“pesquisa do padrão de metilação dos geneslivros01.livrosgratis.com.br/cp085050.pdf ·...
TRANSCRIPT

UNIVERSIDADE DE SÃO PAULO
FACULDADE DE MEDICINA DE RIBEIRÃO PRETO
“Pesquisa do Padrão de Metilação dos genes
GRB10, MEST e da Região 11p15 na Síndrome de
Silver-Russell”.
Jaqueline Carvalho de Oliveira
RIBEIRÃO PRETO
2009

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

UNIVERSIDADE DE SÃO PAULO
FACULDADE DE MEDICINA DE RIBEIRÃO PRETO
“Pesquisa do Padrão de Metilação dos genes
GRB10, MEST e da Região 11p15 na Síndrome de
Silver-Russell”.
Jaqueline Carvalho de Oliveira
Dissertação Apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para Obtenção do Título de Mestre em Ciências.
Área de Concentração: Genética
Orientadora: Profa. Dra. Ester Silveira Ramos.
RIBEIRÃO PRETO
2009

AUTORIZO A DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE.
FICHA CATALOGRÁFICA
Preparada pela Biblioteca Central do Campus Administrativo de Ribeirão Preto/USP
Oliveira, Jaqueline Carvalho de
Análise do Padrão de Metilação dos genes GRB10, MEST e da Região 11p15 na Síndrome de Silver-Russell.
90 p.:il. ; 30cm
Dissertação de Mestrado apresentada à Faculdade Medicina de Ribeirão Preto/USP. Área de concentração: Genética.
Orientadora: Ramos, Ester Silveira.
1. Síndrome de Silver-Russell 2. Metilação 3. GRB10 4. MEST
5. 11p15

FOLHA DE APROVAÇÃO
Jaqueline Carvalho de Oliveira Pesquisa do Padrão de Metilação dos genes GRB10, MEST e da Região 11p15 na Síndrome de Silver-Russell
Dissertação Apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para Obtenção do Título de Mestre em Ciências. Área de Concentração: Genética
Aprovado em:____/____/____
Banca Examinadora Profª Dra Ester Silveira Ramos Instituição:__________________________Assinatura____________________ Prof Dr Instituição:__________________________Assinatura____________________ Prof Dr Instituição:__________________________Assinatura____________________

Dedico aos meus pais, por todo amor e dedicação. Por sempre terem acreditado em mim e me incentivado.

AGRADECIMENTOS
Primeiramente a Deus por ter me permitido chegar até aqui, sempre me
ajudando a vencer os obstáculos.
A Profª.dra. Ester Silveira Ramos, pela oportunidade e confiança depositada
em mim. Por toda orientação e apoio essenciais a esse trabalho e à minha
formação.
Aos médicos do Ambulatório de Genética do HCFMRP, principalmente ao Dr.
Jair Huber, por todo o envolvimento no trabalho e ajuda inestimável durante a coleta
das amostras.
Às famílias dos pacientes que prontamente aceitaram participar da pesquisa,
alguns precisando se locomover até o ambulatório de genética exclusivamente para
a doação das amostras.
Ao apoio financeiro: Fundação de Amparo à Pesquisa do Estado de São
Paulo (FAPESP), Fundação de Amparo ao Ensino Pesquisa e assistência do
Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto (FAEPA),
Conselho Nacional de Desenvolvimento de Pesquisa (CNPq) e Cordenação de
Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Ao Prof. Dr. Raysildo Lôbo e a Profa. Dra. Lúcia Regina Martelli, pela
convivência e incentivo.
Aos membros da Banca Examinadora, por terem aceitado participar desta
avaliação.
Ao doutor Marcus Vinícius de Matos Gomes, pela ajuda nesse trabalho, às
padronizações das primeiras metodologias utilizadas e valiosas discussões sobre as
outras metodologias e sobre os resultados.

À Karine Coelho pela grande ajuda na parte prática desse trabalho, muitas
vezes dividindo comigo as tarefas para que tudo fosse realizado mais rapidamente.
À Lisandra Cristina Caetano da Silva, pela ajuda nas reações de PCR em
tempo-real e também por aguentar minhas lamentações quando nada dava certo.
À Adriane Araújo de Oliveira, pela contribuição nas reações e também por ter
buscado algumas amostras urgentes no meio da tarde.
Ao doutor Álvaro Fabrício Lopes Rios pela grande ajuda e pelas discussões
sobre as metodologias utilizadas.
Às meninas do bloco C que sempre estavam dispostas à ajudar no que fosse
preciso: Paula Lumy Takegushi, Adriana Renzi, Andrea Martins da Silva, Hélida
Magalhães, Flávia Gaona de Oliveira Gennaro, Cristiana Libardi Miranda, Francielle
Marques Araújo, Juliana Meola e também ao Murilo Racy Soares.
À todos os amigos do bloco C ainda não citados: Marcelo Rodrigues,
Fernanda Prado Elias, Flávia Cappelo Donabela, Jeferson Nomelini, Juliana Fabricia
Cuzzi, Ana Carolina Laus, Daniel Blassioli Dentillo, Luciana Caricati Veiga Castelli,
Maria Silvina Juchniuk de Vozzi, Pedro Alejandro Vozzi, Rita de Cássia Pessotti,
Juliana Serafim, Mariana , Ciro Silveira e Pereira.
Aos técnicos do bloco C: Sílvio, Reginaldo e Marli, pela colaboração nos
experimentos e preparo das soluções de laboratório e também ao Luis Bezerra,
Paulo Bezerra e Marco Conrado pela colaboração nos assuntos de informática.
A Profa. Dra. Chong Ae Kim e Profa. Dra. Angela Maria Vianna Morgante
pelas amostras do pacientes provenientes da Universidade de São Paulo, também
analisadas nesse trabalho.
As secretárias do departamento de genética, Susie, Maria Aparecida e Silvia,
pela atenção e convivência nos últimos anos.

A todos meus amigos de Ribeirão Preto, Londrina e Cornélio Procópio, por
fazerem parte da minha vida e permitirem que as etapas da vida sejam mais
prazerosas.
Ao meu namorado, Paulo Emílio Ferreira e Alvarenga, pela ajuda diretamente
nesse trabalho e por estar sempre ao meu lado.
À minha irmã, Simone Carvalho de Oliveira, por todo apoio e pela nossa
amizade, por mesmo distante estar sempre presente.
Aos meus pais, Jusevaldo Amorim de Oliveira e Ivani Carvalho A. de Oliveira,
por tudo que sempre fizeram por mim. Por tudo que foi investido, pelo incentivo e por
sempre me mostrarem que era capaz e que com eles podia contar.

RESUMO
OLIVEIRA, J. C. Pesquisa do Padrão de Metilação dos Genes GRB10, MEST e da Região 11p15 na Síndrome de Silver-Russell. 2009. 71 f. Dissertação (Mestrado em Biologia – Genética). Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo. Ribeirão Preto. 2009. A síndrome de Silver-Russell (SSR) é caracterizada por retardo no crescimento intra-
uterino associado a várias dismorfias características, sendo clínica e geneticamente
heterogênea, com relatos de diferentes cromossomos e genes potencialmente
envolvidos em sua etiologia. Porém, apenas os cromossomos 7 e 11 têm-se
mostrado consistentemente implicados na SSR, ambos contendo regiões envolvidas
com imprinting genômico. Este último é um mecanismo epigenético no qual os dois
alelos de um determinado gene são expressos diferencialmente dependendo da
origem parental. Os genes MEST, GRB10, mapeados no cromossomo 7, e a região
11p15 estão sob regulação do imprinting genômico e são sugeridos na etiologia da
SSR pelo envolvimento no crescimento fetal. No presente trabalho, foram avaliados
o perfil de metilação nas regiões diferencialmente metiladas dos genes MEST e
GRB10, além da análise de metilação de duas regiões controladoras de imprinting
na região 11p15, H19DMR e KvDMR, por metodologias qualitativas (Análise de
restrição combinada com bissulfito e PCR específica para a metilação) e quantitativa
(Digestão Enzimática Sensível à Metilação Associada à PCR em Tempo Real).
Foram selecionados 15 pacientes com SSR além de cinco casos analisados
separadamente (um indivíduo com SSR filho de casal consaguíneo e com irmão
apresentando fenótipo de hipercrescimento, três irmãos com SSR de uma mesma
família e um indivíduo com cariótipo 45,X[25]/46,XX[75] e características típicas da
SSR e da síndrome de Turner). Foi observada a hipermetilação para paciente,
6,7% (1/15), na região diferencialmente metilada do gene MEST, hipometilação para

20% (3/15) dos pacientes na H19DMR, não sendo encontrada alteração na região
da KvDMR, para o grupo analisado. No entanto, hipermetilação dessa última região
foi encontrada para o paciente filho de casal consanguíneo e nos três irmãos,
sugerindo o envolvimento também dessa região na etiologia da SSR. Não foi
possível correlacionar estatisticamente os resultados laboratoriais e a gravidade dos
achados clínicos, entretanto as metodologias utilizadas se mostraram eficientes na
confirmação diagnóstica da SSR, trazendo benefícios diretos para o diagnóstico
mais rápido, além da contribuição para estudos posteriores que poderão auxiliar o
aconselhamento genético das famílias com portadores de SSR e desenvolvimento
de novos tratamentos.
Palavras-chave: metilação; Síndrome de Silver Russell; MEST; GRB10; KvDMR;
H19DMR.
(Versão parcial, a original sem cortes será disponibilizada após publicação).

ABSTRACT
OLIVEIRA, J. C. Methylation pattern at the GRB10 and MEST genes and the 11p15 region in patients with Silver-Russell syndrome. 2009. 71f. Dissertation (Master degree in Biology – Genetics). Medicine School of Ribeirão Preto, University of São Paulo. Ribeirão Preto. 2008.
The Silver-Russell syndrome (SRS) is characterized by intrauterine growth
retardation, which is associated with characteristic dimorphic features. The SRS is a
clinically and genetically heterogeneous. Nevertheless, only chromosome 7 and 11
have been consistently implicated in patients with strict clinical diagnosis of SRS.
Genomic imprinting is a epigenetic phenomenon where the two alleles of certain
genes are expressed differentially, depending on the sex of the parent from whom
each was inherited. The MEST and the GRB10 genes (mapped in the chromosome
7) and the 11p15 region are imprinted and they are suggested in SRS etiology due to
their involvement in fetal growth. In this study, it was analyzed the methylation
pattern in the differential methylated regions in GRB10 and MEST genes and in the
imprinting control regions of 11p15, H19DMR and KvDMR, through qualitative
methods (Combined Bisulfite Restriction Analysis and Methylation Sensitive PCR)
and quantitative (Digestion Sensitive Associated with Real-Time PCR). It was
selected 15 patients with SRS besides five special cases (a individual SRS born of a
consanguineous crossing and a brother with increase of growth, three brothers with
SRS, an individual with 45,X[25]/46,XX[75] cariotype and features of Turner
syndrome and SSR). It was observed a hipermethylation in a patients, 6,7%
(1/15), in MEST gene region, partial hipomethylation in 20% (3/15) patients in
H19DMR. It was been detected partial hipermethylation in KvDMR region in the SRS
born of a consanguineous crossing and in the three brothers, suggesting the role of
this region in etiology of SRS. It was not possible to relate the laboratorial results with

the clinical findings, however, the utilized methods showed to be efficient tools for
diagnosing the SRS, allowing faster diagnosis. Another benefit is the support for
further research that can improve the genetic counseling for families with SRS
patients and the new possibilities of treatment.
Key-words: methylation; Silver-Russell syndrome; MEST; GRB10; KvDMR;
H19DMR.

ABREVIATURAS E SIGLAS
% porcentagem °C grau celsius A Adenina C Citosina CDKN1C cyclin-dependent kinase inhibitor 1C (inibidor de quinase dependente de ciclina 1C) CFTR cystic fibrosis transmembrane conductance regulator ct ciclo threshold - número do ciclo no qual o nível de fluorescência emitido pela amostra amplificada atingiu o nível arbitrário CTCF fator de ligação CCCTC DMR differential methylated region (região diferencialmente metilada) DMRs differential methylated regions (regiões diferencialmente metiladas) DNA deoxyribonucleic acid (ácido desoxirribonucléico) DNMT enzima DNA metiltransferases DNMT1 DNA metiltransferases 1 DNMT2 DNA metiltransferases 2 DNMT3 DNA metiltransferases 3 DUP dissomia uniparental G Guanina GRB10 growth factor receptor-binding protein 10 (proteína ligante de receptor de fator de crescimento 10) H19DMR região diferencialmente metilada H19 HCl ácido clorídrico ICR imprinting control region (região controladoras de imprinting)

ICRs imprinting control regions (regiões controladoras de imprinting) IGF insulin-like growth factor (fator de crescimento semelhante à insulina) IGF1 insulin-like growth factor 1 (fator de crescimento semelhante à insulina 1) IGF1R insulin-like growth factor I receptor (receptor de fator de crescimento semelhante à insulina 1) IGF2 insulin-like growth factor 2 (fator de crescimento semelhante à insulina 2) IGFBP insulin-like growth factor binding protein (proteína ligante de IGF) IR insulin receptors (receptor de insulina) kb kilobases KCl Cloreto de potássio KCNQ1 potassium channel, voltage-gated, kqt-like subfamily, member 1 KCNQ1OT1 KCNQ1-overlapping transcript 1 LIT1 long QT intronic transcript 1 mDUP7 dissomia uniparental materna do cromossomo 7 MEST mesoderm-specific transcript MgCl2 Cloreto de Magnésio mM milimolar MPLP multiplex ligation dependent probe analysis ncRNA RNA não-condificante OMIM online mendelian inheritance in man p braço curto do cromossomo PCR polymerase chain reaction (reação em cadeira da polimerase) PEG1 paternally expressed gene 1 (gene paternalmente expresso 1)

pH potencial hidrogeniônico PHLDA2 pleckstrin homology-like domain, family a, member 2 q braço longo do cromossomo RNA oxyribonucleic acid (ácido ribonucléico) SBW síndrome de Beckwith-Wiedemann SSR síndrome de Silver-Russell T Timina t translocação tRNA RNA transportador U Uracila uL microlitro UTR untranslated region (região não traduzida) µg micrograma

LISTA DE ILUSTRAÇÕES
Figura 1
Características clínicas encontradas em paciente com síndrome de SSR...........................................................................................................
03
Figura 2 Ideograma do cromossomo 7 humano...................................................... 10
Figura 3 Ideograma do cromossomo 11 humano.................................................... 12
Figura 4
Representação esquemática do mapa gênico da região cromossômica 11p15.........................................................................................................
14
Figura 5
Seqüência próxima à região do gene GRB10 analisada, GenBank AC004920..................................................................................................
25
Figura 6
Seqüência próxima à região do gene MEST analisada, GenBank NT_007933................................................................................................
26
Figura 7 Seqüência próxima à região H19DMR analisada, GenBank U50731....... 30
Figura 8 Seqüência próxima à região KvDMR1 analisada, GenBank U90095........ 32
Figura 9
Perfil de metilação do gene GRB10 pelo método COBRA em amostras de indivíduos saudáveis (controle)............................................................
34
Figura 10
Perfil de metilação do gene GRB10 pelo método COBRA em amostras de pacientes com síndrome de Silver-Russell...........................................
34
Figura 11
Perfil de metilação do gene MEST pelo método COBRA em amostras de indivíduos saudáveis (controle)...........................................................
35
Figura 12
Perfil de metilação do gene MEST pelo método COBRA em amostras de pacientes com síndrome de Silver-Russell...........................................
35
Figura 13
Metilação da H19DMR pelo método COBRA em amostras de pacientes com Silver-Russel......................................................................................
36
Figura 14 Gráfico de amplificação em tempo-real da H19DMR................................ 37
Figura 15
Comparação entre a média de porcentagem de metilação da H19DMR no grupo controle e no grupo de pacientes com síndrome de Silver-Russel........................................................................................................
38

Figura 16
Perfil de metilação da KvDMR pelo método MS-PCR em indivíduos saudáveis (controle)..................................................................................
40
Figura 17
Perfil de metilação da KvDMR pelo método MS-PCR em amostras de pacientes com síndrome de Silver-Russell................................................
40
Figura 18 Gráfico de amplificação em tempo-real da KvDMR................................... 42
Figura 19
Comparação entre a média de porcentagem de metilação da KvDMR no grupo controle e no grupo de pacientes com síndrome de Silver-Russell.......................................................................................................
42

LISTA DE TABELAS
Tabela 1
Resultados da H19DMR obtidos pelo método DESM-RT em amostras controle................................................................................................... 38
Tabela 2
Resultados da H19DMR obtidos pelo método DESM-RT em pacientes com Silver-Russell.................................................................................. 39
Tabela 3
Resultados da H19DMR obtidos pelo método DESM-RT em casos particulares............................................................................................. 39
Tabela 4
Resultados da KvDMR obtidos pelo método DESM-RT em amostras controle................................................................................................... 43
Tabela 5
Resultados da KvDMR obtidos pelo método DESM-RT em pacientes com Silver-Russell.................................................................................. 44
Tabela 6
Resultados da KvDMR obtidos pelo método DESM-RT em casos particulares............................................................................................. 44
Tabela 7
Resultado molecular e características clínicas dos pacientes com Silver-Russell.......................................................................................... 46

