desenvolvimento de métodos para quantificação de … · 2017-11-02 · the benzylpenicillin...
TRANSCRIPT

Desenvolvimento de métodos para quantificação de benzilpenicilina em medicamentos
veterinários e seus resíduos em leite bovino e caprino por UFLC-DAD
Alessandra Miranda Cabral
Tese de Doutorado Natal/RN, dezembro de 2015

Alessandra Miranda Cabral
Desenvolvimento de métodos para quantificação de benzilpenicilina em medicamentos
veterinários e seus resíduos em leite bovino e caprino por UFLC-DAD
Tese de Doutorado apresentada ao Programa de Pós-
Graduação em Química da Universidade Federal do Rio
Grande do Norte, em cumprimento às exigências para
obtenção do título de Doutora em Química.
Orientadora: Profa Drª Maria de Fátima Vitória de Moura
Co-Orientador: Profo Drº. Djalma Ribeiro da Silva
NATAL/RN
2015

Divisão de Serviços Técnicos
Catalogação da Publicação na Fonte. UFRN
Biblioteca Setorial do Instituto de Química
Cabral, Alessandra Miranda.
Desenvolvimento de métodos para quantificação de benzilpenicilina em medicamentos
veterinários e seus resíduos em leite bovino e caprino por UFLC-DAD / Alessandra Miranda
Cabral. – Natal, RN, 2015.
237 f. : il.
Orientadora: Maria de Fátima Vitória de Moura.
Co-orientador: Djalma Ribeiro da Silva.
Tese (Doutorado em Química) - Universidade Federal do Rio Grande do Norte. Centro
de Ciências Exatas e da Terra. Programa de Pós-Graduação em Química.
1. Benzilpenicilina – Tese. 2. Medicamentos Veterinários – Tese. 3. Leite – Tese. 4.
Cromatografia Líquida Ultrarrápida Acoplada a Arranjo de Fotodiodos – Tese. I. Moura,
Maria de Fátima Vitória de. II. Da Silva, Djalma Ribeiro. III. Universidade Federal do Rio
Grande do Norte. IV. Título.
RN/UFRN/BSE- Instituto de Química CDU 543.062:636.09 (043.3)


Dedico este trabalho
Ao Deus Criador do Universo,
A minha mãe,
Ao meu irmão.

AGRADECIMENTOS
Queria encontrar, oh Deus, palavras e expressões de louvor para te agradecer por tantas
vitórias, assim agradeço ao teu filho Jesus Cristo pelo presente da Vida e por ter me
proporcionado sabedoria e vigor para continuar minha caminhada, pois como disse João no
capítulo 1 versículo 3 da Bíblia Sagrada “Todas as coisas foram feitas por ele, e sem ele nada
do que foi feito se fez. ”
Agradeço à minha Mãe Maria de Fátima Miranda pelo apoio, incentivo, carinho, fé e
amor que não conhecem limites. Ao meu irmão Alciney Miranda pela fé em minha vocação
para Química. Ao meu Pai Antônio Dantas pelo incentivo e esforço.
Durante todo tempo dos meus estudos na pós-graduação, estive trabalhando ao lado de
pessoas que devo agradecimentos:
À Professora Maria de Fátima Vitória de Moura pela sua orientação, incentivo e
supervisão acadêmica. Ao Professor Djalma Ribeiro da Silva, pela orientação.
A equipe do Instituto de Química e Programa de Pós-Graduação da Universidade
Federal do Rio Grande do Norte pela criação de um ambiente agradável, e de fácil transmissão
de conhecimento.
A todos os meus Queridos Professores que me ensinaram o valor da Ciência no Brasil.
A toda equipe técnica da Central Analítica que me auxiliaram nos períodos que
prolonguei as análises nos laboratórios de Cromatografia, Espectroscopia e Preparo de amostra.
A todos meus colegas pela boa convivência dentro dos laboratórios e em ambientes de
estudo.

"O investigador sofre decepções, os longos meses passados em uma direção errada, os
fracassos. Mas as falhas também são úteis, porque, se bem analisadas, podem levar ao
sucesso. E para o pesquisador não há alegria comparável à de uma descoberta, ainda que
pequena. ”
Alexander Fleming, Escócia (1943)
“Eu sei que tudo quanto Deus faz durará eternamente; nada se lhe deve acrescentar, e nada se
lhe deve tirar; e isto faz Deus para que haja temor diante dele. ”
Eclesiastes 3:14.

RESUMO
A Benzilpenicilina (PENG) é usada como princípio ativo em medicamentos veterinários, para
aumentar a produtividade pecuária, devido suas propriedades terapêuticas. Porém, quando são
de má qualidade e utilizados indiscriminadamente, resultam em resíduos nos alimentos
expostos ao consumo humano, principalmente, no leite que é essencial para dieta das
populações de crianças e idosos. Assim, é imprescindível que se desenvolvam novos métodos
capazes de detectar os resíduos nesse alimento, em níveis que são nefastos a saúde humana, a
fim de contribuir com a segurança alimentar dos consumidores e colaborar com as agências
reguladoras numa fiscalização eficiente. Neste trabalho foram desenvolvidos métodos para o
controle de qualidade dos medicamentos. Também foram validadas metodologias para
identificação e quantificação de resíduos do antibiótico no leite bovino e caprino. Para isso, o
controle analítico foi realizado em duas etapas. Na primeira, os grupos das amostras dos
medicamentos I, II, III, IV e V foram caracterizadas, individualmente, por Espectroscopia no
infravermelho médio (4000 – 600 cm-1). Além disso, 37 amostras, distribuídas nesses grupos,
foram analisadas por Espectroscopia no UV VIS NIR e Cromatografia liquida ultra-rápida
acoplada a arranjo linear de fotodiodos (UFLC-DAD). Os resultados das caracterizações
indicaram semelhanças, entre padrão de referência PENG e amostras, principalmente em
regiões de 1724 a 1818 cm-1 de ν C=O que indica presença de amidas primárias característica de
PENG. O método utilizando UFLC-DAD apresentou R de 0,9991. LD de 7,384 × 10-4 µg mL-
1. LQ de 2,049 × 10-3 µg mL-1. A análise mostrou que 62,16% das amostras apresentaram pureza
≥ 81,21%. O método utilizando espectroscopia na região do UV VIS NIR apresentou erro médio
≤ 8 – 12% entre as medidas de referência e experimental, indicando ser boa alternativa para
determinação rápida de PENG. Na segunda etapa, foi desenvolvido um método para extração e
isolamento da PENG por adição de tampão McIlvaine, utilizado para precipitação total das
proteínas em pH 4,0. Os resultados mostraram excelentes valores de recuperação de PENG,
sendo de 92,05% para amostras de leite bovino (método 1). Enquanto que, para amostras de
leite caprino (método 2) a recuperação de PENG foi de 95,83%. Os métodos por UFLC-DAD
foram validados em concordância com o limite máximo de resíduos (LMR) de 4 µg Kg-1
normatizado pela CAC/GL16. A validação do método 1 indicou R de 0,9975. LD de 7,246 ×
10-4 µg mL-1. LQ de 2,196 × 10-3 µg mL-1. A aplicação do método 1 mostrou que 12% das
amostras analisadas apresentavam concentração de resíduos de PENG > LMR. O método 2
indicou R de 0,9995. LD 8,251 × 10-4 µg mL-1. LQ de 2,5270 × 10-3 µg mL-1. A aplicação do
método mostrou que 15% das amostras estavam acima do tolerável. A análise comparativa entre
os métodos apontou melhores condições de validação para amostras LCP, devido à redução do
efeito de matriz, pois o tcalculado < ttabelado, ocasionado pelo aumento da recuperação do analito na
matriz. Desta forma, todos os procedimentos desenvolvidos apresentaram simplicidade,
rapidez, seletividade, reduzido tempo de análise e consumo de reagentes e solventes tóxicos,
sobretudo se comparados às metodologias já estabelecidas.
Palavras Chaves: Benzilpenicilina. Medicamentos Veterinários. Leite. Cromatografia Líquida
Ultrarrápida Acoplada a Arranjo de Fotodiodos.

ABSTRACT
The Benzylpenicillin (PENG) have been as the active ingredient in veterinary medicinal
products, to increase productivity, due to its therapeutic properties. However, one of unfortunate
quality and used indiscriminately, resulting in residues in foods exposed to human
consumption, especially in milk that is essential to the diet of children and the ageing. Thus, it
is indispensable to develop new methods able to detect this waste food, at levels that are toxic
to human health, in order to contribute to the food security of consumers and collaborate with
regulatory agencies in an efficient inspection. In this work, were developed methods for the
quality control of veterinary drugs based on Benzylpenicillin (PENG) that are used in livestock
production. Additionally, were validated methodologies for identifying and quantifying the
antibiotic residues in milk bovine and caprine. For this, the analytical control was performed
two steps. At first, the groups of samples of medicinal products I, II, III, IV and V, individually,
were characterized by medium infrared spectroscopy (4000 – 600 cm-1). Besides, 37 samples,
distributed in these groups, were analyzed by spectroscopy in the ultraviolet and near infrared
region (UV VIS NIR) and Ultra Fast Liquid Chromatograph coupled to linear arrangement
photodiodes (UFLC-DAD). The results of the characterization indicated similarities, between
PENG and reference standard samples, primarily in regions of 1818 to 1724 cm-1 of ν C=O that
shows primary amides features of PENG. The method by UFLC-DAD presented R on 0.9991.
LOD of 7.384 × 10-4 µg mL-1. LOQ of 2.049 × 10-3 µg mL-1. The analysis shows that 62.16%
the samples presented purity ≥ 81.21%. The method by spectroscopy in the UV VIS NIR
presented medium error ≤ 8 – 12% between the reference and experimental criteria, indicating
is a secure choice for rapid determination of PENG. In the second stage, was acquiring a method
for the extraction and isolation of PENG by the addition of buffer McIlvaine, used for
precipitation of proteins total, at pH 4.0. The results showed excellent recovery values PENG,
being close to 92.05% of samples of bovine milk (method 1). While samples of milk goats
(method 2) the recovery of PENG were 95.83%. The methods for UFLC-DAD have been
validated in accordance with the maximum residue limit (LMR) of 4 µg Kg-1 standardized by
CAC/GL16. Validation of the method 1 indicated R by 0.9975. LOD of 7.246 × 10-4 µg mL-1.
LOQ de 2.196 × 10-3 µg mL-1. The application of the method 1 showed that 12% the samples
presented concentration of residues of PENG > LMR. The method 2 indicated R by 0.9995.
LOD 8.251 × 10-4 µg mL-1. LOQ de 2.5270 × 10-3 µg mL-1. The application of the method
showed that 15% of the samples were above the tolerable. The comparative analysis between
the methods pointed better validation for LCP samples, because the reduction of the matrix
effect, on this account the tcalculs < ttable, caused by the increase of recovery of the PENG. In this
mode, all the operations developed to deliver simplicity, speed, selectivity, reduced analysis
time and reagent use and toxic solvents, particularly if compared to the established
methodologies.
Keywords: Benzylpenicillin. Medicinal Products for Veterinary Use. Milk. Ultra-Fast Liquid
Chromatography Coupled to an Arrangement of Photodiodes.

LISTA DE ABREVIATURAS E SIGLAS
ABAM Abamectina
ACN Acetonitrila
AIST National Institute of Advanced Industrial Science and Technology
AMDUCA The Animal Medicinal Drug Use Clarification Act
ANVISA Agência Nacional de Vigilância Sanitária
AOAC The association of Analytical Communities
ATR attenuated total reflectance
CAS Chemical Abstracts Service
CPVS Compêndio de Produtos Veterinários
CVMP Committee for Medicinal Products for Veterinary Use
CCRVDF Codex Committee on Residues of Veterinary Drugs in Foods
DOX Doxiciclina
EDSPLS derivative spectra partial least squares
EFPE Universidade Federal de Pernambuco
ELISA Enzyme Linked Immuno Sorbent Assay
EMA European Medicines Agency
EMBRAPA Empresa Brasileira de Pesquisa Agropecuária
ENR Enrofloxacina
EURACHEM A focus for analytical chemistry in Europe
FAO Food and Agriculture Organization of the United Nations
FIOCRUZ Fundação Oswaldo Cruz
FS Fator de Segurança
FTIR Fourier transform infrared spectroscopy
GEN Gentamicina
HPLC High-performance liquid chromatography
IDA Ingestão Diária Aceitável
IF Infravermelho
IUPAC International Union of Pure and Applied Chemistry
IVERM Ivermectina
JECFA Expert Committee on Food Additives
LMR Limite Máximo de Resíduos

LB Amostras de Leite Bovino
LCP Amostras de Leite Caprino
MAPA Ministério da Agricultura Pecuária e Abastecimento do Brasil
MERCOSUL Mercado Comum do Sul
MIR Medium infrared spectroscopy
NIR Near infrared spectroscopy
NOEL no observed effects levels
OMS Organização mundial da saúde
OTC Oxitetraciclina
OTCDII Diidrato de Oxitetraciclina
PCA Análise por Componentes Principais
PCR Principal Components Regression
PENG PROC Benzilpenicilina Procaína
PENG Benzilpenicilina
PLS Partial Least Squares
QuEChERS Quick Easy Cheap Effective Rugged Safe
RMSEC Root Mean Square Error of Calibration
RMSEP Root Mean Squares Error of Prediction
SDBR-IR Spectral Database for Organic Compounds
SDX Sulfadoxina
SDZ Sulfadizina
SINDAN Sindicato Nacional da Indústria de Produtos para Saúde Animal
SMZ Sulfametoxina
TC Tetraciclina
TMP Trimetroprina
UEM Universidade Estadual do Maranhão
UFLC-DAD Ultra Fast Liquid Chromatograph coupled to linear arrangement photodiodes
UFRJ Universidade Federal do Rio de Janeiro
UFRPE Universidade Federal Rural de Pernambuco
UFRS Universidade Federal do Rio Grande do Sul
UFV Universidade Federal de Viçosa
UNESP Universidade Estadual Paulista
USP Universidade de São Paulo
UPLC Ultra-Performance Liquid Chromatography

UV ultravioleta
VICH International Cooperation on Harmonisation of Technical Requirements for
Registration of Veterinary Medicinal Products
VISAs Vigilância Sanitária
WHO World Health Organization

LISTA DE TABELAS
Tabela 2.1- Correlação entre os LMRs admitidos em principais subprogramas de
monitoramento e Controle dos Resíduos de Antibióticos em Leite........ 46
Tabela 3.1- Sumário dos métodos de preparo de amostras de leite para
determinação de resíduos de antibióticos utilizando HPLC-DAD.......... 69
Tabela 3.2- Descrição dos antibióticos questionados nos estudos recentes e
principais técnicas empregadas em diferentes matrizes alimentares…. 76
Tabela 3.3- Resumo das principais Normatizações e órgãos reguladores e
Aplicação de Controle de Contaminantes............................................. 86
Tabela 3.4- Comparação dos requisitos de validação preconizados para
quantificação dos resíduos em alimentos.............................................. 89
Tabela 4.1- Descrição dos medicamentos analisados, segundo CPV/SIDAN......... 95
Tabela 4.2- Amostragem dos medicamentos (misturas) para identificação e
quantificação.......................................................................................... 96
Tabela 4.3- Planejamento das condições do sistema cromatográfico para PENG.... 99
Tabela 4.4- Procedimento de modelagem para quantificação de PENG em
medicamentos de uso veterinário........................................................... 106
Tabela 4.5- Descrição das amostras de leite bovino................................................. 111
Tabela 4.6- Descrição das amostras de leite caprino................................................ 112
Tabela 4.7- Planejamento comparativo das condições do sistema cromatográfico
para PENG para análise das amostras controle de leite bovino (LB) e
leite caprino (LCP)................................................................................. 115
Tabela 5.1- Atribuição das bandas espectrais de absorção no FTIR para
antibióticos............................................................................................ 127
Tabela 5.2- Desempenho do método para quantificação de PENG em
medicamentos veterinários..................................................................... 132
Tabela 5.3- Precisão e exatidão do método para quantificação de PENG em
medicamentos veterinários, expressos pelo coeficiente de variação
(%CV).................................................................................................... 133
Tabela 5.4- Robustez do método para quantificação da PENG em medicamentos
veterinários, expresso pelo coeficiente de variação (%CV).................. 134

Tabela 5.5- Cálculo do teor de PENG nos medicamentos analisados por
cromatografia......................................................................................... 137
Tabela 5.6- Características dos modelos de calibração para determinação de PENG
em medicamentos................................................................................... 146
Tabela 5.7- Critérios utilizados para validação do método.....................................
149
Tabela 5.8- Desempenho dos métodos para quantificação de PENG em amostras
de leite bovino e caprino, expresso pela recuperação (%)..................... 155
Tabela 5.9- Comparação do desempenho dos métodos para quantificação de
PENG amostras de leite bovino e caprino............................................... 168
Tabela 5.10- Comparação da precisão e exatidão do Método para quantificação de
PENG em amostras de leite bovino e caprino, expresso pelo
coeficiente de variação (%CV)............................................................... 169
Tabela 5.11- Avaliação comparativa da robustez do método para quantificação de
resíduos de PENG em amostras de leite bovino e caprino, expresso
pelo coeficiente de variação (%CV)....................................................... 171

LISTA DE FIGURAS
Figura 2.1- Estrutura geral das penicilinas, com seus diferentes R............................ 31
Figura 2.2- Reação para formação do núcleo 6-APA................................................ 32
Figura 2.3- Degradação da Penicilina com abertura do anel por ação de enzima em
meio ácido (1) e hidrólise da amina da cadeia lateral em meio ácido (2) 33
Figura 2.4- Estrutura Química da PENG................................................................... 34
Figura 2.5- Frequência para recomendação do uso de antibióticos no Brasil............ 37
Figura 2.6- Tendência de Crescimento da cadeia produtiva dos medicamentos
usados para sanidade animal................................................................... 38
Figura 3.1- Ilustração da rotação óptica de um composto quiral biologicamente
ativo........................................................................................................ 54
Figura 3.2- Visualização de um dos Centros Quirais da Benzilpenicilina (A),
imagem em 3D (B)................................................................................. 55
Figura 3.3- Emprego dos diferentes detectores para análise por fármacos a base de
penicilinas.............................................................................................. 62
Figura 3.4- Frequências dos trabalhos científicos aplicados ao desenvolvimento de
novos métodos para quantificação de resíduos de antibióticos em leite,
durante período de 2010 a 2012.............................................................. 73
Figura 3.5- Tendência do avanço da literatura científica no período de 1983 a 2016
(fev) para quantificação de resíduos de penicilina, em função da
nacionalidade dos pesquisadores............................................................ 79
Figura 3.6- Citação do Brasil e aplicação de métodos para controle, em função do
número de publicação e países................................................................ 82
Figura 3.7- Distribuição do número de publicações em função da filiação dos
autores.................................................................................................... 83
Figura 4.1- Ilustração do Sistema UFLC-DAD......................................................... 93
Figura 4.2- Ilustração para testes com amostras externas.......................................... 107
Figura 4.3- Esquema resumido para testes com amostras externas........................... 108
Figura 4.4- Esquema simplificado do preparo das amostras de leite antes (A) e após
tratamento (B)......................................................................................... 114
Figura 4.5- Estratégia para validação dos métodos bioanalíticos.............................. 120

Figura 5.1- Espectros no UV de amostra contendo PENG e STR em cinco níveis
de concentração...................................................................................... 123
Figura 5.2- Espectros FTIR para grupos dos antibióticos das amostras e PENG
padronizado............................................................................................ 126
Figura 5.3- Perfil cromatográfico da penicilina (PENG) referente ao padrão
certificado............................................................................................... 129
Figura 5.4- Resultados do planejamento das condições cromatográficas.................. 130
Figura 5.5- Curva analítica para quantificação de PENG em medicamentos............. 131
Figura 5.6- Perfil cromatográfico das amostras de medicamentos em grupos........... 135
Figura 5.7- Espectros da PENG certificado e das amostras comerciais de
medicamentos......................................................................................... 138
Figura 5.8- Gráficos dos loadings para M1 – M12.................................................... 139
Figura 5.9- Gráficos dos loadings para M13 – M24.................................................. 140
Figura 5.10- Gráficos dos Scores M1 – M12............................................................... 142
Figura 5.11- Gráficos dos Scores M13 – M24............................................................. 143
Figura 5.12- Representação do erro médio entre os valores de referência e o método
UV VIS NIR para análise quantitativa das amostras Puras (E1 – E24)..... 147
Figura 5.13- Representação do erro médio entre os valores de referência e o método
UV VIS NIR para análise quantitativa das amostras tratadas................... 148
Figura 5.14- Cromatogramas obtidos para amostra controle LB (branco) e amostra
fortificada com PENG............................................................................ 151
Figura 5.15- Cromatogramas obtidos para amostra LCP controle (branco) e amostra
fortificada com PENG............................................................................ 152
Figura 5.16- Estudo da variação de tempo de retenção e área do pico da PENG.......... 157
Figura 5.17- Resultados das condições para análise de PENG em amostras controle
de leite bovino........................................................................................ 158
Figura 5.18- Resultados das condições para análise de PENG em amostras controle
de leite caprino........................................................................................ 159
Figura 5.19- Otimização da operação das bombas em função da pressão e dos
tempos de retenção da PENG.................................................................. 160
Figura 5.20- Perfil cromatográfico de PENG nas amostras controle de leite LB e
LCP fortificadas..................................................................................... 161

Figura 5.21- Perfil cromatográfico de PENG cromatogramas das amostras controle
de leite LB e LCP fortificadas em níveis com seus respectivos brancos.. 162
Figura 5.22- Efeito da concentração de PENG e perfis das curvas para o método 1.... 163
Figura 5.23- Efeito da concentração de PENG e perfis das curvas para o método 2.... 164
Figura 5.24- Curva analítica para amostras de leite LB fortificadas para estudo da
linearidade.............................................................................................. 166
Figura 5.25- Curva analítica para amostras de leite LCP fortificadas para estudo da
linearidade.............................................................................................. 167
Figura 5.26- Análise de leite bovino quanto à presença de resíduos de PENG............ 172
Figura 5.27- Análise de leite caprino quanto à presença de resíduos de PENG........... 173

SUMÁRIO
CAPÍTULO 1
INTRODUÇÃO E OBJETIVOS
1 INTRODUÇÃO E OBJETIVOS................................................................ 22
1.1 INTRODUÇÃO............................................................................................. 22
1.2 OBJETIVOS.................................................................................................. 25
1.2.1 Objetivos Gerais........................................................................................... 25
1.2.2 Objetivos Específicos.................................................................................... 25
CAPÍTULO 2
ANTIBIÓTICOS: Penicilina e Resíduos
2 ANTIBIÓTICOS: Penicilinas e Resíduos................................................. 28
2.1 DESCOBERTA DA PENICILINA.............................................................. 28
2.2 PENICILINAS: Grupos e Propriedades....................................................... 31
2.3 PRODUTOS VETERINÁRIOS: Designação e Faturamento....................... 36
2.4 OCORRÊNCIA, RISCO E CONTROLE DOS RESÍDUOS: Segurança
Alimentar...................................................................................................... 40
CAPÍTULO 3
MÉTODOS OFICIAIS E INDICADORES DOS AVANÇOS DA
LITERATURA CIENTÍFICA
3 MÉTODOS OFICIAIS E INDICADORES DOS AVANÇOS DA
LITERATURA CIENTÍFICA.................................................................. 49
3.1 REVISÃO DOS MÉTODOS APLICADOS AO CONTROLE DE
QUALIDADE DE TERAPÊUTICOS PARA USO VETERINÁRIO.......... 49
3.1.1 Determinação da rotação óptica de um antibiótico.................................. 53
3.1.2 Espectroscopia para identificação e quantificação de antibióticos em
terapêuticos de uso veterinário.................................................................. 57
3.1.2.1 Importância da Região no Ultravioleta e Infravermelho Próximo................. 58

3.1.2.2 Importância da aplicação da Espectroscopia no Infravermelho Médio......... 60
3.1.3 Considerações sobre a Aplicação dos Métodos Cromatográficos no
Controle de Qualidade de Formulações.................................................... 61
3.2 ANÁLISES DOS RESÍDUOS DE ANTIBIÓTICOS................................... 63
3.2.1 Preparo da amostra para quantificação de resíduos de antibióticos...... 65
3.2.1.1 Critérios para Preparo da Amostra................................................................ 67
3.2.2 Métodos Bioanalíticos e Testes Rápidos.................................................... 71
3.2.3 Emprego das Técnicas para Quantificação e Confirmação dos Resíduos
de Antibióticos.............................................................................................. 72
3.2.4 Plano de Controle e Monitoramento de Resíduos de Antimicrobianos
executados no Brasil................................................................................... 80
3.3 ESTRATÉGIAS PARA VALIDAÇÃO DOS MÉTODOS.......................... 85
3.3.1 Características de um Método Validado................................................... 88
CAPÍTULO 4
METODOLOGIA
4 METODOLOGIA...................................................................................... 92
4.1 DESENVOLVIMENTO DE MÉTODO PARA IDENTIDADE E
QUALIDADE DOS MEDICAMENTOS A BASE DE
BENZILPENICILINA................................................................................... 92
4.1.1 Reagentes e Solventes.................................................................................. 92
4.1.2 Instrumentação............................................................................................. 92
4.1.3 Outros materiais para Cromatografia........................................................ 93
4.1.4 Amostras de medicamentos......................................................................... 94
4.1.5 Espectroscopia no infravermelho médio.................................................... 97
4.1.6 Análises por Cromatografia líquida ultrarrápida acoplada em arranjo
linear de fotodiodos...................................................................................... 98
4.1.6.1 Preparo da solução padrão de PENG.............................................................. 98
4.1.6.2 Preparo de amostras........................................................................................ 98
4.1.6.3 Condições das análises cromatográficas......................................................... 99
4.1.6.4 Validação do Método...................................................................................... 100
4.1.6.4.1 Especificidade/Seletividade............................................................................ 100

4.1.6.4.2 Faixa de Trabalho e construção da curva analítica........................................ 100
4.1.6.4.3 Linearidade.................................................................................................... 101
4.1.6.4.4 Precisão.......................................................................................................... 101
4.1.6.4.5 Exatidão......................................................................................................... 101
4.1.6.4.6 Limite de detecção.......................................................................................... 102
4.1.6.4.7 Limite de quantificação.................................................................................. 102
4.1.6.4.8 Robustez......................................................................................................... 102
4.1.6.5 Identificação de PENG nas amostras.............................................................. 103
4.1.6.6 Cálculo das concentrações de PENG em amostras de medicamentos............ 103
4.1.7 Análise na região do UV VIS NIR................................................................ 104
4.2 DESENVOLVIMENTO DE MÉTODO PARA IDENTIFICAÇÃO E
QUANTIFICAÇÃO DE PENG EM LEITE BOVINO E CAPRINO............. 109
4.2.1 Reagentes e Solventes................................................................................... 109
4.2.2 Instrumentação............................................................................................. 109
4.2.3 Outros materiais para Cromatografia........................................................ 109
4.2.4 Amostragem.................................................................................................. 110
4.2.4.1 Amostras de leite controle............................................................................... 110
4.2.4.2 Amostras de leite comerciais (externas).......................................................... 110
4.2.4.2.1 Amostras de leite bovino comercial (LB)........................................................ 110
4.2.4.2.2 Amostras de leite caprino comercial (LCP)................................................... 112
4.2.5 Preparo da solução padrão de PENG.......................................................... 113
4.2.6 Preparo da solução tampão McIlvaine pH 4............................................... 113
4.2.7 Preparo e limpeza das amostras de leite bovino e caprino.......................... 114
4.2.8 Condições para análise cromatográfica...................................................... 115
4.2.9 Validação do método.................................................................................... 115
4.2.9.1 Construção da curva Analítica........................................................................ 115
4.2.9.2 Linearidade..................................................................................................... 116
4.2.9.3 Recuperação................................................................................................... 116
4.2.9.4 Precisão (Repetabilidade)............................................................................... 116
4.2.9.5 Exatidão.......................................................................................................... 117
4.2.9.6 Limite de detecção (LD) ................................................................................ 117
4.2.9.7 Limite de quantificação (LQ) ......................................................................... 118
4.2.9.8 Seletividade..................................................................................................... 118

4.2.9.9 Robustez........................................................................................................ 118
4.2.9.10 Efeito da matriz............................................................................................. 118
4.2.9.11 Estratégias para validação.............................................................................. 120
4.2.10 Cálculo das Concentrações em amostras de leite comerciais..................... 121
CAPÍTULO 5
RESULTADOS E DISCUSSÃO
5 RESULTADOS E DISCUSSÃO................................................................ 123
5.1 DESENVOLVIMENTO DE MÉTODO PARA IDENTIDADE E
QUALIDADE DOS MEDICAMENTOS A BASE DE
BENZILPENICILINA....................................................................................... 123
5.1.1 Testes para identidade dos medicamentos.................................................. 123
5.1.1.1 Condições dos testes....................................................................................... 123
5.1.2 Análise por espectroscopia no infravermelho médio................................. 125
5.1.3 Quantificação de PENG por UFLC-DAD...................................................... 129
5.1.3.1 Estudo das condições de análise..................................................................... 129
5.1.3.2 Validação do método...................................................................................... 131
5.1.3.3 Identificação de PENG em amostras de medicamentos.................................. 135
5.1.3.4 Cálculo do teor de pureza dos medicamentos................................................. 136
5.1.4 Análise de PENG em medicamentos por UV VIS NIR............................... 138
5.1.4.1 Atribuição das bandas no UV VIS NIR e relevância para modelagem dos
dados.............................................................................................................. 138
5.1.4.2 Desempenho dos modelos de acordo com 17760/2009/EC e determinação
das concentrações de PENG........................................................................... 145
5.2 DESENVOLVIMENTO DE MÉTODO PARA IDENTIFICAÇÃO E
QUANTIFICAÇÃO DE PENG EM LEITE BOVINO E CAPRINO ............ 149
5.2.1 Otimização do preparo das amostras de leite controle bovino e caprino 151
5.2.1.1 Avaliando a recuperação do método.............................................................. 153
5.2.2 Estudo das condições cromatográficas para aplicação em amostras de
leite................................................................................................................. 157
5.2.3 Validação do método.................................................................................... 161
5.2.3.1 Correlação entre amostras de leite bovino (LB) e caprino (LCP).................... 161

5.3 CONSIDERAÇÕES FINAIS PARA QUALIDADE E SEGURANÇA DO
LEITE.................................................................................................................... 174
CAPÍTULO 6
CONCLUSÕES
6 CONCLUSÕES............................................................................................ 178
REFERÊNCIAS
REFERÊNCIAS........................................................................................... 182
APÊNDICES.................................................................................................. 216
ANEXOS........................................................................................................ 236

CAPÍTULO 1
INTRODUÇÃO E OBJETIVOS

22
1 INTRODUÇÃO E OBJETIVOS
1.1 INTRODUÇÃO
Medicamentos veterinários associados a Benzilpenicilina (PENG), são amplamente
utilizados a níveis terapêuticos para o controle de mastites em bovinos e caprinos. Porém, essas
substâncias podem ser acumuladas nos tecidos animais ou até mesmo transferidas para os
produtos alimentares em forma de resíduos, que em níveis acima do permitido, constituem um
risco potencial à saúde dos consumidores (AGUILERA-LUIZ et al., 2008; VARGAS
MAMANI; REYES REYES; RATH, 2009). Assim, a primeira etapa para prevenir a formação
de resíduos, é a utilização de medicamentos de qualidade e seu correto emprego, com relação a
sua eficiência, aplicabilidade, segurança e custo. Esses fatores requerem a aplicação das Boas
Práticas de uso, pois são importantes para promoção da qualidade dos alimentos (FILHO et al.,
2012).
Assim, a qualidade do leite reflete na sua valorização, enquanto matéria-prima, em toda
sua cadeia produtiva, pois é um dos produtos alimentícios mais importantes da agropecuária,
em razão da sua estratégia no balanceamento das dietas dos humanos e de seu relevante papel
socioeconômico. Segundo Albright, Tuckey e Woods (1961) e Chambers (1998) o leite
contaminado por resíduos é considerado adulterado e impróprio para o consumo, acarretando
prejuízos econômicos e tecnológicos na indústria de laticínios.
No Brasil, o Programa Nacional de Melhoria da Qualidade do Leite (PNQL), tem como
principal objetivo promover a melhoria da qualidade deste alimento, e consequentemente
garantir a saúde da população. Logo, as principais ações promovidas pelo órgão competente
são a detecção de fraudes e estímulos à produção de alimentos seguros (MAPA, 2011).
Segundo projeção do Instituto Brasileiro de Geografia e Estatística (IBGE), em 2014
foram produzidos, no Brasil, 37 milhões de litros de leite. Atrelado a essa produção, o
tratamento com antibióticos de doenças infecciosas desempenha um papel muito importante
garantindo o crescimento do setor (BRITO; NOBRE; FONSECA, 2009). Segundo estatísticas
do Sindicato Nacional da Indústria de Produtos para Saúde Animal (SINDAN), o faturamento
dos produtos veterinários chegou a 4,5 milhões de reais, 17% são de antimicrobianos.
A ausência de um adequado monitoramento pode resultar em altas concentrações de
resíduos no leite, principalmente se não forem utilizados de acordo com as indicações, e se não
for respeitado o período mínimo de eliminação do resíduo (SCHENCK; CALLERY, 1998). As
consequências da permanência dos resíduos pode causar prejuízos para o processo produtivo,

23
pois pode causar intolerância aos antibióticos, hipersensibilidade às penicilinas, reações
alérgicas e interferência no processo industrial dos derivados do leite, inviabilizando a produção
destes, em escala industrial, causando sérios prejuízos econômicos como, por exemplo, inibindo
fermentos lácteos que são culturas de microrganismos para produção de iogurtes, queijos e
outros derivados (BERENDSEN et al., 2010; BOHM; STACHEL; GOWIK, 2009; BOISON et
al., 1996; CHAMBERS, 1998; JONES; SEYMOUR, 1988; RAISON-PEYRON et al., 2001;
SEYMOUR; JONES; MCGILLIARD, 1988; SHERIDAN et al., 2008).
Por estas razões, as legislações no âmbito internacional como Codex Alimentarius, EC
(European Commission), FDA (Food and Drug Administration), inclusive no Brasil pelo
MAPA (Ministério da Agricultura, Pecuária e Abastecimento do Brasil), limitam a quantidade
de antibióticos no leite e adotam métodos que garantam a disponibilidade de um alimento
seguro. Porém, para que esses critérios venham a ser satisfeitos é preciso que sejam
normalizados, cumpram requisitos e gerem bons resultados. Segundo Paschoal et al (2008)
estes procedimentos devem ser realizados como garantia da qualidade das medições químicas,
através da comparabilidade, rastreabilidade e confiabilidade. Entretanto, para o monitoramento
acontecer, é necessário estabelecer métodos analíticos simples e de confiança na identificação
e a determinação de resíduos, pois muitas vezes as análises efetuadas são no campo suspeito de
contaminação (PUCHADES; CHA, 2010). Com objetivo de satisfazer essas exigências, muitos
métodos cromatográficos em sistemas diversos têm sido elaborados e publicados para na
determinação de antibióticos betalactâmicos e macrolídeos em amostras de alimentos,
utilizando sistema HPLC-UV (BAILÓN-PÉREZ et al., 2009; GARCÍA-MAYOR et al., 2015;
SAMANIDOU et al., 2015). Alguns métodos utilizando espectrômetros tem sido reportados
(ALI AHMED; ELBASHIR; ABOUL-ENEIN, 2015; KUKUSAMUDE et al., 2012). Além
disso, incluem-se vários métodos preparativos de amostras quando são analisados
multirresíduos utilizando UPLC-MS (CEPURNIEKS et al., 2015; CHIESA et al., 2015; HAN
et al., 2015). A aplicação de detectores de fluorescência é muito comum para determinação de
tetraciclinas (MAIA; RATH; REYES, 2008; NI; LI; KOKOT, 2011; PENNACCHIO et al.,
2016; SEIFRTOVÁ et al., 2009; SPISSO et al., 2007).
Alguns métodos de rastreabilidade tem sido adotados para detecção desses resíduos,
como os microbiológicos (NOUWS, 1999), imune ensaios (HORMAZÁBAL; YNDESTAD,
2001; THORSEN et al., 2006), e adicionalmente, os cromatográficos, especialmente com
detector UV (FJ; PS, 1998; HERRERA-HERRERA et al., 2011).

24
O presente trabalho tem por objetivo desenvolver metodologias válidas, com base nas
normas legais em vigor, para o controle de qualidade dos medicamentos de uso veterinário. Os
testes de identidade pela caracterização por espectroscopia na região do infravermelho médio.
Também, propor metodologias e modelos capazes de identificar e quantificar PENG utilizando
UFLC-DAD e espectroscopia na região do UV VIS NIR. Integrado a essas análises, desenvolver
métodos limpos e verdes para a extração, isolamento e boa recuperação da PENG. Além disso,
validar metodologia para a identificação e quantificação dos resíduos de PENG por UFLC-
DAD baseado no LMR de 4µg Kg-1 para leite bovino e caprino, e consequentemente testar os
métodos nas amostras de leite pasteurizado.

25
1.2 OBJETIVOS
1.2.1 Objetivos Gerais
Desenvolver métodos para quantificação de penicilinas contida em medicamentos
antimicrobianos, de uso veterinário, cuja composição esteja associada à Benzilpenicilina
Potássica (PENG K+), Benzilpenicilina Procaína (PENG PROC), Dihidroestreptomicina
(DHSTR), Estreptomicina (STR), Oxitetraciclina (OTC), Tetraciclina (TC), Enrofloxacina
(ENR), Sulfadoxina (SDX), Trimetoprina (TMP), Sulfametazina (SMZ) e Sulfato de
Gentamicina (GEN). Além de propor análises para identidade e controle de qualidade dos
antibióticos comercialmente distribuídos, todos registrados pelo MAPA. Entretanto, o principal
objetivo deste trabalho é desenvolver um método válido, conforme normas legais em vigor,
para identificar e quantificar resíduos de PENG em amostras de leite de origem bovina e
caprina. Além disso, são propostas condições ótimas, limpas e verdes para preparo de amostras
e leitura do resíduo de antibiótico sem prejuízo aos parâmetros de validação.
1.2.2 Objetivos Específicos
Realizar testes de qualidade dos medicamentos veterinários a base de Benzilpenicilina,
utilizando os métodos descritos nas Farmacopeias Brasileiras e Internacional.
Interpretar espectros dos medicamentos na região do infravermelho médio para
comprovar as principais bandas de absorção e vibrações características das moléculas em
estudo, comparando-as com os padrões registrados no banco de dados espectrais de compostos
orgânicos, bem como do padrão PENG certificado.
Desenvolver um método válido para identificação e quantificação desses terapêuticos,
utilizando análise cromatográfica em sistema UFLC-DAD. Aplicar os modelos em amostras
comerciais.
Desenvolver modelos multivariados utilizando espectroscópica na região do UV VIS
NIR para quantificação de PENG em medicamentos. Testar os modelos em amostras externas.
Validar método por cromatografia liquida de alta eficiência em sistema UFLC-DAD
para quantificação dos resíduos do antimicrobiano estabelecendo valores para seletividade,
robustez e sensibilidade. Considerando desde a metodologia de coleta e preparação das
amostras de leite caprino e bovino, bem como a metodologia para extração, preparo de soluções

26
das amostras, com redução significativa de reagentes e solventes tóxicos, mantendo a boa
recuperação do analito.
Aplicar o método válido para verificar a conformidade de algumas amostras de leite
bovino e caprino ultrapasteurizado quanto à presença de resíduos de Benzilpenicilina acima do
LMR de 0,004 µg mL-1. Testar as algumas amostras expostas ao consumo humano e
comercializadas em Natal/RN com origem em oito localidades do Brasil.

CAPÍTULO 2
ANTIBIÓTICOS: Penicilinas e Resíduos

28
2 ANTIBIÓTICOS: Penicilinas e Resíduos
2.1 DESCOBERTA DA PENICILINA
A introdução das sulfonamidas na década de 1930 e Benzilpenicilina na década de 1940
revolucionou completamente a medicina, através da redução da morbidade e mortalidade
provocadas por doenças infecciosas (HILDING, 1998). Os óbitos causados por infecções
durante a Primeira Guerra Mundial (1914 – 1918) impeliram Alexander Fleming,
bacteriologista em Londres, a descobrir substância capaz de matar ou inibir o crescimento de
bactérias (BERENDSEN et al., 2013; HARE, 1982; LIGON, 2004). O próprio Fleming relatou
no seu trabalho, em 1928, que enquanto estudava variantes de estafilococos, observou que uma
das culturas havia sido contaminada por um bolor, e que este provocara o desaparecimento das
bactérias na sua vizinhança (FLEMING, 1929). Fleming e seu colega, Dr. Pryce, identificaram
o fungo como sendo do género do Penicillium, e demonstraram que este produzia uma
substância responsável pelo efeito bactericida – a penicilina. Verificando o efeito desta
substância em diferentes culturas, foi possível constatar quais as espécies de bactérias sensíveis
à ação das penicilinas, que se provou serem apenas organismos aeróbios, gram-positivas não
produtoras de betalactamases (STEFFEE, 1992). Após a descoberta, Fleming, continuou a
estudar a penicilina em seu laboratório, mas não com aquele entusiasmo inicial. Ele nunca
desenvolveu um composto clinicamente útil, embora, em 1929, ele sugeriu que poderia ter
importantes aplicações clínicas, porque ele era um bacteriologista e não um químico. Logo,
Fleming não tentou purificar a penicilina. Foi Florey que desenvolveu a penicilina e a tornou
clinicamente aceita (ROSENTHAL, 1984). Estudos seguintes foram realizados para aplicações
clínicas (ABRAHAM; OXFD; INFIRMARY, 1941) e produção em larga escala.
Apesar de todo reconhecimento, alguns pesquisadores, como por exemplo, Ligon 2004
defendem a tese de que a história da função dos fungos e micologia, no desenvolvimento da
medicina tem sido bastante negligenciada. Logo, discute a história da micologia médica,
referindo-se a ao evento como uma “investigação fascinante”, pois os antibióticos são
disponíveis por causa do estudo dos micróbios (LIGON, 2004). Mas, pode-se afirmar que
Alexander Fleming passou um tempo considerável, durante os meados dos anos 1930,
investigando as infecções bacterianas e os micróbios.

29
A descoberta da penicilina, por Alexandre Fleming, ainda é tema de muitas discussões
no meio científico, por exemplo, Ali et al (2014) apresenta questões tal como: O que poderia
ter acontecido se Fleming não tivesse descoberto a penicilina? Chegando-se à conclusão de que:
o arsenal médico disponível, hoje, teria sido muito diferente e poderia ter se baseado,
unicamente, em variedades desenvolvidas de sulfonamidas ou agentes antibacterianos
sintéticos. Mas o fato é que, essa descoberta, conferiu a Fleming a oportunidade de salvar
milhões de vidas, ganhar um Prêmio Nobel em Fisiologia e tornar-se um dos mais honrados
cientistas da história (HARE, 1982, 1983; ROSENTHAL, 1984).
Quanto aos métodos de isolamento da penicilina a partir do fungo, não permitiram a
produção da penicilina em larga escala, pois a estrutura química não era conhecida, isso
inviabilizou a síntese. Nesta época Dorothy Hodgkin estava envolvida em projeto militar
desenvolvidos por químicos orgânicos renomados. Mas foi em 1941 que Hodgkin determinou
a estrutura da penicilina, utilizando cristalografia de Raios X (EDWALD, 2014; VARGAS,
2012).
Quanto à sulfonamidas, agentes antibacterianos relativamente eficazes, continuam a ser
utilizados na medicina moderna (BIAŁK-BIELIŃSKA et al., 2011; NEELAMKAVIL et al.,
2009). As primeiras sulfonamidas como medicamento foi utilizada durante meados de 1930, e
imediatamente teve um grande impacto no tratamento de infecções bacterianas, principalmente
septicemia e febre puerperal. Durante a idade de “ouro dos antibióticos” essa substância foi
usada mais amplamente. Sem dúvida, muito eficazes; estes compostos. Vale ressaltar que, um
grande compêndio do uso de antibióticos tem sido registrado para uso clínico das sulfonamidas.
Porém, a situação poderia ter sido diferente, se as sulfonamidas não tivessem sido
desenvolvidas, impediria o avanço dos antibióticos durante 1960, uma vez que a indústria
farmacêutica poderia ter argumentado que as sulfonamidas estavam desempenhando um bom
trabalho, logo havia pouca necessidade de se investir na extração de outros agentes
antibacterianos. No entanto, tinha sido feito um acordo para descobrir outros antibióticos e
desenvolvê-los no campo da biotecnologia. Se o compromisso não existisse, a indústria
farmacêutica não teria sido capaz de contar com a grande quantidade de financiamento do
Governo Britânico, principalmente pelo senso crítico de urgência em tempo de guerra, uma vez
que penicilina tinha sido descoberta em momento oportuno (HERSBACH, 1983).

30
Contrariamente, durante a Segunda Guerra Mundial, os alemães e seus parceiros
produziriam quantidades relativamente pequenas de penicilina, certamente nunca o suficiente
para satisfazer as suas necessidades militares; como resultado, eles tiveram que contar com as
sulfonamidas muito menos eficazes (WAINWRIGHT, 2004). Ainda durante Segunda Guerra
Mundial, o microbiologista russo naturalizado americano, Selman Waksman, vencedor do
Prêmio Nobel de Medicina de 1952, anunciou a descoberta da estreptomicina (WAKSMAN,
1952; ZUMLA; NAHID; COLE, 2013). A descoberta desse antibiótico levou à domesticação
da tuberculose. A estreptomicina foi igualmente eficaz no tratamento de várias outras doenças
infecciosas.

31
2.2 PENICILINAS: Grupos e Propriedades
Os Antibióticos são extremamente importantes para os seres humanos. Além disso, são
utilizados para sanidade animal, principalmente dos produtores de alimentos. Quimicamente,
são constituídos de vários grupos de diferentes funções orgânicas e atividades nos organismos.
No momento atual, estão identificadas quase 40 penicilinas, algumas são naturais, outras
semissintéticas. A composição da penicilina é constituída por um núcleo comum: o 6-APA
(Figura 2.1), que é responsável pela atividade dos compostos.
Figura 2.1-Estrutura geral das penicilinas, com seus diferentes R
OH
O
N
S
CH3
CH3
O
NHR
O
CH3
O CH3
CH3
NH2
CH3
O CH3
N
O
CH3CH3
R2
R1
Penicilina G
Penicilina V
Ampicilina (R = H)
Amoxicilina (R = OH) Nafcilina Oxacicilina (R1 = R2 = H)
Cloxacilina (R1 = Cl, R2 = H)
Dicloxacilina (R1 = R2 = Cl)
Ácido 6 - Aminopenicilínico (6-APA)
R
R
R
RR
Desenhada usando software ChemSketch ACD, 2015
Este núcleo é composto por anel tiazolidínico e por um anel betalactâmico, que está
ligado a um grupo amina iniciando a cadeia lateral nas penicilinas. A presença do grupo
carboxílico confere a todas as penicilinas – natureza ácida. Entretanto, as aminopenicilinas
possuem um grupo básico (amina), o que as torna moléculas anfotéricas. Eles são resistentes ao
ácido e pode ser administrada por via oral. Essas substâncias incluem ampicilina e amoxicilina.

32
As penicilinas podem ser classificadas em quatro grupos principais, com base na sua
capacidade bactericida.
A síntese de cada uma dessas penicilinas está relacionada com a cadeia lateral R, obtida
durante processos de fermentação. O radical R dá o nome da penicilina de acordo com a
apresentação contida na
, onde estão apresentados alguns exemplos. Além disso, a cadeia lateral determina as
características antibacterianas e farmacológicas de cada uma das substâncias.
A presença do anel betalactâmico é responsável pela designação geral destes antibióticos
e provém da ligação de dois aminoácidos: cisteína e valina (Figura 2.2).
Figura 2.2-Reação para formação do núcleo 6-APA
NH2CH3
O SH
+
CH3
OH
CH3
CH3
O
OH
O
N
S
CH3
CH3
O
NH2
Cisteína Valina
Desenhada usando software ChemSketch ACD, 2015
Quando as estruturas se unem, forma-se o núcleo bicíclico 6-APA (FREITAS;
BARBOSA; RAMOS, 2013). A Benzilpenicilina (penicilina G) foi o primeiro betalactâmico
purificado para utilização clínica a partir de culturas de Penicillium.
O sistema cíclico de quatro membros é muito reativo. A ligação da amida está sujeita a
uma forte tensão, que o torna sujeito ao ataque de reagentes nucleófilos, resultando a abertura
do anel (FREITAS et al., 2013).
Quanto à atividade antibacteriana, para que essa substância seja eficaz, é necessário que
seu grupo se mantenha estável. A ação metabólica ou ocorrência de reações nessa parte da
molécula ocasiona a perda da sua atividade.
Algumas penicilinas se hidrolisam com muita facilidade quando sofrem ataque de
ácidos, íons metálicos e bases, sendo desativadas principalmente sob a ação de enzimas
betalactamases, presentes naturalmente em microrganismos.

33
A acidez gástrica pode hidrolisar a amida da cadeia lateral e provocar a abertura do anel,
apresenta-se a degradação das penicilinas.
A Figura 2.3 mostra dois prováveis processos de hidrólise, no 1 ocorre a reação química
de quebra de ligação química da penicilina G em meio ácido, formando o ácido penicilóico,
este não apresenta atividade terapêutica, o que torna a administração por via oral inadequada.
O mesmo processo ocorre em 2, porém sob a ação de betalactamases.
Figura 2.3-Degradação da Penicilina com abertura do anel por ação de enzima em meio ácido (1) e hidrólise da
amina da cadeia lateral em meio ácido (2)
OH
O
N
S
CH3
CH3
O
NHR
O
OH
O
NH
S
CH3
CH3NHR
O
OH O
OH
O
NH
S
CH3
CH3NH2
OH O
+ R OH
1
2
Desenhada usando software ChemSketch ACD, 2015
Em contraponto, a penicilina V (fenoximetilpenicilina) apresenta uma maior resistência
ao ataque ácido, e pode ser administrada por via oral. Porém, a sua atividade é muito reduzida,
sendo apenas consideradas em bactérias gram-negativas.

34
A única penicilina natural é a penicilina G, mas a modificação de sua estrutura produziu
penicilina V. Esses antibióticos têm atividade semelhantes aos aminopenicilinas, mas tem maior
eficácia contra bactérias gram-negativas, e não estão incluídas na legislação relativa a produtos
veterinários (BABINGTON et al., 2012).
As penicilinas podem ser classificadas em seis grupos com base na sua atividade. A
PENG está apresentada na Figura 2.4, cujo registro no CAS é 113-98-4 e pKa [HA] é 2,7.
Figura 2.4-Estrutura Química da PENG
OH
O
N
S
CH3
CH3
O
NHO
Desenhada usando software ChemSketch ACD, 2015
A nomenclatura da PENG dada pela IUPAC é (2S,6R) -3,3-dimethyl-7-oxo-6-
[(phenylacetyl)amino]-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid. A fórmula
molecular é C16H18N2O4S. A massa molecular é 334, 390 g moL-1 quando isolada.
Quanto ao espectro, ele pode ser estendido, uma vez que apresentam um grupo ácido
carboxílico ou éster de cadeia lateral variável (SCHATZ, 1984).
A PENG é o antibiótico mais indicado para tratar infecções causadas por organismos
gram-positivos (exceto estafilococos resistentes e enterococos). Ela é administrada via
intramuscular, pois a sua absorção ocorre lentamente.
Quanto à ação nos organismos, as betalactâmicos são completamente ionizadas no
plasma. Essa substância não atravessa membranas biológicas, mas são amplamente distribuídas
nos fluidos extracelulares. A eliminação é geralmente através dos rins.
As penicilinas de largo espectro como a ampicilina, que é ativa contra bactérias gram-
negativas, também são amplamente utilizadas em gado para tratar infecções.

35
Quando em forma de sal, a Benzilpenicilina potássica (PENG K+), é descrita como um
pó cristalino; inodoro ou com fraco odor característico, muito solúvel em água; praticamente
insolúvel em éter R, dextrorotatória e apresenta absorção no UV de 200 a 220 nm (MACNEIL,
1998; THE INTERNATIONAL PHARMACOPOEIA, 2015a).
Para estudar os compostos, quanto a sua atividade, devem-se determinar os parâmetros
que conferem suas características, sendo a sua dissociação expressa pela Equação 1.
HA ⇆ H+ + A− (1)
Dentre as características, podem citar a constante ácida do composto (pKa) que é
representado pelo Equação 2.
Ka =
[H+][A−]
[HA]
(2)
O pKa é descrito pela a Equação 3 que melhor expressa o Ka.
pKa = − log Ka (3)
Por convenção, o ácido fraco com pKa baixo, será pouco ionizável, e uma base fraca
com pKa baixo será altamente ionizável no pH do sangue. Segundo a equação de Henderson-
Hasselbalch, o pKa corresponde ao valor de pH ocorre quando [HA] = [A-], ou quando a espécie
se encontra cinquenta porcento ionizada.
pKa = pH + log[HA]
[A−] (4)
Em termos biológicos o pKa é importante para determinar como a molécula se
comportará em meio aquoso nos componentes do tecido.

36
2.3 PRODUTOS VETERINÁRIOS: Designação e Faturamento.
O artigo 2º do Decreto 1.662/95 do MAPA define os produtos da indústria veterinária,
como toda substância química, biológica, biotecnológica ou preparação manufaturada, cuja
administração seja aplicada de forma individual ou coletiva, direta ou misturada com os
alimentos, destinada à prevenção, ao diagnóstico, à cura ou ao tratamento das doenças dos
animais, incluindo os aditivos, suplementos, melhoradores de produção animal, antissépticos,
desinfetantes de uso ambiental ou equipamentos, pesticidas e todos produtos que, utilizados nos
animais e/ou no seu habitat, protejam, restaurem ou modifiquem suas funções orgânicas e
fisiológicas (MAPA, 2012). Neste contexto, os produtos citados no Decreto e comercializados
estão listados no compêndio de produtos veterinários (CPV), incluindo antimicrobianos,
medicamentos antifúngicos 51 e antiprotozoários, sendo que 14 são formulações a base de
benzilpenicilina e associadas a misturas que contenham a estreptomicina.
O objetivo principal dos medicamentos veterinários é de melhorar a sanidade dos
animais produtores de alimentos controlando diversas doenças (BELOTI et al., 2012), dentre
elas estão as mastites que é uma inflamação e infecção da glândula mamária, em geral
provocada pela presença de microrganismos, promovendo alterações na composição do leite,
aumentando as taxas de células somáticas e danificando irreversivelmente o tecido secretor
(PYÖRÄLÄ, 2003). Dentre os agentes etiológicos identificados desta doença estão:
Staphylococcus coagulase positiva e negativa, Streptococcus spp, Escherichia coli,
Micrococcus spp e Pasteurella spp (ANTANAITIS et al., 2015; GREEN et al., 2002; HOGAN;
SMITH, 2012; LAGO et al., 2011; PYÖRÄLÄ, 2003; ROBERSON, 2012). Essa infecção gera
graves prejuízos econômicos, que podem provocar grandes perdas na atividade leiteira,
prejudicando a qualidade do leite, descarte deste alimento, diminuindo a produção, aumentando
custos com tratamento, assistência veterinária, aumento de mão-de-obra, e causando a
eliminação involuntária de animais no rebanho, (ANTANAITIS et al., 2015; ROBERSON;
WARNICK; MOORE, 2004; SÁNCHEZ; CONTRERAS; CORRALES, 1999), além de ser um
problema de saúde pública (GREEN, 2012; KROGH; ENEVOLDSEN, 2014), por isso é
imprescindível, o bom controle dessas infecções por tratamentos com antibióticos específicos
para cada agente infeccioso.
Foi somente em 1950 que um grupo de cientistas descobriu que as adições de
antibióticos ao tratamento de gado poderiam melhorar a produtividade pecuária, pois
controlavam as infecções bacterianas. Em 2001, Union of Concerned Scientists constatou que
90% da utilização total de antimicrobianos nos Estados Unidos foram para fins não terapêuticos
37
na produção agrícola (HARE, 1999). Desta forma, o uso de antibióticos na agricultura tem
alcançado picos de crescimento, estes fatores têm gerado grandes preocupações por parte das
regulações. Como resultado, as empresas farmacêuticas desenvolveram formulação de dose
baixa de produtos antibióticos que poderiam ser adicionados à ração diária.
As indicações dos produtos utilizados na década de 1960 eram principalmente para
controle de Mastites. Atualmente, os antibióticos mais utilizados são os betalactâmicos, por
exemplo, penicilinas ou cefalosporina, tetraciclinas, fluroquinolona, aminoglicosídeos,
macrolídeos e sulfonamidas. A maioria do tratamento de gado leiteiro é por infusão
intramamária de antibióticos quando o controle é de mastites.
Os medicamentos mais frequentes nos tratamentos de bovinos e caprinos para infecções
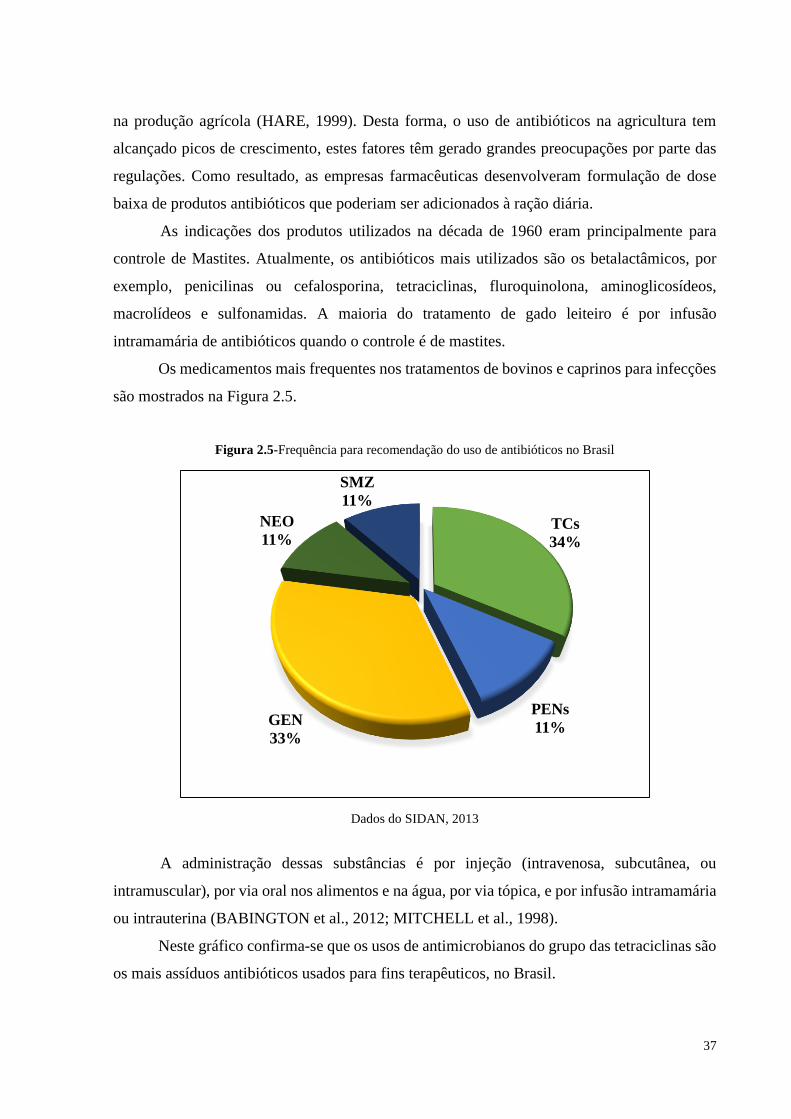
são mostrados na Figura 2.5.
Figura 2.5-Frequência para recomendação do uso de antibióticos no Brasil
Dados do SIDAN, 2013
A administração dessas substâncias é por injeção (intravenosa, subcutânea, ou
intramuscular), por via oral nos alimentos e na água, por via tópica, e por infusão intramamária
ou intrauterina (BABINGTON et al., 2012; MITCHELL et al., 1998).
Neste gráfico confirma-se que os usos de antimicrobianos do grupo das tetraciclinas são
os mais assíduos antibióticos usados para fins terapêuticos, no Brasil.
TCs
34%
PENs
11%GEN
33%
NEO
11%
SMZ
11%
38
É importante citar que, a indústria farmacêutica veterinária é idêntica à farmacêutica
humana. Muitas indústrias de produtos veterinários são fabricantes de medicamento para uso
humano. De maneira que, o crescimento da produtividade do mercado do leite, exige que
produtores usem uma criação seletiva para manutenção da saúde animal, uma vez que garantirá
a prospecção da qualidade da matéria-prima e uma possível aceitação por parte dos
consumidores. Neste ponto, o Brasil é um dos cinco maiores mercados veterinários em todo o
mundo. O setor vem apresentando crescimento, principalmente, ao aumento das exportações de
produtos veterinários, uma vez que o Brasil é um centro de produção importante para as
multinacionais. (SINDAN, 2014).
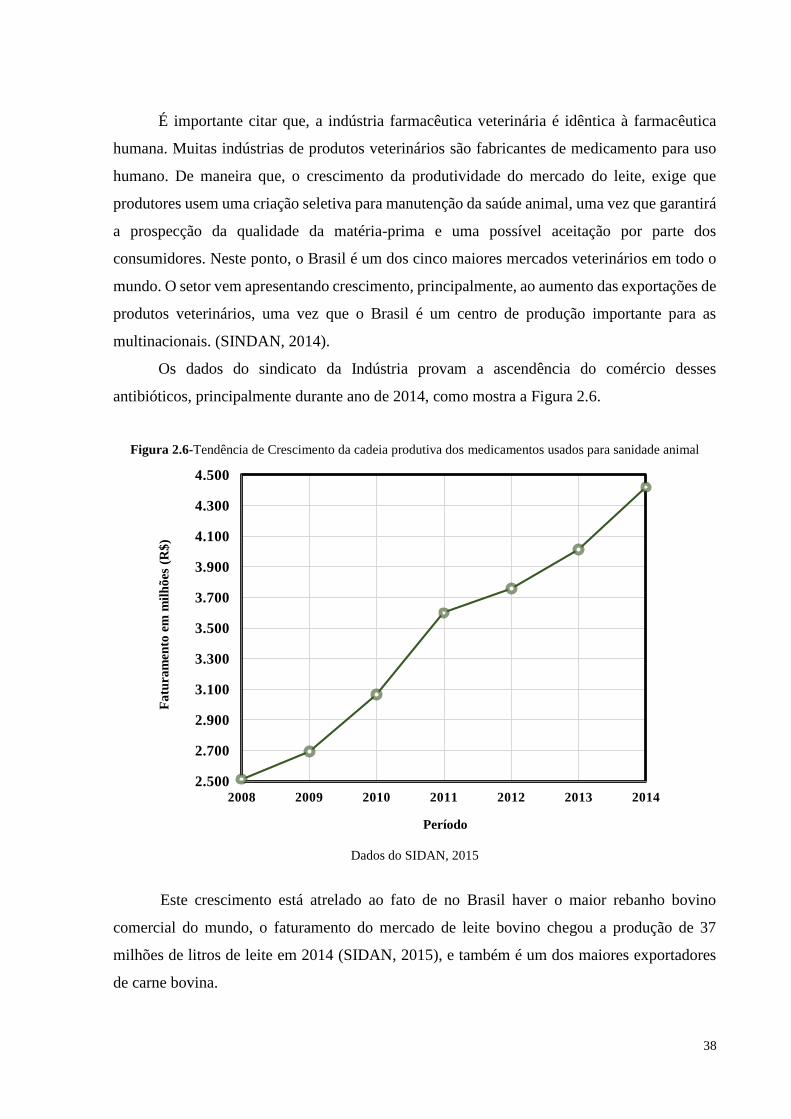
Os dados do sindicato da Indústria provam a ascendência do comércio desses
antibióticos, principalmente durante ano de 2014, como mostra a Figura 2.6.
Figura 2.6-Tendência de Crescimento da cadeia produtiva dos medicamentos usados para sanidade animal
Dados do SIDAN, 2015
Este crescimento está atrelado ao fato de no Brasil haver o maior rebanho bovino
comercial do mundo, o faturamento do mercado de leite bovino chegou a produção de 37
milhões de litros de leite em 2014 (SIDAN, 2015), e também é um dos maiores exportadores
de carne bovina.
2.500
2.700
2.900
3.100
3.300
3.500
3.700
3.900
4.100
4.300
4.500
2008 2009 2010 2011 2012 2013 2014
Fa
tura
men
to e
m m
ilh
ões
(R
$)
Período

39
A influência da produção de leite bovino no crescimento do mercado de antimicrobianos
é nítida, segundo indicadores do IBGE, o Estado do Rio Grande do Norte atingiu produção
67105 mil litros de leite industrializado e cru (IBGE, 2015) esses dados são do primeiro
trimestre de 2015.
O crescimento na produção da cadeia produtiva do leite exige que produtores usem uma
criação seletiva para manutenção da saúde animal, uma vez que garantirá a prospecção da
qualidade da matéria-prima e uma possível aceitação por parte dos consumidores. Esses fatores
contribuem para que os produtores estejam interessados em aumentar a produtividade,
acelerando o ganho de peso do animal e diminuindo o tempo de abate. Entretanto, para isso
acontecer o animal deve ser bem tratado, o que garante a sanidade do rebanho.
Além disso, um dos motivos para tal crescimento é a necessidade de prevenir o controle
da febre aftosa (CAPANEMA et al., 2007). Segundo o SINDAN, os principais clientes da
indústria veterinária são: Médicos Veterinários, Pecuaristas de Corte e Leite, Avicultores,
Suinocultores, Equinoculturas, Revendedores, e Produtores Rurais.
Essa participação de medicamentos para bovinos no faturamento da indústria veterinária
do país está muito associada ao tratamento de febre aftosa (SINDAN, 2014).
No âmbito internacional, atualmente, 100 mil substâncias químicas diferentes são
registadas na União Europeia (UE), dos quais 30 mil produtos são comercializados em
quantidades superiores a 1 tonelada. Entre eles, os compostos farmacêuticos tornaram-se uma
preocupação crescente nos últimos anos, uma vez que foram identificadas como contaminantes
ambientais emergentes (REEVES, 2011)

40
2.4 OCORRÊNCIA, RISCO E CONTROLE DOS RESÍDUOS: Segurança Alimentar.
O uso de substâncias com finalidades terapêuticas em animais produtores de alimentos
requer, de uma maneira permanente, investigação da persistência dos seus possíveis resíduos e
dos seus valores limitantes nos produtos, de forma que não constituam risco à saúde dos
consumidores (BERENDSEN et al., 2010; GILBERT; GARDNER; BIZEC, 2012; REGASSA
et al., 2015). Uma vez que, todas as vias de administração de antibióticos em animais produtores
de alimentos podem resultar no aparecimento destes resíduos nos alimentos, como por exemplo,
carne, ovos e leite.
A permanência dos resíduos no leite pode apresentar consequências danosas, desde a
intolerância aos antibióticos a interferências no processo industrial dos derivados do leite
(HARTLÉN, 1996; PAYNE et al., 2006). Para se ter ideia, em 1958 já se tinha registros de
consequências dos resíduos de penicilina, Vickers e Bagratuni (1958) relatam que a incidência
de sensibilidade a esses antibióticos chegava a atingir 5% da população mundial, sendo que de
12 a 15% apresentavam alergias múltiplas. Os pesquisadores argumentaram que os prejuízos
eram causados pela introdução da ordenha mecânica dos animais em estágio de lactação, pois
o tratamento com penicilina procaína era diretamente na glândula mamária, o resultado do
procedimento era: antibiótico no leite.
Todavia a grande problemática, de acordo com os pesquisadores Pengov e Kirbs (2009),
é que os veterinários da União Europeia estão autorizados a prescrever medicamentos, mas
depois não são obrigados a garantir que resíduos não entrem na cadeia alimentar. Logo, a
ausência de monitoramento e controle de qualidade desses fármacos resulta em altas
concentrações de resíduos no leite humano, que põe em perigo a saúde do ser humano (OLIVER
et al., 2004), principalmente quando a exposição é prolongada, pois podem induzir formação
de patógenos resistentes. (CHANG et al., 2014). A possibilidade de resistência aos antibióticos
pode ser evitada, se tomadas medidas necessárias para controlar as dosagens corretas do
medicamento. Considerando que, a contaminação direta, mais uma vez, pode ser causada pela
administração intramamária, intramuscular e intravenosa.
Contudo, para proteger os consumidores contra os riscos relacionados com os resíduos
de medicamentos, as regras legais, em muitos países, estabelecem limites de máximo de
resíduos (LMR) (COX; POPKEN, 2006; PERRON et al., 2015). Várias medidas são tomadas
a fim de proteger a saúde pública, logo as substâncias farmacologicamente ativas são
classificadas, com base na avaliação científica de sua segurança, na maioria dos países
europeus. Um exemplo são os betalactâmicos que são amplamente aplicados na agricultura,

41
sendo a principal razão de falhas, uma vez que são administrados para satisfazer requisitos de
controle leiteiro, quando são utilizadas como substâncias inibidoras, alguns estudos mostram a
ocorrência de resíduos dessas substâncias no leite (BABINGTON et al., 2012;
THAVARUNGKUL et al., 2007; YAMAKI et al., 2004). Assim, o aumento do uso de
antibióticos potentes ou em dosagens altas (uma prática ilegal na comunidade europeia), a
gestão agrícola é importante para ajudar a minimizar o uso de antimicrobianos, No entanto,
muitas vezes, é uma medida de precaução, não uma obrigatoriedade (AGENCY, 2013;
GOWIK, 2009).
Em 2013 nos Estados Unidos, o FDA emitiu restrições para uso de antibióticos, em
práticas agrícolas da região americana. Medidas para inspeções da conformidade, fiscalizações
e investigações criminais, como forma de controle de resíduos em alimentos de origem animal,
foram estabelecidas. O motivo pelo qual o FDA proibiu o uso de alguns antibióticos, é que
muitas dessas substâncias utilizadas em animais também são usadas para tratar os seres
humanos (USFDA, 2013). Logo a presença dos resíduos constitui-se um risco, pois não seria
possível, na rotina realizada pela legislação que protege a saúde do ser humano, controlá-los
(CHANG et al., 2014).
Para promover o crescimento ou para ajudar o produtor de forma eficiente na produção
de alimentos, alguns antimicrobianos são usados em baixas doses, sendo empregados,
rotineiramente, na ração ou na água de aves, suínos e bovinos, o objetivo é de convertê-los
principalmente em ganho de peso. Segundo Nisha (2008), as utilizações de aditivos alimentares
podem melhorar a eficiência alimentar de 17% em bovinos de corte, 10% em cordeiros, 15%
em aves e 15% em suínos.
De acordo com o Instituto de Saúde Animal, esses eventos podem estar associados ao
aumento substancial dos medicamentos na prática agrícola, contribuindo com cerca de 30% a
70% do peso do animal (KUEHN, 2010). Ainda, de acordo, com o órgão americano, entre os
antimicrobianos utilizados para promoção de crescimento, alguns apresentam maior uso que as
penicilinas e tetraciclinas. Nos Estados Membros da AMDUCA é proibido o uso de
cloranfenicol, furazolidona, sulfonamidas (sulfadimetoxina, sulfabromometazina e
sulfametoxipiridazina) em gado leiteiro no período de lactação e de quinolonas e glicopeptídeos
em animais produtores de alimentos. As inquietações são constantes, pois na maioria das vezes,
essa utilização terapêutica não é supervisionada por um médico veterinário, e sim esses
produtos são comprados livremente, no balcão pelos produtores. Muito embora, o uso

42
terapêutico de um número muito pequeno de antimicrobianos tem sido proibido, devido
principalmente às preocupações com a saúde pública.
Outras nações já proíbem algumas práticas para administração de antibióticos em
produtores de alimentos. A Dinamarca proibiu o uso de antimicrobianos em suínos e frangos
no final de 1990, a União Europeia proibiu o uso de algumas substâncias para promover o
crescimento do gado em 2006, uma delas é o cloranfenicol. No Canadá, este fenicol, seus sais,
derivados, bem como os compostos de nitrofuranos, foi coibida a venda. Na Austrália, o uso de
cloranfenicol, nitrofuranos (incluindo furazolidona e nitrofurazona), Fluroquinolones,
gentamicina, estreptomicina, sulfonamidas e vários não é permitida. De igual modo, o Reino
Unido vetou o uso de penicilina e tetraciclinas para a promoção do crescimento no início de
1970; outros países europeus seguiram o exemplo logo depois, tal como a Suécia que impediu
a utilização de todos os antibióticos para promover o crescimento, em 1986 (BARRETO et al.,
2012; HEALTH CANADA, 2012; KUEHN, 2010; REEVES, 2011; REZK et al., 2015;
VARGAS MAMANI; REYES REYES; RATH, 2009; VERA-CANDIOTI; OLIVIERI;
GOICOECHEA, 2010), Enquanto isso, no Brasil, o cloranfenicol tem sido banido (MAPA,
2003; REZENDE et al., 2012; REZENDE; FILHO; ROCHA, 2012). Todas essas restrições são
baseadas em ocorrências durante períodos de produção de alimentos, por exemplo, segundo
dados da prevalência de resíduos de antibióticos em vacas leiteiras de 2013 a 2014, nos Estados
Unidos, foram registrados 24,3% de permanência de penicilina em leite, e 10,02% de
sulfonamidas, esses dados são oficiais do Relatório de Programas Nacionais Trimestrais (FSIS,
2011). Consequentemente, as medidas tomadas para redução do uso de agentes antimicrobianos
na agricultura resultaram na supressão de alguns produtos.
Diante deste quadro e apesar do setor de produtos veterinários apresentarem ascenção
econômica (comprovado pelo item 2.3 deste Capítulo), afirma-se que os riscos do inadequado
uso de antibióticos em gado leiteiro, para prevenir e tratar infecções microbianas como mastite,
recebeu uma maior atenção por parte da saúde pública da maioria dos países emergentes, pois
é nitida a consciência de que essas substâncias podem acarretar na presença de resíduos e
metabólitos nos gêneros alimentícios, causando uma série de complicações, como infecções
difíceis de tratar, reações alérgicas nos seres humanos, pois são tóxicos pela presença de
bactérias resistentes aos antibióticos, podendo atingir o meio ambiente direta ou indiretamente
(BERENDSEN et al., 2010; BOHM; STACHEL; GOWIK, 2009; CHEN, G. L., AND FANG,
2011; KABIR et al., 2004; LOPEZ et al., 2008; OLIVER et al., 2004; SALMOND; WELCH,
2008; SAMANIDOU; NISYRIOU, 2008; SHERIDAN et al., 2008; WEBER et al., 2005).

43
Ainda há relatos que agentes antimicrobianos, na alimentação independentemente da
dosagem, podem ser potencialmente cancerígenos (KOESUKWIWAT, 2007b). No Brasil, para
garantir a segurança alimentar humana, o MAPA limita a quantidade de antibióticos no leite,
através da Portaria Ministerial nº 574, de 06 de dezembro de 1998 e Instrução Normativa nº 8,
de 29 de abril de 2010 e a de nº 42, de 20 de dezembro de 1999 que tratam do Plano Nacional
de Controle de Resíduos em Produtos de Origem Animal (PNCRC) que consiste em
monitoramento constante visando evitar violações dos limites máximos (LMR) destas
substâncias (MAPA, 1999; MAPA 2010). Essa limitação por LMR depende do composto e da
matriz afetada. De igual modo, no âmbito internacional, as legislações, como Codex
Alimentarius, EC (European Commission), FDA (Food and Drug Administration) também
limitam a quantidade de antibióticos no leite e adotam métodos que garantam a disponibilidade
de um alimento seguro.
O limite máximo de resíduos e contaminantes em alimentos é estabelecido com base na
análise toxicológica e farmacocinética da substância química, ou seja, do risco ou perigo que
ela possa representar para a saúde humana (ALIMENTARIUS, 2006; VICH, 2011) e é
calculado pela Equação 5. Esse risco toxicológico de resíduos, leva em consideração, a
probabilidade do aparecimento de um efeito adverso e a sua magnitude, que é calculado através
da IDA (mg Kg-1) do medicamento presente no produto de origem animal, ao qual se incorpora
um fator de segurança da ordem de até 100 vezes. Este fator é acionado visando atender às
demandas geradas pela variação individual de sensibilidade do consumidor às substâncias
químicas (idade, sexo, raça, estado de higidez, etc.).
LMR =IDA
FS
(5)
A IDA, por sua vez, é calculada a partir do valor NOEL de uma substância química, ao
qual se incorpora, também, um fator de segurança da ordem de 100 expresso pela Equação 6.
Entende-se por NOEL a maior dose de uma substância química que, se usada, não produz
efeitos adversos (FAO, 2013).
IDA =
NOEL P (60Kg)
FS (6)

44
Onde IDA é o valor de ingestão diária, FS é o fator de segurança e NOEL é o efeito não
observado. Sendo que, os valores deste último, advêm da avaliação crítica e sistemática da
literatura científica mundial a respeito da toxicidade das substâncias químicas. São, assim,
analisados considerando a oncogenicidade, genotoxicidade, teratogenicidade, toxicidade
reprodutiva, fetotoxicidade, toxicidade sistêmica aguda, prolongada e crônica (hepática, renal,
cardíaca, muscular, etc.) e os que promovem alergias. Em casos específicos dos
antimicrobianos, são exigidos ainda alguns ensaios ditos especiais e ligados a possíveis efeitos
dos mesmos sobre a microflora do trato gastrintestinal humano (FAO, 2013).
De maneira que, ao fixar-se o LMR, levam-se em conta riscos importantes das
substâncias químicas para a saúde pública, como, por exemplo, aqueles ligados à
oncogenicidade ou genotoxicidade, sendo que o limite de substâncias que alteram o material
genético deve ser zero, isto é, resíduos destes compostos não devem ser encontrados em
alimentos. No caso de antimicrobianos destinados à produção animal, exigem-se ainda para
análise de risco a realização de alguns testes ditos especiais. Assim, além do cálculo da IDA,
embasada em análises de toxicidade (como especificado acima), é também mandatário que se
busquem por possíveis efeitos destes agentes sobre a microflora do trato gastrintestinal humano;
mais que isto, exige-se a determinação da dose a partir da qual este antimicrobiano teria
condições de selecionar linhagens resistentes de bactérias.
Para a análise dos LMR de um antimicrobiano destinado à produção animal são
considerados dois valores de IDA: um dito toxicológico e derivado de cálculos de toxicidade e
outro, dito microbiológico (EMEA, 2002). Para fixar os valores desse limite máximo, a
Comissão do Codex Alimentarius analisa detalhadamente os níveis residuais do mesmo na
carcaça dos animais tratados, conforme a posologia recomendada para a medicação. Para esta
finalidade usam-se, geralmente, medicamentos marcados radiotivamente, medindo-se as
concentrações tissulares dos mesmos (músculo, gordura, fígado, rins e ovo ou leite) horas após
a interrupção do tratamento. Estima-se a quantidade real de resíduos de medicamentos
veterinários que pode ser ingerida pelo ser humano, antes de fixar os LMR (CODEX
ALIMENTARIUS, 2001). A União Europeia usa o mesmo tipo de procedimento para fixar
períodos de carência dos produtos destinados aos animais de produção. Assim, conhecendo-se
a quantidade real de resíduos torna-se possível deliberar sobre os períodos de carência para o
medicamento em análise (CHRISTODOULOU; SAMANIDOU, 2007; KANTIANI et al.,
2009; KARAGEORGOU; SAMANIDOU, 2011; PRUDEN, 2009; RAMBLA-ALEGRE et al.,
2011a; THORSEN et al., 2006).

45
Obviamente que, o estabelecimento de LMR e de períodos de carência para os
antimicrobianos não assegura um “risco zero” para os mesmos. Até porque “risco zero não é
risco, é certeza e certeza em ciência não existe”. No entanto, o uso de medidas conservadoras
como, por exemplo, o estabelecimento da IDA e LMR diminui consideravelmente a
possibilidade de que efeitos adversos venham a ocorrer em consumidores que ingerem
concentrações residuais de antimicrobianos nos gêneros de origem animal, ou seja, as
concentrações devem estar abaixo dos LMR fixados para os alimentos (NATIONS;
ORGANIZATON, 2000).
O intervalo de segurança é o tempo necessário para que o resíduo de risco toxicológico
atinja uma concentração segura, tal como definido pela tolerância, ou seja, é o intervalo de
tempo que o medicamento seja eliminado do organismo do animal, até que o momento do abate
ou ordenha seja permitido (a responsabilidade de executar essa medida é do produtor). Para tal,
a OMS estabelece a tolerância como sendo a concentração para que o a substância química
permaneça o tecido, até que a parte comestível seja considerada segura para consumo humano.
Logo, as tolerâncias são fixadas com base em extensos estudos toxicológicos de perigos
potenciais desse consumo (FAO/WHO, 1968; JOHNSTON, 1992; NISHA, 2008; WHO, 2006).
Desta forma, considera-se a detecção de níveis baixos de resíduos no leite, como sendo de
grande importância para a indústria, e para os agricultores; com a responsabilidade de garantir
que o leite contaminado não é expelido no meio ambiente (CODEX ALIMENTARIUS, 2009;
KANG’ETHE et al., 2005; KNECHT et al., 2004; NICOLICH; WERNECK-BARROSO;
MARQUES, 2006).
É importante citar que mesmo em baixas doses, mas por longos períodos, os antibióticos,
podem resultar em resistentes a microrganismos por isso que, nos Estados Unidos, o FDA e
órgãos estaduais monitorizam leite e outros alimentos para resíduos de drogas. Os antibióticos
aprovados pela FDA para o tratamento intramamária da mastite em vacas leiteiras estão listados
na Tabela 2.4, incluindo seus limites aos máximos permitidos.
No Brasil o PNCRC (já citado) visa garantir a saúde do consumidor, por meio de um
monitoramento da presença de resíduos de medicamentos veterinários e contaminantes
ambientais, em produtos de origem animal (carnes, leite, pescado e mel).

46
Um dos principais objetivos do monitoramento do MAPA é integrar o esforço destinado
à melhoria da produtividade e da qualidade dos alimentos e, secundariamente, proporcionar ao
país condições de se adequar, do ponto de vista sanitário, às regras do comércio internacional
de alimentos, preconizadas pela Organização Mundial do Comércio (OMC) e outros órgãos,
como FAO e Organização Mundial da Saúde (OMS) (MINISTÉRIO DA AGRICULTURA
PECUÁRIA E ABASTECIMENTO, 2007, 2011). Na Tabela 2.1 é apresentado um quadro
comparativo dos valores máximos de resíduos, baseados nos organismos legais, tais dados são
recomendados pelo MAPA e ANVISA. Os compostos analisados são apresentados.
Tabela 2.1- Correlação entre os LMRs admitidos em principais subprogramas de monitoramento e Controle
dos Resíduos de Antibióticos em Leite
Antibióticos LMR (µg Kg-1)
MAPA/PNCRC PamVet Codex FDA/FAO/JECFA EC/EMEA Japan
ABAM 10 NE NE NE NE 20
IVERM 10 NE 10 NE 15 10
PENG e PENV 4 4 4 4 4 4
CTO NE NE 100 100 NE 100
AMP 4 4 NE NE 4 20
TC 100 100 100 100 100 NE
OTC 100 100 100 100 100 NE
CTC 100 100 100 100 100 NE
DOX 100 NE 100 100 100 NE
SDX 100 NE 25 NE 100 60
SMZ 100 100 NE NE 100 NE
TMP 50 100 NE NE 50 50
STR NE NE 200 200 200 200
DHSTR NE 100 200 200 200 200
NEO NE 500 1500 1500 (bovinos) 1500 500
GEN NE NE 200 200 (bovinos) 100 200
ENR 100 40 NE NE 100 50
CLF 0,30 0 NE NE 10 NE
NE: não estabelecido neste no período correspondente
O Codex Alimentarius estabelece o LMR para leite segundo as espécies produtoras,
neste caso levou-se em consideração as de bovinos e caprinos. A comissão europeia também
utiliza o mesmo critério, porém a cada período é estabelecido limite de acordo com a ocorrência
de determinado resíduo. Esses limites são preconizados conforme a exposição de cada
população aos contaminantes, sendo cada país responsável por executar e elaborar os
subprogramas para monitoramento. Os valores não estabelecidos referem-se a dados que não
foram possíveis obter com os cálculos da IDA, por exemplo, o JECFA/FDA argumenta que não
obteve informações necessárias para avaliar a carcinogenicidade do cloranfenicol, bem como
os efeitos sobre a reprodução, principalmente porque o composto é genotóxico, assim não foram

47
obtidos valores de IDA e consequentemente do limite máximo. Apesar dessas dificuldades os
valores LMR, dentro dos subprogramas de monitoramento de resíduos, são renovados, a
exemplo do PamVet/ANVISA que teve sua última atualização em 2006.
Neste contexto, um desafio enfrentado pelos programas de monitoramento é de controlar
um número elevado de amostras de leite quanto à presença dos mais importantes resíduos dos
antimicrobianos, usando um método rápido e confiável, dentre eles os mais utilizados, para
rastreio, são os microbiológicos e imunológicos. Já os métodos cromatográficos são destinados
à determinação quantitativa de fármacos e/ou metabólitos nessas matrizes biológicas, sendo
classificados como métodos Bioanalíticos (ANVISA, 2003).
Algumas aplicações são concernentes à cromatografia liquida (BALIZS; HEWITT,
2003; RAMBLA-ALEGRE et al., 2011a, 2011b; RIEDIKER; DISERENS; STADLER, 2001;
STOLKER; BRINKMAN, 2005), eletroforese capilar (LARA et al., 2006), cromatografia
liquida acoplada a arranjo de fotodiodos UV, (BAILÓN-PÉREZ et al., 2009;
CHRISTODOULOU; SAMANIDOU, 2007), detecção fluorimétrica (RODRÍGUEZ-DÍAZ et
al., 2006) eletroquimioluminescência (GUO; GAI, 2011) e métodos espectroscópicos (NOUR
EL-DIEN et al., 2010) que vêm sendo mais recomendados para detecção simultânea de
antibióticos no leite. Todos os métodos, citados acima, são validados para que tenha a finalidade
de detectar os compostos, abaixo dos seus limites admissíveis, e avaliados de acordo com a
Diretiva do Conselho 2002/657/CE (EUROPEAN COMISSION, 1993).
O funcionamento desses programas é um desafio para muitas nações (GORRIS, 2005;
JACXSENS et al., 2010; KANG’ETHE et al., 2005; KUSSAGA et al., 2014; MA et al., 2012;
RAKOTOHARINOME et al., 2013; REDDING et al., 2014). Kang´ethe et al (2005) mostraram
os riscos de consumir leite comercializado contendo resíduos de antimicrobianos, para isso
foram realizados testes em 110 amostras de leite pasteurizados e 854 não pasteurizado, os
pesquisadores detectaram 16% das amostras continham resíduos de antibióticos em leite que
estavam sendo comercializado no sul do Quênia no continente africano, foi relatado um índice
de exposição até cinco vezes maior que o normal durante um mês.
Neste Contexto, é muito comum, em países pobres e tropicais, a ocorrência de resíduos
de antibióticos em alimentos, pois os respectivos orgãos legisladores, na maioria das vezes,
desconhecem a extensão de tais problemas, além de não haver programas para controlar a
qualidade dos alimentos. Além disso, pode acontecer de não haver proibição da venda desses
produtos contaminados. Desta forma, a ocorrência dos resíduos de antimicrobianos pode ser
atribuída a não observância dos períodos para sua eliminação, antes da venda dos alimentos.

CAPÍTULO 3
MÉTODOS OFICIAIS E INDICADORES DOS AVANÇOS DA
LITERATURA CIENTÍFICA

49
3 MÉTODOS OFICIAIS E INDICADORES DOS AVANÇOS DA LITERATURA
CIENTÍFICA
3.1 REVISÃO DOS MÉTODOS APLICADOS AO CONTROLE DE QUALIDADE DE
TERAPÊUTICOS PARA USO VETERINÁRIO.
Para que um medicamento veterinário venha ser distribuído, este deve obedecer alguns
requisitos de qualidade, isto é, os produtores e fornecedores devem garantir dados legítimos,
que compreendem a veracidade das propriedades físico-químicas, principalmente quanto a
descrição física, tais como: a solubilidade em solventes comuns (água, álcoois, clorofórmio,
acetona,), ao pH em meio aquoso (pH 1 a 8, a dose / volume de solubilidade), a distribuição de
tamanho de partícula, aos valores de pKa, as máximas absorção no ultravioleta (UV), a
absortividade molar, ao ponto de fusão, ao índice de refração (para um líquido) e ao coeficiente
de partição (FINGLETON, 2004; WETZSTEIN, 2000). Desta forma, a natureza química dos
medicamentos é descrita, pois contêm parâmetros que determinam o seu comportamento
farmacocinético (REEVES, 2011). Outra razão para tal averiguação deve-se ao fato de que, os
fármacos podem absorver impurezas, nas várias etapas do seu desenvolvimento, principalmente
no transporte e armazenamento. Esta influência pode causar riscos a sanidade animal e gerar
resíduos, nos produtos da cadeia produtiva (SIDDIQUI; ALOTHMAN; RAHMAN, 2013).
Por estas razões, os métodos desenvolvidos, neste trabalho, desempenham uma função
importante na averiguação da identidade dos antibióticos, pois as informações conferem a
certeza que esses produtos são eficazes (LIU; HU, 2011; PENNACCHIO et al., 2016;
WONGTANGPRASERT; PALAGA; KOMOLPIS, 2011; WUETHRICH; HADDAD;
QUIRINO, 2015).
As normas seguidas para qualidade das formulações injetáveis dos antibióticos
consistem de: medidas de pH, rotação óptica, absorção em ultravioleta visível e análise dos
espectros no infravermelho (VICH GL39, 2006). Os procedimentos para avaliação são
embasados nas normas 01/2008: 50300 e 2009/9/EC (EUROPEAN PHARMACOPOEIA
COMMISSION, 2008), além dos procedimentos oficiais descritas em Farmacopeia Japonesa
(JP XVI, 2007). Esta Farmacopeia tem sido uma das mais completas e versáteis, em termos de
aplicação dos métodos aos antibióticos, aceitas pela comunidade internacional. As prescrições
contidas nas Farmacopeias Brasileiras são respeitadas, a fim de garantir a confiabilidade dos
métodos desenvolvidos para amostras do Brasil (BRASIL, 2010).
Este trabalho considera a denominação pela União Internacional de Química Pura e
Aplicada (IUPAC), Chemical Abstracts Service (CAS), isto é, tem-se atenção à classificação

50
terapêutica, farmacológica, e a identidade quanto à fórmula estrutural, molecular, peso
molecular, grau de impureza, composição qualitativa e quantitativa, ponto de fusão, ponto de
ebulição, pressão de vapor, solubilidade na água e em solventes orgânicos a temperatura
determinada, densidade, rotação óptica, índice de refração e formulação do produto são
importantes como descreve Lawrence (2008) em seu trabalho.
A quantificação de resíduos de PENG em amostras complexas consiste em uma tarefa
difícil, pois se exige, sempre, um método válido que seja eficaz e robusto, porque as moléculas
de betalactâmicos, como é o caso da penicilina, apresentam natureza polar, anfotérica e são
suscetíveis às degradações (CASTRO-PUYANA; CREGO; MARINA, 2010; CHEN et al.,
2008; NOVÁKOVÁ; VLČKOVÁ, 2009; PÉREZ; RODRÍGUEZ; CRUCES-BLANCO, 2007).
Para minimizar esses efeitos, foram realizadas análises de identidade dos grupos de antibióticos,
que podem gerar resíduos em quantidades acima do LMR no leite.
Os valores de pKa são utilizados para delinear as propriedades físico-químicas dos
fármacos, pois o grau de ionização destes dissolvidos, está relacionado com seu valor intrínseco,
e a partir do pH da solução são planejadas as principais condições experimentais. Logo, o grau
de ionização para um fármaco controla a solubilidade, taxa de dissolução, cinéticas de reação,
complexação e as ligações com as proteínas (PRANKERD, 2007; REEVES, 2011).
As autoridades reguladoras exigem que sejam realizados testes de risco para novos
medicamentos e agentes antimicrobianos (produtos formulados) antes da autorização para
comercialização (KORAKIANITI; REKKAS, 2011; MANDENIUS et al., 2009; WHO, 2012).
Além disso, são realizados exames laboratoriais, que asseguram o tempo de prateleira, além da
verificação do tempo de armazenamento e condições de transporte adequadas (BARBER,
2005).
Essa qualidade é reafirmada pelas atividades técnicas e gerência do sistema de
padronização, que inclui a avaliação de produto, documentação até a qualidade do laboratório
realizador das medições (BAERT; DE SPIEGELEER, 2010; FENT; WESTON; CAMINADA,
2006; ORGANIZATON, 2007). Portanto, a finalidade da garantia da qualidade no apoio
farmacêutico é ajudar a certificar que o medicamento seja seguro, eficaz e adequado para o
“paciente”.

51
Alguns trabalhos mostram a importância do controle de qualidade para segurança e
eficácia dos produtos, um exemplo é o trabalho de Mazumder, Bhattacharya e Yadav (2011)
que oferecem uma ampla visão do conceito de gestão na indústria que leva à melhoria dos
atributos desses produtos.
A avaliação do risco considera o processo de fabricação (os perfis toxicológicos das
impurezas resultantes da síntese são de interesse particular), pureza e composição para garantir
a conformidade e o padrão de qualidade. Segundo os métodos descritos pela Farmacopeia
Japonesa (JP XVI, 2007), a composição do medicamento deve ser regida pela pureza mínima
do ingrediente ativo, a proporção de isômeros e de diastereisômeros (se for o caso), e a
concentração máxima de impurezas, incluindo aqueles de preocupação toxicológica.
A avaliação do risco global, conduzida pelas autoridades reguladoras, garante que
drogas antimicrobianas de várias naturezas, e para diferentes lotes de mesma manufatura, têm
perfis que são aceitáveis em termos de eficácia e segurança para sanidade animal, e
consequentemente a saúde pública e ambiental (ALTUNAIJI et al., 2012; DIOP et al., 2009;
EUROPEAN COMMISSION 37/2010/EC, 2009; HALEEM et al., 2014; JP XVI, 2007;
MANN, 2005; MONTGOMERY, 2009; NICOLAS, 2007; ZUCCATO et al., 2006).
Sabendo que, o progresso científico levou ao desenvolvimento de numerosos compostos
sintéticos, é imperativo desenvolver métodos analíticos para determinar essas substâncias tanto
na fase manufatura, e principalmente controlar de qualidade das formulações farmacêuticas e
sua determinação nos organismos. Para este fim, os métodos citados neste trabalho fornecem
uma boa estratégia para garantia da correta dosagem e avaliação da estabilidade dos
medicamentos.
As necessidades do controle de qualidade exigem também que as técnicas empregadas
sejam relativamente rápidas, a fim de minimizar o tempo de inatividade do produto. Como
consequência novos métodos têm sido desenvolvidos para quantificação de antibióticos em
medicamentos, um exemplo é o trabalho de Tan et al (2013) que aplicam um bioensaio
quantitativo para eritromicina em amostras obtidos por fermentação (Saccharopolyspora
erythraea). Os autores usaram como métodos de referência o HPLC e colorimétrico e a fim de
avaliar a exatidão dos métodos. Os resultados indicaram que o método é adequado, pois é
comparável aos resultados dos tradicionais. Segundo os autores foram alcançados um alto
rendimento para os compostos isolados, além disso, o custo foi bem reduzido e aplicável à
análise geral dos antibióticos.

52
Neste contexto, novas técnicas como a espectrometria de absorção atômica, eletroforese,
espectrofluorimetria, cromatografia líquida, espectrometria de massas, espectrometria de
luminescência, voltametria e polarografia têm sido utilizados para a determinação de compostos
farmacêuticos (DA SILVA et al., 2013; GUMUS; FILIK; DEMIRATA, 2005;
OZKORUCUKLU; SAHIN; ALSANCAK, 2011). A respeito disso, Siddiqui, Alothman e
Rahman (2013) destacam uma variedade de técnicas analíticas utilizadas na análise desses
produtos farmacêuticos.
Porém, algumas questões têm sido levantadas sobre tais métodos, em 2005, Görög
(2005) faz uma comparação entre os métodos das farmacopeias europeias e americanas,
avaliando-os de forma crítica. Os autores indagam o que justifica o crescente aumento dos testes
de impureza e dos métodos de ensaio na caracterização da qualidade de um fármaco. Os
pesquisadores discutem a maneira com que são realizados os cálculos dos teores de princípio
ativo a partir dos resultados dos ensaios.
Mas o fato é que os métodos cromatográficos têm sido amplamente utilizados e
recomendados. No entanto, estas técnicas necessitam de equipamentos complexos e de alto
custo, utilização de solventes, preparo de amostras, procedimentos intensivos e trabalhosos.
Apesar disso, a eficiência destas técnicas de separação depende, exclusivamente, do bom
planejamento das condições para identificação e determinação, usando cromatografia, uma vez
que são métodos oficiais de grande aceitação.
Aliado a crescente evolução das novas práticas, os métodos espectroscópicos têm sido
muito reportados para determinação simultânea dos compostos ativos. A aplicação desta técnica
é considerada uma ferramenta capaz de realizar análise quantitativa de misturas complexas de
forma precisa e exata, porém esse bom desempenho depende de um método de referência bem
consolidado (GERVAIN et al., 2011; KHOPTYAR et al., 2013; VETUSCHI et al., 2005).
Há de se falar que nas últimas décadas, a espectroscopia ganhou rapidamente aplicação
no domínio da análise farmacêutica, para superar o problema de interferência, devido a
substâncias que não são compostos de interesse, geralmente presentes nos fármacos
(KAWATA, 2007). Esta técnica associada à regressão multivariada e processamento de dados
tem recebido atenção, pelas indústrias de medicamentos, a fim de realizar medidas rápidas, sem
consumo de reagentes e principalmente diminuindo custos com mão de obra. (ALVARENGA
et al., 2008; BLANCO et al., 2006; CLARKE, 2004; KREMER, 2002; MARK, 1989;
STORME-PARIS et al., 2010; XIE et al., 2010).

53
3.1.1 Determinação da rotação óptica de um antibiótico
A Polarimetria consiste em um dos métodos para averiguação da qualidade dos
medicamentos (BRITISH PHARMACOPOEIA, 1983; JP XVI, 2007). A pureza óptica do
fármaco é determinada pela medição da rotação especifica [𝛼]𝐷200
e subsequente comparação com
os valores de referência, uma vez que cada composto, biologicamente, ativo tem sua quiralidade
(GILPIN; GILPIN, 2011). Essas avaliações são baseadas no pH, concentração da solução (g
mL-1), Temperatura (ºC) e absorção no ultravioleta visível (nm).
As propriedades dos antibióticos, enantiômeros tais como: ponto de fusão e índice de
refração é semelhante, mas difere na atividade óptica, esta apresenta capacidade de girar o plano
de polarização, quando é atravessado por um feixe de luz polarizada, evidencia a atividade do
fármaco, por isso tem-se executado esta medida (ISHII et al., 2008).
De acordo com método 01/2008: 20207 descrita na Farmacopeia Europeia, a rotação
óptica é a propriedade exibida por substâncias quirais, capazes de girar o plano de polarização
da luz polarizada. Substâncias (+) apresentam efeito dextrorotatoria, no qual o plano é girado
no sentido horário, enquanto que compostos (-) apresentam efeito levogiro no qual o plano de
luz polarizada é girado no senti anti-horário. A detecção é baseada na interação entre um centro
quiral no analito, e o incidência da radiação eletromagnética magnética polarizada
(EUROPEAN PHARMACOPOEIA COMMISSION, 2008).
Por convenção a rotação óptica para um líquido é descrita pelas Equação 7.
[∝𝑚]𝜆𝑡 = 0,1745 [∝]𝜆
𝑡 (7)
Onde: α é o ângulo de rotação, quando a 20 ºC, pode ser expressa pela Equação 8.
[∝𝑚]𝐷20 =
∝
𝑙. 𝑝20
(8)
Onde: l é a comprimento do tubo, p20 é a densidade do liquido. Para cálculo com
concentração de solução a Equação 9 é considerada.
[∝𝑚]𝐷20 =
1000 ∝
𝑙. 𝑐 (9)

54
De modo que a rotação óptica específica de um líquido é o ângulo α de rotação, expresso
em graus (°), do plano de polarização no comprimento de onda da linha D do sódio (λ = 589,3
nm) medido a 20 ° C no líquido a ser examinada, calculada com referência a uma camada de 1
dm e dividido pela densidade expressa em gramas por centímetro cúbico.
Quanto ao instrumento, o polarímetro é um dispositivo, simples que mede de forma não
destrutiva as propriedades ópticas de moléculas, através da detecção de mudanças de rotação
do plano da luz linearmente polarizada. Embora, as ondas de luz possam oscilar em planos ao
longo de três dimensões, a orientação rotacional desses planos é definida principalmente pelos
campos elétricos e magnéticos. O aparelho é constituído por dois prismas de Nicol, um atuando
como polarizador e o outro como analisador (LIMA, 1997). As medidas realizadas no
polarímetro apresentam capacidade de os produtos das reações químicas afetarem tais
orientações, sendo alteradas as estruturas moleculares dos compostos opticamente ativos. Logo,
o valor da rotação óptica observada é o resultado da forma e da concentração de qualquer
mistura de moléculas quirais presentes na amostra (LESNEY, 2004). A Figura 3.1 mostra o
plano de luz polarizada da amostra girado no sentido horário.
Figura 3.1-Ilustração da rotação óptica de um composto quiral biologicamente ativo
Adaptado de Prentice-Hall, Inc. A Pearson Company, 1995 – 2002. (Chiral Compound in Plane-Polarized Light)
Disponível em: http://wps.prenhall.com/wps/media/objects/724/741576/chapter_05.html
As rotações observadas de enantiômeros são opostas em cada direção, pois um dos
estereoisômeros rotaciona a luz polarizada numa direção dos ponteiros do relógio, denominado
dextrogiro, na ilustração acima, por exemplo, α pode ser -90º ou + 270º em vez de + 90º.
A desvantagem desta técnica é na quantificação dos compostos, pois para isso necessita-
se de quantidade de amostra na ordem de mg, o que para aplicação de antibióticos para
medicamentos é viável, mas não para análises de resíduos (LIMA, 1997). Assim, os métodos
empregados para quantificação dos fármacos nos medicamentos (GERGELY, 1989; S, 1985)
Métodos têm sido proposto para aperfeiçoar as medidas no instrumento, Kankare et al
(1986) demonstra quais as melhores condições para aumentar o desempenho do aparelho. Os

55
principais requisitos é que o limite da relação sinal/ruído que geralmente é alcançado em níveis
mais baixos de energia. Alguns métodos são mais sensíveis, por exemplo, Ng Linder (2003)
identifica os seis isômeros análogos da tetraciclina. De acordo com os pesquisadores cada
análogo apresenta uma rotação específica, entretanto todos os estes são semelhantes
estruturalmente, mas apresentam diferenças significativas nas rotações.
É fato, muitos compostos são, terapeuticamente, eficazes, pois são quirais, uma vez que
a estereoespecificidade está diretamente relacionada com a assimetria molecular sendo
determinante para sua atuação nos organismos (BARREIRO; FERREIRA; COSTA, 1997;
STALCUP, 2010). Os diastereisômeros e enantiômeros quirais têm significativamente
diferentes atividades biológicas. Um exemplo é a Benzilpenicilina que exibe três centros quirais
(oito formas opticamente ativas isto é 8 estereoisômeros): somente o estereoisômero 3S:5R:6R
apresenta atividade, na Figura 3.2 é apresentada um dos centros quirais da penicilina G. A
conformação dos esteriisômeros foram calculados usando o software ChemSkech. A Estrutura
foi otimizada pelo software.
Figura 3.2-Visualização de um dos Centros Quirais da Benzilpenicilina (A), imagem em 3D (B)
1 OH2
O3
4N5
6 S7
8 CH39
CH31011
12O13
NH14
15
161718
19
20
21
22
O23
S
*
R (A)
(B)
(C)
Desenhada usando software ChemSketch ACD, 2015
Embora os isômeros R e S apresentem os mesmos substituintes, esses dois enantiômeros
formam diferentes relações espaciais. Assim, somente em um isômero pode residir toda a
atividade farmacológica, no caso, os demais podem ser considerados impurezas, por serem
inativos.

56
Contrariamente dois isômeros são capazes de apresentar atividade farmacológica
qualitativa e quantitativa quase idêntica (ISLAM; MAHDI; BOWEN, 1997; LU, 2007;
RENTSCH, 2002). De acordo com Leffingwe (2003), 56% dos medicamentos, atualmente em
uso, são compostos quirais, e 88% destas drogas sintéticas quirais são utilizados
terapeuticamente como misturas racêmicas. Infelizmente, existem muitos enantiómeros, no
qual a estereoespecificidade inferem no metabolismo e efeitos farmacodinâmicos não são
conhecidos (RENTSCH, 2002). Ainda de acordo com Capozziello e Lattanzi (2003) a maioria
das propriedades das moléculas é invariante à reflexão.
Quando examinadas em um ambiente aquiral, os enantiômeros são idênticos em muitos
aspectos, tais como: na solubilidade, na densidade, no ponto de fusão, no tempo de retenção
cromatográfico e no comportamento espectroscópico (UEDA e al., 2007).
Quanto às propriedades de rotação eles mudam seu sinal (CAPOZZIELLO;
LATTANZI, 2003), porém é muito mais difícil produzir um único estereoisômero, embora
possa ser pouco complexo a obtenção de grandes quantidades quando entra em atividade o
Penicillum (HERSBACH, 1983), sendo outros compostos quirais provenientes de espécies
biológicas difíceis, ou mesmo impossíveis sua síntese química (BURNS, 1986;
ELMARROUNI et al., 2010; ISHIBASHI et al., 2005; UEDA et al., 2007).

57
3.1.2 Espectroscopia para identificação e quantificação de antibióticos em terapêuticos
de uso veterinário.
Neste trabalho foram utilizadas as regiões de UV e IF para identificação dos antibióticos
e confirmação de suas absorções. Com relação à espectrofotometria no UV é uma técnica para
medir o grau de absorção de luz entre os comprimentos de onda 200 nm a 800 nm, para
investigar a identidade e pureza das substâncias (JP XVI, 2007; SÁNCHEZ ROJAS; BOSCH
OJEDA, 2009; SPINELLI; GONZALEZ, 2007).
As medidas geralmente são realizadas quando luz monocromática passa por uma
substância em solução, a proporção de luz transmitida é I e a intensidade da luz incidente é I0,
expressas em transmitância; transmitância expressa em percentagem é denominada transmissão
T, e o logaritmo do inverso da transmitância são nomeados de absorbância (EUROPEAN
PHARMACOPOEIA COMMISSION, 2008; JP XVI, 2007; MOAN, 2001).
Nestas medidas, o espectro de absorção das substâncias presente na solução é
característico, dependendo da sua estrutura química.
Esta técnica tem sido aplicada para vários sistemas químicos, como produtos
farmacêuticos, alimentos, cosméticos e amostras ambientais (SHEN, 2011; VETUSCHI et al.,
2005; ZEITLER et al., 2007), bem como o tratamento de dados espectrofotométricos têm sido
abordadas, com o objetivo de extrair uma maior quantidade de informação analítica, a partir de
espectros de bandas não resolvidas (SÁNCHEZ ROJAS; BOSCH OJEDA, 2009).
Os recentes avanços em detectores sensíveis de fibra óptica de eletrônica de alta
velocidade e softwares poderosos tem-se destacado nesses trabalhos. Uma vez que, a utilização
de fibras ópticas, como orientadora da luz, permite uma grande modularidade e flexibilidade na
criação de um sistema de medição óptica. Estes novos espectrômetros usam dispositivos que
oferecem excelente desempenho e amplas aplicações da espectroscopia de UV a NIR. Esses
instrumentos são considerados de baixo custo e de ótima robustez, esses parâmetros têm
revolucionado a detecção na espectroscopia, devido à sua combinação única de sensibilidade
excepcional e de alta velocidade na coleta dos espectros.

58
3.1.2.1 Importância da Região no Ultravioleta e Infravermelho Próximo
Por um longo tempo, absorções na região do infravermelho próximo (NIR) na região de
750 – 2500 nm foram consideradas de interpretação complexa, contrariando o fato de que essas
absorções são harmônicas e apresentam bandas vibracionais do tipo C – H, N – H e S – H., e
como tais, não só são muito menos intensas, mas também é complexa a atribuição das principais
características dos grupos funcionais presentes nas amostras, como é realizado no
infravermelho médio (HOSHI, 2007; MARIN; MOORE, 2011; SIESLER et al., 2008;
WORKMAN, 1996).
Neste aspecto, Plugge e Van Der Vlie (1992) apresentam vantagens para aplicações
quantitativas são elas: as amostras podem ser medidas como tal na refletância ou de
transmissão, sem qualquer preparação e (adição de solventes ou qualquer outro diluente). Desta
forma, o NIR despontou como uma ferramenta útil em diferentes campos industriais onde se
exijam matérias primas de qualidade. Seu uso em análise farmacêutica é bastante comum como
método de rotina.
Vários comentários têm sido propostos incluindo variadas aplicações, tais como: a
identificação de matérias-primas, a determinação do tamanho de partícula e dos principais
componentes (BLANCO; PEGUERO, 2010; DUNN et al., 1998; LUYPAERT; MASSART;
VANDER HEYDEN, 2007; LYON et al., 2002; SARRAGUÇA; LOPES, 2009; SCAFI;
PASQUINI, 2001a).
As técnicas expectroscopias na região do UV VIS NIR, também tem sido utilizada para
determinações quantitativas de compostos específicos em matrizes complexas, como, por
exemplo, uma preparação farmacêutica (BARTON et al., 2002). As etapas podem incluir a
realização de procedimentos tradicionais como espectrometria no UV e emprego de métodos
colorimétricos (KARPIŃSKA; MIKOŁUĆ; PIOTROWSKA-JASTRZEBSKA, 1998). Porém
somente podem ser válidas se fundamentadas em métodos referência oficialmente
reconhecidos.

59
As principais fases para desenvolver o método incluem etapas como: divisão dos dados
em grupos de calibração (ou treino) e definição de um conjunto de dados. Neste aspecto, é
importante que o conjunto da calibração cubra a maior parte da variabilidade que é esperada a
partir de amostras externas (BODSON et al., 2006; SCAFI; PASQUINI, 2001b). Para isso os
espectros NIR das amostras de calibração devem ser matematicamente relacionados com a
propriedade de interesse (por exemplo, concentração). Através da utilização de ferramentas
quimiométricas, é possível construir os modelos, porém é necessário planejar as condições das
variáveis, a fim de chegar a um melhor ajuste entre a medida de referência das amostras de
calibração, e consequentemente dos valores previstos pelo modelo que são testados para
comprovação do desempenho (AGELET; HURBURGH, 2010; SAUER; KIM, 2011; SMALL,
2006; WORKMAN, 2007; ZHANG et al., 2015).
Logo, a aplicação do NIR para análise quantitativa exige o emprego de técnicas
multivariadas na calibração e cuidadosa seleção de amostras representativas, a fim de construir
modelos que representem a realidade, assim a regressão por quadrados parciais (PLS) tem sido
muito utilizada para desenvolvê-los com capacidade de previsão e adequada seleção de
variáveis (ABDI, 2003; BASTIEN; VINZI; TENENHAUS, 2005; RUDNITSKAYA et al.,
2013; STENBERG et al., 2010; TENENHAUS et al., 2005), uma vez que essas abordagens têm
sido ferramentas de simples execução, e principalmente rápidas na construção dos modelos de
calibração PLS.
Algumas das aplicações estão focadas na quantificação de medicamentos (GRÜNHUT
et al., 2008; KHOSHAYAND et al., 2008; TER LAAK; GEBBINK; TOLLS, 2006). Por
exemplo, Ali, Sherazi e Mahesar 2012 que desenvolveram um método para a quantificação de
eritromicina em comprimidos, usando espectroscopia em FTIR para análise de rotina. Neste
trabalho não houve preparo de amostra, exceto a formação de “sedimento” para análise (ALI;
SHERAZI; MAHESAR, 2012). Enquanto que os pesquisadores Li et al (2015), apresentaram
um método de modelagem baseado na técnica EDSPLS. Segundo os autores o procedimento
gerou resultados de predição muito robustos, pois a partir de experimentos com dois conjuntos
de dados, foram construídos bons modelos de calibração, sem aumentar a complexidade do
ponto de vista do analista, sendo um método adequado para validação externa. Esta ferramenta
é aplicada em espectros no NIR e MIR.

60
3.1.2.2 Importância da aplicação da Espectroscopia no Infravermelho Médio
A espectrofotometria na região do infravermelho médio é utilizada também como um
teste de identificação. O espectro de infravermelho é único para qualquer dado composto
químico com a exceção de isômeros óticos que possuem espectros idênticos em solução. Apesar
de a espectroscopia de ressonância magnética nuclear (RMN) e espectrometria de massas (MS)
serem amplamente utilizados na indústria farmacêutica para a identificação de substâncias
medicamentosas, esta técnica pode ser aliada no fornecimento informação adicional quanto às
estruturas, tal como a presença de grupos funcionais característicos (STUART, 2004;
WHARTON, 2000).
Os dados obtidos por FTIR apresentam concordância com os obtidos por um método de
referência com base, por exemplo, em espectrometria de ultravioleta (BERTHOMIEU;
HIENERWADEL, 2009; HSU, 1997). Os procedimentos desenvolvidos oferecem uma boa
alternativa para identificação dos compostos (AMARIE; GANZ; KEILMANN, 2009; MOROS;
GARRIGUES; DE LA GUARDIA, 2007; ZEITLER et al., 2007). Outros procedimentos têm
sido relatados para quantificação de grupos fenólicos em matrizes alimentares (KYRALEOU
et al., 2015).

61
3.1.3 Considerações sobre a Aplicação dos Métodos Cromatográficos no Controle de
Qualidade de Formulações.
Os métodos cromatográficos permitem detecção, separação, identificação e
quantificação de impurezas orgânicas que podem estão presentes nos fármacos. Esses métodos
são seletivos e produzem resultados confiáveis (DEMIRALAY et al., 2012; SIDDIQUI;
ALOTHMAN; RAHMAN, 2013).
A cromatografia liquida de alta eficiência é uma técnica avançada que utiliza a separação
de uma mistura complexa presentes em sistemas químicos e/ou biológicos. Nesta técnica a
escolha do modo de detecção é fundamental para garantir que todos os componentes sejam
detectados (ALVAREZ-MUÑOZ et al., 2015; LIN et al., 2015).
Neste método a amostra é injetada em uma coluna, preparada com uma fase estacionária
e adequada para o meio de um líquido, este é usado como fase móvel e passa através da coluna,
de modo a separar a mistura dos seus componentes, fazendo uso da diferença de capacidade de
retenção (JP XVI, 2007). A mistura injetada na coluna é eluída com a fase móvel e estacionária,
e apresenta característica que representa a relação da distribuição de massa para cada
componente definido pela Equação 10.
K𝑝 = AFE
AFM
(10)
Onde: Kp coeficiente de partição, AFE quantidade do analito na fase estacionária e AFE
são a quantidade do analito na fase móvel.
Os sistemas HPLC disponíveis são muito uteis para controle de qualidade dos
medicamentos, tanto nas indústrias como para órgãos legais competentes (MORETA; TENA;
KANNAN, 2015). Entretanto, as limitações de HPLC incluem os custos das colunas, solventes
e ausência da reprodutibilidade desta em longo prazo.
Um dos detectores amplamente utilizados para HPLC é detector de UV, que é capaz de
monitorizar vários comprimentos de onda simultaneamente (SIDDIQUI; ALOTHMAN;
RAHMAN, 2013). Entre os detectores de LC o de fluorescência (FL) é um dos mais sensíveis,
segundo Ulu e Tuncel, 2012 sua sensibilidade chega a ser 10 vezes maior que o de UV quando
aplicados a materiais absorventes (ULU; TUNCEL, 2012).
Os detectores FL são amplamente utilizados como uma vantagem na medição de
espécies fluorescentes, sendo muito empregados para amostras de vitaminas e de resíduos de

62
antibióticos do grupo das sulfonamidas e quinolonas (AŠPERGER et al., 2014; HE; BLANEY,
2015; PENNACCHIO et al., 2016). As frequências da aplicação de diferentes tipos de detector
em HPLC são apresentadas na Figura 3.3. Neste quadro é exposto as principais aplicações,
durante período de 2009 – 2016, em análises de fármacos a base de penicilina, utilizando
sistema mais diversos sistemas compostos por HPLC.
Figura 3.3-Emprego dos diferentes detectores para análise por fármacos a base de penicilinas
Adaptado da base de dados Scopus 2015
Um dos detectores mais utilizados para análise simultânea é o de massas (DÍAZ-BAO
et al., 2015; REZK et al., 2015). Recentemente, Rodriguez et al (2015) desenvolveram um
método utilizando HPLC-MS/MS para avaliar os principais componentes em amostras de
medicamentos comerciais de uso humano a base de gentamicina. Os pesquisadores planejaram
as condições do sistema cromatográfico, de modo que o método exibiu excelente estabilidade
e linearidade. O desempenho do método foi avaliado através da análise de lotes de gentamicina
comerciais.
Algumas aplicações estão sendo reportadas para quantificação dos antibióticos em
matrizes muito complexas, como o sangue, utilizando sistemas UPLC-MS/MS. Os grupos de
terapêuticos mais expostos a discussão são os betalactâmicos, pois necessitam de controle e
monitorização nas concentrações plasmáticas (CARLIER et al., 2015). Outros estudos sobre os
efeitos dos compostos tóxicos como alterações fisiológicas ou químicas induzidas no corpo de
um mamífero têm sido reportados para quantificação inclusive de amoxicilina, utilizando
detector massas (SKOV et al., 2015).
MS/MS
34%
UV
33%
FL
11%
UV-FL-MS
22%

63
3.2 ANÁLISES DOS RESÍDUOS DE ANTIBIÓTICOS
A falta de um controle quanto à presença de resíduos, em algumas regiões, pode ser
atribuída aos custos das análises. Em muitos casos, esses recursos estão além do alcance da
maioria dos países de baixa renda. De forma que, as técnicas de bioensaio para determinação
rápida de resíduos de antibióticos têm sido aplicadas, devido ao custo e simplicidade de análises
(KNECHT et al., 2004; LAMAR; PETZ, 2007; SHERER, 2006).
Todavia, devido ao risco de falsos resultados, é necessário confirmar e quantificar esses
resíduos por métodos precisos e exatos. Visto que as metodologias relatadas não fazem
distinção entre as várias classes de antibióticos, apenas fornecem resultados semiquantitativos
(KINSELLA et al., 2009), por isso métodos cromatográficos são destinados à determinação
quantitativa de fármacos e/ou metabólitos em matrizes biológicas, sendo classificados como
bioanalíticos, visto que são ferramentas potencializadas que asseguram produtos enquadrados
nas determinações legais, já que as agências governamentais reguladoras exigem métodos que
respeitem os aspectos qualitativos e quantitativos, estabelecidos pelas agências locais de
vigilância sanitária (ANVISA, 2005; NICOLICH; WERNECK-BARROSO; MARQUES,
2006).
A cromatografia liquida (BALIZS; HEWITT, 2003; RAMBLA-ALEGRE et al., 2011a,
2011b; RIEDIKER; DISERENS; STADLER, 2001; STOLKER; BRINKMAN, 2005),
eletroforese capilar (LARA et al., 2006), cromatografia liquida acoplada a arranjo de fotodiodos
UV, (CHRISTODOULOU; SAMANIDOU, 2007; GARCÍA-MAYOR et al., 2006), detecção
fluorimétrica (RODRÍGUEZ-DÍAZ et al., 2006), eletroquimioluminescência (GUO; GAI,
2011) e métodos espectroscópicos (NOUR EL-DIEN et al., 2010) foram mais recomendados
para detecção simultânea de antibióticos no leite. Embora, a detecção por espectrometria de
massas seja considerada inadequada para uso rotineiro, em alguns laboratórios por seu alto
custo. Outros procedimentos utilizando HPLC-UV (ANDERSEN et al., 2005; CINQUINA et
al., 2003a; OKA; ITO; MATSUMOTO, 2000) e espectrometria de massas (BRUNO et al.,
2002) são reportados, para matrizes como leite e ovos.
Algumas aplicações para análises de sulfonamidas e tetraciclinas têm sido citadas (CUI
et al., 2006; GAMBA et al., 2009; HERMO et al., 2008; HOOF et al., 2005; KOESUKWIWAT;
JAYANTA; LEEPIPATPIBOON, 2007a, 2007b; LI et al., 2010; MARAZUELA; MORENO-
BONDI, 2004; RAMIREZ et al., 2003; UDALOVA; DMITRIENKO; APYARI, 2015).
Spisso et al (2007) validam metodologia usando HPLC acoplada à espectroscopia de
fluorescência, para confirmação de resíduos de tetraciclinas, o método teve bom desempenho.

64
Contudo, a cromatografia liquida de alta eficiência acoplada a espectrometria de massas
(HPLC-MS) tem sido reportada para análise de rotina e triagem. Boyd et al (2007)
determinaram cloranfenicol e fenbendazole (aquele têm sido banido na maioria dos países) em
leite bovino, mel, urina e plasma. O limite de detecção (1000 – 4000 mg kg-1) estava acima do
permitido pela legislação europeia. Ainda no mesmo período, Raviolo et al (2007) identificaram
e quantificaram seis sulfonamidas (sulfacetamida de sódio, sulfamethizole, sulfaguanidina,
sulfametazina, sulfatiazol e sulfametoxazol) em leite pasteurizado, justificando o desempenho
do método pela redução do tempo de análise e valores de LMR abaixo dos relatados pela
legislação.
Após três anos, Berendsen et al (2010) confirmaram a presença de albendazol,
sulfametazine, fenilbutazona, difloxacina, espiramicina, tetraciclina, oxitetraciclina em leite
bovino integral e desnatado, destacando que apesar da desvantagem do efeito da matriz, os
limites de detecção estiveram entre 0,1 e 5,2 mg L-1. Uma vez que a análise de resíduos em
matriz complexa como o leite, exige uma investigação mais assídua quanto a natureza
quantitativa da amostra principalmente com relacão ao as proteína e gordura, este último
parâmetro tem sido enfatizado por Cabral, Moura e Da Silva (2012) quando avaliaram a
qualidade do leite em pó comercial e construíram modelos para determinação rápida de
gorduras totais. Esses parâmetros interferem nos procedimentos preparativos das amostras
(FLURER, 1999).
Aguilera-Luiz et al (2008) aplicaram a cromatografia liquida de ionização por
eletroespray para determinação de 12 antibióticos das classes sulfonamidas, macrolídeos e
tetraciclinas em leite integral e desnatado adquiridos do comércio local. Em seguida, Wang e
Li (2009) desenvolveram um método analítico para detecção rápida de clorafenicol, tianfenicol,
tetraciclina, oxitetraciclina, clortetraciclina, metacicline, doxiciclina, cefoperazona, ceftriaxona
e cefaclor em leite pasteurizado. Sendo que Vargas Mamani et al (2009) validaram um método
para confirmação de tetraciclina, oxitetraciclina, clortetraciclina, sulfametazina,
sulfaquinoxaline, sulfametoxazol e clorafenicol verificando que estavam abaixo do LMR em
leite bovino tipo A, B e C. comercializados no Brasil.

65
3.2.1 Preparo da amostra para quantificação de resíduos de antibióticos
O preparo da amostra interfere em todas as etapas seguintes de um ensaio analítico, e,
por conseguinte, é determinante para a identificação, confirmação, e quantificação correta dos
analitos (MATUSZEWSKI; CONSTANZER; CHAVEZ-ENG, 2003a; ZUBATA et al., 2002).
Ela inclui tanto o isolamento quanto a pré-concentração do composto de interesse, presentes
nas mais diversificadas matrizes, a partir da mesma os analitos são favoráveis à separação e
detecção. Esse procedimento analítico para amostras leva em torno de 70% do tempo de análise
total. Considerando que, são preferidos os métodos cromatográficos na análise de moléculas
orgânicas, a clean-up, a separação e isolamento do analito emprega a extração (líquido-líquido)
e extração em fase sólida. Com relação à análise cromatográfica ultra-rápida, esta envolve
várias etapas, o que demanda tempo e trabalho intensivo. Por estas razões, novas técnicas de
preparação de amostra têm sido desenvolvidas (BARROSO et al., 2012; DINIS-OLIVEIRA et
al., 2010; DUAN et al., 2011; REEVES, 2011).
Uma análise da literatura científica mostra, exatamente, 535 artigos (análise de resíduos
de medicamentos veterinários) publicados no período compreendido entre 2009 – 2016 (inicío),
sendo que procedimentos envolvendo extração líquido representam 61,49%, do total. As
abordagens incluídas, neste dado, são: extração líquido-líquido, micro-extração líquido-líquido,
extração em fase sólida, neste último estão incluídos os processos à base de absorventes como
micro-extração em fase sólida.
Porém, houve muitas mudanças na abordagem do preparativo de amostras causado,
principalmente, pela aplicação recente da espectrometria de massas. Desta forma, atualmente,
há uma tendência a se concentrar mais nas extrações e procedimentos de limpeza, a fim de
abranger um número razoável de antibióticos detectados presentes em matrizes alimentares
(BERENDSEN et al., 2010, 2013; CHEN et al., 2008; GUO et al., 2014; IGLESIAS et al.,
2012; JING et al., 2009; MACAROV et al., 2012; NEBOT et al., 2012a, 2012b; STOLKER et
al., 2010; STOLKER; BRINKMAN, 2005; YU et al., 2014), pois as técnicas acopladas a
detector MS permitem o simples uso dos métodos clean-up, seguida de remoção eficaz dos
interferentes, uma vez que estes podem afetar o desempenho do instrumento (KAUFMANN;
BUTCHER; MADEN, 2012; MADUREIRA et al., 2012; MIRANDA; RODRÍGUEZ;
GALÁN-VIDAL, 2009; TAKA; BARAS; CHAUDHRY BET, 2012; VAN ROYEN;
DUBRUEL; DAESELEIRE, 2014).

66
Ultimamente, tem-se uma direção importante, embora lenta, para aplicação das técnicas
de preparo de amostras automatizadas, que são poderosas ferramentas para a limpeza da
amostra, concentração e separação de analitos. Metodologias como esta, possibilitam uma
redução no tempo dos procedimentos, melhores limites de detecção e quantificação, precisão,
reprodutibilidade, e principalmente redução de custos (DONG; ZHANG, 2014; GAUDIN et
al., 2014; KANTIANI et al., 2009; KNECHT et al., 2004; PRAGST, 2007; STOLKER et al.,
2010).
Neste contexto, Chen et al (2008) apresenta comentários de extrema relevância,
mostrando a evolução dos métodos de preparo de amostras, os autores destacam 481 referências
a respeito das técnicas mais promissoras. Os pesquisadores incluem nos seus comentários todos
os tipos de amostras, incluindo as células, órgãos, tecidos e corpos maiores. Além disso, são
apresentadas estratégias para a integração de vários métodos complexos para preparo de
amostras contendo proteínas e estudos proteômica.
A respeito disso, Fumes et al (2015) argumenta que a análise dos compostos, químicos
presentes em concentrações muito baixas, em matrizes complexas, tais como: pesticidas e
antibióticos em carnes, leite, ovos dentre outros de origem animal, requer um preparo de
amostra adequado, que envolva um isolamento do analito e determinação qualitativa,
quantitativa e até a confirmação. Os autores apresentam os avanços e tendências dos métodos
verdes utilizados para técnicas de micro extração.
Enquanto que, Rejczak e Tuzimski (2015) apresentam uma revisão no qual são
abordadas as aplicações do método QuEChERS (rápidos, fáceis, baratos, eficientes, robustos e
seguros). Esta técnica envolve extração líquido-líquido, particionamento utilizando acetonitrila,
seguida de purificação por extração em fase sólida. Os autores relatam que QuEChERS foi
introduzido para a análise de resíduos de pesticidas em frutas e legumes, isto é, alimentos com
alto teor de umidade, porém tem sido empregado para análise de pesticidas, medicamentos
veterinários, micotoxinas, hidrocarbonetos aromáticos policíclicos, corantes, filtros UV,
bifenóis e hormônios. As aplicações são em matrizes variadas até mesmo de origem animal
com o leite (AGUILERA-LUIZ et al., 2008; FILHO et al., 2012; KARAGEORGOU;
SAMANIDOU, 2011; MADUREIRA et al., 2012). Para executar tais procedimentos, em
concordância com os programas de controle de resíduos, tem-se atenção a natureza da amostra
analisada.

67
Atualmente, os resíduos de drogas veterinárias relatados incluem matrizes como fígado,
rim, músculo, gorduras, leite, ovos, mel e carnes. Os valores de LMR observados em gêneros
alimentícios de origem animal devem, obrigatoriamente, está abaixo do valor máximo. Reeves
et al (2011) e Reyes-Herrera et al (2005) afirma que matrizes desta natureza devem ser
avaliadas logo após o abate do animal. Um exemplo é o trabalho de Ruyck et al (2001) que
aperfeiçoa um método quantitativo utilizando LC-MS/MS para determinar o flubendazole e
seus metabolitos. Segundo os autores os componentes de foram extraídos, com acetato de etilo
após a alcalinização das amostras de músculo suíno.
Em outras matrizes são mostradas as dificuldades dos procedimentos, por exemplo, têm-
se registros que o resíduo de antibióticos é monitorado com frequência em ração animal e água
potável (KUKUSAMUDE et al., 2012; LI et al., 2013), porém na ração o método de preparo
das amostras é muito complexo, pois são matrizes que contém um alto teor de proteínas e
carboidratos, alguns exemplos são citados (BERENDSEN et al., 2010; BORRÀS et al., 2011;
GONZÁLEZ DE LA HUEBRA; VINCENT; VON HOLST, 2007; REDDING et al., 2014;
VAN HOLTHOON et al., 2010; WANG et al., 2008; WEI et al., 2011).
3.2.1.1 Critérios para Preparo da Amostra
Siqueira (2011) ressalta que o objetivo do preparo de amostras é isolar o analito de
interesse de uma matriz complexa. Desta forma, esta fase preparativa da amostra envolve os
procedimentos de extração, inclusive etapas de purificação (clean up), sendo adequada para
concentrar os analitos, em um nível apropriado, para detecção. Assim, o tratamento realizado,
previamente, nas amostras envolve remoção das proteínas por: precipitação, ultrafiltração e
diálise. O processo de desproteineização inclui: altas temperaturas, alteração da força iônica ou
osmótica ou pH, agentes orgânicos de precipitação e enzimas proteolíticas.
Os procedimentos podem ser alocados em etapas, assim, em uma primeira fase, realiza-
se a preparação das soluções, contendo os analitos de interesse, seguida de tratamento da
amostra controlando fatores como: ácido, agitação, centrifugação e filtração do sobrenadante
(ANVISA, 2005; EUROPEAN COMMISSION, 2010; KINSELLA et al., 2009).
Simplificadamente, as metodologias devem obedecer a um planejamento experimental, assim
os parâmetros são testados, a fim de obter o melhor extrato, a ser levado ao cromatógrafo, bem
como melhor mistura solvente, e o modo de eluição (se isocrático ou não), consequetemente os
parâmetros inerentes à análise cromatográfica, como velocidade de eluição, terão valores
ótimos. A segunda etapa é a escolha dos padrões de antimicrobianos, esta leva em consideração:

68
a utilização do medicamento, que pode deixar resíduos; dose, tempo de utilização, espectro de
utilidade na medicina veterinária/potencial de exposição ao consumidor e disponibilidade de
metodologia analítica reconhecida pelas organizações.
As etapas citadas garantem o bom desempenho dos métodos bioanalíticos, porém, há
relatos de que os problemas principais estão associados com a instabilidade de fármacos e
metabólitos, isto é, os antibióticos, em matrizes biológicas, podem ser afetados pela temperatura
de armazenamento, exposição a enzimas, pH, anticoagulantes e ciclos de congelamento e
descongelamento. Além disso, a volubilidade pode ocorrer durante qualquer um dos numerosos
passos da metodologia (DÍAZ-BAO et al., 2015; GOTO et al., 2005). Entretanto, um dos
principais objetivos consiste na remoção de qualquer efeito de matriz (BULDINI et al, 2002;
LALLJIE et al, 2007; CHEN et al, 2008). Certamente, os avanços dos processos de separação
e de técnicas de detecção aliados a procedimentos adequados garantem a eliminação desses
interferentes.
Durante muito tempo, a extração líquido-líquido combinado ou não com extração em
fase sólida foram empregadas, em diversos trabalhos (ANDERSEN et al., 2005; BOGIALLI;
DI CORCIA, 2009; CHRISTODOULOU; SAMANIDOU, 2007; SCHLÜSENER; BESTER;
SPITELLER, 2003), bem como tendências para o isolamento de medicamentos veterinários
também foram reportadas (KINSELLA et al., 2009; RIDGWAY; LALLJIE; SMITH, 2007). A
saber, de Sørensen e Snor (2000) que realizaram extração em fase sólida para determinação de
resíduos como cefazolina, cefoperazona, cefquinoma e ceftiofur em leite cru. Enquanto que
Holthoon et al (2010) analisaram o grupo das penicilinas em matrizes diferentes, relatando que
o procedimento de derivatização é um método eficaz, na prevenção da degradação dos resíduos
dos grupos betalactâmicos. De maneira mais ampla, Nebot et al (2012b) descreveram
metodologia para confirmação de sete agentes antimicrobianos em leite, três corticosteróides e
um antifúngico, o método foi validado, com base na Decisão 2002/657/CE da Comissão
Européia. Ele mostrou aplicabilidade e confiabilidade, justificado pelo número de amostras
(100) e coeficientes de regressão entre 0,980 e 0,998.
Diante dessa avaliação, o preparo de amostras tem sido determinante para validação de
métodos bioanalíticos. Uma vez que o HPLC continua a ser um método de escolha, pois tem
sido indicado para separar compostos com elevado e baixo peso molecular, sobretudo com
diferentes polaridades, sendo aplicados a inúmeras matrizes. Infelizmente, alguns os métodos
de preparo de amostra convencionais prejudicam a resolução dos picos, além de ser demorado.

69
De modo que, os métodos de preparo de amostras devem ser eficientes. Alguns são
apresentados na Tabela 3.1.
Tabela 3.1-Sumário dos métodos de preparo de amostras de leite para determinação de resíduos de antibióticos
utilizando HPLC-DAD.
Compostos Extração e Clean-up Referência
Tetraciclinas
Precipitação de proteínas totais com solução Mcvalline.
Diluição do sobrenadante, e centrifugação a 4000 rpm e 5 °C
durante 10 minutos. O sobrenadante foi submetido a extração
em fase sólida, adição de ácido tricloroacético e metanol
seguido de evaporação de solvente e filtração em filtro de
nylon de 0,22 µm.
(NAVRÁTILOVÁ;
BORKOVCOVÁ;
DRAČKOVÁ, 2009)
Penicilinas
Homogeneização das amostras (5g), adição de ácido
clorídrico em pH ajustado de 4,5 – 5 e mistura de
acetona/clorofórmio, agitação da solução durante 2 min e
centrifugação a 2500 rpm durante 10 min, evaporação do
solvente em rotavapor e diluição em água.
(SEN; FILAZI, 2014)
Sulfonamidas
Fortificação das amostras e agitação durante 5 minutos,
diluição das amostras e adição de ácido perclórico,
centrifugação a 10.000 rpm 25 ºC durante 10 min para
precipitação de proteína e filtração do sobrenadante em filtro
de nylon 0,45 µm.
(SHAO et al., 2014)
Aminoglicosídeo
A 50 ml de leite cru (ovelha e cabra) é adicionado HCl 2M e
ajustado o pH para 4,6, repouso à temperatura ambiente
durante 15 minutos, centrifugação a 2000 rpm durante 20
min. Diluição de 5mL do sobrenadante em tampão fosfato pH
de pH 6,7), seguida de filtração por membrana Millipore de
0,22 µm.
(ROMERO et al., 1996)
A 5 mL de leite é adicionada ácido tricloroacético 10%,
seguida de homogeneização das amostras em vortex durante
10 minutos a 3500 rpm a - 4 ºC, por fim, filtração do
sobrenadante em filtro nylon de 0,22 µm
(GHIDINI et al., 2007)
Macrolídeos
Homogeneização de 1mL de leite, fortificação com mistura
de padrão, adição de solução tampão fosfato e acetonitrila
para precipitação de proteínas. Agitação e centrifugação a
3500 rpm durante 10 min a 25 ºC, remoção da gordura pela
adição de NaOH 1M e 5mL de acetato de etila e
centrifugação nas mesmas condições durante 30 min, e
filtração da fase aquosa.
(GARCÍA-MAYOR et
al., 2006)
Fenicois
Adição de acetonitrila em leite, agitação em vortex e
sonicação durante 10 min, centrifugação a 3500 rpm durante
10 min, em seguida evaporação em banho de água a 35 º C e
dissolução dos resíduos. Tratamento com método MSPD
QuEChERS e homogeneização da amostra durante 10 min
em ultrassom seguido de adição de acetona 1%, água,
metanol e acetonitrila
(KARAGEORGOU;
SAMANIDOU, 2011)
Quinolonas
5g de leite e adição de 2,5 mL de ácido tricloroacético em
20% de metanol, centrifugação da amostra a 1500g durante
10 min, adição de tampão fosfato pH 7,4 agitação e
centrifugação durante 15 min, seguida de extração em fase
sólida C18, eluição em 1% de ácido tricloroacético e
acetonitrila.
(CINQUINA et al.,
2003b)

70
Este sumário tem sido incluído, a fim de tornar os procedimentos aplicados, neste
trabalho, adequadamente escolhidos, considerando, o tempo de extração, seletividade, o
número de etapas, o consumo de solvente e toxidade dos reagentes. Uma vez que tais métodos
têm sido desenvolvidos, para se melhorar a recuperação do analito. A exemplo de Fennell et al
(1995) que desenvolveram um método simples para extração das gentamicinas, a partir de
amostras de tecido e de leite. Estas foram fortificadas com o padrão e adicionado tampão de
pH 8,8, a solução foi homogeneizada e centrifugada as condições foram: 17000 rpm a 4 ºC
durante 30 min. Para aquelas, os pesquisadores realizaram infusão intramámaria nos animais e
coletaram o leite durante 48 horas, as condições de extração foram semelhantes. O método
mostrou recuperação > 90%.
Enquanto que, atualmente, Zhon et al (2009) aplicaram polímeros molecularmente
impressos magnéticos (MMIPs), como material de adsorção associado a extração em fase sólida
(MSPE). Os pesquisadores determinaram, simultaneamente, seis macrolídeos em amostras de
carne de porco, peixe e camarão, utilizando sistema HPLC-UV. As recuperações dos extratos
foram de 89,1%. Já Wang et al (2015), desenvolveram um método para quantificação de
betalactâmicos, macrolídeos, tetraciclina, sulfonamidas, e quinolonas, presentes em leite
bovino. O método empregado foi o QuEChERS, sendo que a recuperação dos analitos estiveram
entre 86,09 – 115,99%. Entretanto, os detalhes de procedimentos para rotina são mais
complexos, em trabalhos em sistema com detector MS, um exemplo é Aguilera-luiz et al (2008)
que realizaram extração com base, também, no QuEChERS. Os pesquisadores determinaram
sulfonamidas, macrolídeos e tetraciclinas em leite, garantindo recuperação de 70 – 110%. Ao
passo que, Ortelli et al (2009) quantificaram 150 resíduos de antimicrobianos veterinários,
utilizando UPLC-TOF/MS. Esses procedimentos consistiram em desprotenização total com
adição de solução ácido metanoico/ACN, seguida de ultrafiltração, evaporação e centrifugação,
porém, as recuperações dos analitos foram muito baixos devido à perda importante durante a
ultrafiltração. Turnipseed et al (2008) rastreiam e confirma presença de resíduos
betalactâmicos, sulfonamidas, tetraciclinas, quinolonas e macrolideos, utilizando sistema LC-
MS/MS. Os procedimentos foram realizados pela extração com ACN, ácido fórmico, clean-up
e filtração, o método garantiu recuperação > 70%, exceto para tetraciclinas que foi entre 50 –
70%, enquanto as penicilinas < 50%.

71
3.2.2 Métodos Bioanalíticos e Testes Rápidos
Esses testes são classificados como imunoensaio ou enzimáticos, conforme o seu
objetivo e princípio. As medições rápidas fornecem resultados qualitativos ou
semiquantitativos. Eles são testes portáteis e podem ser aplicados in situ. Além disso, eles
fornecem resultados, em tempo real, de acordo com a necessidade da análise. Geralmente, são
obtidos em um período de 30 min e não há necessidade de preparo de amostras (MAGALHÃES
et al., 2012; REEVES, 2011).
Ensaios microbiológicos, com base na inibição do crescimento de micro-organismos,
são relatados. Eles são específicos para os antibióticos, mas requerem longos períodos de
incubação. A exemplo de Peng et al (2013) que utilizam o método ELISA para a detecção de
antibióticos betalactâmicos, em diferentes matrizes, incluindo leite. Os pesquisadores
desenvolveram os testes como alternativa para ensaios semiquantitativo garantindo
recuperações entre 53,27 – 128,29% (ZENG K, ZHANG J, WANG Y, 2013). Enquanto que,
De Alburquerque Fernandes et al (2013), com objetivo de avaliar a ocorrências de resíduos
tetraciclinas, em amostras de leite pasteurizado, empregaram método de rastreio utilizando um
kit para triagem (Delvotest® SP-NT).
Não obstante, metodologias são aplicadas, para melhorar a quantificação da penicilina
G, utilizando Fluorescência. Este tipo de procedimento tem competido com os imunoensaios.
De maneira que, os pesquisadores argumentam que os métodos tradicionais aplicando HPLC-
DAD, HPLC-MS/MS, HPLC-FL apresentam algumas limitações tais como: demandam tempo,
requer a preparação de amostras complexas, manutenção dos equipamentos e exigem pessoal
treinado (PENNACCHIO et al., 2016).
Assim, alguns métodos imunoessaio (testes microbiológicos e biossensores) são
comentados em artigos de revisão, dentre eles está o trabalho de Puchades e Cha (2010), no
qual apresentam a evolução dos principais métodos de rastreio, especialmente resíduos de
antibióticos presentes em amostras de alimentos. Os autores concluem que os imunoensaios
com detecção por fluorometria são muito bem aceitos, por causa do tempo para identificação,
com vantagem dos níveis serem mais baixos, do que os LMRs estabelecidos pelas legislações.
Os métodos screening são empregados, sobretudo, quando há necessidade de monitorar
elevado número de amostras, este tipo de procedimento são mais preferidos, devido a
capacidade de rastreio e custos, relativamente, baixos (BITTENCOURT et al., 2012; HOFF et
al., 2012; PIKKEMAAT et al., 2009; STOLKER et al., 2010; WEAVER et al., 2013).

72
3.2.3 Emprego das Técnicas para Quantificação e Confirmação dos Resíduos de
Antibióticos
Recentemente, entre períodos de 2010 a 2012, a literatura científica revelou em torno de
300 artigos publicados, dos quais 20,66 % são relacionados a leite e derivados. Quanto ao
método de detecção, 52,33 % utilizam espectrometria de massas para confirmação; dentre estes,
pode-se citar os trabalhos de Léon et al (2012) que utiliza HPLC-MS/MS para rastreio rápido
e simultâneo de substâncias veterinárias ilegais e proibidas. Com objetivos semelhantes,
Gomez-Pérez et al (2012) desenvolveram um banco de dados análise simultânea 350 pesticidas
e drogas veterinárias, utilizando HPL-MS/MS.
Neste sentido, algumas publicações tratam da determinação de antimicrobianos
permitidos, mas com ressalva no LMR, dentre eles estão análises de sulfonamidas e
tetraciclinas, algumas aplicações são relatadas (CUI et al., 2006; GAMBA et al., 2009; HERMO
et al., 2008; HOOF et al., 2005; KOESUKWIWAT; JAYANTA; LEEPIPATPIBOON, 2007b;
LI et al., 2010; MARAZUELA; MORENO-BONDI, 2004; RAMIREZ et al., 2003;
RODRIGUEZ; MORENO-BONDI; MARAZUELA, 2011).
O avanço dos processos de separação e de técnicas de detecção tem se desenvolvido,
concomitantemente, aliados a procedimentos adequados de preparação de amostras, que visam
garantir a eliminação dos interferentes da matriz (BULDINI; RICCI; SHARMA, 2002; CHEN
et al., 2008; RIDGWAY; LALLJIE; SMITH, 2007). A saber, de Tsai et al (2009) que
desenvolveram um método para determinação simultânea de tetraciclina, clortetraciclina e
doxitetraciclina. Os autores mostraram otimos resultados, pois os parâmetros da curva tais
como: linearidade, LD, LQ foram condizentes com a validação e LMR.
Em 2010, Tsai et al (2010) desenvolveram um método para coprecipitação de foi
desenvolvido para determinação de tetraciclinas em amostra de água e leite utilizando HPLC
DAD. O método de extração consistiu em formar precipitados de hidróxido de magnésio e
separá-los por centrifugação e, em seguida, aplicaram uma etapa de dissolução por adição de
ácido. Segundo os pesquisadores o preparativo das amostras mostraram resultados satisfatórios
em condições ótimas, refletindo nos parâmetros da curva e recuperação do analito.
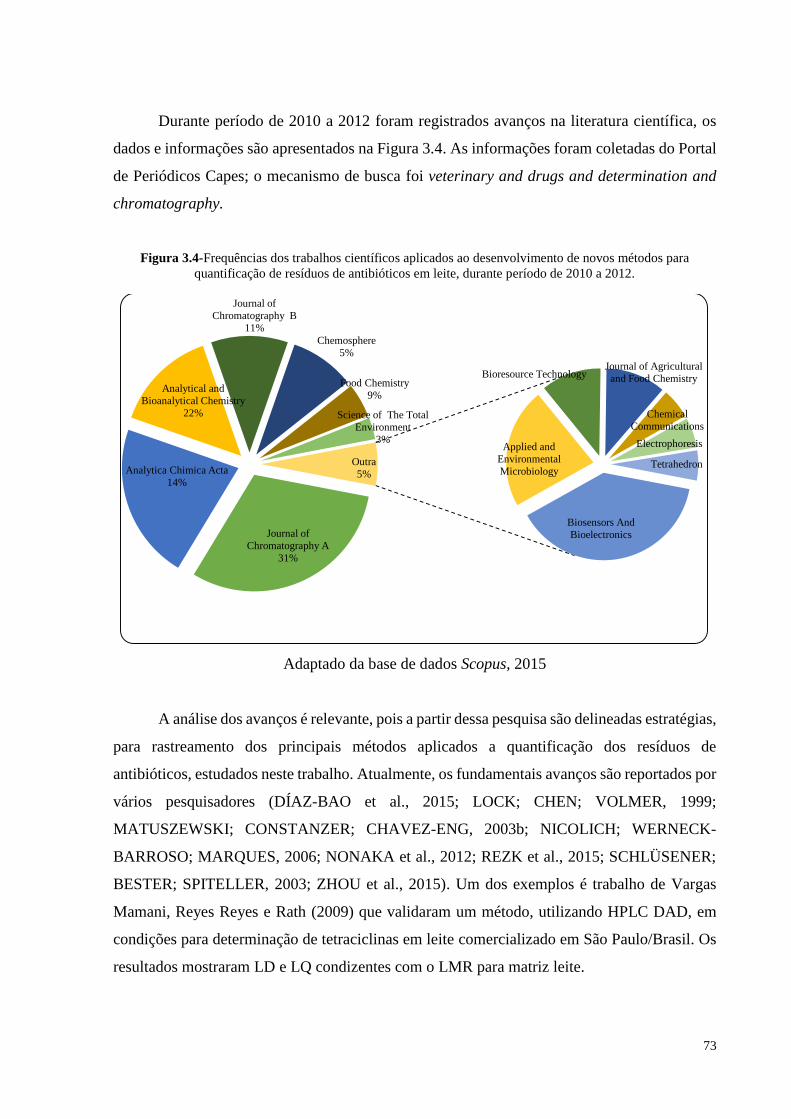
73
Durante período de 2010 a 2012 foram registrados avanços na literatura científica, os
dados e informações são apresentados na Figura 3.4. As informações foram coletadas do Portal
de Periódicos Capes; o mecanismo de busca foi veterinary and drugs and determination and
chromatography.
Figura 3.4-Frequências dos trabalhos científicos aplicados ao desenvolvimento de novos métodos para
quantificação de resíduos de antibióticos em leite, durante período de 2010 a 2012.
Adaptado da base de dados Scopus, 2015
A análise dos avanços é relevante, pois a partir dessa pesquisa são delineadas estratégias,
para rastreamento dos principais métodos aplicados a quantificação dos resíduos de
antibióticos, estudados neste trabalho. Atualmente, os fundamentais avanços são reportados por
vários pesquisadores (DÍAZ-BAO et al., 2015; LOCK; CHEN; VOLMER, 1999;
MATUSZEWSKI; CONSTANZER; CHAVEZ-ENG, 2003b; NICOLICH; WERNECK-
BARROSO; MARQUES, 2006; NONAKA et al., 2012; REZK et al., 2015; SCHLÜSENER;
BESTER; SPITELLER, 2003; ZHOU et al., 2015). Um dos exemplos é trabalho de Vargas
Mamani, Reyes Reyes e Rath (2009) que validaram um método, utilizando HPLC DAD, em
condições para determinação de tetraciclinas em leite comercializado em São Paulo/Brasil. Os
resultados mostraram LD e LQ condizentes com o LMR para matriz leite.
Journal of
Chromatography A
31%
Analytical and
Bioanalytical Chemistry
22%
Analytica Chimica Acta
14%
Journal of
Chromatography B
11%
Food Chemistry
9%
Chemosphere
5%
Science of The Total
Environment
3%
Biosensors And
Bioelectronics
Applied and
Environmental
Microbiology
Bioresource TechnologyJournal of Agricultural
and Food Chemistry
Chemical
Communications
Electrophoresis
TetrahedronOutra
5%

74
Outras aplicações têm sido descritas para análises, empregando procedimentos variados,
sendo classificados como pré-diagnóstico e análise quantitativa, tal como trabalho de
Navrátilová, Borkovcová e Dračková (2009) que desenvolveram método com objetivo de
detectar resíduos de tetraciclinas em leite bovino cru. Ao analisar grandes quantidades de leite
em tanques de armazenamentos. Os pesquisadores utilizaram kits para testes rápidos (citados
no item 3.2.2), cocomitantemente, com HPLC para confirmação dos resíduos. O sistema
cromatográfico utilizado, pelos pesquisadores, foi Waters em coluna C8 (3.9 × 150 mm, 4 μm).
Os resultados foram satisfatórios (rotina de indústria), uma vez que os valores apontados foram,
em sua maioria, menores que o LMR.
Em 2009, Shariati, Yamini e Esrafili (2009) desenvolveram um método para
preconcentração dos resíduos de tetraciclinas, utilizando microextração, sendo compatível com
as matrizes biológicas. Enquanto que, Bailón-Pérez et al (2009) desenvolveram um método
usando HPLC-DAD, para determinação de Penicilina G e V, o método foi aplicado em amostras
de água, leite bovino e humano e carnes. Os métodos também envolveu fase sólida,
concentração e clean up, o desempenho mostrou LD 0,80 – 1,40 µg L-1 compatível com
regulamentos por EC 2377/90. Ao passo que, Rodrigues el al (2010) apresentaram mesmo
método, porem envolvendo a extração e limpeza por dispersão, seguida de isolamento dos
analitos, em metanol acidificado. Assim as tetraciclinas eluídas foram determinadas,
simultaneamente, por meio de análise por injeção em fluxo com detecção por UV. Segundo os
pesquisadores, por comparação, os resultados entre HPLC e MSPE-SPE-HPLC não
apresentaram diferenças significativas.
Em 2011, Kishida (2011) elaboraram um método para determinação de tetraciclinas,
com base na preparação de amostra, que consistiu apenas na ultrafiltração e centrifugação. As
análises foram realizadas em sistema LC-10ADvp da Shimadzu acoplada à coluna Agilent (150
× 4.6 mmID). Os pesquisadores relataram vantagens, no que se refere a não requerer
preparação, principalmente, pela eliminação de reagentes tóxicos, sendo de simples execução
para o monitoramento de rotina em laboratórios.
Em 2013, Yang, Yang e Yan (2013) empregaram adsorvente em zeólita para extração
em fase sólida. O método apresentou vantagens quanto a simplicidade, rápidez e sensibilidade
para determinação de OTC, TC e CTC na água e em leite. As vantagens do adsorvente foram
descritas como: elevada área superficial, porosidade nanoescala permanente e boa estabilidade.
Comprovando este fato, Lv et al (2014) relata que esse tipo de extração tem sido uma ferramenta
poderosa para a separação eficiente e rápida, com base na preconcentração dos resíduos, em

75
amostras de leite. O sistema cromatográfico empregado, pelos pesquisadores, foi o HPLC-DAD
da Shimadzu aliado a coluna C18 (250 × 4.6 mm, 5 mm).
Anteriomente, Kantiani, Farré e Barceló (2009) fizeram uma revisão dos principais
métodos analíticos, que foram desenvolvidos, na última década, com enfoque nos resíduos das
penicilinas. Os pesquisadores incluem descrição das metodologias para detecção de penicilina
em amostras de leite e ração animal. São incluídas abordagens microbiológicas, e
cromatográficas, além dos biossensores.
Diante do grande compêndio de métodos, Bilandzi et al (2011a) validaram metodologia
para identificação rápida de penicilina G, durante três anos, utilizando ensaios microbiológicos
(protocolado pela comissão europeia 2002/657/EC). Posteriomente, os pesquisadores
confirmaram a presença desses antibióticos acima do limite máximo de resíduos, utilizando
HPLC DAD em 12 mg kg-1. Apesar dos resultados evidentes, os pesquisadores não
consideraram os dados um risco toxicológico, pois não afetavam a saúde pública.
Com relação a reportações e aplicações em amostras reais e biológicas, tem-se o trabalho
de Cámara et al (2013), que validaram método, utilizando HPLC DAD para determinação de
resíduos betalactâmicos, em leite de ovelha. Os resultados foram suficientes, pois as
recuperações dos analitos estiveram entre 79 e 96%.
Recentemente na Turquia, Sen e Filazi (2014) desenvolveram um método também em
HPLC DAD para determinação de resíduos de penicilinas, em amostras de leite comerciais.
Porém, o preparo das amostras consistiu em extração por desproteinização, ajustando o pH para
4,5 – 5, seguido por adição de mistura de acetona/clorofórmio, centrifugação e limpeza em filtro
de nylon 0,45 µm. Os resultados, também, foram muitos satisfatórios, abaixo do aceitável pelas
normas legais europeias.
Portanto, os métodos citados, neste capítulo, incluem, geralmente, procedimentos em
três etapas, como: ajuste das condições HPLC-detector (fonte de ionização, analisador,
aquisição dos dados), preparação das amostras e extração dos analitos, conforme os
procedimentos operacionais normatizados pelas legislações. As principais aplicações são
reportadas pela Tabela 3.2 pelos trabalhos mais atuais entre períodos (2007 – 2014).

76
Ta
bel
a 3
.2-D
escr
ição
do
s an
tib
ióti
cos
qu
esti
on
ado
s n
os
estu
do
s re
cen
tes
e p
rin
cip
ais
técn
icas
em
pre
gad
as e
m d
ifer
ente
s m
atri
zes
alim
enta
res
Gru
po
A
brev
iação
F
orm
ula
mo
lecu
lar,
mass
a m
ola
r e
CA
S
Sín
tese
E
stru
tura Q
uím
ica
T
écn
ica
Refe
rên
cia
Bet
alact
âm
ico
s P
EN
G
C1
6H
18N
2O
4S
33
4,3
9 g
/mo
l
61
-33
-6
Pen
icil
liu
m
Fig
ura
2.5
H
PL
C-M
S/M
S
(V
AN
HO
LT
HO
ON
et
al.
, 2
01
0)
Tet
raci
clin
as
TC
C2
2H
24N
2O
8
44
4,4
4g/m
ol
60
-54
-8
Str
epto
myc
es
gen
us
OH
OH
OH
OO
OH
OH
O
N
NH
2
CH
3
CH
3C
H3 H
H
HP
LC
-MS
/MS
(J
ING
et
al.,
20
09
)
OT
C
C2
2H
24N
2O
9
46
0,4
3 g
/mo
l
79
-57
-2
Str
epto
myc
es
rim
osu
s
OH
OH
OH
OH
OO
OH
OH
O
N
NH
2
CH
3C
H3
CH
3
HH
HP
LC
-DA
D
(PÉ
RE
Z-S
ILV
A e
t al
., 2
01
2)
CT
C
C2
2H
23C
lN2O
8
47
8,8
8g/m
ol
57
-62
-5
Str
epto
myc
es
au
reo
faci
ens
Cl
OH
OH
OH
OO
OH
OH
O
N
NH
2
CH
3C
H3
CH
3
HH
HP
LC
-MS
/MS
(M
AS
TR
AN
TO
NI
et a
l., 2
00
7)
DO
X
C2
2H
22N
2O
8
44
4,4
35
g m
oL
-1
56
4-2
5-0
Str
epto
myc
es
gen
us
OH
OH
OH
O
OH
OO
OH
N
NH
2
CH
3
CH
3C
H3
HH
HP
LC
-MS
/MS
(KA
RA
GE
OR
GO
U e
t al
., 2
01
4;
MIR
AN
DA
; R
OD
RÍG
UE
Z;
GA
LÁ
N-V
IDA
L, 2
00
9)
Dia
min
op
irim
idin
a
TM
P
C1
4H
18N
4O
3
29
0,3
2 g
moL
-1
73
8-7
0-5
Sin
téti
co
Mu
taçã
o
cro
mo
ssô
mic
a
CH
3
ON
NN
H2
NH
2O
CH
3
OC
H3
HP
LC
-DA
D
(BIL
AN
DŽ
IĆ e
t al
., 2
01
1a)
Su
lfo
nam
idas
SM
Z
C1
0H
11N
3O
3S
25
3,2
79
g m
oL
-1
72
3-4
6-6
Sin
téti
co
S
OO
O
NH
N
NH
2
CH
3
H
PL
C-M
S/M
S
(NE
BO
T e
t al
., 2
01
2b;
VE
RA
-
CA
ND
IOT
I; O
LIV
IER
I;
GO
ICO
EC
HE
A, 2
01
0)
SD
X
C1
2H
14N
4O
4S
31
0,3
3 g
moL
-1
24
47
-57
-6
Sin
téti
co
SO
O
O
ON
H
NN
NH
2C
H3
CH
3

77
Ta
bel
a 3
.2-D
escr
ição
do
s an
tib
ióti
cos
qu
esti
on
ado
s n
os
estu
do
s re
cen
tes
e p
rin
cip
ais
téc
nic
as e
mp
reg
adas
em
dif
eren
tes
mat
rize
s al
imen
tare
s (c
on
tin
ua
ção
)
Gru
po
A
brev
iação
F
orm
ula
mo
lecu
lar,
mass
a m
ola
r e
CA
S
Sín
tese
E
stru
tura Q
uím
ica
T
écn
ica
R
efe
rên
cia
Am
inog
lico
síd
eo
ST
R
C2
1H
39N
7O
12
58
1, 5
7
57
-92
-1
Str
epto
myc
es
gri
séu
s
NH
NH
OH
OH
O
OH
NH
2
NH
NH
2
NH
O OO
H
O
NH
OH
OH
OH
O
HP
LC
-MS
/MS
(GR
EM
ILO
GIA
NN
I;
ME
GO
UL
AS
; K
OU
PP
AR
IS,
20
10
)
DH
ST
R
C2
1H
41N
7O
12
58
3,5
90
12
8-4
6-1
NH
NH
OH
OH
O
OH
NH
2
NH
NH
2
NH
O OO
H
O
NH
OH
OH
OH
OH
HP
LC
-MS
/MS
Imu
no
ensa
ios
(GR
EM
ILO
GIA
NN
I;
ME
GO
UL
AS
; K
OU
PP
AR
IS,
20
10
; X
U e
t al
., 2
01
1)
GE
N
C2
1H
43N
5O
7
14
03
-66
-3
47
7,5
95
Mic
rom
on
osp
ora
pu
rpu
rea
O
NH
2
NH
OH
O
NH
2
NH
2
O
CH
3
CH
3
O
OH
NH
OH
CH
3
CH
3
H
HP
LC
-MS
/MS
(I
SH
II e
t al
., 2
00
8)
NE
O
C2
3H
52N
6O
25S
3
90
8,8
8
14
04
-04
-2
Str
epto
myc
etes
Sem
i-si
nté
tico
OH
NH
2
OH
NH
2
O
OOH
OH
O
O
NH
2N
H2
OH
NH
2
OH
OH
NH
2
OO
HP
LC
-MS
/MS
(K
AU
FM
AN
N;
BU
TC
HE
R;
MA
DE
N, 2
01
2)

78
Ta
bel
a 3
.2-D
escr
ição
do
s an
tib
ióti
cos
qu
esti
on
ado
s n
os
estu
do
s re
cen
tes
e p
rin
cip
ais
técn
icas
em
pre
gad
as e
m d
ifer
ente
s m
atri
zes
alim
enta
res
(co
nti
nua
ção
)
Gru
po
A
brev
iação
F
orm
ula
mo
lecu
lar,
mass
a m
ola
r e
CA
S
Sín
tese
E
stru
tura Q
uím
ica
T
écn
ica
R
efe
rên
cia
Mac
roli
deo
s
AB
AM
C9
5H
142O
28
Sen
do
,
C4
8H
72O
14 (
B1
a) 8
0%
C4
7H
70O
14 (
B1
b)
20
%
71
751
-41
-2
Mic
rom
on
osp
ora
pu
rpu
rea
OO
O
OH
OO
O
H
H
O
O
OO
O
H
HR
H
OH
HOH
B1
a: R
= -
CH
2C
H3
B1
b:
R =
-C
H3
HP
LC
-MS
/MS
(A
GU
ILE
RA
-LU
IZ e
t al
., 2
00
8)
IVE
RM
C9
5H
146O
28
Sen
do
,
C4
8H
74O
14 (
B1
a) 8
0%
C4
7H
72O
14 (
B1
b)
20
%
17
36
, 1
58
94
70
288
-86
-7
OO
O
O OH
OH
O
O
OOO
OOO
H
R
HH
H
H
B
1a:
R
= -
CH
2C
H3
B1
b:
R =
-C
H3
HP
LC
-MS
/MS
(I
SH
II e
t al
., 2
00
8)
Fen
icois
C
LL
C1
1H
12C
l 2N
2O
5
32
3,1
3g m
oL
-1
56
-75
-7
Str
epto
my
ces
ven
ezu
elae
Cl
Cl
OH
OH
O
O-
O
NH
N+
H
HP
LC
-DA
D
(VA
RG
AS
MA
MA
NI;
RE
YE
S
RE
YE
S;
RA
TH
, 2
00
9a)
Qu
ino
lonas
EN
R
C1
9H
22F
N3O
3
35
9,3
95
93
106
-60
-9
Sin
téti
co
F
OO
OH
NN
NC
H3
UP
LC
–M
S/M
S
(HA
N e
t al
., 2
01
5)

79
Para uma compreensão geral, durante período de 1983 a 2016 (início), foram registrados
progressos na literatura cientifica apresentados pelos Gráficos da Figura 3.5. Os dados são
coletados da Base de dados Scopus, a pesquisa foi realizada no ano de 2015.
Figura 3.5-Tendência do avanço da literatura científica no período de 1983 a 2016 (fev) para quantificação de
resíduos de penicilina, em função da nacionalidade dos pesquisadores.
Adaptado da base de dados Scopus, 2015
Os dados e esclarecimentos são apresentados nos gráficos. consideraram a relação
publicação e país de origem dos autores (A), bem como a tendência dos trabalhos durante
períodos citados (B). As informações foram coletadas do Portal de Periódicos Capes; o
mecanismo de busca tem sido antibiotics in Milk by HPLC. Os dados mostram claramente que
os principais publicadores estão em países mais emergentes.

80
3.2.4 Plano de Controle e Monitoramento de Resíduos de Antimicrobianos executados
no Brasil
No Brasil, os Laboratórios do Ministério da Agricultura Pecuária e Abastecimento
(Lanagros) atuam na realização de programas de vigilância, desenvolvem métodos analíticos,
proporcionam assessoramento técnico na elaboração de diretrizes e normativas e realizam
negociação internacional. Os Lanagros trabalham, segundo regulamentação ISO 17025, sendo
preparados para responder a surtos e crises, relacionadas à presença de resíduos e contaminantes
em alimentos, com disponibilidade, rapidez e confiabilidade que requer uma emergência (DE
QUEIROZ MAURICIO; LINS, 2012).
O Plano Nacional de Controle de Resíduos e Contaminantes (PNCRC) assegura que o
produto que chega ao consumidor, tanto interno como externo, está livre de resíduos de
substâncias perigosas, em níveis não aceitáveis. Para isto, a qualidade e segurança do leite são
atestadas, pelas análises realizadas na Rede Nacional de Laboratórios Agropecuários. No Brasil
há seis Lanagros localizados nas cinco regiões principais do país, estando no Sudeste (Pedro
Leopoldo/MG e Campinas/SP), no Sul (Porto Alegre/RS), no Centro-Oeste (Goiânia/GO), no
Nordeste (Recife/PE) e no Norte (Belém/PA). Desta forma, o PNCRC funciona do seguinte
modo: os contaminantes a serem analisados por técnicas de identificação quantificação e
confirmação são selecionados, de acordo com seu risco em potencial e pela disponibilidade das
metodologias. Todas as informações são registradas no cronograma de monitoramento e
renovadas anualmente. A saber, que os Limites Máximos de Resíduos de medicamentos
veterinários são harmonizados no MERCOSUL e seguem a Resolução GMC nº. 54/2000. Para
compostos cujos valores de LMR não estão estabelecidos pelo MERCOSUL, considerem-se os
valores da Codex Alimentarius (BRASIL., 2010).
Em 21 de Julho de 2015 o MAPA divulgou os subprogramas PNCRC para carnes, leite,
pescado, mel e ovos. Esta publicação foi mostrada na Instrução Normativa nº 13, exposta no
Diário Oficial da União. Os subprogramas estabelecem metas de análise de resíduos e
contaminantes nos alimentos de origem animal (MAPA, 2015). Esse monitoramento é tão
importante para o Brasil que, o plano amplia o rastreio de substâncias em ovos e lácteos, o que
garantir maior qualidade desses produtos e também permitir a exportação para determinados
mercados externos rigorosos (União Europeia) quanto ao rastreio de contaminantes nesses
alimentos.

81
Ainda com relação aos programas de monitoramento, no Brasil, também, tem-se o
Programa de Análise de Resíduos de Medicamentos Veterinários em Alimentos de Origem
Animal (PamVet), este foi estabelecido pela ANVISA, em 2002, a fim de complementar as
ações executadas pelo MAPA. Conforme determina a Lei n. 9.782, de 26 de janeiro de 1999 no
Art. 8º, § 1º, inciso II, o PamVet foi desenvolvido com o objetivo de operacionalizar sua
competência legal de controlar e fiscalizar resíduos de medicamentos veterinários em alimentos
(ANVISA, 2003). O principal objetivo é analisar os alimentos que são de consumo direto a
população brasileira, isto é criar um banco de dados sobre resíduos da ocorrência dos compostos
em alimentos, no Brasil, disponibilizando as informações à sociedade (ANVISA, 2005). Para
isso, a coleta das amostras é realizada em pontos de vendas e analisadas em laboratórios
credenciados pelas Vigilâncias sanitárias estaduais (VISA).
É necessário mencionar que, as técnicas de amostragem seguem as prescrições da Codex
Alimentarius, descritos nos guias de amostragem (CODEX ALIMENTARIUS, 1993). Sendo o
leite um alimento é o mais consumido pela população brasileira e de grande importância para
as crianças e idosos, a primeira matriz de análise pelo PamVet, foi este alimento mais exposto
a saúde pública.
3.2.4.1 Correlação entre os Métodos Aplicados no Controle de Resíduos no Brasil e as
Produções Científicas
Cada laboratório possui programas de análises, que utilizam métodos cromatográficos
válidos para detecção de contaminantes. Para resíduos de Avermectinas, as análises são
realizadas utilizando CLAE-FL e CLAE-EM/EM, executadas no Lanagro de MG. Alguns
resultados têm sido publicados, como é o caso de uma edição especial na Revista “Food
Additives and Contaminants”, nos quais são apresentados os principais resultados das análises
realizadas nos Laboratórios credenciados.
Alguns métodos Bioanalíticos são aplicados como testes rápidos. Tem-se registro do
trabalho de Hoff et al (2012) que aplicam o método FAST e Premium® test, utilizando uma
placa de Agar Muller-Hinton, contendo esporos de Bacillus megaterium, para inibição do
crescimento bacteriano, o teste revela positivo quando há presença dos resíduos. Segundo os
pesquisadores da Lanagro/RS o método Screening foi primeiro utilizado para rastrear amostras
de rim. Enquanto que O Premium® test tem sido aplicado para fluidos e tecidos.

82
Recentemente, os pesquisadores, da Universidade do Paraná, discutem o método de
rastreio de cloranfenicol, para análise de leite cru proveniente de uma indústria. Os
pesquisadores utilizaram o método ELISA (SIFUENTES DOS SANTOS et al., 2015).
Ainda de acordo com Trombete, Santos e Souza (2014), estudos relatam que é mais
provável a contaminação, por sulfonamidas e tetraciclinas no leite comercializado no país.
As Informações obtidas pela base de dados, Scopus, confirmam a tendência dos avanços
dos programas do Brasil para controle resíduo. Os dados foram coletados entre o período de
1990 a 2015. O mecanismo de busca tem sido “Antibiotics milk Brazil”.
Assim, os fatores apresentados na Figura 3.6, reflete em grandes preocupações com a
saúde dos consumidos, quanto a presença de resíduos.
Figura 3.6-Citação do Brasil e aplicação de métodos para controle, em função do número de publicação e países
Adaptado da base de dados Scopus, 2015
Em 2005, Farina et al (2005) discutiram de forma comparativa as abordagens, para
controle de qualidade no leite para exportação. Os autores focaram no setor de processamento
de laticínios na Argentina e no Brasil, mostrando que entre década de 50 e 90 houve um avanço
no desenvolvimento de normas e programas, para garantir a segurança e higiene deste alimento.
Em 2012, os pesquisadores da Lanagro/MG, Nonaka et al (2012) descrevem a
importância das diretrizes, estabelecidas pela Codex Alimentarius para o PNCRC. Segundo
Argentina
2%
Colombia
1%
República
Dominicana
1%
Equador
1%
Guatemala
1%
Alemanha
4%
Espanha
4%
Suíça
4%
Estados Unidos
4%
Brazil
78%
Outra
6%

83
eles, em 2008 a 2009, foram investigados antibióticos dos grupos dos aminoglicosídeo, em
diversas matrizes animal, para tal foram utilizados testes rápidos e métodos cromatográficos
LC-MS/MS e UPLC-MS/MS. O monitoramento envolveu amostras provenientes de
matadouros e produtores.
Rezende et al (2012) desenvolveram um método, utilizando sistema UPLC-MS/MS,
para quantificação de resíduos betalactâmicos e tetraciclinas, presentes em músculo bovino, de
acordo com o regulamento 2002/657/CE. Os resultados foram satisfatórios, pois apresentaram
recuperações de analito entre 90 a 110%.
Na Figura 3.7 são apresentadas as filiações dos pesquisadores que desenvolveram
trabalhos na área de controle de resíduos. Os principais dados no Brasil foram pesquisados para
os anos de 1990 a 2015.
Figura 3.7-Distribuição do número de publicações em função da filiação dos autores
Adaptado da base de dados Scopus, 2015
Assim, a busca pela melhoria da qualidade dos alimentos expostos ao consumo humano,
está de forma presente, principalmente, nos artigos de pesquisa proveniente de instituições
Acadêmicas. Dentre a distribuição proporcional das instituições acadêmicas, os principais
artigos publicados destacam-se em 16 % desenvolvido por pesquisadores da UNESP. O que
mais chama atenção, nesta busca, até o presente momento, é a ausência de publicações
EMBRAPA
6%UFV
6% Instituto de
Zootecnia SP
6%
UFRJ
8%
UFRS
8%
FIOCRUZ
8%
UFPE
8%
UFRPE
10%
UEM
10%
USP
14%
UNESP
16%

84
realizadas no Estado do Rio Grande do Norte, uma vez que este possui um aumento
significativo de produção e exportação de alimentos de origem animal.

85
3.3 ESTRATÉGIAS PARA VALIDAÇÃO DOS MÉTODOS
A validação do método de análise visa diminuir ou controlar os fatores, que levam a
imprecisão ou inexatidão de um dado gerado (CHASIN et al., 1998). Assim, o procedimento
analítico tem como meta demonstrar que o mesmo é adequado aos objetivos propostos, ou seja,
que os parâmetros de desempenho avaliados atendem aos critérios de aceitação preconizados
(ANVISA, 2003). De maneira que, para um método ser válido, deve ter-se garantia da
confiabilidade das medidas analíticas, através de sua precisão, exatidão e robustez. Os guias de
orientação emitidos por legislações como a União de Química Pura e Aplicada (IUPAC) e
Eurachem, CAC, FDA, EC e MAPA compõe um cenário de organizações, que estabelecem
critérios e normas, para validação dos métodos analíticos, dos mais simples (testes rápidos) aos
mais robustos (cromatográficos).
O principal requisito para que métodos analíticos válidos sejam aplicados, no controlo
de resíduos de antibióticos, em alimentos: é que sejam incluídas nas normas e aprovadas, como
medida regulatória do medicamento, usados em produtores de alimentos. Geralmente, os
procedimentos são desenvolvidos e validados apenas para um composto, dependendo da
natureza do resíduo. Entretanto, para as autoridades responsáveis pela execução dos programas,
métodos individuais de resíduos, são muitas vezes limitados, pois sempre há vários
medicamentos da mesma classe, disponível para uso. Logo, são estabelecidos para análises de
rotinas.
Numerosas organizações são fontes de informação, porém apresentam exigências
diferentes, por isso a escolha de uma fonte apropriada de orientação, a seguir, é importante. As
fontes de orientação podem ser agrupadas em fontes científicas independentes, como a IUPAC,
Eurachem, e AOAC Internacional, e os elaborados pelas autoridades reguladoras nacionais e
regionais e organizações internacionais com interesse no estabelecimento de normas
internacionais reguladoras nacionais e práticas, tais como: CAC e o VICH, enquanto que para
organizações nacionais regulamentares sobre métodos incluem a EC, FDA e o MAPA. As
orientações fornecidas por organizações científicas independentes são, geralmente, aplicáveis
a uma grande variedade de procedimentos analíticos.

86
A função de cada organização no cenário atual de desenvolvimento de métodos de
validação, para o controle de contaminantes, em alimentos expostos ao consumo, são
apresentadas na Tabela 3.3 das seguintes laudas deste trabalho.
Tabela 3.3-Resumo das principais Normatizações e órgãos reguladores e Aplicação de Controle de
Contaminantes
Autoridade Competências Base Legal Aplicação
Técnica Referência
Codex
Alimentarius
Estabelece LMR, com base na
toxicidade e farmacocinética de
cada substância.
CAC/GL56-2005
CAC/MRL02-
2009
HPLCDAD
HPLC-FL
HPLC-MS
Método
enzimático
(IBARRA et al.,
2011; SANG et al.,
2013)
FDA
Estabelece LMR, com base na
toxicidade e farmacocinética de
cada substância.
57245–57248
HPLC-FL
HPLC-UV
HPLC-
MS/MS
(BOGIALLI et al.,
2007; MOATS;
HARIK-KHAN,
1995; MUSSER et
al., 2001)
CVMP
Apresenta Guias para validação
de método rápida para resíduos de
medicamentos veterinários
2002/657/CE HPLC-
MS/MS
(IGLESIAS et al.,
2012; NEBOT et
al., 2012a, 2012b)
MAPA
Estabelece programas e planos de
análises de resíduos de
antimicrobianos, em diversos
alimentos expostos ao consumo
humano um dos programas é o
PNCRC e o PamVet
Instrução
normativa nº 9, de
10 de abril de
2008
HPLC-UV
HPLC-MS
(NONAKA et al.,
2012; VARGAS
MAMANI; REYES
REYES; RATH,
2009)
Neste contexto, as instituições propostas pela FAO (como Codex) são muito
importantes, pois países menos desenvolvidos (como já citado neste trabalho), não têm a
capacidade, recursos e, em alguns casos, o conhecimento técnico para enfrentar os desafios do
comércio internacional de alimentos é indisponível. Sabendo que, é de extrema importância
reforçar as medidas de qualidade e segurança alimentar, a fim de melhorar a competitividade
no mercado internacional. Desta forma, a FAO implanta alguns projetos de assistência técnica
para melhorar a qualidade de alimentos (MIYAGISHIMA et al., 1995).

87
Com relação a isso, Boutrif (2003) e Horton (2001) relata que essa assistência é
constituída por: apreciações dos programas existentes de controle de alimentos nacional,
assistência no estabelecimento de legislação alimentar, inspeção dos alimentos, treinamento nas
boas práticas de fabricação, certificado de inspeção de qualidade, análise de alimentos, sistemas
de garantia de qualidade e de gestão em laboratório.
Assim, o lema do Codex é que os consumidores podem confiar na segurança e qualidade
dos produtos alimentares importados, uma vez que seguem padrões de qualidade legítimos e
oficiais (ELLIS, 2008; FAO, 2003; SECRETARIAT OF THE CODEX ALIMENTARIUS
COMMISSION, 2011). Essa Organização baseia-se em evidências científicas, valores
econômicos e sociais. Os documentos fornecem modelos para nações individuais, mas não são
vinculadas as políticas nacionais, isto é não é proveniente de nenhuma lei, instituída por
autoridades nacionais (RANDELL; WHITEHEAD, 1997; SOMOGYI et al., 2011). Nesta
coordenação existem três comitês nomeados responsáveis, pelas validações dos métodos de
resíduos, dentre eles está o de resíduos de medicamentos veterinários (CCRVDF)
(CRAWFORD; KUGLER, 1986). O objetivo é determinar prioridades para a consideração de
resíduos, recomendar LMR, desenvolver códigos de boas práticas, a considerar métodos de
amostragem e análise para a determinação de resíduos s em alimentos (DEMORTAIN, 2012).
A escolha dos critérios de adequação é estabelecida pelos guias de validação emitidos
por órgãos oficiais. De maneira que, a validação consistir, especialmente, no cálculo das figuras
de mérito tais como: precisão, exatidão, limite de detecção, limite de quantificação,
sensibilidade e robustez do método (ANVISA, 2009).
Em 2005, Stolker e Brinkman (2005) revisaram os principais métodos utilizados, para
as classes dos antibióticos entre eles os betalactâmicos. Chegando-se a conclusão que o HPLC-
MS/MS tem sido a técnica preferida, na maioria dos casos de confirmação dos resíduos. Os
autores ainda afirmam que, apesar de existirem muitas ferramentas analíticas, para atender as
demandas dos programas de fiscalização de amostras contaminadas, há várias questões
levantadas, no intuito de minimizar os problemas encontrados nos procedimentos de extração,
no que se diz respeito a recuperação dos compostos. Contrariamente, Repizo et al (2012)
enfatiza que a detecção, por ionização em espectrometria de massas apresenta desvantagem,
pois é suscetível a efeito de matriz. Como a resposta do padrão por espectrometria de massas
em cromatografia líquida pode diferir nos resultados, os pesquisadores, desenvolveram uma
estratégia para minimizar estes efeitos, comparando o sinal do analito, em matrizes dos analitos,
com o sinal de solvente puro, sendo possível gerar curvas analíticas referentes às matrizes puras.

88
3.3.1 Características de um Método Validado
Indiscutivelmente, as orientações e guias de validação estão em constante avanço,
porém, são as preconizações, descritas, que garantem o bom planejamento das condições para
validação das metodologias analíticas.
Alguns comentários têm sido reportados pela bibliografia científica para diversas
aplicações. A exemplo de Dadgar at al (1995) que fazem uma explanação sobre o estudo da
seletividade nos inter e intra-ensaios, onde são discutidos os tipos de interferentes, que podem
influenciar o estudo destas figuras, desde a presença de substâncias endógenas, produtos de
degradação e até mesmo a contaminação por interferentes do ar e ambiente. Com relação aos
parâmetros da curva analítica, os pesquisadores afirmam que o limite de quantificação deve ser
referenciado a partir do limite de detecção, sendo o LQ três vezes maior que o LD. Ainda,
segundo Chasin et al. (1998), o LD é a menor concentração ou quantidade de uma substância
que pode ser identificada, por um procedimento analítico com um nível de confiança
especificado sendo, estatisticamente, diferenciada do ruído. Van de Merbel (2008) mostra as
desvantagens para determinação quantitativa dos compostos endógenos, em amostras
biológicas, por técnicas cromatográficas e enfatiza o efeito de matriz. Neste ponto, Cassiano et
al.(2009) e Matuszewski, Constanzer e Chavez-et al (2003b) enfatizam que esse efeito ocorre
quando substâncias inerentes à matriz biológica concorrem com os compostos de interesse,
comprometendo os a eficiência dos métodos Bioanalíticos, tais como: HPLC/fluorescência,
HPLC/UV, e HPLC-MS/MS.
A ANVISA (2003) propõe um guia de validação, onde é expresso o cálculo das figuras
de mérito, o desenvolvimento de metodologia tem sido aceito em todo o país. Inicialmente, para
Shabir (2003), a robustez de um método consiste na capacidade de não ser afetado por pequenas
variações, deliberado pelos parâmetros dos procedimentos. De maneira que, esse parâmetro, na
cromatografia, é avaliado, variando-se as condições como: solvente orgânico, pH do tampão na
fase móvel, força iônica, colunas e temperatura. Shah et al (2000), mostra que uma validação
bem-sucedida, reúne critérios específicos de aceitação para a exatidão e precisão.
A Tabela 3.4 apresenta as principais características de um método bem executado, sendo
que esta descrição. Além disso, as instruções, sobre as figuras de mérito, presentes nos manuais
da Emea (2012a), Codex Alimentarius (2014), ICH (2005) e trabalhos de Bressole, Bromet-
Petit e Audran (1996).

89
Já o guia do FDA (2011) confirma que a avaliação da robustez deve ser considerada,
durante toda a fase de desenvolvimento do método, em estudo, mostrando a confiabilidade de
uma análise, considerando as variações em parâmetros do método.
Tabela 3.4-Comparação dos requisitos de validação preconizados para quantificação dos resíduos em alimentos
Características Norma legal praticada pelas organizações
CAC/GL 72-2009 2002/657/EC VICH GL 49
Estabilidade do
analito
É o estudo de
armazenamento por um
período que se estende até,
90 dias além do tempo
esperado, para o rastreio,
quantificação e
confirmação do analito.
Sempre que possível, usa-
se amostras reais. Quando
não, é utilizado a matriz
fortificada com a
substância. Análises são
realizadas a cada 1, 2, 4 e
20 semanas, as amostras
armazenadas a -20 ºC.
Controle do tempo de
armazenamento e suas
condições. Esta etapa é parte
do processo de validação.
Realização de um estudo de
estabilidade da matriz. As
amostras devem ser
fortificadas (3 dias).
Teste de
robustez
O teste utilizando o
controle dos pontos
críticos. Variando-se as
condições analítica tais
como: nos volumes de
reagentes,
concentrações, pH, tempo
de retenção, qualidade
reagente e lotes.
Avaliação dos métodos
incluem espécies, matrizes
e abertura de amostras
diferentes. A importância
das é avaliada, utilizando a
abordagem de Youden.
Induzir situações de
modificações ao longo do
tempo. As mudanças podem
incluir: lotes de reagentes,
composição e volume,
número de extrações, marca
coluna.
Curva Analítica
É a primeira etapa que
avalia a resposta dos
padrões, diante faixa de
trabalho. Estas
concentrações (mínimo de
cinco branco). Deve
recobrir o LMR
Para construção da curva
deve ser utilizada, pelo
menos, cinco níveis
(incluindo o zero).
Definindo o intervalo de
concentração, indicando a
equação da curva,
É determinada por toda a
faixa de trabalho da
concentração do analito na
matriz. A execução depende
de três tipos de metodologia:
padrões de solvente/tampão,
matriz de controle e adição
de padrões.
Linearidade
É determinada pela análise
de regressão linear
admitindo a resposta
linear.
NF
É por equação linear,
definida por relação
concentração versus a
resposta 5 níveis de
concentrações.
Teste de
robustez
O teste utilizando o
controle dos pontos
críticos. Variando-se as
condições nos volumes de
reagentes, concentrações,
pH, tempo de retenção,
qualidade reagentes.
Avaliação dos métodos
incluem espécies, matrizes
e abertura de amostras
diferentes. A importância
das é avaliada por Youden.
Induzir situações de
modificações, que podem
incluir: lotes de reagentes,
composição e volume,
número de extrações, marca
coluna analítica e lotes e
eluição HPLC
Sensibilidade
É a concentração mais
baixa à qual o analito pode
ser detectado, dentro de
limites estatísticos.
Calculado com adição de
padrão interno.
Simultaneamente com a
robustez.
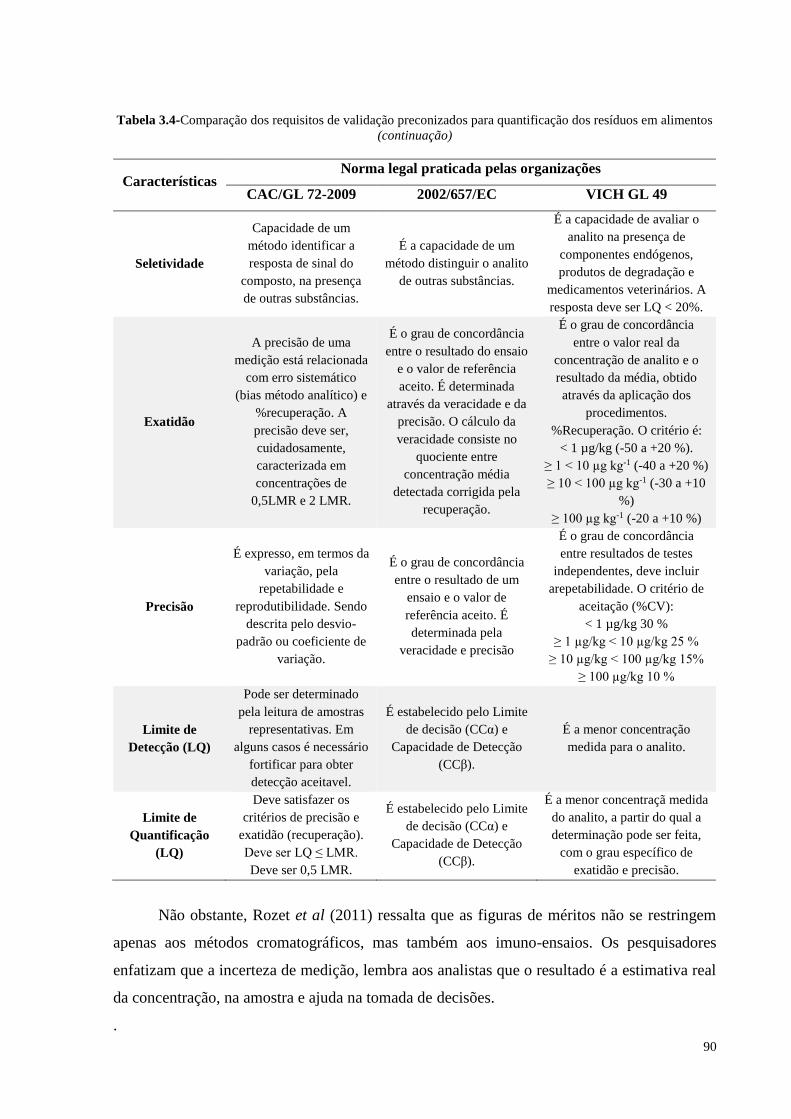
90
Tabela 3.4-Comparação dos requisitos de validação preconizados para quantificação dos resíduos em alimentos
(continuação)
Características Norma legal praticada pelas organizações
CAC/GL 72-2009 2002/657/EC VICH GL 49
Seletividade
Capacidade de um
método identificar a
resposta de sinal do
composto, na presença
de outras substâncias.
É a capacidade de um
método distinguir o analito
de outras substâncias.
É a capacidade de avaliar o
analito na presença de
componentes endógenos,
produtos de degradação e
medicamentos veterinários. A
resposta deve ser LQ < 20%.
Exatidão
A precisão de uma
medição está relacionada
com erro sistemático
(bias método analítico) e
%recuperação. A
precisão deve ser,
cuidadosamente,
caracterizada em
concentrações de
0,5LMR e 2 LMR.
É o grau de concordância
entre o resultado do ensaio
e o valor de referência
aceito. É determinada
através da veracidade e da
precisão. O cálculo da
veracidade consiste no
quociente entre
concentração média
detectada corrigida pela
recuperação.
É o grau de concordância
entre o valor real da
concentração de analito e o
resultado da média, obtido
através da aplicação dos
procedimentos.
%Recuperação. O critério é:
< 1 µg/kg (-50 a +20 %).
≥ 1 < 10 µg kg-1 (-40 a +20 %)
≥ 10 < 100 µg kg-1 (-30 a +10
%)
≥ 100 µg kg-1 (-20 a +10 %)
Precisão
É expresso, em termos da
variação, pela
repetabilidade e
reprodutibilidade. Sendo
descrita pelo desvio-
padrão ou coeficiente de
variação.
É o grau de concordância
entre o resultado de um
ensaio e o valor de
referência aceito. É
determinada pela
veracidade e precisão
É o grau de concordância
entre resultados de testes
independentes, deve incluir
arepetabilidade. O critério de
aceitação (%CV):
< 1 µg/kg 30 %
≥ 1 µg/kg < 10 µg/kg 25 %
≥ 10 µg/kg < 100 µg/kg 15%
≥ 100 µg/kg 10 %
Limite de
Detecção (LQ)
Pode ser determinado
pela leitura de amostras
representativas. Em
alguns casos é necessário
fortificar para obter
detecção aceitavel.
É estabelecido pelo Limite
de decisão (CCα) e
Capacidade de Detecção
(CCβ).
É a menor concentração
medida para o analito.
Limite de
Quantificação
(LQ)
Deve satisfazer os
critérios de precisão e
exatidão (recuperação).
Deve ser LQ ≤ LMR.
Deve ser 0,5 LMR.
É estabelecido pelo Limite
de decisão (CCα) e
Capacidade de Detecção
(CCβ).
É a menor concentraçã medida
do analito, a partir do qual a
determinação pode ser feita,
com o grau específico de
exatidão e precisão.
Não obstante, Rozet et al (2011) ressalta que as figuras de méritos não se restringem
apenas aos métodos cromatográficos, mas também aos imuno-ensaios. Os pesquisadores
enfatizam que a incerteza de medição, lembra aos analistas que o resultado é a estimativa real
da concentração, na amostra e ajuda na tomada de decisões.
.

CAPÍTULO 4
METODOLOGIA

92
4 METODOLOGIA
Todas as análises foram realizadas nas dependências da Universidade Federal do Rio
Grande do Norte no Instituto de Química em Laboratórios da Central Analítica. As
metodologias foram aplicadas, exclusivamente, para fins Acadêmicos, satisfazendo todo o
cronograma para a execução desta Tese.
4.1 DESENVOLVIMENTO DE MÉTODO PARA IDENTIDADE E QUALIDADE DOS
MEDICAMENTOS A BASE DE BENZILPENICILINA
4.1.1 Reagentes e Solventes
Acetona P A ACS (C3H6O), Álcool Etílico (C3H6O) da Synth (São Paulo, Brasil).
Os reagentes para análise em UFLC-DAD foram grau analítico. A acetonitrila foi
fornecida pela Merck (Darmstadt, Alemanha) com pureza de 99,9%. Água foi purificada do
sistema Millipore.
O padrão analítico certificado VENTRANAL™ foi a Benzilpenicilina Potássica
(C16H17KN2O4S) adquirido da Sigma Aldrich (Steinheim, Alemanha), conforme certificado de
análise (ANEXO I).
4.1.2 Instrumentação
Balança analítica de precisão Marte (Minas Gerais, Brasil). Aparelho de para banhos
ultrassônicos foi o USC1600 Unique Group (São Paulo, Brasil).
Espectrômetro no infravermelho de bancada da Perkin Elmer Spectrum 100 FT-IR
(Waltham, Massachusetts, Estados Unidos). As análises foram realizadas em FTIR. Os dados
espectrais foram recolhidos em 4000 a 650 cm-1 em software Spectrum 10. As medidas foram
processadas em ATR com cristal de seleneto de zinco (ZnSe)
Espectrômetro na região do UV e NIR da Cary 5000 UV-Vis-NIR da Agilent
Technologies (Santa Clara, Califórnia, Estados Unidos).
As análises cromatográficas foram realizadas utilizando sistema UFLC-DAD Shimadzu
(Kyoto, Japão) acoplado com detector de UV 254 nm, apresentado na Figura 4.1.

93
Figura 4.1-Ilustração do Sistema UFLC-DAD
O autor, 2015
Esse aparelho é constituído por um compartimento (rack) com capacidade para 61
amostras (A), injetor por loop com injeção máxima de 100 µL (B), bombas reguladoras de
pressão para fase A sistema LC- 20AD (C), Bombas reguladoras de pressão para fase B sistema
LC – 20AD (D), Forno da coluna com máxima temperatura de 40 ºC (E), Detectores por arranjo
de fotodiodos módulo CBM – 20 A (F), Frascos para fase móvel (G).
4.1.3 Outros materiais para Cromatografia
Coluna de fase reversa C18 (2,0 mm × 30 mm ID) Shim-pack XR-ODS (Kyoto, Japão).
Filtro de seringa nylon (13 mm × 0,45 µm) código 2204513500 adquiridos da Nova Analítica
Importação e Exportação LTDA (São Paulo, Brazil). Vials de âmbar de 2mL adquiridos
também da Nova Analítica código 10100209200.
As padronizações das soluções foram realizadas em balão volumétrico de 10 mL e 50
mL. Micropipetas de volumes máximo de 20 µL e 100 µL.

94
4.1.4 Amostras de medicamentos
As amostras foram adquiridas em comércios de produtos veterinários, localizados em
Natal/RN, no período entre 2013 – 2015. A escolha das amostras foi satisfeita, segundo a
utilização na agricultura brasileira para sanidade de animais produtores de leite (bovinos e
caprinos).
As amostras foram de doze marcas diferentes com origem em dois laboratórios do
Brasil. Um total de 37 amostras de medicamento foram utilizadas para calibração do método.
Todos os medicamentos utilizados para análises são registrados no CPV/SIDAN. Os
terapêuticos estão em conformidade com o Decreto nº 5.053, de 22 de abril de 2004 no Capítulo
VI Art. 39 quando trata do registro dos produtos de uso veterinário.
Assim, os produto comercializados a base de penicilina, seguiram as informações de
obrigatoriedades, principalmente, quanto a rotulagem que são: nome completo, legenda USO
VETERINÁRIO, descrição dos ingredientes ativos, indicação, doses por espécie do animal,
advertências, condições de armazenamento, período de carência, quando for o caso declaração
de receita, nome do órgão que registrou, número e data de registro, nome e CNPJ do
estabelecimento detentor do registro, nome e número do registro técnico do profissional,
codificação definida pelo MAPA, fabricação e vencimento.

95
A Tabela 4.1 apresenta as informações principais dos terapêuticos, tais como:
concentração, indicação e período para eliminação em animais lactantes (período de carência).
Tabela 4.1-Descrição dos medicamentos analisados, segundo CPV/SIDAN
Grupos Descrição da Fórmula Carência
(horas) Indicação Reg. MAPA
G1
PENG BEN (1.200.000 UI), PENG
PROC (600.000 UI), PENG K+
(600.000 UI), DIISTR (500 mg) STR
(500 mg) em solução de 6 mL
120
Mastitis causadas por
Streptococcus spp., Bacillus spp.,
Escherichia spp., Staphylococcus
spp.,
1.804 em
09/08/62
G2
PENG PROC (1.500.000 UI), PENG
K+ (1.500.000 UI) DIISTR (1.250 mg),
STR (1.250 mg) em solução 15 mL
96
Mastitis causadas por
Streptococcus spp., Bacillus spp.,
Escherichia spp., Staphylococcus
spp
6.330 em
13/03/98
G3 OTC (0,2 g mL-1) 96
Pneumonia (Pasteurella,
Corynebacterium, Klebsiella,
Haemophilus e Bordetella)
0801 em
29/11/78
G4 OTC (0,2 g ml) 96 Pneumonia causadas por
Pasteurella
1.630 em
04/08/60
G5 OTCDII 0,3 g ml NA
Pneumonia causadas por
Pasteurella, Corynebacterium,
Klebsiella
4834 em
09/09/94
G6 TC (2,00 g)
Ac. Ascorb (1,50 g) NA
Infecções Pulmonares causadas
por Mycoplasma sp
3681/91 em
16/04/1991
G7 IVERM (0,0225 g mL-1) ABAM
(0,0125 g mL-1) NA
Gastrintestinais e pulmonares
causadas por nematódeos 8691/03
G8 SMZ (0,020,0 g mL)
TMP (0,04,0 g mL)
72 (para
bovinos)
Respiratórias causadas por
Haemophilus sp, Bordetella sp, 5.366/95
G9 SDX (0,20,0 g mL)
TMP (0,04,0 g mL) 24
Mastites causadas por
Enterobacter aerogenes,
Staphylococcus aureus
7570/00
G10 GEN 3 mg mL-1 24 Metrites e endometrites
(Staphylococcus spp)
3.821 em
11/10/91
G11 ENR (0,05g mL-1) 120
Escherichia coli, Proteus spp.
Clostridium perfringes 6.665 em
5/01/99
G12 DOX 0,046 g mL-1 NA Clostridium tetani 8.528 em
10/04/2003
NA: lactantes não devem ser tratadas quando o leite for destinado ao consumo humano.

96
As amostras contendo PENG foram separadas e quantificadas por Polarimetria,
Cromatografia e UV VIS NIR. As informações das amostras são apresentadas na Tabela 4.2.
Tabela 4.2-Amostragem dos medicamentos (misturas) para identificação e quantificação
Amostra Grupos
Identificação Quantificação
Cromatogramas Espectros IF
médio Cromatografia
UV VIS
NIR
1 G1 A I A1 A1
2 G1 B I A2 A2
3 G2 B II A3 A3
4 G2 B II A4 A4
5 G2 B II A5 A5
6 G2 B II A6 A6
7 G2 B II A7 A7
8 G2 B II A8 A8
9 G2 B II A9 A9
10 G2 B II A10 A10
11 G2 B II A11 A11
12 G2 B II A12 A12
13 G2 B II B13 B13
14 G3, G4, G5, G6, G8, G9,
G10 e G7 D
I, II, III, IV, V e
VI B14 B14
15 G2 B II B15 B15
16 G2 B II B16 B16
17 G2 B II B17 B17
18 G3, G4, G5, G6, G8, G9,
G10 e G7 D
I, II, III, IV, V e
VI B18 B18
19 G3, G4, G5, G6, G8, G9,
G10 e G7 D
I, II, III, IV, V e
VI B19 B19
20 G2 B II B20 B20
21 G1 e G3 C I e II B21 B21
22 G1 e G4 C I e IV B22 B22
23 G1 e G5 C I B23 B23
24 G1 A VI B24 B24
25 G1 B VI B25 B25
26 G2 B VI B26 B26
27 G2 B VI B27 B27
28 G2 B VI B28 B28
29 G2 B VI B29 B29
30 G2 B VI B30 B30
31 G2 B VI B31 B31
32 G2 B VI B32 B32
33 G3, G4, G5, G6, G8, G9,
G10 e G7 C
I, II, III, IV, V e
VI B33 B33
34 G3, G4, G5, G6, G8, G9,
G10 e G7 C
I, II, III, IV, V e
VI B34 B34
35 G3, G4, G5, G6, G8, G9,
G10 e G7 C
I, II, III, IV, V e
VI B35 B35
36 G3, G4, G5, G6, G8, G9,
G10 e G7 C
I, II, III, IV, V e
VI B36 B36
37 G3, G4, G5, G6, G8, G9,
G10 e G7 C
I, II, III, IV, V e
VI B37 B37

97
4.1.5 Espectroscopia no infravermelho médio
As medidas foram realizadas seguindo o método 01/2008:202224 da European
Pharmacopoea (2008) e trabalho de Sivakesava e Irudayaraj (2002) para caracterização de
antibióticos por FTIR IV.
As amostras foram preparadas em concentração padrão de 2 mg mL-1 em balão
volumétrico de 10 mL. As soluções foram armazenadas em frascos de âmbar e refrigeradas a -
4 ºC.
Na execução das medidas as amostras foram dispostas no acessório de refletância
atenuada total (ATR), onde ocorre a transferência da radiação infravermelha que foi direcionada
para o cristal. Em seguida, o cristal foi, cuidadosamente, limpo com acetona entre sucessivas
medições, e colecionado espectros de branco, para garantir a ausência de resíduos.
As amostras de medicamentos contendo PENG foram preparadas, simultaneamente,
com o espectro de referência padrão.
Os dados espectrais foram coletados em software Spectrum 10. As análises estatísticas
foram realizadas em software OriginProLab 9.0.

98
4.1.6 Análises por Cromatografia líquida ultrarrápida acoplada em arranjo linear de
fotodiodos
4.1.6.1 Preparo da solução padrão de PENG
A solução estoque de PENG 300 μg mL-1 foi preparada por diluição em água ultrapura
Millipore em balão volumétrico de 10 mL, devidamente aferido. A solução foi sonicada durante
15 min e filtradas em filtro nylon millipore, armazenada em frasco de âmbar e mantida
refrigerada a - 20 °C. As soluções trabalho foram preparadas, diariamente, antes das medidas
cromatográficas, na faixa de 0,001 a 0,1 µg mL-1.
4.1.6.2 Preparo de amostras
As amostras medicamentosas foram preparadas segundo procedimentos descritos pelas
Farmacopeias Americana, Brasileira, Europeia, Japonesa e Britânica (BRASIL, 2010;
BRITISH PHARMACOPOEIA, 1983; EUROPEAN PHARMACOPOEIA COMMISSION,
2008; JP XVI, 2007; USP XXVIII). Este procedimento foi adotado para garantir a
aplicabilidade dos resultados gerados por este trabalho.
Todas as soluções injetáveis de medicamentos foram preparadas a partir da diluição com
água Millipore, exceto as amostras contendo Abamectina e Ivermectina insolúveis na fase polar,
sendo preparadas em solvente apolar, acetonitrila. Todas as amostras foram microfiltradas em
filtro de seringa nylon Millipore. Amostras contendo Penicilina (PENG) na sua composição
foram sonicadas durante 15 min.
As amostras padronizadas do grupo 1 e 2 das penicilinas (com referência em PENG K+)
foram preparadas em concentrações teóricas de PENG BENZ (4 – 0,0017µg mL-1), PENG
PROC e PENG K+ (0,71 – 0,0088 µg mL-1), DHSTR e STR (0,54 – 0,0069 µg mL-1). As
amostras do grupo 3, 4, 5 e 6 das tetraciclinas foram elaboradas com faixa de OTC (0,5 – 0,01
µg mL-1). As amostras do grupo 7 macrolídeos ABAM (0,5 – 0,01 µg mL-1) e IVERM (0,25 –
0,005 µg mL-1). As amostras do grupo 8 e 9 das sulfonamidas SMZ e SDX (0,53 – 0,0004 µg
mL-1), TMP (0,10 – 0,0009 µg mL-1). As amostras do grupo 10 aminoglicosídeo GEN (0,5 –
0,01 µg mL-1).

99
4.1.6.3 Condições das análises cromatográficas
Os procedimentos ocorriam sempre com previa desgaseificação das bombas A e B e
limpeza do solvente. A temperatura foi mantida em 25 ºC. A detecção no UV, nessas condições
ocorria, conforme a absorção máxima de cada família de antimicrobianos descritos na Tabela
4.1 pelos grupos das amostras isoladas (apresentadas nos APÊNDICES I – VI) sendo que para
G1 é de 264 nm; G2 de 264 nm; G3, G4, G5 e G12 de 203, 266 e 358 nm; G6 de 220, 370 e
450 nm; G7 de 190. 220 e 280 nm; G8 de 232 e 265 nm; G9 de 220, 254 e 270 mm; G10 de
254 nm; G11 de 190, 252, 257 e 264 nm. Porém o comprimento de onda para a análise foi
selecionado λmax de 220, 252, 254, 257 e 264 nm.
As medidas foram registradas em software LCsolution da Shimadzu (Kyoto, Japão). Os
tratamentos estatísticos foram realizados em software Effi Validation 4.0 (República Tcheca,
EU) versão gratuita.
As análises das melhores condições para identificação de PENG (0,1 µg mL-1) foram
realizadas segundo a descrição da Tabela 4.3.
Tabela 4.3-Planejamento das condições do sistema cromatográfico para PENG
Parâmetros Condição 1 Condição 2 Condição 3
Tempo da Corrida (min) 10 10 10
Tempo de Retenção (min) 4,431 4,327 4,222
Composição da Fase Móvel (H2O/ACN) 90/10 80/20 70/30
Injection Volume (µL) 10 10 10
Fluxo (mL min-1) 0,01 0,02 0,04
Pico da Area (mAU) 2020 2019 1866
A composição da fase móvel foi constituída por H2O millipore 90% e Na2HPO4 a 1% e
fase B ACN 10% para condição 1.
Em seguida, submeteu-se o método a pequenas variações na composição da fase móvel,
no fluxo e na pressão nas bombas A e B, simultaneamente, para verificar a robustez.

100
4.1.6.4 Validação do Método
A validação dos procedimentos seguiu as recomendações do Guia de Validação da
Emea (2006) sendo a norma ICH Q2 (R1) para medicamentos. Os procedimentos para validação
foram realizados por adição de padrão externo PENG. Os cálculos dos parâmetros de validação
foram realizados usando o software Effi Validation.
4.1.6.4.1 Especificidade/Seletividade
Para garantir a identidade de uma substância foram realizados testes de pureza para
comprovação do rendimento do método desenvolvido. O rendimento foi calculado por Equação
11.
%𝑅 =[𝑃𝐸𝑁𝐺]𝑒𝑥𝑝𝑒𝑟𝑖𝑚𝑒𝑛𝑡𝑎𝑙
[𝑃𝐸𝑁𝐺]𝑡𝑒ó𝑟𝑖𝑐𝑎
× 100 (11)
Este cálculo foi realizado em seis níveis de concentração sendo 0,001, 0,002, 004, 0,01,
0,02 e 0,04 µg mL-1 em 15 réplicas, obtendo-se as médias, desvio, coeficiente de variação
(%CV) e erros.
4.1.6.4.2 Faixa de Trabalho e construção da curva analítica
A adição do padrão externo foi realizada com PENG K+ descrito no item 4.1.4.
O modelo para identificação e quantificação da PENG é determinado pela construção
de curva analítica para determinação da concentração desconhecida de PENG em faixa de
trabalho de 0,001 a 0,1 µg mL-1.
As soluções padrões contendo 0,001; 0,002; 0,005; 0,01; 0,02; 0,04; 0,06; 0,08 e 0,1 µg
mL-1 de PENG foram preparadas e injetadas 10 µL, em triplicata, na coluna cromatográfica.
A curva analítica foi construída em função da área do pico de PENG e concentração (µg
mL -1) do padrão PENG.

101
4.1.6.4.3 Linearidade
A linearidade foi determinada com base nos parâmetros da curva analítica, sendo
aceitável quando R ≥ 0,999
A determinação da pureza através do ponto de calibração foi realizada preparando-se a
amostra em solução aquosa com concentração definida no item 4.1.4.
4.1.6.4.4 Precisão
A precisão foi determinada pela relação entre as concentrações teóricas e experimentais
obtidas pela Equação da curva analítica. O cálculo foi obtido pela repetitividade dos resultados.
Calculou-se a concentração do analito com base no sinal obtido pela área do pico (mAU).
Calculou-se as concentrações médias, desvios padrão e os coeficientes de variação para cada
nível de concentração.
Os critérios de aceitação foram obtidos baseado no CGL/SDA que normatiza coeficiente
de variação de 35% para C < 1 mg/kg, 30% para 1 mg/kg ≤ C < 10 mg/kg, 20% para 10mg/kg
≤ C < 100mg/kg, 15% para 100 mg/kg ≤ C < 1000 mg/kg, 10% para 1000 mg/kg ≤ C < 10000
mg/ kg, 7,3 % para 10 mg/kg ≤ C < 100 mg/kg, 5,3% para 100 mg/kg ≤ C < 1000 mg/kg, 3,7%
para 1000 mg/kg ≤ C < 10000 mg/ kg, 2,7% para10 g/kg ≤ C < 100 g/kg e 2% para 100 g/kg ≤
C < 1000 g/kg.
4.1.6.4.5 Exatidão
A exatidão foi determinada pela repetabilidade dos dados e expressos pelos coeficientes
de variação (%CV) em cada nível de repetição sendo 0,001, 0,002, 0,004, 0,01, 0,02 e 0,04 µg
mL-1. Foi expressa pela variância, o desvio padrão e coeficiente de variação da série de
medições.

102
4.1.6.4.6 Limite de detecção
A padronização das soluções em níveis de concentração de 0,001 – 0,1 µg mL-1 foi
preparada e injetada 10 µL em triplicata na coluna cromatográfica. A concentração foi
determinada correspondente ao sinal que equivalente a três vezes o ruído instrumental.
4.1.6.4.7 Limite de quantificação
O LQ foi determinado que é equivalente a 3,3 vezes o LD. O limite de quantificação de
um procedimento analítico foi determinado pela mais baixa quantidade de analito na amostra
determinada quantitativamente com precisão e exatidão. Nesta etapa, o LQ foi usado em para
determinar as impurezas bem como os produtos de degradação.
4.1.6.4.8 Robustez
A robustez foi determinada realizando variações nos parâmetros do método. Os
procedimentos foram mostrados na Tabela 4.3.

103
4.1.6.5 Identificação de PENG nas amostras
As amostras selecionadas para identificação da PENG foram apresentadas isoladamente
na Tabela 4.1. Porém são apresentadas as informações na Tabela 4.2 correspondentes as
amostras para análise no UV VIS NIR.
A identificação foi realizada com base no branco constituída pela fase móvel, foi
realizada a correlação entre as amostras.
4.1.6.6 Cálculo das concentrações de PENG em amostras de medicamentos
Para o cálculo das concentrações de PENG nas amostras foi utilizada a Equação 12.
𝐴𝑆
𝐴𝑅
= 𝐶𝑆
𝐶𝑅
(12)
Onde, As é a área do pico do PENG na amostra, Ar é a área do pico do padrão PENG,
Cr é a concentração do PENG na amostra e Cr é a concentração do padrão PENG.
Quando os parâmetros são rearranjados tem-se a Equação 14, no qual Cs é concentração
do analito da amostra determinada pela Equação 13.
𝐶𝑆 = 𝐶𝑅 × 𝐴𝑆
𝐴𝑅
(13)
Admitindo-se o LD e LQ do método desenvolvido, com base na Equação da curva
analítica.

104
4.1.7 Análise na região do UV VIS NIR e tratamentos de dados
As condições para medidas foram: velocidade de varredura de 109,091 cm-1 min-1 em
região de 200 a 2300 nm. Os dados espectrais foram coletados em software CaryWin foram
convertidos para software 32 GRAMAS (Galactic Industries Corporation, Salem, NH),
analisados usando software Unscrambler® X 10.3 (CAMO, Noruega) e plotados e apresentados
em software OriginPro 9 (OriginLab, Guangzhou, China). Os tratamentos matemáticos e
estatísticos foram: correção da linha de base, normalização, alisamento Savitsky Golay,
correção multiplicativa da luz (MSC) e cálculos derivativos.
A correção de linha de base (baseline) foi realizada pelo cálculo das médias em cada
variável do espectro, de maneira que os valores individuais foram substituídos pelas médias
espectrais.
Todos os dados espectrais foram normalizados em torno da média, usando a Equação
14 descrita, no qual a matriz (37 × 2100), que corresponde a 37 linhas referente as amostras e
2100 colunas referentes as variáveis expressas em comprimentos de onda (nm).
𝑋(𝑖, 𝑘) = 𝑋(𝑖, 𝑘)
|�̅� (𝑖,∎ )| (14)
A equação foi utilizada admitindo-se uma matriz para primeiro método (37 × 2100),
onde o número de linhas (i) representam as amostras e colunas (k) as variáveis espectrais
(comprimentos de onda).
Os espectros passaram por transformações constituída pelo cálculo do algoritmo de
Savitsky Golay que foi calculado e testado por uma Equação polinomial de 1ª e 2ª ordem, em
cada segmento foi obtida uma curva que substituíram os valores originais, testado em janela de
31 pontos.
Outra correção realizada foi a correção multiplicativa da luz (MSC) que consistiu em
construção de linha de regressão para cada espectro das amostras, que foi expressa pelo valor
médio para cada comprimento de onda, utilizada para corrigir os valores em cada espaço
amostras. O cálculo baseava-se na Equação 15.
𝑀𝑛𝑜𝑣𝑎 𝑚𝑎𝑡𝑟𝑖𝑧(𝑖, 𝑘) = 𝑀(𝑖, 𝑘) − 𝑎
𝑏 (15)

105
Na Equação a e b representam os coeficientes da regressão.
Foram calculadas as derivadas de primeira e segunda ordem, as Equação 16 corresponde
a primeira ordem.
dA
dλ= a + 2cx (16)
Quando calculados derivadas de segunda ordem foi utilizada a Equação 17.
d2A
dλ2= 2C (17)
Nestas etapas do procedimento foi aplicado o método derivativo testando o número de
pontos nas janelas e avaliado os gráficos de loadings para determinação das variáveis que
apresentaram maiores influência no modelo.
Os métodos de regressão utilizados para calibração dos modelos foram o PCR e PLS,
estes utilizam as variáveis originais, tendo como critério como número de amostras deve ser
maior que o variáveis de respostas neste caso no comprimento de onda.
Para construção dos modelos no PCR foram utilizados os dados da matriz X (i) e
encontrados as combinações lineares para as variáveis. A Equação 18 descreveu o modelo.
X = TP𝑇 + E (18)
Onde X é a matriz original, E são os resíduos das análises. A resposta do variou de
acordo com o modelo utilizado, sendo que segundo a Equação 19 quando foi obtido o PLS.
𝑦 = 𝑇𝑃𝑇
𝑋 (19)
Neste modelo o critério foi Xcal, X val, Xprev, sendo que se fixou Xprev, enquanto que Xcal
foi a resposta do modelo, desta forma foi realizada a relação descrita pela Equação 20.
y = [1, Xcal] (20)

106
Na Tabela 4.4 são apresentadas a rotina para modelagem dos dados em duas regiões
espectral, uma compreende o UV ao NIR, a segunda compreende a região apenas do NIR. As
analises foram realizadas nos medicamentos puros (E1 – E37) em segunda etapa foram
analisadas as amostras tratadas (E1 – E37), incluindo as amostras para validação externa.
Tabela 4.4-Procedimento de modelagem para quantificação de PENG em medicamentos de uso veterinário.
Modelos Condições implementadas
Tratamentos Regressão Região Espectral
Medicamentos Puros com CPENG (0,01 – 0,3 mg mL-1)
M1 T1
PCR
200 – 2300
M2 T1+ 1st 31 pontos
M3 T1+ 2st 31 pontos
M4 T1
PLS M5 T1+ 1st 31 pontos
M6 T1+ 2st 31 pontos
M7 T1
PCR
750 – 2300
M8 T1+ 1st 31 pontos
M9 T1+ 2st 31 pontos
M10 T1
PLS M11 T1+ 1st 31 pontos
M12 T1+ 2st 31 pontos
Medicamentos Tratados com CPENG (0,001 – 0,03 mg mL-1)
M13 T1
PCR
200 – 2300
M14 T1+ 1st 31 pontos
M15 T1+ 2st 31 pontos
M16 T1
PLS M17 T1+ 1st 31 pontos
M18 T1+ 2st 31 pontos
M19 T1
PCR
750 – 2300
M20 T1+ 1st 31 pontos
M21 T1+ 2st 31 pontos
M22 T1
PLS M23 T1+ 1st 31 pontos
M24 T1+ 2st 31 pontos

107
É necessário enfatizar que, os melhores modelos foram escolhidos de acordo com R, o
erro padrão de calibração (SEC), e repetabilidade para o conjunto de dados de calibração.
O desempenho do modelo de calibração foi avaliado por validação externa. Na Figura
4.2 são apresentadas as estratégias para seleção das amostras.
Figura 4.2-Ilustração para testes com amostras externas
Os valores do erro quadrático médio de validação cruzada são calculados utilizando a
Equação 21. Assim, foi utilizada a validação cruzada (LOOCV).
RMSEC = √1
N1
∑(CR − CP)2
N1
i=1
(21)
A precisão da validação foi verificada pelos valores de RMSEP e R. O erro quadrático
médio da previsão adicionados modelos descritos pela Equação 22.
RMSEP = √1
Nt
∑(CR − CP)2
Nt
i=1
(22)
Nas Equações 22 e 23 N1 e Nt são o número de amostras usados na validação cruzada e
validação externa, respectivamente. CR e CP são os valores de concentração de referência e
previsão, respectivamente.
1 2 3 4 5 6 7 8 91
01
11
21
31
41
51
61
71
81
92
02
12
22
32
42
52
62
72
82
93
03
13
23
33
43
53
63
73
83
94
05
75
94
14
24
34
44
54
64
74
84
95
05
15
25
35
45
65
55
8
0,00
0,01
0,02
0,03
0,04
Validação Externa (36%)
[M]
mg m
L-1
Amostras
Calibração (64%)

108
Os modelos de calibração foram desenvolvidos com espectros originais e Pretratados
conforme. Na Figura 4.3 são apresentadas as principais etapas realizadas para calibração do
método e validação externa com as 24 amostras puras e tratadas.
Figura 4.3- Esquema resumido para testes com amostras externas
O autor, 2015
Nestas etapas O número de variáveis e/ou componentes foi determinado, com base na
soma dos resíduos previstos e correlação (R).

109
4.2 DESENVOLVIMENTO DE MÉTODO PARA IDENTIFICAÇÃO E
QUANTIFICAÇÃO DE PENG EM LEITE BOVINO E CAPRINO
4.2.1 Reagentes e Solventes
Os reagentes utilizados foram: fosfato de potássio dibásico (K2HPO4) P.A ACS Vetec
(São Paulo, Brasil) da Sigma Aldrich, Sódio, ácido cítrico (C6H8O7) P.A ACS, ácido
etilenodiaminotetracético (C10H16N2O8), fosfato de potássio dibásico anidro (K2HPO4) ACS
P.A, hidróxido de sódio (NaOH) PA ACS, ACS
Todos os reagentes empregados para análise por UFLC-DAD foram descritos no item
4.1.1.
4.2.2 Instrumentação
Os instrumentos para pesagens e dissolução dos reagentes foram descritos no item 4.1.2.
Foram utilizados outros instrumentos para preparo das amostras: Centrífuga Refrigerada
de Bancada Excelsa 4 Modelo 280R Fanem (São Paulo, Brasil), pHmetro digital MPA-210
Tecnopon (São Paulo, Brasil). As amostras foram agitadas em Agitador de tubos Phoenix P56
(São Paulo, Brasil).
4.2.3 Outros materiais para Cromatografia
Os procedimentos para identificação dos compostos foram alcançados, usando coluna
de fase reversa C18 (2,0 mm × 30 mm ID) Shim-pack XR-ODS (Kyoto, Japão). As amostras e
padrão PENG K+ foram microfiltradas em filtro de seringa nylon (13 mm × 0.22 mm) código
2202213500C adquirido da Analítica (São Paulo, Brasil). As padronizações das soluções foram
realizadas por aferição, em micropipetas de volumes máximo de 20 µL.

110
4.2.4 Amostragem
4.2.4.1 Amostras de leite controle
Amostras de leite bovino e caprino ausentes de resíduos de PENG foram usadas na
preparação da curva analítica e cálculo dos parâmetros de validação.
4.2.4.2 Amostras de leite comerciais (externas)
A amostragem foi realizada conforme técnicas do programa de regulação para controle
de resíduos veterinários em alimentos CAC/GL 16 – 1993. Esta norma preconiza que a seleção
das amostras é realizada aleatoriamente. Amostras de leite pasteurizado foram adquiridos de
supermercados localizados em Natal/RN, sendo 24 de leite bovino LB e 20 de leite caprino
LCP.
O procedimento para coleta e acondicionamento do leite até laboratório, onde foram
realizados os procedimentos de abertura, foi determinado por etapas, na 1ª cada amostra foi
homogeneizada durante, aproximadamente 3 min; na 2ª foi coletado 500 mL do leite e
acondicionados em frasco de polipropileno de 1º uso sem qualquer inscrição, isto e apenas
designação como descrita na Tabela 4.2 e 4.3, não completando todo o volume disponível do
frasco; na 3ª as amostras foram congeladas de forma a atingir o centro geométrico da amostra
a temperatura de -20 ºC.
4.2.4.2.1 Amostras de leite bovino comercial (LB)
As amostras LB analisadas foram de 11 marcas distintas de origem em seis regiões
diferentes do país comercializadas na cidade do Natal. As amostras são classificadas como
integral e desnatado. As informações principais de amostras são descritas na Tabela 4.5.
As descrições quanto à procedência das amostras foram coletadas das embalagens e
confirmadas pelo Banco de Dados do MAPA disponível para consulta pública dos
estabelecimentos produtores com base no selo de inspeção Federal (Sifs).

111
Tabela 4.5-Descrição das amostras de leite bovino
Amostras Origem*
Algumas das Informações Declaradas por Rotulagem dos
produtos Data da abertura
das amostras (%) Lipídios (%) Proteínas Estabilizantes
LB1 Fortaleza/CE 8 11 citrato de Sódio 29/07/2014
LB2 Fortaleza/CE 0 8 citrato de Sódio 30/07/2014
LB3 MG 11 8
Citrato de sódio,
tripolifosfato de sódio e
fosfato trissódico
07/02/2014
LB4 MG 0 8 citrato de sódio 08/02/2014
LB5 Jaborandi/BA 14 9 citrato de Sódio 29/07/2014
LB6 Jaborandi/BA 5 14 citrato de Sódio 30/07/2014
LB7 Garanhuns/ PE 6 6 citrato de Sódio 29/07/2014
LB8 Garanhuns/ PE 0 6 citrato de Sódio 30/07/2014
LB9 Governador
Valadares/MG 1 8
citrato de sódio e fosfato
de sódio 29/07/2014
LB10 Governador
Valadares/MG 1 8
citrato de sódio e fosfato
de sódio 30/07/2014
LB11 Goiânia/GO 11 8
citrato de sódio,
tripolifosfato de sódio,
pirofosfato ácido de sódio
e fosfato monossódico
29/07/2014
LB12 Goiânia/GO 0 9
citrato de sódio,
tripolifosfato de sódio,
pirofosfato ácido de sódio
e fosfato monossódico
30/07/2014
LB13 Patos de
Minas/MG 11 8 citrato de Sódio 29/07/2014
LB14 Patos de
Minas/MG 0 8 citrato de Sódio 30/07/2014
LB15 Itambuiá/GO 11 8
citrato de sódio,
monofosfato de sódio,
difosfato de sódio e
trifosfato de sódio
29/07/2014
LB16 Itambuiá/GO 0 8
citrato de sódio,
monofosfato de sódio,
difosfato de sódio e
trifosfato de sódio
30/07/2014
LB17 Corumbaíba/GO 11 8
citrato de sódio, trifosfato
de sódio, monofosfato
monossódico e difosfato
dissódico
29/07/2014
LB18 Corumbaíba/GO 2 8
citrato de sódio, trifosfato
de sódio, monofosfato
monossódico e difosfato
dissódico
30/07/2014
LB19 Ponta Grossa/PR 11 8
citrato de sódio,
monofosfato de sódio,
difosfato de sódio e
trifosfato de sódio
29/07/2014
LB20 Ponta Grossa/PR 0 8
citrato de sódio,
monofosfato de sódio,
difosfato de sódio e
trifosfato de sódio
30/07/2014
LB21 Macaiba/RN 11 8 NA 29/07/2014
LB22 Macaiba/RN 0 6 NA 30/07/2014
LB23 Muribeca/SE 11 9
tripolifosfato de sódio,
pirofosfato de sódio e
fosfato monossódico
07/02/2014
LB24 Muribeca/SE 0 9
tripolifosfato de sódio,
pirofosfato de sódio e
fosfato monossódico
07/02/2014
*Todas as informações contidas quanto à origem referem-se às usinas de beneficiamento das respectivas amostras de leite,
NA e não se aplica.

112
4.2.4.2.2 Amostras de leite caprino comercial (LCP)
As amostras LCP analisadas foram de duas marcas distintas e origem em duas regiões
diferentes do país. Na Tabela 4.6 são apresentadas as amostras de leite caprino.
Tabela 4.6-Descrição das amostras de leite caprino
Amostras Origem*
Algumas das Informações Declaradas por Rotulagem dos
produtos Data da abertura
das amostras (%) Lipídios (%) Proteínas Estabilizantes
LCP1 Porto Alegre/RS 9 1
Monofosfato de sódio,
difosfato de sódio e
trifosfato de sódio
29/07/2014
LCP2 Porto Alegre/RS 9 1 29/07/2014
LCP3 Porto Alegre/RS 9 1 29/07/2014
LCP4 Porto Alegre/RS 9 1 29/07/2014
LCP5 Porto Alegre/RS 9 1 29/07/2014
LCP6 Porto Alegre/RS 9 1 29/07/2014
LCP7 Porto Alegre/RS 9 1 29/07/2014
LCP8 Porto Alegre/RS 9 1 29/07/2014
LCP9 Porto Alegre/RS 9 1 29/07/2014
LCP10 Porto Alegre/RS 9 1 29/07/2014
LCP11 Macuco/RJ 13 8
Citrato de sódio e/ou
difosfato de sódio
29/07/2014
LCP12 Macuco/RJ 13 8 29/07/2014
LCP13 Macuco/RJ 13 8 29/07/2014
LCP14 Macuco/RJ 13 8 29/07/2014
LCP15 Macuco/RJ 13 8 29/07/2014
LCP16 Macuco/RJ 13 8 29/07/2014
LCP17 Macuco/RJ 13 8 29/07/2014
LCP18 Macuco/RJ 13 8 29/07/2014
LCP19 Macuco/RJ 13 8 29/07/2014
LCP20 Macuco/RJ 13 8 29/07/2014
*Todas as informações contidas quanto à origem referem-se às usinas de beneficiamento das respectivas amostras de leite,
NA e não se aplica
As amostras apresentadas na Tabela 4.6 estão com teor gordura > 9%, segundo as
recomendações dos fabricantes são produtos integrais.

113
4.2.5 Preparo da solução padrão de PENG
A solução estoque de PENG 300 μg mL-1 foi preparada por diluição com água ultrapura
Millipore em balão volumétrico de 10 mL, devidamente aferido. Esta solução foi filtrada em
filtro Millipore. Em seguida a solução foi sonicada durante 15 min, armazenada em frasco
âmbar e refrigerada a - 20 °C.
As soluções foram preparadas, diariamente, antes das medidas cromatográficas seguida
de procedimento de filtração. A faixa de trabalho utilizada foi de 0,002 – 0,1 µg mL-1.
4.2.6 Preparo da solução tampão McIlvaine pH 4
O tampão Mcllvaine pH 4 foi preparado em balão volumétrico de 1000 mL (aferido) a
partir da mistura de 100 mL de ácido cítrico 0,1 mol L-1, 62,5 mL de fosfato de potássio dibásico
anidro (K2HPO4) 0,2 mol L-1 e 3 g de ácido etilenodiaminotetracético (C10H16N2O8). O ajuste
de pH foi realizado com solução de hidróxido de sódio padronizado a 0,1 mol L-1.

114
4.2.7 Preparo e limpeza das amostras de leite bovino e caprino
O método desenvolvido não utiliza extração por solvente, apenas aplicação de solução
tampão McIlvaine adaptada, seguida de agitação e centrifugação, de acordo com a norma
CAC/GL 16. Os procedimentos adotados para extração e isolamento do analito são descritos na
Figura 4.4.
Figura 4.4-Esquema simplificado do preparo das amostras de leite antes (A) e após tratamento (B)
O autor, 2015
O método envolveu precipitação total das proteínas. Inicialmente, 5 g de amostra
equivalente a 15 mL de leite foi diluída em tampão Mcllvaine pH 4, a mistura foi agitada em
vórtex. Em seguida, foi centrifugada a - 4 ºC a 500 rpm durante 10 min. A fase aquosa foi
filtrada e recolhida para repetição do processo até a precipitação da proteína total seguido de
diluição em balão volumétrico de 50 mL. Em seguida a fase aquosa foi diluída em água
Millipore e filtrada em filtro de nylon (13 mm × 0,22 µm).

115
4.2.8 Condições para análise cromatográfica
As condições instrumentais utilizadas seguem as descrições do item 4.1.6.3, que trata
da corrida cromatográfica do padrão certificado PENG 300 µg mL-1. A fase móvel foi
constituída por H2O millipore 90% e Na2HPO4 a 1% e fase B ACN 10% para condição 1.
O planejamento das condições para análise de leite bovino foi realizado pela adição de
padrão PENG 300 µg mL-1 as amostras controle. Os dados seguem descritos pela Tabela 4.7.
Tabela 4.7-Planejamento comparativo das condições do sistema cromatográfico para PENG para análise das
amostras controle de leite bovino (LB) e leite caprino (LCP)
Parâmetros Condição 1 Condição 2 Condição 3
Tempo da Corrida (min) 10 10 10
Composição da Fase Móvel (H2O/ACN) 90/10 80/20 70/30
Injection Volume (µL) 10 10 10
Fluxo (mL min-1) 0,01 0,02 0,04
4.2.9 Validação do método
A linearidade, recuperação, precisão, exatidão, limite de detecção (LD) e quantificação
(LQ), seletividade, robustez e efeito da matriz foram calculadas para validar todo o processo,
de acordo com a Comissão Europeia 2002/657/CEE e Guia de Validação da ANVISA para
quantificação de resíduos de antibióticos (BARRETO et al., 2012; CHRISTODOULOU;
SAMANIDOU, 2007; DUBREIL-CHÉNEAU et al., 2014; KARAGEORGOU;
SAMANIDOU, 2011; LOPES et al., 2011; REPIZO et al., 2012).
O método foi validado as amostras controle fortificadas com a solução e adição de
padrão interno PENG 300 μg mL-1.
4.2.9.1 Construção da curva Analítica
A curva analítica foi construída, em triplicata, relacionando a concentração de PENG
(μg mL-1) versus área do pico de PENG. As amostras LB e LCP controles foram fortificadas
(separadamente) em oito níveis de concentração sendo 0,002; 0,005; 0,01; 0,02; 0,04; 0,06; 0,08
e 0,1 µg mL-1. A construção desta curva baseou-se no LMR de 0,004 μg mL-1.
O padrão interno PENG foi adicionado em concentração de 300 μg mL-1 em cada um
dos níveis de concentração descritos.

116
4.2.9.2 Linearidade
A análise da linearidade e sensibilidade foi realizada com base na regressão linear dos
dados expressos pela curva analítica.
A linearidade foi avaliada pelo coeficiente de correlação (R) da curva analítica para a
matriz leite fortificado em dez e oito níveis de calibração 0,002 – 0,1 µg mL-1 para LB e LCP,
respectivamente. Sendo obtida correlação entre as matrizes e os brancos das amostras. O critério
para aceitação o método ser linear é correlação (R) > 0,999, conforme recomendações da
Comissão Europeia 2002/657/EC.
4.2.9.3 Recuperação
O cálculo da recuperação foi realizado a partir das matrizes de leite controle fortificada.
Inicialmente 15 alíquotas de cada amostra LB e LCP foram fortificadas em três grupos de 0,002;
0,005 e 0,01 µg mL-1 obedecendo critérios da 2002/657/CEE cujos valores devem ser 0,5LMR;
1LMR e 1,5LMR. Em seguida, calculou-se a concentração presente em cada amostra. A
Equação 23 foi utilizada para cálculo para cálculo de recuperação:
%Recuperação = CM
CR
× 100 (23)
Onde CM e CR são os valores de concentração medidas e de referência do padrão,
respectivamente.
Os critérios para aceitação dos valores de concentração seguiram a CAC/GL 16. Para C
≤ 10 µg mL-1 deve ser de 50 – 120%, para C ≥ 1≤ 10 µg mL-1 deve ser de 70 – 110%, para C ≥
1≤ 100 µg mL-1 deve ser de 80 – 120% e para C ≥ 100 µg mL-1 deve ser 80 – 110%.
4.2.9.4 Precisão (Repetabilidade)
Preparou-se um grupo de amostras fortificadas com PENG de modo a obter
concentrações de 0,002; 0,005 e 0,01 µg mL-1, no qual cada nível de análise foi realizada com
seis réplicas. Foram preditas as concentrações, calculando o desvio padrão e coeficiente de
variação (%CV). Calculou-se a concentração média global para cada amostra fortificada.
O critério de aceitação da precisão é definido pela repetabilidade. Desta forma, para
LMR ≤ 10 µg mL-1 o intervalo deve ser de 60 – 120% com variação de – 50 a 20%, para LMR

117
≥ 1≤ 10 µg mL-1 deve ser de 70 – 110% com variação de – 40 a 20%, para LMR ≥ 1≤ 100 µg
mL-1 deve ser de 80 – 110% com variação de – 30% a 20% e para LMR ≥ 100 µg mL-1 deve
ser de – 20 a 10%.
4.2.9.5 Exatidão
Para avaliar a exatidão do método, os estudos de recuperação foram realizados em seis
níveis de concentração (0,001, 0,002 e 0,004, 0,01, 0,02 e 0,04 µg mL-1). A repetabilidade
(precisão intradia) foi calculada nos mesmos níveis de concentração dos estudos de
recuperação. A exatidão foi calculada usando a Equação 24 e expressa em %CV.
% Exatidão = Cpredita
CReferência
× 100 (24)
Os estudos de recuperação foram realizados em níveis de (0,002, 0,005 e 0,01 µg mL-1)
pela adição das amostras fortificadas em branco em cada nível.
4.2.9.6 Limite de detecção (LD)
O limite de detecção foi estimado utilizando o desvio padrão a partir dos dados de
regressão linear da curva analítica, segundo os critérios incluídos no guia ICH Q2(R1) da Emea
(2006). O LD foi calculado pela Equação 25.
LD =(3,3 x SD)
m (25)
Onde SD é o desvio padrão do branco que deve ser ≤ 20% para resíduos; m é a inclinação
da curva analítica. A aceitabilidade da precisão é definida por LQ < MRL (VARGAS
MAMANI; REYES REYES; RATH, 2009).

118
4.2.9.7 Limite de quantificação (LQ)
O limite de quantificação foi determinado pelo cálculo descrito pela Equação 26,
utilizando também a resposta da inclinação da curva e desvio padrão das medidas do branco.
LQ =(10 x SD)
m (26)
4.2.9.8 Seletividade
A seletividade do método foi avaliada através da análise de amostras de controle
(branco). Além disso, mostrou-se que houve precauções para garantir a resolução, e pureza dos
picos cromatográficos. Os testes apresentados na Tabela 4.7 e detecção no comprimento de
onda específico demonstrou que o pico é atribuído a um só componente.
4.2.9.9 Robustez
A avaliação da robustez foi realizada durante o estudo da otimização das condições
cromatográficas. Na análise foi avaliada se as medidas eram suscetíveis a variações nas
condições cromatográficas. As condições 1, 2 e 3 são descritas na Tabela 4.3 do item 4.1.6.3.
4.2.9.10 Efeito da matriz
O efeito da matriz foi determinado utilizando os cálculos para cada nível i da
concentração (fortificação) comparando-se amostras não-matrizadas e matrizadas (ANVISA,
2003), expresso pelo teste F da Equação 27.
Fcalc,i = SDi,1
2
SDi,22 (27)
Onde 𝑆𝑖,12 𝑒 𝑆𝑖,2
2 são as variâncias das réplicas das amostras não-matrizadas (branco) e
matrizadas (fortificadas), em cada nível de concentração, com a maior variância no numerador.
Uma vez calculados os valores de F, adotou-se um nível de significância α= 0,05 (5%)
ou nível de confiança 1 – α = 0,95 (95%).

119
As variâncias desse nível de concentração foram consideradas iguais, isto é, se a matriz
não tivesse efeito importante sobre a precisão do procedimento, nesse nível de fortificação, i
seria considerado.
Logo, se o t calculado for menor que o tcrítico α,ν, concluiu-se que a matriz não afeta o
ensaio no i nível de fortificação. Os valores para teste foram calculados seguindo a Equação 28.
tcalc,i = |x̅i,1 − x̅i,2|
√s2 (1
ni,1+
1ni,2
)
(28)
O SD dos dois grupos de análises foi agrupado e a igualdade das médias dos dois
conjuntos de amostras foi testada com a distribuição t de Student. O desvio foi definido pela
Equação 29.
s2 = (n1 − 1)s1
2 + (n2 − 1)s22
(n1 + n2 − 2) (29)
Para que ser aceito que a matriz não afete as medidas o cálculo do t-student deve mostrar
que o tcalculado< ttabelado.

120
4.2.9.11 Estratégias para validação
As Estratégias para a validação completa (KARNES; SHIU; SHAH, 1991) seguem a
recomendação de Hartmann et al (1998). Na Figura 4.5 são apresentados o esquema de
validação.
Figura 4.5-Estratégia para validação dos métodos bioanalíticos
Adaptado de Hartmann et al (1998)
O desempenho do método empregado foi verificado segundo a recuperação e precisão.
Os principais requisitos obedecidos foram: quanto à curva analítica calibração que conteve no
mínimo cinco níveis de concentração; para cálculo de recuperação e precisão foram

121
determinadas em três níveis de concentração segundo o LMR, cada nível foi analisado, em seis
réplicas de acordo com Guia de Validação (ANVISA, 2003).
4.2.10 Cálculo das Concentrações em amostras de leite comerciais
A partir da validação e cálculo dos parâmetros foi possível obter a Equação e modelo
que define o método, desta forma foi calculado a concentração de resíduos de PENG ([PENG])
nas amostras de leite pasteurizado tipo bovino (LB) e caprino (LCP). Considerando o método
e a recuperação das amostras.
O cálculo foi realizado obedecendo a Equação 30.
𝐴𝑝𝑖𝑐𝑜 = 𝑚[𝑃𝐸𝑁𝐺] + 𝑏 (30)
Onde APICO é a área do pico, m é a inclinação e b intercepto da curva analítica. Quando
calculado a concentração a Equação foi rearranjada para Equação 31 da curva analítica.
[𝑃𝐸𝑁𝐺] = 𝐴𝑝𝑖𝑐𝑜
𝑚− 𝑏 (31)
O critério para classificação foi baseado no LMR e concentração dos resíduos. Quando
a [PENG] < LMR e [PENG] = LMR ou [PENG] ≤ LMR as amostras são classificadas como
conformes. Quando [PENG] > LMR as amostras são classificadas como não conforme.

CAPÍTULO 5
RESULTADOS E DISCUSSÃO

123
5 RESULTADOS E DISCUSSÃO
5.1 DESENVOLVIMENTO DE MÉTODO PARA IDENTIDADE E QUALIDADE DOS
MEDICAMENTOS A BASE DE BENZILPENICILINA
5.1.1 Testes para identidade dos medicamentos
A proposta desta etapa é desenvolver um modelo para controle de qualidade dos
medicamentos veterinários, segundo as preconizações das Farmacopeias Brasileira (2004) e
British Pharmacopea (1983).
5.1.1.1 Condições dos testes
Para realização das medidas instrumentais foram analisadas as condições de pH,
concentração na solução e absorção da PENG em medicamentos associados a STR e DHSTR.
Na Figura 5.1 são apresentados espectros no UV das amostras contendo penicilina e
estreptomicina.
Figura 5.1-Espectros no UV de amostra contendo PENG e STR em cinco níveis de concentração
200 250 300 350 400 450 500 550 600
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Ab
s
Comprimento de Onda (nm)
PENG1: 3 mg mL-1
PENG2: 2,7 mg mL-1
PENG3: 2,4 mg mL-1
PENG4: 2,1 mg mL-1
PENG5:1,8 mg mL-1
Através da comparação do espectro da amostra, no intervalo de comprimento de onda
especificado, com o espectro de referência e/ou espectro de referência padrão foi possível
identificar PENG em 200 nm e DHSTR em 290 nm, estes resultados estão condizentes com
alguns trabalhos (BOSCH OJEDA; SANCHEZ ROJAS, 2004, 2013; UPSTONE, 2000).

124
Os testes de solubilidade confirmam que os medicamentos contendo PENG são solúveis
em água, por isso a solução é preparada pela simples dissolução seguida de sonicação, a fim de
promover a protonação do sal da PENG e diminuição da formação de depósitos.
Os testes do pH das soluções indicam que grupo das penicilinas que apresentam pH
experimental entre 6 e 7, na faixa de concentração referenciado, tem influência na capacidade
de absorção na região do UV, como mostram os resultados (BEAUSSE, 2004).
Segundo Grenfell e Means (1945) a rotação específica da PENG é + 298, conferindo
um caráter dextrorotatória à substância. As absorções no IV para esses compostos são
condizentes com a norma 1272/2000/EC da União Europeia, exceto quando esses compostos
estão isolados há um erro de ± 10. Com relação ao pH nas mesmas condições a PENG isolada
condiz com a norma 1907/2000/EC, porém quando estão associadas a STR e DHSTR formando
MIXPENG o pH aumenta ocasionado pelo deslocamento do equilíbrio.
A leitura do ângulo de rotação específica, [𝛼]𝐷200
evidencia a pureza dos enantiômeros.
De maneira que, sob condições de pH 5 – 7, as penicilinas apresentam capacidade de girar o
plano de luz para a direita, por isso são dextrogiras. Logo, segundo Prankerd (2007) a estrutura
básica da Benzilpenicilina lhe confere pKa de [HA] 2,7, esse fator altera o curso do fármaco no
organismo, e influenciam na formação de resíduos.

125
5.1.2 Análise por espectroscopia no infravermelho médio.
A espectroscopia na região do infravermelho é utilizada, como teste de identificação dos
medicamentos. O espectro de infravermelho fornecem medidas dos compostos químicos, com
a exceção dos isomeros ópticos. Apesar disto, esta técnica tem sido muito aplicada e
recomendada para controle de qualidade (BRASIL, 2010; BRITISH PHARMACOPOEIA,
1983; EUROPEAN PHARMACOPOEIA COMMISSION, 2008; JP XVI, 2007).
Considerando que as medidas no IV dos medicamentos fornecem impressão digital de
todos os principais componentes do PENG, ABAM/IVERM, GEN, OTC e SMZ, os espectros
das amostras foram comparados com o espectro da substância de referência preparada,
concomitantemente, com espectro padrão. Além disso, os dados são adquiridos
simultaneamente com os de referência, reconhecidos pelas normatizações, apresentados na
Tabela 5.1.
Sabe-se que existem bancos de dados, que mostram informações espectrais, a respeito
das substâncias terapêuticas, como é o caso dos antibióticos. No entanto, não existem tais
bancos sem quaisquer erros ou enganos. Assim, foram realizadas medidas no FTIR, segundo as
preconizações das Farmacopeias legítimas, pois garantem a veracidade das medidas
Assim, os testes desenvolvidos mostram o melhor produzido para a identificação dos
compostos, se comparados bancos de dados internacionalmente disponíveis, como é caso do
SDBS organizado pelo Instituto nacional de avanços na ciência e tecnologia do Japão (AIST).
Na Figura 5.2 são apresentados os espectros da solução padronizada PENG e dos
medicamentos isolados e utilizados em associação com a PENG que são OTC, SMZ/TMP,
SDX/TMP, ABAM/IVERM, GEN e PENG/STR.
As bandas de absorção são atribuídas na região de 4000 a 667 cm-1. As regiões de maior
frequência estão entre 3333 a 2941 cm-1 e está relacionada à deformação axial de O – H
associado de água.

126
Figura 5.2-Espectros FTIR para grupos dos antibióticos das amostras e PENG padronizado
4000 3500 3000 2500 2000 1500 1000
60
80
100
60
80
100
60
80
100
60
80
100
60
80
100
60
80
100
60
80
100
4000 3500 3000 2500 2000 1500 1000
Número de Onda (cm-1
)
SDX/TMP
%T
GEN
PENG/STR
(I)
PENG padronizado
ABAM/IVERM
SMZ/TMP
(V)
(V)
(IV)
(III)
(II)
OTC
Na Tabela 5.1 indicam as semelhanças entre o padrão e os grupos funcionais presentes
nos medicamentos.
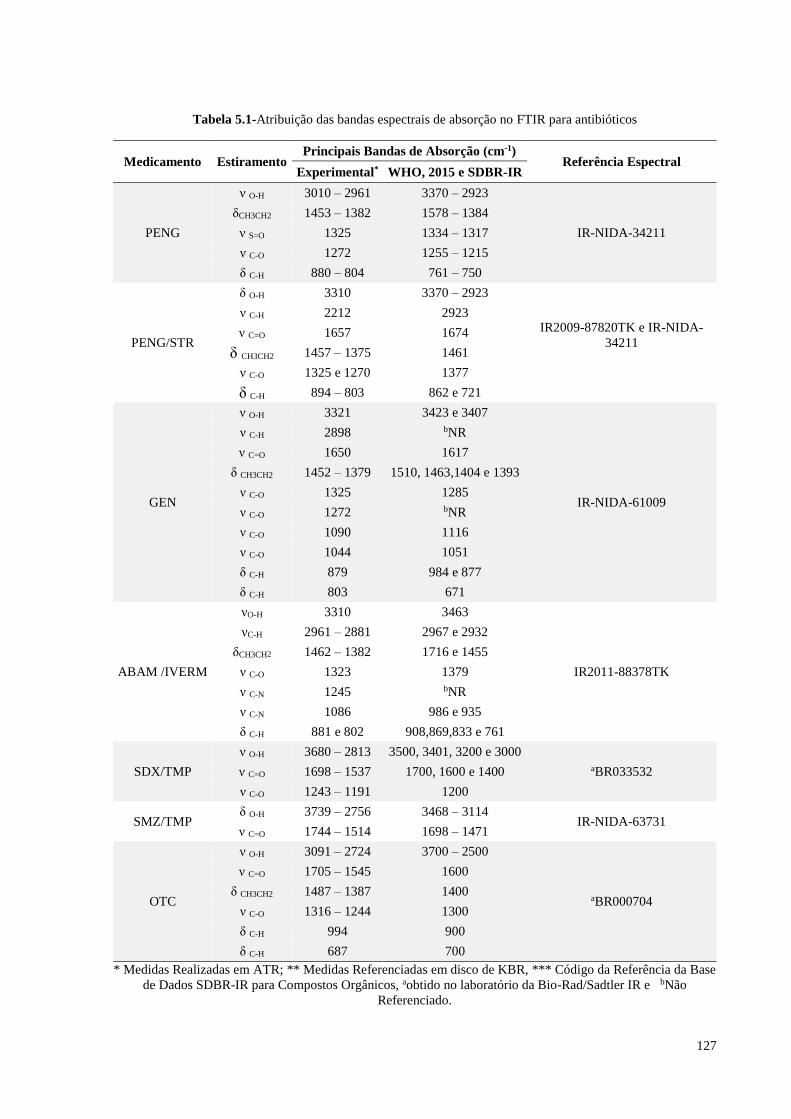
127
Tabela 5.1-Atribuição das bandas espectrais de absorção no FTIR para antibióticos
Medicamento Estiramento Principais Bandas de Absorção (cm-1)
Referência Espectral Experimental* WHO, 2015 e SDBR-IR
PENG
ν O-H 3010 – 2961 3370 – 2923
IR-NIDA-34211
δCH3CH2 1453 – 1382 1578 – 1384
ν S=O 1325 1334 – 1317
ν C-O 1272 1255 – 1215
δ C-H 880 – 804 761 – 750
PENG/STR
δ O-H 3310 3370 – 2923
IR2009-87820TK e IR-NIDA-
34211
ν C-H 2212 2923
ν C=O 1657 1674
δ CH3CH2 1457 – 1375 1461
ν C-O 1325 e 1270 1377
δ C-H 894 – 803 862 e 721
GEN
ν O-H 3321 3423 e 3407
IR-NIDA-61009
ν C-H 2898 bNR
ν C=O 1650 1617
δ CH3CH2 1452 – 1379 1510, 1463,1404 e 1393
ν C-O 1325 1285
ν C-O 1272 bNR
ν C-O 1090 1116
ν C-O 1044 1051
δ C-H 879 984 e 877
δ C-H 803 671
ABAM /IVERM
νO-H 3310 3463
IR2011-88378TK
νC-H 2961 – 2881 2967 e 2932
δCH3CH2 1462 – 1382 1716 e 1455
ν C-O 1323 1379
ν C-N 1245 bNR
ν C-N 1086 986 e 935
δ C-H 881 e 802 908,869,833 e 761
SDX/TMP
ν O-H 3680 – 2813 3500, 3401, 3200 e 3000
aBR033532
ν C=O 1698 – 1537 1700, 1600 e 1400
ν C-O 1243 – 1191 1200
SMZ/TMP δ O-H 3739 – 2756 3468 – 3114
IR-NIDA-63731 ν C=O 1744 – 1514 1698 – 1471
OTC
ν O-H 3091 – 2724 3700 – 2500
aBR000704
ν C=O 1705 – 1545 1600
δ CH3CH2 1487 – 1387 1400
ν C-O 1316 – 1244 1300
δ C-H 994 900
δ C-H 687 700
* Medidas Realizadas em ATR; ** Medidas Referenciadas em disco de KBR, *** Código da Referência da Base
de Dados SDBR-IR para Compostos Orgânicos, aobtido no laboratório da Bio-Rad/Sadtler IR e bNão
Referenciado.

128
Os resultados indicam semelhanças, entre padrão de referência PENG e amostras de
ABAM/IVERM, GEN e PENG As similaridades estão, principalmente, na região de 2857 a
3300cm-1, que mostra presença de O – H associado. A região entre 3030 a 2770 cm-1 é
denominada banda dos alcanos, aparece bem fina.
Quanto ao espectro PENG (padronizado) foram utilizadas, a fim de evidenciar
carbonilas de amida e os cromóforos nos antimicrobianos, à base de penicilina. Estes dados são
característicos, em regiões de 1724 a 1818 cm-1 de ν C=O que denota presença de Amidas
primárias, bem como regiões de 1538 a 1776 cm-1 que é correspondente a ν C-O refletindo a
presença de ácido carboxílico, éteres, álcoois primários de secundários. As bandas entre 1538
a 1724 cm-1 são referentes à amida e aparecem no espectro de todos os compostos, em alguns
casos, as intensidades desloca a banda, porém não altera os resultados, confirmando as
vibrações do grupo carbonila. A região entre 1613 a 1724 cm-1 é referente a estiramento da
ligação das carbonilas, ν C=O, associado a amidas secundárias que aparecem em anéis de
lactamas, a absorção está sujeita a efeitos de conjugação e sofrem deslocamentos para
frequências inferiores que alteram o valor de 1923 a 2380 cm-1, para SDX/TMP, SMZ/TMP e
OTC. As regiões entre 1388 a 1515cm-1 estão presentes em todos os compostos e são específicas
para vibrações de δ CH3CH2. As bandas de 1086 e 1244 cm-1 referentes à ν C-N que distingui aminas
alifáticas de aromáticas, respectivamente. As regiões entre 800 e 885 cm-1, de vibração da
ligação dupla de C = C fora do plano (R1 (R2) C = C) e R1CH = CHR2, indica a formação de
isômeros cis e trans, respectivamente. Em 802 cm-1 aparece uma banda atribuída a presença de
anel benzeno orto substituído, no espectro referente a ABAM/IVERM. Na região espectral entre
2755 e 3740 cm-1 de SMZ/TMP e SDX/TMP aparecem bandas que apontam a formação de
quelato, pelo estiramento δ O-H.

129
5.1.3 Quantificação de PENG por UFLC-DAD.
Esta análise foi selecionada para quantificação da PENG nos medicamentos, pois é
estabelecida com referência pelos organismos legais de controle de qualidade e apresentam
vantagens, principalmente, relacionado ao tempo, a confiabilidade, a sensibilidade, a
especificidade, ao consumo reduzido de solvente e robustez na identificação de grupos ativos
de fármacos.
5.1.3.1 Estudo das condições de análise
Para o estudo das melhores condições de identificação da PENG foram preparadas
soluções padrão e realizado a leitura dos cromatogramas em 220, 252, 257 e 264 nm para
identificar o betalactâmico como é mostrado na Figura 5.3.
Figura 5.3-Perfil cromatográfico da penicilina (PENG) referente ao padrão certificado
0 2 4 6 8 10
0
200
400
600
800
1000
1200
264 nm
257 nm
252 nm220 nm
I. P
(m
AU
)
T.R (min)
A identificação da PENG foi realizada por método isocrático descrita no cromatograma
e a solução padrão foi preparada com ausência de solução tampão. A fase móvel apresentou
melhor composição na condição 1. O fluxo foi de 0,1 mL min-1, devido o diâmetro do poro da
coluna, onde ocorre a separação ser bem reduzido de 2,0 mm. A detecção ocorreu em quatro
comprimentos de onda selecionados previamente, assim foram identificados por bandas de

130
absorção no ultravioleta e referenciados pela International Pharmacopoeia (2015b) para
análises de penicilinas e trabalho de Cámara et al (2013). Os betalactâmicos têm afinidade por
colunas de fase reversa, por isso foi utilizada uma C18.
As análises em condições planejadas o valor máximo considerado foi de 220 nm, pois
apresentavam intensidade e resolução da PENG condizentes. Sabendo que, o bom desempenho
deste método deve-se muito ao estudo preliminar das características físico-químicas e
composição das substâncias, pois parâmetros como o pH influenciam na interação dos
compostos com a fase móvel e estacionária da coluna como é descrito por Hoff et al (2012),
Luo et al (2015) e Zeng et al (2014). Logo, o estudo influenciou na escolha dos antibióticos
que mais se adequavam a separação de penicilinas em corridas de 10 min.
Neste procedimento considerou-se que os parâmetros de desempenho da coluna de
separação, tais como: número de pratos teóricos, fator de retenção, resolução e fator de simetria
refletem nas melhores condições para análise. A Figura 5.4 apresenta a correlação entre os
parâmetros de otimização do método (resolução e seletividade) para cada condição testada.
Figura 5.4-Resultados do planejamento das condições cromatográficas
1,6411,538
1,434
1,7591,863
1,968
1 2 3
0
1
2
3
CC
Pa
râm
etro
s
Condições
Resolução
Seletividade
C
Para todas as condições utilizadas o resultado de resolução do pico da PENG é aceitável.
Segundo as monografias Europeias e Americana o fator de simetria deve estar entre 0,8 e 1,5;
os números de pratos devem ser maiores que 2000 para HPLC; fator de retenção deve ser maior
que 2 (EUROPEAN COMISSION, 1993; EUROPEAN COMMISSION, 2006; LOPES et al.,
2011).

131
As melhores condições de identificação da PENG foram alcançadas, tais como: a
minimização dos efeitos de interferentes e sobreposição de picos, a minimização da adsorção
da penicilina e do padrão externo na coluna cromatográfica, por formação de picos de
simétricos comprovado pelo fator de simetria e cálculos da robustez. Estes fatores foram
descritos pelos pesquisadores Vargas Mamani, Reyes Reys e Rath (2009).
5.1.3.2 Validação do método
Segundo o Guia de Validação da Emea (2006), os procedimentos analíticos devem
descrever, detalhadamente, as etapas necessárias para executar cada teste.
Assim, os dados foram obtidos com nove níveis de concentração, sendo a faixa de
trabalho para PENG de 0,001 a 0,01 µg mL-1 são submetidos a regressão linear, o resultado é
apresentado na Figura 5.5. Estes dados oferecem resultados analíticos quando testados em
amostras externas mostram o conteúdo real de PENG presente nas amostras.
Figura 5.5-Curva analítica para quantificação de PENG em medicamentos
Os resultados são comparados ao padrão aceitável de precisão, por adição de padrão
descrito por Mantovani et al (2012) cujas condições experimentais descritas devem apresentar
curvas lineares entre 95,99 e 99,99% para PENG.
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,09 0,10
200
400
600
800
1000
1200
1400
1600
1800
2000
A.P
(m
AU
)
[PENG] (g mL-1
)

132
Na Tabela 5.2 são apresentados os resultados dos parâmetros calculados para validação
do método.
Tabela 5.2-Desempenho do método para quantificação de PENG em medicamentos veterinários
Parâmetros de Validação Resultados para PENG K+
Linearidade (R) 0,9991
LD (µg mL-1) 7,384 × 10-4
LQ (µg mL-1) 2,0941 × 10-3
Rendimento médio (%) 72,51%
Equação APico = 2059× CPENG + 93,43
Eq: Equação que representa a curva analítica: LD: limite de detecção, LQ: limite
de quantificação.
Para cada 10 µL da amostra e padrão injetado foram avaliados os erros das medidas que
foram de 10,54 a 15,14 %. Esses valores estão dentro do limite aceitável de 20 %, segundo a
VICH GL40 da Emea (1999). A resposta do detector foi registrada em toda faixa ampla do UV,
porém os cromatogramas das amostras são apresentados em 220 nm, uma vez que este valor
tem sido considerado para quantificação de PENG, em diferentes matrizes, inclusive em
métodos desenvolvidos por Babington et al(2012), Bailón-Pérez et al (2009), Rambla-Alegre
et al (2011b) e da JP XVI (2007).
A capacidade do sistema cromatográfico foi investigada, com objetivo de resolver os
picos PENG de suas principais misturas. Durante a amostragem, os compostos com diferentes
formulações de PENG, foram armazenados ao abrigo da luz, do calor e consequente oxidação,
por isso as ótimas condições dos sinais analíticos são garantidas. A especificidade demonstrou
que a OTC interferiu na resolução do pico da PENG, porém este fator não prejudica a
seletividade do método, pois as resoluções dos picos estão no intervalo de dois, como
recomendado por manual de validação do ICH (2005).
Segundo Épshtein (2004), a determinação do LQ, a partir da curva analítica, é um dos
requisitos para aceitabilidade da reprodutibilidade do método. Sendo assim, o LD e LQ refletem
na sensibilidade do método desenvolvido, pois estão condizentes com a curva analítica e
apresentam relação sinal/ruído bastante reduzido.

133
A precisão e exatidão do método foi determinada por ensaio intradia. Os resultados de
precisão são apresentados na Tabela 5.3.
Tabela 5.3-Precisão e exatidão do método para quantificação de PENG em medicamentos veterinários,
expressos pelo coeficiente de variação (%CV)
Adição padrão externo (µg mL-1) Precisão e Exatidão (n=3 × 15)
0,0010 14,48
0,0020 15,52
0,0040 14,51
0,0100 16,55
0,0200 10,35
0,0400 11,46
A especificidade do método foi determinada por observação de interferentes nos
componentes, presentes nas formulações (medição dos brancos). Os resultados dos testes têm
sido comparados com os resultados do padrão de referência. Confirmando que, os antibióticos
associados à PENG não interferem no desempenho do método.

134
Os resultados para robustez são apresentados na Tabela 5.4, variando-se as três
condições.
Tabela 5.4-Robustez do método para quantificação da PENG em medicamentos veterinários, expresso pelo
coeficiente de variação (%CV)
Adição de padrão externo (µg mL-1) Robustez (%CV)
Condição 1 Condição 2 Condição 3
0,001 14,48 14,54 14,61
0,002 15,52 15,56 15,59
0,004 14,51 14,17 14,18
0,01 16,55 16,56 16,57
0,02 10,35 10,36 10,36
0,04 11,46 11,47 11,47
Os resultados mostraram que o método é aplicável para determinar PENG, na presença
dos produtos de degradação, sendo confiável para a rotina, pois foram variadas as condições de
análises e o método apresentou-se robusto.

135
5.1.3.3 Identificação de PENG em amostras de medicamentos
As amostras de medicamentos descritas na Tabela 4.2 foram selecionadas para
identificação de PENG nos medicamentos. Os perfis cromatográficos dos grupos de
medicamentos são apresentados na Figura 5.6.
Figura 5.6-Perfil cromatográfico das amostras de medicamentos em grupos
0 2 4 6 8 10
0
1000
2000
3000
4000
Tempo de Retenção (min)
Extraído - 220nm, 4nm (1) (Branco)mAU
0 2 4 6 8 10
0
1300
2600
3900
5200
2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5 6,0
0
270
540
810
1080
Extraído - 220 nm, 4nm (1)
TR (min)
PENG
mAU
(A)Extraído - 220 nm, 4nm (1)
TR (min)
PENG
mAU
0 2 4 6 8 10
0
1300
2600
3900
5200
3,0 3,5 4,0 4,5 5,0 5,5 6,00,0
9,4
18,8
28,2
TR (min)
PENG
Extraído - 220 nm, 4nm (1)mAU
DHSTR
STR
(B)
TR (min)
PENG
Extraído - 220 nm, 4nm (1)mAU
0 2 4 6 8 10
0
400
800
1200
1600
1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0
110,4
112,8
115,2
117,6
120,0
TR (min)
PENGExtraído - 220 nm, 4 nmmAU
(C)
TR (min)
OTC
PENG
Extraído - 220 nm, 4 nmmAU
0 2 4 6 8 10
0
1200
2400
3600
4800
3,4 3,6 3,8 4,0 4,2 4,4 4,6 4,8 5,0 5,2 5,4 5,6
8,2
12,3
16,4
20,5
TR (min)
PENG
mAU
(D)
TR (min)
PENG GENT ABAM
OTC
SMZ
mAU
O estudo dos cromatogramas mostrou que resolução dos cromatogramas em todos os
níveis é alta, entre 2,33 a 9,67, cobrindo uma faixa ampla de concentração.

136
Os ensaios de adequação, para cada parâmetro foram realizados. Os espectros das
amostras foram registrados, separadamente, para confirmação dos picos, onde ocorrem as
máximas absorções no UV. A solução padrão de PENG K+ foi injetada seis vezes e as áreas dos
picos foram registradas.
A Figura 5.6B mostra o perfil do cromatograma do grupo de amostra, contendo PENG,
evidenciando as principais associações de STR e DHSTR. O tempo de retenção (TR) PENG foi
de 3,17 min.
Os picos cromatográficos da PENG apresentaram boa resolução, expondo, inicialmente,
que PENG está livre de interferência de impurezas (Figura 5.6B). A Figura 5.6.C apresenta
cromatogramas para PENG, contendo OTC, em tempos de retenção de 0,82 e 2,82 min,
respectivamente. Os picos apresentaram boa resolução para OTC, enquanto PENG tem sido
sobreposto. O mesmo ocorre com na Figura 5.6.D, porém o tempo de retenção ocorre em 4,11
min para PENG e SMZ, OTC, GEN e ABAM está em 1,29, 1,66, 5,74 e 7,74, respectivamente.
Além disso, essa ralação prova que o grupo de amostra, contendo mistura de PENG e
estreptomicinas, apresentou resolução de pico equivalente a 3,31. Assim, as estreptomicinas,
também, influenciaram na retenção de PENG 1,61 min, em comparação com padrão (Figura
5.3).
5.1.3.4 Cálculo do teor de pureza dos medicamentos
A Tabela 5.5 apresenta os resultados das análises quantitativas de PENG em
medicamentos. O método apresentou 72,51% de rendimento. A substância foi detectada
aplicando o método com LD de 7,384 × 10-4 µg mL-1.

137
Tabela 5.5-Cálculo do teor de PENG nos medicamentos analisados por cromatografia
Amostras
Concentração predita pelo método
(µg mL-1) com rendimento de
72,51%
Concentração Referência
(µg mL-1)
Pureza de PENG
predita pelo método
(%)
B35 0,00053 0,001 52,58
B33 0,00065 0,001 64,52
A9 0,01460 0,020 73,02
B34 0,00074 0,001 73,98
A8 0,00768 0,010 76,75
B13 0,00077 0,001 76,75
B16 0,00154 0,002 76,75
A10 0,01535 0,020 76,77
A12 0,03071 0,040 76,77
A6 0,00768 0,010 76,79
B26 0,01537 0,020 76,86
B29 0,03074 0,040 76,86
B30 0,03078 0,040 76,95
B18 0,00211 0,002 79,04
B14 0,00081 0,001 81,21
B17 0,00162 0,002 81,21
B22 0,00812 0,010 81,22
B25 0,01628 0,020 81,37
B28 0,03255 0,040 81,38
B36 0,00083 0,001 83,36
B32 0,00084 0,001 83,82
B37 0,00086 0,001 86,39
A2 0,00089 0,001 88,89
A5 0,00889 0,010 88,88
A7 0,00889 0,010 88,91
A11 0,03556 0,040 88,91
A4 0,00178 0,002 88,96
B20 0,00363 0,004 90,64
B21 0,00943 0,010 94,26
B23 0,00943 0,010 94,26
B19 0,00384 0,004 95,94
A1 0,00096 0,001 96,44
A3 0,00194 0,002 96,80
B15 0,00972 0,010 97,16
B24 0,00972 0,010 97,17
B31 0,00097 0,001 97,16
B27 0,01957 0,020 97,87
Os resultados mostraram que as amostras B33 e B35 apresentaram teores muito abaixo
do que o referenciado estando entre 53 – 65%. As amostras A9, B34, A8, B13, B16, A10, A12,
A6, B26, B29, B30 e B18 apresentaram pureza de 73 – 80%. As 12 amostras B14, B17, B22,
B25, B28, B36, B32, B37, A2, A5, A7, A11 e A4 apresentaram pureza de 81 – 89%. Enquanto
que dez amostras sendo B20, B21, B23, B19, A1, A3, B15, B24, B31 e B27 apresentaram teores
aceitáveis de 90 – 98%. Estes resultados indicam que apenas duas amostras não se adequam
aos critérios de aceitabilidade.

138
5.1.4 Análise de PENG em medicamentos por UV VIS NIR
5.1.4.1 Atribuição das bandas no UV VIS NIR e relevância para modelagem dos dados
As bandas foram atribuídas pela comparação dos grupos de medicamentos, à base de
PENG, das amostras puras e pretratadas. As regiões espectrais utilizadas, para construção dos
modelos, são determinantes no fornecimento da informação estrutural da PENG, por isso
considerou-se o mínimo de interferência nas soluções. Os espectros são apresentados na Figura
5.7.
Figura 5.7-Espectros da PENG certificado e das amostras comerciais de medicamentos
230 460 690 920 1150 1380 1610 1840 2070 23000.000
0.011
0.022
0.033
0.044
0.055
0.066
0.077
0.088
0.099
Amostras
PENG (padronização) 1mg mL-1
Ab
s
Comprimento de Onda (nm)
A região de 200 a 2300 nm foi selecionada, para construir os modelos finais, baseados
na aplicação da biblioteca utilizada pelos trabalhos recentes, principalmente o de Chong et al
(2009a). A região correspondente ao 2º Overtone em 1100 – 1200 nm refere-se a vibrações de
CH, CH2 CH3, = CH e ArH. Duas regiões de 1300 a 1600 nm correspondem ao 1º Overtone
referente a vibrações de O – H e N – H, enquanto que em 1650 – 1800 e 1877 a 2000 nm são
específicos para CH, CH2CH3, =CH e ArH. Por fim, em 2000 a 2300nm, mais próxima do
infravermelho médio, são observadas combinações vibracionais de O – H, N – H e C – H. Os
estudos anteriores para construção de um modelo levaram em consideração faixas produzidos
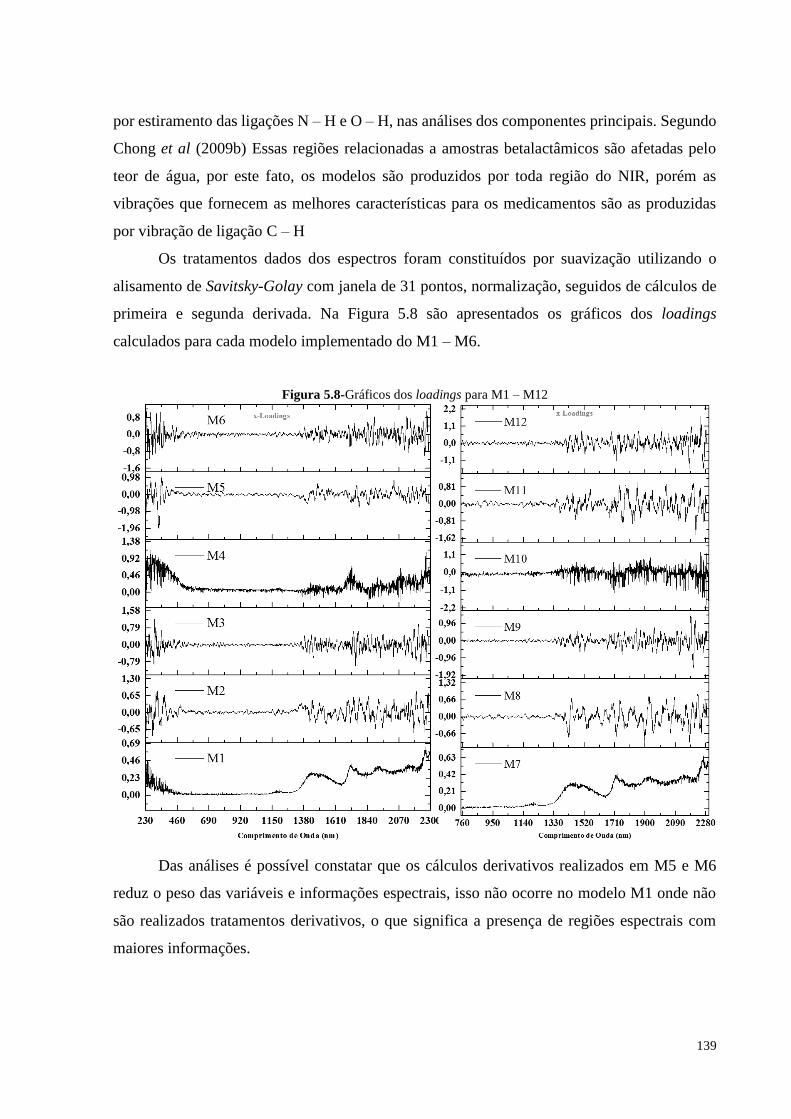
139
por estiramento das ligações N – H e O – H, nas análises dos componentes principais. Segundo
Chong et al (2009b) Essas regiões relacionadas a amostras betalactâmicos são afetadas pelo
teor de água, por este fato, os modelos são produzidos por toda região do NIR, porém as
vibrações que fornecem as melhores características para os medicamentos são as produzidas
por vibração de ligação C – H
Os tratamentos dados dos espectros foram constituídos por suavização utilizando o
alisamento de Savitsky-Golay com janela de 31 pontos, normalização, seguidos de cálculos de
primeira e segunda derivada. Na Figura 5.8 são apresentados os gráficos dos loadings
calculados para cada modelo implementado do M1 – M6.
Figura 5.8-Gráficos dos loadings para M1 – M12
Das análises é possível constatar que os cálculos derivativos realizados em M5 e M6
reduz o peso das variáveis e informações espectrais, isso não ocorre no modelo M1 onde não
são realizados tratamentos derivativos, o que significa a presença de regiões espectrais com
maiores informações.

140
Assim, as análises multivariadas foram realizadas, cuidando para que não houvesse
resultados falso-positivos, e/ou o analito fosse confundido com outros produtos. Neste aspecto,
os outliers podem ser gerados por uma amostra que seja diferente do conjunto das amostras
calibradas. O funcionamento do incorreto do instrumento promove falhas no método de
referência, erros de transcrição ou até mesmo erro do analista. Logo, este resultado pode não
ser considerado falso positivo, mas exerce influência no modelo, aumentando o erro do método.
Na Figura 5.9 são apresentados os gráficos dos loadings para modelos do M7 – M12,
onde são analisadas as variáveis que mais influenciam na modelagem quando se aplica os
tratamentos matemáticos descritos anteriormente.
Figura 5.9-Gráficos dos loadings para M13 – M24
As análises dos gráficos dos loadings mostraram que aplicação de tratamentos em
espectros dos modelos M13 –16 e M19 – M23 mostram informações nas regiões 1300 – 2280
nm.
Diante disto, recentemente, uma versão da Farmacopeia Europeia tem normatizado pela
EMEA/CHMP/CVMP/QWP/17760/2009 descrito em Emea (2012b) a quantificação das
substâncias terapêuticas, utilizando a região do NIR. Esta análise quantitativa foi revisada,
segundo o desempenho analítico do método, considerando a validação pelos cálculos de

141
precisão, linearidade, precisão, especificidade e robustez. Deste modo, o cumprimento dos
critérios tornam aceitos os procedimentos, pois podem ser comparados aos cromatográficos,
enquanto quantitativos como reporta Hartauer e Guillory (1989).
O método desenvolvido na região próximo do visível, permite a análise rápida dos
compostos, contidos nos medicamentos, e que estão envolvidos em toda cadeia do processo de
qualidade (CHONG et al., 2009a, 2009b; HARTAUER; GUILLORY, 1989; LIU; HU, 2011;
MBINZE et al., 2015; SIVAKESAVA; IRUDAYARAJ, 2002). A principal vantagem é a
eliminação dos reagentes tóxicos das medidas analíticas, pela construção de modelos preditivos
para determinação rápida da PENG.
Segundo os autores Liu e Hu (2011) a análise dos espectros da amostra, com base nos
espectros de referência, para identificar e extrair a informação espectral geram ótimos
resultados preditivos. A saber, de Chong et al (2009a) que desenvolveram e aplicaram um
modelo nessas regiões espectrais para identificação de 26 tipos de cefalosporina (grupo de
antibióticos beta lactâmicos), em medicamentos injetáveis. Desta forma, diante desses ótimos
resultados da literatura, o presente trabalho apresenta nova forma de construção do modelo para
determinação de PENG presente nos medicamentos em uma região mais ampla comparada aos
trabalhos citados. As análises mostram que são aptos para análise rápida, sobretudo a respeito
da variabilidade das amostras a base de penicilinas.
Cada medida espectral pode conter fontes de variabilidade associada às influências
externas, tais como: lotes, local e diferença nos tempos de armazenamento. Para minimizar
esses efeitos, os espectros foram coletados em dias diferentes (dia 1, dia 2 e dia 3) e obtidos
médias e desvios. Retifica-se que, a escolha das amostras, para calibração e validação externa,
seguiu a recomendação da EMEA/CVMP/961/01 da Emea (2012b), para quantificação de
PENG nessas regiões espectrais. Na Figura 5.10 são apresentados os scores obtidos pela análise
dos componentes principais.

142
Figura 5.10-Gráficos dos Scores M1 – M12

143
Figura 5.11-Gráficos dos Scores M13 – M24

144
Esses resultados são legitimados principalmente pela amostragem, pois constituiu uma
etapa onde controlou-se as condições para obtenção das medidas. A análise por PCA mostrou
que não houve agrupamento entre as amostras utilizando todos os 24 modelos.
Logo, as amostras de medicamentos são selecionadas, conforme sua concentração de
PENG rotulada e quantificada pelos métodos de referências, principalmente o UFLC-DAD.
Adicionalmente, é realizada análise comparativa com trabalhos atuais e métodos normatizados,
para que não haja anomalias.

145
5.1.4.2 Desempenho dos modelos de acordo com 17760/2009/EC e determinação das
concentrações de PENG
Neste trabalho, a Quimiometria tradicional é fundamental, pois envolve o conjunto de
dados de calibração que utiliza a técnica de validação cruzada, para avaliar a adequação do
modelo aos parâmetros
As amostras selecionadas para validação externa, que foram excluídas do processo de
modelagem, têm sido incluídas para validar os modelos ideais. É importante citar que, o erro
quadrático médio da calibração (RMSEC) e a raiz quadrada média do erro da validação cruzada
(RMSECV) são correlacionados ao número de fatores (componentes principais e/ou variáveis
latentes) e utilizadas para determinação do melhor número de variáveis para modelagem dos
dados. Como RMSEC é sempre decrescente quando se aumenta o número de fatores, ele
fornece a estimativa mais otimista do desempenho do modelo de calibração PLS, isto é, mostra
a capacidade de ajustar-se aos dados observados no conjunto de calibração. Então, além de um
conjunto externo de dados, não envolvidos no grupo de amostras calibradas, esse procedimento
é utilizado, para previsão externa, do conjunto de dados que não fazem parte desse sistema.
O desempenho do método foi averiguado pelo cálculo (RMSEP). Para desenvolver os
modelos de Regressão, contidos na Tabela 5.6, foram considerados três grupos comuns às
amostras isoladas. Os espectros de calibração colecionados, a partir da média, foram utilizados
para gerar os espectros de referência.
As atribuições das bandas espectrais, do Padrão de Referência (PENG), são
minimizadas as condições de interferências dos outros componentes. Os resultados
apresentados na Tabela 5.6 mostraram que o processamento mais adequado é constituído por:
normalização da área, correção da linha de base e por tratamentos de primeira derivada.(DE
BLEYE et al., 2012).

146
Tabela 5.6-Características dos modelos de calibração para determinação de PENG em medicamentos.
Modelos Calibração (Validação Cruzada) Validação Externa
PC R RMSEC EQ1 R RMSEP EQ2
Modelos para Amostra Puras (E)
M1 10 0,5092 0,0589 0,2593 CR + 0,0545 0,1627 0,0825 0,0806 CR + 0,0706
M2 10 0,4195 0,0622 0,1760 CR + 0,0606 0,6341 0,0525 0,3128 CR + 0,0444
M3 10 0,3138 0,0651 0,0984 CR + 0,0663 0,3846 0,0622 0,1139 CR + 0,1139
M4 10 0,2754 0,0766 0,0758 CR + 0,0777 0,2461 0,0747 0,0681 CR + 0,0834
M5 1 0,4054 0,0626 0,1643 CR + 0,0614 0,4265 0,0613 0,0776 CR + 0,0675
M6 10 0,3489 0,0642 0,1218 CR + 0,0646 0,2932 0,0623 0,0777 CR + 0,0665
M7 4 0,8494 0,0421 0,7215 CR + 0,0234 0,7967 0,0461 0,6341 CR + 0,0374
M8 6 0,9952 0,0067 0,9905 CR + 0,0007 0,9948 0,0064 0,9962 CR + 0,0005
M9 5 0,996 0,0061 0,9921 CR + 0,0006 0,9954 0,0063 1,0045 CR + 0,0008
M10 5 0,8408 0,0431 0,7069 CR + 0,0246 0,7385 0,0516 0,5918 CR + 0,0416
M11 5 0,9903 0,0094 0,9807 CR + 0,0014 0,9912 0,0098 0,9442 CR + 0,0080
M12 6 0,9975 0,0047 0,9951 CR + 0,00036 0,9963 0,0056 0,9808 CR + 0,0031
Modelos para Amostras Diluídas (E)
M13 2 0,6803 0,0084 0,4629 CR + 0,0037 0,8105 0,0094 0,4113 CR + 0,0032
M14 2 0,6342 0,0088 0,4022 CR + 0,0041 0,7324 0,0104 0,3437 CR + 0,0032
M15 6 0,6998 0,0082 0,4897 CR + 0,0035 0,6511 0,0107 0,4004 CR + 0,0041
M16 3 0,8178 0,0066 0,6689 CR + 0,0022 0,9232 0,0054 0,8289 CR + 0,0018
M17 1 0,6619 0,0086 0,4382 CR + 0,0038 0,7763 0,0098 0,3980 CR + 0,0029
M18 1 0,7205 0,0079 0,5191 CR + 0,0033 0,7987 0,0094 0,4267 CR + 0,0031
M19 1 0,6638 0,0086 0,4406 CR + 0,0038 0,8119 0,0096 0,3806 CR + 0,0039
M20 3 0,7974 0,0069 0,6358 CR + 0,0025 0,9105 0,0059 0,7676 CR + 0,0028
M21 5 0,8126 0,0066 0,6603 CR + 0,0023 0,9814 0,0029 0,8815 CR + 0,0015
M22 2 0,8034 0,0068 0,6456 CR + 0,0024 0,9119 0,0059 0,7538 CR + 0,0024
M23 1 0,7061 0,0081 0,4986 CR + 0,0034 0,8552 0,0087 0,4599 CR + 0,0034
M24 3 0,8417 0,0062 0,7083 CR + 0,0021 0,9841 0,0026 0,9316 CR + 0,0011
Os resultados da Tabela 5.6 apresentam valores baixos de RMSEP, em ambos os
modelos PCR e PLS em regiões que compreendem o UV VIS NIR (200 a 2300 nm), indicando
que os métodos têm sido adequados para quantificação de PENG.
Na Figura 5.12 são apresentadas as variações do erro entre as concentrações referência
e as medidas nas 24 amostras do conjunto de amostras puras e externas pelo método UV VIS
NIR.
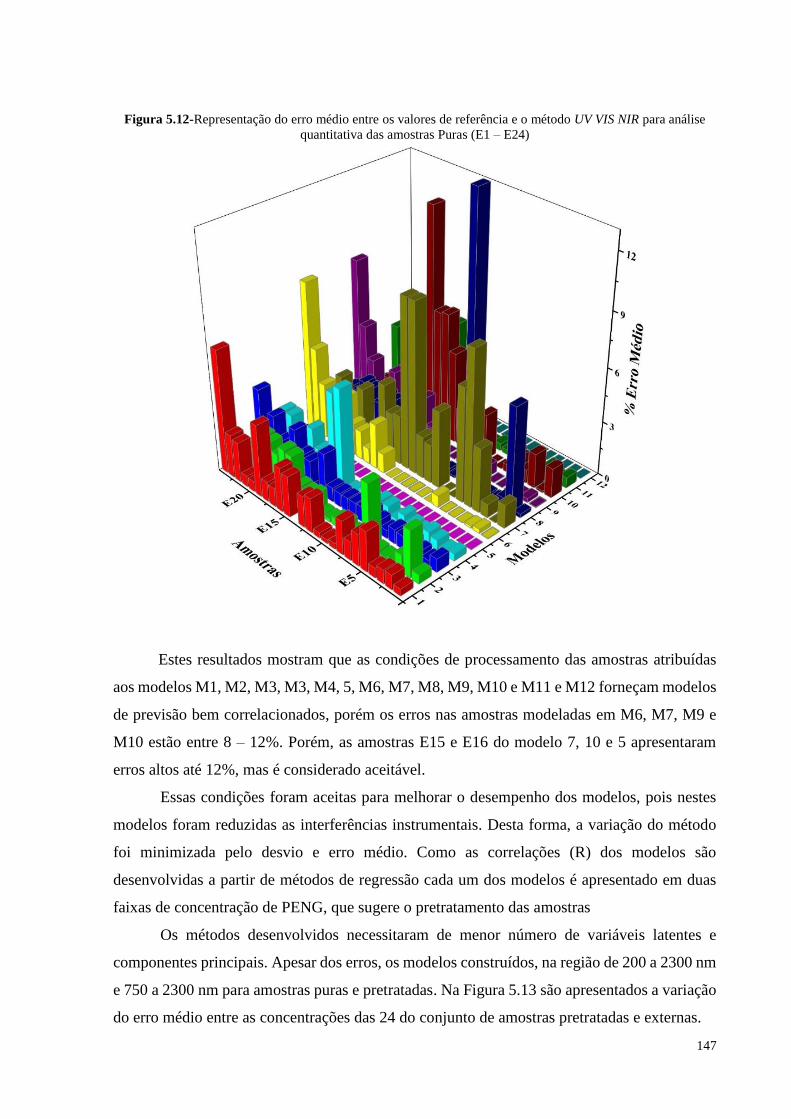
147
Figura 5.12-Representação do erro médio entre os valores de referência e o método UV VIS NIR para análise
quantitativa das amostras Puras (E1 – E24)
Estes resultados mostram que as condições de processamento das amostras atribuídas
aos modelos M1, M2, M3, M3, M4, 5, M6, M7, M8, M9, M10 e M11 e M12 forneçam modelos
de previsão bem correlacionados, porém os erros nas amostras modeladas em M6, M7, M9 e
M10 estão entre 8 – 12%. Porém, as amostras E15 e E16 do modelo 7, 10 e 5 apresentaram
erros altos até 12%, mas é considerado aceitável.
Essas condições foram aceitas para melhorar o desempenho dos modelos, pois nestes
modelos foram reduzidas as interferências instrumentais. Desta forma, a variação do método
foi minimizada pelo desvio e erro médio. Como as correlações (R) dos modelos são
desenvolvidas a partir de métodos de regressão cada um dos modelos é apresentado em duas
faixas de concentração de PENG, que sugere o pretratamento das amostras
Os métodos desenvolvidos necessitaram de menor número de variáveis latentes e
componentes principais. Apesar dos erros, os modelos construídos, na região de 200 a 2300 nm
e 750 a 2300 nm para amostras puras e pretratadas. Na Figura 5.13 são apresentados a variação
do erro médio entre as concentrações das 24 do conjunto de amostras pretratadas e externas.

148
Figura 5.13-Representação do erro médio entre os valores de referência e o método UV VIS NIR para análise
quantitativa das amostras tratadas
Estes resultados mostraram que as condições de processamento das amostras atribuídas
nos modelos M13, M14, M15, M16, M17, M18, M19, M20, M21, M22, M23 e M24 forneceram
modelos de previsão bem correlacionados, uma vez que os erros entre as amostras são menores
que 9%.
Com base nos resultados de ambos os métodos apresentados tanto a calibração,
validação cruzada e validação externa importante, pois na literatura científica há relatos das
aplicações dos modelos multivariados na região (750 a 2500 nm), para controle de qualidade
das soluções injetáveis a base de gentamicinas, dos comprimidos e das cápsulas dos
medicamentos (BROAD et al., 2000; PARISOTTO et al., 2005).
Os modelos mostram que a validação externa, do conjunto composto por 24 modelos de
medicamentos injetáveis, apresentou bom desempenho. Assim, são cumpridos os critérios das
diretrizes Q2R1 da ICH (2005).
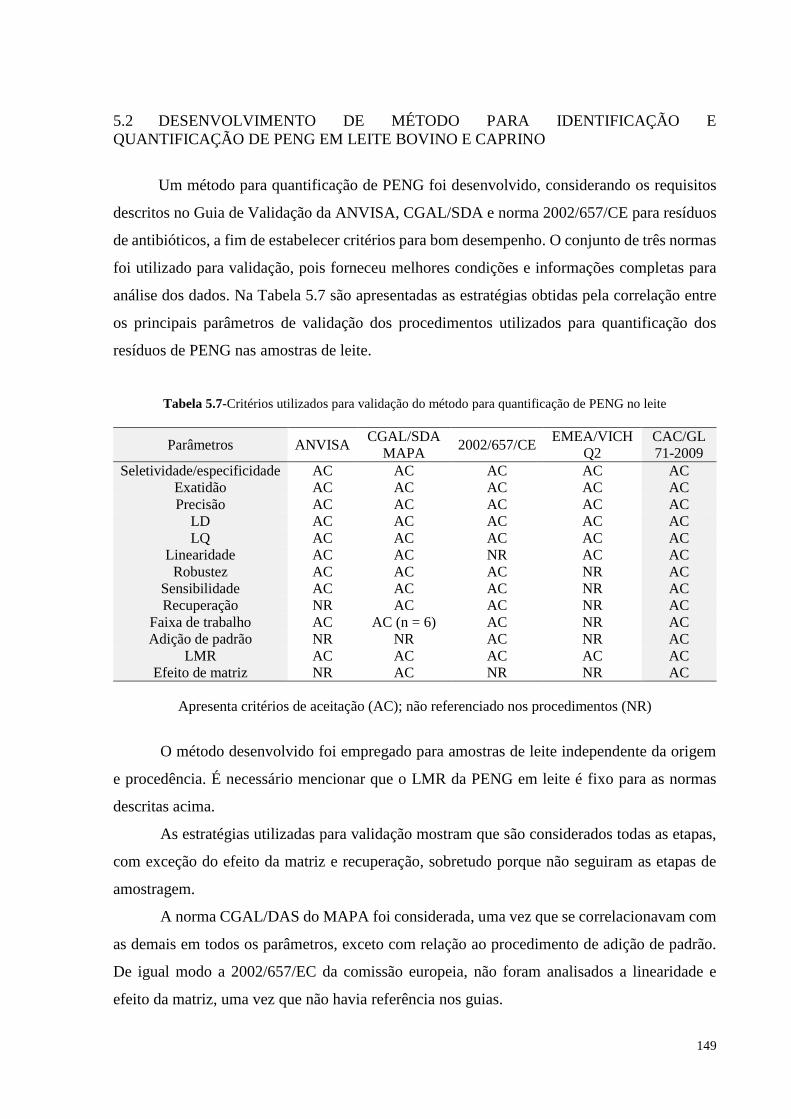
149
5.2 DESENVOLVIMENTO DE MÉTODO PARA IDENTIFICAÇÃO E
QUANTIFICAÇÃO DE PENG EM LEITE BOVINO E CAPRINO
Um método para quantificação de PENG foi desenvolvido, considerando os requisitos
descritos no Guia de Validação da ANVISA, CGAL/SDA e norma 2002/657/CE para resíduos
de antibióticos, a fim de estabelecer critérios para bom desempenho. O conjunto de três normas
foi utilizado para validação, pois forneceu melhores condições e informações completas para
análise dos dados. Na Tabela 5.7 são apresentadas as estratégias obtidas pela correlação entre
os principais parâmetros de validação dos procedimentos utilizados para quantificação dos
resíduos de PENG nas amostras de leite.
Tabela 5.7-Critérios utilizados para validação do método para quantificação de PENG no leite
Parâmetros ANVISA CGAL/SDA
MAPA 2002/657/CE
EMEA/VICH
Q2
CAC/GL
71-2009
Seletividade/especificidade AC AC AC AC AC
Exatidão AC AC AC AC AC
Precisão AC AC AC AC AC
LD AC AC AC AC AC
LQ AC AC AC AC AC
Linearidade AC AC NR AC AC
Robustez AC AC AC NR AC
Sensibilidade AC AC AC NR AC
Recuperação NR AC AC NR AC
Faixa de trabalho AC AC (n = 6) AC NR AC
Adição de padrão NR NR AC NR AC
LMR AC AC AC AC AC
Efeito de matriz NR AC NR NR AC
Apresenta critérios de aceitação (AC); não referenciado nos procedimentos (NR)
O método desenvolvido foi empregado para amostras de leite independente da origem
e procedência. É necessário mencionar que o LMR da PENG em leite é fixo para as normas
descritas acima.
As estratégias utilizadas para validação mostram que são considerados todas as etapas,
com exceção do efeito da matriz e recuperação, sobretudo porque não seguiram as etapas de
amostragem.
A norma CGAL/DAS do MAPA foi considerada, uma vez que se correlacionavam com
as demais em todos os parâmetros, exceto com relação ao procedimento de adição de padrão.
De igual modo a 2002/657/EC da comissão europeia, não foram analisados a linearidade e
efeito da matriz, uma vez que não havia referência nos guias.

150
Os critérios da Emea não foram relatados para robustez, sensibilidade, recuperação e
faixa de concentração, pois não são reportados os critérios de aceitação.
Quanto as referências contidas no guia para validação da Codex Alimentarius pela
norma CAC/GL 71-2009 são admitidos todos os cálculos dos parâmetros, pois apresentam
justificativas para aceitação das matrizes Alimentarius com suspeita de contaminação por
antimicrobianos.
É necessário enfatizar para construção do método de quantificação dos resíduos de
PENG considerou-se a análise do tempo de retenção, área dos picos e concentração de PENG
em amostras fortificadas.

151
5.2.1 Otimização do preparo das amostras de leite controle bovino e caprino
Esta etapa foi primordial para o bom desempenho do método. O preparo de amostra foi
eficaz, robusto e simples, além de utilizar mínimas quantidades de solventes e reagentes tóxicos,
a fim de garantir a segurança dos procedimentos.
Como as etapas visam a eliminação de interferentes, extração e concentração adequada
da PENG são apresentados na Figura 5.14 os cromatogramas referentes as amostras controle
Figura 5.14-Cromatogramas obtidos para amostra controle LB (branco) e amostra fortificada com PENG
0
300
600
900
1200
1500
1800
0 1 2 3 4 5 6 7 8 9 10
0
300
600
900
1200
1500
1800
mAU
Branco da amostra controle LB
T.R (min)
mAU
[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
Amostra fortificada 0,002 g mL-1
com PENG
O principal objetivo da análise da extração dos componentes interferentes foi de
proporcionar a identificação e quantificação dos possíveis resíduos de PENG, em matrizes
biológicas e complexas, dentro dos limites estabelecidos pelos termos legais para PENG no
leite de 0,004 µg mL-1, uma vez que preparar amostras de origem animal, como é o caso do
leite, é uma difícil tarefa, podendo ser até exaustiva, principalmente devido as quantidades de
macromoléculas.
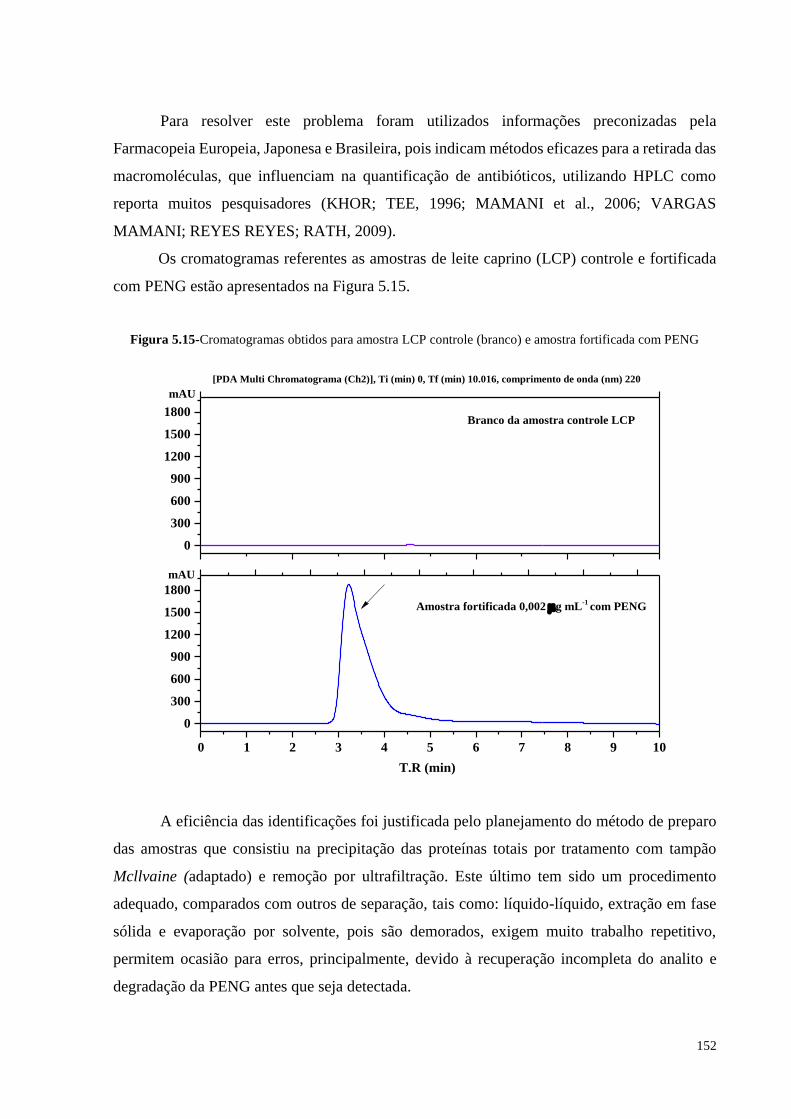
152
Para resolver este problema foram utilizados informações preconizadas pela
Farmacopeia Europeia, Japonesa e Brasileira, pois indicam métodos eficazes para a retirada das
macromoléculas, que influenciam na quantificação de antibióticos, utilizando HPLC como
reporta muitos pesquisadores (KHOR; TEE, 1996; MAMANI et al., 2006; VARGAS
MAMANI; REYES REYES; RATH, 2009).
Os cromatogramas referentes as amostras de leite caprino (LCP) controle e fortificada
com PENG estão apresentados na Figura 5.15.
Figura 5.15-Cromatogramas obtidos para amostra LCP controle (branco) e amostra fortificada com PENG
0
300
600
900
1200
1500
1800
0 1 2 3 4 5 6 7 8 9 10
0
300
600
900
1200
1500
1800
mAU
Branco da amostra controle LCP
T.R (min)
[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
mAU
Amostra fortificada 0,002 g mL-1
com PENG
A eficiência das identificações foi justificada pelo planejamento do método de preparo
das amostras que consistiu na precipitação das proteínas totais por tratamento com tampão
Mcllvaine (adaptado) e remoção por ultrafiltração. Este último tem sido um procedimento
adequado, comparados com outros de separação, tais como: líquido-líquido, extração em fase
sólida e evaporação por solvente, pois são demorados, exigem muito trabalho repetitivo,
permitem ocasião para erros, principalmente, devido à recuperação incompleta do analito e
degradação da PENG antes que seja detectada.

153
5.2.1.1 Avaliando a recuperação do método
Uma das maiores dificuldades para extrair e isolar a PENG das amostras comerciais de
leite está associada com a presença de proteínas e lipídios (macromoléculas). Segundo
informações fornecidas pelos fabricantes, em 200 mL de leite: as proteínas totais correspondem
a 14% para amostras de leite bovino integral e 0 a 5% para amostras de leite desnatado. Em
amostras de leite integral bovino o teor de proteínas é mantido, enquanto que o teor de lipídios
varia de 0 a 5%. Para leite caprino integral os teores de lipídios variam de 9 – 13% e proteínas
de 1 – 8%.
É importante ressaltar que, as quantidades de macromoléculas dependem de inúmeros
fatores como raça, genética, alimentação do animal produtor e condições climáticas onde são
criados, como reporta alguns pesquisadores Reeves et al, 2011 e Nebot et al (2012b), por isso
o preparo das amostras de leite cru (controle) dos procedimentos apresentaram maior
complexidade e as amostras não passaram por processamento, pois as repetições das etapas
foram maiores em 3 vezes para cada amostragem.
As amostras de leite de cabra possuem propriedades diferentes com relação a fração
lipídica. Porém, o teor total de gorduras no leite em média é semelhante ao encontrado em
outras espécies de ruminantes, estes fatores tornam as dificuldades similares.
Como este trabalho tem como objetivo construir modelos válidos para determinar os
resíduos de PENG em amostras expostas a comercialização, considera-se uma média desses
valores. Logo, segundo instrução normativa nº 62 de 2011 que dispõe de regulamento técnico
para leite cru e refrigerado bovino, em 100g de leite no mínimo 3g deve ser de lipídio total e no
mínimo 2,9 g deve ser de proteína total, esses valores são específicos para leite de vaca.
Para minimizar esta dificuldade, as etapas preparativas das amostras de controle e
amostras de LB e LCP (comerciais) foram cuidadosamente planejadas. Inicialmente, foi
escolhido o método desenvolvido por Wang e Yuan-Qian (2009) para determinação de
betalactâmicos em amostras de leite por UPLC, pois foram encontrados ótimos resultados de
recuperação para PENG em amostras que apresentaram 52,2 – 68,3%; 70,1 – 81% e 76,2 –
101% em níveis de concentração de 0,1; 0,5 e 2,5 µg mL-1, respectivamente. Estes valores
expressam um grande esforço para o planejamento das condições, pois eles têm grande
influência na recuperação do analito.

154
Assim, o método original para extração e isolamento do analito, empregado pelos
pesquisadores citados acima e recomendados pelas Farmacopeias, foi modificado de forma a
escolher e incluir agentes precipitantes à solução de McIlvaine, tendo cuidado em corrigir e
manter o pH em 4, uma vez que a PENG se degrada rapidamente em condições muito ácidas e
alcalinas antes que seja identificado ou quantificada por cromatografia.
Os resultados deste método indicaram que o preparo das amostras de leite apresenta
qualidade na remoção das Proteínas Totais, uma vez que 6% da matéria proteica foi removida,
assim como os lipídios totais que ficaram na fase apolar sobrenadante, enquanto que a fase
aquosa tem sido microfiltradas em filtro com diâmetro de 13mm e poro de 0,22 µm para
remoção das impurezas que se injetadas na coluna cromatográfica diminui a vida da mesma.
Outra razão para o emprego das condições foi devido à natureza iônica das penicilinas
que tendem a ionizar-se, ficando carregadas negativamente, por causa dos baixos valores de
pKa do grupo carboxílico (COOH). As etapas de extração e isolamento foram otimizadas para
evitar a hidrólise ácida da PENG, a fim de evitar a quebra do anel betalactâmico e
consequentemente degradação do composto.
Para manter a recuperação e aceitabilidade, os métodos físicos empregados foram de
baixa rotação para centrifugação de 500 rpm a uma temperatura de -4 ºC. Para reforçar a
influência do pH Kukusamude et al (2012) estudaram o efeito do pH entre 2 – 8 usando tampão
fosfato 10 mmol L-1. Os resultados obtidos pelos pesquisadores revelaram que o pH da solução
tem forte efeito sobre a extração das penicilinas. Os pesquisadores relatam que a eficiência de
extração permaneceu constante quando variou o pH de 2 a 6, houve um aumento significativo
quando se elevou o pH a 8, resultando numa maior eficiência de extração.

155
Os resultados na Tabela 5.8 confirmam o melhor desempenho do método Mcllvaine
adaptado para boa recuperação do analito, comparado aos dos pesquisadores Jing et al (2009)
e Shi et al (2011).
Tabela 5.8-Desempenho dos métodos para quantificação de PENG em amostras de leite bovino e caprino,
expresso pela recuperação (%).
Nível de concentração (µg mL1) Método 1 Leite bovino (LB) Método 2 Leite Caprino (LCP)
0,002 89,71 ± 10,11 95,01 ± 4,256
0,005 86,28 ± 4,852 96,64 ± 2,369
0,010 99,25 ± 6,671 95,02 ± 2,936
Os resultados apresentados evidenciam a eficiência do preparo das amostras controle,
uma vez que as recuperações ≥70% são aceitáveis como indica a norma CAC/GL 16 que para
concentrações de analito ≤ 0,01 µg mL-1 a recuperação em intervalo de 50 -120% com erro de
até 20% é aceito. Os erros apresentados em cada nível de concentração também são aceitáveis
até 20%. Para o método 1 o erro é de 9,304 e para o método 2 é de 1,201. Esses valores
diferentes para cada conjunto de amostras de leite bovino e caprino deve-se ao fato deste
apresentar pouca variabilidade nos teores totais de proteínas e lipídios como mostra a Tabela
4.5 e 4.6, respectivamente.
É importante citar que para o cálculo das concentrações reais de PENG em amostras
comerciais admitiu-se a média das recuperações nos três níveis de concentração (C) de
0,5LMR; LMR e 1,5LMR, isto é, quando C < LMR, C = LMR e C ≥ LMR.
Outra vantagem deste método é eliminação de solventes e produtos químicos tóxicos,
que são perigosos, não só para o analista, mas também para o meio ambiente. Comparando- se
com o trabalho de Sen e Filazi (2014) que desenvolveram um método para a quantificação de
penicilina por adição de reagentes e solventes tóxicos, neste caso os pesquisadores usaram
tratamento ácido e extração por solvente, seguido por centrifugação. O método proposto
apresentou recuperação de 96,47 – 104,86% para cloxacilina (CLO) e 97,76 – 104,86% para
dicloxacilina (DICLO) em leite bovino.

156
Furusawa (2000) validou um método, utilizando adição de solvente (etanol), seguido
por reação com mercúrio e cloreto, obtendo a recuperações de 86 % para PENG, os
pesquisadores não indicaram o tipo de leite analisado. Desta forma, comparando este último
trabalho com o método desenvolvido para preparo de amostra, os % Recuperações do novo
método > método Furusawa, com melhores vantagens de serem mais limpos e verdes.
Assim, o método desenvolvido foi acessível, considerando o desempenho e extração do
analito de interesse. É necessário relembrar que os procedimentos adotados se basearam nas
recomendações Farmacopeias (BRASIL, 2010; USP XXVIII, 2007; 2011). Optou-se pela
utilização de uma solução tamponada, em conformidade com as condições prescritas na
Farmacopeia internacional, e dentro das expectativas apresentadas por Bildini, Ricci e Sharma
(2002) quando fazem uma revisão dos recentes avanços dos métodos comentados em 2002.
O método desenvolvido apresenta muitas vantagens, tais como: confiabilidade,
sensibilidade, especificidade, reduzido consumo de solventes , que será refletido nos resultados
de robustez na identificação de grupos, principalmente porque o composto apresenta atividades
farmacológicas conforme mencionado por Gumustas et al (2013) e Maquio Fiorentino et al
(2014), comparados ao trabalho de Sen e Filazi (2014) que desenvolveram um método para a
quantificação de penicilina no leite bovino usando HPLC-DAD e C18 em coluna de fase reversa
(250 mm × 4.6 × 3.5 µm). Apesar de ótimos resultados, o tempo de execução foi de 55 minutos,
o analito apresentou eluição entre 25 e 45 min. O pesquisador Furusawa (2000), também,
desenvolveu um método de validação, usando HPLC-DAD, com tempo de análise 40 min e
recuperação entre 86,4 e 91, 2%.

157
5.2.2 Estudo das condições cromatográficas para aplicação em amostras de leite
A vantagem deste método é a facilidade de: atingir o ponto de equilíbrio e
homogeneidade na adsorção, pois a composição da fase móvel não prejudicou a
reprodutibilidade na eluição.
O planejamento das condições em três estágios 1, 2 e 3 evidencia a resolução dos
cromatogramas no modo isocrático.
Na Figura 5.16 são apresentadas as respostas dos tempos de retenção (R.T) e área do
pico da PENG para planejamento dos procedimentos para leite bovino (LB) e de caprino (LCP).
Figura 5.16-Estudo da variação de tempo de retenção e área do pico da PENG
84
112
140
168
196
224
252
280
C1 C2 C3
3,06
3,09
3,12
3,15
3,18
3,21
3,24 T.R LB
T.R LCP
T.R
(m
in)
Condições
A.P LB
A.P LCP
A.P
(m
AU
)
A análise das respostas foi semelhante com erros médio entre amostras LB e LCP.
Quando analisadas os tempos de retenção para amostras de LB os valores estiveram entre 3,081
– 3,195 min, enquanto que amostras LCP estiveram em 3,096 – 3,186 min. Quando analisadas
as áreas dos picos houveram diferenças em 34,766% para amostras LB com erro de ± 0,0987%.
As medidas apresentaram um intervalo de confiança de 95%, aceitável.

158
Nos dados apresentados, a variação dos tempos de retenção e resolução são necessários,
pois esses têm relatado fluxos para fase móvel muito baixos, principalmente, porque o diâmetro
dos poros, da coluna ser pequeno.
Na Figura 5.17 são apresentados os resultados da resolução dos picos e seletividades
obtidos para as condições correspondentes aos métodos 1.
Os resultados são apresentados também o ajuste para as condições de leitura de amostras
controle de leite bovino (branco) fortificada.
Esses resultados de resolução forneceram uma medida quantitativa da eficiência da
coluna em identificar a PENG.
Figura 5.17-Resultados das condições para análise de PENG em amostras controle de leite bovino
0,1952 0,1949
0,1801
0,2092 0,2089
0,1934
1 2 3
0,0
0,1
0,2
0,3
CC
Pa
râm
etro
s
Condições
Resolução
Seletividade
C
A análise apresentada mostra que o método desenvolvido que apresenta melhores
vantagens consiste de: eluição por isocrático, fase móvel constituída de H2O: ACN (90:10, v/v)
e fluxo de 0,1 mL min-1 referente a condição 1, pois apresenta maior seletividade.
Com relação a seletividade dos métodos, ao mudar a polaridade da fase móvel há
variação na seletividade da separação.

159
A análise mostra que a composição da fase móvel tem grande influência na seletividade
e eficiência da eluição da PENG. Assim, a mudança das condições para ambos os métodos 1 e
2 muda os parâmetros cromatográficos. Logo, a identificação da PENG é conduzida
isotermicamente, isto é, a temperatura no forno da coluna que foi mantida em 40º C, pois a
PENG em 106 ºC – 110 ºC sofre decomposição.
Na Figura 5.18 são apresentados os resultados da resolução dos picos e seletividades
obtidos para as condições do método 2, correspondentes ao ajuste de amostras controle de leite
caprino (branco) fortificada como PENG nas mesmas condições utilizadas no método 1, com
objetivo de comparar o desempenho dos métodos.
Figura 5.18-Resultados das condições para análise de PENG em amostras controle de leite caprino
0,06770,0636
0,0587
0,07260,0771
0,0711
1 2 3
0,00
0,05
0,10
CCC
Pa
râm
etro
s
Condições
Resolução
Seletividade
A coluna de fase reversa, e composição de fase móvel utilizada e propostas, estão muito
adaptadas para quantificação de resíduos de penicilinas, no leite, conforme relata Bilandžić et
al (2011b). Apesar da polaridade moderada deste tipo de coluna, em 80% dos casos, a fase
reversa, é mais utilizada em compostos não iônicos. Essa aplicação, quase universal, permite a
análise de substâncias hidrossolúveis, iônicas, e até as lipofílicas, como reportam os
pesquisadores Rambla-alegre et al (2011b), Vera-Candioti, Olivieri e Goicoechea (2010) e
Wang e Li (2009) em seus trabalhos.
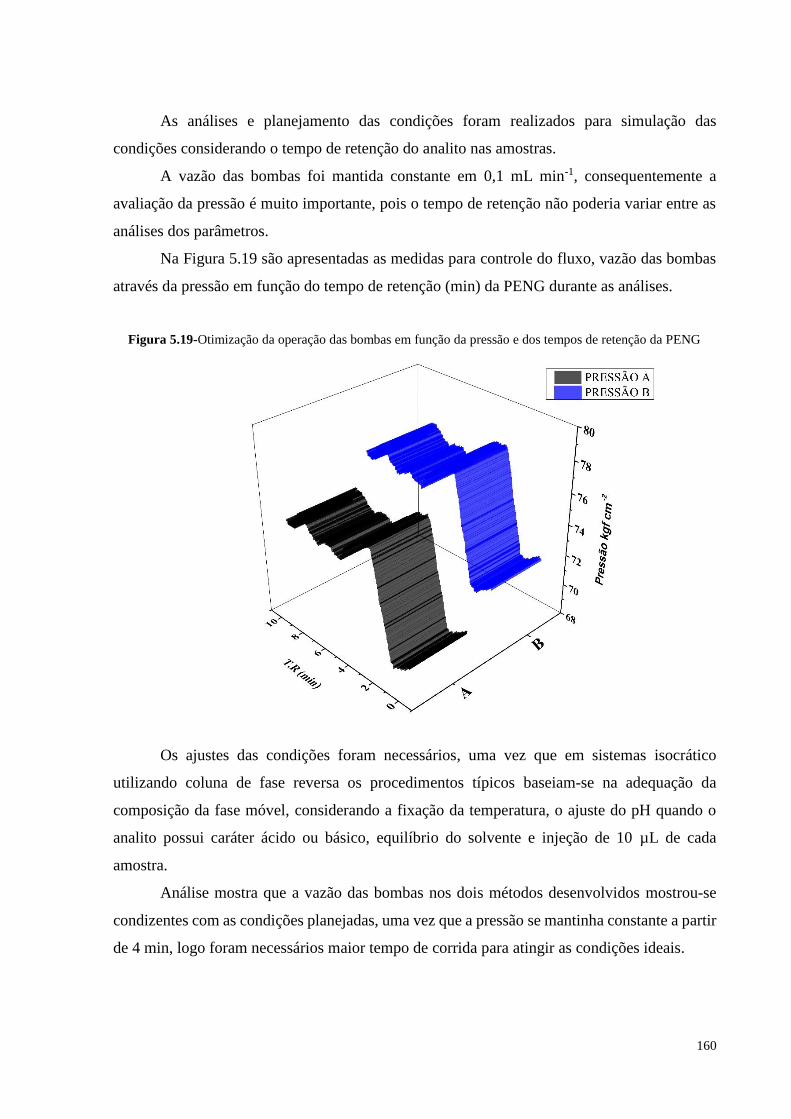
160
As análises e planejamento das condições foram realizados para simulação das
condições considerando o tempo de retenção do analito nas amostras.
A vazão das bombas foi mantida constante em 0,1 mL min-1, consequentemente a
avaliação da pressão é muito importante, pois o tempo de retenção não poderia variar entre as
análises dos parâmetros.
Na Figura 5.19 são apresentadas as medidas para controle do fluxo, vazão das bombas
através da pressão em função do tempo de retenção (min) da PENG durante as análises.
Figura 5.19-Otimização da operação das bombas em função da pressão e dos tempos de retenção da PENG
Os ajustes das condições foram necessários, uma vez que em sistemas isocrático
utilizando coluna de fase reversa os procedimentos típicos baseiam-se na adequação da
composição da fase móvel, considerando a fixação da temperatura, o ajuste do pH quando o
analito possui caráter ácido ou básico, equilíbrio do solvente e injeção de 10 µL de cada
amostra.
Análise mostra que a vazão das bombas nos dois métodos desenvolvidos mostrou-se
condizentes com as condições planejadas, uma vez que a pressão se mantinha constante a partir
de 4 min, logo foram necessários maior tempo de corrida para atingir as condições ideais.

161
5.2.3 Validação do método
5.2.3.1 Correlação entre amostras de leite bovino (LB) e caprino (LCP)
Os resultados obtidos para identificação da PENG foram possíveis, pois houve a
minimização da adsorção do analito na coluna cromatográfica, justificada pela formação de
picos simétricos e bem resolvidos em todos os níveis de concentração apresentados usando
método de 95,99 % de recuperação.
Na Figura 5.20 são apresentados os cromatogramas das amostras do leite bovino
fortificada, em níveis de 0,002 – 0,1 µg mL-1. O tempo de retenção (T.R) médio da PENG é de
4,33 com intervalo de confiança de 95 % e desvio padrão médio de ± 0,0216.
Figura 5.20-Perfil cromatográfico de PENG nas amostras controle de leite LB e LCP fortificadas
0 1 2 3 4 5 6 7 8 9 10
0
125
250
375
500
625
750
875
1000
1125
0 1 2 3 4 5 6 7 8 9 10
0
6
12
18
24
T.R(min)
mAU[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
LB2
T.R(min)
Branco
LB1
LB2
LB3
LB4
LB5
LB6
LB7
LB8
LB9
LB10
mAU[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
As condições cromatográficas descritas são selecionadas pelo bom desempenho do
método, pois os erros experimentais são aceitáveis, considerando a normatização CAC/GL16
da Códex Alimentarius (1993).
Quanto a análise, a linha base dos cromatogramas refere-se ao branco das amostras de
leite controle. Os erros experimentais do sinal analítico estão entre 3,0833 – 7,9692 % para leite
bovino (método 1), esses valores são aceitáveis, considerando o Regulamento 37/2010/EC da
European Commission (2009).
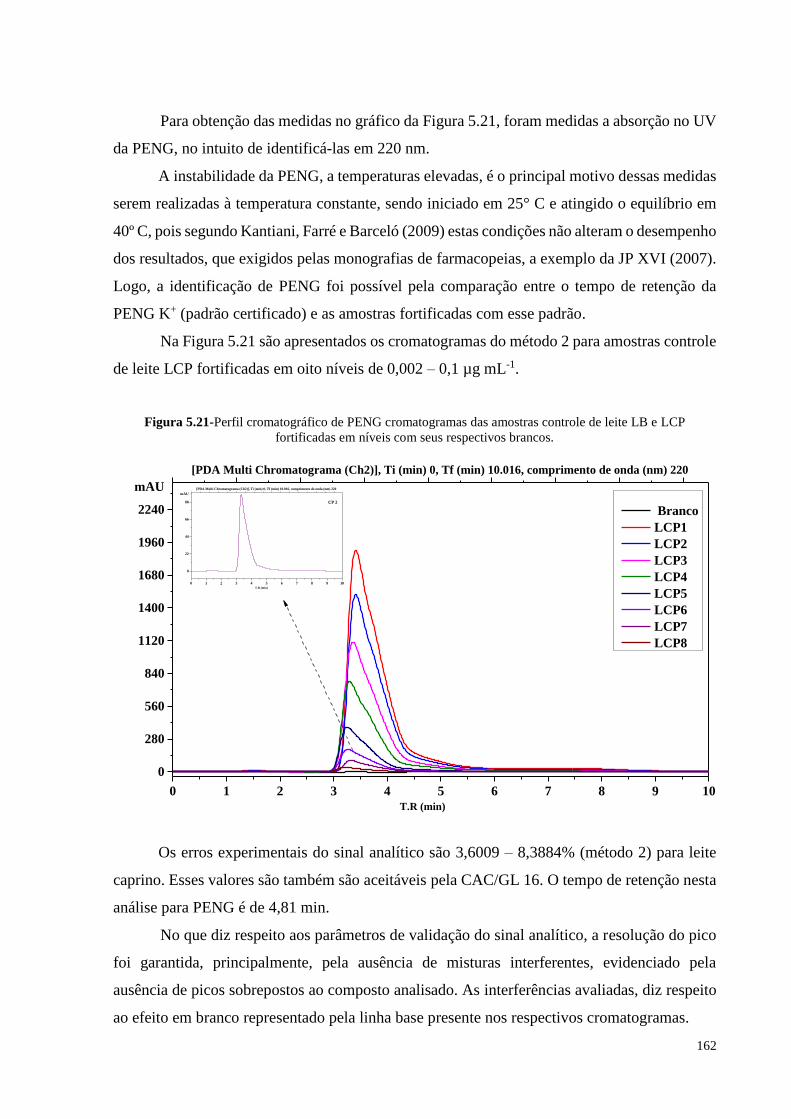
162
Para obtenção das medidas no gráfico da Figura 5.21, foram medidas a absorção no UV
da PENG, no intuito de identificá-las em 220 nm.
A instabilidade da PENG, a temperaturas elevadas, é o principal motivo dessas medidas
serem realizadas à temperatura constante, sendo iniciado em 25° C e atingido o equilíbrio em
40º C, pois segundo Kantiani, Farré e Barceló (2009) estas condições não alteram o desempenho
dos resultados, que exigidos pelas monografias de farmacopeias, a exemplo da JP XVI (2007).
Logo, a identificação de PENG foi possível pela comparação entre o tempo de retenção da
PENG K+ (padrão certificado) e as amostras fortificadas com esse padrão.
Na Figura 5.21 são apresentados os cromatogramas do método 2 para amostras controle
de leite LCP fortificadas em oito níveis de 0,002 – 0,1 µg mL-1.
Figura 5.21-Perfil cromatográfico de PENG cromatogramas das amostras controle de leite LB e LCP
fortificadas em níveis com seus respectivos brancos.
0 1 2 3 4 5 6 7 8 9 10
0
280
560
840
1120
1400
1680
1960
2240
T.R (min)
Branco
LCP1
LCP2
LCP3
LCP4
LCP5
LCP6
LCP7
LCP8
mAU
0 1 2 3 4 5 6 7 8 9 10
0
22
44
66
88
T.R (min)
CP 2
mAU
[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
[PDA Multi Chromatograma (Ch2)], Ti (min) 0, Tf (min) 10.016, comprimento de onda (nm) 220
Os erros experimentais do sinal analítico são 3,6009 – 8,3884% (método 2) para leite
caprino. Esses valores são também são aceitáveis pela CAC/GL 16. O tempo de retenção nesta
análise para PENG é de 4,81 min.
No que diz respeito aos parâmetros de validação do sinal analítico, a resolução do pico
foi garantida, principalmente, pela ausência de misturas interferentes, evidenciado pela
ausência de picos sobrepostos ao composto analisado. As interferências avaliadas, diz respeito
ao efeito em branco representado pela linha base presente nos respectivos cromatogramas.

163
Os resultados indicaram que não há diferença significativa (nível de confiança de 95%),
entre os resultados deste trabalho e os recomendados pelos métodos oficiais, pois seguem
critérios de aceitação.
A seletividade (Especificidade/Aplicabilidade) do método foi avaliada por amostras
representativas do branco (amostras matrizadas) de leite bovino e caprino, respectivamente.
Logo, a análise demonstrou minimização dos interferentes que gerasse resultado falso-positivo,
na identificação. A correlação foi obtida por brancos e amostras controle fortificadas.
Com relação aos efeitos das matrizes foram avaliados separadamente o leite bovino e
caprino. Na Figura 5.22 estão apresentadas as correlações entre esses efeitos calculados pela
concentração ([PENG]) versus a área do pico (AP) nos oito níveis de concentração.
Figura 5.22-Efeito da concentração de PENG e perfis das curvas para o método 1
0,00 0,02 0,04 0,06 0,08 0,10
0
1000
2000
3000
4000
Amostra 2L Branca Fortificada
Amostra 2L Branca
Padrão PENG
Efe
ito
na
A.P
Efeito de [PENG]
O cálculo do t-student mostrou que a matriz não afeta as medidas, uma que o tcalculado foi
de 4,402 e ttabelado foi de 4,785. Logo o tcalculado< ttabelado.
As amostras em branco apresentaram correlação com a adição de padrão PENG em
todos os níveis, como indica a análise dos efeitos da área do pico versus concentração de PENG.

164
Na Figura 5.23 são apresentadas as correlações entre esse efeito calculados pela
concentração [PENG] versus a área do pico (AP) em oito níveis para o método 2.
Os valores encontrados para a área da curva para amostras fortificadas foram mais
intensos do que aqueles observados na curva do padrão PENG. Este fato é explicado pois a
matriz leite, proporciona determinada ionização, quando comparada apenas com a fase móvel.
Com base nestes dados, a recuperação foi calculada considerando-se os valores
encontrados na curva da amostra fortificada, pois representam a intensidade da substância a
analisar, avaliando o efeito de matriz, mas sem prejuízo às etapas de preparo das amostras.
Desta forma, o cálculo do t-student mostrou que não há efeito de matriz nas medidas,
uma que o tcalculado foi de 1,958. Logo o tcalculado< ttabelado.
Figura 5.23-Efeito da concentração de PENG e perfis das curvas para o método 2
0,00 0,02 0,04 0,06 0,08 0,10
0
1000
2000
3000
4000
Amostra LCP Branca Fortificada
Amostra LCP Branca
Padrão PENG
Efe
ito
na
A.P
Efeito de [PENG]
Analisando comparativamente as amostras LB e LCP, tem-se que o efeito da matriz se
mostrou mais acentuado em amostras de leite bovino. Este fato é justificado pela melhor
recuperação para o método 2 (amostras LCP).
O desempenho do método para quantificação da PENG foi verificado através do estudo
de validação, conforme os critérios de descritos pela Tabela 5.7. Desta forma, o método foi
validado, de acordo com os parâmetros: precisão, linearidade, limite de quantificação e
especificidade, pois o sucesso destes depende do bom planejamento seguindo as
recomendações por normas legais (EMEA, 2006; ICH, 2005; MEDICAL, 2011; SHAH et al.,

165
2000; US FOOD AND DRUG ADMINISTRATION, 1996) já descritas no capítulo 3 deste
trabalho.
Nesse processo, os fatores que influenciaram foram: pH da solução para precipitação
total dos interferentes, e as condições cromatográficas. O objetivo de verificá-los foi de avaliar
possíveis alterações nos procedimentos adotados. Para comprovar a importância de pequenas
alterações do pH da solução tampão, definiu-se pH de 4 a 8, foi observado que, nesta faixa,
houve alterações dos resultados, se comparados com método já estabelecido.
Os métodos de validação propostos, neste trabalho, seguiram duas etapas: a primeira
consistiu na validação inicial, no laboratório, com a padronização de todas as soluções
envolvidas; a segunda etapa fundamenta-se na transferência e preparo das amostras, em
condições reais, e em fortificação das matrizes, que foi acelerado por uma sequência de
injeções, sendo averiguado seu desempenho pela recuperação do analito.
Desta forma, a padronização foi realizada, de modo que o método apresentasse
veracidade nos testes externos quando aplicados em ensaios interdia, porém em condições
semelhantes, para alcançar resultados comparáveis.
Sabendo que a aceitação dos critérios de validação, sendo normatizados de acordo com
CAC/GL 72-2009, descreve o grau dos dados produzidos a partir das medições, logo os
resultados aqui apresentados, provam pelos cálculos dos parâmetros, que o método é apto para
ser utilizado na quantificação de resíduos de PENG. Além disso, os critérios são aceitos, a fim
de garantir que os métodos sejam capazes de fornecer resultados robustos.
A linearidade foi determinada pela avaliação do coeficiente de regressão linear (R),
medido para PENG pela satisfação dos critérios. Os resultados seguiram a prescrição da
Decisão 2002/657/EC adotados por trabalhos recentes (BERENDSEN et al., 2013; BOHM;
STACHEL; GOWIK, 2009; CRISTOFANI et al., 2009; GAUDIN et al., 2014;
KARAGEORGOU; SAMANIDOU, 2011; LARA et al., 2006).

166
O novo método 1 para amostras de leite LB é apresentado pela Figura 5.24, os valores
são bem correlacionados para curva analítica dos modelos usados para quantificação da PENG.
Este gráfico foi construído, através da representação da área de pico, em função da
concentração da adição de padrão interno (0,002 – 0,1 µg mL-1).
Figura 5.24-Curva analítica para amostras de leite LB fortificadas para estudo da linearidade
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,09 0,10
0
520
1040
1560
2080
2600
3120
3640
4160
4680
Áre
a d
o P
ico (
mA
U)
[resíduos PENG] (g mL-1)
LM
R
As concentrações foram medidas oito vezes, em cada nível (n = 8), para o leite bovino;
o LD instrumental foi calculado como 3,3 vezes o desvio padrão (DP) da área do pico (ou
proporção entre as áreas dos picos com o IS) do analito, em três réplicas da solução de menor
concentração padrão para cada composto definido pela faixa de 0,002; 0,005;0,01 µg mL-1,
dividido pelo declive da curva de calibração; o LQ foi calculado pelo quociente entre 10 × SD
e inclinação da curva analítica.
Os dados quantitativos estão abaixo dos limites legais, de LMR. Como a quantificação
requer dependência entre a resposta medida e a concentração de analito, já conhecida, a
linearidade da resposta cromatográfica foi testada com curvas usando pontos de calibração em
concentrações incluídas nos critérios de aceitação para 0,5LMR, LMR 1,50LMR.

167
O método 2 para amostras de leite LCP é apresentado na Figura 5.25, ele indica valores
bem correlacionados para curva analítica dos modelos para quantificação da PENG. A curva
analítica foi plotada, em função da concentração versus área do pico. Os resultados da regressão
linear indicaram correlação R condizente com os critérios estabelecidos pelas legislações.
Figura 5.25-Curva analítica para amostras de leite LCP fortificadas para estudo da linearidade
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,07 0,08 0,09 0,10
0
520
1040
1560
2080
2600
3120
3640
4160
4680
Áre
a d
o P
ico
(m
AU
)
[Resíduos de PENG] (g mL-1)
LM
R
Este gráfico também foi construído, através da representação da área de pico, em função
da concentração da adição de padrão interno (0,002 – 0,1 µg mL-1).
Os resultados do ajuste linear para ambos os métodos 1 e 2 indicam excelente
desempenho da linearidade para leite caprino, pois a linearidade apresenta critério de
aceitabilidade, onde o coeficiente de correlação (R) deve ser ≥0,99.
O estudo da recuperação para PENG foi realizada dividindo-se a inclinação da curva
analítica, correspondente a cada matriz investigada. As recuperações absolutas foram
analisadas pela comparação, entre as amostras fortificadas.

168
Na Tabela 5.9 são apresentados os resultados comparativos dos parâmetros de validação
para amostras LB e LCP.
Tabela 5.9-Comparação do desempenho dos métodos para quantificação de PENG amostras de leite bovino e
caprino
Parâmetros Resultados da Validação
LB LCP
LD (µg mL-1) 7,246 × 10-4 8,251 × 10-4
LQ (µg mL-1) 2,196 × 10-3 2,500 × 10-3
%Recuperação 92,05 95,83
Equação APico = 40103 × CPENG + 168,7 APico = 4425 × CPENG ˗ 44,87
R 0,9975 0,9995
Os resultados correspondentes ao desempenho de ambos os métodos (LD e LQ) são
mostrados na Tabela 5.10. Nesta etapa do método, são considerados os valores de Recuperação
≥ 70%, recomendados pela Decisão 2002/657 / CE da European Comission (1993). As médias
de recuperação de PENG são expressas em três níveis 0,002, 0,005 e 0,01 µg mL-1.

169
A precisão do método é verificada por sua repetabilidade, usando injeções, em três níveis
em 15 repetições, para cada amostra, conforme resultados da Tabela 5.10.
Tabela 5.10-Comparação da precisão e exatidão do Método para quantificação de PENG em amostras de leite
bovino e caprino, expresso pelo coeficiente de variação (%CV).
CPENG (µg mL-1) Precisão do Método CV (%)
LB LCP
0,002 9,411 4,665
0,005 6,186 3,486
0,01 10,78 4,129
Os resultados foram satisfatórios a partir de dados do intra-ensaio, e as recuperações
médias para amostras de leite caprino foram maiores do que 95,83%, com coeficientes de
variação (%CV) entre 3,4861 e 4,6647; para leite bovino estes valores foram de 92,05 % com
coeficientes de variação (%CV) entre 9,41 e 10,78, os valores são aceitáveis pelos critérios
Q2R1 da Emea (2006). Essa diferença entre os parâmetros está associada ao efeito da matriz,
como já mencionado anteriormente.

170
Os picos de PENG estão em conformidade com a relação sinal/ruído > 5). Os valores
de LQ estão em concordância com o LMR (0,004 µg mL-1).
A recuperação média satisfaz os critérios de aceitação da Codex Alimentarius (1993),
e o valor LQ estava abaixo do LMR, em conformidade com a CGAL/DAS, isto é, muito inferior
à concentração da substância detectada.
Os métodos desenvolvidos são de simples execução e capazes de quantificar este
antibiótico, em concentrações inferiores às preconizadas pelo MAPA, pois o LQ é inferior ao
LMR para resíduos de PENG para métodos 1 e 2.
A seletividade foi examinada pela análise das amostras matrizadas e em branco. Além
disso, a ausência de picos sobrepostos, acima da relação sinal/ruído, com os tempos de retenção
de PENG, mostrou que o método é livre de substâncias interferentes.
A curva analítica foi ajustada pela adição do padrão, nas concentrações citadas, os
mesmos procedimentos de fortificação, para avaliar a precisão e exatidão foram mantidos, em
ambas as amostras.
Os resultados indicaram que o método é apto para quantificação dos resíduos de PENG
presentes em amostras leite (tipo bovino e caprino).
Comparando-se os resultados com a literatura científica, temos que, Han et al (2015)
consideraram o método desenvolvido como seletivo e rápido, pois foi capaz de determinar,
simultaneamente, 38 resíduos de antibióticos veterinários em leite, dentre ele a PENG,
utilizando UPLC-MS/MS. O método desenvolvido pelos pesquisadores apresentaram
recuperações entre 68 – 118% para resíduos de PENG e precisão intradia, expresso em (%CV),
inferior a 15%, o LQ entre 0,03 – 10 µg kg-1. Se o método desenvolvido for comparado aos
resultados obtidos, pelos pesquisadores, citados acima, que apresenta LQ e valores de
recuperações acima de 70%, e sabendo que a técnica utilizada foi UFLC-DAD, considera-se
que resultados, no presente trabalho, foram bem satisfatórios para aplicação em amostras reais
de leite ultrapasterizado bovino e caprino.
O bom desempenho do método é justificado, também, pelas condições ótimas do preparo
das amostras, que envolve extração dos resíduos de PENG, através de desproteinização,
utilizando tampão McIlvaine pH 4, agitação, centrifugação e limpeza da amostra. Assim foi
possível obter boa recuperação do analito. A saber, de Cámara et al (2013) que utiliza a
precipitação de proteínas totais associado a solvente ACN, centrifugação, seguido de extração
em fase sólida, obtendo níveis para recuperação de betalactâmicos entre 79% a 96%, e (%CV)
entre 0,5% e 4,9%, o LQ, estiveram na faixa de 3,4 – 8,6 µg kg-1, que são inferiores LMR

171
estabelecidos pela União Europeia para os betalactâmicos estudados no leite. Assim os
pesquisadores concluiram que os resultados descritos, tornavam o método adequado para a
análises de rotina, aplicado a amostras de leite de ovelha. Logo por comparação, os resultados
apresentados, no presente trabalho, tem sido análogos aos métodos avançados e divulgados,
recentemente. A saber, de Jank et al (2012), pesquisadores da Lanagro de Porto Alegre/RS, que
desenvolveram método de validação para quantificação dos resíduos betalactâmicos, dentre eles
PENG, em leite bovino, utilizando uma extração líquido-líquido rápido (LLE) para a preparação
de amostras, seguido por LC-MS/MS, sendo que os resultados mostraram recuperação de
54,2% para PENG e LD de 4,7 µg L-1.
A robustez do método descreve a estabilidade dos procedimentos adotados para análises
cromatográficas de PENG. A robustez foi avaliada mediante a análise do coeficiente de
variação (%CV) em cada nível de concentração de 0,002; 0,005 e 0,01 µg mL-1, cada um desses
níveis nas amostras (controle) foram analisadas em três condições cromatográficas diferentes.
Na Tabela 5.11, estão inseridos os resultados que comprovam a robustez do método.
Tabela 5.11-Avaliação comparativa da robustez do método para quantificação de resíduos de PENG em
amostras de leite bovino e caprino, expresso pelo coeficiente de variação (%CV).
Adição de Padrão Robustez do Método CV (%)
LB LCP
CPENG (µg mL-1) C1 C2 C3 C1 C2 C3
0,002 9,411 10,87 10,88 4,664 4,259 4,263
0,005 6,679 6,684 6,688 3,486 3,487 3,489
0,01 10,78 9,302 9,305 4,129 4,098 4,099
C corresponde às condições
Nestes resultados mudanças na composição da fase móvel, bem como variação do fluxo
dos solventes nas bombas e pressão do sistema como modo isocrático alterou minimamente os
valores de tempo de retenção da PENG e da área do pico.
Pode-se observar que para análises das condições 1 (C1) foram apresentadas menos
variação, pois os erros entre as medidas foram reduzidos para método 1 o erro médio foi de ±
1,187%, enquanto que para o método 2 o erro médio foi ± 0,9629. Quando analisados nas três
condições todas apresentaram erros inferiores a 20%, conferindo ao método robustez, pois os
valores são aceitáveis pelas normas CGAL/DAS.

172
A Figura 5.26 apresenta os resultados obtidos da análise de 24 amostras LB tipo integral
e desnatado quanto a presença de resíduos de PENG.
Figura 5.26-Análise de leite bovino quanto à presença de resíduos de PENG L
B1
6
LB
2
LB
1
LB
3
LB
4
LB
5
LB
6
LB
9
LB
10
LB
11
LB
12
LB
13
LB
14
LB
15
LB
17
LB
19
LB
20
LB
21
LB
22
LB
23
LB
24
LB
18
LB
7
LB
8
0,000
0,002
0,004
0,006
0,008
0,010
0,012
0,014
[PENG] > LMR (12%)
[PE
NG
]
g m
L-1
[PE
NG
]< L
MR
(8%
)
[PENG] LMR (79%)
Conforme (87 %) Não conforme (12 %)
Amostras LB UHT analisadas
>
Aplicando Método 1: LD de 1, 79 × 10-6 µg mL-1 para LMR de 0,004 µg mL-1
Analisando os dados do gráfico foi verificado que apenas 12,5% das amostras de leite
bovino analisadas estavam não conforme, isto é, com concentração de PENG muito acima do
tolerado pela legislação chegando à 0,0116; 0,013 e 0,014 µg mL-1, respectivamente. O erro
médio das medidas é de ±1,001%.
Estes resultados indicam que as boas práticas de uso de antimicrobianos em animais
produtores de leite estão sendo seguidas, observando o período de carência do medicamento e
descarte do leite, bem como a administração intramuscular correta das penicilinas.

173
A Figura 5.27 apresenta os resultados obtidos da análise de 24 amostras LCP tipo
integral quanto a presença de resíduos de PENG.
Figura 5.27-Análise de leite caprino quanto à presença de resíduos de PENG
CP
17
CP
18
CP
15
CP
16
CP
20
CP
2
CP
5
CP
1
CP
3
CP
4
CP
8
CP
11
CP
14
CP
7
CP
9
CP
6
CP
10
CP
19
CP
13
CP
12
0,000
0,002
0,004
0,006
0,008
0,010
0,012
0,014
[PE
NG
]
g m
L-1
Não Conforme (15 %)
[PENG] > LMR (15 %)
[PENG] < LMR (40 % )
[PENG] < LMR (50 %)
Amostras CP UHT analisadas
Conforme (90 %)
Aplicando Método 2: LD de 2,67 × 10-6para LMR de 0,004 µg mL-1
Analisando os dados do gráfico foi verificado que apenas 15% das amostras de leite
caprino analisadas apresentavam-se não conforme, com concentração de PENG muito acima
do tolerado pela legislação chegando à 0,0116; 0,013 e 0,014 µg mL-1, respectivamente. O erro
médio das medidas é de ± 0,2963%.
Quando comparados os resultados obtidos pelo método 1 e 2, pode-se afirmar que as
condições para boas práticas no uso de antibióticos a base de benzilpenicilina são similares,
uma vez que os erros entre os valores medidos e de referência são próximos.

174
5.3 CONSIDERAÇÕES FINAIS PARA QUALIDADE E SEGURANÇA DO LEITE
A presença de resíduos PENG, nos gêneros alimentícios, constitui um risco potencial
para a saúde humana. Atualmente, os resíduos de antibióticos são reconhecidos como um
problema ambiental, além de reduzirem a qualidade do leite e causar prejuízos econômicos para
o mercado de produção leiteira, isto é, gerando perdas importantes de renda monetária em
matérias-primas exportadas. Para resolver este problema, são desenvolvidos métodos oficiais
validados e revalidados. O desempenho dos métodos tem sido comparável ao
MET/LRM/PL/020 preconizado pelo MAPA, que trata da análise de multiresíduos de
betalactâmicos, em músculo utilizando UPLC-MS/MS.
Os procedimentos devem assegurar a execução dos testes de qualidade dos
medicamentos descritos na Instrução Normativa de n° 4, de 19 de fevereiro de 2008 que aprova
as Normas Técnicas para a Fiscalização da Produção, Controle, Comercialibzação, Modo de
Utilização de Produtos de uso veterinários destinados a diagnosticar Doenças dos Animais
(GILBERT; GARDNER; BIZEC, 2012; MAPA, 2012). Consequentemente, os métodos levam
em consideração, que formulações injetáveis para os animais produtores de alimentos expostos
ao consumo humano têm sido mais complicadas do que para humanos, principalmente, pela
tolerância no local da aplicação, pois essas injeções podem, também, impactar a saúde humana,
pela segurança alimentar. Assim, os procedimentos estabelecidos fornecem a garantia da
ausência de resíduos de PENG no leite, conforme os limites aceitáveis pelo MAPA.
Apesar da comercialização, dos produtos veterinários, ser livre segundo Art.64 requisito
III Decreto nº 5.053, de 22 de abril de 2004 (MAPA, 2012), o conhecimento da qualidade dos
medicamentos, comercialmente distribuídos, para tratamentos de infecções nos bovinos e
caprinos, é essencial, pois os veterinários no Brasil são autorizados a prescrever medicamentos,
de acordo com a necessidade dos tratamentos, mas não estão obrigados a assegurar que os
resíduos não entrem na cadeia alimentar. No caso dos tratamentos de mastites são, geralmente,
intramusculares em ovelhas leiteiras e os antibióticos concebidos são autorizados. Porém,
devido às diferenças entre espécies de bovinos e caprinos, os dados disponíveis não podem ser
utilizados com os mesmos métodos de injeção e dosagens. Mas o fato é que, há uma grande
disponibilidade de produtos alimentícios em todo comércio, de leite e laticínios, e apesar dessas
diferenças, as normas legais devem ser, obrigatoriamente, asseguradas.
O presente trabalho desenvolveu métodos sensíveis e confiáveis utilizando UFLC-DAD.
Os procedimentos apresentaram bons desempenhos, sendo adequados para aplicação na rotina
de laboratórios, que executam os métodos de validação. Logo, o processo para tratamento

175
necessitou de controle de todas as variáveis, que influenciam, potencialmente, na adsorção da
PENG na coluna cromatográfica a exemplo do pH natural do meio contendo os analitos. Desta
forma, o processo de extração foi planejado utilizando a padronização das soluções, que podiam
ser incompatíveis com as amostras reais, devido a uma diferença acentuada do pH, por isso que
várias estratégias têm sido desenvolvidas para melhor recuperação da PENG.
A quantificação dos resíduos de PENG, através do controle de qualidade do leite servido
na mesa do consumidor do estado do RN foi a principal meta deste trabalho. De maneira que,
chegaram-se à conclusão de que o UFLC-DAD associado às análises complementadas neste
trabalho é uma ferramenta poderosa para controle de qualidade dos medicamentos e do leite.
Quanto às análises dos espectros no infravermelho médio, este teste é classificado como
de identidade e qualidade. Utilizou-se leitura e interpretação dos espectros no FTIR para
verificar as principais bandas de absorção dos compostos presentes nos medicamentos. Todas
as amostras registradas isoladamente, inclusive as de interesse, apresentavam espectros padrão
experimental e valores do padrão referenciados pelo SDBS. Quanto às absorções que
representam os compostos, os resultados indicaram semelhanças entre padrão de referência
PENG e amostra de ABAM/IVERM, GEN e PENG. Os espectros PENG (padronizado) tem
sido de muita importância, como referência, para averiguação da presença do carbonilas de
amida e cromóforos nos medicamentos à base deste terapêutico. O teste de identidade tem sido
aplicado com sucesso, exceto quanto à ausência de informações da presença de isômeros.
As medidas cromatográficas foram realizadas com as amostras individuais, seguida das
amostras contendo PENG nas soluções. Os resultados para quantificação de PENG em 37 de
amostras (externas) de medicamentos, em melhores condições de robustez, apresentaram
valores condizentes com os indicados na rotulagem de cada medicamento, pois o CV era de
10,4319%, admitindo-se erro aceitável de ≤ 20% para traços, segundo preconização
CAC/GL16. Os resultados foram satisfatórios, pois os parâmetros da curva indicaram R 0,9991,
LD 7,384 × 10-4µg mL-1 e LQ 2,094 × 10-3µg mL-1. O rendimento do método foi de 72,51%.
Fazendo uma correlação entre os dados obtidos no sistema cromatográfico, em questão, e
resultados das análises espectrais multivariadas, sem dúvida, a Cromatografia líquida de alta
eficiência, apesar da redução do número de amostras é uma técnica mais aceita e estabelecida,
conhecida pelas legislações internacionais, pois emprega dados reais e experimentais, podendo
até ser estabelecida com método de referência para técnicas mais recentes. Logo, se confirma a
consolidação da HPLC para quantificação dos antimicrobianos, pois apresenta seletividade,
robustez e sensibilidade.

176
A segunda etapa deste trabalho é para validação de metodologia de identificação e
quantificação de resíduos de PENG, em leite bovino e caprino. Também foram propostas novas
metodologias de extração e preparo de amostras com redução significativa de reagentes e
solventes tóxicos, mantendo a boa recuperação do analito.
O método desenvolvido para amostras LB apresentou LD e LQ condizentes com o LMR
e a curva analítica. A aplicação do método mostrou que de 24 amostras comercias analisadas,
12% foram consideradas não conforme, pois o [PENG] > LMR.
O método desenvolvido para amostras LCP apresentou LD e LQ condizentes com o
LMR e a curva analítica. A aplicação do método mostrou que de 20 amostras comercias
analisadas 15% foram consideradas não conforme, pois o [PENG] > LMR.
Quando comparados com os resultados das mais recentes publicações (apresentados no
Capítulo 3 desta tese), este trabalho apresenta procedimentos simples, seletividade,
sensibilidade e redução, importante, no consumo de reagentes tóxicos.
A análise dos recentes programas de monitoramento no Brasil indicou que são
frequentes as preocupações das normas legais do Brasil, quanto à presença de contaminantes
em alimentos, pois são elaborados programas de controle e monitoramento de resíduos, esses
dados foram mostrados no Capítulo 3 no item 3.2.4. Assim, as aplicações das Lanagros são
inúmeras conforme a necessidade, pois esses contaminantes comprometem, em determinada
quantidade, a saúde da população brasileira. Isto pode ser comprovado pela edição especial, em
2012, na Revista Food Additives & Contaminants: Part A no volume 29, indexado no Qualis
da Capes, que tratam da importância dos laboratórios oficiais e análises realizadas nos mesmos.
Porém, observa-se que as publicações, em periódicos, indexados na capes, quanto à
presença de PENG no leite, utilizando análise por HPLC-DAD, tem sido reduzido de 1983 a
2016 (início), pois os pesquisadores filiados a instituições brasileiras não estão no rancking dos
que mais publicam trabalhos usando esta técnica, comprovados por pesquisas realizadas
presentes na Figura 3.6, considerando que o Brasil é um dos maiores de produtores produtos
veterinários, destacando-se os tratamentos à base de penicilinas na produção leiteira.

CAPÍTULO 6
CONCLUSÕES

178
6 CONCLUSÕES
As conclusões deste trabalho são mostradas em dois blocos. No item 6.1 são
apresentadas as conclusões referentes aos resultados obtidos para medicamentos à base de
benzipenicilinas e que integram a parte 1 deste trabalho. Os resultados dos modelos para
quantificação dos medicamentos veterinários, e caracterizações por métodos e técnicas que
asseguram a verificação da qualidade desses terapêuticos foram alcançados.
Diante do panorama dos programas de monitoramento dos resíduos, em alimentos pelas
normatizações legais no âmbito internacional e nacional, têm sido frequentes as preocupações
da legislação brasileira, quanto à presença de contaminantes em alimentos, pois são tomadas
medidas para elaborações de subprogramas de controle e monitoramento de resíduos, em
alimentos de origem animal. Desta forma, a segunda etapa, deste trabalho, foi realizada a
validação de metodologia para a identificação e quantificação de resíduos de PENG, em leite
bovino e caprino, no qual os resultados satisfatórios e aptos para detecção desses resíduos,
comparados com os das mais recentes publicações, pois apresentou simplicidade, redução de
reagentes tóxicos, seletividade e sensibilidade. Um exemplo é o Trabalho 8, disponível em
Apêndice IV. Assim, no item 6.2 apresentaram-se os resultados para o desenvolvimento do
método para quantificação de PENG em amostras de leite ultrapasteurizado bovino e caprino e
que integram a parte 2.
6.1 DESENVOLVIMENTO DE MÉTODOS PARA IDENTIDADE E QUALIDADE DOS
MEDICAMENTOS A BASE DE BENZILPENICILINA
Verificou-se a partir da coleta das amostras que os medicamentos de uso veterinário
estão disponíveis para livre comercialização, isto é, alguns medicamentos registrados pelo
MAPA não apresentam obrigatoriedade de receituário médico veterinário, conforme Decreto
nº 5.053, de 22 de abril de 2004 do MAPA.
As análises dos espectros no infravermelho médio mostraram absorções que
representavam os compostos a base de PENG e indicou semelhanças entre padrão de referência
PENG e amostra de ABAM/IVERM, GEN e PENG.
Os espectros de PENG (padronizado) tem sido de muita importância, como referência,
para averiguação da presença do carbonilas de amida e cromóforos nos medicamentos à base
deste terapêutico.

179
Foram desenvolvidos métodos para controlar a qualidade das formulações injetáveis,
quanto à identificação de princípios ativos, dentre eles a PENG presentes nas marcas dos
produtos, utilizando sistema UFLC-DAD.
Os métodos validados mostraram resultados satisfatórios. Os trabalhos contidos nos
APÊNDICES I e II comprovam aceitação deste tipo de método. As produções evidenciaram a
identificação dos principais compostos ativos, em soluções injetáveis de pesticidas (usados para
controle de neomáteos em bovinos).
Os resultados da aplicação do método mostraram que as amostras A33 e A35
apresentaram teores muito abaixo do que o referenciado estando entre 53 – 65%. As amostras
A9, B34, A8, B13, B16, A10, A12, A6, B26, B29, B30 e B18 apresentaram teores de 73 – 80%.
As amostras B14, B17, B22, B25, B28, B36, B32, B37, A2, A5, A7, A11 e A4 apresentaram
teores entre 81 – 89%. Enquanto que dez amostras nomeadas como B20, B21, B23, B19, A1,
A3, B15, B24, B31 e B27 apresentaram teores aceitáveis de 90 – 98%. Os resultados foram
obtidos através do método com rendimento de 72,51%, LD 7,384 × 10-4 µg mL-1e LQ de 2,0941
× 10-3 µg mL-1.
Integrado a esses estudos quantitativos, foram construídos modelos no UV VIS NIR. Os
modelos desenvolvidos para amostras puras foram eficientes para quantificação de PENG em
95% de confiança. O erro médio do método para determinação em amostras puras foi de ±
1,337%, enquanto que o erro para determinação em amostras tratadas foi de ± 1,035%.
Comparando-se os resultados obtidos no sistema cromatográfico e os resultados das
análises espectrais multivariadas mostram, sem dúvida, que a Cromatografia líquida de alta
eficiência, apesar da redução do número de amostras, mostrou-se mais eficiente.
6.2 DESENVOLVIMENTO DE MÉTODO PARA QUANTIFICAÇÃO DE PENG EM
AMOSTRAS DE LEITE BOVINO E CAPRINO
No preparo das amostras controle, obtiveram-se ótimas valores de recuperação, ficando
superiores a 70% para amostras de leite bovino e caprino. Porém, as recuperações foram
melhores para quantificação principalmente pela reduzida variedade de leite. Além disso, os
erros dos métodos foram inferiores a 20% estando de acordo com as normas legais para análise
de resíduos de antimicrobianos.
As condições cromatográficas foram adequadas para análises das amostras de leite
bovino e caprino com tempo de corrida para cada medida em 10 min.

180
Os parâmetros calculados para validação do método 1 e método 2 foram eficientes, uma
vez que o LD e LQ foram condizentes com LMR de PENG para o leite.
A partir das análises usando o método 1 validado foi verificado que 87 % de 24 amostras
externas comerciais estavam em conformidade e 12% eram não conforme, com erro de ±
1,001%.
O método 2 indicou diferenças significativas entre as amostras. De 20 amostras de leite
caprino analisadas 90% estavam entre em conformidade com o LMR e 15% não conforme com
limite de tolerância de resíduos de PENG muito acima do permitido no leite, com erro de ±
0,2963%.
A análise comparativa mostrou que as amostras de leite bovino com maior frequência
de resíduos de PENG estão acima do tolerado pelas legislações.

REFERÊNCIAS

182
REFERÊNCIAS
ABDI, H. Partial Least Squares ( PLS ) Regression . In: Encyclopedia of Social Sciences
Research Methods. [s.l: s.n.]. p. 1–7.
ABRAHAM, E. P.; OXFD, D. P.; INFIRMARY, R. FUTHER OBSERVATIONS ON
PENICILLIN. The Lancet, v. 238, n. 6155, p. 177–189, 1941.
AGELET, L. E.; HURBURGH, C. R. A Tutorial on Near Infrared Spectroscopy and Its
CalibrationCritical Reviews in Analytical Chemistry, 2010.
AGENCY, E. M. Substances considered as not falling within the scope of Regulation ( EC )
No . 470 / 2009 1 , with regard to residues of veterinary medicinal products in foodstuffs of
animal origin. v. 44, n. May 2009, p. 1–5, 2013.
AGUILERA-LUIZ, M. M. et al. Multi-residue determination of veterinary drugs in milk by
ultra-high-pressure liquid chromatography-tandem mass spectrometry. Journal of
chromatography. A, v. 1205, n. 1-2, p. 10–6, 26 set. 2008.
ALBRIGHT, J. L.; TUCKEY, S. L.; WOODS, G. T. Antibiotics in Milk—A Review.
Journal of dairy science, v. 44, n. 5, p. 779–807, maio 1961.
ALI AHMED, S. M.; ELBASHIR, A. A.; ABOUL-ENEIN, H. Y. New spectrophotometric
method for determination of cephalosporins in pharmaceutical formulations. Arabian
Journal of Chemistry, v. 8, n. 2, p. 233–239, 2015.
ALI, M.; SHERAZI, S. T. H.; MAHESAR, S. A. Quantification of erythromycin in
pharmaceutical formulation by transmission Fourier transform infrared spectroscopy.
Arabian Journal of Chemistry, v. 7, n. 6, p. 1104–1109, 2012.
ALI, S. et al. What if Fleming had not discovered penicillin ? Saudi Journal of Biological
Sciences, v. 21, n. 4, p. 289–293, 2014.
ALIMENTARIUS, C. COMPENDIUM OF METHODS OF ANALYSIS IDENTIFIED
AS SUITABLE TO SUPPORT CODEX MRLs DEVELOPED BY THE CODEX
COMMITTEE ON RESIDUES OF VETERINARY DRUGS IN FOODS 1 This
Compendium is made publicly available and it is to be regularly updated by the Codex
Committee, 2006.
ALTUNAIJI, S. M. et al. Antibiotics for whooping cough (pertussis)Evidence-Based
Child Health, 2012.
ALVARENGA, L. et al. Tablet identification using near-infrared spectroscopy (NIRS) for
pharmaceutical quality control. Journal of Pharmaceutical and Biomedical Analysis, v. 48,
n. 1, p. 62–69, 2008.
ALVAREZ-MUÑOZ, D. et al. Multi-residue method for the analysis of pharmaceuticals and
some of their metabolites in bivalves. Talanta, v. 136, p. 174–182, 2015.

183
AMARIE, S.; GANZ, T.; KEILMANN, F. Mid-infrared near-field spectroscopy. Optics
express, v. 17, n. 24, p. 21794–21801, 2009.
ANDERSEN, W. C. et al. Determination of tetracycline residues in shrimp and whole milk
using liquid chromatography with ultraviolet detection and residue confirmation by mass
spectrometry. Analytica Chimica Acta, v. 529, n. 1-2, p. 145–150, jan. 2005.
ANTANAITIS, R. et al. Efficient diagnostics and treatment of bovine mastitis according to
herd management parameters. Veterinarija ir Zootechnika, v. 69, n. 91, p. 3–10, 2015.
ANVISA. Programa Nacional de Análise de Resíduos de Medicamentos Veterinários em
Alimentos Expostos ao Consumo - PAMVet. p. 1–9, 2003.
ANVISA. Programa de Análise de Resíduos de Medicamentos Veterinários em Alimentos de
Origem Animal - PAMVet - Relatório 2002 / 2003 - Monitoramento de Resíduos em Leite
Exposto ao Consumo ( 1o e 2o anos de atividades ). p. 1–31, 2005.
ANVISA. Programa de análise de resíduos de medicamentos veterinários em alimentos de
origem animal-PAMVET. relatório 2006-2007-Monitoramento de Resíduos em Leite Exposto
ao Consumo. Anvisa, p. 76, 2009.
AŠPERGER, D. et al. HPLC-DAD-FLD determination of veterinary pharmaceuticals in
pharmaceutical industry wastewater with precolumn derivatization using fluorescamine.
Chromatographia, v. 77, n. 15-16, p. 1059–1066, 2014.
BABINGTON, R. et al. Current bioanalytical methods for detection of penicillins. Analytical
and Bioanalytical Chemistry, v. 403, n. 6, p. 1549–1556, 2012.
BAERT, B.; DE SPIEGELEER, B. Quality analytics of internet pharmaceuticals. Analytical
and Bioanalytical Chemistry, v. 398, n. 1, p. 125–136, 2010.
BAILÓN-PÉREZ, M. I. et al. Trace determination of 10 beta-lactam antibiotics in
environmental and food samples by capillary liquid chromatography. Journal of
chromatography. A, v. 1216, n. 47, p. 8355–61, 20 nov. 2009.
BALIZS, G.; HEWITT, A. Determination of veterinary drug residues by liquid
chromatography and tandem mass spectrometry. Analytica Chimica Acta, v. 492, n. 1-2, p.
105–131, set. 2003.
BARBER, N. Regulating Pharmaceuticals in Europe: Striving for Efficiency, Equity and
Quality. Quality & safety in health care, v. 14, n. 3, p. 227–228, 2005.
BARREIRO, E. J.; FERREIRA, V. F.; COSTA, P. R. R. Substâncias enantiomericamente
puras (SEP): A questão dos fármacos quirais. Quimica Nova, v. 20, n. 6, p. 647–656, 1997.
BARRETO, F. et al. Determination and confirmation of chloramphenicol in honey, fish and
prawns by liquid chromatography–tandem mass spectrometry with minimum sample
preparation: validation according to 2002/657/EC Directive. Food Additives &
Contaminants: Part A, v. 29, n. 4, p. 550–558, 2012.

184
BARROSO, M. et al. Role of microextraction sampling procedures in forensic toxicology.
Bioanalysis, v. 4, n. 14, p. 1805–1826, 2012.
BARTON, F. E. et al. Theory and principles of near infrared spectroscopy a. Spectroscopy
Europe, v. 14, n. 1, p. 12–15, 2002.
BASTIEN, P.; VINZI, V. E.; TENENHAUS, M. PLS generalised linear regression.
Computational Statistics & Data Analysis, v. 48, n. 1, p. 17–46, 2005.
BEAUSSE, J. Selected drugs in solid matrices: A review of environmental determination,
occurrence and properties of principal substances. TrAC - Trends in Analytical Chemistry,
v. 23, n. 10-11, p. 753–761, 2004.
BELOTI, V. et al. IMPACTO DA IMPLANTAÇÃO DE BOAS PRÁTICAS DE HIGIENE
NA ORDENHA SOBRE A QUALIDADE MICROBIOLÓGICA E FÍSICO-QUÍMICA DO
LEITE CRU REFRIGERADO Impact of good hygiene practices in milking over
microbiological and physicochemical quality of raw refrigerated milk. Revista Instituto de
Lacticinios Cândido Tostes, v. 67, n. 388, p. 5–10, 2012.
BERENDSEN, B. et al. Evidence of natural occurrence of the banned antibiotic
chloramphenicol in herbs and grass. Analytical and bioanalytical chemistry, v. 397, n. 5, p.
1955–63, jul. 2010.
BERENDSEN, B. J. A et al. Comprehensive analysis of ß-lactam antibiotics including
penicillins, cephalosporins, and carbapenems in poultry muscle using liquid chromatography
coupled to tandem mass spectrometry. Analytical and bioanalytical chemistry, v. 405, n.
24, p. 7859–74, 2013.
BERTHOMIEU, C.; HIENERWADEL, R. Fourier transform infrared (FTIR) spectroscopy.
Photosynthesis Research, v. 101, n. 2-3, p. 157–170, 2009.
BIAŁK-BIELIŃSKA, A. et al. Ecotoxicity evaluation of selected sulfonamides.
Chemosphere, v. 85, n. 6, p. 928–933, 2011.
BILANDŽIĆ, N. et al. Veterinary drug residues determination in raw milk in Croatia. Food
Control, v. 22, n. 12, p. 1941–1948, dez. 2011a.
BILANDŽIĆ, N. et al. Veterinary drug residues determination in raw milk in Croatia. Food
Control, v. 22, n. 12, p. 1941–1948, dez. 2011b.
BITTENCOURT, M. S. et al. High-throughput multiclass screening method for antibiotic
residue analysis in meat using liquid chromatography-tandem mass spectrometry: a novel
minimum sample preparation procedure. Food Additives & Contaminants: Part A, v. 29, n.
4, p. 508–516, 2012.
BLANCO, M. et al. A process analytical technology approach based on near infrared
spectroscopy: Tablet hardness, content uniformity, and dissolution test measurements of intact
tablets. Journal of Pharmaceutical Sciences, v. 95, n. 10, p. 2137–2144, 2006.

185
BLANCO, M.; PEGUERO, A. Analysis of pharmaceuticals by NIR spectroscopy without
a reference methodTrAC - Trends in Analytical Chemistry, 2010.
BODSON, C. et al. Comparison of FT-NIR transmission and UV-vis spectrophotometry to
follow the mixing kinetics and to assay low-dose tablets containing riboflavin. Journal of
Pharmaceutical and Biomedical Analysis, v. 41, n. 3, p. 783–790, 2006.
BOGIALLI, S. et al. A simple and rapid confirmatory assay for analyzing antibiotic residues
of the macrolide class and lincomycin in bovine milk and yoghurt: hot water extraction
followed by liquid chromatography/tandem mass spectrometry. Rapid Communications in
Mass Spectrometry, v. 21, n. 2, p. 237–246, 2007.
BOGIALLI, S.; DI CORCIA, A. Recent applications of liquid chromatography-mass
spectrometry to residue analysis of antimicrobials in food of animal origin. Analytical and
bioanalytical chemistry, v. 395, n. 4, p. 947–66, out. 2009.
BOHM, D. A; STACHEL, C. S.; GOWIK, P. Multi-method for the determination of
antibiotics of different substance groups in milk and validation in accordance with
Commission Decision 2002/657/EC. Journal of chromatography. A, v. 1216, n. 46, p.
8217–23, 13 nov. 2009.
BOISON, J. O. et al. Determination of trimethoprim and sulphadoxine residues in porcine
tissues and plasma. Canadian journal of veterinary research = Revue canadienne de
recherche vétérinaire, v. 60, n. 4, p. 281–7, out. 1996.
BORRÀS, S. et al. Analysis of antimicrobial agents in animal feed. TrAC Trends in
Analytical Chemistry, v. 30, n. 7, p. 1042–1064, jul. 2011.
BOSCH OJEDA, C.; SANCHEZ ROJAS, F. Recent developments in derivative
ultraviolet/visible absorption spectrophotometry. Analytica Chimica Acta, v. 518, n. 1-2, p.
1–24, 2004.
BOSCH OJEDA, C.; SANCHEZ ROJAS, F. Recent applications in derivative
ultraviolet/visible absorption spectrophotometry: 2009-2011. A review. Microchemical
Journal, v. 106, p. 1–16, 2013.
BOUTRIF, E. The new role of Codex Alimentarius in the context of WTO/SPS agreement.
Food Control, v. 14, n. 2, p. 81–88, 2003.
BOYD, B. et al. Development of an improved method for trace analysis of chloramphenicol
using molecularly imprinted polymers. Journal of chromatography. A, v. 1174, n. 1-2, p.
63–71, 7 dez. 2007.
BRASIL. Farmacopeia Brasileira Volume 1. [s.l: s.n.]. v. 1
BRESSOLLE, F.; BROMET-PETIT, M.; AUDRAN, M. Validation of liquid
chromatographic and gas chromatographic methods. Applications to pharmacokinetics.
Journal of chromatography. B, Biomedical applications, v. 686, n. 1, p. 3–10, 8 nov. 1996.
BRITISH PHARMACOPOEIA. British Pharmacopoeia, 1983.

186
BROAD, N. W. et al. Non-invasive determination of ethanol, propylene glycol and water in a
multi-component pharmaceutical oral liquid by direct measurement through amber plastic
bottles using Fourier transform near-infrared spectroscopy. The Analyst, v. 125, n. 11, p.
2054–2058, 2000.
BRUNO, F. et al. An original approach to determining traces of tetracycline antibiotics in
milk and eggs by solid-phase extraction and liquid chromatography/mass spectrometry.
Rapid communications in mass spectrometry : RCM, v. 16, n. 14, p. 1365–76, jan. 2002.
BULDINI, P. L.; RICCI, L.; SHARMA, J. L. Recent applications of sample preparation
techniques in food analysis. Journal of chromatography. A, v. 975, n. 1, p. 47–70, 25 out.
2002.
BURNS, T. D. Recent instrumental advances in pharmaceutical analysis. Analytical
Proceedings, v. 23, n. 3, p. 81–90, 1986.
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. DETERMINING TOTAL
LIPID AND MOISTURE CONTENT IN POWDERED MILK, COMMERCIALIZED
IN BRAZIL USING NEAR INFRARED SPECTROSCOPIC ANALYSIS COMBINED
WITH THE CONSTRUCTION OF MULTIVARIATE MODELSWorld Congress of
Food Science and Technology, 2012. Disponível em:
<http://iufost.org.br/sites/iufost.org.br/files/anais/10832.pdf>
CÁMARA, M. et al. An HPLC-DAD method for the simultaneous determination of nine β-
lactam antibiotics in ewe milk. Food chemistry, v. 141, n. 2, p. 829–34, 15 nov. 2013.
CAPANEMA, L. et al. Panorama da indústria farmacêutica veterinária. BNDES Setorial, v.
25, p. 157–174, 2007.
CAPOZZIELLO, S.; LATTANZI, A. Geometrical approach to central molecular chirality: A
chirality selection rule. Chirality, v. 15, n. 3, p. 227–230, 2003.
CARLIER, M. et al. Ultrafast quantification of β-lactam antibiotics in human plasma using
UPLC–MS/MS. Journal of Chromatography B, v. 978-979, p. 89–94, 2015.
CASSIANO, N. et al. Validação em métodos cromatográficos para análises de pequenas
moléculas em matrizes biológicas. Química Nova, v. 32, n. 4, p. 1021–1030, 2009.
CASTRO-PUYANA, M.; CREGO, A. L.; MARINA, M. L. Recent advances in the analysis
of antibiotics by CE and CEC. Electrophoresis, v. 31, n. 1, p. 229–50, jan. 2010.
CEPURNIEKS, G. et al. The development and validation of a rapid method for the
determination of antimicrobial agent residues in milk and meat using ultra performance liquid
chromatography coupled to quadrupole – Orbitrap mass spectrometry. Journal of
Pharmaceutical and Biomedical Analysis, v. 102, p. 184–192, 2015.
CHAMBERS, J. V. A review of the US Grade A raw milk supply , its quality , marketing and
pricing. Dairy Technology, v. 51, n. 4, p. 108–112, 1998.

187
CHANG, Q. et al. Antibiotics in agriculture and the risk to human health: how worried should
we be? Evolutionary Applications, n. Levy 1997, p. n/a–n/a, 2014.
CHASIN et al. VALIDAÇÃO DE MÉTODOS EM ANÁLISES TOXICOLÓGICAS: UMA
ABORDAGEM GERAL. Revista Brasileira de Toxicologia, v. 11, n. 1, p. 1–6, 1998.
CHEN, Y. et al. Sample preparation. Journal of chromatography. A, v. 1184, n. 1-2, p.
191–219, 14 mar. 2008.
CHEN, G. L., AND FANG, Y. . The LC-MS/MS Methods for the Determination of Specific
Antibiotics Residues in Food Matrices. In: Methods in Molecular Biology- Mass
Spectrometry in Food Safety. 1o. ed. [s.l.] Wiley, 2011. p. 309–355.
CHIESA, L. et al. Determination of veterinary antibiotics in bovine urine by liquid
chromatography–tandem mass spectrometry. Food Chemistry, v. 185, p. 7–15, 2015.
CHONG, X. M. et al. Construction of a universal model for non-invasive identification of
penicillins for injection using near-infrared diffuse reflectance spectroscopy. Vibrational
Spectroscopy, v. 51, n. 2, p. 313–317, 2009a.
CHONG, X. M. et al. Construction of a universal model for non-invasive identification of
cephalosporins for injection using near-infrared diffuse reflectance spectroscopy. Vibrational
Spectroscopy, v. 49, n. 2, p. 196–203, 2009b.
CHRISTODOULOU, E. A; SAMANIDOU, V. F. Multiresidue HPLC analysis of ten
quinolones in milk after solid phase extraction: validation according to the European Union
Decision 2002/657/EC. Journal of separation science, v. 30, n. 15, p. 2421–9, out. 2007.
CINQUINA, A. L. et al. Short communication Validation of a high-performance liquid
chromatography method for the determination of oxytetracycline , tetracycline ,
chlortetracycline and doxycycline in bovine milk and muscle. Journal of Chromatography
A, v. 987, p. 227–233, 2003a.
CINQUINA, A. L. et al. Determination of enrofloxacin and its metabolite ciprofloxacin in
goat milk by high-performance liquid chromatography with diode-array detection
Optimization and validation. Journal of Chromatography A, v. 987, p. 221–226, 2003b.
CLARKE, F. Extracting process-related information from pharmaceutical dosage forms using
near infrared microscopy. Vibrational Spectroscopy, v. 34, n. 1, p. 25–35, 2004.
CODEX ALIMENTARIUS. CODEX GUIDELINES FOR THE ESTABLISHMENT OF
A REGULATORY PROGRAMME FOR CONTROL OF VETERINARY DRUG
RESIDUES IN FOODS CAC/GL 16-1993, 1993.
CODEX ALIMENTARIUS. PROGRAMA CONJUNTO FAO/OMS SOBRE NORMAS
ALIMENTARIAS, 2001.
CODEX ALIMENTARIUS. GUIDELINES FOR THE DESIGN AND
IMPLEMENTATION OF NATIONAL REGULATORY FOOD SAFETY

188
ASSURANCE PROGRAMME ASSOCIATED WITH THE USE OF VETERINARY
DRUGS IN FOOD PRODUCING ANIMALS CAC/GL 71-2009, 2009.
CODEX ALIMENTARIUS. GUIDELINES FOR THE DESIGN AND
IMPLEMENTATION OF NATIONAL REGULATORY FOOD SAFETY
ASSURANCE PROGRAMME ASSOCIATED WITH THE USE OF VETERINARY
DRUGS IN FOOD PRODUCING ANIMALS CAC/GL 71-2009, 2014.
COX, L. A.; POPKEN, D. A. Quantifying potential human health impacts of animal antibiotic
use: Enrofloxacin and macrolides in chickens. Risk Analysis, v. 26, n. 1, p. 135–146, 2006.
CRAWFORD, L.; KUGLER, E. G. Codex committee on veterinary drug residues.
Regulatory toxicology and pharmacology : RTP, v. 6, n. 4, p. 381–384, 1986.
CRISTOFANI, E. et al. A confirmatory method for the determination of tetracyclines in
muscle using high-performance liquid chromatography with diode-array detection. Analytica
chimica acta, v. 637, n. 1-2, p. 40–6, 1 abr. 2009.
CUI, X. et al. Simultaneous determination of seventeen glucocorticoids residues in milk and
eggs by ultra- performance liquid chromatography / electrospray tandem mass spectrometry.
Rapid communications in mass spectrometry, v. 20, n. 15, p. 2355–2364, 2006.
DA SILVA, I. S. et al. Fast simultaneous determination of trimethoprim and
sulfamethoxazole by capillary zone electrophoresis with capacitively coupled contactless
conductivity detection. Journal of separation science, v. 36, n. 8, p. 1405–9, abr. 2013.
DADGAR, D. et al. Application issues in bioanalytical method validation, sample analysis
and data reporting. Journal of pharmaceutical and biomedical analysis, v. 13, n. 2, p. 89–
97, fev. 1995.
DE ALBUQUERQUE FERNANDES, S. A. et al. Daily ingestion of tetracycline residue
present in pasteurized milk: a public health problem. Environmental Science and Pollution
Research, v. 21, n. 5, p. 3427–3434, 17 nov. 2013.
DE BLEYE, C. et al. Critical review of near-infrared spectroscopic methods validations in
pharmaceutical applications. Journal of Pharmaceutical and Biomedical Analysis, v. 69, p.
125–132, 2012.
DE QUEIROZ MAURICIO, A.; LINS, E. S. The National Agricultural Laboratories of Brazil
and the control of residues and contaminants in food. Food Additives & Contaminants:
Part A, v. 29, n. 4, p. 482–489, 2012.
DE RUYCK, H. et al. Determination of flubendazole and its metabolites in eggs and poultry
muscle with liquid chromatography - Tandem mass spectrometry. Journal of Agricultural
and Food Chemistry, v. 49, n. 2, p. 610–617, 2001.
DEMIRALAY, E. Ç. et al. Determination of chromatographic and spectrophotometric
dissociation constants of some beta lactam antibiotics. Journal of Pharmaceutical and
Biomedical Analysis, v. 71, p. 139–143, 2012.

189
DEMORTAIN, D. Enabling global principle-based regulation: The case of risk analysis in the
Codex Alimentarius. Regulation and Governance, v. 6, n. 2, p. 207–224, 2012.
DÍAZ-BAO, M. et al. Fast HPLC-MS/MS Method for Determining Penicillin Antibiotics in
Infant Formulas Using Molecularly Imprinted Solid-Phase Extraction. Journal of analytical
methods in chemistry, v. 2015, p. 959675, jan. 2015.
DINIS-OLIVEIRA, R. J. et al. Collection of biological samples in forensic toxicology.
Toxicology mechanisms and methods, v. 20, n. 7, p. 363–414, 2010.
DIOP, A. et al. Quality control of antibiotics used in Senegal. Medecine tropicale : revue du
Corps de sante colonial, v. 69, n. 3, p. 251–254, 2009.
DONG, M. W.; ZHANG, K. Trends in Analytical Chemistry Ultra-high-pressure liquid
chromatography ( UHPLC ) in method development. Trends in Analytical Chemistry, v. 63,
p. 21–30, 2014.
DUAN, C. et al. Recent developments in solid-phase microextraction for on-site sampling
and sample preparationTrAC - Trends in Analytical Chemistry, 2011.
DUBREIL-CHÉNEAU, E. et al. Confirmation of 13 sulfonamides in honey by liquid
chromatography – tandem mass spectrometry for monitoring plans : Validation according to
European Union Decision 2002 / 657 / EC. Journal of Chromatography A, v. 1339, n. 37,
p. 128–136, 2014.
DUNN, J. F. et al. BOLD MRI vs. NIR spectrophotometry. Will the best technique come
forward? Advances in experimental medicine and biology, v. 454, p. 103–113, 1998.
EDWALD, P. P. 50 Years Ago 100 Years Ago. Nature, v. 505, n. 23, p. 489, 2014.
ELLIS, R. L. Development of veterinary drug residue controls by the Codex Alimentarius
Commission: a review. Food additives & contaminants. Part A, Chemistry, analysis,
control, exposure & risk assessment, v. 25, n. 12, p. 1432–1438, 2008.
ELMARROUNI, A. et al. Expedient synthesis of a stereoisomer of acremolide B. Journal of
Organic Chemistry, v. 75, n. 24, p. 8478–8486, 2010.
EMEA. S Pecifications : T Est P Rocedures and a Cceptance C Riteria for B Iotechnological /
B Iological P Roducts. n. March, p. 1–20, 1999.
EMEA. Position Paper on the establishment of MRLs for milk, 2002.
EMEA. (R1), ICH Topic Q 2 Procedures:, Validation of Analytical Methodology, Text.
EMEA, v. 2, n. November 1994, p. 1–15, 2006.
EMEA. Guideline on bioanalytical method validationEMA Guideline, 2012a.
EMEA. Guideline on the use of Near Infrared Spectroscopy ( NIRS ) by the pharmaceutical
industry and the data requirements for new submissions and variations Guideline on the use of

190
Near Infrared Spectroscopy ( NIRS ) by the pharmaceutical industry and the data. Committee
for Human Products, v. 44, n. January, p. 1–28, 2012b.
ÉPSHTEIN. Structure of Chemical Compounds , Methods of Analysis and Process Control
Validation of Hplc Techniques. Pharmaceutical Chemistry Journal, v. 38, n. 4, p. 212–228,
2004.
EUROPEAN COMISSION. Commission Decision of 14 April 1993 laying down the
methods to be used for detecting residues of substances having a hormonal or a
thyrostatic action. Database of European Law, 1993. Disponível em: <http://eur-
lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1993:118:0064:0074:EN:PDF>
EUROPEAN COMMISSION. Commission Directive 2006/130/EC of 11 December 2006
implementing. Official Journal of the European Union, p. 15–16, 2006.
EUROPEAN COMMISSION. COMMUNITY REFERENCE LABORATORIES
RESIDUES ( CRLs ) GUIDELINES FOR THE VALIDATION OF SCREENING
METHODS ( INITIAL VALIDATION AND TRANSFER ) Table of Contents, 2010.
EUROPEAN COMMISSION 37/2010/EC. Jornal Oficial da União Europeia L 342/59
REGULAMENTO (CE) N. Journal Oficial of Europea, 2009.
EUROPEAN PHARMACOPOEIA COMMISSION. European Pharmacopoea, 2008.
FAO. FAO/WHO Codex Alimentarius Commission - report of the twenty-sixth session,
Rome, 30 June - 7 July 2003. Report of the Session of the FAO/WHO Codex Alimentarius
Commission, p. 143 pp., 2003.
FAO. Residue evaluation of certain veterinary drugs - Joint FAO/WHO Expert
Committee on Food Additives 78th meeting 2013. [s.l: s.n.].
FAO/WHO. SPECIFICATIONS FOR THE IDENTITY AND PURITY OF FOOD
ADDITIVES AND THEIR TOXICOLOGICAL EVALUATION SOME
ANTIBIOTICS, 1968.
FARINA, E. M. M. Q. et al. Private and public milk standards in Argentina and Brazil. Food
Policy, v. 30, n. 3, p. 302–315, 2005.
FDA. Guidance for Industry Process Validation : General Principles and Practices
Guidance for Industry Process Validation : General Principles and Practices, 2011.
Disponível em:
<http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidanc
es/UCM070336.pdf>
FENNELL, M. A. et al. Gentamicin in Tissue and Whole Milk: An Improved Method for
Extraction and Cleanup of Samples for Quantitation on HPLC. Journal of Agricultural and
Food Chemistry, v. 43, n. 7, p. 1849–1852, 1995.
FENT, K.; WESTON, A. A.; CAMINADA, D. Ecotoxicology of human
pharmaceuticalsAquatic Toxicology, 2006.

191
FILHO, N. F. et al. Within-laboratory validation of a multiresidue method for the analysis of
98 pesticides in mango by liquid chromatography-tandem mass spectrometry. Food
Additives & Contaminants: Part A, v. 29, n. 4, p. 641–656, 2012.
FINGLETON, J. LEGISLATION FOR VETERINARY DRUGSFAO, 2004.
FJ, S.; PS, C. Chromatographic methods of analysis of antibiotics in milk. Journal of
chromatography. A, v. 812, p. 99–109, 1998.
FLEMING, A. On the antibacterial action of cultures of a penicillium, with special reference
to their use in the isolation of B. influenzae. British Journal of Experimental Pathology, v.
10, n. 3, p. 226–236, 1929.
FREITAS, A. et al. Liquid chromatography: Review on the last developments on the
detection of antibiotics in food-producing animals. In: Liquid Chromatography: Principles,
Technology and Applications. [s.l: s.n.]. p. 99–140.
FREITAS, A.; BARBOSA, J.; RAMOS, F. Development and validation of a multi-residue
and multiclass ultra-high-pressure liquid chromatography-tandem mass spectrometry
screening of antibiotics in milk. International Dairy Journal, v. 33, n. 1, p. 38–43, 2013.
FSIS, U. UNITED STATES National Residue Program Scheduled Sampling Plans Office
of Public Health Science, 2011.
FUMES, B. H. et al. Recent advances and future trends in new materials in sample
preparation. TrAC Trends in Analytical Chemistry, 2015.
FURUSAWA, N. Rapid liquid chromatographic determination of residual penicillin G in
milk. Fresenius’ journal of analytical chemistry, v. 368, n. 6, p. 624–626, 2000.
GAMBA, V. et al. Development and validation of a confirmatory method for the
determination of sulphonamides in milk by liquid chromatography with diode array detection.
Analytica chimica acta, v. 637, n. 1-2, p. 18–23, 1 abr. 2009.
GARCÍA-MAYOR, M. A. et al. Liquid chromatography-UV diode-array detection method
for multi-residue determination of macrolide antibiotics in sheep’s milk. Journal of
Chromatography A, v. 1122, n. 1-2, p. 76–83, 2006.
GARCÍA-MAYOR, M. A. et al. Occurrence of erythromycin residues in sheep milk.
Validation of an analytical method. Food and Chemical Toxicology, v. 78, p. 26–32, 2015.
GAUDIN, V. et al. Evaluation and validation of biochip multi-array technology for the
screening of six families of antibiotics in honey according to the European guideline for the
validation of screening methods for residues of veterinary medicines. Food additives &
contaminants. Part A, Chemistry, analysis, control, exposure & risk assessment, v. 31, n.
10, p. 1699–711, jan. 2014.
GERGELY, A. A review of the application of chiroptical methods to analytical chemistry.
Journal of pharmaceutical and biomedical analysis, v. 7, n. 5, p. 523–541, 1989.

192
GERVAIN, J. et al. Near-infrared spectroscopy: A report from the McDonnell infant
methodology consortiumDevelopmental Cognitive Neuroscience, 2011.
GHIDINI, S. et al. Residues of aminoglycosides in milk: Confirmatory analysis. Veterinary
Research Communications, v. 31, n. SUPPL. 1, p. 365–367, 2007.
GILBERT, J.; GARDNER, V.; BIZEC, B. LE. Editorial:Brazilian Ministry of Agriculture ,
Livestock and Food Supply ( MAPA ): strategies to tackle chemical food safety. Food
Additives & Contaminants . Part A, v. 29, n. 4, p. 81, 2012.
GILPIN, R. K.; GILPIN, C. S. Pharmaceuticals and related drugs. Analytical Chemistry, v.
83, n. 12, p. 4489–4507, 2011.
GÓMEZ-PÉREZ, M. L. et al. Comprehensive qualitative and quantitative determination of
pesticides and veterinary drugs in honey using liquid chromatography-Orbitrap high
resolution mass spectrometry. Journal of chromatography. A, v. 1248, p. 130–8, 27 jul.
2012.
GONZÁLEZ DE LA HUEBRA, M. J.; VINCENT, U.; VON HOLST, C. Sample preparation
strategy for the simultaneous determination of macrolide antibiotics in animal feedingstuffs
by liquid chromatography with electrochemical detection (HPLC-ECD). Journal of
Pharmaceutical and Biomedical Analysis, v. 43, n. 5, p. 1628–1637, 2007.
GÖRÖG, S. The sacred cow: The questionable role of assay methods in characterising the
quality of bulk pharmaceuticals. Journal of Pharmaceutical and Biomedical Analysis, v.
36, n. 5, p. 931–937, 2005.
GORRIS, L. G. M. Food safety objective: An integral part of food chain management. Food
Control, v. 16, n. 9 SPEC. ISS., p. 801–809, 2005.
GOTO, T. et al. High-throughput analysis of tetracycline and penicillin antibiotics in animal
tissues using electrospray tandem mass spectrometry with selected reaction monitoring
transition. Journal of Chromatography A, v. 1100, n. 2, p. 193–199, 2005.
GOWIK, P. The validation of methods for regulatory purposes in the control of
residuesJournal of Chromatography A, 2009.
GREEN, M. Dairy herd health. [s.l: s.n.].
GREEN, M. J. et al. Influence of dry period bacterial intramammary infection on clinical
mastitis in dairy cows. Journal of dairy science, v. 85, n. 10, p. 2589–99, out. 2002.
GREMILOGIANNI, A. M.; MEGOULAS, N. C.; KOUPPARIS, M. A. Hydrophilic
interaction vs ion pair liquid chromatography for the determination of streptomycin and
dihydrostreptomycin residues in milk based on mass spectrometric detection. Journal of
chromatography. A, v. 1217, n. 43, p. 6646–51, 22 out. 2010.
GRENFELL, C. T.; MEANS, J. A.; BROWN, V. Studies on the naturally an assay method for
Penicillin G. The Journal of Biological Chemistry, v. 179, p. 527–535, 1945.

193
GRÜNHUT, M. et al. Flow-batch technique for the simultaneous enzymatic determination of
levodopa and carbidopa in pharmaceuticals using PLS and successive projections algorithm.
Talanta, v. 75, n. 4, p. 950–958, 2008.
GUMUS, G.; FILIK, H.; DEMIRATA, B. Determination of bismuth and zinc in
pharmaceuticals by first derivative UV-Visible spectrophotometry. Analytica Chimica Acta,
v. 547, n. 1, p. 138–143, 2005.
GUMUSTAS, M. et al. UPLC versus HPLC on drug analysis: Advantageous, applications
and their validation parameters. Chromatographia, v. 76, n. 21-22, p. 1365–1427, 2013.
GUO, L. et al. Simultaneous Determination of Florfenicol and Diclazuril in Compound
Powder by RP-HPLC-UV Method. Journal of Chemistry, v. 2014, p. 1–5, 2014.
GUO, Z.; GAI, P. Development of an ultrasensitive electrochemiluminescence inhibition
method for the determination of tetracyclines. Analytica chimica acta, v. 688, n. 2, p. 197–
202, 4 mar. 2011.
HALEEM, R. M. et al. Quality in the pharmaceutical industry - A literature review. Saudi
Pharmaceutical Journal, 2014.
HAN, R. W. et al. Simultaneous determination of 38 veterinary antibiotic residues in raw milk
by UPLC–MS/MS. Food Chemistry, v. 181, p. 119–126, 2015.
HARE, D. Antimicrobial resistance. The Canadian Veterinary Journal, v. 40, n. 10, p.
693–694, 1999.
HARE, R. History : New light on the history of penicillin by. Medical History, v. 26, n.
August 2012, p. 1–24, 1982.
HARE, R. The scientific activities of Alexander Fleming, other than the discovery of
penicillin. Medical history, v. 27, n. 4, p. 347–372, 1983.
HARTAUER, K. J.; GUILLORY, J. K. Quantitative Fourier transform-infrared/attenuated
total reflectance (FT-IR/ATR) analysis of trimethoprim and sulfamethoxazole in a
pharmaceutical formulation using partial least squares. Pharmaceutical research, v. 6, n. 7,
p. 608–611, 1989.
HARTLÉN, J. Environmental consequences of using residues. Waste Management, v. 16, n.
1-3, p. 1–6, 1996.
HARTMANN, C. et al. Validation of bioanalytical chromatographic methodsJournal of
Pharmaceutical and Biomedical Analysis, 1998.
HE, K.; BLANEY, L. Systematic optimization of an SPE with HPLC-FLD method for
fluoroquinolone detection in wastewater. Journal of Hazardous Materials, v. 282, p. 96–
105, 2015.

194
HEALTH CANADA. Health Products and Food Branch Inspectorate Standard for the
Fabrication , Control and Distribution of Antimicrobial Agents for Use on
Environmental Surfaces and Certain Medical Devices ( GUI-0049 )Canada, 2012.
HERMO, M. P. et al. Improved determination of quinolones in milk at their MRL levels using
LC-UV, LC-FD, LC-MS and LC-MS/MS and validation in line with regulation 2002/657/EC.
Analytica chimica acta, v. 613, n. 1, p. 98–107, 14 abr. 2008.
HERRERA-HERRERA, A. V et al. Determination of quinolone residues in infant and young
children powdered milk combining solid-phase extraction and ultra-performance liquid
chromatography-tandem mass spectrometry. Journal of chromatography. A, v. 1218, n. 42,
p. 7608–14, 21 out. 2011.
HERSBACH, G. J. M. Penicillin production. Antonie van Leeuwenhoek, v. 49, n. 1, p. 93–
94, 1983.
HILDING, D. A. Discovery of penicillin. Mayo Clinic proceedings. Mayo Clinic, v. 73, n.
1, p. 98, 1998.
HOFF, R. et al. Bioactivity-based screening methods for antibiotics residues: a comparative
study of commercial and in-house developed kits. Food Additives & Contaminants: Part A,
v. 29, n. 4, p. 577–586, 2012.
HOGAN, J.; SMITH, K. L. Managing Environmental Mastitis. Veterinary Clinics of North
America - Food Animal Practice, v. 28, n. 2, p. 217–224, 2012.
HOOF, N. VAN et al. Validation of a liquid chromatography–tandem mass spectrometric
method for the quantification of eight quinolones in bovine muscle, milk and aquacultured
products. Analytica Chimica Acta, v. 529, n. 1-2, p. 265–272, jan. 2005.
HORMAZÁBAL, V.; YNDESTAD, M. SIMULTANEOUS DETERMINATION OF
CHLORAMPHENICOL AND KETOPROFEN IN MEAT AND MILK AND
CHLORAMPHENICOL IN EGG , HONEY ,. v. 24, n. 16, p. 2477–2486, 2001.
HORTON, L. R. Risk analysis and the law: international law, the World Trade Organization,
Codex Alimentarius and national legislation. Food additives and contaminants, v. 18, n. 12,
p. 1057–1067, 2001.
HOSHI, Y. Functional near-infrared spectroscopy: current status and future prospects.
Journal of Biomedical Optics, v. 12, n. 6, p. 062106, 2007.
HSU, C. P. S. Infrared Spectroscopy. In: Handbook of Instrumental Techniques for
Analytical Chemistry. [s.l: s.n.]. p. 247–284.
IBARRA, I. S. et al. Magnetic solid phase extraction based on phenyl silica adsorbent for the
determination of tetracyclines in milk samples by capillary electrophoresis. Journal of
chromatography. A, v. 1218, n. 16, p. 2196–202, 22 abr. 2011.
IBGE. Indicadores IBGE para 2015, 2015.

195
ICH. Validation of a analytical Procedures : text and methodology Q2(R1)Guidance,
2005.
IGLESIAS, A. et al. Detection and quantitative analysis of 21 veterinary drugs in river water
using high-pressure liquid chromatography coupled to tandem mass spectrometry.
Environmental science and pollution research international, v. 19, n. 8, p. 3235–49, 2012.
ISHIBASHI, F. et al. Synthesis and algicidal activity of (+)-cyanobacterin and its
stereoisomer. Bioscience, biotechnology, and biochemistry, v. 69, n. 2, p. 391–396, 2005.
ISHII, R. et al. Multi-residue quantitation of aminoglycoside antibiotics in kidney and meat
by liquid chromatography with tandem mass spectrometry. Food additives & contaminants.
Part A, Chemistry, analysis, control, exposure & risk assessment, v. 25, n. 12, p. 1509–
19, dez. 2008.
ISLAM, M. R.; MAHDI, J. G.; BOWEN, I. D. Pharmacological importance of stereochemical
resolution of enantiomeric drugs. Drug safety : an international journal of medical
toxicology and drug experience, v. 17, n. 3, p. 149–165, 1997.
JACXSENS, L. et al. Food safety performance indicators to benchmark food safety output of
food safety management systems. International Journal of Food Microbiology, v. 141, n.
SUPPL., 2010.
JANK, L. et al. β-lactam antibiotics residues analysis in bovine milk by LC-ESI-MS/MS: a
simple and fast liquid–liquid extraction method. Food Additives & Contaminants: Part A,
v. 29, n. 4, p. 497–507, 2012.
JING, T. et al. Determination of trace tetracycline antibiotics in foodstuffs by liquid
chromatography-tandem mass spectrometry coupled with selective molecular-imprinted solid-
phase extraction. Analytical and bioanalytical chemistry, v. 393, n. 8, p. 2009–18, abr.
2009.
JOHNSTON, A. M. Evaluation of certain veterinary drug residues in food. Food Control, v.
3, n. 4, p. 220, 1992.
JONES, G. M.; SEYMOUR, E. H. Cowside antibiotic residue testing. Journal of dairy
science, v. 71, n. 6, p. 1691–9, jun. 1988.
JP XVI. Pharmacopeia JaponesaPharmacopeia Japonesa, 2007.
KABIR, J. et al. Veterinary drug use in poultry farms and determination of antimicrobial drug
residues in commercial eggs and slaughtered chicken in Kaduna State, Nigeria. Food
Control, v. 15, n. 2, p. 99–105, mar. 2004.
KANG’ETHE, E. K. et al. Investigation of the risk of consuming marketed milk with
antimicrobial residues in Kenya. Food Control, v. 16, n. 4, p. 349–355, abr. 2005.
KANKARE, J. J.; STEPHENS, R. The influence of optical design on the signal to noise
characteristics of polarimeters. Talanta, v. 33, n. 7, p. 571–576, 1986.

196
KANTIANI, L. et al. Fully automated analysis of beta-lactams in bovine milk by online solid
phase extraction-liquid chromatography-electrospray-tandem mass spectrometry. Analytical
chemistry, v. 81, n. 11, p. 4285–95, 1 jun. 2009.
KANTIANI, L.; FARRÉ, M.; BARCELÓ, D. Analytical methodologies for the detection of
β-lactam antibiotics in milk and feed samples. TrAC Trends in Analytical Chemistry, v. 28,
n. 6, p. 729–744, jun. 2009.
KARAGEORGOU, E. et al. Ultrasound-assisted dispersive extraction for the high pressure
liquid chromatographic determination of tetracyclines residues in milk with diode array
detection. Food chemistry, v. 150, p. 328–34, 1 maio 2014.
KARAGEORGOU, E. G.; SAMANIDOU, V. F. Development and validation according to
European Union Decision 2002/657/EC of an HPLC-DAD method for milk multi-residue
analysis of penicillins and amphenicols based on dispersive extraction by QuEChERS in
MSPD format. Journal of separation science, p. 1893–1901, 4 jul. 2011.
KARNES, H. T.; SHIU, G.; SHAH, V. P. Validation of bioanalytical methods.
Pharmaceutical research, v. 8, n. 4, p. 421–426, 1991.
KARPIŃSKA, J.; MIKOŁUĆ, B.; PIOTROWSKA-JASTRZEBSKA, J. Application of
derivative spectrophotometry for determination of coenzyme Q10 in pharmaceuticals and
plasma. Journal of Pharmaceutical and Biomedical Analysis, v. 17, n. 8, p. 1345–1350,
1998.
KAUFMANN, A; BUTCHER, P.; MADEN, K. Determination of aminoglycoside residues by
liquid chromatography and tandem mass spectrometry in a variety of matrices. Analytica
chimica acta, v. 711, p. 46–53, 20 jan. 2012.
KAWATA, S. Instrumentation for Near-Infrared Spectroscopy. In: Near-Infrared
Spectroscopy. [s.l: s.n.]. p. 43–73.
KHOPTYAR, D. et al. Broadband photon time-of-flight spectroscopy of pharmaceuticals and
highly scattering plastics in the VIS and close NIR spectral ranges. Optics express, v. 21, n.
18, p. 20941–53, 2013.
KHOR, S.; TEE, E. S. Development of a HPLC method for the simultaneous determination of
several B-vitamins and ascorbic acid. Malaysian journal of nutrition, v. 2, n. 1, p. 49–65,
mar. 1996.
KHOSHAYAND, M. R. et al. Simultaneous spectrophotometric determination of
paracetamol, ibuprofen and caffeine in pharmaceuticals by chemometric methods.
Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, v. 70, n. 3, p.
491–499, 2008.
KINSELLA, B. et al. Current trends in sample preparation for growth promoter and
veterinary drug residue analysis. Journal of chromatography. A, v. 1216, n. 46, p. 7977–
8015, 13 nov. 2009.

197
KISHIDA, K. Simplified extraction of tetracycline antibiotics from milk using a centrifugal
ultrafiltration device. Food Chemistry, v. 126, n. 2, p. 687–690, maio 2011.
KNECHT, B. G. et al. Automated microarray system for the simultaneous detection of
antibiotics in milk. Analytical chemistry, v. 76, n. 3, p. 646–54, 1 fev. 2004.
KOESUKWIWAT, U.; JAYANTA, S.; LEEPIPATPIBOON, N. Validation of a liquid
chromatography-mass spectrometry multi-residue method for the simultaneous determination
of sulfonamides, tetracyclines, and pyrimethamine in milk. Journal of chromatography. A,
v. 1140, n. 1-2, p. 147–56, 26 jan. 2007a.
KOESUKWIWAT, U.; JAYANTA, S.; LEEPIPATPIBOON, N. Solid-phase extraction for
multiresidue determination of sulfonamides, tetracyclines, and pyrimethamine in Bovine’s
milk. Journal of chromatography. A, v. 1149, n. 1, p. 102–11, 11 maio 2007b.
KORAKIANITI, E.; REKKAS, D. Statistical thinking and knowledge management for
quality-driven design and manufacturing in pharmaceuticals. Pharmaceutical Research, v.
28, n. 7, p. 1465–1479, 2011.
KREMER, M. Pharmaceuticals and the developing world. The journal of economic
perspectives, v. 16, n. 4, p. 67–90, 2002.
KROGH, M. A; ENEVOLDSEN, C. Evaluation of effects of metritis management in a
complex dairy herd health management program. Journal of dairy science, v. 97, n. 1, p.
552–61, 2014.
KUEHN, B. M. FDA targets antibiotic use in livestock. JAMA : the journal of the
American Medical Association, v. 304, n. 4, p. 396, 2010.
KUKUSAMUDE, C. et al. High performance liquid chromatography for the simultaneous
analysis of penicillin residues in beef and milk using ion-paired extraction and binary water –
acetonitrile mixture. Talanta, v. 92, p. 38–44, 2012.
KUSSAGA, J. B. et al. Food safety management systems performance in African food
processing companies: A review of deficiencies and possible improvement strategies.
Journal of the Science of Food and Agriculture, v. 94, n. 11, p. 2154–2169, 2014.
KYRALEOU, M. et al. Diffuse reflectance Fourier transform infrared spectroscopy for
simultaneous quantification of total phenolics and condensed tannins contained in grape
seeds. Industrial Crops and Products, v. 74, p. 784–791, 2015.
LAGO, A et al. The selective treatment of clinical mastitis based on on-farm culture results: I.
Effects on antibiotic use, milk withholding time, and short-term clinical and bacteriological
outcomes. Journal of dairy science, v. 94, n. 9, p. 4441–56, set. 2011.
LAMAR, J.; PETZ, M. Development of a receptor-based microplate assay for the detection of
beta-lactam antibiotics in different food matrices. Analytica chimica acta, v. 586, n. 1-2, p.
296–303, 14 mar. 2007.

198
LARA, F. J. et al. Multiresidue method for the determination of quinolone antibiotics in
bovine raw milk by capillary electrophoresis-tandem mass spectrometry. Analytical
chemistry, v. 78, n. 22, p. 7665–73, 15 nov. 2006.
LAWRENCE, Y. X. Pharmaceutical quality by design: Product and process development,
understanding, and control. Pharmaceutical Research, v. 25, n. 4, p. 781–791, 2008.
LEFFINGWELL, J. C. Chirality & bioactivity I.: pharmacology. Leffingwell Reports, v. 3,
n. 1, p. 1–27, 2003.
LEÓN, N. et al. Wide-range screening of banned veterinary drugs in urine by ultra high liquid
chromatography coupled to high-resolution mass spectrometry. Journal of chromatography.
A, v. 1258, p. 55–65, 5 out. 2012.
LESNEY, M. S. APPLICATIONS A Polarizing Point of View. Instruments & Applications,
p. 21–22, 2004.
LI, C. et al. Rapid simultaneous determination of dexamethasone and betamethasone in milk
by liquid chromatography tandem mass spectrometry with isotope dilution. Journal of
chromatography. A, v. 1217, n. 3, p. 411–4, 15 jan. 2010.
LI, Y. et al. Simultaneous extraction and determination of sulfadiazine and sulfamethoxazole
in water samples and aquaculture products using [ Bmim ] BF4(NH4) 3C6H5O7 aqueous
two-phase system coupled with HPLC. Journal of the Iranian Chemical Society, v. 10, n. 2,
p. 339–346, 2013.
LI, Z. et al. An improved ensemble model for the quantitative analysis of infrared spectra.
Chemometrics and Intelligent Laboratory Systems, v. 146, p. 211–220, 2015.
LIGON, B. L. Penicillin: Its Discovery and Early Development. Seminars in Pediatric
Infectious Diseases, v. 15, n. 1, p. 52–57, 2004.
LIMA, V. L. E. OS FÁRMACOS E A QUIRALIDADE: UMA BREVE ABORDAGEM.
Quimica Nova, v. 20, n. 6, p. 657–663, 1997.
LIN, L. H. et al. An LC-MS/MS method for determination of novel fungicide pyraoxystrobin
in rat plasma and tissues: Toxicokinetics and tissue distribution study. Talanta, v. 136, p.
183–189, 2015.
LIU, Y. Y.; HU, C. Q. Construction of predictive models for gentamicin contents in aqueous
solution using specific near infrared spectral regions. Vibrational Spectroscopy, v. 55, n. 2,
p. 241–249, 2011.
LOCK, C. M.; CHEN, L.; VOLMER, D. A. Rapid analysis of tetracycline antibiotics by
combined solid phase microextraction/high performance liquid chromatography/mass
spectrometry. Rapid communications in mass spectrometry : RCM, v. 13, n. 17, p. 1744–
54, jan. 1999.

199
LOPES, R. P. et al. Development and validation (according to the 2002/657/EC regulation) of
a method to quantify sulfonamides in porcine liver by fast partition at very low temperature
and LC-MS/MS. Analytical Methods, v. 3, n. 3, p. 606, 2011.
LOPEZ, M. I. et al. Multiclass determination and confirmation of antibiotic residues in honey
using LC-MS/MS. Journal of agricultural and food chemistry, v. 56, n. 5, p. 1553–9, 12
mar. 2008.
LU, H. Stereoselectivity in drug metabolism. Expert opinion on drug metabolism &
toxicology, v. 3, n. 2, p. 149–158, 2007.
LUO, Z. et al. Molecularly imprinted polymer cartridges coupled to liquid chromatography
for simple and selective analysis of penicilloic acid and penilloic acid in milk by matrix solid-
phase dispersion. Food and Chemical Toxicology, n. June, p. 1–10, 2015.
LUYPAERT, J.; MASSART, D. L.; VANDER HEYDEN, Y. Near-infrared spectroscopy
applications in pharmaceutical analysis. Talanta, v. 72, n. 3, p. 865–883, 2007.
LV, Y.-K. et al. Molecularly Imprinted Solid-Phase Extraction of Tetracyclines Residue from
Milk Using Internal-Surface Reversed-Phase Hybrid Composite Packing Materials. Asian
Journal of Chemistry, v. 26, n. 12, p. 3541–3544, 2014.
LYON, R. C. et al. Near-infrared spectral imaging for quality assurance of pharmaceutical
products: analysis of tablets to assess powder blend homogeneity. AAPS PharmSciTech, v.
3, n. 3, p. E17, 2002.
MA, H. et al. The evolution of productivity performance on China’s dairy farms in the new
millennium. Journal of Dairy Science, v. 95, n. 12, p. 7074–7085, 2012.
MACAROV, C. A. et al. Multi residue determination of the penicillins regulated by the
European Union, in bovine, porcine and chicken muscle, by LC-MS/MS. Food Chemistry, v.
135, n. 4, p. 2612–2621, 2012.
MACNEIL, J. Procaine Benzylpenicillin, 1998.
MADUREIRA, F. D. et al. A multi-residue method for the determination of 90 pesticides in
matrices with a high water content by LC–MS/MS without clean-up. Food Additives &
Contaminants: Part A, v. 29, n. 4, p. 665–678, 2012.
MAGALHÃES, C. G. et al. Test, a microbiological screening test with solvent extraction, for
the detection of antimicrobial residues in poultry muscles. Food Additives & Contaminants:
Part A, v. 29, n. 4, p. 535–540, 2012.
MAIA, P. P.; RATH, S.; REYES, F. G. R. Determination of oxytetracycline in tomatoes by
HPLC using fluorescence detection. Food Chemistry, v. 109, n. 1, p. 212–218, jul. 2008.
MAMANI, M. C. V. et al. Simultaneous determination of tetracyclines in pharmaceuticals by
CZE using experimental design. Talanta, v. 70, n. 2, p. 236–43, 15 set. 2006.

200
MANDENIUS, C. F. et al. Quality-by-design for biotechnology-related pharmaceuticals.
Biotechnology Journal, v. 4, n. 5, p. 600–609, 2009.
MANN, J. Antibiotics: Actions, Origins, ResistanceNatural Product Reports, 2005.
MANTOVANI, L. et al. Stability-indicating RP-HPLC method for analysis of the antibiotic
doripenem in pharmaceutical formulation—comparison to UV spectrophotometry and
microbiological assay. Acta Chromatographica, v. 24, n. 3, p. 367–382, 2012.
MAPA. INSTRUÇÃO NORMATIVA No 9, DE 27 DE JUNHO DE 2003MAPA, 2003.
MAPA. INSTRUÇÃO NORMATIVA No 62, DE 29 DE DEZEMBRO DE 2011, 2011.
MAPA. Legislação relacionada aos produtos de uso veterinário. [s.l: s.n.].
MAPA. INSTRUÇÃO NORMATIVA 13, DE 15 DE JULHO DE 2015 Subprograma de
Monitoramento e Subprograma Exploratório do Plano Nacional de Controle de
Resíduos e Contaminantes - PNCRCDiário Oficial da União, 2015.
MARAZUELA, M. .; MORENO-BONDI, M. . Multiresidue determination of
fluoroquinolones in milk by column liquid chromatography with fluorescence and ultraviolet
absorbance detection. Journal of Chromatography A, v. 1034, n. 1-2, p. 25–32, abr. 2004.
MARIN, T.; MOORE, J. Understanding Near-Infrared SpectroscopyAdvances in
Neonatal Care, 2011.
MARK, H. Chemometrics in near-infrared spectroscopy. Analytica Chimica Acta, v. 223, n.
C, p. 75–93, 1989.
MASQUIO FIORENTINO, F. A. et al. Development and Validation of A Stability-Indicating
Mekc Method for Determination of Flucloxacillin Sodium in Capsules. Current Analytical
Chemistry, v. 10, n. 1, p. 149–157, 2014.
MATUSZEWSKI, B. K.; CONSTANZER, M. L.; CHAVEZ-ENG, C. M. Strategies for the
assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS.
Analytical Chemistry, v. 75, n. 13, p. 3019–3030, 2003a.
MATUSZEWSKI, B. K.; CONSTANZER, M. L.; CHAVEZ-ENG, C. M. Strategies for the
assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS.
Analytical chemistry, v. 75, n. 13, p. 3019–30, 1 jul. 2003b.
MAZUMDER, B.; BHATTACHARYA, S.; YADAV, A. Total quality management in
pharmaceuticals: A review. International Journal of PharmTech Research, v. 3, n. 1, p.
365–375, 2011.
MBINZE, J. K. et al. Development, validation and comparison of NIR and Raman methods
for the identification and assay of poor-quality oral quinine drops. Journal of
Pharmaceutical and Biomedical Analysis, v. 111, p. 21–27, 2015.

201
MEDICAL, I. C. OF. Advances in validation, risk and uncertainty assessment of bioanalytical
methods. Journal of pharmaceutical and biomedical analysis, v. 55, n. 4, p. 848–58, 25
jun. 2011.
MINISTÉRIO DA AGRICULTURA PECUÁRIA E ABASTECIMENTO. Instrução
normativa no 9, de 30 de março de 2007. Plano nacional de controle de resíduos em
produtos de origem animal, 2007.
MINISTÉRIO DA AGRICULTURA PECUÁRIA E ABASTECIMENTO. Instrução
normativa no 62, de 29 de dezembro de 2011. Regulamento técnico de produção,
identidade e qualidade de Leite tipo A, 2011.
MIRANDA, J. M.; RODRÍGUEZ, J. A; GALÁN-VIDAL, C. A. Simultaneous determination
of tetracyclines in poultry muscle by capillary zone electrophoresis. Journal of
chromatography. A, v. 1216, n. 15, p. 3366–71, 10 abr. 2009.
MITCHELL, J. M. et al. Antimicrobial drug residues in milk and meat: causes, concerns,
prevalence, regulations, tests, and test performance. Journal of food protection, v. 61, n. 6,
p. 742–756, 1998.
MIYAGISHIMA, K. et al. Food safety and public health. Food Control, v. 6, n. 5, p. 253–
259, 1995.
MOAN, J. Visible Light and UV Radiation. In: Radiation at home, outdoors and in the
workplace. [s.l: s.n.]. p. 69–85.
MOATS, W. A.; HARIK-KHAN, R. Rapid HPLC Determination of Tetracycline Antibiotics
in Milk. Journal of Agricultural and Food Chemistry, v. 43, n. 4, p. 931–934, abr. 1995.
MONTGOMERY, D. Introduction to statistical quality control. [s.l: s.n.].
MORETA, C.; TENA, M.-T.; KANNAN, K. Analytical method for the determination and a
survey of parabens and their derivatives in pharmaceuticals. Environmental Research, v.
142, p. 452–460, 2015.
MOROS, J.; GARRIGUES, S.; DE LA GUARDIA, M. Quality control Fourier transform
infrared determination of diazepam in pharmaceuticals. Journal of Pharmaceutical and
Biomedical Analysis, v. 43, n. 4, p. 1277–1282, 2007.
MUSSER, J. M. et al. Potential for milk containing penicillin G or amoxicillin to cause
residues in calves. Journal of dairy science, v. 84, n. 1, p. 126–133, 2001.
NATIONS, F. A. A. O. O. T. U. (JECFA);; ORGANIZATON, W. H. (WHO).
PROCEDURES FOR RECOMMENDING MAXIMUM RESIDUE LIMITSRESIDUES
OF VETERINARY DRUGS IN FOODJECFA, , 2000.
NAVRÁTILOVÁ, P.; BORKOVCOVÁ, I.; DRAČKOVÁ, M. Occurrence of Tetracycline,
Chlortetracyclin, and Oxytetracycline Residues in Raw Cow’s Milk. Czech Journal of Food
Sciences, v. 27, n. 5, p. 379–385, 2009.

202
NEBOT, C. et al. Development of a multi-class method for the identification and
quantification of residues of antibiotics, coccidiostats and corticosteroids in milk by liquid
chromatography–tandem mass spectrometry. International Dairy Journal, v. 22, n. 1, p. 78–
85, jan. 2012a.
NEBOT, C. et al. A sensitive and validated HPLC–MS/MS method for simultaneous
determination of seven coccidiostats in bovine whole milk. Food Control, v. 27, n. 1, p. 29–
36, set. 2012b.
NEELAMKAVIL, S. F. et al. The discovery of azepane sulfonamides as potent 11??-HSD1
inhibitors. Bioorganic and Medicinal Chemistry Letters, v. 19, n. 16, p. 4563–4565, 2009.
NG, K.; LINDER, S. W. HPLC separation of tetracycline analogues: comparison study of
laser-based polarimetric detection with UV detection. Journal of chromatographic science,
v. 41, n. 9, p. 460–466, 2003.
NI, Y.; LI, S.; KOKOT, S. Simultaneous voltammetric analysis of tetracycline antibiotics in
foods. Food Chemistry, v. 124, n. 3, p. 1157–1163, 2011.
NICOLAS, A. Contrôle de qualité des antibiotiques dans la Pharmacopée Européenne :
évolution récente dans le cas des aminosides. Annales Pharmaceutiques Francaises, v. 65,
n. 3, p. 174–182, 2007.
NICOLICH, R. S.; WERNECK-BARROSO, E.; MARQUES, M. A. S. Food safety
evaluation: Detection and confirmation of chloramphenicol in milk by high performance
liquid chromatography-tandem mass spectrometry. Analytica Chimica Acta, v. 565, n. 1, p.
97–102, abr. 2006.
NISHA, A. Antibiotic residues-a global health hazard. Veterinary World, v. 1, n. 12, p. 375–
377, 2008.
NONAKA, C. K. V. et al. Occurrence of antimicrobial residues in Brazilian food animals in
2008 and 2009. Food Additives & Contaminants: Part A, v. 29, n. 4, p. 526–534, 2012.
NOUR EL-DIEN, F. A. et al. Extractive spectrophotometric determination of sulphonamide
drugs in pure and pharmaceutical preparations through ion-pair formation with
molybdenum(V) thiocyanate in acidic medium. Journal of Advanced Research, v. 1, n. 3, p.
215–220, jul. 2010.
NOUWS, J. F. M. Suitability of the charm HVS and a microbiological multiplate system for
detection of residues in raw milk at EU maximum residue levels SUITABILITY OF THE
CHARM HVS AND A MICROBIOLOGICAL MULTIPLATE SYSTEM FOR DETECTION
OF RESIDUES IN RAW MILK AT EU MAXIMU. The Veterinary Quarterly, v. 21, n.
January 2013, p. 21–27, 1999.
NOVÁKOVÁ, L.; VLČKOVÁ, H. A review of current trends and advances in modern bio-
analytical methods: Chromatography and sample preparation. Analytica Chimica Acta, v.
656, n. 1-2, p. 8–35, 2009.

203
OKA, H.; ITO, Y.; MATSUMOTO, H. Chromatographic analysis of tetracycline antibiotics
in foods. Journal of chromatography. A, v. 882, n. 1-2, p. 109–33, 16 jun. 2000.
OLIVER, S. P. et al. Efficacy of Extended Ceftiofur Intramammary Therapy for Treatment of
Subclinical Mastitis in Lactating Dairy Cows. Journal of Dairy Science, v. 87, n. 8, p. 2393–
2400, 2004.
ORGANIZATON, W. H. (WHO). Quality assurance of pharmaceuticals. Quality Assurance,
v. 2, n. 1, p. 1–7, 2007.
ORTELLI, D. et al. Comprehensive fast multiresidue screening of 150 veterinary drugs in
milk by ultra-performance liquid chromatography coupled to time of flight mass
spectrometry. Journal of Chromatography B: Analytical Technologies in the Biomedical
and Life Sciences, v. 877, n. 23, p. 2363–2374, 2009.
OZKORUCUKLU, S. P.; SAHIN, Y.; ALSANCAK, G. Determination of sulfamethoxazole
in pharmaceutical formulations by flow injection system/HPLC with potentiometric detection
using polypyrrole electrode. Journal of the Brazilian Chemical Society, v. 22, n. 11, p.
2171–2177, 2011.
PARISOTTO, G. et al. Análise exploratória aplicada no estudo de medicamentos contendo
piroxicam. Revista Brasileira de Ciências Farmacêuticas, v. 41, n. 4, p. 499–505, 2005.
PASCHOAL, R. et al. VALIDAÇÃO DE MÉTODOS CROMATOGRÁFICOS PARA A
DETERMINAÇÃO DE RESÍDUOS DE MEDICAMENTOS VETERINÁRIOS EM
ALIMENTOS. Química Nova, v. 31, n. 5, p. 1190–1198, 2008.
PAYNE, M. A. et al. Extralabel use of penicillin in food animals. Journal of the American
Veterinary Medical Association, v. 229, n. 9, p. 1401–1403, 2006.
PENG, J. et al. Development of a direct ELISA based on carboxy-terminal of penicillin-
binding protein BlaR for the detection of β-lactam antibiotics in foods. Analytical and
Bioanalytical Chemistry, v. 405, n. 27, p. 8925–8933, 2013.
PENGOV, A.; KIRBIS, A. Risks of antibiotic residues in milk following intramammary and
intramuscular treatments in dairy sheep. Analytica Chimica Acta, v. 637, n. 1-2, p. 13–17,
2009.
PENNACCHIO, A. et al. A novel fluorescence polarization assay for determination of
penicillin G in milk. Food Chemistry, v. 190, p. 381–385, 2016.
PÉREZ, M. I. B.; RODRÍGUEZ, L. C.; CRUCES-BLANCO, C. Analysis of different beta-
lactams antibiotics in pharmaceutical preparations using micellar electrokinetic capillary
chromatography. Journal of pharmaceutical and biomedical analysis, v. 43, n. 2, p. 746–
52, 17 jan. 2007.
PÉREZ-SILVA, I. et al. Determination of oxytetracycline in milk samples by polymer
inclusion membrane separation coupled to high performance liquid chromatography.
Analytica chimica acta, v. 718, p. 42–6, 9 mar. 2012.

204
PERRON, G. G. et al. Fighting microbial drug resistance: a primer on the role of evolutionary
biology in public health. Evolutionary Applications, v. 8, n. 3, p. 211–222, 2015.
PIKKEMAAT, M. G. et al. Comparison of three microbial screening methods for antibiotics
using routine monitoring samples. Analytica Chimica Acta, v. 637, n. 1-2, p. 298–304, 2009.
PLUGGE, W.; VAN DER VLIES, C. The use of near infrared spectroscopy in the quality
control laboratory of the pharmaceutical industry. Journal of Pharmaceutical and
Biomedical Analysis, v. 10, n. 10-12, p. 797–803, 1992.
PRAGST, F. Application of solid-phase microextraction in analytical toxicology. Analytical
and Bioanalytical Chemistry, v. 388, n. 7, p. 1393–1414, 2007.
PRANKERD, R. J. Critical Compilation of pKa Values for Pharmaceutical Substances.
[s.l: s.n.]. v. 33
PRUDEN. Production and Transport of Antibiotics from CAFOs. In: Hormones and
Pharmaceuticals Generated by Concentrated Animal Feeding Operations Emerging
Topics in Ecotoxicology. [s.l: s.n.]. p. 63–70.
PUCHADES, R.; CHA, C. Fast screening methods to detect antibiotic residues in food
samples. Trends in Analytical Chemistry, v. 29, n. 9, p. 1038–1049, 2010.
PYÖRÄLÄ, S. Indicators of inflammation in the diagnosis of mastitis. Veterinary Research,
v. 34, n. 5, p. 565–578, 2003.
RAISON-PEYRON, N. et al. Anaphylaxis to beef in penicillin-allergic patient. Allergy, v.
56, n. 8, p. 796–7, ago. 2001.
RAKOTOHARINOME, M. et al. Prevalence of antimicrobial residues in pork meat in
Madagascar. Tropical Animal Health and Production, p. 1–7, 2013.
RAMBLA-ALEGRE, M. et al. Quinolones control in milk and eggs samples by liquid
chromatography using a surfactant-mediated mobile phase. Analytical and bioanalytical
chemistry, v. 400, n. 5, p. 1303–13, maio 2011a.
RAMBLA-ALEGRE, M. et al. Application of a liquid chromatographic procedure for the
analysis of penicillin antibiotics in biological fluids and pharmaceutical formulations using
sodium dodecyl sulphate/propanol mobile phases and direct injection. Journal of
chromatography. A, v. 1218, n. 30, p. 4972–81, 29 jul. 2011b.
RAMIREZ, A. et al. High-performance thin-layer chromatography-bioautography for
multiple antibiotic residues in cow’s milk. Journal of chromatography. B, Analytical
technologies in the biomedical and life sciences, v. 784, n. 2, p. 315–22, 5 fev. 2003.
RANDELL, A. W.; WHITEHEAD, A. J. Codex Alimentarius: food quality and safety
standards for international trade. Revue scientifique et technique (International Office of
Epizootics), v. 16, n. 2, p. 313–321, 1997.

205
RAVIOLO, M. A. et al. Determination of sulfonamides in milk after precolumn derivatisation
by micellar liquid chromatography. Analytica chimica acta, v. 593, n. 2, p. 152–6, 19 jun.
2007.
REDDING, L. E. et al. The use of antibiotics on small dairy farms in rural Peru. Preventive
Veterinary Medicine, v. 113, n. 1, p. 88–95, 2014.
REEVES, T. . Chemical Analysis of Antibiotic Residues in Food. In: WANG, J.; MACNEIL,
J. D.; KAY, J. F. (Eds.). . Chemical Analysis of Antibiotic Residues in Food age. Hoboken,
NJ, USA: John Wiley & Sons, Inc., 2011. p. 1–60.
REGASSA, T. H. et al. Antibiotic Use in Animal Production : Environmental Concerns.
University of Nebraska, v. 196, p. 1–9, 2015.
REJCZAK, T.; TUZIMSKI, T. A review of recent developments and trends in the
QuEChERS sample preparation approach. Open Chemistry, v. 13, n. 1, p. 980–1010, 2015.
RENTSCH, K. M. The importance of stereoselective determination of drugs in the clinical
laboratory. Journal of Biochemical and Biophysical Methods, v. 54, n. 1-3, p. 1–9, 2002.
REPIZO, L. M. et al. A novel and rapid method for determination of natamycin in wines
based on ultrahigh-performance liquid chromatography coupled to tandem mass spectrometry:
validation according to the 2002/657/EC European decision. Analytical and bioanalytical
chemistry, v. 402, n. 2, p. 965–73, jan. 2012.
REYES-HERRERA, I. et al. Concentrations of antibiotic residues vary between different
edible muscle tissues in poultry. Journal of food protection, v. 68, n. 10, p. 2217–2219,
2005.
REZENDE, C. P. et al. Optimisation and validation of a quantitative and confirmatory
method for residues of macrolide antibiotics and lincomycin in kidney by liquid
chromatography coupled to mass spectrometry. Food Additives & Contaminants: Part A, v.
29, n. 4, p. 587–595, 2012.
REZENDE, D. R.; FILHO, N. F.; ROCHA, G. L. Simultaneous determination of
chloramphenicol and florfenicol in liquid milk, milk powder and bovine muscle by LC–
MS/MS. Food Additives & Contaminants: Part A, v. 29, n. 4, p. 559–570, 2012.
REZK, M. R. et al. Multi-residues determination of antimicrobials in fish tissues by HPLC-
ESI-MS/MS method. Journal of chromatography. B, Analytical technologies in the
biomedical and life sciences, v. 978-979, p. 103–10, 26 jan. 2015.
RIDGWAY, K.; LALLJIE, S. P. D.; SMITH, R. M. Sample preparation techniques for the
determination of trace residues and contaminants in foods. Journal of chromatography. A,
v. 1153, n. 1-2, p. 36–53, 15 jun. 2007.
RIEDIKER, S.; DISERENS, J. M.; STADLER, R. H. Analysis of beta-lactam antibiotics in
incurred raw milk by rapid test methods and liquid chromatography coupled with electrospray
ionization tandem mass spectrometry. Journal of agricultural and food chemistry, v. 49, n.
9, p. 4171–6, set. 2001.

206
ROBERSON, J. R. Treatment of clinical mastitis. The Veterinary clinics of North America.
Food animal practice, v. 28, n. 2, p. 271–88, 2012.
ROBERSON, J. R.; WARNICK, L. D.; MOORE, G. Mild to moderate clinical mastitis:
efficacy of intramammary amoxicillin, frequent milk-out, a combined intramammary
amoxicillin, and frequent milk-out treatment versus no treatment. Journal of dairy science,
v. 87, n. 3, p. 583–592, 2004.
RODRIGUEZ, E.; MORENO-BONDI, M. C.; MARAZUELA, M. D. Multiresidue
determination of fluoroquinolone antimicrobials in baby foods by liquid chromatography.
Food Chemistry, v. 127, n. 3, p. 1354–1360, ago. 2011.
RODRIGUEZ, J. A. et al. Determination of tetracyclines in milk samples by magnetic solid
phase extraction flow injection analysis. Microchimica Acta, v. 171, n. 3-4, p. 407–413, 12
set. 2010.
RODRÍGUEZ-DÍAZ, R. C. et al. Determination of fluoroquinolones in milk samples by
postcolumn derivatization liquid chromatography with luminescence detection. Journal of
agricultural and food chemistry, v. 54, n. 26, p. 9670–6, 27 dez. 2006.
RODRIQUEZ, M. et al. Fast determination of underivatized gentamicin C components and
impurities by LC-MS using a porous graphitic carbon stationary phase. Analytical and
Bioanalytical Chemistry, 2015.
ROMERO, C. et al. Detection of cow’s milk in ewe's or goat's milk by HPLC.
Chromatographia, v. 42, n. 3-4, p. 181–184, 1996.
ROSENTHAL, S. A. Alexander Fleming, the Man and the Myth. The Yale journal of
biology and medicine, v. 57, n. 4, p. 718, 1984.
ROZET, E. et al. Advances in validation, risk and uncertainty assessment of bioanalytical
methods. Journal of Pharmaceutical and Biomedical Analysis, v. 55, n. 4, p. 848–858,
2011.
RUDNITSKAYA, A. et al. Assessment of bitter taste of pharmaceuticals with multisensor
system employing 3 way PLS regression. Analytica Chimica Acta, v. 770, p. 45–52, 2013.
S, Y. SIGNAL-TO-NOISE OPTIMIZATION IN POLARIMETRY. Talanta, v. 32, n. 12, p.
1097–1100, 1985.
SALMOND, G.; WELCH, M. Antibiotic resistance: adaptive evolution. The Lancet, v. 372,
n. December 2008, p. S97–S103, dez. 2008.
SAMANIDOU, V. et al. Analytica Chimica Acta Fast extraction of amphenicols residues
from raw milk using novel fabric phase sorptive extraction followed by high-performance
liquid chromatography-diode array detection. Analytica Chimica Acta, v. 855, p. 41–50,
2015.
SAMANIDOU, V.; NISYRIOU, S. Multi-residue methods for confirmatory determination of
antibiotics in milk. Journal of separation science, v. 31, n. 11, p. 2068–90, jun. 2008.

207
SÁNCHEZ, A.; CONTRERAS, A.; CORRALES, J. C. Parity as a risk factor for caprine
subclinical intramammary infection. Small Ruminant Research, v. 31, n. 3, p. 197–201,
1999.
SÁNCHEZ ROJAS, F.; BOSCH OJEDA, C. Recent development in derivative
ultraviolet/visible absorption spectrophotometry: 2004-2008. A review. Analytica Chimica
Acta, v. 635, n. 1, p. 22–44, 2009.
SANG, Z. Y. et al. CODEX-compliant eleven organophosphorus pesticides screening in
multiple commodities using headspace-solid phase microextraction-gas chromatography-mass
spectrometry. Food Chemistry, v. 136, n. 2, p. 710–717, 2013.
SARRAGUÇA, M. C.; LOPES, J. A. Quality control of pharmaceuticals with NIR: From lab
to process line. Vibrational Spectroscopy, v. 49, n. 2, p. 204–210, 2009.
SAUER, C. W.; KIM, J. H. Human milk macronutrient analysis using point-of-care near-
infrared spectrophotometry. Journal of perinatology, v. 31, n. 5, p. 339–343, 2011.
SCAFI, S. H. F.; PASQUINI, C. Identification of counterfeit drugs using near-infrared
spectroscopy. Analyst, v. 126, n. 12, p. 2218–2224, 2001a.
SCAFI, S. H. F.; PASQUINI, C. Identification of counterfeit drugs using near-infrared
spectroscopy. Analyst, v. 126, n. 12, p. 2218–2224, 2001b.
SCHATZ, M. Skin testing and incremental challenge in the evaluation of adverse reactions to
local anesthetics. The Journal of allergy and clinical immunology, v. 74, n. 4 Pt 2, p. 606–
616, 1984.
SCHENCK, F. J.; CALLERY, P. S. Chromatographic methods of analysis of antibiotics in
milk. Journal of chromatography. A, v. 812, n. 1-2, p. 99–109, 3 jul. 1998.
SCHLÜSENER, M. P.; BESTER, K.; SPITELLER, M. Determination of antibiotics such as
macrolides, ionophores and tiamulin in liquid manure by HPLC-MS/MS. Analytical and
Bioanalytical Chemistry, v. 375, n. 7, p. 942–947, 2003.
SECRETARIAT OF THE CODEX ALIMENTARIUS COMMISSION. Codex Alimentarius
Commission Procedural Manual. [s.l: s.n.].
SEIFRTOVÁ, M. et al. An overview of analytical methodologies for the determination of
antibiotics in environmental waters. Analytica Chimica Acta, v. 649, n. 2, p. 158–179, 2009.
SEN, F.; FILAZI, A. Detection of penicillin residues in cow milk using high performance
liquid chromatography with UV-diode Array Detection. Journal Animal and Veterinary
Advances, 2014.
SEYMOUR, E. H.; JONES, G. M.; MCGILLIARD, M. L. Persistence of residues in milk
following antibiotic treatment of dairy cattle. Journal of dairy science, v. 71, n. 8, p. 2292–6,
ago. 1988.

208
SHABIR, G. A. Validation of high-performance liquid chromatography methods for
pharmaceutical analysis: Understanding the differences and similarities between validation
requirements of the US Food and Drug Administration , the US Pharmacopeia and the
International Con. Journal of Chromatography A, v. 987, n. 1-2, p. 57–66, 2003.
SHAH, V. P. et al. Bioanalytical method validation--a revisit with a decade of progress.
Pharmaceutical research, v. 17, n. 12, p. 1551–7, dez. 2000.
SHAO, M. et al. Ionic liquid-based aqueous two-phase system extraction of sulfonamides in
milk. Journal of Chromatography B, v. 961, p. 5–12, 2014.
SHARIATI, S.; YAMINI, Y.; ESRAFILI, A. Carrier mediated hollow fiber liquid phase
microextraction combined with HPLC-UV for preconcentration and determination of some
tetracycline antibiotics. Journal of chromatography. B, Analytical technologies in the
biomedical and life sciences, v. 877, n. 4, p. 393–400, 1 fev. 2009.
SHEN, Y. C. Terahertz pulsed spectroscopy and imaging for pharmaceutical applications: A
review. International Journal of Pharmaceutics, v. 417, n. 1-2, p. 48–60, 2011.
SHERER, J. T. Pharmaceuticals in the environment. American Journal of Health-System
Pharmacy, v. 63, n. 2, p. 174–178, 2006.
SHERIDAN, R. et al. Analysis and occurrence of 14 sulfonamide antibacterials and
chloramphenicol in honey by solid-phase extraction followed by LC/MS/MS analysis.
Journal of agricultural and food chemistry, v. 56, n. 10, p. 3509–16, 28 maio 2008.
SHI, Y.-J. et al. Sorption and biodegradation of tetracycline by nitrifying granules and the
toxicity of tetracycline on granules. Journal of hazardous materials, v. 191, n. 1-3, p. 103–
9, 15 jul. 2011.
SIDDIQUI, M. R.; ALOTHMAN, Z. A.; RAHMAN, N. Analytical techniques in
pharmaceutical analysis: A review. Arabian Journal of Chemistry, abr. 2013.
SIESLER, H. et al. Near-infrared spectroscopy: principles, instruments, applications. [s.l:
s.n.].
SIFUENTES DOS SANTOS, J. et al. Chloramphenicol residues found in milk processed in
Northern Parana, Brazil. Journal für Verbraucherschutz und Lebensmittelsicherheit, p.
2–4, 2015.
SINDAN. MERCADO DE PRODUTOS VETERINÁRIOS. Disponível em:
<http://www.sindan.org.br/sd/base.aspx?controle=8>.
SIQUEIRA, M. E. P. B. Fundamentos do Preparo de Amostra. In: GUANABARA (Ed.). .
Toxicologia Analítica. 1. ed. Rio de Janeiro: [s.n.]. p. 135–141.
SIVAKESAVA, S.; IRUDAYARAJ, J. Rapid determination of tetracycline in milk by FT-
MIR and FT-NIR spectroscopy. Journal of dairy science, v. 85, n. 3, p. 487–93, mar. 2002.

209
SKOV, K. et al. LC–MS analysis of the plasma metabolome—A novel sample preparation
strategy. Journal of Chromatography B, v. 978-979, p. 83–88, 2015.
SMALL, G. W. Chemometrics and near-infrared spectroscopy: Avoiding the pitfalls. TrAC -
Trends in Analytical Chemistry, v. 25, n. 11, p. 1057–1066, 2006.
SOMOGYI, A. et al. Scientific issues related to Codex Alimentarius goals: A review of
principles, with examples. Regulatory Toxicology and Pharmacology, v. 60, n. 1, p. 161–
164, 2011.
SØRENSEN, L. K.; SNOR, L. K. Determination of cephalosporins in raw bovine milk by
high-performance liquid chromatography. Journal of chromatography. A, v. 882, n. 1-2, p.
145–51, 16 jun. 2000.
SPINELLI, S.; GONZALEZ, C. UV-Visible Spectrophotometry of Water and
Wastewater. [s.l: s.n.]. v. 27
SPISSO, B. F. et al. Validation of a high-performance liquid chromatographic method with
fluorescence detection for the simultaneous determination of tetracyclines residues in bovine
milk. Analytica chimica acta, v. 581, n. 1, p. 108–17, 2 jan. 2007.
STALCUP, A M. Chiral separations. Annual review of analytical chemistry (Palo Alto,
Calif.), v. 3, p. 341–363, 2010.
STEFFEE, C. H. Alexander Fleming and penicillin. The chance of a lifetime? North
Carolina medical journal, v. 53, n. 6, p. 308–310, 1992.
STENBERG, B. et al. VISIBLE AND NEAR INFRARED SPECTROSCOPY IN SOIL
SCIENCE. In: Advances in Agronomy, Vol 107. [s.l: s.n.]. v. 107p. 163–215.
STOLKER, A. A M. et al. Fully automated screening of veterinary drugs in milk by turbulent
flow chromatography and tandem mass spectrometry. Analytical and bioanalytical
chemistry, v. 397, n. 7, p. 2841–9, ago. 2010.
STOLKER, A. A. M.; BRINKMAN, U. A. T. Analytical strategies for residue analysis of
veterinary drugs and growth-promoting agents in food-producing animals—a review. Journal
of Chromatography A, v. 1067, n. 1-2, p. 15–53, mar. 2005.
STORME-PARIS, I. et al. Challenging Near InfraRed Spectroscopy discriminating ability for
counterfeit pharmaceuticals detection. Analytica Chimica Acta, v. 658, n. 2, p. 163–174,
2010.
STUART, B. H. Infrared Spectroscopy: Fundamentals and Applications. 1. ed. [s.l.]
Wiley, 2004. v. 8
TAKA, T.; BARAS, M. C.; CHAUDHRY BET, Z. F. Validation of a rapid and sensitive
routine method for determination of chloramphenicol in honey by LC–MS/MS. Food
Additives & Contaminants: Part A, v. 29, n. 4, p. 596–601, 2012.

210
TAN, J. et al. A high-throughput screening strategy for accurate quantification of
erythromycin. Journal of the Taiwan Institute of Chemical Engineers, v. 44, n. 4, p. 538–
544, 2013.
TENENHAUS, M. et al. PLS path modeling. Computational Statistics and Data Analysis,
v. 48, n. 1, p. 159–205, 2005.
TER LAAK, T. L.; GEBBINK, W. A.; TOLLS, J. Estimation of soil sorption coefficients of
veterinary pharmaceuticals from soil properties. Environmental toxicology and chemistry /
SETAC, v. 25, n. 4, p. 933–941, 2006.
THAVARUNGKUL, P. et al. Detecting penicillin G in milk with impedimetric label-free
immunosensor. Biosensors and Bioelectronics, v. 23, n. 5, p. 688–694, 2007.
THE INTERNATIONAL PHARMACOPOEIA. Monographs: Pharmaceutical substances:
Benzathine benzylpenicillin (Benzathini benzylpenicillinum), 2015a. Disponível em:
<http://apps.who.int/phint/en/p/docf/>
THE INTERNATIONAL PHARMACOPOEIA. Monographs: Pharmaceutical substances:
Benzylpenicillin potassium (Benzylpenicillinum kalicum). Disponível em:
<http://apps.who.int/phint/en/p/docf/>.
THORSEN, H. et al. Determination of chloramphenicol residues in meat, seafood, egg,
honey, milk, plasma and urine with liquid chromatography-tandem mass spectrometry, and
the validation of the method based on 2002/657/EC. Journal of chromatography. A, v.
1118, n. 2, p. 226–33, 23 jun. 2006.
TROMBETE, F. M.; SANTOS, R. R. DOS; SOUZA, A. L. R. Antibiotic residues in Brazilian
milk: a review of studies published in recent years. Revista chilena de nutrición, v. 41, n. 2,
p. 191–197, 2014.
TSAI, W.-H. et al. Dispersive solid-phase microextraction method for sample extraction in
the analysis of four tetracyclines in water and milk samples by high-performance liquid
chromatography with diode-array detection. Journal of chromatography. A, v. 1216, n. 12,
p. 2263–9, 20 mar. 2009.
TSAI, W.-H. et al. Determination of tetracyclines in surface water and milk by the
magnesium hydroxide coprecipitation method. Journal of chromatography. A, v. 1217, n. 3,
p. 415–8, 15 jan. 2010.
TURNIPSEED S , ANDERSEN W C, KARBIWNYK M C, MADSON M R, M. K. Multi-
class, multi-residue liquid chromatography/ tandem mass spectrometry screening and
confirmation methods for drug residues in milk. Rapid communications in mass
spectrometry : RCM, v. 22, n. 10, p. 1467–1480, 2008.
UDALOVA, A. Y.; DMITRIENKO, S. G.; APYARI, V. V. Methods for the separation,
preconcentration, and determination of tetracycline antibiotics. Journal of Analytical
Chemistry, v. 70, n. 6, p. 661–676, 2015.

211
UEDA, M. et al. First total synthesis of penmacric acid and its stereoisomer. Tetrahedron
Letters, v. 48, n. 5, p. 841–844, 2007.
ULU, S. T.; TUNCEL, M. Determination of Bupropion Using Liquid Chromatography with
Fluorescence Detection in Pharmaceutical Preparations , Human Plasma and Human Urine.
Journal of Chromatographic Science, v. 25, n. 50, p. 433–439, 2012.
UPSTONE, S. Ultraviolet/visible light absorption spectrophotometry in clinical chemistry.
Encyclopedia of Analytical Chemistry, p. 1699–1714, 2000.
US FOOD AND DRUG ADMINISTRATION. Guidance for industry: Q2B validation of
analytical procedures: methodologyRockville, MD: Nov, 1996. Disponível em:
<http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM128049.pdf>
USFDA. Consumer Updates - Phasing Out Certain Antibiotic Use in Farm Animals. US
Dept. of Health and Human Services, n. December, p. 1–2, 2013.
VAN DE MERBEL, N. C. Quantitative determination of endogenous compounds in
biological samples using chromatographic techniques. TrAC Trends in Analytical
Chemistry, v. 27, n. 10, p. 924–933, nov. 2008.
VAN HOLTHOON, F. et al. Quantitative analysis of penicillins in porcine tissues, milk and
animal feed using derivatisation with piperidine and stable isotope dilution liquid
chromatography tandem mass spectrometry. Analytical and bioanalytical chemistry, v. 396,
n. 8, p. 3027–40, abr. 2010.
VAN ROYEN, G.; DUBRUEL, P.; DAESELEIRE, E. Development and evaluation of a
molecularly imprinted polymer for benzylpenicillin detection and clean-up in milk. Journal
of agricultural and food chemistry, 2014.
VARGAS, M. D. Dorothy Crowfoot Hodgkin: A Life Dedicated to Science. Revista Virtual
de Química, v. 4, n. 1, p. 85–100, 2012.
VARGAS MAMANI, M. C.; REYES REYES, F. G.; RATH, S. Multiresidue determination
of tetracyclines, sulphonamides and chloramphenicol in bovine milk using HPLC-DAD. Food
Chemistry, v. 117, n. 3, p. 545–552, 2009.
VERA-CANDIOTI, L.; OLIVIERI, A. C.; GOICOECHEA, H. C. Development of a novel
strategy for preconcentration of antibiotic residues in milk and their quantitation by capillary
electrophoresis. Talanta, v. 82, n. 1, p. 213–21, 30 jun. 2010.
VETUSCHI, C. et al. Determination of hydrochlorothiazide and irbesartan in pharmaceuticals
by fourth-order UV derivative spectrophotometry. IIFarmaco, v. 60, n. 8, p. 665–670, 2005.
VICH. Guidance for Industry VICH GL49 R ESIDUE K INETICS OF V ETERINARY
D RUGS IN F OOD -P RODUCING A NIMALS : V ALIDATION OF, 2011. Disponível
em:
<http://www.fda.gov/downloads/AnimalVeterinary/GuidanceComplianceEnforcement/Guida
nceforIndustry/UCM207942.pdf>

212
VICH GL39. Specifications: Test Procedures And Acceptance Criteria For New
Veterinary Drug Substances And New Medicinal Products: Chemical
SubstancesVeterinary Medicine, 2006.
VICKERS H R, BAGRATUNI L, A. S. Dermatitis caused by penicillin in milk. The Lancet,
v. 271, n. 7016, p. 351–352, 1958.
WAINWRIGHT, M. Hitler’s penicillin. Perspectives in biology and medicine, v. 47, n. 2, p.
189–198, 2004.
WAKSMAN, S. A. Streptomycin : background , isolation , properties , and utilization.
Eccleiasticus, v. 12, n. 4, p. 370–387, 1952.
WANG, L. et al. Determination of oxytetracycline, tetracycline and chloramphenicol
antibiotics in animal feeds using subcritical water extraction and high performance liquid
chromatography. Analytica Chimica Acta, v. 619, n. 1, p. 54–58, 2008.
WANG, L.; LI, Y.-Q. Simultaneous Determination of Ten Antibiotic Residues in Milk by
UPLC. Chromatographia, v. 70, n. 1-2, p. 253–258, 26 maio 2009.
WANG, Y. et al. Development of a Method for the Analysis of Multiclass Antibiotic
Residues in Milk Using QuEChERS and Liquid Chromatography–Tandem Mass
Spectrometry. Foodborne Pathogens and Disease, v. 12, n. 8, p. 693–703, 2015.
WEAVER, A. A. et al. Paper analytical devices for fast field screening of beta lactam
antibiotics and antituberculosis pharmaceuticals. Analytical Chemistry, v. 85, n. 13, p.
6453–6460, 2013.
WEBER, C. C. et al. Broad-spectrum protein biosensors for class-specific detection of
antibiotics. Biotechnology and bioengineering, v. 89, n. 1, p. 9–17, 5 jan. 2005.
WEI, R. et al. Occurrence of veterinary antibiotics in animal wastewater and surface water
around farms in Jiangsu Province, China. Chemosphere, v. 82, n. 10, p. 1408–1414, 2011.
WETZSTEIN, M. Use and Storage of Veterinary Drugs on the Dairy Farm. Dairy Programs
of Ministry of Agriculture, Fisheries & Food, v. 410, p. 95–97, 2000.
WHARTON, C. W. Infrared spectroscopy of enzyme reaction intermediates. Natural
product reports, v. 17, n. 5, p. 447–453, 2000.
WHO. The use of stems in the selection of International Nonproprietary Names ( INN ) for
pharmaceutical substances (WHO/EMP/QSM/2009.3). WHO Drug Information, p. 1–6,
2006.
WHO. Quality assurance for pharmaceuticalsManagemente sciences for health, 2012.
WONGTANGPRASERT, T.; PALAGA, T.; KOMOLPIS, K. Development of
Oxytetracycline Test Kit Using Enzyme-linked Immunosorbent Assay Technique.
International Proceedings of Chemical, Biological and Environmental Engineering, v.
13, p. 6–10, 2011.

213
WORKMAN, J. NIR spectroscopy calibration Basics. In: Handbook of Near-Infrared
Analysis. [s.l: s.n.]. p. 123 – 149.
WORKMAN, J. J. Interpretive Spectroscopy for Near Infrared. Applied Spectroscopy
Reviews, v. 31, n. 3, p. 251–320, 1996.
WUETHRICH, A.; HADDAD, P. R.; QUIRINO, J. P. Green Sample Preparation for Liquid
Chromatography and Capillary Electrophoresis of Anionic and Cationic Analytes. Analytical
Chemistry, v. 87, p. 4117–4123, 2015.
XIE, Y. et al. Near-infrared spectroscopy quantitative determination of Pefloxacin mesylate
concentration in pharmaceuticals by using partial least squares and principal component
regression multivariate calibration. Spectrochimica Acta - Part A: Molecular and
Biomolecular Spectroscopy, v. 75, n. 5, p. 1535–1539, 2010.
XU, X. et al. Ionic liquid-based microwave-assisted dispersive liquid-liquid microextraction
and derivatization of sulfonamides in river water , honey , milk , and animal plasma.
Analytica Chimica Acta, v. 707, p. 92–99, 2011.
YAMAKI, M. et al. Occurrence of antibiotic residues in milk from Manchega ewe dairy
farms. Journal of dairy science, v. 87, n. 10, p. 3132–3137, 2004.
YANG, X.-Q.; YANG, C.-X.; YAN, X.-P. Zeolite imidazolate framework-8 as sorbent for
on-line solid-phase extraction coupled with high-performance liquid chromatography for the
determination of tetracyclines in water and milk samples. Journal of chromatography. A, v.
1304, p. 28–33, 23 ago. 2013.
YU, W. et al. Rapid and sensitive analysis of cyclobenzaprine by LC–MS/MS: Application to
a pharmacokinetic study of cyclobenzaprine in dog. Asian Journal of Pharmaceutical
Sciences, v. 9, n. 2, p. 117–122, 2014.
ZEITLER, J. A. et al. Terahertz pulsed spectroscopy and imaging in the pharmaceutical
setting--a review. The Journal of pharmacy and pharmacology, v. 59, n. 2, p. 209–223,
2007.
ZENG, A. et al. Determination of azithromycin in raw materials and pharmaceutical
formulations by HPLC coupled with an evaporative light scattering detector. Asian Journal
of Pharmaceutical Sciences, v. 9, n. 2, p. 107–116, 2014.
ZENG K, ZHANG J, WANG Y, W. Z. Development of a Rapid Multi-residue Assay for
Detecting β-lactams Using Penicillin Binding Protein 2x. Biomedical Environmental
Science, v. 26, n. 2, p. 100–109, 2013.
ZHANG, Y. et al. Predicting apple sugar content based on spectral characteristics of apple
tree leaf in different phenological phases. Computers and Electronics in Agriculture, v.
112, p. 20–27, 2015.
ZHOU, Y. et al. Rapid and selective extraction of multiple macrolide antibiotics in foodstuff
samples based on magnetic molecularly imprinted polymers. Talanta, v. 137, p. 1–10, 2015.

214
ZUBATA, P. et al. A new HPLC method for azithromycin quantitation. Journal of
Pharmaceutical and Biomedical Analysis, v. 27, n. 5, p. 833–836, 2002.
ZUCCATO, E. et al. Pharmaceuticals in the environment in Italy: causes, occurrence, effects
and control. Environmental science and pollution research international, v. 13, n. 1, p.
15–21, 2006.
ZUMLA, A.; NAHID, P.; COLE, S. T. Advances in the development of new tuberculosis
drugs and treatment regimens. Nature reviews. Drug discovery, v. 12, n. 5, p. 388–404,
2013.

APÊNDICES

216
APÊNDICE I
PUBLICAÇÃO CIENTÍFICA EM EVENTO INTERNACIONAL E REVISTA DE
CIÊNCIA ABERTA
ARTIGO 1
CABRAL, A. M. et al. The original method for identification of abamectin and ivermectin in
pharmaceutical formulations used in producing animals, using liquid chromatography coupled
to ultra efficiency photodiodes: Risk assessment of pesticide residues in dairy products.
ScienceOpen Posters, p. 1–2, 2014.DOI: 10.14293/P2199-8442.1.SOP-CHEM.PCAOVO.v1
https://www.scienceopen.com/document_file/f6c0bb25-ae42-44d1-99aa-
0e9e5bb51332/ScienceOpen/Poster_Cabral.pdf
TRABALHO 1
CABRAL, A. M. et al. Original method for identification of abamectin and ivermectin in
pharmaceutical formulations used in producing animals, using liquid chromatography coupled
to ultra efficiency photodiodes: Risk assessment of pesticide residues in dairy products. ACS
National Meeting and Exposition, v. 248, p. 258, 2014. http://acselb-529643017.us-west-
2.elb.amazonaws.com/chem/248nm/program/view.php

217
APÊNDICE II
RESUMO EXPANDIDO EM CONGRESSO NACIONAL
TRABALHO 2
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. Uma Abordagem original
sobre aplicação da cromatografia liquida de alta eficiência acoplada a arranjo fotodiodos para
identificação de oxitetraciclina em medicamentos veterinários. Congresso Brasileiro de
Quimica, v. 53, p. 3379, 2013. http://www.abq.org.br/cbq/2013/trabalhos/4/3379-16313.html
TRABALHO 3
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. Identificação de
gentamicina em medicamento veterinário utilizando cromatografia líquida de ultra eficiência
acoplada a fotodiodos. Reunião Anual da SBQ, v. 37, p. 1, 2009.
http://www.sbq.org.br/37ra/cdrom/resumos/T1364-1.pdf
TRABALHO 4
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. Identificação de
Benzilpenicilina G Benzatina em formulações injetáveis: Um diagnóstico preliminar para
análise dos resíduos β-lactâmicos em leite. Congresso Brasileiro de Quimica, v. 53, p. 3385,
2013. http://www.abq.org.br/cbq/2013/trabalhos/10/index.html
TRABALHO 5
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. IDENTIFICAÇÃO
CROMATOGRÁFICA DE OXITETRACICLINA, TRIMETOPRIM, GENTAMICINA,
SULFADOXINA E BENZILPENICILINA G BENZATINA UTILIZADOS EM BOVINOS
PRODUTORES DE LEITE. Congresso Brasileiro de Quimica, v. 54, p. 4959, 2014.
http://www.abq.org.br/cbq/2014/trabalhos/10/4959-16313.html

218
APÊNDICE III
RESUMO EXPANDIDO EM CONGRESSO NACIONAL
TRABALHO 6
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. IDENTIFICAÇÃO DE
TETRACICLINA EM MEDICAMENTOS VETERINÁRIOS A BASE DE ÁCIDO
ASCÓRBICO UTILIZANDO CROMATOGRAFIA LÍQUIDA DE RÁPIDA EFICIÊNCIA
ACOPLADA A ARRANJO DE FOTODIODOS. Congresso Brasileiro de Quimica, v. 54, p.
5287, 2014. http://www.abq.org.br/cbq/2014/trabalhos/4/5287-16313.html
TRABALHO 7
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. COMPLEXATION OF
OXYTETRACYCLINE WHITH OF DRUGS VETERINARY : IDENTIFICATION IN GEL
ELETROFORESIS. XIX ENAAL e V Congresso Latino Americano de Analistas de
Alimentos, p. 146, 2015. http://www.abq.org.br/cbq/2014/trabalhos/4/5287-16313.html

219
APÊNDICE IV
RESUMO EXPANDIDO EM EVENTO LATINO AMERICANO
TRABALHO 8
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. PERFORMANCE METHOD
LIQUID CHROMATOGRAPHY FOR DETERMINATION OF PENICILINN G RESIDUES
IN COW MILK. XIX ENAAL e V Congresso Latino Americano de Analistas de Alimentos,
v. 5, p. 36, 2015.
http://media.wix.com/ugd/7225cb_16d11a7f0ca7499fa5d5ad7b1f5a624e.pdf
TRABALHO 9
CABRAL, A. M.; DE MOURA, M. DE F. V.; DA SILVA, D. R. COMPLEXATION OF
OXYTETRACYCLINE WHITH OF DRUGS VETERINARY : IDENTIFICATION IN GEL
ELETROFORESIS. XIX ENAAL e V Congresso Latino Americano de Analistas de
Alimentos, p. 146, 2015.
http://media.wix.com/ugd/7225cb_16d11a7f0ca7499fa5d5ad7b1f5a624e.pdf

220
APÊNDICE IV
TRABALHOS ACEITOS EM EVENTOS INTERNACIONAIS
TRABALHO 9
CABRAL, A. M. et al. Screening of Penicillin and Aminoglycoside in Therapeutic Drugs Used
in Milk - producing Animals Employing Ultrahigh Performance Liquid Chromatography Diode
Array Detector. World Congress of Food Science & Technology, v. 17, p. 4.7.6, 2014.
http://iufost2014.org/files/IUFoST-detailed-program-2014-08-01.pdf
TRABALHO 10
CABRAL, A. M. et al. New Method Development for Quality Control of Veterinay Drugs the
Basis of Sulfadoxin and Trimethoprim to Guarantee Milk Safety. World Congress of Food
Science & Technology, v. 17, p. 308, 2014. http://iufost2014.org/files/IUFoST-detailed-
program-2014-08-01.pdf

221
APÊNDICE V
ARTIGOS ACEITOS PARA PUBLICAÇÃO
Alessandra Miranda Cabral, Maria de Fátima Vitória de Moura, Djalma Ribeiro da Silva.
Trends of Analysis of Antibiotic Residues in Milk: A Review. Science Journal of Analytical
Chemistry. Special Issue: Analytical Methods for Control & Guarantee Safety Foods. Vol. X,
No. X, 2016, pp. XX-XX. doi: 10.11648/j.XXXX.2016XXXX.XX
Alessandra Miranda Cabral, Maria de Fátima Vitória de Moura, Djalma Ribeiro da Silva.
Development of Method for Quantification of Benzylpenicillin in Goat and Cow Milk by
UFLC-DAD. Science Journal of Analytical Chemistry. Vol. x, No. x, 2016, pp. x-x. doi:
10.11648/j.xxx.xxxxxxxx.xx

222
APÊNDICE VI
CORRIDAS CROMATOGRÁFICAS DOS MEDICAMENTOS UTILIZADOS PARA
CALIBRAÇÃO DOS MODELOS
CORRIDA 1: AVALIAÇÃO DA RESOLUÇÃO DO PICO PENG NA PRESENÇA DE OTC

223
APÊNDICE VI
CORRIDAS CROMATOGRÁFICAS DOS MEDICAMENTOS UTILIZADOS PARA
CALIBRAÇÃO DOS MODELOS (Continuação)
CORRIDA 2. VISUALIZAÇÃO DOS TEMPOS DE RETENÇÃO DE SMZ E TMP

224
APÊNDICE VI
CORRIDAS CROMATOGRÁFICAS DOS MEDICAMENTOS UTILIZADOS PARA
CALIBRAÇÃO DOS MODELOS (Continuação)
CORRIDA 3: VISUALIZAÇÃO DE TEMPO DE RETENÇÃO DE SDX E TMP

225
APÊNDICE VI
CORRIDAS CROMATOGRÁFICAS DOS MEDICAMENTOS UTILIZADOS PARA
CALIBRAÇÃO DOS MODELOS (Continuação)
CORRIDA 4: VISUALIZAÇÃO D ETEMPO DE RETENÇÃO DE GEN NA PRESENÇA
DE BMX E BEN
226
APÊNDICE VI
CORRIDAS CROMATOGRÁFICAS DOS MEDICAMENTOS UTILIZADOS PARA
CALIBRAÇÃO DOS MODELOS (Continuação)
CORRIDA 5: VISUALIZAÇÃO DO TEMPO DE RETENÇÃO DE PENG EM
MEDICAMNETO ISOLADO (MODO GRADIENTE)

227
APÊNDICE VII
CROMATOGRAMAS DOS MEDICAMENTOS UTILIZADOS PARA CALIBRAÇÃO
DOS MODELOS CONSIDERANDO AS ABSORÇÕES NO UV
CROMATOGRAMA 1: PERFIL CROMATOGRÁFICO DE ABAM/IVERM
CONSIDERANDO LMR EM LEITE (MODO ISOCRÁTICO)

228
APÊNDICE VII
CROMATOGRAMAS DOS MEDICAMENTOS UTILIZADOS PARA CALIBRAÇÃO
DOS MODELOS CONSIDERANDO AS ABSORÇÕES NO UV
AVALIAÇÃO 1: PERFIL CROMATOGRÁFICO MIX DE PENG, SMZ, GEN E OTC COM
EM NÍVEIS DE INJEÇÃO 1uL

229
APÊNDICE VII
CROMATOGRAMAS DOS MEDICAMENTOS UTILIZADOS PARA CALIBRAÇÃO
DOS MODELOS CONSIDERANDO AS ABSORÇÕES NO UV (Continuação)
AVALIAÇÃO 1: PERFIL CROMATOGRAFICO MIX DE PENG, SMZ, GEN E OTC COM
EM NÍVEIS DE INJEÇÃO 2 uL
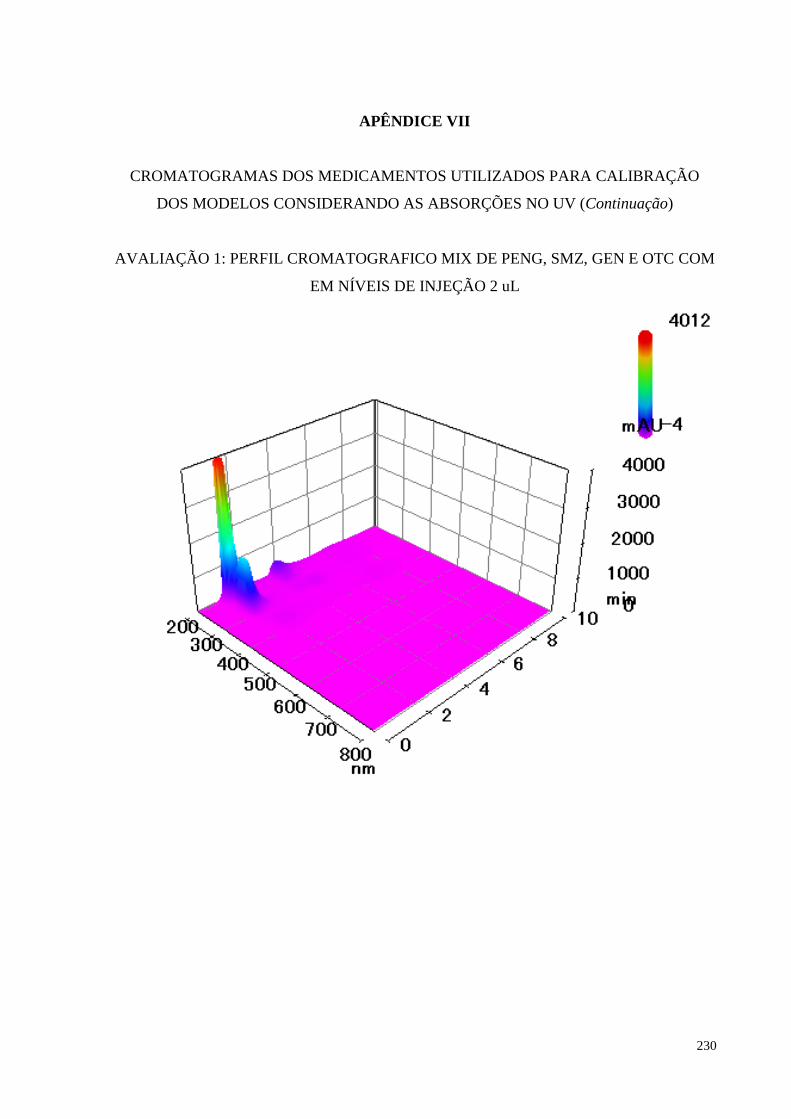
230
APÊNDICE VII
CROMATOGRAMAS DOS MEDICAMENTOS UTILIZADOS PARA CALIBRAÇÃO
DOS MODELOS CONSIDERANDO AS ABSORÇÕES NO UV (Continuação)
AVALIAÇÃO 1: PERFIL CROMATOGRAFICO MIX DE PENG, SMZ, GEN E OTC COM
EM NÍVEIS DE INJEÇÃO 2 uL
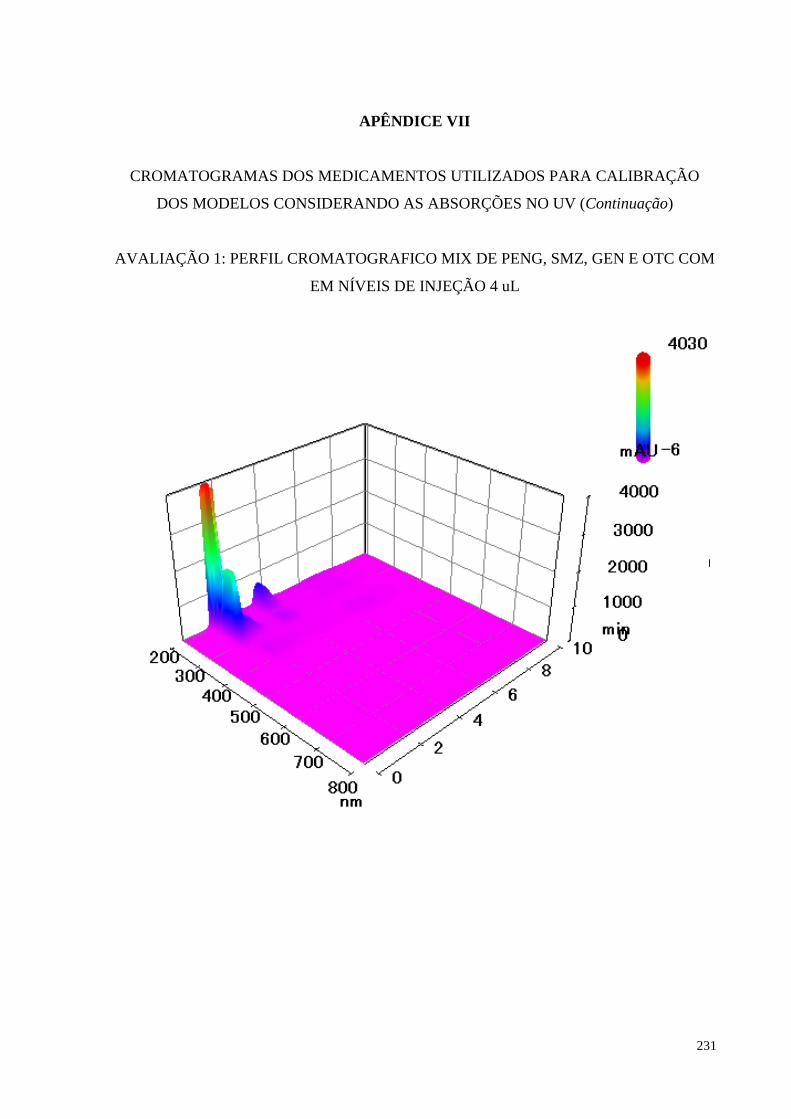
231
APÊNDICE VII
CROMATOGRAMAS DOS MEDICAMENTOS UTILIZADOS PARA CALIBRAÇÃO
DOS MODELOS CONSIDERANDO AS ABSORÇÕES NO UV (Continuação)
AVALIAÇÃO 1: PERFIL CROMATOGRAFICO MIX DE PENG, SMZ, GEN E OTC COM
EM NÍVEIS DE INJEÇÃO 4 uL
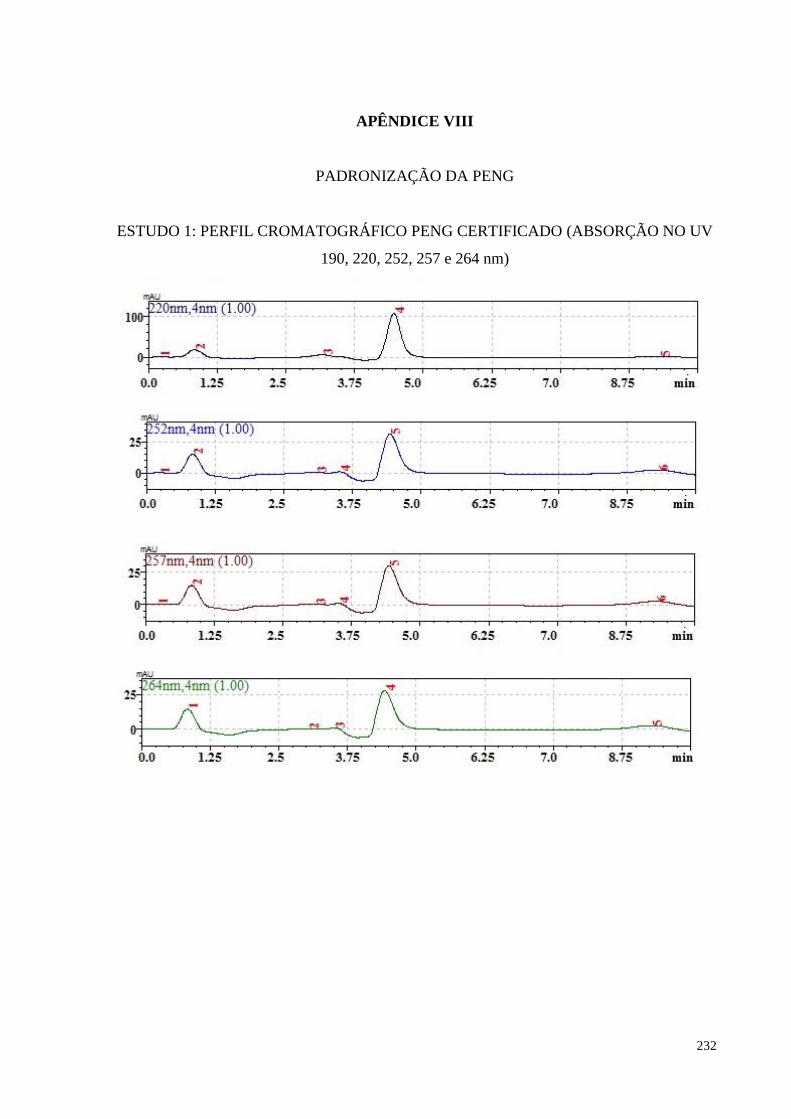
232
APÊNDICE VIII
PADRONIZAÇÃO DA PENG
ESTUDO 1: PERFIL CROMATOGRÁFICO PENG CERTIFICADO (ABSORÇÃO NO UV
190, 220, 252, 257 e 264 nm)
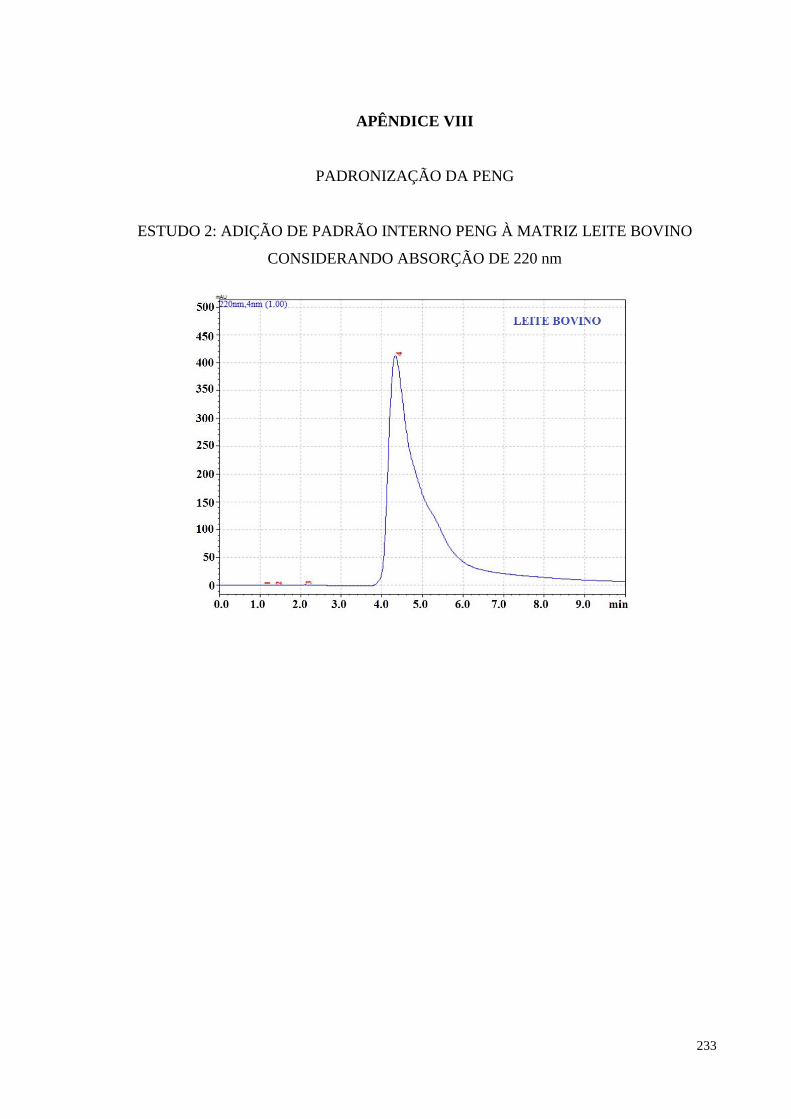
233
APÊNDICE VIII
PADRONIZAÇÃO DA PENG
ESTUDO 2: ADIÇÃO DE PADRÃO INTERNO PENG À MATRIZ LEITE BOVINO
CONSIDERANDO ABSORÇÃO DE 220 nm
234
APÊNDICE VIII
PADRONIZAÇÃO DA PENG (continuação)
ESTUDO 3: ADIÇÃO DE PADRÃO INTERNO PENG À MATRIZ LEITE CAPRINO
CONSIDERANDO ABSORÇÃO DE 220 nm

ANEXOS

236
ANEXO I
Certificado de análise do padrão PENG K+

237
ANEXO I (continuação)
Certificado de análise do padrão PENG K+