renata de uzÊda vital avaliaÇÃo da atividade de ... vital.pdf · figura 4.6 – taxa de consumo...
TRANSCRIPT

UNIVERSIDADE FEDERAL FLUMINENSE
ESCOLA DE ENGENHARIA
DEPARTAMENTO DE ENGENHARIA QUÍMICA E DE PETRÓLEO
GRADUAÇÃO EM ENGENHARIA QUÍMICA
RENATA DE UZÊDA VITAL
AVALIAÇÃO DA ATIVIDADE DE CATALISADORES DE PRATA SOBRE
DIFERENTES SUPORTES NA REAÇÃO DE EPOXIDAÇÃO DO ETILENO
NITERÓI
2016

RENATA DE UZÊDA VITAL
Trabalho de conclusão de curso apresentado à
Coordenação do Curso de Engenharia Química
da Universidade Federal Fluminense, como
requisito parcial para a obtenção do Grau de
Bacharel em Engenheira Química.
Orientadora: Profª. Drª. RITA DE CÁSSIA COLMAN SIMÕES
NITERÓI
2016
“AVALIAÇÃO DA ATIVIDADE DE CATALISADORES DE PRATA SOBRE
DIFERENTES SUPORTES NA REAÇÃO DE EPOXIDAÇÃO DO ETILENO”



iv
AGRADECIMENTOS
Aos meus familiares, por apoiarem minhas escolhas e me incentivarem durante todo o
percurso dessa segunda graduação.
A minha mãe Sandra, por ser minha grande amiga e torcedora.
Ao meu namorado André, por todo seu amor e conselhos.
A Doutora Rita de Cássia Colman pela confiança no meu trabalho e por toda sua
orientação. Serei eternamente grata.
A toda equipe de profissionais, colegas e amigos do Laboratório de Catálise do INT,
com quem tive imenso prazer em trabalhar. Pelos momentos divertidos que tivemos em 2014,
permanecem como grandes memórias para realização de mais esse trabalho.
Ao Prof. Dr. Ricardo Cassela e toda equipe do LESPA, na Universidade Federal
Fluminense, pelas análises de espectrometria de absorção atômica.
A todos os meus amigos pelo apoio e compreensão da minha ausência em momentos
importantes. E, claro, pelos ótimos momentos de diversão e risadas.
Uma dedicação especial aos amigos da Shell, presentes diariamente ou semanalmente,
foram muito importantes para meu crescimento profissional e pessoal. Obrigada por todo
conhecimento que me transmitiram.
A banca examinadora por aceitar o convite.

v
SUMÁRIO
LISTA DE FIGURAS .............................................................................................................. vii
LISTA DE TABELAS ............................................................................................................ viii
LISTA DE ESQUEMAS ........................................................................................................... ix
LISTA DE ABREVIATURAS ................................................................................................... x
RESUMO .................................................................................................................................. xi
ABSTRACT ............................................................................................................................. xii
Capítulo 1 – INTRODUÇÃO ..................................................................................................... 1
Capítulo 2 – REVISÃO BIBLIOGRÁFICA .............................................................................. 4
2.1 – Mecanismos de Reação .................................................................................................. 4
2.1.1 – Mecanismo Eletrofílico-Nucleofílico ...................................................................... 4
2.1.2 – Mecanismo de Modificação com Oxigênio Subsuperficial .................................... 6
2.1.3 – Mecanismo do OMC ............................................................................................... 7
2.2 – Efeito da Natureza do Metal ........................................................................................ 12
2.3 – Efeito do Tipo de Suporte na Reação de Epoxidação .................................................. 15
Capítulo 3 – MATERIAIS E MÉTODOS ................................................................................ 21
3.1 – REAGENTES E EQUIPAMENTOS ........................................................................... 21
3.1.1 – Reagentes para a preparação dos suportes e catalisadores .................................... 21
3.1.2 – Gases para a caracterização e teste dos catalisadores ........................................... 21
3.2 – SÍNTESE DOS SUPORTES ....................................................................................... 21
3.2.1 – MgO ...................................................................................................................... 22
3.2.2 – ZrO2 ....................................................................................................................... 22
3.2.3 – α-Al2O3 .................................................................................................................. 22
3.3 – PREPARAÇÃO DOS CATALISADORES ................................................................ 23
3.4 – CARACTERIZAÇÃO DOS SUPORTES E CATALISADORES .............................. 24
3.4.1 – Espectroscopia de Absorção Atômica (AAS) ....................................................... 24

vi
3.4.2 – Análise Textural (BET e BJH) .............................................................................. 24
3.4.3 – Difratometria de Raios X (DRX) .......................................................................... 24
3.4.4 – Dessorção de Oxigênio à Temperatura Programada (TPD de O2) ........................ 25
3.5 – TESTES CATALÍTICOS ............................................................................................ 27
Capítulo 4 – RESULTADOS E DISCUSSÃO ......................................................................... 29
4.1 – CARACTERIZAÇÃO DOS CATALISADORES ...................................................... 29
4.1.1 - Espectroscopia de Absorção Atômica (AAS) ........................................................ 29
4.1.2 – Análise Textural .................................................................................................... 29
4.1.3 – Difratometria de Raios X (DRX) .......................................................................... 32
4.1.4 – Dessorção de Oxigênio a Temperatura Programada (TPD de O2) ........................ 34
4.2 – TESTES CATALÍTICOS ............................................................................................ 37
Capítulo 5 – CONCLUSÃO ..................................................................................................... 44
Capítulo 6 – REFERÊNCIAS BIBLIOGRÁFICAS ................................................................ 45
APÊNDICE A .......................................................................................................................... 50
APÊNDICE B ........................................................................................................................... 51
APÊNDICE C ........................................................................................................................... 55

vii
LISTA DE FIGURAS
Figura 2.1 – Energias de ativação para a rota direta na superfície Ag2O(001) e para o
mecanismo OMC na Ag(111)................................................................................................... 11
Figura 2.2 – Mecanismo da epoxidação do etileno sobre a prata mássica, sendo oxigênio
eletrofílico (Oel) e nucleofílico (On) ......................................................................................... 12
Figura 2.3 – Epoxidação direta nas superfícies óxidos de Ag2O, Cu2O e Au2O. ..................... 14
Figura 3.1 – Rampa de temperatura para calcinação dos catalisadores de prata. ..................... 23
Figura 3.2 – Rampa de aquecimento do procedimento de TPD de O2 dos catalisadores de
prata. ......................................................................................................................................... 25
Figura 4.1 – Isotermas de adsorção e dessorção de nitrogênio dos catalisadores Ag/MgO e
Ag/ZrO2 e dos respectivos suportes.......................................................................................... 29
Figura 4.2 – Distribuição de tamanho de poros das amostras Ag/ZrO2 e ZrO2. ...................... 31
Figura 4.3 – Difratogramas dos suportes MgO, α-Al2O3 e ZrO2 .............................................. 32
Figura 4.4 – Difratogramas dos catalisadores Ag/MgO, Ag/α-Al2O3 e Ag/ZrO2 .................... 33
Figura 4.5 – Perfil de TPD de O2 dos catalisadores Ag/MgO, Ag/α-Al2O3 e Ag/ZrO2. .......... 34
Figura 4.6 – Taxa de consumo de etileno em função da temperatura de reação na avaliação do
efeito do suporte ....................................................................................................................... 39
Figura 4.7 – Seletividade ao óxido de etileno em função da temperatura de reação na
avaliação do efeito do suporte .................................................................................................. 40
Figura 4.8 – Rendimento do óxido de etileno em função da temperatura de reação na
avaliação do efeito do suporte. ................................................................................................. 40

viii
LISTA DE TABELAS
Tabela 2.1 – Efeito do suporte na oxidação do etileno. ............................................................ 16
Tabela 2.2 – Variações na desativação e seletividade ao C2H4O para catalisadores de prata em
diferentes suportes .................................................................................................................... 18
Tabela 2.3 – Características dos suportes e catalisadores, e suas atividades na epoxidação do
etileno a diferentes temperaturas. ............................................................................................. 20
Tabela 4.1 – Teor metálico de prata nos catalisadores, obtido por AAS. ................................ 29
Tabela 4.2 – Área específica (SBET), volume de poros (Vp) e diâmetro médio de poros (dp) dos
catalisadores e suportes utilizados. ........................................................................................... 31
Tabela 4.3 – Diâmetro de cristalito da prata (dAg,DRX) nos catalisadores, obtido por DRX. .... 34
Tabela 4.4 – Quantidade de oxigênio dessorvido dos perfis de TPD de O2 dos catalisadores. 36
Tabela 4.5 – Dispersão da prata (DAg), área ativa (Sativa) e tamanho de cristalito da prata
(dAg,TPD), obtidos por análises de TPD de O2. .......................................................................... 36
Tabela 4.6 – Comportamento catalítico dos catalisadores suportados de prata: taxa (r) de
consumo de etileno e de formação dos produtos e TOF........................................................... 37
Tabela 4.7 – Atividade e seletividade de catalisadores de Ag suportados em α-Al2O3 e SiO2 da
literatura. ................................................................................................................................... 38
Tabela 4.8 – TOF de formação de óxido de etileno listados a partir de avaliação da literatura
.................................................................................................................................................. 42

ix
LISTA DE ESQUEMAS
Esquema 1.1 – Mecanismo molecular proposto para a reação de formação de óxido de etileno
.................................................................................................................................................... 2
Esquema 2.1 – Esquema geral das reações de oxidação parcial e total proposto por Grant e
Lambert. ...................................................................................................................................... 5
Esquema 2.2 – Mecanismo da reação de oxidação total. ........................................................... 5
Esquema 2.3 – Mecanismo da reação de oxidação ao EO. ........................................................ 5
Esquema 2.4 – Mecanismo da reação de epoxidação proposto por Van Santen e Kuipers ....... 6
Esquema 2.5 – Mecanismo da reação de oxidação total proposto por Van Santen e Kuipers. .. 7
Esquema 2.6 – Esquema geral de reação proposto a partir do intermediário OMC................... 8
Esquema 2.7 – Mecanismo de representação da epoxidação do etileno através do
intermediário OMC. ................................................................................................................... 8
Esquema 2.8 – Geometrias apresentadas no mecanismo do OMC na superfície de Ag(111).. 10

x
LISTA DE ABREVIATURAS
AA – Acetaldeído
AAS – Espectroscopia de Absorção Atômica
BET – Brunauer-Emmet-Teller
DRX – Difração de Raios-X
EDC – Dicloreto de Etileno (Ethylene Dichloride)
EO – Óxido de Etileno
OMC – Complexo Oxametalaciclo (Oxametallacycle)
PET – Politereftalato de Etileno (Polyethyleneterephthalate)
TPD – Dessorção a Temperatura Programada (Temperature Programmed Dessorption)
TPR – Redução a Temperatura Programada (Temperature Programmed Reduction)
XPS – Espectroscopia Fotoeletrônica de Raios X (X-ray Photonelectron Spectroscopy)

xi
RESUMO
O óxido de etileno (EO) é um intermediário muito importante na fabricação de uma
série de produtos petroquímicos tais como etileno glicol, surfactantes, anticongelantes,
adesivos, lubrificantes e solventes. Atualmente, sua produção excede 20 milhões de ton/ano,
representando cerca de 40-50% do valor total de substâncias químicas orgânicas produzidas
mundialmente. Industrialmente, esse epóxido é obtido através da oxidação parcial do etileno
usando ar ou oxigênio sobre catalisadores de Ag suportados em α-Al2O3, um suporte de baixa
área superficial. Apesar desse processo ter sido desenvolvido por volta da década de 40, uma
questão prática importante que não tem sido sistematicamente investigada é o papel do
suporte na catálise do óxido de etileno. A singularidade da alumina como suporte na obtenção
de catalisadores altamente seletivos foi atribuída a sua baixa área superficial. No entanto,
existem outros suportes de área superficial igualmente baixa que resultaram em catalisadores
não seletivos ao EO. Algumas evidências na literatura indicam que os suportes podem ser
ativos na conversão do óxido de etileno a acetaldeído e a CO2. Além disso, a natureza do
suporte pode influenciar na distribuição dos tamanhos de partícula de prata e,
consequentemente, na atividade e seletividade ao óxido de etileno. Assim, o objetivo desse
trabalho é comparar o efeito do suporte no desempenho dos catalisadores de prata suportados
em α-Al2O3, ZrO2 e MgO. Os três materiais possuem área superficial bem distintas, além de
características neutra, ácida e básica, respectivamente. Os catalisadores foram caracterizados
por medidas de área específica, difração de raios X e dessorção a temperatura programada de
O2. O catalisador Ag/α-Al2O3 se mostrou o mais promissor na produção seletiva de óxido de
etileno dentre os estudados, apresentando rendimentos desse produto de até 18% e
seletividade de até 28%. O catalisador Ag/MgO também se mostrou seletivo ao EO em todas
as temperaturas de reação, porém com seletividades (até 18%) e rendimentos (até 14%)
inferiores ao catalisador de Ag/α-Al2O3. Já o catalisador Ag/ZrO2 demonstrou ser muito ativo,
promovendo somente a oxidação total do etileno a CO2 e H2O, observada também pelo
desprendimento de calor e consequente aumento da temperatura durante a reação. A
seletividade ao EO praticamente nula para esse catalisador foi atribuída à presença de sítios
ácidos no suporte que catalisam a reação de oxidação total, por correlação com a literatura.
Palavras-chave: Epoxidação do etileno, catalisadores suportados, prata, α-Al2O3 e MgO.

xii
ABSTRACT
Ethylene oxide (EO) is an important intermediate in the manufacture of a number of
petrochemical products such as ethylene glycol, surfactants, antifreeze, adhesives, lubricants
and solvents. Currently, its production exceeds 20 million tons/year, accounting for about 40
to 50% of the total amount of organic chemicals produced worldwide. Industrially, this
epoxide is obtained by partial oxidation of ethylene by using air or oxygen over Ag catalysts
supported on α-Al2O3, a support of low surface area. Although this process has been
developed around the 40's, an important practical question that hasn’t been systematically
investigated is the role of support in the catalysis of ethylene oxide. The uniqueness of
alumina as a support in obtaining highly selective catalysts was attributed to its low surface
area. However, there are also other low surface area supports which resulted in non-selective
catalysts to EO production. Some evidence in the literature indicates that supports can be
active in the conversion of ethylene oxide to acetaldehyde and CO2. Furthermore, the nature
of the support can influence the size distribution of silver particles and, hence, the activity and
selectivity to ethylene oxide. The objective of this study was to compare the effect of support
in the performance of the silver catalysts supported on α-Al2O3, ZrO2 and MgO. These three
materials have very different surface area when compared to each other, besides neutral, acid
and basic characteristics, respectively. The catalysts were characterized by measurements of
specific surface area, X-ray diffraction and temperature programmed desorption of O2.
Among catalysts studied, the Ag/α-Al2O3 was the most promising in selective production of
ethylene oxide, showing yields up to 18% and selectivity to ethylene oxide of up to 28%. The
catalyst Ag/MgO was also selective to EO in all reaction temperatures studied, however with
lower selectivity (up to 18%) and yield to EO (up to 14%). Furthermore, the catalyst Ag/ZrO2
proved to be too active, only promoting total oxidation of ethylene to CO2 and H2O, also
noted by the release of heat and consequent increasing in temperature during the reaction. The
practically zero selectivity shown for this catalyst was attributed to the presence of acid sites
on the support that catalyse the full oxidation reaction, when comparing with the literature.
Keywords: Ethylene epoxidation, supported catalysts, silver, α-Al2O3 e MgO.

1. INTRODUÇÃO 1
Capítulo 1 – INTRODUÇÃO
A reação de epoxidação do etileno é um tópico importante para ser estudado tanto pela
perpectiva acadêmica, como industrial. Academicamente, ela representa uma das reações de
oxidação parcial mais fundamentais, e é uma das reações catalíticas mais estudadas pela
ciência de superfície. A compreensão da epoxidação catalítica fornece informações valiosas
sobre como catalisadores heterogêneos permitem que uma molécula metaestável, tal como um
epóxido, seja sintetizado preferencialmente sobre o produto termodinamicamente mais
estável, o CO2 (GREINER, 2015). De um ponto de vista industrial, o epóxido de etileno é
uma matéria-prima importante para a produção de uma variedade de materiais e produtos
químicos valiosos. Atualmente, sua produção mundial excede 20 milhões de ton/ano,
colocando a síntese desse intermediário químico oxigenado na lista de substâncias químicas
mais produzidas em todo o mundo (MONNIER et al., 2015)
Aproximadamente 70% do óxido de etileno produzido no mundo é convertido a
etileno glicol, usado como anti-congelante para motores (VAN SANTEN e ÖZBEK, 2013). É
muito utilizado também na produção de tensoativos como, por exemplo, alquilfenóis
etoxilatos (APEs) não aniônicos e como matéria-prima na produção de fibra poliéster ou ainda
de poli(tereftalato de etileno), o PET. Além disso, uma série de importantes produtos do
petróleo e intermediários químicos são derivados do EO, sendo usado extensivamente em
aplicações como eletrônicos, lavagens/tingimentos, pesticidas, têxteis, produção de papel,
automóveis e refino do petróleo. Em 2012 sua utilização mundial foi distribuída da seguinte
forma: 25% no Oriente Médio, 16% na China e 12% nos Estados Unidos. O resto da Ásia é
responsável por 25% da capacidade mundial.
Em 1931, Theodore Lefort desenvolveu um catalisador à base de prata capaz de
promover a oxidação direta do etileno com ar ou oxigênio a óxido de etileno, que substituiu o
processo com glicol cloridrina previamente usado para esse fim (LEFORT, 1931). Desde
1937, quando a Union Carbide abriu sua primeira planta de EO, a oxidação parcial e seletiva
do etileno foi efetuada usando um catalisador à base de Ag conforme sugerido por Lefort
(MONNIER et al., 2015). O mecanismo da reação de oxidação seletiva do etileno geralmente
aceito é mostrado no Esquema 1.1. A reação de formação do acetaldeído age como um
intermediário instável, levando a combustão total. As reações de formação do EO (k1),
oxidação direta ao acetaldeído (k2) e sua oxidação total ao CO2 (k4) são catalisadas pela prata.

1. INTRODUÇÃO 2
Já a reação de isomerização do óxido de etileno a acetaldeído (k3) ocorre predominantemente
na superfície do suporte.
Esquema 1.1 – Mecanismo molecular proposto para a reação de formação de óxido de etileno.
A reação de formação do EO é moderamente exotérmica, enquanto que as reações de
oxidação total do etileno e do óxido de etileno são fortemente exotérmicas (equações 1-3)
(WESTERTERP e BORMAN, 1995).
C2H4 + 1/2 O2 → C2H4O (∆H = - 105 kJ/mol) (1)
C2H4 + 3 O2 → 2 CO2 + 2 H2O (∆H = - 1323 kJ/mol) (2)
C2H4O + 5/2 O2 → 2 CO2 + 2 H2O (∆H = -1218 kJ/mol) (3)
Apesar da termodinâmica favorecer fortemente a formação dos produtos da oxidação
total, a produção seletiva do óxido de etileno na superfície da prata sugere que a reação é
controlada cineticamente.
Nesse processo, a prata exibe propriedades catalíticas únicas na oxidação seletiva do
etileno ao EO preferencialmente à oxidação completa, podendo atingir seletividades
moderadas em catalisadores não promovidos. Os catalisadores de Ag tradicionalmente usados
são suportados em α-Al2O3 e podem apresentar uma seletividade de até 20% para partículas
de Ag com um diâmetro inferior a 30 nm. A seletividade pode ser melhorada aumentando-se
o diâmetro de partícula, atingindo aproximadamente 50% para um diâmetro médio de 1 µm
(VERYKIOS et al., 1980). Na década de 60, a descoberta de compostos promotores como sais
de césio e moderadores gasosos (p. ex. 1,2-diclorometano) levou a um aumento da
seletividade ao óxido de etileno em até cerca de 70%. Atualmente, o uso de catalisadores de
prata altamente promovidos permite uma seletividade de 90% (LINIC e CHRISTOPHER,
2008). Embora o processo de epoxidação direta seja relativamente eficiente, dada a grande
escala de produção de epóxido, mesmo uma melhoria de 1% na seletividade vale cerca de 200
milhões de dólares por ano para os produtores.

1. INTRODUÇÃO 3
Embora exista na literatura uma série de estudos sobre esse processo e o catalisador
empregado, existem alguns pontos que ainda não estão claramente defendidos como o papel
do suporte e a influência do tamanho de partícula no mecanismo dessa reação. A natureza do
suporte pode influenciar na distribuição dos tamanhos de partícula de prata e,
consequentemente, na atividade e seletividade ao óxido de etileno. Nesse sentido, a
singularidade da α-Al2O3 como suporte de catalisadores altamente seletivos ao óxido de
etileno foi atribuída justamente à sua baixa área superficial (< 1 m2/g). No entanto, esses
catalisadores possuem rendimentos relativamente baixos para óxido de etileno, como
resultado da baixa dispersão das partículas de prata sobre a α-Al2O3. Catalisadores de prata
suportados em SiO2 de alta área superficial (até 350 m2/g) foram reportados como sendo mais
ativos e tão seletivos na produção de óxido de etileno (30-70% de seletividade ao EO) quanto
os suportados em α-alumina de baixa área. Seletividades ao EO de 30-60%, similar à obtida
nos catalisadores comerciais, foram observadas nesses catalisadores suportados em SiO2 com
diâmetro de prata de 3-7 nm (HARRIOT, 1971). Partículas menores de prata (em torno de 2
nm) foram consideradas responsáveis por seletividades ao EO praticamente nulas (WU e
HARRIOT, 1975). Na década de 80, Verykios e colaboradores (1988) investigaram o efeito
de diferentes suportes na atividade e seletividade das reações de epoxidação e combustão do
etileno. Foi observado que os catalisadores de Ag suportados em SiO2, α-Al2O3, TiO2 (Rutilo),
ZrO2 e SiC apresentaram seletividades ao EO entre 19-65%. No entanto, para os catalisadores
suportados em γ-Al2O3, MgO, TiO2 (Anatase), Nb2O5, Y2O3 e V2O5 não houve formação de
EO. De acordo com os autores, estes resultados foram atribuídos aos diferentes graus de
dispersão da prata, já que a reação de epoxidação do etileno é conhecida por ser sensível ao
tamanho de cristalito da prata.
Nesse sentido, o objetivo desse trabalho é estudar o efeito do suporte no desempenho
dos catalisadores de prata suportados em α-Al2O3, MgO e ZrO2 na reação de epoxidação do
etileno em diferentes temperaturas (498-548 K). As propriedades dos materiais foram
investigadas por medida de área superficial específica, difratometria de raios X e dessorção de
O2 a temperatura programada.

2. REVISÃO BIBLIOGRÁFICA 4
Capítulo 2 – REVISÃO BIBLIOGRÁFICA
2.1 – Mecanismos de Reação
Na literatura há um grande número de estudos experimentais e teóricos focados nos
mecanismos de reação (GRANT e LAMBERT, 1985;VAN SANTEN e KUIPERS, 1987;
AVDEEV et al., 2000; VAN SANTEN et al., 2013), os quais são baseados principalmente em
sistemas modelo sob baixas pressões de oxigênio onde a natureza da prata é essencialmente
metálica. As primeiras investigações sistemáticas do mecanismo da epoxidação do etileno na
superfície da prata levaram em consideração, majoritariamente, qual a espécie de oxigênio
ativa nessa reação. Inicialmente, estudos cinéticos concluíram que a forma molecular seria a
mais ativa. No entanto, mais tarde foi observado que a espécie atômica era mais favorável à
oxidação total do etileno. Então, qual é a forma de oxigênio efetivamente ativa na epoxidação
do etileno? Dentre os inúmeros estudos propostos na literatura, no presente trabalho são
apresentados alguns dos mecanismos mais aceitos.
2.1.1 – Mecanismo Eletrofílico-Nucleofílico
A primeira investigação do mecanismo utilizando uma superfície de monocristais de
Ag(111) foi reportada por Grant e Lambert (1985). Nesse trabalho foi demonstrado que
apesar do oxigênio molecular estar presente nessa superfície, não reagiu com o etileno para
formação de epóxido de etileno, contrariando os estudos de mecanismos que precederam o de
Grant e Lambert (1985). Além disso, esse foi um dos primeiros experimentos a indicar que a
presença de oxigênio subsuperficial (dissolvido) é necessária para a adsorção de oxigênio
capaz de formar epóxido.
Segundo os autores, o oxigênio dissolvido torna a superfície ativa para a reação de
oxidação ao EO mas também aumenta a atividade para formação de CO2, sendo esse último
efeito associado à oxidação consecutiva do próprio óxido de etileno. Além disso, o oxigênio
subsuperficial pode participar diretamente na produção de CO2 (mas não na formação de
C2H4O), já que o rendimento dessa reação continua a aumentar mesmo após todo consumo do
oxigênio atômico O(a). No Esquema 2.1, é possível observar um esquema geral dessas
reações proposto no trabalho.
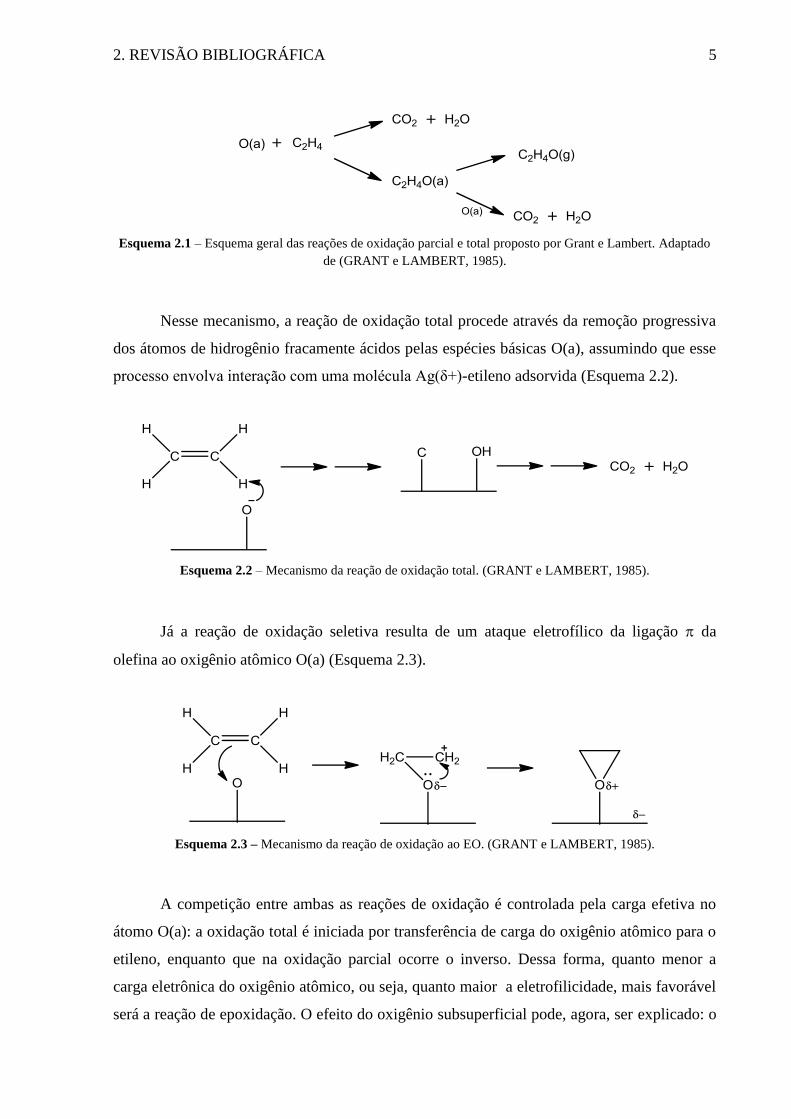
2. REVISÃO BIBLIOGRÁFICA 5
Esquema 2.1 – Esquema geral das reações de oxidação parcial e total proposto por Grant e Lambert. Adaptado
de (GRANT e LAMBERT, 1985).
Nesse mecanismo, a reação de oxidação total procede através da remoção progressiva
dos átomos de hidrogênio fracamente ácidos pelas espécies básicas O(a), assumindo que esse
processo envolva interação com uma molécula Ag(δ+)-etileno adsorvida (Esquema 2.2).
Esquema 2.2 – Mecanismo da reação de oxidação total. (GRANT e LAMBERT, 1985).
Já a reação de oxidação seletiva resulta de um ataque eletrofílico da ligação da
olefina ao oxigênio atômico O(a) (Esquema 2.3).
Esquema 2.3 – Mecanismo da reação de oxidação ao EO. (GRANT e LAMBERT, 1985).
A competição entre ambas as reações de oxidação é controlada pela carga efetiva no
átomo O(a): a oxidação total é iniciada por transferência de carga do oxigênio atômico para o
etileno, enquanto que na oxidação parcial ocorre o inverso. Dessa forma, quanto menor a
carga eletrônica do oxigênio atômico, ou seja, quanto maior a eletrofilicidade, mais favorável
será a reação de epoxidação. O efeito do oxigênio subsuperficial pode, agora, ser explicado: o

2. REVISÃO BIBLIOGRÁFICA 6
oxigênio mássico compete com o atômico na retirada de elétrons do metal. Dessa forma, a
carga negativa no átomo O(a) é reduzida fazendo com que a reação de epoxidação consiga
competir com a oxidação total.
2.1.2 – Mecanismo de Modificação com Oxigênio Subsuperficial
Van Santen e Kuipers (1987) propuseram um mecanismo baseado em novos dados da
adsorção de oxigênio em monocristais de prata e excluíram completamente o conceito da
participação do oxigênio molecular na reação de epoxidação do etileno. De acordo com os
autores, uma fase superficial do óxido AgO, na qual podem ser encontradas espécies Ag+, é
formada sob condições catalíticas. A presença dessa camada subsuperficial de átomos
eletronegativos é um pré-requisito para a criação de sítios onde átomos de oxigênio
eletrofílicos são adsorvidos. Esses cátions encontrados na superfície do AgO podem modelar
tais sítios. Em outras palavras, o oxigênio subsuperficial possui um efeito ativador sobre o
oxigênio nucleofílico em uma superfície de prata pura, tornando-o eletrofílico através da
remoção de elétrons.
De acordo com o Esquema 2.4, a reação de epoxidação ocorre a partir do oxigênio
eletrofílico, ligado fracamente ao átomo de prata de alta carga positiva. Um dos átomos de
carbono é atacado eletrofilicamente por esse átomo de oxigênio eletrofílico. Como resultado,
há transferência de carga do etileno para a superfície do AgO.
Esquema 2.4 – Mecanismo da reação de epoxidação proposto por Van Santen e Kuipers. Adaptado de (VAN
SANTEN e KUIPERS, 1987).
A etapa limitante da reação não-seletiva do etileno é a ruptura da ligação C-H.
Portanto, a reação de oxidação total (Esquema 2.5) é iniciada pela reação preferencial do

2. REVISÃO BIBLIOGRÁFICA 7
oxigênio ligado fortemente a íons prata de baixa carga (Ag+) com um dos átomos de
hidrogênio da molécula de etileno.
Esquema 2.5 – Mecanismo da reação de oxidação total proposto por Van Santen e Kuipers. Adaptado de (VAN
SANTEN e KUIPERS, 1987).
Apesar de simples, o conceito de formação do oxigênio eletrofílico através do
mecanismo se contradiz com dados espectroscópicos. Nesse sentido, a ligação Ag-Oδ-
-Ag
pode ser atribuída ao oxigênio atômico observado a 300 < T < 600 K, no entanto, a natureza
da ligação Ag=Oδ+
ainda não foi identificada como atômica. A forma fracamente ligada
dificilmente pode existir em temperaturas superiores a 600 K, presentes nos catalisadores
reais (AVDEEV et al., 2000; PONEC e BOND, 1995). Além disso, a possibilidade da
transformação de oxigênio nucleofílico em eletrofílico sobre a superfície de prata sem que
haja uma reconstrução superficial, mesmo na presença das espécies AgO modificadores, é um
pouco duvidosa (CARVALHO, 2005). Esse mecanismo foi o primeiro a ressaltar a influência
da presença do oxigênio subsuperficial no aumento da eletrofilicidade desta espécie de
oxigênio.
2.1.3 – Mecanismo do OMC
Inicialmente, acreditou-se que a reação de epoxidação do etileno seguisse um esquema
triangular, com ambos etileno e óxido de etileno contribuindo na formação de CO2 e H2O.
Porém, nos anos 2000, através de cálculos de mecânica quântica e de técnicas
espectroscópicas, Barteau e colaboradores (BARTEAU e LINIC, 2002; TYSOE et al., 2001)
reportaram a descoberta de um intermediário superficial, o complexo oxametalociclo (OMC).
A partir desse intermediário foi proposto um novo esquema geral de reação (Esquema 2.6) por
dois caminhos distintos: um seletivo, que resulta na formação do óxido de etileno e outro não-
seletivo, gerando acetaldeído que subsequentemente se decompõe em CO2.

2. REVISÃO BIBLIOGRÁFICA 8
Esquema 2.6 – Esquema geral de reação proposto a partir do intermediário OMC.
O complexo OMC é formado na superfície da prata através de um mecanismo
Langmuir-Hinshelwood (L-H) por reação entre o oxigênio atômico superficial e o etileno
adsorvido, conforme mostrado no Esquema 2.7. As etapas desse mecanismo são a adsorção
dissociativa de oxigênio (I), a adsorção de etileno (II), uma reação entre o oxigênio e o etileno
adsorvidos na formação do intermediário OMC (III) que, em seguida, se decompõe em
epóxido de etileno (IVa) ou acetaldeído (IVb), o segundo conduz indo à combustão total
indesejada.
Esquema 2.7 – Mecanismo de representação da epoxidação do etileno através do intermediário OMC.
Através desses estudos (BARTEAU e LINIC, 2003) foi possível concluir que o OMC
é o precursor comum de ambas as reações em paralelo. E de acordo com os autores, o
mecanismo apresenta duas etapas cinéticas importantes, a adsorção dissociativa do oxigênio e
a reação superficial de formação do OMC. A etapa que controla a taxa de reação é limitada
pelas condições reacionais utilizadas no processo. Em condições em que a alimentação é rica
em etileno, a etapa limitante é a adsorção do oxigênio e a taxa aumenta de acordo com o

2. REVISÃO BIBLIOGRÁFICA 9
aumento da pressão parcial de oxigênio. Nesse regime, a taxa de reação permanece constante
com o aumento da pressão parcial de etileno, e a seletividade ao óxido de etileno aumenta. O
etileno se comporta como um sítio bloqueador, agindo também na reação de combustão
completa, inibindo-a (OYAMA, 2008). Na presença de cargas enriquecidas com oxigênio, a
etapa limitante é a reação de formação do OMC e um aumento da taxa de reação é observado
aumentando-se a pressão parcial de etileno. Nesse regime, a pressão parcial de oxigênio tem
um efeito acentuado em ambas as taxas de reação e seletividade. Esse fato foi atribuído à
formação de oxigênio subsuperficial, que reduz tanto a energia de ativação para adsorção de
O2 quanto a decomposição do OMC à acetaldeído (MAVRIKAKIS et al., 2005; VAN DEN
HOEK et al., 1989).
Já de acordo com a termodinâmica da reação, a seletividade do produto depende das
barreiras de ativação relativas à formação de óxido de etileno (EO) ou acetaldeído (AA) por
meio da decomposição do intermediário OMC. Até o momento, estudos computacionais na
superfície da Ag(111) e Ag(100) (LINIC e CHRISTOPHER, 2008; BARTEAU e LINIC,
2003) reportaram barreiras de ativação (EOE e EAA) para formação desses produtos que não
são tão distintas entre si. Na literatura são reportadas energias de ativação para a formação de
EO na faixa de 54-88 kJ/mol. Esse fato está de acordo com a seletividade ao epóxido de
etileno de 50% observada nos catalisadores de prata não promovidos (VAN SANTEN e
ÖZBEK, 2013).
Em uma superfície com alta cobertura de oxigênio, a adsorção do etileno na prata é
impedida, mudando o mecanismo de formação do OMC de Langmuir-Hinshelwood (L-H)
para Eley-Rideal (E-R) (VAN SANTEN et al., 2012). Dessa forma, no mecanismo
apresentado no Esquema 2.7, a etapa (II) irá corresponder à reação direta entre um átomo de
oxigênio adsorvido e uma molécula de etileno não adsorvida, formando o OMC.
Investigações em superfícies de Ag(111) altamente oxigenadas (Esquema 2.8)
mostraram a formação de óxidos superficiais no catalisador, porém nenhuma mudança
significativa na reatividade do intermediário OMC foi reportada, contanto que uma vacância
superficial estivesse disponível para estabilizar esse complexo.

2. REVISÃO BIBLIOGRÁFICA 10
Esquema 2.8 – Geometrias apresentadas no mecanismo do OMC na superfície de Ag(111). Adaptado de (VAN
SANTEN e ÖZBEK, 2013).
Na superfície do Ag2O(001) a adsorção do etileno em uma dessas vacâncias de O é
mais exotérmica do que na superfície de Ag(111) (VAN SANTEN et al., 2011). Isso ocorre
porque os dois íons parcialmente positivos de Ag na superfície do óxido são mais
eletrofílicos. Nesse caso, a formação do OMC na superfície oxidada também é mais estável
que na superfície metálica, devido a uma adsorção mais intensa do etileno. Por outro lado,
estudos na superfície Ag2O(001) mostraram que na ausência dessas vacâncias (alta cobertura
de oxigênio), não há formação do OMC e a formação do óxido de etileno é direta, já que o
etileno não pode interagir com um átomo de prata superficial. Na Figura 2.1, é possível
observar que o oxigênio atômico superficial interage diretamente com a ligação C=C,
resultando em uma forma não-ativada do epóxido (EO(ads)) e libera 174 kJ/mol. A dessorção
desse epóxido adsorvido requer 73 kJ/mol. As energias de ativação das etapas responsáveis
pelo controle da taxa de ambas as reações são similares e estão de acordo com os valores
experimentais relatados de 70 kJ/mol. Deve ser notado que na superfície oxidada com uma
alta cobertura de oxigênio, não há uma interação direta entre o etileno e os íons de prata
superficiais e o mecanismo de reação é do tipo Eley-Rideal (E-R). Esse fato evita a ativação
da ligação C-H e a formação do acetaldeído. Essa ausência de vacâncias de oxigênio deve
promover uma maior seletividade, já que a reação não-seletiva não ocorre. De acordo com os
trabalhos propostos por Van Santen e colaboradores (2011), é provável que esse seja
exatamente o papel moderador dos compostos promotores clorados adicionados ao
catalisador.
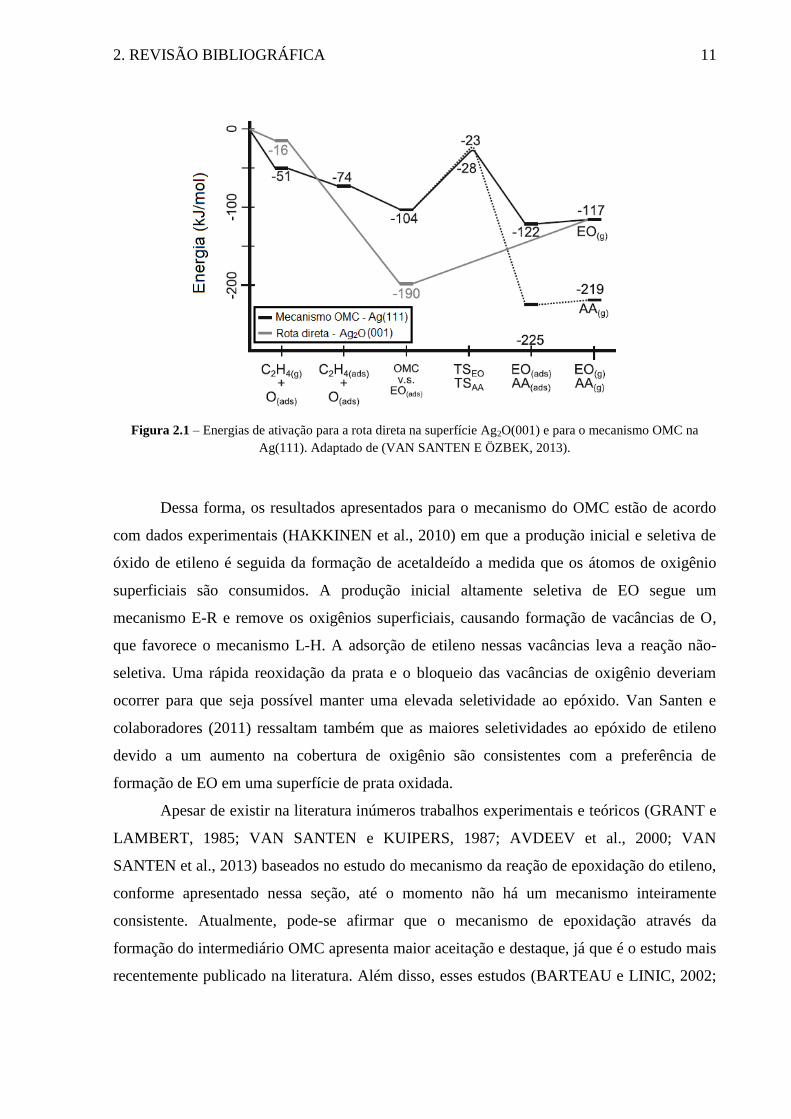
2. REVISÃO BIBLIOGRÁFICA 11
Figura 2.1 – Energias de ativação para a rota direta na superfície Ag2O(001) e para o mecanismo OMC na
Ag(111). Adaptado de (VAN SANTEN E ÖZBEK, 2013).
Dessa forma, os resultados apresentados para o mecanismo do OMC estão de acordo
com dados experimentais (HAKKINEN et al., 2010) em que a produção inicial e seletiva de
óxido de etileno é seguida da formação de acetaldeído a medida que os átomos de oxigênio
superficiais são consumidos. A produção inicial altamente seletiva de EO segue um
mecanismo E-R e remove os oxigênios superficiais, causando formação de vacâncias de O,
que favorece o mecanismo L-H. A adsorção de etileno nessas vacâncias leva a reação não-
seletiva. Uma rápida reoxidação da prata e o bloqueio das vacâncias de oxigênio deveriam
ocorrer para que seja possível manter uma elevada seletividade ao epóxido. Van Santen e
colaboradores (2011) ressaltam também que as maiores seletividades ao epóxido de etileno
devido a um aumento na cobertura de oxigênio são consistentes com a preferência de
formação de EO em uma superfície de prata oxidada.
Apesar de existir na literatura inúmeros trabalhos experimentais e teóricos (GRANT e
LAMBERT, 1985; VAN SANTEN e KUIPERS, 1987; AVDEEV et al., 2000; VAN
SANTEN et al., 2013) baseados no estudo do mecanismo da reação de epoxidação do etileno,
conforme apresentado nessa seção, até o momento não há um mecanismo inteiramente
consistente. Atualmente, pode-se afirmar que o mecanismo de epoxidação através da
formação do intermediário OMC apresenta maior aceitação e destaque, já que é o estudo mais
recentemente publicado na literatura. Além disso, esses estudos (BARTEAU e LINIC, 2002;

2. REVISÃO BIBLIOGRÁFICA 12
VAN SANTEN et al., 2013) apresentam fortes evidências de formação desse intermediário
superficial de reação com base em cálculos teóricos espectroscópicos e de mecânica quântica.
2.2 – Efeito da Natureza do Metal
Até o presente, a prata é o único metal eficiente para ser usado como catalisador na
reação de oxidação do etileno à epóxido de etileno. Inicialmente, sua singularidade é
explicada em relação a seletividade inicial da reação, que é determinada pela interação entre a
superfície do metal e o oxigênio adsorvido e pela fraca habilidade do catalisador em ativar a
ligação C-H do etileno. Outros metais de transição usados na reação de oxidação do etileno,
como Pd, Pt e Ni, são conhecidos por ativar facilmente ligações C-H e conduzir à combustão
completa.
Bal’zhinimaev (1999) enumerou diversas características peculiares da prata
relacionadas às suas propriedades eletrônicas e estrututurais. De acordo com o trabalho
proposto, essas propriedades podem ser afetadas por uma mudança no tamanho das partículas
de prata. Mudanças no estado de valência e o aparecimento de defeitos na superfície da prata
podem ser observados pelo aumento da energia de ativação provocado por uma diminuição no
tamanho destas partículas. Nesse sentido, a prata pode quimissorver dissociativamente o
oxigênio molecular para formar uma camada de óxido de prata superficial que é responsável
pela estabilização desses defeitos formados na superfície da prata (Figura 2.2). De acordo com
o autor, a epoxidação do etileno ocorre nos defeitos da rede cristalina onde estão localizadas
as espécies de oxigênio eletrofílico (Oel). Já as espécies de oxigênio nucleofílico (On),
responsáveis pela reação de oxidação total, estão localizadas nas superfícies metálicas planas.
Figura 2.2 – Mecanismo da epoxidação do etileno sobre a prata mássica, sendo oxigênio eletrofílico (Oel) e
nucleofílico (On). Adaptado de (BAL’ZHINIMAEV, 1999)

2. REVISÃO BIBLIOGRÁFICA 13
Atualmente, diversos estudos na literatura estão comparando a atividade e seletividade
do Au e Cu como outros possíveis metais a serem usados na obtenção do epóxido de etileno.
Chavadej e colaboradores (2007) estudaram a reação em catalisadores suportados de Ag-Au e
observaram que o ouro age como um agente diluente na superfície da prata, criando novos
sítios ativos de Ag que favoreceram a adsorção do oxigênio molecular. Eles também
investigaram catalisadores de Au/TiO2, reportando uma maior seletividade ao epóxido mas
uma menor conversão quando comparado ao catalisador de prata. Em catalisadores de
Au/CeO2 foi observado que a reação de oxidação total foi favorecida mesmo a baixas
temperaturas. Três estudos computacionais (LOPEZ et al, 2005; ILLAS et al., 2006; ILLAS e
TORRES, 2006) baseados na superfície do Au(111) e Cu(111) relataram que a reação de
epoxidação ocorre através de um mecanismo de formação do intermediário OMC, como
observado na superfície da Ag(111). As menores barreiras de ativação para formação do
óxido de etileno (EO) e do acetaldeído (AA) ocorrem na superfície Ag(111). E as maiores
barreiras de ativação ocorreram na superfície do Cu(111), mas o cobre é essencialmente mais
seletivo, ou seja, a energia de ativação para formação do EO é menor do que para o AA. Já na
superfície da Ag(111) e do Au(111) foi observado o oposto, sendo o Au o metal menos
seletivo à formação de epóxido de etileno dentre os estudados. Apesar da maior seletividade
apresentada na superfície do Cu, a produção de EO é endotérmica e pode provocar a
decomposição do produto. Outro problema inerente ao cobre é a formação de camadas de
óxido de cobre que podem desativar o catalisador.
Estudos recentes propuseram que óxidos superficiais são a fase ativa para a reação de
oxidação parcial do etileno e mostraram que a presença desse oxigênio subsuperficial
aumenta a seletividade do catalisador. Em um primeiro trabalho, Fellah e colaboradores
(2011) mostraram que a natureza eletrofílica dos átomos de oxigênio na superfície de
Ag2O(001) faz deles muito seletivos à reação de epoxidação. Foi demonstrada a possibilidade
de um mecanismo direto de formação do EO sob altos regimes de O2, sem formação do
intermediário OMC, que possui uma baixa energia de ativação. Mais tarde, através de estudos
computacionais (VAN SANTEN et al., 2011) esse mesmo grupo comparou a reação de
epoxidação na superfície dos óxidos Au2O, Ag2O e Cu2O para avaliar a reatividade e
seletividade ao epóxido nessas estruturas de óxidos superficiais. Foi utilizado o mesmo plano
cristalino (001) e uma alta cobertura de O2. Na Figura 2.3, é demonstrado que o oxigênio
atômico superficial interage diretamente com a ligação C=C, resultando na formação do óxido
de etileno não-ativado (EO(ads)) que não inclui a formação do intermediário OMC, como
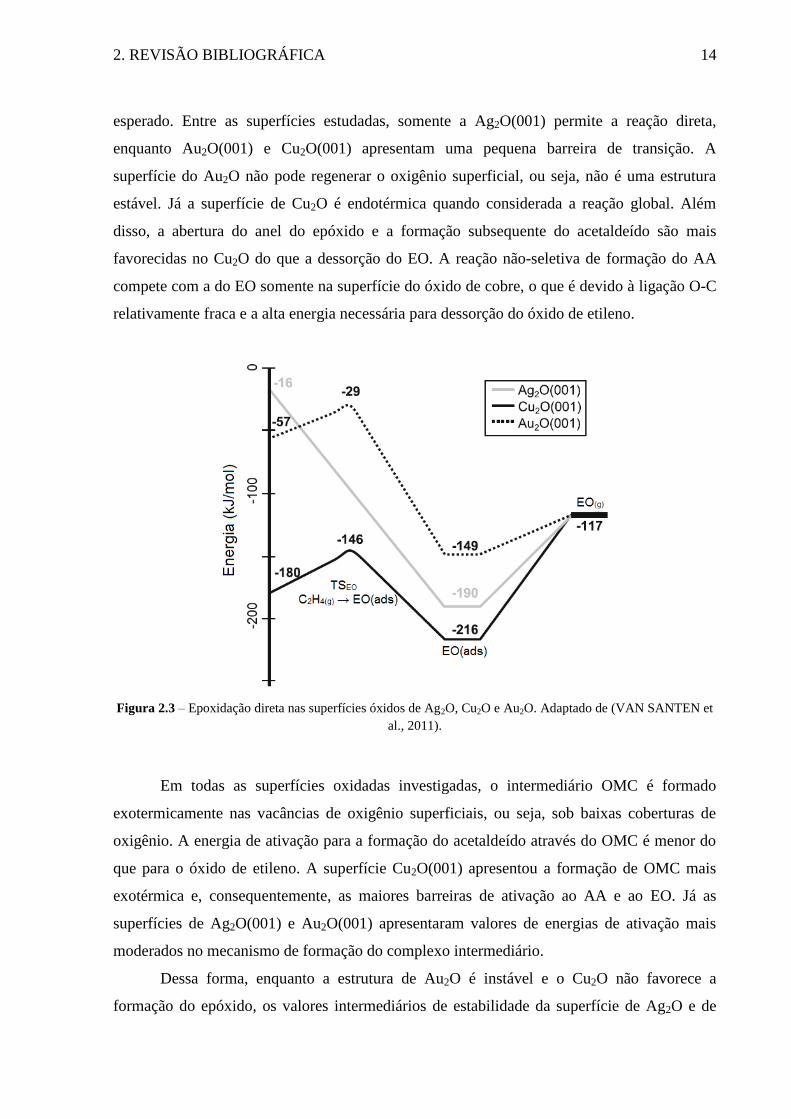
2. REVISÃO BIBLIOGRÁFICA 14
esperado. Entre as superfícies estudadas, somente a Ag2O(001) permite a reação direta,
enquanto Au2O(001) e Cu2O(001) apresentam uma pequena barreira de transição. A
superfície do Au2O não pode regenerar o oxigênio superficial, ou seja, não é uma estrutura
estável. Já a superfície de Cu2O é endotérmica quando considerada a reação global. Além
disso, a abertura do anel do epóxido e a formação subsequente do acetaldeído são mais
favorecidas no Cu2O do que a dessorção do EO. A reação não-seletiva de formação do AA
compete com a do EO somente na superfície do óxido de cobre, o que é devido à ligação O-C
relativamente fraca e a alta energia necessária para dessorção do óxido de etileno.
Figura 2.3 – Epoxidação direta nas superfícies óxidos de Ag2O, Cu2O e Au2O. Adaptado de (VAN SANTEN et
al., 2011).
Em todas as superfícies oxidadas investigadas, o intermediário OMC é formado
exotermicamente nas vacâncias de oxigênio superficiais, ou seja, sob baixas coberturas de
oxigênio. A energia de ativação para a formação do acetaldeído através do OMC é menor do
que para o óxido de etileno. A superfície Cu2O(001) apresentou a formação de OMC mais
exotérmica e, consequentemente, as maiores barreiras de ativação ao AA e ao EO. Já as
superfícies de Ag2O(001) e Au2O(001) apresentaram valores de energias de ativação mais
moderados no mecanismo de formação do complexo intermediário.
Dessa forma, enquanto a estrutura de Au2O é instável e o Cu2O não favorece a
formação do epóxido, os valores intermediários de estabilidade da superfície de Ag2O e de

2. REVISÃO BIBLIOGRÁFICA 15
sua força de ligação com o oxigênio denotam a singularidade da prata como um catalisador
para a epoxidação do etileno. A interação da superfície Ag2O(001) com o oxigênio molecular
é forte o suficiente para reoxidar a superfície da Ag, gerando o óxido. Por outro lado, ela é
fraca o suficiente para prevenir a abertura do anel do epóxido e a ativação de ligações C-H. O
mesmo é valido para a superfície de Ag metálica, que é forte o suficiente para dissociar o O2 e
formar o intermediário OMC, e moderada o suficiente para evitar as reações paralelas
indesejadas.
2.3 – Efeito do Tipo de Suporte na Reação de Epoxidação
O papel do suporte dos catalisadores utilizados na epoxidação do etileno é um fator
importante que não tem sido investigado sistematicamente. No processo industrial são
utilizados catalisadores comerciais suportados em α-alumina de baixa área superficial (< 1
m2/g) com altos teores de prata (10-15% p/p). Esses altos teores de metal são necessários para
fornecer uma alta área superficial ativa apesar de se obter uma baixa dispersão. A natureza do
suporte pode influenciar tanto na dispersão metálica quanto na distribuição dos tamanhos de
partícula de prata. Nesse sentido, a singularidade da α-Al2O3 como suporte na produção de
catalisadores altamente seletivos foi atribuída justamente à sua baixa área superficial. No
entanto, esses catalisadores possuem rendimentos relativamente baixos de produção do óxido
de etileno, como resultado da baixa dispersão das partículas de prata sobre esses suportes de
α-Al2O3 comerciais. Se um maior grau de dispersão metálico pudesse ser alcançado, seria
possível a obtenção da mesma área ativa com uma quantidade significativamente menor de
prata. Como resultado, o metal seria utilizado de forma mais eficiente e o processo seria mais
econômico. Um alto grau de dispersão do metal pode, geralmente, ser conseguido através do
uso de um suporte de elevada área superficial.
Outro fator importante a ser notado é a existência de evidências na literatura que
indicam que a reação competitiva de formação do acetaldeído, que leva à combustão
consecutiva, é conhecida por ser sensível à acidez do suporte catalítico. Técnicas de
microscopia eletrônica demonstraram que a prata tem a forma de partículas esféricas na
superfície do suporte de alumina, deixando a maior parte da superfície do suporte descoberta.
A proporção entre a área superficial de prata e a área de suporte tende a diminuir em suportes
de área superficial elevada, e qualquer efeito de reação no próprio suporte torna-se mais
importante e acentuado.
2. REVISÃO BIBLIOGRÁFICA 16
Existem alguns trabalhos sobre o efeito do suporte na reação de oxidação parcial do
etileno. Em um estudo da década de 70 (HARRIOT, 1971), catalisadores de prata suportados
em SiO2 de alta área superficial foram apresentados como sendo mais ativos e seletivos na
produção de óxido de etileno quanto os suportados em α-alumina de baixa área (T = 453 a
523 K; P = 1 atm). Uma seletividade de 30-60%, similar à obtida nos catalisadores
comerciais, foi observada para prata suportada em sílica não-porosa e sílica-gel tratada
termicamente com áreas de 60-300 m2/g. Foram obtidas seletividades e atividades catalíticas
elevadas nos catalisadores com partículas de prata com diâmetro de 3-7 nm. Partículas
menores de prata levaram a uma baixa seletividade ao EO.
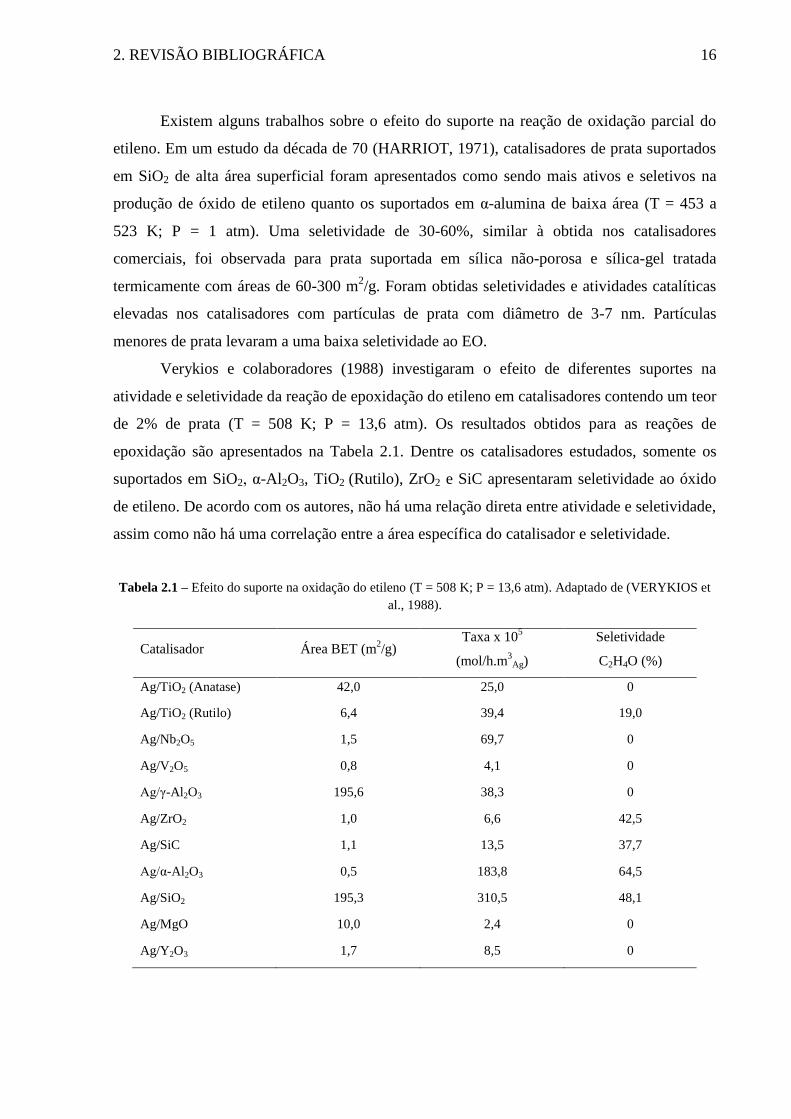
Verykios e colaboradores (1988) investigaram o efeito de diferentes suportes na
atividade e seletividade da reação de epoxidação do etileno em catalisadores contendo um teor
de 2% de prata (T = 508 K; P = 13,6 atm). Os resultados obtidos para as reações de
epoxidação são apresentados na Tabela 2.1. Dentre os catalisadores estudados, somente os
suportados em SiO2, α-Al2O3, TiO2 (Rutilo), ZrO2 e SiC apresentaram seletividade ao óxido
de etileno. De acordo com os autores, não há uma relação direta entre atividade e seletividade,
assim como não há uma correlação entre a área específica do catalisador e seletividade.
Tabela 2.1 – Efeito do suporte na oxidação do etileno (T = 508 K; P = 13,6 atm). Adaptado de (VERYKIOS et
al., 1988).
Catalisador Área BET (m2/g)
Taxa x 105
(mol/h.m3
Ag)
Seletividade
C2H4O (%)
Ag/TiO2 (Anatase) 42,0 25,0 0
Ag/TiO2 (Rutilo) 6,4 39,4 19,0
Ag/Nb2O5 1,5 69,7 0
Ag/V2O5 0,8 4,1 0
Ag/γ-Al2O3 195,6 38,3 0
Ag/ZrO2 1,0 6,6 42,5
Ag/SiC 1,1 13,5 37,7
Ag/α-Al2O3 0,5 183,8 64,5
Ag/SiO2 195,3 310,5 48,1
Ag/MgO 10,0 2,4 0
Ag/Y2O3 1,7 8,5 0

2. REVISÃO BIBLIOGRÁFICA 17
Uma possível explicação para os fenômenos observados estaria relacionada aos
diferentes graus de dispersão da prata em uma série de suportes, já que a reação de
epoxidação do etileno é conhecida por ser sensível ao tamanho de cristalito da prata. Além
disso, os autores observaram uma menor atividade para o catalisador suportado em sílica, com
pequenos cristalitos de prata, quando comparado aos maiores cristalitos presentes no
catalisador suportado em α-alumina. Em reações posteriores de isomerização e combustão do
óxido de etileno, materiais como γ-Al2O3, SiO2, MgO, SiC, TiO2, ZrO2 e V2O5, exibiram uma
atividade significativa que se mostrou ser proporcional a acidez superficial desses suportes. O
envenenamento dos sítios ácidos superficiais por impregnação com um sal alcalino resultou
na supressão da atividade de isomerização e redução da atividade de oxidação.
Na década de 90, Seyedmonir e colaboradores (1990) prepararam catalisadores bem
dispersos utilizando os supores η-Al2O3, TiO2 e SiO2, e estudaram a seletividade e atividade
para a formação do epóxido de etileno na presença e ausência de C2H4Cl2 (EDC – dicloreto de
etileno) e CO2, comparando o resultado com catalisador de baixa dispersão Ag/α-Al2O3 (T =
503 a 547 K; P = 4,5 atm). Na presença de 0,5 ppm de EDC, as seletividades dos catalisadores
Ag/η-Al2O3 e Ag/TiO2 foram muito baixas (cerca de 10%) em comparação com uma
seletividade de aproximadamente 60% apresentada pelo catalisador Ag/α-Al2O3. Esse
resultado foi atribuído à presença de reações secundárias de oxidação que ocorrem nestes
suportes. Em contraste, na ausência de EDC e CO2 a 523 K, foram obtidas seletividades de
17% e 55% para Ag/SiO2 com cristalitos de prata de 4,4 e 7,6 nm, respectivamente. E nas
mesmas condições, o catalisador Ag/α-Al2O3 obteve seletividade de 23% com cristalitos de 1
μm.
Cant e colaboradores (1991) prepararam catalisadores de prata suportados em α-
alumina, titânia e diferentes tipos de sílica (não porosa e porosas com diferentes áreas
específicas) (T = 512 K; P = 1 atm). Como é possível observar na Tabela 2.2, os catalisadores
suportados nas sílicas porosas (Davison 62 e SMR) exibiram desativação e seletividade
similares ao catalisador suportado em sílica não porosa (Cabosil). Os catalisadores suportados
em TiO2 (Anatase) e γ-Al2O3 apresentaram seletividades aproximadamente nulas ao EO, e
desativação similar aos catalisadores Ag/SiO2. De acordo com os autores, estes resultados
podem ser explicados pelo baixo teor metálico destes catalisadores. O catalisador Ag/α-Al2O3
não demonstrou desativação com o tempo, e sua seletividade foi similar a dos catalisadores
Ag/SiO2. Os resultados apresentados são similares aos reportados por Harriot, Verykios e
Seyedmonir. Todos os catalisadores suportados em materiais de alta área específica, com
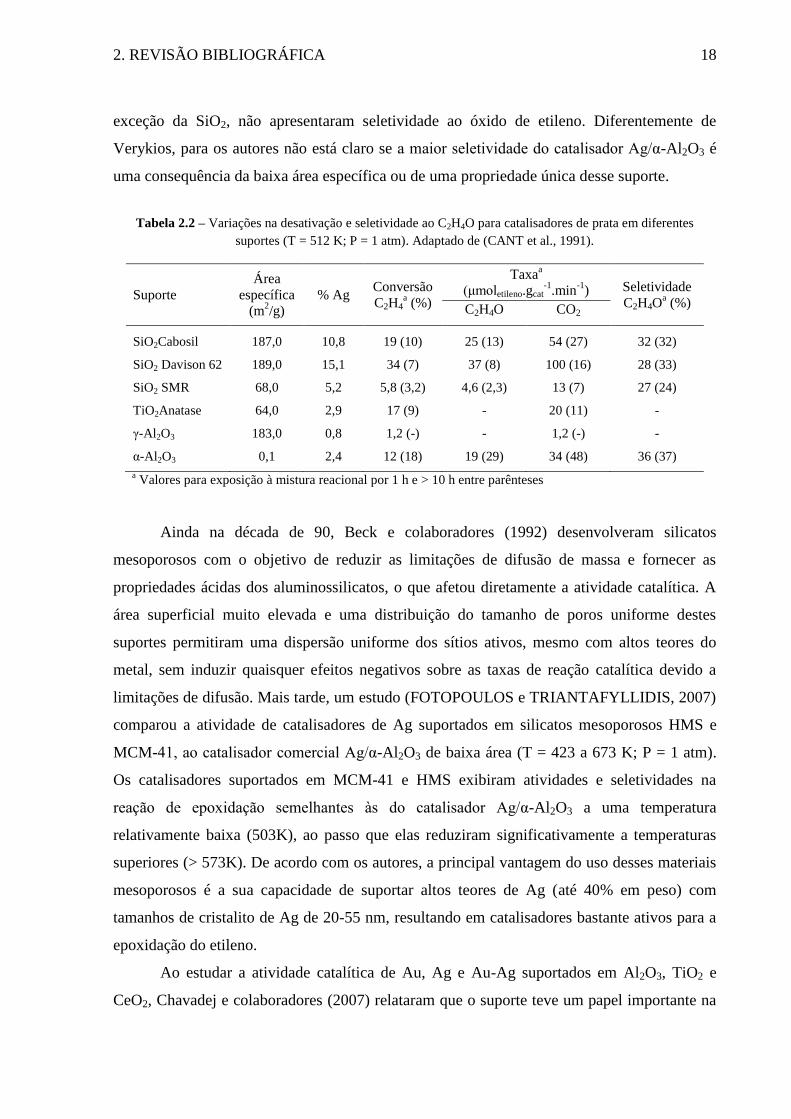
2. REVISÃO BIBLIOGRÁFICA 18
exceção da SiO2, não apresentaram seletividade ao óxido de etileno. Diferentemente de
Verykios, para os autores não está claro se a maior seletividade do catalisador Ag/α-Al2O3 é
uma consequência da baixa área específica ou de uma propriedade única desse suporte.
Tabela 2.2 – Variações na desativação e seletividade ao C2H4O para catalisadores de prata em diferentes
suportes (T = 512 K; P = 1 atm). Adaptado de (CANT et al., 1991).
Suporte
Área
específica
(m2/g)
% Ag Conversão
C2H4a (%)
Taxaa
(μmoletileno.gcat-1
.min-1
) Seletividade
C2H4Oa (%)
C2H4O CO2
SiO2Cabosil 187,0 10,8 19 (10) 25 (13) 54 (27) 32 (32)
SiO2 Davison 62 189,0 15,1 34 (7) 37 (8) 100 (16) 28 (33)
SiO2 SMR 68,0 5,2 5,8 (3,2) 4,6 (2,3) 13 (7) 27 (24)
TiO2Anatase 64,0 2,9 17 (9) - 20 (11) -
γ-Al2O3 183,0 0,8 1,2 (-) - 1,2 (-) -
α-Al2O3 0,1 2,4 12 (18) 19 (29) 34 (48) 36 (37)
a Valores para exposição à mistura reacional por 1 h e > 10 h entre parênteses
Ainda na década de 90, Beck e colaboradores (1992) desenvolveram silicatos
mesoporosos com o objetivo de reduzir as limitações de difusão de massa e fornecer as
propriedades ácidas dos aluminossilicatos, o que afetou diretamente a atividade catalítica. A
área superficial muito elevada e uma distribuição do tamanho de poros uniforme destes
suportes permitiram uma dispersão uniforme dos sítios ativos, mesmo com altos teores do
metal, sem induzir quaisquer efeitos negativos sobre as taxas de reação catalítica devido a
limitações de difusão. Mais tarde, um estudo (FOTOPOULOS e TRIANTAFYLLIDIS, 2007)
comparou a atividade de catalisadores de Ag suportados em silicatos mesoporosos HMS e
MCM-41, ao catalisador comercial Ag/α-Al2O3 de baixa área (T = 423 a 673 K; P = 1 atm).
Os catalisadores suportados em MCM-41 e HMS exibiram atividades e seletividades na
reação de epoxidação semelhantes às do catalisador Ag/α-Al2O3 a uma temperatura
relativamente baixa (503K), ao passo que elas reduziram significativamente a temperaturas
superiores (> 573K). De acordo com os autores, a principal vantagem do uso desses materiais
mesoporosos é a sua capacidade de suportar altos teores de Ag (até 40% em peso) com
tamanhos de cristalito de Ag de 20-55 nm, resultando em catalisadores bastante ativos para a
epoxidação do etileno.
Ao estudar a atividade catalítica de Au, Ag e Au-Ag suportados em Al2O3, TiO2 e
CeO2, Chavadej e colaboradores (2007) relataram que o suporte teve um papel importante na

2. REVISÃO BIBLIOGRÁFICA 19
reação de epoxidação de etileno (T = 493 a 543 K; P = 35,5 atm). O melhor desempenho na
epoxidação do etileno foi observado no catalisador suportado em α-Al2O3 com teor ótimo de
Ag de 13,18% (em peso), enquanto que nos catalisadores Ag/TiO2 ou Ag/CeO2 apenas a
oxidação total foi observada.
Em um estudo mais recente, Chavadej e colaboradores (2012) sintetizaram
catalisadores com diferentes teores de Ag suportados em α-Al2O3 (0,109 m2/g), Al2O3 C (85–
115 m2/g), Al2O3 acidificada, SiO2 (75–105 m
2/g), TiO2 e SrTiO3 sintetizado por um processo
sol-gel. Esses catalisadores foram investigados na epoxidação do etileno em diferentes
temperaturas de reação (T = 498 a 563 K; P = 1,7 atm). Na Tabela 2.3, estão apresentados
alguns dos resultados obtidos nesse trabalho. De acordo com a tabela é possível observar que
um aumento da temperatura de reação de 498 K para 563 K provoca um pequeno aumento de
conversão do etileno e uma grande diminuição da seletividade ao óxido de etileno, com
exceção do catalisador Ag/SrTiO3, em que a seletividade ainda permaneceu muito elevada. O
rendimento ao C2H4O, por sua vez, aumentou até uma determinada temperatura para todos os
catalisadores. Em uma temperatura mais elevada o rendimento diminuiu consideravelmente,
já que é promovida praticamente a oxidação completa nessas condições. Em comparação com
os demais catalisadores, o catalisador 17,16% Ag/SrTiO3 exibiu uma atividade catalítica
superior, com maior rendimento ao óxido (4,5%) e seletividade de até 99% na temperatura de
548 K. Deve-se ressaltar que os autores não apresentaram a definição da seletividade utilizada
nos cálculos de tais seletividades altíssimas ao EO. Em temperaturas inferiores, esse
catalisador não produziu CO2, e de acordo com os autores, esse fato pode estar relacionado
aos resultados apresentados no TPD de O2 e de C2H4 que demonstram altas temperaturas de
dessorção, favorecendo a reação de epoxidação frente à de oxidação completa em
temperaturas menores do que 548 K. Esse catalisador também apresentou o maior tamanho de
partícula de Ag, o que deveria favorecer a reação de epoxidação (OYAMA et al., 2005). O
catalisador Ag/Al2O3,Acid apresentou um rendimento ao C2H4O comparável ao do Ag/SrTiO3 e
uma seletividade ligeiramente maior em temperaturas inferiores a 523 K, o que
provavelmente seria resultante de sua acidez apropriada que deveria suprimir a formação de
CO2 (GAO e SHI, 2010). No entanto, esse catalisador possui o menor tamanho de partícula de
prata.

2. REVISÃO BIBLIOGRÁFICA 20
Tabela 2.3 – Características dos suportes e catalisadores, e suas atividades na epoxidação do etileno a diferentes
temperaturas (6% O2 e 6% C2H4 com balanço de He; T = 498 a 563 K; P = 1,7 atm), (CHAVADEJ et al., 2012).
Suporte
Área
Específica
(m2/g)
Tamanho de
partícula de
Ag (nm)
Temperatura
de reação (K)
Conversão
C2H4 (%)
Seletividade
C2H4O (%)
Rendimento
C2H4O (%)
α-Al2O3 0.1 49,8
498
523
548
563
1,7
2,7
3,6
4,6
98,1
87,5
77,4
38,5
1,7
2,3
2,8
1,8
Al2O3 C 98.3 30,6
498
523
548
563
0,8
1,4
3,9
4,9
96,5
89,9
86,7
27,0
0,8
1,2
3,4
1,3
Al2O3,Acid 123.0 27,9
498
523
548
563
2,1
3,3
4,0
4,5
99,2
96,4
64,9
48,2
2,1
3,2
2,6
2,2
SiO2 90 86.9 41,5
498
523
548
563
1,9
3,6
4,4
5,0
84,3
82,1
86,4
17,3
1,6
3,0
3,8
0,9
SrTiO3 4.4 52,1
498
523
548
563
0,9
2,3
4,6
4,8
97,4
97,7
99,0
51,1
0,9
2,2
4,5
2,5
TiO2 36.7 43,2
498
523
548
563
1,0
1,3
2,5
3,0
76,8
72,9
69,7
10,2
0,8
0,9
1,8
0,3
Fazendo uma análise de todos os trabalhos citados, os resultados indicam que a reação
de epoxidação do etileno depende de vários fatores: da temperatura de reação; do teor de
prata; do tamanho de cristalito da prata; da área específica e da acidez do suporte. Além disso,
é possível observar que a área do suporte influencia diretamente no tamanho de partícula de
prata do catalisador. A atividade do catalisador parece ser influenciada diretamente pelo
suporte utilizado. Os catalisadores Ag/TiO2 e Ag/SiO2, por exemplo, apresentam rendimentos
e seletividades bem distintos apesar do tamanho de partícula similar. Conforme reportado por
Chavadej, ainda existem catalisadores muito promissores na obtenção do óxido de etileno
para serem investigados. Por estes motivos, existe ainda uma grande motivação em entender o
papel do suporte e em desenvolver novos catalisadores com suportes diferentes que ofereçam
um melhor desempenho catalítico para a epoxidação do etileno.

3. MATERIAIS E MÉTODOS 21
Capítulo 3 – MATERIAIS E MÉTODOS
3.1 – REAGENTES E EQUIPAMENTOS
A seguir são listados todos os reagentes usados na síntese dos suportes e catalisadores,
assim como os gases utilizados nas sínteses, caracterizações e nos testes catalíticos.
3.1.1 – Reagentes para a preparação dos suportes e catalisadores
NH4OH (Proquímios)
Na2CO3 (Vetec)
Mg(NO3)2.6H2O (Sigma-Aldrich)
NaOH (Vetec)
Zr(NO3)3 (Sigma-aldrich)
Alumina comercial PURALOX (Condea)
AgNO3 (Sigma-Aldrich)
SiC (Sigma-Aldrich)
3.1.2 – Gases para a caracterização e teste dos catalisadores
Ar sintético 20% O2 em N2 (Air Liquide)
Oxigênio ultrapuro 99,99% (Linde)
Nitrogênio ultrapuro 99,99% (Linde)
Nitrogênio líquido (White Martins)
Hidrogênio ultrapuro 99,99% (Linde)
Hélio ultrapuro 99,99% (Linde)
Mistura gasosa 50% Etileno em N2 (Linde)
3.2 – SÍNTESE DOS SUPORTES
No presente trabalho serão usados MgO, ZrO2 e α-Al2O3 como suportes para
catalisadores de prata. Tais materiais foram escolhidos por possuírem propriedades bastante
distintas entre si como, por exemplo, acidez e área específica. Dessa forma, será possível
avaliar o desempenho catalítico de catalisadores que possuem diferentes características.

3. MATERIAIS E MÉTODOS 22
3.2.1 – MgO
O óxido de magnésio, MgO, foi obtido através do método de coprecipitação. Em um
béquer foi adicionado 200 mL de uma solução aquosa de 30,5 g de Na2CO3, permanecendo
sob agitação mecânica até completa dissolução. Em seguida, adicionou-se gota-a-gota uma
solução de 96,1 g de Mg(NO3)2.6H2O (Sigma-Aldrich) em 200 mL de água destilada. O pH
da mistura resultante foi controlado em 10,0 com 100 mL de solução aquosa de 27,4 g de
NaOH. Após completa adição da solução de nitrato de magnésio a agitação foi mantida por
aproximadamente 24 horas. Em seguida, o precipitado foi filtrado a quente até pH = 7,0 e
seco em estufa a 383 K, por 24 horas. O material foi calcinado a 973 K (taxa de aquecimento
de 5 K/min), por 5 horas, sob fluxo de ar sintético de 50 mL/min.
3.2.2 – ZrO2
O suporte ZrO2 foi sintetizado a partir da adição de 100 mL de uma solução de
Zr(NO3)3 (Sigma-Aldrich) em 160 mL de solução de NH4OH, mantendo-se uma agitação
vigorosa por aproximadamente 30 min. O precipitado branco resultante foi filtrado a vácuo e
colocado em estufa a 383 K, por 24 horas, para secagem. O material foi calcinado a 773 K
(taxa de aquecimento de 5 K/min), por 6 horas, sob fluxo de ar sintético de 50 mL/min.
A composição de cada fase cristalina da ZrO2 foi calculada a partir das alturas (h) e
larguras à meia altura (β) dos picos característicos das fases monoclínica e tetragonal
conforme as equações (1) e (2) apresentadas abaixo (SU et al., 2000; KHAODEE et al.,
2008):
% fase monoclínica= ∑ (h.β) fase monoclínica
∑ (h.β) fases tetragonal e monoclínica x 100 (1)
% fase tetragonal = ∑ (h.β) fase tetragonal
∑ (h.β) fases tetragonal e monoclínica x 100 (2)
3.2.3 – α-Al2O3
O suporte α-alumina foi obtido a partir da calcinação de alumina comercial
PURALOX (Condea) a 1473 K, por 5 horas, com uma taxa de aquecimento de 10 K/min.

3. MATERIAIS E MÉTODOS 23
3.3 – PREPARAÇÃO DOS CATALISADORES
O catalisador suportado em α-Al2O3 foi preparado através do método de impregnação
úmida. Em um balão de 100 mL, adicionou-se uma solução aquosa de AgNO3 (15% em peso
de Ag) ao suporte. A mistura foi mantida sob agitação lenta por aproximadamente 1 hora. Em
seguida, foi feita a evaporação do solvente sob vácuo em um rotaevaporador. O sólido
resultante foi seco em estufa a 343 K, por 12 horas.
Os demais catalisadores foram preparados através do método de impregnação seca. Os
suportes foram impregnados com uma solução aquosa de AgNO3 (Sigma-Aldrich), utilizado
como sal precursor, de modo a se obter 15% p/p de prata no catalisador. A solução foi
adicionada gota a gota ao suporte até que fosse atingido o ponto úmido. A secagem do
material resultante foi feita em estufa a 343 K, por 12 horas.
O tratamento térmico seguiu metodologia empregada industrialmente (CARVALHO,
2005), com calcinação sob fluxo de ar sintético (30 mL/min) a 623 K, por 4 horas, para os
catalisadores suportados em MgO, ZrO2 e α-Al2O3 (taxa de aquecimento lenta de 3 K/min).
Foram feitos dois patamares intermediários a 473 K e a 533 K, por 2 horas cada. A rampa de
temperatura está representada na Figura 3.1.
0 100 200 300 400 500 600
300
350
400
450
500
550
600
650
533 K, 2h
623 K, 4h
Tem
pera
tura
(K
)
Tempo (min)
473 K, 2h
Figura 3.1 – Rampa de temperatura para calcinação dos catalisadores de prata.

3. MATERIAIS E MÉTODOS 24
3.4 – CARACTERIZAÇÃO DOS SUPORTES E CATALISADORES
Todas as caracterizações dos suportes e catalisadores descritas a seguir foram obtidas
por uma parceria com o Laboratório de Catálise (LACAT), da Divisão de Catálise e Processos
Químicos (DCAP) do Instituto Nacional de Tecnologia (INT), com exceção da caracterização
por espectrospia de absorção atômica.
3.4.1 – Espectroscopia de Absorção Atômica (AAS)
A composição química dos catalisadores foi determinada através da técnica de
espectrometria de absorção atômica com chama ar/acetileno. As amostras foram analisadas
em um equipamento Varian, modelo AA240FS, no Laboratório de Espectroanalítica Aplicada
(LESPA) da Universidade Federal Fluminense. Inicialmente, foi feito um pré-tratamento das
amostras que incluiu aquecimento a 373 K, por 30 min, e solubilização de 150 mg de cada
amostra com HNO3, resultando em soluções de 2,0 ppm.
3.4.2 – Análise Textural (BET e BJH)
A fim de avaliar as propriedades texturais dos suportes e catalisadores, foram obtidas
as isotermas de adsorção por fisissorção de nitrogênio, e determinados os valores de área
específica, volume e diâmetro de poros. As análises foram realizadas utilizando um
equipamento ASAP 2020 da Micromeritics. Inicialmente, as amostras foram secas em estufa a
373 K, por 24 horas. Posteriormente, as amostras foram submetidas a um pré-tratamento no
próprio aparelho, que consistiu no aquecimento de 200 mg de amostra a 623 K,
permanecendo nessa temperatura até que fosse atingido o vácuo de 9 µmHg. Após o
tratamento, a amostra foi novamente pesada para a determinação de sua massa real. A área
específica dos materiais foi calculada através do método de Brunnauer-Emmett-Teller (BET).
O volume e diâmetro médio de mesoporos foram determinados pelo método de Barret-Joyner-
Halenda (BJH).
3.4.3 – Difratometria de Raios X (DRX)
Os difratogramas das amostras foram obtidos utilizando-se um difratômetro Rigaku,
modelo Miniflex, com radiação CuK ( = 0,1542nm). Os dados foram coletados numa faixa
de 2θ entre 20 e 80º, usando-se uma velocidade de varredura de 0,02º/passo e um tempo de

3. MATERIAIS E MÉTODOS 25
contagem de 1 segundo/passo. Foi utilizada a equação de Scherrer, apresentada abaixo, para o
cálculo do tamanho de cristalito da espécie Ag metálica:
d = (k .)/(.cos)
Onde: d = diâmetro médio da partícula
k = constante de proporcionalidade = 0,9 (partículas esféricas)
comprimento de onda da radiação = 0,1542 nm para a fonte de Cu
largura à meia altura do pico (em rad)
3.4.4 – Dessorção de Oxigênio à Temperatura Programada (TPD de O2)
A dispersão da Ag, área ativa e o tamanho de cristalito da prata nos catalisadores
foram determinados por dessorção de oxigênio à temperatura programada. Esses
experimentos foram feitos em uma unidade acoplada a um espectrômetro de massas.
Inicialmente, 250 mg de cada amostra foi submetida a um aquecimento de 303 K até 523 K
(taxa de aquecimento de 10 K/min) sob fluxo de oxigênio puro de 30 mL/min, permanecendo
nesse patamar por 1 hora. Em seguida, as amostras foram resfriadas sob o mesmo fluxo de
oxigênio puro até 303 K. Após esse pré-tratamento, foi feita uma purga com He por 30 min e
as amostras foram aquecidas da temperatura ambiente até 823 K (taxa de aquecimento de 20
K/min). A rampa de aquecimento está representada na Figura 3.2.
Figura 3.2 – Rampa de aquecimento do procedimento de TPD de O2 dos catalisadores de prata.
0 100 200 300 400 500 600
300
350
400
450
500
550
600
650
533 K, 2h
623 K, 4h
Te
mp
era
tura
(K
)
Tempo (min)
473 K, 2h

3. MATERIAIS E MÉTODOS 26
A dispersão da prata (DAg), área ativa (Sativa) e o tamanho de cristalito da prata
(dAg,TPD) (SEYEDMONIR et al., 1990) foram calculados de acordo com as equações 3, 4 e 5,
respectivamente:
DAg = ρsítios, sup (TPD de O2)
ρsítios, total (AAS) (3)
SAtiva = ρsítios, sup . Nav . f
L (4)
dAg,TPD = 1,3
DAg (5)
Sendo: ρsítios, sup = Apico . nloop
ApulsoO2 . mcat
e ρsítios, total = % Ag
MMAg
Onde: D𝐴𝑔 = dispersão da prata (%)
Sativa = área ativa (m2)
dAg,TPD = tamanho de cristalito da prata obtido por TPD de O2 (nm)
ρsítios, sup = densidade de sítios superficial, obtida por TPD de O2 (mol.gcat-1
)
ρsítios, total = densidade de sítios total, obtida por AAS (mol.gcat-1
)
Nav = número de Avogadro (6,023 x 1023
)
f = fator estequiométrico (2 Ag/O2)
L = número de átomos superficiais por m2 (1,15 x 10
19 átomos de Ag.m
-2)
Apico = área do pico de O2 dessorvido do catalisador
nloop = número de moles de O2 em um loop
Apulso = área do pulso de O2 puro
mcat = massa de catalisador (g)
% Ag = teor de prata, obtido por AAS
MMAg = massa molar da prata metálica (107,87 g.mol-1
)

3. MATERIAIS E MÉTODOS 27
3.5 – TESTES CATALÍTICOS
Todos os testes catalíticos da reação de epoxidação do etileno foram conduzidos em
um reator de vidro de leito fixo sob pressão atmosférica. O reator contendo o catalisador foi
colocado em um forno equipado com um controlador de temperatura.
Os catalisadores (30 mg) foram diluídos em SiC (100 mg) e submetidos a um pré-
tratamento com uma mistura 20% H2/He a 623 K, por 1 hora. Após pré-tratamento, os
catalisadores foram resfriados até 473 K com uma purga de He puro. Para condução da reação
utilizou-se uma razão de C2H4/O2 igual a 0,5 na carga de alimentação, sendo a composição
total da mistura gasosa de 6% C2H4; 12% O2; 54% N2 e 28% He. A vazão total da carga de
alimentação foi de 42 mL/min e as temperaturas de reação usadas foram 498, 523 e 548 K.
Foi feito aquecimento gradativo dos catalisadores após exposição à mistura reacional com
uma taxa de aproximadamente 5 K/min, partindo-se inicialmente da temperatura de 473 K.
As composições dos gases de alimentação e dos produtos foram analisadas em um
cromatógrafo a gás Agilent, modelo 6890N, equipado com detectores de condutividade
térmica (TCD) e de ionização de chama (FID), e colunas Porapak-Q e 20%
Carbowax/Silicoport. A formação de acetaldeído não foi quantificada já que nas condições
utilizadas nesse estudo esse produto foi decomposto diretamente a dióxido de carbono. Foram
utilizados dados experimentais com uma média de erro menor do que 5% para avaliação
correta do desempenho catalítico.
Os catalisadores suportados em α-Al2O3 e MgO apresentaram melhor desempenho
catalítico e foram avaliados também nos testes de estabilidade na temperatura de 498 K, por
24 horas.
A taxa de consumo do etileno (rEt), taxa de formação de EO (r EO), taxa de formação
de CO2 (rCO2), seletividade ao EO (SEO), o rendimento do EO, TOF de consumo de etileno
(TOFEt) e os TOFs de formação dos produtos (TOFEO e TOFCO2) foram calculados de acordo
com as equações 6, 7, 8, 9, 10, 11, 12 e 13 respectivamente:
r Et = QEt . (nEt
0 − nEts )
mAg . nEt0 (6)
r EO = QEt . nEO
mAg . nEt0 (7)

3. MATERIAIS E MÉTODOS 28
rCO2=
QEt . nCO2
mAg . nEt0 (8)
SEO(%) = rEO
rCO2
∗ 100 (9)
REO(%) = nEO
nEt0 − nEt
s ∗ 100 (10)
TOFEt = rEt
ρsítios, sup (TPD de O2) x 60 (11)
TOFEO = rEO
ρsítios, sup (TPD de O2) x 60 (12)
TOFCO2=
rCO2
ρsítios, sup (TPD de O2) x 60 (13)
Onde: rEt = taxa de consumo de etileno (mol.min-1
.gAg-1
)
r EO = taxa de formação de óxido de etileno (mol.min-1
.gAg-1
)
rCO2 = taxa de formação de dióxido de carbono (mol.min
-1.gAg
-1)
QEt = vazão molar de etileno (mol/min)
nEt0 = mols de etileno na entrada
nEts = mols de etileno na saída
nEO = mols de óxido de etileno
nCO2 = mols de dióxido de carbono
SEO = seletividade ao óxido de etileno (%)
REO = rendimento de óxido de etileno (%)
TOFEt = TOF de consumo de etileno (s-1
)
TOFEO = TOF de formação de óxido de etileno (s-1
)
TOFCO2 = TOF de formação de dióxido de carbono (s
-1)
4. RESULTADOS E DISCUSSÃO 29
Capítulo 4 – RESULTADOS E DISCUSSÃO
4.1 – CARACTERIZAÇÃO DOS CATALISADORES
4.1.1 - Espectroscopia de Absorção Atômica (AAS)
A Tabela 4.1 apresenta os resultados da análise de AAS dos catalisadores de prata
suportados em diferentes materiais. Esses resultados demonstram que os teores de prata
metálica nos diferentes catalisadores se aproximaram do valor nominal desejado (15%).
Tabela 4.1 – Teor metálico de prata nos catalisadores, obtido por AAS.
Catalisadores % Ag
Ag/α-Al2O
3 12,1
Ag/ZrO2 12,6
Ag/MgO 13,2
4.1.2 – Análise Textural
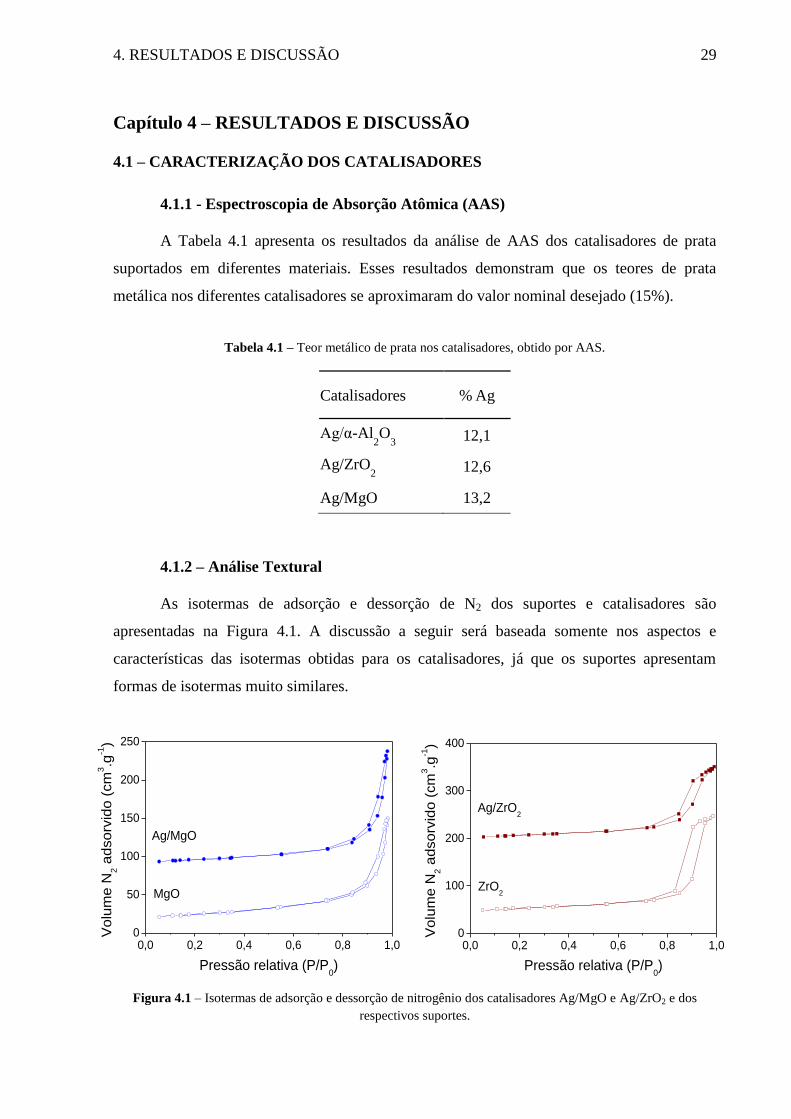
As isotermas de adsorção e dessorção de N2 dos suportes e catalisadores são
apresentadas na Figura 4.1. A discussão a seguir será baseada somente nos aspectos e
características das isotermas obtidas para os catalisadores, já que os suportes apresentam
formas de isotermas muito similares.
Figura 4.1 – Isotermas de adsorção e dessorção de nitrogênio dos catalisadores Ag/MgO e Ag/ZrO2 e dos
respectivos suportes.
0,0 0,2 0,4 0,6 0,8 1,00
50
100
150
200
250
MgO
Vo
lum
e N
2 a
dso
rvid
o (
cm
3.g
-1)
Pressão relativa (P/P0)
Ag/MgO
0,0 0,2 0,4 0,6 0,8 1,00
100
200
300
400
ZrO2
Volu
me N
2 a
dsorv
ido (
cm
3.g
-1)
Pressão relativa (P/P0)
Ag/ZrO2

4. RESULTADOS E DISCUSSÃO 30
O catalisador de prata suportado em MgO apresentou isoterma de adsorção do tipo IV,
que é caracterizada pelo ponto de inflexão em baixas pressões relativas e um patamar em altas
pressões relativas, que corresponde ao preenchimento de todos os poros, com adsorvido no
estado líquido. Esse tipo de isoterma é observado em sólidos contendo mesoporos ou
materiais mesoporosos ordenados de geometria que permite a ocorrência de condensação
capilar a pressões relativas inferiores à unidade (por exemplo, poros cilíndricos, cônicos, em
forma de tinteiro, entre outros). Sua isoterma apresentou um ponto de inflexão não bem
definido (FIGUEIREDO e RIBEIRO, 2007; CONDON, 2006). Apesar da possível presença
de mesoporos, nessa isoterma é observado loop de histerese do tipo H3 muito discreto, que é
caracterizado por dois ramos assintóticos quase verticais em pressões relativas próximas da
unidade, e está associado à agregados não rígidos de partículas em forma de placa, originando
poros em fenda. Nesse tipo de poro não há condensação capilar e, portanto, o material apenas
retém o adsorvido pelo mecanismo de adsorção em multicamadas. Portanto, histereses do tipo
III não exibem nenhuma limitação de adsorção a altas pressões relativas, indicando que o
adsorvente não possui uma estrutura mesoporosa (ERTL et al., 1997; SING, 1982). Dessa
forma, não é aconselhado derivar a distribuição de tamanho de poro a partir desse tipo de
isoterma.
O catalisador de prata suportado em ZrO2 também apresentou isoterma de adsorção do
tipo IV, com um ponto de inflexão não bem definido. Segundo a classificação da IUPAC, o
loop de histerese do tipo H1 é caracterizado por dois ramos quase paralelos da isoterma, e
normalmente está associado a materiais porosos constituídos por aglomerados rígidos de
partículas esféricas com uma distribuição de mesoporos estreita e relativamente uniforme.
Além disso, os ramos praticamente paralelos das isotermas de adsorção e de dessorção serão
mais verticais quanto mais estreita for a distribuição de tamanho dos mesoporos. Nessa
isoterma o loop de histerese observado a pressões relativas superiores a 0,9 indica a presença
de pseudo-mesoporos que são formados pelo empacotamento das partículas (SING, 1982).
A distribuição de poros do catalisador Ag/ZrO2 e do respectivo suporte são
apresentadas na Figura 4.2. Para calcular a distribuição de poros a partir das isotermas de
fisissorção utilizou-se o método BJH. Ao adotar a abordagem desse método é necessário
assumir certas limitações como, por exemplo, o fato da equação de Kelvin ser aplicável
somente na região de mesoporos (2 a 50 nm). Ou seja, as regiões de microporos e macroporos
não devem ser utilizadas no cálculo de distribuição de poros por esse método. Dessa forma,
não são apresentadas as curvas de distribuição dos catalisadores e suportes que, a partir das

4. RESULTADOS E DISCUSSÃO 31
isotermas, não exibiram características mesoporosas. Também é necessário destacar que as
curvas de distribuição foram calculadas a partir do ramo de dessorção.
Figura 4.2 – Distribuição de tamanho de poros das amostras Ag/ZrO2 e ZrO2.
As curvas da Figura 4.2 apresentaram uma distribuição de poros na faixa de 2 a 30 nm
para os materiais Ag/ZrO2 e ZrO2, e confirmaram a presença de mesoporos nas estruturas dos
materiais, conforme havia sido observado a partir das isotermas. Os materiais ZrO2 e
Ag/ZrO2, possuem em sua estrutura mesoporos com diâmetro médio de aproximadamente 15
e 14 nm, respectivamente.
As propriedades texturais dos suportes e dos catalisadores de prata são apresentadas na
Tabela 4.2.
Tabela 4.2 – Área específica (SBET), volume de poros (Vp) e diâmetro médio de poros (dp) dos catalisadores e
suportes utilizados.
Materiais Amostras SBET (m²/g)
Vp (cm3/g) dp (nm)
Catalisadores
Ag/α-Al2O
3 < 10 - -
Ag/ZrO2 58 0,25 14,0
Ag/MgO 41 - -
Suportes
α-Al2O
3 < 10 - -
ZrO2 65 0,33 14,2
MgO 58 - -
0 10 20 30 40 50 60 70 80 90 100
ZrO2
Volu
me d
e p
oro
s (
cm
3.g
-1)
Diâmetro de poros (nm)
Ag/ZrO2
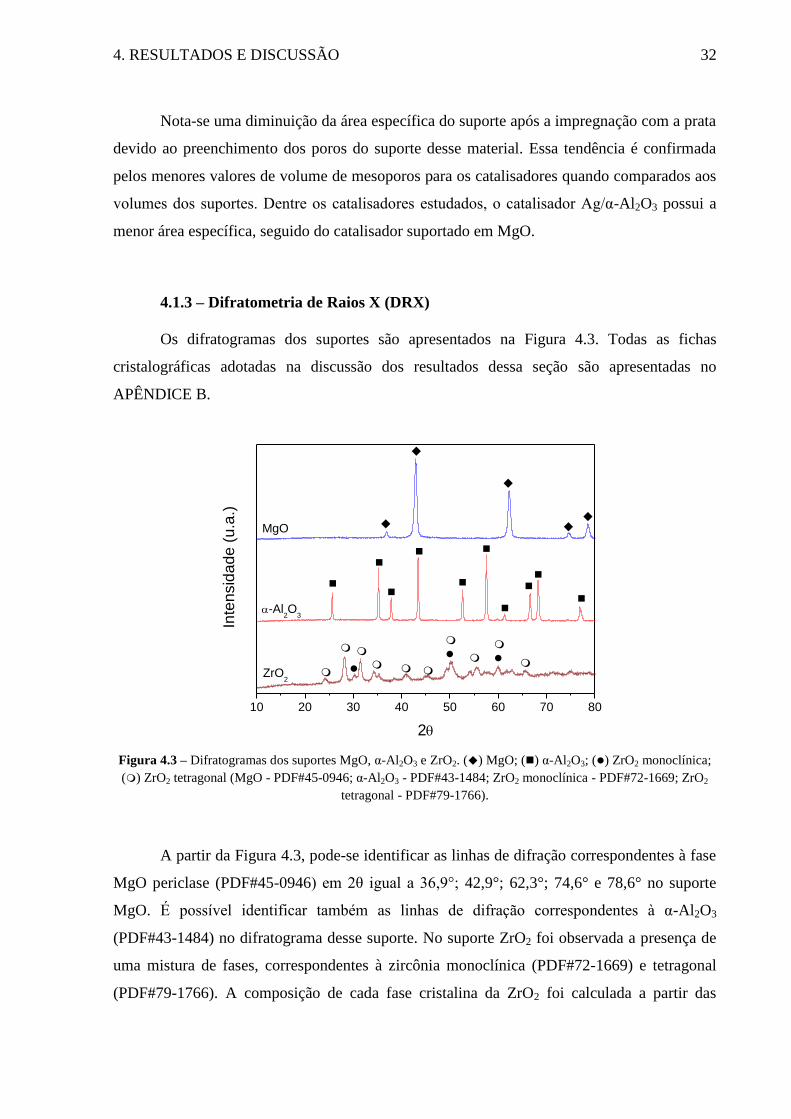
4. RESULTADOS E DISCUSSÃO 32
Nota-se uma diminuição da área específica do suporte após a impregnação com a prata
devido ao preenchimento dos poros do suporte desse material. Essa tendência é confirmada
pelos menores valores de volume de mesoporos para os catalisadores quando comparados aos
volumes dos suportes. Dentre os catalisadores estudados, o catalisador Ag/α-Al2O3 possui a
menor área específica, seguido do catalisador suportado em MgO.
4.1.3 – Difratometria de Raios X (DRX)
Os difratogramas dos suportes são apresentados na Figura 4.3. Todas as fichas
cristalográficas adotadas na discussão dos resultados dessa seção são apresentadas no
APÊNDICE B.
Figura 4.3 – Difratogramas dos suportes MgO, α-Al2O3 e ZrO2. () MgO; () α-Al2O3; () ZrO2 monoclínica;
() ZrO2 tetragonal (MgO - PDF#45-0946; α-Al2O3 - PDF#43-1484; ZrO2 monoclínica - PDF#72-1669; ZrO2
tetragonal - PDF#79-1766).
A partir da Figura 4.3, pode-se identificar as linhas de difração correspondentes à fase
MgO periclase (PDF#45-0946) em 2θ igual a 36,9°; 42,9°; 62,3°; 74,6° e 78,6° no suporte
MgO. É possível identificar também as linhas de difração correspondentes à α-Al2O3
(PDF#43-1484) no difratograma desse suporte. No suporte ZrO2 foi observada a presença de
uma mistura de fases, correspondentes à zircônia monoclínica (PDF#72-1669) e tetragonal
(PDF#79-1766). A composição de cada fase cristalina da ZrO2 foi calculada a partir das
10 20 30 40 50 60 70 80
-Al2O
3
ZrO2
Inte
nsid
ad
e (
u.a
.)
2
MgO

4. RESULTADOS E DISCUSSÃO 33
alturas (h) e larguras à meia altura (β) dos picos característicos das fases monoclínica (2 =
28,2°e 31,5°) e tetragonal (2 = 30,2°), conforme as equações (1) e (2) apresentadas no item
3.2.2. De acordo com os resultados dessas equações aplicadas ao difratograma da amostra
ZrO2 nota-se que a fase predominante na estrutura da zircônia é a fase monoclínica
(aproximadamente 87%).
Os difratogramas dos catalisadores estão apresentados na Figura 4.4. Nos
difratogramas de todos os catalisadores são observadas as principais linhas de difração
correspondentes à prata metálica (PDF#04-0783) em 2θ igual a aproximadamente 38º, 44º,
64º e 77º. Essas linhas são relativas aos respectivos planos cristalinos (111), (200), (220) e
(311). Foram detectadas, também, as fases cristalinas relativas aos suportes como pode ser
observado nos difratogramas dos suportes puros. Como é possível observar pelas linhas de
difração bem definidas e estreitas dos suportes e da prata metálica, os catalisadores suportados
em MgO e α-Al2O3 possuem estruturas altamente cristalinas. Já o catalisador suportado em
ZrO2 exibe estrutura menos cristalina.
Figura 4.4 – Difratogramas dos catalisadores Ag/MgO, Ag/α-Al2O3 e Ag/ZrO2. (- - - -) Ag metálica; () MgO;
() α-Al2O3; () ZrO2 monoclínica e () ZrO2 tetragonal (Ag- PDF#04-0783).
Os diâmetros médios dos cristalitos de Ag0 foram calculados a partir da equação de
Scherrer, considerando a linha de difração de maior intensidade em 2θ igual a
aproximadamente 38°, referente ao plano cristalino (111) da prata metálica. Os resultados
estão apresentados na Tabela 4.3.
10 20 30 40 50 60 70 80
2
Ag/ZrO2
Ag/-Al2O
3
Ag/MgO
Inte
nsid
ad
e (
u.a
.)
Ag/ZrO2
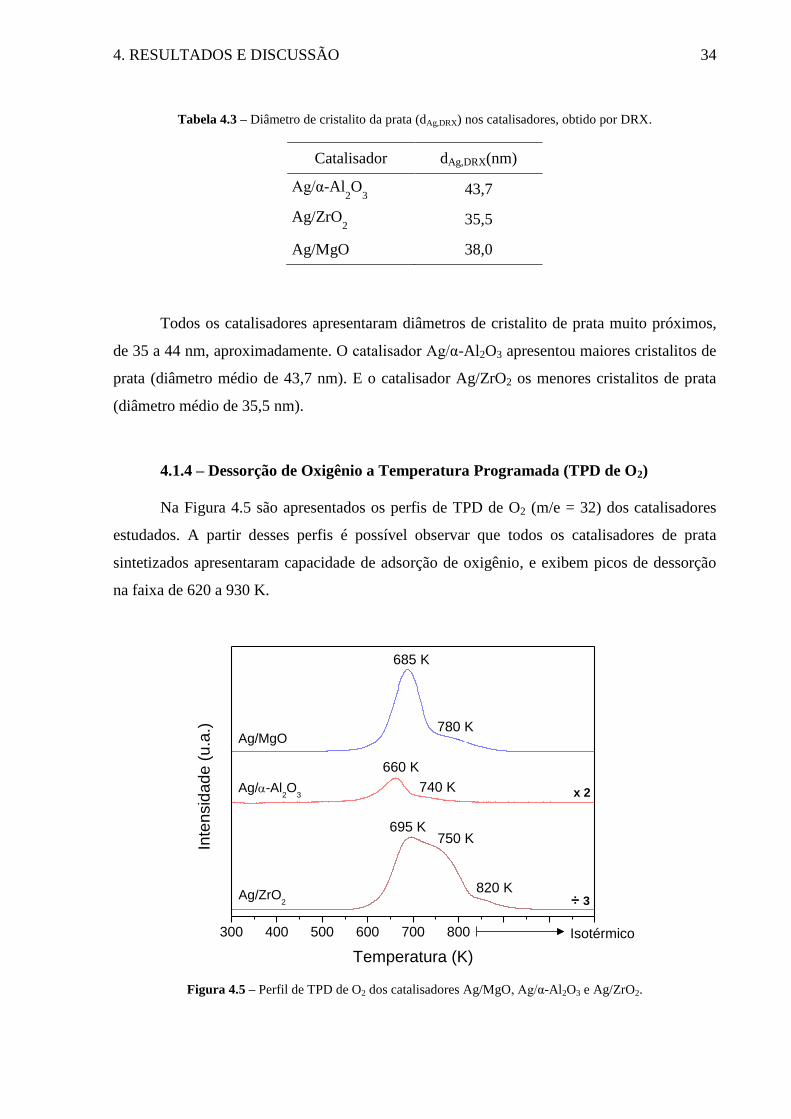
4. RESULTADOS E DISCUSSÃO 34
Tabela 4.3 – Diâmetro de cristalito da prata (dAg,DRX) nos catalisadores, obtido por DRX.
Catalisador dAg,DRX(nm)
Ag/α-Al2O
3 43,7
Ag/ZrO2 35,5
Ag/MgO 38,0
Todos os catalisadores apresentaram diâmetros de cristalito de prata muito próximos,
de 35 a 44 nm, aproximadamente. O catalisador Ag/α-Al2O3 apresentou maiores cristalitos de
prata (diâmetro médio de 43,7 nm). E o catalisador Ag/ZrO2 os menores cristalitos de prata
(diâmetro médio de 35,5 nm).
4.1.4 – Dessorção de Oxigênio a Temperatura Programada (TPD de O2)
Na Figura 4.5 são apresentados os perfis de TPD de O2 (m/e = 32) dos catalisadores
estudados. A partir desses perfis é possível observar que todos os catalisadores de prata
sintetizados apresentaram capacidade de adsorção de oxigênio, e exibem picos de dessorção
na faixa de 620 a 930 K.
Figura 4.5 – Perfil de TPD de O2 dos catalisadores Ag/MgO, Ag/α-Al2O3 e Ag/ZrO2.
300 400 500 600 700 800 900 1000 1100
x 2740 K
660 K
Ag/-Al2O
3
÷ 3Ag/ZrO2
Inte
nsid
ad
e (
u.a
.)
Temperatura (K)
Isotérmico
695 K750 K
820 K
Ag/MgO
685 K
780 K

4. RESULTADOS E DISCUSSÃO 35
Nos perfis de todos os catalisadores estudados foram observados picos de dessorção na
faixa de 620-690 K. Os perfis dos catalisadores suportados em MgO e α-Al2O3 apresentam
ombros discretos em torno de 780 e 740K, respectivamente. Já o catalisador suportado em
ZrO2 apresentou um pico de dessorção de O2 intenso em torno de 740 K, e nota-se ainda a
presença de um pico/ombro em torno de 820 a 860 K.
Estudos de TPD de O2 em superfícies modelo de monocristais de Ag(110) reportaram
picos de dessorção de O2 em temperaturas iguais a 200 K e 590 K, que foram atribuídos ao
oxigênio molecular e ao oxigênio atômico, respectivamente (BARTEAU e MADIX, 1980).
Na superfície da Ag (111) foi observado um pico em 579 K, atribuído ao oxigênio atômico
(CAMPBELL, 1985). Bal’zhinimaev (1999) observou dois picos no perfil de TPD de O2 em
temperaturas próximas a 580 e a 690 K. Por comparação com estudos de XPS, Bal’zhinimaev
(1999) atribuiu o pico em menores temperaturas ao oxigênio nucleofílico nos sítios de Ag em
terraços, e o pico em maiores temperaturas ao oxigênio eletrofílico, ou seja, o oxigênio
efetivamente epoxidante presente nos defeitos ou degraus da prata.
Ao realizar um estudo baseado na interação do oxigênio com uma folha cristalina de
prata em diferentes temperaturas, Bukhtiyarov e colaboradores (1994) identificaram o
aparecimento de uma forma de oxigênio dissolvida na matriz da prata (oxigênio
subsuperficial) em análises de TPD em temperaturas superiores a 420 K. No experimento
realizado na temperatura de 520 K, a dessorção dessa espécie foi observada a
aproximadamente 620 K. Com o aumento da temperatura para 620 K, foi reportado um
deslocamento do pico de dessorção dessa espécie de oxigênio para 750 K. Em um estudo de
TPD de O2 em catalisador Ag/α-Al2O3, Muhler e colaboradores (2002) identificaram um pico
de dessorção em aproximadamente 542 K e uma análise mais detalhada revelou ombros
discretos em 570 K. Segundo os autores, os picos em temperaturas mais elevadas indicam a
presença de um segundo tipo de sítio de adsorção que corresponde, provavelmente, ao
oxigênio subsuperficial, que resulta da difusão das espécies oxigênio para o interior da
partícula de Ag.
Na Tabela 4.4 estão listadas as quantidades de oxigênio dessorvido que foram
calculadas através da decomposição das áreas obtidas dos perfis de TPD de O2 (apresentados
no APÊNCIDE C).
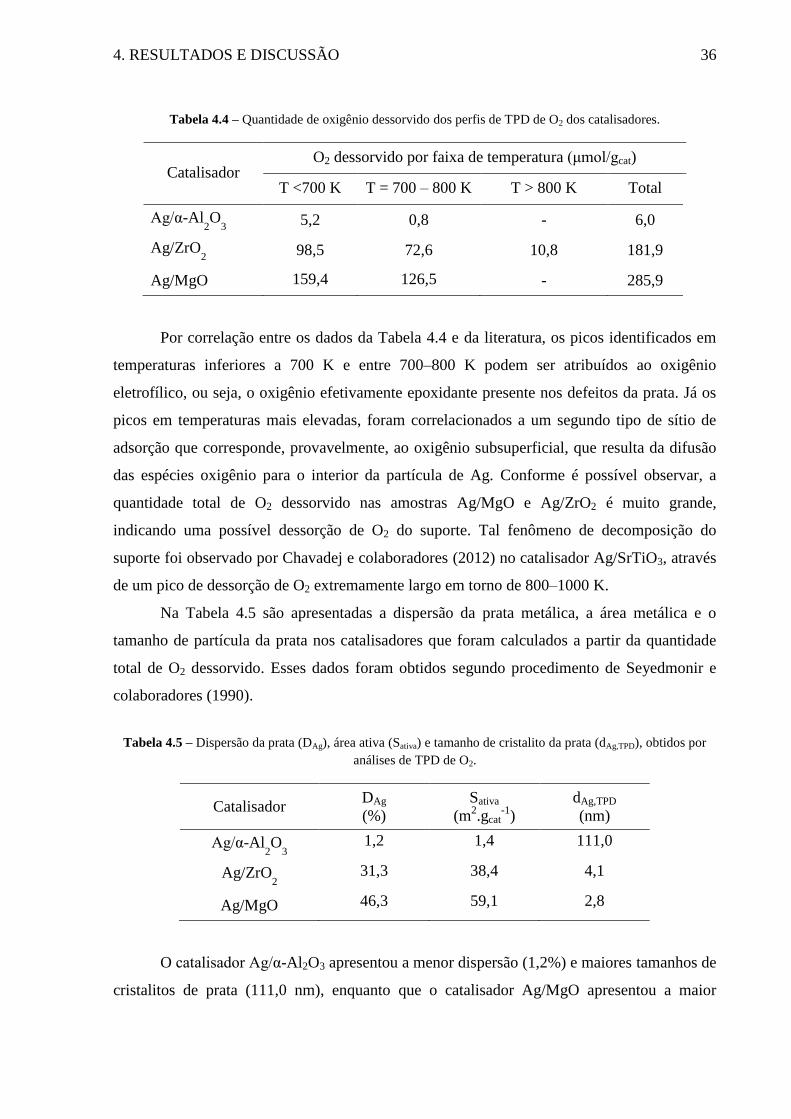
4. RESULTADOS E DISCUSSÃO 36
Tabela 4.4 – Quantidade de oxigênio dessorvido dos perfis de TPD de O2 dos catalisadores.
Catalisador O2 dessorvido por faixa de temperatura (μmol/gcat)
T <700 K T = 700 – 800 K T > 800 K Total
Ag/α-Al2O
3 5,2 0,8 - 6,0
Ag/ZrO2 98,5 72,6 10,8 181,9
Ag/MgO 159,4 126,5 - 285,9
Por correlação entre os dados da Tabela 4.4 e da literatura, os picos identificados em
temperaturas inferiores a 700 K e entre 700–800 K podem ser atribuídos ao oxigênio
eletrofílico, ou seja, o oxigênio efetivamente epoxidante presente nos defeitos da prata. Já os
picos em temperaturas mais elevadas, foram correlacionados a um segundo tipo de sítio de
adsorção que corresponde, provavelmente, ao oxigênio subsuperficial, que resulta da difusão
das espécies oxigênio para o interior da partícula de Ag. Conforme é possível observar, a
quantidade total de O2 dessorvido nas amostras Ag/MgO e Ag/ZrO2 é muito grande,
indicando uma possível dessorção de O2 do suporte. Tal fenômeno de decomposição do
suporte foi observado por Chavadej e colaboradores (2012) no catalisador Ag/SrTiO3, através
de um pico de dessorção de O2 extremamente largo em torno de 800–1000 K.
Na Tabela 4.5 são apresentadas a dispersão da prata metálica, a área metálica e o
tamanho de partícula da prata nos catalisadores que foram calculados a partir da quantidade
total de O2 dessorvido. Esses dados foram obtidos segundo procedimento de Seyedmonir e
colaboradores (1990).
Tabela 4.5 – Dispersão da prata (DAg), área ativa (Sativa) e tamanho de cristalito da prata (dAg,TPD), obtidos por
análises de TPD de O2.
Catalisador DAg
(%)
Sativa
(m2.gcat
-1)
dAg,TPD
(nm)
Ag/α-Al2O
3 1,2 1,4 111,0
Ag/ZrO2 31,3 38,4 4,1
Ag/MgO 46,3 59,1 2,8
O catalisador Ag/α-Al2O3 apresentou a menor dispersão (1,2%) e maiores tamanhos de
cristalitos de prata (111,0 nm), enquanto que o catalisador Ag/MgO apresentou a maior

4. RESULTADOS E DISCUSSÃO 37
dispersão da prata (46,3%) e menores tamanhos de cristalitos de prata (2,8 nm). Na literatura
foram estudados catalisadores suportados em α-alumina com teores de prata em torno de 15%
com valores de dispersão da Ag bastante semelhantes ao obtido nesse trabalho, em torno de
1,5% (SCHMAL et al., 2007; SEYEDMONIR et al., 1990). Em relação aos demais
catalisadores, não há na literatura dados de dispersão ou tamanho de partícula de prata para
comparação dos resultados obtidos. Deve-se ressaltar que para os catalisadores suportados em
MgO e ZrO2 os valores de dispersão, área ativa e tamanho de cristalito da prata foram
superestimados, já que na quantidade total de O2 dessorvido dessas amostras há a contribuição
da quantidade de O2 proveniente do suporte.
Há uma diferença significativa entre os valores obtidos para os diâmetros de cristalito
por TPD de O2 e por DRX. Acredita-se que essa diferença pode ser explicada pela baixa
sensibilidade da técnica de difração de raios X, em que partículas inferiores a 2 nm e
superiores a 50 nm não são detectadas. Ou seja, caso os catalisadores não possuam uma
distribuição de tamanho de cristalito estreita, o cálculo do diâmetro de cristalito pela equação
de Scherrer não é preciso.
4.2 – TESTES CATALÍTICOS
O efeito da natureza do suporte no desempenho de catalisadores de prata na reação de
epoxidação do etileno foi investigado empregando-se uma série de suportes com propriedades
distintas, os resultados obtidos são listados na Tabela 4.6.
Tabela 4.6 – Comportamento catalítico dos catalisadores suportados de prata: taxa (r) de consumo de etileno e
de formação dos produtos e TOF (C2H4/O2 = 0,5; vazão = 42 mL/min e mcat = 100 mg).
Catalisadores T
(K)
XEt
(%)
r x 10-4
(mol.min-1
.gAg-1
)
TOF x 10-2
(s-1
) SEO
(%)
REO
(%) C2H4 EO CO2 C2H4 EO CO2
Ag/α-Al2O
3
498
523
548
3,5
7,3
13,7
3,0
6,3
11,8
0,5
1,0
1,5
1,9
4,2
8,3
9,4
19,3
36,1
1,7
3,2
4,7
6,0
12,8
25,6
28,2
24,7
18,5
18,0
16,4
13,4
Ag/ZrO2
498
523
548
8,7
64,5
60,6
23,8
176
165
0,6
0,07
0,1
24,5
180
168
2,7
20,1
19,0
≈ 0
2,8
20,6
19,2
2,5
-
-
2,5
-
-
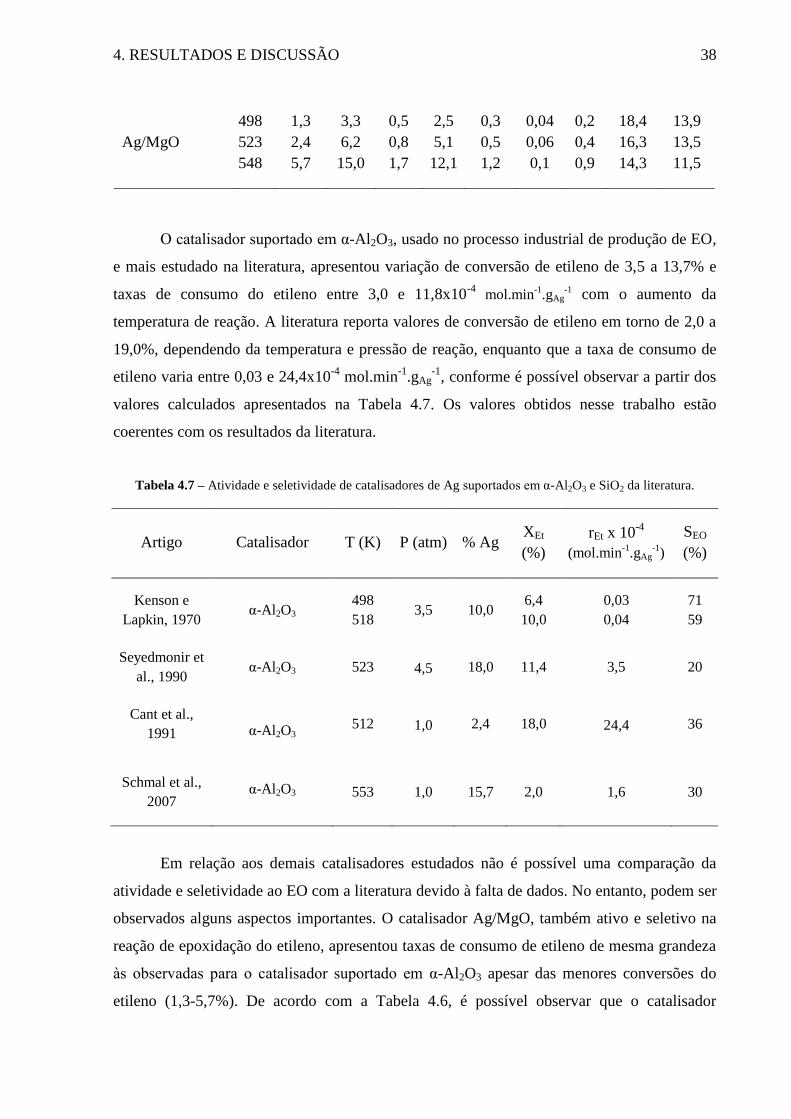
4. RESULTADOS E DISCUSSÃO 38
Ag/MgO
498
523
548
1,3
2,4
5,7
3,3
6,2
15,0
0,5
0,8
1,7
2,5
5,1
12,1
0,3
0,5
1,2
0,04
0,06
0,1
0,2
0,4
0,9
18,4
16,3
14,3
13,9
13,5
11,5
O catalisador suportado em α-Al2O3, usado no processo industrial de produção de EO,
e mais estudado na literatura, apresentou variação de conversão de etileno de 3,5 a 13,7% e
taxas de consumo do etileno entre 3,0 e 11,8x10-4
mol.min-1
.gAg-1 com o aumento da
temperatura de reação. A literatura reporta valores de conversão de etileno em torno de 2,0 a
19,0%, dependendo da temperatura e pressão de reação, enquanto que a taxa de consumo de
etileno varia entre 0,03 e 24,4x10-4
mol.min-1
.gAg-1
, conforme é possível observar a partir dos
valores calculados apresentados na Tabela 4.7. Os valores obtidos nesse trabalho estão
coerentes com os resultados da literatura.
Tabela 4.7 – Atividade e seletividade de catalisadores de Ag suportados em α-Al2O3 e SiO2 da literatura.
Artigo Catalisador T (K) P (atm) % Ag XEt
(%)
rEt x 10-4
(mol.min-1
.gAg-1
)
SEO
(%)
Kenson e
Lapkin, 1970 α-Al2O3
498
518 3,5 10,0
6,4
10,0
0,03
0,04
71
59
Seyedmonir et
al., 1990 α-Al2O3 523 4,5 18,0 11,4 3,5 20
Cant et al.,
1991 α-Al2O3 512 1,0 2,4 18,0 24,4 36
Schmal et al.,
2007 α-Al2O3 553 1,0 15,7 2,0 1,6 30
Em relação aos demais catalisadores estudados não é possível uma comparação da
atividade e seletividade ao EO com a literatura devido à falta de dados. No entanto, podem ser
observados alguns aspectos importantes. O catalisador Ag/MgO, também ativo e seletivo na
reação de epoxidação do etileno, apresentou taxas de consumo de etileno de mesma grandeza
às observadas para o catalisador suportado em α-Al2O3 apesar das menores conversões do
etileno (1,3-5,7%). De acordo com a Tabela 4.6, é possível observar que o catalisador
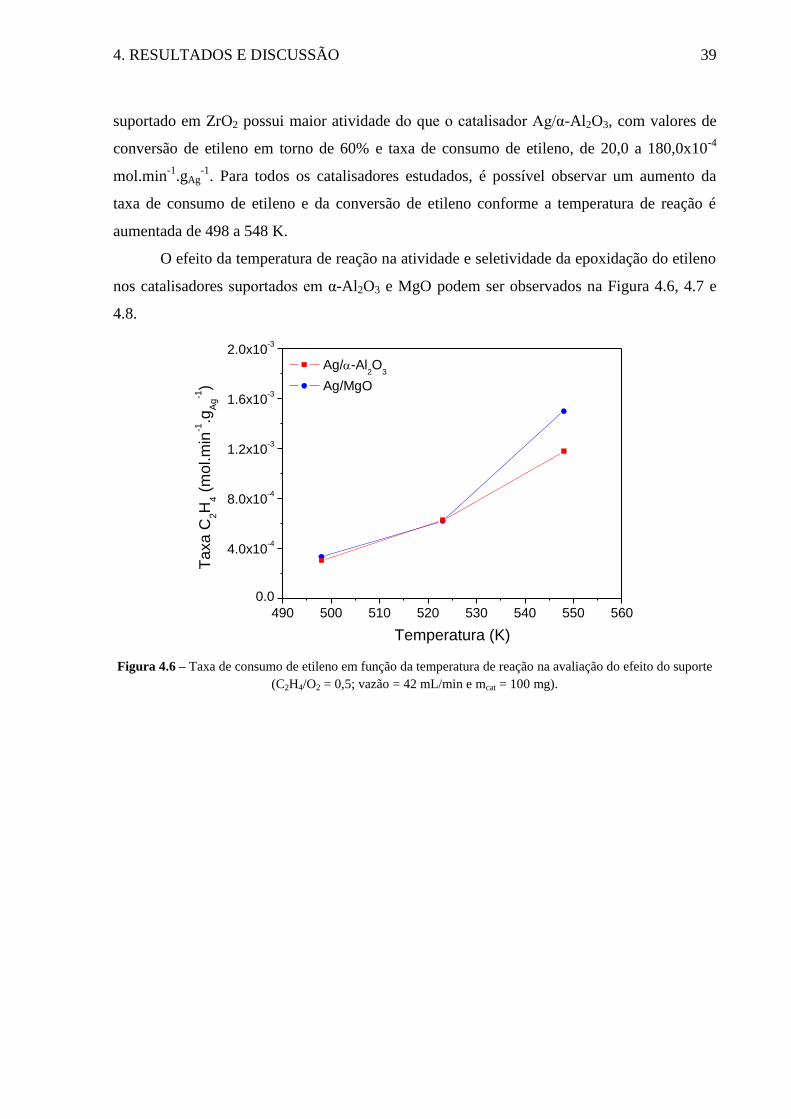
4. RESULTADOS E DISCUSSÃO 39
suportado em ZrO2 possui maior atividade do que o catalisador Ag/α-Al2O3, com valores de
conversão de etileno em torno de 60% e taxa de consumo de etileno, de 20,0 a 180,0x10-4
mol.min-1
.gAg-1
. Para todos os catalisadores estudados, é possível observar um aumento da
taxa de consumo de etileno e da conversão de etileno conforme a temperatura de reação é
aumentada de 498 a 548 K.
O efeito da temperatura de reação na atividade e seletividade da epoxidação do etileno
nos catalisadores suportados em α-Al2O3 e MgO podem ser observados na Figura 4.6, 4.7 e
4.8.
Figura 4.6 – Taxa de consumo de etileno em função da temperatura de reação na avaliação do efeito do suporte
(C2H4/O2 = 0,5; vazão = 42 mL/min e mcat = 100 mg).
490 500 510 520 530 540 550 560
0.0
4.0x10-4
8.0x10-4
1.2x10-3
1.6x10-3
2.0x10-3
Ta
xa
C2H
4 (
mo
l.m
in-1.g
Ag
-1)
Temperatura (K)
Ag/-Al2O
3
Ag/MgO
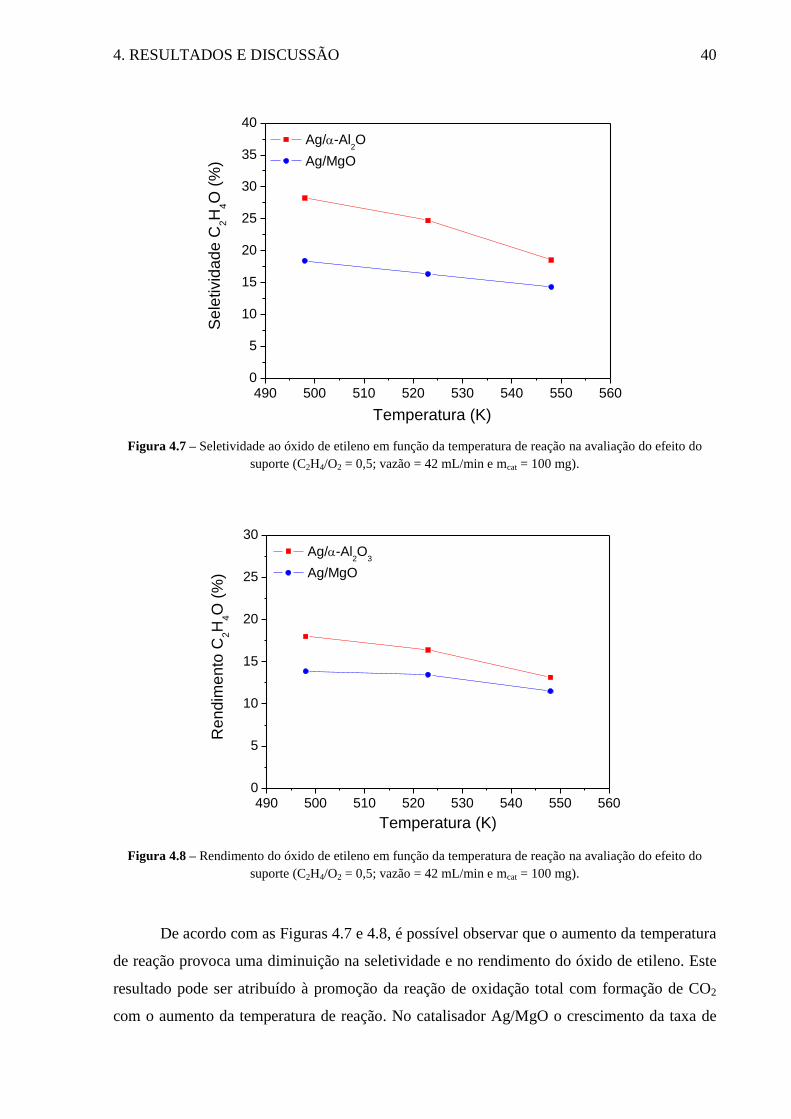
4. RESULTADOS E DISCUSSÃO 40
Figura 4.7 – Seletividade ao óxido de etileno em função da temperatura de reação na avaliação do efeito do
suporte (C2H4/O2 = 0,5; vazão = 42 mL/min e mcat = 100 mg).
Figura 4.8 – Rendimento do óxido de etileno em função da temperatura de reação na avaliação do efeito do
suporte (C2H4/O2 = 0,5; vazão = 42 mL/min e mcat = 100 mg).
De acordo com as Figuras 4.7 e 4.8, é possível observar que o aumento da temperatura
de reação provoca uma diminuição na seletividade e no rendimento do óxido de etileno. Este
resultado pode ser atribuído à promoção da reação de oxidação total com formação de CO2
com o aumento da temperatura de reação. No catalisador Ag/MgO o crescimento da taxa de
490 500 510 520 530 540 550 5600
5
10
15
20
25
30
35
40
Ag/-Al2O
Ag/MgO
Se
letivid
ad
e C
2H
4O
(%
)
Temperatura (K)
490 500 510 520 530 540 550 5600
5
10
15
20
25
30
Ag/-Al2O
3
Ag/MgO
Re
nd
ime
nto
C2H
4O
(%
)
Temperatura (K)

4. RESULTADOS E DISCUSSÃO 41
consumo de etileno apresenta uma maior variação de 523 K (6,2x10-4
mol.min-1
.gAg-1
) para
548 K (15,0x10-4
mol.min-1
.gAg-1
). O catalisador Ag/α-Al2O3 se mostrou mais seletivo ao EO,
promovendo também uma reação com maior rendimento dentre os catalisadores estudados.
Na menor temperatura de reação (498 K), foi reportada a maior seletividade ao óxido de
etileno (28%) e o maior rendimento (18%). Na literatura, são reportadas seletividades ao EO
em torno de 20-30% em catalisadores de Ag/α-Al2O3 para reação à pressão atmosférica. Cant
e colaboradores (1991) reportaram seletividades ao EO em torno de 36% para o catalisador
2% Ag/α-Al2O3 em condições de 1,0 atm e 523 K (Tabela 4.7). Chavadej e colaboradores
(2012) reportaram seletividades ao EO muito altas, em torno de 77-98% para o catalisador
suportado em α-Al2O3 para reações de 498-548 K e a 1,7 atm, enquanto que o aumento da
temperatura para 563 K provocou uma diminuição drástica da seletividade para 38%
(CHAVADEJ et al, 2012). Deve-se destacar, no entanto, que os artigos apresentam cálculo de
seletividade a partir da divisão de concentração do EO pela concentração dos produtos (EO e
CO2), enquanto que em nosso estudo a seletividade é calculada a partir das taxas de formação
dos produtos. Ademais, muitos trabalhos (p.ex. Chavadej e colaboradores, 2012) não
apresentam a definição utilizada nos cálculos de seletividade. Além disso, na literatura
geralmente não são apresentados dados de rendimento do EO. Chavadej e colaboradores
(2012) reportam rendimentos em torno de 3% para um catalisador suportado em α-Al2O3,
valores muito inferiores aos rendimentos apresentados no presente trabalho. No entanto, uma
comparação direta não pode ser feita já que os autores não apresentam a fórmula utilizada no
cálculo desses valores.
No catalisador suportado em ZrO2, é possível observar a partir da Tabela 4.6
seletividades muito baixas ao EO. O aumento da temperatura de reação provoca o aumento
drástico da taxa de consumo de etileno, sendo observada uma forte liberação de calor (± 20-
30 K de diferença da temperatura ajustada), característica de reações exotérmicas. Dessa
forma, esse catalisador apresentou elevada atividade e seletividade na formação do CO2, e
uma atividade muito baixa na reação de produção do EO. Diferentemente dos dados
observados no presente trabalho, Verykios e colaboradores (1988) observaram para uma
seletividade ao EO de 45% para o catalisador 2% Ag/ZrO2 na reação de epoxidação a 508 K.
Conforme é possível observar a partir da Tabela 4.6, o catalisador suportado em α-
Al2O3 apresenta TOF de formação de EO na ordem de 2,0 a 5,0 x 10-2
s-1
. Deve ser notado
que os TOF de formação de EO aqui obtidos possuem uma ordem de magnitude 10 vezes
maior do que a maioria das reações em pressões mais elevadas da literatura, conforme é

4. RESULTADOS E DISCUSSÃO 42
possível ver por comparação com os valores listados na Tabela 4.8. De acordo com esse fato,
os catalisadores sintetizados aqui são mais ativos. Além disso, TOFs em torno de 0,04x10-2
a
0,1x10-2
s-1
foram observados no catalisador Ag/MgO com pequenas partículas de prata (3
nm), ou seja, valores 100 vezes menores do que os observados no catalisador Ag/α-Al2O3 com
partículas de Ag maiores.
Tabela 4.8 – TOF de formação de óxido de etileno listados a partir de avaliação da literatura. (Adaptado de
Seyedmonir et al, 1990)
Artigo Catalisador T (K) P (atm) DAg
(%)
TOFEO
(s-1
)
Campbell,
1985
Ag(110)
Ag (111) 490 - -
2,5
1,9
Grant e
Lambert,
1985
Ag (111) 490 - - 0,1
Verykios et
al., 1988 Ag/α-Al2O3 513 15,0 0,9–2,6 1,0-7,0 x 10
-3
Seyedmonir et
al., 1990 18% Ag/α-Al2O3 523 4,5 0,1 14,9x10
-2
O aumento da temperatura de reação provoca um aumento nos valores dos TOFs de
formação de ambos os produtos, no caso dos catalisadores seletivos ao óxido de etileno (Ag/
α-Al2O3 e Ag/MgO). Nestes catalisadores o aumento de ambos os TOFs de formação dos
produtos são mais ou menos proporcionais. Por exemplo, para o catalisador suportado em α-
Al2O3 há um aumento de 3 vezes para o TOFEO e de 4 vezes para o TOFCO2. Essa mesma
proporcionalidade não é observada para o catalisador não seletivo (Ag/ZrO2), em que o
aumento abrupto do TOF de formação de CO2 não acompanha o aumento do TOFEO. Na
verdade, o aumento de temperatura acarreta na diminuição do TOFEO até um valor nulo para
esse catalisador.
A partir desses resultados, é possível observar que não há uma correlação entre a área
específica do catalisador e seletividade ao produto desejado, como normalmente é proposto na
literatura para diversos catalisadores. De acordo com a literatura (VERYKIOS et al., 1988),

4. RESULTADOS E DISCUSSÃO 43
uma possível explicação para os fenômenos observados pode estar baseada nas diferentes
dispersões dos catalisadores, já que essa reação é conhecida por ser sensível ao tamanho de
cristalito da prata. Altas seletividades ao EO podem ser observadas geralmente em
catalisadores que apresentem cristalitos de prata extremamente grandes suportados em óxidos
anfóteros e de baixa área. De fato, no catalisador suportado em óxidos anfótero (α-Al2O3), que
possui maiores cristalitos de prata, foram obtidas as maiores seletividades ao óxido de etileno
em comparação com os demais materiais estudados. No entanto, essa tendência não é
observada nos óxidos com características ácidas ou básicas como, por exemplo, o catalisador
suportado em ZrO2 que possui alta dispersão da Ag (31,3%) e não é seletivo ao EO, enquanto
que o catalisador suportado em MgO com maior dispersão (46,3%) é seletivo ao óxido de
etileno. De acordo com a literatura (SEYEDMONIR et al., 1990; VERYKIOS et al., 1988), a
presença de sítios ativos no suporte que catalisam a oxidação total do etileno, provavelmente,
devido à reação secundária de oxidação do epóxido de etileno, é uma das razões para baixas
seletividades. Essa tendência é demonstrada na seletividade ao EO nula obtida para o
catalisador suportado em ZrO2, que possui sítios ácidos superficiais. Já o catalisador
suportado em MgO, que não possui em sua composição sítios ácidos provenientes do suporte,
se mostrou seletivo ao EO em todas as temperaturas estudadas. Portanto, catalisadores com
dispersões similares podem apresentar comportamentos divergentes na reação de epoxidação
de etileno. Esse fato pode estar relacionado a uma interação entre a partícula da Ag e a
superfície do suporte que pode afetar, pelo menos parcialmente, o estado de oxidação da
prata.

5. CONCLUSÃO 44
Capítulo 5 – CONCLUSÃO
Foi feita uma comparação e avaliação do desempenho de catalisadores teor de prata de
15% suportados em α-Al2O3, ZrO2 e MgO, visando um estudo do efeito do suporte e da
temperatura na reação de epoxidação do etileno. Os catalisadores sintetizados apresentaram
características bem distintas de dispersão e, por sua vez, de diâmetro de cristalito da prata.
Todos os catalisadores apresentaram um aumento na taxa de consumo de etileno e uma
diminuição da seletividade e rendimento ao EO com o aumento da temperatura. O catalisador
suportado em ZrO2 demonstrou alta atividade catalítica na reação indesejada de formação de
CO2, que pode estar associada a presença de sítios ativos no suporte que catalisam a oxidação
total do etileno, provavelmente devido à reação secundária de oxidação do epóxido de etileno.
Somente os catalisadores suportados em α-Al2O3 e MgO foram seletivos na produção do
óxido de etileno.
Dentre os catalisadores seletivos, o catalisador Ag/α-Al2O3 é o mais promissor na
produção seletiva de óxido de etileno dentre os estudados, apresentando rendimentos desse
produto de até 18% e seletividade ao óxido de etileno de até 28%. Para ambos catalisadores
seletivos, os maiores rendimentos e seletividades ao óxido se deram na menor temperatura de
reação (498 K), diminuindo ligeiramente com o aumento da temperatura. Na reação com o
catalisador Ag/α-Al2O3 foram observados TOFs de formação de epóxido de etileno com uma
ordem de magnitude 10 vezes maior do que os propostos na literatura, demonstrando um
catalisador mais ativo. No caso dos catalisadores seletivos ao óxido de etileno, o aumento da
temperatura de reação provoca um aumento nos valores dos TOFs de formação de ambos os
produtos, tendência não observada para o catalisador Ag/ZrO2. Além disso, TOFs 100 vezes
menores foram observados para as pequenas partículas de prata (3 nm) no catalisador
Ag/MgO, quando compara-se com as partículas de Ag maiores no catalisador suportado em α-
Al2O3 (111 nm).
Comparando-se os catalisadores com propriedades ácidas (Ag/ZrO2) ou básicas
(Ag/MgO) foi possivel observar que o catalisador suportado em MgO, que não possui em sua
composição sítios ácidos provenientes do suporte, se mostrou seletivo ao EO em todas as
temperaturas estudadas. Enquanto o catalisador suportado em ZrO2, que possui sítios
superficiais ácidos, demonstrou seletividade ao EO nula. Conforme explicitado anteriormente,
uma interação entre a partícula da Ag e a superfície do suporte pode afetar, pelo menos
parcialmente, o estado de oxidação da prata.

6. REFERÊNCIAS BIBLIOGRÁFICAS 45
Capítulo 6 – REFERÊNCIAS BIBLIOGRÁFICAS
AKELLA, LAKS M.; LEE, HONG H.Selectivity Characterization of Ethylene Oxidation
Reactions: Oxygen Chemisorption. Journal of Catalysis, v. 86, p. 465-472, 1984.
AVDEEV, VASILII I.; BORONIN, ANDREI I.; KOSCHEEV, SERGEI V.; ZHIDOMIROV,
GEORGII M. Quasimolecular stable forms of oxygen on silver surface. Theoretical analysis
by density functional theory method. Journal of Molecular Catalysis A: Chemical. v. 154, p.
257-270, 2000.
BAL’ZHINIMAEV, B. S. Ethylene Epoxidation over Silver Catalysts. Kinetics and Catalysis,
v. 40, n. 6, p. 195-810, 1999.
BARTEAU, Mark A.; LINIC, Suljo. Construction of a reaction coordinate and a microkinetic
model for ethylene epoxidation on silver from DFT calculations and surface science
experiments. Journal of Catalysis, v. 214, p. 200-212, 2003.
BARTEAU, Mark A.; LINIC, Suljo. Formation of a Stable Surface Oxametallacycle that
Produces Ethylene Oxide. Journal of the American Chemical Society, v.124, p. 310-317,
2002.
BARTEAU, M. A.; MADIX, R. J. The Adsorption of Molecular Oxygen Species on Ag
(110). Surface Science, v. 97, p. 101-110, 1980.
BECK, J. S.; VARTULI, J. C.; ROTH, W. J.; LEONOWICZ, M. E.; KRESGE, C. T.;
SCHMITT, K. D.; CHU, C. T-W.; OLSON, D. H.; SHEPPARD, E. W.; MCCULLEN, S. B.;
HIGGINS, J. B.; SCHLENKERT, J. L. A New Family of Mesoporous Molecular Sieves
Prepared with Liquid Crystal Templates. Journal of the American Chemical Society, v. 114, p.
10834-10843, 1992.
BUKHTIYAROV, V. L.; BORONIN, A. I.; SAVCHENKO, V. I. Stages in the Modification
of a Silver Surface for Catalysis of the Partial Oxidation of Ethylene. I. Action of Oxygen.
Journal of Catalysis, v. 150, p. 262-267, 1994.
CAMPBELL, Charles T. The Selective Epoxidation of Ethylene Catalyzed by Ag(111): A
Comparison with Ag(110). Journal of Catalysis, v. 94, p. 436-444, 1985.
CANT, Noel W.; YONG, You-Sing; KENNEDY, Eric M. Oxide catalysed reactions of
ethylene oxide under conditions relevant to ethylene epoxidation over supported silver.
Applied Catalysis, v. 76, p. 31-48, 1991.

6. REFERÊNCIAS BIBLIOGRÁFICAS 46
CARVALHO, Marta Cristina N. Amorim. Estudo da Fase Ativa do Catalisador de Prata para
a Oxidação de Etileno. Rio de Janeiro, 2005. Tese (Doutorado em Engenharia Química),
Universidade Federal do Rio de Janeiro.
CHAVADEJ, Sumaeth; CHONGTERDTONNSKULA, Atiporn; SCHWANK, Johannes W.
Schwank. Effects of oxide supports on ethylene epoxidation activity over Ag-based catalysts.
Journal of Molecular Catalysis A: Chemical, v. 358, p. 58– 66, 2012.
CHAVADEJ, Sumaeth; ROJLUECHAI, Siriphong; SCHWANK, Johannes W.; MEEYOO,
Vissanu. Catalytic activity of ethylene oxidation over Au, Ag and Au–Ag catalysts: Support
effect. Catalysis Communications, v. 8, p. 57–64, 2007.
CONDON, James B. Surface Area and Porosity Determinations by Physisorption –
Measurements and Theory, 1st Edition, UK: Elsevier, 2006, 274 p.
ERTL, G.; KNÖZINGER, H; WEITKAMP, J. Handbook of Heterogeneous Catalysis,
Volume 2, Germany: VCH Verlagsgesellschaft mdH, 1997, 910 p.
FELLAH, Mehmet F.; VAN SANTEN, Rutger A.; ONAL, Isik. Epoxidation of Ethylene by
Silver Oxide (Ag2O) Cluster: A Density Functional Theory Study. Catalysis Letters, v. 141, p.
762-771, 2011.
FIGUEIREDO, José Luís; RIBEIRO, Fernando Ramôa. Catálise Heterogênea, 2ª Edição,
Lisboa: Fundação Calouste Gulbenkian, 2007, 352 p.
FOTOPOULOS, Apostolos P.; TRIANTAFYLLIDIS, Kostas S. Ethylene epoxidation on Ag
catalysts supported on non-porous, microporous and mesoporous silicates. Catalysis Today, v.
127, p. 148-156, 2007.
GAO, Zhiming; SHI, Yingxiao. Suppressed formation of CO2 and H2O in the oxidative
coupling of methane over La2O3/MgO catalyst by surface modification. Journal of Natural
Gas Chemistry, v. 19, p. 173–178, 2010.
GRANT, ROBERT B.; LAMBERT, RICHARD M.A. Single Crystal Study of the Silver-
Catalysed Selective Oxidation and Total Oxidation in Ethylene. Journal of Catalysis, v. 92, p.
364-375, 1985.
HAKKINEN, Hannu; FRONDELIUS, Pentti; HONKALA, Karoliina. Formation of Gold(I)
Edge Oxide at Flat Gold Nanoclusters on an Ultrathin MgO Film under Ambient Conditions.
Angewandte Chemie International Edition, v. 49, p. 7913-7916, 2010.
ILLAS, Francesc; LOPEZ, Nuria; TORRES, Daniel. A theoretical study of coverage effects
for ethylene epoxidation on Cu(111) under low oxygen pressure. Journal of Catalysis, v. 243,
p. 404-409, 2006.

6. REFERÊNCIAS BIBLIOGRÁFICAS 47
ILLAS, Francesc; TORRES, Daniel. On the Performance of Au(111) for Ethylene
Epoxidation: A Density Functional Study. The Journal of Physical Chemistry B, v. 110, p.
13310-13313, 2006.
KENSON, ROBERT E.; LAPKIN, M. Kinetics and Mechanism of Ethylene
Oxidation.Reactions of Ethylene and Ethylene Oxide on a Silver Catalyst. The Journal of
Physical Chemistry, v. 74, n. 7, p. 1493-1502, 1970.
KHAODEE, Watcharapong;JONGSOMJIT, Bunjerd;PHASERTHDAM, Piyasan; GOTO,
Shigeo; ASSABUMRUNGRAT, Suttichai. Impact of temperature ramping rate during
calcinations on characteristics of nano-ZrO2 and its catalytic activity for isosynthesis. Journal
of Molecular Catalysis A: Chemical, v. 280, p. 35–42, 2008.
LEFORT, T. E. French patent 729952, Societe Francaise de Catalyse Generale, 1931.
LINIC, Suljo; CHRISTOPHER, Phillip. Engineering selectivity in heterogeneous catalysis:
Ag nanowires as selective ethylene epoxidation catalysts. Journal of the American Chemical
Society, v. 130, p. 11264–11265, 2008.
LOPEZ, Nuria; TORRES, Daniel; ILLAS, Francesc; LAMBERT Richard M. Why Copper Is
Intrinsically More Selective than Silver in Alkene Epoxidation: Ethylene Oxidation on
Cu(111) versus Ag(111). Journal of the American Chemical Society, v. 127, p. 10774-10775,
2005.
MAVRIKAKIS, Manos; XU, Ye; GREELEY, Jeff. Effect of subsurface oxygen on the
reactivity of the Ag(111) surface. Journal of the American Chemical Society, v. 127, p.
12823-12827, 2005.
MUHLER, M.; BUSSER, G.; HINRICHSEN O. The temperature-programmed desorption of
oxygen from an alumina-supported silver catalyst. Catalysis Letters, v. 79, p. 49-54, 2002.
OYAMA, Ted S. Rates, Kinetics, and Mechanisms of Epoxidation: Homogeneous,
Heterogeneous, and Biological Routes. Mechanisms in Homogeneous and Heterogeneous
Epoxidation Catalysis, p. 3-99, 2008.
OYAMA, TED S.; LU, Jinqing; BRAVO-SUÁREZ, Juan J.; TAKAHASHI, Atsushi;
HARUTA, Masatake. In situ UV–vis studies of the effect of particle size on the epoxidation
of ethylene and propylene on supported silver catalysts with molecular oxygen. Journal of
Catalysis, v. 232, p. 85–95, 2005.
PONEC, Vladimir; BOND, Geoffrey C. Catalysis by Metals and Alloys. Studies in Surface
Science and Catalysis, v. 95, p. 1-734, 1995.

6. REFERÊNCIAS BIBLIOGRÁFICAS 48
SCHMAL, M.; CARVALHO, Marta C. N. A.; PASSOS, Fabio B. Study of the active phase
of silver catalysts for ethylene epoxidation. Journal of catalysis, v. 248, p. 121-129, 2007.
SEYEDMONIR, S. R.; PLISCHKE, J. K.; VANNICE, M. A.; YOUNG, H. W. Ethylene
Oxidation over Small Silver Crystallites. Journal of Catalysis, v. 123, p. 534-549, 1990.
SING, K. S. W. Reporting physisorption data for gas/solid systems with special reference to
the determination of surface area and porosity. Pure and Applied Chemistry, v. 54, p. 2201-
2218, 1982.
SU, Caili; LI, Junrong; HE, Dehua; CHENG, Zhengxing; ZHU, Qiming. Synthesis of
isobutene from synthesis gas overnanosize zirconia catalysts. Applied Catalysis A: General,v.
202, p. 81–89, 2000.
TYSOE, W. T.; STACCHIOLA, D.; WU, G.; KALTCHEV, M. A reflection–absorption
infrared spectroscopic study of the adsorption of ethylene and ethylene oxide on oxygen-
covered Ag(111). Surface Science, v. 486, p. 9-23, 2001.
VAN DEN HOEK; P. J.; BAERENDS, E. J.; VAN SANTEN, R. A. Ethylene Epoxidation on
Ag(110): The Role of Subsurface Oxygen. Journal of Physical Chemistry. v. 93, p. 6469-
6475, 1989.
VAN SANTEN, R. A; ÖZBEK, M. O. The Mechanism of Ethylene Epoxidation Catalysis.
Catalysis Letters, v. 143, p. 131–141, 2013.
VAN SANTEN, R. A; ÖZBEK, M. O; ONAL, I. Effect of Surface and Oxygen Coverage on
Ethylene Epoxidation. Topics in Catalysis, v.55, p. 710–717, 2012.
VAN SANTEN, R. A; ÖZBEK, M. O; ONAL, I. Why silver is the unique catalyst for
ethylene epoxidation. Journal of Catalysis, v.284, p. 230–235, 2011.
VAN SANTEN, R. A.; KUIPERS, H. P. C. E. The Mechanism of Ethylene Epoxidation.
Advances in Catalysis, v. 35, p. 265-321, 1987.
VERYKIOS, Xenophon; PITCHAI, Rangasamy; LEE, James K. Support Participation in
Chemistry of Ethylene Oxidation on Silver Catalysts. Applied Catalysis, v. 44, p. 223-237,
1988.
VERYKIOS, Xenophon; STEIN, Fred P.; COUGHIAN, Robert W. Influence of Metal
Crystallite Size and Morphology on Selectivity and Activity of Ethylene Oxidation Catalyzed
by Supported Silver. Journal of Catalysis, v. 66, p. 368-382, 1980.
WESTERTERP, K. R.; BORMAN, P. C. An Experimental Study of the Kinetics of the
Selective Oxidation of Ethene over a Silver on a-Alumina Catalyst. Industrial Engineering
Chemistry Research, v. 34, p. 49-58, 1995.

6. REFERÊNCIAS BIBLIOGRÁFICAS 49
WU, James C.; HARRIOT, Peter. The Effect of Crystallite Size on the Activity and
Selectivity of Silver Catalysts. Journal of Catalysis, v. 39, p. 395-402, 1975.

APÊNDICE 50
APÊNDICE A
Cálculos para o preparo dos catalisadores
Tomando por base a síntese de 10,0 g do catalisador com 15,0% em peso de prata
suportado em SiO2.
% (𝑝
𝑝) =
𝑚𝐴𝑔
𝑚𝐴𝑔 + 𝑚𝑆𝑢𝑝𝑜𝑟𝑡𝑒𝑥 100
Dados:
. Massa molar do AgNO3 = 169,87 g/mol
. Massa atômica Ag = 107,87 u
Logo, a massa molar de nitrato de prata será:
m (Ag) = 15% de 10,0 g = 1,5 g
Ag(NO3) - Ag
169,87 g - 107,87 g
x - 1,5 g x = 1,18 g de Ag(NO3)
Portanto, msuporte = 10,0 - mAg = 10,0 – 1,5 = 8,5 g

APÊNDICE 51
APÊNDICE B
As fichas cristalográficas adotadas na discussão dos resultados estão apresentadas
abaixo:

APÊNDICE 52

APÊNDICE 53

APÊNDICE 54
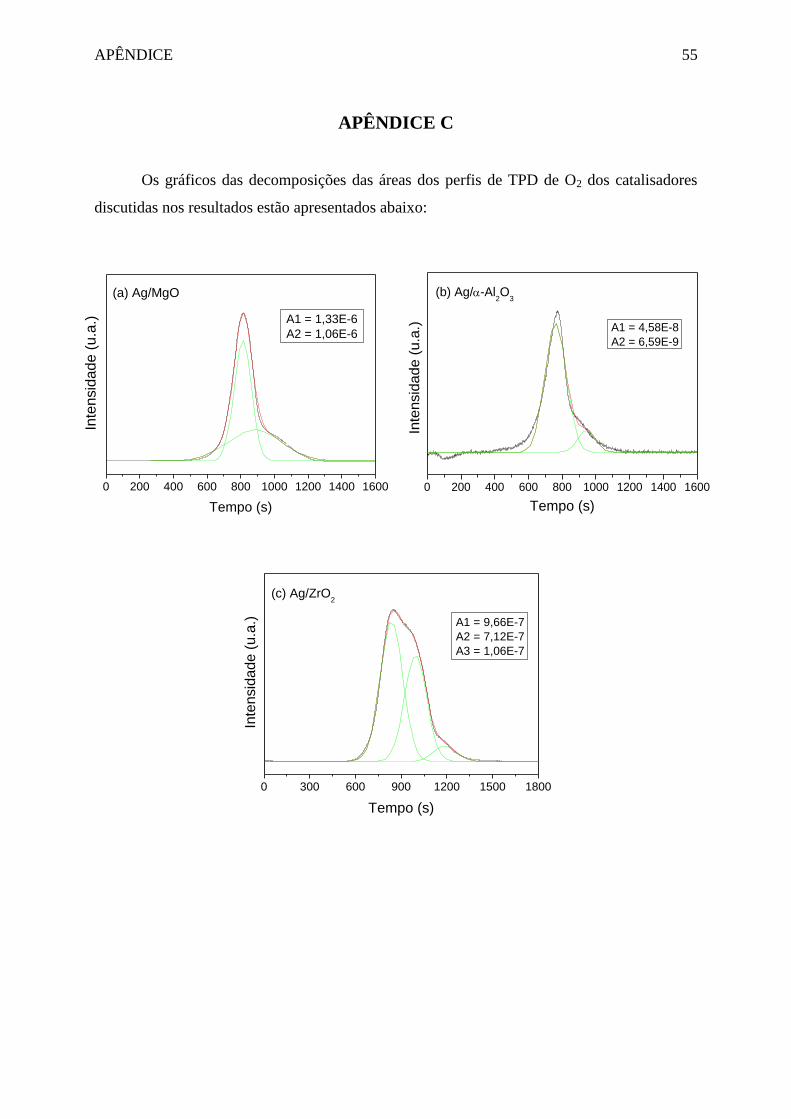
APÊNDICE 55
APÊNDICE C
Os gráficos das decomposições das áreas dos perfis de TPD de O2 dos catalisadores
discutidas nos resultados estão apresentados abaixo:
0 200 400 600 800 1000 1200 1400 1600
(a) Ag/MgO
Inte
nsid
ad
e (
u.a
.)
Tempo (s)
A1 = 1,33E-6
A2 = 1,06E-6
0 200 400 600 800 1000 1200 1400 1600
(b) Ag/-Al2O
3
Inte
nsid
ade (
u.a
.)
Tempo (s)
A1 = 4,58E-8
A2 = 6,59E-9
0 300 600 900 1200 1500 1800
(c) Ag/ZrO2
Inte
nsid
ade (
u.a
.)
Tempo (s)
A1 = 9,66E-7
A2 = 7,12E-7
A3 = 1,06E-7