SUMÁRIO
1. INTRODUÇÃO.................................................................................................... 01 1.1. SÍNDROME DE SILVER-RUSSEL.................................................................. 02 1.2. ASPECTOS GENÉTICOS............................................................................... 04 1.3. EPIGENÉTICA E IMPRINTING GENÔMICO.................................................. 07 1.4. GENE GRB10.................................................................................................. 09 1.5. GENE MEST ................................................................................................... 11 1.6. REGIÃO 11p15................................................................................................ 11 2. OBJETIVOS........................................................................................................ 16 2.1. OBJETIVO GERAL.......................................................................................... 17 2.2. OBJETIVOS ESPECÍFICOS............................................................................ 17 3. CASUÍSTICA...................................................................................................... 18 3.1. PACIENTES..................................................................................................... 19 4. MATERIAL E MÉTODOS................................................................................... 20 4.1. ANÁLISE MOLECULAR................................................................................... 21 4.1.1. Coleta de Sangue Periférico...................................................................... 21 4.1.2. Extração de DNA......................................................................................... 21 4.1.3. Modificação do DNA com Bissulfito de Sódio.........................................
22
4.1.4. Identificação de Ilhas CpGs e Desenhos de Primers.............................. 23 4.1.5. Análise de Metilação .................................................................................. 24 4.1.5.1. Análise do padrão de metilação do gene GRB10...................................... 24 4.1.5.2. Análise do padrão de metilação do gene MEST........................................
26
4.1.5.3. Análise do Padrão de Metilação da H19DMR............................................ 27

4.1.5.3.1. PCR Análise Combinada de Bissulfito e Restrição Enzimática ( (COBRA)........................................................................................................ 27 4.1.5.3.2. Digestão Enzimática Sensível à Metilação Associada à PCR em T Tempo Real (DESM-RT)................................................................................ 28 4.1.5.4. Análise do Padrão de Metilação da KvDMR1..................................................... 30 4.1.5.4.1. PCR específica para a metilação (MS-PCR)................................. 30 4.1.5.4.2. Digestão Enzimática Sensível à Metilação Associada à PCR em T Tempo Real (DESM-RT)................................................................................ 31 5 RESULTADOS.................................................................................................... 33 5.1. ANÁLISE DO PADRÃO DE METILAÇÃO DO GENE GRB10........................ 34 5.2. ANÁLISE DO PADRÃO DE METILAÇÃO DO GENE MEST........................... 35 5.3. ANÁLISE DO PADRÃO DE METILAÇÃO DA H19DMR.................................. 36 5.3.1. COBRA......................................................................................................... 36 5.3.2. DESM-RT...................................................................................................... 36 5.4. ANÁLISE DO PADRÃO DE METILAÇÃO DA KVDMR1.................................. 40 5.4.1. MS-PCR........................................................................................................ 40 5.4.2. DESM-RT...................................................................................................... 41 5.5. CORRELAÇÃO DOS ACHADOS CLÍNICOS E MOLECULARES................... 45 6. DISCUSSÃO................................................................................................... 48 7. CONCLUSÃO.................................................................................................... 61 8. REFERÊNCIAS.................................................................................................. 63

1. INTRODUÇÃO1. INTRODUÇÃO1. INTRODUÇÃO1. INTRODUÇÃO

1. Introdução
2
1.1. SÍNDROME DE SILVER-RUSSELL
A síndrome de Silver-Russell (SSR; OMIM 180860) foi independentemente
descrita por Silver et al. (1953) e Russell (1954), recebendo, somente em 1961, essa
denominação (BLACK, 1961).
Essa síndrome é caracterizada por retardo no crescimento intra-uterino que
persiste no período pós-natal e apresenta incidência estimada entre 1:50.00 e
1:10.000 nascimentos (ANDERSON et al., 2002; MONK et al., 2002), não havendo
relação com sexo ou origem étnica (WEE, 2007).
As alterações “maiores” (ou principais) encontradas na maioria dos pacientes
e que caracterizam a síndrome são: baixo peso ao nascimento, baixa estatura
resultante de retardo do crescimento pós-natal e face triangular característica
(HITCHINS et al., 2001).
No entanto, algumas outras alterações “menores” auxiliam no diagnóstico e
são frequentemente observadas como a clinodactilia e/ou o encurtamento do quinto
dedo de mãos, a macrocefalia aparente e a assimetria corporal. Também podem
estar presentes alterações de orelha, rima labial com inclinação para baixo, ponte
nasal proeminente e filtro naso-labial bem demarcado, hipotonia e hipotrofia
muscular, atraso motor e neuropsicomotor, alterações dentárias, alteração de voz
(“esganiçada”), prega palmar única (ou simiesca), sindactilia, campodactilia,
artrogripose distal, manchas café-com-leite, puberdade precoce, alterações genitais
incluindo hipospádia, hérnia inguinal, atraso de fala/linguagem e dificuldade de
alimentação nos primeiros meses de vida (EGGERMANN et al., 1997; PRICE et al.,
1999; HITCHINS et al., 2001; ROSSI et al., 2006) (Figura 1).

1. Introdução
3
Figura 1. Características clínicas encontradas em paciente com síndrome de SSR. (A) Face triangular, macrocefalia aparente, orelhas displásicas, (B) clinodactilia do 5° dedo da mão.
É observado um risco de hipoglicemia na infância especialmente se a
alimentação não é frequente, o que pode ser agravado pela falta de apetite e a
absorção reduzida de caloria também característicos desses pacientes (PERKINS,
HOANG-XUAN, 2002; AZCONA, STANHOPE, 2005).
Não há consenso em relação ao déficit cognitivo nos pacientes com SSR,
mas Lai et al., em 1994, relataram que aproximadamente metade das crianças com
a síndrome tem uma diminuição significativa em suas habilidades cognitivas. Price et
al. (1999) e Noeker et al. (2004) também mostraram uma moderada, mas
significativa, alteração cognitiva. Azcona e Stanhope, em 2005, sugeriram que a
hipoglicemia estaria relacionada à falha no desenvolvimento intelectual dos
pacientes com SSR.
A B

1. Introdução
4
Outras características associadas com a síndrome são tendência para
sudorese excessiva e palidez durante as primeiras semanas de vida, especialmente
na cabeça e na parte superior do tronco, a deformidade de Sprengel, anomalias
renais, defeitos gastrintestinais, defeitos cardíacos, micrognatia, esclera azulada,
fechamento retardado da fontanela anterior e neoplasias como craniofaringioma,
seminoma testicular, carcinoma hepático e tumor de Wilms (PRICE et al., 1999;
ANDERSON et al., 2002; PERKINS, HOANG-XUAN, 2002;).
Não há critérios diagnósticos estritos para a SSR, entretanto a presença de
três características “maiores” simultaneamente a uma ou mais características
“menores” confirmatórias são geralmente requeridas para um diagnóstico positivo.
Mas isso não é obrigatório, existindo pacientes diagnosticados com muitas
características “menores”, porém, sem nenhuma das “maiores” (HITCHINS et al.,
2001; NOGUEIRA et al., 2001).
O diagnóstico pode ser subjetivo, sendo influenciado pela experiência clínica
do investigador. Além do mais, o fenótipo do paciente com SSR adulto é menos
evidente ao comparado à infância. Portanto um diagnóstico laboratorial seria
importante para confirmar o clínico (SCHÖNHERR et al., 2006).
1.2. ASPECTOS GENÉTICOS
Vários estudos têm evidenciado a heterogeneidade etiológica da SSR sendo
a maioria dos casos esporádicos, atribuídos a mutações novas dominantes (OUNAP
et al., 2004; ROSSIGNOL, 2006). No entanto, em revisão realizada a partir de 197
casos, 19% apresentava mais de um afetado na família (DUCAN et al., 1990;

1. Introdução
5
OUNAP et al., 2004) e diferentes modelos de herança têm sido sugeridos através de
estudos baseados em histórico familiar.
Uma herança autossômica recessiva foi sugerida pela verificação de muitas
famílias com mais de um filho afetado gerados por pais fenotipicamente normais e
também pela presença de afetados nascidos de casamentos consanguíneos
(GOUDA et al. 1996). Transmissões autossômicas dominantes ou ligadas ao
cromossomo X também foram sugeridas (OUNAP et al., 2004; PARTINGTON,
1986). Adicionalmente, o relato de gêmeos monozigóticos discordantes
fenotipicamente em relação à síndrome abre a discussão para o envolvimento de
fatores ambientais e fatores genéticos pós-zigóticos (HITCHINS et al., 2001).
Com relação às anomalias cromossômicas estruturais, a maioria dos
pacientes com SSR apresenta um cariótipo normal. Entretanto, para alguns
pacientes foi relatada a ocorrência de alterações em diferentes cromossomos
correlacionados ao fenótipo desta síndrome. Entre as anomalias citadas, existe a
translocação balanceada envolvendo o cromossomo 17q25 em dois pacientes com
características típicas de SSR, um com a translocação t(17;20)(q25;q13) e o outro
t(1;17)(q31;q25)( WAKELING et al., 1998).
Para o cromossomo 8, foi encontrada uma deleção paternalmente herdada de
8q11-13 em um paciente com SSR (SCHINZEL at al., 1994) e para o cromossomo
18 verificou-se a deleção completa do braço curto. Há também outro caso relatado
com trissomia do 18 em mosaico (CHAUVEL, MOORE, HASLAM, 1975; SCHINZEL
et al., 1994).
Tem sido relatada a presença de anel no cromossomo 15 ou deleções distais
de 15q em pacientes com características de SSR. No braço curto do cromossomo 15
está localizado o IGF1R (receptor de fator de crescimento semelhante à insulina I),

1. Introdução
6
pertencente a família de fatores de crescimento semelhante à insulina (IGF). Essa
família inclui a insulina, o IGF1 e o IGF2, seus receptores e seis proteínas ligantes
de IGF (IGFBPs), que desempenham um papel fundamental na regulação do
crescimento fetal. Por esse papel, o IGF1R foi candidatado na etiologia da SSR
nesses pacientes com alteração do cromossomo 15, entretanto estudos moleculares
mais aprofundados falharam em mostrar mutações nesse gene em pacientes com
SSR (HITCHINS et al., 2001).
A verificação de dois pacientes com SSR que também possuíam, em
homozigose, uma mesma mutação no gene CFTR causador da fibrose cística
proveniente de mães portadoras, sugeriu o envolvimento da dissomia uniparental
materna do cromossomo 7 (mDUP7) na etiologia da síndrome (KOTZOT et al.,
1995). Trabalhos posteriores confirmaram o importante papel dessa alteração,
sendo descrita atualmente como presente em aproximadamente 5-10% dos casos
(BINDER et al., 2008).
A partir de 2005, três trabalhos demonstraram relação da SSR com alterações
do padrão de metilação da região 11p15 em alta frequência (31-50%) das crianças
afetadas pela síndrome (EGGERMANN et al., 2005; GICQUEL et al., 2005; BLIEK et
al., 2006;).
Apesar da variedade de alterações genéticas em diferentes regiões, apenas
os cromossomos 7 e 11 têm sido considerados, até os últimos anos, candidatos
consistentes para a possível utilização nas triagens clínicas, ambos com evidências
do envolvimento do imprinting genômico.

1. Introdução
7
1.3. EPIGENÉTICA E IMPRINTING GENÔMICO
O imprinting genômico é um processo de controle da transcrição gênica no
qual apenas um dos alelos é expresso, de acordo com a sua origem parental. A
base molecular é epigenética, isto é, modificação herdável na expressão do gene
sem mudanças na sequência do DNA (WOLFFE; MATZKE, 1999).
Os mecanismos epigenéticos podem incluir modificações como metilação do
DNA, modificações nas histonas (metilação, acetilação, ubiquitinação e fosforilação)
e alteração na conformação da cromatina (WEKSBERG; SHUMAN; SMITH, 2005;
DELEVAL; WAGSCHAL; FEIL, 2006).
A metilação do DNA, modificação epigenética melhor caracterizada, consiste
na adição de um radical metil (CH3) no carbono 5 de Citosina geralmente seguida
por Guanina (dinucleotídeo CpG), catalisada por enzimas DNA metiltransferases
(DNMTs) (PAULSEN; FERGUNSONSMITH, 2001). Em mamíferos foram
identificadas três classes principais de DNMTs: DNMT1, DNMT2 e DNMT3
(BESTOR, 2000). A DNMT1, também denominada metiltransferase de manutenção,
está envolvida no processo de metilação que ocorre durante a divisão celular
mitótica, copiando o padrão de metilação da fita original. A DNMT2, diferentemente
das demais metiltransferases, parece apresentar uma atividade metiltransferase de
tRNA (BESTOR, 2000; JELTSCH et al., 2006). A terceira classe das DNMTs está
relacionada à aquisição de metilação de DNA de novo (DNA não-metilado
previamente) (BOURC’HIS et al.,2001; HATA et al., 2002).
Existem vários mecanismos segundo os quais a metilação do DNA pode atuar
no controle da expressão gênica. O mais simples é através de um silenciamento
direto do promotor, havendo a não transcrição do alelo metilado. A metilação do

1. Introdução
8
DNA pode interferir diretamente na sequência de ligação de fatores trans-atuantes
como ocorre com a proteína CTCF (fator de ligação CCCTC), um fator de transcrição
com domínios dedos-de-zinco envolvido na formação de barreiras e isolamentos de
cromatina, o que modula a expressão e/ou inativação de genes específicos (BELL;
FELSENFELD, 2000; HOLMGREN et al., 2001; RAND et al., 2004, MONK et al,
2008).
Um outro mecanismo envolve o silenciamento, dependente de metilação, de
um RNA não-codificante (ncRNA) especificamente em um dos alelos parentais.
Consequentemente, o ncRNA transcrito é expresso apenas a partir do alelo não
metilado e, através do recrutamento de proteínas, mantém modificações repressivas
nas histonas, levando ao silenciamento de genes próximos (MONK et al., 2008) .
As modificações epigenéticas envolvidas na regulação do imprinting
genômico ocorrem em sequências especializadas chamadas de regiões
controladoras de imprinting (imprinting control regions ou ICRs) responsáveis,
geralmente, pela regulação de um grupo de genes agrupados em domínios
cromossômicos. Essas regiões, de aproximadamente 1-2 kb, são marcadas
diferencialmente nos alelos herdados paterna ou maternamente e, como
consequência, são ativadas em apenas uma das duas cópias parentais, regulando
através de atividade em cis a expressão de um dos alelos especificamente (LEWIS;
REIK, 2006).
Esses padrões epigenéticos são mantidos nos tecidos somáticos,
independentemente do sexo do embrião, mas são apagados e refeitos durante a
gametogênese, baseados no padrão sexual da transmissão (LI, 2002). Falhas
nesses processos podem resultar em alterações da expressão dos genes regulados
por imprinting.

1. Introdução
9
Outro fator importante que pode resultar na desregulação do mecanismo de
imprinting é a dissomia uniparental (DUP), fenômeno onde dois cromossomos
homólogos são herdados do mesmo progenitor (KOTZOT et al., 2008).
A DUP pode ocorrer principalmente através de dois mecanismos. A fusão de
um gameta dissômico com um normal monossômico e subsequente perda de um
cromossomo extra durante a divisão mitótica, evento denominado “resgate da
trissomia”, ou a fusão de um gameta nulissômico com um normal dando origem a um
zigoto monossômico, seguida por uma duplicação mitótica do cromossomo
existente, denominada “duplicação mitótica” (HITCHINS et al., 2001).
1.4. GENE GRB10
O gene GRB10 (growth factor receptor-binding protein 10) codifica uma
proteína adaptadora que interage tanto com o receptor de insulina (IR) quanto com o
receptor de fator de crescimento semelhante à insulina 1 (IGF1R) (YAMAMOTO et
al., 2008).
O IGF1R tem afinidade a dois importantes reguladores de crescimento
embrionário, o IGF1 e o IGF2, sendo que este último também atua via IR e, portanto,
vias susceptíveis a interferência da GRB10. Além disso, a GRB10 deve estar
envolvida na regulação da homeostase de glicose, que é regulada pelo IR
(YAMAMOTO et al., 2008).
Trabalhos em camundongos com silenciamento do gene GRB10 mostram um
fenótipo de hipercrescimento embrionário (CHARALAMBOUS, et al. 2003) sendo
que, ao contrário, a sua hiperexpressão desencadeia um retardo de crescimento

1. Introdução
10
(SHIURA et al., 2005). Essas evidências sugerem um importante papel negativo do
GRB10 no crescimento celular.
Em camundongos esse gene está mapeado no cromossomo 11 e apresenta
expressão materno-específica. Já em humanos, localizado em 7p11.2–p13, o gene
GRB10 é expresso paternalmente no cérebro fetal e têm expressão bialélica em
outros tecidos como coração, pulmão, pele, rim entre outros, exceto pela expressão
materna de uma isoforma (γ 1) em músculo esquelético (BLAGITKO et al., 2000).
Os dados obtidos em camundongos e a identificação de pacientes com
duplicação da região 7p11.2-p13 sugeriram a região como candidata a apresentar
um papel importante na etiologia da SSR (MONK et al., 2002). Entretanto, a
expressão bialélica na grande maioria dos tecidos e a não descrição de mutações
específicas nesse gene são contrários à responsabilização isolada do gene, o que
requer maiores estudos.
Figura 2: Ideograma do cromossomo 7 humano. Regiões candidatas na etiologia da SSR analisadas nesse trabalho, gene MEST e GRB10. (p) braço curto, (q) braço longo.

