he most asia
TRANSCRIPT

1
INTRODUÇÃO
Hemostasia é o processo pelo qual o corpo espontaneamente deixa de sangrar e mantém o
sangue no estado fluído dentro do compartimento vascular. A hemostasia contribui para a
homeostase pela tendência do corpo em manter a coagulação e o sangramento em equilíbrio.
(FIGURA 1).
FIGURA 1: Hemostasia: um sistema em equilíbrio
A hemostasia ocorre em três estágios: a hemostasia primária, a hemostasia secundária e a
fibrinólise. Durante a hemostasia primária, as plaquetas interagem com o subendotélio do vaso
injuriado e com outras plaquetas por meio de interações plaqueta-plaqueta. Um aglomerado de
plaquetas se forma com estas interações, e o plug hemostático formado é conhecido como plug
hemostático primário. O plug hemostático primário temporariamente assegura o sangramento,
mas ele é frágil e facilmente se desaloja da parede do vaso sangüíneo. Subseqüentemente, fitas
insolúveis de fibrina se depositam sobre o plug hemostático primário para torná-lo forte e estável
e para permitir o reparo da parede vascular sem posterior perda de sangue. A formação de fibrina
se resulta da atividade da hemostasia secundária. A fibrina é formada por uma série de complexas
reações bioquímicas de proteínas plasmáticas solúveis, chamadas fatores de coagulação, a medida
que eles vão se associando aos vasos sangüíneos injuriados e às plaquetas do plug plaquetário. O
plug ou coágulo que se forma é então chamado de plug hemostático secundário (FIGURA 2).

2
FIGURA 2: O sangramento ocorre após uma injúria do vaso sangüíneo. O sistema hemostático é
ativado para evitar a perda excessiva de sangue. A hemostasia ocorre em dois estágios. Durante a
hemostasia primária as plaquetas se agregam no local da injúria (plug hemostático primário).
Durante a hemostasia secundária, a fibrina se forma em volta do plug de plaquetas para formar o
plug de fibrina-plaqueta (plug hemostático secundário)
Após reparo da injúria vascular, componentes adicionais do sistema hemostático atuam
para romper a rede de fibrina e remover o coágulo num processo conhecido como fibrinólise.

3
Todas as fases e componentes do sistema hemostático são controlados por inibidores bioquímicos
e fisiológicos.
Entretanto, potenciais riscos estão associados com essa rápida hemostasia localizada. Por
exemplo: diante de um desequilíbrio poderá haver um sangramento excessivo ou uma trombose.
Desde que o processo da hemostasia envolve o consumo de plaquetas e fatores da coagulação, há
também limites quanto ao grau da injúria vascular que pode ser reparada. Se a área injuriada dos
vasos excede a capacidade das plaquetas e dos fatores da coagulação em promover a selagem e
em impedir o sangramento, complicações aparecem, e sem alguma forma de tratamento o
equilíbrio hemostático não é atingindo.
FUNÇÃO DO SISTEMA VASCULAR
O sistema vascular consiste de três tipos de vasos sangüíneos: artérias, veias e capilares.
Estrutura dos Vasos Sangüíneos:
A estrutura de todos os vasos sangüíneos é similar (FIGURA 3). Ela consiste de uma
cavidade central, o lúmen, através do qual o sangue flui. O lúmen é circundado por uma
monocamada de células endoteliais que separa o sangue dos tecidos subjacente. Elas fornecem
um ambiente protetor para os elementos celulares do sangue e os constituintes solúveis do
plasma. A superfície luminal das células endoteliais possui uma fina camada composta de
proteínas e de substâncias ricas em carboidratos (mucopolissacarídeos). Esta camada é chamada
de glicocalix. A superfície aluminal está ligada a uma membrana basal que consiste de uma única
forma de colágeno, o colágeno tipo IV, embebidas na matriz protéica. As camadas de tecido
abaixo da membrana basal variam em espessura e composição dependendo do tamanho e do tipo
do vaso sangüíneo. Essas camadas abaixo da superfície aluminal das células endoteliais formam
o subendotélio.
A FIGURA 3 mostra um corte transversal dos vasos. Nesta figura pode-se observar que as
camadas das artérias e das veias são distintas. A camada mais interna, a túnica íntima, é composta
de monocamadas de células endoteliais, de membrana basal e de tecido conectivo que as mantém
juntas e, nas artérias existe uma membrana elástica interna. A camada média, túnica média, é
mais espessa nas artérias do que nas veias. Nas artérias as células de músculo liso predominam e
são circundadas por tecido conectivo frouxo consistindo primariamente de fibras de elastina,
fibras de colágeno, fibras reticulares e proteoglicanas. Nas veias há somente umas poucas células
de músculo liso, menos fibras de elastina e uma matrix similar de tecido conectivo. A túnica

4
adventícia, a camada externa, é mais espessa nas veias do que nas artérias. Nessa camada há uns
poucos fibroblastos embutidos no colágeno e no tecido conectivo. Os fibroblastos sintetizam e
secretam as fibras e outros componentes da matriz.
FIGURA 3- Estrutura e Função dos Vasos Sangüíneos: comparação entre arteríolas, Vênulas e
Capilares – as células endoteliais, as células do músculo liso e os fibroblastos das arteríolas e das
vênulas sintetizam e secretam os componentes da matriz subendotelial do tecido conectivo e as
proteínas da membrana basal. Os capilares têm muito pouco tecido conectivo e são compostos
basicamente de células endoteliais e membrana basal.
Colágeno tipo1 Fibras de Elastina

5
Capilares:
Os capilares (FIGURA 3) compõem a maior área superficial de todos os tipos de vaso
sangüíneos, embora, individualmente, eles são os menores. Eles têm aproximadamente 5 a 10m
de diâmetro, o suficiente para passar uma única célula sangüínea. A luz (lúmen) de um capilar é
formada de uma única célula endotelial. O tecido abaixo da membrana basal de um capilar é
escasso e não contém células de músculo liso.
Veias:
Os vasos que aproximam ou deixam os capilares vão se tornando cada vez maior em
diâmetro à mediada que eles se aproximam do coração. As veias que imediatamente deixam os
capilares são as vênulas (FIGURA 3). As vênulas possuem diâmetro de aproximadamente 20 a
200m e são os principais locais da atividade hemostática após uma injúria traumática. As
vênulas são compostas de endotélio e membrana basal circundada por uma camada de tecido
conectivo extracelular com uns poucos fibroblastos e algumas células de músculo liso. O tecido
conectivo contém fibras de colágeno tipo I.
O fluxo sangüíneo nas veias é lento comparado com o das artérias, mas as válvulas
existentes nas veias de diâmetro intermediário e maior previnem o retorno e mantém o sangue
fluindo em direção ao coração.
Artérias:
O sangue é bombeado do coração para os tecidos através das artérias. O lúmen das artérias
torna-se progressivamente menor para formar os vasos pré-capilares, referidos como arteríolas
(FIGURA 3). As arteríolas são também um dos principais locais da hemostasia, são similares às
veias em relação a sua estrutura, exceto que uma membrana definitiva de tecido elástico envolve
a membrana basal.
Função dos Vasos Sangüíneos:
Após uma injúria, os vasos lesados iniciam a hemostasia. A primeira resposta dos vasos à
injúria é a constrição para reduzir o fluxo de sangue para a área injuriada e por conseqüência
evitar o extravasamento de sangue. A vasoconstrição ocorre imediatamente e perdura por um
período de tempo muito curto. Ela é, em parte, provocada por fatores neurogênicos e, em parte,
por várias moléculas regulatórias que interagem com receptores presentes na superfície das
células dos vasos sangüíneos. As moléculas incluem a serotonina e a tromboxana A2, ambas são

6
produtos da ativação plaquetária, e a endotelina que é produzida pelas células endoteliais. Por
outro lado, as células endoteliais sintetizam e secretam a prostaglandina I2 (PGI2). A PGI2
provoca vasodilatação das arteríolas. A vasodilatação aumenta o fluxo sangüíneo na área
injuriada e causa vermelhidão na pele.
Também após a injúria, as células endoteliais das vênulas contraem produzindo “gaps”
(espaços) entre elas. O fluido do plasma escapa para os tecidos e causa inchaço (edema). Este
fenômeno é chamado de permeabilidade vascular aumentada.
Fisiologicamente, a superfície das células endoteliais é carregada negativamente e repele as
proteínas circulantes e as plaquetas, que também são carregadas negativamente.
Bioquimicamente, uma ampla variedade das substâncias que são sintetizadas e secretadas pelas
células endoteliais contribui para um ambiente não reativo. Exemplos são o sulfato de heparana,
um mucopolissacarídeo do glicocalix, e a trombomodulina, uma proteína de membrana da célula
endotelial, ambas inibidoras da formação da trombina. A PGI2 por provocar vasodilatação dos
vasos sangüíneos é uma inibidora da ativação das plaquetas. Adicionalmente, as células
endoteliais sintetizam o ativador do plasminogênio tecidual (tPA) e um inibidor do tPA chamado
de inibidor do ativador do plasminogênio (PAI). As células endoteliais também produzem e
processam uma glicoproteina chamada fator de von Willwbrand (vWf). O vWf é produzido
pelas células endoteliais e armazenado em estruturas chamadas corpúsculos de Weible-Palade. O
vWf no subendotélio liga as fibras de colágeno na matrix extracelular e fornece um suporte para a
ligação das plaquetas no estágio inicial da formação do coágulo. A membrana da célula endotelial
também contém tromboplastina, que é exposta durante a injúria vascular e é responsável pela
ativação de uma das vias que leva a formação da fibrina durante a hemostasia secundária.
Quando a injúria endotelial ocorre, as plaquetas e as proteínas da coagulação (fatores da
cascata da coagulação), presentes no plasma, são expostos ao tecido subendotelial. As interações
entre os componentes do vaso e os componentes do plasma levam a formação do plug
hemostático.
PLAQUETAS NA HEMOSTASIA
O segundo componente principal do sistema hemostático são as plaquetas. As plaquetas
apresentam uma forma discóide, são células anucleadas e possuem um diâmetro de
aproximadamente 2 a 3m. A concentração normal das plaquetas no sangue é de 150 a
450x103/mm
3.

7
Produção das Plaquetas:
As plaquetas são produzidas na medula óssea a partir da CFU-GEMM (Stem cell
formadora de colônias para granulócitos, Eritrócitos megacariócitos e monócitos). Uma
concentração adequada de plaquetas no sangue periférico é mantida por um processo regulatório.
Dois tipos de fatores de regulação já foram descritos. Alguns atuam sobre a Stem Cell células
progenitoras, enquanto outros exercem influência sobre as células mais maduras da linhagem
megacariocítica. A Interleucina-3 (IL-3) e o fator estimulante de crescimento para granulócitos e
monócitos (GM-CSF) são fatores de crescimento que atuam de maneira sinérgica para estimular
as Stem Cells a diferenciar em células progenitoras megacariocíticas e também para induzir a
proliferação das mesmas.
O segundo tipo de fator de crescimento é chamado de trombopoietina. A trombopoietina
induz a maturação dos megacariócitos. Isso influencia diretamente o número de plaquetas que são
produzidas e a velocidade de liberação destas células para o sangue periférico.
Estágios de Desenvolvimento do Megacariócito:
As Stem Cell ao serem estimuladas pela IL-3 e pelo GM-CSF se diferenciam em
megacarioblastos. Os megacarioblastos seguem uma seqüência de maturação que é diferente das
outras linhagens celulares da medula óssea. No caso das plaquetas os megacarioblastos sofrem
uma maturação nuclear antes da maturação citoplasmática se iniciar. O processo de maturação
nuclear consiste de uma série de endomitoses ou ploidia. Em cada endomitose, o conteúdo de
DNA da célula é duplicado, mas a divisão celular não ocorre. O conteúdo celular, ou o nível de
ploidia, das células pode ser de 4N a 32N, ou até mesmo maior. O estágio 8N é o primeiro
estágio reconhecível no esfregaço de medula óssea e é nível de ploidia mais comum de ser
encontrada. A maturação citoplasmática pode começar no nível de ploidia igual a 8N ou maior.
Quatro estágios de desenvolvimento dos megacariócitos foram descritos de acordo com a
morfologia observada nos esfregaços corados por Romanowsky (FIGURA 4). As características
morfológicas de cada estágio de maturação estão descritas abaixo:
Estágio I: Os megacarioblastos têm um escasso citoplasma basofílico. Nenhum grânulo é
visível. O núcleo é usualmente redondo e o nucléolo pode ser visível. Os megacarioblastos
possuem um diâmetro de 6 a 24m.
Estágio II: Os promegacariócitos contêm grânulos azurófilos visíveis. O núcleo é lobulado
e pode aparecer endentado ou na forma de “ferradura de cavalo”. O nucléolo desaparece. Os

8
promegacariócitos possuem um diâmetro de 14 a 30m. O sistema de membrana citoplasmática
começa a desenvolver neste estágio, mas ele pode ser visto somente com microscopia eletrônica.
FIGURA 4: Seqüência de maturação das plaquetas. A Stem Cell comissionada (CFU-meg) sob a
influência de fatores humorais (GM-CSF e IL-3) diferencia em megacarioblasto 2N. A maturação
nuclear ocorre por meio de uma série de endomitoses. A maturação citoplasmática estimulada pela
trombopoietina, ocorre nas células na fase de ploidia 8N ou superior.
Estágio III: Os megacariócito são caracterizados pelo seu grande tamanho, 16 a 56m de
diâmetro e um grande número de grânulos e demarcação de membranas. O núcleo tem múltiplos
lobos e a quantidade de citoplasma é maior em relação aos estágios II e I.
Estágio IV: O núcleo é compactado, mas lobulado. O citoplasma é abundante. Os grânulos
são organizados dentro de regiões que são separadas por uma demarcação de membrana. As
células no estágio IV possuem 20 a 60m de diâmetro e são chamadas de megacariócitos
maduros.

9
Liberação das Plaquetas:
O mecanismo de liberação das plaquetas não é completamente entendido, mas acredita-se
que as plaquetas são liberadas em grupos chamados de proplaquetas. As proplaquetas são
liberadas para o sinusóide da medula óssea, onde elas são convertidas em plaquetas e liberadas
para o sangue periférico. O núcleo dos megacariócitos permanece na medula óssea, sendo
degradados e removidos pelas células do sistema retículo endotelial.
Aproximadamente cinco dias são requeridos para um megacarioblasto de desenvolver em
plaqueta. Dois terços das plaquetas que são liberadas no sangue periférico circula na corrente
sangüínea e um terço são seqüestradas pelo baço. O tempo de vida médio das plaquetas no
sangue periférico é de aproximadamente 9,5 dias.
Estrutura da Plaqueta:
As plaquetas não ativadas na circulação apresentam uma forma discóide com a superfície
lisa. Ao contrário das hemácias e dos leucócitos as plaquetas possuem várias aberturas
semelhantes aos furos de uma esponja. Estas aberturas são canais membranosos que estendem até
o fundo do citoplasma da plaqueta.
Após uma injúria, ocorrem uma série de mudanças que afetam a morfologia e a bioquímica
da plaqueta. As mudanças causam a ativação das plaquetas e somente após esse fenômeno é que
as mesmas são capazes de formar o tampão hemostático primário.
A ultraestrutura da plaqueta é dividida em quatro regiões ou zonas arbitrárias: a zona
periférica, a zona estrutural, a zona organelar, e os sistemas de membrana (FIGURA 5)
FIGURA 5: Diagrama das características ultra-estruturais da plaqueta não ativada

10
Zona Periférica:
A zona periférica da plaqueta consiste de uma membrana citoplasmática, tendo o seu lado
externo coberto com uma camada felpuda e a sua porção interna revestida por uma fina região
submembranosa entre a zona periférica e a próxima camada.
A superfície externa coberta com a camada felpuda é também chamada de glicocalix. Ele é
composto de várias glicoproteínas, proteínas, e mucopolissacarídeos que são provavelmente
adsorvidos do plasma. Estão incluídos no glicocalix os fatores V, VIII e fibrinogênio da cascata
da coagulação. O glicocalix também é encontrado na membrana superficial dos canais internos.
Algumas das proteínas da membrana superficial da plaqueta são receptores para substâncias que
estimulam a ativação da plaqueta.
A membrana citoplasmática da plaqueta é composta de uma bicamada fosfolipídica e
proteínas integrais. O arranjo assimétrico dos fosfolipídeos é um fator importante na função das
plaquetas ativadas. As proteínas integrais apresentam em sua composição açúcares e por isso são
chamadas de glicoproteínas. Elas são abreviadas em “Gp” e são numeradas em algarismos
romanos de I a IX de acordo com a migração eletroforética de cada glicoproteína. Entre estas
glicoproteínas quatro são de extrema importância para a função da plaqueta, as quais receberão
um maior enfoque.
Glicproteína Ib (FIGURA 6a) é receptor para o fator de von Willebrand (vWf). Ela está
complexada com a Gp IX na membrana, mas a função da Gp IX é desconhecida. A GpIb possui
duas cadeias, chamadas (alfa) e (beta). A cadeia é grande e contém sítios de ligação para o
vWf, trombina, ristocetina (usada nos testes de agregação plaquetária), e auto-anticorpos
produzidos em pessoas sensíveis à quinidina. Cada plaqueta contém aproximadamente 25.000
moléculas de GpIb.
O complexo glicoproteina IIb/IIIa (FIGURA 6b) é um receptor para o fibrinogênio. Ele
também liga a outras proteínas adesivas, tais como o vWf, a trombospondina, a vitronectina e a
fibronectina. Há aproximadamente 50.0000 moléculas de GpIIb/IIIa por plaqueta. A GpIIb tem
duas cadeias, a cadeia (alfa) e (beta)O lado citoplasmático das duas proteínas estão
associados com a actina do citoesqueleto da plaqueta. O complexo GpIIb/IIIa está oculto na
plaqueta não ativada e aparecem somente quando as plaquetas são ativadas. Ele é requerido para
o processo de agregação plaquetária.
O ácido aracdônico, um ácido graxo não insaturado, é o principal componente da porção
fosfolipídica da membrana. Ele é um precursor dos muitos estimuladores que provocam
agregação plaquetária e a constrição dos vasos.
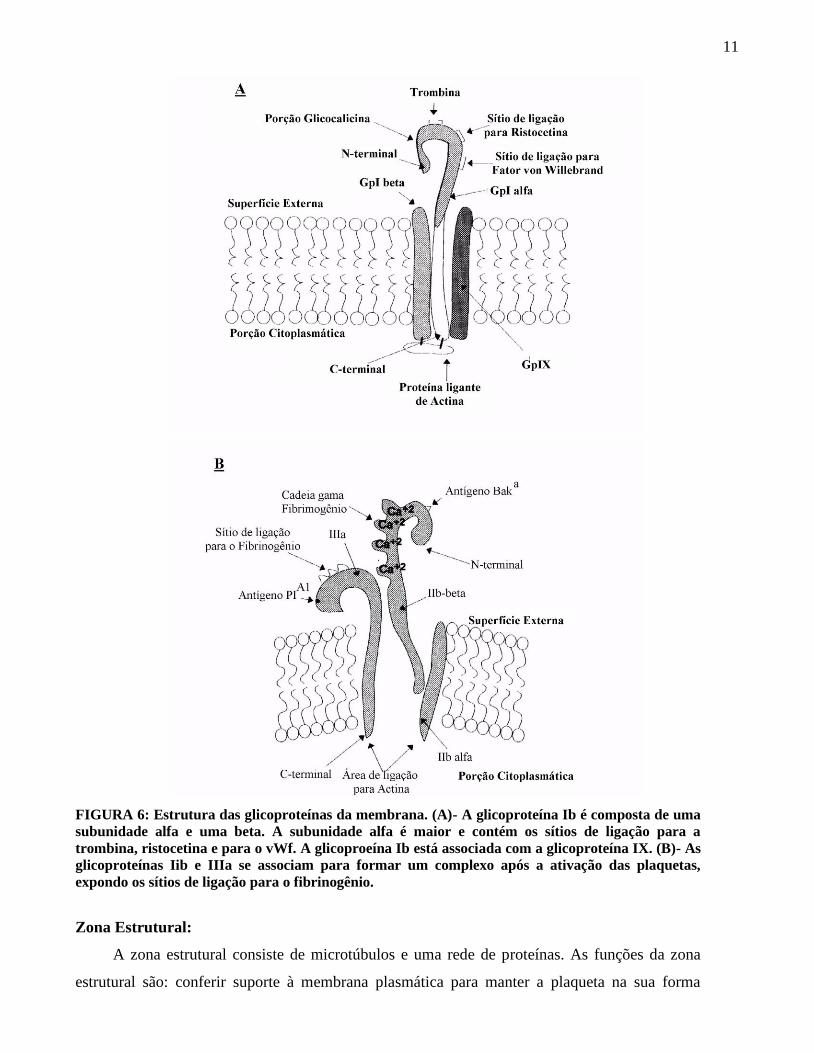
11
FIGURA 6: Estrutura das glicoproteínas da membrana. (A)- A glicoproteína Ib é composta de uma
subunidade alfa e uma beta. A subunidade alfa é maior e contém os sítios de ligação para a
trombina, ristocetina e para o vWf. A glicoproeína Ib está associada com a glicoproteína IX. (B)- As
glicoproteínas Iib e IIIa se associam para formar um complexo após a ativação das plaquetas,
expondo os sítios de ligação para o fibrinogênio.
Zona Estrutural:
A zona estrutural consiste de microtúbulos e uma rede de proteínas. As funções da zona
estrutural são: conferir suporte à membrana plasmática para manter a plaqueta na sua forma

12
discóide e para proporcionar mudança de forma quando a plaqueta é ativada. Os microtúbulos são
compostos de uma proteína chamada tubulina. Eles são importantes para manter a plaqueta em
sua forma discóide.
A rede de proteínas consiste de actina, de proteína ligante de actina e de várias outras
proteínas estruturais (FIGURA 7). A actina é a proteína mais abundante nas plaquetas e
representam 15 a 20% do total de proteína encontrada na plaqueta. A actina apresenta duas
formas, G ou globular e F ou filamentosa. A forma F consiste de várias moléculas G
polimerizadas. A proteína ligante de actina está ligada à porção citoplasmática no complexo
GpIb/IX e ancora a actina à membrana. A actina também faz parte de uma rede de suporte ao
longo de todo o citoplasma. No citoplasma ela está associada com a miosina e outras proteínas
contráteis.
FIGURA 7- Citoesqueleto e rede de citoesqueleto na plaqueta não ativada. O citoesqueleto é
composto de actina F e proteína ligante de actina associada com as glicoproteínas integrais da
bicamada fosfolipídica. A rede de citoesqueleto no citoplasma consiste primariamente de actina F e
actina G, de proteína ligante de actina e de miosina que estão distribuídas aleatoriamente no
citoplasma da plaqueta.
Zona Organelar:
A zona organelar fica abaixo da camada de microtúbulos e consiste de mitocrôndrias,
partículas de glicogênio, e no mínimo três tipos de grânulos dispersos dentro do citoplasma:
corpúsculos densos, grânulos alfa, e grânulos lisosomais. Os grânulos densos contêm os seguintes

13
mediadores não protéicos da função plaquetária e da hemostasia: ADP, ATP, e outros
nucleotídeos, íons cálcio e serotonina. O ADP nos corpúsculos densos é conhecido como ADP
não metabólico ou ADP do “Pool” de estoque para distinguir do ADP metabólico encontrado no
citoplasma. O ADP metabólico fornece energia para o metabolismo normal da plaqueta, enquanto
que o ADP de estoque é importante para as reações de agregação plaquetária.
Os grânulos alfa são os mais numerosos de todos os três tipos de grânulos. Os grânulos alfa
contêm proteínas hemostáticas, tais como: o vWf, fator V e fibrinogênio e proteínas com funções
variadas, incluindo fatores de crescimento e o inibidor do ativador do plasminogênio (PAI-1).
Os grânulos lisosomais contêm várias enzimas hidrolíticas e são similares aos lisosomas
encontrados nas outras células.
As plaquetas também contêm enzimas da via glicolítica, do ciclo de Kreb´s, e da síntese e
degradação do fibrinogênio.
Sistema de Membrana:
Um tipo de membrana chamado sistema canicular aberto conectado à superfície é a
membrana que circunda os canais que vão da superfície da plaqueta para o interior da plaqueta.
Um segundo tipo de membrana é o sistema tubular denso, que tem origem no retículo
endoplasmático rugoso. É um dos locais de estoque para os íons cálcio. Os canais do sistema
tubular denso não conectam com a superfície da plaqueta.
Os dois sistemas de membrana, o sistema canicular aberto e o sistema tubular denso, se
fundem em várias áreas do citoplasma da plaqueta.
Função das Plaquetas:
As plaquetas estão envolvidas em vários aspectos da hemostasia, tais como: vigilância da
continuidade do vaso sangüíneo, formação do tampão hemostático primário, formação do tampão
hemostático secundário e cicatrização do tecido injuriado. A função das plaquetas em manter a
continuidade dos vasos sangüíneos parece controvérsia, mas tem sido demonstrado que estas
células aderem nos pequenos “gaps” (aberturas) provocadas pela separação das células
endoteliais. As plaquetas aderem às fibras de colágeno do subendotélio que se expõe por meio
dos “gaps” e previnem o extravasamento sangüíneo.
Quando a injúria vascular ocorre e há uma quebra na continuidade da parede dos vasos, as
plaquetas reagem para formar um agregado conhecido como tampão plaquetário primário. O

14
sangramento pára porque as aberturas nos vasos são mecanicamente fechadas por uma massa de
plaquetas.
Após a formação do tampão hemostático primário, os fosfolipídeos da membrana das
plaquetas agregadas ativam reações de superfície para a formação de fibrina. A fibrina estabiliza
o tampão hemostático primário e a rede de fibrina em conjunto com as plaquetas é chamada de
tampão hemostático secundário.
As plaquetas secretam mediadores que ajudam a selar os tecidos injuriados. O fator de
crescimento derivado das plaquetas, um mitogênio estocado nos grânulos alfa, estimula as células
do músculo liso e possivelmente os fibroblastos a multiplicar e substituir as células que foram
lesadas pela injúria.
PARTICIPAÇÃO DAS PLAQUETAS NA HEMOSTASIA PRIMÁRIA
Formação do Tampão Hemostático Primário:
As plaquetas são discóides e inertes na circulação quando o endotélio vascular está íntegro.
A injúria no vaso sangüíneo provoca uma mudança no ambiente. As plaquetas respondem à
mudança se tornando ativadas. O tampão hemostático primário é o resultado da transformação
das plaquetas inativas para plaquetas ativadas. O tampão hemostático primário se forma numa
seqüência específica de etapas que são chamadas de adesão, ativação, agregação e secreção
(FIGURA 8).
FIGURA 8: Diagrama das plaquetas formando o tampão hemostático primário. A injúria vascular
estimula as plaquetas a aderir ao colágeno subendotelial. Após a adesão as plaquetas mudam de
forma, se agregam e secreta o conteúdo de seus grânulos. Plaquetas adicionais se tornam ativadas
pelas substâncias secretadas e se agregam para formar uma barreira no local da lesão.

