vacinologiae engenharia de vacinas - universidade federal ... · dos meios de cultura ou das...
TRANSCRIPT

UNIVERSIDADE FEDERAL DE PELOTAS
Centro de Desenvolvimento Tecnológico – CDTec
Curso de Graduação em Biotecnologia
Vacinologia e Engenharia de Vacinas
Política e Regulação de Soros e Vacinas
Daiane [email protected]

ETAPAS DE DESENVOLVIMENTO
TECNOLÓGICO DE VACINAS
~= 10-20 anos
Descoberta Estudos Experimentais
Estudos Clínicos
Scale-up Estudos ClínicosFase I
Estudos ClínicosFase II
Estudos ClínicosFase III
Registro Produto
-- 1 -
Experimentais
- 2 -
Clínicos
- 3 - - 4 -
Fase I
- 5 -
Fase II
- 6 -
Fase III
- 7 -- 8 - - 9 -
Fase IV
- 10 -Características: multidisciplinar; longo período de maturação; alto risco de investimento.

Registro de medicamentosRegistro de medicamentos
Bases legais

Lei 6.360/76.
� Nenhum produto, inclusive o importado,
poderá ser industrializado, exposto à venda ou
entregue ao consumo antes de ser registradoentregue ao consumo antes de ser registrado
no Ministério da Saúde.

Lei dispõem, além do registro, sobre... � Da Autorização das Empresas e do Licenciamento dos Estabelecimentos� Da Responsabilidade Técnica� Da Rotulagem e Publicidade� Das Embalagens
Dos Meios de Transporte� Dos Meios de Transporte� Das Infrações e Penalidades� Da Fiscalização� Do Controle de Qualidade dos Medicamentos� Dos Órgãos de Vigilância Sanitária� Das Disposições Finais e Transitórias
Lei 6.360/76
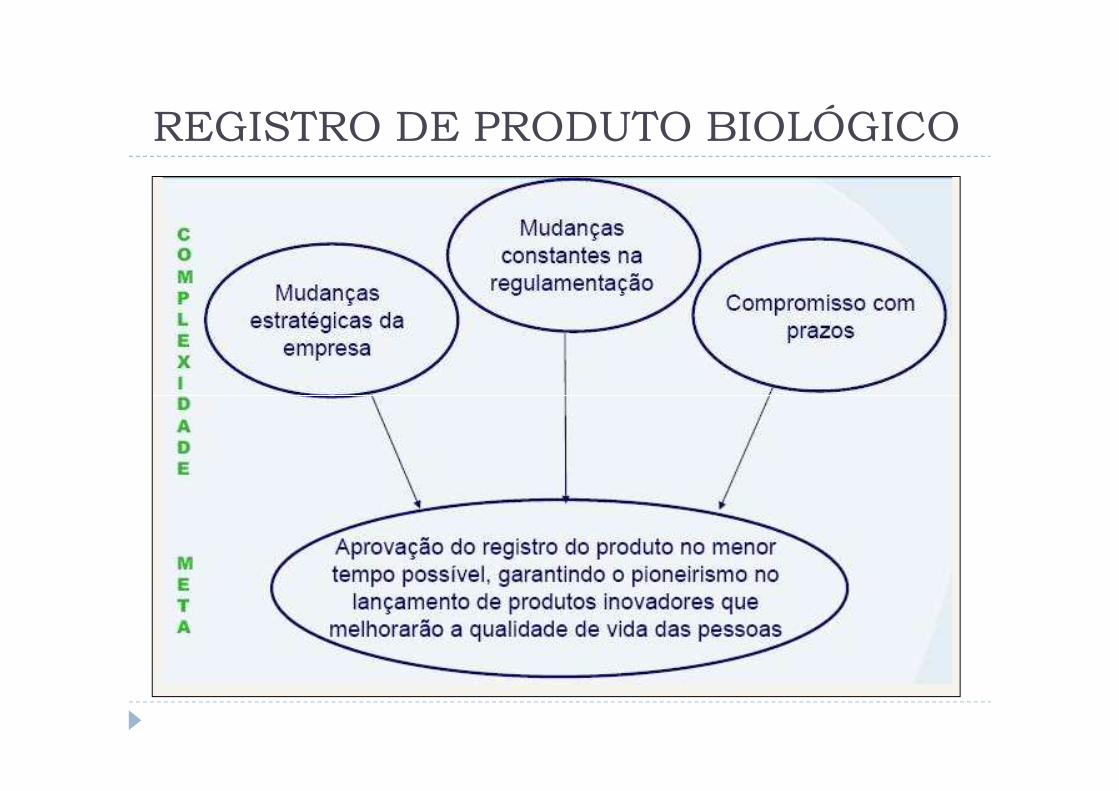
REGISTRO DE PRODUTO BIOLÓGICO

