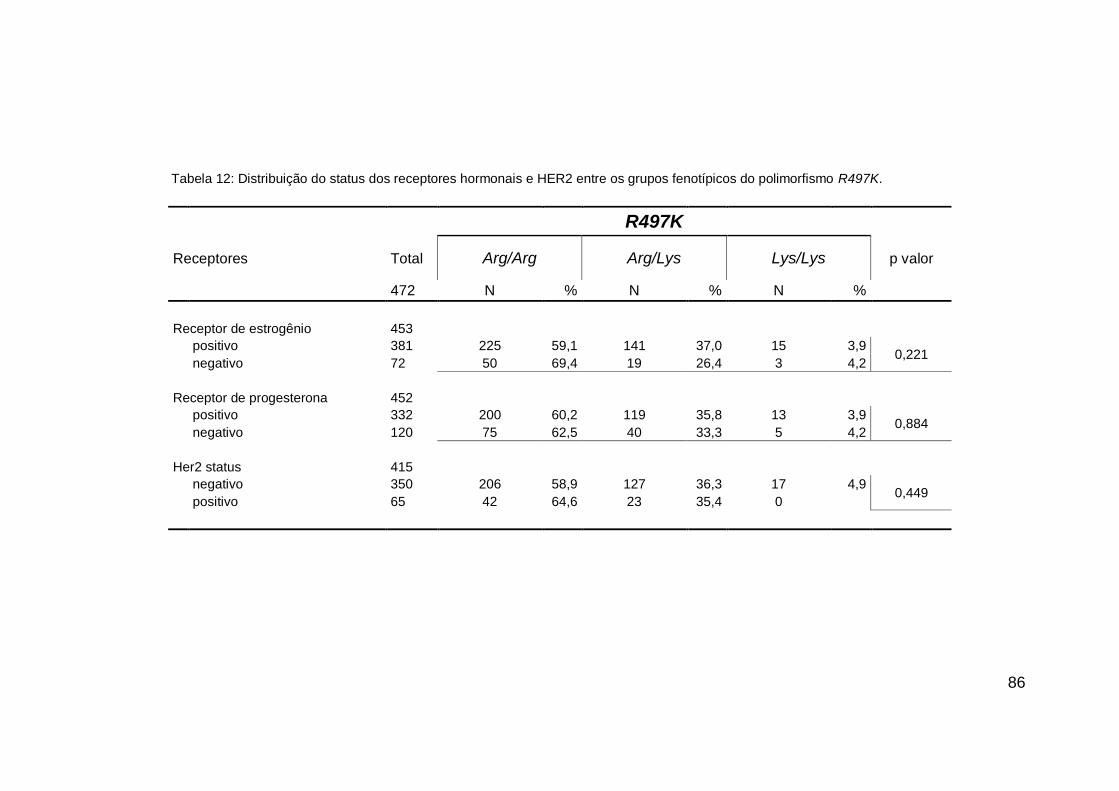
86
R497K
Receptores Total Arg/Arg Arg/Lys Lys/Lys p valor
472 N % N % N %
Receptor de estrogênio 453
positivo 381 225 59,1 141 37,0 15 3,9 0,221
negativo 72 50 69,4 19 26,4 3 4,2
Receptor de progesterona 452
positivo 332 200 60,2 119 35,8 13 3,9 0,884
negativo 120 75 62,5 40 33,3 5 4,2
Her2 status 415
negativo 350 206 58,9 127 36,3 17 4,9 0,449
positivo 65 42 64,6 23 35,4 0
Tabela 12: Distribuição do status dos receptores hormonais e HER2 entre os grupos fenotípicos do polimorfismo R497K.
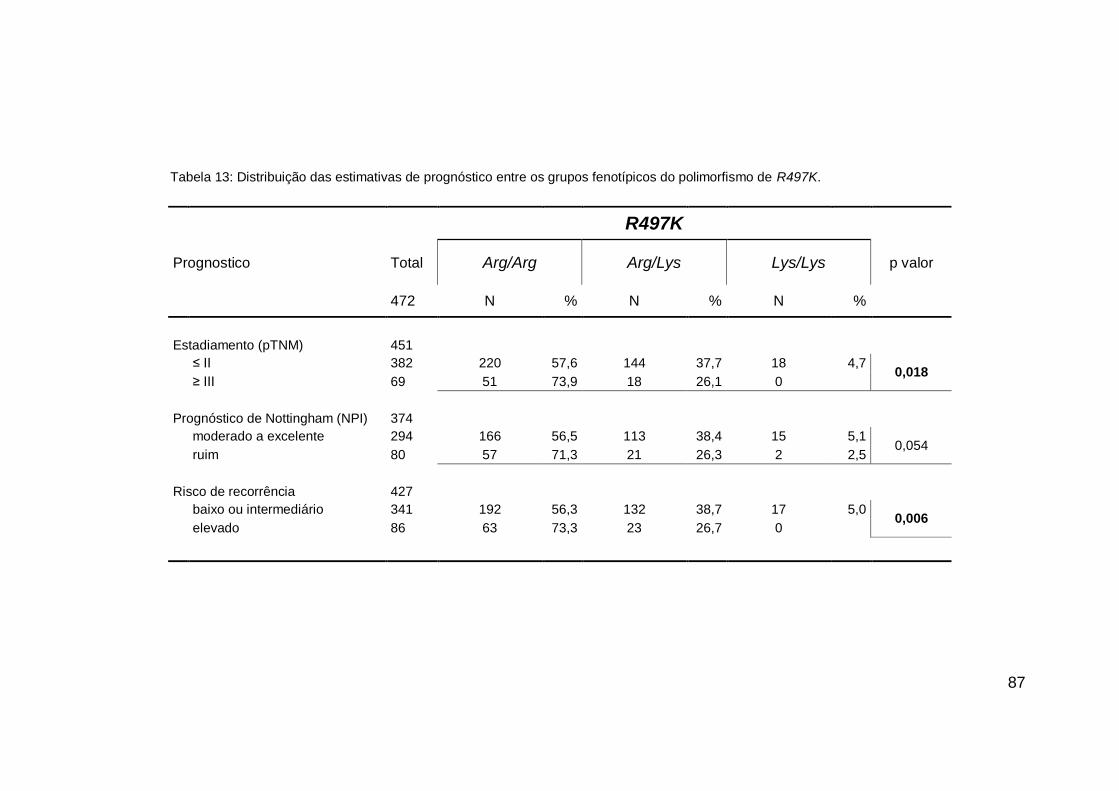
87
R497K
Prognostico Total Arg/Arg Arg/Lys Lys/Lys p valor
472 N % N % N %
Estadiamento (pTNM) 451
≤ II 382 220 57,6 144 37,7 18 4,7 0,018
≥ III 69 51 73,9 18 26,1 0
Prognóstico de Nottingham (NPI) 374
moderado a excelente 294 166 56,5 113 38,4 15 5,1 0,054
ruim 80 57 71,3 21 26,3 2 2,5
Risco de recorrência 427
baixo ou intermediário 341 192 56,3 132 38,7 17 5,0 0,006
elevado 86 63 73,3 23 26,7 0
Tabela 13: Distribuição das estimativas de prognóstico entre os grupos fenotípicos do polimorfismo de R497K.

88
A única variável independente que mostrou associação com a
distribuição dos grupos fenotípicos foi o status linfonodal. Entre as pacientes
com status linfonodal N2 ou N3 houve menor prevalência do alelo variante para
Lisina (Lys).
Na tabela 13, a distribuição dos grupos fenotípicos gerados pelo
genótipo do polimorfismo R497K é avaliada em função das variáveis de
estimativa de prognóstico. Entre as pacientes com doença em estádio ≥ III ou
consideradas com alto risco de recorrência houve menor ocorrência do alelo
variante para Lisina (Lys).
No gráfico 4, observamos a tendência da diminuição de status linfonodal
pN2/pN3 em função dos genótipos R497K (p = 0,002). A proporção de status
pN2/pN3 em relação ao status pN0/pN1 foi significativamente menor entre as
pacientes Arg/Lys quando comparadas às pacientes com o genótipo/fenótipo
de referência Arg/Arg com OR (IC 95%) = 0,468; (0,258 ~ 0,850). Entre as 18
pacientes homozigotas para o genótipo/fenótipo variante (Lys/Lys) não houve
ocorrência de status linfonodal pN2/pN3, não sendo possível determinar a
razão de chances nesse caso. Entretanto, tal ausência da categoria de pior
prognóstico pN2/pN3 entre as pacientes Lys/Lys reforça a magnitude de
associação entre o status linfonodal e o polimorfismo R497K. De acordo com
esta noção, o estadiamento e a estimativa de risco de recorrência, que são
variáveis afetadas pelo status linfonodal, também influenciaram
significativamente a distribuição dos genótipos/fenótipos R497K (tabela 14). O
prognóstico de Nottingham pareceu influenciar a distribuição dos
genótipos/fenótipos R497K, embora tal associação não tenha alcançado
significância estatística (p = 0,054).
As variáveis que demonstraram ter um valor de p menor do que 0,10
para a diferença entre as distribuições fenotípicas de R497K foram
selecionadas para avaliação das razões de chance considerando os
genótipos/fenótipos variantes de forma agrupada (Arg/Lys + Lys/Lys) para
comparação com o genótipo/fenótipo de referência (Arg/Arg). A tabela 14
mostra que as pacientes com genótipos/fenótipos variantes possuem menos
chance de ter status linfonodal pN2/pN3 do que as pacientes Arg/Arg (OR =
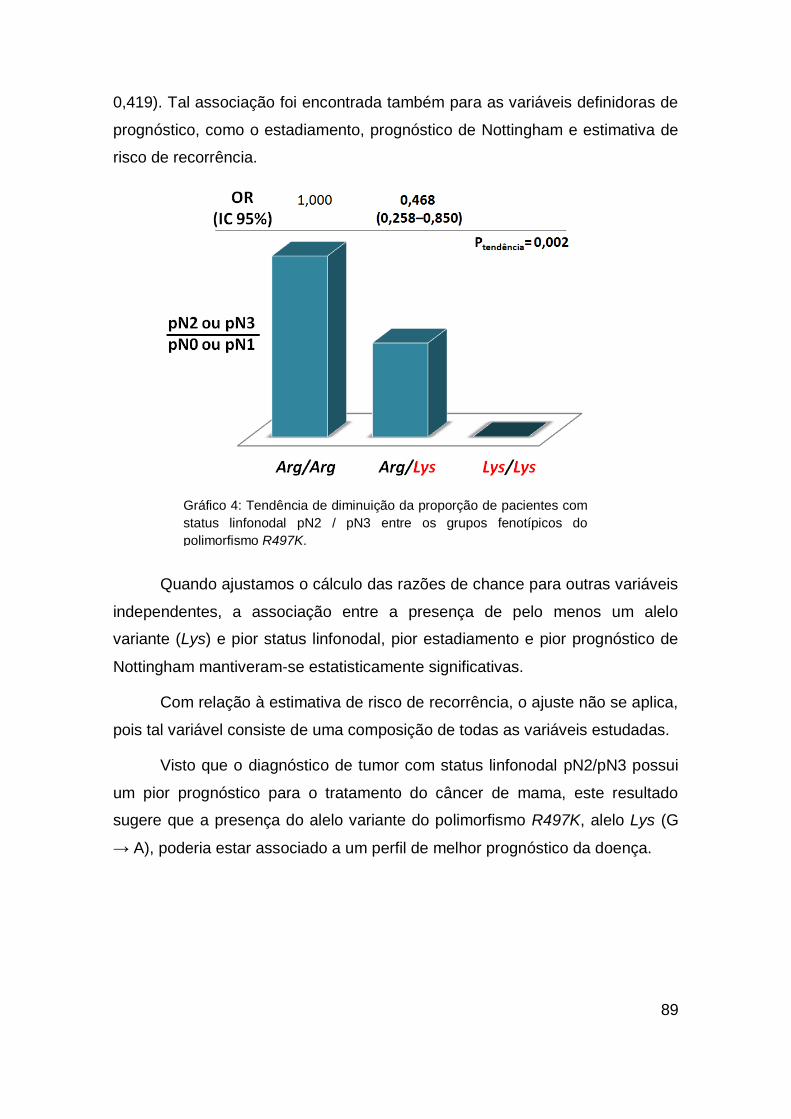
89
0,419). Tal associação foi encontrada também para as variáveis definidoras de
prognóstico, como o estadiamento, prognóstico de Nottingham e estimativa de
risco de recorrência.
Quando ajustamos o cálculo das razões de chance para outras variáveis
independentes, a associação entre a presença de pelo menos um alelo
variante (Lys) e pior status linfonodal, pior estadiamento e pior prognóstico de
Nottingham mantiveram-se estatisticamente significativas.
Com relação à estimativa de risco de recorrência, o ajuste não se aplica,
pois tal variável consiste de uma composição de todas as variáveis estudadas.
Visto que o diagnóstico de tumor com status linfonodal pN2/pN3 possui
um pior prognóstico para o tratamento do câncer de mama, este resultado
sugere que a presença do alelo variante do polimorfismo R497K, alelo Lys (G
→ A), poderia estar associado a um perfil de melhor prognóstico da doença.
Gráfico 4: Tendência de diminuição da proporção de pacientes com
status linfonodal pN2 / pN3 entre os grupos fenotípicos do
polimorfismo R497K.
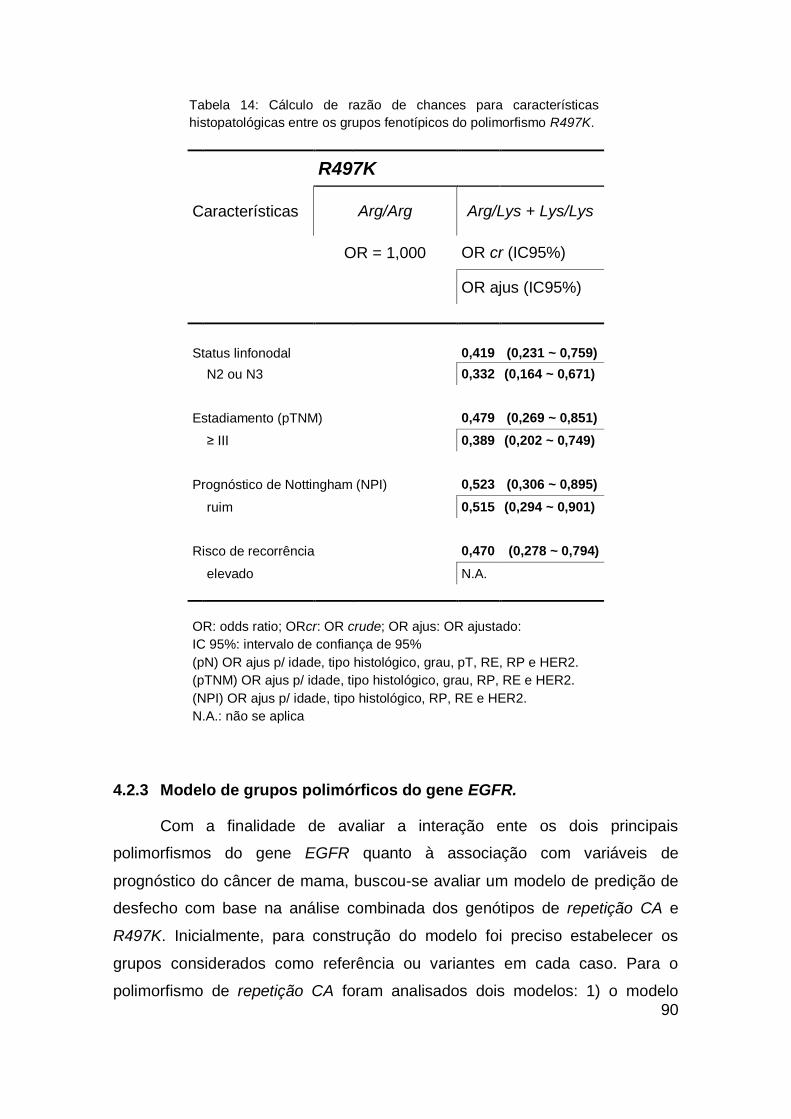
90
R497K
Características Arg/Arg Arg/Lys + Lys/Lys
OR = 1,000 OR cr (IC95%)
OR ajus (IC95%)
Status linfonodal 0,419 (0,231 ~ 0,759)
N2 ou N3 0,332 (0,164 ~ 0,671)
Estadiamento (pTNM) 0,479 (0,269 ~ 0,851)
≥ III 0,389 (0,202 ~ 0,749)
Prognóstico de Nottingham (NPI) 0,523 (0,306 ~ 0,895)
ruim 0,515 (0,294 ~ 0,901)
Risco de recorrência 0,470 (0,278 ~ 0,794)
elevado N.A.
OR: odds ratio; ORcr: OR crude; OR ajus: OR ajustado:
IC 95%: intervalo de confiança de 95%
(pN) OR ajus p/ idade, tipo histológico, grau, pT, RE, RP e HER2.
(pTNM) OR ajus p/ idade, tipo histológico, grau, RP, RE e HER2.
(NPI) OR ajus p/ idade, tipo histológico, RP, RE e HER2.
N.A.: não se aplica
4.2.3 Modelo de grupos polimórficos do gene EGFR.
Com a finalidade de avaliar a interação ente os dois principais
polimorfismos do gene EGFR quanto à associação com variáveis de
prognóstico do câncer de mama, buscou-se avaliar um modelo de predição de
desfecho com base na análise combinada dos genótipos de repetição CA e
R497K. Inicialmente, para construção do modelo foi preciso estabelecer os
grupos considerados como referência ou variantes em cada caso. Para o
polimorfismo de repetição CA foram analisados dois modelos: 1) o modelo
Tabela 14: Cálculo de razão de chances para características
histopatológicas entre os grupos fenotípicos do polimorfismo R497K.