15
Adesão das Plaquetas:
Quando o endotélio é lesado e o sangramento ocorre, as plaquetas escapam dos vasos
sangüíneos e atingem os tecidos subendoteliais. Elas imediatamente aderem aos componentes do
subendotélio. As fibras de colágeno são componentes de extrema importância neste processo. As
superfícies subendoteliais possuem componentes que em condições normais as plaquetas não são
expostas. A adesão das plaquetas ao colágeno ocorre somente com a ajuda do vWf e da GpIb da
membrana da plaqueta.
O vWF, que é sintetizado pelas células endoteliais, é estocado e secretado por estas células
na área subendotelial e no plasma. No subendotélio, o vWf é adsorvido nas fibras de colágeno. O
vWf também é encontrado nos grânulos alfa das plaquetas. A FIGURA 9 mostra que a molécula
do vWf é uma glicoproteina grande e possui uma série de 2 a 50 subunidades idênticas. Cada
subunidade tem receptores pra o colágeno e para a GpIb. Em situações de normalidade, o vWF
das plaquetas não é incorporado ao “Pool” plasmático circulante. O vWF, produzido pela célula
endotelial, forma no plasma um complexo não-covalente com o Fator VIII coagulante (FVIII:C).
FIGURA 9: O complexo Fator VIII/vWF é composto de uma proteína procoagulante que é
codificada por um gene localizado no cromossoma X (Fator VIII) e por um multímeros que atua na
adesão plaquetária. Para uma adequada adesão plaquetária, os complexos formados pelos
multímeros maiores são necessários.
Essa ligação além de conferir estabilidade ao FVIII:C, pelo aumento da sua sobrevida, ela
também protege o referido fator contra a inativação proteolítica e assim potencializa a sua

16
atividade como co-fator da cascata da coagulação. A outra grande função do vWF é a de
promover a adesividade plaquetária ao subendotélio. Estas funções do vWF decorrem da sua
peculiar estrutura, já que ela é formada por multímeros de diversos tamanhos (pequenos,
intermediários e grandes). Os multímeros de maior tamanho são os que propiciam a ligação das
plaquetas ao subendotélio, enquanto que os de menor tamanho são os que, preferencialmente, se
ligam ao FVIII:C.
Quando a adesão da plaqueta ocorre, o vWf se liga ao colágeno e à GpIb presente na
membrana da plaqueta, tornando uma ponte de conexão entre a plaqueta e as fibras de colágeno.
(FIGURA 10). Como as ligações continuam as plaquetas vão formando um verdadeiro “zipper”
pela ligação de seus receptores às várias proteínas de adesão que estão presentes na matrix do
tecido conectivo. Muitas plaquetas se aderem ao subendotélio formando uma monocamada de
plaquetas com a finalidade de cobrir a superfície de colágeno.
FIGURA 10: Adesão das plaquetas ao subendotélio por intermédio do Fator de von willebrand,
após a injúria do vaso sangüíneo. A glicoproteína IB está envolvida no processo de adesão das
plaquetas ao subendotélio vascular.
Ativação das Plaquetas:
A adesão das plaquetas às fibras de colágeno via o vWf desengatilha uma série de
mudanças morfológicas e funcionais conhecidas como ativação plaquetária. A ativação é um
processo complicado que não é totalmente entendido ainda. Sabe-se que no processo de ativação
ocorre mudança no metabolismo bioquímico, na forma, nos receptores de superfície e na

17
orientação dos fosfolipídeos da membrana. Somente as plaquetas ativadas são capazes de dar
continuidade às etapas subseqüentes do processo de formação do tampão hemostático primário.
O agente que induz a ativação da plaqueta é chamado de agonista. Cada agonista liga a um
receptor específico da plaqueta e causa uma série de reações no citoplasma da plaqueta. As
mudanças bioquímicas se iniciam quando o vWf se liga ao colágeno e à GpIb presente na
membrana da plaqueta. Quando isso ocorre, as enzimas na membrana se tornam ativadas e
clivam de maneira específica os fosfolipídeos de membrana. Os produtos resultantes são
segundos mensageiros que entram no citoplasma da plaqueta e transfere o sinal para as estruturas
internas da célula. Muitas reações são subseqüentemente ativadas pelos segundos mensageiros.
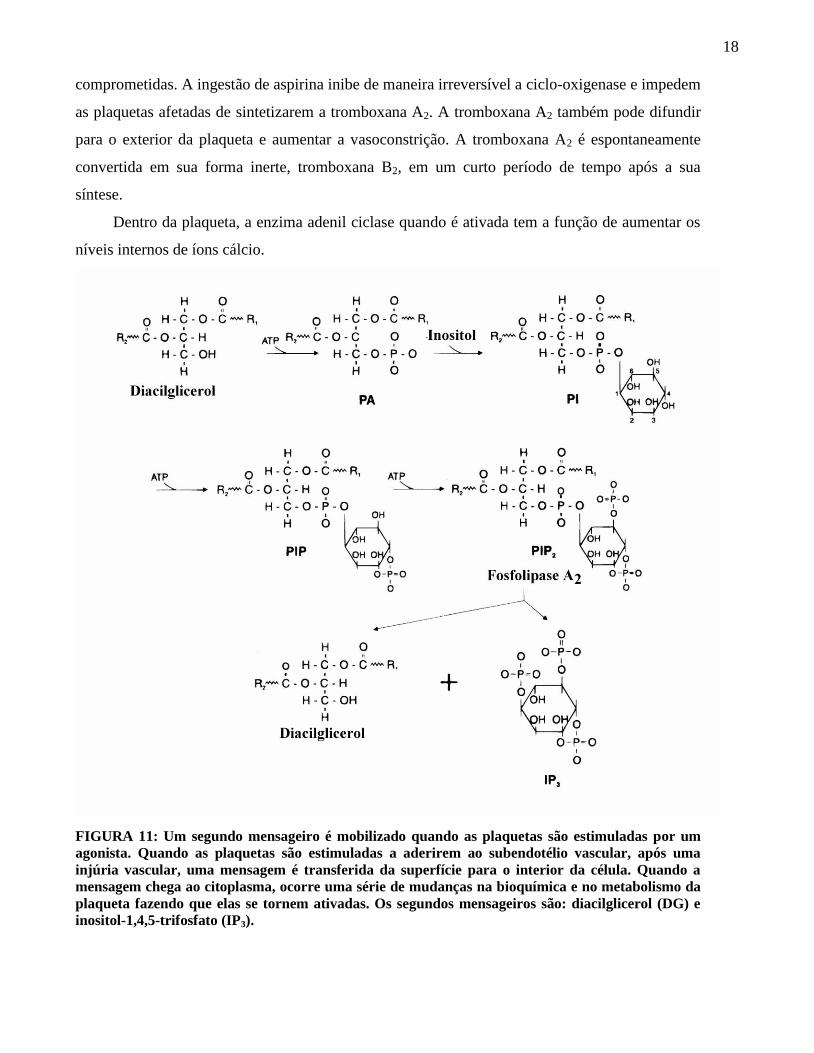
Os segundos mensageiros são produtos de três enzimas de membrana: a fosfolipase C, a
fosfolipase A2 e a adenil ciclase. Os produtos de todas as três enzimas causam um rápido
movimento intracelular de íons cálcio dos locais de estoque, sistema tubular denso, e também do
lado externo, plasma, para o citoplasma da plaqueta. As plaquetas não ativadas têm baixos níveis
de íons cálcio no citoplasma. Muitos sistemas que são lentos nas plaquetas ativadas se tornam
rápidos na presença de íons cálcio.
O substrato para a fosfolipase C é um fosfolípede derivado do fosfatidilinositol (PI),
presente na porção interna da bicamada. Um ou dois grupos fosfatos podem ser enzimaticamente
adicionados ao PI via ATP nas posições 4 e 5 do inositol para formar o fosfoinositol-4-
monofosfato e o fosfoinositol-4,5-bifosfato (PIP2), também chamado de difosfatidilinositol e
trifosfatidilinositol, respectivamente.
A fosfolipase C ao romper o PIP2 forma dois produtos, o inositol-1,4,5-trifosfato (IP3) e o
diacilglicerol. O IP3 estimula uma série de reações que resulta na liberação de íons cálcio do
sistema tubular denso. O diacilglicerol ativa as proteínas kinase C, que irá fosforilar outras
proteínas. As proteínas fosforiladas pela Kinase C ativa a secreção dos grânulos e o aparecimento
dos receptores para o fibrinogênio, a GpIIb/IIIa (FIGURA 11).
A Fosfolipase A2 é estimulada pelo aumento da concentração intracitoplasmática de íons
cálcio. A fosfolipase A2 atua sobre o fosfatilinositol e a fosfatidilcolina liberando o ácido
aracdônico. O ácido aracdônico é um ácido graxo monoinsaturado e um precursor de uma
variedade de substâncias regulatórias. Na plaqueta, a tromboxana A2 é sintetizada a partir do
ácido aracdônico pela enzima ciclo-oxigenase e tromboxana A2 sintetase (FIGURA 12).
Dentro da plaqueta, a tromboxana A2 estimula a secreção dos grânulos plaquetários. A
secreção plaquetária não ocorre de maneira normal se a síntese da tromboxana A2 estiver
bloqueada e conseqüentemente as etapas subseqüentes da função plaquetária ficarão seriamente
18
comprometidas. A ingestão de aspirina inibe de maneira irreversível a ciclo-oxigenase e impedem
as plaquetas afetadas de sintetizarem a tromboxana A2. A tromboxana A2 também pode difundir
para o exterior da plaqueta e aumentar a vasoconstrição. A tromboxana A2 é espontaneamente
convertida em sua forma inerte, tromboxana B2, em um curto período de tempo após a sua
síntese.
Dentro da plaqueta, a enzima adenil ciclase quando é ativada tem a função de aumentar os
níveis internos de íons cálcio.
FIGURA 11: Um segundo mensageiro é mobilizado quando as plaquetas são estimuladas por um
agonista. Quando as plaquetas são estimuladas a aderirem ao subendotélio vascular, após uma
injúria vascular, uma mensagem é transferida da superfície para o interior da célula. Quando a
mensagem chega ao citoplasma, ocorre uma série de mudanças na bioquímica e no metabolismo da
plaqueta fazendo que elas se tornem ativadas. Os segundos mensageiros são: diacilglicerol (DG) e
inositol-1,4,5-trifosfato (IP3).

19
FIGURA 12: Síntese das prostaglandinas nas plaquetas e nas células endoteliais durante a formação
do tampão de plaquetas.
A ativação continua com a mudança da forma da plaqueta quando os níveis internos de
cálcio atingem um limiar. A mudança de forma é a transformação de uma forma discóide para
uma forma esférica contendo prolongamentos, chamados pseudópodes, em sua superfície.
(FIGURA 13). Esta mudança de forma envolve as proteínas da zona estrutural, incluindo as
proteínas do citoesqueleto, a actina e a miosina da malha citoplasmática e os microtúbulos. O
resultado da mudança de forma é que cada plaqueta passa a ter uma maior área superficial de
membrana para as reações bioquímicas e uma maior chance de contato com outras plaquetas.
Além disso, a ativação das plaquetas provoca o aparecimento do receptor GpIIb/IIIa ao qual o
fibrinogênio se liga. Esse receptor é oculto nas plaquetas não ativadas e aparece logo após a
ativação das plaquetas por algum agonista. As plaquetas são capazes de ligar ao fibrinogênio
somente após o aparecimento do receptor GpIIb/IIIa. O íon cálcio é necessário para a ligação do
fibrinogênio à plaqueta.

20
FIGURA 13: Mudança de forma da plaqueta após estímulo por um agonista. Ocorre o
aparecimento de pseudópodes sobre a superfície da plaqueta e uma rede intracitoplasmática de
actina e miosina. Ocorre mudanças bioquímicas e degradação enzimática de fosfolípedes de
membrana para a formação de segundos mensageiros, exposição dos receptores glicoproteína
IIb/IIIa para a ligação de fibrinogênio e secreção dos grânulos.
Nas plaquetas não ativadas a glicoproteina IIb e a glicoproteina IIIa são entidades
separadas. Muitos autores descrevem a natureza do aparecimento do receptor GpIIb/IIIa como
uma fusão das duas glicoproteínas após a estimulação por um agonista (FIGURA 14)
FIGURA 14: As etapas iniciais da agregação plaquetária. Após estímulo por um agonista, os
receptores glicoproteína IIb/IIIa para a ligação de fibrinogênio são expostos sobre a superfície da
plaqueta. O fibrinogênio forma uma ponte entre duas ou mais plaquetas.

21
Um outro aspecto da ativação é uma mudança na superfície da membrana plaquetária que
permite as proteínas formadoras de fibrina (fatores da cascata de coagulação) ligarem a ela. Essa
função é conhecida como atividade procoagulante da plaqueta. As proteínas são ligadas em
complexos que permite a correta orientação das enzimas e das moléculas de substrato para a
formação da fibrina.
Agregação das Plaquetas:
Após as plaquetas serem ativadas, a formação do tampão hemostático primário continua
com uma fase conhecida como agregação plaquetária. A agregação plaquetária é a ligação de
uma plaqueta na outra. Novas plaquetas que se aproximam ao local do sangramento se tornam
ativadas pelo contato com agonistas, tais como o ADP, que são liberados pelas células endoteliais
lesadas e pelas plaquetas previamente ativadas. Com a ativação, as novas plaquetas mudam de
forma e os receptores GpIIb/IIIa se tornam expostos. As novas plaquetas ativadas então se ligam
àquelas que estão aderidas às fibras de colágeno.
A agregação ocorre em duas fases, primária e secundária. Durante a agregação primária as
plaquetas se aderem frouxamente umas às outras e se o estímulo pelo agonista for fraco, a
agregação primária se desfaz. Já a agregação secundária requer um maior período de tempo para
acontecer e começa somente quando as plaquetas iniciam a liberação do seu ADP de estoque,
como também a liberação de outros conteúdos presentes em seus grânulos e a sintetizar
tromboxana A2. As substâncias liberadas atuam como agonistas para dar continuidade ao
processo de estimulação. Se as plaquetas são incapazes de liberar ADP e/ou sintetizar
tromboxana A2, a agregação secundária não ocorre e por conseqüência elas se desagregam.
O fibrinogênio e o cálcio extracelular são necessários para a agregação ocorrer. Ambos são
constituintes plasmáticos. Ambos são liberados pelas plaquetas a partir de seus grânulos internos
de estocagem com o intuito de atingir altas concentrações na área injuriada. Uma única molécula
de fibrinogênio é capaz de se ligar em duas plaquetas por causa da sua estrutura molecular. O
fibrinogênio é uma molécula trinodular e composta por duas metades idênticas. A molécula
possui três pares de cadeias polipeptídicas, sendo duas cadeias alfa, duas cadeias beta, e duas
cadeias gama. Estas cadeias estão unidas por três ligações de sulfeto, uma ligação entre as cadeias
alfa e duas entre as cadeias gama (FIGURA 15). As duas metades do fibrinogênio são unidas na
porção amino-terminal das três cadeias, formando um nódulo central chamado de domínio central
ou domínio E. As regiões carboxi-terminal das cadeias beta e gama em conjunto com uma curta
seqüência da cadeia alfa formam os dois nódulos terminais, um em cada extremidade da

22
molécula. Esses nódulos são também referidos como domínios terminais ou domínio D. Uma
molécula de fibrinogênio pode ligar aos receptores GpIIb/IIIa de duas plaquetas diferentes pelos
seus sítios de ligação nas cadeias alfa e gama de seus domínios D (FIGURA 15).
Aproximadamente 16 a 80.000 moléculas de fibrinogênio estão ligadas em cada plaqueta ativada.
Durante um curto intervalo de tempo, a ligação do fibrinogênio é reversível, mas após cerca de
10 a 30 minutos ela se torna irreversível.
As glicoproteínas: vWf, trombospondina, fibronectina e as proteínas de adesão celular
também se ligam ao receptor GpIIb/IIIa da plaqueta.
FIGURA 15: O fibrinogênio é uma molécula de estrutura trinodular composta de três pares de
cadeias polipeptídicas (A, B e ) unidas entre si por ligações de sulfeto. Os três nódulos são
denominados domínios D e E, sendo dois domínios D nas duas extremidades e o domínio E na região
central da molécula. A trombina remove os peptídeos pequenos, A e B das cadeias e para
formar os monômeros de fibrina.
Secreção (liberação) pelas Plaquetas:
Após a adesão, mudança de forma e agregação primária, as plaquetas começam a
descarregar o conteúdo de seus grânulos na área circundante. O processo é conhecido como
secreção ou liberação. A secreção é dependente de energia e requer ATP. Isso ocorre
gradualmente por um período de tempo e antes ou concomitante com a agregação secundária.

23
A secreção ocorre em dois caminhos. Por um mecanismo, o sistema canicular aberto se
funde com a membrana dos grânulos. O conteúdo é então secretado pelo sistema canicular aberto.
Por um caminho alternativo, as membranas de alguns grânulos se fundem umas com as outras e
por fim com a membrana plasmática da plaqueta para a secreção de seu conteúdo.
Alguns das substâncias secretadas são agonistas que estimulam o aparecimento dos
receptores de membrana. O aparecimento dos receptores aumenta os níveis internos de íons
cálcio e com isso aumenta a taxa de conteúdo secretado.
Tampão Hemostático Primário:
As plaquetas quando ativadas se agregam para formar uma barreira que sela a injúria e
previne a perda de sangue. A barreira formada é chamada de tampão hemostático primário. O
tempo para o sangramento cessar depende da extensão e da profundidade da injúria, como
também do tamanho do vaso sangüíneo. Injúrias em que somente capilares e pequenos vasos são
afetados, usualmente cessam o sangramento dentro de 10 minutos.
PLAQUETAS E A HEMOSTASIA SECUNDÁRIA:
O tampão hemostático primário é relativamente instável e é facilmente desalojado. O
tampão plaquetário é estabilizado e firmemente ancorado à parede do vaso pelo processo da
hemostasia secundária. A hemostasia secundária começa com a formação de fibrina em volta das
plaquetas agregadas. Posteriormente a massa de fibrina-plaqueta se contrai para formar um
coágulo mais firme e coesivo. Essa contração é chamada de retração do coágulo. As plaquetas
participam da formação da rede de fibrina e da retração do coágulo.
Atividade Procoagulante das Plaquetas:
A ativação das plaquetas resulta na exposição de sítios de ligação para os fatores da cascata
de coagulação envolvidos na formação de fibrina sobre a superfície plaquetária. A ligação dos
fatores da cascata de coagulação aos receptores específicos presentes na membrana da plaqueta
fornece uma orientação correta da molécula para as reações enzimáticas ocorrer e para a
formação de trombina. A capacidade das plaquetas estimuladas em catalisar o processo da
cascata de coagulação pelo fornecimento da superfície fosfolipídica é conhecida como fator 3 da
plaqueta ou atividade procoagulante da plaqueta.

24
HEMOSTASIA SECUNDÁRIA
A hemostasia secundária ocorre quando as proteínas solúveis no plasma, conhecidas como
fatores da coagulação, se interagem em uma série de reações enzimáticas bastante complexas
para converter o fibrinogênio, uma proteína solúvel no plasma, em fibrina, proteína insolúvel. As
reações ocorrem em cascata de tal maneira que os fatores inativos da coagulação, os zimogênios,
são seqüencialmente ativados em enzimas (FIGURA 16).
FIGURA 16: Representação esquemática da ativação dos fatores na cascata da coagulação. Os
fatores circulam como proteínas inertes (zimogênios) até serem ativados a uma enzima por clivagem
proteolítica.
Cada zimogênio serve primeiramente como um substrato e posteriormente como uma
enzima. O substrato final da cascata é o fibrinogênio e quando atua como substrato para enzima
final, a trombina, o fibrinogênio é convertido em fibrina. A ativação da cascata começa quando
os zimogênios são expostos às camadas subendoteliais dos vasos sangüíneos. Todas as reações
enzimáticas exceto a última (formação de fibrina a partir do fibrinogênio) requerem uma
superfície fosfolipídica fornecida pela membrana das plaquetas ativadas e dos vasos injuriados. A

25
necessidade de uma superfície é importante para que haja uma localização das reações e da
formação de fibrina no local da injúria.
Todas as enzimas da cascata são serino-proteases, exceto o Fator XIII que é uma
transamidase. As reações originais são amplificadas muitas vezes pela presença dos cofatores e
da ativação positiva por feedback.
INTERAÇÕES E CONTROLE DA HEMOSTASIA
Os monômeros de fibrina se polimerizam formando uma malha em volta do tampão
hemostático primário semelhante a uma rede, produzindo uma barreira física estável para não
deixar escapar o sangue. Ambas as hemostasias primária e secundária são necessárias para a
formação normal do coágulo. Deficiências na hemostasia primária usualmente produz pequenos
pontos hemorrágicos por baixo da pele, chamados de petéquias e sangramento nas mucosas. Por
outro lado, deficiências na hemostasia secundárias produzem ecmoses, grandes hematomas e
hemorragias mais sérias nas juntas e nas cavidades do corpo.
O processo de formação da fibrina é um processo muito bem controlado, de tal forma que a
formação do coágulo é limitado ao vaso injuriado para evitar o espalhamento da ativação da
coagulação. A atividade proteolítica dos fatores ativados é limitada pelos inibidores naturais ou
reguladores. O processo de formação da fibrina é também controlado pelo feedback negativo; ou
seja, a trombina em grandes quantidades destrói os cofatores da cascata de coagulação de maneira
limitante à velocidade da sua própria produção.
Depois que o coágulo de fibrina desempenhou o seu papel de bloquear o extravasamento
sangüíneo, o vaso injuriado começa a ser reparado e a fibrina é digerida pela plasmina, uma
enzima do sistema fibrinolítico. A plasmina normalmente circula como uma proteína inerte, o
plasminogênio, e é ativada pelas serina-proteases da cascata de coagulação e pelo ativador do
plasminogênio tecidual (tPA), um fator liberado pelo endotélio do vaso sangüíneo injuriado.
FATORES DA CASCATA DA COAGULAÇÃO
Os fatores da cascata da coagulação são designados por algarismos Romanos, de I a XIII,
de acordo com a sua ordem de descoberta e não de acordo com a seqüência de reação em que eles
atuam. Cada fator tem um ou mais nomes. O fator VI é agora considerado a forma ativada do
fator V. Quando um Fator se torna ativado, ele tem uma atividade enzimática e a letra “a” é
adicionada como sufixo ao algarismo romano. Por exemplo, o Fator XII após ativação passa a ser

26
chamado de Fator XIIa. Há várias exceções desta terminologia. O Fator II (protrombina) em sua
forma ativada é preferencialmente conhecida como trombina e não Fator IIa, e quando o
fibrinogênio (Fator I) é clivado pela trombina ele é preferencialmente chamado de fibrina. A
fibrina é o produto final da casta da coagulação e não tem nenhuma atividade enzimática. A
tromboplastina tecidual e o cálcio não têm nenhuma forma ativada.
Fonte e Características dos Fatores da Cascata da Coagulação:
Os fatores da cascata da coagulação são sintetizados no fígado. O plasminogênio, do
sistema fibrinolítico, e os inibidores de protease também são sintetizados no fígado. Na doença
hepática severa, a concentração dessas proteínas pode estar marcadamente diminuída.
O Fator VIII é um complexo macromolecular composto de duas proteínas distintas: uma
porção pequena com atividade procoagulante (VIII:C) e uma porção multimérica grande que
serve para ligar as plaquetas ao colágeno (o fator de von Willebrand, vWf). A síntese do fator
VIII:C ocorre no fígado e a do vWF nas células endoteliais e nos megacariócitos.
De acordo com as propriedades físicas, os fatores da coagulação podem ser divididos em
três grupos: gruo da protrombina, grupo do fibrinogênio e o grupo dos fatores de contato
(TABELA 1).
Tabela 1- Grupos dos Fatores da Cascata da Coagulação de acordo com as Características Físicas
GRUPO DE CONTATO GRUPO DA PROTROMBINA GRUPO DO FIBRINOGÊNIO
Característica: requer o contato
com uma superfície para
ativação
XII
XI
Precalicreína
HMWK
Característica: requer Vitamina
K para a síntese funcional e
necessita de Ca2+
para ligar à
superfície fosfolipídica.
II
VII
IX
X
Característica: molécula de alto
peso molecular e ausente no
soro (consumidos durante a
coagulação).
I
V
VIII
XIII
As principais características dos fatores da cascata da coagulação estão descritas na
TABELA 2

27
Tabela 2- Algumas Características dos Fatores da Coagulação
Observações: Todas as informações colocadas nesta tabela são de origem humana.
CTP: componente da tromboplastina plasmática; ATP: antecedente da tromboplastina plasmática;
FSF: Fator estabilizante da fibrina; HMWK: cininogênio de alto peso molecular.
ATIVIDADE
Fator* Nome Descritivo
Bioquímica Local de Síntese
Meia-vida Biológica
Soro Adsorvido do Plasma por
BaSO4
Função [ ] Plasma (mg/L)
I Fibrinogênio Glicoproteina
multimérica Fígado
72-120
horas Ausente Inalterada
Precursor da fibrina da via
comum
2.500 -
3.000
II Protrombina Glicoproteina
multimérica Fígado (Vit.K)
67-106
horas Ausente Ausente
Pro-enzima e precursor da
trombina na via comum 70 - 150
V Pro-acelerina Glicoproteina
multimérica Fígado
12-36
horas Ausente Inalterada Cofator da via comum 4 - 14
VII Pro-convertina Glicoproteina
monomérica Fígado (Vit.K)
4-6
horas Aumentada Ausente Pro-enzima da via extrínsica 0,5
VIII-vWF Glicoproteina
multimérica Ausente Inalterada
Cofator da via intrínsica;
atua na adesão plaquetária 7
VIII:C Anti-hemofílico Glicoproteina
monomérica Fígado
10-14
horas Ausente Inalterada Cofator da via intrínsica 0,1
VWf Glicoproteina
multimérica
Endotélio
Megacariócito
22-40
horas Presente Inalterada Atua na adesão plaquetária ?
IX CTP Glicoproteina
monomérica Fígado (Vit.K)
18-40
horas Aumentada Ausente Pro-enzima da via intrínsica 4
X Fator de Stuart Glicoproteina
dimérica Fígado (Vit.K)
24-60
horas Inalterada Ausente Proenzima da via comum 10
XI ATP Glicoproteina
dimérica Fígado
48-84
horas Inalterada Lig. Diminuída Proenzima da via intrínsica 2 – 7
XII Fator Hageman Glicoproteina
monomérica ?
52-60
horas Inalterada Inalterada Proenzima da via intrínsica 27 – 45
XIII FSF Glicoproteina
multimérica
Fígado
Megacariócito
72-168
horas Diminuída Inalterada Proenzima da via comum 1
Précalicreína Précalicreína -globulina
monomérica Fígado ? Inalterada ?
Cofator da via intrínsica;
sistema cininas 50
HMWK HMWK -globulina
monomérica Fígado ? Inalterada ?
Cofator da via intrínsica;
sistema cininas 70-90

28
Grupo da Protrombina:
O grupo da protrombina inclui os Fatores II, VII, IX e X. Esses fatores têm uma massa
molecular de 50.000 a 100.000 Daltons e migram na eletroforese junto com a -globulina ou com
a -globulina. Íons cálcio são necessários para a ligação dos fatores a uma superfície rica em
ácido fosfolipídico (superfície negativa), onde os fatores são ativados em enzima.
A vitamina K tem uma função muito importante para que estes fatores sejam sintetizados
de maneira funcional. Desta forma, os fatores do grupo da protrombina são também conhecidos
como fatores dependentes de vitamina K. A vitamina K é encontrada em alguns óleos vegetais e
em folhosos (brócolis, couve-flor, alface,...). Ela também é sintetizada no intestino por várias
bactérias. A vitamina K é lipossolúvel e por isso é absorvida no trato gastrointestinal somente na
presença dos sais biliares. A vitamina K é necessária para a adição de um grupo carboxila (-
COOH) ao carbono dos resíduos de ácido glutâmico (-carboxilação) na porção amino-terminal
da cadeia polipeptídica (FIGURA 17). Essa modificação pós-traducional fornece um receptor
crítico para os íons cálcio, o qual proporciona a ligação do fator à superfície fosfolipídica. Assim
na ausência de vitamina K, os fatores são sintetizados no fígado e podem ser encontrados no
plasma, mas eles são totalmente não funcionais porque eles não têm grupos carboxilas (– COOH)
necessários para a sua ligação à superfície fosfolipídica.
FIGURA 17: Atuação da Vitamina K na -carboxilação dos Fatores II, VII, IX e X da Cascata da
Coagulação, e o mecanismo de ação dos antagonistas da Vitamina K.