Aspectos gerais
� REGISTRO DE MEDICAMENTOS
� VALIDADE: 5 anos. Poderá ser revalidado por períodos iguais esucessivos, mantido o número do registro inicial
� REVALIDAÇÃO: 6 meses antes de expirar a validade.� REVALIDAÇÃO: 6 meses antes de expirar a validade.
� LEGITIMIDADE: publicação no DOU
� CANCELAMENTO DE REGISTRO: iniciar novo processo
� INSPEÇÃO: requisitos de BPF (Boas Práticas de Fabricação)
� Lei 6.360/76.

Evolução da Legislação de Registro de Medicamentos
� Mesmos critérios para diferentes categorias de
medicamentos
- Criação da ANVISA em 1999 (Lei 9.782/99)
� Medicamentos Genéricos – Lei 9.787/99
� Hemoderivados – RDC 46/00
� Produtos Biológicos – RDC 80/02 (primeiro marco normativo)
� Produtos Biológicos – RDC 315/05 (resolução vigente)
RDC (Resolução da Diretoria Colegiada)

Principais modificações no registro de
produtos biológicos implementadas pela
RDC 315/05:
� • Descrição de todas as etapas do processo de fabricação do princípioativo e do produto biológico a granel
� • Documentação de validação dos procedimentos de transporte;
Hemoderivados:� • Hemoderivados: relatório dos processos de controle de qualidade doplasma e a respectiva documentação de validação;
� • No caso de medicamento biológico, pode-se apresentar estudosclínicos de não-inferioridade, como demonstração da atividade terapêuticae segurança;
� • Documentação de produção e controle de qualidade de 3 lotesconsecutivos do princípio ativo e produto terminado.

O que é registrado como produto biológico no Brasil?
� Vacinas
� Soros Hiperimunes
� Hemoderivados
� Biomedicamentos: derivados de fluidos biológicos ou de
tecidos de origem animal, procedimentos biotecnológicostecidos de origem animal, procedimentos biotecnológicos
� Anticorpos monoclonais
� Medicamentos contendo microorganismos vivos, atenuados ou
mortos
� Probióticos
� Alérgenos

O que são produtos biológicos?
�Medicamentos obtidos a partir de fluidos biológicosou de tecidos de origem animal
� Medicamentos obtidos por procedimentos� Medicamentos obtidos por procedimentosbiotecnológicos

Resolução da Diretoria Colegiada – RDC
nº. 315/05
� Dispõe sobre o regulamento técnico de Registro,
Alterações Pós-Registro e Revalidações dos Produtos
BiológicosTerminados.

Legislação Específica
� Portaria 174/96 - SOROS Antivenenos, antitóxicos e antirábicos
� RDC 46/00 – HEMODERIVADOS
� RDC 210/03 –VALIDAÇÃO E BOAS PRÁTICAS DE FABRICAÇÃO
� RDC 233/05 – PRODUTOS ALERGÊNICOS
� RDC 234/05 – CONTROLE DE QUALIDADE� RDC 234/05 – CONTROLE DE QUALIDADE
� RDC 274/04 – GANGLIOSÍDEOS
� RDC 323/03 – MEDICAMENTOS PROBIÓTICOS
� RDC 315/05 – REGISTRO e ALTERAÇÕES
� RDC 350/05 – IMPORTAÇÃO
� RDC 47/09 – DIZERES DE BULA
� RDC 71/09 – DIZERES DE ROTULAGEM

