estudo comparativo dos metabolitos do exemestano num ... · farmacêuticos, do laboratório de...
TRANSCRIPT

AANNDDRREEIIAA IISSAABBEELL GGOONNÇÇAALLVVEESS LLOOPPEESS
Estudo comparativo dos metabolitos do exemestano num modelo
celular de cancro da mama estrogénio-dependente
Dissertação do 2º Ciclo de Estudos Conducente
ao Grau de Mestre em Toxicologia Analítica Clínica e Forense
Trabalho realizado sob a orientação de
Professora Doutora Natércia Teixeira
Professora Doutora Georgina Correia-da-Silva
Outubro, 2012

ii
Declarações
É autorizada a reprodução parcial desta dissertação apenas para efeitos de investigação,
mediante declaração escrita do interessado, que a tal se compromete.

iii
Comunicações em Painel (“Poster”) em Encontros Científicos
Lopes, A., Amaral, C., Varela, C., Roleira, F.M., Tavares da Silva, E., Correia da
Silva, G., e Teixeira, N. Biological Effects of Exemestane Metabolites In An Estrogen-
receptor-positive Aromatase-overexpressing Breast Cancer Cell Line. Apresentado no 5º
Encontro de Investigação Jovem da Universidade do Porto, Fevereiro 2012, Porto,
Portugal.
Lopes, A., Amaral, C., Varela, C., Roleira, F.M., Tavares da Silva, E., Correia da
Silva, G., e Teixeira, N. Comparative study of exemestane metabolites in breast cancer
cell model. Apresentado no XXI Porto Cancer Meeting 2012, Abril 2012, Porto, Portugal.

iv

v
Agradecimentos
À Prof. Doutora Natércia agradeço, desde o início, a disponibilidade e a atenção.
Agradeço o apoio, a exigência, a partilha do saber e as valiosas contribuições para este
trabalho. Acima de tudo, obrigada por me acompanhar nesta jornada e por estimular o
meu interesse pelo conhecimento.
Agradeço à Prof. Doutora Georgina, que sempre manteve a porta aberta, a prontidão e
disponibilidade. Obrigada pelos conhecimentos transmitidos e capacidade de estímulo ao
longo de todo o trabalho. Agradeço também a paciência na hora das fotografias e o seu
olhar crítico sobre os resultados. A sua boa disposição é contagiante!
À Professora Maria de Lourdes pela atenção que sempre demonstrou desde o início do
mestrado. Obrigada pela preocupação demonstrada nestes dois anos para podermos
chegar ao fim desta etapa tranquilamente.
Agradeço ainda ao grupo de investigação do Prof. Doutor Elisiário Tavares do Centro de
Estudos Farmacêuticos, do Laboratório de Química Farmacêutica da Faculdade de
Farmácia da Universidade de Coimbra pela síntese dos compostos e pela disponibilidade
demonstrada.
Aos meus colegas de laboratório que tão bem me acolheram! Obrigada ao Bruno, à
Marta, à Mariana! À Sandra, ao Henrique, ao João e à Filomena obrigada pelas
gargalhas e palhaçadas. Um agradecimento particular ao David e à Susana pela
disponibilidade nos pequenos percalços. Agradeço também ao Daniel e à Dona Casimira
por todo o apoio prestado.
Um obrigado especial à Cristina por todo o apoio, mesmo! Obrigada pelas explicações e
pela paciência, pelo incentivo e compreensão. Nem sempre temos a sorte de encontrar
no nosso caminho alguém que nos identifiquemos. Obrigada pelas palavras de ânimo e
pelos sorrisos! Foi muito bom partilhar a câmara contigo!
Às minhas companheiras do número 93, à Marina e à Helena, por me terem tirado de
casa quando mais precisava e por ouvirem sempre os meus desabafos. Obrigada por me
fazerem ver a vida de outra perspetiva. Mas, acima de tudo, obrigada pela amizade!
Obrigada às amigas que estão longe, mas sempre presentes. Obrigada Flor, Sandra,
Carina, Neca e Tico por sempre se preocuparem comigo, e por me fazerem crer que a
amizade preenche uma vida!

vi
Jorge, obrigada por me fazeres ver que o dia nem sempre é tão cinzento. Obrigada pela
compreensão, encorajamento, carinho e amizade. Por muitas vezes que te agradeça
nunca será suficiente!
Agradeço à minha família, que sempre compreendeu os momentos em que não estive
presente durante todo o meu percurso académico e que sempre me apoiou e encorajou!
Um obrigado especial aos meus avós, que sempre se preocuparam e perguntavam
“Então, ainda falta muito?!”.
Ao meu irmão que permaneceu a meu lado durante as noites de estudo e trabalho
durante estes anos e que teimava em não ceder ao sono!
Aos meus pais, que nunca duvidaram e questionaram as minhas escolhas, que sempre
me apoiaram! Foi convosco que aprendi que com trabalho e humildade se chega a
qualquer lado, e o que sou hoje, sou graças a vocês. Obrigado pelo amor, pelo carinho,
pela confiança! Obrigada por tornarem tudo possível!

vii
Resumo
O cancro da mama é a patologia maligna mais frequente nas mulheres em todo o mundo.
A terapia dos cancros da mama estrogénio-dependentes em mulheres pós-menopáusicas
através da inibição da enzima aromatase, que catalisa a conversão de androgénios a
estrogénios, tem sido muito bem-sucedida. Assim, os inibidores da aromatase (AIs) são
uma alternativa ao tratamento com o tamoxifeno ou com o fulvestrant.
O exemestano, um AI esteróide de terceira geração, é um inativador irreversível da
enzima aromatase frequentemente utilizado na segunda linha terapêutica do cancro da
mama estrogénio-dependente. No entanto, apesar dos estudos epidemiológicos de
segurança clínica e toxicidade, permanecem por elucidar as suas vias de metabolização
bem como a identificação dos seus metabolitos. Este trabalho pretende avaliar os efeitos
celulares de metabolitos do exemestano, sintetizados no Centro de Estudos
Farmacêuticos, do Laboratório de Química Farmacêutica da Faculdade de Farmácia da
Universidade de Coimbra, numa linha celular humana de cancro da mama estrogénio-
dependente, que sobreexpressa a aromatase (MCF-7aro). Foram determinados os IC50 e
a avaliação dos efeitos biológicos demonstrou a presença de condensação de cromatina,
formação de blebbs de membrana e formação de vesículas acidas (AVOs) para as
concentrações mais elevadas. Verificou-se a diminuição da viabilidade e proliferação
celular, sendo o composto C33 o mais eficiente. Por outro lado, todos os compostos
conduziram à retenção de ciclo celular na fase G0/G1. Avaliou-se ainda os efeitos dos
compostos C32, C33 e 17-βHE na viabilidade das células LTEDaro, uma linha celular que
mimetiza as células resistentes à terapia hormonal, demonstrando que, contrariamente
ao exemestano, os seus metabolitos conduzem à diminuição da viabilidade celular de
forma dose-dependente. No entanto, tendo em vista a elucidação do papel da autofagia
na indução de morte ou sobrevivência das células cancerígenas sensíveis (MCF-7aro) e
das células resistentes à terapia hormonal (LTEDaro), avaliaram-se os efeitos dos
compostos C32, C33 e 17-βHE na viabilidade celular na presença ou ausência de um
inibidor da autofagia (3-MA) sugerindo que, como no caso do exemestano, a autofagia
não está envolvida nos mecanismos de sobrevivência das células MCF-7aro. No entanto,
a autofagia parece estar associada aos mecanismos de morte celular nas células
hormono-resistentes. Estas observações são importantes para elucidar os efeitos
biológicos induzidos pelos metabolitos do exemestano nas células mamárias malignas,
resistentes ou não, ao tratamento com o exemestano.
Palavras-chave: metabolitos do exemestano, aromatase, cancro da mama, estrogénio,
autofagia, apoptose.

viii

ix
Abstract
Breast cancer is the most common malignancy in women worldwide. The breast cancer
estrogen-dependent therapy in pos-menopausal woman by the inhibition of the enzyme
aromatase, that is responsible for catalysing the last step in the conversion of androgens
to estrogens, has been very successful. The aromatase inhibitors (AIs) have proven to be
good alternatives to the tamoxifen or fulvestrant treatment.
Exemestane, a third generation steroidal AI, is an irreversible inactivator of aromatase. It
is frequently used as second-line therapy for breast cancer estrogen-dependent. Despite
its epidemiological studies of clínical safety and toxicity the metabolic pathways and
identification of its metabolites remain to be elucidate. In this work, we evaluated the
cellular effect of exemestano metabolites, synthesized by Centro de Estudos
Farmacêuticos, do Laboratório de Química Farmacêutica da Faculdade de Farmácia da
Universidade de Coimbra, in an estrogen-dependent human breast cancer cell line, that
overexpress high levels of aromatase (MCF-7aro). It was determined the IC50 of
compounds and evaluated the biological effects in this cell line. It was observed formation
of chromatin condensation, membrane blebbs and the presence of acidic vesicles (AVOs)
in the highest concentrations. Exemestane metabolites induced a decline in cell
proliferation and viability, being compound C33 the most potent. On the other hand,
exemestane metabolites induced cell cycle arrest in G0/G1 phase. It was also evaluated
the effects of compounds C32, C33 e 17-βHE on the cell line LTEDaro, that over-expresse
aromatase and mimics the late stage of acquired resistance. It was demonstrated that,
contrary to exemestane, the metabolites lead to a decrease in cell viability in a dose-
dependent manner. However, in order to elucidate the role of autophagy in the induction
of cell death or survival of cancer cells sensitive (MCF-7aro) and cells resistant to
hormone therapy (LTEDaro), it was evaluated the effects of compounds C32, C33 and 17-
βHE on cell viability in the presence or absence of an inhibitor of autophagy (3-MA). Our
data suggest that autophagy is not involved with mechanisms of cell survival, as with
exemestane, in the hormone sensitive cell line. However, it appears that autophagy is
associated to cell death in the hormone resistant cell line. These observations are
important to elucidate the biological effects induced by the exemestano metabolites in
cells sensitive or resistant to exemestano therapy.
Keywords: exemestane metabolites, aromatase, breast cancer, estrogen, autophagy,
apoptosis.

x

xi
Índice
Declarações............................................................................................................ ii
Comunicações em Painel (“Poster”) em Encontros Científicos ............................... iii
Agradecimentos ...................................................................................................... v
Resumo ................................................................................................................. vii
Abstract ................................................................................................................. ix
Índice ..................................................................................................................... xi
Lista de imagens .................................................................................................. xiii
Lista de tabelas .................................................................................................... xiv
Lista de abreviaturas ............................................................................................ xv
Capítulo I: Introdução ........................................................................................................ 3
1.1. Cancro da mama estrogénio-dependente ........................................................... 5
1.1.1. Estrogénios .................................................................................................. 6
1.1.2. Recetores de estrogénio .............................................................................. 6
1.1.3. Vias de sinalização ...................................................................................... 8
1.1.4. Síntese de estrogénios .............................................................................. 11
1.1.5. Aromatase.................................................................................................. 13
1.2. Terapia endócrina ............................................................................................. 15
1.2.1. Modeladores e inativadores dos recetores de estrogénio ........................... 15
1.2.2. Inibidores da aromatase ............................................................................. 16
1.3. Exemestano ...................................................................................................... 18
1.3.1. Farmacocinética e farmacodinâmica .......................................................... 21
1.3.2. Metabolitos do exemestano ....................................................................... 22
1.3.3. Efeitos biológicos ....................................................................................... 24
1.3.4. Eficácia terapêutica .................................................................................... 25
1.3.5. Segurança e efeitos secundários ............................................................... 28
1.4. Resistência à terapia endócrina ........................................................................ 31
1.4.1. ER e os co-reguladores .......................................................................... 31
1.4.2. Crosstalk entre ER e recetores de tirosina cinases (RTK) ...................... 35

xii
1.4.3. Regulação do ciclo e morte celular ......................................................... 37
1.4.4. Autofagia e apoptose ............................................................................. 39
Objetivos ..................................................................................................................... 43
Capítulo II: Materiais e Métodos ...................................................................................... 45
2.1. Materiais .......................................................................................................... 47
2.2. Compostos ........................................................................................................ 47
2.3. Culturas celulares ............................................................................................ 48
2.3.1. MCF7-aro ................................................................................................... 48
2.3.2. LTEDaro ..................................................................................................... 48
2.4. Determinação do IC50 nas células MCF-7aro .................................................... 49
2.5. Estudos morfológicos ........................................................................................ 49
2.5.1. Coloração de Giemsa ................................................................................. 50
2.5.2. Coloração de Hoechst ................................................................................ 50
2.5.3. Coloração com laranja de acridina ............................................................ 50
2.6. Viabilidade e proliferação celular....................................................................... 51
2.7. Ciclo celular ...................................................................................................... 51
2.8. Análise estatística ............................................................................................. 52
Capítulo III: Resultados ................................................................................................... 53
3.1. Determinação do IC50 nas células MCF-7aro .................................................... 55
3.2. Estudos morfológicos ........................................................................................ 56
3.3. Análise da viabilidade e proliferação celular nas células MCF-7aro .................. 61
3.4. Ciclo celular ...................................................................................................... 65
3.5. Avaliação da presença de vesículas ácidas por coloração com laranja de
acridina .................................................................................................................... 67
3.6. Análise da viabilidade celular nas células LTEDaro .......................................... 68
3.7. Efeito do inibidor da autofagia na viabilidade celular ......................................... 70
Capítulo IV: Discussão e Conclusões ............................................................................. 73
Capítulo V: Referências Bibliográficas ............................................................................ 81

xiii
Lista de imagens
Figura 1. Domínios estruturais do ERα e o ERβ. .............................................................. 7
Figura 2. Via de sinalização dos ER ................................................................................. 8
Figura 3. Via não genómica ou de sinalização esteróide iniciada na membrana (MISS). . 9
Figura 4. Representação esquemática das várias vias de sinalização intracelulares do
estrogénio.. ..................................................................................................................... 10
Figura 5. Biossíntese de estrogénios a partir do colesterol. ............................................ 12
Figura 6. Estrutura terciária da aromatase isolada de placenta humana. ........................ 14
Figura 7. Inibidores da aromatase (AIs).. ........................................................................ 17
Figura 8. Estrutura química do exemestano. .................................................................. 18
Figura 9. Mecanismo de inibição da aromatase pelo exemestano .................................. 18
Figura 10. Mamografia do mesmo tumor mamário antes e após 3 meses de tratamento
com exemestano (25 mg/dia) .......................................................................................... 19
Figura 11. Principais vias metabólicas envolvidas no metabolismo do exemestano. ...... 22
Figura 12. Metabolitos do exemestano.. ......................................................................... 23
Figura 13. 1α,2α-epoxi-6-metilenandrost-4-ene-3,17-diona ............................................ 24
Figura 14. Relação entre o ERα e ERβ.. ........................................................................ 33
Figura 15. Vias de sinalização dos recetores de estrogénio (ER).. ................................. 36
Figura 16. Regulação do ciclo celular. ............................................................................ 38
Figura 17. Regulação da autofagia. ................................................................................ 40
Figura 18. Compostos em estudo ................................................................................... 47
Figura 19. Determinação do IC50 dos compostos C32, C33 e 17-βHE em células MFC-7aro.
........................................................................................................................................ 55
Figura 20. Células MCF-7aro tratadas com testosterona a 1 nM, examinadas por
microscopia de contraste de fase, coloração de Giemsa e coloração de Hoechst, após 72
horas ............................................................................................................................... 56
Figura 21. Efeito do composto C32 (10 e 15 µM) na morfologia celular, verificada por
microscopia de contraste de fase, coloração de Giemsa e coloração de Hoechst.. ......... 57
Figura 22. Efeito do composto C33 (0,5; 1; 2,5 e 5 µM) na morfologia celular, verificada
por microscopia de contraste de fase.. ............................................................................ 58
Figura 23. Efeito do composto C33 na morfologia celular, verificada por coloração de
Giemsa e coloração de Hoechst. ..................................................................................... 59
Figura 24. Efeito do composto 17-βHE (10 e 15 µM) na morfologia celular, verificada por
microscopia de contraste de fase, coloração de Giemsa e coloração de Hoechst .......... 60

xiv
Figura 25. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular, avaliado pelo
ensaio de MTT. ............................................................................................................... 62
Figura 26. Efeito dos compostos C32, C33 e 17-βHE na síntese de DNA. ......................... 63
Figura 27. Efeito dos compostos C32, C33 e 17-βHE na integridade da membrana celular.
....................................................................................................................................... 64
Figura 28. Histograma representativo do efeito dos compostos C32, C33 e 17-βHE no ciclo
celular das células MCF-7aro.. ........................................................................................ 66
Figura 29. Coloração de laranja de acridina das células MCF-7aro, tratadas com
testosterona a 1 nM e com os compostos C32, C33 e 17-βHE, durante 144 horas.. .......... 67
Figura 30. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células
LTEDaro, avaliado pelo ensaio de MTT. ......................................................................... 69
Figura 31. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células
MCF-7aro, na presença ou ausência de 3-MA. ............................................................... 70
Figura 32. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células
LTEDaro, na presença ou ausência de 3-MA. ................................................................. 71
Lista de tabelas
Tabela 1. Classificação dos inibidores da aromatase. .................................................... 16
Tabela 2. Ensaios clínicos do exemestano realizados em diferentes linhas de tratamentos
em mulheres pré e pós-menopausicas ........................................................................... 25
Tabela 3. Efeito dos compostos C32, C33 e 17-βHE no ciclo celular das células MCF-7aro.
....................................................................................................................................... 65

xv
Lista de abreviaturas
17-βHE 17β-hidroexemestano
3-MA 3-metiladenina
4-OHA Formestano
AIs Inibidores da aromatase
Akt Proteína cinase B
AMPK Proteína cinase AMP
AP1 Proteína ativadora 1
AR Recetores de androgénio
AREG Proteína anfiregulina
ARHI Proteína G relacionada com a Ras
AVOs Vesículas ácidas
C32 6α/β-spirooxiranandrosta-1,4-dieno-3,17-diona
C33 1α,2α-epoxi-6-metilenandrost-4-ene-3,17-diona
CDK Proteínas cinases
COX-2 Ciclooxigenase-2
DBD Domínio de ligação de DNA
DHEA Desidroepiandrosterona
DHEAS Desidroepiandrosterona sulfato
DHT 5α-dihidrotestosterona
DMSO Dimetilsulfóxido
DRAM Damage-regulated autophagy modelator
EDTA Ácido etilenodiaminotetracético
EGFR Recetor do fator de crescimento epidérmico
EORTC European Organisation for Research and Treatment of Cancer
ER Recetores de estrogénio
ERE Elemento de resposta ao estrogénio
GFR Recetores de fatores de crescimento
HDL Lipoproteínas de alta densidade

xvi
HER Recetor epidérmico humano
HER2 Recetor epidérmico humano 2
IES Intergroup Exemestane Study
IGF-1 Fator de crescimento semelhante à insulina do tipo I
IGFR Recetor do fator de crescimento semelhante à insulina
IL-11 Interleucina 11
LBD Domínio de ligação de ligando
LDH Lactato desidrogenase
LDL Lipoproteínas de baixa densidade
LH Hormona luteinizante
LTED Long Term Estrogen Deprivation
MA Mesgestrol acetato
MAPK Proteína cinase ativada por mitogénios
MEM Meio de cultura Eagle’s
MISS Via de sinalização esteróide iniciada na membrana
MNAR Modulador da ação não genómica do recetor de estrogénio
mTOR Mammalian target of rapamycin
MTT 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio
NCOA3 Recetor nuclear co-ativador 3
NEM Meio de cultura sem vermelho de fenol
NF-ĸB Fator nuclear ĸB
NISS VIA de sinalização iniciada no núcleo
NSABP B33 National Surgical Adjuvant Breast and Bowel Project B33
PGE2 Prostaglandina E2
PI Iodeto de Propídio
PI3K Fosfatidilinositol-3 cinase
PINP Peptídeo de pró-colagénio tipo I
PKA Proteína cinase A
PKC Proteína cinase C
PR Recetores de progesterona

xvii
PTEN Proteína supressora de tumor homóloga à tensina
PTH Hormona paratiroide
PUMA p53 modeladora da apoptose
RB Proteína do retinoblastoma
ROS Espécies reativas de oxigénio
SBF Soro bovino fetal
SBF-CT SBF inativado pelo calor e tratado com carvão ativado
SEM Erro padrão da média
SERDs Inativadores seletivos dos recetores de estrogénio
SERMs Modeladores seletivos dos recetores de estrogénio
SP1 Proteína específica 1
T Testosterona
TEAM The Tamoxifen Exemestane Adjuvant Multinational
TKR Recetores de crescimento
TKR Recetores Tirosina cinase
TNF-α Fator de necrose tumoral α

xviii

EESSTTUUDDOO CCOOMMPPAARRAATTIIVVOO DDOOSS MMEETTAABBOOLLIITTOOSS DDOO EEXXEEMMEESSTTAANNOO NNUUMM
MMOODDEELLOO CCEELLUULLAARR DDEE CCAANNCCRROO DDAA MMAAMMAA EESSTTRROOGGÉÉNNIIOO--DDEEPPEENNDDEENNTTEE

Capítulo I: Introdução
2

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
3
Capítulo I: Introdução

Capítulo I: Introdução
4

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
5
1.1. Cancro da mama estrogénio-dependente
O cancro da mama é a patologia maligna mais frequente nas mulheres, no entanto a taxa
de mortalidade tem vindo a diminuir devido ao desenvolvimento de rastreios e
tratamentos mais adequados e eficazes. Estima-se que cerca de 60 a 75% dos tumores
mamários são estrogénio-dependentes [1]. As hormonas esteróides (estrogénio e
progesterona) estão envolvidas, respetivamente, na regulação do crescimento ductal e do
desenvolvimento alveolar na glândula mamária normal, podendo estar implicadas no
desenvolvimento e progressão do cancro da mama [2].
Os ovários são a principal fonte de estrogénios circulantes nas mulheres pré-
menopáusicas. Após a menopausa, os ovários cessam a produção de estrogénios, sendo
a síntese de hormonas esteróides assegurada por outros tecidos, nomeadamente, o
tecido mamário, ocorrendo a aromatização dos androgénios circulantes, androstenediona
e testosterona, a estrogénios, estrona e estradiol, respectivamente. Esta reação é
catalisada pela enzima aromatase, um dos passos essenciais, na biossíntese de
estrogénios. A síntese periférica de estrogénios conduz a um aumento da concentração
de estrogénios in situ. De facto, dois terços dos cancros da mama apresentam atividade
aromatásica e os níveis de estrogénios nos tecidos malignos são mais elevados do que
nos tecidos mamários normais [3].
A presença de biomarcadores no cancro tornou ainda mais eficaz a identificação dos
tipos de tumores, bem como, a escolha de tratamentos específicos. Os marcadores mais
analisados são os recetores de estrogénio (ER), os recetores de progesterona (PR) e o
recetor epidérmico humano 2 (HER2). No entanto, verificam-se diferenças na expressão
destes marcadores. Esta heterogeneidade intratumoral pode conduzir a uma
classificação errada do tipo de tumor, ou a tratamentos ineficientes, sendo, por isso,
necessário mais estudos [4].
O tratamento deste tipo de carcinomas mamários está associado à terapia endócrina que
inclui os modeladores seletivos dos recetores de estrogénio (SERMs) [5], com atividade
antagonista parcial ou total, os inativadores seletivos dos recetores de estrogénio
(SERDs) [6] e os inibidores da aromatase (AIs) [7].

Capítulo I: Introdução
6
1.1.1. Estrogénios
Os estrogénios estão envolvidos no desenvolvimento de vários órgãos e tecidos, tais
como os ossos, cérebro, sistema cardiovascular e diversos tecidos do trato urogenital [8].
Desempenham também um papel importante na regulação da secreção de
gonadotrofinas, na ovulação, desenvolvimento das características sexuais femininas e na
regulação da síntese de lipoproteínas [9]. No entanto, fatores hormonais como os
estrogénios, a progesterona, a prolactina e outras hormonas podem estimular o
crescimento do tecido epitelial da mama, podendo, deste modo, devido ao crescimento
exacerbado, acumular lesões no DNA, aumentando, assim, o risco de cancro da mama
[10]. A enzima aromatase presente no tecido adiposo da mama, promove a síntese de
estrogénios, existindo, assim, concentrações locais mais elevadas do que as existentes
na corrente sanguínea [11].
1.1.2. Recetores de estrogénio
As ações biológicas dos estrogénios são mediadas pelos seus ER específicos, ERα e
ERβ. Estes recetores pertencem à superfamília de recetores nucleares. O ERα e o ERβ
são codificados por diferentes genes, o ESR1 e ESR2, respetivamente [12].
Estes recetores têm cinco domínios estruturais semelhantes, classificados de A a F,
representados na figura 1. O domínio A/B, na região N-terminal, é altamente variável na
sua sequência e está envolvido nas interações proteína-proteína. Esta baixa
percentagem de homologia (17%) pode indicar que o ERα e o Erβ interagem de forma
distinta com as proteínas. O domínio C é altamente conservado e é responsável pela
ligação ao DNA (DBD). A região D, que contém um sinal de localização nuclear, é
importante para a estrutura tridimensional dos recetores. O domínio E está envolvido nas
ligações ao ligando (LBD) e encontra-se associado à dimerização e interação com co-
fatores. Por fim, a região C-terminal (domínio F) é altamente variável e está envolvida na
transativação dos recetores [13-15].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
7
Figura 1. Domínios estruturais do ERα e o ERβ. Adaptado de [14].
Os ER encontram-se amplamente distribuídos pelo organismo. Estes recetores têm um
papel importante no desenvolvimento de vários órgãos, como os pulmões, o cólon e o
sistema vascular [14]. No entanto, a sua abundância relativa pode variar consoante as
regiões do próprio órgão [15]. Assim, como os ERα e ERβ estão envolvidos na regulação
do crescimento celular (proliferação e diferenciação), a sua desregulação pode conduzir a
uma proliferação exacerbada das células tumorais, bem como influenciar o processo de
metastização. Embora estes recetores estejam presentes em cerca de metade dos
cancros da mama, pensa-se que a sua expressão pode perder importância com o
desenvolvimento e progressão do tumor [16].
Através de estudos moleculares, o mecanismo de ação dos ER trouxe novos horizontes
no que toca ao aumento de eficácia nos tratamentos. De facto, a determinação dos níveis
destes recetores no cancro da mama é uma ferramenta essencial para prever a
progressão da doença, a escolha do tratamento, bem como a elucidação dos
mecanismos associados à resistência de fármacos [2]. Alguns estudos demonstraram o
aumento da razão ERα/ERβ nos cancros da mama quando comparada com tumores
benignos ou tecidos normais, sugerindo que o ERα está mais associado com a
carcinogénese, enquanto que o ERβ atua como protetor contra a atividade mitogénica
dos estrogénios nas lesões malignas [17].
Os mecanismos e as diferentes vias de transcrição mediadas pelos ER envolvem
múltiplos genes, no entanto a terapia envolvendo o uso de antagonistas (SERDs) ou
modeladores (SERMs) destes recetores pode trazer enormes benefícios no tratamento
desta patologia.

