reduÇÃo e acetilaÇÃo de nitroaromÁticos como...
TRANSCRIPT

UNIVERSIDADE TECNOLOGICA FEDERAL DO PARANA
COORDENACAO DE QUIMICA
CURSO DE BACHARELADO EM QUIMICA
MARCELO VICTOR RAGASSI
REDUCAO E ACETILACAO DE NITROAROMATICOS
COMO PROPOSTA DE REACOES NA AGUA
VERMELHA (RED WATER)
TRABALHO DE CONCLUSAO DE CURSO
PATO BRANCO
2016

MARCELO VICTOR RAGASSI
REDUCAO E ACETILACAO DE NITROAROMATICOS
COMO PROPOSTA DE REACOES NA AGUA
VERMELHA (RED WATER)
Trabalho de Conclusao de Curso, apresentado a
Comissao de Diplomacao do Curso de Bacharelado
em Quımica da Universidade Tecnologica Federal
do Parana (UTFPR), Campus Pato Branco, como
requisito parcial para a obtencao do tıtulo de Bacharel
em Quımica.
Orientador: Dr. Davi Costa Silva
Pato Branco
2016

TERMO DE APROVACAO
O trabalho de diplomacao intitulado REDUCAO E ACETILACAO DE NITROA-
ROMATICOS COMO PROPOSTA DE REACOES NA AGUA VERMELHA
(RED WATER) foi considerado APROVADO de acordo com a ata da banca exami-
nadora No 4.1.2016-B de 2016.
Fizeram parte da banca os professores:
Dr. Davi Costa Silva
Dr. Marcio Barreto Rodrigues
Dra. Sirlei Dias Teixeira

AGRADECIMENTOS
A Deus, por estar sempre presente e guiar o meu caminho.
A minha famılia, por tudo que fizeram por mim e, em momento algum, deixaram de
acreditar em mim.
A minha namorada Vanessa, que esteve comigo em todos os momentos e sempre me
apoiou nas minhas decisoes.
Ao professor e orientador Davi Costa Silva, pela orientacao, dedicacao e amizade que
foi de grande importancia para a conclusao deste trabalho.
Ao professor Cesar Augusto Refosco Yednak, pelo incentivo, amizade e apoio durante
todo o perıodo que cursei a graduacao. E, tambem, por me apresentar o LATEX.
A todos os amigos que nao foram citados, mas tambem nao foram esquecidos.
A Central de Analises da UTFPR campus Pato Branco, pela disponibilidade de espaco,
reagentes e equipamentos.
A IMBEL pela disponibilizacao das amostras.

“Nao vos amoldeis as estruturas deste mundo,
mas transformai-vos pela renovacao da mente,
a fim de distinguir qual e a vontade de Deus:
o que e bom, o que Lhe e agradavel, o que e perfeito”.
(Bıblia Sagrada, Romanos 12, 2)

RESUMO
RAGASSI, M. V. Reducao e acetilacao de nitroaromaticos como proposta de reacoes naagua vermelha (RED WATER). 2016. 49 f. Trabalho de conclusao de curso (Graduacaoem Bacharelado em Quımica) - Universidade Tecnologica Federal do Parana. Pato Branco,2016.
Os nitroaromaticos podem ser reduzidos para originar aminas aromaticas atraves do usode acido e um metal zero-valente. Essas aminas sao usadas como ponto de partida paraa sıntese de intermediarios de varias substancias, dentre elas, farmacos e cristais lıquidos.As industrias belicas que produzem TNT geram, dentre outros, a agua vermelha comoresıduo. Esta, contem formas assimetricas de TNT’s que, neste trabalho, sao recuperadose reduzidos em meio aquoso. Este trabalho pode ser dividido em duas etapas: uma emque foi utilizado apenas reagentes puros, e na outra, a agua vermelha. Na primeira etapa,fez-se a acetilacao e nitracao da anilina, e, em seguida, a nitroacetanilida foi “desprote-gida” para sintetizar um composto azo. Cada intermediario formado foi caracterizado porinfravermelho e cromatografia gasosa acoplada a espectrometro de massas. Na segundaetapa, fez-se a recuperacao e reducao dos nitroaromaticos presentes na agua vermelha.Devido a dıficuldade de extracao dos produtos da reacao na agua vermelha, optou-se pelareacao ¨ONE POT¨ em meio aquoso e, entao, caracterizou-se via CCD.
Palavras-chave: Nitroaromaticos. Reducao. Aminas aromaticas. Acetilacao. Agua
Vermelha.

ABSTRACT
RAGASSI, M. V. Reduction and acetylation nitroaromatic as proposed reactions in theRed Water. 2016. 49 f. Completion of coursework (Chemistry Undergraduate Baccalau-reate) – Technological Federal University of Parana. Pato Branco, 2014.
Nitroaromatic can be reduced to yield aromatic amines through the use of acid and zero-valent metal. These amines are used as starting point for the synthesis of intermediatesof various substances such pharmaceuticals and liquid crystals. The defense industriesthat produce TNT generate, among others, the red water as residue. This contains asym-metric forms of TNT’s that in this work, are recovered and reduced in aqueous medium.This work can be divided into two stages: one in which used was only pure reagents, andin the other the red water. n the first step was taken acetylation and nitration of ani-line, and then the nitroacetanilide was “deprotected” for synthesizing an azo compound.Each formed intermediate was characterized by infra red and gas chromatography coupledwith mass spectrometry. In the second step, the recovery and reduction of nitroaromaticpresent in the red water was made. Due of the difficulty of extracting the reaction prod-ucts in the red water, we opted for the reaction one pot in aqueous medium and thencharacterized by TLC.
Keywords: Nitroaromatic. Reduction. Aromatic amines. Acetylation. Red Water.

LISTA DE FIGURAS
Figura 1 – Molecula de nitrobenzeno. . . . . . . . . . . . . . . . . . . . . . . . . . 15
Figura 2 – Molecula de TNT. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Figura 3 – Diagrama esquematico apresentando os possıveis caminhos para a reducao
do nitrobenzeno. As setas para baixo (reacoes I-III e VI-VIII) indicam
reacoes de reducao e as setas para a direita (reacoes IV-V) indica con-
densacao. O caminho predominante de reducao do grupo nitro para
amino equivalente pode ocorrer sequencialmente passando pelo nitro-
sobenzeno e fenilhidroxilamina (reacoes I-III). . . . . . . . . . . . . . . 18
Figura 4 – Procedimento para a obtencao de um composto azo a partir do nitro-
benzeno. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Figura 5 – Molecula de um cristal lıquido do tipo 4-(alcoxi)-4’-nitroazobenzeno 5a-b. 21
Figura 6 – Esquema da sıntese do paracetamol (4), da fenacetina (5) e da dulcina
(7) a partir do nitrobenzeno (1) . . . . . . . . . . . . . . . . . . . . . . 21
Figura 7 – Porcao do espectro eletromagnetico mostrando a relacao entre a ra-
diacao do infravermelho vibracional e a de outros tipos de radiacao. . . 22
Figura 8 – Exemplo das formas de vibracao do tipo estiramento e dobramento. . . 23
Figura 9 – Processo de extracao. Em “A”, e introduzido o solvente 1 que contem
uma mistura de moleculas (brancas e pretas). Em “B”, o solvente
2 (sombreado), apos a agitacao, contem a maior parte das moleculas
brancas, as quais foram extraıdas para o novo solvente. Em “C”, com
a remocao da fase inferior, tem-se a mistura parcialmente separada. . . 26
Figura 10 – Mecanismo da reacao de sıntese da acetanilida a partir da anilina . . . 32
Figura 11 – Espectro de infravermelho obtido para a acetanilida sintetizada. . . . . 33
Figura 12 – Cromatograma da acetanilida. . . . . . . . . . . . . . . . . . . . . . . . 33
Figura 13 – Espectro de massas da acetanilida. . . . . . . . . . . . . . . . . . . . . 34
Figura 14 – Mecanismo da reacao de sıntese da p-nitroacetanilida a partir da ace-
tanilida . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Figura 15 – Espectro de infravermelho da p-nitroacetanilida. . . . . . . . . . . . . . 35
Figura 16 – Cromatograma da amostra de p-nitroacetanilida. . . . . . . . . . . . . 36
Figura 17 – Espectro de massas da amostra da p-nitroacetanilida. . . . . . . . . . . 37
Figura 18 – Mecanismo da reacao de sıntese da p-nitroanilina a partir da p-nitroacetanilida 37
Figura 19 – Espectro de infravermelho da p-nitroanilina. . . . . . . . . . . . . . . . 38
Figura 20 – Cromatograma da amostra de p-nitroanilina. . . . . . . . . . . . . . . . 39
Figura 21 – Espectro de massas da amostra da p-nitroanilina. . . . . . . . . . . . . 39
Figura 22 – Fragmentacao para a anilina apos o impacto de eletrons sofrido no
espectrometro de massas. . . . . . . . . . . . . . . . . . . . . . . . . . 40

Figura 23 – Fragmentacao para o nitrobenzeno apos o impacto de eletrons sofrido
no espectrometro de massas. . . . . . . . . . . . . . . . . . . . . . . . . 40
Figura 24 – Reacao de formacao do ıon nitrosonio. . . . . . . . . . . . . . . . . . . 40
Figura 25 – Reacao de formacao do p-(4-nitrobenzenoazo)-fenol . . . . . . . . . . . 41
Figura 26 – Espectro de infravermelho do p-(4-nitrobenzenoazo)-fenol. . . . . . . . 42
Figura 27 – Imagem da amostra com caracterısticas de emulsao. A foto da esquerda
mostra a visao lateral, e a da direita, a superior. . . . . . . . . . . . . . 43
Figura 28 – Imagem da cromatoplaca (CCD) com a amostra padrao de acetanilida
a esquerda e com a amostra derivada da agua vermelha a direita. . . . 44

LISTA DE TABELAS
Tabela 1 – Proporcoes de acidos nıtrico e sulfurico utilizadas durante a producao
de TNT pelo processo classico. . . . . . . . . . . . . . . . . . . . . . . 16
Tabela 2 – Agentes secantes comuns e suas caracterısticas. . . . . . . . . . . . . . 27
Tabela 3 – Perfil de nitroaromaticos encontrados em uma fracao aquosa de agua
vermelha residual. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42

SUMARIO
1 INTRODUCAO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2 OBJETIVOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.1 OBJETIVO GERAL . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.2 OBJETIVOS ESPECIFICOS . . . . . . . . . . . . . . . . . . . . . . . . . 14
3 REFERENCIAL TEORICO . . . . . . . . . . . . . . . . . . . . . . . 15
3.1 NITROCOMPOSTOS AROMATICOS . . . . . . . . . . . . . . . . . . . . 15
3.1.1 2,4,6-Trinitrotolueno . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.2 REDUCAO E ACETILACAO DE NITROAROMATICOS . . . . . . . . . . 17
3.3 OBTENCAO DE CRISTAIS LIQUIDOS E FARMACOS A PARTIR DE DE-
RIVADOS DE NITROAROMATICOS. . . . . . . . . . . . . . . . . . . . . 19
3.4 AS TECNICAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
3.4.1 Espectroscopia no Infravermelho com Transformada de Fourier . . . . . . . 22
3.4.2 Cromatografia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
3.4.3 Extracao por Solvente . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
4 METODOLOGIA . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4.1 ENSAIOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4.1.1 Sıntese da Acetanilida . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4.1.2 Sıntese da p-nitroacetanilida . . . . . . . . . . . . . . . . . . . . . . . . . 28
4.1.3 Sıntese da p-nitroanilina . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4.1.4 Sıntese do p-(4-nitrobenzenoazo)-fenol . . . . . . . . . . . . . . . . . . . . 29
4.2 TESTES UTILIZANDO A AGUA VERMELHA . . . . . . . . . . . . . . . 29
4.2.1 Obtencao e reducao dos nitroaromaticos . . . . . . . . . . . . . . . . . . . 29
4.2.2 Processo de extracao . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.2.3 Derivatizacao das aminas em agua . . . . . . . . . . . . . . . . . . . . . . 30
4.2.4 Analise por CCD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.3 ANALISES POR INFRAVERMELHO . . . . . . . . . . . . . . . . . . . . . 31
4.4 CONDICOES CROMATOGRAFICAS . . . . . . . . . . . . . . . . . . . . 31
5 RESULTADOS E DISCUSSAO . . . . . . . . . . . . . . . . . . . . . 32
5.1 ENSAIOS COM REAGENTES PUROS . . . . . . . . . . . . . . . . . . . 32
5.1.1 Acetanilida . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
5.1.2 p-nitroacetanilida . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
5.1.3 p-nitroanilina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36

5.1.4 p-(4-nitrobenzenoazo)-fenol . . . . . . . . . . . . . . . . . . . . . . . . . 40
5.2 RESULTADOS DOS PROCEDIMENTOS USANDO COMO MATERIAL
DE PARTIDA A AGUA VERMELHA . . . . . . . . . . . . . . . . . . . . 42
6 CONSIDERACOES FINAIS . . . . . . . . . . . . . . . . . . . . . . . 45
REFERENCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46

12
1 INTRODUCAO
As aguas provenientes das industrias de explosivos do tipo 2,4,6-trinitrotolueno
(TNT) sao efluentes altamente perigosos e toxicos, sendo classificados pela Agencia de
Protecao Ambiental dos EUA como poluentes de elevado potencial impactante. A des-
carga da agua residual de TNT sem tratamento provoca severa poluicao em aguas e solos
(ZHANG; ZHAO; YE, 2011). Logo, os processos industriais sao responsaveis pela maior
parte da contaminacao e dependendo da eficacia no processo de tratamento do resıduo,
menor sera a contaminacao de solos e aguas (RODRIGUES; SILVA; PAVIA, 2009).
O desafio de recuperar e utilizar moleculas organicas, e tantos outros compostos
que satisfacam grande parte das necessidades da sociedade moderna, permanece atual, e
e nesse contexto que enquadra-se este trabalho, por meio de tentativas utilizando reacoes
conhecidas da sıntese organica e metodologias simples na recuperacao destes compostos
presentes em agua, obtendo alguns compostos que possam ser uma fonte significativa
para utilizacao como intermediarios na sıntese de farmacos, cristais lıquidos, pesticidas
biodegradaveis, antibioticos e outros.
Metodos rapidos e seletivos de reducao e acetilacao de compostos nitro aromaticos
em agua pode ser uma alternativa de recuperacao e preparacao de amino aromaticos
derivados presentes em aguas residuais, principalmente a agua vermelha. A reducao
nitrocompostos pode ser mediada por palha de aco e acido acetico em ultrassom, e a
acetolise, por anidrido acetico e acido acetico.
O presente trabalho tem por finalidade verificar reacoes fundamentais de acetilacao
e nitracao, alem de otimizar e recuperar aminas aromaticas da agua vermelha para veri-
ficacao de processos de aproveitamento do resıduo objetivando maior rentabilidade junta-
mente com a reducao da agressao ao meio ambiente. Este trabalho encontra-se organizado
da seguinte forma: o capıtulo 2 apresenta os objetivos do trabalho. No capıtulo 3, e feita
uma abordagem teorica a respeito de nitroaromaticos. Neste contexto, sao apresentadas
as formas de reducao e acetilacao dos nitroaromaticos, e o processo de obtencao de cristais
lıquidos e farmacos a partir de seus derivados. Para encerrar o capıtulo, sera apresentada
os princıpios de algumas tecnicas usadas durante o trabalho. O capıtulo 4 e dedicado
a descricao das metodologias utilizadas tanto para os ensaios com reagentes puros como
para os testes utilizando a agua vermelha. Os resultados e discussao sao apresentados no
capıtulo 5. Este, ficou dividido em duas partes. Na primeira parte, os mecanismos das
reacoes iniciadas com reagentes puros sao mostrados na forma de figuras. Encontra-se,
tambem, os resultados das analises de infravermelho e cromatografia gasosa acoplada ao
espectrometro de massas, seguidos de suas respectivas interpretacoes. Na segunda parte
esta contida a avaliacao dos processos realizados com a agua vermelha. As consideracoes
finais e algumas possıveis extensoes deste trabalho sao discutidas no capıtulo 5. Todo

Capıtulo 1. INTRODUCAO 13
o estudo desenvolvido foi realizado na Central de Analises e no Laboratorio de Quımica
Organica da UTFPR, campus Pato Branco - Parana.

14
2 OBJETIVOS
2.1 OBJETIVO GERAL
Reduzir rapida e seletivamente nitroaromaticos em meio aquoso e preparar como
amino derivados em sıntese organica.
2.2 OBJETIVOS ESPECIFICOS
X Sintetizar a acetanilida;
X Sintetizar e reduzir a p-nitroacetanilida;
X Caracterizar os compostos aromaticos via espectrofotometria de infravermelho e
espectrometria de massas;
X Realizar reacoes de recuperacao, reducao e derivatizacao de nitroaromaticos presen-
tes na agua vermelha.

15
3 REFERENCIAL TEORICO
3.1 NITROCOMPOSTOS AROMATICOS
Sao chamados nitrocompostos aromaticos aqueles que apresentam como grupo
funcional o NO2 ligado a uma estrutura aromatica. O nitrobenzeno e um exemplo de ni-
trocomposto. Este, e uma substancia lıquida nas condicoes ambiente, apresenta coloracao
amarelada, toxidez e e muito empregado como solvente de substancias organicas.
Figura 1 – Molecula de nitrobenzeno.
Dentre os componentes desse grupo, os nitrocompostos aromaticos, ou apenas
nitroaromaticos, sao os que tem recebido grande atencao nos ultimos tempos devido a
muitas aplicacoes que estes compostos apresentam, bem como tambem os enormes riscos
que podem apresentar ao meio ambiente e aos seres vivos. Eles sao utilizados na fa-
bricacao de poliamidas, pesticidas, agrotoxicos, explosivos e farmacos como paracetamol,
antibioticos e drogas anticancerıgenas, devido suas propriedades de inibir a replicacao do
DNA (SHABBIR et al., 2015). Estudos comprovam que estes compostos sao “candidatos
potenciais a agentes terapeuticos” (PAULAI; SERRANO; TAVARES, 2009). Essa ativi-
dade biologica e resultado das mudancas de estabilidade desses compostos, por meio da
interacao entre o nitrocomposto e seu alvo na biofase, isto quando tratado em nanoescala
e resultado do grupo nitro ligado ao anel aromatico possuir grande forca retiradora de
eletrons e, consequentemente, diminuir a densidade eletronica do anel aromatico (YAN et
al., 2005). Em outras palavras, o grupo nitro e suscetıvel de reducao.
A industria quımica tem, entao, grande dependencia dos compostos nitroaromaticos,
isto porque eles sao a unica fonte comercial de aromaticos com nitrogenio ligado ao anel,
logo, se a mesma quer produzir um produto que tenha esta caracterıstica, o ponto de par-
tida da sıntese e um nitroaromatico, como por exemplo a manufatura da anilina que e uma
das duas maiores utilizacoes destes compostos (RICKERT, 1985), seguida da producao
de explosivos.

Capıtulo 3. REFERENCIAL TEORICO 16
3.1.1 2,4,6-Trinitrotolueno
O TNT e dentre os nitroaromaticos o mais conhecido devido a sua grande aplicacao
na industria de explosivos. Seus serios problemas ambientais sao causados quando seus
descartes sao despejados indevidamente no meio ambiente.
Figura 2 – Molecula de TNT.
Consoante Falone e Vieira (2004), foi com a I Guerra Mundial que deu-se inıcio
a producao, estocagem e disposicao de explosivos nas instalacoes militares, sendo inten-
sificado na II Guerra Mundial. Ainda de acordo com os mesmos autores, os explosivos
sao compostos organicos que apresentam nitrogenio em sua estrutura e, quando oxidados,
formam pequenas moleculas gasosas, sendo elas N2, H2O e CO2. O grau de instabilidade
desses compostos permite que, sob acao de uma forte mudanca de temperatura, choque
eletrico ou impactos mecanicos, se decomponham rapida e espontaneamente liberando
grande quantidade de energia na forma de calor e gases, a elevada temperatura e pressao.
Para a formacao do TNT e realizada a nitracao em 3 etapas do tolueno usando-
se uma mistura de acido nıtrico e sulfurico (RODRIGUES et al., 2007), como mostrado
na Tabela 1. O tolueno possui como grupo ativador uma metila, dessa forma passa a
orientar o substituinte, na primeira etapa, nas posicoes orto ou para em relacao a si
mesmo (SOLOMONS; FRYHLE, 2005a).
Tabela 1 – Proporcoes de acidos nıtrico e sulfurico utilizadas durante a producao de TNTpelo processo classico.
1◦ Estagio 2◦ Estagio 3◦ Estagio
HNO3 (%) 28 32 49H2SO4 (%) 56 61 49H2O (%) 16 7 2
Fonte: RODRIGUES et al., 2007.
A conclusao da terceira etapa de nitracao leva, entao, a producao do TNT, for-
mando tambem varios subprodutos como: cinzas ou resıduos minerais; isomeros as-
simetricos; produtos de oxidacao lateral como acido trinitrobenzoico, nitrofenois e tetra-
nitrometano; produtos de oxidacao do xileno e benzeno, por estes muitas vezes estarem
como impurezas no tolueno (RODRIGUES et al., 2007).

Capıtulo 3. REFERENCIAL TEORICO 17
O TNT bruto e purificado por meio de lavagens com agua e sulfito de sodio,
gerando dois tipos de resıduos: a agua amarela (AA) e a agua vermelha (AV). Na agua
amarela encontra-se, em geral, tracos de TNT, resıduos de acido e tolueno, enquanto que
na agua vermelha encontra-se dinitrotolueno e mononitrotolueno, alem de derivados do
nitrobenzeno (LUDWICHK et al., 2015).
3.2 REDUCAO E ACETILACAO DE NITROAROMATICOS
Os compostos nitroaromaticos tem por caracterıstica apresentar uma grande re-
sistencia a hidrolise, oxidacao biologica e ataque quımico, dessa forma, torna-se difıcil
o tratamento desta classe de compostos (RODRIGUES, 2005). Isso acontece porque o
grupo NO2 apresenta uma carga parcial positiva no atomo diretamente ligado ao anel,
com isso retira eletrons do carbocation em desenvolvimento no processo de uma reacao
e aumenta, consequentemente, a carga positiva no anel (SOLOMONS; FRYHLE, 2005a),
blindando-o ao ataque oxidativo.
Existem diversos estudos feitos sobre a reducao destes compostos (DONG et al.,
2010; AGRAWAL; TRATNYEK, 1996; LIU; ZHU; YE, 2012; THOMAS; HERNANDEZ;
KUO, 2008), objetivando diminuir os impactos ambientais causados pelos compostos ni-
trados provenientes dos efluentes das industrias belicas. Os produtos obtidos apos esse
processo de reducao dos nitroaromaticos sao aminas aromaticas, que apresentam carac-
terısticas menos explosivas (KEUM; LI, 2004) e mais biodegradaveis (OH et al., 2005;
SUN et al., 2010) do que aqueles primeiros.
Uma das maneiras de reduzir nitroaromaticos e utilizando ferro zero-valente (Fe0),
em meio acido. Este processo produz intermediarios, dentre os quais sao o nitrosobenzeno
e a N-fenil-hidroxiamina (DONG et al., 2010). As reacoes descritas nas equacoes (3.1) -
(3.4) e a Figura 3 ilustram este processo.
C6H5NO2 + Fe0 + 2H+ → C6H5NO + Fe2+ + H2O; (3.1)
C6H5NO + Fe0 + 2H+ → C6H5NHOH + Fe2+; (3.2)
C6H5NHOH + Fe0 + 2H+ → C6H5NH2 + Fe2+ + H2O. (3.3)
Dessa forma, a equacao geral para o processo pode ser escrita como:
C6H5NO2 + 3Fe0 + 6H+ → C6H5NH2 + 3Fe2+ + 2H2O. (3.4)
Durante o processo de reducao, o ferro zero-valente (Fe0), metalico, e rapidamente
oxidado para o ıon ferroso (Fe+2), o que causa uma dissolucao do metal, a corrosao.

Capıtulo 3. REFERENCIAL TEORICO 18
Figura 3 – Diagrama esquematico apresentando os possıveis caminhos para a reducao donitrobenzeno. As setas para baixo (reacoes I-III e VI-VIII) indicam reacoes dereducao e as setas para a direita (reacoes IV-V) indica condensacao. O caminhopredominante de reducao do grupo nitro para amino equivalente pode ocorrersequencialmente passando pelo nitrosobenzeno e fenilhidroxilamina (reacoesI-III).
Fonte: AGRAWAL; TRATNYEK, 1996.
Na eletroquımica, a corrosao do metal e representada, quando em condicoes anaerobias
(AGRAWAL; TRATNYEK, 1996), pela meia reacao anodica:
Fe0 + 2H+ Fe2+ +H2. (3.5)
A figura Figura 3 mostra na reacao IV a formacao do azoxibenzeno, que e um pro-
duto de condensacao do nitrosobenzeno com a fenilhidroxiamina, apresenta rendimentos
consideraveis no processo de reducao do grupo nitro presente no nitrosobenzeno (PIZZO-
LATTI; YUNES, 1990). Os produtos seguintes dessa via sintetica apresentam reducoes
mais complicadas ate a formacao da anilina. (AGRAWAL; TRATNYEK, 1996).
De acordo com Souza e Peralta-Zamora (2005) a propria la de aco comercial pode
ser a unica fonte de ferro metalico para reacoes de reducao, uma vez que o ferro zero-
valente reduz substratos organicos mediante a transferencia de 2 eletrons, ja que o ferro
apresenta um grande poder redutivo (E0 Fe0/Fe2+ 0,440 V). Nao obstante, o emprego
de outros metais, alem do ferro, como por exemplo o zinco e o estanho na reducao de
compostos nitroaromaticos, tambem mostram bons resultados, uma vez que ha indıcios
de uso industrial desses metais para a sıntese de aminas (MORRISON; BOYD, 2002).
Contudo, o rendimento obtido quando e feita a utilizacao do ferro metalico e muito alto
(LYONS; SMITH, 1927).
Metodos utilizando-se de bacterias e fungos isolados do solo (MCCORMICK et

Capıtulo 3. REFERENCIAL TEORICO 19
al., 1976) e o uso de diferentes tipos de plantas (PODLIPNa; POSPısILOVa; VANeK,
2015) para a reducao de TNT e outros nitroaromaticos tambem apresentam resultados
bastantes satisfatorios.
O processo de acetilacao de alcoois, tiois e aminas e frequentemente empregado
nas metodologias de sıntese organica por ser de grande eficiencia e de baixo custo para
a “protecao” destes grupos em processos sinteticos de varias etapas (GREENE; WUTS,
2005). Dessa forma, torna estas substancias mais resistentes a oxidacoes e menos reativas
em reacoes de substituicoes aromaticas. As aminas podem ser acetiladas de diversas for-
mas, como por exemplo atraves de reacao com anidrido acetico, cloreto de acila ou acido
acetico glacial. Nao obstante, estudos tem mostrado rotas alternativas para a acetilacao
de aminas aromaticas, como mostra Saikia et al., (2016), que fez uso da acetonitrila, e ou-
tros autores que estudaram uma metodologia utilizando anidrido acetico e NaHSO4·SiO2
(THIRUPATHI, 2007).
Pacheco et al. (2007) mostrou que e possıvel realizar a transformacao de nitroa-
romaticos em aminas e acetamidas utilizando raızes tuberosas de Arracacia xanthorrhiza
e Beta vulgaris e um microorganismo.
Borges et al. (2005) utilizou varias reacoes de acetilacao da anilina descritas em
livros de quımica organica experimental, fazendo testes em fase aquosa nao catalisada
e catalisada por bases inorganicas. Em seu trabalho, o procedimento considerado mais
vantajoso foi a acetilacao sob baixas temperaturas e em meio aquoso com anidrido acetico
e acetato de sodio como base, obtendo ate 90% de rendimento.
Para acetilacao de aminas tambem sao incentivados a fazer uso de acido tioacetico.
A vantagem deste metodo reside no facto de que a acetilacao da anilina e homologos,
neste caso, ocorre em baixas temperaturas. A reacao ocorre com a liberacao de sulfeto
de hidrogenio.
3.3 OBTENCAO DE CRISTAIS LIQUIDOS E FARMACOS A PAR-
TIR DE DERIVADOS DE NITROAROMATICOS.
A obtencao das aminas aromaticas e um dos caminhos para a formacao dos azocom-
postos, que podem ser usados como intermediarios na sıntese de cristais lıquidos (CLs).
Azocompostos compostos sao caracterizados por uma ligacao dupla entre atomos de ni-
trogenios, mas diferentemente das aminas nao apresentam carater basico. As arilaminas
primarias quando reagem com acido nitroso formam como produto sais de arenodiazonio,
estes quando mantidos a baixas temperaturas apresentam certa estabilidade (MORRI-
SON; BOYD, 2002):
ArNH2 +NaNO2 + 2HX −→ ArN+ ≡ N : X− +NaX + 2H2O (3.6)

Capıtulo 3. REFERENCIAL TEORICO 20
em que o primeiro termo a esquerda da seta na reacao e uma arilamina primaria e o
primeiro a direita da seta, o sal de arenodiazonio. O ıon diazonio pode ser escrito como
um hıbrido de ressonancia:
Ar-N+ ≡ N: ←→ Ar-N=N+.
A reacao de formacao do sal arenodiazonio envolve um acido fraco e instavel,
o acido nitroso (HNO2). Este, e preparado in situ, atraves do uso de nitrito de sodio
(NaNO2) juntamente com uma solucao aquosa de acido forte (SOLOMONS; FRYHLE,
2005b). Para ilustrar essa reacao, pode ser feito o uso do acido sulfurico:
H2SO4 + 2NaNO2(aq) −→ 2HONO(aq) +Na2SO4(aq). (3.7)
Os sais de arenodiazonio sao merecedores de grande atencao, pois podem ser prepa-
rados por quase todas aminas aromaticas primarias, alem de reagirem com varias classes
de compostos.
Quando em condicoes apropriadas os sais de diazonio regem com certos compostos
aromaticos e geram produtos conhecidos por azo compostos, cuja formula geral e escrita
como:
Ar −N = N − Ar′.
Por serem eletrofilos fracos, os ıons arenodiazonio sao capazes de reagir com com-
postos aromaticos altamente reativos, como por exemplo o fenol, e desta reacao ocorre
a producao de compostos azo. Quando essa substituicao eletrofılica aromatica ocorre,
denomina-se de reacao de acoplamento diazo, que quando acontece entre derivados de
fenois e da anilina, observa-se o direcionamento com grande exclusividade na posicao
para, quando esta encontra-se disponıvel, se nao, o acoplamento ocorre na posicao orto.
Em geral, acoplamentos com fenois ocorrem em solucoes um tanto alcalinas e com aminas
em solucoes fracamente acidas (MORRISON; BOYD, 2002).
Um outro metodo para a obtencao de um composto azo ocorre a partir do proprio
nitrobenzeno (BIGELOW; ROBINSON, 1942). A Figura 4 ilustra este procedimento.
Figura 4 – Procedimento para a obtencao de um composto azo a partir do nitrobenzeno.
Fonte: BIGELOW; ROBINSON, 1942.
Nesse procedimento o nitrocomposto e colocado para reagir, sob aquecimento, com
po de zinco e hidroxido de sodio em uma solucao de metanol.

Capıtulo 3. REFERENCIAL TEORICO 21
Alguns exemplos mais comuns de compostos azo sao os corantes, como o vermelho
de metila, carmoisina e a benzidina.
A sıntese de um cristal lıquido pode ser iniciada a partir de um nitroazobenzeno.
Cristiano et al. (2014), traz uma metodologia de sıntese em multiplas etapas para a
obtencao de um CL do tipo 4-(alcoxi)-4’-nitroazobenzeno 5a-b (Figura 5).
Figura 5 – Molecula de um cristal lıquido do tipo 4-(alcoxi)-4’-nitroazobenzeno 5a-b.
Fonte: CRISTIANO et al., 2014.
Os nitroaromaticos tambem se mostram presentes na sıntese de farmacos. O pa-
racetamol e fenacetina sao dois analgesicos que podem ser sintetizados a partir do nitro-
benzeno. A Figura 6 mostra um esquema para a sıntese do paracetamol, da fenacetina e,
tambem, da dulcina, que e um edulcorante.
Figura 6 – Esquema da sıntese do paracetamol (4), da fenacetina (5) e da dulcina (7) apartir do nitrobenzeno (1)
Fonte: BAPTISTELLA; GIACOMINI; IMAMURA, 2003.
O processo de sıntese do paracetamol inicia-se com a reducao do nitrobenzeno
utilizando zinco metalico como catalisador e apos duas etapas obtem-se o paracetamol.
Em seguida, atraves da reacao de Williamson, obtem-se a fenacetina. A dulcina e obtida
quando a fenacetina e submetida a hidrolise acida e, posteriormente, tratada com ureia.

Capıtulo 3. REFERENCIAL TEORICO 22
3.4 AS TECNICAS
Esta secao tem por objetivo fazer uma breve introducao sobre as tecnicas que fo-
ram utilizadas no desenvolvimento deste trabalho. Primeiramente sera abordada a tecnica
de infravermelho com transformada de Fourier (IVTF), em seguida sera explicado alguns
conceitos sobre a cromatografia, focando a cromatografia em camada fina e a cromato-
grafia gasosa acoplada com espectrometro de massas, e, para finalizar, sera exposto um
metodo classico de extracao, a extracao lıquido-lıquido, tambem chamada de extracao por
solvente.
3.4.1 Espectroscopia no Infravermelho com Transformada de Fourier
A absorcao de frequencias de radiacao eletromagnetica na regiao do infravermelho
e observada em quase todos os compostos que apresentam ligacoes covalente, podendo ser
de natureza organica ou inorganica (PAVIA et al., 2013).
A luz visıvel se encontra na regiao do espectro eletromagnetico em que o compri-
mento de onda (λ) comeca em 400 nm e vai ate 800 nm (1 nm = 10−9 m). A regiao do
infravermelho dentro desse espectro esta localizada em comprimentos de onda maiores do
que a luz visıvel e menores do que as ondas de radio. A maior parte do interesse dentro
da quımica organica esta limitada entre 4000 e 400 cm−1 (SILVERSTEIN et al., 2006).
A Figura 7 mostra a relacao do espectro do infravermelho vibraciona,l que abrange a
faixa de 2,5µm ate 15µm (1µm = 10−6 m), com outras regioes peculiares do espectro
eletromagnetico.
Figura 7 – Porcao do espectro eletromagnetico mostrando a relacao entre a radiacao doinfravermelho vibracional e a de outros tipos de radiacao.
Fonte: PAVIA et al., 2013.
Quando as moleculas absorvem radiacao eletromagnetica, ou seja, energia, elas sao

Capıtulo 3. REFERENCIAL TEORICO 23
excitadas, e passam de um estado de menor energia para um de maior energia. O processo
de absorcao de radiacao infravermelha e como qualquer outro processo de absorcao, ele
e quantizado. A absorcao de radiacao no infravermelho so acontecera quando houver
ligacoes com momento de dipolo que variam em funcao do tempo, isto quer dizer que nem
todas as ligacoes em uma molecula serao capazes de absorver energia nessa faixa (PAVIA
et al., 2013).
As vibracoes que as moleculas apresentam podem ser de dois tipos: estiramento e
flexao ou dobramento. Esta caracteriza-se, basicamente, pela varicao do angulo de ligacao
entre os atomos e aquela e um movimento ritmado ao longo do eixo da ligacao, onde a
distancias entre os atomos diminuem e aumentam.
Figura 8 – Exemplo das formas de vibracao do tipo estiramento e dobramento.
Fonte: PAVIA et al., 2013.
Para a interpretacao dos espectros e muito comum o uso de uma tabela de cor-
relacao, em que mostram o tipo de vibracao e a frequencia em cm−1 que elas ocorrem.
3.4.2 Cromatografia
A cromatografia representa um papel importante nos metodos mais modernos
de analise, isto porque apresenta grande facilidade de realizar separacao, identificacao
e quantificacao de substancias quımicas. Ela pode atuar por si mesma ou em conjunto
com outras tecnicas, como por exemplo a espectrometria de massas (COLLINS; BRAGA;
BONATTO, 1997).
Segundo a definicao, cromatografia e um metodo fısico-quımico de separacao que
tem como fundamento a migracao diferencial dos componentes de uma mistura entre duas
fases que mantem contato ıntimo. Uma dessas fases leva o nome de fase estacionaria,
justamente por se manter imovel. A outra fase, que se move atraves dessa primeira, e a
fase movel.
No inıcio do seculo XX, Mikhael Semenovich Tswett fez uso dos termos “cromato-
grafia”, “cromatograma ” e “metodo cromatografico” em dois de seus trabalhos, nos quais
descrevia processos de separacao (COLLINS, 2009). A origem dessas palavras utilizadas
por Tswett e grega, “chrom” significa cor e “graphe”, escrever. Embora o processo nao
dependa da cor, Tsweet em seus trabalhos no ano de 1906, meticulosamente deixou claro

Capıtulo 3. REFERENCIAL TEORICO 24
que a separacao nao era dependente da cor e sim das interacoes das substancias, coloridas
ou nao, com a fase estacionaria atraves do processo de adsorcao (COLLINS, 2009).
Com o desenvolvimento e aperfeicoamento da tecnica de cromatografia esta passou
a ser utilizada de diversas formas. Por isso, faz-se necessario uma classificacao das dife-
rentes modalidades de cromatografia. Os criterios mais comuns sao aqueles relacionados
a tecnica empregada, ao mecanismo de separacao e as varias fases utilizadas.
A cromatografia em camada delgada (CCD) envolve uma fase estacionario solida
que pode ser sılica, alumina acida, basica ou neutra, celulose (quando a cromatografia e
realizada em papel), poliamidas e “sephadex”, e, uma fase movel lıquida. Logo, e uma
tecnica de adsorcao lıquido-solido e a separacao ocorre pela diferenca de afinidade dos
componentes presentes em uma mistura pela fase estacionaria.
A CCD por ser um metodo simples e economico e predominantemente escolhida
para acompanhar reacoes organicas (DEGANI; CASS; VIEIRA, 1998). Nesta tecnica
tem-se como parametro o fator de retencao (Rf), o qual e definido pela razao entre a
distancia percorrida pela substancia em questao e a distancia percorrida pela fase movel.
Os Rf que se encontram entre 0,4 e 0,6 sao considerados ideais, pois as substancias nao
ficaram totalmente retidas na fase movel e nem na fase estacionaria.
Em geral, neste tipo de cromatografia o material adsorvente encontra-se depositado
sobre vidro, alumınio ou filme plastico. As amostras a ser analisadas devem ser aplicadas
aproximadamente a 1 cm da base inferior da placa com o auxılio de um capilar (DEGANI;
CASS; VIEIRA, 1998).
Apos o processo de eluicao, como a maioria dos compostos organicos sao incolores, e
necessario que se faca o uso de reveladores para a analise dos resultados. Para tal, existem
processos destrutivos e nao destrutivos. Os metodos mais comuns nao destrutivos sao a
utilizacao de placas com a fase estacionaria fluorescente e o uso de iodo (DEGANI; CASS;
VIEIRA, 1998). No primeiro caso utiliza-se luz ultravioleta para revelar os compostos e no
segundo caso a placa e colocada em um cuba ou um recipiente contendo iodo molecular e
este por sofrer sublimacao complexa-se com compostos insaturados e a placa contendo as
substancias a serem analisadas mostrara pontos amarronzados. Ja os processos destrutivos
tem como base a oxidacao dos compostos sobre a placa.
A cromatografia a gas (CG) e uma das tecnicas de maior utilidade para a separacao
e analise de substancias volateis que nao se decompoem. Dentre os modos de utilizacao
destacam-se o teste de pureza de um composto e a separacao dos componentes de uma
mistura. Nao obstante, em alguns casos ela serve para identificar compostos. Neste tipo
de cromatografia a amostra e inserida no equipamento atraves de um sistema de injecao
que a coloca em uma coluna contendo a fase estacionaria. Com o uso de temperaturas
adequadas no local da injecao a amostra e vaporizada e de acordo com as propriedades da
fase estacionaria e da amostra, as substancias ficam retidas por um determinado tempo e
alcancam a saıda da coluna em diferentes tempos. Neste ponto, um detector adequado faz

Capıtulo 3. REFERENCIAL TEORICO 25
a deteccao, quantificacao e em alguns casos ate mesmo a identificacao destas substancias.
De maneira geral, uma corrente de gas (fase movel) passa com fluxo contınuo pela coluna
e assim que amostra vaporizada e inserida nessa corrente de gas ela e arrastada atraves
da coluna. Esse gas, o gas de arraste, deve ser um gas inerte.
O alto poder de resolucao da cromatografia gasosa torna possıvel a analise de de-
zenas de substancias e sua sensibilidade, chegando ate mesmo a detectar cerca de 10−12 g,
e um dos principais motivos de seu uso ser bastante acentuado (COLLINS; BRAGA; BO-
NATTO, 1997). Esta tecnica permite analises qualitativas e quantitativas. As analises
qualitativas baseiam-se na velocidade que cada componente passa pela coluna, utilizando
como parametro o tempo de retencao (Tr). O tempo de retencao de uma substancia e
o tempo gasto desde o momento em que a amostra e injetada, ate o momento em que o
maior numero de moleculas da substancia sai do sistema cromatografico e e detectado.
A analise quantitativa relaciona-se com a area formada sob os picos no cromatograma,
uma vez que o sinal enviado pelo detector e proporcional a concentracao das substancias
presentes na amostra.
Uma variante recente da cromatografia a gas e aquela que utiliza como detector
o espectrometro de massa acoplado (PAVIA et al., 2009). Logo que a amostra deixa
a coluna ela e introduzida na camara de ionizacao do espectrometro. Nesta camara as
moleculas contidas no gas sao transformadas em ıons. Estes sao acelerados e atravessam
o analisador de massas. Dessa forma, cada fracao que elui da coluna tem seu espectro
de massa determinado. Esse tipo de equipamento, em geral, contem uma biblioteca de
padroes de espectro de massas na memoria do computador, portanto, se os compostos ja
forem conhecidos eles podem ser identificados atraves de comparacao.
3.4.3 Extracao por Solvente
Quando um soluto de um solvente migra para outro solvente ocorre um processo
denominado de extracao. Isto acontece porque esse soluto apresenta maior afinidade pelo
segundo solvente do que pelo primeiro, em outras palavras, devido a sua polaridade. Uma
caracterıstica importante deste processo e que os dois solventes devem ser imiscıveis, de
modo que formem duas fases ou camadas.
Na Figura 9 e possıvel ver um exemplo de extracao. Nesse processo muitas vezes
utiliza-se o funil de separacao. Nele e adicionado o primeiro solvente contendo uma
mistura de moleculas. Em seguida adiciona-se o segundo solvente. Entao, fecha-se o funil
e inicia-se um processo de agitacao, nao muito vigorosa para evitar, quando possıvel,
a formacao de emulsao, e, logo apos, o funil e colocado em repouso para que ocorra a
separacao das fases. A extracao termina quando uma das fases e coletada abrindo-se a
torneira do funil.
O processo de agitacao faz com que o soluto se distribua entre as duas fases lıquidas.
Uma vez que estas fases se separam ocorre um equilıbrio, tal que a razao das concentracoes

Capıtulo 3. REFERENCIAL TEORICO 26
Figura 9 – Processo de extracao. Em “A”, e introduzido o solvente 1 que contem umamistura de moleculas (brancas e pretas). Em “B”, o solvente 2 (sombreado),apos a agitacao, contem a maior parte das moleculas brancas, as quais foramextraıdas para o novo solvente. Em “C”, com a remocao da fase inferior, tem-sea mistura parcialmente separada.
Fonte: Pavia et al., 2009.
do soluto em cada camada torna-se uma constante. O coeficente de distribuicao (K) e o
nome dado a esta constante e e definido como
K =C2
C1
,
em que C1 e C2 sao as concentracoes no equilıbrio, expressas em gramas por litro ou
miligramas por mililitro do soluto X no solvente 1 e no solvente 2, respectivamente. E
importante deixar claro que essa relacao independe das quantidades dos dois solventes
misturados, dependendo apenas da natureza dos solventes utilizados (PAVIA et al., 2009).
A primeira extracao nao ira separar toda a mistura, ou seja, nem todo soluto
deixara o solvente 1 e passara para o solvente 2, isto pode ser observado na Figura 9
quando ainda encontra-se moleculas brancas no solvente removido no bequer e moleculas
pretas no solvente que continua no funil. Por isso, e aconselhavel realizar varias extracoes
utilizando pequenas quantidades de solvente do que apenas uma com grande quantidade
de solvente.
Quando um dos solventes e organico e o outra e a agua, o primeiro pode absor-
ver pequenas quantidades desta outra, mesmo a solubilidade da agua no solvente sendo
pequena. Um metodo para remover a agua e fazendo o uso de um agente secante, um
sal anidro capaz de reter agua. Para isso, o agente secante e colocado diretamente na
solucao, onde ele adiciona moleculas de agua e torna-se hidratado (PAVIA et al., 2009).
A Tabela 2 apresenta os agentes secantes mais comuns e suas caracterısticas quanto a

Capıtulo 3. REFERENCIAL TEORICO 27
forma hidratada, a capacidade de agua removida por unidade de agente secante, a intei-
reza, que relaciona a quantidade de agua que fica em equilıbrio com o agente secante, a
velocidade de acao e, traz tambem, seus usos mais corriqueiros.
Tabela 2 – Agentes secantes comuns e suas caracterısticas.
Acidez Hidratado Capacidade Inteireza Velocidade Uso
Sulfato demagnesio
Neutro MgSO4·7H2O Alto Medio Rapido Geral
Sulfato desodio
Neutro Na2SO4·7H2O Alto Baixo Medio Geral
Cloreto decalcio
Neutro CaCl2·2H2O eCaCl2·6H2O
Baixo Alto Rapido Hidrocar-bonetose Halo-genetos
Sulfato decalcio
Neutro CaSO4 · 12H2O Baixo Alto Rapido Geral
Carbonatode potassio
Basico K2CO3 ·112H2O e
K2CO3·2H2OMedio Medio Medio Aminas,
Esteres,Cetonas
Fonte: PAVIA et al., 2009.
Em geral, para grande escala utiliza-se de um Erlenmeyer como recipiente para
efetuar o procedimento de secagem. Ja quando a operacao e em pequena escala, e comum
o uso de um frasco conico ou mesmo de um tubo de ensaio.

28
4 METODOLOGIA
4.1 ENSAIOS
Esta secao ira tratar dos procedimentos de sıntese da acetanilida, da p-nitroacetanilida,
da p-nitroanilina e do p-(4-nitrobenzenoazo)-fenol, que foram utilizados como testes pre-
liminares para subsequentes reacoes de acetilacao envolvendo a agua vermelha. Estas
sınteses foram realizadas seguindo a metodologia proposta por Cristiano et al., (2014).
4.1.1 Sıntese da Acetanilida
Em um bequer de 100 mL, foram adicionados 3,5 mL (0,04 mol) de anilina, 1,1 g
(0,01 mol) de acetato de sodio e 4,0 mL (0,07 mol) de acido acetico. Sob agitacao constante,
anidrido acetico (5,0 mL, 0,05 mol) foi adicionado, em pequenas porcoes. Apos 10 min, a
mistura resultante foi vertida sobre 120 mL de agua gelada. Os cristais formados foram
coletados por filtracao a vacuo e colocados em dessecador ate o dia seguinte.
4.1.2 Sıntese da p-nitroacetanilida
Em um bequer de 50 mL, foram adicionados 3,34 g (0,025 mol) de acetanilida,
4,6 mL (0,08 mol) de acido acetico glacial e 9,6 mL de H2SO4. A solucao resultante foi
resfriada entre 0 e 5 ◦C, em banho de gelo. Separadamente, em outro bequer, foi prepa-
rada uma solucao nitrante, composta de 2,2 mL (0,05 mol) de HNO3 e 1,4 mL de H2SO4.
Esta solucao foi resfriada entre 0 e 5 ◦C e, em seguida, adiciona-se lentamente sobre a
solucao acida de acetanilida, com agitacao constante e evitando que a temperatura no
meio reacional ultrapassasse 10 ◦C. Apos adicao, a mistura foi deixada em repouso por
30 min a temperatura ambiente. Posteriormente, esta foi vertida sobre uma mistura de
gelo/agua (70 mL) e o precipitado amarelo foi filtrado, sob vacuo. O produto bruto foi
recristalizado em etanol e deixado no dessecador.
4.1.3 Sıntese da p-nitroanilina
Em balao de fundo redondo de 50 mL, equipado com condensador, foram adicio-
nados 0,635 g (3,48 mmol) de p-nitroacetanilida e 5 mL de uma solucao aquosa 70% de
H2SO4. A mistura resultante foi refluxada por 25 min. Apos esfriar, o meio reacional
foi vertido sobre 30 mL de agua fria em um bequer. A solucao avermelhada foi filtrada
com papel pregueado para eliminar impurezas solidas presentes. Para precipitar a p-
nitroanilina, o pH do meio foi ajustado para 13 com adicao cuidadosa de solucao de

Capıtulo 4. METODOLOGIA 29
NaOH 6 mol/L. Os cristais foram coletados por filtracao a vacuo, lavando-os com agua
fria. O solido amarelo escuro foi deixado no dessecador.
4.1.4 Sıntese do p-(4-nitrobenzenoazo)-fenol
Como o rendimento da etapa anterior nao foi o esperado, deu-se continuidade
com a sıntese apenas para verificar a formacao do p-(4-nitrobenzenoazo)-fenol. Dessa
forma a adicao dos reagentes nao respeitou a estequiometria da reacao. O procedimento
seguido foi: em um bequer de 100 mL adicionou-se a pequena quantia de p-nitroanilina
formada na etapa anterior (menos de 0,5 g), 2 mL de H2SO4 e 10 mL de agua. Resfriou-se
a solucao resultante ate 5 ◦C e com o auxılio de um bastao de vidro a solucao foi mantida
sob agitacao constante. Uma solucao gelada de NaNO2 (0,69 g) em 2 mL de agua foi
adicionada lentamente, tomando cuidado para que a temperatura do meio reacional nao
ultrapassasse o limite de 10 ◦C. Uma solucao de 0,94 g de fenol em 5 mL de NaOH(aq)
1 mol/L foi adicionada na suspensao formada e foi mantida a 5 ◦C em um bequer de
100 mL. Manteve-se uma agitacao constante por 5 minutos, sob banho de gelo. Os cristais
formados foram coletados por filtracao a vacuo.
4.2 TESTES UTILIZANDO A AGUA VERMELHA
Apos verificacao dos processos sinteticos utilizados na secao 4.1, foram realizados
alguns testes com a agua vermelha, que foi gentilmente cedida pela Industria de Mate-
rial Belico do Brasil (IMBEL), objetivando obter nitroaromaticos, reduzi-los e realizar o
procedimento de acetilacao em meio aquoso.
4.2.1 Obtencao e reducao dos nitroaromaticos
Em um bequer de 50 mL foram colocados 10 mL da amostra da agua vermelha.
Em uma capela, este bequer foi colocado sobre uma chapa de aquecimento e manteve-se
a temperatura de 80 ◦C para que toda a agua presente fosse evaporada. Em seguida,
em um bequer de 250 mL colocou-se 1,9139 g do resıduo solido-pastoso obtido da etapa
anterior juntamente com 31,5 mL de acido acetico 5% e 2,9446 g de palha de aco. A reacao
ocorreu com o auxılio de ultrassom por aproximadamente 120 minutos. Apos este perıodo
foi realizada uma filtracao, para retirar a palha de aco presente no meio, fazendo algumas
lavagens com pequenas porcoes de acido acetico 5% quando necessario.
4.2.2 Processo de extracao
X Primeira Tentativa:

Capıtulo 4. METODOLOGIA 30
No procedimento de extracao das aminas aromaticas formadas anteriormente em
meio aquoso, utilizou-se uma mistura de eter etılico/hexano (1:1) e o metodo de extracao
por solvente (subsecao 3.4.3), onde utilizou-se de um funil de separacao de 125 mL. A
amostra e o solvente foram colocados no funil de separacao na proporcao de 1:1 e agitados
por 2 minutos, abrindo a valvula do funil algumas vezes durante o processo, para que
liberar os gases formados, e esperou-se decantar para coletar a fase organica.
X Segunda Tentativa:
No funil de separacao, foram misturados na proporcao de 1:1 tolueno e a amostra.
Realizou-se o mesmo procedimento descrito na Primeira Tentativa.
X Terceira Tentativa:
No processo de extracao foi usada a Solucao Tampao de Kolthoff , que foi preparada
com 50 mL de carbonato de sodio 0,1 mol/L, 10 mL de solucao de HCl 0,1 mol/L e 40 mL
de agua destilada (MORITA; ASSUMPcaO, 1995).
Na tentativa de extrair os nitroaromaticos reduzidos na etapa anterior, utilizou-se
o procedimento de extracao lıquido-lıquido, da seguinte forma: para 20 mL da amostra,
adicionou-se, em um funil de separacao de 125 mL, 50 mL da solucao tampao de Kolthoff
juntamente com 30 mL de diclorometano e agitou-se por dois minutos, fazendo algumas
pausas para abrir a valvula do funil para liberar os gases formados. Esperou-se ate que
houvesse a separacao das fases e coletou-se a fase organica, a mais densa. Em seguida,
foram adicionados mais 30 mL de diclorometano e repetiu-se o processo. Esse procedi-
mento foi reproduzido por quatro vezes. Na amostra extraıda foi adicionado uma pequena
quantidade de sulfato de magnesio anidro para reter a agua residual e submeteu a filtracao.
4.2.3 Derivatizacao das aminas em agua
Foi feita a derivatizacao de 1,22 g da amostra extraıda utilizando o mesmo procedi-
mento para a sıntese da acetanilida, descrito na subsecao 4.1.1, com algumas adaptacoes
conforme necessario. As quantidade de acetato de sodio, acido acetico, anidrido acetico
utilizados foram 0,55 g, 2,0 mL, 2,5 mL, respectivamente.
4.2.4 Analise por CCD
Em uma placa com dimensoes aproximadas de 10 cm x 5 cm, com a fase estacionaria
sendo sılica, aplicou-se a amostra acetilada juntamente com a acetanilida preparada na
subsecao 4.1.1, que foi usada como padrao. Para a aplicacao, solubilizou-se a amostra
acetilada em metanol e a acetanilida em acetato de etila. A eluicao ocorreu em um frasco

Capıtulo 4. METODOLOGIA 31
de vidro, simulando uma cuba cromatografica, e, como eluente utilizou-se uma mistura
de 1:1 de acetato de etila e hexano. O revelador utilizado foi o iodo.
4.3 ANALISES POR INFRAVERMELHO
Para a determinacao das funcoes organicas e caracterizacao dos materiais obtidos
em cada etapa de sıntese, foi utilizado neste trabalho um espectrometro de infravermelho
(IV) da marca Perkin Elmer, modelo Frontier, utilizando a tecnica de RTA (Refletancia
Total Atenuada), que de acordo com Pavia et al. (2013) e o melhor metodo para obter
espectros de amostras solidas. A faixa de transmitancia utilizada e compreendida entre
400 a 4000 cm−1 e resolucao de 2 cm−1 com um numero de acumulacoes de 32 varreduras
para cada espectro.
4.4 CONDICOES CROMATOGRAFICAS
Para a realizacao da analise em CG/MS, o equipamento e suas condicoes foram as
seguintes: Cromatografo Gasoso (CG) Varian 430, com coluna VF-5 ms (30 m x 0,25 mm
D.I x 0,25µm de filme) acoplado a um detector de massas de baixa resolucao. Cro-
matografo Gasoso Varian 431/CP-3800 acoplado a um detector de massa Varian 210 ion
trap MS, faixa de aquisicao: 50-250 u; energia de ionizacao: 70 eV; modo de aquisicao
normal (impacto de eletrons; transferline coluna/MS a 200 ◦C; ıon trap a 170 ◦C. O forno
foi programado para uma temperatura inicial de 50 ◦C (1 min), 60 ◦C 3 ◦C/min (4,33 min),
240 ◦C 3,5 ◦C/min (14,5 min). O gas de arraste helio (6,0 analıtico) manteve o fluxo cons-
tante igual a 1,2 mL/min.

32
5 RESULTADOS E DISCUSSAO
Neste capıtulo serao apresentados os resultados obtidos pra cada processo sintetico
descrito no Capıtulo 4. Devido a alta solubilidade e difıcil extracao de nitros e aminas
aromaticas presentes na agua vermelha, optou-se pelos testes preliminares com reagentes
conhecidos para subsequentes reacoes de reducao e acetilacao em meio aquoso.
5.1 ENSAIOS COM REAGENTES PUROS
5.1.1 Acetanilida
A sıntese da acetanilida, tambem denominada por N-fenilacetamida, e ilustrada
na Figura 10:
Figura 10 – Mecanismo da reacao de sıntese da acetanilida a partir da anilina
Como pode ser observado, ocorre um processo de acetilacao, com o intuito de “pro-
teger” o grupo funcional amino. As aminas acetiladas sao menos susceptıveis a oxidacoes,
portanto, menos reativas. Uma maneira de reverter o processo e obter novamente a anilina
e atraves da hidrolise acida ou basica. Para que a reacao de protecao ocorra, e necessario
um meio tamponado, ou seja, uma solucao que e capaz de manter o pH do meio constante
e, assim, dar condicoes para que a reacao ocorra (MORRISON; BOYD, 2002). O acido
acetico e o acetato de sodio fazem o papel de solucao tampao para esta reacao, e dessa
forma impedem que a anilina seja protonada.
Uma vez preparada a acetanilida, esta foi caracterizada por espectroscopia de
infravermelho e cromatografia gasosa acoplada ao detector de massas. O espectro de
infravermelho da acetanilida, mostrado na Figura 11, apresentou um pico em 3292 cm−1,
sendo caracterıstica de vibracoes de estiramento de N-H. Em 1662 cm−1 e notado um
pico intenso, o qual foi atribuıdo ao grupamento carbonila (C=O) presente na estrutura
das amidas. Este pico, em geral, aparece como o mais intenso do espectro (PAVIA et al.,
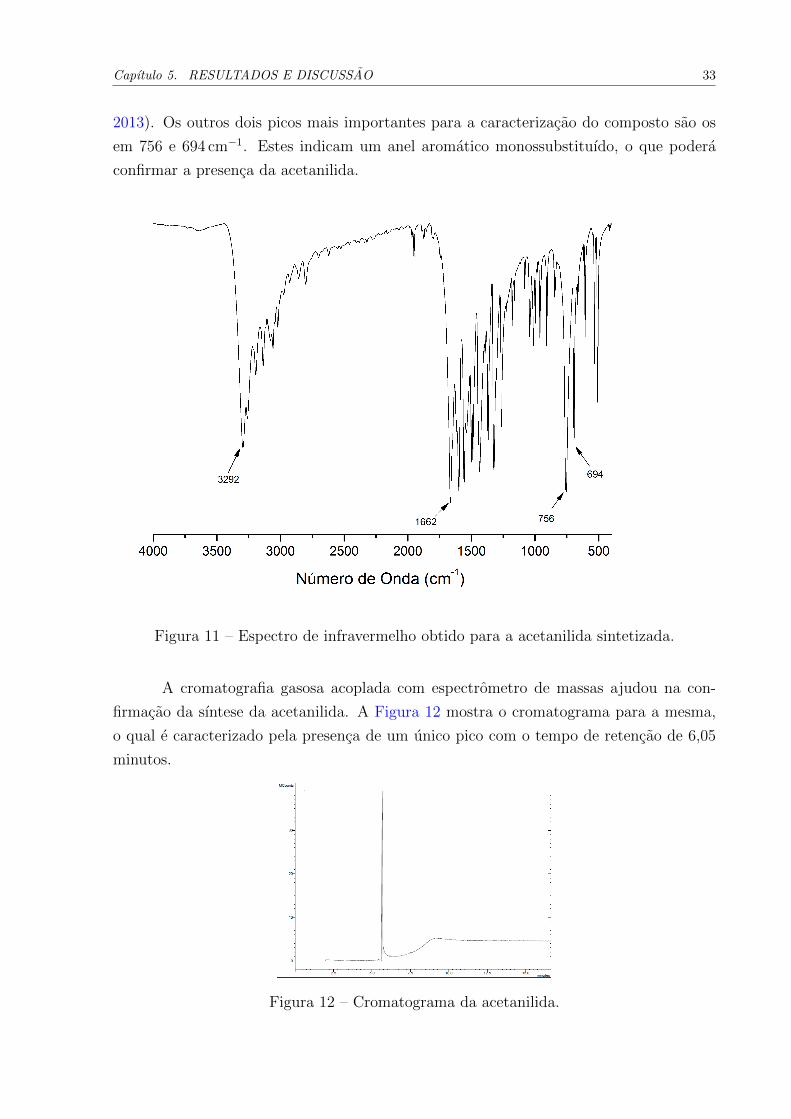
Capıtulo 5. RESULTADOS E DISCUSSAO 33
2013). Os outros dois picos mais importantes para a caracterizacao do composto sao os
em 756 e 694 cm−1. Estes indicam um anel aromatico monossubstituıdo, o que podera
confirmar a presenca da acetanilida.
Figura 11 – Espectro de infravermelho obtido para a acetanilida sintetizada.
A cromatografia gasosa acoplada com espectrometro de massas ajudou na con-
firmacao da sıntese da acetanilida. A Figura 12 mostra o cromatograma para a mesma,
o qual e caracterizado pela presenca de um unico pico com o tempo de retencao de 6,05
minutos.
Figura 12 – Cromatograma da acetanilida.

Capıtulo 5. RESULTADOS E DISCUSSAO 34
O espectro de massas da amostra (Figura 13), onde espectro superior e o do
composto da amostra e o inferior o padrao obtido atraves da biblioteca NIST, apresentou
como pico base m/z = 93 (fragmento de maior intensidade) e outros fragmentos com m/z
= 135 (ıon molecular), 94, e 92. A comparacao do espectro da amostra com o padrao
permite confirmar a sıntese da acetanilida.
Figura 13 – Espectro de massas da acetanilida.
5.1.2 p-nitroacetanilida
O mecanismo de nitracao eletrofılica aromatica, demonstrado na Figura 14, ajuda
a compreensao da sıntese da p-nitroacetanilida a partir da acetanilida . Na etapa 1 ocorre
a formacao do ıon nitronio, que e uma molecula altamente instavel. O ıon nitronio, na
etapa 2, reage com o anel aromatico na posicao para, uma vez que o grupo amina acetilado
na posicao 1 do anel atua como ativador do anel aromatico. Por fim, na etapa 3, o ıon
hidrogenossulfato recupera um hidrogenio e reconstitui-se na forma de acido sulfurico, o
catalisador da reacao.
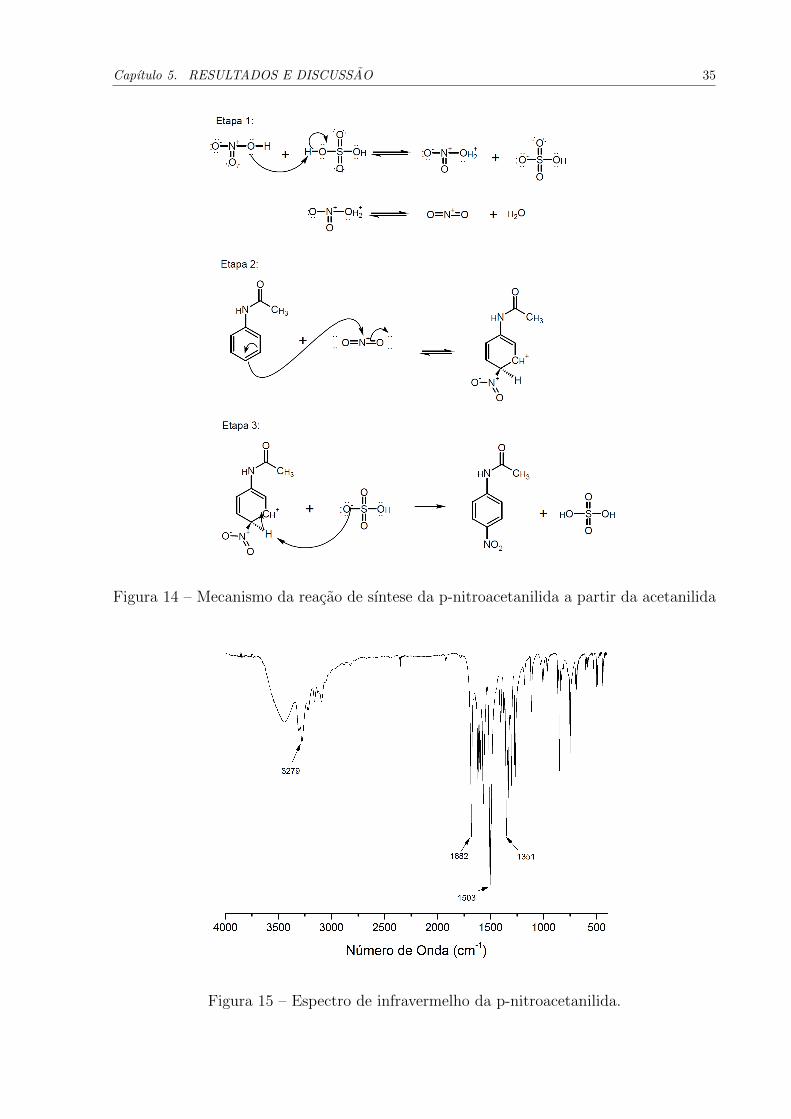
Na Figura 15 tem-se o espectro de infravermelho para a amostra da p-nitroacetanilida.
Em 3279 cm−1 encontra-se a vibracao de estiramento N-H, a qual esta levemente deslocada
em relacao ao espectro da acetanilida (Figura 11). Outras vibracoes importantes obser-
vadas neste espectro e que caracterizam a presenca do grupo nitro sao aquelas em 1503 e
em 1351 cm−1. A primeira e devido a vibracao assimetrica de estiramento do grupo NO2
e a segunda refere-se ao estiramento simetrico deste grupo. O pico referente a vibracao
da carbonila foi observado levemente deslocado, em relacao ao espectro da acetanilida, e
aparece em 1662 cm−1.
O resultado obtido da analise por cromatografia gasosa, mostrado na Figura 16,
apresentou um pico no tempo de retencao em 7,78 minutos referente a p-nitroacetanilida.
Podem ser verificados no cromatograma outros picos em outros tempos de retencao, o
Capıtulo 5. RESULTADOS E DISCUSSAO 35
Figura 14 – Mecanismo da reacao de sıntese da p-nitroacetanilida a partir da acetanilida
Figura 15 – Espectro de infravermelho da p-nitroacetanilida.

Capıtulo 5. RESULTADOS E DISCUSSAO 36
que sugere a formacao de minoritaria de outros compostos, uma reacao nao completa ou
decomposicao da coluna do cromatografo.
Figura 16 – Cromatograma da amostra de p-nitroacetanilida.
Atraves da analise do espectrometro de massas, encontrou-se como pico base a
razao m/z = 138 e o pico do ıon molecular em m/z=180. Outras razoes m/z relevantes
encontradas foram: 108, 92, 80 e 65. Quanto ao pico em m/z = 138, ele e referente saıda
do grupo acetil (m/z = 43) e a adicao de um hidrogenio ao atomo de nitrogenio, sendo
este o fragmento mais estavel. Em m/z = 92 tem-se a saıda do grupo nitro e originando
um cation composto pela molecula de benzeno ligada a um NH. A Figura 17 ilustra o
espectro de massas da p-nitroacetanilida. A biblioteca NIST mostrou uma probabilidade
de 85% de chance do composto ser a p-nitroacetanilida.
5.1.3 p-nitroanilina
Com a confirmacao da sıntese da p-nitroacetanilida, o processo de desprotecao do
grupo amina foi iniciado, ou seja, o processo de desacetilacao. Este, envolveu aqueci-
mento para que a amida, presente na estrutura da molecula, sofresse hidrolise acida. O
mecanismo da reacao de desacetilacao e mostrado na Figura 18.
O mecanismo ilustrado na Figura 18, mostra na primeira etapa a protonacao do
oxigenio do grupo carboxila e, consequentemente, a ativacao do carbono do mesmo grupo.
A agua que esta no meio atua como nucleofilo reage com o carbono da carboxila. Em
seguida, o nitrogenio ligado ao carbono carboxılico, por meio de um rearranjo, captura um

Capıtulo 5. RESULTADOS E DISCUSSAO 37
Figura 17 – Espectro de massas da amostra da p-nitroacetanilida.
Figura 18 – Mecanismo da reacao de sıntese da p-nitroanilina a partir da p-nitroacetanilida
hidrogenio ligado ao oxigenio positivo, ficando com carga positiva. Dessa forma, a ligacao
entre esse nitrogenio e o carbono se rompe atraves de uma quebra de ligacao heterolıtica.
O espectro de infravermelho para a amostra de p-nitroanilina e mostrado na Fi-

Capıtulo 5. RESULTADOS E DISCUSSAO 38
gura 19.
Figura 19 – Espectro de infravermelho da p-nitroanilina.
Em relacao a Figura 15, e possıvel notar que na regiao de maior numero de onda
e onde ocorrem as principais alteracoes do espectro. Em 3458 cm−1 e 3367 cm−1, sao
encontrados as absorcoes referentes as aminas primarias. Outra absorcao que ajuda a
confirmar o resultado positivo da sıntese e aquela em 1613 cm−1. Esta, e devido a vibracao
de flexao (do tipo “tesoura”) da ligacao N-H das aminas primarias, uma vez que a faixa
que abrange 1640 ate 1560 cm−1 esta relacionada com este tipo de vibracao e apresenta
bandas um pouco mais largas e de media intensidade para as aminas primarias (PAVIA
et al., 2013).
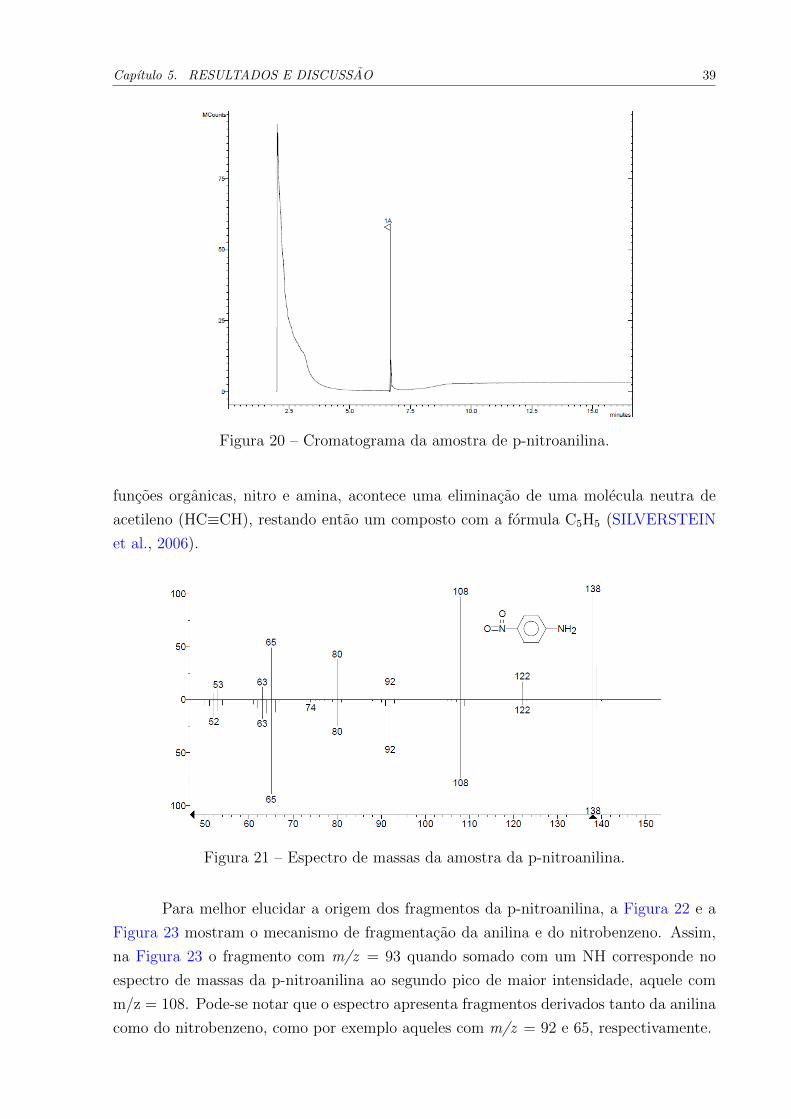
A analise cromatografica da amostra (Figura 20) apresentou um pico com tempo
de retencao igual a 6,9 minutos.
Quando analisado pelo espectrometro de massas, esse pico apresentou, de acordo
com a biblioteca NIST, 79,4% de probabilidade de ser o composto esperado. No espectro
de massa mostrado na Figura 21, e possıvel notar os picos com m/z igual a: 138, 122, 108,
92 e 65. O pico em 138, o pico base, e o possıvel pico do ıon molecular, uma vez que a p-
nitroanilina possui uma massa molecular de 138,12 g.mol−1. Em m/z = 122 tem-se a saıda
do grupo NH2 do ıon molecular. O pico em m/z = 108 e o segundo de maior intensidade,
consequentemente refere-se ao segundo produto da fragmentacao que e mais estavel. Este
e devido a saıda do NO presente na estrutura do ıon molecular. Quanto ao pico em m/z
= 92, ele ocorre com a saıda da funcao nitro. Ja para m/z=65, apos a perda das duas
Capıtulo 5. RESULTADOS E DISCUSSAO 39
Figura 20 – Cromatograma da amostra de p-nitroanilina.
funcoes organicas, nitro e amina, acontece uma eliminacao de uma molecula neutra de
acetileno (HC≡CH), restando entao um composto com a formula C5H5 (SILVERSTEIN
et al., 2006).
Figura 21 – Espectro de massas da amostra da p-nitroanilina.
Para melhor elucidar a origem dos fragmentos da p-nitroanilina, a Figura 22 e a
Figura 23 mostram o mecanismo de fragmentacao da anilina e do nitrobenzeno. Assim,
na Figura 23 o fragmento com m/z = 93 quando somado com um NH corresponde no
espectro de massas da p-nitroanilina ao segundo pico de maior intensidade, aquele com
m/z = 108. Pode-se notar que o espectro apresenta fragmentos derivados tanto da anilina
como do nitrobenzeno, como por exemplo aqueles com m/z = 92 e 65, respectivamente.

Capıtulo 5. RESULTADOS E DISCUSSAO 40
Figura 22 – Fragmentacao para a anilina apos o impacto de eletrons sofrido no es-pectrometro de massas.
Figura 23 – Fragmentacao para o nitrobenzeno apos o impacto de eletrons sofrido noespectrometro de massas.
5.1.4 p-(4-nitrobenzenoazo)-fenol
O processo de diazotacao de uma amina primaria envolve uma serie de etapas.
Quando colocado na presenca de um acido forte, o acido nitroso se dissocia formando ıons
NO+. Essa reacao e mostrada na Figura 24.
Figura 24 – Reacao de formacao do ıon nitrosonio.
Os ıons +NO juntamente com o nitrogenio presente na amina, reagem e dao origem
ao ıon N-nitrosamonio como um intermediario, em seguida esse intermediario perde um
proton e forma a N-nitrosamina. Esta, por meio de tautomerizacao, transforma-se em um
diazoidroxido. Contudo, na presenca de acido, o diazoidroxido perde uma agua e origina
o ıon diazonio (SOLOMONS; FRYHLE, 2005b).
A reacao de acoplamento diazo entre a p-nitroanilina e o fenol em que ocorre a
formacao do p-(4-nitrobenzenoazo)-fenol e mostrada na Figura 25.
Os fenois acoplam-se mais facilmente com os cations de arenodiazonio em solucoes
com carater pouco alcalino, isto porque nestas condicoes o fenol apresenta-se, em uma
consideravel quantidade, como ıons fenoxidos, que sao mais reativos frente uma reacao de
substituicao eletrofılica (MORRISON; BOYD, 2002). No entanto quando o pH e maior

Capıtulo 5. RESULTADOS E DISCUSSAO 41
Figura 25 – Reacao de formacao do p-(4-nitrobenzenoazo)-fenol
que 10, o sal de arenodiazonio comeca a reagir com o proprio ıon hidroxido para formar
um diazoidroxido, que nao e muito reativo (SOLOMONS; FRYHLE, 2005b).
O espectro de infravermelho para a amostra (Figura 26) apresenta algumas evidencias
da sıntese do p-(4-nitrobenzenoazo)-fenol. A absorcao em 3340 cm−1 sugere a presenca
de OH proveniente de um fenol. Esta banda e caracterizada pela vibracao de estiramento
O-H o que ocasiona um banda ampla (PAVIA et al., 2013). A absorcao em 1233 cm−1
ajuda a confirmar a presenca de um OH, uma vez que esta e caracterıstica da vibracao
de estiramento C-O quando o composto e um fenol. Outras duas bandas de absorcao de
grande importancia neste espectro sao aquelas em 1599 cm−1 e 1468 cm−1 que sao refe-
rentes ao estiramento do anel aromatico (C=C) que muito frequente ocorrem em pares.
Em 3044 cm−1 encontra-se a vibracao relacionada ao movimento de estiramento do car-
bono sp2 com um hidrogenio (=C-H), presente em aneis aromaticos. Os dois ultimos
picos de maior intensidade, proximos de 750 e 680 cm−1, tambem sao caracterısticos de
aneis aromaticos. Quanto as vibracoes devido a presenca do grupo NO2, estas possivel-
mente encontram-se sobrepostas. A ligacao que caracteriza os compostos azo, N=N, e
verificada pela presenca da vibracao de deformacao N=N em 1364 cm−1, caracterıstica de
azobenzenos assimetricos (GUP; GIZIROGLU; KıRKAN, 2007).
Nao foram realizadas analises cromatograficas devido ao equipamento encontrar-se
fora de operacao.

Capıtulo 5. RESULTADOS E DISCUSSAO 42
Figura 26 – Espectro de infravermelho do p-(4-nitrobenzenoazo)-fenol.
5.2 RESULTADOS DOS PROCEDIMENTOS USANDO COMO
MATERIAL DE PARTIDA A AGUA VERMELHA
Uma analise da agua vermelha realizada por Calegari et al. (2015) revelou o per-
fil dos nitroaromaticos presentes em uma fracao aquosa de agua vermelha residual. O
resultado dessa analise e apresentado na Tabela 3.
Tabela 3 – Perfil de nitroaromaticos encontrados em uma fracao aquosa de agua vermelharesidual.
Composto % Total
2,6-Dinitrotolueno (2,6-DNT) 11,602,4-Dinitrotolueno (2,6-DNT) 28,093,5-Dinitrotolueno (3,5-DNT) 0,362,4,6-Trinitrobenzeno (TNB) 2,412,4,6-Trinitrotolueno (TNT) 1,024-Amino-2,6-Dinitrotolueno 8,64
Fonte: CALEGARI et al., 2015.

Capıtulo 5. RESULTADOS E DISCUSSAO 43
A amostra de 10 mL de agua vermelha, apos 4 horas na chapa de aquecimento,
apresentou-se com certo aspecto pastoso, e rendeu 2,1554 g de possıveis nitroaromaticos.
Em seguida, foi realizado o processo de reducao em meio acidificado e com uso de ultra-
som. O ultra-som, por gerar muito mais energia no sistema, traz resultados melhores
do que o uso de um simples agitador. Como as aminas aromaticas que se formaram
encontravam-se em meio aquoso, onde apresentam grande solubilidade, no processo de
extracao foram enfrentadas algumas dificuldades.
Na primeira tentativa de extracao nao houve solubilizacao das aminas na mistura
de solventes organicos, possivelmente porque a mistura de solvente era muito apolar.
Na segunda tentativa, o uso do tolueno foi devido a presenca do anel benzenico que
aparece tanto na estrutura das aminas formadas quanto no proprio tolueno. Como na
agua vermelha os compostos sao derivados do tolueno, a amostra reduzida deve conter
aminas que sao tambem derivadas do tolueno, e assim ocorrer uma interacao entre o
solvente usado na extracao e essas aminas. Neste caso tambem nao houve solubilizacao
da amostra no solvente.
Na terceira tentativa de extracao obteve-se uma emulsao, como pode ser verificado
na Figura 27.
Figura 27 – Imagem da amostra com caracterısticas de emulsao. A foto da esquerdamostra a visao lateral, e a da direita, a superior.
As emulsoes por serem termodinamicamente instaveis precisam de energia para
ser formarem. No caso da emulsao formada na terceira tentativa de extracao, a energia
fornecida foi atraves da agitacao realizada pra que houvesse a interacao entre o solvente
e as aminas na fase aquosa. Na imagem mostrada na Figura 27, pode-se perceber que a
emulsao ja havia comecado a retornar para o estado estavel, onde a fase organica encontra-
se separada da aquosa, uma vez que nao foram introduzidos no sistema qualquer agente
emulsificante para aumentar a estabilidade cinetica desta emulsao.
Com o intuito de verificar a formacao produtos de reducao dos nitroaromaticos
presentes na agua vermelha e separa-los da fase aquosa, foi realizado o processo de ace-
tilacao da amostra, cujo mecanismo pode ser visto na Figura 10. Neste caso, porem,

Capıtulo 5. RESULTADOS E DISCUSSAO 44
pode haver um, dois ou ate tres sıtios para que o processo de acetilacao possa acorrer,
diferentemente da anilina, que e formada pela presenca apenas de um grupo NH2.
Mediante o processo de derivatizacao foi observado uma separacao de fase no meio
reacional, como era esperado, mas nao houve formacao de cristais. Com esse resultado
nao foi possıvel proceder com a filtracao a vacuo, como no caso da acetanilida. Uma parte
da amostra foi coletada, aquela que continha a fase organica, e adicionou-se ao meio um
agente secante, o sulfato de magnesio anidro, para retirar a agua residual. Retirou-se uma
alıquota, que foi analisada pela tecnica de cromatografia em camada fina.
A Figura 28 mostra o resultado da analise cromatografica apos a revelacao. Ob-
servando a “corrida” das amostras na Figura 28, nota-se que a amostra de acetanilida
percorreu uma distancia de 5,4 cm, e a amostra analisada, 5,0 cm. Alem da mancha
em 5,0 cm, nota-se uma faixa muito intensa que se estende desde 5,4 cm ate 8,0 cm, que
foi mesma distancia percorrida pela fase movel. De acordo com estes resultados, o Rf
calculado para a acetanilida foi de 0,675 e para a amostra foi de 0,625. Como ja menci-
onado na subsecao 3.4.2, os Rf ideais encontram-se entre 0,4 e 0,6, logo os Rf calculados
nao distanciam-se muito da idealidade. Tendo em vista a proximidade destes valores, e
possıvel considerar a formacao de compostos acetilados na amostra analisada, aquela a
direita na Figura 28. Contudo, nao se pode desconsiderar a presenca de outros compostos
na amostra.
Figura 28 – Imagem da cromatoplaca (CCD) com a amostra padrao de acetanilida aesquerda e com a amostra derivada da agua vermelha a direita.
Esse resultado sugere, pois, que apos o processo de reducao e acetilacao houveram
modificacoes na estrutura da amostra, ou seja, os sıtios onde apresentavam o grupo NH2
foram, possivelmente, acetilados.

45
6 CONSIDERACOES FINAIS
Neste trabalho foram realizadas reacoes de sıntese organica envolvendo processo
de acetilacao, nitracao eletrofılica, desacetilacao e acoplamento diazo, alem da utilizacao
de tecnicas de purificacao como extracao lıquido-lıquido e cristalizacao. O trabalho foi
realizado em duas situacoes: uma que envolvia reagente puros e outra, a agua vermelha.
Na primeira situacao foi possıvel verificar os mecanismos das reacoes e caracterizar os pro-
dutos formados, verificando o exito das reacoes. Neste caso nao houveram dificuldades
nas etapas sinteticas, nas purificacoes e na etapa de caracterizacao. Mesmo quando foi
verificada a baixa probabilidade (26%) do composto sintetizado ser a acetanilida, a etapa
seguinte com a alta possibilidade (85%) do composto formado ser a p-nitroacetanilida
e o sucesso nas subsequentes sınteses, possibilitaram a confirmacao da obtencao da ace-
tanilida. Para a segunda situacao, quando o material de partida foi a agua vermelha,
houve grande dificuldade de obter os resultados desejados. Parte desta dificuldade e de-
vida as caracterısticas da propria agua vermelha, e outra parte, a falta de equipamentos
para a caracterizacao dos materiais na fase inicial e apos cada reacao, pois os mesmos
encontravam-se em estado inoperacional.
A proposta de utilizar metodos rapidos e seletivos de reducao e acetilacao de
compostos nitroaromaticos em agua vermelha pode ser verificada quando analisado os
resultados via CCD, pois os resultados foram proximos para as amostras analisadas. Este
trabalho pode ser estendido e otimizado para que os nitroaromaticos presentes na agua
vermelha possam ser recuperados, tratados e ate mesmo utilizados, e, assim, contribuir
positivamente com o meio ambiente.

46
REFERENCIAS
AGRAWAL, A.; TRATNYEK, P. G. Reduction of nitro aromatic compounds byzero-valent iron metal. Environmental Science & Technology, v. 30, n. 1, p.153–160, 1996. Disponıvel em: 〈http://dx.doi.org/10.1021/es950211h〉. Acesso em: 16mar. 2016.
BAPTISTELLA, L. H. B.; GIACOMINI, R. A.; IMAMURA, P. M. Sıntese dosanalgesicos paracetamol e fenacetina e do adocante dulcina: um projeto para quımicaorganica experimental. Quımica Nova na Escola, v. 26, n. 2, p. 284–286, 2003.
BIGELOW, H. E.; ROBINSON, D. B. Azobenzene. Organic Syntheses, v. 22, p. 28,1942. Disponıvel em: 〈http://www.orgsyn.org/demo.aspx?prep=CV3P0103〉. Acesso em:30 set. 2015.
BORGES, A. D. L. et al. Sıntese de sulfadiazina e sulfadiazina de prata em escalasemi-micro: pratica experimental em sıntese de farmacos. Quımica Nova, scielo,v. 28, p. 727 – 731, 08 2005. Disponıvel em: 〈http://www.scielo.br/scielo.php?script=sci arttext&pid=S0100-40422005000400030&nrm=iso〉. Acesso em: 03 mai. 2016.
CALEGARI, M. A. et al. Identficacao, caracterizacao e recuperacao de aminasaromaticas presentes na agua vermelha. Synergismus Scyentifica UTFPR, v. 10,p. 1–7, 2015.
COLLINS, C.; BRAGA, G. L.; BONATTO, P. S. Introducao a MetodosCromatograficos. 7. ed. Campinas: Unicamp, 1997.
COLLINS, C. H. Michael Tswett e o “nascimento” da cromatografia. ScientiaChromatographica, v. 1, n. 1, 2009.
CRISTIANO, R. et al. Sıntese de cristais lıquidos derivados do nitroazobenzeno: umaproposta de sıntese multi-etapas aplicada as aulas de quımica organica experimental.Quımica Nova, v. 37, n. 1, p. 181–185, 2014.
DEGANI, A. L. G.; CASS, Q. B.; VIEIRA, P. C. Cromatografia: um breve ensaio.Quımica Nova na Escola, n. 7, p. 21–25, 1998.
DONG, J. et al. Effects of ph and particle size on kinetics of nitrobenzene reduction byzero-valent iron. Journal of Environmental Sciences, v. 22, n. 11, p. 1741 – 1747,2010. ISSN 1001-0742. Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S1001074209603144〉. Acesso em: 10 mar. 2016.
FALONE, S. Z.; VIEIRA, E. M. Adsorcao/dessorcao do explosivo tetril em turfa e emargissolo vermelho amarelo. Quımica Nova, v. 27, n. 6, p. 849–854, 2004.
GREENE, T. W.; WUTS, P. G. M. Protective Groups in Organic Synthesis. 2. ed.New York: Wiley, 2005.
GUP, R.; GIZIROGLU, E.; KıRKAN, B. Synthesis and spectroscopic propertiesof new azo-dyes and azo-metal complexes derived from barbituric acid andaminoquinoline. Dyes and Pigments, v. 73, n. 1, p. 40 – 46, 2007. Disponıvel em:

REFERENCIAS 47
〈http://www.sciencedirect.com/science/article/pii/S0143720805003323〉. Acesso em: 22mai. 2016.
KEUM, Y.-S.; LI, Q. X. Reduction of nitroaromatic pesticides with zero-valent iron.Chemosphere, v. 54, n. 3, p. 255 – 263, 2004. ISSN 0045-6535. Disponıvel em:〈http://www.sciencedirect.com/science/article/pii/S0045653503007823〉. Acesso em: 16mar. 2016.
LIU, G.-h.; ZHU, S.-N.; YE, Z. Reduction in the acute toxicity of explosive wastewatercontaining toxic nitroaromatic compounds by a nanoscale zerovalent iron pretreatmentprocess. Water, Air, & Soil Pollution, v. 223, n. 8, p. 5049–5055, 2012. ISSN1573-2932. Disponıvel em: 〈http://dx.doi.org/10.1007/s11270-012-1257-7〉. Acesso em:16 mar. 2016.
LUDWICHK, R. et al. Characterization and photocatalytic treatability of red waterfrom brazilian tnt industry. Journal of Hazardous Materials, v. 293, p. 81–86, Ago2015.
LYONS, R. E.; SMITH, L. T. Die reduktion von nitroverbindungen mit eisen undloslichen chloriden. Berichte der deutschen chemischen Gesellschaft (A and BSeries), WILEY-VCH Verlag, v. 60, n. 1, p. 173–182, 1927. ISSN 1099-0682. Disponıvelem: 〈http://dx.doi.org/10.1002/cber.19270600132〉. Acesso em: 21 mar. 2016.
MCCORMICK, N. G. et al. Microbial transformation of 2,4,6-trinitrotoluene and othernitroaromatic compounds. Applied and Environmental Microbiology, n. 949-958,p. 226–228, 1976.
MORITA, T.; ASSUMPcaO, R. M. V. Manual de Solucoes, Reagentes e Solventes.2. ed. Sao Paulo: Edgard Blucher, 1995.
MORRISON, R. T.; BOYD, R. N. Organic Chemistry. 6. ed. New Delhi: Prentice-Hall,Inc, 2002. p. 733-742, 763-777.
OH, S.-Y. et al. Zero-valent iron pretreatment for enhancing the biodegradabilityof {RDX}. Water Research, v. 39, n. 20, p. 5027 – 5032, 2005. ISSN 0043-1354.Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S0043135405005762〉.Acesso em: 16 mar. 2016.
PACHECO, A. de O. et al. Biotransformations of nitro-aromatic compounds toamines and acetamides by tuberous roots of arracacia xanthorrhiza and beta vulgarisand associated microorganism (Candida guilliermondii). Enzyme and MicrobialTechnology, v. 42, 2007. Disponıvel em: 〈http://gen.lib.rus.ec/scimag/index.php?s=10.1016/j.enzmictec.2007.08.001〉. Acesso em: 03 mai. 2016.
PAULAI, F. A. R.; SERRANO, S. H. P.; TAVARES, L. C. Aspectos mecanısticos dabioatividade e toxicidade de nitrocompostos. Quımica Nova, scielo, v. 32, p. 1013 –1020, mar. 2009. ISSN 0100-4042. Disponıvel em: 〈http://www.scielo.br/scielo.php?script=sci arttext&pid=S0100-40422009000400032〉. Acesso em: 10 mar. 2016.
PAVIA, D. L. et al. Quımica Organica Experimental: tecnicas em pequena escala.2. ed. Porto Alegre: Bookman, 2009. 887 p.

REFERENCIAS 48
PAVIA, D. L. et al. Introduction to Spectroscopy. 5. ed. Stamford: CengageLearning, 2013. 785 p.
PIZZOLATTI, M. G.; YUNES, R. A. Azoxybenzene formation from nitrosobenzene andphenylhydroxylamine. a unified view of the catalysis and mechanisms of the reactions.J. Chem. Soc., Perkin Trans. 2, The Royal Society of Chemistry, p. 759–764, 1990.Disponıvel em: 〈http://dx.doi.org/10.1039/P29900000759〉. Acesso em: 21 mar. 2016.
PODLIPNa, R.; POSPısILOVa, B.; VANeK, T. Biodegradation of 2,4-dinitrotoluene bydifferent plant species. Ecotoxicology and Environmental Safety, v. 112, p. 54 – 59,2015. ISSN 0147-6513. Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S0147651314003467〉. Acesso em: 13 mar. 2016.
RICKERT, D. E. Toxicity of nitroaromatic compounds. New York: HemispherePublishing Corporation, 1985. (Chemical Industry Institute of toxicology series).
RODRIGUES, M. B. Tratamento de efluentes proveniente da fabricacao de TNTde uma industria de explosivos utilizando preocessos redutivos e oxidativosavancados. 2005. 133 f. Tese (Doutorado em Biotecnologia Industrial) — Faculdade deEngenharia Quımica de Lorena, Lorena.
RODRIGUES, M. B. et al. Caracterizacao fısica, quımica e ecotoxicologica de efluenteda industria de fabricacao de explosivos. Quımica Nova, v. 30, n. 7, p. 1623–1627, Ago2007.
RODRIGUES, M. B.; SILVA, F. T. da; PAVIA, T. C. B. Characterization of wastewaterfrom the brazilian tnt industry. Journal of hazardous materials, v. 164, n. 1, p.385–388, 2009.
SAIKIA, U. P. et al. Selective n-acetylation of aromatic amines using acetonitrileas acylating agent. Tetrahedron Letters, 2 2016. Disponıvel em: 〈http://gen.lib.rus.ec/scimag/index.php?s=10.1016/j.tetlet.2016.01.108〉. Acesso em: 03mai. 2016.
SHABBIR, M. et al. Synthesis, biological and electrochemical evaluation of novelnitroaromatics as potential anticancerous drugs. Bioelectrochemistry, v. 104, p. 85– 92, 2015. ISSN 1567-5394. Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S1567539415000316〉. Acesso em: 10 mar. 2016.
SILVERSTEIN, R. M. et al. Identificacao Espectrometrica de CompostosOrganicos. 4. ed. Rio de Janeiro: LTC, 2006.
SOLOMONS, G. T. W.; FRYHLE, C. B. Quımica Organica. 8. ed. Rio de Janeiro:LTC, 2005. v. 1.
SOLOMONS, G. T. W.; FRYHLE, C. B. Quımica Organica. 8. ed. Rio de Janeiro:LTC, 2005. v. 2.
SOUZA, C.; PERALTA-ZAMORA, P. Degradacao de corantes reativos pelo sistemaferro metalico/peroxido de hidrogenio. Quımica Nova, n. 2, p. 226–228, 2005.

REFERENCIAS 49
SUN, H. et al. A novel integrated active capping technique for the remediationof nitrobenzene-contaminated sediment. Journal of Hazardous Materials,v. 182, n. 1–3, p. 184 – 190, 2010. ISSN 0304-3894. Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S0304389410007533〉. Acesso em: 16 mar.2016.
THIRUPATHI, B. D. P. A highly selective and efficient acetylation of alcoholsand amines with acetic anhydride using NaHSO4·SiO2 as a heterogeneous catalyst.Journal of Molecular Catalysis A: Chemical, v. 269, 2007. Disponıvel em:〈http://gen.lib.rus.ec/scimag/index.php?s=10.1016/j.molcata.2006.12.029〉.
THOMAS, J. M.; HERNANDEZ, R.; KUO, C.-H. Single-step treatment of 2,4-dinitrotoluene via zero-valent metal reduction and chemical oxidation. Journal ofHazardous Materials, v. 155, n. 1–2, p. 193 – 198, 2008. ISSN 0304-3894. Disponıvelem: 〈http://www.sciencedirect.com/science/article/pii/S0304389407016767〉. Acesso em:11 mar. 2016.
YAN, X.-F. et al. Quantitative structure–activity relationships of nitroaromatics toxicityto the algae (scenedesmus obliguus). Chemosphere, v. 59, n. 4, p. 467 – 471, 2005.ISSN 0045-6535. Disponıvel em: 〈http://www.sciencedirect.com/science/article/pii/S0045653505002390〉. Acesso em: 10 mar. 2016.
ZHANG, M.; ZHAO, Q.; YE, Z. Organic pollutants removal from 2, 4, 6-trinitrotoluene(tnt) red water using low cost activated coke. Journal of Environmental Sciences,v. 23, n. 12, p. 1962–1969, 2011.