91
baseado no tamanho de cada alelo, definidos como curto (CA ≤ 16) ou longo
(CA > 16), ou 2) o modelo baseado na soma dos dois alelos individuais,
caracterizando dois genótipos, definidos como ∑CAn < 36 ou ∑CAn ≥ 36. Para
o polimorfismo R497K, o genótipo/fenótipo predominante Arg/Arg foi
considerado como referência para o modelo, enquanto os genótipos/fenótipos
contendo Lys em hetrozigose ou homozigose foram considerados variantes.
Foram avaliados a seguir dois modelos preditivos de interação entre os
polimorfismos EGFR, EGFR16 e EGFR35, cuja diferença reside na forma de
definição dos genótipos de repetição CA, seguindo, respectivamente, o modelo
curto/longo para os alelos individuais ou o ∑CAn.
4.2.3.1 Modelo EGFR16
Neste modelo, os genótipos dos polimorfismos do gene EGFR foram
agrupados como Referência16 (Ref16) e Variante16 (Var16), conforme descrito no
quadro 7.
Grupos EGFR
R497K Repetição CA
Referência
16 Arg/Arg
curto/curto ou curto/longo
Variante
16
Arg/Lys ou Lys/Lys
longo/longo
Quadro 7: Definição dos grupos genotípicos de
referência e variante do modelo EGFR16
.

92
4.2.3.2 Modelo EGFR35
Neste modelo, os genótipos dos polimorfismos do gene EGFR foram
agrupados como Referência35 (Ref35) e Variante35 (Var35) de acordo com o
quadro 8.
Grupos EGFR35
R497K Repetição CA
Referência
35 Arg/Arg ∑CAn < 36
Variante
35
Arg/Lys ou Lys/Lys
∑CAn ≥ 36
4.2.3.3 Aplicação do modelo EGFR16
A tabela 15 mostra as variáveis clinico-patológicas que apresentaram
associação significativa com algum dos polimorfismos EGFR quando
analisados de forma independente. Aqui, o modelo EGFR16 é usado para
avaliação de associação com tais variáveis selecionadas. Com exceção do
status de RP, todas as demais variáveis afetaram significativamente a
distribuição dos grupos genotípicos do modelo EGFR16.
Analisando a variável histopatológica que apresentou diferença na
distribuição dos polimorfismos, o status linfonodal se apresentou
estatisticamente diferente, e assim, observando as razões de chance, o grupo
Var16/Var16 é o único que difere do grupo referência.
Quadro 8: Definição dos grupos genotípicos de
referência e variante do modelo EGFR35
.
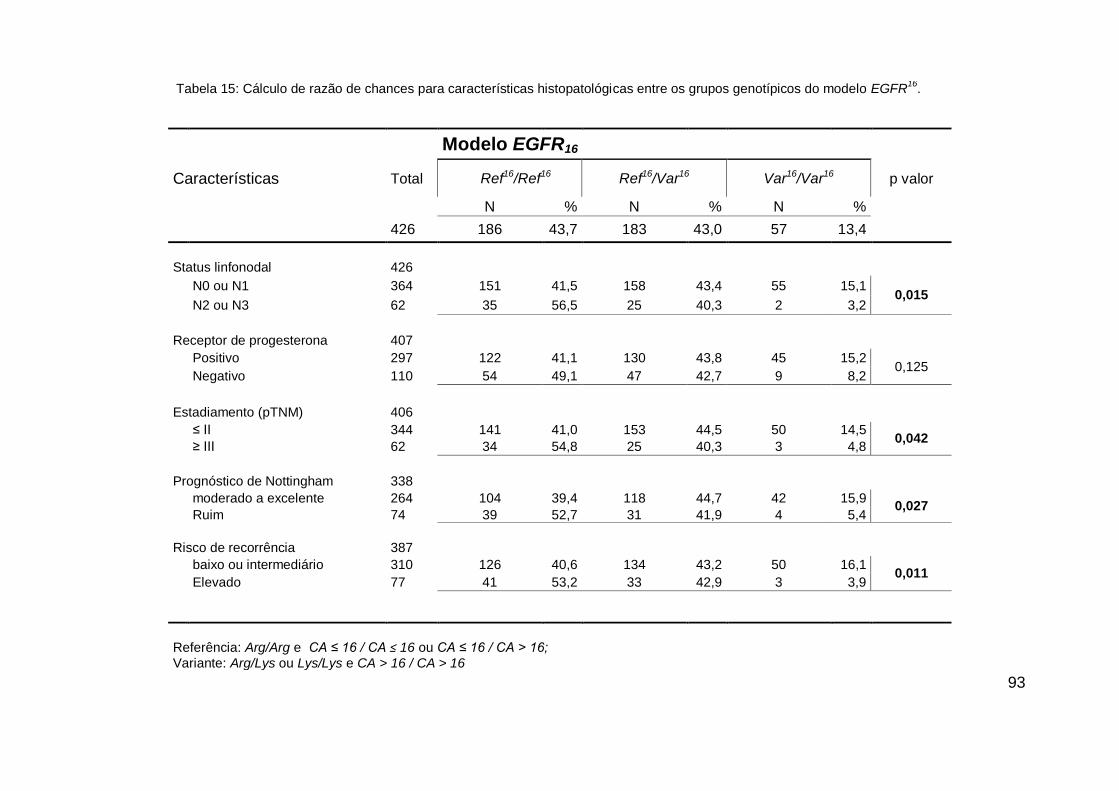
93
Modelo EGFR16
Características Total Ref16/Ref16 Ref16/Var16 Var16/Var16 p valor
N % N % N %
426 186 43,7 183 43,0 57 13,4
Status linfonodal 426
N0 ou N1 364 151 41,5 158 43,4 55 15,1 0,015
N2 ou N3 62 35 56,5 25 40,3 2 3,2
Receptor de progesterona 407
Positivo 297 122 41,1 130 43,8 45 15,2 0,125
Negativo 110 54 49,1 47 42,7 9 8,2
Estadiamento (pTNM) 406
≤ II 344 141 41,0 153 44,5 50 14,5 0,042
≥ III 62 34 54,8 25 40,3 3 4,8
Prognóstico de Nottingham 338
moderado a excelente 264 104 39,4 118 44,7 42 15,9 0,027
Ruim 74 39 52,7 31 41,9 4 5,4
Risco de recorrência 387
baixo ou intermediário 310 126 40,6 134 43,2 50 16,1 0,011
Elevado 77 41 53,2 33 42,9 3 3,9
Referência: Arg/Arg e CA ≤ 16 / CA ≤ 16 ou CA ≤ 16 / CA > 16;
Variante: Arg/Lys ou Lys/Lys e CA > 16 / CA > 16
Tabela 15: Cálculo de razão de chances para características histopatológicas entre os grupos genotípicos do modelo EGFR16
.

94
Na tabela 16, observamos a tendência da diminuição de status linfonodal
pN2/pN3 em função dos genótipos EGFR16 (p = 0,005). A proporção de status
pN2/pN3 em relação ao status pN0/pN1 foi significativamente menor entre as
pacientes Var16/Var16 quando comparadas às pacientes Ref16/Ref16 com OR (IC
95%) = 0,156; (0,036 ~ 0,674).
A tabela 17 mostra o cálculo das razões de chance para o genótipo
EGFR16 Var/Var, que reúne os dois polimorfismos, em comparação com os
demais genótipos, aqui considerados em conjunto.
Os resultados indicam que o genótipo EGFR16 Var/Var está associado a
uma menor ocorrência de mais de 3 metástases linfonodais e que está
associado a índices de melhor prognóstico de Nottingham bem como ao menor
risco de recorrência. Tais associações se mantêm após o ajuste para outras
variáveis independentes.
Tabela 16: Tendência de diminuição da proporção de pacientes com status linfonodal
pN2 / pN3 entre os grupos genotípicos do modelo EGFR16
.
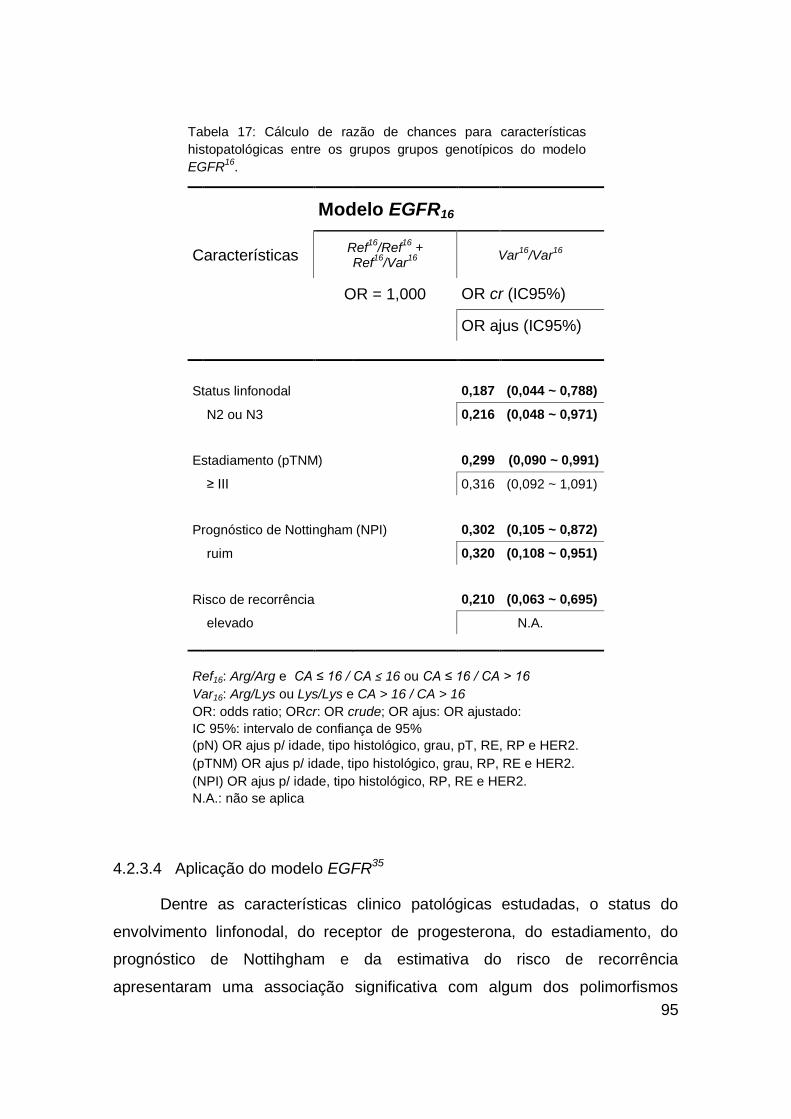
95
Modelo EGFR16
Características Ref
16/Ref
16 +
Ref16
/Var16
Var
16/Var
16
OR = 1,000 OR cr (IC95%)
OR ajus (IC95%)
Status linfonodal 0,187 (0,044 ~ 0,788)
N2 ou N3 0,216 (0,048 ~ 0,971)
Estadiamento (pTNM) 0,299 (0,090 ~ 0,991)
≥ III 0,316 (0,092 ~ 1,091)
Prognóstico de Nottingham (NPI) 0,302 (0,105 ~ 0,872)
ruim 0,320 (0,108 ~ 0,951)
Risco de recorrência 0,210 (0,063 ~ 0,695)
elevado N.A.
Ref16: Arg/Arg e CA ≤ 16 / CA ≤ 16 ou CA ≤ 16 / CA > 16
Var16: Arg/Lys ou Lys/Lys e CA > 16 / CA > 16
OR: odds ratio; ORcr: OR crude; OR ajus: OR ajustado:
IC 95%: intervalo de confiança de 95%
(pN) OR ajus p/ idade, tipo histológico, grau, pT, RE, RP e HER2.
(pTNM) OR ajus p/ idade, tipo histológico, grau, RP, RE e HER2.
(NPI) OR ajus p/ idade, tipo histológico, RP, RE e HER2.
N.A.: não se aplica
4.2.3.4 Aplicação do modelo EGFR35
Dentre as características clinico patológicas estudadas, o status do
envolvimento linfonodal, do receptor de progesterona, do estadiamento, do
prognóstico de Nottihgham e da estimativa do risco de recorrência
apresentaram uma associação significativa com algum dos polimorfismos
Tabela 17: Cálculo de razão de chances para características
histopatológicas entre os grupos grupos genotípicos do modelo
EGFR16
.

96
estudados. A distribuição destas características entre os grupos genotípicos do
modelo EGFR35 foram comparadas. Na tabela 18, observamos que o status do
envolvimento linfonodal, o RP, o prognóstico de Nottihgham e a estimativa do
risco de recorrência mostraram uma distribuição estatisticamente diferente
entre os grupos do modelo EGFR35. A incidência de pacientes com status RP
negativo pareceu diminuir de acordo com a ocorrência do grupo de genótipos
variante35.
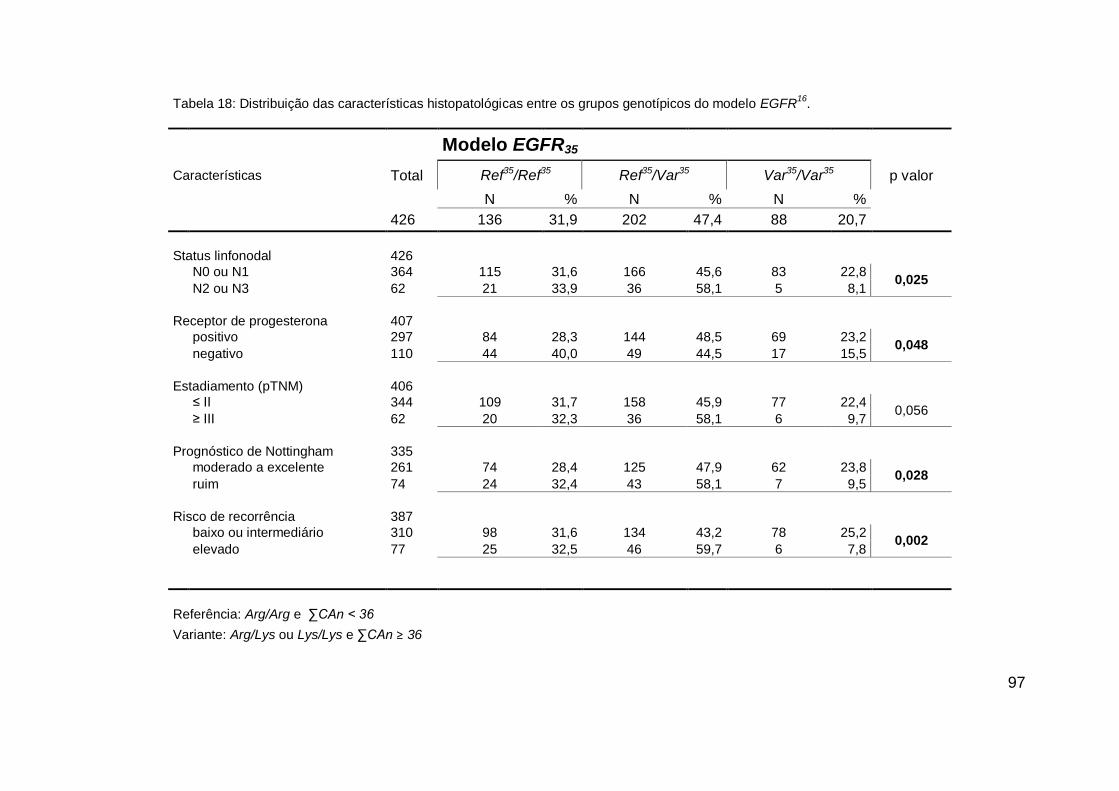
97
Modelo EGFR35
Características Total Ref35/Ref35 Ref35/Var35 Var35/Var35 p valor
N % N % N %
426 136 31,9 202 47,4 88 20,7
Status linfonodal 426
N0 ou N1 364 115 31,6 166 45,6 83 22,8 0,025
N2 ou N3 62 21 33,9 36 58,1 5 8,1
Receptor de progesterona 407
positivo 297 84 28,3 144 48,5 69 23,2 0,048
negativo 110 44 40,0 49 44,5 17 15,5
Estadiamento (pTNM) 406
≤ II 344 109 31,7 158 45,9 77 22,4 0,056
≥ III 62 20 32,3 36 58,1 6 9,7
Prognóstico de Nottingham 335
moderado a excelente 261 74 28,4 125 47,9 62 23,8 0,028
ruim 74 24 32,4 43 58,1 7 9,5
Risco de recorrência 387
baixo ou intermediário 310 98 31,6 134 43,2 78 25,2 0,002
elevado 77 25 32,5 46 59,7 6 7,8
Referência: Arg/Arg e ∑CAn < 36
Variante: Arg/Lys ou Lys/Lys e ∑CAn ≥ 36
Tabela 18: Distribuição das características histopatológicas entre os grupos genotípicos do modelo EGFR16
.

98
Na tabela 19, observamos que não houve tendência de diminuição da
ocorrência de pacientes com status linfonodal pN2 / pN3 dentre os grupos do
modelo EGFR35 (p = 0,084). Porém, as razões de chance envolvendo a
incidência de pacientes com status RP negativo foram estatisticamente
diferentes entre o grupo Ref35/Var35 e Var35/Var35.
Também na tabela 19, observamos que houve uma tendência de
diminuição da ocorrência de pacientes com status RP negativo dentre os
grupos do modelo EGFR35 (p = 0,015). Porém, as razões de chance
envolvendo a incidência de pacientes com status RP negativo não parecem
diferir entre os grupos.
Dentre as características estudadas na tabela 18, o status do
envolvimento linfonodal, do RP, do estadiamento, do prognóstico de
Nottingham e da estimativa de risco de recorrência foram as a variáveis que
demonstraram ter um valor de p menor que 0,10 para a diferença entre as
distribuições dos grupos deste modelo. Para cálculo de odds ratio, os grupos
foram dicotomizados em pacientes Ref35/Ref35 e pacientes Ref35/Var35 versus
pacientes Var35/Var35. A tabela 20 mostra que as pacientes com genótipo
Var35/Var35 têm por volta de 70% a menos de chance de ter status linfonodal
pN2 / pN3 do que pacientes Ref35/Ref35 ou Ref35/Var35, com um OR (IC95%) =
0,297 (0,115 ~ 0,765).
Tabela 19: Tendência de diminuição da proporção de pacientes com status linfonodal pN2
/ pN3 entre os grupos genotípicos do modelo EGFR35
.
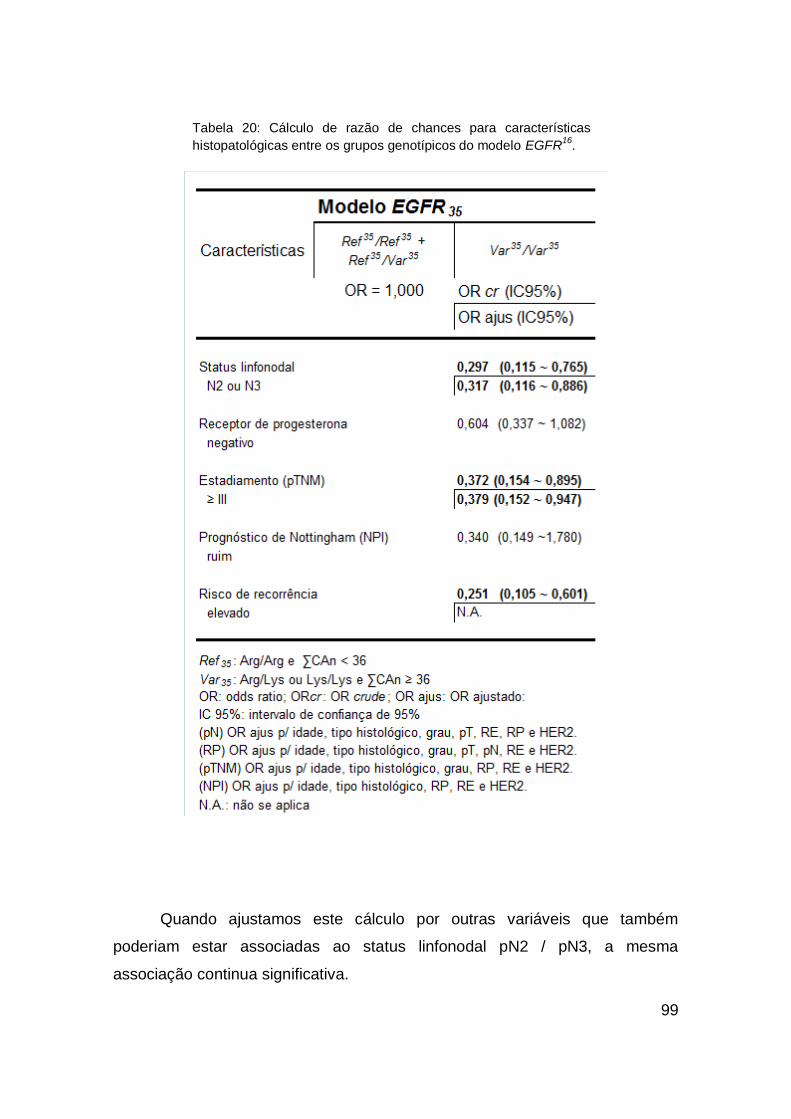
99
Quando ajustamos este cálculo por outras variáveis que também
poderiam estar associadas ao status linfonodal pN2 / pN3, a mesma
associação continua significativa.
Tabela 20: Cálculo de razão de chances para características
histopatológicas entre os grupos genotípicos do modelo EGFR16
.

100
Ainda observando a mesma tabela, verificamos que das variáveis
definidoras de prognóstico, apenas o estadiamento apresentou associação
entre as pacientes do grupo Var35/Var35 e perfis de melhor prognóstico. Quando
o ajuste do cálculo de odds ratio foi realizado, esta associação com o
estadiamento permaneceu estatisticamente significativa. Finalmente,
observamos que as pacientes com genótipo Var35/Var35 têm por volta de 75% a
menos de chance de ter um risco elevado de recorrência do que as pacientes
Ref35/Ref35 ou Ref35/Var35, com um OR (IC95%) = 0,251 (0,105 ~ 0,601).

101
5 DISCUSSÃO
O câncer de mama é uma doença heterogênea e de fisiologia complexa
e ainda não totalmente compreendida. A biologia do processo de
carcinogênese se apresenta de maneiras variadas em pacientes com a mesma
doença. Durante o processo de diferenciação e proliferação celular, diversas
moléculas efetoras e receptoras realizam cruciais processos de transmissão da
informação pelos tecidos e todos os genes que codificam estas proteínas
possuem versões polimórficas cuja frequência e distribuição muitas vezes são
distintas entre as populações (Tan 2008). Assim, estas variações genéticas têm
se mostrado possíveis biomarcadores de prognóstico do câncer (Azzato et al.
2010).
Dentre os genes notadamente relacionados com a carcinogênese (os
oncogenes), está o gene EGFR (Miller et al. 1994; Wykosky et al. 2011). O
gene EGFR possui variações polimórficas cujas distribuições são
desconhecidas na população brasileira. O perfil genético da população
brasileira, por ter sofrido alguns processos de miscigenação em sua história,
apresenta uma variabilidade que não permite sua identificação de
ancestralidade pela cor da pele (Pimenta et al. 2006; Suarez-Kurtz 2010).
Neste estudo, pudemos avaliar as distribuições genotípicas de dois
polimorfismos numa população de mulheres com câncer de mama. Analisando
a distribuição do polimorfismo de repetição CA, os alelos com 16 repetições CA
apresentaram uma frequência de 0,425 (IC95% = 0,398 ~ 0,458) entre as
pacientes. Este achado possui semelhança com os resultados publicados
sobre este polimorfismo em população norte-americana e de outros continentes
(Liu et al. 2003) (Gráfico 5). Estudos em populações orientais mostram maior
frequência de alelos longos, de 20 repetições CA do que entre os ocidentais
(Buerger et al. 2004; Nomura et al. 2007)
O polimorfismo R497K apresentou frequência do alelo variante Lys de
0,218 (IC95% = 0,192 - 0,245), resultado que não difere das frequências
relatadas para pacientes europeus e norte-americanos (Gonçalves et al. 2008;
McKibbin et al. 2010). Populações orientais apresentam aparentemente maior

102
frequência do alelo Lys do que populações ocidentais (Sasaki et al. 2009;
Huang et al. 2009).
Os polimorfismos do gene EGFR aqui estudados já foram investigados
em alguns trabalhos (Agelopoulos et al. 2008; Zhou et al. 2009). No caso do
polimorfismo de repetição CA, estudos com pacientes com câncer de mama
não possuem tamanho amostral amplo e não definem com clareza uma
associação causal entre os tamanhos das repetições dos dinucleotídeos e a
atividade do gene (Jami et al. 2008; Lin et al. 2011). Alguns autores que
publicaram resultados de ensaios in vitro apresentam a hipótese de que os
alelos maiores possuem menor atividade transcricional e assim, apresentam
quantidade de transcrito inferior às células com alelos menores (Liu et al.
2007).
Diante dos dados publicados compilados no quadro 2, e a diversidades
alélica encontrada para este polimorfismo, o agrupamento genotípico para este
polimorfismo foi testado de duas formas. Uma, levando em conta o número de
repetições em cada alelo por homozigose ou heterozigose. Os alelos curtos,
formados pelo alelo mais frequente (16) e os outros menores; e os alelos
longos formados pelos alelos maiores que 16 repetições CA, onde o alelo 20
CA, foi o mais frequente. A outra, levando em conta a soma do número de
Gráfico 5: Distribuição dos genótipos 16/16, 16/20 e 20/20 do polimorfismo de repetição CA em diferentes etnias. A) Frequência dos genótipos no presente estudo, representando a população brasileira. B) Estudo de avaliação da distribuição deste polimorfismo em caucasianos, afro-americanos, chineses e asiáticos não-chineses publicado por Liu et al.
(2003).