1. Introdução
11
1.5. GENE MEST
O gene MEST (mesoderm-specific transcript), também denominado PEG1
(paternally expressed gene 1), está localizado em humanos na região 7q31 (Figura
1) e apresenta expressão restrita ao alelo paterno durante a fase embrionária
(RIESEWIJK et al., 1997).
A função específica do gene é desconhecida, embora se tenha verificado a
presença de motivos encontrados em enzimas alfa/beta hidrolases, enzimas
envolvidas em atividades lipolíticas, o que sugere a relação com essa família
(RIESEWIJK et al., 1998).
A verificação de camundongos com retardo de crescimento pré e pós-natal
devido ao silenciamento do gene e pacientes com SSR com mDUP7 específica da
região 7q31 indicam o gene MEST como candidato na etiologia da doença
(LEFEBVRE et al., 1998; HANNULA et al., 2001).
Entretanto, alguns trabalhos não relacionaram alterações específicas do gene
com o desenvolvimento da síndrome (RIESEWIJK et al., 1998, SCHÖHERR et al,
2008) sendo ainda incerto o papel do MEST na etiologia da SSR.
1.7. REGIÃO 11p15
A região 11p15 possui dois centros reguladores de imprinting, a ICR1 e a
ICR2. A primeira, também chamada H19DMR, é mapeada 2kb acima (upstream) do
gene H19. Esse gene, maternalmente expresso, codifica um RNA não-codificante de
função ainda desconhecida, sendo sugerido que atue como supressor tumoral
(GABORY et al., 2006; YOSHIMIZU et al., 2008).

1. Introdução
12
Figura 3: Ideograma do cromossomo 11 humano. Em destaque a região 11p15, analisada nesse trabalho. (p) braço curto, (q) braço longo.
Nesse domínio também está presente o gene IGF2 cujo alelo expresso é o
paterno e codifica um fator de crescimento semelhante à insulina, sendo um dos
principais reguladores do crescimento fetal e pós-natal (DELAVAL et al., 2006).
Níveis aumentados da expressão em camundongos causam hipercrescimento fetal
(EGGENSCHWILER et al., 1997) e a expressão reduzida está associada ao
hipocrescimento (DECHIARA et al., 1990).
Estudos em camundongos mostraram que o alelo não metilado materno na
ICR1 é ligado a muitas cópias da proteína CTCF (VERONA; MANN; BARTOLOMEI,
2003). Essa ligação cria um limite de cromatina, que atua como inibidor do gene
IGF2 e acentuador da expressão do H19. Como consequência, o IGF2 não é
expresso a partir do alelo materno. No alelo paterno, por ação da enzima DNMT1,
há a metilação do DNA e não há a ligação da CTCF, permitindo o acesso do
enhancer ao IGF2. Durante as etapas iniciais do desenvolvimento, a metilação do
DNA paterno se estende da ICR até o promotor do H19, guiando a repressão do
alelo paterno. Em resumo, a manutenção da diferença de metilação do DNA na ICR

1. Introdução
13
garante que o gene IGF2 seja expresso apenas a partir do alelo paterno e o H19, do
materno (VERONA; MANN; BARTOLOMEI, 2003; MOSS, WALLRATH, 2007).
O outro domínio de imprinting da região é um longo agrupamento regulado
pela ICR2, também denominado de KvDMR1, maternalmente metilada e situada em
um íntron do gene KCNQ1 (WEBESBERG; SHUMAN; SMITH, 2005). Essa ICR
também compreende o promotor do gene KCNQ1OT1 (ou LIT1) que se localiza
dentro da sequência do gene KCNQ1, mas com transcrito antisense. O gene
KCNQ1OT1 expressa, a partir do alelo paterno, um longo RNA não-codificante de
função ainda desconhecida potencialmente envolvido na regulação do KCNQ1
(WEBESBERG; SHUMAN; SMITH, 2005)(figura 4).
O KCNQ1, também conhecido como KvLQT1, codifica um canal de potássio e
apenas o alelo materno é expresso (LEE et al., 1999). Alterações na expressão
desse gene foram descritas em doenças cardíacas (NEYROUD et al., 1999) e seu
envolvimento em alterações de crescimento ainda não é conhecido. Ao lado desse,
muitos outros genes do domínio têm silenciamento paterno sendo um deles o
CDKN1C (cyclin-dependent kinase inhibitor), também chamado de P57kip2. Este
gene codifica um inibidor de quinase dependente de ciclina, sendo regulador
negativo de proliferação celular e crescimento que pode também atuar como
supressor tumoral (FEINBERG, 1999). Um outro gene silenciado paternalmente no
domínio é o PHLDA2, importante no crescimento placentário (DELAVAL;
WAGSCHAL; FEIL, 2006).

1. Introdução
14
Figura 4: Representação esquemática do mapa gênico da região cromossômica 11p15. As caixas pretas representam os genes silenciados, as azuis os genes expressos paternalmente e em vermelho os expressos maternalmente. As caixas amarelas representam as regiões diferencialmente metiladas (DMRs). (ICR) centros reguladores de imprinting. As setas ligadas às caixas indicam a orientação da transcrição, os círculos azuis indicam a metilação e o arco rosa, a proteína CTCF.
Alterações no padrão de metilação nas ICRs da região 11p15 como,
hipermetilação e silenciamento do H19 materno e ativação de ambos os alelos do
IGF2 (WEKSBERG; SHUMAN; SMITH, 2005) e perda da metilação da KvDMR1
associada com a expressão bialélica do LIT1 (LEE et al., 1999), estão relacionadas
com a síndrome de Beckwith-Wiedemann (SBW). Essa última caracteriza-se por
hipercrescimento intra-uterino e pós-natal, macroglossia, alterações de parede
abdominal e assimetria corpórea (GOMES et al., 2007).
Com relação à SSR, inicialmente quatro estudos evidenciaram com
alterações do padrão de imprinting da região 11p15, demonstrando hipometilação na
H19DMR em alta frequência (31-50%) nas crianças afetadas (EGGERMANN et al.,
2005; GICQUEL et al., 2005; BINDER et al., 2006; BLIEK et al., 2006). Em 2007,
Schönherr et al. verificaram também alteração na KvDMR1 em pacientes com SSR.

1. Introdução
15
Os dados da literatura apontam para o envolvimento do padrão de metilação
nos domínios de 11p15 na etiologia da SSR, entretanto, a verdadeira relação entre
as alterações epigenéticas e a doença permanece incerta.

2222.... OBJETIVOS OBJETIVOS OBJETIVOS OBJETIVOS

2. Objetivos
17
2.1. OBJETIVO GERAL
Avaliar a influência do perfil epigenético dos genes GRB10, MEST e da região
11p15 no desenvolvimento da síndrome de Silver-Russell.
2.2. OBJETIVOS ESPECÍFICOS
• Analisar o padrão de metilação na região diferencialmente metilada da
porção 5´UTR do gene GRB10;
• Analisar o padrão de metilação na região diferencialmente metilada da
porção 5´UTR do gene MEST;
• Analisar o padrão de metilação na região diferencialmente metilada do
H19 (H19DMR);
• Analisar o padrão de metilação na região diferencialmente metilada
KvDMR1;
• Correlacionar os resultados laboratoriais aos achados clínicos dos
pacientes portadores de SSR;
• Avaliar a aplicação das metodologias utilizadas para a confirmação
diagnóstica da SSR.

3.3.3.3. CASUÍSTICACASUÍSTICACASUÍSTICACASUÍSTICA

3. Casuística
19
3.1. PACIENTES
A amostra foi selecionada a partir de pacientes atendidos pela equipe médica
do Ambulatório de Aconselhamento Genético do Hospital das Clínicas da Faculdade
de Medicina de Ribeirão Preto - Universidade de São Paulo (HC-FMRP-USP)
mediante aprovação da Comissão de Normas Éticas e Regulamentares deste
hospital (Processo HCRP n° 1497 / 2008).
Foram analisadas amostras de 10 indivíduos controles voluntários saudáveis,
15 portadores da síndrome de Silver-Russell clássicos (SR1 a SR15) atendidos pela
equipe médica do Ambulatório de Aconselhamento Genético do Hospital das
Clínicas da Faculdade de Medicina de Ribeirão Preto - Universidade de São Paulo
(HC-FMRP-USP).
Adicionalmente, foram realizadas análises moleculares em um indivíduo com
fenótipo claro de SSR, atendido no HC-FMRP-USP, filho de pais consanguíneos e
com dois irmãos não SSR, porém com malformações congênitas (SR16).
Em três pacientes com fenótipo claro de SSR e irmãos, todos de uma mesma
família, atendidos no campus São Paulo da Universidade de São Paulo (irSR1,
irSR2, irSR3).
Também foi analisado um indivíduo com cariótipo condizente com síndrome
de Turner, 45,X[25]/46,XX[75], porém com caracteres não condizentes com a
alteração cromossômica e relacionada à SSR (ST1), atendido no HC-FMRP-USP.
Os critérios para o diagnóstico da SSR foram a presença de baixa estatura de
origem pré-natal associada à pelo menos duas outras características clínicas como
aparente macrocefalia, face triangular, manchas café-com-leite, assimetria corpórea,
quinto dedo de mãos curtos e/ou com clinodactilia.

4. MATERIAL E MÉTODOS4. MATERIAL E MÉTODOS4. MATERIAL E MÉTODOS4. MATERIAL E MÉTODOS

4. Material e Métodos
21
4.1. ANÁLISE MOLECULAR
As análises moleculares foram realizadas no Laboratório do Grupo de
Epigenética e Reprodução do Departamento de Genética da Faculdade de Medicina
de Ribeirão Preto – USP.
4.1.1. Coleta de Sangue Periférico
Para a análise molecular foram coletados 5-10 mL de sangue periférico de
cada paciente em tubo estéril tipo Vacutainner contendo o anticoagulante EDTA. O
material foi obtido após esclarecimento aos familiares sobre a pesquisa e assinatura
do termo de consentimento livre-esclarecido.
4.1.2. Extração de DNA
Amostras de DNA foram extraídas utilizado o método de extração e
precipitação em NaCl (OLERUP;ZETTERQUIST, 1992, modificado).
Ao volume de 0,5mL de sangue foi adicionado 1,0mL de Tampão de Lise
[Sacarose 0,32M; Tris HCl 12mM (pH 7,5); MgCl2 5,0M; Triton X 1,0%] em
microtubos de 1,5mL. As amostras foram centrifugadas a 13000rpm por 20
segundos, o sobrenadante descartado e o procedimento repetido até a obtenção de
pellet branco. O pellet foi ressuspendido em 80µL de Tampão de Proteinase K (5X)
[NaCl 0,375M; EDTA ( pH8,0) 0,12 M], 8,0µL de proteinase K (20ng/mL), 10µL de
SDS 20% e 283,3µL de água destilada e incubado à 55°C por 16 horas. Foram

4. Material e Métodos
22
adicionados 120µL de NaCl 5M e após agitação por 15 segundos a amostra foi
centrifugada a 13000rpm por 8 minutos. O sobrenadante foi transferido para outro
microtubo onde foi adicionado 1,0mL de etanol à 20°C, homogeneizado e
centrifugado por 10 minutos à 13000rpm a uma temperatura de 4°C. O
sobrenadante foi descartado e o pellet restante desidratado. O pellet foi
ressuspendido em 50µL de água deionizada.
4.1.3. Modificação do DNA com Bissulfito de Sódio
O tratamento do DNA com bissulfito de sódio tem por finalidade converter in
vitro as Citosinas presentes na molécula de DNA em Uracila, porém, esta conversão
não ocorre quando as Citosinas encontram-se metiladas. Através dessa reação, é
possível a análise do padrão de metilação com base na presença de Citosinas que
permanecem no DNA após a modificação com bissulfito de sódio (Citosinas
metiladas) (FROMMER et al., 1992; CLARK et al., 1994; HERMAN et al.,1996).
Na execução do protocolo de modificação do DNA com bissulfito de sódio,
inicialmente, 1µg de DNA genômico foi diluído em 70µL de água ultra pura (Milli Q)
estéril. A seguir, foram adicionados 8µL de NaOH 3M para a desnaturação do DNA e
as amostras foram incubadas a 37°C por 15 minutos seguido de 94°C por 5 minutos.
Terminado o tempo de incubação, as amostras foram imediatamente
colocadas em gelo e, a cada tubo, acrescentou-se 1mL de uma solução constituída
por bissulfito de sódio 2,7M, hidroquinona 0,6mM e NaOH 0,36M.
A seguir as amostras foram incubadas a 55°C, no escuro, por 5 horas
(utilizou-se 100µL de óleo mineral para evitar a evaporação das amostras) e

4. Material e Métodos
23
posteriormente purificadas a partir do kit de purificação Wizard DNA Clean –up
System (Promega).
Terminada a purificação, ao DNA foi adicionado 5µL de NaOH 3M e incubado
a 37°C por 15 minutos. A seguir, o DNA foi novamente precipitado em 5µL de
Acetato de Sódio 3M pH 5,2 e 100µL de etanol absoluto e incubado a –20°C
overnight. No dia seguinte, as amostras foram centrifugadas a 13000rpm por 20
minutos e lavadas com etanol 70% seguido de centrifugação a 13000rpm por 5
minutos. O sobrenadante foi então desprezado, e após secar, o DNA modificado foi
resuspendido em 30Μl de água ultra pura (Milli Q) estéril.
4.1.4. Identificação de Ilhas CpGs e Desenhos de Primers
Previamente às análises moleculares foi realizada uma minuciosa revisão
bibliográfica para a escolha de regiões que melhor representassem sítios
regulatórios e DMRs experimentalmente comprovadas.
Também foi utilizado o software Methprimer (LI;DAHIYA, 2002) para a
identificação de ilhas CpGs e para a construção dos primers específicos, com
ajustes realizados através do programa GeneRunner V.3.05 (Hasting Software Inc.),
sempre evitando a formação de estruturas secundárias como dímeros e loops.
A presença de polimorfismos e sequências repetitivas foi excluída utilizando
os programas BLAST SNP Sequence (disponível em
http://www.ncbi.nlm.nih.gov/SNP/snpblastByChr.html) e RepeatMasker Open-3.0
(SMIT; HUBLEY; GREEN, 1996), respectivamente.

4. Material e Métodos
24
4.1.5. Análise de Metilação
4.1.5.1. Análise do padrão de metilação do gene GRB10
Para a análise de metilação na DMR próxima ao gene GRB10, utilizou-se o
Método de Análise Combinada de Bissulfito e Restrição Enzimática (COBRA –
Combined Bisulfite Restriction Analysis).
O método COBRA consistiu-se na análise do padrão de metilação de DNA,
previamente modificado por bissulfito de sódio, com auxílio de digestão enzimática.
O DNA modificado foi amplificado com primers que não discriminam os alelos
metilado e não metilado, pois não contém em sua sequência dinucleotídeos CGs.
Posteriormente, o fragmento resultante da amplificação foi submetido à restrição
enzimática com a enzima BstUI que reconhece e cliva a sequência CGCG, de modo
que a digestão somente ocorreu quando as Citosinas estavam metiladas (não foram
convertidas em Uracilas pelo bissulfito de sódio).
De acordo com a sequência no GenBank AC004920, foram utilizados os
seguintes primers: hGRB10-S= 5´ TATTTAGTTTTYGAGGGATTGGG 3´ e hGRB10-
AS= 5´ AAAATTAAACAACAAAAAACCCCC 3´ para o DNA modificado. Os
nucleotídeos sublinhados são os correspondentes às Citosinas que foram
modificadas para Timinas. No primer sense, hGRB10-S, está anotada uma base Y,
correspondente a iguais proporções de ambas as bases pirimidinas (C e T) uma vez
que a presença do nucleotídeo CG não pôde ser evitada e é susceptível à metilação.

4. Material e Métodos
25
42301 aaagcgggcc aacgccgcct ctggggacgc catccgggcg agggtgggat gccgcgccac
42361 cgcccgagcg tgcccggggg ctcccagcgc catcaccacg caggtgcccg ggggcccctc
42421 cgcggagccg gctgccgccc ggctgcaata ctcagcctcc gagggactgg gcgctcctga
42481 ggagcgggag gacgacggag cagcgcccgg tcctggagca caacaggaat cccaggacca
42541 aacccatgtg acgcggccac gccccaacgc ccggcggggg ccctctgctg cccaatcccc
42601 ggatccctgg ttctcatggc aaccctgaag aaccccttgt tccctccact gcctcctctc
42661 ctcccccaca gcccccccgc ctgatgggca aggacgcgca cactggccac tctgacgatg
42721 ccgaaagccc tccatgtcta cccgctacgc cgcattcgca gacaggcggg ggacatcgcg
42781 gccgcggcaa gctagagatg ccgcctgctc gagcaacctt ccacctgcag ggccggccag
Figura 5: Sequência próxima à região do gene GRB10 analisada, GenBank AC004920. Verde: primers utilizados. Maiúsculas e em negrito: sítio de clivagem da enzima BstUI.
Para a amplificação do fragmento correspondente, o meio de reação
constituiu-se de KCl 50 mM; MgCl2 1,5 mM; Tris HCl (pH 8,4) 10 mM; dNTP (0,1mM
de cada nucleotídeo); primer 0,2 mM (de cada); Taq DNA Polimerase 1U; DNA
genômico 0,1µg em volume final de 25 uL.
As reações foram realizadas em aparelho termociclador programável T-
gradiente (Biometra) seguindo protocolo que se consistiu de uma etapa inicial de
95°C por 10 minutos (hot start), seguido de 40 ciclos de 95°C por 45 segundos, 58°C
por 45 segundos e 72°C por 45 segundos.
Após verificada a presença de fragmento amplificado em gel de poliacrilamida
10% corado com nitrato de prata (0,2%), 1ug do produto de PCR foi submetido à
digestão com 5U da enzima BstUI e incubado em 5mM NaCl, 1mM Tris-HCl, 1mM
de MgCl2, 0,1mM DTT, pH 7,9 (Buffer 2, 1x), overnight à 60°C.
Os produtos das digestões foram visualizados por eletroforese em gel de
poliacrilamida 10% corado com nitrato de prata (0,2%).
Uma digestão completa foi esperada em caso de metilação em ambos os
alelos, devido à manutenção do sítio de restrição após a modificação com bissulfito
de sódio (CGCG se tornaria UGUG). Para toda reação de modificação por bissulfito
de sódio, foi incluído também uma amostra de espermatozóide (padrão

4. Material e Métodos
26
conhecidamente hipometilado para a região) para controle de modificação completa
em todas as Citosinas não metiladas.
4.1.5.2. Análise do padrão de metilação do gene MEST
Para a análise de metilação da DMR do gene MEST (GenBank NT_007933),
utilizou-se o método COBRA.
A amplificação do fragmento correspondente ocorreu em meio com KCl 50
mM; MgCl2 1,5 mM; Tris HCl (pH 8,4) 10 mM; dNTP (0,1mM de cada nucleotídeo);
primer 0,2 mM (de cada); Taq DNA Polimerase 1U; DNA genômico 0,1µg em volume
final de 25 uL.
Foram utilizados os primers: hMEST/PEG1- S 5´ TGTTGGTTAGTTTTGTA
TGGTTG 3´ e hMEST/PEG1- AS 5´ CCCAAAAACAACCCCAACAC 3´. A
amplificação ocorreu, após um hot start, em 40 ciclos de 95°C por 45 segundos,
58°C por 45 segundos e 72°C por 45 segundos, termociclador T-gradiente
(Biometra).
5881 tgccgcggca accagcacac cccggcacct cctctgcggc agctgcgcct cgcaagcgca
5941 gtgccgcagc gcacgccgga gtggctgtag ctgcccggcg cggcgccgcc ctgcgcgggc
6001 tgtgggctgc gggctgcgcc cccgctgctg gccagctctg cacggctgcg ggctctgcgg
6061 cgcccggtgc tctgcaacgc tgcggcgggc ggcatgggat aacgcggcca tggtgcgccg
6121 agatcgcctc cgcaggtgag tgtgcggtgg gaacgagggg gtgtggctgg cggccctggg
6181 actagggcgc aggcgagcgg aggactgtgt gcccgtgtcc gagctggggc tgcctctggg
6241 cgaaaactct accgacaggc ggcacgcatt ccgcgcccgc tctgcctact tgaggagggg
6301 gtgtcactcc tgcccgcaat ggaatgttca gaacgcggga cctccttggg ttaggatttc
Figura 6: Sequência próxima à região do gene MEST analisada, GenBank NT_007933. Verde: primers utilizados. Maiúsculas e em negrito: sítio de clivagem da enzima BstUI.

4. Material e Métodos
27
O fragmento amplificado foi submetido à digestão com 5U da enzima BstUI,
overnight à 60°C.
Os produtos das digestões foram visualizados por eletroforese em gel de
poliacrilamida 10% corado com nitrato de prata (0,2%).
Também foi realizada a modificação de DNA por bissulfito de sódio de
amostra de espermatozóide (padrão conhecidamente hipometilado para a região)
em toda reação, para controle de modificação completa.
4.1.5.3. Análise do Padrão de Metilação da H19DMR
A análise da região H19DMR foi realizada através de dois métodos. O método
COBRA e o método Digestão Enzimática Sensível à Metilação Associada à PCR em
Tempo Real (DESM-RT).
4.1.5.3.1. Análise Combinada de Bissulfito e Restrição Enzimática (COBRA)
Para o método COBRA, foram utilizados os 0,2 mM de cada primer:P1A: 5´
GTATAGTATATGGGTATTTTTGGAGGTTTT 3´ e P1B: 5´ TAAATATCCTATTCCCA
AATAACCC 3´ em meio com KCl 50 mM; MgCl2 1,5 mM; Tris HCl (pH 8,4) 10 mM;
dNTP (0,1mM de cada nucleotídeo); primer 0,2 mM (de cada); Taq DNA Polimerase
1U; DNA genômico 0,1µg em volume final de 25 uL. O fragmento foi amplifica em 50
ciclos de 95°C por 45 seg, 61°C por 45 seg e 72° por 45 seg.

4. Material e Métodos
28
Do produto amplificado, 1ug foi submetido à digestão com 5U da enzima
BstUI e incubada em 5mM NaCl, 1mM Tris-HCl, 1mM de MgCl2, 0,1mM DTT, pH 7,9
(Buffer 2, 1x), overnight à 60°C e visualizados por eletroforese em gel de
poliacrilamida 30% corado com nitrato de prata (0,2%).
Como controle de eficiência da modificação por bissulfito de sódio, foi
amplificado um fragmento a partir de DNA genômico (primers H19 UPH19II: 5’-
CAATGAGGTGTCCCAGTTCCA-3’ e H19 UPH19III: 5´-CACATAAGTAGGCGTG
ACTTGA-3´. Esse fragmento amplificado, não metilado, foi incluído em todas as
reações por bissulfito de sódio, sendo usada como controle de hipometilação. Já
amostras de espermatozóide humano, conhecidamente hipermetiladas para a
região, foi utilizada como controle de digestão total.
Para excluir a presença de alterações na sequência de DNA nos sítios
analisados, o que poderia falsear os resultados de metilação, na metodologia
COBRA, o fragmento amplificado a partir do DNA genômico (primers UPHII e
UPHIII) foram também digeridos com a BstUI em todas as amostras analisadas.
4.1.5.3.2. Digestão Enzimática Sensível à Metilação Associada à PCR em
Tempo Real (DESM-RT)
Pela metodologia de PCR em tempo real associada à digestão com enzimas
sensíveis à metilação foi obtido, de forma quantitativa, o padrão de metilação na
região H19DMR a partir da comparação da amplificação de DNA não digerido com
DNA digerido com a enzima HpaII, enzima capaz de clivar apenas os alelos não
metilados (GOMES et al., 2007). Para a comparação foi utilizado o número do ciclo

4. Material e Métodos
29
no qual o nível de fluorescência emitido pelas amostras amplificadas atingiu o nível
arbitrário - ct (threshold). De acordo com o princípio de amplificação exponencial das
amostras (ou seja, o dobro de produto a cada ciclo) a porcentagem de DNA metilado
foi calculada pela fórmula (1/2)d - nd, sendo d a média do ct do DNA digerido e nd a
média do ct do DNA não digerido.
A digestão de 0,2 ug de DNA genômico foi realizada em meio contendo com 1
mM Bis Tris Propano-HCl, 1 mM MgCl2, 0,1 mM DTT (pH 7.0) com 10 U da enzima
HpaII (NE- New England) à 37°C, overnight, seguido por uma etapa de 60°C à 20
minutos para inativação. O material não digerido recebeu o mesmo tratamento
apenas retirando-se a enzima.
Todas as amostras foram também digeridas separadamente pela enzima
MspI, com mesmo sítio de restrição da HpaII porém não sensível à metilação, para
excluir alterações na sequência de DNA.
Foi utilizado o sistema de detecção de sequência StepOne™ Real-Time PCR
(Applied Biosystems, Foster City, CA) com 5 ul SYBR Green PCR Master Mix
(Applied Biosystems) e 0,4 uMol de cada primer: H19 UPH19II: 5’-
CAATGAGGTGTCCCAGTTCCA-3’ e H19 Real T-2: 5’-CTATGAGTGTCCTATTCCC
-3’.
Cada amostra foi quantificada em duplicata ou triplicata, com desvio padrão
máximo estipulado em 0,2. Todo resultado foi confirmado pelo menos duas vezes,
com uma nova digestão e nova amplificação em uma placa de reação diferente da
primeira análise.
As condições de ciclagem foram: 95°C for 10 min e 35 ciclos (95°C por 15 seg
e 61°C por 60 seg), seguidos por análise de dissociação realizada de 60° a 95°,
variando 0,1°C a cada segundo.

4. Material e Métodos
30
1081 tgcactgttg aaggttgggg agatgggagg agatactagg ggaacaatga ggtgtcccag
1141 ttccatggat gatggggatc tcggccctag tgtgaaaccc ttctcgcagg gtctctggca
1201 ggcacagagc ccgggggctc ttgcatagca catgggtatt tctggaggct tctcttcggt
1261 ctcaccgcct ggatggcacg gaattggttg tagttgtgga atcggaagtt gcgcgcggcg
1321 gcagtgcagg ctcacacatc acagcccgag cccgccccaa ctggggttcg cccgtggaaa
1381 cgtcccgggt cacccaagcc acgcgtcgca gggttcacgg gggtcatctg ggaataggac
1441 actcatagga gccgcaccag atcttcaggt cgggcattat ccacagcccc gtggccccgg
1501 gtcacactcc gagggcttca gtgtcatggc ctgggactca agtcacgcct acttatgtga
1561 tgatcacagt gtgttccacc aaaatcttac attttccaca tctatcccag agcacagctc
Figura 7: Sequência próxima à região H19DMR analisada, GenBank U50731. Cinza: região dos primers utilizados no método COBRA. Maiúsculas e em negrito: sítios de clivagem das enzimas BstUI e HpaII. Negrito: primers utilizados no método de Digestão Enzimática Associada à PCR em Tempo Real. Sublinhado: primers utilizados para amplificação da região H19 usada como controle de modificação.
4.1.5.4. Análise do Padrão de Metilação da KvDMR1
Para análise molecular do padrão de metilação da KvDMR também foram
utilizadas duas metodologias: a PCR específica para a metilação (MS-PCR –
methylation specific PCR) e a DESM-RT.
4.1.5.4.1. PCR específica para a metilação (MS-PCR)
O princípio da MS-PCR consiste na amplificação específica de fragmentos
metilados e não-metilados de uma determinada região do DNA modificada por
bissulfito de sódio (HERMAN et al.,1996). Para tal, utilizam-se primers específicos
capazes de discriminarem e hibridarem apenas a um tipo de fragmento.
Após a modificação por bissulfito de sódio as Citosinas não-metiladas
convertidas em Uracilas são reconhecidas pela Taq Polimerase como Timina, devido
à similaridade estrutural. Dessa forma, para a amplificação do fragmento metilado foi
utilizado o primer denominado “M” (hibridação em fragmento que contém Citosinas
não convertidas – CG) e para a amplificação do fragmento não metilado foi utilizado
o primer “U” (hibridação em fragmento que contém timinas - TG).

4. Material e Métodos
31
Ambas as reações realizaram-se em KCl 50 mM; MgCl2 1,5 mM; Tris HCl (pH
8,4); 10 mM; dNTP (0,1mM de cada nucleotídeo); primer 0,2 mM (de cada); Taq
DNA Polimerase 1U e 0,1 µg de DNA genômico, no aparelho termociclador
programável T-gradiente (Biometra).
A ciclagem para amplificação do fragmento M consistiu de uma fase de
denaturação do DNA a 94° C por 5 min e de 50 ciclos de 94°C por 1 min; 59,3°C por
1min e 30 seg e 72°C por 1 minuto, seguido por uma fase final de extensão de 72°C
por 10 min, com os primers: M1A: 5’ TTTTTTCGGTTAATGATAGGATACG 3’ e M1B:
5’ TCTACCTAAAAACTACGACAACGCT 3’
Para o fragmento U foi realizada uma denaturação a 94°C por 5 min, seguido
de 50 ciclos de 94°C por 1 min; 56,2°C por 1min e 30 seg e 72°C por 1 min, seguido
por uma fase final de extensão de 72°C por 10 min e os primers utilizados foram:
U2A: 5’ TTTTTTGGTTAATGATAGGATATGG 3’ e U2B: 5´ TCTACCTAAAAACTA
CAACAACACT 3’.
Os produtos de amplificação foram visualizados em gel de poliacrilamida
(30%) corado com nitrato de prata (0.2%).
4.1.5.4.2. Digestão Enzimática Sensível à Metilação Associada à PCR em
Tempo Real (DESM-RT)
Na análise por DESM-RT na KvDMR, também foi realizada digestão com a
enzima HpaII, comparando-a ao material não digerido.
A digestão de 0,2 ug de DNA genômico ocorreu em meio contendo com 1 mM
Bis Tris Propano-HCl, 1 mM MgCl2, 0,1 mM DTT (pH 7.0) com 5 U da enzima HpaII

4. Material e Métodos
32
(NE- New England) à 37°C, overnight, seguido por uma etapa de 60°C à 20 minutos
para inativação.
Também foi utilizado o sistema de detecção de sequência StepOne™ Real-
Time PCR (Applied Biosystems, Foster City, CA) com 5 ul SYBR Green PCR Master
Mix (Applied Biosystems) e 10pmol de cada primer (sense: 5’-
GTGCCTCTCAGCGTGGTCC-3’; antiscoense: 5’- AACCACGATGACTGACGCA C -
3’).
Cada amostra foi quantificada em duplicata ou triplicata, com desvio padrão
máximo estipulado em 0,2. Todo resultado foi confirmado pelo menos mais uma vez,
com uma nova digestão e nova amplificação em uma placa de reação diferente da
primeira análise.
As condições de ciclagem foram: 95°C for 10 min e 35 ciclos (95°C por 15 seg
e 65,4°C por 45 seg), seguido por análise de dissociação realizada de 60° a 95°,
variando 0,1°C a cada segundo.
Amplificação do material digerido com MspI, para controle, também foi
realizada em todas as amostras.
67201 gtggtgctca gggacgacgg gtgggggcag ggagggcggc ggctcgcgcc acctcacacc
67261 cagccagtgc ctcatgcggc gctggacccg ctgggccaat ctgagcccgg gtggcatcaa
67321 aaccagactc tttcggccaa tgacaggaca cggcacatca ctttccgcac ccagccaatc
67381 cgtgcagcag cccgccgcaa gccttcccct gctgccgccc aatcagcagg tggggggcgg
67441 tcgccacgtc ggcagcggcg ggggcagtcg gagcgctgcc gcagtctcca ggcagaacgg
67501 tcgccgcgtc gcctcagcac ggacctccag ggagctcctc agcaagatcc tgccagggcg
67561 cccctcagcg cgattctgcc ggggtgcctc tcagcgtggt cctccccggg gctcctcagc
67621 acgattctcc cggtgcgccc ctcagcgcgg tcctcctcgg tgcgtcagtc atcgtggttc
67681 tccccggcgc gcccctcggc gcggttctcc tcggggctcc tcagcgcggc gctcttctgg
67741 gggctcctcg gcgcagttct ccccggggac tcctcggcgc cgttctcctc ggggcacccg
Figura 8: Sequência próxima à região KvDMR1 analisada, GenBank U90095. Cinza: região dos primers utilizados no método MS-PCR. Negrito: primers utilizados no método de Digestão Enzimática Associada à PCR em Tempo Real. Maiúsculas e em negrito: sítio de clivagem da enzima HpaII. Em negrito e sublinhado, Citosinas analisadas.

5555. . . . RESULTADOSRESULTADOSRESULTADOSRESULTADOS

5. Resultados
34
5.1. ANÁLISE DO PADRÃO DE METILAÇÃO DO GENE GRB10
A análise do padrão de metilação do gene GRB10 pelo método COBRA
demonstrou metilação monoalélica (normal) para todos os controles e todos os
pacientes com SSR (Figuras 9 e 10).
L ESP C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 BR
Figura 9: Perfil de metilação do gene GRB10 pelo método COBRA em amostras de indivíduos saudáveis (controle). Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (C1 a C10) - amostras dos controles, com padrão normal de metilação. (ESP) – amostra de espermatozóide, controle hipometilado , (BR) – controle negativo da amplificação. L ESP SR1 SR2 SR3 SR4 SR5 SR6 SR7 SR8 SR9 SR10 BR
Figura 10: Perfil de metilação do gene GRB10 pelo método COBRA em amostras de pacientes com Silver-Russell. Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (SR1 a SR10) – amostras dos pacientes, padrão normal de metilação. (ESP) – amostra de espermatozóide, controle hipometilado , (BR) – controle negativo da amplificação.
151pb
46 pb
105pb
46 pb
105pb
151pb

5. Resultados
35
5.2. ANÁLISE DO PADRÃO DE METILAÇÃO DO GENE MEST
A análise do padrão de metilação do gene MEST evidenciou um padrão
indicativo de hipermetilação para apenas um paciente com SSR (1/15, 6,7%), sendo
encontrado padrão monoalélico normal de metilação para os demais pacientes e
amostras controle (Figuras 11 e 12).
L ESP C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 BR
Figura 11: Perfil de metilação do gene MEST pelo método COBRA em amostras de indivíduos saudáveis (controle). Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (C1 a C10) - amostras dos controles, com padrão normal de metilação. (ESP) – amostra de espermatozóide, controle hipometilado , (BR) – controle negativo da amplificação. L ESP SR1 SR2 SR3 SR4 SR5* SR6 SR7 SR8 SR9 SR10 BR
Figura 12: Perfil de metilação do gene MEST pelo método COBRA em amostras de pacientes com síndrome de Silver-Russell. Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (SR1 a SR4 e SR6 a SR10) - amostras dos pacientes, padrão normal de metilação. (SR5*) - amostra de paciente com hipermetilação. (ESP) – amostra de espermatozóide, controle hipometilado , (BR) – controle negativo da amplificação.
79 pb
131pb
79 pb
210pb
131pb
210pb

5. Resultados
36
5.3. ANÁLISE DO PADRÃO DE METILAÇÃO DA H19DMR
5.3.1. COBRA
Foi observado padrão condizente com a metilação monoalélico para todas as
amostras analisadas (Figura 13).
L HIP ESP SR1 SR2 SR3 SR4 SR5 SR6 SR7 SR8 SR9 SR10 BR
Figura 13: Perfil de metilação da H19DMR pelo método COBRA em amostras de pacientes com síndrome de Silver-Russell. Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (SR1 a SR10) – amostras dos pacientes, padrão normal de metilação. (HIP) – fragmento amplificado com primers UPHII e UPHII posteriormente modificado por bissulfito de sódio – controle hipometilado, (ESP) - amostra de espermatozóide, controle hipermetilado, (BR) – controle negativo da amplificação.
5.3.2. DESM-RT
A análise da H19DMR pelo método DESM-RT permite a quantificação da
porcentagem de metilação.
Para os 10 indivíduos controle, obteve-se média de metilação igual a 40% ±
(Tabela 1). A partir desse grupo foram considerados níveis normais de metilação os
valores entre a média mais duas vezes o valor do desvio padrão, tanto para mais ou
para menos, ou seja, foram consideradas porcentagens normais entre 22 e 58%.
223pb
92pb 89pb
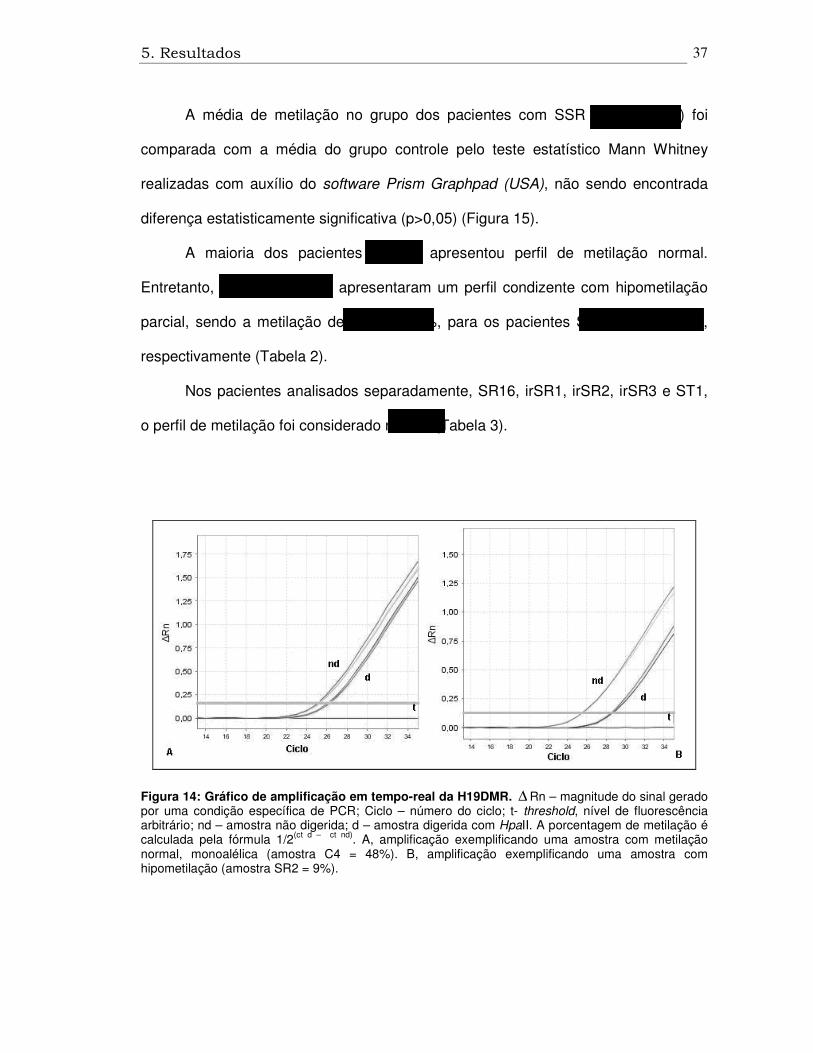
5. Resultados
37
A média de metilação no grupo dos pacientes com SSR (31% ± 0,11) foi
comparada com a média do grupo controle pelo teste estatístico Mann Whitney
realizadas com auxílio do software Prism Graphpad (USA), não sendo encontrada
diferença estatisticamente significativa (p>0,05) (Figura 15).
A maioria dos pacientes (12/15) apresentou perfil de metilação normal.
Entretanto, três deles (20%) apresentaram um perfil condizente com hipometilação
parcial, sendo a metilação de 9, 12 e 19%, para os pacientes SR2, SR8 e SR13,
respectivamente (Tabela 2).
Nos pacientes analisados separadamente, SR16, irSR1, irSR2, irSR3 e ST1,
o perfil de metilação foi considerado normal (Tabela 3).
Figura 14: Gráfico de amplificação em tempo-real da H19DMR. ∆ Rn – magnitude do sinal gerado por uma condição específica de PCR; Ciclo – número do ciclo; t- threshold, nível de fluorescência arbitrário; nd – amostra não digerida; d – amostra digerida com HpaII. A porcentagem de metilação é calculada pela fórmula 1/2(ct d – ct nd). A, amplificação exemplificando uma amostra com metilação normal, monoalélica (amostra C4 = 48%). B, amplificação exemplificando uma amostra com hipometilação (amostra SR2 = 9%).

5. Resultados
38
Figura 15: Comparação entre a média de porcentagem de metilação da H19DMR no grupo controle e no grupo de pacientes com síndrome de Silver-Russell. % de Metilação – porcentagem de metilação obtida pelo DESM-RT, CTL - amostras controles, SRS – amostras de pacientes com SSR.
Tabela 1: Resultados da H19DMR obtidos pelo método DESM-RT em amostras controle.
Amostra % de Metilação
C1 41%
C2 38%
C3 28%
C4 48%
C5 30%
C6 50%
C7 51%
C8 44%
C9 27%
C10 36%
Porcentagem metilação das amostras controle C1, C2, C3, C4, C5, C6, C7, C8, C9 e C10.

5. Resultados
39
Tabela 2: Resultados da H19DRM obtidos pelo método DESM-RT em pacientes com síndrome de Silver-Russell.
Amostra % de Metilação Perfil
SR1 41% Monoalélico
SR2 9% HIPOMETILADO
SR3 31% Monoalélico
SR4 45% Monoalélico
SR5 39% Monoalélico
SR6 35% Monoalélico
SR7 25% Monoalélico
SR8 12% HIPOMETILADO
SR9 30% Monoalélico
SR10 44% Monoalélico
SR11 35% Monoalélico
SR12 33% Monoalélico
SR13 19% HIPOMETILADO
SR14 32% Monoalélico
SR15 38% Monoalélico
Porcentagem metilação dos pacientes com SSR. SR1, SR2, SR4, SR5, SR6, SR7, SR9, SR10, SR11, SR12, SR14, SR15 – perfil normal, monoalélico, de metilação, SR3, SR8, SR13 – perfil hipometilado.
Tabela 3: Resultados da H19DRM obtidos pelo método DESM-RT em casos particulares. Amostra % de Metilação Perfil
SR16 36% Monoalélico
irSR1 39% Monoalélico
irSR2 43% Monoalélico
irSR3 38% Monoalélico
ST1 44% Monoalélico
Porcentagem metilação dos pacientes SSR16, irSR1, irSR2, irSR3, ST1 – perfil normal, monoalélico, de metilação.

5. Resultados
40
5.4. ANÁLISE DO PADRÃO DE METILAÇÃO DA KVDMR1
5.4.1. MS-PCR
Observou-se um perfil condizente com metilação monoalélica normal para
todas as amostras analisadas (Figura 16 e 17).
C1 C2 C3 C4 C5 C6 C7 . L M U M U M U M U M U M U M U br
Figura 16: Perfil de metilação da KvDMR pelo método MS-PCR em indivíduos saudáveis (controle). Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (C1 a C7) – amostras dos controles, com padrão normal de metilação. (M) – reação utilizando primers M1A e M1B, (U) – reação utilizando primers U2A e U2B. (br) – controle negativo da amplificação. SR1 SR2 SR3 SR4 . SR5 SR6 SR7 SR8 . L M U M U M U M U M U M U M U M U br
Figura 17: Perfil de metilação da KvDMR pelo método MS-PCR em amostras de pacientes com síndrome de Silver-Russell. Eletroforese em gel de poliacrilamida 10% visualizado por impregnação com nitrato de prata. (L) - marcador molecular (Ladder 100bp, Promega). (SR1 a SR8) – amostras dos pacientes, com padrão normal de metilação. (M) – reação utilizando primers M1A e M1B, (U) – reação utilizando primers U2A e U2B. (br) – controle negativo da amplificação.
169pb
169pb

5. Resultados
41
5.4.2. DESM-RT
Analisando quantitativamente o perfil de metilação da região KvDMR,
constatamos que os 10 indivíduos controle apresentaram uma média de metilação
de 49% ± 0,07 (Tabela 3). A partir desse grupo foram considerados níveis normais
de metilação os valores entre 35 e 63%.
Entre os 15 pacientes analisados, a média de metilação foi de 45% ± 3%, não
apresentando diferença estatisticamente significante quando comparado ao grupo
controle (p>0,05 – teste (método Mann Whitney, software Prism Graphpad (USA)
(Figura 19). Sendo que todos apresentaram perfil condizente de metilação normal,
monoalélico (tabela 4). O paciente ST1 também apresentou perfil normal (Tabela 6).
Já os pacientes com histórico de consanguinidade se mostraram parcialmente
hipermetilados. O paciente SR16 apresentou 68% de metilação nessa região e foi
verificado, ainda, para os três irmãos de 72%, 74% e 65% de metilação (Tabela 6).
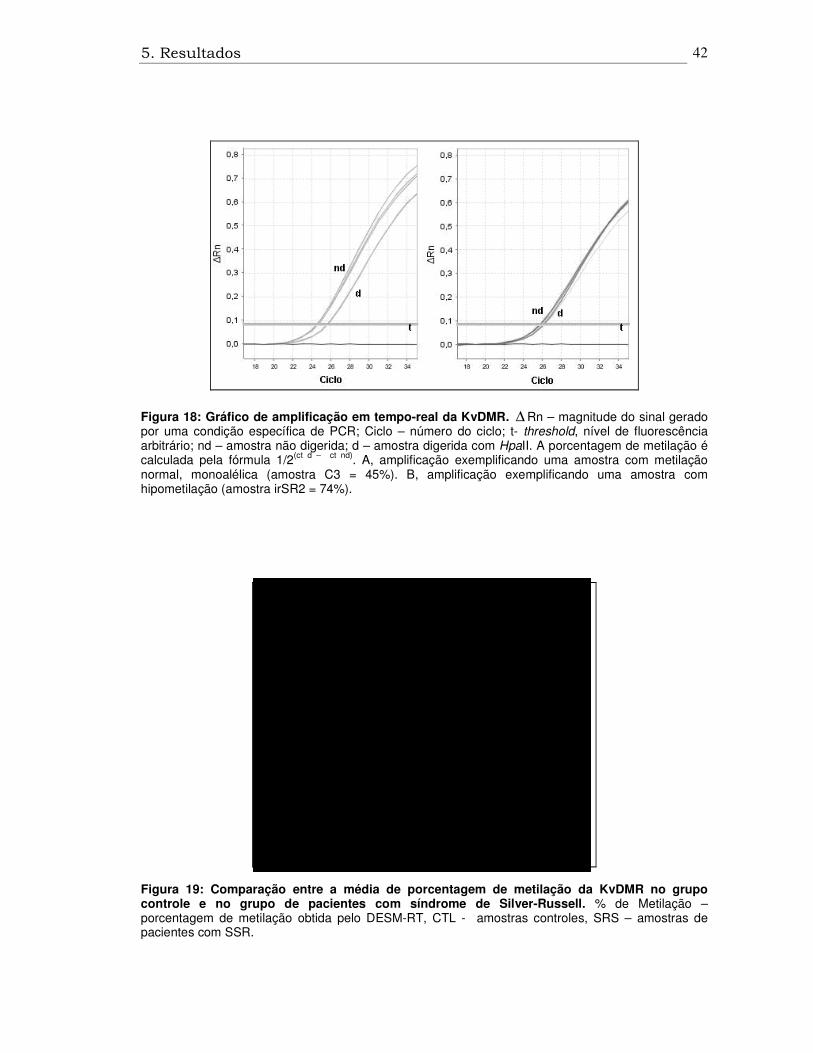
5. Resultados
42
Figura 18: Gráfico de amplificação em tempo-real da KvDMR. ∆ Rn – magnitude do sinal gerado por uma condição específica de PCR; Ciclo – número do ciclo; t- threshold, nível de fluorescência arbitrário; nd – amostra não digerida; d – amostra digerida com HpaII. A porcentagem de metilação é calculada pela fórmula 1/2(ct d – ct nd). A, amplificação exemplificando uma amostra com metilação normal, monoalélica (amostra C3 = 45%). B, amplificação exemplificando uma amostra com hipometilação (amostra irSR2 = 74%).
Figura 19: Comparação entre a média de porcentagem de metilação da KvDMR no grupo controle e no grupo de pacientes com síndrome de Silver-Russell. % de Metilação – porcentagem de metilação obtida pelo DESM-RT, CTL - amostras controles, SRS – amostras de pacientes com SSR.

5. Resultados
43
Tabela 4: Resultados da KvDMR obtidos pelo método DESM-RT em amostras controle.
Amostra % de Metilação
C1 58%
C2 53%
C3 45%
C4 60%
C5 56%
C6 42%
C7 44%
C8 41%
C9 42%
C10 45%
Porcentagem metilação das amostras controle C1, C2, C3, C4, C5, C6, C7, C8, C9 e C10.
5. Resultados
44
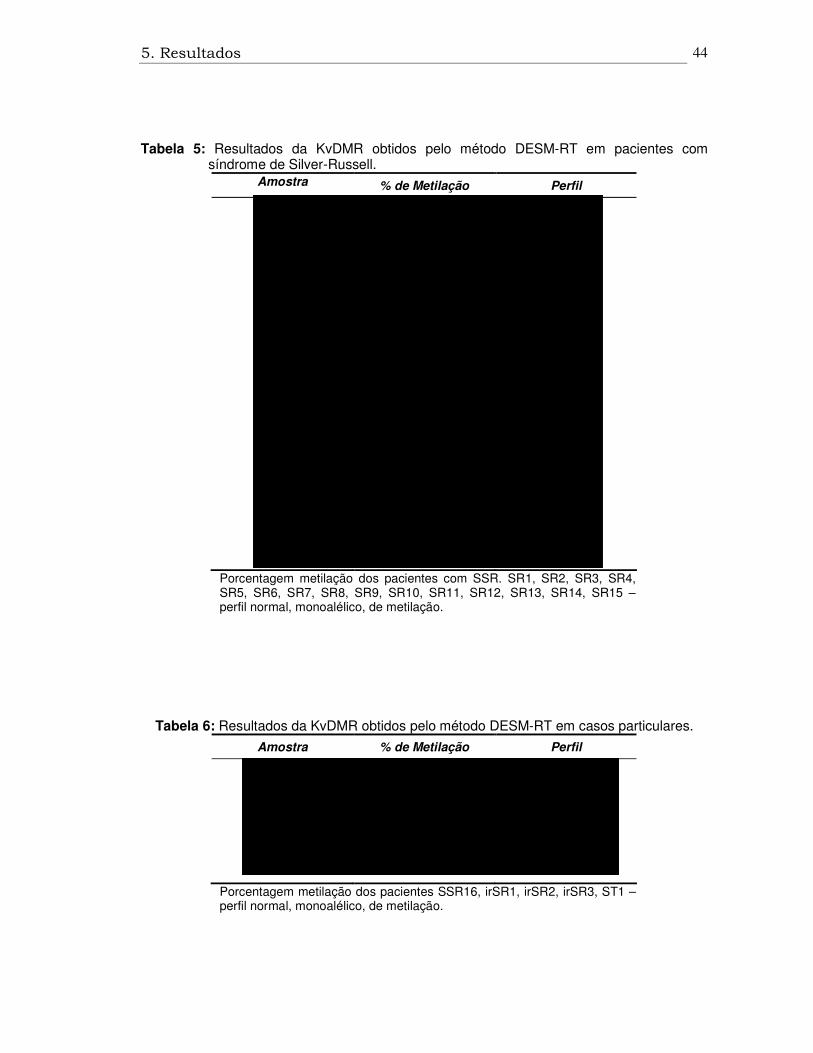
Tabela 5: Resultados da KvDMR obtidos pelo método DESM-RT em pacientes com síndrome de Silver-Russell.
Amostra % de Metilação Perfil
SR1 41% Monoalélico
SR2 48% Monoalélico
SR3 49% Monoalélico
SR4 46% Monoalélico
SR5 41% Monoalélico
SR6 44% Monoalélico
SR7 44% Monoalélico
SR8 49% Monoalélico
SR9 38% Monoalélico
SR10 48% Monoalélico
SR11 49% Monoalélico
SR12 45% Monoalélico
SR13 47% Monoalélico
SR14 43% Monoalélico
SR15 42% Monoalélico
Porcentagem metilação dos pacientes com SSR. SR1, SR2, SR3, SR4, SR5, SR6, SR7, SR8, SR9, SR10, SR11, SR12, SR13, SR14, SR15 – perfil normal, monoalélico, de metilação.
Tabela 6: Resultados da KvDMR obtidos pelo método DESM-RT em casos particulares.
Amostra % de Metilação Perfil
SR16 68% HIPERMETILADO
irSR1 72% HIPERMETILADO
irSR2 74% HIPERMETILADO
irSR3 65% HIPERMETILADO
ST1 43% Monoalélico
Porcentagem metilação dos pacientes SSR16, irSR1, irSR2, irSR3, ST1 – perfil normal, monoalélico, de metilação.

