apostila laboratório quimica i , engenharia ambiental ufms
DESCRIPTION
Apostila da matéria de lab de quimica 1 de engenharia ambiental da ufmsTRANSCRIPT

1
Centro de Ciências Exatas e Tecnologia (CCET) Cursos de Química
2012
Apostila dos
Experimentos
LAB. QUÍMICA 1 - Engenharia Ambiental

2
Laboratório de Química 1- DQI
PROFESSORES: Dr. Amilcar Machulek Jr. (PO1- Engenharia Ambiental) Dr. Silvio C. de Oliveira (PO2- Engenharia Ambiental)

3
Laboratório de Química 1- DQI
ÍNDICE 1. INTRODUÇÃO AO TRABALHO EM UM LABORATÓRIO......................................... 2. SEGURANÇA NO LABORATÓRIO.................................................................................... 2.1 NORMAS BÁSICAS DE SEGURANÇA NO LABORATÓRIO................................................ 2.2 DESCARTE DE REJEITOS (RESÍDUOS)............................................................................ 2.3 ACIDENTES COMUNS EM LABORATÓRIO E PRIMEIROS SOCORROS..................................... 3. EXPERIÊNCIAS.................................................................................................................. EXPERIÊNCIA Nº 1 – VISITA À OFICINA DE VIDRARIA E MANUSEIO DE UM BICO DE BUNSEN............................................... EXPERIÊNCIA Nº 2 – TÉCNICAS DE PESAGEM E UTILIZAÇÃO DE BALANÇAS – ALGARISMOS SIGNIFICATIVOS, PRECISÃO E EXATIDÃO.................. EXPERIÊNCIA Nº 3 – MEDIDAS APROXIMADAS E PRECISAS DE VOLUME...................... EXPERIÊNCIA Nº 4 – MANUSEIO DO HANDBOOK OF CHEMISTRY AND PHYSICS E DO MERCK INDEX……………………………………...................... EXPERIÊNCIA Nº 5 – MÉTODOS USUAIS DE PURIFICAÇÃO DE SUBSTÂNCIAS E DE SEPARAÇÃO DOS COMPONENTES DE MISTURA............................... EXPERIÊNCIA Nº 6 – SEPARAÇÕES CROMATOGRÁFICAS................................................ EXPERIÊNCIA Nº 7 – TIPOS DE REAÇÕES QUÍMICAS EM SOLUÇÕES AQUOSAS................ EXPERIÊNCIA Nº 8 – PONTO DE FUSÃO.......................................................................... EXPERIÊNCIA Nº 9 – ÁGUA DE HIDRATAÇÃO.................................................................... EXPERIÊNCIA Nº 10 – CINÉTICA QUÍMICA.......................................................................... REFERÊNCIAS .......................................................................................................... TABELA PERIÓDICA ........................................................................................................
04 05 05 07 08 09 09 16 21 25
30 33 36
40 47 49
53 55

4
Laboratório de Química 1- DQI
1 INTRODUÇÃO AO TRABALHO NUM LABORATÓRIO
O Laboratório Químico é um lugar de experimentação onde os acadêmicos terão a oportunidade de aprender Química de um ponto de vista que nunca poderiam atingir por intermédio de livros, demonstrações ou filmes; é a possibilidade de alcançar maior compreensão da Química e a oportunidade de ver e trabalhar com as próprias mãos. Para atingir esses objetivos, são necessárias qualidades tais como dedicação, interesse, curiosidade, pontualidade, disciplina, etc.
A significação dos resultados obtidos dependerá muito do cuidado com que se desenvolverão as operações de laboratório. Boa técnica é mais do que uma questão de habilidade manual; requer uma atenção total aos propósitos essenciais da experiência. Técnicas de Química Experimental não são objetivos, mas sim os instrumentos que nos permitem atingir a meta final, de extrair informações úteis a partir de observações pessoais.
Aprender o manuseio de compostos e a manipulação de aparelhos é obviamente uma parte essencial à educação dos profissionais das Áreas de Ciências Exatas e Biológicas. Para ajudar o desenvolvimento de boas técnicas, várias sugestões são apresentadas:
- Nunca começar uma experiência sem antes compreendê-la totalmente; isto significa estudar o experimento antes de entrar no laboratório.
- Esmero é muito importante para uma boa técnica. Descuidar ao manusear compostos químicos e aparelhos, pode não somente levar a maus resultados, como também é perigoso. Há geralmente uma razão de como e porque cada operação é desenvolvida como descrita na literatura, embora a razão, a princípio, possa não ser óbvia para o estudante iniciante.
As aulas de laboratório têm por finalidade fazer com que você compreenda os princípios fundamentais da Química, através de métodos científicos elaborados, habilitando-o no manuseio correto e cuidadoso de drogas, aparelhos e utensílios.
Observe que o laboratório químico contém as seguintes características de segurança aos que nele trabalham.
Janelas amplas de ambos os lados que possibilitam boa ventilação do ambiente; Portas em dois locais distintos, que abram para fora (facilitam a saída em caso de emergência),
sendo uma das portas grande (dupla) para possibilitar a entrada de equipamentos; ♦Lava-olhos e chuveiro – dispositivos para uso em emergências; ♦Extintores de incêndio próximos ao laboratório. ♦Salas anexas para aparelhagem (balanças, aparelhos para ponto fusão, dentre outros); ♦Ampla iluminação e ♦Bancadas revestidas com material que permita fácil limpeza.
⇒ TRABALHO EM EQUIPE
Todos os trabalhos serão realizados por equipes de dois ou mais alunos. Compreenda, pois, o
seu papel e colabore para que os trabalhos realizados sejam o resultado de um esforço conjunto. Na solução de problemas surgidos esforce-se ao máximo para resolvê-los, consultando o professor sempre que for preciso. Procure estar presente na hora marcada para o início das aulas e evite saídas desnecessárias durante os trabalhos de laboratório.
⇒ RELATÓRIO DAS AULAS PRÁTICAS
É muito importante que o estudante tenha o seu caderno de laboratório para anotar todos os dados, observações e resultados obtidos em determinada experiência.
Todo profissional, no exercício de sua atividade, necessita se comunicar, seja sob a forma escrita ou oral. A elaboração de relatórios de aulas práticas consiste num treinamento de comunicação. O enfoque a ser dado a um relatório não é apenas o de responder a um questionário ou escrever

5
Laboratório de Química 1- DQI
aleatoriamente sobre o trabalho realizado; deve, porém, ser encarado como uma comunicação sobre uma atividade prática realizada, dirigida não apenas ao professor, mas a qualquer leitor que se interesse pelo assunto.
Antes de iniciar a elaboração de um relatório, é necessário pensar no assunto a ser relatado, analisar os aspectos importantes que devam ser abordados e planejar uma seqüência lógica de exposição. Com esta análise preliminar estarão sendo definidos os aspectos essenciais do trabalho a serem mencionados.
Para algumas aulas práticas realizadas, a critério do professor, deverá ser entregue um breve
relatório contendo: Nome do Curso; Nome da Disciplina; Nome do(a) Professor(a); Nome dos alunos; Titulo da prática executada; Introdução: Breve histórico sobre o processo de que trata o relatório. Situa o leitor sobre o
assunto a ser exposto; Resumo do Procedimento: Descrição breve dos procedimentos que serão utilizados. Fornece
informações básicas sobre a técnica empregada; Resultado(s) Obtido(s): Descrição dos dados colhidos na experiência, de preferência, quando
oportuno, em tabelas e/ou gráficos. Deverão constar, também, os cálculos necessários para a obtenção dos resultados. Todas as equações químicas envolvidas no processo deverão ser representadas;
Respostas às perguntas feitas (quando houver); Críticas, observações, dificuldades encontradas: A critério do acadêmico poderão ser feitas
criticas e observações sobre os resultados obtidos, possíveis causas de erros, sugestões para o emprego de outros métodos, etc. Poderão ser relatados, também, problemas ocorridos durante o processo de execução do experimento;
Conclusões: Análise dos resultados em função dos objetivos propostos. Poucas frases bem elaboradas para encerrar o trabalho.
Referências: Ao final de todo trabalho escrito ou oral, devem ser citados os autores que forneceram subsídios para sua confecção.
2 SEGURANÇA NO LABORATÓRIO
2.1 NORMAS BÁSICAS DE SEGURANÇA NO LABORATÓRIO
A segurança no laboratório é uma responsabilidade que deve ser assumida por professores, monitores e alunos. No recinto do laboratório não é permitida brincadeiras ou atitudes que possam provocar danos para si ou outras pessoas. Apesar disso, os laboratórios de química não são necessariamente lugares perigosos embora muito dos perigos estejam associados a eles. Acidentes são, na maioria das vezes, causados por falta de cuidado, ignorância e desinteresse pelo assunto.
Embora não seja possível enumerar todas as causas de possíveis acidentes num laboratório, existem alguns cuidados que são básicos e que, se observados, ajudam a evitá-los. São eles:
•No laboratório é OBRIGATÓRIO o uso do jaleco e de óculos de segurança (para quem
não usa óculos de grau). Procure manter seu jaleco limpo e, por questão de higiene, não utilize-o em locais sociais públicos, tais como restaurantes, bancos, etc.
•É PROIBIDO comer, beber ou fumar no laboratório; •Evite trabalhar sozinho no laboratório, a presença de outras pessoas será sempre uma valiosa
ajuda em caso de acidentes;

6
Laboratório de Química 1- DQI
•Prepare-se antes de tentar realizar os experimentos. Procure ler e entender os roteiros experimentais; consulte a literatura especializada. Em caso de dúvidas, discuta o assunto com o professor antes de tentar fazer o experimento;
•Utilize sempre que necessário materiais que possam garantir maior segurança no trabalho tais como: luvas, pinça, óculos (obrigatório), jaleco (obrigatório) etc.
•Conserve sempre limpos os equipamentos, vidrarias e sua bancada de trabalho. Evite derramar líquidos, mas se o fizer, limpe o local imediatamente;
•Gavetas e portas dos armários devem ser mantidas sempre fechadas quando não estiverem sendo utilizadas;
•Ao término do período de laboratório, lave o material utilizado, limpe sua bancada de trabalho, seu banco, a pia e outras áreas de uso em comum. Verifique se os equipamentos estão limpos e desligados e os frascos reagentes fechados;
•Lave suas mãos freqüentemente durante o trabalho prático, especialmente se algum reagente químico for respingado. Ao final do trabalho, antes de deixar o laboratório, lave as mãos;
•Leia com atenção os rótulos dos frascos de reagentes químicos para evitar pegar o frasco errado. Certifique-se de que o reagente contido no frasco é exatamente o citado no roteiro experimental;
•Nunca torne a colocar no frasco, o reagente não utilizado. Não coloque objeto algum nos frascos de reagentes, exceto o conta-gotas de que alguns são providos;
•Evite contato físico com qualquer tipo de reagente químico. Tenha cuidado ao manusear substâncias corrosivas como ácidos e bases - use a CAPELA;
•A diluição de ácidos concentrados deve ser feita adicionando-se o ácido, lentamente, com agitação constante, sobre a água - com essa metodologia adequada, o calor gerado no processo de mistura, é absorvido e dissipado no meio. NUNCA proceda ao contrário (água sobre o ácido);
•Nunca deixe frascos contendo reagentes químicos inflamáveis próximos à chama; •Não deixe nenhuma substância sendo aquecida por longo tempo sem supervisão; •Não jogue nenhum material sólido dentro das pias ou ralos. O material inútil (rejeito) deve
ser descartado de maneira apropriada; •Quando for testar um produto químico pelo odor, não coloque o frasco sobre o nariz.
Desloque os vapores que se desprendem do frasco com a mão para a sua direção; •Use a CAPELA para experiências que envolvem o uso ou liberação de gases tóxicos ou
corrosivos; •Não aqueça tubos de ensaio com a extremidade aberta voltada para si mesmo ou para alguém
próximo. Sempre que possível o aquecimento deve ser feito na CAPELA; •Não deixe recipientes quentes em lugares em que possam ser pegos inadvertidamente.
Lembre-se de que o vidro quente tem a mesma aparência do vidro frio; •Não pipete de maneira alguma, líquidos corrosivos ou venenosos, por sucção, com a boca.
Procure usar sempre a “pêra de sucção” para pipetar. •O bico de Bunsen deve permanecer aceso somente quando estiver sendo utilizado; •Não trabalhe com material imperfeito; •Em caso de acidentes, comunique o professor imediatamente. Ele deverá decidir sobre a
gravidade do acidente e tomar as atitudes necessárias; •Em caso de possuir alguma alergia, estar grávida ou em qualquer outra situação que possa ser
afetado quando exposto a determinados reagentes químicos, comunique o professor logo no primeiro dia de aula;
•Em caso de incêndio este deverá ser abafado imediatamente com uma toalha ou, se necessário, com o auxilio do extintor de incêndio apropriado;
•Comunique o professor, monitor ou técnico sempre que notar algo anormal no laboratório; •Faça apenas as experiências indicadas pelo professor. Caso deseje tentar qualquer
modificação do roteiro experimental discuta com o professor antes de fazê-lo. 2.2 DESCARTE DE REJEITOS (RESÍDUOS)

7
Laboratório de Química 1- DQI
Até há pouco tempo, os laboratórios descartavam seus rejeitos (resíduos) sem os cuidados
necessários; solventes voláteis eram evaporados (lançados para a atmosfera), sólidos eram descarregados em lixo comum e, líquidos e soluções, eram descartados na pia. Essas práticas não são recomendadas e, atualmente, existe uma preocupação maior no descarte de rejeitos químicos. Existem regras estabelecidas para o descarte de rejeitos, especialmente os perigosos; no entanto, muitas vezes são difíceis e de custo elevado para serem implementadas. Assim, na prática, procura-se, sempre que possível, minimizar a quantidade de resíduos perigosos gerados nos laboratórios de ensino.
Alguns procedimentos são adotados nesse sentido, como por exemplo: Redução da escala (quantidade de sustância) de produtos químicos usados nos experimentos; Substituição de reagentes perigosos por outros menos perigosos; Conversão dos resíduos para uma forma menos perigosa através de reação química, antes do
descarte; Redução dos volumes a serem descartados (concentrando as soluções ou separando os
componentes perigosos por precipitação); Recuperação dos reagentes para novamente serem utilizados. Instruções para descarte dos resíduos são fornecidas junto com as experiências. Quando os
resíduos gerados na experiência não forem perigosos, poderão ser descartados na pia de acordo com as seguintes instruções:
Soluções que podem ser jogadas na pia devem ser antes diluídas com água, ou jogar a solução
vagarosamente acompanhada de água corrente; Sais solúveis podem ser descartados como descrito em 1. Pequenas quantidades de solventes orgânicos solúveis em água (ex: metanol ou acetona)
podem ser diluídos antes de serem jogados na pia. Grandes quantidades desses solventes, ou outros que sejam voláteis, não devem ser descartados dessa maneira. No caso, tentar recuperá-los.
Soluções ácidas e básicas devem ter seu pH ajustado na faixa de 2 a 11 antes de serem
descartadas. Em caso de pequenos volumes dessas soluções (por exemplo, 10 mL ou pouco mais), essas podem ser diluídas e descartadas.
Em caso de dúvida, perguntar ao professor como proceder o descarte. Algumas orientações básicas: RESÍDUO INSOLÚVEL NÃO PERIGOSO: Papel, cortiça, areia, podem ser, descartados
em um cesto de lixo comum do laboratório. Alumina, sílica gel, sulfato de sódio, sulfato de magnésio e outros, devem ser embalados para evitar a dispersão do pó e descartados em lixo comum. Se esses materiais estiverem contaminados com resíduos perigosos, deverão ser manuseados de outra forma.
RESÍDUOS SÓLIDOS SOLÚVEIS NÃO PERIGOSOS: Alguns compostos orgânicos
(exemplo o ácido benzóico) podem ser dissolvidos com bastante água e descarregados no esgoto. Podem, também, ser descartados junto com resíduos insolúveis não perigosos. Caso estejam contaminados com materiais mais perigosos deverão ser manuseados de outra forma.
RESÍDUOS LÍQUIDOS ORGÂNICOS NÃO PERIGOSOS: Substâncias solúveis em água
podem ser descartadas no esgoto. Por exemplo, etanol pode ser descartado na pia do laboratório; 1-butanol, éter etílico e a maioria dos solventes e compostos que não são miscíveis em água, não podem ser descartados dessa maneira. Líquidos não miscíveis com a água deverão ser colocados em recipientes apropriados para líquidos orgânicos, para posterior tratamento.

8
Laboratório de Química 1- DQI
RESÍDUOS PERIGOSOS GENÉRICOS: Neste grupo estão incluídas substâncias como hexano, tolueno, aminas (anilina, trietilamina), amidas, ésteres, ácido clorídrico e outros. Deve-se ter especial atenção para as incompatibilidades, ou seja, algumas substâncias não podem ser colocadas juntas no mesmo recipiente devido à reação entre elas. Por exemplo, cloreto de acetila e dietilamina reagem vigorosamente; ambos são reagentes perigosos e seus rejeitos devem ser mantidos em recipientes separados. Compostos halogenados como 1-bromobutano, cloreto de t-butila e outros, também devem ser guardados em recipientes separados dos demais compostos.
ÁCIDOS E BASES INORGÂNICAS FORTES: Devem ser neutralizados, diluídos e então
descartados. AGENTES OXIDANTES E REDUTORES: Oxidar os redutores e reduzir os oxidantes
antes do descarte. O professor dará informações de como proceder. Esses são alguns exemplos de procedimentos de descarte de rejeitos produzidos no
Laboratório Químico. É prática comum, antes de iniciar em experimento, buscar na literatura especializada informações sobre os efeitos tóxicos das substâncias que serão utilizadas e os cuidados necessários para manuseio e descarte das mesmas.
2.3 ACIDENTES COMUNS EM LABORATORIO E PRIMEIROS SOCORROS I. Queimaduras Causadas pelo calor - quando leves, aplicar pomada de Picrato de Butesina e, quando graves,
devem ser cobertas com gaze esterilizada, previamente umedecida com solução aquosa de bicarbonato de sódio 5%.
Causadas por ácidos - deve-se lavar imediatamente a região com bastante água durante pelo
menos 5 minutos. Em seguida, tratar com solução de bicarbonato de sódio a 5% e lavar novamente com água. Secar o local e aplicar Merthiolate.
Causadas por bases - proceder como em b, aplicando solução de ácido acético 1%. II. Ácidos nos olhos – Deve-ser lavar com bastante água durante aproximadamente 15
minutos e aplicar solução de bicarbonato de sódio 1%. III. Bases nos olhos – Proceder como em II e aplicar solução de ácido bórico 1%. IV. Intoxicação por gases – Remover a vítima para um ambiente arejado e deixar descansar.
Em caso de asfixia fazer respiração artificial. V. Ingestão de substâncias tóxicas – Recomenda-se beber muita água e em seguida beber: Um copo de solução de bicarbonato de sódio 1% ou leite de magnésia, em caso de ingestão de
ácidos; Um copo de solução de ácido cítrico ou ácido acético a 2%, em caso de ingestão de bases.

9
Laboratório de Química 1- DQI
3 EXPERIÊNCIAS
EXPERIÊNCIA No 1 – VISITA À OFICINA DE VIDRARIA E MANUSEIO DE UM BICO DE BUNSEN
1 Oficina de Vidraria Observação do trabalho de um técnico de vidraria na confecção de aparelhos de vidro
utilizados em laboratório químico 2 Uso do bico de Bunsen Há vários tipos de bicos de gás usados em laboratório, tais como: bico de Bunsen, bico de
Tirril, bico de Mecker, etc. Todos, entretanto, obedecem ao mesmo princípio de funcionamento: o gás combustível é introduzido em uma haste vertical, onde há uma abertura para a entrada de ar atmosférico, sendo queimado na sua parte superior. Tanto a vazão do gás como a entrada de ar podem ser controlados de forma conveniente.
Como se vê na Figura 1a, com o regulador de ar primário parcialmente fechado, distinguimos
três zonas de chama. Abrindo-se registro de ar, dá-se entrada de suficiente quantidade de O2 (do ar), dando-se na
região intermediária combustão mais acentuada dos gases, formando, além do CO, uma maior quantidade de CO2 e H2O, tornando assim a chama quase invisível.
As reações químicas básicas da combustão são: 2H2 + O2(ar) → 2H2O 2C + O2(ar) → 2CO 2CO+ O2(ar) → 2CO2 O bico de Bunsen é usado para a quase totalidade de aquecimentos efetuados em laboratório,
desde os de misturas ou soluções de alguns graus acima da temperatura ambiente, até calcinações, feitas em cadinhos, que exigem temperaturas de cerca de 6000C. Procedimentos mais avançados de laboratório podem requerer mantas com aquecimento elétrico, chapas elétricas, banhos aquecidos eletricamente, maçaricos oxiacetilênicos, fornos elétricos e outros.
Para se aquecerem bequer, erlenmeyer, balões etc., não se deve usar diretamente o bico de
Bunsen; estes aquecimentos são feitos através da tela de amianto, cuja função é deixar passar o calor uniformemente e não permitir que passe a chama.
Para acender o bico do gás, proceda da seguinte maneira: ♦Feche completamente a entrada de ar no bico; ♦Abra lentamente a válvula do gás e aproxime a chama de um fósforo lateralmente, obtendo
uma chama grande e luminosa, de cor amarela. ♦Abra vagarosamente a entrada de ar de modo que a chama fique completamente azul.
Verifique os diferentes tipos de chama na Figura 2a; ♦Caso a chama se apague ou haja combustão no interior do tubo, feche a entrada do gás e
reinicie as operações anteriores. O gás combustível é geralmente o gás de rua ou o G.L.P. (gás liquefeito de petróleo). O comburente, via de regra, é o ar atmosférico.

10
Laboratório de Química 1- DQI
Figura 1a – Queimador de gás (Bico de Bunsen)
Zona externa: Violeta pálida, quase invisível, onde os gases fracamente expostos ao ar sofrem combustão completa, resultando em CO2 e H2O. Esta zona é chamada de zona oxidante (Temperaturas de 1560-1540ºC).
Zona intermediaria: Luminosa, caracterizada por combustão incompleta, por deficiência do suprimento de O2. O carbono forma CO, o qual se decompõe pelo calor, resultando diminutas partículas de C (carbono) que, incandescentes, dão luminosidade à chama. Esta zona é chamada de zona redutora (Temperaturas abaixo de 1540ºC).
Zona interna: Limitada por uma “casca” azulada contendo os gases que ainda não sofreram combustão – mistura carburante (Temperaturas em torno de 300ºC).
.
Figura 1b – Diferentes chamas obtidas com um bico de Bunsen

11
Laboratório de Química 1- DQI
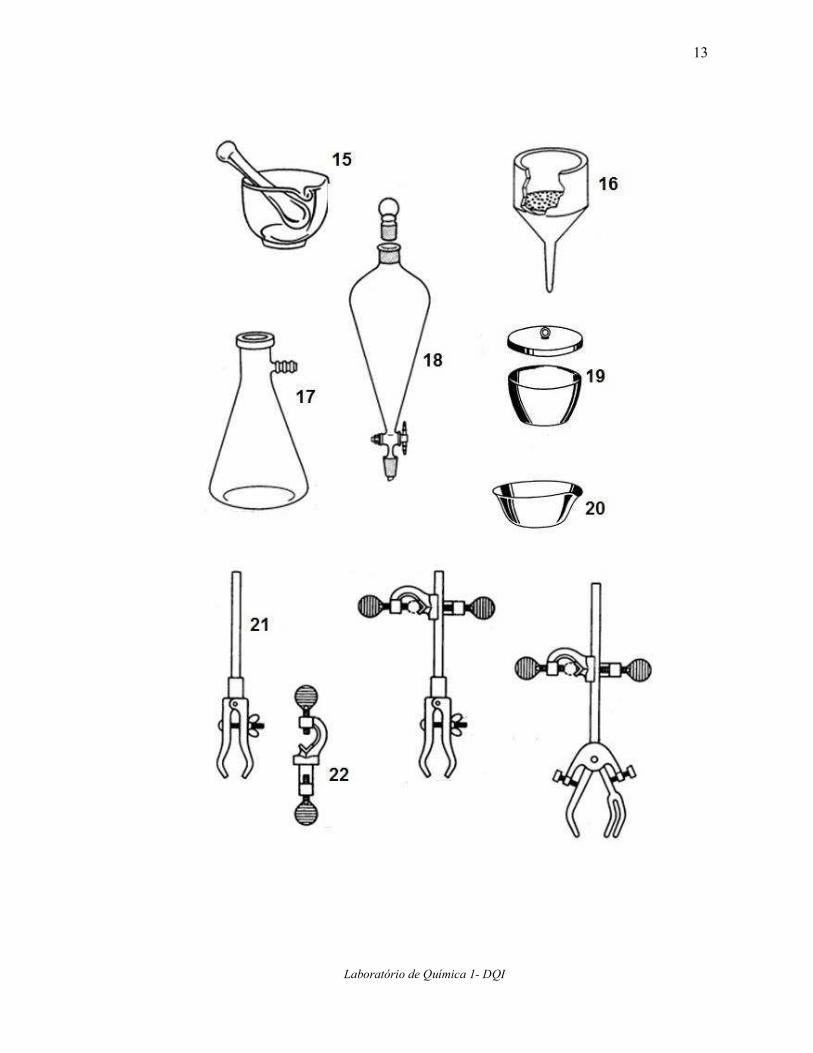
3. Reconhecendo os equipamentos básicos de laboratório Observe com atenção as explicações do professor a anote o nome do equipamento e sua
função na tabela a seguir.

12
Laboratório de Química 1- DQI
13
Laboratório de Química 1- DQI

14
Laboratório de Química 1- DQI
Nome Função
1
2
3
4
5
6

15
Laboratório de Química 1- DQI
Nome Função
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29

16
EXPERIÊNCIA No 2 – TÉCNICAS DE PESAGEM E UTILIZAÇÃO DE BALANÇAS – ALGARISMOS SIGNIFICATIVOS, PRECISÃO E EXATIDÃO I Introducão I.1 Utilização de Balanças
Uma das mais comuns e importantes operações de laboratório é a determinação de massa ou “pesagem”. O termo pesagem se refere à medida de massa de um corpo que é feita por comparação com massas conhecidas, com a utilização de balanças. Há uma grande variedade de balanças de laboratório, desde as mais grosseiras até as de mais alta sensibilidade. É comum se encontrar, por exemplo, balanças de escala tripla, para determinação de massas até centenas de gramas, com precisão de ± 0,1 g ou ± 0,01 g, e balanças analíticas, para carga máxima de 160 g, com precisão de ± 0,0001 g e até com 5 casas decimais.
Balanças de plataforma: Utilizadas para pesagem de 0,1g a centenas de gramas.
Balanças Elétricas/Eletrônicas: A cada dia, as balanças estão se modernizando, tornando-se mais exatas e de manejo mais simplificado. Atualmente, as balanças eletrônicas têm escala digital, fornecendo o peso instantaneamente, sem necessidade de manipular botões. Têm precisão mais apurada.
Cuidados Gerais com Balanças de Laboratórios O manejo de qualquer balança requer cuidados especiais por ser um instrumento de alto custo e de grande sensibilidade. São eles:
♦Não remova os pratos, nem os troque com os de outra balança. Mantenha a balança no seu lugar;
♦Não coloque na balança nenhuma substância que não esteja à temperatura ambiente; ♦Mantenha a balança em local onde a vibração, mudanças bruscas de temperatura ou de
umidade e movimento do ar sejam mínimos;

17
Laboratório de Química 1- DQI
♦Conserve a balança sempre limpa, retirando qualquer respingo, partículas ou poeira de seus pratos com uma escova especial;
♦Nunca coloque qualquer objeto diretamente sobre a balança. Líquidos e sólidos, em pó ou granulado, devem ser mantidos em algum recipiente seco, previamente pesado (tarado) e à temperatura ambiente. Se, durante a pesagem, o material for passível de interagir com a atmosfera (evaporação, oxidação, absorção de umidade), o frasco deve ser fechado. Para sólidos que não requerem proteção da atmosfera e que sejam inertes, a pesagem é feita colocando-se sobre os pratos, uma folha de papel adequado;
♦Toda transferência de substância e/ou de pesos, deve ser feita somente quando os pratos estivem travados;
♦Execute todas as operações com movimentos suaves e cuidadosos;
♦Use pinças e espátulas; nunca use os dedos para manusear os objetos e substâncias que estão sendo pesadas;
♦Ao terminar seu trabalho, remova todos os pesos e objetos da balança. Mantenha-a coberta ou fechada. No caso de balanças elétricas, tenha a certeza de que ela esteja desligada. I.2 Notação Científica, Algarismos Significativos, Erro de Uma Medida, Precisão e Exatidão NOTAÇÃO CIENTÍFICA, é também denominada por padrão ou notação em forma exponencial, é uma forma de escrever números que acomoda valores demasiado grandes ou pequenos para serem convenientemente escritos em forma convencional. O uso desta notação está baseado nas potências de 10.
Um número escrito em notação científica segue o seguinte modelo:
m x 10e onde m é denominado mantissa e e a ordem de grandeza
A mantissa SEMPRE será um valor em módulo entre 1 e 10. Exemplo: O valor 0,0000000586 em notação científica torna-se 5,86 (avanço de 8 casas) → 5,86 . 10-8. ALGARISMOS SIGNIFICATIVOS são os algarismos exatos e o duvidoso em uma medida. A contagem dos algarismos significativos é feita do primeiro algarismo diferente de zero, da esquerda para a direita. As potências de base 10 não contam como algarismos significativos. Exemplo:

18
Laboratório de Química 1- DQI
Quando você realizou as medidas com a régua milimetrada (Fig.2.1) do espaço S, você colocou duas casas decimais. é correto o que você fez? Sim, porque você considerou os algarismos significativos. O que são os algarismos significativos? Quando você mediu o valor de S = 5,81 cm com a régua milimetrada você teve certeza sobre os algarismos 5 e 8, que são os algarismos corretos (divisões inteiras da régua), sendo o algarismo 1 avaliado denominado duvidoso. Consideramos algarismos significativos de uma medida os algarismos corretos mais o primeiro duvidoso.
Algarismos significativos = algarismos corretos + primeiro algarismo duvidoso
5,81 5,8 1
Sempre que apresentamos o resultado de uma medida, este será representado pelos algarismos significativos. Veja que as duas medidas 5,81cm e 5,83m não são fundamentalmente diferentes, porque diferem apenas no algarismo duvidoso. Regras para arredondamento de números ao ajustar os Algarismos Significativos Para efetuar um arredondamento de um número, poderemos considerar duas situações distintas: • Se o algarismo a suprimir for inferior a 5, mantém-se o algarismo anterior. Exemplo: 3,234 → 3,23 • Se o algarismo a suprimir for igual ou superior a 5, acrescenta-se uma unidade ao algarismo anterior. Exemplo: 4,38 → 4,4 PRECISÃO E EXATIDÃO Precisão é o grau de variação de resultados de uma medição. Representa a dispersão de resultados entre ensaios independentes , repetidos de uma mesma amostra. Não é o mesmo que exatidão, que se refere à conformidade com o valor real. A precisão é avalidada pelo desvio-padrão de uma série de repetições da mesma análise. O desvio-padrão (DP) pode ser definido como:

19
Laboratório de Química 1- DQI
Em que: DP = Desvio Padrão Xi = valor individual de cada medida (X1, X2, X3... Xn)
= média aritmética dos valores Xi
n = número de medidas E o Coeficiente de Variação (CV) ou desvio padrão relativo percentual como:
CV = DP x 100 X Exatidão é o grau de concordância entre o valor médio obtido dos resultados e o valor de referência aceito como verdadeiro. Pode ser expresso como Erro Relativo Percentual (ER) ou Valor Percentual de Acerto. ER = Vmedido – Vref x 100,00 Vref
Exemplo: Vmedido = 1,02 mg/kg ; Vref = 1,15 mg.kg ER = 11,3% Exatidão = 100,00 – 11,3 = 88,7%
X
DP =

20
II Procedimento Experimental Efetue as operações de pesagem dadas a seguir. Pesagem de um sólido. Pese 1 béquer de 50 mL em duas balanças: semi analítica (0,01 mg) e analítica (0,0001 mg) em triplicatas. Pese 1,00 g de amostra dentro do bequer, considerando o valor médio da massa do béquer. Anote o valor com três algarismo significativo. Retire o béquer do prato da balança, zere novamente a balança e peso o conjunto. Repita o procediemnto mais uma vez. Leve o conjunto (bequer + amostra) a uma balança analítica, anote o valor massa com cinco algarismo significativo, e determine a massa da amostra pela diferença: massa da amostra = massa (bequer + amostra) – massa do bequer Tabela 1: Comparação de medidas de pesagens do béquer Balanças
Bequer vazio m DP CV
Resultado (valor médio±±±±DP m1 m2 m3
semi- analitica Analitica Tabela 2: Comparação das massas da amostra em duas balança Balanças
amostra m DP CV
Resultado (valor médio±±±±DP m1 m2 m3
semi- analitica Analitica

21
Laboratório de Química 1- DQI
EXPERIÊNCIA No 3 – MEDIDAS APROXIMADAS E PRECISAS DE VOLUMES INTRODUÇÃO 3.4.1. INTRODUÇÃO De um modo geral, para medidas aproximadas de volumes de líquidos, usam-se cilindros graduados ou provetas, enquanto, para medidas precisas, usam se pipetas, buretas e balões volumétricos, que constituem o chamado material volumétrico.
Aparelhos volumétricos são calibrados pelo fabricante e a temperatura padrão de calibração é de 20oC. Em trabalhos de laboratório, as medidas de volume aproximadas são efetua-das na quase totalidade dos casos com provetas graduadas, e de modo muito grosseiro com béqueres com escala e, as medidas volumétricas chamada precisas, com aparelhos volumétricos
APARELHOS VOLUMÉTRICOS: A prática de análise volumétrica requer a medida de volumes líquidos com elevada precisão. Para efetuar tais medidas são empregados vários tipos de aparelhos, que podem ser classificados em duas categorias: a)Aparelhos calibrados para dar escoamento a determinados volumes. b)Aparelhos calibrados para conter um volume do líquido. Na primeira classe (a) estão contidas as pipetas e as buretas e, na segunda (b), estão incluídos os balões volumétricos. A medida de volumes líquidos com qualquer dos referidos aparelhos está sujeita a uma série de erros devidos às seguintes causas: a)Ação da tensão superficial sobre superfícies líquidas. b)Dilatações econtrações provocadas pelas variações detemperatura. c)Imperfeita calibração dos aparelhos volumétricos. d)Erros de paralaxe.
A leitura de volume de líquidos claros (transparentes) deve ser feita pela parte inferior, ou seja, na tangente ao menisco, estando a linha de visão do operador perpendicular à escala graduada e a de líquidos escuros pela parte superior. BALÕES VOLUMÉTRICOS: Os balões volumétricos são balões defundo chato e gargalo comprido calibrados para conter determinados volumes líquidos. Os balões volumétricos são providos de rolhas esmerilhadas e de polietileno. O traço de referência marcando o volume pelo qual o balão volumétrico foi calibrado é gravado sobre a meia altura do gargalo. A distância entre o traço de referência e a boca do gargalo deve ser relativamente grande para permitir a fácil agitação do líquido , quando, depois de completado o volume até a marca, se tem de homogeneizar uma solução. O traço de referência é gravado sob a forma de linha circular, tal que, por ocasião da observação, o plano tangente à superfície inferior do menisco tem que coincidir com o plano do círculo de referência.

22
Laboratório de Química 1- DQI
FIGURA: Métodoapropriadoparaleituradomenisco. Os balões volumétricos são construídos para conter volumes variados; os mais usados são os de 50, 100, 200, 500, 1000 e 2000 mL, e são especialmente usados na preparação de soluções de concentração conhecida. Para se preparar uma solução e mum balão volumétrico, transfere-se ao mesmo os soluto ou a solução a se rdiluída. Adiciona-se a seguir solvente até cercade ¾ da capacidade total do balão, mistura se os componentes e deixa se em repouso até atingir a temperatura ambiente. Adiciona-se solvente até “acertar o menisco”, isto é, até o nível do líquido coincidir com a marca no gargalo. As últimas porções de solvente devem ser adicionadas com um conta gotas, lentamente, e não devem ficar gotas presas no gargalo. Fecha-se bem o balão e vira-se o mesmo de cabeça para baixo, várias vezes, agitando-o, para homogeneizar o seu conteúdo. PIPETAS: Existem dois tipos de pipetas: pipetas volumétricas ou de transferência, construídas para dar escoamento a um determinado volume líquido, que são mais precisas, e as pipetas graduadas ou cilíndricas, que possuem escalas permitindo escoar volumes variáveis de líquidos. Entre os dois tipos de pipetas há ainda uma sub divisão, que se refere ao modo de escoamento: total e parcial. Escoamento total apresenta duas faixas estreitas acima do código de cor (indicativo do volume da pipeta) e escoamento parcial a presenta uma faixa estreita logo acima da faixa de código de cor. As pipetas volumétricas são constituídas por um tubo de vidro com um bulbo na parte central. O traço de referência é gravado na parte do tubo acima do bulbo.A extremidade inferior é afilada e o orifício deve ser ajustado demodo que o escoamento não se processe rápido demais, o que faria com que pequenas diferenças de tempo de escoamento ocasionassem erros apreciáveis. As pipetas volumétricas são construídas com as capacidades de 1, 2, 5, 10, 20, 50, 100 e 200mL, sendo de uso mais freqüente as de 25 e 50 mL. As pipetas graduadas consistem de um tubo de vidro estreito e, geralmente graduadas em 0,1mL. São usadas para medir pequenos volumes líquidos. Para se encher uma pipeta, coloca-se a ponta no líquido e faz-se sucção tomando o cuidado de manter a ponta da mesma sempre abaixo do nível da solução ou líquido. Caso contrário,ao se fazer a sucção, o líquido alcança a pêra de borracha ou a boca. Quando o líquido é inócuo (água por ex.) não há perigo de se pipetar com a boca, des de que se tome certos cuidados para que não ocorra contaminação pela saliva. A sucção deve ser feita até o líquido ultrapassar o traço de referência. Feito isto, tapa-se a pipeta como dedo indicador (ligeiramente úmido) e deixa-se escoar o líquido lentamente até o traço de referência (zero). O ajuste deve ser feito de maneira a evitar erros de paralaxe. Para escoar os líquidos, deve-se colocar a pipeta na posição vertical, com a ponta encostada na parede do recipiente que vai receber o líquido; levanta-se o dedo indicador até que o líquido escoe totalmente. Espera-se 15 ou 20 segundos e retira-se a gota aderida à ponta da pipeta. Buretas - As buretas servem para dar escoamento a volumes variáveis de líquidos. São constituídas de tubo de vidro uniformemente calibrados, graduados em 0,1mL. São providas de dispositivos permitindo o fácil controle de escoamento. O dispositivo consiste de uma torneira de vidro ou de

