universidade federal da bahia faculdade de medicina
TRANSCRIPT

I
UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de fevereiro de 1808
Monografia
Larissa Fonseca Borges Lopes
Salvador (Bahia)
Novembro, 2015

II
FICHA CATALOGRÁFICA
(elaborada pela Bibl. SONIA ABREU, da Bibliotheca Gonçalo Moniz: Memória da Saúde Brasileira/SIBI-UFBA/FMB-UFBA)
Lopes, Larissa Fonseca Borges L864 Avaliação clínica das síndromes de microdeleção investigadas pela hibridização fluorescente in situ em Salvador (Bahia, Brasil) / Larissa Fonseca Borges Lopes. Salvador: LFB Lopes, 2015. viii, 72 fls. Professor orientador: Angelina Xavier Acosta. Monografia como exigência parcial e obrigatória para Conclusão de Curso de Medicina da Faculdade de Medicina da Bahia (FMB), da Universidade Federal da Bahia (UFBA).
1. Deficiência intelectual. 2. Genética. 3. Técnica de FISH. I. Acosta, Angelina Xavier. II. Universidade Federal da Bahia. Faculdade de Medicina da Bahia. III. Título.
CDU – 575

III
UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA DA BAHIA
Fundada em 18 de fevereiro de 1808
Monografia
Avaliação clínica das síndromes de microdeleção
investigadas pela hibridização fluorescente in situ em
Salvador (Bahia, Brasil)
Larissa Fonseca Borges Lopes
Professor orientador: Angelina Xavier Acosta
Monografia de Conclusão do
Componente Curricular MED-
B60/2015.1, como pré-requisito
obrigatório e parcial para conclusão do
curso médico da Faculdade de Medicina
da Bahia da Universidade Federal da
Bahia, apresentada ao Colegiado do
Curso de Graduação em Medicina.
Salvador (Bahia)
Novembro, 2015

IV
Monografia: Avaliação clínica das síndromes de microdeleção investigadas
pela hibridização fluorescente in situ em Salvador (Bahia, Brasil), de Larissa
Fonseca Borges Lopes.
Professor orientador: Angelina Xavier Acosta
COMISSÃO REVISORA:
Angelina Xavier Acosta (Presidente, Professor orientador), Professora do
Departamento de Pediatria da Faculdade de Medicina da Bahia da Universidade
Federal da Bahia.
Acácia Fernandes Lacerda de Carvalho, Professora do Departamento de Biologia
Geral do Instituto de Biologia da Universidade Federal da Bahia.
Manuel Lessa Ribeiro Neto, Doutorando do Curso de Doutorado do Programa de
Pós-graduação em Ciências da Saúde (PPgCS) da Faculdade de Medicina da Bahia da
Universidade Federal da Bahia.
Membro suplente
Luciana Mattos Barros Oliveira, Professora do Departamento de Biorregulação
do Instituto de Ciências da Saúde da Universidade Federal da Bahia.
TERMO DE REGISTRO ACADÊMICO:
Monografia avaliada pela Comissão Revisora, e julgada
apta à apresentação pública no IX Seminário Estudantil de
Pesquisa da Faculdade de Medicina da Bahia/UFBA, com
posterior homologação do conceito final pela coordenação
do Núcleo de Formação Científica e de MED-B60
(Monografia IV). Salvador (Bahia), em ___ de
_____________ de 2015.

V
“Nós temos o poder de deixar a corrente passar através
de nós e nos utilizar para produzir a luz no mundo.”
(Madre Teresa de Calcutá)

VI
Ao meu núcleo familiar sempre
presente: Cláudia, Valci,
Andressa, Aurora e Lucas,
pessoas de importância singular,
cujas existências contribuíram
todos os dias para ser quem sou e
chegar onde cheguei.

VII
EQUIPE Larissa Fonseca Borges Lopes, Faculdade de Medicina da Bahia/UFBA. Correio-e:
Professor orientador: Angelina Xavier Acosta, Faculdade de Medicina da
Bahia/UFBA;
Esmeralda Santos Alves, Bióloga, Serviço de Genética Médica do HUPES;
Joanna Goes Castro Meira, Geneticista, Serviço de Genética Médica do HUPES;
Marcela Câmara Machado Costa, Neurologista, Serviço de Genética Médica do
HUPES;
Graziela Paz de Souza, Secretária do Serviço de Genética Médica do HUPES;
Inis Leahy Sala Santos, Psicóloga, Mestranda em Saúde da Criança e do Adolescente
– Programa de Pós-graduação em Medicina e Saúde;
Aruanã Mairê Maia Fontes, Psicóloga, Mestranda em Saúde da Criança e do
Adolescente – Programa de Pós-graduação em Medicina e Saúde;
Camila Schlang Cabral da Silveira, Estudante da Faculdade de Medicina da
Bahia/UFBA;
Isabella Fernanda Silva Ferreira, Estudante da Faculdade de Medicina da
Bahia/UFBA;
Acácia Fernandes Lacerda de Carvalho, Instituto de Biologia/UFBA; e
Renata Lúcia Leite Ferreira de Lima, Instituto de Biologia/UFBA
INSTITUIÇÕES PARTICIPANTES
UNIVERSIDADE FEDERAL DA BAHIA
Faculdade de Medicina da Bahia (FMB)
Complexo Hospitalar Universitário Professor Edgard Santos (COM-HUPES)
Instituto de Biologia (IBio)
FONTES DE FINANCIAMENTO
1. Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) – número do
grant 402025/2010-5; e
2. Recursos próprios.

VIII
AGRADECIMENTOS
À minha Professora orientadora, Doutora Angelina Xavier Acosta, por quem nutro
profunda admiração. Agradeço pelas inestimáveis oportunidades acadêmicas que
me foram concedidas, pela orientação profissional constante e pela marcante
competência, um verdadeiro exemplo a seguir em minha vida de futura médica.
À Doutora Acácia Fernandes Lacerda de Carvalho e ao Doutorando Manuel Lessa
Ribeiro Neto, membros da Comissão Revisora, pela disponibilidade e pelas
contribuições valiosas para o aprimoramento desta Monografia.
Às Doutoras Joanna Goes Castro Meira e Marcela Câmara Machado Costa,
médicas do serviço de Genética Médica, pela ajuda no ambulatório e discussão de
casos, ambas contribuindo substancialmente para o melhor andamento da pesquisa.
À bióloga Esmeralda Santos Alves, pela ajuda com os dados da pesquisa, em especial
os resultados laboratoriais. Agradeço pela constante disponibilidade e conhecimento
compartilhado.
À biomédica Paula Brito Corrêa, pela contribuição nos ensinamentos sobre as técnicas
laboratoriais deste estudo.
À minha colega Camila Schlang Cabral da Silveira, pela fundamental colaboração no
levantamento de dados da pesquisa e por partilhar comigo tantos conhecimentos e
experiências acadêmicas.
À secretária Graziela Paz de Souza, cujo trabalho foi essencial para a organização e
andamento do projeto.
Às psicólogas Inis Leahy Sala Santos e Aruanã Mairê Maia Fontes, que realizaram
as avaliações neuropsicológicas na amostra, permitindo a inclusão e classificação
dos pacientes com deficiência intelectual.
À equipe do Instituto de Biologia, que colaborou com resultados de exames
laboratoriais.
A todos os pacientes participantes e seus familiares, sem os quais este estudo não seria
possível.

1
SUMÁRIO
ÍNDICE DE FLUXOGRAMA, GRÁFICO E TABELAS 2
I. RESUMO 3
II. OBJETIVOS 4
III. FUNDAMENTAÇÃO TEÓRICA 5
IV. METODOLOGIA 10
IV.1. Casuística 10
IV.2. Considerações Éticas 11
V. RESULTADOS 12
VI. DISCUSSÃO 24
VII. CONCLUSÕES 30
VIII. SUMMARY 31
IX. REFERÊNCIAS BIBLIOGRÁFICAS 32
X. ANEXOS 36
• ANEXO I: Parecer do CEP 36
• ANEXO II: Parecer adendo CEP 39
• ANEXO III: Termo de Consentimento Livre e Esclarecido 41
• ANEXO IV: Ficha clínica 45
• ANEXO V: Ficha clínica para Síndrome de Angelman 50
• ANEXO VI: Ficha clínica para Síndrome de Prader Willi 52
• ANEXO VII: Ficha clínica para Síndrome de Williams-Beuren 53
• ANEXO VIII: Ficha clínica para Síndrome de Rubinstein-Taybi 55
• ANEXO IX: Ficha clínica para Síndrome de Miller-Dieker 56
• ANEXO X: Ficha clínica para Síndrome de Smith-Magenis 57
• ANEXO XI: Ficha clínica para Síndrome de Langer-Giedion 58
• ANEXO XII: Ficha clínica para Síndrome de Wolf-Hirschhorn 60
• ANEXO XIII: Ficha clínica para Síndrome de Cri-du-Chat 61
• ANEXO XIV: Ficha clínica para Síndrome de Sotos 63
• ANEXO XV: Ficha clínica para Síndrome Velocardiofacial 64

2
ÍNDICE DE FLUXOGRAMA, GRÁFICO E TABELAS
FLUXOGRAMA
FLUXOGRAMA 1. Evolução dos resultados da pesquisa em relação à amostra
inicial.
12
GRÁFICO
GRÁFICO 1. Resultados da FISH, em porcentagem. 13
TABELAS
TABELA 1. Caracterização clínica dos pacientes com suspeita de Síndrome de
Prader-Willi. 14
TABELA 2. Caracterização clínica dos pacientes com suspeita de Síndrome de
Williams. 16
TABELA 3. Caracterização clínica dos pacientes com suspeita de Síndrome de
Smith-Magenis.
18
TABELA 4. Caracterização clínica do paciente com suspeita de Síndrome de
Sotos. 20
TABELA 5. Caracterização clínica do paciente com suspeita de Síndrome de
Angelman.
21
TABELA 6. Caracterização clínica da paciente com suspeita de Síndrome
Velocardiofacial.
22
TABELA 7. Caracterização clínica da paciente com suspeita de Síndrome de
Langer-Giedion. 23

3
I. RESUMO
AVALIAÇÃO CLÍNICA DAS SÍNDROMES DE MICRODELEÇÃO
INVESTIGADAS PELA HIBRIDIZAÇÃO FLUORESCENTE IN SITU
EM SALVADOR (BAHIA, BRASIL). Fundamentação Teórica: A deficiência
intelectual (DI) é uma condição neuropsiquiátrica comum, que atinge até 3% da população
jovem. A etiologia da DI pode ter origem ambiental, genética ou multifatorial, sendo as
causas genéticas responsáveis por até 47,1% dos casos. As microdeleções têm incidência de 1
em 1.000 recém-nascidos, sendo, portanto, frequentes; além disso não podem ser detectadas
na análise citogenética convencional. Para sua detecção, são necessárias técnicas laboratoriais
citomoleculares, como a hibridização fluorescente in situ (FISH). Objetivos: Descrever a
caracterização clínica e sua comparação com o resultado laboratorial obtido dos pacientes em
investigação diagnóstica para síndromes de microdeleção através da FISH. Metodologia: O
desenho da pesquisa é um estudo descritivo, que utiliza como base uma série de casos. Os
pacientes com DI isolada ou sindrômica com suspeita de causa genética foram triados e
aqueles que obedeceram os critérios de inclusão foram selecionados para o estudo. Formulada
a suspeita diagnóstica de síndrome de microdeleção, foi preenchida uma ficha específica e
realizada coleta de material biológico (sangue) para investigação laboratorial pela FISH.
Resultados: Dos 106 pacientes incluídos com DI, 15 realizaram FISH, sendo que 4
apresentaram resultados alterados e 11 normais. Discussão: Dos pacientes que realizaram
FISH neste estudo, 26,7% (4/15) obtiveram resultado alterado. Há casos de clínica sugestiva
para determinada síndrome, que deveriam continuar a investigação diagnóstica para
confirmação através de uma técnica mais ampla que a FISH, como MLPA ou técnicas de
hibridação genômica, e também há pacientes que, após reavaliação clínica, pode-se pensar em
outra suspeita diagnóstica. Conclusões: A FISH é uma boa técnica diagnóstica para detectar
síndromes de microdeleção cuja clínica do paciente seja altamente sugestiva, caso contrário
recomenda-se técnicas de hibridação genômica ou MLPA como primeiro exame diagnóstico.
Palavras chaves: 1. Deficiência intelectual, 2. Genética, 3. Técnica de FISH.

4
II. OBJETIVOS
Principal
Descrever a caracterização clínica e sua comparação com o resultado laboratorial obtido dos
pacientes em investigação diagnóstica para as síndromes de microdeleção através da FISH,
atendidos no Serviço de Genética Médica do Complexo Hospitalar Universitário Professor
Edgard Santos (COM-HUPES), vinculado à Universidade Federal da Bahia (UFBA).
Secundários
1. Descrever principais indicações clínicas para a realização da FISH em pacientes com DI.
2. Estimar a frequência das síndromes de microdeleção entre os pacientes com DI avaliados
pela FISH.

5
III. FUNDAMENTAÇÃO TEÓRICA
A deficiência intelectual (DI) é uma condição neuropsiquiátrica comum, que atinge cerca de
1% da população jovem, porém as estimativas podem variar até cerca de 3% de prevalência(1).
Segundo o censo demográfico realizado pelo Instituto Brasileiro de Geografia e Estatística
(IBGE), em 2010, há mais de 2,6 milhões de habitantes com DI, o que corresponde a 1,4% da
população, sendo 1,4 milhões do sexo masculino e 1,2 milhões do sexo feminino(2). Acredita-
se que a DI é mais comum no sexo masculino devido a grande quantidade de mutações
ligadas ao cromossomo X(1).
Classifica-se a DI através do quociente de inteligência (QI), sendo este menor que 70.
Segundo a Classificação Internacional de Doenças (CID-10), considera-se retardo mental leve
quando o paciente obtiver QI entre 50 e 69, DI moderado entre 35 e 49, DI grave com
amplitude aproximada de QI entre 20 e 40 e retardo mental profundo quando o QI for abaixo
de 20(3).
A etiologia da DI pode ser ambiental, genética ou multifatorial, sendo as causas genéticas
responsáveis por cerca de 17,4% a 47,1% dos casos(4). Entre as causas genéticas de DI, as
cromossomopatias têm grande destaque e podem muitas vezes ser diagnosticadas através do
cariótipo. As microdeleções não são detectadas pela citogenética convencional (que detecta
alterações de 5 a 10 Mb), pois geralmente são causadas por deleções de 2 a 4 Mb de DNA(5).
Para detectá-las, então, pode-se fazer uso de outras técnicas citomoleculares, como a
hibridização fluorescente in situ (FISH), que utiliza sondas específicas para identificar a
microdeleção em questão(5). Além desta técnica, existem também a amplificação de múltiplas
sondas dependentes de ligação (MLPA), que identifica microdeleções e microduplicações em
genes específicos(6), e técnicas de hibridação genômica, como a hibridização genômica
comparativa em array (aCGH), que é uma técnica de alta resolução capaz de triar o genoma
para detectar microdeleções, microduplicações e demais anomalias referentes à quantidade de
material(5).
A citogenética convencional é capaz de diagnosticar 3,7% dos casos de DI(7). Já a FISH é
responsável por identificar 6% dos casos, enquanto técnicas de hibridação genômica como a
aCGH possuem capacidade de diagnóstico em 15% dos casos(8).
Acredita-se que as síndromes de microdeleção são responsáveis por aproximadamente cerca
de 5% dos casos de DI sem causa definida(9) e têm incidência de até 1 em 1.000 recém-
nascidos, sendo, portanto, frequentes(5). As síndromes de microdeleção possuem um amplo

6
fenótipo, dependendo da extensão da deleção, e podem ser associadas ou não à DI. Muitas
destas síndromes são caracterizadas por possuírem associação de DI com outras anomalias
congênitas e, dentre as que podem ser investigadas por FISH, estão: Síndrome de Angelman,
Síndrome de Prader-Willi, Síndrome de Williams-Beuren, Síndrome de Rubinstein-Taybi,
Síndrome de Miller-Dieker, Síndrome de Smith-Magenis, Síndrome de Langer-Giedion,
Síndrome de Wolf-Hirschhorn, Síndrome de Cri-du-Chat, Síndrome de Sotos e Síndrome
Velocardiofacial(5, 9).
A Síndrome de Angelman é caracterizada principalmente pela DI, convulsões, alterações no
eletroencefalograma (EEG) e dismorfias faciais, como microcefalia, hipoplasia maxilar,
macrostomia e prognatismo. O paciente pode apresentar, até os 6 meses de idade, dificuldade
para se alimentar e hipotonia. Depois, poderá ser notado um atraso do desenvolvimento
neuropsicomotor. Com 1 ano de idade, poderão ser notadas mais características da síndrome
na criança, como ausência de fala, dismorfias, riso inapropriado, ataxia e convulsões.
Normalmente, as crianças apresentam uma personalidade alegre, hiperatividade, dificuldades
para dormir, sensibilidade aumentada ao calor e atração pela água. A expectativa de vida
destes pacientes parece ser normal. A síndrome é causada, em 60% a 75% dos casos, por uma
deleção no cromossomo 15 materno, mais especificamente no 15q11-q13. Cerca de 2% a 5%
dos casos são causados por dissomia uniparental do cromossomo 15, que pode ser composta
por dois cromossomos homólogos de um genitor, ou por uma duplicação de um cromossomo,
ou por uma mistura destas duas composições(10).
A Síndrome de Prader-Willi, assim como a Síndrome de Angelman, também é causada por
uma deleção no cromossomo 15q11-q13, porém paterno ao invés do materno. Este
mecanismo corresponde a cerca de 75% a 80% dos casos, mas ela também pode ser causada,
em 20% a 25% dos casos, por dissomia uniparental materna. A síndrome se caracteriza, do
nascimento até os 2 anos de idade, por hipotonia, sucção deficiente, sonolência e choro
diminuído. Dos 2 aos 6 anos de idade, a criança apresenta também um atraso global do
desenvolvimento. Dos 6 aos 12 anos de idade, somam-se a estes sintomas prévios a hiperfagia
e obesidade(11, 12).
Outras características da síndrome são DI, compulsões e irritabilidade, dificuldades
cognitivas, motoras e de linguagem, disfunção hipotalâmica levando ao hipogonadismo e à
baixa estatura. Normalmente, a puberdade é incompleta ou tardia. A síndrome apresenta
dismorfias faciais características, como estreitamento da cabeça entre as têmporas, olhos

7
amendoados, estrabismo, lábio superior fino, ponte nasal estreita e comissura labial para
baixo. Os critérios maiores para diagnosticar a síndrome são as dismorfias faciais citadas,
atraso do desenvolvimento, dificuldades para se alimentar na infância, hipogonadismo,
hipotonia central na infância e rápido ganho de peso de 1 a 6 anos de idade. Já os critérios
menores são: movimentos fetais diminuídos ou letargia na infância, miopia ou esotropia,
hipopigmentação, mãos estreitas, baixa estatura (quando comparada aos familiares),
distúrbios do sono, mãos e pés pequenos, dificuldade para articular o discurso, saliva viscosa,
problemas comportamentais e escoriação neurótica.(11, 12).
A Síndrome de Williams-Beuren, conhecida também apenas como Síndrome de Williams, é
causada por uma deleção no cromossomo 7q11.23. Entre os achados clínicos mais comuns
dos portadores desta síndrome, estão: baixa estatura, estrabismo, DI, dismorfias faciais e
odontológicas, anormalidades cardíacas (principalmente vasculopatia com estenose aórtica
supravalvular), renais e oftalmológicas, alterações neurológicas e cognitivas, com atraso da
linguagem, além de personalidade amigável. Entre as dismorfias faciais, as mais comuns são
filtro longo, narina antevertida e boca grande com lábios grossos(13, 14).
A Síndrome de Rubinstein-Taybi é caracterizada por diversas anomalias congênitas além de
DI. Entre as anomalias, destaca-se a baixa estatura, microcefalia, pododáctilos grandes,
polegares largos, sobrancelhas arqueadas, cílios longos, fissura palpebral oblíqua para baixo,
ponte nasal larga, palato ogival, nariz pontiagudo, alterações dentárias e micrognatia. A
expressão facial característica é uma careta ou sorriso com os olhos quase fechados. Durante a
infância, normalmente os pacientes cursam com dificuldade para haver ganho ponderal,
porém na puberdade pode haver sobrepeso. Podem estar presentes na síndrome também
dificuldade de concentração e mudanças de humor repentinas. É uma síndrome esporádica,
que pode ser causada por uma deleção no cromossomo 16p13.3 ou por mutações nas proteínas
CBP (proteína de ligação da CREB) e p300 (proteína de ligação associada a E1A)(15).
A Síndrome de Miller-Dieker é caracterizada pela lisencefalia, condição em que o cérebro
apresenta diminuição ou ausência das circunvoluções, sendo, portanto, um “cérebro liso”.
Além disso, o paciente pode apresentar narinas antevertidas, mandíbula pequena, filtro longo,
lábio superior proeminente, malformações cardíacas, renais, digitais e na vascularização da
retina. Em cerca de 90% dos pacientes, a síndrome é causada pela deleção no cromossomo
17p13.3, onde se localiza o gene Lis1, afetando assim o desenvolvimento do telencéfalo. A

8
minoria dos pacientes pode ter desenvolvido a síndrome por causas infecciosas ou vasculares
durante a gestação(16).
A Síndrome de Smith-Magenis, que tem como causa a deleção do cromossomo 17p11.2 em
90% dos casos, é uma síndrome esporádica caracterizada por DI e anomalias congênitas
múltiplas. Os portadores, em geral, possuem anomalias esqueléticas, oculares e craniofaciais,
atraso do desenvolvimento e da fala, distúrbios do sono, hipotonia, perda auditiva, infecções
otológicas, problemas cardíacos e renais e, ocasionalmente, fenda lábio-palatina. Cerca de
10% dos pacientes portadores desta síndrome possuem mutação no gene RAI1 (ácido
retinóico induzido 1)(17).
A Síndrome de Langer-Giedion, também conhecida como Síndrome Trico-rino-falangeana
tipo II, é uma síndrome esporádica causada por deleção no cromossomo 8, na região q24.11-
q24.13, em 50% dos casos. Os pacientes podem apresentar nariz bulboso em formato de pêra,
cabelos finos, micrognatia, hipotricose, filtro largo, crescimento retardado e anomalias nas
epífises das mãos, que podem gerar braquidactilia, clinodactilia e contraturas dos dedos. Há
também a presença de exostoses ósseas múltiplas, microcefalia e DI(18, 19).
A Síndrome de Wolf-Hirschhorn é causada pela deleção na região distal do braço curto do
cromossomo 4, mais especificamente a região 4p16.3. É uma síndrome mais comum no sexo
feminino, que se caracteriza principalmente por uma face típica, conhecida como “face do
guerreiro grego”, atraso do crescimento intrauterino, atraso do desenvolvimento
neuropsimotor, déficit cognitivo, epilepsia, EEG alterado, hipotonia e dificuldade para se
alimentar. Estes sinais estão presentes na maioria dos portadores da síndrome, porém outros
também podem ser comuns, como alterações dermatológicas, esqueléticas, do sistema nervoso
central, imunodeficiências, alteração da dentição e problemas cardíacos(20).
A Síndrome de Cri-du-Chat, conhecida também como Síndrome do miado do gato, recebeu
este nome pelo fato dos portadores apresentarem um choro parecido com o miado de um gato
nos primeiros anos de vida, secundário a alterações na laringe. Esta síndrome é causada, na
maioria das vezes, por uma deleção total ou parcial no braço curto do cromossomo 5, na
região 5p15.1-5p15.3. Os portadores da síndrome podem apresentar também, no período
neonatal, alterações neurológicas, baixo peso ao nascer, atraso do desenvolvimento
neuropsicomotor, disfagia, sucção pobre, hipotonia, refluxo gastroesofágico e nasal e
episódios de asfixia. Entre as manifestações craniofaciais mais comuns estão microcefalia,
assimetria facial, face em lua cheia (substituída na adolescência ou vida adulta por uma face

9
alongada e estreita), hipertelorismo, epicanto, comissura labial para baixo, ponte nasal larga,
filtro curto e microretrognatia mandibular(21).
A Síndrome de Sotos é caracterizada principalmente pelo crescimento excessivo pré e pós-
natal, atraso do desenvolvimento neuropsicomotor, idade óssea avançada, macrodolicocefalia,
testa alta e curvada, hipertelorismo, fissura palpebral oblíqua para baixo, mandíbula
proeminente, pés e mãos grandes, palato ogival e rubor facial. A altura e o peso tendem a
normalizar na puberdade. A maioria dos casos são esporádicos e causados por mutação no
gene NSD1 ou microdeleção do cromossomo 5q35(22, 23).
A Síndrome Velocardiofacial é uma das síndromes com anomalias múltiplas mais comuns
em humanos e possui um fenótipo variado, envolvendo diversos órgãos. Há indivíduos com
expressão leve da síndrome, sendo próximos do normal, enquanto outros podem ter formas
graves e limitantes. Esta doença é causada por uma microdeleção no cromossomo 22, na
região 22q11.2. Os portadores comumente possuem defeito cardíaco congênito, problemas
imunes, dificuldades na alimentação, fenda palatina, atraso do desenvolvimento
neuropsicomotor e problemas psiquiátricos(24).
Considerando que, no Brasil, muitos serviços de Genética Médica ainda são incompletos,
por não serem vinculados a programas gerais de assistência médica e por serem baseados na
demanda espontânea, esses programas ainda não tiveram, em sua maioria, uma utilização
significativa pela população e, consequentemente, grande impacto em políticas de saúde(25).
Dessa forma, pode-se inferir que o subdiagnóstico genético ainda é frequente, principalmente
nas regiões do país carentes destes serviços, uma vez que o diagnóstico depende de exames de
melhor resolução, que muitas vezes não são tão acessíveis. As síndromes de microdeleção, em
seu conjunto, são frequentes e responsáveis por muitos casos de DI tidos como causa
indefinida, uma vez que são subdiagnosticadas. Logo, é preciso que sejam melhores
estudadas, pois possuem quadro clínico diverso e necessitam de uma boa caracterização
clínica que oriente o melhor exame a ser utilizado, a fim de que seus diagnósticos sejam
otimizados.

10
IV. METODOLOGIA
IV.1. Casuística
O desenho da pesquisa é um estudo descritivo, que utiliza como base uma série de casos. A
pesquisa desenvolvida foi uma parte de um projeto maior, intitulado “Implantação de uma
Rede de Investigação Genética da Deficiência Mental nas Regiões Norde/Nordeste/Centro-
Oeste no Âmbito do SUS”, aprovado pelo CEP (ANEXO I), e minha participação foi
oficializada por meio de um Parecer adendo do CEP (ANEXO II). O projeto se baseou
inicialmente em uma triagem clínica realizada pelo médico geneticista, no Ambulatório de
Genética Médica do COM-HUPES/UFBA, com inclusão dos casos de DI isolada ou
sindrômica de etiologia desconhecida, com suspeita de causa genética. Foi aplicado então o
Termo de Consentimento Livre e Esclarecido (TCLE – ANEXO III) para os responsáveis
pelos pacientes, quando desejavam participar do estudo, além de assinarem uma autorização
para fotografar o paciente. Em seguida, os dados do paciente foram preenchidos em uma ficha
clínica padronizada (ANEXO IV) e foi solicitado o cariótipo, realizado no Laboratório de
Genética Médica (LGM) do COM-HUPES. Os casos de Síndrome de Down não fizeram parte
do projeto. Além disso, um psicólogo do serviço realizou a avaliação neuropsicológica dos
pacientes, utilizando o instrumento SON-R 6-40, um teste não verbal de inteligência. Esta
avaliação classifica o nível de DI (leve, moderado ou grave), ou mesmo exclui a suspeita de
DI para determinado paciente (QI maior que 70).
Critérios de inclusão: para os pacientes com cariótipo normal e características sugestivas de
alguma das síndromes de microdeleção listadas (Síndrome de Angelman, Síndrome de
Prader-Willi, Síndrome de Williams-Beuren, Síndrome de Rubinstein-Taybi, Síndrome de
Miller-Dieker, Síndrome de Smith-Magenis, Síndrome de Langer-Giedion, Síndrome de
Wolf-Hirschhorn, Síndrome de Cri-du-Chat, Síndrome de Sotos e Síndrome
Velocardiofacial), foi preenchida uma ficha com os critérios clínicos da síndrome em questão
(ANEXO V a ANEXO XV), para posterior realização da FISH. Foram incluídos na pesquisa
106 pacientes portadores de DI isolada ou sindrômica de etiologia desconhecida, com suspeita
de causa genética, sendo uma amostra de conveniência, a partir da qual se fizeram as suspeitas
das síndromes de microdeleção com realização da FISH, no total de 15 pacientes, todos com
DI sindrômica. As variáveis avaliadas nos pacientes com suspeita de síndrome de

11
microdeleção foram: idade, sexo, dismorfias, anomalias congênitas, consanguinidade,
recorrência familiar, procedência e QI (nível de DI).
Para a realização da FISH, houve a coleta de 5ml de sangue em anti-coagulante do paciente.
Foram utilizadas sondas comerciais específicas para cada síndrome de microdeleção. A
análise de 20 núcleos ou metáfases foi feita utilizando-se microscópios de fluorescência
Olympus do HUPES. Sendo identificado apenas um sinal de hibridação para a sonda
específica, o resultado é dado como positivo, confirmando a suspeita para aquela síndrome de
microdeleção. Ao identificar dois sinais de hibridação para a sonda específica, o resultado é
considerado negativo e os pacientes seguem investigando a etiologia, sugerindo-se, em alguns
casos, avaliação genética por hibridação genômica. Por fim, foi estimada a frequência de
síndromes de microdeleção dentre a amostra estudada.
IV.2. Considerações Éticas
Os dados da pesquisa foram colhidos através do projeto “Implantação de uma Rede de
Investigação Genética da Deficiência Mental nas Regiões Norte/Nordeste/Centro-Oeste no
Âmbito do SUS”, aprovado pelo CEP (ANEXO I) com o cadastro 13/11, em 19/05/2011.
Minha participação no projeto foi oficializada por meio de um parecer adendo do CEP
(ANEXO II).
Uma vez estabelecida a suspeita de DI de causa genética para os pacientes do Ambulatório
de Genética Médica do COM-HUPES/UFBA, foi aplicado o TCLE (ANEXO III) aos
responsáveis pelos pacientes, que concordaram em participar. Foram feitas fotografias dos
pacientes cujos responsáveis assinaram uma autorização para este fim.

12
V. RESULTADOS
Inicialmente, foram incluídos na pesquisa 119 pacientes com suspeita de DI isolada ou
sindrômica de etiologia desconhecida. Entretanto, 13 pacientes foram excluídos dessa amostra
inicial por não se encaixarem nos critérios de inclusão: sete pacientes possuíam QI maior que
70, 1 paciente apresentava idade mental compatível com a cronológica e atraso do
desenvolvimento neuropsicomotor justificado por surdez, 4 pacientes possuíam DI secundária
e 1 paciente possuía Síndrome de Down (mosaicismo revelado após realização de cariótipo
ampliado). Sendo assim, a amostra incluída na pesquisa foi de 106 pacientes.
Entre os 106 pacientes, 18 tiveram suspeita inicial para síndromes de microdeleção, porém 3
deles não realizaram a coleta do material, havendo perda de seguimento. Sendo assim, 15
pacientes foram submetidos à FISH, tendo 11 apresentado resultado da FISH normal e 4
alterados (FLUXOGRAMA 1).
FLUXOGRAMA 1: Evolução dos resultados da pesquisa em relação à amostra inicial.
Dos 15 pacientes que realizaram a FISH, 5 tiveram suspeita para Síndrome de Prader-Willi,
4 para Síndrome de Williams, 2 para Síndrome de Smith-Magenis, 1 para Síndrome de Sotos,
1 para Síndrome de Angelman, 1 para Síndrome Velocardiofacial e 1 para Síndrome de
Langer-Giedion. Oito pacientes eram do sexo feminino e 7 do sexo masculino. A idade dos
119 pacientes
Total
106 pacientes
Incluídos
18 pacientesSuspeita de
microdeleção
15 pacientes
Realizaram FISH
11 pacientes
FISH normal
4 pacientes
FISH alterada
3 pacientes
Não realizaram FISH
88 pacientesSem indicação de
FISH
13 pacientes
Excluídos
13
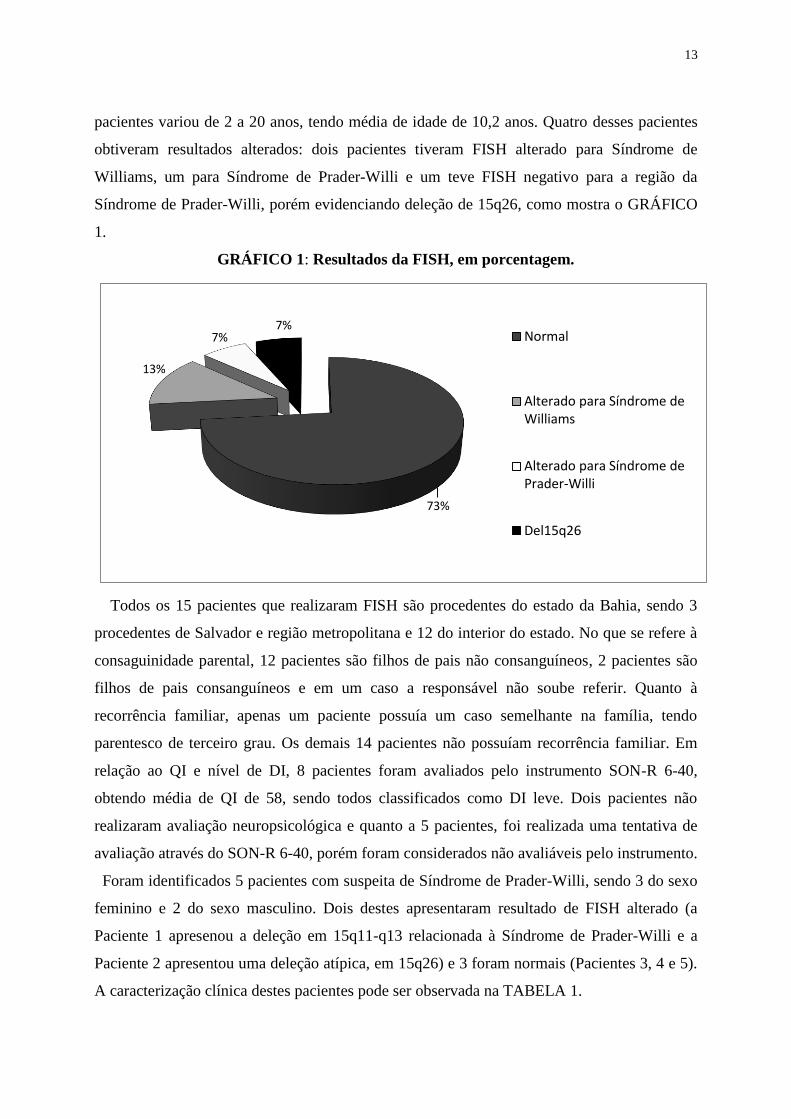
pacientes variou de 2 a 20 anos, tendo média de idade de 10,2 anos. Quatro desses pacientes
obtiveram resultados alterados: dois pacientes tiveram FISH alterado para Síndrome de
Williams, um para Síndrome de Prader-Willi e um teve FISH negativo para a região da
Síndrome de Prader-Willi, porém evidenciando deleção de 15q26, como mostra o GRÁFICO
1.
GRÁFICO 1: Resultados da FISH, em porcentagem.
Todos os 15 pacientes que realizaram FISH são procedentes do estado da Bahia, sendo 3
procedentes de Salvador e região metropolitana e 12 do interior do estado. No que se refere à
consaguinidade parental, 12 pacientes são filhos de pais não consanguíneos, 2 pacientes são
filhos de pais consanguíneos e em um caso a responsável não soube referir. Quanto à
recorrência familiar, apenas um paciente possuía um caso semelhante na família, tendo
parentesco de terceiro grau. Os demais 14 pacientes não possuíam recorrência familiar. Em
relação ao QI e nível de DI, 8 pacientes foram avaliados pelo instrumento SON-R 6-40,
obtendo média de QI de 58, sendo todos classificados como DI leve. Dois pacientes não
realizaram avaliação neuropsicológica e quanto a 5 pacientes, foi realizada uma tentativa de
avaliação através do SON-R 6-40, porém foram considerados não avaliáveis pelo instrumento.
Foram identificados 5 pacientes com suspeita de Síndrome de Prader-Willi, sendo 3 do sexo
feminino e 2 do sexo masculino. Dois destes apresentaram resultado de FISH alterado (a
Paciente 1 apresenou a deleção em 15q11-q13 relacionada à Síndrome de Prader-Willi e a
Paciente 2 apresentou uma deleção atípica, em 15q26) e 3 foram normais (Pacientes 3, 4 e 5).
A caracterização clínica destes pacientes pode ser observada na TABELA 1.
73%
13%
7%7%
Normal
Alterado para Síndrome deWilliams
Alterado para Síndrome dePrader-Willi
Del15q26
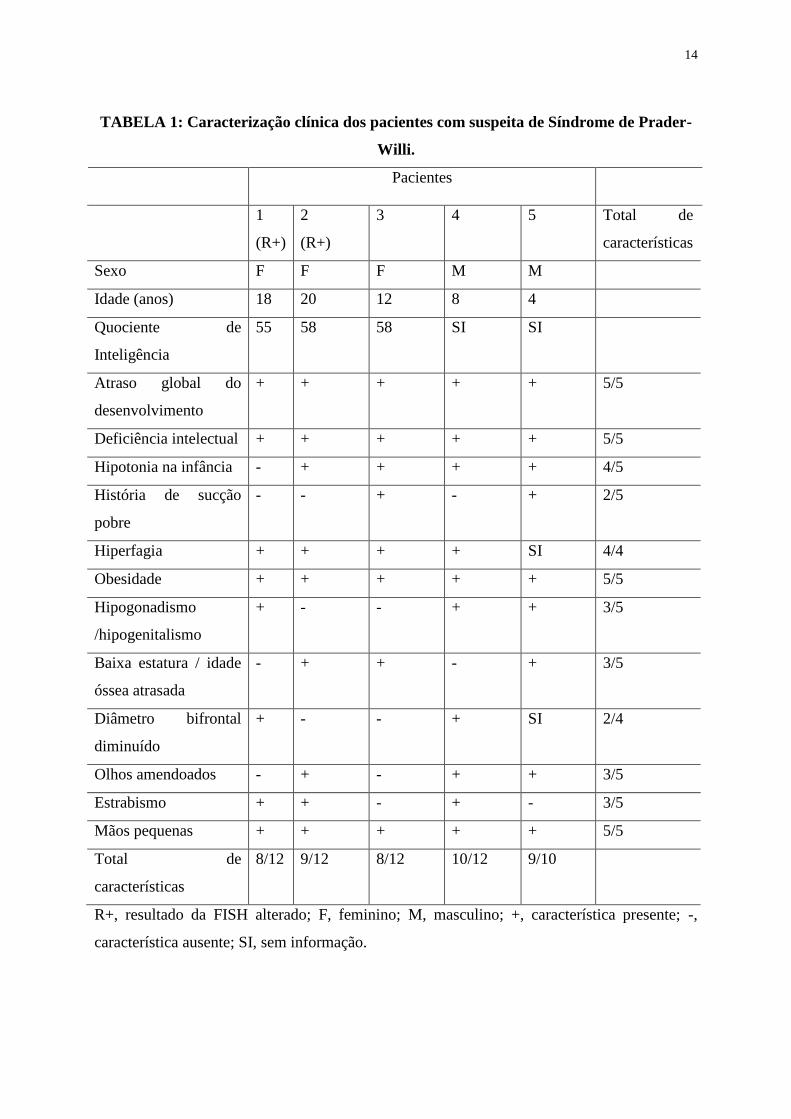
14
TABELA 1: Caracterização clínica dos pacientes com suspeita de Síndrome de Prader-
Willi.
Pacientes
1
(R+)
2
(R+)
3 4 5 Total de
características
Sexo F F F M M
Idade (anos) 18 20 12 8 4
Quociente de
Inteligência
55 58 58 SI SI
Atraso global do
desenvolvimento
+ + + + + 5/5
Deficiência intelectual + + + + + 5/5
Hipotonia na infância - + + + + 4/5
História de sucção
pobre
- - + - + 2/5
Hiperfagia + + + + SI 4/4
Obesidade + + + + + 5/5
Hipogonadismo
/hipogenitalismo
+ - - + + 3/5
Baixa estatura / idade
óssea atrasada
- + + - + 3/5
Diâmetro bifrontal
diminuído
+ - - + SI 2/4
Olhos amendoados - + - + + 3/5
Estrabismo + + - + - 3/5
Mãos pequenas + + + + + 5/5
Total de
características
8/12 9/12 8/12 10/12 9/10
R+, resultado da FISH alterado; F, feminino; M, masculino; +, característica presente; -,
característica ausente; SI, sem informação.

15
Foram identificados 4 pacientes com suspeita de Síndrome de Williams, sendo 1 do sexo
feminino e 3 do sexo masculino. Dois destes apresentaram resultado de FISH alterado
(Pacientes 6 e 7) e 2 foram normais (Pacientes 8 e 9). A caracterização clínica destes
pacientes pode ser observada na TABELA 2.

16
TABELA 2: Caracterização clínica dos pacientes com suspeita de Síndrome de
Williams.
Pacientes
6
(R+)
7
(R+)
8 9 Total de
características
Sexo F M M M
Idade (anos) 8 7 2 7
Quociente de Inteligência 55 NA NA NA
Atraso do desenvolvimento neuropsicomotor /
DI
+ + + + 4/4
Retardo do crescimento e ganho de peso < 5º
percentil
- + - - 1/4
Personalidade excessivamente amigável + + - - 2/4
Ansiedade + + + - 3/4
Hipersensibilidade ao som SI + + - 2/3
Estreitamento bitemporal SI - - + 1/3
Epicanto / Ponte nasal plana + - + + 3/4
Estrabismo + + - - 2/4
Nariz curto / Anteversão de narinas SI + + + 3/3
Filtro curto - - + - 1/4
Bochechas cheias - + + - 2/4
Dentes pequenos e espaçados - + - - 1/4
Boca grande + - - + 2/4
Testa larga + + - - 2/4
Ponta nasal bulbosa + + + + 4/4
Hipoplasia malar SI + - - 1/3
Lábios grossos e proeminentes + + + + 4/4
Maloclusão SI + - - 1/3
Mandíbula pequena SI + + - 2/3
Problemas cardiovasculares - - + - 1/4
CONTINUA

17
TABELA 2 (Continuação)
Anormalidade de tecido conectivo SI - - - 0/3
Hipercalcemia / hipercalciúria SI - - - 0/3
Total de características 9/14 15/22 11/22 7/22
R+, resultado da FISH alterado; F, feminino; M, masculino; +, característica presente; -,
característica ausente; SI, sem informação; NA, não avaliável pelo SON-R.
Foram identificadas duas pacientes com suspeita de Síndrome de Smith-Magenis, sendo
ambas do sexo feminino, e ambos resultados da FISH foram normais. A caracterização clínica
dessas pacientes pode ser observada na TABELA 3.
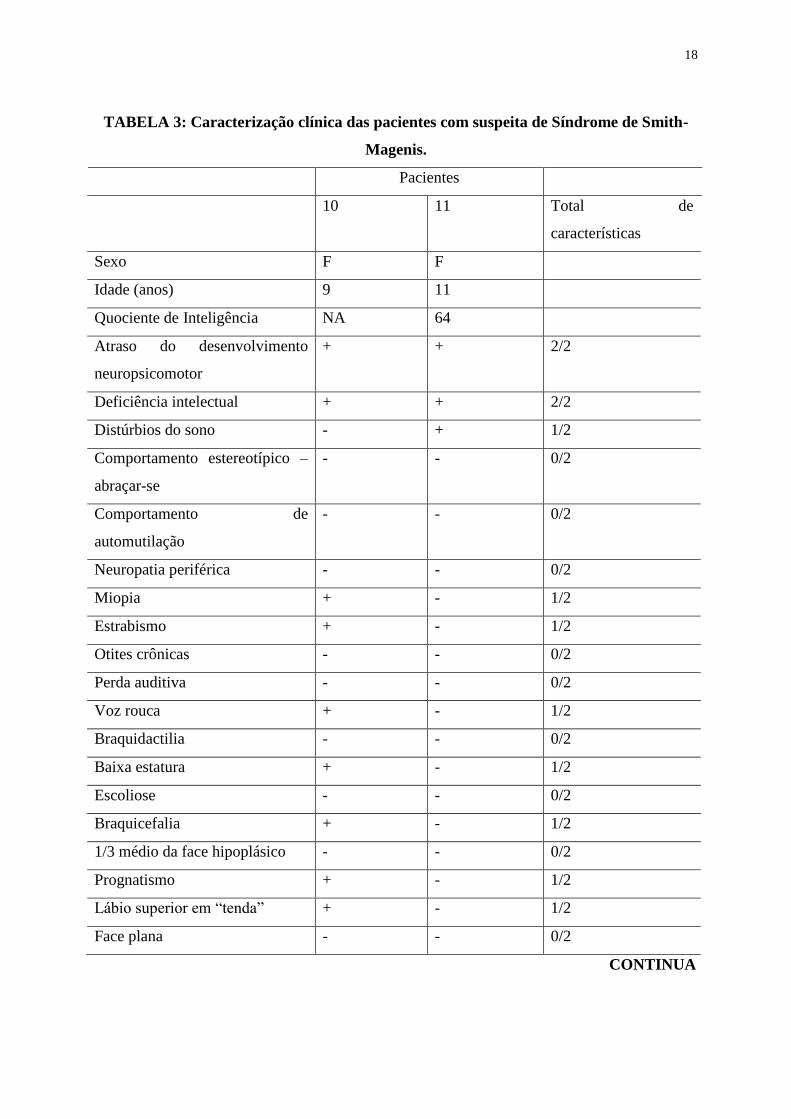
18
TABELA 3: Caracterização clínica das pacientes com suspeita de Síndrome de Smith-
Magenis.
Pacientes
10 11 Total de
características
Sexo F F
Idade (anos) 9 11
Quociente de Inteligência NA 64
Atraso do desenvolvimento
neuropsicomotor
+ + 2/2
Deficiência intelectual + + 2/2
Distúrbios do sono - + 1/2
Comportamento estereotípico –
abraçar-se
- - 0/2
Comportamento de
automutilação
- - 0/2
Neuropatia periférica - - 0/2
Miopia + - 1/2
Estrabismo + - 1/2
Otites crônicas - - 0/2
Perda auditiva - - 0/2
Voz rouca + - 1/2
Braquidactilia - - 0/2
Baixa estatura + - 1/2
Escoliose - - 0/2
Braquicefalia + - 1/2
1/3 médio da face hipoplásico - - 0/2
Prognatismo + - 1/2
Lábio superior em “tenda” + - 1/2
Face plana - - 0/2
CONTINUA

19
TABELA 3 (Continuação)
Total de características 9/19 3/19
F, feminino; +, característica presente; -, característica ausente; NA, não avaliável pelo SON-
R.
Foi avaliado um paciente com suspeita de Síndrome de Sotos, do sexo masculino, sendo o
resultado de FISH normal. A caracterização clínica desse paciente está descrita na TABELA
4.

20
TABELA 4: Caracterização clínica do paciente com suspeita de Síndrome de Sotos.
Paciente
12
Sexo M
Idade (anos) 12
Quociente de Inteligência 62
Atraso do desenvolvimento neuropsicomotor +
Deficiência intelectual +
Macrocefalia -
Proeminência malar +
Cabelo esparso em região frontotemporal +
Frontal alto e proeminente +
Fenda palpebral oblíqua para baixo +
Rosto estreito e alongado -
Mandíbula pequena e proeminente -
Cabeça em formato de pêra invertida +
Hipotonia e coordenação pobre +
Altura e/ou perímetro cefálico 2 ou mais
desvios-padrão acima da média
-
Total de características 8/12
M, masculino; +, característica presente; -, característica ausente.
Foi avaliado um paciente com suspeita de Síndrome de Angelman, do sexo masculino, sendo
o resultado de FISH normal. A caracterização clínica desse paciente está descrita na TABELA
5.

21
TABELA 5: Caracterização clínica do paciente com suspeita de Síndrome de Angelman.
Paciente
13
Sexo M
Idade (anos) 9
Quociente de Inteligência NA
Atraso do desenvolvimento neuropsicomotor +
Desenvolvimento progressivo (sem perda de
habilidades)
-
Deficiência intelectual +
Comprometimento da fala +
Hipotonia -
Distúrbios do equilíbrio +
Distúrbios do sono +
Riso frequente -
Convulsões +
Microcefalia SI
EEG anormal +
Prognatismo -
Língua protusa +
Boca grande +
Dentes espaçados +
Estrabismo +
Pele hipocrômica, cabelos e olhos claros +
Total de características 12/16
M, masculino; +, característica presente; -, característica ausente; NA: não avaliável pelo
SON-R; SI, sem informação.
Foi avaliada uma paciente com suspeita de Síndrome Velocardiofacial, do sexo feminino,
sendo o resultado de FISH normal. A caracterização clínica desta paciente é mostrada na
TABELA 6.

22
TABELA 6: Caracterização clínica da paciente com suspeita de Síndrome
Velocardiofacial.
Paciente
14
Sexo F
Idade (anos) 13
Quociente de Inteligência 55
Atraso do desenvolvimento neuropsicomotor +
Deficiência intelectual +
Defeito cardíaco congênito +
Anomalia de palato -
Imunodeficiência SI
Hipocalcemia SI
Anomalias gastrointestinais, problemas de
alimentação e para engolir, constipação
SI
Perda auditiva SI
Anomalias renais SI
Deficiência de hormônio do crescimento
(GH)
SI
Anomalias de laringe, traqueia e esôfago SI
Doenças autoimunes +
Total de características 4/5
F, feminino; +, característica presente; -, característica ausente; SI, sem informação.
Foi identificada uma paciente com suspeita de Síndrome de Langer-Giedion, do sexo
feminino, sendo o resultado de FISH normal. A caracterização clínica desta paciente é
descrita na TABELA 7.

23
TABELA 7: Caracterização clínica da paciente com suspeita de Síndrome de Langer-
Giedion.
Paciente
15
Sexo F
Idade (anos) 13
Quociente de Inteligência 57
Atraso do desenvolvimento neuropsicomotor +
Deficiência intelectual +
Nariz proeminente em forma de pêra +
Orelhas proeminentes -
Microcefalia -
Lábio superior fino +
Baixa estatura -
Pele redundante na infância -
Aumento do número de nevos -
Cabelo fino e esparso -
Epífises falangeanas em formato de cone -
Articulações hiperextensíveis -
Múltiplas exostoses +
Quirodactilia +
Perda auditiva +
Total de características 7/16
F, feminino; +, característica presente; -, característica ausente.

24
VI. DISCUSSÃO
A DI possui uma notável heterogeneidade em suas etiologias e, consequentemente, em suas
manifestações clínicas. No estudo realizado por Cabarca et al.(26), 23,8% dos casos de DI
foram explicados por causas genéticas, principalmente em pacientes com DI leve a moderada,
sendo a segunda causa mais frequente do problema, pois as causas ambientais foram
responsáveis por 36,4% da DI. Entretanto, 23% da amostra permaneceu sem diagnóstico
específico, sendo que a maioria possuía estudos normais de neuroimagem, metabólicos e
citogenética convencional. Neste grupo considerável de pacientes sem diagnóstico, técnicas
como FISH, MLPA e aCGH poderiam ser valiosas para sua elucidação. Contudo, há limitação
ao acesso dessas técnicas genéticas de última geração, apesar da importância desses
estudos(26).
As síndromes de microdeleção possuem diversos sinais e sintomas, havendo normalmente
associação com atraso do desenvolvimento e anomalias congênitas variadas. Elas são
detectadas a partir de técnicas como FISH, MLPA e aCGH, pois possuem extensões menores
que a capacidade de detecção do cariótipo, que tem uma resolução limitada. A FISH consegue
superar essas limitações, uma vez que possui maior resolução que a citogenética
convencional, promovendo análise de uma região específica, tendo como uma desvantagem o
fato de não analisar o genoma completo(27). A MLPA, por sua vez, permite a detecção de
anormalidades como deleções e duplicações em até 50 sequências genômicas ao mesmo
tempo. Já a aCGH é capaz de identificar perdas e ganhos de material em todo o genoma,
configurando-se como uma técnica de alta resolução(28). Porém, a FISH e a MLPA são
consideradas técnicas mais acessíveis financeiramente e, portanto, geralmente são usadas
como métodos de triagem, como primeiro ou segundo passo(27, 28).
No presente estudo, 26,7% (4/15) dos pacientes que realizaram FISH obtiveram resultado
alterado, sendo um número bastante superior ao encontrado por Halder et al.(27): 7,6%
(23/301). O estudo de Halder et al. levanta como possível fator que leva à baixa positividade
do teste a acurácia diminuída da clínica dos pacientes, que pode acontecer principalmente em
amostras constituída por bebês e crianças, fase em que as dismorfias faciais podem não ser
bem identificadas, levando a uma indicação inapropriada da FISH(27). Este estudo teve média
de idade dos casos de 10,2 anos, enquanto os casos de Halder et al. tiveram média de idade de
4,47 anos, sendo uma possível razão para a maior positividade do presente estudo.

25
A FISH é considerada mais limitada que técnicas como a MLPA em casos sem uma
caracterização clínica altamente sugestiva de determinada síndrome. Apesar de ambas as
técnicas possuírem concordância entre seus resultados, a MLPA torna-se vantajosa para
substituir a FISH na triagem da prática clínica, por ser mais ampla(29). Sabe-se que as
microdeleções frequentemente ocorrem concomitante com outras deleções e duplicações, que,
portanto, a FISH não irá detectar. Por isso, sugere-se que a MLPA seja utilizada como
primeiro teste de triagem nos pacientes com DI e cariótipo normal, considerando-se que é
uma técnica relativamente simples e de baixo custo(29), utilizando posteriormente FISH para
confirmação diagnóstica, triagem dos familiares, diagnóstico pré-natal, ou aCGH, caso
necessário(27, 30).
Realizou-se investigação para Síndrome de Prader-Willi (SPW) em 5 pacientes neste estudo,
havendo 2 resultados alterados e 3 normais. Todos os pacientes investigados apresentaram
obesidade, mãos pequenas, atraso do desenvolvimento neuropsicomotor e DI, sendo que os 3
pacientes que realizaram teste de QI foram classificados como DI leve. Além disso, a
hiperfagia foi uma característica presente em todos os 4 pacientes em que a informação foi
obtida. A hipotonia na infância esteve presente em 4 dos 5 pacientes.
Em um estudo feito por Kim et al.(31), 93,3% dos pacientes com SPW possuíam DI ou atraso
do desenvolvimento, todos apresentavam dismorfias faciais (como olhos amendoados e lábio
superior fino), 73,3% teve dificuldade de alimentação na infância e 66,7% apresentou
hipotonia neonatal ou na infância. Outra característica marcante na amostra desses autores foi
que 75% dos pacientes possuía criptorquidismo, indicando que essa possa ser uma
caraterística importante a ser pesquisada na síndrome.
Todos os 5 pacientes deste estudo obtiveram uma caracterização clínica sugestiva da
síndrome, pois todos se encaixaram em mais de 66,6% das características pesquisadas.
Entretanto, a FISH somente confirmou a SPW em 1 paciente. A segunda paciente com FISH
alterada não apresentou a del15q11-q13, tendo demonstrado uma deleção em 15q26, porém
com clínica compatível com SPW. Deve-se ter em mente, entretanto, que a microdeleção
paterna é responsável por 75 a 80% dos casos de SPW, e que portanto a síndrome pode ser
causada por outros mecanismos moleculares, como dissomia uniparental materna e defeitos
no imprinting(12). Capelli et al.(32) descreveram uma paciente com deleção de 15q26.1 com
clínica compatível com a Síndrome de Angelman (atraso do desenvolvimento,
comportamento autista, epilepsia e dismorfias faciais), porém sem alteração na região típica

26
de Prader-Willi/Angelman. A fim de haver melhor avaliação da extensão da deleção e/ou
concomitância com outras alterações, a paciente foi encaminhada para realização de array.
Os 3 pacientes que tiveram resultado normal foram encaminhados para realização de novos
estudos, sendo que um deles, após reavaliação clínica, teve nova suspeita com posterior
confirmação diagnóstica: pan-hipopituitarismo.
Ao todo, 4 pacientes realizaram FISH para Síndrome de Williams (SW), sendo 2 resultados
alterados, confirmando a doença, e 2 normais. Todos os pacientes possuíam atraso do
desenvolvimento neuropsicomotor e/ou DI, mas apenas um realizou o teste de QI, sendo
classificado como DI leve. Além disso, todos os 4 possuíam ponta nasal bulbosa e lábios
grossos e proeminentes, e todos os 3 que pesquisaram a característica nariz curto/anteversão
de narinas tiveram esse dado positivo. Há dados que foram presentes exclusivamente nos
pacientes com resultado de FISH alterado, como a personalidade excessivamente amigável,
estrabismo e testa larga, indicando que essas características podem ser mais específicas e
importantes para diferenciar os prováveis pacientes com a síndrome.
Antonell et al.(14) encontraram que 100% dos pacientes com SW em seu trabalho possuíam
face e personalidade típicas e 96% possuía hiperacusia, indicando a importância da
hipersensibilidade ao som ser pesquisada nos pacientes suspeitos. Os sintomas
gastrointestinais estiveram presentes em 89% dos pacientes e os problemas cardiovasculares
em 66%. Entretanto, nenhum dos pacientes com FISH alterado no presente estudo apresentou
problemas cardiovasculares.
Ambos os pacientes com FISH confirmando a SW tiveram uma boa caracterização clínica,
uma vez que tiveram mais de 64% das características clínicas pesquisadas presentes. Já os
pacientes que tiveram FISH normal tiveram até 50% das características presentes. Esses 2
pacientes com FISH normal foram reavaliados clinicamente e encaminhados para realização
de aCGH. A suspeita de SW foi afastada para ambos e para um deles foi feita suspeita de
Síndrome de Carpenter, posteriormente.
Neste estudo, duas pacientes realizaram investigação para Síndrome de Smith-Magenis
(SSM) e ambas tiveram resultado normal. Quanto ao QI, uma delas foi considerada não
avaliável pelo SON-R e a outra foi classificada como tendo DI leve. Ambas apresentaram
atraso do desenvolvimento e DI; contudo, nenhuma das duas apresentou comportamento
estereotípico, automutilação, neuropatia periférica, otites crônicas, perda auditiva,
braquidactilia, escoliose, 1/3 médio da face hipoplásico e face plana.

27
De acordo com o trabalho de Elsea et al.(17), 100% dos pacientes estudados apresentou DI
variável e mais de 90% possuía braquicefalia, terço médio da face hipoplásico, hipotonia,
atraso motor e na fala, distúrbios do sono e anormalidades dentárias, sendo essas
provavelmente características muito importantes para o diagnóstico de SSM. Além disso, 70 a
90% possuía lábio superior “em tenda” e comportamento de automutilação, mais de 80%
possuía braquidactilia e voz rouca e 80 a 90% possuía otites crônicas.
A partir da comparação feita com a caracterização clínica do trabalho de Elsea et al., pode-se
inferir que as pacientes do presente estudo não possuíam uma clínica bastante sugestiva de
SSM, repercutindo no resultado normal da FISH. Uma das pacientes teve 47,3% das
características pesquisadas presentes e a outra paciente teve apenas 15,7%. Considerando o
quadro, fez-se necessária uma nova avaliação clínica de ambas e nova formulação
diagnóstica, sendo ambas encaminhadas para realização de aCGH.
Foi avaliado um paciente com suspeita de Síndrome de Sotos (SS) e o resultado da FISH foi
normal. Este paciente possui DI leve e foram consideradas presentes 66,6% das características
pesquisadas. A SS é uma das síndromes de crescimento excessivo mais comuns, sendo que
90% dos indivíduos com a doença possuem altura e/ou perímetro cefálico mais que 2 desvios-
padrão acima da média, havendo idade óssea avançada em 74% a 100% dos casos(22).
Portanto, apesar do paciente do presente estudo possuir a maioria das características
pesquisadas, ele não possui uma característica considerada importante para o diagnóstico da
síndrome, que é a alteração citada na altura e/ou perímetro cefálico.
No estudo de Leventopoulos et al.(33), concluiu-se que 86% dos pacientes com SS possuía
crescimento excessivo, 97% tinha atraso do desenvolvimento, 76% tinha idade óssea
avançada e 47% tinha anomalias cerebrais detectadas em exame de ressonância magnética.
Considerando as diferenças clínicas do paciente com a literatura e o resultado normal da
FISH, a suspeita diagnóstica foi reavaliada e excluída, sendo o paciente encaminhado para
realização de aCGH.
Neste estudo, foi avaliado um paciente para Síndrome de Angelman (SA), que possui DI e
atraso do desenvolvimento neuropsicomotor. O QI do paciente não pôde ser estimado, pois
foi considerado não avaliável pelo SON-R. O resultado da FISH foi normal, apesar de ter
havido uma boa caracterização clínica, com 75% das características pesquisadas presentes.
Deve-se ressaltar, entretanto, que assim como a SPW, a SA possui diferentes mecanismos
moleculares de origem. Cerca de 60 a 75% dos casos são causados pela microdeleção materna

28
em 15q11-q13, existindo também outros mecanismos como dissomia uniparental paterna,
mutações no centro de imprinting e mutação no gene UBE3A(10).
Há 4 sinais considerados consistentes para a SA, que são a presença de DI funcionalmente
severa, distúrbio de movimento/balanço, discurso prejudicado, fenótipo comportamental
(facilmente excitável, feliz, riso fácil, hipermotricidade)(34). O paciente do presente estudo
apresenta 2 desses sinais, que são a DI e dificuldade na fala. Entre os sinais tidos como
frequentes da SA, estão as convulsões (ocorrendo entre 80 a 95% dos casos), EEG anormal,
distúrbios do sono, microcefalia, problemas gastrointestinais, fascinação pela água, hábito de
colocar objetos na boca e pronação dos tornozelos(34), sendo que os 3 primeiros encontram-se
presentes e os demais não foram pesquisados no paciente.
De acordo com o quadro apresentado, faz-se necessária uma nova pesquisa de dados clínicos
do paciente e prosseguir com uma nova investigação laboratorial, a fim de que possa-se
melhor avaliar se a suspeita deve ser mantida ou descartada.
Uma paciente obteve suspeita de Síndrome Velocardiofacial (SVCF), com resultado de
FISH normal. Ela apresenta DI leve, classificada de acordo com o QI, atraso do
desenvolvimento neuropsicomotor, doença cardíaca congênita e doença autoimune. Apesar de
possuir 80% das características pesquisadas presentes, foram pesquisadas apenas 5
características, deixando de avaliar, portanto, 7 quesitos.
Cerca de 70% dos casos de SVCF possuem doença cardíaca congênita, dado que pode ter
contribuído para levantar a suspeita de SVCF. Entretanto, uma característica comum da
síndrome é a anomalia de palato, que não está presente na paciente deste estudo, além de
anomalias vasculares e de faringe, que não foram pesquisadas. Para pacientes com SVCF, é
preciso que seja feita uma avaliação clínica aprofundada, pois muitos dos sinais e sintomas
apresentados na síndrome podem ser comuns a outras doenças também(24). A paciente foi,
posteriormente, encaminhada para realização de aCGH.
No presente estudo, uma paciente realizou investigação para a Síndrome de Langer-Giedion
(SLG), sendo o resultado da FISH normal. Ela possui DI leve, atraso do desenvolvimento,
nariz proeminente em forma de pêra, lábio superior fino, múltiplas exostoses, quirodactilia e
perda auditiva. Foram positivas apenas 43,7% das características clínicas pesquisadas, o que
pode ter levado ao resultado negativo.
Devidayal et al.(18) descreveram o caso de uma paciente com SLG confirmada, que possuía
cabelos, sobrancelhas e cílios finos e esparsos, nariz bulboso, filtro longo, lábio superior fino,

29
mandíbula hipoplásica, dentes irregulares, cabeça pequena e orelhas grandes e protrusas e
exostoses nos ossos longos. Além dessas características, podem estar associadas a essa
síndrome outros sinais e sintomas como DI, microcefalia, perda auditiva, fenda palatina,
miopia, epilepsia, atraso do crescimento e articulações hiperextensíveis. Sendo assim,
considerando que a paciente não possui características clínicas suficientes para fortalecer a
suspeita de SLG, foi realizado encaminhamento para realização de aCGH.
Sendo assim, após avaliação dos casos descritos, pode-se inferir que há casos de clínica
sugestiva para determinada síndrome, que deveriam portanto continuar a investigação
diagnóstica para confirmação através de uma técnica mais ampla que a FISH, como MLPA ou
hibridação genômica, e também há pacientes que, após reavaliação clínica, pode-se pensar em
outra suspeita diagnóstica. A boa descrição clínica faz-se necessária em todos os casos, pois,
além de conduzir a suspeita, orienta inclusive o melhor método diagnóstico, uma vez que a
FISH, em geral, resolve apenas os casos de clínica muito sugestiva. A detecção das síndromes
de microdeleção nos pacientes com DI e/ou atraso do desenvolvimento faz-se essencial, uma
vez que permite o adequado manejo do distúrbio e ajuda as famílias através do
aconselhamento genético(29).

30
VII. CONCLUSÕES
1. A FISH é uma técnica diagnóstica adequada para detectar síndromes de microdeleção cuja
clínica do paciente seja altamente sugestiva e para confirmação de casos.
2. Para casos em que a caracterização clínica não foi bastante sugestiva, não recomenda-se a
utilização da FISH como primeiro exame diagnóstico e sim outras técnicas como hibridação
genômica e MLPA.
3. Deve-se ter em mente o uso racional de técnicas laboratoriais em Genética Médica,
visando a mais adequada para cada caso em questão, além de realizar uma boa avaliação
clínica para orientar o melhor método.

31
VIII. SUMMARY
CLINICAL EVALUATION OF MICRODELETION SYNDROMES
INVESTIGATED BY FLUORESCENCE IN SITU HYBRIDIZATION IN
SALVADOR (BAHIA, BRASIL). Background: Intellectual disability (ID) is a
common neuropsyquiatric disorder that affects up to 3% of young population. ID’s etiology
can be environmental, genetics or both, and the genetic causes are responsible for up to 47,1%
of the cases. Microdeletions can be considered frequent, as they occur with 1 out of 1,000
newborns. Furthermore, they cannot be detected by conventional cytogenetics analysis. In
order to detect them, molecular techniques like fluorescence in situ hybridization (FISH) are
necessary. Objective: To describe clinical characterization and its comparison to laboratory
results in patients who are going through diagnostic research for microdeletion syndromes by
FISH. Methodology: This is a descriptive study in which patients with isolated or syndromic
ID of unknown etiology with suspicion of genetic cause were submitted to clinical and
laboratory screening that progressed or not with suspicion of microdeletion syndromes. To
confirm the diagnosis, FISH has been done. Results: Out of 106 patients with ID included, 15
were submitted to FISH. Of those patients, 4 had altered results and 11 had normal ones.
Discussion: Considering the patients submitted to FISH in this study, 26,7% (4/15) had
altered result. There are cases with suggestive clinic characterization for some syndromes that
should be further investigated to confirm the diagnosis using a more extensive technique than
FISH, such as MLPA or genomic hybridization. Moreover, there are patients who should be
investigated for other suspects, after a new clinic evaluation. Conclusions: FISH is a good
diagnostic technique to detect microdeletion syndromes when the clinical characterization is
highly suggestive. If it is not, it is recommended to use genomic hybridization techniques or
MLPA as the first diagnostic exam.
Key words: 1. Intellectual disability, 2. Genetics, 3. FISH technique.

32
IX. REFERÊNCIAS BIBLIOGRÁFICAS
1. Vasconcelos MM. Retardo mental. J. Pediatr. 2004 Abr; 80 (2): 71-82.
2. Instituto Brasileiro de Geografia e Estatística - IBGE - 2010. Censo Demográfico.
Brasil; 2010. Disponível em:
ftp://ftp.ibge.gov.br/Censos/Censo_Demografico_2010/Caracteristicas_Gerais_Religia
o_Deficiencia/tab1_3.pdf
3. Organização Mundial da Saúde. CID-10 - Classificação Estatística Internacional de
Doenças e Problemas Relacionados à Saúde. 10ª rev. São Paulo: Universidade de São
Paulo; 1997. Disponível em:
http://www.datasus.gov.br/cid10/V2008/WebHelp/f70_f79.htm
4. Stevenson RE, Massaey PS, Schroer RJ, McDermott S, Richter B. Preventable
Fraction of Mental Retardation: analisis based on individuals with severe mental
retardation. Ment Retard. 1996; 34 (1): 82-8.
5. Fan YS, editor. Molecular Cytogenetics: Protocols and Applications (Methods in
Molecular Biology). 1. ed. New Jersey: Humana Press; 2002.
6. Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA Assay in the Molecular
Diagnosis of Gene Copy Number Alterations in Human Genetic Diseases. Int J Mol
Sci. 2012; 13: 3245-76.
7. Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice
parameter: evaluation of the child with global developmental delay: report of the
Quality Standards Subcommittee of the American Academy of Neurology and The
Practice Committee of the Child Neurology. Neurology. 2003 Fev; 60 (3): 367-80.
8. Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, et al. Microarray
based comparative genomic hybridisation (array-CGH) detects submicroscopic
chromosomal deletions and duplications in patients with learning disability/mental
retardation and dysmorphic features. J Med Genet. 2004 Abr; 41 (4): 241-8.

33
9. Cho EH, Park YN, Cho JH, Kang YS. Comparing Two Diagnostic Laboratory Tests
for Several Microdeletions Causing Mental Retardation Syndroms: Multiplex
Ligation-Dependent Amplification vs Fluorescent In Situ Hybridization. Korean J Lab
Med. 2009 Fev; 20 (1): 71-6.
10. Van Buggenhout G, Fryns JP. Angelman syndrome. Eur J Hum Genet. 2009; 17:
1367-73.
11. Wattendorf DJ, Muenke M. Prader-Willi Syndrome. Am Fam Physician. 2005 Set; 72
(5): 827-30.
12. Grechi E, Cammarata B, Mariani B, Di Candia S, Chiumello G. Prader Willi
Syndrome: Clinical Aspects. J Obes. 2012, Out.
13. Francke U. Williams-Beuren syndrome: genes and mechanisms. Hum Mol Genet.
1999; 8 (10): 1947-54.
14. Antonell A, del Campo M, Flores R, Campuzano V, Pérez-Jurado LA. Síndrome de
Williams: aspectos clínicos y bases moleculares. Rev Neurol. 2006; 42 (1): 69-75.
15. Hennekam RCM. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006; 14: 981-5.
16. Escalera GI, Marina MLC, del Valle FM, Guardia NM, Rodríguez L, Martínez-
Fernández ML. Síndrome de Miller-Dieker. An Pediatr (Barc). 2009 Mar; 70 (3): 304-
6.
17. Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008; 16: 412-21.
18. Deividayal, Marwaha RK. Langer-Giedion Syndrome. Indian Pediatr. 2006; 43: 174-
5.
19. Carvalho JF. Síndrome Trico-Rino-Falangeana: Primeiro Caso Brasileiro. Acta
Reumatol Port. 2009; 34: 125-7.

34
20. Blanco-Lago R, Málaga I, García-Peñas JJ, García-Ron A. Síndrome de Wolf-
Hirschhorn. Serie de 27 pacientes: características epidemiológicas y clínicas. Situación
actual de los pacientes y opinión de sus cuidadores respecto al proceso diagnóstico.
Rev Neurol. 2013; 57: 49-56.
21. Rodríguez-Caballero Á, Torres-Lagares D, Rodríguez-Pérez A, Serrera-Figallo MA,
Hernández-Guisado JM, Machuca-Portillo G. Cri du chat syndrome: A critical review.
Med Oral Patol Oral Cir Bucal. 2010 Mai; 15 (3): 473-8.
22. Baujat G, Cormier-Daire V. Sotos Syndrome. Orphanet J Rare Dis. 2007; 2:36.
23. Juneja A, Sultan A. Sotos syndrome. J Indian Soc Pedod Prev Dent. 2011 Dez; 29 (6):
48-51.
24. Shprintzen RJ. Velo-Cardio-Facial Syndrome: 30 Years of Study. Dev Disabil Res
Rev. 2008; 14 (1): 3-10.
25. Horovitz DDG, Llerena JC, Mattos RA. Atenção aos defeitos congênitos no Brasil:
panorama atual. Cad. Saúde Pública. 2005 Jul-Ago; 21 (4): 1055-64.
26. Cabarcas L, Espinosa E, Velasco H. Etiología del retardo mental en la infancia:
experiencia en dos centros de tercer nivel. Biomédica. 2013 Jul-Set; 33 (3): 402-10.
27. Halder A, Jain M, Chaudhary I, Gupta N, Kabra M. Fluorescence in situ hybridization
(FISH) using non-commercial probes in the diagnosis of clinically suspected
microdeletion syndromes. Indian J Med Res. 2013 Jul; 138: 135-142.
28. Weise A, Mrasek K, Klein E, Mulatinho M, Llerena Jr. JC, Hardekopf D, et al.
Microdeletion and Microduplication Syndromes. J Histochem Cytochem. 2012 Mai;
60 (5): 346-58.

35
29. Boggula VR, Shukla A, Danda S, Hariharan SV, Nampoothiri S, Kumar R, et al.
Clinical utility of multiplex ligation-dependent probe amplification technique in
identification of aeriology of unexplained mental retardation: A study in 203 Indian
patients. Indian J Med Res. 2014 Jan; 139 (1): 66-75.
30. Khouzani HL, Kariminejad A, Zamani G, Ghalandary M, Bozorgmehr B, Amirsalari
S, et al. Investigation of microdeletions in syndromic intelectual disability my MLPA
in Iranian population. Arch Iran Med. 2014; 17 (7): 471-474.
31. Kim YJ, Cheon CK. Prader-Willi syndrome: a single center’s experience in Korea.
Korean J Pediatr. 2014; 57 (7): 310-316.
32. Capelli LP, Krepischi ACV, Gurgel-Giannetti J, Mendes MFS, Rodrigues T, Varela
MC, et al. Deletion of the RMGA and CHD2 genes in a child with epilepsy and
mental deficiency. Eur J Med Genet. 2012 Fev; 55 (2): 132-4.
33. Leventopoulos G, Kitsiou-Tzeli S, Kritikos K, Psoni S, Mavrou A, Kanavakis E, et al.
A Clinical Study of Sotos Syndrome Patients With Review of the Literature. Pediatr
Neurol. 2009; 40 (5): 357-364.
34. Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin
Genet. 2014 Mai; 7: 93-104.

36
X. ANEXOS
ANEXO I: Parecer do CEP

37

38

39
ANEXO II: Parecer adendo CEP com minha inclusão no projeto

40

41
ANEXO III: Termo de Consentimento Livre e Esclarecido
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
INFORMAÇÕES PARA OS PACIENTES, RESPONSÁVEIS E FAMILIARES
Você está sendo convidado a participar voluntariamente do projeto de pesquisa “Implantação
de uma Rede de Investigação Genética da Deficiência Mental nas Regiões
Norte/Nordeste/Centro-Oeste no Âmbito do SUS”. Leia atentamente as informações a seguir
antes de dar o seu consentimento. No caso de não entender bem peça mais esclarecimento e só
assine após ter certeza de ter esclarecido todas as suas dúvidas.
JUSTIFICATIVA E OBJETIVOS DO ESTUDO
Algumas pessoas apresentam atraso do desenvolvimento (para sentar, andar e/ou falar) e
dificuldade para aprender desde o nascimento devido defeitos que ocorreram na formação do
cérebro. Sabemos que em cerca de metade dessas pessoas a dificuldade para aprender pode ter
sido causada por alterações na constituição genética (DNA), outra metade pode ter sido por
problemas na gestação ou no parto de causa ambiental, e ainda, em alguns casos ainda não
conseguimos determinar a causa. O atraso do desenvolvimento pode apresentar diferentes
formas entre as pessoas podendo ser leve, moderado ou grave. Nós estamos fazendo um
estudo para tentar conhecer quais são essas causas e também para verificar se essas pessoas
apresentam problemas em outras partes do corpo além da dificuldade para aprender.
De que forma posso auxiliar neste estudo?
Você pode auxiliar neste estudo autorizando a participação e publicação de dados da consulta,
no que diz respeito aos exames médicos da pessoa, ao exame físico, aos exames realizados
como os bioquímicos, citogenéticos e de biologia molecular (caso tenham sido realizados
durante a investigação diagnóstica), e de fotografias quando for para mostrar alguma
característica relevante.
Quais os ricos e limites que podem ser encontrados nos exames?
Para fazer este exame é necessário tirar um pouco de sangue. O único incomodo será a picada
da agulha.

42
O paciente não terá nenhum dano físico na realização deste exame. Não é necessário estar em
jejum, nem tomar nenhum medicamento. Os procedimentos são iguais ao exame de sangue de
rotina.
É raro, mas é possível, que por problemas técnicos, o exame forneça resultados inconclusivos
sendo necessário repetir o exame.
Os resultados obtidos para essa pesquisa são absolutamente confidenciais, portanto serão
comunicados somente à pessoa ou responsável e ao profissional médico que acompanha a
pessoa. A comunicação dos resultados a terceiros só poderá ser realizada mediante
autorização do interessado.
Como será feita esta pesquisa?
As pessoas que atendem ao objetivo do estudo serão convidadas para participar da pesquisa.
Receberão uma cópia deste documento que deverá ser lido, entendido e assinado. Os
participantes serão atendidos pelo médico que fará algumas perguntas, examinará e se
necessário poderá solicitar alguns exames.
Quais os benefícios e malefícios deste estudo?
Não haverá nenhuma vantagem direta, tal como remuneração, com a participação neste
estudo, porém, os resultados poderão ajudar no entendimento sobre a deficiência mental,
direcionando médicos, educadores, terapeutas e outros profissionais no tratamento do paciente
melhorando a saúde e condição de vida.
O que vai ser feito com o material e os dados coletados de cada paciente?
O material e a ficha-protocolo com resultados dos exames dos pacientes serão armazenados
no Serviço de Genética Médica do Hospital Universitário Prof. Edgard Santos - Universidade
Federal da Bahia (UFBA). As amostras não serão oferecidas a outros centros ou laboratórios e
serão registradas por números, para evitar identificação dos pacientes. A identificação dos
pacientes será mantida em sigilo absoluto e estará sob a guarda dos pesquisadores
responsáveis.

43
A pessoa ou responsável legal poderá escolher entre ser informado ou não dos resultados do
estudo; aqueles que se interessarem em saber sobre os resultados obtidos com o presente
estudo serão orientados pela médica geneticista.
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
INFORMAÇÕES PARA OS PACIENTES, RESPONSÁVEIS E FAMILIARES
Eu,____________________________________________________, manifesto o meu
consentimento com envolvimento do meu filho no projeto de pesquisa intitulado:
“Implantação de uma Rede de Investigação Genética da Deficiência Mental nas Regiões
Norte-Nordeste-Centro Oeste no Âmbito do SUS.”
A natureza e objetivo do projeto de pesquisa, descritos na folha de informação em anexo,
foram explicadas a mim. Eu compreendo e concordo em participar.
- Eu compreendo que meu filho poderá não ter benefício direto por participar do estudo;
- Eu entendo que os possíveis riscos e/ou efeitos adversos, desconfortos e inconveniências,
como foi destacado na folha de informações, foram explicados a mim;
- Eu compreendo que, apesar das informações obtidas no estudo poderem ser publicadas, elas
serão confidenciais e meu filho não será identificado a partir delas;
- Eu compreendo que posso retirar meu filho do estudo em qualquer etapa e que isto não irá
afetar os cuidados médicos ou quaisquer outros aspectos da relação recebidos pelo meu filho;
- Eu compreendo que não haverá pagamento para meu filho por participar deste estudo;
- Eu tive a oportunidade de discutir a participação de meu filho neste projeto de pesquisa com
um membro da família ou amigo, e/ou tive a oportunidade de ter um membro da família ou
amigo presente enquanto o projeto de pesquisa estava sendo explicado pelo pesquisador;
- Eu estou ciente de que devo guardar uma cópia do Termo de Consentimento, depois de
assinado, e da folha de informações;
- Eu concordo que o material coletado (sangue) de meu filho seja utilizado no projeto acima.
Salvador, ______ de _____________ de ________
_________________________________________
Nome do Paciente

44
_________________________________________
Assinatura do responsável
_________________________________________
Assinatura da testemunha
_________________________________________
Assinatura do pesquisador

45
ANEXO IV: Ficha clínica

46

47

48

49

50
ANEXO V: Ficha clínica para Síndrome de Angelman
Nome do paciente:______________________________________________________________
Prontuário: _______________________ Data: ____/____/____
Protocolo clínico para diagnóstico da Síndrome de Angelman
Critérios diagnósticos Dados +
História do desenvolvimento e achados laboratoriais*
1. Pré-natal e história obstétrica normais, com perímetro cefálico normal e ausência de
defeitos congênitos. Dificuldades de alimentação podem estar presentes no recém-
nascido e na criança.
2. Atraso no desenvolvimento evidente aos 6-12 meses de idade, por vezes associado à
hipotonia truncal. Movimentos instáveis dos membros e / ou aumento do sorrindo
podem ser evidentes.
3. Desenvolvimento atrasado, porém progressivo (sem perda de habilidades)
4. Perfis laboratoriais (químico, metabólico e hematológico) normais.
5. Cérebro estruturalmente normal à ressonância magnética ou tomografia
computadorizada (pode ter atrofia cortical leve ou desmielinização).
Características clínicas
A. Consistentes (100%)
1. Atraso no desenvolvimento, funcionalmente severo.
2. Distúrbio do equilíbrio, geralmente marcha atáxica e / ou movimentos trêmulos dos
membros. Distúrbio de movimento pode ser leve. Pode não aparecer a ataxia típica,
mas pode estar presente balanço para frente, instabilidade, falta de jeito, ou
movimentos espasmódicos rápidos.
3. Padrão de comportamento: qualquer combinação de riso frequente e / ou sorriso;
atitude feliz aparente; personalidade facilmente excitável, muitas vezes com
movimentos de batimento das mãos ou acenos; comportamento hipermotor.
4. Comprometimento da fala, com nenhum ou uso mínimo de palavras; habilidades de
comunicação receptiva e não verbal superior à verbal.
B. Frequentes (mais que 80%)
1. Microcefalia.
2. Convulsões, geralmente antes dos 3 anos.
3. EEG anormal.
C. Associadas (20%–80%)
1. Occipital plano.

51
*Esses
acha
dos
são úteis como critérios de inclusão, mas os desvios não devem excluir o diagnóstico.
Fonte: American Journal of Medical Genetics 140A:413–418 (2006).
2. Sulco occipital.
3. Língua protusa.
4. Língua empurrando os dentes; distúrbios de sucção / deglutição.
5. Problemas de alimentação e/ou hipotonia truncal na infância.
6. Prognatismo.
7. Boca larga, dentes espaçados.
8. Salivação (baba) frequente.
9. Mastigação excessiva.
10. Estrabismo.
11. Pele hipocrômica, cabelos e olhos claros em comparação com a família.
12. Exacerbação dos reflexos tendinosos profundos na extremidade inferior.
13. Braço flexionado na deambulação.
14. Pronação do pé durante a marcha ou posicionamento em valgo.
15. Aumento da sensibilidade ao calor
16. Ciclos circadianos anormais e diminuição da necessidade de sono.
17. Fascínio por água; fascínio por itens dobráveis como papel ou plástico.
18. Comportamentos alimentares anormais.
19. Obesidade (crianças mais velhas)
20. Escoliose
21. Constipação

52
ANEXO VI: Ficha clínica para Síndrome de Prader Willi
Nome do
paciente:______________________________________________________________
Prontuário: _______________________ Data:
____/____/____
Protocolo clínico para diagnóstico da Síndrome de Prader-Willi
Fonte: PEDIATRICS Vol. 108 No. 5 November 1, 2001
Idade de
avaliação Critérios suficientes para exame molecular Dados +
Até 2 anos 1. Hipotonia com sucção pobre.
1 a 6 anos 1. História de hipotonia com sucção pobre.
2. Atraso global do desenvolvimento.
6 a 12 anos
1. História de hipotonia com sucção pobre (hipotonia
frequentemente persiste).
2. Atraso global do desenvolvimento.
3. Hábito de comer excessivamente (hiperfagia; obsessão com
alimentos) com obesidade central, se não controlada.
A partir dos
13 anos
1. Déficit cognitivo, geralmente retardo mental leve.
2. Hábito de comer excessivamente (hiperfagia; obsessão com
alimentos) com obesidade central, se não controlada.
3. Hipogonadismo hipotalâmico e / ou problemas de
comportamento típicos (incluindo acessos de raiva e
características de transtorno obsessivo-compulsivo).

53
ANEXO VII: Ficha clínica para Síndrome de Williams-Beuren
Nome:_________________________________________________________
Prontuário:_______________
Idade: ___________ Data:______________
Protocolo para diagnóstico clínico da Síndrome de Williams
Crescimento (evidência passada ou presente) Se presentes 3 de 5, marque 1 ponto ____
Comportamento e Desenvolvimento Se presentes 3 de 6, marque 1 ponto ____
Aspectos faciais Se presentes 8 de 17, marque 3 pontos ____
Problemas cardiovasculares
Por ecocardiografia (a) Se presente 1 de 2, marque 5 pontos ____
Problemas cardiovasculares (b) Se presente 1 de 3, marque 1 ponto ____
Anormalidade do tecido conectivo Se presentes 2 de 6, marque 2 pontos ____
Obesidade
[ ]Nascimento pós-termo (>41 semanas) [ ] Cólica prolongada: irritabilidade > 4 meses
[ ] Retardo de crescimento e ganho de peso < 5º percentil
[ ] Vômitos ou refluxo gastroesofágico [ ] Obstipação crônica
[ ]Personalidade excessivamente amigável [ ] Atraso no crescimento ou retardo mental
[ ] Hipersensibilidade ao som [ ] Problemas visoespaciais
[ ] Ansiedade [ ] Retardo na iniciação da fala, seguido por discurso
excessivo
[ ] Estreitamento bitemporal [ ] Testa larga
[ ] Dobras epicânticas ou ponte nasal plana [ ] Plenitude periorbital
[ ]Estrabismo (presente ou passado) [ ]Padrão iridiano de rendilhado estrelado
[ ] Nariz curto ou anteversão das narinas [ ] Ponta nasal bulbosa
[ ] Bochechas cheias [ ] Hipoplasia malar (ossos maxilares planos)
[ ] Filtro curto [ ] Lábios grossos e proeminentes
[ ] Dentes pequenos e bastante espaçados [ ]Maloclusão
[ ] Boca grande [ ] Mandíbula pequena
[ ] Lóbulos das orelhas proeminentes
[ ] Estenose aórtica supravalvar [ ] Estenose periférica de artéria pulmonar
[ ] Outra doença cardíaca congênita [ ] Hipertensão
[ ] Murmúrio cardíaco
[ ] Voz rouca [ ] Pescoço longo ou ombros inclinados
[ ] Hérnia inguinal [ ] Limitação ou liberdade excessiva de articulação
[ ] Divertículos intestinais/vesicais [ ] Prolapso retal

54
Estudos do cálcio Se presentes 1 de 2, marque 2 pontos _____
Total de pontos: ________
*Se o total de pontos for menor que 3 o diagnóstico de S. de Williams é improvável. Se o
escore for ≥ 3, o estudo de FISH deve ser considerado. Pacientes com Síndrome de Williams
tiveram escore médio de 9 (SD ± 2,86).
*Fonte: Committee on Genetics; American Academy of Pediatrics: Health care supervision
for children with Williams syndrome. Pediatrics. 2002;109:329.”
Diagnóstico clínico:
A Síndrome de Williams é caracterizada pela presença de fácies dismórfica, doença
cardiovascular, geralmente estenose aórtica supravalvar, deficiência mental um perfil
cognitivo característico e hipercalcemia idiopática.
Teste genético
A base para o diagnóstico da Síndrome de Williams é a detecção da deleção de genes
contígua na região crítica da Síndrome de Williams-Beuren (WBsCR), que envolve o gene da
elastina, ELN. Mais de 99% das pessoas com o diagnóstico clínico da SWB tem essa deleção
de genes contígua, o que pode ser detectado usando o teste de FISH (fluorescent in situ
hybridization) ou análise de mutação marcada.
[ ] Hipercalcemia [ ] Hipercalciúria

55
ANEXO VIII: Ficha clínica para Síndrome de Rubinstein-Taybi
Síndrome de Rubinstein-Taybi
Tabela diagnóstica com achados clínicos da Síndrome de Rubinstein-Taybi
Fissuras palpebrais oblíquas para baixo
Cílios longos
Sobrancelhas bastante arqueadas
Nariz proeminente com columela abaixo da asa nasal
Principais características Sorriso com aparência de careta
Palato arqueado
Leve micrognatia
Hálux largos e dedos dos pés grandes
Deficiência mental
Retardo no crescimento
Defeitos cardíacos congênitos
Anormalidades estruturais do trato geniturinário
Atraso na fala
Comportamento autista
Dificuldade de alimentação neonatal
Outras características Obstrução do ducto lacrimal
Estrabismo
Erro de refração
Tumores
Obesidade na infância e/ou adolescência
Teste genético: anormalidades cromossômicas são ocasionalmente observadas na rotina de
testagem citogenética. CREBBP e EP300 são os únicos genes associados com a SRT
atualmente conhecidos. Análise de FISH para CREBBP, detecta microdeleções em
aproximadamente 10% dos indivíduos com SRT. Análise seqüencial, detecta mutações em
CREBBP em outros 30%-50% dos indivíduos afetados. Mutações em EP300 são identificadas
em aproximadamente 3% dos indivíduos com SRT.

56
ANEXO IX: Ficha clínica para Síndrome de Miller-Dieker
Síndrome de Miller-Dieker
Achados clínicos da Síndrome de Miller-Dieker
Lisencefalia
Encefalopatia epilética
Heterotopia subcortical de banda
Convulsões
Fronte proeminente
Escavação bitemporal
Principais características Nariz curto com narinas antevertidas
Lábio superior proeminente com face vermelha
voltada
para baixo
Mandíbula pequena
Hérnia inguinal
Dificuldade de alimentação
Hipotonia leve
Testes genéticos: A síndrome de Miller-Dieker é causada tanto por pequenas deleções,
visíveis à citogenética, como microdeleções, detectáveis por FISH, envolvendo 17p13.3.
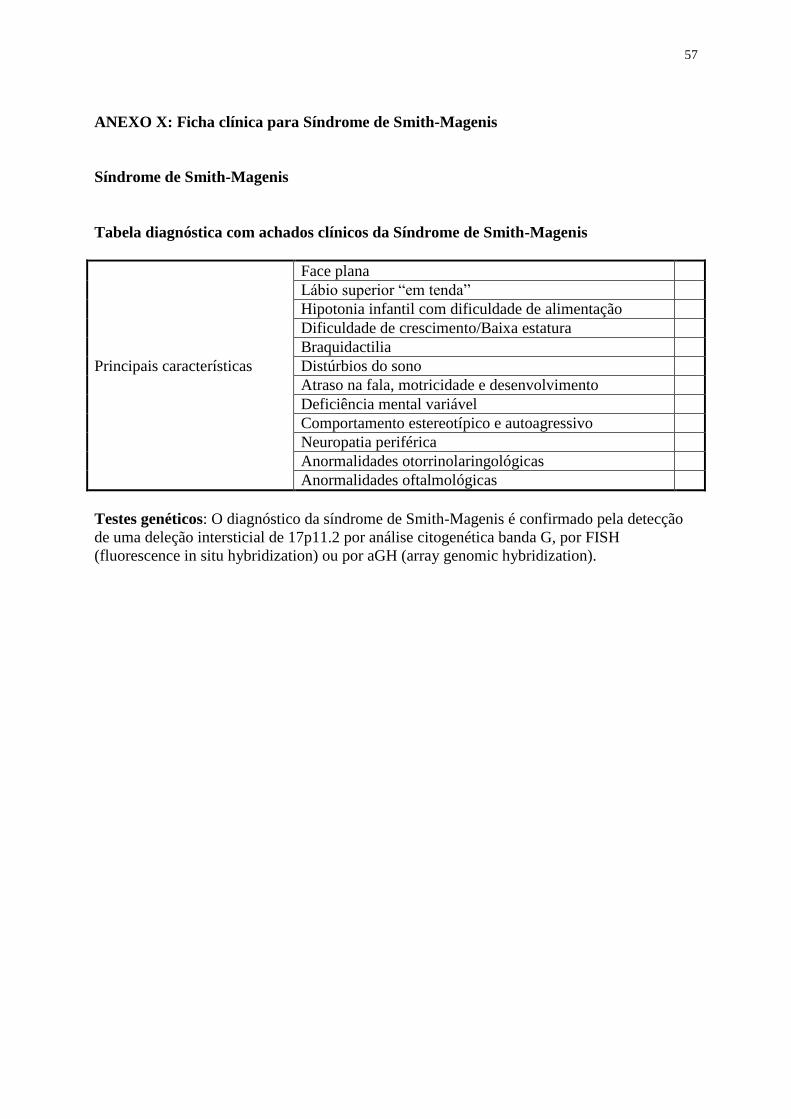
57
ANEXO X: Ficha clínica para Síndrome de Smith-Magenis
Síndrome de Smith-Magenis
Tabela diagnóstica com achados clínicos da Síndrome de Smith-Magenis
Face plana
Lábio superior “em tenda”
Hipotonia infantil com dificuldade de alimentação
Dificuldade de crescimento/Baixa estatura
Braquidactilia
Principais características Distúrbios do sono
Atraso na fala, motricidade e desenvolvimento
Deficiência mental variável
Comportamento estereotípico e autoagressivo
Neuropatia periférica
Anormalidades otorrinolaringológicas
Anormalidades oftalmológicas
Testes genéticos: O diagnóstico da síndrome de Smith-Magenis é confirmado pela detecção
de uma deleção intersticial de 17p11.2 por análise citogenética banda G, por FISH
(fluorescence in situ hybridization) ou por aGH (array genomic hybridization).

58
ANEXO XI: Ficha clínica para Síndrome de Langer-Giedion
Nome do
paciente:______________________________________________________________
Prontuário: _______________________ Data:
____/____/____
Protocolo para diagnóstico clínico de Síndrome de Langer-Giedion
Marque x ao lado dos achados positivos à anamnese e ao exame físico.
Fácies típica
Nariz proeminente em forma de pera.
Orelhas proeminentes.
Microcefalia.
Lábio superior fino.
Retardo de crescimento
Baixa estatura.
Pele e fâneros
Pele redundante na infância.
Aumento do número de nevos.
Cabelo fino e esparso.
Acometimento
osteoarticular
Epífises falangeanas em formato de
cone.
Articulações hiperextensíveis.
Múltiplas exostoses.
Quirodactilia.
Retardo mental
Atraso dos marcos do
desenvolvimento.
Desenvolvimento da linguagem lento.
Perda auditiva.
Fonte: 1) MARQUES, AS. MIOT, HÁ. MIOT, LDB. MARQUES, MEA. Você conhece esta
síndrome?. An Bras Dermatol 2005; 80(1): 85-8.
2)http://www.omim.org/entry/150230?search=number%3A%28150230%20OR%20604386%2
0OR%20608177%29&highlight=150230%20604386, com acesso em 27 de outubro de 2011.
Diagnóstico clínico
O diagnóstico clínico é firmado com os achados de fácies típica, acometimento
osteoarticular, retardo mental, atraso do crescimento e alterações de pele e fâneros.
Teste genético

59
A confirmação do diagnóstico clínico deve ser feita pela pesquisa de deleção no
cromossomo 8 em teste citogenético, seguido de FISH caso haja discordância entre suspeita
clínica e exame de cariótipo.

60
ANEXO XII: Ficha clínica para Síndrome de Wolf-Hirschhorn
Nome do
paciente:______________________________________________________________
Prontuário: _______________________ Data:
____/____/____
Protocolo para diagnóstico clínico de Síndrome de Wolf-Hirschhorn
Marque x ao lado dos achados positivos à anamnese e ao exame físico.
Fácies típica
“Capacete de guerreiro grego” (raiz do
nariz contínua com a testa).
Microcefalia.
Testa alta com glabela proeminente.
Hipertelorismo.
Epicanto.
Sobrancelhas muito arqueadas.
Filtro pequeno.
Boca voltada para baixo.
Micrognatismo.
Orelhas malformadas.
Deficiência de crescimento
Deficiência de crescimento pré-natal.
Retardo do crescimento pós-natal.
Atraso do
desenvolvimento/
incapacidade intelectual
Hipotonia e subdesenvolvimento
muscular.
Convulsões
História de convulsão.
Fonte: http://www.ncbi.nlm.nih.gov/books/NBK1183/, com acesso em 13 de outubro de
2011.
Diagnóstico clínico
O diagnóstico de Síndrome de Wolf-Hirschhorn é sugerido pela presença de fácies
típica, deficiência de crescimento, retardo psicomotor e história de convulsões. A confirmação
depende da detecção de deleção da região crítica de Wolf-Hirschhorn (cromossomo 4p16.3).
Teste genético
FISH detecta mais de 95% das deleções de 4p16.3 na Síndrome de Wolf-Hirschhorn,
enquanto análise citogenética o faz em 50% a 60% dos casos, nos quais a deleção é maior.
Essas duas técnicas isoladas não revelam bem rearranjos cromossômicos complexos
associados à deleção, sendo indicado aCGH quando necessária a caracterização completa.

61
ANEXO XIII: Ficha clínica para Síndrome de Cri-du-Chat
Nome do
paciente:______________________________________________________________
Prontuário: _______________________ Data:
____/____/____
Protocolo para diagnóstico clínico de Síndrome de Cri du Chat
Marque x ao lado dos achados positivos à anamnese e ao exame físico.
Fácies típica
Microcefalia.
Rosto redondo.
Ponte nasal larga.
Hipertelorismo.
Epicanto.
Fissuras palpebrais voltadas para
baixo.
Boca voltada para baixo.
Baixa implantação das orelhas.
Micrognatia.
Filtro pequeno.
Má oclusão dentária.
Choro característico
Choro semelhante a miado de gato.
Dermatóglifos anormais
Prega única nas mãos.
Características neonatais
Baixo peso ao nascimento.
Asfixia.
Cianose.
Sucção prejudicada.
Hipotonia.
Atraso do
desenvolvimento
psicomotor/ retardo de
crescimento
Retardo de crescimento pré-natal e
pós-
natal.
Corpo delgado.
Pés e mãos pequenos.
Desenvolvimento verbal lento.
Atraso dos marcos do
desenvolvimento.

62
Retardo mental.
Fonte: MAINARDI, PC. Cri du Chat syndrome Orphanet Journal of Rare Diseases
2006, 1:33.
Diagnóstico clínico
O diagnóstico é feito com base no achado de fácies típica, dermatóglifos anormais,
hipotonia e choro semelhante a miado de gato. O diagnóstico pode ser confirmado por teste de
cariótipo. Se houver caso suspeito com resultado normal de cariótipo, FISH deve ser
realizado.
Teste genético
Teste citogenético é o exame de primeira escolha para a pesquisa de deleção no
braço curto do cromossomo 5 (5p-) , mas pode resultar falso negativo, nesses casos FISH é o
método mais indicado para confirmar o diagnóstico clínico. PCR e aCGH permitem definir
pontos de quebra e microrrearranjos quando FISH não é preciso o suficiente .

63
ANEXO XIV: Ficha clínica para Síndrome de Sotos
Nome do
paciente:______________________________________________________________
Prontuário: _______________________ Data:
____/____/____
Protocolo para diagnóstico clínico de Síndrome de Sotos
Marque x ao lado dos achados positivos à anamnese e ao exame físico.
Fácies típica
*Critério diagnóstico mais
específico da síndrome.
Proeminência malar.
Cabelo esparso em região frontotemporal.
Frontal alto e proeminente.
Fendas palpebrais inclinadas para baixo.
Rosto estreito e alongado.
Mandíbula pequena e proeminente.
Cabeça com formato de pera invertida.
Dificuldade de
aprendizagem
Atraso do início dos marcos do
desenvolvimento e da capacidade motora.
Hipotonia e coordenação pobre.
Atraso da linguagem.
Prejuízo intelectual (variável de leve a
grave).
Crescimento exuberante Altura e/ou perímetro cefálico 2 ou mais
desvios-padrão acima da média.
Fonte: http://www.ncbi.nlm.nih.gov/books/NBK1479/, com acesso em 13 de outubro de 2011.
Diagnóstico clínico
O diagnóstico clínico de Síndrome de Sotos pode ser feito se o paciente atende a
critérios de fácies típica, dificuldade de aprendizagem e crescimento exuberante, estando
esses três tipos de alterações concomitantemente presentes em 90% dos casos. Para pacientes
que são casos suspeitos, mas não preenchem os três aspectos anteriormente citados, é
necessário teste genético para confirmação.
Teste genético
Mutação no gene NSD1 é reconhecidamente causa de Síndrome de Sotos e pode ser
detectadas por FISH ou outros métodos analíticos de deleção/duplicação, como MLPA. A
maioria dos pacientes não apresenta alteração ao teste citogenético, havendo raros casos com a
translocação 5q35.

64
ANEXO XV: Ficha clínica para Síndrome Velocardiofacial
Síndrome Velocardiofacial
Tabela diagnóstica com achados clínicos da Síndrome Velocardiofacial
Defeito cardíaco congênito
Anomalia de palato
Face típica
Dificuldade de aprendizado
Imunodeficiência
Hipocalcemia
Principais características Anomalias gastrointestinais, problemas de alimentação e
para engolir, constipação
Perda auditiva
Anomalias renais
Deficiência de hormônio do crescimento (GH)
Anomalias de laringe, traqueia e esôfago
Doenças autoimunes
Testes genéticos: O diagnóstico é confirmado quando há microdeleção de 22q11.2, por FISH,
MLPA ou CMA (chromosomal microarray).