5. Resultados
45
5.5. CORRELAÇÃO DOS ACHADOS CLÍNICOS E MOLECULARES
Com relação aos aspectos clínicos, verificamos que o peso médio das
crianças ao nascimento foi de 1881 gramas, com desvio padrão de 634g. Todos os
pacientes analisados apresentaram retardo de crescimento pré e pós-natal (Tabela
7). Não foi possível obter o prontuário de dois pacientes analisados molecularmente
(SR2 e SR4), sendo, portanto, excluídos desta parte da análise.
O paciente (SR5) que apresentou hipermetilação do gene MEST possuía face
triangular, macrocefalia aparente, clinodactilia do 5° dedo, alterações de orelha, rima
labial com inclinação para baixo, leve atraso no desenvolvimento neuropsicomotor e
necessidade de ensino especial. Esse paciente não apresentou assimetria corporal
e também não há relato de alterações genitais.
Entre os pacientes (SR8 e SR13) com hipometilação da H19DMR analisados,
o caso SR8 apresentava macrocefalia aparente, face triangular, alterações
dentárias, clinodactilia do 5° dedo e uma discreta assimetria corpórea. O caso SR13,
também apresentou face triangular, aparente macrocefalia e clinodactilia, porém
com ausência de assimetria corpórea evidente.
Devido ao pequeno número de pacientes com alterações moleculares, não foi
possível uma análise estatística refinada para relacionar os achados laboratoriais
aos clínicos.
Os três pacientes irmãos analisados, oriundos do Hospital das Clínicas de
São Paulo (Faculdade de Medicina da USP, São Paulo) estão sendo publicados
separadamente por esse grupo, não sendo descritos com detalhes nesse trabalho.
Entretanto, o indivíduo SR16, com hipermetilação da KvDMR, apresentou peso e
comprimento ao nascimento de 2,400g e 45cm, retardo de crescimento pós-natal,

5. Resultados
46
macrocefalia aparente, face triangular, alterações dentárias, orelhas alteradas,
clinodactilia, assimetria corpórea, manchas café-com-leite, além de cisto renal à
direita, fístula sacro-coccígea, epífese do 2° ao 5° dedos tão largas quanto à diáfise
e musculatura hipotrófica.
O paciente com cariótipo condizente com a síndrome de Turner era do sexo
feminino, 45,X[25]/46,XX[75], e apresentava algumas características próprias dessa
alteração, como cabelo com implantação baixa em tridente, algumas que podem ser
compatíveis com ambas as doenças como baixo peso pós e pré-natal. Entretanto, a
paciente apresentava características típicas de SSR, como macrocefalia aparente,
face pequena e clinodactilia de 5° dedo de mãos.
Tabela 7. Resultado molecular e características clínicas dos pacientes com síndrome de
Silver-Russell.
amostra MEST H19 P.N. (g)
C.N. (cm)
R.C.pós-natal
Face Triang
Macrocef Aparente
Clinod 5°
SR1 2.250 42 x x x x
SR3 2.600 46 x x x x
SR5 x 2.600 46 x x x x
SR6 1.680 ND x x x
SR7 1.630 42 x x x
SR8 x 2300 45 x x x x
SR9 1380 35 x x x
SR10 760 34,5 x x x x
SR11 900 ND x x x x
SR12 1.500 39 x x x x
SR13 x 2.150 44 x x x x
SR14 2.000 43 x x x x
SR15 2.700 47 x x x
Aspectos moleculares e clínicos dos pacientes SR1, SR2, SR3, SR4, SR5, SR6, SR7, SR8, SR9, SR10, SR11, SR12, SR13, SR14, SR15. P.N. = peso ao nascimento, C.N. = comprimento ao nascimento, R.C.pós-natal = retardo de crescimento pós-natal, Face Triang = face triangular, Macrocef. Aparente = macrocefalia aparente, Clinod. 5° = clinodactilia do quinto dedo da mão.

5. Resultados
47
Continuação da Tabela 7. Alteração molecular e características clínicas dos pacientes com síndrome de Silver-Russell. amostra Sindact
II-III Assimet M Café c
Leite Alt de Orelha
Alt Dent
Dif Alimen
Déficit Cognit
Aum Sudor
R Lab Baixo
Alt de Voz
Atraso Fala
Outro
SR1 x x x x hérnia gástrica
SR3 x x
SR5 x x x x x hipotonia
SR6 x x alt genitais
SR7
SR8 x x
SR9 x x x x x x hipotonia
SR10 x x x prega simiesca
SR11 x x
SR12 x x x x x x campodactilia
SR13 x x alt. gastrointest.
SR14 x x x x x
SR15 x x x x
Aspectos clínicos dos pacientes SR1, SR2, SR3, SR4, SR5, SR6, SR7, SR8, SR9, SR10, SR11, SR12, SR13, SR14, SR15. Sindact II-III = sindactilia entre segundo e terceiro pododáctilos, Assimet = assimetria corporal, M Café c Leite = manchas café-com-leite, Alt de Orelha = alterações de orelha, Alt Dent = alterações dentárias, Dif Alimen = dificuldade de alimentação, R Lab baixo = rima labial com inclinação para baixo, Alt de Voz = alterações de voz, Atraso Fala = atraso de fala, Outro = Outras alterações.

6666. . . . DISDISDISDISCUSSÃOCUSSÃOCUSSÃOCUSSÃO

6. Discussão
49
Trabalhos anteriores descreveram o papel da DUP materna completa do
cromossomo 7 na etiologia da SSR, a qual tem sido responsabilizada por cerca de
10% dos casos (KOTZOT et al., 1995; EGGERMMAN et al., 1997; HANULLA et al.,
2002). A análise molecular da maioria dos trabalhos foi realizada por marcadores
moleculares do tipo microssatélite, tendo como desvantagem a necessidade de
amostras de DNA dos pais dos pacientes, geralmente de difícil acesso e com
dificuldades em relação à paternidade real, além da exigência de análise de
múltiplos loci.
Kim et al., em 2004, descreveram uma metodologia com a qual era possível
analisar a DUP pelo perfil de metilação em DMRs de dois genes que sofrem
imprinting e que são candidatos a um papel importante na etiologia da SSR, o
GRB10 e o MEST. Por meio desse ensaio pode-se obter informação adicional do
status de metilação específica desses genes. Entretanto, esse teste é ineficaz para
detecção de DUPs segmentares não relacionadas a esses genes, também citadas
em alguns casos (PRICE et al., 1999; MONK et al., 2002).
A análise da DUP do cromossomo 7 no presente trabalho foi realizada a partir
da pesquisa proposta por Kim. Entretanto, foram desenhados novos primers, sendo
escolhidas regiões já descritas como DMRs e testadas para a detecção de DUP
(AURNAULD et al., 2003; KIM et al., 2004). O padrão alterado de metilação não
encontrado simultaneamente nos genes MEST e GRB10 sugere a ausência de
mDUP7 completa, demonstrando que essa alteração não era o principal mecanismo
responsável pela SSR nos pacientes analisados.
Com relação especificamente ao gene GRB10, o grande interesse em
candidatá-lo na etiologia da síndrome iniciou-se com a descrição de dois pacientes

6. Discussão
50
com duplicação da região 7p11-p13 (JOYCE et al. 1999; MONK et al. 2000), sendo
também escolhido por estar sujeito ao imprinting genômico e por apresentar, em
estudos com camundongos e in vitro, um efeito supressor de crescimento
(YOSHIHASHI et al., 2000).
O padrão de metilação na DMR do gene GRB10 no presente trabalho
mostrou-se normal em todos os controles e pacientes. Outros grupos, buscando
mutações no gene ou duplicações da região, também não conseguiram relacionar o
desenvolvimento da síndrome com alterações específicas do GRB10. Por exemplo,
a duplicação descrita 7p11-p13 não foi mais encontrada em 55 pacientes com SSR
posteriormente analisados (JOYCE et al., 1999; MONK et al., 2000; MARTINEZ et
al., 2001), sendo também frustrada a busca de mutações pontuais nos éxons do
GRB10 (BLAGITKO et al., 2000; HITCHINS et al., 2001)
Apenas Yoshihashi et al., em 2000, conseguiram encontrar uma mesma
mutação pontual no GRB10 em dois pacientes. Entretanto, o fato dessa mutação ser
detectada em apenas dois de 163 pacientes sugere que essa mutação não é a
grande responsável pelo desenvolvimento da doença. Essa mutação pode constituir-
se na verdade em um raro polimorfismo restrito à população japonesa, sendo
necessários estudos funcionais para elucidar seu real papel.
O gene PEG1/MEST também tem sido descrito como candidato na etiologia
da SSR, principalmente devido a duas principais linhas de evidências. Entre essas, o
gene foi o primeiro descrito a estar sujeito ao imprinting genômico no cromossomo 7,
além de ter sua expressão confinada ao alelo paterno, o que condiz com a idéia de
que estaria inativado nos pacientes com mDUP7. A outra evidência vem de estudos

6. Discussão
51
em camundongos onde o silenciamento específico do alelo paterno resultou em
embriões viáveis, mas com crescimento retardado (RIESEWIJK et al., 1998).
Através da análise de metilação na região 5´UTR do gene MEST foi
identificado um paciente (SR5) com hipermetilação da região, o que representa 6,7%
(1/15). Isso condiz com a hipótese de que a baixa expressão do gene estaria ligada
ao desenvolvimento da síndrome, podendo ser também responsabilizada por parte
dos fenótipos encontrados nos pacientes com mDUP7.
Entretanto, a maioria dos trabalhos não relatou alterações específicas do
gene MEST, como visto por Riesewijk et al., em 1998, os quais não encontraram
mutações condizentes com uma possível alteração de função na região promotora,
nos éxons e nem nas ligações íntron-éxon em 49 pacientes com SSR, além de
comprovarem um padrão normal de metilação na região 5´ do gene em 35
pacientes. Kobayashi et al., em 2001, não encontraram mutações na sequência do
gene, verificaram um perfil normal de metilação, além de uma expressão normal em
linfócitos e Meyer et al., em 2003, que buscaram mutações no gene MEST1T1,
identificado como um RNA não-codificante possivelmente envolvido na regulação do
MEST, verificaram a ausência de mutações especificamente ligadas aos 46
pacientes com SSR analisados.
Contrariamente, Hanulla et al., em 2001, descreveram um paciente com
mDUP específica da região 7q31-qter. Essa observação, conjuntamente com o
resultado de metilação verificado no paciente SR5, sugere que o gene MEST teria
papel na etiologia da síndrome.
O paciente SR5 possui alteração de metilação especificamente no gene
MEST, sendo normal para o gene GRB10. Como proposto por Kim et al., em 2004,

6. Discussão
52
apenas a alteração de metilação nas duas regiões seria compatível com a mDUP7
completa, geralmente encontrada nos pacientes com SSR, sugerindo a não
existência dessa alteração no paciente. Porém, não podemos excluir, embora menos
provável, a possibilidade de ter ocorrido uma dissomia e posteriormente uma
alteração de metilação na região do GRB10, o que resgataria o perfil normal
apresentado. Ainda não foi possível concluir se o paciente tem uma alteração
específica de metilação ou uma mDUP7 segmentar da região, como descrito por
Hanulla et al. (2001). O que não diminui a possibilidade que essa hipermetilação do
gene MEST tenha um efeito fenotípico, semelhante ao que teria nos portadores de
DUP7 completa.
Porém, devido ao número de relatos que não responsabilizaram o gene
MEST isoladamente e que dos pacientes analisados apenas um possuiu a alteração,
sugere-se que o gene deva estar envolvido na etiologia da síndrome, mas em uma
minoria de casos e/ou em associação com outros localizados no próprio
cromossomo 7 e em outros cromossomos.
A relação na literatura do cromossomo 11 com a SSR iniciou-se com a
publicação de por Gicquel et al., em 2005. Esses pesquisadores começaram a
estudar essa relação devido ao forte envolvimento da região 11p15 no controle de
crescimento fetal. Em suas análises, esses pesquisadores examinaram o perfil de
metilação na KvDMR1, no promotor do H19 e na H19DMR em sangue periférico de
nove indivíduos, sendo que cinco desses apresentaram uma perda de metilação
parcial nos domínios do H19 e todos apresentaram um perfil normal na KvDMR1. Os
casos com hipometilação também foram analisados com relação ao perfil na
IGF2DMR2, apresentando igual perda de metilação parcial. Para avaliar a real
diminuição de expressão devida à perda de metilação, foi analisado o nível de RNA

6. Discussão
53
do IGF2 em fibroblastos de dois pacientes, confirmando-a. A expressão bialélica do
H19 foi também verificada em um indivíduo heterozigoto para um polimorfismo na
região, confirmando o relaxamento da regulação do imprinting em pacientes
parcialmente hipometilados.
Posteriormente Ergermman et al (2006) chegaram às mesmas conclusões
observando perda de metilação parcial na H19DMR em 16 de 51 (31%) pacientes e
perfil de metilação normal na porção centromérica de 11p15. Ainda em 2006, outro
trabalho confirmou a hipometilação parcial da ICR1, sendo detectada em cinco de 25
pacientes (20%) com características de SSR e não encontrando essa alteração em
grupos de pacientes com atraso de crescimento isolado e com outras dismorfias não
associadas à SSR (SCHÖNHERR et al., 2006). A partir de então, outros trabalhos
vêm confirmando o papel da H19DMR na etiologia da SSR e descrevendo a
alteração como presente em alta frequência (20 a 65%) (YAMAZAWA et al, 2008),
inclusive relatando uma ausência de metilação em alguns casos (BLIEK et al.,
2006).
A ausência de metilação da região não foi verificada em nossos estudos.
Esse dado foi sugerido pelo perfil normal de metilação H19DMR através do método
COBRA, um ensaio qualitativo e que mostrou a presença de alelos metilados nas
amostras, sendo posteriormente confirmada pela análise quantitativa, sendo a
metilação mais baixa observada de 9% (SR2). Porém, a ausência de metilação
apenas foi descrita por Bliek et al., em 2006, sendo que os resultados de
hipometilação parcial verificada no presente trabalho estão em acordo com a grande
maioria dos relatos (GICQUEL et al., 2005; NETCHINE et al., 2007; BRUCE et al.,
2009).

6. Discussão
54
A perda parcial de metilação foi a alteração molecular mais encontrada na
população estudada, em concordância com a literatura. A frequência de pacientes
com hipometilação descrita aqui (20%), foi igual aos dados de Schönherr (2006),
porém foram também já descritas frequências mais altas como 31, 40 e até 64%
(ERGGERMAN et al.,2006; NETCHINE et al., 2007; BARTHOLDI et al., 2008). As
diferenças nas taxas de metilação verificadas podem refletir a metodologia
empregada, uma vez que a maioria das vezes foi utilizado Southern blot e
recentemente têm sido descritas metodologias mais sensíveis como a MLPA e a
PCR em tempo real (ZESCHNIGK ET AL., 2008; EGGERMANN ET AL., 2008).
Entretanto, as diferenças de frequência não podem ser totalmente justificadas por
essa abordagem visto que a maior já descrita, 64%, foi verificada por Netchine et al.
(2008) utilizando Southern blot e nas nossas análises, com frequência mais baixa,
foi utilizada a DESM-RT, com PCR em tempo real.
A DESM-RT utilizada nesse trabalho foi descrita pela primeira vez por nosso
grupo (GOMES et al, 2007). Posteriormente, Bruce et al., 2008, utilizando a mesma
metodologia, confirmaram a aplicabilidade da técnica e descreveram 62% pacientes
com SSR como tendo hipometilação da H19DMR, frequência muito mais alta da que
por nós relatada (BRUCE et al, 2008; BRUCE et al, 2009).
Outra possibilidade para explicar as diferenças de frequência seria o critério
diagnóstico utilizado. Alguns trabalhos podem ter apenas incluído em seu grupo de
estudos os casos mais característicos, descritos como mais severos e mais
relacionados especificamente a alteração da H19DMR (BINDER et al., 2008;
BRUCE et al., 2009), já outros podem ter incluído pacientes com algumas poucas
características de SSR, esses citados como tendo baixa frequência de alteração de

6. Discussão
55
metilação (NETCHINE et al., 2008). Por isso, um consenso pré-definido para o
diagnóstico clínico da SSR seria útil para estudos futuros.
Com relação à KvDMR, seu envolvimento na SSR ainda é incerto, ao
contrário do que ocorre nos pacientes com a SBW, dos quais a grande maioria dos
casos (aproximadamente 50%) se deve à alteração nessa região (BLIEK et al.,
2001; DEBAUN et al., 2002; GOMES; RAMOS, 2003; GOMES; SANTOS; RAMOS,
2005; WEKSBERG et al., 2005). Na SSR, essa parece não ter tanta importância,
visto o número de trabalhos que não encontraram alterações (GICQUEL et al., 2005;
ERGERMMAN et al., 2006; NETCHINE et al., 2007).
O atual trabalho confirma essa verificação pela ausência de alteração no
grupo dos 15 pacientes com SSR analisados, entretanto, a análise dos casos
particulares demonstrou uma hipermetilação parcial leve, mas significativa, para os
três irmãos com fenótipo de SSR (irSR1, irSR2, irSR3) e o paciente filho de pais
consanguíneos (SR16).
O envolvimento da KvDMR na SSR foi sugerido pela verificação de pacientes
com duplicação materna da região 11p15. Em 2002, Fisher et al. descreveram essa
duplicação em três pacientes com atraso de crescimento, sendo também encontrada
a mesma duplicação em dois pacientes em um trabalho realizado por Eggermann et
al., em 2005, analisando 46 pacientes.
O nosso trabalho verificou que os pacientes com alteração na KvDMR
apresentaram perfil normal de metilação na H19DMR, sendo condizente com um
único relato descrito de uma duplicação restrita à ICR2 (SCHÖNHERR et al., 2007),
o que enfatiza o possível papel da região no desenvolvimento da SSR. Porém, não
pudemos concluir se a alteração encontrada é devida à duplicação de DNA ou a um
distúrbio especificamente de metilação.

6. Discussão
56
Pela porcentagem normal de metilação na H19DMR (entre 36 e 43%),
sugere-se que esses pacientes não possuem mDUP da região 11p15, entretanto,
Bullman et al.(2008), descreveram um paciente SSR com leve aumento de metilação
na KvDMR através de um ensaio quantitativo de MS-PCR. Esse, inicialmente,
possuía uma quantificação de metilação na H19DMR não descrita como alterada,
porém, estudos mais aprofundados, com MS-MLPA ( multiplex ligation dependent
probe analysis), demonstraram se tratar de mosaicismo de mDUP da região 11p15.
Portanto, não podemos excluir essa possibilidade em nosso trabalho.
Embora sem a verificação de que o aumento de metilação na KvDMR
observado nesse trabalho era devido ou não a uma duplicação como descrito por
Schönherr et al., 2007, a uma DUP como relatado por Bullman et al.(2008) ou a um
distúrbio específico de metilação, sugere-se o envolvimento dessa região na
etiologia da SSR.
Interessantemente, todos os pacientes descritos neste trabalho com alteração
na KvDMR possuem casos condizentes com herança recessiva, (SR16 – filho de
pais consanguíneos; irSR1, irSR2, irSR3 – irmãos, filhos de pais fenotipicamente
sem a síndrome). Para o paciente SR16, a alteração de metilação pode ser devida a
alguma mutação recessiva de genes regulatórios, o que teria levado à
hipermetilação apresentada pelo indivíduo. Já nos casos irSRs, existe a
possibilidade de estar ocorrendo uma alteração nos mecanismos de metilação de
um dos gametas, o que causaria o distúrbio nos três indivíduos, por exemplo, uma
falha de apagamento completo da marcação epigenética nos espermatozóides do
pai. Uma alteração cromossômica também não pode ser descartada. Contudo,
mesmo sem o entendimento dos mecanismos que permitiriam o processo, a
transmissão de defeitos epigenéticos também pode ser considerada, principalmente

6. Discussão
57
a partir do relato de Bartholdi et al. (2008) no qual foi descrito transmissão vertical de
pai para filha da hipometilação da H19DMR, demonstrando que há um risco de
recorrência de epimutações. Esses achados têm importantes implicações no
aconselhamento genético, uma vez que se acredita ser baixo o risco de recorrência
de alterações epigenéticas, subestimando os riscos. Entretanto, estudos posteriores
se fazem necessários para o melhor entendimento do processo.
Os níveis de hipermetilação descritos neste trabalho (de 65 a 74%) e também
o baixo mosaicismo descrito por Bartlholdi (18%), além do número de relatos que
não relacionaram a KvDMR com a SSR contrastam com as frequências de alteração
da mesma região na SBW. Existe a possibilidade de que essa alteração esteja
presente em maior frequência nos pacientes com SSR, entretanto, devido a razões
técnicas ou alterações em outras células, não leucócitos, esses números sejam
subestimados. Alternativamente, o aumento da metilação em altos níveis ou em
alguns tecidos específicos poderia ser incompatível com a vida.
A partir dos dados aqui encontrados foi verificada alteração em três regiões
distintas, possivelmente envolvidas na etiologia da SSR. Entretanto, a maioria dos
pacientes apresentou perfil normal de metilação, sugerindo que existam ainda outras
regiões a serem responsabilizadas por fenótipos característicos da SSR.
Enfatizando a heterogeneidade genética da SSR, também foi descrito um
indivíduo com síndrome de Turner e dismorfias condizentes com a SSR (ST1). Os
pacientes com síndrome de Turner apresentam, na maioria dos casos, apenas um
cromossomo X em algumas de suas células. Sabe-se que fenótipos decorrentes de
doenças com padrão de herança ligada ao X recessiva podem ser mais
frequentemente observados em meninas portadoras dessa síndrome por estarem os
genes em cópia única, como nos meninos. A partir dessa verificação, existe a

6. Discussão
58
possibilidade de que a paciente (ST1) possua alguma mutação ligada ao X
responsável pelo fenótipo de SSR.
A relação do cromossomo X com a SSR já foi sugerida em 1986, com a
descrição de uma família com baixa estatura, pigmentações anormais e dismorfias
faciais, sugerindo herança ligada ao X (PARTINGTON, 1986). Também já foi
descrito em 1972 um paciente com cariótipo 45,X/46,XY e achados clínicos de SSR
(TULINIUS, TRYGGVASON, HAUKSDOTTIR, 1972). Esses relatos, adicionalmente
à paciente descrita no presente trabalho, demonstram o envolvimento de outros
genes ainda não conhecidos na etiologia da SSR e sugerem que algum esteja
localizado no cromossomo X.
Além dos aspectos moleculares, a relação direta entre as alterações
epigenéticas e o fenótipo não é clara. Alguns trabalhos têm tentado encontrar essa
ligação.
Netchine et al. (2007) mostraram que peso e comprimento ao nascimento e
índice de massa corpórea pós-natal foram significativamente menores no grupo de
pacientes com hipometilação da H19. Esse trabalho também relatou o aumento de
assimetria corpórea e macrocefalia relativa nesse grupo de pacientes. Bartholdi et al.
(2008) pontuando pacientes com suspeita de SSR de 0 (mais brando) a 15 (mais
severo), de acordo com o número de características secundárias presentes,
descreveram que 41 de 42 pacientes com alteração de metilação na H19DMR
apresentavam pontuação maior ou igual a 8, relacionando a alteração com a
gravidade do fenótipo. A média de pontuação nos pacientes com alteração na
H19DMR, foi de 13. Já nos 10 pacientes com DUP7 a média foi de 8 pontos. Porém,
nos pacientes diagnosticados sem nenhuma das alterações moleculares analisadas,

6. Discussão
59
a média de pontuação foi 9, sendo também descrito um aumento de assimetria nos
pacientes com alteração da H19.
O número de pacientes com alterações moleculares em nosso trabalho foi
baixo para um estudo mais aprofundado relacionado ao fenótipo. Entretanto,
verificamos que os dois pacientes com alteração da H19DMR e fenótipo analisado
apresentaram quadro característico, ambos com macrocefalia e um deles com
assimetria corpórea. Todavia essas características também foram frequentemente
encontradas em outros pacientes com ausência da alteração molecular.
A partir do trabalho de Bartholdi et al. (2008), sugere-se que os portadores de
mDUP7 tenham fenótipo mais brando, entretanto, o paciente descrito nesse trabalho
com alteração no gene MEST apresentou várias características “menores” da SSR,
o que contrasta com essa idéia. Porém, esse dado é de apenas um paciente, o que
impede relacionarmos a alteração molecular com o fenótipo.
Independente da relação exata da alteração molecular e do fenótipo, é nítido
que na etiologia da SSR diferentes cromossomos podem levar a características
fenotípicas semelhantes. Estudos futuros deverão explorar como ocorre essa
interação funcional entre regiões distintas para se compreender melhor a regulação
do complexo mecanismo de crescimento humano.
O presente estudo confirma a possibilidade de utilização das quatro regiões
analisadas na prática clínica, auxiliando no melhor diagnóstico desses pacientes,
visto que aproximadamente 35% dos portadores de SSR apresentaram uma
alteração molecular detectada. Como as alterações podem se apresentar na forma
de mosaicos acredita-se que essa porcentagem pode ser aumentada com a
pesquisa adicional de outros tecidos, melhorando ainda mais a detecção dessas
anomalias.

6. Discussão
60
Benefícios diretos do diagnóstico mais rápido e com maior credibilidade
podem ser obtidos a partir de nosso trabalho, além da contribuição para estudos
posteriores que poderão auxiliar no aconselhamento genético das famílias com
portadores de SSR e desenvolvimento de novos tratamentos.

7777. . . . CONCLUSÃOCONCLUSÃOCONCLUSÃOCONCLUSÃO

7. Conclusão
62
• O padrão de metilação na região diferencialmente metilada do gene
GRB10 não estava envolvido na etiologia da SSR nos pacientes analisados;
• Foi verificada hipermetilação na amostra de um paciente (6,7%) na
região diferencialmente metilada do gene MEST;
• O padrão de metilação na H19DMR foi parcialmente hipometilado em
20% dos pacientes;
• Um perfil de hipermetilação na KvDMR1 foi detectado em alguns
pacientes analisados separadamente, mostrando o envolvimento dessa região na
etiologia da SSR;
• Nenhuma das alterações observadas foi detectada simultaneamente
em um mesmo paciente.
• Não foi possível fazer uma análise estatística correlacionando uma
determinada alteração molecular e o desenvolvimento de um quadro fenotípico mais
acentuando ou a alguma característica específica;
• A aplicação das metodologias utilizadas mostrou-se eficiente na
confirmação diagnóstica da SSR, uma vez que aproximadamente 35% dos
portadores de SSR apresentaram a alteração molecular detectável.

8. REFERÊNCIAS8. REFERÊNCIAS8. REFERÊNCIAS8. REFERÊNCIAS

8. Referências
64
ANDERSON J, VISKOCHIL D, O'GORMAN M, GONZALES C. Gastrointestinal complications of Russell-Silver syndrome: a pilot study. Am J Med Genet, v. 113 (1), p. 15-19, 2002.
ARNAUD P, MONK D, HITCHINS M, GORDON E, DEAN W, BEECHEY CV, PETERS J, CRAIGEN W, PREECE M, STANIER P, MOORE GE, KELSEY G. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum Mol Genet, v. 12, p. 1005-1019, 2003.
AZCONA C, STANHOPE R. Hypoglycaemia and Russell-Silver syndrome. J Pediatr Endocrinol Metab, v. 18(7), p. 663-670, 2005.
BARTHOLDI D, KRAJEWSKA-WALASEK M, ÕUNAP K, GASPAR H, CHRZANOWSKA KH, ILYANA H, KAYSERILI H, LURIE LW, SCHINZEL A, BAUMER A. Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell Syndrome (SRS): Results from a large cohort of patients with SRS and SRS-like phenotypes. J Med Genet. 2008.
BELL, A.C., FELSENFELD, G. Methylation of a CTCF dependent boundary controls imprinted expression of the Igf2 gene. Nature, v. 405, p. 482-485, 2000.
BESTOR, T.H. The DNA methyltransferases of mammals. Hum Mol Genet, v. 9, p. 2395-2402, 2000.
BINDER G, SEIDEL AK, MARTIN DD, SCHWEIZER R, SCHWARZE CP, WOLLMANN HA, EGGERMANN T, RANKE MB. The Endocrine Phenotype in Silver-Russell Syndrome Is Defined by the Underlying Epigenetic Alteration. J Clin Endocrinol Metab, v. 93 (4), p.1402–1407, 2008.
BINDER G, SEIDEL AK, WEBER K, HAASE M, WOLLMANN HA, RANKE MB, EGGERMANN T. IGF-II Serum Levels Are Normal in Children with Silver- Russell Syndrome Who Frequently Carry Epimutations at the IGF2 Locus. J Clin Endoc Metab, v. 91 (11), p. 4709–4712, 2006.
BLACK J. Low birth weight dwarfism. Arch Dis Child, v. 36, p. 633-644, 1961.
BLAGITKO N, MERGENTHALER S, SCHULZ U, WOLLMANN HA, CRAIGEN W, EGGERMANN T, ROPERS HH, KALSCHEUER VM. Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum Mol Genet, v. 9 (11), p. 1587-1595, 2000.
BLIEK J, TERHAL P, VAN DEN BOGAARD MJ, MAAS S, HAMEL B, SALIEB- BEUGELAAR G, SIMON M, LETTEBOER T, VAN DER SMAGT J, KROES H, MANNENS M. Hypomethylation of the H19 Gene Causes Not Only Silver- Russell Syndrome (SRS) but Also Isolated Asymmetry or an SRS-Like Phenotype. Am J Hum Genet, v. 78, p. 604–614, 2006.

8. Referências
65
BLIEK, J., MAAS, S.M., RUIJTER, J.M., HENNEKAM, R.C., ALDERS, M., WESTERVELD, A., MANNENS, M.M. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum Mol Genet, v. 1, p. 467-476, 2001.
BOURC’HIS, D., XU, G.L., LIN, C.S., BOLLMAN, B. BESTOR, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science, v. 294, p. 2536-2539, 2001.
BRUCE S, HANNULA-JOUPPI K, PELTONEN J, KERE J, LIPSANEN-NYMAN M. Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethylation associates with SRS phenotype severity and genital and skeletal anomalies. J Clin Endocrin Metab, v. 94(2), p. 579-587, 2009.
BRUCE S, HANNULA-JOUPPI K, PELTONEN J, KERE J, LIPSANEN-NYMAN M. Restriction Site–Specific Methylation Studies of Imprinted Genes with Quantitative Real-Time PCR. Clin Chemistry, v. 54 (3), p. 1-9, 2008.
BULLMAN H, LEVER M, ROBINSON DO, MACKAY DJG, HOLDER SE, WAKELING EL. Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver_Russell syndrome. J Med Genet, v. 45, p. 396- 399, 2008.
CHARALAMBOUS, M., SMITH, F.M., BENNETT, W.R., CREW, T.E., MACKENZIE, F., WARD, A. Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc Natl Acad Sci, v. 100, p. 8292–8297, 2003.
CHAUVEL PJ, MOORE CM, HASLAM RHA. Trisomy-18 mosaicism with features of Russell-Silver syndrome. Dev Med Child Neurol, v. 17, p. 220–224, 1975.
DEBAUN, M.R., NIEMITZ, E.L., MANEIL, D.E., BRANDENBURG, S.A. LEE, M.P., FEINBERG, A.P. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet, v. 70, p. 604-611, 2002.
DECHIARA TM, EFSTRATIADIS A, ROBERTSON EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II disrupted by targeting. Nature, v. 345, p. 78–80, 1990.
DELAVAL K, WAGSCHAL A, FEIL R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. BioEssays, v. 28, p. 453–459, 2006.
DUNCAN PA, HALL JG, SHAPIRO LR, VIBERT BK. Threegeneration dominant transmission of the Silver-Russell syndrome.Am J Med Genet, v. 35, p. 245- 250, 1990.