REGISTRO DE PRODUTO BIOLÓGICO Arcabouço normativo
RDC 315/05
RDC 46/00HEMODERIVADOS
RDC 140/03 BULA
RDC 333/03RDC 168/02ROTULAGEM
RDC 233/05ALERGÊNICOS
Lei 6360/76Dec. 79094/77
RDC 315/05REGISTRO
RDC 323/03PROBIÓTICOS
RDC 274/04GANGLIOSÍDEOS
ROTULAGEM
RDC 210/03BOAS PRÁTICAS DE FABRICAÇÃO
Portaria 174/96SOROS Antivenenos, antitóxicos e antirábicos
RDC 350/05IMPORTAÇÃO
RDC 234/05CONTROLE QUALIDADE

Validação
� Ato documentado que atesta que qualquer procedimento,processo, equipamento, material, operação ou sistemarealmente conduza aos resultados esperados.
� RDC 210/03, ANVISA (agência reguladora vinculada ao Ministério daSaúde do Brasil)Saúde do Brasil)
� É regulamentação

RDC 210/03
Principalmente direcionada para validações de:
Comparada à RDC 134/01, a RDC 210/03 é mais exigente quanto a documentação e caracterização do
Sistema e Gerenciamento de Qualidade de cada empresa.
Principalmente direcionada para validações de:
�Processos
�Métodos Analíticos
�Áreas e Equipamentos
�Sistemas de água e ar
�Limpeza

Documentos GeraisDocumentos Gerais-Burocracia-
Registro de Produto Biológico

DOSSIÊ
� Documentação legal
� Documentação técnica
� Relatório de experimentação terapêutica

Documentação Legal
� Boas Práticas de Fabricação
� Taxa de Fiscalização de Vigilância Sanitária
Porte da empresa� Porte da empresa
� Autorização de Funcionamento de Empresa (AFE) +
Alvará Sanitário
� Certidão de Regularidade Técnica

Documentação Técnica
� Relatório técnico
� Bula + rotulagem
� Estudos de estabilidade� Estudos de estabilidade
� Relatório de produção e controle de qualidade
� Certificado de BPF

Certificado de Boas Práticas de Fabricação(CBPF)
� Enviar cópia do CBPF válido, expedido pela Anvisa, referente àempresa detentora do registro, no caso de produtos fabricadosno Brasil ou importados.
� Enviar cópia do CBPF válido expedido pela Anvisa, referente à� Enviar cópia do CBPF válido expedido pela Anvisa, referente àempresa fabricante do(s) -------, no caso de produtos fabricadosno Brasil ou importados.
� Enviar cópia do CBPF expedido pela Autoridade Sanitáriado país de fabricação do(s) -----------, conforme RDC315/2005.

Comprovante de Registro
� Comprovante de Registro no país de fabricação do Produto
Biológico Terminado ou do país que registrou o produto de
acordo com o item 7 do Capítulo II da RDC 315/05,
acompanhado da respectiva bula original e traduzida, aprovadaacompanhado da respectiva bula original e traduzida, aprovada
pela Autoridade Sanitária Competente do país que concedeu o
registro.

Código de Barra
� Enviar código de barra para TODAS AS
APRESENTAÇÕES (Registro e Renovação)

Relatório Técnico
� Cuidados de armazenagem e procedimentos utilizadosdurante o transporte
� Formas de acondicionamento e condições a seremmantidas para garantir a qualidade do produto.
� Documentação de validação dos procedimentos de� Documentação de validação dos procedimentos detransporte do produto biológico em sua embalagemprimária e do produto biológico terminado.
� Anexar uma declaração da empresa de que oarmazenamento e transporte atendem aos requisitos dacadeia de frio (produto termolábil).

