análise genômica - unesp: câmpus de botucatu · genoma ou de um genoma completo métodode...
TRANSCRIPT

9/7/2013
1
Análise
Genômica
Guilherme T Valente ([email protected])Centro de Biologia e Engenharia GenéticaUniversidade Estadual de Campinas - UNICAMPCampinas, SP
Uma overview sobre processamento de high throughput sequencing

9/7/2013
2
Sequenciamento de ácidos nucleicos Processo que
determina a sequência de nucleotídeos de uma porção do
genoma ou de um genoma completo
Método de Maxam-Gilbert (1977) Método de Sanger (1977)
Primeiros Métodos First generation sequence
Géis e capilar

9/7/2013
3
Método de Sanger é o mais difundido até os dias atuais
1989 Primeiro gene humano (proteína mutada gera a fibrose cística);
1995 O genoma de Haemophilus influenzae é o primeiro genoma a ser sequenciado;
1996 O genoma de Saccharomyces cerevisiae é o primeiro genoma eucarionte a ser
sequenciado;
1998 O genoma de C. elegans é o primeiro genoma de um pluricelular a ser
sequenciado;
2001 - Primeiro rascunho da sequência do genoma humano é publicado;
2003 - 99% do genoma humano foi sequenciado pelo Projeto do Genoma Humano.
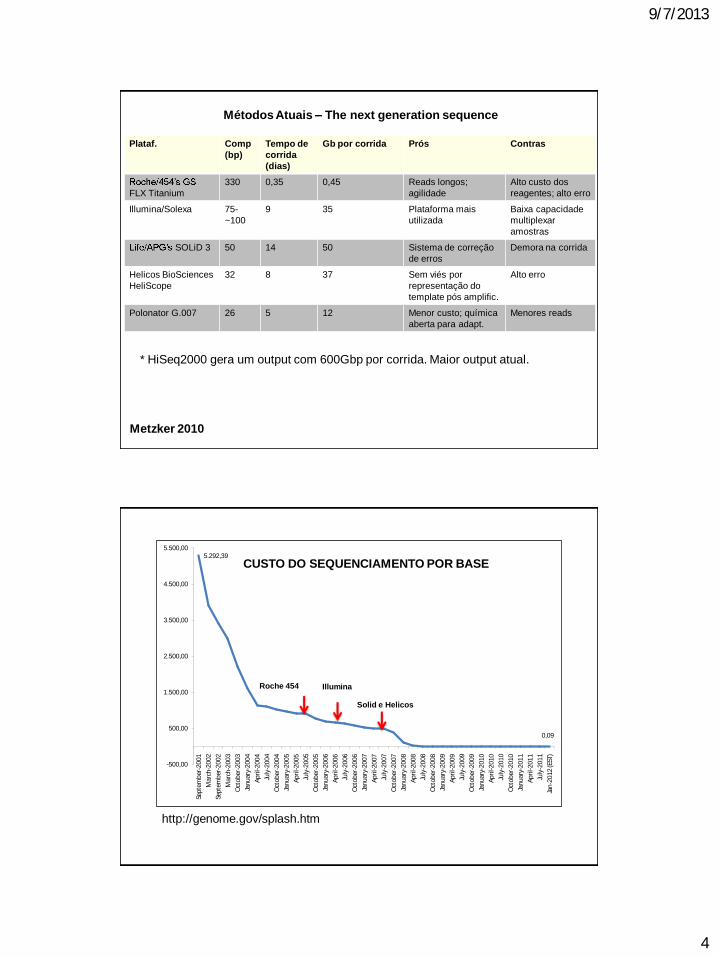
Métodos Atuais The next generation sequence
1 - melhor custo-benefício para projetos high throughput sequencing;
2 custo por bp muito menor;
3 sequenciamento muito mais rápido e eficiente (>40 Gbase/corrida).
4 versatilidade Genomas, expressão gênica, diagnóstico, gen. população,
epigenética, metagenômica, entre outros
9/7/2013
4
Métodos Atuais The next generation sequence
Plataf. Comp
(bp)
Tempo de
corrida
(dias)
Gb por corrida Prós Contras
FLX Titanium
330 0,35 0,45 Reads longos;
agilidade
Alto custo dos
reagentes; alto erro
Illumina/Solexa 75-
~100
9 35 Plataforma mais
utilizada
Baixa capacidade
multiplexar
amostras
SOLiD 3 50 14 50 Sistema de correção
de erros
Demora na corrida
Helicos BioSciences
HeliScope
32 8 37 Sem viés por
representação do
template pós amplific.
Alto erro
Polonator G.007 26 5 12 Menor custo; química
aberta para adapt.
Menores reads
Metzker 2010
* HiSeq2000 gera um output com 600Gbp por corrida. Maior output atual.
5.292,39
0,09
-500,00
500,00
1.500,00
2.500,00
3.500,00
4.500,00
5.500,00
Sep
tem
ber
-20
01
Mar
ch-2
00
2
Sep
tem
ber
-20
02
Mar
ch-2
00
3
Oct
ob
er-2
00
3
Jan
uar
y-2
00
4
Ap
ril-
20
04
July
-20
04
Oct
ob
er-2
00
4
Jan
uar
y-2
00
5
Ap
ril-
20
05
July
-20
05
Oct
ob
er-2
00
5
Jan
uar
y-2
00
6
Ap
ril-
20
06
July
-20
06
Oct
ob
er-2
00
6
Jan
uar
y-2
00
7
Ap
ril-
20
07
July
-20
07
Oct
ob
er-2
00
7
Jan
uar
y-2
00
8
Ap
ril-
20
08
July
-20
08
Oct
ob
er-2
00
8
Jan
uar
y-2
00
9
Ap
ril-
20
09
July
-20
09
Oct
ob
er-2
00
9
Jan
uar
y-2
01
0
Ap
ril-
20
10
July
-20
10
Oct
ob
er-2
01
0
Jan
uar
y-2
01
1
Ap
ril-
20
11
July
-20
11
Jan
-20
12
(EST
)
http://genome.gov/splash.htm
CUSTO DO SEQUENCIAMENTO POR BASE
Roche 454 Illumina
Solid e Helicos