23
Laboratório de Química 1- DQI
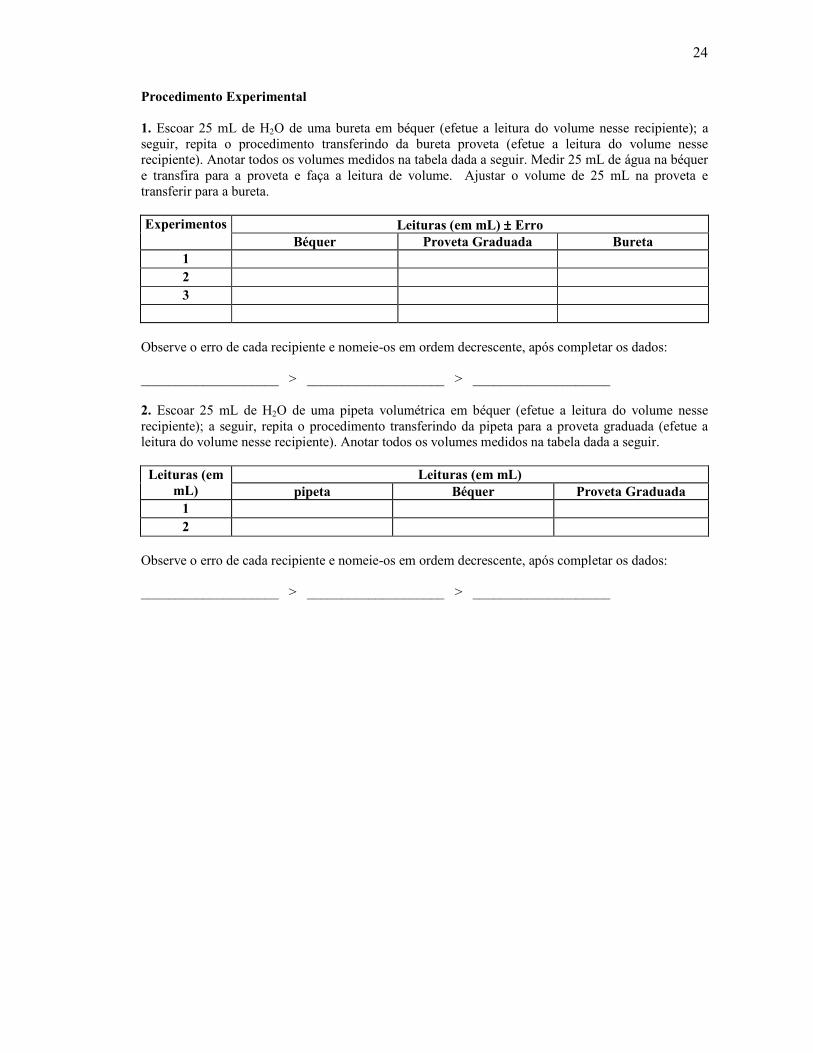
polietileno entre o tubo graduado e a ponta afilada da bureta FIGURA 3.9. Sobre as buretas é conveniente saber que: • As buretas podem ser dispostas em suportes universais contendo mufas. • As buretas de uso mais constante são as de 50 mL, graduadas em décimos de 0,1 mL. Também são muito usadas as de 25 mL. • Nos trabalhos de escala semimicro, são freqüentemente usadas buretas de 5 e 10 mL graduadas em 0,01 ou 0,02. • Para o uso com soluções que possam sofrer o efeito da luz, são recomendadas buretas de vidro castanho (âmbar). • As torneiras das buretas devem ser levemente lubrificadas para que possam ser manipuladas com mais facilidade. Serve para este fim uma mistura de partes iguais de vaselina e cera de abelhas; misturas especiais são encontradas no comércio. • A ponta da bureta deve ser estreita, para que somente possa sair, aproximadamente 50 mL em uns 60 segundos, estando a torneira totalmente aberta. As buretas são usadas na análise volumétrica, de acordo com as seguintes recomendações: a) A bureta limpa e vazia é fixada a um suporte na posição vertical. b) Antes de usar o reagente, deve-se agitar o frasco que o contém, pois não é raro haver na parte superior do mesmo gotas de água condensada. c) A bureta é lavada duas vezes com porções de 5 mL do reagente em questão, que são adicionadas por meio de um funil; cada porção é deixada escoar completamente antes da adição da seguinte. d) Enche-se então, a bureta até um pouco acima do zero da escala e remove-se o funil. e) Abre-se a torneira para encher a ponta ou expulsar todo o ar e, deixa-se escoar o líquido, até que a parte inferior do menisco coincida exatamente com a divisão zero. Quando se calibra a bureta (acerto do zero) deve-se tomar o cuidado de eliminar todas as bolhas de ar que possam existir. As buretas (nos seus mais diversos modelos) são utilizadas para se realizarem TITULAÇÕES, que são procedimentos comuns em laboratório para se determinar (ou confirmar) a concentração de soluções. Para tal procedimento coloca-se o frasco erlenmeyer que vai receber o líquido sob a bureta e deixa-se o líquido escoar gota a gota, geralmente a uma velocidade não superior a 10mL por minuto. Controla-se a torneira da bureta com a mão esquerda (FIGURA 3.10). Após o escoamento da quantidade necessária de líquido, espera-se 10 a 20 segundos e lê-se o volume retirado.
24
Procedimento Experimental 1. Escoar 25 mL de H2O de uma bureta em béquer (efetue a leitura do volume nesse recipiente); a seguir, repita o procedimento transferindo da bureta proveta (efetue a leitura do volume nesse recipiente). Anotar todos os volumes medidos na tabela dada a seguir. Medir 25 mL de água na béquer e transfira para a proveta e faça a leitura de volume. Ajustar o volume de 25 mL na proveta e transferir para a bureta.
Experimentos Leituras (em mL) ±±±± Erro
Béquer Proveta Graduada Bureta 1 2 3
Observe o erro de cada recipiente e nomeie-os em ordem decrescente, após completar os dados: ____________________ > ____________________ > ____________________ 2. Escoar 25 mL de H2O de uma pipeta volumétrica em béquer (efetue a leitura do volume nesse recipiente); a seguir, repita o procedimento transferindo da pipeta para a proveta graduada (efetue a leitura do volume nesse recipiente). Anotar todos os volumes medidos na tabela dada a seguir. Leituras (em
mL) Leituras (em mL)
pipeta Béquer Proveta Graduada 1 2
Observe o erro de cada recipiente e nomeie-os em ordem decrescente, após completar os dados: ____________________ > ____________________ > ____________________

25
Laboratório de Química 1- DQI
EXPERIÊNCIA No 4 - MANUSEIO DO HANDBOOK OF CHEMISTRY AND PHYSICS E DO MERCK
INDEX
Manuseio do Handbook – Noções
O Handbook é um livro para consulta, que congrega uma infinidade de informações sobre várias áreas da ciência tais como: Matemática, Física, Química, Astronomia, História da Ciência, dentre outras. Ele é dividido por seções, A,B,C etc., sendo que cada seção tem paginação própria e no inicio de cada seção há explicações de como se utilizar o seu conteúdo. Para ilustrar o trabalho com o Handbook escolheu-se a Seção C do Handbook, que se inicia com regras de nomenclatura dos compostos orgânicos, uma vez que é necessário saber o nome do composto químico para poder encontrá-lo. Como todos os dados são tabelados e as informações são muitas, houve a necessidade de se criar códigos e símbolos para compactar, ao máximo, num espaço mínimo. Dessa forma, antes de se iniciar as tabelas, sempre há textos explicativos e tabelas de símbolos e abreviações sendo tudo organizado na ordem alfabética. A procura no Handbook também pode ser feita através do índice que se encontra no final do livro. Tendo-se a palavra ou nome, o índice o remeterá à seção e páginas corretas.
Existem, também, alguns Handbooks que são especializados para algumas áreas da ciência, tais como: Handbook de Química Analítica, dentre outros. Manuseio do Merck Index O Merck Index, como o Handbook, é um livro para consulta que possui uma série de informações sobre alguns milhares de compostos, não utilizando tabelas, mas sim uma seqüência de nomes de compostos em ordem alfabética numerada que contem as informações, como mostra um exemplo a seguir. Também aqui, no início do livro, há um texto explicativo e tabela de símbolos e abreviações utilizadas. Para procurar um determinado composto, pode-se utilizar também, o índice de nomes ou de fórmulas que se encontra no final do livro. O Merck Index tem a vantagem sobre o Handbook pelo fato de ter usos e cuidados das substâncias. CRC Handbook
OF Chemistry and Physics
A Ready Reference Book of Chemical and Physical Data
Editor
ROBERT C. WEAST, Ph.D. Vice President, Research, Consolidated Natural Gas Service Company, Inc. Formerly Professor of Chemistry at Case Institute of Technology

26
Laboratório de Química 1- DQI
In collaboration with a large number of professional chemists and physicists Whose assistance is acknowledged in the list of general collaborators and in Connection with the particular tables or sections involved.
TABLE OF CONTENTS SECTION A MATHEMATICAL TABLES A-1 SECTION B THE ELEMENTS AND INORGANIC COMPOUNDS B-1 SECTION C ORGANIC COMPOUNDS C-1 SECTION D GENERAL CHEMICAL D-1 SECTION E GENERAL PHYSICAL CONSTANTS E-1 SECTION F MISCELLANEOUS F-1

27
Laboratório de Química 1- DQI
................................./ .
28
Laboratório de Química 1- DQI
THE MERCK INDEX AN ENCYCLOPEDIA OF CHEMICALS, DRUGS, AND BIOLOGICALS
TWELFTH EDITION Susan Budavari, Editor Maryadele J. O’Neil, Senior Associate Editor Ann Smith, Associate Editor Patricia E., Heckelman, Assistant Editor Joanne F. Kinneary, Assitance Editor Published by Merck Research Laboratories Division of MERCK & CO., INC. Whitehouse Station, NJ
29
Laboratório de Química 1- DQI

30
Laboratório de Química 1- DQI
EXPERIÊNCIA No 5 – MÉTODOS USUAIS DE PURIFICAÇÃO DE SUBSTÂNCIAS E DE SEPARAÇÃO DOS COMPONENTES DE MISTURAS 5.1 Introdução 5.1.1 Métodos Usuais de Purificação de Substâncias.
Vários métodos têm sido empregados para a purificação de substâncias. Destaca-se para purificação de compostos líquidos, os vários tipos de destilação; para purificação de compostos sólidos utiliza-se comumente a recristalização e, em alguns casos, a sublimação. 5.1.2 Métodos Usuais de Separação dos Componentes de uma Mistura
As misturas são comuns em nosso dia a dia; como exemplo temos as bebidas, os combustíveis e a própria terra em que pisamos. Poucos materiais são encontrados puros. Qualquer material feito de duas ou mais substâncias, que não são combinadas quimicamente, é uma mistura.
O isolamento dos componentes puros, a partir de uma mistura, requer a separação de um componente do outro e vários métodos têm sido desenvolvidos com essa finalidade. Alguns se baseiam nas diferenças de propriedades físicas dos componentes da mistura; outros, nas diferenças de propriedades químicas ou de características químicas. Algumas das técnicas mais simples de separação dos componentes de misturas são: sublimação, extração, decantação, filtração e evaporação. 5.1.3 Destilação
Existem alguns tipos de destilação, tais como: destilação simples, destilação fracionada, destilação por arraste a vapor, dentre outros. O processo de destilação, independentemente do tipo, tem sido muito empregado para purificação de substâncias (Emprego 1) e para separação dos componentes de misturas (Emprego 2). O processo de destilação se baseia na combinação sucessiva dos processos de vaporização e de condensação. 5.1.3.1 Destilação Simples O processo de destilação simples é um dos métodos mais comuns de purificação de líquidos. O esquema usado é o descrito na Figura 5a e a destilação pode ocorrer num sistema fechado ou semi-aberto.
Figura 5a – Aparelhagem utilizada em destilação simples.

31
Laboratório de Química 1- DQI
A destilação é um método muito simples: um líquido é levado à ebulição tornando-se vapor, o qual é, então, condensado e retorna ao estado liquido: O liquido é coletado e armazenado. Os líquidos, ao serem aquecidos, têm a energia cinética de suas moléculas gradativamente aumentada, fazendo com que algumas alcancem energia suficiente para escapar da fase líquida e passar para a fase vapor.
O vapor acima do líquido exerce uma pressão chamada de pressão de vapor. Quanto maior o número de moléculas que adquirem energia suficiente para escapar para a fase vapor, maior será a pressão de vapor dessas moléculas. Quando a pressão de vapor se iguala a pressão externa (pressão atmosférica) ocorre a ebulição. A temperatura em que ocorre a ebulição de um líquido é o ponto de ebulição. O liquido no frasco de destilação é aquecido à ebulição, o vapor alcança o condensador que está resfriado pela água corrente, condensa e retorna à fase liquida. Se a mistura tem um componente de baixo ponto de ebulição (uma substância volátil com uma alta pressão de vapor), ele destilará primeiro e pode ser coletado primeiro. Os compostos de ponto de ebulição mais altos (substâncias com baixa pressão de vapor) permanecem no frasco de destilação. Estes compostos só poderão ser destilados se a temperatura do sistema for aumentada.
Quando a destilação é realizada à pressão atmosférica, em sistema aberto, o líquido destila no seu ponto de ebulição "normal". Entretanto, quando a ebulição ocorre em um sistema fechado, é possível mudar o ponto de ebulição do liquido pela redução da pressão no sistema; se a pressão é reduzida, utilizando-se uma bomba à "vácuo", o ponto de ebulição do liquido é reduzido.
Assim, líquidos sensíveis ao calor, que se decompõem quando atingem o ponto de ebulição, à pressão atmosférica, destilam com mínima ou nenhuma decomposição à pressão reduzida. Por exemplo, anilina, líquido orgânico, de fórmula C6H5NH2 pode ser destilada a 184
oC (760mmHg) ou a 68oC (10 mmHg). 5.3.1.2 Uso da destilação simples para separação dos componentes de uma mistura 1. Nesse experimento, uma mistura de sal a água será separada por destilação. A água, volátil, será separada do sal, NaCl, não volátil. A pureza da água destilada será demonstrada por teste químico especifico para os íons Na+ e Cl-. 2. Montar, com muito cuidado (vidraria muito cara), um aparelho como mostrado na Figura 5a. O kit contendo todas as peças será obtido com o professor. 3. Usar um frasco de fundo redondo, de 100 mL, como frasco de destilação. Adicionar a esse frasco, 50 mL da mistura sal e água, já preparada, e algumas pérolas de ebulição, para evitar ebulição violenta. 4. Ligar, vagarosamente, a água que irá resfriar o condensador, de modo que todo o condensador fique cheio de água. O fluxo de água corrente deve ser pequeno para que as mangueiras não desconectem do condensador. Ajustar o bulbo de mercúrio do termômetro abaixo da junção do condensador com o frasco de destilação, como mostra a Figura 5a. 5. Aquecer o balão de destilação suavemente até que o líquido entre em ebulição (ferva) e os vapores se desprendam. Esses vapores irão entrar no condensador, se tornarão líquidos e serão coletados num frasco apropriado. O aquecimento será feito com mantas de aquecimento. 6. Coletar a água destilada até que aproximadamente metade da mistura tenha destilado. Anotar a temperatura dos vapores neste ponto. Desligar o aquecimento e deixar o sistema voltar à temperatura ambiente. 7. Proceder os testes para o íon cloreto (Teste A) e para o íon sódio (Teste B) tanto para o líquido do frasco de destilação como para o líquido do frasco coletor.