Bula e rotulagem
Bula (RDC 140/03)Enviar modelo de texto debula, seguindo a seqüência doconteúdo:
I) Identificação do medicamento;
II) Informações ao paciente;
III) Informações técnicas aos
� Adequar as embalagens dosmedicamentos, conforme RDC333/2003
� A incapacidade técnica de se
Rotulagem (RDC 333/03)
III) Informações técnicas aosprofissionais de saúde;
IV) Dizeres legais, conforme art. 2°da RDC 140/2003.
As informações contidas no texto debula devem estar de acordo com abula aprovada pela AutoridadeSanitária do país de origem, como Relatório Técnico do produto ecom os estudos clínicos(indicações e reações adversas).
� A incapacidade técnica de sefazer constar todas as informaçõesna embalagem primária, previstasna RDC 333/03, deve serjustificada a ANVISA

Bula
"A exposição a níveis elevados de mercúrio metálico, inorgânicos ou orgânicos podem danificar
permanentemente o cérebro, rins e feto em cérebro, rins e feto em desenvolvimento. Efeitos sobre o funcionamento do cérebro pode resultar em irritabilidade, timidez,
tremores, mudanças nos problemas de visão ou audição e memória. "

Composição de uma vacina • líquido de suspensão: água destilada ou solução salina
fisiológica, podendo conter proteínas e outros componentes origináriosdos meios de cultura ou das células utilizadas no processo de produçãodas vacinas
• conservantes, estabilizadores e antibióticos: pequenasquantidades de substâncias antibióticas para evitar o crescimento dequantidades de substâncias antibióticas para evitar o crescimento decontaminantes (bactérias e fungos)
• estabilizadores (nutrientes): são adicionados a vacinasconstituídas por agentes infecciosos vivos atenuados
• adjuvantes: compostos utilizados para aumentar o poderimunogênico de algumas vacinas, amplificando o estímulo provocado poresses agentes imunizantes

Vacinas veterinárias
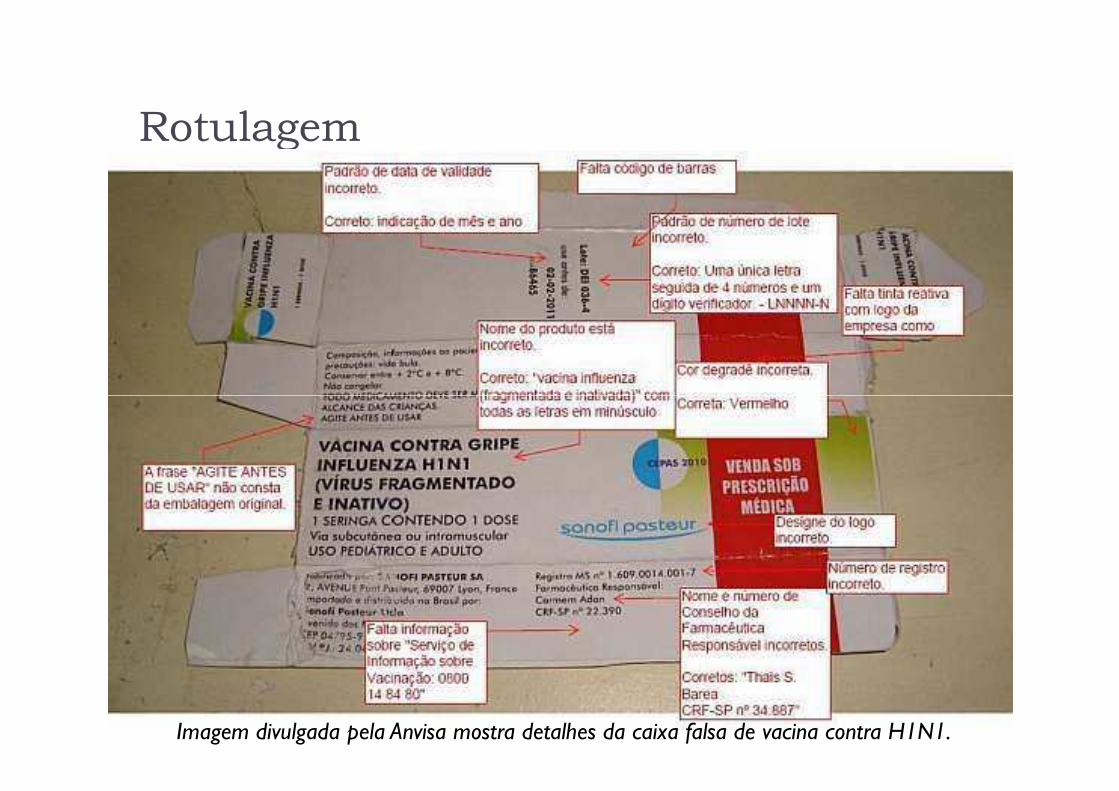
Rotulagem
Imagem divulgada pela Anvisa mostra detalhes da caixa falsa de vacina contra H1N1.

Os dados devem estar organizados nas seguintes seções:
Estudos pré-clínicos:
� TOXICOLOGIA GERAL
Relatório de experimentação terapêutica
� TOXICOLOGIA ESPECÍFICA: SUB-AGUDA, CRONICA, REPRODUTIVA (quandoaplicável)
� ESTUDOS FARMACODINÂMICOS (INCLUINDO ESTUDOSMICROBIOLOGICOS)
� ATIVIDADE MUTAGÊNICA
� POTENCIAL ONCOGÊNICO

Os dados devem estar organizados nas seguintes seções:
Estudos Clínicos
� a) Estudos Clínicos Fase I
Relatório de experimentação terapêutica
a) Estudos Clínicos Fase I
� b) Estudos Clínicos Fase II
� c) Estudos Clínicos Fase III
� ESTUDOS EM POPULAÇOES ESPECIAIS – quando aplicável

Centro de Pesquisas René Rachou – FIOCRUZ (Belo Horizonte, MG)


Relatório Pós-Comercialização
� A empresa deve enviar os estudos de Pós-comercialização
(relatório de farmacovigilância) atualizados, realizados nos
países onde já há registro e comercialização do produto
(REGISTRO E RENOVAÇÃO)(REGISTRO E RENOVAÇÃO)
� Estudos realizados no Brasil, em qualquer uma das fases,
deverão ser apresentados acompanhados de declaração do
estágio atual da pesquisa pelo grupo responsável

Estudo de Estabilidade
� Descrição dos estudos de estabilidade do Produto Biológico Terminado,
compatíveis com o prazo de validade solicitado:
� Mínimo 03 lotes do produto,
� Concentração,
� Forma farmacêutica,
� Acondicionamento primário e
� Condições ambientais ideais para armazenamento e transporte doproduto
� Estabilidade após reconstituição (se aplicável)
� Os dados dos estudos de estabilidade devem ser apresentados sob a forma
de tabelas e gráficos.
� Critérios internacionalmente estabelecidos: MERCOSUL e OMS.
� Referências complementares: EMEA, ICH e FDA.

Produção e Controle de Qualidade
� Documentação de produção e controle de qualidade de 3
lotes consecutivos, do Princípio Ativo na solicitação de
registro de Medicamento Biológico, no caso, de Medicamento
Biológico Novo de pelo menos 1 lote.Biológico Novo de pelo menos 1 lote.
� Documentação de produção e controle de qualidade de 3
lotes consecutivos, do Produto BiológicoTerminado.

Vacinas� Informação administrativo-legal: lista dos países onde o
produto já foi registrado e o resumo das características deaprovação;
� Avaliação do risco do meio ambiente (se aplicável)
� Diferença no processo de produção entre lotes clínicos� Diferença no processo de produção entre lotes clínicospilotos e lotes de produção (se aplicável)
� Demonstrar compatibilidade entre os componentes dasvacinas combinadas ou conjugadas
� Programas de estudo de estabilidade posterior ao registro
� Vacinas combinadas e administração concomitante comoutras vacinas

Vacinas
Considerações especiais:
� Vacinas atenuadas: avaliação de possível “sheeding” viral
� Toxicidade de novas substâncias incorporadas a � Toxicidade de novas substâncias incorporadas a formulação: adjuvantes, conservantes, aditivos
� Adjuvantes: especificação, origem e estudos de farmacodinâmica e segurança

Licenciamento de ImportaçãoPrincipais Causas de Exigência
� A empresa deve enviar documento (p.ex. fluxograma de
produção) relacionando o(s) número(s) do(s) lote(s) do
princípio ativo com os lotes do produto acabado
� Documentação prevista na RDC 68/2003 atualizada e
quadros Q1 e Q2 corretamente preenchidos

