universidade federal do parÁ prÓ-reitoria de … · 2 resumo do relatÓrio anterior entre os...
TRANSCRIPT

UNIVERSIDADE FEDERAL DO PARÁ
PRÓ-REITORIA DE PESQUISA E PÓS-GRADUAÇÃO
DIRETORIA DE PESQUISA
PROGRAMA INSTITUCIONAL DE BOLSAS DE INICIAÇÃO CIENTÍFICA
RELATÓRIO TÉCNICO-CIENTÍFICO
Período: 08/09 a 07/10
( ) Parcial
( X ) Final
IDENTIFICAÇÃO DO PROJETO
Título do Projeto de Pesquisa (ao qual está vinculado o Plano de Trabalho): APLICAÇÃO DE
TÉCNICAS DE SIMULAÇÃO COMPUTACIONAL NO PLANEJAMENTO DE
BIOMOLÉCULAS COM AÇÃO ANTIMALÁRICA
Nome do Orientador: Prof. Dr. Cláudio Nahum Alves
Titulação do Orientador: Doutorado
Faculdade: Faculdade de Química
Unidade: Instituto de Ciências Exatas e Naturais
Laboratório: Laboratório de Planejamento e Desenvolvimento de Fármacos
Título do Plano de Trabalho: ESTUDO TEÓRICO DE INIBIDORES DA ENZIMA GAPDH
DE Trypanosoma cruzi ATRAVÉS DE DOCKING MOLECULAR, DINÂMICA
MOLECULAR E QM/MM.
Nome do Bolsista: José Rogério de Araújo Silva
Tipo de Bolsa: ( X ) PIBIC/CNPq
( ) PIBIC/UFPA
( ) PIBIC/INTERIOR
( ) PIBIC/FAPESPA
( ) PARD
( ) PARD – Renovação
( ) Bolsistas PIBIC do edital CNPq 001/2007

2
RESUMO DO RELATÓRIO ANTERIOR
Entre os principais gases de efeito estufa estão: o dióxido de carbono (CO2) e o
metano (CH4), sendo que este último com uma ação mais agressiva na atmosfera, requerendo
assim uma maior atenção em relação com o seu controle. Aproximadamente 70% do total de
metano são de origem biológica produzida através da metanogênese. A Metil Coenzima M
Redutase (MCR) é atuante na etapa final da metanogênese. Neste trabalho foi utilizado o
método de modelagem por homologia e dinâmica molecular (DM) para se obter a estrutura
tridimensional da MCR. A estrutura 3D gerada foi modelada por homologia com MCRα de
Methanopyrus klanderi, com identidade de 70%. O alto valor de identidade há entre as
estruturas molde e homóloga estão de acordo com o valor de RMSD (Root Mean Square
Deviation) obtido de 0,06 Å. A média da porcentagem de resíduos localizados em regiões
favoráveis (regiões em que os ângulos e energia residual são compatíveis) das estruturas 3D
deste trabalho foi de 84%, indicando uma boa qualidade estereoquímica. Para os sistemas
foram realizados 10 ns de DM, o target apresentou uma maior variação no RMSD em relação
ao template, esta diferença deve-se ao fato de o target ser um fragmento do template,
apresentando, assim, maior grau de mobilidade em relação ao template. A estrutura 3D do
template apresenta uma estabilidade estrutural a partir de ~4ns de DM, enquanto o target
alcança esta estabilidade a partir de ~7ns de DM. A modelagem por homologia apresentou
resultados satisfatórios quanto à construção do modelo 3D de MCR, principalmente em
relação ao alto grau de identidade e a qualidade estereoquímica do modelo obtido. A DM foi
utilizada como ferramenta para refinar a estrutura de MCR obtida por homologia. Desta
forma, o modelo obtido poderá ser usado em estudos, como: docking, mecanismo de reação e
mecanismo enzimático.

3
INTRODUÇÃO
Alguns protozoários parasitas da família Trypanosomatidae são responsáveis por uma
grande variedade de graves doenças tais como a tripanossimíase americana ou
esquizotripanose, também conhecida como doença de Chagas. Mais ou menos 100 milhões de
pessoas – um quarto do total de habitantes da América Latina – estão em áreas de risco de
contrair esta doença e estima-se que entre 16 e 18 milhões de pessoas estão infectadas
(MOREL, 1999; FRANCO-PAREDES et al., 2009).
A doença de Chagas é causada por infecção pelo parasita Trypanosoma cruzi, que é
transmitido por insetos da espécie Triatoma infestans (popularmente conhecido no Brasil
como “barbeiro”). Em alguns casos a transmissão ocorrer por transfusões sanguíneas ou
congenitamente por mães infectadas (URBINA et al.,1999).
Não há fármacos recomendados para o tratamento da doença de Chagas apesar de ter
sido descrita sua etiologia em 1909, pelo médico sanitarista Carlos Chagas. Os medicamentos
disponíveis para o tratamento paliativo da doença são o Nifurtimox e o Benzonidazol (Fig. 1)
e apresentam efeitos colaterais severos, tais como: anorexia, distúrbios gastrintestinais e
erupções cutâneas (CHAGAS, 1909; PINK et al.,2005).
Figura 1: Estruturas 2D dos fármacos Nifurtimox e Benzonidazol, respectivamente.
Uma moderna abordagem para o desenvolvimento de fármacos para alvos
direcionados envolve a identificação de enzimas que participam em parte do caminho
metabólico essencial de patógenos (BARRETT et al., 1999). Desta forma, entre os alvos
enzimáticos contra o T. cruzi encontrados estão as enzimas tripanotiona redutase, farnesil
transferase, gliceraldeído-3-fosfato desidrogenase, DNA topoisomerase, di-hidrofolato
redutase e farnesilpirofosfato sintase (DIAS, 2000; COURA et al., 2002; DARDONVILLE,
2005, COURA, 2003).

4
Figura 2: Estrutura 3D da enzima GAPDH de T. cruzi (código pdb 1QXS).
Dentre estes alvos, a enzima gliceraldeído-3-fosfato desidrogenase (GAPDH) (Fig. 2),
que é um homotretâmero de massa molecular aproximadamente 156 kDa, cuja as
propriedades físico químicas são conservadas na maioria das espécies (SEYDOUX et al.,
1973), foi selecionada para este trabalho.
Os parasitas da família Trypanosomatidae são altamente dependentes da via glicolítica
para a produção de ATP (adenosina trifosfato) (OPPERDOES, 1987; OPPERDOES et al.,
2001) e a modelagem in silico desta via mostrou que a reação é catalisada pela enzima
glicolítica gliceraldeído-3-fosfato desidrogenase (gGAPDH) (BAKKER et al., 1999;
BAKKER et al., 2000), que catalisa a fosforilação oxidativa do gliceraldeído-3-fosfato em
1,3-difosfoglicerato (DPG) na presença de fosfato inorgânico e NAD+ como coenzima (Fig.
3) (SOUZA et al., 1998).
Figura 3: Representação esquemática da reação catalisada pela enzima GAPDH.
A enzima GAPDH também desempenha funções importantes em outros processos
celulares. Classificada como uma enzima multifuncional, a GAPDH desempenha funções em
diversos processos celulares como apoptose, no transporte e fusão de membrana nuclear,
formação de microtúbulos, transporte nuclear de RNA, replicação e reparo de DNA, controle
de expressão de gene (ROSES et al., 1996; SIROVER, 1999). Além de evidências que

5
indicam a participação da GAPDH em algumas doenças neurodegenerativas (e.g., Alzheimer
e Parkinson), câncer de próstata e distúrbios metabólicos (ROSES et al., 1996; TATTON et
al., 2003; WALDMEIER; TATTON, 2004; CARLILE et al., 2000; BURKE et al., 1996).
JUSTIFICATIVA
O Trypanosoma cruzi é o parasita causador da doença de Chagas (CHAGAS, 1909).
A doença de Chagas é uma das patologias de mais larga distribuição no continente americano.
É conhecida a existência de vetores da doença desde o sul dos Estados Unidos à Argentina.
São mais de cem espécies responsáveis pela transmissão natural da infecção pelo T. cruzi,
intervindo diretamente na sua veiculação no ambiente domiciliar ou participando na
manutenção da enzootia chagásica. Estima-se que sejam de 16 a 18 milhões os indivíduos
infectados e de aproximadamente oitenta milhões a população em risco de contaminação na
América Latina (FRANCO-PAREDES et al., 2009). Descoberta em 1909, pelo cientista
brasileiro Carlos Chagas, esta doença deve merecer atenção especial do governo e dos
segmentos da população, pois, embora seja difícil sua cura ela, pode e deve ser prevenida,
evitando-se que as novas gerações se contaminem. (SUCAM, 1989).
O peso relativo da doença de Chagas, com respeito a outras enfermidades
transmissíveis endêmicas na América Latina e no Caribe, e medido por “Anos de Vida
Ajustados à Incapacidade – AVAI – só é superado pelo conjunto das enfermidades diarréicas,
infecções respiratórias e AIDS (SILVEIRA et al., 2002) e equivale a, aproximadamente,
cinco vezes a soma de malária, esquistossomose e leishmanioses (SILVEIRA et al., 2002).
Devido aos reduzidos investimentos da indústria farmacêutica em pesquisas que
tenham por objetivo o desenvolvimento de novos compostos bioativos para o tratamento
dessa doença torna-se claro a necessidade de pesquisas que levem ao desenvolvimento de
novos fármacos nesta área, conduzindo projetos que tenham como objetivo a descoberta de
novos inibidores potentes e específicos de enzimas chaves do metabolismo do T. cruzi
(SOUZA et al., 1998; URBINA; DOCAMPO, 2003)
Na tentativa de controlar a atual situação, o Brasil desenvolveu práticas estratégias
que visam prevenir a mortalidade em decorrência da doença de Chagas. Novas drogas têm
sido desenvolvidas, objetivando as associações terapêuticas eficazes contra a infecção. Assim,
a busca de novos produtos mais ativos e com baixa toxidade possibilitará um melhor manejo
terapêutico desta infecção. Desta forma, neste estudo utilizou-se técnicas de simulação
computacional, tais como: Docagem Molecular (“molecular docking”), Dinâmica Molecular

6
(DM) e QM/MM, para ajudar no planejamento de novos compostos bioativos capazes de
apresentar elevada atividade anti tripanomicida e com baixa toxidade ao organismo humano.
OBJETIVOS
Objetivo geral
Elucidação da interação da enzima GAPDH de T. cruzi com os inibidores HOP e ácido
fosfônico através de estudos de Docagem Molecular, Dinâmica Molecular e QM/MM.
Objetivos Específicos
Planejar novos compostos bioativos utilizando como métodos docagem molecular,
DM e QM/MM;
Determinar a flexibilidade dos resíduos de aminoácidos (oriundos da enzima GAPDH
de T. cruzi) responsáveis pelo reconhecimento molecular dos inibidores através de
estudos de dinâmica molecular e QM/MM;
Determinar a energia de interação entre os resíduos de aminoácidos e dos inibidores
que serão estudados através de metodologia QM/MM;
Realizar cálculos de energia livre e interação por resíduo.
MÉTODOS E FUNDAMENTAÇÃO TEÓRICA
OBTENÇÃO DA ESTRUTURA DA ENZIMA NO PDB
Inicialmente foi obtida a estrutura com a qual os cálculos serão realizados. A
estrutura inicial foi localizada no Protein Data Bank (PDB), com o código 1QXS (LADAME
et al., 2003). Este PDB apresenta a estrutura base para os inibidores: ácido 3-hidróxi-2-oxo-4-
fosfonóxi-butilfosfônico (HOP) e ácido 4-butilfenil-amino-metileno-fosfônico (Fig. 4),
denominado ácido fosfônico. A estrutura cristalográfica da GAPDH de T. cruzi foi utilizada
para esse estudo.

7
Figura 4: Estruturas 2D dos inibidores estudados: HOP e Ácido Fosfônico, respectivamente.
A estrutura do inibidor HOP e seus derivados utilizados neste estudo são oriundos da
própria cristalografia da enzima. Já o ácido fosfônico é uma molécula que está sendo estudada
no Instituto de Química de São Carlos, no grupo do Prof. Carlos Montanari - grupo de
Química Medicinal de Produtos Naturais (NEQUIMED) – o qual temos uma parceria
estabelecida.
DOCAGEM MOLECULAR
A docagem molecular (do inglês “molecular docking”) consiste na predição da
estrutura de um complexo intermolecular formado entre duas ou mais moléculas constituintes
(SOUSA et al., 2006), em outras palavras, na predição da conformação bioativa de uma
pequena molécula (ligante) no sítio de ligação de uma macromolécula (enzima ou receptor)
seguido de avaliação (pontuação) e classificação do modo de ligação proposto (GUIDO et al.,
2008; KITCHEN et al., 2004). Pioneira durante o início dos anos 80 (KUNTZ et al., 1982), a
docagem molecular continua sendo um campo de pesquisas vigorosas, tornando-se uma
ferramenta útil na descoberta de fármacos, e um componente primário em muitos sofwares
utilizados no processo de planejamento de fármacos (KITCHEN et al., 2004). De forma
especial, a docagem proteína-ligante ocupa um lugar de destaque no campo geral de docagem
molecular, por suas aplicações na medicina (MUEGGE; RAREY, 2001). Neste sentido, esta
ferramenta foi utilizada para a elucidação da conformação estrutural do inibidor ácido
fosfônico no sítio de ligação da enzima GAPDH de T. cruzi, bem como a determinação deste
sítio.
Os sofwares utilizados para realizar os estudos de docagem molecular descritos neste
trabalho foram: AutoDock 4.2 (com a função de busca baseada no algoritmo genético
Lamarckiano incorporada a uma flexibilidade limitada do receptor)(MORRIS et al., 1998;
HUEY et al., 2007; MORRIS et al., 2009) e AutoDock Vina versão 1.0.3 (com uma variedade

8
de funções de otimização global estocástica, incluindo algoritmos genéticos, também
utilizando uma porção flexível do receptor) (TROTT; OLSON, 2009). Além do
AutoDockTools4 versão 1.4.5 (MORRIS et al., 1998; MORRIS et al., 2009), utilizado na
preparação das estruturas (proteína-ligante) para os procedimentos da docagem.
AutoDock
O programa AutoDock (MORRIS et al., 1998) possui licença livre para acadêmicos,
além de possuir uma boa precisão e alta versatilidade (SOUSA et al., 2006). Tais fatores
fazem com que este programa seja muito utilizado, e bem citado na literatura (SOUSA et al.,
2006).
O AutoDock utiliza o algoritmo genético Lamarckiano (LGA – “Lamarckian genetic
algorithm”) (MORRIS et al., 1998), mas engloba também o método de simulação Monte
Carlo (METROPOLIS et al., 1953, KIRKPATRICK et al., 1983) e um algoritmo genético
tradicional (HOLLAND, 1975). Entretanto os dois últimos métodos não são tão eficientes e
confiáveis como o LGA (SOUSA et al., 2006). O programa também utiliza uma função de
quinto termo baseada no campo de força AMBER (WEINER et al., 1984), e que compreende
também, um termo de dispersão 6-12 de Lennard-Jones, um termo direcional 10-12 de ligação
de hidrogênio, um potencial eletrostático coulômbico, um termo entrópico e um termo de
dessolvatação intermolecular (HUEY et al., 2006, SOUSA et al., 2006). O fator de escalo
para cada um desses cinco termos está calibrado, empiricamente, por um conjunto de 30
complexos proteína-ligante estruturalmente conhecidos (SOUSA et al., 2006).
A versão utilizada neste estudo, versão 4.2, engloba os métodos utilizados nas
versões anteriores em conjunto com a possibilidade de flexibilidade de parte do receptor
(MORRIS et al., 2009). Sendo este um fator importante, pois os métodos de docagem que
levam em consideração a flexibilidade de parte da enzima, geralmente, geram melhores
resultados (OSTERBERG et al., 2002; ERICKSON et al., 2004).
AutoDock Vina
O AutoDock Vina é um novo programa para docagem molecular e virtual screening
(TROTT; OLSON, 2009). Esta versão atinge, aproximadamente, duas ordens de magnitude
acelerada, quando comparada com o AutoDock4.2 e suas versões anteriores. Ao mesmo,
melhorando significativamente, a precisão do modo de ligação dos ligantes (TROTT;
OLSON, 2009).

9
No desenvolvimento do AutoDock Vina, uma variedade de funções de otimização
global estocástica foram abordadas (TROTT; OLSON, 2009), incluindo algoritmos genéticos
(HOLLAND, 1975), otimização de átomos (EBERHART; KENNEDY, 1995), método de
simulação (KIRKPATRICK et al., 1983), e outros, combinados com vários procedimentos de
otimização local e “truques” especiais que aceleram a otimização (TROTT; OLSON, 2009).
Os procedimentos operacionais realizados com a versão 1.0.3, do AutoDock Vina, são
semelhantes aos procedimentos realizados com o AutoDock 4.2. Também foram utilizados
resíduos flexíveis no processo de docagem.
DINÂMICA MOLECULAR E QM/MM
PREPARAÇÃO DOS SISTEMAS
O primeiro passo depois de adquirido o modelo da enzima e docagem do ácido
fosfônico é adicionar hidrogênios a estrutura, considerando o estado de protonação dos
aminoácidos a um pH 7,0. Como os valores padrões de pKa podem ser modificados por
efeitos locais do entorno protéico (ANTOSIEWICZ et al., 1994), deve-se realizar cálculos de
pKa para os distintos estados de protonação dos resíduos ionizáveis, para recalcular o pKa
para esses resíduos pretendemos usar o cluter method (ANTOSIEWICZ et al., 1994) e o
software Propka (LI et al., 2005).
A etapa seguinte é colocar o sistema dentro de uma caixa de água, tendo como centro
de massa o inibidor, pretendemos usar uma caixa cúbica de água de 125 Å com moléculas
TIP3, a caixa cúbica deve estar previamente relaxada. Para que o tempo de cálculo seja menor
pretendemos congelar todos os resíduos que estejam a uma distância maior do que 20 Å do
centro de massa.
Os inibidores serão descritos pelo método semiempírico AM1 (DEWAR et al., 1993),
estando por tanto dentro da região QM. Para realizar cálculos a um nível quântico mais
elevado, pretendemos usar o método B3LYP (KIM; JORDAN, 1994) com a base 6-31g*.
Toda proteína e moléculas TIP3 serão tratadas mediante o campo de força OPLS-AA/TIP3
(JORGENSEN et al., 1983), todos esses cálculos serão realizados utilizando o “programa”
DYNAMO (FIELD, 1999; FIELD et al., 2000).
PROGRAMAS UTILIZADOS
Como comentado anteriormente, para realizar as simulações de DM usaremos o
“programa” DYNAMO (FIELD, 1999; FIELD et al., 2000). DYNAMO na verdade é uma

10
livraria escrita em linguagem Fortran, que é uma potente e moderna linguagem de
programação (FIELD, 1999; FIELD et al., 2000). DYNAMO nos permitirá realizar cálculos
AM1, usar o campo de força OPLS-AA/TIP3, realizar cálculos de energia, realizar otimização
de geometrias, realizar pesquisa de estruturas de transição e calcular caminhos de reação
(FIELD, 1999; FIELD et al., 2000). DYNAMO tem a vantagem de ser gratuito e possuir a
arquitetura aberta (FIELD, 1999; FIELD et al., 2000) o que oferece a liberdade de se poder
escrever programas para cálculos de energia de interação e realizar otimização de geometrias
a nível ab initio. Para visualizar as estruturas usaremos o programa VMD (VMD, 2009), que é
um programa que oferece a vantagem de ser gratuito e ter boa condição gráfica. VMD é um
programa de visualização molecular em três dimensões, é muito usado para visualizar
sistemas biológicos como proteínas (VMD, 2009). Tal programa nos permite ler arquivos nos
formatos pdb e trj, o que nos permite visualizar as trajetórias obtidas com cálculos de
dinâmicas molecular.
MÉTODOS HIBRÍDOS QM/MM
Em reações químicas de interesse biológico é imprescindível introduzir nos cálculos
computacionais o efeito do meio, seja o solvente ou um envoltório protéico, já que o meio
pode modificar significativamente a estrutura e a reatividade dos compostos. Por tanto, torna-
se importante dispor de um método capaz de descrever a interação química e as interações do
sistema químico com o meio. Por esse motivo foi escolhido o método híbrido QM/MM
(WARSHEL; LEVITT, 1976; KOLLMAN et al., 2001), onde se combina a mecânica
quântica com a mecânica molecular.
Primeiramente, o sistema estudado em três partes, uma região QM (trata-se mediante
os métodos de mecânica quântica), uma região MM (trata-se mediante os campos de força da
mecânica molecular clássica) e uma região de contorno.
A região QM inclui todos os átomos envolvidos diretamente na reação química, ou
seja, os átomos que participam da formação e rupturas das ligações. Os átomos desta região
são representados como elétrons e núcleos, e as superfícies de potencial associadas a eles são
determinadas pela aproximação de Born-Oppenheimer (BORN; OPPENHEIMER, 1927), de
modo que a energia é função das posições dos núcleos.
A zona MM contém o resto dos átomos do sistema. Estes são descritos classicamente,
e suas interações são determinadas mediante funções de energia potencial empírica, de modo
que esses átomos não podem estar envolvidos no processo de formações ou ruptura de

11
ligações. Os átomos MM interagem com os átomos QM envolvidos na reação, de modo que
seus movimentos e interações influem sobre os átomos QM, e vice-versa. Por último, se
encontra a região denominada de contorno, que corresponde a um conjunto de restrições a
serem aplicadas.
Representação esquemática da divisão de um sistema na região QM,
região MM e região de contorno.
CÁLCULO DA ENERGIA DE INTERAÇÃO
A energia para um sistema QM/MM pode ser obtida por:
(1)
onde 0H é o hamiltoniano no vácuo para a parte QM selecionada, é a função de onda
polarizada, Emm representa a energia do campo de força e MM/QMV é a união entre o operador
do subsistema QM e MM e inclui um termo eletrostático e de Van der Waals.
A equação 1 pode ser escrita numa forma diferente a fim de definir a energia de
interação QM/MM. De forma eficaz, a energia total pode ser escrita como a soma da energia
QM do subsistema no vácuo, a energia de interação QM/MM e a energia MM:
(2)
onde 0 é a função de onda padrão não polarizada (fase gasosa) do subsistema QM. A energia
de interação (EQM/MM) pode ser mais adiante decomposta considerando que o operador de
junção entre o subsistemas QM e MM inclui um termo eletrostático e de Van der Waals. Esta
última contribuição é normalmente avaliada usando a expressão de Lennard-Jones (JONES,
1924):

12
(3)
onde Rij é a distância entre os centros de interação QM e MM. Como esta expressão não
envolve coordenada eletrostática este termo de energia não precisa ser incluído na avaliação
SCF da função de onda do subsistema QM.
Após a obtenção dos resultados para energia de interação pretendemos comparar os
resultados teóricos com os dados biológicos (SHAW et al., 1996; MCCLAIN et al., 2002).
CÁLCULOS DE ENERGIA LIVRE
Um dos problemas centrais do campo da biologia computacional é determinação
quantitativa da afinidade absoluta do binding nos mais diversos e complexos sistemas.
Predizendo a energia livre de ligação entre o ligante e a proteína pode-se obter um valor
prático para identificação de novas moléculas que podem ligar-se nos alvos receptores
atuando como agentes terapêuticos (WLODAWER, 2002). Um dos métodos para cálculo de
energia livre é o Free Energy Pertubation (FEP), que é um método usado para calcular
energia livre de diferentes sistemas. O método FEP quando combinado com dinâmica
molecular pode ser usado para estudar a interação enzima-inibidor (JARMULA et al., 2003;
UDIER-BLAGOVIC et al., 2004). Pretendemos realizar cálculos de FEP utilizando o
programa DYNAMO modificado e comparar os resultados obtidos com os dados de atividade
biológica. Caso ocorra correlação entre os dados, poderemos propor um modelo de um novo
composto baseado nos valores de energia livre.
Para avaliar a Energia Livre Eletrostática foi introduzido um parâmetro eletrostático λ
na equação da energia de interação do sistema QM/MM, logo a equação da Energia Livre
vem descrita como:
MM
vdW
MMQM
MMQM
MMQM
MMe
MMoMMQM EE
r
qZ
r
qHE /
,,
/ˆ
O cálculo da diferença de energia livre entre duas janelas sucessivas é executado por
meio de métodos de energia livre de perturbação (FEP), que para casos não polarizáveis
reduzem os campos de força clássicos:

