universidade de são paulo (vfrod6xshulrugh$julfxowxud³ … · dade federal de são joão del-rei...
TRANSCRIPT

Universidade de São Paulo
Escola Superior de Agricultura “Luiz de Queiroz”
Mapeamento comparativo de QTLs entre sorgo sacarino e cana-de-açúcar
para caracteres bioenergéticos
Guilherme da Silva Pereira
Tese apresentada para obtenção do título de Doutor em
Ciências. Área de concentração: Genética e
Melhoramento de Plantas
Piracicaba
2015

Guilherme da Silva Pereira
Licenciado e Bacharel em Ciências Biológicas
Mapeamento comparativo de QTLs entre sorgo sacarino e cana-de-açúcar para
caracteres bioenergéticos
Orientador:
Prof. Dr. ANTONIO AUGUSTO FRANCO GARCIA
Tese apresentada para obtenção do título de Doutor em
Ciências. Área de concentração: Genética e
Melhoramento de Plantas
Piracicaba
2015

Dados Internacionais de Catalogação na Publicação
DIVISÃO DE BIBLIOTECA - DIBD/ESALQ/USP
Pereira, Guilherme da Silva Mapeamento comparativo de QTLs entre sorgo sacarino e cana-de-açúcar para
caracteres bioenergéticos / Guilherme da Silva Pereira. - - Piracicaba, 2015. 85 p. : il.
Tese (Doutorado) - - Escola Superior de Agricultura “Luiz de Queiroz”, 2015.
1. Sorghum bicolor 2. Saccharum × officinarum 3. Mapeamento de múltiplos QTLs 4. Análise de ligação 5. Sintenia I. Título
CDD 633.174 P436m
“Permitida a cópia total ou parcial deste documento, desde que citada a fonte – O autor”

3
A Deus,
pelos dons que, por certo, são dEle:
ofereço.
Aos meus pais, Geraldo e Maristela,
por todo empenho na educação e amor na criação dos filhos:
dedico.

4

5
AGRADECIMENTOS
Nesses quatro anos, inúmeras instituições e pessoas fizeram-se indispensáveis para o
desenvolvimento deste trabalho em particular e para o meu próprio crescimento pessoal e
profissional. Na tentativa de citar a todos, concedendo-lhes os devidos méritos, talvez eu
me faça enfadonho. No entanto, considerando os agradecimentos não apenas uma obrigação,
mas, verdadeiramente, um modesto reconhecimento por toda contribuição recebida, penso que
é válido fazê-los apropriadamente.
Inicialmente, gostaria de expressar minha gratidão pela oportunidade de cursar o dou-
torado à Escola Superior de Agricultura “Luiz de Queiroz” (ESALQ) e ao Programa de Pós-
Graduação em Genética e Melhoramento de Plantas da ESALQ, assim como pelas bolsas con-
cedidas pela Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), pelo
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) e, finalmente, pela
Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP – processo no 12/25236-4).
A percepção de que este trabalho não é de uma pessoa só, mas, sim, de toda uma equipe,
consolida-se rapidamente no decorrer do texto. Por isso, faltam palavras para agradecer devida-
mente ao Prof. Dr. Antonio Augusto Franco Garcia, que, por ter aceitado me orientar, propiciou
tudo isto. Tendo me confiado este trabalho e me entusiasmado com sua maneira de ver a ciência
e de fazer pesquisa, o Augusto me ensinou mais do que conteúdos de genética e estatística. É
mérito dele, portanto, não só eu ter me capacitado a atuar sobre tópicos diversos da área, como
também ter percebido que com esforço sempre podemos ir além. Sua capacidade de atrair
pessoal competente e altruísta faz do Laboratório o ambiente ideal para nosso desenvolvimento
acadêmico, profissional e pessoal.
Alguns colegas e ex-colegas do Laboratório contribuíram diretamente com este trabalho;
foram eles: Luciano Da Costa E Silva e Maria Marta Pastina nas etapas de mapeamento de
QTLs que envolveram o uso do OneQTL, e Gabriel Margarido e Marcelo Mollinari nos respec-
tivos trabalhos envolvendo o Tassel-GBS e o SuperMASSA e nas enriquecedoras discussões ao
longo deste trabalho. Os demais, indiretamente e cada um ao seu modo, também tiveram suas
contribuições. É o caso das trocas de ideias sobre diversos assuntos em genética e estatística
com Rodrigo Gazaffi, Renato Rodrigues, Edjane Freitas, Adriana Cheavegatti, João Ricardo
Bachega e Carina Anoni, marcadamente, e do sempre animado e estimulante convívio com
Graciela Sobierajski, Rodrigo Amadeu, Maria Izabel Cavassim, Rafael Tassinari, Luís Felipe
Ferrão, Marianella Quezada, Letícia Lara, Amanda Avelar, Danilo Cursi e Fernando Correr.

6
Conhecê-los e ter a amizade de vocês, pessoal, já faria este doutorado valer a pena! Ter con-
vivido com a Adriana, em especial, durante esses quatro anos, me trouxe inúmeros benefícios
pessoais e profissionais, por ser sempre tão centrada e acolhedora.
Este trabalho reuniu dados e informações de dois grupos de pesquisa distintos e, ob-
viamente, sem a existência de tais parcerias, seria impossível realizá-lo. Assim, pela parceria
envolvendo a população de mapeamento de sorgo sacarino, gostaria de agradecer ao Dr. Ju-
randir Vieira de Magalhães, da Embrapa Milho e Sorgo, Sete Lagoas-MG, e aos seus alunos e
demais pesquisadores e técnicos envolvidos. O Jurandir não só me co-orientou, contribuindo
com o inteiro desenvolvimento da pesquisa, como também propiciou diversos avanços. E, pela
parceria envolvendo a população de mapeamento de cana-de-açúcar, meus agradecimentos à
Profa. Dra. Monalisa Sampaio Carneiro, da Universidade Federal de São Carlos (UFSCar),
Araras-SP, e à Profa. Dra. Anete Pereira de Souza, da Universidade Estadual de Campinas
(Unicamp), Campinas-SP, e aos seus respectivos alunos e equipe técnica.
Sendo também parte dos trabalhos de doutorado de Vander Fillipe de Souza, da Universi-
dade Federal de São João del-Rei (UFSJ), Sete Lagoas-MG, e de Thiago Willian Balsalobre, da
Unicamp, gostaria de agradecê-los imensamente por trabalharem na obtenção e disponibilização
dos dados e na discussão dos resultados das respectivas populações de mapeamento de sorgo
sacarino e de cana-de-açúcar. Muito obrigado também pela amizade de vocês e por comparti-
lharem seus conhecimentos sobre as culturas comigo. Este trabalho, claramente, também é de
vocês.
Também registro minha eterna gratidão aos professores dos programas de pós-graduação
da ESALQ em Genética e Melhoramento de Plantas e em Estatística e Experimentação Agronô-
mica pelos conhecimentos compartilhados durante disciplinas e cursos e pela imensa contribui-
ção na minha formação acadêmica. Meu muito obrigado se estende a todos os funcionários
da ESALQ, e, em especial, a Léia, Rogério, Wilma, Fernando, Macedônio, Natálio, Carlinhos,
Valdir, Berdan, do Departamento de Genética; a S. Antonio e Marcos, da portaria do prédio; a
Lucas, do Serviço de Pós-Graduação; a Glória, da Biblioteca; e a Patrícia, do Ponto de Apoio
da FAPESP.
Obrigado aos atuais e antigos moradores da República, Mateus Figueiredo, Endson Nu-
nes, Diego Velasquez, Sanzio Barrios, Rodrigo Marques, Rubén Díaz, Thiago Aragão, Mateus
Vicente, Hugo Rosa, Otávio Carneiro, Rafael Tassinari, Yuri Caires, Lucas Santos, Stevan
Bordignon, Gustavo Martins, Luís Felipe Ferrão e Tomaz Andrade pela parceria e amizade,

7
e à D. Elza e à D. Mônica, por cuidarem de nós. E a todos os amigos que fiz em Piracicaba-SP,
por terem tornado minha permanência na cidade ainda mais agradável; desculpem-me por não
me arriscar a citá-los aqui, sob o prejuízo de esquecer alguém...
Finalmente, agradeço imensamente ao Dr. Paulo Augusto Vianna Barroso e à Dra. Lucia
Vieira Hoffmann, ambos da Embrapa Algodão, Goiânia-GO, e à Profa. Dra. Maria Lucia Car-
neiro Vieira, da ESALQ, pela amizade, conselhos e estímulos à carreira científica. Obrigado
aos meus pais, Geraldo e Maristela, e aos meus irmãos, Gustavo e Júnior, e demais familiares e
amigos de Campina Grande-PB ou espalhados pelo Brasil, pelo apoio e incentivo aos estudos e
pela compreensão devida às ausências.

8

9
SUMÁRIO
RESUMO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
ABSTRACT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
1 INTRODUÇÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2 MATERIAL E MÉTODOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.1 Materiais Vegetais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.1.1 Sorgo sacarino . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.1.2 Cana-de-açúcar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.2 Análise Fenotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.2.1 Sorgo sacarino . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.2.2 Cana-de-açúcar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.2.3 Correlações genotípicas e herdabilidades . . . . . . . . . . . . . . . . . . . . . . 27
2.3 Análise Genotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.3.1 Sorgo sacarino . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.3.2 Cana-de-açúcar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.3.2.1 Marcadores baseados em GBS . . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.3.2.2 Marcadores baseados em géis . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.3.2.3 Construção de mapa genético . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.4 Mapeamento de QTLs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3 RESULTADOS E DISCUSSÃO . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.1 Sorgo Sacarino . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.1.1 Análise fenotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.1.2 Análise genotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3.1.3 Mapeamento de QTLs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
3.2 Cana-de-Açúcar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.2.1 Análise fenotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.2.2 Análise genotípica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.2.2.1 Estratégias de descoberta de polimorfismos utilizando GBS . . . . . . . . . . . 48
3.2.2.2 Estimação de ploidia e dosagem alélica . . . . . . . . . . . . . . . . . . . . . 49
3.2.2.3 Marcadores baseados em géis . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
3.2.2.4 Mapa genético . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
3.2.3 Mapeamento de QTLs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60

10
3.3 Mapeamento Comparativo de QTLs . . . . . . . . . . . . . . . . . . . . . . . . . . 62
4 CONSIDERAÇÕES FINAIS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
REFERÊNCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
APÊNDICES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79

11
RESUMO
Mapeamento comparativo de QTLs entre sorgo sacarino e cana-de-açúcar paracaracteres bioenergéticos
Sorgo sacarino e cana-de-açúcar são duas importantes gramíneas com fins potencial-mente bioenergéticos. No entanto, apesar do conhecido relacionamento evolutivo, os genomasdessas espécies diferem em complexidade e tamanho. O sorgo, Sorghum bicolor, é diploide,com número básico de cromossomos igual a dez, os quais totalizam ∼726 Mb já sequenciadas.Já a cana cultivada, Saccharum × officinarum, é um autopoliploide com frequente aneuploidia,e apresenta genoma monoploide estimado em ∼1 Gb. Provavelmente, decorre deste fato, e doscruzamentos interespecíficos que originaram as variedades atuais, a relativa dificuldade em serealizar estudos genéticos em cana, e, como consequência, em se incrementar os trabalhos demelhoramento na espécie. Nesse contexto, a possibilidade de integrar estudos de mapeamentoentre sorgo e cana torna-se viável dado o emprego de metodologias apropriadas. O presentetrabalho objetivou mapear e comparar QTLs para caracteres agro-industriais nos genomas deambas as espécies, baseando-se no relacionamento evolutivo existente entre elas. Para tanto,foram utilizadas duas populações de mapeamento. A população de sorgo sacarino foi consti-tuída por 223 RILs genotipadas por mais de cem mil marcadores baseados em GBS fisicamentemapeados contra o genoma da espécie. A população de cana-de-açúcar constituiu-se de umaprogênie F1 segregante com 153 indivíduos genotipados por 500 marcadores baseados em géis(SSR e TRAP) e 7.049 marcadores baseados em GBS, segregando em dose única. Esses mar-cadores possibilitaram a construção de um mapa genético informativo e saturado, com 993marcadores distribuídos ao longo de 223 grupos de ligação, totalizando 3.682,05 cM. Ambas aspopulações foram avaliadas para quatro caracteres de interesse bioenergético: altura de colmos,toneladas de colmos ou de massa verde por hectare, e porcentagens de pol de caldo e de fibra.Modelos mistos foram utilizados para a análise dos dados fenotípicos, evidenciando a existênciade interação genótipo-ambiente a partir da estruturação de matrizes de variâncias-covariânciasgenéticas. As médias ajustadas conjunta e marginalmente foram utilizadas na descoberta deQTLs. Para este fim, modelos de mapeamento de múltiplos intervalos uni- e multivariadosforam utilizados e determinaram a descoberta de 53 e 36 regiões contendo QTLs para as po-pulações de sorgo e cana, respectivamente, para o conjunto dos quatro caracteres. Os genomasforam comparados utilizando os marcadores baseados em GBS de cana com informação posi-cional em relação ao genoma do sorgo. Um total de 16 regiões sintênicas identificadas entreas espécies possibilitaram inferências a respeito do controle evolutivamente conservado doscaracteres relacionados. Mais oito regiões foram adicionadas a estas após análise de marcadoresindividualmente para a população de cana. A descoberta dessas regiões subjacente à variaçãode caracteres bioenergéticos sugere aplicações na clonagem de genes e na seleção assistida pormarcadores, beneficiando os programas de melhoramento de ambas as espécies.
Palavras-chave: Sorghum bicolor; Saccharum × officinarum; Mapeamento de múltiplos QTLs;Análise de ligação; Sintenia

12

13
ABSTRACT
Comparative QTL mapping between sweet sorghum and sugarcane for bioenergy traits
Sweet sorghum and sugarcane are two important grasses for bioenergy purposes. Howe-ver, despite their known evolutionary relationship, the genomes of these species differ in com-plexity and size. Sorghum bicolor is a diploid species, with basic chromosome number of tenand ∼726 Mb completely sequenced, whereas Saccharum × officinarum has a autopolyploidgenome with frequent aneuploidy and monoploid size estimated at ∼1 Gb. Therefore, geneticstudies and breeding in sugarcane is challenging. In this context, the possibility of integratingmapping studies between sorghum and sugarcane becomes feasible given the recent develop-ment of appropriate methodologies. In this work, we aimed to map and compare QTLs forbioenergy traits in both species. To do this, two mapping populations were used. The populationof sorghum consisted of 223 RILs genotyped by more than one hundred thousand GBS-basedmarkers, which were physically mapped against the species genome. The population of sugar-cane is an F1 segregating progeny with 153 individuals genotyped by 500 gel-based (SSR andTRAP) and 7,049 GBS-based single-dose markers. These markers allowed the constructionof an informative and dense genetic map with 993 markers belonging to 223 linkage groupsand spanning 3,682.05 cM. Both populations were evaluated for four bioenergy traits: stalkheight, prodution in tons per hectare, and percentages of pol and fiber. Mixed models were usedto analyze phenotypic data and showed genotype-by-environment interaction on their geneticvariance-covariance structures. Joint and marginal adjusted means were used for QTL disco-very. Toward this end, univariate and multivariate multiple interval mapping models were used,and a total of 53 and 36 QTLs were found for sorghum and sugarcane, respectively. Comparisonof the genomes were based on GBS markers in sugarcane with relative sorghum chromosomeinformation. A total of 16 syntenic regions were identified between the species, allowing in-ferences in relation to evolutionary conserved control of the related traits. In addition, eightregions were also identified by considering single marker analyses. The discovery of QTLsunderlying such bioenergy traits may suggest further applications in gene cloning and markerassisted selection for both sweet sorghum and sugarcane species.
Keywords: Sorghum bicolor; Saccharum × officinarum; Multiple QTL mapping; Linkageanalysis; Synteny

14

15
1 INTRODUÇÃO
Sorgo sacarino e cana-de-açúcar são duas importantes matérias-prima de bioetanol,
energia verde do século XXI. O desenvolvimento de veículos flex (os quais podem utilizar tanto
etanol quanto gasolina como combustível) e as mudanças climáticas causadas pelo efeito estufa,
por exemplo, garantem a expansão da demanda mundial pelo álcool (AMORIM et al., 2011).
Considerando essa crescente demanda, há exigência de aumento na produção por unidade de
área e no teor de sacarose do caldo da cana-de-açúcar, cultura mais bem estabelecida nesse
cenário. Alternativamente, tem-se buscado implementar a expansão de outras culturas bio-
energéticas, como o sorgo sacarino. Ainda, quantidades superiores de fibra nas duas espécies
podem ser potencialmente destinadas à geração de etanol de segunda geração. Nesse contexto, o
melhoramento de plantas tem um importante papel no sentido de aumentar a produção e permitir
a expansão dessas culturas, por realizar seleção de genótipos que apresentem as características
agronômicas e industriais requeridas. O entendimento da genética dos caracteres de herança
poligênica, como são a maioria dos caracteres agro-industriais, é parte fundamental do processo
de melhoramento.
Dentre os cereais mais cultivados no mundo, o sorgo fica atrás apenas de trigo, arroz,
milho e cevada. De acordo com a Food and Agricultural Organization of the United States
(FAO), 42 milhões de hectares foram destinados para a produção de sorgo em todo o mundo,
tendo-se produzido mais de 61 milhões de toneladas em 2013; nesse ano, o Brasil produziu
cerca de dois milhões de toneladas em mais de 770 mil hectares plantados (FAO, 2014). A cana-
de-açúcar, por sua vez, está entre as mais importantes espécies tropicais cultivadas, sendo fonte
de sacarose e de vários subprodutos oriundos do processamento de seu caldo e bagaço, como
etanol e celulose. Em todo o mundo, foram destinados 24 milhões de hectares para o plantio
de cana, tendo-se colhido mais de 1,7 bilhão de toneladas em 2013; o Brasil é o maior produtor
mundial, tendo produzido 739 milhões de toneladas em 9,8 milhões de hectares plantados na
safra 2012/2013 (FAO, 2014).
O sorgo, Sorghum bicolor (L.) Moench, é uma gramínea autógama de genoma pequeno
(∼726 mega bases – Mb) e diploide (x = 10, 2n = 2x = 20). Essa espécie é caracterizada
pela elevada resistência a estresses abióticos (como o hídrico e o salino), e por possuir o me-
tabolismo C4 de fixação de carbono, sendo bastante eficiente no acúmulo de biomassa. Dentre
os sorgos cultivados, variedades foram selecionadas não apenas para produção de grãos (sorgo
granífero), mas também para produção de fibra (sorgo biomassa), forragem (sorgo forrageiro)

16
e açúcar (sorgo sacarino). O último tipo é caracterizado por genótipos que produzem colmos
de estatura elevada que acumulam açúcares (principalmente sacarose). No Brasil, o cultivo
de sorgo sacarino tem sido considerado como interessante complemento à cultura da cana-de-
açúcar no período de renovação do canavial. Em diversos aspectos, sorgo sacarino demonstra-se
vantajoso em termos operacionais quando comparado à cana-de-açúcar. Por exemplo, o cultivo
é realizado a partir de sementes, o ciclo vegetativo é relativamente curto (∼4 meses), há geração
de grãos para alimentação animal, é eficiente no uso do nitrogênio, e apresenta boa resistência
hídrica e boa adaptabilidade a solos com salinidade acima do normal (ZEGADA-LIZARAZU;
MONTI, 2012). Além disso, o processo de obtenção do etanol de primeira e segunda gerações
a partir sorgo sacarino é semelhante ao da cana, e o bagaço também pode ser aproveitado como
fonte de energia para as caldeiras no processo de produção.
Saccharum × officinarum designa cientificamente a cana-de-açúcar autopoliploide cul-
tivada (CHEAVEGATTI-GIANOTTO et al., 2011), com tamanho de genoma monoploide es-
timado em ∼1 Gb (SOUZA et al., 2011). Basicamente, duas espécies do gênero Saccharum
constituem os genomas das cultivares atuais: a “cana-nobre” S. officinarum L. (x = 10,
2n = 8x = 80) de colmos grossos e suculentos, com alto conteúdo de sacarose e boas
características gerais para industrialização, e a cana selvagem S. spontaneum L. (x = 8,
2n = 5-16x = 40-128), com elevada adaptabilidade a diversas condições ambientais (GRIVET;
ARRUDA, 2002). Por serem alógamas e sexualmente compatíveis, os primeiros trabalhos
de melhoramento da cana envolveram cruzamentos entre essas duas espécies, seguidos de
retrocruzamentos com S. officinarum, caracterizando a nobilização (MATSUOKA; GARCIA;
ARIZONO, 2005). Em decorrência desse processo, os materiais atuais apresentam um signi-
ficativo acréscimo de complexidade genômica em relação às espécies originais, resultando em
um genoma caracterizado pelo elevado grau de ploidia com frequente aneuploidia (D’HONT
et al., 1998; GRIVET; ARRUDA, 2002). Essa peculiaridade tem sido apontada como desa-
fiadora, pois dificulta os estudos genéticos e a aplicação de metodologias mais modernas no
melhoramento da espécie.
Sorgo sacarino tem sido geneticamente estudado a partir do uso de marcadores mo-
leculares que visaram, por exemplo, acessar diversidade (ALI et al., 2007; RITTER et al.,
2007), a qual tem se mostrado razoavelmente útil do ponto de vista do melhoramento. Além
disso, marcadores possibilitaram a construção de mapas genéticos e o mapeamento de locos
de caracteres quantitativos (do inglês, quantitative trait loci – QTLs) (RITTER et al., 2008;

17
SHIRINGANI; FRISCH; FRIEDT, 2010; SHIRINGANI; FRIEDT, 2011; GUAN et al., 2011),
assim como a realização de mapeamento associativo (MURRAY et al., 2009). Estudos genéticos
para a cana também têm sido desenvolvidos visando, sobretudo, reduzir o tempo de seleção de
cultivares. Corroborados pelo histórico do melhoramento, estudos citogenéticos revelaram que
a composição do genoma das cultivares comerciais de cana modernas é 70-80% de S. offici-
narum, 10-20% de S. spontaneum e 5-17% de cromossomos recombinantes (D’HONT et al.,
1998; GRIVET; ARRUDA, 2002). Ferramentas moleculares e modelos genético-estatísticos
capazes de acessar tamanha complexidade têm sido buscados. Aqui, pode ser citado o uso de
marcadores moleculares em estudos de diversidade (LIMA et al., 2002), construção de mapas
genéticos (GARCIA et al., 2006), mapeamento de QTLs baseado em mapas de ligação (PINTO
et al., 2009; PASTINA et al., 2012), e mapeamento associativo (RABOIN et al., 2006).
De fato, uma das grandes aplicações dos marcadores moleculares é na construção de
mapas genéticos. Isto porque populações segregantes podem ser genotipadas para inúmeros
locos e ter o genoma mapeado com base no desequilíbrio de ligação exibido entre esses mar-
cadores. De modo geral, a natureza da espécie a ser investigada é que determina os tipos
de população e de marcadores a serem utilizados no estudo. Plantas que formam linhagens
por sucessivas autofecundações, fixando um alelo para cada loco podem constituir populações
convencionais de mapeamento, quais sejam: retrocruzamentos, populações F2 e linhagens en-
dogâmicas recombinantes (do inglês, recombinant inbred lines – RILs); este é o caso do sorgo.
No entanto, para muitas espécies é difícil ou impossível a obtenção de linhagens homozigóti-
cas, e as análises de mapeamento são realizadas em populações F1 segregantes, derivadas do
cruzamento de genitores não-endogâmicos; isto se observa para cana-de-açúcar, por exemplo.
Evidentemente, espécies muito conhecidas do ponto de vista genético, como o sorgo, dispõem
de diversas metodologias para acessar o polimorfismo a nível de DNA e podem, inclusive,
possuir o genoma sequenciado, possibilitando o mapeamento físico dos marcadores. Por outro
lado, a dificuldade na obtenção e na análise dos dados moleculares de espécies de genomas
maiores e mais complexos, como é o caso da cana-de-açúcar, pode caracterizar desafios em
estudos dessa natureza, nos quais a densidade de marcadores é fundamental.
O genoma do sorgo encontra-se completamente sequenciado (PATERSON et al., 2009),
fazendo da espécie um modelo atrativo para genômica funcional de gramíneas C4. Por isso, in-
clusive, o sorgo tem se beneficiado amplamente de plataformas de sequenciamento da próxima
geração (do inglês, next generation sequencing – NGS) visando a descoberta e genotipagem

18
de polimorfismos de base única (do inglês, single nucleotide polymorphisms – SNPs) e de
polimorfismos baseados em inserção-deleção (indels). A metodologia de genotipagem por
sequenciamento (do inglês, genotyping-by-sequencing – GBS) tem colaborado com estudos
na espécie (MURRAY et al., 2009; MORRIS et al., 2013). Já para cana, iniciativas recentes
estão buscando sequenciar o genoma (SOUZA et al., 2011), o qual já dispõe de grande número
de etiquetas de sequências expressas (do inglês, expressed sequence tags – ESTs) do projeto
SUCEST (VETTORE et al., 2003), a partir das quais se derivaram importantes marcadores para
os trabalhos de mapeamento da espécie. Recentemente, a genotipagem de SNPs em espécies
de genoma complexo, como a cana, tem possibilitado estudos genéticos ainda mais avançados
(MOLLINARI, 2012). Porém, em detrimento da genotipagem de SNPs utilizando NGS, o uso
dessa tecnologia tem-se limitado a alguns importantes projetos adicionais de sequenciamento
em cana, como o do seu transcriptoma (CARDOSO-SILVA et al., 2014) e do seu genoma metil-
filtrado (GRATIVOL et al., 2014).
Mapas genéticos prestam-se a diversos tipos de aplicações, com destaques para o em-
prego em estudos evolutivos e de sintenia, no mapeamento de QTLs, dando suporte para clona-
gem posicional de genes e seleção assistida por marcadores, como foi observado em exemplos
anteriores. Ainda, mesmo no contexto dos inúmeros projetos existentes para sequenciamento
de genomas, os mapas têm sido frequentemente requeridos, pois servem como arcabouço para
ancoragem de mapas físicos e montagem e revisão de genomas (HAHN; ZHANG; MOYLE,
2014; BARTHOLOME et al., 2014; DEOKAR et al., 2014). Mapear QTLs, especificamente,
significa caracterizá-los quanto a número, posição e efeitos a partir da inferência em todo o
genoma sobre as relações entre o genótipo e o fenótipo de caracteres quantitativos. No contexto
do melhoramento de plantas, além de possibilitar conhecimento sobre interação entre QTLs
e dos QTLs com o ambiente, o mapeamento de QTLs constitui um importante passo para a
caracterização da arquitetura genética de locos responsáveis por caracteres com padrão contínuo
de variação fenotípica, como são a maioria dos caracteres de importância agronômica.
Para tanto, além de marcadores moleculares, as adequadas experimentação em campo
e análise dos dados fenotípicos são mandatórias. Modelos mistos, que utilizam metodologia
de estimação baseada em máxima verossimilhança restrita (do inglês, restricted maximum like-
lihood – REML), têm sido empregados nesse cenário por proporcionar apropriada representação
das complexas estruturas de variância-covariâncias (VCOV) originadas dos padrões de corre-
lação entre ambientes. Importante no cenário do melhoramento de plantas, essa abordagem

19
proporciona, em última análise, um melhor entendimento do fenômeno da interação genótipo-
ambiente (MALOSETTI; RIBAUT; VAN EEUWIJK, 2013), por exemplo.
Em sorgo sacarino, diversos estudos têm buscado locos responsáveis por caracteres
bioenergéticos, como aqueles relacionados a teor de açúcar (RITTER et al., 2008; SHIRIN-
GANI; FRISCH; FRIEDT, 2010; GUAN et al., 2011) e a qualidade de fibra (SHIRINGANI;
FRIEDT, 2011). Nesses trabalhos, foram utilizadas populações experimentais derivadas de
cruzamentos entre sorgo granífero e sorgo sacarino. Marcadores baseados em microssatélites
ou sequências simples repetidas (do inglês, simple sequence repeats – SSRs), e na identificação
de polimorfismos de tamanho de fragmentos amplificados (do inglês, amplified fragment length
polymorphisms – AFLPs) e de SNPs foram tipicamente utilizados na construção dos mapas.
Essas populações estabeleceram importantes passos para a descoberta de QTLs, dado o emprego
de diversas metodologias genético-estatísticas, como análise de marcas individualmente (do
inglês, single marker analysis – SMA), mapeamento por intervalo (do inglês, interval mapping
– IM), e mapeamento por intervalo composto (do inglês, composite interval mapping – CIM).
Mapeamento associativo para brix e altura também já foi realizado para painel de 125 indivíduos
(maioria sacarino) (MURRAY et al., 2009). Desafortunadamente, modelos de mapeamento de
múltiplos intervalos (do inglês, multiple interval mapping – MIM) ainda não foram relatados
para sorgo sacarino, desprezando o potencial aumento na precisão e no poder de detecção dos
QTLs (SILVA; ZENG, 2010).
Para cana-de-açúcar, apesar dos problemas mencionados anteriormente em relação à sua
genética, tem-se obtido conspícuo progresso em termos de obtenção de marcadores, construção
de mapas genéticos e análise de QTLs. Brevemente, os marcadores comumente utilizados
baseavam-se na identificação de polimorfismos de tamanho de fragmento de restrição (do inglês,
restriction fragment length polymorphisms – RFLPs), AFLPs e SSRs. Os primeiros mapas de
cana foram elaborados segundo metodologia conhecida como duplo pseudocruzamento-teste
(GRATTAPAGLIA; SEDEROFF, 1994), e modelos de SMA e IM também foram empregados
para o mapeamento de caracteres agronômicos e de resistência a doenças nos mapas resultantes
para cada um dos genitores, separadamente, como em diversos exemplos revisados por Pastina
et al. (2010). Atualmente, metodologias para construção de mapas integrados (WU et al.,
2002a, 2002b) e modelos de CIM e de MIM estão disponíveis para populações F1 segregantes
com exemplos de aplicação em cana (GARCIA et al., 2006; PASTINA et al., 2012; GAZAFFI
et al., 2014). Além disso, técnica de genotipagem quantitativa de SNPs para poliploides comple-