32
Laboratório de Química 1- DQI
8. Desmontar, com muito cuidado (vidraria muito cara) toda o sistema de destilação e colocar todas as peças para lavagem, em lugar especialmente destinado a esse objetivo. Teste A. Identificação do Íon Cloreto (Cl-) Colocar em 2 tubos de ensaio, limpos e secos, 2 mL do líquido do frasco coletor e 2 mL do líquido do frasco de destilação. Adicionar a cada tubo, 5 gotas de solução de nitrato de prata (AgNO3); observar e anotar o que acontece. Íons prata reagem com íons cloreto formando um precipitado branco de cloreto de prata. Ag+ + Cl- → AgCl(s) (precipitado branco). Teste B. Identificação do Íon Sódio (Na+)
Colocar em 2 tubos de ensaios, limpos e secos, 2 mL do liquido do frasco coletor e 2 mL do líquido do frasco de destilação. Limpeza do Fio de Níquel: mergulhar um fio de níquel no ácido nítrico (HNO3) concentrado e a seguir colocá-lo na chama do bico de Bunsen até que cor amarela da chama desapareça. Mergulhar este fio limpo no líquido do frasco coletor e a seguir levá-lo à chama; anotar as observações. Repetir o procedimento de limpeza do fio de niquel e, então, mergulhá-lo. no líquido do frasco de destilação; levá-lo à chama e observar a cor da chama; anotar as observações. Íons Na+ produzem uma chama amarela brilhante quando aquecidos pelo bico de Bunsen (vide Figura 5b).
Figura 5b - Íons Sódio aquecidos pelo bico de Bunsen. Observe a chama amarela.

33
EXPERIENCIA 6 - SEPARAÇÃO DE MISTURAS - CROMATOGRAFIA I. Introdução A separação de uma mistura de diversos componentes com propriedades físicas e químicas similares, na maioria das vezes não pode ser realizada por métodos como a destilação. Atualmente, dispomos de um conjunto de técnicas que se constitui em uma das ferramentas mais utilizadas nos laboratórios de química. Trata-se da CROMATOGRAFIA. Tsweet, botânico russo, descreveu (por volta de 1906) a separação de mistura de pigmentos de plantas por meio de um processo que denominou cromatografia. (khroma = cor e graphein = gráfico, desenho). Embora na maioria das vezes, os solutos cromatografados não produzam bandas coloridas, o termo cromatografia é aplicado a qualquer separação empregando o mesmo princípio que o método descrito por Tswett. No caso da cromatografia de solutos incolores, após a separação é necessário a aplicação de um reagente químico ou da ação de luz ultravioleta ( ou outro agente físico) para localizar as bandas ou pontos onde se encontram os solutos separados. A cromatografia é muito utilizada para análises qualitativa e quantitativa de misturas de solutos e para o isolamento de componentes de misturas. As separações cromatográficas baseiam-se em processos de adsorção e de partição. O sistema cromatográfico é composto de uma fase móvel e uma fase estacionária. As separações de solutos dependem de uma distribuição desses entre a fase móvel e a fase estacionária. As separações cromatográficas podem ser realizadas : SOBRE PAPEL - as fibras de celulose servem de suporte (FIGURAS 6a e 6b) . Devido aos grupos alcóolicos das unidades de glucose da celulose, há formação de ligações de hidrogênio com a água (umidade atmosférica). A água retida no papel é que atua como fase estacionária. Isso significa que somente compostos polares podem ser separados sobre papel. A separação dos componentes sobre papel ocorre por partição dos solutos entre a fase móvel e a fase estacionária. Os solutos mais solúveis na fase móvel migram mais e aqueles mais solúveis na água (fase estacionária) ficam mais tempo retidos no papel e portanto, migram menos.
FIGURA 6a – Cromatografia em papel.
FIGURA 6b – Forma de utilização da cromatografia em tira de papel.

34
Laboratório de Química 1- DQI
Considerando a migração de um dado componente em um sistema cromatográfico, temos a migração do soluto em um cromatograma (Figura 6c): O movimento de um soluto sobre papel ou camada delgada pode ser matematicamente expresso pelo valor de Rf, (fator de retenção), que é calculado pelo quociente entre a distância percorrida pelo soluto (em cm) e a distância percorrida pelo solvente (ou eluente) . Então: Rf = distância percorrida pelo soluto (em cm) distância percorrida pelo eluente (em cm)
FIGURA 6c - Migração de um soluto em um cromatograma (papel ou camada fina).
Se ocorrer variações na temperatura, ou na composição do solvente ou na fase estacionária, os valores de Rf se alterarão. Os valores de Rf quando comparados com aqueles de substância padrão, auxiliam na identificação dos componentes presentes em uma mistura. EM CAMADA DELGADA - a fase estacionária pode ser um sólido como alumina, sílica gel, celulose, ou outros, distribuídos sob a forma de uma fina camada sobre um suporte. O suporte pode ser uma placa de vidro, de alumínio, ou de poliamida. As separações. No caso de alumina e sílica gel, as separações dos componentes de uma mistura ocorrem por processos de adsorção-desorção desses, entre a fase móvel e a fase estacionária. EM COLUNA - o fundamento das separações são os mesmos já citados, apenas que a fase estacionária é colocada em um tubo de vidro e a fase móvel, é passada continuamente para eluição dos componentes da mistura. Se todas as condições são mantidas constantes, os valores de Rf permanecem constantes.
FIGURA 6d – Cromatografia em coluna.

35
Laboratório de Química 1- DQI
II. Objetivos Separar misturas de compostos em papel e sobre giz. III. Procedimentos 3.1 - Cromatografia sobre giz Utilizar um pedaço de giz de superfície e base uniformes. Aplicar em ponto ou linha, tinta de caneta hidrocor (escolher a cor). Mergulhar o giz em vidro contendo o eluente (acetona : água 10:1 v/v). O nível do eluente no vidro não deve atingir a altura do ponto de aplicação das amostras. Observar o desenvolvimento do cromatograma e retirá-lo quando o eluente chegar próximo ao topo do giz. 3.2 - Cromatografia sobre papel circular Utilizar uma folha de papel de filtro W.1. Cortar de forma adequada e aplicar a(s) amostra(s) no centro do papel. Utilizar placas de petri como cuba cromatográfica e a mistura acetona : água (9:1 v/v) como eluente. A tira de papel resultante do corte deve ficar mergulhada no eluente. Observar o desenvolvimento do cromatograma e retirar o papel quando o eluente estiver próximo da borda do papel. 3.3 - Cromatografia em tira de papel Uma tira de papel será fornecida pelo professor. Marcar a linha base, que terá altura de 1 cm e aplicar a amostra com capilar afilado. Em béquer de 150 mL, adicionar um pouco da mistura eluente (acetona : H2O 9:1 v/v) e a seguir posicionar a tira de papel. Deixar o cromatograma se desenvolver até aproximadamente 0,5 cm do final da tira de papel. 3.4 – Cromatografia utilizando instrumental - Cromatografia em fase gasosa e cromatografia líquida de alta eficiência

36
EXPERIÊNCIA No 7 – Tipos de Reações Químicas em Soluções Aquosas I Introdução
Muitas reações que você vai encontrar no Laboratório de Química se passam em solução aquosa. Os químicos estão interessados nessas reações, não apenas por serem o caminho de chegada a produtos úteis, mas também porque são as reações que ocorrem nos vegetais e animais da Terra. Vamos examinar alguns padrões comuns das reações para ver quais podem ser as respectivas "forças motrizes"; em outras palavras, como se pode saber que, ao se misturarem duas substâncias químicas, haverá reação entre elas e a formação de um ou mais compostos novos? 1.1 Classificação das reações químicas quanto as forças motrizes Quatro tipos importantes de processos provocam a ocorrência de reações, quando os reagentes se misturam em solução aquosa. 1° TIPO: Reações de Precipitação As reações de precipitação são aquelas em que os íons se combinam em solução para formar um produto de reação insolúvel. Exemplo: Equação geral: Pb(NO3)2 (aq) +2 KI (aq) → Pbl2 (s) + 2 KNO3 (aq) Equação Iônica Líquida: Pb2+ (aq) + 2 l
- (aq) → Pbl2 (s) (sólido amarelo) 2° TIPO: Reações Ácido-Base As reações ácido-base, são aquelas em que os íons H+ e OH- combinam-se para formar água. Exemplo: Equação geral: HNO3 (aq) + KOH (aq) → KNO3 (aq) + HOH (l) Equação iônica líquida: H+ (aq) + OH
- (aq) → H2O
(Esta é a equação iônica líquida de todas as reações entre ácidos fortes e bases) 3o TIPO: Reações com Desprendimento de Gás As reações com desprendimento de gás são aquelas em que os reagentes se combinam em solução para formar um produto de reação que se desprende na forma de gás. Como exemplos mais comuns tem-se as reações envolvendo, principalmente, carbonatos de metais e ácidos, com formação do ácido

37
Laboratório de Química 1- DQI
carbônico, H2CO3, como produto o qual, na maioria das vezes, se decompõe em H2O e CO2. O dióxido de carbono é o gás que se vê borbulhar durante a reação. Exemplo: Equação geral: NiCO3 (s) + 2 HNO3 (aq) → Ni(NO3)2 (aq) + H2CO3 (aq) H2CO3 (aq) → CO2 (g) + H2O Equação iônica líquida: NiCO3 (s) + 2 H
+ (aq) → Ni2+ (aq) + CO2 (g) + H2O (l)
4o TIPO: Reações de Oxidação–Redução (Oxi–Redução) As reações de oxidação–redução são aquelas em que o processo importante é a transferência de elétrons de uma substância para outra. Exemplo: Equação geral: Cu (s) + 2 AgNO3 (aq) → Cu(NO3)2 (aq) + 2 Ag (s) Equação iônica líquida: Cu (s) + 2 Ag
+ (aq) → Cu2+ (aq) + 2 Ag (s) “Forças Motrizes” Responsáveis pelas Reações em Soluções Aquosa Tipo de Reação Força Motriz Precipitação Formação de composto insolúvel Ácido – base :Neutralização Formação de um sal e Água Desprendimento de gás Evolução de gás, insolúvel em água, como o CO2 Oxidação – redução (redox) Transferência de elétrons Estes tipos de reações são, em geral, fáceis de serem reconhecidos; uma reação pode ter mais de uma força motriz. 1.2 Classificação das reações químicas quanto aos seguintes itens: I) LIBERAÇÃO OU ABSORÇÃO DE CALOR As reações podem ser classificadas quanto `a absorção ou liberação de calor em: ENDOTÉRMICAS, quando ocorrem com a absorção de calor do meio ambiente, e EXOTÉRMICAS, quando liberam calor para o meio ambiente. II) QUANTO À VELOCIDADE As reações podem ser classificadas em RÁPIDAS ou INSTANTÂNEAS e LENTAS quando levam horas, meses ou anos para ocorrer. III) QUANTO À REVERSIBILIDADE

38
Laboratório de Química 1- DQI
As reações podem ser REVERSIVEIS, quando não se completam e podem ocorrer no sentido inverso pela variação da concentração de reagentes e produtos, temperatura, etc. e IRREVERSIVEIS, quando ocorrem completamente. II. Procedimento Experimental
Tomar cerca de 14 tubos de ensaio, numerá-los, para efetuar cada uma das reações químicas descritas a seguir. Observe todas as soluções dos reagentes desse experimento, contidas em frascos conta-gotas colocadas sobre a bancada do laboratório. Leia com atenção o rótulo de cada solução, antes de misturar os reagentes. Procure seguir as instruções abaixo anotando as mudanças detalhadamente em seu caderno de laboratório. Para cada reação use 10 gotas de solução, exceto quando houver outra especificação. Observe o que ocorre nas reações: precipitação, desprendimento de gás, mudança de coloração, aquecimento ou resfriamento do tubo, etc.
Coloque em um tubo de ensaio, ácido clorídrico diluído + solução de nitrato de prata. Observe.
Filtre a mistura obtida no item 1 pela utilização de um pequeno funil de vidro contendo papel de filtro dobrado, sobre o tubo de ensaio Nº 2; após a filtração deixe o sistema montado no mesmo local, de modo que o resíduo obtido no papel de filtro (Cloreto de Prata) fique exposto à luz; depois de algum tempo observe a mudança de sua coloração.
Coloque em um tubo de ensaio, solução de sulfato de cobre + solução de hidróxido de sódio. Observe.
Coloque em um tubo de ensaio, solução de nitrato de chumbo + solução de sulfato de sódio. Observe.
Coloque em um tubo de ensaio, solução de nitrato de chumbo + solução de iodeto de potássio. Se nada for observado à frio, aqueça com cuidado e observe.
Coloque em um tubo de ensaio, solução de nitrato de chumbo + solução de ácido clorídrico
diluído. Se nada for observado, acrescente 2 gotas de H Cl concentrado, que está na CAPELA.
Coloque em um tubo de ensaio, 2 mL (40 gotas) de ácido clorídrico 4 M + 2 mL (40 gotas) de hidróxido de sódio 4 M. Observe.
Coloque em um tubo de ensaio,
aproximadamente 1 grama (uma ponta de espátula) de óxido de cálcio (cal viva) e adicione água. Agite e espere decantar. Transfira o líquido sobrenadante para outro tubo de ensaio. Adicione duas gotas de fenolftaleína.

39
Laboratório de Química 1- DQI
Com o auxilio de uma pinça metálica, queime um pedaço de magnésio metálico – CUIDADO: AO QUEIMAR O Mg VOCÊ DEVE EVITAR OLHAR DIRETAMENTE PARA A CHAMA BRILHANTE. Coloque o metal + o pó branco formado num tubo de ensaio e adicione algumas gotas de água e, em seguida, duas gotas de fenolftaleína. Coloque em um tubo de ensaio, um pedaço de zinco metálico e adicione aproximadamente 10 gotas de acido clorídrico diluído. Se nada for observado, acrescente 2 gotas de H Cl concentrado, que está na CAPELA
Coloque em um tubo de ensaio aproximadamente 40 gotas de solução de nitrato de prata e mergulhe um fio de cobre bem fino enrolado ou em espiral. Observe.
Coloque em um tubo de ensaio, um pouco de bicarbonato de sódio sólido e adicione gotas de acido clorídrico diluído. Observe.
Coloque em um tubo de ensaio, aproximadamente 10 gotas de solução de iodato de potássio + 10 gotas de solução de iodeto de potássio e uma gota de acido clorídrico diluído. Observe.
Coloque em um tubo de ensaio, aproximadamente 10 gotas de permanganato de potássio e 3 a 5 gotas de solução de acido sulfúrico diluído. Aqueça a mistura suavemente e acrescente miligramas de oxalato de sódio sólido. Agite e aqueça novamente, se necessário. Observe.
Escreva a equação química balanceada correspondente a cada reação. Escreva a equação
iônica quando for caso.
40
Laboratório de Química 1- DQI
EXPERIÊNCIA No 8 – Ponto de Fusão
I Introdução
Propriedades Físicas As substâncias químicas possuem propriedades físicas distintas, permitindo que, na análise de
uma amostra desconhecida, a comparação das propriedades físicas desta com dados da literatura possa conduzir a uma identificação.
As propriedades físicas geralmente listadas nos Handbooks de química são: cor, formacristalina (se sólido), índice de refração (se líquido), densidade, solubilidade em vários solventes, ponto de fusão, ponto de ebulição e características de sublimação. Quando um novo composto é isolado ou sintetizado, essas propriedades quase sempre acompanham o registro na literatura.
A transição de uma substância do estado sólido para líquido e para gasoso e a operação inversa, representa mudanças físicas. Isto significa que há uma mudança na forma ou no estado da substância sem qualquer alteração na composição química. Água sofre mudanças de estado, de gelo para água líquida e depois vapor, entretanto, a composição das moléculas nos três estados permanece H2O.
H2O(s) H2O(l) H2O(g) gelo líquido vapor
II Ponto de Fusão e Calibração de Termômetros
Ponto de fusão de um sólido cristalino é a temperatura na qual o sólido começa a se tornar líquido sob pressão de 1atm; é o intervalo de temperatura de fusão do primeiro ao último cristal da substância. Para substâncias puras, a mudança de estado sólido para líquido é bem definida (dentro de 0,5ºC), sendo, portanto, a temperatura de fusão, valiosa para fins de identificação. O ponto de fusão é influenciado pela presença de outras substâncias e constitui um critério importante de pureza. Se o líquido puro for resfriado, ocorrerá solidificação à mesma temperatura, portanto, o ponto de fusão e de congelamento são idênticos.
O ponto de fusão de substâncias puras ocorre numa faixa muito estreita de temperatura e constitui um critério de pureza dos sólidos. Impurezas causam o abaixamento do ponto de fusão e o alargamento da faixa de fusão. Ex: o ácido benzóico puro tem PF=122,13ºC; quando se obtém um PF = 121 – 122ºC é considerado impuro. A adição de uma outra substância à substância pura faz com que o ponto de fusão decresça, conforme o gráfico da FIGURA 1.
FIGURA 1: Mostra a diminuição do Ponto de Fusão do α-naftol à medida que se adiciona naftaleno.

41
Laboratório de Química 1- DQI
A – C → curva que mostra o ponto de fusão decrescente conforme aumenta a % de naftaleno. B – C → representa o ponto de fusão do naftaleno com α-naftol como ‘impureza’. C → temperatura eutética (61,0ºC), onde a composição da mistura é: Xnaft. = 0,605 e Xα-naft. = 0,395 . (X = fração molar)
O ponto eutético (C) é a temperatura limite, abaixo da qual nenhum líquido pode existir, ou seja, temperatura abaixo de 61ºC resulta apenas na solidificação completa dos dois componentes (solução sólida). O ponto eutético refere-se ao ponto de fusão de uma mistura eutética de duas ou mais substâncias, ou seja, uma mistura sólido – sólido a determinada composição que se comporta como uma substância pura e, portanto, tem ponto de fusão bem definido. Assim, o ponto de fusão também pode ser utilizado para a identificação de substâncias, quando associado a outras técnicas.
III Experimental
1 – Calibração de Termômetros
Os termômetros mais comuns e baratos, com freqüência apresentam erros nas medidas, que podem ser eliminados, ou pelo menos minimizados, fazendo-se a calibração desses termômetros pela imersão completa da coluna de mercúrio no vapor ou no líquido. Como nas determinações cotidianas a coluna toda não é circundada pelo vapor nem completamente imersa no líquido, a parte exposta ao ambiente do laboratório, não se expande tanto quanto o bulbo de mercúrio, originando leituras menores que a da temperatura real. O erro devido a isso é pequeno a 100ºC, mas apreciável a temperaturas maiores, ou seja, de 3 – 5º a 200ºC e de 6 – 10 º a 250ºC.
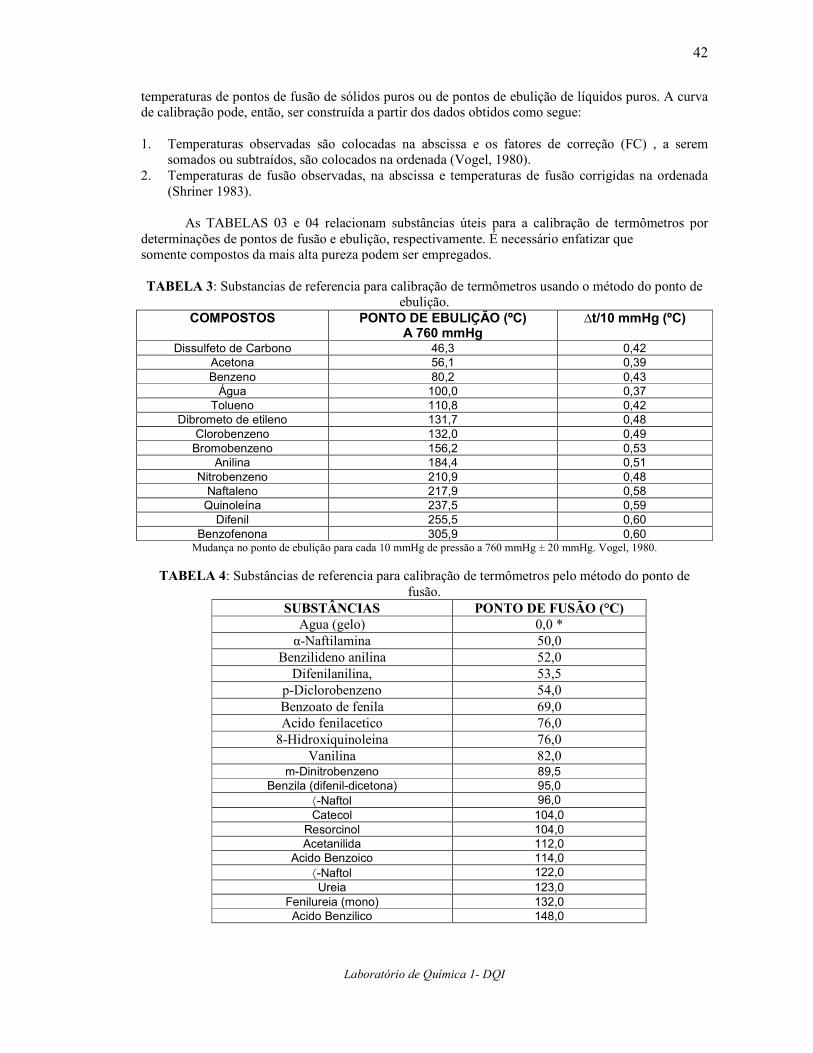
Esse erro introduzido nas medidas devido à parte exposta da coluna de mercúrio pode ser corrigido adicionando-se um Fator de Correção (FC) como segue: K = coeficiente aparente de expansão do mercúrio no vidro (tabelado). N = comprimento (em graus) da parte do termômetro não aquecida à temperatura do bulbo, isto é, o
comprimento da coluna exposta. T1 = temperatura observada. T2 = temperatura real da parte da coluna de mercúrio exposta determinada por meio de um
termômetro auxiliar ( posicionado ao lado do termômetro a ser calibrado ) cujo bulbo deve se posicionar à metade da haste exposta.
Valores de K para vidro normal e de borosilicato (Pyrex) estão mostrados nas
TABELAS 01 e 02, respectivamente.
TABELA 1: Valores de K para vidro normal
TEMPERATURA (°C) 0 a 150 200 250 300 VALORES DE K (x10 –4) 1,58 1,59 1,61 1,64
TABELA 2: Valores de K para vidro borosilicato (Pyrex)
TEMPERATURA (°C) 100 150 200 250 300 350 400 450 VALORES DE K (x10 –4) 1, 64 1,65 1,67 1,70 1,74 1,78 1,83 1,88
Além do erro devido à parte exposta, os termômetros químicos comuns de baixo custo estão sujeitos a erros, devido a irregularidades no diâmetro interno do tubo, e algumas vezes, às graduações da escala que podem não ser muito precisas. É, portanto, essencial testar o termômetro em várias
FC (°C) = K N ( T1 – T2 )
42
Laboratório de Química 1- DQI
temperaturas de pontos de fusão de sólidos puros ou de pontos de ebulição de líquidos puros. A curva de calibração pode, então, ser construída a partir dos dados obtidos como segue:
1. Temperaturas observadas são colocadas na abscissa e os fatores de correção (FC) , a serem
somados ou subtraídos, são colocados na ordenada (Vogel, 1980). 2. Temperaturas de fusão observadas, na abscissa e temperaturas de fusão corrigidas na ordenada
(Shriner 1983).
As TABELAS 03 e 04 relacionam substâncias úteis para a calibração de termômetros por determinações de pontos de fusão e ebulição, respectivamente. É necessário enfatizar que somente compostos da mais alta pureza podem ser empregados. TABELA 3: Substancias de referencia para calibração de termômetros usando o método do ponto de
ebulição. COMPOSTOS PONTO DE EBULIÇÃO (ºC)
A 760 mmHg ∆t/10 mmHg (ºC)
Dissulfeto de Carbono 46,3 0,42 Acetona 56,1 0,39 Benzeno 80,2 0,43 Água 100,0 0,37 Tolueno 110,8 0,42
Dibrometo de etileno 131,7 0,48 Clorobenzeno 132,0 0,49 Bromobenzeno 156,2 0,53
Anilina 184,4 0,51 Nitrobenzeno 210,9 0,48 Naftaleno 217,9 0,58 Quinoleína 237,5 0,59 Difenil 255,5 0,60
Benzofenona 305,9 0,60 Mudança no ponto de ebulição para cada 10 mmHg de pressão a 760 mmHg ± 20 mmHg. Vogel, 1980.
TABELA 4: Substâncias de referencia para calibração de termômetros pelo método do ponto de
fusão. SUBSTÂNCIAS PONTO DE FUSÃO (°C)
Agua (gelo) 0,0 * α-Naftilamina 50,0
Benzilideno anilina 52,0 Difenilanilina, 53,5
p-Diclorobenzeno 54,0 Benzoato de fenila 69,0 Acido fenilacetico 76,0 8-Hidroxiquinoleina 76,0
Vanilina 82,0 m-Dinitrobenzeno 89,5
Benzila (difenil-dicetona) 95,0 ⟨-Naftol 96,0 Catecol 104,0
Resorcinol 104,0 Acetanilida 112,0
Acido Benzoico 114,0 ⟨-Naftol 122,0 Ureia 123,0
Fenilureia (mono) 132,0 Acido Benzilico 148,0

43
Laboratório de Química 1- DQI
s-Difeniltiureia 150,0 Acido salicilico 154,0 * Hidroquinona 159,0
p-Toluilureia (mono) 170,0 Acido succinico 181,0
* Estes compostos não são convenientes para uso no método da chapa de aquecimento. Vogel, 1980
O ponto zero e melhor determinado com uma mistura intima de água destilada e gelo puro feito de agua destilada.
III.2 – Objetivos
1 – Determinar o ponto de fusão de vários compostos orgânicos puros. 2 – Identificar as substâncias por meio dos pontos de fusão obtidos. 3 – Calibrar termômetros comuns de laboratório.
III.3 – Materiais e Reagentes
. Tubos capilares . Ácido α-Fenilacético . Tubo de vidro para socar . Ácido o Benzóico . Borrachas para prender o capilar . Ácido Maleico . Vidro de relógio pequeno . Ácido Salicílico . Termômetro de 250°C . Ácido Succínico . Tubo de Thiele . Ácido Fumárico . Óleo para banho . Acetanilida . Suporte de ferro . α-Naftol . Espátula . Ácido α-Fenilacetico . Garras . Fósforos
III.4 – Procedimento Experimental
1 – A amostra desconhecida será fornecida pelo professor; anotar o número da amostra. Colocar aproximadamente 0,1 g da amostra em um vidro de relógio e cuidadosamente, triturá-la com a espátula.
2 – Selar uma das extremidades do tubo capilar para ponto de fusão, conforme FIGURA 2.
FIGURA 2 - Preparo de um tubo capilar para ponto de fusão.
3 – Empacotar o sólido no tubo selado conforme FIGURA 3: 3.1 – Pressionar a extremidade aberta do capilar, verticalmente, sobre a amostra, tal que uma
pequena quantidade da amostra será forçada a entrar no capilar, FIGURA 3 A e B.
3.2 – Inverter a posição do capilar para que a amostra caia no fundo do capilar. Bater com cuidado, o capilar sobre uma superfície plana para que toda amostra se deposite no fundo do capilar (~1,2 mm), FIGURA 3 C.

44
Laboratório de Química 1- DQI
3.3 – Se no passo 4.3.2 não se conseguir depositar toda a amostra, utilizar um tubo de vidro
auxiliar para ajudá-lo a empacotar a amostra no capilar.
FIGURA 3: Empacotando um tubo capilar.
4 – Prender o tubo capilar ao termômetro por meio de anéis de borracha e alinhar o bulbo de
mercúrio conforme a FIGURA 4. Esse termômetro fornecerá a temperatura T1.
FIGURA 4: Adaptação do tubo capilar ao termômetro.
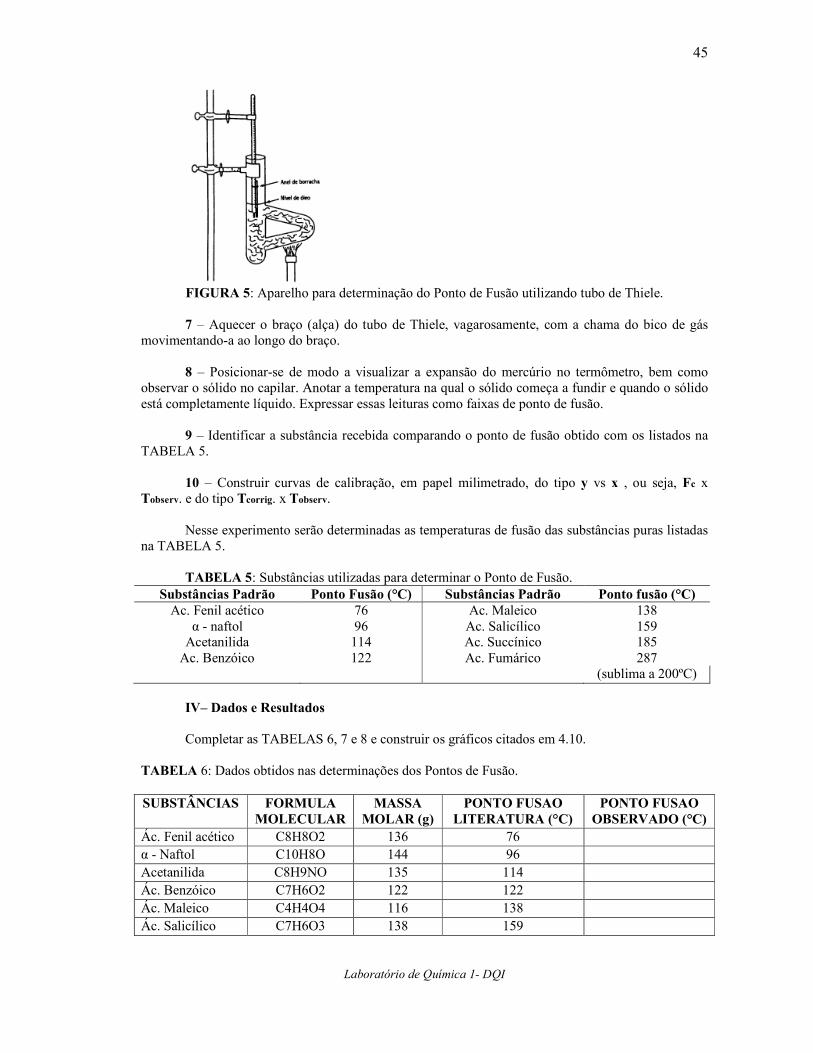
5 – Prender o tubo de Thiele a um suporte e enchê-lo (até certa altura) com óleo silicone ou
óleo mineral. Introduzir no tubo de Thiele o termômetro atado ao capilar, preso a um suporte poruma garra, conforme mostra a FIGURA 5.
6 – Adaptar um termômetro auxiliar, ao lado do termômetro que está mergulhado no óleo, tal
que o bulbo desse termômetro auxiliar fique aproximadamente à metade da haste exposta do termômetro do banho de óleo. O termômetro auxiliar fornecerá T2, que deverá ser lida para cada determinação de ponto de fusão.
45
Laboratório de Química 1- DQI
FIGURA 5: Aparelho para determinação do Ponto de Fusão utilizando tubo de Thiele.
7 – Aquecer o braço (alça) do tubo de Thiele, vagarosamente, com a chama do bico de gás
movimentando-a ao longo do braço.
8 – Posicionar-se de modo a visualizar a expansão do mercúrio no termômetro, bem como observar o sólido no capilar. Anotar a temperatura na qual o sólido começa a fundir e quando o sólido está completamente líquido. Expressar essas leituras como faixas de ponto de fusão.
9 – Identificar a substância recebida comparando o ponto de fusão obtido com os listados na TABELA 5.
10 – Construir curvas de calibração, em papel milimetrado, do tipo y vs x , ou seja, Fc x Tobserv. e do tipo Tcorrig. x Tobserv.
Nesse experimento serão determinadas as temperaturas de fusão das substâncias puras listadas na TABELA 5.
TABELA 5: Substâncias utilizadas para determinar o Ponto de Fusão.
Substâncias Padrão Ponto Fusão (°C) Substâncias Padrão Ponto fusão (°C) Ac. Fenil acético 76 Ac. Maleico 138
α - naftol 96 Ac. Salicílico 159 Acetanilida 114 Ac. Succínico 185 Ac. Benzóico 122 Ac. Fumárico 287
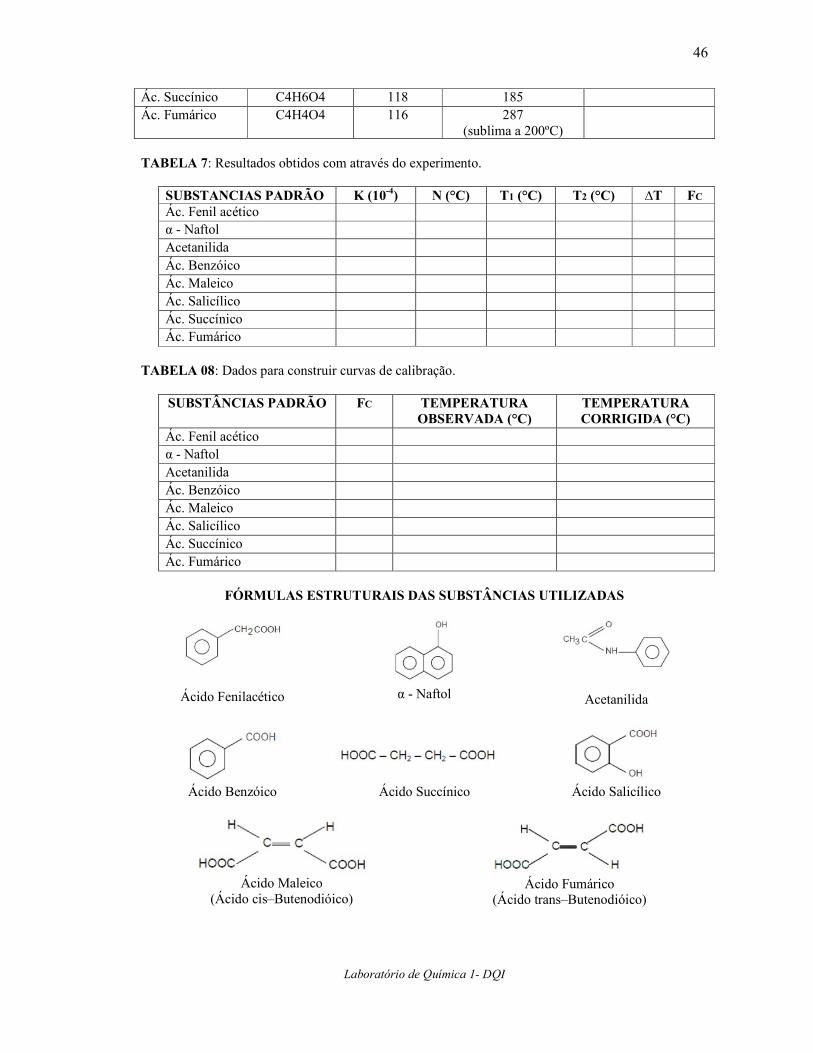
(sublima a 200ºC) IV– Dados e Resultados Completar as TABELAS 6, 7 e 8 e construir os gráficos citados em 4.10.
TABELA 6: Dados obtidos nas determinações dos Pontos de Fusão. SUBSTÂNCIAS
FORMULA
MOLECULAR MASSA
MOLAR (g) PONTO FUSAO
LITERATURA (°C) PONTO FUSAO OBSERVADO (°C)
Ác. Fenil acético C8H8O2 136 76 α - Naftol C10H8O 144 96 Acetanilida C8H9NO 135 114 Ác. Benzóico C7H6O2 122 122 Ác. Maleico C4H4O4 116 138 Ác. Salicílico C7H6O3 138 159
46
Laboratório de Química 1- DQI
Ác. Succínico C4H6O4 118 185 Ác. Fumárico C4H4O4 116 287
(sublima a 200ºC)
TABELA 7: Resultados obtidos com através do experimento.
SUBSTANCIAS PADRÃO K (10-4) N (°C) T1 (°C) T2 (°C) ∆T FC Ác. Fenil acético α - Naftol Acetanilida Ác. Benzóico Ác. Maleico Ác. Salicílico Ác. Succínico Ác. Fumárico
TABELA 08: Dados para construir curvas de calibração.
SUBSTÂNCIAS PADRÃO FC
TEMPERATURA OBSERVADA (°C)
TEMPERATURA CORRIGIDA (°C)
Ác. Fenil acético α - Naftol Acetanilida Ác. Benzóico Ác. Maleico Ác. Salicílico Ác. Succínico Ác. Fumárico
FÓRMULAS ESTRUTURAIS DAS SUBSTÂNCIAS UTILIZADAS
Ácido Fenilacético
α - Naftol
Acetanilida
Ácido Benzóico
Ácido Succínico
Ácido Salicílico
Ácido Maleico
(Ácido cis–Butenodióico)
Ácido Fumárico
(Ácido trans–Butenodióico)

47
EXPERIÊNCIA No 09 – Água de Hidratação I. Introdução Alguns compostos quando aquecidos não fundem, mas sofrem decomposição. Na
decomposição o composto pode se “quebrar” irreversivelmente, ou reversivelmente, em duas ou mais substancias. Se o processo e reversível, uma recombinação leva a obtenção do composto original. Hidratos são exemplos de compostos que não fundem com o aquecimento, mas se decompõem de forma reversível. Ex: Na2SO4.10H2O. Os produtos da decomposição são sal anidro e água. O hidrato original pode ser regenerado pela adição de água ao sal anidro.
Os hidratos contem água como uma parte constituinte da estrutura cristalina do composto.
Quando o sal cristaliza a partir de uma solução aquosa, o número de moléculas de H2O ligados ao
íon metálico é característico de cada metal e em uma proporção definida. Assim, quando sulfato de
cobre cristaliza a partir de uma solução aquosa, forma-se o sal azul de sulfato de cobre hidratado, CuSO4.xH2O. Como mostrado pela fórmula, x moléculas de água de hidratação estão ligadas ao íon
cobre II no sulfato.
A fórmula não indica como as moléculas de água estão arranjadas no retículo cristalino e são
necessários estudos da estrutura para se determinar isto. Em todos os casos, a remoção de água por
aquecimento provoca modificações no retículo, transformando-o em pó não cristalino. A água é
perdida na forma de vapor. Por ex: os cristais azuis de CuSO4.xH2O podem se tornar brancos com a
perda das moléculas de água a 250°C.
CuSO4.XH2O → CuSO4 + XH2O
Alguns sais anidros são capazes de se tornarem hidratados pela exposição à umidade
ambiente.Esses sais são chamados de higroscópicos e podem ser usados na química como agentes
secantes. Alguns sais são tão bom secantes e são capazes de absorver tanto a umidade do ambiente que eles são capazes de se dissolverem! Cloreto de Cálcio é um exemplo desse sal que é chamado de
deliqüescente.Como muitos hidratos contém água em quantidade estequiométrica, é possível
determinar a razão molar de H2O para sal, como também a % de água no hidrato:
%H2O = [ massa H2O perdida(g) / massa hidrato(g) ] × 100
II – Objetivos
1 – Observar o comportamento de um hidrato. 2 – Determinar a % de H2O no hidrato problema. 3 – Determinar a razão de água para sal no hidrato problema.
III – Materiais e Reagentes
. Hidrato Tubo de ensaio . Cloreto de cálcio anidro Garra para tubo de ensaio . Espátula Bico de gás . Balança Papel alumínio . Fósforo Almofariz com pistilo . Vidro de relógio Estante para tubo de ensaio
IV– Procedimento Experimental
1 – Propriedades do CaCl2 anidro

48
Laboratório de Química 1- DQI
1.1 – Com uma espátula, transferir uma pequena quantidade de CaCl2 anidro para um vidro de relógio.
1.2 – Deixar o vidro de relógio em repouso por um tempo, enquanto se trabalha com a determinação da composição do hidrato item 4.2. O que aconteceu com o sólido CaCl2?
2 – Determinação da composição do hidrato 2.1 – Pesar um tubo de ensaio perfeitamente limpo e seco. Anotar o peso. 2.2 – Pesar o mesmo tubo com aproximadamente 1,5g do hidrato previamente pulverizado.
Anotar o peso. 2.3 – Aquecer o tubo suavemente por 5 minutos tomando o cuidado para que vapor d’água não
se condense nas paredes do tubo.
2.4 – Aumentar a chama e continuar o aquecimento por mais 15 minutos. Deve-se evitar o
aquecimento muito intenso e o desprendimento de fumaça. 2.5 – Desligar a chama, cobrir o tubo com papel alumínio e deixá-lo esfriar à temperatura
ambiente apoiado em um suporte adequado. 2.6 – Remover a cobertura do tubo e pesar. Anotar o peso (m4). 2.7 – Repetir o aquecimento por aproximadamente 5min. e resfriar . 2.8 – Remover a cobertura e pesar. Anotar o peso (m5). 2.9 – Observar os dois valores obtidos de m4 e m5. Se estes valores coincidem ou apresentam
uma diferença mínima (0,01), pode-se encerrar o aquecimento e passar para os cálculos. Caso contrário, repetir o aquecimento e pesagem.
V – Dados e Resultados Massa do tubo ................................................................. m1 = .............................. Massa do tubo + hidrato ................................................. m2 = ............................... Massa do hidrato ( m2 – m1 ) ......................................... m3 =................................ Massa do tubo + sal anidro (1) ....................................... m4 =................................ Massa do tubo + sal anidro (2) ....................................... m5 = ............................... Diferença entre as massas m4 e m5 ............................................................................ Média entre as massas m4 e m5 ..................................... m6 = ............................... Massa de água perdida ( m2 – m6 ) ............................... m7 = ............................... Massa de sal anidro ( m3 – m7 ) .................................... m8 = ............................... Número de mols de água no hidrato ........................................................................... Dar a fórmula do hidrato ............................................................................................ Dar a razão de água para sal no hidrato ..................................................................... Dar a razão de água para sal no hidrato .....................................................................

