universidade de mogi das cruzes alex martins …livros01.livrosgratis.com.br/cp028061.pdf · •...
TRANSCRIPT

1
UNIVERSIDADE DE MOGI DAS CRUZESALEX MARTINS MACHADO
CONSTRUÇÃO DE BACULOVÍRUS RECOBINANTES CONTENDO OGENE DA NUCLEOPROTEÍNA DO HANTAVIRUS ARARAQUARA
Mogi das Cruzes, SP2007

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
UNIVERSIDADE DE MOGI DAS CRUZESALEX MARTINS MACHADO
CONSTRUÇÃO DE BACULOVÍRUS RECOBINANTES CONTENDO OGENE DA NUCLEOPROTEÍNA DO HANTAVIRUS ARARAQUARA
Dissertação de mestrado apresentada aUniversidade de Mogi das Cruzes paraobtenção do título de Mestre pelo cursode Pós-Graduação em Biotecnologia.
Profº Orientador: Dr. José Luiz Caldas Wolff
Mogi das Cruzes, SP2007

3

4
Dedicatória
Gostaria de fazer esta dedicatória primeiramente aos meus pais, Celso
e Celma, pelo seu apoio incondicional e seus ensinamentos que me levaram a ser quem sou e
a alcançar mais esta conquista.
Também dedico especialmente a minha filha Mitzy que com muito
carinho, amor e paciência soube entender e apoiar este desafio

5
Agradecimento
Pelo Dom da vida, por ter me guiado e dirigido durante todo este período e por ter
feito este sonho possível, agradeço a Deus.
Agradeço a todos os professores doutores convidados para participarem da banca de
defesa: Prof. Dr. Ronaldo de Carvalho Araújo, Prof. Dr. Alexandre Wagner Silva Hilsdorf,
Prof. Dr. Wellington Luiz de Araújo, Prof. Dr. Luis Tadeu Moraes Figueiredo, Prof. Dr.
Carlos Augusto Pereira e Prof. Dr. Ronaldo Zucatelli.
Agradeço também a todos os docentes do programa de pós-graduação
em biotecnologia pelos ensinamentos e amizade.
Aos professores: Dr. Alexandre Wagner Silva Hilsdorf (Laboratório de
Genética de peixes e Aqüicultura); Dr. Ronaldo de Carvalho Araújo e Dr. Jorge Luiz
Pesquero (Laboratório de Animais transgênicos e Proteínas Recombinantes); Dra. Claudia
Bincoletto Trindade (Laboratório de Imunotoxicologia); Dr. Wellington Luiz de Araújo e Dr.
João Lúcio de Azevedo (Laboratório de Genética de Microorganismos); Dra. Regina Lúcia
Batista da Costa de Oliveira e Dr. Luiz Roberto Nunes (Laboratório de Genômica funcional);
Dra. Iseli Lourenço Nantes (Laboratório de Análises de estruturas e funções de
hemoproteínas) e Dr. Luis Tadeu Moraes Figueiredo (Laboratório de Virologia USP-RP)
agradeço por terem gentilmente aberto as portas dos seus laboratórios para que pudesse
realizar experimentos importantes do meu projeto.
A todos os meus amigos, fora do âmbito de trabalho, e familiares, que
seria impossível lista-los aqui, agradeço pela ajuda, amizade, carinho e principalmente pela
paciência, pois muitas vezes, não pude estar presente junto a eles.

6
Agradeço finalmente a todo o pessoal da Universidade de Mogi das
Cruzes: alunos, técnicos, professores e funcionários dos diversos laboratórios desta
instituição. Os nomes destes amigos estão listados aqui:
• César Henrique Yokomizo (Laboratório de Virologia Molecular)
• Fernanda de Oliveira (Laboratório de Virologia Molecular)
• Renata Martins (Técnica Laboratório de Virologia Molecular)
• Corina Macedo Vieira (Laboratório de Virologia Molecular)
• Luis Jungers de Barros (Técnico Laboratório de Virologia Molecular)
• Bruno Ramos Pellegrini (Laboratório de Virologia Molecular)
• Karina Cunha (Ex-integrante Laboratório de Virologia Molecular)
• Bruna Visniauskas (Ex-integrante Laboratório de Virologia Molecular)
• Alessandra Machado (Ex-integrante Laboratório de Virologia Molecular)
• Juliana Viana (Laboratório de Genética de Peixes e aqüicultura)
• Fabiana Iervolino (Laboratório de Genética de Peixes e aqüicultura)
• Tatiane Ivy Okasaky (Laboratório de Genética de Peixes e aqüicultura)
• Paulo Henrique de Mello (Laboratório de Genética de Peixes e aqüicultura)
• André Robson Justino da Silva (Laboratório de Genética de Peixes e aqüicultura)
• Angela Aparecida Moreira (Laboratório de Genética de Peixes e aqüicultura)
• Marizilda Magro (Laboratório de Genética de Peixes e aqüicultura)
• Sarah Lívia da Silva F. Matta (Laboratório de Genética de Peixes e aqüicultura)
• Fernanda Gallinaro Pessoa (Laboratório de Genética de Peixes e aqüicultura)
• Maria Cristina Pires Brum (Laboratório de Genética de Microorganismos)
• Emy Tiyo Mano (Laboratório de Genética de Microorganismos)
• Aline Aparecida Camargo das Neves (Laboratório de Genética de Microorganismos)

7
• Fernanda Alves Caravieri (Laboratório de Genética de Microorganismos)
• Marilia P. Bixillia (Laboratório de Genética de Microorganismos)
• Flávia Mendes da Cunha Holanda (Laboratório de Genética de Microorganismos)
• Christiano Marcello Vaz Barbosa (Laboratório de Imunotoxicologia)
• Carlos Rocha Oliveira (Laboratório de Imunotoxicológia)
• João Davison (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Sandro Soares (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Viviane Calixto (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Deborah Aiame (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Anderson Haro (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Amanda Carlos Castilho (Laboratório de Animais transgênicos e Proteínas
recombinantes)
• Milton Moraes (Laboratório de Animais transgênicos e Proteínas recombinantes)
• Renata Mesquita (Secretária Pós-Graduação)
• Luciana Gonçalves Soares (Secretária Pós-Graduação)
• Neilce Ribeiro Prado (Secretária NIB)
• Priscila de Oliveira Luiz (Técnica Laboratório de Genômica estrutural e funcional)
• Daniela Martins Fausto (Técnica Laboratório Multidisciplinar de Bioquímica)
Em fim, se eu esqueci de alguém me perdoem, foi devido à urgência em
escrever esta dissertação.

8
"A melhor de todas as coisas é aprender. O dinheiropode ser perdido ou roubado, a saúde e força podem falhar, mas o que você dedicou à suamente é seu para sempre."
Louis L'Amour

9
RESUMO
A hantavirose é uma das zoonose que vem preocupando as autoridades sanitárias de todoo mundo. Sua ocorrência se deve principalmente a distúrbios ecológicos e é transmitida aohomem através de inalação de partículas virais contida na excreta de roedores. São conhecidas2 doenças humanas distintas causadas pelo hantavírus: a febre hemorrágica com síndromerenal (FHSR) e a Síndrome Pulmonar e Cardiovascular (SPCVH). Os hantavírus são víruspertencentes à família Bunyaviridae, esféricos, envelopados, com diâmetro de 80 a 120 nm eprojeções glicoproteínas na sua superfície. Possuem genoma de RNA, com polaridadenegativa, fita simples e trissegmentado. O diagnóstico da infecção é realizado pela detecçãode anticorpos IgM contra Sin nombre vírus, através de testes ELISA. Este é um vírusencontrado principalmente nos EUA e a reação em ELISA se dá por reação cruzada, já que oshantavírus brasileiros são outros. A produção de antígenos nativos é um problema devidoprincipalmente ao alto risco envolvido na manipulação do hantavírus. Por esta razão osantígenos produzidos pela técnica do DNA recombinante estão se tornando padrão. O geneque codifica a Nucleoproteína viral é frequentemente usado para este propósito e váriossistemas de expressão já foram usados na tentativa de expressão desta proteína. O gene danucleoproteína, do Araraquara Hantavírus, foi inserido em plasmídeos e expressado em E.coli. Estudos anteriores indicam que as proteínas expressas em Sistema de Baculovírus podemser mais adequadas que a proteína produzida em E. coli. O objetivo deste trabalho foi produzirbaculovirus recombinantes contendo o gene da nucleoproteína do Araraquara Hantavírusutilizando para isso o vetores de transferência pSyn contendo o gene de interesse. Foiconstruído dois baculovírus recombinantes, um capaz de poduzir a proteína N junto a umacauda de histidina e outro, uma variante do baculovírus anterior, que é capaz de produzir anucleoproteína junto a uma seqüência de exportação. Após obtenção dos baculovírusrecombinantes estes foram purificados através de diluição seriada em 96 poços obtendo assimos vírus purificados. Esta purificação foi comprovada através de reação da PCR a qualtambém permitiu verificar o gene de interesse no genoma dos vírus recombinantes. Após apurificação dos vírus, estoques virais foram produzidos e servirão para uma futura expressão epurificação da proteína N do Hantavírus Araraquara.
Palavras-Chaves: Hantavírus Araraquara, Diagnóstico Hantavirose, ELISA nucleoproteína,Sistema de Expressão de Baculovírus.

10
ABSTRACT
The Araraquara hantavirus is the new member of Hantavirus genus, familyBunyaviridae and is the etiologic agent hantavirus pulmonary syndrome (SPCVH) in Brazil.The diagnostic of that disease is based in serologic tests that be the high importance ofhantavirus infection confirmation and for to epidemiologic studies. The production of nativeantigen presents two limitations: lower title in cell culture and high risk of contamination. Inagreement those factors, antigens are produce by the DNA recombinant technique becamestandard in this test. The N gene, that encoding the virus nucleoprotein is commonly use forthe production recombinant antigen. The gene product is important antigenic protein, a timethat high titles of IgM, directed to her, are produces in acute phase of infection, and still thatlower, titles of IgG are find for long periods after the viral infection. The objective of thiswork was the recombinant baculovirus construction that expression of Araraquara hantavirusnucleoprotein (N) fusion that one poly-Hys (6XHys) in N-terminal position. For that the Ngene, was isolated by rt-PCR technique from extract patient’s RNA. This product was clonedin pSynXIVVI+X3 transference vector and multiplied in E. coli cells. The transference vectorwas co-transfect between the Autographa californica multicapsid nucleopolyedrovirus(AcMNPV) DNA in insect cells (SF9). The recombinant virus produced was purificatedtrough serial dilutions. After the purification, it was made a new insect cells infection and 5days later was calculate of MOI and viral stock production.
Keywords: Araraquara Hantavírus, Serodiagnosis of Hantavírus, ELISA, Baculovirusexpression system.

11
LISTA DE TABELAS
Tabela 1 Relação entre os principais Hantavírus, doenças causadas e seusreservatórios naturais.
26
Tabela 2 Distribuição dos casos confirmados de SPCVH segundo período deocorrência e estado. Brasil 2005.
33
Tabela 3 Oligonucleotídeos utilizados para amplificar o gene N com cauda deHistidina.
48
Tabela 4 Oligonucleotídeos utilizados para amplificar a seqüência sinal deexportação do gene GP67.
49
Tabela 5 Oligonucleotídeos desenhados para sequenciar o gene danucleoproteína.
53
Tabela 6 Oligonucleotídeos desenhados para sequenciar o cassete dos vetoresde transferência.
53
Tabela 7 Relação entre baculovírus recombinantes e primers utilizados paraverificação do inserto no seu genoma.
56

12
LISTA DE ILUSTRAÇÕES
Figura 1 Morfologia esquemática dos Hantavírus 21
Figura 2 Distribuição geográfica dois diversos Hantavírus identificados nasAméricas
23
Figura 3 Esquema de infecção por Hantavírus 24
Figura 4 Reservatório naturais dosa Hantavírus no Brasil 27
Figura 5 Ciclo replicativo dos Hantavírus 28
Figura 6 Micrografia de um Baculovírus nucleopoliedrovírus 39
Figura 7 Esquema de produção dos baculovírus recombinantes. 42
Figura 8 Mapa do vetor de transferência pSynXIVVIX3 46
Figura 9 Seqüência do gene da nucleoproteína com cauda de histidina comlocalização dos primers utilizados para amplificação.
48
Figura 10 Seqüência do sinal de exportação do gene GP67 com localização dosprimers utilizados para amplificação.
49
Figura 11 Fluxograma mostrando a produção dos vetores de transferência pSyn-Hist-N e pSyn-Exp-Hist-N.
54
Figura 12 Blast-N da seqüência obtida através de seqüênciamento do pET-N-Ara 60
Figura 13 Blast-X da seqüência obtida através de sequenciamento do pET-N-Ara 61
Figura 14 Eletroforese em gel de agarose 1,0% mostrando os fragmentos deHistidina com nucleoproteína e seqüência sinal de exportação.
61
Figura 15 Eletroforese em gel de agarose 1,0% mostrando o tamanho final dosvetores de transferência recombinantes pSyn-HIst-N e pSyn-Exp-Hist-N
63
Figura 16 Fluxograma mostrando o tamanho dos fragmentos amplificados,plasmídeos pCRII e votores de transferência pSyn-HIst-N e pSyn-Exp-Hist-N.
64
Figura 17 Esquema pSyn-HIst-N 66
Figura 18 Esquema pSyn-Exp-Hist-N 68
Figura 19 Fotografias de células SF9 não infectadas, infectadas com vSyngal ecom Baculovírus recombinantes.
69

13
Figura 20 Eletroforese em gel de agarose 1,0% para verificar o inserto dosfragmentos no interior do genoma dos baculovírus recombinantes.
70
Figura 21 Diluição seriada Hist-N e Exp-Hist-N 71
Figura 22 Eletroforese em gel de agarose 1,0% mostrando a inserção dos genes deinteresse no genoma dos baculovírus recombinantes e a ausência dogene da betagalactosidase
72

14
LISTA DE ABREVIAÇÕES
6xHis Cauda de Histidina com 6 aminoácidos de Histidina
AcNPV Autographa californica nucleopoliedrovirus
ANAJ Vírus Anajatuba
AND Vírus Andes
ARA Vírus Araraquara
BAY Vírus Bayon
BCC Vírus Black Creeck Canal
BLAST Basic local Alignment Search Tool
BV Budded vírus
CAS Vírus Castelo dos Sonhos
DNA Ácido Desoxirribonucléico
ELISA Enzyme-linked immunosorbentassays
Exp Sinal de Exportação do gene da GP67
FSHR Febre Hemorrágica com Síndrome Renal
GPC Complexo Precursor de Glicoproteínas
IFA Imunofluorescência indireta
IgG Imunoglobulina G
IgM Imunoglobulina M
JUQ Vírus Juquitiba
LEC Vírus Lechiguanas
LN Vírus Laguna Negra
MOI Multiplicity of Infection
NY Vírus New York

15
ORF Finder Open Reading Frame
ORN Vírus Oran
PCR Reação em Cadeia de Polimerase
pCRII-Exp-Hist-N Plasmídeo de clonagem (pCRII) com sinal de exportação,nucleoproteína e cauda de histidina.
pCRII-Hist-N Plasmídeo de clonagem (pCRII) com a nucleoproteína e cauda dehistidina.
PDV Polyhedra-derived vírus
pET-N-Ara Vetor de expressão pET-TOPO 200 D contendo o gene N doAraraquara Hantavírus
pSyn-Exp-Hist-N Vetor de transferência (pSyn) contendo o sinal de exportação,nucleoproteína e cauda de histidina.
pSyn-Hist-N Vetor de transferência (pSyn) contendo a nucleoproteína e cauda dehistidina.
RIME Vírus Rio Mearim
RNA Ácido Ribonucléico
RT-PCR Reação em cadeia de polimerase com transcrição reversa.
SF9 Células de ovário de Spodoptera frugiperda
SMC Sítio de Múltipla Clonagem
SN Vírus Sin nombre
SPCVH – SPCH Síndrome Pulmonar e Cardiovascular por Hantavírus

16
SUMÁRIO
1. Introdução......................................................................................................................... 181.1 Histórico................................................................................................................. 181.2 Morfologia e Genética dos Hantavírus.................................................................. 191.3 Classificação dos Hantavírus................................................................................. 211.4 Transmissão............................................................................................................ 241.5 Reservatórios Naturais dos Hantavírus.................................................................. 251.6 Replicação dos Hantavírus..................................................................................... 271.7 Hantaviroses........................................................................................................... 29
1.7.1 Patogênese da SCPVH............................................................................... 291.7.2 Manifestações Clínicas............................................................................... 30
1.8 Imunologia das Hantaviroses ................................................................................ 311.9 Epidemiologia das Hantaviroses............................................................................ 321.10 Diagnóstico das Hantaviroses.............................................................................. 341.11 Produção de Antígenos Recombinantes para testes diagnósticos........................ 351.12 Proteínas Recombinantes..................................................................................... 371.13 Sistema de Expressão de Baculovírus.................................................................. 38
1.13.1 Baculovírus.............................................................................................. 381.13.2 Expressão Gênica nos Baculovírus.......................................................... 401.13.3 Utilização dos Baculovírus como Sistema de Expressão ........................ 40
2. Justificativa ...................................................................................................................... 433. Objetivo............................................................................................................................. 444. Método.............................................................................................................................. 45
4.1 Vetores, vírus e Linhagem Celular utilizada.......................................................... 454.1.1 Vetor de clonagem pET-N-Ara................................................................. 454.1.2 Vetor de Transferência pSynXIVVIX3..................................................... 454.1.3 Plasmídeo de clonagem pCRII................................................................... 474.1.4 Vírus vSyngalVI......................................................................................... 474.1.5 Linhagem celular utilizada......................................................................... 47
4.2 Desenho de oligonucleotídeos para síntese dos fragmentos utilizados nopreparo dos vetores de transferência ........................................................................... 47
4.2.1 Primers para o gene N com poliHistidina (6xHis)..................................... 484.2.2 Primers para o sinal de exportação do gene GP67..................................... 49
4.3 Síntese dos fragmentos e clonagem....................................................................... 504.3.1 Síntese da região do Gene N com poliHistidina (6xHis) para inserçãoem plasmídeos..................................................................................................... 504.3.2 Síntese do sinal de exportação do gene da GP67....................................... 50
4.4 Construção dos vetores de transferência ............................................................... 504.4.1 Clonagem do gene Hist-N no vetor de transferênciapSynXIVVIX3.................................................................................................... 504.4.2 Clonagem da seqüência de exportação (Exp) no vetor de transferênciapSyn-Hist-N........................................................................................................ 51
4.5 Purificação dos vetores de transferência: pSyn-Hist-N e pSyn-Exp-Hist-N................................................................................................................................... 514.6 Sequencimento dos vetores de transferência ......................................................... 52
4.6.1 Sequenciamento do pET-N-Ara................................................................. 524.6.2 Sequenciamento dos vetores de transferência recombinantes................... 53
4.7 Preparo do DNA do vSyngal VI-........................................................................... 56

17
4.8 Construção dos Baculovírus recombinantes através do processo de co-transfecção.................................................................................................................... 564.9 Confirmação da inserção do gene de interesse ..................................................... 574.10 Purificação dos Baculovírus recombinantes........................................................ 58
4.10.1 Diluição seriada em 96 poços.................................................................. 584.11 Verificação da pureza dos Baculovírus Recombinantes...................................... 58
4.12 Cálculo do “Multiplicity of Infection” (MOI) dos estoques virais .................... 595. Resultados e Discussões.................................................................................................... 60
5.1 Sequenciamento do pET-N-Ara............................................................................. 605.2 Montagem dos vetores de transferência recombinantes......................................... 625.3 Sequenciamento e Análise das regiões críticas dos vetores de transferênciarecombinantes.............................................................................................................. 65
5.3.1 pSyn-Hist-N............................................................................................... 665.3.2 pSyn-Exp-Hist-N........................................................................................ 67
5.4 Obtenção dos baculovírus recombinantes.............................................................. 695.5 Verificação do inserto no genoma dos baculovírus recombinantes....................... 715.6 Purificação dos baculovírus recombinantes e Cálculo do MOI............................. 725.7 Verificação da pureza dos baculovírus recombinantes.......................................... 735.8 Cálculo do MOI e produção dos estoques virais.................................................... 74
Conclusões e Sugestões......................................................................................................... 75Referências Bibliográficas.................................................................................................... 76Anexos................................................................................................................................... 88Anexo 1 – Reação de PCR.................................................................................................... 89Anexo 2 – Kit de Purificação GFX....................................................................................... 89Anexo 3 – Clonagem de Fragmentos em Plasmídeos........................................................... 90Anexo 4 – Transformação .................................................................................................... 90Anexo 5 – Extração Kit Qiagen............................................................................................ 90Anexo 6 – Extração de DNA de “Budded virus”.................................................................. 91Anexo 7 – Extração de DNA de poliedros virais.................................................................. 91

18
1. INTRODUÇÃO
1.1HISTÓRICO
Os hantavírus estão presentes no continente Asiático, e a moléstia com febre e fenômenos
hemorrágicos, ocasionada pelos mesmos, tem sido descrita na literatura chinesa desde o início
do século X (LEE, 1989).
A hantavirose foi novamente descrita em 1913 na União Soviética, Coréia e China (vírus
Hantaan) e em 1934, milhares de casos foram confirmados no continente Europeu,
principalmente na Escandinávia e Leste Europeu (vírus Puumala e Dobrava) (apud:
COLLIER & OXFORD 2000)
Em 1950, durante a guerra da coréia 3200 soldados americanos foram vitimados pela
febre hemorrágica com síndrome renal associado ao vírus Hantaan, ficando conhecida como
febre hemorrágica Coreana (GARMENDIA 2004, EARLE 1954)
Ainda que muitos estudos sobre esta patologia foram realizados somente em 1975, um
cientista coreano chamado Lee, isolou o vírus de tecidos de um roedor silvestre Apodemus
agrarius habitante do vale (taan) as margens do rio Han, na Coréia do Sul (LEE 1989; LEE et
al 1978). No mesmo ano, Lee confirmou a relação entre o hantavírus isolado e os quadros de
febre hemorrágica ocorridos anteriormente (LEE 1989).
Nas Américas, a primeira observação da presença de hantavirus foi em 1984, no norte dos
Estados Unidos, onde após captura do roedor silvestre Microtus pensilvanicuss observou-se
presença de anticorpos contra hantavírus no soro deste animal (YANAGIHARA et al 1985).
Entretanto, somente em 1993 aconteceu a primeira epidemia de Síndrome Pulmonar e
Cardiovascular por Hantavírus (SPCVH) que vitimou milhares de índios navajos da região de
Four Corners (NICHOL et al 1993).
Estudos sorológicos dos roedores predominantes nesta região e pessoas contaminadas
possibilitaram constatar a presença de um novo Hantavírus, o qual foi denominado Sin
Nombre vírus (SN) (NICHOL et al 1993)
Após a epidemia nos Estados Unidos, causada pelo SN, várias outras regiões, tanto da
América do Norte, Centro e Sul apresentaram casos de SPCVH .

19
No Brasil, o primeiro caso confirmado desta patologia ocorreu em 1993 na cidade de
Juquitiba, estado de São Paulo. Seu agente etiológico foi estudado a partir de análises
filogenéticas do genoma destes vírus, realizadas em 1999 e recebeu o nome de Vírus Juquitiba
(JUQ) fazendo referência a cidade onde apareceu o primeiro caso (JOHNSON et al 1999;
MONROE et al 1999, VASCONCELOS et al 1997).
Em 1995 e 1996 foram diagnosticados novos casos de SPCVH no Brasil através de testes
sorológicos de ELISA. Um caso foi relatado na região do Vilarejo de Castelo dos Sonhos,
estado de Mato Grosso o qual após análises filogenéticas realizadas por Johnson 1999 foi
denominado como uma nova linhagem viral, Vírus Castelo dos sonhos (CAS) (JOHNSON et
al 1999; SUZUKI et al 2004).
Em 1996, dois casos foram descritos na região sudoeste do estado de São Paulo,
especificamente nas cidades de Araraquara e Franca. Esta linhagem foi denominada vírus
Araraquara (ARA) (JOHNSON et al 1999; SUZUKI et al 2004).
Após este período, muitos novos casos têm surgido em todo o Brasil, predominantemente
na região sul e sudeste onde as práticas agrícolas e o desmatamento da vegetação nativa são
mais intensos.
Em 2003, foram isolados dois novos hantavírus a partir de tecidos de roedores silvestre
capturados na região norte do país. Estes novos hantavírus foram denominados vírus
Anajatuba (ANAJ) e vírus Rio Mearim (RIME) e fazem parte de um mesmo grupo
filogenético, podendo ser apresentados como uma única espécie (ROSA et al 2005).
1.2 MORFOLOGIA E GENÉTICA DOS HANTAVÍRUS
Os agentes do gênero Hantavirus são vírus esféricos, envelopados, com diâmetro de 73 a
150 nm e projeções glicoproteicas na sua superfície (G1 e G2) de aproximadamente 7 nm
fixadas em um envelope lipídico de duas camadas (LEDNICKY et al 2003; KANERVA et al
1998) (Figura 1)
Estas glicoproteínas G1 e G2 são do tipo I transmembrânicas, e possuem a função
específica de interatuar com os receptores celulares para permitir a entrada do vírus nas
múltiplas células a qual infecta (Macrófagos, plaquetas, células endoteliais, entre outras)
(GAVRILOVSKAYA et al 1999).

20
Estes vírus possuem um genoma de fita simples de RNA, com polaridade negativa e
trissegmentado, com seqüências nucleotídicas complementares nas extremidades 3’ e 5’ o que
permite que o RNA viral se mantenha circular dentro do virion (PLYUSNIN et al 1996;
MAES et al 2004). Esta complementariedade é altamente conservada no gênero Hantavírus e
sua função deve estar relacionada com a replicação viral.
Os segmentos do genoma viral são denominados: L (Large – Grande) que possui
aproximadamente 6500 nucleotídeos e codifica uma RNA polimerase dependente de RNA
(240 a 260 Kda) responsável por todos os passos de transcrição e replicação do genoma viral.
O segmento M (Médium – Médio) com aproximadamente 3600 a 3800 nucleotídeos que
codifica um precursor poliproteíco (GPC) que será clivado formando as duas glicoproteínas
(G1 e G2). Finalmente temos o segmento S (Small – pequeno) com tamanho que varia de
1300 a 2100 nucleotídeos e é responsável pela codificação da proteína nucleocapsídica viral
(NICHOL et al 1993; PLYUSNIN 2002).

21
Figura 1: Morfologia esquemática dos Hantavírus.Observamos que a partícula viral possui tamanho médiode 100 nm e projeções glicoproteícas no envelope viral; O material genético .RNA de fita simples seencontra circular e envolto por um polímero protéico de formado pela nucleoproteína viral.Fonte: KAPRAF (20002)
1.3 CLASSIFICAÇÃO DOS HANTAVÍRUS
Os hantavirus são um gênero pertencente à família Bunyaviridae e possuem cerca de 30
genótipos diferentes, dos quais, 20 genótipos são realmente patogênicos ao homem
(ELLIOTT et al 2000; VAPALAHTI et al 1996).
São conhecidas 2 doenças humanas distintas causadas pelo hantavírus: a febre
hemorrágica com síndrome renal (FHSR), que ocorre principalmente na Ásia e Europa, e a
síndrome pulmonar e cardiovascular (SPCVH), que ocorre nas Américas (VERITY et al
2000; KANERVA et al 1998).

22
A FHSR apresenta múltiplos agentes etiológicos, entre eles se destacam os vírus Hantaan,
Seoul, Dobrava, Puumala, entre outros. Já na SPCVH, própria das Américas os principais
vírus envolvidos são: vírus Sin nombre (SN), New York (NY), Bayou (BAY), Black Creeck
Canal (BCC), Laguna Negra (LN), Andes (AND), Lechiguanas (LEC), Oran (ORN),
Juquitiba (JUQ), Araraquara (ARA) e Castelo dos Sonhos (CAS), entre outros.
A distribuição destes vírus na América é bastante específica sendo que grupos virais
presentes em uma região mantêm diferenças genômicas e de reservatório com os outros
grupos (SILVA-VERGARA et al 2002) (Figura 2).

23
Figura 2: Distribuição geográfica dos diversos hantavirus identificados nas Américas. Podemos observarque no Brasil se destacam 3 genótipos virais diferentes: Vírus Juquitiba, Vírus Araraquara e VírusCastelo dos Sonhos.Fonte: FERREIRA (2003)
Como vimos anteriormente, os hantavirus recebem seu nome através da cidade ou
localização onde foi isolada mais a palavra vírus (VAN REGENMORTEL 2000).

24
1.4 TRANSMISSÃO
Em contraste com outros vírus da família Bunyaviridae os hantavirus não possuem
transmissão arbovírica, mas via aerossóis provenientes da excreta (urina, fezes e saliva) de
pequenos mamíferos cronicamente infectados principalmente roedores (Figura 3) Estes se
mantêm assintomáticos e levam consigo o vírus por meses e até anos (PLYUSNIN et al
1996; SIMMONS & RILEY 2002).
A infecção humana, portanto, depende do contato do homem com roedores silvestres
cronicamente infectados. Este contato pode ocorrer principalmente através das atividades
agrícolas, como plantação e armazenamento de cereais, demolição de construções rurais,
monoculturas de grãos, entre outras (FERREIRA 2003).
Figura 3: Processo de infecção por Hantavírus. O aumento das chuvas leva a uma aumento na disponibilidadede alimentos. Como conseqüência há um aumento na quantidade de roedores silvestres que podem conter ohantavírus,. Estes roedores não apresentam sintomatologia alguma, e quando defecam eliminam partículas viraisnas fezes. Os aerossóis produzidos por estas secreções, contendo as partículas virais, quando inalados podeminfectar ao ser humano.Fonte: DERETSKY (2005)

25
O crescente desmatamento da vegetação típica, para realização de monoculturas, tem
propiciado a imigração destes roedores, antes estritamente silvestres, para regiões peri-
urbanas, onde a abundância de alimentos gera condições de sobrevivência fazendo com que a
hantavirose já não seja estritamente uma doença rural (FIGUEIREDO 2003).
Outras formas de transmissão, menos freqüentes, incluem mordidas de roedores
contaminados, bem como ingestão de alimentos contendo partículas virais provenientes da
urina ou fezes de roedores (PETER et al 1998; SIMPSON 1998).
Uma outra possibilidade de contaminação, somente descrita uma vez, é a interhumana. Na
Argentina, em 2002, foram confirmados 3 casos de infecção por hantavirose transmitida de
forma interpessoal. Isto foi posteriormente confirmado já que o único fator de risco destes
indivíduos foi o contato com um paciente infectado (PINNA et al 2004)
1.5 RESERVATÓRIOS NATURAIS DOS HANTAVÍRUS
No gênero Hantavírus, os roedores atuam como seus reservatórios naturais. Acredita-se
que vírus e roedores tenham co-evoluído de forma íntima, já que existe especificidade entre
cada hantavírus e um roedor-reservatório numa determinada região geográfica (PLYUSNIN
2002)
Na Europa, os animais das subfamílias Murinae e Arvicolinae particularmente os
pertencentes aos gêneros Apodemus e Clethrionomys são os principais reservatórios de
hantavírus causador da FHSR (CDC 2000; CDC1993).
Nas Américas, a grande maioria dos roedores que transmitem a hantavirose pertence à
subfamília Sigmodontinae. Nos Estados Unidos, os transmissores mais importantes são os
Peromyscus maniculatus e P. leucopus. Na Argentina, onde várias variedades virais já foram
descritas, os principais roedores são os do gênero Oligoryzomys, em particular, o O.
flavescens e o O. longicaudatus (TISCHLER et al 2005).
No Brasil, destacam-se como principais roedores-reservatórios o Bolomys lasiurus (Rato
do Mato) e o Oligoryzomis nigripes (Rato do arroz) (Figura 4) (SUZUKI et al 2004).
Na tabela 1 podemos observar a relação entre os principais hantavírus e seus reservatório
naturais, bem como sua distribuição geográfica.

26
É possível que outros roedores não mencionados também sejam reservatórios naturais de
hantavírus, já que muitas genótipos encontrados infectando seres humanos ainda não se
conhece reservatório.
Tabela 1. Relação entre os principais hantavírus, suas doenças causadas e seus reservatório naturais.
Espécie Doença Reservatório Distribuição
Geográfica.
Hantaan FHSR Apodemus agrarius China, Rússia,
Coréia.
Dobrava FHSR Apodemus flavicolis Bálcãs
Seoul FHSR Rattus norvegicus Cosmopolita
Puumala FHSR Clethionomys glareolus Europa,
Escandinávia
Prospect Hill ND Lemmus sibiricus Estados Unidos
Tula ND Lemmus sibiricus Estados Unidos
Pergamino ND Akodon azarae América Central
Sin Nombre SPCVH Peromyscus maniculatus Estados Unidos
New York SPCVH Peromyscus leucopus Estados Unidos
Andes vírus SPCVH Oligoryzomys longycaudatus Argentina, Chile
Maciel ND Necromys benefactus Argentina Central
Oran ND Oligoryzomys longycaudatus Argentina
Lechiguanas SPCVH Oligoryzomys flavencens Argentina
Bermejo SPCVH Oligoryzomys chacoensis Paraguay,
Argentina
Laguna Negra SPCVH Calomys laucha Paraguay, Bolívia
Rio Mármore ND Oligoryzomys microtis Bolívia
Araraquara SPCVH Bolomys laziurus Brasil
Castelo dos
Sonhos
SPCVH ND Brasil
Rio Mearim ND Oligoryzomys fornersi BrasilFonte: MORELLI (2005). - ND: Não Descrito

27
Figura 4: Reservatórios naturais de Hantavirus no Brasil. Se destacam o Bolomys lasiurus (Rato do RaboPeludo) e os pertencentes ao gênero Oligoryzomys sp (Rato do Arroz)Fonte: GARCIA et al (2001)
1.6 REPLICAÇÃO DOS HANTAVÍRUS
A ligação dos Hantavírus as células alvo acontece pela ligação das glicoproteínas (G1 e
G2) virais com receptores de integrinas. Os hantavírus considerados não patogênicos ligam-se
a um tipo específico de integrinas chamadas de B1, já os patogênicos ligam-se a integrinas
B3, que são receptores de células do sistema imune, plaquetas, entre outras
(GAVRILOVSKAYA et al 1999; RAYMOND et al 2005)
Após a ligação aos receptores a partícula viral penetra por endocitose e o envelope
lipídico funde-se ao endossomo ocorrendo à liberação do RNA e da polimerase viral, que
começa imediatamente a transcrição das ribonucleoproteínas produzindo o mRNAs
(GAVRILOVSKAYA et al 1999; RAYMOND et al 2005).
Nesta fase do ciclo o vírus utiliza cap-primers obtidos dos mRNAs do hospedeiro
presentes no citoplasma celular das células infectadas (HUTCHINSON et al 1996).
Os mRNAs derivados dos segmentos S e L são traduzidos por ribossomos livres
formando a proteína N de aproximadamente 54 Kda e a RNA polimerase dependente de RNA
de aproximadamente 260 Kda (HUTCHINSON et al 1996).
O mRNA do segmento M é traduzido por ribossomos ligados a membrana, formando
um precursor poliproteíco que será posteriormente clivado por peptidases do hospedeiro,
formando as glicoproteínas G1 e G2 (Figura 5) (GAVRILOVSKAYA et al 1998).

28
As proteínas G1, G2 e N são imunogênicas para o hospedeiro infectado, levando a
produção de anticorpos neutralizantes contra epitopos destas proteínas (GARCIN et al 1995;
RAYMOND et al 2005).
Concomitante ao processo de transcrição e tradução das proteínas virais, a replicação
genômica dos hantavírus acontece. Esta se inicia quando a RNA polimerase dependente de
RNA muda sua função de transcriptase para replicase resultando na cópia de todo o RNA
viral (GARCIN et al 1995).
Após a replicação do material genômico os virions são formados por brotamento no
citoplasma para o interior de vesículas do aparelho de golgi onde há uma alta concentração
das proteínas G1 e G2.
Devido a que os Hantavírus não possuem proteína de matriz, para a ligação do
envelope e das ribonucleoproteínas, é provável que ocorra interação direta entre os
ribonucleocapsídeos e as proteínas do envelope viral localizadas no lumem das vesículas.
Após o brotamento os virions são transportados à superfície da célula e secretados por
exocitose (Figura 5) (HUTCHINSON et al 1996).
Figura 5: Replicação dos Hantavírus. 1. Ligação da partícula viral a célula-alvo através dasglicoproteínas e receptores de membrana denominados integrinas. 2. Entrada via endocitose mediada porinteração glicorpoteínas receptor. 3. Fusão da membrana do endossomo e liberação dosribonucleocapsídios e da RNA polimerase no citoplasma. 4. Transcrição primária. 5. Tradução dasproteínas virais. 6. Replicação do RNAv via cRNA intermediário. 7. Montagem dos virions no aparelhode golgi. 8. Brotamento das novas partículas virais. 9. Saída por exocitose.

29
1.7 HANTAVIROSES
Duas patologias estão associadas à infecção por hantavírus em humanos: a febre
hemorrágica com síndrome renal (FHSR), que ocorre principalmente na Ásia e Europa e a
Síndrome pulmonar e cardiovascular (SCPH), que ocorre nas Américas (PADULA et al
2000A; FIGUEIREDO et al 2001).
Após a inalação das partículas virais ocorre um período de incubação que varia de 3 a
33 dias e após este período sinais clínicos são desenvolvidos (JEOR 2004).
1.7.1 Patogênese da SCPVH.
A patogenia da SPCVH tem sido bastante estudada nos últimos anos (RAYMOND et al
2005). A via respiratória é a porta de acesso do vírus ao organismo, e as células endoteliais
dos pequenos vasos os principais alvos.
A patogenia não esta diretamente relacionada a infecção destas células mas sim a uma
resposta imune exacerbada contra os agentes, com grande produção de citocina (MAKELA et
al 2004; SUNDSTROM et al 2001; KHAIBOULLINA et al 2000).
A seriedade da patologia em questão é desencadeada com a agressão do sistema imune as
células infectadas, ocorrendo extravasamento de líquidos, edema pulmonar com insuficiência
respiratória (FIGUEIREDO et al 1999).
As plaquetas também são infectadas pelo vírus, estas são destruídas levando a um quadro
de trombocitopenia que leva a ocorrência de quadros hemorrágicos (GAVRILOVSKAYA et
al 1998).
Junto ao extravasamento de líquido nos pulmões, há uma infiltração massiva de células
TCD8 para este órgão. Estas células, são ativadas pela presença do antígeno e produzem
citocinas que estimularão macrófagos locais a produziram um fator de ativação plaquetária e
leucotrienos que aumentam a permeabilidade vascular aumentando ainda mais o edema
pulmonar, levando a uma insuficiência respiratória (GAVRILOVSKAYA et al 1998).

30
O choque na SPCVH não tem um mecanismo fisiopatológico bem definido, porém a
hipovolemia causada pelo extravasamento de líquidos adjunto com um processo inflamatório
do miocárdio pode levar ao desencadeamento do mesmo (GAVRILOVSKAYA et al 1998).
1.7.2 Manifestações Clínicas
A síndrome pulmonar e cardiovascular por Hantavirus (SCPVH), é uma doença
emergente com descrição crescente de casos no Brasil. O período de incubação desta doença
pode chegar a 5 semanas e infecções subclínicas ou oligossintomáticas são comuns.
(BHARADWAJ et al 2000; YOUNG et al 2000; JEOR 2004).
A fase inicial da doença (3 a 6 dias) caracteriza-se por sintomas como febre, milagias,
náuseas, diarréias, cefaléia, vômitos, dor abdominal, dor torácica, sudorese e vertigem.
Existe uma grande dificuldade em reconhecer precocemente a SPCVH já que os sintomas
são muito parecidos com outras patologias com dengue, leptospirose, febre amarela, entre
outras (RIDEL et al 2004; HAMIDON & SAADIAH 2003).
Com o inicio da fase cardiopulmonar a doença progride rapidamente aparecendo sintomas
como tosse e dispnéia. Caracteriza-se pela progressiva infiltração de liquido e proteínas no
interstício e alvéolos pulmonares, levando a taquipnéia, hipoxemia e taquicardia. A
hipotensão pode evoluir para o choque, com grave depressão miocardica podendo levar o
individuo a óbito por falência respiratória (VERITY et al 2003, FIGUEIREDO et al 2001).
Nesta fase, 30 a 70% dos pacientes apresentam transtornos hemorrágicos compatíveis
com o extravasamento de líquidos podendo levar a insuficiência renal, necessitando de diálise
transitória.
Até o ano de 2005, 43% dos que contraiam a SPCVH, no Brasil, morriam.
A covalescência dos pacientes com SPCH, especialmente daqueles que necessitaram
de intubação e ventilação mecânica costuma ser prolongada. A avaliação tardia tem mostrado
evidencia de seqüelas como fadiga crônica e restrição da função pulmonar (FIGUEIREDO et
al 2001). Segundo Figueiredo (2001), os indivíduos acometidos por SPCH que foram tratados
precocemente não mostram nenhuma seqüela e todos os testes pulmonares mostraram
resultados dentro dos limites da normalidade.

31
1.8 IMUNOLOGIA DAS HANTAVIROSES
Após inalação das partículas com vírus, este é fagocitado por células dendríticas ou
macrófagos da mucosa de vias aéreas e alvéolo, que migram a linfonodos regionais
apresentando antígenos a células T e ativando-as intensamente (MAES et al 2004).
Grandes quantidades de células T ativadas são liberadas em sangue periférico e
tecidos, sendo descritas como imunoblastos ou linfócitos atípicos. Também, células do
endotélio capilar pulmonar são infectadas. Para tanto, o hantavírus utiliza como receptor de
membrana 3 integrinas abundantes nestas células (LUNDKVIST et al 1993, MAES et al
2004).
A infecção da célula endotelial inibe sua capacidade migratória em vitronectina, o que
altera funcionalmente a barreira capilar-alvéolo. Também, as células endoteliais infectadas
produzem quimiocinas que atraem e estimulam a ação de células CD8+ citotóxicas e
mononucleares. Estas, por sua vez, liberam citocinas pró-inflamatórias incluindo TNF- e
IFN- , indutoras de óxido nítrico que produz vasodilatação local (LUNDKVIST et al 1994,
MAES et al 2004).
Assim, as citocinas pró-inflamatórias, atuam de forma sinérgica à alteração de barreira
capilar estimulando o extravasamento de líquido ao interstício e edema pulmonar. Plaquetas,
que possuem 3 integrinas, se infectam e são destruídas participando do processo vascular,
bem como de fenômenos hemorrágicos que ocorrem na SPCVH. O TNF- , também, atua
deprimindo a função miocárdica, que leva ao choque cardiogênico acompanhante do quadro
(LUNDKVIST et al 1993, MAES et al 2004).
Correlaciona-se o nível de ativação das células CD8+ com gravidade da SPCVH. Os
casos fatais possuem teores muito elevados destas células no sangue periférico. Por outro
lado, vigorosa atuação de anticorpos neutralizantes está associada à redução na carga viral e à
cura da doença, bem como à proteção contra re-infecções por hantavirus (SIMPSON 1998;
SIMMONS et al 2002).

32
1.9 EPIDEMIOLOGIA DAS HANTAVIROSES
No Brasil, os conhecimentos sobre a SPCH avançaram muito nos últimos anos. Os
primeiros pacientes portadores de SCPH foram diagnosticados em 1993, sendo que até o final
de 2005 um total de 626 casos já haviam sido notificados, sendo que aproximadamente 43,6%
destes morreram (SANTOS et al 2005; LUNA et al 2005; FUNASA 2005) (Tabela 2).
Observamos que a maioria das infecções ocorreram nos estados de Paraná (19,80%), Minas
Gerais (18,69%), Santa Catarina (17,89%), São Paulo (14,05%), Mato Grosso (7,98%) e Rio
Grande do Sul (7,02%) onde as atividades agropecuárias constituem o trabalho de milhões de
brasileiros (Tabela 2) (SANTOS et al 2005; LUNA et al 2005; FUNASA 2005)
Podemos observar também alguns estados que não apresentavam casos descritos da
doença a alguns anos atrás (Goiás, Distrito Federal, Amazonas, entre outros) começam a
manifestar seus primeiros casos, o que indica um aumento no número de pessoas infectadas e
também um aumento na notificação desta doença (Tabela 2).
Acreditamos ainda, que exista um número de casos ainda maior do que apresentados
aqui, já que como descrito anteriormente a sintomatologia é muito semelhante a outras
doenças o que poderia levar ao não relato da patologia como hantavírose.
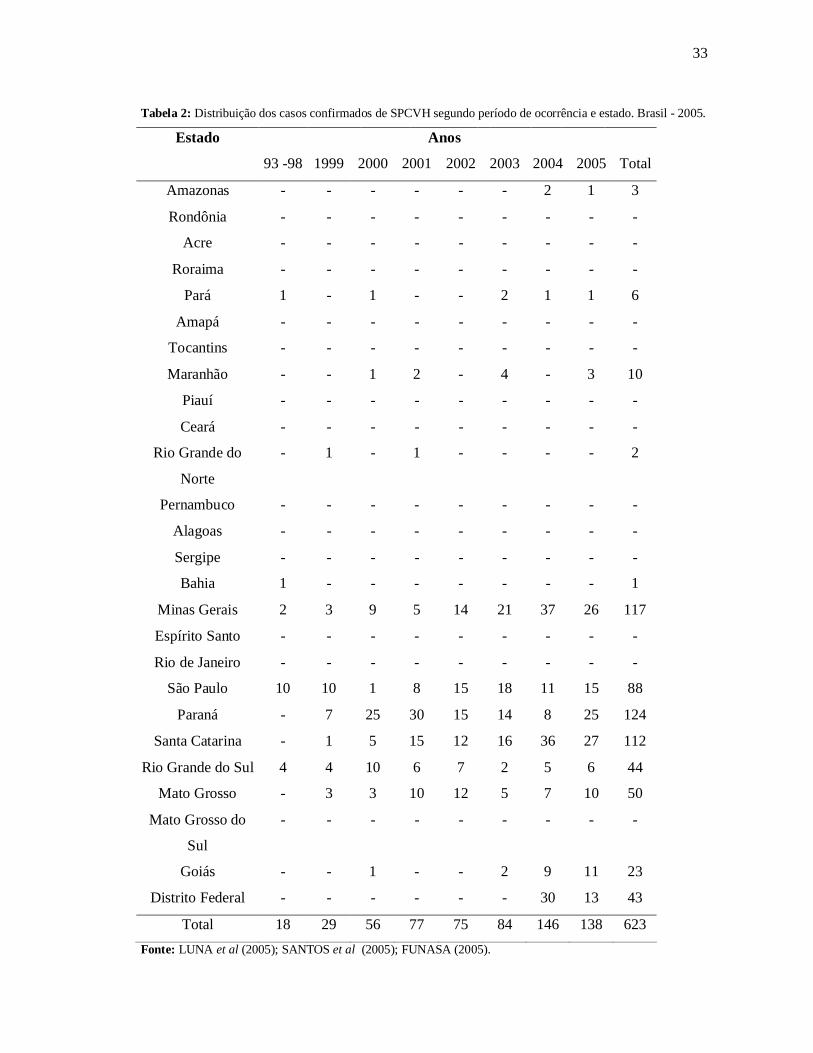
33
Tabela 2: Distribuição dos casos confirmados de SPCVH segundo período de ocorrência e estado. Brasil - 2005.
Estado Anos
93 -98 1999 2000 2001 2002 2003 2004 2005 Total
Amazonas - - - - - - 2 1 3
Rondônia - - - - - - - - -
Acre - - - - - - - - -
Roraima - - - - - - - - -
Pará 1 - 1 - - 2 1 1 6
Amapá - - - - - - - - -
Tocantins - - - - - - - - -
Maranhão - - 1 2 - 4 - 3 10
Piauí - - - - - - - - -
Ceará - - - - - - - - -
Rio Grande do
Norte
- 1 - 1 - - - - 2
Pernambuco - - - - - - - - -
Alagoas - - - - - - - - -
Sergipe - - - - - - - - -
Bahia 1 - - - - - - - 1
Minas Gerais 2 3 9 5 14 21 37 26 117
Espírito Santo - - - - - - - - -
Rio de Janeiro - - - - - - - - -
São Paulo 10 10 1 8 15 18 11 15 88
Paraná - 7 25 30 15 14 8 25 124
Santa Catarina - 1 5 15 12 16 36 27 112
Rio Grande do Sul 4 4 10 6 7 2 5 6 44
Mato Grosso - 3 3 10 12 5 7 10 50
Mato Grosso do
Sul
- - - - - - - - -
Goiás - - 1 - - 2 9 11 23
Distrito Federal - - - - - - 30 13 43
Total 18 29 56 77 75 84 146 138 623Fonte: LUNA et al (2005); SANTOS et al (2005); FUNASA (2005).

34
1.10 DIAGNOSTICO DAS HANTAVIROSES
O diagnóstico das hantaviroses baseia-se essencialmente na realização de testes
sorológicos, já que o isolamento viral não se mostra factível na prática clínica diária (PETERS
et al 1998; LUNDKVIST et al 1994; KALLIO-KOKKO et al 1993).
O teste imunoenzimático (ELISA), que separa anticorpos das classes IgM e IgG; é o
mais utilizado para este tipo de diagnóstico. Outros testes sorológicos disponíveis abarcam a
imunofluorescência indireta IFA, a neutralização, hemaglutinação passiva e westernblot que
não são freqüentemente utilizados devido ao seu alto custo (KSIAZEK et al 1995; PADULA
et al 2000B; GROEN et al 1989).
A imunohistoquímica é outra técnica utilizada para o diagnóstico das Hantaviroses.
Esta é aplicada aos tecidos com a finalidade de identificar antígenos virais, sendo utilizada em
casos fatais, nos quais não se podem obter amostras de soro. Diagnósticos retrospectivos
também podem ser realizados através do uso dessa técnica (HEISKE et al 1999; FERREIRA
2003). Segundo Morelli et al (2004) o PCR_RT (PCR com transcrição reversa) é outra
técnica plausível de utilização. Esta pode identificar o RNA viral em amostras de sangue ou
tecidos provenientes de casos suspeitos ja nos primeiros 7 a 10 dias de doença (De PAULA et
al 2002; SOARES et al 2005). Embora os primers utilizados sejam sensíveis para
amplificação e provenientes de seqüências genéticas obtidas de vírus encontrados em tecidos
humanos ou de roedores, existem diferenças significativas entre os vírus isolados de cada
região ou país, diminuíndo a sensibilidade da técnica como uso padrão no diagnóstico das
hantaviroses (GARIN et al 2001).
No Brasil o diagnóstico da infecção por hantavirus é confirmado pela detecção de
anticorpos IgM contra o vírus Sin Nombre (SN) ou vírus Andes (ADN), através de testes
sorológicos ELISA, utilizando para isto uma nucleoproteína recombinante produzida pelo
Center for Desease Control (CDC, US) ou produzida pelo Intistuto Carlos Malbran em
Buenos Aires, Argentina. Estes testes são de grande importância para a confirmação da
infecção por hantavirus e para estudos de avaliação epidemiológica desta importante virose
(PADULA et al 2000B; MORELI et al 2004). Mesmo assim estes testes sorológicos só são
realizados em 3 grandes centros (Institutos Adolfo Lutz, Evandro Chagas e Oswaldo Cruz)
devido principalmente a dificuldade de obtenção dos antígenos que não são produzidos no
país (MORELI et al 2004).

35
Como podemos observar este teste sorológico com antígeno de Sin Nombre vírus não
é totalmente específico para os hantavírus brasileiros e dá-se por reação cruzada, ou seja,
capacidade dos anticorpos IgM contra nucleoproteína de hantavírus brasileiros ligarem
inespecificamente a nucleoproteína do Sin Nombre vírus.
Devido a isto, se torna necessário o uso de genótipos Brasileiros, aumentando
significativamente a sensibilidade e especificidade dos testes sorológicos além de diminuir o
custo já que não seria mais necessária a importação de material para realização destes kits
diagnósticos (PADULA et al 2000B; SUZUKI et al 2004; PADULA et al 2000A).
1.11 PRODUÇÃO DE ANTÍGENOS PARA TESTES DIAGNÓSTICOS
A produção de antígenos nativos para os testes sorológicos apresenta várias limitações,
entre elas o alto risco envolvido na manipulação do hantavirus, para o qual é necessários
laboratórios com nível de biossegurança 3 (VAPALAHTI et al 1996; KSIAZEK et al 1995;
BILLECOQ et al 2003). Outras limitações para o uso dos antígenos nativos são os baixos
títulos obtidos em cultura de células, dificuldade de adaptação do vírus às células, crescimento
lento (3 a 10 dias), e mutações nas seqüências de nucleotídeos geradas pelas sucessivas
passagens em cultura de células. Estas mutações geram proteínas defeituosas que podem
interferir na replicação viral e atenuação da virulência, diminuindo cada vez mais a produção
destes em culturas de células (TEMONEM et al 1993; ELLIOTT et al 1994; LUNDKVIST et
al 1994).
Devido às razões anteriormente citadas, os antígenos de hantavirus produzidos pela
técnica do DNA recombinante estão tornando-se padrão nos testes sorológicos (KSIAZEK et
al 1995;BILLECOCQ et al 2003; KORAKA et al 2000). O gene N, que codifica a
nucleoproteína viral, é freqüentemente usado para a produção de antígenos recombinantes
(ELGH et al 1996; GOTT et al 1997; XU et al 2002). Isto se deve ao fato desta proteína ser
um dos principais antígenos na infecção, sendo que altos títulos de imunoglobulina M anti-
nucleoproteína são detectados principalmente na fase aguda da infecção viral a partir dos
primeiros dias de infecção (GOTT et al 1997; XU et al 2002). A imunoglobulina G começa a
aparecer já na fase aguda da infecção e estende sua permanência até depois da fase de

36
convalescença, sendo que décadas após a infecção, ainda há, embora com baixos títulos, IgG
direcionadas à nucleoproteína (SETTERGREN et al 1991, HUJJAKA et al 2003).
Vários sistemas de expressão já foram utilizados para a síntese da nucleoproteína
recombinante de hantavírus não Brasileiros. Por exemplo, o gene N inserido em plasmídeos
expressa de forma eficiente à proteína em células de E. coli (TAKAKURA et al 2003;
JONSSON et al 2001; KALLIO-KOKKO et al 2000; HJELLE et al 1997). Este gene N
também foi inserido através de plasmídeos específicos em células de Sacharomyces cerevisae
produzindo quantidades significativas da proteína recombinante (SCHMIDT et al 2005A;
RAZANSKIENE et al 2004; SCHMIDT et al 2005B).
Outros métodos para a produção de proteína recombinante tem sido descritos nos
últimos anos, entre eles, a produção de um peptídeo sintético com os 84 aa iniciais da
nucleoproteína (LI et al 2002). Também existem relatos de expressão desta proteína em
células de mamíferos (Vero E6) a qual mostrou ser um eficaz antígeno em testes sorológicos
(LI et al 2006; KALLIO-KOKKO et al 2001).
O gene da nucleoproteína também foi inserido no genoma de Baculovírus, a partir do
qual altas concentrações de proteína N recombinante foram obtidas em células de inseto em
cultura (YOSHIMATSU et al 1993; MORII et al 1998; KARIWA et al 2003; HUJAKKA et
al 2001; SIROLA et al 2004; KALLIO-KOKKO et al 1998).
Em 2005, Moreli e colaboradores produziram uma proteína N recombinante de
Araraquara Hantavírus em E. coli no laboratório de Virologia da Faculdade de Medicina de
Ribeirão Preto (MORELI 2005). O gene desta proteína, isolado a partir do RNA viral extraído
do soro de pacientes da região de Ribeirão Preto, foi clonado no plasmídeo pET Directional
TOPO® (Invitrogen, USA) e multiplicado em células E. coli BL21 DE3 (Invitrogen, USA). A
expressão da proteína N recombinante foi confirmada por imunobloting utilizando-se
anticorpos contidos no soro de pacientes que tiveram a síndrome cardiopulmonar pelo
Hantavírus. Nestes ensaios verificou-se que a proteína expressa reagia com o soro dos
pacientes que tiveram a síndrome cardiopulmonar e não com o soro controle negativo
(MORELI 2005). A proteína recombinante purificada em colunas de níquel após
solubilização em uréia tem se mostrado bastante adequada nos testes realizados até o
momento com soro de pacientes infectados com hantavirus e em estudos sorológicos com
roedores selvagens (MORELLI 2005).
Estudos realizados anteriormente por Sjolander (1997), comparando métodos
sorológicos para detecção do Puumala vírus (um dos 30 genótipos de Hantavirus) indicam
que a proteína recombinante N, produzidas em células de inseto utilizando sistema de

37
expressão de Baculovirus, pode ser mais adequada que a proteína produzida em E. coli. Neste
trabalho foi mostrado que a proteína produzida no baculovírus foi tão eficiente quanto
antígenos nativos do hantavirus na capacidade de detectar anticorpos específicos do tipo IgG.
Já, a proteína N recombinante produzida em E. coli mostrou ser menos sensível
provavelmente devido a resíduos contaminantes de E. coli mesmo após várias purificações.
Assim, há sempre um risco maior de se gerar resultados falsos positivos nos testes
diagnósticos produzidos com estas proteínas. (SJOLANDER et al 1997; VAPALAHTI et al
1996).
Recentemente, um teste comercial foi desenvolvido na Escandinávia, por Hujakka e
colaboradores (2001), este utiliza a proteína N produzida em células de inseto a partir de
baculovírus. Estes trabalhos sugerem que há vantagens na produção da proteína N utilizando
o sistema de expressão de baculovírus, já que este não necessita de complicados processos de
purificação o que simplifica sua produção em larga escala, diminui os custos e também reduz
as chances de falsos positivos nos testes fabricados a partir destas proteínas.
1.12 PROTEÌNAS RECOMBINANTES
A produção de proteínas recombinantes em grande escala já abrange os substitutos de
hormônios, mediadores intercelulares humanos e proteínas virais potencialmente usadas na
vacinação, imunomodulação e diagnóstico em humanos e animais (PHILLIPS et al 2005;
KOST et al 2005).
A produção de proteínas recombinantes envolve uma série de passos, iniciando-se com
a identificação e caracterização genética da proteína e das suas propriedades bioquímicas, e a
definição dos requisitos estruturais para a sua atividade funcional, como o processamento pós-
traducional, glicosilação, formação de heterodímeros, etc (PHILLIPS et al 2005; KOST et al
2005).
A produção propriamente dita envolve a escolha do vetor de expressão, a escolha de
células em função da complexidade de processamento molecular necessário para a obtenção
da proteína funcional, o aperfeiçoamento do bioprocesso de produção, e a separação,
purificação e preparo para o uso médico ou diagnóstico da proteína (PHILLIPS et al 2005).

38
O uso médico de mediadores hormonais, fatores de crescimento e citocinas está em
franca expansão, em função de avanços consideráveis alcançados na compreensão de vias de
sinalização intercelular e intracelular (KOST et al 2005).
Uma nova geração de proteínas recombinantes virais está encontrando aplicação
diagnósticas tanto em seres humanos como em animais e estão tornando-se padrão na maioria
dos testes imunodiagnósticos (PHILLIPS et al 2005).
Uma atenção particular deverá ser dada à expressão de proteínas em sistemas
alternativos, além de sistemas tradicionais que usam microrganismos ou células animais. Os
sistemas mais complexos devem permitir a expressão simultânea de mais de um gene com
obtenção de proteínas heterodiméricas (PHILLIPS et al 2005; KOST et al 2005).
A expressão de proteínas humanas, virais e de outros organismos em animais
superiores é muito promissora, devendo levar à produção de complexos moleculares
fisiologicamente ativos, com um rendimento produtivo muito mais importante, a baixo custo
(PHILLIPS et al 2005; KOST et al 2005).
1.13 SISTEMA DE EXPRESSÃO DE BACULOVÍRUS
1.13.1 Baculovirus
Os baculovirus compreendem o maior grupo de vírus de insetos conhecido. Estudos
mostram mais de 700 espécies de artrópodes infectados por baculovírus, sendo que a ordem
Lepidóptera possui a maior quantidade de isolados (O’ REILLY et al 1992; TANADA &
KAYA 1993)
Pertencem os baculovírus a família Baculoviridae, e possuem um genoma de DNA
circular de fita dupla, que pode variar entre 70 a 210 Kb. Possui um capsídeo protéico que
constitui a unidade infectiva dos vírus chamada de nucleocapdsídeo (MOSCARDI 1998)
Durante seu ciclo infectivo, os baculovírus podem assumir 2 formas distintas, as quais
possuem além de diversas morfologias diferentes funções. Uma das formas é a chamada BV
(“Budded vírus”) que não possui o corpo de inclusão característico dos baculovírus, e os PDV
(“Polyhedra-derived vírus”) que possuem o corpo de inclusão tendo em seu interior uma ou
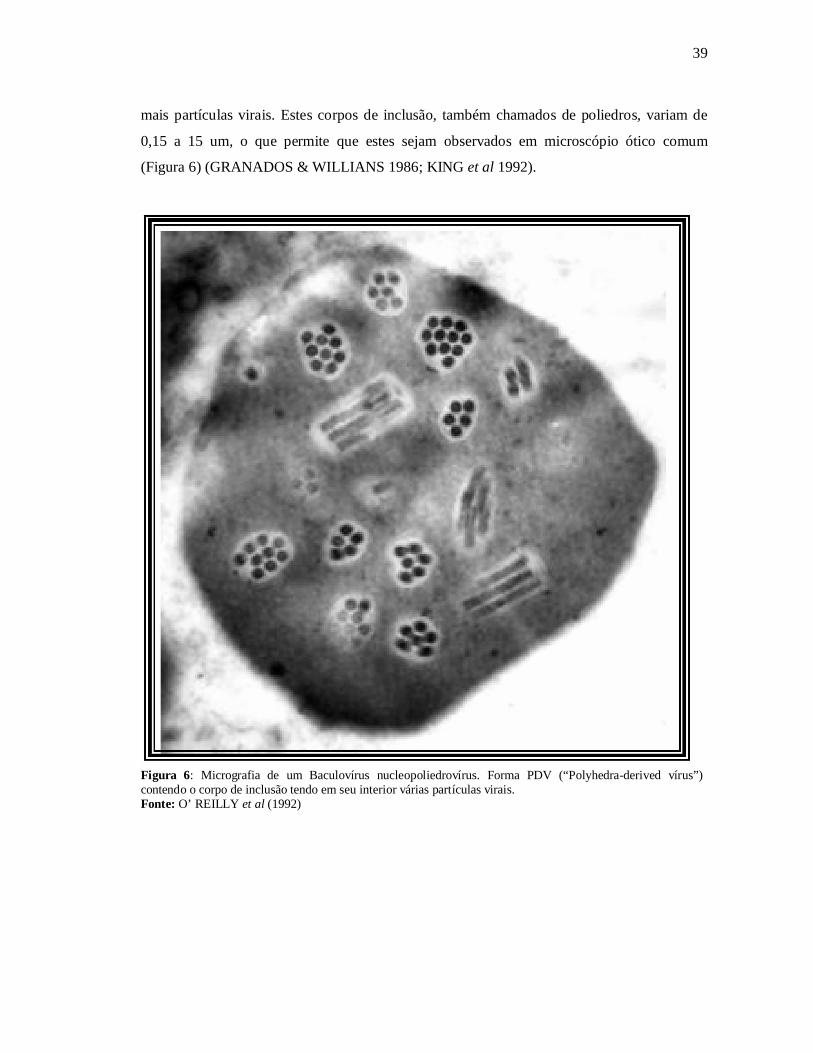
39
mais partículas virais. Estes corpos de inclusão, também chamados de poliedros, variam de
0,15 a 15 um, o que permite que estes sejam observados em microscópio ótico comum
(Figura 6) (GRANADOS & WILLIANS 1986; KING et al 1992).
Figura 6: Micrografia de um Baculovírus nucleopoliedrovírus. Forma PDV (“Polyhedra-derived vírus”)contendo o corpo de inclusão tendo em seu interior várias partículas virais.Fonte: O’ REILLY et al (1992)

40
1.13.2 Expressão Gênica nos Baculovírus.
Uma vez iniciado o processo de infecção, seja em lagartas ou cultura de células, a
expressão gênica nos baculovírus ocorre em forma ordenada, conhecida como expressão em
cascata (O’REILLY et al 1992). Esta expressão é dividida em 2 fases conhecidas como 1.
Fase Imediata (“early”) e 2. Fase Tardia (“late”).
A fase imediata pode ser subdividida em duas subfases: 1. Inicial imediata onde os genes
expressos não dependem da replicação do DNA viral sendo que a maioria possui funções
transregulatórias e 2. Inicial tardia, que se caracteriza pela dependência de proteínas virais e
estão diretamente relacionados com a replicação do genoma viral (O’REILLY et al 1992;
CARSON et al 1991A).
A fase tardia se caracteriza pela completa dependência da expressão dos genes da fase
imediata e da replicação do genoma viral. Estes genes estão intimamente relacionados a
montagem e oclusão viral (O’REILLY et al 1992; CARSON et al 1991B).
Nesta fase 2 proteínas são altamente expressas: a poliedrina que é a principal proteína
formadora do corpo de inclusão e a proteína p10 que esta envolvida no processo de oclusão
(O’REILLY et al 1992; CARSON et al 1991B).
1.13.3 Utilização dos Baculovírus como sistema de expressão – Sistema de
Expressão utilizado.
A observação de que alguns genes eram hiperexpressos no estágio final do processo
infectivo e o maior conhecimento do genoma e morfologia dos baculovírus, bem como sua
fácil manipulação resultaram na utilização destes vírus como sistemas de expressão de
proteínas exógenas (KOST et al 2005; O’REILLY et al 1992)
Outra característica que possibilitou sua utilização neste propósito foi que o
nucleocapsídeo viral é capaz de comportar grandes quantidades de DNA adicional
possibilitando inserir longos e múltiplos insertos no seu genoma (BERGER et al 2004).
Este sistema oferece grandes vantagens quando comparado a outros sistemas de expressão
de proteínas heterólogas. Dentre estas vantagens se destacam: a) produção de altos níveis de
proteína recombinante, b) fortes promotores de fase tardia que promovem a produção da

41
proteína sem afetar o ciclo infectivo do vírus, c) capacidade de clonagem de grandes insertos,
d) simplicidade de manipulação, e) modificações pos-traducionais, entre outras (O’REILLY
et al 1992; KING et al 1992; PHILLIPS et al 2005; KOST et al 2005)
Para a utilização deste sistema, utilizam-se células de inseto, que por serem eucariotos
superiores, são capazes de realizar as modificações pós-traducionais gerando proteínas
estruturalmente idênticas ou muito semelhantes às encontradas "in vivo".
As células de inseto mais comumente usadas são células epiteliais do ovário da lagarta
Spodoptera frugiperda, linhagens conhecidas como Sf9 e Sf21, além de células ovo de
Trichoplusia ni (FOLGUERAS-FLATSCHART et al 2000)
O método para produção de baculovírus recombinantes é realizado utilizando vetores de
transferência que são unidades replicativas que contêm regiões flanqueadoras do gene a
substituir no genoma dos baculovírus. Estes vetores de transferência também contêm fortes
promotores de fase tardia, como por exemplo, o promotor da poliedrina modificado, um sítio
de múltipla clonagem, onde o gene de interesse será inserido e o gene da poliedrina, que
servirá como gene marcador de recombinação homologa (WANG et al 1991).
Após a produção dos vetores de transferência realiza-se uma co-transfecção do DNA viral
e do vetor de transferência em células de inseto. Esta co-transfecção é realizada utilizando
lipossomos, que irão se fundir a membrana plasmática das células de inseto, permitindo que o
DNA viral e vetor de transferência sejam interiorizados ao citoplasma celular (Figura 7)
(WANG et al 1991).
Já no interior das células acontecerá a recombinação homóloga entre os genes de interesse
mais o gene da poliedrina com gene a substituir, formando assim os baculovírus
recombinantes (Figura 7) (O’REILLY et al 1992; WANG et al 1991).
Os vírus utilizados neste processo são geralmente os AcNPV (Autographa californica
nucleopoliedrovirus) modificados que possuem em seu genoma o gene da beta-galactosidase
e não possuem o gene da poliedrina que é a proteína formadora do corpo de oclusão. Dessa
forma, a seleção dos recombinantes é facilitada, já que somente aqueles que produzirem
oclusão serão considerados vírus recombinantes, já que receberam, além do gene de interesse,
o gene da poliedrina (Figura 7) (O’REILLY et al 1992; WANG et al 1991).
Esta técnica permite uma purificação facilitada dos recombinantes podendo ser realizada
através de diluições seriadas das amostras (O’REILLY et al 1992).

42
Figura 7: Produção dos baculovírus recombinantes. Os vetores de transferência são colocadosjunto ao DNA viral (digerido com Bsu36I) em um lipossomos. Este se fundirá a membranaplasmática das células de inseto SF9, fazendo com que penetrem no interior da célula. Já nointerior acontecerá a recombinação homóloga, entre o gene de interesse e o gene da betagalactosidase, que não é essencial a replicação do vírus. O gene da poliedrina também é inserido eserve como gene marcador, pois os vírus recombinantes produziram vírus oclusos.

43
2 JUSTIFICATIVA
A Hantavirose é uma das zoonoses que vem preocupando as autoridades sanitárias de todo
o mundo. No Brasil há um aumento significativo de casos de hantavirose, com uma taxa de
mortalidade que chega a 40%.
O diagnóstico da doença apenas por sinais clínicos é bastante difícil devido
principalmente à similaridade entre os sintomas clínicos da hantavirose com outras doenças.
Devido a isso se faz necessário o diagnóstico diferencial através de sorologia, o que somente é
realizado em grandes centros de referência, principalmente devido ao alto custo deste teste.
A proteína usada para estes testes é a de Sin nombre vírus, que não é um hantavírus
brasileiro, e deve ser importada dos EUA e Argentina. Outra problemática é que os testes que
utilizam esta proteína recombinante funcionam por reação cruzada, o que pode comprometer
o diagnóstico.
Como vemos faz-se necessário um teste diagnóstico utilizando proteínas recombinantes de
hantavírus brasileiros.
Esta produção trará grandes vantagens, como: diminuição do custo de produção, maiores
suprimentos da proteína, maior especificidade e sensibilidade nos testes diagnósticos e
independência tecnológica.
No último ano, a proteína N de Araraquara hantavírus, foi produzida experimentalmente
em E. coli, mas estudos mostram uma melhor antigenicidade da proteína N quando utilizado o
sistema de expressão de baculovírus.
Devido a este cenário, propomos a construção de 2 baculovírus recombinantes para uma
futura produção da proteína N, sendo que estes contem modificações e inserções que poderão
facilitar uma futura purificação desta proteína recombinantes.

44
3 OBJETIVO
OBJETIVO GERAL:
• Produzir os baculovírus recombinantes que sejam capazes de expressar a proteína
N de Araraquara Hantavírus.
OBJETIVOS ESPECÍFICOS:
• Construir vetores de transferência pSyn contendo o gene N da Araraquara
Hantavírus.
• Inserir uma cauda de poli-Histidina na região upstream do gene da Nucleoproteína.
• Inserir a seqüência sinal de exportação do gene da GP67 na região upstream da
cauda de poli-Histidina.
• Purificar e produzir um estoque viral para uma futura produção da proteína N
recombinante.

45
4 MÉTODO
4.1 VETORES, VÍRUS E LINHAGEM CELULAR UTILIZADA.
4.1.1 Vetor de clonagem pET-N-Ara
O vetor pET-N-Ara nos foi gentilmente cedido pelo Dr. Marcos Moreli, do laboratório
de Virologia da Faculdade de Medicina de Ribeirão Preto.
O gene N do Araraquara Hantavírus foi isolado a partir do soro de pacientes que
adquiriram a síndrome pulmonar cardiovascular utilizando a técnica RT-PCR (PCR com
transcrição reversa) que a partir de uma molécula de RNA produz cópias de DNA. O produto
do RT-PCR foi inserido no vetor de expressão pET Directional TOPO® (Invitrogen USA).
(MORELI 2005)
O vetor pET Directional TOPO® (Invitrogen USA) contendo o gene N do Araraquara
Hantavírus foi seqüenciado no nosso laboratório com o intuito de confirmar a seqüência de
nucleotídeos deste gene.
4.1.2 Vetor de transferência pSynXIVVIX3
Este vetor (figura 8) nos foi gentilmente cedido pelo Prof. Dr. Bergman Ribeiro, da
Universidade de Brasília (UnB).
É utilizado especificamente para o processo de recombinação homóloga com o
genoma do baculovírus recombinante vSyngal VI+.

46
Este processo se dá devido a presença das regiões flanqueadoras do gene da
Betagalactosidase presente no vetor, e a presença deste gene no vírus recombinante
anteriormente citado.
Este vetor possui um sítio de múltipla clonagem localizado entre os nucleotídeos 784 e
820 no qual o gene de interesse é inserido ficando sob o controle transcricional de um
promotor altamente ativo na fase tardia da infecção (Wang et al., 1991)
Figura 8: Mapa do vetor de transferência pSynXIVVIX3. Observamos a presença de umsítio de múltipla clonagem (SMC) localizado entre os nucleotídeos 784 e 820. Este sítio éregulado pelo promotor modificado da poliedrina (PsynXIVPpolh). Também observamos apresença do gene da poliedrina localizado a partir dos nucleotídeos 1739.Fonte: O’REILLY et al., (1992)
O vetor também possui o gene da poliedrina completo que neste caso funciona como
gene marcador, ou seja, a inserção do gene recombinante no vírus resulta na também inserção
da poliedrina podendo distinguir os vírus recombinantes dos não recombinantes.

47
4.1.3 Plasmídeo pCRII
O TA Cloning Kit pCR 2.1 (Invitrogen USA) é um plasmídeo que propicia a inserção
direta de produtos de PCR. Este passo nos permite ter o gene de interesse em plasmídeo tanto
para estoque do mesmo como para digestão com enzimas de restrição específicas para
liberação do fragmento e posterior inserção em um vetor de transferência.
4.1.4 Vírus vSyngal VI–
O baculovírus utilizado neste trabalho foi o Autographa califórnica
nucleopolyhedrovirus (AcNPV) recombinante, denominado vSyngal VI- (Wang, et al., 1991).
Este recombinante expressa a proteína beta-galactosidase e não expressa a proteína do
poliedro viral, a poliedrina.
Este vírus também nos foi cedido pelo Dr. Bergman Ribeiro da Universidade de
Brasília (UnB).
4.1.5 Linhagem celular utilizada.
A linhagem celular utilizada neste trabalho foi células de inseto SF-9 (células epiteliais
ovarianas de Spodoptera frugiperda) derivadas dos tecidos ovarianos de larvas adultas. O
meio de cultura utilizado foi o meio SF- 900 (Invitrogen USA), que não utiliza soro bovino
fetal.
4.2 DESENHO DE OLIGONUCLEOTIDEOS PARA A SÍNTESE DOS
FRAGMENTOS UTILIZADOS NO PREPARO DOS VETORES DE
TRANSFERÊNCIA
Os vetores de transferência são necessários para a construção dos baculovírus
recombinantes. Neste trabalho, os vetores de transferência foram obtidos pela inserção do
gene N, amplificados por PCR, no plasmídeo pSyn.

48
4.2.1 Primers para o gene N com a poliHistidina (6xHis)
Após o seqüenciamento do pET-N-Ara confirmou-se a presença de uma região de
poli Histidina (6xHis) antes da localização do gene N.
O desenho dos oligonucleotideos, realizado com base nas seqüências obtidas, foi
produzido tendo como ponto inicial o códon de iniciação da região da poli histidina (Figura
9).
Neste oligonucleotideo foi inserido o sítio de reconhecimento da enzima de restrição
Bgl II (Primer Forward). Outro oligonucleotideo foi desenhado a partir do “Stop códon” da
nucleoproteína na posição 1284pb. Neste oligonucleotideo foi incorporado o sítio de enzima
de restrição correspondente ao da enzima XmaI (Primer Reverse) (Tabela 3).
Tabela 3: Oligonucleotideos utilizados para amplificação do gene N com Cauda de Histidina
Nome do Primer Seqüência
Forward Histidina 5’ GAAGA TCT ATG CGG GGT TCT CAT CAT 3’
Reverso Histidina 5 TAACCC GGG TCA CAG CTT TAA GGG TCC 3’
Figura 9: Seqüência do gene da nucleoproteína com cauda de Histidina. Localização dos primers forward ereverso da nucleoproteína, utilizados para a síntese desta seqüência.

49
4.2.2 Primers para o sinal de exportação do gene gp67
O gene da GP67A, contido no plasmídeo pAcGP67A (Pharmigen, USA), foi
seqüenciado em nosso laboratório. A partir da seqüência obtida, oligonucleotideos foram
desenhados com o intuito de amplificar a seqüência do sinal de exportação deste gene (Figura
10). No primer forward, localizado a partir do códon inicial do sinal de exportação, foi
inserido o sítio de enzima de restrição XhoI e no primer reverse, localizado a partir do códon
final do sinal de exportação o sítio correspondente a enzima Bgl II (Tabela 4).
Tabela 4: Oligonucleotideos utilizados para amplificação da seqüência sinal de exportação do gene GP67.
Nome do Primer Seqüência
Forward Sinal
Exportação
5’CCG CTC GAG ATG CTA CTA GTA AAT CAG TCA
CACCAA GGC 3’
Reverso Sinal
Exportação
5’ GA AGA TCT CGC AAA GGC AGA ATG CGC CGC
CGC CGC CAA 3’
Figura 10: Seqüência do sinal de exportação do gene GP67 retirado do plasmídeo pAcGP67A comlocalização dos primers forward e reverso, utilizados para a síntese desta seqüência.

50
4.3 SINTESE DOS FRAGMENTOS E CLONAGEM
4.3.1 Síntese da região do gene N com a poliHistidina (6xHis) para inserção em
plasmídeos
Foi realizada uma reação de PCR (Anexo 1) usando como DNA molde o pET-N-Ara e
os oligonucleotideos Forward Histidina e Reverso Nucleoproteína (Tabela d). O fragmento
amplificado foi purificado através de kit GFX de purificação (Anexo 2) (Armsham USA) e
posteriormente clonado no plasmídeo pCRII (Anexo 3) (Invitrogen USA) seguindo as
instruções do fabricante. A este plasmídeo denominamos pCRII-Hist-N.
4.3.2 Síntese do sinal de exportação do gene gp67 para inserção em plasmídeos
Foi realizada uma reação de PCR (Anexo 1) usando como DNA molde o plasmídeo
pAcGP67A (Pharmigen, USA) e os primers específicos Forward Sinal de Exportação e
Reverso Sinal de Exportação anteriormente desenhados (Tabela e)
O fragmento foi purificado em kit GFX de purificação (Anexo 2) (Armsham USA) e
posteriormente clonado no plasmídeo pCRII (Anexo 3) seguindo as instruções do fabricante.
A este plasmídeo denominamos pCRII-Exp-Hist-N
4.4 CONSTRUÇÃO DOS VETORES DE TRANSFERÊNCIA
4.4.1.-. Clonagem do gene Hist-N no vetor de transferência PsynXIVVIX3.
O plasmídeo pCRII contendo o gene da nucleoproteína com Histidina (pCRII-Hist-N)
foi digerido com enzimas de restrição, BglII (Biolabs) e XmaI (Biolabs) para liberação do
fragmento de interesse. Realizou-se uma eletroforese em gel de agarose 0,8% para confirmar
o fragmento que foi posteriormente recortado do gel e purificado usando o kit GFX de
Purificação (Anexo 2) (Amersham USA).

51
O fragmento foi ligado no vetor de transferência pSynXIVVIX3 (Wang et al.,1991) que
tinha sido previamente digerido com as mesmas enzimas.
A este vetor de transferência denominamos pSyn-Hist-N e a confirmação da ligação do
gene Hist-N ao pSyn XIVVIX3 foi realizada através de uma reação de PCR (Anexo 1) usando
primers específicos. (Tabela 3).
4.4.2 Clonagem da seqüência de exportação (Exp) no vetor de transferência Psyn-His-N.
A seqüência sinal de exportação (Exp) clonada no plasmideo pCRII (Invitrogen USA) foi
digerida com enzimas de restrição, BglII (Biolabs) e XhoI (Invitrogen) para liberação do
fragmento. Realizou-se uma eletroforese em gel de agarose 0,8% para confirmar o fragmento
que foi posteriormente recortado do gel e purificado usando o kit GFX de Purificação (Anexo
2) (Amersham USA).
O fragmento foi clonado no vetor de transferência pSyn-Hist-N anteriormente produzido,
que tinha sido previamente digerido com as mesmas enzimas.
Para confirmar a ligação da seqüência exportadora (Exp) ao pSyn-Hist-N procedeu-se a
uma reação de PCR (Anexo 1) com primers específicos para a seqüência exportadora (Tabela
4).
4.5 PURIFICAÇÃO DOS VETORES DE TRANSFERÊNCIA: PSYN-
HIST-N e PSYN-EXP-HIST-N.
A pureza dos vetores de transferência e do DNA viral é crítica para o sucesso da co-
transfecção em células de inseto.
Devido a estas características de pureza para utilização dos plasmídeos, 1 ul de cada vetor
de transferência: pSyn-Hist-N, pSyn-Exp-Hist_N foram transformados em células

52
competentes (E. coli DH5α) (Anexo 4) e os recombinantes selecionados pela resistência a
ampicilina propiciada pelo vetor pSynXIVVIX3.
Cada vetor foi extraído da E. coli transformada utilizando o Kit de Extração (Anexo 5)
(Qiagen USA). Este tipo de extração gera um DNA suficientemente puro para utilização na
técnica de co-transfecção.
Realizou-se uma eletroforese em gel de agarose 0,8% para confirmação dos tamanhos
finais dos vetores e a qualidade dos mesmos.
4.6 SEQUENCIAMENTO DOS VETORES
A região do cassete do vetor pET-N-Ara bem como
dos vetores de transferência foram seqüenciados. Este cassete é composto pela região
promotora, cauda de histidina, gene N, sinal de Terminação, poliA e sinal de exportação
(presente somente em vetor de transferência).
4.6.1 Sequenciamento do pET-N-Ara
O seqüenciamento do pET-N-ARA foi realizado segundo o método de interrupção de
cadeia (SANGER et al., 1977) utilizando-se o kit BigDye v.3 (Applied Biosystems). Para isto
utilizou-se os primers universais T7 promoter e T7 Reverse que se localizam flanqueando o
sítio de múltipla clonagem do pET Directional TOPO® (Invitrogen USA). Após a reação, o
material foi analisado no equipamento do seqüenciador automático ABI Prism 377 Sequencer
(Applied Biosystems) do Laboratório de Virologia Molecular e posteriormente analisado pelo
programa BLAST (ALTSCHUL et al 1990) que compara a seqüência gerada a um banco de
dados (Genbank) e afere similaridade com outras seqüências, permitindo verificar a
integridade da seqüência e sua similaridade com outras espécies.
Este procedimento possibilitou confirmar a seqüência do gene N do Araraquara
Hantavírus e a região da poliHistidina (6xHistidina) presente antes da localização do gene N.

53
4.6.2 Sequenciamento dos vetores de transferência recombinantes.
O cassete dos vetores de transferência pSyn-Hist-N, pSyn-Exp-Hist_N foram
seqüenciados pela técnica de interrupção de cadeia (SANGER et al., 1977) utilizando-se o kit
BigDye v.3 (Applied Biosystems) no seqüenciador ABI Prism 377 Sequencer (Applied
Biosystems) para verificação dos insertos, seqüência dos fragmentos e integridade das
moléculas.
Para isto construiu-se um olinucleotideo com o intuito de seqüenciar todo o inserto.
(Tabela 5).
Tabela 5: Oligonucleotídeo desenhado para seqüenciar o gene da nucleoproteína
Nome do Primer Seqüência
Intermediário Nucleoproteína 5’ AGA CAG CAG ACT GGA AG 3’
Para o seqüenciamento dos 2 vetores de transferência foram utilizados também
oligonucleotideos do vetor de transferência pSynXIVVIX3 que estão localizados nas regiões
flanqueadoras do sítio de múltipla clonagem. Estes oligonucleotideos foram gentilmente
cedidos pelo Prof. Dr. Bergman Ribeiro da Universidade de Brasília (UnB) (Tabela 6).
Tabela 6: Oligonucleotídeos desenhados para seqüenciar o cassete dos vetores de transferência
Nome do Primer Seqüência
PSyn Forward 5’ GGG CCA AGC TTG GCG TTA TTG 3’
Psyn Reverso 5’ TCT GTA AAT CAA CAA CGC ACA G 3’
As seqüências geradas através do sequenciamento foram analisadas primeiramente
pelo próprio ABI Prism 377 Sequencer (Applied Biosystems) e posteriormente montadas
através do programa Códon Code Aligner. Este programa utiliza o sistema Phed/Pharp para
aferir a qualidade de cada base seqüenciada e tem a capacidade de remover tanto as
seqüências provenientes do vetor como as seqüências de baixa qualidade, um processo
chamado de "trimagem". Após este processo as seqüências são montadas através de
alinhamento de seqüências similares para obtenção da totalidade dos fragmentos
seqüenciados.

54
Após passarem pela trimagem, as seqüências obtidas foram analisadas através do
programa ORF Finder (Open Reading Frame Finder) que procura e analisa janelas de leitura,
possibilitando visualizar as possíveis proteínas que podem ser formadas a partir da seqüência
utilizada. Depois de esta análise, foi realizado também um BLAST a partir dos resultados do
ORF Finder. Neste programa as seqüências são comparadas a um banco de dados (Genbank) e
afere similaridade com outras seqüências, permitindo verificar a integridade da seqüência
clonada.
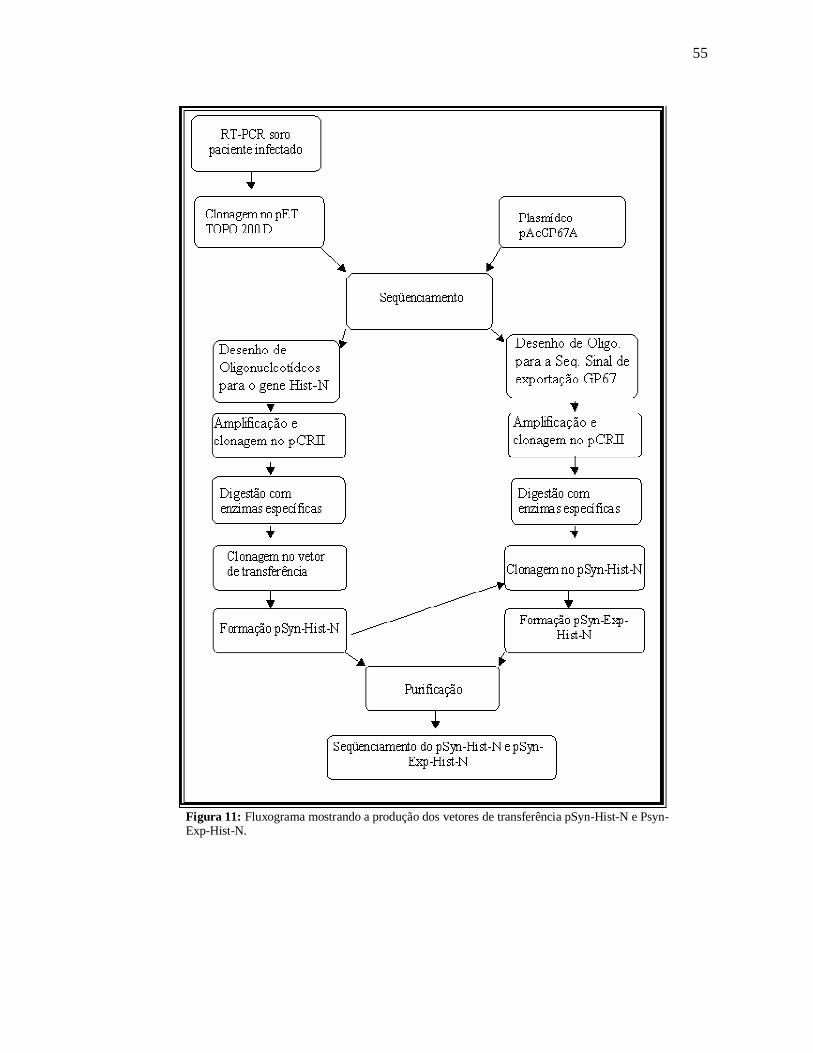
55
Figura 11: Fluxograma mostrando a produção dos vetores de transferência pSyn-Hist-N e Psyn-Exp-Hist-N.

56
4.7 PREPARO DO DNA DO VSYNGAL VI-.
O sobrenadante de células de inseto (SF9) infectadas com o vSyngal VI- (6 a 7 dias
após infecção) foi coletado e o DNA dos “budded vírus” (forma não oclusa dos baculovírus),
presentes neste sobrenadante, foi extraído por técnica de extração de “budded vírus” (Anexo
6) e concentrado em coluna Centricon 30,000 NMWL (Amicon-USA). Posteriormente o
DNA foi digerido com enzima Bsu36I (BioLabs), purificado com fenol-cloroformio e
novamente concentrado em coluna Centricon 30,000 NWM
D (Amicon-USA).
4.8 CONSTRUÇÃO DOS BACULOVÍRUS RECOMBINANTES
ATRAVÉS DO PROCESSO DE CO-TRANSFECÇÃO
Células SF-9 (1x106 células ou 50 a 70% de densidade) foram transferidas para uma
placa de 60mm de diâmetro e deixado overnight a 27ºC para formação de monocamada de
células. Após isto, 0,5 ug de DNA do vSyngal linearizado com Bsu36I e 1 ug de um dos 2
plasmídeos recombinantes foram diluídos em 0,5 mL de meio SF-900 (Invitrogen USA),
misturados a 50 ul de solução de lipofectin (Gibco-BRL) e incubados por 15 minutos a
temperatura ambiente.
O meio de cultura das células foi substituído por 1 ml da mistura de DNAs/lipossomos
e a placa de 60mm incubada por 3 horas. Neste período os lipossomos irão se fundir a
membrana plasmática das células propiciando a entrada do DNA viral e vetor de transferência
ao citoplasma celular, onde acontecerá o processo de recombinação homóloga.
Após esse período a mistura é trocada por meio SF-900 novo e as células incubadas a
27ºC por 5 a 7 dias. Como vimos, dentro das células de inseto ocorrera recombinação
homóloga entre as regiões flanqueadoras do gene de interesse, no vetor de transferência, e as
regiões homólogas no genoma do vírus vSyngal VI-, que flanqueia o gene da beta-
galactosidase. Desta forma o vírus recombinante terá o gene de interesse no lugar do gene da
beta-galactosidase e também o gene da poliedrina completo, pois este faz parte do vetor de

57
transferência pSynXIVVIX3. Desta forma o recombinante produzira corpos de oclusão, o que
facilitará sua identificação. Já o vírus vSyngal VI- não recombinante não produzira corpos de
oclusão, pois não carrega consigo o gene da poliedrina, único constituinte do poliedro.
(PHARMINGEN,1999)
Após a formação de poliedros nas células o meio de cultura (sobrenadante) que
contêm as formas não oclusas de baculovírus (“budded vírus”) recombinantes e não
recombinantes foi utilizado para infectar novas células. Os baculovírus recombinantes
produzidos foram denominados vSyn-Hist-N e vSyn-Exp-Hist N.
As células infectadas com presença de poliedros no núcleo foram utilizados para
extração do DNA dos vírus recombinantes com o propósito de confirmar a inserção do gene
de interesse no genoma dos baculovírus vSyngal VI- recombinantes.
4.9 CONFIRMAÇÃO DA INSERÇÃO DO GENE DE INTERESSE
Para confirmar a inserção dos genes no genoma dos baculovírus recombinantes
selecionados foi realizada uma extração de DNA dos poliedros virais como descrito por
O’Reilly et al 1992 (Anexo 7). Com este DNA foram realizadas reações de PCR (Anexo 1)
com oligonucleotideos específicos para os genes: N com Histidina e com sinal de exportação
(Tabela 7).
Tabela 7: Relação entre baculovirus recombinantes e primers utilizados para verificação do inserto no seugenoma.
Baculovírus recombinante Primers
vBac-Hist-N • Primer Forward Histidina
• Primer Reverse Nucleoproteína
vBac-Exp-Hist-N • Primer Forward Sinal de Exportação
• Primer Reverse Nucleoproteína

58
4.10 PURIFICAÇÃO DOS BACULOVÍRUS RECOMBINANTES
4.10.1 Diluição seriada em 96 poços
Com o sobrenadante das células infectadas (5 a 7 dias após a transfecção) foi realizada
uma diluição seriada do virus em placas de 96 poços contendo células SF9. A identificação
dos vírus recombinantes se deu pela presença de corpos de oclusão no núcleo das células
infectadas. Foram selecionadas as maiores diluições que produziram corpos de oclusão no
núcleo das células infectadas e o sobrenadante destas usado novamente em outra diluição
seriada. Este processo foi repetido 3 vezes para cada vírus recombinante.
4.11 VERIFICAÇÃO DA PUREZA DOS VBACS RECOMBINANTES
O sobrenadante de células de inseto infectadas (5 a 7 dias após a infecção) foi
utilizado para obtenção dos vírus não oclusos (budded vírus) dos quais foi extraído o DNA
pela técnica descrita por O’Reilly et al 1992 (Anexo 6).
A extração do DNA genômico foi confirmada através de gel de agarose 0,8%.
O DNA genômico de vBac-Hist-N e vBac-Exp-Hist-N foram usados como molde em
uma reação de PCR (Anexo 1) para verificação da pureza dos recombinantes. Foram
utilizados os primers específicos para o gene Hist-N epara Exp-Hist-N. Também foram
utilizados oligonucleotideos desenhados para o gene da beta-galactosidase, já que este é o
gene substituído na recombinação homóloga.
Assim, se o PCR confirmar fragmentos do gene da beta-galactosidase isto sugere que
os baculovirus recombinantes não estão totalmente purificados, sendo necessárias outras
passagens em diluição seriada em 96 poços.

59
4.12 CÁLCULO DO “MULTIPLICITY OF INFECTION” (M.O.I) E
CRIAÇÃO DOS ESTOQUES VIRAIS.
Foram utilizados os materiais das placas de diluição tanto de vBac-Hist-N e vBac-
Exp-Hist-N seriada anteriormente descritas para a produção de um estoque. A partir deste
estoque foi calculada a concentração de partículas virais por ml. Estas concentrações foram
posteriormente igualadas através de uma diluição da maior concentração para facilitar e
manter um mesmo M.O.I. para as duas amostras.
A partir destes valores o M.O. I foi calculado utilizando a fôrmula descrita por
O’Reilly et al 1992:
M.O.I = Nº Vírus / Nº células

60
5 RESULTADOS E DISCUSSÃO
5.1 SEQUENCIAMENTO DO pET-N-ARA.
O pET-N-ARA proveniente do laboratório de Virologia da faculdade de Medicina da
USP Ribeirão Preto foi seqüenciado através de primers universais T7 Forward e T7 Reverse.
Obtivemos, através da técnica de interrupção de cadeia (SANGER et al 1977), uma
seqüência de aproximadamente 1500 pb, correspondente ao gene da nucleoproteína do
Araraquara hantavirus, cauda de histidina e regiões flanqueadoras do sítio de múltipla
clonagem (SMC) presentes neste plasmídeo.
Esta seqüência foi analisada através do programa Basic Local Alignment Search Tool
(BLAST-N) (ALTSCHUL et al., 1990) que localiza regiões similares entre seqüências,
compara seqüências de nucleotídeos e calcula um valor estatístico de similaridade auxiliando
a verificação da família a que pertence o gene (NCBI) (Figura 12).
Ao aplicar este programa à seqüência obtida, resultaram em uma similaridade de 82%
com os vírus Oran vírus, Andes vírus e Bermejo vírus. Estes resultados sustenta os
encontrados por Moreli et al 2004, Silva –Vergara et al 2002 e Figueiredo et al 2003, que
mostram que os principais vírus Brasileiros mostram uma alta similaridade com os vírus
encontrados na Argentina principalmente com o Andes vírus.

61
Figura 12: BLAST- N da seqüência obtida através do sequenciamento do pET-N-ARA. Mostra altasimilaridade da seqüência encontrada como os hantavirus: Oran vírus, Andes vírus e Bermejo vírus.
A seqüência obtida também foi analisada no BLAST-X que realiza basicamente o mesmo
processo que o BLAST-N mais comparando através das seqüências de aminoácidos,
traduzindo primeiramente o gene para depois compara-lo.
Esta análise, como se previa, mostrou alta similaridade, 95% com o Maciel Hantavirus e
94 e 93% com os Hantavirus: Bermejo vírus e Lechiguanas vírus correspondentemente. Estes
resultados corroboram com os obtidos por Bolhman (2002) que mostra alta similaridade entre
a nucleoproteína dos principais vírus encontrados no Brasil com os vírus encontrados na
argentina.
Podemos observar através dos resultados obtidos que há uma maior similaridade
quando comparadas as seqüências de aminoácidos ao invés das seqüências de nucleotídeos
como afirmou Plyusnin (2002) e Padula (2000).
Observamos que ao comparar as seqüências tanto de aminoácidos como de
nucleotídeos não há comparação com o próprio Araraquara Hantavírus. Isto se deve a que este
vírus foi relatado recentemente e a seqüência do gene da nucleoproteína depositada no
GenBank (NCBI) não esta totalmente disponível.

62
Figura 13: BLAST- X da seqüência obtida através do sequenciamento do pET-N-ARA. Mostra altasimilaridade da nucleoproteína do Hantavírus Araraquara com a dos hantavirus: Vírus Maciel, Vírus Bermejo,e Vírus Lechiguanas.
5.2 MONTAGEM DOS VETORES DE TRANSFERENCIA
RECOMBINANTES
Através do sequenciamento do pET-N-ARA oligonucleotideos desenhados
propiciaram a amplificação, através de reação de PCR, do gene da nucleoproteína com cauda
de Histidina.
Esta amplificação gerou um fragmento de aproximadamente 1400pb (Figura 13).
Primers desenhados para a seqüência do sinal de exportação amplificaram este gene
usando como DNA molde o plasmídeo pAcGP67A (Pharmigen USA) em reação de PCR.
Este fragmento apresentou aproximadamente 120 pb (Figura 13).
A eletroforese em gel de agarose 1.0% mostra os fragmentos amplificados através de
reação de PCR, para visualização e confirmação de seus tamanhos.

63
Figura 14: Gel de agarose 1,0% mostrando osfragmentos de Histidina com nucleoproteína e Seqüênciado sinal de exportação do gene GP67 amplificados comprimers específicos. 1: Marcador DNA λ Hind III (5 ul)2: Gene Nucleoproteína Histidina 3: Seqüência deexportação do gene GP67.
Estas seqüências amplificadas, após serem clonadas (Anexo 3) no plasmídeo pCRII
que possui 4.0Kb e selecionadas as ligantes através de reação de PCR (Anexo 1) com primers
específicos, geraram os seguintes plasmídeos recombinantes: pCRII-Hist-N com
aproximadamente 5,4Kb e pCRII-Exp com aproximadamente 4,12 Kb.
Após a digestão destes plasmídeos com enzimas de restrição especificas, os
fragmentos gerados foram clonados no vetor de transferência pSynXIVVIX3 (5,8Kb) gerando
assim os vetores recombinantes. Estes vetores recombinantes denominados pSyn-Hist-N e
pSyn-Exp-Hist-N tinham um tamanho aproximado de 7,2Kb e 7,32Kb correspondentemente
(Figura 14).
A eletroforese em gel de agarose a 1,0% mostra o tamanho final dos vetores
recombinantes, bem como sua integridade e concentração (Figura 14)

64
Figura 15: Tamanho final dos vetores de transferênciarecombiantes, pSyn-Hist-N e pSyn-Exp-Hist-N. Estesvetores foram digeridos com enzima de restrição XmaIpara visualização do seu tamanho. 1: Marcador DNA λHind III (5 ul) 2: pSyn-Hist-N 3: pSyn-Exp-Hist-N.

65
Figura 16: Fluxograma mostrando o tamanho dos fragmentos amplificados, plasmídeos pCRII montados evetores de transferência, pSyn-Hist-N e pSyn-Exp-Hist-N. 1. Amplificação do gene da Nucleoproteína comcauda de Histidina, utilizando como molde o pET-N-ARA e primers específicos. 2. Produção de um fragmentode 1400 pb referentes a nucleoproteína com cauda de Histidina. 3. Clonagem do fragmento no plasmídeo declonagem pCRII (4.0Kb). 4. Digestão do pCRII-Hist-N com enzimas de restrição específicas e clonagem dofragmento no vetor de transferência pSyn (5,8 Kb). 5. Produção do vetor de transferência pSyn-Hist-N. 6.Amplificação da seqüência sinal de exportação do gene GP67 utilizando como molde o plasmídeo pAcGP67A.7. Produção de um fragmento de 120 pb. 8. Clonagem do fragmento no plasmídeo de clonagem pCRII (4.0Kb).9. Digestão do vetor de transferência pSyn-Hist-N com enzimas de restrição específicas. 10 Clonagem daseqüência sinal de exportação. 11. Produção do vetor de transferência pSyn-Exp-Hist-N
5.3 SEQUENCIAMENTO E ANALISE DAS REGIÕES CRÍTICAS DOS
VETORES DE TRANSFERENCIA RECOMBINANTES
Todos os vetores de transferência foram seqüenciados no laboratório de Virologia
Molecular da Universidade de Mogi das Cruzes. Foi utilizado o método de interrupção de
cadeia (SANGER et al 1977), Kit Big Dye v.3 e seqüenciador ABI Prism 377 Sequencer
(applied Biosystem).

66
Este sequenciamento propiciou verificar a integridade dos fragmentos clonados,
mutações e frame de leitura, sendo as seqüências analisadas e montadas pelo programa códon
code aligner e posteriormente pelos programas ORF FINDER e BLAST (NCBI).
As análises realizadas pelo programa ORF FINDER propiciou verificar a janela de
leitura da proteína e analisar a proteína a ser formada. Verificou-se também a seqüência
inicial da proteína, bem como a final e sua inserção no vetor de transferência pSyn.
A análise realizada pelo programa BLAST mostrou as mesmas similaridades
encontradas quando comparada a seqüência do pET-N-ARA.
Após estas análises, os dois vetores de transferência foram esquematizados junto com
a ligação dos fragmentos no plasmídeo para uma melhor compreensão de sua conformação.
5.3.1 pSyn-Hist-N
Vetor de transferência contendo o gene N do hantavirus Araraquara junto com uma
cauda de Histidina (6xHis) inserido no Sítio de Múltipla Clonagem (SMC) entre as enzimas
de restrição Bgl II e XmaI. (Figura 16)
O propósito de utilizar esta seqüência de polihistidina, e de facilitar a posterior
purificação destas proteínas em coluna de níquel. Estudos anteriores realizados por Bornhorst
et al 2000, mostra que a coluna de níquel é capaz de purificar de 90 a 95% das proteínas
ligadas a Histidina presentes em uma amostra. Além desta característica esta seqüência não
interfere na conformação e atividade da proteína já que possui um sítio de proteinase Xa que
possibilita remover a polihistidina após a purificação se necessário. Esta remoção não altera
as características das proteínas e muitas vezes nem se faz necessária (SCHAFER et al 2002A;
SCHAFER et al 2002B).
Trabalhos anteriores realizados por Sirola (2003) e Hujakka (2001) entre outros,
mostram uma purificação facilitada utilizando coluna de níquel. A presença da cauda de
histidina propiciou este tipo de purificação, sendo que altas concentrações de proteína
recombinante foram purificadas por este método. Nestes trabalhos a proteína N foi expressa e
purificada em coluna de níquel através do método desnaturante com 6M de uréia.
Esta desnaturação propicia que a proteína exponha o sítio de polihistidina (6xHis)
facilitando sua ligação na coluna de níquel, podendo assim ser purificada.

67
A análise do sequenciamento deste vetor mostrou integridade do gene N e da cauda de
Histidina. Na análise do ORF Finder, que visualiza as possíveis janelas de leitura, gerou os
seguintes dados: Frame +1, De:13 a 1407, Tamanho 1395 pb correspondente à nucleoproteína
com cauda de histidina.
Análise de Blast mostrou similaridade com os hantavirus: Oran vírus, Andes vírus e
Bermejo vírus como ocorrido na analise realizada com o pET-N-ARA.
Figura 17: pSyn-Hist-N esquematizado. Na parte superior podemos observar a conformação do plasmídeo,tamanho, o gene da poliedrina (gene marcador) e o ponto de inserção do gene da nucleoproteína com caudade histidina que foi no SMC entre as enzimas Bgl II e XmaI. Na parte inferior podemos observar maisdetalhadamente os pontos de inserção do gene. Observamos também a localização do promotor modificadoda poliedrina PpolhXIV na região upstream da cauda de histidina.
5.3.2 pSyn-Exp-Hist-N
Vetor de transferência recombinante que contém o gene da nucleoproteína do
Araraquara Hantavirus junto com a cauda de histidina (6xHis) inserido no SMC entre as

68
enzimas de restrição Bgl II e XmaI. Também possui a seqüência correspondente ao sinal de
exportação do gene GP67 (Figura 17).
A seqüência sinal de exportação presente neste vetor recombinante tem o propósito de
facilitar a purificação desta proteína, já que será exportada para o meio de cultura, diminuindo
assim a possibilidade de formar agregados insolúveis (KATAGIRI et al., 2002). Outra
facilidade é a presença da cauda de histidina que permitirá a purificação em coluna de níquel
(BORNHORST et al 2000).
A purificação de proteínas com cauda de histidina secretadas diretamente para o meio de
cultura pode apresentar alguns problemas. O meio de cultura usado para células de inseto tem
um pH ácido (6,0 a 6,5) e também contém substancias que podem inibir a ligação da proteína
com 6xHis no complexo níquel-agarose (BORNHORST et al 2000).
Alguns aminoácidos como a glutamina, glicina e histidina presentes em altas
concentrações no meio de crescimento das células podem competir com as proteínas
recombinantes para a ligação na matriz Ni-NTA (Qiagen Usa).
Devido a isto a diálise em membrana com tampão TBS (pH 8,0) do meio de cultura
geralmente provê condições ótimas de análise em Ni-NTA (COLIGAN et al 1995).
Trabalhos realizados por Murphy (1993) mostrou um alto nível de expressão da
proteína recombinante GP120 de HIV-1 sendo que 70% desta proteína foi exportada para o
meio de cultura.
Em trabalhos realizados por Oers, (2001) e Golden (1998) mostram que a utilização do
sinal de exportação de baculovirus mostra ser mais eficaz na secreção de proteínas
recombinantes quando comparados com sinais de genes de mamíferos e outros sinais de
exportação presentes em baculovirus, entre eles o egt.
Nestes trabalhos o sinal de exportação de GP67 mostrou ser bastante eficaz e quando
comparados a outras seqüências sinais, observou-se que além de ser bastante utilizado é
bastante funcional. Nos trabalhos, realizados por Golden 1998, mostrou que a seqüência sinal
de GP67 só é menos funcional que a seqüência sinal do gene ativador de tecidos
plasminogênicos, mas mesmo assim mostrou um alto nível de exportação da proteína
recombinante, cerca de 60%.

69
Figura 18: pSyn-Exp-Hist-N esquematizado. Na parte superior podemos observar a conformação do plasmídeo,tamanho, o gene da poliedrina (gene marcador) e o ponto de inserção do gene da nucleoproteína com cauda dehistidina que foi no SMC entre as enzimas Bgl II e XmaI. Também podemos observar a inserção da seqüênciasinal de exportação inserida entre as enzimas XhoI e Bgl II. Na parte inferior podemos observar maisdetalhadamente os pontos de inserção dos genes. Observamos também a localização do promotor modificado dapoliedrina PpolhXIV na região upstream do sinal de expostação.
A análise do sequenciamento deste vetor mostrou integridade do gene N com de cauda
de Histidina e seqüência sinal. Na análise do ORF Finder, que visualiza as possíveis janelas
de leitura, gerou os seguintes dados: Frame +1, De 7 a 1521, Tamanho1515pb correspondente
à nucleoproteína com cauda de histidina e seqüência sinal de GP67.
Análise de Blast mostrou similaridade com os hantavirus: Oran vírus, Andes vírus e
Bermejo vírus como ocorrido na analise realizada com o pET-N-ARA. Isto confirma a correta
construção do vetor de transferência.
5.4 OBTENÇÃO DOS BACULOVIRUS RECOMBINANTES
Após 5 a 7 dias da co-transfecção do vetor de transferência junto ao DNA do vSyngal,
as células Sf9 foram observadas em microscópio ótico (40X) para visualização dos efeitos
citopáticos, aumento de núcleo, vacúolos e grânulos citoplasmáticos e poliedros no interior do
núcleo (Figura 18).

70
Figura 19: Células Sf9 em monocamada. Aumento em 40X 1. Células sem infecção. Observamos que possuemum tamanho uniforme sem grandes variações. 2. Células com infecção, mas sem produção de poliedros, apenasefeitos citopáticos (2A). Estas células apresentam um aumento do seu volume celular e do núcleo, além depresença de vacúolos no interior celular.. 3. Células com infecção e poliedros no interior do núcleo (3A). Estascélulas apresentam-se maiores que as sem infecção, com aumento do núcleo, sendo que em muitos casos quasenão se distingue o citoplasma.
Também foram contadas as células infectadas com poliedros aparentes no núcleo e
células infectadas sem poliedros, com o intuito de estimar a porcentagem de recombinantes
gerados.
Trabalhos realizados com sistemas de baculovirus comerciais mostraram uma taxa de
recombinação homologa de até 90%, já na utilização de baculovirus não comerciais chega a
40% (IKONOMOU et al., 2003; DOVERSKOG et al., 2000).
Em nosso trabalho estimamos uma porcentagem de recombinação homóloga de
aproximadamente 35% utilizando baculovirus não comercial o que corrobora com os
trabalhos mencionados anteriormente.
A grande diferença entre a utilização de baculovírus comerciais e não comerciais se dá
principalmente devido a que os baculovírus comerciais são altamente purificados e possuem
tecnologias mais desenvolvidas que facilitam grandemente o processo de recombinação
homologa.
O meio de cultura (Sobrenadante) contendo os “budded vírus” (forma não oclusa dos
vírus) foram utilizados para infectar novas células e estas após 5 a 7 dias foram novamente
observadas mostrando uma quantidade maior de celulas infectadas e com maior quantidade de
poliedros no interior dos núcleos.

71
5.5 VERIFICAÇÃO DO INSERTO NO GENOMA DOS BACULOVIRUS
RECOMBINANTES
As células infectadas com poliedros presentes no núcleo foram recolhidas para
verificar se os fragmentos de interesse estavam inseridos no genoma dos baculovirus
recombinantes. Este teste foi realizado utilizando DNA genômico dos recombinantes e
primers específicos.
A eletroforese em gel de agarose 1,0% mostra o gene da nucleoproteína com cauda de
histidina amplificados a partir do baculovirus recombinante vBAc-Hist-N e o gene da
nucleoproteína com cauda de histidina e sinal de exportação amplificados a partir do vBac-
Exp-Hist-N. Esta eletroforese confirma a inserção dos fragmentos no genoma dos
baculovirus (Figura 19).
Figura 20: Eletroforese em gel de agarose 1.0% para verificaçãodo inserto dos fragmentos de interesse no genoma dosbaculovirus recombinantes: vBAc-Hist-N e vBac-Exp-Hist-N. 1:Marcador DNA λ Hind III (5 ul) 2: vBac-Hist-N 3: vBac-Exp-Hist-N.

72
5.6 PURIFICAÇÃO DOS BACULOVIRUS RECOMBINANTES
Como vimos anteriormente à taxa de recombinação homóloga em nosso sistema de
expressão foi de 35%. Devido a isto a purificação em placas de 96 poços (96 well) através de
diluição seriada se fez necessária. Após 3 passagens pelas placas com diluições sucessivas,
que variavam entre 101 e 1010 virus/ml, a última diluição com presença de poliedros for
recolhida.
Assim tanto o baculovirus recombinante vBAc-His-N como o vBac-Exp-Hist-N foram
purificados através de 3 passagens em diluições seriadas (Figura 20). Este material foi
novamente colocado em células SF9 produzindo um estoque do vírus obtendo um título de
1x107 vírus/ml para o vBac-Hist e 1x108 vírus/ml para o vBac-Exp-Hist-N.A partir O vBac-
Exp-Hist-N foi diluído para manter 1x107 que é a mesma diluição de vBac-Hist-N.
Figura 21: Diluição seriada Hist-N (Linhas A,B,C e D) e Exp-Hist-N (Linhas E, F, G e H). Foram realizadasdiluições seriadas que variavam de 101 (Coluna 1) a 1010 (Coluna 10). As colunas 11 e 12 foram usadas comocontroles negativos. Foram escolhidas as maiores diluições a apresentarem poliedros.

73
5.7 VERIFICAÇÃO DA PUREZA DOS BACULOVIRUS
RECOMBINANTES
O DNA dos baculovirus recombinantes purificados serviu de molde em uma reação de
PCR com promers específicos para o gene Hist-N e Exp-Hist-N bem como para o gene da
Betagalactosidase.
Esta amplificação tinha o intuito de verificar a presença do gene da betagalactosidase,
já que este é o gene trocado no processo de recombinação homóloga. Assim, a presença deste
gene sugere a não purificação dos baculovirus recombinantes.
A eletroforese em gel de agarose 1,0% mostra o gene de Hist-N e Exp-Hist-N
amplificados comprovando novamente sua inserção no genoma e a ausência de amplificação
do gene da betagalactosidase sugerindo assim a purificação dos recombinantes. Como
controle positivo foi utilizado o vSyngalVI- que possui o gene da betagalactosidase (Figura
21).
Figura 22: eletroforese em gel de agarose 1,0% mostra o gene de Hist-N e Exp-Hist-Namplificados comprovando novamente sua inserção no genoma e a ausência deamplificação do gene da betagalactosidase. Como controle positivo mostra o gene dabetagalactosidase amplificado a partir do DNA do vSyngalVI-, vírus recombinante quepossui este gene. 1: Marcador DNA λ Hind III (5 ul) 2: Hist-N (vBac-Hist-N), 3: Geneda Betagalactosidase (vBac-Hist-N), 4: Exp-Hist-N (vBac-Exp-Hist-N), 5: Gene daBetagalactosidase (vBac-Exp-Hist-N), 6: Gene da Betagalactosidase (vSyngalVI-).

74
5.8 CÁLCULO DO M.O.I E PRODUÇÃO DE ESTOQUES VIRAIS.
A partir destes resultados e a contagem de células por mililitro (2x106 células/ml) foi
calculado o M.O.I que foi de 5, tanto para os baculovirus recombinantes vBac-Hist-N como
para o vBAc-Exp-Hist-N. Este cálculo baseou-se na fórmula descrita por O’ Reilly 1992.
Trabalhos realizados por O’Reilly em 1992 mostra que para uma efetiva e abundante
produção de proteína recombinante, o M.O.I dos baculovírus recombiantes devem variar entre
0,2 e 10. Assim mantivemos o M.O.I em 5 com o intuito de facilitar uma futura produção da
proteína recombinante.
Após o cálculo do M.O.I, os dois baculovírus recombinantes foram multiplicados,
mantendo o M.O.I em 5, para produção de um estoque viral, visando a produção e purificação
das proteínas recombinantes.

75
CONCLUSÕES E SUGESTÕES
A partir dos resultados obtidos neste trabalho podemos concluir que o sistema de
expressão de baculovirus é uma boa opção para expressão de proteína N de Hantavirus, já que
neste sistema é possivel adicionar seqüências e recursos que possibilitam uma purificação
facilitada da proteína recombinante.
Podemos observar que os baculovirus receberam o gene da nucleoproteína e que
possuem potencial para expressão da proteína N recombinante que servirá como uma
ferramenta diagnóstica da hantavirose no Brasil.
Faz-se necessária a produção e purificação da proteína bem como estudos de
antigenicidade desta proteína, o que possibilitará uma futura utilização desta em testes
diagnósticos.

76
REFERÊNCIAS BIBLIOGRAFICAS
ALTSCHUL S. F., GISH W., MILLER W., MYERS E. W., LIPMAN D. J. Basic Localalignment search tool. Journal Molecular Biology 1990; 215(3):403-410.
BERGER I., FLTZGERALD D. J., RICMOND T. J. Baculovirus expression system forheterologous multiprotein complexes. Nature Biotechnology 2004; 22: 1583-1587.
BHARADWAJ M., BOTTEN J., TORREZ-MARTINEZ N., HJELLE B. Rio Marmoré vírus:genetic characterization of a newly recognized hanatvirus of the pygmy rice rat,Oligoryzomys microtis, from Bolivia. American Journal Tropical Medicine and Hygiene.1997; 57(3): 368-374.
BILLECOCQ A., COUDRIER D., BOUE F., COMBES B., ZELLER H., ARTOIS M.,BOULOY M. Expression of the nucleoprotein of the Puumala virus from the recombinantSemlike Forest virus replicon: characterization and use as a potential diagnostic tool. ClinicalDiagnostic Laboratory Immunology. 2003 Jul;10(4):658-663.
BORNHORST J. A., FALKE J. J. Purification of proteins using polyhistidine affinity tags.Methods in Enzimology 2000; 326:245-254.
CARSON D. D., SUMMERS M. D., GUARINO L. A. Transiet expression of the Autographacalifornica nuclear polyhedrosis virus immediate-early gene, IE-N is regulated by three viralelements. Journal of Virology 1991B; 65:945-951.
CARSON, D.D.; SUMMERS, M.D.; GUARINO, L.A. Molecular analysis of a baculovirusregulatory gene. Virology 1991A;182:279-286.
CDC - CENTERS FOR DISEASE CONTROL AND PREVENTION. Hantavirus pulmonarysyndrome. Panama 1999 – 2000 MMWR 49:205-207.
CDC - CENTERS FOR DISEASE CONTROL AND PREVENTION: Hantavirus pulmonarysyndrome, United States 1993 MMWR 42:421-424.
COLLIER L., OXFORD J. Human Virology. Oxford University Press. Second Edition. USA2000.
COLIGAN J. E., DUNN B. M., PLOEGH H. L., SPEICHER D. W., WINGFIELD P. T.Current protocols in protein science. 1995 Vol. 1. John Witney and Sons, New York.

77
De PAULA S. O., PIRES NETO R. J., CORREA J. A., ASSUMPCAO S. R., COSTA M. L.,LIMA D. M., FONSECA B. A. The use of reverse transcription-polymerase chain reaction(RT_PCR) for the rapid detection and identification of dengue virus in an endemic region: avalidation study. Transactions of the Royal Society of Tropical Medicine and Hygiene2002; 96(3): 266-269.
DERETSKY Z. National Science Foundation. Disponível em:http://www.nsf.gov/news/special_reports/ecoinf/solved.jsp Data : 21 setembro 2006.
DOVERSKOG M., BERTRAM E. LJUNGGREN J. , OHMAN L., SENNERSTAM R.,HAGGSTROM L. Cell cycle progression in serum-free cultures of Sf9 insect cells;modulation by conditioned medium factors and implications for proliferation andproductivity. Biotechnology Progress. 2000;16:837-846.
EARLE D. P. Analysis of sequencial physiologic derangements in epidemic hemmorragicfever, with a commentary on management. American Journal Medicine 1954; 16(5): 690-709.
ELGH F., LUNDKVIST A., ALEXEYEV A. O., STENLUD H., AVXIC-ZUPANC T.,HJELLE B., LEE W. H., SMITH J. K., VAINIONPÃÃ R., WIGER D., WADELL G., JUTOP. Serological Diagnosis of Hantavirus infections by an Enzyme-linked immunosorbent assaybased on detection of Immunoglobulin G and M responses to recombinant nucleocapsidproteins of five viral serotypes. Journal Clinical Microbiology 1997; 35(5):1122-1130.
ELLIOT L. H., KSIAZEK T. G., ROLLIN P. E. Isolation of the causative agent of hantaviruspulmonary syndrome. American Journal Tropical Medicine and Hygiene. 2000; 51(1):102-108.
ELLIOT L. H., KSIAZEK T. G., ROLLIN P. E. Isolation of the causative agent of hantaviruspulmonary syndrome. American Journal Tropical Medicine and Hygiene 1994; 51(1): 102-108.
FERREIRA M. S. Hantaviroses. Revista Sociedade Brasileira de Medicina Tropical. 2003;36(1):81-96.
FIGUEIREDO L. T., MORELI M. L., ALMEIDA V. S., FELIX P. R., BRUNO J. C.,FERREIRA I. B., MANCANO F. D. Hantavirus pulmonary syndrome (HPS) in Guariba SP,Brazil. Report of 2 cases. Revista Instituto Medicina Tropical São Paulo 1999; 41(2): 131 -137.

78
FIGUEIREDO M. L. T., CAMPOS M. G., RODRIGUES B. F. Síndrome pulmonar ecardiovascular por Hantavírus: aspectos epidemiológicos, clínicos, do diagnóstico laboratoriale do tratamento. Revista Sociedade Brasileira de Medicina Tropical 2001;34(1)32-47.
FIGUEIREDO M. T., SOUZA M. L. T., CARVALHO R. C. R., BARBAS V. C. S.,PINCELLI P. M. Síndrome pulmonar e cardiovascular por Hantavírus. Jounal Pneumologia2003 Set/Out;29(5):309-323.
FOLGUERAS-FLATSCHART AV, FLATSCHART RB, RESENDE M, SOGAYAR MC.Early detection of productive baculovirus DNA transfection. Biotechniques 2000; 29:430-434.
FUNASA. Ministério da Saúde, Fundação Navional de Saúde. Hantavirose no Brasil. InfomeEpidemiológico do SUS. 2005; 13(5):1-10. Disponível em:http://bvsms.saude.gov.br/bvs/periodicos/inf_epi_2005_5completo.pdf. Retirado em: 20 deOutubro de 2006.
GARCIA M., MARTINS S. L. Zoonose – Hantavirus 2001. Disponível em:http://www.mgar.vet.br/zoonoses/aulas/aula_hantavirose.htm Retirado em: 21 de setembro de2006
GARCIN D., LEZZI M., DOBBS M., ELLIOTT R. M., SCHMALJOHN C., KANG C. Y.,KOLAKOFSKY D. The 5’ ends of Hantaan virus (Bunyavirus) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. Journal Virology 1995; 69(9): 5754-5762.
GARIN D., PEYREFITTE C., CRANCE J. M., LE FAOU A., JOUAN A., BOULOY M.Highly sensitive Taqman PCR detection of Puumala hantavirus. Microbes and infection2001; 3(9): 739-745.
GARMENDIA M. M., Síndrome pulmonar por Hantavirus, uma enfermedad transmitida porroedores. Revista Digital Del Centro Nacional de Investigaciones agropecuarias deVenezuela. Número Especial 2004. Disponible em:www.ceniap.gov.ve/ceniaphov/articulos/me1arti/molina-m/arti/molina-m.htm Acesso em: 17de outubro 2006.
GAVRILOVSKAYA I. N., BROWN E. J., GINSBERG N. H., MACKOW E. R. Cellularentry of hantavirus wivh cause hemorrhagic fever with renal syndrome is mediated by beta 3integrins. Journal Virology, 1999; 73(5): 3951-3959.

79
GAVRILOVSKAYA I. N., SHEPLEY M., SHAW R., GINSBERG M. H., MACKOW E. R.Beta 3 Integrins mediate the cellular entry of hantavirus that cause respiratory failure. ProcessNational Academy Science USA, 1998; 95(12): 7074-7079.
GOLDEN A., AUSTEN D. A., SCHRAVENDIJK M. R., SULLIVAN B. J., KAWASAKI E.S., OSBURNE M. S. Effect of promoters and signal sequence on the production of secretedHIV-1 gp 120 protein in the baculovirus system. Protein Expression And Purification1998;14(1):8-12.
GOTT P., ZOLLER L., DARAI., BAUTZ E. K. A major domain of hantavirus is located onthe aminoproximal site of the viral nucleocapsid protein. Virus genes 1997; 14(1):31-40.
GRANADOS R. R., WILLIAMS K. A. In vivo infection and replication of baculoviruses. In:GRANADOS R. R. FEDERICI B. A. (Eds). The biology of baculoviruses. Boca Raton:CRC, 1986; 1:89-108
GROEN J., VAN DER G. G., HOOFD G., OSTERHAUS A. Comparison ofimmunofluorescence and ezyme-linked immunosorbent assays for the serology of Hantaanvirus infections. Journal Virology Methods 1989; 23(2): 195-203.
HAMIDON B. B., SAADIAH S. Seoul Hantavirus infection mimicking dengue fever.Medical Journal Malaysia 2003; 58 (5): 786-787.
HEISKE A., ANHEIER B., PILASKI J., KLENK H. D., GRONE H. J., FELDMANN H.Polymerase chain reaction detection Puumala virus RNA in formaldehyde-fixed biopsymaterial. Kidney Institute 1999; 55(5): 2062-2069.
HJELLE B., TORREZ-MARTINEZ N., KOSTER F. T. Hantavirus pulmonary syndrome-related virus from Bolivia. Lancet 1996; 34(8993): 57.
HUJAKKA H., KOISTINEN V., EERIKAINEN P., KURONEN I., MONONEN I.,PARVIAINEN M., LUNDKVIST A., VAHERI A., NARVANEN A., VAPALAHTI O. NewImmunochromatographic rapid test for diagnosis of acute Puumala virus infection. JournalClinical Microbiology 2001; 39(6):2146-2150.
HUJAKKA H., KOISTINEN V., KURONEN I., EERIKAINEN P., PARVIAINEN M.,LUNDKVIST A., VAHERI A., VAPALAHTI O., NARVANEN A. Diagnostic rapid test foracute hantavirus infections: specific tests for Hantaan, Dobrava and Puumala virus versus ahantavirus combination test. Journal of Virology Methods 2003; 108:117-122.

80
HUTCHINSON K. L., PETERS C. J., NICHOL S. T. Sin nombre virus mRNA synthesis.Virology 1996; 224(1):139-149.
IKONOMOU L., SCHNEIDER Y. L., AGATHOS S. N. Insect cell culture for industrialproduction of recombinant proteins. Application Microbiol Biotechnology 2003; 62:1-20.
JEOR S. C. Three-week incubation period for hantavirus infection. Pediatric Infect Dis J.2004; 23(10):974-975.
JOHNSON A. M., SOUZA L. T., FERREIRA I. B., PEREIRA L. E., KSIAZEK T. G.,ROLLIN P. E., PETERS C. J., NICHOL S. T. Genetic investigation of a novel hantavirusescausing fatal HPS in Brazil. Journal Medical Virology 1999; 59(4): 527-535.
JONSSON B. C., GALLEGOS J., FERO P., SEVERSON W., XU X., SCHMALJOHN S. C.Purification and characterization of the Sin Nombre virus nucleocapsid protein expressed inEscherichia coli. Protein expression and purification 2001; 23:134-141.
KALLIO-KOKKO H., LEVEELAHTI R., BRUMMER-KORVENKONTIO M.,LUNDKVIST A., VAHERI A., VAPAHTI O. Human immune response to Puumala virusglycoproteins and nucleocapsid protein expressed in mammalian cells. Journal of MedicalVirology 2001; 65: 605-613.
KALLIO-KOKKO H., LUNDKVIST A., PLYUSNIN A., AVSIC-ZUPANC T., VAHERI A.,VAPALAHTI O. Antigenic properties and diagnostic potential of recombinant Dobrava virusnucleocapsid protein. Journal of Medical Virology 2000; 61:206-274.
KALLIO-KOKKO H., VAPAHTI O., HEDMAN K., BRUMMER-KORVENKONTIO M.,VAHERI A. Puumala virus antibody and immunoglobulin G avidity assays based on arecombinant nucleocapsid antigen. Journal Clinical Microbiology 1993; 31(3):677-680.
KALLIO-KOKKO H., VAPALAHTI O., LUNDKVIST A., VAHERI A. Evaluation ofPuumala virus IgG and IgM enzyme immunoassays based en recombinant baculovirus-expressed nucleocapsid protein for early nephropathia epidemica diagnosis. Clinical andDiagnostic Virology 1998;10:83-90.
KANERVA M., MUSTONEN J., VAHERI A. Patogenesis of Puumala and other hantavirusinfections. Reviews in medical virology 1998;8:67-98.

81
KARIWA H., TANABE H., MIZUTANI T., KON Y., LOKUGANAGE K.,LOKUGAMAGE N., IWASA M. A., HAGIYA T., ARAKI K., YOSHIMATSU K.,ARIKAWA J., TAKASHIMA I. Synthesis of Seoul virus RNA and structural proteins incultured cells. Archives Virology 2003; 148(9): 1671-1685.
KATAGIRI Y., INGHAM C. K. Enhanced production of Green Fluorescent Fusion Proteinsin a Baculovirus expression system by addition of secretion signal. BioTechniques 2002;33:24-26.
KHAIBOULLINA S. F., NETSKI D. M., KRUMPE P., ST JEOR S. C. Effects of tumornecrosis factor alpha on sin nombre virus infection in vitro. Journal Virology 2000; 74(24):11966-11971.
KING L. A., POSSEE R. D. The Baculovirus expression system: A laboratory guide.Chapman & Hall. First edition. USA1992.
KORAKA P., AVSIC-ZUPANC T., OSTERHAUS A. D., GROEN J. Evaluation of twocommercially available immunoassays for the detection of hantavirus antibodies in serumsamples. Journal Clinical Virology 2000; 17(3): 189-196.
KOST T. A., CONDREAY J. P., JARVIS D. L. Baculovirus as versatile vectors for proteinexpression in insect and mammalian cells. Nature Biotechnology 2005; 23(5):567-575.
KSIAZEK T. G., PETERS C. J., ROLLIN P. E., ZAKI S., NICHOL S., SPIROPOULOU C.,MORZUNOV S., FELDMANN H., SANCHEZ A., KHAN A. S. Identification of a newNorth American hantavirus that causes acute pulmonary insufficiency. American JournalTropical Medicine and Hygiene 1995; 52(2): 117-123.
LEDNICKY J. A. Hantaviruses a short review. Archives Pathology Laboratory Medicine2003; 127(1): 30-35.
LEE H. W. Hemorrhagic fever with renal syndrome in Korea. Reviews of infectious diseases1989; 11(4): 864-876.
LEE H. W., LEE P. W., LAHDEVIRTA J., BRUMMER-KORVENTKONIO M. Aetiologicalrelation between Korean haemorrhgaic fever and nephropathia epidemica. Lancet 1979;1(8109): 186-187.
LI G., PAN L., MOU D., CHEN Y. Characterization of truncated hantavirus nucleocapsidprotein and their application for serotyping. J. of Med. Virology 2006; 78: 926-932.

82
LI Z., BAI X., BIAN H. Serologic Diagnosis of Hantaan vírus infection based on a prptideantigen. Clinical Chemistry 2002; 48(4):645-647.
LUNA E. J. A., ELKHOURY M. R. Desafios para a vigilancia e controle da síndromecardiopulmonar por hantavirus (SCPH). Boletim Eletrônico Sociedade Brasileira deMedicina Tropical. N.1. Fevereiro 2005. Disponível em:www.sbmt.org/boletin/1/1desafios.pdf Retirado em: 21 de setembro de 2006.
LUNDKVIST A., HORLING J., NIKLASSON B. The humoral response to Puumala virusinfection (NE)investigated viral protein specific imunoassays. Archives Virology1993;130(1-2):121-130.
LUNDKVIST A., NIKLASSON B. Haemorrhagic fever with renal syndrome and otherhantavirus infections. Reviews Medical Virology 1994;4:177-184.
MAES P., CLEMENT J., GAVRILOVSKAYA I., VAN RANST M. Review: Hantavirus:Immunology, treatment and Prevention. Viral Immunology 2004;17(4):481:497.
MAKELA S., MUSTONEN J., LA-HOUHALA I., HURME M., KOIVISTO A. M.,VAHERI A., PASTERNACK A. Urinary excretion of interleuukin-6 correlates withproteinúria in acute Puumala hantavirus-induced nephritis. American. Journal KidneyDisease 2004; 43(5): 809-816.
MONROE M. C., MORZUNOV S. P., JOHNSON A. M., BOWEN M. D., ARTSOB H.,YATES T., PETERS C. J., ROLLIN P. E., KSIAZEK T. G., NICHOL S. T. Genetic diversityand distribution of Peromyscus-borne hantaviruses in North American. Emergency InfectionDisease 1999; 5(1): 75-86
MORELI M. L., SOUZA R. L. M., FIGUEIREDO L. T. M. Detection of Brazilian hantavirusby reverse transcription polymnerase chain reaction amplification os N gene in patients withhantavirus cardiopulmonary syndrome. Memórias Instituto Oswaldo Cruz 2004; 99(6):633-638.
MORELLI L. M. Diagnóstico de Hantavírus por detecção genômica com estudo filogenéticoe produção de uma proteína N recombinante. Tese Apresentada para Título de Doutor.Faculdade de Medicina de Ribeirão Preto. Universidade de São Paulo. 2005. p.145.
MORII M., YOSHIMATSU K., ARIKAWA J., ZHOU G., KARIWA H., TAKASHIMA I.Antigenic characterization of Hantaan and Seoul virus nucleocapsid proteins expressed by

83
recombinant baculovirus: application of a truncated protein, lacking an antigenic regioncommom to the two viruses, as a serotyping antigenic. Journal Clinical Microbiology 1998;36(9):2514-2521.
MOSCARDI F. Utilização de vírus entomopatogênicos em campo. IN: ALVES S. B. (Ed).Controle microbiano de inseto. Piracicaba: FEALQ, 1998: 509-539.
MURPHY C. I., MCINTIRE J. R., DAVIS D. R., HODGDON H., SEALS J. R., YOUNG E.Enhanced expression, secretion, and large-scale purification of recombinant HIV-1 gp 120 ininsect cells using the baculovirus egt and p67 signal peptides. Protein expression andpurification 1993; 4:349-357.
NICHOL S. T., SPIROPOULOU C. F., MORZUNOV S., ROLLIN P. E., KSIASKEK T. G.,FELDMANN H. Genetic indentification of Hantavirus associated with an outbreak of acuterespiratory illness. Science 1993;262:914-917.
OERS M. M., THOMAS A. A. M., MOORMANN R. J. M., VLAK J. M. Secretory pathwaylimits the enhanced expression of classical swine fever virus E2 glycoprotein in insect cells.Journal of Biotechnology 2001; 86:31-38
O’REILLY D. R., MILLER L. K., LUCKOV V. A. Baculovirus expression vectors. Alaboratory Manual. Cold Spring Harbor, Cold Spring Harbor laboratory, 1992.
PADULA P. J., COLAVECCHIA S. B., MARTINEZ V. P., DELLA VALLE M. O. G.,EDELSTEIN A., MIGUEL S. D. L., RUSSI J., RIQUELME J. M., COLUCCI N.,ALMIRÓN M., RABINOVICH R. D. Genetic diversity, distribution, and serological featuresof hantavirus infection in five countries in south america. Journal Clinical Microbiology2000A; 38(8):3029-3035.
PADULA P. J., ROSSI S., DELLA VALLE M. O., MARTINEZ P. V., COLAVECCHIA S.B., EDELSTEIN A., MIGUEL S. D., RABINOVICH R. D., SEGURA E. L. Developmentand evaluation of a solid-phase enzyme immunoassays based on Andes hantavirusrecombinant nucleoprotein. Journal Medical Microbiology 2000B; 49:149-155.
PETERS C. J., HPS in the Americas. In: CCHELD W. M., CRAIG W. A., HUGHES J. M.Emergin Infectious 2. Washington DC, ASM Press; 1998, 17-64.
PHARMINGEN. Baculovirus: Expression Vector System. Instruction Manual. Sextaedição. Pharmingen. 1999.

84
PHILLIPS B., ROTMANN D., WICKI M., MAYR L. M., FORSTNER M. Time reductionand process optimization of the baculovirus expression system for more efficient recombinantprotein production in insect cells. Protein expression and purification 2005; 42:211-218.
PINNA D. M., MARTINEZ V. P., BELLOMO C. M., LOPEZ C., PADULA P. Newepidemiologic and molecular evidence of person to person transmission of hantavirus AndesSout. Medicina (Buenos Aires) 2004; 64(1): 43-46.
PLYUSNIN A. Genetics of hantaviruses: implications to taxonomy. Archives Virology 2002;147:665-682.
PLYUSNIN A., VAPALAHTI O., VAHERI A. Hantaviruses: genome structure, expressionand evolution. Journal of General Virology 1996; 77:2677-2687.
RAYMOND T., GORBUNOVA E., GAVRILOVSKAYA I. N., MACKOW E. R. Pathogenichantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent(alpha) v (beta) 3 integrin conformers. Process National Academy Science USA 2005;102(4): 1163-1168.
RAZANSKIENE A., SCHMIDT J., GELDMACHER A., RITZI A., NIEDRIG M.,LUNDKVIST A., KRÜGER H. D., MEISEL H., SASNAUSKAS K., ULRICH R. Highyields of stable and highly pure nucleocapsid proteins os different hantavirus can be generatedin the yeast Saccharomyces cerevisiae. Journal of Biotechnology 2004; 11:319-333.
RIDEL G. M., LUIS I. R., TEJA J. Emerging and reemerging diseases: a health problem inthe Américas. Revista Panamericana de Salud Pública 2004; 15(4): 285-287. ROSA E. S., MILLS J. N., PADULA P. J., ELKHOURI M. R., KSIAZEK T. G., MENDESW. S., SANTOS E. D., ARAUJO G. C., MARTINEZ V. P., ROSA J. F., EDELSTEIN A.,VASCONCELOS P. F. Vector Borne Zoonotic Disease 2005; 5(1): 11-19.
SANGER, D. L.; NICKLEN, S.; COULSON, A. R. DNA sequencing with chain terminatinginhibitors. Proceedings National Academy Science. 1977; 74:5463-5467.
SANTOS E. D., GARRETT D. O. Epidemiologia e Serviços de saúde. 2005;14(1):1
SCHAFER F., ROMER U., EMMERLICH M., LUBENOW H., STEINERT K. Automatedhigh-throughput purification of 6xHIs-Tagged proteins. Journal of BiomolecularTechniques 2002; 13:131-142.

85
SCHAFER F., SCHAFER A., STEINERT K. A highly specific system for efficientenzymatic removal of tags from recombinant proteins. Journal Biomolecular Technology.2002; 13: 158-160.
SCHMIDT J., JANDRIG B., KLEMPA B., YOSHIMATSU K., ARIKAWA J., MEISEL H.,NIEDRIG M., PITRA C., KRÜGER H. D., ULRICH R. Nucleocapsid protein of cell culture-adapted Seoul virus strain 80-39: Analysis of its encoding sequence, expression in yeast andimmuno-reactivity. Virus Genes 2005A; 30(1):37-48.
SCHMIDT J., MEISEL H., HJELLE B., KRUGER D. H., ULRICH R. Development andevaluation of serological assays for detection of human hantavirus infections caused by SinNombre virus. Journal of Clinical Virology 2005B; 33:247-253.
SETTERGREN B., ALHM C., JUTO P., NIKLASSON B. Specific Puumala IgG virus half acentury after haemorrhagic fever with renal syndrome. Lancet. 1991 Jul.6;338(8758):66.
SILVA-VERGARA L. M., COSTA J. J. C., BARATA H. C., CURI M. G. V., TIVERON J.G. C., TEIXEIRA C. A. Hantavirus pulmonary syndrome in Uberaba, Minas Gerais, Brazil.Memórias Instituto Oswaldo Cruz 2002;97(6):783-787.
SIMMONS J. H., RILEY L. K. Hantaviruses: an overview. Comparative medicine. 2002;52(2): 97-110.
SIMPSON S. Q. Hantavirus pulmonary syndrome. Heart Lung 1998; 27(1): 51-57.
SIROLA H., KALLIO-KOKKO R. E., KOISTINEN V., KURONEN I., LUNDKVIST A.,VAHERI A., VAPALAHTI O., HENTTONEN H., NARVANEN A. Rapid field test fordetection of hantavirus antibodies in roedents. Epidemiology Infection 2004; 132:549-553.
SJOLANDER B. K., ELGH F., KALLIO-KOKKO H., VAPALAHTI O., HAGGLUND M.,PALMCRANTZ V., JUTO P., VAHERI A., NIKKLASSON B., LUNDKVIST A. Evaluationof serological Methods for diagnosis of Puumla hantavirus infection (NE). Journal ClinicalMicrobiology 1997 Dec:3264-3268.
SOARES P. B., DEMETRIO C., SANFILIPPO L., KAWANOTO A. H., BRENTANO L.,DURIGON E. L. Standardization of a duplex RT_PCR for the detection of Inflenza A andNewCastle disease virus im migratory birds. Journal Virology Methods 2005; 123(2): 125-130.

86
SUNDSTROM J. B., McMULLAN L.K., SPIROPOULOU C. F., HOOPER W. C., ANSARIA. A., PETERS C. J., ROLLIN P. E. Hantavirus infection induces the espression of RANTESand IP-10 without causing increased permeability in human lung microvascular endothelialcells. Journal Virology 2001; 75(13): 6070-6085.
SUZUKI A., BISORDI I., LEVIS S., GARCIA J., PEREIRA L. E., SOUZA R. P.,SUGAHARA T. K. N., PINI N., ENRIA D., SOUZA LTM. Identifying Rodent HantavirusReservoirs, Brazil. Emerging Infectious Diseases 2004 10(12):43-50.
TAKAKURA A., GOTO K., ITOH T., YOSHIMATSU K., TAKASHIMA I., ARIKAWA J.Establishment of an Enzyme-linked immunosorbent assay for detection of HantavirusAntibody of rats using a recombinant of nucleocapsid protein expressed in Escherichia coli.Experience Animal 2003;52(1):25-30.
TANADA Y., KAYA H. Y. Insect pathology. New York: Academic, 1993. 666p.
TEMONEM M., VAPALAHTI O., HOLTHOFER H., BRUMMER-KORVENKONTIO M.,VAHERI A., LANKINEN H. Susceptibility of human cells to Puumala virus infection.Journal General Virology 1993; 74(1): 515-518.
TISCHLER N. D., GALENO H., ROSEMBLATT M., VALENZUELA P. D. T. Human androdent humoral immune responses to Andes vírus structural proteins. Virology 2005;334:319-326.
VAN REGENMORTEL M. H. V. Introduction to the species concept in virus taxonomy. In:VAN REGENMORTEL M. H. V., FAUQUET C. M., BISHOP D. H. L., CARSTEN E. B.,ESTES M. K., LEMON S. M., MANILOFF J., MAYO M. A., MCGEOCH D. J., PRINGLEC. R., WICKNER R. B. (eds) Virus Taxonomy. VIIth report of the internationalcommittee on taxonomy of viruses. Academic Press. San Diego 2000: 3-16.
VAPALAHTI O., LUNDKVIST A., KALLIO-KOKKO H., PAUKKU K., JULKUNEN I.,LANKINEN H., VAHERI A. Antigenic properties and diagnostic potential of Puumala virusnucleocapsid protein expressed in insect cells. Journal Clinical Microbiology 1996 Jan;34(1):119-125.
VASCONCELOS M. I., LIMA V. P., IVERSSON L. B., ROSA M. D., da ROSA A. P., daROSA E. S., PEREIRA L. E., NASSAR E., KATZ G., MATIDA L. H., ZAPAROLI M. A.,FERREIRA J. J., PETERS C. J. Hantavirus pulmonary syndrome in the rural área ofJuquitiba, São Paulo metropolitan área, Brazil. Revista Instituto Medicina Tropical SãoPaulo. 1997; 39(4): 237-238.

87
VERITY R., PRASAD E., GRIMSRUD K., ARTSOB H., DREBOT M., MIEDZINSKI L.,PREIKSAITIS J. Hantavirus Pulmonary Syndrome in Northern Alberta, Canada: Clinical andLaboratory Findings for 19 cases. Clinical Infecctious Diseases 2000; 31:942-946.
VERITY R., PRASAD E., GRIMSRUD K., ARTSOB H., DREBOT M., MIEDZINSKI L.,PREIKSAITIS J. Hantavirus Pulmonary Syndrome in Northern Alberta, Canada: Clinical andLaboratory Findings for 19 cases. Clinical Infecctious Diseases. 2000; 31:942-946.
WANG X., OOI B. G., MILLER L. K. Baculovirus vectors for multiple gene expression andfor occluded virus production. Gene. 1991;100:131-137.
XU X., SEVERSON W., VILLEGAS N., SCHMALJOHN C. S., JONSSON C. B. The RNAbinding domain of the Hantaan virus N protein maps to a central, conserved region. J. ofVirology 2002; 76(7):3301-3308.
YANAGIHARA R., CHIN C. T., WEISS M. B., GAJDUSEK D. C., DIWAN A. R.,POLAND J. B., KLEEMAN K. T., WILFERT C. M., MEIKLEJOHN G., GLEZEN W. P.Serological evidence of Haantan virus infection in the United States. American JournalTropical Medicine and Hygiene 1985; 34(2): 396-399.
YOSHIMATSU K., ARIKAWA J., Li H., KARIWA H., HASHIMOTO N. SUZUKI N.Western blotting using recombinant Haantan virus nucleocapsid protein expressed insilkworm as a serological confirmation of hantavirus infection in human sera. JournalVeterinary Medicine Science 1996; 58(1): 71-74.
YOUNG J. C., HANSEN G. R., GRAVES T. K., DEASY M. P., HUMPHREY J. G., FRITZC. L., GORHAM K. L., KHAN A. S., KSIAZEK T. G., METZGER K. B., PETERS C. J.The incubation period of hantavirus pulmonary syndrome. American Journal TropicalMedicine and Hygiene 2000; 62(6): 714-717.

88
ANEXOS

89
ANEXO 1 - Reação de PCR.
Para a realização das reações de PCR, contidas neste trabalho foram utilizados:1. Enzima Taq Polimerase (Biotools 1U/uL)2. DNTPs (25 mM)3. DNA amostra4. Oligonucleotideos específicos (10pmol)5. Reaction Buffer 10X com 2 mM de MgCl2
Foi utilizado o seguinte programa de PCR:
1. 94ºC por 5minutos.2. 94ºC por 20 segundos.3. 50ºC por 20 segundos.4. 72ºC por 1 minuto5. Repetir 30 vezes a partir da fase 2.6. 72ºC por 30 segundos.7. Manter 4ºC.
ANEXO 2 Kit de Purificação GFX
Procedeu-se ao seguinte protocolo, seguindo instruções do fabricante:1. Pesar o microtubo vazio2. Cortar o fragmento do gel, colocar no tubo e pesar (descontar o peso do tubo vazio)3. Para cada 10 mg der gel adicionar 10 µl de capture Buffer4. Agitar e deixar no banho-maria a 65°C por 15 minutos ou até dissolver o gel5. Dar um spin6. Colocar uma coluna GFX para cada tubo coletor7. Transferir o conteúdo dos tubos na coluna8. Esperar 1 minuto em temperatura ambiente9. Centrifugar por 1minuto a 13000 rpm10. Descartar o conteúdo do tubo coletor e colocar a coluna de volta neste mesmo tubo11. Adicionar 500 µl de Wash Buffer12.Centrifugar por 1 minuto em temperatura ambiente.13.Descartar o conteúdo do tubo coletor e colocar a coluna de volta neste mesmo tubo14.Adicionar 500 µl de Wash Buffer15.Centrifugar por 1 minuto em temperatura ambiente.16.Descartar o conteúdo do tubo coletor e colocar a coluna de volta neste mesmo tubo17.Centrifugar por 1 minuto em temperatura ambiente a coluna vazia.18.Eluir com 30 a 50 ul de Água MilliQ.

90
ANEXO 3 Clonagem de fragmentos em plasmídeos
Para a realização dasclonagens, contidas neste trabalho foram utilizados:1. Enzima T4 DNA Ligase (Invitrogen - 1U/ul)2. DNA Ligase Buffer (5X)3. Plasmídeo Linearizado ou vetores de transferência.4. DNA
Deixou-se a 16ºC overnight.
ANEXO 4 - Transformação
Procedeu-se ao seguinte protocolo para realização das transformações:
1. Inocular 7,5 µl de cada amostra nas células competentes2. Deixar 30 minutos no gelo3. Colocar no banho-maria por 1’:45” a 47 °C4. Acrescentar 250 µl de meio líquido LB5. Incubar as amostras no shaker por 1 hora a 37° C a 250 rpm.6. Colocar 75 µl de cada amostra nas placas com meio LB completo (IPTG, XGAL e
AMP)7. As placas devem ser levadas para o shaker overnight a 37° C
ANEXO 5 Extração Kit Qiagen
Este protocolo é projetado para purificação de até 20ug de DNA plasmídial de altareprodução.
1. Resuspender o pellet bacteriano em 250 ul de Buffer P1.2. Adicionar 250 ul de Buffer P23. Adicionar 350 ul de Buffer N34. Centrifugar por 10 minutos a 14000 rpm.5. Colocar sobrenadante na coluna QIAprep6. Centrifugar por 30 a 60 segundos.7. Enxaguar com 750 ul de Buffer PE8. Centrifugar por 30 a 60 segundos.9. Descartar o liquido e centrifugar por mais 1 minuto.10. Eluir em 30 a 50 uL de Água MilliQ.

91
ANEXO 6 Extração de DNA de Budded Vírus
Este protocolo baseia-se no descrito por O’Reilly 1992.1. Retirar o meio da placa de cultura2. Centrifugar em baixa rotação (1000 a 2000 rpm) por 5 minutos.3. Retirar o sobrenadante e coloca-lo em eppendorfs. (eppendorf de 1,5 mL)4. Centrifugar a 20.000x g por 30 minutos a 4ºC.5. Descartar o sobrenadante e ressuspender o pellet em 100 uLde disruption buffer. (cada
eppendorf).6. Juntar todo o volume dos eppendorfs em um novo eppendorf de 1,5 mL.7. Adicionar Proteinase K para ter uma concentração de 500 ug/mL8. Colocar no banho-maria a 55ºC overnight9. Fazer precipitação com 500 ul de Fenol por 2x10. Fazer a precipitação com 500 ul de Clorofórmio por 2x.11. Juntar todo o volume em um falcon de 15 ml e completar com água milli Q.12. Passar pela coluna Centricon ( ) todo o volume13. Lavar 2 a 4 vezes com água milli Q.14. Obter o volume preso na coluna.
ANEXO 7 Extração de DNA de poliedros virais
Este protocolo baseia-se no descrito por O’Reilly 1992.1. Spin nas células com poliedros por 10 min a 4000 rpm2. Adicionar 1 mL de solução protease/SDS feira na hora (500 microgramas/ml e SDS
(0,1% final)3. Incubar a 55 C por 2 horas com agitação4. Spin na microcentrífuga de mesa ( 10 K RPM, 10 minutos)5. Descartar sobrenadante6. Resuspender precipitado em 0,5 mL de 0,2 M NaCO27. Deixar ON com agitação leve a temperatura ambiente8. centrifugar a 10 000 RPM por 10 min.;9. guardar sobrenadante (A)10. resuspender precipitado em 0,25 mL de 0,2 M NaCO211. agitar por 30 min. a 37 C12. Centrifugar em microcentrífuga de mesa (10 K RPM), 10 min.;13. -remover sobrenadante e adicionar a amostra "A"14. neutralizar sobrenadante com 1/4 Vol de 1 M Tris HCl pH 7.015. guardar material na geladeira ent re 2 e 4 horas16. Centrifugar a 20.000 x g por 1 hora17. descartar o precipitado18. Para cada 500 µl de sobrenadante adicionar: 10 µl de SDS (10%) e 10 µl de protease19. Incubar a 55ºC overnight20. Extração fenol/ clorofórmio21. Concentrar em Centricon com grande volume de água.

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo