kelen cristina ribeiro malmegrim de fariaslivros01.livrosgratis.com.br/cp071827.pdf · doenças...
TRANSCRIPT

KELEN CRISTINA RIBEIRO MALMEGRIM DE FARIAS
Avaliação da reconstituição imunológica em paciente s com
diabete melito do tipo 1 e esclerose múltipla após
transplante autólogo de células tronco hematopoétic as
Ribeirão Preto
2006

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

KELEN CRISTINA RIBEIRO MALMEGRIM DE FARIAS
Avaliação da reconstituição imunológica em paciente s com
diabete melito do tipo 1 e esclerose múltipla após
transplante autólogo de células tronco hematopoétic as
Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Imunologia Básica e Aplicada Orientador: Prof. Dr. Júlio César Voltarelli
Ribeirão Preto
2006

AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER
MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A
FONTE.
FICHA CATALOGRÁFICA
Farias, Kelen Cristina Ribeiro Malmegrim de
Avaliação da reconstituição imunológica em pacientes com diabete melito do tipo 1 e esclerose múltipla após transplante autólogo de células tronco hematopoéticas. Ribeirão Preto, 2006. 286 p.
Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de concentração: Imunologia Básica e Aplicada. Orientador: Voltarelli, Júlio César
1. Transplante de células tronco hematopoéticas 2. Doenças auto-imunes 3. Reconstituição imunológica 4. Diabete melito do tipo 1 5. Esclerose múltipla 6. Repertório de células T

FOLHA DE APROVAÇÃO
Kelen Cristina Ribeiro Malmegrim de Farias
Avaliação da reconstituição imunológica em pacientes com diabete melito do tipo 1 e esclerose
múltipla após transplante autólogo de células tronco hematopoéticas
Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Imunologia Básica e Aplicada
Aprovado em:
Banca Examinadora
Prof. Dr. __________________________________________________________________________
Instituição: _______________________________ Assinatura:________________________________
Prof. Dr. __________________________________________________________________________
Instituição: _______________________________ Assinatura:________________________________
Prof. Dr. __________________________________________________________________________
Instituição: _______________________________ Assinatura:________________________________
Prof. Dr. __________________________________________________________________________
Instituição: _______________________________ Assinatura:________________________________
Prof. Dr. __________________________________________________________________________
Instituição: _______________________________ Assinatura:________________________________

Aos meus pais, Ronaldo e Mariléa
Ao meu marido, Cléver

Aos pacientes transplantados,
Doutores na arte da vida,
que incansáveis, tornam o desejo de luta maior que o próprio medo
que sedentos de vida, assumem por ela todos os riscos,
o risco mesmo de perdê-la.
Com vocês, estamos sempre aprendendo
que a luta, nem sempre seguida de vitória,
é o que importa e torna preciosa nossa efêmera existência.
Aos que vitoriosos alcançaram o objetivo de viver,
Aos que persistentes continuam lutando,
Aos que perderam a batalha, mas engrandeceram seu espírito
e deixaram em nossa memória a lembrança de sua coragem.
(Autor desconhecido)

AGRADECIMENTOS
Ao Prof. Dr. Júlio César Voltarelli pela orientação, confiança e oportunidade de trabalhar em sua linha
de pesquisa. Admiro muito a maneira como conduz sua pesquisa clínica e sua equipe do TMO.
Aproveito a oportunidade para parabenizar-lhe pelos avanços e sucessos conquistados nos últimos
quatro anos nos ensaios clínicos de transplante autólogo de células tronco em doenças auto-imunes.
À diretoria científica e administrativa do Centro Regional de Hemoterapia de Ribeirão Preto do HC-
FMRP-USP, em cujos laboratórios de pesquisa esse trabalho foi desenvolvido.
Ao coordenador do Centro de Terapia Celular (CEPID da FAPESP), Prof. Dr. Marco Antônio Zago,
pela oportunidade de trabalhar nesse centro de referência em pesquisa, o qual tem obtido muito
sucesso pela sua capacidade de liderança e empenho na formação dos pesquisadores.
Ao Programa de Pós-graduação em Imunologia Básica e Aplicada, do Departamento de Imunologia e
Bioquímica da FMRP-USP, pelos excelentes professores e pela qualidade de formação que é
oferecida. Agradeço em especial à Ana, que sempre me ajudou com muita atenção, eficiência e
carinho em todos os momentos em que precisei.
À FAPESP, Finep e CNPq pelo apoio financeiro, imprescindível para a realização deste trabalho.
Ao Laboratório de Citometria de Fluxo do Centro Regional de Hemoterapia de Ribeirão Preto do HC-
FMRP-USP. À Patrícia e Fabiana pela dedicação, presteza e ajuda, que foram imprescindíveis para a
realização deste trabalho. Agradeço pelas centenas de tubinhos marcados, “passados” e analisados,
e também pelos conselhos e discussões sobre os experimentos. Mas, principalmente, agradeço pela
amizade sincera, carinho, conversas, pelos ótimos momentos que passamos e pelas inúmeras
risadas que demos juntas nesses últimos quatro anos!
Ao Laboratório de Biologia Celular do Centro Regional de Hemoterapia de Ribeirão Preto do HC-
FMRP-USP. À Maristela, Ane Rose e Karina pela ajuda na realização deste trabalho, pela discussão
dos experimentos, e principalmente, pela amizade, apoio e carinho nesses últimos anos. Agradeço
especialmente à Aline, minha estagiária, pela amizade, carinho, dedicação e ajuda imensurável nos
experimentos nesse último um ano e meio.

Ao Laboratório de Biologia Molecular do Centro Regional de Hemoterapia de Ribeirão Preto do HC-
FMRP-USP. À Dra. Simone e Luciene pela ajuda na realização dos experimentos. Agradeço também
à Carmen pela amizade e por sempre me ajudar com muita atenção e eficiência na parte burocrática
nos momentos em que precisei.
À equipe médica da Unidade de Transplante de medula Óssea do Hospital das Clínicas da FMRP-
USP, em especial à Dra. Maria Carolina de Oliveira, Dra. Beatriz Stracieri, Dra. Daniela Moraes, Dra.
Belinda Simões e ao Dr. Fabiano Pieroni, pela atenção e presteza no acompanhamento dos
pacientes, encaminhamento das amostras e no fornecimento dos dados clínicos.
À equipe de enfermagem da Unidade de Transplante de medula Óssea do Hospital das Clínicas da
FMRP-USP pela atenção, dedicação, paciência e presteza na coleta das amostras dos pacientes.
À equipe médica do Setor de Doenças Neuromusculares, em especial à Dra. Doralina Brum e ao Dr.
Amilton Barreira, pela colaboração nesse projeto, atenção e presteza no acompanhamento dos
pacientes com esclerose múltipla.
À equipe médica da Divisão de Endocrinologia e Metabolismo do HC-FMRP-USP, em especial ao Dr.
Eduardo Couri e ao Dr. Milton César Foss, pela colaboração nesse projeto, atenção e presteza no
acompanhamento dos pacientes com diabete melito do tipo 1.
Ao Prof. Dr. Jorge Kalil, Prof. Dra. Luiza Guilherme, Prof. Dra. Verônica Coelho e Dra. Kellen Faé, do
Laboratório de Imunologia do Instituto do Coração da FM-USP, pela oportunidade que me deram de
poder desenvolver parte de meu trabalho de doutorado em um grupo de excelência em pesquisa em
Imunologia no Brasil, iniciando-me no estudo do repertório de linfócitos T. Agradeço em especial à
Dra. Kellen pela colaboração nesse trabalho, pelos ensinamentos e explicações, pela ajuda na
realização dos experimentos e na discussão dos resultados. Agradeço pelo carinho e cuidado no
período em que trabalhei em seu laboratório. Agradeço também a todos os outros companheiros do
laboratório que me receberam com carinho, e de alguma forma contribuíram para realização desta
parte do meu trabalho.
Aos pacientes que participaram deste estudo pela compreensão e paciência. Espero ter contribuído,
um pouco que seja, para o entendimento dos mecanismos de ação do transplante autólogo de células

tronco em doenças auto-imunes, para que no futuro essa nova abordagem terapêutica para o
tratamento de doenças auto-imunes possa ser melhorada.
A todos meus companheiros dos laboratórios de pesquisa do Hemocentro que de diferentes formas
contribuíram para este trabalho. Agradeço pela amizade, força e ajuda nos experimentos. Em
especial, agradeço à minha amiga Keikinho pela amizade e carinho, por me escutar tantas vezes, por
compartilhar sonhos e esperanças, e pela ajuda incondicional.
Aos meus “amigos da imuno” (vocês sabem quem são...) pela amizade e carinho sinceros, pela força,
conselhos e apoio nos momentos em que precisei, pelos bons momentos compartilhados em nossas
saídas e encontros, e também pelas nossas conversas sobre nossa paixão que é a Imunologia. Foi
muito bom conviver com vocês nesses últimos quatro anos!
Aos meus queridos pais, Ronaldo e Mariléa, pelo amor e apoio incondicionais que sempre recebi para
ir em busca de meus sonhos. Obrigada por compreenderem minha ausência nesses últimos tempos.
Saibam que cada conquista minha é um triunfo de vocês.
Ao meu querido Cléver, meu amor, minha referência... Agradeço a Deus, sempre, por ter colocado
você em minha vida. Obrigada pela compreensão, carinho, apoio e ajuda incondicionais,
principalmente nesses últimos tempos. Essa conquista também é sua, assim como a sua (lembra?)
também foi minha, e tenho certeza de que outros tantos sonhos nós conquistaremos juntos.
A Deus pela minha existência, pelo amor incondicional, pela saúde, força e capacidade que me dá
todos os dias.

“The marvelous richness of human experience would lose
something of rewarding joy if there were no limitations to
overcome. The hilltop hour would not be half so wonderful if
there were no dark valleys to traverse”.
Helen Keller (1880 – 1968)

RESUMO
Farias, K.C.R.M. Avaliação da reconstituição imunológica em pacient es com diabete melito do
tipo 1 e esclerose múltipla após transplante autólo go de células tronco hematopoéticas. 2006.
286p. Tese (Doutorado) – Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo,
Ribeirão Preto, 2006.
Ensaios clínicos têm demonstrado que a imunoablação seguida de transplante autólogo de
células tronco hematopoéticas (TACTH) é capaz de suprimir a atividade inflamatória em pacientes
com doenças auto-imunes (DAIs) e pode induzir remissões clínicas prolongadas nesses pacientes,
mas o mecanismo de ação do TACTH ainda não é bem esclarecido. O racional do TACTH em DAIs
baseia-se na idéia de que a imunoablação intensa possa eliminar as células auto-reativas e que o
novo sistema imune reconstituído dos precursores hematopoéticos possa restabelecer tolerância. O
objetivo deste trabalho foi avaliar a reconstituição imunológica em pacientes com diabete melito tipo 1
(DM, N=11) e pacientes com esclerose múltipla (EM, N=18), seqüencialmente após o TACTH. A
reconstituição imunológica observada nos pacientes com DM (um ano de seguimento pós-
transplante) e nos pacientes com EM (dois anos de seguimento pós-transplante), foi caracterizada por
mecanismos periféricos timo-independentes. Após o transplante, houve uma predominância de
células T de memória central, memória efetora e também de células T efetoras diferenciadas,
principalmente de linfócitos T CD8+. Essas células provavelmente se originam da expansão
homeostática periférica de linfócitos T de memória residuais que sobreviveram ao regime de
condicionamento ou foram re-infundidos com as células tronco no momento do transplante. Os
números de linfócitos T CD4+ e CD8+ naive, incluindo as células T CD4+CD45RA+CD31+ recém-
imigrantes do timo, não recuperaram os níveis basais durante o período pós-transplante analisado.
Após o TACTH, houve uma predominância de células T CD4+ e CD8+ produtoras de citocinas do
padrão TH1 (INF-γ e TNF-α). Por outro lado, foi observado um aumento da porcentagem de células T
CD4+ e CD8+ produtoras de citocinas do padrão TH2 (IL-4, IL-5 e IL-10) no pré-condicionamento e em
alguns períodos após o TACTH. Análises espectrais do repertório da cadeia Vβ dos receptores de
células T (TCRs), por TCRBV CDR3 spectratyping, identificaram quatro padrões básicos de
reconstituição do repertório. O padrão que consistiu na reconstituição da diversidade a partir de um

repertório pré-transplante diverso, foi o mais dominante em todos os pacientes analisados. Para
algumas famílias Vβ foi observado um padrão de reconstituição que consistiu na recuperação da
diversidade a partir de um repertório pré-transplante restrito, o que sugere um aumento da
diversidade do repertório de células T após o transplante. Foram observadas mudanças na
composição do repertório de células T após o TACTH, evidenciadas por alterações qualitativas e
quantitativas dos picos de CDR3 das famílias Vβ, que poderiam explicar a indução da remissão da
doença auto-imune observada na maioria dos pacientes. Foi observada uma rápida reconstituição de
células T CD4+CD25high e um aumento na expressão do gene Foxp3, marcador molecular específico
para células T reguladoras CD4+CD25high, na maioria dos pacientes avaliados. Esses resultados
sugerem uma melhora de mecanismos reguladores que podem contribuir para o restabelecimento da
tolerância imunológica nos pacientes com DM e EM submetidos ao TACTH.
Palavras-chave: Transplante de células tronco hematopoéticas, Doenças auto-imunes,
Reconstituição imunológica, Diabete melito do tipo 1, Esclerose múltipla, Repertório de células T.

ABSTRACT
Farias, K.C.R.M. Analysis of immune reconstitution in type 1 diabete s and multiple sclerosis
patients following hematopoeitic stem cell transpla ntation. 2006. 286p. Thesis (Doctoral) –
Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, 2006.
Clinical trials have indicated that autologous hematopoietic stem cell transplantation (AHSCT) can
persistently suppress inflammatory disease activity in a subset of patients with autoimmune diseases
(AIDs), but the mechanism of action of the AHSCT has not yet been totally elucidated. The rationale
for HSCT in autoimmune diseases has been the notion that intensive immune depletion could
eliminate autoreactive immune cells irrespective of antigenic specificity and that regenerating the
immune system from hematopoietic precursors could reestablish tolerance. The goal of this work was
to evaluate the immune reconstitution in patients with type 1 diabetes mellitus (DM1; N=11) and
multiple sclerosis (MS; N=18) who received AHSCT. The immune reconstitution observed in the DM1
patients (one year follow-up) and in the MS patients (two years follow-up) was characterized by
peripheral thymic-independent mechanisms. After transplantation, there was a predominance of
central-memory T cells, effector-memory T cells and differentiated-effector T cells, mainly of the CD8+
T cell subset. These cells probably originate from peripheral homeostatic proliferation of residual
memory T cells that have survived the conditioning chemotherapy or were reinfused with the HSC
graft. The numbers of naive CD4+ and CD8+ T cells, including the recent-thymic emigrants
CD4+CD45RA+CD31+, did not revert to baseline levels during follow-up. After transplant, there was a
predominance of TH1 cells, mainly CD8+ T cells, producing INF-γ e TNF-α. In contrast, it was observed
an increased percentage of CD4+ and CD8+ T cells producing TH2 cytokines (IL4, IL-5 and IL-10) at
pre-conditioning and at some time points after AHSCT. Analysis of the T cell receptor Vβ repertoire by
TCRBV CDR3 spectratyping identified four basic patterns of repertoire reconstitution. The pattern that
consisted of reconstitution of diversity from a normally diverse repertoire was the most dominant in the
analyzed patients. For some Vβ families were observed a pattern that consisted of recovery of
diversity from a restricted repertoire, suggesting increased repertoire diversity after AHSCT. There
were changes in the composition of the T cell repertoire post-AHSCT, evidenced by qualitative and
quantitative alterations in the CDR3 peaks, which might explain the induction of the remission of the

autoimmune disease, observed for the majority of the patients. A rapid reconstitution of CD4+CD25high
T cells and an increased expression of the Foxp3 gene, a specific molecular marker for regulatory
CD4+CD25high T cells, were observed in the majority of the analyzed patients. These results suggest
an improvement of the regulatory mechanisms, which might contribute to reestablishment of
immunological tolerance in the DM1 and MS patients submitted to the AHSCT.
Keywords: Hematopoeitic stem cell transplantation, Auto-immune diseases, Immune reconstitution,
Type 1 diabetes, Multiple Sclerosis, T cell repertoire.

LISTA DE ILUSTRAÇÕES
Figura 1. Reconstituição de leucócitos totais, linfócitos, monócitos e granulócitos em pacientes com
diabete melito do tipo 1 após o TACTH......................................................................................72
Figura 2. Reconstituição de linfócitos T e B em pacientes com diabete melito tipo 1 após o TACTH..73
Figura 3. Reconstituição de linfócitos T, células NK, células NKT-like, e de células dendríticas em
pacientes com diabete melito tipo 1 após o TACTH....................................................................74
Figura 4. Reconstituição de linfócitos T CD4 naive, de memória, de memória efetora e efetores
diferenciados em pacientes com diabete melito tipo 1 após o TACTH ........................................75
Figura 5. Reconstituição de linfócitos T CD8 naive, de memória, de memória efetora e efetores
diferenciados em pacientes com diabete melito tipo 1 após o TACTH ........................................76
Figura 6. Expressão de Fas e FasL em linfócitos T CD4+ ou TCD8+, ou em linfócitos B reconstituídos
em pacientes com diabete melito tipo 1 após o TACTH..............................................................77
Figura 7. Expressão de marcadores de ativação celular em linfócitos T CD4+, T CD8+ e linfócitos B
reconstituídos, e reconstituição de linfócitos T CD4+ recém-imigrantes do timo, em pacientes com
diabete melito tipo 1 após o TACTH...........................................................................................78
Figura 8. Reconstituição de linfócitos T CD4+CD25+ e linfócitos T CD4+CD25high em pacientes com
diabete melito tipo 1 após o TACTH...........................................................................................79
Figura 9. Expressão de CTLA-4 e GITR em linfócitos T CD4+CD25high em pacientes com diabete
melito tipo 1 antes e após o TACTH...........................................................................................80
Figura 10. Reconstituição de leucócitos totais, linfócitos, monócitos e granulócitos em pacientes com
esclerose múltipla após o TACTH ..............................................................................................88
Figura 11. Reconstituição de linfócitos T e B em pacientes com esclerose múltipla após o TACTH...89
Figura 12. Reconstituição de linfócitos T, células NK, células NKT-like, e de células dendríticas em
pacientes com esclerose múltipla após o TACTH.......................................................................90
Figura 13. Reconstituição de linfócitos T CD4 naive, de memória, de memória efetora e efetores
diferenciados em pacientes com esclerose múltipla após o TACTH............................................91
Figura 14. Reconstituição de linfócitos T CD8 naive, de memória, de memória efetora e efetores
diferenciados em pacientes com esclerose múltipla após o TACTH............................................92
Figura 15. Expressão de Fas e FasL em linfócitos T CD4+ ou TCD8+, ou em linfócitos B reconstituídos
em pacientes com esclerose múltipla após o TACTH.................................................................93

Figura 16. Expressão de marcadores de ativação celular em linfócitos T CD4+, T CD8+ e linfócitos B
reconstituídos, e reconstituição de linfócitos T CD4+ recém-imigrantes do timo, em pacientes com
esclerose múltipla após o TACTH ..............................................................................................94
Figura 17. Reconstituição de linfócitos T CD4+CD25+ e linfócitos T CD4+CD25high em pacientes com
esclerose múltipla após o TACTH ..............................................................................................95
Figura 18. Expressão de CTLA-4 e GITR em linfócitos T CD4+CD25high em pacientes com esclerose
múltipla antes e após o TACTH..................................................................................................96
Figura 19. Porcentagem de células T CD4+ e T CD8+ produtoras de citocinas intracelulares do padrão
TH1 em pacientes com diabete melito tipo 1 pré- e pós-TACTH................................................100
Figura 20. Porcentagem de células T CD4+ e T CD8+ produtoras de citocinas intracelulares do padrão
TH2 em pacientes com diabete melito tipo 1 pré- e pós-TACTH................................................101
Figura 21. Porcentagem de células T CD4+ e T CD8+ produtoras de citocinas intracelulares do padrão
TH1 em pacientes com esclerose múltipla pré- e pós-TACTH...................................................104
Figura 22. Porcentagem de células T CD4+ e T CD8+ produtoras de citocinas intracelulares do padrão
TH2 em pacientes com esclerose múltipla pré- e pós-TACTH...................................................105
Figura 23. Padrões de reconstituição do repertório da cadeia Vβ do TCR pós-TACTH nos pacientes
com diabete melito do tipo 1.....................................................................................................114
Figura 24. Mudanças qualitativas na composição do repertório da cadeia Vβ do TCR após o TACTH
nos pacientes com diabete melito do tipo 1. .............................................................................116
Figura 25. Padrões de reconstituição do repertório da cadeia Vβ do TCR pós-TACTH nos pacientes
com esclerose múltipla.............................................................................................................127
Figura 26. Mudanças na composição do repertório da cadeia Vβ do TCR após TACTH nos pacientes
com esclerose múltipla.............................................................................................................129
Figura 27. Expressão gênica de Foxp3 em células mononucleares do sangue periférico de pacientes
com diabete melito do tipo após o TACTH................................................................................133
Figura 28. Expressão gênica de Foxp3 em células mononucleares do sangue periférico de pacientes
com esclerose múltipla após o TACTH.....................................................................................134
Figura A.1. Análise de subpopulações linfócitárias do sangue periférico..........................................187
Figura A.2. Análise de subpopulações de células T do sangue periférico ........................................188
Figura A.3. Análise de subpopulações de células T do sangue periférico ........................................189

Figura A.4. Análise de células T naive, efetoras e de memória, e de células dendríticas do sangue
periférico..................................................................................................................................190
Figura A.5. Análise de populações de células T reguladoras CD4+CD25+ do sangue periférico......191
Figura A.6. Análise de subpopulações linfócitárias do sangue periférico..........................................192
Figura B.1. Produção de citocinas intracelulares do padrão TH1 e TH2 por subpolulações linfócitárias
................................................................................................................................................193
Figura B.2. Produção de citocinas intracelulares do padrão TH1 e TH2 por subpolulações linfócitárias
................................................................................................................................................194
Figura B.3. Análise da expressão de CD69 por células T CD3+ estimuladas com PMA e Inonomicina
................................................................................................................................................195
Figura C.1. Método de TCRBV CDR3 Spectratyping.......................................................................196
Figura C.2. Imagem de um gel de seqüenciamento.........................................................................197
Figura H.1. Reação de Real time PCR para genes GAPDH (A) e Foxp3 (B)....................................212
Figura I.1. Repertório da cadeia Vβ do RCT no paciente com diabete melito do tipo 1 LSM (DM1), pré
e pós-TACTH...........................................................................................................................214
Figura J.1. Repertório da cadeia Vβ do RCT no paciente com diabete melito do tipo 1 ALSR (DM2),
pré e pós-TACTH.....................................................................................................................219
Figura K.1. Repertório da cadeia Vβ do RCT no paciente com diabete melito do tipo 1 WSL (DM3),
pré e pós-TACTH.....................................................................................................................224
Figura L.1. Repertório da cadeia Vβ do RCT no paciente com diabete melito do tipo 1 MGB (DM4),
pré e pós-TACTH.....................................................................................................................229
Figura M.1. Repertório da cadeia Vβ do RCT no paciente com diabete melito do tipo 1 RFLS (DM5),
pré e pós-TACTH. ..................................................................................................................233
Figura N.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla GG (EM1), pré e
pós-TACTH..............................................................................................................................238
Figura O.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla SSS (EM3), pré e
pós-TACTH..............................................................................................................................243
Figura P.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla DFG (EM4), pré e
pós-TACTH..............................................................................................................................248

Figura Q.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla ARJTA (EM5), pré e
pós-TACTH..............................................................................................................................252
Figura R.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla SHGE (EM6), pré e
pós-TACTH..............................................................................................................................257
Figura S.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla MFM (EM8), pré e
pós-TACTH..............................................................................................................................261
Figura T.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla GG (EM1), pré e
pós-TACTH..............................................................................................................................266
Figura U.1. Repertório da cadeia Vβ do RCT no paciente com esclerose múltipla OLP (EM10), pré e
pós-TACTH..............................................................................................................................270
Figura V.1. Repertório da cadeia Vβ do RCT nos indivíduos-controle saudáveis DN12 e DN20.......274
Figura W.1. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 ALSR (DM2), pré- e pós-TACTH .........................................................278
Figura W.2. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 LSM (DM1) (A) e WLS (DM3) (B) pré- e pós-TACTH...........................279
Figura W.3. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 MGB (DM4) (A) e RLFS (DM5) (B) pré- e pós-TACTH.........................280
Figura X.1. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
esclerose múltipla GG (DM1) (A) e SSS (EM3) (B) pré- e pós-TACTH .....................................281
Figura X.2. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
esclerose múltipla DFG (EM4) (A) e ARJTA (EM5) (B) pré- e pós-TACTH...............................282
Figura X.3. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico do paciente com
esclerose múltipla SHGE (EM6) (A) e MFM (EM8) (B) pré- e pós-TACTH ...............................283
Figura Y.1. Freqüência das famílias Vβ do RCT em linfócitos T do sangue periférico dos indivíduos-
controle saudáveis DN12 e DN20 ............................................................................................285

LISTA DE TABELAS
Tabela 1. Pacientes com esclerose múltipla submetidos ao TACTH..................................................38
Tabela 2. Pacientes com diabete melito do tipo 1 submetidos ao TACTH..........................................39
Tabela 3. Seqüência dos primers utilizados na amplificação das famílias Vβ do TCR........................48
Tabela 4. Tamanho esperado do produto de PCR da amplificação VB-CB (em pares de bases) para o
segmento CDR3 de 10 aminoácidos ..........................................................................................50
Tabela 5. Avaliação clínica pós-TACTH nos pacientes com esclerose múltipla..................................60
Tabela 6. Avaliação clínica pós-TACTH nos pacientes com diabete melito do tipo 1..........................61
Tabela 7. Análise da diversidade do repertório Vβ do TCRs dos pacientes com diabete melito do tipo
1 pré e seqüencialmente após o TACTH. .................................................................................111
Tabela 8. Freqüência das famílias Vβs do TCR com expansões relevantes em linfócitos T do sangue
periférico dos pacientes com diabete melito do tipo 1, pré e seqüencialmente após o TACTH..117
Tabela 9. Análise da diversidade do repertório Vβ dos TCRs dos pacientes com esclerose múltipla
pré e seqüencialmente após o TACTH.....................................................................................121
Tabela 10. Freqüência das famílias Vβs do TCR com expansões relevantes em linfócitos T do sangue
periférico dos pacientes com esclerose múltipla, pré e seqüencialmente após o TACTH. .........131
Tabela D.1. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com diabete melito do tipo 1, pré- e pós-transplante ..............198
Tabela D.2. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com diabete melito do tipo 1, pré- e pós-transplante ..............199
Tabela D.3. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com diabete melito do tipo 1, pré- e pós-transplante ..............200
Tabela D. 4. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e pacientes com diabete melito do tipo 1 pré- e pós-transplante, para cada
subpopulação celular analisada ...............................................................................................201
Tabela D.5. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e pacientes com diabete melito do tipo 1 pré- e pós-transplante, para cada
subpopulação celular analisada ...............................................................................................202