20
xos foi estabelecida com ênfase em cana (GARCIA et al., 2013), e mostram-se promissoras na
construção de mapas genéticos integrados (MOLLINARI, 2012), agora, considerando dosagem
alélica dos marcadores (SERANG; MOLLINARI; GARCIA, 2012).
Para este fim, Garcia et al. (2013) analisaram SNPs obtidos utilizando moderna tecnolo-
gia baseada em espectrometria de massa (Sequenom iPLEXTM MassARRAY R©) (OETH et al.,
2006), capaz de informar sobre as proporções relativas entre os alelos. Na ausência de artefatos
técnicos criando vieses para um ou ambos os alelos, a razão entre as quantificações de cada alelo
informa sobre a ploidia do loco e a dose de cada alelo em um indivíduo. A análise consistiu
na estimação da ploidia e classificação dos indivíduos nos clusters esperados dada a ploidia
utilizando um modelo gráfico Bayesiano implementado em um software livre denominado
SuperMASSA (SERANG; MOLLINARI; GARCIA, 2012; MOLLINARI; SERANG, 2015).
Esta metodologia leva em conta as frequências esperadas de classes genotípicas considerando
modelo de Equilíbrio de Hardy-Weinberg para paineis de diversidade ou, alternativamente,
modelo de segregação para progênies de irmãos-completos. Uma avaliação de 987 e 249
marcadores SNP para um painel de diversidade e para uma população de mapeamento de cana-
de-açúcar, respectivamente, revelou que os níveis de ploidia variavam de loco para loco, mais
provavelmente, entre 6 e 14. Adicionalmente, para os marcadores da população biparental,
apenas cerca de 30% do total de marcadores estariam segregando em dose única (GARCIA et
al., 2013).
Estudos genômicos comparativos, por sua vez, só são possíveis devido à existência de re-
giões genômicas (eventualmente genes) que se mantiveram relativamente estáveis em conteúdo
de nucleotídeos ao longo dos tempos evolutivos, proporcionando uma base útil para inferir a cor-
respondência inclusive entre genomas distantemente relacionados, assim como entre genes que
divergiram bastante ou foram realocados (TANG et al., 2008). Por isso, sucesso tem sido obtido
nos estudos de genomas de plantas pertencentes à mesma família. No entanto, o nível em que
os genes nos cromossomos correspondem em conteúdo (sintenia) e em ordem (colinearidade)
difere marcadamente entre os táxons, devido à retenção diferencial de genes ou famílias gênicas,
às duplicações de sequências repetitivas e à mobilidade causada pelos elementos transponíveis
(TANG et al., 2008). Sabidamente, a determinação dos graus de sintenia e colinearidade entre
as espécies relacionadas de plantas modelos e cultivadas é valiosa na aplicação da informação
genômica no melhoramento de culturas (RAHMAN; PATERSON, 2010).
A genômica comparativa tornou-se um campo promissor de pesquisa dados os projetos

21
genomas finalizados ou em desenvolvimento no sentido de proporcionar respostas requeridas
sobre organização e evolução dos genomas de espécies de interesse. Além disso, há grandes
chances de se transferir e integrar informações sobre localização e ação de genes interespecifi-
camente (SORRELS, 2006). Antes da era genômica, essas comparações também eram possíveis
graças a existência de marcadores compartilhados entre os mapas de ligação das espécies estu-
dadas. Hoje em dia, a disponibilidade de ambos (mapas e sequências) torna possível estudos
mais detalhados. A partir daí, inúmeros estudos ajudaram a elucidar o elevado grau de sintenia
entre genomas de gramíneas, por exemplo, e forneceram insights sobre as contribuições de
transposons, duplicação de genes, e de sequências não codificadoras conservadas ao longo da
evolução do genoma de plantas (BENNETZEN; FREELING, 1997).
No caso do sorgo e da cana-de-açúcar, comparação entre mapas genéticos baseados
em RFLPs revelaram um elevado nível de sintenia (GRIVET et al., 1994; DUFOUR et al.,
1997; GUIMARÃES; SILLS; SOBRAL, 1997; MING et al., 1998). Além disso, com base no
nível de sintenia existente entre as gramíneas, já foi possível identificar genes evolutivamente
conservados associados ao controle de conteúdo de açúcar (SINGH et al., 2011) e de resistência
à ferrugem (LE CUNFF et al., 2008) em cana-de-açúcar, por exemplo. Adicionalmente, Ming et
al. (2002) alinhou grupos de ligação de duas diferentes populações de cana com mapa de sorgo
para auxiliar na avaliação de QTLs afetando caracteres relacionados a açúcar mapeados na
espécie. Usando essa abordagem, 62 QTLs de cana para esses caracteres puderam ser inferidos
em nove cromossomos de sorgo.
Como foi visto, o sorgo tornou-se uma espécie modelo para análises genômicas funcio-
nais e estruturais em gramíneas (CALVIÑO; MESSING, 2012), por ser uma espécie diploide de
genoma relativamente pequeno e de elevada endogamia, e por ser geneticamente mais próximo
da cana-de-açúcar e do milho, espécies das quais divergiu recentemente, a cerca de 8–9 milhões
de anos (JANNOO et al., 2007). Por outro lado, as cultivares modernas de cana-de-açúcar
são derivadas de cruzamentos entre duas espécies de Saccharum, seguidos de alguns ciclos de
retrocruzamentos e seleção, e caracterizam-se por serem alógamas com genoma autopoliploide
frequentemente aneuploide (D’HONT et al., 1998). No entanto, apesar da relativa dificuldade
em se trabalhar com a genética da espécie, esforços têm se acumulado no sentido de mapear seu
genoma por meio de marcadores, e utilizar esses mapas na descoberta de locos que controlem
caracteres quantitativos.
O mapeamento comparativo entre sorgo sacarino e cana-de-açúcar tornou-se uma ferra-

22
menta atrativa pois as espécies pertencem à mesma família botânica (Andropogoneae), possuem
similaridade de caracteres fenotípicos avaliados, e dispõem de recursos genômicos suficientes
(genoma ou marcadores moleculares baseados em sequências). Caracteres agronômicos, tais
como altura e produção de biomassa, e industriais, como os caracteres relacionados ao conteúdo
de açúcar e fibra, são de grande interesse dos melhoristas de ambas as espécies. Para tanto,
mapeamento de QTLs nessas espécies com base em marcadores que forneçam ampla cobertura
dos genomas e em metodologias genético-estatísticas adequadas torna-se requerimento indis-
pensável em estudos dessa natureza.
Nesse sentido, o presente trabalho objetivou mapear QTLs em duas populações – uma
de sorgo sacarino e outra de cana-de-açúcar – e compará-los com base nas correlações entre
os genomas dessas espécies. Para tanto, fenótipos de interesse bioenergético foram avaliados
em campo e marcadores moleculares baseados em GBS, principalmente, foram genotipados
em ambas as populações. Aqui, buscou-se também estabelecer estratégias de análise dos dados
fenotípicos e moleculares para as duas espécies a partir da utilização de adequadas ferramentas e
metodologias de genética-estatística e bioinformática da atualidade. Até onde é conhecido, este
é o único estudo desta natureza envolvendo sorgo sacarino. Além disso, procurou-se utilizar
germoplasma adaptado às condições do Brasil, para as duas espécies.

23
2 MATERIAL E MÉTODOS
2.1 Materiais Vegetais
2.1.1 Sorgo sacarino
A população experimental de RILs de sorgo sacarino originou-se do cruzamento entre
as cultivares ‘BR501-Brandes’ (genitor feminino) e ‘BR505-Wray’ (genitor masculino) e é
formada por 272 indivíduos obtidos pelo método descendente de única semente (do inglês,
single seed descent – SSD). Genitores e população fazem parte do programa de melhoramento
da Embrapa Milho e Sorgo, Sete Lagoas, Estado de Minas Gerais. Os genitores são bastante
constrastantes, principalmente para caracteres relativos a conteúdo de açúcar, e se referem a
germoplasma adaptado às condições do Brasil.
2.1.2 Cana-de-açúcar
A população F1 segregante de cana-de-açúcar foi obtida a partir do cruzamento entre as
variedades ‘SP80-3280’ (genitor feminino) e ‘RB835486’ (genitor masculino) e é constituída
por 153 indivíduos. Genitores e população fazem parte do programa de melhoramento de cana-
de-açúcar da Universidade Federal de São Carlos (UFSCar), Araras, Estado de São Paulo, que
participa da Rede Interinstitucional de Desenvolvimento do Setor Sucroalcooleiro (RIDESA).
Os genitores são variedades amplamente cultivadas no país por serem bastante produtivas.
‘SP80-3280’ (‘SP71-1088’ × ‘H57-5028’) foi umas das variedades sequenciadas por projetos
de sequenciamento de transcriptoma da cana (VETTORE et al., 2003; CARDOSO-SILVA et al.,
2014) e tem tido todo o genoma sequenciado pela iniciativa brasileira (SOUZA et al., 2011).
Essa variedade apresenta resistência ao carvão (Ustilago scitaminea) e à ferrugem marrom
(Puccinia melanocephala), enquanto ‘RB835486’ (‘L60-14’ × ?) é suscetível a essas duas
doenças fúngicas.
2.2 Análise Fenotípica
Para ambas as espécies, diversos caracteres fenotípicos foram avaliados conforme Ma-
nual de Instruções do Conselho dos Produtores de Cana-de-Açúcar, Açúcar e Álcool do Estado
de São Paulo (CONSECANA-SP, 2006). Neste trabalho, quatro caracteres foram considerados:
altura média de colmos (ALT, em m), produção de massa verde (PMV, em ton · ha−1) para sorgo
ou toneladas de colmos por hectare (TCH, em ton · ha−1) para cana, quantidade de sacarose ou

24
pol do caldo (POL, em %) e conteúdo de fibra (FIB, em %). A variável PMV difere da variável
TCH por levar em consideração o peso dos colmos e das folhas, enquanto TCH só se refere
ao peso dos colmos. Modelos fenotípicos lineares de efeitos mistos, descritos a seguir, foram
utilizados na obtenção dos componentes de variância e das médias ajustadas, servindo como
base para os cálculos de herdabilidades e de correlações genotípicas e para o mapeamento de
QTLs.
2.2.1 Sorgo sacarino
Os experimentos de campo da população de mapeamento de sorgo sacarino foram con-
duzidos no município de Sete Lagoas, Estado de Minas Gerais, em área pertencente à Embrapa
Milho e Sorgo, por três anos agrícolas consecutivos (2011, 2012 e 2013). O delineamento
experimental foi estabelecido no esquema de látices quadrados 15 × 15, repetidos três vezes,
com cada repetição totalizando 223 indivíduos da população mais os dois genitores. As parcelas
foram compostas por duas linhas de 5 m de comprimento, espaçadas em 0,70 m.
As análises dos caracteres fenotípicos para os múltiplos anos de avaliação foram reali-
zadas no software GenStat (v. 16.1) (VSN INTERNATIONAL, 2014) via REML considerando
o seguinte modelo misto:
yi jkm = µ + am + rk(m) + b j(km) + tim + εi jkm (1)
em que yi jkm é a observação do indivíduo i no bloco j, repetição k e ano m; µ é a média geral
fixa; am é o efeito fixo do m-ésimo ano (m = 1, . . . ,M; M = 3); rk(m) é o efeito fixo da k-
ésima repetição (k = 1, . . . ,K; K = 3) no ano m; b j(km) é o efeito aleatório do j-ésimo bloco
( j = 1, . . . , J; J = 15) na repetição k e ano m; tim é o efeito ora fixo ora aleatório do i-ésimo
tratamento (i = 1, . . . , I; I = 225) no ano m; e εi jkm é o erro aleatório. O termo tim foi separado
em dois grupos, sendo gim o efeito aleatório dos genótipos da população (i = 1, . . . , Ig; Ig =
223), e cim o efeito fixo dos genitores da população (i = Ig + 1, . . . , Ig + Ic; Ic = 2). Para
genótipos da população, foi assumido que o vetor g = (g11, . . . , gIgM)′ tem uma distribuição
normal multivariada com vetor de médias zero e matriz de variâncias-covariâncias (VCOV)
genética G = GM ⊗ IIg , i.e. g ∼ N(0,G), em que GM = GAM×M é a matriz de VCOV para o
fator de agrupamento sobrescrito anos (A) de dimensões M × M, ⊗ representa o produto direto
de Kronecker, e IIg é uma matriz identidade de dimensões Ig × Ig. Para resíduos, foi assumido

25
ε ∼ N(0,R), em que ε = (ε1111, . . . , εIJKM)′ e R = RM ⊗ RK ⊗ II·J é a matriz de VCOV residual,
sendo RM = RAM×M a matriz de VCOV residual para anos (A) e RK = RR
K×K a matriz de VCOV
residual para repetições (R).
As matrizes GM, RM e RK foram examinadas para diferentes estruturas de interesse
biológico e estatístico, e os modelos foram comparados utilizando os critérios de informação
de Akaike (do inglês, Akaike information criterion – AIC) (AKAIKE, 1974) e Bayesiano (do
inglês, Bayesian information criterion – BIC) (SCHWARZ, 1978), conforme realizado por
Pastina et al. (2012). A estratégia adotada para a definição das matrizes foi a de, inicialmente,
selecionar a matriz GM, e, em seguida, selecionar as matrizes RM e RK , nesta ordem. Estruturas
de VCOV genéticas consideraram, para diferentes anos, variâncias genéticas homogêneas e
covariâncias nulas (identidade – ID) ou não-nulas e iguais (uniforme – UNIF), ou variâncias
genéticas heterogêneas com covariâncias nulas (diagonal – DIAG) ou não-nulas e diferentes
(auto-regressiva de 1a ordem – AR1; simétrica composta heterogênea – CSHet; não-estruturada
– UNST). As matrizes AR1, CSHet e UNST diferem em complexidade e estimabilidade, por
conterem certas restrições que exigem a estimação de mais ou menos parâmetros (Tabela 1).
Também, estruturas de VCOV residuais foram selecionadas com o objetivo de permitir even-
tuais heteroscedasticidade e dependência entre as observações para os fatores de agrupamentos
anos e repetições, próprios do delineamento experimental utilizado.
Tabela 1 – Descrição e número de parâmetros (nPAR) dos modelos examinados para as matrizes devariância-covariância genética GM na análise fenotípica de população de mapeamentode sorgo sacarino
Matrizes nPAR Descrição
GM = GAM×M
ID 1 Matriz identidade (ou de variância genética homogênea)UNIF 2 Matriz uniformeDIAG M Matriz diagonal (ou de variância genética heterogênea)AR1 M+1
2 Matriz auto-regressiva de 1a ordemCSHet M + 1 Matriz simétrica composta com variância genética heterogêneaUNST M(M+1)
2 Matriz não-estruturadaNota: GM considerou diferentes estruturas para os efeitos genéticos dentro de M anos (A)
Finalmente, após ajuste dos modelos de VCOV da parte aleatória do modelo para cada
caráter, o efeito fixo de interação entre anos e genitores foi testado utilizando a estatística de
Wald e mantido em cada modelo quando estatisticamente significativo (p < 0,05).

26
2.2.2 Cana-de-açúcar
Para cana-de-açúcar, outro modelo foi utilizado pois, além de a população ter sido
plantada em duas localidades, acrescentado mais um fator à análise, a experimentação para
a espécie considera particularidades adicionais. A primeira delas refere-se à tomada de dados
no tempo (cortes), a partir de um primeiro experimento implantado. A segunda particularidade
reside no próprio delineamento, o qual deve ser próprio para experimentos muito grandes, como
este aqui avaliado, em decorrência da utilização de parcelas de tamanhos superiores.
Os experimentos de campo da população de mapeamento de cana-de-açúcar foram
conduzidos nos municípios de Araras e Ipaussu, Estado de São Paulo, por três anos agrícolas
consecutivos (2010, 2011 e 2012), cada ano representando consecutivos cortes (canas planta,
soca e ressoca). O delineamento experimental foi estabelecido nos dois locais no esquema de
blocos aumentados de Federer (FEDERER; RAGHAVARAO, 1975), repetidos três vezes, sendo
cada repetição composta por nove blocos incompletos com 27 indivíduos mais três tratamentos
comuns (os dois genitores e a variedade ‘RB867515’). As parcelas foram compostas por duas
(em Araras) ou três (em Ipaussu) linhas de 3 m de comprimento, espaçadas em 1,5 m. Dentre
os indivíduos avaliados em cada repetição, 90 não pertenciam à população de mapeamento,
por terem sido identificados como contaminantes em análises moleculares com SSRs (dados
não apresentados). Portanto, para fins de ajuste de modelo, eles não foram considerados como
parte do componente aleatório referente a genótipos da população. Além disso, enquanto ALT
e TCH foram avaliados para as três repetições, POL e FIB foram avaliados apenas para duas
delas. Uma última particularidade referiu-se às ausências de avaliações de ALT e TCH no
segundo corte em Araras e de POL e FIB no segundo corte em ambos os locais por conta de
eventuais problemas durante a condução dos experimentos.
As análises dos caracteres fenotípicos para múltiplos locais e cortes de avaliação foram
realizadas no software GenStat (v. 16.1) (VSN INTERNATIONAL, 2014) via REML conside-
rando o seguinte modelo misto:
yi jkmn = µ + ln + am + rk(mn) + b j(kmn) + timn + εi jkmn (2)
em que yi jkmn é a observação do indivíduo i no bloco j, repetição k, corte m e local n; µ é a média
geral fixa; ln é o efeito fixo do n-ésimo local (n = 1; N = 2); am é o efeito fixo do m-ésimo
ano ou corte (m = 1, . . . ,M; M = 2 ou M = 3 dependendo do local); rk(mn) é o efeito fixo da

27
k-ésima repetição (k = 1, . . . ,K; K = 2 ou K = 3 dependendo do caráter) no corte m e local
n; b j(kmn) é o efeito aleatório do j-ésimo bloco ( j = 1, . . . , J; J = 9) na repetição k, corte m
e local n; timn é o efeito ora fixo ora aleatório do i-ésimo indivíduo (i = 1, . . . , I; I = 156) no
corte m e local n; e εi jkmn é o erro aleatório. O termo timn foi separado em dois, sendo gimn o
efeito aleatório dos tratamentos regulares (genótipos da progênie, i = 1, 2, . . . , Ig; Ig = 153), e
cimn o efeito fixo dos tratamentos comuns (testemunhas, i = Ig + 1, . . . , Ig + Ic; Ic = 3). Para
genótipos da progênie, foi assumido que o vetor g = (g111, . . . , gIgMN)′ tem uma distribuição
normal multivariada com vetor de médias zero e matriz de VCOV genética G = GP ⊗ IIg , i.e. g
∼ N(0,G), em que P = M · N, ⊗ representa o produto direto de Kronecker, e IIg é uma matriz
identidade de dimensões Ig × Ig. Várias estruturas de VCOV para GP (Tabela 2) foram exami-
nadas e comparadas via AIC e BIC, conforme realizado por Pastina et al. (2012). Estruturas de
VCOV genéticas usaram uma combinação fatorial de locais e cortes como diferentes ambientes
(E), i.e. GP = GEP×P, ou, alternativamente, o produto direto de matrizes de VCOV genéticas para
locais (L) e cortes (A), i.e. GP = GLN×N ⊗ GA
M×M. Para resíduos, foi assumido ε ∼ N(0,R), em
que ε = (ε11111, . . . , εIJKMN)′ e R = RP⊗RK⊗II·J a matriz de VCOV residual, sendo as matrizes
RP = REP×P (combinação fatorial) ou RP = RL
N×N⊗RAM×M (produto direto) e RK = RR
K×K também
examinadas para diferentes estruturas.
Dado o ajuste dos modelos de VCOV da parte aleatória do modelo para cada caráter, os
efeitos fixos das interações entre locais, anos e tratamentos comuns foram testados utilizando a
estatística de Wald e mantidos nos modelos quando estatisticamente significativos (p < 0,05).
2.2.3 Correlações genotípicas e herdabilidades
Correlações genotípicas foram calculadas como coeficiente de Pearson entre as médias
ajustadas para cada caráter e testadas assumindo nível global de significância de 0,05 utilizando
o pacote do R (R CORE TEAM, 2014) denominado psych (REVELLE, 2014), que também foi
utilizado para desenhar os gráficos de dispersão entre os pares de caracteres.
Para a população de cana-de-açúcar, herdabilidades em sentido amplo a nível de plantas
(H2plantas) foram computadas utilizando as estimativas dos componentes de variância dos modelos
ID para estruturas de VCOV dos efeitos genéticos e residuais por meio da razão σ̂2G/σ̂
2P, em que
σ̂2G é a variância genética e σ̂2
P é a variância fenotípica total para cada caráter. Calculou-se a
razão σ̂2G/σ̂
2P̄ como uma medida aproximada de herdabilidades no sentido amplo ao nível de
médias (H2médias), em que σ̂2
P̄ é a variância fenotípica entre médias genotípicas para cada caráter

28
Tabela 2 – Descrição e número de parâmetros (nPAR) dos modelos examinados para a matriz de variância-covariância genética GP na análise fenotípica de população de mapeamento de cana-de-açúcar
Matriz GP nPAR Descrição
GP = GEP×P
ID 1 Matriz identidade (ou de variância genéticahomogênea)
UNIF 2 Matriz uniformeDIAG P Matriz diagonal (ou de variância genética
heterogênea)CSHet P + 1 Matriz simétrica composta com variância genética
heterogêneaUNST P(P+1)
2 Matriz não-estruturadaGP = GL
N×N ⊗GAM×M
UNST ⊗ ID N(N+1)2 + 1 Matrizes não-estruturada e identidade para locais e
cortes, respectivamenteUNST ⊗ UNIF N(N+1)
2 + 2 Matrizes não-estruturada e uniforme para locais ecortes, respectivamente
UNST ⊗ DIAG N(N+1)2 + M Matrizes não-estruturada e diagonal para locais e
cortes, respectivamenteUNST ⊗ AR1 N(N+1)+2(M+1)
2 − 1 Matrizes não-estruturada e auto-regressiva de 1a
ordem para locais e cortes, respectivamenteUNST ⊗ CSHet
N(N+1)2 + M + 1 Matrizes não-estruturada e simétrica composta para
locais e cortes, respectivamenteUNST ⊗ UNST N(N+1)+M(M+1)
2 − 1 Matrizes não-estruturadas para locais e cortesNota: GP considerou diferentes estruturas para os efeitos genéticos dentro de P = M ·N combinações de local-corte(ambientes, E), ou N locais (L) e M cortes (A)
obtida utilizando-se a média harmônica do número de ambientes como denominador da estima-
tiva de variância da interação genótipo-ambiente, e a média harmônica do número de repetições
em todos os ambientes como denominador da estimativa de variância residual (HOLLAND;
NYQUIST; CERVANTES-MARTÍNEZ, 2003). Para a população de sorgo sacarino, como a
variância genética é exclusivamente aditiva, i.e. σ2G = σ2
A, as herdabilidades em sentido restrito
foram calculadas de maneira semelhante e denominadas, por convenção, como h2plantas e h2
médias.
As estimativas dos componentes de variância para essa população também foram obtidas a partir
de modelos ID para estruturas de VCOV dos efeitos genéticos e residuais.

29
2.3 Análise Genotípica
2.3.1 Sorgo sacarino
O procedimento de genotipagem por sequenciamento (GBS) foi realizado pelo Institute
for Genomic Diversity (Cornell University, Ithaca, NY, EUA) de acordo com o protocolo des-
crito em detalhes por Elshire et al. (2011) em plataforma HiSeqTM 2000 (Illumina R© Inc., San
Diego, CA, EUA). Três bibliotecas, cada uma com 95 indivíduos mais um controle negativo,
foram construídas a partir da digestão de DNA genômico dos indivíduos com a enzima ApeKI,
que reconhece sítio de cinco bases (sendo uma degenerada) e é sensível à metilação. Além do
par de genitores da população presentes uma vez em cada biblioteca, as duas primeiras bibli-
otecas continham 91 indivíduos da população de mapeamento mais dois controles positivos,
e a terceira biblioteca continha os 90 indivíduos da população restantes mais três controles
positivos.
O pipeline Tassel-GBS (GLAUBITZ et al., 2014) implementado no software Tassel (v.
4.3.8) foi utilizado para descoberta de SNPs e indels. Para tanto, tags de GBS de 64 pb foram
alinhadas contra os dez cromossomos do genoma do sorgo (v. 2.1; ∼658 Mb) (PATERSON et
al., 2009). As tags utilizadas no mapeamento deveriam estar presentes, no mínimo, três vezes no
conjunto das três bibliotecas (argumento -c do MergeMultipleTagCountPlugin) do Tassel-
GBS. Chamadas de genótipos foram armazenadas em arquivos do tipo hapmap. Dados perdidos
(incluindo bases degeneradas) foram imputados utilizando o software Npute (ROBERTS et al.,
2007). Neste programa, foram testados tamanhos de janela variando de 5 a 150 pb dentro de
cada cromossomo; aquelas janelas que forneciam maior acurácia foram, então, selecionadas
para imputação. Finalmente, os dados imputados foram filtrados para uma frequência alélica
mínima (do inglês, minor allele frequency – MAF) de 0,40.
2.3.2 Cana-de-açúcar
Dois tipos de marcadores foram utilizados na análise genotípica da população de mape-
amento de cana-de-açúcar. Primeiramente, serão apresentadas as metodologias de obtenção e
análise de marcadores quantitativos baseados em GBS, ou seja, daqueles capazes de caracterizar
dosagem alélica dos locos genotipados, e, depois, de marcadores qualitativos, baseados em géis,
que informam apenas sobre ausência e presença das marcas, impossibilitando a caracterização
de dosagem alélica.

30
2.3.2.1 Marcadores baseados em GBS
O procedimento de GBS para a população de cana foi realizada seguindo os mesmos
procedimentos utilizado para a população de sorgo (ELSHIRE et al., 2011), com modificações
adicionais. Os genitores da população foram repetidos três vezes e constituíram uma primeira
biblioteca. Cinquenta e seis indivíduos da progênie foram sequenciados a partir de uma segunda
biblioteca, e os 95 indivíduos restantes foram sequenciados a partir de uma terceira biblioteca.
Dois indivíduos da população de mapeamento não foram incluídos. Cada indivíduo pertencente
a uma biblioteca fazia parte de uma reação de plex 96 (incluindo um controle negativo, cada).
Visando obter maior contagens de leituras (profundidade), as bibliotecas foram restringidas
pela enzima PstI, que reconhece sítio de seis bases e é parcialmente sensível à metilação.
Adicionalmente, as bibliotecas foram corridas em duas lanes distintas de HiSeqTM 2000.
Foi utilizado o pipeline Tassel-GBS para a descoberta de SNPs e indels. Como este
pipeline requer um genoma de referência e o sequenciamento do genoma de cana ainda não está
finalizado (SOUZA et al., 2011), foram utilizadas as seguintes pseudo-referências: (i) genoma
metil-filtrado da cana-de-açúcar (∼674 Mb arranjadas em 1.109.444 scaffolds) (GRATIVOL et
al., 2014), (ii) genoma do sorgo (v. 2.1; ∼726 Mb arranjadas em 10 cromossomos e 1.600 scaf-
folds restantes) (PATERSON et al., 2009), (iii) transcriptoma via RNA-Seq da cana-de-açúcar
(∼780 Mb arranjadas em 119.768 scaffolds) (CARDOSO-SILVA et al., 2014), e (iv) sequências
do projeto SUCEST (∼152 Mb arranjadas em 237.954 sequências) (VETTORE et al., 2003);
as respectivas abreviações (“mf”, “sb”, “rna” e “est”) foram utilizadas na nomenclatura dos
marcadores, acrescidas da identificação do cromossomo, scaffold ou contig e da posição do
polimorfismo da referência. O algoritmo Bowtie 2 (v. 2.2.1) (LANGMEAD; SALZBERG,
2013) foi usado para mapear as tags de GBS de 64 pb contra cada referência; as tags utilizadas
no mapeamento deveriam estar presentes, no mínimo, cinco vezes no conjunto das seis lanes
(argumento -c do MergeMultipleTagCountPlugin). Contagens exatas das leituras alelo-
específicas (profundidades) foram armazenadas em arquivos do tipo variant call format (VCF).
Para este fim, o tipo de variável Java do DiscoverySNPCallerPlugin do Tassel-GBS foi
modificado de byte para short visando permitir o armazenamento de um valor máximo de
32.767 contagens. Adaptações para lidar com o tipo de variável short foram necessárias para
os plugins subsequentes do pipeline também. As modificações no pipeline do Tassel-GBS
foram desenvolvidas no Laboratório de Bioinformática Aplicada à Bioenergia, da ESALQ, e
gentilmente cedidas pelo Prof. Gabriel R. A. Margarido.

31
Estimação de ploidias e dosagens
O pipeline Tassel-GBS gerou dados (D) de contagens alelo-específicas na forma D =
{(x1, y1), (x2, y2), . . . , (xn, yn)} para cada loco bialélico para os indivíduos i = 1, 2, . . . , n, os
quais puderam ser visualizados em gráficos de dispersão e foram analisados no software Super-
MASSA (SERANG; MOLLINARI; GARCIA, 2012; MOLLINARI; SERANG, 2015). Como
um primeiro critério de controle de qualidade, locos com mais de 25% de dados perdidos foram
excluídos. Adicionalmente, foram eliminados das análises os locos cujas contagens médias de
leituras para o alelo de referência eram inferior a 50. Finalmente, dados individuais com coor-
denadas radiais ri =
√x2
i + y2i menores que 0,10 ·max(r1, r2, . . . , rn) também foram removidos.
Para cada loco, foram testadas todas as ploidias pares variando de 2 a 20, tendo sido
selecionada aquela que retornou a maior verossimilhança (GARCIA et al., 2013). Dados dos
genitores repetidos foram considerados nas análises por fornecerem restrições adicionais du-
rante a estimação. Seguindo a recomendação de Mollinari e Serang (2015) para encontrar a
solução de máxima posteriori (do inglês, maximum a posteriori – MAP) para as estimativas
dos parâmetros do modelo gráfico (SERANG; MOLLINARI; GARCIA, 2012), o argumento
naive posterior report threshold foi mantido em zero. Em seguida, os valores de
probabilidades individuais a posteriori dada a ploidia selecionada (entre 6 e 14) também foram
calculadas variando o threshold de 0 a 1, a uma razão de progressão aritmética de 0,05. Esse
recurso possibilitou reconhecer o threshold máximo que permitiu alguma atribuição individual
a determinado cluster. Somente as ploidias variando de 6 a 14 foram consideradas porque,
em estudo anterior, elas foram apontadas como mais prováveis no genoma da cana e também
exibiram maiores proporções de marcadores em dose única (do inglês, single-dose markers –
SDMs) (GARCIA et al., 2013).
Como posterior critério de controle de qualidade, foram selecionadas apenas SDMs
cujas medianas de todas as probabilidades individuais a posteriori foram maior que 0,80. As
saídas do SuperMASSA foram recodificadas no software R para propósito de mapeamento
substituindo-se os respectivos alelos de referência e alternativos por a e b. Locos com padrões
de genótipos duplicados dentro e entre referências foram inspecionados sobre as chamadas de
genótipos recodificadas. Aqui, apenas uma chamada foi levada para a análise de ligação, sendo
armazenada a relativa informação de redundância. Um gráfico circular foi usado para sumarizar
os resultados de locos duplicados dentro e entre referências usando o pacote do R circlize (GU
et al., 2014).

32
2.3.2.2 Marcadores baseados em géis
Um total de 120 marcadores SSRs foram genotipados, sendo 98 e 16 derivados das
respectivas bibliotecas gênicas, nomeadas de SCA (PINTO et al., 2004, 2006), SCB (MAR-
CONI et al., 2011), SCC (OLIVEIRA et al., 2009) e IISR (SINGH et al., 2013), e genômicas,
nomeadas de SMC (CORDEIRO; TAYLOR; HENRY, 2000) e CIR (RABOIN et al., 2006), de
cana-de-açúcar. Adicionalmente, seis marcadores SSRs originaram-se a partir de bibliotecas
gênicas de sorgo, nomeadas de SB, Xtxp, CNL e SvPEPCAA (BROWN et al., 1996; KONG;
DONG; HART, 2000; WANG et al., 2005). Ainda, polimorfismos de amplificação de regiões-
alvo (do inglês, target region amplification polymorphism – TRAP) foram avaliados utilizando
a combinação de um de três primers degenerados, nomeados de ARB1, ARB2 e ARB3 (LI;
QUIROS, 2001), com um de três primers fixos, desenhados a partir das sequências dos genes
sucrose phosphate synthase (SuPS) (ALWALA et al., 2006; CRESTE et al., 2010), caffeic acid
3-O-methyltransferase (COMT) e cinnamoyl-CoA reductase (CCR) (ANDRU et al., 2011). Para
ambas as técnicas, amplicons foram obtidos via PCR conforme os desenvolvedores, depois
separados em géis de poliacrilamida desnaturantes e, finalmente, corados por prata. A nomen-
clatura para esses locos envolveu as siglas das bibliotecas ou primers utilizadas na literatura,
acrescidas de um numeral indicando a posição da banda no gel.
2.3.2.3 Construção de mapa genético
Alelos codominantes foram codificados como a e b enquanto alelos nulos foram codifi-
cados como o e tratados como recessivos, conforme notação de Wu et al. (2002a). Marcadores
codominantes baseados em GBS acessaram segregação de três tipos de cruzamentos: ‘B3.7’
(ab × ab), ‘D1.10’ (ab × aa) e ‘D2.15’ (aa × ab). Adicionalmente, marcadores dominantes
baseados em géis acessaram mais três tipos de cruzamentos, que são aqueles usualmente uti-
lizados em mapas integrados de cana-de-açúcar: ‘C.8’ (ao × ao), ‘D1.13’ (ao × oo) e ‘D2.18’
(oo × ao). ‘D1’ e ‘D2’ referem-se a cruzamentos nos quais o loco marcador é heterozigoto (e,
portanto, informativo) somente para ‘SP80-3280’ ou para ‘RB835486’, respectivamente, ambos
segregando na razão de 1:1. ‘B3’ e ‘C’ referem-se a cruzamentos nos quais o loco é heterozigoto
(e simétrico) para ambos os genitores, segregando nas razões de 1:2:1 e 3:1, respectivamente.
Como o SuperMASSA permite predizer o(s) genótipo(s) parental(is) perdido(s) através dos
dados da população, todos os marcadores do tipo ‘B3’ puderam ser recuperados. Por outro
lado, marcadores dos tipos ‘D1’ e ‘D2’ somente foram recuperados quando existiam dados para

33
um genitor, pelo menos; na completa ausência de dados para os genitores, esse marcadores
foram descartados.
Para marcadores baseados em géis, testes de qui-quadrado (χ2) foram realizados de
acordo com as segregações esperadas inferidas por meio dos genótipos parentais. Para um nível
de significância global de 0,05, os p-valores foram corrigidos usando razão de falsa descobertas
(do inglês, false discovery rate – FDR) para testes não-dependentes (BENJAMINI; YEKUTI-
ELI, 2001) como implementado na função p.adjusted nativa do software R; locos distorcidos
foram excluídos das análises de ligação. Já para marcadores baseados em GBS, a segregação
já foi considerada quando da estimação de ploidia e dosagem pelo software SuperMASSA
(SERANG; MOLLINARI; GARCIA, 2012).
A análise de ligação foi realizada sobre os marcadores em dose-única não-duplicados e
filtrados para dados perdidos e distorção de segregação utilizando o pacote do R denominado
OneMap (v. 2.0-4) (MARGARIDO; SOUZA; GARCIA, 2007). Grupos de cosegregação ou de
ligação (GLs) foram obtidos considerando LOD score > 9,0 e fração de recombinação < 0,1
calculados a partir de testes de dois-pontos. Em seguida, ordenação dos marcadores foi obtida
utilizando busca exaustiva de ordem para grupos com até cinco marcadores (função compare),
ou ordenando um subconjunto informativo de cinco marcadores como um framework inicial
sobre o qual novos marcadores eram consecutivamente posicionados (função order.seq). Em
seguida, os marcadores inicialmente não-ligados foram novamente testados sobre os grupos
ordenados usando a função try.seq. Nessa etapa, outros marcadores considerados desligados
também foram testados: (i) marcadores alocados nos extremos dos grupos de ligação então
ordenados que distavam em mais de 20 centiMorgans (cM) do marcador mais próximo, e
(ii) marcadores pertencentes a grupos muito pequenos (com tamanhos inferiores a 1 cM ou
contendo apenas dois locos). A ordem nos grupos com mais de cinco marcadores foi, final-
mente, refinada considerando a saída da função ripple, que aplica algoritmo de mesmo nome
(LANDER; GREEN, 1987) dentro de uma janela deslizante de cinco marcadores (MOLLINARI
et al., 2009). Gráficos do tipo heat maps foram inspecionados visualmente, e correção manual
foi feita quando necessária ao longo da construção do mapa.
Grupos de hom(e)ologia (GHs) foram definidos de acordo com as referências genômicas
de cana-de-açúcar compartilhadas pelos marcadores baseados em GBS. Marcadores SSR basea-
dos em géis também foram utilizados para estabelecer conexões entre os grupos de ligação, uma
vez que poderiam gerar diferentes alelos compartilhando o mesmo par de primers. Finalmente,

34
grupos de sintenia (GSs) com sorgo reuniram os GLs com marcadores GBS originados quando
da utilização do genoma de sorgo como referência nas etapas de descoberta e genotipagem de
marcadores baseados em GBS utilizando o pipeline Tassel-GBS. Os grupos de ligação foram
desenhados no software MapChart (v. 2.2) (VOORRIPS, 2002), considerando as distâncias de
mapa estimadas no OneMap pela função de Kosambi.
2.4 Mapeamento de QTLs
A busca por QTLs em ambas as populações baseou-se em modelos de mapeamento de
múltiplos intervalos (do inglês, multiple interval mapping – MIM) univariado (KAO; ZENG;
TEASDALE, 1999) e multivariado (SILVA; WANG; ZENG, 2012). Esses modelos, atualmente
implementados no pacote do R denominando OneQTL (v. 1.0-1), estão disponíveis para avali-
ação de populações derivadas de genitores endogâmicos (caso das RILs de sorgo sacarino) e de
genitores não-endogâmicos (caso da população F1 segregante de cana-de-açúcar). O OneQTL
foi elaborado no Laboratório de Genética-Estatística, da ESALQ, onde o presente trabalho se
desenvolveu, sendo gentilmente cedido pelo Dr. Luciano Da Costa E Silva. O pacote será
liberado em breve.
Primeiro, as médias ajustadas conjunta e marginalmente para cada ambiente (anos, no
caso do sorgo, e combinações local-corte, no caso da cana) foram utilizadas em modelo MIM
univariado, permitindo a detecção de QTLs nas médias (conjuntas e marginais) de cada ambi-
ente, separadamente. Depois, as médias ajustadas marginalmente foram utilizadas em modelo
MIM multivariado, permitindo a detecção adicional de eventuais QTLs em interação com o
ambiente. A seguir, será descrito brevemente o modelo MIM multivariado para populações F1
segregantes que serve, com alguns detalhes adicionalmente descritos, para as análises univaria-
das e de populações de RILs, inclusive.
O pacote OneQTL tem implementado o seguinte modelo linear de múltiplos QTLs para
múltiplos ambientes (SILVA; WANG; ZENG, 2012; MARGARIDO, 2011):
yei = µe +
R∑r=1
(per xpir + qer xqir + der xdir) + εei (3)
em que yei é o fenótipo do indivíduo i (i = 1, 2, . . . , Ig) no ambiente e (e = 1, . . . , E), µe é
o intercepto para cada ambiente e, per e qer são os efeitos genéticos aditivos do QTL r (r =
1, 2, . . . ,R QTLs) dos genitores P e Q, respectivamente, e der é o efeito genético de dominância

35
do QTL r no ambiente e, e εi ∼ N(0,ΣE) é o resíduo; xpir e xqir são as variáveis indicadoras
dos genótipos do QTL r no indivíduo i, e xdir = xpir xqir, e correspondem às probabilidades
condicionais para os contrastes ortogonais definidos no cruzamento entre dois genitores P e Q,
com os sobrescritos de 1 a 4 indicando até quatro diferentes alelos (GAZAFFI et al., 2014).
Assim:
xpir =
1 se P1Q3
1 se P1Q4
−1 se P2Q3
−1 se P2Q4
xqir =
1 se P1Q3
−1 se P1Q4
1 se P2Q3
−1 se P2Q4
xdir = xpir xqir
Ao se considerar uma população de RILs obtida a partir de duas linhagens P e Q, com
os sobrescritos 1 e 2 indicando seus respectivos alelos, e xpir = 0, o modelo de mapeamento
acima reduz-se a um único efeito aditivo (qer, no caso) com a variável indicadora recebendo os
seguintes valores:
xqir =
1 se P1P1
−1 se Q2Q2
Nas populações aqui estudadas, convencionou-se que P representaria os genitores fe-
mininos (‘BR501-Brandes’ e ‘SP80-3280’ para as respectivas populações de cana e sorgo),
e Q representaria os genitores masculinos (‘BR505-Wray’ e ‘RB835486’ para as respectivas
populações de cana e sorgo).
Finalmente, se e = E = 1, o modelo de mapeamento torna-se univariado.
A construção do modelo de múltiplos QTLs baseou-se em busca forward de QTLs
putativos ao se testar a significância de seus efeitos principais sobre cada posição no genoma.
No caso do sorgo, essas posições corresponderam às próprias marcas, e, no caso da cana, essas
posições foram atribuídas por uma grade de busca de 1 cM, utilizando tamanhos de janelas
(do inglês, window size) de 1 quilo bases (kb) e 10 cM, respectivamente. As posições dos
QTLs no modelo foram refinadas a cada três ciclos de inclusão de QTLs. Finalmente, as
permanências dos efeitos principais foram testadas via eliminação backward. Nos modelos
multivariados, coeficientes de regressão aparentemente não-relacionados (do inglês, seemingly
unrelated regressions) (ZELLNER, 1962) resultantes da eliminação backward tiveram seus
efeitos estimados e foram marcados como não-significativos, ao invés de mantê-los com efeitos
nulos.
Para o mapeamento em sorgo sacarino, foram utilizados os níveis de significância

36
genome-wide baseados em score (ZOU; ZENG, 2008; SILVA; ZENG, 2010) de 0,20 e 0,05
nas respectivas etapas de entrada (busca forward) e saída (eliminação backward) de QTLs
em cada modelo. Já para o mapeamento em cana-de-açúcar, diversos níveis de significância
baseados em score para busca forward foram testados sob uma grade que variou de 0,10 a 0,90,
a uma razão de progressão aritmética de 0,10. Nos casos em que os modelos progressivamente
parametrizados apresentavam indícios de problemas numéricos (não convergência), o processo
de busca forward era abortado e o menor valor de significância permitindo a adição de um maior
número de QTLs era escolhido. O nível de significância utilizado na eliminação backward
foi estabelecido como um quarto do valor utilizado na busca forward. Níveis de significância
superiores a 0,20 foram permitidos no mapeamento desta população por conta da existência de
diversos fatores limitantes quando da descoberta de regiões sugestivas para controle dos carac-
teres quantitativos na espécie. Dentre esses fatores, são enfatizadas a complexidade do genoma
da cana e as abordagens apenas aproximadas a partir de espécies diploides para construção de
mapas de ligação e para mapeamento de QTLs.
A aproximação de Haley-Knott (HALEY; KNOTT, 1992) foi utilizada para o ajuste
do modelo e o método da máxima verossimilhança foi adotado para a estimação dos efeitos
genéticos e do componente de variância residual. Também foram obtidos os valores de LOD
scores associados a todas as posições pesquisadas, assim como os intervalos de confiança LOD−
1,5 em relação às posições mais prováveis dos QTLs. Adicionalmente, p-valores empíricos
para os efeitos de QTLs no modelo foram obtidos via reamostragem baseada em score (SILVA;
WANG; ZENG, 2012).
Adicionalmente, análise de marcadores individualmente (SMA) foi implementada no
software R para os locos marcadores de cana-de-açúcar. Assim, as médias ajustadas conjuntas
e marginais foram utilizadas como variáveis independentes, enquanto os genótipos dos marca-
dores recodificados (0 para a, 1 para ab e 2 para b) foram utilizados como variável dependente
em modelos de regressão simples. Os p-valores dos efeitos dos marcadores individuais sobre
os caracteres foram acessados via teste t e considerados significativos quando p < 0,01.
Locos resultantes dos mapeamentos uni- e multivariados de QTLs para as populações
de sorgo sacarino e cana-de-açúcar foram agrupados, dentro de cada população, com base
na proximidade com que apareceram nos cromossomos ou nos GLs, respectivamente. Nesse
sentido, foram utilizados uma janela de 10 Mb para agrupar os QTLs de sorgo, tamanho médio
pelo qual se estendiam os efeitos das marcas, e os intervalos de confiança para agrupar os

37
QTLs de cana. Evidentemente, no caso do sorgo, dois QTLs distintos para uma mesma análise
mapeados dentro da janela de 10 Mb foram considerados diferentes, e foram eventualmente
agrupados com QTLs mais próximos à esquerda ou à direita, dentro dessa janela.
Finalmente, as posições dos QTLs dos caracteres similares avaliados nas populações de
mapeamento de cada espécie foram comparadas tomando por base os marcadores da cana e as
regiões cromossômicas do sorgo em que os testes para existência de QTLs foram significativos.
Inferências a respeito da conservação de locos que controlam caracteres de herança complexa
foram realizadas com base nessas observações. A comparação entre os QTLs mapeados nas
duas populações foi realizada considerando o alinhamento dos GLs e dos marcadores não
mapeados da cana contra o genoma do sorgo utilizando, exclusivamente, as informações de
SNPs e indels derivados de GBS, os quais já indicavam diretamente sua posição no genoma
do sorgo. O genoma do sorgo utilizado foi o mesmo que serviu de referência para mapear os
polimorfismos na população de sorgo sacarino.

38

39
3 RESULTADOS E DISCUSSÃO
Para ambas as populações de sorgo sacarino e de cana-de-açúcar, foram realizadas aná-
lises de dados fenotípicos e genotípicos. Fazem parte da “análise fenotípica” a análise via
modelos mistos dos dados fenotípicos, a obtenção de componentes de variância e herdabilida-
des, e o cálculo de correlações entre caracteres. Já a “análise genotípica” contém a descrição
dos dados de marcadores obtidos para ambas as populações, assim como as classificações para
ploidias e dosagens dos marcadores quantitativos e a construção do mapa genético para cana,
especificamente. Finalmente, também são apresentados os resultados dos mapeamentos de
QTLs para as populações, separadamente e, então, esses resultados são comparados.
3.1 Sorgo Sacarino
3.1.1 Análise fenotípica
Avaliações fenotípicas em campo foram obtidas e analisadas para três anos agrícolas
(2011, 2012 e 2013) para a população de sorgo sacarino. Os dados foram obtidos para quatro
caracteres de interesse bioenergético: altura de colmos (ALT, em m), produção de massa verde
(PMV, em ton · ha−1), pol de caldo (POL, em %) e conteúdo de fibra (FIB, em %).
Considerando a abordagem de modelos mistos, inicialmente estabeleceu-se a parte ale-
atória do modelo completo a partir da seleção das mais apropriadas matrizes de VCOV. Nesta
seleção, adotou-se os valores de AIC para comparação das matrizes de VCOV genéticas, en-
quanto os valores de BIC foram principalmente considerados na comparação de matrizes de
VCOV residuais. Esta escolha determinou a possibilidade de revelar o máximo de informação
sobre eventual diferença entre as variâncias genéticas ao longo dos anos, assim como sobre a
ocorrência de importantes covariâncias entre os mesmos. Isto se justifica pelo fato de o BIC
penalizar a inclusão de parâmetros em relação ao AIC (PASTINA et al., 2012). Por exemplo,
para os caracteres PMV e FIB, modelos não-estruturados (UNST) foram selecionados para as
matrizes de VCOV genéticas GM, enquanto para ALT, matriz simétrica composta heterogênea
(CSHet) foi preferida (Tabela 3). Essa heteroscedasticidade significa, em linhas gerais, que há
interação genótipo-ambiente (MALOSETTI; RIBAUT; VAN EEUWIJK, 2013), cujo entendi-
mento é importantíssimo no contexto dos programas de melhoramento de espécies vegetais.
Apenas para o caráter POL, matriz com variâncias homogêneas e covariâncias idênticas (uni-
forme – UNIF) foi selecionada. Para a composição das matrizes de VCOV residuais, diversas

40
estruturas foram selecionadas, sendo aquelas menos complexas geralmente preferidas pelo BIC.
Tabela 3 – Modelos examinados para diferentes matrizes de variância-covariância para efeitos genéti-cos dentro de M anos (GA
M×M) na análise fenotípica de população de mapeamento de sorgosacarino
Caráter ALT PMV POL FIBnPAR
Estrutura AIC BIC AIC BIC AIC BIC AIC BIC
GM = GAM×M
ID 51,8 68,6 14435,5 14452,2 8263,6 8280,4 7433,5 7450,2 1UNIF −154,5 −132,1 14375,3 14397,7 8107,8 8130,1 7374,6 7397,0 2DIAG 37,4 65,3 14339,7 14367,7 8264,8 8292,7 7335,2 7363,1 3AR1 −145,9 −123,6 14374,3 14396,7 8116,7 8139,1 7369,6 7391,9 2CSHet −180,6 −147,0 14269,1 14302,6 8107,9 8141,4 7262,6 7296,2 4UNST −179,3 −134,6 14266,5 14311,2 8111,5 8156,2 7258,3 7303,0 6Notas: nPAR é o número de parâmetros para as matrizes identidade (ID), uniforme (UNIF), diagonal (DIAG),auto-regressiva de 1a ordem (AR1), simétrica composta (CSHet) e não-estruturada (UNST); em negrito, osmenores valores para os critérios de informação de Akaike (AIC) e Bayesiano (BIC)
Mantendo-se as estruturas de VCOV selecionadas, foram testadas as significâncias dos
efeitos de interação entre anos e genitores. Para todos os caracteres, essas interações foram
não-significativas (p > 0,05) e, portanto, foram retiradas do modelo final. Esta proposta de
análise prevê que as significâncias de efeitos fixos podem variar dadas as estruturas aleatórias
selecionadas no então modelo de médias completo sob análise (GALECKI; BURZYKOWSKI,
2013).
Foram obtidas importantes informações sobre as correlações existentes entre as variáveis
analisadas (Figura 1). A maioria das correlações foram elevadas e significativas, e pelo menos
a correlação significativa entre ALT e POL também já havia sido verificada (RITTER et al.,
2008). Correlações negativas envolveram o caráter FIB, sendo a relação oposta com POL não-
significativa, como Ming et al. (2002) observaram em uma das populações de mapeamento
de cana-de-açúcar estudadas por eles. Já as significativas correlações negativas de FIB com
caracteres de produção, como ALT e PMV, representa um desafio para o melhoramento de
sorgo voltada para a produção de biomassa, por exemplo. Correlações significativas e positivas
indicam que seleção indireta pode ser praticada; no entanto, essa seleção pode ficar prejudi-
cada quando dois caracteres de interesse apresentam correlação negativa. O entendimento da
arquitetura genética desses caracteres pode auxiliar na tomada de decisões em programas de
melhoramento.
As estimativas de componentes de variância e de herdabilidades são mostradas na Tabela

41
ALT20 30 40
0,71*** ***0,46
11 13 15
2.2
2.8
***−0,36
2030
40 PMV**0,30 **−0,22
POL
46
811
−0,12ns
2.2 2.8
1113
15
4 6 8 11
FIB
Figura 1 – Histogramas das médias ajustadas para cada caráter (na diagonal), gráficos de dispersão(abaixo da diagonal) e valores de correlações genéticas (acima da diagonal) entre pares decaracteres avaliados em população de mapeamento de sorgo sacarino; p-valores foram cor-rigidos para um nível de significância global a 5% (∗∗p < 0,01, ∗∗∗p < 0,001 e ns é não-significativo)
4. Os coeficientes de herdabilidade a nível de média classificam-se de moderados a altos,
variando de 0,592 para FIB a 0,813 para ALT. Esses valores mostram que boa parte da variação
fenotípica observada pode ser atribuída a diferenças ao nível genotípico, e são semelhantes
àqueles citados na literatura (RITTER et al., 2008; SHIRINGANI; FRISCH; FRIEDT, 2010).
Os coeficientes de variação podem ser considerados adequados para o experimento, variando
de 6,3% para ALT a 25,8% para PMV.
Tabela 4 – Estimativas de médias, componentes de variâncias genética (σ2G) e fenotípica
(σ2P), e coeficientes de variação (CV, em porcentagem) e de herdabilidade
(h2) aos níveis de plantas e de médias para caracteres avaliados em populaçãode mapeamento de sorgo sacarino derivada do cruzamento entre as cultivares‘Brandes’ (P1) e ‘Wray’ (P2)
Caráter Médias P1 P2 σ̂2G σ̂2
P CV (%) h2plantas h2
médias
ALT 2,69 2,92 2,46 0,040 0,087 6,3 0,462 0,813PMV 27,47 33,12 21,81 19,050 93,000 25,8 0,205 0,603POL 7,25 10,51 3,99 1,693 4,670 19,8 0,363 0,762FIB 12,78 12,41 13,14 0,538 2,651 9,8 0,203 0,592

42
3.1.2 Análise genotípica
Leituras curtas foram obtidas a partir do sequenciamento de três bibliotecas de plex 96
produzidas a partir da digestão pela enzima ApeKI de DNA genômico de 272 RILs da população
de mapeamento de sorgo e de três replicatas dos genitores. Foram produzidas mais de 520
milhões de leituras no total, ou 173 milhões de leituras por lane, em média. Do total, cerca de
470 milhões (∼90%) de leituras, ou 1,6 milhão de leituras por amostra, em média, continham
uma sequência barcode e o sítio remanescente da ação da enzima. Posteriormente, 3.834.717
tags de 64 pb foram identificadas e, então, alinhadas contra o genoma do sorgo. A razão total
de alinhamento utilizando Bowtie 2 foi de 79,95%, resultante das 1.880.627 (49,04%) tags
alinhadas exatamente uma vez e das 1.185.194 (30,91%) alinhadas mais de uma vez. Um total
de 768.896 (20,05%) de tags não se alinharam, portanto.
O número total de variantes bialélicas detectadas na população de mapeamento de
sorgo sacarino utilizando o pipeline Tassel-GBS foi de 461.241 e, dependendo do cromos-
somo, variou de 31.969 (cromossomo 8) a 71.878 (cromossomo 1). Deste total, SNPs foram
mais frequentes que indels a uma razão de 3,0:1. Em relação aos SNPs, transversões (subs-
tituições purina-pirimidina) foram identificadas 1,3 vezes mais que transições (substituições
purina-purina ou pirimidina-pirimidina), quando o contrário (mais transições) era esperado. No
entanto, essa expectativa deve ser observada mais rigorosamente em genomas completamente
sequenciados. Isto porque esse viés pode estar relacionado à relativa facilidade de desaminação
de resíduos de citosinas metiladas em regiões adjacentes a guaninas (regiões CpG), levando à
transição para timinas (KELLER; BENSASSON; NICHOLS, 2007). Como aqui foi utilizada
enzima sensível à metilação para construção das bibliotecas, acredita-se que, ao se evitar essas
regiões, indiretamente também evitou-se a detecção de um maior número de transições. Infeliz-
mente, estudos envolvendo GBS falham em contabilizar a razão entre transições-transversões
propriamente, apesar de seu potencial uso na conferência da qualidade do sequenciamento, por
exemplo.
Antes de realizar qualquer filtragem dos dados, genótipos ausentes foram imputados
utilizando o software Npute (ROBERTS et al., 2007), e determinou a existências de diferentes
tamanhos de janelas de imputação entre os cromossomos, variando de 12 (cromossomo 7) a 18
(cromossomo 4) bases e fornecendo uma acurácia média de 98,4%. Finalmente, utilizando uma
MAF de 0,40, 360.950 locos foram excluídos, permanecendo 100.291 locos. Em média, cada
cromossomo da população continha 10 mil marcadores, variando de 4.086 (cromossomo 8) a

43
14.326 (cromossomo 2). No cenário de uma população experimental de autógama típica, como
é o sorgo, esses números podem ser considerados excelentes. De fato, blocos de desequilíbrio
relativamente grandes podem ser evidenciados pela cosegregação de inúmeros SNPs, indicando
que alguns deles são redundantes.
3.1.3 Mapeamento de QTLs
Um total de 53 QTLs foram descobertos utilizando as abordagens de MIM uni- e mul-
tivariado para os quatro caracteres avaliados na população de mapeamento de sorgo sacarino.
O resumo dessas análises estão apresentados na Tabela 5. Os resultados detalhados em relação
aos efeitos ao longo dos anos e suas significâncias estatísticas estão presentes nos Apêndices A
e B (Tabelas 12 e 13).
Ambos os caracteres agronômicos ALT e PMV tiveram 12 QTLs detectados cada, os
quais se distribuíram ao longo de oito cromossomos do sorgo (exceto cromossomos 5 e 7 para
ALT, e cromossomos 1 e 5 para PMV). Para ALT, os seis QTLs detectados na análise multivari-
ada foram confirmados nas análises univariadas; já para PMV, a análise univariada acrescentou
mais duas regiões, além das outras quatro também detectadas na análise multivariada. Para
os dois caracteres, a existência de QTLs específicos para determinados anos ou para a média
dos três anos foram adicionalmente evidenciados. Interessantemente, dentre os dois caracteres
industriais, POL contribuiu com a descoberta de 17 QTLs, enquanto FIB, com a descoberta
de 12 QTLs, também distribuídos ao longo de oito cromossomos (exceto cromossomos 2 e 4
para POL, e cromossomos 5 e 7 para FIB). De fato, entre os caracteres avaliados, POL revelou-
se o mais fenotipicamente constrastante entre os genitores, gerando, por consequência, ampla
variação de fenótipos entre as RILs. Ou seja, por construção, a população de mapeamento é
amplamente favorável ao mapeamento para este caráter. Para POL, as análises uni- e multiva-
riadas acrescentaram, exclusiva e respectivamente, seis e duas novas regiões ao total de nove
QTLs evidenciados nas duas análises. Já para FIB, em adição aos oito QTLs compartilhados
por ambas as estratégias, cada qual indicou a existência de controle genético para esse caráter
em mais duas regiões distintas.
O caráter ALT já havia sido investigado em trabalho anterior (RITTER et al., 2008)
utilizando as estratégias de SMA e CIM, por meio das quais evidências para a existência de
efeitos principais nos cromossomos 1, 3 e 6 também foram encontradas. Em adição, os autores
apontam QTL no cromossomo 5. Aqui, também aparecem putativos locos controlando o caráter

44
Tabela 5 – Cromossomos (Cr.) e posições (em mega bases – Mb) dos QTLs detecta-dos em população de mapeamento de sorgo sacarino utilizando modelosmapeamento de múltiplos intervalos (MIM)
Caráter QTL Cr. Posição (Mb) Caráter QTL Cr. Posição (Mb)
ALT 1 1 62,6-67,8 POL 4 3 13,5-15,92 2 26,3 5 3 51,4-57,83 3 61,3 6 3 64,9-68,14 3 68,3 7 5 2,9-4,85 4 54,2 8 5 4,8-5,66 6 18,4 9 6 43,2-46,27 6 42,2-42,6 10 6 52,5-53,58 8 45,1 11 7 48,39 9 2,6-10,1 12 8 14,0
10 9 47,8-50,4 13 8 37,011 10 11,1 14 8 53,412 10 43,3-55,7 15 9 2,9-9,5
PMV 1 2 4,9 16 9 10,12 2 62,7 17 10 5,13 3 21,3 FIB 1 1 59,2-61,04 4 1,3 2 2 1,75 4 57,0-57,3 3 3 60,66 6 17,5 4 3 68,1-69,27 6 40,1-41,8 5 4 1,4-9,68 7 60,0 6 4 65,19 8 5,4 7 6 34,9
10 9 5,4 8 6 46,2-47,611 10 12,9-13,6 9 8 37,0-37,412 10 38,3-52,5 10 9 5,3
POL 1 1 3,9-6,6 11 10 2,52 1 51,4-63,1 12 10 56,4-58,53 3 0,9-1,8
nos cromossomos 2, 4, 8, 9 e 10.
Outro exemplo interessante deste conjunto de resultados é a região terminal do cromos-
somo 3, com efeitos importantes (vide LOD conjunto da análise multivariada, LODc) para os
caracteres relacionados a açúcar, como aqueles das regiões POL-6 (LODc = 19,45) e FIB-2
(LODc = 13,50). Esse controle genético para conteúdo de açúcar em sorgo já foi investigado
em estudos anteriores. Guan et al. (2011), trabalhando com população de RILs derivada do
cruzamento entre ‘Shihong137’ (sorgo granífero) e ‘L-Tian’ (sorgo sacarino) e mapa com 118
marcadores SSRs, encontraram quatro QTLs para sólidos solúveis totais ou brix (nos cromosso-
mos 1, 2, 3 e 7), sendo aquele do cromossomo 3 estável para os dois ambientes avaliados. Além

45
disto, painel de sorgo, majoritariamente sacarino, detectou região genômica que é, potencial-
mente, idêntica àquela aqui detectada (MURRAY et al., 2009). Finalmente, Ritter et al. (2008)
também obtiveram QTLs para POL, com destaque para aqueles existentes nos cromossomos
1, 5, 6 e 10, sendo que o cromossomo 3 também aparece para o caráter conteúdo de açúcar.
Adicionalmente, regiões sugestivas, como aquelas evidenciadas nos cromossomos 8 e 9, até
então não relatadas, devem ser melhor investigadas, visando à adição de mais informações
sobre a arquitetura genética desse caráter.
3.2 Cana-de-Açúcar
3.2.1 Análise fenotípica
Como para a população de sorgo sacarino, foram realizadas avaliações fenotípicas em
campo para a população de cana-de-açúcar para os seguintes caracteres: altura de colmos (ALT,
em m), toneladas de colmos por hectare (TCH, em ton · ha−1), pol de caldo (POL, em %) e
conteúdo de fibra (FIB, em %). Esses experimentos foram realizados em dois locais e durante
três anos consecutivos (2010, 2011 e 2012).
As estruturas mais apropriadas para as matrizes de VCOV genética foram seleciona-
das de acordo com os valores de AIC (Tabelas 6); nesses casos, os valores de BIC foram
completamente concordantes. A matriz de VCOV que considera a estratégia de agrupamento
por combinação local-corte foi preferida para o caráter ALT, enquanto produtos de matrizes
considerando os agrupamentos naturais de locais e cortes foram preferidos pelos demais ca-
racteres (TCH, POL e FIB), situações nas quais menos parâmetros necessitaram ser estimados.
Diversas estruturas para os caracteres não puderam ser ajustadas porque não foi possível estimar
os parâmetros de variância-covariância das matrizes via REML, e aparecem como NAs na
Tabela 6. Possivelmente, o desbalanceamento causado pela ausência de cortes para POL e FIB
também pode ter impedido a seleção de matrizes mais econômicas na parametrização, como
a auto-regressiva de 1a ordem (AR1), quando não-estruturadas (UNST) foram selecionadas.
Para todos os caracteres, foi possível observar a heterogeneidade de efeitos genéticos entre os
níveis dos fatores local e corte, sejam eles agrupados em ambientes ou não. De modo geral,
esse comportamento era esperado. Em Pastina et al. (2012), resultados semelhantes foram
observados para TCH, FIB e POL.
Para o caráter ALT, que melhor ajustou matriz de VCOV genética como combinação
fatorial de local-corte, a matriz de VCOV residual também foi avaliada utilizando ambientes

46
Tabela 6 – Modelos examinados para diferentes matrizes de variância-covariância para efeitos genéticosdentro de P = M · N combinações local-corte (GE
P×P) ou de N locais (GLN×N) e M cortes
(GAM×M) na análise fenotípica de população de mapeamento de cana-de-açúcar
Caráter ALT TCHnPAR
POL FIBnPAR
Estrutura AIC BIC AIC BIC AIC BIC AIC BIC
GP = GEP×P
ID 1507,0 1525,9 41149,3 41168,1 1 7348,4 7365,4 6325,7 6342,7 1UNIF 1339,3 1364,4 40915,5 40940,6 2 NA NA NA NA -DIAG 1430,5 1474,5 41127,6 41171,6 5 7348,0 7382,0 6311,7 6345,6 4CSHet 1285,2 1335,4 40902,1 40952,4 6 NA NA NA NA -UNST NA NA NA NA - 7110,7 7178,7 NA NA 10GP = GL
N×N ⊗GAM×M
UNST ⊗ ID 1361,1 1392,5 36746,6 36778,0 4 7261,8 7290,1 6192,9 6221,2 4UNST ⊗ UNIF 1301,1 1338,8 NA NA 5 NA NA NA NA -UNST ⊗ DIAG 1347,0 1391,0 36749,6 36793,6 6 7263,6 7297,6 6190,3 6224,3 5UNST ⊗ AR1 1298,4 1336,1 36511,5 36549,2 5 7119,3 7153,2 6069,5 6103,5 5UNST ⊗ CSHet NA NA NA NA - NA NA NA NA -UNST ⊗ UNST NA NA NA NA - 7109,1 7148,8 6050,5 6090,1 6Notas: nPAR é o número de parâmetros para as matrizes identidade (ID), uniforme (UNIF), diagonal (DIAG), auto-regressiva de 1a ordem (AR1), simétrica composta (CSHet) e não-estruturada (UNST) ou produto direto delas; emnegrito, os menores valores para os critérios de informação de Akaike (AIC) e Bayesiano (BIC); NA significa quenão foi possível ajustar o modelo especificado, usando o software GenStat
(E) como fator de agrupamento. Nesse caso, a matriz REP×P ajustou uma estrutura uniforme
(UNIF). Já para os caracteres TCH, POL e FIB, que melhor ajustaram matrizes de VCOV
genética como produto direto de matrizes, as matrizes R de VCOV residuais também foram
avaliadas utilizando locais (L) e cortes (A) como fatores de agrupamento, separadamente. Em
relação às matrizes RLN×N para locais, foram selecionadas matrizes identidade (ID) para POL
e FIB, e matriz UNST para TCH. Para as matrizes RAM×M para cortes, matrizes UNIF foram
selecionadas para TCH e POL, enquanto matriz simétrica composta (CSHet) foi selecionada
para FIB. Finalmente, matrizes diagonais (DIAG) foram selecionadas para RRK×K em todos os
casos (dados não apresentados).
Posteriormente, para cada caráter investigado, a seleção da estrutura de média (parte fixa
do modelo) considerando as matrizes selecionadas anteriormente e utilizando a estatística de
Wald determinou que os efeitos de interação entre cortes e testemunhas para TCH, POL e FIB,
e entre locais e testemunhas para FIB foram não-significativas (p > 0,05); já ALT apresentou
todos os efeitos de interação estatisticamente significativos. Os efeitos não-significativos foram
retirados do modelo e, em seguida, as médias ajustadas (conjunta e marginalmente) foram
obtidas.

47
As dispersões das médias ajustadas entre os pares de caracteres avaliados evidenciaram
correlações não-significativas (Figura 2). Essa ausência de correlação contraria em parte a
tendência observada para dados relatados para a cultura (HOARAU et al., 2002; AITKEN et
al., 2008), muito embora mesmo a ausência de correlação entre POL e FIB, por exemplo, já
tenha sido verificada (MING et al., 2002).
ALT70 90 120 11.0 13.0
2.1
2.4
7090
120
TCH
POL
1618
202.1 2.411
.013
.0
16 18 20
FIB
−0,06ns
−0,06ns
0,11ns
0,02ns0,15ns
−0,05ns
Figura 2 – Histogramas das médias ajustadas para cada caráter (na diagonal), gráficos de dispersão(abaixo da diagonal) e valores de correlações genéticas (acima da diagonal) entre pares decaracteres avaliados em população de mapeamento de cana-de-açúcar; p-valores foram corri-gidos para um nível de significância global a 5% (ns é não-significativo)
As estimativas das variâncias genética (σ̂2G) e fenotípica (σ̂2
P) para cada caráter, bem
como os coeficientes de herdabilidade (H2), foram obtidas a partir dos modelos de ID para os
efeitos aleatórios genéticos e residuais (Tabela 7). Os coeficientes de herdabilidade a nível de
médias classificam-se como altos, variando de 0,781 para ALT a 0,844 para FIB. Esses valores
mostram que boa parte da variação fenotípica observada pode ser atribuída a diferenças ao
nível genotípico, e são comparáveis àqueles observados na literatura (HOARAU et al., 2002;
AITKEN et al., 2008). Em comparação à população de sorgo sacarino, as herdabilidades dos
caracteres avaliados na população de cana-de-açúcar foram relativamente maiores. Parte disto
é justificado pelo tipo de delineamento, com vários locais, cortes e repetições para cana, o que
aumenta a qualidade do experimento e diminui a variância residual. A qualidade do experimento
pode ser também verificada pelos valores razoáveis de coeficientes de variação, cujos valores
foram de 6,4% para POL a 23,5% para TCH. Outra possível explicação refere-se ao menor
número de QTLs segregando nesta população, já que os genitores não são muito divergentes.

48
Tabela 7 – Estimativas de médias, componentes de variâncias genética (σ2G) e fenotípica (σ2
P),e coeficientes de variação (CV, em porcentagem) e de herdabilidade (H2) aosníveis de plantas e de médias para caracteres avaliados em população de mapea-mento de cana-de-açúcar derivada do cruzamento entre as variedades ‘SP80-3280’(P1) e ‘RB835486’ (P2)
Caráter Médias P1 P2 σ̂2G σ̂2
P CV (%) H2plantas H2
médias
ALT 2,31 2,36 2,24 0,017 0,090 11,7 0,193 0,781TCH 93,61 95,84 85,48 582,000 2120,000 23,5 0,275 0,836POL 18,44 18,63 18,85 0,889 2,273 6,4 0,391 0,837FIB 12,26 12,04 12,34 0,592 1,465 7,7 0,404 0,844
3.2.2 Análise genotípica
3.2.2.1 Estratégias de descoberta de polimorfismos utilizando GBS
Leituras curtas foram obtidas a partir do sequenciamento de bibliotecas de plex 96 pro-
duzidas a partir da digestão pela enzima PstI de DNA genômico de 151 indivíduos da população
de mapeamento de cana e de três replicatas dos genitores. Mais de 1,6 bilhão de leituras foram
produzidas no total, com 268 milhões leituras por lane, em média. Cerca de 600 milhões de
leituras (∼41% do total), ou 4,2 milhões de leituras por amostra, em média, continham uma
sequência barcode e o sítio de corte remanescente da ação da enzima. Como alguns indivíduos
das primeira e segunda bibliotecas não pertenciam à população de mapeamento, leituras extras
contendo barcodes não foram identificadas. De cada biblioteca duplamente sequenciada, ∼2
milhões, ∼106 milhões e ∼222 milhões de leituras com barcodes foram identificadas, em média.
Do total de leituras com barcodes, mais de 3,1 milhões de tags foram levadas para
alinhamento contra quatro pseudo-referências diferentes. Três das quatro referências foram
originadas de bibliotecas de DNA ou RNA de cana-de-açúcar. O genoma metil-filtrado da cana
(GRATIVOL et al., 2014) resultou na maior taxa de alinhamento, com 87,94% (2.729.457) de
tags alinhadas. Em relação às referências baseadas em RNA, 38,53% (1.195.723) e 23,89%
(741.537) das tags foram alinhadas contra transcriptoma obtido via RNA-Seq (CARDOSO-
SILVA et al., 2014) e sequências do projeto SUCEST (VETTORE et al., 2003), respectivamente.
Entretanto, suas taxas de alinhamento não-único diferiram bastante (Tabela 8), em acordo com
os achados de Vicentini et al. (2012) sobre a redundância existente na base de dados do SU-
CEST. Finalmente, a referência do genoma do sorgo teve um total de 42,29% (1.312.661) de
tags alinhadas (Tabela 8).

49
Tabela 8 – Alinhamento utilizando Bowtie 2 de 3.103.708 tags de GBS em população de mapeamentode cana-de-açúcar em valores absolutos e relativos (entre parênteses). O total de tagsalinhadas é a soma dos alinhamentos único e não-único
Pseudo-referênciasTags
não-alinhadasTags alinhadas
Total Alinhamentoúnico
Alinhamentonão-único
Genoma metil-filtradoda cana
374.251(12,06%)
2.729.457(87,94%)
1.823.623(58,76%)
905.834(29,18%)
Genoma do sorgo 1.791.047(57,71%)
1.312.661(42,29%)
1.060.705(34,17%)
251.956(8,12%)
Transcriptoma viaRNA-Seq da cana
1.907.985(61,47%)
1.195.723(38,53%)
1.099.697(35,43%)
96.026(3,10%)
Sequências do projetoSUCEST
2.362.171(76,11%)
741.537(23,89%)
256.450(8,26%)
485.087(15,63%)
De 39.058 a 151.755 variantes bialélicas foram identificadas dependendo da refe-
rência (Tabela 9) utilizando o pipeline Tassel-GBS. Para todas as referências, SNPs foram
mais frequentes que indels a uma razão de 2,6:1, em média, próxima daquela encontrada
para a população de sorgo. Em relação aos SNPs, transições (substituições purina-purina
ou pirimidina-pirimidina) foram identificadas 1,4 vezes mais que transversões (substituições
purina-pirimidina), concordando, em parte, com a razão esperada para análise de genoma com-
pleto. Como foi relatado, a enzima PstI utilizada é apenas parcialmente sensível à metilação.
Todos esses dados iniciais, entretanto, estavam inflados por dados perdidos. Como não
há metodologia de imputação de dados perdidos no cenário de poliploides complexos como
a cana, cerca de 12% dos marcadores com mais de 25% de dados perdidos foram excluídos
nas chamadas de cada referência (Tabela 9). Além disso, locos com baixa profundidade ou
ambíguos também estavam amplamente presentes. Então, ∼64% de locos sub-representados
também foram excluídos considerando uma contagem média mínima de 50 leituras para o alelo
de referência. Embora um elevado número de locos tenha sido removido, de 8.885 a 38.378
de marcadores polimórficos com profundidades elevadas e filtrados para genótipos perdidos
foram levados para análise no software SuperMASSA. Redundâncias remanescentes foram
investigadas somente após essa análise.
3.2.2.2 Estimação de ploidia e dosagem alélica
Níveis de ploidia variando de 2 a 20 foram avaliadas no software SuperMASSA utili-
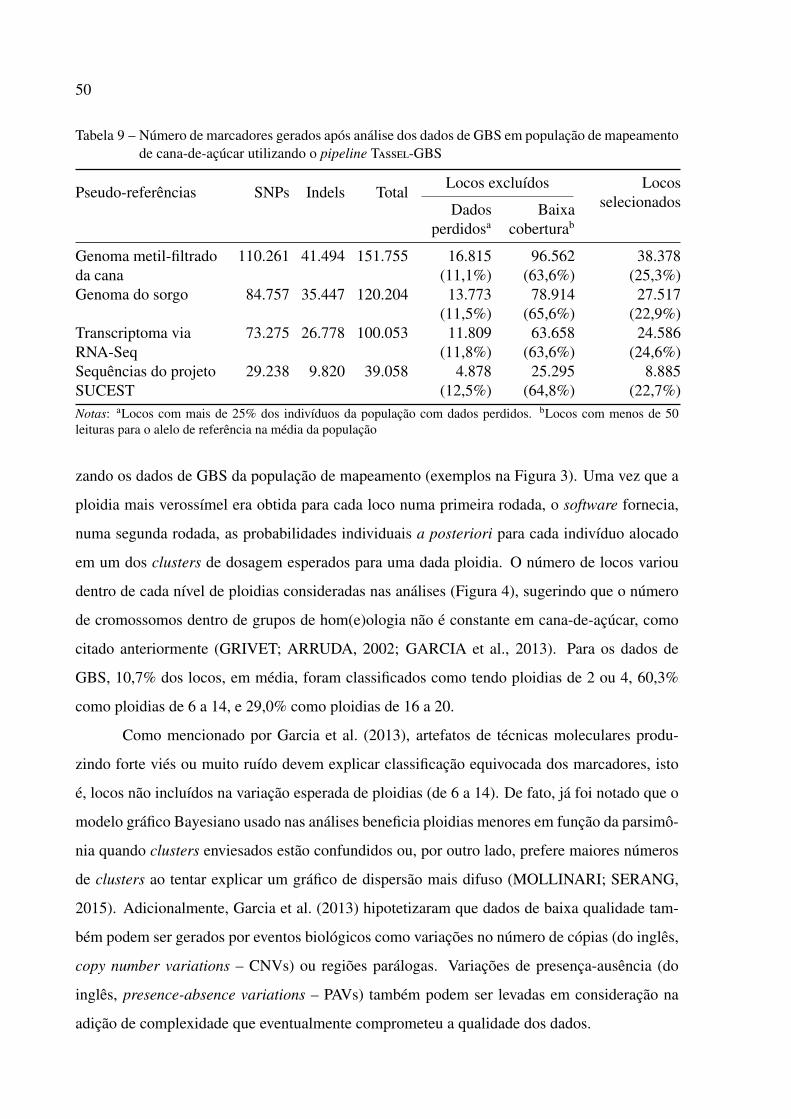
50
Tabela 9 – Número de marcadores gerados após análise dos dados de GBS em população de mapeamentode cana-de-açúcar utilizando o pipeline Tassel-GBS
Pseudo-referências SNPs Indels TotalLocos excluídos Locos
selecionadosDadosperdidosa
Baixacoberturab
Genoma metil-filtradoda cana
110.261 41.494 151.755 16.815(11,1%)
96.562(63,6%)
38.378(25,3%)
Genoma do sorgo 84.757 35.447 120.204 13.773(11,5%)
78.914(65,6%)
27.517(22,9%)
Transcriptoma viaRNA-Seq
73.275 26.778 100.053 11.809(11,8%)
63.658(63,6%)
24.586(24,6%)
Sequências do projetoSUCEST
29.238 9.820 39.058 4.878(12,5%)
25.295(64,8%)
8.885(22,7%)
Notas: aLocos com mais de 25% dos indivíduos da população com dados perdidos. bLocos com menos de 50leituras para o alelo de referência na média da população
zando os dados de GBS da população de mapeamento (exemplos na Figura 3). Uma vez que a
ploidia mais verossímel era obtida para cada loco numa primeira rodada, o software fornecia,
numa segunda rodada, as probabilidades individuais a posteriori para cada indivíduo alocado
em um dos clusters de dosagem esperados para uma dada ploidia. O número de locos variou
dentro de cada nível de ploidias consideradas nas análises (Figura 4), sugerindo que o número
de cromossomos dentro de grupos de hom(e)ologia não é constante em cana-de-açúcar, como
citado anteriormente (GRIVET; ARRUDA, 2002; GARCIA et al., 2013). Para os dados de
GBS, 10,7% dos locos, em média, foram classificados como tendo ploidias de 2 ou 4, 60,3%
como ploidias de 6 a 14, e 29,0% como ploidias de 16 a 20.
Como mencionado por Garcia et al. (2013), artefatos de técnicas moleculares produ-
zindo forte viés ou muito ruído devem explicar classificação equivocada dos marcadores, isto
é, locos não incluídos na variação esperada de ploidias (de 6 a 14). De fato, já foi notado que o
modelo gráfico Bayesiano usado nas análises beneficia ploidias menores em função da parsimô-
nia quando clusters enviesados estão confundidos ou, por outro lado, prefere maiores números
de clusters ao tentar explicar um gráfico de dispersão mais difuso (MOLLINARI; SERANG,
2015). Adicionalmente, Garcia et al. (2013) hipotetizaram que dados de baixa qualidade tam-
bém podem ser gerados por eventos biológicos como variações no número de cópias (do inglês,
copy number variations – CNVs) ou regiões parálogas. Variações de presença-ausência (do
inglês, presence-absence variations – PAVs) também podem ser levadas em consideração na
adição de complexidade que eventualmente comprometeu a qualidade dos dados.

51
200 400 600 8000
●
●
●●
●
●
●
● ●●
●
●
●
●
●
●
●
●
●●
● ●
●●
●
●● ●
●
●
●
●●
●●
●
●
●
●●
●
●
●
●
●●
●
●
● ●
●
●
● ●
●
●●
●
●
●
●
●
●●
●
●●
●
●
●●
●●●
●●
●
●
●●
●
●
●●
●●
●●
●
●
●●
●
●
●
● ●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●●●●● ●● ●● ●● ●● ●● ● ● ●● ●
10b:0a9b:1a
8b:2a7b:3a 6b:4a
5b:5a
4b:6a
3b:7a
2b:8a
1b:9a
0b:10a0
200
400
Alelo de referência (a)
Alel
o al
tern
ativo
(b)
Prob. indiv.
●
0.250.500.751.00
Genótipo●
●
●
●
●
aaaaaaaaaaaaaaaaaaabaaaaaaaabbaaaaaaabbbaaaaaabbbb
Genótipo●
●
●
●
●
●
●
●
aaaaaaaaaaabaaaaaaaaaabbaaaaaaaaabbbaaaaaaaabbbbaaaaaaabbbbbaaaaaabbbbbbaaaaabbbbbbbaaaabbbbbbbb
Prob. indiv.
●
0.50.60.70.8
●
●
●●●●●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●
●●
●
●
●
●
●
● ●
●
●●●
●
●●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●
● ●●
●●●
●
●
●●
●
●
●
●
●●
●
●
●
● ● ●●●
●
●
●●
●
●
● ●●
●●
●
●
●●
●●
●
●
●
●●
●
●●
●
●
●●
●●
●●●
●
●
●
●●
●
●
●
●●
●
●●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
12b:0a
11b:1a10b:2a
9b:3a8b:4a 7b:5a 6b:6a
5b:7a
4b:8a
3b:9a
2b:10a
1b:11a
0b:12a0
250
500
250 500 750Alelo de referência (a)
Alel
o al
tern
ativo
(b)
0
●●● ●●
●●
●
● ●
●
●●
●
●●
●●●
●
●●
●●
●●
●●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
● ●
●
●●
●
●
●
●●● ●●
●
●
●●
●
●
●
●
●
●
● ●
●
●
●
●
●● ●
●
● ●
● ●
●
● ●●
●
●●
●
● ●●
●
●
● ●●●●●
●
●●
●●
●●
●
●
●
●
● ●
●
●●●
●
●
●●●
●●
●●
●
●●● ●
●
●●●
●●
●
●● ● ●●
●
●●● ●●
●
●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●
● ●
●
●●● ● ●
●
●
●
● ●●●
●
●
● ●
●
● ●
●● ●
●
●
●
●
●
●●
●
● ●●●
●
●
●
●
●
●● ●
●
●
●●
●●
●●
●●
●
8b:0a
7b:1a6b:2a 5b:3a
4b:4a
3b:5a
2b:6a
1b:7a
0b:8a0
200
400
0 200 400 600Alelo de referência (a)
Alel
o al
tern
ativo
(b)
Prob. indiv.
●
0.250.500.751.00
Genótipo●
●
●
aaaaaaaaaaaaaaabaaaaaabb
●●●
●
●● ●
●●
●
●●
● ●
●●●● ●
●●
●●●
● ●● ●
●
●
●●
●
●
●●
●
●
●●●
●
●
●
●
●●●
●●●●
●
●
●
●
●●
●●
●
●● ●●
●● ●
●
●●
● ●●●
●●
●
●●
●●
●
● ●● ●●
●●●
●●●●●
●
●
●
●
●●
●
●●
●
●●● ●● ●●
●
●
●●
●●
●
●●
●●
● ●
● ●
●
●●●
●●
● ●
●
●
●●
●
●
●
●
●●●
●
●
●
●
●
●
●●
●●●●
●
●
●
●
●●
●●
●
●
● ●●
●
●
●
●
●
● ●●
●
●●
●
●
●
●●
●
● ●●
●
●
●
●
●●●●●
●
●
● ●
●
●
● ●
●
●●● ●●
6b:0a5b:1a 4b:2a
3b:3a
2b:4a
1b:5a
0b:6a0
2000
0 2000 4000Alelo de referência (a)
Alel
o al
tern
ativo
(b)
Genótipo●
●
aaaaaaaaaaab
Prob. indiv.
●
0.250.500.751.00
C) SCBFAD1089H10_156 (Ploidia 10; Prob. = 0,83) D) SCBFLR1060D02_354 (Ploidia 12; Prob. = 0,77)
B) SCACCL6008F10_430 (Ploidia 8; Prob. = 0,99)A) SCACLR2022G02_123 (Ploidia 6; Prob. = 0,99)
Figura 3 – Exemplos de marcadores baseados em GBS em população de mapeamento de cana-de-açúcarclassificados pelo software SuperMASSA. Probabilidades superiores a 0,80 caracterizarambons locos como em A, B e C que foram bem classificados nas ploidias 6, 8 e 10, respectiva-mente; loco D foi classificado na ploidia 12, mas com probabilidade inferior a 0,80
Aqui, foram usados os mesmos critérios ad-hoc de Garcia et al. (2013) para classificar
cada loco em uma categoria de qualidade baseada na probabilidade a posteriori para cada
ploidia: categoria “A” incluiu locos com a probabilidade a posteriori maior que 0,80, categoria
“B” reuniu locos cuja soma das duas maiores probabilidades a posteriori foi maior ou igual a
0,80 e, finalmente, categoria “C” incluiu todos os outros casos. A categoria “A” representou,
em média, 60,4% dos locos para todas as ploidias (Figura 4), o qual foi menor que os 77,6%
dos dados baseados em Sequenom previamente estudados (GARCIA et al., 2013). De fato,
esperava-se que os dados de contagem de leituras de GBS trouxessem alguma piora devido à
ampla cobertura genômica ou aos eventuais artefatos técnicos. Apesar dessa redução, todavia,
grande parte dos locos puderam ser posteriormente explorados para propósitos de mapeamento.
Para fins de construção de mapa genético, foram selecionados os locos classificados
nas categorias “A” e ploidias variando de 6 a 14, representando 40,7% locos, em média, do total
analisado no SuperMASSA ou de 3.433 a 15.906 marcadores dependendo da referência (Tabela

52
20(73.4)
20
18
16
1412
10
8 6
4
20(14.7)
14
12
10(11.7)
86(26.7)
4(14.8)
20(75.9)
1814
12 10 8
20(11.5)
18(9.4)
16
14
12(10.5)
10(17.2)
8(19.6)
6(14.2)
4(5.5)
20(13.2)
18
16
14
12(5.8)
10(13.6)
8(18.7)
6(26.5)
4(16.0)
2
20(74.9)
1620(12.4)
18
16(8.3)
14
12(10.2)
10(16.9)
8(16.8)
6(12.0)
4
20(15.1)
16
14
12(6.7)
10(15.0)
8(18.0)
6(24.7)
4(13.0)
2
20(71.9)
12
20(10.8)
18(8.6)
16(6.5)
14(7.4)
12(11.6)
10(18.0)
8(18.7)
6(14.0)
4
20(12.6)
16
14
12(6.0)10
(15.0)
8(19.6)
6(25.8)
4(13.9)
2
Metil-filtrado Sorgo RNA-Seq SUCEST
A (61.8%) B (25.6%) A (61.7%) A (59.1%)
C (12.6%) B (26.7%) C (11.6%) C (13.7%)B (27.2%)
C (13.7%
)A
(59.1%)
B (27.2%
)
Figura 4 – Níveis de ploidias com maiores probabilidades a posteriori para cada referência utilizada nomapeamento de tags de GBS em população de mapeamento de cana-de-açúcar. Equivalendoàs áres dos retângulos e entre parênteses, a proporção relativa de cada ploidia ou categoriasde qualidade. Categorias “A” e “B” incluíram locos com a maior ou a soma das duas maioresprobabilidades a posteriori superiores ou igual a 0,80, respectivamente, enquanto categoria“C” incluiu todos os outros casos
10). Adicionalmente, esses locos de boa qualidade e ploidia razoável foram caracterizados
quanto à dosagem. Marcadores em doses única (do inglês, single dose markers – SDMs) e múl-
tipla (do inglês, multiple dose markers – MDMs) foram igualmente representados pelos dados
de GBS, com cerca de 50% cada, em média. Garcia et al. (2013) encontraram uma baixa pro-
porção (30,5%) de marcadores segregando em dose única na população experimental de cana-
de-açúcar que estudaram. Ou seja, a maioria dos marcadores segregaram, de fato, em doses
superiores, as quais ocorrem quando as configurações nos genitores diferem dos cruzamentos
aaaaaaab × aaaaaaab (segregação 1:2:1) e aaaaaaaa × aaaaaaab ou aaaaaaab × aaaaaaaa
(segregação 1:1), por exemplo, no caso octaploide. Assim, quaisquer cruzamentos de genó-
tipos com doses superiores e, portanto, diferentes dos casos nuliplex (aaaaaaaa) e simplex
(aaaaaaab), provaram-se ser mais frequentes.
Como um critério de controle final de qualidade dos dados de GBS, foram selecionados
somente SDMs cujas medianas de todas as probabilidades a posteriori individuais foram mai-
ores que 0,80. Esse critério ad-hoc objetivou garantir que os locos usados no mapeamento
genético tivessem, pelo menos, 50% dos indivíduos com elevada probabilidade (superior a
0,80) de estarem em determinados clusters para uma dada ploidia. Dependendo do número
de clusters, SDMs foram classificados como segregando nas razões 1:2:1 (três clusters) ou 1:1

53
(dois clusters). Em média, a percentagem que representaram cada classe de segregação foram
16,4% e 83,6%, respectivamente.
Tabela 10 – Locos selecionados com boa qualidade (categoria “A”) e ploidia razoável (de 6 a 14) classi-ficados como marcadores em doses única (SDMs) e múltiplas (MDMs) a partir de GBS empopulação de mapeamento de cana-de-açúcar. SDMs selecionados foram adicionalmentecaracterizados quanto ao padrão de segregação na população de mapeamento
Referência TotalDosagem SDMsa
selecionadosPadrão de segregação
MDMs SDMs 1:2:1 1:1
Genoma metil-filtradoda cana
15.906 7.014(44,1%)
8.892(55,9%)
5.266 912(17,3%)
4.354(82,7%)
Genoma do sorgo 11.789 5.784(49,1%)
6.005(50,9%)
3.433 605(17,6%)
2.828(82,4%)
Transcriptoma da canavia RNA-Seq
9.808 4.959(50,6%)
4.849(49,4%)
2.869 469(16,4%)
2.400(83,6%)
Sequências do projetoSUCEST
3.433 1.736(50,6%)
1.697(49,4%)
983 141(14,3%)
842(85,7%)
Nota: aSDMs com mediana de todas as probabilidades a posteriori individuais > 0,80
A redundância de SDMs selecionados foi inspecionada após filtragem de ploidia e qua-
lidade e com os genótipos já codificados como a, ab e b. Comparavelmente, todas as referências
mostraram níveis bastante similares de redundância dentro e entre elas (Figura 5). Por exemplo,
basicamente o mesmo nível geral de 22,7% para redundância dentro de referências foi encon-
trado. Somente 84 marcadores foram atribuídos igualmente para todas as quatro referências,
representando 3,8% de cada. Já entre quaisquer três referências, 13,7% dos locos foram dados
como ambíguos. Finalmente, redundância foi amplamente verificada entre referências pareadas,
representando 20,2% de cada referência. Interessantemente, as referências forneceram, em
média, 39,6% de novos locos, cada. Mantendo somente uma única chamada para cada marcador
redundante, um total de 7.049 locos de GBS foram levados ao mapeamento.
Finalmente, é importante mencionar que apesar de este número (7.049) ser apenas ∼7%
do número útil de locos marcadores obtidos via GBS em sorgo (100.291), este tipo de genoti-
pagem já se mostra promissora para cana-de-açúcar, considerando as laboriosas genotipagens
em gel ou em Sequenom, as quais fornecem, geralmente, uma menor quantidade de dados.
Além disto, GBS, até onde é sabido, foi utilizado pela primeira vez em cana neste trabalho e
revelou-se uma opção com melhor custo-benefício quando comparado a outras metodologias
de genotipagem de SNPs em geral.
3.2.2.3 Marcadores baseados em géis

54
Metil-filtrado
Sorgo
RNA-Seq
SUCEST
400
800
1200
1600
2000
2400
280032003600
4000
4400
4800
5200
400800
1200
1600
2000
2400
2800
3200
400 800 1200 16002000
2400
2800
400800
Figura 5 – Gráfico circular mostrando redundância entre marcadores em dose-única obtidos a partir dequatro referências usadas para mapear tags de GBS de população de mapeamento de cana-de-açúcar. Regiões em vermelho representam redundância dentro de cada referência, enquantoregiões em verde, laranja e azul representam, respectivamente, redundância entre quatro, trêse duas referências. Regiões remanescentes em cinza representam locos exclusivos para cadareferência
Um total de 999 fragmentos polimórficos foram gerados pelos marcadores SSRs e
TRAPs, sendo 451 (45,2%) e 548 (54,8%) marcadores testados para as segregações de 3:1 e
1:1, respectivamente. Do total, 499 (49,9%) marcadores desviaram desses tipos esperados de
segregação; assim, os 500 locos não-distorcidos foram levados à análise de ligação. Oliveira et
al. (2007) e Pinto et al. (2009), por exemplo, trabalhando com populações segregantes de cana-
de-açúcar e marcadores de mesma natureza, também relataram proporções semelhantes de mar-
cadores distorcidos. Esse elevado número de marcadores distorcidos no cenário de genotipagem
qualitativa em poliploides é esperado e bem conhecido. De fato, há inúmeras maneiras de locos
com doses superiores segregarem (RIPOL et al., 1999); no entanto, marcadores baseados em
géis apresentam eficácia irrelevante em acessar tal informação. Já SNPs, por serem bialélicos e
codominantes, conseguem distinguir indivíduos heterozigóticos. Em cana-de-açúcar, todavia, a

55
proporção dos alelos e a ploidia variam de loco para loco (GRIVET; ARRUDA, 2002; GARCIA
et al., 2013).
Os SNPs com segregação esperada de 1:2:1, na verdade, contêm indivíduos heterozigó-
ticos com genótipos que variam de abbbbbbb (1 a’s : 7 b’s) até aaaaaaab (7 a’s : 1 b’s), no caso
de um loco octaploide, por exemplo. Para marcadores dominantes, como os SSRs, todas essas
classes de heterozigotos ficam confundidas com aquela do alelo dominante (presença de banda),
dando origem à segregação esperada de 3:1. Isto justificaria, inclusive, a elevada distorção de
segregação de marcadores baseados em géis até então observada, uma vez que genotipagem
qualitativa falha em caracterizar precisamente as doses alélicas. Para o caso dos SNPs, essas
considerações sobre a segregação, por sua vez, já foram incorporadas ao modelo de populações
F1 segregantes de chamada de genótipos do SuperMASSA e, por isso, as segregações dos locos
selecionados não foram testadas.
3.2.2.4 Mapa genético
O mapa genético para a população de cana-de-açúcar foi construído a partir da análise
no software OneMap de 7.549 marcadores no total, sendo 7.049 marcadores baseados em GBS
e 500 marcadores baseados em géis. Para cana-de-açúcar, esse total de marcadores pode ser
considerado bastante satisfatório. Deste total, foram alocados no mapa 993 (13,1%) marcado-
res, dos quais 934 provieram de GBS e os 59 restantes representavam regiões microssatélites
genotipadas; nenhum loco TRAP foi mapeado nesta análise. Quanto às classes de segregação,
foram mapeados 254, 15, 518 e 206 marcadores dos tipos ‘B3’, ‘C’, ‘D1’ e ‘D2’, respectiva-
mente. Os marcadores distribuíram-se ao longo de 223 grupos de ligação (GLs) e cobriram
um total de 3.682,05 cM (Figura 6), determinando uma elevada densidade média de 3,71 cM
quando comparado a outros trabalhos (PASTINA et al., 2010). Os tamanhos dos GLs variaram
de 1,06 cM (GL 70) a 235,67 cM (GL 46), com uma média de 16,51 cM; 56 GLs apresentaram
tamanhos menores que 2 cM, 95 GLs exibiram tamanhos maiores ou iguais a 2 cM e menores
que 10 cM, e os 72 GLs restantes tinham tamanhos maiores ou iguais a 10 cM. Distâncias
superiores a 15 cM entre pares de marcadores consecutivos foram observadas em pouco mais
de 10% dos casos, representando pouquíssimos gaps.
Apesar de o aproveitamento das marcas ter sido relativamente baixo, esses números
demonstram que, para cana-de-açúcar, este mapa pode ser considerado denso e saturado. A
taxa de aproveitamento de 13,1% refere-se, principalmente, ao cuidadoso descarte de marcado-

56
res redundantes e à exclusão de locos distantemente alocados quando da primeira ordenação.
Recentemente, Aitken et al. (2014) desenvolveram um mapa para cana baseado em marcadores
do tipo Diversity Array Technology (DArT), com uma densidade aproximada (4,3 cM), mas
um número superior de marcadores (2.267); neste caso, nenhum tratamento foi realizado para
evitar o mapeamento de marcadores redundantes, bem conhecidos para as plataformas de ge-
notipagem em larga escala. Adicionalmente, o presente trabalho incluiu marcadores do tipo
‘B3’, o que, dentre os mapas de cana existentes na literatura, apenas o esboço apresentado por
Marconi (2011) contaram com o acréscimo desse tipo mais informativo de marcadores, obtidos
a partir da genotipagem quantitativa do Sequenom. Essa classe de segregação em populações
F1 segregantes contribui importantemente com a integração dos grupos de ligação, a qual, em
cana, era limitada aos marcadores do tipo ‘C’, dominantes e, portanto, menos informativos (WU
et al., 2002a).
De modo geral, poucas inversões na ordem dos marcadores em relação ao genoma do
sorgo foram observadas, como, por exemplo, nos GLs 1, 2, 71, 83 e 117; nesta avaliação,
pequenas inversões locais foram desconsideradas porque o tamanho da população limita a cor-
reta estimação de fração de recombinação entre marcadores muito próximos. Considerações
semelhantes a estas foram apresentadas por Guimarães, Sills e Sobral (1997) e Aitken et al.
(2014), por exemplo.
A Figura 6 exibe a distribuição dos GLs agrupados em 18 grupos de hom(e)ologia
(GHs), obtidos a partir das origens genômicas comuns dos locos mapeados em diferentes GLs.
O número de GLs alocados em GHs variou de dois a cinco, sendo o maior GH contendo 44
marcadores (GH 11), e o menor contendo sete marcadores (GH 18). Ainda, os GLs foram reu-
nidos em grupos de sintenia (GSs) com sorgo, baseando-se na distribuição dos locos oriundos
de GBS usando como referência o genoma dessa espécie; aqui, a numeração dos GSs refere-
se ao cromossomo do sorgo do qual os locos se originaram. O número de GLs em cada GS
variou de nove (para o GS 6) a 21 (para o GS 1). De fato, o maior número de locos (53) de cana
originados do alinhamento contra o genoma do sorgo foi obtido para o cromossomo 1, enquanto
o menor número de locos (18) estava disponível para o cromossomo 6. Eventualmente, GSs
apresentavam locos originados de outros cromossomos que não daquele da maioria dos locos.
Por exemplo, locos originários do cromossomo 8 do sorgo também foram encontrados nos GS
2, 3 e 9. O GS 8, por sua vez, também evidenciou locos representantes dos cromossomos 1,
2, 3 e 4 do sorgo. Finalmente, nos GS 3 e 9, locos originários dos cromossomos 1 e 7 e dos

57
cromossomos 5 e 8 foram detectados, respectivamente.
Muito embora as qualidades dos mapas de cana atuais sejam limitadas pelas metodolo-
gias de genotipagem e mapeamento que desconsideram as múltiplas doses, algo já se especulou
sobre a evolução do genoma de Saccharum quando comparado ao de sorgo. Aitken et al. (2014)
observaram que eventos de translocação ou, ainda, fusões entre porções cromossômicas teriam
ocorrido em cana entre regiões equivalentes no sorgo como, por exemplo, entre os pares de cro-
mossomos 2–8 e 5–6, o que teria determinado a redução do número básico de cromossomos de
x = 10 em sorgo para x = 8 em S. officinarum. De fato, as hipóteses envolvendo translocações
seguem alguma tendência nos estudos comparativos sorgo-cana. Por exemplo, além da translo-
cação entre os cromossomos 2–8, já anteriormente relatado por Dufour et al. (1997), Ming et al.
(1998) identificaram outros putativos eventos cromossômicos em cana, como aqueles entre os
pares de cromossomos 2–4, 6–7 e 7–8, correspondentes em sorgo. Essa variação, identificadas
ora em genótipos das espécies, ora em híbridos, parece guardar uma certa especificidade dos
indivíduos avaliados, de tal modo que definir, até o momento, um padrão de evolução geral para
o gênero permanece indeterminado. Estudos dessa natureza (baseados em mapa de ligação) só
apresentarão o devido respaldo genético-estatístico quando da utilização de mapas baseados na
informação de doses múltiplas dos marcadores moleculares.

58
mf169280_4530.00
mf107365_1630:rna66593_107917.25
sb1_7123462833.72
mf6005_4200sb1_6946857053.50
sb1_70966878:SCRUFL3064D01_612mf4471_369769.22
mf139503_870:sb1_73150959:rna93460_274684.90
GL 1sb1_56663420.00
mf167018_570:sb1_4518545:rna76920_38516.92mf187608_48930.91sb1_879499431.01mf163283_68242.91
mf37718_902:sb1_1252358761.48
GL 2SCMCRT2105C02_5630.00rna70357_1480.79rna87007_113913.90
mf88388_535:sb1_17903786:rna73930_68529.51mf19957_1893mf19957_1913mf19957_1917mf19957_1899
48.74
GL 3mf21975_19070.00
sb1_3163177719.43sb1_31631788sb1_3163177820.13
mf157160_75831.98mf60753_201542.62mf60753_201343.33
GL 4 GS 1
mf130222_16:sb1_92051700.00
mf94307_1830:sb1_1028494415.53mf94307_1834:sb1_10284940mf94307_1831:sb1_1028494317.88
mf94307_1842:sb1_1028493223.18mf10516_2074:sb1_11491234:rna85024_402338.36
GL 5
mf134350_155:sb1_9184184:rna84395_10770.00
sb1_918419319.34
mf62649_24629.62
GL 6
sb1_601246650.00sb1_601246819.08sb1_6012468210.25
GL 7mf90402_2018:rna62695_1552sb1_265796360.00
mf90402_20219.97
GL 8sb1_56376790.00sb1_56376763.66mf137319_11235.07
GL 9
rna80099_135sb1_122110260.00
mf111813_19663.13
GL 10SCJLAM1062A07_5090.00SCJLAM1062A07_5060.37sb1_144559813.05
GL 11SCPIRT3023C11_4880.00mf124912_1103:sb1_31631838:rna91673_8600.70SCPIRT3023C11_4872.82
GL 12 sb1_38520340.00sb1_38520221.18mf131701_5751.95mf131701_5692.73
GL 13 mf775_18110.00mf775_18220.38sb1_613565381.94sb1_613565542.33sb1_613565392.71
GL 14
mf4847_1669:sb1_611263030.00mf4847_1648:sb1_611262821.13mf4847_1651:sb1_611262852.27
GL 15
sb1_33002930.00sb1_33002941.45mf97741_20372.17
GL 16mf57100_839:sb1_24294765:rna41137_1110.00sb1_242947831.42mf57100_855:rna41137_942.13
GL 17 sb1_566768910.00sb1_566769090.39sb1_566768760.79sb1_566769181.98
GL 18sb1_571794610.00mf249421_1760.72rna91551_37281.45
GL 19mf3236_150:sb1_536918390.00SCAGFL1090E04_3650.71rna54097_51.43
GL 20rna87586_8570.00mf129219_3930.36sb1_84908961.07
GL 21
mf39868_13700.00
sb4_9379sb4_938019.48
sb4_934230.68sb4_936532.27
GL 60sb4_525223090.00
mf170396_190:rna84921_21517.72mf170396_185:rna84921_21018.44mf257741_115:rna90037_247527.62
GL 61mf65289_17450.00
sb4_6674042913.65
sb4_6001292124.80
GL 62mf103825_20170.00rna93080_1385.40sb4_1732798.26
GL 63mf171030_1810.00mf171030_1800.39sb4_4524455sb4_45244034.93
GL 64sb4_615024050.00sb4_615024110.74sb4_615024131.48sb4_61502406sb4_615024042.21
sb4_61502383sb4_615023973.69
GL 65sb4_608540520.00mf138957_559:SCSGFL1082C12_410.72mf138957_516:SCSGFL1082C12_841.07sb4_608540091.78
GL 66
sb4_525653510.00sb4_52565299sb4_525653160.83
sb4_525653121.67
GL 67mf30676_1567:sb4_620997640.00mf30676_1558:sb4_62099756mf30676_1562:sb4_620997690.77
mf30676_1566:sb4_620997651.54
GL 68sb4_553859400.00sb4_55385923sb4_55385921sb4_55385945
0.70
sb4_553859351.41
GL 69sb4_579499870.00sb4_579499880.71mf131190_981:rna109043_2421.06
GL 70
GS 4
GS 2
GH 1sb2_732509370.00
sb2_6770847415.17sb8_175513728.50sb8_175517728.89mf162663_77633.99
SCEPRZ3085A05_49552.05rna36509_31654.90
GL 26sb2_648505630.00sb2_648506088.50
sb2_6429561827.64mf21649_2398:rna77227_94828.35
GL 27
mf22998_1819:sb2_45124456:rna61362_140.00
sb2_936374419.20sb2_9363737mf25477_208319.57
GL 28sb2_618828380.00sb2_618828533.87sb2_618828699.73rna89739_220111.21
GL 29sb2_3921697:rna23776_2470.00sb2_3921699:rna23776_230rna23776_2515.91
GL 30rna85603_2350.00sb2_657345970.37rna85603_2415.65
GL 31mf156030_349:rna80420_5430.00mf156030_340:sb2_62761717:rna80420_5341.43sb2_627617473.57mf156030_370:rna80420_5644.29
GL 32
sb2_88892160.00sb2_88892030.80sb2_88892101.59sb2_88892152.79
GL 33mf131707_782:rna77205_1590.00sb2_765509920.71sb2_765510172.13
GL 34sb2_595318990.00mf124769_1818:rna70639_4930.70SCJFFL1C04F08_4962.11
GL 35rna64404_3560.00mf26733_297:SCJLRT1020F05_2411.01sb2_25888381.99
GL 36
mf1902_5201:sb2_60552980.00mf1902_52220.75sb2_60553191.50
GL 38mf23383_1362:sb2_712262040.00mf23383_1360:sb2_712262020.72mf23383_1361:sb2_712262031.45
GL 39sb2_24520560.00sb2_2452096sb2_24520841.43
GL 40
mf140205_336:sb2_702998910.00mf140205_338:sb2_702998930.82sb2_702998851.64
GL 37
mf252507_1510.00
mf126424_1540:sb2_72296849:rna28296_10717.02mf297207_259mf297207_27527.20
rna95936_16427.89rna95936_18028.58sb2_66149510mf63081_231:rna59405_74347.46
GL 22
sb2_722968840.00sb2_722968570.72mf126424_1505:rna28296_721.44
GL 23
mf43680_1882:rna87845_18760.00
sb2_7620146113.65
mf135308_198:rna87035_96425.05
rna36483_8136.48
mf28963_593:rna71428_89851.03
GL 24sb2_699012880.00sb2_699012850.69rna36483_9810.69
GL 25
GH 2
sb4_50271830.00
mf152767_34:sb3_4681905915.74
sb3_754682933.91
rna90267_78749.74
mf148614_22:sb2_74689463:rna63124_33967.49
mf22704_278685.30rna75825_47797.54mf116093_1952106.44mf209_8523sb7_31444384120.45
mf48310_1950:rna31514_183136.03rna94083_4829mf298625_214145.60
mf23282_1264151.93mf131549_185155.04mf118315_112157.05mf263837_508161.91sb1_29954416sb1_29954417169.51
mf125302_409179.25rna38643_345186.05rna63025_162189.09sb8_11128174SCQGLV1017E09_203191.96
mf134709_228202.97rna66129_815217.89mf25719_1557235.67
GL 46
SCCCRT3007D12_3980.00
mf14674_2118.27mf14674_2819.00
SCSFAM1074E10_28737.26mf16592_376646.70
mf19574_311158.15SCBGLR1118F03_32969.37sb3_7260737070.96
mf15382_3593:rna114756_6888.91
GL 47
sb3_8202819:rna88371_780:SCQGLB2043C04_1620.00
mf248692_52219.36mf248692_49026.95
SCB63-742.98SCB63-246.91
sb3_545448863.81
GL 48mf254640_4590.00
mf138324_67117.66sb3_6659976918.47
GL 49mf246042_62:rna93296_28140.00sb3_56913329.91sb3_569135610.27sb3_569138010.63
GL 50
rna34626_189sb3_725538200.00
sb3_725538155.98
GL 51
sb3_55933875mf94888_14990.00
mf94888_14950.75mf94888_14961.49mf94888_15032.24sb3_559338722.99sb3_559338763.36sb3_559338603.73sb3_559338534.48sb3_559338665.22
GL 52
sb3_541826sb3_5418040.00
rna79964_14653.66
GL 53mf181373_1970.00sb3_630801281.44sb3_630801363.40
GL 54rna84713_16870.00mf24938_2197:sb3_61684818:rna84713_17370.76rna84713_16863.06
GL 55
rna51833_2240.00sb3_535985122.26mf11846_19623.01
GL 56sb3_13926600sb3_13926569sb3_13926603
0.00
mf59691_1832:rna26746_82mf59691_1822:rna26746_751.55
GL 57sb3_60020720.00sb3_60020940.78sb3_60020901.55
GL 58sb3_31624110.00sb3_31624100.35sb3_31624120.69sb3_31624161.38
GL 59
mf161288_935:sb3_710131600.00
sb1_780073214.56
SCC132-629.06
IISR95-546.31SCA07-154.49
mf23980_653:rna50826_832:SCRLFL8050C11_15371.23
GL 41
mf229549_3010.00mf229549_2820.71
mf24082_241:sb1_7645431:rna71461_58420.22mf95450_36529.65SCB14-436.00
IISR95-647.69
rna8915_6164.09
GL 42
mf161288_908:sb3_71013188mf161289_60.00
mf161288_909:sb3_710131830.71mf161288_912:sb3_710131841.43rna8915_762.17mf25124_261320.89mf25124_2612mf25124_261421.82
GL 43
mf173375_331:sb3_66175806:rna67765_4890.00mf173375_332:rna67765_4880.70sb3_7147926313.54
mf5834_3222:sb3_70642404:rna71341_94326.54mf202254_62034.75
mf122751_58756.27
GL 44
sb3_661758050.00sb3_661757751.41mf173375_299:rna67765_5222.82
GL 45
GS 3
GH 3
GH 4
mf164660_3530.00
mf301119_48419.34
rna8917_35432.01
sb5_6191696645.07mf72170_9054.34
sb5_5738716971.30
GL 75mf13004_11980.00
mf20099_3409:rna26005_15912.55
sb5_149103123.13
GL 76mf187824_657mf187824_6560.00
sb5_540576610.30
GL 77sb5_613223900.00sb5_613223990.78mf19346_6203.99mf19346_6045.54rna81267_16987.89
GL 78sb5_485335760.00sb5_485335840.74mf74936_1372.21
GL 79
rna82296_3800.00rna82296_3820.73sb5_591940871.51
GL 80SCCCFL4094B12_666mf91913_1896:sb5_51126366:rna56836_4680.00
mf91913_1903:sb5_51126359:rna56836_4610.71mf91913_1854:rna56836_5101.43
GL 81sb5_122188000.00sb5_122187960.87sb5_122187941.31
GL 82
CIR12-10.00sb5_410760714.50rna93602_173027.20mf20704_179728.78rna93602_170529.17sb5_410758229.96sb5_2327923235.83mf20704_169447.12
GL 71mf186657_5730.00
mf77182_198914.22
mf186657_54125.81CIR12-331.41
GL 72CIR12-50.00rna67380_196710.10sb5_192241910.53mf68117_47210.97mf68117_49611.40rna67380_194312.26sb5_192244313.13
GL 73
rna87132_16110.00CIR12-62.13SCB258-88.79
GL 74
GS 5GH 5
Figura 6 – Mapa genético de cana-de-açúcar exibindo a distribuição dos grupos de ligação (GLs) aolongo dos grupos de hom(e)ologia (GHs) e de sintenia com o sorgo (GSs). Marcadores cor-respondendo a cada cromossomo do sorgo foram destacados em cores diferentes (continua)

59
SCMCFL5007H09_5300.00mf128725_768:rna87743_4750.75
mf70623_178717.12
mf71727_80530.80
SCB190-247.83SCA53-1256.18
SCA53-1466.68
GL 149
mf53133_20430.00
SCA53-717.25rna90367_97820.09
GL 150
SCA53-40.00IISR88b-83.92SCB190-36.09SCB190-47.85SCRUFL1115F07_51919.71
GL 151
mf21505_16230.00IISR88b-18.60SCB259-214.25
GL 152
mf144619_10920.00rna79698_909:SCEQLR1050H07_1741.44SCEQLR1050H07_162rna79698_896SCEQLR1050H07_161rna79698_897
11.08
rna79698_880SCEQLR1050H07_14511.85
GL 153rna79698_879SCEQLR1050H07_1440.00
SCEQLR1050H07_1631.82
GL 154
rna79698_845SCEQLR1050H07_1120.00
mf173918_2011.68
GL 155
mf162072_2970.00SCA61-59.27CIR14-413.21
SCC22-528.58
GL 156
rna91193_34180.00rna91193_33660.35SCA61-68.68
GL 157
sb6_540729390.00
mf199486_43417.45mf269343_59128.12mf269343_59228.93rna82667_111238.06mf82223_280:rna88090_185:SCEZHR1085D06_6749.38
mf105220_65:sb6_61885140:rna67771_13963.96
GL 85mf25152_18900.00
rna85834_52311.25
mf259257_35:sb6_5989170822.30
sb6_5561058237.85
mf133566_536:sb6_53739371:rna67333_880:SCJFRZ2025B05_49954.43
GL 86
mf169609_363:sb6_56756053:rna91834_30550.00
mf29338_212912.92rna67575_78723.07
GL 87sb6_610334070.00sb6_610333946.60sb6_610333936.95
GL 88sb6_558180570.00mf9329_1462.36mf9329_1423.94mf9329_1434.73
GL 89
sb6_548023660.00sb6_548023670.77rna84162_5043.10
GL 90sb6_528210290.00sb6_52821016:rna91909_23170.74rna91909_23201.47
GL 91
mf101931_19740.00mf101931_19751.52
SCB45-1517.09
mf66100_2040:rna76662_57630.24mf193065_406:rna91599_57738.51
sb6_6084316054.38
mf138081_984:sb6_58911526:rna92992_550574.29
sb6_5342415593.80mf21210_3491105.62rna65009_82109.73SCEQRT1031B01_43110.67mf130399_989:rna91800_3110122.76sb6_61974231124.21
GL 83mf96855_108:rna89108_195:SCJFRT2060G09_1210.00
SCB45-111.18mf52262_851:rna62110_4720.03
GL 84 GS 6
GH 6
rna6636_2450.00rna6636_2340.42mf213219_629.81mf134347_151:rna85085_32418.02mf134347_161:rna85085_33418.71sb8_505875831.85sb8_505870333.22rna89226_22734.59sb8_259626946.77
sb1_49067072:rna84746_22566.84
mf98396_3285.80
sb3_66052657104.70
GL 116
sb8_546925210.00rna19848_58511.33rna19848_58412.69mf89788_1742:rna11183_5723.51
mf129609_497:sb8_45219409:rna53230_47635.54sb8_4846797048.99mf1831_65063.83sb8_5482088263.93sb8_5482090264.03
GL 117
rna83196_1540.00mf298048_1541.58rna66703_4346.96mf259847_4648.57SCC21-613.41mf266703_60:sb8_236517223.04SCSFFL4015F07_61625.28SCSFFL4015F07_599SCSFFL4015F07_57139.50
GL 118mf260672_3170.00mf260672_3190.76sb8_549990124.04
GL 119
rna86526_765sb8_550447790.00
rna86526_7421.53rna86526_7573.82
GL 120
mf225694_1650.00mf225694_1820.38sb8_320496021.92
GL 121
rna104430_1220.00rna104430_1474.94rna104430_1485.39rna104430_1326.30rna104430_146sb2_28015127.64
sb2_28015118.75sb2_28015099.86IISR46b-629.20SCC96-135.06sb8_4906560454.46mf124476_599:rna77118_134455.90mf108148_190371.70mf108148_190172.41rna116131_13184.75rna116131_12185.37sb2_5255003986.02mf22144_274:rna77309_2394102.34mf22144_299:rna77309_2419102.69SCQSAD1059D04_643mf137176_704:rna91243_1488117.91
SCQSAD1059D04_598118.57mf273365_290:rna59310_367130.18SCQGLB2041C03_184141.98
GL 106mf211484_5540.00
sb8_5386734213.00rna94088_7117.67mf17498_330327.68SCC84-437.68SCC84-744.25CIR24-148.84mf59476_3558.98
GL 107
SCC96-20.00
SCB58-0612.33
SCB58-0124.09
CNL170-340.90
GL 108
rna94088_137rna94088_1270.00
SCSBRT3036H07_372.00
SCSBRT3036H07_6518.56
GL 109
SCB65-40.00CIR24-49.32SCC96-511.28SCB374-313.24
GL 110
rna35352_950.00mf148031_160:sb8_458213381.22mf22378_14029.10SCB330-211.13SCB285-415.27mf144424_184:sb4_64604262:rna85911_164629.56
GL 111
SCA09-140.00CIR36-14.09SCB330-88.87
GL 112sb8_45821370mf148031_1920.00
mf148031_1892.21
GL 113
mf298048_1140.00mf298048_1150.87mf298048_1091.74
GL 114mf117247_70:sb8_29496080.00rna90411_5390.39rna90411_5520.78mf117247_83:sb8_29496211.17
GL 115
GS 8
GH 12 GH 13GH 11
SCJLFL1048B12_150.00mf82770_569:rna75593_23215.91sb7_900492814.13mf82770_522:rna75593_236814.85mf82770_536:rna75593_235415.57sb7_900494216.29sb7_900497517.02
GL 92
IISR199-50.00sb7_90049243.69mf82770_518:rna75593_23724.43
GL 93
mf259069_282:rna74147_2570.00mf119068_848.13
sb7_5783777222.26
sb7_5728203937.06
GL 101mf224601_3690.00
sb7_6290503115.74
mf11972_3078:sb7_63328647:rna85308_45328.24
GL 102SCSGLV1011E12_1170.00sb7_551834900.77mf20974_3307:rna90238_16865.33
GL 103sb7_628164020.00sb7_628164520.71sb7_628164542.48
GL 104mf300799_295:sb7_4977200:rna90695_8350.00mf300799_350:sb7_4977145:rna90695_8900.38mf300799_348:sb7_4977147:rna90695_8881.15
GL 105
mf14092_35880.00
SCSGAM2076A12_44420.00rna89943_130526.09sb7_129495033.55
GL 94
rna89943_12890.00rna89943_12711.43sb7_12949842.86sb7_12949664.29
GL 95
rna73797_1790.00
mf189055_558mf189055_54419.84
mf5308_2995:sb7_6283327231.32SCB208-437.70
sb7_6359177350.01
GL 96
SCB208-70.00SCB208-6SCB208-53.71
GL 97
sb5_527789560.00
mf88506_237:rna90068_56415.10
mf228309_36226.63
mf246837_96:rna30410_33236.98mf106282_195048.05mf175158_11459.92mf175158_7160.77mf175158_11561.61mf175158_11664.12mf9343_132:rna89611_8371.97mf17470_1855:rna71385_104189.03
GL 98mf228309_3730.00sb4_103886210.72sb7_2791053mf17470_380015.52
mf17470_379924.41
GL 99
mf9343_910.00rna89611_1255.91sb7_86352246.64
GL 100
GH 9
GS 7
GH 10GH 7
GH 8
mf333_87720.00
rna69406_7313.16
mf268853_60430.17
mf70727_202743.63sb9_208249346.34sb5_1723774658.32
GL 124mf1498_2066:rna54162_4230.00rna47586_452mf271387_6016.34
SCJFFL3C06H03_32718.80mf137131_97421.04sb9_5799071625.16sb9_57990663sb9_57990665sb9_57990667
35.97
mf118637_7751.39
GL 125sb9_572485730.00mf187016_576:rna91581_959:SCJLFL3014H06_277mf187016_564:rna91581_9474.67
GL 126mf36186_1261:sb9_424728750.00mf36186_12503.68mf36186_12494.39
GL 127
mf25869_1318:sb9_49839063:SCEZRZ3051D09_140.00mf25869_13192.82mf25869_13163.52
GL 128
mf185387_660:rna92287_3094:SCJFST1012B01_4320.00mf185387_661:rna92287_3095:SCJFST1012B01_4330.72sb9_471385051.45sb9_471385042.17
GL 129mf282153_297:sb9_474917180.00sb9_474917001.51sb9_474916991.89
GL 130mf64272_3320.00sb9_542113761.09sb9_542113921.45mf64272_3251.82
GL 131mf20169_31140.00sb9_67015711.46sb9_67016131.82
GL 132
mf7015_38190.00
sb9_212971519.74
mf176614_73:sb8_81469731.89mf56267_7540.16
mf2389_10858.00rna86401_59358.73
GL 122
mf2398_1883:rna105287_1670.00mf2398_1865:rna105287_1490.74mf2398_1867:rna105287_1511.48
GL 123
GS 9
GH 14
mf145829_7660.00
SCB243-511.21
mf20333_326sb10_5784961925.40
GL 136SCACLR2022G02_125SCACLR2022G02_1230.00
sb10_554845286.58
GL 137sb10_122515710.00sb10_122515680.38mf148405_789:rna84272_8201.13mf148405_792:rna84272_8231.51SCACCL6008F10_4304.97SCACCL6008F10_4335.34
GL 138sb10_40284330.00sb10_40284180.70sb10_40284391.41mf22094_25314.23
GL 139sb10_547370410.00rna60045_2090.92mf123870_15061.84SCSGST3119G01_623.74
GL 140
mf63079_420:sb10_1569397:rna80668_16280.00mf63079_441:rna80668_16492.84mf63079_440:rna80668_16483.55
GL 141SCSBAD1085B09_5830.00SCSBAD1085B09_545sb10_16176280.35
sb10_16175903.49
GL 142
sb10_52459938rna73201_4310.00
mf39519_15983.03
GL 143SCRLFL1009B10_7000.00sb10_62975060.80mf118126_19892.25
GL 144
mf102591_1349:rna46052_48sb10_518394990.00
sb10_518395022.11
GL 145sb10_12120879mf24759_1581:rna93305_12990.00
sb10_121209090.95sb10_121208841.36sb10_121209031.77
GL 146mf5311_1337:sb10_65599310.00mf5311_1332:sb10_6559936mf5311_1331:sb10_65599330.75
sb10_65599781.51
GL 147rna81133_890.00sb10_5994690mf23869_9021.47
GL 148
mf44188_67:SCQGST3154C02_1020.00
rna89011_230416.51rna103356_4920.04mf27574_227121.56rna74863_16436.40rna78920_239mf242090_501:sb10_3101695:SCVPFL1131C02_48347.15
GL 133
SCJLFL1050E02_4840.00
mf27574_225110.53
rna89011_224420.81
GL 134
mf44188_101:SCQGST3154C02_680.00sb10_18460580.79mf44188_88:SCQGST3154C02_811.59
GL 135
GS 10
GH 15
GH 16
GH 17
GH 18
Figura 6 – Mapa genético de cana-de-açúcar exibindo a distribuição dos grupos de ligação (GLs) aolongo dos grupos de hom(e)ologia (GHs) e de sintenia com o sorgo (GSs). Marcadores corres-pondendo a cada cromossomo do sorgo foram destacados em cores diferentes (continuação)

60
SCQGFL3058A08_1130.00
SCJFRT2059D09_103617.84mf120196_201528.39mf120196_197728.77mf120196_1946:rna12237_319:SCUTSD1028G03_45239.40
rna92600_38656.99mf280385_9358.73
GL 158mf27787_19750.00
SCEZRT2021H09_21316.31mf148018_22317.85mf148333_34127.33
mf64185_203646.45
GL 159SCA68-10.00
SCA26-213.82
SCB52-125.49
SCA26-640.46
GL 160rna77255_2740.00rna77255_3140.78
mf23388_3021:sbsuper_2967_81318.03mf47373_94:rna61315_50818.04mf47373_79:rna61315_53322.88CIR23-435.72
GL 161mf180162_2610.00
rna58431_36215.93
rna57550_27234.81
GL 162SCUTHR1063C12_3230.00SCUTHR1063C12_2922.19mf126486_60411.27mf126486_60112.03rna72383_15425.93
GL 163
mf131849_4070.00mf131849_403:rna78607_588rna78607_6071.45
rna78607_593rna78607_60214.03
rna78607_59015.48
GL 164mf216115_1400.00CIR32-113.03mf1009_73239.32
GL 165rna15183_280.00rna15183_321.58rna15183_35rna15183_43rna15183_33rna15183_48
4.73
rna15183_506.30
GL 166mf173642_1650.00mf173642_185mf173642_1606.19
GL 167mf2160_19190.00mf2160_19383.83mf2160_19396.12
GL 168mf13122_3432mf13122_34570.00
rna8524_234:SCCCCL7C02A06_226rna8524_257:SCCCCL7C02A06_2014.48
rna8524_269:SCCCCL7C02A06_1915.34
GL 169
mf124850_230:rna74092_188:SCJFSB1014H04_5430.00mf124850_234:rna74092_192:SCJFSB1014H04_547mf124850_232:rna74092_190:SCJFSB1014H04_5451.48
mf124850_237:rna74092_195:SCJFSB1014H04_5505.17
GL 170mf86666_1510.00mf86666_1360.73mf86666_1481.46mf86666_1392.92mf86666_1705.11
GL 171mf20652_30600.00rna20961_24.28rna20961_15.06
GL 172rna75417_6440.00rna75417_6434.27rna75417_6454.97
GL 173mf56117_1430.00rna43594_1772.10mf56117_1193.13rna43594_1534.53
GL 174mf159535_4580.00mf159535_4860.74mf159535_4704.42
GL 175mf147639_660mf147639_628mf147639_634
0.00
rna70994_5252.22rna70994_5242.94rna70994_5274.38
GL 176
mf9631_4060.00mf9631_4553.76mf9631_4424.13
GL 177mf106436_19260.00mf106436_19111.49mf106436_19024.00
GL 178mf137130_7590.00mf137130_802mf137130_8012.11
mf137130_8033.95
GL 179mf290774_2000.00mf290774_1910.84mf290774_2423.41mf290774_2443.83
GL 180SCJLRZ3074B03_3080.00rna88469_533:SCJLRZ3074B03_3610.93SCJLRZ3074B03_3103.74
GL 181mf171029_8650.00mf171029_8610.73mf171029_8601.10mf171029_8591.46mf171029_8492.59mf171029_8483.33
GL 182mf88115_1752mf88115_1739mf88115_1763mf88115_1779
0.00
mf88115_17643.45
GL 183
mf133464_13450.00mf133464_13440.83mf133464_13723.31
GL 184rna78819_830.00rna78819_41rna78819_402.42
rna78819_593.22
GL 185mf121346_249:rna49630_7060.00SCJFFL3C03D06_51.52SCJFFL3C03D06_453.03
GL 186rna25641_327rna25641_3260.00
rna25641_3233.00
GL 187mf235706_5820.00rna82715_1220.72SCJLLR1105F05_2852.95
GL 188mf74707_1692mf74707_16900.00
SCUTST3129G07_522.18SCUTST3129G07_532.90
GL 189mf132449_7100.00rna37006_364rna37006_3691.45
rna37006_3842.87
GL 190
mf58011_1340.00mf58011_1350.71mf58011_1461.43mf58011_1402.86
GL 191mf56960_1900.00mf56960_1530.70mf56960_1882.11mf56960_1852.82
GL 192SCSGAD1143C06_7210.00SCSGAD1143C06_7240.90SCSGAD1143C06_7332.04SCSGAD1143C07_22.79
GL 193SCUTFL3075C10_2270.00SCUTFL3075C10_2170.72SCUTFL3075C10_2192.57
GL 194rna6396_1540.00rna6396_1690.82mf65519_19862.50
GL 195rna118191_1620.00mf242733_1161.53mf242733_1262.27
GL 196rna82166_28680.00rna82166_29191.22rna82166_29182.44
GL 197mf138317_3740.00mf138317_382mf138317_3610.75
mf138317_3721.50mf138317_3662.26
GL 198
mf118920_1904:rna76795_440.00mf118920_1885:rna76795_251.10mf118920_1909:rna76795_491.83mf118920_1910:rna76795_502.20
GL 199rna91851_5860.00rna91851_5820.73rna91851_5851.46rna91851_5872.19
GL 200rna76842_13350.00rna76842_12950.72mf127162_7501.45mf127162_7902.17
GL 201rna92142_32520.00rna92142_3250mf50676_101:SCEQRT2090H04_5052.16
GL 202mf145392_6420.00mf145392_6400.72mf145392_6341.44mf145392_6432.15
GL 203rna90832_21780.00rna90832_21711.60rna90832_21701.99
GL 2040.000.390.771.93
mf153107_406mf153107_404mf153107_402mf153107_410
GL 205
rna66308_2150.00rna66308_2251.16rna66308_2241.93
GL 206mf149100_3570.00mf149100_3550.39mf149100_3541.93
GL 207rna107958_330.00rna107957_3950.46mf68587_20191.37mf68587_20201.80
GL 208rna90457_17820.00rna90457_17660.85rna90457_1774rna90457_17650.86
rna90457_17711.79
GL 209mf214894_1460.00mf214894_144mf214894_1070.80
mf214894_1311.61
GL 210mf41970_6630.00mf41970_658mf41970_6521.60
GL 211
mf269642_3010.00mf269642_321mf269642_322mf269642_320
0.79
mf269642_3241.58
GL 212
mf52975_20850.00mf52975_20830.78mf52975_20671.53
GL 213
mf298305_1450.00mf298305_1540.75mf298305_1371.50
GL 214SCEQRT2101H10_920.00SCEQRT2101H10_1490.74SCEQRT2101H10_1481.48
GL 215mf284236_2410.00mf284236_2510.73mf284236_2521.46
GL 216mf238211_1010.00mf238211_1220.73mf238211_1151.45
GL 217
mf160760_127mf160760_114mf160760_113
0.00
rna42720_609rna42720_584rna42720_606
0.36
mf160760_131rna42720_602rna42720_590
0.71
mf160760_1431.44
GL 218
rna17505_124mf123864_17760.00
mf123864_1793rna17505_1411.42
GL 219
mf127973_1301:SCEPCL6021D04_3030.00SCEPCL6021D04_310SCEPCL6021D04_3081.39
GL 220
mf18436_500.00mf18436_680.37SCSFFL3092B07_1880.75SCSFFL3092B07_2051.12
GL 221rna74937_900.00rna74937_760.36rna74937_831.07
GL 222mf166881_980.00mf166881_940.71mf166881_1041.07
GL 223
Figura 6 – Mapa genético de cana-de-açúcar exibindo a distribuição dos grupos de ligação (GLs) aolongo dos grupos de hom(e)ologia (GHs) e de sintenia com o sorgo (GSs). Marcadores corres-pondendo a cada cromossomo do sorgo foram destacados em cores diferentes (conclusão)
3.2.3 Mapeamento de QTLs
Para os quatro caracteres avaliados na população de mapeamento de cana-de-açúcar
(ALT, TCH, POL e FIB), um total de 36 QTLs foram detectados utilizando as abordagens de
MIM uni- e multivariado. Os QTLs estão listados de forma resumida na Tabela 11 e QTLs em
comum nas duas análises já estão agrupados com base nos intervalos de confiança dos mesmos.
Detalhadamente, os Apêndices C e D (Tabelas 14 e 15) trazem os efeitos e suas respectivas
significâncias dos QTLs para as análises, separadamente.
Caracteres agronômicos ALT e TCH resultaram em um maior número de QTLs quando
comparados aos caracteres industriais POL e FIB. As análises para ALT e TCH determinaram
a possível existência de nove e 15 QTLs, respectivamente. Já as análises para POL resultaram
em quatro QTLs, e, para FIB, em oito. Interessantemente e de um modo geral, cada uma das
abordagens uni- e multivariada agregaram informações uma em relação à outra. Assim, por

61
Tabela 11 – Grupos de ligação (GLs) e posições (em centiMorgans – cM) dos QTLsdetectados em população de mapeamento de cana-de-açúcar utilizandomodelos mapeamento de múltiplos intervalos (MIM)
Caráter QTL GL Posição (cM) Caráter QTL GL Posição (cM)
ALT 1 1 53,5 TCH 10 122 2,02 24 1,0-51,0 11 131 0,03 43 21,8 12 133 36,44 46 181,0 13 163 0,05 83 58,0 14 167 4,0-6,06 112 0,0 16 207 0,4-1,07 117 35,5 POL 1 4 43,38 133 20,0 2 6 29,69 222 0,0 3 26 22,0-28,5
TCH 1 1 33,7-75,0 4 97 3,72 5 17,9 FIB 1 6 4,0-5,03 7 0,0 2 9 1,04 42 20,2 3 26 18,05 43 0,0-16,0 4 50 10,66 64 3,0-4,9 5 60 7,0-9,07 71 40,0 6 124 2,0-6,08 72 31,4 7 150 14,0-15,09 88 6,6 8 167 0,0
exemplo, a um único QTL em comum entre as análises (no GL 24) para ALT acrescentaram-se
cinco e três diferentes regiões exclusivas a partir das respectivas análises uni- e multivariada.
Para TCH, os dois QTLs em comum (nos GLs 1 e 167) apareceram entre nove e quatro QTLs
adicionais originados das análises uni- e multivariadas, respectivamente. Para POL, a análise
multivariada não acrescentou informações, pois os dois QTLs (nos GLs 4 e 6) já haviam sido
descobertos nas análises separadas. E, finalmente, para FIB, nenhum dos QTLs coincidiram
entre as análises.
Da análise de marcadores individualmente, foram obtidas um total de 198 associações
significativas (p < 0,01) para as médias conjuntas dos quatro caracteres avaliados, sendo 56 para
ALT, 38 para TCH, 57 para POL e 47 para FIB. Efeitos dos marcadores sobre as médias mar-
ginais para cada ambiente resultaram no mesmo conjunto de marcadores significativos basica-
mente (dados não mostrados), e foram desconsiderados nas avaliações subsequentes. Também,
apenas marcadores com informação genômica adicional relacionada ao genoma do sorgo foram
listados no Apêndice E (Tabela 16). Assim, para cada caráter restaram, respectivamente, 31, 17,
24 e 27 marcadores contendo informação útil na etapa de comparação posterior. Mapeamento

62
de QTLs baseado em análise de marcadores individualmente tem implementação simples e
rápida, e não requer que o locos estejam posicionados no mapa de ligação. No entanto, as
estimativas dos efeitos são demasiadamente enviesadas pela existência pelo desequilíbrio de
ligação não considerado porque a maioria dos marcadores simplesmente falha em agrupar-se
no mapa. Além disso, a análise não permite separar efeitos aditivos e dominantes em população
F1 segregante. Modernamente, essas avaliações prestam-se como uma análise exploratória, mas
tem sido bastante utilizada no contexto dos estudos genéticos em cana-de-açúcar.
Pastina et al. (2010) revisaram inúmeros trabalhos de mapeamento de QTLs e listaram
seus principais achados, sendo os mesmos quase sempre limitados a mapas pouco saturados
e metodologia de mapeamento baseada na análise de marcas individualmente. Além disso,
a utilização de marcadores anônimos (sem informação genômica), dificulta comparação entre
trabalhos. Já em comparação ao mapeamento na população de sorgo sacarino aqui trabalhada,
menos regiões associadas a caracteres quantitativos utilizando MIM foram encontradas na po-
pulação de cana-de-açúcar. Além da limitação dos modelos de construção de mapas genéticos
e de mapeamento de QTLs considerando doses únicas apenas, a análise em cana-de-açúcar
baseou-se em uma população com genitores relativamente pouco constrastantes.
Marcadores segregando em doses únicas são resultados de tipos muito específicos de
cruzamentos e, como observado neste trabalho e na literatura (GARCIA et al., 2013), evidências
recentes afirmam que esse tipo de segregação não corresponde à maioria daquelas passíveis de
detecção em uma população de cana. Portanto, a busca por QTLs fica, em adição, restrita
à probabilidade de os alelos dos QTLs também estarem segregando em dose única ao longo
genoma e de estarem em desequilíbrio de ligação com as regiões então amostradas por esses
marcadores. Futuramente, a construção de mapas genéticos considerando doses superiores e
a utilização modelos de mapeamento de QTLs adequados possibilitará um conhecimento mais
refinado da arquitetura genética dos caracteres quantitativos na espécie.
3.3 Mapeamento Comparativo de QTLs
As origens em comum dos marcadores baseados em GBS e a avaliação de caracteres
similares em ambas as populações possibilitaram a comparação dos QTLs mapeados. Este
alinhamento baseou-se nos marcadores SNPs e indels de cana originados a partir da utilização
do genoma do sorgo como referência quando da descoberta e genotipagem desses polimorfis-
mos utilizando o pipeline Tassel-GBS. Aqui, os GLs de cana com QTLs mapeados utilizandos

63
modelos MIM uni- e multivariados foram alinhados em relação aos cromossomos do sorgo
considerando os GSs a que pertenciam (Figura 7). Em adição, marcadores analisados sepa-
radamente, com efeitos significativos sobre as médias conjuntas dos caracteres também foram
alinhados aos cromossomos do sorgo de acordo com suas posições relativas à referência (Figura
8).
Nas figuras, é possível observar, inicialmente, a distribuição relativa dos marcadores
SNPs e indels ao longo do genoma do sorgo, com algumas regiões em branco, não cobertas por
marcadores. Esses setores sub-representados estariam, possivelmente, relacionadas às regiões
centroméricas e para-centroméricas, dentre outras, evitadas por conta da sensibilidade à meti-
lação da enzima ApeKI utilizada (ELSHIRE et al., 2011). Também, regiões repetitivas, como
são as centroméricas, costumam alinhar-se pobremente às referências, causando-lhes um viés
de representação.
Interessantemente, os conectores entre os cromossomos do sorgo e os GLs de cana
revelaram potenciais locais conservados evolutivamente envolvidos no controle genético dos
caracteres em comum avaliados nas duas populações (Figura 7). Aqui, essas regiões foram
listadas na ordem sorgo/cana com que foram nomeadas anteriormente (Tabelas 5 e 11). Para
ALT, três QTLs em regiões sintênicas puderam ser evidenciados: ALT-1/ALT-1 no GS 1, ALT-
4/ALT-3 no GS 3 e ALT-8/ALT-7 no GS 8. Com evidência indiretas, também é possível destacar
ALT-11/ALT-8 no GS 10 e, ainda, aparecem nos mesmos GSs: ALT-2/ALT-2 (GS 2) e ALT-
7/ALT-5 (GS 6). Entre PMV e TCH, três correspondências genômicas foram encontradas em
relação a PMV-4/TCH-6 no GS 4 e PMV-10/TCH-10 no GS 9; também mais distantemente
mapeados estão PMV-6,PMV-7/TCH-9 no GS 6 e PMV-11/TCH-12 no GS 10. No caso de POL,
próxima relação evolutiva parece existir para POL-1/POL-2 no GS 1, e, no mesmo GS, poderia
ser especulada uma relação entre POL-2/POL-1. Finalmente, para FIB, há evidência de con-
servação para o par de locos FIB-5/FIB5 no GS 4 e FIB-10/FIB-6 no GS 9; além destes, estão
presentes nos mesmos GSs: FIB-1/FIB-1,FIB-2 (GS 1) e FIB-2/FIB-3 (GS 2). Assim, dentre o
total de 89 QTLs mapeados nas populações de sorgo e cana, 34 (38,2%) estão envolvidos em 16
regiões putativas de conservação de controle genético para os quatro caracteres avaliados, sendo
oito regiões fisicamente mais próximas aos marcadores em comum do que as oito restantes.
Estas últimas regiões listadas podem ter apresentado complicações na comparação em função
de rearranjos cromossômicos após divergência entre as espécies.
Já os marcadores com efeitos significativos sobre as médias conjuntas dos caracteres

64
PMV-8 POL-11
Cr. 7
ALT-1 POL-1
POL-2
FIB-1
Cr. 1
mf169280_453
mf107365_1630:rna66593_1079
sb1_71234628
mf6005_4200
sb1_69468570sb1_70966878:S
CRUFL3064D
01_612m
f4471_3697
mf139503_870:sb1_73150959:rna93460_2746
ALT-1
TCH-1
GL
1
mf21975_1907
sb1_31631777sb1_31631788sb1_31631778m
f157160_758m
f60753_2015m
f60753_2013
POL-1
GL
4
mf130222_16:sb1_9205170
mf94307_1830:sb1_10284944
mf94307_1834:sb1_10284940
mf94307_1831:sb1_10284943
mf94307_1842:sb1_10284932
mf10516_2074:sb1_11491234:rna85024_4023
TCH-2
GL
5
mf134350_155:sb1_9184184:rna84395_1077
sb1_9184193
mf62649_246
POL-2 FIB-1
GL
6
sb1_60124665sb1_60124681sb1_60124682
TCH-3
GL
7
sb1_5637679sb1_5637676
mf137319_1123
FIB-2
GL
9
mf43680_1882:rna87845_1876
sb2_76201461
mf135308_198:rna87035_964
rna36483_81
mf28963_593:rna71428_898
ALT-2
GL
24
sb2_73250937
sb2_67708474sb8_1755137sb8_1755177
mf162663_776
SCEPRZ3085A
05_495rna36509_316
POL-3
FIB-3
GL
26
ALT-2 PMV-1PMV-2 FIB-2
Cr. 2
ALT-3ALT-4 PMV-3 POL-3POL-4POL-5
POL-6
FIB-3FIB-4
Cr. 3
mf161288_908:sb3_71013188
mf161289_6
mf161288_909:sb3_71013183
mf161288_912:sb3_71013184
rna8915_76m
f25124_2613m
f25124_2612m
f25124_2614
ALT-3
TCH-5
GL
43
sb4_5027183
mf152767_34:sb3_46819059
sb3_7546829
rna90267_787
mf148614_22:sb2_74689463:rna63124_339
mf22704_2786
rna75825_477m
f116093_1952m
f209_8523sb7_31444384
mf48310_1950:rna31514_183
rna94083_4829m
f298625_214m
f23282_1264m
f131549_185m
f118315_112m
f263837_508sb1_29954416sb1_29954417m
f125302_409rna38643_345rna63025_162sb8_11128174
SCQ
GLV
1017E09_203
mf134709_228
rna66129_815m
f25719_1557
ALT-4
GL
46
mf246042_62:rna93296_2814
sb3_5691332sb3_5691356sb3_5691380
FIB-4
GL
50
ALT-5
PMV-4PMV-5
FIB-5FIB-6
Cr. 4
mf39868_1370
sb4_9379sb4_9380sb4_9342sb4_9365
FIB-5
GL
60
mf171030_181
mf171030_180sb4_4524455sb4_4524403
TCH-6
GL
64
POL-7
POL-8
Cr. 5
CIR
12-1sb5_4107607
rna93602_1730m
f20704_1797rna93602_1705
sb5_4107582sb5_23279232m
f20704_1694TCH-7
GL
71
ALT-6ALT-7
PMV-6PMV-7POL-9
POL-10 FIB-7FIB-8
Cr. 6
mf101931_1974
mf101931_1975
SCB45-15
mf66100_2040:rna76662_576
mf193065_406:rna91599_577
sb6_60843160
mf138081_984:sb6_58911526:rna92992_5505
sb6_53424155m
f21210_3491rna65009_82
SCEQ
RT1031B
01_43m
f130399_989:rna91800_3110sb6_61974231
ALT-5
GL
83
sb6_61033407sb6_61033394sb6_61033393
TCH-9
GL
88
ALT-8 PMV-9POL-12POL-13POL-14
FIB-9
Cr. 8
sb8_54692521rna19848_585rna19848_584
mf89788_1742:rna11183_57
mf129609_497:sb8_45219409:rna53230_476
sb8_48467970m
f1831_650sb8_54820882sb8_54820902
ALT-7
GL
117
ALT-9ALT-10
PMV-10
POL-15
POL-16
FIB-10
Cr. 9
mf7015_3819
sb9_2129715
mf176614_73:sb8_814697
mf56267_75
mf2389_108
rna86401_593
TCH-10
GL
122
mf333_8772
rna69406_73
mf268853_604
mf70727_2027sb9_2082493
sb5_17237746
FIB-6
GL
12
4
mf64272_332
sb9_54211376sb9_54211392m
f64272_325 GL
13
1TCH-11
ALT-11ALT-12
PMV-11PMV-12
POL-17
FIB-11FIB-12 Cr. 10
mf44188_67:S
CQ
GST3154C
02_102
rna89011_2304rna103356_49m
f27574_2271rna74863_164rna78920_239m
f242090_501
:sb10_3101695
:SCVPFL1131
C02_48
3
ALT-8TCH-12
GL
13
3
0246810121416182022242628303234363840424446485052545658606264666870727476
(Mb)
0246810121416182022242628303234363840424446485052545658606264(M
b)
Figura7
–A
linhamento
dosgrupos
deligação
(GL
s)de
cana-de-açúcarem
relaçãoaos
cromossom
os(C
r.)de
sorgo.B
arrasverticais
delimitam
picosde
QT
Ls
detectadosemam
bososmapeam
entosdem
últiplosintervalosuni-em
ultivariados.Marcadoresde
cana-de-açúcarcorrespondendoa
cadacrom
ossomo
dosorgo
foramdestacados
emcores
diferentes.Réguas
lateraisestão
mega
bases(M
b)

65
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58 60 62 64 66 68 70 72 74 76 (Mb)
ALT-1POL-1
POL-2
FIB-1Cr.
1sb
1_30
7723
5
TCH
sb1_
6035
7211
TCH
sb1_
7645
431:
mf2
4082
_241
:rna
7146
1_58
4
POL
sb1_
9236
816
POL
sb1_
1641
7625
:mf1
5545
9_26
4
POL
sb1_
1641
7644
:mf1
5545
9_24
5
POL
sb1_
3148
8584
POL
sb1_
3162
4046
:rna
1173
39_1
89
POL
sb1_
1869
6270
FIB
sb1_
50799
049:
mf1
0280
9_26
:rn
a7691
0_20
9:SCM
CSD20
59B
01_30
6
FIB
sb1_
7339
8501
:mf1
6444
2_75
4
FIB
sb1_
5267
1762
ALT
sb1_
1445
5981
ALT
sb1_
1221
9024
ALT
sb1_
4188
246:
mf1
6342
1_39
4:rn
a984
20_1
33
ALT
ALT-2PMV-1 PMV-2FIB-2Cr.
2
sb2_
22609
350:
mf1
1974
5_84
7:rn
a8027
8_84
9
ALT
sb2_
68475
923:
mf2
3247
_584
:rn
a6999
3_35
1
ALT
sb2_
7455
650
TCH
sb2_
5002
6760
TCH
sb2_
6008
2918
POL
sb2_
6054
7042
POL
sb2_
6120
2411
POL
sb2_
6205
7915
:mf4
5156
_460
POL
sb2_
6533
0382
:mf1
0732
5_50
POL
sb2_
6784
9128
POL
sb2_
6817
295
FIB
sb2_
5224
2660
FIB
sb2_
6054
7040
FIB
sb2_
6357
7008
FIB
sb2_
71421
489:
mf1
9659
9_40
6:rn
a8049
7_17
02:
SCQ
GAM
210
8C09
_244
FIB
ALT-3 ALT-4PMV-3POL-3 POL-4 POL-5
POL-6
FIB-3 FIB-4
Cr.
3
sb3_
3902
31:m
f104
21_2
34:rna
7995
7_29
7
ALT
sb3_
5418
65:rna
7996
4_14
21
ALT
sb3_
1928
213
ALT
sb3_
6126
551
ALT
sb3_
7546
835
ALT
sb3_
9286
412
ALT
sb3_
7333
7885
ALT
sb3_
8750
650
TCH
sb3_
5211
7257
TCH
sb3_
7212
8809
TCH
sb3_
2978
336
POL
sb3_
5788
3510
:mf1
9116
_195
5
POL
sb3_
5418
04
FIB
sb3_
5418
26
FIB
sb3_
5691
216
FIB
sb3_
1477
1406
FIB
sb3_
1825
9258
:mf1
4542
9_19
6:rn
a898
60_5
10
FIB
sb3_
5892
4723
FIB
sb3_
6828
7506
FIB
sb3_
7204
6711
:mf1
1840
0_18
10
FIB
sb3_
7204
6712
:mf1
1840
0_18
11
FIB
ALT-5
PMV-4 PMV-5
FIB-5 FIB-6Cr.
4
sb4_
9399
ALT
sb4_
4976
3421
ALT
sb4_
6365
4242
ALT
sb4_
6460
4253
ALT
sb4_
1732
79
TCH
sb4_
7119
89
TCH
sb4_
61731
192:
mf4
575_
1833
:rna7
8723
_91
POL
sb4_
9380
FIB
sb4_
59449
990:
mf3
033_
3849
:rna4
9437
_175
FIB
sb4_
6038
7435
FIB
POL-7
POL-8
Cr.
5
sb5_
1723
7746
ALT
sb5_
4853
3705
ALT
sb5_
4853
3707
ALT
sb5_
61994
091:
mf6
4351
_190
0:rn
a6861
2_53
9
ALT
sb5_
4806
6020
TCH
sb5_
14386
234:
mf1
3178
3_82
2:SCRUFL1
022E
03_4
14
POLsb
5_29
034
328:
mf1
4690
3_96
8:rn
a476
53_
53FIB
sb5_
5014
5700
:mf1
7681
7_63
2
FIB
sb5_
5476
0011
FIB
ALT-6 ALT-7
PMV-6 PMV-7 POL-9
POL-10FIB-7 FIB-8Cr.
6
sb6_
4503
2977
ALT
sb6_
7821
63:m
f600
82_4
3
TCH
sb6_
4503
2977
TCH
sb6_
6056
0180
:rna
9354
4_97
0
POL
sb6_
5730
5087
FIB
PMV-8POL-11Cr.
7
sb7_
5354
4684
:rna
8026
9_11
00
ALT
sb7_
7580
104:
mf3
440_
5522
TCH
sb7_
56993
024:
mf1
3213
0_27
9:rn
a926
58_
443
TCH
sb7_
3717
230:
mf1
0121
5_19
80
FIB
ALT-8PMV-9 POL-12 POL-13 POL-14
FIB-9
Cr.
8
sb8_
3123
434
ALT
sb8_
1112
8174
ALT
sb8_
5298
195:
mf1
2931
7_11
84
TCH
sb8_
5688
896
FIB
sb8_
5386
7342
FIB
ALT-9 ALT-10
PMV-10
POL-15
POL-16
FIB-10
Cr.
9
sb9_
4917
2170
ALT
sb9_
4917
2185
ALT
sb9_
4553
6069
TCH
sb9_
1087
507
POL
sb9_
4492
9840
POL
ALT-11 ALT-12
PMV-11 PMV-12
POL-17
FIB-11 FIB-12
Cr.
10
sb10
_1156
537:
mf1
7153
9_60
1:rn
a7750
0_45
8:SCJF
FL1C
08G
04_21
8
ALT
sb10
_115
6542
ALT
sb10
_326
0857
ALT
sb10
_326
0858
ALT
sb10
_899
9958
TCH
sb10
_5265
0429
:mf1
4695
7_2
46:
rna8
857
5_13
70:S
CJL
FL1
051F
07_6
36
POL
sb10
_562
9589
5
POL
sb10
_5654
0062
:rna
15848
_178
:SCBG
LB20
13E05
_99
POL
sb10
_601
3129
5
POL
sb10
_601
3129
6
POL
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58 60 62 64 (Mb)
Figu
ra8
–A
linha
men
todo
sm
arca
dore
sco
mef
eito
ssi
gnifi
cativ
osem
popu
laçã
ode
cana
-de-
açúc
arem
rela
ção
aos
crom
osso
mos
(Cr.)
deso
rgo.
Mar
cado
res
deca
na-d
e-aç
úcar
corr
espo
nden
doa
cada
crom
osso
mo
doso
rgo
fora
mde
stac
ados
emco
res
dife
rent
es.R
égua
sla
tera
ises
tão
meg
aba
ses
(Mb)

66
estiveram presentes em regiões identificadas anteriormente e, em adição, detectaram outros
locais de possível controle genético conservado (Figura 8). O número de marcadores ali-
nhados variou de quatro (no cromossomo 7) a 21 (no cromossomo 3). Do mesmo modo,
combinações de QTLs e marcadores são listadas na ordem sorgo/cana para destacar o rela-
cionamento; um sinal de “+” foi adicionado para o caso de existir mais de um marcador
significativo para o caráter na mesma região. Para ALT, aparece, como possível confirma-
ção, ALT-2/sb2_22609350, ALT-4/sb3_73337885 e ALT-7/sb6_45032977, e, como novidades,
ALT-5/sb4_49763421 e ALT-10/sb9_49172170+. No caso de PMV e TCH, aparecem PMV-
4/sb4_173279+, PMV-7/sb6_45032977 e PMV-11/sb10_8999958 para regiões já identificadas,
e PMV-1/sb2_7455650, PMV-8/sb7_56993024 e PMV-9/sb8_5298195 como novos putativos
locais de sintenia. No que se refere ao controle de POL, apenas um local já havia sido indicado
(POL-1/sb1_7645431+), enquanto ficaram evidentes, pela primeira vez, POL-3/sb3_2978336,
POL-5/sb3_57883510 e POL-15/sb9_1087507. E, finalmente, para FIB, já se conhecia a re-
gião FIB-5/sb4_9380, assim como FIB-2/sb2_6817295, FIB-3/sb3_58924723 e FIB-4/sb3_-
68287506+ já haviam sido citados. Desse modo, marcadores que, separadamente, exibiram
efeitos significativos sobre a variação dos quatro caracteres contribuíram, adicionalmente, com
mais oito regiões que possivelmente compartilham o controle genético desses caracteres nas
duas espécies. Além disso, houve confirmação para 11 regiões citadas na análise anterior.
Surpreendentemente, nenhum QTL para POL-6 foi detectado nos GLs de cana perten-
centes ao GS 3, tampouco marcadores individuais relacionados ao cromossomo 3 do sorgo
apresentaram correspondência. Na população de sorgo sacarino, este QTL é bastante relevante
para explicação da variabilidade fenotípica do caráter POL. Como já foi discutido, a fixação
de alelos desta região na população de cana, já determinando o elevado conteúdo de açúcar
conhecido nas variedades parentais, possivelmente limitou a detecção de QTLs. De todo modo,
foi possível observar a capacidade de interligar os dois conjuntos genômicos por meio de
sequências de marcadores evolutivamente compartilhadas, tornando viável estudos genômicos
posteriores visando à caracterização dessas regiões interespecificamente.

67
4 CONSIDERAÇÕES FINAIS
Neste trabalho, populações de mapeamento de sorgo sacarino e de cana-de-açúcar foram
caracterizadas fenotípica e genotipicamente. Os fenótipos referiram-se aos caracteres agro-
industriais altura de colmos, toneladas de colmos ou de massa verde por hectare, e porcentagens
de pol de caldo e de fibra, importantes em culturas bioenergéticas. Em adição, as populações
também foram caracterizadas por meio de marcadores moleculares, com ênfase em SNPs e
indels, originados a partir de técnica de genotipagem por sequenciamento. Obviamente, ambos
os tipos de dados exigiram ferramentas apropriadas no sentido de explorar ao máximo suas
informações.
A abordagem de modelos mistos via REML foi utilizada para tratar os dados fenotí-
picos por combinar formulação adequeada aos complexos experimentos genéticos, método de
estimação de parâmetros apropriado e estruturação das matrizes de variâncias-covariâncias dos
efeitos aleatórios. Assim sendo, foi possível atribuir aos efeitos genéticos e residuais diferen-
tes estruturas, cada qual mais plausível aos carateres analisados sob determinado contexto de
delineamento e de população estudada. Matrizes exibindo variâncias genéticas heterogêneas,
por exemplo, foram selecionadas de um modo geral, exemplificando claramente a violação de
pressupostos dos modelos de análise de variância comumente utilizados no melhoramento e,
adicionalmente, enfatizando a existência não negligenciável da interação genótipo-ambiente.
Ainda, dados de sequenciamento da próxima geração de bibliotecas de GBS para cada
população foram avaliados quanto à performance na etapa simultânea de descoberta e genoti-
pagem de polimorfismos derivados de substituições de bases e de inserções-deleções. Basica-
mente, a técnica constituiu-se como uma importante estratégia na tentativa de refinar estudos
genéticos em ambas as espécies. Primeiro, porque GBS apareceu ineditamente dentre trabalhos
relacionados ao mapeamento de QTLs em populações de sorgo sacarino e em cana-de-açúcar,
conferindo saturação e informatividade genética aos genomas. Segundo, porque forneceu,
para cana, a possibilidade de investigar, com uma maior abrangência genômica, a questão
relacionada à dosagem alélica de locos em uma população biparental utilizando o software
SuperMASSA e a elaboração de denso e saturado mapa genético com auxílio do pacote do
R OneMap, para o qual metodologia para incoporação de marcadores em doses superiores é
prevista. E, finalmente, porque subsidiou a comparação entre os genomas das duas espécies,
sob o pressuposto de considerável conservação entre suas sequências genômicas.
De fato, detectar QTLs para caracteres relacionados a produção e açúcar em popu-

68
lações de mapeamento de espécies com fins bioenergéticos constitui uma valiosa etapa para
compreensão do controle genético subjacente à variação fenotípica útil ao melhoramento. Nos
casos das populações de sorgo sacarino e cana-de-açúcar aqui utilizadas, ambas se beneficiaram
das estratégias de mapeamento de múltiplos intervalos uni- e multivariadas para a descoberta
de QTLs implementadas no pacote do R OneQTL. Além disso, a tradicional metodologia de
análise de marcas individualmente acrescentou novas descobertas para a população de cana-de-
açúcar. Este trabalho destaca-se, sobretudo, pela utilização de fenótipos comparáveis entre as
espécies, o que é raramente encontrado na literatura. Nesse sentido, cana-de-açúcar beneficiou-
se importantemente do relacionamento evolutivo próximo com sorgo, uma vez que não possui
espécie diploide relativa, como em batata. Ainda, pode-se atribuir a esse relacionamento a
possibilidade de verificar a consistência em mapas de populações diferentes de cana-de-açúcar.
Uma vez que ambas as populações estão ligadas a instituições brasileiras mantenedoras
de importantes programas de melhoramento para as espécies, há plena possibilidade de incorpo-
ração dessas descobertas convenientemente em seus trabalhos de pesquisa e de desenvolvimento
de tecnologias e de cultivares ou variedades. Ou seja, com base na comparação dos genomas, o
reconhecimento de várias regiões de potencial controle evolutivamente conservado demonstrou
evidências de plausível utilização de estratégias visando a clonagem de genes ou de seleção
assistida por marcadores.

69
REFERÊNCIAS
AITKEN, K. S.; HERMANN, S.; KARNO, K.; BONNETT, G. D.; MCINTYRE, L. C.;JACKSON, P. A. Genetic control of yield related stalk traits in sugarcane. Theoretical andApplied Genetics, Berlin, v. 117, n. 7, p. 1191–1203, 2008.
AITKEN, K. S.; MCNEIL, M. D.; HERMANN, S.; BUNDOCK, P. C.; KILIAN, A.;HELLER-USZYNSKA, K.; HENRY, R. J.; LI, J. A comprehensive genetic map of sugarcanethat provides enhanced map coverage and integrates high-throughput Diversity ArrayTechnology (DArT) markers. BMC Genomics, London, v. 15, p. 152, 2014.
AKAIKE, H. A new look at the statistical model identification. Transactions on AutomaticControl, Notre Dame, v. 19, n. 6, p. 716–723, 1974.
ALI, M. L.; RAJEWSKI, J. F.; BAENZIGER, P. S.; GILL, K. S.; ESKRIDGE, K. M.;DWEIKAT, I. Assessment of genetic diversity and relationship among a collection of US sweetsorghum germplasm by SSR markers. Molecular Breeding, Berlin, v. 21, n. 4, p. 497–509,2007.
ALWALA, S.; KIMBENG, C. A.; GRAVOIS, K. A.; BISCHOFF, K. P. TRAP, a new tool forsugarcane breeding: comparison with AFLP and coefficient of parentage. Journal AmericanSociety Sugar Cane Technologists, Belle Glade, v. 26, p. 62–86, 2006.
AMORIM, H. V.; LOPES, M. L.; OLIVEIRA, J. V. C.; BUCKERIDGE, M. S.; GOLDMAN,G. H. Scientific challenges of bioethanol production in Brazil. Applied Microbiology andBiotechnology, Berlin, v. 91, n. 5, p. 1267–1275, 2011.
ANDRU, S.; PAN, Y.-B.; THONGTHAWEE, S.; BURNER, D. M.; KIMBENG, C. A. Geneticanalysis of the sugarcane (Saccharum spp.) cultivar ‘LCP 85-384’. I. Linkage mapping usingAFLP, SSR, and TRAP markers. Theoretical and Applied Genetics, Berlin, v. 123, n. 1, p.77–93, 2011.
BARTHOLOME, J.; MANDROU, E.; MABIALA, A.; JENKINS, J.; NABIHOUDINE, I.;KLOPP, C.; SCHMUTZ, J.; PLOMION, C.; GION, J.-M. High-resolution genetic maps ofEucalyptus improve Eucalyptus grandis genome assembly. The New Phytologist, Cambridge,Early View, p. 1–14, 2014.
BENJAMINI, Y.; YEKUTIELI, D. The control of the false discovery rate in multiple testingunder dependency. The Annals of Statistics, Hayward, v. 29, n. 4, p. 1165–1188, 2001.
BENNETZEN, J. L.; FREELING, M. The unified grass genome: synergy in synteny. GenomeResearch, Cold Spring Harbor, v. 7, p. 301–306, 1997.
BROWN, S. M.; HOPKINS, M. S.; MITCHELL, S. E.; SENIOR, M. L.; WANG, T. Y.;DUNCAN, R. R.; GONZALEZ-CANDELAS, F.; KRESOVICH, S. Multiple methods for theidentification of polymorphic simple sequence repeats (SSRs) in sorghum [Sorghum bicolor(L.) Moench]. Theoretical and Applied Genetics, Berlin, v. 93, p. 190–198, 1996.
CALVIÑO, M.; MESSING, J. Sweet sorghum as a model system for bioenergy crops. CurrentOpinion in Biotechnology, London, v. 23, n. 3, p. 323–329, 2012.

70
CARDOSO-SILVA, C. B.; COSTA, E. A.; MANCINI, M. C.; BALSALOBRE, T. W. A.;CANESIN, L. E. C.; PINTO, L. R.; CARNEIRO, M. S.; GARCIA, A. A. F.; SOUZA, A. P.;VICENTINI, R. De novo assembly and transcriptome analysis of contrasting sugarcanevarieties. PLoS ONE, San Francisco, v. 9, n. 2, p. 1–10, 2014.
CHEAVEGATTI-GIANOTTO, A.; ABREU, H. M. C.; ARRUDA, P.; BESPALHOK-FILHO,J. C.; BURNQUIST, W. L.; CRESTE, S.; DI-CIERO, L.; FERRO, J. A.; FIGUEIRA, A.V. O.; FILGUEIRAS, T. S.; GROSSI-DE-SA, M. F.; GUZZO, E. C.; HOFFMANN, H. P.;LANDELL, M. G. A.; MACEDO, N.; MATSUOKA, S.; REINACH, F. C.; ROMANO, E.;SILVA, W. J.; SILVA-FILHO, M. C.; ULIAN, E. C. Sugarcane (Saccharum × officinarum): areference study for the regulation of genetically modified cultivars in Brazil. Tropical PlantBiology, Berlin, v. 4, n. 1, p. 62–89, 2011.
CONSELHO DOS PRODUTORES DE CANA-DE-AÇÚCAR, AÇÚCAR E ÁLCOOL DOESTADO DE SÃO PAULO – CONSECANA-SP. Manual de Instruções. 5. ed. Piracicaba:CONSECANA-SP, 2006. 112 p.
CORDEIRO, G. M.; TAYLOR, G. O.; HENRY, R. J. Characterisation of microsatellite markersfrom sugarcane (Saccharum sp.), a highly polyploid species. Plant Science, Shannon, v. 155,n. 2, p. 161–168, 2000.
CRESTE, S.; ACCORONI, K. A. G.; PINTO, L. R.; VENCOVSKY, R.; GIMENES, M. A.;XAVIER, M. A.; LANDELL, M. G. A. Genetic variability among sugarcane genotypesbased on polymorphisms in sucrose metabolism and drought tolerance genes. Euphytica,Wageningen, v. 172, n. 3, p. 435–446, 2010.
DEOKAR, A. A.; RAMSAY, L.; SHARPE, A. G.; DIAPARI, M.; SINDHU, A.; BETT, K.;WARKENTIN, T. D.; TAR’AN, B. Genome wide SNP identification in chickpea for use indevelopment of a high density genetic map and improvement of chickpea reference genomeassembly. BMC Genomics, London, v. 15, n. 1, p. 708, 2014.
D’HONT, A.; ISON, D.; ALIX, K.; ROUX, C.; GLASZMANN, J. C. Determination of basicchromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes.Genome, Ottawa, v. 41, p. 221–225, 1998.
DUFOUR, P.; DEU, M.; GRIVET, L.; D’HONT, A.; PAULET, F.; BOUET, A.; LANAUD, C.;GLASZMANN, J. C.; HAMON, P. Construction of a composite sorghum genome map andcomparison with sugarcane, a related complex polyploid. Theoretical and Applied Genetics,Berlin, v. 94, n. 3–4, p. 409–418, 1997.
ELSHIRE, R. J.; GLAUBITZ, J. C.; SUN, Q.; POLAND, J. A.; KAWAMOTO, K.; BUCKLER,E. S.; MITCHELL, S. E. A robust, simple genotyping-by-sequencing (GBS) approach for highdiversity species. PLoS ONE, San Francisco, v. 6, n. 5, p. 1–9, 2011.
FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS – FAO.FAOSTAT Analysis. 2014. Disponível em: <http://faostat3.fao.org/home/index.html>. Acessoem: 14 nov. 2014.
FEDERER, W. T.; RAGHAVARAO, D. On augmented designs. Biometrics, Washington,v. 31, n. 1, p. 29–35, 1975.

71
GALECKI, A.; BURZYKOWSKI, T. Linear Mixed-Effects Models Using R: a step-by-stepapproach. New York: Springer, 2013. 556 p.
GARCIA, A. A. F.; KIDO, E. A.; MEZA, A. N.; SOUZA, H. M. B.; PINTO, L. R.; PASTINA,M. M.; LEITE, C. S.; SILVA, J. A. G.; ULIAN, E. C.; FIGUEIRA, A. V.; SOUZA, A. P.Development of an integrated genetic map of a sugarcane (Saccharum spp.) commercialcross, based on a maximum-likelihood approach for estimation of linkage and linkage phases.Theoretical and Applied Genetics, Berlin, v. 112, n. 2, p. 298–314, 2006.
GARCIA, A. A. F.; MOLLINARI, M.; MARCONI, T. G.; SERANG, O. R.; SILVA, R. R.;VIEIRA, M. L. C.; VICENTINI, R.; COSTA, E. A.; MANCINI, M. C.; GARCIA, M.O. S.; PASTINA, M. M.; GAZAFFI, R.; MARTINS, E. R. F.; DAHMER, N.; SFORCA,D. A.; SILVA, C. B. C.; BUNDOCK, P.; HENRY, R. J.; SOUZA, G. M.; VAN SLUYS,M.-A.; LANDELL, M. G. A.; CARNEIRO, M. S.; VINCENTZ, M. A. G.; PINTO, L. R.;VENCOVSKY, R.; SOUZA, A. P. SNP genotyping allows an in-depth characterisation of thegenome of sugarcane and other complex autopolyploids. Scientific Reports, London, v. 3,n. 3399, p. 1–10, 2013.
GAZAFFI, R.; MARGARIDO, G. R. A.; PASTINA, M. M.; MOLLINARI, M.; GARCIA,A. A. F. A model for quantitative trait loci mapping, linkage phase, and segregation patternestimation for a full-sib progeny. Tree Genetics & Genomes, Berlin, v. 10, n. 4, p. 791–801,2014.
GLAUBITZ, J. C.; CASSTEVENS, T. M.; LU, F.; HARRIMAN, J.; ELSHIRE, R. J.; SUN, Q.;BUCKLER, E. S. Tassel-GBS: a high capacity genotyping by sequencing analysis pipeline.PLoS ONE, San Francisco, v. 9, n. 2, p. 1–11, 2014.
GRATIVOL, C.; REGULSKI, M.; BERTALAN, M.; MCCOMBIE, W. R.; SILVA,F. R.; ZERLOTINI-NETO, A.; VICENTINI, R.; FARINELLI, L.; HEMERLY, A. S.;MARTIENSSEN, R. A.; FERREIRA, P. C. G. Sugarcane genome sequencing by methylationfiltration provides tools for genomic research in the genus Saccharum. The Plant Journal,Oxford, v. 79, p. 162–172, 2014.
GRATTAPAGLIA, D.; SEDEROFF, R. Genetic linkage maps of Eucalyptus grandis andEucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers.Genetics, Austin, v. 1137, p. 1121–1137, 1994.
GRIVET, L.; ARRUDA, P. Sugarcane genomics: depicting the complex genome of animportant tropical crop. Current Opinion in Plant Biology, London, v. 5, n. 2, p. 122–127,2002.
GRIVET, L.; D’HONT, A.; DUFOUR, P.; HAMON, P.; ROQUEST, D. Comparative genomemapping of sugar cane with other species within the Andropogoneae tribe. Heredity, London,v. 73, p. 500–508, 1994.
GU, Z.; GU, L.; EILS, R.; SCHLESNER, M.; BRORS, B. circlize implements and enhancescircular visualization in R. Bioinformatics, Oxford, v. 30, n. 19, p. 2811–2812, 2014.
GUAN, Y.-A.; WANG, H.-L.; QIN, L.; ZHANG, H.-W.; YANG, Y.-B.; GAO, F.-J.; LI, R.-Y.;WANG, H.-G. QTL mapping of bio-energy related traits in Sorghum. Euphytica, Wageningen,v. 182, n. 3, p. 431–440, 2011.

72
GUIMARÃES, C. T.; SILLS, G. R.; SOBRAL, B. W. S. Comparative mapping ofAndropogoneae: Saccharum L. (sugarcane) and its relation to sorghum and maize.Proceedings of the National Academy of Sciences of the United States of America,Washington, v. 94, p. 14261–14266, 1997.
HAHN, M. W.; ZHANG, S. V.; MOYLE, L. C. Sequencing, assembling, and correcting draftgenomes using recombinant populations. G3, Bethesda, v. 4, n. 4, p. 669–679, 2014.
HALEY, C. S.; KNOTT, S. A. A simple regression method for mapping quantitative trait lociin line crosses using flanking markers. Heredity, London, v. 69, p. 315–324, 1992.
HOARAU, J.-Y.; GRIVET, L.; OFFMANN, B.; RABOIN, L.-M.; DIORFLAR, J.-P.; PAYET,J.; HELLMANN, M.; D’HONT, A.; GLASZMANN, J.-C. Genetic dissection of a modernsugarcane cultivar (Saccharum spp.). II. Detection of QTLs for yield components. Theoreticaland Applied Genetics, Berlin, v. 105, n. 6-7, p. 1027–1037, 2002.
HOLLAND, J. B.; NYQUIST, W. E.; CERVANTES-MARTÍNEZ, C. T. Estimating andinterpreting heritability for plant breeding: an update. In: JANICK, J. (Ed.). Plant BreedingReviews. New Jersey: John Wiley & Sons, 2003. v. 22, cap. 2, p. 9–112.
JANNOO, N.; GRIVET, L.; CHANTRET, N.; GARSMEUR, O.; GLASZMANN, J. C.;ARRUDA, P.; D’HONT, A. Orthologous comparison in a gene-rich region among grassesreveals stability in the sugarcane polyploid genome. The Plant Journal, Oxford, v. 50, n. 4, p.574–585, 2007.
KAO, C.-H.; ZENG, Z.-B.; TEASDALE, R. D. Multiple interval mapping for quantitative traitloci. Genetics, Austin, v. 152, n. 3, p. 1203–1216, 1999.
KELLER, I.; BENSASSON, D.; NICHOLS, R. A. Transition-transversion bias is not universal:a counter example from grasshopper pseudogenes. PLoS Genetics, San Francisco, v. 3, n. 2, p.185–191, 2007.
KONG, L.; DONG, J.; HART, G. E. Characteristics, linkage-map positions, and allelicdifferentiation of Sorghum bicolor (L.) Moench DNA simple-sequence repeats (SSRs).Theoretical and Applied Genetics, Berlin, v. 101, p. 438–448, 2000.
LANDER, E. S.; GREEN, P. Construction of multilocus genetic linkage maps in humans.Proceedings of the National Academy of Sciences of the United States of America,Washington, v. 84, p. 2363–2367, 1987.
LANGMEAD, B.; SALZBERG, S. L. Fast gapped-read alignment with Bowtie 2. NatureMethods, New York, v. 9, n. 4, p. 357–359, 2013.
LE CUNFF, L.; GARSMEUR, O.; RABOIN, L. M.; PAUQUET, J.; TELISMART, H.;SELVI, A.; GRIVET, L.; PHILIPPE, R.; BEGUM, D.; DEU, M.; COSTET, L.; WING,R.; GLASZMANN, J. C.; D’HONT, A. Diploid/polyploid syntenic shuttle mapping andhaplotype-specific chromosome walking toward a rust resistance gene (Bru1) in highlypolyploid sugarcane (2n ≈ 12x ≈ 115). Genetics, Austin, v. 180, n. 1, p. 649–60, 2008.
LI, G.; QUIROS, C. F. Sequence-related amplified polymorphism (SRAP), a new markersystem based on a simple PCR reaction: its application to mapping and gene tagging inBrassica. Theoretical and Applied Genetics, Berlin, v. 103, p. 455–461, 2001.

73
LIMA, M. L. A.; GARCIA, A. A. F.; OLIVEIRA, K. M.; MATSUOKA, S.; ARIZONO,H.; SOUZA JR, C. L.; SOUZA, A. P. Analysis of genetic similarity detected by AFLP andcoefficient of parentage among genotypes of sugar cane (Saccharum spp.). Theoretical andApplied Genetics, Berlin, v. 104, n. 1, p. 30–38, 2002.
MALOSETTI, M.; RIBAUT, J.-M.; VAN EEUWIJK, F. A. The statistical analysis ofmulti-environment data: modeling genotype-by-environment interaction and its genetic basis.Frontiers in Physiology, Lausanne, v. 4, p. 1–17, 2013.
MARCONI, T. G. Mapa funcional em cana-de-açúcar utilizando marcadores molecularesbaseados em SSR e SNP. 2011. 160 p. Tese (Doutorado em Genética e Biologia Molecular)— Universidade de Campinas, 2011.
MARCONI, T. G.; COSTA, E. A.; MIRANDA, H. R. C. A. N.; MANCINI, M. C.;CARDOSO-SILVA, C. B.; OLIVEIRA, K. M.; PINTO, L. R.; MOLLINARI, M.; GARCIA,A. A. F.; SOUZA, A. P. Functional markers for gene mapping and genetic diversity studies insugarcane. BMC Research Notes, London, v. 4, n. 1, p. 264, 2011.
MARGARIDO, G. R. A. Mapeamento de QTLs em múltiplos caracteres e ambientes emum cruzamento comercial de cana-de-açúcar usando modelos mistos. 2011. 107 p. Tese(Doutorado em Genética e Melhoramento de Plantas) — Escola Superior de Agricultura “Luizde Queiroz”, Universidade de São Paulo, Piracicaba, 2011.
MARGARIDO, G. R. A.; SOUZA, A. P.; GARCIA, A. A. F. OneMap: software for geneticmapping in outcrossing species. Hereditas, Lund, v. 144, n. 3, p. 78–79, 2007.
MATSUOKA, S.; GARCIA, A. A. F.; ARIZONO, H. Melhoramento da cana-de-açúcar. In:BORÉM, A. (Ed.). Melhoramento de Espécies Cultivadas. 2. ed. Viçosa: UFV, 2005. p.205–251.
MING, R.; LIU, S.; LIN, Y.; SILVA, J.; WILSON, W.; BRAGA, D.; DEYNZE, A. V.;WENSLAFF, T. F.; WU, K. K.; MOORE, P. H.; BURNQUIST, W.; SORRELLS, M. E.;IRVINE, J. E.; PATERSON, A. H. Detailed alignment of Saccharum and sorghumchromosomes: comparative organization of closely related diploid and polyploid genomes.Genetics, Austin, v. 150, p. 1663–1682, 1998.
MING, R.; WANG, Y.-W.; DRAYE, X.; MOORE, P. H.; IRVINE, J. E.; PATERSON, A. H.Molecular dissection of complex traits in autopolyploids: mapping QTLs affecting sugar yieldand related traits in sugarcane. Theoretical and Applied Genetics, Berlin, v. 105, n. 2-3, p.332–345, 2002.
MOLLINARI, M. Desenvolvimento de um modelo para construção de mapas genéticosem autopoliploides, com aplicações em cana-de-açúcar. 2012. 98 p. Tese (Doutorado emGenética e Melhoramento de Plantas) — Escola Superior de Agricultura “Luiz de Queiroz”,Universidade de São Paulo, Piracicaba, 2012.
MOLLINARI, M.; MARGARIDO, G. R. A.; VENCOVSKY, R.; GARCIA, A. A. F. Evaluationof algorithms used to order markers on genetic maps. Heredity, v. 103, n. 6, p. 494–502, 2009.
MOLLINARI, M.; SERANG, O. Quantitative SNP genotyping of polyploids withMassARRAY and other platforms. In: BATLEY, J. (Ed.). Plant Genotyping: methods inmolecular biology. New York: Springer, 2015. v. 1245, cap. 17, p. 215–241.

74
MORRIS, G. P.; RAMU, P.; DESHPANDE, S. P.; HASH, C. T.; SHAH, T.; UPADHYAYA,H. D.; RIERA-LIZARAZU, O.; BROWN, P. J.; ACHARYA, C. B.; MITCHELL, S. E.;HARRIMAN, J.; GLAUBITZ, J. C.; BUCKLER, E. S.; KRESOVICH, S. Population genomicand genome-wide association studies of agroclimatic traits in sorghum. Proceedings of theNational Academy of Sciences of the United States of America, Washington, v. 110, n. 2, p.453–458, 2013.
MURRAY, S. C.; ROONEY, W. L.; HAMBLIN, M. T.; MITCHELL, S. E.; KRESOVICH,S. Sweet sorghum genetic diversity and association mapping for brix and height. The PlantGenome, Madison, v. 2, n. 1, p. 48, 2009.
OETH, P.; BEAULIEU, M.; PARK, C.; KOSMAN, D.; DEL MISTRO, G.; VAN DENBOOM, D.; JURINKE, C. iPLEXTM Assay: increased plexing efficiency and flexibility forMassARRAY R© system through single base primer extension with mass-modified terminators.Sequenom Application Note, San Diego, v. 8876, n. 006, p. 1–12, 2006. Disponível em:<www.sequenom.com>. Acesso em: 12 dez. 2014.
OLIVEIRA, K. M.; PINTO, L. R.; MARCONI, T. G.; MARGARIDO, G. R. A.; PASTINA,M. M.; TEIXEIRA, L. H. M.; FIGUEIRA, A. V.; ULIAN, E. C.; GARCIA, A. A. F.; SOUZA,A. P. Functional integrated genetic linkage map based on EST-markers for a sugarcane(Saccharum spp.) commercial cross. Molecular Breeding, Berlin, v. 20, n. 3, p. 189–208,2007.
OLIVEIRA, K. M.; PINTO, L. R.; MARCONI, T. G.; MOLLINARI, M.; ULIAN, E. C.;CHABREGAS, S. M.; FALCO, M. C.; BURNQUIST, W.; GARCIA, A. A. F.; SOUZA, A. P.Characterization of new polymorphic functional markers for sugarcane. Genome, Ottawa,v. 52, p. 191–209, 2009.
PASTINA, M. M.; MALOSETTI, M.; GAZAFFI, R.; MOLLINARI, M.; MARGARIDO, G.R. A.; OLIVEIRA, K. M.; PINTO, L. R.; SOUZA, A. P.; VAN-EEUWIJK, F. A.; GARCIA,A. A. F. A mixed model QTL analysis for sugarcane multiple-harvest-location trial data.Theoretical and Applied Genetics, Berlin, v. 124, n. 5, p. 835–849, 2012.
PASTINA, M. M.; PINTO, L. R.; OLIVEIRA, K. M.; SOUZA, A. P.; GARCIA, A. A. F.Molecular mapping of complex traits. In: HENRY, R. J.; KOLE, C. (Ed.). Genetics, Genomicsand Breeding of Sugarcane. Enfield: Science Publishers, 2010. cap. 7, p. 117–148.
PATERSON, A. H.; BOWERS, J. E.; BRUGGMANN, R.; DUBCHAK, I.; GRIMWOOD,J.; GUNDLACH, H.; HABERER, G.; HELLSTEN, U.; MITROS, T.; POLIAKOV, A.;SCHMUTZ, J.; SPANNAGL, M.; TANG, H.; WANG, X.; WICKER, T.; BHARTI, A. K.;CHAPMAN, J.; FELTUS, F. A.; GOWIK, U.; GRIGORIEV, I. V.; LYONS, E.; MAHER,C. A.; MARTIS, M.; NARECHANIA, A.; OTILLAR, R. P.; PENNING, B. W.; SALAMOV,A. A.; WANG, Y.; ZHANG, L.; CARPITA, N. C.; FREELING, M.; GINGLE, A. R.; HASH,C. T.; KELLER, B.; KLEIN, P.; KRESOVICH, S.; MCCANN, M. C.; MING, R.; PETERSON,D. G.; RAHMAN, M.; WARE, D.; WESTHOFF, P.; MAYER, K. F. X.; MESSING, J.;ROKHSAR, D. S. The Sorghum bicolor genome and the diversification of grasses. Nature,London, v. 457, n. 7229, p. 551–556, 2009.
PINTO, L. R.; GARCIA, A. A. F.; PASTINA, M. M.; TEIXEIRA, L. H. M.; BRESSIANI,J. A.; ULIAN, E. C.; BIDOIA, M. A. P.; SOUZA, A. P. Analysis of genomic and functionalRFLP derived markers associated with sucrose content, fiber and yield QTLs in a sugarcane(Saccharum spp.) commercial cross. Euphytica, Wageningen, v. 172, n. 3, p. 313–327, 2009.

75
PINTO, L. R.; OLIVEIRA, K. M.; MARCONI, T.; GARCIA, A. A. F.; ULIAN, E. C.;SOUZA, A. P. Characterization of novel sugarcane expressed sequence tag microsatellites andtheir comparison with genomic SSRs. Plant Breeding, Berlin, v. 125, p. 378–384, 2006.
PINTO, L. R.; OLIVEIRA, K. M.; ULIAN, E. C.; GARCIA, A. A. F.; SOUZA, A. P. Surveyin the sugarcane expressed sequence tag database (SUCEST) for simple sequence repeats.Genome, Ottawa, v. 47, p. 795–804, 2004.
R CORE TEAM. R: a language and environment for statistical computing. Vienna: RFoundation for Statistical Computing, 2014. Disponível em: <http://www.r-project.org>.Acesso em: 14 jan. 2014.
RABOIN, L.-M.; OLIVEIRA, K. M.; LE CUNFF, L.; TELISMART, H.; ROQUES, D.;BUTTERFIELD, M.; HOARAU, J.-Y.; D’HONT, A. Genetic mapping in sugarcane, a highpolyploid, using bi-parental progeny: identification of a gene controlling stalk colour and a newrust resistance gene. Theoretical and Applied Genetics, Berlin, v. 112, n. 7, p. 1382–1391,2006.
RAHMAN, M.-U.; PATERSON, A. H. Comparative genomics in crop plants. In: JAIN, S. M.;BRAR, D. S. (Ed.). Molecular Techniques in Crop Improvement. 2. ed. Dordrecht: SpringerNetherlands, 2010. cap. 2, p. 23–61.
REVELLE, W. psych: procedures for psychological, psychometric, and personality research.Evanston, 2014. Disponível em: <http://cran.r-project.org/package=psych>. Acesso em: 12nov. 2013.
RIPOL, M.; CHURCHILL, G. A.; SILVA, J. A. G.; SORRELLS, M. Statistical aspects ofgenetic mapping in autopolyploids. Gene, Amsterdam, v. 235, n. 1–2, p. 31–41, 1999.
RITTER, K. B.; JORDAN, D. R.; CHAPMAN, S. C.; GODWIN, I. D.; MACE, E. S.;MCINTYRE, C. L. Identification of QTL for sugar-related traits in a sweet × grain sorghum(Sorghum bicolor L. Moench) recombinant inbred population. Molecular Breeding, Berlin,v. 22, n. 3, p. 367–384, 2008.
RITTER, K. B.; MCINTYRE, C. L.; GODWIN, I. D.; JORDAN, D. R.; CHAPMAN, S. C.An assessment of the genetic relationship between sweet and grain sorghums, within Sorghumbicolor ssp. bicolor (L.) Moench, using AFLP markers. Euphytica, Wageningen, v. 157,n. 1-2, p. 161–176, 2007.
ROBERTS, A.; MCMILLAN, L.; WANG, W.; PARKER, J.; RUSYN, I.; THREADGILL, D.Inferring missing genotypes in large SNP panels using fast nearest-neighbor searches oversliding windows. Bioinformatics, Oxford, v. 23, n. 13, p. i401–i407, 2007.
SCHWARZ, G. Estimating the dimension of a model. The Annals of Statistics, Hayward, v. 6,n. 2, p. 461–464, 1978.
SERANG, O.; MOLLINARI, M.; GARCIA, A. A. F. Efficient exact maximum a posterioricomputation for bayesian SNP genotyping in polyploids. PLoS ONE, San Francisco, v. 7, n. 2,p. 1–13, 2012.
SHIRINGANI, A. L.; FRIEDT, W. QTL for fibre-related traits in grain × sweet sorghum as atool for the enhancement of sorghum as a biomass crop. Theoretical and Applied Genetics,Berlin, v. 123, n. 6, p. 999–1011, 2011.

76
SHIRINGANI, A. L.; FRISCH, M.; FRIEDT, W. Genetic mapping of QTLs for sugar-relatedtraits in a RIL population of Sorghum bicolor L. Moench. Theoretical and Applied Genetics,Berlin, v. 121, n. 2, p. 323–336, 2010.
SILVA, L. D. C. E.; WANG, S.; ZENG, Z.-B. Multiple trait multiple interval mapping ofquantitative trait loci from inbred line crosses. BMC Genetics, London, v. 13, n. 67, p. 1–24,2012.
SILVA, L. D. C. E.; ZENG, Z.-B. Current progress on statistical methods for mappingquantitative trait loci from inbred line crosses. Journal of Biopharmaceutical Statistics,Abingdon, v. 20, n. 2, p. 454–481, 2010.
SINGH, R. K.; JENA, S. N.; KHAN, S.; YADAV, S.; BANARJEE, N.; RAGHUVANSHI, S.;BHARDWAJ, V.; DATTAMAJUMDER, S. K.; KAPUR, R.; SOLOMON, S.; SWAPNA, M.;SRIVASTAVA, S.; TYAGI, A. K. Development, cross-species/genera transferability of novelEST-SSR markers and their utility in revealing population structure and genetic diversity insugarcane. Gene, Amsterdam, v. 524, n. 2, p. 309–329, 2013.
SINGH, R. K.; SINGH, R. B.; SINGH, S. P.; SHARMA, M. L. Identification of sugarcanemicrosatellites associated to sugar content in sugarcane and transferability to other cerealgenomes. Euphytica, Wageningen, v. 182, n. 3, p. 335–354, 2011.
SORRELS, M. E. Applications of comparative genomics to crop improvement. In: LAMKEY,K. R.; LEE, M. (Ed.). Plant Breeding: the Arnel R. Hallauer International Symposium. Iowa:Blackwell, 2006. cap. 12, p. 171–181.
SOUZA, G. M.; BERGES, H.; BOCS, S.; CASU, R.; D’HONT, A.; FERREIRA, J. E.;HENRY, R.; MING, R.; POTIER, B.; VAN SLUYS, M.-A.; VINCENTZ, M.; PATERSON,A. H. The sugarcane genome challenge: strategies for sequencing a highly complex genome.Tropical Plant Biology, Berlin, v. 4, p. 145–156, 2011.
TANG, H.; BOWERS, J. E.; WANG, X.; MING, R.; ALAM, M.; PATERSON, A. H. Syntenyand collinearity in plant genomes. Science, New York, v. 320, n. 5875, p. 486–8, 2008.
VETTORE, A. L.; SILVA, F. R.; KEMPER, E. L.; SOUZA, G. M.; SILVA, A. M.; FERRO,M. I. T.; HENRIQUE-SILVA, F.; GIGLIOTI, E. A.; LEMOS, M. V. F.; COUTINHO, L. L.;NOBREGA, M. P.; CARRER, H.; FRANCA, S. C.; BACCI-JUNIOR, M.; GOLDMAN,M. H. S.; GOMES, S. L.; NUNES, L. R.; CAMARGO, L. E. A.; SIQUEIRA, W. J.; VANSLUYS, M.-A.; THIEMANN, O. H.; KURAMAE, E. E.; SANTELLI, R. V.; MARINO,C. L.; TARGON, M. L. P. N.; FERRO, J. A.; SILVEIRA, H. C. S.; MARINI, D. C.; LEMOS,E. G. M.; MONTEIRO-VITORELLO, C. B.; TAMBOR, J. H. M.; CARRARO, D. M.;ROBERTO, P. G.; MARTINS, V. G.; GOLDMAN, G. H.; OLIVEIRA, R. C.; TRUFFI, D.;COLOMBO, C. A.; ROSSI, M.; ARAUJO, P. G.; SCULACCIO, S. A.; ANGELLA, A.;LIMA, M. M. A.; ROSA-JUNIOR, V. E.; SIVIERO, F.; COSCRATO, V. E.; MACHADO,M. A.; GRIVET, L.; DI-MAURO, S. M. Z.; NOBREGA, F. G.; MENCK, C. F. M.; BRAGA,M. D. V.; TELLES, G. P.; CARA, F. A. A.; PEDROSA, G.; MEIDANIS, J.; ARRUDA, P.Analysis and functional annotation of an expressed sequence tag collection for tropical cropsugarcane. Genome Research, Cold Spring Harbor, v. 13, n. 12, p. 2725–2735, 2003.

77
VICENTINI, R.; DEL BEM, L. E. V.; VAN SLUYS, M. A.; NOGUEIRA, F. T. S.;VINCENTZ, M. Gene content analysis of sugarcane public ESTs reveals thousands of missingcoding-genes and an unexpected pool of grasses conserved ncRNAs. Tropical Plant Biology,Berlin, v. 5, n. 2, p. 199–205, 2012.
VOORRIPS, R. E. MapChart: software for the graphical presentation of linkage maps andQTLs. The Journal of Heredity, Washington, v. 93, n. 1, p. 77–78, 2002.
VSN INTERNATIONAL. GenStat for Windows 16th Edition. Hemel Hempstead: VSNInternational, 2014. Disponível em: <genstat.co.uk>. Acesso em: 12 nov. 2014.
WANG, M. L.; BARKLEY, N. A.; YU, J.-K.; DEAN, R. E.; NEWMAN, M. L.; SORRELLS,M. E.; PEDERSON, G. A. Transfer of simple sequence repeat (SSR) markers from majorcereal crops to minor grass species for germplasm characterization and evaluation. PlantGenetic Resources, Cambridge, v. 3, n. 1, p. 45–57, 2005.
WU, R.; MA, C.-X.; PAINTER, I.; ZENG, Z.-B. Simultaneous maximum likelihood estimationof linkage and linkage phases in outcrossing species. Theoretical Population Biology, Berlin,v. 61, n. 3, p. 349–363, 2002.
WU, R.; MA, C.-X.; WU, S. S.; ZENG, Z.-B. Linkage mapping of sex-specific differences.Genetic Research, Oxford, v. 79, p. 85–96, 2002.
ZEGADA-LIZARAZU, W.; MONTI, A. Are we ready to cultivate sweet sorghum as abioenergy feedstock? A review on field management practices. Biomass and Bioenergy,Oxford, v. 40, p. 1–12, 2012.
ZELLNER, A. An efficient method of estimating seemingly unrelated regressions and tests foraggregation bias. Journal of the American Statistical Association, New York, v. 57, n. 298,p. 348–368, 1962.
ZOU, W.; ZENG, Z.-B. Statistical methods for mapping multiple QTL. International Journalof Plant Genomics, Nasr City, v. 2008, p. 1–8, 2008.

78

79
APÊNDICES

80

81
APÊNDICE A – QTLs resultantes do mapeamento de múltiplos intervalos (MIM) univa-
riado em população de sorgo sacarino
Tabela 12 – Estimativas dos efeitos aditivos e os respectivos cromossomos (Cr.) e posições(em mega bases – Mb) dos QTLs mapeados em população de sorgo sacarino paraas médias marginais e conjuntas de três anos de avaliação
Caráter QTL Cr. Posição(Mb) LOD (DP)a Anob
1 2 3 Conjunta
ALT 1 1 62,6-67,8 8,13 (0,59) −0,063∗∗∗ 0,083∗∗∗ 0,055∗∗∗ 0,072∗∗∗2 2 26,3 4,17 −0,040∗∗∗3 3 61,3 4,09 −0,042∗∗∗4 3 68,3 3,46 −0,039∗∗∗5 4 54,2 4,66 −0,045∗∗∗6 6 18,4 7,49 0,081∗∗∗7 6 42,6 7,41 0,054∗∗∗8 8 45,1 5,33 −0,047∗∗∗9 9 10,1 7,93 −0,060∗∗∗
10 9 47,8-50,4 8,71 (2,00) −0,057∗∗∗ −0,078∗∗∗ −0,068∗∗∗ −0,074∗∗∗11 10 11,1 5,72 −0,049∗∗∗12 10 44,4-55,7 7,42 (1,77) −0,059∗∗∗ −0,092∗∗∗ −0,048∗∗∗ −0,062∗∗∗
PMV 1 2 4,9 5,41 −1,302∗∗∗3 3 21,3 9,06 −1,098∗∗∗4 4 1,3 5,41 1,297∗∗∗5 4 57,0-57,3 5,76 (0,55) −2,013∗∗∗ −1,283∗∗∗6 6 17,5 13,44 (1,17) 3,485∗∗∗ 1,934∗∗∗7 6 41,8 13,34 1,346∗∗∗8 7 60,0 6,36 −0,906∗∗∗
10 9 5,4 4,06 −1,003∗∗∗11 10 12,9-13,6 9,06 (1,68) −2,939∗∗∗ −0,952∗∗∗ −1,884∗∗∗12 10 44,6-52,5 5,81 (1,68) −1,575∗∗∗ 2,196∗∗∗ 1,188∗∗∗
POL 1 1 3,9-6,6 11,16 (3,91) −0,496∗∗∗ −0,386∗∗∗ −0,449∗∗∗ −0,401∗∗∗2 1 51,4 4,69 −0,213∗∗∗3 3 0,9-1,8 5,44 (0,04) −0,294∗∗∗ −0,230∗∗∗4 3 13,5-15,9 8,22 (4,04) −0,288∗∗∗ −0,333∗∗∗5 3 51,4 7,24 −0,402∗∗∗6 3 64,9-67,6 18,65 (10,37) −0,804∗∗∗ −0,479∗∗∗ −0,480∗∗∗ −0,550∗∗∗7 5 2,9 6,79 −0,342∗∗∗8 5 4,8-5,6 8,55 (2,42) −0,316∗∗∗ −0,455∗∗∗ −0,332∗∗∗9 6 46,2 8,11 (0,95) 0,423∗∗∗ 0,270∗∗∗
10 6 52,5-52,7 8,80 0,447∗∗∗ 0,310∗∗∗11 7 48,3 7,16 0,278∗∗∗12 8 14,0 5,62 −0,295∗∗∗14 8 53,4 8,51 0,297∗∗∗15 9 3,0-9,5 8,15 (2,81) −0,313∗∗∗ −0,474∗∗∗16 9 10,1 12,26 (2,55) −0,489∗∗∗ −0,384∗∗∗17 10 5,1 5,77 −0,302∗∗∗
FIB 1 1 59,8-61,0 6,65 (0,79) 0,144∗∗∗ 0,223∗∗∗2 2 1,7 7,21 −0,142∗∗∗3 3 60,6 3,91 −0,110∗∗∗4 3 68,2-69,2 11,51 (6,73) 0,333∗∗∗ 0,289∗∗∗ 0,249∗∗∗ 0,242∗∗∗5 4 1,4-8,0 7,88 (3,63) −0,326∗∗∗ −0,187∗∗∗ −0,169∗∗∗6 4 65,1 7,59 (1,87) −0,363∗∗∗ −0,196∗∗∗8 6 47,6 4,86 −0,266∗∗∗9 8 37,0-37,4 11,17 (2,91) 0,372∗∗∗ 0,205∗∗∗ 0,260∗∗∗ 0,290∗∗∗
11 10 2,5 5,62 (0,51) −0,127∗∗∗ −0,206∗∗∗ −0,194∗∗∗12 10 56,5-58,5 8,08 (2,43) 0,326∗∗∗ 0,178∗∗∗ 0,191∗∗∗
Notas: aLOD é a média dos LODs obtidos das análises individuais; para os QTLs co-localizados,desvio-padrão (DP) foi calculado. bEfeitos significativos quando ∗∗∗p < 0,001

82
APÊNDICE B – QTLs resultantes do mapeamento de múltiplos intervalos (MIM) multi-
variado em população de sorgo sacarino
Tabela 13 – Estimativas dos efeitos aditivos e os respectivos cromossomos (Cr.) e po-sições (em mega bases – Mb) dos QTLs mapeados em população de sorgosacarino para as médias marginais de três anos de avaliação
Caráter QTL Cr. Posição(Mb) LODca LODi (DP)b Anoc
1 2 3
ALT 1 1 67,7 9,30 5,52 (2,62) 0,061∗∗∗ 0,078∗∗∗ 0,036∗∗
4 3 68,3 10,68 2,60 (3,14) −0,025∗ −0,020 −0,059∗∗∗
7 6 42,2 18,42 5,91 (5,10) 0,020 0,097∗∗∗ 0,057∗∗∗
9 9 2,6 5,93 3,39 (1,69) −0,047∗∗∗ −0,061∗∗∗ −0,027∗∗
10 9 47,8 9,66 6,76 (1,78) −0,067∗∗∗ −0,061∗∗∗ −0,065∗∗∗
12 10 43,3 11,30 4,28 (3,47) −0,041∗∗∗ −0,085∗∗∗ −0,031∗
PMV 2 2 62,7 5,43 0,69 (0,52) −0,208 0,952∗ 0,2973 3 21,3 7,94 3,17 (3,62) −0,451 −1,172∗∗ −0,961∗∗∗
7 6 40,1 19,98 8,11 (6,60) 0,478 3,109∗∗∗ 1,326∗∗∗
8 7 60,0 6,81 3,26 (1,98) −0,895∗∗∗ −1,302∗∗ −0,826∗∗∗
9 8 5,4 5,62 1,05 (1,73) −0,193 −0,017 0,600∗∗∗
12 10 38,3 7,68 5,55 (1,07) −1,278∗∗∗ −2,179∗∗∗ −0,895∗∗∗
POL 1 1 6,5 10,93 7,65 (1,91) −0,390∗∗∗ −0,340∗∗∗ −0,468∗∗∗
2 1 63,1 6,74 2,16 (2,15) 0,265∗∗∗ 0,204∗∗ 0,0124 3 15,9 7,88 3,79 (3,39) −0,386∗∗∗ −0,220∗∗ −0,189∗
5 3 57,8 6,10 2,38 (2,61) −0,151∗ −0,117 −0,380∗∗∗
6 3 68,1 19,45 10,58 (7,49) −0,614∗∗∗ −0,423∗∗∗ −0,317∗∗∗
8 5 4,8 7,85 4,19 (2,67) −0,330∗∗∗ −0,316∗∗∗ −0,162∗
9 6 43,2 8,36 1,71 (2,66) −0,033 0,308∗∗∗ 0,07610 6 53,5 8,62 5,00 (2,51) 0,325∗∗∗ 0,200∗∗ 0,378∗∗∗
13 8 37,0 5,86 0,91 (0,93) −0,176∗∗ 0,033 0,12615 9 2,9 7,54 3,64 (2,80) −0,237∗∗∗ −0,369∗∗∗ −0,146∗
16 9 10,1 10,31 6,97 (2,31) −0,338∗∗∗ −0,311∗∗∗ −0,467∗∗∗
FIB 1 1 59,2 5,93 2,25 (1,98) 0,028 0,095∗∗∗ 0,157∗∗∗
4 3 68,1 13,50 10,09 (3,22) 0,322∗∗∗ 0,214∗∗∗ 0,272∗∗∗
5 4 9,6 7,29 4,43 (2,22) −0,181∗∗ −0,150∗∗∗ −0,175∗∗∗
6 4 65,1 9,32 5,25 (3,91) −0,380∗∗∗ −0,136∗∗∗ −0,084∗
7 6 34,9 12,29 1,42 (0,83) −0,139∗∗ −0,093∗∗∗ 0,073∗
8 6 46,2 7,92 1,98 (2,29) −0,080 0,056∗ 0,178∗∗∗
9 8 37,0 13,85 10,95 (2,38) 0,414∗∗∗ 0,217∗∗∗ 0,238∗∗∗
10 9 5,3 10,31 3,22 (2,89) 0,007 −0,116∗∗∗ −0,192∗∗∗
11 10 2,5 6,75 3,80 (2,30) −0,175∗∗ −0,103∗∗∗ −0,209∗∗∗
12 10 56,4 6,75 4,78 (2,10) 0,321∗∗∗ 0,136∗∗∗ 0,123∗∗
Notas: aLODc é o valor de LOD conjunto. bLODi é a média dos LODs individuais obtidosnas análises multivariadas, para a qual desvio-padrão (DP) foi calculado. cEfeitos significativosquando ∗p < 0,05, ∗∗p < 0,01 e ∗∗∗p < 0,001

83
APÊNDICE C – QTLs resultantes do mapeamento de múltiplos intervalos (MIM) univa-
riado em população de cana-de-açúcar
Tabela 14 – Estimativas dos efeitos aditivos (p e q) e de dominância (d) e os respectivos grupos de ligação(GLs) e posições (em centiMorgans – cM) dos QTLs mapeados em população de cana-de-açúcar para as médias marginais e conjuntas de cinco combinações de locais e cortes
Caráter QTL GL Posição(cM) LOD (DP)a Ef. Local-Corteb
1-1 1-3 2-1 2-2 2-3 Conjunta
ALT 2 24 1,0 1,82 (0,44) q 0,022∗∗ 0,022∗∗ 0,022∗∗ 0,030∗∗ 0,023∗∗ 0,022∗∗d −0,040∗ −0,033∗
4 46 181,0 5,36 (0,01) p 0,209∗∗∗ 0,240∗∗∗ 0,208∗∗∗ 0,236∗∗∗ 0,240∗∗∗ 0,240∗∗∗5 83 58,0 2,47 (0,05) d 0,034∗∗∗ 0,034∗∗∗ 0,034∗∗∗ 0,034∗∗∗ 0,034∗∗∗ 0,034∗∗∗6 112 0,0 2,85 (0,14) p −0,028∗∗∗ −0,029∗∗∗ −0,028∗∗∗ −0,027∗∗∗ −0,029∗∗∗ −0,029∗∗∗8 133 20,0 2,45 (0,15) p 0,028∗∗ 0,028∗∗ 0,028∗∗ 0,030∗∗∗ 0,028∗∗ 0,028∗∗9 222 0,0 8,99 (0,29) p −0,033∗ −0,033∗ −0,033∗ −0,029∗ −0,033∗ 0,086∗∗∗
d 0,085∗∗∗ 0,086∗∗∗ 0,086∗∗∗ 0,084∗∗∗ 0,086∗∗∗
TCH 1 1 69,2-75,0 3,20 (0,74) p −5,372∗∗∗ −3,947∗∗ −3,828∗∗∗ −3,266∗∗ −4,256∗∗∗q 6,331∗∗ 5,459∗∗ 3,947∗∗ 4,520∗∗ 4,683∗∗d 5,082∗ 4,315∗ 4,822∗
2 5 17,9 3,47 p 2,401∗q −3,437∗∗∗
5 43 0,0-16,0 2,48 (0,29) q 5,048∗∗ 2,505∗ 2,683∗d −4,465∗ −1,957∗ −2,166∗
6 64 3,0-4,9 1,74 (0,15) p −3,692∗∗ −3,340∗∗ −2,951∗∗ −3,179∗∗7 71 40,0 2,27 q −3,219∗∗9 88 6,6 1,91 (0,18) p 2,961∗ 3,260∗∗
d 4,150∗10 122 2,0 2,30 q −4,561∗
d 5,689∗∗11 131 0,0 4,31 (0,50) p 2,958∗ 3,412∗ 2,964∗∗ 3,107∗∗ 3,106∗
d −7,221∗∗∗ −5,231∗ −5,418∗∗ −4,824∗∗ −6,085∗∗13 163 0,0 5,83 (0,66) q 4,091∗∗∗ 4,463∗∗∗ 3,986∗∗∗ 3,547∗∗∗ 4,339∗∗∗
d −7,468∗∗∗ −6,525∗∗∗ −6,393∗∗∗ −6,745∗∗∗ −7,019∗∗∗14 167 6,2 2,08 (0,83) p −2,187∗ −2,808∗ −2,337∗
d 3,007∗∗ 2,072∗ 3,343∗∗ 2,272∗15 207 0,4-1,0 4,39 (1,21) d 8,490∗∗∗ 10,701∗∗∗ 6,550∗∗ 8,408∗∗∗ 9,925∗∗∗ 9,129∗∗∗
POL 1 4 43,3 4,75 (0,44) p −0,308∗∗∗ −0,287∗∗∗ −0,257∗∗∗ −0,239∗∗∗ −0,283∗∗∗2 6 29,6 4,08 (0,33) q −0,264∗∗∗ −0,245∗∗∗ −0,203∗∗∗ −0,189∗∗∗ −0,233∗∗∗
d 0,214∗∗ 0,199∗∗ 0,197∗∗ 0,184∗∗ 0,215∗∗3 26 22,0-28,5 2,62 (0,15) q 0,295∗∗∗ 0,272∗∗∗ 0,239∗∗∗ 0,221∗∗∗ 0,262∗∗∗4 97 3,7 1,25 (0,02) d 0,118∗ 0,110∗ 0,130∗
FIB 1 6 4,0-5,0 3,39 (0,11) p −0,272∗∗∗ −0,228∗∗∗ −0,236∗∗∗d 0,206∗ 0,170∗ 0,169∗
2 9 1,0 2,33 (0,02) p 0,180∗∗∗ 0,149∗∗5 60 7,0-9,0 2,80 (0,15) p −0,281∗∗∗ −0,230∗∗∗ −0,256∗∗∗ −0,208∗∗∗ −0,233∗∗6 124 2,0-6,0 4,45 (1,50) p 0,259∗∗ 0,211∗∗ 0,175∗∗ 0,143∗∗ 0,213∗∗
d 0,441∗∗∗ 0,364∗∗∗ 0,265∗∗ 0,222∗∗ 0,365∗∗∗7 150 14,0-15,0 3,41 (0,13) p 0,201∗∗ 0,165∗∗ 0,170∗∗
q −0,190∗ −0,153∗ −0,162∗8 167 0,0 3,26 (0,01) q 0,129∗ 0,110∗
d −0,267∗∗∗ −0,222∗∗∗
Notas: aLOD é a média dos LODs obtidos nas análises univariadas; para os QTLs co-localizados, desvio-padrão(DP) foi calculado. bEfeitos significativos quando ∗p < 0,05, ∗∗p < 0,01 e ∗∗∗p < 0,001

84
APÊNDICE D – QTLs resultantes do mapeamento de múltiplos intervalos (MIM) multi-
variado em população de cana-de-açúcar
Tabela 15 – Estimativas dos efeitos aditivos (p e q) e de dominância (d) e os respectivos grupos de ligação(GLs) e posições (em centiMorgans – cM) dos QTLs mapeados em população de cana-de-açúcar para as médias marginais de cinco combinações de locais e cortes
Caráter QTL GL Posição(cM) LODca LODi (DP)b Ef. Local-Cortec
1-1 1-3 2-1 2-2 2-3
ALT 1 1 53,5 2,61 0,44 (0,00) p −0,014 −0,014 −0,014 −0,014 −0,0142 24 51,0 1,76 1,20 (0,00) p 0,045 0,045∗ 0,045∗ 0,045∗ 0,0453 43 21,8 3,34 0,44 (0,01) p −0,015 −0,015 −0,015 −0,015 −0,0157 117 35,5 4,46 0,01 (0,00) p −0,002 −0,002 −0,002 −0,001 −0,001
TCH 1 1 33,7 4,50 0,59 (0,16) p −2,093 −1,602 −1,798 −1,761 −1,4233 7 0,0 6,02 0,76 (0,09) q −3,128 −3,474 −2,876 −2,709 −3,182
d 0,201 1,334 0,468 0,731 1,4484 42 20,2 8,66 0,30 (0,14) p −2,622 −2,167 −1,54 −1,495 −1,266
q −0,218 −0,022 −0,028 −0,061 0,1998 72 31,4 7,50 0,80 (0,22) p 2,286∗ 2,474∗ 1,435 1,725 1,671
12 133 36,4 7,08 0,16 (0,11) p −1,787 −0,952 −1,431 −0,856 −0,738q 1,849 1,458 1,564 1,159 1,275d 1,758 1,444 1,716 1,741 1,461
14 167 4,0 12,61 1,24 (0,35) d 3,372∗ 4,05∗∗ 2,212∗∗ 3,018 2,843
POL 1 4 43,3 5,22 4,24 (0,06) p −0,315∗∗∗ −0,294∗∗∗ −0,246∗∗∗ −0,229∗∗∗
2 6 29,6 4,66 2,58 (0,19) q −0,265∗∗∗ −0,246∗∗∗ −0,191∗∗∗ −0,177∗∗∗
d 0,228∗ 0,212∗ 0,185∗∗ 0,173∗∗
FIB 3 26 18,0 4,04 0,09 (0,09) p 0,05 0,037 −0,006 −0,011q −0,017 −0,009 0,037 0,037
4 50 10,6 5,40 3,40 (0,69) p −0,167∗ −0,143∗ −0,126 −0,108d −0,011 −0,008 0,039 0,032
Notas: aLODc é o valor de LOD conjunto. bLODi é a média dos LODs individuais obtidos nas análises multi-variadas, para a qual desvio-padrão (DP) foi calculado. cEfeitos significativos quando ∗p < 0,05, ∗∗p < 0,01 e∗∗∗p < 0,001

85
APÊNDICE E – Marcadores significativos resultantes da análise de marcas individual-
mente em população de cana-de-açúcar
Tabela 16 – Estimativas dos efeitos aditivos individuais dos marcadores de cana-de-açúcar relacionadosao genoma do sorgo sobre as médias conjuntas de quatro caracteres
Caráter Marcador Efeito Caráter Marcador Efeito Caráter Marcador Efeito
ALT sb1_4188246 −0,056∗∗ TCH sb2_7455650 12,238∗∗ POL sb9_44929840 −0,521∗∗
sb1_12219024 −0,059∗∗ sb2_50026760 −12,443∗∗ sb10_52650429 −0,366∗∗
sb1_14455981 −0,068∗∗ sb3_8750650 11,263∗∗ sb10_56295895 −0,401∗∗
sb1_52671762 0,066∗∗ sb3_52117257 11,352∗∗ sb10_56540062 0,480∗∗
sb2_22609350 −0,060∗∗ sb3_72128809 12,069∗∗ sb10_60131295 0,520∗∗∗
sb2_68475923 −0,056∗∗ sb4_173279 16,728∗∗ sb10_60131296 0,527∗∗∗
sb3_390231 0,070∗∗ sb4_711989 11,076∗∗ FIB sb1_18696270 0,333∗∗
sb3_541865 −0,069∗∗ sb5_48066020 11,319∗∗ sb1_50799049 0,331∗∗
sb3_1928213 −0,064∗∗ sb6_782163 −11,525∗∗ sb1_73398501 −0,383∗∗
sb3_6126551 −0,079∗∗ sb6_45032977 −13,507∗∗ sb2_6817295 −0,399∗∗
sb3_7546835 −0,073∗∗ sb7_7580104 13,795∗∗ sb2_52242660 0,468∗∗
sb3_9286412 −0,070∗∗ sb7_56993024 −15,794∗∗ sb2_60547040 0,298∗∗
sb3_73337885 −0,064∗∗ sb8_5298195 −10,093∗∗ sb2_63577008 0,361∗∗
sb4_9399 0,077∗∗ sb9_45536069 −11,525∗∗ sb2_71421489 0,323∗∗
sb4_49763421 0,068∗∗ sb10_8999958 −12,116∗∗ sb3_541804 −0,353∗∗
sb4_63654242 0,086∗∗ POL sb1_7645431 0,407∗∗ sb3_541826 −0,352∗∗
sb4_64604253 −0,057∗∗ sb1_9236816 −0,532∗∗∗ sb3_5691216 −0,306∗∗
sb5_17237746 −0,078∗∗ sb1_16417625 0,395∗∗ sb3_14771406 0,349∗∗
sb5_48533705 0,059∗∗ sb1_16417644 0,391∗∗ sb3_18259258 0,398∗∗
sb5_48533707 0,063∗∗ sb1_31488584 −0,411∗∗ sb3_58924723 0,346∗∗
sb5_61994091 −0,068∗∗ sb1_31624046 0,409∗∗ sb3_68287506 −0,344∗∗
sb6_45032977 −0,084∗∗∗ sb2_60082918 0,467∗∗∗ sb3_72046711 −0,328∗∗
sb7_53544684 −0,063∗∗ sb2_60547042 0,345∗∗ sb3_72046712 −0,357∗∗
sb8_3123434 −0,070∗∗ sb2_61202411 −0,401∗∗ sb4_9380 0,419∗∗
sb8_11128174 −0,247∗∗ sb2_62057915 0,403∗∗ sb4_59449990 0,380∗∗
sb9_49172170 −0,063∗∗ sb2_65330382 −0,403∗∗ sb4_60387435 0,378∗∗
sb9_49172185 −0,062∗∗ sb2_67849128 0,373∗∗ sb5_29034328 0,390∗∗
sb10_1156537 −0,059∗∗ sb3_2978336 −0,378∗∗ sb5_50145700 −0,358∗∗
sb10_1156542 −0,064∗∗ sb3_57883510 0,461∗∗ sb5_54760011 0,378∗∗
sb10_3260857 −0,077∗∗∗ sb4_61731192 −0,362∗∗ sb6_57305087 −0,546∗∗
sb10_3260858 −0,077∗∗∗ sb5_14386234 0,454∗∗ sb7_3717230 −0,358∗∗
TCH sb1_3077235 10,692∗∗ sb6_60560180 −0,388∗∗ sb8_5688896 0,364∗∗
sb1_60357211 13,391∗∗ sb9_1087507 −0,507∗∗ sb8_53867342 −0,327∗∗
Notas: Efeitos significativos quando ∗∗p < 0,01 e ∗∗∗p < 0,001