genes supressores...
TRANSCRIPT

Genes supressores tumoraisGENES SUPRESSORES DE TUMOR (antioncogenes):
controlam (inibem) a divisão celular FUNÇÃO NORMAL
NA CÉLULA
GENES
SUPRESSORES
TUMORAIS
GENES
SUPRESSORES
TUMORAIS
Causas?
Hereditária
Vírus
Agentes químicos
Irradiações
Mutações pontuais
•proliferação anormal
das células
•formação do tumor

A maioria deles atua como MUTAÇÃO RECESSIVA de perda de função (deve haver
mutação nos 2 alelos)
•Apenas 1% é hereditário
•Gene supressor de tumor: 20 síndromes s
•Funções:
-controle da divisão celular, diferenciação, morte
celular programada e reparo do DNA


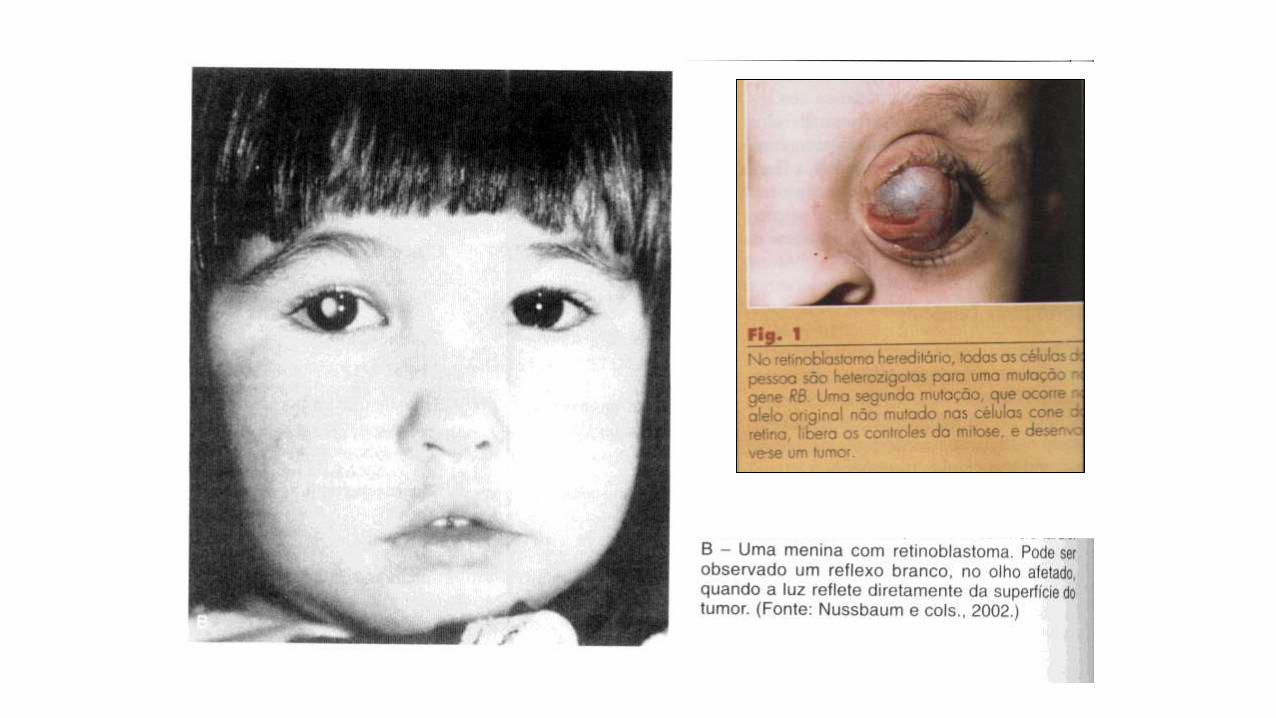
Ex. RETINOBLASTOMA
-Rb1: pode bloquear o ciclo
celular entre G1 e S,
impedindo a duplicação do
DNA
-tumor maligno raro da
retina
-maioria esporádica
-leva a morte se não tratado
-1/20.000 nascimentos
-manifestação no primeiro
ano de vida
RB1
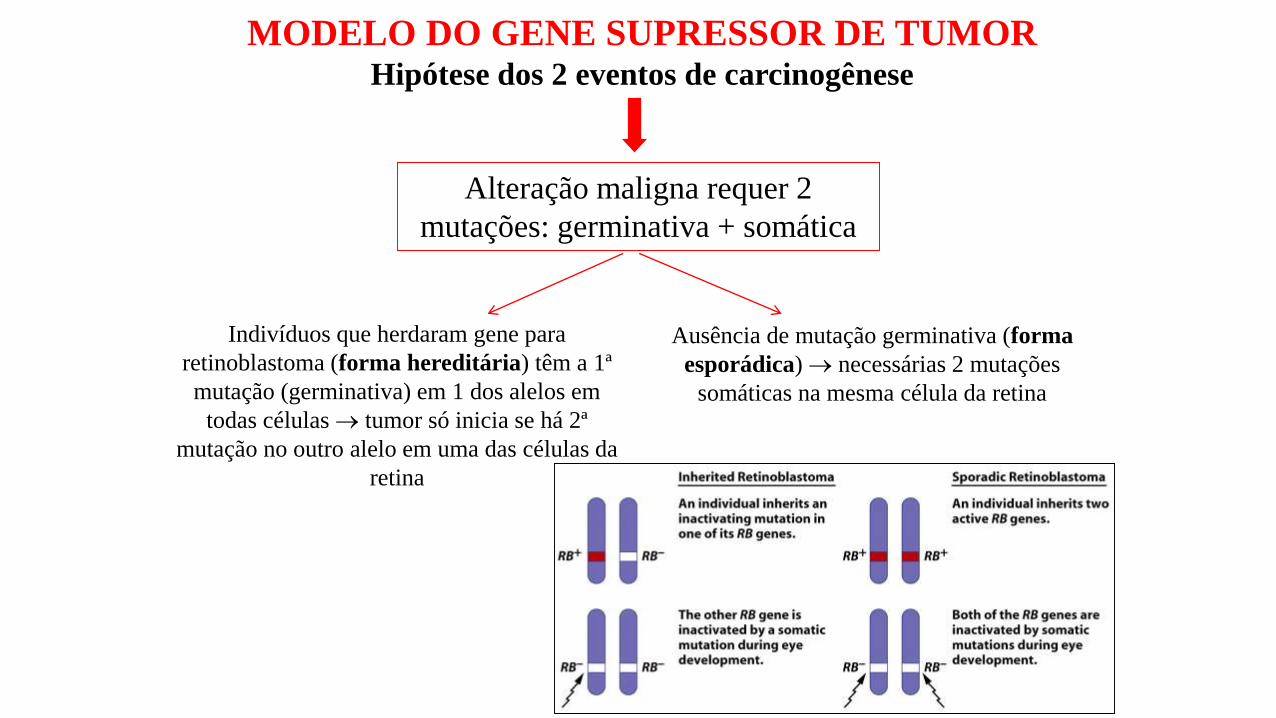
MODELO DO GENE SUPRESSOR DE TUMORHipótese dos 2 eventos de carcinogênese
Alteração maligna requer 2
mutações: germinativa + somática
Indivíduos que herdaram gene para
retinoblastoma (forma hereditária) têm a 1ª
mutação (germinativa) em 1 dos alelos em
todas células tumor só inicia se há 2ª
mutação no outro alelo em uma das células da
retina
Ausência de mutação germinativa (forma
esporádica) necessárias 2 mutações
somáticas na mesma célula da retina
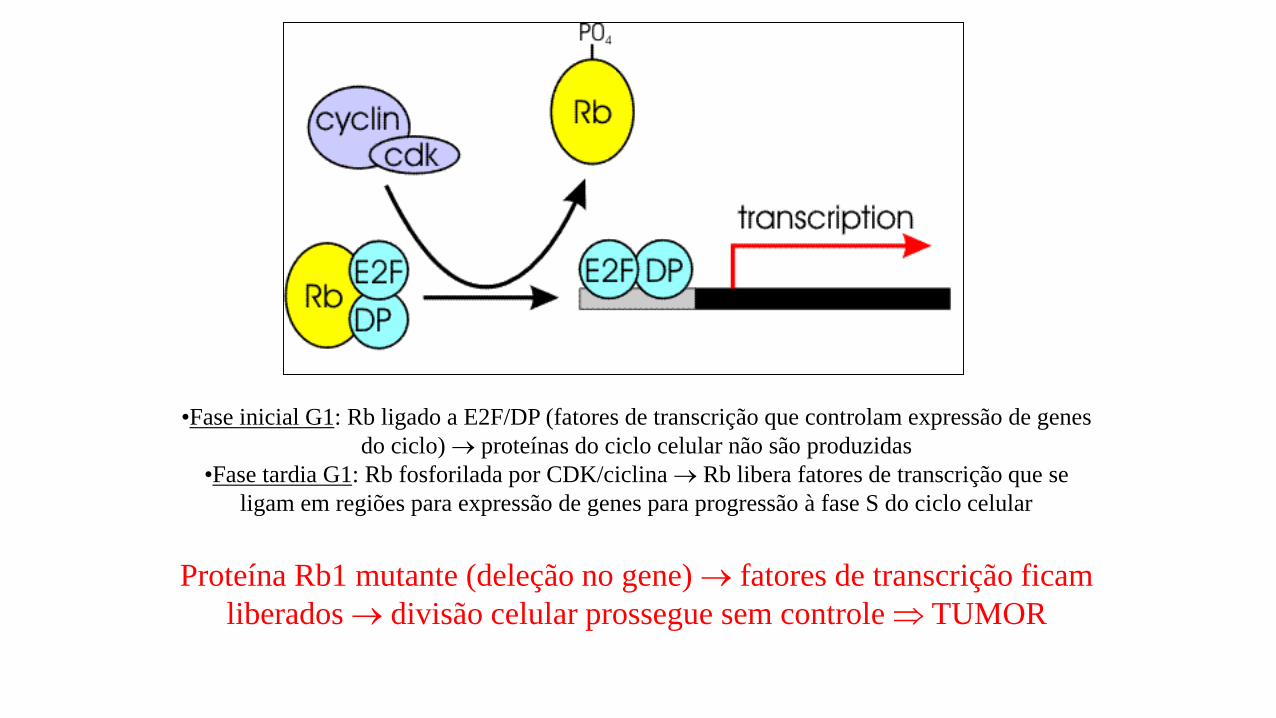
•Fase inicial G1: Rb ligado a E2F/DP (fatores de transcrição que controlam expressão de genes
do ciclo) proteínas do ciclo celular não são produzidas
•Fase tardia G1: Rb fosforilada por CDK/ciclina Rb libera fatores de transcrição que se
ligam em regiões para expressão de genes para progressão à fase S do ciclo celular
Proteína Rb1 mutante (deleção no gene) fatores de transcrição ficam
liberados divisão celular prossegue sem controle TUMOR

-40% forma hereditária
-mutação somática na célula da retina:
perda da função do alelo normal
TUMOR bilateral
-início precoce
-60% esporádicos (não
hereditário)
-2 alelos mutados RB1 em
uma célula da retina
TUMOR unilateral
-início tardio



P53:-fator de transcrição inativado nas formas esporádicas de muitos cânceres
-presente em 50% dos tipos de cânceres
•Dano ao DNA fosforilação de p53 forma estável e ativa estimula a
transcrição de genes que param ciclo celular permite reparo ao DNA mutado ou
ativa genes que causam apoptose à célula danificada
•Ausência de p53 em algumas células não há “freio” no ciclo celular
progressão celular mutações adicionais CÂNCER
Células com DNA
modificado se
dividem
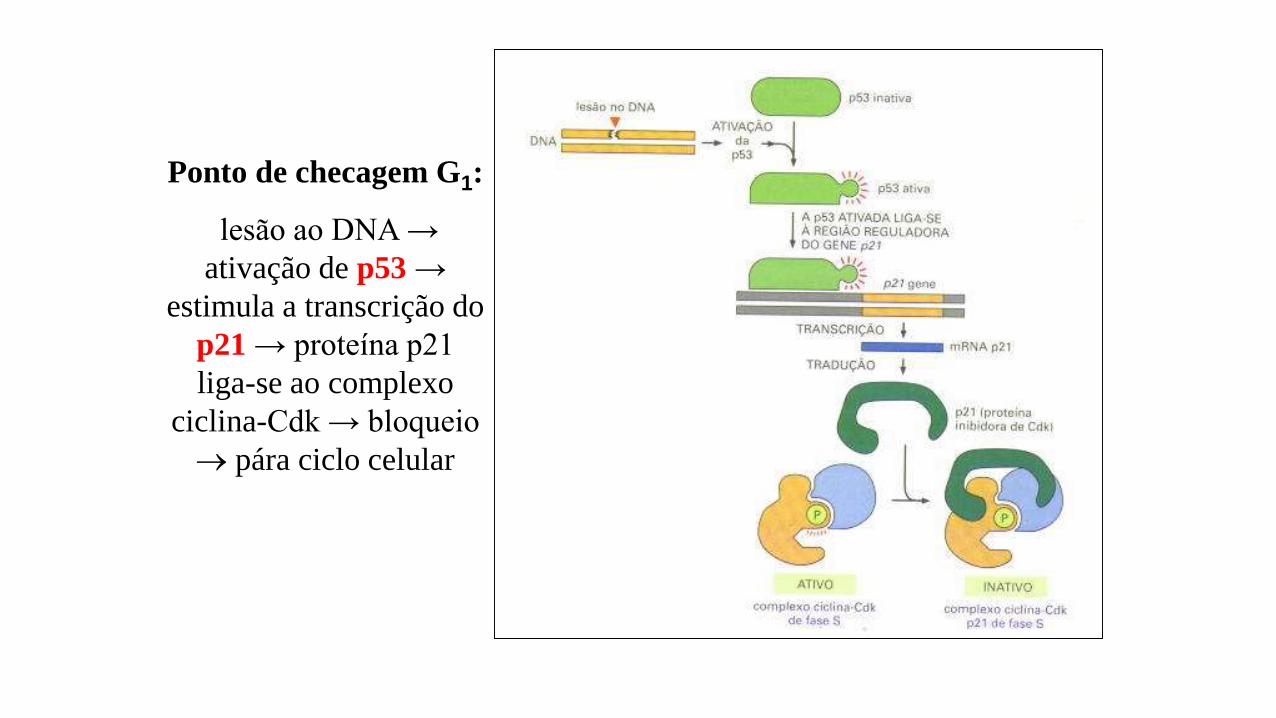
Ponto de checagem G1:
lesão ao DNA →
ativação de p53 →
estimula a transcrição do
p21 → proteína p21
liga-se ao complexo
ciclina-Cdk → bloqueio
pára ciclo celular

Síndrome de Li-Fraumeni•História familiar de formas ≠s de câncer
•Idade jovem
•Gene candidato p53 inativado em muitos destes cânceres
•síndrome autossômica dominante de câncer hereditário
•câncer ósseo, câncer cerebral, câncer de mama, leucemia e câncer de pulmão

Sequenciamento nucleotídico p53:• Mutação no éxon 5 – nucleotídeo 437• Triptofano TGG → códon parada TAG

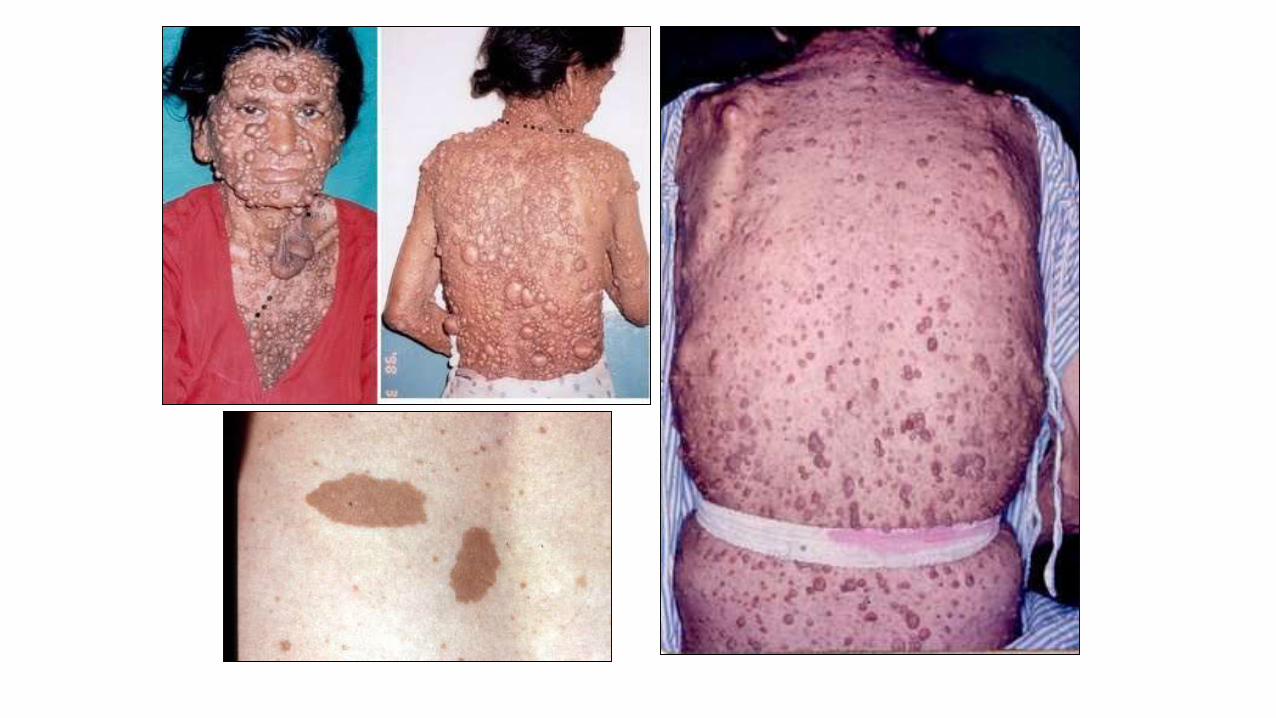
NF1
Neurofibromatose 1•Gene normal funciona na
regulação da divisão celular
•Falha na regulação do crescimento
das células normais origem dos
neurofibromas
•Anomalias neurológicas,
musculoesqueléticas,
oftalmológicas, dermatológicas

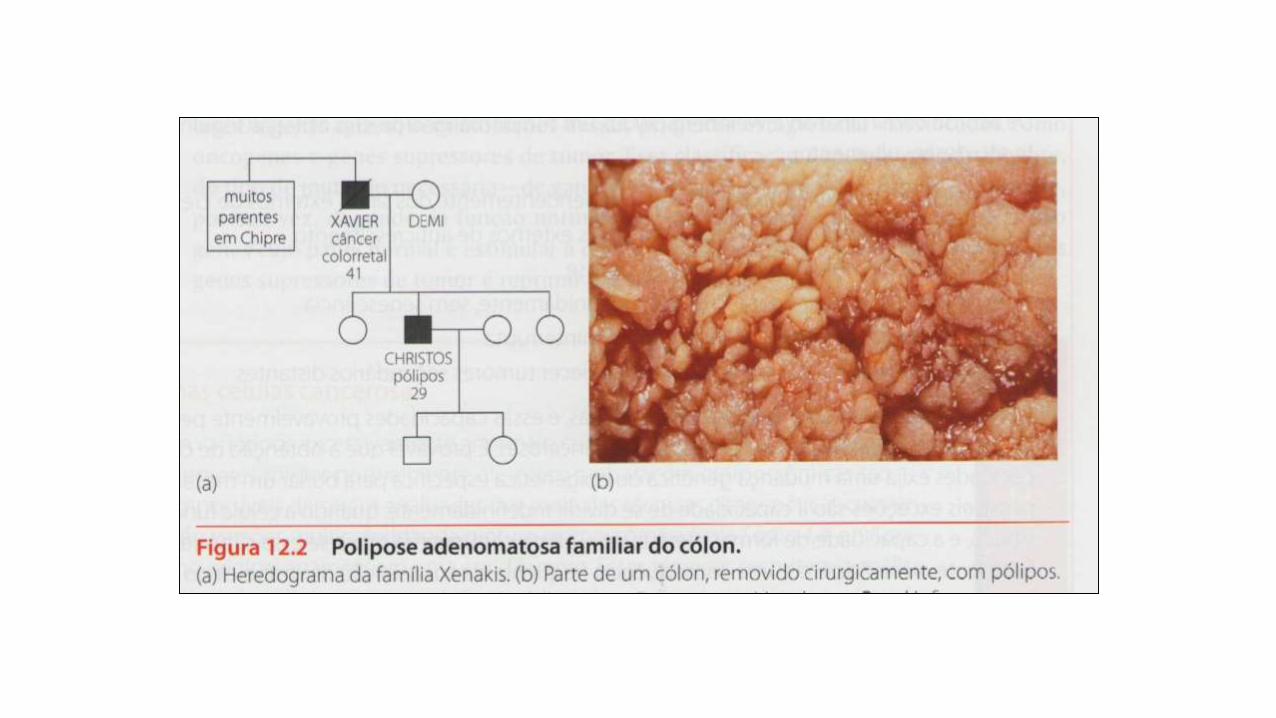
APC
Mucosa de cólon apresentando
vários pólipos
Polipose de cólon familiar
•Câncer colorretal
•Desenvolvimento de numerosos pólipos benignos,
que podem tornar-se malignos
•Remoção do cólon
•APC: controla a proliferação e diferenciação de
células epiteliais do intestino (mecanismo ainda pouco
conhecido)
•mutação em APC: célula sem “freio”, divisão celular
continua sem verificação
•Tumores benignos mutação em APC
•Tumores malignos APC + vários outros genes

BRCA1 e BRCA2
Câncer hereditário de mama e ovário
•70-80% de câncer de mama familiar 10-25 vezes mais chance de
desenvolver câncer de mama e ovário do que outras famílias
•BRCA1 – reparo ao DNA danificado por radiação
• abundantes na fase S do ciclo celular: reparo de quebras
• BRCA1 ou BRCA2 defeituosos – desenvolvimento de câncer de mama ou
ovário
•Pacientes com mutações nestes genes possuem instabilidade cromossômica e
mutações frequentes em outros genes supressores de tumor
•Tratamento: exames frequentes, mastectomia bilateral total

(+) : portadores
(-): não-portadores
Br: mama
Ov: ovário
Lu: pulmão
Pa: pâncreas
Pr: próstata
d: idade ao morrer
Bmx: mastectomia
Família segregando a mutação BRCA2: C3590G

•Risco aumentado em até 3 vezes se um parente de 1°
grau é afetado
•Risco aumentado de 10 vezes se mais de um parente
de 1° grau é afetado
•Riscos aumentados mais ainda se o diagnóstico de
câncer do parente de 1° grau afetado for antes dos 40
anos


MUTAÇÕES
►Mutações de ganho de função:
Proteína mutante adquire novas características ativação de ONCOGENES proteína codificada (oncoproteína)geralmente tem sua função aumentada.
► Mutações de perda de função:Perda de função de ambos os alelos de um gene (por
mutação, deleção) proteína resultante não é sintetizada, ou é
sintetizada em uma taxa pequena (GENE SUPRESSOR DE
TUMOR).

Efeito dominante
Efeito recessivo
Aa
aa

CONSIDERAÇÕES FINAISO câncer como uma doença genética
• Acúmulo de lesões não corrigidas de DNA
• Maior tempo de exposição a cancerígenos Maior
chance de mutação
• Redução da “vigilância” imunológica
Aumento da incidência de câncer
Envelhecimento

Com 50 anos 1 em 54
A chance de uma mulher ter câncer de mama
aumenta com a idade
Com 30 anos 1 em 2.212
Com 40 anos 1 em 235
Com 60 anos
Com 70 anos
Com 80 anos 1 em 10
1 em 23
1 em 14
CÂNCER DE MAMAEstatística