pós graduação em gestão da
TRANSCRIPT


Pós Graduação em Gestão da
Qualidade - Aula 7

Autodiagnóstico nas Áreas de
Utilidades
Andrea Geyer

UTILIDADES

UTILIDADES
Água e Vapor
• FB 6ª Edição (pg. 787-799), PIC/S Aide Memoire, WHO TRS 1033 Annex 3, USP <1231>
Aquecimento, Ventilação e Ar Condicionado (AVAC) - Heating, Ventilation & Air Conditioning (HVAC)
• FB 6ª Edição (pg. 702-710), PIC/S Aide Memoire, WHO TRS 961 Annex 5 e 1019 Annex 2, USP <1116> , ISO 14644, WHO Environmental Monitoring of Clean Rooms (Vaccines)
Gases (ex. Ar comprimido e Nitrogênio)
• ISO 8573, USP, PIC/S Aide Memoire, ISPE GPG Process Gases
UTILIDADES
Plano Mestre de Validação
(PMV)
Qualificação (QP, QI, QO,
QD)Monitoramento
Calibração e Manutenção Preventiva
Documentação (POPs e
Registros)

UTILIDADESIN 47/19 - Qualificação e Validação
• Art. 5º Como parte de um sistema de gerenciamento de risco à qualidade, as decisões sobre o escopo e a extensão da qualificação e validação devem se basear em uma avaliação de risco justificada e documentada das instalações, equipamentos, utilidades e processos.
• Art. 7º Todas as atividades de qualificação e validação devem ser planejadas e devem levar em consideração o ciclo de vida útil das instalações, equipamentos, sistemas, utilidades, processos e produtos.
• Art. 25. As atividades de qualificação devem considerar todos os estágios, desde o desenvolvimento inicial das especificações de requisitos do usuário, até o fim do uso do equipamento, instalação, utilidade ou sistema.
RDC 301/19
• Art. 243. Os seguintes documentos devem estar prontamente disponíveis para o Departamento de Controle de Qualidade:
• VI - dados de monitoramento ambiental (ar, água e outras utilidades), quando necessário;
IN 35/19
• Art. 84. Todos os equipamentos, tais como esterilizadores, sistemas de tratamento e filtração de ar, filtros de ventilação e de gases, sistemas de tratamento, geração, armazenamento e distribuição de água, devem estar sujeitos à qualificação e manutenção preventiva.

ÁGUA E VAPOR

ÁGUA
• Os processos de purificação; armazenamento e distribuição devem garantir que as especificações farmacopeicas sejam atendidas, mantidas e controladas adequadamente.
• Os requisitos de qualidade da água dependerão de sua finalidade e emprego, e a escolha do sistema de purificação destina atender ao grau de pureza estabelecido.
• O usuário é responsável pela seleção do tipo de água adequado aos seus objetivos, bem como pelos controles e verificações necessários, em intervalos que garantam a manutenção da qualidade desejada.
• Deve assegurar que o sistema apresente desempenho adequado e capacidade para fornecer água com o nível de qualidade estabelecido para atender aos parâmetros especificados nas monografias individuais.
FB 6ª Ed

FB 6ª Ed. Pg. 792
*Dependendo da aplicação, pode ser esterilizada, sem necessariamente atingir o limite de endotoxinas bacterianas estabelecido para a Água para injetáveis.
*

APLICAÇÕES – NÃO ESTÉREIS
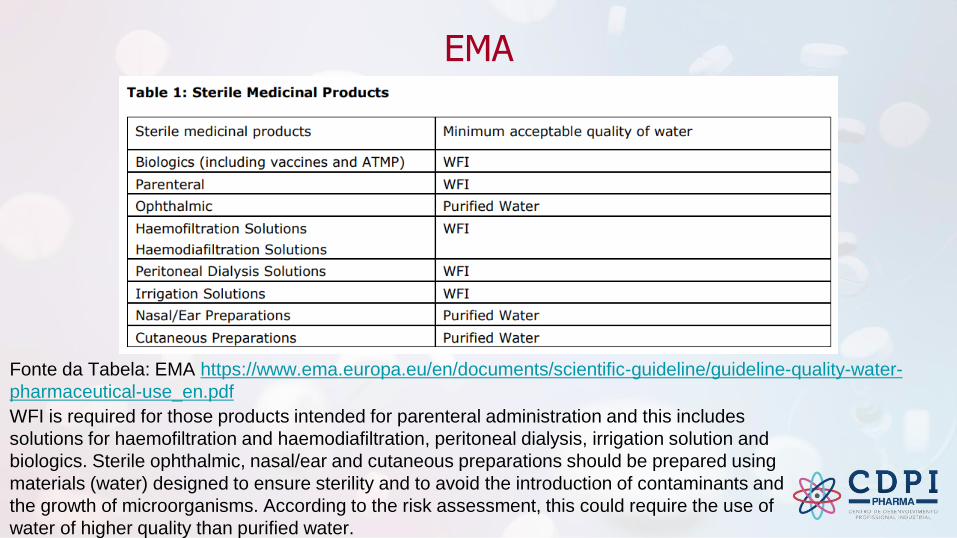
Fonte da Tabela: EMA https://www.ema.europa.eu/en/documents/scientific-
guideline/guideline-quality-water-pharmaceutical-use_en.pdf
Purified Water which satisfies the test for endotoxins described in Ph. Eur. monograph 0008 may be used in the manufacture of dialysis solutions.
Fonte da Tabela: EMA https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-water-
pharmaceutical-use_en.pdf
EMA
*
WFI is required for those products intended for parenteral administration and this includes
solutions for haemofiltration and haemodiafiltration, peritoneal dialysis, irrigation solution and
biologics. Sterile ophthalmic, nasal/ear and cutaneous preparations should be prepared using
materials (water) designed to ensure sterility and to avoid the introduction of contaminants and
the growth of microorganisms. According to the risk assessment, this could require the use of
water of higher quality than purified water.
USP <1231>

LIMPEZA EQUIPAMENTOS – ÁGUA
FORMULAÇÃO
NÃO ESTÉRIL
No mínimo último enxague das superfícies que
entram em contato com o produto devem ser
realizadas com água purificada.
FORMULAÇÃO
ESTÉRIl
No mínimo último enxague das superfícies que
entram em contato com o produto devem ser
realizadas com água de qualidade injetável.

ÁGUA POTÁVEL (FB)
O ponto de partida para qualquer processo de purificação de água para fins farmacêuticos é a água potável
Deve atender às especificações da legislação brasileira
• Portaria de Consolidação n°5, de 28 de setembro de 2017 anexo XX
A água potável é empregada, normalmente, nas etapas iniciais de procedimentos de limpeza e como fonte de obtenção de água de mais alto grau de pureza. Pode ser utilizada, também, na climatização térmica de alguns aparatos e na síntese de ingredientes intermediários.
O controle deve ser periódico para garantir que o sistema de purificação utilizado esteja apropriado para as condições da fonte de alimentação e que não houve alteração na qualidade da água fornecida.

Água para uso farmacêutico
Água Purificada (AP ou PW)
• É obtida por uma combinação de sistemas de purificação, em uma sequência lógica, tais como: múltipla destilação; troca iônica; osmose reversa; eletrodeionização; ultrafiltração, ou outro processo capaz de atender, com a eficiência desejada, aos limites especificados para os diversos contaminantes.
Água para Injetáveis (API ou WFI)
• O processo de purificação de primeira escolha é a destilação, em equipamento cujas paredes internas sejam fabricadas em metal apropriado, como o aço inox AISI 316L, vidro neutro ou quartzo. Alternativamente, a API pode ser obtida por processo equivalente ou superior à destilação para a remoção de contaminantes químicos e micro-organismos, desde que seja validado e monitorado quanto aos parâmetros estabelecidos.

SISTEMAS DE PURIFICAÇÃO DE
ÁGUA
• Os processos de obtenção empregam
operações unitárias sequenciais – os
estágios de purificação – que estão voltados
à remoção de determinados contaminantes
e à proteção de estágios de purificação
subsequentes.
• O projeto de instalação de um sistema de
purificação de água deve considerar a
qualidade da água de fornecimento e da
água desejada ao final, a vazão necessária,
a distância entre o sistema de produção e
os pontos de uso, o traçado da tubulação e
conexões, o material empregado,
facilidades de assistência técnica e
manutenção e os instrumentos adequados
para o monitoramento.
Água para uso farmacêutico
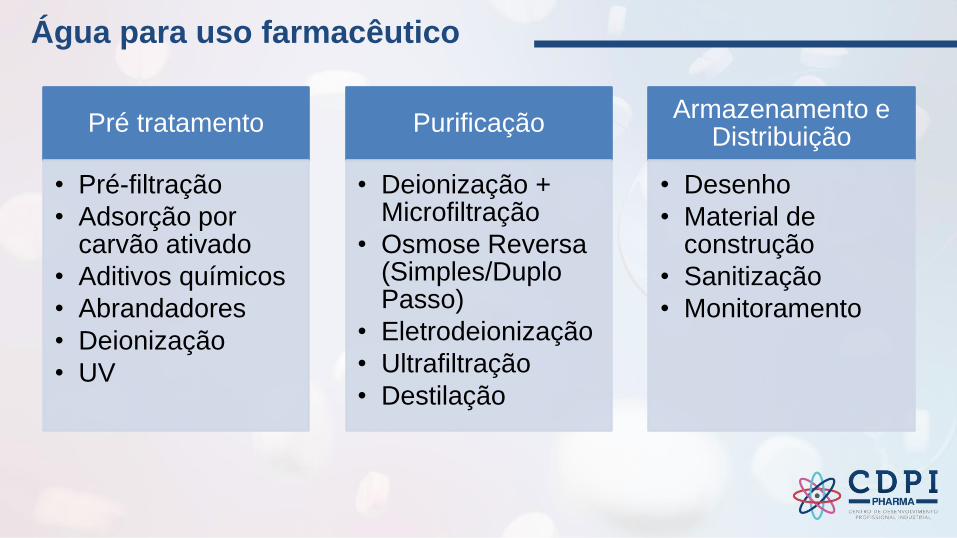
Pré tratamento
• Pré-filtração
• Adsorção por carvão ativado
• Aditivos químicos
• Abrandadores
• Deionização
• UV
Purificação
• Deionização + Microfiltração
• Osmose Reversa (Simples/Duplo Passo)
• Eletrodeionização
• Ultrafiltração
• Destilação
Armazenamento e Distribuição
• Desenho
• Material de construção
• Sanitização
• Monitoramento
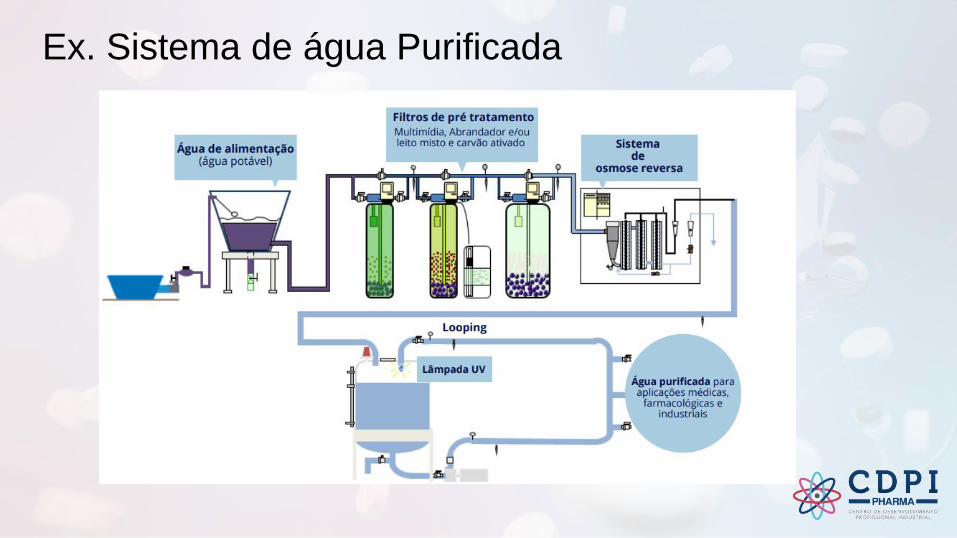
Ex. Sistema de água Purificada

Pré-filtração
Também conhecida como filtração de profundidade ou filtração inicial, destina-se a remover contaminantes particulados na faixa de tamanho entre 5 e 10 μm, essencialmente para proteger as tecnologias subsequentes, utilizando filtros de areia ou combinação de filtros.
• Areia
• Multimídia
• Cartucho
As medidas de controle envolvem o monitoramento de pressão e fluxo durante o uso e retrolavagem, higienização e substituição de meios filtrantes. Uma preocupação importante do projeto é o dimensionamento do filtro para evitar canalização ou perda do meio resultante de taxas de fluxo de água inadequadas, bem como dimensionamento adequado para minimizar retrolavagem excessivamente frequente ou substituição do filtro de cartucho.