Capítulo I: Introdução
8
1.1.3. Vias de sinalização
Como outros membros da família dos recetores nucleares, os ER, após ligação ao
estrogénio, regulam a transcrição de genes por ligação direta ao DNA. No entanto,
respostas mais rápidas (em segundos ou minutos) podem também ocorrer caso o
estrogénio se ligue a ER membranares ou a outros recetores membranares que não são
nem estrutural nem geneticamente relacionados com os ER, levando à ativação de
diferentes vias de transdução de sinal. Estes mecanismos de ação podem ser
classificados como via genómica e via não-genómica.
Na via genómica ou de sinalização iniciada no núcleo (NISS), os estrogénios ligam-se
aos seus recetores, ocorrendo a formação de homo ou heterodímeros de ER. Esta via
pode ocorrer sob duas formas, a via clássica e a via não clássica (figura 2).
Figura 2. Via de sinalização dos ER: NISS (sinalização esteróide iniciada no núcleo). Na via clássica, A) o
ER liga-se diretamente ao DNA, na região ERE, recrutando co-reguladores. Na via não clássica, B) a
regulação e transcrição génica é assegurada por interações com outras proteínas. Adaptado de [18].
Na via clássica, o complexo nuclear liga-se à região do elemento de resposta ao
estrogénio (ERE), resultando no recrutamento de proteínas coreguladoras (co-
activadores ou co-repressoras). Esta ligação resulta na ativação ou supressão da
expressão de genes. Na via não clássica os ER regulam a transcrição génica através de
interações com outras proteínas reguladas por fatores de transcrição, como o complexo
Fos-Jun, que se ligam a sequências de DNA alternativas (AP-1). Os ER interagem com

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
9
estes fatores, promovendo a ligação ao DNA e consequente ativação do processo de
transcrição [18].
Na via não-genómica, ou de sinalização esteróide iniciada na membrana (MISS), os
estrogénios atuam nos ER localizados perto ou na membrana plasmática ou através das
proteínas associadas à membrana dependentes de estrogénio (figura 3). A resposta da
via não-genómica resulta na ativação de vias de sinalização que levam ao aumento dos
níveis de cálcio ou de óxido nítrico e à ativação de múltiplas cascatas de cinases,
incluindo a proteína cinase ativada por mitogénios (MAPK), fosfatidilinositol-3 cinase
(PI3K), proteína cinase A (PKA) e a proteína cinase C (PKC). Estas induzem a
transcrição de genes envolvidos em múltiplos processos, como proliferação,
diferenciação e apoptose [12].
Figura 3. Via não genómica ou de sinalização esteróide iniciada na membrana (MISS) [19].
A via genómica e não genómica não são totalmente independentes, verificando-se uma
convergência de múltiplas respostas desencadeadas por estas vias, designado por
mecanismo de crosstalk (figura 4). A ativação da via genómica conduz a um aumento da
transcrição de genes importantes na via de ativação do recetor do fator de crescimento
epidérmico (EGFR). Consequentemente, o ER ativo, e através da interação com
moléculas sinalizadoras intermediárias, tais como Shc e o modulador da ação não
genómica do ER (MNAR), pode conduzir à ativação de recetores tirosina cinase (TKR),
como o EGFR, o recetor do fator de crescimento semelhante à insulina (IGFR), recetores
acoplados à proteína G, o que pode resultar na indução da atividade da via EGFR/2 e de
fatores como a HER2 o c-Scr. A família de proteínas HER é constituída por recetores de
fatores de crescimento epidérmicos incluindo o EGFR (HER1), ErbB2 (HER2), ErbB3

Capítulo I: Introdução
10
(HER3) e o ErbB4 (HER4). À exceção do HER2, todos estes recetores têm ligandos
específicos, que após a ligação promovem a formação de homo ou heterodímeros com
consequente ativação de tirosinas cinases. Assim, as proteínas EGFR estão envolvidas
em múltiplas vias de sinalização como a PI3K/Akt/mTOR, a Ras/Raf/Mek e a STATS, que
desempenham um papel importante na proliferação, angiogénese e diferenciação [5, 20].
O HER3 contém vários locais de ligação para a proteína PI3K. Por sua vez, o HER2
encontra-se sobreexpresso em 20-25% dos cancros da mama estrogénio-dependentes. A
ativação destas tirosinas cinases leva à ocorrência de fosforilações, levando à ativação
de várias vias de sinalização envolvendo a PI3K e à perda da atividade da proteína
supressora de tumor homóloga à tensina (PTEN) [21].
Figura 4. Representação esquemática das várias vias de sinalização intracelulares do estrogénio. A via 1 representa a via clássica, atuando os ER como factores de transcrição nucleares. A via 2 envolve ER e proteínas membranares (via não genómica) e a via 3 apresenta a ligação de estrogénio ao recetor GPR30/GPER, que leva à transdução de sinal através de cinases citoplasmáticas. As vias de sinal 2 e 3 são vias de resposta rápida, comparativamente à via 1. Adaptado de [8].
De facto, a via PI3K/Atk/mTOR é umas vias mais importantes no desenvolvimento do
cancro, pois regula a proliferação, o crescimento e a sobrevivência celular (figura 4).
Nesta via, os fatores de crescimento ligam-se aos EGFR, ativando a PI3K [22]. Uma vez
ativada interage com o mediador central desta via, a serina/treonina cinase Akt que
estimula a síntese proteica e o crescimento através da ativação da serina/treorina cinase

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
11
mTOR. Esta, por sua vez, pode exercer feedback positivo na Akt, amplificando a ativação
desta via. A desregulação desta via pode ocorrer pela mutação na proteína PI3K,
amplificação da Akt e da disfunção do gene supressor de tumor PTEN.
Por outro lado, as proteínas cinases fosforilam os ER nucleares que recrutam os seus co-
activadores e outros fatores de transcrição, conduzindo ao aumento da expressão
genética, e consequentemente proliferação celular. Todos os produtos resultantes destas
vias aumentam a ativação dos TKRs por fatores de crescimento, completando a atividade
dos ER e o seu crosstalk com os recetores de fatores de crescimento e ativação de
cinases [23]. Por sua vez, outros estudos demonstraram que o crosstalk entre o ER e a
via HER1/2 parece ser determinante no desenvolvimento de resistência de novo e de
resistência adquirida ao tratamento do cancro da mama [18, 24], desempenhando um
papel importante na regulação da proliferação celular bem como na inibição da apoptose
[25].
1.1.4. Síntese de estrogénios
A síntese de estrogénios ocorre em locais distintos nas mulheres pré e pós-
menopáusicas. As células teca dos ovários são a principal fonte de estrogénio na mulher
antes da menopausa. O estrogénio é libertado para a corrente sanguínea exercendo a
sua ação nos tecidos alvo (tecido adiposo e epitelial, endotélio vascular, ossos e cérebro)
[11, 26]. No entanto, na pós-menopausa, há uma diminuição da síntese de estrogénios
pelos ovários, passando a ocorrer a sua síntese essencialmente no tecido adiposo.
Durante a gravidez a síntese ocorre essencialmente na placenta.
O colesterol é a molécula percursora das hormonas esteróides (figura 5). A partir deste
percursor inicia-se a síntese de estrogénios mediada por enzimas altamente específicas
como enzimas citocromo P450, desidrogenases esteróides e redutases [26].
Os principais precursores androgénicos dos estrogénios são a androstenediona, a
desidroepiandrosterona (DHEA) e a desidroepiandrosterona sulfato (DHEAS), cujas
concentrações circulantes são mais elevadas que as dos esteróides, constituindo assim
um grande reservatório de precursores [26]. Por fim, a aromatase (enzima da família do
citocromo P450) cataliza a conversão de androgénios a estrogénios [27].

Capítulo I: Introdução
12
Figura 5. Biossíntese de estrogénios a partir do colesterol. 3β-HSB: 3β-hidroxiesteróide desidrogenase-∆5,4
-
isomerase; 17β-HSB: 17β-hidroxiesteróide desidrogenase. Adaptado de [28].
A biossíntese de estrogénios ocorre em três passos. A primeira e a segunda hidroxilação
ocorrem no grupo 19-metil. O terceiro passo envolve a clivagem da ligação C10-C19,
conduzindo à aromatização do anel A [29]. Existe também outra via relacionada com a
síntese de estrogénios no tecido mamário e no endométrio, que tem como finalidade a
inativação de estrogénios. A via da sulfatase é caracterizada pela conversão de sulfato
de estrona em estrona pela enzima sulfatase e pela sua biotransformação a derivados
sulfatados inativos por ação das sulfotransferases [30].
Os estrogénios sintetizados nos tecidos periféricos atuam essencialmente nesses
tecidos, de forma autocrina, não ocorrendo libertação para o fluido intersticial, nem para a
corrente sanguínea [31]. Não é, assim, possível correlacionar os níveis sanguíneos com a
atividade hormonal que ocorre nos tecidos alvo, dado que estes podem atingir níveis
elevados no local de síntese e de ação.
A síntese in situ de estrogénios, especialmente em mulheres pós-menopáusicas, tem um
papel importante na patogénese e desenvolvimento do cancro da mama estrogénio-
dependente. No entanto, a sobre-expressão da enzima aromatase, envolvida na síntese
destas hormonas, também parece estar associada ao desenvolvimento mais agressivo
desta patologia e ao aumento de recorrências a curto e médio prazo [32]. Alguns autores
defendem, ainda, que os estrogénios podem ser transformados em metabolitos
genotóxicos e/ou estar envolvidos em ciclos de oxidação-redução, conduzindo à
formação de espécies reativas de oxigénio (ROS), que podem levar a lesões no DNA,
causando mutações [33].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
13
1.1.5. Aromatase
A aromatase pertence à superfamília do citocromo P450 que é constituída por 460
elementos distribuídos por 74 famílias. É uma hemoproteína expressa em vários tecidos
do organismo, como ovários, placenta, cérebro e tecido adiposo [34]. A aromatase é uma
proteína glicosilada com 58 kDa [35]. Localiza-se na membrana do reticulo
endoplasmático e é constituída por duas proteínas, o citocromo P450 e a flavoproteína,
NADPH-citocromo P450 reductase [36]. Este complexo enzimático é responsável pela
catalisação das reações que levam à formação do anel aromático característico dos
estrogénios [37, 38]. A aromatase é codificada pelo gene CYP19A1, com 10 exões e
aproximadamente 120 kb, localizado no braço mais curto do cromossoma 15 (15q21)
[35].
A regulação da aromatase em cada tecido é muito complexa envolvendo promotores
associados a enhancers e a supressores específicos. No cancro da mama, há um
aumento do mRNA transcrito pela regulação do promotor I.4. Este aumento poderá ser
devido à maior libertação de fatores de necrose tumoral α (TNF-α) e de interleucinas 11
(IL-11) por parte das células epiteliais malignas [39]. No entanto, nem todos os transcritos
da aromatase são devidos ao promotor I.4 sendo os outros resultantes da ação dos
promotores II e I.3. Ao contrário do promotor I.4, os promotores II e I.3 possuem TATA
boxes distantes por 215 bp, o que pode indicar que estes promotores partilham os
mesmos fatores de regulação [39]. Os promotores II e I.3 podem ainda ser ativados pela
prostaglandina E2 (PGE2), sintetizada e excretada pelas células malignas [39, 40],
conduzindo ao aumento da expressão da enzima e consequente atividade quando
comparada à atividade desta enzima no tecido mamário normal. Este processo é a
principal razão para o aumento da síntese de estrogénios neste tecido podendo conduzir
ao desenvolvimento de cancro da mama. Muitos tumores expressam ainda grandes
quantidades de ciclooxigenase-2 (COX-2) e de PGE2. Estas estimulam a expressão da
aromatase, estando demonstrado que os inibidores da COX-2 também inibem a
transcrição da aromatase [35]. De facto, cerca de 80-90% da transcrição da aromatase
no cancro da mama é assegurada por estes promotores, podendo ainda haver
transcrição mediada pelo promotor I.7 [40]. Este promotor existe nas células epiteliais e é
sobre-expresso no cancro da mama [35], podendo conduzir à sobre-expressão da
aromatase na mama. Por outro lado, estudos comprovaram que a expressão da
aromatase é mais elevada nos adipócitos que se encontram perto das células
cancerígenas do que nas células epiteliais e no estroma da mama, havendo, assim,
acumulação de estrogénios devido à sua maior produção in situ [41].

Capítulo I: Introdução
14
A estrutura da aromatase permaneceu desconhecida durante anos, dificultando deste
modo o conhecimento do seu mecanismo de ação. No entanto, muito recentemente foi
possível determinar a estrutura da aromatase presente na placenta humana, por
cristalografia (figura 6) [42]. A estrutura terciária da aromatase consiste em 12 hélices α
principais (identificadas de A a L) e 10 folhas β (numeradas de 1 a 10) [43]. O centro
catalítico da enzima é constituído pelos resíduos Ile305, Ala306, Asp309 e Thr310 da
hélice I, Phe221 e Trp224 da hélice F, Ile133 e Phe134 do loop B-C, Leu372 e Val373 da
hélice K e do loop β3, Met374 da folha β3 e da Leu477 e Ser478 do loop β8-β9 [42]. Os
loops entre as hélices B’ e C, β7 e β8, β9 e β10 fazem parte do local ativo da enzima [43].
Figura 6. Estrutura terciária da aromatase isolada de placenta humana. A azul-escuro está representado o
terminal amina e a vermelho o terminal carboxilo. As hélices α principais estão identificadas de A a L e as folhas β numeradas de 1 a 10. A ligação da androstenediona está também representada com a cor magenta [43].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
15
1.2. Terapia endócrina
O cancro da mama apresenta diversos estados clínicos que variam com a evolução
característica de cada tumor e a reação ao tratamento. É portanto, necessário, adaptar
cada plano de tratamento a cada paciente, aumentando a taxa de sobrevivência, sem
interferir com a qualidade de vida do doente.
A terapêutica do cancro da mama pode ser assegurada por tratamentos locais, como a
cirurgia e radioterapia, e por terapias sistémicas, tais como a quimioterapia e terapia
hormonal. O desenvolvimento da terapia endócrina para tratamento do cancro da mama
é um dos mais notáveis progressos na história da terapêutica do cancro [44].
Nas mulheres pré-menopaúsicas, a biossíntese de estrogénio pode ser bloqueada por
ablação dos ovários, por cirurgia ou radioterapia, ou pelo tratamento com agonistas dos
recetores da hormona luteinizante (LH), responsável pela ação da aromatase nos
ovários. Os modeladores seletivos dos ER (SERMs), como o tamoxifeno [45], ou os
inativadores seletivos dos ER (SERDs), como o fulvestrant [46] também são terapêuticas
eficazes. Nas mulheres pós-menopáusicas, para além da terapêutica com os SERMs e
SERDs, os inibidores da aromatase (AIs) de terceira geração são também propostos,
conduzindo à redução dos níveis de estrogénios in situ.
1.2.1. Modeladores e inativadores dos recetores de estrogénio
Os SERMs atuam como antagonistas ou agonistas do ER, inibindo ou ativando os ER.
Um dos fármacos desta categoria, mais utilizado mundialmente, é o tamoxifeno, descrito
como sendo um fármaco essencial no tratamento do cancro da mama. O tamoxifeno é
administrado geralmente como terapia de primeira linha. Atua nos tecidos mamários,
ligando-se aos ER permitindo a sua dimerização e ligação ao ERE, recrutando co-
repressores e impedindo, assim, a transcrição de genes. Inicialmente foi utilizado para
tratamento de cancro da mama em estado avançado, sendo posteriormente utilizado na
terapia adjuvante. Mais recentemente foi aprovado como fármaco para reduzir o risco de
desenvolvimento de tumores mamários em fase inicial em mulheres pré e pós-
menopáusicas. Apesar da sua eficiência clínica nos tumores mamários, o tamoxifeno
também modela o perfil lípido devido à sua atividade anti-estrogénica, verificando-se uma
redução de lipoproteínas de baixa densidade e mantendo ou aumentando as

Capítulo I: Introdução
16
lipoproteínas de alta densidade. No entanto, apresenta também alguns efeitos
secundários, nomeadamente o aumento do risco de eventos tromboembólicos, de
carcinoma do endométrio e de outras patologias malignas [47].
Os SERDs ligam-se aos ER impedindo a sua dimerização, acelerando a degradação
destes, e conduzindo, assim, à inativação da via de sinalização iniciada por estas
hormonas [48]. O SERD fulvestrant é bem tolerado, sendo eficaz no tratamento de
cancros da mama estrogénio-dependentes [49], no entanto, também se verificam efeitos
secundários como náuseas, vómitos e dores de estômago [50].
1.2.2. Inibidores da aromatase
Os AIs atuam de forma distinta, inibindo a função da enzima aromatase responsável pela
conversão de androgénios a estrogénios, diminuindo, desta forma, a síntese de
estrogénios. Estes fármacos caraterizam-se pela sua estrutura química sendo
classificados em AIs esteróides e não-esteróides. Os AIs esteróides, ou do tipo I, são
derivados da androstenediona e ligam-se irreversivelmente à aromatase. Por sua vez, os
AIs não esteróides, ou do tipo II, são inibidores competitivos da aromatase (tabela 1) [1].
Tabela 1. Classificação dos inibidores da aromatase (AIs).
Esteróides (Tipo I) Não Esteróides (Tipo II)
Primeira Geração Testolactona Aminoglutetimida
Segunda Geração Formestano Fadrozol
Terceira Geração Exemestano Anastrozol e Letrozol
O desenvolvimento dos AIs levou à descoberta de fármacos mais eficazes e mais
potentes. De facto, a terceira geração de AIs esteróides, como o exemestano, e não
esteróides, como o anastrozol e letrozol, é a mais potente e demonstra um espetro de
ação mais reduzido, cujos efeitos secundários resultantes da terapia são menores, sendo
atualmente a mais utilizada nas mulheres pós-menopaúsicas (figura 7). Paralelamente,
estudos clínicos demonstraram que o uso de AIs no tratamento de cancro da mama
apresenta uma maior eficácia comparativamente ao tratamento com o tamoxifeno [51].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
17
Figura 7. Inibidores da aromatase (AIs). A terceira geração de AIs atua exclusivamente sobre a aromatase,
tendo efeitos colaterais minimos. A potência dos AIs é determinada pela percentagem de inibição no organismo. 4-OHA (4-hidroxiandrostenediona ou Formestano®).
Estudos, utilizando a linha celular de cancro da mama MCF-7, revelaram que os AIs de
terceira geração induzem uma retenção de ciclo celular na fase G0/G1 e morte celular, por
apoptose. A via de sinalização apoptótica induzida envolve as proteínas supressoras de
tumor p53 e a p21 e a ativação de caspases, havendo diminuição da expressão da Bcl-2
e da ciclina D1 [52].
Os AIs podem ser administrados durante um longo período, embora o risco de
desenvolvimento de resistência a estes fármacos não possa ser ignorado [53]. O
tratamento com AIs de terceira geração leva ao decréscimo dos níveis séricos de
estrogénios de cerca de 81%-94% para o anastrozol, 88%-98% para o letrozol e 52-72%
para o exemestano [44]. Esta diminuição está associada à perda de massa óssea,
ocorrência de eventos cardiovasculares, complicações ginecológicas e alterações do
sistema músculo-esquelético [54-57].

Capítulo I: Introdução
18
1.3. Exemestano
O exemestano (6-metilenandrosta-1,4-dieno-3,17-diona) pertence à classe dos AIs
esteróides, atuando como o substrato natural da aromatase, a androstenediona (figura 8).
O exemestano liga-se covalentemente à enzima, inativando-a irreversivelmente.
Figura 8. Estrutura química do exemestano.
Estudos, relacionando a estrutura e função do exemestano, sugerem que após ligação ao
local ativo da aromatase, este é convertido em intermediários reativos que se ligam
irreversivelmente à enzima, causando a sua inativação. A interação entre o exemestano e
a aromatase é feita em três etapas. A primeira consiste na ligação do fármaco à enzima.
Na segunda, o exemestano é convertido a um intermediário que se liga à enzima,
resultando, assim, num complexo que conduz à inativação irreversível da aromatase [29,
58]. Por último, ocorre a degradação da enzima por proteossomas (figura 9) [58].
Figura 9. Mecanismo de inibição da aromatase pelo exemestano [58].
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
19
O exemestano é altamente específico para a enzima, e embora se verifique uma
diminuição significativa de estrogénios circulantes, não se verificam alterações na síntese
de corticosteróides ou de aldosterona a nível adrenal, nem nos níveis plasmáticos de 17-
hidroxiprogesterona e de dehidroepiandrostenediona (DHEA) [59]. Estudos envolvendo
mulheres com cancro da mama em estado avançado, mostraram que uma dose de
exemestano de 10 ou 25 mg suprime os níveis de estradiol, estrona e de estrona sulfato
(cerca de 85% a 95% comparativamente aos níveis basais) após 12 a 13 semanas de
tratamento [60]. Uma dose única de 25 mg de exemestano, induz um decréscimo máximo
dos níveis de estrogénios circulantes dois a três dias após a ingestão, persistindo durante
quatro a cinco dias [61]. Este valor mínimo de atividade da aromatase persistiu durante
cinco dias, com tomas diárias de 25 mg do fármaco. Verificou-se também, com a dose
mais baixa de 5 mg, 50% de inibição dos níveis de estrogénios séricos e dos níveis
urinários de estrona [59].
Estudos in vivo demonstraram que o exemestano é altamente específico e um potente
inibidor da aromatase, levando a profundas alterações endócrinas no ambiente tumoral,
bem como nos tecidos adjacentes. Estes efeitos verificam-se na redução do volume do
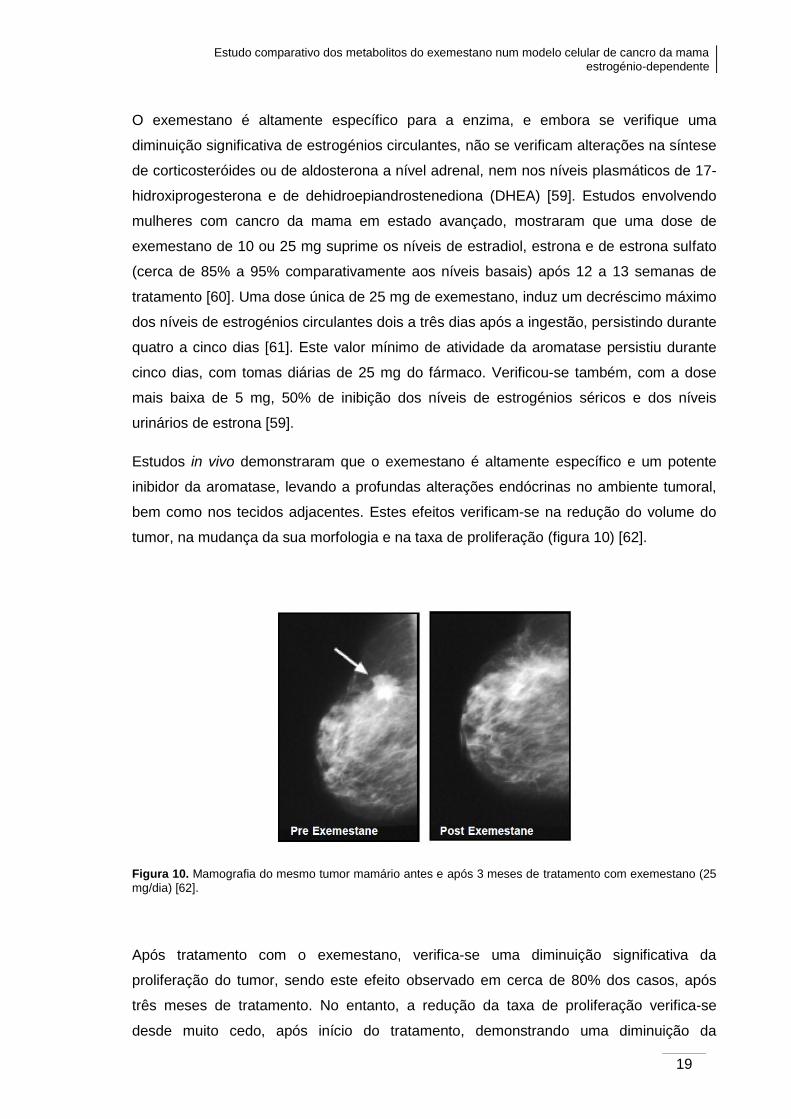
tumor, na mudança da sua morfologia e na taxa de proliferação (figura 10) [62].
Figura 10. Mamografia do mesmo tumor mamário antes e após 3 meses de tratamento com exemestano (25
mg/dia) [62].
Após tratamento com o exemestano, verifica-se uma diminuição significativa da
proliferação do tumor, sendo este efeito observado em cerca de 80% dos casos, após
três meses de tratamento. No entanto, a redução da taxa de proliferação verifica-se
desde muito cedo, após início do tratamento, demonstrando uma diminuição da

Capítulo I: Introdução
20
expressão do Ki67, um marcador da proliferação celular, após 10 a 14 dias de terapêutica
[63].
O exemestano em baixas doses tem, comparativamente ao 17β-estradiol, efeitos
estrogénicos em baixas doses, verificando-se uma fraca interação com o ER α [64]. Este
AI demonstra também uma ínfima afinidade para o recetor de androgénio (0,28%
comparativamente à dihidrotestosterona). Doses diárias de 25 mg de exemestano não
produziram nenhum efeito nos níveis circulantes de androstenediona, sulfato de
desidroepiandrosterona (DHEAS) ou 17-hidroxiprogesterona, nem foram associadas aos
ligeiros decréscimos dos níveis circulantes de testosterona. Aumentos dos níveis basais
de testosterona e androstenediona têm sido observados após tomas de doses iguais ou
superiores a 200 mg [61]. Por sua vez, um estudo sugere que durante o tratamento com
o exemestano, a atividade androgénica intratumoral é mais elevada nos doentes com
cancro da mama comparativamente a pacientes sem terapia com exemestano [27, 65].
De facto, verificam-se maiores concentrações de 5α-dihidrotestosterona (DHT), um
metabolito resultante da transformação da testosterona, nos tecidos mamários
cancerígenos tratados com exemestano. A DHT é sintetizada in situ a partir da
testosterona nos tecidos mamários cancerígenos, sendo o nível intratumoral de DHT
condicionado pela quantidade do seu percursor, a testosterona. A expressão da
aromatase está inversamente associada às concentrações intratumorais de DHT nos
carcinomas mamários sem terapia, suprimindo a síntese de DHT a partir da
androstenediona, indicando, assim, que a aromatase é um regulador negativo da
produção de DHT no cancro da mama, através da redução da concentração da
testosterona. Por outro lado, a razão DHT/testosterona sugere que os níveis da atividade
da 5α-redutase, enzima responsável pela conversão de testosterona a DHT, é similar
aquando o tratamento com exemestano. Assim, a inibição da aromatase está relacionada
com o aumento das concentrações locais de DHT. Este efeito, mediado pelo
exemestano, pode ocorrer devido ao bloqueio da síntese de estrogénios, que conduz à
formação de mais percursores androgénicos, ou através da ativação da expressão de
genes específicos dependentes de androgénios [65].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
21
1.3.1. Farmacocinética e farmacodinâmica
O exemestano é administrado oralmente e a sua farmacocinética foi estudada em
mulheres menopáusicas saudáveis bem como em pacientes com cancro da mama
avançado. A distribuição e metabolismo do exemestano foram estudados com o auxílio
de um composto marcado radioactivamente. Após ingestão, o exemestano é absorvido
no trato gastrointestinal (aproximadamente 42%), sendo que os seus níveis plasmáticos
aumentam cerca de 40% após um pequeno-almoço rico em gorduras [61].
Posteriormente é distribuído pelos tecidos, ligando-se em cerca de 90% às proteínas
plasmáticas (maioritariamente à albumina e à α1-glicoproteína ácida). O seu pico de
concentração plasmática verifica-se duas horas após ingestão, sendo o seu tempo de
semi-vida aproximadamente 24 horas. No entanto, embora permaneça sem explicação, o
exemestano parece ser absorvido mais rapidamente em mulheres com cancro da mama
do que em mulheres saudáveis (1,2 horas para doentes com cancro da mama e 2,9
horas para mulheres saudáveis) [61]. Os níveis plasmáticos do principal metabolito do
exemestano, 17-beta-hidroexemestano, são dez vezes menores do que os níveis do
exemestano [59].
O exemestano é eliminado através da urina e das fezes, detetando-se quantidades do
fármaco inalterado inferiores a 1% da dose ingerida [66]. Após tomas repetidas, a
clearence do fármaco é 45% menor nas mulheres com cancro da mama do que em
mulheres pós-menopáusicas saudáveis [61, 67].
Pouco se conhece sobre as vias de metabolização do exemestano e as enzimas
envolvidas e metabolitos resultantes. No entanto, sabe-se que a CYP3A4, CYP2B6,
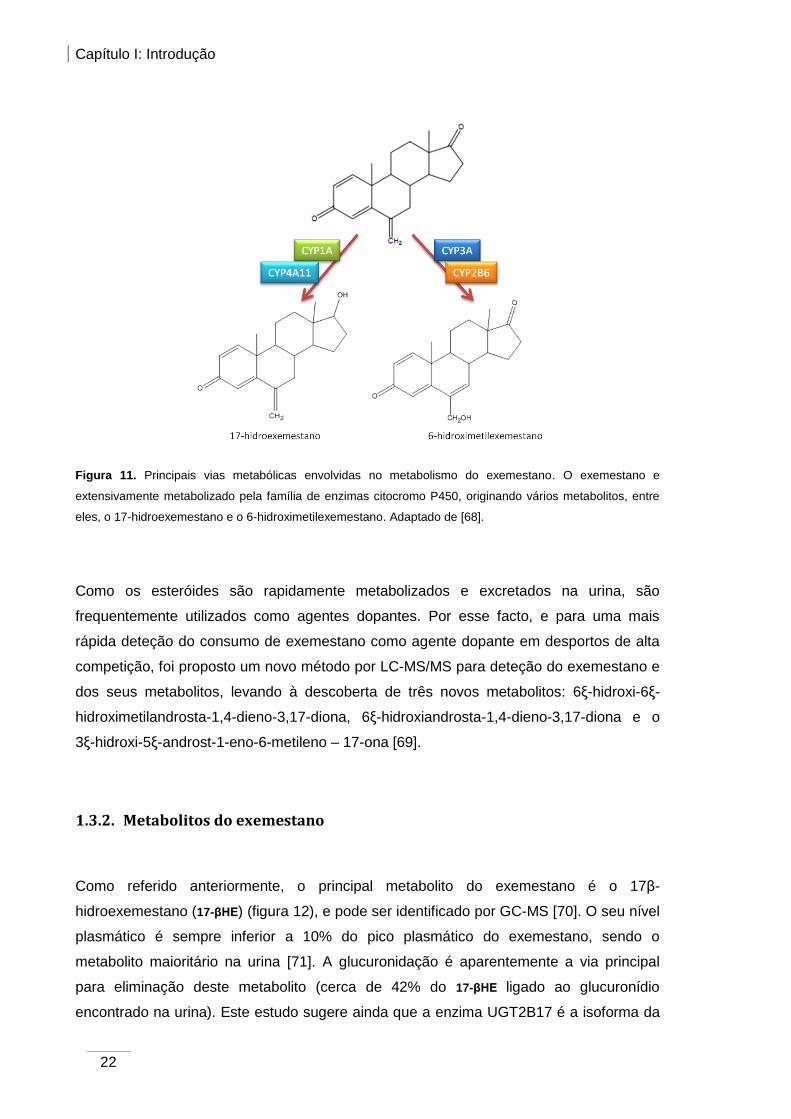
CYP1A e CYP4A11 estão envolvidas na transformação do exemestano (figura 11). O
exemestano é metabolizado a 17-hidroexemestano, através da redução do grupo
carbonilo na posição 17, pelas enzimas CYP4A11 e CYP1A1/2, no entanto, a formação
deste metabolito é mediada primeiramente por aldocetoredutases. Por sua vez, a
formação do 6-hidroximetilexemestano é catalisada essencialmente pela CYP3A, devido
à oxidação do grupo metileno na posição 6 [68]. A formação destes dois metabolitos varia
amplamente entre as amostras de fígado humano. Contrariamente à CYP3A, as CYP1A,
CYP2B6 e CYP4A11 estão praticamente ausentes do fígado, portanto a atividade destas
isoenzimas devido a polimorfismos genéticos e interações com outros medicamentos
e/ou xenobióticos pode alterar a biodisponibilidade do exemestano [68].
Capítulo I: Introdução
22
Figura 11. Principais vias metabólicas envolvidas no metabolismo do exemestano. O exemestano e
extensivamente metabolizado pela família de enzimas citocromo P450, originando vários metabolitos, entre
eles, o 17-hidroexemestano e o 6-hidroximetilexemestano. Adaptado de [68].
Como os esteróides são rapidamente metabolizados e excretados na urina, são
frequentemente utilizados como agentes dopantes. Por esse facto, e para uma mais
rápida deteção do consumo de exemestano como agente dopante em desportos de alta
competição, foi proposto um novo método por LC-MS/MS para deteção do exemestano e
dos seus metabolitos, levando à descoberta de três novos metabolitos: 6ξ-hidroxi-6ξ-
hidroximetilandrosta-1,4-dieno-3,17-diona, 6ξ-hidroxiandrosta-1,4-dieno-3,17-diona e o
3ξ-hidroxi-5ξ-androst-1-eno-6-metileno – 17-ona [69].
1.3.2. Metabolitos do exemestano
Como referido anteriormente, o principal metabolito do exemestano é o 17β-
hidroexemestano (17-βHE) (figura 12), e pode ser identificado por GC-MS [70]. O seu nível
plasmático é sempre inferior a 10% do pico plasmático do exemestano, sendo o
metabolito maioritário na urina [71]. A glucuronidação é aparentemente a via principal
para eliminação deste metabolito (cerca de 42% do 17-βHE ligado ao glucuronídio
encontrado na urina). Este estudo sugere ainda que a enzima UGT2B17 é a isoforma da

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
23
UGT responsável por esta reação [72]. O 17-βHE, tal como o exemestano, possui
atividade anti-aromatásia, no entanto, é menos potente apresentando um IC50 em
microssomas da placenta de 69 nM [71] e um IC50 de 2,3 ± 0,83 µM em células que
sobreexpressam a enzima aromatase (HEK293-aro) [72]. Tal como o exemestano, tem
atividade androgénica, apresentando uma afinidade para os recetores de androgénios
cerca de 100 vezes superior ao exemestano. Simultaneamente, o 17-βHE, para além da
inibição da aromatase, também previne a perda óssea e não influencia os níveis séricos
do colesterol [73, 74].
O metabolito 6α/β-spirooxiranandrosta-1,4-dieno-3,17-diona, referido neste trabalho como
composto 32 (figura 12), foi inicialmente descrito por Buzzetti et al. [71], apresentando um
valor de IC50, em microssomas de placenta humana, de 206 nM. Este metabolito é
referido em vários estudos como um composto intermediário do metabolismo do
exemestano, resultando da epoxidação do grupo metileno na posição 6, estando também
envolvido na síntese de outros metabolitos [69, 75]. Por sua vez, o metabolito 6-
hidroximetilexemestano ou 6-hidroximetilandrosta-1,4,6-trieno-3,17-diona (figura 12),
também foi descrito inicialmente por estes autores, apresentando um valor de IC50 de 560
nM [71].
Figura 12. Metabolitos do exemestano. A) 17β-hidroexemestano; B) 6-hidroximetilexemestano e C) 6α/β-
spirooxiranandrosta-1,4-dieno-3,17-diona.
Recorrendo à técnica de GC/MS foi possível, então, identificar outros metabolitos do
exemestano: 6ξ-hidroxi-6ξ-hidroximetilandrosta-1,4-dieno-3,17-diona, 6ξ-hidroxiandrosta-
1,4-dieno-3,17-diona e o 3ξ-hidroxi-5ξ-androst-1-eno-6-metileno – 17-ona. O perfil de
excreção destes metabolitos foi determinado, verificando-se a excreção máxima entre
quatro a seis horas após a toma de 25 mg de exemestano [69].

Capítulo I: Introdução
24
Embora novos estudos sobre o metabolismo e metabolitos do exemestano estejam a ser
desenvolvidos, muito ainda permanece por esclarecer. Com base nas vias de
metabolização propostas, novas moléculas são projetadas como intermediários. A
molécula 1α,2α-epoxi-6-metilenandrost-4-ene-3,17-diona, referida neste trabalho como
composto 33, parece ser, também, um composto intermediário na via de metabolização do
exemestano (figura 13) [69, 75, 76].
Figura 13. 1α,2α-epoxi-6-metilenandrost-4-ene-3,17-diona, possível metabolito do exemestano.
1.3.3. Efeitos biológicos
Poucos são os estudos relativos aos efeitos biológicos do exemestano nas células. O
exemestano tem um IC50 de 27 nM em microssomas de placenta humana [71] e de 1,4 ±
0,42 µM em células HEK293-aro, que expressam a enzima aromatase [72].
Os estudos, conduzidos no nosso laboratório, numa linha celular de cancro da mama
recetor de estrogénio positivo que sobre-expressa a aromatase (MCF-7aro), indicam que
após tratamento durante 6 dias o exemestano induz a formação de blebs membranares,
fragmentação e condensação da cromatina e vacuolização do citoplasma [77]. Tal como
os AIs não esteroides de terceira geração [52], o exemestano também promove retenção
do ciclo celular na fase G0/G1, acompanhado de uma diminuição da viabilidade e
proliferação celular. No entanto, para longos tempos de tratamento, o exemestano
conduz à retenção do ciclo em G2/M, efeito este, que se encontra associado à ocorrência
de apoptose. Verifica-se ainda, após o tratamento com o exemestano, o aumento da
atividade das caspases 9 e 7, formação de ROS bem como a presença de vesículas
ácidas (AVOs), indicando a ocorrência de apoptose e de autofagia [77]. Este estudo
demonstra ainda que a mitocôndria desempenha um papel importante na morte celular
por apoptose e que a autofagia poderá desempenhar um papel de pró-sobrevivência das
células MCF-7aro, protegendo as células da morte celular por apoptose. Outros estudos

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
25
sugerem que o papel da autofagia nas células malignas do cancro da mama está
associado à menor frequência da ocorrência da apoptose e ao prolongamento da
sobrevivência destas células [78-80].
1.3.4. Eficácia terapêutica
Cada vez é mais evidente que o uso de AIs é equivalente ou superior à terapia com o
tamoxifeno. No entanto, é menos claro o papel da terapia combinada com um AI e o
tamoxifeno. De facto, o tamoxifeno e o exemestano são extensivamente metabolizados
pelas enzimas P450, podendo existir interações farmacocinéticas quando combinados.
No entanto, um estudo clínico demonstrou que a combinação dos dois fármacos foi bem
tolerada durante 8 semanas, não sendo necessário alterações no ajustamento das doses
administradas [81].
A eficácia clínica do exemestano foi avaliada nas diversas fases de desenvolvimento do
cancro da mama estrogénio-dependente, bem como nas diferentes linhas de tratamento,
tendo sido comparado com tamoxifeno e outros AIs de terceira geração (tabela 2).
Tabela 2. Ensaios clínicos do exemestano realizados em diferentes linhas de tratamentos em mulheres pré e
pós-menopausicas. Abreviaturas: TAM, tamoxifeno, EXE, exemestano, ST, sem tratamento, MA, mesgestrol acetato.
Estratégia de Tratamento Estudo Protocolo Referências
Terapia alternada
IES TAM vs. TAM → EXE [82]
TEAM EXE vs TAM → EXE
TAM vs EXE [59]
Terapia sequencial NSABP B33 EXE vs. ST [83]
Primeira linha EORTC EXE vs. TAM [84]
Terceira linha EXE vs. TAM → MA [85]
Na procura de alternativas ao tratamento com o tamoxifeno, os AIs de terceira geração
ganharam importância ao demonstrarem ser clínicamente mais eficazes que o
tamoxifeno. De facto, os AIs são mais efetivos na taxa de resposta global e no benefício
clínico, não se verificando diferenças significativas nos efeitos secundários, excluindo os
efeitos tromboembólicos e as complicações ginecológicas [86].

Capítulo I: Introdução
26
O estudo desenvolvido pela European Organisation for Research and Treatment of
Cancer (EORTC), consta de duas fases (II e III), tendo como base a comparação do
tratamento com o exemestano ou com o tamoxifeno. Enquanto que na fase II foi avaliada
a eficácia e a segurança clínica do exemestano, na fase III, foi determinado o tempo
médio sem progressão da doença na primeira linha de tratamento do cancro da mama
hormono-dependente mestastizado. O exemestano demonstrou ser eficaz e bem
tolerado, conduzindo a um aumento da taxa de resposta ao tratamento com o
exemestano (46%) comparativamente ao tamoxifeno (31%). Os resultados revelam ainda
um aumento do tempo médio sem progressão da doença com exemestano (11,8 meses
contra os 8,1 meses após tratamento com tamoxifeno) [84]. Outros estudos de fase II
demonstraram a eficácia do exemestano na primeira linha de terapia para o cancro da
mama com metástases [87]. No entanto, estudos futuros serão necessários para
esclarecer a eficácia do exemestano como primeira linha de tratamento especialmente
com a combinação de outros fármacos, como agentes angiogénicos e inibidores da
HER2, podendo o exemestano ser considerado uma opção para a primeira linha de
tratamento para o cancro estrogénio-dependente [88].
O estudo IES (Intergroup Exemestane Study) foi desenhado de modo a comparar a
terapia com o tamoxifeno durante 5 anos e a terapia alternada de tamoxifeno com
exemestano, após 2 a 3 anos de terapêutica com tamoxifeno, num total de 5 anos de
tratamento, em pacientes pós-menopáusicas com cancro da mama estrogénio-
dependente. Os resultados desse estudo revelaram que a terapia com tamoxifeno e
exemestano, após 2 a 3 anos de tratamento apenas com tamoxifeno, está associada ao
aumento significativo do benefício clínico, do tempo de sobrevivência sem a doença e à
diminuição da ocorrência de metastases. Verificou-se também uma diminuição no risco
de ocorrência de cancro da mama contralateral, cancro do endométrio e outros cancros
primários [82]. Quando os resultados do estudo IES foram revelados, foi projetado outro
estudo, The Tamoxifen Exemestane Adjuvant Multinational (TEAM), com o objetivo de
avaliar o exemestano comparativamente à terapia alternada de tamoxifeno seguido de
exemestano. Neste estudo não se verificaram diferenças no tempo sem progressão da
doença, nem na taxa de sobrevivência, nem no tempo de ocorrência. No entanto,
comparando apenas o tratamento com o exemestano ou com o tamoxifeno nos dois
primeiros anos do estudo, verifica-se o aumento no tempo de ocorrências e na diminuição
do risco de recaída [59]. No entanto, nos estudos IES e TEAM verifica-se uma redução
de 3% de recaídas associado ao tratamento com exemestano comparativamente à
terapêutica com o tamoxifeno [89].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
27
O projeto NSABP B33 (National Surgical Adjuvant Breast and Bowel Project B33), que
comparou a eficácia do tratamento com o exemestano comparativamente a um grupo
sem tratamento, após 4 a 5 anos de terapia com o tamoxifeno, revelou o aumento do
tempo de sobrevivência sem a doença e de remissão do tumor com tratamento com
exemestano contra um grupo placebo após cinco anos de terapia com tamoxifeno [83].
O tratamento com exemestano no cancro da mama mestastizado demonstrou ser uma
alternativa quando há a falha do tratamento com outros fármacos. Outros estudos vieram
comprovar a eficácia do exemestano como terceira linha de tratamento em cancro da
mama, avaliando o benefício clínico do exemestano após falha do tratamento com
tamoxifeno e com mesgestrol acetato (MA). Estes estudos demonstraram o aumento da
resposta em 13% bem como em 30% de benefício clínico, com duração de resposta de 9
e 8 meses comparativamente ao tamoxifeno e ao MA. Além disso, os pacientes tratados
com exemestano tem 23% menor risco de morte, comparativamente aos pacientes
sujeitos ao tratamento com MA. Verifica-se também um aumento significativo no tempo
de progressão do tumor e no tempo de falha do tratamento, demonstrando também um
aumento do tempo de sobrevivência [85]. Outro estudo verificou um aumento no tempo
médio de sobrevivência após o tratamento e uma diminuição de peso nos pacientes
tratados com o exemestano [90]. Foi também avaliada a ingestão de AIs de terceira
geração comparativamente ao MA após tratamento com o tamoxifeno, demonstrando que
os AIs possuem uma eficácia e uma segurança superior quando comparado com o MA
[91].
Todos estes estudos (EORTEC, IES, TEAM e NSABP B33) demonstram a eficácia do
exemestano na primeira linha de tratamento bem como após terapêutica com o
tamoxifeno, verificando-se benefícios no tempo de sobrevivência sem a doença e no
tempo de recorrência [59].
Os AIs de terceira geração são, de facto, extremamente potentes e eficientes, e os seus
resultados clínicos sugerem que possuem maior potencial do que o tamoxifeno na terapia
contra o cancro da mama [92]. No entanto, verificam-se diferenças a nível de mecanismo
de ação e de eficácia clínica neste grupo de fármacos [93]. Apesar de o tratamento com
AIs ser altamente eficiente, verificam-se diferenças significativas após o tratamento com
AIs esteróides e não esteróides. Estas divergências podem estar relacionadas com a
natureza das moléculas [94], efeitos androgénicos, diferenças de potência de inibição de
cada AI [95] bem como das diferentes doses administradas. No entanto, tem havido uma
maior frequência do uso de exemestano como primeira linha da terapia hormonal bem
como o seu uso após falha do tratamento com outros AIs.

Capítulo I: Introdução
28
Um dos estudos demonstrou a segurança e a eficácia do exemestano na terceira ou
quarta linha de tratamento com cancro da mama mestastizado após tratamento com AIs
não-esteróides. O exemestano levou a um aumento da resposta de 4,8% contra o
tratamento com os AIs não-esteróides. Também se verificou que o exemestano é mais
eficaz em pacientes com doença em tecidos moles (50%) relativamente ao tratamento
com anastrozol e o letrozol. Verifica-se também um aumento em cerca de 24% de
benefício clínico durante mais de um ano em mulheres com cancro da mama com
tratamento com tamoxifeno e um AI não esteróide. Deste modo, o tratamento com
exemestano pode ser uma boa alternativa para o cancro da mama cujas distintas terapias
hormonais não resultaram [74]. Nos pacientes que não responderam ao tratamento com
AIs não esteroides e foram submetidos à terapêutica com o exemestano, verificou-se um
decréscimo na recorrência da doença e uma diminuição não significativa na mortalidade
[96].
1.3.5. Segurança e efeitos secundários
Comparativamente ao tamoxifeno, os pacientes tratados com AIs de terceira geração
revelaram uma menor incidência de cancro endometrial, tromboembolismo venoso e
eventos cerebrovasculares. Em contrapartida demonstraram uma maior incidência de
fraturas e um aumento da ocorrência de eventos cardiovasculares. No entanto, não se
verificou o aumento do risco cardiovascular durante a terapia com AIs comparativamente
com o grupo placebo (sem tratamento), mas verifica-se um aumento desse risco
comparativamente ao tamoxifeno [59, 97].
Devido à diminuição dos níveis de estrogénios nas mulheres pós-menopáusicas através
da terapia hormonal, seria de esperar efeitos relacionados com a menopausa. Estes
efeitos são classificados em categorias (inexistentes, leves, moderados ou graves),
sendo os mais frequentes os afrontamentos, a transpiração, mudanças de humor,
sintomas ginecológicos e musculares, como artralgia e decréscimo da densidade óssea.
Estudos demonstraram o aumento de cãibras musculares e artralgia para o tratamento
com o exemestano comparativamente com o tratamento com o tamoxifeno [59]. No
entanto, outros autores defendem que a ocorrência de artralgia ou mialgia ou de sintomas
menopáusicos durante o tratamento está relacionada com o aumento significativo da taxa
de sobrevivência [98]. Outro estudo revela que uma maior incidência da síndrome do
túnel cárpico (patologia que afeta o nervo mediano da mão e pulso) está relacionada com

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
29
o tratamento do cancro da mama comparativamente ao tamoxifeno, embora a sua
frequência seja baixa [99].
A terapia endócrina está também associada à perda de massa óssea. No entanto, o
aumento de fraturas ósseas é mais acentuado no tratamento com AIs não esteróides,
comparativamente ao tamoxifeno [100]. De facto, a forma como os estrogénios exercem
os seus efeitos a nível ósseo permanece por esclarecer, no entanto estima-se que sejam
mediados pelos seus recetores. A modelação da atividade de citoquinas pelos
estrogénios parece exercer um papel importante no metabolismo ósseo nas mulheres
pós-menopáusicas, mediando a atividade dos osteoclastos e osteoblastos, resultando
numa diminuição da ocorrência de apoptose dos osteoclastos e num decréscimo da
atividade dos osteoblastos [57, 101]. Para além disso, a reabsorção de cálcio no trato
gastrointestinal e no rim pode estar condicionada, levando à estimulação da hormona
paratiroide (PTH) [102, 103]. Por sua vez, o aumento dos níveis séricos da PTH estimula
os osteoclastos, conduzindo, deste modo, à perda de massa óssea [101]. Estes efeitos
são verificados a nível celular através da interferência nas interações entre os
osteoclastos, osteoblastos e osteócitos, interpondo-se na remodelação do osso, levando
à perda de massa óssea e aumentando a probabilidade de ocorrência de fraturas. Nos
casos de tratamento inicial do cancro da mama, a perda óssea pode ser revertida
recorrendo a terapias auxiliares [104]. Portanto, é necessário a administração de terapias
complementares, como bisfosfonatos [105] ou suplementos de cálcio e de vitamina D
[101], tentando minimizar este efeito verificado no tratamento a longo prazo com os AIs
[106, 107]. No entanto, estudos recentes demonstraram que o exemestano pode estar
associado a um efeito protetor a nível ósseo. Este efeito é devido às propriedades
androgénicas do exemestano e do seu principal metabolito, o 17-βHE [95, 108]. Estudos
pré-clínicos sugerem que o exemestano pode estimular a formação de massa óssea. O
exemestano conduz à melhoria do metabolismo e força óssea em ratos
ovariectomizados, tal como o 17-hidroexemestano [109]. Para além disso, outro estudo
demonstrou que há um aumento significativo dos níveis séricos da fração N-
terminal do peptídeo de pró-colagénio tipo I (PINP), um marcador da formação óssea
[110]. Em ratos, a testosterona parece estar relacionada com a inibição da formação de
osteoclastos através da ação da hormona paratiróide que atua nos recetores de
androgénio (AR) [111]. No entanto, no homem, apenas os osteoblastos possuem AR.
Deste modo, o exemestano pode exercer benefícios a nível ósseo nestas células, através
das suas propriedades androgénicas [112].
Comparações indiretas entre AIs de terceira geração sugerem ainda que o exemestano
pode levar a uma maior incidência do aumento de peso comparativamente com o

Capítulo I: Introdução
30
anastrozol ou o letrozol devido às suas propriedades androgénicas [113]. Para além
disso, quando administrado em doses elevadas, o exemestano pode conduzir ao
desenvolvimento de acne [114]. Por outro lado, os efeitos mais frequentes induzidos pelo
exemestano são as náuseas, o cansaço e afrontamentos (7,5% a 10,8% dos pacientes)
comparativamente a AIs não esteróides [74].
A diminuição dos níveis de estrogénios associada à terapia hormonal apresenta também
impactos a nível do perfil lipídico levando a um aumento do risco de doenças
cardiovasculares. De facto, a terapia com AIs induz variações dos níveis circulantes de
colesterol total, lipoproteínas de alta densidade (HDL), lipoproteínas de baixa densidade
(LDL) e triglicéridos. A correlação entre as alterações hormonais e o perfil lipídico está
envolvida no desenvolvimento de doenças cardiovasculares, como o enfarte do
miocárdio, embora o seu mecanismo permaneça por elucidar [54].
Comparativamente ao tamoxifeno, o exemestano não apresenta efeitos protetores no
perfil lípido, verificando-se uma diminuição dos níveis de HDL nos pacientes com
tratamento a longo prazo [115]. No entanto, outro estudo demonstrou que o exemestano
apresenta efeitos benéficos nos níveis de triglicéridos, em relação ao tamoxifeno, durante
um ano de tratamento, não apresentando alterações nos outros parâmetros lipídicos
[116].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
31
1.4. Resistência à terapia endócrina
A resistência à terapia endócrina traduz-se na não-resposta dos pacientes à terapia,
podendo designar-se resistência de novo, quando os tumores são resistentes ao
tratamento endócrino, ou resistência adquirida, quando se tornam resistentes à terapia,
limitando, assim, a eficácia deste tratamento. Torna-se portanto necessário a elucidação
e compreensão dos mecanismos moleculares característicos desta condição. A utilização
de linhas celulares que mimetizam o ambiente dos cancros da mama resistentes ou não
(células LTEDaro e MCF-7aro) tem demonstrado ser fundamental para a elucidação dos
mecanismos associados à resistência, bem como no desenvolvimento e avaliação de
novos fármacos [117].
O mecanismo de resistência é dependente do fármaco usado na terapia hormonal. De
facto, para além das diferenças entre os mecanismos de resistência observadas entre o
tamoxifeno e os AIs, verificam-se também divergências entre AIs esteróides e não
esteróides. Estas podem estar relacionadas com a natureza das moléculas e a sua
interação com o local ativo da enzima [94] e com os efeitos androgénicos [95]. Um estudo
com as células MCF-7aro que se tornaram resistentes, revelou uma expressão génica
diferente para cada AI, sugerindo mecanismos de resistência distintos [95].
1.4.1. ER e os co-reguladores
A desregulação de vários aspetos envolvidos na atividade e sinalização dos ERs é um
dos mecanismos da resistência endócrina, conduzindo as células tumorais a vias
alternativas de proliferação e de sobrevivência. Estudos recentes sobre o perfil de
expressão génica dos ER demonstraram haver uma grande heterogeneidade de ER no
cancro da mama, levando à identificação molecular de dois subtipos distintos: o cancro
ER luminal A e o ER luminal B. O cancro da mama tipo luminal A é caraterizado pela alta
expressão dos ER e dos genes envolvidos na via clássica, com uma taxa baixa de
proliferação, um comportamento menos agressivo e uma alta taxa de resposta à terapia
endócrina. Por sua vez, o cancro da mama tipo luminal B está associado à baixa
expressão de ER e dos genes relacionados com os ER, tendo uma elevada taxa de
proliferação celular, um comportamento mais agressivo e uma menor sensibilidade à
terapia endócrina [118].