8. Referências
66
EGGENSCHWILER J, LUDWIG T, FISHER P, LEIGHTON PA, TILGHMAN SM, EFSTRATIADIS A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi- Behmel syndromes. Genes Dev, v. 11, p. 3128–3142, 1997.
EGGERMANN T, MEYER E, OBERMANN C, HEIL I, SCHÜLER H, RANKE MB, EGGERMANN K, WOLLMANN HA. Is maternal duplication of 11p15 associated with Silver-Russell syndrome? J Med Genet, v. 42, p. 26-30, 2005.
EGGERMANN T, SCHÖNHERR N, EGGERMANN K, BUITING K, RANKE MB, WOLLMANN HA, BINDER G. Use of multiplex ligation-dependent probe amplification increases the detection rate for 11p15 epigenetic alterations in Silver–Russell syndrome. Clin Genet, v. 73, p. 79–84, 2008.
EGGERMANN T, SCHÖNHERRN N, MEYER E, OBERMANN C, MAVANY M, EGGERMANN K, RANKE MB, WOLLMANN HA. Epigenetic mutations in 11p15 in Silver-Russell syndrome are restricted to the telomeric imprinting domain. J Med Genet. v.43, p. 615-616, 2006.
EGGERMANN T, WOLLMANN HA, KUNER R, EGGERMANN K, ENDERS H, KAISER P, RANKE MB. Molecular studies in 37 Silver-Russell syndrome patients: frequency and etiology of uniparental disomy. Hum Genet, v. 100, p. 415–419, 1997.
FISHER AM, THOMAS NS, COCKWELL A, STECKO O, KERR B, TEMPLE IK, CLAYTON P. Duplications of chromosome 11p15 of maternal origin result in a phenotype that includes growth retardation. Hum Genet, v. 111 (3), p. 290- 296, 2002.
FEINBERG AP. Imprinting of a genomic domain of 11p15 and loss of imprinting in cancer: an introduction. Cancer Res, v. 59, p. 1743–1746, 1999.
FROMMER, M., MCDONALD, L.E., MILLAR, D.S., COLLIS, C.M., WATT, F., GRIGG, G.W., MOLLOY, O.L., PAUL, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA, v. 89, p. 1827-1831, 1992.
GABORY A, RIPOCHE MA, YOSHIMIZU T, DANDOLO L. The H19 gene: regulation and function of a non-conding RNA. Cytogenet Genome Res, v. 113 (1-4), p.188-193, 2006.
GICQUEL C, ROSSIGNOL S, CABROL S, HOUANG M, STEUNOU V, BARBU V, DANTON F, THIBALDI N, MERRER M, BURGLEN L, BERTRAN AN, NETCHINE I, BOUC Y. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet, v. 37, p. 1003–1007, 2005.

8. Referências
67
GOMES MV, GOMES CC, PINTO W JR, RAMOS ES. Methylation pattern at the KvDMR in a child with Beckwith-Wiedemann syndrome conceived by ICSI. Am J Med Genet A, v. 143 (6), p. 625-629, 2007.
GOMES, M.V., RAMOS, E.S. Beckwith-Wiedemann and Isolated Hemihyperplasia. S P Med Journ, v. 121, p. 133-138, 2003.
GOMES, M.V., SANTOS, A.S., RAMOS, E.S. H19DMR methylation analysis in patients with Beckwith-Wiedemann syndrome and isolated hemihyperplasia. Genet Mol Biol, v. 28, p. 210-213, 2005.
GOUDA SA, BASTAKI L, AL-AWADI SA, AL-MAZIDI Z, AL-GHANIM M, SABR MA, FARAQ TI. Silver-Russell syndrome in Bedouin sibs: autosomal recessive inheritance confirmed. Am J Hum Genet, v. 59, p. 509, 1996.
HANNULA K, LIPSANEN-NYMAN , KRISTO P KAITILA I, SIMOLA KO, LENKO HL, APANAINEN P, HOLMBERG C, KERE J. Genetic Screening for Maternal Uniparental Disomy of Chromosome 7 in Prenatal and Postnatal Growth Retardation of Unknown Cause. Pediatrics, v. 109 (3), p. 441 – 448, 2002.
HANNULA K, LIPSANEN-NYMAN M, KONTIOKARI T, KERE J. A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver-Russell syndrome delimits a candidate gene region. Am. J. Hum. Genet, v. 68, p. 247-253, 2001.
HATA, K., Okano, M., Lei, H., Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development, v. 129, p. 1983-1993, 2002.
HERMAN, J.G., GRAFF, J.R., MYOHANSEN, S., NELKIN, B.D., BAYLIN, S.B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A, v. 93, p. 9821-9826, 1996.
HITCHINS MP, STANIER P, PREECE MA, MOORE GE. Silver-Russell syndrome: a dissection of the genetic aetiology and candidate chromosomal regions. J Med Gene, v. 38, p. 810-9, 2001.
HOLMGREN, C., KANDURI, K., DELL, G., WARD, A., MUKHOPADHYAY, R., KANDURI, M., LOBANEKOV, V., OHLSSON, R. Cpg methylation regulates the Igf2/H19 insulator. Curr Biol, v. 11, p. 1128-1130, 2001.
JELTSCH, A., NELLEN, W., LYKO, F. Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci, v. 31, p. 306- 308, 2006.
JOYCE CA, SHARP A, WALKER JM, BULLMAN H, TEMPLE KI. Duplication of 7p12.1-p13, including GRB10 and IGFBP1,in a mother and daughter with f eatures of Silver-Russell syndrome. Hum Genet, v. 105, p. 273–280, 1999.

8. Referências
68
KIM Y, KIM SS, KIM G, PARK S, PARK IS, YOO HW. Detection of maternal uniparental disomy at the two imprinted genes on chromosome 7, GRB10 and PEG1/MEST, in a Silver–Russell syndrome patient using methylation- specific PCR assays. Clin Genet, v. 67, p. 267–269, 2004.
KOBAYASHI S, UEMURA H, KOHDA T, NAGAI T, CHINEN Y, NARITOMI K, KINOSHITA EI, OHASHI H, IMAIZUMI K, TSUKAHARA M, SUGIO Y, TONOKI H, KISHINO T, TANAKA T, YAMADA M, TSUTSUMI O, NIIKAWA N, KANEKO-ISHINO T, ISHINO F. No evidence of PEG1/MEST gene mutations in Silver-Russell syndrome patients. Am J Med Genet, v. 104 (3), p. 225-231, 2001.
KOTZOT D, SCHMITT S, BERNASCONI F, ROBINSON WP, LURIE IW, ILYINA H, MÉHES K, HAMEL BC, OTTEN BJ, HERGERSBERG M. Uniparental disomy 7 in Silver-Russell syndrome and primordial growth retardation. Hum Mol Genet, v. 4 (4), p. 583-587,1995
KOTZOT D. Maternal uniparental disomy 7 and Silver-Russell syndrome - clinical update and comparison with other subgroups. Eur J Med Genet, v. 51 (5), p. 444-451, 2008.
LAI KC, SKUSE D, STANHOPE R, HINDMARSH P. Cognitive abilities of children with the Russell-Silver syndrome. Arch Dis Child, v. 71, p.490– 496, 1994.
LEE MP, DEBAUN MR, MITSUYA K, GALONEK HL, BRANDENBURG S, OSHIMURA M, FEINBERG AP. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith–Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci USA, v. 96, p. 5203–5208, 1999.
LEFEBVRE L, VIVILLE S, BARTON SC, ISHINO F, KEVERNE EB, SURANI MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet, v. 20, p.163-169, 1998.
LEWIS A, REIK W. How imprinting centres work. Cytogenet Genome Res, v. 113 (1-4), p. 81-89, 2006.
LI E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet, v. 3 (9), p. 662-73, 2002.
LI, L.C., DAHIYA, R. Methprimer: designing primers for methylation PCRs. Bioinformatics, v. 18, p. 1427-1431, 2002.
MARTINEZ MJ, BINKERT F, SCHINZEL A, KOTZOT D. No evidence of dup(7)(p11.2p13) in Silver-Russell syndrome. Am J Med Genet, v. 99 (4), p. 335-337, 2001.

8. Referências
69
MEYER E, WOLLMANN HA, EGGERMANN T. Searching for genomic variants in the MESTIT1 transcript in Silver-Russell syndrome patients. J Med Genet, v. 40(5), p.65, 2003.
MONK D, BENTLEY L, HITCHINS M, MYLER RA, CLAYTON-SMITH J, ISMAIL S, PRICE SM, PREECE MA, STANIER P, MOORE GE. Chromosome 7p disruptions in Silver Russell syndrome: delineating an imprinted candidate gene region. Hum Genet, v. 111 (4-5), p. 376-387, 2002.
MONK D, WAKELING EL, PROUD V, HITCHINS M, ABU-AMERO SN, STANIER P, PREECE MA, MOORE GE. Duplication of 7p11.2-p13, including GRB10, in Silver-Russell syndrome. Am J Hum Genet, v. 66 (1), p. 36-46, 2000.
MONK D, WAGSCHAL A, ARNAUD P, MÜLLER PS, PARKER-KATIRAEE L, BOURC'HIS D, SCHERER SW, FEIL R, STANIER P, MOORE GE. Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing. Genome Res, v. 18 (8), p. 1270-1281, 2008.
MOSS TJ, WALLRATH LL. Conections between epigenetic gene silencing and human disease. Mutation Research, v. 1-2 (618), p. 163-174, 2007.
NETCHINE I, ROSSIGNOL S, DUFOURG MN, AZZI S, ROUSSEAU A, PERIN L, HOUANG M, STEUNOU V, ESTEVA B, THIBAUD N, DEMAY MCR, DANTON F, PETRICZKO E, BERTRAND AM, HEINRICHS C, CAREL JC, LOEUILLE GA, PINTO G, JACQUEMONT ML, GICQUEL C, CABROL S, BOUC YL. 11p15 Imprinting Center Region 1 Loss of Methylation Is a Common and Specific Cause of Typical Russell-Silver Syndrome: Clinical Scoring System and Epigenetic-Phenotypic Correlations. J Clin Endoc Metabol, v. 92(8), p. 3148–3154, 2007.
NEYROUD N, RICHARD P, VIGNIER N, DONGER C, DENJOY I, DEMAY L, SHKOLNIKOVA M, PESCE R, CHEVALIER P, HAINQUE B, COUMEL P, SCHWARTZ K, GUICHENEY P. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ Res, v. 84 (3), p. 290-297, 1999.
NOEKER M, WOLLMANN HA. Cognitive development in Silver–Russell syndrome: a siblingcontrolled study. Dev Med & Child Neuro, v. 46, p. 340–346, 2004.
NOGUEIRA AMM, COSTEIRA MJT, MOREIRA HPS, ANTUNES HA. Síndrome de Russel-Silver. An Esp Pediatr, v. 54, p. 591-594, 2001.
OLERUP, O., ZETTERQUIST, H. HLA-DR typing by PCR amplification with sequence-specific primers (PCR-SSP) in 2 hours: an alternative to serological DR typing in clinical practice including donor-recipient matching in cadaveric transplantation. Tissue Antigens, v. 39, p. 225- 235, 1992.

8. Referências
70
OUNAP K, REIMAND T, MAGI ML, BARTSCH O. Two sisters with Silver-Russell phenotype. Am J Med Genet, v. 131(3), p. 301-306, 2004.
PARTINGTON MW. X-linked short stature with skin pigmentation: evidence for heterogeneity of the Silver-Russell syndrome. Clin Genet, v. 29, p. 151-156, 1986.
PAULSEN, M., FERGUNSON-SMITH, A.C. Dna methylation in genomic imprinting, development, and disease. J Pathol, v. 195, p. 97-110, 2001.
PERKINS RM, HOANG-XUAN MT. The Russell-Silver syndrome: a case report and brief review of the literature. Pediatr Dermatol, v. 19 (6), p. 546-549, 2002.
PRICE SM, STANHOPE R, GARRETT C, PREECE MA, TREMBATH RC. The spectrum of Silver-Russell syndrome: a clinical criteria and molecular genetic study and new diagnostic. J. Med. Genet, v. 36, p. 837-842, 1999.
RAND, E., BEN-PORATH, I., KESHET, I., CEDAR, H. CTCF elements direct allelespecific undermethylation at the imprinted H19 locus. Curr Biol, v. 14, p. 1007-1012, 2004.
RIESEWIJK AM, BLAGITKO N, SCHINZEL AA, HU L, SCHULZ U, HAMEL BC, ROPERS HH, KALSCHEUER VM. Evidence against a major role of PEG1/MEST in Silver-Russell syndrome. Eur J Hum Genet. v. 6 (2), p. 114- 120, 1998.
RIESEWIJK AM, HU L, SCHULZ U, TARIVERDIAN G, HÖGLUND P, KERE J, ROPERS HH, KALSCHEUER VM.: Monoallelic expression of human PEG1/MEST is paralleled by parent- specific methylation in fetuses. Genomics, v. 42, p. 236–244, 1997.
ROSSI, N.F, UEDA KH, RICHIERI-COSTA A, GIACHETI CM. Rev CEFAC, v.8 (4), p. 548-556, 2006.
ROSSIGNOL S. Silver-Russell syndrome and its genetic origins. J Endocrinol Invest, v. 29 (1), p. 9-10, 2006.
RUSSELL A. A syndrome of ‘intrauterine dwarfism’ recognized at birth with cranio-facial dysostosis, disproportionally short arms, and other anomalies. Proc R Soc Med, v. 47, p. 1040-1044, 1954.
SCHINZEL AA, ROBINSON WP, BINKERT F, FANCONI A. An interstitial deletion of proximal 8q (q11-q13) in a girl with Silver-Russell syndrome- like features. Clin Dysmorphol, v. 3, p. 63-69, 1994.
SCHÖHERR N, JAGER S, RANKE MB, WOLLMANN HA, BINDER G, EGGERMANN T. No evidence for isolated imprinting mutations in the

8. Referências
71
PEG1/MEST locus in Silver-Russell patients. Eur J Med Genet, v. 51, p. 322-324, 2008.
SCHÖNHERR N, MEYER E, EGGERMANN K, RANKE MB, WOLLMANN HA, EGGERMANN T. (Epi)mutations in 11p15 significantly contribute to Silver– Russell syndrome: but are they generally involved in growth retardation? Eur J Med Genet, v. 49, p. 414–418, 2006.
SCHÖNHERR N, MEYER E, ROOS A, SCHMIDT A, WOLLMANN HA, EGGERMANN T. The centromeric 11p15 imprinting centre is also involved in Silver–Russell syndrome. J Med Genet, v. 44, p. 59-63, 2007.
SHIURA, H., MIYOSHI, N., KONISHI, A., WAKISAKA-SAITO, N., SUZUKI, R., MUGURUMA, K., KOHDA, T., WAKANA, S., YOKOYAMA, M., ISHINO, F., AND KANEKO-ISHINO, T. Meg1/Grb10 overexpression causes postnatal growth retardation and insulin resistance via negative modulation of the IGF1R and IR cascades. Biochem. Biophys. Res. Commun. v. 329, p. 909–916, 2005.
SILVER H, KIYASU W, GEIRGE J, DEAMER W. Syndrome of congential hemihypertrophy, short stature, and elevated urinary gonadotrophins. Pediatr, v. 12, p. 368–376, 1953.
TULINIUS H, TRYGGVASON K, HAUKSDOTTIR H. 45, X-46, XY chromosome mosaic with features of the Russell-Silver syndrome: a case report with a review of the literature. Dev Med Child Neurol, v. 14 (2), p. 161-172, 1972.
VERONA RI, MANN MR, BARTOLOMEI MS. Genomic imprinting: intricacies of epigenetic regulation in clusters. Ann Rev Cell Dev Biol, v. 19, p. 237– 259, 2003.
WAKELING EL, ABU-AMERO SN, STANIER P, PREECE MA, MOORE GE. Human EGFR, a candidate gene for the Silver-Russell syndrome, is biallelically expressed in a wide range of fetal tissues. Eur J Hum Genet, v. 6, p. 158-164, 1998.
WEE, SA. Russell-Silver syndrome. Dermatol Online J, v. 13 (1), p. 16, 2007.
WEKSBERG R, SHUMAN C, SMITH AC. Beckwith–Wiedemann Syndrome. Am J Med Genet, v. 137(c), p. 12–23, 2005.
WOLFFE AP, MATZKE MA. Epigenetics: regulation through repression. Science, v. 286 (5439), p. 481-486, 1999.
YAMAMOTO Y, ISHINO F, KANEKO-ISHINO T, SHIURA H, UCHIO-YAMADA K, MATSUDA J, SUZUKI O, SATO K. Type 2 diabetes mellitus in a non- obese mouse model induced by Meg1/Grb10 overexpression. Exp Anim, v. 57 (4), p. 385-395, 2008.

8. Referências
72
YAMAZAWA K, KAGAMI M, NAGAI T, KONDOH T, ONIGATA K, MAEYAMA K, HASEGAWA T, HASEGAWA Y, YAMAZAKI T, MIZUNO S, MIYOSHI Y, MIYAGAWA S, HORIKAWA R, MATSUOKA K, OGATA T. Molecular and clinical findings and their correlations in Silver-Russell syndrome: implications for a positive role of IGF2 in growth determination and differential imprinting regulation of the IGF2–H19 domain in bodies and placentas. J Mol Med, v. 86, p. 1171–1181, 2008.
YOSHIHASHI H,MAEYAMA K, KOSAKI R, OGATA T, TSUKAHARA M, GOTO Y, HATA J, MATSUO N, SMITH R, KOSAKI K. Imprinting of human GRB10 and its mutations in two patients with Russell-Silver syndrome. Am J Hum Genet, v. 67, p. 476-483, 2000.
YOSHIMIZU T, MIROGLIO A, RIPOCHE MA, GABORY A, VERNUCCI M, RICCIO A, COLNOT S, GODARD C, TERRIS B, JAMMES H, DANDOLO L. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A, v. 105 (34), p. 12417-12422, 2008.
ZESCHNIGK M, ALBRECHT B, BUITING K, KANBER D, EGGERMANN T, BINDER G, GROMOLL J, PROTT EC, SELAND S, HORSTHEMKE B. IGF2/H19 hypomethylation in Silver–Russell syndrome and isolated hemihypoplasia. Eur J Hum Genet, v. 16, p. 328–334, 2008.
(Versão parcial, a original sem cortes será disponibilizada após publicação).
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo