apresentação do powerpointamericasoncologia.com.br/wp-content/uploads/2013/11/disturbios... ·...
TRANSCRIPT

DISTURBIOS PLAQUETÁRIOS
CARLOS EDUARDO PIZZINO
1

- Os distúrbios plaquetários são classificados como: • Desordens númericas (trombocitopenia e trombocitose) • Desordens funcionais - Plaquetas são fragmentos celulares provenientes dos megacariócitos - Valores normais: 150 mil – 450 mil/mcL (sendo 2,5% da população normal – trombocitopenia leve sem doença) - Produção diária: 35 – 50 mil/mcL por dia (sendo 1/3 sequestradas baço e 2/3 circulantes) - Duração: 8 -10 dias - Função das plaquetas: hemostasia primária
INTRODUÇÃO

HEMATOPOIESE

MEGACARIOPOIESE

Megacariócitos (geram 1000-5000 plaquetas/cada)

HEMOSTASIA PRIMÁRIA: -Adesão Plaquetária • Lesão do endotélio / exposição do colágeno • Interação da Glicoproteína (Gp) Ia/IIa (plaqueta) – colágeno • Ligação do fator Von Willebrand – Gp Ib (plaqueta) -Ativação e Agregação Plaquetária • Modificação da forma • Degranulação (liberação de ADP, cálcio) • Capacidade de se ligar umas as outras (Gp IIb/IIIa) • Liberação de Tromboxane A2 – retroalimentação positiva • Exposição de fosfolipídeos essenciais para a reação de coagulação
FORMAÇÃO DO TROMBO PLAQUETÁRIO

Hemostasia Primária

TROMBOCITOPENIA

DEFINIÇÃO: Valores inferiores a 150 mil/mcL (mm3) MECANISMOS DE TROMBOCITOPENIA: -Pseudotrombocitopenia -Destruição acelerada -Diminuição da produção medular / Trombopoiese ineficaz -Distribuição anormal -Diluicional

PSEUDOTROMBOCITOPENIA (ARTEFATUAL)
CLUMP PLAQUETÁRIO (EDTA)
AGLUTININAS PLAQUETÁRIAS
SATELITISMO PLAQUETÁRIO
PARAPROTEINEMIA
Pseudotrombocitopenia

Grumos plaquetários (EDTA)

Satelitismo plaquetário

Diminuição da produção plaquetária
Hipoplasia de células tronco hematopoiéticas
Quimioterápicos / Radioterapia
Anemia Aplástica
Álcool
Quadro pós viral (Rubéola, Varicela, Sarampo, EBV, HCV, Parvovírus)
HIV
Doenças Congênitas (Sd. Trombocitopenia com ausência de rádio –TAR, Trombocitopenia amegacariocítica, síndrome de Bernard-Soulier, síndrome de May-Heglin )

Substituição da medula óssea
Leucemias
Tumores metastáticos (Próstata, Mama, Linfoma)
Mielofibrose
Trombopoiese Ineficaz
Deficiência de B12 e folato
Síndromes mielodisplásicas

Aumento da destruição periférica
Por atividade imunológica
Purpura Trombocitopênica Imunológica Primária (PTI)
Neoplasias (Leucemia Linfóide Crônica, Linfoma)
Doenças imunológicas (Lupus Eritematoso Sistêmico, Poliarterite nodosa, Síndrome Antifosfolipídeo)
Infecção (Mononucleose por EBV, CMV, HIV, Dengue, HCV, H. pylori)
Medicamentos ( Heparina, Quinidina, Sulfas, Valproato)
Destruição Aloimune (Pós transfusões, Pós Transplante, Neonatal)

Não Imunológicos
Coagulação Intravascular Disseminada
Púrpura Trombocitopênica Trombótica / Síndrome Hemolítico-Urêmica
Síndrome HELLP (complicação da Eclampsia)
Hemangioma Cavernoso Gigante (Kasabach-Merritt)
Bypass Cardiopulmonar (Cardiopatia Cianótica Congênita)

Distribuição Anormal - HIPERESPLENISMO
% Plaquetas Marcadas após 2 horas de infusão

Diluicional
-Gestação
-Transfusões Maciças


MANIFESTAÇÕES CLÍNICAS:
-Assintomáticos -Sangramento cutâneo-mucoso • Epistaxe • Gengivorragia • Bolhas hemorrágicas em mucosa oral • Petéquias / Púrpuras / Equimoses -Sangramento gastrointestinal -Sangramento genito-urinário (Hematúria/Menorragia/Metrorragia) -Sangramento em Sistema Nervoso Central (SNC) -Sangramentos após procedimentos invasivos
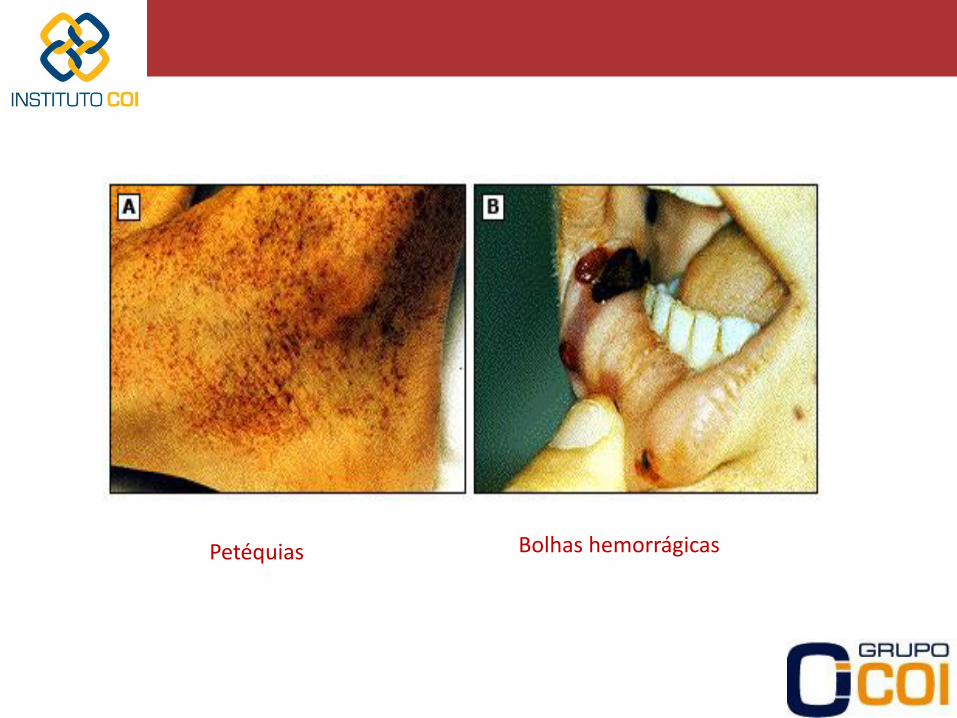
Petéquias Bolhas hemorrágicas
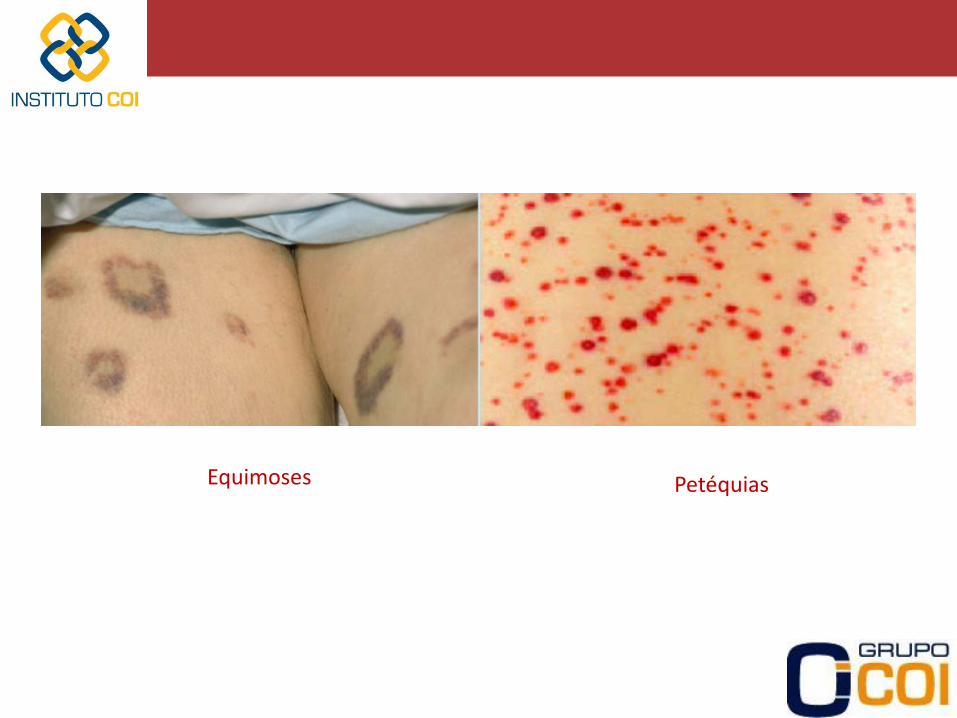
Equimoses Petéquias

Gengivorragia Sangramento SNC

DIAGNÓSTICO: -Anamnese (História Clínica) • Uso de medicamentos recentes • Infecção viral recente • Doença hematológica (Leucemias , Mielodisplasia) • História familiar de sangramento ou trombocitopenia • Gravidez • Transfusões recentes • Doenças não hematológicas (Sepse, Choque anafilático, Transfusões maciças, Hepatopatia) • Estado nutricional


- Hemograma completo (no citrato) • Contagem plaquetária Graus de trombocitopenia: Trombocitopenia leve: >100.000 plaquetas/mm3 Trombocitopenia moderada: 50.000-100.000 plaquetas/mm3 Trombocitopenia grave: <50.000 plaquetas/mm3 • Alterações nas outras séries (Anemia, Leucopenia ou Leucocitose)

- Análise do sangue periférico • Presença de artefatos (grumos plaquetários)

• Anormalidades morfológicas nas plaquetas (Plaquetas gigantes nas síndromes de Bernard-Soulier e May-Heglin)

• Presença de blastos (indicativo de leucemia aguda)

• Presença de leucoeritroblastose (invasão medular por neoplasia/tuberculose/ fibrose)

• Sinais de mielodisplasia (ex. neutrófilos hiposegmentados –pseudo Pelger-Huet)

• Presença de neutrófilos plurisegmentados e macrovalócitos (def. B12 e folato)

• Presença de esquizócitos (indicativo de hemólise microangiopática – Purpura Trombocitopênica Trombótica/Hemolítico Urêmica)

- Marcadores de hemólise (no caso de megaloblastose/anemia hemolítica) • Elevação de LDH • Queda da haptoglobina • Elevação dos reticulócitos • Elevação das Bilirrubinas com predomínio da forma indireta - Função renal - Dosagem de vitamina B12 e ácido fólico - Sorologias (HIV, HCV, HBV)

- Hepatograma completo (hepatopatia crônica como causa de hiperesplenismo) - FAN (marcadores de doença imunológica como Lúpus) - Coagulograma (TAP, PTT, Fibrinogênio – para descartar CIVD / Hepatopatia) - US de abdome: evidência de hepatopatia/esplenomegalia

- Aspirado e Biópsia de Medula Óssea • Útil para verificar a presença de megacariócitos numericamente aumentados
ou normais (aumento da destruição de plaquetas) ou ausência (diminuição da produção de plaquetas)
• Utilizado para trombocitopenia isolada no idoso (PTI x Mielodisplasia) • Verificar presença de infiltração medular (infecção, neoplasias) ou fibrose • Para diagnóstico de Aplasia Medular (se outras citopenias associadas)
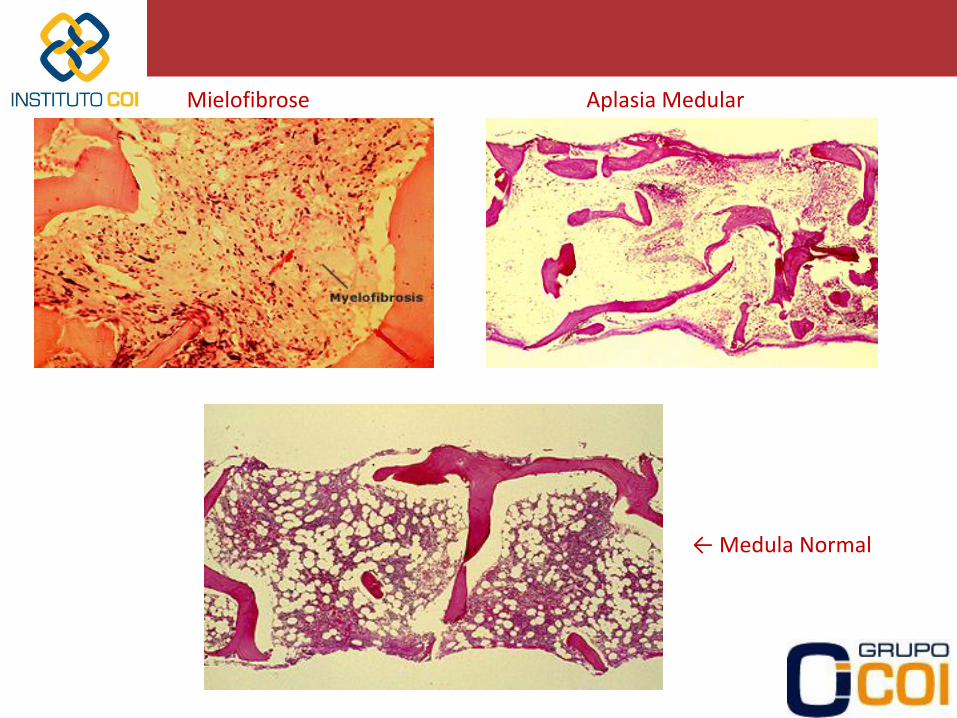
← Medula Normal
Aplasia Medular Mielofibrose

Megaloblastos
Neoplasia sólida

TROMBOCITOPENIA GESTACIONAL -Assintomática / Leve -Nenhuma história prévia (excetuando-se em gestações anteriores) -Final da gestação -Sem trombocitopenia fetal -Resolução espontânea após parto -Geralmente maior que 70000 plaquetas/mm3 -Efeito dilucional

TROMBOCITOPENIA INDUZIDA POR HEPARINA (HIT) INTRODUÇÃO: - Geralmente ocorre 5 a 10 dias após o inicio do uso da heparina - Duas formas – tipo I ou não imunológica e a tipo II ou imunológica - Incidência global de 2,6% dos pacientes expostos a heparina por mais de 4
dias (sendo 0,5% -HBPM e 1 a 4% Hep. não fracionada) - Fatores que influenciam esta incidência: • Uso de heparina não fracionada • Pacientes cirúrgicos / mulheres

Duas formas de HIT

FISIOPATOLOGIA - Formação de anticorpos contra o complexo heparina-fator-4 plaquetário

MANIFESTAÇÕES CLÍNICAS E DIAGNÓSTICO - Queda em mais de 50% do nível plaquetário basal em 5 a 10 dias - Raramente atingem contagem inferiores a 20 mil (mediana de 60mil) – logo
sangramentos são infrequentes - HIT relatada com doses habituais de heparina e doses baixas utilizadas em
hemodiálise, irrigação de cateteres vasculares ou procedimentos de revascularização
- TROMBOSE VENOSA E/OU ARTERIAL • Liberação de fatores pró-coagulantes provocados pela ativação plaquetária • Ligação dos anticorpos ao heparan-sulfato do endotélio – levando a ativação endotelial • Mortalidade de 25% - TVP/TEP - Necrose cutânea nos sítios de aplicação da heparina subcutânea

- Na suspeita clínica de HIT: • Testes Imunológicos – Detectam anticorpos circulantes (IgG, IgA e IgM), anti complexo Heparina - PF4 Alta sensibilidade > 97% Especificidade limitada 74 – 86%, também detectam Ac. Anti PF4 –heparina em pacientes sem TIH Valor preditivo negativo é alto (maior que 95%) • Teste Funcional – avalia ativação plaquetária e detecta Acs. Heparina dependente, capazes de ligação e ativação dos receptores Fc nas plaquetas Sensibilidade > 90% (dependendo da experiência do Labo) Especificidade 77 – 100% (dependendo do contexto clínico de exposição a heparina) Teste de liberação de Serotonina marcada com C (14) de plaquetas ativadas Sensibilidade: 88 – 100 % e Especificidade: 89 – 100 % (teste pouco disponível) Valor preditivo positivo alto (89- 100%), Valor preditivo negativo ( 81%)

TRATAMENTO - Suspensão da heparina - Se trombose presente: • outros anticoagulantes inibidores da trombina por 6 meses • paciente renal – argatroban doses convencionais • paciente hepatopata – fondaparinux • paciente renal / hepatopata – argatroban dose reduzida • warfarin pode ser usado se plaquetas acima de 150 mil após medicações acima - Se trombose ausente – observação x anticoagulação por 3 meses

ALGORITMO

PURPURA TROMBOCITOPÊNICA TROMBÓTICA / SÍNDROME HEMOLÍTICO URÊMICA
INTRODUÇÃO
- Microangiopatia trombótica (MAT) - quadro histológico gerado por alterações endoteliais em capilares e arteríolas que levam à trombose microvascular, o que resulta em anemia hemolítica microangiopática e trombocitopenia

CAUSAS DE MICROANGIOPATIA TROMBÓTICA
PÚRPURA TROMBOCITOPÊNICA TROMBÓTICA
SÍNDROME HEMOLÍTICA URÊMICA
COAGULAÇÃO VASCULAR DISSEMINADA
HIPERTENSÃO MALIGNA
ECLÂMPSIA
VASCULITE (LES/POLIARTERITE NODOSA/SAAF / ESCLERODERMIA)
HEMANGIOMA CAVERNOSO
REJEIÇÃO DE TRANSPLANTE RENAL
TRANSPLANTE DE MEDULA ÓSSEA

ACHADOS CLÍNICOS E LABORATORIAIS - Anemia hemolítica microangiopática:
Anemia hemolítica intravascular com Coombs direto negativo e fragmentação (esquizócitos) na hematoscopia

- Trombocitopenia - Insuficiência renal: aumento de escórias nitrogenadas, podem ocorrer
proteinúria e hematúria. - Febre
- Alterações neurológicas: presente em aproximadamente 30% dos casos,
manifesta-se geralmente por confusão mental e cefaleia, podendo ocorrer convulsão e coma.
PÊNTADE CLÁSSICA Anemia Microangiopática + trombocitopenia + Insuficiência renal
+ Febre + Alterações neurológicas MORTALIDADE – 90% SE PRESENTES

- Determinação da atividade da protease do FvW – ADAMTS13: • Enzima de clivagem do FvW, encontra-se com sua atividade reduzida nos casos de PTT idiopática ou congênita. Exame de difícil acesso e de validade questionável, uma vez que o resultado não é imediato e portanto não deve ser aguardado para decisão terapêutica.

CLASSIFICAÇÃO / TRATAMENTO CATEGORIA CLÍNICA FISIOPATOLOGIA TRATAMENTO
PTT idiopática
- Descartar CIVD ou qualquer outra causa secundária associada.
- Faixa etária de 30 a 50 anos
- Formas graves : Pêntade
Ocorre por uma deficiência auto-imune da ADAMTS13.
80% dos casos responde a plasmaférese e pode se beneficiar de imunossupressão.
PTT secundária
- Associada a condições como câncer, infecção, TMO quimioterapia ou uso de drogas como ciclosporina, quinidina e ticlopidina
Mecanismo normalmente desconhecido
Deve-se tratar a condição de base e suspender os medicamentos que estiverem envolvidos

CATEGORIA CLÍNICA FISIOPATOLOGIA TRATAMENTO
PTT congênita
Semelhante à PTT, síndrome rara podendo ter sua primeira manifestação em qualquer faixa etária
Causada por uma mutação no gene da ADAMTS13, o que leva a uma deficiência em sua atividade
Apenas com a infusão de plasma
SHU
- Insuficiência renal aguda, geralmente oligúrica
- Precedida por uma quadro de enterocolite hemorrágica
- crianças com idade inferior a 5 anos
Infecção por E. coli O157:H7
Auto-limitada após tratamento de suporte intensivo

CATEGORIA CLÍNICA FISIOPATOLOGIA TRATAMENTO
SHU atípica
- Não é precedida pelo
quadro diarreico
Ativação inadequada do sistema complemento
Semelhante à SHU

Suspeita diagnóstica = Iniciar Plasmaférese2
Suspeitou de PTT idiopática? Iniciar prednisona 1mg/kg ou metilprednisolona 125mg IV 12/12h3
Descobriu causa secundária? Suspender a plasmaférese e tratar a doença de base
Suspeitou de SHU? Manter somente com plasmaférese
Avaliar resposta
Resposta parcial, transitória ou nova manifestação neurológica? - Metilprednisolona 1g/dia 3 dias - Rituximabe 375mg/m2 1x/semana por 4 doses1 - Intensificar plasmaférese (2x/dia)
Resposta completa? (plaquetas > 150 mil por 48h) - Parar plasmaférese - Manter corticoide - Manter CVC
Contagem plaquetária normal por 1-2 semanas? - Remover CVC - Retirada lenta do corticóide
Recaída? - Reiniciar plasmaférese diária - Reiniciar corticoide - Rituximabe
TRATAMENTO

PURPURA TROMBOCITOPENICA
IMUNOLÓGICA (PTI)

INTRODUÇÃO -Doença adquirida mediada por anticorpos, caracterizada por trombocitopenia isolada (<100.000/mm3) sem qualquer doença subjacente e/ou desencadeante.
MECANISMO A trombocitopenia é decorrente da destruição de plaquetas, mediada por anticorpos, e da produção plaquetária menos eficaz.

INCIDÊNCIA
Maior nas mulheres entre 30 e 60 anos
• PTI aguda da infância tem incidência de 4,0-5,3/100.000/ano
• PTI crônica da infância tem incidência de 0,46/100.000/ano
• PTI crônica do adulto tem incidência de 5,8-6,6/100.000/ano
CLASSIFICAÇÃO
Recém-diagnosticada
Persistente (3-12 meses de duração)
Crônica (12 meses)

FISIOPATOLOGIA

MAIOR DESTRUIÇÃO:
Plaquetas revestidas por auto-anticorpos são fagocitadas por macrófagos e fagócitos esplênicos e hepáticos, que expressam receptores Fc
Os antígenos plaquetários correspondem principalmente às glicoproteínas IIb/IIIa e Ib/V/IX, mas também a GP Ia/IIa
Fagocitose plaquetária por células apresentadoras de antígenos
Auto-anticorpos produzidos por linfócitos B, com estimulação concomitante de linfócitos T-auxiliadores

Pesquisa de anticorpos antiplaquetas:
Baixa sensibilidade (50%) e especificidade (70%)
Ou seja:
Nem sempre positiva
Pode ser positiva em pacientes sem PTI

MENOR PRODUÇÃO DE PLAQUETAS:
Soro de pacientes com PTI inibe o crescimento de megacariócitos em cultura, demonstrando-se a menor produção plaquetária (1980)
Pequeno aumento das concentrações plasmáticas de trombopoetina

Níveis de Trombopoietina

CLASSIFICAÇÃO
- Trombocitopenia imune primária
- PTI secundária
Doenças autoimunes: LES, SAF, doença de Graves, síndrome de Evans
Doenças linfoproliferativas: LLC, LNH, macroglobulinemia de Waldenström
Doenças infecciosas: HCV, H.pylori, HIV, dengue
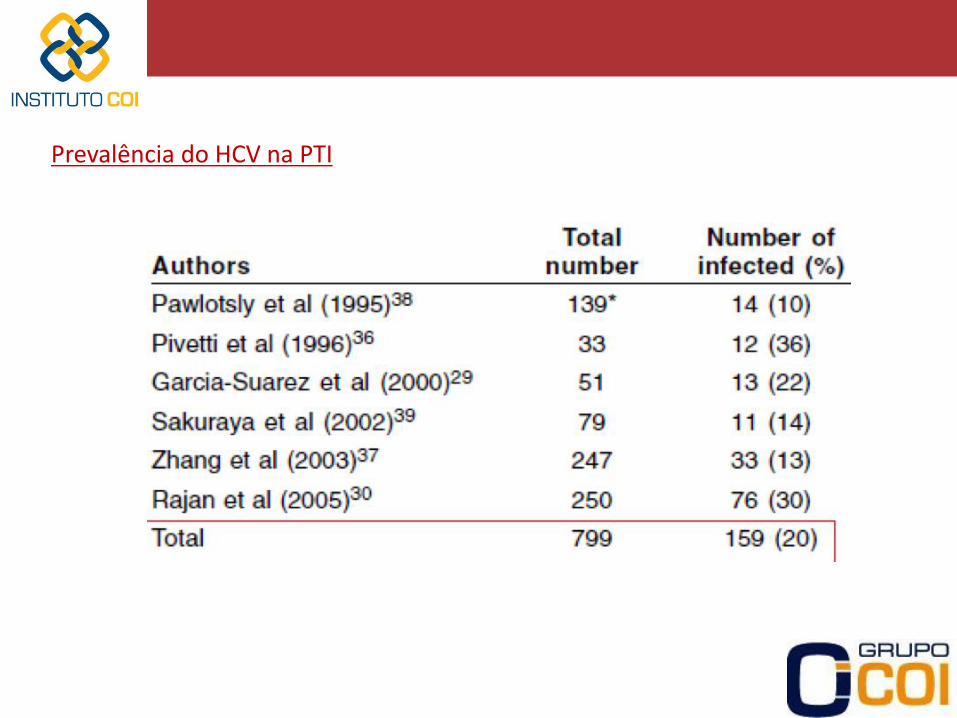
Prevalência do HCV na PTI

MANIFESTAÇÕES CLÍNICAS
- Paciente em bom estado geral
- Exame físico normal, exceto pela presença de sangramento
- Hemograma normal, exceto por trombocitopenia isolada
Graus de trombocitopenia:
Trombocitopenia leve: >100.000 plaquetas/mm3
Trombocitopenia moderada: 50.000-100.000 plaquetas/mm3
Trombocitopenia grave: <50.000 plaquetas/mm3
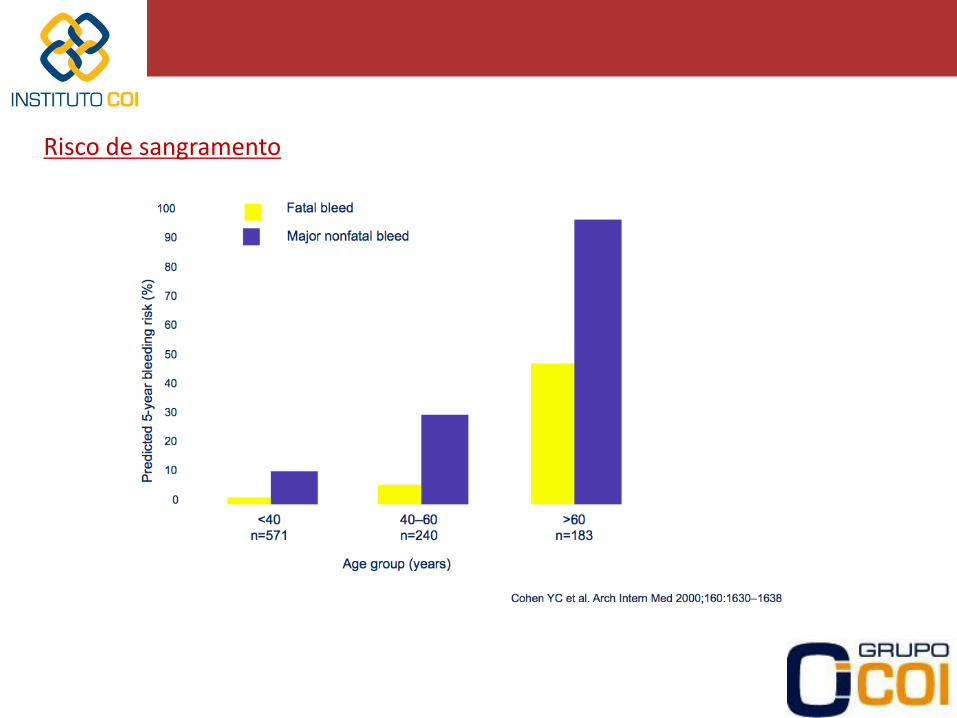
Risco de sangramento
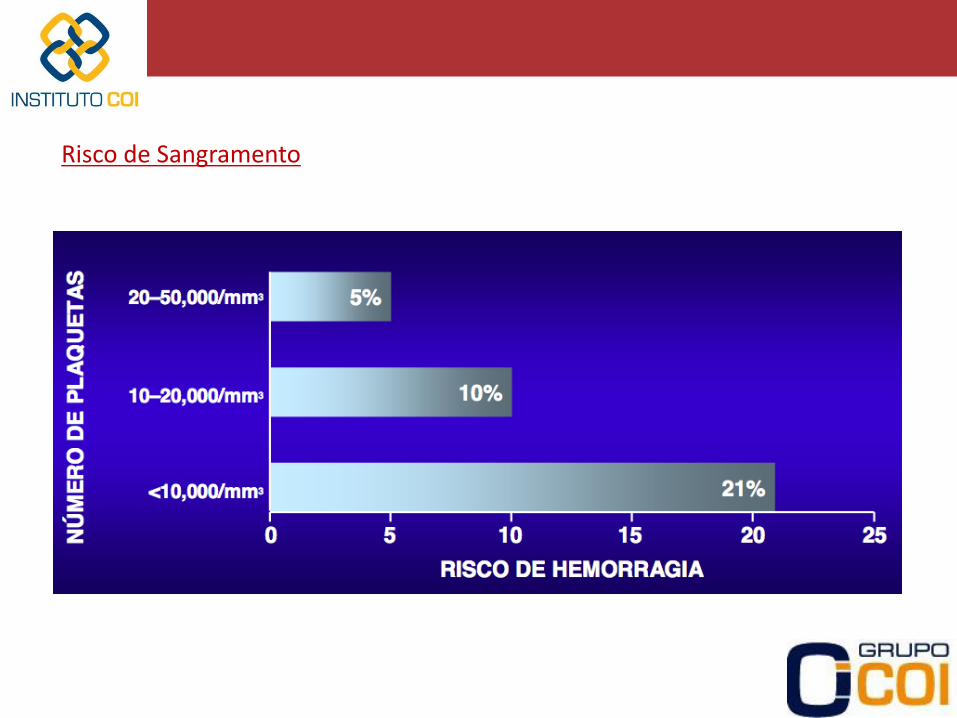
Risco de Sangramento

EQUIMOSES PETÉQUIAS

DIAGNÓSTICO: - Baseia-se na exclusão de outras causas de trombocitopenia (Sangue
periférico, hepatograma, US de abdomen, dosagem de B12, folato) - Pesquisar causas secundárias: • FAN, anticorpo antifosfolipídeos, anticoagulante lúpico • Sorologias para HCV, HIV, Dengue • EDA com pesquisa para H.pylori - Aspirado de medula óssea • no caso de idosos (diferenciar de Mielodisplasia) • suspeita de outra doença hematológica
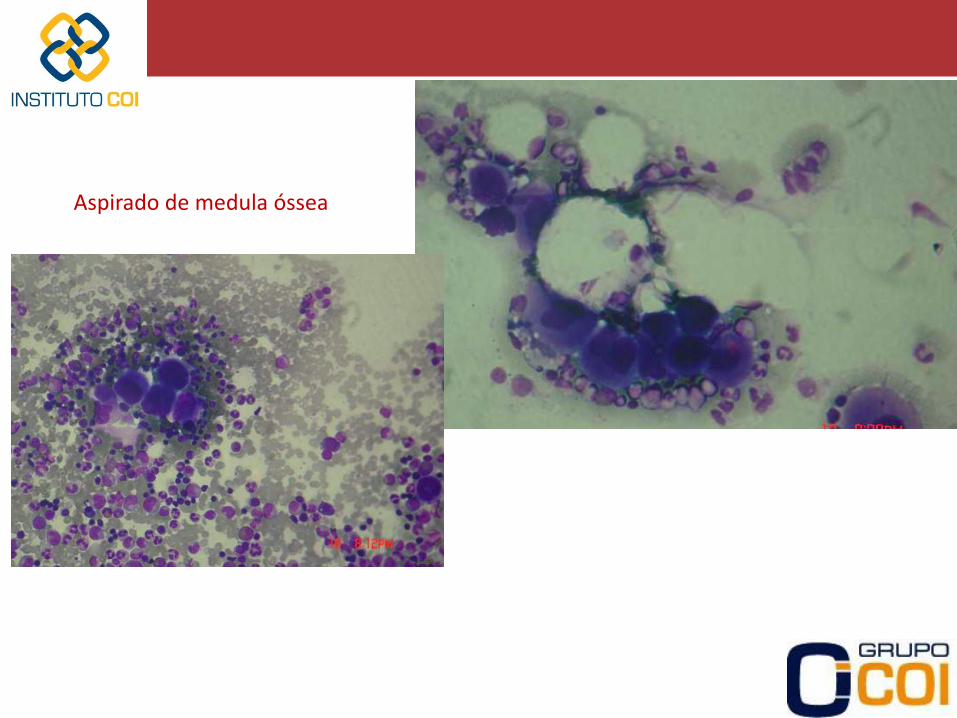
Aspirado de medula óssea

Crianças X Adultos

TRATAMENTO
- OBJETIVOS
Redução da produção de autoanticorpos
Aumento da sobrevida plaquetária
Aumento da produção de plaquetas

IIndicação de tratamento:
- Trombocitopenia moderada a grave que estão sangrando ou em risco para tal;

Visando reduzir a produção de autoanticorpos:
Corticosteróides (prednisona, prednisolona, dexametasona, metil-prednisolona)
Imunoglobulina
Rituximabe
Soro anti-D
Azatioprina
Vincristina
Ciclofosfamida
Ciclosporina A
Danazol
Dapsona
Micofenolato mofetil
Quimioterapia combinada

Visando aumentar a sobrevida plaquetária:
Imunoglobulina
Esplenectomia
Plaquetas incubadas com vincristina

Visando aumentar a produção plaquetária:
Corticosteróides
Agentes trombopoiéticos – Eltrombopag / Romiplastin

Tratamento Inicial: Glicocorticóides como agente único; - Regimes utilizados: • Prednisona 1mg/Kg/dia via oral (regime empregado pela maioria dos hematologistas); A maioria dos adultos responde a este esquema em duas semanas; • Dexametasona 40mg/dia via oral ou venosa por 4 dias repetidos a cada 14 a 28 dias. Número total de ciclos variando de 1 a 6. Sendo que o GINEMA sugere máximo benefício com 3 ciclos. (Blood 2007; 109:1401)

Uso de Imunoglobulina intravenosa (IVIG e anti-D) - Agentes capazes de elevar a contagem plaquetária em alguns dias, porém com efeitos durando algumas semanas; - Anti-D é efetivo somente em pacientes Rh positivos; - Doses preconizadas: • IVIG – 1g/Kg por dia administrado em dois dias; • Anti-D – 50 a 75 mcg/Kg por dia uma única vez;
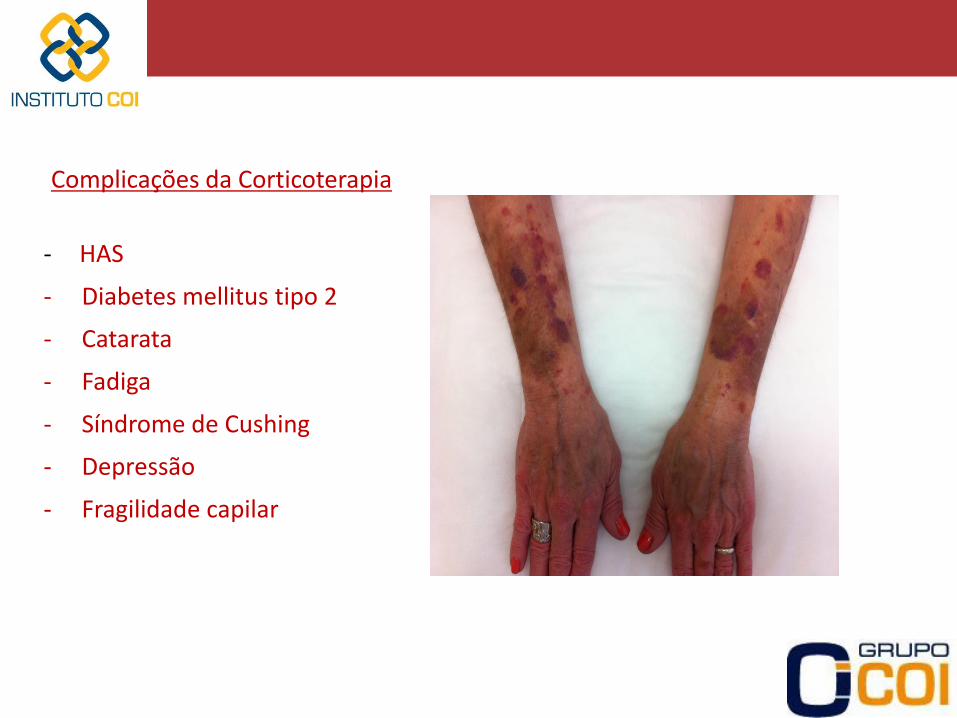
Complicações da Corticoterapia - HAS
- Diabetes mellitus tipo 2
- Catarata
- Fadiga
- Síndrome de Cushing
- Depressão
- Fragilidade capilar

Tratamento da PTI refratária: Definição: - Trombocitopenia grave e persistente; - Necessidade de terapias para sustentar ou aumentar as contagens plaquetárias

Esplenectomia

Ação dos agonistas da trombopoietina


DISTÚRBIOS FUNCIONAIS
PLAQUETÁRIOS
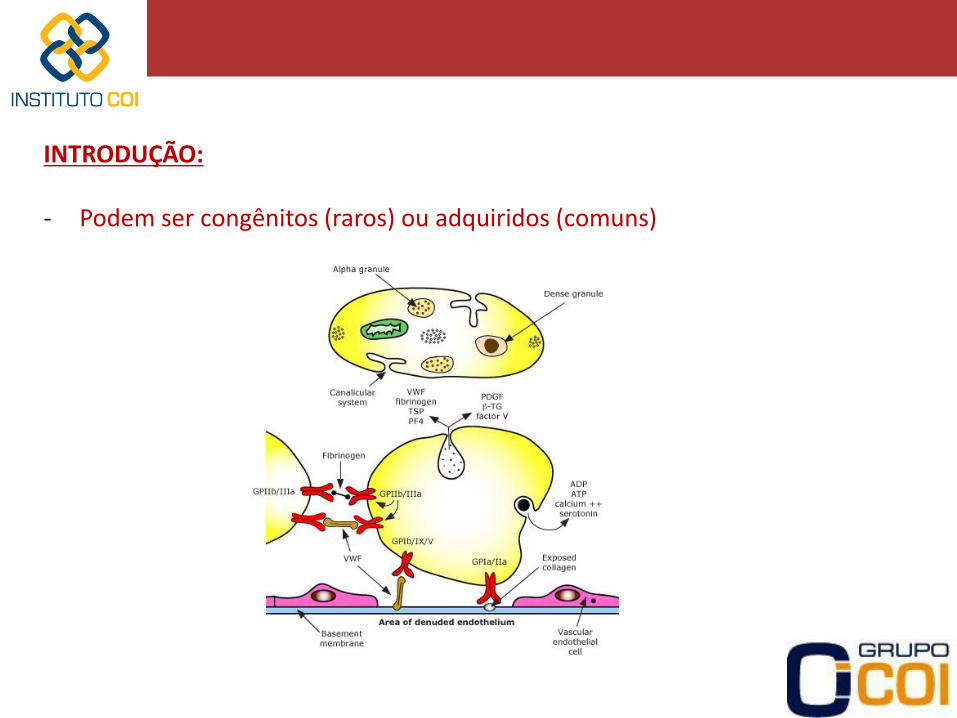
INTRODUÇÃO: - Podem ser congênitos (raros) ou adquiridos (comuns)

ETIOLOGIA
HEREDITÁRIAS ADQUIRIDAS
Doença de Von Willebrand Síndrome de Von Wilebrand adquirida
Síndrome Bernard Soulier Inibidores da COX (AAS/AINEs)
Deficiência de grânulos Antagonistas do ADP (Clopidogrel)
Síndrome de Wiskott-Aldrich Inibidores da GpIIB/IIIA (Tirofiban)
Trombastenia de Glanzmann Síndromes mieloproliferativas
Disfibrinogemia Uremia
Hipofibrinogemia Bypass cardiopulmonar
Paraproteinemias

MÉTODOS UTILIZADOS NO DIAGNÓSTICO DOS DISTÚRBIOS FUNCIONAIS PLAQUETÁRIOS - Hemograma e exame do sangue periférico • podem ter associação com contagens anormais plaquetárias • evidência de doença mieloproliferativa, mielodisplásica ou desordens plasmocitárias • presença de plaquetas gigantes ou pequenas

Rouleaux no mieloma múltiplo

• Anormalidades morfológicas nas plaquetas (Plaquetas gigantes nas síndromes de Bernard-Soulier e May-Heglin)

Tempo de sangramento
• Teste de screening para função plaquetária
• Não tem muita acurácia
• Não é válido quando há trombocitopenia
• Tem valor se estiver bem alargado (>10 min) diante de uma plaquetometria normal
• Nesta situação sugere: disfunção plaquetária ou então doença de Von Willebrand

Teste de Agregação Plaquetária • Utiliza-se diversos agonistas para aferir a ativação e agregação in vitro

DESORDENS ADQUIRIDAS: - Agentes Antiplaquetários • mais comum circunstância clínica • podem também estar associado a trombocitopenia em 2 a 13% dos casos → AAS – inibidor irreversível da COX (tanto 1 quanto 2) – bloqueando a produção de tromboxane A2 e levando inibição da agregação e vasoconstricção Dose antiagregante – 50 a 100mg/dia Risco pequeno de sangramento, a não ser nos casos de combinação com clopidogrel ou warfarin →Anti-inflamatórios não esteroidais (AINEs)- inibidor reversível da COX-1 Risco de sangramento gastrointestinal alto

Síntese de prostaglandinas

→ Dipiridamol – mecanismo de ação incerto
Associação com AAS baixa dose reduz em 37% o risco de recorrência de AVC
→ Clopidogrel e Ticlopidina – antagonistas do ADP
→ Antagonistas dos receptores GpIIb/IIIa (abciximab/eptifibatide/ tirofiban)
Utilizado em síndrome coronariana de alto risco
Podem levar a trombocitopenia imunomediada

Ação das medicações antiplaquetárias

- Doença renal com uremia • Decorrente do acúmulo de toxinas nitrogenadas que diminuem a adesividade, agregação e ativação plaquetária • Diagnóstico de exclusão – descartar lesões estruturais, heparina utilizada em diálise. - Paraproteinemias • pacientes com mieloma múltiplo ou macroglobulinemia de Waldenstrom podem ter disfunção plaquetária • Sangramentos neste pacientes podem ser provenientes também da hiperviscosidade ou doença de Von Willebrand - Desordens mieloproliferativas • Apesar de cursarem com trombocitose na maioria dos casos, anormalidades na agregação e degranulação das plaquetas tem sido demonstrados.
- Bypass cardio pulmonar

DESORDENS HEREDITÁRIAS: - TROMBASTENIA DE GLANZMANN • Condição autossômica recessiva • Deficiência da GpIIb/IIIa (fundamental para ligação ao fibrinogênio) • Tempo de sangramento prolongado + Ausência de clumps plaquetários + plaquetas não agregantes + plaquetas numericamente normais • Manifestações hemorrágicas moderada a graves em crianças < 5anos • Tratamento: Transfusões de plaquetas de repetição

-DESORDENS DAS PLAQUETAS GIGANTES
• Englobam três entidades: síndrome Bernard-Soullier (def. glicoproteína Ib), síndrome das plaquetas cinzas (deficiência de granulos alfa) e síndrome de May-Hegglin (inclusões neutrofílicas anormais, doença de Von Willebrand IIB associada) • Síndromes raras • Trombocitopenia inferiores a 50000/mm3 • Tratadas com transfusões de plaquetas

ALGORITMO DESORDENS HEREDITÁRIAS

TROMBOCITOSE

IIIIIIIIIIIIIIIIIIIIIIIIIIII INTRODUÇÃO
- Contagem maior que 450-500 mil plaquetas/mm3 - Cerca de 2,5% da população sem doença possuem plaquetas superiores a 450
mil - Duas questões a serem respondidas a cerca da trombocitose: • Fenômeno reacional ou marcador de doença hematológica clonal (neoplásica ou autônomo)? • Qual é o risco imediato para o paciente e como isto deve ser manejado?

DEFINIÇÕES:
TROMBOCITOSE REACIONAL→ ausência de desordem mielodisplásica / mieloproliferativa crônica em pacientes que apresentam condições que a justifiquem TROMBOCITOSE AUTÔNOMA→ presença de desordem mielodisplásica / mieloproliferativa crônica TROMBOCITOSE ESPÚRIA (PSEUDOTROMBOCITOSE)→ condições que geram uma falsa elevação das plaquetas. Ex.: Crioglobulinemia mista Fragmentos citoplasmáticos de pacientes com leucoses ou linfomas ou anemia hemolíticas

ETIOLOGIA:
CONDIÇÕES HEMATOLÓGICAS NÃO MALIGNAS
PERDA AGUDA DE SANGUE
ANEMIA HEMOLÍTICA AGUDA
ANEMIA FERROPRIVA
TRATAMENTO DA DEFICIÊNCIA DE B12
EFEITO REBOTE DO TRATAMENTO DA PTI (PURPURA TROMBOCIT. IMUNOL.)

CONDIÇÕES NEOPLÁSICAS
NEOPLASIAS SÓLIDAS METASTÁTICAS
LINFOMA
REBOTE APÓS USO DE AGENTES MIELOSUPRESSORES
DOENÇAS INFLAMATÓRIAS CRÔNICAS E AGUDAS
DESORDENS REUMATOLÓGICAS E VASCULITES
DOENÇA INFLAMATÓRIA INTESTINAL
DOENÇA CELÍACA
SÍNDROME POEMS (MIELOMA OSTEOESCLERÓTICO)

DANO TECIDUAL
QUEIMADURAS
INFARTO DO MIOCÁRDIO
TRAUMA SEVERO
ESPLENECTOMIA OU ASPLENIA FUNCIONAL
PÓS-OPERATÓRIO
PANCREATITE AGUDA
INFECÇÕES
INFECÇÕES CRÔNICAS
TUBERCULOSE

REAÇÃO MEDICAMENTOSA
VINCRISTINA
CORTICÓIDES
ÁCIDO TRANSRETINÓICO
HEPARINA DE BAIXO PESO
TROMBOPOIETINA E AGONISTAS
OUTRAS CAUSAS
EXERCÍCIOS
FALÊNCIA RENAL
SÍNDROME NEFRÓTICA
REAÇÕES ALÉRGICAS

MANIFESTAÇÕES CLÍNICAS: - Assintomáticos - Distúrbio vasomotor (cefaléia, sintomas visuais, dor torácica atípica,
disestesia acral e eritromelalgia) - Trombose - Sangramentos - Esplenomegalia

AVALIAÇÃO INICIAL: - Repetição do exame - Anamnese • História recente de trauma ou cirurgia • Esplenectomia • Sintomas sugestivos de infecção / inflamação • História passada ou presente de sangramento, trombose ou deficiência de ferro • Diagnóstico prévio de doença crônica hematológica • Perda de peso, fadiga ou sintomas que indique neoplasia

- Marcadores inflamatórios e/ou infecciosos • Proteína C reativa (normal nos casos de trombocitose autônoma) • VHS • Fibrinogênio • Ferritina (tb para diag. de ferropenia) - Hematoscopia • Corpúsculo de Howell-Jolly • Hemácias em alvo • Leucoeritroblastose • Hemácias em lágrima • Leucocitose reativa (desvio a esquerda)

Anemia Ferropriva

Corpúsculos de Howell-Jolly e hemácias em alvo (Asplenia funcional ou cirúrgica)

Corpúsculos de Dohle e granulações grosseiras (situações de infecção)

- Aspirado e Biópsia de Medula Óssea • Quando for descartado trombocitose reativa ou persistir dúvidas → investigar trombocitose autônoma • Estudos citogenéticos e moleculares para determinação de processo hematológico primário Investigação para: • LMC (Leucemia Mielóide Crônica) → Pesquisa de Cromossomo Philadelphia → Pesquisa do produto de fusão bcr-abl • Policitemia vera → Elevação de massa eritrocitária / saturação de O2 normal → Baixa concentração de eritropoietina → Presença da mutação da JAK-2 em 90% dos casos • Mielofibrose primária → Leucoeritroblastose / Hemácias em lágrima → Fibrose medular (aumento da reticulina)

• Síndromes mielodisplásicas → Síndrome do 5q- → Anemia refratária com sideroblasto em anel • Trombocitemia Essencial → Diagnóstico de exclusão ↓

MANEJO CLÍNICO: -Trombocitose reacional → Tratar causa / Hidroxiuréia - Sangramento • Descontinuar antiagregantes plaquetários (AAS/AINES) • Excluir a possibilidade de CIVD associado e deficiência de fatores da coagulação (principalmente V) → fazer plasma em caso de presença -Trombose • Se localização atípica → sugere trombocitose autônoma • Aferese de plaquetas se mais de 800 mil plaquetas • Pesquisa de trombofilias associadas • Terapia anticoagulante → 3 meses na ausência de trombofilias → 6 ou mais meses caso contrário -Distúrbios vasomotores • AAS em baixa dose (100mg/dia)

Trombocitose
Repetir hemograma
Anamnese e exame físico
Avaliação de sangue
periférico
Espúrio (fragmentos
de células)
Parar investigação
Corpúsculo de
Howell-Jolly Hemácia em alvo
Hipoesplenismo, asplenia
Hemácia em
lágrima, leucoeritro-
blastose
Avaliar síndrome
Mieloproli-ferativa
Aspirado e biópsia de
Medula óssea
Citogenética
Avaliar JAK-2 e BCR-ABL
Anomalia pseudo-
Peuger-Hüet
Avaliar síndrome
mielodisplásica
Granulações grosseiras
corpúsculo de Döhle
Reacional
Marcadores inflamatórios
(PCR, VHS)
Normais
Seguir investigação
Aumentados
Trombocitose reacional
Algoritmo:
ALGORITMO
