engenharia química - técnico lisboa · a acetalisação com acetona é um dos muitos processos...
TRANSCRIPT

Valorização da glicerina
Produção de Solketal
Mariana Simões Marçal
Dissertação para obtenção do grau de mestre em
Engenharia Química
Orientadora: Professora Doutora Ana Paula Vieira Soares Pereira Dias
Júri
Presidente: Professor Doutor Carlos Manuel Faria de Barros Henriques
Orientadora: Professora Doutora Ana Paula Vieira Soares Pereira Dias
Vogal: Professor Doutor Jaime Filipe Borges Puna
Dezembro 2015

ii

iii
RESUMO
O biodiesel produzido por esterificação de óleos vegetais é apontado como um combustível
renovável viável para a substituição do diesel de origem fóssil. A viabilidade económica da sua
produção exige que se valorize o subproduto maioritário (10% mássico), a glicerina.
A acetalisação com acetona é um dos muitos processos referidos na literatura para o
aproveitamento da glicerina proveniente da produção de biodiesel. O produto resultante, solketal, tem
inúmeras aplicações, tais como cosmética, indústria farmacêutica, como aditivo ao diesel de forma a
reduzir o ponto de congelamento, melhorar as propriedades lubrificantes, aumentar o poder
antidetonante (octanagem) e como aditivo ao jet fuel, o que reduz a inflamabilidade do combustível.
A reacção entre a glicerina e a acetona exige a presença de um catalisador.
Estudou-se a acetalisação da glicerina com acetona na presença de catalisadores ácidos. A
reacção foi conduzida à pressão atmosférica e à temperatura de ebulição da acetona. Testaram-se os
catalisadores preparados por desaluminação ácida da montmorilonite KSF e à base de SnO2 e
SnO2/SO42-
depositados em sílica. A amberlyst 15 foi usada como catalisador ácido de referência.
A acidez dos catalisadores preparados foi avaliada por desidratação do metanol a dimetil éter
em fase gasosa.
Nas condições testadas, os resultados mostram que a acidez superficial é uma condição
necessária para o bom desempenho catalítico. Para catalisadores muito ácidos (SnO2/SO42-
) ocorre a
hidrólise do solketal formado.
Palavras-chave: solketal, glicerol, acetalisação, montmorilonite, sílica, SnO2

iv
ABSTRACT
Biodiesel produced by esterification of vegetable oils is pointed as a viable renewable fuel to
replace petroleum diesel. The economic viability of biodiesel production requires the valorization of the
major byproduct (10% w/w) glycerine.
The acetalization with acetone is one of the many processes refered in literature for the
glycerine valorization from biodiesel production. The resulting product, solketal, is a versatil chemical
havingseveral applications, costmetic, pharmaceutical industry, as diesel additive to reduce the
freezing point, inhance the lubricant properties, increase the antiknock power and as a flammability
reducing agent to jet fuel.
The reaction between glycerine and acetone requires the presence of a catalytst.
The glycerin acetalization with acetone was carried out over acid catalysts at the acetone
reflux temperature and normal pressure. The tested catalysts were prepared by acid dealumination of
montmorillonite KSF and silica supported tin catalysts modified with SO42-
species. The commercial
amberlyst 15 (wet) was used as standard acid catalyst.
The surface acidity of the synthetized catalysts was evaluated by methanol to dimethylether
dehydration in vapor phase.
In the tested conditions the surface acidity of the prepared catalysts played an important role
on the studied catalytic process. Nevertheless the catalyst with the strongest
acidity (SnO2/SO42) promoted undesired the hydrolysis of the formed solketal.
Keywords: solketal, glycerol, acetalisation, montmorilonite, silica, SnO2

v
AGRADECIMENTOS
Em primeiro lugar, quero agradecer à orientadora da tese de mestrado, Ana Paula Dias, por todo o
apoio, esforço paciência e disponibilidade. Gostei muito de trabalhar com uma pessoa de enorme
simpatia, que sempre nos tratou como mais do que simples alunos e orientandos.
As análises termogravimétricas foram realizadas na termobalança do professor Francisco Pereira.
Quero mostrar a minha gratidão pela cedência e por toda a sua atenção.
Agradeço também à professora Joana Neiva Correia pela disponibilidade na utilização do
infravermelho.
À engenheira Maria Isabel Leiria, um muito obrigada pela paciência e ajuda nas manhãs e tardes
passadas no laboratório
A todos os meus amigos que sempre me apoiaram e aturaram ao longo da vida e do curso, André
Palma, Sara Bernardo, Ângelo Sousa, Joana Leitão, Pedro Silva, Rita Costa, Sofia Pinto, Miguel
Dionísio, Joana Bouçadas, Leonor Rocha e Filipa Fernandes, muito obrigada pela amizade.
Um especial obrigada aos meus amigos e colegas do laboratório de catálise heterogénea, João
Rafael, André Aguilar, Albertina Soares e Inês Lino. Sem a vossa companhia teria sido muito mais
difícil concluir a tese.
Finalmente, quero agradecer à minha família, pai, mãe, irmão e avó.

vi
Índice
1. INTRODUÇÃO .................................................................................................................................... 1
2. ESTADO DA ARTE ............................................................................................................................. 2
2.1 COMBUSTIVEIS FÓSSEIS E RENOVÁVEIS ................................................................................... 2
2.1.1 Fontes não renováveis ............................................................................................................... 2
2.1.1.1.Carvão ................................................................................................................................. 2
2.1.1.2 Petróleo ............................................................................................................................... 3
2.1.1.3 Gás natural .......................................................................................................................... 4
2.1.2 Fontes Renováveis ..................................................................................................................... 5
2.1.2.1 Hídrica ................................................................................................................................. 5
2.1.2.2 Eólica ................................................................................................................................... 6
2.1.2.3 Solar .................................................................................................................................... 7
2.1.2.4 Geotérmica .......................................................................................................................... 8
2.1.2.5 Ondas .................................................................................................................................. 8
2.1.2.6 Biomassa ............................................................................................................................. 9
2.1.3 Enquadramento Global ............................................................................................................ 10
2.1.4 Mercado energético em Portugal ............................................................................................. 12
2.2 BIOMASSA E BIODIESEL .............................................................................................................. 14
2.2.1 Produção de biodiesel .............................................................................................................. 14
2.1.1.1 Matéria-Prima para a produção de biodiesel .................................................................... 15
2.2.2. Breve história do biodiesel ...................................................................................................... 16
2.2.3 Vantagens e desvantagens ...................................................................................................... 16
2.2.4 Mercado .................................................................................................................................... 17
2.2.5 Perspectiva futura ..................................................................................................................... 19
2.3 GLICEROL ...................................................................................................................................... 20
2.3.1 Breve História ........................................................................................................................... 21
2.3.2 Propriedades ............................................................................................................................ 21
2.3.3. Produção (natural e sintética) ................................................................................................. 22
2.4 REACÇÕES DO GLICEROL PARA APLICAÇÃO INDUSTRIAL.................................................... 23
2.4.1 Hidrogenólise ............................................................................................................................ 23
2.4.2 Eterificação ............................................................................................................................... 24
2.4.3 Esterificação ............................................................................................................................. 24

vii
2.4.4 Oxidação .................................................................................................................................. 25
2.4.5 Desidratação ............................................................................................................................ 26
2.4.6 Carbonato de glicerol ............................................................................................................... 26
2.4.7 Clorinação ................................................................................................................................ 27
2.4.8 Acetalisação ............................................................................................................................. 27
2.5 CATALISADORES REFERIDOS NA LITERATURA ....................................................................... 28
2.5.1 Oxihidróxido de Nióbio ............................................................................................................. 29
2.5.2 Purolite PD206 ......................................................................................................................... 30
2.5.3 Heteropoliácidos imobilizados em sílica................................................................................... 30
2.5.4 Zeólito-Beta .............................................................................................................................. 31
2.5.5 Compósitos Carbono-Sílica ...................................................................................................... 31
2.5.6 Ácido p-toluenosulfónico .......................................................................................................... 32
2.5.7 Amberlyst-15 ............................................................................................................................ 32
2.5.8 Nanopartículas de Níquel em nanotubos de carbono .............................................................. 33
2.5.9 Óxidos de TiO2-SiO2 ................................................................................................................. 33
2.5.10 Silicatos com metais incorporados ......................................................................................... 34
2.5.11 SnO2 preparado com molibdénio e tungsténio....................................................................... 34
2.5.12 Espuma celular mesoporosa modificada ............................................................................... 34
2.5.13 Níquel-zircónio com suporte de carbono activado ................................................................. 35
2.5.14 Óxido de ferro com suporte de aluminosilicato ...................................................................... 36
2.5.15 Membrana de zeólito .............................................................................................................. 36
2.5.16 Sílica modificada com ácido sulfónico.................................................................................... 36
2.5.17 Zircónia e catalisador com zircónia como precursor .............................................................. 37
2.5.18 ZrO2-SiO2 ................................................................................................................................ 37
2.5.19 Fosfato de molibdénio com suporte SBA-15 .......................................................................... 38
2.5.20 Zn, Cu, Ni e Co com suporte de M-AlPO4 .............................................................................. 38
2.5.21 Tabela resumo ........................................................................................................................ 38
3.MÉTODOS E MATERIAIS.................................................................................................................. 41
3.1 PREPARAÇÃO DE CATALISADORES .......................................................................................... 41
3.1.1 Montmorilonite com tratamento ácido ...................................................................................... 41
3.1.2 Deposição de SnO2 em sílica ................................................................................................... 42

viii
3.1.3 Deposição de SnO2/SO42-
, em sílica ........................................................................................ 43
3.2 CARACTERIZAÇÃO DE CATALISADORES .................................................................................. 43
3.2.1 Infravermelho com transformadas de Fourier (FTIR) ............................................................... 43
3.2.2 Desidratação do metanol a dimetil éter (DME) ........................................................................ 44
3.3 COMPORTAMENTO CATALÍTICO ................................................................................................ 45
3.3.1 Descrição da instalação ........................................................................................................... 45
3.3.2 Técnica Operatória ................................................................................................................... 46
3.3.3 Análise do efluente do reactor .................................................................................................. 47
4.RESULTADOS EXPERIMENTAIS DA CARACTERIZAÇÃO DE CATALISADORES ....................... 48
4.1 Infravermelho com transformada de Fourier (FTIR) ................................................................... 48
4.2 Desidratação do metanol a dimetil éter (DME) ........................................................................... 55
5. COMPORTAMENTO CATALÍTICO .................................................................................................. 57
5.1 Espectroscopia de Infravermelho com transformada de Fourier ................................................ 57
5.1.1 Glicerina................................................................................................................................ 57
5.1.2 Acetona................................................................................................................................. 58
5.1.3 Solketal ................................................................................................................................. 58
5.2 Termogravimetria ........................................................................................................................ 62
6. CONCLUSÕES ................................................................................................................................. 68
7. BIBLIOGRAFIA .................................................................................................................................. 69
8. ANEXOS ............................................................................................................................................ 75
8.1 Termogravimetria ........................................................................................................................ 75
8.2 Transformada de Fourier Infravermelho...................................................................................... 79
8.2.1 Catalisadoresapósreacção ................................................................................................... 79
8.2.2 Amostras............................................................................................................................... 89

ix
ÍNDICE DE FIGURAS
Figura 1 - Diferentes componentes obtidos através do petróleo.............................................................3
Figura 2 - Parque eólico, Sines, Portugal................................................................................................7
Figura 3 - Diferentes matérias-primas, biomassa....................................................................................9
Figura 4 - Biomassa e sua utilização….................................................................................................10
Figura 5 - Produção de energia primária por tipo de energia (Mtoe)….................................................11
Figura 6 - Emissões de CO2 (Mt) por tipo de fonte de energia….........................................................12
Figura 7 - Evolução do balanço energético….......................................................................................12
Figura 8 - Evolução da dependência energética…...............................................................................12
Figura 9 - Consumo de energia primária por fonte energética ….........................................................12
Figura 10 - Processo de produção de biodiesel por transesterificação por via alcalina........................15
Figura 11 - Percentagem de redução de emissões de poluentes em B20 e B100…............................17
Figura 12 - Produção mundial de biodiesel….......................................................................................18
Figura 13 - Produção de biodiesel na Europa…...................................................................................18
Figura 14 - Produção de biodiesel em Portugal …...............................................................................19
Figura 15 - Perspectiva de produção e comércio de biodiesel mundial até 2020….............................20
Figura 16 - Mercado do glicerol (em percentagem), 2002….................................................................20
Figura 17 - Transesterificação de um triglicérido com metanol….........................................................22
Figura 18 - Reacção de produção de 1,2-propanodiol…......................................................................24
Figura 19 - Eterificação do glicerol com isobuteno…….........................................................................24
Figura 20 - Reacção de glicerol com ácido acético…...........................................................................25
Figura 21 - Produtos da oxidação do glicerol …...................................................................................26
Figura 22 - Produção de acroleína através da desidratação de glicerol …….......................................26
Figura 23 - Síntese do carbonato de glicerol com dimetilcarbonato…..................................................27
Figura 24 - Clorinação de glicerol……...................................................................................................27
Figura 25 - Reacção de acetalisação de glicerol com aldeídos…….....................................................27
Figura 26 - Mecanismo proposto para a reacção do glicerol com acetona envolvendo catalisador
ácido……...............................................................................................................................................28
Figura 27 - Esquema de tratamento ácido da Montmorilonite…….......................................................41
Figura 28 - Esquema das reacções de oxidação e desidratação do metanol ……...............................44

x
Figura 29 - Representação esquemática dos produtos da reacção de oxidação do metanol como
função do carácter ácido-básico dos centros activos catalíticos……....................................................44
Figura 30 - Fotografia da instalação montada.......................................................................................45
Figura 31 - Gráfico obtido na termogravimetria, para uma reacção de acetalisação............................47
Figura 32 – Espectro FTIR de catalisador frescoamberlyst..................................................................48
Figura 33 - Espectros de FTIR dos catalisadores frescos de KSF e KSF com tratamento ácido……..49
Figura 34 - Espectros de FTIR dos catalisadores de KSF e KSF com tratamento ácido…………...... 50
Figura 35 - Espectros de FTIR para catalisadores frescos de SnO2/SiO2............................................51
Figura 36 - Espectro de FTIR de catalisadores de amberlyst, após reacção....................................... 51
Figura 37 - Espectro de FTIR de montmorilonite, após reacção……………….................................... 52
Figura 38 - Espectro de FTIR para KSF com tratamento ácido de H2SO4, após reacção....................53
Figura 39 – Espectro de FTUR para KSF com tratamento ácido de HCl 1,52%, após reacção...........53
Figura 40 - Espectro de FTIR para KSF com tratamento ácido de HCl 4,3%, após reacção...............54
Figura 41 - Espectro de FTIR para KSF com tratamento ácido de HCl 9% 200ºC 1h, após reacção...54
Figura 42 - Espectro de FTIR para SnO2/SiO2 e seu tratamento ácido, após reacção.........................55
Figura 43 - Rendimento de reacção de oxidação de metanol a DME, a 200 e 215 ºC.........................56
Figura 44 - Espectro de FTIR de glicerina.............................................................................................57
Figura 45 - Espectro de FTIR de acetona.............................................................................................58
Figura 46 - Espectro de FTIR de solketal..............................................................................................58
Figura 47 - Espectro de FTIR de amostra finall de solketal 7 e 9.........................................................59
Figura 48- Espectro de FTIR de amostra final de solketal 7 e 9, entre 950 e 1150 cm-1
......................60
Figura 49 - Espectro de FTIRde amostra final de solketal 7 e 9, entre 1050 e 1190 cm-1
....................60
Figura 50 - Espectro FTIRde amostra final de solketal 7 e 9, entre 1100 e 1330 cm-1
.........................61
Figura 51 - Espectro FTIR de amostra final de solketal 7 e 9, entre 3000 e 3800 cm-1
........................61
Figura 52 -Termograma para mistura reaccional com conversão nula.................................................62
Figura 53 - Termograma para solketal2................................................................................................63
Figura 54 - Termograma para solketal4................................................................................................63
Figura 55 - Termograma para solketal8................................................................................................63
Figura 56 - Termograma para solketal7................................................................................................64
Figura 57 - Termograma para solketal9................................................................................................64
Figura 58 - Termograma para solketal20..............................................................................................65

xi
Figura 59 - Termograma para solketal14..............................................................................................65
Figura 60 - Termograma para solketal19..............................................................................................66
Figura 61 - Termograma para solketal21..............................................................................................66
Figura 62 – Termograma para solketal3...............................................................................................75
Figura 63 – Termograma para solketal5...............................................................................................75
Figura 64 – Termograma para solektal6...............................................................................................76
Figura 65 – Termograma para solketal10.............................................................................................76
Figura 66 – Termograma para solketal12.............................................................................................76
Figura 67 – Termograma para solketal13.............................................................................................77
Figura 68 – Termograma para solketal 15............................................................................................77
Figura 69 – Termograma para solketal16.............................................................................................77
Figura 70 – Termograma para solketal17.............................................................................................78
Figura 71 – Termograma para solketal18.............................................................................................78
Figura 72 – Termograma para solketal19.............................................................................................78
Figuras 73 e 74 - - Espectro de FTIR para KSF com tratamento ácido de HNO3 15% 200ºC 2h, após
reacção..................................................................................................................................................79
Figuras 75 e 76 - Espectro de FTIR para KSF com tratamento ácido de H3PO4 8% 200ºC 2h, após
reacção..................................................................................................................................................80
Figuras 77 e 78 - - Espectro de FTIR para KSF com tratamento ácido de HCl 3,8% 26ºC 1h, após
reacção..................................................................................................................................................81
Figuras 79 e 90 - Espectro de FTIR para KSF com tratamento ácido de HCl 9% 27ºC 1h, após
reacção..................................................................................................................................................82
Figuras 91 e 92 - Espectro de FTIR para KSF com tratamento ácido de HCl9% 200ºC 2h, após
reacção..................................................................................................................................................83
Figuras 93 e 94 – Espectro de FTIR para KSF com tratamento ácido de H2SO4, após reacção........84
Figuras 95 e 96 - Espectro de FTIR para KSF com tratamento ácido de HCl 1,52% 26ºC 1h, após
reacção..................................................................................................................................................85
Figuras 97 e 98 - Espectro de FTIR para KSF com tratamento ácido de HCl4,3% 26ºC 1h, após
reacção..................................................................................................................................................86
Figuras 99 e 100 – Espectro de FTIR para KSF com tratamento ácido de HCl9% 200ºC 1h, após
reacção..................................................................................................................................................87
Figuras 101 e 102 - Espectro de FTIR para SnO2/SiO2 e SnO2-SO42-/SiO2, após reacção……......88
Figura 103 – Espectros de FTIR do efluente reaccional das reacções com montmorilonite KSF, entre
700 e 1800 cm-1...................................................................................................................................89

xii
Figura 104 – Espectros de FTIR do efluente reacional das reacções com amberlyst, entre 700 e 1800
cm-1.......................................................................................................................................................89
Figura 105 – Espectros de FTIR do efluente reaccional das reacções de montmorilonite com
tratamento ácido de HCl 9%, entre 700 e 1800 cm-1...........................................................................90
Figura 106 – Espectros de FTIR do efluente reaccional das reacções de montmorilonite com
tratamento ácido de HCl 1,52%, 3,8% e 4,3%, entre 700 e 1800 cm-1................................................90
Figura 107 – Espectros de FTIR do efluente reaccional das reacções de montmorilonite com
tratamento ácido de HNO3, H3PO4 e H2SO4 entre 700 e 1800 cm-1.................................................91

xiii
ÍNDICE DE ACRÓNIMOS
OPEP - Organização dos Países Exportadores de Petróleo
GEE - Gases de efeito de estufa
B100 - Biodiesel puro
B20 - Mistura com 20% de biodiesel e 80% de diesel de petróleo
P - Pressão
T - Temperatura
FTIR - Fourier TransformInfraredSpectroscopy
DME - Dimetil éter

xiv
ÍNDICE DE TABELAS
Tabela 1 - Vantagens e desvantagens do uso de carvão……................................................................3
Tabela 2 - Vantangens e desvantagens do uso de petróleo…...............................................................4
Tabela 3 - Vantagens e desvantagens do uso de gás natural….............................................................4
Tabela 4 - Vantagens e desvantagens de energia hídrica………...........................................................6
Tabela 5 - Vantagens e desvantagens do uso de energia eólica…........................................................7
Tabela 6 - Vantagens e desvantagens do uso de energia solar……......................................................8
Tabela 7 - Vantagens e desvantagens da energia geotérmica…...........................................................8
Tabela 8 - Vantagens e desvantagens do uso de energia das onda......................................................9
Tabela 9 - Vantagens e desvantagens do uso de biomassa …............................................................10
Tabela 10 - Percentagens de produção de energia primária por tipo de energia…..............................11
Tabela 11 - Percentagem de consumo de energia primária por fonte de energia…….........................13
Tabela 12 - Vantagens e desvantagens do biodiesel............................................................................16
Tabela 13 - Temperaturas de ebulição do glicerol a diferentes pressões.............................................21
Tabela 14 - Tabela resumo dos catalisadores referidos na literatura...................................................39
Tabela 15 - Valores para a preparação de catalisadores com Montmorilonite.....................................42
Tabela 16 - Condições experimentais para as diferentes reacções......................................................46
Tabela 17 - Propriedades físicas de reagentes e produtos de reacção................................................48
Tabela 18 - Bandas e ligações correspondentes do espectro de glicerina...........................................57
Tabela 19 - Bandas e ligações correspondentes do espectro de infravermelho de acetona................58
Tabela 20 - Bandas e ligações correspondentes do espectro de infravermelho de solketal................59
Tabela 21 - Reacções que apresentaram conversão, por termogravimetria........................................62
Tabela 22 - Valores de conversão para cada reacção, por termogravimetria.......................................67

1
1. INTRODUÇÃO
Com a crescente preocupação em relação ao ambiente, relacionada principalmente com as
alterações climáticas e as emissões dos gases de efeito de estufa, têm sido realizados cada vez mais
esforços no sentido de atenuar a situação. Esta atenuação passa, maioritariamente, por substituir a
utilização de combustíveis fósseis por combustíveis renováveis, incluindo a biomassa. O biodiesel,
proveniente da biomassa, pode ser utilizado como alternativa ao diesel, proveniente do petróleo. O
aumento da produção de biodiesel resulta também numa produção de glicerina, o que faz com que
haja maior produção de glicerina do que aquela que o mercado consegue absorver.
Sendo assim, existe a necessidade de valorizar este produto,de forma a que a produção de
biodiesel seja lucrativa. Nesse sentido, foram desenvolvidos vários processos tais como esterificação,
eterificação, hidrogenólise, oxidação, desidratação e acetalisação da glicerina.
No presente trabalho, tem-se como objectivo estudar novos catalisadores para a produção de
solketal através da acetalisação da glicerina. Este produto pode ser usado por exemplo, como aditivo
em diesel de forma a reduzir o ponto de congelamento, melhorar as propriedades lubrificantes,
aumentar o poder antidetonantee reduzir a viscosidade. É também usado em jet fuel, de forma a
alterar a sua estrutura química e composição, o que reduz a inflamabilidade do combustível ao
aumentar o tamanho das gotículas [1] [2] [3] [4].

2
2. ESTADO DA ARTE
2.1 COMBUSTIVEIS FÓSSEIS E RENOVÁVEIS
2.1.1 Fontes não renováveis
As fontes não renováveis são fontes provenientes de recursos naturais mas que se
encontram em quantidades finitas, isto é, não se renovam [5].
A revolução industrial, no século XVIII, e a "grande aceleração" após a segunda-guerra
levaram a melhorias no desenvolvimento social e económico, mas criaram também muitos problemas
a nível ambiental principalmente para o futuro [6].
As fontes de energia não renovável provêm de:
Carvão;
Petróleo;
Gás Natural.
2.1.1.1.Carvão
O carvão foi o principal abastecedor de energia do processo de industrialização e, sendo
assim, no século 19 passou-se de uma produção de 10 para 1000 ton por ano [7].Este é o recurso
fóssil mais abundante no mundo, com reservas que se estimam poder durar 300-500 anos, se a sua
taxa actual de consumo se mantiver [8].
O carvão pode ter várias composições, dependendo da sua fonte, com 25 a 97% de carbono,
2 a 6% de hidrogénio, 2 a 20% de oxigénio, pequenas quantidades de azoto, enxofre e distintos
minerais [9].
As duas maiores preocupações ambientais relacionadas com o carvão são a libertação de
grandes quantidades de CO2 e SO2 (chuva ácida) para a atmosfera, provenientes da sua queima e
exploração [10].
Na tabela 1 apresentam-se algumas vantagens e desvantagens do uso de carvão:

3
Tabela 1 - Vantagens e desvantagens do uso de carvão [10]
Vantagens Desvantagens
Grandes reservas Impacto ambiental muito elevado
Elevado rendimento da rede energética Ameaça à saúde humana, quando é
queimado
Tecnologia bem desenvolvida Poluição da água
Tecnologias avançadas para redução da
poluição do ar
Emissões de partículas radioactivas e tóxicas
para a atmosfera
Baixo custo Elevadas emissões de CO2 quando produzido
e queimado
2.1.1.2 Petróleo
A exploração intensiva de petróleo foi iniciada em 1853 com a descoberta do processo de
destilação em querosene através do petróleo (Ignacy Lukasiewicz). Na segunda metade do século
XIX fizeram-se algumas descobertas que potenciaram o uso de petróleo e o tornaram na primeira
fonte de energia: método de extracção de petróleo do subsolo (Edwin Drake) e motor de combustão
interna (Nikolaus Otto) [7].
O petróleo pode ser utilizado como matéria-prima para a produção de vários químicos e
materiais sintéticos como o plástico, mas é para a produção de combustíveis que é mais usado [8].
Os componentes são removidos numa coluna de destilação tendo em conta os seus pontos
de ebulição. Os componentes mais voláteis, com menor número de carbonos e com ponto de
ebulição mais baixo são removidos no topo da coluna (figura 1) [10].
Figura 1 - Diferentes componentes obtidos através do petróleo

4
O uso de gasolina e diesel no sector dos transportes corresponde a 43% das emissões
globais de CO2, actualmente [10].
Na tabela 2 estão algumas das vantagens e desvantagens da utilização de petróleo.
Tabela 2 - Vantangens e desvantagens do uso de petróleo [10]
Vantagens Desvantagens
Elevado rendimento energético É necessário encontrar substitutos nos
próximos 50 anos
Fácil transporte entre países Poluição do ar
Baixo uso do território de exploração Os preços não encorajam a procura de
alternativas
Tecnologia bem desenvolvida Pode causar poluição da água
Sistema de distribuição eficiente A sua queima liberta CO2
2.1.1.3 Gás natural
O gás natural começou a ser usado em 1950 e é uma mistura de 50 a 90% de metano.
Contém também pequenas quantidades dos gases pesados de hidrocarbonetos, propano e butano.
Na sua exploração, o propano e o butano são liquefeitos e removidos como GPL (Gás de petróleo
liquefeito). O resto do gás (maioritariamente metano) é purificado e de seguida bombeado para
gasodutos de distribuição pressurizados [10].
As perspectivas das suas reservas é que só acabem num espaço de 62-125 anos, mantendo
a taxa de consumo actual, o que é ligeiramente melhor do que o tempo de reservas para o petróleo
[10].
Tabela 3 - Vantagens e desvantagens do uso de gás natural [10]
Vantagens Desvantagens
Baixo custo Liberta CO2 quando queimado
Elevado rendimento da rede energética Altamente explosivo como LNG (gás natural liquefeito)
Menos poluição atmosférica que os
outros combustíveis fósseis
É necessário um sistema de gasodutos.
Menor emissão de CO2 do que outros
combustíveis fósseis
Pode ocorrer fuga de metano dos gasodutos e é um dos
gases causadores do efeito de estufa
Facilmente transportável por pipeline
Baixo uso do solo

5
2.1.2 Fontes Renováveis
As fontes renováveis são fontes de energia que também provém de recursos naturais, no
entanto são inesgotáveis. A origem das fontes de energia é:
Hídrica
Eólica
Solar
Geotérmica
Ondas
Biomassa
Dada a preocupação ambiental com as alterações climatéricas devido à libertação de gases
poluentes para a atmosfera tais como grandes quantidades de vapor de água, dióxido de carbono
(responsável pelo efeito de estufa), óxidos de azoto (NOx), óxidos de enxofre (SO2) e hidrocarbonetos
(HC) (responsáveis não só pela poluição do ar, mas também pela formação de chuvas ácidas, smog
fotoquímico e concentrações elevadas de ozono troposférico) [10] e ainda a limitação das reservas
dos combustíveis fósseis, existe um grande impulso para que ocorra uma transição de energias não-
renováveis para energias renováveis.
Com as energias renováveis é possível obter uma série de benefícios, incluindo [11]:
Redução de impactos ambientais, incluindo emissões de gases responsáveis pelo efeito de
estufa;
Segurança energética;
Desenvolvimento económico estratégico, incluindo desenvolvimento rural;
Acesso a recursos energéticos off-grid.
2.1.2.1 Hídrica
É obtida a partir do curso de água e pode ser aproveitada por um desnível ou queda de água,
através da energia cinética de uma massa de água [10].

6
Tabela 4 - Vantagens e desvantagens de energia hídrica[10] [12].
Vantagens Desvantagens
Elevada eficiência (80%) Elevados custos e tempo para a construção
Electricidade de baixo custo Elevado impacto ambiental relacionado com a
área necessária para criar o reservatório
Elevado tempo de vida Elevadas emissões de CH4 devido ao
decaimento de biomassa
Não existe emissão de CO2 em locais de
clima temperado
Diminuição do transporte de sedimentos
naturais a jusante das comportas
Pode ser um meio de controlar as cheias a
jusante das comportas
Provoca a erosão dos solos
Fonte de água para agricultura durante todo o
ano
Um período de seca pode afectar a produção
de energia
Esta fonte de energia não é, de todo, a solução para substituir os combustíveis fósseis visto que, tem
uma capacidade limitada de expansão comparada com outras energias renováveis, devido à grande
área que é precisa para construir barragens e ainda porque as estimativas da capacidade global
hidroeléctrica apontam para valores de energia da ordem de 2,5x1019
J, valor este que é três vezes
menor queo valor de produção de energia eléctrica prevista para 2020 [7].
2.1.2.2 Eólica
Energia que provém do movimento do vento. A sua produção é feita através de turbinas que
usam o movimento do vento para gerar energia eléctrica (figura 2) [10].

7
Figura2 - Parque eólico, Sines, Portugal
É vista como uma energia promissora que cresceu rapidamente em muitos países
desenvolvidos. De notar também que o seu limite de produção é de 7,2x1019
J, o que equivale ao
total de produção de electricidade projectada para 2020 [7].
Apesar da elevada tecnologia já existente nos parques eólicos há ainda possibilidade de
desenvolvimento no que diz respeito à criação de parques offshore (fora da costa), onde existem
menos impactos ambientais nomeadamente relacionados com rotas migratórias e radares e onde a
velocidade do vento é superior [10].
Tabela 5 - Vantagens e desvantagens do uso de energia eólica [10]
Vantagens Desvantagens
Elevada eficiência São necessários ventos constantes
Custos moderados Sistema de backup necessário quando o
vento é fraco
Impactos ambientais muito baixos Grandes áreas necessárias
Não existe emissão de CO2 Poluição visual e sonora
Fácil expansão e construção Pode interferir nos voos migratórios das aves
Projecto on e offshore Contém plásticos provenientes da indústria do
petróleo
2.1.2.3 Solar
Aproveita a luz do sol que é captada por painéis solares. Estes transformam posteriormente a
energia em eléctrica (solar fotovoltaica) ou térmica (solar activa ou passiva), que permite aquecer os
edifícios e as águas sanitárias.
É muito pouco provável que a energia solar substitua a energia proveniente dos combustíveis
fósseis nas próximas décadas devido não só à sua baixa eficiência, na ordem dos 10 a 30%, como
também à localização limitada e a necessidade de elevados investimentos iniciais [7].

8
Dada a energia solar fotovoltaica ser a utilizada para a produção de energia eléctrica, a tabela
6, de vantagens e desvantagens, prende-se apenas com este tipo de energia.
Tabela 6 - Vantagens e desvantagens do uso de energia solar [10]
Vantagens Desvantagens
Rede energética moderada Baixa eficiência
Não existe emissão de CO2 Custo inicial elevado
Impacto ambiental moderado Necessita de um sistema de backup ou de
armazenamento
Construção rápida Uso elevado do solo
Trabalha em dias de menos sol Precisa de acesso constante aos raios
solares
Fácil expansão
2.1.2.4 Geotérmica
É a energia obtida através do calor proveniente do interior da Terra.
O potencial anual estimado para a produção de energia eléctrica é pouco mais de metade do
potencial da energia produzida através de parques eólicos. No entanto, caso haja inovações
tecnológicas, a sua exploração pode tornar-se mais competitiva [7].
Tabela 7 - Vantagens e desvantagens da energia geotérmica[10]
Vantagens Desvantagens
Emissões de CO2 menores que em
combustíveis fósseis
Elevado custo
Impacto ambiental moderado Impacto sonoro
Baixa área necessária Escassez de zonas aplicáveis
Alta eficiência Algumas emissões de CO2
2.1.2.5 Ondas
As ondas que são formadas e transportadas pelo vento transportam uma enorme quantidade
de energia cinética.
Esta energia terá uma contribuição mínima na produção de energia mundial devido aos
elevados custos e não existência de áreas apropriadas [7].

9
Tabela 8 - Vantagens e desvantagens do uso de energia das ondas[12]
Vantagens Desvantagens
Fiabilidade Possível corrosão
Não poluente Custos de manutenção elevados
Difícil prevenção da acumulação de
vegetação marinha
2.1.2.6 Biomassa
Várias matérias-primas podem dar origem à biomassa: plantas (madeira e agricultura) e
resíduos animais(figura 3)[10].
Figura 3 - Diferentes matérias-primas, biomassa
A biomassa pode ser usada para queima directa ou através da sua transformação dar origem
a biocombustíveis líquidos e gasosos (figura 4) [10].

10
Figura 4 - Biomassa e sua utilização [10]
De notar que, a queima directa de biomassa liberta CO2, e é por isso necessário, que a sua
taxa de utilização não seja superior à plantação de novas plantas.
Os biocombustíveis líquidos podem ajudar a substituir a gasolina e o diesel e dominar a
indústria da bioenergia [10].
Tabela 9 - Vantagens e desvantagens do uso de biomassa [10]
Vantagens Desvantagens
Reduz emissões de hidrocarbonetos Aumenta as emissões de NOx
Não há libertação de CO2 para a atmosfera
caso a colheita seja feita de modo sustentável
Libertação de CO2 se for usada de modo não
sustentável
A plantação pode ajudar a recuperar solos
degradados
Mais caro que o diesel convencional
Reduz emissões de CO Plantações de biomassa podem competir com
as de agricultura
Pode reduzir a dependência das importações Desflorestação
2.1.3 Enquadramento Global
Nos dias de hoje, a qualidade de vida dos países desenvolvidos é em grande parte suportado
pelos combustíveis fósseis. A utilização destes está muito enraizada, o que deixa pouca margem de
para que novas fontes de produção de energia possam surgir.
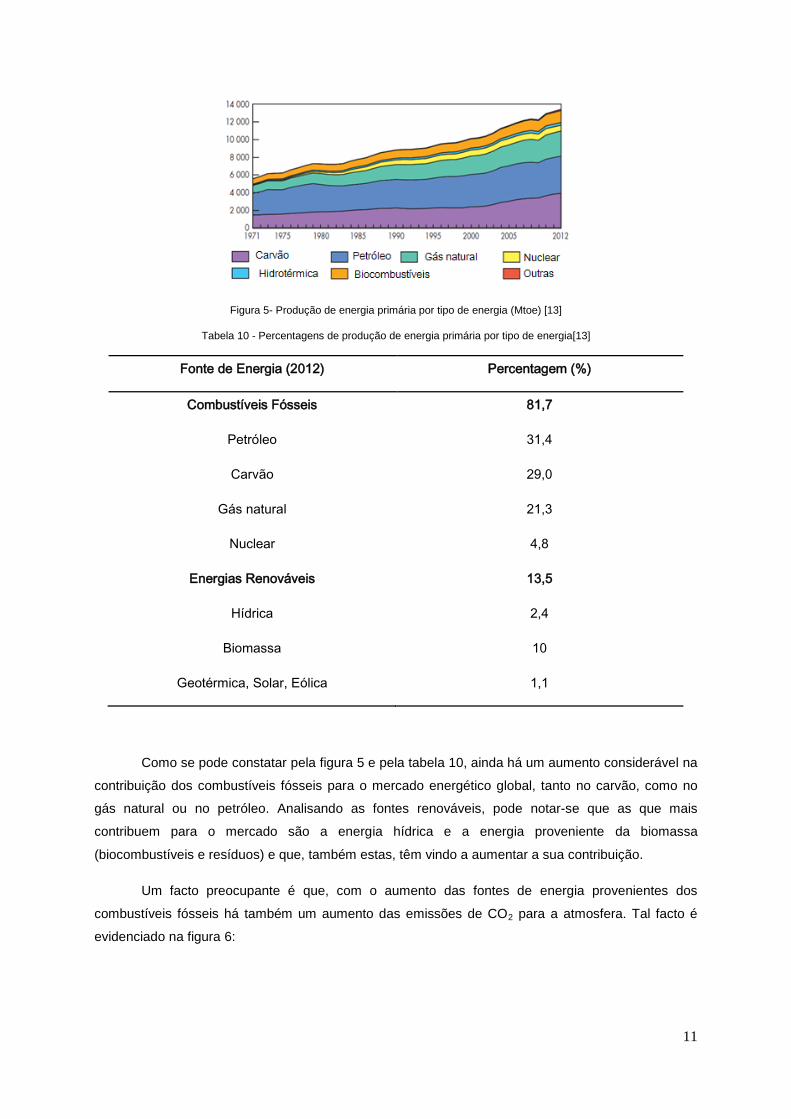
11
Figura 5- Produção de energia primária por tipo de energia (Mtoe) [13]
Tabela 10 - Percentagens de produção de energia primária por tipo de energia[13]
Fonte de Energia (2012) Percentagem (%)
Combustíveis Fósseis 81,7
Petróleo 31,4
Carvão 29,0
Gás natural 21,3
Nuclear 4,8
Energias Renováveis 13,5
Hídrica 2,4
Biomassa 10
Geotérmica, Solar, Eólica 1,1
Como se pode constatar pela figura 5 e pela tabela 10, ainda há um aumento considerável na
contribuição dos combustíveis fósseis para o mercado energético global, tanto no carvão, como no
gás natural ou no petróleo. Analisando as fontes renováveis, pode notar-se que as que mais
contribuem para o mercado são a energia hídrica e a energia proveniente da biomassa
(biocombustíveis e resíduos) e que, também estas, têm vindo a aumentar a sua contribuição.
Um facto preocupante é que, com o aumento das fontes de energia provenientes dos
combustíveis fósseis há também um aumento das emissões de CO2 para a atmosfera. Tal facto é
evidenciado na figura 6:
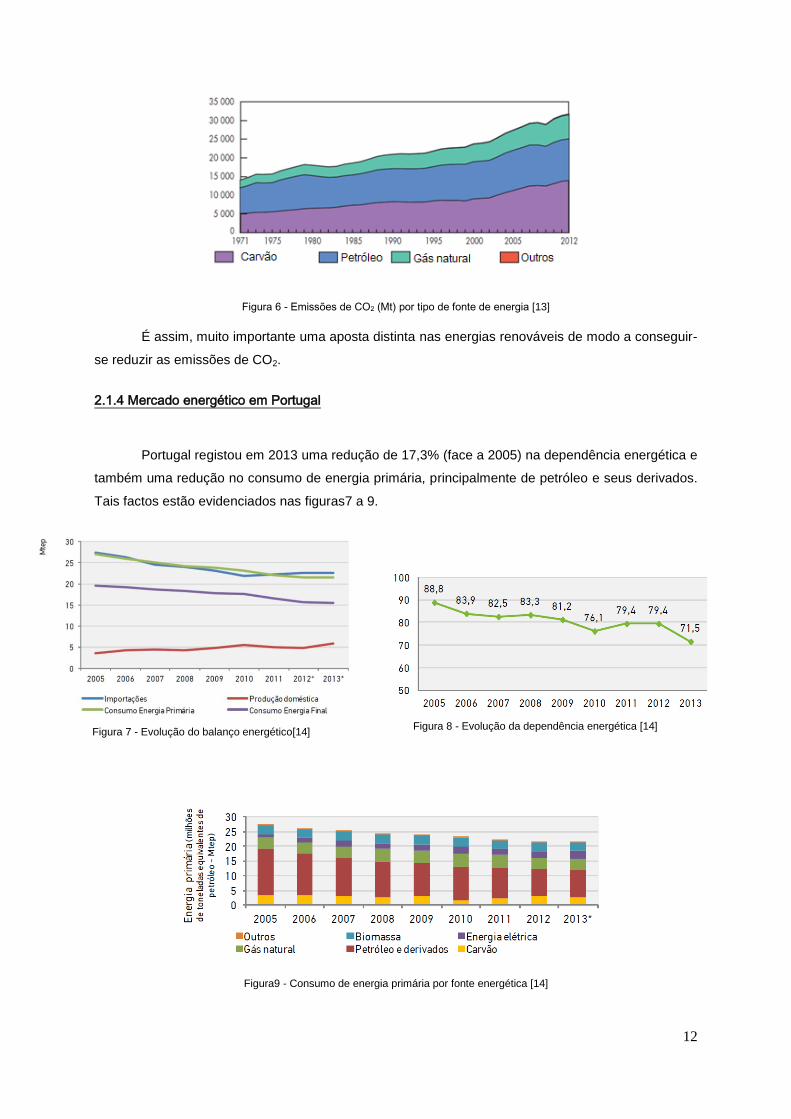
12
Figura 6 - Emissões de CO2 (Mt) por tipo de fonte de energia [13]
É assim, muito importante uma aposta distinta nas energias renováveis de modo a conseguir-
se reduzir as emissões de CO2.
2.1.4 Mercado energético em Portugal
Portugal registou em 2013 uma redução de 17,3% (face a 2005) na dependência energética e
também uma redução no consumo de energia primária, principalmente de petróleo e seus derivados.
Tais factos estão evidenciados nas figuras7 a 9.
Figura9 - Consumo de energia primária por fonte energética [14]
Figura 7 - Evolução do balanço energético[14] Figura 8 - Evolução da dependência energética [14]

13
Tem sido feito um esforço para obter uma menor dependência energética, diminuindo as
importações, principalmente de petróleo e seus derivados, e fazendo uma aposta forte nas energias
renováveis (produção doméstica). Aproveita-se assim as excelentes condições apresentadas pelo
país, tais como rede hidrográfica densa, exposição solar média anual elevada e grande costa
marítima [14].
Para 2013, o consumo de energias primárias pode ver-se na tabela 11:
Tabela 11 - Percentagem de consumo de energia primária por fonte de energia[15]
Fonte de energia Percentagem (%)
Combustíveis Fósseis 75,1
Petróleo 45,1
Carvão 12,4
Gás Natural 17,6
Energias Renováveis 24,9
Solar 0,2
Geotérmica 0,1
Hídrica 6,0
Eólica 4,8
Biomassa 13,8
O petróleo continua a ser a fonte de energia maioritária para consumo, sendo superior ao
total de energias renováveis. De entre as energias renováveis a que contribui mais é a biomassa
seguida da energia hídrica. Portugal tem ainda uma grande margem de aproveitamento dos seus
recursos para a produção de mais energia renovável, prevendo-se assim que cresça nos próximos
anos aliando preocupações ambientais com uma menor dependência energética.

14
2.2 BIOMASSA E BIODIESEL
Como foi referido no capítulo anterior, devido às crescentes preocupações ambientais e às
limitadas reservas de combustíveis fósseis, têm sido feitos cada vez mais esforços no sentido de
aumentar a utilização de combustíveis renováveis, com particular atenção dada à biomassa e aos
biocombustíveis. Esta é a maneira de se atingir um crescimento económico sustentável e harmonizar
a co-existência entre humanos e o ambiente.
Existem várias fontes de biomassa tais como biomassa florestal e biomassa
agrícola.Podemos, no entanto, também dividir a biomassa em três tipos: sólida, gasosa e líquida, em
que[10][7] [12]:
Biomassa sólida: Produtos e resíduos agrícolas (vegetais e animais), resíduos florestais,
industriais e urbanos.
Biomassa gasosa: O combustível gasoso é chamado biogás.
Biomassa líquida: Pode existir como bioetanol, biodiesel e metanol.
Os biocombustíveis estão diferenciados em duas gerações de acordo com a matéria-prima
que lhes dá origem. Os biocombustíveis de 1ª geração são obtidos através de bens de consumo
alimentar e envolvem o uso de tecnologia convencional, enquanto que os de 2ª geração são obtidos
através de matérias-primas não alimentares, tais como madeira e partes não comestíveis dos
vegetais, envolvendo tecnologia avançada de forma a remover os açúcares úteis da biomassa fibrosa
e lenhosa [7].
O biodiesel é um substituto do diesel e pode ser usado na sua totalidade (100%) ou em
mistura diesel-biodiesel. Quando usado em mistura recebe o nome de B2, B4, B25 em que o número
corresponde à sua percentagem usada na mistura.
2.2.1 Produção de biodiesel
O processo mais comum para a produção do biodiesel é a transesterificação (figura 10). Esta
transesterificação dá-se na presença de um catalisador homogéneo básico (NaOH ou KOH), ácido,
enzimático (lipase) ou heterogéneo. Os triglicéridos presentes nos óleos vegetais ou animais reagem
com o álcool (metanol ou etanol) e formam o biodiesel [78].

15
Figura 10 - Processo de produção de biodiesel por transesterificação por via alcalina [78]
Num balanço de massa feito à produção de biodiesel, por cada 100 kg de óleos ou gorduras
usadas, são precisos 10-15 kg de álcool (metanol ou etanol) para produzir 100-105 kg de biodiesel e
10 kg de glicerol [16].
Sendo assim, com o objectivo de se produzir biodiesel também se produzem grandes
quantidades de glicerina. Esta produção de glicerina é superior ao consumo actual, e é então
necessário encontrar váriosprodutos comerciais para lhe acrescentar mercado. Um desses valores
comerciais é o uso de glicerina para produção de aditivos para combustíveis, estudado neste
trabalho.
2.1.1.1 Matéria-Prima para a produção de biodiesel
Algumas das matérias-primas utilizadas são algodão, amendoim, colza, girassol, mamona,
soja, canola, pinhão manso e ainda micro-algas, sendo que destas, em Portugal, as mais utilizadas
são girassol, colza, soja e palma[17][18].
Pode-se notar que algumas composições dos óleos (matérias-primas) são mais adequados
do que outros, tendo em conta [19]:
Tamanho da cadeia de cada radical
Presença ou ausência de insaturações
Posição das insaturações.

16
O tamanho da cadeia de radicais fornece maior viscosidade, o que pode influenciar
negativamente o rendimento da reacção e dificultar a separação da glicerina. Afecta também o
número de cetano, a lubricidade, o ponto de névoa e o ponto de entupimento.
Também pode haver produção de biodiesel através de óleos alimentares usados. O preço é
um factor muito importante na produção de biodiesel, e como o preço do óleo alimentar usado é 2 a 3
vezes mais barato do que os óleos vegetais virgens, esta é uma matéria-prima com grande potencial
[20].
2.2.2. Breve história do biodiesel
Para mostrar a evolução do biodiesel ao longo dos anos, mostra-se de seguida as mais
importantes datas [21]:
1900 - Primeiro ensaio de um motor movido a óleos vegetais, por Rudolf Diesel.
1939-1945 - Uso de combustíveis obtidos a partir de óleos vegetais em "frota de guerra".
1970 - Aumenta o interesse no biodiesel, fruto de um choque petrolífero
1991 - A Comunidade Europeia aprova uma redução de impostos em 90% para
biocombustíveis, incluindo biodiesel.
2.2.3 Vantagens e desvantagens
Na tabela 12 encontram-se alguns aspectos positivos e negativos relativos ao biodiesel, em
comparação com outros combustíveis de origem fóssil [21][22]:
Tabela 12 - Vantagens e desvantagens do biodiesel
Vantagens Desvantagens
Fonte renovável Maior viscosidade do combustível
Reduz emissões de gases com efeito de
estufa
Impacto nos preços dos alimentos
Pressão no sentido de diminuir preços do
petróleo
Esgotamento do solo e desflorestação
Menos inflamável que o diesel convencional Aumenta emissões de NOx
Diminui a dependência do petróleo Maior custo de produção do que o diesel
convencional
Pode ser produzido por um maior número de
países
Problemas de armazenamento e estabilidade
devido à presença de grupos oxigenados
Tem um maior índice de cetano e melhor
lubrificação do que o dieselconvecional, o que
melhora a eficiência do motor e o ciclo de
vida de operação.
Só tem benefícios ambientais se a biomassa
for produzida de forma sustentável

17
Como pode ser evidenciado na tabela 12, uma das vantagens é a redução de emissões de
gases que contribuem para o efeito de estufa. Na figura 11, podem ver-se as percentagens de
redução de emissões de gases poluentes em B100 e B20, em relação ao diesel convencional:
Figura 11 - Percentagem de redução de emissões de poluentes em B20 e B100[18]
Há uma redução significativa de emissões de gases que provocam efeito de estufa,
maioritariamente de CO2, com uma redução de 78% em relação ao diesel convencional, para B100.
No entanto, há um aumento de emissões de NOx.
2.2.4 Mercado
Mundial
Em relação à produção mundial de biodiesel, pela figura 12 pode ver-se que tem vindo a
crescer anualmente, impulsionada pelos altos preços do petróleo, busca de redução de dependência
de combustíveis fósseis e problemas ambientais tais como o efeito de estufa, inseridos no protocolo
de Kyoto (redução de emissões de GEE).
Os maiores produtores mundiais em 2012 eram Estados Unidos da América, Argentina,
Brasil, Alemanha, França e Indonésia [23].

18
Figura 12 - Produção mundial de biodiesel[23]
Europa
Na Europa, a utilização de biodiesel começou em 1991 com subsídios para a produção
agrícola não-alimentar. A legislação europeia requer o uso de biocombustíveis misturados com
combustíveis fósseis, estabelecendo um mínimo de 5,75% (valor para o ano de 2010) de quota de
mercado para o biodiesel[23]. Até 2020, existe também uma Directiva Comunitária que obriga à
substituição de 10% dos combustíveis tradicionais por alternativos, no sector dos transportes [14]. De
modo a conseguir atingir os objectivos mínimos, muitos estados-membros da União Europeia estão a
aplicar isenções fiscais sobre os combustíveis.
A produção europeia de biodiesel pode ser vista na figura 13. Entre 2011 e 2012 houve uma
quebra de produção que corresponde a uma quebra de consumo de gasóleo nos mesmos anos [15]:
Figura 13 - Produção de biodiesel na Europa[23]
0
50
100
150
200
250
300
350
400
450
500
2000 2002 2004 2006 2008 2010 2012
Mil
hare
s d
e B
arr
is /
dia
Anos
0
50
100
150
200
2000 2002 2004 2006 2008 2010 2012
Mil
ha
res d
e B
arr
is /
an
o
Anos

19
Portugal
Portugal está incluído na Directiva Comunitária que obriga à substituição de 10% dos
combustíveis tradicionais por alternativos até 2020.
Através de dados da Direcção Geral de Energia e Geologia, verifica-se que em Portugal a
produção de biodiesel tem vindo a aumentar. Também teve uma quebra nos anos de 2011 e 2012,
semelhante à quebra europeia, devido à diminuição de consumo de gasóleo nos mesmos anos, mas
desde então tem vindo a crescer.A figura14mostra esse facto:
Figura 14 - Produção de biodiesel em Portugal[15]
Encontram-se em Portugal várias fábricas de biodiesel, entre elas a da Iberol, Prio
Biocombustíveis, Torrejana, Sovena e Biovegetal.As primeiras duas fábricas têm produção de
100kton/ano e como matérias-primas óleo de soja, colza, girassol e palma[24].
2.2.5 Perspectiva futura
Produção
Uma das grandes desvantagens do biodieselé o seu preço de produção. Têm sido estudadas
várias alternativas tais como envolver lipases (enzimas) como catalisadores. Estas podem ser
excelentes opções porque aumentam a pureza do biodiesel e permitem a separação da glicerina.
Têm também outras vantagens como menor preço e menores problemas no processo [26].
Mercado
Através da figura 15, pode ver-se que está previsto um aumento na produção de biodiesel
mundial até 2020.
0E+0
5E+4
1E+5
2E+5
2E+5
3E+5
3E+5
4E+5
4E+5
2006 2008 2010 2012 2014
To
n
Anos

20
Figura 15 - Perspectiva de produção e comércio de biodiesel mundial até 2020 (Bln)[27]
De notar que, com o aumento da produção de biodiesel, há também um aumento de
produção de glicerol.
2.3 GLICEROL
O Glicerol também conhecido como glicerina ou 1,2,3-propanotriol é um composto orgânico
que pertenceaogrupo dos álcoois. O termo glicerol refere-se ao composto puro e o termo glicerina é
aplicado a uma solução comercial de glicerol e água, em que o principal componente é glicerol.O
glicerol em crude é obtido com 70-80% de pureza e é purificado até 95,5-99%.
A glicerina é muito utilizada principalmente na alimentação, indústria farmacêutica e
cosmética (figura 16)[28].
Figura 16 - Mercado do glicerol (em percentagem), 2002[28]
11%
14%
11%
10% 8%
6% 2%
2%
2%
18%
16%
Alimentação
Poliéster
Outros
Triacetina
Resina alquídica
Tabaco
Detergentes
Celofane
Explosivos
Farmacêuticos
Produtos de cuidado pessoal

21
2.3.1 Breve História
O Glicerol foi descoberto em 1779 pelo químico sueco Carl WilhelmScheele. Este ao fazer um
tratamento a um óleo natural (azeite) com materiais alcalinos reparou na formação de um líquido
transparente e adocicado[28].
Uma maior importância dada à glicerina aconteceu em 1823, quando o químico Chevreul
notou que a glicerina é formada através da saponificação de óleos, ao descobrir uma nova forma de
converter a gordura animal em ácidos gordos e glicerina[29].
Só em 1866 é que a glicerina se tornou industrialmente e economicamente importante,
quando Alfred Nobel inventou a dinamite. A dinamite pode ser formada através do nitro glicerol e foi
muito usada para a construção de túneis, canais e estradas [29].
2.3.2 Propriedades
Devido às suas inúmeras propriedades físicas e químicas, a glicerina tem uma grande
quantidade de utilizações. É incolor, inodoro e muito viscoso. De seguida, apresentam-se algumas
das suas propriedades mais relevantes [28].
Temperatura de ebulição
A temperatura de ebulição da glicerina é 290ºC à pressão atmosférica (760 mmHg). De
seguida estão alguns valores de temperatura de ebulição a pressões baixas[29]
Tabela 13 - Temperaturas de ebulição do glicerol a diferentes pressões
P (mmHg) T (ºC) P (mmHg) T (ºC)
5 152 30 190,9
10 166,1 40 198
20 181,3
Viscosidade
A viscosidade do glicerol a 20ºC é 1,5 Pa/s, que é um valor alto em relaçãoao que seria
expectável, devido às ligações de hidrogénio [28]
Solubilidade
Tem características de solubilidade semelhantes às da água e dos álcoois alifáticos simples
devido ao grupos hidroxilos. É completamente miscível em água. Em relação à sua solubilidade com
acetona, é solúvel até 5% em peso em acetona a temperatura ambiente. É completamente insolúvel
em ácidos gordos, hidrocarbonetos e solventes clorinados [29].

22
Sabor
A glicerina tem um sabor doce, no entanto é menos doce do que a sucrose [29].
2.3.3. Produção (natural e sintética)
O glicerol natural é obtido na conversão de gorduras e óleos em ácidos gordos e ésteres
metílicos de ácidos gordos, enquanto que o glicerol sintético é obtido através de propeno. Existem
outros tipos de produção mas que não têm muita aplicabilidade industrial que são a fermentação de
açúcar ou hidrogenação de hidratos de carbono [29].
Em relação à produção natural de glicerol existem vários processos:
Separação de gorduras
Através da separação de gorduras ou hidrólise sem a adição de alcalinos. Este método é para
a obtenção de ácidos gordos e posteriormente álcoois gordos. A glicerina é então obtida na água
doce. Esta glicerina é chamada de crude de saponificação [30].
Transesterificação
Consiste na esterificação de óleos e gorduras em ésteres metílicos.
As gorduras reagem com metanol na presença de um catalisador alcalino para produção de
ésteres de metilo e glicerol. Estes produtos são separados com água. Os ésteres metílicos têm como
utilização a sua redução a álcoois gordos correspondentes ou alternativa ao diesel como biodiesel, já
referido anteriormente [28].
Figura 17 - Transesterificação de um triglicérido com metanol[28]
Saponificação
É um processo muito tradicional que consiste na utilização de processos contínuos para
produzir sabão.
Neste processo as gorduras são fervidas juntamente com soda cáustica (NaOH) e sal.

23
No final do processo obtém-se duas fases distintas:
Camada superior (sabão)
Camada inferior (glicerol, água, sal e o excesso de soda cáustica)
A camada inferior pode sofrer uma evaporação [30].
A produção de glicerol a partir de propileno foi posta em prática em 1948 e são conhecidos
três processos que envolvem os seguintes intermediários:
Cloreto de alilo - Epiclorohidrina
Acroleína - Álcool Alílico - Glicidol
Óxido de propeno - Álcool Alílico - Glicidol
No entanto, só a primeiro método de produção é que tem importância industrial [30].
2.4 REACÇÕES DO GLICEROL PARA APLICAÇÃO INDUSTRIAL
Devido a uma crescente produção de glicerol, o número de químicos e novos produtos tendo
como matéria-prima o glicerol tem vindo a aumentar nos últimos anos. Este desenvolvimento de
novos produtos torna a produção de biodiesel mais sustentável.
Estes derivados do glicerol podem ser usados em variados campos tais como combustíveis,
produtos farmacêuticos, químicos, detergentes e nas indústrias automóveis e de construção [28].
De seguida mostram-se algumas das reacções que têm glicerol como matéria-prima.
2.4.1 Hidrogenólise
Os principais produtos da reacção são o 1,2 propanodiol e o 1,3 propanodiol. O primeiro
produto tem propriedades anti-congelantes e é usado para a produção de fármacos, produtos
alimentares, cosméticos, detergentes e tintas[28]. O segundo produto é usado na produção de fibras
sintéticas [77].
Esta reacção dá-se na presença de catalisadores metálicos, tais como Cu, Ni e Pd, em
solução ou em fase gasosa [77][28].

24
Figura 18 - Reacção de produção de 1,2-propanodiol[28]
Noutras condições, a hidrogenólise da glicerina pode também dar origem a n-propanol,
isopropanol, propeno (através da sua polimerização dá origem a polipropileno e posteriormente a
plásticos) e propano [77].
2.4.2 Eterificação
A reacção de eterificação da glicerina pode ser elaborada com isobuteno, tert-butanol, álcoois
como o metanol e o etanol ou álcool benzílico. A adição de éteres de glicerol em combustíveis diesel
tem efeitos positivos tais como reduzir a quantidade de fumos e partículas, óxidos de carbono e
compostos carbonílicos presentes no tubo de escape e também reduzir o ponto de névoa do diesel e
a viscosidade do biodiesel[77][28]. Na figura 19 está a reacção de produção de mono, di e tri-éteres
provenientes da reacção entre o glicerol e o isobuteno.
Figura 19 - Eterificação do glicerol com isobuteno[28]
2.4.3 Esterificação
A esterificação de glicerol ocorre ao reagir glicerol com ácidos carboxílicos. Estes podem ser
usados como emulsificantes na indústria alimentar, cosmética e têxtil[28].

25
Figura 20 - Reacção de glicerol com ácido acético [28]
Uma das reacções mais importantes é a reacção da glicerina com ácido acético, com a
formação de mono (MAG), di (DAG) e triacetina (TAG). A triacetina é usada na indústria de cosmética
e do tabaco e recentemente tem sido considerada como aditivo para o biodiesel[77].
2.4.4 Oxidação
A oxidação selectiva do glicerol tem um interesse particular porque produz derivados
oxigenados do glicerol com elevada importância comercial[28].
Os diferentes compostos formados, que se podem ver na figura21, dependem da hidroxila
que sofre a oxidação. Por exemplo, caso seja a hidroxila primária dá origem a gliceraldeído e caso
seja a hidroxila central forma-se dihidroxiacetona[77][28].
Estes compostos têm várias finalidades tais como: o gliceraldeído é um intermediário para o
metabolismo de hidratos de carbono, o ácido glicérico é muito usado na indústria farmacêutica e a
dihidroxiacetona é utilizada em bronzeadores artificiais e na produção de polímeros [77][28].

26
Figura 21 - Produtos da oxidação do glicerol[28]
2.4.5 Desidratação
Dois químicos importantes são retirados desta reacção: acroleína e 3-hidroxipropanal (3-
HPA). A acroleína é usada como herbicida e pode originar outros produtos, tais como propano,
plásticos e ácido acrílico (usado como absorvente em fraldas). O 3-HPA dá origem, entre outros, a
acroleína, ácido malónico, acrilamida e pode ser usada para terapêutica na indústria farmacêutica e
como conservante em produtos alimentares.
Figura 22 - Produção de acroleína através da desidratação de glicerol[28]
2.4.6 Carbonato de glicerol
Tem como fórmula química 4-hidroximetil-1,3-dioxolan-2-ona. A sua finalidade é ser usado
como biolubrificante e também como solvente para plásticos e resinas[28].

27
Figura 23 - Síntese do carbonato de glicerol com dimetilcarbonato[28]
2.4.7 Clorinação
Com a clorinaçãohá formação de 1,3-dicloropropanol que é muito importante na síntese de
epiclorohídrina. Esta é utilizada na formação de resinas, na indústria farmacêutica, purificação de
água e indústria alimentar.
Figura 24 - Clorinação de glicerol[28]
2.4.8 Acetalisação
Reacções de acetalisação de glicerol com aldeídos formam, em geral, anéis de 5 ou 6
membros que têm como utilização, por exemplo, ser aditivos para o biodiesel. O aditivo melhora as
propriedades de fluxo a frio, nas quais se inclui o ponto de névoa [77].
Figura 25 - Reacção de acetalisação de glicerol com aldeídos[77]

28
Existe também a reacção de acetalisação de glicerina com acetona, que é a reacção
estudada neste trabalho, que promove a produção de solketal. O solketal pode ser usado na indústria
farmacêutica, cosmética, em tintas cujo solvente é água e como aditivo em dieselde forma a diminuir
a formação de goma, rezudir o ponto de congelamento, melhorar as propriedades lubrificantes,
aumentar o poder antidetonante (octanagem) e reduzir também a viscosidade.É também usado em
jet fuel, de forma a alterar a sua estrutura química e composição, o que reduz a inflamabilidade do
combustível ao aumentar o tamanho das gotículas[1][2][3][4]. Esta reacção pode ser vista, com um
pouco mais de detalhe, na figura 26.
Figura 26 - Mecanismo proposto para a reacção do glicerol com acetona envolvendo catalisador ácido[31]
2.5 CATALISADORES REFERIDOS NA LITERATURA
Os catalisadores mais adequados para a catálise da reacção entre o glicerol e a acetona têm
características específicas tais como ser um catalisador ácido, com elevada área específica, elevado
número de centros activos, ser hidrofóbico, reduzindo a interferência das moléculas de água na
superfície do catalisador, visto que estas desfavorecem a produção de solketal, e ter boa estabilidade
térmica.
No final do capítulo está um quadro resumo com os valores mais importantes para cada
catalisador.

29
2.5.1 Oxihidróxido de Nióbio
Os catalisadores que têm como base o nióbio são muito atractivos para catálise heterogénea
pois têm diversas propriedades muito particulares e a habilidade de actuar como precursor e como
suporte, o que é atribuído à sua elevada acidez, propriedades redox e fotosensitividade. O óxido de
nióbio mostra boa tolerância à água e exibe excelente actividade catalítica em comparação com
outros sólidos ácidos. Para a síntese do catalisador oxihidróxido de nióbio usa-se o precursor cloreto
de nióbio e procede-se à alteração da superfície por hidrofobização parcial dos grupos
surfactantes[32][33].
Foram realizados ensaios com glicerina dessalinizada, comercial ou crude. Obtiveram-se
melhores valores para a glicerina dessalinizada para os catalisadores HY-340 (ácido nióbico
comercial) com 55% de conversão e S4-2D (catalisador com surfactante e feito em agitação
constante durante 48h) com 43% de conversão, em relação aos catalisadores S4-SS (catalisador
sem surfactante e com agitação constante durante 12h) e S4 (catalisador com surfactante e feito com
agitação constante durante 12h). Este, S4-2D, obtém melhores resultados porque tem elevada área
específica, maior TOF e maior número de centros activos. A selectividade para este mesmo
catalisador, S4-2D é de 95% após1h de reacção. A conversão do glicerol aumenta quando aumenta a
razão molar acetona:glicerol e, para 4:1, obtém-se um valor de 73%.
Para a glicerina comercial ou crude, os valores são mais baixos devido à presença de NaCl e
à elevada viscosidade.
Como conclusão deste catalisador, nota-se que a maior hidrofobização baixa o número de
centros activos, mas os grupos hidrofóbicos têm um papel importante ao actuar na interface
acetona/glicerina, reduzindo a interferência das moléculas de água na superfície do catalisador,
favorecendo a produção de solketal [32].
Noutro estudo com oxihidróxido de nióbio usaram-se diferentes precursores que foram
hidrofobizados com cetiltrimetilamónio de bromo. O catalisador deoxihidróxido de nióbio e os
catalisadores 2 e 3 de oxihidróxido de nióbio hidrofóbico mas com precursores diferentes. Ser
anfipático é importante porque dá versatilidade ao catalisador, mais especificamente dando-lhe a
possibilidade de que a reacção em que actua envolva tanto solventes polares como não polares e
mesmo sistemas bifásicos. Sendo estes catalisadores tanto hidrofóbicos como hidrofílicos, ocupando
interface entre os líquidos, foram os que obtiveram melhores resultados. Tal facto está associado com
os grupos surfactantes, que facilitam as interacções entre a acetona e a glicerina. O melhor
catalisador tem como precursor o oxalato de amónio e alguma acidez superficial. Neste caso, para 60
min de reacção, obteve-se 65% de conversão e 95% de selectividade[33].

30
2.5.2 Purolite PD206
A Purolite PD206 funciona como catalisador heterogéneo e como resina de troca iónica. É
maioritariamente usada na purificação de biodiesel para remoção de contaminantes tais como o
glicerol. Para que a reacção se dê mais facilmente, é usado etanol como co-solvente, aumentando
assim a solubilidade do glicerol em acetona.
As condições óptimas para se obter 95% de rendimento e 100% de conversão são 20ºC, 120
bar e acetona/glicerina numa razão molar de 5:1, com 0,77g de catalisador e 0,1 mL/min de caudal.
Dada a alta pressão de vapor da acetona, ao aumentar a temperatura a quantidade de
acetona no líquido diminui porque é vaporizada. Consequentemente a concentração de acetona
como reagente na superfície do catalisador diminui e diminui a eficiência das interacções entre a
acetona e o catalisador. Sendo assim, a temperatura pode desfavorecer a formação de solketal. De
modo idêntico, ao aumentar a pressão, a solubilidade do glicerol em acetona aumenta e permite que
a reacção se dê eficientemente. O aumento da razão molar acetona/glicerina aumenta tanto a
selectividade como a conversão devido à remoção de água por parte da acetona. A acetona serve
assim como solvente subcrítico que avança o processo em temperaturas baixas e o seu excesso
remove a água do sistema. Em relação ao caudal, com o seu aumento os reagentes parecem não ter
tempo para interagir de modo eficaz, havendo redução da selectividade. Esta aumenta como
acréscimo de catalisador[34].
2.5.3 Heteropoliácidos imobilizados em sílica
Os heteropoliácidos são catalisadores heterogéneos bastante activos em catálise ácida. Têm,
no entanto, algumas desvantagens tais como baixa estabilidade térmica, baixa área superficial e
problemas de separação. Para reverter a situação usa-se sílica como suporte de modo a imobilizar os
ácidos.
Foram realizados vários catalisadores com diferentes heteropoliácidos tais como ácido
tungstofórico, ácido tungstosílico, ácido molibdofosfórico e ácido molibdosílico. De entre estes o que
obteve melhores resultados foi o ácido tungstofórico por ser o mais ácido mais forte, com conversão
ao fim de 3h de 97% e selectividade 98%. A conversão aumenta com a temperatura e com a razão
molar acetona/glicerol, sendo que a selectividade é mantida constante.A conversão aumenta também
com o acréscimoda quantidade de catalisador e otempo requerido para chegar ao equilíbrio diminui
com a quantidade de catalisador devido ao aumento do número de centros activos. A selectividade
também não é alterada[35].

31
2.5.4 Zeólito-Beta
O Zeólito é um aluminosilicato importante devido ao elevado número de centros activos,
elevada estabilidade térmica e selectividade perante moléculas desejadas com tamanha do poro
regular.
Foram realizados ensaios com zeólito-beta de menor tamanho cristalino e de maior tamanho
cristalino. O zeólito H-β-1 sofre uma dealuminação com concentrações de 0,01 (H-β-1A ) e 0,05M (H-
β-1B) de ácido oxálico. Ao zéolito H-β-1 que sofre uma troca iónica com iões de Cobre dá-se o nome
de Cu/H-β. O mesmo catalisador mas na forma Na-β (Cu-β) foi realizado. Foi também preparado um
zeólito H-β de maior tamanho cristalino na forma Na (β-2).
A dealuminação com ácido oxálico provoca uma diminuição da conversão de glicerol devido à
também diminuição dos centros activos fortemente ácidos. No entanto, a selectividade mantém-se.
A conversão de glicerol diminui ao fazer a troca de iões com Cu2+
e ao converter a forma
protónica em forma Na, há um aumento da conversão devido ao aumento de centros activos ácidos.
Em relação ao tamanho dos poros, o zeólito com menor tamanho cristalino obteve melhores
resultados, devido a ter umadifusividade mais rápida.
Os melhores resultados foram 86% de conversão e 98,5% de selectividade[36].
2.5.5 Compósitos Carbono-Sílica
Neste catalisador existe carbonização e sulfonação simultânea da glicose na presença de
sílica orgânica, em que a glicose funciona como fonte de carbono e precursor na formação do
material compósito de sílica-carbono. Deve-se referir que o que maioritariamente influencia a reacção
de produção de solketal é a larga área superficial específica, a superfície hidrofóbica e o número de
centros activos.
Foram preparados vários catalisadores de modo a observar a mesoporosidade: por hidrólise
com ácido sulfúrico seguido de tratamento hidrotérmico e carbonização (HSCS) e por hidrólise com
ácido sulfúrico seguido de tratamento térmico e carbonização (SCS). Os catalisadores foram feitos
com diferentes razões glucose/sílica.
As maiores conversões e selectividades foram obtidas para os catalisadores que foram
sujeitos a um tratamento hidrotérmico, o que é explicado pelo facto de o tratamento hidrotérmico
facilitar a formação de maior densidade ácida total e maiores mesoporos. De notar também que,
somente o tamanho dos mesoporosnão é responsável pela actividade catalítica.Os melhores valores
deconversão e selectividade obtidos foram de 82% e 99% respectivamente [37].

32
2.5.6 Ácido p-toluenosulfónico
O ácido p-toluenosulfónico é preparado pela sulfonação do tolueno e muito usado como
catalisador ácido.
Numa reacção com razão molar 2:1, com 1%(w/w) de catalisador e 12h de reacção, a
conversão é 54,9%. O valor da conversão aumenta ao aumentar a quantidade de acetona na
reacção, sendo que os 100% nunca são obtidos devido ao equilíbrio e à presença de água na
reacção[38].
Usando 6% (w/w) de catalisador e mantendo a temperatura a 70-80ºC, obteve-se, após 20
minutos de reacção, uma conversão superior a 70%, que chega aos 90% após os 40 minutos [39].
2.5.7 Amberlyst-15
Amberlyst-15 tem poliestireno com grupo sulfónico fortemente ácido e, sendo assim, é usado
como catalisador ácido em catálise heterogénea.
Foram realizados vários ensaios com diferentes tipos de glicerina (variando na matéria-prima
que a formou) a 70-80ºC. Para glicerina de mamona obteve-se uma conversão de 15%, fruto da
presença de água que enfraquece os centros activos ácidos do catalisador e do metanol
contaminante que promove reacções paralelas. Para a glicerina de soja a conversão obtida foi de
45%, notando-se que a presença da base é um factor importante para a reacção proceder. Para a
glicerina de palma a conversão foi de 12%, facto justificado apenas pela presença de contaminantes
que podem ter afectado e influenciado a reacção.
Pode-se assim concluir que a glicerina proveniente de diferentes oleoginosas possui
contaminantes tais como metanol ou etanol, água, ácidos graxos, sais e bases que interferem na
reacção, quer seja enfraquecendo os centros activos como promovendo reacções paralelas [40].
Foram também realizados ensaios para comparação entre Amberlyst-15 e zeólito-beta, tendo
em conta impurezas na glicerina (metanol, água e cloreto de sódio).
Para o glicerol puro, Amberlyst-15 obteve maior conversão, 95%, enquanto o zeólito teve
90%. Ao adicionar metanol, a conversão baixa, porque este compete com os centros activos, para
76% no Amberlyst-15 e 86% para o zeólito. A adição de água influencia também a conversão e de
forma mais acentuada porque a acidez do catalisador baixa e sendo assim, a conversão passa a 79%
para Amberlyst-15 e 81% para o zeólito. O mesmo acontece na presença de NaCl que provoca a
neutralização dos centros activos, sendo que para o Amberlyst-15 se obtêm valores de 71% e para o
zeólito 69%.

33
No geral, o que acontece é o mesmo para ambos os catalisadores, no entanto notam-se
diferenças nos seus valores, sendo o zeólito o que apresenta melhores números, muito em facto
devido às suas características hidrofóbicas, sendo assim mais resistente à água [41].
2.5.8 Nanopartículas de Níquel em nanotubos de carbono
Para este catalisador as nanopartículas de níquel são suportadas em nanotubos de carbono,
formando um catalisador sólido, selectivo e ácido.
A reacção foi realizada numa atmosfera de azoto para evitar que ocorressem reacções de
oxidação. Os catalisadores preparados tinham diferentes percentagens de Níquel na amostra e, ao
aumentar a quantidade de níquel no catalisador aumenta a conversão e desaparecem produtos
secundários, devido à presença de centros activos metálicos e à sua forte acidez. Isto explica a
performance selectiva do Níquel na formação de produtos cíclicos. Esta conversão é de 96% com
selectividade de 72% para Ni(1,8). No entanto, ao aumentar a quantidade de Níquel a partir destes
1,8 (% de níquel em massa), a conversão começa a baixar devido à agregação de espécies de
níquel, devido à saturação das ligações metal-suporte e ao aumento das interacções metal-metal,
dentro e fora dos nanotubos baixando o número de centros activos acessíveis[42].
2.5.9 Óxidos de TiO2-SiO2
Os óxidos de TiO2-SiO2 oferecem ligações Si-O-Ti que são centros activos ácidos e são
preparados pela técnica sol-gel.
Numa reacção com razão molar 4:1, a 70ºC, durante 3 horas e com 2,2% (w/w) de catalisador
pode ver-se que a conversão de glicerol é muito baixa para apenas TiO2 e aumenta com a razão Si/Ti
até um máximo de 86,5%. A acetalisação ocorre nos centros activos ácidos especialmente em
espécies ácidas de Bronstead, mais especificamente neste caso como centros activos
proporcionados por moléculas de água adsorvidas por ligações Ti-O-Si localizadas em óxidos Ti-Si. A
selectividade máxima é também 86,5%, sendo a formação do solketal apenas controlada pela
cinética, e assim promovida pelo catalisador mais apropriado.
É também importante referir o efeito da calcinação no catalisador. A calcinação com
temperaturas muito elevadas leva à aglomeração de espécies de TiOx e a calcinação a temperaturas
baixas dificilmente leva a uma maior interacção entre o TiOx e o SiO2, o que enfraquece a capacidade
de absorver água dos centros activos. Sendo assim, uma temperatura de calcinação apropriada é
uma vantagem para a produção de centros activos ácidos e para a sua acidez.
Tal como em outros catalisadores, maior tempo de reacção leva a uma maior conversão e
maior selectividade, a conversão aumenta também com o aumento da razão acetona/glicerina porque
pouca quantidade de acetona é desvantajosa para a dispersão do glicerol. Tanto a conversão como a
selectividade aumentam com o aumento da temperatura da reacção.

34
Em condições óptimas, a conversão é de 95% e a selectividade de 90%. Essas condições
óptimas são 2,2% (w/w) de catalisador, razão molar 4:1, 3 horas de reacção e a 90ºC[43].
2.5.10 Silicatos com metais incorporados
Neste caso, os catalisadores são silicatos em que os átomos de metais incorporados na rede
de sílica funcionam como centros ácidos de Lewis. Os diferentes catalisadores contêm zircónio (Zr-
TUD-1), háfnio (Hf-TUD-1) e estanho (Sn-MCM-41). Estes catalisadores microporosos foram
estudadospela combinação entre os seus centros activos, poros acessíveis e largos e elevada área
superficial específica.
A reacção teve como condições razão molar 1:1, 80ºC, com tempo de reacção de 6 horas e
com 3% (w/w) de catalisador.
Foi obtida selectividade completa para todos os catalisadores. O melhor catalisador estudado
foi o Hf-TUD-1 com valor de conversão de 52%. Isto acontece não só devido ao maior número de
centros activos, mas também aos poros largos e elevada área superficial que optimizam a difusão
dos reagentes e produtos e o contacto com os centros activos, enquanto que a superfície
relativamente hidrofóbica favorece a remoção de água, minimizando a hidrólise do solketal [44].
2.5.11 SnO2 preparado com molibdénio e tungsténio
Neste catalisador, procede-se à adição de iões de molibdénio e tungsténio ao estanho. A
adição de iões de molibdénio foi a que melhorou significativamente a actividade catalítica devido a ter
maior superfície dos centros activos ácidos, maior área superficial específica, propriedades redox e
melhor efeitos de rede.
A conversão aumenta significativamente com a adição de molibdénio e tungsténio, existindo
uma relação linear entre a área superficial e a quantidade de centros activos. A conversão foi então
de 15% para Sn (SnO2 puro), 55% para WSn (SnO2 e tungsténio) e 61% para MSn (SnO2 e
molibdénio). A selectividade foi de 96% para todos os catalisadores[45].
2.5.12 Espuma celular mesoporosa modificada
A modificação da espuma celular foi feita com 3-mercaptopropiltrimetoxisilano (MPTMS). A
espuma celular é hidrofílica (MP-MCF), o que faz com que as moléculas de água geradas na reacção
sejam absorvidas pela espuma, evitando assim o problema da reversibilidade da reacção devido à
presença de água. Para melhorar a oxidação do MPTMS foram adicionados os metais Nb (MP-
NbMCF) e Ta (MP-TaMCF). Foi também preparado um catalisador com espécies fosfóricas (MP-
PTaMCF).
Foram realizados ensaios com várias razões molares acetona/glicerina e várias temperaturas.

35
O aumento da temperatura da reacção provoca evaporação da acetona, revertendo a
produção de solketal e, sendo assimaltas temperaturas só seriam benéficas usando reactores
autoclave. Sem catalisador, a produção de solketal apenas acontece a partirde 70ºC.
O excesso de acetona tem o efeito de promover a reacção, praticamente duplicando a
conversão quando se duplica a razão molar acertona/glicerina.
A selectividade é muito pouco alterada, no entanto há variações na conversão. O catalisador
que obtém melhores valores de concentração é o de Nióbio, com valores de 80% de conversão e
97% de selectividade. A modificação de MCF's com Nb e Ta é importante para a acetalisação porque
se dá um aumento da acidez de Bronstead através da oxidação e há um maior tamanho dos poros
das células. A adição de fósforo aumenta a acidez mas diminui o tamanho dos poros[31].
2.5.13 Níquel-zircónio com suporte de carbono activado
Para a preparação deste catalisador foi usado níquel-zircónio tendo como suporte carbono
activado, sendo que as experiências foram realizadas em condições de azoto.
As condições da reacção são 4% (w/w) de catalisador, com temperatura entre 25-65ºC,
reacção de 4 horas e com várias razões molares de acetona/glicerina.
Ao analisar os resultados, a conversão aumenta com o aumento da percentagem de Níquel
num catalisador do tipo Ni/AC. Um aumento do Níquel provoca maior número de centros activos na
superfície do catalisador, o que promove a conversão. Para 5% Ni/AC a conversão é de 98% e
selectividade de 86%, com razão molar 8:1 e a 45ºC.
No entanto, para Zr/AC os valores de conversão são mais baixos do que para Ni/AC, devido
às limitações difusionais internas e à distribuição não-uniforme dos agregados metálicos no suporte
de AC.
Numa mistura de ambos, Zr e Ni, o catalisador com maior actividade catalítica foi o 5%Ni-
1%Zr/AC com conversão completa e selectividade de 74%.
Para testar as diferentes condições foram realizados ensaios com diferentes quantidades de
catalisador, razão molar acetona/glicerina, tempo de reacção e temperatura de reacção. A conversão
aumenta com maior quantidade de catalisador devido a maior número de centros activos ácidos, com
maior quantidade de acetona deslocando a reacção para a direita e com o aumento do tempo de
reacção devido à quebra e formação de novas ligações pelas moléculas. Esta maior quantidade de
acetona promove também o aumento do anel de 6 membros devido ao aumento da polaridade da
reacção. A temperatura ideal obtida foi 45ºC, sendo que, no entanto, abaixo desta se obtêm melhores
valores de selectividade. Em relação ao tempo de reacção evidencia-se que o solketal é formado
após 1 hora de reacção, enquanto que o anel de 6 carbonos apenas se forma depois do solketal [3].

36
2.5.14 Óxido de ferro com suporte de aluminosilicato
Este catalisador apresenta melhorias nas propriedades ácidas e maior número de centros
activos. Com a adição do óxido de ferro ao aluminosilicato (Al-SBA-15) há um aumento da acidez
superficial.
Obtém-se 6% de conversão sem a adição de óxido de ferro e 51% com a adição de óxido de
ferro, sendo que a selectividade é de 99% para ambos os catalisadores.
Sendo assim, a presença das partículas do óxido de ferro foram muito importantes para a
conversão do glicerol, aumentando-a em 46%[46].
2.5.15 Membrana de zeólito
Para a reacção de produção de solketalfoi usada uma membrana de zeólito para remoção
contínua da água que desfavorece a sua produção. Usualmente usa-se um sistema de extracção de
líquido para a remoção da água, usando como líquidos o clorofórmio ou a nafta, visto terem pontos de
ebulição mais baixos que a acetona. Também pode ser usada destilação reactiva para a remoção da
água. O problema de se utilizar membranas é que estas não são estáveis em condições ácidas. Para
evitar este problema foi usada separação por vapor de modo a que a membrana não fique submersa
no líquido, mas em contacto com o vapor.
A síntese foi feita com montmorilonite e a membrana é de zeólito NaA.
Com 10% (w/w) de catalisador montmorilonite e 24 horas de reacção, para razão molar 5:1
obteve-serendimento de 60% sem membrana e 100% com membrana [47].
2.5.16 Sílica modificada com ácido sulfónico
A sílica modificada com ácido sulfónico é um material que apresenta grande área superficial,
largos poros, boa estabilidade térmica, com possibilidade de controlo da superfície
hidrofóbica/hidrofílica e boas concentrações de centros activos ácidos.
Foram realizados 3 catalisadores, com ácido propilsulfónico (Pr-SBA-15), ácido
arenesulfónico (Ar-SBA-15 e ácido arenesulfónicohidrofobizado. Estes catalisadores foram
comparados com catalisadores comerciais com ácido sulfónico tais como Amberlyst-15.
O melhor catalisador foi o comercial Amberlyst-15 com conversão de 85,1%. De entre os
catalisadores preparados obteve melhores resultados o Ar-SBA-15 com 82,5% devido a ter maior
acidez de mais centros activos ácidos. De notar que a conversão chega aos 80% em apenas 30
minutos de reacção. Seria de esperar que o catalisador hidrofobizado tivesse melhores resultados
afectando a concentração de moléculas de água, no entanto tal não acontece porque este catalisador

37
tem menor área superficial disponível e porque a hidrofobização pode afectar a concentração polar de
outras espécies sem ser a água [48].
2.5.17 Zircónia e catalisador com zircónia como precursor
A zircónia é um óxido de zircónio. Os óxidos de metais oferecem várias vantagens tais como
serem estáveis, poderem ser regenerados e serem activos.
Foram realizados vários catalisadores com ião sulfato, molibdato e tungstato. Os ensaios
realizados tiveram como condições 90 min, variando a razão molar acetona/glicerina, temperatura e
quantidade de catalisador.
A maior actividade foi obtida para o catalisador de zirconia com ião sulfato devido ao maior
número de centros activos e maior área superficial do catalisador. A conversão do glicerol para este
catalisador foi de 98%. Um aumento da quantidade de acetona aumenta a conversão mas não tem
influencia na selectividade. A conversão também aumenta com maior quantidade de catalisador, que
se deve ao facto de haver maior número de centros activos disponíveis. A selectividade não é
afectada pela quantidade de catalisador [49].
2.5.18 ZrO2-SiO2
Os óxidos de ZrO2-SiO2 providenciam simultaneamente centros ácidos de Bronstead e de
Lewis, devido às ligações Zr-O-Si ou grupos Si-OH terminais.
Foram realizados vários catalisadores variando na razão molar Zr/Si e temperatura de
calcinação e as reacções tiveram como condições acetona/glicerina de 4:1, 70ºC, 3 horas de reacção
e 2,2% (w/w) de catalisador.
A conversão e a selectividade tiveram melhores valores para a razão molar 0,5 Zr/Si, devido
ao maior número de centros activos ácidos e pelo teor de ZrO2. A conversão atinge valores de 90.2%
e a selectividade de 89%.
A temperatura de calcinação melhor foi 500ºC. Uma calcinação com temperaturas muito altas
ou muito baixas pode afectar o número de centros activos. Temperatura baixa é desvantajosa para a
formação das ligações Zr-O-Si e temperaturas altas destroem essas ligações.
Em relação às condições de reacção, a conversão aumenta com o tempo de reacção e com o
aumento da razão molar até 4:1, a partir desse valor desce devido à diminuição da concentração de
catalisador [2].

38
2.5.19 Fosfato de molibdénio com suporte SBA-15
Um catalisador de fosfato de molibdénio que não tenha suporte tem várias desvantagens tais
como baixa área superficial e baixo tamanho dos poros. Sendo assim, usa-se como suporte SBA-15
de forma a obter maior estabilidade e dispersão na fase activa. SBA-15 é um material composto por
sílica que tem grande estabilidade térmica e elevada área superficial.
Foram preparados vários catalisadores com diferentes percentagens de fosfato de
molibdénio. De entre os catalisadores estudados, o que apresenta piores resultados é o SBA-15
devido à sua pouca acidez. Para os catalisadores com fosfato de molibdénio, o melhor para a
reacção é o 40 wt% MoPO/SBA-15, com valores de 100% de conversão e 98% de selectividade. A
conversão vai aumentando até que atinge o pico com o catalisador 40 wt% MoPO/SBA-15 devido ao
aumento de centros activos ácidos, propriedades de textura e porosidade. A selectividade não varia
em todos os catalisadores porque o solketal é termodinamicamente mais estável do que o anel de 6
membros [50].
2.5.20 Zn, Cu, Ni e Co com suporte de M-AlPO4
AlPO4 é um óxido de metal ao qual tem sido dada elevada importância. No entanto, apresenta
algumas desvantagens tais como ter baixa área superficial e o colapso da sua estrutura. Sendo
assim, foram introduzidos os átomos de Zn, Cu, Ni e Co.
O catalisador que obteve melhores resultados foi o M-Ni/AlPO4, com 75,44% de rendimento e
75,12% de selectividade. A maior razão para isto acontecer é que uma maior área superficial
aumenta o número de átomos de Ni expostos na superfície do catalisador, aumentando assim o
número de centros activos ácidos. Outra razão plausível é uma acessibilidade superior por parte da
acetona e da glicerina aos centros activos[51].
2.5.21 Tabela resumo
A tabela 14 obtém um resumo de todos os catalisadores referidos na literatura, incluindo os
seus principais resultados e condições de reacção.

39
Tabela 14 - Tabela resumo dos catalisadores referidos na literatura
Catalisador Treacção
(ºC)
Wcatalisador
(g)
Razão
molar
treacção
(min)
Conversão
(%)
Selectividade
(%)
Rendimento
(%)
Oxihidróxido de Nióbio
HY-340
70 0,2 2:1
60
55 ----- -----
S4 22 ----- -----
S4-2D 43 95 -----
S4-SS 40 ----- -----
S4-2D 4:1 73 ----- -----
Oxalato de Amónio 70 0,2 2:1 60 65 95 -----
Purolite PD206 20 0,77 5:1 ----- ----- 100 95
Heteropoliácidos imobilizados
em sílica
Ácido tungstofosfórico 70 0,2 6:1 180 97 98 -----
Zeólito-β
H-β-1
28 0,12 2:1 60
86 98,5 84,7
H-β-2 38 94,6
H-β-1A 79,6 98,5 78,4
H-β-1B 75,8 98,5 74,6
Cu/H-β 67,5 98
Cu-β 71,5 98,7
Compósitos Carbono-Sílica
SCS 1/0,3
70 0,25 6:1 30
76 95 -----
SCS 1/1 79 76 -----
SCS 1/2 75 90 -----
HSCS 1/0,3 79 98 -----
HSCS 1/1 80 98 -----
HSCS 1/2 82 99 -----
Ácido p-toluenosulfónico ----- ----- 2:1 720 54,9 ----- -----
70 0,3 1:1 40 90 100 -----
Amberlyst-15
Glicerina Mamona 70 0,32 1:1 60 15 ----- -----
Glicerina Soja 70 0,32 1:1 60 45 ----- -----
Glicerina Palma 70 0,32 1:1 60 12 ----- -----
Glicerol Puro 70 0,36 2:1 60 95 ----- -----
Nanopartículas de Níquel
MWCNT
40 0,3 6:1 180
41 84 -----
Ni(=0,6)/MWCNT 63 88 -----
Ni(=1,8)/MWCNT 96 72 -----
Ni(=2,2)/MWCNT 79 87 -----
Óxido TiO2-SiO2 90 2,2 4:1 180 95 90 -----
Sílica com metais incorporados
Zr-TUD-1
80 0,025 1:1 360
46 100 -----
Hf-TUD-1 52 100 -----
Sn-MCM-1 42 100 -----
40
Tabela 14 (continuação) - Tabela resumo dos catalisadores referidos na literatura
SnO22 com Mo e W
Sn
28
-----
1:1 90
15 96 -----
WSn ----- 55 96 -----
MSn ----- 61 96 -----
Espuma celular mesoporosa
MP-MCF
40 0,07 2:1 180
70 97 -----
MP-NbMCF 80 97 -----
MP-TaMCF 77 98 -----
MP-PTaMCF 73 99 -----
Ni-Zr em CA
AC
45 0,2 8:1 180
33 81 -----
5%Ni/AC 98 86 -----
5%Zr/AC 67 63 -----
5%Ni-1%Zr/AC 100 74 -----
Fe em suporte de Al-SBA-15
Al-SBA-15 100 0,05 1:1 480
6 99 -----
Fe/Al-SBA-15 58 99 -----
ZéolitoNaA
Sem membrana ----- 0,7 5:1 1440
----- ----- 60
Com membrana ----- ----- ----- 100
Sílica modificada com ácido
sulfónico
Pr-SBA-15
70 0,25 6:1 30
70 ----- -----
Ar-SBA-15 82,5 ----- -----
Ar-SBA-15 hidrofobizado 80 ----- -----
Amberlyst-15 85,1 ----- -----
Zircónia
ZrO2
28 0,05 6:1 90
10 97 -----
WOx/ZrO2 80 97 -----
MoOx/ZrO2 88 97 -----
SO42-/ZrO2 98 97 -----
ZrO2-SiO2
0,5 Zr/Si
70 2,2 4:1 180
90,2 89 -----
1 Zr/Si 86 87 -----
2 Zr/Si 85 86 -----
Fosfato de molibénio/SBA-15
SBA-15
28 0,05 3:1 120
68 98 -----
20%MoPO/SBA-15 81 98 -----
40%MoPO/SBA-15 100 98 -----
MoPO 64 97 -----
Zn, Cu, Ni, Co/AlPO4
M-AlPO4 80 0,2 8:1 60 ----- ----- 57,37

41
M-Zn/AlPO4 ----- ----- 66,24
M-Cu/AlPO4 ----- ----- 69,37
M-Ni/AlPO4 ----- 75,12 75,44
M-Co/AlPO4 ----- ----- 67,2
3.MÉTODOS E MATERIAIS
3.1 PREPARAÇÃO DE CATALISADORES
3.1.1 Montmorilonite com tratamento ácido
A montmorilonite KSF é sintética e feita através de montmorilonite natural de fórmula química
(Na,Ca)0,33(Al,Mg)2(Si4O10). A sua estrutura cristalina é formada por várias camadas em que cada
camada é constituída por um octaedro de alumina entre duas sílicas tetraédricas. Na
montmorilonitehá centros catalíticos com tanto ácido de Lewis (associado às bordas) como ácido de
Bronstead (associado à região interlamelar), tendo esta também propriedades que lhe permite
funcionar como um catalisador eficiente e permitir uma boa troca de iões. Esta troca de iões é muito
importante porque permite modificar a estrutura química alterando propriedades físico-químicas que
podem ter elevado potencial [52][53].
O tratamento da montmorilonite com ácido permite a troca de catiões da intercamada (Na+ ou
Ca+) por H
+, a remoção de Al
3+ da camada octaédrica e tetraédrica, deixando os grupos de SiO4
tetraédricos intactos, e a redução da capacidade de troca de iões para metade, o que aumenta a
acidez do catalisador. Também ocorrem algumas modificações físicas tais como o aumento das
bordas das plaquetas do pó, mantendo-se fortemente empacotadas no centro, aumento da área
superficial e aumento do diâmetro dos poros[51][53].
Figura 27 - Esquema de tratamento ácido da montmorilonite[54]
Tabela 14 (continuação) - Tabela resumo dos catalisadores referidos na literatura
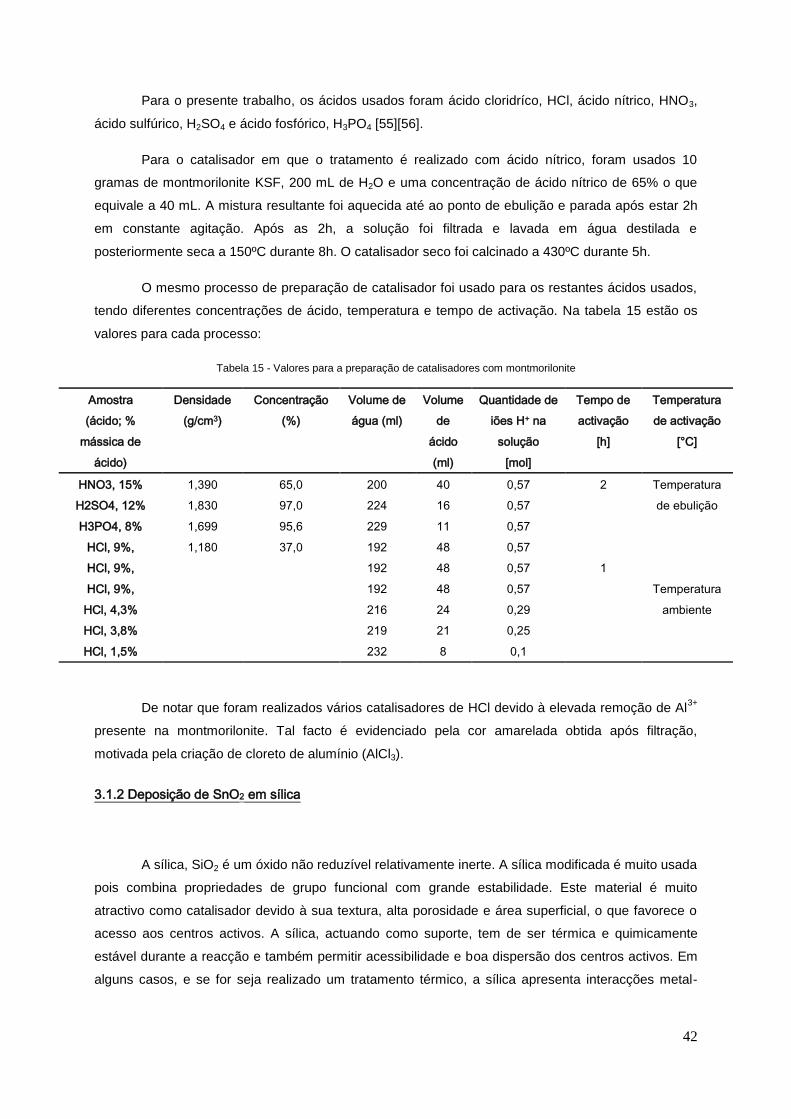
42
Para o presente trabalho, os ácidos usados foram ácido cloridríco, HCl, ácido nítrico, HNO3,
ácido sulfúrico, H2SO4 e ácido fosfórico, H3PO4 [55][56].
Para o catalisador em que o tratamento é realizado com ácido nítrico, foram usados 10
gramas de montmorilonite KSF, 200 mL de H2O e uma concentração de ácido nítrico de 65% o que
equivale a 40 mL. A mistura resultante foi aquecida até ao ponto de ebulição e parada após estar 2h
em constante agitação. Após as 2h, a solução foi filtrada e lavada em água destilada e
posteriormente seca a 150ºC durante 8h. O catalisador seco foi calcinado a 430ºC durante 5h.
O mesmo processo de preparação de catalisador foi usado para os restantes ácidos usados,
tendo diferentes concentrações de ácido, temperatura e tempo de activação. Na tabela 15 estão os
valores para cada processo:
Tabela 15 - Valores para a preparação de catalisadores com montmorilonite
Amostra
(ácido; %
mássica de
ácido)
Densidade
(g/cm3)
Concentração
(%)
Volume de
água (ml)
Volume
de
ácido
(ml)
Quantidade de
iões H+ na
solução
[mol]
Tempo de
activação
[h]
Temperatura
de activação
[°C]
HNO3, 15% 1,390 65,0 200 40 0,57 2 Temperatura
de ebulição H2SO4, 12% 1,830 97,0 224 16 0,57
H3PO4, 8% 1,699 95,6 229 11 0,57
HCl, 9%, 1,180 37,0 192 48 0,57
HCl, 9%, 192 48 0,57 1
HCl, 9%, 192 48 0,57 Temperatura
ambiente HCl, 4,3% 216 24 0,29
HCl, 3,8% 219 21 0,25
HCl, 1,5% 232 8 0,1
De notar que foram realizados vários catalisadores de HCl devido à elevada remoção de Al3+
presente na montmorilonite. Tal facto é evidenciado pela cor amarelada obtida após filtração,
motivada pela criação de cloreto de alumínio (AlCl3).
3.1.2 Deposição de SnO2 em sílica
A sílica, SiO2 é um óxido não reduzível relativamente inerte. A sílica modificada é muito usada
pois combina propriedades de grupo funcional com grande estabilidade. Este material é muito
atractivo como catalisador devido à sua textura, alta porosidade e área superficial, o que favorece o
acesso aos centros activos. A sílica, actuando como suporte, tem de ser térmica e quimicamente
estável durante a reacção e também permitir acessibilidade e boa dispersão dos centros activos. Em
alguns casos, e se for seja realizado um tratamento térmico, a sílica apresenta interacções metal-

43
suporte. Este tratamento térmico provoca redução, o que aumenta a área superficial do catalisador
alterando assim a sua actividade catalítica e a selectividade[57][58][59].
O clorato de estanho tem algumas propriedades atractivas tais como ser um sólido estável,
altamente tolerante a água e pouco corrosivo[60].
Foram então preparadas soluções com 200 mL de H2O e 25 g de SiO2. Deixou-se a solução
dissolver até estar completamente solubilizada e adicionou-se 10,6 g de SnCl2.2H2O. De seguida
deixa-se evaporar até à secura. O preparado final foi seco numa estufa durante toda a noite e
posteriormente calcinado a 430ºC durante 5h.
3.1.3 Deposição de SnO2/SO42-, em sílica
Ao pó obtido anteriormente foi adicionada uma solução de ácido sulfúrico com 2,5 mL de
H2SO4. Tal como o preparado anterior, este foi também evaporado até à secura, colocado numa
estufa para ficar completamente seco e calcinado a 430ºC durante 5h.
3.2 CARACTERIZAÇÃO DE CATALISADORES
3.2.1 Infravermelho com transformadas de Fourier (FTIR)
A espectroscopia FTIR é um método de espectroscopia infravermelho. Estuda a vibração dos
átomos quando estes são sujeitos a uma radiação e mede a quantidade de energia absorvida pela
amostra a cada comprimento de onda. Uma molécula absorve energia sempre que a frequência de
radiação iguala a frequência de vibração, desde que haja uma variação do momento dipolar[61].
Um espectrofotómetro de FTIR tem dois constituintes muito importantes, um interferómetro e
um computador. A luz de uma fonte de infravermelho incide no divisor de feixes do interferómetro.
Este divisor de feixes transmite e reflecte a luz incidente. Os feixes de luz reflectida e transmitida são
enviadas para espelhos e reflectidas para o divisor de feixes onde são recombinadas, deixam o
interferómetro, interagem com a amostra e atingem o detector. O resultado final é um interferograma,
que é o registo do sinal produzido pela combinação das múltiplas frequências possíveis de obter com
a transformada de Fourier[61].
As características de absorção são observadas no espectro de transmissão ou de absorção,
como bandas, que mostra o comprimento de onda a que a amostra absorve radiação.
Os espectros foram obtidos usando o espectrofotómetro BOMEN FTLA200-100 da ABB. Este
equipamento tem um acessório para a reflecção atenuada total (HTAR), da Pike Technologies e um

44
cristal de ZnSe. Sessenta e quatro verificações foram acumuladas em cada espectro de forma a se
obter uma razão sinal-ruído aceitável.
3.2.2 Desidratação do metanol a dimetil éter (DME)
As reacções com metanol podem ser divididas em reacções de desidratação e reacções de
oxidação. A figura 28 representa essas reacções:
Figura 28 - Esquema das reacções de oxidação e desidratação do metanol[62]
De notar que a formação de DME (CH3OCH3) é a única que apenas necessita de um passo, o
de desidratação.
Figura 29 - Representação esquemática dos produtos da reacção de oxidação do metanol em função do carácter ácido-base
dos centrosactivos[61]

45
Através da figura 29 pode notar-se que a formação de DME se deve à presença de centros
ácidos fortes. Sendo assim, a reacção de formação de DME é indicadora da acidez do catalisador[61].
A conversão do metanol e a selectividade em dimetil éter são dadas por:
𝐶𝑜𝑛𝑣𝑒𝑟𝑠ã𝑜𝑀𝑒𝑂𝐻 =𝑛𝑀𝑒𝑂𝐻(𝑖𝑛𝑖𝑐𝑖𝑎𝑖𝑠) − 𝑛𝑀𝑒𝑂𝐻(𝑓𝑖𝑛𝑎𝑖𝑠)
𝑛𝑀𝑒𝑂𝐻 (𝑖𝑛𝑖𝑐𝑖𝑎𝑖𝑠)× 100 𝐸𝑞. 1
𝑆𝑒𝑙𝑒𝑐𝑡𝑖𝑣𝑖𝑑𝑎𝑑𝑒𝐷𝑀𝐸 =𝑛𝐷𝑀𝐸𝑝𝑟𝑜𝑑𝑢𝑧𝑖𝑑𝑜𝑠 × 2
𝑛𝑀𝑒𝑂𝐻(𝑖𝑛𝑖𝑐𝑖𝑎𝑖𝑠) − 𝑛𝑀𝑒𝑂𝐻(𝑓𝑖𝑛𝑎𝑖𝑠)× 100 𝐸𝑞. 2
3.3 COMPORTAMENTO CATALÍTICO
3.3.1 Descrição da instalação
A instalação usada para a realização dos ensaios constitui várias peças fulcrais:
Placa de aquecimento;
Agitador magnético;
Balão de 500 mL;
Condensador;
Termómetro;
Cronómetro.
Na figura 30, pode ver-sea instalação montada,para a realização de um ensaio.
Figura 30 - Fotografia da instalação montada

46
3.3.2 Técnica Operatória
Os ensaios catalíticos eram iniciados pela montagem de toda a instalação. O primeiro passo
era colocar as quantidades desejadas de reagentes, glicerina (99,5% de pureza) e acetona, 10 mL de
solvente de forma a aumentar a solubilidade de acetona em glicerina, e catalisador, no balão de 500
mL. De seguida, colocava-se um set point de 58ºC no termómetro, de forma a, com o condensador,
se realizar um refluxo de acetona. Por fim, adicionava-se o agitador magnético de forma a ter uma
mistura homogénea e em constante agitação.
Quando a mistura reaccional atingia os 58ºC e havia refluxo, era iniciada a contagem do
tempo de reacção.Eram retiradas amostras de mistura reaccional a espaços regulares de tempo, de
15 em 15 minutos até perfazer 2h de reacção, e uma última amostra quando marcasse 2h30.
As condições experimentais para as várias reacções estão na tabela 16:
Tabela 16 - Condições experimentais para as diferentes reacções
Nome Catalisador %
MassaCatalisador
Rácio
molar Solvente
2 Montmorilonite 10 1,5:1 n-Hexano
3 KSF_H3PO4 8% 200ºC 2h 5 2:1 n-Hexano
4 Montmorilonite 10 1:1 n-Hexano
5 Montmorilonite 5 4:1 n-Hexano
6 Montmorilonite 10 4:1 n-Hexano
7 SnO2-SO42-
/SiO2 10 2:1 n-Hexano
8 Montmorilonite 10 2:1 n-Hexano
9 SnO2/SiO2 10 2:1 n-Hexano
10 ---------- ---------- 2:1 Etanol
11 Amberlyst 10 2:1 Etanol
12 Montmorilonite 10 2:1 Etanol
13 KSF_HCl 4,3% 26ºC 1h 5 2:1 n-Hexano
14 KSF_HCl 9% 200ºC 2h 5 2:1 n-Hexano
15 KSF_HCl 1,5% 27ºC 1h 5 2:1 n-Hexano
16 KSF_HNO3 15% 200ºC 2h 5 2:1 n-Hexano
17 KSF_H2SO4 12% 200ºC 2h 5 2:1 n-Hexano
18 Montmorilonite 5 4:1 ----------
19 KSF_HCl 9% 200ºC 1h 5 2:1 n-Hexano
20 KSF_HCl 3,8% 27ºC 1h 5 2:1 n-Hexano
21 KSF_HCl 9% 27ºC 1h 5 2:1 n-Hexano
22 Amberlyst 5 4:1 ----------
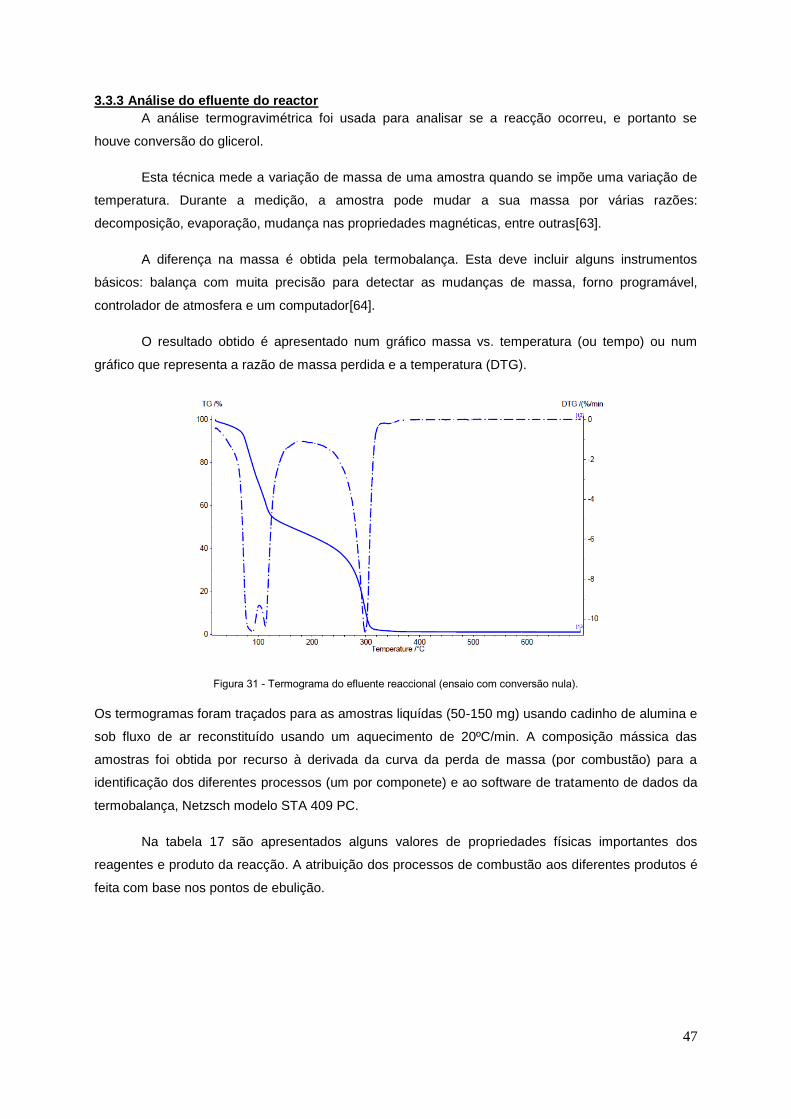
47
3.3.3 Análise do efluente do reactor
A análise termogravimétrica foi usada para analisar se a reacção ocorreu, e portanto se
houve conversão do glicerol.
Esta técnica mede a variação de massa de uma amostra quando se impõe uma variação de
temperatura. Durante a medição, a amostra pode mudar a sua massa por várias razões:
decomposição, evaporação, mudança nas propriedades magnéticas, entre outras[63].
A diferença na massa é obtida pela termobalança. Esta deve incluir alguns instrumentos
básicos: balança com muita precisão para detectar as mudanças de massa, forno programável,
controlador de atmosfera e um computador[64].
O resultado obtido é apresentado num gráfico massa vs. temperatura (ou tempo) ou num
gráfico que representa a razão de massa perdida e a temperatura (DTG).
Figura 31 - Termograma do efluente reaccional (ensaio com conversão nula).
Os termogramas foram traçados para as amostras liquídas (50-150 mg) usando cadinho de alumina e
sob fluxo de ar reconstituído usando um aquecimento de 20ºC/min. A composição mássica das
amostras foi obtida por recurso à derivada da curva da perda de massa (por combustão) para a
identificação dos diferentes processos (um por componete) e ao software de tratamento de dados da
termobalança, Netzsch modelo STA 409 PC.
Na tabela 17 são apresentados alguns valores de propriedades físicas importantes dos
reagentes e produto da reacção. A atribuição dos processos de combustão aos diferentes produtos é
feita com base nos pontos de ebulição.
48
Tabela 17 - Propriedades físicas de reagentes e produtos de reacção[28][65][66][67][68][69]
Temperatura de Ebulição (ºC)
Densidade (g.cm-1
) Viscosidade (Pa.s)
Glicerol 290 1,261 1,5 a 20ºC Acetona 56 0,791 2,95x10
-3 a 25ºC
n-Hexano 68 0,6548 3x10-3
a 25ºC Solketal 190 1,06 1,1x10
-2 a 20ºC
2,2-dimetil-1,3-dioxan-5-ol
204 1,1 --------
4.RESULTADOS EXPERIMENTAIS DA CARACTERIZAÇÃO DE CATALISADORES
4.1 Infravermelho com transformada de Fourier (FTIR)
Obtiveram-se os espectros FTIR para os vários catalisadores, frescos e após reacção. Neste capítulo,
esses espectros são estudados. Note-se que, em anexo, foram colocadas figuras que têm, em
destaque, o intervalo de comprimentos de onda das bandas mais relevantes.
Catalisadores frescos
O primeiro catalisador fresco a ser analisado é o de Amberlyst, uma resina de troca catiónica.
Figura 32– EspectrodeFTIR para catalisador fresco de amberlyst
5001000150020002500300035004000
Ab
so
rvân
cia
(a.a
)
Número de onda (cm-1)

49
O espectro de Amberlyst apresenta 4 bandas de maior intensidade[70]:
3410cm-1
: Ligações -OH relacionadas com grupos hidroxila na superfície do
catalisador, nos grupos -SO3H;
2910 cm-1
: Elongação anti-simétrica de vibração de C-H em grupos –CH2
1630 cm-1
: Vibrações no anel de benzeno e nos grupos C-H no anel de benzeno
estireno;
1110cm-1
: Representa a vibração de elongação simétrica dos grupos -SO3- ;
1030cm-1
:Representa a vibração de elongação anti-simétrica dos grupos -SO3-
Em relação ao espectro dos catalisadores de montmorilonite e catalisadores com tratamento
ácido de montmorilonite.
Figura 33 - Espectro de FTIR dos catalisadores frescos KSF e KSF com tratamento ácido
Começa por analisar-se a montmorilonite pura, sem qualquer tratamento[53][71][55]:
1635 cm-1
: Vibração angular de moléculas de água adsorvidas, H-O-H;
1110 cm-1
: Vibração de ligação Al-OH
1050 cm-1
: Elongação Si-O;
910 cm-1
: Vibração de deformação de Al-OH-Al;
800 cm-1
: Ligação Si-O e Al-O.
680 cm-1
: Ligação Al-O-Si
De forma a analisar melhor os espectros foi usada a equação de Kubelka-Munk. Assume-se
que a luz incidente é difusa, com distribuição isotrópica, as partículas estão distribuídas
60011001600
Ab
so
rvân
cia
(a.a
.)
Número de onda (cm-1)
KSF_H3PO_8%_200C_2h
KSF_HCl_1,52%_26C_1h
KSF_HCl_4,3%_26C_2h
KSF_HCL3,8%_26C_1h
KSF_HCL9%_200C_1h
KSF_HCL9%_200C_2h
KSF_HNO3_15%_200C_2h
KSF_HCL9%_27C_1h
KSF_H2SO4
KSF_raw
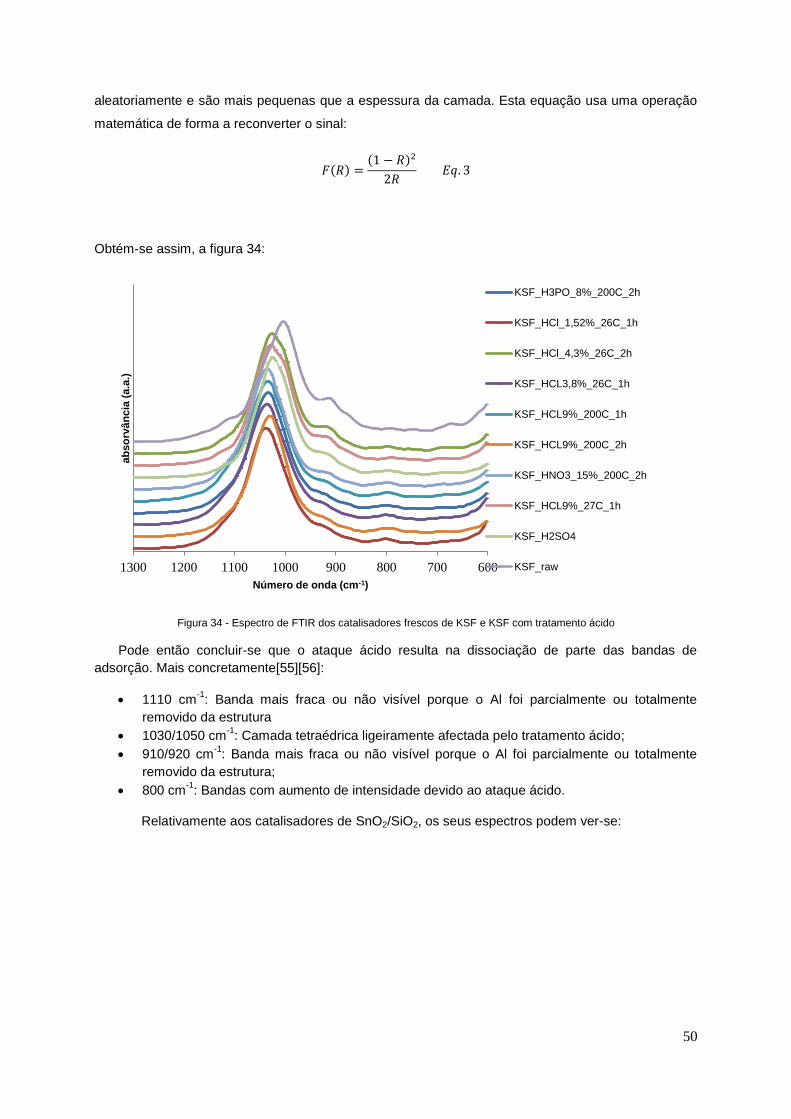
50
aleatoriamente e são mais pequenas que a espessura da camada. Esta equação usa uma operação
matemática de forma a reconverter o sinal:
𝐹(𝑅) =(1 − 𝑅)2
2𝑅 𝐸𝑞. 3
Obtém-se assim, a figura 34:
Figura 34 - Espectro de FTIR dos catalisadores frescos de KSF e KSF com tratamento ácido
Pode então concluir-se que o ataque ácido resulta na dissociação de parte das bandas de
adsorção. Mais concretamente[55][56]:
1110 cm-1
: Banda mais fraca ou não visível porque o Al foi parcialmente ou totalmente
removido da estrutura
1030/1050 cm-1
: Camada tetraédrica ligeiramente afectada pelo tratamento ácido;
910/920 cm-1
: Banda mais fraca ou não visível porque o Al foi parcialmente ou totalmente
removido da estrutura;
800 cm-1
: Bandas com aumento de intensidade devido ao ataque ácido.
Relativamente aos catalisadores de SnO2/SiO2, os seus espectros podem ver-se:
6007008009001000110012001300
ab
so
rvân
cia
(a.a
.)
Número de onda (cm-1)
KSF_H3PO_8%_200C_2h
KSF_HCl_1,52%_26C_1h
KSF_HCl_4,3%_26C_2h
KSF_HCL3,8%_26C_1h
KSF_HCL9%_200C_1h
KSF_HCL9%_200C_2h
KSF_HNO3_15%_200C_2h
KSF_HCL9%_27C_1h
KSF_H2SO4
KSF_raw
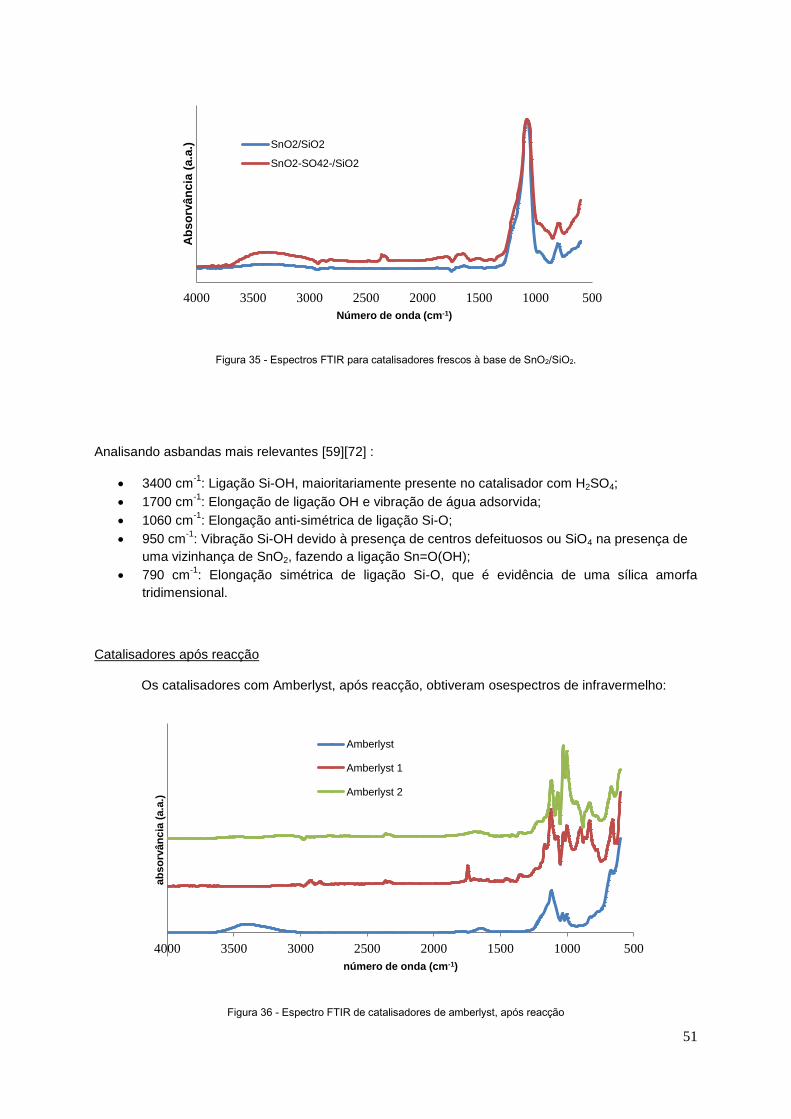
51
Figura 35 - Espectros FTIR para catalisadores frescos à base de SnO2/SiO2.
Analisando asbandas mais relevantes [59][72] :
3400 cm-1
: Ligação Si-OH, maioritariamente presente no catalisador com H2SO4;
1700 cm-1
: Elongação de ligação OH e vibração de água adsorvida;
1060 cm-1
: Elongação anti-simétrica de ligação Si-O;
950 cm-1
: Vibração Si-OH devido à presença de centros defeituosos ou SiO4 na presença de
uma vizinhança de SnO2, fazendo a ligação Sn=O(OH);
790 cm-1
: Elongação simétrica de ligação Si-O, que é evidência de uma sílica amorfa
tridimensional.
Catalisadores após reacção
Os catalisadores com Amberlyst, após reacção, obtiveram osespectros de infravermelho:
Figura 36 - Espectro FTIR de catalisadores de amberlyst, após reacção
5001000150020002500300035004000
Ab
so
rvâ
nc
ia (
a.a
.)
Número de onda (cm-1)
SnO2/SiO2
SnO2-SO42-/SiO2
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Amberlyst
Amberlyst 1
Amberlyst 2
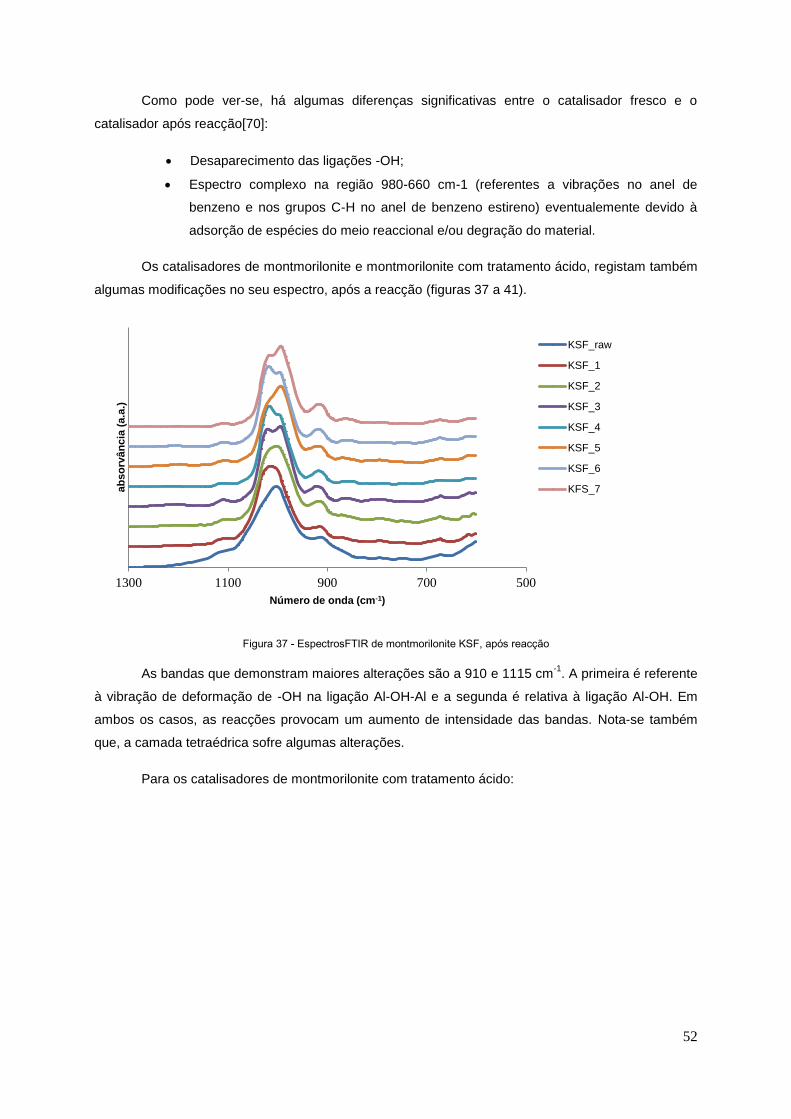
52
Como pode ver-se, há algumas diferenças significativas entre o catalisador fresco e o
catalisador após reacção[70]:
Desaparecimento das ligações -OH;
Espectro complexo na região 980-660 cm-1 (referentes a vibrações no anel de
benzeno e nos grupos C-H no anel de benzeno estireno) eventualemente devido à
adsorção de espécies do meio reaccional e/ou degração do material.
Os catalisadores de montmorilonite e montmorilonite com tratamento ácido, registam também
algumas modificações no seu espectro, após a reacção (figuras 37 a 41).
Figura 37 - EspectrosFTIR de montmorilonite KSF, após reacção
As bandas que demonstram maiores alterações são a 910 e 1115 cm-1
. A primeira é referente
à vibração de deformação de -OH na ligação Al-OH-Al e a segunda é relativa à ligação Al-OH. Em
ambos os casos, as reacções provocam um aumento de intensidade das bandas. Nota-se também
que, a camada tetraédrica sofre algumas alterações.
Para os catalisadores de montmorilonite com tratamento ácido:
50070090011001300
ab
so
rvân
cia
(a.a
.)
Número de onda (cm-1)
KSF_raw
KSF_1
KSF_2
KSF_3
KSF_4
KSF_5
KSF_6
KFS_7

53
Figura 38 - Espectro de FTIR para KSF com tratamento ácido de H2SO4, após reacção
Na reacção com ácido sulfúrico, a alteração mais evidente no catalisador é que após a
reacção há grupos -OH provenientes da água que se formou durante a reacção (banda a 3400 cm-1
).
Nas reacções com ácido nítrico e ácido fosfórico, as alterações são semelhantes às que
aconteceram na reacção com ácido sulfúrico. Os seus espectros estão em anexo.
Para os catalisadores de montmorilonite com tratamento ácido de ácido clorídrico, tendo em
conta as diferentes concentrações:
Figura 39 - Espectro de FTIR para KSF com tratamento ácido de HCl 1,52%, após reacção
Para o catalisador com concentração 1,52%, a alteração mais clara é a 910 cm-1
em que se
forma a banda referente a vibração de deformação de -OH na ligação Al-OH-Al.
O catalisador com concentração 3,8% apresenta apenas a mesma alteração no espectro
(vide Anexo).
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_H2SO4
KSF_H2SO4 1
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_1,52%_26C_1h
HCl 1,5% 1
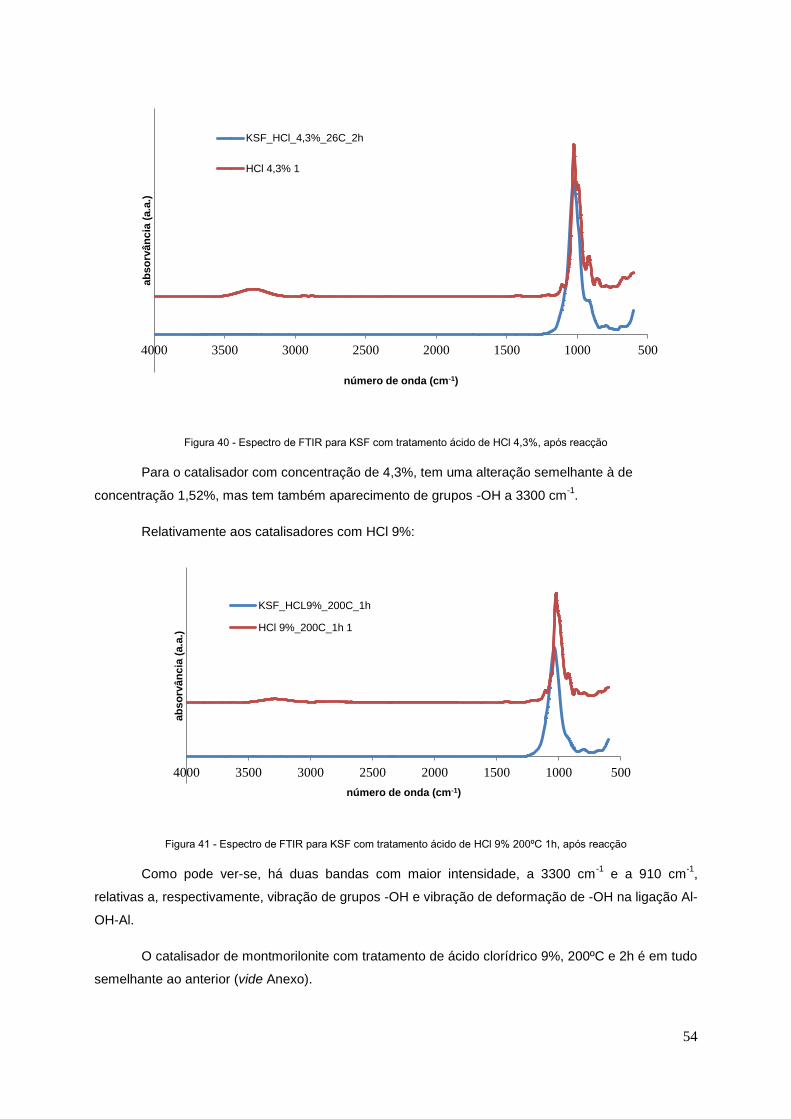
54
Figura 40 - Espectro de FTIR para KSF com tratamento ácido de HCl 4,3%, após reacção
Para o catalisador com concentração de 4,3%, tem uma alteração semelhante à de
concentração 1,52%, mas tem também aparecimento de grupos -OH a 3300 cm-1
.
Relativamente aos catalisadores com HCl 9%:
Figura 41 - Espectro de FTIR para KSF com tratamento ácido de HCl 9% 200ºC 1h, após reacção
Como pode ver-se, há duas bandas com maior intensidade, a 3300 cm-1
e a 910 cm-1
,
relativas a, respectivamente, vibração de grupos -OH e vibração de deformação de -OH na ligação Al-
OH-Al.
O catalisador de montmorilonite com tratamento de ácido clorídrico 9%, 200ºC e 2h é em tudo
semelhante ao anterior (vide Anexo).
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_4,3%_26C_2h
HCl 4,3% 1
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_200C_1h
HCl 9%_200C_1h 1
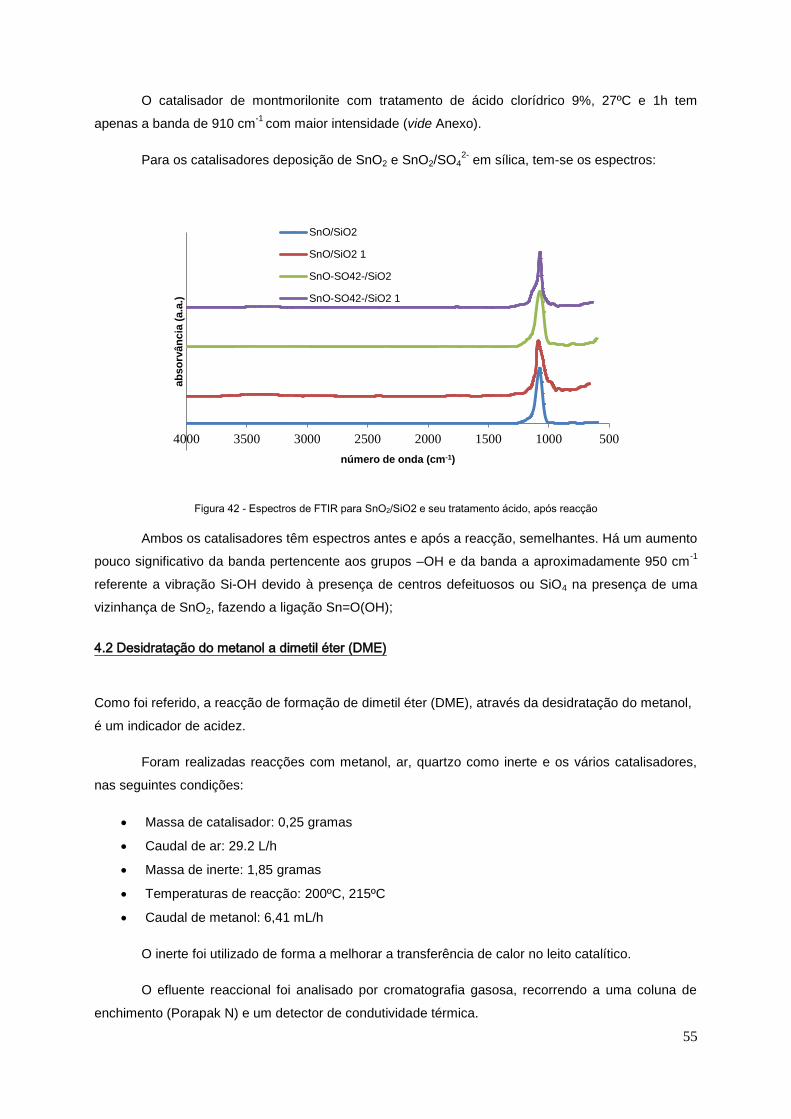
55
O catalisador de montmorilonite com tratamento de ácido clorídrico 9%, 27ºC e 1h tem
apenas a banda de 910 cm-1
com maior intensidade (vide Anexo).
Para os catalisadores deposição de SnO2 e SnO2/SO42-
em sílica, tem-se os espectros:
Figura 42 - Espectros de FTIR para SnO2/SiO2 e seu tratamento ácido, após reacção
Ambos os catalisadores têm espectros antes e após a reacção, semelhantes. Há um aumento
pouco significativo da banda pertencente aos grupos –OH e da banda a aproximadamente 950 cm-1
referente a vibração Si-OH devido à presença de centros defeituosos ou SiO4 na presença de uma
vizinhança de SnO2, fazendo a ligação Sn=O(OH);
4.2 Desidratação do metanol a dimetil éter (DME)
Como foi referido, a reacção de formação de dimetil éter (DME), através da desidratação do metanol,
é um indicador de acidez.
Foram realizadas reacções com metanol, ar, quartzo como inerte e os vários catalisadores,
nas seguintes condições:
Massa de catalisador: 0,25 gramas
Caudal de ar: 29.2 L/h
Massa de inerte: 1,85 gramas
Temperaturas de reacção: 200ºC, 215ºC
Caudal de metanol: 6,41 mL/h
O inerte foi utilizado de forma a melhorar a transferência de calor no leito catalítico.
O efluente reaccional foi analisado por cromatografia gasosa, recorrendo a uma coluna de
enchimento (Porapak N) e um detector de condutividade térmica.
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
SnO/SiO2
SnO/SiO2 1
SnO-SO42-/SiO2
SnO-SO42-/SiO2 1

56
Os valores de rendimento obtidos podem ser observados na figura 43:
Figura 43 - Rendimento de reacção de oxidação de metanol a DME, a 200 e 215 ºC
Através da figura 43, pode evidenciar-se que o aumento de temperatura não afecta de forma
significativa o rendimento, embora apresente constantemente um pequeno aumento.
Deve referir-se também que, em alguns catalisadores, KSF HCl 9% 27ºC 1h, KSFHCl 3,8%,
KSFHCl 1,5% e KSFHNO3 15%, o tratamento ácido torna os catalisadores com maior acidez em
relação à montmoriloniteKSF.
Os catalisadores com melhores valores obtidos de rendimento foram SnO2-SO42-
/SiO2 e
montmorilonite com tratamento ácido de HCl 1,5% e HNO3 15%. Pode então concluir-se que estes
catalisadores têm características ácidas fortes, favoráveis para a reacção de acetona com a glicerina.
É preciso notar que o ensaio não foi feito para o catalisador SnO2/SiO2 porque ocorreu uma
avaria na fita de aquecimento do cromatógrafo. Seria, no entanto, importante perceber a diferença de
acidez entre o catalisador com e sem o tratamento ácido.
0
2
4
6
8
10
12
14
ren
dim
en
to (
%)
catalisadores

57
5. COMPORTAMENTO CATALÍTICO
5.1 Espectroscopia de Infravermelho com transformada de Fourier
Para iniciar a análise dos espectros das amostras colhidas no final de cada reacção, mostram-se os
espectros de glicerina, acetona e solketal.
5.1.1 Glicerina
Figura 44 - Espectro de FTIR de glicerina
As bandas e as suas ligações correspondentes estão na tabela 18[74].
Tabela 18 - Bandas e ligações correspondentes do espectro de glicerina
Banda (cm-1
) Ligação correspondente
3400 O-H 2900 C-H 1400 C-O 1050 C-O 800 C-H
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)

58
5.1.2 Acetona
As suas bandas e ligações estão na tabela 19[74]:
Tabela 19 - Bandas e ligações correspondentes do espectro de infravermelho de acetona
Banda (cm-1
) Ligação correspondente
3000 C-H 1700 C=O 1400 α-CH2 1300 α-CH3 1100 C-C-C
5.1.3 Solketal
Figura 45 - Espectro de FTIR de acetona [73]
Figura 46 - Espectro de FTIR de solketal [73]
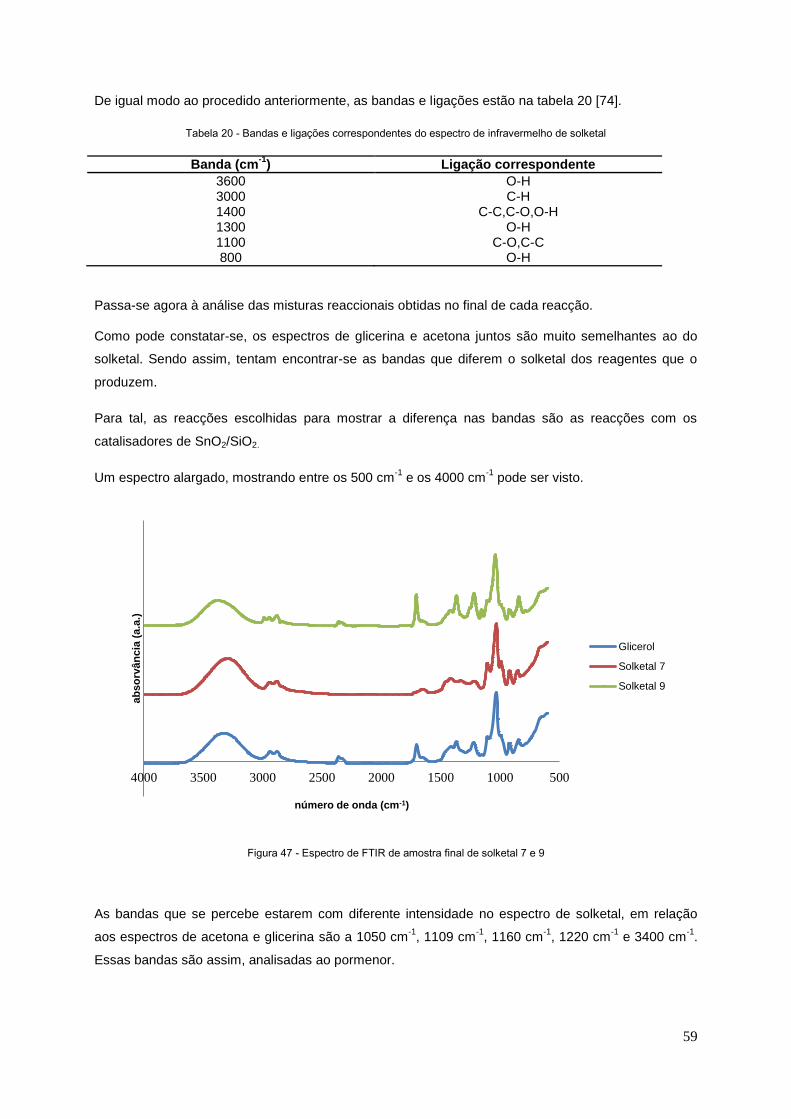
59
De igual modo ao procedido anteriormente, as bandas e ligações estão na tabela 20 [74].
Tabela 20 - Bandas e ligações correspondentes do espectro de infravermelho de solketal
Banda (cm-1
) Ligação correspondente
3600 O-H 3000 C-H 1400 C-C,C-O,O-H 1300 O-H 1100 C-O,C-C 800 O-H
Passa-se agora à análise das misturas reaccionais obtidas no final de cada reacção.
Como pode constatar-se, os espectros de glicerina e acetona juntos são muito semelhantes ao do
solketal. Sendo assim, tentam encontrar-se as bandas que diferem o solketal dos reagentes que o
produzem.
Para tal, as reacções escolhidas para mostrar a diferença nas bandas são as reacções com os
catalisadores de SnO2/SiO2.
Um espectro alargado, mostrando entre os 500 cm-1
e os 4000 cm-1
pode ser visto.
Figura 47 - Espectro de FTIR de amostra final de solketal 7 e 9
As bandas que se percebe estarem com diferente intensidade no espectro de solketal, em relação
aos espectros de acetona e glicerina são a 1050 cm-1
, 1109 cm-1
, 1160 cm-1
, 1220 cm-1
e 3400 cm-1
.
Essas bandas são assim, analisadas ao pormenor.
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 7
Solketal 9

60
Figura 48- Espectro de FTIR de amostra final de solketal 7 e 9, entre 950 e 1150 cm-1
Como pode ver-se, o espectro da glicerina está ligeiramente mais à direita que o de solketal 9, esse
aparentaassim,apresentarsolketal. O espectro de solketal 7 é em tudo semelhante ao de glicerina.
Figura 49 - Espectro de FTIR para amostra final de solketal 7 e 9, entre 1050 e 1190 cm-1
Neste caso, o espectro de solketal 9 apresenta uma diminuição da intensidade na banda, enquanto
que o de solketal 7 parece apresentar até um aumento de intensidade em relação à glicerina.
9501000105011001150
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 7
Solketal 9
10501070109011101130115011701190
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 7
Solketal 9

61
Figura 50 - Espectro de FTIR para amostra final de solketal 7 e 9, entre 1100 e 1330 cm-1
Há o aparecimento de uma banda por volta dos 1160 cm-1
, diferenciando o solketal 9 do solketal 7 e
glicerina. A banda a 1220 cm-1
apresenta maior intensidade para solketal 9.
Figura 51 - Espectro de FTIR de amostra final de solketal 7 e 9, entre 3000 e 3800 cm-1
Neste caso, a banda de solketal apresenta-se mais à esquerda, a 3390 cm-1
enquanto que a de
glicerina está a 3320 cm-1
.
As amostras das reacções que aparentam ter solketal foram solketal 4, 5, 9, 11, 13, 14, 16, 17, 18,
20, 21 e 22.
11001150120012501300
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 7
Solketal 9
300031003200330034003500360037003800
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 7
Solketal 9

62
5.2 Termogravimetria
Antes de começar a analisar os resultados, mostra-se um ensaio de termogravimetriarealizado para
uma reacção que demonstra maioritariamente glicerol puro[75]:
Figura 52 – Termograma para mistura reaccional com conversão nula.
As reacções que apresentaram melhores resultados de conversão estão apresentadas na
tabela 21:
Tabela 21 - Reacções que apresentaram conversão, por termogravimetria
Nome Catalisador Razão molar
Acetona/Glicerina
%
Catalisador Solvente
2 Montmorilonite 1,5:1 10% n-Hexano
4 Montmorilonite 1:1 10% n-Hexano
7 SnO2-SO42-
/SiO2 2:1 10% n-Hexano
8 Montmorilonite 2:1 10% n-Hexano
9 SnO2/SiO2 2:1 10% n-Hexano
14 HCl 9% 200ºC 2h 2:1 10% n-Hexano
19 HCl 9% 200ºC 1h 2:1 10% n-Hexano
20 HCl 3,8% 27ºC 1h 2:1 10% n-Hexano
21 HCl 9% 27ºC 1h 2:1 10% n-Hexano
São os termogramas destes catalisadores que se apresentam, as restantes estão em anexo.
As primeiras reacções a serem analisadas são as relativas ao catalisador
MontmoriloniteKSF,solketal 2,4 e 8.

63
Figura 53 - Termograma para solketal 2
Figura 54 - Termograma para solketal 4
Figura 55 - Termograma para solketal 8
Para as duas primeiras reacções, solketal 2 e solketal 4, não há presença de glicerol e, sendo
assim, a conversão é completa. A banda mais visível é referente a um produto da reacção que não

64
ésolketal, por volta dos 250ºC, e tem variação de massa de 71,35%, 56,47% e 71,17%,
respectivamente, para solketal, 2, 4 e 8. Essa banda podem ser eventuais dimeros ou trimeros. As
bandas anteriores que se podem observar podem ser produtos da hidrólise do solketal em solketal 2
e 8 e água, em solketal 4.
Relativamente as reacções com os catalisadores de SnO2/SiO2, solketal 7 e solketal 9:
Figura 56 - Termograma para solketal 7
Figura 57 - Termograma para solketal 9
A reacção solketal 7 apresenta conversão completa de glicerol, com variação de massa de
46,14% para solketal. O outro produto presente pode ser um produto de hidrólise, que ocorre na
presença de excesso de água e catalisador muito ácido[76]. A reacção solketal 9 apresenta 3 bandas,
a primeira, a 100ºC referente a água, a segunda solketal, por volta de 200ºC com variação de massa
de 36,36% e a terceira glicerol, a 290ºC, com 36,6% de variação de massa.
Para o catalisador de HCl 3,8% 27ºC 1h obteve-se o seguinte ensaio termogravimétrico:

65
Figura 58 - Termograma para solketal 20
Pode ver-se que esta reacção apresenta muita glicerina, portanto a sua conversão não é
completa, notando-se também a presença de solketal. A variação de massa da glicerina e do solketal
é, respectivamente, 36,16% e 15,37%. As outras duas bandas que podem observar-se são alusivas à
água e a produtos de hidrólise.
No que diz respeito aos catalisadores de HCl 9%:
Figura 59 - Termograma para solketal 14
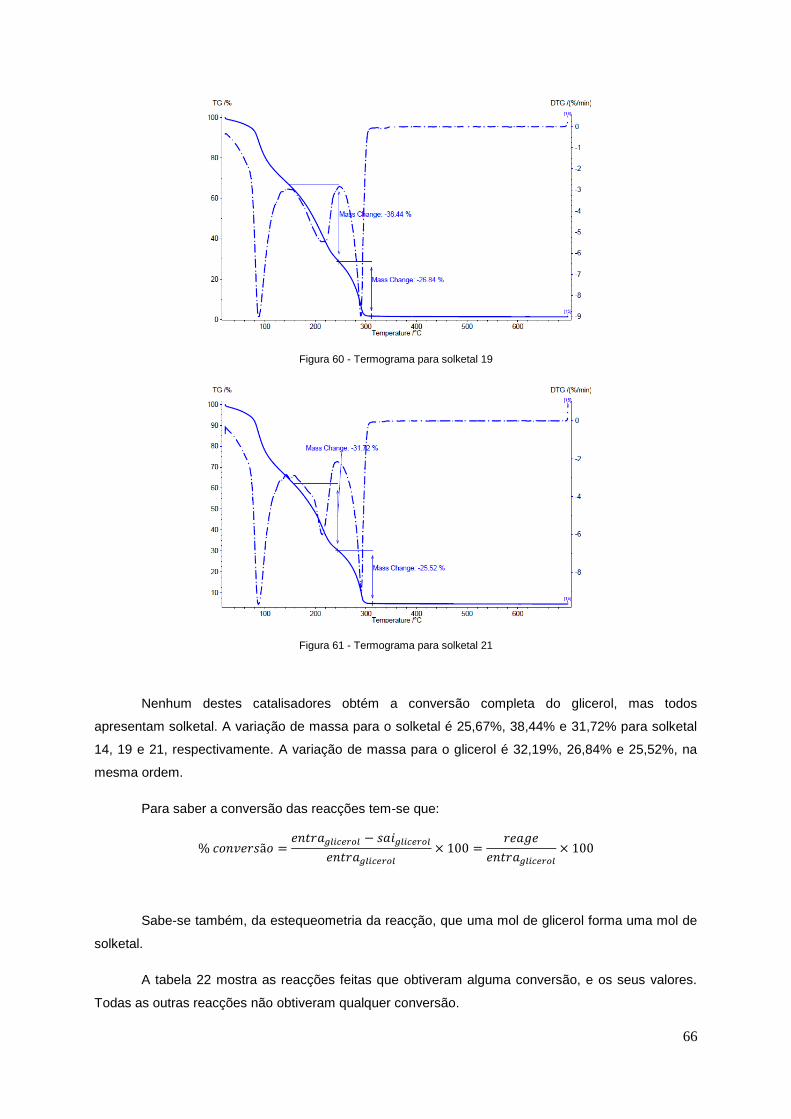
66
Figura 60 - Termograma para solketal 19
Figura 61 - Termograma para solketal 21
Nenhum destes catalisadores obtém a conversão completa do glicerol, mas todos
apresentam solketal. A variação de massa para o solketal é 25,67%, 38,44% e 31,72% para solketal
14, 19 e 21, respectivamente. A variação de massa para o glicerol é 32,19%, 26,84% e 25,52%, na
mesma ordem.
Para saber a conversão das reacções tem-se que:
% 𝑐𝑜𝑛𝑣𝑒𝑟𝑠ã𝑜 =𝑒𝑛𝑡𝑟𝑎𝑔𝑙𝑖𝑐𝑒𝑟𝑜𝑙 − 𝑠𝑎𝑖𝑔𝑙𝑖𝑐𝑒𝑟𝑜𝑙
𝑒𝑛𝑡𝑟𝑎𝑔𝑙𝑖𝑐𝑒𝑟𝑜𝑙
× 100 =𝑟𝑒𝑎𝑔𝑒
𝑒𝑛𝑡𝑟𝑎𝑔𝑙𝑖𝑐𝑒𝑟𝑜𝑙
× 100
Sabe-se também, da estequeometria da reacção, que uma mol de glicerol forma uma mol de
solketal.
A tabela 22 mostra as reacções feitas que obtiveram alguma conversão, e os seus valores.
Todas as outras reacções não obtiveram qualquer conversão.

67
Tabela 22 - Valores de conversão para cada reacção, por termogravimetria
% Variação de massa
Reacção Solketal Glicerina nglicerina(mol) nsolketal (mol) Conversão (%) 2 71,35* 0 0 0,54 100 4 56,74* 0 0 0,43 100 7 46,14 0 0 0,34 100 8 71,17* 7,5 0,08 0,54 87 9 36,36 36,6 0,4 0,28 41
14 25,67 32,19 0,35 0,19 36 19 38,44 26,84 0,29 0,29 50 20 15,37 36,16 0,39 0,12 23 21 31,72 25,52 0,28 0,24 46
* Dímero ou trímero de solketal

68
6. CONCLUSÕES
A indústria de biodiesel produz grandes quantidades de glicerina que o mercado tradicional
não consegue absorver. Por outro lado, o elevado preço das matérias-primas exige que sejam
valorizados todos os sub-produtos do processo de produção de biodiesel, de forma a tornar o preço
de produção do biodiesel, competitivo. A glicerina proveniente do biodiesel pode ser valorizada
recorrendo a diferentes processos. A acetalização com acetona é um dos processos para a
valorização da glicerina. O processo produz solketal que é um químico muito versátil, com aplicações
para a indústria cosmética, farmacêutica, e como aditivio para diesel e combústivel jet.
A reacção a temperatura de ebulição da acetona exige a presença de catalisador e é
facilitada pela presença de um solvente que melhore a miscibilidade dos componentes da mistura
reaccional. Neste contexto, estudou-se a produção de solketal usando catalisadores com carácter
ácido na presença de solvente (n-hexano ou etanol). Os catalisadores foram preparados por
desaluminação na presença de ácidos inorgânicos (HNO3, H2SO4, H3PO4 e HCl) de montmorilonite
KSF. Estudou-se o efeito da natureza do ácido, temperatura no tratamento e duração do tratamento.
Verificou-se que o ácido clorídrico é o mais eficiente para desaluminação. Testaram-se ainda
catalisadores peparados por deposição de SnO2 em sílica modificados e não modificados com SO42-
.
Usou-se o amberlyst 15 como catalisador ácido de referência.
Os catalisadores preparados foram catacterizados por FTIR antes e após reacção. Para as
amostras de montmorilonite submetidas a tratamento ácido, os espectros FTIR confirmam a remoção
parcial de alumínio. As amostras dos mesmos catalisadores após reacção apresentam ligeiras
alterações espectrais devido a algumas espécies de mistura reaccional que permanecem adsorvidas
na superfície.
A acidez superficial dos sólidos sintetizados foi caracterizada reccorendo à reacção de
desidratação do metanol a dimetiléter em fase vapor. O catalisador de SnO2/SiO2 modificado com
SO42-
revelou ser o mais ácido. Por outro lado, o tratamento com HCl e HNO3 da montmorilonite
beneficia a acidez das amostras relativamente à montmorilonite original.
Os dados do comportamento catalítico para a acetalização mostram que a acidez tem um
papel fundamental no processo catalítico, contudo, catalisadores muito ácidos promovem a hidrólise
do solketal formado.

69
7. BIBLIOGRAFIA
[1] C.Fan, C.Xu, C.Liu, Z.Huang, J, Liu, Z.Ye, Ketalization of glycerol with acetone to O-Heterocyclic
compounds over ZrO2-SiO2 solid acid catalysts, Heterocycles, 85 (2012), pp. 2977-2986.
[2] C.Fan, C.Xu, C.Liu, Z.Huang, J, Liu, Z.Ye, Ketalization of glycerol with acetone to O-Heterocyclic
compounds over ZrO2-SiO2 solid acid catalysts, Heterocycles, 85 (2012), pp. 2977-2986.
[3] M.S. Khayoon, B.H. Hameed, Solventlessacetalization of glycerol with acetone to fuel oxygenates
over Ni–Zr supported on mesoporous activated carbon catalyst, Applied Catalysis A: General, 464–
465 (2013), pp.191-199.
[4] H.D. Willauer, R. Ananth, J.B. Hoover, G.W. Mushrush, F.W. Williams. Federal Aviation
Administration Fire Safety. [Online] [Cited: Dezembro 1, 2015.]
http://www.fire.tc.faa.gov/2004Conference/files/fuel/H.%20Willaue_Chemicals_for_reducing_fuel_flam
mabilty.pdf.
[5] M.C. Pereira, Energia e ambiente num mundo com muita gente, Nação e Defesa, nº122 (2009),
pp.25-41.
[6] F.D. Santos, Os desafios ambientais criados pela aceleração do pós-guerra, Nação e Defesa,
nº122 (2009), pp.61-78.
[7] F.D. Santos, The Contemporary Situation, 3, in Humans on Earth: From origins to possible futures,
pp.121, Springer, Portugal, 2010.
[8] R. Curley, in Fossil fuels-Energy: past, present and future, Britannica Educational Publishing, New
York, 2012.
[9] J.F. de Carvalho, Combustíveis Fósseis e Insustentabilidade, Energia, Ambiente e
Sociedade/Artigos, 2014, pp.30-33.
[10] G.T. Miller, S. Spoolman, Energy, 13, in Environmental Science: Problems, concepts and
solutions, pp.279, Thomson Corporation, 2008.
[11]International Energy Agency.[Online] 2015. [Citação: 5 de October de 2015.]
http://www.iea.org/topics/renewables/.
[12] V. Nelson, in Introduction to Renewable Energy, CRC Press Taylor & Francis Group, New
Mexico, 2011.
[13] InternationalEnergyAgency. [Online] [Citação: 2 de Abril de 2015.]
https://www.iea.org/publications/freepublications/publication/key-world-energy-statistics-2014.html.
[14] Agência Portuguesa do Ambiente. [Online] [Citação: 17 de Março de 2015.]
http://www.apambiente.pt.

70
[15] Direcção Geral de Engenharia e Geologia. [Online] [Citação: 12 de Abril de 2015.]
http://www.dgeg.pt/.
[16] L. A.H. Nogueira, Does biodiesel make sense?,Energy, 36 (2011), pp. 3659-3666.
[17] F.Girio, C.Matos, Laboratório Nacional de Engenharia e Geologia. [Online] 24 de Setembro de
2010. [Citação: 31 de Março de 2015.] http://www.lneg.pt/.
[18] C.M. Drapcho, N.P. Nhuan, T.H. Walker, Biodiesel, 6, in Biofuels engineering process technology,
pp.197, The McGraw-Hill Companies, Inc, South Carolina ,2008
[19] R. Dionysio, F. Meirelles, Coordenação Central de Educação à Distância - PUC. [Online]
[Citação: 30 de Março de 2015.] http://web.ccead.puc-
rio.br/condigital/mvsl/Sala%20de%20Leitura/conteudos/SL_combustiveis.pdf.
[20] A. N. Phan, T. M. Phan, Biodiesel production from waste cooking oils, Fuel, 87 (2008), pp. 3490-
3496.
[21] M. Ahmad, M.A. Khan, M. Zafar, S. Sultana, Introduction to Biodiesel, 1, in Practical handbook on
biodiesel production and properties, pp.1, CRC Press Taylor & Francis Group, Islamabad, 2013.
[22] J.C.J. Bart, N. Palmeri, S. Cavallaro, Vegetable oil formulations for utilisation as biofuels, 4 in
Biodiesel science & technology, pp.114, Woodhead Publishing Limited, 2010.
[23] U.S. Energy Information Administration. [Online] [Citação: 20 de Abril de 2015.]
http://www.eia.gov/cfapps/ipdbproject/iedindex3.cfm?tid=79&pid=81&aid=1&cid=regions&syid=2008&
eyid=2012&unit=TBPD.
[24] EUR-Lex: Access to European Union Law. [Online] 7 de Dezembro de 2005. [Citação: 17 de
Março de 2015.] http://eur-lex.europa.eu/legal-
content/PT/TXT/?uri=uriserv:OJ.C_.2006.195.01.0069.01.POR
[25] Energias Renováveis em Portugal. s.l. : Ministério da Economia e da Inovação, 2007.
[26] P.S.Bisen, B.S.Sanodiya, G.S.Thakur, R.K. Baghel, G.B.K.S. Prasad, Biodiesel production with
special emphasis on lipase-catalysed transesterification, Biotechnology Letter, 32 (2010), pp. 1019-
1030
[27] OECD FAO Agricultural Outlook 2011-2020. Brasil : OECD-FAO, 2011.
[28] M. Pagliaro, M. Rossi, The Future of Glycerol, RSC Publishing, Itália, 2010
[29] Glycerine: An overview. s.l. : The Soap and Detergent Association, 1990.
[30] Manufacturing Processes. [Online] [Citação: 17 de Março de 2015.]
http://www.sbioinformatics.com/design_thesis/Glycerol/Glycerol_Methods-2520of-2520Production.pdf.

71
[31] V.Calvino-Casilda, K.Stawicka, M.Trejda, M.Ziolek, M.A.Bañares, Real-time Raman monitoring
and control of the catalytic acetalization of glycerol with acetone over modified mesoporous cellular
foams, The Journal of Physical Chemistry C, 118 (2014), pp. 10780-10791.
[32] T. E. Souza, I. D. Padula, M. M.G. Teodoro, P.Chagas, J. M. Resende, P. P. Souza, L. C.A.
Oliveira, Amphiphilic property of niobium oxyhydroxide for waste glycerol conversion to produce
solketal, Catalysis Today, 254 (2015), pp. 83-89.
[33] M.S.T.E.Souza, M.F.Portilho, P.M.T.G.Souza, P.T.Souza, L.C.A. Oliveira, Modified niobium
oxyhydroxide catalyst: an acetalization reaction to produce bio-additives for sustainable use of waste
glycerol, ChemCatChem, 6 (2014), pp.2961-2969.
[34] M. Shirani, H. S. Ghaziaskar, C. Xu, Optimization of glycerol ketalization to produce solketal as
biodiesel additive in a continuous reactor with subcritical acetone using Purolite® PD206 as catalyst,
Fuel Processing Technology, 124 (2014, pp. 206-211.
[35] P. Ferreira, I.M. Fonseca, A.M. Ramos, J. Vital, J.E. Castanheiro, Valorisation of glycerol by
condensation with acetone over silica-included heteropolyacids, Applied Catalysis B:
Environmental,98 (2010), pp. 94-99
[36] P. Manjunathan, S. P. Maradur, A.B. Halgeri, G. V. Shanbhag, Room temperature synthesis of
solketal from acetalization of glycerol with acetone: Effect of crystallite size and the role of acidity of
beta zeolite, Journal of Molecular Catalysis A: Chemical, 396 (2015), pp. 47-54.
[37] D. Nandan, P. Sreenivasulu, L.N. SivakumarKonathala, M. Kumar, N. Viswanadham, Acid
functionalized carbon–silica composite and its application for solketal production, Microporous and
Mesoporous Materials, 179 (2013), pp. 182-190.
[38] N. Suriyaprapadilok, B. Kitiyanan, Synthesis of Solketal from Glycerol and Its Reaction with
Benzyl Alcohol, Energy Procedia, 9 (2011), pp. 63-69.
[39] C.X.A. Silva, V.L.C. Gonçalves, C.J.A. Mota, Obtenção de aditivos oxigenados para a gasolina a
partir da glicerina de produção de biodiesel, 4º Congresso Brasileiro de P&D em Petróleo e Gás,176-1
(2007)
[40] C.X.A. Silva, C.J.A. Mota, Produção de cetais da glicerina para mistura com combustíveis, 5º
Congresso Brasileiro de P&D em Petróleo e Gás, 244 (2009)
[41] C. X.A. da Silva, C. J.A. Mota, The influence of impurities on the acid-catalyzed reaction of
glycerol with acetone, Biomass and Bioenergy, 35 (2011), pp. 3547-3551.
[42] M.S. Khayoon, A. Abbas, B.H.Hameed, S. Triwahyono, A.A. Jalil, A.T. Harris, A.I. Minett,
Selective acetalization of glycerol with acetone over nickel nanoparticles supported on multi-walled
carbon nanotubes, Catalysis Letters, 144(2014), pp.1009-1015.

72
[43] C.Fan, C.Xu, C.Liu, Z.Huang, J.Liu, Z.Ye, Catalytic acetalization of biomass glycerol with acetone
over TiO2-SiO2 mixed oxides, Reaction Kinetics, Mechanisms and Catalysis,107 (2012), issue 1, pp.
189-202.
[44] L.Li, T.I. Korányi, B.F.Sels, P.P.Pescarmona,Highly-efficient conversion of glycerol to solketal
over heterogeneous Lewis acid catalysts,Green Chemistry, 14 (2012), pp. 1611-1619.
[45] B. Mallesham, P. Sudarsanam, G.Raju, B.M.Reddy, Design of highly efficient Mo and W-
promoted SnO22 solid acids for heterogeneous catalysis: acetalization of bio-glycerol, Green
Chemistry, 15 (2013), pp.478-489.
[46] C. Gonzalez-Arellano, S.De, R.Luque, Selective glycerol transformations to high value-added
products catalysed by aluminosilicate-supported iron oxide nanoparticles,Catalysis Science &
Technology, 4 (2014), 4242-4249
[47] L.Roldán, R.Mallada, J.M.Fraile, J.A.Mayoral, M.Menéndez, Glycerol upgrading by ketalization in
a zeolite membrane reactor, Asia-Pacific Journal of Chemical Engineering, 4 (2009), pp.279-284.
[48] G.Vicente, J.A.Melero, G.Morales, M.Paniagua, E.Martín, Acetalisation of bio-glycerol with
acetone to produce solketal over sulfonic mesostructured silicas, Green Chemistry, 12 (2010), 899-
907
[49] P. S. Reddy, P.Sudarsanam, B.Mallesham, G. Raju, B. M. Reddy, Acetalisation of glycerol with
acetone over zirconia and promoted zirconia catalysts under mild reaction conditions, Journal of
Industrial and Engineering Chemistry, 17(2011), pp.377-381.
[50] S. Gadamsetti, N. P. Rajan, G. Srinivasa Rao, K. V. R. Chary, Acetalization of glycerol with
acetone to bio fuel additives over supported molybdenum phosphate catalysts, Journal of Molecular
Catalysis A: Chemical, 410 (2015),pp. 49-57.
[51] B. Mallesham, P. Sudarsanam, G.Raju, B.M.Reddy, Design of highly efficient Mo and W-
promoted SnO22 solid acids for heterogeneous catalysis: acetalization of bio-glycerol, Green
Chemistry, 15 (2013), pp.478-489.
[52]N.Kaur, D.Kishore, Montmorillonite: An efficient, heterogenous and green catalyst for organic
synthesis, Journal of Chemical and Pharmaceutical Research, 4 (2012), issue 2, pp.991-1015.
[53] A.R. do Nascimento, J.A.B.R.L.R. Alves, M.A.F.Melo, D.M.A. Melo, M.J.B. de Souza, A.M.G.
Pedrosa, Effect of the acid treatment of montmorillonite clay in the oleic acid esterification reaction,
Materials Research, 18 (2015), pp.283-287.
[54] A. Darehkordi, S.M.S. Hosseini, M. Tahmooresi, Montmorillonite modified as an efficient and
environmental friendly catalyst for one-pot synthesis of 3,4-dihydropyrimidine-2(1H) ones, Iranian
Journal of Materials Science & Engineering, 9 (2012), number 3, pp.49-57.

73
[55] M.T. Angaji, A.Z. Zinali, N.T. Qazvini, Study of physical, chemical and morphological alterations of
smectite clay upon activation and funcionalization via the acid treatment, World Journal of Nano
Science and Engineering, 3 (2013), number 4, pp. 161-168
[56] L. Bieseki, F. Bertella, H. Treichel, F.G. Penha, S.B.C Pergher, Acid treatments of
montmorillonite-rich clay for Fe removal using a factorial design method, Materials Research, 16
(2013), pp.1122-1127.
[57] B.K Min, A.K Santra, D.W Goodman, Understanding silica-supported metal catalysts: Pd/silica as
a case study, Catalysis Today, 85 (2003), pp, 113-124.
[58] F.Fakhfakh, L.Baraket, A.Ghorbel, J. M. Fraile, C. I. Herrerías, J. A. Mayoral, Effect of support
properties on the performance of silica-supported bis(oxazoline)–copper chiral complexes, Journal of
Molecular Catalysis A: Chemical, 329 (2010), pp. 21-26.
[59] K. Niknam, M.A. Zolfigol, D. Saberi, H. Molaee, Preparation of silica supported tin chloride: as a
recyclable catalyst for the silylation oh hydroxyl groups with HMDS, Journal of the Chinese Chemical
Society, 56 (2009), pp. 1257-1264.
[60] A.B. Ferreira, A.L. Cardoso, M.J.da Silva, Tin-catalyzed esterification and transesterification
reactions: a review, ISRN Renewable Energy, 12 (2012), pp.1-13.
[61] B.C. Smith, Fundamentals of Fourier Transform Infrared Spectroscopy, CRC Press, 2011.
[62] A.P.V. Soares, M.F. Portela, A. Kiennemann, Methanol selective oxidation to formaldehyde over
iron-molybdate catalysts, Catalysis Review: Science and Engineering, 47:1 (2005), pp. 125-174.
[63] E. Moukhina, Direct analysis in modelatedthermogravimetry, ThermochimicaActa, 576(2014),
pp.75-83
[64] A.G.Barneto, J.A.C., J.E.M.Alfonso, R.S.Serrano, Simultation of the thermogravimetry analysis of
three non-wood pulps, Bioresource Technology, 101(2010), pp. 3220-3229
[65] ChemSpider. [Online] Royal Society of Chemistry. http://www.chemspider.com/.
[66] Chemister. [Online] http://chemister.ru/Database/search-en.php.
[67] Viscopedia. [Online] http://www.viscopedia.com/viscosity-tables/.
[68] Chem Synthesis. [Online] http://www.chemsynthesis.com/base/chemical-structure-14154.html
[69] Drug Future - Chemical Index Database. [Online] http://www.drugfuture.com/chemdata/.
[70] G. Fan, C.Liao, T. Fang, S. Luo, G. Song, Amberlyst 15 as a new and reusable catalyst for the
conversion of cellulose into cellulose acetate, Carbohydrate Polymers, 112, 203-209

74
[71] J. Ravichandran, B. Sivasankar, Properties and catalytic activity of acid-modified montmorillonite
and vermiculite, Clay and Clay Minerals, 45(1997), issue 6, pp.854-858.
[72] H. Yang, C. Du, S. Jin, A. Tang, Preparation and characterization of SnO22 nanoparticles
incorporated into talc porous materials (TPM), Materials Letters, 61, pp. 3736-3739
[73] National Institute of Standards and Technology [Online] http://www.nist.gov/
[74] Michigan State University, Department of Chemistry.[Online] Mario 5, 2013.[Cited: Dezembro 2,
2015.] https://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJmL/Spectrpy/InfraRed/infrared.htm.
[75] M.Z.B.Yunos, Effect of glycerol on performance rice straw/starch based polymer, Journal of
Apllied Sciences, 11, pp. 2456-2459, issue 13
[76] L.P. Ozorio, R. Pianzolli, M.B.S. Mota, C.J.A. Mota, Reactivity of glycerol/acetone ketal (solketal)
and glycerol/formaldehyde acetals towards acid-catalyzed hydrolysis, Journal of the Brazilian
Chemical Society, 23, pp. 931-937, number 5
[77] C.J.A. Mota, C.F.M. Pestana, Co-produtos da produção de biodiesel, Revista Virtual da Química,
2011, 3(5), pp. 416-425.
[78] A.L.Genovese, M.E.M. Udaeta, L.C.R. Galvão, Aspectos energéticos da biomassa como recurso
no Brasil e no Mundo.

75
8. ANEXOS
8.1 Termogravimetria
Figura 62 – Termograma para solketal 3
Figura 63 – Termograma para solketal 5

76
Figura 64 – Termograma para solektal 6
Figura 65 – Termograma para solketal 10
Figura 66 – Termograma para solketal 12
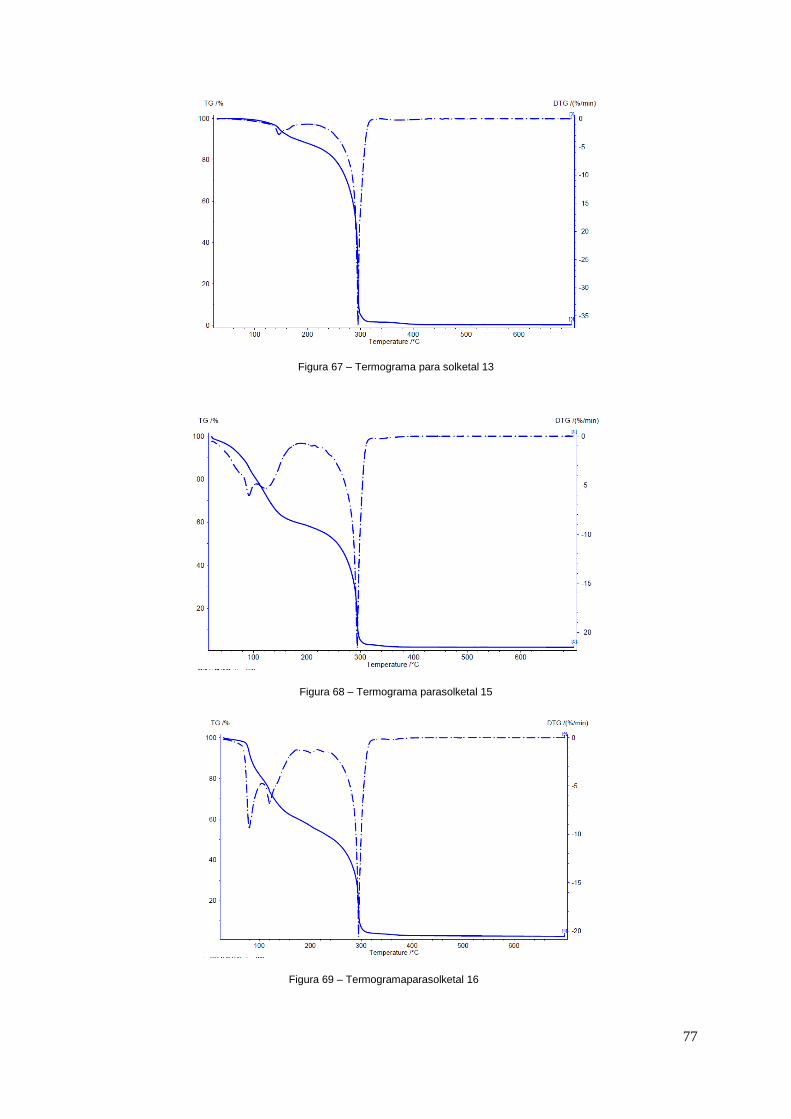
77
Figura 67 – Termograma para solketal 13
Figura 68 – Termograma parasolketal 15
Figura 69 – Termogramaparasolketal 16

78
Figura 70 – Termograma parasolketal 17
Figura 71 – Termogramaparasolketal 18
Figura 72 – Termogramaparasolketal 19

79
8.2 Transformada de Fourier Infravermelho
8.2.1 Catalisadoresapósreacção
Figuras 73 e 74 - - Espectro de FTIR para KSF com tratamento ácido de HNO3 15% 200ºC 2h, após reacção
5001000150020002500300035004000
ab
so
rban
ce (
a.a
.)
wave number (cm-1)
KSF_HNO3_15%_200C_2h
HNO3 1
500700900110013001500
ab
so
rban
ce (
a.a
.)
wave number (cm-1)
KSF_HNO3_15%_200C_2h
HNO3 1
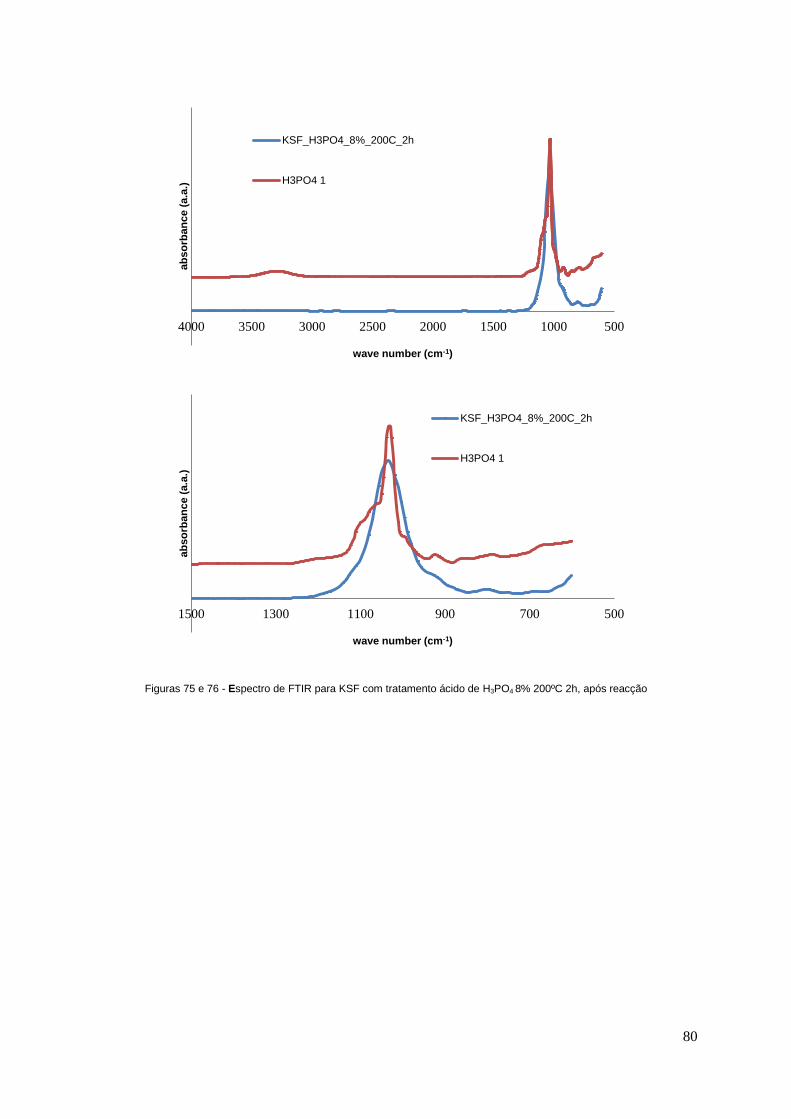
80
Figuras 75 e 76 - Espectro de FTIR para KSF com tratamento ácido de H3PO4 8% 200ºC 2h, após reacção
5001000150020002500300035004000
ab
so
rban
ce (
a.a
.)
wave number (cm-1)
KSF_H3PO4_8%_200C_2h
H3PO4 1
500700900110013001500
ab
so
rban
ce (
a.a
.)
wave number (cm-1)
KSF_H3PO4_8%_200C_2h
H3PO4 1

81
Figuras 77 e 78 - - Espectro de FTIR para KSF com tratamento ácido de HCl 3,8% 26ºC 1h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL3,8%_26C_1h
KSF_HCl 3,8% 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL3,8%_26C_1h
KSF_HCl 3,8% 1

82
Figuras 79 e 90 - Espectro de FTIR para KSF com tratamento ácido de HCl 9% 27ºC 1h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_27C_1h
KSF_HCl 9% 27ºC 1h 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_27C_1h
KSF_HCl 9% 27ºC 1h 1

83
Figuras 91 e 92 - Espectro de FTIR para KSF com tratamento ácido de HCl9% 200ºC 2h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_200C_2h
KSF_HCl 9% 200ºC 2h 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_200C_2h
KSF_HCl 9% 200ºC 2h 1

84
Figuras 93 e 94 – Espectro de FTIR para KSF com tratamento ácido de H2SO4, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_H2SO4
KSF_H2SO4 1
700800900100011001200130014001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_H2SO4
KSF_H2SO4 1
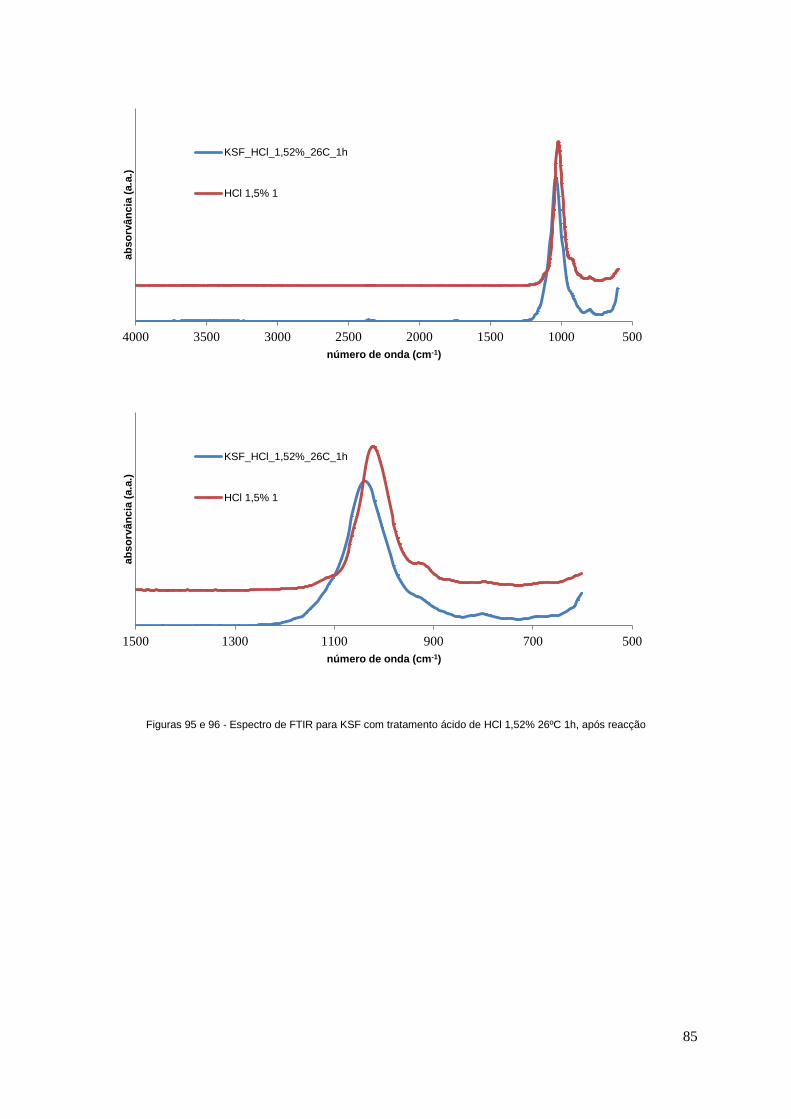
85
Figuras 95 e 96 - Espectro de FTIR para KSF com tratamento ácido de HCl 1,52% 26ºC 1h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_1,52%_26C_1h
HCl 1,5% 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_1,52%_26C_1h
HCl 1,5% 1
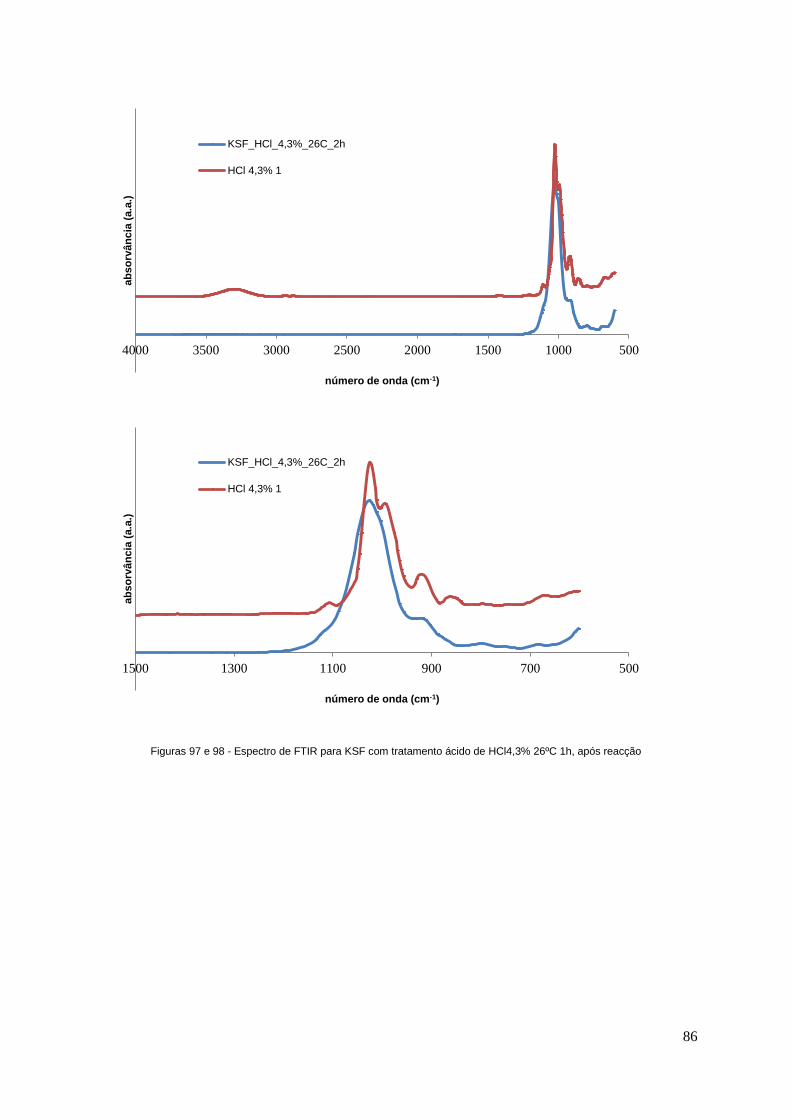
86
Figuras 97 e 98 - Espectro de FTIR para KSF com tratamento ácido de HCl4,3% 26ºC 1h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_4,3%_26C_2h
HCl 4,3% 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCl_4,3%_26C_2h
HCl 4,3% 1
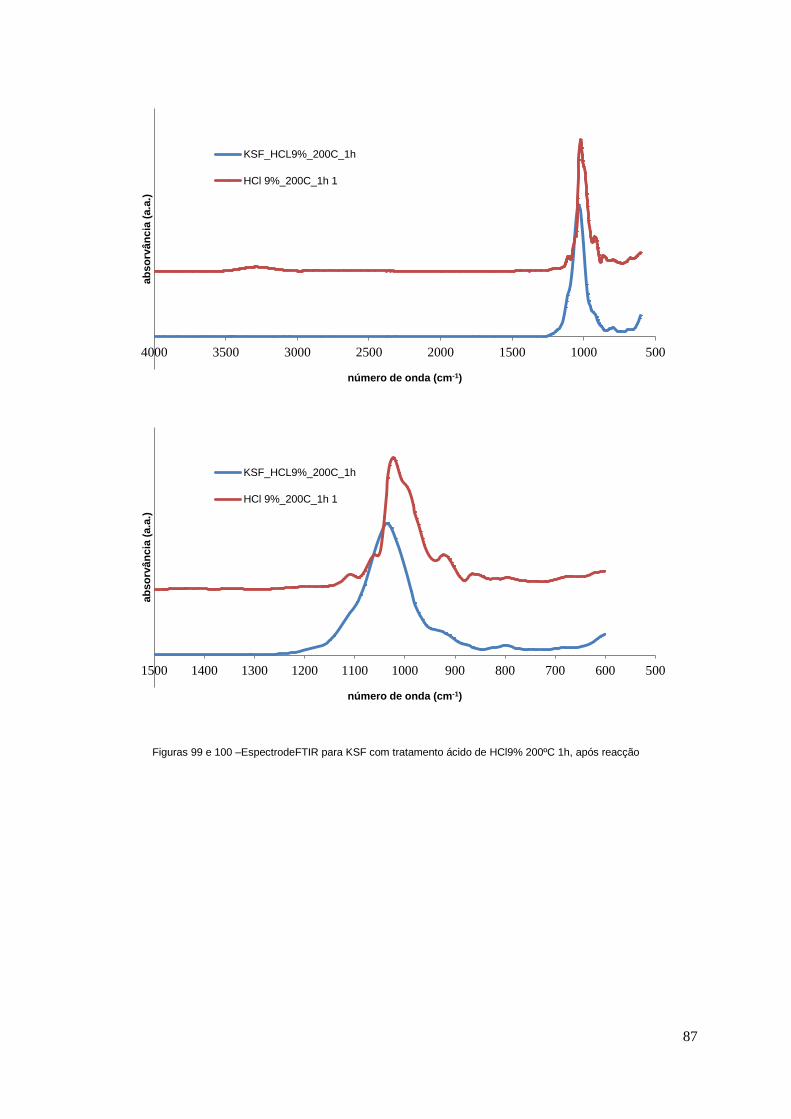
87
Figuras 99 e 100 –EspectrodeFTIR para KSF com tratamento ácido de HCl9% 200ºC 1h, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_200C_1h
HCl 9%_200C_1h 1
500600700800900100011001200130014001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
KSF_HCL9%_200C_1h
HCl 9%_200C_1h 1

88
Figuras 101 e 102 - Espectro de FTIR para SnO2/SiO2 e SnO2-SO42-/SiO2, após reacção
5001000150020002500300035004000
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
SnO2/SiO2
SnO2/SiO2 1
SnO2-SO42-/SiO2
SnO2-SO42-/SiO2 1
500700900110013001500
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
SnO2/SiO2
SnO2/SiO2 1
SnO2-SO42-/SiO2
SnO2-SO42-/SiO2 1

89
8.2.2 Amostras
Figura 103 – Espectros de FTIRdo efluente reaccional das reacções com montmorilonite KSF, entre 700 e 1800 cm-1.
Figura 104 – Espectros de FTIRdoefluentereacional das reacções com amberlyst, entre 700 e 1800 cm-1
7009001100130015001700
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 12
Solketal 18
Solketal 2
Solketal 4
Solketal 6
Solketal 8
Solketal 5
7009001100130015001700
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 11
Solketal 22

90
Figura 105 – Espectros de FTIR do efluente reaccional das reacções de montmorilonite com tratamento ácido de HCl 9%, entre
700 e 1800 cm-1
Figura 106 – Espectros de FTIR do efluente reaccional das reacções de montmorilonite com tratamento ácido de HCl 1,52%,
3,8% e 4,3%, entre 700 e 1800 cm-1
7009001100130015001700
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 19
Solketal 14
Solketal 21
7009001100130015001700
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Glicerol
Solketal 15
Solketal 13
Solketal 20
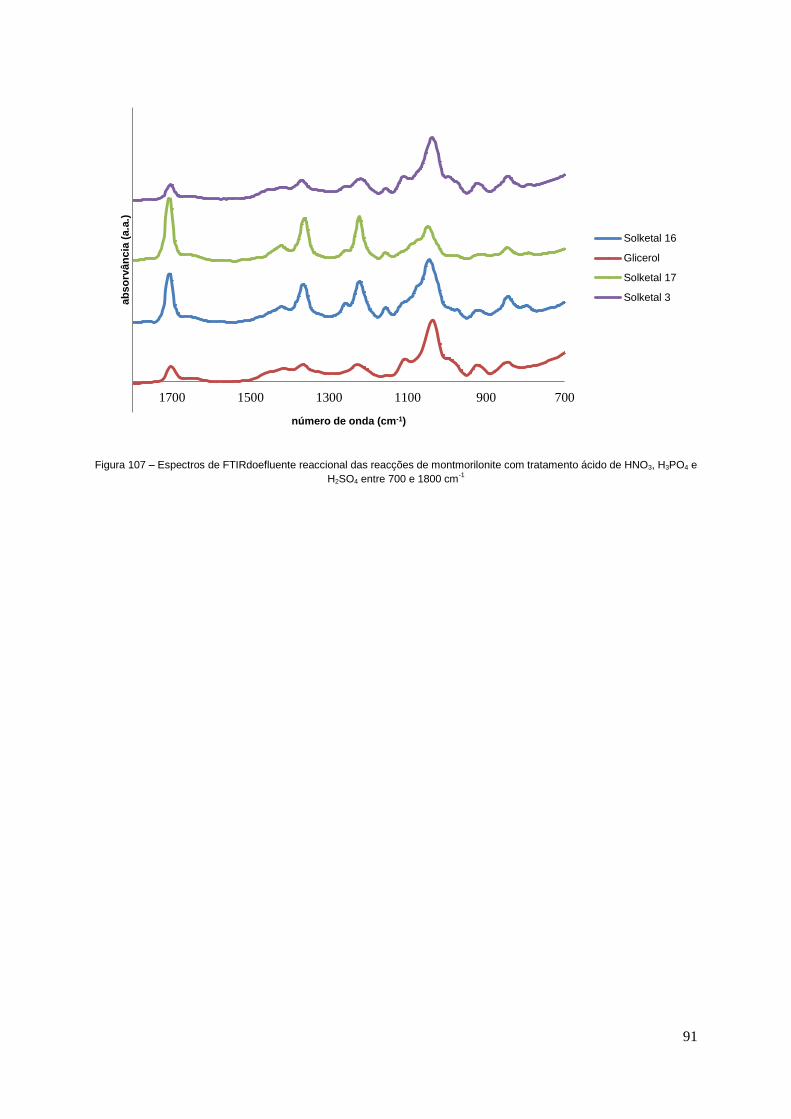
91
Figura 107 – Espectros de FTIRdoefluente reaccional das reacções de montmorilonite com tratamento ácido de HNO3, H3PO4 e
H2SO4 entre 700 e 1800 cm-1
7009001100130015001700
ab
so
rvân
cia
(a.a
.)
número de onda (cm-1)
Solketal 16
Glicerol
Solketal 17
Solketal 3