Tabela E.1. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com esclerose múltipla, pré- e pós-transplante ......................203
Tabela E.2. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com esclerose múltipla, pré- e pós-transplante ......................204
Tabela E.3. Valores de média, desvio padrão e mediana das subpopulações celulares analisadas nos
controles saudáveis e pacientes com esclerose múltipla, pré- e pós-transplante ......................205
Tabela E.4. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e de pacientes com esclerose múltipla pré- e pós-transplante, para cada subpopulação
celular analisada......................................................................................................................206
Tabela E.5. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e de pacientes com esclerose múltipla pré- e pós-transplante, para cada subpopulação
celular analisada......................................................................................................................207
Tabela F.1. Valores da média, desvio padrão e mediana das populações de células T CD4+ ou CD8+
produtoras de citocinas nos controles saudáveis e pacientes com diabete melito do tipo 1, pré- e
pós-transplante ........................................................................................................................208
Tabela F.2. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e de pacientes com diabete melito do tipo 1 pré- e pós-transplante, para as
populações de células T CD4+ ou CD8+ produtoras de citocinas .............................................209
Tabela G.1. Valores da média, desvio padrão e mediana das populações de células T CD4+ ou CD8+
produtoras de citocinas nos controles saudáveis e pacientes com esclerose múltipla, pré- e pós-
transplante...............................................................................................................................210
Tabela G.2. Valores de p encontrados nas análises estatísticas entre os grupos de controles
saudáveis e de pacientes com esclerose múltipla pré- e pós-transplante, para as populações de
células T CD4+ ou CD8+ produtoras de citocinas.....................................................................211
Tabela I.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 LSM (DM1), pré- e pós-TACTH...........................................................215
Tabela J.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 ALSR (DM2), pré- e pós-TACTH .........................................................220

Tabela K.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
diabete melito WSL (DM3), pré- e pós-TACTH.........................................................................225
Tabela L.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 MGB (DM4), pré- e pós-TACTH ..........................................................230
Tabela M.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
diabete melito do tipo 1 RLFS (DM5), pré- e pós-TACTH .........................................................234
Tabela N.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla GG (EM1), pré- e pós-TACTH.....................................................................239
Tabela O.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla SSS (EM3), pré- e pós-TACTH....................................................................244
Tabela P.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla DFG (EM4), pré- e pós-TACTH ...................................................................249
Tabela Q.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com com
esclerose múltipla ARJTA (EM5), pré- e pós-TACTH................................................................252
Tabela R.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla SHGE (EM6), pré- e pós-TACTH.................................................................258
Tabela S.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla MFM (EM8), pré- e pós-TACTH...................................................................262

Tabela T.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla WRS (EM9), pré- e pós-TACTH ..................................................................267
Tabela U.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico do paciente com
esclerose múltipla OLP (EM10), pré- e pós-TACTH..................................................................271
Tabela V.1. Freqüência individual (%) dos segmentos da região CDR3 de cada família Vβ do RCT e
freqüência total (%) de cada família Vβ em linfócitos T do sangue periférico dos indivíduos-
controle saudáveis DN12 e DN20 ............................................................................................275

LISTA DE ABREVIATURAS E SIGLAS
� aa - aminoácido
� APC - do inglês “antigen presenting cell” ou célula apresentadora de antígeno
� ATG - do inglês “anti-thymocyte-globulin” ou globulina anti-linfocitária
� BEAM - BCNU, etoposídeo, aracitin, melfalan
� CD - do inglês "cluster of differentiation", ou grupamento de diferenciação
� CDR - do inglês “complementary determinant region”, ou região determinante de
complementariedade
� CTLA-4 - do inglês “citotoxic T lymphocyte-associated molecule-4”, ou molécula 4 associada ao
linfócito T citotóxico
� DAIs - Doenças auto-imunes
� DEPC - dietilpirocarbonato
� DM - Diabete melito do tipo 1
� DMSO - dimetilsulfóxido
� DNA - do inglês “desoxirobonucleic acid” ou ácido desoxirribonucléico
� dNTP - desoxi-nucleotídeos trifosfato
� EAE - do inglês “experimental autoimmune encephalomyelitis”, ou encefalite auto-imune
experimental
� EDSS - Expanded Disability Status Score
� EDTA - sal di-sódico do ácido etilenodiaminotetracético
� EM - Esclerose Múltipla
� FACS - do inglês "Fluorescence-Activated Cell Sorter"
� FITC - do inglês "fluorescein isothiocyanate" ou isotiocianato de fluoresceína
� G-CSF - do inglês “granulocyte-colony stimulating factor” ou fator estimulador de colônias de
granulócitos
� GM-CSF - do inglês “granulocyte macrophage-colony stimulating factor” ou fator estimulador de
colônias de granulócitos e macrófagos
� HLA - do inglês “Human Leukocyte Antigens” ou antígenos leucocitários humanos
� IFN-γ - do inglês “gamma interferon” ou interferon gama
� IL - interleucina
� LES - Lúpus eritematoso sistêmico
� MBP - do inglês “myelin basic protein” ou proteína básica de mielina
� MHC - do inglês “Major Histocompatibility Complex” ou Complexo Principal de
Histocompatibilidade
� PBMCs - do inglês “peripheral blood mononuclear cells”, ou células mononucleares de sangue
periférico
� PBS - do inglês “phosphate buffer saline”, ou tampão salina fosfato
� PE - "PhycoErythrin" ou ficoeritrina

� PCR - do inglês "polymerase chain reaction" ou reação de polimerização em cadeia
� Pré-cond - pré-condicionamento
� Pré-mob - pré-mobilização
� q.s.p - quantidade suficiente para
� TCR - inglês “T cell receptor”, ou receptor de célula T
� RNA - do inglês “ribonucleic acid” ou ácido ribonucléico
� RNAm - ácido ribonucléico mensageiro
� RPMI - meio Roswell Park Memorial Institute
� SBF - soro bovino fetal
� TA - temperatura ambiente
� TACTH - Transplante autólogo de células tronco hematopoéticas
� TCTH - Transplante de células tronco hematopoéticas
� TH - T "helper” - células T auxiliadoras
� TNF-α - do inglês “tumor necrosis factor alpha” ou fator de necrose tumoral alfa
� Treg - célula T reguladora
� Tris - Tris-hidroximetil aminometano básico
� Tx - transplante
� Vα - região variável da cadeia alfa do receptor de célula T
� Vβ - região variável da cadeia beta do receptor de célula T

SUMÁRIO
1 Introdução....................................................................................................................................1
1.1 Auto-imunidade e doenças auto-imunes.................................................................................2
1.2 Diabete melito do tipo 1..........................................................................................................9
1.3 Esclerose múltipla ................................................................................................................13
1.4 Transplante de células tronco hematopoéticas em doenças auto-imunes .............................17
1.5 Mecanismos de ação do TCTH autólogo em doenças auto-imunes ......................................23
1.6 Repertório do receptor de células T em doenças auto-imunes..............................................28
2 Objetivos ...................................................................................................................................31
2.1 Objetivo geral .......................................................................................................................32
2.2 Objetivos específicos............................................................................................................32
3 Casuística, Material e Métodos ..................................................................................................33
3.1 Delineamento do estudo.......................................................................................................34
3.2 Casuística ............................................................................................................................34
3.3 Controles saudáveis.............................................................................................................37
3.4 Isolamento das células mononucleares do sangue periférico................................................37
3.5 Imunofenotipagem das subpopulações celulares do sangue periférico .................................39
3.6 Detecção de citocinas intracelulares em linfócitos T ativados................................................41
3.7 Extração de RNA pelo método de Trizol ...............................................................................43
3.7.1 Eletroforese de amostras de RNA em gel de agarose sob condições desnaturantes ....44
3.8 Transcrição reversa..............................................................................................................45
3.8.1 Validação da transcrição..............................................................................................45
3.9 Método de TCRBV CDR3 Spectratyping...............................................................................46
3.9.1 Reação de PCR (Vβ-Cβ) para determinação das famílias Vβ do Receptor de Células
(TCR) ....................................................................................................................................46
3.9.2 Reação de elongação Vβ-Cβ (Run-off).........................................................................47
3.9.3 Preparo do gel de seqüenciamento..............................................................................47
3.9.4 Preparo das amostras e aplicação no gel de seqüenciamento......................................48

3.9.5 Perfil da região CDR3 do RTC, cálculo do tamanho da região CDR3 do RTC e da
freqüência das famílias Vβ, pelo método de TCRBV CDR3 Spectratyping ..................................49
3.10 Análise da expressão de Foxp3 por Real Time RT- PCR..................................................51
3.11 Análise estatística ............................................................................................................53
4 Resultados.................................................................................................................................55
4.1 Resultados Clínicos..............................................................................................................56
4.1.1 Pacientes com esclerose múltipla.................................................................................56
4.1.2 Pacientes com diabete melito do tipo 1 ........................................................................57
4.2 Avaliação da reconstituição imunológica após o TACTH.......................................................62
4.2.1 Pacientes com diabete melito do tipo 1 ........................................................................62
4.2.2 Pacientes com esclerose múltipla.................................................................................81
4.3 Avaliação do perfil de citocinas após o TACH.......................................................................97
4.3.1 Pacientes com diabete melito do tipo 1 ........................................................................98
4.3.2 Pacientes com esclerose múltipla...............................................................................102
4.4 Análise da diversidade do repertório de linfócitos T ............................................................106
4.4.1 Pacientes com diabete melito do tipo 1 ......................................................................109
4.4.2 Pacientes com esclerose múltipla...............................................................................119
4.5 Análise da expressão do gene Foxp3 em células mononucleares.......................................132
4.5.1 Pacientes com diabete melito do tipo 1 ......................................................................132
4.5.2 Pacientes com esclerose múltipla...............................................................................133
5 Discussão ................................................................................................................................135
5.1 Avaliação da reconstituição imunológica após o TACTH.....................................................136
5.2 Perfil de citocinas após o TACTH .......................................................................................154
5.3 Diversidade do repertório de células T após o TACTH........................................................157
5.4 Análise da reconstituição de células T reguladoras e da expressão de Fopx3 após o TACTH...
..........................................................................................................................................162
6 Conclusões..............................................................................................................................166
Referências ....................................................................................................................................170
Apêndices e Anexo.........................................................................................................................186

1 Introdução

Introdução 2
1.1 Auto-imunidade e doenças auto-imunes
As doenças auto-imunes (DAIs) constituem um grupo complexo e heterogêneo de doenças
que ocorrem em 3-5% da população geral. As DAIs são caracterizadas pela perda da tolerância
imunológica a antígenos próprios e conseqüente destruição tecidual por células auto-reativas e auto-
anticorpos (revisado por Davidson e Diamond, 2001; Marrack et al., 2001). Nas doenças auto-imunes
órgão-específicas (por exemplo, diabete melito do tipo 1 e esclerose múltipla), as células auto-
reativas e auto-anticorpos são direcionados contra componentes próprios expressos somente num
tecido ou tipo celular específico. Nas doenças auto-imunes sistêmicas (por exemplo, lúpus
eritematoso sistêmico, esclerose sistêmica e artrite reumatóide), as células auto-reativas e auto-
anticorpos são direcionados contra vários auto-antígenos que são usualmente expressos numa
grande variedade de tecidos, estando presentes no núcleo, no citoplasma ou na superfície celular
(revisado por Davidson e Diamond, 2001; Marrack et al., 2001). Critérios para auto-imunidade foram
definidos por Rose e Bona (1993), que postularam que a prova direta da auto-imunidade em
humanos é a capacidade de transferir adotivamente a doença pela transferência de células imunes;
prova indireta é a capacidade de transferir uma doença auto-imune experimental (que mimetiza a
doença auto-imune humana) pela transferência adotiva de células imunes, e a demonstração de que
a doença responde a agentes imunossupressores.
As DAIs constituem uma importante causa de morbidade e mortalidade ao redor do mundo.
Várias dessas doenças são muito difíceis de tratar e impossíveis de curar, pela razão óbvia de que os
alvos da resposta imune, os auto-antígenos, não podem ser eliminados. As DAIs representam um
problema econômico-social relevante, pois afetam freqüentemente adultos jovens, principalmente
mulheres (revisado por Rioux e Abbas, 2005).
Durante vários anos o dogma central da imunologia baseava-se na deleção clonal de células
auto-reativas como único mecanismo de tolerância imunológica para a prevenção da auto-imunidade.
A visão atual sobre tolerância imunológica reconhece que um nível baixo de auto-reatividade é
fisiológico (Dighiero e Rose, 1999) e crucial para o funcionamento normal do sistema imune. Auto-
antígenos ajudam a formar o repertório de linfócitos maduros e a sobrevivência das células T naive e
de células B na periferia requer exposições contínuas a antígenos próprios (Goldrath e Bevan, 1999).
Uma vez que não existem diferenças fundamentais entre as estruturas de antígenos próprios e de
antígenos não-próprios, a idéia é que os linfócitos evoluíram não para distinguir o “próprio” do “não-

Introdução 3
próprio”, com tinha sido proposto anteriormente, mas para responder a antígenos somente em certos
microambientes, geralmente na presença de citocinas e outros fatores inflamatórios. Assim,
atualmente a auto-reatividade não é mais vista mais como patológica, mas sim como fisiológica. Uma
vez que a auto-reatividade é fisiológica, o desafio é entender como isso se torna um processo
patológico e como as células imunes, principalmente células T e B, contribuem para a injúria tecidual
(Davidson e Diamond, 2001).
Portanto, as DAIs se desenvolvem quando linfócitos auto-reativos escapam dos mecanismos
de tolerância imunológica, são ativados e começam a atacar os tecidos próprios. Embora os
mecanismos pelos quais isso ocorre ainda não sejam completamente esclarecidos, acredita-se que o
desenvolvimento de DAIs é resultante de uma interação entre fatores genéticos, fatores ambientais e
eventos estocásticos (revisado por Rioux e Abbas, 2005). As baixas taxas de concordância para as
DAIs entre gêmeos idênticos sugerem uma contribuição fundamental de fatores ambientais para
desenvolvimento da auto-imunidade. Os genes de suscetibilidade descritos são relacionados a
moléculas de HLA, citocinas, moléculas co-estimuladoras, vias de sinalização de apoptose,
receptores de antígenos, células reguladoras, depuração de imunocomplexos, e dentre outros
(revisado por Marrack et al., 2001).
De acordo com a suscetibilidade genética, as DAIs podem ser classificadas como simples ou
complexas. As DAIs simples são causadas por alterações num único gene, tais como as doenças:
ALPS (autoimmune lymphoproliferative syndrome; causada por mutações nos genes Fas ou FasL,
que levam à ausência de apoptose de linfócitos T e B auto-reativos), a IPEX (immune dysregulation,
polyendocrinopathy, enteropathy , X-linked syndrome; causada por mutação do gene Foxp3, que leva
à ausência de geração de células T reguladoras) e a APS-1 (autoimmune polyendocrine syndrome;
causada por mutação do gene AIRE, que leva à expressão diminuída de auto-antígenos no timo,
resultando na seleção negativa defeituosa de células T auto-reativas).
Por outro lado, as DAIs complexas resultam da combinação de vários alelos de
suscetibilidade (de lócus diferentes), fatores ambientais (tais como níveis de hormônios, infecções
virais ou microbianas, dieta) e de eventos estocásticos. Exemplos de DAIs complexas são o lúpus
eritematoso sistêmico, esclerose sistêmica, artrite reumatóide, diabete melito do tipo 1, esclerose
múltipla, doença de Crohn, dentre outras (revisado por Rioux e Abbas, 2005).

Introdução 4
Mecanismos de tolerância imunológica
Tolerância central e periférica
A diversidade dos repertórios de células T e B são gerados durante o desenvolvimento
dessas células no timo e na medula óssea, respectivamente, através de mecanismos de
recombinação somática que envolvem o rearranjo dos genes dos TCRs (T cell receptors, ou
receptores de células T) e dos BCRs (B cell receptors, ou receptores de células B), resultando em
uma diversidade enorme de células T ou B que expressam receptores de diferentes especificidades.
Cada célula T ou B apresenta um único receptor rearranjado entre de bilhões de possibilidades. No
caso das células B, diversidade adicional é gerada pelo processo de hipermutação somática, que
consiste na substituição de nucleotídeos nos genes dos BCRs, e ocorre nos órgãos linfóides
periféricos durante fases tardias da resposta imune.
O repertório de células T é determinado pelos processos de seleção positiva e negativa que
ocorrem no timo, durante os quais os timócitos são selecionados e amadurecem. A afinidade do TCR
por complexos MHC-peptídeo próprio é o parâmetro crucial que determina o destino do timócito e
constitui a base dos processos seleção positiva e negativa. Assim, timócitos cujos TCRs não
reconhecem ou reconhecem com afinidade muito baixa os complexos MHC-peptídeos-próprios,
morrem por negligência, sendo este o destino da maioria dos timócitos. Os timócitos duplo-positivos
que expressam TCRs com baixa afinidade por complexos MHC-peptídeos próprios, apresentados por
células epiteliais do córtex tímico do indivíduo, recebem sinais para sobreviver e se diferenciar em
células T maduras. Esse processo é chamado de seleção positiva. Já, os timócitos duplo-positivos
cujos TCRs apresentam alta afinidade por complexos MHC-peptídeos próprios são selecionados pelo
processo de seleção negativa que será discutido adiante.
Entre 20 a 50% dos TCRs e dos BCRs gerados pela recombinação V(D)J ligam-se com
afinidade potencialmente perigosa à auto-antígenos (revisado por Goodnow et al., 2005). Uma vez
que somente 3-5% da população desenvolve doenças auto-imunes, é notável que esse número
imenso de receptores auto-reativos é tão bem regulado na maioria das pessoas. Esta regulação
deve-se a mecanismos de tolerância imunológica que lidam com as células T e B que expressam
esses receptores auto-reativos com alta afinidade por antígenos próprios. Os mecanismos de
tolerância imunológica pode ser divididos em mecanismos de tolerância central e mecanismos de
tolerância periférica. Na tolerância central, linfócitos imaturos que reconhecem antígenos próprios

Introdução 5
com alta afinidade nos órgãos linfóides centrais (medula óssea para as células B e timo para as
células T) morrem por apoptose (deleção clonal), ou fazem “edição” do receptor, ou são
funcionalmente “inativados” (anergia clonal). Na tolerância periférica, os linfócitos auto-reativos
maduros que encontram auto-antígenos na periferia são mortos por apoptose induzida por ativação,
ou são funcionalmente “inativados” (anergia clonal), ou são controlados por mecanismos supressores
mediados por células T reguladoras (revisado por Walker e Abbas, 2002; Goodnow et al., 2005).
O principal mecanismo de tolerância central é a deleção clonal, que constitui a morte dos
linfócitos que apresentam receptores “proibidos” ou auto-reativos com alta afinidade por antígenos
próprios, por apoptose. Outro mecanismo é a edição do receptor, ou seja, a célula que apresenta um
receptor com alta afinidade por antígenos próprios pode “editar” esse receptor, passando por outra
recombinação V(D)J para apresentar um receptor diferente que não seja auto-reativo. Esse é um dos
principais mecanismos de tolerância no desenvolvimento de células B. O terceiro mecanismo de
tolerância central constitui em mudanças bioquímicas intrínsecas e mudanças na expressão gênica
que reduzem a capacidade da célula de ser ativada através do receptor auto-reativo. Esse fenômeno
é chamado de anergia clonal, ou seja, um estado de não-responsividade ao antígeno específico,
mesmo em condições ótimas de estimulação (revisado por Hogquist et al., 2005).
Em relação às células T, o mecanismo principal da tolerância central é a deleção clonal, ou
seja, a eliminação dos timócitos que expressam TCRs com alta afinidade por antígenos próprios
apresentados por células dendríticas e células epiteliais da medula do timo (deleção clonal). Os
mecanismos de tolerância central por anergia clonal e edição de receptor também ocorrem, mas têm
um papel menos importante. Esses três mecanismos “inativam” ou eliminam células T auto-reativas
de alta afinidade e são considerados os mecanismos de seleção negativa (revisado por Hogquist et
al., 2005). No entanto, vale notar que durante a seleção negativa no timo, algumas células T que
apresentam afinidade alta por auto-antígenos são selecionadas por mecanismos ainda não
completamente esclarecidos, resultando na diferenciação de células T com fenótipo “regulador”
(revisado por Hogquist et al., 2005).
Finalmente, se células T e B auto-reativas evadirem os três mecanismos de tolerância central
descritos acima, fenômeno chamado de “ignorância imunológica”, mecanismos de tolerância
periférica se tornam responsáveis pelo controle das mesmas na periferia (revisado por Walker e

Introdução 6
Abbas, 2002; Goodnow et al., 2005). Dentre eles, vale destacar o mecanismo periférico de supressão
ativa das células auto-reativas por células T reguladoras que será discutido a seguir.
Células T reguladoras
Como discutido anteriormente, o sistema imune desenvolveu vários mecanismos para
estabelecer e sustentar a auto-tolerância imunológica (a ausência de resposta a antígenos próprios),
incluindo eliminação física (deleção clonal) ou inativação funcional (anergia). Existem várias
evidências de que a supressão ativa de células T auto-reativas, mediada por células T, constitui um
outro mecanismo essencial de auto-tolerância (revisado por Shevach, 2000; Maloy e Powrie, 2001;
Coutinho et al., 2001; Sakaguchi, 2004). Embora a idéia de células T que controlam negativamente
respostas imunes não seja nova para os imunologistas, já houve grande controvérsia em relação à
sua existência e se elas constituem uma entidade celular funcionalmente distinta. Além disso, houve
dúvidas de sua importância no controle de desordens imunológicas como as doenças auto-imunes.
Nos últimos anos, entretanto, ressurgiu o interesse pelas células T supressoras ou reguladoras em
várias áreas da imunologia básica e clínica (revisado por Sakaguchi, 2004; Baecher-Allan e Hafler,
2004). Este interesse é parcialmente devido ao melhor entendimento de que o sistema imune normal
produz endogenamente, uma subpopulação de células T que é altamente especializada para função
supressora e que anormalidades no número ou função dessas células podem ser a causa de
doenças auto-imunes ou inflamatórias em animais ou em humanos (revisado por Sakaguchi, 2004).
A co-existência de células T auto-reativas e protetoras foi revelada pela auto-imunidade
sistêmica observada em camundongos linfopênicos após a transferência de células T CD4+ naive, ou
pela proteção contra o desenvolvimento da auto-imunidade conferida pela co-transferência de uma
subpopulação de células T CD4+ que expressa cadeia α do receptor de IL-2 (CD25) (Sakaguchi et al.,
1995). Evidências recentes sugerem que as próprias células T CD4+CD25+ são auto-reativas, e que
esta propriedade tem um papel essencial no desenvolvimento da linhagem de células T reguladoras
(Tregs). Assim, a auto-reatividade pode ser benéfica como parte de um mecanismo celular dedicado
à prevenção da auto-imunidade (revisado por Kronenberg e Rudensky, 2005).
A maioria das Tregs são CD4+ e expressam constitutivamente a molécula CD25 (cadeia α do
receptor de IL-2, IL-2Rα) (Sakaguchi et al., 1995). São produzidas normalmente pelo timo como uma
subpopulação de células T funcionalmente distintas e maduras, e constituem aproximadamente 2-

Introdução 7
10% da população de células T CD4+ (revisado por Sakaguchi, 2004). Além da geração tímica de
células Tregs CD4+CD25+, células T periféricas não-Treg podem adquirir a expressão de Foxp3 e
converter-se em células Tregs in vivo após estimulação antigênica crônica ou em condições de
linfopenia (Apostolou e Boehmer, 2004; Curotto de Lafaille et al., 2004; Walker et al., 2003).
As células Tregs CD4+CD25+ produzem citocinas reguladoras tais como IL-10, IL-4 e TGF-β
(transforming growth factor-β), e também expressam preferencialmente as moléculas CTLA-4
(cytotoxic T lymphocyte antigen-4, regulador negativo da ativação de células T; receptor para CD80 e
CD86), GITR (glucocorticoid-induced TNFR-family related receptor) e PD-1 (programmed death-1).
Entretanto, a identificação de Tregs durante respostas imunes ou em tecidos inflamados é complicada
porque a maioria desses marcadores, inclusive o CD25, são também expressos em células T recém-
ativadas não-reguladoras (revisado por Sakaguchi, 2004). Recentemente, a molécula CD27 foi
identificada como um marcador estável em células Tregs que pode ser usado em conjunto com CD25
para distinguir Tregs de células T efetoras em tecidos inflamados (Ruprecht et al., 2005).
As Tregs CD4+CD25+ são funcionalmente competentes quando isoladas ex vivo, e após
estimulação pelo TCR são capazes de suprimir a proliferação e produção de IL-2 de células T
CD4+CD25- ou células T CD8+ de maneira contato-dependente (revisado por Shevach, 2000;
Sakaguchi, 2004). Experimentos de transferência adotiva usando células marcadas revelaram que as
células Tregs proliferam in vivo e sobrevivem por longos períodos, mostrando sua capacidade de
auto-renovação (Gavin et al., 2002). Vale ressaltar que, após expansão, as células Tregs mantém ou
até aumentam sua capacidade supressora. Estes dados, em combinação com a origem tímica das
células Tregs, sugerem que estas células constituem mesmo uma linhagem celular específica.
Vários mecanismos tentar explicar a supressão mediada por células Tregs in vivo.
Primeiramente, a expressão elevada pelas células Tregs do IL-2R de alta afinidade pode resultar na
competição pela IL-2 com as outras células. No entanto, é improvável que esse seja o principal
mecanismo, pois mesmo na presença de IL-2 exógena, as células Tregs inibem a regulação positiva
de RNAm de IL-2 em células T respondedoras (Thornton et al., 2004). Além da privação de IL-2, duas
citocinas imunossupressoras, IL-10 e TGF-β, foram implicadas num mecanismo supressor ativo
mediado por células Tregs in vivo (revisado por Sakaguchi, 2004). No entanto, outros tipos celulares,
incluindo células T não-reguladoras, produzem essas citocinas.