9/7/2013
5
95.263.072
7.950
0
10.000.000
20.000.000
30.000.000
40.000.000
50.000.000
60.000.000
70.000.000
80.000.000
90.000.000
100.000.000
Sep
tem
ber
-20
01
Mar
ch-2
00
2
Sep
tem
ber
-20
02
Mar
ch-2
00
3
Oct
ob
er-2
00
3
Jan
uar
y-2
00
4
Ap
ril-
20
04
July
-20
04
Oct
ob
er-2
00
4
Jan
uar
y-2
00
5
Ap
ril-
20
05
July
-20
05
Oct
ob
er-2
00
5
Jan
uar
y-2
00
6
Ap
ril-
20
06
July
-20
06
Oct
ob
er-2
00
6
Jan
uar
y-2
00
7
Ap
ril-
20
07
July
-20
07
Oct
ob
er-2
00
7
Jan
uar
y-2
00
8
Ap
ril-
20
08
July
-20
08
Oct
ob
er-2
00
8
Jan
uar
y-2
00
9
Ap
ril-
20
09
July
-20
09
Oct
ob
er-2
00
9
Jan
uar
y-2
01
0
Ap
ril-
20
10
July
-20
10
Oct
ob
er-2
01
0
Jan
uar
y-2
01
1
Ap
ril-
20
11
July
-20
11
Jan
-20
12
(EST
)
Genoma humano U$ 3 bi; ~3 bilhões bp; 11 anos; ~12X
Genoma U$ 2.000,00; ~1 bilhão bp; alguns dias; ~30X
CUSTO DO SEQUENCIAMENTO POR GENOMA
http://genome.gov/splash.htm
Roche 454 Illumina
Solid e Helicos
Illumina HiSeq: 600 Gb

9/7/2013
6
Genoma humano - ~3 bilhões = ~800 Alberts, 10
anos
Genoma de um ciclídeo - ~1 bilhão x 30 = ~30
bilhões = ~8.000 Alberts, alguns dias
Antes e hoje
Objetivo rastrear as variações genéticas do genoma humano.
Objetivo gerar conhecimento sobre o genoma de câncer com o intuito de gerar
tratamentos e diagnósticos
Hoje

9/7/2013
7
Métodos do futuro próximo The third generation sequence
* - Perspectiva para os próximos anos. Promete sequenciar um genoma humano em 15 min.
Vantagens ao NGS sem PCR; reads longos
Plataf. Comp
(bp)
Tempo de
corrida
(dias)
Gb por corrida Prós Contras
Pacific
Biosciences
(target release:
2010)
3.000
(alguns
>20.000)
? ? Reads mais longos Alto erro
NanoPore *100.000 *15 min ? Reads mais longos Alto erro
Metzker 2010; Hayden 2012
-time sequencing technology (SMRT)

9/7/2013
8
History of Genome Sequencing The Sanger Method
The sequencing of DNA molecules began in the 1970s with development of the Maxam-Gilbert
ethod, and later the Sanger method. Originally developed by Frederick Sanger in 1975, most
DNA sequencing that occurs in medical and research laboratories today is performed using
sequencers employing variations of the Sanger method. Termed the chain-termination method, it
involves a reaction where chain-terminator nucleotides are labeled with fluorescent dyes,
combined with fragmented DNA, DNA sequencing primers and DNA polymerase. Each
nucleotide in the DNA sequence is labeled with a different dye color and a chromatogram is
produced, with each color representing a different letter in the DNA code A, T, C, or G.
Advances in sequencing technology and computer programming enabled relatively fast and cost
efficient DNA sequencing. However, sequencing of entire genomes of organisms was difficult
and time consuming. At the time the Human Genome Project was officially started in 1990, it was
thought that sequencing the human genome would take fifteen years. The sequence was
released in 2003, although some gaps still exist. The estimated project cost for the entire Human
Genome Project is around $3 billion, although this figure represents a wide range of scientific
activities that went into the project, not exclusively genome sequencing. Optimally, current
sequencers are able to sequence approximately 2.8 million base pairs per 24 hour period.
However, even the smallest organisms such as bacteria are hundreds of millions of base-pairs
long and the human genome is about 3 billion (3,000,000,000) base pairs. At this rate, using the
most modern Sanger sequencers, it takes almost three years to sequence the human genome.
History of Genome Sequencing
Discovery of DNA structure by Watson and Crick (1953)
1950 1960 1970 1980 1990 2000
Development of Sanger Sequencing by Frederick
Sanger (1977)
Polymerase Chain Reaction (PCR) invented by Kary Mullis
(1983) Human Genome
Project Initiated
454 Life Sciences Corp. is
founded
Sequence of Human Genome Completed
(2003)
Development of Pyrosequencing by
454 Sequencing was used to sequence the following genomes (along with many others): - Barley (plant) - Neanderthal - Helicobacter pylori (H.pylori - bacteria that cause gastric ulcers) - HIV mutant strains
Source: U.S. Department of Energy Office of Science, Systems Biology for Energy and the Environment, Human Genome Project Information Base URL: http://genomics.energy.gov/
2012-2013
Reads >10 kbp
Kahn SD. Science 2011

9/7/2013
9
Genoma
referência
NGS
Reads
Análise dos reads
De novo
DNA/RNA
Bibliotecas

9/7/2013
10
Maiores
problemas dos
sequenciamentos
em larga escala!!!Estocagem
Montagem
Análise
Estocagem

9/7/2013
11
Estocagem
Genoma de referência De novo assemblyX
1 - Perda de cópias de genes surgidos recentementes.
2 Algumas reg. podem não existir no gen. ref.
3 - Problemas na montagem dos reads referentes a elementos
repetitivos (problemas principal nos eucariontes).
Montagem

9/7/2013
12
Análise X- Não existe um programa aplicável a responder todas as perguntas biológicas;
- Programas novos emergem diariamente.
Genoma
referência
NGS
Reads
Análise dos reads
De novo
DNA/RNA
Bibliotecas

9/7/2013
13
Utilizando um genoma de referência
Short-read mappers
- Muito utilizado atualmente nos projetos genomas e transcriptomas;
- Recomendado quando há uma boa referência (genoma ou transcriptoma).
NATURE BIOTECHNO LO GY VOLUME 27 NUMBER 5 MAY 2009 455
to understand why the mapping problems are
computationally difficult, which difficulties
have been overcome and what challenges and
opportunities remain.
Challenges of mapping short reads
The first challenge is a practical one: if the
reference genome is very large, and if we have
billions of reads, how quickly can we align the
reads to the genome? Sequence alignment is a
classic problem in bioinformatics, supported
by a large body of literature describing different
variants for both exact and inexact alignment.
As a practical matter, the task of mapping
billions of sequences to a mammalian-sized
genome calls for extraordinarily efficient algo-
rithms, in which every bit of memory is used
optimally or near optimally.
The second challenge is strategic: if a read
comes from a repetitive element in the refer-
ence, a program must pick which copy of the
repeat the read belongs to. Because this may
be impossible to decide with confidence, the
program may choose to report multiple pos-
sible locations or to pick a location heuristi-
cally. Sequencing errors or variations between
the sequenced chromosomes and the reference
genome exacerbate this problem, because the
In this case, to make sense of the reads, their
positions within the reference sequence must
be determined. This process is known as align-
ing or mapping the read to the reference. In
one version of the mapping problem, reads
must be aligned without allowing large gaps in
the alignment (we describe this in more detail
in the Short-read mappers section below).
A more difficult version of the problem arises
primarily in RNA-Seq, in which alignments are
allowed to have large gaps corresponding to
introns (discussed below in the Spliced-read
mappers section).
These read mapping problems are certainly
not new, and there are many programs that
perform both spliced and unspliced alignment
for the older Sanger-style capillary reads. Even
so, these programs neither scale up to the much
greater volumes of data produced by short-
read sequencers nor scale down to the short
read lengths. Aligning the reads from ChIP-Seq
or RNA-Seq experiments can take hundreds or
thousands of central processing unit (CPU)
hours using conventional software tools such
as BLAST or BLAT. Fortunately, new software
packages designed to meet the computational
challenges of short-read sequencing are quickly
appearing. Before choosing one, it is essential
A new generation of DNA sequencers that can
rapidly and inexpensively sequence billions
of bases is transforming genomic science. These
new machines are quickly becoming the tech-
nology of choice for whole-genome sequencing
and for a variety of sequencing-based assays,
including gene expression, DNA-protein inter-
action, human resequencing and RNA splicing
studies . For example, the RNA-Seq proto-
col, in which processed mRNA is converted to
cDNA and sequenced, is enabling the identifi-
cation of previously unknown genes and alter-
native splice variants; the ChIP-Seq approach,
which sequences immunoprecipitated DNA
fragments bound to proteins, is revealing net-
works of interactions between transcription
factors and DNA regulatory elements4; and
the whole-genome sequencing of tumor cells
is uncovering previously unidentified cancer-
initiating mutations5.
One of the challenges presented by the new
sequencing technology is the so-called read
mapping problem. Sequencing machines made
by Illumina of San Diego, Applied Biosystems
(ABI) of Carlsbad, California, and Helicos
of Cambridge, Massachusetts, produce short
sequences of 25 100 base pairs (bp), called
reads, which are sequence fragments read
from a longer DNA molecule present in the
sample that is fed into the machine. In con-
trast to whole-genome assembly, in which these
reads are assembled together to reconstruct a
previously unknown genome, many of the
next-generation sequencing projects begin
with a known, or so-called reference, genome.
How to map billions of short reads onto genomesCole Trapnell & Steven L Salzberg
Mapping the vast quantities of short sequence fragments produced by next-generation sequencing platforms is a
challenge. What programs are available and how do they work?
Cole Trapnell and Steven L. Salzberg are at the
Center for Bioinformatics and Computational
Biology, University of Maryland, College Park,
Maryland, USA.
e-mail: [email protected] or [email protected]
Table 1 A selection of short-read analysis software
Program Website
Open
source?
Handles ABI color
space?
Maximum read
length
Bowtie http://bowtie.cbcb.umd.edu Yes No None
BWA http://maq.sourceforge.net/bwa-man.shtm l Yes Yes None
Maq http://maq.sourceforge.net Yes Yes 127
Mosaik http://bioinformatics.bc.edu/marthlab/Mosai k No Yes None
Novoalign http://www.novocraft.com No No None
SOAP2 http://soap.genomics.org.c n No No 60
ZOOM http://www.bioinfor.com No Yes 240
PRI M ER
NATURE BIOTECHNO LO GY VOLUME 27 NUMBER 5 MAY 2009 455
to understand why the mapping problems are
computationally difficult, which difficulties
have been overcome and what challenges and
opportunities remain.
Challenges of mapping short reads
The first challenge is a practical one: if the
reference genome is very large, and if we have
billions of reads, how quickly can we align the
reads to the genome? Sequence alignment is a
classic problem in bioinformatics, supported
by a large body of literature describing different
variants for both exact and inexact alignment.
As a practical matter, the task of mapping
billions of sequences to a mammalian-sized
genome calls for extraordinarily efficient algo-
rithms, in which every bit of memory is used
optimally or near optimally.
The second challenge is strategic: if a read
comes from a repetitive element in the refer-
ence, a program must pick which copy of the
repeat the read belongs to. Because this may
be impossible to decide with confidence, the
program may choose to report multiple pos-
sible locations or to pick a location heuristi-
cally. Sequencing errors or variations between
the sequenced chromosomes and the reference
genome exacerbate this problem, because the
In this case, to make sense of the reads, their
positions within the reference sequence must
be determined. This process is known as align-
ing or mapping the read to the reference. In
one version of the mapping problem, reads
must be aligned without allowing large gaps in
the alignment (we describe this in more detail
in the Short-read mappers section below).
A more difficult version of the problem arises
primarily in RNA-Seq, in which alignments are
allowed to have large gaps corresponding to
introns (discussed below in the Spliced-read
mappers section).
These read mapping problems are certainly
not new, and there are many programs that
perform both spliced and unspliced alignment
for the older Sanger-style capillary reads. Even
so, these programs neither scale up to the much
greater volumes of data produced by short-
read sequencers nor scale down to the short
read lengths. Aligning the reads from ChIP-Seq
or RNA-Seq experiments can take hundreds or
thousands of central processing unit (CPU)
hours using conventional software tools such
as BLAST or BLAT. Fortunately, new software
packages designed to meet the computational
challenges of short-read sequencing are quickly
appearing. Before choosing one, it is essential
A new generation of DNA sequencers that can
rapidly and inexpensively sequence billions
of bases is transforming genomic science. These
new machines are quickly becoming the tech-
nology of choice for whole-genome sequencing
and for a variety of sequencing-based assays,
including gene expression, DNA-protein inter-
action, human resequencing and RNA splicing
studies . For example, the RNA-Seq proto-
col, in which processed mRNA is converted to
cDNA and sequenced, is enabling the identifi-
cation of previously unknown genes and alter-
native splice variants; the ChIP-Seq approach,
which sequences immunoprecipitated DNA
fragments bound to proteins, is revealing net-
works of interactions between transcription
factors and DNA regulatory elements4; and
the whole-genome sequencing of tumor cells
is uncovering previously unidentified cancer-
initiating mutations5.
One of the challenges presented by the new
sequencing technology is the so-called read
mapping problem. Sequencing machines made
by Illumina of San Diego, Applied Biosystems
(ABI) of Carlsbad, California, and Helicos
of Cambridge, Massachusetts, produce short
sequences of 25 100 base pairs (bp), called
reads, which are sequence fragments read
from a longer DNA molecule present in the
sample that is fed into the machine. In con-
trast to whole-genome assembly, in which these
reads are assembled together to reconstruct a
previously unknown genome, many of the
next-generation sequencing projects begin
with a known, or so-called reference, genome.
How to map billions of short reads onto genomesCole Trapnell & Steven L Salzberg
Mapping the vast quantities of short sequence fragments produced by next-generation sequencing platforms is a
challenge. What programs are available and how do they work?
Cole Trapnell and Steven L. Salzberg are at the
Center for Bioinformatics and Computational
Biology, University of Maryland, College Park,
Maryland, USA.
e-mail: [email protected] or [email protected]
Table 1 A selection of short-read analysis software
Program Website
Open
source?
Handles ABI color
space?
Maximum read
length
Bowtie http://bowtie.cbcb.umd.edu Yes No None
BWA http://maq.sourceforge.net/bwa-man.shtm l Yes Yes None
Maq http://maq.sourceforge.net Yes Yes 127
Mosaik http://bioinformatics.bc.edu/marthlab/Mosai k No Yes None
Novoalign http://www.novocraft.com No No None
SOAP2 http://soap.genomics.org.c n No No 60
ZOOM http://www.bioinfor.com No Yes 240
PRI M ER
Qual programa?
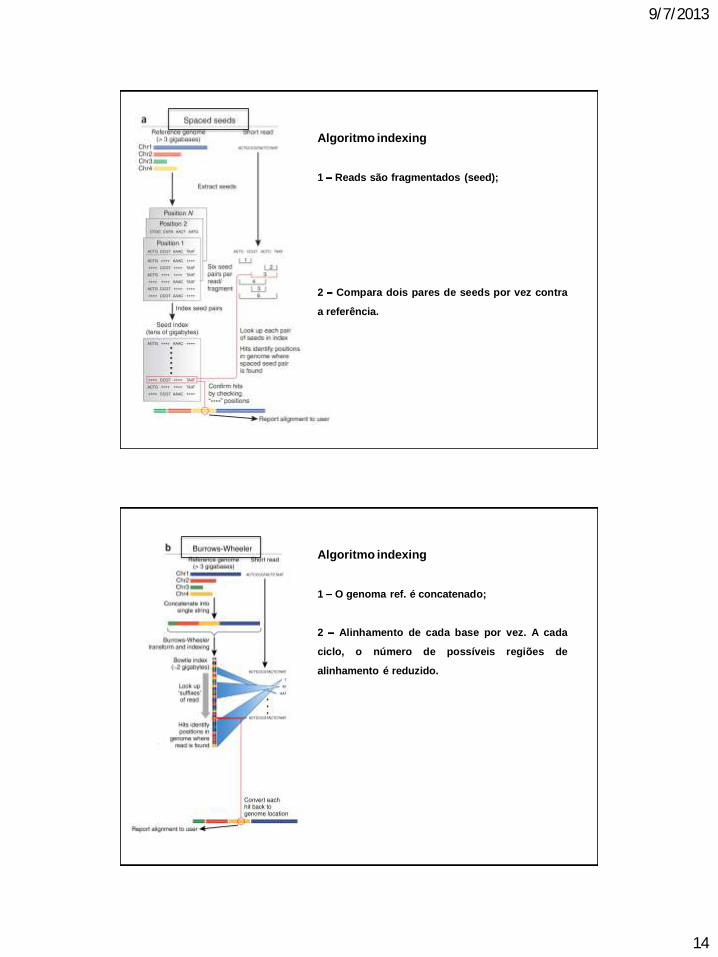
9/7/2013
14
Algoritmo indexing
1 Reads são fragmentados (seed);
2 Compara dois pares de seeds por vez contra
a referência.
Algoritmo indexing
1 O genoma ref. é concatenado;
2 Alinhamento de cada base por vez. A cada
ciclo, o número de possíveis regiões de
alinhamento é reduzido.

9/7/2013
15
456 VOLUME 27 NUMBER 5 MAY 2009 NATURE BIOTECHNO LO GY
Short-read mappers
Such programs as Maq and Bowtie (Table 1)
use a computational strategy known as index-
ing to speed up their mapping algorithms. Like
the index at the end of a book, an index of a
large DNA sequence allows one to rapidly find
shorter sequences embedded within it. Maq is
based on a straightforward but effective strategy
called spaced seed indexing6 (Fig. 1a). In this
strategy, a read is divided into four segments of
equal length, called the seeds. If the entire read
aligns perfectly to the reference genome, then
clearly all of the seeds will also align perfectly.
If there is one mismatch, however, perhaps due
to a single-nucleotide polymorphism (SNP),
then it must fall within one of the four seeds,
but the other three will still match perfectly.
Using similar reasoning, two mismatches will
fall in at most two seeds, leaving the other two
to match perfectly. Thus, by aligning all pos-
sible pairs of seeds (six possible pairs) against
the reference, it is possible to winnow the list of
candidate locations within the reference where
the full read may map, allowing at most two
mismatches. Maqs spaced seed index enables
it to perform this winnowing operation very
efficiently. The resulting set of candidate reads
is typically small enough that the rest of the
read that is, the other two seeds that might
contain the mismatches may be individually
checked against the reference.
Bowtie takes an entirely different approach,
borrowing a technique originally developed
for compressing large files called the Burrows-
Wheeler transform. Using this transform, the
index for the entire human genome fits into
less than two gigabytes of memory (an amount
that is commonly available on todays desktop
and even laptop computers) in contrast to a
spaced seed index, which may require over 50
gigabytes and yet reads can still be aligned
efficiently. Bowtie aligns a read one character
at a time to the Burrows-Wheeler transformed
genome (Fig. 1b). Each successively aligned
new character allows Bowtie to winnow the
list of positions to which the read might map.
If Bowtie cannot find a location where a read
aligns perfectly, the algorithm backtracks
to a previous character of the read, makes a
substitution and resumes the search. In effect,
the Burrows-Wheeler transform enables
Bowtie to conquer the mapping problem by
first solving a simple subproblem align one
character and then building on that solution
to solve a slightly harder problem align two
characters and then continuing on to three
characters, and so on, until the entire read has
been aligned. Bowties alignment algorithm is
substantially more complicated than Maqs, but
Bowties alignment speed is more than 30-fold
faster7.
using traditional alignment algorithms such as
BLAST or BLAT, but such grids are not acces-
sible to everyone. To reduce the computing cost
of analysis for sequencing-based assays and to
make them available to all investigators, we and
others have created a new generation of align-
ment programs capable of mapping hundreds
of millions of short reads on a single desktop
computer. Vendors of sequencing machines
provide specialized mapping software, such as
the ELAND program from Illumina, but in this
article we focus on third-party packages, some of
which are free and open source. These programs
are built on algorithms that exploit features of
short DNA sequencing reads to map millions of
reads per hour while minimizing both process-
ing time and memory requirements.
alignment between the read and its true source
in the genome may actually have more differ-
ences than the alignment between the read
and some other copy of the repeat. The spliced
mapping problem faces this same challenge but
is further complicated by the possible presence
of intron-sized gaps.
DNA sequencers from Illumina, ABI, Roche
(of Basel, Switzerland), Helicos and other compa-
nies produce millions of reads per run. Complete
assays may involve many runs, so an investigator
may need to map millions or billions of reads
to a genome. For example, the recent cancer
genome sequencing project by Ley et al.5 gener-
ated nearly 8 billion reads from 132 sequencing
runs. A large, expensive computer grid might
map the reads from this experiment in a few days
Burrows-Wheeler
Reference genome(> 3 gigabases)
Bowtie index(~2 gigabytes)
Concatenate intosingle str ing
Burrows-Wheelertransform and indexing
ACTCCCGTACTCTAAT
AAT
AT
T
ACTCCCGTACTCTAAT
Convert eachhit back togenome location
Position N
Position 2
Index seed pairs
Position 1
Seed index(tens of gigabytes)
Hits identify positionsin genome wherespaced seed pairis found
Look up each pairof seeds in index
Six seedpairs perread/fragment
Confirm hitsby checking
**** positions
ACTC CCGT ACTC TAAT
Report alignment to user
Chr1Chr2Chr3Chr4
Spaced seeds
Reference genome(> 3 gigabases)
Short read
Extract seeds
ACTCCCGTACTCTAAT
Short read
ACTCCCGTACTCTAAT
Look up
of read
Hits identifypositions in
genome whereread is found
1
2
3
4
5
6
CTGC CGTA AACT AATG
ACTG CCGT AAAC TAAT
ACTG **** AAAC ****
ACTG **** AAAC ****
**** CCGT **** TAAT
ACTG **** **** TAAT
**** **** AAAC TAAT
ACTG CCGT **** ****
**** CCGT AAAC ****
**** CCGT **** TAAT
ACTG **** **** TAAT
**** CCGT AAAC ****
ba
Chr1Chr2Chr3Chr4
Figure 1 Two recent algorithmic approaches for aligning short (20 200-bp) sequencing reads.
(a) Algorithms based on spaced-seed indexing, such as Maq, index the reads as follows: each position
in the reference is cut into equal-sized pieces, called seeds and these seeds are paired and stored
in a lookup table. Each read is also cut up according to this scheme, and pairs of seeds are used as
keys to look up matching positions in the reference. Because seed indices can be very large, some
algorithms (including Maq) index the reads in batches and treat substrings of the reference as queries.
(b) Algorithms based on the Burrows-Wheeler transform, such as Bowtie, store a memory-efficient
representation of the reference genome. Reads are aligned character by character from right to left
against the transformed string. With each new character, the algorithm updates an interval (indicated
by blue beams ) in the transformed string. When all characters in the read have been processed,
alignments are represented by any positions within the interval. Burrows-Wheeler based algorithms can
run substantially faster than spaced seed approaches, primarily owing to the memory efficiency of the
Burrows-Wheeler search. Chr., chromosome.
PRI M ER
Alinha 70-75% dos reads
Desafios:
1 Alinhamento de indels;
2 Ajustamento dos parâmetros;
3 - Mapeamento correto dos TEs ou outros DNAs repetitivos: a cópia
alinhada é realmente pertencente aquela posição?
4 Péssima escolha se não houver uma referência com qualidade.
Problemas?

9/7/2013
16
Montagem de genoma - de novo assembly
Algoritmos Prós Contras
Overlaps Mais sensíveis Mais complexos
Shared K-mers Menos complexos Menos sensíveis
Assemblies de NGS usando teoria de grafos
overlap-layout-consensus assemblers
De Bruijn graph assemblers
Greedy assemblers
Vértices (reads)
Arestas (overlapping)

9/7/2013
17
Leonard Euler 1736
Encontrar um caminho pela cidade que cruze cada ponte somente uma vez
Teoria de grafos(redes)
33
Problema matemático - As sete pontes de Königsberg (ex-capital da Prussia)
O algoritmo baseado em overlaps-layout-consensus (OLC) compara todos os reads
entre si
Vértices (reads)
Arestas (overlapping)
1 Todos os reads são comparados entre si;
2 Reads que se sobrepõem são selecionados;
3 A sequencia consenso é determinada.

9/7/2013
18
O algoritmo baseado em greedy assemblers alinha par-a-par todos os reads e os dois
maiores overlapping são escolhidos.
Algoritmo baseado em De Bruijn graph. Nós representam prefixos e sulfixos de
um alfabeto finito, enquanto as arestas são os overlapping entre essas letras

9/7/2013
19
De Bruijn graph é baseado no problema das sete pontes de Königsberg .
A alta exigência de RAM é devido ao tamanho do grafo.

9/7/2013
20
As sequências repetitivas introduzem um grande número de caminhos válidos
RAM, tempo e acurácia.
A Practical Compariso
n of De Novo Genome Assembly
Software Tools for Next-Generation Sequencing
Technologies
Wenyu Zhang, JiajiaChen, Yang Yang, Yifei Tang, Jin
g Shang, Bairong Shen*
Center for Systems Biology, Soochow University, Suzhou, Jiangsu, China
Abst ract
The advent of next-generation sequencing technologies is accompanied with
the development of many whole-genome
sequence assembly methods and software, especially for denovo fragment assembly. Due to the poor knowledge about the
applicability
and performanceof these softw
are tools, choosing a befitting assembler becomes a tough task. Here, we
provide the information of adaptivityfor each
program, then above all, compare the performanceof eight distin
ct tools
against eight groups of simulated datasets from
Solexa sequencing platform. Considering the computational time,
maximum random access memory (RAM) occu
pancy, assembly accuracy and integrity, our study indicate that strin
g-based
assemblers,overlap-layout-co
nsensus (OLC) assemblers are well-suited for very short reads and longer reads of small
genomes respectively. For large datasets of more than hundred millio
ns of short reads, De Bruijn graph-based assemblers
would be more appropriate. In terms of software implementation, strin
g-based assemblers are superior to graph-based
ones, of whichSOAPdenovo is complex for the creation of configuration file. Our compariso
n study will assist researchers in
selecting a well-su
ited assembler and offer essential information for the improvement of existing assemblers or the
developing of novel assemblers.
Citat ion: Zhang W, Chen J, Yang Y, Tang Y, Shang J, et al. (2011) A Practical Compariso
n of De Novo Genome Assembly Software Tools for Next-Generation
Sequencing Technologies. PLoS ONE 6(3): e17915. doi:10.1371/journal.pone.0017915
Editor: I. King Jordan, Georgia Institute of Technology, United States of America
Received November 20, 2010; Accept ed February 15, 2011; Published March14, 2011
Copyright :2011 Zhang et al. This is an open-acce
ss article distri
buted under the terms of the Creative Commons Attribution License, which
permits
unrestricted use, distri
bution, and reproduction in any medium, provided the original author and source
are credited.
Funding: This work was supported by grants from the National Nature ScienceFoundation of China (31040020) and the National 973 Programs of China
(2007CB947002, 2010CB945600). The funders had no role in study design, data collection and analysis,
decision to publish
, or preparation of the manuscript.
Compet ing Interests: The authors have declared that no competing interestsexist.
* E-mail: [email protected]
Introduct ion
Inrece
nt years,the next-generation sequencing (or deep
sequencing) technologies
have beenevolving rapidly, with
the
potential toaccel
eratebiological and
biomedical research
dramatically
[1]. However,the downstre
amanalysis
of short
reads datasetsafter
sequencing is a tough task; one of the biggest
challenges for the analysis of high throughput sequencing reads is
the whole genome assembly. DNA fragment assem
bly has a long
history since the emergence of the first
generation of sequencing
technologies
[2,3]. The assembly proced
ure becomes especia
lly
difficult when
tackling short and high throughput reads with
different erro
r profiles[4]. Acco
rding to the existence of refe
rence
information, theassembly proced
ure can beclassified asrefe
rence-
guidegenome assembly and denovo
genome assembly, of which
the
former is relatively
toilless with
the aid of reference genome or
proteome information while the laterin more challen
ging. Herein,
we focus on the comparison and evaluation of tools for de novo
assembly of genome sequence.
The genome assemblersgenerally take a file
of short sequence
reads and a fileof quality-value as the input. Since the quality-
value filefor the high throughput short reads is usually
highly
memory-intensive, onlya
fewassemblers
, forexample,
SHARCGS [5], and ALLPATHS-LG[6] adopt it in
the
posterior assembly
process. For the sake of computational
memory saving and convenience of data inquiry, high-through-
put short reads data is always initially formattedto specif
ic data
structure. Curren
tly, existing data stru
cture for this usage can be
predominantly
classified
into two categories: strin
g-basedmodel
and graph-basedmodel. We theref
orecall the corresponding
assemblers as strin
g-basedand graph-based. Strin
g-basedassem
-
blers, implemented with
Greedy-extension algorithm, are mainly
reported for the assem
bly of small genomes [5,7,8,9], while the
latter ones are designed aiming at handling complexgenomes
[10,11,12].
One of the most intractable bottlenecks for practic
al assembly
of next - generation short reads is how toprocess rep
etitive
fragmentsfrom
complicated
genomes,especia
llyeukaryote
genomes.Intuitively, sequencing with
longer reads is a potential
solution, whileit becomes
impractical with
limit current of
sequencing technology. The paired-end (PE) sequencing can, to
someextent, compensate for read length [13]. Several assemblers
,
suchas SSAKE [9], SOAPdenovo [11], AbySS [12], Velvet
[14,15], exploit PE sequencing information to reduce gaps from
assembled contigs. Another
big challenge for the assembly of
short reads is the intensive computational time req
uirement. To
decreasethe tim
e costof the assembly
procedure,thread
parallelization is
implemented
ina couple
of graph-based
assemblers [11,12].
At our lastenumeration, 24 academic
denovo
genome
assemblers,
eachpossessin
g itsown range of applica
tion, are
developedfor short reads datasets from
different sequencing
PLoS ONE | www.plosone.org
1
March2011 | Volume 6 | Issu
e 3 | e17915
A Practical Compariso
n of De Novo Genome Assembly
Software Tools for Next-Generation Sequencing
Technologies
Wenyu Zhang, JiajiaChen, Yang Yang, Yifei Tang, Jing Shang, Bairong Shen*
Center for Systems Biology, Soochow University, Suzhou, Jiangsu, China
Abst ract
The advent of next-generation sequencing technologies is accompanied with
the development of many whole-genome
sequence assembly methods and software, especially for denovo fragment assembly. Due to the poor knowledge about the
applicability
and performanceof these software tools, choosing a befitti
ng assembler becomes a tough task. Here, we
provide the information of adaptivityfor each
program, then above all, compare the performanceof eight distin
ct tools
against eight groups of simulated datasets from
Solexa sequencing platform. Considering the computational time,
maximum random access memory (RAM) occu
pancy, assembly accuracy and integrity, our study indicate that strin
g-based
assemblers,overlap-layout-co
nsensus (OLC) assemblers are well-suited for very short reads and longer reads of small
genomes respectively. For large datasets of more than hundred millio
ns of short reads, De Bruijn graph-based assemblers
would be more appropriate. In terms of software implementation, string-based assemblers are superior to graph-based
ones, of whichSOAPdenovo is complex for the creation of configuration file. Our compariso
n study will assist researchers in
selecting a well-su
ited assembler and offer essential information for the improvement of existing assemblers or the
developing of novel assemblers.
Citat ion: Zhang W, Chen J, Yang Y, Tang Y, Shang J, et al. (2011) A Practical Compariso
n of De Novo Genome Assembly Software Tools for Next-Generation
Sequencing Technologies. PLoS ONE 6(3): e17915. doi:10.1371/journal.pone.0017915
Editor: I. King Jordan, Georgia Institute of Technology, United States of America
Received November 20, 2010; Accept ed February 15, 2011; Published March14, 2011
Copyright:2011 Zhang et al. This is an open-acce
ss article distri
buted under the terms of the Creative Commons Attribution License, which
permits
unrestricted use, distri
bution, and reproduction in any medium, provided the original author and source
are credited.
Funding: This work was supported by grants from the National Nature ScienceFoundation of China (31040020) and the National 973 Programs of China
(2007CB947002, 2010CB945600). The funders had no role in study design, data collection and analysis,
decision to publish
, or preparation of the manuscript.
Compet ing Interests: The authors have declared that no competing interestsexist.
* E-mail: [email protected]
Introduct ion
Inrece
nt years,the next-generation sequencing (or deep
sequencing) technologies
have beenevolving rapidly, with
the
potential toaccel
eratebiological and
biomedical research
dramatically
[1]. However,the downstre
amanalysis
of short
reads datasetsafter
sequencing is a tough task; one of the biggest
challenges for the analysis of high throughput sequencing reads is
the whole genome assembly. DNA fragment assem
bly has a long
history since the emergence of the firstgeneration of sequencing
technologies
[2,3]. The assembly proced
ure becomes especia
lly
difficult when
tackling short and high throughput reads with
different erro
r profiles[4]. According to the existen
ce of reference
information, theassembly proced
ure can beclassified asrefe
rence-
guidegenome assembly and denovo
genome assembly, of which
the
former is relatively
toilless with
the aid of reference genome or
proteome information while the laterin morechallen
ging. Herein,
we focus on the comparison and evaluation of tools for de novo
assembly of genome sequence.
The genome assemblersgenerally take a file
of short sequence
reads and a fileof quality-value as the input. Since the quality-
value filefor the high throughput short reads is usually
highly
memory-intensive, onlya
fewassemblers
, forexample,
SHARCGS [5], and ALLPATHS-LG[6] adopt it in
the
posterior assembly
process. For the sake of computational
memory saving and convenience of data inquiry, high-through-
put short reads data is always initially formattedto specif
ic data
structure. Curren
tly, existing data stru
cture for this usage can be
predominantlyclassifi
edinto two categories
: string-based
model
and graph-basedmodel.
We therefore call the corres
ponding
assemblers as string-based
and graph-based. String-based
assem-
blers, implemented with
Greedy-extension algorithm, are mainly
reported for the assembly of small genomes [5,7,8,9], while the
latter ones are designedaiming at handling complex
genomes
[10,11,12].
One of the most intractable bottlenecks for practic
al assembly
of next - generation short reads is how to processrepetiti
ve
fragmentsfrom
complicated genomes,
especiallyeukaryote
genomes.Intuitively, sequencing with
longer reads is a potential
solution, whileit beco
mesimpractic
al withlimit current of
sequencing technology. The paired-end (PE) sequencing can, to
someextent, compensate for read length [13]. Several assemblers
,
suchas SSAKE [9], SOAPdenovo [11], AbySS [12], Velvet
[14,15], exploit PE sequencing information to reduce gaps from
assembled contigs. Another big challenge for the assembly of
short reads is the intensive computational time requirem
ent. To
decrease
the time costof the assembly
procedure,
thread
parallelization is implem
entedin
a coupleof graph-based
assemblers [11,12].
At our lastenumeration, 24 academic
denovo
genome
assemblers,each
possessing its
own range of application, are
developed for short reads datasets fromdiffer
ent sequencing
PLoS ONE | www.plosone.org
1
March2011 | Volume 6 | Issu
e 3 | e17915
platforms in the last few years. These assembly tools with
corresponding websites and references are listed in Table S1. We
classify and list these assemblersaccording to their data structure
models in Figure 1. In the present study, eight short reads
assemblers, representing four various assembly strategies, were
benchmarked against two types of simulated short reads datasets
derived from four different genomes. Our objective is to gain the
assemblers performance information about computational time
and memory cost, assembly accuracy, completeness and size
distribution of assembled contigs when each assembler is applied
to handle datasets with different data size, then to provide
essential information for researchers in choosing suitable tools
and for computational biologists to develop novel assemblers.
The result indicates that each assembly strategy has its own
range of applicability while PE readsand longer readsare indeed
with the capability to increase the quality of assembled contigs to
some extent, and parallel computing is of great potential in short
reads assembly, with which the computational time is notably
reduced.
Results
At present, mainly three distinct strategies are applied in short
reads assembly. Among them, Greedy-extension is the implemen-
tation of string-based method, while DeBruijn graph and overlap-
layout-consensus (OLC) are two different graph-based approaches.
Each assembly tool issuitable for dataset from specific sequencing
platform.
For each short reads assembly procedure, less computational
time and memory cost is our expectation. The computational
time of the assembly process is determined by both the dataset
complexity and the assembly strategy. The information about
running times, maximum memory occupancies for different
assemblers applied to different datasets is illustrated in Figures 2
and 3. For string-based assembler, the time and memory cost is
approximately proportionate to dataset size, although it is also
affected by the complexity of dataset. Among them, SSAK E
runs in rather less time than other peer assemblers, but the
RAM usage increases dramatically with augmentation of dataset
size. QSRA [7] is developed upon the original VCAK E
algorithm, which indeed reduces the computational time, at
the cost of RAM occupation. SHARCGS runs in comparable
speed as QSRA, however it is highly memory-intensive, even
unable to handle E.coli short reads dataset with our computer
power used in this study. Edena is a typically graph-based
assembly tool, which has two running modes: strict and nonstrict
modes [16]. For the strict mode, fewer but more accurate
contigs are generated, while nonstrict mode acts on the contrary.
Compared with string-based tools, Edena is superior in terms of
time and RAM utilization. Velvet and SOAPdenovo typify
another graph-based method. Similar to Edena, they implement
Figure 1. Overview of de novo short reads assemblers. Programs developed from year of 2005 to 2010 are classified according to the assemblystrategies. Currently, there are mainly four sorts of assemblers, while the other ones are denoted as Different box symbols areutilized to distinguish assemblers that for short reads from different platforms.doi:10.1371/journal.pone.0017915.g001
Comparison of De Novo Genome Assembly Tools
PLoS ONE | www.plosone.org 2 March 2011 | Volume 6 | Issue 3 | e17915

9/7/2013
21
platforms in the last few years. These assembly tools with
corresponding websites and references are listed in Table S1. We
classify and list these assemblersaccording to their data structure
models in Figure 1. In the present study, eight short reads
assemblers, representing four various assembly strategies, were
benchmarked against two types of simulated short reads datasets
derived from four different genomes. Our objective is to gain the
assemblers performance information about computational time
and memory cost, assembly accuracy, completeness and size
distribution of assembled contigs when each assembler is applied
to handle datasets with different data size, then to provide
essential information for researchers in choosing suitable tools
and for computational biologists to develop novel assemblers.
The result indicates that each assembly strategy has its own
range of applicability while PE readsand longer readsare indeed
with the capability to increase the quality of assembled contigs to
some extent, and parallel computing is of great potential in short
reads assembly, with which the computational time is notably
reduced.
Results
At present, mainly three distinct strategies are applied in short
reads assembly. Among them, Greedy-extension is the implemen-
tation of string-based method, while DeBruijn graph and overlap-
layout-consensus (OLC) are two different graph-based approaches.
Each assembly tool issuitable for dataset from specific sequencing
platform.
For each short reads assembly procedure, less computational
time and memory cost is our expectation. The computational
time of the assembly process is determined by both the dataset
complexity and the assembly strategy. The information about
running times, maximum memory occupancies for different
assemblers applied to different datasets is illustrated in Figures 2
and 3. For string-based assembler, the time and memory cost is
approximately proportionate to dataset size, although it is also
affected by the complexity of dataset. Among them, SSAK E
runs in rather less time than other peer assemblers, but the
RAM usage increases dramatically with augmentation of dataset
size. QSRA [7] is developed upon the original VCAK E
algorithm, which indeed reduces the computational time, at
the cost of RAM occupation. SHARCGS runs in comparable
speed as QSRA, however it is highly memory-intensive, even
unable to handle E.coli short reads dataset with our computer
power used in this study. Edena is a typically graph-based
assembly tool, which has two running modes: strict and nonstrict
modes [16]. For the strict mode, fewer but more accurate
contigs are generated, while nonstrict mode acts on the contrary.
Compared with string-based tools, Edena is superior in terms of
time and RAM utilization. Velvet and SOAPdenovo typify
another graph-based method. Similar to Edena, they implement
Figure 1. Overview of de novo short reads assemblers. Programs developed from year of 2005 to 2010 are classified according to the assemblystrategies. Currently, there are mainly four sorts of assemblers, while the other ones are denoted as Different box symbols areutilized to distinguish assemblers that for short reads from different platforms.doi:10.1371/journal.pone.0017915.g001
Comparison of De Novo Genome Assembly Tools
PLoS ONE | www.plosone.org 2 March 2011 | Volume 6 | Issue 3 | e17915
Melhor performance para shot-reads muito pequenos
Melhor performance para datasets muito grandes

9/7/2013
22
Maiores reads = menosbolhas = menos contigs
Testes estatísticos confirmar o qual completo e
contíguo está o genoma/transcriptoma
Teste N50
N50 = bons contigs ou scaffolds = genoma bom
N50 = muitos contigs ou scaffolds pequenos = genoma mal
sequenciado = genoma mal montado
% gaps % bases não sequenciadas (Ns) Ns = genoma bom
% cobertura % do genoma sequenciado. % = genoma bom
A cobertura é baseada em dados citológicos (90%-95% é consi. bom)
9/7/2013
23
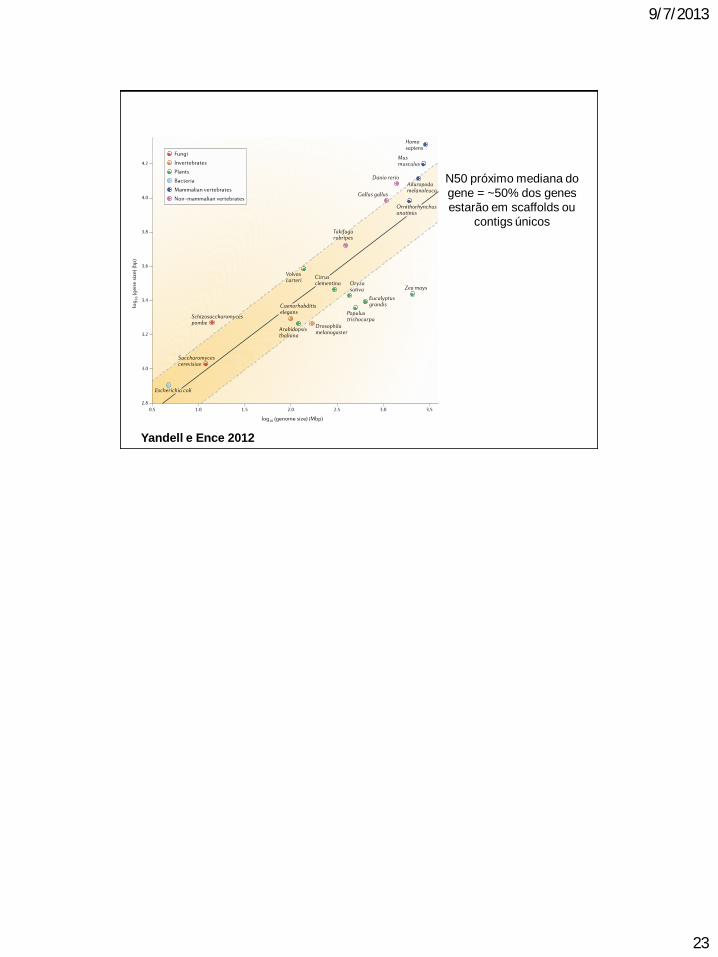
Yandell e Ence 2012
N50 próximo mediana do
gene = ~50% dos genes
estarão em scaffolds ou
contigs únicos