103
repetições dos dois alelos da paciente estudada. As somas de 36 e logo em
seguida de 32 repetições CA foram as mais frequentes. Assim, por este
modelo, as pacientes com somas maiores do que 35 repetições CA foram
agrupadas. Quando analisamos a associação deste polimorfismo com as
características histopatológicas, verificamos que as pacientes com alelos
maiores, ou seja, com maior número de repetições CA, tiveram menor chance
de ter status RP-, tanto quando analisamos pelo modelo de alelos curtos e
longos, tanto pelo modelo das somas.
Não é comum associar o status RP- com algum fenótipo específico no
câncer de mama quando analisado isoladamente, embora a ausência de super
expressão de RP no câncer de mama esteja associada à progressão da
doença, sobretudo em mulheres pós-menopausa (Balleine et al. 1999).
Entender a atividade do RP no contexto da carcinogênese e do tratamento
adjuvante também é um desafio (Lange et al. 1998). Um mecanismo de
crosstalk (intercomunicação) entre as vias de cascata sinalizadora dos
receptores esteróides e dos receptores de fatores de crescimento tem sido
descrito em estudos moleculares (Fox et al. 2010; Hoefnagel et al. 2012).
Existem algumas evidências sugerindo que este crosstalk entre receptores de
estrogênio e receptores da família erbB aciona cascatas de sinalização que
podem levar a uma regulação negativa dos níveis de transcrição de receptores
de progesterona (Massarweh et al. 2008). A regulação transcricional regida
pela via PIK3-Akt-mTOR, disparada pela atividade sinérgica do EGFR e do ER
é descrita como um mecanismo de diminuição dos níveis de RP (Thakkar &
Mehta 2011).
No contexto do presente estudo, partindo da hipótese de que a atividade
transcricional do gene EGFR é inversamente proporcional ao número de
repetições CA, tumores com alelos mais longos estariam associados a menor
atividade do EGFR e, portanto, esta via de regulação negativa do RP não
estaria tão ativa, contribuindo para menor chance de ter status RP-. Este
raciocínio poderia justificar o resultado encontrado de que as pacientes
longo/longo apresentaram menor ocorrência de tumores com status RP- do que
pacientes curto/curto (OR= 0,447; IC95% = 0,238 - 0,836). Assumindo-se que

104
tumores RP- apresentam pior prognóstico, pacientes com alelos maiores
(longo/longo ou ∑CAn > 35) também teriam esta estimativa.
Estudos com outros tipos de câncer parecem corroborar a noção de que
alelos maiores (longo/longo ou ∑CAn > 35) estão associados a melhores
desfechos. Num estudo em câncer de pulmão, Dubey e colaboradores (2006)
demonstraram que pacientes com alelos mais longos (∑CAn > 35), tiveram
melhor sobrevida (p = 0,03) (Dubey et al. 2006). Em outro estudo em câncer de
esôfago, pacientes com alelos mais longos (homozigotos de alelos ≥ 20
repetições CA) tiveram melhor sobrevida (p = 0,045) (Lee et al. 2011). Porém,
no câncer de mama, o status de RP, quando analisado isoladamente, parece
contribuir pouco para o prognóstico.
O status do RP e, principalmente de RE, além de contribuírem para
estimativa dos prognósticos de sobrevida e de risco de recorrência, são
determinantes para predição de resposta à terapia hormonal no câncer de
mama (Massarweh et al. 2008; Harrell et al. 2006). Dentre as pacientes RE+, o
status de RP parece ter também valor preditivo (Bardou et al. 2003). O status
do EGFR também tem sido pesquisado como marcador de resposta à terapia
hormonal e sua maior atividade é descrita como via de resistência (Arpino et al.
2005). É possível que a presença dos polimorfismos EGFR possa influenciar o
status do EGFR e, indiretamente, a resposta terapêutica à terapia hormonal
adjuvante. O presente trabalho não avaliou a associação entre os
polimorfismos EGFR e o padrão de expressão ou o status de atividade do
EGFR.
Analisando os resultados de associação entre o polimorfismo R497K e
variáveis histopatológicas, identificamos que a ocorrência do alelo variante Lys
está associada a menor invasão linfonodal, num modelo de co-dominância, ou
seja, a tendência de se encontrar pacientes com status pN2 ou pN3 parece
diminuir progressivamente em função da ocorrência do alelo variante em
heterozigose ou homozigose (Ptendência = 0,002). Estes dados corroboram os
resultados encontrados no único estudo publicado envolvendo câncer de mama
e o polimorfismo EGFR R497K, segundo o qual pacientes com alelo variante

105
Lys apresentaram menor prevalência de presença de linfonodos positivos (p =
0,030) (Kallel et al. 2009).
Quando pesquisamos sobre os mecanismos que podem estar
associados ao processo de invasão linfonodal, o EGFR parece desempenhar
um fator crucial nas vias de diferenciação celular de formação de um fenótipo
neoplásico mais invasivo e aderente, não só no câncer de mama (Jones et al.
1996; Oonk et al. 2007; Sarkis et al. 2010). Alguns trabalhos em modelos
experimentais sugerem que a via de ativação do EGFR está em crosstalk com
vias de ativação de moléculas como integrinas, metaloproteinases e E-
caderina, responsáveis pela característica celular de invasão tecidual e,
portanto, formação de metástases (Ricono et al. 2010; Georgopoulos et al.
2010; Hudson & Stack 2010).
As quimiocinas e seus receptores também parecem desempenhar papel
crucial no mecanismo de linfangiogênese. Assim, a atividade destas moléculas
nos tumores de mama parece estar associada à ocorrência de metástases
linfonodais (Müller et al. 2001; Cabioglu et al. 2007). Os complexos
mecanismos de sinalização celular relacionados com esta associação não
estão por completo elucidados. Porém, Liu et al. (2010) demonstraram que a
expressão de receptores de quimiocinas como CXCR4 e CCR7 está
estatisticamente associada ao status EGFR positivo em tumores invasivos de
mama (p < 0,01), levando a maior ocorrência de metástases linfonodais nestes
tumores do que naqueles que apresentaram uma baixa expressão destes
receptores (p < 0,001). O status linfonodal é um fator fundamental na definição
de estimativas de prognóstico por notoriamente influenciar o desfecho final da
doença (Chagpar et al. 2011).
Quando analisamos o EGFR sob o ponto de vista da interação com os
seus ligantes, considerando a conformação espacial de seus domínios e o
processo de dimerização, muitas variáveis podem contribuir para o nível de sua
atividade (Shepard et al. 2008). Estudos cristalográficos e de modelagem
molecular buscam entender quais mudanças estruturais podem contribuir para
uma menor afinidade do sítio catalítico com os ligantes (Samaga et al. 2009;
Alvarado et al. 2010).

106
A associação entre o alelo Arg e o status linfonodal positivo parece
sugerir que a forma selvagem do polimorfismo R497K contribua para maior
atividade do EGFR, favorecendo maior capacidade proliferativa e maior poder
invasivo no câncer de mama.
A consequência da troca de um aminoácido Arg por um Lys na atividade
do receptor foi inicialmente descrita nos estudos de Moriai et al. (1994), que
mostraram menor ativação dos fatores de transcrição c-myc, fos e jun em
associação à estrutura da versão variante, possivelmente por atenuada
interação com o ligante e reduzida capacidade de dimerização.
Os resultados encontrados no presente estudo se mostram de acordo
com os achados de Moriai et al. (1994) e de.Brandt et al. 2006, que, de uma
forma geral, propõem que as formas variantes dos polimorfismos EGFR estão
associadas a características de prognóstico favorável para melhor desfecho do
tratamento do câncer de mama. Entretanto, tais estudos prévios têm limitações
em relação ao tamanho amostral e não analisaram os dois polimorfismos em
conjunto. Dessa forma, no presente trabalho, avaliamos modelos de
agrupamento dos genótipos predominantes (considerados de referência) e dos
genótipos variantes para uma análise conjunta do impacto dos dois principais
polimorfismos EGFR sobre fatores prognósticos do câncer de mama.
No modelo EGFR16, a tendência de melhor desfecho de acordo com a
ocorrência das variantes polimórficas se mantém, exceto para a associação
com o status do RP. A associação entre as formas variantes e os parâmetros
de estadiamento ou as estimativas de prognóstico apresentaram maior
magnitude e maior significância estatística do que quando estudadas
separadamente para cada polimorfismo. Este aumento de magnitude na
associação parece traduzir um efeito sinérgico entre os dois plimorfismos, de
tal forma que o grupo variante na análise combinada apresenta chance de alto
risco de recorrência cerca de 80% menor do que o grupo de referência. O
modelo EGFR35 seguiu o mesmo perfil, porém os valores associativos se
apresentaram ligeiramente menores. Estes resultados parecem corroborar os
dados publicados por Zhang et al. (2005), que mostraram que pacientes com

107
genótipo Arg/Arg e dois alelos < 20 repetições CA tiveram pior sobrevida livre
de recorrência do que pacientes com genótipos variantes.
Analisando os estudos a seguir, observamos que a terapia anti-EGFR
em pacientes de câncer de pulmão e colon mostraram potencial influência dos
polimorfismos EGFR. Ma et al. (2009) mostraram que o gefitinibe, um inibidor
tirosina-cinase indicado para o tratamento do câncer de pulmão de células não
pequenas foi mais eficaz em pacientes com pelo menos um alelo ≤16
repetições CA (p < 0,001). De forma semelhante, Sasaki et al. (2009)
mostraram que a presença do alelo Lys em pacientes com câncer de pulmão e
linfonodo positivo resultou em melhor sobrevida em resposta ao tratamento
com gefitinibe (p = 0,004) Pacientes com genótipo selvagem parecem se
beneficiar mais desta terapia do que os pacientes com genótipo variante
(Sasaki et al. 2009). Em câncer de colon metastático, Wang et al. (2007) e
Gonçalves et al. (2008) descreveram que pacientes Lys parecem exibir melhor
sobrevida livre de doença do que os pacientes Arg quando tratados com
cetuximabe, um anticorpo monoclonal anti-EGFR (p < 0,001 e p = 0,042,
respectivamente). Press et al. (2008) e Park et al. (2011) não encontraram
associações entre a resposta ao tratamento com cetuximabe em pacientes com
câncer de colon metastático e o número de repetições CA do gene EGFR. De
todo modo, a investigação sobre a resposta aos antineoplásicos citostáticos
clássicos e a terapia anti-EGFR no câncer de mama merece ser avaliada de
acordo com o perfil genotípico destes polimorfismos.
Com os resultados obtidos neste estudo, é possível perceber que
variações na atividade do EGFR interferem com fatores prognósticos do câncer
de mama. Algumas associações com relação ao impacto deste polimorfismo na
ação de fármacos antineoplásicos foram frutos de testes em modelos in vitro
porém ainda sem definição clara sobre as associações e seus impactos nas
condutas clínicas (Puyo et al. 2008).
Alterações no gene EGFR e nos genes que codificam proteínas de suas
cascatas de sinalização podem notoriamente interferir no prognóstico de alguns
tipos de câncer, como a mutação de do gene KRAS em pulmão e colon
influenciando significativamente na resposta a terapia anti-EGFR (Park et al.