13
dreFMMQM
MMQM
MMe
MMi
MMQM
MMQM
MMe
MMj
r
qZ
r
q
r
qZ
r
q
MMQMElect
ji
,,,,
ln1/
Então, a integração é calculada para todos os valores de λ. Este procedimento pode ser
aplicado tanto para (i→j) quanto para (j→i) contanto que se comparadas mostrem informação
sobre a convergência do processo.
RESULTADOS E DISCUSSÃO
DOCAGEM MOLECULAR
O composto ácido fosfônico foi identificado como um inibidor da enzima GAPDH de
T. cruzi, seu mecanismo de inibição foi determinado experimentalmente como sendo não
competitivo (WIGGERS, 2007). Porém neste trabalho observou-se que este inibidor também
pode possuir um mecanismo de inibição competitivo.
Primeiramente, para docagem molecular do ácido fosfônico, solvente e ligantes (neste
caso o HOP e o NAD+) foram removidos da estrutura 3D da enzima GAPDH utilizando o
programa Chimera UCSF (PETTERSEN et al., 2004; GODDARD et al., 2007), os
hidrogênios polares foram adicionados utilizando o programa AutoDockTools4 (ADT)
(MORRIS et al., 1998). Procedimento semelhante também foi realizado, porém retirando
apenas o solvente e o ligante HOP.
As cargas parciais de Gasteiger-Marsili (GASTEIGER; MARSILI, 1978;
GASTEIGER; MARSILI, 1980) para cada átomo da proteína e do inibidor foram calculadas.
Foram realizados dois procedimentos para busca pelo possível sítio de interação utilizando
uma caixa (com espaçamento de 0.375 Angstroms) que abrangeu toda a região de interação
do inibidor (ácido fosfônico) e o cofator (NAD+), levando ao resultado no qual o sítio de
inibição competitivo, visto que não é incomum encontrar o sítio ativo da GAPDH ocupada
por ligantes (PAVAO et al., 2002; CASTILHO et al., 2003). E o outro procedimento, também
utilizando uma caixa (com espaçamento de 0.375 Angstroms), porém não abrangendo a
região do NAD+. Ambos os procedimentos foram realizados mantendo uma parte da GAPDH
como rígida e o resíduo Thr199 como flexível, pois este resíduo é capaz de interagir com o
ácido fosfônico em ambos os sítios de interação.

14
Quando o inibidor ácido fosfônico foi docado no sítio competitivo da enzima, tanto na
presença como na ausência do cofator NAD+, os melhores resultados foram obtidos
utilizando o programa AutoDock Vina (TROTT; OLSON, 2009) como pode ser visto na
Figura 5.
Figura 5: Mapa de superfície do sítio competitivo da GAPDH com a estrutura do ácido fosfônico complexa. (A)
– ácido fosfônico complexado no sítio competitivo na presença do cofator NAD+. (B) - ácido fosfônico
complexado no sítio competitivo na presença ausência do cofator. As figuras foram obtidas usando o programa
Chimera UCSF.
A docagem do ácido fosfônico realizada no sítio competitivo na presença do cofator
NAD+ levou a obtenção de 3 conformações de menor energia que variam entre -31.38 e -
33.05 kJ/mol, mantendo interações com os resíduos catalíticos Ser195, Tyr196, Thr197,
Thr199, Asp210, Thr226 e Arg249 (PAVAO et al., 2002; LADAME et al., 2003; DE
MARCHI et al., 2004), incluindo também, os resíduos Cys166 e His194 que são essenciais
para a atividade catalítica (SOUZA et al., 1998). Além de todas as conformações formarem
pontes de hidrogênio com os resíduos mais próximos.
A interação com resíduo Asp210 foi observada em todas as conformações obtidas do
ligante ácido fosfônico. Esta é uma importante interação, pois qualquer molécula (ligante) que
interaja especificamente com este resíduo impede a formação de uma ponte salina formada
entre os resíduos Asp210 e a Arg249, ou seja, pode ser um potente inibidor específico
(SOUZA et al., 1998), ver Figura 6. Uma vez que a ponte salina formada entre estes dois
resíduos chamou a atenção para um mecanismo de propagação possivelmente importante para
regular a ligação do substrato ao sítio ativo e a liberação do produto após o término da reação
(SOUZA et al., 1998).

15
Figura 6: Representação gráfica dos resíduos Asp210 e Arg249 que formam a ponte salina. Figura obtida
usando o programa VMD.
No procedimento de docagem realizado no sítio não-competitivo da enzima os
resultados obtidos nos programas AutoDock4.2 (MORRIS et al., 1998; HUEY et al., 2007;
MORRIS et al., 2009) e AutoDock Vina (TROTT; OLSON, 2009) foram semelhantes. Neste
caso, a presença do cofator NAD+ não ocasionou interferências nas conformações do ácido
fosfônico em ambos os programas, pois a caixa que delimitava esse sítio não abrangia a
região do cofator.
Figura 7: Conformações de menores energias docadas na enzima GAPDH no sítio não competitivo, a região
violácea representa a interação do grupo fosfato do ácido fosfônico com os resíduos de Thr199. (A) – Quatro
conformações de menor energia obtidas pelo AutoDock4.2; (B) – Duas conformações de menor energia obtidas
pelo AutoDock Vina. As figuras foram obtidas usando o programa Chimera UCSF.
Como observados na Figura 7 ambos os programas utilizados docaram o ácido
fosfônico em uma região de loop, que compreende os resíduos Thr197 a Thr199. Os valores
de energia encontrados para as conformações geradas pelo AutoDock4.2 variam entre -25.44

16
e -29.37 kJ/mol. Enquanto que os valores de energia encontrados para as conformações
geradas pelo AutoDock Vina variam entre -28.87 e -29.70 kJ/mol.
Nas conformações observadas na Figura 7 o ácido fosfônico interage com os resíduos
Ala198, Thr199, Gln200, Lys201, Asp204, Gly205 e Val206. Ressaltando que em todos os
casos ocorreu interação de hidrogênio entre o ligante e o resíduo Lys201 (Fig. 8).
Figura 8: Representação do sitio de interação não-competitivo, com seus respectivos resíduos. Os resíduos
localizados na parte superior pertencem à cadeia C e os resíduos da parte inferior pertencem à cadeia B. (A) –
Conformação de menor energia para o ácido fosfônico encontrado pelo AutoDock4.2. (B) – Conformação de
menor energia para o ácido fosfônico encontrado pelo AutoDock Vina.
Trabalhos anteriores demonstram que grupos PO4 e SO4 em estruturas cristalográficas
de GAPDH de Trypanosoma brucei, Leishmania mexicana (YUN et al.,2000) e Trypanosoma
cruzi (GHOSH et al., 2004) posicionam-se em regiões próximas aos resíduos Thr197 e
Thr199. Neste estudo o grupo H2PO3 do ácido fosfônico foi posicionado, também em uma
região próxima ao resíduo Thr199 (a uma distância < 4.0 Å), como pode ser visto nas Figuras
6A e 6B.
Os resultados dos softwares utilizados foram semelhantes. Neste sentido foram
selecionados para uma melhor análise os resultados obtidos pelo software AutoDock Vina
(TROTT; OLSON, 2009). Segundo Carlsson e colaboradores (CARLSSON; BOUKHARTA;
AQVIST, 2008), funções de pontuação empíricas estimam a afinidade de ligação a partir de
uma única estrutura do complexo proteína-ligante pode ser usado para filtrar candidatos a
fármacos de um grande banco de dados de compostos. Considerando que as funções de
pontuação empíricas utilizam altas velocidades ao serem executadas, elas tratam as