Identificação de produto: SORO ANTI-RÁBICO
QUADRO
Q1
1. Substância (s) (DCB, DCI, CAS, INCI)Soro anti-rábico
1. Substância (s) (DCB, DCI, CAS, INCI)Soro Fetal Equino Correto
1. Substância (s) (DCB, DCI, CAS, INCI)Polisorbato 80
2. Nome comercial: Tween 80

Lote: XX4488
Identificação da mercadoria (Nome comercial):
Vacina contra Raiva
Identificação da mercadoria (Nome comum ou nome químico):
QUADRO Q2
Identificação da mercadoria (Nome comum ou nome químico):
Vacina contra Raiva
Identificação da empresa fabricante: Al-Queda
País de origem (fabricação): Afeganistão
RDC 68/2003

Licenciamento de importação Produto Biológico
1. Protocolo resumido de produção (OMS)
2. Certificado de análise do princípio ativo
3. Certificado de análise do produto acabado
4. Certificado de liberação do lote pela Autoridade Sanitária do país defabricaçãofabricação
� Documentos técnicos do Hemoderivado utilizado como estabilizante:
a) declaração de origem do plasma utilizado
b) certificado de análise do controle de qualidade do plasma utilizado
c) certificado de liberação da sorologia do plasma utilizado
RDC 350/05

Consultas PúblicasConsultas Públicas
Registro de Produtos Biológicos

Consultas Públicas

Consultas Públicas
� CP 71/2009: Alterações pós-registro de Produtos Biológicos
� CP 72/2009: Estudos de estabilidade de Produtos Biológicos
� CP 49/2010: Registro de Produtos Biológicos Novos e� CP 49/2010: Registro de Produtos Biológicos Novos eProdutos Biológicos
� CP 49/2010: (Revisão da RDC 315/05 ) Dispõe sobre o registro de produtos biológicos novos e produtos biológicos

Consulta Pública nº 49/2010 – Definições
� I – Produto Biológico Novo: é o medicamento biológicoque contém molécula com atividade biológica conhecida,ainda não registrada no Brasil e que tenha passado portodas as etapas de fabricação.
� II – Produto Biológico: o Produto Biológico não novo ouconhecido é o medicamento biológico que contémmolécula com atividade biológica conhecida, já registradano Brasil e que tenha passado por todas as etapas defabricação.

� III – Produto Biológico Comparador: é o produtobiológico já autorizado pela Anvisa com base na submissãode um dossiê completo, e que já tenha sido comercializadono País.
Consulta Pública nº 49/2010 – Definições

Consulta Pública nº 49/2010Vias Regulatórias Possíveis para oRegistro de Produtos Biológicos
� Dossiê completo
� Desenvolvimento individualDesenvolvimento individual
� Desenvolvimento por comparabilidade

Consulta Pública nº 49/2010Vias Regulatórias Possíveis para oRegistro de Produtos Biológicos
DOSSIÊ COMPLETO (produto biológico novo)
� Dados totais sobre desenvolvimento, produção,
controle de qualidade, estudos não clínicos e clínicos
(Fases I, II e III).

Consulta Pública nº 49/2010Vias Regulatórias Possíveis para oRegistro de Produtos Biológicos
DESENVOLVIMENTO INDIVIDUAL (Produto BiológicoNovo e/ou Produto Biológico)
� Não é feita comparação com outro produto biológico járegistrado. Dossiê completo deve ser apresentado.Estudos clínicos de não inferioridade ou estudos clínicosde equivalência, de fase III, poderão ser aceitos paraconfirmação da eficácia e segurança.

Consulta Pública nº 49/2010Vias Regulatórias Possíveis para oRegistro de Produtos Biológicos
DESENVOLVIMENTO POR COMPARABILIDADE(Produto Biológico)
� Deve ser eleito um Produto Biológico comparador. Éutilizado o exercício de comparabilidade em termos dequalidade, eficácia e segurança, entre o produtodesenvolvido para ser comparável e o produto biológicocomparador.

Vias regulatórias possíveis para registro de um produto biológico

Daiane [email protected]