Capítulo I: Introdução
32
A expressão dos ERs é a principal via para a resposta da terapia endócrina, sendo
portanto o biomarcador de referência para a escolha do tratamento endócrino. Assim, a
perda de expressão destes recetores é apontada como primeiro mecanismo para o
desenvolvimento de resistência de novo à terapia endócrina e desenvolvimento de um
fenótipo mais agressivo. A expressão dos ERs é controlada, primeiramente, por
mecanismos epigenéticos e de pós-transcrição, podendo estar relacionada com o
aumento das histonas deacetilases, responsáveis pela condensação de cromatina,
resultando numa estrutura do nucleossoma mais compacta. Por outro lado, a ocorrência
de variantes do ERs por slipcing alternativo foi verificada em tecidos normais e
cancerígenos, indicando que este pode ser um mecanismo de perda de expressão destes
recetores [118].
Geralmente, nos cancros da mama ER- verifica-se o aumento da expressão de EGFR e
HER2, comparativamente aos tumores ER+, podendo, deste modo, a ativação da via dos
fatores de crescimento conduzir à diminuição ou perda da função dos ERs. A ativação
desta via pode ainda levar à repressão da transcrição dos genes codificantes dos ERs,
conduzindo à resistência endócrina [119]. A perda de expressão dos ERs pode ser
responsável pela resistência adquirida ao tamoxifeno. De facto, cerca de 20% dos
tumores com resistência adquirida não expressam ERs, sendo que, posteriormente,
aproximadamente 1/5 desses tumores responde, na segunda linha de tratamento, aos
AIs ou ao fulvestrant. Por outro lado, a resistência adquirida através do tratamento com
fulvestrant pode estar relacionada com a perda de função dos ERs, aumentando a via
EGFR/MAPK [119].
A variação da expressão génica ou dos níveis proteicos dos ERs pode contribuir para a
resistência endócrina e o desenvolvimento de um fenótipo mais agressivo [120]. O ERα é
uma proteína que regula a expressão de vários genes envolvidos na proliferação e
sobrevivência celular, tendo assim um papel importante no desenvolvimento do cancro da
mama, bem como na sua progressão. Foram descritas várias variantes do RNA
codificante dos ERα e ERβ em cancros da mama primários, tendo sido detetadas
mutações pontuais nos ERα em doentes com metastases [17]. As mutações no ERα
podem afetar a resposta dos agentes anti-estrogénios. Estas mutações, apenas ocorrem
em menos de 1% dos cancros da mama, não contribuindo significativamente para a
resistência dos SERMs [119].
Por sua vez, os recetores β, como já foi referido anteriormente, parecem possuir funções
antagonistas às funções dos ERα (figura 14), regulando a transcrição mediada pelo
recetor α, sendo a expressão nos tumores mamários de ER beta associada a um melhor

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
33
prognóstico. A expressão e atividade do ERβ podem estar implicadas na resistência aos
SERMs. Verificaram-se altos níveis destes recetores em células mamárias cancerígenas
tratadas com tamoxifeno. No entanto, outro estudo demonstrou que baixos níveis de ERβ
podem estar relacionados com o desenvolvimento da resistência ao tamoxifeno. Deste
modo, o papel do ER beta na resistência, permanece por elucidar [119].
Figura 14. Relação entre o ERα e ERβ. Enquanto que o ERα diminui a apoptose e aumenta a diferenciação e
proliferação celular, o ERβ tem funções opostas aumentando a apoptose e diminuindo a diferenciação e proliferação celular.
Os ERs exercem a sua ação na expressão génica associados a proteínas reguladoras,
formando o complexo de transcrição. Deste modo, a expressão destas proteínas
influencia a transcrição mediada pelos ERs, pois a sua presença a nível nuclear pode
condicionar os níveis de expressão ou atividade dos ERs, mediando assim, a sua
sinalização. Na maioria dos cancros hormono-dependentes, é a via clássica que contribui
para a proliferação e progressão das células malignas. No entanto, a ativação de outras
vias, como a via não clássica e a via não genómica, poderá ter influência na resistência à
terapia hormonal. Estas vias alternativas podem induzir alterações na expressão de co-
ativadores, co-repressores, recetores tirosina cinases e outras moléculas envolvidas
contribuindo, assim, para a resistência endócrina [118, 121].
Deste modo, estudos realizados com AIs e o tamoxifeno demonstraram o papel relevante
que os ER desempenham na resistência. De facto, em células resistentes ao letrozol,
verificou-se que os ERα são ativos [18]. Sabe-se, que esta atividade pode conduzir à

Capítulo I: Introdução
34
ativação de genes e vias de sinalização que resultam na proliferação celular [122]. Uma
das funções do ERα passa por regular modificações pós-transcrição, como a fosforilação,
metilação e ubiquitinação, influenciando a interação com outras proteínas, como os co-
reguladores transcripcionais e moléculas sinalizadoras no citoplasma como a proteína
ativadora 1 (AP1), proteína especifica 1 (SP1) e o fator nuclear ĸB (NF-ĸB). O aumento
da atividade transcripcional da AP1 e do NF-ĸB está associado à resistência endócrina
[123]. Por outro lado, a sobre-expressão e o aumento da fosforilação de co-ativadores
dos ERα, como o recetor nuclear co-ativador 3 (NCOA3), leva à transcrição de genes
mediada pelo ERα, que confere resistência in vitro, estando também associada à baixa
eficácia do tamoxifeno nos doentes [123]. Os ER e as proteínas co-reguladoras podem
ainda sofrer alterações pós-tradução. Os recetores de fatores de crescimento, como o
EGFR/HER2 e o IGF-1, e cinases envolvidas na resposta ao stress celular (Akt, MAPKs,
PKA, entre outras) regulam múltiplas modificações pós-transcripcionais, influenciando,
deste modo, a atividade dos ER e a sensibilidade das células às várias terapias
endócrinas [120]. No entanto, a resistência também se verifica em células com baixos
níveis de co-repressores, interferindo, deste modo, com a atividade do tamoxifeno [119].

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
35
1.4.2. Crosstalk entre ER e recetores de tirosina cinases (RTK)
A via dos recetores de crescimento é uma alternativa para estimular o crescimento e
proliferação das células malignas, podendo contornar os efeitos que advêm da terapia
endócrina, como a inibição ou modelação da via dos ERs.
Como já foi referido, o crosstalk entre os ER e a HER2/fatores de crescimento é uma via
de sinalização fundamental para o desenvolvimento da resistência à terapia hormonal
[18, 124]. A via genómica e não genómica, mediada pelos ERs, pode ser regulada
também pelos RTK, pois os ERs e os co-ativadores podem ser fosforilados pelos RTK e
por outras cinases intracelulares, como a MAPK e PI3K/Akt, conduzindo à ativação dos
ERs na presença de anti-estrogénios, levando, deste modo, à ativação da via genómica
não clássica [118].
Por outro lado, a desregulação da via PI3K/Atk/mTOR pode conduzir à amplificação
desta via, fomentando, deste modo, a proliferação, o crescimento e a sobrevivência
celular (figura 15). A desregulação desta via pode ocorrer pela mutação na proteína PI3K,
amplificação da Akt e disfunção do gene supressor de tumor PTEN. A perda de função da
PTEN ocorre em aproximadamente 40 a 50% dos cancros da mama e resulta na ativação
da mTOR [20]. Verifica-se ainda que a híper-ativação da sinalização PI3K está associada
à redução de ER e à expressão génica mediada por estes recetores, que, por sua vez,
estão mais associados a tumores mamários do tipo luminal B [118]. Assim, as
modificações desta via estão relacionadas com a resistência à terapia, bem como ao
desenvolvimento de metastases. Atualmente existe um enorme foco no desenvolvimento
de inibidores desta via de sinalização, nomeadamente de inibidores da mTOR [22].
Estudos indicam que o tamoxifeno juntamente com inibidores de mTOR, nomeadamente
a rapamicina, pode reverter os efeitos da resistência associada a esta proteína. Verifica-
se ainda que os efeitos da resistência mediada pela mTOR são independentes dos
mecanismos dos ERα [125]. Embora ainda não haja resultados conclusivos do estudo de
fase III, em que se faz a comparação entre a eficácia do tratamento com o exemestano e
com ou sem o everolimus, um outro inibidor da mTOR, parece ocorrer um aumento do
tempo médio sem progressão da doença [20]. Deste modo, a combinação entre a inibição
da mTOR e a terapia endócrina pode conduzir a um benefício clínico no tratamento de
cancros da mama resistentes à terapia.

Capítulo I: Introdução
36
Figura 15. Vias de sinalização dos recetores de estrogénio (ER). O estrogénio pode ligar-se a ERs nucelares e conduzir à transcrição génica (a). A presença de ERs fora do núcleo celular e associados à membrana
celular auxiliam a resposta dos estrogénios através dos recetores de fatores de crescimento (GFR), como o EGFR, HER2 e o IGF1 (b) e do recrutamento de outras moléculas ativadoras (c), conduzindo à ativação de
múltiplas vias de cinases, como a PI3K/Akt e a Ras/MAPK. Esta ativação leva, por sua vez, à fosforilação de vários co-reguladores que medeiam, nomeadamente, a via dos ERs. Por outro lado, os sinais do microambiente conduzem à ativação das vias relacionadas com o stress celular e das proteínas integrinas. Estas vias, mediadas por cinases, como a cinase de adesão focal (FAK), cinase c-Jun N terminal (JNK) e a MAPK, podem modular componentes envolvidos na transcrição, incluindo os ERs (d). As alterações que
possam ocorrer em qualquer uma destas vias podem conduzir à resistência à terapia endócrina, de modo a contornar a inibição da via dos ERs [120].
A resistência de novo ou adquirida ao tamoxifeno, pode apresentar a sobre-expressão ou
hiper-ativação dos RTKs, conduzindo ao crosstalk entre os ERs e os GFRs, um ciclo
responsável pela proliferação celular e o envolvimento da via genómica e não genómica.
No entanto, os AIs e o fulvestrant são capazes de inibir a atividade dos ERs, continuando
a ser eficazes em tumores ER+ e que sobre-expressam HER2. Os tumores tornam-se
eventualmente resistentes aos estrogénios, desenvolvendo outras vias de sobrevivência,
na ausência de estrogénio. Um estudo demonstrou que o exemestano induz a expressão
do fator de crescimento epidérmico e da proteína anfiregulina (AREG) que é essencial
para a proliferação das células da mama hormono-resistentes (LTEDaro), sendo a
expressão de AREG dependente dos ER. Verificou-se ainda que, neste modelo celular, a
expressão de AREG mediada pelos ER é crucial na resistência ao exemestano [126]. No
entanto, uma das consequências da resistência da terapia endócrina é o aumento da

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
37
sensibilidade ao estrogénio, levando a que as células malignas sejam capazes de
proliferar mesmo na presença de baixos níveis de estrogénios [127]. Estudos em células
LTEDaro demonstram o aumento dos níveis de ER, comparativamente às células
sensíveis MCF-7aro. Assim, a resistência aos AIs pode passar pela sensibilização das
células aos estrogénios, levando à adaptação da resposta mediada pelos ERs, como o
aumento da expressão do HER2 e IGFR1 que, por sua vez, levam à ativação da MAPK e
da fosforilação dos ERs. Este processo sugere a presença do crosstalk entre ER e GFRs
[119].
1.4.3. Regulação do ciclo e morte celular
A sensibilidade ao tratamento endócrino também pode ser condicionada pelos
reguladores, positivos e negativos, do ciclo celular, pois a taxa de proliferação das células
malignas é menor durante o tratamento [123].
A sobre-expressão dos reguladores positivos do ciclo celular, como o MYC e a ciclina E1
e D1 envolvidas na resistência endócrina, ocorre ou por ativação de ciclinas dependentes
de cinases, responsáveis pela regulação na fase G1 ou pela inibição das proteínas
reguladoras do ciclo, a p21 e a p27 [118]. A reduzida expressão, estabilidade ou atividade
da proteína p21 e p27, bem como a inativação da proteína do retinoblastoma (RB), está
associada à baixa eficácia da terapia endócrina. A perda de função da proteína RB, uma
proteína supressora de tumor, foi identificada na maior parte das terapias endócrinas sem
sucesso. Os ER estimulam o crescimento celular através da transcrição de ciclina D1
[128]. Por sua vez, a ciclina D1 promove o ciclo celular através da ligação a proteínas
cinases (CDK4/6), que são importantes na progressão do ciclo celular quando a célula se
encontra na fase G1. Outra das funções das ciclinas D é a fosforilação e inativação da
proteína RB, conduzindo à dissociação do complexo RB/fator de transcrição E2F (figura
16). O fator de transcrição E2F dissociado e em conjunto com outros constituintes
moleculares regula a transição da fase G1 para a fase S do ciclo celular [129].

Capítulo I: Introdução
38
Figura 16. Regulação do ciclo celular. Sinais mitogénicos convergem até ao nível da regulação da ciclina D1
e da sua associação à CDK4/6. As CDK4/6 fosforilam e inativam as proteínas RB supressoras de tumor, conduzindo à dissociação do factor de transcrição E2F, que em conjunto com outros constituintes moleculares, está envolvido na regulação da transição da fase G1 para a fase S do ciclo celular [129].
No cancro da mama, a via de sinalização RB caracterizada pela perda de função da RB
está associada à resistência ao tamoxifeno. Estudos demonstraram que as linhas
celulares resistentes à ação do tamoxifeno respondem à ação dos inibidores da CDK4/6
e demonstram altos níveis de ciclina D1 e de RB fosforilada [130]. Assim, os inibidores da
CDK4/6 inibem a progressão do ciclo celular em grande parte de linhas celulares
resistentes [129].
Múltiplos recetores de fatores de crescimento (GFRs) são ativados por moduladores
específicos, conduzindo à alteração da expressão ou da atividade dos reguladores
negativos do ciclo celular, como a sobre-expressão do HER2 e a hiperativação da Akt e
da cinase Src [120]. A tirosina cinase Src tem um papel importante em várias vias de
sinalização celular regulando a proliferação, diferenciação, sobrevivência, invasão e
angiogénese. Esta cinase interage com múltiplas proteínas celulares, incluindo os ER,
estando assim envolvida na ativação da via não genómica [131]. A sua interação entre o
recetor EGF e os ER leva à estimulação mitogénica nas células cancerígenas MCF-7,
permitindo, assim, que estas proliferem e sobrevivam mesmo na presença do tamoxifeno,
da mesma forma que a expressão da Src atenua a sensibilidade das células ao fármaco.
Também se verificou a resistência ao tamoxifeno nas células malignas que apresentavam
um aumento da atividade da Src. Este aumento de atividade estava associado à

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
39
migração celular e ao desenvolvimento de um fenótipo mais agressivo. Para além disso,
os estrogénios e a Src estão também envolvidos na inibição da p27. No tratamento com
anastrozol verificou-se o aumento da atividade da Src nas células malignas, in vivo e in
vitro, sugerindo a possível importância que os inibidores da Src podem ter na terapia
destes cancros [132].
1.4.4. Autofagia e apoptose
As alterações no balanço entre a proliferação e a morte celular podem afetar a resposta
ao tratamento. O tratamento com agentes anti-estrogénios ou com AIs conduz à ativação
da resposta de stress celular e da via apoptótica nas células cancerígenas da mama. A
apoptose, ou morte celular programado do tipo I, é caraterizada pela condensação de
cromatina, degradação do DNA, fragmentação da célula em corpos apoptóticos e é
mediada pela ativação das caspases [133]. Os mecanismos envolvidos ainda não estão
completamente esclarecidos mas as respostas celulares incluem a regulação dos
membros da família da Bcl-2 e o aumento da ceramida. O crosstalk entre os efeitos
apoptóticos dos anti-estrogénios e a via do TNF mediada pelo PI3K, Akt e NF- ĸB,
conduz à apoptose. Por outro lado, a sobre-expressão de moléculas anti-apoptoticas
como a BCL-XL, e a diminuição da expressão de constituintes pro-apoptóticos como a
BIK, BAK e a caspase 9, podem conduzir à resistência endócrina [123]. No entanto, estas
alterações na atividade destes modeladores pode ser uma das consequências da
ativação da via PI3K/Akt/mTOR, através da sobre-expressão dos RTKs e o aumento da
via não genómica mediada pelos ERs [120]. Estas moléculas podem também ser
moduladas pelas proteínas da membrana e os seus fatores de sinalização, como o fator
NF-ĸB [118].
Mais recentemente, foi sugerido que, em células tumorais da mama resistentes, a
autofagia está envolvida na resistência à terapia endócrina [123]. A autofagia é um
processo catabólico que tem como função degradar proteínas e outros constituintes
celulares, reaproveitando os seus componentes para produção de energia e síntese de
constituintes celulares [134]. Este processo, também conhecido como morte celular
programada do tipo II, é caraterizado pela presença de vesículas citoplasmáticas que
englobam componentes ou constituintes celulares, sendo estes degradados pela via
lisossomal (figura 17) [135]. Por outro lado, a autofagia pode, também, desempenhar um
papel protetor na sobrevivência de células tumorais, tendo um papel importante na
progressão da patologia e desenvolvimento de metastases [136].

Capítulo I: Introdução
40
Figura 17. Regulação da autofagia. A autofagia ocorre a nível basal, mas pode ser induzida em resposta a
nutrientes ou hormonas. A via regulatória mais caraterizada é via PI3K/Atk/mTOR. Adaptado de [137]
A regulação da autofagia é assegurada pela via de sinalização PI3K/Akt/mTOR, mas
existem outras vias que tornam este processo mais complexo [136]. Esta pode ser
ativada por mutações na PI3K, por amplificação da Akt ou por perda da atividade da
PTEN. Após a sua ativação, a autofagia é inibida, estimulando o crescimento e
proliferação celular [136]. Tal como a PTEN, a proteína G relacionada com a Ras
supressora de tumor (ARHI) também pode induzir a autofagia, inibindo a sinalização pelo
PI3K-Atk [138]. Por outro lado, a proteína cinase AMP (AMPK) é ativada nas condições
de alterações dos níveis de energia e de stress celular, aumentando a razão AMP/ATP,
reprimindo a mTOR e ativando a autofagia. No entanto, a AMPK é regulada por outras
proteínas cinases dependentes de cálcio. Assim, alterações na via
AMPK/mTOR/Autofagia, pode contribuir para a progressão tumoral e resistência à terapia
endócrina. A proteína supressora de tumor, a p53, está envolvida na regulação da
autofagia, visto estar associada à resposta de stress celular e à reparação de DNA. Por
um lado, a p53 pode ativar a AMPK, conduzindo à autofagia. Simultaneamente, a p53
pode promover a autofagia através de outras moléculas reguladoras, a DRAM (damage-
regulated autophagy modelator), a Bcl-2 associada à proteína X (Bax) e a p53
modeladora da apoptose (PUMA), proteínas também associadas à indução da apoptose.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
41
Assim, a compreensão do duplo papel da p53 na regulação da autofagia pode ser
determinante para a compreensão de aspetos do metabolismo do cancro, bem como da
resistência associada à terapia endócrina [138]. A beclina 1 está envolvida na formação
do autofagossoma, nos primeiros estadios da autofagia, sendo que a ativação da beclina
1 está envolvida na inibição da tumorogénese [135]. Por sua vez, a Bcl-2, uma proteína
oncogénica, sobre-expressa em 50 a 70% dos cancros da mama, está associada à
resistência à terapia hormonal. A Bcl-2 pode inibir a autofagia, através na inativação da
beclina, podendo, desta forma, potenciar os efeitos oncogénicos mediados pela Bcl-2
[138, 139]. Deste modo, a inibição da autofagia pode atuar sinergicamente com as outras
terapêuticas utilizadas no cancro [136], apostando na modelação do processo autofágico
através da sua maior via regulatória, a PI3K/Akt/MTOR como uma nova estratégia
terapêutica (figura 17) [140].

Capítulo I: Introdução
42

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
43
Objetivos
Atualmente, a frequência do cancro da mama é elevada em todo o mundo. Estima-se que
cerca de 75% dos cancros da mama são dependentes de estrogénios, verificando-se a
presença de recetores de estrogénio (ER+). A terapia através da inibição da aromatase
nos cancros da mama estrogénio-dependentes em mulheres pós-menopáusicas tem sido
muito bem-sucedida. O desenvolvimento de fármacos como os inibidores da aromatase
(AIs) levou à descoberta de moléculas eficazes e mais potentes. De facto, a terceira
geração de AIs é a mais potente e aquela cujo espectro de ação é mais reduzido,
minimizando, deste modo, os efeitos adversos, sendo portanto, a que é mais utilizada
atualmente nas mulheres pós-menopáusicas.
O exemestano é, atualmente, o AI esteróide mais utilizado na segunda linha de
tratamento deste tipo de tumores. Apesar dos inúmeros estudos epidemiológicos e de
segurança clínica desenvolvidos, permanecem por esclarecer os efeitos biológicos deste
fármaco a nível celular. De facto, as suas vias de metabolização bem com os seus
metabolitos continuam a ser alvo de estudo, não estando, ainda, complemente
elucidados. Por outro lado, sabendo-se que o exemestano é rapidamente metabolizado,
permanece por saber se os efeitos induzidos são devidos a este AI ou aos seus
metabolitos. Deste modo, o objetivo deste trabalho é avaliar a atividade bioquímica e os
efeitos biológicos in vitro de três metabolitos do exemestano, o 17β-hidroexemestano,
6α/β-spirooxiranandrosta-1,4-dieno-3,17-diona e 1α,2α-epoxy-6-methylenandrost-4-ene-
3,17-diona, na linha celular de cancro da mama estrogénio-dependente, MCF-7aro.
Para além disso, o desenvolvimento de resistência à terapia hormonal não pode ser
ignorado, sendo necessário continuar a elucidar os mecanismos associados a esta
condição. Deste modo, um outro objetivo deste trabalho é avaliar os efeitos dos
metabolitos do exemestano na viabilidade celular da linha LTEDaro, uma linha celular
que mimetiza as células do cancro da mama estrogénio-dependente resistente à terapia
hormonal.

Capítulo I: Introdução
44

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
45
Capítulo II: Materiais e Métodos

Capítulo II: Materiais e Métodos
46

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
47
2.1. Materiais
O meio de cultura Eagle’s (MEM), soro bovino fetal (SBF), L-glutamina, antibiótico-
antimicótico, geneticina e a tripsina foram adquiridos à Gibco Invitrogen Co. (Paisley,
Scotland, UK). O 17-βHE foi adquirido à @rt Molecule (Poitiers, France).
O azul de tripano, testosterona (T), ácido etilenodiaminotetracético (EDTA),
dimetilsulfóxido (DMSO), carvão ativado, piruvato de sódio, formestano (4-OHA), 3-
metiladenina (3-MA), 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio (MTT) e Hoechst foram
fornecidos pela Sigma-Aldrich Co. (St. Louis, MO, USA). O corante de Giemsa foi
fornecido pela Merck (Damnstadt, Germany) e o meio de montagem Vectashield pelo
Vector Laboratories, Inc. (Burlingame, CA, USA). O líquido de cintilação foi adquirido à
ICN Radiochemicals (Irvine, CA, USA) e o reagente de Bradford à Bio-Rad (Bio-Rad
Labs, Munich, Germany), enquanto que os filtros do cell harvester foram obtidos da
Molecular Devices (Sunnyvale, CA, USA).
2.2. Compostos
Os compostos 32 (6α/β-spirooxiranandrosta-1,4-dieno-3,17-diona) e 33 (1α,2α-epoxy-6-
methylenandrost-4-ene-3,17-diona) foram sintetizados no Centro de Estudos
Farmacêuticos, do Laboratório de Química Farmacêutica da Faculdade de Farmácia da
Universidade de Coimbra. Foram preparadas soluções stock de 20 mM dos compostos
C32, C33 e 17-βHE, em DMSO, e armazenadas a -20˚C. As diluições apropriadas dos
compostos foram preparadas antes de cada ensaio. O formestano e o exemestano foram
usados como compostos de referência.
Figura 18. Compostos em estudo; A) 17β-hidroexemestano (17-βHE); B) 6α/β-spirooxiranandrosta-1,4-dieno-
3,17-diona (composto C32) e C) 1α,2α-epoxy-6-methylenandrost-4-ene-3,17-diona (composto C33).

Capítulo II: Materiais e Métodos
48
2.3. Culturas celulares
2.3.1. MCF7-aro
A linha celular de cancro da mama recetor de estrogénio positivo que sobre-expressa a
aromatase, MCF-7aro, foi obtida pela transfecção estável da linha celular MCF-7 com o
gene da aromatase humana da placenta e cedida pelo Dr. Shiuan Chen (Beckman
Research Institute, City of Hope, Duarte, CA, U.S.A.). As células MCF-7aro foram
mantidas a 37˚C com 5% de CO2 em meio de cultura MEM (Meio Mínimo Essencial de
Eagle) com vermelho de fenol contendo sais de Eagle, piruvato de sódio (1 mmol/L),
glutamina (2 mmol/L), penicilina-estreptomicina-anfotericina B (1%), geneticina G418 (700
ng/ml) e 10% de soro bovino fetal (SBF) inativado pelo calor. Para evitar a interferência
dos esteróides presentes no SBF e o efeito estrogénico do vermelho de fenol, três dias
antes do início da experiência, o meio de cultura foi substituído por um meio sem
vermelho de fenol, contendo 5% de SBF tratado previamente com carvão ativado (SBF-
CT). Todas as experiências foram realizadas com 1nM de testosterona (T), substrato da
aromatase e agente indutor da proliferação celular, e com os compostos C32, C33 e 17-
βHE. Todos os ensaios foram realizados em triplicado, e os resultados representam pelo
menos três experiências realizadas independentemente.
2.3.2. LTEDaro
A linha celular LTEDaro foi obtida da linha celular MCF-7aro. As células LTEDaro são
privadas durante 6 meses de estrogénios (Long Term Estrogen Deprivation),
mimetizando as células do cancro da mama estrogénio-dependente resistente à terapia
hormonal. Esta linha celular foi também cedida pelo Dr. Shiuan Chen. As células
LTEDaro foram mantidas a 37˚C com 5% de CO2 em meio de cultura sem vermelho de
fenol (NEM), contendo piruvato de sódio (1 mmol/L), glutamina (2 mmol/L), penicilina-
estreptomicina-anfotericina B (1%), geneticina G418 (700 ng/ml) e 10% de SBF inativado
pelo calor e tratado com carvão ativado (SBF-CT). Todas as experiências foram
realizadas na ausência de testosterona e com os compostos C32, C33 e 17-βHE. Todos os
ensaios foram realizados em triplicado, e os resultados representam pelo menos três
experiências realizadas independentemente.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
49
2.4. Determinação do IC50 nas células MCF-7aro
A determinação do IC50 dos vários compostos foi avaliada através da quantificação da
água tritiada libertada, de acordo com o método descrito por Thompson e Siiteri, usando
[1β-3H] androstenediona como substrato da enzima aromatase. A avaliação da atividade
da aromatase por esta técnica foi validada previamente por Zhou e colaboradores [141].
As células foram cultivadas em placas de 24 poços, na densidade de 3,5 x 105 células/ml
com meio de cultura MEM. Quando atingida a confluência, as células foram colocadas
em meio MEM sem soro, e incubadas com os inibidores a diferentes concentrações (0,1
a 25 µM), 50 nM de [1β-3H] androstenediona e 500 nM de progesterona, durante uma
hora, a 37˚C com 5% de CO2. A progesterona inibe a ação da enzima 5α-redutase, que
tal como a aromatase, tem a androstenediona como substrato. Após uma hora, a reação
enzimática foi terminada por adição de 300 µl de ácido tricloroacético (20%).
Posteriormente, recolheu-se a mistura para tubos de microcentrífuga com um pellet de
carvão/dextrano, onde permaneceu durante uma hora à temperatura ambiente, sendo de
seguida centrifugada a 15000xg durante 10 min. O sobrenadante foi retirado para outro
tubo contendo carvão/dextrano, de modo a garantir a remoção de todos os resíduos de
androstenediona e de outros componentes esteróides, e centrifugado. Após
centrifugação, retiraram-se 200 µl de sobrenadante para tubos de cintilação, contendo 3
ml de cocktail de cintilação. A leitura foi realizada num contador de cintilação (LS 6500,
Beckman Instruments, CA, U.S.A.).
Para determinação do conteúdo proteico, as células foram incubadas com 500 µl de
NaOH durante a noite, com agitação e à temperatura ambiente. A concentração de
proteínas foi determinada pelo método de Bradford. Neste ensaio, o formestano (5 µM),
um inibidor esteróide da aromatase, foi usado como controlo positivo. Todos os ensaios
foram realizados em triplicado e os resultados representam, pelo menos, três
experiências realizadas independentemente.
2.5. Estudos morfológicos
As alterações morfológicas foram analisadas por microscopia de contraste de fase e
pelas colorações de Giemsa e Hoechst. Para se verificar a presença de vesiculas ácidas,
procedeu-se à coloração com laranja de acridina. Para a realização destes ensaios, as
células foram cultivadas durante 48, 72 e 144 horas e com diferentes concentrações dos
compostos (5 a 15 µM), em placas de 24 poços, sendo a densidade celular de 1,0 x 106
células/ml.

Capítulo II: Materiais e Métodos
50
2.5.1. Coloração de Giemsa
A coloração de Giemsa permite-nos observar alterações morfológicas a nível do núcleo e
citoplasma. Após o tratamento e lavagem com PBS, as células foram fixadas com
metanol durante 30 minutos à temperatura ambiente, sendo posteriormente coradas com
corante de Giemsa, diluído em PBS (1:10), durante 30 minutos. Após lavagem com água
e secagem, as lâminas foram montadas em DPX. As células foram observadas ao
microscópio Eclipse E400, Nikon, equipado com o software de análise de imagem
LeicaQwin.
2.5.2. Coloração de Hoechst
Através da coloração de Hoechst é possível avaliar alterações na morfologia nuclear,
nomeadamente alterações a nível da condensação da cromatina. Após o período de
incubação, as células foram lavadas com PBS e fixadas com paraformaldeído (4% em
PBS) durante 30 min. Após fixação, as células foram incubadas com a solução de
Hoechst 33258 a 0,5 mg/ml em PBS durante 30 min. Após incubação, as lâminas foram
lavadas com PBS, e montadas em meio de montagem Vectashield sendo imediatamente
observadas ao microscópio de fluorescência (Eclipse E400, Nikon, Japan), equipado com
um filtro de excitação com o máximo de transmissão aos 360/400 nm, e processadas
pelo software de imagem Nikon ACT-2U.
2.5.3. Coloração com laranja de acridina
A coloração com laranja de acridina permite analisar a existência de vesículas ácidas
(AVOs). Após os períodos de incubação, as células foram incubadas com o corante
laranja de acridina (0,1 µg/ml) durante 30 minutos. De seguida, o corante foi removido, e
as lâminas montadas em PBS, sendo imediatamente observadas ao microscópio de
fluorescência (Eclipse E400, Nikon, Japan), equipado com um filtro de excitação com o
máximo de transmissão aos 490 nm. A presença de AVOs foi observada através da
fluorescência amarela/laranja/vermelha emitida.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
51
2.6. Viabilidade e proliferação celular
A viabilidade celular foi avaliada pela determinação da atividade mitocondrial recorrendo
ao uso de um sal de tetrazólio, o 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólio (MTT) que é
convertido a um sal de formazano, e pela libertação da enzima latacto desidrogenase
(LDH). As células MCF-7aro e LTEDaro foram cultivadas em placas de 96 poços, sendo
aplicada uma densidade celular de 2,5 x 104 células/ml para o período de incubação de
24, 48 e 72 horas e de 1 x 104 células/ml para o período de incubação correspondente às
144 horas. Posteriormente, as células foram incubadas com diferentes concentrações
dos compostos C32, C33 e 17-βHE (0,01 – 15 µM) durante 24, 48, 72 e 144h, com ou sem
3-metiladenina (3-MA, 1 mM), um inibidor da autofagia. Após a adição do MTT (0,5
mg/ml), a quantidade de azul de formazano formada foi analisada por espectrofotometria
a 540 nm. Por sua vez, a atividade da enzima lactato desidrogenase (LDH) libertada foi
doseada pelo Kit de LDH (Non-Radioactive Cytotoxicity Assay, Promega Corporation) de
acordo com o protocolo fornecido.
Para avaliar a proliferação celular, após o tratamento com cada um dos metabolitos,
realizou-se o ensaio de incorporação de [H3]-timidina. Uma vez que a timidina se
incorpora no DNA durante o processo de replicação, a quantidade de [H]3–timidina
incorporada é um indicador da síntese de DNA e consequentemente de proliferação
celular. Para a realização deste ensaio, a [H3]-timidina (0,5 μCi) foi adicionada a cada
poço, 8 horas antes do final do período de incubação correspondente a cada tratamento.
Após este período, as células foram sujeitas a um ciclo de congelação/descongelação e o
conteúdo celular foi recolhido recorrendo ao cell harvester (Skatron Instruments,
Noruega). Os filtros contendo o DNA foram colocados em tubos de cintilação, aos quais
foi adicionado 1 ml de cocktail de cintilação. A [H3]-timidina incorporada foi determinada
num contador de cintilação (LS 6500, Beckman Instruments, CA, U.S.A.).
2.7. Ciclo celular
As células foram cultivadas em placas de 6 poços, com densidade celular de 1,0 x 106
células/ml. Após 72h de incubação com os diferentes inibidores (5 a 15 µM), o meio foi
retirado, as células não aderentes recolhidas, e procedeu-se à tripsinização.
Posteriormente, as células foram centrifugadas (1200xg, 5 mim, 4˚C) e lavadas com PBS.
Após nova centrifugação, as células foram fixadas com etanol a 70% diluído em PBS
(1:10), sendo mantidas a 4ºC por um período de 2 horas. Após fixação, as células foram,
de novo, centrifugadas e o pellet obtido foi lavado com PBS. As células foram re-
suspendidas em 0,5 ml de PBS contendo 5 μg/ml de iodeto de propídio (PI), 0,1% triton

Capítulo II: Materiais e Métodos
52
X- 100 e 200 μg/ml DNAse-free Rnase A, e mantidas 30 min à temperatura ambiente.
Esta solução permite a degradação do RNA presente nas amostras, a permeabilização
das membranas através do Triton X-100 e a marcação das células com PI, garantindo
deste modo que a análise se refere apenas ao conteúdo de DNA. Esta análise é efetuada
por citometria de fluxo e baseada na aquisição de 10000 células num citómetro, Becton
Dickinson FACSCalibur (San Jose, CA, U.S.A.) equipado com o software CELLQuest
Pró. Os três canais fluorescentes (FL-1, FL-2 e FL-3) foram mantidos na escala linear. Os
efeitos dos compostos foram indicados pela percentagem de células nas fases G1, S e
G2/M do ciclo celular. Após a aquisição, a análise dos resultados foi realizada usando o
programa CELLQuest Pró.
2.8. Análise estatística
O tratamento de dados foi realizado através do programa GraphPad Prism 4 (GraphPad
Software Inc., San Diego, CA, USA). Os resultados são apresentados como Média ± Erro
Padrão da Media (SEM). A diferença significativa entre as médias dos resultados foi
calculada usando os testes t-student e One e Two-Way Analysis of Variance (ANOVA)
com o teste de multicomparação de Bonferroni. As diferenças foram consideradas
significativas para p ≤ 0,05.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
53
Capítulo III: Resultados

Capítulo III: Resultados
54

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
55
3.1. Determinação do IC50 nas células MCF-7aro
A determinação do IC50 dos compostos C32, C33 e 17-βHE foi avaliada nas células MCF-
7aro recorrendo a um método radiométrico, no qual a água tritiada libertada da [1β-3H]
androstenediona, por ação da aromatase, foi usada como índice de formação de
estrogénio. O formestano (4-OHA), um potente inibidor da aromatase, com uma
percentagem de inibição na ordem dos 99%, foi utilizado como inibidor de referência.
Figura 19. Determinação do IC50 dos compostos C32, C33 e 17-βHE em células MFC-7aro. O gráfico
representa a inibição da atividade da aromatase pelos compostos em estudo. Para realização do ensaio, as
células foram incubadas com 50 nM de [1β-3H] androstenediona (1 µCi), com os compostos (0,1 a 25 µM) e
500 nM de progesterona. O formestano a 5 µM foi utilizado como composto de referência. Os ensaios foram
realizados em triplicado e os resultados representam pelo menos três experiências independentes. Os
valores representam a média ± SEM.
Os resultados obtidos indicam que o composto C32 é o composto mais potente com um
IC50 de 0,70 µM. Para o composto C33 e para o 17-βHE, o IC50 é de 0,75 e 4,20 µM,
respetivamente (figura 19). O valor do IC50 do exemestano nas células MCF7-aro é de
0,90 µM.
Capítulo III: Resultados
56
3.2. Estudos morfológicos
De forma a avaliar as possíveis alterações morfológicas induzidas pelos compostos C32,
C33 e 17-βHE em células MFC-7aro, estas foram cultivadas na presença de testosterona a
1 nM, com ou sem os compostos, durante 48, 72 e 144 horas. As células foram
analisadas por microscopia de contraste de fase e coradas pelas colorações de Giemsa e
Hoechst. As células tratadas apenas com testosterona foram consideradas como grupo
controlo (figura 20), não se verificando alterações durante os vários períodos de
incubação.
Figura 20. Células MCF-7aro tratadas com testosterona a 1 nM, examinadas por microscopia de contraste de
fase (A) coloração de Giemsa (B) e coloração de Hoechst (C), após 72 horas de cultura. Ampliação original:
200x.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
57
Após as 48 horas de incubação com os compostos não se verificaram alterações
morfológicas visíveis pela microscopia de contraste de fase, coloração de Giemsa e
Hoechst.
Ao fim de 72 horas de tratamento, com o composto C32, verifica-se através da coloração
de Hoechst, a presença de condensação de cromatina para a concentração mais elevada
(15 µM) (figura 21).
Figura 21. Efeito do composto C32 (10 e 15 µM) na morfologia celular, por microscopia de contraste de fase
(A e B), coloração de Giemsa (C e D) e coloração de Hoechst (E e F). As células MCF-7aro foram cultivadas
na presença do composto C32, durante 72 horas, com 10 µM (A, C e E) e 15 µM (B, D e F). As células
tratadas apresentam condensação de cromatina observada pela coloração de Hoechst ( ). Ampliação
original: 200x.
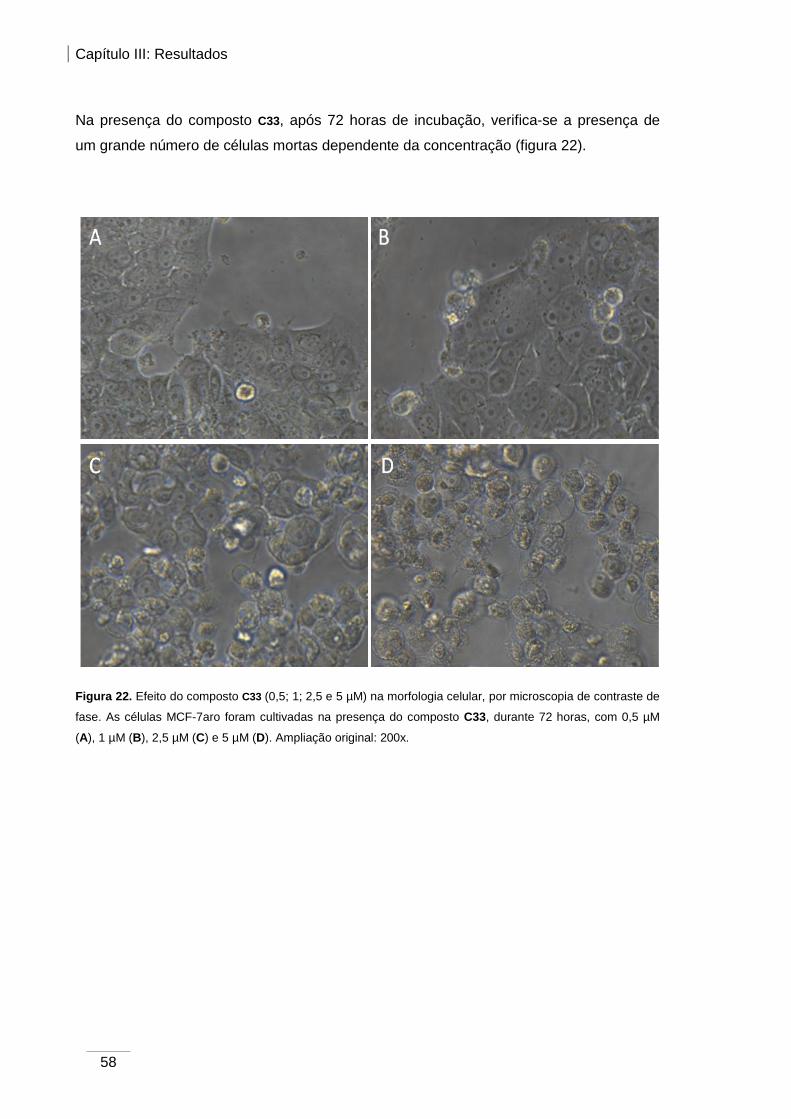
Capítulo III: Resultados
58
Na presença do composto C33, após 72 horas de incubação, verifica-se a presença de
um grande número de células mortas dependente da concentração (figura 22).
Figura 22. Efeito do composto C33 (0,5; 1; 2,5 e 5 µM) na morfologia celular, por microscopia de contraste de
fase. As células MCF-7aro foram cultivadas na presença do composto C33, durante 72 horas, com 0,5 µM
(A), 1 µM (B), 2,5 µM (C) e 5 µM (D). Ampliação original: 200x.
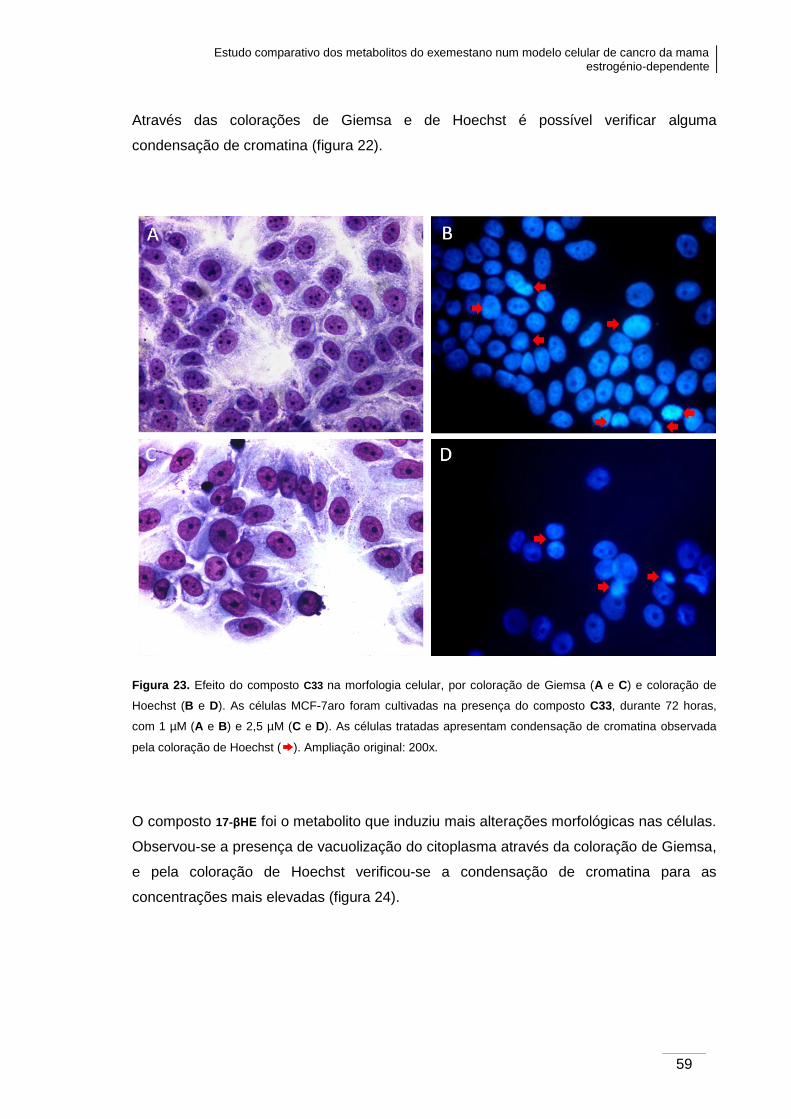
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
59
Através das colorações de Giemsa e de Hoechst é possível verificar alguma
condensação de cromatina (figura 22).
Figura 23. Efeito do composto C33 na morfologia celular, por coloração de Giemsa (A e C) e coloração de
Hoechst (B e D). As células MCF-7aro foram cultivadas na presença do composto C33, durante 72 horas,
com 1 µM (A e B) e 2,5 µM (C e D). As células tratadas apresentam condensação de cromatina observada
pela coloração de Hoechst ( ). Ampliação original: 200x.
O composto 17-βHE foi o metabolito que induziu mais alterações morfológicas nas células.
Observou-se a presença de vacuolização do citoplasma através da coloração de Giemsa,
e pela coloração de Hoechst verificou-se a condensação de cromatina para as
concentrações mais elevadas (figura 24).
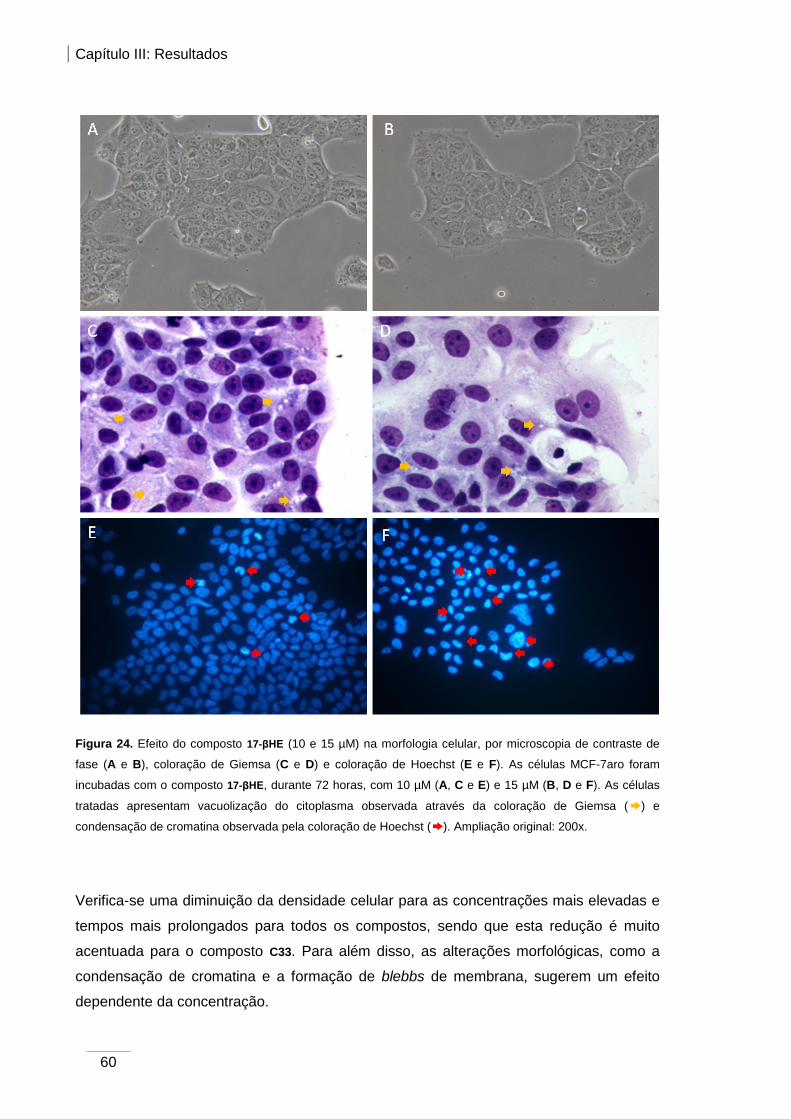
Capítulo III: Resultados
60
Figura 24. Efeito do composto 17-βHE (10 e 15 µM) na morfologia celular, por microscopia de contraste de
fase (A e B), coloração de Giemsa (C e D) e coloração de Hoechst (E e F). As células MCF-7aro foram
incubadas com o composto 17-βHE, durante 72 horas, com 10 µM (A, C e E) e 15 µM (B, D e F). As células
tratadas apresentam vacuolização do citoplasma observada através da coloração de Giemsa ( ) e
condensação de cromatina observada pela coloração de Hoechst ( ). Ampliação original: 200x.
Verifica-se uma diminuição da densidade celular para as concentrações mais elevadas e
tempos mais prolongados para todos os compostos, sendo que esta redução é muito
acentuada para o composto C33. Para além disso, as alterações morfológicas, como a
condensação de cromatina e a formação de blebbs de membrana, sugerem um efeito
dependente da concentração.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
61
3.3. Análise da viabilidade e proliferação celular nas células MCF-7aro
O efeito dos compostos na viabilidade da linha celular MCF-7aro foi determinado pela
conversão de um sal de tetrazólio a sal de formazano, pela mitocôndria (ensaio de MTT).
Os resultados demonstram que há uma redução da viabilidade celular dependente da
concentração, sendo mais acentuada para as maiores concentrações dos compostos em
estudo (figura 25). Após 48 horas de incubação, para a concentração de 1 µM, verifica-se
que os compostos C32, C33 e 17-βHE conduzem a uma redução significativa da viabilidade
celular para 84%, 60% e 91%, enquanto que a 15 µM se verifica uma diminuição para
62%, 14% e 54%, respetivamente. Às 72 horas, os compostos C32 e 17-βHE, para a
concentração de 1 µM, induzem uma redução da viabilidade celular de 15% e 28%, e a
15 µM de 59% e 49%, respetivamente. Por sua vez, o composto C33, a 1 µM, conduz a
um decréscimo na ordem dos 50%, enquanto que para as restantes concentrações (2,5 a
15 µM), a diminuição da viabilidade é superior a 85%. Após as 144 horas de incubação
verifica-se a diminuição da viabilidade celular induzida pelos compostos C32, C33 e 17-βHE,
a 1 µM, para 84%, 20% e 85%, respetivamente. No entanto, a 15 µM, essa redução é
mais acentuada (48% de viabilidade para o C32, 11% para o C33 e 35% para o 17-βHE).
O composto C33 é o mais eficaz, verificando-se uma maior diminuição da viabilidade
celular nos diferentes tempos de incubação comparativamente aos outros compostos em
estudo. Verifica-se, ainda, que nos períodos de incubação de 72 e 144 horas, as
concentrações mais elevadas do composto C33 (2,5 a 15 µM) induzem um decréscimo
acentuado da viabilidade celular, não se verificando diferenças significativas entre as
várias concentrações. Dada a redução acentuada de viabilidade celular induzida pelo C33
nas concentrações testadas (1 a 15 µM), foi também avaliado o seu efeito a
concentrações mais baixas, a 0,1 e 0,5 µM. Deste modo, verificou-se um decréscimo da
viabilidade celular aproximadamente 9 e 20% (48 horas), 10 e 14% (72 horas) e de 28 e
48% (144 horas), respetivamente.
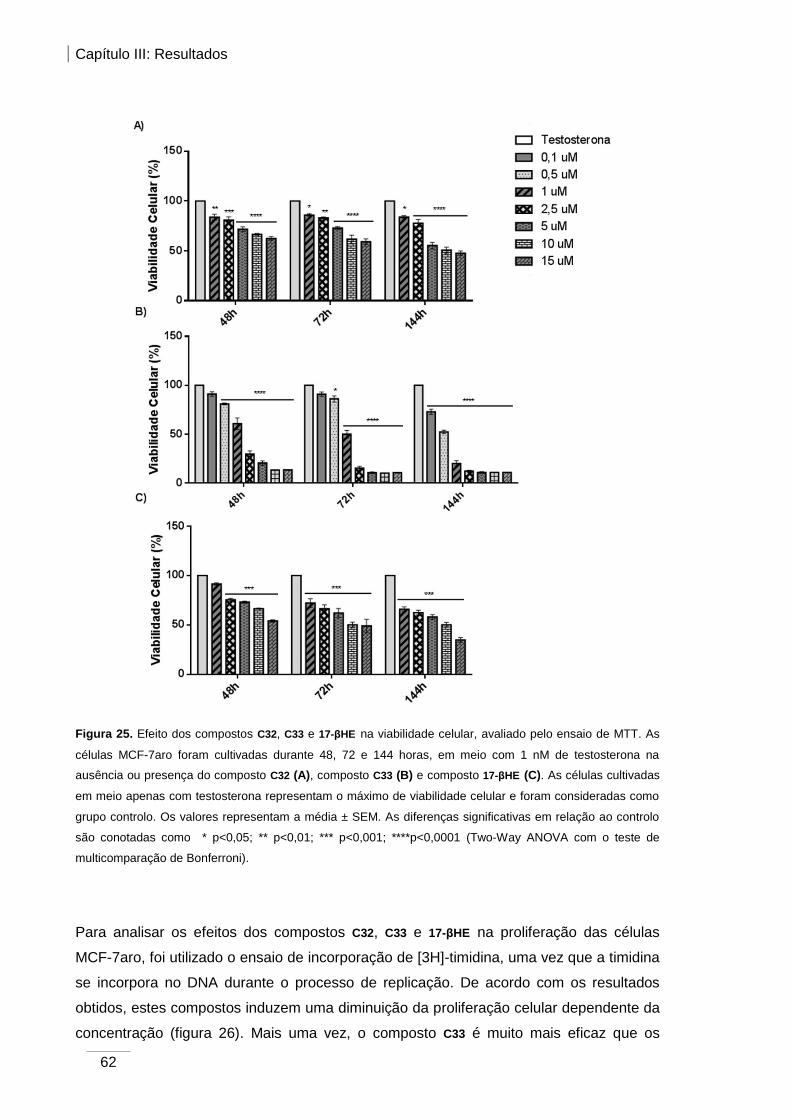
Capítulo III: Resultados
62
Figura 25. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular, avaliado pelo ensaio de MTT. As
células MCF-7aro foram cultivadas durante 48, 72 e 144 horas, em meio com 1 nM de testosterona na
ausência ou presença do composto C32 (A), composto C33 (B) e composto 17-βHE (C). As células cultivadas
em meio apenas com testosterona representam o máximo de viabilidade celular e foram consideradas como
grupo controlo. Os valores representam a média ± SEM. As diferenças significativas em relação ao controlo
são conotadas como * p<0,05; ** p<0,01; *** p<0,001; ****p<0,0001 (Two-Way ANOVA com o teste de
multicomparação de Bonferroni).
Para analisar os efeitos dos compostos C32, C33 e 17-βHE na proliferação das células
MCF-7aro, foi utilizado o ensaio de incorporação de [3H]-timidina, uma vez que a timidina
se incorpora no DNA durante o processo de replicação. De acordo com os resultados
obtidos, estes compostos induzem uma diminuição da proliferação celular dependente da
concentração (figura 26). Mais uma vez, o composto C33 é muito mais eficaz que os

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
63
compostos C32 e 17-βHE, causando uma redução acentuada da síntese de DNA em todas
as concentrações testadas (após 48 horas de incubação verifica-se uma diminuição da
proliferação de 61%, 77%, 93% e 99% para as concentrações de 1; 2,5; 5 e 10 µM,
respetivamente, e às 72 horas de 78%, 81%, 98% e 99%). Às 48 horas, para a
concentração de 1 µM, verifica-se uma diminuição da síntese de DNA para valores de
80% e 71% para os compostos C32 e 17-βHE, respetivamente, enquanto que, para a
concentração de 15 µM, se observa uma redução para valores de 43% e 33%. Às 72
horas, o comportamento é semelhante, induzindo uma diminuição da síntese de DNA
para 64% e 65%, a 1 µM, e de 30% e 27%, a 15 µM, para os compostos C32 e 17-βHE,
respetivamente.
Figura 26. Efeito dos compostos C32, C33 e 17-βHE na síntese de DNA. As células MCF-7aro foram
cultivadas durante 48 e 72h horas, em meio com 1 nM de testosterona na ausência ou presença do composto
C32 (A), composto C33 (B) e do composto 17-βHE (C). As células cultivadas em meio apenas com
testosterona representam o máximo de proliferação celular e foram consideradas como grupo controlo. Os
compostos induzem um decréscimo na proliferação celular dose-dependente. Os valores representam a
média ± SEM. As diferenças significativas em relação ao controlo são conotadas como *** p<0,001 (Two-Way
ANOVA com o teste de multicomparação de Bonferroni).
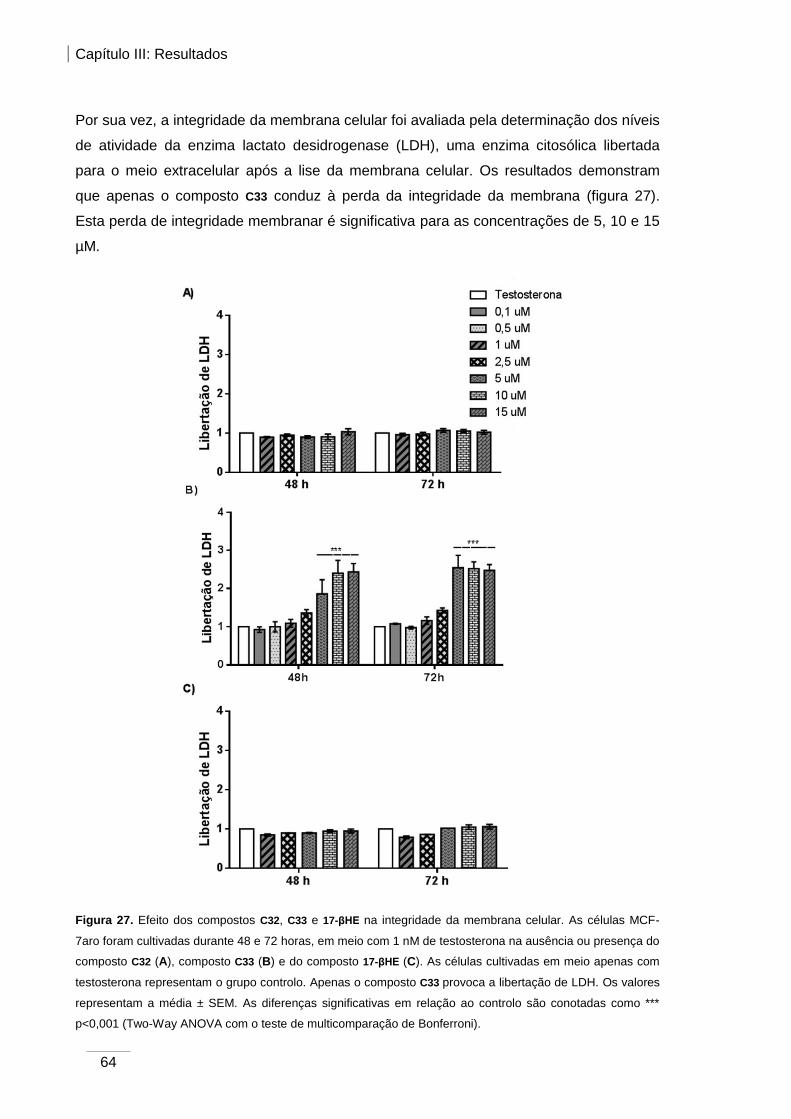
Capítulo III: Resultados
64
Por sua vez, a integridade da membrana celular foi avaliada pela determinação dos níveis
de atividade da enzima lactato desidrogenase (LDH), uma enzima citosólica libertada
para o meio extracelular após a lise da membrana celular. Os resultados demonstram
que apenas o composto C33 conduz à perda da integridade da membrana (figura 27).
Esta perda de integridade membranar é significativa para as concentrações de 5, 10 e 15
µM.
Figura 27. Efeito dos compostos C32, C33 e 17-βHE na integridade da membrana celular. As células MCF-
7aro foram cultivadas durante 48 e 72 horas, em meio com 1 nM de testosterona na ausência ou presença do
composto C32 (A), composto C33 (B) e do composto 17-βHE (C). As células cultivadas em meio apenas com
testosterona representam o grupo controlo. Apenas o composto C33 provoca a libertação de LDH. Os valores
representam a média ± SEM. As diferenças significativas em relação ao controlo são conotadas como ***
p<0,001 (Two-Way ANOVA com o teste de multicomparação de Bonferroni).
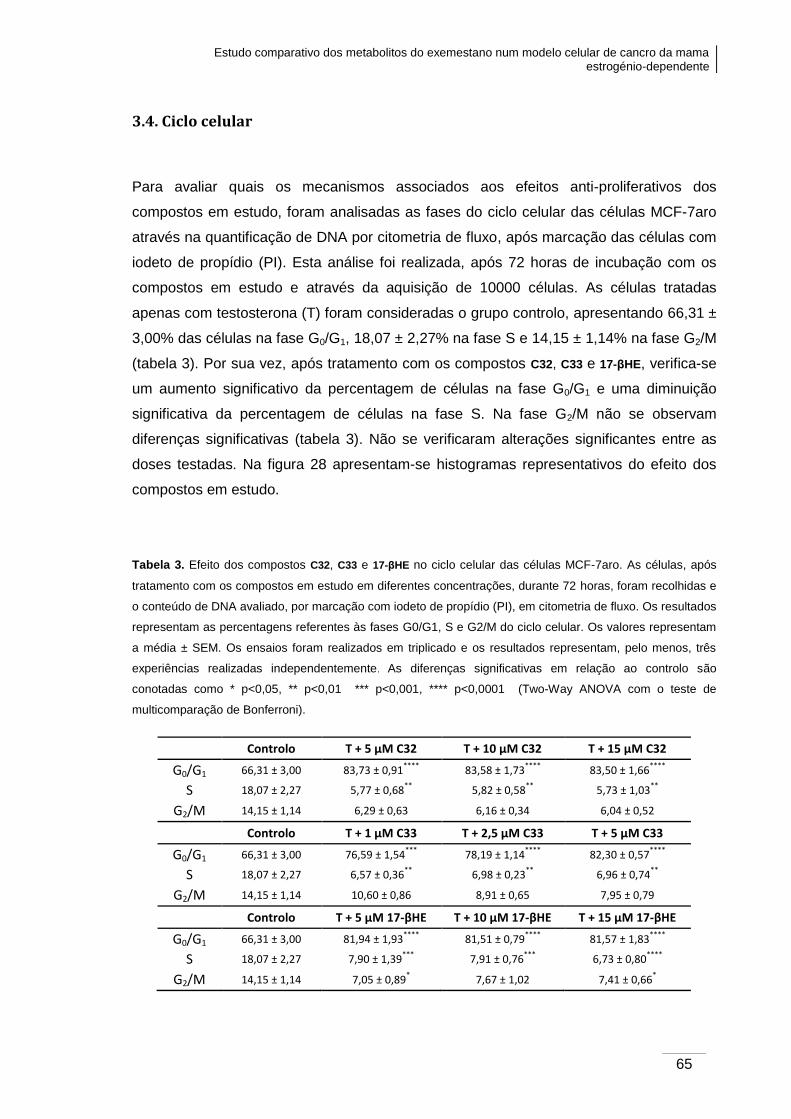
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
65
3.4. Ciclo celular
Para avaliar quais os mecanismos associados aos efeitos anti-proliferativos dos
compostos em estudo, foram analisadas as fases do ciclo celular das células MCF-7aro
através na quantificação de DNA por citometria de fluxo, após marcação das células com
iodeto de propídio (PI). Esta análise foi realizada, após 72 horas de incubação com os
compostos em estudo e através da aquisição de 10000 células. As células tratadas
apenas com testosterona (T) foram consideradas o grupo controlo, apresentando 66,31 ±
3,00% das células na fase G0/G1, 18,07 ± 2,27% na fase S e 14,15 ± 1,14% na fase G2/M
(tabela 3). Por sua vez, após tratamento com os compostos C32, C33 e 17-βHE, verifica-se
um aumento significativo da percentagem de células na fase G0/G1 e uma diminuição
significativa da percentagem de células na fase S. Na fase G2/M não se observam
diferenças significativas (tabela 3). Não se verificaram alterações significantes entre as
doses testadas. Na figura 28 apresentam-se histogramas representativos do efeito dos
compostos em estudo.
Tabela 3. Efeito dos compostos C32, C33 e 17-βHE no ciclo celular das células MCF-7aro. As células, após
tratamento com os compostos em estudo em diferentes concentrações, durante 72 horas, foram recolhidas e
o conteúdo de DNA avaliado, por marcação com iodeto de propídio (PI), em citometria de fluxo. Os resultados
representam as percentagens referentes às fases G0/G1, S e G2/M do ciclo celular. Os valores representam
a média ± SEM. Os ensaios foram realizados em triplicado e os resultados representam, pelo menos, três
experiências realizadas independentemente. As diferenças significativas em relação ao controlo são
conotadas como * p<0,05, ** p<0,01 *** p<0,001, **** p<0,0001 (Two-Way ANOVA com o teste de
multicomparação de Bonferroni).
Controlo T + 5 µM C32 T + 10 µM C32 T + 15 µM C32
G0/G1 66,31 ± 3,00 83,73 ± 0,91****
83,58 ± 1,73****
83,50 ± 1,66****
S 18,07 ± 2,27 5,77 ± 0,68**
5,82 ± 0,58**
5,73 ± 1,03**
G2/M 14,15 ± 1,14 6,29 ± 0,63 6,16 ± 0,34 6,04 ± 0,52
Controlo T + 1 µM C33 T + 2,5 µM C33 T + 5 µM C33
G0/G1 66,31 ± 3,00 76,59 ± 1,54***
78,19 ± 1,14****
82,30 ± 0,57****
S 18,07 ± 2,27 6,57 ± 0,36**
6,98 ± 0,23**
6,96 ± 0,74**
G2/M 14,15 ± 1,14 10,60 ± 0,86 8,91 ± 0,65 7,95 ± 0,79
Controlo T + 5 µM 17-βHE T + 10 µM 17-βHE T + 15 µM 17-βHE
G0/G1 66,31 ± 3,00 81,94 ± 1,93****
81,51 ± 0,79****
81,57 ± 1,83****
S 18,07 ± 2,27 7,90 ± 1,39***
7,91 ± 0,76***
6,73 ± 0,80****
G2/M 14,15 ± 1,14 7,05 ± 0,89* 7,67 ± 1,02 7,41 ± 0,66
*
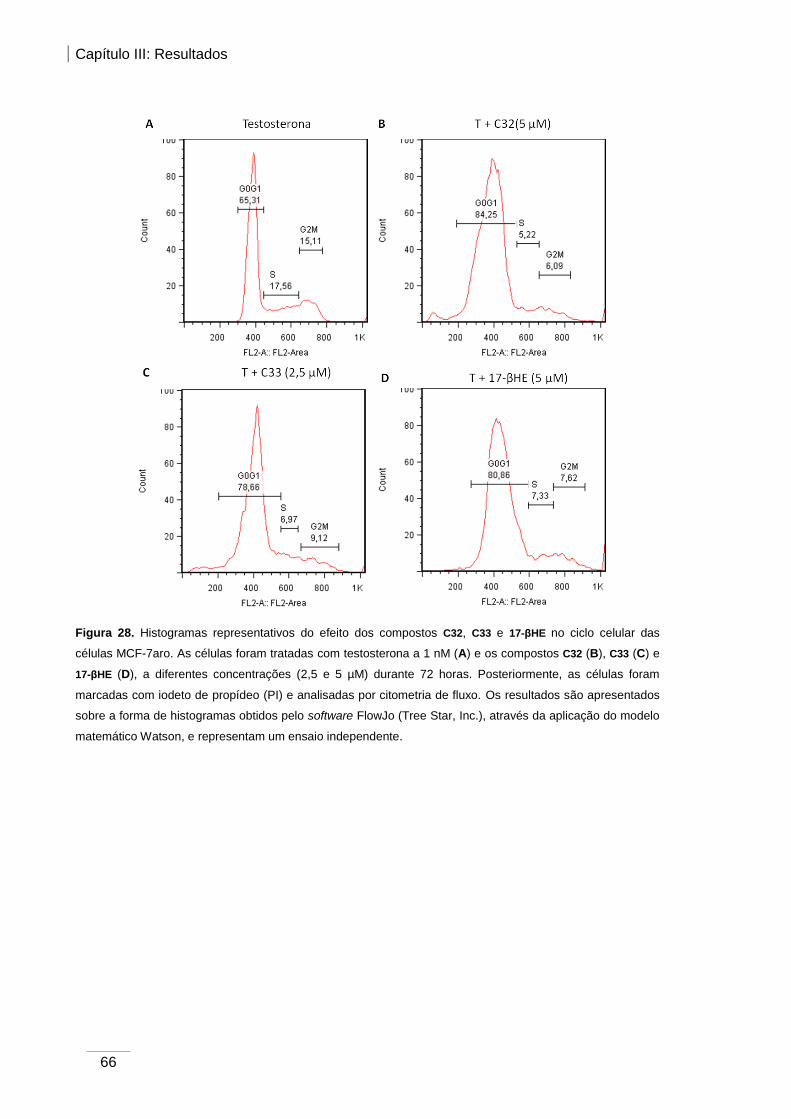
Capítulo III: Resultados
66
Figura 28. Histogramas representativos do efeito dos compostos C32, C33 e 17-βHE no ciclo celular das
células MCF-7aro. As células foram tratadas com testosterona a 1 nM (A) e os compostos C32 (B), C33 (C) e
17-βHE (D), a diferentes concentrações (2,5 e 5 µM) durante 72 horas. Posteriormente, as células foram
marcadas com iodeto de propídeo (PI) e analisadas por citometria de fluxo. Os resultados são apresentados
sobre a forma de histogramas obtidos pelo software FlowJo (Tree Star, Inc.), através da aplicação do modelo
matemático Watson, e representam um ensaio independente.
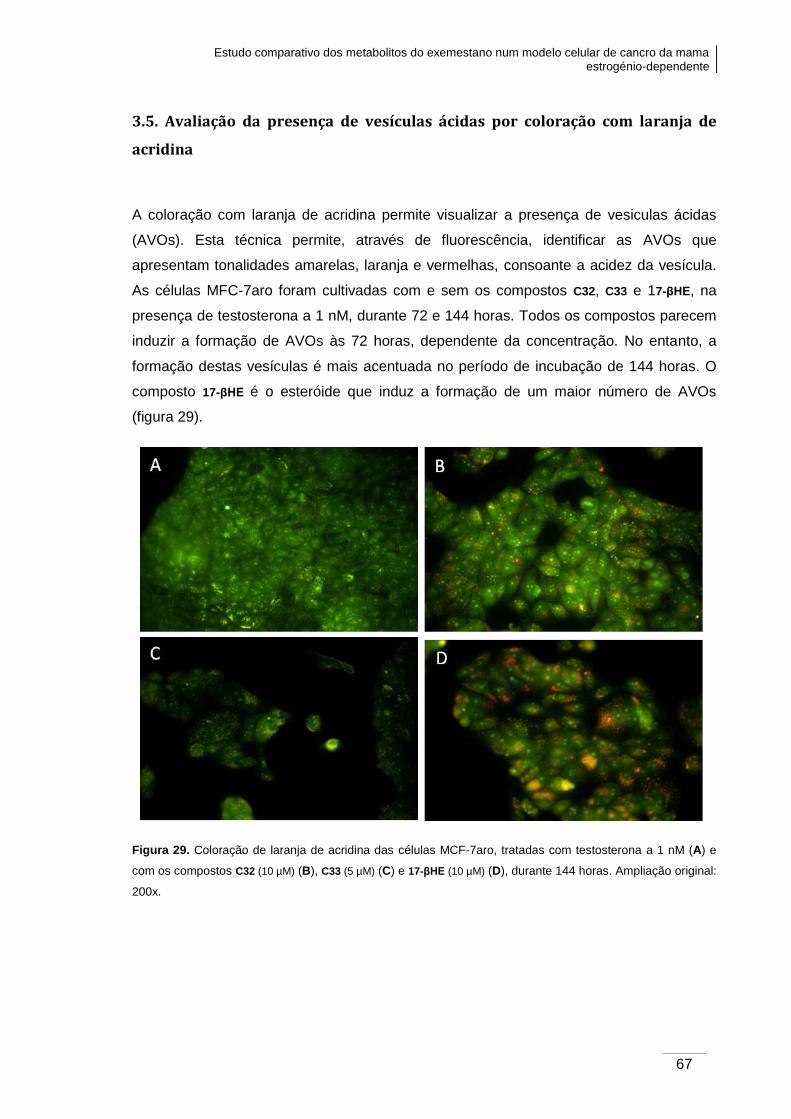
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
67
3.5. Avaliação da presença de vesículas ácidas por coloração com laranja de
acridina
A coloração com laranja de acridina permite visualizar a presença de vesiculas ácidas
(AVOs). Esta técnica permite, através de fluorescência, identificar as AVOs que
apresentam tonalidades amarelas, laranja e vermelhas, consoante a acidez da vesícula.
As células MFC-7aro foram cultivadas com e sem os compostos C32, C33 e 17-βHE, na
presença de testosterona a 1 nM, durante 72 e 144 horas. Todos os compostos parecem
induzir a formação de AVOs às 72 horas, dependente da concentração. No entanto, a
formação destas vesículas é mais acentuada no período de incubação de 144 horas. O
composto 17-βHE é o esteróide que induz a formação de um maior número de AVOs
(figura 29).
Figura 29. Coloração de laranja de acridina das células MCF-7aro, tratadas com testosterona a 1 nM (A) e
com os compostos C32 (10 µM) (B), C33 (5 µM) (C) e 17-βHE (10 µM) (D), durante 144 horas. Ampliação original:
200x.

Capítulo III: Resultados
68
3.6. Análise da viabilidade celular nas células LTEDaro
As células LTEDaro são obtidas através da privação a estrogénios (Long Term Estrogen
Deprivation), mimetizando, deste modo, as células do cancro da mama estrogénio-
dependente resistentes à terapia hormonal. Estudos demonstraram que esta linha celular
é resistente ao tratamento com o exemestano [77].
Assim, o efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células
LTEDaro foi avaliado, recorrendo, uma vez mais, ao ensaio de MTT. Os resultados
demonstraram que há uma redução da viabilidade celular, sendo essa redução mais
acentuada para as maiores concentrações dos compostos em estudo (figura 30). No
entanto, o composto C33, tal como acontece nas células MCF7-aro, parece ser mais
eficaz, comparativamente aos outros esteróides. Às 48 horas de incubação verifica-se um
decréscimo significativo da viabilidade celular induzido pelos compostos C32, C33 e 17-
βHE, a 1 µM, para 93%, 85% e 91%, e a 15 µM para 68%, 18% e 64%, respetivamente.
Às 72 horas, o composto C33 conduz à redução da viabilidade celular para 77%, na
concentração de 1 µM, enquanto que os compostos C32 e 17-βHE reduzem a viabilidade
celular para 89 e 85%, respetivamente. Na concentração de 15 µM, verifica-se a redução
da viabilidade para 65% para o C32, 10% para o C33 e 55% para o 17-βHE.Por fim, às 144
horas, para a concentração de 1 µM verifica-se uma diminuição da viabilidade para 83%,
41% e 69% induzida, respetivamente, pelos compostos C32, C33 e 17-βHE, enquanto que a
15 µM, essa redução é mais acentuada (50% para o C32, 6% para o C33 e 36% para o 17-
βHE).
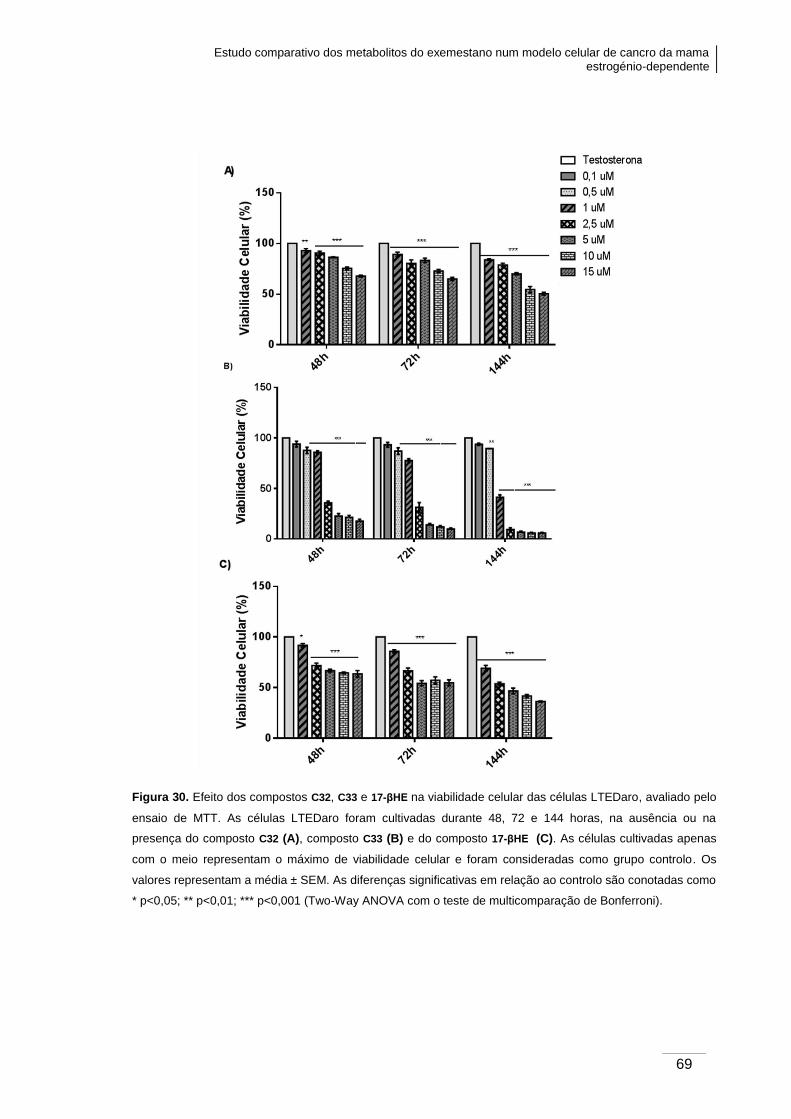
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
69
Figura 30. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células LTEDaro, avaliado pelo
ensaio de MTT. As células LTEDaro foram cultivadas durante 48, 72 e 144 horas, na ausência ou na
presença do composto C32 (A), composto C33 (B) e do composto 17-βHE (C). As células cultivadas apenas
com o meio representam o máximo de viabilidade celular e foram consideradas como grupo controlo. Os
valores representam a média ± SEM. As diferenças significativas em relação ao controlo são conotadas como
* p<0,05; ** p<0,01; *** p<0,001 (Two-Way ANOVA com o teste de multicomparação de Bonferroni).
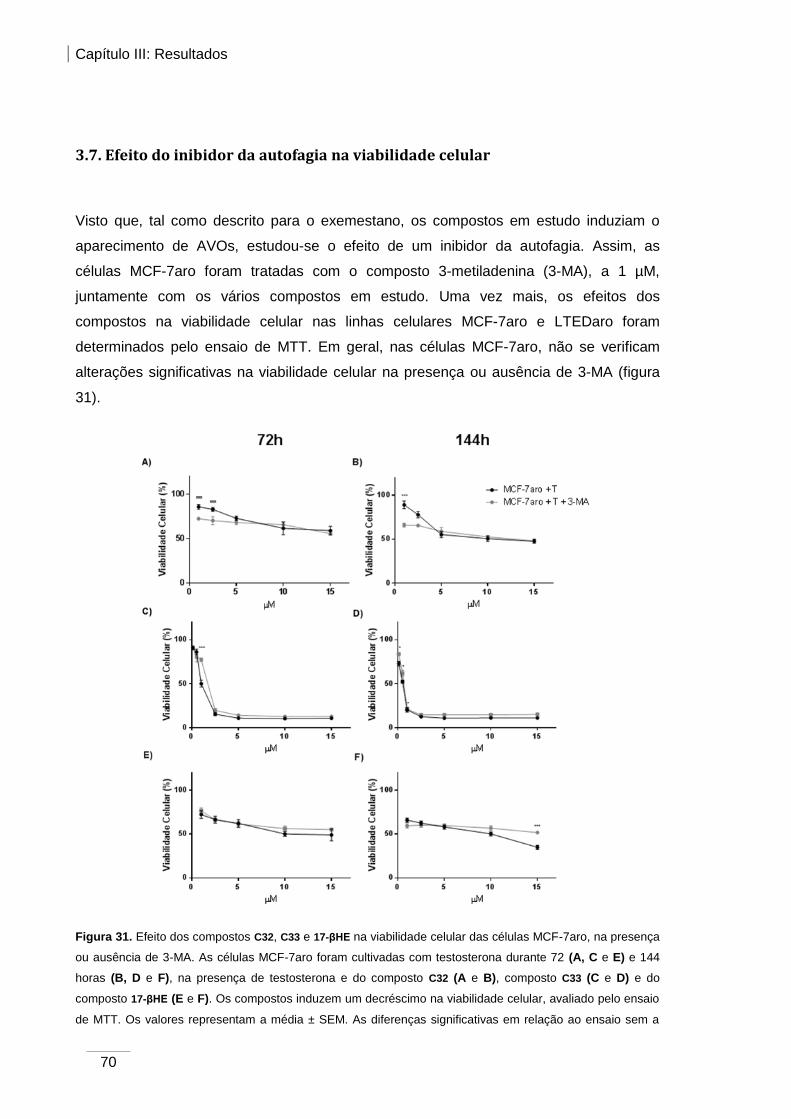
Capítulo III: Resultados
70
3.7. Efeito do inibidor da autofagia na viabilidade celular
Visto que, tal como descrito para o exemestano, os compostos em estudo induziam o
aparecimento de AVOs, estudou-se o efeito de um inibidor da autofagia. Assim, as
células MCF-7aro foram tratadas com o composto 3-metiladenina (3-MA), a 1 µM,
juntamente com os vários compostos em estudo. Uma vez mais, os efeitos dos
compostos na viabilidade celular nas linhas celulares MCF-7aro e LTEDaro foram
determinados pelo ensaio de MTT. Em geral, nas células MCF-7aro, não se verificam
alterações significativas na viabilidade celular na presença ou ausência de 3-MA (figura
31).
Figura 31. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células MCF-7aro, na presença
ou ausência de 3-MA. As células MCF-7aro foram cultivadas com testosterona durante 72 (A, C e E) e 144
horas (B, D e F), na presença de testosterona e do composto C32 (A e B), composto C33 (C e D) e do
composto 17-βHE (E e F). Os compostos induzem um decréscimo na viabilidade celular, avaliado pelo ensaio
de MTT. Os valores representam a média ± SEM. As diferenças significativas em relação ao ensaio sem a
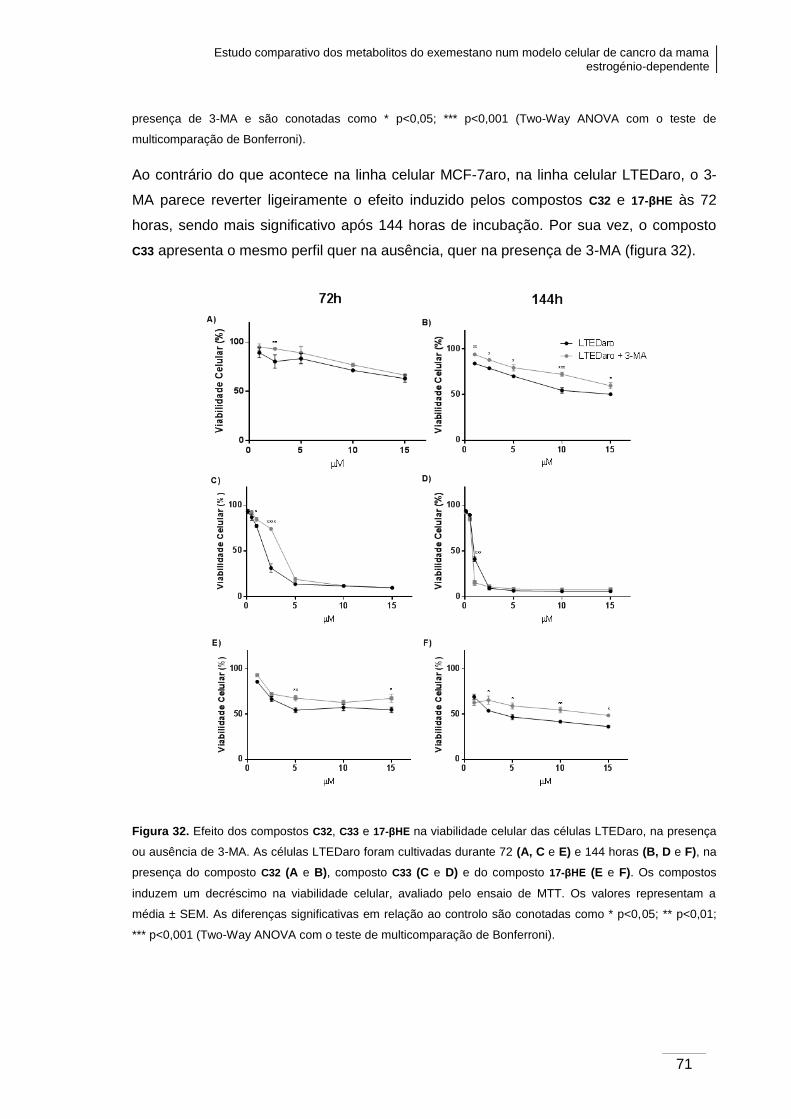
Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
71
presença de 3-MA e são conotadas como * p<0,05; *** p<0,001 (Two-Way ANOVA com o teste de
multicomparação de Bonferroni).
Ao contrário do que acontece na linha celular MCF-7aro, na linha celular LTEDaro, o 3-
MA parece reverter ligeiramente o efeito induzido pelos compostos C32 e 17-βHE às 72
horas, sendo mais significativo após 144 horas de incubação. Por sua vez, o composto
C33 apresenta o mesmo perfil quer na ausência, quer na presença de 3-MA (figura 32).
Figura 32. Efeito dos compostos C32, C33 e 17-βHE na viabilidade celular das células LTEDaro, na presença
ou ausência de 3-MA. As células LTEDaro foram cultivadas durante 72 (A, C e E) e 144 horas (B, D e F), na
presença do composto C32 (A e B), composto C33 (C e D) e do composto 17-βHE (E e F). Os compostos
induzem um decréscimo na viabilidade celular, avaliado pelo ensaio de MTT. Os valores representam a
média ± SEM. As diferenças significativas em relação ao controlo são conotadas como * p<0,05; ** p<0,01;
*** p<0,001 (Two-Way ANOVA com o teste de multicomparação de Bonferroni).

Capítulo III: Resultados
72

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
73
Capítulo IV: Discussão e Conclusões

Capítulo IV: Discussão e Conclusões
74

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
75
O cancro da mama é uma das patologias com maior taxa de mortalidade em todo o
mundo. Estima-se que cerca de 60% das mulheres pré-menopáusicas e 75% das
mulheres pós-menopáusicas possuam cancros da mama dependentes de estrogénios
(ER+) [1, 3, 88]. Os estrogénios desempenham um papel importante no desenvolvimento
e tratamento deste tipo de tumores. Assim, a supressão dos efeitos destas hormonas
esteróides é considerada uma potente arma terapêutica nos cancros da mama
dependentes de estrogénio. Enquanto que os SERMs e os SERDs limitam os efeitos dos
estrogénios através da modelação dos seus recetores, os AIs bloqueiam o ultimo passo
da conversão de androgénios a estrogénios, através da inibição da enzima aromatase.
O exemestano é o AI esteróide mais utilizado na terapêutica do cancro da mama ER+,
que atua inibindo irreversivelmente a enzima aromatase através dos seus intermediários
reativos [29]. Apesar dos estudos epidemiológicos e de segurança clínica, pouco se sabe
sobre os seus efeitos a nível celular, bem como as suas vias de metabolização e os seus
metabolitos. Assim, neste trabalho pretendeu-se elucidar os efeitos biológicos do 17-beta-
hidroxiexemestano (17-βHE), do 6α/β-spirooxiranandrosta-1,4-dieno-3,17-diona (C32), e do
1α,2α-epoxy-6-methylenandrost-4-ene-3,17-diona (C33), in vitro, nas linhas celulares
MF7-7aro e LTEDaro. Os compostos 17-βHE e C32 já foram descritos na literatura como
metabolitos do exemestano [69, 71, 75], enquanto que o C33 é considerado um potencial
metabolito [76].
De modo a verificar se os compostos em estudo seriam, tal como o exemestano,
inibidores da aromatase, determinou-se os respetivos IC50 nas células MCF-7aro. Os
resultados obtidos indicam que os compostos C32 e o C33 são os mais potentes, com um
IC50 de 0,70 e 0,75 µM, respetivamente, enquanto que o composto 17-βHE apresenta um
valor de IC50 bastante superior (4,2 µM). Comparativamente ao exemestano, que
apresenta um IC50 de 0,9 µM em células que expressam a enzima aromatase (MCF-
7aro), verifica-se que os metabolitos C32 e C33 são inibidores mais potentes.
A nível celular, os compostos induziam a condensação de cromatina e formação de
blebbs na membrana, características morfológicas associadas à morte celular
programada tipo I, a apoptose. No entanto, o composto 17-βHE parece ser mais eficaz a
induzir a condensação de cromatina. Para além destas alterações, verificou-se que os
compostos induziam a vacuolização do citoplasma das células MCF-7aro, nas
concentrações e tempos de incubação mais elevados, sendo estas alterações mais
frequentes com o composto 17-βHE. A formação de vesiculas ácidas (AVOs), visível
através da coloração de Giemsa e laranja de acridina é uma característica do processo
de autofagia.

Capítulo IV: Discussão e Conclusões
76
Estas alterações morfológicas induzidas pelos esteróides em estudo são concordantes
com as alterações induzidas pelo exemestano já descritas nas células MCF-7aro, em que
se verifica que este induz formação de blebbing, fragmentação e condensação de
cromatina e vacuolização do citoplasma sendo estas alterações mais visíveis após seis e
nove dias de tratamento, a 15 µM [77].
Dado que as alterações morfológicas sugerem a ocorrência de morte celular, foram
avaliados os efeitos dos compostos C32, C33 e 17-βHE, na viabilidade e proliferação das
células MCF-7aro. Esta linha celular expressa a aromatase em quantidade suficiente para
estimular a proliferação através da conversão da testosterona a estradiol. A testosterona,
por sua vez, estimula o crescimento e proliferação das células MCF-7aro em
concentrações que atingem valores mínimos de 1 nM, e que se encontram dentro da
concentração fisiológica média para esta hormona. Verificou-se que os compostos em
estudo conduzem ao decréscimo da viabilidade e da proliferação de forma dose-
dependente, sendo, no entanto, o composto C33 o mais eficaz, e o único que conduz à
perda de integridade da membrana celular. A diminuição da viabilidade/proliferação
mediada pelos compostos C32, C33 e 17-βHE pode estar associada à retenção do ciclo ou
à indução de morte celular. Quando analisado o ciclo das células MCF-7aro, após
tratamento com os compostos, verifica-se a retenção do ciclo celular na fase G0/G1
bloqueando, assim, a transição da fase G1 para a fase S do ciclo ou prolongando a
permanência das células na fase G1. Por outro lado, estes resultados são concordantes
com a diminuição da incorporação de timidina tritiada no DNA, sugerindo, um decréscimo
da proliferação celular. Dos inibidores em estudo, o C33, mais uma vez, é o composto
mais eficaz.
Comparativamente ao exemestano, verificaram-se diferenças na viabilidade e
proliferação das células MCF-7aro, após 72 horas de incubação [77]. O efeito na
viabilidade celular do exemestano a 5 µM apenas é significativo após nove dias de
incubação, enquanto que os metabolitos em estudo, induzem um decréscimo da
viabilidade a partir das 48 horas. Por outro lado, após o tratamento com os compostos
C32, C33 e 17-βHE verifica-se a diminuição significativa da proliferação celular após 48
horas de incubação, sugerindo que os metabolitos são mais eficientes que o exemestano
na redução da viabilidade/proliferação das células MCF-7aro. No entanto, tal como o
exemestano [77], os metabolitos conduzem à retenção do ciclo na fase G0/G1. O
exemestano e o 17-βHE possuem efeitos androgénicos [64], e, por outro lado, sabe-se que
os androgénios exercem predominantemente efeitos anti-proliferativos nas células do
cancro da mama. Assim, a retenção do ciclo poderá ser devido não só à atividade anti-
aromatásica, levando a uma redução dos estrogénios, mas também, caso do 17-βHE, aos

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
77
efeitos androgénicos, dado que estes ocorrem independentemente da presença de
estrogénios, estando associados a um aumento da proporção de células MCF-7 na fase
G0/G1 [27].
Apesar do desenvolvimento de terapias mais eficientes, a resistência endócrina
associada à terapia hormonal não pode ser ignorada, condicionando, assim, a eficácia do
tratamento. Torna-se portanto necessário continuar a elucidar os mecanismos associados
a esta condição. Recentemente, foi sugerido que a autofagia estaria envolvida na
resistência à terapia endócrina, em células tumorais da mama AI-resistentes [123]. A
autofagia é um processo conhecido de morte celular programada tipo II, caraterizado pela
presença de vesículas citoplasmáticas que englobam constituintes celulares, sendo estes
degradados pela via lisossomal [135]. No entanto, a autofagia pode, também,
desempenhar um papel importante na sobrevivência de células tumorais e na progressão
da patologia e desenvolvimento de metastases [136]. De facto, estudos conduzidos no
nosso laboratório revelaram que o exemestano induz a formação de AVOs, sugerindo o
envolvimento do processo autofágico [77]. Por outro lado, o exemestano não causa
alterações na viabilidade das células hormono-resistentes da mama, LTEDaro, que
mimetizam o ambiente tumoral resistente à terapia. No entanto, o exemestano, na
presença de um inibidor da autofagia, induz uma diminuição da viabilidade destas
células, indicando que a autofagia desempenha um papel importante nos mecanismos de
sobrevivência das células tumorais (dados não publicados).
Assim, estudou-se a eficácia dos metabolitos do exemestano na resistência endócrina
adquirida, avaliando-se os efeitos dos compostos C32, C33 e 17-βHE na viabilidade celular
das células LTEDaro. Contrariamente ao exemestano, que não tem qualquer efeito nas
nestas células [142, 143], os compostos C32, C33 e 17-βHE conduzem a um decréscimo da
viabilidade celular dependente da dose e do tempo, sendo, uma vez mais, o composto
C33 o mais eficaz. No entanto, após 144 horas de tratamento com os esteróides, a 15 µM,
a redução da viabilidade é superior a 50%.
Visto que, o tratamento com o exemestano induzia a ocorrência da autofagia, estando
esta relacionada com a menor frequência da apoptose e sobrevivência das células MCF-
7aro e LTEDaro [77, 144], analisou-se, ainda, o papel que esta poderia desempenhar na
redução da viabilidade celular induzida pelos esteróides C32, C33 e 17-βHE. Deste modo,
foram avaliados os efeitos dos compostos em estudo nas células MCF-7aro e LTEDaro,
na presença de um inibidor da autofagia, o 3-metiladenina (3-MA), que bloqueia a
formação de auto fagossomas, através da inibição da PI3K [145]. Os compostos C32, C33
e 17-βHE na presença do inibidor não induziam alterações na viabilidade das células MCF-

Capítulo IV: Discussão e Conclusões
78
7aro, indicando que a autofagia, ao contrário do que acontece com o exemestano, não
está relacionada com mecanismos de sobrevivência nas células sensíveis à terapia
hormonal. No entanto, nas células hormono-resistentes, os compostos C32 e 17-βHE
conduzem a uma menor perda de viabilidade na presença do 3-MA, ao contrário do
composto C33 que induz, tal como nas células MCF-7aro, o mesmo tipo de resposta, quer
na ausência, quer na presença do 3-MA. Estes resultados são concordantes com a
presença mais frequente de AVOs induzida pelos compostos C32 e 17-βHE, do que com o
C33, às 72 e 144 horas, sugerindo que, neste caso, a autofagia induzida pelos compostos
está relacionada com a morte e não com a sobrevivência, indicando que os metabolitos
induzem um decréscimo da viabilidade celular por mecanismos diferentes do
exemestano. Assim, a eficácia do tratamento com o exemestano poderá estar associada
não só a este, mas também aos seus metabolitos, que parecem atuar por mecanismos
diferentes. No entanto, mais estudos terão de ser realizados para elucidar os
mecanismos envolvidos.
Em suma, tal como o exemestano, os compostos C32, C33 e 17-βHE conduzem a uma
diminuição da viabilidade e proliferação das células MFC-7aro, facto este, que pode estar
relacionado com a retenção do ciclo celular da fase G0/G1. No entanto, tal como acontece
com o exemestano, outros mecanismos podem estar associados, tal como a formação de
espécies reativas de oxigénio (ROS) e ativação de caspases, podendo estes processos
estar envolvidos na apoptose, devido à ativação da via mitocondrial. Deste modo, os
efeitos anti-proliferativos dos compostos C32, C33 e 17-βHE nas células MCF-7aro podem
também estar relacionados com a morte celular por apoptose. De facto, verifica-se
condensação de cromatina e formação de blebbs de membrana após o tratamento com
os compostos em estudo. No entanto, mais estudos são necessários para confirmar o
envolvimento da apoptose nos efeitos anti-proliferativos/viabilidade induzidos por estes
esteróides. Nas concentrações elevadas, os compostos C32, C33 e 17-βHE, induzem o
aparecimento de AVOs, característico do processo autofágico. No entanto, ao contrário
do que acontece com o exemestano, este processo não pode ser considerado um
mecanismo de sobrevivência, dado que a inibição da autofagia não induz a diminuição da
viabilidade das células MCF-7aro mediada pelos compostos C32, C33 e 17-βHE. Por outro
lado, contrariamente ao exemestano, os compostos em estudo conduzem à diminuição
da viabilidade das células LTEDaro. Esta linha celular quando tratada com os metabolitos
C32 e 17-βHE na presença do inibidor da autofagia conduz a um aumento da viabilidade o
que parece indicar que a autofagia está envolvida nos processos de morte neste tipo de
células. No entanto, embora a autofagia pareça estar envolvida na redução da

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
79
viabilidade, mais estudos serão necessários para esclarecer o papel deste processo na
viabilidade das células LTEDaro.
O uso do exemestano no tratamento do cancro da mama estrogénio-dependente
pressupõe a elucidação das suas vias de metabolização, dos metabolitos envolvidos,
bem como das suas consequências a nível celular. Assim, pretende-se que este primeiro
estudo sobre os metabolitos do exemestano conduza ao esclarecimento dos seus efeitos
biológicos, tendo em vista, o desenvolvimento de novos esteróides inibidores da
aromatase, resultando, deste modo, na maior eficácia das terapias hormonais, bem como
no sucesso terapêutico dos cancros da mama hormono-resistentes.

Capítulo IV: Discussão e Conclusões
80

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
81
Capítulo V: Referências Bibliográficas

Capítulo V: Referências Bibliográficas
82

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
83
[1] J. Berry, "Are all aromatase inhibitors the same? A review of controlled clinical trials in breast cancer.," Clin Ther, vol. 27, pp. 1671-84, Nov 2005.
[2] J. P. Thakkar and D. G. Mehta, "A review of an unfavorable subset of breast cancer: estrogen receptor positive progesterone receptor negative.," Oncologist, vol. 16, pp. 276-85, 2011.
[3] L. F. Macedo, G. Sabnis, and A. Brodie, "Aromatase inhibitors and breast cancer.," Ann N Y Acad Sci, vol. 1155, pp. 162-73, Feb 2009.
[4] N. R. Bertos and M. Park, "Breast cancer - one term, many entities?," J Clin Invest, vol. 121, pp. 3789-96, Oct 2011.
[5] L. Orlando, P. Schiavone, P. Fedele, N. Calvani, A. Nacci, P. Rizzo, et al., "Molecularly targeted endocrine therapies for breast cancer.," Cancer Treat Rev, vol. 36 Suppl 3, pp. S67-71, Nov 2010.
[6] N. Platet, A. M. Cathiard, M. Gleizes, and M. Garcia, "Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion.," Crit Rev Oncol Hematol, vol. 51, pp. 55-67, Jul 2004.
[7] V. C. Jordan and A. M. Brodie, "Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer.," Steroids, vol. 72, pp. 7-25, Jan 2007.
[8] S. Nilsson and J. Gustafsson, "Estrogen receptors: therapies targeted to receptor subtypes.," Clin Pharmacol Ther, vol. 89, pp. 44-55, Jan 2011.
[9] L. R. Nelson and S. E. Bulun, "Estrogen production and action.," J Am Acad Dermatol, vol. 45, pp. S116-24, Sep 2001.
[10] J. A. Collins, J. M. Blake, and P. G. Crosignani, "Breast cancer risk with postmenopausal hormonal treatment.," Hum Reprod Update, vol. 11, pp. 545-60, 2005 Nov-Dec 2005.
[11] E. Simpson, M. Jones, S. Davis, and G. Rubin, "Do intracrine mechanisms regulate aromatase expression?," J Steroid Biochem Mol Biol, vol. 69, pp. 447-52, 1999 Apr-Jun 1999.
[12] G. G. Chen, Q. Zeng, and G. M. Tse, "Estrogen and its receptors in cancer.," Med Res Rev, vol. 28, pp. 954-74, Nov 2008.
[13] C. M. Klinge, "Estrogen receptor interaction with estrogen response elements.," Nucleic Acids Res, vol. 29, pp. 2905-19, Jul 2001.
[14] E. K. Shanle and W. Xu, "Selectively targeting estrogen receptors for cancer treatment.," Adv Drug Deliv Rev, vol. 62, pp. 1265-76, Oct 2010.
[15] S. T. Pearce and V. C. Jordan, "The biological role of estrogen receptors alpha and beta in cancer.," Crit Rev Oncol Hematol, vol. 50, pp. 3-22, Apr 2004.
[16] C. Zhao, K. Dahlman-Wright, and J. A. Gustafsson, "Estrogen receptor beta: an overview and update.," Nucl Recept Signal, vol. 6, p. e003, 2008.
[17] S. A. Fuqua, "The role of estrogen receptors in breast cancer metastasis.," J Mammary Gland Biol Neoplasia, vol. 6, pp. 407-17, Oct 2001.
[18] G. Arpino, C. De Angelis, M. Giuliano, A. Giordano, C. Falato, M. De Laurentiis, et al., "Molecular mechanism and clinical implications of endocrine therapy resistance in breast cancer.," Oncology, vol. 77 Suppl 1, pp. 23-37, 2009.
[19] C. Teyssier, M. Le Romancer, S. Sentis, S. Jalaguier, L. Corbo, and V. Cavaillès, "Protein arginine methylation in estrogen signaling and estrogen-related cancers.," Trends Endocrinol Metab, vol. 21, pp. 181-9, Mar 2010.
[20] C. Marquette and L. Nabell, "Chemotherapy-resistant metastatic breast cancer.," Curr Treat Options Oncol, vol. 13, pp. 263-75, Jun 2012.
[21] T. W. Miller, M. Pérez-Torres, A. Narasanna, M. Guix, O. Stål, G. Pérez-Tenorio, et al., "Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer.," Cancer Res, vol. 69, pp. 4192-201, May 2009.

Capítulo V: Referências Bibliográficas
84
[22] C. Villarreal-Garza, J. Cortes, F. Andre, and S. Verma, "mTOR inhibitors in the management of hormone receptor-positive breast cancer: the latest evidence and future directions.," Ann Oncol, May 2012.
[23] L. Björnström and M. Sjöberg, "Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes.," Mol Endocrinol, vol. 19, pp. 833-42, Apr 2005.
[24] G. Arpino, L. Wiechmann, C. K. Osborne, and R. Schiff, "Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance.," Endocr Rev, vol. 29, pp. 217-33, Apr 2008.
[25] E. R. Levin, "Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor.," Mol Endocrinol, vol. 17, pp. 309-17, Mar 2003.
[26] E. R. Simpson, "Aromatization of androgens in women: current concepts and findings.," Fertil Steril, vol. 77 Suppl 4, pp. S6-10, Apr 2002.
[27] T. Suzuki, Y. Miki, K. Takagi, H. Hirakawa, T. Moriya, N. Ohuchi, et al., "Androgens in human breast carcinoma.," Med Mol Morphol, vol. 43, pp. 75-81, Jun 2010.
[28] P. A. Thompson and C. Ambrosone, "Molecular epidemiology of genetic polymorphisms in estrogen metabolizing enzymes in human breast cancer," J Natl Cancer Inst Monogr, pp. 125-34, 2000.
[29] Y. Hong, B. Yu, M. Sherman, Y. C. Yuan, D. Zhou, and S. Chen, "Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase.," Mol Endocrinol, vol. 21, pp. 401-14, Feb 2007.
[30] J. R. Pasqualini, "Estrogen sulfotransferases in breast and endometrial cancers.," Ann N Y Acad Sci, vol. 1155, pp. 88-98, Feb 2009.
[31] H. Sasano, T. Suzuki, Y. Miki, and T. Moriya, "Intracrinology of estrogens and androgens in breast carcinoma.," J Steroid Biochem Mol Biol, vol. 108, pp. 181-5, Feb 2008.
[32] A. Subramanian, M. Salhab, and K. Mokbel, "Oestrogen producing enzymes and mammary carcinogenesis: a review.," Breast Cancer Res Treat, vol. 111, pp. 191-202, Sep 2008.
[33] R. Santen, E. Cavalieri, E. Rogan, J. Russo, J. Guttenplan, J. Ingle, et al., "Estrogen mediation of breast tumor formation involves estrogen receptor-dependent, as well as independent, genotoxic effects.," Ann N Y Acad Sci, vol. 1155, pp. 132-40, Feb 2009.
[34] U. Meinhardt and P. E. Mullis, "The aromatase cytochrome P-450 and its clinical impact.," Horm Res, vol. 57, pp. 145-52, 2002.
[35] I. Czajka-Oraniec and E. R. Simpson, "Aromatase research and its clinical significance.," Endokrynol Pol, vol. 61, pp. 126-34, 2010 Jan-Feb 2010.
[36] S. E. Bulun, D. Chen, M. Lu, H. Zhao, Y. Cheng, M. Demura, et al., "Aromatase excess in cancers of breast, endometrium and ovary.," J Steroid Biochem Mol Biol, vol. 106, pp. 81-96, 2007 Aug-Sep 2007.
[37] E. R. Simpson, M. Misso, K. N. Hewitt, R. A. Hill, W. C. Boon, M. E. Jones, et al., "Estrogen--the good, the bad, and the unexpected.," Endocr Rev, vol. 26, pp. 322-30, May 2005.
[38] Y. Hong, H. Li, Y. C. Yuan, and S. Chen, "Molecular characterization of aromatase.," Ann N Y Acad Sci, vol. 1155, pp. 112-20, Feb 2009.
[39] D. Chen, S. Reierstad, M. Lu, Z. Lin, H. Ishikawa, and S. E. Bulun, "Regulation of breast cancer-associated aromatase promoters.," Cancer Lett, vol. 273, pp. 15-27, Jan 2009.
[40] S. E. Bulun, Z. Lin, H. Zhao, M. Lu, S. Amin, S. Reierstad, et al., "Regulation of aromatase expression in breast cancer tissue.," Ann N Y Acad Sci, vol. 1155, pp. 121-31, Feb 2009.
[41] S. Chen, D. Zhou, T. Okubo, Y. C. Kao, and C. Yang, "Breast tumor aromatase: functional role and transcriptional regulation.," Endocr Relat Cancer, vol. 6, pp. 149-56, Jun 1999.
[42] D. Ghosh, J. Griswold, M. Erman, and W. Pangborn, "Structural basis for androgen specificity and oestrogen synthesis in human aromatase.," Nature, vol. 457, pp. 219-23, Jan 2009.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
85
[43] D. Ghosh, J. Griswold, M. Erman, and W. Pangborn, "X-ray structure of human aromatase reveals an androgen-specific active site.," J Steroid Biochem Mol Biol, vol. 118, pp. 197-202, Feb 2010.
[44] A. U. Buzdar, J. F. Robertson, W. Eiermann, and J. M. Nabholtz, "An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane.," Cancer, vol. 95, pp. 2006-16, Nov 2002.
[45] M. J. Higgins and V. Stearns, "Pharmacogenetics of endocrine therapy for breast cancer.," Annu Rev Med, vol. 62, pp. 281-93, Feb 2011.
[46] D. P. McDonnell and S. E. Wardell, "The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer.," Curr Opin Pharmacol, vol. 10, pp. 620-8, Dec 2010.
[47] J. R. Benson and V. Pitsinis, "Update on clinical role of tamoxifen.," Curr Opin Obstet Gynecol, vol. 15, pp. 13-23, Feb 2003.
[48] A. Valachis, D. Mauri, N. P. Polyzos, D. Mavroudis, V. Georgoulias, and G. Casazza, "Fulvestrant in the treatment of advanced breast cancer: a systematic review and meta-analysis of randomized controlled trials.," Crit Rev Oncol Hematol, vol. 73, pp. 220-7, Mar 2010.
[49] S. R. Johnston, "New strategies in estrogen receptor-positive breast cancer.," Clin Cancer Res, vol. 16, pp. 1979-87, Apr 2010.
[50] "Patient Information - Faslodex," ed. Cheshire, England: AstraZeneca, 2012. [51] S. Gonnelli and R. Petrioli, "Aromatase inhibitors, efficacy and metabolic risk in the
treatment of postmenopausal women with early breast cancer.," Clin Interv Aging, vol. 3, pp. 647-57, 2008.
[52] A. Thiantanawat, B. J. Long, and A. M. Brodie, "Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens.," Cancer Res, vol. 63, pp. 8037-50, Nov 2003.
[53] U. Dutta and K. Pant, "Aromatase inhibitors: past, present and future in breast cancer therapy.," Med Oncol, vol. 25, pp. 113-24, 2008.
[54] B. R. Bird and S. M. Swain, "Cardiac toxicity in breast cancer survivors: review of potential cardiac problems.," Clin Cancer Res, vol. 14, pp. 14-24, Jan 2008.
[55] J. A. Files, M. G. Ko, and S. Pruthi, "Managing aromatase inhibitors in breast cancer survivors: not just for oncologists.," Mayo Clin Proc, vol. 85, pp. 560-6; quiz 566, Jun 2010.
[56] P. Hadji, "Menopausal symptoms and adjuvant therapy-associated adverse events.," Endocr Relat Cancer, vol. 15, pp. 73-90, Mar 2008.
[57] J. Pfeilschifter, "Role of cytokines in postmenopausal bone loss.," Curr Osteoporos Rep, vol. 1, pp. 53-8, Sep 2003.
[58] X. Wang and S. Chen, "Aromatase destabilizer: novel action of exemestane, a food and drug administration-approved aromatase inhibitor.," Cancer Res, vol. 66, pp. 10281-6, Nov 2006.
[59] M. Kittaneh and S. Glück, "Exemestane in the adjuvant treatment of breast cancer in postmenopausal women.," Breast Cancer (Auckl), vol. 5, pp. 209-26, 2011.
[60] D. C. Johannessen, T. Engan, E. Di Salle, M. G. Zurlo, J. Paolini, G. Ornati, et al., "Endocrine and clinical effects of exemestane (PNU 155971), a novel steroidal aromatase inhibitor, in postmenopausal breast cancer patients: a phase I study.," Clin Cancer Res, vol. 3, pp. 1101-8, Jul 1997.
[61] "Aromasin® exemestane tablets [prescribing information]," ed. NY: Pfizer Inc., October 2008.
[62] W. R. Miller and J. M. Dixon, "Endocrine and clinical endpoints of exemestane as neoadjuvant therapy.," Cancer Control, vol. 9, pp. 9-15, 2002 Mar-Apr 2002.
[63] W. R. Miller, "Clinical, pathological, proliferative and molecular responses associated with neoadjuvant aromatase inhibitor treatment in breast cancer.," J Steroid Biochem Mol Biol, vol. 118, pp. 273-6, Feb 2010.

Capítulo V: Referências Bibliográficas
86
[64] S. Masri, K. Lui, S. Phung, J. Ye, D. Zhou, X. Wang, et al., "Characterization of the weak estrogen receptor alpha agonistic activity of exemestane," Breast Cancer Res Treat, vol. 116, pp. 461-70, Aug 2009.
[65] K. Takagi, Y. Miki, S. Nagasaki, H. Hirakawa, Y. Onodera, J. Akahira, et al., "Increased intratumoral androgens in human breast carcinoma following aromatase inhibitor exemestane treatment.," Endocr Relat Cancer, vol. 17, pp. 415-30, Jun 2010.
[66] M. Untch and C. Jackisch, "Exemestane in early breast cancer: a review.," Ther Clin Risk Manag, vol. 4, pp. 1295-304, Dec 2008.
[67] E. A. Ariazi, A. Leitão, T. I. Oprea, B. Chen, T. Louis, A. M. Bertucci, et al., "Exemestane's 17-hydroxylated metabolite exerts biological effects as an androgen.," Mol Cancer Ther, vol. 6, pp. 2817-27, Nov 2007.
[68] L. K. Kamdem, D. A. Flockhart, and Z. Desta, "In vitro cytochrome P450-mediated metabolism of exemestane.," Drug Metab Dispos, vol. 39, pp. 98-105, Jan 2011.
[69] G. e. A. Cavalcanti, B. C. Garrido, F. D. Leal, M. C. Padilha, X. de la Torre, and F. R. de Aquino Neto, "Detection of new urinary exemestane metabolites by gas chromatography coupled to mass spectrometry.," Steroids, vol. 76, pp. 1010-5, 2011 Sep-Oct 2011.
[70] G. e. A. Cavalcanti, B. C. Garrido, F. D. Leal, M. C. Padilha, X. de la Torre, H. M. Pereira, et al., "Analysis of exemestane and 17β-hydroxyexemestane in human urine by gas chromatography/mass spectrometry: development and validation of a method using MO-TMS derivatives.," Rapid Commun Mass Spectrom, vol. 24, pp. 3297-302, Nov 2010.
[71] F. Buzzetti, E. Di Salle, A. Longo, and G. Briatico, "Synthesis and aromatase inhibition by potential metabolites of exemestane (6-methylenandrosta-1,4-diene-3,17-dione)." Steroids, vol. 58, pp. 527-32, Nov 1993.
[72] D. Sun, G. Chen, R. W. Dellinger, A. K. Sharma, and P. Lazarus, "Characterization of 17-dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics.," Pharmacogenet Genomics, vol. 20, pp. 575-85, Oct 2010.
[73] P. E. Goss, "Breast cancer prevention--clinical trials strategies involving aromatase inhibitors.," J Steroid Biochem Mol Biol, vol. 86, pp. 487-93, Sep 2003.
[74] P. E. Lønning, E. Bajetta, R. Murray, M. Tubiana-Hulin, P. D. Eisenberg, E. Mickiewicz, et al., "Activity of exemestane in metastatic breast cancer after failure of nonsteroidal aromatase inhibitors: a phase II trial.," J Clin Oncol, vol. 18, pp. 2234-44, Jun 2000.
[75] G. de Albuquerque Cavalcanti, B. Carius Garrido, F. Dias Leal, M. Costa Padilha, M. Mazzarino, X. de la Torre, et al., "Detection of new exemestane metabolites by liquid chromatography interfaced to electrospray-tandem mass spectrometry.," J Steroid Biochem Mol Biol, vol. 127, pp. 248-54, Nov 2011.
[76] R. Suresh Kumar, M. Narasimha Naidu, K. Srinivasulu, K. Raja Sekhar, M. Veerender, and M. K. Srinivasu, "Development and validation of a stability indicating LC method for the assay and related substances determination of exemestane, an aromatase inhibitor," J Pharm Biomed Anal, vol. 50, pp. 746-52, Dec 2009.
[77] C. Amaral, M. Borges, S. Melo, E. T. da Silva, G. Correia-da-Silva, and N. Teixeira, "Apoptosis and Autophagy in Breast Cancer Cells following Exemestane Treatment," PLoS One, vol. 7, p. e42398, 2012.
[78] M. J. Abedin, D. Wang, M. A. McDonnell, U. Lehmann, and A. Kelekar, "Autophagy delays apoptotic death in breast cancer cells following DNA damage," Cell Death Differ, vol. 14, pp. 500-10, Mar 2007.
[79] W. L. Sun, J. Chen, Y. P. Wang, and H. Zheng, "Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development," Autophagy, vol. 7, pp. 1035-44, Sep 2011.
[80] S. Kanematsu, N. Uehara, H. Miki, K. Yoshizawa, A. Kawanaka, T. Yuri, et al., "Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells," Anticancer Res, vol. 30, pp. 3381-90, Sep 2010.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
87
[81] P. R. Hutson, R. R. Love, T. C. Havighurst, E. Rogers, and J. F. Cleary, "Effect of exemestane on tamoxifen pharmacokinetics in postmenopausal women treated for breast cancer.," Clin Cancer Res, vol. 11, pp. 8722-7, Dec 2005.
[82] R. C. Coombes, E. Hall, L. J. Gibson, R. Paridaens, J. Jassem, T. Delozier, et al., "A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer.," N Engl J Med, vol. 350, pp. 1081-92, Mar 2004.
[83] E. P. Mamounas, J. H. Jeong, D. L. Wickerham, R. E. Smith, P. A. Ganz, S. R. Land, et al., "Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial.," J Clin Oncol, vol. 26, pp. 1965-71, Apr 2008.
[84] R. J. Paridaens, L. Y. Dirix, L. V. Beex, M. Nooij, D. A. Cameron, T. Cufer, et al., "Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group.," J Clin Oncol, vol. 26, pp. 4883-90, Oct 2008.
[85] S. Jones, C. Vogel, A. Arkhipov, L. Fehrenbacher, P. Eisenberg, B. Cooper, et al., "Multicenter, phase II trial of exemestane as third-line hormonal therapy of postmenopausal women with metastatic breast cancer. Aromasin Study Group.," J Clin Oncol, vol. 17, pp. 3418-25, Nov 1999.
[86] H. B. Xu, Y. J. Liu, and L. Li, "Aromatase inhibitor versus tamoxifen in postmenopausal woman with advanced breast cancer: a literature-based meta-analysis.," Clin Breast Cancer, vol. 11, pp. 246-51, Aug 2011.
[87] A. Barnadas, M. Gil, S. González, I. Tusquets, M. Muñoz, A. Arcusa, et al., "Exemestane as primary treatment of oestrogen receptor-positive breast cancer in postmenopausal women: a phase II trial.," Br J Cancer, vol. 100, pp. 442-9, Feb 2009.
[88] S. Glück, "Exemestane as first-line therapy in postmenopausal women with recurrent or metastatic breast cancer.," Am J Clin Oncol, vol. 33, pp. 314-9, Jun 2010.
[89] C. J. van de Velde, D. Rea, C. Seynaeve, H. Putter, A. Hasenburg, J. M. Vannetzel, et al., "Adjuvant tamoxifen and exemestane in early breast cancer (TEAM): a randomised phase 3 trial.," Lancet, vol. 377, pp. 321-31, Jan 2011.
[90] A. U. Buzdar, "A summary of second-line randomized studies of aromatase inhibitors.," J Steroid Biochem Mol Biol, vol. 79, pp. 109-14, Dec 2001.
[91] P. Goss, "Anti-aromatase agents in the treatment and prevention of breast cancer.," Cancer Control, vol. 9, pp. 2-8, 2002 Mar-Apr 2002.
[92] J. Geisler and P. E. Lønning, "Aromatase inhibition: translation into a successful therapeutic approach.," Clin Cancer Res, vol. 11, pp. 2809-21, Apr 2005.
[93] W. R. Miller, T. J. Anderson, D. B. Evans, A. Krause, G. Hampton, and J. M. Dixon, "An integrated view of aromatase and its inhibition.," J Steroid Biochem Mol Biol, vol. 86, pp. 413-21, Sep 2003.
[94] M. Beresford, I. Tumur, J. Chakrabarti, J. Barden, N. Rao, and A. Makris, "A qualitative systematic review of the evidence base for non-cross-resistance between steroidal and non-steroidal aromatase inhibitors in metastatic breast cancer.," Clin Oncol (R Coll Radiol), vol. 23, pp. 209-15, Apr 2011.
[95] W. R. Miller, J. Bartlett, A. M. Brodie, R. W. Brueggemeier, E. di Salle, P. E. Lønning, et al., "Aromatase inhibitors: are there differences between steroidal and nonsteroidal aromatase inhibitors and do they matter?," Oncologist, vol. 13, pp. 829-37, Aug 2008.
[96] I. Obiorah and V. C. Jordan, "Progress in endocrine approaches to the treatment and prevention of breast cancer.," Maturitas, vol. 70, pp. 315-21, Dec 2011.
[97] M. S. Ewer and S. Glück, "A woman's heart: the impact of adjuvant endocrine therapy on cardiovascular health.," Cancer, vol. 115, pp. 1813-26, May 2009.

Capítulo V: Referências Bibliográficas
88
[98] P. Hadji, D. G. Kieback, J. Tams, A. Hasenburg, and M. Ziller, "Correlation of treatment-emergent adverse events and clinical response to endocrine therapy in early breast cancer: a retrospective analysis of the German cohort of TEAM.," Ann Oncol, Mar 2012.
[99] J. S. Mieog, J. P. Morden, J. M. Bliss, R. C. Coombes, C. J. van de Velde, and I. S. Committee, "Carpal tunnel syndrome and musculoskeletal symptoms in postmenopausal women with early breast cancer treated with exemestane or tamoxifen after 2-3 years of tamoxifen: a retrospective analysis of the Intergroup Exemestane Study.," Lancet Oncol, vol. 13, pp. 420-32, Apr 2012.
[100] R. E. Coleman, L. M. Banks, S. I. Girgis, E. Vrdoljak, J. Fox, S. J. Cawthorn, et al., "Reversal of skeletal effects of endocrine treatments in the Intergroup Exemestane Study.," Breast Cancer Res Treat, vol. 124, pp. 153-61, Nov 2010.
[101] L. Folkestad, N. H. Bjarnason, J. K. Bjerregaard, and K. Brixen, "The effect of aromatase inhibitors on bone metabolism.," Basic Clin Pharmacol Toxicol, vol. 104, pp. 3-10, Jan 2009.
[102] N. Miyakoshi, Y. Kasukawa, T. A. Linkhart, D. J. Baylink, and S. Mohan, "Evidence that anabolic effects of PTH on bone require IGF-I in growing mice.," Endocrinology, vol. 142, pp. 4349-56, Oct 2001.
[103] G. Lombardi, C. Di Somma, L. Vuolo, E. Guerra, E. Scarano, and A. Colao, "Role of IGF-I on PTH effects on bone.," J Endocrinol Invest, vol. 33, pp. 22-6, 2010.
[104] A. Brufsky, N. Bundred, R. Coleman, R. Lambert-Falls, R. Mena, P. Hadji, et al., "Integrated analysis of zoledronic acid for prevention of aromatase inhibitor-associated bone loss in postmenopausal women with early breast cancer receiving adjuvant letrozole.," Oncologist, vol. 13, pp. 503-14, May 2008.
[105] N. J. Bundred, "Aromatase inhibitors and bone health.," Curr Opin Obstet Gynecol, vol. 21, pp. 60-7, Feb 2009.
[106] D. M. Reid, "Prevention of osteoporosis after breast cancer.," Maturitas, vol. 64, pp. 4-8, Sep 2009.
[107] C. B. Confavreux, A. Fontana, J. P. Guastalla, F. Munoz, J. Brun, and P. D. Delmas, "Estrogen-dependent increase in bone turnover and bone loss in postmenopausal women with breast cancer treated with anastrozole. Prevention with bisphosphonates.," Bone, vol. 41, pp. 346-52, Sep 2007.
[108] A. M. Cheung, L. Tile, S. Cardew, S. Pruthi, J. Robbins, G. Tomlinson, et al., "Bone density and structure in healthy postmenopausal women treated with exemestane for the primary prevention of breast cancer: a nested substudy of the MAP.3 randomised controlled trial.," Lancet Oncol, vol. 13, pp. 275-84, Mar 2012.
[109] P. E. Goss, S. Qi, A. M. Cheung, H. Hu, M. Mendes, and K. P. Pritzker, "Effects of the steroidal aromatase inhibitor exemestane and the nonsteroidal aromatase inhibitor letrozole on bone and lipid metabolism in ovariectomized rats.," Clin Cancer Res, vol. 10, pp. 5717-23, Sep 2004.
[110] P. E. Goss, P. Hadji, M. Subar, P. Abreu, T. Thomsen, and J. Banke-Bochita, "Effects of steroidal and nonsteroidal aromatase inhibitors on markers of bone turnover in healthy postmenopausal women.," Breast Cancer Res, vol. 9, p. R52, 2007.
[111] Q. Chen, H. Kaji, T. Sugimoto, and K. Chihara, "Testosterone inhibits osteoclast formation stimulated by parathyroid hormone through androgen receptor.," FEBS Lett, vol. 491, pp. 91-3, Feb 2001.
[112] Y. Miki, T. Suzuki, M. Hatori, K. Igarashi, K. I. Aisaki, J. Kanno, et al., "Effects of aromatase inhibitors on human osteoblast and osteoblast-like cells: a possible androgenic bone protective effects induced by exemestane.," Bone, vol. 40, pp. 876-87, Apr 2007.
[113] A. U. Buzdar, "New generation aromatase inhibitors--from the advanced to the adjuvant setting.," Breast Cancer Res Treat, vol. 75 Suppl 1, pp. S13-7; discussion S33-5, Oct 2002.
[114] A. U. Buzdar, "Pharmacology and pharmacokinetics of the newer generation aromatase inhibitors.," Clin Cancer Res, vol. 9, pp. 468S-72S, Jan 2003.

Estudo comparativo dos metabolitos do exemestano num modelo celular de cancro da mama estrogénio-dependente
89
[115] C. Markopoulos, U. Dafni, J. Misitzis, V. Zobolas, E. Tzoracoleftherakis, D. Koukouras, et al., "Extended adjuvant hormonal therapy with exemestane has no detrimental effect on the lipid profile of postmenopausal breast cancer patients: final results of the ATENA lipid substudy.," Breast Cancer Res, vol. 11, p. R35, 2009.
[116] G. Atalay, L. Dirix, L. Biganzoli, L. Beex, M. Nooij, D. Cameron, et al., "The effect of exemestane on serum lipid profile in postmenopausal women with metastatic breast cancer: a companion study to EORTC Trial 10951, 'Randomized phase II study in first line hormonal treatment for metastatic breast cancer with exemestane or tamoxifen in postmenopausal patients'." Ann Oncol, vol. 15, pp. 211-7, Feb 2004.
[117] S. Chen, "An "omics" approach to determine the mechanisms of acquired aromatase inhibitor resistance.," OMICS, vol. 15, pp. 347-52, Jun 2011.
[118] M. Giuliano, R. Schifp, C. K. Osborne, and M. V. Trivedi, "Biological mechanisms and clinical implications of endocrine resistance in breast cancer.," Breast, vol. 20 Suppl 3, pp. S42-9, Oct 2011.
[119] M. Zilli, A. Grassadonia, N. Tinari, A. Di Giacobbe, S. Gildetti, J. Giampietro, et al., "Molecular mechanisms of endocrine resistance and their implication in the therapy of breast cancer," Biochim Biophys Acta, vol. 1795, pp. 62-81, Jan 2009.
[120] C. K. Osborne and R. Schiff, "Mechanisms of endocrine resistance in breast cancer.," Annu Rev Med, vol. 62, pp. 233-47, Feb 2011.
[121] Y. Miyoshi, K. Murase, M. Saito, and K. Oh, "Prediction of hormone sensitivity for breast cancers.," Breast Cancer, vol. 17, pp. 86-91, Apr 2010.
[122] C. Wong and S. Chen, "Heat shock protein 90 inhibitors: new mode of therapy to overcome endocrine resistance.," Cancer Res, vol. 69, pp. 8670-7, Nov 2009.
[123] E. A. Musgrove and R. L. Sutherland, "Biological determinants of endocrine resistance in breast cancer," Nat Rev Cancer, vol. 9, pp. 631-43, Sep 2009.
[124] A. Reid, L. Vidal, H. Shaw, and J. de Bono, "Dual inhibition of ErbB1 (EGFR/HER1) and ErbB2 (HER2/neu)." Eur J Cancer, vol. 43, pp. 481-9, Feb 2007.
[125] M. Beeram, Q. T. Tan, R. R. Tekmal, D. Russell, A. Middleton, and L. A. DeGraffenried, "Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling.," Ann Oncol, vol. 18, pp. 1323-8, Aug 2007.
[126] X. Wang, S. Masri, S. Phung, and S. Chen, "The role of amphiregulin in exemestane-resistant breast cancer cells: evidence of an autocrine loop.," Cancer Res, vol. 68, pp. 2259-65, Apr 2008.
[127] V. Craig Jordan, J. Lewis-Wambi, H. Kim, H. Cunliffe, E. Ariazi, C. G. Sharma, et al., "Exploiting the apoptotic actions of oestrogen to reverse antihormonal drug resistance in oestrogen receptor positive breast cancer patients.," Breast, vol. 16 Suppl 2, pp. S105-13, Dec 2007.
[128] O. W. Prall, E. M. Rogan, and R. L. Sutherland, "Estrogen regulation of cell cycle progression in breast cancer cells.," J Steroid Biochem Mol Biol, vol. 65, pp. 169-74, Apr 1998.
[129] C. A. Lange and D. Yee, "Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer.," Endocr Relat Cancer, vol. 18, pp. C19-24, 2011.
[130] R. S. Finn, J. Dering, D. Conklin, O. Kalous, D. J. Cohen, A. J. Desai, et al., "PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro.," Breast Cancer Res, vol. 11, p. R77, 2009.
[131] P. Fedele, N. Calvani, A. Marino, L. Orlando, P. Schiavone, A. Quaranta, et al., "Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: Where are we now and where are we going?," Crit Rev Oncol Hematol, Apr 2012.
[132] S. Hiscox, P. Barrett-Lee, A. C. Borley, and R. I. Nicholson, "Combining Src inhibitors and aromatase inhibitors: a novel strategy for overcoming endocrine resistance and bone loss.," Eur J Cancer, vol. 46, pp. 2187-95, Aug 2010.

Capítulo V: Referências Bibliográficas
90
[133] R. A. Lockshin and Z. Zakeri, "Apoptosis, autophagy, and more.," Int J Biochem Cell Biol, vol. 36, pp. 2405-19, Dec 2004.
[134] A. C. Kimmelman, "The dynamic nature of autophagy in cancer.," Genes Dev, vol. 25, pp. 1999-2010, Oct 2011.
[135] D. Gozuacik and A. Kimchi, "Autophagy as a cell death and tumor suppressor mechanism.," Oncogene, vol. 23, pp. 2891-906, Apr 2004.
[136] N. Chen and V. Karantza, "Autophagy as a therapeutic target in cancer.," Cancer Biol Ther, vol. 11, pp. 157-68, Jan 2011.
[137] N. Mizushima, B. Levine, A. M. Cuervo, and D. J. Klionsky, "Autophagy fights disease through cellular self-digestion.," Nature, vol. 451, pp. 1069-75, Feb 2008.
[138] S. Chen, S. K. Rehman, W. Zhang, A. Wen, L. Yao, and J. Zhang, "Autophagy is a therapeutic target in anticancer drug resistance," Biochim Biophys Acta, vol. 1806, pp. 220-9, Dec 2010.
[139] A. Eisenberg-Lerner and A. Kimchi, "The paradox of autophagy and its implication in cancer etiology and therapy.," Apoptosis, vol. 14, pp. 376-91, Apr 2009.
[140] S. Ito, N. Koshikawa, S. Mochizuki, and K. Takenaga, "3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition.," Int J Oncol, vol. 31, pp. 261-8, Aug 2007.
[141] D. J. Zhou, D. Pompon, and S. A. Chen, "Stable expression of human aromatase complementary DNA in mammalian cells: a useful system for aromatase inhibitor screening.," Cancer Res, vol. 50, pp. 6949-54, Nov 1990.
[142] S. Masri, S. Phung, X. Wang, and S. Chen, "Molecular characterization of aromatase inhibitor-resistant, tamoxifen-resistant and LTEDaro cell lines.," J Steroid Biochem Mol Biol, vol. 118, pp. 277-82, Feb 2010.
[143] S. Masri, S. Phung, X. Wang, X. Wu, Y. C. Yuan, L. Wagman, et al., "Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor.," Cancer Res, vol. 68, pp. 4910-8, Jun 2008.
[144] A. Cristina, V. Carla, A. Margarida, S. E. T, R. Fernanda, C. Shiuan, et al., "Effects of steroidal aromatase inhibitors on sensitive and resistant breast cancer cells: aromatase inhibition and autophagy," ed, 2012.
[145] Y. Chen, M. B. Azad, and S. B. Gibson, "Methods for detecting autophagy and determining autophagy-induced cell death," Can J Physiol Pharmacol, vol. 88, pp. 285-95, Mar 2010.