o doente com leucemia ou linfoma:xa.yimg.com/kq/groups/21008465/1404056112/name/leucemia... ·...
TRANSCRIPT

O DOENTE COM LEUCEMIA OU LINFOMA
Prof. Cármino Antonio De SouzaTitular de Hematologia-Hemoterapia do Departamento de Clínica Médica Faculdade de Ciências Médicas - Universidade Estadual de Campinas (Unicamp)
LEUCEMIAS
As leucemias são doenças neoplásicas do sistema linfo-hematopoiético. O termo leucemia significa "sangue branco" e esta denominação nasceu da presença de um extrato espesso de glóbulos brancos, habitualmente quase imperceptível, que poderia quase se igualar aos glóbulos vermelhos. Esta terminologia foi ampliada para todas as doenças neoplásicas da célula-tronco, independente da quantidade de células leucêmicas circulantes. Este grupo de doenças é caracterizado pela presença de blastos no sangue periférico e medula óssea e, em termos gerais podem ser denominadas de doenças ou síndromes mielo- ou linfoproliferativas.
Origem celular e conceitos gerais:
As leucemias são doenças clonais pois se originam da transformação de uma única célula que prolifera de modo ilimitado e autônomo. Quando as células têm características fenotípicas (citológicas e moleculares) linfóides se fala em leucemia linfóide ou síndrome linfoproliferativa. Quando as células neoplásicas apresentam características fenotípicas mielóide se fala em leucemias mielóides. Tanto as leucemias linfóides quanto as mielóides se distinguem nos seus diversos sub tipos segundo a linha celular (T ou B para as linfóides e, granulocítica, monocítica, megacariocítica ou eritróide para as mielóides) e o grau de maturação que as células podem atingir.
Epidemiologia e etiopatogenia:
1. Agentes físicos: As radiações ionizantes (RI) são os primeiros agentes leucemogênicos reconhecidos. Inúmeras observações desta ação biológica das RI foram historicamente publicadas:
Crianças expostas a irradiação tímica no período intra-utero; Nas populações de Hiroshima e Nagasaki a incidência de leucemias, principalmente
as agudas, aumentou significativamente em uma mediana de 5-8anos após as explosões;
2. Fumo: Fumantes apresentam maior probabilidade de desenvolverem leucemias principalmente a mielóide aguda;
3. Agentes Químicos: No momento atual não é possível estimar e atribuir com segurança as leucemias agudas causadas por exposição ocupacional. Neste campo, o que é seguro, é a exposição ao benzeno com causa de leucemias. O primeiro caso de leucemia benzeno-associada foi descrito em 1928 e muitos casos anteriores provavelmente já haviam ocorrido. Existem dúvidas se podemos falar em dose "segura" ou "aceitável" de exposição ao benzeno. Entretanto, os estudos mais recentes indicam uma freqüência significativamente aumentada de leucemia aguda, prevalentemente mielóide, com exposições anuais de 10 p.p.m. (parte por milhão) ou mais. Os trabalhadores mais estudados neste campo são os de indústria siderúrgica, refinarias de petróleo, frentistas de postos de gasolina e indústria de calçados. Trabalhadores da produção ou com uso de óxido de etileno têm apresentado maior número de anormalidades citogenéticas e leucemias agudas.

Maior risco de leucemia aguda também tem sido relatado em embalsamadores, técnicos de anatomia, auxiliares de serviços funerários, pintores e gráficos. No âmbito da agricultura, exposição à pesticidas e herbicidas bem como agentes virais têm sido relatados como possíveis causas de leucemia (risco-campo). Um grupo significativo de pacientes que desenvolvem leucemias são aqueles expostos a agentes mielo-tóxicos, particularmente, agentes alquilantes. Se calcula que cerca de 10% dos pacientes curados da Doença de Hodgkin desenvolvam uma segunda neoplasia em 10 anos, sendo bastante freqüente as leucemis agudas.
4. Campos elétricos e magnéticos: Os campos elétricos e magnéticos produzidos por um pequeno eletrodoméstico nem de perto produz o campo de energia da RI. Entretanto, numerosos experimentos demonstram que estes campos promovem a variação de importantes enzimas intra-celulares. Estudos epidemiológicos sugerem que estes campos possam aumentar o risco de neoplasias, em geral, ou leucemias, em particular.
5. Virus: Existem duas classes de retrovírus leucemogênicos: a classe aguda que causam leucemias e sarcomas em animais que contenham oncogenes codificantes e a classe de retrovírus leucemogênicos "crônicos" cujos principais representantes são os do grupo HTLV (HTLV1/2, HIV). Outro vírus que está associado aos linfomas e, eventualmente, a alguns tipo de leucemia é o Epstein Baar vírus (EBV). Classicamente, este vírus está associado à Doença de Hodgkin e ao linfoma de Burkitt.
LEUCEMIAS MIELÓIDES
As leucemias mielóides são neoplasias do sistema hematopoiéticos, com produção anormal de células, maduras ou imaturas, da linhagem mielóide: granulocítica, monocitária, plaquetária e eritrocitária. A origem da neoplasia pode ser a de células progenitoras comissionadas ou pluripotentes, isto é, ainda não orientadas para a linhagem mielóide. Pode apresentar um curso clínico agudo, sub-agudo ou crônico que correspondem, não apenas à duração de instalação e progressão da doença mas, principalmente, as características biológicas diversas. Desse modo, na doença crônica o clone patológico diferencia e amadurece em seus elementos "normais" determinando uma excessiva produção de granulócitos imaturos e maduros (leucemia mielóde crônica), megacariócitos e plaquetas (trombocitemia primária) e, de eritroblastos e eritrócitos (policitemia vera). Na doença aguda, entretanto, a maturação das células é abortiva e a doença é caracterizada por um acúmulo de precursores ou "blastos" (mieloblastos, monoblastos, megacarioblastos e pro-eritroblatos). Quando o número de blastos é menos acentuado, tanto no sangue periférico quanto na medula óssea, podemos estar diante de um grupo de doenças incluídas na síndrome mielodisplásica. A Tabela 1 Apresenta o esquema das diversas leucemias mielóides ou mielo-proliferativas. A Tabela 2 apresenta as principais características biológicas das leucemias mielóides e síndromes mielo-proliferativas. Neste manual abordaremos apenas as doenças mais freqüentes dentro de cada grupo de leucemias e linfomas.

Tabela 1: Esquema de classificação das leucemia mielóides ou síndromes mielo-proliferativas
aguda sub-aguda crônicaLMA SMD LMC
M0 (sem diferenciação) Anemia refratária mielofibroseM1 (sem maturação) ARSA trombocitemia primáriaM2 (com maturação) AR com excesso de blastos Policitemia veraM3 (promielocítica) LMMC
M4 (mielo-monocítica)M5 (monoblástica)M6 ( eritroblástica)
M7 (megacarioblástica)LMA; leucemia mielóide aguda; SMD: síndrome mielodisplásica; LMC: leucemia mielóide crônica; ARSA: anemia refratária com sideroblastos em anel; LMMC: leucemia mielo-monocítica crônica.
Tabela 2 : Principais características biológicas e patogenéticas das leucemias mielóides e síndromes mieloproliferativas
agudas sub agudas crônicasproliferação incontrolável,
autônoma e com pouca diferenciação
parcialmente controlável e com
diferenciação parcial
parcialmente controlada e com
diferenciaçãomaturação muito reduzida ou
ausentereduzida e ineficaz normal
acúmulo de blastos elevado modesto irrelevante (na fase estável)
produção de elementos maduros
fortemente defeituosa
defeituosa aumentada
clínica anemia, infecções, hemorragias
anemia, mais raramente infecções
e hemorragia
hepato e esplenomegalia e
aumento de glóbulos vermelhos e/ou
brancos e/ou plaquetas
LEUCEMIA MIELÓIDE AGUDA
Epidemiologia: As LMA ocorrem em qualquer idade, mas a sua freqüência aumenta de maneira considerável com a idade. A idade mediana de acometimento está entre 50 e 60 anos. Porém, a distribuição e freqüência varia nas diversas áreas geográficas e populações, tanto devido a fatores genéticos como ambientais. É importante recordar que a LMA se divide, sob os planos biológicos, clínico e prognóstico, em três grandes categorias: LMA "primária" ou "de novo" , que são pacientes onde não se pode demonstrar exposição a agentes leucemogênicos ou doenças precedentes; LMA

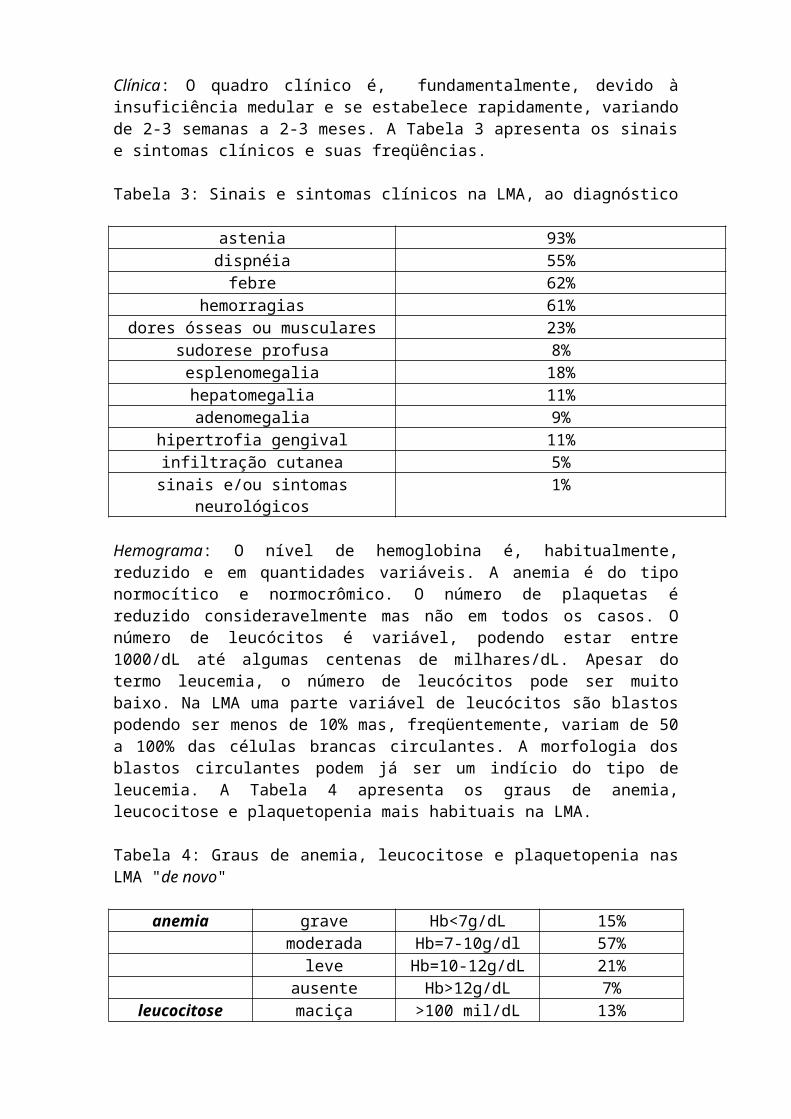
"secundárias à agentes leucemogênicos", particularmente em pacientes tratados com quimio e/ou radioterapia e; LMA "secundária à mielodisplasia". Fisiopatologia: O quadro clínico da LMA depende da: 1- produção defeituosa e insuficiente de células sangüíneas maduras; 2- infiltração dos tecidos e órgãos não hematopoiéticos pelas células leucêmicas e; 3- liberação, em parte pelas células leucêmicas e em parte pelo sistema imunológico, de mediadores químicos (citoquinas) que podem provocar febre, dor, perda de peso, sudorese, etc. Clínica: O quadro clínico é, fundamentalmente, devido à insuficiência medular e se estabelece rapidamente, variando de 2-3 semanas a 2-3 meses. A Tabela 3 apresenta os sinais e sintomas clínicos e suas freqüências.
Tabela 3: Sinais e sintomas clínicos na LMA, ao diagnóstico
astenia 93%dispnéia 55%
febre 62%hemorragias 61%
dores ósseas ou musculares 23%sudorese profusa 8%esplenomegalia 18%hepatomegalia 11%adenomegalia 9%
hipertrofia gengival 11%infiltração cutanea 5%
sinais e/ou sintomas neurológicos 1% Hemograma: O nível de hemoglobina é, habitualmente, reduzido e em quantidades variáveis. A anemia é do tipo normocítico e normocrômico. O número de plaquetas é reduzido consideravelmente mas não em todos os casos. O número de leucócitos é variável, podendo estar entre 1000/dL até algumas centenas de milhares/dL. Apesar do termo leucemia, o número de leucócitos pode ser muito baixo. Na LMA uma parte variável de leucócitos são blastos podendo ser menos de 10% mas, freqüentemente, variam de 50 a 100% das células brancas circulantes. A morfologia dos blastos circulantes podem já ser um indício do tipo de leucemia. A Tabela 4 apresenta os graus de anemia, leucocitose e plaquetopenia mais habituais na LMA.
Tabela 4: Graus de anemia, leucocitose e plaquetopenia nas LMA "de novo"
anemia grave Hb<7g/dL 15%moderada Hb=7-10g/dl 57%
leve Hb=10-12g/dL 21%ausente Hb>12g/dL 7%
leucocitose maciça >100 mil/dL 13%marcante 50-100 mil/dL 17%moderada 10-50 mil/dL 29%ausente <10 mil/dL 41%
plaquetopenia grave <20 mil/dL 23%moderada 20-50 mil/dL 38%
leve 50-100 mil/dL 22%ausente >100 mil/dL 17%

Mielograma: O material é sempre obtido pela aspiração com agulha e, mais raramente, com a biópsia de medula óssea, quando o aspirado é pobre ou a punção é seca. A celularidade é quase sempre aumentada e há o desaparecimento do tecido adiposo. A infiltração blástica deve ser de, no mínimo, 20% para se confirmar o diagnóstico de LMA ou LLA. A classificação mais utilizada no mundo para diferenciação dos diversos tipos de leucemias agudas (LMA nos seus oito tipos; M0 a M7 e LLA nos seus três tipos; L1 a L3), é aquela proposta pelo grupo franco-americano-britânico (FAB). A Tabela 1 apresenta os diversos tipos de LMA na classificação FAB.
Citogenética: Na maior parte das LMA se encontram anormalidades citogenéticas, algumas delas características e com importância prognóstica. As t(8;21) na forma M2, t(15;17) na M3, e inversão do cromossomo 16 na M4 com eosinofilia são as anormalidades associadas ao melhor prognóstico, dentro de terapêuticas modernas e específicas, como no caso da LMA-M3 (promielocítica). Outras anormalidades, particularmente as mais complexas, estão associadas a pior prognóstico. A citogenética tem adquirido, nas últimas décadas, importância prognóstica fundamental, sendo um exame absolutamente necessário ao diagnóstico para a melhor escolha da terapêutica seja quimioterápica ou transplante de medula óssea.
Biologia Molecular: Em muitas leucemias caracterizadas por translocações cromossômicas específicas, é possível, através de técnicas de PCR a amplificação da seqüência de DNA ou RNA quimérico associado ao tumor. A mais relevante destas anormalidades é o característico transcrito quimérico neoplásico PML-RARα, associado à LMA-M3 isto é, a promielocítica. Porém, dezenas de oncogenes estão associados aos diversos tipos de leucemias, tanto agudas como crônicas.
Curso clínico e prognóstico: A probabilidade de cura em longo prazo é da ordem de 25%, com a utilização de terapêutica convencional e em leucemias "de novo". Porém, o prognóstico é fortemente influenciado pela idade do paciente. Quanto mais idoso o paciente, menor a tolerância às complicações da fase de insuficiência medular e a capacidade do organismo de tolerar a terapêutica anti-leucêmica, tanto a quimioterapia quanto o transplante de medula óssea. Além disso, a LMA do idoso é intrinsecamente mais resistente e tem características clínicas e biológicas mais próximas às leucemias secundárias. Fatores biológicos tais como a "massa" tumoral, tipo citológico, imunofenótipo, alterações cariotípicas, dentre outros, podem alterar negativamente a evolução do paciente. A Tabela 5 apresenta alguns dos fatores que podem influir negativamente no prognóstico.
Tabela 5: LMA - características que influenciam negativamente na evolução da LMA.
idade avançadacomprometimento ou insuficiência orgânica fígado, rins, coração, SNC, pulmões,
anormalidades metabólicasmassa tumoral hiperleucocitose
tipo citológico pela "FAB" M5; M6; M7 e M0imunofenótipo leucemia bifenotípica
cariótipo t(9;22) (Ph1+), t(4;11), alterações cromossômicas complexas
Terapêutica: A terapêutica da LMA se baseia na administração de agentes citostáticos com a finalidade de reduzir de maneira substancial (ou mesmo erradicar) a população de
células leucêmicas, permitindo que a célula progenitora normal residual repovoe normalmente a medula óssea. Os tratamentos "standards" são baseados em dois fármacos, que podem ser usados em dose variável, que são a Citarabina (Ara-C) ou os antraciclínicos (daunomicina, idarrubicina, mitoxantrone etc). Os esquemas clássicos são compostos de uma terapêutica de indução da remissão e de dois a quatro ciclos de intensificação ou consolidação. Outras drogas podem ser usadas nesta fase tais como a ciclofosfamida, a 6-tioguanina e o VP-16-213. Nesta fase, 15-20% são primariamente resistentes, 15-20% morrem de complicações, antes de serem avaliados quanto a resposta e, em torno de 60% obtém remissão hematológica. No final desta primeira fase, excluindo-se os pacientes resistentes ou falecidos pela toxicidade da terapia, várias modalidades de abordagem complementar podem ser propostas: nenhum tratamento, continuação da poli-quimioterapia, transplante autólogo de medula óssea e transplante alogênico de medula óssea. A Tabela 6 apresenta os esquemas e opções principais para a abordagem complementar das LMA, após terapêutica de indução.
Tabela 6: opções para a abordagem pós indução da LMA
opção terapêutica sucessiva
aplicação custo % de sobrevivência em 5 anos
nenhuma todos nenhum 10-20%poli-quimioterapia 80% redução da
qualidade de vida e toxicidade
10-30%
transplante autólogo 30-40% mortalidade de 5-10%
30-50%
transplante alogênico
20-25% mortalidade de 10-30%
30-60%
Terapêutica de suporte: A terapêutica de suporte é fundamental para proteger e tratar os pacientes da anemia, infecções e hemorragias. Quanto mais intensa for a terapêutica, incluindo o transplante de medula óssea, mais a terapêutica de suporte deve ser sofisticada e intensa. As transfusões de hemácias, plaquetas e os antibióticas adequados e modernos, são fundamentais à manutenção da vida do paciente nas semanas ou até meses que transcorrerão de falência da medula óssea, até a sua completa recuperação.
LEUCEMIA MIELÓDE CRÔNICA
A leucemia mielóde crônica (LMC) é uma síndrome mielo-proliferativa que apresenta um característico curso clínico bifásico. Esta doença começa como uma doença indolente (fase crônica) caracterizada por uma expansão clonal hematopoiética, mas ainda capaz de completar o processo de maturação de todos os setores do sangue e cuja duração pode ser de anos. Após esta fase ocorre, irremediavelmente, uma verdadeira leucemia aguda, dita "crise blástica", precedida de um curto período de instabilidade hematológica denominada fase acelerada. A hematopoiese clonal da LMC é identificada por um marcador citogenético denominado cromossomo Filadélfia (Ph), gerado pela translocação recíproca dos braços longos dos cromossomos 9 e 22. Esta translocação cria um gene de fusão denominado bcr/abl que exerce uma atividade enzimática proliferativa de tirosino-quinase com ação proliferativa sobre as células hematopoiéticas.

Epidemiologia: A LMC é uma doença relativamente rara, com uma incidência média de 10 casos/milhão de habitantes/ano; discretamente mais freqüente no sexo masculino, rara na criança e com um pico de incidência aos 50 anos.
Clínica: Os sintomas da doença são modestos e inespecíficos e podem ser divididos em dois grupos: a) dependentes da expansão da massa granulocítica e plaquetária, que aumenta o volume do baço que pode comprimir e deslocar órgãos vizinhos causando tensão, dor no hipocôndrio esquerdo, sensação de plenitude pós prandial e, mais raramente, pode levar a pequenos infartos sub capsulares do baço. A leucocitose e plaquetose podem levar a infartos retinianos e do corpo cavernoso do pênis; b) sintomas dependentes da anemia como cansaço, fadiga, perda de peso, febre ou febrícula, dores ósseas ou musculares e sudorese profusa. Todos estes sintomas dependem da massa tumoral, apesar de não haver uma relação linear entre eles. Cerca de 30% dos casos são diagnosticados por acaso em exames hematológicos de rotina. A Tabela 7 apresenta a freqüência de alguns sinais clínicos e dados hematimétricos da LMC.
Laboratório: O hemograma e a análise morfológica do sangue periférico são fundamentais para a definição diagnóstica. O hemograma demonstra leucocitose, desde poucos milhares de leucócitos até centenas de milhares, com contagem diferencial característica, com presença de um número crescente de células a partir das mais jovens (mieloblastos) até os granulócitos. Há, em geral, um número maior de mielócitos que pode variar de 5 a 20%, eosinofilia (1-5%) e basofilia (1-5%). A anemia e a plaquetose ocorre em, aproximadamente, 1/3 dos casos, ao diagnóstico. A Tabela 7 apresenta um sumário dos achados laboratoriais.
Curso clínico e prognóstico:
A evolução natural da LMC ou o tratamento com quimioterapia paliativa (bussulfano ou hidroxiuréia) apresentam um tempo mediano de sobrevivência de 4 a 5 anos. Raros são os pacientes que tem sobrevida superior a 10 anos (<10%) apenas com este enfoque. Muitos elementos clínicos e hematológicos apresentam valor prognóstico, mas bastariam poucos fatores, muitos simples, para calcular o risco e, portanto a expectativa de vida. Assim, a idade, tamanho do baço, número de plaquetas e % de blastos no sangue periférico determinam o índice de Sokal, muito usado em todo o mundo. A única possibilidade terapêutica curativa, no estado da arte atual, é o transplante alogênico de medula óssea. Porém, esta modalidade terapêutica carrega uma alta mortalidade e esbarra na indisponibilidade de doadores para o seu uso mais difuso. A primeira droga que foi capaz de modificar a história natural desta doença em um número significativo de pacientes foi o interferon-α que elevou a mediana de sobrevivência para 6 anos com, aproximadamente, 20-30% de pacientes com normalização do cariótipo obtido pela técnica de citogenética convencional. A partir de 2000, uma nova e importante droga foi introduzida, inicialmente para o tratamento da fase acelerada e crise blástica, e logo após, na grande maioria dos países do mundo, como primeira linha. O Mesilato de Imatinibe (Glivec®) é um inibidor específico do sítio de ação do gene de fusão bcr-abl impedindo sua atividade como tirosino-quinase e, assim, reduz ou impede a atividade proliferativa sobre o tecido mielóide. As respostas hematológica e citogenética são muito rápidas e um percentual não desprezível de pacientes obtém remissão molecular. Apesar disto, algumas importantes dúvidas ainda permanecem quanto ao potencial de cura desta droga mas, ceratmente, estamos diante de um avanço que só será completamente entendido no futuro. Neste capítulo, não abordaremos as outras síndromes mielo-proliferativas (mielofibrose primária, policitemia vera e trombocitemia hemorrágica).

Tabela 7: freqüência de sinais e sintomas clínicos e dados laboratoriais mais relevantes na LMC.
sintomas %ausentes 36presentes 64astenia 46
sintomas abdominais 28perda de peso 26
febre 17dores ósseas ou musculares 9
sudorese 8baço -
não palpável 22<6cm do rebordo costal 38>6cm do rebordo costal 40
fígado -não palpável 32
<6cm do rebordo costal 61>6cm de rebordo costal 7
leucocitose -<20000/dL 3
20-100000/dL 41>100000/dL 56plaquetas -
<150000/dL 9150-500000/dL 60
>500000/dL 31hemoglobina -
<9g/dL 89-11.5g/dL 33>11.5g/dL 59
SÍNDROMES LINFOPROLIFERATIVOS
As síndromes linfoproliferativas incluem todas as doenças neoplásicas dos linfócitos, tanto com expressão leucêmica, linfomatosa (Doença de Hodgkin e linfomas não Hodgkin) e gamopatias monoclonais. As doenças leucêmicas são sistêmicas e são, fundamentalmente, do sangue e medula óssea, enquanto os linfomas acometem prevalentemente os órgãos linfóides periféricos e podem, eventualmente, permanecer confinados aos linfonodos. Neste capítulo deixaremos de lado as gamopatias monoclonais e trataremos das mais freqüentes doenças leucêmicas e linfomatosas. A Tabela 8 apresenta as mais importantes síndromes linfoproliferativas

Tabela 8: divisão clínica das principais síndromes linfoproliferativas.
leucemias agudas leucemia linfóide aguda crônicas leucemia linfóide crônica
leucemia prolinfocítica crônicatrico-leucemia
linfomas doença de Hodgkin linfomas não Hodgkin
LEUCEMIA LINFÓIDE AGUDA
As leucemias linfóides agudas (LLA) se originam de precursores dos linfócitos B ou T ou mesmo de células progenitoras pluripotenciais. A tranformação leucêmica gera um precursor de blasto linfóide (linfoblasto) que substitui a medula hematopoiética, circulam no sangue periférico e infiltram órgãos e tecidos linfóides tais como linfonodos, fígado, sistema nervoso central, testículo, ossos, etc). A LLA é mais freqüente nas crianças e é, sem duvida, a neoplasia mais freqüente no grupo pediátrico.
Fisiopatologia e clínica: como na LMA as bases fisiopatológicas da LLA são:a) supressão da hematopoiese normal e conseqüente anemia, neutropenia e plaquetopenia; b) infiltração de órgãos linfóides e não linfóides com conseqüente síndrome de massa tumoral, principalmente com hepato e esplenomegalia, infiltração múltipla e simétrica de linfonodos e síndrome mediastínica; c) liberação de linfoquinas e mediadores de inflamação causando febre, dores ósseas, musculares e articulares e sudorese profusa. O diagnóstico morfológico é feito através da classificação FAB, com três tipos: L1- pequeno blasto homogêneo, mais freqüente na infância; L2 - blasto heterogêneo, mais freqüente no adulto e L3 - blasto hiper vacuolizado. Dentro de uma rotina mais especializada destas leucemias, deve haver uma melhor identificação das células através de imunofenotipagem. Métodos citogenéticos e moleculares podem ser agregados aos recursos morfológicos no sentido de uma melhor caracterização da LLA e seu prognóstico. Decurso clínico, prognóstico e terapêutica: Alguns dados, ao diagnóstico, têm evidenciado influência negativa ao prognóstico, habitualmente bom, da LLA. A hiper leucocitose (>50000/dL), presença do cromossomo Ph1, imunofenótipo B maduro e leucemia bifenotípica, apresentam mau prognóstico. Além disso, a forma pediátrica tem melhor evolução que a forma do adulto. A Tabela 9 apresenta as principais diferenças entre a LLA da criança e do adulto.
Tabela 9: comparação das LLA da criança e do adulto
criança adultoremissão completa 95% 80%
longa sobrevivência >60% 20%tipo citológico L1 L2
imunofenótipo prevalente Pré-B-comum nenhumimunofenótipo híbrido raros freqüente
cromossomo Ph1 Raro freqüente

O tratamento da LLA é feito em três fases: 1- indução da remissão, onde as drogas fundamentais são a vincristina, daunomicina e prednisona; 2- profilaxia do SNC com quimioterapia intratecal e/ou radioterapia craniana e; 3- intensificação e manutenção da remissão com poli quimioterapia com múltiplas drogas tais como methotrexate, ciclofosfamida, citarabina, dentre outras.
LEUCEMIA LINFÓIDE CRÔNICA
A LLC é uma neoplasia hematológica caracterizada, em 98% dos casos, da proliferação e acúmulo no sangue, medula óssea e tecidos linfáticos de pequenos linfócitos, aparentemente maduros e de derivação B, e sem agentes etiológicos aparentemente envolvidos.
Patogênese: Na maioria das LLC se observa um progressivo acúmulo de linfócitos leucêmicos aprisionados nas fases G0 e G1 de proliferação, funcionalmente incompetentes, com prolongada sobrevivência de até vários anos. Este fato se deve a inibição da morte programada das células (apoptose) devido a uma aumentada expressão do gene bcl-2, devido a hipometilação do DNA.
Epidemiologia: A LLC é muito freqüente em indivíduos de raça branca e é responsável de cerca de 25% de todas as leucemias. Nos indivíduos de raça amarela é quase desconhecida representando menos de 2.5% das leucemias. É uma doença de paciente adultos, com idade mediana de 63 anos, sendo rara entre 30 e 40 anos e quase inexistente abaixo dos 30 anos de idade.
Clínica: Tanto a apresentação como o curso clínico da LLC, são bastante variáveis. É importante dizer desde logo que 25 a 30% dos casos de LLC não apresentam qualquer sinal ou sintoma clínico e o diagnóstico é feito a partir de exames de rotina. Também em um número pequeno, pode haver "falência" parcial da medula óssea com anemia e/ou plaquetopenia. Estas duas anormalidades podem ser de causa auto-imune. Outros sinais e sintomas são mal-estar, cansaço, sudorese noturna e febre. É característico da maioria dos quadros iniciais a linfonodomegalia superficial, não confluentes ou fistulados e presença de hepato e/ou esplenomegalia.
Laboratório: O hemograma , a biópsia de medula óssea e o estudo imunofenotípico do sangue periférico e da medula óssea definem o diagnóstico. Os dados indispensáveis para o diagnóstico são linfocitose periférica superior a 5000/dL e infiltração medular superior a 30% sempre por pequenos linfócitos. Do ponto de vista da imunofenotipagem, os linfócitos da LLC expressam CD5 e antígenos da linhagem B tais como CD19, CD20, CD21 e CD23.
Classificação: As classificações mais aceitas são as classificações de Rai e Binet, que levam em conta dados de diagnóstico, infiltração visceral e grau de falência da medula óssea. A Tabela 10 apresenta a classificação original de Rai e a Tabela 11 as sub divisões por risco segundo o National Cancer Institute (NCI) - Sponsored Working Group para LLC.

Tabela 10: Estadiamento segundo Rai
estádio 0 linfocitose no sangue >5000/dL e na medula óssea >30%
estádio I estádio 0 + linfonodomegaliaestádio II estádio 0 + esplenomegalia +/-
hepatomegaliaestádio III estádio 0 + anemia (Hb<11.0g/dL)estádio IV estádio 0 + plaquetopenia <100000/dL
Tabela 11: Grupos prognósticos segundo o NCI
grupo prognóstico estádiobaixo risco 0
risco intermediário I e IIalto risco III e IV
Curso clínico: A LLC é uma doença caracterizada pelo acúmulo progressivo da população neoplasica linfóide, com o evento habitual de progressão do estádio mais baixo em direção ao mais alto. Cerca de 50% dos pacientes inicialmente no estádio 0 progridem para outros estádios e outros permanecem por longos períodos estáveis. Pacientes que progridem devem ser tratados a luz dos tratamentos mais eficientes, mas um percentual expressivo de pacientes podem não tratar ou permanecer longos períodos sem tratamento e com a doença estável. Outras complicações possíveis são a anemia hemolítica auto-imune e o desenvolvimento de segunda neoplasia, mais freqüente do que na população da mesma idade. Ao contrário do LMC onde a crise blástica é inexorável, a síndrome de Richter ("crise blástica da LLC") é um evento raro (<2%).
Tratamento: Para uma abordagem adequada terapêutica da LLC é necessário ter em conta parâmetros fundamentais que são: idade, estádio clínico e atividade da doença, que pode ser indolente ou agressiva. Nos pacientes com doença limitada ou indolente, sobretudo em idosos, é hábito não tratar até que haja uma progressão evidente. Em geral o objetivo é conter o avanço da doença e isto pode se obter com drogas alquilantes e, mais modernamente, com análogos da purina principalmente a Fludarabina. Por outro lado, pacientes jovens e com doença agressiva e avançada, o tratamento deve ser sempre instituído e, raramente, pode ser considerada a terapia de alta dose (transplante de medula óssea). Eventualmente tratamentos paliativos como radioterapia e esplenectomia podem ser indicados.
LINFOMAS
DOENÇA DE HODGKIN
Há mais de 170 anos, Thomas Hodgkin descreveu pela primeira vez esta entidade mas apenas há cerca de 30 anos é que se pode entender sua natureza, biologia, modo de progressão e abordagem terapêutica.

Etiopatogenia e modo de progressão:
A etiologia da DH não é completamente conhecida. Há um componente familiar em raros casos e a vinculação da doença com a infecção pelo vírus de Epstein Baar pode ser demonstrada em muitos casos, particularmente em áreas endêmicas, tais como a África e mesmo no Brasil. O início da doença é, quase sempre, em linfonodos periféricos; as áreas mais atingidas, por continuidade, são aquelas no curso do ducto torácico, e as vias de disseminação são a linfática e, apenas tardiamente, a hemática.
Clínica: A DH pode acometer todas as idades, mas a freqüência máxima ocorre entre 20 e 30 anos. Os casos em crianças são mais comuns na África e em outros países do terceiro mundo e está, provavelmente, associados à infecção pelo vírus EB. A doença tem maior freqüência no sexo masculino, na proporção de 2:1, aproximadamente. Aproximadamente 50% dos pacientes não apresentam sinais ou sintomas constituicionais ao diagnóstico, e a doença é suspeitada através do crescimento de linfonodos periféricos ou presença de massa mediastinal ou retro-peritonial. Na quase totalidade dos casos tem crescimento lento, e apresenta uma tendência de coalisão e discreta aderência a planos superficiais e profundos, sem anormalidades da pele ou compressão de raízes nervosas. As localizações extra-nodais mais freqüentes são as pulmonares, principalmente nos pacientes com doença com grande massa do mediastino (>10cm), e hepática, principalmente nos que apresentam doença esplênica. O acometimento ósseo pode ser por continuidade do linfonodo ou, o que é mais comum, através da disseminação hematogênica.
Estadiamento: Em 1971 em Ann Arbor foi proposta uma classificação, válida até o momento, que estabeleceu alguns critérios para medir a DH. Em 1989 foi proposto alguns avanços ao estadiamento original (Cotswold) acrescentando o conceito de "grande massa" (bulky disease) e acometimento extra-nodal. A Tabela 12 apresenta a classificação por estádios de Ann Arbor.
Tabela 12: Classificação por estádios - Ann Arbor
estádio I acometimento de um linfonodo, uma cadeia ou uma região linfonodal ou e de um sítio extra-nodal
estádio II acometimento de duas ou mais cadeias de linfonodos localizadas acima ou abaixo do diafragma, com eventual extensão a um órgão extra-nodal contíguo IIe, lesão do baço se acrescenta a letra S
estádio III acometimento de duas ou mais cadeias de linfonodos localizadas acima e abaixo do diafragma, com eventual extensão a um órgão extra-nodal contíguo IIIe, lesão do baço se acrescenta a letra S.
estádio IV Acometimento difuso de um ou mais órgãos extra-linfáticos (vísceras) com ou sem acometimento linfonodal.

Pacientes com sinais ou sintomas constitucionais, isto é, febre, emagrecimento e sudorese noturna, recebem ainda a letra B ao lado do estádio; pacientes sem estes sintomas recebem a letra A. Na realidade brasileira, infelizmente, a maioria dos pacientes é atendida nos Centros especializados e já estão em estádios avançados, isto é, III e IV, e, grande parte deles, apresenta sinais e sintomas constitucionais. O estadiamento é realizado a partir de investigações clínica, propedêutica, radiológica, tomográfica, biópsia de medula óssea e, se necessário, por métodos cintilográficos (gálio ou PET scan), ressonância eletro-magnética, ecografia ou biópsias de outras localizações. O chamado estadiamento cirúrgico com a realização da laparotomia exploradora, muito utilizado na década de 70, hoje está, praticamente, abandonado.
Laboratório: Dentre os exames laboratoriais que se pode fazer, alguns simples como a velocidade de hemossedimentação (VHS), o hemograma e os enzimas hepáticos são de fácil execução e podem dar informações relevantes. Assim, a VHS está freqüentemente elevada nos pacientes que apresentem os sinais e sintomas B. O hemograma é freqüentemente alterado com presença de anemia, em geral normocítica e levemente hipocrômica; leucocitose com neutrofilia podendo haver eosinofilia, e é muito importante a presença de linfopenia. A elevação das enzimas hepáticos pode ser um sinal de acometimento do fígado e/ou do esqueleto.
Histologia: O diagnóstico da DH não pode prescindir de uma biópsia de linfonodo, de preferência removido "inteiro", ou através de biópsia por agulha quando o acesso é difícil como nas trans torácicas ou obtidas por mediastinoscopia ou pequena toracotomia. Os critérios diagnósticos são aqueles estabelecidos na Conferência de Rye em 1966, onde se indica, para o diagnóstico, a obrigatoriedade do encontro da célula de Reed-Sternberg. A partir do encontro desta célula, se divide a DH em quatro tipos histológicos: predominância linfocitária, variantes difusa e nodular; esclerose nodular, tipos I e II; celularidade mista e depleção linfocitária. Não se pode determinar com certeza, até hoje, que os tipos histológicos tenham algum papel no prognóstico da DH. Parece que apenas o tipo de depleção linfocitária, responsável por menos de 5% dos casos de DH, possam ter importância prognóstica. O tipo histológico mais freqüente no primeiro mundo e, provavelmente também no Brasil, é o de esclerose nodular.
Curso clínico e prognóstico: O melhor conhecimento da biologia , a introdução do estadiamento e os avanços na poli quimioterapia e radioterapia, transformaram radicalmente o prognóstico da DH. A sobrevivência nesta doença, após 15 anos, é de 80% sendo que 60% permanecem livres de doença. A Tabela 13 apresenta os faores que isoladamente podem ter impacto prognóstico desfavorável.
Tabela 13: fatores prognósticos desfavoráveis
estádio III e IVsintomas e sinais B
histologia depleção linfocitáriaidade > 60 anos
massa tumoral grande mediastino (bulky), múltiplo acometimento do baço, mais que 4 sítios
nodais acometidos

Tratamento: A DH é, habitualmente, radio- e quimio-sensível. O tratamento da DH é baseado nestas duas armas terapêuticas, isoladas ou associadas. Além disso, pacientes quimio-resistentes ou recidivados, podem ser tratados, como terapia de salvamento com o transplante de medula óssea, principalmente o denominado transplante autólogo. Novas modalidades terapêuticas como os novos tipos de transplante e os anticorpos monoclonais, tais como o anti-CD30, estão em fase de desenvolvimento e experimentação clínica.
LINFOMAS NÃO HODGKIN
Os linfomas não Hodgkin (LNH) são processos neoplásicos complexos e muito heterogêneos de doenças que tendem a reproduzir as características morfológicas, fenotípicas, genotípicas e ainda, uma ou mais etapas dos processos de maturação e transformação dos elementos linfóides. Os linfócitos envolvidos nas doenças podem ser do tipo B ou T em diversas fases de diferenciação. A complexidade deste grupo de doenças se reflete, até hoje, na grande dificuldade de se estabelecer um consenso mundial sobre as entidades existentes e mesmo sobre sua classificação. Apenas na década de 90, após grandes e frustadas tentativas de consenso, é que 19 grupos de hemopatologistas de todo o mundo desenharam a chamada classificação "REAL" (revised european american lymphoma), base para a atual classificação da Organização Mundial de Saúde (OMS). Na verdade, tanto a classificação "REAL" como a "OMS" não são propriamente classificações, mas elencos de doenças com características clínico e patológicas aceitas pelos especialistas em clínica onco-hematológica e hemopatologia. Tendo em vista a extrema heterogeneidade deste grupo de doenças, alguns pressupostos devem ser respeitados tais como: a classificação deve ser histo-geneticamente correta, com capacidade de distingir caracteres morfológicos, clínicos, fenotípicos e moleculares; não pode excluir a Doença de Hodgkin, hoje aceita como um linfoma de derivação B; distingir as forma B e T, tendo em vista suas diferenças epidemiológicas, sintomatológicas, morfológicas e prognósticas; reconhecer entidades (nodais ou extra-nodais) que se originem de órgãos periféricos ao sistema imunológicos; de cada entidade listar as características morfológicas, clínicas, fenotípicas e moleculares; dar idéia do grau de agressividade de cada processo que pode ser influenciado pela cinética tumoral, apoptose, moléculas envolvidas nos mecanismos de resistência etc.; revisar e incluir entidades provisórias. É importante colocar a visão do clínico que deseja reunir várias entidades em grupos baseados, principalmente, na história natural das doenças. Deste modo, podemos dividir os LNH em indolentes, moderadamente agressivos, agressivos e muito agressivos. Neste campo há um aparente paradoxo pois apenas os linfomas agressivos, e raros casos de indolentes são potencialmente curáveis no estado-da-arte atual. A Tabela 14 apresenta a classificação "REAL" assumida pela OMS como a classificação atual de consenso.
Incidência, sexo e idade: Os LNH representam, nos países ocidentais de 3-5% de todos os tumores malígnos e são cerca de 10-15/100000 casos/ano. Porém, a partir de 1970 a incidência dos LNH vem crescendo cerca de 4% acima do crescimento populacional e já representa uma preocupação em termos de saúde pública pelo seu comportamento epidemiológico. Os LNH são raros em idades inferiores a 5 anos e tem sua mediana, levando-se em conta todos os pacientes, de 50 anos. Os homens tem predominância sobre as mulheres. Os chamados linfomas "indolentes" predominam nos idosos (média entre 55 e 60 anos) ao passo que os agressivos predominam nas 3a e 4a décadas.

Tabela 14: classificação REAL para os linfomas malígnos:
NEOPLASIA DE DERIVAÇÃO B
Neoplasia de precursores Bleucemia/linfoma linfoblástico
Neoplasia das células B periféricasLLC/ leucemia prólinfocítica/LNH pequenos linfócitoslinfoma linfo-plasmocítico/imunocitoma/linfoma da zona do mantoLNH centro-foliculares, foliculares (graus I a III)LNH da zona marginal (MALT e monocitóide B)leucemia de células filamentosasplasmocitoma/mieloma múltiplolinfoma difuso de grande célula B (inclui o B de mediastino)linfoma de Burkittlinfoma B de grande célula "Burkitt-like"
NEOPLASIA DE DERIVAÇÃO T/NK
Neoplasia de precursores de célula Tleucemia/linfoma T linfoblástico
Neoplasia das céluals T/NK periféricasLLC/leucemia prolinfocíticaleucemia a grande célula granular micose fungóide/síndrome de Sézarylinfoma T periféricoLNH gama-delta hepato-esplênicoLNH sub cutâneo TLNH angioimunoblásticoLNH angiocêntricoLNH intestinal de célula Tleucemia/linfoma T do adultolinfoma a grande célula T anaplásico, CD30+ (T e null)linfoma a grande célula anaplásico, "Hodgkin's like"
DOENÇA DE HODGKIN (já descrito anteriormente)
Etiopatogenia: Na maioria dos casos de LNH os agentes etiológicos são desconhecidos. Apenas em alguns tipos de LNH pode ser vinculada a etiologia a fatores virais tais como os vírus EBV, HTLVI/II e HIV.

Acometimento e disseminação: A disseminação dos LNH é muito menos previsível que a da DH e estas doenças apresentam um caráter, habitualmente, muito mais sistêmico ao diagnóstico. De fato, a maior parte dos casos de LNH surgem já com múltiplas localizações nodais, acometimento do baço, medula óssea dentre outras sedes extra-nodais. Esta disseminação precoce pode ocorrer em diversos tipos de LNH, mas é muito característico dos linfomas indolentes ou nos linfomas linfoblástico e Burkitt. Neste grupo de doenças também se encontram os linfomas associados às mucosas, cuja tendência é de apresentarem um comportamento mais localizado. São os denominados linfomas tipo MALT.
Clínica: Tomando todos os tipos de LNH, é muito comum que a doença seja oligossintomática ou mesmo assintomática ao diagnóstico. Os chamados sinais e sintomas B (febre, prurido, sudorese e perda de peso), ocorrem apenas em cerca de 20% dos casos e, principalmente, nas formas agressivas. A clínica mais habitual é de linfonodomegalia, podendo haver localizações extra-nodais em 20 a 30% dos casos. Os LNH podem cursar com localizações extra-nodais, muito raras na DH, tais como trato gastro-intestinal, pele, sistema nervoso central, testículo, ossos, glândulas salivares e lacrimais, miocárdio e pericárdio. Podem ainda acometer o fígado e os pulmões. Os LNH podem disseminar-se rapidamente por via hematogênica e o acometimento da medula óssea, com ou sem síndrome leucêmica, está presente em 10 a 30% das séries analisadas. Para a determinação do estádio da doença se utiliza os critérios estabelecidos em Ann Arbor, descrito no capítulo da DH.
Laboratório: O exame hematológico é com freqüência, pouco expressivo. Pode haver anemia, leve leucocitose, ou mesmo leucopenia e linfopenia. Podem ocorrer em diversos tipos histológicos o encontro de células atípicas próprias dos linfomas de base, com típica alteração leucêmica. Os parâmetros biológicos presentes ao diagnósticos podem ser a elevação da desidrogenase lática (LDH) e ß2-microglobulina. Alguns métodos histológicos para avaliar o grau de proliferação podem ser empregados tais como o anticorpo anti Ki-67, PCNA, AgNors etc. Estes marcadores, em alguns estudos demonstram estarem associados ao prognóstico dos LNH, mas não são práticos para uso universal. O estadiamento, a semelhança da Doença de Hodgkin, é realizado a partir de investigação clínico-propedêutica, radiológica, tomográfica, biópsia de medula óssea e, se necessário, por métodos cintilográficos (gálio ou PET scan), ressonância eletro-magnética, ecografia ou biópsias de outras localizações.
Prognóstico: Nos LNH existem fatores prognósticos tradicionais, isto é, os que avaliam a extensão da doença, idade, "performance status", estádio avançado, número de sítios extra-nodais, acometimento do mediastino com grande massa, e a obtenção da remissão completa após a primeira linha de tratamento. Os parâmetros biológicos relevantes são a LDH e ß2-microglobulinas. Nos últimos anos estudos cromossômicos e moleculares têm agregado conhecimento biológico neste campo; algumas destas anormalidades estão associadas a determinados tipos de LNH e algumas com valor prognóstico e de monitoramento terapêutico. Estes imunofenotípicos também podem auxiliar na caracterização da doença e prognóstico, como a aplicação da expressão do gene de resistência a múltiplas drogas e moléculas solúveis. No momento, utilizando terapêutica quimioterápica convencional, 40-50% dos linfomas agressivos são curados e 50 a 60% dos linfomas indolentes têm longa sobrevivência (>10 anos).
Tratamento: Os objetivos gerais do tratamento dos LNH são essencialmente dois: curativo, nos chamados linfomas agressivos ou muito agressivos e paliativo, isto é, conter a doença e manter a boa qualidade de vida, principalmente nos chamados

linfomas indolentes. Para tanto, um arsenal complexo de terapêuticas isoladas ou combinadas podem ser utilizadas dentre de protocolos específicos. As modalidades terapêuticas mais importantes são: radioterapia, em campos envolvidos ou estendidos; quimioterapia, mono- ou poli-quimioterapia; terapias de alta dose denominadas transplantes de medula óssea, autólogo ou alogênico; terapia imunológica com anticorpos monoclonais (hoje disponíveis o anti-CD20 (Mabthera®) e anti-CD52 (Campath®), dentre muitos em desenvolvimento) e cirurgia que, no contexto dos linfomas, é muito utilizada para a definição diagnóstica, sendo ainda uma importante arma na definição do estadiamento. Como já foi dito anteriormente, os LNH são complexos e com dezenas de entidades clínicas que nós não abordaremos em detalhes neste capítulo. Porém, é importante ressaltar que, cada vez mais, a abordagem deste grupo é feita através de protocolos clínicos bem estruturados e que respeitem critérios diagnósticos, prognósticos, e utilização hierárquica das diversas modalidades terapêuticas, oferecendo aos pacientes as melhores oportunidades no sentido da cura e do controle da neoplasia.
BIBLIOGRAFIA
1. Hoffman R: Hematology. Basic principles and practice, Churchill Livingstone, 2nd
edition, New York - USA, 1995 2. Cheson BD et al. National Cancer Institute - Sponsored Working Group Guidelines
for Chronic lymphocitic Leukemia: Revised Guidelines for diagnosis and treatment. Blood, 1996, 87:4990.
3. De Souza CA, Vigorito AC, Ruiz MA, et al: Validation of the EBMT risk score in chronic myeloid leukemia in Brazil and allogeneic transplant outcome. Haematologica (2005);90(2)-232-237.
4. Bailliere's Clinical Haematology. Acute lymphoblastic leukemia. Baillière Tindall (London) 1994, 7:2.
5. Lister TA et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds Meeting. J Clin Oncol 1989, 7:1630.
6. Harris NL et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994,84:1361.
7. Shipp M et al. A predictive model for aggressive non Hodgkin's lymphoma. N Engl J Med , 1993, 329:987.
8. De Vita VT Jr et al. Hodgkin's disease. N Engl J Med, 1993, 326:678.9. Tura ST. Lezioni di Ematologia. Società Editrice Esculapio s.r.l. - Bologna, 1997 10. Hoffbrand AV and Pettit. Essential Haematology. Blackwell Scientific publications,
3rd edition, Oxford - UK, 1994.11. Thomas ED, Blume KG and Forman SJ. Hematopoietic cell transplantation.
Blackwell Scientific publications, 2nd edition, Malden, Massachusetts - USA, 1999.12. Wintrobe MM et al. Clinical Hematology. Williams and Wilkins , 10th edition,
Baltimore, Mariland - USA, 1999.13. Willians WJ et al. Hematology. McGraw-Hill Book Company, 6th edition, New
York - USA, 2001.14. Zago MA, Falcão RP, Pasquini R. Hematologia: fundamentos e prática. Livraria
Atheneu - São Paulo - Brasil, 2001.