17
contribuições de energia livre de ligação, como por exemplo, entropia e solvatação, de uma
forma muito aproximada, o que dificulta a obtenção de conformações precisas dos ligantes
(CARLSSON; BOUKHARTA; AQVIST, 2008), tal como ocorre com os softwares utilizados
neste estudo (TROTT; OLSON, 2009), que determinam o sítio de ligação de uma proteína,
mas pode não determinar tão precisamente a melhor conformação do ligante neste sítio.
Para tal, o inibidor HOP, da própria cristalografia foi docado novamente na proteína
no mesmo sítio com seus respectivos resíduos, conservando suas coordenadas iniciais, o
resultado é mostrado na Figura 8.
Figura 8: Inibidor HOP complexado a proteína. O inibidor com as coordenadas oriundas da cristalografia está
com seus átomos de carbono em verde, enquanto que o inibidor com as coordenadas da docagem é mostrado
com os átomos de carbono em azul. (A) primeira conformação obtida com o AutoDock Vina, menor energia. (B)
oitava conformação obtida com o AutoDock Vina.
Como pode ser observado na Figura 8, o software conseguir determinar o sítio de
ligação do inibidor HOP, porém a determinação da conformação mais próxima aquela
encontrada na cristalografia não foi determinada com a mesma precisão. De acordo com o
programa a conformação de menor energia é a primeira com valor de energia igual à -25.94
kJ/mol (Fig. 8A), porém suas coordenadas não condizem com as coordenadas da
cristalografia, já a oitava conformação com valor de energia igual à -25.10 kJ/mol (Fig. 8B), é
a conformação cujas coordenadas são mais próximas da cristalografia. Neste sentido é
importante de seja realizado um estudo QM/MM para determinar com maior precisão a
conformação mais favorável energeticamente.
Análise das energias por QM/MM para o inibidor ácido fosfônico
Foi realizado um estudo QM/MM para os dois possíveis sítios ligação (competitivo e
não competitivo). Primeiramente, o inibidor em todas as suas conformações foi tratado com o
18
método semiempírico AM1 (DEWAR et al., 1993), a cadeia peptídica e o cofator NAD foram
tratados com o campo de força OPLS-AA (JORGENSEN et al., 1983), utilizando a livraria
DYNAMO (FIELD, 1999; FIELD et al., 2000). As correções dos valores de energia
encontrados pelo método semiempírico AM1 foram realizadas pelo método DFT (KOHN,
1995) utilizando o programa Gaussian03 (FRISCH et al., 2003).
Sítio competitivo
Os valores de energia encontrados para o sítio competitivo são resumidos na Tabela 1,
mostrada abaixo.
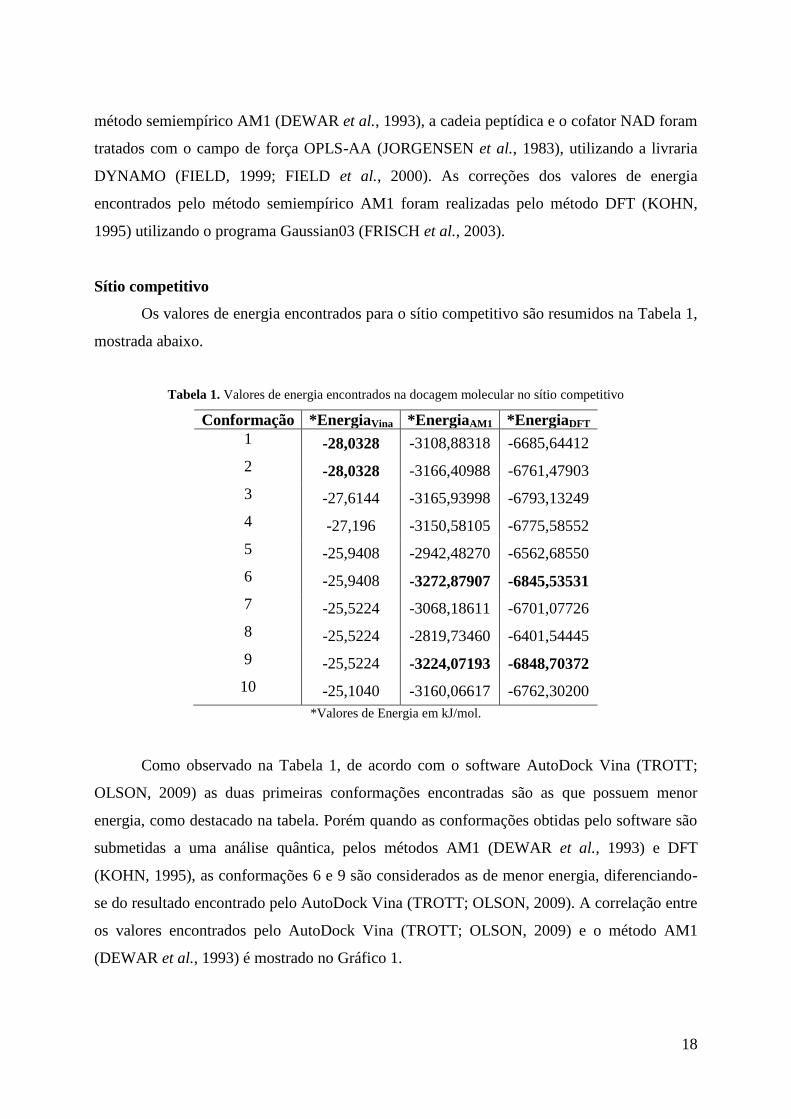
Tabela 1. Valores de energia encontrados na docagem molecular no sítio competitivo
Conformação *EnergiaVina *EnergiaAM1 *EnergiaDFT
1 -28,0328 -3108,88318 -6685,64412
2 -28,0328 -3166,40988 -6761,47903
3 -27,6144 -3165,93998 -6793,13249
4 -27,196 -3150,58105 -6775,58552
5 -25,9408 -2942,48270 -6562,68550
6 -25,9408 -3272,87907 -6845,53531
7 -25,5224 -3068,18611 -6701,07726
8 -25,5224 -2819,73460 -6401,54445
9 -25,5224 -3224,07193 -6848,70372
10 -25,1040 -3160,06617 -6762,30200
*Valores de Energia em kJ/mol.
Como observado na Tabela 1, de acordo com o software AutoDock Vina (TROTT;
OLSON, 2009) as duas primeiras conformações encontradas são as que possuem menor
energia, como destacado na tabela. Porém quando as conformações obtidas pelo software são
submetidas a uma análise quântica, pelos métodos AM1 (DEWAR et al., 1993) e DFT
(KOHN, 1995), as conformações 6 e 9 são considerados as de menor energia, diferenciando-
se do resultado encontrado pelo AutoDock Vina (TROTT; OLSON, 2009). A correlação entre
os valores encontrados pelo AutoDock Vina (TROTT; OLSON, 2009) e o método AM1
(DEWAR et al., 1993) é mostrado no Gráfico 1.
19
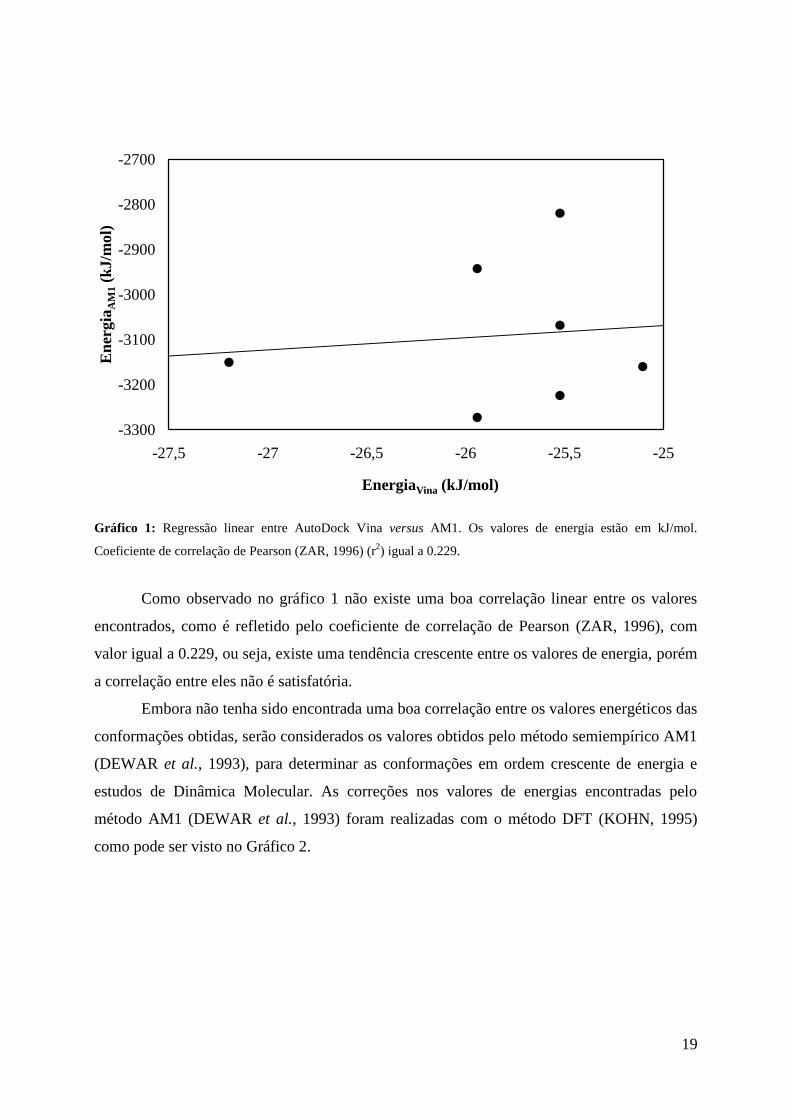
Gráfico 1: Regressão linear entre AutoDock Vina versus AM1. Os valores de energia estão em kJ/mol.
Coeficiente de correlação de Pearson (ZAR, 1996) (r2) igual a 0.229.
Como observado no gráfico 1 não existe uma boa correlação linear entre os valores
encontrados, como é refletido pelo coeficiente de correlação de Pearson (ZAR, 1996), com
valor igual a 0.229, ou seja, existe uma tendência crescente entre os valores de energia, porém
a correlação entre eles não é satisfatória.
Embora não tenha sido encontrada uma boa correlação entre os valores energéticos das
conformações obtidas, serão considerados os valores obtidos pelo método semiempírico AM1
(DEWAR et al., 1993), para determinar as conformações em ordem crescente de energia e
estudos de Dinâmica Molecular. As correções nos valores de energias encontradas pelo
método AM1 (DEWAR et al., 1993) foram realizadas com o método DFT (KOHN, 1995)
como pode ser visto no Gráfico 2.
-3300
-3200
-3100
-3000
-2900
-2800
-2700
-27,5 -27 -26,5 -26 -25,5 -25
En
ergia
AM
1(k
J/m
ol)
EnergiaVina (kJ/mol)
20
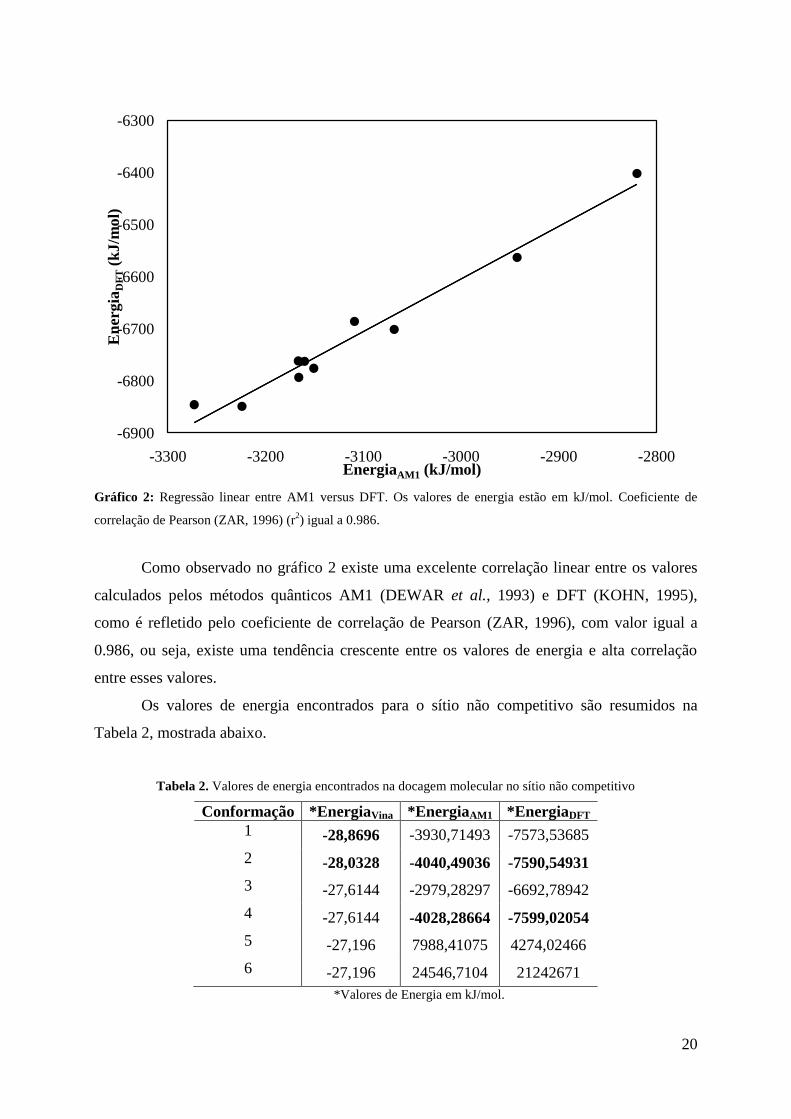
Gráfico 2: Regressão linear entre AM1 versus DFT. Os valores de energia estão em kJ/mol. Coeficiente de
correlação de Pearson (ZAR, 1996) (r2) igual a 0.986.
Como observado no gráfico 2 existe uma excelente correlação linear entre os valores
calculados pelos métodos quânticos AM1 (DEWAR et al., 1993) e DFT (KOHN, 1995),
como é refletido pelo coeficiente de correlação de Pearson (ZAR, 1996), com valor igual a
0.986, ou seja, existe uma tendência crescente entre os valores de energia e alta correlação
entre esses valores.
Os valores de energia encontrados para o sítio não competitivo são resumidos na
Tabela 2, mostrada abaixo.
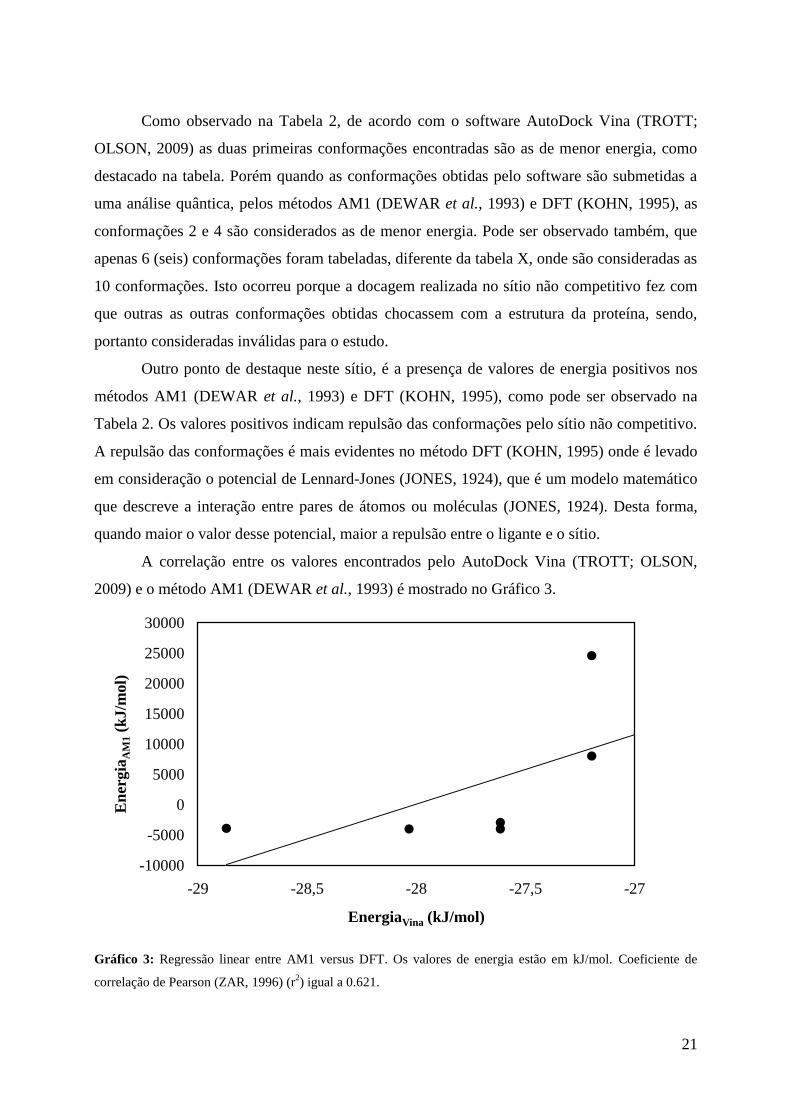
Tabela 2. Valores de energia encontrados na docagem molecular no sítio não competitivo
Conformação *EnergiaVina *EnergiaAM1 *EnergiaDFT
1 -28,8696 -3930,71493 -7573,53685
2 -28,0328 -4040,49036 -7590,54931
3 -27,6144 -2979,28297 -6692,78942
4 -27,6144 -4028,28664 -7599,02054
5 -27,196 7988,41075 4274,02466
6 -27,196 24546,7104 21242671
*Valores de Energia em kJ/mol.
-6900
-6800
-6700
-6600
-6500
-6400
-6300
-3300 -3200 -3100 -3000 -2900 -2800
En
ergia
DF
T(k
J/m
ol)
EnergiaAM1 (kJ/mol)
21
Como observado na Tabela 2, de acordo com o software AutoDock Vina (TROTT;
OLSON, 2009) as duas primeiras conformações encontradas são as de menor energia, como
destacado na tabela. Porém quando as conformações obtidas pelo software são submetidas a
uma análise quântica, pelos métodos AM1 (DEWAR et al., 1993) e DFT (KOHN, 1995), as
conformações 2 e 4 são considerados as de menor energia. Pode ser observado também, que
apenas 6 (seis) conformações foram tabeladas, diferente da tabela X, onde são consideradas as
10 conformações. Isto ocorreu porque a docagem realizada no sítio não competitivo fez com
que outras as outras conformações obtidas chocassem com a estrutura da proteína, sendo,
portanto consideradas inválidas para o estudo.
Outro ponto de destaque neste sítio, é a presença de valores de energia positivos nos
métodos AM1 (DEWAR et al., 1993) e DFT (KOHN, 1995), como pode ser observado na
Tabela 2. Os valores positivos indicam repulsão das conformações pelo sítio não competitivo.
A repulsão das conformações é mais evidentes no método DFT (KOHN, 1995) onde é levado
em consideração o potencial de Lennard-Jones (JONES, 1924), que é um modelo matemático
que descreve a interação entre pares de átomos ou moléculas (JONES, 1924). Desta forma,
quando maior o valor desse potencial, maior a repulsão entre o ligante e o sítio.
A correlação entre os valores encontrados pelo AutoDock Vina (TROTT; OLSON,
2009) e o método AM1 (DEWAR et al., 1993) é mostrado no Gráfico 3.
Gráfico 3: Regressão linear entre AM1 versus DFT. Os valores de energia estão em kJ/mol. Coeficiente de
correlação de Pearson (ZAR, 1996) (r2) igual a 0.621.
-10000
-5000
0
5000
10000
15000
20000
25000
30000
-29 -28,5 -28 -27,5 -27
En
ergia
AM
1(k
J/m
ol)
EnergiaVina (kJ/mol)
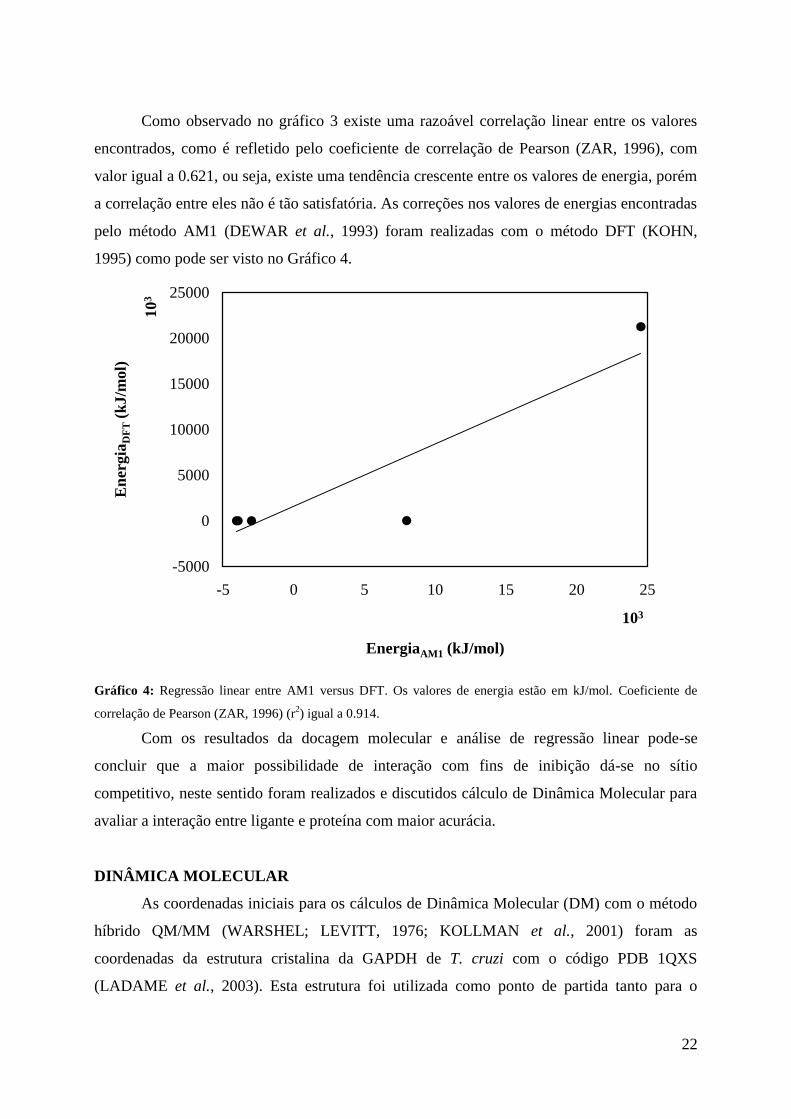
22
Como observado no gráfico 3 existe uma razoável correlação linear entre os valores
encontrados, como é refletido pelo coeficiente de correlação de Pearson (ZAR, 1996), com
valor igual a 0.621, ou seja, existe uma tendência crescente entre os valores de energia, porém
a correlação entre eles não é tão satisfatória. As correções nos valores de energias encontradas
pelo método AM1 (DEWAR et al., 1993) foram realizadas com o método DFT (KOHN,
1995) como pode ser visto no Gráfico 4.
Gráfico 4: Regressão linear entre AM1 versus DFT. Os valores de energia estão em kJ/mol. Coeficiente de
correlação de Pearson (ZAR, 1996) (r2) igual a 0.914.
Com os resultados da docagem molecular e análise de regressão linear pode-se
concluir que a maior possibilidade de interação com fins de inibição dá-se no sítio
competitivo, neste sentido foram realizados e discutidos cálculo de Dinâmica Molecular para
avaliar a interação entre ligante e proteína com maior acurácia.
DINÂMICA MOLECULAR
As coordenadas iniciais para os cálculos de Dinâmica Molecular (DM) com o método
híbrido QM/MM (WARSHEL; LEVITT, 1976; KOLLMAN et al., 2001) foram as
coordenadas da estrutura cristalina da GAPDH de T. cruzi com o código PDB 1QXS
(LADAME et al., 2003). Esta estrutura foi utilizada como ponto de partida tanto para o
-5000
0
5000
10000
15000
20000
25000
-5 0 5 10 15 20 25
En
ergia
DF
T(k
J/m
ol)
10
3
EnergiaAM1 (kJ/mol)
103

23
inibidor HOP, complexado a estrutura, e seus derivados, como para o estudo de interação do
ácido fosfônico no sítio competitivo.
Como os valores de pKa podem ser modificados por efeitos locais do entorno protéico
(ANTOSIEWICZ; McCAMMON; GILSON, 1994), uma atribuição rigorosa dos estados de
protonação de todos os resíduos a um pH 7 foi realizada recalculando os valores padrões de
pKa titulável dos aminoácidos, utilizando o programa propKa (LI; ROBERTSON; JENSEN,
2005). Estes resultados mostraram que os resíduos Asp254 e Asp259 estão protonados. O
resíduo Asp254 forma interage através de ponte de hidrogênio com o resíduo Asp334.
Enquanto que o resíduo Asp259 interage com o grupo N-terminal da Lys328 e o grupo
hidroxila da Thr261.
Análises para o inibidor HOP e seus derivados
O inibidor ácido 3-hidróxi-2-oxo-4-fosfonóxi-butilfosfônico (HOP) oriunda da própria
estrutura cristalográfica, bem como seus derivados (Tabela 3) foram utilizados para a
simulação de DM.
Tabela 3: inibidor HOP com seus derivados e respectivos valores de IC50 e Kd
IC50(µM)* Kd(µM)*
HOP
2000 600
CP5
800 160
CP6
2000 570
CP7
900 300
CP8
700 120
*Valores experimentais obtidos por Ladame e colaboradores (LADAME et al., 2003).
24
Como pode ser observado na Tabela 3, existe uma relação entre IC50 e Kd (Constante
de dissociação), quanto menor o IC50 menor a Kd, ou seja, os inibidores que possuem menor
valor de IC50 são os melhores, pois tendem a não se dissociar dentro da enzima, como é
determinado pela sua respectiva constante de dissociação.
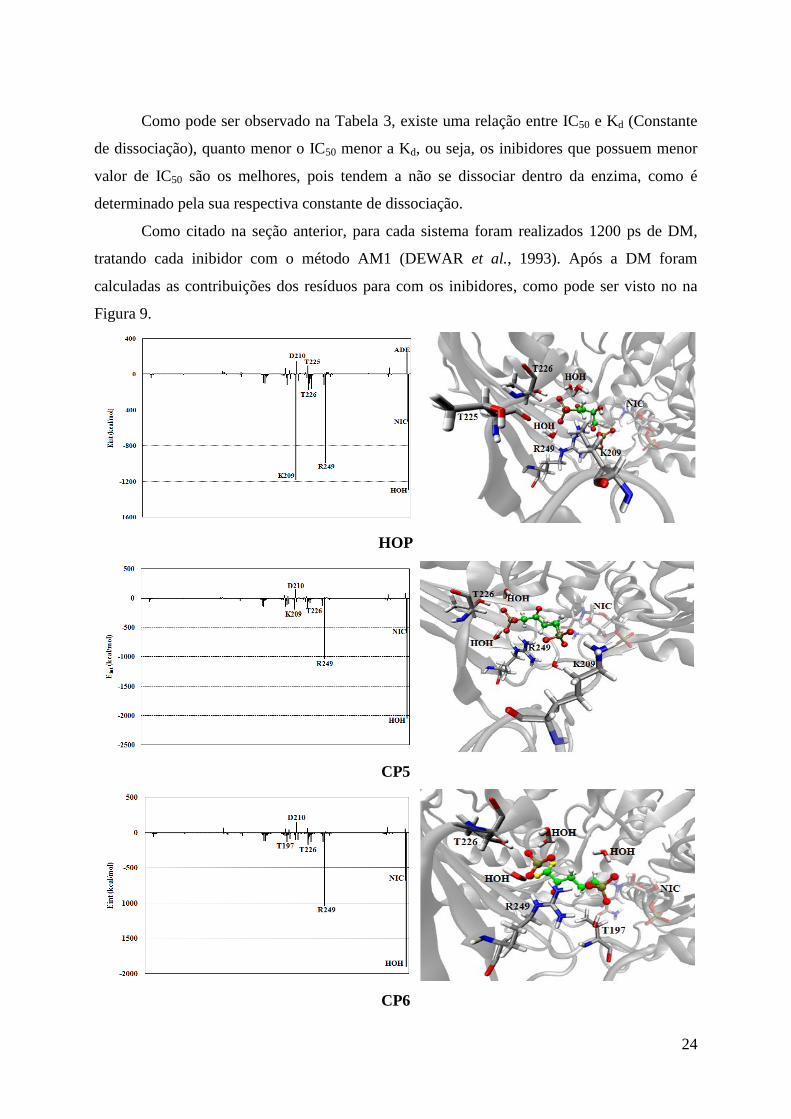
Como citado na seção anterior, para cada sistema foram realizados 1200 ps de DM,
tratando cada inibidor com o método AM1 (DEWAR et al., 1993). Após a DM foram
calculadas as contribuições dos resíduos para com os inibidores, como pode ser visto no na
Figura 9.
HOP
CP5
CP6
25
CP7
CP8
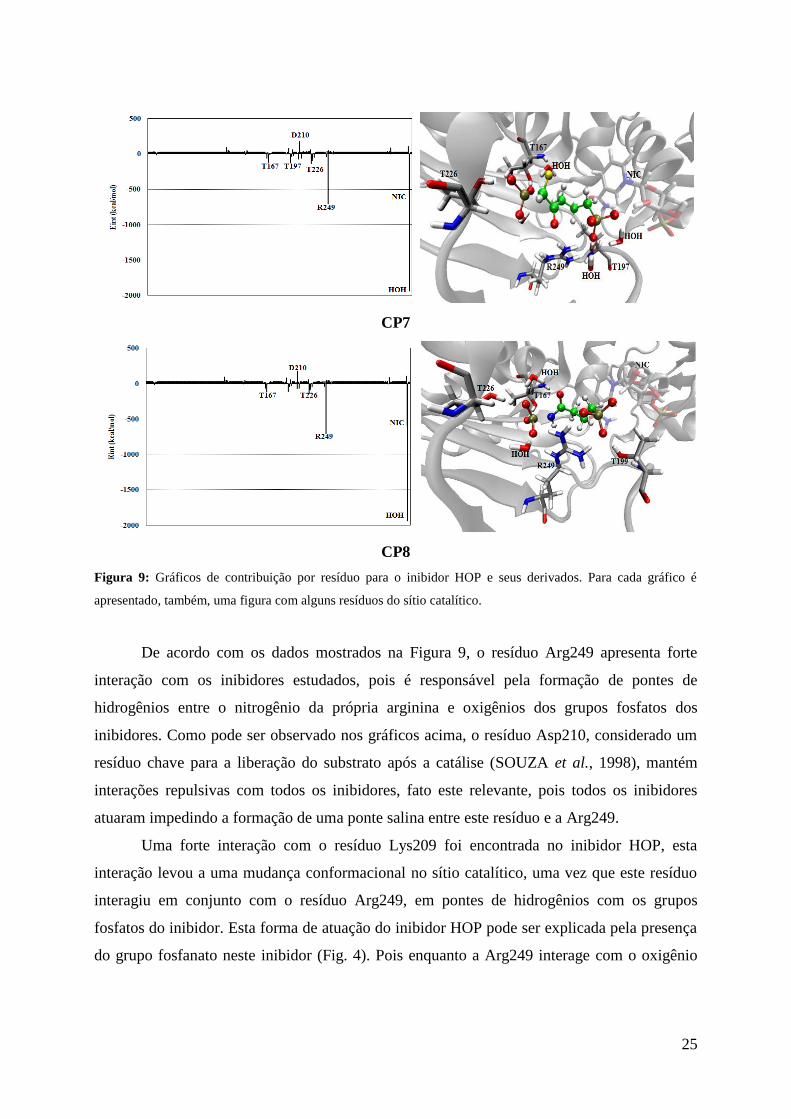
Figura 9: Gráficos de contribuição por resíduo para o inibidor HOP e seus derivados. Para cada gráfico é
apresentado, também, uma figura com alguns resíduos do sítio catalítico.
De acordo com os dados mostrados na Figura 9, o resíduo Arg249 apresenta forte
interação com os inibidores estudados, pois é responsável pela formação de pontes de
hidrogênios entre o nitrogênio da própria arginina e oxigênios dos grupos fosfatos dos
inibidores. Como pode ser observado nos gráficos acima, o resíduo Asp210, considerado um
resíduo chave para a liberação do substrato após a catálise (SOUZA et al., 1998), mantém
interações repulsivas com todos os inibidores, fato este relevante, pois todos os inibidores
atuaram impedindo a formação de uma ponte salina entre este resíduo e a Arg249.
Uma forte interação com o resíduo Lys209 foi encontrada no inibidor HOP, esta
interação levou a uma mudança conformacional no sítio catalítico, uma vez que este resíduo
interagiu em conjunto com o resíduo Arg249, em pontes de hidrogênios com os grupos
fosfatos do inibidor. Esta forma de atuação do inibidor HOP pode ser explicada pela presença
do grupo fosfanato neste inibidor (Fig. 4). Pois enquanto a Arg249 interage com o oxigênio

26
do fosfanato que, está ligado ao átomo de carbono, o resíduo Lys209 interage com os átomos
de oxigênios do mesmo grupo, que estão apenas ligados aos átomos de fósforo.
Para o anel nicotinamida (NIC) do cofator NAD, são apresentadas interações de
hidrogênio com um grupo fosfato dos inibidores. Além disso, águas de cristalização também
apresentaram fortes interações, principalmente, pontes de hidrogênios com os grupos fosfatos,
para a estabilização dos inibidores no sítio catalítico.
Análises para o inibidor ácido fosfônico
Figura 10: Estrutura planar do ácido fosfônico.
Como pode ser visto na Figura 10, o inibidor ácido fosfônico possui dois grupos
fosfatos ligado ao carbono adjacente a um átomo de nitrogênio, sendo caracterizado como um
composto bifosfanato (Fig. 11A), que são análogos de compostos pirofosfanatos (Fig. 11B)
(MARTIN; GRILL, 2000), onde sua atividade biológica de pode ser modificada alterando a
estrutura das duas cadeias laterais no átomo de carbono (MARTIN; GRILL, 2000). Desta
forma, podem-se determinar possíveis estados de protonação para esses grupos. Por isso
determinou-se três possíveis estados de protonação, onde a carga do ácido é alterada de
acordo com a presença átomos de hidrogênios nos grupos fosfatos.
Figura 11: Estruturas químicas de bifosfanatos e pirofosfonatos. (A) Ácido bifosfônico e (B) Ácido
pirofosfórico.
Primeiramente, foi atribuída carga -4 para o inibidor não possuindo hidrogênios nos
grupos fosfatos (Fig. 12A). Depois foi adicionado um hidrogênio a um destes grupos,
atribuindo-se a este carga -3 (Fig. 12B). E por último, foi adicionado um átomo de hidrogênio

27
ao mesmo grupo fosfato, a este se atribuiu carga -2 (Fig. 12C). A intenção da adição de
hidrogênios foi analisar a influência da carga para o sistema e de que forma os resíduos do
sítio catalítico iriam contribuir para as interações com o inibidor.
Figura 12: Estruturas planares do ácido fosfônico em seus respectivos estados de protonação: (A) carga -4; (B)
carga -3; (C) carga -2.
Para cada estado de protonação do ácido fosfônico foram realizados 1200 ps de DM,
tratando o inibidor com o método AM1(DEWAR et al., 1993). Após a DM foram calculadas
as contribuições dos resíduos para com os inibidores, como pode ser visto no na Figura 13.
(A)
(B)

28
(C)
Figura 13: Gráficos de contribuição por resíduo para o inibidor ácido fosfônico em seus estados de protonação.
(A) Carga -4; (B) Carga -3; (C) Carga -2.
Conforme a Figura 13, os estados de protonação estudados do inibidor ácido fosfônico
mantiveram interações com os resíduos catalíticos Thr226, Arg249 e Asp210 (PAVAO et al.,
2002; LADAME et al., 2003; DE MARCHI et al., 2004). Assim como o inibidor HOP e seus
derivados, o ácido fosfônico desfez a ponte salina entre os resíduos Arg249 e Asp210
(SOUZA et al., 1998), fazendo com que o Asp210 fosse afastado do sítio catalítico, enquanto
que a Arg249 manteve interações com os grupos fosfatos do ácido fosfônico. Outra
característica dos estados de protonação estudados foi a interação dos grupos fosfatos com
grupos hidroxilas de resíduos próximos a eles, como: Ser224, Ser247, Thr226 no estado de
protonação com carga -4; Ser247 e Thr226 para os estados de protonação com cargas -3 e -2.
Além de interações de hidrogênios com moléculas de água ao redor dos grupos fosfatos.
Outra interação que foi mantida nos três estados de protonação foi com o resíduo Arg212,
embora este resíduo não faça interações diretas com o inibidor ele contribui para o
deslocamento do Asp210, pois como a ponte salina entre este e a Arg249 é desfeita, a Arg212
interage fortemente com o Asp210 por pontes de hidrogênios.
Embora as interações com os resíduos tenham sido semelhantes nos diferentes estados
de protonação, o estudo por QM/MM foi capaz de mostrar um mecanismo intramolecular para
o inibidor nos estados de protonação com cargas -3 e -2, como pode ser observado nas Figuras
14 e 16.
29
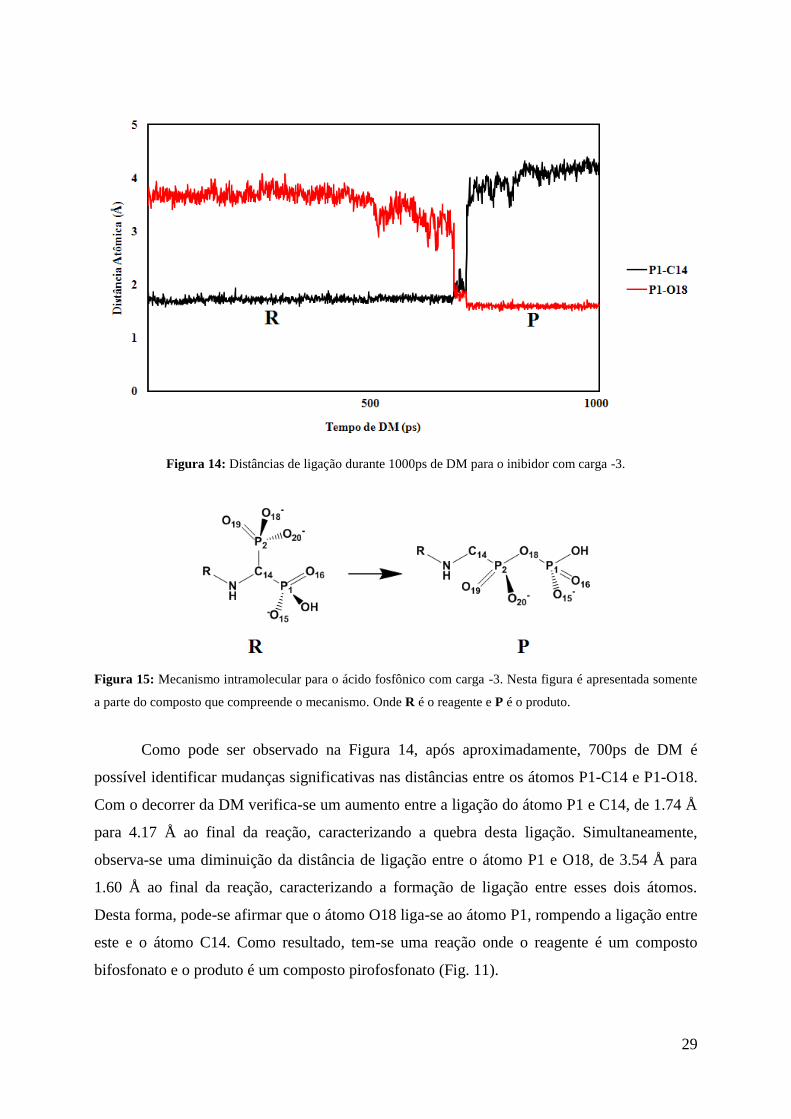
Figura 14: Distâncias de ligação durante 1000ps de DM para o inibidor com carga -3.
Figura 15: Mecanismo intramolecular para o ácido fosfônico com carga -3. Nesta figura é apresentada somente
a parte do composto que compreende o mecanismo. Onde R é o reagente e P é o produto.
Como pode ser observado na Figura 14, após aproximadamente, 700ps de DM é
possível identificar mudanças significativas nas distâncias entre os átomos P1-C14 e P1-O18.
Com o decorrer da DM verifica-se um aumento entre a ligação do átomo P1 e C14, de 1.74 Å
para 4.17 Å ao final da reação, caracterizando a quebra desta ligação. Simultaneamente,
observa-se uma diminuição da distância de ligação entre o átomo P1 e O18, de 3.54 Å para
1.60 Å ao final da reação, caracterizando a formação de ligação entre esses dois átomos.
Desta forma, pode-se afirmar que o átomo O18 liga-se ao átomo P1, rompendo a ligação entre
este e o átomo C14. Como resultado, tem-se uma reação onde o reagente é um composto
bifosfonato e o produto é um composto pirofosfonato (Fig. 11).
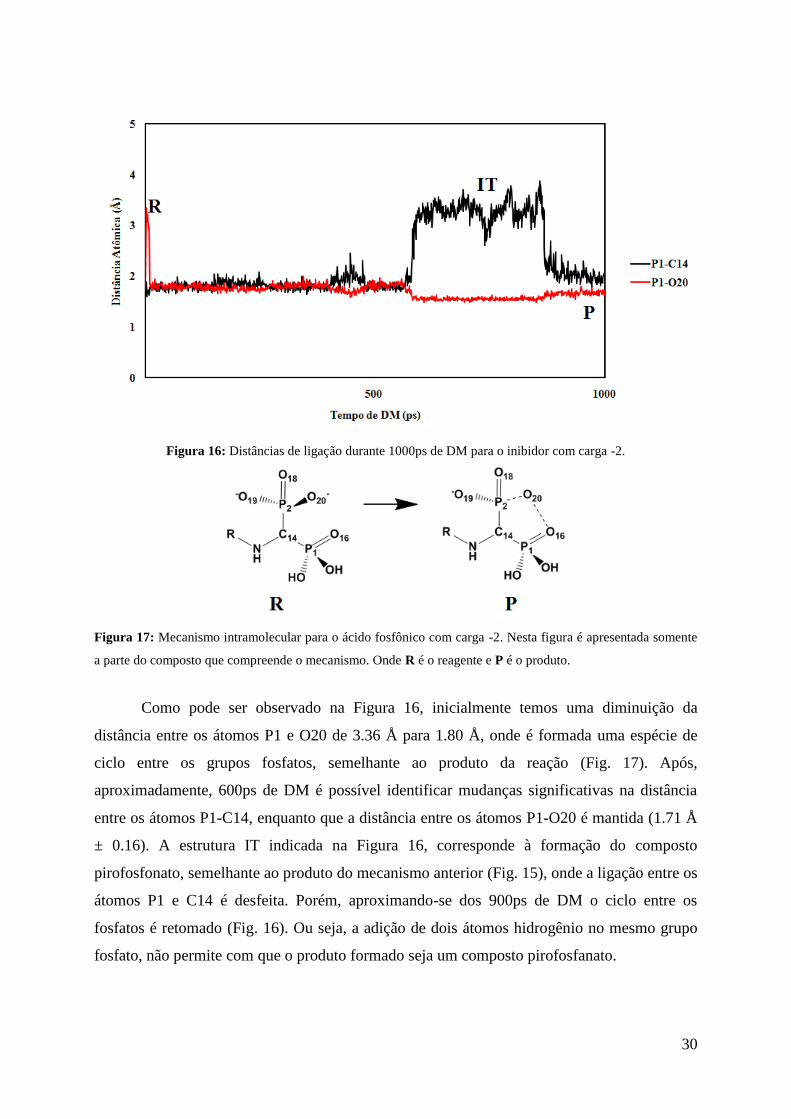
30
Figura 16: Distâncias de ligação durante 1000ps de DM para o inibidor com carga -2.
Figura 17: Mecanismo intramolecular para o ácido fosfônico com carga -2. Nesta figura é apresentada somente
a parte do composto que compreende o mecanismo. Onde R é o reagente e P é o produto.
Como pode ser observado na Figura 16, inicialmente temos uma diminuição da
distância entre os átomos P1 e O20 de 3.36 Å para 1.80 Å, onde é formada uma espécie de
ciclo entre os grupos fosfatos, semelhante ao produto da reação (Fig. 17). Após,
aproximadamente, 600ps de DM é possível identificar mudanças significativas na distância
entre os átomos P1-C14, enquanto que a distância entre os átomos P1-O20 é mantida (1.71 Å
± 0.16). A estrutura IT indicada na Figura 16, corresponde à formação do composto
pirofosfonato, semelhante ao produto do mecanismo anterior (Fig. 15), onde a ligação entre os
átomos P1 e C14 é desfeita. Porém, aproximando-se dos 900ps de DM o ciclo entre os
fosfatos é retomado (Fig. 16). Ou seja, a adição de dois átomos hidrogênio no mesmo grupo
fosfato, não permite com que o produto formado seja um composto pirofosfanato.
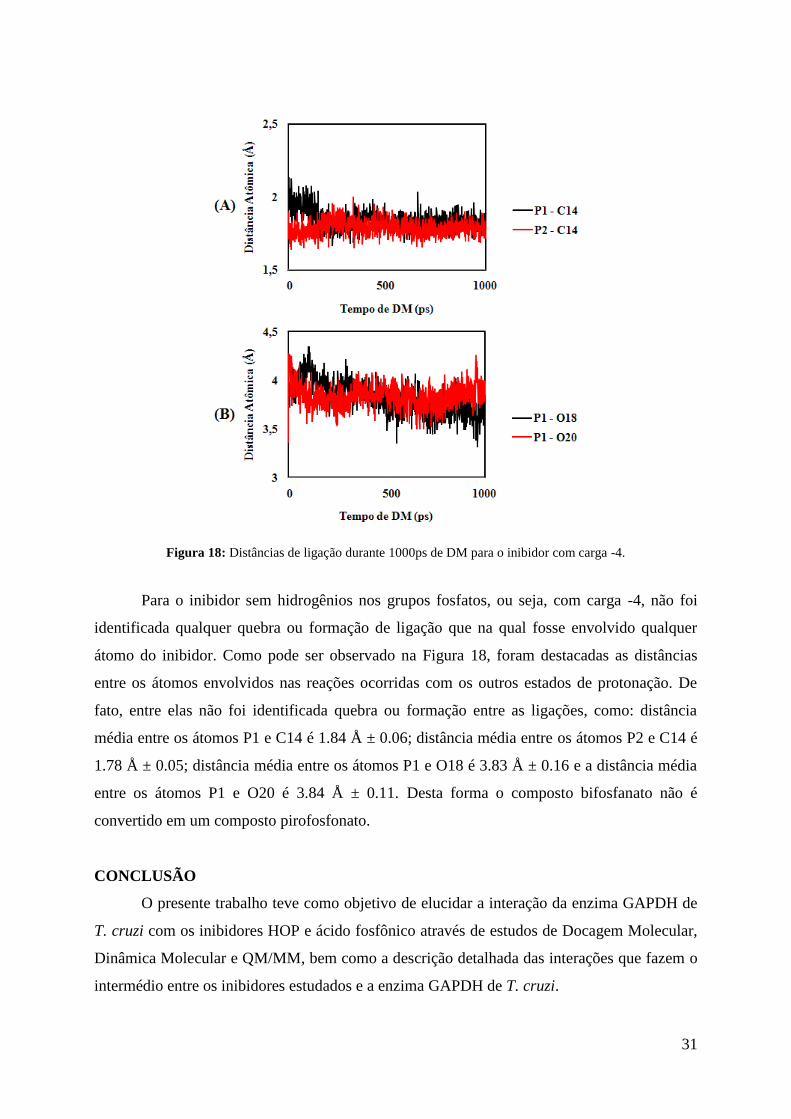
31
Figura 18: Distâncias de ligação durante 1000ps de DM para o inibidor com carga -4.
Para o inibidor sem hidrogênios nos grupos fosfatos, ou seja, com carga -4, não foi
identificada qualquer quebra ou formação de ligação que na qual fosse envolvido qualquer
átomo do inibidor. Como pode ser observado na Figura 18, foram destacadas as distâncias
entre os átomos envolvidos nas reações ocorridas com os outros estados de protonação. De
fato, entre elas não foi identificada quebra ou formação entre as ligações, como: distância
média entre os átomos P1 e C14 é 1.84 Å ± 0.06; distância média entre os átomos P2 e C14 é
1.78 Å ± 0.05; distância média entre os átomos P1 e O18 é 3.83 Å ± 0.16 e a distância média
entre os átomos P1 e O20 é 3.84 Å ± 0.11. Desta forma o composto bifosfanato não é
convertido em um composto pirofosfonato.
CONCLUSÃO
O presente trabalho teve como objetivo de elucidar a interação da enzima GAPDH de
T. cruzi com os inibidores HOP e ácido fosfônico através de estudos de Docagem Molecular,
Dinâmica Molecular e QM/MM, bem como a descrição detalhada das interações que fazem o
intermédio entre os inibidores estudados e a enzima GAPDH de T. cruzi.

32
De acordo com os resultados obtidos por docagem molecular foram identificadas duas
regiões distintas para o mecanismo de inibição do ácido fosfônico complexado a GAPDH.
Porém, auxiliados por análises estatísticas de regressão linear, correlacionando os valores
energéticos fornecidos pelos softwares utilizados (neste caso, AutoDock Vina) e valores
encontrados por métodos quânticos (AM1 e DFT), conclui-se que o sítio de interação do
inibidor ácido fosfônico se dava em apenas um sítio, neste caso o sítio competitivo.
Os estudos de Dinâmica Molecular e QM/MM auxiliaram, de forma satisfatória, na
elucidação das interações exercidas pelos inibidores. Além disso, os estudos de DM foram
fundamentais para a determinação da flexibilidade dos resíduos de aminoácidos responsáveis
pelo reconhecimento molecular dos inibidores sobre investigação, bem como, o mecanismo
existente para o inibidor ácido fosfônico em 2 (dois) dos estados de protonação estudados.
Adicionalmente, estas informações, amparadas pelos ensaios bioquímicos, em parceria com o
grupo NEQUIMED, podem melhor elucidar o mecanismo de inibição imposto pelo ácido
fosfônico e HOP.
Desta forma, a integração de diferentes abordagens tais como Docagem Molecular,
Dinâmica Molecular e QM/MM, forneceram informações muito relevantes que contribuíram
para o entendimento teórico do mecanismo de ação dos diferentes inibidores apresentados
neste trabalho.
REFERÊNCIAS BIBLIOGRÁFICAS
Antosiewicz, J.; McCammon, J. A.; Gilson, M. K. Journal of Molecular Biology 1994, 238,
415-436.
Bakker, B. M.; Michels, P. A. M.; Opperdoes, F. R.; Westerhoff, H. V. Journal of Biological
Chemistry 1999, 274, 14551-14559.
Bakker, B. M.; Westerhoff, H. V.; Opperdoes, F. R.; Michels, P. A. M. Molecular and
Biochemical Parasitology 2000, 106, 1-10.
Barrett, M. P.; Mottram, J. C.; Coombs, G. H. Trends in Microbiology 1999, 7, 82-88.
Born, M.; Oppenheimer, R. Annual Physics (Leipzig) 1927, 84, 457 - 484.
Burke, J. R.; Enghild, J. J.; Martin, M. E.; Jou, Y. S.; Myers, R. M.; Roses, A. D.; Vance, J.
M.; Strittmatter, W. J. Nature Medicine 1996, 2, 347-350.
Carlile, G. W.; Chalmers-Redman, R. M. E.; Tatton, N. A.; Pong, A.; Borden, K. E.; Tatton,
W. G. Molecular Pharmacology 2000, 57, 2-12.

33
Carlsson, J.; Boukharta, L.; Aqvist, J. Journal of Medicinal Chemistry 2008, 51, 2648-2656.
Castilho, M. S.; Pavao, F.; Oliva, G.; Ladame, S.; Willson, M.; Perie, J. Biochemistry 2003,
42, 7143-7151.
Chagas, C. Memórias Do Instituto Oswaldo Cruz 1909, 1, 159 - 218.
Coura, J. R. Ciência e Cultura 2003, 55, 30-33.
Coura, J. R.; de Castro, S. L. Memórias Do Instituto Oswaldo Cruz 2002, 97, 3-24.
Dardonville, C. Expert Opinion on Therapeutic Patents 2005, 15, 1241-1257.
De Marchi, A. A.; Castilho, M. S.; Nascimento, P. G. B.; Archanjo, F. C.; Del Ponte, G.;
Oliva, G.; Pupo, M. T. Bioorganic & Medicinal Chemistry 2004, 12, 4823-4833.
Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P. Journal of the American
Chemical Society 1993, 115, 5348-5348.
Dias, J. C. P. Um Manual Prático para o Clínico Geral Rio de Janeiro, 1989.
Dias, J. C. P. In BRENEZER, Z.; ANDRADE, A. Z. (EDs). Trypanosoma cruzi e Doença de
Chagas; Guanabara, Ed. Rio de Janeiro, 2000; Vol. 1, p 48-74.
Eberhart, R.; Kennedy, J. In Procceedings of the Sixth International Symposium on Micro
Machine and Human Science 1995, p 39.
Erickson, J. A.; Jalaie, M.; Robertson, D. H.; Lewis, R. A.; Vieth, M. Journal of Medicinal
Chemistry 2004, 47, 45-55.
Field, M. J. A Practical Introduction to the Simulation of Molecular Systems Cambridge,
1999.
Field, M. J.; Albe, M.; Bret, C.; Proust-De Martin, F.; Thomas, A. Journal of Computational
Chemistry 2000, 21, 1088-1100.
Franco-Paredes, C.; Bottazzi, M. E.; Hotez, P. J. Plos Neglected Tropical Diseases 2009, 3.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Montgomery, J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.;
Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;
Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.;
Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H.
P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.;
Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma,
K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A.
D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J.
B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.;
Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Laham, A.;

34
Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.;
Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision C.02, 2003.
Gasteiger, J.; Marsili, M. Tetrahedron Letters 1978, 3181-3184.
Gasteiger, J.; Marsili, M. Tetrahedron 1980, 36, 3219-3228.
Ghosh, S.; Chan, J. M. W.; Lea, C. R.; Meints, G. A.; Lewis, J. C.; Tovian, Z. S.; Flessner, R.
M.; Loftus, T. C.; Bruchhaus, I.; Kendrick, H.; Croft, S. L.; Kemp, R. G.; Kobayashi, S.;
Nozaki, T.; Oldfield, E. Journal of Medicinal Chemistry 2004, 47, 175-187.
Goddard, T. D.; Huang, C. C.; Ferrin, T. E. Journal of Structural Biology 2007, 157, 281-287.
Guido, R. V. C.; Oliva, G.; Andricopulo, A. D. Current Medicinal Chemistry 2008, 15, 37-46.
Holland, R. S. Adaptation in Natural and Artificial Systems Ann Arbor, 1975.
Huey, R.; Morris, G. M.; Olson, A. J.; Goodsell, D. S. Journal of Computational Chemistry
2007, 28, 1145-1152.
Jarmula, A.; Cieplak, P.; Les, A.; Rode, W. Journal of Computer-Aided Molecular Design
2003, 17, 699-710.
Jones, J. E. Proceedings of the Royal Society A 1924, 106, 463 - 477.
Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. Journal of
Chemical Physics 1983, 79, 926-935.
Kirkpatrick, S.; Gelatt, C. D.; Vecchi, M. P. Science 1983, 220, 671-680.
Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J. Nature Reviews Drug Discovery 2004,
3, 935-949.
Kohn, W. International Journal of Quantum Chemistry 1995, 56, 229-232.
Kollman, P. A.; Kuhn, B.; Donini, O.; Perakyla, M.; Stanton, R.; Bakowies, D. Accounts of
Chemical Research 2001, 34, 72-79.
Kuntz, I. D.; Blaney, J. M.; Oatley, S. J.; Langridge, R.; Ferrin, T. E. Journal of Molecular
Biology 1982, 161, 269-288.
Ladame, S.; Castilho, M. S.; Silva, C.; Denier, C.; Hannaert, V.; Perie, J.; Oliva, G.; Willson,
M. European Journal of Biochemistry 2003, 270, 4574-4586.
Li, H.; Robertson, A. D.; Jensen, J. H. Proteins-Structure Function and Bioinformatics 2005,
61, 704-721.
Martin, T. J.; Grill, V. Australian Prescriber 2000, 23, 130-132.
McClain, D. A.; Lubas, W. A.; Cooksey, R. C.; Hazel, M.; Parker, G. J.; Love, D. C.;

35
Hanover, J. A. Proceedings of the National Academy of Sciences of the United States of
America 2002, 99, 10695-10699.
Metropolis, N.; Rosenbluth, A. W.; Rosenbluth, M. N.; Teller, A. H.; Teller, E. Journal of
Chemical Physics 1953, 21, 1087-1092.
Morel, C. M. Memorias Do Instituto Oswaldo Cruz 1999, 94, 3-16.
Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson,
A. J. Journal of Computational Chemistry 1998, 19, 1639-1662.
Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson,
A. J. Journal of Computational Chemistry 2009, 30, 2785-2791.
Muegge, I.; Rarey, M. Reviews in Computational Chemistry 2001, 17, 1 - 60.
Opperdoes, F. R. Annual Review of Microbiology 1987, 41, 127-151.
Opperdoes, F. R.; Michels, P. A. M. International Journal for Parasitology 2001, 31, 482-
490.
Osterberg, F.; Morris, G. M.; Sanner, M. F.; Olson, A. J.; Goodsell, D. S. Proteins-Structure
Function and Bioinformatics 2002, 46, 34-40.
Pavao, F.; Castilho, M. S.; Pupo, M. T.; Dias, R. L. A.; Correa, A. G.; Fernandes, J. B.; da
Silva, M.; Mafezoli, J.; Vieira, P. C.; Oliva, G. Febs Letters 2002, 520, 13-17.
Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.;
Ferrin, T. E. Journal of Computational Chemistry 2004, 25, 1605-1612.
Pink, R.; Hudson, A.; Mouries, M. A.; Bendig, M. Nature Reviews Drug Discovery 2005, 4,
727-740.
Roses, A. D.; Burke, J. R.; Vance, J. M.; Strittmatter, W. J. Nature Medicine 1996, 2, 610-
610.
Schmunis, G. A. In Clínica e Terapêutica da Doença de Chagas. Um Manual Prático para o
Clínico Geral; Fiocruz, Ed. Rio de Janeiro, 1997, p 11-24.
Seydoux, F.; Bernhard, S.; Pfenning.O; Payne, M.; Malhotra, O. P. Biochemistry 1973, 12,
4290-4300.
Shaw, P.; Freeman, J.; Bovey, R.; Iggo, R. Oncogene 1996, 12, 921-930.
Silveira, A. C.; Arias, A. R.; Segura, E.; Guillén, G.; Russomando, G.; Schenone, H.; Dias, J.
C. P.; Padilla, J. V.; Lorca, M.; Salvatella, R. In História de uma Iniciativa Internacional.
1901/2001.; Organização Pan-Americana da Saúde: 2002.
Sirover, M. A. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology

36
1999, 1432, 159-184.
Sousa, S. F.; Fernandes, P. A.; Ramos, M. J. Proteins-Structure Function and Bioinformatics
2006, 65, 15-26.
Souza, D. H. F.; Garratt, R. C.; Araujo, A. P. U.; Guimaraes, B. G.; Jesus, W. D. P.; Michels,
P. A. M.; Hannaert, V.; Oliva, G. Febs Letters 1998, 424, 131-135.
SUCAM; Superintendência de Campanhas de Saúde Pública. Ministério da Saúde: Brasília,
1989, p 52.
Tatton, W. G.; Chalmers-Redman, R.; Brown, D.; Tatton, N. Annals of Neurology 2003, 53,
S61-S70.
Trott, O.; Olson, A. J. Journal of Computational Chemistry 2009, 31, 455-461.
Udier-Blagovic, M.; Tirado-Rives, J.; Jorgensen, W. L. Journal of Medicinal Chemistry 2004,
47, 2389-2392.
Urbina, J. A. Memórias Do Instituto Oswaldo Cruz 1999, 94, 349-355.
Urbina, J. A.; Docampo, R. Trends in Parasitology 2003, 19, 495-501.
VMD; 1.8.7 ed.; VMD User's Guide: Urbana, 2009.
Waldmeier, P. C.; Tatton, W. G. Drug Discovery Today 2004, 9, 210-218.
Weiner, S. J.; Kollman, P. A.; Case, D. A.; Singh, U. C.; Ghio, C.; Alagona, G.; Profeta, S.;
Weiner, P. Journal of the American Chemical Society 1984, 106, 765-784.
Wiggers, H. J., Universidade de São Paulo, 2007.
Wiggers, H. J.; Cheleski, J.; Zottis, A.; Oliva, G.; Andricopulo, A. D.; Montanari, C. A.
Analytical Biochemistry 2007, 370, 107-114.
Wlodawer, A. Annual Review of Medicine 2002, 53, 595-614.
Yun, M.; Park, C. G.; Kim, J. Y.; Park, H. W. Biochemistry 2000, 39, 10702-10710.
Zar, J. H. Biostatistical Analysis; Third Edition ed. New Jersey, 1996.
DIFICULDADES
Parametrização dos ligantes (NAD+, HOP e ácido fosfônico);
Manipulação dos programas de docagem;

37
Constantes quedas de energia no Laboratório.
PARECER DO ORIENTADOR: O aluno desenvolveu suas atividades com
responsabilidade, dentro do período previsto no cronograma. Atuando de forma eficiente no
desenvolvimento das tarefas inerentes ao plano de trabalho. Portanto, atribuo conceito
EXCELENTE, para o mesmo.
Data:05/08/2010
_____________________________________
ASSINATURA DO ORIENTADOR
_____________________________________
ASSINATURA DO ALUNO