joÃo paulo de abreu cruz validaÇÃo de procedimento de...
TRANSCRIPT

1
UNIVERSIDADE DE SÃO PAULO
ESCOLA DE ENGENHARIA DE LORENA - EEL/USP
JOÃO PAULO DE ABREU CRUZ
VALIDAÇÃO DE PROCEDIMENTO DE LIMPEZA DE EQUIPAMENTOS MULTIUSOS
NA INDÚSTRIA FARMACÊUTICA.
Declaro que esta monografia foi revisada e encontra-se apta para avaliação e apresentação
perante banca avaliadora.
Data:____/____/2014.
_________________________________________ Orientador: Prof. Prof. MSc. Marcos Villela Barcza
Lorena - SP 2014

2
JOÃO PAULO DE ABREU CRUZ
Validação de procedimento de limpeza de equipamentos multiusos na indústria farmacêutica.
Trabalho de conclusão de curso apresentado à Escola de Engenharia de Lorena da Universidade de São Paulo como requisito parcial para obtenção do título de Engenheiro Industrial Químico.
Área de Concentração: Qualidade e Produtividade. Orientador: Prof. MSc. Marcos Villela Barcza
Lorena - SP 2014

3
Dedicatória
Aos meus pais, Ildeu Cruz e Cassia Regina, e irmãos, Pedro e Mariana, pelo exemplo de vida,
pelos ensinamentos, pela humildade e paciência. Muito obrigado pelo carinho, amor e
companheirismo.
À minha namorada Gislaine, por sempre estar ao meu lado me aconselhando e dando força
para enfrentar o dia da melhor forma possível. Muito obrigado pelo carinho.

4
AGRADECIMENTOS
Ao meu primo, Augusto (in memorian), por me mostrar que a dificuldade é superada com um
sorriso no rosto e que devemos lutar sempre. Obrigado por cada momento ao seu lado,
saudades.
Ao meu avô, João (in memorian), por me mostrar o que significa humildade e bondade.
Obrigado por tudo que passamos juntos.
À minha sobrinha, Maria Clara, por alegrar o meu dia cada vez que a vejo.
Aos companheiros de trabalho, Luiz Daniel, Simão Campos, Anderson Cosmo, Iara Roma,
Juliana Silva, Odair Cardoso, Luiz Fernando, Fabiola Moreno e Darcio Santos, pelo apoio,
ensinamentos e paciência durante minha primeira experiência profissional.
Aos meus amigos de república, Breno Sartori, Pedro Bonfim, Rafael Aquiyama, Murillo
Negreiros, Milton Guimarães, Matheus Fescina, Luis Eduardo, Jackson Araujo, Jhunu,
Araújo, Leonardo Boaretto e Marcelo Rais, por cada dia vivido nesta jornada acadêmica,
fazendo da república uma segunda família.
Aos amigos de infância, Pedro Ourique, Renan Rizzi, João Campos, Vitor Azevedo e Adriano
Longhi, que mesmo longe, me motivaram e acompanharam durante esta caminhada.
Ao Professor MSc. Marcos Villela Barcza, pela sua atenção e disponibilidade demonstrada
durante a orientação deste trabalho.
A todos os amigos e familiares que me incentivaram, inspiraram e fizeram com que eu
concluísse esta jornada.
.

5
“A vontade de se preparar deve ser maior do que a vontade de vencer. Vencer será
consequência da boa preparação”.
Bernardinho

6
RESUMO
CRUZ, J. P. A. Validação de procedimento de limpeza de equipamentos multiusos na
indústria farmacêutica. 2014. Monografia - Escola de Engenharia de Lorena, Universidade de
São Paulo, Lorena, 2014.
Este trabalho apresenta uma estratégia utilizada para a execução de uma validação de limpeza,
aplicando-se os conceitos de boas práticas de fabricação, matriz de pior caso, cálculos de RCL
(Residual Cleaning Limit), técnicas de amostragem e validação de metodologias de
quantificação de resíduo. Os dois produtos desafiados na validação de limpeza são de forma
farmacêutica líquida, constituídos por API’s (Active Pharmaceutical Ingredients) diferentes,
os quais são utilizados no combate de parasiticidas, tais como, carrapatos, bernes e sarnas. O
equipamento avaliado foi uma envasadora de líquidos, composta por tubulações, válvulas,
tanque reservatório e bicos dosadores, que apresentam superfícies de contato com os
produtos. Com os conceitos apresentados neste trabalho, foi possível evidenciar, de forma
documental, que um determinado procedimento de limpeza é robusto, atendendo ao
requerimento exigido pelo MAPA e garantindo a qualidade do produto a ser produzido após a
execução do processo de limpeza.
Palavras-chave: Validação. Matriz. Resíduo. Amostragem. Qualidade.

7
LISTA DE ILUSTRAÇÕES
Figura 1 – Fluxograma geral de Validação de Limpeza. .......................................................... 19
Figura 2 – Fluxograma da área de envase ................................................................................ 35
Figura 3 – Ponto 1 de amostragem (superfície interna do fundo do reservatório) ................... 40
Figura 4 – Ponto 2 de amostragem (superfície interna do tampo do reservatório da
envasadora) ............................................................................................................................... 41
Figura 5 – Ponto 3 de amostragem (superfície interna da parede do reservatório da
envasadora) ............................................................................................................................... 41
Figura 6 – Ponto 4 de amostragem (superfície interna da tubulação de chegada de produto ao
reservatório) .............................................................................................................................. 42
Figura 7 – Ponto 5 de amostragem (superfície interna do manifold do reservatório) .............. 43
Figura 8 – Ponto 6 de amostragem (superfície interna do pistão) ............................................ 43
Figura 9 – Ponto 7 de amostragem (superfície interna da vareta de transferência de produto) 44
Figura 10 – Ponto 8 de amostragem (superfície interna do bico dosador de produto) ............. 45

8
LISTA DE TABELAS
Tabela 1 – Descrição dos termos de solubilidade..................................................................... 22
Tabela 2 – Descrição dos termos de toxicidade. ...................................................................... 22
Tabela 3 – Classificação segundo dose terapêutica .................................................................. 23
Tabela 4 – Vantagens e desvantagens de cada método de amostragem. .................................. 26
Tabela 5 – Tabela de pontuação para identificação do pior caso ............................................. 31
Tabela 6 – Tabela de criticidade de solubilidade do resíduo no solvente de limpeza .............. 32
Tabela 7 – Tabela de criticidade de toxicidade do resíduo....................................................... 32
Tabela 8 – Tabela de criticidade de facilidade operacional de limpeza ................................... 33
Tabela 9 – Tabela de identificação do pior caso ...................................................................... 49
Tabela 10 – Dados para o cálculo do RCL ............................................................................... 52
Tabela 11 – Resultados da inspeção visual após primeira execução do procedimento de
limpeza ..................................................................................................................................... 53
Tabela 12 – Resultados de resíduo de Deltametrina após primeira execução do procedimento
de limpeza ................................................................................................................................. 53
Tabela 13 – Resultados da inspeção visual após segunda execução do procedimento de
limpeza ..................................................................................................................................... 54
Tabela 14 – Resultados de resíduo de Deltametrina após segunda execução do procedimento
de limpeza ................................................................................................................................. 54
Tabela 15 – Resultados da inspeção visual após terceira execução do procedimento de
limpeza ..................................................................................................................................... 55
Tabela 16 – Resultados de resíduo de Deltametrina após terceira execução do procedimento
de limpeza ................................................................................................................................. 56
Tabela 17 – Resultado geral de inspeção visual ....................................................................... 56
Tabela 18 – Resultado geral de quantificação de resíduo ........................................................ 57

9
LISTA DE ABREVIATURAS E SIGLAS
ANVISA Agência Nacional de Vigilância Sanitária
API Active Pharmaceutical Ingredients
BPF Boas Práticas de Fabricação
BPFM Boas Práticas de Fabricação de Medicamentos
CIP Clean In Place
COP Clean Out Place
HPLC High Performance Liquid Cromatography
MAPA Ministério da Agricultura, Pecuária e Abastecimento
NOEL Nível de Efeito Não Observado
OMS Organização Mundial da Saúde
PCP Planejamento e Controle de Produção
RDC Resolução de Diretoria Colegiada
RCL Residual Cleaning Limit
USP United States Pharmacopeial
WHO World Health Organization

10
SUMÁRIO
1. INTRODUÇÃO ................................................................................................................. 13
1.1 Contextualização ......................................................................................................... 13
1.2 Justificativa .................................................................................................................. 14
1.3 Objetivos ..................................................................................................................... 14
1.3.1 Objetivo Geral .......................................................................................................... 14
1.3.2 Objetivos Específicos ............................................................................................... 14
2. REVISÃO BIBLIOGRÁFICA .......................................................................................... 16
2.1 Boas Práticas de Fabricação ........................................................................................ 16
2.2 Validação e Qualificação ............................................................................................. 16
2.3 Validações de Limpeza ............................................................................................... 17
2.4 Elaboração de Procedimentos de Limpeza .................................................................. 20
2.5 Matriz de Pior Caso ..................................................................................................... 21
2.5.1 Solubilidade do resíduo no solvente de limpeza .................................................... 22
2.5.2 Facilidade operacional de limpeza ......................................................................... 22
2.5.4 Toxicidade do resíduo ............................................................................................ 22
2.5.4 Dose terapêutica ..................................................................................................... 23
2.6 Determinação dos Limites de Aceitação ................................................................ 23
2.7 Técnicas de Amostragem ....................................................................................... 25
2.7.1 Amostragem direta (esfregaço pelo swab) ............................................................... 25
2.7.2 Amostragem indireta (rinsagem) .............................................................................. 26
2.7.3 Principais diferenças entre a amostragem direta e indireta ...................................... 26
2.8 Inspeção Visual ...................................................................................................... 27
3. METODOLOGIA ............................................................................................................. 28
3.1 Escopo .................................................................................................................... 28
3.2 Definições e abreviações ........................................................................................ 29
3.3 Produtos desafiados no estudo de validação de limpeza ........................................ 30

11
3.4 Equipamentos envolvidos na limpeza .................................................................... 30
3.5 Matriz de pior caso ................................................................................................. 30
3.5.1 Solubilidade do resíduo no solvente de limpeza .................................................... 31
3.5.2 Toxicidade do ativo ................................................................................................ 32
3.5.3 Facilidade de Limpeza ........................................................................................... 33
3.6 Cálculo do RCL ...................................................................................................... 33
3.7 Estratégia de Validação .......................................................................................... 34
3.7.1 Procedimento de limpeza detalhado (limpeza completa e limpeza simples) ......... 34
3.7.1.1 Procedimento de limpeza completa .................................................................... 35
3.7.1.2 Procedimento de limpeza simples ...................................................................... 37
3.7.2 Tipo de amostragem ............................................................................................... 39
3.7.2.1 Ponto 1: superfície interna do fundo do reservatório ......................................... 39
3.7.2.2 Ponto 2: superfície interna do tampo do reservatório da envasadora ................. 40
3.7.2.3 Ponto 3: superfície interna da parede do reservatório da envasadora................. 41
3.7.2.4 Ponto 4: superfície interna da tubulação de chegada de produto ao reservatório
42
3.7.2.5 Ponto 5: superfície interna do manifold do reservatório .................................... 42
3.7.2.6 Ponto 6: superfície interna do pistão .................................................................. 43
3.7.2.7 Ponto 7: superfície interna da vareta de transferência de produto ...................... 44
3.7.2.8 Ponto 8: superfície interna do bico dosador de produto ..................................... 44
3.7.3 Critério de aceitação ............................................................................................... 45
3.7.3.1 Inspeção Visual .................................................................................................. 45
3.7.3.2 Método de detecção de resíduo .......................................................................... 46
3.7.3.3 Análises microbiológicas .................................................................................... 46
3.7.4 Clean equipment hold time ..................................................................................... 47
3.7.5 Dirty equipment hold time ...................................................................................... 47
4. RESULTADO E DISCUSSÃO ......................................................................................... 48

12
4.3.2 Resultados obtidos após primeira execução do procedimento de limpeza ................ 52
4.3.3 Resultados obtidos após segunda execução do procedimento de limpeza ................ 54
4.3.3 Resultados obtidos após terceira execução do procedimento de limpeza ................. 55
5.3.3 Discussão dos resultados ........................................................................................... 56
5. CONCLUSÃO E SUGESTÕES ........................................................................................... 58
5.1 Conclusão ...................................................................................................................... 58
5.2 Sugestões ....................................................................................................................... 58
REFERÊNCIAS ....................................................................................................................... 59

13
1. INTRODUÇÃO
1.1 Contextualização
Nos dias de hoje, os assuntos qualidade e produtividade caminham juntos, visto que
para buscar espaço no mercado é necessário produzir produtos com menor custo e qualidade
garantida. Para a produção de medicamentos de uso veterinário isso não é diferente e para
garantir a qualidade e melhorar processos produtivos, as empresas farmacêuticas vêm
utilizando conceitos de validação de processos de produção, validação de métodos analíticos,
validação de sistemas computadorizados e validação de procedimentos de limpeza. Em
termos gerais, validação é ato de comprovar, de forma documentada, que qualquer processo,
procedimento ou método é robusto e reproduz os resultados esperados (WHO, 2006).
O objetivo das boas práticas de fabricação inclui a prevenção de contaminação cruzada
entre matérias-primas e produtos farmacêuticos. Portanto a validação de limpeza é
fundamental para evidenciar, de forma documentada, que o procedimento utilizado na
execução de uma limpeza consegue remover os resíduos a níveis pré-determinados de
aceitação, e para isso leva em consideração o tamanho de lote, dosagem, toxicidade e área de
equipamento em contato com o produto (WHO, 2006). Além disso, a validação de limpeza é
um requisito exigido pela Instrução Normativa 13, de 03 de outubro de 2003 do MAPA para
as indústrias farmacêuticas de saúde animal (MAPA,2003).
Para um estudo de validação de limpeza existem diferentes caminhos a serem
seguidos, porém, o caminho escolhido deve definir critérios, parâmetros e metodologias que
são cientificamente justificáveis e que garantam que o procedimento de limpeza atende as
especificações pré-estabelecidas (ANVISA, 2013).
A indústria farmacêutica tem a possibilidade de utilizar um mesmo equipamento, ou
linha de produção, para a fabricação de medicamentos diferentes. Porém, se o número de
produtos que compartilhar o mesmo equipamento for muito elevado, irá resultar em diversas
situações de validação, o que seria inviável se tivesse que validar todas as possibilidades de
combinações entre produto contaminante (produzido antes da execução da limpeza) e produto
subsequente (produzido após execução da limpeza). Assim, um estudo de escolha de um pior
caso pode ser realizado a fim de viabilizar o estudo de validação de limpeza (ANVISA, 2006).
Para definição do pior caso, devem-se considerar todos os possíveis produtos
contaminantes e produtos subsequentes em um equipamento / linha de produção de uso
compartilhado. Além disso, as seguintes informações devem ser conhecidas:

14
• Área do equipamento / linha de produção em contato com o produto;
• Solvente utilizado na execução da limpeza e a solubilidade do mesmo com
resíduo a ser removido;
• Dificuldade de limpeza para cada produto em contato com equipamento.
• O menor valor de RCL obtido a partir dos métodos de cálculo que levam em
consideração: tamanho de lote, dose terapêutica diária, fator de segurança, dose
terapêutica máxima e toxicidade ;
Após avaliação de todos estes fatores, é possível definir o pior caso considerando-se o
menor RCL encontrado. Assim, o estudo de validação de limpeza tem como base desafiar a
pior situação encontrada.
1.2 Justificativa
As indústrias farmacêuticas de saúde animal estão cada vez mais preocupadas com a
proteção dos seus consumidores e pacientes, portanto há uma busca pela ciência para animais
mais saudáveis. Para isso, toda empresa deve alinhar custo com qualidade a fim de atender
adequadamente seu cliente. Para garantir a qualidade dos produtos, um dos itens obrigatórios
é a validação dos procedimentos de limpeza, portanto este trabalho apresenta a execução de
um estudo de validação de limpeza de uma linha de envase, a qual é utilizada para a produção
de dois diferentes produtos antiparasiticidas.
1.3 Objetivos
1.3.1 Objetivo Geral
O objetivo principal deste trabalho foi validar um procedimento de limpeza de equipamentos
multiusos na indústria farmacêutica.
1.3.2 Objetivos Específicos
Para realização do objetivo principal, os seguintes objetivos específicos foram
determinados:
• Definir, através de uma matriz, o pior produto a ser desafiado durante a validação
de limpeza.
• Definir, através de cálculos mais usuais, o RCL para o produto pior caso.
• Definir a técnica de amostragem a ser utilizada na validação de limpeza.

15
• Definir os locais de amostragem da linha de envase.
• Validar efetivamente o procedimento de limpeza da linha de envase.
• Definir tempo de espera de equipamento limpo e tempo de espera de equipamento
sujo.

16
2. REVISÃO BIBLIOGRÁFICA
2.1 Boas Práticas de Fabricação
A Resolução de Diretoria Colegiada (RDC) nº 17, de 16 de abril de 2010, é a
legislação vigente que direciona as Boas Práticas de Fabricação de Medicamentos (BPFM).
Esta resolução substitui a RDC nº 210, de 04 de agosto de 2003 e a Portaria SVS / MS nº 500,
de 9 de outubro de 1997 (DUTRA, 2011).
A empresa farmacêutica deve seguir as BPFM a fim de diminuir os riscos que podem
estar presentes na fabricação de medicamentos. Os riscos podem ocorrer de diferentes formas,
seja ela por contaminação cruzada, contaminação por partículas, troca ou mistura de produto,
mas de qualquer forma todos os riscos devem ser controlados. Para isso seguem alguns
requerimentos que devem ser cumpridos pela indústria farmacêutica (ANIVSA, 2010):
• Pessoal deve ser devidamente qualificado e treinado;
• Instalações e espaços devem ser adequados e estar identificados;
• Procedimentos e instruções devem estar aprovados e vigentes;
• Armazenamento e transporte devem ser adequados;
• Instalações, equipamentos e pessoal qualificado para controle em processo.
A qualificação de equipamentos, qualificação de fornecedores, validação de limpeza,
validação de métodos analíticos, validação de sistemas computadorizados e validação de
processos são requerimentos da RDC nº 17, de 16 de abril de 2010, e estas atividades
auxiliam no controle dos riscos inerentes à produção de medicamentos (RDC17/10, 2010).
2.2 Validação e Qualificação
Validação é ato que comprova, de forma documental, que um determinado processo,
procedimento ou método é robusto e reprodutível, ou seja, leva aos resultados esperados
(WHO, 2006).
Qualificação e Validação são termos com o mesmo significado, porém o termo
“qualificação” é utilizado para equipamentos, sistemas e utilidades, e o termo “validação” é
utilizado para processos, sistemas computadorizados e metodologias analíticas. Porém há a
possibilidade de encontrar o termo “validação” com o sentido de “qualificação” (ANVISA,
2006).

17
A qualificação e validação devem evidenciar que instalações, utilidades, sistemas
computadorizados, equipamentos e processos foram projetados de acordo com as exigências
de BPF (qualificação de projeto), foram instalados conforme projeto (qualificação de
instalação), operam conforme especificações requeridas (qualificação de operação) e
apresentam um desempenho de processo robusto e reprodutível (qualificação de desempenho)
(ANVISA, 2010). Este estudo é realizado quando há novas instalações, novos equipamentos,
novas utilidades ou novos processos; ou quando há mudanças significativas em equipamentos,
metodologias e processos; ou quando a validade (periodicidade) de validação / qualificação
está expirada (WHO, 2006).
Estudos de qualificação e validação devem ser realizados de acordo com um protocolo
aprovado. No protocolo deve contemplar o objetivo do estudo, o local onde será realizado, a
responsabilidade de cada pessoa ou departamento envolvido, a descrição do procedimento a
ser seguido, o equipamento que será desafiado, os padrões e critérios relevantes ao processo
ou produto, o tipo de qualificação / validação e os testes, amostragens e monitoramentos
realizados durante o estudo. Após a execução do protocolo de qualificação / validação deve-se
analisar os resultados encontrados e elaborar um relatório contemplando o título e objetivo do
estudo realizado; detalhes do material, equipamento, processo, procedimentos e métodos
utilizados. Os resultados encontrados durante o estudo devem ser avaliados, analisados e
comparados com os critérios de aceitação pré-determinados. Com todos os resultados
avaliados e analisados deve-se concluir o estudo e demonstrar o status de qualificação /
validação (“Qualificado” / “Validado” ou “Não Qualificado” / “Não Validado”) (WHO,
2006).
2.3 Validações de Limpeza
Validação de limpeza é uma evidência documentada de que um procedimento de
limpeza consegue remover resíduos a níveis pré-determinados, levando em consideração
alguns fatores, tais como: tamanho de lote, dosagem do produto, toxicologia e tamanho do
equipamento (WHO, 2006).
Para um estudo de validação de limpeza existem diferentes caminhos a serem
seguidos, porém, o caminho escolhido deve definir critérios, parâmetros e metodologias que
são cientificamente justificáveis e que garantam que o procedimento de limpeza atende as
especificações pré-estabelecidas (ANVISA, 2013).

18
A indústria farmacêutica tem a possibilidade de utilizar um mesmo equipamento, ou
linha de produção, para a fabricação de medicamentos diferentes. Porém, se o número de
produtos que compartilhar o mesmo equipamento for muito elevado, irá resultar em diversas
situações de validação, o que seria inviável se tivesse que validar todas as possibilidades de
combinações entre produto contaminante (produzido antes da execução da limpeza) e produto
subsequente (produzido após execução da limpeza). Assim, um estudo de escolha de um pior
caso pode ser realizado a fim de viabilizar o estudo de validação de limpeza (ANVISA, 2006).
O estudo de validação de limpeza deve abordar e definir os seguintes assuntos
(ANVISA, 2013):
• Através de cálculos apropriados deve-se definir o critério de aceitação para cada
parâmetro desafiado;
• Elaboração dos procedimentos de limpeza abordando tipo de equipamento,
características do produto (solubilidade, toxicidade, etc) e agente de limpeza;
• Método analítico utilizado para análise de resíduo;
• Elaboração do protocolo de validação de limpeza;
• Plano de testes;
• Elaboração do relatório de validação.
Após três execuções do protocolo de validação, ou seja, após três execuções do
procedimento de limpeza, deve-se elaborar um relatório de validação de limpeza levando em
consideração os resultados encontrados com sua respectiva evidência (dados brutos); a
comparação dos critérios de aceitação pré-determinados com os respectivos resultados
encontrados e avaliação e justificativa de qualquer desvio, caso ocorra. Após conclusão do
estudo validação, qualquer alteração que venha ocorrer no procedimento de limpeza, métodos
analíticos de análise de resíduo, equipamentos, agentes de limpeza e processo de fabricação,
deve ser controlada e se esta mudança for crítica, deve-se revalidar o procedimento de
limpeza (ANVISA, 2006).
O fluxograma a seguir resume as etapas de validação de limpeza conforme figura 1
(NASSANI, 2005):

19
Figura 1 – Fluxograma geral de Validação de Limpeza.
Fonte: NASSANI, 2005.

20
2.4 Elaboração de Procedimentos de Limpeza
Em um estudo de validação de limpeza a primeira análise que deve ser realizada é do
procedimento de limpeza que será desafiado. Muitas empresas elaboram complexos sistemas
de detecção de resíduos e elaborados planos de amostragem sem antes analisar o
procedimento de limpeza em questão, isso pode ocasionar em estratégias inadequadas para o
estudo a ser realizado, resultando em uma perda de tempo e um esforço desnecessário
(ANVISA, 2006).
Antes de iniciar o estudo de validação de limpeza os seguintes itens devem ser
verificados (ANVISA, 2006):
• Deve-se verificar se há algum procedimento escrito e aprovado, com o registro de
treinamento do pessoal responsável por executá-lo;
• Deve-se verificar se no procedimento escrito está detalhando os pontos críticos do
equipamento e a maneira que se deve limpá-los;
• Caso a limpeza seja realizada manualmente, deve-se detalhar o tempo de limpeza,
a quantidade de solvente utilizado, o tipo de detergente necessário e o método de
limpeza empregado.
• Deve-se verificar se o material utilizado na execução da limpeza está padronizado
e se a preparação do detergente está detalhada, contemplando o tipo e a
concentração do detergente utilizado. Após o término da validação a
concentração, composição e marca do detergente utilizado não pode ser alterado,
caso isso ocorra um novo estudo de validação de limpeza deve ser realizado ou
uma justificativa técnica plausível deve ser registrada;
• O procedimento de limpeza deve contemplar o tempo de equipamento sujo, ou
seja, o tempo que o equipamento pode ficar sujo antes da execução do
procedimento de limpeza. Isso se faz necessário, pois o tempo de equipamento
sujo é inversamente proporcional à facilidade de limpeza, ou seja, quanto mais
tempo o equipamento ficar sujo, mais difícil de limpar.
• O procedimento de limpeza deve contemplar o tempo de equipamento limpo, ou
seja, o tempo que o equipamento pode ficar limpo até sua utilização. Isso se faz
necessário, pois quanto mais tempo o equipamento fica exposto ao ambiente, mais
suscetível ele fica à proliferação de micro-organismos.

21
• O procedimento de limpeza deve contemplar o tempo máximo que um
equipamento pode ser utilizado em uma campanha. Muitas vezes, há apenas
limpezas parciais quando lotes estão sendo produzidos em campanha.
Na indústria farmacêutica há diversas técnicas de limpeza, dentre elas: limpeza
manual, CIP (Clean in Place), COP (Clean Out Place), semi - automática e automática. Para
todos os tipos de limpeza deve-se levar em consideração o tempo de limpeza, o número de
ciclos de limpeza, etc (ANVISA, 2013).
2.5 Matriz de Pior Caso
A indústria farmacêutica tem a possibilidade de utilizar um mesmo equipamento, ou
linha de produção, para a fabricação de medicamentos diferentes. Porém, se o número de
produtos que compartilhar o mesmo equipamento for muito elevado, irá resultar em diversas
situações de validação, o que seria inviável se tivesse que validar todas as possibilidades de
combinações entre produto contaminante (produto produzido antes da execução da limpeza) e
produto subsequente (produto produzido após execução da limpeza). Assim, um estudo de
escolha de um pior caso pode ser realizado a fim de viabilizar o estudo de validação de
limpeza (ANVISA, 2006).
O pior caso é definido a partir de uma situação, às vezes subjetiva, em que uma linha
de produção é submetida a um cenário em que há um maior esforço para realização da
limpeza. Para definir um pior caso deve-se considerar o produto contaminante (produto
produzido antes da execução da limpeza) e o produto subsequente (produto que será
produzido após execução da limpeza e que pode ser contaminado pelo produto contaminante)
(ANVISA, 2006).
Para definição do pior caso para o produto contaminante deve-se levar em
consideração a solubilidade do resíduo no solvente de limpeza (quanto menos solúvel mais
difícil de limpar), a facilidade operacional de limpeza (a experiência do operador deve ser
levada em consideração para definição do produto mais difícil de limpar), a toxicidade do
resíduo (quanto mais tóxico pior será o cenário), e dose terapêutica (quanto menor a dose
terapêutica pior será o cenário) (ANVISA, 2006).

22
2.5.1 Solubilidade do resíduo no solvente de limpeza
A tabela 1 indica os termos utilizados para solubilidade de um determinado soluto em
um solvente de interesse.
Tabela 1 – Descrição dos termos de solubilidade
Descrição Quantidade aproximada de volume por 1
parte de peso do soluto
Muito Solúvel Menos do que 1 parte
Altamente Solúvel De 1 a 10 partes
Solúvel De 10 a 30 partes
Moderadamente Solúvel De 30 a 100 partes
Levemente Solúvel De 100 a 1000 partes
Muito levemente Solúvel De 1000 a 10000 partes
Praticamente Insolúvel / Insolúvel 10000 partes ou maior
Fonte: (USP 37, 2014)
2.5.2 Facilidade operacional de limpeza
O critério de facilidade operacional de limpeza também é utilizado para definição de
um pior caso dentro de uma matriz de produtos. Esta avaliação pode ser realizada em forma
de entrevistas com os operadores ou supervisores da área de produção. Por se tratar de uma
informação subjetiva não há justificativa científica para este critério (ANVISA,2013).
2.5.4 Toxicidade do resíduo
A tabela 2 indica os termos utilizados para toxicidade de resíduos levando em
consideração a dose letal para humanos.
Tabela 2 – Descrição dos termos de toxicidade.
Descrição Provável dose oral letal para humanos
(mg / Kg)
Praticamente não tóxico 15000
Levemente tóxico 5000 - 15000
Moderadamente tóxico 500 - 5000
Muito tóxico 50 – 500
Extremamente tóxico 5 - 50

23
Descrição Provável dose oral letal para humanos
(mg / Kg)
Super tóxico < 5
Fonte: ANVISA, 2013
2.5.4 Dose terapêutica
As doses terapêuticas geralmente são avaliadas em uma matriz de pior caso quando a
administração do medicamento em questão é de via oral ou parenteral (ANVISA,2013). A
tabela 3 indica os termos utilizados para doses terapêuticas.
Tabela 3 – Classificação segundo dose terapêutica
Menor dose terapêutica (mg)
>1000 mg
100 – 1000 mg
10 – 99 mg
1 – 9 mg
< 1 mg
Fonte: ANVISA, 2013
Neste tipo de avaliação, quanto menor a dose terapêutica, pior será a situação.
2.6 Determinação dos Limites de Aceitação Existem diferentes métodos de determinação dos limites de aceitação em um estudo de
validação de limpeza. Os métodos mais utilizados na determinação do limite de aceitação são
(ANVISA, 2006):
• 0,1 % da dose limite: determinação do limite de aceitação no produto
subsequente.
A determinação do RCL a partir do cálculo da dose terapêutica diária é realizada
através da fórmula descrita a seguir:
RCL�µg/cm² = 0,01 × MínimaDoseTerapêuticaDiáriaA�mg × 1000 × MenortamanhodelotedoProdutoB�mL
MáximaDoseDiáriaB�mL × ÁreadeEquipamentoemcontatocomoproduto�cm&
0,01 – fator de segurança para medicamento de uso tópico.
1000 – fator de conversão de mg para µg.

24
Mínima Dose Terapêutica Diária A (mg) – Mínima dose diária do produto
contaminante.
Menor tamanho de Lote do Produto B (mL) – É o menor granel (lote) do produto
subsequente.
Máxima Dose Diária B (mL) – É a quantidade total do produto subsequente que o
paciente pode receber.
Área de Equipamento em contato com o produto (cm²) – Área de equipamento
não dedicado em contato com o produto.
A – Produto Contaminante.
B – Produto Subsequente.
• Nível de Efeito Não Observado (NOEL): para determinação do NOEL a seguinte
fórmula deve ser utilizada:
NOEL�mg = dL50 × MassaMédiadoPaciente
2000
NOEL (mg) – Nível de Efeito não observado.
dL50 (mg / Kg) – A dose letal é uma indicação da letalidade de uma dada
substância. Dado que a resistência muda de indivíduo para indivíduo, a dose letal
representa uma dose (normalmente medida em miligramas de substância por
quilograma de massa corporal do indivíduo testado) capaz de matar uma dada
50% dos indivíduos de uma população em teste.
Massa Média do Paciente – Massa média do paciente que irá receber este tipo de
medicamento.
2000 – Constante Empírica.
A determinação do RCL a partir do cálculo do NOEL é realizada através da
fórmula descrita a seguir:
RCL�µg/cm² = NOELA�mg × 1000 × MenortamanhodelotedoProdutoB�mL
MáximaDoseDiáriaB�mL × ÁreadeEquipamentoemcontatocomoproduto�cm&
1000 – fator de conversão de mg para µg.
NOEL A (mg) – Nível de Efeito não observado do produto contaminante.

25
Menor tamanho de Lote do Produto B (mL) – É o menor granel (lote) do produto
subsequente.
Máxima Dose Diária B (mL) – É a quantidade total do produto subsequente que o
paciente pode receber em 24 horas.
Área de Equipamento em contato com o produto (cm²) – Área de equipamento
não dedicado em contato com o produto.
A – Produto Contaminante.
B – Produto Subsequente.
• 10 ppm: neste tipo de cálculo admite-se a concentração de 10 ppm do produto
contaminante no produto subsequente.
Para o cálculo do RCL deve-se utilizar a fórmula descrita a seguir:
RCL�µg/cm² =10�µg/mL × MenortamanhodelotedoProdutoB�mL
ÁreadeEquipamentoemcontatocomoproduto�cm2
Este tipo de cálculo só é utilizado se o método de dose limite e o método baseado
no NOEL apresentarem um RCL maior do que o calculado por este método. Não
há uma explicação científica para usá-lo, ou seja, a lógica da sua utilização foge
dos padrões BPF (ANVISA, 2006).
2.7 Técnicas de Amostragem
Um estudo de validação de limpeza deve ter justificativas técnicas para adotar um
determinado plano de amostragem. Os métodos mais utilizados para se realizar a amostragem
são a amostragem direta (esfregaço pelo swab) e a amostragem indireta (rinsagem) (ANVISA,
2010).
2.7.1 Amostragem direta (esfregaço pelo swab)
A amostragem direta, realizada pelo esfregaço do swab, é realizada em um local
específico do equipamento, ou seja, não representa toda a superfície do equipamento. Para
este tipo de amostragem geralmente amostra-se uma área pequena do equipamento,
utilizando-se um material e um método pré-definido (tipo de swab, solvente utilizado e

26
técnica de amostragem). A realização deste método permite a amostragem de resíduos
insolúveis devido à ação física da técnica (ANVISA, 2010).
2.7.2 Amostragem indireta (rinsagem)
Diferente do método de amostragem direta, este tipo de amostragem cobre toda
superfície do equipamento, incluindo as áreas que o swab não consegue amostrar. É
importante assegurar que o solvente utilizado na realização da amostragem consegue
recuperar quantidades apropriadas de resíduo. Além disso, o volume de solvente utilizado na
rinsagem do equipamento deve ser adequado uma vez que é de conhecimento que grandes
quantidades de solvente pode interferir e levar a conclusões errôneas em relação à quantidade
de resíduo no equipamento (ANVISA, 2010).
2.7.3 Principais diferenças entre a amostragem direta e indireta
A tabela 4 mostra as vantagens e desvantagens de cada método (ANVISA, 2006):
Tabela 4 – Vantagens e desvantagens de cada método de amostragem.
Método Vantagens Desvantagens
Amostragem direta da superfície (swab)
• Resíduos secos e insolúveis podem ser removidos.
• Permite o estabelecimento do nível de contaminação por área, estabelecendo onde o procedimento deve ser melhorado.
• Permite recuperação do contaminante a partir de áreas onde a água de rinsagem teve contato deficiente.
• A área amostrada deve permitir livre acesso ao operador, o que é impraticável em muitos equipamentos.
• O solvente ou o material do swab não deve ser fonte de contaminação adicional ou interferir na metodologia analítica.
• A porcentagem de recuperação do ativo por parte do swab deve ser estabelecida utilizando um estudo de recuperação que demonstra exatamente o procedimento utilizado na prática (mesmo swab, placa com o mesmo tipo de aço do equipamento e definição da área).
• Possível interferência do material de construção do swab deve ser avaliada durante o estudo de validação analítica.
Amostragem indireta da superfície (rinsagem)
• Permite amostragem de grandes áreas.
• Permite amostragem de locais de difícil acesso, como bicos de envase, tubulações e pequenas peças.
• Causa a diluição do contaminante, o que
às vezes compromete ou impossibilita o desempenho da metodologia analítica.
• O contaminante pode não ser solúvel no solvente utilizado.
• O contaminante pode estar ocluído ou aderido em alguma superfície, de modo que a simples rinsagem não é capaz de retirá-lo.

27
Método Vantagens Desvantagens
• A metodologia analítica utilizada deve ser específica para o contaminante, métodos não específicos como a adoção do critério farmacopeico para a água utilizada não são aceitáveis.
• Em alguns casos, como por exemplo, com bicos de envase, as primeiras porções extraídas sempre serão as mais contaminadas. Portanto a uniformização como um todo deve ser realizada.
Fonte: ANVISA, 2006.
2.8 Inspeção Visual
A inspeção visual de equipamentos produtivos é requisito básico para critério de
aceitação em validação de limpeza e no cotidiano da empresa. O “visualmente limpo” pode
ser utilizado como critério único de aceitação, entre lotes de um mesmo produto em uma
campanha ou equipamentos dedicados, desde que comprove que a contaminação
microbiológica está sob controle (ANVISA, 2006).
Uma inspeção visual deve ser realizada em três condições (LEBLANC, 2002):
• Inspeção visual antes da utilização dos equipamentos produtivos.
• Técnica de Monitoramento após a Limpeza.
• Inspeção como critério único de aceitação de um estudo de validação de limpeza,
baseando-se cientificamente.
Para adotar o “visualmente limpo” como critério único de aceitação, durante uma
validação de limpeza, alguns critérios devem ser cumpridos (LEBLANC, 2002):
• Determinar, experimentalmente, o limite de “visualmente limpo”. Alguns
estudiosos estabelecem o limite de “visualmente limpo” de 4,0 µg/cm², porém um
estudo mais completo de definição do limite visual é indicado no caso de
utilização da inspeção visual como único critério de aceitação.
• O limite de “visualmente limpo” determinado deve ser menor que o limite de
resíduo de Limpeza (RCL) pré-determinado.
Atendendo estes critérios, a inspeção visual será mais crítica do que a própria
quantificação do resíduo.

28
3. METODOLOGIA Para os estudos realizados neste trabalho de conclusão de curso de graduação em
Engenharia Industrial Química, da Escola de Engenharia de Lorena, foi utilizada a
metodologia de pesquisa-ação. O trabalho em questão foi executado em novembro de 2013,
em uma indústria farmacêutica de saúde animal, situada no Vale do Paraíba, no estado de São
Paulo.
O fator estimulante para condução deste estudo foi o caráter de qualidade e a
necessidade regulatória de validar todos os procedimentos de limpeza utilizados em
equipamentos multiusos, em uma indústria do ramo farmacêutico veterinário. A partir daí, o
departamento de Technical Services, foi o responsável por criar uma estratégia de validação
para os procedimentos de limpeza da empresa, incluindo o da linha de envase de dois
produtos antiparasiticidas com dois diferentes ingredientes ativos, o qual é o foco principal
deste trabalho. Após executar o plano descrito na estratégia de validação, o objetivo principal
foi validar o procedimento de limpeza, garantindo-se assim sua robustez e reprodutibilidade.
Este trabalho se restringe às particularidades do procedimento de limpeza da linha de
envase desafiada. Qualquer alteração de equipamento, procedimento ou produto deve ser
avaliada, e caso necessário, devem-se realizar adequações na estratégia de validação e por fim
executar um novo estudo de validação.
3.1 Escopo
Para este estudo de validação de limpeza, os seguintes temas foram abordados:
• Definições e abreviações relevantes ao estudo;
• Produtos desafiados no estudo de validação de limpeza;
• Equipamentos envolvidos na limpeza;
• Matriz de pior caso;
• Cálculo de RCL;
• Estratégia de validação;
• Procedimento de limpeza detalhado (limpeza completa e limpeza simples);
• Tipo de amostragem;
• Critérios de aceitação (inspeção visual e resíduo de ativo);
• Tempo de espera de equipamento sujo (dirty equipment hold time);
• Tempo de espera de equipamento limpo (clean equipment hold time);

29
3.2 Definições e abreviações
• Produto contaminante: produto manipulado previamente na respectiva linha de
produção e que poderia vir a contaminar o subsequente.
• Produto subsequente: próximo produto que será fabricado, e que se contaminado,
pode levar o resíduo para o paciente.
• RCL (Residual Cleaning Limit): É o critério de aceitação calculado, levando em
consideração tamanho de lote, toxicidade, dose terapêutica e área de equipamento
em contato com o produto, para o estudo de Validação de Limpeza sob o aspecto
físico - químico. Simboliza o nível de resíduo aceitável do ingrediente ativo do
produto contaminante a ser carreado para o produto subsequente.
• Pior caso: O pior caso é uma situação, às vezes hipotética, onde se estabelece a
pior situação que poderia acontecer em uma linha de produção no que se refere à
criticidade da limpeza. O pior caso é formado pelo produto contaminante e produto
subsequente.
• Resíduo: Substâncias do produto contaminante, deixadas nas superfícies dos
equipamentos, que podem ser carreadas para o produto subsequente.
• Rinsagem: Técnica de amostragem na qual o carreamento das impurezas é feito
através de um solvente ou água.
• Swab: Material absorvente utilizado para amostragem e denomina também a
técnica de amostragem que envolve o esfregaço de uma superfície (do
equipamento) com este material.
• Tempo de espera de equipamento sujo (dirty equipment hold time): Tempo
máximo entre o fim da produção / envase e o início da limpeza.
• Tempo de espera de equipamento limpo (clean equipment hold time): Validade
da limpeza, ou seja, tempo máximo entre o final da limpeza e a utilização dos
equipamentos.
• Método de cálculo de RCL “dose terapêutica”: Método de cálculo de RCL a
partir da máxima dose terapêutica diária do ativo do produto contaminante.
• Método de cálculo de RCL “NOEL” (Nível de efeito não observado): Método
de cálculo de RCL a partir da toxicidade do ativo do produto contaminante.
• Método de cálculo de RCL pelo “10PPM”: Método de cálculo de RCL a partir
da concentração de 10 µg / mL do produto contaminante.

30
3.3 Produtos desafiados no estudo de validação de limpeza
Para facilitar o entendimento deste estudo de validação de limpeza, os produtos
desafiados foram denominados como Produto A e Produto B. O Produto A é utilizado como
carrapaticida, mosquicida, larvicida e sarnicida em bovinos, seu ingrediente ativo é a
Deltametrina e o veículo é denominado como Solvente A para este trabalho. O Produto B é
utilizado como carrapaticida e sarnicida em gatos, seu ingrediente ativo é o Amitraz e o
veículo é denominado como Solvente B para este trabalho.
3.4 Equipamentos envolvidos na limpeza
Os equipamentos dedicados, utilizados neste estudo de validação de limpeza, são os
seguintes:
• Tanque – pulmão de 2000 litros (Tanque 1): tanque-pulmão da linha de
formulação do Produto A.
• Tanque – pulmão de 2000 litros (Tanque 2): tanque-pulmão da linha de
formulação do Produto B.
Os equipamentos não dedicados, utilizado neste estudo de validação de limpeza, são
os seguintes:
• Tanque – reservatório (Tanque 3): tanque reservatório da envasadora, apresenta
contato direto com o Produto A e Produto B.
• Envasadora de líquidos e seus respectivos componentes (bicos dosadores, pistões,
mangueiras, varetas e tubulações): envasadora do Produto A e Produto B.
Apenas os equipamentos de uso não dedicado foram desafiados no estudo de validação
de limpeza, ou seja, o tanque – reservatório e a envasadora de líquidos com seus respectivos
componentes (bicos dosadores, pistões, mangueiras, varetas e tubulações).
3.5 Matriz de pior caso
Para definição do pior caso para o produto contaminante levou-se em consideração os
seguintes parâmetros de avaliação encontrados na literatura: solubilidade do resíduo no
solvente de limpeza (quanto menos solúvel mais difícil de limpar), a facilidade operacional de
limpeza (a experiência do operador foi levada em consideração para definição do produto
mais difícil de limpar) e a toxicidade do resíduo (quanto mais tóxico pior será o cenário).

31
A dose terapêutica, um parâmetro crítico de avaliação que também pode ser
encontrado na literatura, não foi considerada na matriz de pior caso deste estudo, pois este
critério geralmente é utilizado apenas para medicamentos de uso oral ou parenteral. Como se
trata de dois medicamentos de uso tópico, este critério não é aplicável.
Para facilitar a identificação do pior caso, foi criada uma tabela de pontuação, o qual
quantifica cada critério avaliado da seguinte forma:
Tabela 5 – Tabela de pontuação para identificação do pior caso
Produto Princípio
Ativo
Toxicidade Solubilidade Facilidade de
Limpeza Pontuação
Criticidade (1-4)
Criticidade (1-4)
Criticidade (1-4)
Produto A
Produto B
Fonte: próprio autor.
Com todas as informações dos parâmetros avaliados deve-se preencher a coluna
“Pontuação” da seguinte forma:
Pontuação = Criticidade de toxicidade + Criticidade de Solubilidade + Criticidade de facilidade de
limpeza
Caso ocorrer um empate na pontuação entre diferentes produtos deve-se levar em
consideração os critérios de desempate a seguir:
• 1º critério de desempate: produto com ativo mais tóxico (menor dL 50).
• 2º critério de desempate: produto com ativo com maior solubilidade no solvente
de limpeza.
• 3º critério de desempate: produto com maior dificuldade de limpeza.
3.5.1 Solubilidade do resíduo no solvente de limpeza
Para classificar a solubilidade do resíduo no solvente de limpeza verificou-se se o
Solvente A e o Solvente B são altamente solúveis nos resíduos, seguindo a tabela de
classificação de solubilidade (Tabela 1). Para isso, em dois balões volumétricos de 50 mL

32
diferentes, solubilizou-se 2 gramas de Deltametrina em 20 gramas do Solvente A (veículo do
Produto A) e 2 gramas de Amitraz também em 20 gramas do Solvente A. Após devida
homogeneização, ambas as misturas foram deixadas em repouso por 5 minutos e foi
verificada se havia a presença de precipitado. Esta mesma operação foi repetida para o
Solvente B.
Para escolher o solvente de limpeza da linha de envase, levou-se em consideração o
custo e o grau de solubilidade no resíduo. Após a escolha do solvente de limpeza, foi possível
ver a criticidade de solubilidade utilizando-se a tabela 6:
Tabela 6 – Tabela de criticidade de solubilidade do resíduo no solvente de limpeza
Criticidade Descrição Quantidade aproximada de volume por 1
parte de peso do soluto
1 Muito Solúvel Menos do que 1 parte
Altamente Solúvel De 1 a 10 partes
2 Solúvel De 10 a 30 partes
Moderadamente Solúvel De 30 a 100 partes
3 Levemente Solúvel De 100 a 1000 partes
Muito levemente Solúvel De 1000 a 10000 partes
4 Praticamente Insolúvel / Insolúvel 10000 partes ou maior
Fonte: próprio autor.
3.5.2 Toxicidade do ativo
Para a definição da toxicidade de todas as substâncias estudadas considerou-se a dose
letal do ativo de cada produto. A dose letal leva em consideração a via de administração
(tópico, oral, injetável, etc) e diferentes tipos de animais (ratos, camundongos, coelhos, cães,
etc).
Portanto, a fim de quantificar a criticidade de toxicidade, criou-se a tabela 7 baseando-
se na tabela 2, de classificação de toxicidade. Observe a tabela 7:
Tabela 7 – Tabela de criticidade de toxicidade do resíduo
Criticidade Descrição Provável dose oral letal para ratos (mg /
Kg)
1 Praticamente não tóxico 15000
Levemente tóxico 5000 - 15000
2 Moderadamente tóxico 500 - 5000

33
Criticidade Descrição Provável dose oral letal para ratos (mg /
Kg)
Muito tóxico 50 – 500
3 Extremamente tóxico 5 - 50
4 Super tóxico < 5
Fonte: próprio autor.
3.5.3 Facilidade de Limpeza
A fim de quantificar o parâmetro em questão, foi criada a tabela 8, a qual divide a
facilidade de limpeza em 4 níveis de criticidade.
Tabela 8 – Tabela de criticidade de facilidade operacional de limpeza
Criticidade Descrição
1 Fácil
2 Moderadamente Fácil
3 Moderadamente Difícil
4 Difícil
Fonte: próprio autor.
Para definição da criticidade de facilidade de limpeza, o operador de produção
respondeu qual nível de dificuldade se enquadra a limpeza de cada produto desafiado
(Produto A e Produto B), considerando-se as criticidades descritas na tabela 8. Esta entrevista
foi realizada em forma de um questionário simples, no qual o operador de produção indicava
o nível de criticidade para a limpeza de cada produto.
3.6 Cálculo do RCL
Existem diferentes métodos de determinação dos limites de aceitação em um estudo de
validação de limpeza. Os métodos mais utilizados na determinação do limite de aceitação são
(ANVISA, 2006):
• 0,1 % da dose limite ou método “dose terapêutica”;
• Nível de Efeito Não Observado (NOEL) ou método “NOEL”;

34
• 10 ppm.
Para medicamentos de uso tópico, o qual os mesmos devem ser diluídos antes da sua
utilização, não é possível determinar uma mínima dose terapêutica diária exata. Os produtos
desafiados neste estudo de validação apresentam estas características, portanto, o método
“0,1% da dose limite” não foi utilizado para este estudo.
Não há uma explicação científica para o método “10 ppm”, ou seja, a lógica da sua
utilização foge dos padrões BPF (ANVISA, 2006). Portanto, este método também não foi
utilizado para determinação RCL.
Assim, para este estudo de validação, o método mais adequado para cálculo de RCL é
o “Nível de Efeito Não Observado (NOEL)”.
3.7 Estratégia de Validação
Este estudo de validação de limpeza da linha de envase teve como estratégia desafiar e
garantir a eficiência de três execuções do procedimento de limpeza, conforme item 3.7.1, após
o término do envase do produto pior caso. Porém, antes de iniciar as execuções do
procedimento de limpeza, os seguintes itens foram realizados:
• Desenvolveu-se um procedimento de limpeza (limpeza simples e completa);
• Escolheu-se uma técnica de amostragem para verificação de resíduo;
• Definiram-se os testes e os critérios de aceitação;
• Definiu-se uma estratégia de tempo de espera de equipamento limpo (clean
equipment hold time);
• Definiu-se uma estratégia de tempo de espera de equipamento sujo (dirty
equipment hold time);
3.7.1 Procedimento de limpeza detalhado (limpeza completa e limpeza simples)
Para facilitar o entendimento da limpeza simples e limpeza completa observe a figura
2.
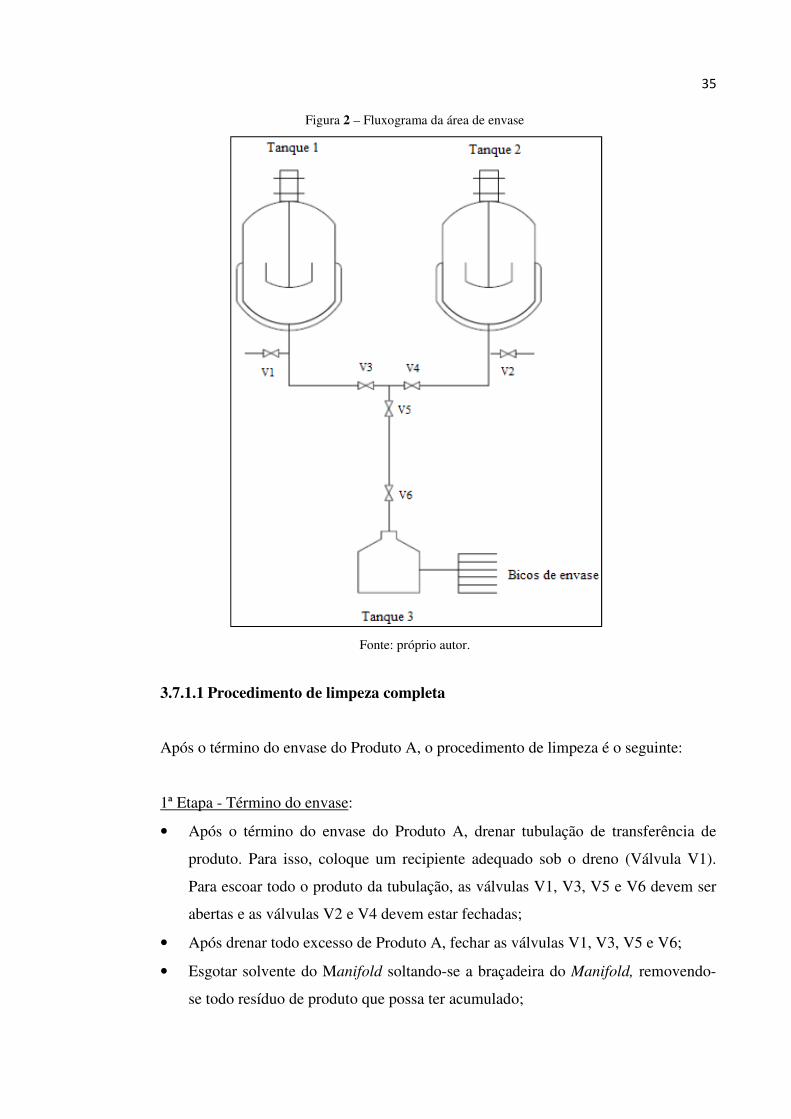
35
Figura 2 – Fluxograma da área de envase
Fonte: próprio autor.
3.7.1.1 Procedimento de limpeza completa
Após o término do envase do Produto A, o procedimento de limpeza é o seguinte:
1ª Etapa - Término do envase:
• Após o término do envase do Produto A, drenar tubulação de transferência de
produto. Para isso, coloque um recipiente adequado sob o dreno (Válvula V1).
Para escoar todo o produto da tubulação, as válvulas V1, V3, V5 e V6 devem ser
abertas e as válvulas V2 e V4 devem estar fechadas;
• Após drenar todo excesso de Produto A, fechar as válvulas V1, V3, V5 e V6;
• Esgotar solvente do Manifold soltando-se a braçadeira do Manifold, removendo-
se todo resíduo de produto que possa ter acumulado;

36
• Com um pano absorvente limpo, retirar o excesso de produto do Manifold e do
reservatório da envasadora (Tanque 3), contemplando a parede interna, o tampo
superior e o sensor de nível.
2ª Etapa - Abertura de válvulas:
• Verificar se o tanque-pulmão (Tanque 1) está limpo;
• Adicionar 40 litros de Solvente A no tanque-pulmão (Tanque 1);
• Abrir válvulas V3, V5 e V6;
3ª Etapa – Transferência do Solvente A para a linha de envase:
• Abrir válvula pneumática da envasadora. Neste momento o solvente de limpeza
será transferido para o reservatório da envasadora (Tanque 3);
• Após transferência de aproximadamente 20 litros deve-se fechar válvula V6 para
interromper a transferência.
4ª Etapa – Limpeza dos componentes da envasadora:
• Posicionar sob os bicos dosadores um recipiente adequado, para que o solvente de
limpeza seja destinado ao seu interior, quando a envasadora for acionada;
• Acionar os bicos de envase. Feito isso, o solvente de limpeza irá passar pelo
manifold, mangueiras, pistões, varetas e bicos dosadores.
• Após término dos 20 litros de solvente de limpeza deve-se desligar os bicos
dosadores.
5ª Etapa: Esgotar solvente de limpeza do manifold:
• Posicionar um balde embaixo do manifold;
• Soltar a braçadeira do manifold, esgotando-se assim todo o solvente de limpeza;
• Após drenar todo o solvente de limpeza deve-se conectar a braçadeira no
manifold.
6ª Etapa: Segunda limpeza:
• Repetir etapas 3, 4 e 5 com o restante do solvente de limpeza (aproximadamente
20 litros) que está no tanque-pulmão (Tanque 1).

37
7ª etapa: Drenar tubulação:
• Com um pana absorvente limpo deve-se retirar o excesso de solvente de limpeza
do manifold e do reservatório da envasadora (Tanque 3);
• Após o término da limpeza, drenar tubulação de transferência de produto. Para
isso, coloque um recipiente adequado sob o dreno (Válvula V1). Para escoar todo
o solvente de limpeza da tubulação, as válvulas V1, V3, V5 e V6 devem ser
abertas e as válvulas V2 e V4 devem estar fechadas;
• Após drenar todo excesso de solvente de limpeza, fechar as válvulas V1, V3, V5 e
V6;
NOTA: Após término do envase do Produto B, deve-se seguir as mesmas etapas,
porém a manobra das válvulas se diferenciam da seguinte forma:
• Onde lê-se V1 deve-se entender V2;
• Onde lê-se V2 deve-se entender V1;
• Onde lê-se V3 deve-se entender V4;
• Onde lê-se V4 deve-se entender V3;
• Onde lê-se Tanque 1 deve-se entender Tanque 2;
• Onde lê-se Produto A deve-se entender Produto B;
3.7.1.2 Procedimento de limpeza simples
A Limpeza Simples não pode ser a única limpeza realizada antes do envase do Produto
B. Esta limpeza só é aceitável caso tenha sido realizado o procedimento descrito no item
3.7.1.1.
A finalidade da limpeza simples não é evitar a contaminação cruzada de ativos, e sim
garantir que não tenha Solvente A na linha de envase quando o Produto B for envasado, visto
que o Solvente A não faz parte da composição do Produto B.
Portanto, antes do envase do Produto B, deve-se realizar a limpeza da seguinte forma:
1ª Etapa - Abertura de válvulas:
• Verificar se o tanque-pulmão (Tanque 2) está limpo;
• Adicionar 20 litros de Solvente B no tanque-pulmão (Tanque 2);
• Abrir válvulas V4, V5 e V6;

38
2ª Etapa – Transferência do Solvente A para a linha de envase:
• Abrir válvula pneumática da envasadora. Neste momento o solvente de limpeza
será transferido para o reservatório da envasadora (Tanque 3);
• Após transferência deve-se fechar válvula V6 para interromper a transferência.
3ª Etapa – Limpeza dos componentes da envasadora:
• Posicionar sob os bicos dosadores um recipiente adequado, para que o solvente de
limpeza seja destinado ao seu interior, quando a envasadora for acionada;
• Acionar os bicos de envase. Feito isso, o solvente de limpeza irá passar pelo
manifold, mangueiras, pistões, varetas e bicos dosadores.
• Após término da transferência do solvente de limpeza deve-se desligar os bicos
dosadores.
4ª Etapa: Esgotar solvente de limpeza do manifold:
• Posicionar um balde embaixo do manifold;
• Soltar a braçadeira do manifold, esgotando-se assim todo o solvente de limpeza;
• Após drenar todo o solvente de limpeza deve-se conectar a braçadeira no
manifold.
5ª etapa: Drenar tubulação:
• Com um pana absorvente limpo deve-se retirar o excesso de solvente de limpeza
do manifold e do reservatório da envasadora (Tanque 3);
• Após o término da limpeza, drenar tubulação de transferência de produto. Para
isso, coloque um recipiente adequado sob o dreno (Válvula V2). Para escoar todo
o solvente de limpeza da tubulação, as válvulas V2, V4, V5 e V6 devem ser
abertas e as válvulas V1 e V3 devem estar fechadas;
• Após drenar todo excesso de solvente de limpeza, fechar as válvulas V2, V4, V5 e
V6;

39
3.7.2 Tipo de amostragem
Um plano de amostragem foi definido baseando-se em justificativas técnicas e
considerando as dificuldades em relação à superfície a ser amostrada. O método escolhido
para este estudo de validação de limpeza foi o método de swab devido aos seguintes fatores:
• Consegue recuperar resíduos secos e insolúveis;
• Permite estabelecer o limite de contaminação por área, indicando onde o
procedimento deve ser melhorado, caso necessário;
• Permite desafiar separadamente os pontos críticos do equipamento (bicos
dosadores, pistões, mangueiras, varetas e tubulações).
• Permite amostrar áreas que o solvente de limpeza tem dificuldade de limpar.
Para a execução da amostragem, o swab foi umedecido em uma solução de Isoctano e
Dioxano, em uma proporção de 80% de Isoctano e 20% de Dioxano, a fim de recuperar
grande parte da Deltametrina que poderia estar depositada na superfície do equipamento. Esta
solução foi escolhida, pois é o solvente de diluição utilizado na preparação de amostras do
método de quantificação de resíduo de Deltametrina, descrito no procedimento validado de
quantificação de resíduos.
Para se obter a melhor recuperação de resíduo de Deltametrina da superfície dos
equipamentos e componentes da envasadora, cada ponto crítico foi amostrado duas vezes,
com diferentes swabs. Esta metodologia de amostragem é requerida, pois o cálculo de
recuperação de resíduo, descrito no método de quantificação de resíduo de Deltametrina,
desenvolvido e validado pelo departamento de Controle de Qualidade, mostrou que se
consegue recuperar 89,75 % de resíduo de Deltametrina quando dois swabs são utilizados
para amostrar um mesmo ponto.
A amostragem dos pontos críticos da linha de envase foi realizada após cada execução
do procedimento de limpeza.
3.7.2.1 Ponto 1: superfície interna do fundo do reservatório
A superfície interna do fundo do reservatório é um local onde há o contato direto com
o produto durante todo o processo de envase. Portanto, para desafiar este ponto, foi amostrada
uma área de 100 cm², representando assim toda a superfície interna do fundo do reservatório
da envasadora. Observe o ponto amostrado na figura 3:

40
Figura 3 – Ponto 1 de amostragem (superfície interna do fundo do reservatório)
Fonte: próprio autor.
3.7.2.2 Ponto 2: superfície interna do tampo do reservatório da envasadora
Durante o processo de enchimento do reservatório da envasadora, há a deposição de
uma pequena parcela do produto no tampo superior. O fato desta superfície não ficar em
contato constante com o produto, permite que o mesmo seque, e dificulte a limpeza. Portanto,
para desafiar este ponto, foi amostrada uma área de 100 cm², representando assim toda a
superfície do tampo superior do reservatório da envasadora. Observe o ponto amostrado na
figura 4:

41
Figura 4 – Ponto 2 de amostragem (superfície interna do tampo do reservatório da envasadora)
Fonte: próprio autor.
3.7.2.3 Ponto 3: superfície interna da parede do reservatório da envasadora
A superfície interna da parede do reservatório é um local onde há o contato direto com
o produto durante todo o processo de envase. Portanto, para desafiar este ponto, foi amostrada
uma área de 100 cm², representando assim toda a superfície interna da parede do reservatório
da envasadora. Observe o ponto amostrado na figura 5:
Figura 5 – Ponto 3 de amostragem (superfície interna da parede do reservatório da envasadora)
Fonte: próprio autor.

42
3.7.2.4 Ponto 4: superfície interna da tubulação de chegada de produto ao reservatório
Este ponto será desafiado com a finalidade de representar toda a tubulação envolvida
no processo de envase de produto. Portanto, foi amostrada uma área de 100 cm²,
representando toda a superfície interna da tubulação compartilhada da linha de envase.
Observe o ponto amostrado na figura 6:
Figura 6 – Ponto 4 de amostragem (superfície interna da tubulação de chegada de produto ao reservatório)
Fonte: próprio autor.
3.7.2.5 Ponto 5: superfície interna do manifold do reservatório
A Superfície interna do manifold é um local onde há o contato direto com o produto
durante todo o processo de envase. Portanto, para desafiar este ponto, foi amostrada uma área
de 100 cm², representando assim toda a superfície interna do manifold. Observe o ponto
amostrado na figura 7:

43
Figura 7 – Ponto 5 de amostragem (superfície interna do manifold do reservatório)
Fonte: próprio autor.
3.7.2.6 Ponto 6: superfície interna do pistão
O pistão possui um formato que dificulta a limpeza, por este motivo é um possível
local de deposição de resíduo. Portanto, para desafiar este ponto, foi amostrada uma área de
100 cm², representando assim toda a superfície interna dos pistões que compõe a envasadora.
Observe o ponto amostrado na figura 8:
Figura 8 – Ponto 6 de amostragem (superfície interna do pistão)
Fonte: próprio autor.

44
3.7.2.7 Ponto 7: superfície interna da vareta de transferência de produto
A vareta de transferência de produto possui um formato que dificulta a limpeza, por
este motivo é um possível local de deposição de resíduo. Por se tratar de uma tubulação de
pequeno diâmetro, foi amostrada uma área de 20 cm², representando-se assim todas as varetas
de transferência de produto. Observe o ponto amostrado na figura 9:
Figura 9 – Ponto 7 de amostragem (superfície interna da vareta de transferência de produto)
Fonte: próprio autor.
3.7.2.8 Ponto 8: superfície interna do bico dosador de produto
O Bico dosador de produto possui um formato que dificulta a limpeza, por este motivo
é um possível local de deposição de resíduo. Por se tratar de uma tubulação de pequeno
diâmetro, foi amostrada uma área de 10 cm², representando-se assim todos os bicos dosadores
da envasadora. Observe o ponto amostrado na figura 10:

45
Figura 10 – Ponto 8 de amostragem (superfície interna do bico dosador de produto)
Fonte: próprio autor.
3.7.3 Critério de aceitação
O estudo de validação de limpeza deve considerar os seguintes testes:
• Inspeção visual;
• Método de detecção de resíduo pior caso;
• Análises microbiológicas
Para cada teste realizado, deve-se ter um critério de aceitação tecnicamente
justificável.
3.7.3.1 Inspeção Visual
A inspeção visual de equipamentos produtivos é requisito básico para critério de
aceitação em um estudo de validação de limpeza e para o cotidiano da empresa. O
“visualmente limpo” pode ser utilizado como critério único de aceitação, entre lotes de um
mesmo produto em uma campanha ou equipamentos dedicados.
Portanto, após o término de cada limpeza, foi realizada a inspeção visual do
reservatório da envasadora, a fim de garantir que não havia impurezas visíveis na superfície

46
de contato com o produto contaminante. A seguir estão os pontos inspecionados durante o
estudo de validação:
• Superfície interna do tampo superior do reservatório da envasadora;
• Superfície interna do fundo do reservatório da envasadora;
• Superfície interna da parede do reservatório da envasadora;
• Sensor de nível do reservatório da envasadora;
3.7.3.2 Método de detecção de resíduo
Para quantificação do resíduo de Deltametrina (pior caso), o departamento de controle
de qualidade desenvolveu e validou um método de análise por HPLC (High Performance
Liquid Cromatography).
O limite de aceitação para resíduo de Deltametrina foi determinado através do cálculo
de RCL, utilizando-se a metodologia de NOEL, conforme mencionado no item 3.6.
3.7.3.3 Análises microbiológicas
O Produto A e o Produto B apresentam como veículos hidrocarbonetos aromáticos. A
complexidade e estabilidade da estrutura química dos hidrocarbonetos aromáticos,
considerando-se a ressonância dos anéis aromáticos, impossibilitam a sua degradação, pois os
micro-organismos não possuem enzimas capazes de reconhecê-los e degradá-los. Além disso,
os produtos ectoparasiticidas em questão, não apresentam quantidades consideráveis de água
em sua composição, e sabendo-se que os micro-organismos necessitam da água para o
carreamento de nutrientes de dentro para fora das células, devido ao diferencial de
concentração (transporte conhecido como osmose), conclui-se que a água é fundamental para
o crescimento microbiano (JACQUES, 2007). Portanto, a falta de “alimento”, devido à
complexidade dos hidrocarbonetos aromáticos e a falta de água, fazem com os equipamentos
que foram avaliados neste estudo de validação, se tornem um ambiente inadequado para a
sobrevivência e o crescimento de micro-organismos. Portanto, análise microbiológica não foi
requerida neste estudo de validação de limpeza.

47
3.7.4 Clean equipment hold time
A avaliação do clean equipment hold time para os equipamentos e componentes da
linha de envase não é necessário, visto que os produtos envasados nesta linha inibem o
crescimento de micro-organismo, conforme mencionado no item 3.7.3.3.
3.7.5 Dirty equipment hold time
O procedimento de limpeza deve contemplar o dirty equipment hold time, ou seja, o
tempo que o equipamento pode ficar sujo antes da execução do procedimento de limpeza. Isso
se faz necessário, pois o tempo de equipamento sujo é inversamente proporcional à facilidade
de limpeza, ou seja, quanto mais tempo o equipamento ficar sujo, mais difícil de limpar. Para
a análise do dirty equipment hold time, foi executado o procedimento de limpeza após três (3)
dias do término do envase do primeiro lote do produto pior caso. Depois da execução desta
limpeza, os pontos críticos foram amostrados e os resultados foram avaliados.

48
4. RESULTADO E DISCUSSÃO
4.1 Escolha do produto pior caso
Para definição do pior caso para o produto contaminante levou-se em consideração a
solubilidade do resíduo no solvente de limpeza, a toxicidade do resíduo e a facilidade
operacional da limpeza.
4.1.1 Solubilidade do resíduo no solvente de limpeza
Os testes de solubilidade realizados no laboratório de controle de qualidade da
empresa demonstraram que tanto o Solvente A quanto o Solvente B são altamente solúveis
em ambos os resíduos (deltametrina e amitraz), visto que a solubilização ocorreu na
proporção de 1 parte do soluto para 10 partes do solvente. Portanto, a criticidade de ambos os
solventes é 1 quando observamos a tabela 6.
A escolha do solvente de limpeza levou em consideração o custo do material, visto
que ambos os solventes tem o mesmo grau de criticidade para solubilidade. Portanto, o
solvente escolhido para realização do procedimento de limpeza foi o Solvente A, já que
apresenta um custo mais baixo quando comparado ao Solvente B.
4.1.2 Toxicidade do resíduo
Para a definição da toxicidade de todas as substâncias estudadas considerou-se a dose
letal do ativo de cada produto. O dL 50 é 31 mg / Kg para a Deltametrina e 800 mg / Kg para
o Amitraz (The Merck Index, 2006). Esses valores levam em consideração administração via
oral e o animal de estudo são os camundongos. Não foi possível encontrar na literatura o dL
50 para administração tópica, portanto, a fim de padronizar, utilizaram-se valores de dL 50
levando em consideração administração via oral, tanto para a Deltametrina como para o
Amitraz.
Portanto, com estes valores encontrados na literatura, é possível classificar, segundo
tabela 7, como extremamente tóxico para a Deltametrina (grau de criticidade 3) e
moderadamente tóxico para o Amitraz (grau de criticidade 2).

49
4.1.3 Facilidade de Limpeza
A entrevista realizada com o operador mais experiente da linha de envase apontou que
ambos os produtos são “moderadamente fácil” de se realizar a limpeza. Portanto, levando-se
em consideração a tabela 8, o grau de criticidade é 2 para os dois produtos desafiados neste
estudo.
4.1.4 Identificação do pior caso
Com base nas informações apresentadas nos itens anteriores é possível preencher a
tabela 5 e definir o produto pior caso. Observe a tabela 9.
Tabela 9 – Tabela de identificação do pior caso
Produto Princípio
Ativo
Toxicidade Solubilidade Facilidade de
Limpeza Pontuação
Criticidade (1-4)
Criticidade (1-4)
Criticidade (1-4)
Produto A Deltametrina 3 1 2 6
Produto B Amitraz 2 1 2 5
Fonte: próprio autor.
Levando-se em consideração os valores apresentados na tabela 9 é possível afirmar
que o Produto A é o produto pior caso, visto que apresentou uma pontuação maior na
metodologia de identificação de pior caso. Observe também que os dois produtos são bem
parecidos quando comparados os parâmetros de avaliação, a única diferença é na toxicidade
do ativo, o qual o Produto A se apresentou mais tóxico, levando assim uma criticidade maior.
Com isso, o estudo de validação foi executado após o envase do Produto A, assim foi possível
desafiar o procedimento de limpeza levando em consideração o pior cenário.
4.2 Cálculos de RCL
Para este estudo de validação, o método mais adequado para cálculo de RCL é o
“Nível de Efeito Não Observado (NOEL)”. Para determinação do RCL a partir deste método
cálculo, deve-se saber o NOEL do produto pior caso (Produto A), máxima dose diária do

50
produto subsequente (Produto B), tamanho mínimo de lote do produto subsequente (Produto
B) e área de equipamento compartilhado.
4.2.3 Máxima dose diária do produto contaminante
Para definir a máxima dose diária para o produto subsequente (Produto B) considerou-
se um pior caso, ou seja, admitiu-se que a máxima dose diária é igual ao volume útil do
frasco, resultando assim na quantidade máxima de resíduo do produto contaminante (Produto
A) no produto subsequente (Produto B). Portanto, a máxima dose diária do Produto B é igual
a 40 mL.
4.2.4 Tamanho mínimo do lote subsequente
Para definir o tamanho mínimo do lote subsequente considerou-se a quantidade
mínima de Produto B envasado na linha produtiva estudada, representando assim a maior
concentração possível do produto contaminante (Produto A) no produto subsequente (Produto
B). Portanto, através de dados do departamento de Planejamento e Controle de Produção
(PCP), foi possível definir que o menor tamanho de lote comercial do produto subsequente
(Produto B) é de 200 litros, ou seja, 200 000 mL.
4.2.4 Área de equipamento compartilhado
Os equipamentos não dedicados, utilizado neste estudo de validação de limpeza, são:
• Tanque – reservatório (Tanque 3);
• Envasadora de líquidos e seus respectivos componentes (bicos dosadores, pistões,
mangueiras, varetas e tubulações).
Para determinar a área total de superfície em contato com o produto para os
equipamentos e componentes listados acima, levou-se em consideração manuais de operação
e especificações da linha de envase. A área total calculada, de superfície em contato com o
produto, foi de 34.660,21 cm².

51
4.2.6 Cálculo de NOEL do produto contaminante
Para determinação do NOEL a seguinte fórmula foi utilizada:
NOEL�mg = dL50 × MassaMédiadoPaciente
2000
Segundo The Merck Index, 4ª Edição, o dL 50 é 31 mg / Kg para a Deltametrina. Esse
valor leva em consideração administração via oral e o animal de estudo são os camundongos.
Observando-se as bulas dos medicamentos, o Produto A é utilizado em animais de grande
porte (equinos e bovinos), já o Produto B é utilizado em animais de pequeno porte (cães e
gatos). Pela diferença de peso dos pacientes, fica difícil estabelecer uma massa média.
Portanto, para considerar um pior caso, admitiu-se uma massa de 1 Kg.
Assim, foi possível calcula o NOEL do produto contaminante (Produto A). Observe a
fórmula a seguir:
NOEL�mg = 31 × 1
2000= 0,0155mg/,-
4.1 Determinação do RCL
A determinação do RCL a partir do cálculo do NOEL é realizada através da fórmula
descrita a seguir:
RCL�μg/cm² = NOELContaminante�mg × 1000 × Menortamanhodelotedoprodutosubsequente�mL
Máximadosediáriadoprodutosubsequente�mL × Áreadeequipamento�cm&
1000 – fator de conversão de mg para µg.
NOEL contaminante (mg) – Nível de Efeito Não Observado do produto contaminante.
Menor tamanho de lote do produto subsequente (mL) – É o menor granel (lote) do
produto subsequente.
Máxima Dose Diária do produto subsequente (mL) – É a quantidade total do produto
subsequente que o paciente pode receber em 24 horas.
Área de Equipamento em contato com o produto (cm²) – Área de equipamento não
dedicado em contato com o produto.

52
Observe os parâmetros de cálculo de RCL na tabela 10:
Tabela 10 – Dados para o cálculo do RCL
Produto Princípio
Ativo Máxima dose diária (mL)
Tamanho mínimo de lote (mL)
Área de Equipamento (cm²)
NOEL (mg)
Produto A Deltametrina - -
34 660,21
0,0155
Produto B Amitraz 40 200 000 -
Fonte: próprio autor.
Sabendo-se que o pior caso é o Produto A e utilizando-se os dados da tabela 10, é
possível calcular o RCL, ou seja, é possível determinar o critério de aceitação para o estudo
de validação de limpeza. Observe o cálculo a seguir:
RCL�µg/cm² = 0,0155 × 1000 × 200000
40 × 34660,21= 2,2360µg/cm²
Portanto, a máxima quantidade permitida de resíduo de deltametrina é de 2,2360
µg/cm² após uma execução do procedimento de limpeza (item 3.7.1.1).
4.3 Execução da validação de limpeza
Para realização deste estudo de validação de limpeza, executaram-se três
procedimentos de limpeza completa (item 3.7.1.1), após o envase do produto pior caso
(Produto A). A primeira execução do procedimento de limpeza foi realizada 72 horas após o
término do envase, avaliando-se assim o dirty equipment hold time. A segunda e a terceira
execução do procedimento de limpeza foram realizadas no mesmo dia do término do envase
do Produto A.
4.3.2 Resultados obtidos após primeira execução do procedimento de limpeza
A seguir estão os dados da execução da limpeza:
• Término do envase: 01/11/2013 (16h 50 min).
• Início da limpeza: 05/11/2013 (07h 10 min).
• Término da limpeza: 05/11/2013 (08h 55min).

53
Os resultados da inspeção visual realizada após a primeira execução do procedimento
de limpeza estão descritos na tabela 11 e os resultados de resíduo de deltametrina nos pontos
críticos da linha de envase estão descritos na tabela 12.
Tabela 11 – Resultados da inspeção visual após primeira execução do procedimento de limpeza
Inspeção Visual do reservatório da envasadora
Data da Inspeção Visual 05.11.2013
Horário da Inspeção Visual 09 h 00 min
O reservatório apresentava Impurezas Visíveis? Não havia impurezas visíveis.
Fonte: Próprio autor.
Tabela 12 – Resultados de resíduo de Deltametrina após primeira execução do procedimento de limpeza
Resultados da análise de quantificação de resíduo de Deltametametrina
Ponto Descrição do ponto Amostrado Área Amostrada
(cm²) Resultado
(≤ 2,2360 µg/cm²)
1 Superfície interna do fundo do Reservatório 100 cm² 0,0131
2 Superfície interna do Tampo Superior do
Reservatório 100 cm² 0,0333
3 Superfície interna da parede do Reservatório 100 cm² 0,0327
4 Superfície interna da tubulação de chegada ao
reservatório 100 cm² 0,0000
5 Superfície interna do Manifold 100 cm² 0,1689
6 Superfície interna do Pistão. 100 cm² 1,3263
7 Superfície interna da Vareta de transferência de
Produto 20 cm² 0,4160
8 Superfície interna do bico dosador de produto 10 cm² 0,2285
Fonte: próprio autor.
Para a análise do dirty equipment hold time, foi executado o procedimento de limpeza
após três dias, quatorze horas e vinte minutos do término de envase do primeiro lote do
Produto A. Os resultados apresentados acima comprovam que o procedimento de limpeza

54
completo (item 3.7.1.1) é eficiente mesmo após 3 dias do término do envase. Portanto pode-se
definir o dirty equipment hold time (tempo de espera de equipamento sujo) de 72 horas, ou
seja, 3 dias corridos.
4.3.3 Resultados obtidos após segunda execução do procedimento de limpeza
A seguir estão os dados da execução da limpeza:
• Término do envase: 13/11/2013
• Início da limpeza: 13/11/2013
• Término da limpeza: 13/11/2013
Os resultados da inspeção realizada após segunda execução do procedimento de
limpeza estão descritos na tabela 13 e os resultados de resíduo de deltametrina nos pontos
críticos da linha de envase estão descritos na tabela 14.
Tabela 13 – Resultados da inspeção visual após segunda execução do procedimento de limpeza
Inspeção Visual do reservatório da envasadora
Data da Inspeção Visual 13.11.2013
Horário da Inspeção Visual 12 h 30 min
O reservatório apresentava Impurezas Visíveis? Não havia impurezas visíveis.
Fonte: próprio autor.
Tabela 14 – Resultados de resíduo de Deltametrina após segunda execução do procedimento de limpeza
Resultados da análise de quantificação de resíduo de Deltametametrina
Ponto Descrição do ponto Amostrado Área Amostrada (cm²)
Resultado (≤ 2,2360 µg/cm²)
1 Superfície interna do fundo do Reservatório 100 cm² 0,0986
2 Superfície interna do Tampo Superior do
Reservatório 100 cm² 0,0000
3 Superfície interna da parede do Reservatório 100 cm² 0,1563

55
Resultados da análise de quantificação de resíduo de Deltametametrina
Ponto Descrição do ponto Amostrado Área Amostrada
(cm²) Resultado
(≤ 2,2360 µg/cm²)
4 Superfície interna da tubulação de chegada ao
reservatório 100 cm² 0,0000
5 Superfície interna do Manifold 100 cm² 0,4566
6 Superfície interna do Pistão. 100 cm² 0,1830
7 Superfície interna da Vareta de transferência de
Produto 20 cm² 0,5128
8 Superfície interna do bico dosador de produto 10 cm² 0,2481
Fonte: próprio autor.
Observa-se que tanto a inspeção visual quanto a verificação de resíduo de deltametrina
apresentaram resultados satisfatórios, ou seja, a segunda execução do procedimento de
limpeza foi eficiente.
4.3.3 Resultados obtidos após terceira execução do procedimento de limpeza
A seguir estão os dados da execução da limpeza:
• Término do envase: 14/11/2013
• Início da limpeza: 14/11/2013
• Término da limpeza: 14/11/2013
Os resultados da inspeção realizada após segunda execução do procedimento de
limpeza estão descritos na tabela 15 e os resultados de resíduo de deltametrina nos pontos
críticos da linha de envase estão descritos na tabela 16.
Tabela 15 – Resultados da inspeção visual após terceira execução do procedimento de limpeza
Inspeção Visual do reservatório da envasadora
Data da Inspeção Visual 14.11.2013
Horário da Inspeção Visual 16 h 15 min
O reservatório apresentava Impurezas Visíveis? Não havia impurezas visíveis.
Fonte: próprio autor.

56
Tabela 16 – Resultados de resíduo de Deltametrina após terceira execução do procedimento de limpeza
Resultados da análise de quantificação de resíduo de Deltametametrina
Ponto Descrição do ponto Amostrado Área Amostrada
(cm²) Resultado
(≤ 2,2360 µg/cm²)
1 Superfície interna do fundo do Reservatório 100 cm² 0,0669
2 Superfície interna do Tampo Superior do
Reservatório 100 cm² 0,0000
3 Superfície interna da parede do Reservatório 100 cm² 0,1403
4 Superfície interna da tubulação de chegada ao
reservatório 100 cm² 0,0513
5 Superfície interna do Manifold 100 cm² 0,7114
6 Superfície interna do Pistão. 100 cm² 0,6682
7 Superfície interna da Vareta de transferência de
Produto 20 cm² 1,1414
8 Superfície interna do bico dosador de produto 10 cm² 0,0000
Fonte: próprio autor.
Observa-se que tanto a inspeção visual quanto a verificação de resíduo de deltametrina
apresentaram resultados satisfatórios, ou seja, a terceira execução do procedimento de limpeza
foi eficiente.
5.3.3 Discussão dos resultados
Nas tabelas 17 e 18 estão todos os dados obtidos no estudo de validação de limpeza:
Tabela 17 – Resultado geral de inspeção visual
Execução Resultado
1ª Execução Não havia impurezas visíveis.
2ª Execução Não havia impurezas visíveis.
3ª Execução Não havia impurezas visíveis.
Fonte: próprio autor.

57
Tabela 18 – Resultado geral de quantificação de resíduo
Descrição do ponto Amostrado Área
Amostrada (cm²)
Resultado (≤ 2,2360 µg/cm²)
1ª Execução 2ª Execução 3ª Execução
Superfície interna do fundo do Reservatório
100 cm² 0,0131 0,0986 0,0669
Superfície interna do Tampo Superior do Reservatório
100 cm² 0,0333 0,0000 0,0000
Superfície interna da parede do Reservatório
100 cm² 0,0327 0,1563 0,1403
Superfície interna da tubulação de chegada ao reservatório
100 cm² 0,0000 0,0000 0,0513
Superfície interna do Manifold 100 cm² 0,1689 0,4566 0,7114
Superfície interna do Pistão. 100 cm² 1,3263 0,1830 0,6682
Superfície interna da Vareta de transferência de Produto
20 cm² 0,4160 0,5128 1,1414
Superfície interna do bico dosador de produto
10 cm² 0,2285 0,2481 0,0000
Fonte: próprio autor.
Conforme tabela 17, nas três inspeções visuais realizadas, não foram detectados
resíduos visíveis na superfície dos equipamentos, ou seja, não foram encontrados resíduos
visíveis na superfície interna do tampo superior, fundo, parede e sensor de nível do
reservatório da envasadora. Isso mostra que o critério de aceitação para inspeção visual foi
atendido, comprovando a eficiência do procedimento de limpeza, descrito no item 3.7.1.1,
para remoção de resíduos detectáveis a olho nu.
Conforme tabela 18, nas três amostragens realizadas após cada execução do
procedimento de limpeza, não foram obtidas quantidades de resíduo de Deltametrina maior
que a especificação calculada pelo método de NOEL (≤ 2,2360 µg / cm²). Isso mostra que o
critério de aceitação para quantificação de resíduo de Deltametrina foi atendido, comprovando
a eficiência do procedimento de limpeza, descrito no item 3.7.1.1, para remoção de resíduo a
níveis seguro pré-determinados. Além disso, é possível afirmar que o procedimento de
limpeza também é eficiente, admitindo-se um tempo de equipamento sujo de 3 dias, visto que
antes da primeira execução, o equipamento ficou com resíduo do Produto A por três dias,
quatorze horas e vinte minutos.

58
5. CONCLUSÃO E SUGESTÕES
5.1 Conclusão
O Produto A foi utilizado como pior caso para este estudo de validação de limpeza da
linha de envase de antiparasitisicdas de uso tópico, apresentando uma toxicidade maior do que
o ativo do Produto B, gerando assim uma pontuação maior nos parâmetros de identificação de
produto pior caso.
De acordo com os resultados obtidos dentro dos limites de aceitação, considera-se
aprovada a validação de limpeza da linha multiuso de envase do Produto A e Produto B. É
importante ressaltar que a metodologia desenvolvida foi específica para esta validação,
necessitando assim de adaptações para utilização em outros equipamentos e substâncias
ativas.
Esta validação gera confiabilidade no procedimento de limpeza, visto que se
conseguiu remover resíduos do produto pior caso (Produto A) a níveis seguros, pré-
determinado com base científica. Com isso há a garantia e a segurança na qualidade dos
produtos fabricados, uma vez que se exclui a possibilidade de contaminação cruzada. Além
disso, esta validação traz o benefício de aumento de produção industrial, já que uma mesma
linha pode ser utilizada para envasar dois tipos de produtos.
5.2 Sugestões
Avaliar a possibilidade de utilizar a metodologia de validação proposta em outras
linhas de envase de produtos líquidos antiparasiticidas de uso tópico.
Definir uma estratégia de monitoramento do procedimento de limpeza desafiado a fim
de evitar revalidações periódicas, visto que o monitoramento iria gerar dados para análise
constante do procedimento de limpeza.
Realizar um novo estudo completo de validação, a fim de aumentar o dirty equipment
hold time, dando flexibilidade ao cotidiano de produção.

59
REFERÊNCIAS
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA, ANVISA. Guias Relacionados à
Garantia de Qualidade. Brasília, DF, 31 de Outubro de 2006. p32.
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA, ANVISA. Resolução RDC Nº 17,
de 16 de Abril de 2010. Ministério da Saúde. 16 de Abril de 2010.
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA, ANVISA. Validação de Limpeza
para Farmoquímicas. Brasília, DF, 2013.
DUTRA, V.C. Boas Práticas de fabricação de Medicamentos. Dossiê Técnico - Rede de
Tecnologia e Inovação do rio de Janeiro – REDETEC . Rio de Janeiro, RJ, 2011.
JACQUES,R.J.S et al. “Biodegradação de hidrocarbonetos aromáticos policíclicos”.
Ciência e Natura, UFSM, 29(1):7-24, 2007.
LEBLANC, D.A. “Visually Clean” as a Sole Acceptance Criterion for Cleaning
Validation Protocols. PDA Journal of Pharmaceutical Science and Technology, San
Antonio, Texas, 2002.
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO. Instrução
Normativa nº 13 - Regulamento de Boas Práticas de Fabricação de Produtos de Uso
Veterinário. Brasília, DF, 03 de Outubro de 2003.
NASSANI, M. Cleaning Validation in the Pharmaceutical industry. Journal of validation
Technology – Institute of Validation Technology, Agosto de 2005.
THE MERCK INDEX, 4ª Edição. Amitraz. United States, 2006.
THE MERCK INDEX, 4ª Edição. Deltamethrin. United States, 2006.

60
UNITED STATES PHARMACOPEIAL CONVETION, USP 37 – NF32. Description and
Solubility. United States, 01 de Agosto de 2014.
WORLD HEALTH ORGANIZATION, WHO. Annex 4: Suplementary guidelines on good
manufacturing practices: validation. Geneva, 2006.