49
Laboratório de Química 1- DQI
EXPERIÊNCIA No 10 – Cinética Química
DETERMINACAO DA LEI DE VELOCIDADE DE PROCESSOS DO COTIDIANO
– ORDEM DE REAÇÃO –
I. Introdução No estudo das reações químicas, dois fatores devem ser considerados: a espontaneidade do
processo (estudo termodinâmico) e a velocidade da reação (estudo cinético). A cinética química estuda as reações do ponto de vista da velocidade com que essas reações
se processam, dos fatores que afetam essa velocidade e do mecanismo pelo qual elas ocorrem. As velocidades das reações químicas são determinadas através de leis empíricas, chamadas
leis de velocidades, deduzidas a partir do efeito da concentração dos reagentes e produtos na velocidade da reação.
As velocidades das reações dependem também de outros fatores como, por exemplo, pressão, temperatura e catalisadores. O efeito da temperatura é particularmente importante, pois permite conclusões a respeito de fatores energéticos envolvidos na reação.
A velocidade de uma reação não depende apenas dos estados inicial e final do sistema (como é o caso, por exemplo, da variação de entalpia), mas, de cada etapa intermediária pela qual o sistema alcança o estado final. Um estudo cinético da reação permite perceber quais as etapas envolvidas, esclarecendo-se assim o “mecanismo” pelo qual a reação se processa.
Considere a reação: A + B → C, e que [A], [B] e [C] sejam as concentrações instantâneas dos componentes A, B e C. A velocidade da reação é a velocidade da formação do produto (C) ou a velocidade de consumo de um dos reagentes (A ou B).
A velocidade do consumo do reagente A é a variação da concentração de A com o tempo: . A velocidade de formação do produto é .
Neste caso a estequiometria indica que a velocidade de formação de C é igual à velocidade de consumo de A ou de B, pois quando uma molécula de A e uma molécula de B são consumidas, forma-se uma molécula de C.
Para a reação: A + 2 B → 3 C + D , a relação entre as velocidades de variação das concentrações dos vários componentes é mais complicada e temos:
=
Freqüentemente verifica-se que a velocidade da reação é proporcional a uma potência da concentração dos reagentes. Por exemplo, seja a reação:
A + B → C ν = k[A][B] , onde cada valor de concentração tem expoente 1.
A constante de proporcionalidade k é denominada constante de velocidade. Ela não depende das concentrações mas sim da temperatura. A constante de velocidade k é a constante de proporcionalidade que relaciona a velocidade às concentrações numa dada temperatura, e deve ser determinada experimentalmente. É um parâmetro importante, pois uma vez conhecido, permite que se calcule a velocidade da reação para qualquer conjunto de concentrações.
A equação que expressa a velocidade da reação em função das concentrações instantâneas das espécies químicas envolvidas é denominada lei de velocidade e é determinada experimentalmente para cada reação.
O expoente que afeta as concentrações das espécies presentes na lei de velocidade é a ordem de reação com relação a este componente. A ordem total da reação é a soma das ordens (expoentes) de todos os componentes contidos na expressão. Por exemplo: ν = k[A]1/2[B] é de ordem ½ com relação a A, de ordem 1 com relação a B e de ordem total 1,5.

50
Laboratório de Química 1- DQI
A ordem de uma reação é um número (expoente num termo de concentração na equação de velocidade) que reflete a influência de um dos reagentes na velocidade da reação.
REAÇÃO DE PRIMEIRA ORDEM
2 N2O5 → 4 NO2 + O2
[N2O5] tem expoente 1 e isto significa que a reação é de primeira ordem em relação ao N2O5,
ou seja, se a concentração do N2O5 for duplicada a velocidade da reação também duplica, ou se a concentração N2O5 for ¼ do valor inicial, a velocidade será ¼ da velocidade inicial.
REAÇÃO DE SEGUNDA ORDEM
2 NO(g) + Cl2(g) → 2 NOCl(g) (a 50ºC)
A reação é de segunda ordem em relação ao NO, de primeira ordem em relação ao Cl2 e de
terceira ordem global. Os dados experimentais mostram que se [Cl2] for mantida constante e a [NO] for duplicada, a velocidade da reação aumenta quatro vezes.
REAÇÃO DE ORDEM ZERO
2 NH3(g) → N2(g) + 3 H2(g) (a 856ºC)
A velocidade da reação é independente da concentração dos reagentes, no caso, amônia.
Reações heterogêneas podem ter ordem zero. Deve ser observado que as ordens em relação aos reagentes podem ser iguais aos coeficientes estequiométricos correspondentes, porém o mais comum é serem diferentes. Há vários métodos de se determinar a ordem da reação:
� Método da Velocidade Inicial. � Método Gráfico. � Método da Meia-Vida.
No presente estudo, será utilizado o método gráfico para a determinação da ordem da reação. As equações que relacionam a concentração com o tempo são diferentes para reações de
primeira e segunda ordem e ordem zero, e uma maneira prática de se determinar a ordem da reação e a constante de velocidade é o método gráfico, onde essas equações são rearranjadas de modo a se obter um gráfico retilíneo, cuja equação é:
ν = k[N2O5]
ν = k[NO]2[Cl2]
ν = k[NH3]0 = k
∆x
∆y
x = tgα = ∆y/∆x
α

51
Laboratório de Química 1- DQI
As equações de velocidade rearranjadas são:
Com os dados de concentração e tempo, constrói-se gráficos para obtenção de uma reta que mostrará a ordem da reação. A reta será obtida quando: ⇒ [A]t vs t for um processo de ordem zero. ⇒ ln[A]t vs t for um processo de primeira ordem. ⇒ 1/[A]t vs t for um processo de segunda ordem.
Expressões mais complexas para equações de velocidade como v = k[A1][A2] ou v = k[A1]2[A2] não dão linhas retas nos gráficos ln[A] ou 1/[A] contra o tempo. Nestes casos é necessário usar outros procedimentos. De maneira resumida temos:
TABELA 1: Propriedades características das reações do tipo A � Produtos.
II – Objetivo
Determinar se dois processos do cotidiano (queima de uma vela e jogo de moedas) seguem uma lei de velocidade de ordem zero, primeira ordem ou segunda ordem. Esses experimentos não representam uma experiência verdadeira de cinética química, mas servem como modelos simplistas para experiências de cinética química mais complexas e rigorosas, desenvolvidas em laboratórios, nos cursos de química. Você vai coletar dados sobre a variação da massa de uma vela de aniversário à medida que ela queima. Estes dados servirão para modelar dados de concentração que seriam coletados num experimento de cinética. Você deverá analisar a dependência dos dados com o tempo e determinar a ordem da reação.
Nessas experiências, os alunos coletam dados devido a variações na quantidade absoluta do objeto presente (massa da vela e número de moedas), ao invés de variações de concentração, e esses dados absolutos se assemelham ao modelo de concentração das experiências de cinética química.
III – Material Utilizado . 1 vela de aniversário pequena e fósforo; . 1 suporte onde a vela possa ser colocada para queimar; . balança; . relógio que marque segundos;

52
Laboratório de Química 1- DQI
IV – Procedimento Experimental
1 – QUEIMA DA VELA 1.1 – Pesar o suporte onde será fixada a vela. 1.2 – Pesar o conjunto suporte mais vela. 1.3 – Subtrair a massa do suporte da massa do conjunto para se obter a massa da vela. 1.4 – Acender a vela e deixar queimar por um minuto. Apagar a vela, pesar o conjunto e
subtrair a massa do suporte. 1.5 – Repetir esta operação a cada um minuto até a vela acabar.
Pode-se fazer uma analogia, comparando essa técnica de coleta de dados ao método de tomar alíquotas de uma reação em andamento.
DADOS COLETADOS � Massa da vela + suporte = _______________________ � Massa do suporte = ____________________________ � Massa da vela a t=0 = __________________________ � Massas coletadas na queima da vela TEMPO (minutos)
MASSA DA VELA (g)
TEMPO (minutos)
MASSA DA VELA (g)
TEMPO (minutos)
MASSA DA VELA (g)
0 8 16 1 9 17 2 10 18 3 11 19 4 12 20 5 13 21 6 14 22 7 15 23 Dados e resultados sobre a queima da vela Tempo (min)
Massa da vela (g)
In massa [massa]-1 Tempo (min)
Massa da vela (g)
In massa
[massa]-1
V - Construir os três gráficos com os dados obtidos e determinar a ordem do processo, a constante de velocidade k e o valor do intercepto da ordenada a t=0.

53
REFERÊNCIAS Abbot, D., Andrews, R.S. “Introduccion a la cromatografia”, 3ª. Ed. Editora Alhambra, Madrid, 1973. Atkins, P. J. “Physical Chemistry”. Oxford University Press, 5ª edição, EUA, 1995. Beran, J.A., “Laboratory Manual For Principles of General Chemistry”, 5a ed., John Wiley & Sons, New York, 1994. Bettelheim, F., Landsberg, J. and Lee, J. “Laboratory Experiments for General,organic and Biochemistry”, 2a. Ed., New York, 1995. Committee on Hazardous Substances in Laboratory – “Prudent Practices for Handling Hazardous Chemical in Laboratories”, National Academic Press, Washington, 1981. Committee on Hazardous Substances in the Laboratory – “Prudent Practices for Disposal of Chemicals from Laboratories”, Academic Press, Washington, 1983. Costa, M.H. & Honda, N.K., “Apostila de Química Geral – Atividades de Laboratório”, Depto Química / UFMS, 2000. CRC Handbook of Chemistry and Physics, 58a /ed. CRC Press, Inc. Florida, 1977. Domingues, X. A. D., “Cromatografia em papel y em capa delgada”, OEA, Washington, 1975. Ebbing, D.D. Química Geral, 5a edição, Livros Técnicos e Científicas, Editora S.A, 1998. Hunt, H.R., and Block, T.F. “Experiments for General Chemistry”, 2a Ed. John Wiley & Sons, 1994. Kotz, J.C. & Treichel, P., Química & Reações Químicas, 3a edição, Livros Técnicos e Científicos, Editora S.A, 1998. Moore, W. J., Físico-Química, 4a edição, Editora Edgard Blucher Ltda, 1972. Pavia, D.L., Lampman, E. M., Kriz, C.S., “Introduction to Organic Laboratory Techniques a Contemporary Approach”, 2nd ed, Saunders College Publishing, New York, 1982. Roberts, JR, J.L., Hollemberg, J.L. and Postma, J.M. “General Chemistry in the Laboratory”, 3a Ed. W.H. Freeman and Conpany, New York, 1991. Sanger, M. J., Wiley Jr., R. A., Richter, E. W. “Rate Law Determination of Everyday Processes” J. Chem. Educ., 79, (agosto de 2002), 989 – 991. Shreve, R.N. and Brink Jr, J.A., “Indústrias de Processos Químicos”, 4a. ed. Ed. Guanabara Dois, Rio de Janeiro, 1977. Silva, R.R., Bocchi, N. and Rocha Filho, R.C., “Introdução à Química Experimental”, Mcgraw-Hill, São Paulo, 1990. Soares, B. C., Souza, N. A, Pires, D.X. “Química Orgânica: teoria e técnicas de preparação, purificação e identificação de compostos orgânicos”, Editora Guanabara Dois, Rio de Janeiro, 1988.

54
Laboratório de Química 1- DQI
Swinehart, J.S., “Organic Chemistry – an Experimental Approach”, Prentice– Hall, INC, Englewood Cliffs, 1969. The Merck Index, 11a. Ed. Merck & Co, Inc. Rahway, 1989. Trindade, D.F., Oliveira, F.P., Banuth, G.S.L., Bispo, J.C. “Química Básica Experimental”, E.P. Parma. Vogel, A, I. “Química Orgânica – Análise Orgânica Qualitativa”, 3a. ed., Ao Livro Técnico SA, R.J.,1978. Zubrick, J.W. “The Organic Chemical Laboratory Survival Manual – A student’s guide to techniques”, 4a Ed. John Wiley & Sons, New York, 1997.
55
Laboratório de Química 1- DQI