29
Grupo do Fibrinogênio:
O grupo do fibrinogênio inclui os Fatores I, V, VIII e XIII. Esse grupo de fatores é também
referido como grupo consumível porque eles são consumidos durante a formação da fibrina e por
conseqüência estão ausentes no soro. Os Fatores I, V, e XIII têm massas moleculares de 300.000
a 340.000 Daltons. O Fator VIII é um complexo macromolecular com uma massa molecular de
aproximadamente 1.200.000 Daltons. Os fatores deste grupo migram na eletroforese junto com a
-globulina ou com a -globulina.
Grupo dos Fatores de Contato:
O grupo de contato inclui os Fatores XI e XII, e também as proteínas précalicreína e
cininogênio de alto peso molecular (HMWK). Esses fatores estão envolvidos na ativação inicial
da via intrínsica e requerem o contato com uma superfície carregada negativamente para se
tornarem ativos. O Fator XI e o Fator XII tem uma massa molecular de 80.000 Daltons e 165.000
Daltons, respectivamente, e migram na eletroforese junto com a -globulina ou com a -
globulina.
COAGULAÇÃO SANGÜÍNEA: TEORIA DA CASCATA
O processo da coagulação sangüínea envolve uma série de reações bioquímicas que
transforma substâncias circulantes solúveis em um gel insolúvel pela conversão do fibrinogênio
solúvel em fibrina. Esse processo requer proteínas plasmáticas (fatores da coagulação),
fosfolípides e cálcio.
A formação da rede de fibrina durante a coagulação sangüínea pode ocorrer por dois
caminhos distintos: a via extrínsica e a via intrínsica. Ambas as vias levam a ativação do fator X
que converge a cascata para uma via comum levando a formação de trombina, a qual irá
converter o fibrinogênio em fibrina. Assim, a formação do Fator Xa, da trombina e da fibrina
compõe a via comum. Conforme o exposto a coagulação sangüínea é então dividida em três vias:
via intrínsica, via extrínsica e via comum. Esta divisão foi feita de acordo com o modo e a
seqüência de ativação dos fatores da coagulação “in vitro”. A FIGURA 18 mostra que cada via
envolve reações entre um grupo específico de fatores da coagulação.

30
FIGURA 18: Fatores da cascata da coagulação das vias intrínsica, extrínsica e comum.
As três vias requerem uma ativação inicial, que leva a uma subseqüente ativação de outros
fatores sob a forma de uma cascata. De acordo com a teoria da cascata, cada fator da coagulação
é convertido em sua forma ativa por um fator precedente que também foi ativado por outras
reações bioquímicas. O cálcio ionizado (Ca2+
) participa como cofator em algumas destas reações.
Como cada reação é promovida por uma reação precedente, a deficiência de algum dos fatores
trará como conseqüência uma coagulação não acontecendo em velocidade normal e, portanto um
tempo maior para a formação do coágulo e um maior tempo de sangramento pelo vaso sangüíneo
que foi lesado.
A via intrínsica é ativada pelo contato das proteínas com as superfícies negativamente
carregadas. No entanto, a via extrínsica é ativada pelo contato do Fator VII com o Fator tissular,
também chamado de Fator III, Fator tecidual ou tromboplastina. As vias intrínsica e extrínsica se
convergem na via comum.
O termo extrínseco é usado porque a ativação desse caminho requer um fator que não
circula no sangue, o Fator III (tromboplastina ou Fator tecidual). O Fator tecidual é uma proteína
integral de membrana encontrada no tecido subendotelial e se torna exposta quando o vaso
sangüíneo é lesado. O Fator tecidual possui uma porção fosfolipídica que é a fonte de fosfolípides
necessários para a ativação da via extrínsica. (FIGURA 19).
Précalicreína HMWK Fator XII Fator XI Fator IX Fator VIII
Fator III
Fator VII
Fator X Fator V
Fator II (protrombina) Fator I (fibrinogênio)
FIBRINA
Via Comum
LENTO RÁPIDO
Via Intrínsica Via Extrínsica

31
FIGURA 19: Visão geral da Cascata da Coagulação
Via Extrínsica
Na via extrínsica, o Fator VII é ativado a Fator VIIa na presença de íons cálcio e Fator
tecidual (Fator III), que se torna exposto após a injúria vascular. Esta via é mais rápida porque ela
requer somente a participação do Fator VIIa, do Fator IV (Ca2+
) e do Fator III (tromboplastina
tecidual) para ativar o Fator X, não necessitando assim da ativação dos fatores XII, XI, IX e
VIII:C.
As FIGURAS 19 e 20 mostram que a via extrínseca fornece um meio para uma produção
muito rápida de pequenas quantidades de trombina necessária à formação de fibrina. Além disso,
a trombina gerada pela via extrínseca pode acelerar a via intrínsica pelo aumento da atividade dos
fatores V e VIII:C.
No laboratório, o Tempo de Protrombina (TP) é o teste usado para monitorar a via
extrínsica.

32
FIGURA 20: Via Extrínsica da cascata da coagulação
Via Intrínsica:
O Fator XII ao entrar em contato com o colágeno subendotelial, fosfolípedes ou
calicreína se torna ativado (FXIIa) para iniciar a formação do coágulo pela via intríseca. O Fator
XIIa é somente parcialmente ativado por esse contato. A precalicreína e o cininogênio de alto
peso molecular (HMWK) são adicionalmente necessários para aumentar ou amplificar os fatores
de contato envolvidos nesta via. (As FIGURAS 19 e 21).
FIGURA 21: Via Intrínsica da Cascata da Coagulação
Especificamente, o Fator XIIa na presença de HMWK converte a precalicreína em
calicreína. A calicreína por “feedback” acelera a ativação do Fator XII em Fator XIIa,
aumentando a atividade da via intrínsica.

33
A ativação do Fator XII atua como um “link” em muitos aspectos do sistema hemostático,
incluindo o sistema fibrinolítico, o sistema cininas e o sistema complemento, conforme
esquematizado na FIGURA 22.
FIGURA 22: Interrelação entre a cascata da coagulação e os sistemas fibrinolítico, cininas e
complemento.
A ativação por contato ocorre na ausência de íons cálcio (Ca2+
) e também envolve a
ativação do Fator XI em Fator XIa na presença de HMWK.
Uma vez formado, o Fator XIIa na presença de HMWK ativa o Fator XI. Vale ressaltar que
o Fator XIIa também é capaz de ativar o Fator XI mesmo na ausência de HMWK, porém a
ativação ocorre de maneira muito lenta. O Fator XIa uma vez presente e na presença de Ca2+
irá
ativar o Fator IX em Fator IXa. O Fator IXa formará um complexo com o Fator VIII:C, Ca2+
e
fosfolipídeos para ativar o Fator X. O Fator Xa formará a trombina necessária para a formação de
fibrina. O complexo consistindo do Fator IXa-Fator VIII:C-fosfolípides-Ca2+
é chamado de
complexo tenase porque ele ativa o Fator X. A FIGURA 23 mostra um esquema do complexo
macromolecular formado pelos fatores IXa, VIII:C, X, fosfolípedes e Ca2+
organizado sobre a
superfície da plaqueta ativada durante a coagulação sangüínea fornece um ambiente protetor que

34
facilita a cascata da coagulação sem a interferência dos anticoagulantes fisiológicos normalmente
presentes no plasma.
FIGURA 23: O complexo formado pela ativação seqüencial da via intrínsica (Fatores IXa, VIII,
Ca2+
) ativa o Fator X. O cofator, Fator VIII:C liga aos fosfolípides (PF3) na superfície da plaqueta e
serve para orientar os Fatores IXa e X a aumentar a formação do Fator Xa. Os Fatores IX e X são
ligados à plaqueta via interações com os íons Ca2+
.
A FIGURA 24 ilustra de maneira esquemática a molécula do Fator VIII. Nesta figura
observa-se que o Fator VIII é composto de vários componentes. A principal porção desta proteína
é o vWf, considerada ser a proteína carreadora do Fator VIII. O vWf não é ativo na cascata da
coagulação. A subunidade protéica menor é o Fator VIII responsável pela atividade coagulante
ou procoaculante e por isso é chamada de Fator VIII:C. O Fator VIII:C é que atua funcionalmente
na cascata da coagulação. A porção vWf do complexo carreia a porção procoagulante VIII:C.
Assim, a porção vWf exibe um efeito estabilizador sobre o Fator VIII:C e o protege da atividade
proteolítica existente no plasma. No laboratório, o teste usado para avaliar a atividade da via
intrínsica é o Tempo de Tromboplastina Parcial Ativado (TTPa).
FIGURA 24: A molécula do Fator VIII (VIII/vWF) é um polímero com múltiplas subunidades
compostas de vWF (vWF:Ag) conectadas a uma pequena unidade coagulante conhecida como
VIII:C.

35
Via Comum:
A via comum se inicia com a ativação do Fator X ou pela via extrínsica ou pela via
intrínsica ou por ambas as vias (FIGURAS 19 e 25). O Fator Xa, na presença do Fator V, de Ca2+
e de fosfolípides (PF3) converte a protrombina em trombina. A trombina uma vez formada irá
desempenhar as seguintes atividades: ativação por “feedback” dos fatores V e VIII; formação dos
monômeros solúveis de fibrina a partir do fibrinogênio; ativação do Fator XIII, que por sua vez
irá estabilizar a rede de fibrina, e indução da agregação plaquetária. A via comum contém os
fatores V, X, II e I, esses fatores podem ser monitorados tanto pelo Tempo de Protrombina (TP)
como pelo Tempo de Tromboplastina Parcial ativado (TTPa). Pela via extrínsica o Fator VIIa na
presença da tromboplastina tecidual e Ca2+
ativará o Fator X e pela via intrísica, o complexo
tenase (Fator IXa, na presença do Fator VIII:C, fosfolípides e Ca2+
) também ativará o Fator X.
FIGURA 25: Via comum da cascata da coagulação
O Fator Xa em conjunto com o Fator V, na presença de Ca2+
e fosfolípedes (PF3)
converterá o Fator II (protrombina) em Fator IIa (trombina). A FIGURA 26 mostra a associação
dos fatores Xa, Va, Ca2+
e PF3 (fosfolípides da membrana da plaqueta). Esta associação é
também chamada de complexo protrombinase porque ele enzimaticamente converte o substrato
protrombina em trombina, enzima ativa na formação dos monômeros de fibrina a partir do
fibrinogênio.
X

36
FIGURA 26: O complexo protrombinase (Fatores Xá, Va, Ca2+
e PF3) ativa a formação de
trombina a partir da protrombina. O Fator V liga aos fosfolípides (PF3) na superfície da plaqueta e
serve para orientar os Fatores Xa e a protrombina a aumentar a formação do Fator Xa. Os Fatores
Xa e protrombina são ligados à plaqueta via interações com os íons Ca2+
.
A trombina atua sobre o fibrinogênio para formar os monômeros de fibrina. A trombina
cliva uma ligação peptídica de cada cadeia alfa e de cada cadeia beta para formar os
fibrinopeptídeos A e B. Após essa clivagem o produto resultante da remoção dos fibrinopeptíeos
A e B é chamado de monômero de fibrina.
No entanto, o coágulo insolúvel de fibrina é formado em três etapas distintas:
1- clivagem hidrolítica das ligações arginina-glicina pela trombina liberando os
fibrinopeptídeos A e B das cadeias alfa e beta, com a formação dos monômeros de fibrina.
2- formação espontânea de um polímero instável de fibrina a partir dos monômeros de
fibrina.
3- estabilização do polímero de fibrina pelo Fator XIIIa.
Com a clivagem do fibrinogênio pela trombina, as cargas negativas presentes em volta do
domínio E, responsáveis pela repulsão eletrostática entre as moléculas de fibrinogênio, são
eliminadas. Como resultado, a carga líquida do domínio central muda de oito cargas negativas
para cinco cargas positivas. Os domínios D, entretanto, retém as suas cargas líquidas negativas.
As mudanças nas forças eletrostáticas desempenham uma função muito importante na
polimerização dos monômeros de fibrina. Os domínios D carregados negativamente de um
monômero espontaneamente se associam com o domínio E carregado positivamente de um outro
monômero para formar um polímero de fibrina (FIGURA 27).

37
FIGURA 27: A trombina cliva os peptídeos A e B do domínio E do fibrinogênio para formar os
monômeros de fibrina. A clivagem proporciona uma troca das cargas negativas por cargas positivas
no domínio E. Isso permite o crescimento espontâneo dos polímeros de fibrina a medida que as
cargas positivas do domínio E se interagem com as cargas negativas dos domínios D de outros
monômeros de fibrina. O polímero é inicialmente formado por pontes de hidrogênio e por
interações iônicas.
A reação final da formação do polímero de fibrina é a estabilização do polímero de fibrina
pelo Fator XIIIa. O Fator XIII é ativado pela trombina. O Fator XIIIa é uma transamidase
dependente de cálcio que é responsável pela catálise da formação de ligações covalentes entre os
resíduos de glutamina e lisina presentes no polímero de fibrina. Essas ligações covalentes
produzem uma rede de fibrina de maior força mecânica e de maior resistência à digestão
proteolítica pela plasmina.
Amplificação da Cascata:
A divisão da cascata da coagulação em via extrínsica e intrínsica está sendo abandonada,
porque a teoria da cascata foi modificada. A literatura descreve que o Fator VIIa da via extrínsica
pode diretamente ativar o Fator IX da via intrínsica. Além disso, o Fator VII pode ser ativado

38
pelos fatores XIIa, IXa, Xa, e pela trombina. De acordo com essa teoria, o Fator VII pode ser a
proteína chave para iniciar a coagulação sangüínea (FIGURA 19).
CONTROLE FISIOLÓGICO DA COAGULAÇÃO
O processo dinâmico de geração de fibrina pelos fatores ativados da coagulação é
normalmente limitado ao local da injúria vascular. Mesmo com uma intensa estimulação da
coagulação sangüínea, tal como ocorre em trauma massivo, a circulação permanece fluída nos
vasos não envolvidos.
Os mecanismos fisiológicos que são responsáveis pelo controle da coagulação são:
a) Fluxo sangüíneo
b) Remoção dos fatores ativados pelo fígado
c) Inibição por feedback
d) Inibidores bioquímicos (anticoagulantes fisiológicos)
e) Sistema Fibrinolítico
Fluxo Sangüíneo:
Um coágulo não se forma se não houver vasocronstrição e ativação dos fatores da cascata
da coagulação. Inicialmente, a formação do coágulo é aumentada pela vasoconstrição que
temporariamente diminui o fluxo sangüíneo no local da área injuriada. A vasoconstrição serve
para forçar as plaquetas e os fatores da coagulação entrar em contato com a superfície
subendotelial que se tornou exposta após a lesão vascular. Essa ativação promove a ativação da
hemostasia primária e secundária. O retorno do fluxo sangüíneo ao normal, após a formação da
rede de fibrina, na região da injúria vascular é importante para limitar a coagulação por meio de
uma diluição contínua dos fatores ativados, presentes no local, e retirando-os para serem
metabolizados.
Remoção dos Fatores Ativados pelo Fígado:
Como o sangue trás os fatores ativados da cascata da coagulação para o fígado, eles são
seletivamente removidos pelos hepatócitos. A plasmina do sistema fibrinolítico (sistema que será
discutido adiante) e os produtos de degradação da fibrina também são removidos pelo fígado.
Inibição por “Feedback”:
Alguns fatores ativados da cascata da coagulação têm potencial para destruir outros fatores
existentes na cascata. A trombina tem habilidade para temporariamente ativar os fatores V e VIII,

39
mas quando a concentração da trombina se torna muito alta, esses fatores são enzimaticamente
destruídos por ela. O Fator Xa primeiro aumenta a atividade do Fator VII e então, por meio de
uma reação com o inibidor do Fator tecidual, inibe a ativação de mais Fator X pelo complexo
Fator VIIa/Fator Tecidual. A coagulação também é controlada indiretamente pelo seu produto
final, a fibrina. A fibrina tem uma afinidade muito forte pela trombina. Uma vez adsorvida ao
complexo de fibrina, a trombina é liberada muito lentamente, limitando a quantidade de trombina
disponível para a formação de mais fibrina. Em adição, os produtos da degradação do polímero
de fibrina pela plasmina atuam inibindo a formação de mais fibrina a partir do fibrinogênio e da
polimerização dos monômeros de fibrina.
Inibidores Bioquímicos:
Os inibidores bioquímicos são proteínas solúveis no plasma que regulam as reações enzimáticas
das serino-proteases por inibir a ativação e a amplificação da cascata da coagulação.
Estes inibidores incluem: a anti-trombina III, o cofator II da Heparina, o inibidor da via do Fator
Tecidual, a proteína C e S, a 2-macroglobulina e a 1-anti-tripsina (FIGURA 28)
Características dos Inibidores:
Anti-trombina III (ATIII):
É uma Glicoproteína de massa molecular de 56kDa, produzida pelo fígado, pelas células
endoteliais e possivelmente pelos megacariócitos. É o inibidor mais importante da coagulação.
Sua função é inibir todas as serino-proteases presentes na cascata da coagulação, ou seja, os
fatores XIIa, XIa, IXa, Xa e IIa, como também a plasmina e a calicreína. A molécula de ATIII
forma um complexo estequiométrico de 1:1 com a TROMBINA. A HEPARINA, uma
glicosaminaglicana, ao se ligar na ATIII promove uma mudança conformacional na molécula
deste inibidor e essa mudança conformacional proporciona um aumento de cerca de 1000x na
velocidade de formação do complexo ATIII-TROMBINA.
Cofator II da Heparina:
É uma glicoproteína de 65kDa e inibe somente a trombina. Devido ao fato da afinidade da
heparina pelo Cofator II da heparina ser menor do que para a ATIII, uma alta concentração de
heparina é necessária para aumentar a inibição da trombina por esse cofator.

40
FIGURA 28: Interrelações entre as células endoteliais, proteína C, proteína S, antitrombina III e o
esquema da cascata da coagulação para a regulação da formação do trombo.
Inibidor da via do Fator Tecidual (TFPI):
É produzido no fígado, pulmões e células endoteliais. Este inibidor possui 3 domínios,
sendo dois com capacidade inibitória: um domínio inibe o complexo Fator VIIa/TF, o outro Inibe
diretamente o Fator Xa e o terceiro não apresenta função conhecida.
Proteína C e S:
A Proteína C é um anticoagulante dependente de vitamina K. Em 1976 ela foi isolada e
suas propriedades físico-químicas descritas. A ativação da proteína C requer a presença de íons
Ca+2
e trombomodulina, uma glicoproteína integral presente na membrana das células endoteliais
(FIGURA 28). A trombomodulina aumenta a ativação da Proteína C pela trombina. A
trombomodulina tem uma alta afinidade pela trombina e forma um complexo de 1:1. Quando
ligada à trombomodulina a trombina não pode clivar o fibrinogênio para formar fibrina, mas se
torna um potente iniciador da anticoagulação por ativar a Proteína C. A Proteína C ativada
rapidamente destrói os fatores Va e VIIIa, providenciando um mecanismo de regulação para a

41
formação de fibrina. A Proteína C ativada também contribui para a fibrinólise porque ela
enzimaticamente inativa o Inibidor da Ativação do Plasminogênio-tipo 1 (PAI-1).
A atividade da Proteína C ativada é potencializada pela Proteína S. A Proteína S também é
dependente de Vitamina K para a sua síntese de maneira funcional. Somente 40% da Proteína S
não ligada estão ativa na hemostasia. O restante (60%) está ligado ao C4b do complemento. A
Proteína S por ter uma alta afinidade para os fosfolípides de membrana, promove a ligação da
Proteína C aos fosfolípides existentes na membrana das plaquetas e das células endoteliais. Com
isso há uma aceleração da inativação dos fatores Va e VIIIa, que também estão adsorvidos na
superfície das plaquetas. A proteína S pode star diminuída nas mulheres que estão em uso de
contraceptivo oral. A deficiência das Proteínas C e S podem estar associada com trombose.
2-Macroglobulina:
- É uma glicoproteína com a função de inibir a trombina, plasmina e calicreína. A ligação
da 2-macroglobulina com a trombina é lenta e resulta numa clivagem proteolítica da 2-
macroglobulina seguida por uma redução na atividade da trombina. A trombina ligada à 2-
macroglobulina continua lentamente a formar fibrina a partir do fibrinogênio. Isso sugere que a
2-macroglobulina pode funcionar primariamente como um mecanismo de “clearance” das
serino-proteases, uma vez que os complexos serino-protease/2-macroglobulina são rapidamente
retirados do plasma.
SISTEMA FIBRINOLÍTICO
O sistema fibrinolítico é ativado em resposta à ativação da cascata de coagulação,
principalmente em resposta à ativação dos fatores de contato. A ativação do sistema fibrinolítico
produz uma enzima proteolítica, chamada plasmina, que é capaz de digerir, por proteólise a
fibrina e o fibrinogênio. Conforme a FIGURA 29, os principais componentes do sistema
fibrinolítico são: plasminogênio, ativadores do plasminogênio, plasmina, fibrina, produtos de
degradação da fibrina (FDP) e os inibidores da ativação do plasminogênio.
Plasminogênio:
O plasminogênio é uma -globulina que possui uma massa molecular de aproximadamente
80.000 Daltons e que circula no sangue sob a forma de um zimogênio. O plasminogênio é

42
sintetizado no fígado e grandes quantidades são adsorvidas no polímero de fibrina durante a
formação do coágulo.
FIGURA 29: O sistema fibrinolítico pode ser ativado pelo ativador do plasminogênio tecidual (tAP),
derivado das células endoteliais ou pelos ativadores intrínsicos e calicreína. A estreptoquinase, um
ativador exógeno, pode também ativar o sistema. A plasmina, o produto final da ativação, controla
a coagulação pela digestão do polímero de fibrina e pela inativação dos Fatores V, VIII e XII. Os
inibidores da ativação do plasminogênio (PAI-1 e PAI-2) e os inibidores da plasmina (2-
antiplasmina e 2-macroglobulina) servem para controlar o sistema fibrinolítico. A proteína C
ativada (PCa) inibe o PAI-1 e pode aumentar a liberação do tPA, aumentando assim a fibrinólise
A plasmina é uma enzima com especificidade tripsina-like e é formada a partir do
plasminogênio por uma série de ativadores que enzimaticamente hidrolisam este zimogênio. A
plasmina pode hidrolisar os fatores V e VIII, como também a fibrina e o fibrinogênio. Se a
plasmina ficar livre no plasma ela pode causar uma degradação proteolítica de muitos fatores da
coagulação, como também degradar componentes do sistema das cininas e do sistema
complemento. Esse processo proteolítico extremamente perigoso é controlado pela rápida
formação do complexo plasmina/2-antiplasmina quando a plasmina se encontra livre na
circulação.
Os ativadores do plasminogênio podem ser encontrados no sangue (ativadores intrínsecos)
e em muitos tecidos (extrínsicos). Além disso, algumas substâncias que normalmente não estão
Ativadores
PLASMINOGÊNIO
PLASMINA
FIBRINA FDP
Inibidores
PAI-1
PAI-2
PAI-3
PCa
t-PA (Extrínsico)
Streptokinase
Intrínsico
2-Antiplasmina
2-Macroglobulina

43
presentes no sangue durante a coagulação e fibrinólise (ativadores exógenos) podem ganhar
acesso à circulação em estados patológicos e ativar a formação de fibrina.
Ativadores Intrínsicos:
Estes ativadores estão presentes no sangue. Eles estão envolvidos com a fase de contato da
via intrínsica. O Fator XIIa interage com o pro-ativador do plasminogênio. Essa interação
promove a conversão do pro-ativador em ativador do plasminogênio que subseqüentemente cliva
o plasminogênio para formar a plasmina.
Ativadores Extrínsicos:
O ativador do plasminogênio tecidual (t-PA) é produzido pelas células do endotélio
vascular. O ativador do plasminogênio tecidual, uma serino-protease, é o ativador mais rápido do
plasminogênio, até mesmo do que os ativadores intrínsicos. Ele tem uma forte afinidade pela
fibrina, com a qual ele forma um complexo t-PA/fibrina do tipo bimolecular. Na presença da
fibrina a eficiência catalítica do t-PA para a ativação do plasminogênio é aumentada cerca de
1000 vezes. Por outro lado, o t-PA não ligado na fibrina tem uma afinidade muito baixa pelo
plasminogênio e, assim, ele não é eficiente na ativação da formação de fibrina a partir do
plasminogênio. O fator Xa e a trombina, como também a bradicinina e a proteína Ca podem
aumentar a liberação do t-PA pelo endotélio vascular. O aumento dos níveis de t-PA pode causar
uma excessiva fibrinólise e pode estar associada com uma tendência a sangramento.
Teoricamente, diante de uma extensiva injúria, o t-PA é liberado na circulação onde ele pode
causar uma fibrinogenólise sistêmica. Esse tipo de injúria, entretanto, está sempre associada com
a coagulação intravascular disseminada (CID) devido à rápida ativação da via extrínsica pela
simultânea exposição da tromboplastina.
A plasmina formada sobre o coágulo de fibrina é lentamente inativada pela 2-
antiplasmina porque a plasmina ligada à fibrina tem o seu sítio de ligação e o seu sítio catalítico
ocupados pela interação com a fibrina. A plasmina livre na circulação é rapidamente inativada
pela 2-antiplasmina porque ela tem o seu sítio catalítico disponível. Assim, o processo
fibrinolítico parece ser desengatilhado e limitado pela fibrina.
Um outro ativador extrínsico do plasminogênio é o ativador do plasminogênio tipo
uroquinase (u-PA). Este ativador está presente na urina, plasma e em muitos tipos celulares. Este
tipo de ativador potencializa a ativação de formação da plasmina a partir do plasminogênio ligado

44
a fibrina por cerca de 10 vezes. Quando o plasminogênio se encontra livre no plasma a ativação
da formação de plasmina por este ativador é mais lenta.
Ativador Exógeno:
A estreptoquinase é uma enzima produzida pelo estreptococos -hemolítico que também
ativa o plasminogênio, por isso ela é usada primariamente como um agente terapêutico para a
dissolução de coágulos.
Inibidores da Ativação do Plasminogênio:
Os Inibidores da Ativação do Plasminogênio tipo 1 (PAI-1) e tipo 2 (PAI-2) são inibidores
rápidos e específicos dos ativadores extrínsicos do plasminogênio.
A lipoproteína A, Lp(a), tem demonstrado inibir o t-PA e o u-PA. A Lp(a) também
compete com a plasmina para ligar à fibrina. Isso desloca a plasmina para o plasma onde ela é
rapidamente inibida pela 2-antiplasmina. A Lp(a) também bloqueia a inibição do t-PA pelo
PAI-1 na presença do fibrinogênio e da heparina. Assim o efeito da Lp(a) sobre a fibrinólise irá
depender da concentração do PAI-1, do t-PA e da Lp(a).
Digestão da Fibrina:
A plasmina é responsável por uma degradação assimétrica e progressiva da rede de fibrina
ou do fibrinogênio, formando fragmentos distintos de proteína, feridos como produtos de
degradação da fibrina ou do fibrinogênio (FDP). Esses fragmentos são rapidamente retirados da
circulação pelo fígado. A detecção dos fragmentos no plasma, acima dos valores de normalidade,
é de grande importância na clínica para auxiliar no diagnóstico de algumas desordens
hemostáticas.
Os produtos de digestão do fibrinogênio pela plasmina são: os fragmentos X, Y, D e E. O
primeiro fragmento formado é o fragmento X mais os pequenos peptídeos. Na próxima reação
proteolítica a plasmina cliva o fragmento X formando então os fragmentos Y e D. Posteriormente
o fragmento Y é digerido enzimaticamente pela plasmina dando origem aos fragmentos D e E. A
degradação dos monômeros de fibrina e do polímero instável de fibrina (polímero formado sem a
atividade do Fator XIIIa) pela plasmina é idêntica à do fibrinogênio. A degradação do polímero
de fibrina estabilizado pelo Fator XIIIa é mais lenta e os produtos são estruturalmente diferentes
por causa das ligações intermoleculares induzidas pelo Fator XIIIa. Os sítios de degradação do
polímero de fibrina pela plasmina são os mesmos vistos na molécula de fibrinogênio, porém

45
devido a presença das ligações intermoleculares existentes no polímero de fibrina, os sítios
podem não ficar disponíveis para a atividade enzimática exercida pela plasmina. É por este
motivo que os produtos de degradação do polímero estável de fibrina completamente diferentes
dos produtos de degradação do fibrinogênio. A FIGURA 30 mostra que os produtos de
degradação do polímero de fibrina são combinações de X, Y, D e E, por exemplo: DD/E,
YD/DY, YY/DXD.
FIGURA 30: Degradação da fibrina pela plasmina. A letra “P” indica os locais de clivagem pela
plasmina no polímero de fibrina e nos fragmentos maiores que vão sendo formados durante o
processo.

46
Os fragmentos de fibrina, quando em altas concentrações no plasma, podem exercer um
efeito anticoagulante sobre o sistema de formação do coágulo. O fragmento X pode ainda reagir
lentamente com a trombina. Essa lenta reação com o fragmento X resulta numa menor
disponibilidade da trombina para reagir com o fibrinogênio. Os fragmentos Y e D inibem a
polimerização dos monômeros de fibrina, e o fragmento E inibe a trombina.
CONTROLE FISIOLÓGICO DA FIBRINÓLISE
Da mesma forma que a cascata da coagulação é inibida por inibidores bioquímicos, a o
sistema fibrinolítico também sofre inibição. A plasmina e o t-PA são serino-proteases cuja ação
deve ser limitada ao local da injúria vascular. Os principais inibidores dessas enzimas são: a 2-
antiplasmina, a 2-macroglobulina, o PAI-1 e o PAI-2.
2-antiplasmina:
A 2-antiplasmina é uma glicoproteina monomérica, sintetizada no fígado e forma um
complexo de 1:1 com a plasmina ou com o plasminogênio. A formação do complexo
plasmina/plasminogênio bloqueia a adsorção do plasminogênio na rede de fibrina. A atividade da
plasmina livre também é inibida. A inibição da plasmina ligada à rede de fibrina pela 2-
antiplasmina é muito lenta, pois os sítios de inibição estão ocupados.
2-macroglobulina:
2-macroglobulina é um inibidor menos eficiente da plasmina do que a 2-antiplasmina.
2-macroglobulina, irá inibir a plasmina somente quando ela encontrar em excesso e livre no
plasma.
PAI-1 e PAI-2:
O PAI-1 é o inibidor priumário do t-PA e do U-PA presente no plasma. Ele também ativa a
proteína C. O PAI-1 no plasma é o mais ativo. O PAI-1 é liberado pelos grânulos alfa das
plaquetas durante a ativação plaquetária. O PAI-2 inibe o t-PA com menor eficiência do que o
PAI-1.
Uma síntese ou uma liberação deficiente do t-PA ou níveis muito aumentados de PAI-1
compromete a fibrinólise e está associado com trombose.

47
INTERRELAÇÃO ENTRE O SISTEMA HEMOSTÁTICO E OS SISTEMAS CININAS E
COMPLEMENTO
A FIGURA 31 mostra que os mecanismos hemostáticos, o sistema complemento e o
sistema das cininas se interrelacionam para bloquear o extravasamento de grandes quantidades de
sangue pela área injuriada do vaso sangüíneo e para impedir a invasão do organismo por
patógenos ou outro antígeno qualquer.
FIGURA 31: Interrelação entre a cascata da coagulação e os sistemas fibrinolítico, cininas e
complemento
Sistema Cininas:
O sistema cininas, importante na inflamação, permeabilidade vascular e quimiotaxia, é
ativado pela cascata da coagulação e pelo sistema fibrinolítico. Nesses sistemas a precalicreína é
ativada a calicreína pelo Fator XIIa e pela plasmina. O sistema cinina está também envolvido na
fase de ativação por contato da via intrínsica da cascata da coagulação. A ativação do Fator XII
em Fator XIIa não ocorre de modo efetivo sem a presença de calicreína e de HMWK. A
calicreína amplifica a ativação do fator XII e o HMWK é um fator essencial na ativação do Fator
XI em Fator XIa. O HMWK é necessário para a cascata da coagulação e para a fibrinólise.
A calicreína é uma enzima que atua sobre os cininogênios de alto peso molecular e de baixo
peso molecular para formar as cininas. As cininas geradas podem incluir a calidina e a
bradicinina. A bradicinina tem a função de: aumentar a permeabilidade vascular; contrair o

48
músculo liso; dilatar os vasos de baixo calibre; induzir a inflamação e a dor, e liberar as
prostaglandinas dos tecidos.
Sistema Complemento:
O sistema complemento é composto de aproximadamente 22 proteínas séricas que,
trabalhando juntas com os anticorpos e fatores da coagulação, desempenham uma importante
função nas reações imunes e alérgicas.
A plasmina ativa o sistema complemento pela clivagem do C3 em C3a e C3b. O C3a é uma
anaflotoxina que causa um aumento da permeabilidade vascular via degranulação dos mastócitos
para a liberação de histamina.
DESORDENS DA HEMOSTASIA
Desordens hereditárias da Função das Plaquetas:
As desordens plaquetárias funcionais estão relacionadas à participação da plaqueta na
formação do trombo hemostático, tanto na hemostasia primária como na secundária (Quadro 1).
A Doença de von Willebrand (vWD) foi incluída porque, apesar de ser um distúrbio plasmático,
altera a função plaquetária. Entretanto, o comentário sobre ela será feito em Distúrbios
Hereditários da Coagulação. Também a afibrinogenemia congênita, apesar de pertencer aos
distúrbios plasmáticos hereditários, foi citada nesse grupo por ser responsável pela alteração da
agregação plaquetária.
Quadro 1: Desordens funcionais das plaquetas
Defeitos de Transdução de Sinais:
- Anormalidade da via do ácido araquidônico (AA):
Diminuição da liberação do AA
Deficiência da ciclo-oxigenase
- Defeitos na interação plaqueta-agonistas:
Defeitos dos receptores: TXA2, colágeno,
ADP, epinefrina
- Defeitos na ativação da proteína G:
Deficiência de G (alfa)q
- Defeitos no metabolismo de fosfoinositol:
Deficiência de PLC-ß2
- Defeitos na mobilização de cálcio
- Defeitos na fosforilação de proteína
- Defeitos na regulação do cito-esqueleto:
Síndrome de Wiskott-Aldrich
Anormalidade da Agregação Plaquetária:
- Afibrinogenemia congênita
- Trombastenia de Glanzmann (anormalidade da Gp
IIb/IIIa)
Deficiência de Substâncias do “Pool” de Armazenamento:
- Deficiência dos corpos densos:
Síndrome de Hermansky-Pudlak
Síndrome de Chediak-Higashi
Trombocitopenia com ausência de rádio
- Deficiência dos grânulos alfa:
Síndrome da plaqueta cinza
Anormalidades Relacionadas a Adesividade da Plaqueta:
- Doença de von Willebrand
- Pseudo von Willebrand (tipo plaquetário semelhante
à vWD)
- Síndrome de Bernard-Soulier (anomalia ou ausência
da GpIb)

49
Esse grupo de distúrbios tem como manifestação clínica hemorragias de diversas intensidades,
freqüentemente mucocutâneas, após cirurgias e traumas.
As desordens plaquetárias congênitas não podem ser distinguidas com bases, apenas, nas
manifestações clínicas. O estudo laboratorial é destaque de importância diagnóstica. Entretanto,
para chegar ao esclarecimento de muitos distúrbios nesse grupo, são imprescindíveis exames
sofisticados, por meio de instrumentos ou de exames bioquímicos que, somente, são efetuados
em laboratórios de coagulação altamente especializados.
O tempo de sangramento (TS) aumentado é freqüentemente visto nesses distúrbios, e no
exame do esfregaço de sangue observam-se freqüentemente plaquetas gigantes. O estudo da
agregação plaquetária, por meio do agregômetro, com o uso de diversos agonistas (ADP,
colágeno, epinefrina e ristocetina) é importante para o diagnóstico desse grupo de distúrbios.
Para compreender melhor os distúrbios relacionados à transdução de sinal, a FIGURA 32
mostra um desenho, esquemático, das principais reações desencadeadas pelos indutores
fisiológicos da agregação plaquetária.
FIGURA 32: desenho esquemático das reações plaquetárias que ocorrem após o estímulo dos indutores de
agregação. CO = ciclo-oxigenase, PKC = proteinaquinase, G = proteínas G, MLC = cadeia leve da miosina,
MLCK = MLCquinase, R = receptor, DAG = diacilglicerol, TK = tirosinoquinase, TS = tromboxane-sintetase,
PAF = fator ativador de plaqueta, IP3 = trifosfato de inositol, PLA2 = fosfolipase A2, PIP2 = bifosfato de
fosfatidil-inositol.
O desenho da FIGURA 32 destaca, apenas, as reações que representam a transdução dos
sinais que chegam à membrana plaquetária, e que são responsáveis pela indução da secreção de
diversas substâncias plaquetárias.

50
Como em outras células, a ativação plaquetária e a secreção, são reguladas pelas alterações
dos níveis de ATP, do influxo de cálcio, da hidrólise dos fosfolipídios da membrana da plaqueta e
da fosforilação de diversas proteínas intracelulares.
Como mostra a FIGURA 32, essas reações têm início quando o efetor se liga a um receptor
específico que, por sua vez, interage com a proteína G. As proteínas G constituem um grupo
heterogêneo de proteínas, integrais de membrana, que têm a propriedade de promover a ligação
dos receptores com as enzimas efetoras intracelulares. A partir da interação, específica, do
agonista com o seu receptor e a proteína G, inicia-se a hidrólise do fosfolipídio da membrana
plaquetária, o fosfatidilinositol-4,5-bifosfato (PIP2). Com isso são gerados diacilglicerol (DAG) e
trifosfato de inositol (IP3). O IP3 ativa o movimento de cálcio no citosol plaquetário que, por sua
vez, estimula a fosfolipase A2 (PLA2). A fosfolipase A2 ativa a produção de tromboxane A2, que
por sua vez promove a fosforilação da cadeia leve da miosina. A miosina, ao interagir com a
actina, promove, por contração, a compressão dos constituintes do citosol plaquetário, inclusive
dos corpos densos e dos grânulos alfa, e com isso leva à secreção dos diversos tipos de
substâncias. A actina e a miosina também promovem a mudança da forma das plaquetas. A DAG
ativa a proteína-quinase C (PKC) que leva a fosforilação de diversos substratos, inclusive da P47,
que contribuem para a secreção plaquetária.
A seguir serão apresentadas as características mais importantes para esclarecer o
diagnóstico dos distúrbios funcionais das plaquetas.
Pseudo vW (P-vW):
É muito semelhante ao tipo 2B da vWD. Pode apresentar aumento do TS (tempo de
sangria), do Fator VIII:C, do vWF(ag) e do vW:RCoF. A agregação plaquetária está aumentada
com baixas concentrações de ristocetina (0.3-0.5mg/ml). Pode haver discreta trombocitopenia.
Ao contrário do tipo vWD 2B onde o defeito está no vWF, aqui há um defeito qualitativo no
receptor plaquetário GpIb-IX para o vWF. Esse defeito torna o referido receptor mais ávido para
os grandes multímeros do vWF. Como conseqüência dessa avidez, os grandes multímeros são
consumidos, deixando disponível, no plasma, somente o vWF de baixo peso molecular .
Síndrome de Bernard-Soulier (SB-S):
Freqüentemente autossômica recessiva e associada à consangüinidade. Pacientes
heterozigotos apresentam 50% da quantidade normal do receptor plaquetário GpIb-IX. Há um
discreto ou até mesmo ausência de sangramento. Casos de SB-S homozigotos são raríssimos. O

51
tempo de sangramento pode estar aumentado e no esfregaço de sangue são vistas plaquetas
gigantes. Pode haver trombocitopenia. A anormalidade mais característica está no teste de
agregação plaquetária que é normal para os diversos agonistas fisiológicos (ADP, epinefrina,
colágeno), mas alterado para a ristocetina. A diferença entre a vWD é que a adição de plasma
normal não corrige o defeito da agregação na SB-S, ao contrário da resposta na vWD. Testes
bioquímicos, tais como: eletroforese em SDS-agarose, auto-radiografia, "imunoblotting" de
lisado plaquetário com anticorpo anti-GpIb-IX, confirmam o diagnóstico da deficiência da
referida glicoproteína.
Afibrinogenemia congênita:
Nesse distúrbio, devido à falta de fibrinogênio para fazer a ligação entre duas plaquetas por
meio de ligações entre as GpIIb-IIIa, a curva de agregação plaquetária terá a mesma característica
observada na Trombastenia de Glanzmann que será comentada a seguir.
Trombastenia de Glanzmann:
A Trombastenia de Glanzmann é uma doença autossômica recessiva que decorre de um
distúrbio funcional da GpIIb-IIIa. Esse distúrbio deriva da mutação nos genes que codificam as
GpIIb e IIIa, com as conseqüentes alterações quantitativa e qualitativa das referidas
glicoproteínas. Manifesta-se clinicamente por sangramento muco-cutâneo, particularmente
epistaxe, púrpura e menorragia. A alteração laboratorial, mais característica, é a intensa redução
ou a ausência da agregação plaquetária, com o emprego do agregômetro, aos diversos agonistas
fisiológicos. A curva característica é a do tipo C mostrada na FIGURA 33 do tópico: Testes
Laboratoriais.
Deficiência de Substâncias do “Pool” de Armazenamento:
Em alguns distúrbios desse grupo o defeito está relacionado à diminuição de substâncias
armazenadas nos corpos densos e nos grânulos alfa (citados no Quadro 1). Na curva de agregação
plaquetária, a segunda parte da curva está ausente (curva B da FIGURA 33 do tópico: Testes
Laboratoriais), a qual corresponde à agregação das plaquetas estimulada pelas substâncias
liberadas durante a ativação plaquetária. Os distúrbios congênitos desse grupo foram
mencionados no Quadro 1.

52
Defeitos dos Sinais de Transdução:
Ao contrário do grupo anterior, nesse grupo, a deficiência de substâncias do “Pool” de
armazenamento, decorre de um distúrbio localizado no mecanismo que é desencadeado quando o
receptor da membrana plaquetária é ativado pelo agonista. O distúrbio pode acontecer em
diversos setores desse mecanismo, desde o momento da interação com o receptor até a produção
dos efetores responsáveis pela liberação das substâncias do “Pool” de armazenamento, bem como
das substâncias formadas por diversas reações (ver FIGURA 32). O teste de agregação
plaquetária mostrará ausência da segunda curva (estímulo endógeno), o mesmo defeito verificado
no grupo anterior. É importante assinalar que o ácido acetil salicílico também interfere na
produção dessa segunda curva da agregação plaquetária, ao inibir a ciclo-oxigenase e diminuir a
produção do tromboxane A2.
Desordem da Interação de Fatores da Coagulação com a Membrana Plaquetária:
A síndrome de Scott é uma desordem hemorrágica rara na qual, as plaquetas e outras
células sangüíneas falham em promover a união de fatores plasmáticos (Va e Xa) com a
membrana celular. O aumento da quantidade da fosfatidilserina, carregada negativamente, na
superfície externa da membrana plaquetária é essencial para a formação da superfície coagulante.
Sabe-se que o defeito, na síndrome de Scott, decorre da incapacidade em mobilizar a
fosfatidilserina da camada interna da membrana celular para a camada externa, em resposta à
elevação do Ca2+
na superfície interna da membrana (Zhou).
TESTES LABORATORIAIS
Como em qualquer setor da medicina, aqui também é imprescindível lançar mão dos três
meios de coleta de dados para chegar a um diagnóstico: história da doença atual (HDA) com a
história pregressa pertinente (HP), história familiar (HF) e exames complementares.
È importante ressaltar que, apesar da importância dos dados coletados na HDA e na HF,
nos distúrbios hemorrágicos, os testes laboratoriais dirigidos para os componentes hemostáticos,
são prioritários. A razão para isso está em que, na maioria das vezes, somente com eles é possível
chegar ao diagnóstico conclusivo, e eles serão importantes para o acompanhamento do paciente
quando houver distúrbio hemorrágico.
Por estas razões, o raciocínio será feito com base nos exames para o estudo da hemostasia e
nos dados obtidos pela HDA e pela HF para nortear as investigações.
Esse tópico será dividido em dois itens:

53
1 - Exames de rotina: o número mínimo necessário de testes para descobrir anormalidade
no mecanismo hemostático, acompanhados da anamnese dirigida para distúrbios hemorrágicos.
2 - Exames empregados para esclarecimento do defeito hemostático. Os exames mais
específicos para caracterizar as doenças e as síndromes hemorrágicas mais freqüentes e mais
raras serão citados quando estas forem abordadas.
1 - Exames de Rotina:
Como pode ser visto, a FIGURA 33, considera como exames de rotina, o tempo de
protrombina (TP), o tempo de cefalina, também conhecido como tempo de tromboplastina parcial
ativado (TTPa), o tempo de sangramento (TS) e a contagem de plaquetas.
FIGURA 33: Na área azul constam os fatores cujo defeito é identificado pelo tempo de
tromboplastina parcial ativado. Na área amarela estão os fatores cujo defeito é identificado pelo
tempo de protrombina. Observar que os fatores X, V, II e I (fibrinogênio) quando alterados,
prolongam os dois testes. HMWK = Cininogênio de alto peso molecular. PK = Pré-calicreina. TF =
Fator tecidual. Pq = Plaqueta. O FXIII está fora do alcance desses testes. Detalhes no texto.
Na FIGURA 33, os fatores que fazem parte da área amarela, quando têm o nível reduzido
ou algum impedimento para o seu funcionamento (anticoagulante ou defeito na estrutura
molecular), alteram o TP. Já os defeitos dos fatores da área azul alteram o TTPa. No entanto,
quando os fatores X, V, II e fibrinogênio, ou seja, os fatores que fazem parte das áreas azul e
amarela, apresentam defeitos, ou seus níveis estão diminuídos no sangue, o TP e o TTPa estarão
alterados.
O FXIII não é atingido pelos dois exames citados; será, posteriormente, abordado
comentários sobre a identificação de sua alteração.
Visto isso, é importante considerar algumas exceções:

54
I - Para cada fator da coagulação, a diminuição do nível somente altera o exame
correspondente, a partir de um determinado limite. Como pode ser visto na literatura, níveis de
15-18% de FVIII:C não alteraram o TTPa do hemofílico estudado. Também foi normal o TTPa
de um hemofílico com FIX de 23%. Nesses casos, dados obtidos na coleta das HP e HF puderam
despertar a suspeita de algum distúrbio no paciente.
II - Como pode ser visto na FIGURA 33, a diminuição do nível de FXIII não leva à
alteração desses dois exames. Nesse caso, suspeitaremos de defeito desse fator quando houver
hemorragia no momento ou nas HP e HF, particularmente quando os pais apresentarem
consangüinidade.
III - Além do TTPa não se alterar com a variação do nível do FXIII, também não se altera
com a diminuição do nível do FVII.
IV - O inverso acontece com os fatores XII, HMWK e PK. A diminuição sangüínea do
nível desses fatores altera o TTPa, porém, não há qualquer possibilidade de sangramento,
espontâneo ou induzido por cirurgia.
V - O nível baixo de fibrinogênio nem sempre altera o TP e o TTPa, entretanto, o tempo de
trombina (TT) e a dosagem do fator podem chamar a atenção para o distúrbio. Entretanto, é
importante lembrar que o TT ainda é normal até o fibrinogênio atingir o nível mínimo de 1000
mg/L. Além dos exames citados, para completar os exames de rotina, o tempo de sangramento
(TS) e a contagem de plaquetas (CPq) também devem ser incluídos.
O TS de maior sensibilidade é aquele em que se faz uma incisão no antebraço com tamanho
e profundidade uniformizados por um molde, ou por meio de um dispositivo que dispara uma
lâmina. Em ambos os casos, as incisões são feitas com um manguito mantendo a pressão em 40
mm de Hg. Dependendo do método, o normal máximo vai de 7 a 10 minutos.
O TS alterado serve para indicar diminuição numérica das plaquetas ou alteração funcional.
Também aqui, temos que ter em mente que níveis de plaquetas até 50.000/µL podem apresentar
TS normal.
O dismorfismo plaquetário no esfregaço sangüíneo, pode oferecer indicio de distúrbio
funcional plaquetário.
Observações sobre os Exames de Rotina:
A avaliação da hemostasia “in vitro” é realizada através de testes que exploram
seletivamente as diferentes vias da coagulação, a saber, o Tempo de Protrombina ou TP, Tempo
de Tromboplastina Parcial Ativada ou TTPa, Tempo de Trombina ou TT e as dosagens dos

55
fatores isolados como o Fibrinogênio, o fator VIII, o IX e assim por diante. A coleta de sangue
para estudo da coagulação deve ser a menos traumática possível, com o mínimo de estase venosa,
e ser sempre feita com material plástico, incluindo seringas e tubos. O uso de frascos contendo
vácuo deve ser evitado, pois este tende a ativar a coagulação e encurtar os tempos.
O anticoagulante usado, é o Citrato de sódio 100 mM pH 6,5, pois este é o ideal para
conservação dos fatores. O Oxalato de sódio também pode ser usado, mas alguns fatores são
menos estáveis neste tipo de anticoagulante. O EDTA não deve ser usado por pode provocar a
ativação das plaquetas. A Heparina não se presta como anticoagulante no estudo da coagulação.
A proporção entre o volume de anticoagulante e o volume de sangue total é muito importante,
pois os testes são baseados no tempo que o plasma leva para coagular depois da adição do cálcio
que anula o efeito do citrato. A proporção padronizada é de 9 partes de sangue total para 1 parte
de Citrato de Sódio 100 mM pH 6,5.
Esta proporção é válida para indivíduos com hematócrito em torno de 45%. Nos casos de
policitemia com Hematócrito maior que 55% a quantidade de anticoagulante deve ser ajustada,
do contrário os tempos serão anormalmente longos.
O sangue total é então centrifugado a 3.000 rpm por 15 minutos para obtenção do plasma
pobre em plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos
testes, e ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da
coagulação. Os testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser
congelado imediatamente para um ensaio posterior.
I – Tempo de Protrombina
Princípio:
O tempo de protrombina ou TP, consiste na determinação do tempo de coagulação de um
PPP, após a adição de tromboplastina (Fator III) e de cálcio, a 37oC. A adição de um excesso de
tromboplastina promoverá a ativação de todo o fator VII contido na amostra de plasma. O Fator
VIIa ativará o fator X, iniciando a via comum da cascata da coagulação. Desta forma, o TP mede
os fatores envolvidos na Via Extrínseca e na Via Comum, sendo absolutamente independente da
Via Intrínseca.
A tromboplastina é um reagente obtido a partir de cérebro humano, de coelho, de boi ou de
macaco. Ele contém o fator tissular, composto por uma parte protéica e uma parte fosfolipídica
que substitui o fator 3 plaquetário (F3P) na ativação dos fatores que se ligam a fosfolipídeos de

56
membrana. O TP é sensível a redução dos fatores: VII, X, V, II e I. O TP é o teste mais sensível
para avaliação da redução dos fatores dependentes de vitamina K (II, VII, IX e X), sendo o teste
usado no controle de paciente em uso de anticoagulante orais.
Procedimentos:
A tromboplastina a ser usada pode ser com ou sem cálcio, dependendo do fabricante. Ela
pode ser apresentada na forma liofilizada, devendo ser reconstituída de acordo com a orientação
do fabricante, obedecendo sempre a condição e o tempo de conservação.
No caso da tromboplastina sem cálcio a realização do teste obedece ao seguinte esquema:
Técnica 1:
Obtenção de Plasma Pobre em Plaquetas:
Centrifugar o sangue total a 3.000 rpm por 15 minutos para obtenção do plasma pobre em
plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos testes, e
ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da coagulação. Os
testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser congelado
imediatamente para um ensaio posterior.
Em um tubo de ensaio pré-aquecido a 37oC:
Adicionar 100 µl de Tromboplastina
Incubar a 37o C por 2 minutos
Adicionar 100 µl de plasma pobre em plaquetas citratado
Incubar a 37o C por 2 minutos
Adicionar 100 µl de CaCl2 0,025 M
Disparar o cronômetro e anotar o tempo que gasto para formar o coágulo.
Para as tromboplastinas que já contém cálcio, o seguinte esquema deve ser obedecido
(técnica 2):
Técnica 2:
Obtenção de Plasma Pobre em Plaquetas:
Centrifugar o sangue total a 3.000 rpm por 15 minutos para obtenção do plasma pobre em
plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos testes, e
ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da coagulação. Os

57
testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser congelado
imediatamente para um ensaio posterior.
Em um tubo de ensaio pré-aquecido a 37oC:
Adicionar 100 µl de plasma pobre em plaquetas citratado
Incubar a 37o C por 2 minutos
Adicionar 200µl de Tromboplastina pré-aquecida a 37oC
Disparar o cronômetro e anotar o tempo que gasto para formar o coágulo.
Expressão dos Resultados:
Os resultados podem ser expressos em segundos, mas esta forma traz o inconveniente da
variação diária do teste. Assim, é sempre necessário realizar o TP de um “Pool” de plasma
normal e comparar o resultado do paciente com o resultado do indivíduo normal, fazendo-se a
relação dos TP paciente sobre o normal.
Exemplo:
TP PACIENTE= 28 segundos TP NORMAL= 14 segundos
Então R= [ TP paciente/TP normal]; R=[28 segundos/14 segundos]; R= 2,0
O TP pode ainda ser expresso em Atividade de Protombina ou AP. Esta forma de
expressar o TP deriva de uma curva realizada com um “Pool” de plasma normal diluído em
diferentes concentrações com salina. Assim, o plasma puro representa 100% de AP, o plasma
diluído ao meio representa 50% de AP, assim por diante. O TP é realizado da maneira habitual
para todas as diluições do plasma e uma curva é construída com os valores obtidos, tal como no
exemplo abaixo:
Exemplo do Cálculo da Atividade Protrombina:
Diluição da amostra (“Pool” de Plasma Normal) para determinar o TP
Diluição Atividade Tempo
Plasma Puro 100% 14,6 segundos
Diluição 1:2 50% 19,2 segundos
Diluição 1:4 25% 26,0 segundos
Diluição 1:8 12,5% 37,9 segundos
Diluição 1:16 6,25% 60,4 segundos
Uma curva é então construída em papel logarítmo, onde se colocam os TP em segundos na
ordenada e os AP em % na abscissa. Pelos pontos pode-se traçar uma reta. Para se ter o AP do

58
paciente, basta interpolar na reta o valor encontrado para o TP, e o resultado pode ser expresso
em atividade de protrombina.
Alguns laboratórios fornecem, como resultado, apenas o TP do paciente, ou apenas a
relação TP paciente/TP normal ou apenas a atividade e fazem referência aos valores normais do
método. Esse modo de informar deixa muito a desejar, e poderá acarretar sérios problemas para o
acompanhamento de pacientes que estão recebendo anticoagulantes orais. A razão para isso é que
os resultados do TP variam muito com a tromboplastina que está sendo empregada no referido
teste.
O TP é o tempo obtido após a adição de cálcio à mistura de tromboplastina com o plasma a
ser estudado. Acontece que a atividade da tromboplastina varia de fabricante para fabricante e,
quando do mesmo fabricante, a atividade varia de partida para partida. Em outras palavras, a
determinação do TP de um plasma dará resultados diferentes se usarmos tromboplastinas de
diferentes origens, ou de diferentes partidas.
Desse modo, com a finalidade de uniformizar os resultados do teste, a Organização
Mundial de Saúde apresenta uma tromboplastina que serve de referência para a padronização das
tromboplastinas empregadas no TP. Assim, com essa tromboplastina padrão, os fabricantes de
tromboplastina podem calcular o índice de sensibilidade internacional (ISI: international
sensitivity index). Os melhores índices são aqueles mais próximos de 1. Com ISI mais afastado
do padrão internacional, haverá maiores diferenças nos tempos de protrombina. O ISI vem
referido no frasco.
Para dar maior sensibilidade ao método, emprega-se a relação entre o tempo de
protrombina do paciente e o tempo de protrombina de uma mistura de plasmas normais (5 ou
mais plasmas). A esse resultado denominamos relação tempo de protrombina do paciente com o
tempo dos plasmas normais. Adicionando-se o ISI a essa relação, teremos o RNI (international
normalized ratio).
Desse modo, empregamos a seguinte fórmula para determinar o INR que tem o ISI como
expoente:
RNI = (TP DO PACIENTE/ TP DA MISTURA DE PLASMAS NORMAIS)ISI
Os valores normais do RNI variam entre 0.9 e 1.2.

59
II- Tempo de Tromboplastina Parcial Ativado
Princípio:
O TTPA consiste na determinação do tempo de coagulação do PPP citratado, após a adição
de um ativador da fase de contato da coagulação (por isso é dito “ativado”), e de um reagente, a
cefalina, que substitui o fosfolipídeo da membrana plaquetária ou FP3, uma vez que se trabalha
com o PPP. O último reagente a ser adicionado é o cálcio, que reverte a ação do citrato.
O ativador da fase de contato pode ser o caolim, o ácido elágico, a celite ou ainda o dextram.
Este ativador quando colocado em excesso vai ativar o Fator XII, na presença de precalicreína e
cininogênio de alto peso molecular. O Fator XIIa vai converter o Fator XI em XIa. O Fator XIa
ativa o fator IX em IXa, e este junto com o fator VIII:C, que atua como cofator, e Ca2+
vai ativar
o Fator X. Esta é a chamada Via Intrínseca da coagulação. O Fator Xa inicia então a Via
Comum, ativando o Fator II na presença de Fator V e do FP3, que no TTPA é representado pela
cefalina. O Fator IIa ou trombina, atua sobre o fibrinogênio, transformando-o em fibrina.
A cefalina é um fosfolipídeo extraído de cérebro de maneira semelhante à tromboplastina, mas
com a diferença que a cefalina não possui atividade de fator tissular, isto é, não é capaz de ativar
o fator VII (por isso ela é chamada tromboplastina parcial), mas é capaz de atuar como uma
superfície de fosfolipídeo, substituindo aquela representado fisiologicamente pela membrana
plaquetária. Assim o TTPa é sensível ao nível dos fatores da Via Instrínseca e da Via Comum:
XII, PK, cininogênio de alto peso molecular, XI, VIII-C, X, V, II e fibrinogênio. Ele é bastante
sensível à presença de heparina, sendo o teste de escolha para monitor a heparinoterapia.
Procedimento:
A cefalina de diferentes procedências geralmente contém o ativador. No caso do celite ou
caolim, o ativador é particulado, o que impede a leitura do TTPa em coagulômetro. O reagente
contendo ácido elágico é homogêneo e permite o uso do coagulômetro. O procedimento do teste
obedece ao seguinte esquema:
Técnica:
Obtenção de Plasma Pobre em Plaquetas:
Centrifugar o sangue total a 3.000 rpm por 15 minutos para obtenção do plasma pobre em
plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos testes, e
ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da coagulação. Os

60
testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser congelado
imediatamente para um ensaio posterior.
Em um tubo de ensaio pré-aquecido a 37oC:
Adicionar 100 µl de PPP citratado
Adicionar 100 µl de cefalina
Incubar a 37o C por 3 a 5 minutos
Adicionar 100 µl de CaCl2 0,025 M
Disparar o cronômetro e anotar o tempo
Expressão dos Resultados:
O resultado deve ser expresso pela relação entre o tempo obtido para o paciente e o tempo
do “Pool” de plasma normal do dia. É considerado normal uma relação TTPa paciente/TTPa
“Pool” normal de até 1,25, o que significa dizer que o TTPa do paciente não deve estar além de
25% acima do TTPa do plasma normal. Os valores em segundos variam com o ativador e a
cefalina utilizados, de modo que a expressão dos resultados em segundos não é recomendada.
III- Tempo de Trombina
Princípio:
O TT consiste no tempo de coagulação do PPP após a adição de baixa concentração de
trombina bovina, que vai transformar diretamente o fibrinogênio em fibrina. O TT vai então
depender da concentração de fibrinogênio e da presença de inibidores da formação de fibrina
como é o caso da ação da heparina e dos produtos de degradação da fibrina (PDF).
Procedimento:
A trombina bovina a ser utilizada deve ter 5U/ml, de modo a fornecer um TT em plasma
normal de 15 a 20 segundos. A solução da trombina deve ser mantida em banho de gelo, pois
essa enzima é muito instável à temperatura ambiente. O procedimento obedece ao seguinte
esquema:

61
Técnica:
Obtenção de Plasma Pobre em Plaquetas:
Centrifugar o sangue total a 3.000 rpm por 15 minutos para obtenção do plasma pobre em
plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos testes, e
ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da coagulação. Os
testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser congelado
imediatamente para um ensaio posterior.
Em um tubo de ensaio pré-aquecido a 37oC:
Adicionar 200 µl de PPP citratado
Incubar a 37oC por 2 minutos
Adicionar 100 µl Trombina 5U/ml pré-aquecida a 37oC por 2 minutos
Disparar o cronômetro e anotar o tempo
Expressão dos resultados:
Os resultados são expressos como a relação entre o TT do paciente e o TT “Pool” de
plasma normal do dia. O valor normal não deve ultrapassar 1,20.
IV- Dosagem do Fibrinogênio
Princípio:
A dosagem do fibrinogênio pelo método de Clauss baseia-se na coagulação do PPP muito
diluído por uma alta concentração de trombina bovina. Nestas condições, o tempo de coagulação
é proporcional à concentração de fibrinogênio, não sofrendo influência da heparina e variando
pouco com a presença de PDF. Como o plasma é diluído, a visualização do coágulo é muito
difícil, sendo necessário o uso de coagulômetro.
Procedimento:
A trombina bovina é preparada dissolvendo-se o conteúdo do frasco com salina para obter
uma concentração de 500U/ml (Solução Estoque). Esta deve ser aliquotada e congelada a - 70o C
ou conservada a – 20o C por pouco tempo. A solução de trabalho é preparada no momento do uso
pela diluição da solução estoque para uma concentração final de 70 U/ml em tampão Veronal. A
solução de trabalho deve ser conservada em banho de gelo, não devendo ser recongelada.
A realização do teste obedece ao seguinte esquema:

62
Técnica:
Obtenção de Plasma Pobre em Plaquetas:
Centrifugar o sangue total a 3.000 rpm por 15 minutos para obtenção do plasma pobre em
plaquetas (PPP). O PPP pode ser transferido para um tubo plástico para realização dos testes, e
ser mantido à temperatura ambiente, pois a refrigeração ativa a fase de contato da coagulação. Os
testes devem ser realizados dentro de pouco tempo, ou então o plasma deve ser congelado
imediatamente para um ensaio posterior.
Em um tubo de ensaio pré-aquecido a 37oC:
Adicionar 200 µl do PPP previamente diluído
Incubar a 37oC por 2 minutos
Adicionar 100µl de Trombina 70 U/ml
Disparar o cronômetro e anotar o tempo
Um plasma padrão, com uma concentração conhecida de fibrinogênio deve ser utilizado
para construir uma curva padrão. A diluição 1:10 corresponde a 100% ou à concentração real do
fibrinogênio do plasma padrão. Por exemplo, ao utilizar o plasma padrão de 22,2g/l de
fibrinogênio, a diluição 1:10 deste plasma corresponde a 2.2 g/l. Assim pode se construir a curva
padrão da seguinte maneira:
Técnica:
Diluição do plasma padrão para a realização da curva padrão:
Diluição [Fibrinogênio] g/l de plasma Tampão Veronal Plasma Padrão
Diluição 1:10 2,22 900µl 100µl
Diluição 1:15 1,48 1.400µl 100µl
Diluição 1:20 1,11 1.900µl 100µl
Diluição 1:25 0,89 2.400µl 100µl
Os tempos obtidos para cada ponto são colocados em um papel logarítmo e a curva é
traçada. Pelo menos três pontos devem estar alinhados. Nas concentrações altas ou baixas, a
curva perde a linearidade, desta forma o tempo gasto para a formação do coágulo deve estar
contido na zona de linearidade.
O plasma do paciente deve ser diluído 1:10, e se o tempo obtido estiver na zona de
linearidade a leitura da concentração do fibrinogênio pode ser feita. Se o tempo obtido estiver a

63
baixo o plasma deve ser diluído 1:15 ou 1:20 até que o tempo esteja dentro da zona de
linearidade, e o resultado deve ser multiplicado pelo fator de diluição.
Composição do Tampão veronal:
Dietil Barbiturato de sódio 11,75 g
NaCl 14,67 g
Água destilada 1000 ml
Acertar o pH para 7,4 com HCl 1 N e acertar o volume com água destilada para 2.000ml.
V- Tempo de Sangramento (TS)
Para a determinação do TS, emprega-se um dispositivo descartável que, ao disparar uma
lâmina, produzirá uma incisão com 6 mm de extensão por 1 mm de profundidade.
O local da incisão é a porção ventral do antebraço, longe de qualquer vaso sangüíneo.
Esse teste é efetuado mantendo-se um manguito no braço com pressão de 40 mm Hg.
O tempo de sangramento normal não deve ultrapassar de 7 a 10 minutos.
No entanto, o tempo de sangria que é realizado na maioria dos laboratórios se baseia na
realização de uma pequena incisão na polpa digital, com uma lanceta estéril, de cerca de 3mm de
profundidade e deixar o sangue fluir espontaneamente. Acionar o cronômetro no momento em
que aparecer a primeira gota de sangue e com um papel de filtro, absorver a cada 15 segundos a
gota de sangue formada, sem pressionar a incisão. Quando cessar o fluxo de sangue, parar o
cronômetro. O intervalo decorrido entre o aparecimento da primeira gota e a última gota
representa o tempo de sangria do paciente. Normalmente, não se obtém mais sangue ao fim de
um a três minutos, ou seja, observam-se no papel de filtro cerca de 4 a 12 gotas, que vão
diminuindo gradativamente de tamanho. Quando o tempo de sangria se encontra prolongado, o
sangue continua fluir além do tempo normal, formando-se gotas durante um certo tempo. O valor
de referência é de 1 a 3 minutos.
Para uma verdadeira reflexão da função das plaquetas "in vivo", o paciente deve evitar o
uso de aspirina ou drogas “aspirina-like” durante 7 dias antes da realização do tempo de sangria,
pois a aspirina e muitas droga “aspirina-like” prolongam o tempo de sangria. Igualmente, uma
contagem de plaquetas deve ser executada antes de realizar o tempo de sangria, pois uma
contagem de plaquetas inferior a 100 x 109/L resultará em um tempo de sangria prolongado. Se
condições acima forem conhecidas, um tempo de sangria prolongado indicará deficiência
orgânica das plaquetas. Possíveis causas de deficiência orgânica das plaquetas incluem:

64
deficiências orgânicas hereditárias e adquiridas, Doença de von Willebrand, afibrinogenemia,
hipofibrinogenemia severa, e algumas desordens vasculares.
VI- Plaquetometria
A contagem de plaquetas é efetuada em contador automático de células ou em câmara de
Neubauer.
Os valores normais variam entre 150.000 - 400.000 /mm3
Diante de algumas situações as seguintes providências devem der tomadas:
I - Havendo alteração de um dos exames de rotina, tem que prosseguir com os testes de
esclarecimento, mesmo que não se identifique hemorragia anormal na HDA, na HP e na HF.
II - Se apenas na HDA, na HP ou na HF houver evidência de sangramento anormal, o
estudo para esclarecimento deverá ser efetuado, mesmo que os exames de rotina estejam normais.
2 - Exames para Esclarecimento do Defeito Hemostático
Sempre que houver alteração em um ou nos dois exames de rotina (TP e TTPa), deve-se
pensar na possibilidade de decréscimo do nível de um ou de vários fatores. Para esclarecer, deve-
se efetuar a dosagem dos fatores.
Os fatores a serem dosados são aqueles que estão relacionados ao exame alterado,
conforme referidos no item anterior.
Em continuação ao esclarecimento do defeito hemostático, caso haja alteração do TP e do
TTPa, e as dosagens dos fatores da via intrínseca e extrínseca, como também dos fatores X, V e
II, estejam normais, deve-se realizar o TT que, alterado, poderá indicar nível baixo de
fibrinogênio. A dosagem desse fator confirmará, ou não, a suspeita.
Caso os exames de rotina, e os de esclarecimento estejam normais, deve-se suspeitar de
anormalidade do FXIII. Para esclarecer é importante efetuar um teste simples de solubilidade do
coágulo de plasma em solução de uréia. Se o nível de FXIII estiver baixo, a ligação covalente
entre os monômeros de fibrina não se fará e, conseqüentemente, a fibrina gerada será solúvel na
solução de uréia. A dosagem do fator confirmará o diagnóstico.
Além de se pensar em nível baixo de fator quando o exame de rotina estiver alterado, é
imprescindível que se afaste a possibilidade da presença de um anticoagulante circulante.
Para tal, é importante efetuar o tempo de recalcificação da mistura do plasma do paciente com
plasma normal, em partes iguais. Caso o tempo da mistura não for maior que 4 segundos do
tempo do plasma normal, pode-se dizer que houve correção e, nesse caso, estará afastada a

65
possibilidade de anticoagulante. Como há anticoagulante com ação tempo-dependente, deve-se
realizar o exame após uma hora da mistura.
Também pode realizar, no lugar do tempo de recalcificação da mistura dos plasmas, o TP
ou o TTPa, escolhendo o alterado ou o mais alterado no exame de rotina.
Prosseguindo o raciocínio, caso os exames de rotina, para coagulação, estejam normais,
mas o TS apresenta-se aumentado, a plaquetometria confirmará, ou não, a trombocitopenia.
Na ausência de trombocitopenia, ou quando o nível da trombocitopenia não justificar a alteração
do TS, deve pesquisar a existência de defeito funcional plaquetário e Doença de von Willebrand.
Para esses esclarecimentos deve-se contar com exames que somente são realizados em
laboratórios especializados no estudo da hemostasia. Vários deles utilizam aparelhos mais
avançados e técnicas bioquímicas, que na maior parte das vezes somente são efetuadas em
laboratórios que se dedicam à pesquisa.
A seguir serão fornecidos maiores detalhes sobre os aparelhos e as técnicas de diagnóstico
usadas mais freqüentemente no diagnóstico dos distúrbios funcionais plaquetários, incluindo a
doença de von Willebrand.
Um novo teste, prático, está sendo empregado para o diagnóstico da vWD - é o tempo de
oclusão. Para tal, é empregado um aparelho, o PFA-100 (Platelet Function Analyzer, Dade
International, Massy, France) para pesquisar defeito de adesividade e agregação plaquetárias,
particularmente na vWD.
O sangue total citratado é aspirado, sob alto "shear rate" (5000-6000 dyn/cm2), através de
um tubo capilar constituído de uma membrana revestida com colágeno e com agonistas
plaquetários (epinefrina e ADP) e com uma abertura central de 150 µm de diâmetro. O tempo
requerido para obter oclusão da abertura, por um trombo plaquetário, é denominado tempo de
oclusão (TO) ["closure time"].
O analizador é bem adaptado para testes de rotina, tendo as vantagens da simplicidade, da
fácil execução, do emprego de variado "shear rate/stress", reprodução do ambiente "in vivo", e
demonstrou grande sensibilidade e especificidade.
Outro exame importante é o estudo da agregação plaquetária por meio do agregômetro.
Nesse exame, pela transmissão de luz, obtém-se um registro da agregação plaquetária com o
emprego de indutores fisiológicos como: colágeno, epinefrina, ADP, trombina, e não fisiológicos
como a ristocetina. A FIGURA 34 mostra a curva de agregação plaquetária produzida pelas
referidas substâncias.

66
Na FIGURA 34 a curva A é normal e do tipo bifásica produzida por indutores fisiológicos
como a epinefrina ou o ADP. As concentrações das substâncias, usadas no teste, podem variar na
dependência da finalidade do estudo. A primeira parte da curva é gerada pela ação direta da
substância empregada; já a segunda parte decorre da ação de substâncias excretadas pelo sistema
canalicular aberto, e derivadas dos corpúsculos densos (reação de liberação) no momento em que
a plaqueta é ativada. Fica fácil entender que se houver deficiência de substâncias do “Pool” de
armazenamento, ou defeito das reações decorrentes da ação do indutor sobre seu receptor na
membrana plaquetária, a segunda parte da curva estará ausente já que não serão liberadas as
substâncias responsáveis pela agregação endógena. A curva característica desse tipo de resposta é
a curva B. Esse tipo de curva é visto nas deficiências de substâncias do “Pool” de armazenamento
e nos distúrbios da transdução de sinais.
FIGURA 34: Registros da agregação plaquetária obtidos com diversos agonistas. A = curva normal
bifásica. B = curva anormal demonstrando ausência da agregação pelo indutor endógeno. C = curva
anormal demonstrando ausência de resposta aos indutores exógeno e endógeno. Detalhes no texto.
Na curva C, a curva é plana em virtude da ausência de resposta aos indutores externos e
internos. Esse tipo de resposta é encontrado na Trombastenia de Glanzmann com o emprego dos
indutores fisiológicos e na doença de von Willebrand com o emprego da ristocetina.
Outros testes, mais específicos para determinados distúrbios, serão comentados no tópico
correspondente.

67
DISTÚRBIOS HEREDITÁRIOS DA COAGULAÇÃO
Doença de von Willebrand (vWD): É uma das mais freqüentes desordens hemorrágicas hereditárias, com uma prevalência de
0.9-1.3% na população em todo o mundo e de pouco menos de 3% nos Estados Unidos. De
grande destaque na vWD é a grande discrepância existente na história das hemorragias entre os
familiares e, até mesmo, no paciente em diferentes períodos. Também não existe relação precisa
entre a intensidade dos fenômenos hemorrágicos e os dados laboratoriais. No estudo de BIRON e
colaboradores, apenas 8 de 30 pacientes, com o tipo 1 da vWD, com redução significante do nível
de vWF(ag), apresentavam hemorragia na HDA ou na HF. Os distúrbios hemostáticos referentes
à vWD estão relacionados às alterações surgidas na síntese do vWF e na conseqüente
anormalidade funcional.
Outro ponto importante que precisa ser ressaltado para entender os testes diagnósticos
dessa doença é o do "shear rate/stress" que será discutido nesta seção devido a sua importância
para a compreensão do assunto.
"Shear rate" corresponde à interface do sangue circulante com a parede vascular. Em outras
palavras, é a velocidade de deslocamento da lâmina de sangue circulante, mais próxima à parede
do vaso. As plaquetas dessa camada de sangue são, justamente, aquelas que entrarão em contato
com o subendotélio exposto após a lesão do endotélio.
"Shear stress" representa a força de colisão das plaquetas entre si e da plaqueta com uma
superfície, por exemplo, a fibrila de colágeno. No fenômeno de "shear rate" as plaquetas são
arrastadas em uma superfície; já no "shear stress" elas se chocam com a superfície.
A importância disso está em que a atividade de ligação do vWF aumenta com o aumento do
"shear rate/stress".
Um novo teste, prático, está sendo empregado para o diagnóstico da vWD - é o tempo de
oclusão. Para tal, é empregado um aparelho, o PFA-100 (Platelet Function Analyzer, Dade
International, Massy, France) para pesquisar defeito de adesividade e agregação plaquetárias,
particularmente na vWD. A explicação sobre este teste já foi dada na seção “Exames para
Esclarecimento do Defeito Hemostático”.
Com o que já foi exposto até agora, é possível interpretar os testes, os quais estão
apresentados no Quadro 2, para o esclarecimento diagnóstico da vWD.
Suspeita-se de vWD quando, além de hemorragia apresentada pelo paciente,
particularmente, em cirurgia e pela mucosa, houver relato de sangramento em familiares e
aumento do tempo de sangramento com plaquetometria normal.

68
Se ao efetuar os exames apresentados no Quadro 2, observar diminuição do nível de
FVIII:C, diminuição proporcional do nível de vWF(ag), alteração da curva de agregação com
ristocetina e baixo percentual da atividade da ristocetina como cofator há uma confirmar do
diagnóstico da doença de von Willebrand. A realização do teste de avaliação do tempo de
oclusão, realizado no analizador PFA-100, mostrará um tempo de oclusão (TO) ampliado, o que
colabora para a confirmação do diagnóstico. Confirmado o diagnóstico, deve-se partir para a
determinação do tipo da vWD por meio do estudo dos multímeros do vWF usando a eletroforese
em SDS-agarose.
Quadro 2: Testes laboratoriais para diagnóstico da doença de von Willebrand
Dosagem do FVIII:C:
É a dosagem do FVIII:C que se encontra acoplado ao vWF
Avaliação do vWF(ag):
Trata-se da fração antigênica da molécula que é avaliada pela imunoeletroforese
quantitativa usando-se anticorpo específico (técnica de Laurel). Normal = 50-150%.
Recentemente, têm sido empregados testes de maior sensibilidade como o de ELISA e o de
citometria de fluxo.
Avaliação do vW:R CoF:
É a avaliação da atividade da ristocetina como cofator. Verifica-se a agregação de
plaquetas normais, fixadas em formol, pela ristocetina e plasma do paciente. A ristocetina é
empregada em concentração padrão e o plasma do paciente em quantidades diversas.
Compara-se com a aglutinação usando-se quantidades diversas de plasma normal. Se houver
defeito relativo ao vWF do paciente, a agregação com a ristocetina será anormal. Normal =
60-150%
RIPA:
RIPA (aggregation of platelet-rich plasma with ristocetin). É o registro, no
agregômetro, da agregação plaquetária usando-se plasma do paciente rico em plaquetas, com
adição de ristocetina em quantidade de 1.2 mg/ml De acordo com a pesquisa, quantidades
diversas podem ser empregadas
Estudo dos multímeros do vWF:
Determinação dos multímeros por meio da eletroforese de SDS-agarose (sulfato de dodecyl
sódico-agarose)
Estudo dos multímeros do vWF:
Determinação dos multímeros por meio da eletroforese de SDS-agarose (sulfato de dodecyl
sódico-agarose)
Tempo de oclusão:
Tempo de oclusão determinado pelo analizador PFA-100, conforme discutido
anteriormente.

69
Com a eletroforese, pode-se determinar as características dos multímeros, podendo então
caracterizar a vWD nos tipos apresentados no Quadro 3.
Quadro 3: Tipos da doença de von Willebrand
Tipos da vWD Principais características
Doença Tipo I Autossômica dominante. Cerca de 70-80% dos pacientes estão nesse grupo.
Diminuição discreta ou moderada do vWF no plasma, porém, estruturalmente
normal. Há diminuição paralela do vWF(ag), FVIII-C e da Atividade
Ristocetina-cofator
Doença Tipo II
Subtipos:
2A, 2B, 2C, 2M,
2N
Cerca de 15-20% dos pacientes estão nesse grupo. Alteração qualitativa da
molécula de vWF com níveis normais ou pouco diminuidos do referido fator:
2A: Autossômica dominante. Decréscimo quantitativo, parcial, dos multímeros
da molécula de vWF, de grande e intermediário tamanhos. Há diminuição da
interação com as plaquetas. A quantidade do vWF(ag) e do FVIII-C estão
normais ou próximos do normal. A atividade da ristocetina como cofator está
bastante reduzida.
2B: Autossômica dominante. Há diminuição da proporção dos grandes
multímeros e aumento da proporção dos pequenos multímeros. Nesse tipo, a
interação do vWF com a plaqueta está aumentada levando à agregação
intravascular. As plaquetas agregadas são afastadas rapidamente da circulação o
que leva à trombocitopenia intermitente. Os níveis do FVIII-C e do vWF são
variáveis. A agregação das plaquetas pela ristocetina está aumentada, mesmo em
baixas doses dessa substância.
2C: Autossômica recessiva. Decréscimo da proporção dos grandes multímeros,
porém qualitativamente anormais. Aumento dos pequenos multímeros. A
atividade da ristocetina como cofator está diminuida e em desproporç ão com a
redução do vWF.
2M: Tem alteração laboratorial semelhante a do tipo 2A. Os grandes multímeros
não estão diminuidos, mas o vWF interage pouco com as plaquetas. Há
diminuição da atividade do vWF, mas estão normais o vWF(ag) e do FVIII-C.
2N: Autossômica recessiva. A principal característica desse tipo é intensa
diminuição da afinidade do vWF pelo FVIII-C. Conseqüentemente, o FVIII-C
não fica mais protegido da rápida proteólise, o que resulta na diminuição de
cerca de 5% do nível desse fator em relação ao nível normal. O vWF(ag) e o
vWR-CoF estão freqüentemente normais.
Doença Tipo III Autossômica recessiva. Total deficiência do vWF, porém, sem alteração
estrutural da molécula. Esse tipo apresenta uma incidência de 1 em 1 milhão de
pessoas. Caracteriza-se pela ausência da agregação plaquetária pela ristocetina
(RIPA), intenso decréscimo do nível do vWF(ag), indetectável atividade da
ristocetina como cofator e nível de FVIII-C muito baixo.

70
Diante do que foi exposto pode-se dizer que a doença de von Willebrand se constitui em
uma verdadeira armadilha para o médico, já que as observações que apresentamos a seguir
podem afastar a hipótese de uma diátese hemorrágica na avaliação pré-operatória:
1- Seus principais sintomas são hemorragias vistas, freqüentemente, em outras patologias
ou sem causa determinada, como epistaxe recorrente, menorragia, pós-extração dentária, pós
amidalectomia, alguns dias após o parto.
2- O paciente pode não apresentar qualquer história pregressa de sangramento.
3- Pode não haver história de hemorragia nos membros consangüíneos da família.
4- Os exames pré-operatórios para pesquisa de distúrbio hemostático podem ser normais.
Verifica-se também a existência de discrepância muito grande nos dados dos diversos
autores sobre a prevalência da doença. É fácil deduzir que essa discrepância decorre da
dificuldade em se pensar na doença pelas razões expostas nos itens anteriores, e pela
indisponibilidade de maiores recursos diagnósticos laboratoriais em muitos paises.
Adendo: Síndrome de von Willebrand Adquirida (vWS)
Apesar de ser um distúrbio adquirido, é importante comentá-lo nesse item, em virtude da
semelhança, clínica e laboratorial, com a vWD que é um distúrbio hereditário. A vWS, como na
vWD, decorre de alterações quantitativas e/ou qualitativas da molécula do vWF.
È um distúrbio muito raro, o que torna mais difícil sua diferenciação com o correspondente
hereditário que, como dissemos, chega a 1% da população mundial e a pouco menos de 3% da
população dos Estados Unidos. A vWS reproduz várias alterações laboratoriais vistas na vWD,
tais como prolongamento do tempo de sangramento, diminuição do FVIII:C, da ristocetina como
cofator (vWF:R CoF), do vWF(ag) e da aglutinação plaquetária induzida pela ristocetina (RIPA).
Já que o estudo laboratorial não é suficente para fazer a diferenciação entre a vWD e a vWS, é
importante constatar a inexistência de história pregressa de hemorragia no paciente, e de
alterações dos exames característicos da vWD nos familiares.
A vWS tem sido descrita em associação com: gamopatias monoclonais, linfomas, tumor de
Wilms, mieloma, doença cardíaca congênita, lupus eritematoso sistêmico, angiodisplasia,
desordens convulsivas tratadas pelo ácido valpróico e hipotireoidismo. Cerca de um terço dos
casos publicados estão associados à gamopatia monoclonal, mas na maioria dos casos o
mecanismo responsável pela vWS não está claramente definido. Segundo alguns autores, o vWF
é removido rapidamente do sangue por mecanismos diversos: 1- auto-anticorpos específicos que
inativam o vWF, 2- anticorpos inespecíficos que formam imuno-complexos circulantes com o

71
vWF o que favorece seu afastamento pelos macrófagos, 3- absorção do vWF por células malígnas
e 4- por degradação proteolítica do vWF. Será adicionado um 5º mecanismo, o da absorção
seletiva de multímeros, de alto e intermediário peso molecular, em pacientes com doença
cardíaca, particularmente com defeito do septo ventricular ou da válvula. Nesse último caso,
havendo deficiência dos referidos multímeros, o distúrbio assemelha-se ao tipo hereditário vWD
2B.
Deficiência de outros fatores da coagulação:
Como foi exposto anteriormente, a partir dos testes de rotina para o estudo da hemostasia,
podemos desconfiar da redução do nível de um ou de vários fatores da coagulação. Para
esclarecimento, partiremos para a dosagem do fator com base no teste de rotina alterado.
A dosagem do fator da coagulação é de fácil execução, estando ao alcance de qualquer
laboratório.
Em função da gravidade da deficiência dos fatores VIII e IX da cascata da coagulação,
serão enfatizadas as conseqüências que o paciente poderá apresentar na deficiência destes fatores,
como também a abordagem terapêutica.
Deficiência do Fator VIII:
A deficiência do Fator VIII caracteriza uma patologia conhecida como Hemofilia A. A
hemofilia A é a desordem hereditária mais comum da cascata da coagulação sangüínea. È uma
desordem de sangramento ligada ao sexo e que tem sido documentada há muitos séculos atrás. O
gene que expressa o Fator VIII está localizado no cromossoma X, assim os filhos serão
hemofílicos e as filhas serão obrigatoriamente portadoras do traço hemofílico. As mulheres que
apresentam um gene anormal geralmente não exibem sangramento, pois elas possuem dois
cromossomas X e um único gene normal é capaz de produzir quantidades suficientes de Fator
VIII:C funcional.
A severidade das manifestações clínicas está diretamente relacionada com o grau de
deficiência do Fator.
a) Doença Leve: Os níveis do Fator VIII:C estão acima de 0,05U/ml (valor normal:
1U/ml), geralmente estes pacientes são assintomáticos. Corre risco quando submetidos a um
trauma cirúrgico muito extenso.

72
b) Doença Moderada: Os níveis do Fator VIII:C estão entre 0,02-0,05U/ml. Neste caso os
pacientes são freqüentemente assintomáticos, mas ocasionalmente sangramentos podem ocorrer
sem nenhuma causa definida.
c) Doença Severa: Os níveis do Fator VIII:C estão abaixo de 0,01U/ml. Neste caso o
paciente apresenta doença hemorrágica grave, com sangramentos espontâneos, que requer uma
terapia constante.
O Tempo de Tromboplastina Parcial Ativado (TTPa) será anormal, ou seja, prolongado e o
Tempo de sangramento mais o Tempo de Protrombina (TP) serão normais.
Para uma análise específica deve ser feita a dosagem do Fator VIII:C. Níveis < 0,01U/ml
(<1% do normal) são encontrados nos pacientes com doença severa.
Aproximadamente 10% a 15 %dos pacientes com hemofilia A desenvolvem anticorpos
anti-Fator VIII:C. Esses anticorpos são do tipo IgG e por isso são capazes de neutralizar o Fator
VIII:C a 37oC.
Atualmente, existem muitas formas de tratamento para a hemofilia A. Dentre elas algumas
merecem destaque:
1- Concentrados de plasma humano são amplamente utilizados.
2- Um grande número de hemofílicos é tratado com concentrados de Fator VIII altamente
purificados e liofilizado. O crioprecipitado é uma rica fonte de Fator VIII, mas não é o produto de
escolha devido ao risco de transmissão de doenças, principalmente a Hepatite C e a AIDS.
Atualmente, com as técnicas de biologia molecular está sendo possível a clonagem do Fator VIII
e com isso reduzir incidência da transmissão das doenças virais.
3- Pacientes que tem desenvolvido anticorpos anti-Fator VIII são tratados com
concentrados de Fator VIII de origem suína.
4- Análogos da Vasopresina estimula a liberação de Fator VIII dos grânulos densos,
aumentando efetivamente os níveis deste fator por cerca de 3 a 4 vezes. Porém em pacientes com
níveis < 0,05U/ml este procedimento pode não ser eficiente.
Um nível de 30% da atividade normal é o nível terapêutico ideal para manter a hemostasia.
A presença de anticorpos anti-Fator VIII pode ser diagnosticada quando estudos de mistura
com plasma normal não corrige o TTPa prolongado.
Deficiência do Fator IX:
A deficiência de Fator IX é conhecida como hemofilia B e é uma desordem de sangramento
recessiva ligada ao sexo. Mulheres com a deficiência são completamente raras, mas elas podem

73
ser transportadoras. Embora os sintomas clínicos são completamente similares, o sangue de um
paciente com hemofilia A não normaliza o tempo de coagulação do sangue de um paciente com
hemofilia B.
Os níveis da atividade do Fator IX é que determina a severidade da doença, com as
deficiências mais severas tendo menos de 1% da atividade, e as deficiências moderadas com 5%
a 25% da atividade. Na hemofilia B severa, há uma total ausência do Fator IX, demonstrada pela
ausência da atividade coagulante e antigênica. Alguns pacientes com Hemofilia B sintetizam uma
variante não funcional da molécula do Fator IX, enquanto outros não possuem atividade
antigênica identificada no plasma e conseqüentemente possuem uma ausência verdadeira da
síntese da molécula. Deficiência adquirida de Fator IX pode ser vista nos pacientes com doença
hepática ou com deficiência de vitamina K e nos pacientes que estão em uso de anticoagulante
oral.
O tratamento dos pacientes com deficiência severa a moderada de Fator IX consiste de
infusões regulares de plasma fresco congelado, concentrados do complexo protrombina que
contem os Fatores II, VII, IX e X ou concentrados de Fator IX. A complicação mais séria nos
pacientes com hemofilia B é o desenvolvimento de anticorpos anti-fator IX. Esses são
extremamente difíceis de tratar e ocorre em aproximadamente 10% dos pacientes com esta
deficiência.
3- Dosagem de Fatores da Cascata da Coagulação:
A dosagem de fatores pode ser feita individualmente, utilizando-se um plasma deficiente no
fator que ser quer determinar.
O princípio do método é o seguinte: um plasma completamente deficiente em fator VIII,
por exemplo, o plasma de um hemofílico A, tem um TTPa muito prolongado. Entretanto, ele
contém níveis normais dos demais fatores, de modo que a adição de um plasma normal vai
encurtar o TTPa proporcionalmente à concentração de fator VIII presente neste plasma normal.
Desta forma, pode-se construir um curva relacionando o valor do TTPa em segundos, com a
concentração do fator em porcentagem.
A dosagem de qualquer fator individualmente baseia-se nesse princípio, o plasma deficiente
no fator em questão pode ser obtido de individuo congenitamente deficiente, como é o caso dos
hemofílicos, ou pode ser obtido artificialmente. O melhor método para obtenção de plasma
artificialmente deficiente em um fator é a imunoadsorção, que consiste na passagem do plasma
por uma coluna onde esteja fixado um anticorpo específico contra o fator que se quer retirar. O

74
plasma eluído da coluna contém todos os fatores com exceção do fator que ficou retido na coluna
pelo anticorpo.
Princípio:
Consiste em usar um plasma imunoadsorvido, deficiente apenas em fator VIII ou em fator
IX ou ainda em fator XI, e verificar a capacidade de diferentes diluições de um “Pool” de plasma
normais em corrigir o TTPa desta mistura, construindo-se então uma curva.
Reagentes:
1 – “Pool” de plasmas normais, colhidos em citrato de sódio. O plasma pode ser fresco de
preferência, mas pode também ter sido congelado. Lembrar que congelamento a –20oC não é
suficiente para prevenir a perda de atividade do fator VIII no decorrer do tempo. É interessante
que o plasma normal para a curva tenha o mesmo tratamento que o plasma a ser testado. Se
utilizar plasma congelado, verificar se ele está totalmente descongelado à 37oC, por um tempo
não inferior a 10 minutos.
2 – Plasma deficiente em fator VIII por imunoadsorção (Lab. Dade). Reconstituir o
conteúdo do frasco com 1 ml de água destilada (esterilizada). Deixar sob agitação lenta (agitador
magnético) pelo menos por 15 minutos para que o conteúdo fique bem dissolvido. Fazer um
“Pool”, se vários frascos de plasmas deficientes forem usados, misturando o conteúdo de todos os
frascos em um só. Manter em geladeira ou em banho de gelo, e nestas condições o reagente é
estável por 8 horas.
3 – Tampão de Owren, pH 7,35, guardado a 4oC.
4 – Reagente para realização do TTPa:
Cefalina: podendo ser usada a ativada com ácido elágico (Cefamat-Biolab), para permitir a
leitura em coagulômetro. Cloreto de Cálcio 0.025 M.
Os reagentes 3 e 4 devem permanecer à 37oC.
Procedimento:
1 – Preparação da curva padrão:
O “Pool” de plasmas normais é diluído em tampão de Owren e são mantidas em banho de
gelo.

75
Diluição do “Pool” de Plasma normal para construção da curva padrão
Tubo Atividade Diluição PPP Tampão
1 100% 1:5 400µl 1.600µl
2 50% 1:10 1000µl do tubo 1 1000µl
3 25% 1:20 1000µl do tubo 2 1000µl
4 12,5 1:40 1000µl do tubo 3 1000µl
5 6,25% 1:80 1000µl do tubo 4 1000µl
6 3,125% 1:160 1000µl do tubo 5 1000µl
7 1,56% 1:320 1000µl do tubo 6 1000µl
2 – Colocar nos tubos de ensaio, no coagulômetro, nesta ordem:
100µl do plasma deficiente;
100µl do Cefamat;
100µl da diluição a ser testada;
Incubar exatamente por 5 minutos, ao fim dos quais pipetar:
100µl de CaCl2 0,025M.
Anotar o tempo de coagulação.
3 – Os plasmas dos pacientes a serem testados são diluídos como para 100% de atividade,
ou seja, uma diluição de 1:5.
Caso o tempo seja muito curto, o diluição pode ser maior, para que o tempo caia dentro da
curva.
4 – As determinações devem ser feitas em duplicata ou triplicata, registrando-se a média
dos valores obtidos.
5 – Os resultados são colocados em papel di-log.
A curva é traçada, e o valor para os pacientes é lido na curva.
6 – Não deixar o plasma deficiente com o Cefamat por muito tempo, não mais do que meia
hora. Muito cuidado com as pipetagens, para não contaminar os reagentes.

76
Dosagem do Fator V:
Princípio:
A dosagem do fator V obedece ao mesmo princípio. O plasma deficiente em fator V pode
ser obtido por imunoadsorção (Lab. Dade), ou ser preparado no laboratório, através do
envelhecimento do plasma normal. E então verificada a capacidade de um plasma corrigir o TP
de um plasma deficiente em fator V.
Reagentes:
1– “Pool” de plasmas normais, colhidos em citrato de sódio. O plasma pode ser congelado
ou fresco. O fator V perde atividade com o tempo, quando congelado a – 20o C.
2– Tampão de Owren, como usado para dosagem do fator VIII.
3– O plasma deficiente em Fator V é obtido da seguinte maneira:
a) colher sangue total em oxalato de sódio, 1.34%, na proporção de 1 volume de
anticoagulante para 9 volumes de sangue. O Fator V é menos estável em oxalato do que em
citrato de sódio.
Centrifugar o sangue para obter o PPP. Verificar o TP e o TTPa de cada plasma.
b) fazer um “Pool” dos plasmas normais e colocar em tubo plástico e absolutamente limpo,
em banho-maria à 37o C.
c) incubar durante 24 h e verificar o TP. Que deve estar entre 60 e 80 segundos.
d) aliquotar este plasma em alíquotas de 1ml e este será o plasma deficiente em fator V.
e) se o TP em 24 h for menor do que 60 segundos, incubar ainda algumas horas. Se for
maior do que 80 segundos, desprezar o plasma.
4 – Reagente para realização do TP:
Tromboplastina:
Usar o reagente comercial (Replastin).
Reconstituir o frasco com 1 ml de água destilada e esperar 5 minutos para estabilização.
Esta preparação é estável por 21 dias à 4oC. Fazer um “Pool” se vários frascos forem utilizados.
CaCl2 0,025M.
Procedimento:
1 – Preparação da curva padrão:
77
Um “Pool” de plasma normais, colhidos em citrato de sódio, é diluído em tampão de Owren e as
diluições são mantidas em banho de gelo.
Diluição do “Pool” de Plasma Normal:
Tubo Atividade Diluição PPP Tampão
1 100% 1:10 200µl 1.800µl
2 50% 1:20 1000µl do tubo 1 1000µl
3 25% 1:40 1000µl do tubo 2 1000µl
4 12,5 1:80 1000µl do tubo 3 1000µl
5 6,25% 1:160 1000µl do tubo 4 1000µl
2 – O plasma a testar é diluído 1:10, como para o tubo 1 da curva padrão.
3 – Pipetar no tubo de ensaio, no coagulômetro, na seguinte seqüência:
100µl do plasma deficiente;
100µl da Tromboplatina;
100µl da diluição previamente aquecida à 37oC;
Incubar por 30 segundos;
100µl de CaCl2;
Anotar o TP
4 – Construir a curva em papel di-log, interpolar os resultados dos plasmas a serem testados.
Pesquisa de Anticoagulante Circulante:
O anticoagulante circulante ou inibidor da coagulação, é representado por um anticorpo,
geralmente da classe IgG, que interfere no processo da coagulação, podendo ser detectado nos
testes “in vitro”. A natureza deste inibidor é variável, podendo aparecer em pacientes portadores
de deficiências congênitas de alguns fatores, como a hemofilia A, por exemplo, onde a presença
do anticorpo é secundária à exposição do indivíduo a uma proteína que ele não produz.
O inibidor pode ser também de natureza auto-imune, como é o caso do anticorpo anti-
fosfolipídeo, também conhecido como anticoagulante lúpico. Este anticorpo é dirigido contra
fosfolipídeos, o que faz com que ele interfira com o reagente utilizado nos teste “in vitro” como a

78
Cefalina e a Tromboplastina, prolongando os tempos de coagulação, embora não haja inibição da
coagulação “in vivo”.
A detecção destes inibidores obedece ao mesmo princípio geral.
Existe o prolongamento do tempo de coagulação pela presença do inibidor, o que não é
corrigido pela adição de plasma normal. Isso serve para diferenciar um prolongamento do tempo
de coagulação por causa da deficiência de um determinado fator, pois neste caso, o tempo seria
plenamente corrigido pela adição de um plasma normal.
Os inibidores contra os fatores da Via Intrínseca ou Comum são melhores avaliados pelo
TTPa. A pesquisa deve ser feita sempre que houver prolongamento do TTPa, isto é, quando a
relação Plasma Doente/Plasma Normal for maior que 1,25. Faz se então uma mistura em partes
iguais do plasma do doente com o plasma normal, repetindo-se o TTPa. No caso de deficiência
de fator, o TTPa da mistura deve ser totalmente corrigido, caindo para um valor menor do que
1,25. No caso da presença de um inibidor o TTPa da mistura permanecerá maior do que 1,25.
Existem alguns inibidores, como é o caso do inibidor do fator VIII que ocorre após imunização
em hemofílicos ou como em um autoanticorpo em algumas doenças auto-imunes, que tem uma
ação lenta e progressiva. Nestes casos, pode ocorrer a correção imediata do TTPa a despeito da
presença do inibidor. Por este motivo é importante a realização do TTPa imediatamente após se
fazer a mistura do plasma do doente com o plasma normal em parte iguais (M1) e também após a
incubação desta mistura por 1 ou 2 horas à 37oC (M2), o que permitirá que a ação inibidora seja
evidenciada.
Exemplo do Resultado de uma Pesquisa de Deficiência de fator Anticoagulante circulante:
TTPa Doente/TTPa normal: 1.76, 1.79 e 1.81
TTPa M1 (Plasma Doente + Plasma Normal sem incubação)/TTPa Normal: 1,12; 1,65 e 1.23
TTPa M2 (Plasma Doente + Plasma Normal incubado/2 horas a 37oC)/TTPa Normal: 1,24; 1,68 e 1,70
MONITORAMENTO DA ANTICOAGULOTERAPIA
O tratamento anticoagulante pode ser feito com a administração de heparina em dose alta
por via intravenosa, na fase inicial, seguida da administração profilática de drogas
anticoagulantes orais, os anti-vitamina K (AVK), por tempo prolongado. Qualquer um desses
tratamentos deve ser adequadamente monitorizado por meio de testes laboratoriais destinados a
avaliação da heparinemia e ao controle do tratamento com anti-vitamina K.

79
Monitoramento da heparinemia:
A heparina é um mucopolissacaríde formado pela polimerização de várias unidades de
ácido urônico e glicosamina, com PM variando de 3.000 a 40.000 D. A ação anticoagulante da
heparina se faz através da sua interação com a antitrombina III (ATIII), um inibidor natural que
bloqueia fatores da coagulação ativados, especialmente a Trombina e o fator Xa. A ATIII é um
inibidor de ação lenta e progressiva, entretanto, quando ela está ligado à heparina, a velocidade
de inibição é enormemente aumentada e o efeito anticoagulante é imediato. O uso da heparina
como tratamento anticoagulante é feito pela sua administração intravenosa, em altas doses, o que
é capaz de alterar testes laboratoriais usados para monitorização do efeito de uma determinada
dose. O uso profiláctico em dose baixa por via subcutânea, não leva a níveis plasmáticos capazes
de alterar testes de laboratório, de modo que a monitorização da dose não pode ser feita pelos
testes convencionais. O monitoramento da dose da heparina pode ser feito por vários testes: Os
Testes globais, os que avaliam a ação da heparina na cascata da coagulação como um todo: (a)
Tempo de Coagulação ou TC. É um teste sensível quando realizado de forma adequada, à 37oC e
de forma padronizada; (b) Tempo de Coagulação Ativado TCA. É realizado com sangue total na
presença de Celite, sendo mais sensível que o TC, especialmente em altas concentrações de
heparina; (c) Tempo de Tromboplastina Parcial Ativada ou TTPa. É o teste que melhor se
correlaciona com a heparinemia. É realizado com o plasma pobre em plaquetas, sendo o teste de
escolha para monitorar a heparinemia; (d) Tempo de Trombina ou TT. É um teste sensível, mas
não mantém uma correlação tão linear, especialmente em concentrações mais altas de heparina; e
(e) Testes Específicos: são testes que avaliam a ação da heparina diretamente sobre as enzimas
que ela inibe, através de substratos cromogênicos. Eles podem estimar a atividade antitrombina
ou anti-IIa e a atividade anti–Xa da heparina.
A utilização do TTPa para monitorar a heparina deve ser padronizado para cada reagente, e
em cada laboratório. Esta padronização baseia-se na construção de uma curva de heparinemia “in
vitro”, a partir de um “Pool” de plasmas normais, no intuito de se avaliar a sensibilidade do
reagente a um determinado nível de heparina no plasma. A escolha da heparinemia para garantir
uma boa anticoagulação é baseada em dados empíricos, e está em torno de 0,3 a 0,4 unidades de
heparina por ml de plasma. A construção da curva permite saber, para um dado reagente, o
quanto se deve prolongar o TTPa do paciente em relação ao normal, para que a heparinemia
eficiente seja atingida.
A curva de heparinemia é construída avaliando-se o TTPa de um “Pool” de plasmas
normais ao qual foram adicionadas concentrações fixas de heparina. São feitas as seguintes
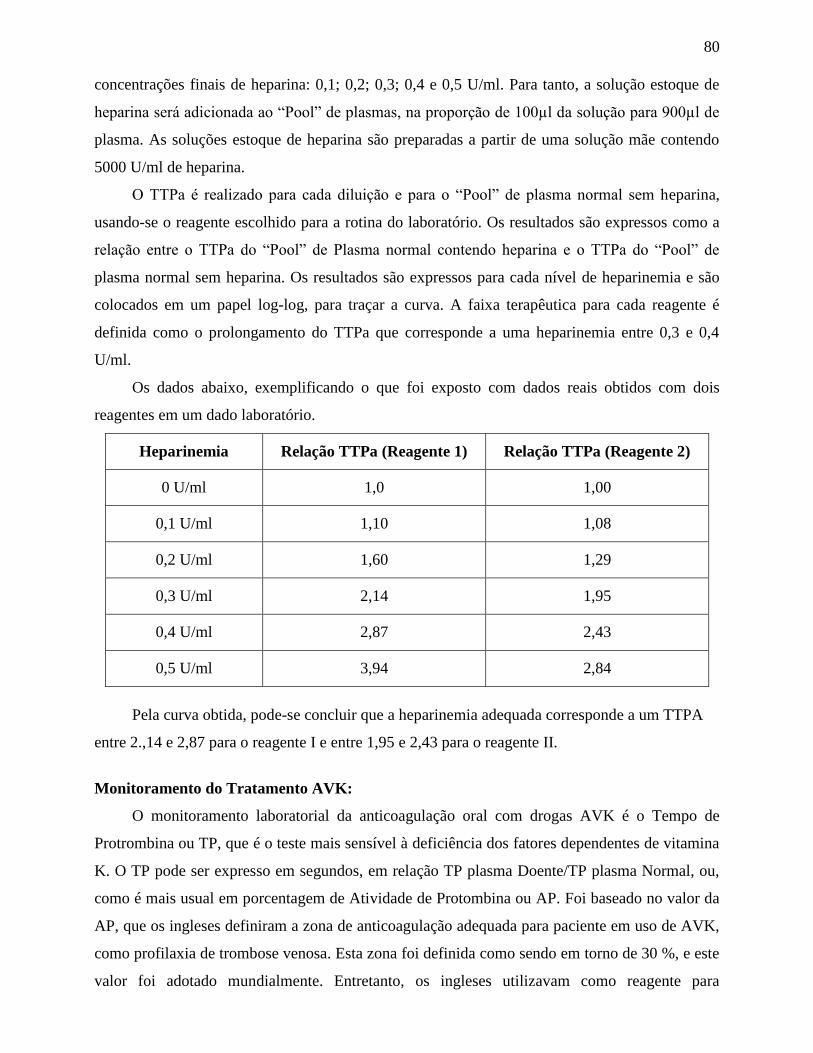
80
concentrações finais de heparina: 0,1; 0,2; 0,3; 0,4 e 0,5 U/ml. Para tanto, a solução estoque de
heparina será adicionada ao “Pool” de plasmas, na proporção de 100µl da solução para 900µl de
plasma. As soluções estoque de heparina são preparadas a partir de uma solução mãe contendo
5000 U/ml de heparina.
O TTPa é realizado para cada diluição e para o “Pool” de plasma normal sem heparina,
usando-se o reagente escolhido para a rotina do laboratório. Os resultados são expressos como a
relação entre o TTPa do “Pool” de Plasma normal contendo heparina e o TTPa do “Pool” de
plasma normal sem heparina. Os resultados são expressos para cada nível de heparinemia e são
colocados em um papel log-log, para traçar a curva. A faixa terapêutica para cada reagente é
definida como o prolongamento do TTPa que corresponde a uma heparinemia entre 0,3 e 0,4
U/ml.
Os dados abaixo, exemplificando o que foi exposto com dados reais obtidos com dois
reagentes em um dado laboratório.
Heparinemia Relação TTPa (Reagente 1) Relação TTPa (Reagente 2)
0 U/ml 1,0 1,00
0,1 U/ml 1,10 1,08
0,2 U/ml 1,60 1,29
0,3 U/ml 2,14 1,95
0,4 U/ml 2,87 2,43
0,5 U/ml 3,94 2,84
Pela curva obtida, pode-se concluir que a heparinemia adequada corresponde a um TTPA
entre 2.,14 e 2,87 para o reagente I e entre 1,95 e 2,43 para o reagente II.
Monitoramento do Tratamento AVK:
O monitoramento laboratorial da anticoagulação oral com drogas AVK é o Tempo de
Protrombina ou TP, que é o teste mais sensível à deficiência dos fatores dependentes de vitamina
K. O TP pode ser expresso em segundos, em relação TP plasma Doente/TP plasma Normal, ou,
como é mais usual em porcentagem de Atividade de Protombina ou AP. Foi baseado no valor da
AP, que os ingleses definiram a zona de anticoagulação adequada para paciente em uso de AVK,
como profilaxia de trombose venosa. Esta zona foi definida como sendo em torno de 30 %, e este
valor foi adotado mundialmente. Entretanto, os ingleses utilizavam como reagente para

81
determinação do TP, uma Tromboplastina preparada com o cérebro humano, que é bastante
sensível à deficiência dos fatores dependentes de Vitamina K. Os autores americanos utilizavam
uma Tromboplastina derivada de cérebro de coelho, que é menos sensível, o que faz com que as
zonas terapêuticas sejam diferentes para cada reagente considerado. Isto ficou evidente quando os
resultados foram comparados, pois os americanos, adotando a zona terapêutica de 30%,
administravam doses maiores de AVK e tinham mais complicações hemorrágicas. Na verdade, a
AP de 30% obtida com a Tromboplastina de cérebro humano corresponde a uma AP de cerca de
45% com a Tromboplastina de cérebro de coelho. Esta constatação demonstrou a necessidade de
ser padronizar os resultados obtidos com reagentes de origens diferentes, de modo a se
estabelecer um modo de expressão comum e utilizável por todo mundo. Esta padronização é feita
através da determinação do “Indice de Sensibilidade Internacional” ou ISI de cada reagente.
O seu cálculo é feito para cada lote, realizando-se o TP de plasmas de pacientes em tratamento
AVK estável, usando a Tromboplastina a testar e a Tromboplastina de referência, que é a
Tromboplastina inglesa. Isto permite a construção de uma curva e o cálculo do ISI é feito usando-
se a inclinação desta reta. Na prática é o fabricante da Tromboplastina quem realiza esta
padronização e o ISI, calculado para cada lote, é fornecido na bula do reagente. Uma vez
conhecido o ISI, pode-se calcular o chamado RNI “Índice Normatizado Internacional”. Este valor
corresponde à relação entre o TP do plasma doente e o TP do plasma normal, caso houvesse
utilizado a Tromboplastina de referência. Assim, qualquer que seja a sensibilidade do reagente
utilizado, o nível de anticoagulação, avaliado pelo RNI é sempre o mesmo.
O Cálculo do INR é feito pela seguinte fórmula:
RNI = (TP DO PACIENTE/ TP DO “POOL” DE PLASMAS NORMAIS)ISI
Por exemplo:
Se o TP do doente foi de 23,4 segundos, e o TP do normal foi de 14,6 segundos, e se o ISI
do lote da Tromboplastina for de 1,53, o RNI será igual a 2,0. Este valor significa que o TP do
doente é duas vezes maior o TP do normal, caso se tivesse utilizado a Tromboplastina de
referência. A expressão do resultado em RNI cabe ao laboratório que realiza o teste. Atualmente,
os reagentes disponíveis no mercado são padronizados em relação à Tromboplastina de
referência, e o ISI está impresso em cada lote. Com esta padronização pode-se definir um nível
de anticoagulação desejada para cada situação clínica, e o ajuste da dose de anticoagulante oral,
deve ser feito baseado nisso. O nível adequado foi determinado através de estudos clínicos e
pode ser resumido na seguinte tabela 3.

82
Tabela 3: O nível adequado de RNI de acordo com cada condição Clínica.
Alvo Faixa
Prevenção de trombose venosa 2,52 a 3,0
Prevenção de trombose venosa recorrente 3,02 a 4,0
Prevenção de tromboembolismo arterial 3,53 a 4,5
O RNI acima de 5,0 está associado a um risco muito grande de sangramento.
É importante se Ter em mente que a expressão dos resultados em INR só tem sentido quando se
trata de pacientes em uso de drogas AVK. A expressão dos resultados do TP em outras situações
clínicas deve ser feita pelo AP ou pela relação TP doente/TP normal. Isto porque a padronização
é feita baseada na diferença de sensibilidade dos reagentes à deficiência específica dos fatores
vitamina K dependentes.
DISTÚRBIO HEMOSTÁTICO POR VENENO DE SERPENTE
Coagulação intravascular disseminada e fibrinólise em pacientes picados por serpentes
venenosas. Os venenos de serpente mostram atividades fisiopatológicas do tipo coagulante,
proteolítica, hemolítica e neurotóxica.
Somente as atividades relacionadas à ação dos venenos no mecanismo hemostático é que
serão comentadas e para facilitar a exposição será apresentada uma subdivisão, de forma bastante
resumida na FIGURA 35.
As ações do veneno podem estar relacionadas com:
a) Relacionado com o mecanismo da coagulação:
- Atuação direta sobre o FX
- Atuação direta sobre a protrombina (gerando trombina)
- Atuação direta sobre o fibrinogênio (produzindo fibrina)
b) Relacionado com a plaqueta:
- Consumo de plaquetas pela ativação da coagulação
- Atuação direta sobre as plaquetas
c) Relacionado com o mecanismo fibrinolítico:
- Secundária à formação da fibrina (fibrinólise de trombo)
- Ativação direta do mecanismo fibrinolítico (fibrinólise primária)

83
d) Relacionado com o vaso:
- Lesão da parede vascular (proteólise das proteínas da membrana basal)
FIGURA 35: Diferentes modos de ação dos venenos de serpente no mecanismo hemostático. A =
ativando o FX, B = ativando o FII, C = atuando diretamente sobre o fibrinogênio, D = agregando as
plaquetas, E = ativando diretamente o mecanismo fibrinolítico, F = lesando o vaso sangüíneo
(proteólise)
Um mesmo veneno pode agir em pontos diversos do mecanismo hemostático e com
intensidade variável, dependendo da característica do tipo do veneno e da sua dose.
Existia muita contradição acerca do modo de ação do veneno, isso se deve ao fato de que
diferentes concentrações foram usadas nas pesquisas in vitro e in vivo. O melhor modo para se
entender a ação dos venenos de serpente é analisar o típico veneno coagulante e proteolítico, o
veneno da Bothrops jararaca. O veneno botrópico ao infundir lentamente na circulação
desfibrina o sangue, tanto pela geração de trombina como pela ação direta sobre o fibrinogênio.
Se a infusão é rápida e a dose total é pequena, essa desfibrinação é precedida de
hipercoagulabilidade que perdura por alguns minutos. Se a dose é elevada, e se for via venosa, há
morte rápida por coagulação intravascular maciça dentro de minutos, seguida de liquefação do
sangue pela fibrinólise posterior. Se a dose injetada for superior a 0.4 mg/kg de peso corporal,
ocorre coagulação imediata e maciça, produzindo morte em poucos minutos. Assim, se
compreende que o encontro de sangue incoagulável em indivíduo picado por serpente, é indício
de que penetrou na circulação uma dose razoável de veneno (igual ou superior a 0.1 mg/kg).

84
Importante trabalho foi desenvolvido por Nahas e cols. com os venenos das seguintes serpentes:
Echis colorata, Bothrops atrox, Bothrops jararaca, Crotalus terrificus, Akistrodon rhodostoma,
Trimeresurus purpureomaculata, Bitis gabonica e Vipera russelli. Os resultados permitiram
definir a intensidade e os diversos pontos de ação no mecanismo da coagulação conforme
demonstra o Quadro 4. Esses autores evidenciaram, ao usar o veneno de E. colorata, uma ação
fibrinogenolítica rápida e potente, ou seja, ativação direta do mecanismo fibrinolítico.
Posteriormente, foi isolado uma fração fibrinolítica desse veneno.
Quadro 4: Ação dos venenos de alguns tipos de serpentes segundo Nahas e cols. (10).
(*) = Ativação do mecanismo fibrinolítico
TIPO DE SERPENTE ATIVAÇÃO
DO FATOR X
ATIVAÇÃO DO
FATOR II
AÇÃO DIRETA SOBRE
O FATOR I
V. russelli ++++ 0 0
B. atrox +++ 0 + *
B. jararaca ++ 0 ++ *
E. colorata ++ ++++ 0 *
A. rhodostoma + 0 ++++ *
C. terrificus 0 0 +
T. purpureomaculata 0 0 + *
B. gabonica 0 0 0
Pacientes picados por E. carinatus apresentam trombocitopenia (mínimo de 24.000/mm³ de
sangue), fibrinogênio em 5 mg/100ml, prolongamento do tempo de protrombina, anemia (volume
globular mínimo de 23%), hemácias fragmentadas, presença de monômeros de fibrina e queda
dos fatores V e FVIII. Nestes pacientes a resposta da heparina foi rápida com bloqueio da
coagulação e da agregação plaquetária. Desta forma, pode-se concluir que a agregação
plaquetária dependeu da ação da trombina e que as alterações sangüíneas foram conseqüência da
ação direta do veneno sobre a protrombina.
Estudos mostram que a incoagulabilidade do sangue produzida pela A. rhodostoma, deve-
se, principalmente, à ação direta do veneno sobre o fibrinogênio com liberação apenas do
fibrinopeptídio A. Estudos verificaram que a fração polipeptídica desse veneno proporciona,
inicialmente, a queda do fibrinogênio no plasma com formação de fibrina e sua posterior lise.
Fato idêntico parece ocorrer com os venenos da C. adamanteus e da E. colorata.

85
Estudos sobre a ação do veneno da E. colorata em cobaias mostraram que a desfibrinação
se dá por dois modos: ativação direta do mecanismo fibrinolítico na circulação (fibrinólise
primária) e desencadeamento da fibrinólise no trombo de fibrina (fibrinólise secundária). Se
pequena quantidade de veneno é injetada, esse último mecanismo é o mais importante para a
desfibrinação e, por ser a hipercoagulabilidade de pouca intensidade, nenhum trombo é
encontrado. Desta forma, pode-se observar que a lesão da parede vascular produzida pelo veneno
é de importância primária e que os distúrbios da coagulação contribuem para agravar a
hemorragia. As serpentes venenosas existentes no Brasil pertencem a 4 gêneros: Crotalus
(cascavel), Bothrops (jararaca), Micrurus (corais venenosas) e Lachesis (surucucu).
Por ordem de freqüência as causadoras de acidentes são: B. jararaca, C. durissus terrificus,
B. jararacussu, B. alternata, B. neuwiedi e B. atrox. As picadas de corais venenosas são muito
raras. No Hospital Vital Brasil, do Instituto Butantan, em São Paulo, cerca de 90% dos
atendimentos por picadas de serpentes venenosas referem-se ao gênero Bothrops. A mortalidade
dos indivíduos picados por este tipo de serpente, quando não recebem antiveneno, é de cerca de
8% caindo para 0.768% com a soroterapia. A percentagem das picadas por cascavel é de 8.59%
com 72% de mortalidade para os não tratados com antiveneno e 11.49% para os que receberam
esse tratamento.
Tratamento:
1 - Afastamento da causa
Consiste na neutralização da ação do veneno e na tentativa de diminuir a quantidade do
mesmo, através de sucção no local da picada nos primeiros minutos.
a) Casos com sintomatologia discreta: 50 unidades por via sub-cutânea.
b) Casos com sintomatologia intensa: 50 unidades por via sub-cutânea e 50 unidades por
via venosa.
c) Casos de máxima gravidade: 150 a 300 unidades como dose total, sendo 70 unidades por
via sub-cutânea e o restante por via venosa.
Existem dois graus extremos de sintomatologia que servem para avaliar a gravidade do
caso e orientar a terapêutica com antiveneno:
A) Benigno: depois de duas horas, sangue coagulável. Pequena reação local. Não há risco
de vida nem de necrose.
B) Grave: sangue incoagulável. Reação local moderada ou intensa. Possibilidade de
necrose local. Risco de vida por choque com colapso periférico dentro dos primeiros três dias e,

86
também, por hemorragia renal intensa, ou derrame no sistema nervoso, ou acidente vascular
cerebral.
2 - Reposição dos fatores e das plaquetas
Após terapêutica com antiveneno, a regeneração do fibrinogênio ocorre em poucas horas,
tanto no homem como nos animais de experiência. Se houver necessidade de elevar os fatores da
coagulação e das plaquetas, seguir as mediadas apresentada no tratamento geral da coagulação
intravascular disseminada.
3 - Anticoagulação e antifibrinólise
Como mostrado anteriormente, os venenos de serpentes podem formar trombina e/ou atuar
diretamente sobre o fibrinogênio levando à formação de uma fibrina que difere daquela formada
pela trombina. Pela ação do veneno, dá-se a liberação apenas do fibrinopeptídio A. Esse tipo de
monômero sofre somente polimerização longitudinal e é mais suscetível à ação lítica da
plasmina.
É fato conhecido que a heparina não tem capacidade de bloquear a ação direta do veneno
sobre o fibrinogênio (11). Quando o veneno gera trombina, a heparinização pode ser seguida da
elevação dos fatores e das plaquetas no sangue. Há estudos que mostram a incapacidade de
bloquear in vivo, por meio da heparina, a síndrome de desfibrinação produzida pelo veneno da B.
jararaca que além de atuar diretamente sobre o fibrinogênio, gera trombina após ativar o Fator X.
Assim, a heparinoterapia não deve ser usada na picada de serpente.
Como conseqüências da ação direta do veneno sobre o fibrinogênio temos a
desfibrinogenação e a circulação de produtos de degradação devido à lise da fibrina formada.
Pode-se considerar ambas alterações como antagônicas à hipercoagulabilidade o que acarretam
certa proteção ao organismo.
Por outro lado, a severidade do quadro clínico não está relacionada à alteração sangüínea e
sim a outros efeitos do veneno que são prontamente neutralizados pelo antiveneno.
DISTÚRBIO HEMOSTÁTICO NAS SEPSES
Introdução
Ainda não existe consenso sobre a definição de sepse. Entretanto, em 1991, as sociedades
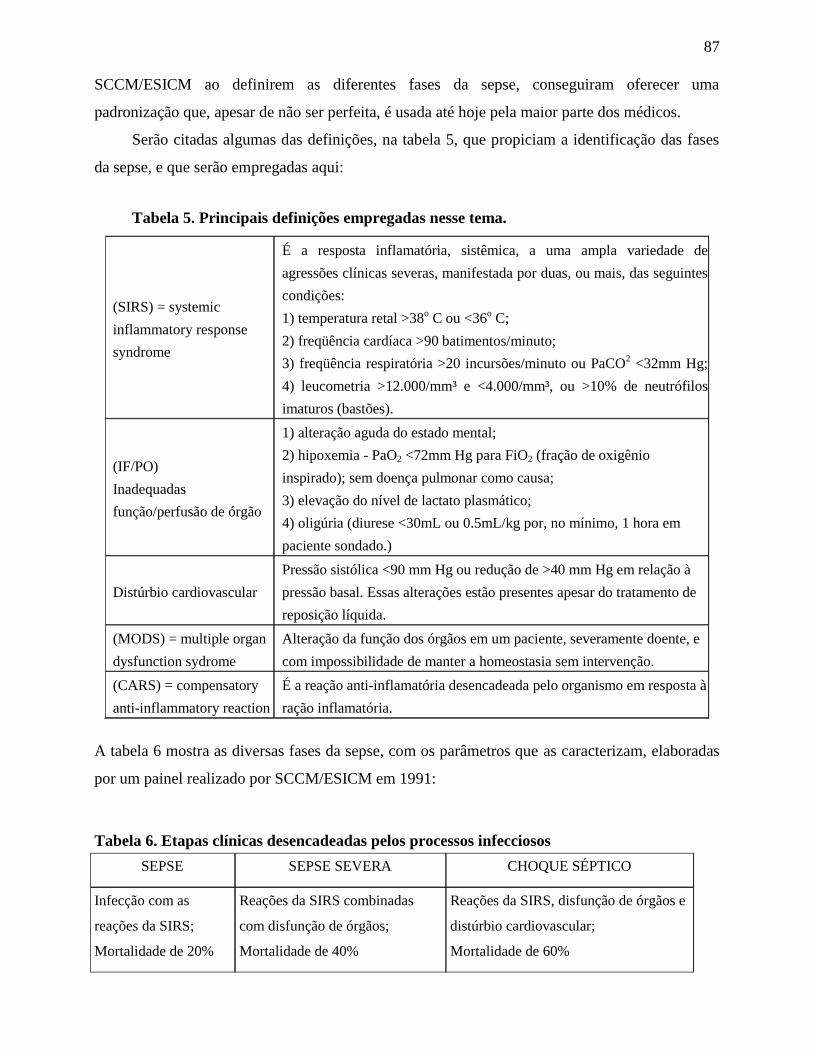
87
SCCM/ESICM ao definirem as diferentes fases da sepse, conseguiram oferecer uma
padronização que, apesar de não ser perfeita, é usada até hoje pela maior parte dos médicos.
Serão citadas algumas das definições, na tabela 5, que propiciam a identificação das fases
da sepse, e que serão empregadas aqui:
Tabela 5. Principais definições empregadas nesse tema.
(SIRS) = systemic
inflammatory response
syndrome
É a resposta inflamatória, sistêmica, a uma ampla variedade de
agressões clínicas severas, manifestada por duas, ou mais, das seguintes
condições:
1) temperatura retal >38o C ou <36
o C;
2) freqüência cardíaca >90 batimentos/minuto;
3) freqüência respiratória >20 incursões/minuto ou PaCO2 <32mm Hg;
4) leucometria >12.000/mm³ e <4.000/mm³, ou >10% de neutrófilos
imaturos (bastões).
(IF/PO)
Inadequadas
função/perfusão de órgão
1) alteração aguda do estado mental;
2) hipoxemia - PaO2 <72mm Hg para FiO2 (fração de oxigênio
inspirado); sem doença pulmonar como causa;
3) elevação do nível de lactato plasmático;
4) oligúria (diurese <30mL ou 0.5mL/kg por, no mínimo, 1 hora em
paciente sondado.)
Distúrbio cardiovascular
Pressão sistólica <90 mm Hg ou redução de >40 mm Hg em relação à
pressão basal. Essas alterações estão presentes apesar do tratamento de
reposição líquida.
(MODS) = multiple organ
dysfunction sydrome
Alteração da função dos órgãos em um paciente, severamente doente, e
com impossibilidade de manter a homeostasia sem intervenção.
(CARS) = compensatory
anti-inflammatory reaction
É a reação anti-inflamatória desencadeada pelo organismo em resposta à
ração inflamatória.
A tabela 6 mostra as diversas fases da sepse, com os parâmetros que as caracterizam, elaboradas
por um painel realizado por SCCM/ESICM em 1991:
Tabela 6. Etapas clínicas desencadeadas pelos processos infecciosos
SEPSE SEPSE SEVERA CHOQUE SÉPTICO
Infecção com as
reações da SIRS;
Mortalidade de 20%
Reações da SIRS combinadas
com disfunção de órgãos;
Mortalidade de 40%
Reações da SIRS, disfunção de órgãos e
distúrbio cardiovascular;
Mortalidade de 60%

88
Fisiopatologia:
Até os meados da década de oitenta, aceitava-se que as alterações que acompanham a sepse
eram produzidas, diretamente, por substâncias liberadas pelas bactérias. Em 1985, alguns autores
sugeriram que essas alterações decorriam das reações inflamatórias produzidas pelo organismo,
quando agredido pelas substâncias liberadas pelas bactérias. Em 1997, alguns pesquisadores
passaram a defender uma nova teoria, mostrando que as múltiplas alterações apresentadas pelas
diversas fases da sepse eram conseqüência, não só da reação de defesa do organismo, como
também da ação de substâncias que medeiam essas defesas, e da combinação de ambas.
Na tabela 7, está apresentada a lista parcial de moléculas com atividade pró-inflamatórias e
anti-inflamatórias.
Tabela 7: Moléculas pró e anti-inflamatórias MCP = Proteína quimiotractante de monócito; PAI = Inibidor do ativador do plasminogênio.
Moléculas pró-inflamatórias Moléculas anti-inflamatórias
TNF-alfa
IL-1 beta
IL-2
IL-6
IL-8
IL-15
Neutrofilo elastase
IFN-gama
Proteinoquinase
MCP-1
MCP-2
Fator inibidor da
leucemia (Fator-D)
Tromboxane
Fator ativador das plaquetas
Moléculas solúveis de adesão
Neuropeptídios vasoativos
Fosfolipase A2
Tirosinoquinase
PAI-1
Geração de radicais livres
Neopterin
CD14
Prostaciclina
Prostaglandina
IL-1 ra
IL-4
IL-10
IL-13
Receptor de IL-1 TipoII
Fator de transformação de
crescimento-beta Epinefrina
Receptores TNF-alfa solúveis
Receptor antagonismo-
Leucotriene B1
CD14 recombinte soluvel
Lipopolissacarídio ligado à
proteína
Para organizar o nosso comentário sobre fisiopatologia, apresentamos na FIGURA 36 a
seqüência das diversas etapas a partir da eliminação de substâncias pelas bactérias, até atingir a
disfunção de órgãos e, por fim, a falência total.
Como mostra a FIGURA 36, na etapa 1 estão as principais substâncias, liberadas pelas
bactérias, e que vão atuar nas células (etapa 2) responsáveis pela produção dos mediadores pró e
anti-inflamatórios (etapa 3). São esses mediadores, citados na Tabela 7, que vão levar a uma série
de reações (etapa 4), comentada anteriormente. Como pode ser visto na etapa 4, a
hipercoagulabilidade e a inibição da fibrinólise, vieram juntar-se às reações pró e anti-
inflamatórias.

89
FIGURA 36: As diversas etapas que se seguem após a ação das bactérias sobre os tecidos,
terminando com a disfunção e/ou falência dos órgãos.
A partir da segunda metade da década de noventa, tem sido dada maior importância aos
distúrbios da coagulação e da fibrinólise, como os responsáveis pela disfunção e falência dos
órgãos nas sepses.
Em seqüência, na etapa 5, tem como resultante: trombo vascular, reação tecidual, aumento
da permeabilidade vascular, quimiotaxia e ativação celulares.
Na etapa 6 pode-se observar o resultado final dos distúrbios mencionados anteriormente, a
disfunção de órgãos até a falência total.
Distúrbio da coagulação e da fibrinólise nas sepses.
Como foram mostrados no tópico sobre Hemostasia, os mecanismos da coagulação,
anticoagulação, fibrinólise e antifibrinólise, são mantidos em equilíbrio nos estados de
normalidade. Para facilitar o entendimento da interação desses mecanismos, mostramos na
FIGURA 37 os fatores da coagulação que fazem parte da via extrínseca e os seus inibidores.

90
FIGURA 37. Em azul os fatores da coagulação e em vermelho os anticoagulantes; SH = Sulfato de
heparam; TFPI = Inibidor da via do fator tecidual; TF = Fator tecidual; ATIII= Antitrombina III;
TM= Trombomodulina; PC= Proteína C; PS= Proteína S e IIa= Trombina.
A ATIII ao formar complexos com vários fatores da coagulação, particularmente com os
fatores II, IX, X e XI, os inativa. Essa inativação é potencializada pela heparina e pelo sulfato de
heparam (molécula semelhante à heparina que está presente na superfície da célula endotelial).
A PC torna-se uma protease ativa ao formar um complexo com a TM (proteína presente na
superfície da célula endotelial) e o FIIa (trombina), sendo auxiliada pelo co-fator PS. A PCa
promove a proteólise dos fatores V e VIII inibindo, desse modo, a geração de trombina.
O TFPI forma um complexo com o TF e o FVII que inibe o FX. Entretanto, nas sepses, esse
equilíbrio é quebrado com a exacerbação da hipercoagulabilidade e a conseqüente formação de
trombos na microcirculação.
As alterações responsáveis por esse desequilíbrio são constituídas por:
a) Ativação do mecanismo de coagulação: observações clínicas têm demonstrado que, nas
sepses, o mecanismo de coagulação é ativado pelo caminho extrínseco e não pelo intrínseco.
Desse modo, a ação de citocinas procoagulantes, citadas anteriormente, promovem a exposição
do fator tecidual (TF) pela célula endotelial. A exposição do TF após a injúria dos tecidos, tem
início a exacerbação da coagulação até a geração da trombina que é a responsável pela formação
do trombo de fibrina.
b) Hipoativação dos mecanismos de anticoagulação: há indícios de que a anticoagulação
está diminuída nas sepses, colaborando desse modo para o incremento da hipercoagulabilidade.
Esmon e Esmon mostram que o complexo formado pela trombomodulina, proteína C e proteína S

91
torna-se hipoativo quando o endotélio vascular sofre injúria. Por outro lado, a antitrombina III
tem seus níveis reduzidos em muitos pacientes com sepse. Há suspeitas de que o TFPI esteja
reduzido nos estados sépticos, o que colaboraria para a instalação da hipercoagulabilidade.
c) Supressão da fibrinólise: decorrente da ação de citocinas, a atividade do PAI-1 está
aumentada, acarretando a diminuição do ativador do plasminogênio tecidual (tPA). A diminuição
da atividade fibrinolítica associada à diminuição da anticoagulação concorreriam para a
facilitação da instalação de trombos de fibrina. É importante ressaltar que há uma relação direta
entre as diversas fases da sepse e a intensidade da formação dos trombos e da reação tecidual. A
fase mais avançada está relacionada à intensa trombose dos vasos de pequeno calibre e,
conseqüentemente, à maior disfunção de órgãos ou à falência total.
Diagnóstico Laboratorial dos Distúrbio da Hemostasia nas Sepses
Logicamente, é de se esperar que as alterações laboratoriais dependam do estágio em que se
encontra a sepse. Com freqüência, na fase inicial da sepse, é comum não encontrarmos alterações
nos testes para estudo da hemostasia.
Entretanto, o D-dímero, teste que acusa lise de fibrina, já pode ser positivo nas fases
iniciais, sem contudo haver alteração dos demais testes para a hemostasia. Esse teste é de grande
sensibilidade, porém, não é específico para acusar presença de trombo na microcirculação, já que
a lise de fibrina extravascular, decorrente do derrame hemorrágico, torna o teste positivo.
A quase totalidade dos pacientes com sepse avançada apresenta elevação do nível do D-dímero.
O teste da lise de euglobulina, para estudo da atividade fibrinolítica na circulação
sangüínea, com freqüência, é normal ou está ampliado (hipoatividade fibrinolítica sistêmica).
Os níveis de Proteína C, Proteína S e de Antitrombina III podem cair no início da instalação
da sepse e suas alterações estão relacionadas à severidade da infecção, servindo, inclusive, como
marcadores de mal prognóstico.
É na fase de choque séptico, quando a coagulação intravascular disseminada pode estar
presente, e os microtrombos estão disseminados, que vamos encontrar grandes alterações nos
testes para estudo da hemostasia. Além dos já mencionados, há aumento do PT, PTT, monômero
de fibrina, e estão reduzidos os níveis do fibrinogênio plasmático e das plaquetas.
Terapêutica dos distúrbios hemostáticos.
A terapêutica do choque não é pertinente a este tema.
Será comentado, exclusivamente, sobre o tratamento dos distúrbios da hemostasia no
paciente com sepse. Entretanto, fica ressaltado que o combate ao microorganismo causador da

92
infecção e aos demais distúrbios por ele causado, bem como a rapidez no início do tratamento,
são imprescindíveis para se obter sucessos. A ativação do mecanismo da coagulação, com falha
dos mecanismos de anticoagulação e de fibrinólise e a conseqüente formação de microtrombose,
sendo as principais alterações nas sepses, têm levado os pesquisadores a tentar a correção desses
distúrbios. Mas, como veremos, essa conduta não tem trazido resultados animadores e os poucos
trabalhos que mostram algum benefício, ainda requerem comprovação. Mesmo assim serão
apresentados alguns comentários sobre as opções de tratamento nas sepses:
1) O emprego da forma recombinante do inibidor da via do fator tecidual (TFPI), parece
reduzir a mortalidade, particularmente, dos pacientes com sepse severa. Os estudos com esse
produto estão em andamento.
2) A Anti-trombina III (AT-III), que tem a propriedade de inibir a trombina, pode
influenciar diretamente a permeabilidade capilar através da indução de prostaciclina (PGI2). O
emprego terapêutico da AT-III tem mostrado resultados promissores, embora um estudo com
mais de 2300 pacientes não confirmaram bons resultados. O emprego da Proteina C (PC), que
ajuda a interromper o ciclo da coagulação e da inflamação, também tem sido um meio terapêutico
promissor.
3) A Proteina C ativada (PCa), que inativa os fatores da coagulação Va e VIIIa, bloqueia a
geração de trombina, neutraliza o PAI-1 e aumenta o tPA, e tem também ação anti-inflamatória,
tem sido intensamente pesquisada na sua forma recombinante. Também com esse produto os
resultados têm sido animadores. Em suma, até agora não há qualquer tratamento,
comprovadamente efetivo, dirigido para os distúrbios hemostáticos nas sepses.
COAGULAÇÃO INTRAVASCULAR DISSEMINADA:
Introdução:
No capítulo sobre hemostasia, foi demonstrado que o mecanismo hemostático é constituído
da interação dos sistemas de coagulação, de fibrinólise, de anticoagulação e de atividades
relacionadas à parede dos vasos sangüíneos. A quebra do equilíbrio dinâmico entre esses
componentes leva às diáteses hemorrágicas.
É fácil entender o mecanismo fisiopatológico de alguns distúrbios hemorrágicos em virtude
da simplicidade da alteração que os causam. Por exemplo, nas hemofilias A e B, a razão para o
distúrbio está na deficiência dos fatores VIII e IX, respectivamente. Na púrpura
trombocitopênica, a diminuição do número das plaquetas no sangue explica o transtorno.

93
Entretanto, na coagulação intravascular disseminada (CID) as alterações fisiopatológicas não são
tão simples assim. Desta forma, uma explicação mais detalhada será dada:
Na CID os quatro componentes citados apresentam-se alterados, porém, com variação do
distúrbio em cada componente, na dependência da etiologia de cada caso. É importante assinalar
que a CID manifesta-se clinicamente de dois modos distintos: com microtrombose em diversos
órgãos e a conseqüente hipofunção ou falência total dos mesmos e, com hemorragias em diversas
partes do corpo e a conseqüente hipovolemia, podendo chegar ao choque. Na primeira
circunstância, as alterações decorrem do predomínio da ativação do mecanismo da coagulação
sobre o da fibrinólise, e na segunda, o predomínio do mecanismo fibrinolítico é o responsável
pelo distúrbio.
É importante salientar que a CID não é uma doença e sim uma síndrome, já que os
distúrbios citados são secundários a diferentes causas, apesar de que em raros casos, não se
consegue identificar qualquer etiologia, particularmente na fibrinólise isolada (fibrinólise
primária idiopática).
Fisiopatologia:
Na quase totalidade das vezes, a manifestação clínica, decorrente da microtrombose
generalizada, é conseqüência da ativação do mecanismo da coagulação que tem inicio pelo
caminho extrínseco, às custas do fator tecidual (FT). Esse fator fica exposto quando a célula
endotelial é ativada pela trombina, ou por outras substâncias; ou quando os constituintes da
parede vascular e/ou o tecido perivascular entram em contato com o sangue após lesão do
endotélio.
Por outro lado, o nível de FT pode ficar elevado na circulação ao ser liberado por diversos
tipos de células malignas, como as da leucemia promielocítica. As células malignas, também,
podem liberar outros tipos de substâncias com atividade tromboplástica.
Nas infecções, o nível de FT, no sangue, pode estar elevado devido à ação de: endotoxina,
fator de necrose tumoral (TNF), interleucina-1 e de outras substâncias, geradas nos processos
inflamatórios, sobre a célula endotelial.
O FT fica livre no sangue circulante quando a sua concentração ultrapassa o nível de seu
inibidor específico, o inibidor da via do fator tecidual (TFPI = tissue factor pathway inhibitor).
Como foi mostrado no capítulo Hemostasia, a ativação da coagulação pelo FT conduzirá à
geração de trombina, que é o agente principal da formação dos microtrombos de fibrina.

94
È claro que em casos patológicos a trombina é gerada em quantidade superior à do estado
fisiológico, o que impossibilita sua neutralização pelos inibidores. Por outro lado, a insuficiência
do mecanismo fibrinolítico defensivo, contribui para a falta de remoção da fibrina na
microcirculação. Também colabora para a deposição de fibrina na microcirculação, o aumento do
principal inibidor da fibrinólise, o inibidor do ativador do plasminogênio tipo 1 (PAI-1).
Em algumas situações, o início da ativação da coagulação pode se dar pelo caminho intrínseco. O
FXII pode ser ativado pela endotoxina, complexo antígeno-anticorpo, êmbolos de gordura e
queimaduras.
A manifestação clinica por hemorragia decorre, principalmente, quando a ativação do
mecanismo fibrinolítico supera a da coagulação.
O mecanismo fibrinolítico é ativado, fisiologicamente, sempre que a coagulação é ativada.
Esse tipo de ativação é denominado de fibrinólise defensiva. Ela é defensiva porque promove a
lise da fibrina formada indevidamente, ou do trombo hemostático quando sua função estiver
completada. Entretanto, em casos patológicos, a fibrinólise pode ser ativada excessivamente, na
circulação, ao invés de se fazer, apenas, nos locais onde se forma o trombo hemostático. Nesse
caso, a fibrinólise, ao invés de trazer beneficio para o organismo, promoverá alterações
patológicas provenientes do excesso de plasmina circulante.
Como conseqüência da exacerbação do excesso de plasmina na circulação teremos a
diminuição do nível de diversas proteínas que tomam parte na coagulação e na fibrinólise.
Os mais atingidos, quando o processo é agudo, são: FV, FVII, FVIII, FIX, FX, FXIII,
fibrinogênio (FI), antitrombina III (AT-III), proteína C (PC), alfa2-antiplasmina (alfa2-AP) e
plasminogênio, porém, somente nos processos agudos. Além da queda do nível dos fatores
mencionados, outra razão para a hemorragia é a destruição, pela plasmina, do trombo
hemostático no momento de sua formação. Também causa transtorno, para a formação do trombo
hemostático, a produção de produtos de degradação do fibrinogênio e da fibrina pela plasmina
(PDFs). Os PDFs comprometem a agregação plaquetária. È importante destacar que, ao contrário
do que se pensa, a maior razão para a queda do nível de algumas das proteínas, citadas
anteriormente, que tomam parte no mecanismo hemostático, não decorre da ação da trombina, e
sim da lise pela plasmina.
Por outro lado, nos processos agudos, devido à ação da trombina sobre as plaquetas, a
trombocitopenia é um achado freqüente. A exacerbação do mecanismo fibrinolítico não é
responsável por essa alteração. Todas as referências, acima, relacionam-se à CID de intensidades
moderada e intensa (CID aguda). Entretanto, quando o processo é de pouca intensidade e de

95
evolução lenta, e com poucas complicações, a CID é dita crônica. Nesse caso, como o consumo
(ou lise) dos fatores e de plaquetas é pouco intenso, a exacerbação, compensatória, da produção
dos elementos atingidos, promove a elevação de seus níveis. Esse fenômeno é conhecido como
rebote. Enquanto nas CIDs de evolução aguda a hemorragia é um sintoma freqüente, nas de
evolução crônica destacam-se os fenômenos microtrombóticos.
No Quadro 5 apresentamos as diversas causas que freqüentemente estão associadas às CIDs
aguda e crônica.
Diagnóstico laboratorial:
Fazem parte do diagnóstico laboratorial de CID dois grupos de exames: os exames que
podem levar à suspeita do distúrbio e os que confirmam o diagnóstico.
Exames que, quando alterados, levam à suspeição de CID:
Plaquetometria, tempo de protrombina (TP), tempo de tromboplastina parcial ativado
(TTPa) e dosagem do fibrinogênio.
Desse modo, quando houver alteração desses exames, e estiver presente uma das doenças
referidas no Quadro 5, deve-se pensar em CID e, para esclarecimento, deve-se realizar os
exames, que serão mencionados a seguir.
Exames que, quando alterados, levam à confirmação de CID:
Dosagem dos fatores da coagulação V, VII, VIII, IX, X, XIII e proteína C: A queda do
nível sangüíneo de alguns desses fatores é observada nas CIDs agudas e nas crônicas, verifica-se,
mais freqüentemente, elevação devido ao fenômeno de rebote.
Dosagem da Antitrombina III (AT-III): O nível da AT-III decresce devido à sua ação
neutralizadora sobre a grande quantidade de trombina gerada.
Dosagem dos produtos de degradação da fibrina e do fibrinogênio (FDP): É um teste
simples que mede a aglutinação de partículas de látex. Apesar da baixa especificidade, é de alta
sensibilidade, estando elevado em 85-100% dos pacientes com ativação dos mecanismos de
coagulação e de fibrinólise.
Teste do D-dímero: Enquanto o teste dos FDP, quando positivo, indica que tanto a fibrina
quanto o fibrinogênio foram degradados, o teste do D-dímero é específico para indicação de que,
apenas, a fibrina foi degradada enzimaticamente.

96
Quadro 5: Causas relacionadas às CIDs crônica e aguda.
AGUDAS:
- Infecção (bactéria, virus, fungo, etc)
- Doença maligna (leucemia, metástase não hematológica, etc.)¤ Obstétrica (descolamento
prematuro de placenta, embolia de líquido amniótico, degeneração gordurosa aguda da gravidez,
eclampsia)
- Trauma
- Queimadura
- Veneno de serpente
- Transfusão de sangue incompatível
- Falência hepática aguda
CRÔNICAS:
- Maligna (leucemias, tumores sólidos)
- Obstétrica (feto morto retido)
- Hematológica (síndromes mieloproliferativas, hemoglobinúria paroxística noturna, síndrome
hemolítica urêmica)
- Vascular (artrite reumatóide, doença de Raynaud)
- Inflamatória (colite ulcerativa, doença de Crohn, sarcoidose)
- Aneurisma aórtico
- Hemangioma gigante (síndrome de Kasabach-Merritt)
- Rejeição aguda de transplante renal
No teste D-dímero é empregado um anticorpo anti-domínio D-D do monômero de fibrina.
È, justamente, nesse domínio que se dá a união entre dois monômeros de fibrina, o que o torna
específico para acusar a presença de trombina. Apesar da alta especificidade para a presença da
trombina, sua sensibilidade é inferior a do teste dos FDP.
Pesquisa de monômeros de fibrina: Em estado de normalidade o monômero de fibrina
sofre polimerização e forma o polímero de fibrina no trombo hemostático. Quando é produzido
monômero de fibrina na circulação, ele forma complexos com fibrinogênio e/ou com produtos de
degradação da fibrina e do fibrinogênio (PDF). Ao identificamos esses complexos, concluímos
que há coagulação exacerbada em alguma parte do organismo, o que implica em dizer que pode
depender da coagulação intravascular disseminada ou da localizada (trombo), ou de coagulação
extravascular.
O teste para identificação da presença dos monômeros de fibrina, na circulação, é realizado
adicionando-se protamina, ou etanol, ao plasma do paciente. Após a adição dessas substâncias, o

97
monômero de fibrina é liberado do complexo e se une a outros monômeros (polimerização). Com
isso, forma-se uma tênue rede de fibrina.
O teste de protamina, que é mais fiel, acusa de 20-50 µg/ml de monômero, e de 50-100
µg/ml de PDF de grande tamanho. Esse teste é simples e de grande sensibilidade.
Dosagem de fibrinopeptídio A (FPA). Pode ser empregado o método de ELISA ou de
radioimunoensaio. Quando a trombina atua sobre o monômero de fibrina, libera fibrinopeptídios.
Desse modo, o nível elevado de FPA é sinal de ativação do mecanismo da coagulação.
Dosagem dos fragmentos 1+2 da protrombina (PF 1+2). Altos níveis desses fragmentos,
medidos pelo método ELISA, indicam que o FXa atuou sobre a protrombina. O nível desses
fragmentos está elevado em mais de 90% dos pacientes com CID.
Ressaltamos que um único exame não é diagnóstico de CID mas, dentro das possibilidade
de cada laboratório, o emprego de alguns dos testes mencionados anteriormente, poderão nos
conduzir ao diagnóstico desse distúrbio.
O "Scientific and Standardization Committee" da "International Society on Thrombosis and
Haemostasis" elaborou um algoritmo com escores, para a classificação da CID, o qual está
reproduzido no Quadro 6.
Quadro 6: Escores para a classificação da CID
1 Avaliação do risco: O paciente apresenta uma desordem que seja reconhecida como associada
com a CID?
Caso negativo - não usar esse algoritmo. Caso positivo - prosseguir:
2 Solicitar os testes globais de coagulação: plaquetometria, tempo de protrombina (TP),
fibrinogênio, monômeros solúveis de fibrina ou produtos de degradação d0 polímero de fibrina
(PDF/D-dímero)
3 Escore global dos resultados dos testes de coagulação:
Plaquetometria................................................................................ ..
( > 100 = 0, < 100 = 1; < 50 = 2 )
Marcadores relativos à fibrina (PDF/D-dímero).............................
(ausência de aumento: 0; aumento moderado: 2; aumento intenso: 3)
Tempo de protrombina prolongado..................................................
(< 3 segundos = 0; >3 seg. e <6 seg. = 1; > 6 seg. = 2)
Nível de fibrinogênio...........................................................................
(>1.0 g/l = 0; < 1.0 g/l = 1)
4 Cálculo do escore total......................................................................... (Soma dos escores do item 3)
5 Interpretação do escore total do item 4:
Se maior ou menor do que 5, é compatível com CID. Repetir o escore diariamente.
Se menor do que 5, sugestivo (não afirmativo) de CID declarada. Repetir o escore a cada 1 ou 2 dias.

98
Tratamento:
Destacamos quatro pontos importantes no tratamento da CID:
1- Combate à causa,
2 - prevenção e/ou tratamento do choque,
3 - reposição dos componentes sangüíneos e
4- combate da hipercoagulabilidade e da hiperfibrinólise.
É fácil entender que as medidas a serem adotadas em cada item dependerão de cada tipo de
processo e de seu grau de intensidade. Serão abordados os detalhes, de alguns desses itens,
quando for discutir os principais distúrbios. Isso também pode ser visto no tema "Distúrbio
Hemostático por Veneno de Serpente".
1- Combate à causa:
Como foi visto, a CID, salvo raras exceções, tem sempre uma causa identificável (ver
Quadro 5). Está comprovado que sem combater a causa, é praticamente impossível o sucesso
terapêutico, mesmo com as medidas de amparo ao paciente que serão comentadas nos itens
seguintes.
Combatida a causa, o distúrbio desaparecerá. O tempo de recuperação varia de acordo com
o tipo do responsável pelo processo. Assim, quando a CID tiver sido provocada pelo
descolamento prematuro de placenta, a normalização levará algumas horas após o esvaziamento
do útero. Já no caso de septicemia, será de alguns dias já que a resposta ao tratamento da infecção
é mais demorada.
2 - Prevenção e/ou tratamento do choque.
O desenvolvimento desse item não é pertinente ao tema. Para detalhes sobre esse assunto,
consultar os textos de medicina intensiva. Entretanto, é óbvio que sem a prevenção e/ou
tratamento do choque, bem como os cuidados para impedir ou tratar a falência dos diversos
órgãos, o resultado estará fadado ao insucesso.
3- Reposição dos componentes sangüíneos.
A reposição dos glóbulos vermelhos deve ser feita por meio de concentrado de hemácias
sempre que o hematócrito cair a níveis críticos. Cada unidade de concentrado eleva o hematócrito
em cerca de 3%. Caso se lance mão do sangue total estocado, deve-se ter em mente que será
infundindo um material pobre em alguns fatores da coagulação (principalmente FV e FVIII) e em

99
plaquetas. Por esse motivo, deve-se orientar na reposição desses elementos que apresentarem
níveis baixos no sangue, sempre baseados nos estudos laboratoriais. Considerar, também, que a
reposição desses elementos não se fará com a mesma facilidade observada nas patologias por
deficiência de síntese, já que estão sendo consumidos ou hidrolisados, apesar de que quando
circulando, recebem proteção dos inibidores fisiológicos. Por outro lado, não esquecer que o
sangramento não depende, somente, dos níveis baixos dos fatores e das plaquetas; a plasmina em
excesso, pela sua ação lítica, dificulta ou impede a formação do trombo hemostático e os
produtos de degradação do fibrinogênio e da fibrina interferem na função plaquetária. Desse
modo, lançar mão dos concentrados de plaqueta, do crioprecipitado que é rico em FVIII,
fibrinogênio e Antitrombina III (AT-III), do concentrado de AT-III e do plasma fresco congelado
que fornecerá diversos fatores da coagulação. O concentrado de plaquetas deve ser infundido,
apenas, quando o paciente estiver sangrando, ou seja, quando as plaquetas estiverem abaixo de
50.000/l de sangue, ou quando houver indicação de algum procedimento invasivo. A quantidade
a ser infundida é de uma bolsa de concentrado, obtida de cada doador, para cada 10 kg de peso.
Cada bolsa infundida eleva as plaquetas no sangue em 10.000-12.000/mm3. No caso de
plaquetoferese, 1 bolsa de concentrado de plaquetas corresponde à quantidade de 6 bolsas de
doadores diversos. Já os concentrados de fatores da coagulação têm sido pouco indicados; por um
lado porque repõem apenas alguns fatores e, por outro lado, porque podem conter traços de
substâncias com ação tromboplástica, o que incrementaria a hipecoagulabilidade. Para repor
diversos fatores devemos lançar mão do plasma fresco (6 unidades em 24 horas).
O plasma fresco congelado também pode ser infundido na quantidade de 16 ml/kg de peso
corporal, via intravenosa, para o adulto, quando a relação do TTPa (TTPa do doente/TTPa
normal) for >1,5. Para a criança a dose é de 10-15 ml/kg de peso corporal. Essas quantidades
aumentam os fatores da coagulação no sangue em cerca de 10-20%, e devem ser repetidas a cada
8 horas.
O crioprecipitado que contém 80-100 U de FVIII, 100-300 mg de fibrinogênio, de
fibronectina e de fator von Willebrand é mais indicado para reposição de fibrinogênio. O que
mais preocupa no seu emprego é a impossibilidade da inativação do HIV.
Como o fibrinogênio é um fator estável, ele é facilmente encontrado no plasma e no sangue
total, mesmo quando estocados em condições adequadas de conservação. A diminuição do nível
do fibrinogênio no sangue só preocupa quando ele fica inferior a 100 mg/dl.
O concentrado de AT-III tem sido empregado, mas faltam estudos científicos que
comprovem sua utilidade. Teoricamente, como o nível de AT-III cai, freqüentemente, na CID,

100
seu emprego seria útil para bloquear a hipercoagulabilidade, pela sua ação anticoagulante, direta,
no mecanismo de coagulação e como cofator para a ação da heparina. A dose indicada é de
100U/kg de peso corporal via intravenosa durante 3 horas, seguida de infusão contínua de
100U/kg de peso corporal/dia.
4 - Combate da hipercoagulabilidade e da hiperfibrinólise.
O emprego de substâncias antifibrinolíticas deve ser feito com muito cuidado e em casos
específicos. Isso porque, havendo deposição de fibrina nos vasos de pequeno calibre, o bloqueio
da fibrinólise impedirá sua remoção; ainda mais que em determinados casos, como por exemplo,
nas septicemias, a atividade fibrinolítica está diminuída.
Alguns trabalhos têm mostrado sua utilidade quando esse distúrbio é produzido pela
leucemia promielocítica e pelas doenças malignas não hematológicas disseminadas.
O ácido aminocapróico é um antifibrinolítico muito usado e a dose recomendada é de 5-
10g, lentamente por via intravenosa, seguida por 2-4 g/hora via intravenosa, sem exceder a 30
g/dia.
O ácido tranexâmico é um potente antiplasmínico não indicado nos trabalhos americanos.
Temos usado esse produto na dose de 250-500 mg, lentamente na veia, a cada 6 horas.
Resumindo o exposto, o mecanismo fibrinolítico constitui uma defesa do organismo contra
a hipecoagulabilidade e seu bloqueio pode trazer sérias conseqüências. Deve-se, pois, usá-lo com
precaução e certos de sua indicação quando sua exaltação estiver trazendo sérias conseqüências
para o paciente.
A heparina é o medicamento mais usado para tentar bloquear a hipercoagulabilidade,
entretanto, seu uso tem sido motivo de controvérsias por várias razões: os trabalhos não têm
comprovado bons resultados nas CIDs agudas como acontece nas crônicas e, a dose ideal não foi
determinada como nos processos trombóticos.
Na realidade, o êxito do tratamento, na maioria dos casos, é devido, principalmente, ao
combate à causa e ao suporte ao doente do que propriamente ao uso do anticoagulante. Por outro
lado, devido aos diversos graus de intensidade na ativação do mecanismo da coagulação pelas
substâncias tromboplásticas, com a conseqüente geração de quantidades variáveis de trombina, a
padronização da dose de heparina fica muito difícil de ser realizada.

101
BIBLIOGRAFIA
1- BAJZAR, L. Activatable fibrinolysis inhibitor and an antifibrinolytic pathway. Arterioscler.
Thromb. Vasc. Biol. 20: 2511-2518; 2000.
2- DAVEY, M.G.; LUSCHER, E.F.; Actions of thrombin and other coagulant and proteolytic
enzymes on blood platelets. Nature. 216: 857-858; 1967.
3- DAVIE, E.W.; FUJIKAWA, K.; and KISIEL, W. The coagulation cascade: initiation,
maintenance, and regualtion. Biochemistry. 30: 10363-10370; 1991.
4- de FOUW, N.J.; van HINSBERGH, V.W.M.; de JONG, Y.F.; HAVERKATE, F.; BERTINA,
R.M. The interaction of activated protein C and thrombin with the plasminogen activator
inhibitor released from human endothelial cells. Thromb. Haemost. 57: 176-182; 1987.
5- DIVILANSKY, A. AND BIRAN, H. Hypofibrinogenemia after Echis colorata Bite in Man.
Acta Haematol. (Kbh) 49:123-130;1973.
6- FEINSTEIN, D.I. Diagnosis and management of disseminated intravascular coagulation: the
role of heparin therapy. Blood 60: 284-287; 1982.
7- FLEURY, V.; ANGLES-CANO, E. Characterization of the binding of plasminogen to fibrin
surfaces: the role of carboxi-terminal lysines. Biochemistry. 30: 7630-7638; 1991.
8- HALL, S.W.; NAGASHIMA, M.; ZHAO, L.; MORSER, J.; LEUNG, L.L.K. Thrombin
interacts with thrombomodulin, protein C, and thrombin-activatable fibrinolysis inhibitor
via specific and distinct domains. J. Biol. Chem. 274: 25510-25516; 1999.
9- HARMENING, D.M. - Clinical Hematology and Fundamentals of Hemostasis, 3nd
Ed. – F.A.
Davis Company - Philadelphia - New York-1998
10- HIRSH, J.; DALEN, J.E,; ANDERSON, D.R.; POLLER, L.; BUSSEY, H.; ANSELL, J.; ET
AL. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal
therapeutic range. Chest. 119: 8S-21S; 2001
11- KRISHNASWAMY, S.; FIELD, K.A.; EDGINGTON, T.S.; MORRISSEY, J.H.; MANN,
K.G. Role of the membrane surface in the activation of human coagulation factor X. J.
Biol. Chem. 267: 26110-26120; 1992.
12- LEVI, M.; TEN CATE, H. Disseminated intravascular coagulation. N. Engl. J. med. 341:
586-592; 1999.
13- McKENZIE, S.B.- Textbook of hematology, 2nd
Ed. –Willians & Wilkins – USA-1996
14- NEMERSON, Y. and REPKE, D. Tissue factor accelerates the activation of coagulation
factor VII: the role of a bifunctional coagulation cofator. Thromb. Res. 40: 351-358;
1985.

102
15- NORMAN, B.; WALLEN, P.; RAMBY, M. Fibrinolysis mediated by tissue plasminogen
activator disclosure of kinetic transition. Eur. J. Biochem. 149:193-200; 1985.
16- STIENE-MARTIN, E.A.; LOTSPEICH-STEININGER, C.A.; KOEPKE, J.A. - Clinical
Hematology: Principles, Procedures, Correlations, 2nd
Ed. – Lippicott-Raven Publishers –
Philadelphia - New York-1998.