108
2011; Milano et al. 2008). Formas mutantes do receptor são frequentemente
encontradas em células neoplásicas de alguns tumores (Nomura et al. 2007). A
forma mutante mais comum é o EGFRVIII, resultado da deleção dos exons 2 a
7, que pode estar expresso em gliobastoma multiforme, câncer de pulmão,
próstata, ovário e mama (Gupta et al. 2010).
Mutações em K-Ras e em EGFR não parecem ocorrer frequentemente
no câncer de mama nem ter associação com prognóstico (Sánchez-Muñoz et
al. 2010; Nieto et al. 2007; Dahan et al. 2011). Diferentemente das mutações,
os polimorfismos são variações que ocorrem naturalmente na espécie humana.
No contexto do câncer de mama, os polimorfismos do gene EGFR podem ter
algum valor prognóstico e devem ser avaliados como potenciais biomarcadores
na construção de modelos preditivos de resposta do câncer de mama.
Outras inúmeras variáveis genéticas e epigenéticas podem também
interferir na expressão e na atividade do EGFR (Franco-Hernandez et al. 2007;
Jovanovic et al. 2010), bem como na atividade de inúmeras vias de sinalização
do processo de carcinogênese, interferindo na indicação e na resposta ao
tratamento antineoplásico do câncer de mama (Bouchalova et al. 2010;
Thompson et al. 2011). A elaboração de modelos que reúnam fatores clinico-
patológicos, genéticos, hábitos de vida e o histórico familiar da paciente têm
sido elaborados por inúmeros pesquisadores e agências científicas no mundo
todo (Relling et al. 2010; Parisi et al. 2010).
O presente trabalho faz parte de um projeto chamado “Polimorfismos
genéticos e evolução clínica, resposta terapêutica e reações adversas em
pacientes submetidas ao tratamento do câncer de mama”, que envolve o
acompanhamento de pacientes diagnosticadas com câncer de mama no
Hospital do Câncer III / INCA. Dentre os objetivos deste projeto, está a
construção de um modelo preditor de sobrevida e recorrência de metástases
no tratamento do câncer de mama. A análise da contribuição das variações
alélicas normais do genoma humano nos desfechos do tratamento é parte da
estratégia para construção deste modelo.
As associações encontradas neste estudo, devem ser melhor
investigadas, pela quantificação de RNAm do gene EGFR em tumores destas

109
pacientes e avaliação da expressão do EGFR nestes tumores por ensaios de
imunohistoquímica (Suo et al. 2002). Porém, os resultados encontrados aqui já
sugerem que estas variações genéticas podem ser biomarcadores de
prognóstico do câncer de mama, e quando aplicados juntos a outras
plataformas de perfil de assinatura gênica tumoral poderiam acrescentar poder
de predição e de determinação de condutas terapêuticas mais eficazes, com o
objetivo de gerar melhor qualidade de vida às pacientes e menores custos em
políticas de saúde (Brandt et al. 2006; Spano et al. 2008;; Williams et al. 2006).

110
6 CONCLUSÕES
Os polimorfismos do gene EGFR selecionados para o presente estudo
apresentam freqüências alélicas em brasileiros semelhantes às relatadas para
europeus e americanos, incluindo dados de afro-americanos.
Os polimorfismos do gene EGFR apresentam associação com fatores
prognósticos do câncer de mama, particularmente com a presença de
metástases linfonodais e parecem ter uma interação sinérgica nesta
associação.
A associação entre os polimorfismos do gene EGFR e a estimativas de
prognóstico se mantiveram mesmo quando ajustadas para outras variáveis
independentes, sugerindo que a combinação dos grupos genotípicos
identificados como variantes possa contribuir como um biomarcador na
construção de modelos preditivos de evolução do câncer de mama.
Com base nos dados de associação entre os polimorfismos estudados e
fatores prognósticos do câncer de mama, e considerando o papel funcional
estabelecido para este receptor em processos tumorais, propomos que os
grupos genotípicos EGFR sejam avaliados como biomarcadores de resposta
terapêutica aos antineoplásicos citostáticos clássicos e a terapias anti-EGFR
no câncer de mama.

111
7 BIBLIOGRAFIA
Aaronson, S. A. (1991) Growth Factors and Cancer. Science. n. 254, p. 1146-1152.
Abreu, E. D., Koifman, S. (2002) Fatores prognósticos no câncer da mama feminina, Revista Brasileira de Cancerologia; 48(1).
Adjuvant! Online – Decision making tools for health care professionals [Online]. Acessado em 20 de setembro de 2011. Disponivel em: http://www.adjuvantonline.com
Adly, S., Hewedi, I. H., & Mokhtar, N. M. (2010). Clinicopathologic significance of molecular classification of breast cancer: relation to nottingham prognosis index. Journal of the Egyptian National Cancer Institute, 22(4), 209-15.
Aebi S. Gelber S, Castiglione-Gertsch M, et al. (2000) Is chemotherapy alone adequate therapy for young women with oestrogen-receptor positive breast cancer? Lancet 355: 1869-74.
Agelopoulos, Konstantin, Kersting, C., et al. (2007). Egfr amplification specific gene expression in phyllodes tumours of the breast. Cellular oncology : the official journal of the International Society for Cellular Oncology, 29(6), 443-51.
Agelopoulos, K, Buerger, H., & Brandt, B. (2008). Allelic imbalances of the egfr gene as key events in breast cancer progression--the concept of committed progenitor cells. Current cancer drug targets, 8(5), 431-45.
Alberts, B. Johnson, B., Walter, P., et al. Molecular Biology of the Cell. 4ed. Nova Iorque: Garland Sciences, 2004.
Allred, D. C. (2010). Issues and updates: evaluating estrogen receptor-alpha, progesterone receptor, and HER2 in breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc, 23 Suppl 2(S2), S52-9.
Alvarado, D., Klein, D. E., & Lemmon, M. A. (2010). Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell, 142(4), 568-79.
Amador, M. L., Oppenheimer, D., Perea, S., Maitra, A., Cusatis, G., Cusati, G., Iacobuzio-Donahue, C., et al. (2004). An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer research, 64(24), 9139-43.
Anders, C. K., Hsu, D. S., Broadwater, G, et al. (2008). Young age at diagnosis correlates with worse prognosis and defines a subset of breast

112
cancers with shared patterns of gene expression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 26(20), 3324-30.
Andre, F., Berrada, N., & Desmedt, C. (2010). Implication of tumor microenvironment in the resistance to chemotherapy in breast cancer patients. Current opinion in oncology, 22(6), 547-51.
Arpino, G., Weiss, H., Lee, A. V., et al (2005). Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. Journal of the National Cancer Institute, 97(17), 1254-61
Arriagada, R., Le, M. G., Dunant, A., et al. (2006). Twenty-five years of follow-up in patients with operable breast carcinoma: correlation between clinicopathologic factors and the risk of death in each 5-year period. Cancer, 106(4), 743-50.
ASCO/CAP (2007) Guideline recommendations for HER2 Testing in Breast Cancer. Arch Pathol Lab Med. 131:18-43.
Avraham, R., & Yarden, Y. (2011). Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nature reviews. Molecular cell biology, 12(2), 104-17.
Azzato, E. M., Pharoah, P. D. P., Harrington, P., et al. (2010). A genome-wide association study of prognosis in breast cancer. Cancer epidemiology, biomarkers & prevention, 19(4), 1140-3.
Balleine, R. L., Earl, M. J., Greenberg, M. L., & Clarke, C. L. (1999). Absence of progesterone receptor associated with secondary breast cancer in postmenopausal women. British journal of cancer, 79(9-10), 1564-71.
Bandrés, E., Barricarte, R., Cantero, C., et al. (2007). Epidermal growth factor receptor (EGFR) polymorphisms and survival in head and neck cancer patients. Oral oncology, 43(7), 713-9.
Bardou, V.-J., Arpino, G., Elledge, R. M., et al. (2003). Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. Journal of clinical oncology 21(10), 1973-9.
Barnadas, A. (2010). Neoadjuvant treatment in young women with breast cancer. Breast cancer research and treatment, 123 Suppl , 35-8.
Beasley, G. M., & Olson, J. a. (2010). What’s New in Neoadjuvant Therapy for Breast Cancer? Advances in Surgery, 44(1), 199-228.

113
Berruti, A., Generali, D., Kaufmann, M., et al. (2011). International expert consensus on primary systemic therapy in the management of early breast cancer: highlights of the Fourth Symposium on Primary Systemic Therapy in the Management of Operable Breast Cancer, Cremona, Italy (2010). Journal of the National Cancer Institute. Monographs, 2011(43), 147-51.
Berry DA, Cirrincione C, Henderson IC, et al. (2006) Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA. Apr 12;295(14):1658-67.
Bertucci, F., Nasser, V., Granjeaud, S., et al. (2002). Gene expression profiles of poor-prognosis primary breast cancer correlate with survival. Human molecular genetics, 11(8), 863-72.
Bilancia, D., Rosati, G., Dinota, A, et al. (2007). Lapatinib in breast cancer. Annals of oncology : 18 Suppl 6(Supplement 6), vi26-30.
Blamey RW, Davies CJ, Elston CW, et al. (1979) Prognostic factors in breast cancer -- the formation of a prognostic index. Clin Oncol. Sep;5(3):227-36.
Blows, F. M., Driver, K. E., Schmidt, M. K., et al. (2010). Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS medicine, 7(5).
Bos, P. D., Zhang, X. H.-F., Nadal, C., et al. (2009). Genes that mediate breast cancer metastasis to the brain. Nature, 459(7249).
Bouchalova, K., Cizkova, M., Cwiertka, K., et al. (2009). Triple negative breast cancer--current status and prospective targeted treatment based on HER1 (EGFR), TOP2A and C-MYC gene assessment. Biomedical papers of the Medical Faculty of the University Palacký, Olomouc, Czechoslovakia, 153(1), 13-7.
Bouchalova, K., Cizkova, M., Cwiertka, K., et al. (2010). Lapatinib in breast cancer - the predictive significance of HER1 (EGFR), HER2, PTEN and PIK3CA genes and lapatinib plasma level assessment. Biomedical papers of the Medical Faculty of the University Palacký, Olomouc, Czechoslovakia, 154(4), 281-8.
Boyle P, Levin B, editors. World cancer report 2008. Lyon: IARC Press; 2008. 510 p.
Bradley C, Given C, Roberts C. (2002) Race, socioeconomic status, and breast cancer treatment and survival. J Natl Cancer Inst 2002;94(7):490-6.
Brand, T. M., Iida, M., Li, C., et al. (2011). The Nuclear Epidermal Growth Factor Receptor Signaling Network and its Role in Cancer. Discovery Medicine 12(66), 419-432.

114
Brandt, B., Hermann, S., Straif, K., et al. (2004). Advances in Brief Modification of Breast Cancer Risk in Young Women by a Polymorphic Sequence in the egfr Gene. Cancer, 7-12.
Brandt, B., Meyer-Staeckling, S., Schmidt, H., et al. (2006). Mechanisms of egfr gene transcription modulation: relationship to cancer risk and therapy response. Clinical cancer research 12(24), 7252-60.
BREAST CANCER STUDY GROUP (BCSG). Polychemotherapy for early breast cancer. An overview of randomizes trials Early Breast Cancer Trialist’ Collaborative Group. A summary of key results. Onkologie 2002; 25: 143-150.
Buerger, H, Schmidt, H., Beckmann, A, et al. (2001). Genetic characterisation of invasive breast cancer: a comparison of CGH and PCR based multiplex microsatellite analysis. Journal of clinical pathology, 54(11), 836-40.
Buerger, H, Gebhardt, F., Schmidt, H, et al. (2000). Length and Loss of Heterozygosity of an Intron 1 Polymorphic Sequence of egfr Is Related to Cytogenetic Alterations and Epithelial Growth Factor Receptor Expression Advances in Brief Length and Loss of Heterozygosity of an Intron 1 Polymorphic Sequence Cancer Research, 60; 854-857.
Buerger, H., Packeisen, J., Boecker, A., et al. (2004). Allelic length of a CA dinucleotide repeat in the egfr gene correlates with the frequency of amplifications of this sequence--first results of an inter-ethnic breast cancer study. The Journal of pathology, 203(1), 545-50.
Buisine, M.-P., Wacrenier, A., Mariette, C., et al. (2008). Frequent mutations of the CA simple sequence repeat in intron 1 of EGFR in mismatch repair-deficient colorectal cancers. World journal of gastroenterology : WJG, 14(7), 1053-9.
Buzdar, A. et al. (2001) Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol. Jul 15;19(14):3357-66.
Cabioglu, N., Gong, Y., Islam, R., et al. (2007). Expression of growth factor and chemokine receptors: new insights in the biology of inflammatory breast cancer. Annals of oncology18(6), 1021-9.
Campbell, H. E., Gray, A. M., Harris, A. L., et al. (2010). Estimation and external validation of a new prognostic model for predicting recurrence-free survival for early breast cancer patients in the UK. British journal of cancer, 103(6), 776-86.
Carter CL, Allen C, Henson DE. (1989) Relation of tumor size, lymph node status and survival in 24740 breast cancer cases. Cancer 63:181-7.

115
Carvalho, S. T., Stiepcich, M. M., Fregnani, J. H., et al. (2011). Evaluation of prognostic factors in stage IIA breast tumors and their correlation with mortality risk. Clinics, 66(4), 607-612.
Chagpar, A. B., Camp, R. L., & Rimm, D. L. (2011). Lymph node ratio should be considered for incorporation into staging for breast cancer. Annals of surgical oncology, 18(11), 3143-8. doi:10.1245/s10434-011-2012-9
Chi, D. D., Hing, A. V., Helms, C. et al. (1992) Two chromosome 7 dinucleotide repeat polymor- phisms at gene loci epidermal growth factor receptor (EGFR) and pro alpha 2 (I) collagen (COL1A2). Hum. Mol. Genet. 1, 135.
Chorev, M., & Carmel, L. (2012). The function of introns. Frontiers in genetics, 3(April), 55. doi:10.3389/fgene.2012.00055
Chotteau-Lelièvre, A., Révillion, F., Lhotellier, V., , et al. (2004). Prognostic value of ERM gene expression in human primary breast cancers. Clinical cancer research, 10(21), 7297-303.
Choura, M., Frikha, F., Kharrat, N., Aifa, S., & Rebaï, A. (2010). Investigating the function of three non-synonymous SNPs in EGFR gene: structural modelling and association with breast cancer. The protein journal, 29(1), 50-4.
Ciardiello, F., Troiani, T., Caputo, F., et al. (2006). Phase II study of gefitinib in combination with docetaxel as first-line therapy in metastatic breast cancer. British journal of cancer, 94(11), 1604-9.
Cohen, A. L., Soldi, R., Zhang, H., et al. (2011). A pharmacogenomic method for individualized prediction of drug sensitivity. Molecular systems biology, 7(513), 513.
Cui X, Schiff R, Arpino G et al. (2005) Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol 2005; 23:7721–7735.
Cuzick, J., Sestak, I., Baum, M., Buzdar, A., Howell, A., Dowsett, M., & Forbes, J. F. (2010). Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. The lancet oncology, 11(12), 1135-41.
Da Silva, L., Simpson, P. T., Smart, C. E., et al. (2010). HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast cancer research : BCR, 12(4).
Dahan, L., Norguet, E., Etienne-Grimaldi, M.-C. et al. (2011). Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC cancer, 11(1), 496.

116
Danova, M., Delfanti, S., Manzoni, M., & Mariucci, S. (2011). Tissue and soluble biomarkers in breast cancer and their applications: ready to use? Journal of the National Cancer Institute. Monographs (43), 75-8.
Dapic, V., Carvalho, M. A. & Monteiro, A. N. (2005) Breast cancer susceptibility and the DNA damage response. Cancer Control Mar;12(2):127-39.
Darwin, C. R. (1859) On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London: John Murray. [1st edition]
Di Fiore PP, et al. (1987) Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 51(6):1063–1070.
Di Saverio, S., Gutierrez, J., & Avisar, E. (2008). A retrospective review with long term follow up of 11,400 cases of pure mucinous breast carcinoma. Breast cancer research and treatment, 111(3), 541-7
Dong, L. M., Brennan, P., Karami, S., Hung, R. J., Menashe, I., Berndt, S. I., Yeager, M., et al. (2009). An analysis of growth, differentiation and apoptosis genes with risk of renal cancer. PloS one, 4(3), e4895.
Dubey, S., Stephenson, P., Levy, D. E., et al. (2006). EGFR dinucleotide repeat polymorphism as a prognostic indicator in non-small cell lung cancer. Journal of thoracic oncology, 1(5), 406-12.
EARLY BREAST CANCER TRIALISTS’ COLLABORATIVE GROUP http://www.ctsu.ox.ac.uk/~ebctcg/ [Online] acessado em 7 de abril de 2012.
EARLY BREAST CANCER TRIALISTS’ COLLABORATIVE GROUP (2012) Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials Lancet; 379: 432-44
EARLY BREAST CANCER TRIALISTS’ COLLABORATIVE GROUP (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomized trials. Lancet 365: 1687–1717.
Easton, D. F. et al. (1993) Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. Am J Hum Genet, 1993 v.52, n.4, p.678-701.
Easton, D. F., Pooley, K. A., Dunning, A. M., et al. (2009). UKPMC Funders Group Genome-wide association study identifies novel breast cancer susceptibility loci. Public Health, 447(7148), 1087-1093.

117
Edwards, J. G., Swinson, D. E. B., Jones, J. L., et al. (2006). EGFR expression: associations with outcome and clinicopathological variables in malignant pleural mesothelioma. Lung cancer (Amsterdam, Netherlands), 54(3), 399-407.
Ellegren H. (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet. Jun;5(6):435-45.
Elston CW, Ellis IO. (1991) Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology Nov;19(5):403-10.
Endres, N. F., Engel, K., Das, R., et al. (2011). Regulation of the catalytic activity of the EGF receptor. Current opinion in structural biology, 21(6), 777-84.
Etienne-Grimaldi, M.-C., Pereira, S., Magné, N., et al. (2005). Analysis of the dinucleotide repeat polymorphism in the epidermal growth factor receptor (EGFR) gene in head and neck cancer patients. Annals of oncology, 16(6), 934-41. doi:10.1093/annonc/mdi189
Fedi, P., Tronick, S.R., and Aaronson, S.A (1997) Growth factors In Cancer Medicine, Baltimore, MD: Williams and Wilkins, pp. 41–64.
Finak, G., Sadekova, S., Pepin, F., et al. (2006). Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast cancer research : BCR, 8(5)
Fisher, B. et al. (1997) Effect of properative chemotherapy on local-regional disease in women with operable breast cancer: finds from National Sirurgical Adjuvant Breast and Bowel Project B-18. J Clin Oncol; 2483-93
Fisher, B., Costantino, J. P., Wickerham, D. L., et al. (2005). Tamoxifen for the Prevention of Breast Cancer : Current Status of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. Cancer, 97(22).
Fitzgibbons, P. L., Page, D. L., Weaver, D., et al. (2000). Prognostic Factors in Breast Cancer College of American Pathologists Consensus Statement 1999. Arch Pathol lab Med; 124: 966-78.
Flynn, J. F., Wong, C., & Wu, J. M. (2009). Anti-EGFR Therapy: Mechanism and Advances in Clinical Efficacy in Breast Cancer. Journal of oncology, Volume 2009.
Ford, D. et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet., v.62, p.676–689. 1998.

118
Fox, E. M., Andrade, J., & Shupnik, M. A. (2010). Novel Actions of Estrogen to Promote Proliferation: Integration of Cytoplasmic and Nuclear Pathways. Steroids. 74(7), 622-627.
Franco, J. et al. (1997) Tratamento cirúrgico do câncer de mama. In: Franco J. Mastologia - Formação do especialista. Rio de Janeiro Atheneu 1° edição
Franco-Hernandez, C., Martinez-Glez, V., Arjona, D., et al. (2007). EGFR sequence variations and real-time quantitative polymerase chain reaction analysis of gene dosage in brain metastases of solid tumors. Cancer genetics and cytogenetics, 173(1), 63-7.
Fryback, D. G., Stout, N. K., Rosenberg, M. A, et al. (2006). The Wisconsin Breast Cancer Epidemiology Simulation Model. Journal of the National Cancer Institute. Monographs, 53726(36), 37-47.
Fumagalli, D., & Sotiriou, C. (2010). Treatment of pT1N0 breast cancer: multigene predictors to assess risk of relapse. Annals of oncology, 21 Suppl 7(Supplement 7)
Galea MH, Blamey RW, Elston CE, Ellis IO (1992) The Nottingham Prognostic Index in primary breast cancer. Breast Cancer Res Treat, 22:207-219.
Gebhardt, F., Zänker, K. S., & Brandt, B. (1999). Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. The Journal of biological chemistry, 274(19), 13176-80.
Geiger, A. M., Fischberg, G. M., Chen, W., & Bernstein, L. (2004). Stroke Risk and Tamoxifen Therapy for Breast Cancer. October, 96(20).
Georgopoulos, N. T., Kirkwood, L. a, Walker, D. C., & Southgate, J. (2010). Differential regulation of growth-promoting signalling pathways by E-cadherin. PloS one, 5(10), e13621.
Gilmer, T. M., Cable, L., Alligood, K., et al. (2008). Impact of common epidermal growth factor receptor and HER2 variants on receptor activity and inhibition by lapatinib. Cancer research, 68(2), 571-9
Goldhirsch A, Glick JH, Gelber RD, et al. Panel members. Meeting highlights: international expert consensus on the primary therapy of early breast cancer 2005. Ann Oncol. 2005 Oct;16(10):1569-83. Epub 2005 Sep 7.
Goldhirsch, A, Ingle, J. N., Gelber, R. D., et al. (2009). Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Annals of oncology 20(8)

119
Gomes, R.; Skaba, M. M. V. F. & Vieira, R. J. S. (2002) Reinventando a vida: a proposta para uma abordagem sócio-antropológica do câncer de mama feminina. Cadernos de Saúde Pública, v.18, n.5, p.197-204.
Gonçalves, A., Esteyries, S., Taylor-Smedra, B., Lagarde, A., Ayadi, M., Monges, G., Bertucci, F., et al. (2008). A polymorphism of EGFR extracellular domain is associated with progression free-survival in metastatic colorectal cancer patients receiving cetuximab-based treatment. BMC cancer, 8, 169
Gori, S., Sidoni, A, Colozza, M., Ferri, et al. (2009). EGFR, pMAPK, pAkt and PTEN status by immunohistochemistry: correlation with clinical outcome in HER2-positive metastatic breast cancer patients treated with trastuzumab. Annals of oncology 20(4), 648-54.
Greenlee, R.T. et al. (2000) Cancer statistics, 2000. CA Cancer J Clin; 50(1):7-33
Guimarães, J.R.Q, coordenador. Manual de Oncologia Vol 1, São Paulo; 2008. BBS Editora 3ª edição.
Gupta, P., Han, S.-Y., Holgado-Madruga, M., et al (2010). Development of an EGFRvIII specific recombinant antibody. BMC biotechnology, 10, 72. doi:10.1186/1472-6750-10-72
Haley, J. D., & Waterfield, M. D. (1991). Contributory effects of de novo transcription and premature transcript termination in the regulation of human epidermal growth factor receptor proto-oncogene RNA synthesis. The Journal of biological chemistry, 266(3), 1746-53.
Han, S.-W., Jeon, Y. K., Lee,., et al. (2007). Intron 1 CA dinucleotide repeat polymorphism and mutations of epidermal growth factor receptor and gefitinib responsiveness in non-small-cell lung cancer. Pharmacogenetics and genomics, 17(5), 313-9.
Hanahan, D., Weinberg, R. A., & Francisco, S. (2000). The Hallmarks of Cancer Review University of California at San Francisco. Hormone Research, 100, 57-70.
Hanstein, B., Djahansouzi, S., Dall, P., Beckmann, M. W., & Bender, H. G. (2004). Insights into the molecular biology of the estrogen receptor define novel therapeutic targets for breast cancer. European journal of endocrinology, 150(3), 243-55.
Harrell, J. C., Dye, W. W., Allred, D. C., et al. (2006). Estrogen Receptor Positive Breast Cancer Metastasis : Altered Hormonal Sensitivity and Tumor Aggressiveness in Lymphatic Vessels and Lymph Nodes. Cancer Research, (18), 9308-9315.

120
Harris JR, Lippman ME, Morrow M et al (2004) Diseases of the breast. Philadelphia, Lippincott Williams & Wilkins 3rd edn
Haybittle JL, Blamey RW, Elston CW, et al (1982). A prognostic index in primary breast cancer. British journal of cancer. Mar; 45(3):361-6.
Henry, N. L., Stearns, V., Flockhart, D. A.,et al. (2008). Drug Interactions and Pharmacogenomics in the Treatment of Breast Cancer and Depression. Psychiatry: Interpersonal and Biological Processes, (October), 1251-1255.
Hess, K.R. et al. (2003) Estrogen Receptors and distinct patterns of breast relapse. Breast Cancer Res Ttreat; 78: 105-18.
Hind, D., Ward, S., Nigris, E. D., et al. (2007). Hormonal therapies for early breast cancer. Health Technology Assessment 11: No. 26
Hirakawa, S. (2009). From tumor lymphangiogenesis to lymphvascular niche. Cancer, 100(6), 983-989.
Hoefnagel, L. D. C., Moelans, C. B., Meijer, S. L., et al. (2012). Prognostic value of estrogen receptor α and progesterone receptor conversion in distant breast cancer metastases.
Hoeijmakers, H.J.J. (2001) Genome maintenance mechanisms for preventing cancer. Nature; 411: 366-374,.
Horwitz KB, McGuire WL. Specifi c progesterone receptors in human breast cancer. Steroids 1975 ; 25 : 497 – 505.
Huang, C.-L., Yang, C.-H., Yeh, K.-H., et al. (2009). EGFR intron 1 dinucleotide repeat polymorphism is associated with the occurrence of skin rash with gefitinib treatment. Lung cancer (Amsterdam, Netherlands), 64(3), 346-51.
Hudson, L. G., & Stack, M. S. (2010). EGF-receptor regulation of matrix metalloproteinases in epithelial ovarian carcinoma, Future Oncol. April ; 5(3)
Huober, J., von Minckwitz, G., Denkert, C., et al. (2010). Effect of neoadjuvant anthracycline-taxane-based chemotherapy in different biological breast cancer phenotypes: overall results from the GeparTrio study. Breast cancer research and treatment, 124(1), 133-40.
Hutter RV The role of the pathologist in breast cancer management. Cancer. 1990 Sep 15;66(6 Suppl):1363-72. Review.
Instituto Nacional de Câncer; BRASIL. Ministério da Saúde. Estimativa 2006: incidência de câncer no Brasil / Instituto Nacional de Câncer. – Rio de Janeiro:INCA, 2005.

121
Instituto Nacional de Câncer ; BRASIL. Ministério da Saúde. Estimativa 2010: incidência de câncer no Brasil / Instituto Nacional de Câncer. – Rio de Janeiro: INCA, 2009.
Instituto Nacional de Câncer BRASIL Ministério da Saúde Coordenação de Assistência. Serviço de Oncologia Clínica. Serviço de oncologia clínica: rotinas internas do INCA / Instituto Nacional de Câncer. - Rio de Janeiro: INCA, 2011.
Instituto Nacional de Câncer José Alencar Gomes da Silva. BRASIL Ministério da Saúde. Coordenação Geral de Ações Estratégicas. Coordenação de Prevenção e Vigilância. Estimativa 2012 : incidência de câncer no Brasil / Instituto Nacional de Câncer José Alencar Gomes da Silva, Coordenação Geral de Ações Estratégicas, Coordenação de Prevenção e Vigilância. – Rio de Janeiro : INCA, 2011.
International Union Against Cancer (UICC) (1958) Clinical stage classification and presentation of results, malignant tumours of the breast and larynx. Paris, Committee on Clinical Stage Classification and Applied Statistics.
International Union Against Cancer (UICC), (2002) TNM Classification of Malignant Tumours - 6th ed. Edited by L.H. Sobin and Ch. Wittekind. John Wiley & Sons -
Irigaray P, Newby JA, Clapp R, Hardell L, Howard V, Montagnier L, Epstein S, Belpomme D. (2007) Lifestyle-related factors and environmental agents causing cancer: an overview. Biomed Pharmacother,; n. 61, v. 10, p. 640-58.
Jacobsen, B. M., Schittone, S. A, Richer, J. K., & Horwitz, K. B. (2005). Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Molecular endocrinology 19(3), 574-87.
Jacobsen, B. M., & Horwitz, K. B. (2012). Progesterone receptors, their isoforms and progesterone regulated transcription. Molecular and cellular endocrinology 2012 Jun 24;357(1-2):18-29
Jahanzeb, M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer. 2008 Aug;8(4):324-33.
Jami, M. S., Hemati, S., Salehi, Z., & Tavassoli, M. (2008). Association between the length of a CA dinucleotide repeat in the EGFR and risk of breast cancer. Cancer investigation, 26(4), 434-7.
Jacquin, J. P., et al. (2012) Docetaxel-containing adjuvant chemotherapy in patients with early stage breast cancer. Consistency of effect independent of nodal and biomarker status: a meta-analysis of 14 randomized clinical trials. Breast Cancer Res Treat. Jan 24

122
Ji, R.-cheng. (2006). Lymphatic endothelial cells , tumor lymphangiogenesis and metastasis : New insights into intratumoral and peritumoral lymphatics. Cancer, 677-694.
Jimeno, A., & Hidalgo, M. (2006). Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochimica et biophysica acta, 1766(2), 217-29.
Jones, J. L., Royall, J. E., & Walker, R. a. (1996). E-cadherin relates to EGFR expression and lymph node metastasis in primary breast carcinoma. British journal of cancer, 74(8), 1237-41.
Jorissen, R. (2003). Epidermal growth factor receptor: mechanisms of activation and signalling. Experimental Cell Research, 284(1), 31-53.
Jovanovic, J., Rønneberg, J. A., Tost, J., & Kristensen, V. (2010). The epigenetics of breast cancer. Molecular oncology, 4(3), 242-54.
Kallel, I., Rebai, M., Khabir, A., Farid, N. R., & Rebaï, A. (2009). Genetic polymorphisms in the EGFR (R521K) and estrogen receptor (T594T) genes, EGFR and ErbB-2 protein expression, and breast cancer risk in Tunisia. Journal of biomedicine & biotechnology, 2009, 753683.
Kallergi, G., Agelaki, S., Kalykaki, A., et al. (2008). Phosphorylated EGFR and PI3K/Akt signaling kinases are expressed in circulating tumor cells of breast cancer patients. Breast cancer research, 10(5), R80.
Kashi, Y., King, D., & Soller, M. (1997). Simple sequence repeats as a source of quantitative genetic variation. Trends in genetics: TIG, 13(2), 74-8.
Kelsey, J. et al. (1996) Methods in Observational Epidemiology. New York: Editora Oxford.
Kelsey, J. L., & Berkowitz, G. S. (1988). Breast Cancer Epidemiology. New York, 5615-5623.
Kersting, C., Packeisen, J., Leidinger, B., et al. (2006). Pitfalls in immunohistochemical assessment of EGFR expression in soft tissue sarcomas. Journal of clinical pathology, 59(6), 585-90.
Key, T. Verkasalo, P. Banks, E. (2001) Epidemiology of breast cancer. Lancet Oncol; 2:133-140.
Lambe, M. et al. (1996) Age at first and last birth, and risk of breast cancer: a population-2. based study in Sweden. Breast Cancer Res Treat. 38(3):305-11.

123
Lange, C. A., Richer, J. K., Shen, T., & Horwitz, K. B. (1998). Convergence of Progesterone and Epidermal Growth Factor Signaling in Breast Cancer. The journal of biological chemistry 273(47), 31308-31316.
Lannin, D.R. et al. (1998) Influence of socioeconomic and cultural factors on racial differences in late-stage presentation of breast cancer. JAMA; 279(22):1801-7
Lee AH, Pinder SE, Macmillan RD, et al. (2006) Prognostic value of lymphovascular invasion in women with lymph node negative invasive breast carcinoma. Eur J Cancer Feb;42(3):357-62.
Lee, B. L., Liedke, P. E. R., Barrios, C. H,. et al (2012). Review Breast cancer in Brazil : present status and future goals. Lancet Oncol; 13: e95–102
Lee, J.-M., Yang, S.-Y., Yang, P.-W., et al. (2011). Polymorphism in epidermal growth factor receptor intron 1 predicts prognosis of patients with esophageal cancer after chemoradiation and surgery. Annals of surgical oncology, 18(7), 2066-73.
Leite, M. (1999) Tratamento do câncer de mama - Radioterapia. . In: Chaves I et al. Mastologia: Aspectos Multidisciplinares. Belo Horizonte: Editora Medsi,.
Leong, A. S.-Y., & Zhuang, Z. (2011). The changing role of pathology in breast cancer diagnosis and treatment. Pathobiology : journal of immunopathology, molecular and cellular biology, 78(2), 99-114.
Levin, E. R. (2003). Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Molecular endocrinology (Baltimore, Md.), 17(3), 309-17.
Lewin B. ( 2001) Genes VII. Editora ARTMED. São Paulo, SP. Pág. 39-42.
Lin, Y.-L., Wang, W.-Y., Lin, J.-C., Wong, Y.-K., & Chien, Y.-C. (2011). The status of EGFR CA SSR1 is a potential prognostic factor for patients with oral squamous cell carcinoma. Oral oncology, 47(6), 482-6.
Liu, G., Gurubhagavatula, S., Zhou, W., Wang, Z., et al. (2008). Epidermal growth factor receptor polymorphisms and clinical outcomes in non-small-cell lung cancer patients treated with gefitinib. The pharmacogenomics journal, 8(2), 129-38.
Liu, W., Innocenti, F., Chen, P., et al. (2003). Interethnic difference in the allelic distribution of human epidermal growth factor receptor intron 1 polymorphism. Clinical cancer research: 9(3), 1009-12.
Liu, W., Wu, X., Zhang, W., et al. (2007). Relationship of EGFR mutations, expression, amplification, and polymorphisms to epidermal growth factor

124
receptor inhibitors in the NCI60 cell lines. Clinical cancer research, 13(22 Pt 1), 6788-95. doi:10.1158/1078-0432.CCR-07-0547
Liu, Y., Ji, R., Li, J., et al. (2010). Correlation effect of EGFR and CXCR4 and CCR7 chemokine receptors in predicting breast cancer metastasis and prognosis. Journal of experimental & clinical cancer research : CR, 29, 16.
Lurje, G., Nagashima, F., Zhang, W, et al. (2008). Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clinical cancer research, 14(23), 7884-95.
Ma, F., Sun, T., Shi, Y., et al. (2009). Polymorphisms of EGFR predict clinical outcome in advanced non-small-cell lung cancer patients treated with Gefitinib. Lung cancer (Amsterdam, Netherlands), 66(1), 114-9.
Macmahon, B. et al. (1970) Age at first birth and breast cancer risk. Bull World Health Organ. 3;43(2):209-21.
Marchant, D.J. (1997) Risk factors. British Journal of Cancer, 55, 207-211.
Marzolini C, Paus E, Buclin T, Kim RB. (2004) Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther, n. 75, p.13-33.
Massarweh, S., Osborne, C. K., Creighton, C. J, et al. (2008). Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer research, 68(3), 826-33.
Masuda, H., Masuda, N., Kodama, Y., et al. (2011). Predictive factors for the effectiveness of neoadjuvant chemotherapy and prognosis in triple-negative breast cancer patients. Cancer chemotherapy and pharmacology, 67(4), 911-7.
Masui H, Kawamoto T, Sato JD, et a (1984) Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res 44:1002-1007.
Matsubara, D., Ishikawa, S., Sachiko, O. et al (2010). Co-activation of epidermal growth factor receptor and c-MET defines a distinct subset of lung adenocarcinomas. The American journal of pathology, 177(5), 2191-204. American Society for Investigative Pathology.
Mauri, D., Polyzos, N. P., Salanti, G., et al. (2008). Multiple-treatments meta-analysis of chemotherapy and targeted therapies in advanced breast cancer. Journal of the National Cancer Institute, 100(24), 1780-91.

125
McKibbin, T., Zhao, W., Tagen, M., et al. (2010). Epidermal growth factor receptor polymorphisms and risk for toxicity in paediatric patients treated with gefitinib. European journal of cancer 46(11), 2045-51.
Meche, A., Cîmpean, A. M., & Raica, M. (2009). Immunohistochemical expression and significance of epidermal growth factor receptor (EGFR) in breast cancer. Romanian journal of morphology and embryology, 50(2), 217-21.
Mendelsohn J. (2002) Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol; 20:1s-13s.
Mendelsohn, J., & Baselga, J. (2003). Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. Journal of clinical oncology, 21(14), 2787-99. doi:10.1200/JCO.2003.01.504
Miki, Y. et al. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science, 1994.; v.266, n. 5182, p. 66-71.
Milano, G., Etienne-Grimaldi, M.-C., Dahan, L., et al. (2008). Epidermal growth factor receptor (EGFR) status and K-Ras mutations in colorectal cancer. Annals of oncology, 19(12), 2033-8.
Mill, C. P., Chester, J. A., & Ii, D. J. R. (2010). EGFR may couple moderate alcohol consumption to increased breast cancer risk. Breast Cancer (London) (1): 31–38.
Miller MC, Mohrenweiser HW, Bell DA. (2001) Genetic variability in susceptibility and response to toxicants. Toxicol. Lett.; 120:269-280.
Miller, D. L., el-Ashry, D., Cheville, A L., et al (1994). Emergence of MCF-7 cells overexpressing a transfected epidermal growth factor receptor (EGFR) under estrogen-depleted conditions: evidence for a role of EGFR in breast cancer growth and progression. Cell growth & differentiation 5(12), 1263-74.
Modjtahedi, H., & Essapen, S. (2009). Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anti-cancer drugs, 20(10), 851-5.
Moore, G.E. Cancer: 100 Different Diseases The American Journal of Nursing, 1966 vol. 66, No. 4 p 749
Moriai T, Kobrin MS, Korc M. (1993) Cloning of a variant epidermal growth factor receptor. Biochem Biophys Res Commun.Mar 31;191(3):1034-9.
Moriai, T., Kobrin, M. S., Hope, C., et al (1994). A variant epidermal growth factor receptor exhibits altered type alpha transforming growth factor

126
binding and transmembrane signaling. Proceedings of the National Academy of Sciences of the United States of America, 91(21), 10217-21.
Moroni, M., Veronese, S., Benvenuti, S., et al. (2005). Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. The lancet oncology, 6(5), 279-86.
Müller, A, Homey, B., Soto, H., (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature, 410(6824), 50-6.
Murphy, C. G., & Modi, S. (2009). HER2 breast cancer therapies: a review Biologics : targets & therapy, 3, 289-301.
Nakamura, R., Sakakibara, M., Nagashima, T., & Sangai, T. (2009). Accumulation of regulatory T cells in sentinel lymph nodes is a prognostic predictor in patients with node-negative breast cancer. European Journal of Cancer, 45(12), 2123-2131.
NATIONAL CANCER INSTITUTE BETHESDA, MD. [Online]. acesso em 04 nov 2008 Disponível em: http://www.cancer.gov/cancertopics/factsheet/Risk/BRCA
National Center for Biotechnology Information (NCBI) [Online]. acessado em junho de 2009. Disponivel em: http://www.ncbi.nlm.nih.gov/pubmed/
Nie, Q., Yang, X.-N., An, S.-J., et al. (2011). CYP1A1*2A polymorphism as a predictor of clinical outcome in advanced lung cancer patients treated with EGFR-TKI and its combined effects with EGFR intron 1 (CA)n polymorphism. European journal of cancer (Oxford, England : 1990), 47(13), 1962-70.
NIH Consens (2000) Adjuvant therapy for breast cancer. NIH Consens
Statement. Nov 1-3;17(4):1-35.
Nieto, Y., Nawaz, F., Jones, R. B., Shpall, E. J., & Nawaz, S. (2007). Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. Journal of clinical oncology, 25(28), 4405-13.
Nomura, M., Shigematsu, H., Li, et al. (2007). Polymorphisms, mutations, and amplification of the EGFR gene in non-small cell lung cancers. PLoS medicine, 4(4), e125.
Ochi, N., Matsuo, Y., Sawai, H., Yasuda, A., & Takahashi, H. (2007). Vascular Endothelial Growth Factor-C Secreted by Pancreatic Cancer Cell Line Promotes Lymphatic Endothelial Cell Migration in an In Vitro Model of Tumor Lymphangiogenesis. Cancer, 34(4), 444-451.

127
Olayioye, M. A., Neve, R. M., Lane, H. A., & Hynes, N. E. (2000). The ErbB signaling network : receptor heterodimerization in development and cancer. EMBO Journal, 19(13).
Oonk, M. H. M., de Bock, G. H., van der Veen, D. J., et al (2007). EGFR expression is associated with groin node metastases in vulvar cancer, but does not improve their prediction. Gynecologic oncology, 104(1), 109-13. doi:10.1016/j.ygyno.2006.07.035
Paci, E. et al. (1996) Long-term sequelae of breast cancer surgery. Tumori; 82: 321-4.
Paech K, Webb P, Kuiper GG, et al. (1997) Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. Sep 5;277(5331):1508-10.
Parisi, F., González, A. M., Nadler, Y., et al (2010). Benefits of biomarker selection and clinico-pathological covariate inclusion in breast cancer prognostic models. Breast cancer research : BCR, 12(5), R66.
Park, J. H., Han, S.-W., Oh, D.-Y., et al. (2011). Analysis of KRAS, BRAF, PTEN, IGF1R, EGFR intron 1 CA status in both primary tumors and paired metastases in determining benefit from cetuximab therapy in colon cancer. Cancer chemotherapy and pharmacology, 68(4), 1045-55.
Partridge, B. A. H., Wang, P. S., Winer, E. P., & Avorn, J. (2003). Nonadherence to Adjuvant Tamoxifen Therapy in Women With Primary Breast Cancer. Cancer, 21(4), 602-606.
Perera, F.P. Environment and cancer: Who are susceptible? Science, 1997. 278:1068-1073,
Peto, J. (2001). Cancer epidemiology in the last century and the next decade. Nature, 411. 17 (May) p390.
Piato, J.R.M. (2001) Linfonodo Sentinela no Carcinoma Infiltrativo Inicial de Mama: Estudo de sua Localização e de sua Capacidade Preditiva em Relação ao Estado da Axila. Rev. Bras. Ginecol. Obstet., vol.24, n.1, pp. 69-69.
Pimenta, J. R., Zuccherato, L. W., Debes, A. A., et al. (2006). Color and genomic ancestry in Brazilians: a study with forensic microsatellites. Human heredity, 62(4), 190-5. doi:10.1159/000096872
Pinder, S.E. Ellis, I.O. (2003) The diagnosis and management of pre-invasive breast disease ductal carcinoma in situ (DCIS) and atpical ductal hyperplasia (ADH) – current definitions and classification. Breast cancer Res, 5: 254-257.

128
Pirmohamed M, Aithal GP, Behr E, et al., (2011) The Phenotype Standardization Project: Improving Pharmacogenetic Studies of Serious Adverse Drug Reactions. Clinical Pharmacology & Therapeutics, p. 784-785
Polli, J. W., Olson, K. L., Chism, J. P., et al. (2009). An Unexpected Synergist Role of P-Glycoprotein and Breast Cancer Resistance Protein on the Central Nervous System Penetration of the Tyrosine Kinase Inhibitor Lapatinib quinazolinamine ; GW572016 Pharmacology, 37(2), 439-442.
Press, O. A, Zhang, W., Gordon, M. A, et al. (2008). Gender-related survival differences associated with EGFR polymorphisms in metastatic colon cancer. Cancer research, 68(8), 3037-42.
Preston-Martins S, Pike MC, Ross RK et al. Increased cell division as a cause of human cancer. Cancer Res 1990; 50, 7415-7421.
Puyo, S., Le Morvan, V., & Robert, J. (2008). Impact of EGFR gene polymorphisms on anticancer drug cytotoxicity in vitro. Molecular diagnosis & therapy, 12(4), 225-34.
Qian, C., Berghuis, B., Tsarfaty, G., Bruch, et al. (2006). Preparing the “Soil”: The Primary Tumor Induces Vasculature Reorganization in the Sentinel Lymph Node before the Arrival of Metastatic Cancer Cells. Cancer Res 2006; 66: (21) 10635.
Rakha, E. A, Reis-Filho, J. S., & Ellis, I. O. (2010). Combinatorial biomarker expression in breast cancer. Breast cancer research and treatment, 120(2), 293-308.
Rego, R. L., Foster, N. R., Smyrk, T. C., et al. (2010). Prognostic effect of activated EGFR expression in human colon carcinomas: comparison with EGFR status. British journal of cancer, 102(1), 165-72.
Relling, M. V., Altman, R. B., Goetz, M. P., & Evans, W. E. (2010). Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. The lancet oncology, 11(6), 507-9.
Richardson, L.C. et al. (2001) Early-stage breast cancer treatment among medically underserved women diagnosed in a national screening program, 1992-1995. Breast Cancer Res Treat; 69(2):133-42.
Ricono, J. M., Huang, M., Barnes, L. A., et al. (2010). Specific crosstalk between EGFR and integrin αvβ5 promotes carcinoma cell invasion and metastasis. Cancer, 69(4), 1383-1391.
Rosell, R., Carcereny, E., Gervais, R., et al. (2012). Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a

129
multicentre, open-label, randomised phase 3 trial. The lancet oncology, 13(3), 239-246.
Sainsbury JR, Farndon JR, Needham GK, Malcolm AJ, Harris AL. (1987) Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet. Jun 20;1(8547):1398-402.
Samaga, R., Saez-Rodriguez, J., Alexopoulos, L. G., et al. (2009). The logic of EGFR/ErbB signaling: theoretical properties and analysis of high-throughput data. PLoS computational biology, 5(8), p1
Sánchez-Muñoz, A., Gallego, E., de Luque, V., et al. (2010). Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC cancer, 10, 136.
Sanger, F., Nicklen, S., Coulson, A. R. (1977) DNA sequencing with chainterminating inhibitors. Proceedings of the National Academy of Sciences; USA, 74: 5463-5467.
Sanpaolo, P., Barbieri, V., Pedicini, P., & Fusco, V. (2011). Her-2 prognostic value in very early-stage breast cancer: a single-institution retrospective analysis. Medical oncology (Northwood, London, England), 0-6.
Sarkis, S. A, Abdullah, B. H., Abdul Majeed, B. A, & Talabani, N. G. (2010). Immunohistochemical expression of epidermal growth factor receptor (EGFR) in oral squamous cell carcinoma in relation to proliferation, apoptosis, angiogenesis and lymphangiogenesis. Head & neck oncology, 2, 13.
Sasaki, H., Okuda, K., Shimizu, S., et al. (2009). EGFR R497K polymorphism is a favorable prognostic factor for advanced lung cancer. Journal of cancer research and clinical oncology, 135(2), 313-8.
Sato JD, Kawamoto T, Le AD, et a (1983) Biological effect in vitro of monoclonal antibodies to human EGF receptors. Mol Biol Med 1:511-529.
Schoppmann SF, Bayer G, Aumayr K, et al. (2004) Austrian Breast and Colorectal Cancer Study Group. Prognostic value of lymphangiogenesis and lymphovascular invasion in invasive breast cancer. Ann Surg. Aug;240(2):306-12.
Shepard, H. M., Brdlik, C. M., & Schreiber, H. (2008). Signal integration : a framework for understanding the efficacy of therapeutics targeting the human EGFR family, The Journal of Clinical Investigation 118(11).
Siena, S., Sartore-Bianchi, A., Di Nicolantonio, F., et al (2009). Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. Journal of the National Cancer Institute, 101(19), 1308-24.

130
Silverstein, M.J. et al. (1996) The van Nuys prognostic index for ductal carcinoma in situ. Breast J ; 2:38-40.
Simon SD, B.J.a.B.C., et al. (2009), The AMAZONE project of the Brazilian breast cancer study group (GBECAM). San Antonio Breast Cancer Symposium. 12(11).
Smith, I. C. et al. (2002) Neo adjuvant chemotherapy in breas câncer significantly anhanced response with docetaxel J Clin Oncol; 20 : 1456-66
Soerjomataram, I., Louwman, M. W. J., Ribot, J. G., et al. (2008). An overview of prognostic factors for long-term survivors of breast cancer. Breast cancer research and treatment, 107(3), 309-30. doi:10.1007/s10549-007-9556-1
Sørlie T, Perou CM, Tibshirani R, et al. (2001). Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications Proc Natl Acad Sci U S A. 2001 Sep 11;98(19):10869-74.
Sotiriou C, Neo SY, McShane LM. et al. (2003) Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. Sep 2;100(18):10393-8.
Spano, J. P., Milano, G., Vignot, S., & Khayat, D. (2008). Potential predictive markers of response to EGFR-targeted therapies in colorectal cancer. Critical reviews in oncology/hematology, 66(1), 21-30.
Spurdle, A. B. et al. (2011) Common genetic variation at BARD1 is not associated with Breast cancer risk in BRCA1 or BRCA2 mutation carriers. Cancer epidemiology Biomarkers Prevention, 20 (5) 1032-1038
Stingl, J. (2011). Estrogen and progesterone in normal mammary gland development and in cancer. Hormones & cancer, 2(2), 85-90.
Suarez-Kurtz, G. (2010). Pharmacogenetics in the brazilian population. Frontiers in pharmacology, 1(October), 118.
Sueoka-Aragane, N., Imai, K., Komiya, K., et al. (2008). Exon 19 of EGFR mutation in relation to the CA-repeat polymorphism in intron 1. Cancer science, 99(6), 1180-7.
Suo, Z., Risberg, B., Kalsson, M. G. (2002). EGFR family expression in breast carcinomas. c-erbB-2 and c-erbB-4 receptors have different effects on survival. The Journal of pathology, 196(1), 17-25. doi:10.1002/path.1003
Tan, P. (2008). Germline polymorphisms as modulators of cancer phenotypes. BMC medicine, 6, 27.

131
Tavassoli, F. A., Devilee, P. (2003) Tumors of the breast and female genital organs. Pathology and genetics. World Health Organization. Lyon: IARC Press.
Thakkar, J. P., & Mehta, D. G. (2011). A review of an unfavorable subset of breast cancer: estrogen receptor positive progesterone receptor negative. The oncologist, 16(3), 276-85.
The National Institutes of Health Consensus Development Conference: Adjuvant Therapy for Breast Cancer. Bethesda, Maryland, USA. November 1-3, 2000. Proceedings. J Natl Cancer Inst Monogr. 2001;(30):1-152. Review.
Thompson & Thompson; Nussbaum, RL; Mcinnes RL; Willard, H.F.(2002) Genética Médica. Rio de Janeiro, RJ. Editora Guanabara Koogan S.A – 6ªedição.
Thompson, P. a, Brewster, A. M., Kim-Anh, D., et al. (2011). Selective genomic copy number imbalances and probability of recurrence in early-stage breast cancer. PloS one, 6(8),
Tidow, N., Boecker, A., Schmidt, H., et al. (2003). Advances in Brief Distinct Amplification of an Untranslated Regulatory Sequence in the egfr Gene Contributes to Early Steps in Breast Cancer Development. Cancer, 1172-1178.
Tischkowitz, M., Brunet, J.-S., Bégin, L. R., et al. (2007). Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC cancer, 7, 134.
Tiseo, M., Rossi, G., Capelletti, M.,., et al. (2010). Predictors of gefitinib outcomes in advanced non-small cell lung cancer (NSCLC): study of a comprehensive panel of molecular markers. Lung cancer (Amsterdam, Netherlands), 67(3), 355-60. Elsevier Ireland Ltd.
Travis, R. C., & Key, T. J. (2003). Oestrogen exposure and breast cancer risk. Breast Cancer Research, 239-247.
Valero, V. (2002) Primary chemotherapy with docetaxel for the management of breast cancer. Oncology; 16:35-43
Veronesi, U. et al. (1990) Quadrantectomy versus lumpectomy for small size breast cancer Eur J Cancer ; 26:671
Viani, G. A. et al. (2007) Adjuvant trastuzumab in the treatment of her-2-positive early breast cancer: a meta-analysis of published randomized trials. BMC Cancer Aug 8;7:153
Vicini, F. A. et al. (2002) Optimizing breast cancer treatment efficacy with intensity-modulated radiotherapy Int J Radiat Oncol Biol Phys. Dec 1;54 (5): 1336-44.

132
Voduc, K. D., Cheang, M. C. U., Tyldesley, S., et al (2010). Breast cancer subtypes and the risk of local and regional relapse. Journal of clinical oncology, 28(10), 1684-91.
Voet D, Voet G, Charlotte WP. (2000) Fundamentos da Bioquímica Porto Alegre; Artmed.
von Minckwitz, G., Kümmel, S., Vogel, P.,et al (2008). Intensified neoadjuvant chemotherapy in early-responding breast cancer: phase III randomized GeparTrio study. Journal of the National Cancer Institute, 100(8), 552-62.
Walker RA (2003) Prognostic and Predictive Factors in Breast Cancer. 1st edition. New York: Informa Health Care; 2003.
Wang, W.-S., Chen, P.-M., Chiou, T.-J., et al. (2007). Epidermal growth factor receptor R497K polymorphism is a favorable prognostic factor for patients with colorectal carcinoma. Clinical cancer research, 13(12), 3597-604.
Wang, Y., Yin, Q., Yu, Q., et al., (2011). A retrospective study of breast cancer subtypes: the risk of relapse and the relations with treatments. Breast cancer research and treatment, 130(2), pp.489-98.
Weinberg RA. (2008) A biologia do câncer. Porto Alegre: Artmed 864p.
Williams, C., Brunskill, S., Altman, D., et al. (2006). Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy. Health technology assessment (Winchester, England), 10(34), iii-iv, ix-xi, 1-204.
Wilson S, Jones L, Coussens C & Hanna K (eds.) (2002) Cancer and the Environment: Gene- Environment Interactions. Washington, DC: National Academy Press.
Wong, S. Y., & Hynes, R. O. (2007). Tumor – Lymphatic Interactions in an Activated Stromal Microenvironment. Journal of Cellular Biochemistry, 850
World Health Organization – WHO (2010) [Online] Disponível em http://www.who.int/en/. Acesso em 29 de novembro de 2010.
Wykosky, J., Fenton, T., Furnari, F., et al (2011). Review EGFR: A Driver of Oncogenesis. Chinese Journal of Cancer, 5-12.
Xu, H., Yu, Y., Marciniak, D., et al. (2005). Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Molecular cancer therapeutics, 4(3), 435-42.
Yenidunya, S., Bayrak, R., & Haltas, H. (2011). Predictive value of pathological and immunohistochemical parameters for axillary lymph node metastasis in breast carcinoma. Diagnostic pathology, 6(1), 18.

133
Yonemori, K., Tsuta, K., Shimizu, C., et al. (2010). Immunohistochemical expression of HER1, HER3, and HER4 in HER2-positive breast cancer patients treated with trastuzumab-containing neoadjuvant chemotherapy. Journal of surgical oncology, 101(3), 222-7.
Yoshida, K., Nakachi, K., Imai, K., et al. (2009). Lung cancer susceptibility among atomic bomb survivors in relation to CA repeat number polymorphism of epidermal growth factor receptor gene and radiation dose. Carcinogenesis, 30(12), 2037-41.
Yu, H., Gong, X., Luo, X., et al. (2008). Co-Expression of EGFRVIII with erbB-2 enhances tumorigenesis. Cancer biology & Therapy, 7:11; 1818-1828.
Zhang, W., Park, D. J., Lu, B., et al. (2005). Epidermal growth factor receptor gene polymorphisms predict pelvic recurrence in patients with rectal cancer treated with chemoradiation. Clinical cancer research, 11(2 Pt 1), 600-5.
Zhang, Y., Banerjee, S., Wang, Z., et al. (2006). Antitumor activity of epidermal growth factor receptor-related protein is mediated by inactivation of ErbB receptors and nuclear factor-kappaB in pancreatic cancer. Cancer research, 66(2), 1025-32.
Zhou, B., Rao, L., Peng, Y., et al. (2009). Epidermal growth factor receptor gene polymorphisms, R497K, but not (CA)n repeat, is associated with dilated cardiomyopathy. Clinica chimica acta, 403(1-2), 184-7
Zhou Q, Cheung YB, Jada SR, et al. (2006) EGFR Intron 1 polymorphism in Asian Populations and its correlation with EGFR gene expression and amplification in breast tumor tissues. Cancer Biol Ther. Nov;5(11):1445-9.
Zwaans, B. M. M., & Bielenberg, D. R. (2007). Potential therapeutic strategies for lymphatic metastasis. Microvasc Res. 2007 ; 74(2-3).

134
ANEXO I

135

136

137

138

139

140
ANEXO III

141

142
ANEXO IV