Introdução 8
Estudos in vitro mostraram que as células Tregs suprimem respostas de células T CD4+ ou T
CD8+ por um mecanismo contato-dependente, mas independente de IL-10 e TGF-β (revisado por
Shevach, 2000). Foi sugerido que esse mecanismo envolve a sinalização “reversa” pelo crosslinking
de moléculas B7 na superfície de células dendríticas ou de células T, após ligação à CTLA-4 (o
receptor de alta afinidade para as moléculas B7 que é expresso em níveis elevados pelas células
Tregs). Em células dendríticas, o crosslinking das moléculas B7 leva à indução de indoleamine-2,3-
dioxygenase (IDO), resultando na depleção local de triptofano (Mellor et al., 2004). Em células T, as
conseqüências bioquímicas da ligação das moléculas B7 pelo CTLA-4 expresso nas células Tregs
são desconhecidas. Foi mostrado também que, em camundongos e humanos, células Tregs ativadas
são capazes de matar células T-alvo pela via dependente da perforina e granzima (Grossman et al.,
2004). Em resumo, apesar do acúmulo de informação sobre os mecanismos moleculares da função
supressora das células Tregs CD4+CD25+, estes ainda não foram totalmente identificados e
esclarecidos (revisado por Kronenberg e Rudensky, 2005).
Embora a expressão de CD25 venha sendo essencial para o isolamento e enumeração das
células Tregs CD4+CD25+, seu emprego como um marcador fenotípico de células Tregs CD4+CD25+
durante respostas imunes é muito limitado, pois células T CD4+ e T CD8+ recém-ativadas, não-
reguladoras, expressam transientemente a molécula CD25. A busca por um marcador específico de
células Tregs em camundongos resultou na identificação do fator de transcrição Foxp3, que é
expresso em células Tregs mas não em células T recentemente ativadas. Foi mostrado que as
células Tregs CD4+CD25+ expressam especificamente o gene regulador Foxp3, que codifica um fator
de transcrição chamado de forkhead transcription factor 3, que controla seu desenvolvimento e
função (Hori et al., 2003; Fontenot et al., 2003; Khattri et al., 2003). A expressão retroviral do gene
Foxp3 em células T periféricas CD4-CD25+ de camundongos ou humanos resultou na aquisição da
função supressora por essas células. Portanto, a expressão de Foxp3 pode ser usada como um
marcador molecular específico para células Tregs CD4+CD25+ (revisado por Kronenberg e Rudensky,
2005).
Alguns anos antes da identificação do Foxp3 como marcador específico de células Tregs,
mutações no gene Foxp3 (localizado no cromossomo X) tinham sido identificadas como causa de
uma desordem auto-imune fatal observada em pacientes (denominada IPEX, immune dysregulation,
polyendocrinopathy, enteropathy, X-linked syndrome) e em camundongos mutantes que desenvolvem

Introdução 9
espontaneamente esta doença auto-imune (denominados scurfy mice). A doença se manifesta em
homens, mas não em mulheres heterozigotas. No entanto, aproximadamente 50% das células T
dessas mulheres não expressam Foxp3 (Bennett et al., 2001; Brunkow et al., 2001).
Durante o desenvolvimento das células T, timócitos com alta afinidade por complexos MHC-
peptídeo próprio podem vir a regular positivamente o gene Foxp3 em resposta ao aumento do tempo
ou da força de sinalização pelo TCR, em combinação com a sinalização por CD28 e com outros
sinais desconhecidos. Após a indução de Foxp3, timócitos se transformam numa linhagem de células
Tregs e têm função de manter as respostas de células T sob controle, desse modo prevenindo a auto-
imunidade (revisado por Fontenot e Rudensky, 2005; Kronenberg e Rudensky, 2005).
Embora outras populações de células T potencialmente supressoras tenham sido descritas
(células NKT, TR1, TH3 e outras), seus mecanismos de geração e função ainda não estão bem
esclarecidos, bem como suas funções supressoras ainda não foram realmente comprovadas
(revisado por Bach, 2003). Desse modo, atualmente, os pesquisadores têm se concentrado no estudo
das funções das células Tregs CD4+CD25+Foxp3+. Existem várias evidências atuais de que as células
Tregs CD4+CD25+Foxp3+ atuam na supressão da ativação imune, funcionando como mediadores
críticos da homeostasia imune e da auto-tolerância (Fontenot e Rudensky, 2005). Assim, tem sido
mostrado que a população de células Tregs CD4+CD25+Foxp3+ é ativamente engajada no controle de
uma variedade de respostas imunes fisiológicas e patológicas e podem ser exploradas não somente
para a prevenção de ou tratamento de doenças auto-imunes, mas também para a indução de
tolerância a antígenos não-próprios (tolerância a transplantes), controle negativo de respostas imunes
aberrantes (como alergias) e aumento da defesa do hospedeiro (por exemplo, na imunidade tumoral e
imunidade microbiana) (revisado por Sakaguchi, 2004; Sakaguchi, 2005).
1.2 Diabete melito do tipo 1
O diabete melito do tipo 1 (DM), é uma doença auto-imune órgão-específica mediada por
células T de padrão TH1, e caracterizada pela destruição seletiva de células β pancreáticas
produtoras de insulina. A doença clínica manifesta-se somente após destruição de aproximadamente
80-90% das células β. O processo destrutivo das células β leva à falta do hormônio insulina, que
resulta num estado de hiperglicemia, devido ao aumento da produção hepática de glicose e
diminuição da captura de glicose da circulação. Na ausência de insulina, também ocorre um aumento

Introdução 10
da quebra de gordura e da oxidação de ácidos graxos, que resulta numa excessiva produção de
cetonas. Se não tratados, os distúrbios metabólicos levam progressivamente à depressão do sistema
nervoso central, coma e morte. Assim, pacientes necessitam de tratamento permanente com insulina
exógena para sobrevivência. Algumas complicações crônicas graves são os problemas vasculares
que levam à insuficiência renal, cegueira, doença cardíaca e úlceras crônicas. A taxa de destruição
das células β varia de paciente para paciente, mas tende a ser mais agressiva em crianças e
adolescentes. O diabete melito do tipo 1 desenvolve-se mais freqüentemente durante a infância e
adolescência, mas pode aparecer na idade adulta também (revisado por Notkins et al., 2002).
O DM é tratado pela terapia convencional com insulina ou terapia intensiva com insulina
(conhecida como ITT). Ensaios clínicos de tratamentos com ciclosporina, azatioprina/corticóides, ou
anticorpo monoclonal anti-CD3, visando à preservação funcional das células β, vêm sendo realizados
com resultados significativos, mas insuficientes para aplicação clínica rotineira (revisado por Palmer
et al, 2004). Mais recentemente, o transplante de ilhotas pancreáticas vem sendo proposto como
alternativa terapêutica para o DM.
A suscetibilidade ao DM é determinada por fatores genéticos e ambientais. A importância da
herança genética do DM foi determinada por estudos de incidência da doença em famílias
acometidas. O risco de desenvolvimento de DM em parentes de indivíduos diabéticos de primeiro
grau é de 5-6%, comparado com o risco de 0,4% na população geral. Além disso, a taxa de
concordância para a doença é muito maior em gêmeos monozigóticos (30-40%) do que em gêmeos
dizigóticos (6%) (revisado por Notkins e Lernmark, 2001; Notkins, 2002; Kelly et al., 2003). Embora
esta observação seja indicativa da grande contribuição genética para o risco de desenvolvimento de
DM, a relativa baixa taxa de concordância entre gêmeos idênticos sugere que os genes de
suscetibilidade têm baixa penetrância, ou seja, nem todos os indivíduos de alto risco para DM irão
desenvolver a doença.
Os primeiros genes de suscetibilidade para DM encontrados foram os genes que codificam
para o sistema HLA, localizados no cromossomo 6p21. Dentre os alelos de HLA que conferem
suscetibilidade estão o DRB1 (que codificam as moléculas DR3 e DR4) e os alelos DQA1 e DQB1
(que codificam as moléculas DQ2 e DQ8, respectivamente). Estudos subseqüentes demonstraram
uma associação entre o DM e a região do gene da insulina no cromossomo 11p. Embora outros lócus
de suscetibilidade tenham sido descritos posteriormente, estudos demonstraram que o lócus IDDM1

Introdução 11
(situado na região dos genes do HLA) é o principal determinante genético do risco de
desenvolvimento de DM, responsável por 42% da herança genética familiar de DM. Por outro lado, o
lócus IDDM2 (região do gene da insulina) contribui com 10% da suscetibilidade genética (revisado por
Notkins et al., 2002; Kelly et al., 2003).
A discordância entre gêmeos idênticos reflete a geração de diferentes repertórios de células
imunes através dos rearranjos randômicos dos genes que codificam os receptores das células T e B,
e a ocorrência de eventos estocásticos ou de mutações somáticas. Além disso, indica a importância
dos fatores não-genéticos ou ambientais no desenvolvimento do DM. A importância dos fatores
ambientais é apoiada pela variação sazonal da incidência do DM, com a maioria dos casos ocorrendo
entre o outono ou inverno, e a variação geográfica da incidência da doença. Os fatores ambientais
relacionados ao o risco de desenvolvimento do DM incluem infecções virais (principalmente pelos
vírus coxsackie B4 e da rubéola), dieta na infância, vacinação, influências climáticas, toxinas e
estresse (Knip e Akerblom , 1999).
O estágio precoce do DM é caracterizado por uma insulite, devida à infiltração das ilhotas
pancreáticas por células mononucleares imunes, incluindo linfócitos T e B, monócitos, células
dendríticas e células NK (Itoh et al, 1993). Embora ela possa refletir uma resposta inflamatória normal
em resposta à injúrias teciduais causadas por infecções virais por exemplo, foi mostrado que o
infiltrado celular contribui diretamente para a destruição das células β pancreáticas (revisado por
Roep, 2003). Macrófagos e células dendríticas são as primeiras células a infiltrarem as ilhotas
pancreáticas. Além de apresentaram antígenos das células β aos linfócitos, uma vez ativados
produzem citocinas pró-inflamatórias (IL-1β, IL-6, TNF-α e INF-β) e óxido nítrico, e contribuem
também para a destruição das células β (revisado por Mathis et al., 2001). A detecção de células T
auto-reativas contra auto-antígenos presentes nas células β pancreáticas, que incluem a insulina, o
GAD (glutamic acid decarboxylase) e a IA-2 (protein tyrosine phosphatase 2), no sangue periférico de
indivíduos diabéticos recém-diagnosticados, demonstrou que mecanismos de auto-imunidade estão
envolvidos na destruição das células β (Naquet et al., 1988; Atkinson et al., 1992; Hawkes et al.,
2000).
Estudos em animais demonstraram que as células T têm um papel essencial na patogênese
do DM. Os camundongos NOD (Non-Obese Diabetic) desenvolvem espontaneamente diabete
insulino-dependente e compartilham várias características imunológicas e patológicas com a doença

Introdução 12
humana (revisado por Anderson e Bluestone, 2005). O desenvolvimento da doença nesses animais
requer a presença de ambas células T CD4+ e CD8+. Camundongos NOD sem timo ou NOD/scid
(severe combined immundeficiency) não desenvolvem insulite ou diabete (Ogawa et al., 1985;
Christianson et al., 1993). Além disso, células T isoladas de camundongos NOD são capazes de
transferir a doença para animais não-diabéticos e acelerar o início da diabete em camundongos NOD
neonatos (Wicker et al., 1986; Bendelac et al., 1987). Embora ambas as células T CD4+ TH1 e células
T CD8+ sejam importantes na patogênese do DM, trabalhos mostraram que as células T CD8+
citotóxicas são as principais células efetoras na destruição das células β pancreáticas.
Citocinas do padrão TH1 produzidas pelas células T CD4+ e também por T CD8+ são essenciais
para patogênese da DM, pois potencializam respostas imunes mediadas por células, incluindo a
ativação de macrófagos. Dois mecanismos celulares de destruição das células β foram descritos, o
mecanismo associado ao reconhecimento (1) e o mecanismo associado à ativação (2) (revisado por
Mathis et al., 2001; Roep et al., 2003; Jun et al., 2002). No mecanismo (1) a célula T CD8+ é ativada
diretamente pelo reconhecimento de antígenos das células β apresentados por moléculas de MHC de
classe I presentes nas próprias células β pancreáticas. A ativação das células T CD8+
conseqüentemente provoca a morte das células β por contato célula-célula através das vias de
sinalização por Fas/FasL (Itoh et al., 1997) ou perforina/granzima (Kagi et al., 1997). De acordo com o
segundo mecanismo (2), as células T CD4+ ou T CD8+ reconhecem antígenos das células β
indiretamente, apresentados por APCs (células dendríticas ou macrófagos) no contexto de moléculas
de MHC de classe II ou classe I, respectivamente. A ativação resultante das células T leva à morte
das células β mediada por células T CD8+ citotóxica (através de receptores de superfície Fas/FasL ou
TNF-α/TNF-R), à liberação de citocinas pró-inflamatórias e mediadores de morte celular pelas células
T, à ativação de macrófagos com conseqüente aumento de sua capacidade citotóxica, e à ativação
das células β e estimulação da produção de mediadores de morte célula por elas mesmas (revisado
por Mathis et al., 2001; Roep et al., 2003; Jun et al., 2002).
A etiologia auto-imune do DM é também demonstrada pela presença de auto-anticorpos
circulantes específicos contra antígenos das células β pancreáticas. Estes auto-anticorpos são
detectados em 85-90% dos indivíduos com DM ao diagnóstico. Ainda não está claro se eles
participam diretamente da destruição das células β ou aparecem secundariamente devido à liberação

Introdução 13
de auto-antígenos das ilhotas danificadas por outros componentes do sistema imune. Não obstante,
os auto-anticorpos são ótimos marcadores da patogênese do DM. O aparecimento deles precede o
início da doença clínica, freqüentemente em vários meses a vários anos (revisado por Notkins, 2002),
o que explica o fato dos pacientes apresentarem auto-anticorpos contra vários antígenos das células
β no momento em que os sintomas clínicos se tornam aparentes. Desse modo, a presença de vários
auto-anticorpos pode ser usada como um marcador sensível para prever os riscos de
desenvolvimento do DM, embora alguns indivíduos positivos para auto-anticorpos não desenvolvam a
doença auto-imune (Bingley et al., 1997).
1.3 Esclerose múltipla
A esclerose múltipla (EM) é uma doença auto-imune inflamatória, mediada por células T de
padrão TH1, que afeta especificamente o sistema nervoso central (SNC). A EM, assim como outras
DAIs, geralmente se manifesta em adultos jovens, acometendo principalmente mulheres.
Em seu início, a EM é clinicamente caracterizada como EM surto-remissiva (EM-SR,
observada em 85-90% dos pacientes) ou EM progressiva primária (EM-PP, observada em 10-15%
dos pacientes). As recaídas (ou “ataques”, ou “surtos”) são tipicamente subagudas, uma vez que os
sintomas se desenvolvem durante algumas horas a vários dias, persistem por vários dias a semanas,
e depois se dissipam gradualmente. Os surtos são provavelmente causados pela entrada de células T
auto-reativas ativadas (anti-mielina) no SNC, causando inflamação aguda associada à edema. A
capacidade dos esteróides de rapidamente interromper os sintomas da EM, sugere que o edema
agudo e sua subseqüente resolução correspondem à recaída clínica (surto) e à remissão,
respectivamente.
A progressão dos pacientes com EM-SR é muito variável. Em geral, se não forem tratados
aproximadamente 50% dos pacientes necessitarão de apoio para caminhar dentro de dez anos após
o início da EM. No início da doença, as lesões que capturam gadolíneo por ressonância magnética
nuclear (RMN) são freqüentes e consistentes com o influxo de células auto-reativas ativadas no SNC,
causando uma perturbação da barreira hemato-encefálica. O aumento na freqüência dos surtos e
recuperação inadequada nos primeiros anos da doença clínica predizem uma deterioração mais
rápida. A observação de múltiplas lesões que capturam gadolíneo por RMN também prediz um curso
mais grave da doença. (revisado por Hafler, 2004; Hemmer et al, 2002; Compston e Coles, 2002).

Introdução 14
Entretanto, com o tempo, a capacidade de recuperação após os surtos diminui e a
incapacidade neurológica começa a piorar. Aproximadamente 40% dos pacientes com EM-SR param
de apresentar os surtos e desenvolvem uma desordem neurodegenerativa progressiva secundária,
associada à inflamação crônica do SNC, conhecida como EM progressiva secundária (EM-PS). A
evolução da forma progressiva secundária da doença é associada com uma diminuição significante
da freqüência de lesões que capturam gadolíneo e do volume do parênquima cerebral (atrofia
cerebral). Enquanto no início, a forma EM-SR era sensível à imunossupressão, à medida que o tempo
passa, a resposta da EM à imunoterapia diminui e pode desaparecer em formas tardias da EM-PS.
Atualmente, ao invés de ser considerada como uma doença com duas formas, a EM vem
sendo considerada uma doença contínua, mas caracterizada inicialmente por eventos inflamatórios
agudos, e posteriormente pela indução secundária de um processo neurodegenerativo, refratário à
imunoterapia. Já a forma primária progressiva é caracterizada desde o início pela ausência de surtos,
mas por um declínio gradual da incapacidade neurológica. Clinicamente, essa forma de EM está
associada à falta de resposta a qualquer forma de imunoterapia (revisado por Hafler, 2004; Hemmer
et al, 2002; Compston e Coles, 2002).
O conceito da EM como uma doença auto-imune inflamatória deriva do fato que a EM
responde a tratamentos imunomoduladores ou imunosupressores. Glucocorticóides são
administrados em altas doses durante as exacerbações clínicas da doença e atuam reduzindo a
inflamação, o edema e produção de citocinas pró-inflamatórias. O tratamento com INF-β ou com
Glatiramer-acetate (GA, copolymer-1, Cop-1) constituem terapias aprovadas para a EM-SR. Ademais,
vários agentes quimioterapêuticos que apresentam efeitos imunossupressores mais intensos e
duradouros, tais como a ciclofosfamida, mixantrone e azatioprina, são usados em estágios mais
avançados da doença, ou seja, na transição de EM-SR para EM-PS, ou em pacientes que não
respondem às terapias aprovadas (revisado por Hemmer et al, 2002; Hafler, 2004; Sospedra e Martin,
2005).
A etiologia da EM ainda não foi esclarecida, mas com base no conhecimento atual, acredita-
se que a doença se desenvolve em indivíduos geneticamente suscetíveis e requer o
desencadeamento por fatores ambientais. A EM é altamente prevalente em caucasianos (0,05 -
0,15%), mas raramente observada em asiáticos e africanos. Parentes de primeiro grau de indivíduos
afetados têm um risco aproximadamente 20-50 vezes maior de desenvolver de doença (2-5%), e a

Introdução 15
taxa de concordância entre gêmeos idênticos varia entre 20-35% em diferentes estudos (25% nos
estudos mais recentes) (revisado por Hemmer et al, 2002; Sospedra e Martin, 2005). Indivíduos
suscetíveis apresentam um ou mais genes de suscetibilidade na região do HLA no cromossomo
6p21, que são responsáveis por 10-60% do risco genético para EM (Haines et al., 1998).
Similarmente a outras DAIs mediadas por células T, os genes do HLA que conferem suscetibilidade à
EM são o HLA-DR, o HLA-DQ, o haplótipo HLA-DR15 em caucasianos (DRB1*1501, DRB5*0101,
DQA1*0102, DQB1*0602), e outros DRs em populações etnicamente mais distantes (Haines et al.,
1998; Dyment et al., 2004).
Entre os fatores ambientais relacionados à etiologia da EM estão os agentes infecciosos
virais ou bacterianos (principalmente o herpesvírus 6, o Epstein-Barr vírus e a bactéria Chlamydia
pneumoniae), além de influências hormonais, comportamentais, climáticas e geográficas (revisado
por Coo e Aronson, 2004). Dois principais mecanismos foram propostos para explicar como infecções
virais ou bacterianas poderiam induzir a EM (bem como outras DAIs): (1), mimetismo molecular,
ativação de células auto-reativas pela reatividade cruzada entre antígenos próprios e não-próprios;
(2), bystander activation, assume que células auto-reativas sejam ativadas em decorrência de
eventos inflamatórios não-específicos que ocorrem durante as infecções. Uma terceira proposta seria
que as infecções induziriam a EM através da combinação desses dois mecanismos descritos
(revisado por Sospedra e Martin, 2005).
Rivers et al. (1933) mostraram que a injeção de suspensões de medula espinhal ou cérebro
em macacos saudáveis induzia uma doença similar à EM, levando à hipótese de que a EM tratava-se
de uma doença auto-imune. Várias décadas depois, pesquisadores mostraram que a injeção de
componentes protéicos da bainha de mielina, juntamente com um adjuvante em camundongos naive
suscetíveis, causava encefalomielite, que atualmente é conhecida como EAE (experimental
autoimmune encephalomyelitis, ou encefalomielite auto-imune experimental) (Pettinelli e McFarlin,
1981; Ben-Nun e Cohen, 1982). Trabalhos mostraram que a EAE era induzida em camundongos
naive pela transferência adotiva de células T anti-mielina, mas não pela transferência de auto-
anticorpos. Esse fato levou os pesquisadores a concluírem que a EM constitui uma doença auto-
imune mediada por células T.
As células T CD4+ TH1 auto-reativas apresentam um papel central na patogênese da EM
devido a várias observações: (1), células T CD4+ contribuem para o infiltrado de células inflamatórias

Introdução 16
encontradas no SNC e líquor, em pacientes com EM; (2), o risco genético para EM é conferido em
grande parte por moléculas HLA-DR e HLA-DQ; (3), a produção de auto-anticorpos, ativação de
células T auto-reativas CD8+ e vários outros aspectos das respostas adaptativa e inata são, pelo
menos em parte, controladas por células T CD4+ TH1 (revisado por Hemmer et al, 2002; Sospedra e
Martin, 2005). No contexto de funções efetoras, as células T CD8+ são mais diretamente envolvidas
na injúria do SNC do que as células T CD4+, pelas seguintes razões, dentre outras: (1), exceto para a
microglia, nenhuma célula residente do SNC expressa MHC de classe II; (2), expansões oligoclonais
proeminentes de células T CD8+ são encontradas no líquor e no SNC de pacientes com EM
(Jacobsen et al, 2002); (3), a expressão de MHC de classe I é induzida em neurônios danificados. Em
resumo, ambas respostas de células T CD4+ e T CD8+ contribuem para patogênese da EM, embora
em diferentes estágios e com funções distintas (revisado por Hemmer et al, 2002; Sospedra e Martin,
2005).
A MBP (myelin basic protein), a PLP (proteolipid protein) e a MOG (myelin oligodendrocyte
glycoprotein) constituem os principais auto-antígenos alvos de células T auto-reativas e de auto-
anticorpos em pacientes com EM. Embora a MBP seja a proteína da mielina mais estudada, ela
constitui a segunda proteína mais abundante da mielina (30-40%), após a PLP (revisado por Hemmer
et al, 2002; Sospedra e Martin, 2005).
A observação de que os níveis de imunoglobulinas encontram-se elevados no líquido
cefalorraquidiano de pacientes com EM, mas não no soro, indica uma produção local por células B.
Além disso, as imunoglobulinas apresentam uma distribuição oligoclonal, ou seja, são produzidas por
células B em expansão clonal (Qin et al., 1998). Essas observações sugerem um papel importante
para as células B e auto-anticorpos na patogênese da EM. Além de células B servirem como APCs
para células T auto-reativas, as células B auto-reativas produzem auto-anticorpos anti-mielina que
podem ser encontrados em áreas de desmielinização ativa no SNC do paciente com EM (Genain et
al, 1999). Os auto-anticorpos podem causar desmielinização pela opsonização da mielina para
fagocitose ou via ativação do sistema complemento (revisado por Hemmer et al, 2002; Sospedra e
Martin, 2005).

Introdução 17
1.4 Transplante de células tronco hematopoéticas em doenças auto-imunes
A maioria dos pacientes com DAIs tem uma expectativa de vida quase normal. No entanto,
alguns pacientes sofrem com formas de auto-imunidade graves e progressivas, resistentes a terapias
convencionais. O transplante de células tronco hematopoéticas (TCTH) tem sido nos últimos anos
uma alternativa terapêutica para pacientes com essas DAIs graves e refratárias aos tratamentos
convencionais.
O TCTH envolve a administração de células tronco hematopoéticas (CTHs), que têm a
capacidade de auto-renovação e de originar todos os tipos celulares maduros do sistema
hematopoético, imunológico e possivelmente alguns tipos celulares não-hematopoéticos (Kondo et
al., 2003). O receptor é preparado para o transplante por um tratamento imunossupressor potente,
geralmente quimioterapia ou radioterapia, seguido pela infusão de células hematopoéticas autólogas
(TCTH autólogo; células coletadas do próprio paciente antes da imunossupressão) ou de células
hematopoéticas alogênicas (TCTH alogênico; células coletadas de um doador compatível), para
restaurar o sistema hematopoético e imunológico do receptor rapidamente, evitando citopenias
prolongadas (Burt et al., 2002; Popat et al., 2005; Hough et al., 2005; Sykes e Nikolic, 2005).
Foi mostrado que este procedimento consegue curar DAIs em modelos animais e há alguns
anos tem sido explorado em vários ensaios clínicos de faseI/II (Sykes e Nikolic, 2005; Popat et al.,
2005; Hough et al., 2005). Até agora, o TCTH autólogo tem sido preferido ao TCTH alogênico devido
à elevada toxicidade e grande potencial de rejeição observados em transplantes alogênicos. Além
disso, o risco da ocorrência de GVHD (“Graft-Versus-Host-Disease”) em TCTH alogênicos, que é
devido ao ataque de células T alogênicas do doador contra alo-antígenos do receptor, é relacionado a
taxas de mortalidade e morbidade significantes.
A iniciativa de se empregar imunossupressão em altas doses seguida de TCTH autólogo no
tratamento de DAIs partiu de dois conjuntos de evidências. O primeiro derivou da observação de
casos isolados de pacientes que apresentavam concomitantemente doença auto-imune e neoplasia
hematológica ou aplasia de medula óssea e que, após serem submetidos a transplante de medula
óssea alogênico para tratamento da doença hematológica, obtiveram remissões prolongadas da
doença auto-imune, sem recaídas da mesma (Nelson et al.,1997). Vale ressaltar que o transplante
autólogo não-manipulado, nesta mesma situação, não produziu os mesmos resultados benéficos em
alguns estudos, provavelmente devido à re-inoculação das células auto-reativas com o enxerto (Euler

Introdução 18
et al., 1996). O segundo conjunto de evidências originou-se de estudos de auto-imunidade em
modelos animais, que discutiremos a seguir.
Modelos animais de auto-imunidade e TCTH
Estudos em modelos animais estabeleceram a base racional para o transplante de células
tronco hematopoéticas em DAIs, mas também demonstraram que a suscetibilidade para DAIs parece
residir nas células hematopoéticas. O transplante de células da medula óssea de camundongos NZB
(lupus-prone) em linhagens de camundongos não-suscetíveis letalmente irradiados, induziu síndrome
lúpica nesses camundongos (Morton e Siegel, 1974). A transferência de DAIs por meio de células
hematopoéticas foi subseqüentemente confirmada para várias doenças, através de vários modelos
animais de lúpus eritematoso sistêmico (LES), encefalomielite auto-imune experimental (EAE), artrite
induzida por adjuvante, síndrome anti-fosfolípide e diabete melito do tipo 1 (Ikehara et al., 1990).
Por outro lado, resistência a DAI pode ser também transferida para linhagens de camundongos
suscetíveis por meio de células hematopoéticas. Em camundongos, o transplante de células
hematopoéticas alogênicas de linhagens de animais não-suscetíveis preveniram o desenvolvimento
de LES, diabete tipo 1, EAE e outras DAIs (Ikehara et al., 1985; Ikehara et al., 1998). Em humanos, o
papel das células hematopoéticas e das células estromais no desenvolvimento da patologia auto-
imune não é muito esclarecido. No entanto, alguns relatos clínicos sugerem uma contribuição crítica
de células tronco hematopoéticas defectivas no desenvolvimento da patologia auto-imune (Bargetzi et
al., 1997; Lampeter etal., 1998).
Infelizmente, estudos em animais mostram que a prevenção do início da auto-imunidade é
muito mais fácil do que a reversão da doença auto-imunes já estabelecida. Até 2004, por exemplo,
mais de 195 métodos para prevenção ou retardo do início de diabete tipo 1 em camundongos NOD
foram identificados (Roep et al., 2004). No entanto, somente poucas abordagens terapêuticas foram
efetivas na reversão de diabete tipo 1 e LES já estabelecidos, sendo uma delas o TCTH alogênico
(Ikehara et al., 1989; Yasumizu et al., 1987).
Os estudos que revelaram o potencial da imunossupressão em altas doses e do TCTH para o
tratamento da auto-imunidade foram realizados em vários modelos experimentais diferentes de
doenças auto-imunes (van Bekum, 2000a; van Bekum, 2000b; van Bekum, 2002), pois não existia um
consenso em relação a quais modelos seriam os mais representativos da doença auto-imune

Introdução 19
humana. Existem dois tipos distintos de modelos experimentais de doenças auto-imunes, os modelos
de doenças auto-imunes hereditárias ou espontâneas e os modelos de doenças auto-imunes
induzidas por auto-antígenos (van Bekkum, 2002). Nas doenças da primeira categoria, os fatores
genéticos predominam na patogênese da doença, e os sintomas desenvolvem com a idade na
maioria ou em todos dos membros da linhagem. Esses modelos incluem síndromes lúpus-like em
camundongos, diabete medito insulino-dependente em camundongos e ratos, e uma síndrome
complexa de artrite/colite/dermatite em ratos transgênicos para HLA-B27.
Na segunda categoria, as doenças auto-imunes não se desenvolvem espontaneamente, mas
requerem indução pela imunização com antígenos de tecidos específicos (por exemplo, antígenos de
cérebro, no caso da encefalomielite auto-imune experimental (EAE), que constitui um modelo da EM
humana; antígenos teciduais como o colágeno, no caso da artrite induzida por colágeno; ou
antígenos bacterianos como do Mycobacterium tuberculosis, no caso da artrite induzida por
adjuvantes). Foi demonstrado em experimentos de cruzamento, que a suscetibilidade e a resistência
à indução da doença auto-imunes, são amplamente determinadas geneticamente, por isso as
doenças auto-imunes só podem ser induzidas em linhagens específicas de camundongos. Entretanto,
influências ambientais também são envolvidas nas formas de doenças auto-imunes induzidas (van
Bekkum, 2003). Uma vez que a etiologia das doenças auto-imunes humanas complexas é
multifatorial, como discutido anteriormente, onde os fatores dominantes são genéticos tanto quanto
ambientais, a visão atual é que as doenças auto-imunes induzidas representem modelos de doenças
mais realísticos para investigações pré-clínicas.
No caso de DAIs espontâneas, o TCTH singênico (isto é, TCTH de um doador geneticamente
idêntico), realizado em animais jovens, não conseguiu prevenir a auto-imunidade clínica, entretanto, o
TCTH alogênico de uma linhagem resistente para outra suscetível, preveniu a doença, e em alguns
casos, quando realizado logo após o início da doença, melhorou as manifestações auto-imunes
(Yasumizu et al, 1987). Esses resultados levaram alguns pesquisadores a concluir que a auto-
imunidade decorre de alterações nas células tronco hematopoéticas (Good e Ikehara, 1997).
Em contraste, estudos em modelos de doenças auto-imunes induzidas realizados por van
Bekkum e colaboradores (2000) mostraram uma resposta terapêutica de transplante singênicos ou
pseudoautólogos no tratamento dessas doenças. Em modelos animais o TCTH singênico (isto é,
TCTH de um doador geneticamente idêntico) pode ser usado ao invés de TCTH autólogo. Por

Introdução 20
exemplo, a recuperação da paresia induzida pela EAE foi mais rápida em animais tratados com
irradiação corpórea total e transplante singênico comparada à recuperação observada em animais
não-tratados, embora transplantes alogênicos fossem melhores na prevenção de recaídas
espontâneas ou induzidas pela imunização com mielina (van Gelder e van Bekkum, 1995; van Gelder
et al., 1993).
Os resultados dos estudos de van Bekkum e colaboradores (2003) sugeriram que o sucesso do
transplante de células tronco autólogo depende da completa erradicação dos componentes efetores
da doença auto-imune, ou seja, das células T efetoras ativadas e de memória. Quando traduzidos
para a clínica, os dados experimentais indicaram que os pacientes deveriam ser submetidos a um
regime de condicionamento (ou imunossupressão) que induzisse o máximo de linfoablação e que o
enxerto fosse depletado de células T (van Bekkum, 2003). Quanto maior foi a intensidade da terapia
de condicionamento usada, maior foi o sucesso do auto-TCTH na EAE (van Bekkum, 2003; Burt et
al., 1998a).
Foi mostrado também que o período no qual o transplante é realizado é muito importante. Por
exemplo, no tratamento de EAE em camundongos SLJ, o transplante autólogo somente foi efetivo se
realizado precocemente no início da doença, antes de ocorrerem injúrias significativas do tecido alvo
Nesse caso, quase reversão completa foi conseguida, mas nenhum efeito do transplante autólogo foi
observado em estágios mais avançados da doença (Burt et al., 1998a).
Em resumo, enquanto o TCTH alogênico mostrou-se muito efetivo em reverter auto-imunidade
em todos os modelos animais estudados, o TCTH autólogo não foi capaz de reverter com sucesso,
diabete melito do tipo 1 e LES. No entanto, surpreendentemente, níveis variáveis de remissão foram
conseguidos após o TCTH autólogo em vários modelos experimentais de DAIs, como miastenia
gravis, artrite induzida por adjuvante e EAE (van Bekkum, 2003).
Assim, essas várias observações em modelos animais de TCTH autólogos ou alogênicos
sugeriram que ambas abordagens seriam benéficas para a terapia de pacientes com DAIs refratárias,
embora o TCTH autólogo seja mais seguro. Não obstante, os estudos em animais indicam que a
resistência à DAIs reside na célula tronco hematopoética e pode ser transferida por ela (Ikehara et al.,
1985; Ikehara et al., 1990; Ikehara et al., 1998). Desse modo, o transplante de células tronco
hematopoéticas alogênicas poderia resultar na possível cura da DAI, e além disso, na tolerância a

Introdução 21
antígenos do doador, permitindo a aceitação de outros tecidos do doador (por exemplo, ilhotas
pancreáticas), sem o uso de terapia imunossupressora crônica.
Além disso, a maior eficácia do TCTH alogênico em alguns modelos animais de DAIs sugere
que células T alogênicas do doador sejam capazes de eliminar linfócitos auto-reativos do receptor, e
portanto mediar a forma benéfica de imunoterapia chamada de GVA (“graft-versus autoimmunity”).
Porém, não existe razão para esperar que essas respostas imunes sejam específicas para linfócitos
auto-reativos. Após o TCTH alogênico, podem ocorrer concomitantemente respostas imune deletérias
do enxerto contra o hospedeiro (“graft-versus-host response”). Desse modo, dadas as complicações
como GVHD e falha de enxertia após o TCTH alogênico e os resultados positivos do TCTH autólogo
em modelos animais descritos acima, o TCTH autólogo é a abordagem mais lógica para o resgate
hematopoético e vem sendo amplamente utilizado em ensaios clínicos.
Mais recentemente, vários grupos têm utilizado protocolos de condicionamento não-
mieloablativos ou em doses reduzidas em TCTH alogênicos, para indução de tolerância ao
transplante e restabelecimento da auto-tolerância em modelos animais de DAIs (revisado por Sykes e
Nikolic, 2005). Embora estes avanços recentes têm tornado o TCTH alogênico menos tóxico, ele
ainda constitui um procedimento de risco que muito provavelmente não irá substituir os tratamentos
atuais para formas de DAIs menos graves e tratáveis.
Ensaios clínicos de TCTH autólogo em doenças auto-i munes
Com base em todos os fatores discutidos acima, o TCTH autólogo é atualmente preferido ao
TCTH alogênico em ensaios clínicos. Nos últimos anos, vários ensaios clínicos de imunossupressão
em altas doses seguida de TACTH foram iniciados em pacientes com esclerose múltipla (EM),
esclerose sistêmica, artrite reumatóide, lúpus eritematoso sistêmico e miastenia gravis (Burt et al.,
1998b, Burt et al., 2003; Popat et al., 2005; Hough et al., 2005; Tyndall et al., 2005).
Esses ensaios têm utilizado regimes de condicionamento de intensidade variável, variáveis
tipos de depleção de células T (in vivo ou ex vivo) e diferentes fontes de células tronco e protocolos
de mobilização. Para obtenção das células para o TACTH, antes do condicionamento as células
tronco são mobilizadas da medula para o sangue periférico, o que permite a coleta dessas células
sem uso de anestesia geral. A maioria dos protocolos usa o G-CSF (granulocyte colony-stimulating
factor) e/ou ciclofosfamida, uma droga mielossupressora mas que leva a uma expressiva mobilização

Introdução 22
das células tronco. O protocolo do TACTH permite que tratamentos imunoablativos intensos sejam
usados nos pacientes com DAIs refratárias aos tratamentos convencionais.
Dados obtidos desses ensaios clínicos sugerem que o TACTH é viável e pode levar a remissão
das DAIs (Burt et al., 2003; Popat et al., 2005; Hough et al., 2005; Tyndall et al., 2005). Embora os
resultados variem de doença para doença, mais de um terço dos pacientes tem apresentado
remissões completas e prolongadas, freqüentemente sem necessidade de uso de drogas
imunossupressoras (Tyndall et al., 2005). Em algumas doenças, no entanto, recaídas são
relativamente freqüentes. No entanto, dos pacientes que recaem, vários passam a responder aos
tratamentos convencionais que não eram efetivos antes do transplante.
Desde 1996, ao redor de 1000 transplantes já foram realizados mundialmente em vários
ensaios clínicos de fase I/II. A toxicidade e mortalidade relacionada ao transplante diminuiu ao longo
do tempo com o aumento da experiência na aplicação desse tratamento, melhor seleção de pacientes
e modificações nos protocolos de tratamento. Ensaios clínicos randomizados de fase III que irão
comparar o TACTH com a terapia convencional, foram recentemente iniciados nos Estados Unidos e
Europa (Popat et al., 2005; Hough et al., 2005; Tyndall et al., 2005). Idealmente, esses resultados
irão ajudar a esclarecer se os benefícios do TACTH devem-se somente à depleção temporária os
linfócitos auto-reativos pela imunossupressão intensa ou ao restabelecimento de mecanismos de
regulação imune que limitam a auto-imunidade. Além disso, estes estudos fornecerão prova de
eficácia, que é necessária para uma aplicação mais ampla do TACTH no tratamento de DAIs.
Os pacientes com EM constituem o maior grupo de pacientes com doenças auto-imunes que já
foram tratados com TACTH até o momento (Burt et al., 2005). Estudos clínicos de fase I e II têm sido
conduzidos em pacientes com EM progressiva primária e secundária. A mobilização das células
tronco hematopoéticas é realizada com ciclofosfamida combinada com fator estimulador de colônia de
granulócitos (G-CSF) ou somente com G-CSF. O primeiro e maior grupo de transplantes foi realizado
em 85 pacientes com EM, cujos resultados foram registrados no EBMT (European Group for Blood
and Marrow Transplantation). Neste ensaio clínico, foi utilizado o regime de condicionamento BEAM
(BCNU, etoposídeo, aracitin, melfalan). Cerca de 6% dos pacientes faleceram devido a fatores
tóxicos, foi observada melhora neurológica em 21% dos pacientes e uma sobrevida livre de
progressão em 74% dos pacientes com doença progressiva secundária ou primária (Tyndall et al.,
2005).

Introdução 23
Nesse estudo, as lesões inflamatórias agudas do tecido nervoso, detectadas utilizando a
imagem de ressonância magnética nuclear (RMN) por captura de gadolíneo, foram significativamente
reduzidas, demonstrando a eficácia do TACTH no tratamento da EM. No entanto, foi observado que a
deterioração da função neuronal pode continuar no caso de doença em fase avançada. Um estudo
randomizado de fase III, denominado ASTIMS (Autologous Stem Cell Transplantation International
Multiple Sclerosis trial), foi iniciado na Europa em 2004 e irá avaliar aproximadamente 200 pacientes
ao longo de vários anos após o TACTH utilizando a RMN para avaliação da função neuronal dos
pacientes (Tyndall et al., 2005).
No Brasil, o TACTH em DAIs vem sendo realizado desde 2001 em alguns centros, mas
principalmente na Unidade de Transplante de Medula Óssea do Hospital das Clínicas da Faculdade
de Medicina de Ribeirão Preto da Universidade de São Paulo e no Hospital Israelita Albert Einstein
em São Paulo. Pacientes portadores de formas graves e progressivas de esclerose múltipla e
doenças reumáticas (lúpus eritematoso sistêmico, esclerose sistêmica e arterite de Takayasu),
refratárias às terapias convencionais, são tratados com terapia de imunossupressão em altas doses
seguida pelo TACTH (Voltarelli e Ouyang, 2003; Voltarelli et al., 2004b; Voltarelli et al., 2005a;
Voltarelli et al., 2005b). Desde o início de 2004, o TACTH está sendo realizado como tratamento de
pacientes com diabete melito do tipo 1 recém-diagnosticados, exclusivamente no Brasil (Voltarelli et
al., 2004a; Voltarelli, 2004c; Voltarelli, 2006). O objetivo do TACTH nesses pacientes é de preservar a
quantidade residual de células β das ilhotas pancreáticas (Burt et al., 2002a; Voltarelli, 2004c), uma
vez que quando a doença clínica é diagnosticada já houve a perda de aproximadamente 80-90% das
células β pancreáticas (Notkins, 2002). O TACTH é realizado nos pacientes dentro de no máximo seis
semanas após o diagnóstico. Entre os vários critérios de inclusão, um dos mais importantes, além do
diagnóstico recente, é positividade para anticorpos anti-GAD65 ao diagnóstico.
1.5 Mecanismos de ação do TCTH autólogo em doenças auto-imunes
Conforme discutido anteriormente, as DAIs são determinadas por fatores genéticos e
ambientais. Se e quando um indivíduo geneticamente predisposto desenvolverá uma doença auto-
imune, orgão-específica ou sistêmica, dependerá do contato com agentes ambientais, infecciosos ou
não-infecciosos (revisado por Marrack et al., 2001; Davidson e Diamond, 2001). Estudos
retrospectivos de pacientes que migraram de áreas de alta incidência para áreas de baixa incidência

Introdução 24
de DAIs sugerem que existe um período latente, caracterizado por um atraso de vários anos entre o
contato com o agente ambiental hipotético e o início da doença clínica (revisado por Openshaw et al.,
2002). Esses conceitos de fatores ambientais e período latente foram importantes para a constituição
da base racional do TCTH autólogo como tratamento de DAIs.
Conforme proposto por Openshaw et al. (2002), se somente a predisposição genética for
suficiente para o desenvolvimento de auto-imunidade clínica, então a predisposição para a doença
reside exclusivamente nas células tronco hematopóeticas, e o melhor resultado que pode ser
esperado do TCTH autólogo seria um efeito anti-inflamatório temporário conferido pela
imunossupressão em altas doses. Em contraste, se exposições a fatores ambientais específicos em
períodos críticos são importantes, então a terapia de imunossupressão em altas doses seguida pelo
TCTH autólogo poderia levar a uma “mudança cronológica” (“time-shift” ou “re-setting”) da doença
auto-imune clínica para um período mais precoce, análogo ao período latente, restaurando a auto-
tolerância no paciente. Esta hipótese pressupõe que respostas a re-exposição às circunstâncias
ambientais que levaram ao desencadeamento da auto-imunidade serão suficientemente diferentes,
de modo que auto-imunidade clínica não irá reaparecer (Tyndall e Koike, 2002; Openshaw et al.,
2002).
Com base nessa primeira hipótese proposta, outras três hipóteses não mutuamente
exclusivas, foram formuladas para explicar o mecanismo de ação do TACTH em DAIs (Burt et al.,
2002b; Muraro e Martin, 2003): (1) a imunoablação intensa elimina a resposta imune patogênica, (2) o
TACTH reconstitui um sistema imune novo e tolerante, (3) as células tronco hematopoéticas
promovem reparo tecidual. No entanto, apesar do rápido acúmulo de experiência clínica durante os
últimos dez anos, dados sobre o mecanismo de ação do TACTH em doenças auto-imunes humanas
eram muito escassos até 2004, quando os primeiros estudos mais elaborados de reconstituição
imunológica começaram a aparecer na literatura (Sun et al., 2004; Muraro et al., 2005; Farge et al.,
2005; de Kleer et al., 2005; Kötter et al., 2005). Atualmente, esses trabalhos apresentam informações
que começam a explicar os mecanismos de ação do TACTH baseando-se em observações da
reconstituição imunológica nos pacientes.
Atualmente, o mecanismo mais aceito e que vem sendo demonstrado nos trabalhos recentes é
a “reprogramação” do sistema imune após o TACTH, e constitui uma junção das duas primeiras
hipóteses citadas anteriormente (Burt et al., 2002b; Muraro e Martin, 2003). De acordo com esse

Introdução 25
mecanismo, primeiramente a imunossupressão em altas doses elimina as células B e T auto-reativas
de memória. Em seguida, baseando-se no fato de que fatores ambientais desconhecidos tenham um
papel essencial no desencadeamento da DAI, é possível que a eliminação do repertório de células B
e T pré-existente permita que o sistema imune do paciente reinicie do zero, gerando novas células B
e T que sejam auto-tolerantes após o TACTH (revisado por Sykes e Nikolic, 2005).
Porém, não está claro se as células T e B de memória são completamente removidas do
sistema hematopoético, linfóide e de órgãos alvos, por qualquer que seja o regime de
condicionamento. Assim, a imunoablação incompleta pode ser responsável pelas elevadas taxas de
recaída precoce observada em alguns ensaios clínicos de TACTH (Tyndall et al, 2005). Não obstante,
há evidências de melhora e remissão da DAI após TACTH mesmo com imunoablação incompleta
(revisado por Muraro e Douek, 2006).
A manutenção de reações auto-imunes depende de múltiplas populações celulares e existem
várias evidências de alterações em número e função de algumas populações celulares após o
TACTH. Por exemplo, a quimioterapia intensa induz uma redução significativa e prolongada de
células T CD4+, que é caracterizada pela inversão da razão CD4+:CD8+, predominância de células T
de memória e regeneração de células T predominantemente por mecanismos timo-independentes
(revisado por Guillaume et al., 1998; Jameson et al., 2002). Estas células T reconstituídas podem
exibir suscetibilidade aumentada à apoptose e funções alteradas (Hakim et al., 1997; Singh et al.,
1999; Muraro et al., 2005). Assim, a imunoablação intensa e a reconstituição imunológica alterada
podem levar à remissão clínica da DAI nos primeiros meses após o TACTH.
Um estudo recente (Muraro et al., 2005) mostrou pela primeira vez que a remissão clínica
observada em pacientes com EM após o TACTH, que é acompanhada pela supressão da atividade
inflamatória, não é decorrente somente da imunossupressão que esses pacientes recebem, mas de
mudanças qualitativas profundas no sistema imunológico que demonstram uma renovação do
compartimento de células T nesses pacientes. Dois anos após o TACTH, os pacientes apresentaram
uma diversidade clonal mais ampla do repertório do TCR e uma extensa renovação das
especificidades clonais comparadas com o pré-transplante, que é devida à produção de novas células
T naive pelo timo desses pacientes.
No entanto, concomitante ao reaparecimento de novas células T naive, dois outros trabalhos
mostraram a reconstituição tímica de células T auto-reativas após o TACTH. Openshaw et al. (2000)

Introdução 26
mostraram que a resposta proliferativa de células mononucleares à MBP (myelin basic protein) ou
peptídeos imunodominantes da MBP estava significantemente suprimida em pacientes com EM pelo
menos até 20 meses pós-TACTH, no entanto houve uma mudança no padrão de reconhecimento
antigênico após o transplante. Mais recentemente, Sun et al. (2004) demonstraram o reaparecimento
de células T auto-reativas anti-mielina por volta de 12 meses após o TACTH também em pacientes
com EM, concomitante à reconstituição de novas células T naive produzidas pelo timo. Vale ressaltar
que os pacientes com EM desses estudos continuaram em remissão clínica da doença mesmo com o
reaparecimento de células T auto-reativas anti-mielina, demonstrando que a remissão da doença
após o transplante não deve-se somente à eliminação das células auto-reativas patogênicas mas
também a outras mudanças determinadas pelo TACTH.
Traynor et al. (2000) mostraram que ocorrem mudanças significativas no padrão de citocinas
em pacientes com lúpus eritematoso sistêmico (LES) após o TACTH. A expressão de interleucina-4
(IL-4), citocina do tipo TH2, foi encontrada aumentada em linfócitos de pacientes com lúpus
eritematoso sistêmico antes do transplante e normalizou após o TACTH. Por outro lado, os níveis de
interferon-γ (IFN-γ), que estavam abaixo do normal nesses pacientes antes do transplante,
aumentaram para níveis normais após o TACTH. Num estudo mais recente, de Kleer et al. (2005)
mostraram evidências que sugerem uma “reprogramação” de células auto-reativas em crianças com
artrite juvenil idiopática (AJI) após o TACTH. Usando APCs artificiais (lipossomas conjugados com
complexos MHC-peptídeo) para isolar células auto-reativas contra um peptídeo da Hsp60 (heat shock
protein 60; auto-antígeno importante na AJI), eles demonstraram que o TACTH induz a mudança do
fenótipo pró-inflamatório (caracterizado por elevados níveis de RNAm de IFN-γ e T-bet) das células
auto-reativas pré-transplante para um fenótipo tolerante pós-transplante (caracterizado por elevados
níveis de RNAm de IL-4 e GATA-3).
Dois trabalhos recentes mostraram uma restauração de células T reguladoras (Tregs)
CD4+CD25high após o TACTH. Verda et al. (2005) mostraram um aumento significante do número
absoluto de células T CD4+CD25high em pacientes com doença de Crohn após TCTH não-
mieloablativo. Em outro estudo recente, de Kleer et al. (2005) mostraram também a restauração da
freqüência de células Tregs CD4+CD25high em crianças com artrite juvenil idiopática (AJI) após o
TACTH, de números significantemente reduzidos antes do TACTH (comparando-se aos de indivíduos
saudáveis) para níveis normais após o transplante. Os autores sugerem que esta recuperação deve-

Introdução 27
se à proliferação homeostática das células CD4+CD25high durante os primeiros meses da
reconstituição imunológica, e à produção tímica de novas células Tregs CD4+CD25high que expressam
Foxp3 numa etapa posterior da reconstituição. Estes trabalhos indicam que a restauração das células
Tregs CD4+CD25high poderia contribuir para o restabelecimento da homeostasia imune, indução da
tolerância imune e remissão da DAI após o TACTH.
Os conceitos de homeostasia imune e proliferação homeostática são importantes para o
entendimento dos mecanismos de reconstituição imunológica que serão discutidos nesse trabalho. A
homeostasia imune é um processo auto-regulador que visa à manutenção da estabilidade do sistema
imunológico. Nesse contexto, a homeostasia de células T refere-se à manutenção/preservação do
número de células T ao longo do tempo. Isto pode ser conseguido pela simples sobrevivência das
células ou por um balanço entre morte e proliferação celular. A proliferação homeostática ou
expansão homeostática constitui um dos mecanismos homeostáticos que controlam o tamanho e
composição da população de células T maduras. A proliferação homeostática ocorre como
conseqüência de uma deficiência periférica de linfócitos (linfopenia) em resposta a fatores
homeostáticos, que incluem citocinas (IL-7 e IL-15) e sinais provenientes da estimulação por
complexos MHC-peptídeo próprio (Jameson, 2002; Muraro e Douek, 2006).
Em resumo, os trabalhos recentes descritos acima substanciam a indução da “regeneração” de
um sistema imune novo e tolerante como o racional do TACTH em DAIs, e provêem a base racional
para os ensaios clínicos, futuros ou em andamento, dessa estratégia terapêutica em DAIs.
Além da restauração da auto-tolerância, há a hipótese de que o TACTH poderia também levar
à regeneração de tecidos que são destruídos pela DAI. Teoricamente, isto poderia ser possível pela
regeneração tecidual por células tronco endógenas uma vez que a auto-imunidade fosse revertida, ou
pela diferenciação de células tronco hematopoéticas (CTHs) ou de outras células tronco re-infundidas
no paciente no transplante, em tecidos não-hematopoéticos. Após o TCTH alogênico em
camundongos NOD no início da doença, foi mostrado que ocorre uma regeneração endógena de
ilhotas pancreáticas (Zorina et al., 2003). Foi também mostrado que células tronco transplantadas
promoveram a regeneração endógena pancreática (Mathews et al., 2004; Hess et al, 2003).
Entretanto, estudos em camundongos NOD após TCTH alogênico para diabete já estabelecida
mostraram que a regeneração endógena das ilhotas não consegue manter normoglicemia e sugerem
que o transplante de ilhotas seja necessário para corrigir a doença (Nikolic et al, 2004).

Introdução 28
Recentemente, vários grupos demonstraram que CTHs podem diferenciar-se em células hepáticas,
células do miocárdio, dos rins e em outros tipos de células não-hematopoéticas, incluindo células das
ilhotas pancreáticas (Zulewski et al., 2001; Jiang et al., 2002), mas é possível que estes estudos
reflitam fusão celular ao invés de diferenciação a partir de CTHs infundidas no transplante (Terada et
al., 2002). Portanto, o potencial das CTHs para a regeneração tecidual após o TACTH em pacientes
com DAIs é ainda controverso e consiste uma área de intensa investigação.
1.6 Repertório do receptor de células T em doenças auto-imunes
O receptor de células T (TCR) é um heterodímero de membrana, formado por cadeias α e β
ou por cadeias γ e δ. Cada cadeia α, β, γ ou δ é formada por um domínio variável e um domínio
constante. O TCR αβ é formado por duas cadeias polipeptídicas com domínios variáveis (Vα e Vβ) e
domínios constantes (Cα e Cβ). Análises da estrutura tridimensional do TCR αβ mostraram que os
domínios variáveis Vα e Vβ formam três alças que interagem com o complexo MHC/peptídeo (Garcia
et al., 1996). Estas alças correspondem às Regiões Determinantes de Complementaridade (CDR). Os
genes Vα e Vβ codificam as regiões CDR1 e CDR2, e a região CDR3 é gerada pela recombinação
somática dos segmentos V e J para Vα, e os segmentos V, D e J para Vβ, sendo esta a região de
maior variabilidade e está envolvida no reconhecimento dos peptídeos apresentados pelas moléculas
de MHC (Davies e Bjorkman, 1988; Garcia et al., 1999; Goldrath et al., 1999). A região CDR3 varia
em extensão dependendo da adição ou remoção de nucleotídeos nas ligações VD e DJ durante o
processo de recombinação. Assim, a diversidade do repertório de TCR resulta do rearranjo de
diferentes segmentos gênicos, de sua ligação imprecisa, da adição de nucleotídeos durante o
processo de recombinação e da combinação de diferentes cadeias α e β ou γ e δ (Davis e Bjorkman,
1988). Teoricamente, o limite da diversidade do repertório do TCR seria de mais de 1 x 1013
especificidades. No entanto, não pode haver mais especificidades de TCR do que o número de
células T totais num indivíduo. Assim, foi mostrado que a diversidade em camundongos é de 1-2 x
108 especificidades diferentes, e em humanos é de 1 x 1012 (Nikolich-Zugich et al., 2004).
Atualmente, o repertório do TCR pode ser analisado por citometria de fluxo, pelo uso de
anticorpos monoclonais anti-Vβ ou Vα específicos ou por técnicas de PCR (Reação em Cadeia da
Polimerase) e Real Time PCR. Essas técnicas permitem a identificação e quantificação de famílias

Introdução 29
preferencialmente expandidas. Alternativamente, o repertório do TCR pode ser analisado pela técnica
de Immunoscope ou CDR3 Length Spectratyping. Considerando que o padrão de distribuição dos
picos de CDR3 da cadeia Vβ do TCR numa população policlonal normal de células T segue uma
curva de Gauss, esta técnica permite detectar variações no padrão de distribuição desses segmentos
numa dada população celular. Assim, qualquer expansão clonal anormal no repertório pode ser
detectada como picos que se desviam da distribuição gaussiana normal dos segmentos de CDR3
(Pannetier, 1993; Pannetier, 1995).
Vários estudos indicam que pacientes com DAIs apresentam restrição (skewing) do repertório
de linfócitos T, com expressão anormal de algumas famílias Vβ. Essas anormalidades são causadas
por expansões oligoclonais de algumas populações de células T, que são instáveis e
preferencialmente observadas no início das DAIs. Essas perturbações do repertório de células T
naive podem contribuir para a predisposição do indivíduo ao desenvolvimento da DAI (revisado por
Davidson e Diamond, 2001; Sospedra e Martin, 2005).
Holbrook et al. (1996) mostraram que linfócitos do sangue periférico de pacientes com lúpus
eritematoso sistêmico apresentam restrições do repertório da cadeia Vβ do TCR. Igualmente, em
pacientes com doença de Crohn, as células T encontradas nos segmentos do trato intestinal afetados
apresentam restrição do repertório, mas não as encontradas nos segmentos não-envolvidos (Posnett
et al., 1990).
Mais recentemente, Gran et al (1998) mostraram anormalidades do repertório de células T
isoladas do sangue periférico de pacientes com EM, com expansões significantes das famílias Vβ9,
Vβ1, Vβ11 e Vβ22. Em outro estudo, expansões oligoclonais de células T foram encontradas no
líquido cefalorraquidiano de pacientes com EM (Gestri et al., 2001). Muraro et al. (2002) mostraram
que pacientes com EM-SR apresentam expansões de linfócitos circulantes expressando
determinadas famílias Vβ mais freqüentemente do que em controles normais, além disso 80% essas
expansões são oligoclonais e correlacionam-se significantemente com atividade inflamatória da EM
detectada por RMN e com respostas imunes contra a MBP. Matsumoto et al. (2003) também
mostraram a presença de células T em expansões oligoclonais, particularmente expressando a
família Vβ5.2, no sangue periférico de pacientes com EM. Em outro estudo recente em pacientes com
EM, foi mostrado um aumento da reatividade à mielina e da secreção de IFN-γ por células T CD4+ e T

Introdução 30
CD8+ com distribuição alterada dos picos de CDR3 (Laplaud et al., 2004). Em pacientes com diabete
melito do tipo 1 recém-diagnosticado, Luppi et al (2000) demonstraram uma restrição do repertório do
TCR, com expressão preferencial da família Vβ7.
Interessantemente, Traynor et al. (2002) mostraram, pela primeira vez, que o TACTH poderia
causar mudanças na composição do sistema imune e levar à normalização do repertório de células T
em pacientes com DAIs. Os autores analisaram a diversidade do repertório de células T em pacientes
com lúpus eritematoso sistêmico submetidos à terapia de imunossupressão em altas doses seguida
pelo TACTH, e mostraram que o repertório de células T que era restrito nesses pacientes antes do
transplante apresentou uma distribuição normal (comparável a de um indivíduo controle saudável)
após o TACTH.

2 Objetivos

Objetivos 32
2.1 Objetivo geral
Avaliar a reconstituição imunológica em pacientes com as doenças auto-imunes diabete melito
do tipo 1 e esclerose múltipla, submetidos à terapia de imunossupressão em altas doses seguida pelo
transplante autólogo de células tronco hematopoéticas.
2.2 Objetivos específicos
Avaliar, previamente e seqüencialmente após o transplante:
1. Diversas subpopulações celulares do sistema imune no sangue periférico.
2. O perfil de produção de citocinas intracelulares por linfócitos T do sangue periférico.
3. A diversidade de linfócitos T do sangue periférico, por meio do estudo do repertório da cadeia
Vβ do receptor de células T.
4. A expressão do gene Foxp3 em células mononucleares do sangue periférico.

3 Casuística, Material e Métodos

Casuística, Materiais e Métodos 34
3.1 Delineamento do estudo
Tratou-se um estudo prospectivo conduzido com o objetivo de se estudar a reconstituição
imunológica em pacientes com doenças auto-imunes (diabete melito do tipo 1 e esclerose múltipla)
submetidos ao tratamento com imunossupressão em altas doses seguida de transplante autólogo de
células tronco hematopoéticas. Este trabalho foi desenvolvido no Centro de Terapia Celular (CEPID
FAPESP) do Centro Regional de Hemoterapia do Hospital das Clínicas da Faculdade de Medicina de
Ribeirão Preto da Universidade de São Paulo (HC-FMRP-USP) em colaboração com a Unidade de
Transplante de Medula Óssea (TMO) do HC-FMRP-USP, o Setor de Doenças Neuromusculares e a
Divisão de Endocrinologia e Metabolismo do HCFMRP.
3.2 Casuística
Foram incluídos nesse estudo pacientes com diabete melito do tipo 1 e esclerose múltipla, que
foram submetidos ao tratamento com imunossupressão em altas doses seguida de transplante
autólogo de células tronco hematopoéticas, na Unidade de TMO do Hospital das Clínicas da FMRP-
USP, de dezembro de 2002 a dezembro de 2005. Foram estudados onze pacientes com diabete
melito do tipo 1 e dezoito pacientes com esclerose múltipla transplantados.
As Tabelas 1 e 2 sumarizam as principais características dos pacientes, dados clínicos de
antes do transplante e dados sobre o procedimento do transplante. Os protocolos de transplante
específicos para essas doenças auto-imunes foram aprovados pelo Comitê de Ética do Hospital das
Clínicas da FMRP-USP (Diabetes melito do tipo 1 - Proc. 10095/02, Esclerose múltipla - Proc.
2693/2001) e pela Comissão Nacional de Ética em Pesquisa (Diabetes melito do tipo 1 - RG 7160,
Esclerose múltipla - RG 2944), e os pacientes assinaram o termo de consentimento livre e
esclarecido. Nesses protocolos, estão definidos os critérios de seleção dos pacientes.
Os pacientes com esclerose múltiplos submetidos ao TACTH, apresentavam formas
progressivas da doença (Poser et al, 1983), refratárias ao tratamento com corticosteróides e IFN-β.
Entre os dezoito pacientes, quatorze tinham a forma de esclerose múltipla progressiva secundária
(EM-PS), três apresentavam esclerose múltipla progressiva primária (EM-PP) e uma paciente tinha a
forma surto-remissiva da doença (EM-SR) (Hafler et al., 2004; Compston e Coles, 2002).
Os pacientes com diabete melito do tipo 1 submetidos ao TACTH foram pacientes recém-
diagnosticados. O TACTH foi realizado nesses pacientes dentro de no máximo seis semanas após o

Casuística, Materiais e Métodos 35
diagnóstico. Com exceção do primeiro paciente, que foi diagnosticado após episódio de cetoacidose
diabética, foram excluídos do protocolo todos pacientes que já tinham apresentado episódios de
cetoacidose diabética antes do diagnóstico. Todos os pacientes apresentavam anticorpos anti-GAD65
positivos ao diagnóstico.
O esquema do tratamento de imunossupressão em altas doses seguida de TACTH constituiu-
se de quatro fases: fase de mobilização das células tronco hematopoéticas, fase de condicionamento
(imunossupressão), infusão das células tronco hematopoéticas (ou transplante propriamente dito) e o
seguimento do paciente após o transplante.
Fase de mobilização das células tronco hematopoética s. As células tronco hematopoéticas
foram mobilizadas da medula óssea para o sangue periférico nesses pacientes com ciclofosfamida e
G-CSF (granulocyte-colony stimulating factor). A ciclofosfamida (2 g/m2) foi infundida em duas doses
(12 em 12 horas) e o G-CSF (10 � g/Kg/dia) foi administrado em duas doses (12 em 12 horas), 24
horas após última dose de ciclofosfamida e continuado até o término da coleta das CTH por
leucoaférese. O início da coleta foi determinado pela contagem de leucócitos e de células CD34+ no
sangue periférico: leucócitos >1000/mm3 e células CD34+ >10 células/mm3. A aférese foi realizada em
uma ou duas vezes até atingir o valor mínimo de 3,0 x 106 CD34+/kg de peso. Foram coletadas entre
3,4-25,1 x 106 células CD34+/kg (média 8,5x106/kg) dos pacientes com EM, e entre 5,8-23,1 x 106
células CD34+/kg (média 10.6 x 106/kg) dos pacientes com DM. Após a coleta, as células tronco
hematopoéticas foram criopreservadas em solução crioprotetora contendo 10% de dimetilsulfóxido e
armazenadas em nitrogênio líquido até o momento do transplante.
Fase de condicionamento. Os regimes de condicionamento foram diferentes para as duas
doenças auto-imunes transplantadas (Tabelas 1 e 2 ). Os pacientes com diabete melito do tipo 1
receberam ciclofosfamida e globulina anti-timocitária (ATG, antithymocyte globulin) de coelho. A
ciclofosfamida foi administrada em quatro doses de 50 mg/kg/dia nos dias -6, -5, -4 e -3 antes do
transplante. A ATG de coelho foi administrada na dose de 0,5 mg/dia no dia -6, e na dose de 1 mg/dia
nos dias -5, -4, -3 e -2.
Os onze primeiros pacientes com esclerose múltipla (pacientes 1 - 11) receberam um regime
de condicionamento constituído de poliquimioterapia denominado BEAM (BCNU, 300 mg/m2 no dia -
6; etoposide, 200 mg/m2 nos dias -5 a -2; ARA-C, 200 mg/m2 nos dias -5 a -2; e melphalan, 200
mg/m2 nos dias -5 a -1) associado a ATG de cavalo (15 mg/kg/dia nos dias -5, -3, -1, +1, +2 e +3

Casuística, Materiais e Métodos 36
(Tabela 1 ). O segundo grupo de pacientes com esclerose múltipla (pacientes 12 - 18) foi submetido
ao mesmo regime de condicionamento dos pacientes com diabete melito do tipo 1, constituído de
ciclofosfamida e ATG de coelho.
Infusão das células tronco hematopoéticas. A infusão das células tronco hematopoéticas
ocorreu no dia 0 (data do transplante) e G-CSF (5 � g/Kg/day) foi administrado do dia +5 até que a
contagem de neutrófilos atingisse valor >1,000/mm3. Na maioria do pacientes, foi infundida a
quantidade total de células CD34+ coletada.
A enxertia da medula após o TACTH foi definida como: 1, enxertia de leucócitos: primeiro dia
entre três dias consecutivos em que o número de neutrófilos foi >500 células/µl; 2, enxertia de
plaquetas: primeiro dia em que o número de plaquetas foi >20.000, sem que o paciente tenha
recebido infusão de plaquetas nos últimos 7 dias.
Seguimento do paciente após o transplante. Os pacientes permaneceram internados na
Unidade de TMO do HC-FMRP-USP até a ocorrência da enxertia e em seguimento ambulatorial
intensivo (de diário a semanal) no Hospital-Dia até aproximadamente 60 dias após o transplante.
Após esse período, os pacientes retornaram às suas casas, e compareceram a consultas periódicas
de acompanhamento no ambulatório da Unidade de TMO do HC-FMRP-USP.
Foram colhidas amostras de sangue periférico dos pacientes nos seguintes períodos: pré-
mobilização (ou basal, ou pré-transplante), pré-condicionamento (amostras são colhidas poucas
horas antes do início do condicionamento), e dias D+60, D+100, D+180, D+270, D+360, D+540,
D+720 pós-transplante. As coletas das amostras de sangue periférico foram adaptadas aos retornos
dos pacientes ao hospital após o transplante. Portanto, nem sempre a data das coletas coincidiram
exatamente com as datas programadas citadas anteriormente. Além disso, nem todos os pacientes
possuem amostras coletadas de todos os períodos citados acima, devido principalmente ao estado
clínico do paciente ou ao não comparecimento do paciente aos retornos determinados. O primeiro
paciente com esclerose múltipla (GG) foi transplantado antes do início desse estudo, portanto não
foram coletadas amostras de pré-mobilização, pré-condicionamento e dos primeiros meses após o
TACTH desse paciente.

Casuística, Materiais e Métodos 37
3.3 Controles saudáveis
As amostras de sangue periférico de indivíduos-controle saudáveis foram obtidas de doadores
de sangue de repetição do Centro Regional de Hemoterapia de Ribeirão Preto do Hospital das
Clínicas da FMRP-USP, sob consentimento informado. Foram colhidas amostras de sangue periférico
de 10 doadores com idade menor que 30 anos (Grupo 1; que foi usado para comparação com o
grupo dos pacientes com diabete melito do tipo 1) e de 10 doadores com idade entre 30 - 55 anos
(Grupo 2; que foi usado para comparação com o grupo de pacientes com esclerose múltipla).
3.4 Isolamento das células mononucleares do sangue periférico
Amostras de 50-70 ml sangue periférico de pacientes pré ou pós-TACTH, colhidas na presença
de heparina sódica (14UI/ml), foram separadas por centrifugação em gradiente de densidade, técnica
descrita por Boyum (1974). As amostras foram diluídas 1:2 em tampão fosfato salina (PBS), aplicadas
cuidadosamente sobre o gradiente de Ficoll Hypaque (d=1,077, Amersham-Pharmacia, Uppsala,
Suécia), e centrifugadas a 500g por 30 minutos a temperatura ambiente (TA) para obtenção da
camada de células mononucleares. As células presentes na interfase plasma-Ficoll Hypaque foram
cuidadosamente coletadas e lavadas duas vezes com PBS, ressuspensas em RPMI 1640 (GIBCO-
BRL, Life Technologies, New York, USA) contendo 10% soro bovino fetal (Hyclone, Logan, USA) e
contadas em câmara de Newbauer. Para congelamento, 5x106 células mononucleares foram
ressuspensas em solução de congelamento gelada, constituída de soro bovino fetal (Hyclone, Logan,
USA) + 10% de DMSO (Dimetilsulfóxido, Sigma-Aldrich, Steinhein, Alemanha) gelada. As células
foram aliquotadas em tubos de congelamento de 1 ml gelados e colocadas em freezer a -20°C por 30
minutos, transferidas para freezer -80°C overnight e depois para um tanque de nitrogênio líquido.

Casuística, Materiais e Métodos 38
Tabela 1. Pacientes com esclerose múltipla submetidos ao TACTH.
PPAACCIIEENNTTEE SSEEXXOO// IIDDAADDEE ((AANNOOSS))
TTIIPPOO EEMM
EEDDSSSS PPRRÉÉ--
TTAACCTTHH RRMMNN PPRRÉÉ--TTAACCTTHH RREEGGIIMMEE CCOONNDDIICCIIOONNAAMMEENNTTOO
DDAATTAA TTAACCTTHH
TTEEMMPPOO PPÓÓSS--TTAACCTTHH ((MMEESSEESS))
1. GG M/50 PS 5,5 Ausência BEAM +ATG cavalo 21.08.02 40
2. SAF F/48 PS 6,0 Ausência BEAM +ATG cavalo 29.01.03 OB
3. SSS M/37 PS 6,5 Ausência BEAM +ATG cavalo 19.05.03 31
4. DFG M/51 PS 6,5 Ausência BEAM +ATG cavalo 16.06.03 30
5. ARJTA M/51 PS 6,5 Presença BEAM +ATG cavalo 04.10.03 27
6. SHGE F/42 PS 6,5 Ausência BEAM +ATG cavalo 19.02.04 22
7. RMVL F/37 PS 6,0 Ausência BEAM +ATG cavalo 22.03.04 OB
8. MFM F/42 PS 7,0 ND BEAM +ATG cavalo 02.06.04 18
9. WRS M/21 PP 6,5 Presença BEAM +ATG cavalo 22.07.04 17
10. OLP M/52 PP 6,5 Ausência BEAM +ATG cavalo 09.09.04 15
11. LST M/28 PS 6,5 Ausência BEAM +ATG cavalo 03.11.04 OB
12. SHC M/45 PS 6,5 Ausência Ciclofosfamida + ATG coelho 11.05.05 7
13. MAFC F/48 SR 4,5 Ausência Ciclofosfamida + ATG coelho 10.06.05 6
14. RPS M/54 PP 6,5 Ausência Ciclofosfamida + ATG coelho 21.06.05 6
15. CMBM F/36 PS 6,5 Ausência Ciclofosfamida + ATG coelho 24.08.05 4
16. LFL F/34 PS 6,5 Ausência Ciclofosfamida + ATG coelho 23.08.05 4
17. LBSL F/42 PS 6,0 Ausência Ciclofosfamida + ATG coelho 20.10.05 2
18. CC M/50 PS 6,5 Ausência Ciclofosfamida + ATG coelho 04.11.05 1
TACTH, Transplante autólogo de células tronco hematopoéticas; EM, esclerose múltipla; PS, esclerose múltipla progressiva secundária; PP, esclerose múltipla primária progressiva; SR, esclerose múltipla surto-remissiva; EDSS, Expanded Disability Status Score (Anexo 1 ); RMN, Ressonância Magnética Nuclear; Ausência, ausência de lesões de atividade inflamatória por RMN; Presença, presença de lesões de atividade inflamatória por RMN; BEAM, BCNU, etoposídeo, aracitin, melfalan; ATG, Antithimocyte globulin, G-CFS, Granulocyte-colony stimulating factor; OB, óbito; ND, não determinado. Regime de mobilização: Ciclofosfamida (2g/m2) + G-CSF (10 � g/Kg/dia). Regime de condicionamento: BEAM + ATG cavalo (6x 15mg/kg/dia), ou Ciclofosfamida (4 x 50 mg/Kg/dia) + ATG coelho (4,5 mg).
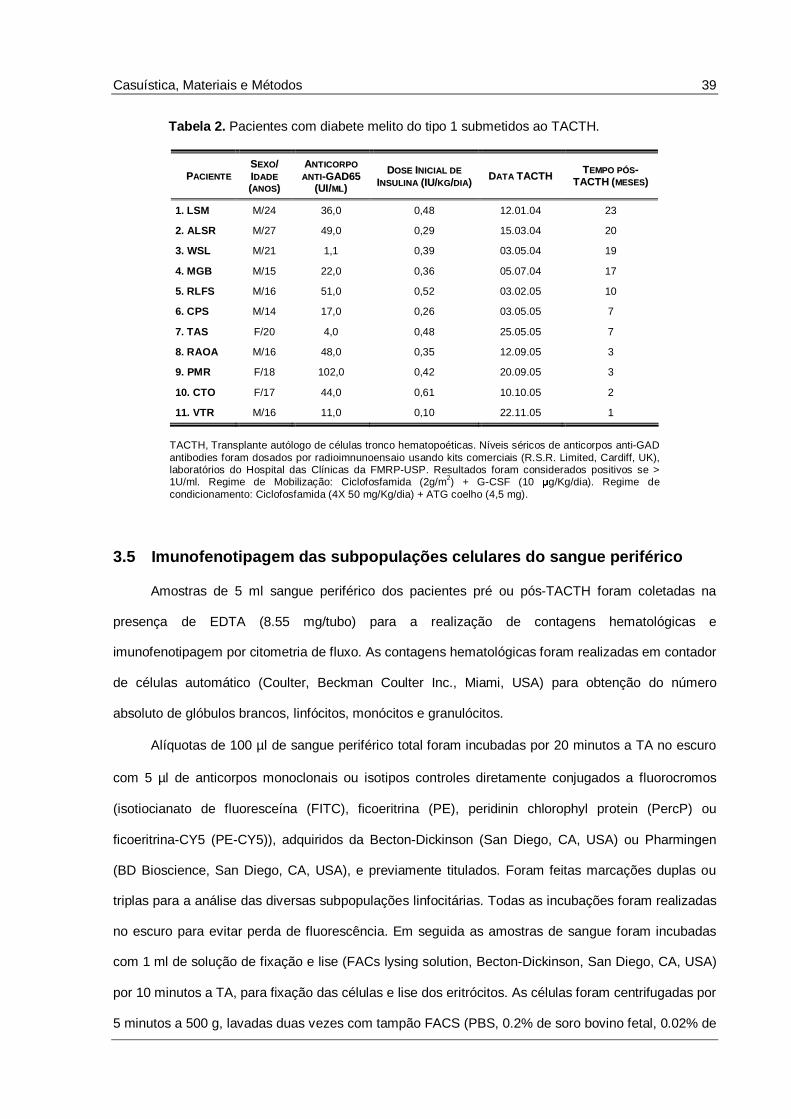
Casuística, Materiais e Métodos 39
Tabela 2. Pacientes com diabete melito do tipo 1 submetidos ao TACTH.
PPAACCIIEENNTTEE SSEEXXOO// IIDDAADDEE ((AANNOOSS))
AANNTTIICCOORRPPOO AANNTTII--GGAADD6655
((UUII//MMLL))
DDOOSSEE IINNIICCIIAALL DDEE IINNSSUULLIINNAA ((IIUU//KKGG//DDIIAA))
DDAATTAA TTAACCTTHH TTEEMMPPOO PPÓÓSS--
TTAACCTTHH ((MMEESSEESS))
1. LSM M/24 36,0 0,48 12.01.04 23
2. ALSR M/27 49,0 0,29 15.03.04 20
3. WSL M/21 1,1 0,39 03.05.04 19
4. MGB M/15 22,0 0,36 05.07.04 17
5. RLFS M/16 51,0 0,52 03.02.05 10
6. CPS M/14 17,0 0,26 03.05.05 7
7. TAS F/20 4,0 0,48 25.05.05 7
8. RAOA M/16 48,0 0,35 12.09.05 3
9. PMR F/18 102,0 0,42 20.09.05 3
10. CTO F/17 44,0 0,61 10.10.05 2
11. VTR M/16 11,0 0,10 22.11.05 1
TACTH, Transplante autólogo de células tronco hematopoéticas. Níveis séricos de anticorpos anti-GAD antibodies foram dosados por radioimnunoensaio usando kits comerciais (R.S.R. Limited, Cardiff, UK), laboratórios do Hospital das Clínicas da FMRP-USP. Resultados foram considerados positivos se > 1U/ml. Regime de Mobilização: Ciclofosfamida (2g/m2) + G-CSF (10 � g/Kg/dia). Regime de condicionamento: Ciclofosfamida (4X 50 mg/Kg/dia) + ATG coelho (4,5 mg).
3.5 Imunofenotipagem das subpopulações celulares do sangue periférico
Amostras de 5 ml sangue periférico dos pacientes pré ou pós-TACTH foram coletadas na
presença de EDTA (8.55 mg/tubo) para a realização de contagens hematológicas e
imunofenotipagem por citometria de fluxo. As contagens hematológicas foram realizadas em contador
de células automático (Coulter, Beckman Coulter Inc., Miami, USA) para obtenção do número
absoluto de glóbulos brancos, linfócitos, monócitos e granulócitos.
Alíquotas de 100 µl de sangue periférico total foram incubadas por 20 minutos a TA no escuro
com 5 µl de anticorpos monoclonais ou isotipos controles diretamente conjugados a fluorocromos
(isotiocianato de fluoresceína (FITC), ficoeritrina (PE), peridinin chlorophyl protein (PercP) ou
ficoeritrina-CY5 (PE-CY5)), adquiridos da Becton-Dickinson (San Diego, CA, USA) ou Pharmingen
(BD Bioscience, San Diego, CA, USA), e previamente titulados. Foram feitas marcações duplas ou
triplas para a análise das diversas subpopulações linfocitárias. Todas as incubações foram realizadas
no escuro para evitar perda de fluorescência. Em seguida as amostras de sangue foram incubadas
com 1 ml de solução de fixação e lise (FACs lysing solution, Becton-Dickinson, San Diego, CA, USA)
por 10 minutos a TA, para fixação das células e lise dos eritrócitos. As células foram centrifugadas por
5 minutos a 500 g, lavadas duas vezes com tampão FACS (PBS, 0.2% de soro bovino fetal, 0.02% de

Casuística, Materiais e Métodos 40
azida sódica), ressuspensas em 200 µl de tampão FACS e analisadas imediatamente no citômetro de
fluxo FACSort (Becton-Dickinson, San Diego, CA, USA).
As seguintes subpopulações linfocitárias foram analisadas: CD3+ (linfócitos T totais),
CD3+CD4+ (linfócitos T helper ou auxiliares), CD3+CD8+ (linfócitos T citotóxicos), CD19+ (linfócitos B),
CD3-CD16+,56+ (células “natural killer”, NK), CD3+CD16+,56+ (células NKT-like), CD3+CD69+,
CD3+HLADR+ e CD3+CD25+ (marcadores de ativação celular em linfócitos T), CD19+CD38+
(marcadores de ativação celular em linfócitos B), CD4+CD45RA+CD31+ (linfócitos T auxiliares virgens,
recém-imigrados do timo), CD4+(CD8+)CD27+CD45RO- (linfócitos T naive), CD4+(CD8+)CD27+
CD45RO+ (linfócitos T de memória central), CD4+(CD8+)CD27-CD45RO+ (linfócitos T de memória
efetora), CD4+(CD8+)CD27-CD45RO- (linfócitos T efetores diferenciados), CD4+CD25high (células T
reguladoras), CD4+CD25highCTLA-4+ (células T reguladoras que expressam CTLA-4 ou CD152),
CD4+CD25highGITR+ (células T reguladoras que expressam GITR), CD3+TCRαβ+ e CD3+TCRγδ+
(expressão de receptor de célula T αβ ou γδ), CD3+CD4+Fas+, CD3+CD8+Fas+, CD3+CD19+Fas+,
CD3+CD4+FasL+, CD3+CD8+FasL+ (expressão de moléculas relacionadas à apoptose, Fas (CD95) e
FasL (CD178) em linfócitos T e B).
Populações de monócitos CD45+CD14+ e CD14+CD25+ também foram analisadas para
verificar e excluir contaminação de monócitos na gate de linfócitos. Foram analisadas também duas
subpopulações de células dendríticas do sangue periférico, as células dendríticas linfóides (LIN-
CD11c-HLADR+) e células dendríticas mielóides (LIN-CD11c+HLADR+). LIN (lineage) corresponde a
uma mistura de anticorpos contra vários antígenos de superfície característicos de algumas linhagens
celulares (os quais são ausentes em células dendríticas): CD3, CD19, CD20, CD14, CD45, CD34,
CD16, CD56. Controles isotípicos (IgG1-FITC, IgG2a-PE e IgG1-PercP) foram incluídos em todos os
experimentos para determinação de marcações inespecíficas.
Durante a aquisição das células, foi desenhada uma gate na população de linfócitos (R1),
estabelecida com base nos parâmetros de tamanho (FSC) e granularidade (SSC). Foram adquiridos
20.000 eventos/amostra. As análises foram realizadas utilizando-se o software Cellquest (Becton-
Dickinson, San Diego, CA, USA). As células mortas foram excluídas por tamanho e granularidade. As
diversas subpopulações celulares foram analisadas usando-se dois ou três tipos de fluorocromos. No
caso de marcações duplas, os eventos da gate de linfócitos (R1) foram analisados para marcação
com os diferentes anticorpos por dot plots de fluorescência 1 (FL1, FITC - isotiocianato de

Casuística, Materiais e Métodos 41
fluoresceína) versus fluorescência 2 (FL2, PE - ficoeritrina). Ou então, em outro tipo de análise de
marcação dupla, foi selecionada uma determinada subpopulação por uma gate R2 (por exemplo,
FL1), e a marcação de fluorescência 2 (FL2, PE) nessa gate R2 foi representada por um histograma
de FL2.
No caso de marcações triplas, foram feitos dot plots de FL1 versus FL2, em seguida foi
desenhada uma gate R2 na população duplo-positiva. A marcação de fluorescência 3 (FL3, PercP ou
PE-CY5) nessa gate R2 foi representada por um histograma de FL3. Ou então, em outro tipo de
análise de marcações triplas, foi selecionada uma determinada subpopulação por uma gate R2 (por
exemplo, FL1), e foram feitos dot plots de FL1 versus FL2 para análise dos eventos da gate R2.
Para análise das subpopulações de células dendríticas do sangue periférico, foi feito um dot
plot de FL3 versus FL2 (HLADR-Percp versus LIN-FITC), em seguida foi desenhada uma gate R1 na
população de células HLADRhighLIN-. Em seguida, a marcação de fluorescência na gate R1 foi
analisada por um dot plot de FL3 versus FL2 (HLADR-Percp versus CD11c-PE). As células
dendríticas linfóides são LIN-CD11c-HLADR+ (gate R3) e as células dendríticas mielóides são LIN-
CD11c+HLADR+ (gate R2).
Os resultados das análises de imunofenotipagem foram expressos em porcentagem de células
positivas ou valores absolutos que foram calculados multiplicando-se o valor da porcentagem obtida
pelo número de linfócitos/µl, que foi determinado pelas contagens hematológicas da amostra de
sangue periférico. Exemplos dessas análises de experimentos de imunonofenotipagem por citometria
de fluxo encontram-se representadas no Apêndice A.
3.6 Detecção de citocinas intracelulares em linfóci tos T ativados
A produção de citocinas por linfócitos T CD4 (CD3+CD8-) ou CD8 (CD3+CD8+) ativados foi
detectada por citometria de fluxo, realizando-se marcação intracelular das citocinas produzidas pelos
linfócitos após ativação com um estímulo policlonal (Jung et al., 1993; Masher et al., 1999). A
padronização do ensaio (concentração dos reagentes, tempo de incubação) foi feita utilizando-se
células de indivíduos-controle saudáveis. Células mononucleares do sangue periférico foram isoladas
e criopreservadas como descrito anteriormente. Para o ensaio de produção de citocinas intracelulares
as células mononucleares foram descongeladas, lavadas duas vezes com RPMI 1640 (GIBCO-BRL,
Life Technologies, New York, USA) contendo 10% de soro bovino fetal (Hyclone, Logan, USA) e

Casuística, Materiais e Métodos 42
ressuspensas nesse meio de cultura (2x106 células/ml). Em seguida, as células foram estimuladas
com 25 ng/ml de PMA (forbol-miristato-acetato; Sigma-Aldrich, Steinhein, Alemanha) e 1 µg/ml de
Ionomicina (Sigma-Aldrich, Steinhein, Alemanha), na presença ou não de Brefeldina A (Sigma-
Aldrich, Steinhein, Alemanha) por 6 horas. A Brefeldina A (10 µg/ml) foi adicionada à cultura nas
últimas 4 horas do ensaio. A Brefeldina A é um metabólito fúngico que interfere com o transporte
vesicular do retículo endoplasmático rugoso para o complexo de Golgi, servindo como um inibidor do
transporte de proteínas, e desse modo promovendo o acúmulo das citocinas sintetizadas de novo no
complexo de Golgi.
Foram feitos três tipos de amostras: 1, controle não-estimulado (células + Brefeldina A), usado
para determinar o nível de síntese residual de citocinas devido à ativação in vivo; 2, amostra
estimulada (células + Brefeldina A + PMA + Ionomicina); 3, controle da ativação (células + PMA +
Ionomicina), usado para determinar o nível de ativação celular por meio da expressão do marcador de
ativação precoce CD69 (Nakamura et al., 1989), cuja expressão de superfície é significantemente
inibida na presença de Brefeldina A. O cálculo da resposta específica das células ao estímulo
policlonal foi calculado da seguinte maneira: (AE - ICAE) - (CNE - ICCNE), onde AE = amostra
estimulada; ICAE = isotipo controle da amostra ativada; CNE = controle não-estimulado; ICCNE =
isotipo controle do controle não-estimulada.
Após incubação de 6 horas com os estímulos, as células (0,5x106 células/amostra) foram
incubadas com 200 µl de 0.5 M EDTA por 15 minutos a TA, e em seguida marcadas com anticorpos
monoclonais, previamente titulados, contra os antígenos de superfície (CD3/CD8 ou CD3/CD69) ou
isotipos controles, por 20 minutos a TA. Os anticorpos são diretamente conjugados a fluorocromos, e
foram adquiridos da Becton-Dickinson (San Diego, CA, USA) ou Pharmingen (BD Biosciences, San
Diego, CA, USA). Todas as incubações foram realizadas no escuro para evitar perda de
fluorescência. Após a marcação, as células foram lavadas com tampão FACS, centrifugadas por 5
minutos a 500 g, ressuspensas em 1 ml de solução de fixação e lise (FACs lysing solution, BD, San
Diego, CA, USA), incubadas por 10 minutos a TA e congeladas a -80°C overnight. Em seguida, as
células rapidamente descongeladas, centrifugadas por 5 minutos a 500g, ressupensas em 500 µl de
solução de permeabilização (FACS permeabilizing solution, BD, San Diego, CA, USA) e incubadas
por 10 minutos a TA. Após lavagem com tampão FACS, as células foram incubadas com anticorpos
anti-citocinas (IL-2, IFN-γ, TNF-α, IL-4, IL-5 e IL-10) ou isotipos controles intracelulares diretamente

Casuística, Materiais e Métodos 43
conjugados a fluorocromos por 30 minutos a TA. As células foram então lavadas novamente,
ressuspensas em 200 µl de tampão FACS e analisadas no citômetro de fluxo FACSort (Becton-
Dickinson, San Diego, CA, USA). Controles isotípicos (IgG1-FITC, IgG2a-PE e IgG1-Percp) foram
incluídos em todos os experimentos para determinação de marcação inespecífica.
Durante a aquisição das células, foi desenhada uma gate na população de linfócitos (R1),
estabelecida com base nos parâmetros de tamanho (FSC) por granularidade (SSC). Foram
adquiridos 20.000 eventos/amostra. As análises foram realizadas utilizando-se o software Cellquest
(Becton-Dickinson, San Diego, CA, USA). Os eventos da gate de linfócitos (R1) foram analisados por
dot plots de fluorescência 1 (FL1, anti-CD8-FITC) versus fluorescência 3 (FL3, anti-CD3-PercP). Em
seguida, foram desenhadas uma gate R2 na população duplo-positiva (CD3+CD8+, de linfócitos T
CD8+) e uma gate R3 na população CD3+CD8- (que contém linfócitos T CD4+ em sua grande
maioria). Esse tipo de marcação foi escolhido pois a expressão da molécula CD4 é drasticamente
diminuída em linfócitos ativados com PMA e ionomicina. A expressão de CD8 também diminui, no
entanto em proporções bem menores. A marcação de fluorescência 2 (FL2, anti-citocinas-PE) na gate
R2 foi representada por um histograma de FL2. Exemplos dessas análises de experimentos de
citocinas intracelulares por citometria de fluxo encontram-se representadas no Apêndice B.
3.7 Extração de RNA pelo método de Trizol
A extração do RNA das células mononucleares do sangue periférico foi realizada pelo método
descrito por Chomzynski et al. (1987), o qual utiliza solução de Trizol (GIBCO BRL Life Technologies,
Grand Island, NY, USA) para isolamento de RNA total. Trizol é uma solução monofásica de fenol e
isotiocianato de guanidina que rompe a célula mantendo a integridade do RNA. Partindo de uma
suspensão de aproximadamente de 5x106 células mononucleares resuspensas em 200 µl de solução
fisiológica previamente tratada com DEPC (Dietilpirocarbonato; Sigma-Aldrich, Steinhein, Alemanha),
adicionou-se 1ml de Trizol. Esta solução foi homogeneizada e então incubada por 5 minutos a
temperatura ambiente, para permitir completa dissociação de complexos de nucleoproteínas. Em
seguida adicionou-se 0,2 ml de clorofórmio para cada ml de trizol, seguido de agitação vigorosa por
aproximadamente 15 segundos. O material foi posteriormente incubado por 2 a 3 minutos à
temperatura ambiente e centrifugado por 15 minutos a 1120 g a 4oC.

Casuística, Materiais e Métodos 44
Após esta centrifugação ocorre a separação da solução em três fases (aquosa, interface e
orgânica). A fase aquosa foi transferida para um novo tubo contendo 0,5 ml de álcool isopropílico
para cada ml de trizol, para a precipitação do RNA. Este tubo foi homogeneizado por inversão,
seguido de incubação por 10 minutos ou overnight a -80°C e então centrifugado novamente a 1120 g
por 15 minutos a 4oC. O sobrenadante foi removido e o “pellet” de RNA lavado com etanol 75%
diluído em água DEPC seguido de centrifugação a 640 g por 5 minutos a 4oC. O RNA assim obtido foi
deixado secar por 5 minutos para evaporação do etanol e então ressuspendido em 10µl de água
DEPC. Este material foi mantido a -80oC até o momento da transcrição. Após extração, o RNA obtido
foi quantificado através de leitura em espectrofotômetro (Beckman, DU530, Fullerton, CA, USA) nos
comprimentos de onda (λ) de 260 e 280 nm. O grau de pureza da amostra foi verificado através da
análise da relação entre 260 e 280nm, sendo considerada uma boa extração aquela que apresentou
valores entre 1,8 a 2,0. Para o cálculo da concentração da amostra considerou-se que a densidade
ótica (DO) igual a 1 corresponde a 40 µg de RNA /ml no comprimento de onda de 260 nm.
3.7.1 Eletroforese de amostras de RNA em gel de aga rose sob condições
desnaturantes
Após a quantificação das amostras de RNA foi preparado um o gel sob condições
desnaturantes para visualização da integridade do RNA. Um volume total de 70 ml de agarose
fundida foi misturado a 20 ml de formaldeído 37% e 22 ml de tampão de migração MOPS 5X (20,6g
de MOPS dissolvidos em 800 mL de acetato de sódio 50mM, pH 7,0 ajustado com NaOH 2N e
adicionado 10 ml de EDTA 0,5M pH 8,0; volume final ajustado para 1000 ml). Durante o tempo de
solidificação do gel, as amostras de RNA (4,5 µl de solução de RNA, com cerca de 3 µg RNA) foram
desnaturadas em 2,0 µl de tampão MOPS 5X, 3,5 µl de formaldeído 37% (Merck, Alemanha) e 10,0 µl
de formamida, por 15 minutos a 65°C. Após esse tempo, as amostras foram colocadas
imediatamente no gelo, sendo adicionados 1,0 µl de brometo de etídeo diluído 3:1 (solução estoque
10mg/mL) e 2,0 µl de dye loading solution (1/10 do volume). Após eletroforese a 80 V durante 90
minutos, as bandas de RNA foram visualizadas em transiluminador UV.

Casuística, Materiais e Métodos 45
3.8 Transcrição reversa
Após constatação da qualidade e pureza do RNA, a transcrição reversa do RNA para cDNA foi
realizada utilizando um kit comercial (Superscript III First Strand Synthesis System for RT-PCR,
Invitrogen, Carlsbad, CA, USA). Foram adicionados num tubo de 0,2 ml: 2µg de RNA total (em 2µl),
1µl de oligo(dT)20 (50 µM/µl), 1µl 10 mM dNTP mix e água DEPC q.s.p. 8 µl. Esta mistura foi incubada
a 65oC por 5 minutos em termociclador para remoção de estruturas secundárias, e então colocada
em gelo por pelo menos 1 minuto. Em seguida foi preparada a mistura de síntese de cDNA: 2 µl
tampão para PCR 10X (200 mM Tris-HCl pH 8.4, 500 mM KCl), 4 µl 25 mM MgCl2, 2 µl 0,1 M DTT,
1µl RNaseOUTTM (40U/µL), e 1 µl da enzima transcriptase reversa SuperScript III RT (200 U/µl).
Foram adicionados 10 µl dessa mistura de síntese à mistura de RNA/primer. Os componentes da
reação foram gentilmente misturados e centrifugados brevemente, incubados por 50 minutos a 50°C
e a reação foi terminada a 85°C por 5 minutos e colocada em gelo. Em seguida, o tubo de reação foi
centrifugado brevemente e incubado por 20 min com 1 µl de RNase H, e o cDNA foi estocado a -
20oC.
3.8.1 Validação da transcrição
O gene da β-actina tem sido comumente utilizado como controle da reação de transcrição do
RNA em cDNA. Para esta reação foram utilizados 2 µl do cDNA transcrito, 2,5 µl de tampão para
PCR 10X, 1,0 µl de 1,5 mM MgCl2, 3 µl de 10 mM dNTP mix, 1 µl de cada primer (10 µM), 0,3 µl de
Taq polimerase (5 unidades/µl) e água DEPC q.s.p. 25µl. Os reagentes foram misturados e
centrifugados brevemente, colocados em termociclador pré-aquecido a 94oC, e submetidos a um
passo de desnaturação inicial por 2 minutos a 94oC, seguido por 30 ciclos de PCR (desnaturação a
94oC por 40 segundos, anelamento a 55oC por 1 minuto e extensão a 72oC por 1 minuto,
completados por uma extensão final de 10 minutos a 72oC). Em seguida, o produto da reação foi
mantido a 4°C e carregado em gel de agarose 1,5% corado com brometo de etídeo (0,5µg/ml) e
submetido à corrida eletroforética a 100 V durante 30 minutos. O tamanho esperado do produto de
PCR é de 353 pb.

Casuística, Materiais e Métodos 46
3.9 Método de TCRBV CDR3 Spectratyping
Este método permite analisar a diversidade do repertório de linfócitos T e detectar de
populações celulares em expansão clonais (Pannetier et al., 1993; Pannetier et al.,1995). Esta técnica
é iniciada por uma amplificação por PCR dos segmentos Vβ-Cβ do TCR utilizando primers
específicos para cada uma das famílias Vβ. O produto desta reação é submetido à outra amplificação
por PCR que inclui um primer adicional marcado com fluorocromo. Nesta reação amplifica-se o
segmento gênico que contém a região CDR3 (do inglês Complementarity-Determining Regions) do
TCR, uma vez que esta região está localizada entre os segmentos V-D-J. Em seguida, estes
segmentos amplificados por PCR são separados em gel de seqüenciamento e, com o auxílio de um
seqüenciador automático e do software Gene MapperTM (Applied Biosystem), é possível calcular o
tamanho (em pares de bases) dos diferentes segmentos (picos) de CDR3, o número de picos de
CDR3, estimar a freqüência de cada família Vβ no repertório do TCR, e analisar a clonalidade e
distribuição (gaussiana ou não-gaussiana) dos picos na região CDR3. Um esquema do método de
TCRBV CDR3 Spectratyping é mostrado na Figura C.1 (Apêndice C ).
3.9.1 Reação de PCR (V ββββ-Cββββ) para determinação das famílias V ββββ do Receptor de
Células (TCR)
Esta reação de PCR possibilita a amplificação do segmento gênico Vβ-Cβ do TCR. Para um
volume final de reação de 50µl, foram utilizados 4% de cDNA transcrito, tampão 10X (500mM KCl e
100mM Tris-HCl), 2mM MgCl2, 200µM dNTP, 0,5µM do primer CB e 0,5µM do primer � Vβ específico,
1 U Taq polimerase e água qsp 50µl. Esta reação foi processada em termociclador por 1 minuto e 30
segundos a 94oC, seguidos de 40 ciclos de 30 segundos a 94o C, 45 segundos a 60o C, 45 segundos
a 72oC, completados com uma extensão final de 4 minutos a 72o C. A Tabela 3 mostra a seqüência
dos primers utilizados para a amplificação das 24 famílias Vβ, bem como o tamanho esperado do
produto de PCR. O produto de amplificação foi carregado em gel de agarose 1,5% corado com
brometo de etídeo (0,5µg/ml) e submetido à corrida eletroforética de 100 V por 1 h 30 min.

Casuística, Materiais e Métodos 47
3.9.2 Reação de elongação V ββββ-Cββββ (Run-off )
Inicialmente, foi realizada uma reação de PCR amplificando o segmento gênico Vβ-Cβ do TCR
conforme protocolo descrito acima e o produto de PCR assim gerado foi utilizado para a reação de
elongação. Esta reação foi realizada utilizando primer Cβ interno marcado com fluorocromo (FAM) [5´
CAC AGC GAC CTC GGG TGG G 3´]. Para a reação de elongação utilizou-se tampão 10x contendo
500mM KCl e 100mM Tris-HCl, 3mM MgCl2, 200µM dNTP, 0,5µM primer CB marcado, 0,2U Taq
polimerase e água qsp 8µL, seguido da adição separadamente de 2µL do produto de PCR de cada
uma das 24 famílias Vβ. Esta reação foi então processada em termociclador por 1 minuto e 30
segundos a 94oC, seguidos de 8 ciclos de 30 segundos a 94oC, 45 segundos a 60oC, 45 segundos a
72oC, completados com uma extensão final de 45 minutos a 72oC.
3.9.3 Preparo do gel de seqüenciamento
A análise do tamanho (pares de bases) dos diferentes segmentos (picos) de CDR3
amplificados pelas reações de PCR foi realizada em gel de seqüenciamento (6% de poliacrilamida, 8
M de uréia e 1x tampão Tris-Borato-EDTA (TBE)). As placas de vidro foram lavadas com detergente
não abrasivo e enxaguadas com água corrente e depois com água destilada. Antes do acoplamento
das placas no suporte de migração, as mesmas foram limpas novamente com água destilada quente
para completa remoção de resíduos. Para uma solução de 50 ml de gel foram pesados 18g de uréia e
1,3 a 1,4g da resina Amberlite (Sigma-Aldrich, Steinhein, Alemanha), acrescidos de 24,5 ml de água
destilada e 5,2 ml de solução 40% de acrilamida/bis-acrilamida 19/1 (BioRad, Hercules, CA, USA).
Esta solução foi colocada em agitação leve até dissolução completa da uréia. Em seguida, esta
solução foi filtrada em filtro de 0,22 µm contendo em sua parte inferior 5 ml de tampão Tris-Borato-
EDTA (TBE 10x). O volume da solução foi completado para 50 ml com água destilada. Por último,
acrescentou-se 250 µl de solução de persulfato de amônia 10% e 35µl de TEMED (Amresco, Solon,
Ohio, USA) e homogeneizou-se lentamente. Com o auxílio de uma seringa, o gel foi carregado nas
placas de vidro e utilizado após completa polimerização (aproximadamente 1h).

Casuística, Materiais e Métodos 48
Tabela 3. Seqüência dos primers utilizados na amplificação das famílias Vβ do TCR
FFAAMMÍÍLLIIAA
VVBB SSEEQQÜÜÊÊNNCCIIAASS DDOOSS PPRRIIMMEERRSS VVBB--CCBB
((PPAARREESS DDEE BBAASSEESS))
EESSPPEECCIIFFIICCIIDDAADDEESS AAMMPPLLIIFFIICCAADDAASS
VB1 5’ CCGCACAACAGTTCCCTGACTTGC 3’ 322 VB1S1
VB2 5’ GGCCACATACGAGCAAGGCGTCGA 3’ 360 VB2S1, S2
VB3 5’ CGCTTCTCCCTGGATTCTGGAGTCC 3’ 299 VB3S1
VB4 5’ TTCCCATCAGCCGCCCAAACCTAA 3’ 325 VB4S1
VB5 5’ AAGCTCTGAGCTGAATGTGAACGCC 3’ 299 VB5S1, S2, S3, S5, S6, S7, S8
VB6 5’ CTCTGAAGATCCAGCGCACAGAGC 3’ 292 VB6S1, S3, S4, S5, S7, S11, S14
VB7 5’ CCTGAATGCCCCAACAGCTCTCTC 3’ 320 VB7S1, S2, S3
VB8 5’ CCATGATGCGGGGACTGGAGTTGC 3’ 406 VB8S1, S2, S3
VB9 5’ TTCCCTGGAGCTTGGTGACTCTGC 3’ 279 VB9S1, S2
VB10 5’ CCACGGAGTCAGGGGACACAGCAC 3' 277 VB10S1
VB11 5’ TGCCAGGCCCTCACATACCTCTCA 3’ 276 VB11S1
VB12 5’ TGTCACCAGACTGGGAACCACCAC 3’ 449 VB12S2, S3, S4
VB13 5’ CACTGCGGTGTACCCAGGATATGA 3’ 456 VB13S1, S2, S3, S4, S6, S7, S8, S9
VB14 5’ GGGCTCGGCTTAAGGCAGACCTAC 3’ 398 VB14S1
VB15 5’ CAGGCACAGGCTAAATTCTCCCTG 3’ 311 VB15S1
VB16 5’ GCCTGCAGAACTGGAGGATTCTGG 3’ 279 VB16S1
VB17 5’ TCCTCTCACTGTGACATCGGCCCA 3’ 295 VB17S1
VB18 5’ CTGCTGAATTTCCCAAAGAGGGCC 3’ 322 VB18S1
VB19 5’ TCTCAATGCCCCAAGAACGCACCC 3’ 320 VB19S1
VB20 5’ TGCCCCAGAATCTCTCAGCCTCCA 3’ 337 VB20S1
VB21 5’ TCCAGCCTGCAAAGCTTGAGGACT 3’ 283 VB21S1, S3, S4
VB22 5’ GATCCGGTCCACAAAGCTGG 3’ 287 VB22S1
VB23 5’ TGAACTGAACATGAGCTCCTTGG 3’ 295 VB23S1
VB24 5’ GACATCCGCTCACCAGGCCTG 3’ 288 VB24S1
CB 5’ CGGGCTGCTCCTTGAGGGGCTGCG 5’
3.9.4 Preparo das amostras e aplicação no gel de se qüenciamento
O produto de PCR da reação de run-off foi diluído (1:10) em água. Em seguida 2µl desta
diluição, acrescidos de 1� µl de marcador de peso molecular (Gene Scan™ - 500 Rox standard,
Applied Biosystems) e 2µl de tampão para carregar amostra (loading buffer - formamida, acrescida de
blue dextan, 50 mg/ml e EDTA, 25 mM na proporção 5:1). Esta solução foi colocada em termociclador

Casuística, Materiais e Métodos 49
a 950C por 2 minutos para completa desnaturação do produto de PCR. As amostras foram mantidas
em gelo até o momento da aplicação no gel de seqüenciamento, no qual foram carregados 5µl de
amostra/poço. Os produtos de PCR da reação de run-off foram submetidos à corrida eletroforética por
2 h 30 min em um seqüenciador de DNA automático (ABI 377 Prism DNA Sequencer, Applied
Biosystem, Foster City, CA, USA).
3.9.5 Perfil da região CDR3 do RTC, cálculo do tama nho da região CDR3 do RTC e da
freqüência das famílias V ββββ, pelo método de TCRBV CDR3 Spectratyping
Considerando que a região CDR3 está compreendida entre os resíduos de aminoácidos 95-
106 da cadeia variável β do TCR (Pannetier et al., 1993; Pannetier et al., 1995) e que a posição dos
primers VB e CB utilizados são fixas, o tamanho observado para o produto de PCR VB-CB marcado
depende do tamanho da junção V(D)J de cada TCR específico. O tamanho (em pares de bases) do
segmento CDR3 com 10 resíduos de aminoácidos para cada família Vβ foi estimado previamente
(Pannetier et al., 1993; Pannetier et al., 1995), como mostrado na Tabela 4 .
Estes valores são utilizados para o cálculo de cada segmento de CDR3 pelo software Gene
Mapper™ (Applied Biosystem, Foster City, CA, USA), sendo comparados com um marcador de peso
molecular conhecido (Gene Scan™ - 500 Rox standard, Applied Biosystems, Foster City, CA, USA)
que é carregado juntamente com as amostras a serem analisadas na corrida eletroforética. Este
marcador de peso molecular é conjugado com o fluorocromo denominado “Rox”, diferente do
fluorocromo denominado “Fam” utilizado na reação de run-off para os segmentos VB-CB. Isto
possibilita a discriminação de diferentes tamanhos (em pares de bases) detectados pelos raios lasers
emitidos pelo seqüenciador automático. Como mencionado anteriormente, o pico correspondente a
10 resíduos de aminoácidos é definido por um número de pares de bases conhecido. Assim, como
cada 3 pares de base correspondem a 1 resíduo de aminoácido, é possível portanto definir o número
de aminoácidos de cada segmento de CDR3 para uma determinada família Vβ. No entanto, a
definição exata do tamanho e seqüência dos segmentos de CDR3 é possível somente após o
seqüenciamento desta região.
A distribuição dos segmentos de CDR3 pode apresentar um perfil policlonal normal com
distribuição como uma Curva de Gauss, com segmentos variando de 4 a 16 resíduos de aminoácidos,
sendo os tamanhos de 8 a 10 resíduos de aminoácidos os mais freqüentes. Ou ainda, pode
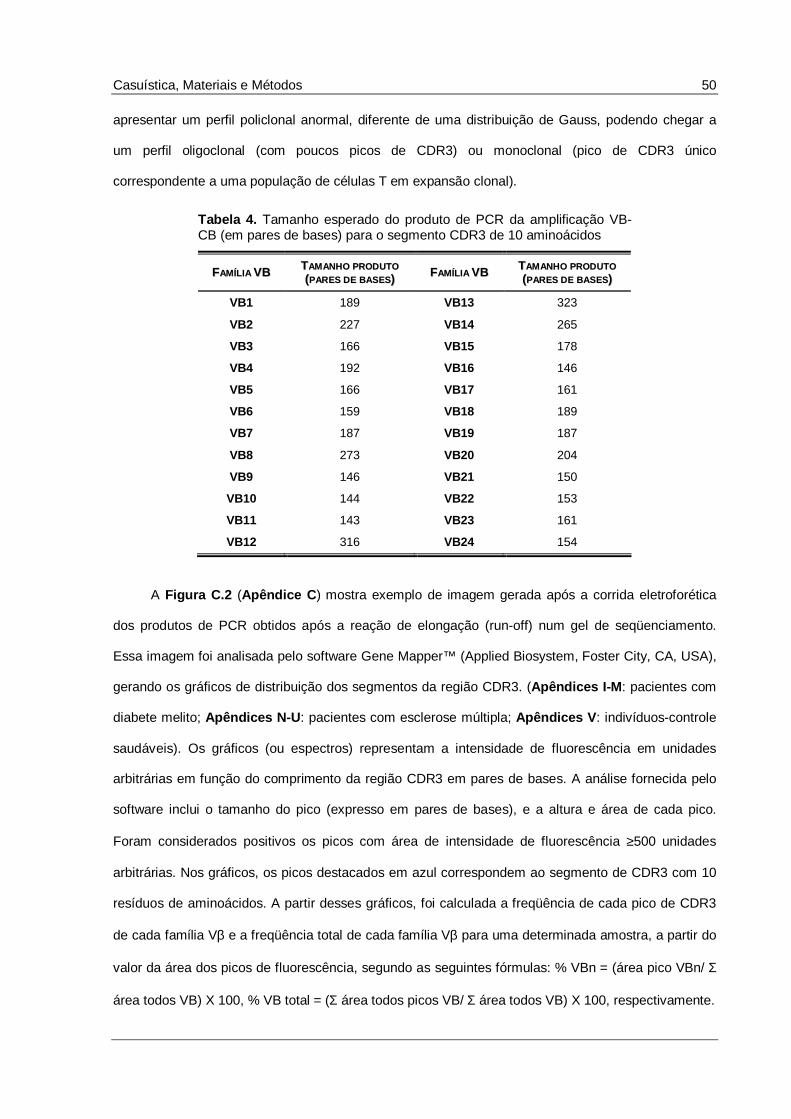
Casuística, Materiais e Métodos 50
apresentar um perfil policlonal anormal, diferente de uma distribuição de Gauss, podendo chegar a
um perfil oligoclonal (com poucos picos de CDR3) ou monoclonal (pico de CDR3 único
correspondente a uma população de células T em expansão clonal).
Tabela 4. Tamanho esperado do produto de PCR da amplificação VB-CB (em pares de bases) para o segmento CDR3 de 10 aminoácidos
FFAAMMÍÍLLIIAA VVBB TTAAMMAANNHHOO PPRROODDUUTTOO
((PPAARREESS DDEE BBAASSEESS)) FFAAMMÍÍLLIIAA VVBB TTAAMMAANNHHOO PPRROODDUUTTOO
((PPAARREESS DDEE BBAASSEESS))
VB1 189 VB13 323
VB2 227 VB14 265
VB3 166 VB15 178
VB4 192 VB16 146
VB5 166 VB17 161
VB6 159 VB18 189
VB7 187 VB19 187
VB8 273 VB20 204
VB9 146 VB21 150
VB10 144 VB22 153
VB11 143 VB23 161
VB12 316 VB24 154
A Figura C.2 (Apêndice C ) mostra exemplo de imagem gerada após a corrida eletroforética
dos produtos de PCR obtidos após a reação de elongação (run-off) num gel de seqüenciamento.
Essa imagem foi analisada pelo software Gene Mapper™ (Applied Biosystem, Foster City, CA, USA),
gerando os gráficos de distribuição dos segmentos da região CDR3. (Apêndices I-M : pacientes com
diabete melito; Apêndices N-U : pacientes com esclerose múltipla; Apêndices V : indivíduos-controle
saudáveis). Os gráficos (ou espectros) representam a intensidade de fluorescência em unidades
arbitrárias em função do comprimento da região CDR3 em pares de bases. A análise fornecida pelo
software inclui o tamanho do pico (expresso em pares de bases), e a altura e área de cada pico.
Foram considerados positivos os picos com área de intensidade de fluorescência ≥500 unidades
arbitrárias. Nos gráficos, os picos destacados em azul correspondem ao segmento de CDR3 com 10
resíduos de aminoácidos. A partir desses gráficos, foi calculada a freqüência de cada pico de CDR3
de cada família Vβ e a freqüência total de cada família Vβ para uma determinada amostra, a partir do
valor da área dos picos de fluorescência, segundo as seguintes fórmulas: % VBn = (área pico VBn/ Σ
área todos VB) X 100, % VB total = (Σ área todos picos VB/ Σ área todos VB) X 100, respectivamente.

Casuística, Materiais e Métodos 51
Em nosso trabalho, a diversidade do repertório da cadeia Vβ do TCR foi analisada com base no
complexity score descrito por Wu et al. (2000). O complexity score é um sistema de pontuação que
quantifica as mudanças no repertório Vβ do TCR ao longo do tempo.
De acordo com esse sistema, primeiramente a complexidade dentro de cada família Vβ é
determinada pela contagem do número de picos. Então as famílias são classificadas com uma
pontuação de 0 a 8, baseada no grau de complexidade (ou diversidade). A complexidade normal é
caracterizada por uma distribuição gaussiana dos diferentes tamanhos de transcritos para uma
determinada família, e pela quantidade de 8-10 picos para cada família Vβ. A pontuação 0 é dada se
a família é ausente, a pontuação 1 é atribuída se a família Vβ apresentar somente um único pico
monoclonal, a pontuação 2 é dada se a família apresentar um perfil biclonal, a pontuação 3 é dada se
a família apresentar 3 picos, e assim por diante. Finalmente, a pontuação 8 é atribuída a um
spectratype de 8 picos ou mais, com uma aparência diversa, complexa e policlonal. Assim, o
complexity score geral é calculado pela soma dos complexity scores de cada família Vβ. No nosso
trabalho, o complexity score máximo é de 192 (8 X 24 famílias Vβ).
3.10 Análise da expressão de Foxp3 por Real Time RT- PCR
A análise da expressão do gene Foxp3 nas células mononucleares do sangue periférico dos
pacientes, antes e após o transplante, foi feita por Real Time RT-PCR. As reações foram realizadas
em placas de 96 poços, utilizando os reagentes SYBR-Green PCR Master Mix (Applied Biosystems,
Foster City, CA, USA) e o equipamento 7500 Real Time PCR system (Applied Biosystems, Foster
City, CA, USA). O RNA foi extraído das células mononucleares dos pacientes pelo método de Trizol e
o cDNA foi sintetizado e validado para a amplificação do gene da β-actina. O gene endógeno GAPDH
(Glyceraldehyde-3-phosphate dehydrogenase) foi utilizado como gene de referência para normalizar
as reações de PCR para a quantidade e qualidade de RNA total usada nas reações de transcrição
reversa.
A determinação da intensidade de fluorescência na reação foi feita pelo cálculo do ∆Rn (∆Rn =
Rn+ - Rn-), onde Rn+ = intensidade de emissão do SYBR-Green intensidade de emissão do ROX em
um dado momento da reação, e Rn- = intensidade de emissão do SYBR-Green intensidade de
emissão do ROX, antes da amplificação. O composto ROX é utilizado como controle interno passivo,
pois a fluorescência que emite tem intensidade constante durante toda a reação, enquanto que o a

Casuística, Materiais e Métodos 52
fluorescência emitida pelo SYBR-Green aumenta à medida que este se liga nas duplas fitas de DNA.
Durante os ciclos iniciais da reação, não há acúmulo de produtos de amplificação e os valores de ∆Rn
permanecem na linha de base (fluorescência do ROX > SYBR-Green). Na fase logarítmica da reação
ocorre acúmulo dos produtos de amplificação e a ∆Rn ultrapassa a linha de base. Para a
quantificação relativa, após a reação foi estabelecido um valor de ∆Rn, que é uma linha de corte
(threshold) para cada curva de amplificação de um dado par de primers, definida acima da
fluorescência inespecífica. O número do ciclo em que a ∆Rn da amostra cruza o threshold
corresponde ao Ct (cycle threshold) da amostra. O valor de Ct é preditivo da quantidade de RNAm
alvo presente na amostra.
Os primers utilizados nas reações de amplificação foram desenhados pelo programa Primer
Express (Applied Biosystems, EUA). Na padronização das reações de real time RT-PCR, os cDNAs e
primers foram titulados e foi calculada a eficiência da amplificação para o gene endógeno (GAPDH) e
para gene alvo (Foxp3). Os pares de primers utilizados foram: GAPDH (5’-TGG TCT CCT CTG ACT
TCA-3’, 5’-AGC CAA ATT CGT TGT CAT-3’); Foxp3 (5’-GAG AAG GGC AGG GCA CAA T-3’, 5’-GTG
CCA TGC AGG CCC ACC-3’). O produto de amplificação do gene GAPDH é 117 bp e do gene Foxp3
de 101 bp. Os primers foram usados na concentração final de 200 nM. As reações foram preparadas
em triplicatas num volume final de 10 µl. A reação de real time RT-PCR foi iniciada a 95ºC por 10
minutos, seguida de 40 ciclos de 15 segundos a 95ºC e 1 minuto a 60ºC, de acordo com o manual de
instruções do fabricante ABI PRISM 7500. O máximo coeficiente de variação permitido entre as
triplicatas foi de 1%, caso contrário o experimento foi repetido.
A especificidade dos primers foi avaliada pela curva de dissociação dos produtos de PCR, que
pode detectar amplificações inespecíficas, incluindo primer-dimers (Apêndice H ). Para isso, após a
reação, a placa foi submetida a um segundo programa: 95°C por 1 minuto, 60°C por 1 minuto e 95°C
por 1 minuto. A curva de dissociação consiste na monitorização da fluorescência das amostras em
relação ao aumento de temperatura. A fluorescência das amostras decresce com o aumento da
temperatura, pois à medida que as pontes de hidrogênio, que mantém as duplas fitas unidas se
rompem (devido ao aumento de temperatura), o SYBR-Green é liberado. A fluorescência é emitida
somente quando o DNA está em dupla fita. Assim, quando observamos somente um pico de
fluorescência em uma dada temperatura significa que houve amplificação de um produto específico.
Esta temperatura é a temperatura de anelamento ou melting point (Tm) do produto de amplificação.

Casuística, Materiais e Métodos 53
A normalização e quantificação relativa da expressão gênica foram realizadas pelo método de
2-∆∆CT (Livak et al., 2001), onde CT = threshold cicle (ciclo da reação em que a fluorescência da
amostras excede o threshold), e ∆∆CT = (CT, alvo - CT, endógeno) pós-transplante - (CT, alvo - CT, endógeno) pré-
transplante. Usando o método de 2-∆∆CT, os dados são representados como diferença (em vezes) na
expressão gênica, que foi normalizada para um gene endógeno de referência e é relativa a um
controle ou calibrador. Em nossos experimentos, o calibrador usado foi o valor basal ou pré-
transplante. Para o calibrador, o ∆∆CT é igual a zero e 20 é igual a um, assim mudança na expressão
gênica relativa ao período pré-transplante é igual a um. Para os períodos pós-transplante, o valor de
2-∆∆CT indica a diferença (em vezes) na expressão gênica relativa ao período pré-transplante.
O método 2-∆∆Ct para cálculo da expressão gênica assume que a eficiência de amplificação do
gene alvo e do gene de referência é igual a 2, ou seja, 100%. Para o cálculo da eficiência foi utilizada
a equação E = 10(-1/slope), onde E corresponde à eficiência e slope corresponde ao coeficiente de
angulação da curva (Bustin, 2000; Pfaffl, 2001). Para cada gene estudado foi realizada uma reação
com diluições seriadas de amostra de cDNA (1/5 a 1/1250) e o primers de interesse. Os valores de Ct
obtidos foram plotados em gráfico para cálculo do slope e da eficiência. A eficiência de amplificação
do gene GAPDH foi igual a 2,07 e do gene Foxp3 foi igual a 2,05. A correlação entre a eficiência das
amplificações foi de 0,998. O Apêndice H mostra as curvas de amplificação, as curvas de
dissociação, as curvas de amplificação com o threshold e o cálculo da eficiência de amplificação dos
genes estudados.
3.11 Análise estatística
A significância estatística das mudanças imunológicas foi avaliada longitudinalmente por
modelos de efeitos mistos. Os modelos lineares de efeitos mistos (efeitos aleatórios e fixos) são
utilizados na análise de dados onde as respostas de um mesmo indivíduo estão agrupadas e a
suposição de independência entre observações num mesmo grupo não é adequada (Schall et al.,
1991). Foi utilizado o procedimento PROC MIXED do software SAS (SAS/STAT® User’s Guide,
Version 8.02, SAS Institute Inc., 1999, Cary, Carolina do Norte, EUA). Os box-plots foram preparados
utilizando o software estatístico MINITAB Release 14 (Minitab Inc., USA). As extremidades das caixas
indicam o primeiro e terceiro quartis, as linhas dentro das caixas indicam a mediana, os pontos dentro
das caixas a média, e as barras se estendem do valor máximo ao mínimo. Os valores "máximos" e

Casuística, Materiais e Métodos 54
"mínimos" são calculados, respectivamente, pelas expressões: Upper limit = Q3 + 1.5 (Q3 - Q1) e
Lower limit = Q1- 1.5 (Q3 - Q1), onde Q1 é o primeiro quartil e Q3 é o terceiro quartil. Qualquer valor
fora desses limites são os outliers, acusado nos gráficos com um círculo cheio. Os asteriscos no eixo
X indicam significância estatística (p<0,05).
As diferenças entre os valores dos indivíduos saudáveis versus pré-mobilização, e entre os
valores pré-mobilização versus todos os outros seguimentos pós-transplante, foram consideradas
estatisticamente significantes quando p valor menor que 5% (p <0,05). Tabelas com todos os valores
de média, desvio padrão e mediana dos resultados, assim como o valor de p, encontram-se nos
Apêndices D-G (Apêndices D e F: resultados pacientes com diabete melito do tipo 1; Apêndices F e
G: resultados pacientes com esclerose múltipla). No texto em Resultados, todos os valores absolutos
(células/µl) ou porcentagem (%) foram apresentados como média ± desvio padrão.

Referências

Referências 171
Referências *
American Diabetes Association. Diagnosis and Classification of Diabetes. Diabetes Care 2004; 27 (Suppl I): S5-S10.
Anderson BE, McNiff JM, Matte C, Athanasiadis I, Shlomchik WD, Shlomchik MJ. Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood 2004, 104(5):1565-1573.
Anderson MS, Bluestone JA.The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005, 23:447-85.
Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004, 199(10):1401-1408.
Apostolou I, von Boehmer H.In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004, 199(10):1401-8.
Arpinati M, Green CL, Heimfeld S, Heuser JE, Anasetti C. Granulocyte-colony stimulating factor mobilizes T helper 2-inducing dendritic cells. Blood 2000, 95(8):2484-2490.
Ashton-Rickardt PG, Tonegawa S. A differential-avidity model for T-cell selection. Immunol Today 1994, 15(8):362-366.
Atkinson MA, Kaufman DL, Campbell L, Gibbs KA, Shah SC, Bu DF, Erlander MG, Tobin AJ, Maclaren NK. Response of peripheral-blood mononuclear cells to glutamate decarboxylase in insulin-dependent diabetes. Lancet 1992, 339(8791):458-459.
Autran B, Leblond V, Sadat-Sowti B, Lefranc E, Got P, Sutton L, Binet JL, Debre P. A soluble factor released by CD8+CD57+ lymphocytes from bone marrow transplanted patients inhibits cell-mediated cytolysis. Blood 1991, 77(10):2237-2241.
Baars PA, Ribeiro Do Couto LM, Leusen JH, Hooibrink B, Kuijpers TW, Lens SM, van Lier RA. Cytolytic mechanisms and expression of activation-regulating receptors on effector-type CD8+CD45RA+CD27- human T cells. J Immunol 2000, 165(4):1910-1917.
Bach JF. Regulatory T cells under scrutiny. Nat Rev Immunol. 2003, 3(3):189-198.
Bargetzi MJ, Schonenberger A, Tichelli A, Fried R, Cathomas G, Signer E, Speck B, Gratwohl A. Celiac disease transmitted by allogeneic non-T cell-depleted bone marrow transplantation. Bone Marrow Transplant. 1997, 20(7):607-609.
Barthlott T, Kassiotis G, Stockinger B. T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003, 197(4):451-60.
Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med. 1987, 166(4):823-832.
Bendelac A, Rivera MN, Park SH, Roark JH. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol. 1997, 15:535-562.
* De acordo com:
International Committee of Medical Journal Editors (Vancouver Style) – Grupo de Vancouver.

Referências 172
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001, 27(1):20-21.
Ben-Nun A, Cohen IR. Experimental autoimmune encephalomyelitis (EAE) mediated by T cell lines: process of selection of lines and characterization of the cells. J Immunol. 1982, 129(1):303-308.
Berzins SP, Godfrey DI, Miller JF, Boyd RL. A central role for thymic emigrants in peripheral T cell homeostasis. Proc Natl Acad Sci USA. 1999, 96(17):9787-9791.
Berzins SP, Uldrich AP, Sutherland JS, Gill J, Miller JF, Godfrey DI, Boyd RL. Thymic regeneration: teaching an old immune system new tricks. Trends Mol. Med. 2002, 8(10):469-476.
Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes 1997, 46(11):1701-1710.
Bluestone JA and Abbas AK. Natural versus adaptive regulatory T cells. Nature Rev Immunol 2003, 3(3):253-257.
Bougnères F, Landais P, Boisson C, Carel JC, Frament N, Boitard C, Chaussain JL, Bach JF. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes 1990, 39(1):1264-1272.
Boyum, A. Separation of blood leukocytes, granulocytes and lymphocytes. Tissue Antigens 1974, 4(4):269-274.
Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101(7):2711-2720.
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001, 27(1):68-73.
Burt RK, Cohen B, Rose J, Petersen F, Oyama Y, Stefoski D, Katsamakis G, Carrier E, Kozak T, Muraro PM, Martin R, Hintzen R, Slavin S, Karussis D, Haggiag S, Voltarelli JC, Ellison GW, Jovanovic B, Popat U, McGuirk J, Statkute L, Verda L, Haas J, Arnold R. Hematopoietic stem cell transplantation for multiple sclerosis. Arch Neurol 2005, 62(6): 860-864.
Burt RK, Kenyon N, Voltarelli JC, Kaufman DB. Hematopoietic stem cell transplantation as treatment for type 1 diabetes. In: Burt RK and Marmont A (eds). Stem cell therapy for autoimmune diseases. Landes Biociences, Georgetown, Texas, USA, 2004.
Burt RK, Oyama Y, Traynor A, Kenyon NS. Hematopoietic stem cell therapy for type 1 diabetes: induction of tolerance and islet cell neogenesis. Autoimmun Rev. 2002a, 1(3):133-138.
Burt RK, Padilla J, Begolka WS, Canto MC, Miller SD. Effect of disease stage on clinical outcome after syngeneic bone marrow transplantation for relapsing experimental autoimmune encephalomyelitis. Blood 1998a, 91(7):2609-2616.
Burt RK, Slavin S, Burns WH, Marmont AM. Induction of tolerance in autoimmune diseases by hematopoeitic stem cell transplantation: getting closer to a cure? Blood 2002b, 99(3):768-784.
Burt RK, Traynor AE, Craig R, Marmont AM. The promise of hematopoietic stem cell transplantation for autoimmune diseases. Bone Marrow Transplantation 2003, 31(7):521-524.
Burt RK, Traynor AE, Pope R, Schroeder J, Cohen B, Karlin KH, Lobeck L, Goolsby C, Rowlings P, Davis FA, Stefoski D, Terry C, Keever-Taylor C, Rosen S, Vesole D, Fishman M, Brush M, Mujias S,

Referências 173
Villa M, Burns WH. Treatment of autoimmune disease by intense immunosuppressive conditioning and autologous hematopoietic stem cell transplantation. Blood 1998b, 92(10): 3505-3514.
Burt RK, Traynor AE. Hematopoietic stem cell transplantation: a new therapy for autoimmune disease. Oncologist 1999; 4(1):77-83.
Bustin, SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000, 25: 169-193.
Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000, 164(11):5782-5787.
Chen TC, Cobbold SP, Fairchild PJ, Waldmann H. Generation of anergic and regulatory T cells following prolonged exposure to a harmless antigen. J Immunol. 2004, 172(10):5900-5907.
Chklovskaia E, Nowbakht P, Nissen C, Gratwohl A, Bargetzi M, Wodnar-Filipowicz A. Reconstitution of dendritic and natural killer-cell subsets after allogeneic stem cell transplantation: effects of endogenous flt3 ligand. Blood 2004, 103(10):3860-3868.
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal.Biochem 1987, 162(1):156-159.
Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 1993, 42(1):44-55.
Compston A, Coles A. Multiple sclerosis. Lancet 2002, 359(9313):1221-1231.
Coo H, Aronson KJ. A systematic review of several potential non-genetic risk factors for multiple sclerosis. Neuroepidemiology 2004, 23(1-2):1-12.
Cook JJ, Hudson I, Harrison LC, Dean B, Colman PG, Werther GA, Warne GL, Court JM. Double-blind controlled trial of azathioprine in children with newly diagnosed type I diabetes. Diabetes 1989, 38(6): 779-783.
Cozzo C, Larkin J 3rd, Caton AJ. Cutting edge: self-peptides drive the peripheral expansion of CD4+CD25+ regulatory T cells. J Immunol. 2003, 171(11):5678-5682.
Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004, 173(12):7259-7268.
Davidson A, Diamond B. Autoimmune diseases. N Engl J Med 2001; 345(5):340-350.
Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334(6181):395-402.
de Kleer I, Vastert B, Klein M, Teklenburg G, Arkesteijn G, Yung GP, Albani S, Kuis W, Wulffraat N, Prakken B. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood 2006, 107(4):1696-1702.
de Kleer I, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, de Jager W, Pugayung G, Giannoni F, Rijkers G, Albani S, Kuis W, Prakken B. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. 2004, 172(10):6435-6443.
Dighiero G, Rose NR. Critical self-epitopes are key to the understanding of self-tolerance and autoimmunity. Immunol Today. 1999, 20(9):423-428.

Referências 174
Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker LJ, Koup RA. Changes in thymic function with age and during the treatment of HIV infection. Nature 1998, 396(6712):690-695.
Douek DC, Vescio RA, Betts MR, Brenchley JM, Hill BJ, Zhang L, Berenson JR, Collins RH, Koup RA. Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet. 2000, 355(9218):1875-1881.
Douek DC. The contribution of the thymus to immune reconstitution after hematopoietic stem-cell transplantation. Cytotherapy 2002, 4(5):425-426.
Dyment DA, Ebers GC, Sadovnick AD. Genetics of multiple sclerosis. Lancet Neurol. 2004, 3(2):104–110
Effros RB, Pawelec G. Replicative senescence of T cells: does the Hayflick Limit lead to immune exhaustion? Immunol. Today 1997, 18(9):450–454.
Elliott RB, Crossley JR, Berryman CC, James AG. Partial preservation of pancreatic �-cell function in
children with diabetes. Lancet 1981, 2(8247): 631-632.
Encinas JA, Kuchroo VK. Mapping and identification of autoimmunity genes. Curr Opin Immunol 2000; 12(6):691-697.
Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 1999, 11(2):173-181.
Euler HH, Marmont AM, Bacigalupo A, Fastenrath S, Dreger P, Hoffknecht M, Zander AR, Schalke B, Hahn U, Haas R, Schmitz N. Early recurrence or persistence of autoimmune diseases after unmanipulated autologous stem cell transplantation. Blood 1996;88(9):3621-3625.
Eyrich M, Leiler C, Croner T, Lang P, Schumm M, Mascher B, Schilbach K, Klingebiel T, Handgretinger R, Niethammer D, Schlegel PG. Impaired T-cell activation and cytokine productivity after transplantation of positively selected CD34+ allogeneic hematopoietic stem cells. Hematol J. 2004, 5(4):329-340.
Fallen PR, Duarte RF, McGreavey L, Potter M, Ethell M, Prentice HG, Madrigal JA, Travers PJ. Identification of non-naïve CD4+CD45RA+ T cell subsets in adult allogeneic haematopoietic cell transplant recipients. Bone Marrow Transplant 2003; 32(6):609-616.
Fallen PR, McGreavey L, Madrigal JA, Potter M, Ethell M, Prentice HG, Guimarães A, Travers PJ. Factors affecting reconstitution of the T cell compartment in allogeneic haematopoietic cell transplant recipients. Bone Marrow Transplant 2003; 32(10):1001-1014.
Farge D, Henegar C, Carmagnat M, Daneshpouy M, Marjanovic Z, Rabian C, Ilie D, Douay C, Mounier N, Clave E, Bengoufa D, Cabane J, Marolleau JP, Gluckman E, Charron D, Toubert A. Analysis of immune reconstitution after autologous bone marrow transplantation in systemic sclerosis. Arthritis Rheum. 2005, 52(5):1555-1563.
Fassas A, Kimiskidis VK. Stem cell transplantation for multiple sclerosis: what is the evidence? Blood Rev. 2003, 17(4):233–240.
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22(3):329-341.
Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005, 6(4):331-337.

Referências 175
Franzke A, Piao W, Lauber J, Gatzlaff P, Konecke C, Hansen W, Schmitt-Thomsen A, Hertenstein B, Buer J, Ganser A. G-CSF as immune regulator in T cells expressing the G-CSF receptor: implications for transplantation and autoimmune diseases. Blood 2003, 102(2):734-739.
Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 1996, 274(5285):209-219.
Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999, 17:369-397.
Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol. 2002, 3(1):33-41.
Gbadamosi J, Buhmann C, Tessmer W, Moench A, Haag F, Heesen C. Effects of mitoxantrone on multiple sclerosis patients’ lymphocyte subpopulations and production of immunoglobulin, TNF-alpha and IL-10. Eur. Neurol. 2003, 49(3):137-141.
Ge Q, Hu H, Eisen HN, Chen J. Different contributions of thymopoiesis and homeostasis-driven proliferation to the reconstitution of naive and memory T cell compartments. Proc. Natl. Acad. Sci. USA 2002, 99(5):2989-2994.
Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5(2):170-175
Gestri D, Baldacci L, Taiuti R, Galli E, Maggi E, Piccinni MP, Vergelli M, Massacesi L. Oligoclonal T cell repertoire in cerebrospinal fluid of patients with inflammatory diseases of the nervous system. J Neurol Neurosurg Psychiatry. 2001, 70(6):767-772.
Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain 2000, 123(Pt 3):508-518.
Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 1999, 11(2):183-190.
Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature 1999, 402(6759):255-262.
Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis driven proliferation. J. Exp. Med. 2000, 192(4):557–564.
Good R, Ikehara S. Preclinical investigations that subserve efforts to employ bone marrow transplantation for rheumatoid or autoimmune diseases. J Rheumatol Suppl. 1997, 48:5-12.
Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005, 435(7042):590-597.
Gorochov G, Debre P, Leblond V, Sadat-Sowti B, Sigaux F, Autran B. 1994. Oligoclonal expansion of CD8+CD57+ T cells with restricted T-cell receptor beta chain variability after bone marrow transplantation. Blood 1994, 83(2):587-595.
Gorski J, Yassai M, Zhu X, Kissela B, Keever C, Flomenberg N. Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 spectratyping: correlation with the immune status. J. Immunol.1994, 152(10):5109-5119.
Gran B, Gestri D, Sottini A, Quiros Roldan E, Bettinardi A, Signorini S, Primi D, Ballerini C, Taiuti R, Amaducci L, Massacesi L. Detection of skewed T-cell receptor V-beta gene usage in the peripheral blood of patients with multiple sclerosis. J Neuroimmunol. 1998, 85(1):22-32.

Referências 176
Gratwohl A, Tyndall A. From autoimmunity to stem cell transplantation. Clinical Reviews in Hematology/Oncology 1999; 30(2): 159-172.
Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21(4):589-601.
Guillaume T, Rubinstein DB, Symann M. Immune reconstitution and immunotherapy after autologous hematopoietic stem cell transplantation. Blood 1998; 92(5):1471-1490.
Hafler DA. Multiple sclerosis. J. Clin. Invest. 2004, 113(6):788–794.
Hahn BH. The potential role of stem cell transplantation in patients with systemic lupus erythematosus. J Rheumatol 1997; 24 (Suppl 48): 89-93.
Haines JL, Terwedow HA, Burgess K, Pericak-Vance MA, Rimmler JB, Martin ER, Oksenberg JR, Lincoln R, Zhang DY, Banatao DR, Gatto N, Goodkin DE, Hauser SL. Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The Multiple Sclerosis Genetics Group. Hum Mol Genet. 1998, 7(8):1229-1234.
Hakim FT, Cepeda R, Kaimei S, Mackall CL, McAtee N, Zujewski J, Cowan K, Gress RE. Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood 1997, 90(9):3789-3798.
Hakim FT, Memon SA, Cepeda R, Jones EC, Chow CK, Kasten-Sportes C, Odom J, Vance BA, Christensen BL, Mackall CL, Gress RE. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005, 115(4):930-939.
Hamann D, Baars PA, Hooibrink B, van Lier RW. Heterogeneity of the human CD4+ T-cell population: two distinct CD4+ T-cell subsets characterized by coexpression of CD45RA and CD45RO isoforms. Blood 1996; 88(9):3513-3521.
Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ Tcells. J Exp Med 1997, 186(9): 1407–1418.
Haraguchi K, Takahashi T, Hiruma K, Kanda Y, Tanaka Y, Ogawa S, Chiba S, Miura O, Sakamaki H, Hirai H. Recovery of Valpha24+ NKT cells after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004, 34(7):595-602.
Harrison LC, Colman PG, Dean B, Baxter R, Martin FI. Increase in remission rate in newly diagnosed type l diabetic subjects treated with azathioprine. Diabetes 1985, 34(12):1306-1308.
Hawkes CJ, Schloot NC, Marks J, Willemen SJ, Drijfhout JW, Mayer EK, Christie MR, Roep BO. T-cell lines reactive to an immunodominant epitope of the tyrosine phosphatase-like autoantigen IA-2 in type 1 diabetes. Diabetes 2000, 49(3):356-366.
Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci. 2002, 3(4):291-301.
Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Eng J Med 2002, 346(22):1692-1698.
Herrmann MM, Gaertner S, Stadelmann C, van den Brandt J, Boscke R, Budach W, Reichardt HM, Weissert R. Tolerance induction by bone marrow transplantation in a multiple sclerosis model. Blood 2005, 106(5):1875-1883.
Hess D, Li L, Martin M, Sakano S, Hill D, Strutt B, Thyssen S, Gray DA, Bhatia M. Bone marrow-derived stem cells initiate pancreatic regeneration. Nature Biotech 2003, 21(7):763-770.

Referências 177
Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nat Rev Immunol. 2005, 5(10):772-82.
Holbrook MR, Tighe PJ, Powell RJ. Restrictions of T cell receptor β chain repertoire in the peripheral blood of patients with systemic lupus erythematosus. Ann Rheum Dis 1996; 55(9): 627-631.
Hough RE, Snowden JA., Wulffraat NM. Haematopoietic stem cell transplantation in autoimmune diseases: a European perspective. Br. J. Haematol 2005, 128(4):432-459.
Hug A, Korporal M, Schroder I, Haas J, Glatz K, Storch-Hagenlocher B, Wildemann B. Thymic export function and T cell homeostasis in patients with relapsing remitting multiple sclerosis. J. Immunol. 2003, 171(1):432–437.
Ikehara S, Good RA, Nakamura T, Sekita K, Inoue S, Oo MM, Muso E, Ogawa K, Hamashima Y. Rationale for bone marrow transplantation in the treatment of autoimmune diseases. Proc. Natl Acad. Sci. USA 1985, 82(8), 2483-2487, 1985.
Ikehara S, Kawamura M, Takao F, Inaba M, Yasumizu R, Than S, Hisha H, Sugiura K, Koide Y, Yoshida TO, Ida T, Imura H, Good RA. Organ-specific and systemic autoimmune diseases originate from defects in haematopoietic stem cells. Proc. Natl Acad. Sci. USA 1990, 87(21):8341-8344.
Ikehara S, Yasumizu R, Inaba M, Izui S, Hayakawa K, Sekita K, Toki J, Sugiura K, Iwai H, Nakamura T, Muso E, Hamashima Y, Good RA. Long-term observations of autoimmune-prone mice treated for autoimmune disease by allogeneic bone marrow transplantation. Proc. Natl Acad. Sci. USA 1989, 86(9):3306-3310.
Ikehara S. Bone marrow transplantation for autoimmune diseases. Acta Haematol. 1998, 99(3):116-132.
Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M, Kawata S, Tarui S,Kono N, Matsuzawa Y. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993, 92(5):2313-2322.
Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, Makino S, Nagata S, Kono N, Matsuzawa Y. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1997, 186(4):613-618.
Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, Schock S, Oertel WH, Sommer N, Hemmer B. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 2002, 125(Pt 3):538-550.
Jaeckel E, Kretschmer K, Apostolou I, von Boehmer H. Instruction of Treg commitment in peripheral T cells is suited to reverse autoimmunity. Semin Immunol. 2006, 18(2):89-92.
Jameson SC. Maintaining the norm: T-cell homeostasis. Nat Rev Immunol. 2002, 2(8):547-556.
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418(6893):41-49.
Jun HS, Khil LY, Yoon JW. Role of glutamic acid decarboxylase in the pathogenesis of type 1 diabetes. Cell Mol Life Sci. 2002, 59(11):1892-1901.
Jun HX, Jun CY, Yu ZX. In vivo induction of T-cell hyporesponsiveness and alteration of immunological cells of bone marrow grafts using granulocyte colony-stimulating factor. Haematologica 2004, 89(12):1517-24.

Referências 178
Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med. 1997, 186(7):989-997.
Kared H, Masson A, Adle-Biassette H, Bach JF, Chatenoud L, Zavala F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4(+)CD25(+) regulatory T-cells. Diabetes. 2005, 54(1):78-84.
Kayser C, Alberto FL, da Silva NP, Andrade LE. Decreased number of T cells bearing TCR rearrangement excision circles (TREC) in active recent onset systemic lupus erythematosus. Lupus 2004, 13(12):906-911.
Kelly MA, Rayner ML, Mijovic CH, Barnett AH. Molecular aspects of type 1 diabetes. Mol Pathol. 2003, 56(1):1-10.
Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005, 352(25):2598-2608.
Kimmig S, Przybylski GK, Schmidt CA, Laurisch K, Mowes B, Radbruch A, Thiel A. Two subsets of naïve T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med 2002; 195(6):789-794.
King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004, 117(2):265-77.
Klangsinsirikul P, Russell NH. Peripheral blood stem cell harvests from G-CSF-stimulated donors contain a skewed Th2 CD4 phenotype and a predominance of type 2 dendritic cells. Exp Hematol. 2002, 30(5):495-501.
Knip M, Akerblom HK. Environmental factors in the pathogenesis of type 1 diabetes mellitus. Exp Clin Endocrinol Diabetes. 1999, 107(Suppl 3):S93-100.
Koehne G, Zeller W, Stockschlaeder M, Zander AR. Phenotype of lymphocyte subsets after autologous peripheral blood stem cell transplantation. Bone Marrow Transplant. 1997, 19(2):149-156.
Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000, 97(16):9203-9208.
Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003, 21:759-806.
Kotter I, Daikeler T, Einsele H, Koch S, Lochmann L, Kanz L, Loffler J. Relapse of autoimmune diseases after autologous T cell depleted stem cell transplantation may be triggered by T cells recently emigrated from the thymus. Ann Rheum Dis. 2005, 64(12):1787-9.
Kronenberg M, Rudensky A. Regulation of immunity by self-reactive T cells. Nature 2005, 435(7042):598-604.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale. Neurology 1983, 33:1444-1452.
Lampeter EF, McCann SR, Kolb H. Transfer of diabetes type 1 by bone marrow transplantation. Lancet 1998, 351(9102):568-569.

Referências 179
Laplaud DA, Ruiz C, Wiertlewski S, Brouard S, Berthelot L, Guillet M, Melchior B, Degauque N, Edan G, Brachet P, Damier P, Soulillou JP. Blood T-cell receptor beta chain transcriptome in multiple sclerosis. Characterization of the T cells with altered CDR3 length distribution. Brain 2004, 127(Pt 5):981-995.
Laylor R, Dewchand H, Simpson E, Dazzi F. Engraftment of allogeneic hematopoietic stem cells requires both inhibition of host-versus-graft responses and 'space' for homeostatic expansion. Transplantation 2005, 79(11):1484-91.
Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆C
T method. Methods 2001, 25:402-408.
Luppi P, Zanone MM, Hyoty H, Rudert WA, Haluszczak C, Alexander AM, Bertera S, Becker D, Trucco M. Restricted TCR V beta gene expression and enterovirus infection in type I diabetes: a pilot study. Diabetologia 2000, 43(12):1484-1497.
Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Magrath IT, Wexler LH, Dimitrov DS, Gress RE. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood 1997, 89(10):3700–3707.
Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Horowitz ME, Magrath IT, Shad AT, Steinberg SM, Wexler LH, Gress RE. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. 1995, 332(3):143-149.
Mackall CL, Granger L, Sheard MA, Cepeda R, Gress RE. T-cell regeneration after bone marrow transplantation: differential CD45 isoform expression on thymic-derived versus thymic-independent progeny. Blood 1993, 82(8):2585-2594.
Malphettes M, Carcelain G, Saint-Mezard P, Leblond V, Altes HK, Marolleau JP, Debre P, Brouet JC, Fermand JP, Autran B. Evidence for naive T-cell repopulation despite thymus irradiation after autologous transplantation in adults with multiple myeloma: role of ex vivo CD34+ selection and age. Blood 2003, 101(5):1891-1897.
Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nature medicine 2001; 7(8):899-905.
Mathews V, Hanson PT, Ford E, Fujita J, Polonsky KS, Graubert TA. Recruitment of bone marrow-derived endothelial cells to sites of pancreatic beta-cell injury. Diabetes 2004, 53(1):91-98.
Mathis D, Vence L, Benoist C. Beta-Cell death during progression to diabetes. Nature 2001, 414(6865):792-798.
Matsumoto Y, Yoon WK, Jee Y, Fujihara K, Misu T, Sato S, Nakashima I, Itoyama Y. Complementarity-determining region 3 spectratyping analysis of the TCR repertoire in multiple sclerosis. J Immunol. 2003, 170(9):4846-4853.
Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004, 4(10):762-774.
Mollet L, Sadat-Sowti B, Duntze J, Leblond V, Bergeron F, Calvez V, Katlama C, Debre P, Autran B. CD8hi+CD57+ T lymphocytes are enriched in antigen-specific T cells capable of down-modulating cytotoxic activity. Int Immunol 1998, 10(3):311-323.
Monteiro J, Batliwalla F, Ostrer H, Gregersen PK. Shortened telomeres in clonally expanded CD28-CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J. Immunol. 1996, 156(10):3587-3590.
Morley JK, Batliwalla FM, Hingorani R, Gregersen PK. Oligoclonal CD8+ T cells are preferentially expanded in the CD57+ subset. J. Immunol. 1995, 154(11):6182-6190.

Referências 180
Morton JI, Siegel BV. Transplantation of autoimmune potential. Development of antinuclear antibodies in H-2 histocompatible recipients of bone marrow from New Zealand Black mice. Proc. Natl Acad. Sci. USA 1974, 71(6): 2162-2165..
Mosmann TR, Li L, Sad S. Functions of CD8 T-cell subsets secreting different cytokine patterns. Semin Immunol. 1997, 9(2):87-92.
Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J. Immunol. 2000, 165(4):1733-1737.
Muraro PA and Martin R. Immunological questions on hematopoietic stem cell transplantation for multiple sclerosis. Bone Marrow Transplant 2003, 32:S41–S44.
Muraro PA, Bonanni L, Mazzanti B, Pantalone A, Traggiai E, Massacesi L, Vergelli M, Gambi D. Short-term dynamics of circulating T cell receptor V beta repertoire in relapsing-remitting MS. J Neuroimmunol. 2002, 127(1-2):149-159.
Muraro PA, Cassiani Ingoni R, Martin R. Hematopoietic stem cell transplantation for multiple sclerosis: current status and future challenges. Curr. Opin. Neurol. 2003, 16(3):299–305.
Muraro PA, Douek DC, Packer A, Chung K, Guenaga FJ, Cassiani-Ingoni R, Campbell C, Memon S, Nagle JW, Hakim FT, Gress RE, McFarland HF, Burt RK, Martin R. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J. Exp. Med. 2005, 201(5):805-816.
Muraro PA, Douek DC. Renewing the T cell repertoire to arrest autoimmune aggression. Trends Immunol. 2006, 27(2):61-67.
Naquet P, Ellis J, Tibensky D, Kenshole A, Singh B, Hodges R, Delovitch TL. T cell autoreactivity to insulin in diabetic and related non-diabetic individuals. J Immunol. 1988, 140(8):2569-2578.
Nelson JL, Torrez R, Louie FM, Choe OS, Storb R, Sullivan KM. Pre-existing autoimmune disease in patients with long-term survival after allogeneic marrow transplantation. J. Rheumatol. 1997; 24 (Suppl 48): 23-29.
Nikolic B, Takeuchi Y, Leykin I, Fudaba Y, Smith RN, Sykes M. Mixed hematopoietic chimerism allows cure of autoimmune diabetes through allogeneic tolerance and reversal of autoimmunity. Diabetes 2004, 53(2):376-383.
Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004, 4(2):123-132.
Notkins AL, Lernmark A. Autoimmune type 1 diabetes: resolved and unresolved issues. J Clin Invest 2001, 108(9): 1247-1252.
Notkins AL. Immunologic and genetic factors in type 1 diabetes. J Biol Chem. 2002, 277(46):43545-43548.
Ogawa M, Maruyama T, Hasegawa T, Kanaya T, Kobayashi F, Tochino Y, . et al. The inhibitory effect of neonatal thymectomy on the incidence of insulitis in non-obese diabetes (NOD) mice. Biomed. Res. 1985, 6:103–106.
Openshaw H, Nash RA, McSweeney PA. High dose immunosuppression and hematopoeitic stem cell transplantation in autoimmune disease: clinical review. Biol Blood Marrow Transplant 2002; 8:233-248.
Palmer JP, Fleming GA, Greenbaum CJ, Herold KC, Jansa LD, Kolb H, Lachin JM, Polonsky KS, Pozzilli P, Skyler JS, Steffes MW. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve
�-cell function. Diabetes 2004; 53(1): 250-264.

Referências 181
Pan L, Delmonte J Jr, Jalonen CK, Ferrara JL. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces seveity of experimental graft-versus-host disease. Blood 1995, 86(12):4422-9.
Pannetier C, Cochet M, Darche S, Casrouge A, Zoller M, Kourilsky P. The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc.Natl.Acad.Sci.USA 1993, 90(9):4319-4323.
Pannetier C, Even J, Kourilsky P. T-cell repertoire diversity and clonal expansions in normal and clinical samples. Immunol.Today 1995, 16(4):176-181.
Peggs KS, Mackinnon S. Immune reconstitution following haematopoietic stem cell transplantation. Br J Haematol. 2004, 124(4):407-420.
Peggs KS, Verfuerth S, Pizzey A, Khan N, Moss P, Goldstone AH, Yong K, Mackinnon S. Reconstitution of T-cell repertoire after autologous stem cell transplantation: influence of CD34 selection and cytomegalovirus infection. Biol. Blood Marrow Transplant. 2003, 9(3):198-205.
Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol. 1981, 127(4):1420-1423.
Pfaffl, MW. A new mathematical model for relative quantification for real time RT-PCR. Nucleic Acids Res. 2001, 29: 2002-2007.
Popat U, Krance R. Haematopoietic stem cell transplantation for autoimmune disorders: the American perspective. Br. J. Haematol 2004, 126(5):637-649.
Posnett DN, Schmelkin I, Burton DA, August A, McGrath H, Mayer LF. T cell antigen receptor V gene usage. Increases in V beta 8+ T cells in Crohn's disease. J Clin Invest. 1990, 85(6):1770-1776.
Pulendran B, Banchereau J, Burkeholder S, Kraus E, Guinet E, Chalouni C, Caron D, Maliszewski C, Davoust J, Fay J, Palucka K. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J Immunol. 2000, 165(1):566-572.
Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest. 1998, 102(5):1045-1050.
Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR. �-cell function in newly-onset type 1 diabetes
and immunomodulation with a heatshock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet 2001, 358(9295):1749-1753.
Reimer P, Kunzmann V, Wilhelm M, Weissbrich B, Kraemer D, Berghammer H, Weissinger F. Cellular and humoral immune reconstitution after autologous peripheral blood stem cell transplantation (PBSCT). Ann. Hematol. 2003, 82(5):263–270.
Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature 2005, 435(7042):584-589.
Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J. Exp. Med. 1933, 58:39–53.
Roep BO, Atkinson M. Animal models have little to teach us about type 1 diabetes: 1. In support of this proposal. Diabetologia 2004, 47(10):1650-1656.
Roep BO. The role of T-cells in the pathogenesis of Type 1 diabetes: from cause to cure. Diabetologia 2003, 46(3):305-321.

Referências 182
Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today. 1993, 14(9):426-430.
Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005. 201(11):1793-803.
Rutella S, Zavala F, Danese S, Kared H, Leone G. Granulocyte colony-stimulating factor: a novel mediator of T cell tolerance. J Immunol. 2005, 175(11):7085-91.
Sadat-Sowti B, Debre P, Mollet L, Quint L, Hadida F, Leblond V, Bismuth G, Autran B. An inhibitor of cytotoxic functions produced by CD8+CD57+ T lymphocytes from patients suffering from AIDS and immunosuppressed bone marrow recipients. Eur J Immunol. 1994, 24(11):2882-2888.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995, 155(3):1151-1164.
Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004, 22:531-562.
Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005, 6(4):345-352.
Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004, 22:745-63.
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401(6754):708-712.
Schall R. Estimation in generalized linear models with random effects. Biometrika 1991, 78(4):719-727.
Schlenke P, Sheikhzadeh S, Weber K, Wagner T, Kirchner H. Immune reconstitution and production of intracellular cytokines in T lymphocyte populations following autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 2001; 28(3):251-257.
Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61(12):4756-60.
Selin LK, Lin MY, Kraemer KA, Pardoll DM, Schneck JP, Varga SM, Santolucito PA, Pinto AK, Welsh RM. Attrition of T cell memory: selective loss of LCMV epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity 1999, 11(6):733–742.
Selin LK, Welsh RM. Plasticity of T cell memory responses to viruses. Immunity. 2004, 20(1):5-16.
Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nature Rev Immunol 2002, 2(6):389-400.
Silverstein J, Maclaren N, Riley W, Spillar R, Radjenovic D, Jonhson. Immunosuppression with azathioprine and prednisone in recent-onset insulin-dependent diabetes mellitus. N Engl J Med 1988, 319(10): 599-604.
Singh RK, Varney ML, Buyukberber S, Ino K, Ageitos AG, Reed E, Tarantolo S, Talmadge JE. Fas-FasL-mediated CD4+ T-cell apoptosis following stem cell transplantation. Cancer Res. 1999, 59(13):3107–3111.

Referências 183
Sloand EM, Kim S, Maciejewski JP, Van Rhee F, Chaudhuri A, Barrett J, Young NS. Pharmacologic doses of granulocyte colony-stimulating factor affect cytokine production by lymphocytes in vitro and in vivo. Blood 2000,95(7):2269-2274.
Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005, 23:683-747.
Sun W, Popat U, Hutton G, Zang YCQ, Krance R, Carrum G, Land GA, Heslop H, Brenner M, Zhang JZ. Characteristics of T-cell receptor repertoire and myelin-reactive T cells reconstituted from autologous haematopoietic stem-cell grafts in multiple sclerosis. Brain 2004, 127(5):996-1008.
Sykes M, Nikolic B. Treatment of severe autoimmune disease by stem cell transplantation. Nature 2005, 435(7042):620-627.
Tanchot C, Le Campion A, Martin B, Leaument S, Dautigny N, Lucas B. Conversion of naive T cells to a memory-like phenotype in lymphopenic hosts is not related to a homeostatic mechanism that fills the peripheral naive T cell pool. J. Immunol. 2002, 168(10):5042–5046.
Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416(6880):542-545.
Theofilopoulos AN, Kono DH. The genes of systemic autoimmunity. Proc Assoc Am Physicians 1999; 111(3):228-240.
Thiel A, Alexander T, Schmidt CA, Hiepe F, Arnold R, Radbruch A, Verda L, Burt RK. Immune reconstitution after hematopoietic stem cell transplantation. In: Burt RK Marmont A (eds). Stem cell therapy for autoimmune diseases. Landes Biociences, Georgetown, Texas, USA, 2004, 206-222.
Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004, 172(11):6519-6523.
Touraine JL, Roncarolo MG, Raudrant D, Bacchetta R, Golfier F, Sembeil R, Gebuhrer L. Induction of transplantation tolerance in humans using fetal cell transplants. Transplant Proc. 2005, 37(1):65-66.
Traynor AE, Schroeder J, Rosa RM, Cheng D, Stefka J, Mujais S, Baker S, Burt RK. Treatment of severe systemic lupus erythematosus with high-dose chemotherapy and hematopoietic stem cell transplantation: a phase I study. Lancet 2000; 356(9231): 701-707.
Tyndall A, Gratwohl A. Immune ablation and stem cell therapy in autoimmune disease: clinical experience. Arthritis Res 2000, 2(4):276-280.
Tyndall A, Koike T. High-dose immunoablative therapy with hematopoietic stem cell support in the treatment of severe autoimmune: current status and future direction. Inter Med 2002; 41(8):608-612.
Tyndall A, Saccardi R. Hematopoietic stem cell transplantation in the treatment of severe autoimmune disease - results from phase I/II studies, prospective randomized trials and future directions. Clin. Exp. Immunol 2005, 141(1):1-9.
van Bekkum DW. Autologous stem cell transplantation for treatment of autoimmune diseases. Stem Cells 1999; 17(3): 172-178.
van Bekkum DW. Experimental basis for the treatment of autoimmune diseases with autologous hematopoietic stem cell transplantation. Bone Marrow Transplant 2003, 32(Suppl 1):S37-S39.
van Bekkum DW. Immune ablation and stem cell therapy in autoimmune disease: experimental basis for autologous stem cell transplantation. Arthritis Res 2000, 2(4):281-284.
van Bekkum DW. Stem cell transplantation in experimental models of autoimmune disease. J Clin Immunol 2000; 20(1):10-16.

Referências 184
van Bekkum, DW. Experimental basis of hematopoietic stem cell transplantation for treatment of autoimmune diseases. J Leuk Biol 2002, 72(4):609-620.
van Gelder M, Kinwel-Bohre EP, van Bekkum DW. Treatment of experimental allergic encephalomyelitis in rats with total body irradiation and syngeneic BMT. Bone Marrow Transplant. 1993, 11(3):233-241.
van Gelder M, van Bekkum DW. Treatment of relapsing experimental autoimmune encephalomyelitis in rats with allogeneic bone marrow transplantation from a resistant strain. Bone Marrow Transplant. 1995, 16(3):343-351.
van Laar JM. Immune ablation and stem-cell therapy in autoimmune disease: immunological reconstitution after high-dose immunosuppression and haematopoietic stem-cell transplantation. Arthritis Res. 2000, 2(4):270–275.
Vasconcelos ZF, Dos Santos BM, Farache J, Palmeira TS, Areal RB, Cunha JM, Barcinski MA, Bonomo A. G-CSF-treated granulocytes inhibit acute graft-versus-host disease. Blood. 2006, 107(5):2192-9.
Vasconcelos ZF, Santos BM, Costa ES, Lima M, Tabak DG, Bouzas LF, Azevedo WM, Barcinski MA, Bonomo A. T-lymphocyte function from peripheral blood stem-cell donors is inhibited by activated granulocytes. Cytotherapy 2003;5(4):336-45.
Verda L, Oyama Y, Craig R, Statkute L, Krosnjar N, Quigley K, Burt RK. Restoration of the CD4+CD25 bright regulatory cell subset in patients with Crohn’s disease following autologous non-myeloablative stem cell transplantation (NST). Blood 2005, 106(11):2193.
Voltarelli JC, Couri CE, Stracieri ABPL, Moraes DA, Oliveira MCB, Pieroni F, Coutinho MA, Malmegrim KCR, Simões BP, Foss MC, Burt RK. High dose immunosuppression and autologous hematopoietic stem cell transplantation in early onset type 1 diabetes mellitus. Submitted 2006b.
Voltarelli JC, Couri CEB, Oliveira MCB, Stracieri ABPL, Moraes DA, Godoi DF, Coutinho MA, Malmegrim KCR, Foss-Freitas MC, Foss MC, Simões BP, Squiers EC, Burt RK. Autologous hematopoietic stem cell transplantation for type 1 diabetes mellitus. Blood 2004a, 104(11):5224.
Voltarelli JC, Couri CEB, Oliveira MCB, Stracieri ABPL, Moraes DA, Coutinho MA, Malmegrim KCR, Foss-Freitas MC, Simões BP, Foss MC, Burt RK. Autologous hematopoietic stem cell transplantation for type 1 diabetes mellitus. Bone Marrow Transplant. 2006a, 37(S1):26.
Voltarelli JC, Oliveira MC, Stracieri AB, Godoi DF, Moraes DA, Coutinho MA, Malmegrim KCR, Santos AC. Hematopoietic stem cell transplantation for refractory Takayasu’s arteritis. Rheumatology 2004b, 43(10):1308-1309.
Voltarelli JC, Ouyang J. Hematopoietic stem cell transplantation for autoimmune diseases in developing countries: current status and future prospectives. Bone Marrow Transplant. 2003, 32 (S 1):S69-S71.
Voltarelli JC, Stracieri ABPL, Oliveira MCB, Godoi DF, Moraes DA, Pieroni F, Malmegrim KCR, Coutinho MA, Simoes BP. Transplante de células tronco hematopoéticas em doenças reumáticas. Parte 1: Experiência internacional. Rev Bras Reumatol. 2005a, 45: 229-241.
Voltarelli JC, Stracieri ABPL, Oliveira MCB, Godoi DF, Moraes DA, Pieroni F, Malmegrim KCR, Coutinho MA, Simões BP, Massumoto C, Hamerschlak N, Scheinberg M, Ferreira E, Coutinho M, Ostronoff M, Sturaro D, Dulley F. Transplante de células tronco hematopoéticas em doenças reumáticas - parte 2: experiência brasileira perspectivas futuras. Revista Brasileira de Reumatologia 2005b, 45: 301-312.

Referências 185
Voltarelli JC. Transplante de células tronco hematopoéticas no diabete melito do tipo 1. Rev Bras Hematol Hemoter. 2004c, 26(1):43-45.
Walker LS, Abbas AK.The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002, 2(1):11-9.
Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest. 2003, 112(9):1437-1443.
Weekes MP, Carmichael AJ, Wills MR, Mynard K, Sissons JG. Human CD28-CD8+ T cells contain greatly expanded functional virus-specific memory CTL clones. J Immunol. 1999, 162(12):7569-7577.
Weinberg K, Blazar BR, Wagner JE, Agura E, Hill BJ, Smogorzewska M, Koup RA, Betts MR, Collins RH, Douek DC. Factors affecting thymic function after allogeneic hematopoietic stem cell transplantation. Blood 2001, 97(5):1458-1466.
Weiner HL, Cohen JA. Treatment of multiple sclerosis with cyclophosphamide: critical review of clinical and immunologic effects. Mult Scler. 2002, 8(2):142-154.
Wicker LS, Miller BJ, Mullen Y. Transfer of autoimmune diabetes mellitus with splenocytes from nonobese diabetic (NOD) mice. Diabetes 1986, 35(8):855–60.
Wills MR, Carmichael AJ, Weekes MP, Mynard K, Okecha G, Hicks R, Sissons JG. Human virus-specific CD8+ CTL clones revert from CD45ROhigh to CD45RAhigh in vivo: CD45RAhighCD8+ T cells comprise both naive and memory cells. J Immunol. 1999, 162(12):7080-7087.
Woodland DL, Dutton RW. Heterogeneity of CD4(+) and CD8(+) T cells. Curr Opin Immunol. 2003, 15(3):336-342.
Wu CJ, Chillemi A, Alyea EP, Orsini E, Neuberg D, Soiffer RJ, Ritz J.Reconstitution of T-cell receptor repertoire diversity following T-cell depleted allogeneic bone marrow transplantation is related to hematopoietic chimerism. Blood. 2000, 95(1):352-9.
Wu Z, Bensinger SJ, Zhang J, Chen C, Yuan X, Huang X, Markmann JF, Kassaee A, Rosengard BR, Hancock WW, Sayegh MH, Turka LA. Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med. 2004, 10(1):87-92.
Yasumizu R, Sugiura K, Iwai H, Inaba M, Makino S, Ida T, Imura H, Hamashima Y, Good RA, Ikehara S. Treatment of type 1 diabetes mellitus in non-obese diabetic mice by transplantation of allogeneic bone marrow and pancreatic tissue. Proc. Natl Acad. Sci. USA 1987, 84(18):6555-6557.
Zorina TD, Subbotin VM, Bertera S, Alexander AM, Haluszczak C, Gambrell B, Bottino R, Styche AJ, Trucco M. Recovery of the endogenous beta cell function in the NOD model of autoimmune diabetes. Stem Cells 2003, 21(4):377-388.
Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001, 50(3):521-33.
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo