registro de medicamentos - farmácia unisa 2008 · técnica (câmara técnica de medicamentos...
TRANSCRIPT

REGISTRO DE MEDICAMENTOS
Deborah Masano Cavaloti
Manira Georges Soufia
1

2
No Brasil, os medicamentos são registrados na Anvisa, por sua Gerência-Geral de
Medicamentos (GGMED) que inclui:
• Gerência de Medicamentos Novos,
• Pesquisa e Ensaios Clínicos (GEPEC),
• Gerência de Medicamentos Similares (GEMES),
• Gerência de Medicamentos Genéricos (GEMEG),
• Gerência de Medicamentos Isentos, Fitoterápicos e Homeopáticos(GMEFH),
• Unidade de Produtos Biológicos e Hemoderivados (UPBIH),
• Unidade de Produtos Controlados (UPROC) e a
• Unidade de Farmacovigilância (UFARM).
Como a Anvisa avalia o registro de medicamentos novos no Brasil
Brasília, 20 de janeiro de 2005

3
Novos medicamentos, portanto,Novos medicamentos, portanto,
podem ser registrados em podem ser registrados em
diferentes partes da diferentes partes da
GGMEDGGMED

4
O termo “medicamento novo”, sem outra adjetivação,
é, na prática, utilizado para se referir a medicamentos
novos com princípios ativos sintéticos e semi-
sintéticos, associados ou não, que são os avaliados
pela GEPEC.
Quando se utiliza o termo “medicamento novo” sem
outro complemento não se está referindo, portanto,
a produtos biológicos, fitoterápicos, homeopáticos,
medicamentos ditos “específicos”, medicamentos
isentos de registro, e nem tampouco a cópias
(genéricos e similares).

5
A avaliação de um dossiê de registro
costuma ser dividida em três partes:
• análise farmacotécnica
• análise de eficácia, e
• análise de segurança.

A análise farmacotécnica inclui a verificação de todas asetapas da fabricação do medicamento desde:
• Aquisição dos materiais,
• Produção,
• Controle de qualidade,
• Liberação,
• Estocagem,
• Expedição de produtos terminados e os controles relacionados.
Essa análise é feita por técnicos da própria Anvisa,
em geral farmacêuticos.
6

ANÁLISE
FARMACOTÉCNICA
Pode ser dividida em duas etapas: a
primeira delas consistindo em se conferir
se toda a documentação exigida para o
registro consta no processo; a segunda da
análise propriamente dita do dossiê de
registro.
7

8
O dossiê é composto:O dossiê é composto:
a)a) PorPor umauma parteparte documental,documental, compostacomposta porpor::
FormuláriosFormulários dede petiçãopetição dede registro,registro, ComprovanteComprovante dede recolhimentorecolhimento dada taxataxa dede
fiscalização,fiscalização, LicençaLicença dede FuncionamentoFuncionamento dada empresa,empresa, CertificadoCertificado dede
ResponsabilidadeResponsabilidade Técnica,Técnica, NotificaçãoNotificação dada produçãoprodução dede loteslotes--pilotopiloto (para(para
produtosprodutos nacionais),nacionais), CertificadoCertificado dede BoasBoas PráticasPráticas dede FabricaçãoFabricação ee
Controle (BPFC) emitido pela Controle (BPFC) emitido pela AnvisaAnvisa. .
b) Pelo Relatório Técnico, b) Pelo Relatório Técnico,
QueQue devedeve conterconter todastodas asas informaçõesinformações referentesreferentes aosaos estudosestudos clínicosclínicos ee aa
parteparte farmacotécnicafarmacotécnica.. ÉÉ nana análiseanálise farmacotécnicafarmacotécnica queque sãosão avaliadasavaliadas aa bulabula
(quanto(quanto àà suasua estrutura),estrutura), aa estabilidadeestabilidade dodo produto,produto, asas informaçõesinformações técnicastécnicas
do(s)do(s) princípio(s)princípio(s) ativo(s),ativo(s), aa farmacodinâmica,farmacodinâmica, aa farmacocinética,farmacocinética, todatoda aa
produçãoprodução dodo medicamentomedicamento ee todotodo oo controlecontrole dede qualidadequalidade dodo produtoproduto..

ANÁLISE DE EFICÁCIA E
SEGURANÇA
As avaliações de eficácia e segurança foram, durante
vários anos, feitas por câmaras técnicas constituídas
por um número variável de especialistas, em geral
médicos e professores universitários de reconhecida
experiência em diversas áreas da Medicina, que se
reuniam periodicamente para discutir os processos de
registro de novos medicamentos, assim como de
inclusões e alterações pós-registro.
9

A análise de eficácia e segurança era feita somente apóso término da análise farmacotécnica, por uma câmaratécnica (Câmara Técnica de Medicamentos – CATEME),
em reuniões mensais.
Atualmente a CATEME tem outras funções, passando adiscutir questões de ordem mais geral ligadas amedicamentos, o que tem servido para orientar oposicionamento da Anvisa frente a elas. O número deprocessos de registro submetidos à avaliação dessa
Câmara foi reduzido drasticamente.
10

INDEPENDÊNCIA DA ANVISA EM
RELAÇÃO A SEUS CONSULTORES
• A GEPEC não é no momento auto-suficiente para avaliar aeficácia e segurança dos medicamentos novos a ela submetidospara registro. O papel hoje desempenhado pelos consultoresad hoc é imprescindível.
• A GEPEC tem avaliado os pareceres de seus consultores e, muitoembora freqüentemente os tenha acatado, isso nem sempreacontece, em parte porque pareceres de dois consultoresdiferentes sobre um mesmo produto nem sempre sãoconcordantes e, em casos mais raros, por discordar do(s)parecer(es). Quando a Agência tem parecer diverso do de seusconsultores, a divergência fica documentada no processo.Respaldando ou não as recomendações de seus consultores, aAnvisa explicita em seus pareceres os motivos das tomadas dedecisão, contribuindo, pois, para a transparência do processo.
11

RENOVAÇÃO DE REGISTRO
12
Relação dos documentos gerais necessários:
1) Formulário de Petição devidamente preenchido;
2) Via original do comprovante de recolhimento da taxa de
fiscalização de vigilância sanitária ou de isenção, quando
for o caso;
1) Certificado de Responsabilidade técnica, atualizado,
emitido pelo Conselho Regional de Farmácia;

4) Apresentar documento comprobatório de venda no período devigência do registro, e os números das notas fiscais e a relação deestabelecimentos compradores em um máximo de 3 (três) notaspor forma farmacêutica e concentração. Poderá ser apresentadadeclaração referente às apresentações comerciais nãocomercializadas das quais a empresa tenha interesse em manter oregistro, desde que pelo menos uma apresentação daquela formafarmacêutica e concentração tenha sido comercializada. Poderá,ainda, ser apresentado comprovante de exportação no caso deprodutos registrados exclusivamente para esse fim.
SOMENTE PARA LABORATÓRIOS OFICIAIS:
Quando não houver a produção do medicamento no referido períodoapresentar justificativa da sua não comercialização;
13

5) Última versão de bula impressa que acompanha o produto em suasembalagens comerciais;
6) Apresentar listagem que contemple todas as alterações e/ou inclusõespós-registro ocorridas durante o último período de validade do registrodo produto, acompanhados de cópia do DOU, ou na ausência, cópia doprotocolo da(s) petição(ões) correspondente(s);
7) Para produtos importados apresentar os respectivos laudos de trêslotes importados nos últimos três anos do controle de qualidade físico-químico, químico, microbiológico e biológico, de acordo coma formafarmacêutica, realizado pelo importador no Brasil.
14

LEI 6.360 / 1976LEI 6.360 / 1976
REGISTRO
Segundo o Art. 12 da Lei 6.360/1976, “Nenhum dos produtos de que
trata esta Lei, inclusive os importados, poderá ser industrializado,
exposto à venda ou entregue ao consumo antes de registrado no
Ministério da Saúde.”
RENOVAÇÃO DO REGISTRO
Art 12...
§ 1º “O registro a que se refere este artigo terá validade por 5 (cinco)
anos e poderá ser reavaliado por períodos iguais e sucessivos,
mantido o número de registro inicial.” 15

LEI 6.360 / 1976LEI 6.360 / 1976
PÓS-REGISTRO
Art. 13
“Qualquer modificação de fórmula, alteração de elementos de
composição ou de seus quantitativos, adição, subtração ou
inovação introduzida na elaboração do produto, dependerá de
autorização prévia e expressa do Ministro da Saúde e será
desde logo averbada no registro.”
16

PREENCHIMENTO DE
FORMULÁRIOS
• Formulário de Petição 1 (FP1)
• Formulário de Petição 2 (FP2)
• Alteração de registro
17

Destinado à colocação darazão social da empresasolicitante que estápleiteando registro doproduto
Destinado à colocação donúmero de autorização defuncionamento da empresa. Aúltima quadrícula da direita édestinada à colocação dodígito verificador
Não preencherDestinado à colocação docódigo da classe terapêutica,bem como a descrição, porextenso, da referida classeterapêutica. Tab. 1
(ANVISA)
Não preencher
Não preencher
Destinado à colocação do princípio ativo
Não preencher
F DADOS RELACIONADOS A FÓRMULA
14 N.º
APRES
15.F.
FISICA/
FARMAC.
16.
COMPONE
NTES DA
FÓRUMLA
17.
CÓDIGO
DA DCB
18. TIPO
19
CONCENT
RAÇÃO
QUANT/VO
LUME
20. UNID.
DE
DEMONST
RAÇAO
DA
FÓRMULA
01 COMFUROSEMI
DA 0612013/05 05 40 MG COM
LACTOSE 3540360/16 16 53 MG COM
AMIDO DE
MILHO 0203165/16 16 61MG COM
TALCO 0229083/16 16 5,5 MG COM
ESTEARAD
O DE
MAGNÉSI
O
0306437/16 16 0,5 MG COM

(Uso exclusivo do órgão
da Vigilância Sanitária)
Destinado à informação dos assuntosobjeto da petição, podendo serapresentados um máximo de 4 (quatro).Cada assunto deve ser apresentado atravésdo código específico, acompanhado darespectiva descrição. Tab. 1
Destinado à colocação darazão social do fabricante
Destinado à colocação donúmero de autorização defuncionamento da empresa.Destinados à informação do
nome do município e da siglada Unidade Federada ondeestá instalado o fabricante.

Destinado a assinalar o destino doproduto (institucional,industrial/profissional, comercial oude uso restrito a hospitais).
Destinado à colocação do númerode dias, meses ou anos de validadedo produto e à marcação daquadrícula de tempocorrespondente.
Destinado à colocação doprincípio ativo do produto
Destinado à informação relativa à apresentação do produto,obedecendo a seguinte ordem de preenchimento: concentração,forma farmacêutica, via de administração, tipo de embalagemexterna, tipo de acondicionamento, x quantidade/peso /volumede formas farmacêuticas por acondicionamento.
Destinado à colocação do código e adescrição da forma física. Tab. 2
Destinado à colocação docódigo e da descrição darestrição de uso ou devenda do produto. Tab. 3
Destinado à colocação docódigo e da descrição doscuidados de conservação.Tab. 4
Destinado àcolocação docódigo e dadescrição doacondicionamento(embalagemprimária). Tab. 5
Destinado à colocação do códigoe da descrição da embalagemexterna do produto. Tab. 6

21
A ser assinado, e carimbado, pelo
representante legal e técnico responsável da empresa.
Reservado ao órgão de
vigilância sanitária

(ANVISA)
Destinado à colocação do mês edo ano de vencimento do registrodo produto (só em caso deregistro já concedido.
Campo E: Destinados à colocação dosdados referentes ao produto, cujaapresentação e fórmula são similaresà que se pretende na solicitação, ouseja, nome da empresa, número doregistro e nome do produto.

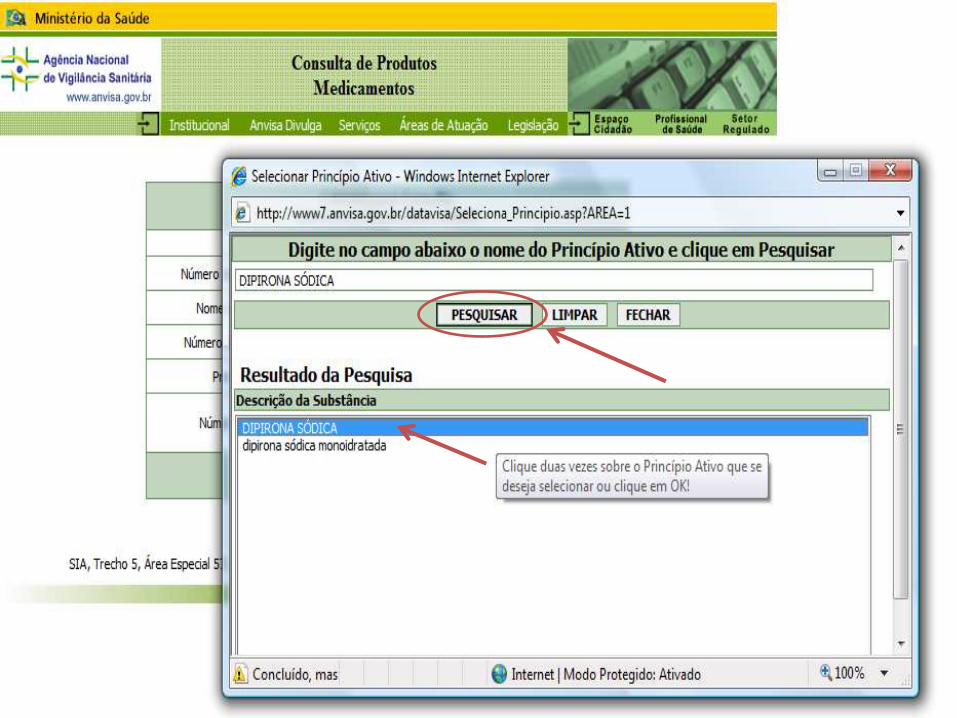
CONSULTA DE MEDICAMENTOS REGISTRADOS



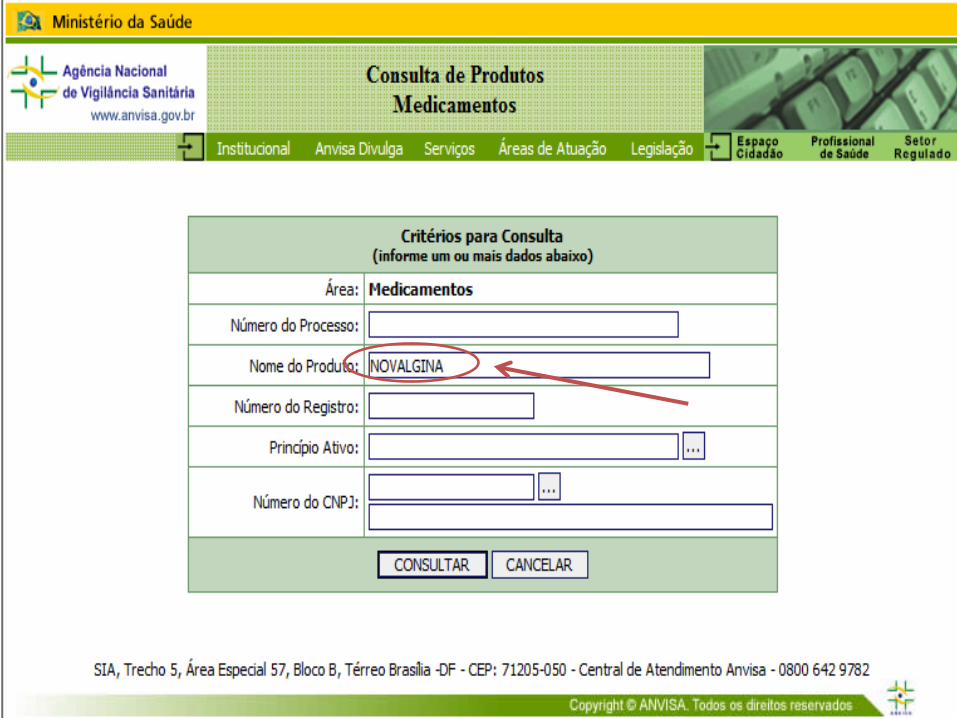



CONSULTA POR PRINCÍPIO ATIVO