Adsorção por carvão vegetal ativado
Remove agentes oxidantes por redução química,
em especial o cloro livre, que afeta outras
tecnologias baseadas em membrana, como a
osmose reversa ou a ultrafiltração.
Propicia o crescimento bacteriano e a formação de
biofilme, o que implica na necessidade de
sanitização do próprio carvão ativado, com vapor
direto ou água quente, por exemplo, e do controle
de partículas e contagem microbiana de seu
efluente
As medidas de controle podem envolver o
monitoramento de taxas de fluxo de água e
pressões diferenciais, higienização com água
quente ou vapor, retrolavagem, teste de
capacidade de adsorção e substituição frequente
do leito de carvão

Tratamento com aditivos químicos
Proteção de outras tecnologias (osmose reversa)
• Controle de microrganismos (ozônio, cloro)
• Remover sólidos suspensos (agentes floculantes)
• Remoção de cloro (metabissufito)
• Ajustar o pH
• Remover carbonatos e amônia
Precisam ser removidos em algum estágio posterior de purificação e não podem deixar resíduo na água final

Abrandadores
Resinas regeneráveis de troca iônica, que capturam os íons cálcio e magnésio, e liberam íons sódio na água
Proteção de tecnologias sensíveis à incrustação, como a osmose reversa.
Controlar a contagem microbiana, com regeneração frequente, recirculação ou outras formas de redução de contagem microbiana, para evitar a formação de biofilme
• UV
• Filtração
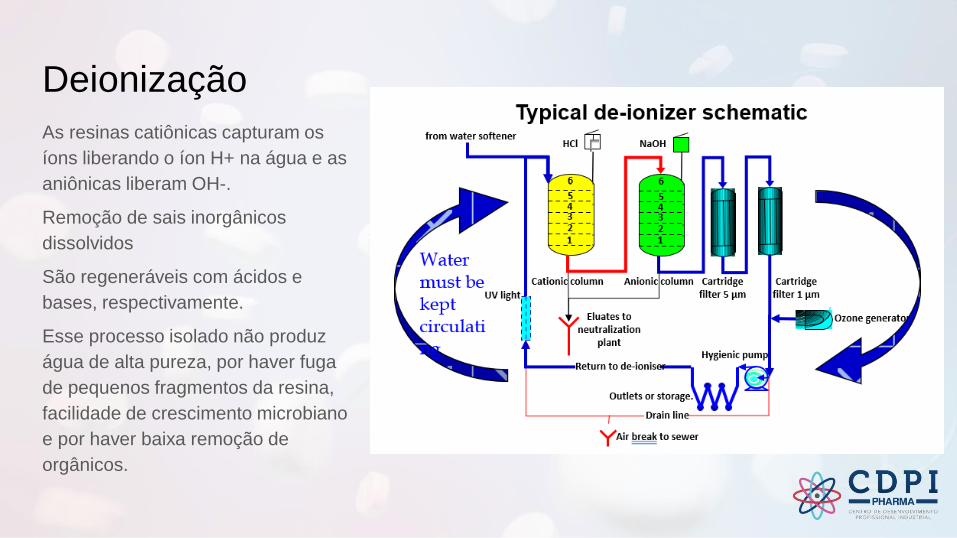
Deionização
As resinas catiônicas capturam os
íons liberando o íon H+ na água e as
aniônicas liberam OH-.
Remoção de sais inorgânicos
dissolvidos
São regeneráveis com ácidos e
bases, respectivamente.
Esse processo isolado não produz
água de alta pureza, por haver fuga
de pequenos fragmentos da resina,
facilidade de crescimento microbiano
e por haver baixa remoção de
orgânicos.

Lâmpada UV
A radiação UV é utilizada em sistemas de purificação de
água em dois comprimentos de onda: 185 nm e 254 nm,
que promovem dois efeitos:
• 185 nm e 254 nm – Oxidação de compostos orgânicos
e consequente redução de sua concentração
• 254 nm – Ação germicida - reduzir a contagem
microbiana.
Destruição de Ozônio
➢ Monitorar a intensidade da lâmpada e considerar a
profundidade/espessura do leito e o fluxo de água no
local da radiação.
➢ Troca periódica (controle do número de horas ou
radiação emitida)
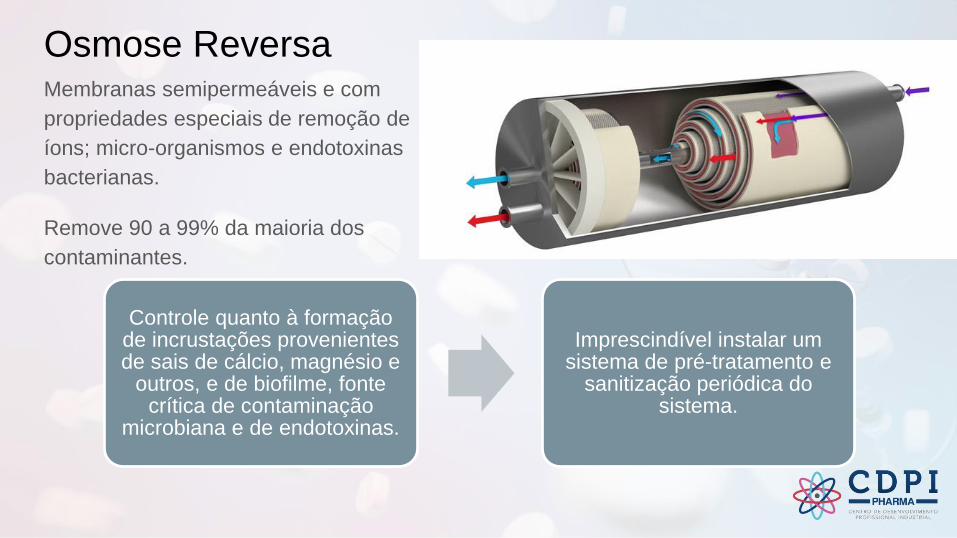
Osmose ReversaMembranas semipermeáveis e com
propriedades especiais de remoção de
íons; micro-organismos e endotoxinas
bacterianas.
Remove 90 a 99% da maioria dos
contaminantes.
Controle quanto à formação de incrustações provenientes de sais de cálcio, magnésio e
outros, e de biofilme, fonte crítica de contaminação
microbiana e de endotoxinas.
Imprescindível instalar um sistema de pré-tratamento e
sanitização periódica do sistema.
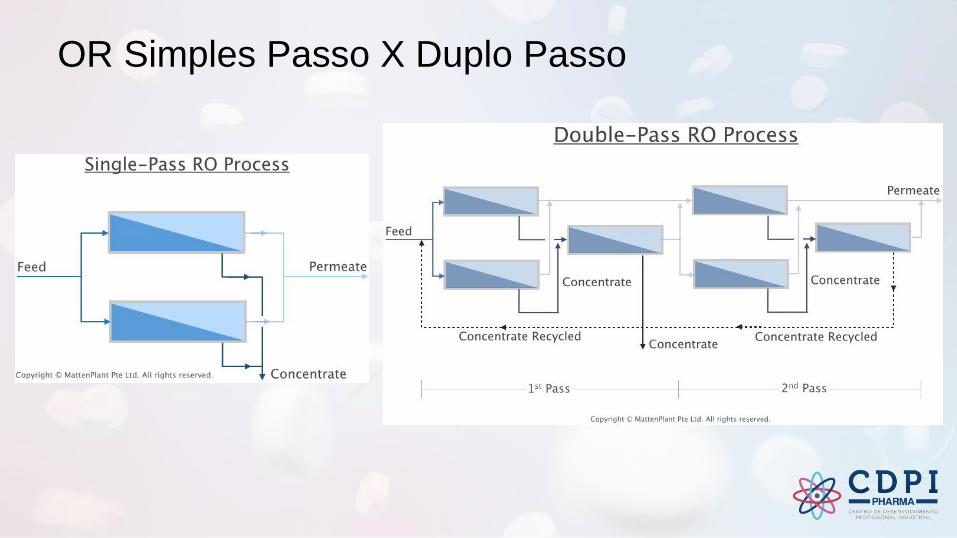
OR Simples Passo X Duplo Passo
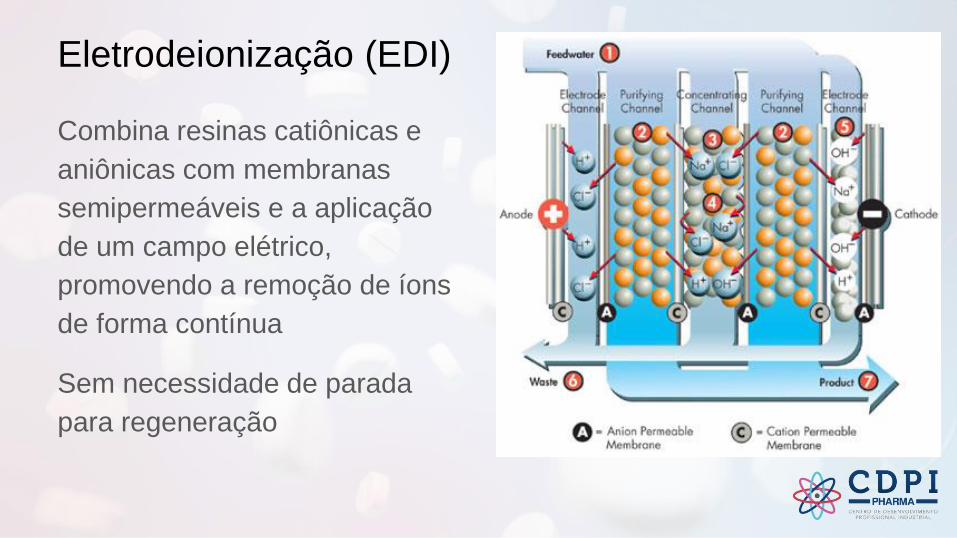
Eletrodeionização (EDI)
Combina resinas catiônicas e
aniônicas com membranas
semipermeáveis e a aplicação
de um campo elétrico,
promovendo a remoção de íons
de forma contínua
Sem necessidade de parada
para regeneração

Microfiltração
Filtro 0,22 μm
Podem ser usados após operações unitárias que tendem a liberar microrganismos
Não devem ser usados nos anéis de distribuição ou pontos de uso

Destilação
➢ Vaporização da água e posterior condensação;
➢ Era requisitado pela Farmacopeia Europeia
para WFI;
➢ Retira íons, microorganismos, material
orgânico, outras impurezas;
➢ Retira Solventes Orgânicos;
➢ Alto custo.
➢ Tipos de Destiladores:
➢ Simples
➢ Multiefeito / Multicoluna
➢ Compressão de vapor

Destilação
Multicoluna Compressão de Vapor
Destilação
Concentração de silicatos é crítica
Possibilidade de carreamento de compostos
voláteis no condensado
Fundamental o controle da água potável de entrada

Água para Injetáveis (WFI)
• A tecnologia de destilação é consagrada pelo seu longo histórico de confiabilidade e pode ser validada para produção de água para injetáveis.
A operação unitária final para obtenção de água para injetáveis é limitada à destilação ou outro
processo equivalente ou superior
• A ultrafiltração colocada em uma sequência após outras tecnologias de purificação de contaminantes químicos pode ser adequada para a produção de água para injetáveis, se demonstrar a mesma eficácia e confiabilidade da destilação na validação.
Para produzir a água para injetáveis, há novas e promissoras aplicações validáveis devido ao
desenvolvimento de novos materiais para tecnologias como osmose reversa e
ultrafiltração, que permitem operar e sanitizarem temperatura mais elevada, possibilitando
uma redução microbiana mais efetiva.

Ultrafiltração
Remoção de endotoxinas (filtros na faixa de
10 000 Da)
Membrana especial com a propriedade de
reter moléculas conforme o seu peso
molecular e estereoquímica.
Essa tecnologia pode ser usada em uma
etapa final ou intermediária do sistema de
purificação, desde que validada
Requer um pré-tratamento, um controle
adequado das condições operacionais e
procedimentos apropriados de limpeza e
sanitização, para manter a qualidade da água
conforme o estabelecido.

➢ O método escolhido de purificação da água, ou sequência de etapas de
purificação, deve ser apropriado à aplicação em questão.
➢ Os seguintes itens devem ser considerados ao selecionar o método de
tratamento da água:
I - a especificação da qualidade da água;
II - o rendimento ou eficiência do sistema de purificação;
III - a qualidade da água de alimentação e as alterações sazonais; e
IV - a confiabilidade e a robustez dos equipamentos de tratamento de água em
funcionamento.
ÁGUA PARA USO FARMACÊUTICO

BREAK

ARMAZENAMENTO E DISTRIBUIÇÃO
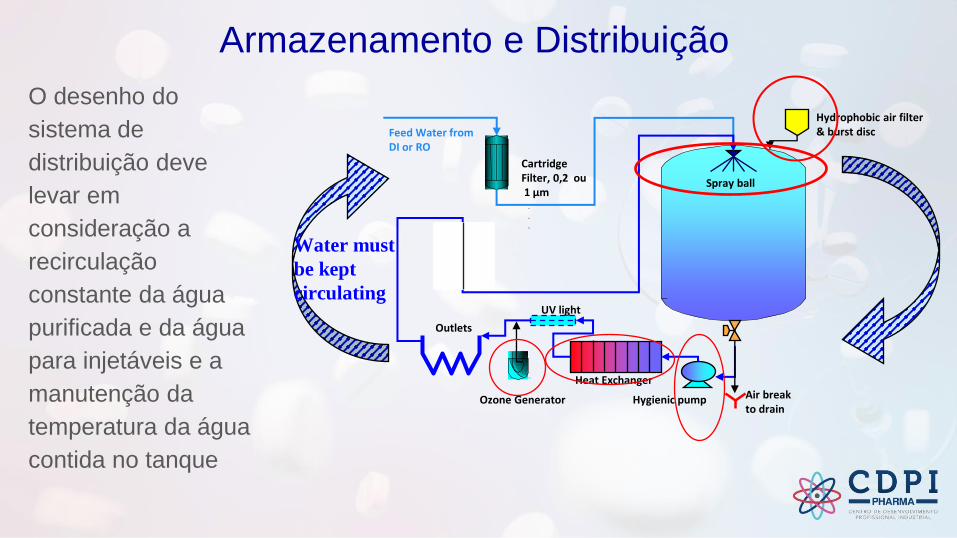
Armazenamento e Distribuição
O desenho do
sistema de
distribuição deve
levar em
consideração a
recirculação
constante da água
purificada e da água
para injetáveis e a
manutenção da
temperatura da água
contida no tanque
Water must
be kept
circulating
Spray ball
CartridgeFilter, 0,2 ou1 µm
Air breakto drain
Outlets
Hygienic pump
UV light
Feed Water fromDI or RO
Heat Exchanger
Ozone Generator
Hydrophobic air filter& burst disc

➢ O espaço entre a superfície da água e a tampa do reservatório é uma área de
risco em que gotas de água e ar podem entrar em contato em temperaturas que
incentivam a proliferação de microorganismos;
➢ Os reservatórios devem ser configurados para evitar zonas mortas em que
possa haver contaminação microbiológica;
➢ Filtros de ventilação são colocados em reservatórios para permitir que o nível
interno de líquido flutue. Os filtros devem reter bactérias, devem ser
hidrofóbicos e devem ser configurados idealmente para permitir teste de
integridade no local. Testes offline também são aceitáveis;
Controle da contaminação:

Vent Filter – Filtro de Ar
• Permite a variação do nível de líquido
• Retém bactérias
• Hidrofóbico
• Teste de integridade

Spray Ball
• Permite que água entre em contato com
toda a superfície do tanque
• Operação
• Sanitização

Bomba Sanitária
• Desenho apropriado
• Prevenir a contaminação do
sistema
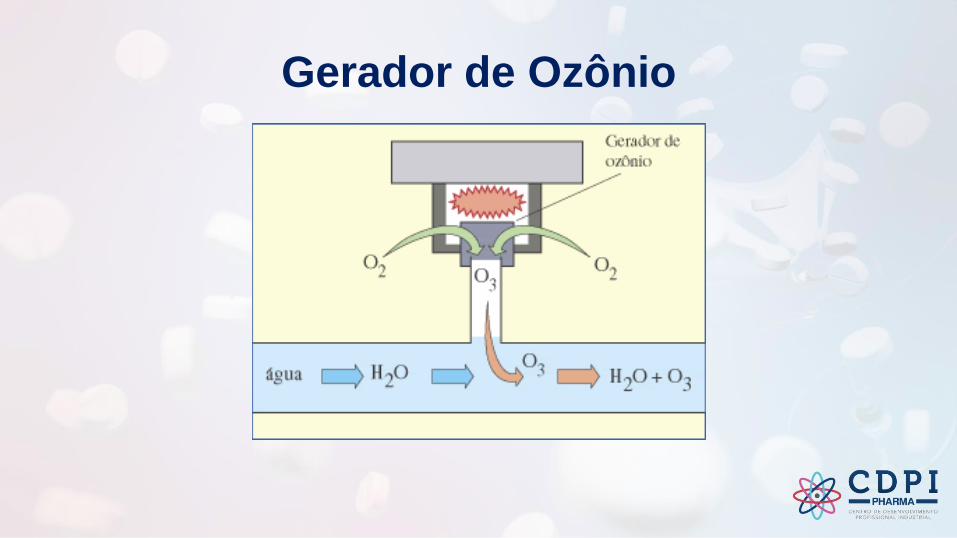
Gerador de Ozônio
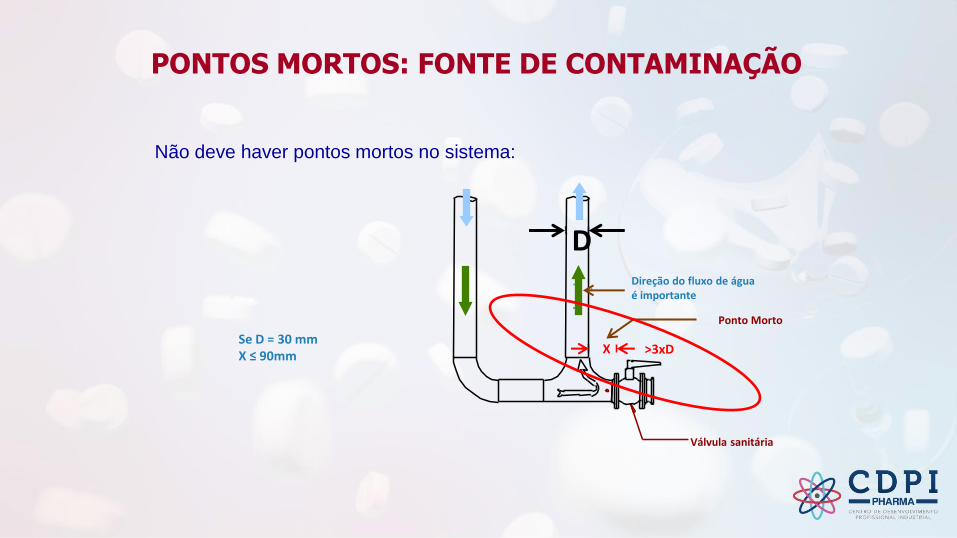
Não deve haver pontos mortos no sistema:
Se D = 30 mmX ≤ 90mm
Ponto Morto
>3xD
Direção do fluxo de água é importante
Válvula sanitária
D
X
PONTOS MORTOS: FONTE DE CONTAMINAÇÃO
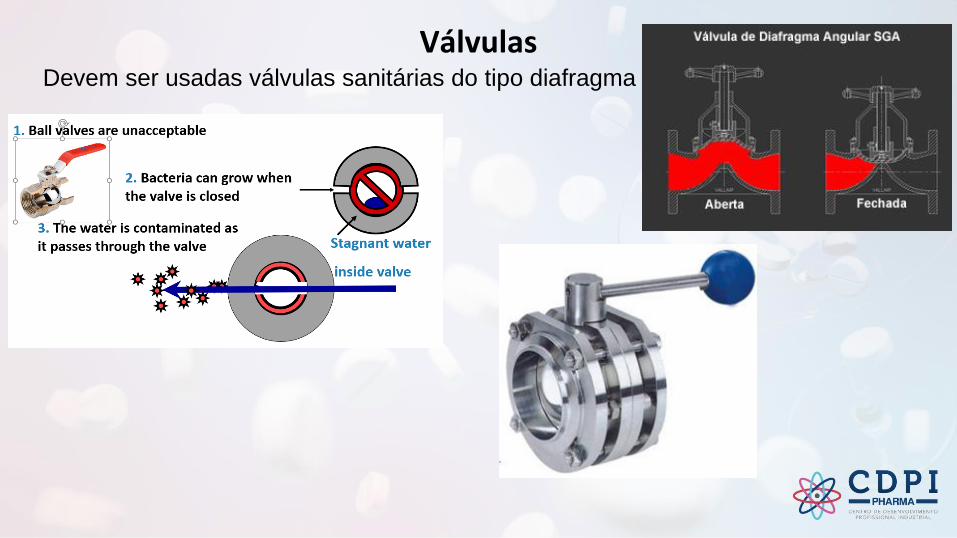
VálvulasDevem ser usadas válvulas sanitárias do tipo diafragma

➢ A distribuição de água purificada e de água para injetáveis deve ser realizada
utilizando preferencialmente um anel de circulação contínua.
➢ A proliferação de contaminantes dentro do tanque de armazenamento e do
anel de distribuição deve ser controlada.
➢ A filtração não deve ser utilizada nos anéis de distribuição ou em pontos de
uso para controlar a biocontaminação. Tais filtros podem mascarar a
contaminação do sistema.
➢ As bombas de circulação devem ter desenho sanitário que evitem a
contaminação do sistema.
Sistemas de distribuição

➢ Os materiais que entram em contato com a água para uso
farmacêutico, incluindo a tubulação, válvulas e armações,
lacres, diafragmas e instrumentos devem ser selecionados para
satisfazer os seguintes objetivos:
I – compatibilidade (temperatura e substâncias químicas)
II - prevenção de vazamento
III - resistência à corrosão: passivação após a instalação inicial ou
após modificação (limpeza após)
IV - acabamento interno liso
V – soldagem
VI - desenho de flanges ou juntas: desenho higiênico ou sanitário.
VII – documentação
Materiais de Construção

Sistema de armazenamento e distribuição
Materiais de Construção
• Aço Inox 316L
• Fluoreto de polivinilideno (PVDF),
• Polipropileno (PP)
• Perfluoroalkoxy (PFA)
• PVC
• Altamente susceptível à contaminação
• Não resiste a sanitização
• Pode contaminar a água com agentes químicos residuais
• Aço Inox 304L
• Não possui resistência adequada

Aço 316 – Possui Molibdênio;
Aço 304 – Não possui Molibdênio;
Molibdênio – Maior resistencia contra corrosão;
“L” – Significa low carbon – Baixo teor de carbono – Maior
proteção contra corrosão;
O aço inox de “boa qualidade”deve estar presente quando
em contato com água WFI e PW.
Materiais de construção
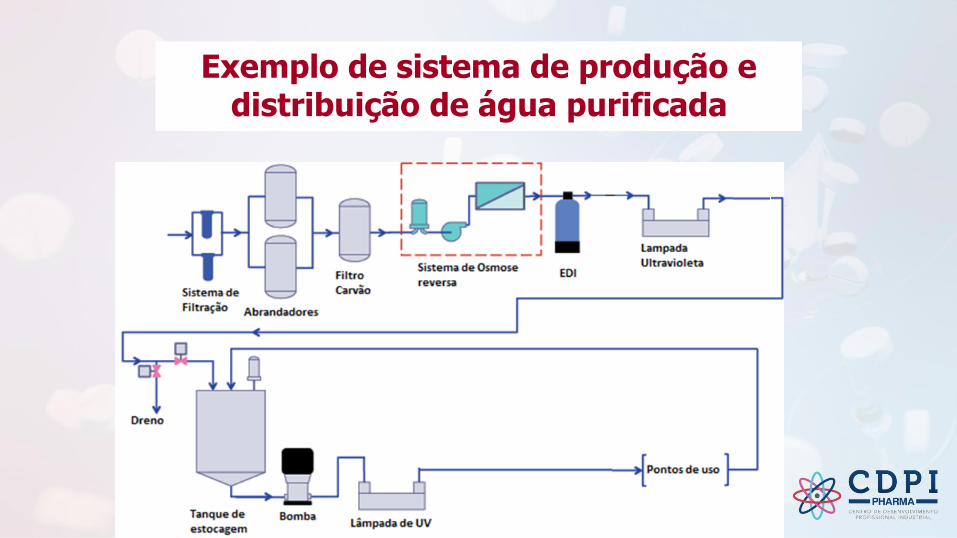
Exemplo de sistema de produção e distribuição de água purificada

VAPOR

Vapor puro
IN 35/2019 - Art. 131. Deve-se assegurar que o vapor usado para a esterilização seja de qualidade adequada e não contenha um nível de aditivos que possa causar contaminação dos materiais a serem esterilizados.
• O condensado deve cumprir com a especificação de água WFI para TOC, Condutividade e endotoxinas.
• Além disso, os seguintes parâmetros devem ser testados no vapor utilizado na esterilização de materiais que entram em contato direto com o produto:
• Gases não condensáveis (≤3,5%) – ar e CO2 quando presentes no vapor podem atuar como isolantes térmicos e também podem impactar na temperatura do vapor saturado.
• Vapor superaquecido (não superior a 125ºC) – A presença de vapor superaquecido, devido a sua alta energia potencial, pode danificar a carga, seja pelo derretimento dos frascos plásticos ou pela carbonização dos papéis utilizados como envoltórios, quando da condensação deste durante o ciclo de autoclavação sobre a carga.
• Secura (>0.95 w/w*) – A presença de vapor sem o nível apropriado de umidade pode comprometer o nível de garantia de esterilidade da carga, uma vez que o vapor molhado não entrega o mesmo nível de energia para a carga.
Referência técnica: EN 285:2015 Sterilization. Steam sterilizers
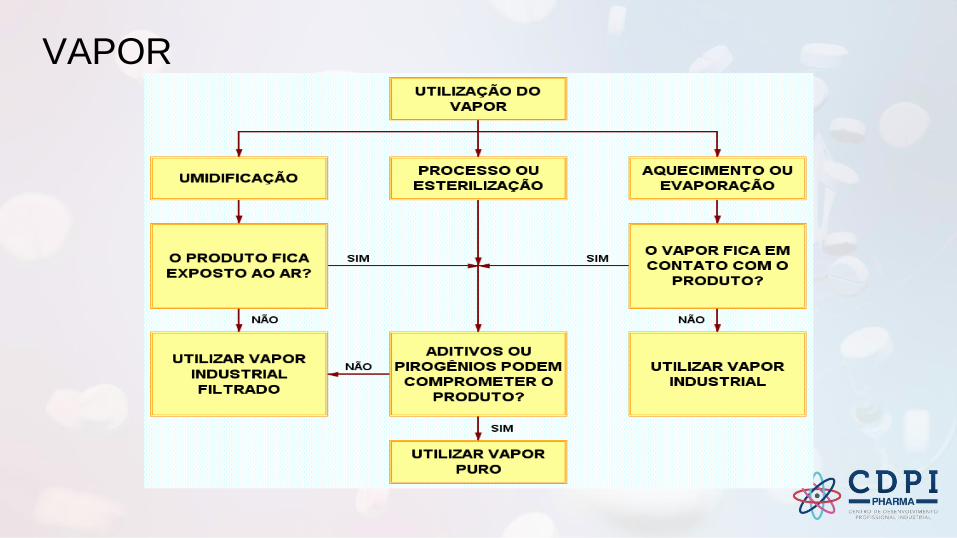
VAPOR

Vapor puro
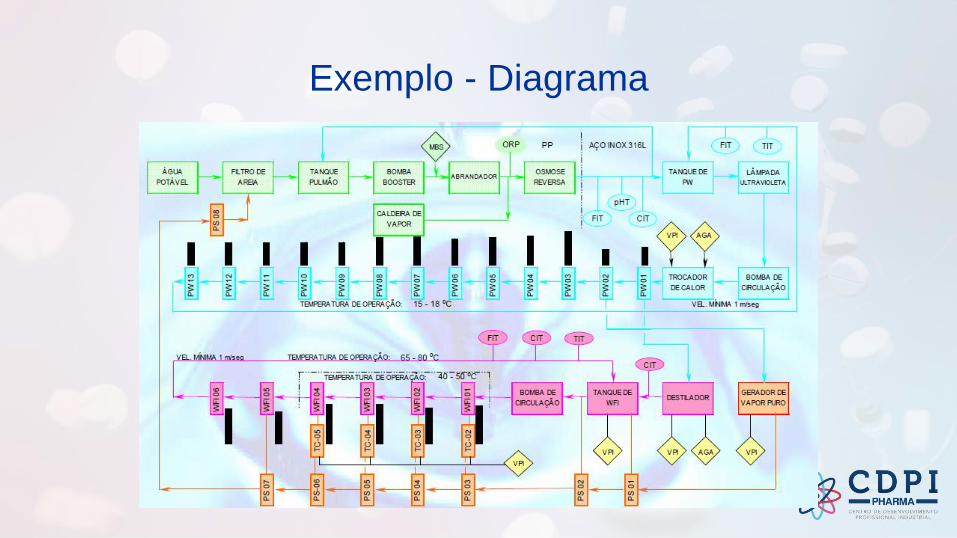
Exemplo - Diagrama

SISTEMA DE ÁGUA
QUALIFICAÇÃO E MANUTENÇÃO
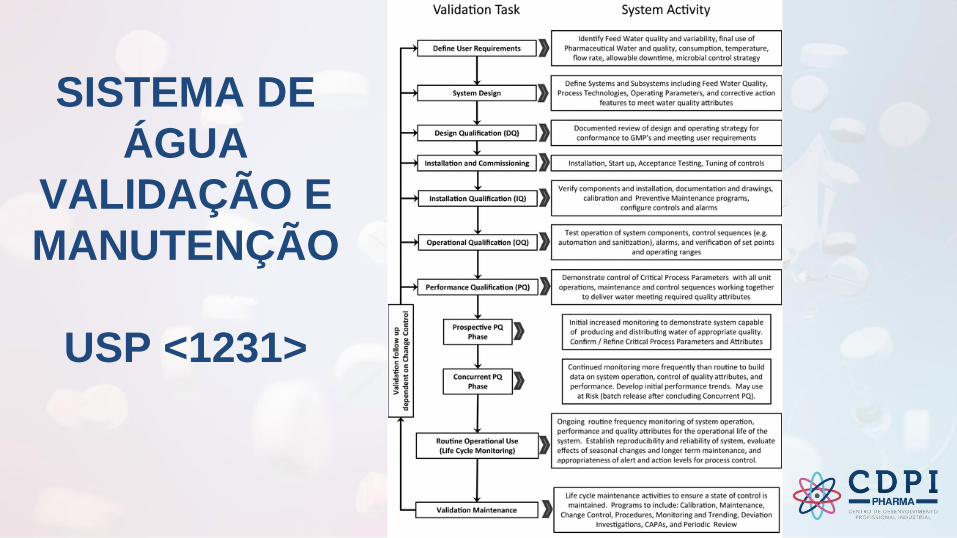
SISTEMA DE
ÁGUA
VALIDAÇÃO E
MANUTENÇÃO
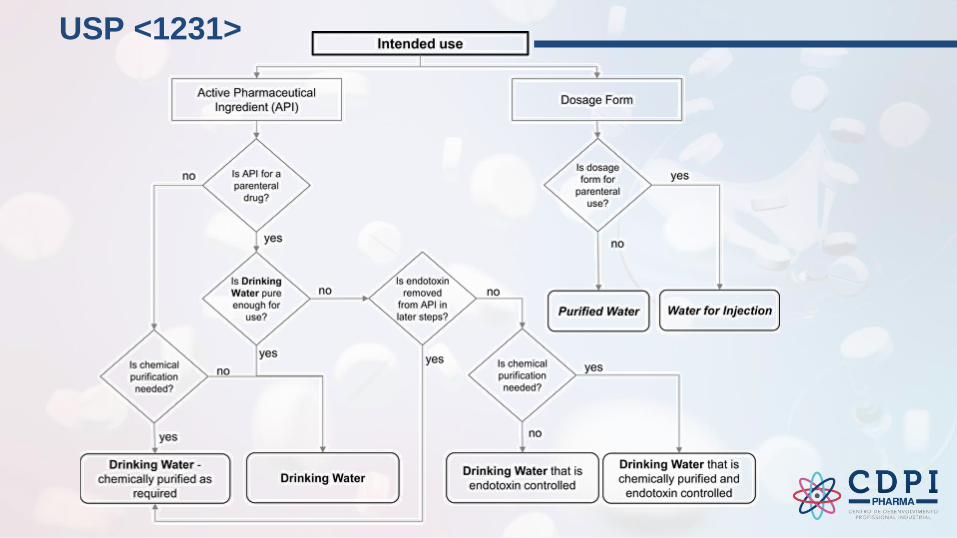
USP <1231>
User requirements specification (URS) and design qualification (DQ)
Especificação dos Requisistos do usuário (ERU) e Qualificação de Projeto (QP)
ERU - identificar os elementos de projeto, operação, manutenção e
qualidade necessários para produzir o tipo de água desejado a partir da fonte de água disponível,
incluindo sua variabilidade de atributos prevista. Os elementos
essenciais de qualidade precisam ser incorporados neste estágio e
quaisquer riscos de BPF mitigados a um nível aceitável.
QP - A revisão das especificações, projeto do sistema, componentes,
funções e operação deve ser realizada para demonstrar que o
sistema está em conformidade com as BPF e verificar se o projeto
atende aos requisitos do usuário.
Installation Qualification (IQ)Qualificação de Instalação (QI)
Confirmar que o sistema foi instalado e
documentado corretamente:
• verificação dos componentes,
tubulação, instalação e qualidade da
solda;
• documentação das especificações de
todos os componentes do sistema
presentes (certificados e manuais);
• verificar se os desenhos representam
com precisão a configuração final do
sistema
Preparação para testes operacionais,
incluindo calibração de instrumentos,
configuração de níveis de alarme e
ajuste de parâmetros operacionais (por
exemplo, taxa de fluxo, pressão).

Operational Qualification (OQ)
Qualificação de Operação (QO)
Verificar se o sistema está operando de forma confiável:
•alarmes
•Sequências de controle (sistemas de descarte)
•verificações funcionais de equipamento
•verificação de faixas de operação (desafios)
Os POPs para todos os aspectos da operação do sistema de água, manutenção, uso da água, amostragem e teste de água, etc. devem estar implementados e o treinamento do operador deve ser concluído.
Na conclusão do OQ, o sistema de água deve demonstrar que os componentes estão operacionais e o sistema está produzindo água adequada.

Performance Qualification (PQ)
Qualificação de Desempenho (QD)
• A fim de demonstrar a confiabilidade e robustez de um sistema e seu desempenho, uma abordagem de três fases deve ser usada para validação, cobrindo pelo menos um ano de operação.
• Testes na fonte de água (água potável) devem ser incluídas na validação e como parte do monitoramento de rotina para verificação da influência de variações sazonais
USP <1231>
WHO TRS 1033 Annex 3

Fase 1
USP - 2– 4 weeks / WHO - at least two weeks
• amostrar, testar e monitorar a água de alimentação
• amostrar, testar e monitorar após cada etapa da purificação
• amostrar, testar e monitorar cada ponto de uso e amostragem, incluindo o final do ciclo de distribuição
• verificação das faixas de operação
• operação, limpeza e manutenção
• procedimentos de sanitização e faixas operacionais
• demonstrar a produção e distribuição consistente de água de qualidade e quantidade exigidas;
• estabelecer níveis de alerta e ação provisórios
• procedimento de falha
A água não deve ser utilizada na fabricação nesta fase

Fase 2
USP - 2– 4 weeks / WHO - at least two weeks
Mesmo plano de amostragem da Fase 1 (USP somewhat reduced level that will still give adequate data on system performance)
A água pode ser utilizada na fabricação desde que a Fase 1 tenha conclusão satisfatória (USP - products may be released only after water quality attributes have been determined to be acceptable and this validation phase has been completed)
A fase 2 deve ainda:
•demonstrar operação consistente do sistema dentro das faixas estabelecidas;
•demonstrar produção consistente e distribuição de água na quantidade e qualidade necessárias quando o sistema é operado de acordo com os POPs.

Fase 3
•A Fase 3 deve seguir a fase 2, garantindo que a duração da Fase I, 2 e 3 cobrem pelo menos 12 meses.
•Pontos de amostragem, frequência e testes podem ser reduzidos de acordo com um plano de rotina
•Os dados devem ser submetidos à análise de tendências (ex. trimestralmente)
•Uma revisão do sistema deve ser realizada após a conclusão da Fase 3 como parte da avaliação da capacidade de desempenho do sistema.
•Ações apropriadas deve ser adotadas quando necessário
•A água pode ser utilizada durante esta fase.
•Os dados e informações obtidos durante a Fase 3 devem demonstrar o desempenho confiável do sistema durante este período de tempo cobrindo as diferentes estações.

Monitoramento
Art. 112. A tubulação de água purificada e água para injetáveis e, se for o caso, de qualquer outro tipo de água, deve ser sanitizada de acordo com procedimentos escritos que contenham detalhes sobre os limites de contaminação microbiológica, bem como as medidas a serem adotadas.
RDC 301/19
Art. 100.As fontes de água, os equipamentos de tratamento de água e a água tratada devem ser monitorados regularmente quanto à contaminação química e biológica e, quando apropriado, para endotoxinas.
Parágrafo único. Devem ser mantidos registros dos resultados do monitoramento e de qualquer ação realizada.
IN 35/19 - Estéreis
Art. 9° A qualidade química e microbiológica da água utilizada na produção deve ser especificada e monitorada.
Art. 10 Procedimentos específicos de manutenção do sistema de água devem ser estabelecidos, a fim de evitar o risco da proliferação microbiana.
Art. 11 Após qualquer sanitização química dos sistemas de água, um procedimento validado de rinsagemdeve ser seguido para assegurar que o agente sanitizante tenha sido efetivamente removido.
IN 41/19 - Líquidos, Cremes ou Pomadas

Monitoramento contínuo do sistema
O monitoramento de rotina deve seguir o plano de amostragem definido na fase 3, qualquer modificação deve ser avaliada por meio do sistema de controle de mudanças
Amostragem deve ser feita de acordo com o uso da água na rotina (mangueiras, drenagem, etc)
Combinação de instrumentos online e offline
Parâmetros como fluxo, pressão, temperatura devem ser monitorados online - bem como condutividade e TOC, quando possível
Testes offline periódicos para confirmar os resultados do teste online
Outros parâmetros podem ser monitorado por meio de testes offline

Monitoramento contínuo do sistema
Os resultados para os atributos de qualidade identificados devem ser submetidos a análise estatística em intervalos definidos para identificar tendências (ex. mensal, trimestral e anual)
• Os resultados devem estar dentro de limites de controle definidos (ex. 3 sigma)
• Níveis de alerta e ação devem ser estabelecidos com base nos dados históricos (USP)
• Tendências adversas e resultados fora do limite devem ser investigados para a causa raiz, seguida das ações corretivas e preventivas adequadas
• Contaminação microbiana de água WFI: microrganismo deve ser identificado
Água Purificada - Plano de Amostragem (USP)
Não há frequência de amostragem prescrita para pontos do sistema de Água
Purificada, então as frequências típicas de
amostragem variam de diária a mensal, com amostragem
ocorrendo em algum lugar do sistema pelo menos em
intervalos semanais
Uma análise de risco deve ser realizada para determinar o
plano de amostragem para um sistema de Água Purificada.
Água Para Injetáveis- Plano de Amostragem (USP)
Em geral, espera-se que a amostragem de água para teste de endotoxina microbiana e bacteriana ocorra diariamente em algum lugar do sistema, com cada ponto sendo amostrado periodicamente, com base em uma avaliação
de risco
As expectativas regulatórias para os planos de amostragem do sistema de distribuição de Água para Injeção são mais prescritivas porque o controle microbiano deve ser muito mais rigoroso, pois está relacionado ao atributo
da endotoxina bacteriana.
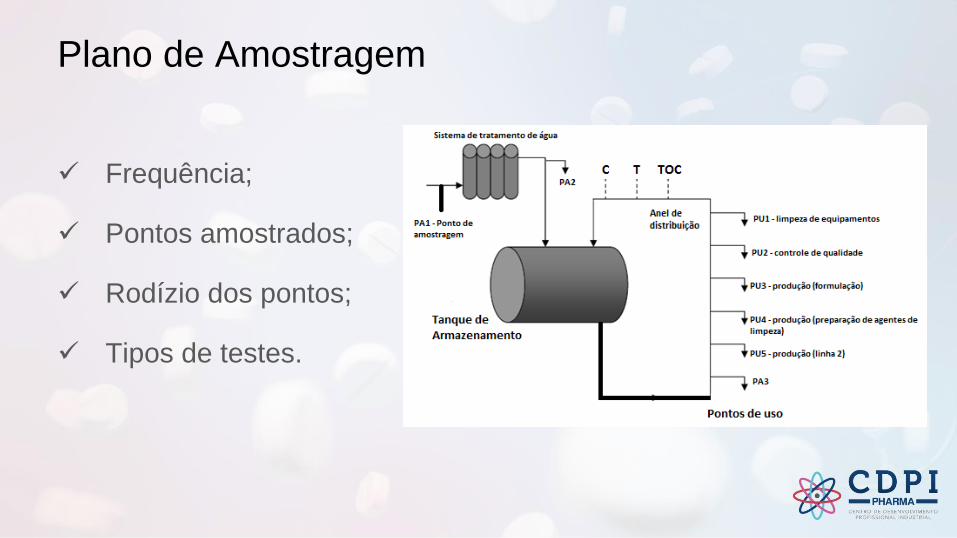
Plano de Amostragem
✓ Frequência;
✓ Pontos amostrados;
✓ Rodízio dos pontos;
✓ Tipos de testes.

Sanitização
As etapas de sanitização requerem validação para demonstrar a capacidade de reduzir e manter a contaminação microbiana em níveis aceitáveis.
A validação dos métodos térmicos deve incluir um estudo de distribuição de calor para demonstrar que as temperaturas de sanitização são alcançadas em todo o sistema
A frequência de rotina de higienização deve ser apoiada pelos resultados do monitoramento microbiano do sistema.
As conclusões derivadas da análise de tendências dos dados microbiológicos devem ser utilizadas como mecanismo de alerta para a necessidade de manutenção extraordinária
A frequência de rotina de sanitização deve ser estabelecida de tal forma que o sistema opere em um estado de controle microbiológico e não exceda regularmente os Níveis de Alerta e Ação

Sanitização
RDC 301 - Art. 112. A tubulação de água purificada e água para injetáveis e, se for o caso, de qualquer outro tipo de água, deve ser sanitizada de acordo com procedimentos escritos que contenham detalhes sobre os limites de contaminação microbiológica, bem como as medidas a serem adotadas.
• §1ºOs sistemas não devem ser operados além de sua capacidade projetada.
• §2ºA água para injetáveis deve ser produzida, armazenada e distribuída de forma a impedir o crescimento microbiano, usando de alternativas como, por exemplo, circulação constante a uma temperatura superior a 70°C.
IN 35 - Art. 83.Os sistemas de tratamento e distribuição de água devem ser projetados, construídos e mantidos de forma a garantir uma produção confiável de água de qualidade adequada.

Sanitização - WFI
Outras tecnologias de prevenção da contaminação microbiana podem ser usadas, tais como ozonização, luz ultravioleta.
Entretanto, é necessário a comprovação de produção confiável de água de qualidade injetável.
• Qualificação/Validação
• Monitoramento abrangente
• Limites de alerta/ação
Água para injetáveis com recuperação de microrganismos constante não é aceitável

Manutenção Preventiva
Um programa de manutenção preventiva deve ser estabelecido para garantir que o sistema de água permaneça em estado de controle
Registros de manutenção devem ser mantidos
O programa de manutenção deve levar em consideração:
• frequência definida para os elementos do sistema, por exemplo filtros, instrumentos, medidores
• programa de calibração
• POPs para tarefas específicas
• controle e armazenamento de peças sobressalentes
• manutenção preventiva e plano e instruções de manutenção, incluindo limpeza após manutenção
• revisão e aprovação dos sistemas para uso após a conclusão do trabalho
• registro e revisão de problemas e falhas durante a manutenção
Controle de mudanças
Operações unitárias e componentes do sistema
Parâmetros de operação
Sanitização do sistema
Procedimentos laboratoriais (incluindo amostragem)
Controle de Mudanças

Controle de mudanças
Nem todas as mudanças exigem validação, mas o impacto da mudança nos parâmetros do processo e atributos de qualidade deve ser identificado, avaliado e corrigido - pode resultar em uma validação seletiva para demonstrar o estado controle do sistema e a capacidade de manter os atributos de qualidade da água.
Certas atividades de calibração e manutenção preventiva podem ser consideradas tarefas de rotina se não afetarem a operação do sistema ou a qualidade da água. A substituição de componentes usando peças exatamente iguais geralmente não afeta a operação ou o controle do sistema.
A substituição de componentes por aqueles que não são peças exatamente iguais, mas têm especificações funcionais semelhantes pode ser realizada por meio de uma avaliação de risco documentada no sistema de controle de mudanças.

Revisão Periódica
Os sistemas de água devem ser revisados em intervalos periódicos (ex. anualmente). A revisão deve incluir:
•Mudanças desde a última revisão
•Tendências de desempenho e qualidade (capabilidade)
•Falhas e histórico de alarmes
•Investigações
•Resultados fora das especificações e fora dos limites;
•Revisão dos Limites de alerta e ação;
•Conformidade com os requisitos atuais de BPF
•Atualização da documentação
•Histórico de manutenção e calibração
•Registros (livros de registro e dados eletrônicos)
•Adequação do sistema informatizado (trilha de auditoria, usuários e privilégios)
A revisão pode resultar em ajustes nos processos operacionais ou de sanitização, planos de calibração ou manutenção ou planos de monitoramento, testes adicionais ou na repetição de certas tarefas de qualificação (requalificação).

Water systems
6 Utilities
6.7 Water treatment plant and distribution systems should be designed, constructed and maintained to minimize the risk of particulates, microbial contamination/proliferation and pyrogens (e.g. sloping of piping to provide complete drainage and theavoidance of dead legs), and prevent the formation of biofilms to ensure a reliable source of water of an appropriate quality.
Where filters are included in the system, special attention should be given to the monitoring and maintenance of these filters. Water produced should comply with the current monograph of the relevant Pharmacopeia.
6.8 Water systems should be qualified to maintain the appropriate levels of physical, chemical and microbial control, taking seasonal variation into account.
6.15 WFI systems should include continuous monitoring systems such as Total Organic Carbon (TOC) and conductivity, (unless justified otherwise) as these may give a better indication of overall system performance than discrete sampling. Sensor locations should be based on risk and the outcome of qualification.
PIC/S Draft Annex I 2020

Steam used as a direct sterilizing agent
6 Utilities
79
6.16 Feed water to a pure steam (clean steam) generator should be appropriately purified. Pure steam generators should be designed, qualified and operated in a manner to ensure that the quality of steam produced meets defined chemical and endotoxin levels.
6.17 Steam used as a direct sterilizing agent should be of suitable quality and should not contain additives at a level which could cause contamination of product or equipment. For a pure steam generator supplying pure steam used for the direct sterilization of materials or product-contact surfaces (e.g. porous hard-good autoclave loads), steam condensate should meet the current monograph for WFI of the relevant Pharmacopeia.
A suitable sampling schedule should be in place to ensure that representative pure steam samples are obtained for analysis on a regular basis. Other aspects of the quality of pure steam used for sterilization should be assessed periodically against validated parameters. These parameters should include the following: non-condensable gases, dryness value (dryness fraction) and superheat.
PIC/S Draft Annex I 2020

ALMOÇO

SISTEMA DE AR

SISTEMAS DE AR
Não estéreis
RDC 301/2019
Art. 79. As áreas de produção devem ser efetivamente ventiladas, com instalações de tratamento do ar apropriadas aos produtos manipulados, incluindo temperatura e, onde necessário, umidade e filtração, às operações realizadas e ao ambiente externo.
Estéreis
IN 35/2019
Art. 6ºAs áreas limpas para a fabricação de medicamentos estéreis são classificadas de acordo com as características exigidas do ambiente.
Parágrafo único. Cada operação de fabricação requer um nível de limpeza ambiental adequado no estado operacional, a fim de minimizar os riscos de contaminação do medicamento ou dos materiais que estão sendo trabalhados por material particulado ou microbiológico.

Não estéreis
O grau necessário de limpeza do ar em
instalações de fabricação de não estéreis
normalmente pode ser alcançada sem o
uso filtros HEPA, desde que o ar não seja
recirculado ou no caso de uma instalação
de produto único.
Muitas instalações de produtos não
estéreis são capazes de atender aos
requisitos de Grau D ou ISO 8 em repouso
(mas não é necessário classificar).
Uma avaliação de risco deve ser realizada
para determinar as condições exigidas e a
extensão da validação requerida.
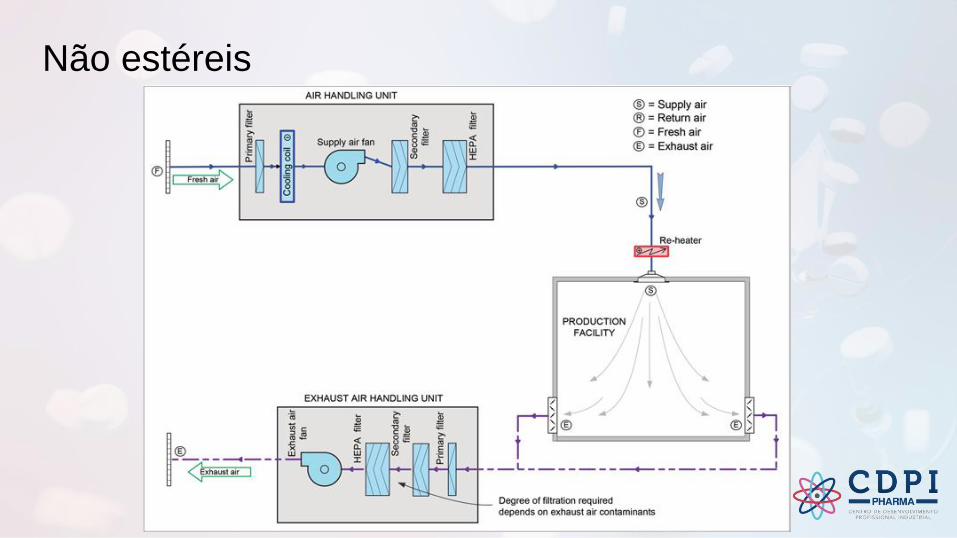
Não estéreis

Não estéreis
• Diferencial de Pressão
• O diferencial de pressão deve ser de magnitude suficiente para garantir a contenção e prevenção da reversão do fluxo (5 Pa a 20 Pa)
• Exaustores/Retorno
Contenção de pós
• Quando apropriado, a temperatura e a umidade relativa devem ser controladas, monitoradas e registradas, para garantir a conformidade com os requisitos pertinentes aos materiais e produtos e fornecer um ambiente confortável para o operador
Temperatura e Umidade

Não estéreis
Diferencial
de Pressão
✓ As portas devem abrir para o lado da pressão mais alta (ajuda a manter a porta
fechada)
✓ A localização do fornecimento e do retorno ou do exaustão deve facilitar as direções
adequadas de fluxo de ar em uma área
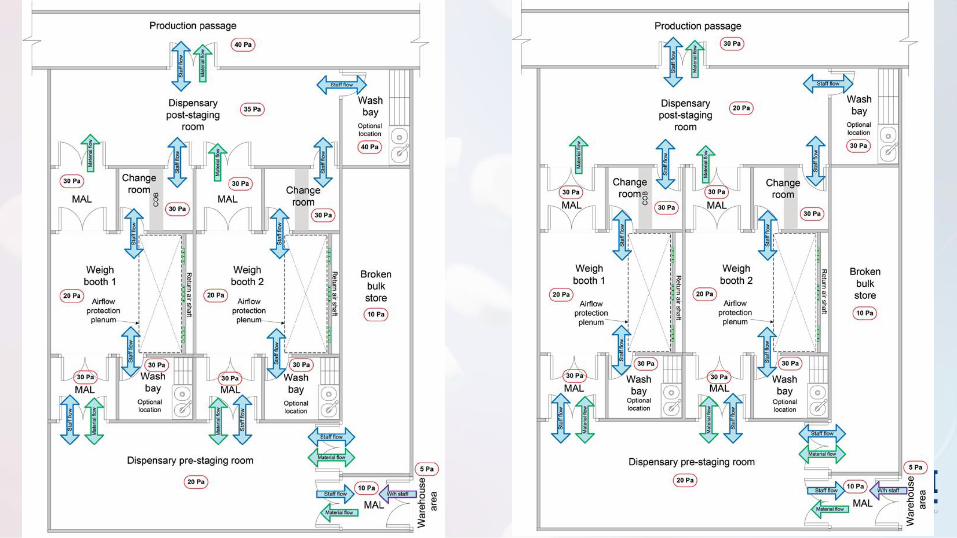
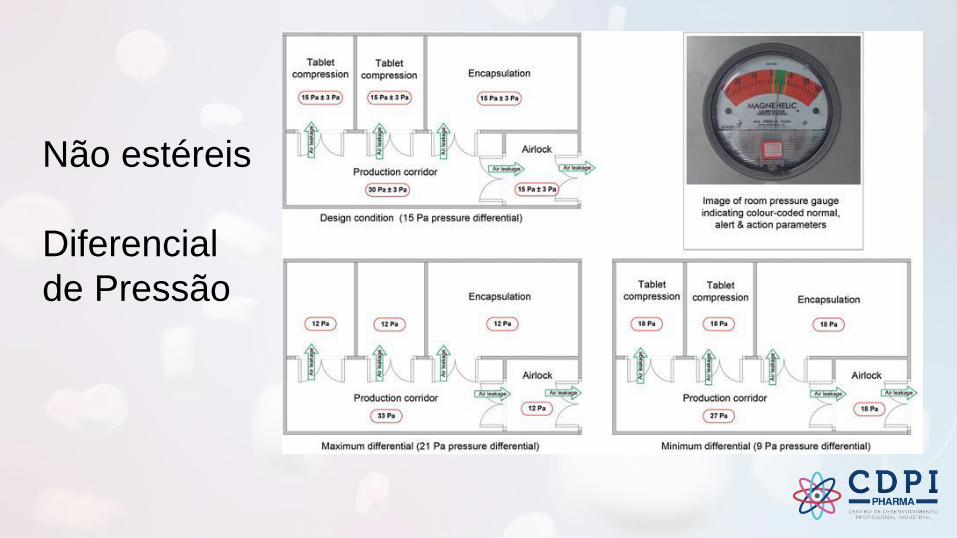
Não estéreis
Diferencial
de Pressão

Não estéreis – Qualificação /Requalificação
temperatura e umidade relativa
fluxo de ar / número de
trocas / recuperação
Diferenciais de pressão
Padrão de fluxo de ar
Velocidades de fluxo de ar
unidirecionais
Testes de vazamento do
filtro HEPA
Testes de vazamento em
dutos
Contagens microbiológicas

Manutenção Preventiva
Documentação atualizada dos sistemas HVAC, incluindo manuais de operação e manutenção, desenhos esquemáticos, procedimentos e registros.
Reparos, manutenção e manutenção preventiva (incluindo limpeza, substituição de componentes, alterações, qualificação) devem ser executados de acordo com os procedimentos.
Registros devem ser mantidos por um tempo apropriado.

SISTEMA DE AR
ESTÉREIS

Produtos Estéreis
IN 35/2019
OperaçõesX
Grau de Limpeza
Art. 6ºAs áreas limpas para a fabricação de medicamentos estéreis são classificadas de acordo com as características exigidas do ambiente. Parágrafo único. Cada operação de fabricação requer um nível de limpeza ambiental adequado no estado operacional, a fim de minimizar os riscos de contaminação do medicamento ou dos materiais que estão sendo trabalhados por material particulado ou microbiológico

Classificação das áreas limpas e dos equipamentos que fornecem ar limpo
Art. 9º As salas limpas e os equipamentos que fornecem ar limpo devem ser classificados de acordo com a versão vigente da norma ISO 14644-1 seguindo os métodos de ensaio da ISO 14644-3.
≥0,5μm ISO 5; ≥5,0 μm ISO M(20; 5μm);
em repouso e operação
≥ 0,5μm ISO 5; ≥ 5,0 μm ISO M(29; 5μm) em
repouso; ISO 7 em operação
ISO 7 em repouso / ISO 8 em operação
ISO 8 em repouso
• Padronização de Nomenclatura nos relatórios de Certificação.
• Para partículas de 5μm, áreas Grau A não mais serão ISO Classe 4,8 em repouso e operação.
• Para partículas de 5μm, áreas Grau B não serão ISO Classe 5 em repouso
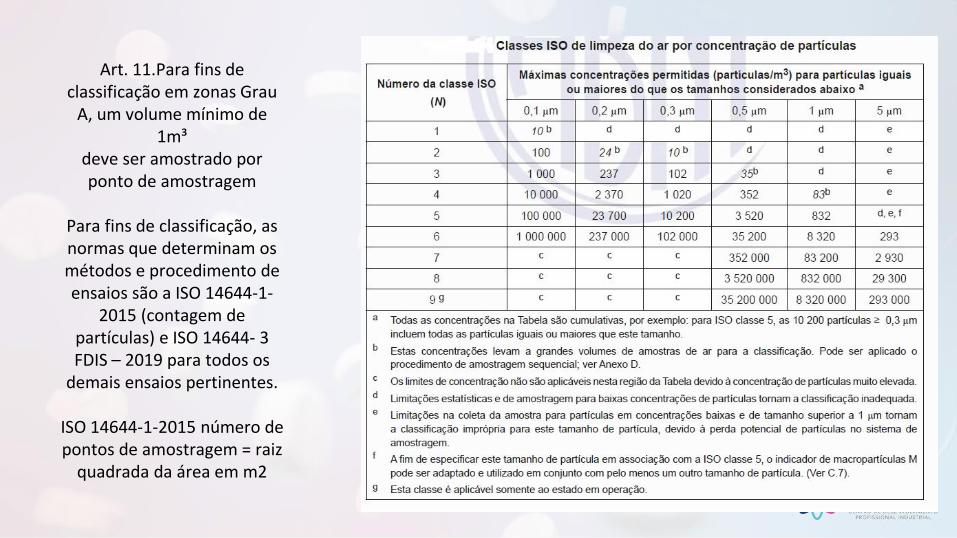
Art. 11.Para fins de classificação em zonas Grau
A, um volume mínimo de 1m³
deve ser amostrado por ponto de amostragem
Para fins de classificação, as normas que determinam os métodos e procedimento de ensaios são a ISO 14644-1-
2015 (contagem de partículas) e ISO 14644- 3 FDIS – 2019 para todos os
demais ensaios pertinentes.
ISO 14644-1-2015 número de pontos de amostragem = raiz
quadrada da área em m2
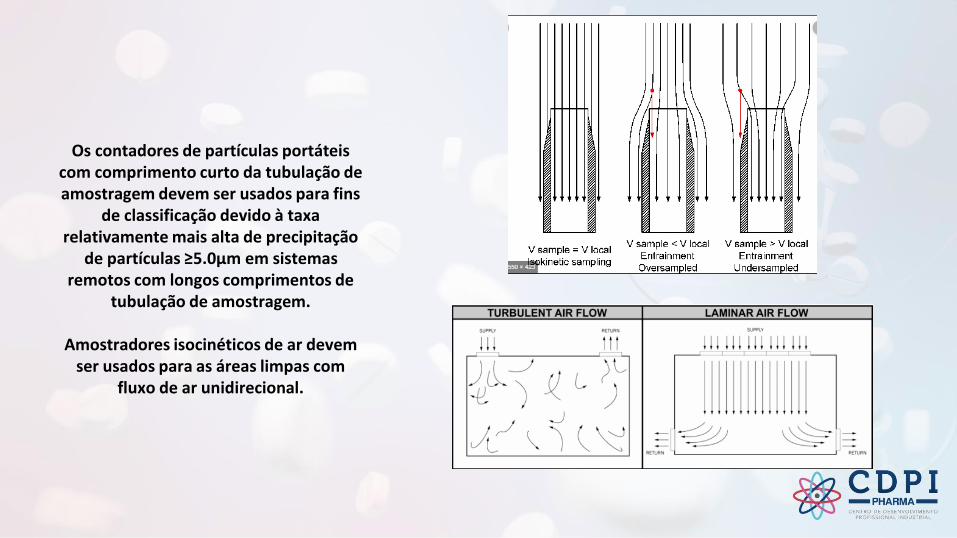
Os contadores de partículas portáteis com comprimento curto da tubulação de amostragem devem ser usados para fins
de classificação devido à taxa relativamente mais alta de precipitação
de partículas ≥5.0μm em sistemas remotos com longos comprimentos de
tubulação de amostragem.
Amostradores isocinéticos de ar devem ser usados para as áreas limpas com
fluxo de ar unidirecional.

Art. 8º Na fabricação de medicamentos estéreis quatro graus de limpeza podem ser distinguidos: I - Grau A: (…) Os sistemas de fluxo de ar unidirecional devem fornecer uma velocidade de ar homogênea na faixa de 0,36 a 0,54 m/s (valor de referência) medida na posição de trabalho das estações de trabalho com fluxo de ar
unidirecional abertas. A manutenção do padrão de fluxo de ar unidirecional deve ser demonstrada e validada. Um fluxo de ar unidirecional e com velocidades mais baixas pode ser usado em isoladores e caixas com luva;
• 0,45±20%m/s na posição de trabalho (valor de referência)
• Pode ser substituído pelo preconizado pela ISO 14644-3 -0,45±20%m/s a 15-30cm da superfície do filtro desde que em algum momento passado esta medição tenha assegurado um perfil adequado de fluxo unidirecional na posição de trabalho por meio dos estudos de visualização do fluxo de ar (smoke test)

Art. 9ºAs salas limpas e os
equipamentos que
fornecem ar limpo devem
ser
classificados de acordo
com a versão vigente da
norma ISO 14644-1
seguindo os métodos
de ensaio da ISO 14644-3.
A norma ISO 14644-2
provê informações sobre
os testes para
demonstração
da conformidade contínua
com o grau de limpeza
estabelecido.
Teste Procedimento para o
teste
Frequência
Teste de contagem de
partículas
ISO 14644-1, anexo A A, B - 6 meses
C, D - 12 meses
Diferencial de pressão ISO 14644-3, anexo B1 12 meses
Volume de ar (trocas) ISO 14644-3, anexo B2 12 meses
Velocidade do fluxo de
ar (fluxo unidirecional
ou contenção
ISO 14644-3, anexo B2 12 meses
Teste de vazamento de
filtros
ISO 14644-3, anexo B7 24 Meses
Verificação de
vazamento/contenção
ISO 14644-3, anexo B8 24 Meses
Recuperação ISO 14644-3, anexo B4 24 Meses
Visualização do fluxo de
ar
ISO 14644-3, anexo B3 24 Meses
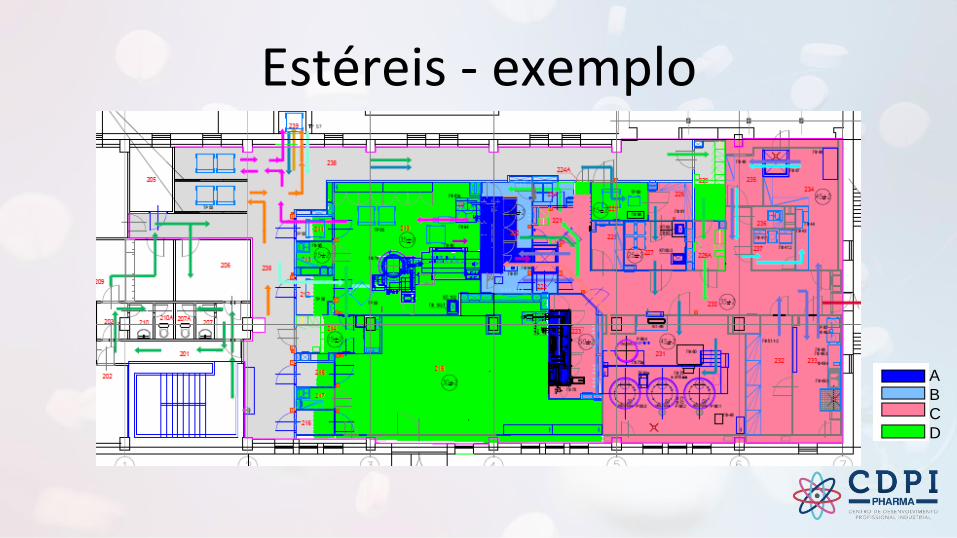
Estéreis - exemplo
A
B
C
D

Cleanroom Classification X Cleanroom Qualification
Cleanroom classification – A method of assessing the level
of air cleanliness against a specification for a cleanroom
or clean air equipment by measuring the non-viable
airborne particulateconcentration (Reference for
the classification of the cleanrooms and clean air
devices can be found in the ISO 14644 series of
standards).
Cleanroom qualification – A method of assessing the level of compliance of a classified
cleanroom or clean air equipment with its intended use (The classification of a
cleanroom or clean air equipment is part of its
qualification).
PIC/S Draft Annex I 2020 - 4. Premises
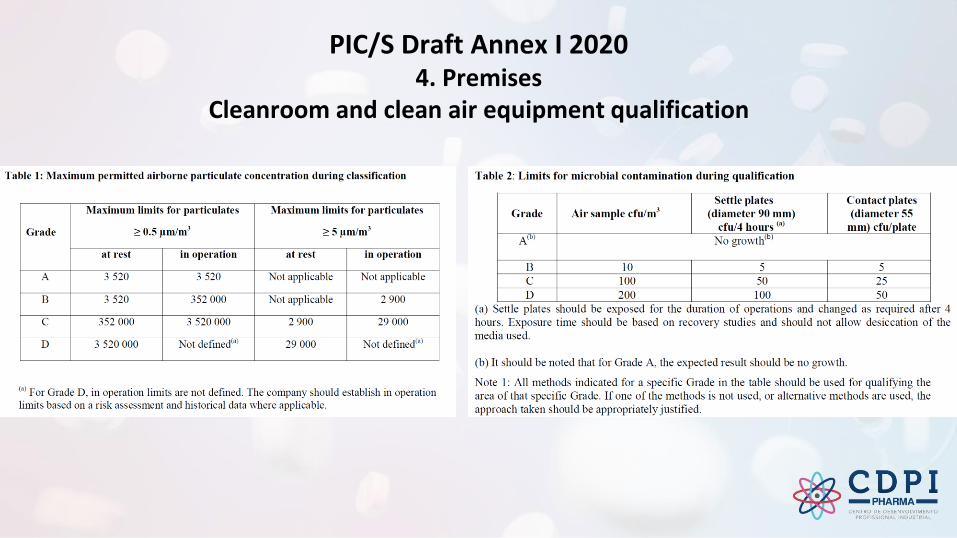
PIC/S Draft Annex I 20204. Premises
Cleanroom and clean air equipment qualification
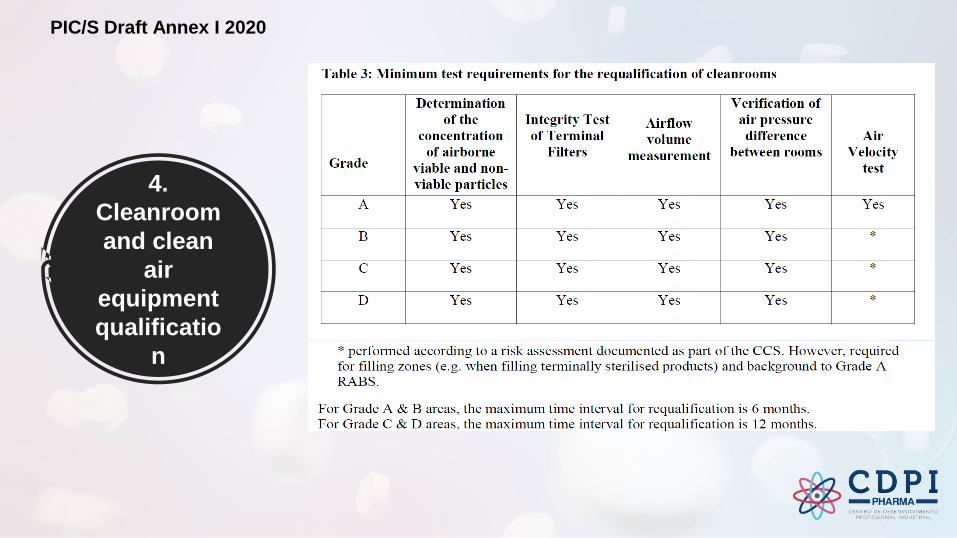
4.
Cleanroom
and clean
air
equipment
qualificatio
n
PIC/S Draft Annex I 2020
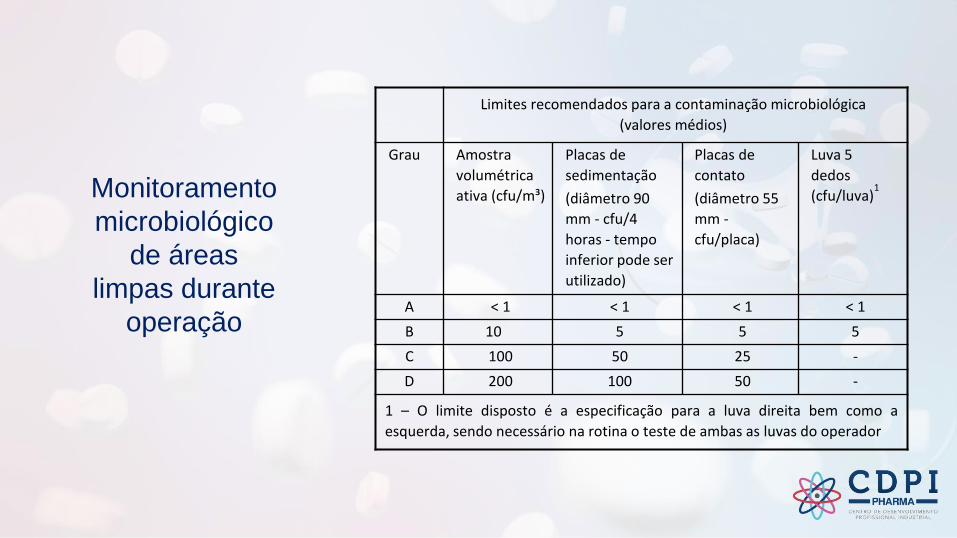
Monitoramento
microbiológico
de áreas
limpas durante
operação
Limites recomendados para a contaminação microbiológica
(valores médios)
Grau Amostra
volumétrica
ativa (cfu/m³)
Placas de
sedimentação
(diâmetro 90
mm - cfu/4
horas - tempo
inferior pode ser
utilizado)
Placas de
contato
(diâmetro 55
mm -
cfu/placa)
Luva 5
dedos
(cfu/luva)1
A < 1 < 1 < 1 < 1
B 10 5 5 5
C 100 50 25 -
D 200 100 50 -
1 – O limite disposto é a especificação para a luva direita bem como a
esquerda, sendo necessário na rotina o teste de ambas as luvas do operador

Monitoramento das áreas limpas e dos
equipamentos que fornecem ar limpo
Art. 9º - Parágrafo único. A classificação deve claramente distinguir-se do monitoramento ambiental das operações em processo.
Art. 20.As salas limpas e os equipamentos que fornecem ar limpo devem ser monitorados rotineiramente em operação.
Parágrafo único. Os pontos de amostragem para monitoramento devem ser estabelecidos com base em um estudo formal de análise de risco e nos resultados obtidos durante a classificação das salas limpas ou equipamentos que fornecem ar limpo.
Art. 21.Para as áreas de Grau A, o monitoramento de partículas deve ser realizado ao longo de toda a duração dos processos críticos, incluindo a montagem do equipamento, exceto quando justificado pela presença de contaminantes no processo que danificariam o contador de partículas ou representariam um perigo, como por exemplo organismos vivos e riscos radiológicos, onde nesses casos, o monitoramento ao longo das operações de preparação do equipamento, antes das situações impeditivas, deve ser realizado.

Monitoramento ambiental (USP)
A localização e o movimento de pessoal dentro da sala limpa se correlacionam com o risco de contaminação ao ambiente e aos processos conduzidos nesse ambiente.
Os locais de amostragem devem ser selecionados de forma que avaliem o impacto da movimentação de pessoal e do trabalho dentro da área, particularmente intervenções e manipulações dentro da zona crítica.
A rota mais provável de contaminação é o transporte pelo ar, portanto os locais mais críticos são aqueles localizados perto de materiais estéreis expostos.
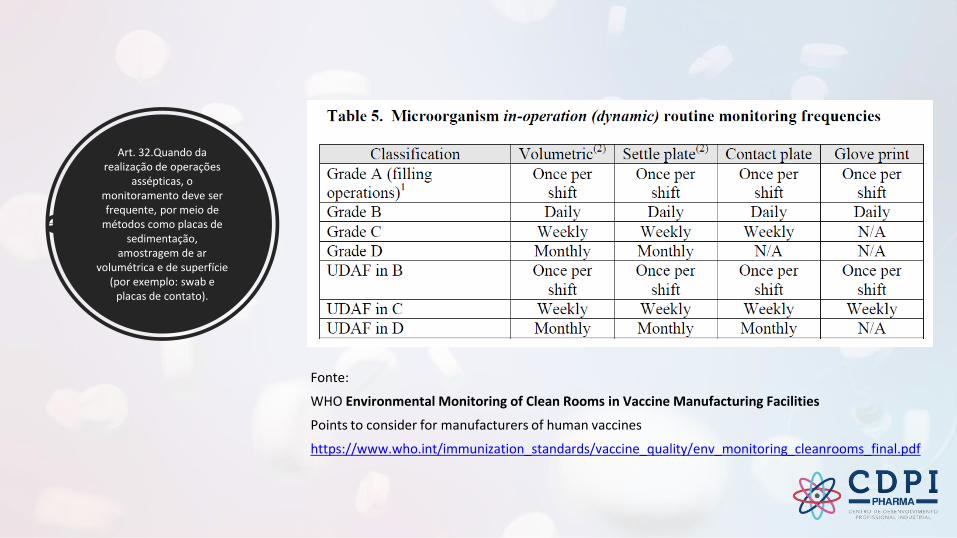
Art. 32.Quando da realização de operações
assépticas, o monitoramento deve ser frequente, por meio de
métodos como placas de sedimentação,
amostragem de ar volumétrica e de superfície
(por exemplo: swab e placas de contato).
Fonte:
WHO Environmental Monitoring of Clean Rooms in Vaccine Manufacturing Facilities
Points to consider for manufacturers of human vaccines
https://www.who.int/immunization_standards/vaccine_quality/env_monitoring_cleanrooms_final.pdf

Art. 22.Um sistema similar de monitoramento deve ser
utilizado para áreas Grau B, contudo a frequência de
amostragem pode ser reduzida.
§1ºA extensão do monitoramento da área Grau B
correlaciona-se com a efetividade da segregação
desta com a área Grau A que circunda.
Quanto mais efetiva for a separação desta em relação a área Grau A, menor o risco e menor pode ser a frequência de amostragem
Contagem pelo procedimento de monitoramento de partículas, via contadores portáteis.

Art. 36.Devem ser estabelecidos limites adequados de alerta e ação para os resultados do
monitoramento microbiológico e de partículas.
Parágrafo único. Se esses limites forem excedidos, os procedimentos operacionais devem
descrever as ações corretivas.
O uso dos limites máximos recomendados no quadro do art. 35 não é adequado como limites de alerta ou de ação, sem a comprovação estatística prévia, de que estes representam o estado de controle da área.
FB - Quando o nível microbiológico especificado para um ambiente controlado for excedido, revisão da documentação e investigação devem ocorrer. A investigação deve incluir a revisão da documentação de manutenção da área; da documentação de desinfecção; dos parâmetros físicos ou operacionais inerentes, tais como, mudanças na temperatura ambiental e umidade relativa e o estágio de treinamento dos funcionários envolvidos. Em seguida à investigação, as ações adotadas podem incluir o reforço no treinamento das pessoas para enfatizar o controle microbiológico do ambiente; a amostragem adicional em frequência aumentada; a desinfecção adicional; os testes adicionais de produto; a identificação do contaminante microbiano e sua possível fonte e a reavaliação e revalidação dos atuais procedimentos operacionais padronizados, se necessário. Com base na revisão da investigação e nos resultados dos testes, o significado do nível microbiológico excedido e a aceitabilidade das operações ou produtos processados sob aquela condição podem ser definidos. Toda investigação e justificativa das ações devem ser documentadas e fazer parte do sistema de gerenciamento da qualidade.

IN 35/2019
Art. 73. §2ºA última antecâmara destinada à paramentação deve, no estado em repouso, ter o mesmo grau da área a qual fornece acesso.
Art. 75.O fornecimento de ar filtrado deve manter uma pressão positiva e um fluxo de ar em relação às áreas circundantes de grau de limpeza inferior, sob todas as condições operacionais, varrendo a área efetivamente.
§1ºAs salas adjacentes de diferentes graus devem ter um diferencial de pressão de 10 a 15 pascais (valor de referência).
Art. 76.Deve ser demonstrado que os padrões de fluxo de ar não representam um risco de contaminação.
Art. 77.Deve ser assegurado que os fluxos de ar não distribuem partículas a partir de fontes geradoras, como pessoas, operações ou máquinas, para áreas de maior risco ao produto. (Teste de fumaça)
Art. 78.Um sistema de alarmes deve ser fornecido com o fim de indicar falhas no suprimento de ar.
Art. 79.Os indicadores de diferencial de pressão devem estar instalados nas áreas onde esta medida for importante.

PIC/S Draft
Annex I 2020
9. Viable and
non-viable
environmental &
process
monitoring
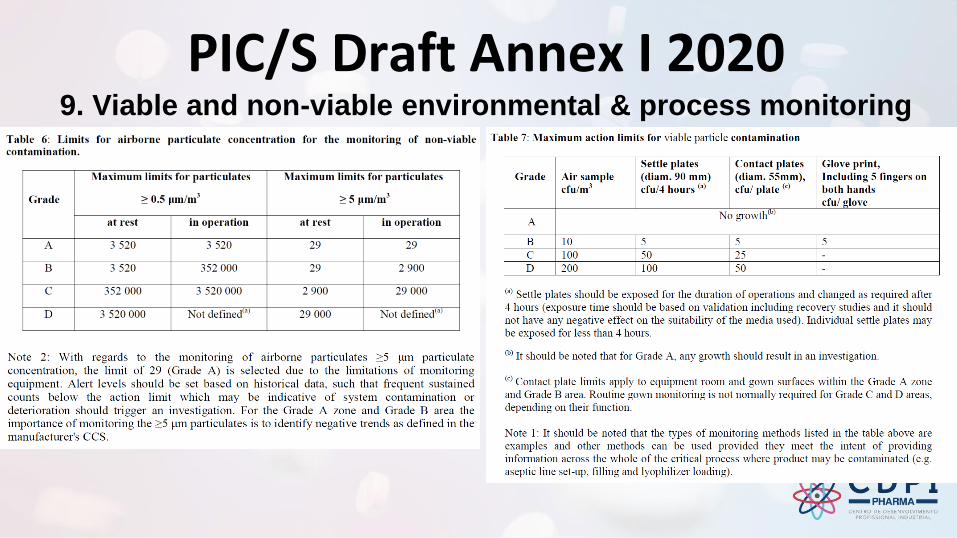
PIC/S Draft Annex I 2020 9. Viable and non-viable environmental & process monitoring

Environmental monitoring
9.4 Risk assessments should be performed in order to establish a comprehensive environmental monitoring program, i.e. sampling locations, frequency of monitoring, monitoring method used and incubation conditions.
These risk assessments should be conducted based on detailed knowledge of; the process inputs and final product, the facility, equipment, specific processes, the operations involved, historical monitoring data, monitoring data obtained during qualification and knowledge of typical microbial flora isolated from the environment.
Consideration of other information such as air visualization studies should also be included.
These risk assessments should be reviewed regularly in order to confirm the effectiveness of the site’s environmental monitoring program. The monitoring program should be considered in the overall context of the trend analysis and the CCS for the site.
PIC/S Draft Annex I 2020

Environmental monitoring9.8
Appropriate alert levels and action limits should be set for the results of viable and non-viable particle monitoring. Alert levels should be established based on results of cleanroom qualification tests or trend data and should be subject to periodic review.
9.9
Alert levels for Grade A (non-viable particles only) Grade B, Grade C and Grade D should be set such that adverse trends (e.g. a numbers of events or individual events that indicate a deterioration of cleanliness) are detected and addressed.
9.12
If action limits are exceeded, operating procedures should prescribe a root cause investigation, an assessment of the potential impact to product and requirements for corrective and preventive actions. If alert levels are exceeded, operating procedures should prescribe assessment and follow up, which should include consideration of an investigation and/or corrective actions to avoid any further deterioration of the environment.
PIC/S Draft Annex I 2020

Environmental and personnel monitoring-viable particles
9.31 Microorganisms detected in Grade A zone and Grade B area should be identified to species level and the potential impact of such microorganisms on product quality (for each batch implicated) and overall state of
control should be evaluated.
9.32 Personnel gloves (and any part of the gown that may potentially have direct impact
on the product sterility (e.g. the sleeves if these enter a critical zone) should be
monitored for viable contamination after critical operations and on exit from the
cleanroom. Other surfaces should be monitored at the end of an operation.
PIC/S Draft Annex I 2020

BREAK

GASES

GASES
Contato direto com o produto
Ar comprimido
Nitrogênio
Oxigênio
Dióxido de carbono
Argônio

Gases
• Qualificação geradorProdução onsite
• Controle de matéria-primaCilindros
GASES
Desenho Qualificação Monitoramento
Manutenção e calibração
Operação

GASES - DESENHO
• contato com o produto ou com o “equipamento de processo”
• tipo de produto - não estéril / estéril (esterilizados terminalmente, procedimentos assépticos)
Uso
• ISPE
Adequação dos materiais de construção
• Não estéreis (0,45 ou 1,2µm)
• Estéreis (0,2 µm)
Prevenção de contaminação (filtros)
Diagramas da linha (tubulação, fluxo, válvulas, filtros, salas)

GASES - QUALIFICAÇÃO
QP, QI, QO e QD
• Limites de contaminantes sólidos, água e óleo
• Capacidade
• Queda de pressão dos filtros
• Operação de alarmes
• Identificação dos componentes

GASES - QUALIFICAÇÃO
QP, QI, QO e QD
• Limites de contaminantes sólidos, água e óleo
• Capacidade
• Queda de pressão dos filtros
• Operação de alarmes

GASES - QUALIFICAÇÃO
Qualificação de desempenho• ISPE GPG Process Gases
• Inicial - 3 dias no gerador e em cada ponto de uso (especificação completa)
• Requalificação – anual (1 dia)

GASES - MONITORAMENTO
Testes de vazamento
Testes de integridade dos filtros
Controle de pressão
CQ - óleo, água, partículas, biocarga
• Análise de risco – plano de amostragem
• Ponto “Pior caso”
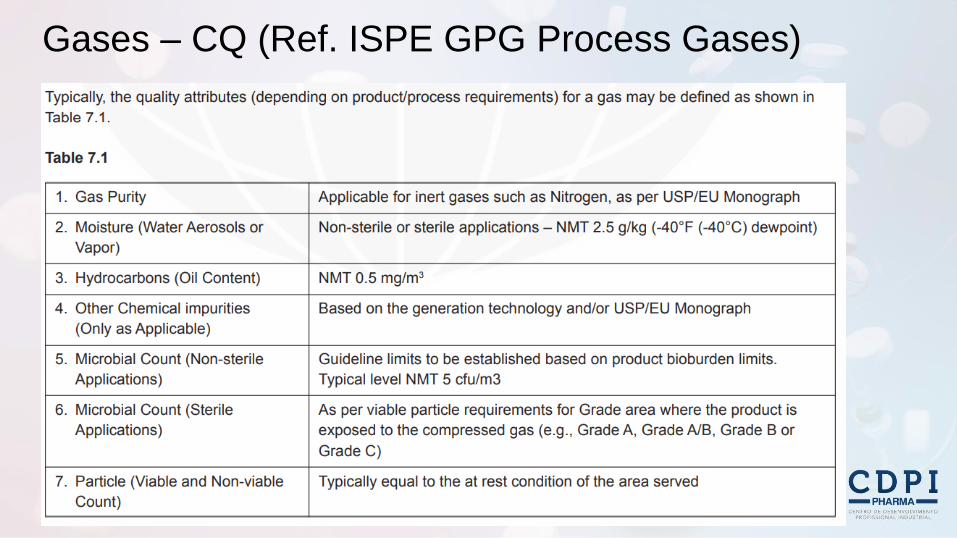
Gases – CQ (Ref. ISPE GPG Process Gases)

GASES - MANUTENÇÃO
Programa de manutenção
• Troca de filtros
Programa de calibração
Registros
Avaria /Emergência
• Interação entre manutenção não planejada e requalificação

GASES - OPERAÇÃO
POPs
Desvios e ações corretivas
Controle de Mudanças
Limpeza / sanitização/ esterilização
Logbook - parâmetros de monitoramento, incidentes, trocas de filtro, períodos de desligamento, limpeza/sanitização, manutenção

Gases – Estéreis (IN 35/2019)
Art. 84. Todos os equipamentos, tais como esterilizadores, sistemas de tratamento e filtração de ar, filtros de ventilação e de gases, sistemas de tratamento, geração, armazenamento e distribuição de água, devem estar sujeitos à qualificação e manutenção preventiva.
Art. 111.Os gases não combustíveis devem ser filtrados por filtros de retenção de micro-organismos.
Art. 155.A integridade dos filtros críticos de gases e de respiro deve ser confirmada após o uso.
Art. 156.A integridade de outros filtros deve ser confirmada em intervalos apropriados.
USP ⟨1229.15⟩ STERILIZING FILTRATION OF GASES
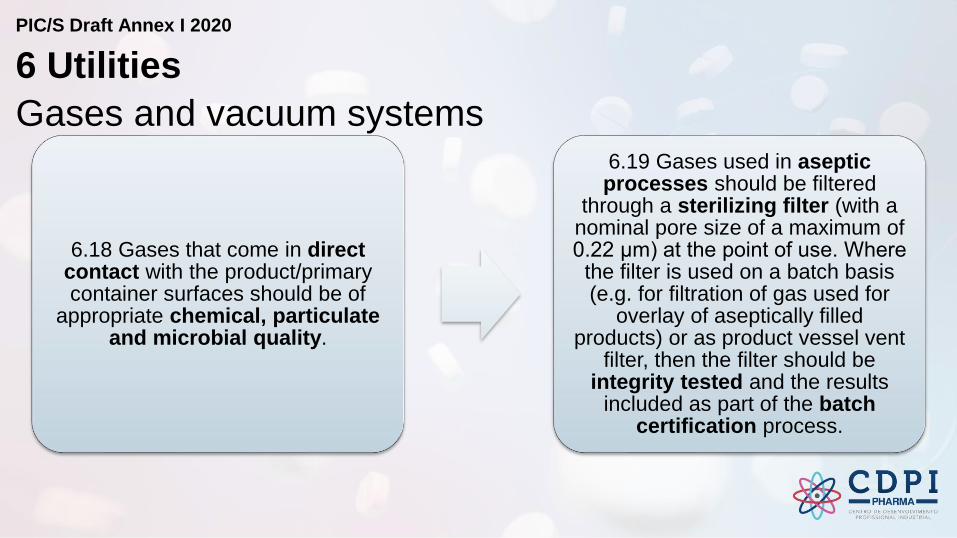
Gases and vacuum systems
6.18 Gases that come in direct contact with the product/primary container surfaces should be of
appropriate chemical, particulate and microbial quality.
6.19 Gases used in aseptic processes should be filtered
through a sterilizing filter (with a nominal pore size of a maximum of 0.22 μm) at the point of use. Where
the filter is used on a batch basis (e.g. for filtration of gas used for
overlay of aseptically filled products) or as product vessel vent
filter, then the filter should be integrity tested and the results
included as part of the batch certification process.
6 Utilities
PIC/S Draft Annex I 2020

Informações de contato sobre o palestrante,
email, numero de whatsap, site
OBRIGADO!