desenvolvimento e validaÇÃo de mÉtodos para...
TRANSCRIPT

UNIVERSIDADE ESTADUAL DE CAMPINAS
INSTITUTO DE QUÍMICA
BRUNO MOLERO DA SILVA
“DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODOS PARA
DETERMINAÇÃO DE COMPOSTOS FENÓLICOS EM CEVADA
EMPREGANDO CROMATOGRAFIA LÍQUIDA DE ULTRA ALTA
EFICIÊNCIA ACOPLADA À ESPECTROMETRIA DE MASSAS
SEQUENCIAL”
CAMPINAS 2017

BRUNO MOLERO DA SILVA
“DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODOS PARA DETERMINAÇÃO DE COMPOSTOS FENÓLICOS EM CEVADA EMPREGANDO
CROMATOGRAFIA LÍQUIDA DE ULTRA ALTA EFICIÊNCIA ACOPLADA À ESPECTROMETRIA DE MASSAS SEQUENCIAL”
Tese de Doutorado apresentada ao Instituto de
Química da Universidade Estadual de Campinas como
parte dos requisitos exigidos para a obtenção do título de
Doutor em Ciências.
Orientador(a): Prof(a). Dr(a). Carla Beatriz Grespan Bottoli
ESTE EXEMPLAR CORRESPONDE À VERSÃO FINAL DA TESE DEFENDIDA PELO ALUNO BRUNO MOLERO DA SILVA, E ORIENTADA PELA PROF(A). DR(A). CARLA BEATRIZ GRESPAN BOTTOLI.
CAMPINAS
2017



Dedico este trabalho...
Aos meus pais, Ataides e Marinês que me deram oportunidade e incentivo para realizar meus estudos. Vocês me deram o presente que dinheiro nenhum pode comprar nesse mundo material; que foi a educação e os exemplos de dignidade e honestidade que tive desde criança. Tenho muito orgulho de vocês! Também dedico este trabalho à minha querida avó Matilde Toral Ortega pelo carinho e ensinamentos deixados, que me deram força para seguir nos momentos mais difíceis (in memorium).

“Nada na vida deve ser temido, somente compreendido. Agora é hora de compreender mais para temer menos!”
Marie Curie

AGRADECIMENTOS
À Deus pelo amor incondicional e aos protetores de luz pela oportunidade desta conquista em minha vida. À Profa. Dra. Carla Beatriz Grespan Bottoli pela orientação, liberdade de trabalho, ensinamentos e otimismo na rotina diária. Agradeço pelo voto de confiança e por permitir o desenvolvimento desta pesquisa juntamente com o meu trabalho. À minha família: meus pais Ataides e Marinês pela compreensão e apoio em todos os momentos da minha vida. À minha irmã Laíze e meu cunhado Rudnei que sempre torceram por mim. Ao meu querido sobrinho Rafael que é a alegria da nossa família. À minha amada noiva Gabriela pelo incomparável carinho, compreensão e paciência comigo. Você é a mulher da minha vida! Te amo!! Aos meus familiares, tios, tias e primos (especialmente ao Fábio por enviar as amostras de cevada comercializadas em Belo Horizonte) pelo carinho e amizade. Em especial ao meu tio Dorival que sempre me inspirou e incentivou pela sua forma racional de encarar a vida mesmo quando surgem inesperados desafios. Aos colegas de laboratório do LabCrom: Jordana, Carla Grazielli, Fabiana, Camila, Mariana, Cláudio, Luana, Karen, Julie e Mariane pela amizade e bons momentos compartilhados. Obrigado galera! Aos colegas do laboratório de Cromatografia Gasosa: Mayra, Soraia, Jadson, Paloma, Leandro, Bruna e Sandra. Em especial às guerreiras no trabalho Gabriela Salazar e Paula Lima pela verdadeira amizade e companheirismo. À Ms. Lucília responsável pelo LabCrom pela amizade e por ser uma pessoa tão solícita. À Priscila que é responsável técnica do laboratório de Espectrometria de Massas pelo treinamento no equipamento, disponibilidade, experiência e amizade. Aos grandes amigos de Palmital que sempre estiveram do meu lado. Agradecimento especial ao Bruno Orlandi, Bruno Ireno, Tunico e Tugu.
Ao meu amigo Everson Biazon que enviou as amostras de Curitiba pela disposição em ajudar-me.
À Daiane Cobianchi por enviar as amostras de cevada de Florianópolis meu sincero agradecimento.

Ao amigo Fabricio Ferreira, aluno de Doutorado do IQ-Unicamp pela disponibilidade de discussão de temas relevantes em Química Analítica.
À Dra. Fernanda Oliveira Lima pelos ensinamentos e treinamento no equipamento de Eletroforese Capilar. Aos funcionários do Restaurante Universitário da Unicamp que tanto me ajudaram com as refeições durante esses anos. Ao Professor Dr. Ronaldo Pilli por permitir o uso do rotaevaporador de seu laboratório. Aos meus alunos da rede pública estadual das escolas por onde passei e em especial à E.E. Professor Celso Henrique Tozzi de Jaguariúna-SP. Aos amigos do SKK de Palmital-SP pela amizade de longos anos e pela alegria de sempre!! Aos secretários da CPG do IQ, Isabela, Miguel e Bel pela constante disposição em ajudar-me. Aos pais da Gabi, Inês e Nelson, pela amizade e finais de semana descontraídos em Mogi Guaçu. Aos Professores do IQ- Unicamp pelas disciplinas que cursei e em especial à Profa. Dra Isabel Jardim pela atenção e ensinamentos. À todos aqueles que, de maneira direta ou indireta contribuíram para a realização deste trabalho.

RESUMO
A cevada é um cereal com alto valor nutricional e baixo custo que pode servir como
uma excelente fonte de antioxidantes. Para quantificar os compostos fenólicos em
cevada é necessária a escolha de técnicas de preparo de amostras adequadas,
principalmente por se tratar de uma matriz complexa. Neste trabalho, vários extratos
de cevada foram preparados usando diferentes técnicas de extração como refluxo,
agitação com aquecimento, com e sem atmosfera inerte, avaliação da hidrólise ácida
ou básica e o método QuEChERS (Quick, Easy, Cheap, Effective, Rugged e Safe).
Os parâmetros de extração dos extratos etanólicos realizados com agitação sob
atmosfera inerte e em refluxo foram otimizados a partir de um planejamento fatorial
completo e os fatores avaliados foram técnica de extração, temperatura e tempo de
extração. As respostas foram obtidas pela caracterização dos extratos por ionização
por eletrospray no modo negativo (ESI(-)-MS/MS), a partir da intensidade do sinal
analítico e o número de compostos fenólicos identificados. A quantificação dos
compostos fenólicos em grãos de cevada foi realizada empregando os métodos por
refluxo e QuEChERSe a técnica de cromatografia líquida de ultra alta eficiência
acoplada à espectrometria de massas sequencial (UHPLC-MS/MS). Os métodos
foram validados de acordo com as recomendações da Agência Brasileira de
Vigilância Sanitária (ANVISA) e na avaliação de algumas amostras comerciais de
grãos de cevada, notou-se que as concentrações dos compostos fenólicos catequina
e epicatequina foram as mais elevadas. Já os ácidos cafeico e protocatecóico
tiveram as concentrações mais baixas. Estas diferenças podem ocorrer devido à
variação sazonal, assim como as condições de armazenamento dos grãos na
comercialização.

ABSTRACT
Barley is a cereal with high nutritional value and low cost that can serve as an
excellent source of antioxidants. In order to quantify phenolic compounds in barley, it
is necessary to choose suitable sample preparation techniques, mainly because it is
a complex matrix. In this work, several barley extracts were prepared using different
extraction techniques such as reflux, heating with stirring, with and without an inert
atmosphere, evaluation of acid or basic hydrolysis and the QuEChERS (Quick, Easy,
Cheap, Effective, Rugged and Safe) method. The extraction parameters of the
ethanolic extracts carried out under agitation using an inert atmosphere and at reflux
were optimized from a complete factorial design. The factors evaluated were
extraction technique, temperature and extraction time. The responses were obtained
by the characterization of the extractions by electrospray MS in the negative mode
[ESI (-) - MS / MS], based on the intensity of the analytical signal and the number of
phenolic compounds identified. Quantification of phenolic compounds in barley grains
was performed using the reflux and QuEChERS methods, followed by ultra high
efficiency liquid chromatography coupled to sequential mass spectrometry (UHPLC-
MS / MS). The methods were validated according to the recommendations of the
Brazillian Health Regulatory Agency (ANVISA). When some commercial samples of
barley grains were evaluated, the concentrations of the phenolic compounds catechin
and epicatechin were the highest. Caffeic and protocatecoic acids had the lowest
concentrations. These differences can occur due to the seasonal variation, as well as
the storage conditions of the grains during the commercialization.

LISTA DE FIGURAS Figura 1: Estrutura do grão de cevada...................................................................22
Figura 2: Algumas fontes de EROs e mecanismos de defesa...............................24
Figura 3: Esqueleto de base a partir dos quais os compostos fenólicos de origem
vegetal são derivados.............................................................................................25
Figura 4: Representação das etapas das principais versões do método QuEChERS
a) original; b) acetato; e c) citrato.............................................................................36
Figura 5: Esquema ilustrativo de um equipamento de eletroforese capilar onde R1 e
R2, e1 e e2 são os reservatórios e eletrodos, respectivamente. F representa a fonte
de alta tensão, D é o detector, C é o computador para obtenção dos dados e EOF
representa o fluxo eletrosmótico, que é gerado após ser aplicado o
potencial..................................................................................................................42
Figura 6: Esquema de um analisador triplo quadrupolo operando no modo SRM
onde Q1 e Q3 são quadrupolos de transmissão e Q2 um quadrupolo onde ocorre a
fragmentação dos íons...........................................................................................48
Figura 7: Mecanismo da evaporação do íon e da explosão coulômbica...............51
Figura 8: Estruturas químicas dos compostos fenólicos em estudo......................62
Figura 9: Fluxograma referente ao procedimento do método QuEChERS...........67
Figura 10: Fragmentação do padrão do ácido ferúlico no modo negativo
empregando infusão direta.....................................................................................73
Figura 11: Fragmentação do padrão do ácido p-cumárico no modo negativo
empregando infusão direta.....................................................................................73
Figura 12: Fragmentação do padrão da catequina no modo negativo empregando
infusão direta..........................................................................................................74
Figura 13: Fragmentação do padrão de epicatequina no modo negativo empregando
infusão direta..........................................................................................................74
Figura 14: Fragmentação do padrão do ácido cafeico no modo negativo
empregando infusão direta.....................................................................................75
Figura 15: Fragmentação do padrão sintético de rutina no modo negativo
empregando infusão direta.....................................................................................75
Figura 16: Fragmentação do padrão sintético do ácido vanílico no modo negativo
empregando infusão direta.........................................................................................76

Figura 17: Fragmentação do pico de razão m/z 193 (ácido ferúlico) na fração do
extrato A no modo negativo empregando infusão direta........................................77
Figura 18: Fragmentação do pico de razão m/z 193 (ácido ferúlico) na fração do
extrato C1 no modo negativo empregando infusão direta.........................................79
Figura 19: Fragmentação do pico de razão m/z 193 (ácido ferúlico) na fração do
extrato C2 no modo negativo empregando infusão direta......................................79
Figura 20: Fragmentação do pico de razão m/z 163 (ácido p-cumárico) na fração do
extrato C1 no modo negativo empregando infusão direta.........................................80
Figura 21: Fragmentação do pico de razão m/z 163 (ácido p-cumárico) na fração do
extrato C2 no modo negativo empregando infusão direta......................................80
Figura 22: Fragmentação do pico de razão m/z 289 (catequina) na fração do extrato
C1 no modo negativo empregando infusão direta.....................................................81
Figura 23: Fragmentação do pico de razão m/z 289 (catequina) na fração do extrato
C2 no modo negativo empregando infusão direta..................................................81
Figura 24: Fragmentação do pico de razão m/z 289 (epicatequina) na fração do
extrato C2 no modo negativo empregando infusão direta.........................................82
Figura 25: Fragmentação do pico de razão m/z 193 (ácido ferúlico) na fração do
extrato C3 no modo negativo empregando infusão direta......................................83
Figura 26: Fragmentação do pico de razão m/z 179 (ácido cafeico) na fração do
extrato C3 no modo negativo empregando infusão direta......................................84
Figura 27: Fragmentação do pico de razão m/z 179 (ácido cafeico) na fração do
extrato C4 no modo negativo empregando infusão direta.........................................84
Figura 28: Fragmentação do pico de razão m/z 289 (catequina) na fração do extrato
C3 no modo negativo empregando infusão direta.....................................................85
Figura 29: Fragmentação do pico de razão m/z 289 (catequina) na fração do extrato
C4 no modo negativo empregando infusão direta.....................................................85
Figura 30: Fragmentação do pico de razão m/z 289 (epicatequina) na fração do
extrato C3 no modo negativo empregando infusão direta.........................................86
Figura 31: Fragmentação do pico de razão m/z 289 (epicatequina) na fração do
extrato C4 no modo negativo empregando infusão direta.........................................86
Figura 32: Fragmentação do pico de razão m/z 163 (ácido p-cumárico) na fração do
extrato C3 no modo negativo empregando infusão direta......................................87

Figura 33: Fragmentação do pico de razão m/z 163 (ácido p-cumárico) na fração do
extrato C4 no modo negativo empregando infusão direta.........................................87
Figura 34: Fragmentação do pico de razão m/z 609 (rutina) na fração do extrato C4
no modo negativo empregando infusão direta........................................................88
Figura 35: Fragmentação do pico de razão m/z 167 (ácido vanílico) na fração do
extrato C4...............................................................................................................88
Figura 36: ESI no modo positivo dos íons precursores da mistura contendo 11
padrões de compostos fenólicos com voltagens do cone avaliadas em (A) 60, (B) 45,
(C) 30, (D) 20, (E) 10 V...........................................................................................91
Figura 37: ESI no modo negativo da mistura contendo 11 padrões de compostos
fenólicos com voltagem do cone avaliada em 30 V................................................92
Figura 38: Cromatograma da mistura contendo 11 padrões a 3 µg mL-1 com ESI no
modo negativo. Condições cromatográficas: coluna Waters Acquity UPLC BEH C-18
1.7 μm (50 × 2,1 mm d.i.); Fase móvel: A= H2O(0,1% ácido fórmico) e B= ACN
(0,1% ácido fórmico). Gradiente: 3% B por 1 min; 3% a 15% B em 4 min; 15% a 40%
em 3 min; 40% a 3% em 1 min. Vazão: 0,350 mL min-1. Modo de Ionização: ESI-
(ionização por eletronebulização no modo negativo, 30 kV). Volume de injeção: 2
μL. Detecção: espectrômetro de massas com analisador
triploquadrupolar.....................................................................................................95
Figura 39: Cromatograma da mistura contendo 11 padrões a 3 µg mL-1 com ESI no
modo negativo com adição de NH4OH 0,025%. Condições cromatográficas: coluna
Waters Acquity UPLC BEH C-18 1.7 μm (50 × 2,1 mm d.i.); Fase móvel: A= H2O
(0,025% NH4OH) e B= ACN (0,1% ácido fórmico). Gradiente: 3% B por 1 min; 3% a
15% B em 4 min; 15% a 40% em 3 min; 40% a 3% em 1 min. Vazão: 0,350 mL min-1.
Modo de Ionização: ESI-(ionização por eletronebulização no modo negativo, 30 kV).
Volume de injeção: 2 μL. Detecção: espectrômetro de massas com analisador
triploquadrupolar.....................................................................................................96
Figura 40: Cromatograma de íons totais dos compostos fenólicos presentes numa
solução de 50 µg mL-1 em metanol e determinação por UHPLC-MS/MS em 11
canais de aquisição, no modo SRM. Identificação dos picos: A e B: Catequina e
Epicatequina; C: ácido clorogênico; D: ácido vanílico; E: ácido cafeico; F: ácido p-
cumárico; G: ácido ferúlico; H: ácido sinápico; I: rutina; J: ácido protocatecóico e L:
miricetina.................................................................................................................98

Figura 41: Cromatogramadeidentificação do ácido ferúlico no extrato A. Condições
cromatográficas: coluna Waters Acquity UPLC BEH C-18 1.7 μm (50 × 2,1 mm d.i.);
Fase móvel: A= H2O (0,1% ácido fórmico) e B= ACN (0,1% ácido fórmico).
Gradiente: 3% B por 1 min; 3% a 15% B em 4 min; 15% a 40% em 3 min; 40% a 3%
em 1 min. Vazão: 0,350 mL min-1. Modo de Ionização: ESI-(ionização por
eletronebulização no modo negativo, 30 kV). Volume de injeção: 2 μL. Detecção:
espectrômetro de massas com analisador triploquadrupolar.....................................99
Figura 42: Cromatograma deidentificação de ácidos ferúlico, p-cumárico e cafeico;
catequina e epicatequina obtidos para o extrato C1. Condições cromatográficas da
Figura 41....................................................................................................................99
Figura 43: Cromatograma deidentificação dos ácidos ferúlico, p-cumárico e cafeico;
catequina e epicatequina no extrato C2. Condições cromatográficas da Figura
41..............................................................................................................................100
Figura 44: Cromatograma deidentificação de ácidos ferúlico, p-cumárico e cafeico;
catequina e epicatequina no extrato C3. Condições cromatográficas da Figura
41..............................................................................................................................101
Figura 45: Cromatograma do extrato C4 otimizado com ESI no modo negativo.
Condições cromatográficas da Figura 41.................................................................102
Figura 46: Superfície de resposta dos fatores A: tempo de extração ×
temperatura; B: técnica de extração × temperatura e C: técnica de extração ×
tempo para a epicatequina......................................................................................104
Figura 47: Superfície de resposta dos fatores A: tempo de extração ×
temperatura; B: técnica de extração × temperatura e C: técnica de extração ×
tempo para a catequina...........................................................................................106
Figura 48: Superfície de resposta dos fatores A: tempo de extração ×
temperatura; B: técnica de extração × temperatura e C: técnica de extração ×
tempo para o ácido cafeico......................................................................................108
Figura 49: Superfície de resposta dos fatores A: tempo de extração ×
temperatura; B: técnica de extração × temperatura e C: técnica de extração ×
tempo para o ácido ferúlico.....................................................................................110
Figura 50: Superfície de resposta dos fatores A: tempo de extração ×
temperatura; B: técnica de extração × temperatura e C: técnica de extração ×
tempo para o ácido p-cumárico...............................................................................112

Figura 51: Cromatogramas obtidos por UHPLC-MS/MS para separação da mistura
de compostos fenólicos do extrato C4 fortificado (3 µg mL-1).Condições
cromatográficas da Figura 41..................................................................................114
Figura 52: Curvas analíticas para os ácidos clorogênico, sinápico, ferúlico e vanílico,
cafeico, p-cumárico e protocatecóico além de rutina, catequina, epicatequina e
miricetina preparadas em solvente e na matriz pelo método de extração em
refluxo.......................................................................................................................117
Figura 53: Efeito matriz (%) para os compostos fenólicos em cevada usando
UHPLC-MS/MS. (1) catequina; (2) epicatequina; (3) ácido clorogênico; (4) ácido
vanílico; (5) ácido cafeico; (6) ácido p-cumárico; (7) ácido ferúlico; (8) ácido sinápico;
(9) rutina; (10) ácido protocatecóico; (11) miricetina................................................118
Figura 54: Resíduos da regressão linear obtida para os compostos fenólicos
analisados por UHPLC-MS/MS para a curva analítica construída na matriz...........121
Figura 55: Cromatogramas do extrato fortificado com 3 µg mL-1 obtidos pelo método
QuEChERS..............................................................................................................126
Figura 56: Curvas analíticas para os ácidos clorogênico, sinápico, ferúlico e vanílico,
cafeico, p-cumárico e protocatecóico além de rutina, catequina, epicatequina e
miricetina preparadas em solvente e na matriz pelo método
QuEChERS..............................................................................................................129
Figura 57: Percentual do efeito matriz (%) empregando QuEChERS acetato para
extração dos compostos fenólicos em cevada usando UHPLC-MS/MS. (1) catequina;
(2) epicatequina; (3) ácido clorogênico; (4) ácido vanílico; (5) ácidocafeico; (6) ácido
p-cumárico; (7) ácido ferúlico; (8) ácido sinápico; (9) rutina; (10) ácido
protocatecóico; (11) miricetina.................................................................................130
Figura 58: Resíduos da regressão linear obtida para os compostos fenólicos
analisados por UHPLC-MS/MS para a curva analítica construída na matriz pelo
método QuEChERS.................................................................................................132
Figura 59: Cromatogramas adquiridos no modo SRM do extrato de cevada por
QuEChERS e análise por UHPLC-MS/MS para amostra A6..................................139

LISTA DE TABELAS Tabela 1: Produção de cevada em 2005: área de colheita e produção..................21
Tabela 2: Ensaios para um planejamento fatorial 23 para otimização da extração de
compostos fenólicos em cevada..............................................................................69
Tabela 3:Presença de compostos fenólicos em diferentes extratos.......................89
Tabela 4: Compostos fenólicos analisados com ionização no modo positivo.......92
Tabela 5: Melhores condições de operação do espectrômetro de massas para a
análise dos compostos fenólicos.............................................................................93
Tabela 6: Parâmetros do espectrômetro de massas para a análise dos compostos
fenólicos..................................................................................................................93
Tabela 7: Figuras de mérito para a validação do método de extração otimizado.
LQm: Limite de quantificação do método. LQi: Limite de quantificação do
instrumento...........................................................................................................122
Tabela 8: Concentração dos compostos fenólicos na amostra de cevada BRS lagoa
225........................................................................................................................123
Tabela 9: Eficiência de extraçãodos compostos fenólicos no método por
refluxo...................................................................................................................124
Tabela 10: Figuras de mérito da validação referente ao método QuEChERS. LOQm:
Limite de quantificação do método. LOQi: Limite de quantificação do
instrumento...........................................................................................................133
Tabela 11: Concentração dos compostos fenólicos na amostra de cevada.......134
Tabela 12: Eficiência de extraçãodos compostos fenólicos no
métodoQuEChERS...............................................................................................135
Tabela 13: Comparação entre as técnicas de preparo de amostras
empregadas..........................................................................................................135
Tabela 14: Concentração de compostos fenólicos em grãos de cevada avaliada pelo
método por refluxo e pelo método QuEChERS.....................................................137
Tabela 15: Comparação entre a diferença nas médias do método por refluxo e
método QuEChERS...............................................................................................138
Tabela 16: Concentração de compostos fenólicos em amostras comerciais de grãos
cevada provenientes de diferentes regiões do Brasil............................................140

LISTA DE ACRÔNIMOS E ABREVIATURAS ANVISA- Agência Nacional de Vigilância Sanitária.
CE- Eletroforese capilar, do inglês “Capillary electrophoresis”.
CV- Coeficiente de variação.
C18- Octadecil
GC- Cromatografia Gasosa
ERO- Espécies reativas de oxigênio
ESI- Ionização por eletrospray, do inglês “Electrospray ionization”.
FM- Fase móvel.
GCB- Carbono grafitizado, do inglês, “Graphitized Carbon Black”.
HPLC- Cromatografia líquida de alta eficiência, do inglês “High Performance Liquid
Cromatography”.
LD- Limite de detecção.
LQ- Limite de quantificação.
MS- Espectrometria de massas, do inglês “Mass Spectrometry”.
MS/MS- Espectrometria de massas sequencial.
m/z- razão massa/carga.
n- número de replicatas.
PSA- amina primária secundária, do inglês, Primary Secondary Amine.
PTFE- politetrafluoretileno.
QuEChERS: rápido, fácil, barato, efetivo, robusto e seguro, do inglês, “Quick, Easy,
Cheap, Effective, Robust and Safe”.
SRM- Monitoramento de reações selecionadas, do inglês “Selected reaction
monitoring”.
TOF- Analisador por tempo de voo, do inglês “Time of flight”.
UHPLC- Cromatografia líquida de ultra alta eficiência, do inglês “Ultra High
Performance Liquid Chromatography”.
UHPLC-MS/MS- Cromatografia líquida de ultra alta eficiência acoplada à
espectrometria de massas sequencial, do inglês “Ultra High Performance Liquid
Chromatography with tandem mass spectrometry”.



20
1.INTRODUÇÃO
1.1. Cevada
A cevada (Hordeum vulgare L.) é um cereal muito antigo, sendo alguns
fragmentos deste grão foram encontrados no Irã em aproximadamente 7900 a.C,
antes mesmo do trigo.
A composição da cevada inclui proteínas, vitaminas, minerais e fibras,
sendo um cereal bastante consumido no mundo [1]. É o cereal que melhor se adapta
a baixas temperaturas e cresce em terras expostas à luz, bem escoadas e porosas e
se adaptam melhor em solos com pH acima de 6,0, pois o pH baixo retarda o
crescimento da sua raiz [2]. O grão pode ser plantado no solo sem arar e por esta
razão pode ser cultivado em lotes minúsculos.
A cevada tem sido cultivada no Brasil desde a década de 1930 e, como
consequência dos esforços do melhoramento genético e do desenvolvimento de
técnicas de manejo, a cultura foi difundida pelo sul do Brasil, onde se localizam as
áreas com locais mais adequados, em termos de clima e solo, para o plantio deste
cereal [3].
A cevada tem sidoo quarto cereal mais consumido no mundo de acordo
com a Organização das Nações Unidas para alimentação e Agricultura[4].
No entanto, apenas cerca de 2% do total tem sido usado para
alimentação humana com o consumo do cereal e, o restante é direcionado para
alimentação animal e para a indústria de cerveja [5]. Nos últimos anos, no Brasil,
houve um grande interesse da sua utilização na alimentação humana, devido ao seu
alto valor nutricional e também ao seu baixo custo. Mesmo assim, mais de 95% da
cevada ainda é cultivada para fins cervejeiros [6].
O grão de cevada é constituído por casca, aleurona, endosperma,
escutelo e embrião. A casca é formada pela palha, que é eliminada por uma fina
camada, a aleurona, que recobre o endosperma, e o embrião, que corresponde a
apenas 7% do grão. O endosperma é constituído basicamente de amido e proteína,
sendo responsável pela formação de enzimas durante a malteação, enquanto que o
embrião é o local das atividades vitais do grão [7].
21
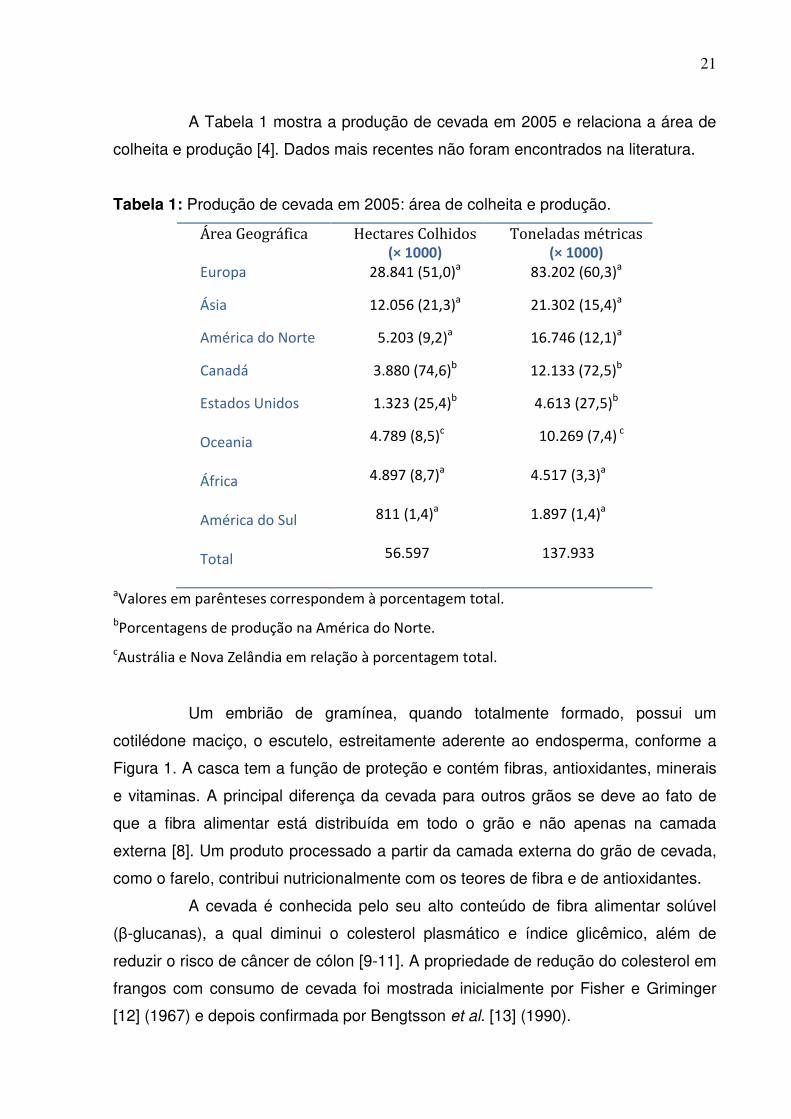
A Tabela 1 mostra a produção de cevada em 2005 e relaciona a área de
colheita e produção [4]. Dados mais recentes não foram encontrados na literatura.
Tabela 1: Produção de cevada em 2005: área de colheita e produção.
Área Geográfica Hectares Colhidos (× 1000)
Toneladas métricas (× 1000)
Europa 28.841 (51,0)a 83.202 (60,3)a
Ásia 12.056 (21,3)a 21.302 (15,4)a
América do Norte 5.203 (9,2)a 16.746 (12,1)a
Canadá 3.880 (74,6)b 12.133 (72,5)b
Estados Unidos Oceania África América do Sul Total
1.323 (25,4)b
4.789 (8,5)c
4.897 (8,7)a
811 (1,4)a
56.597
4.613 (27,5)b
10.269 (7,4) c
4.517 (3,3)a
1.897 (1,4)a
137.933
aValores em parênteses correspondem à porcentagem total.
bPorcentagens de produção na América do Norte.
cAustrália e Nova Zelândia em relação à porcentagem total.
Um embrião de gramínea, quando totalmente formado, possui um
cotilédone maciço, o escutelo, estreitamente aderente ao endosperma, conforme a
Figura 1. A casca tem a função de proteção e contém fibras, antioxidantes, minerais
e vitaminas. A principal diferença da cevada para outros grãos se deve ao fato de
que a fibra alimentar está distribuída em todo o grão e não apenas na camada
externa [8]. Um produto processado a partir da camada externa do grão de cevada,
como o farelo, contribui nutricionalmente com os teores de fibra e de antioxidantes.
A cevada é conhecida pelo seu alto conteúdo de fibra alimentar solúvel
(β-glucanas), a qual diminui o colesterol plasmático e índice glicêmico, além de
reduzir o risco de câncer de cólon [9-11]. A propriedade de redução do colesterol em
frangos com consumo de cevada foi mostrada inicialmente por Fisher e Griminger
[12] (1967) e depois confirmada por Bengtsson et al. [13] (1990).

22
Figura 1: Estrutura do grão de cevada. Adaptado de [9]
Em 1975, Trowell et al. [14] fomentaram a hipótese de que a dieta rica em
fibras pode ter um papel importante no controle do colesterol, no entanto, essas
investigações foram centradas na fibra de trigo. Em 1996, Rimm et al. [15]
conduziram um estudo com mais de 50.000 profissionais de saúde e concluíram que
havia uma relação inversa entre ingestão de fibras e infarto do miocárdio. Dentre os
três principais alimentos que contribuem para o total de fibras, estão: vegetais, frutas
e cereais. A fibra de cereais foi a mais fortemente associada com a prevenção de
doenças do coração.
1.2. Antioxidantes
Antioxidantes são compostos que reagem com substâncias reativas como
os radicais livres presentes, por exemplo, no corpo humano. Eles atuam em
diferentes níveis na proteção dos organismos com mecanismo de defesa contra os
radicais livres, além de impedir a sua formação, principalmente pela inibição das
reações em cadeia com ferro e o cobre.
Estudos comprovam que os grãos de cereais contêm compostos
antioxidantes [16, 17].

23
Os antioxidantes em alimentos são caracterizados como compostos que
podem retardar ou inibir as reações de oxidação provocadas pelas espécies reativas
de oxigênio (ERO), as quais são compostos químicos resultantes da ativação ou
redução do oxigênio molecular. Dentre as principais espécies reativas de oxigênio
estão o ânion superóxido, o peróxido de hidrogênio (H2O2), o radical peroxila, o
radical hidroxila, o oxigênio singlete e o radical peroxinitrito [18].
De modo geral, as reações de oxidação são de natureza radicalar com
propagação em cadeia envolvendo o oxigênio e outras substâncias, normalmente
envolvendo os lipídios e proteínas, que levam à formação dos radicais livres. As
reações ocorrem em três estágios, iniciação, propagação e terminação.
O radical livre pode ceder o elétron desemparelhado, oxidando-se, ou
receber um elétron, reduzindo-se. Na tentativa de se estabilizar quimicamente, os
radicais livres propiciam reações em cadeia que terminam alterando a conformação,
a estrutura e as funções de diversos componentes celulares [19].
O estresse oxidativo, resultante da formação e ação das espécies reativas
de oxigênio, está relacionado com o envelhecimento celular, desenvolvimento de
câncer, doenças neurológicas, doenças cardiovasculares e diabetes [20]. Além
disso, a exposição a alguns agentes agressores externos, como a poluição
ambiental, o fumo do tabaco, toxinas, entre outros, podem propiciar a acumulação
de radicais livres no organismo. Os radicais livres podem ser originados através de
fontes endógenas e exógenas e esses são metabolizados no organismo pelo
sistema de antioxidantes enzimáticos (por ação das enzimas superóxido dismutase
(SOD), glutationa peroxidase (GPx e catalase) ou, também, podem ser neutralizados
através de substâncias antioxidantes não enzimáticas (como tocoferóis, ácido
ascórbico, carotenos, entre outros) [21, 22]. Verificam-se algumas fontes de
espécies reativas de oxigênio (EROs) na Figura 2.

24
Figura 2: Algumas fontes de EROs e mecanismos de defesa [23].
Os antioxidantes atuam em diferentes níveis na proteção dos organismos
como no mecanismo de defesa contra os radicais livres, cuja função é impedir a sua
formação, principalmente pela inibição das reações em cadeia com ferro e cobre. Os
antioxidantes são capazes de interceptar os radicais livres gerados pelo
metabolismo celular ou por fontes exógenas, impedindo o ataque sobre os lipídios,
os aminoácidos das proteínas, a dupla ligação dos ácidos graxos poli-insaturados e
as bases do DNA, evitando a formação de lesões e perda da integridade celular [24].
1.2.1. Compostos Fenólicos
Dentre as diversas classes de antioxidantes de ocorrência natural, os
compostos fenólicos têm recebido muita atenção nos últimos anos. Esses
compostos englobam uma vasta gama de substâncias, as quais possuem pelo
menos um anel aromático com um ou mais substituintes hidroxílicos. Os
intermediários formados pela ação dos compostos fenólicos são relativamente
estáveis, devido à ressonância do anel aromático presente na estrutura dessas

25
substâncias [25]. O número de hidroxilas presentes nos compostos fenólicos, assim
como a sua localização, têm influência na sua ação como antioxidante [26].
A ação desses compostos como antioxidante ocorre devido ao
mecanismo de doação de hidrogênio e/ ou transferência de elétrons para o radical
livre, sendo assim chamados de sequestradores de radicais livres. Portanto, os
compostos fenólicos podem ser definidos como moléculas capazes de diminuir ou
previnir a oxidação de outras moléculas, podendo atuar em alimentos ou em
sistemas biológicos [18].
Os compostos fenólicos se enquadram em diversas categorias como
fenóis simples, ácidos fenólicos (derivados dos ácidos benzóico e cinâmico),
cumarinas, flavonóides, estilbenos, taninos condensados e hidrolisáveis, lignanas e
ligninas [27]. A Figura 3 representa as estruturas gerais dos compostos fenólicos.
Figura 3: Esqueleto de base a partir dos quais os compostos fenólicos de origem vegetal são derivados. Adaptado de Stalikas [28].

26
Figura 3: Esqueleto de base a partir dos quais os compostos fenólicos de origem vegetal são derivados. Adaptado de Stalikas (continuação) [28].
Os principais grupos de compostos fenólicos encontrados nos cereais são
os flavanóides e derivados hidroxicinâmicos [29].
Nos cereais, os compostos fenólicos podem ser encontrados na forma
livre, solúvel ou conjugados, tanto no endosperma, como no gérmen e nas frações
de farelo [30]. A maior parte destes compostos encontra-se na forma conjugada,
esterificada aos polissacarídeos das paredes celulares [31].
Os compostos fenólicos solúveis encontram-se compartimentalizados
dentro dos vacúolos celulares [32] e estão na forma livre (aglicona) ou conjugada,
enquanto os fenólicos insolúveis encontram-se ligados à estruturas da parede
celular, esterificados com arabinose ou resíduos de galactose dos componentes
pécticos ou hemicelulósicos [33]. Os ácidos fenólicos livres representam a menor
parte dos compostos fenólicos e são solúveis em soluções aquosas-orgânicas, tais
como metanol, etanol ou acetona [34]. Os compostos fenólicos conjugados
frequentemente estão sob a forma de ésteres e amidas, raramente ocorrendo como
glicosídeos. Eles incluem compostos de baixa massa molar, solúveis em água,
presentes no citosol, ou formas lipossolúveis, associadas às ceras da superfície dos
cereais [35].

27
De forma geral, os compostos fenólicos são abundantes nos cereais e
entre eles incluem-se: ácido ferúlico, ácido p- cumárico, ácido cafeico, ácido vanílico
e ácido protocatecóico, presentes comumente nas camadas mais externas dos
grãos [36].
Vários efeitos benéficos à saúde têm sido atribuídos aos compostos
fenólicos presentes nos alimentos. Estes compostos estão presentes em uma
grande variedade de gêneros alimentícios e bebidas, incluindo frutas, chás, café e
vinho tinto [37]. Estudos realizados correlacionam aos mesmos propriedades
antioxidantes, anti-inflamatórias, anti-microbiana, anti-carcinogênica e atividade
antiviral, principalmente contra vírus envelopados, além de agirem melhorando a
permeabilidade capilar, podendo auxiliar no tratamento de alguns distúrbios
circulatórios [38,39].
1.3. Técnicas de preparo de amostras para determinação de
compostos fenólicos
Vários métodos de preparação de amostras têm sido desenvolvidos para
determinar compostos fenólicos em uma grande variedade de amostras, desde a
filtração em membrana de 0,45 µm de espessura [28,40,41] até procedimentos mais
elaborados como extração acelerada por solvente, do inglês Accelerated Solvent
Extraction (ASE) [17]. As técnicas de preparo de amostras mais difundidas para
determinação de compostos fenólicos são: extração sólido-líquido [9,42], extração
em fase sólida, do inglês Solid Phase Extraction (SPE) [19,43], microextração em
fase sólida, do inglês Solid Phase Micro Extraction (SPME) [44], sonicação, entre
outras [17, 28].
Por causa da grande diversidade de compostos fenólicos com diferenças
de polaridade, acidez, número de grupos hidroxilas, anéis aromáticos e níveis de
concentração, diversos procedimentos de tratamentos das amostras podem ser
empregados para identificar e quantificar compostos fenólicos.
Para avaliação de matrizes complexas como cereais, a presença de
interferentes pode encobrir o sinal analítico dos compostos em estudo e, portanto
um tratamento prévio neste tipo de amostras é fundamental para fazer a limpeza

28
(clean-up) para redução dos materiais co-extraídos da amostra e para reduzir o
limite de detecção do método e evitar resultados imprecisos e inexatos [45].
De modo geral, as amostras de cereais são inicialmente trituradas,
passando pelo processo de maceração, aumentando a superfície de contato quando
um solvente for adicionado à amostra. Vários fatores são considerados relevantes
no processo de extração, dentre eles: a natureza do solvente, a dissolução das
substâncias extraíveis, a temperatura, o pH e o tempo de extração.
Os compostos fenólicos raramente estão presentes nos vegetais em sua
forma livre, mas comumente, apresentam-se em sua forma glicosilada [46]. Assim,
para extração de compostos fenólicos em grãos, pode-se optar pela escolha de
solventes orgânicos como etanol, metanol, acetona e acetato de etila em mistura
com água para extração de compostos fenólicos solúveis (CFS).
Para determinadas sementes ou matérias primas onde os compostos
fenólicos encontram-se na forma glicosilada (ligados a carboidratos), torna-se
necessário uma etapa adicional com o uso de ácidos para promover a hidrólise[18].
No caso da cevada, dentre os diversos métodos utilizados no preparo de
amostras para identificação e/ou determinação de compostos fenólicos, destacam-
se a extração por refluxo [39], agitação mecânica [47], ultrassom, [27, 16, 48],
hidrólise ácida ou hidrólise básica [40, 41, 49, 50] e extração assistida por micro-
ondas [51].
Um tratamento por explosão a vapor tem sido proposto como um dos
métodos mais promissores para separar os principais constituintes dos materiais
lignocelulósicos, seguido de extração com metanol, e foi aplicado para extração de
compostos fenólicos em cevada. O pré-tratamento com explosão a vapor atua tanto
química como fisicamente na estrutura do material lignocelulósico e está baseado no
contato direto da biomassa com vapor saturado à alta pressão por um determinado
tempo de residência no reator, seguido de descompressão rápida à condição
atmosférica (explosão) [52]. Ao longo deste processo, as ligações químicas que
mantêm os componentes macromoleculares da fitobiomassa fortemente associados
são em parte quebradas, de forma que, no momento da descompressão, o material
é desfibrado com facilidade e assim reduzido a partículas menores. Gong et al. [52]
empregaram este processo em diferentes temperaturas variando entre 210 a 250°C
durante 30 s para avaliação dos compostos fenólicos em cevada. A técnica de
Cromatografia Líquida de Alta Eficiência (HPLC), do inglês, High Performance Liquid

29
Cromatography com detecção por UV foi usada para avaliar o teor de ácido p-
cumárico e ácido ferúlico antes e depois da explosão com vapor. Os resultados
mostraram que o total de compostos fenólicos solúveis e a capacidade antioxidante
total aumentaram com a utilização da explosão a vapor com maiores tempos de
extração [52].
Uma metodologia baseada na liofilização das amostras de sementes,
folhas e caules de cevada coletadas em diferentes regiões de Portugal passou por
um processo de maceração à temperatura ambiente, seguida de extração com água
em banho de ultra-som e centrifugação durante 10 min para posterior análise em
HPLC/UV. Foram determinados uma grande diversidade de compostos fenólicos,
incluindo flavonóides como isoorientina, isovitexina, seus glicosídeos eácido ferúlico
[16]. Destaca-se neste trabalho a concentração de 2150,8 mg kg-1 para isorientina
nas folhas de cevada.
Diferentes tipos de solventes extratores foram avaliados por sonicação
durante 1 h em 3 variedades de cevada chinesa para avaliar o efeito da mistura de
solventes na atividade antioxidante, capacidade de extração e seletividade de
compostos fenólicos em cevada através da análise individual e do teor de
compostos fenólicos totais. Os extratos com 80% de acetona mostraram a maior
capacidade de extração para catequina e ácidos ferúlico, cafeico, vanílico, p-
cumárico, 80% de metanol para epicatequina e ácido siríngico e 100% de água para
os ácidos protocatecóico e gálico. Os autores concluíram que a extração com 80%
de acetona é a mais recomendada para determinação de compostos fenólicos livres
em cevada [48]. Embora neste trabalho os autores tenham ressaltado o uso da
acetona, sabe-se que o etanol também é um poderoso solvente extrator de
compostos fenólicos [39, 42].
Dvorakova et al. [47] testaram duas proporções de misturas de solventes,
metanol/água ou acetona/água na razão 70:30 v/v, usando agitador mecânico a 250
rpm para a extração de compostos fenólicos. Em seguida, os extratos foram
centrifugados durante 5 min. O sobrenadante foi coletado e evaporado até a secura.
Posteriormente, a amostra foi extraída com acetato de etila e concentrada por
evaporação para posterior análise por HPLC acoplada à Espectrometria de Massas
com ionização por eletrospray (HPLC-ESI-MS). Catequina e prodelfinidina B3 foram
os principais compostos encontrados e a acetona: água 70:30 v/v mostrou maior
capacidade de extração dos compostos fenólicos.

30
Bonoli et al. [53] avaliaram a eficiência para recuperar compostos
fenólicos em cevada pela extração com solventes pressurizados, empregando a
técnica de Extração Acelerada por Solvente, usando diferentes misturas como
etanol-água 4:1 (v/v); metanol-água 4:1 (v/v); acetona-água 4:1 (v/v) e posterior
hidrólise ácida ou básica. As recuperações das extrações foram avaliadas após a
hidrólise ácida ou hidrólise alcalina e determinadas pela técnica de Eletroforese
Capilar, do inglês, Capillary Electrophoresis (CE) e análises espectrofotométricas.
Acetona aquosa e etanol aquoso extraíram elevadas concentrações de catequinas e
taninos hidrolisados. O aumento na extração dos compostos fenólicos,
especialmente ácidos hidroxicinâmicos, ocorreu quando o tempo de digestão por
hidrólise básica foi prolongado. O estudo concluiu que compostos fenólicos livres
podem ser perdidos no processo de desengorduramento numa etapa prévia à
hidrólise.
Gallegos-Infanteet al. [54] reportaram um estudo no qual os grãos de
cevada foram processados e submetidos à remoção de gordura com hexano
seguido de extração dos compostos fenólicos com acetona/água 70:30 v/v, num
agitador mecânico à temperatura ambiente. O teor de compostos fenólicos totais foi
avaliado pelo método de Folin-Ciocalteau e pelo método de 2,2-difenil-1-picrilidrazil
(DPPH). Os resultados mostraram que o aquecimento dos extratos e a torrefação da
cevada aumentou o teor de compostos fenólicos em comparação com os extratos
sem o processamento, mas a germinação reduziu o teor de compostos fenólicos
[54]. Os autores reportaram que o aumento na concentração de compostos fenólicos
apóso aquecimento dos extratos e torrefação pode ser associado à liberação dos
mesmos a partir da decomposição dos constituintes celulares em função do
tratamento térmico. Similarmente, Siddhuraju & Becker [55] também observaram a
diminuição do teor de compostos fenólicos após a germinação e o aumento após o
cozimento ou ao assar o grão de feijão.
Entre os vários solventes extratores relatados na literatura para a
extração de compostos fenólicos em cevada, vale ressaltar a eficiência do etanol
como solvente extrator para este tipo de análise. A variabilidade na capacidade
antioxidantefoi determinada por diferentes métodos independentes para a avaliação
da atividade antioxidante, incluindo os testes de DPPH,
Ressonância paramagnética eletrônica (RPE) e análise térmica diferencial, do inglês
Differential thermal analysis (DTA) após extração com etanol 65 %, sob aquecimento

31
a 80 °C. Destaca-se que a DTA é uma técnica rápida para investigação da
capacidade antioxidante nos cereais e foi utilizada para a determinação da
estabilidade à oxidação das culturas selecionadas como trigo, cevada e milho [42].
Em pesquisa recente, compostos fenólicos como a rutina, ácidos cafeico
e ferúlico foram identificados e quantificados em diferentes cultivares de cevada
brasileira em extração com mistura de solvente hidroetanólico 80:20 v/v e banho
ultra-sônico para avaliar os fatores climáticos no conteúdo de compostos fenólicos
[56]. Os autores concluíram que uma menor incidência solar, maior índice
pluviométrico e uma menor temperatura média foram considerados fatores
favoráveis no aumento do conteúdo de fenólicos totais e de rutina nas cultivares de
cevada de diferentes safras.
Em outro trabalho, os compostos fenólicos foram extraídos da casca de
cevada utilizando a extração com fluido pressurizado, usando etanol ou o líquido
iônico 2-metil-hidroxietil acetato de amônio. Os melhores resultados para extração
de compostos fenólicos (189,1 ± 3,1 mg/g de casca) foram obtidos com o líquido
iônico pressurizado. Já para extração de carboidratos (450,3 ± 7,8 mg/g de casca),
os melhores resultados foram com o etanol pressurizado [57]. Concluiu-se que
dentre as variáveis avaliadas para otimizar a extração usando fluido pressurizado
para determinação de carboidratos e compostos fenólicos, a temperatura foi
considerada a variável mais significativa quando comparada com a razão do fluxo e
concentração do etanol, parâmetros avaliados na etapa de otimização [57].
Um procedimento muito comum na determinação de compostos fenólicos
em cevada é a hidrólise. A hidrólise alcalina e a hidrólise ácida são bastante citadas
nos procedimentos de preparo de amostras de compostos fenólicos em alimentos
[17, 28]. A hidrólise ácida e a saponificação são os meios mais comuns de liberar os
ácidos fenólicos, ainda que possam se decompor sob estas condições. O método de
hidrólise ácida envolve o tratamento do extrato, como por exemplo com o ácido
clorídrico em refluxo [28]. Já a hidrólise básica consiste no tratamento da amostra
com solução contendo um álcali, normalmente hidróxido de sódio. As principais
variáveis investigadas na hidrólise química são a concentração do ácido/base,
tempo de hidrólise e temperatura. Neste contexto, alguns trabalhos são citados na
literatura destacando a importância desse tipo de procedimento na determinação de
compostos fenólicos.

32
Após a hidrólise alcalina do extrato metanólico de cevada, 7 compostos
fenólicos foram separados por HPLC/UV. Para isso, 1 g de cevada foi hidrolisado
com 50 mL de NaOH 2 mol L-1 sob atmosfera de nitrogênio, por 4 h. A mistura foi
acidificada com HCl 2 mol L-1 até pH 1 e extraída 3 vezes com acetato de etila. Três
compostos, o ácido trans-ferúlico, o ácido trans-cumárico e o ácido cis-ferúlico,
foram quantificados [58].
Em 2008, outro trabalho realizado por Kvasnicka et al. [59] relataram que
submetendo as amostras de cevada pulverizadas dispersas em solução alcalina de
NaOH ou solução ácida de HCl, foi possível hidrolisar seus compostos fenólicos,
facilitando a separação da matriz heterogênea que compõe seus grãos. Após a
hidrólise, uma extração fazendo uso de um solvente como metanol auxiliado por
ultrassom pôde ser eficaz para posterior análise por CE e HPLC. Foi possível a
determinação da concentração do ácido ferúlico de 43,4 mg 100 g-1 em uma das
amostras de cevada por CE e 43,0 mg 100 g-1 por HPLC.
Com duas etapas de hidrólises alcalinas e outra com extração
hidroetanólica, seguidos por análise por cromatografia líquida e detecção por
espectrometria de massas com ionização por eletrospray e analisadores por tempo
de voo e quadrupolo, foi possível identificar o ácido p-cumárico, ácido gálico e
isoquercitrina nos grãos de cevada. A extração com etanol 50% atingiu o maior
conteúdo fenólico livre em comparação com água e etanol absoluto. Notou-se
também que a elevação da temperatura até 80 °C aumentou os teores fenólicos [41].
Diferentes solventes extratores foram avaliados para obter a composição
de compostos fenólicos por métodos espectrofotométricos com absorção em 280,
320 e 370 nm pelo método de Folin-Ciocalteu. Foram utilizados o metanol, etanol e
acetona misturados com água na proporção de 4:1 (v/v) para extração de compostos
fenólicos livres. A mistura etanólica forneceu a maior recuperação dos compostos
fenólicos. Também foi realizada a comparação entre a hidrólise alcalina e a hidrólise
ácida. Notou-se que a concentração de ácidos fenólicos tais como cinâmico, ferúlico
e cumárico aumentou quando o tempo de digestão na hidrólise alcalina foi
prolongado, sendo que o ácido ferúlico foi o mais abundante nas amostras de
cevada. Houve um decréscimo na concentração de compostos fenólicos quando a
hidrólise ácida foi utilizada [53].
Em outro trabalho, algumas amostras de cevada foram submetidas à
hidrólise com solução de 3% de H2SO4 por 15 min a 130°C na razão líquido/ sólido

33
de 8:1 g/g. Os sólidos foram separados por filtração, lavados com água, secos com
ar e a remoção das ligninas foi feita com acetato de etila. O fracionamento dos
extratos foi realizado com diferentes razões de metanol:água. O processo de
fracionamento usado permitiu a obtenção de extratos com diferentes atividades
antioxidantes [60].
Em 2006, Freitas [61] observou o aumento do efeito antioxidante de forma
proporcional ao aumento da concentração de polifenóis totais em diferentes tipos de
cerveja. Para determinar os polifenóis totais foi utilizado o método de Folin-
Ciocalteu. Os conteúdos fenólicos nas cervejas analisadas variaram de 249,73 a
808,58 mg/L. A cerveja escura de trigo apresentou os maiores valores de polifenóis
totais, seguida das cervejas escuras de cevada, as cervejas claras de trigo e as
cervejas claras de cevada.
1.3.1. QuEChERS
Em 2003, Anastassiades e colaboradores relataram um atraente método
para preparo de amostras denominado QuEChERS (rápido, fácil, barato, efetivo,
robusto e seguro, do inglês, quick, easy, cheap, effective, rugged and safe) com o
objetivo de superar as limitações práticas das técnicas de preparo de amostra de
multirresíduos de agrotóxicos disponíveis, introduzindo um novo procedimento de
preparo de amostras para explorar as possibilidades oferecidas pela instrumentação
analítica moderna [62]. Este método cobre um grande escopo de analitos, incluindo
analitos polares, moderadamente polares e apolares em várias matrizes. O
procedimento envolve uma etapa única de extração da amostra com acetonitrila,
seguido de um particionamento líquido-líquido pela adição de sulfato de magnésio
anidro e cloreto de sódio. A remoção de água e a purificação são realizadas
simultaneamente com extração em fase sólida dispersiva (d-SPE, do inglês
Dispersive Solid Phase Extraction), usando sulfato de magnésio e um sorvente de
amina primária secundária (PSA, do inglês Primary Secundary Amine).
A utilização de acetonitrila como solvente possibilita a extração de uma
menor quantidade de co-extrativos lipofílicos provenientes da amostra, como por
exemplo, ceras, gorduras e pigmentos. Por outro lado, a acetonitrila possibilita uma
ampla faixa de extração de compostos com diferentes polaridades e, quando

34
acidificada permite eficazes recuperações de agrotóxicos que geralmente
apresentam problemas de estabilidade [62]. Além disso, a acetonitrila é mais
adequada para LC-MS/MS do que acetona e acetato de etila.
Na etapa de partição, a adição de sais para promover o efeito salting out
proporciona melhores percentuais de recuperação para os analitos polares, uma vez
que os sais diminuem a solubilidade destes compostos na fase aquosa, bem como a
quantidade de água na fase orgânica e vice-versa [63]. A escolha do MgSO4 no
desenvolvimento do método QuEChERS foi devido à maior capacidade de remover
água quando comparado com outros sais. Além disso, sua hidratação se trata de
uma reação exotérmica, tendo como resultado o aquecimento entre 40 e 45 °C da
amostra, favorecendo a extração, especialmente dos compostos apolares [63].
O procedimento de agitação manual ou com auxílio do agitador tipo vórtex
possui várias vantagens em relação à agitação mecânica, tais como possibilidade de
realizar a extração a campo; a extração ocorre em um único frasco fechado; rapidez,
uma vez que não há necessidade de lavagem do homogeneizador no intervalo entre
as extrações. Portanto, no método QuEChERS, a agitação pode ser manual ou
utilizando vórtex [62].
A etapa de limpeza ou clean-up é fundamental para aumentar a
confiabilidade dos resultados e promover robustez ao sistema cromatográfico.
A d-SPE proposta por Anastassiades et al. [64] está relacionada com um
procedimento muito simples para ser empregado na limpeza de diferentes extratos
destinado à análise cromatográfica. Na proposta original agita-se o extrato (1 mL)
com pequena quantidade de sorvente PSA (25 mg). A função deste sorvente é de
remover açúcares, ácidos graxos, ácidos orgânicos e pigmentos [65]. A agitação tem
como objetivo a distribuição uniforme do sorvente e tende a favorecer a etapa de
limpeza do extrato. Após a centrifugação, uma alíquota do extrato final é retirada
para análise. Assim, o sorvente atua como um filtro químico, retendo os coextrativos
da matriz. A d-SPE busca alcançar um extrato final com menor quantidade de
interferentes, aliada a um menor custo quando comparada com outras técnicas
convencionais [63, 66].
O método QuEChERS apresenta algumas vantagens sobre os métodos
tradicionais de preparo de amostras para determinação de resíduos de agrotóxicos,
dentre eles podem-se citar: altos porcentuais de recuperação (> 85%) para um
grande número de compostos de diferentes polaridades e volatilidades.

35
A desvantagem do método QuEChERS está na relação massa de
amostra/volume de extrato final que é de 1 g por 1 mL. Este valor é baixo quando
comparado com os obtidos por outros métodos que utilizam uma etapa de
concentração, apresentando relação amostra/ extrato final de 2 a 5 g por 1 mL [63] .
Portanto, se a matriz não é uma fonte de ruídos nas análises isto pode conduzir, no
método QuEChERS, a valores de limite de quantificação (LQ) mais elevados, para o
mesmo volume de injeção. No entanto, considerando a alta detectabilidade das
técnicas cromatográficas disponíveis atualmente como Cromatografia Gasosa
acoplada à espectrometria de massas sequencial (GC-MS/MS) e Cromatografia
Líquida acoplada à espectrometria de massas sequencial (LC-MS/MS), o método
QuEChERS é adequado e constitui o estado da arte para determinação de
agrotóxicos.
1.3.2. Diferentes versões do método QuEChERS
Mesmo com a versão original do método que forneceu excelentes
resultados para diferentes tipos de amostras, algumas aplicações mostraram que
certas substâncias químicas apresentavam problemas de estabilidade e/ ou
recuperação dependendo do pH da matriz [66]. Em 2005, de acordo com Lehotay et
al. [67], a adição de uma etapa de tamponamento foi a primeira modificação no
método QuEChERS, com o objetivo de obter recuperação entre 70 e 120%.
O método QuEChERS e suas várias versões modificadas têm sido
utilizado com sucesso para a extração de agrotóxicos de uma variedade de frutas e
vegetais. Anastassiades e Lehotay [64] e Lehotay et al. [67] modificaram o método
QuEChERS original, não tamponado, passando a utilizar um tampão acetato e
citrato, respectivamente, para evitar a degradação de certos agrotóxicos sob
condições alcalinas. Na Figura 4 é possível diferenciar as três diferentes versões
deste método.

36
Figura 4: Representação das etapas das principais versões do método QuEChERS a) original; b) acetato; e c) citrato [62].
Em 2005, Lehotay et al. [67] desenvolveram o método QuEChERS
acetato, no qual o efeito tamponante é decorrente da adição de acetato de sódio.
No método QuEChERS acetato, a extração é efetuada com acetonitrila contendo 1%
(v/v) de ácido acético (HAc) e efeito tamponante (pH=4,8) é decorrente da adição de
acetato de sódio.
Em 2007 este método foi adotado como método oficial da Association of
Official Analytical Chemists (AOAC) para a determinação de resíduos de agrotóxicos
em alimentos [68].
Em 2007, Anastassiades et al. propuseram o desenvolvimento do método
QuEChERS citrato, que utiliza uma mistura de citrato de sódio di-hidratado e sesqui-
hidratado como responsáveis pelo efeito tamponante (pH=5,0-5,5) [69]. A partir
disso, o Comité Européen Committee de Normalisation (CEN), oficializou o método
“QuEChERS citrato” como método de referência na União Européia [70].
Outra modificação muito relevante na etapa de limpeza foi o uso de uma
maior quantidade de PSA na etapa de d-SPE em amostras de cereais com o
objetivo de remover, de forma mais eficiente, os ácidos graxos co-extraídos,

37
açúcares e pigmentos [71]. Além do uso de PSA, o carbono grafitizado também
pode ser usado na etapa de limpeza para redução do teor de pigmentos nos extrato
provenientes de amostras vegetais a partir da adição de uma pequena quantidade
de carvão ativado ou carbono grafitizado. Este possui uma grande área superficial e
contém grupos altamente polares na superfície com alto potencial para formação de
ligações de hidrogênio.
Aplicações diversas do método QuEChERS são citadas na literatura. Em
2012, Hackbart et al. usaram o método QuEChERS nas versões original e acetato
para determinação de ocratoxina e citrinina em arroz e farelo de arroz [72]. As
condições otimizadas ocorreram quando foi utilizada a maior quantidade de água (20
mL) e menor quantidade de terra diatomácea (200 mg).
Em 2005, a partir dos excelentes resultados obtidos para frutas e legumes
com o método QuEChERS, levou Lehotay et al. [67] a fazer a avaliação da
determinação de resíduos de agrotóxicos em matrizes que contivessem até 20% de
gordura (leites e ovos). Eles concluíram que alimentos que contêm de 2 a 20% de
gordura também podem conter resíduos de agrotóxicos tanto lipofílicos como
hidrofílicos, e portanto, seria fundamental desenvolver métodos analíticos capazes
de determinar analitos com uma ampla faixa de polaridade. Dentre os alimentos
incluem o leite, ovos, nozes, milho, soja, trigo, cevada entre outros grãos, peixes,
aves, carne de porco, carne bovina e abacate.
Assim, a eficiência da limpeza dos extratos de matrizes gordurosas como
leite, carnes, leite e abacate usando o método QuEChERS foi avaliada empregando
sorventes como PSA, carbono grafitizado, do inglês, graphitized carbon black (GCB)
e ciclo(hetero)alquenila (B38) na etapa de d-SPE [73].
Uma modificação bastante importante na etapa d-SPE foi a adição do
sorvente octadecilsilano (C18) para propiciar uma limpeza mais efetiva de amostras
com elevados teores de gordura [66].
Alguns trabalhos realizados com amostras com elevado teor de gordura,
como o leite bovino, empregaram C18 e PSA na etapa de limpeza [73]. Neste estudo
foram avaliados diferentes sorventes na etapa de limpeza por d-SPE como C18 e
PSA em combinação com MgSO4 anidro. Esta combinação de sorventes também foi
aplicada com sucesso na determinação de agrotóxicos em carne e gordura bovina
por cromatografia gasosa acoplada à espectrometria de massas (GC-MS). Os
percentuais de recuperação obtidos para os agrotóxicos em carne e gordura bovina

38
em diferentes concentrações variaram de 70 a 129%, com estimativa de desvio
padrão ≤ 27%. Em 2009, Prestes et al. determinaram 19 agrotóxicos em amostras
de leite, abacate e ovos [74].
O elevado número de trabalhos que aplica este tipo de preparo de
amostra na determinação de resíduos de agrotóxicos em alimentos demonstra
claramente as vantagens desta técnica que é rápida, fácil, econômica, efetiva,
robusta e segura, como seu próprio nome afirma.
Embora o método QuEChERS seja muito explorado para análise de
agrotóxicos em diversos tipos de matrizes, ainda há pouco relatos na literatura sobre
seu uso na determinação de compostos fenólicos.
Recentemente, Fontana & Bottini [75] determinaram 9 compostos
fenólicos em vinhos produzidos na Argentina empregando o método QuEChERS. A
partir das condições otimizadas, as amostras contendo 5 mL de vinho (previamente
acidificado com 1% de ácido fórmico) foram extraídas usando 2,5 mL de acetonitrila.
Para etapa de separação 1,5 g de NaCl e 4 g de MgSO4 foram adicionados. Em
seguida, 1 mL do sobrenadante da etapa de partição foi submetido à etapa de
limpeza utilizando d-SPE com uma combinação de 150 mg de CaCl2, 50 mg de PSA
e 50 mg de C18 como sorventes. Uma alíquota do extrato foi analisada por
HPLC/UV. Os resultados encontrados para as figuras de mérito foram satisfatórios e
o método proporcionou limites de quantificação variando de 0,03 a 0,26 µg mL-1
entre os analitos. A precisão intermediária ficou abaixo de 12% para todos os
analitos em amostras de vinho tinto e branco.
1.4. Planejamento experimental no preparo de amostras
A quimiometria é a ciência que aplica estatística por meio de
planejamentos de experimentos (fatoriais, de misturas ou mistos) e tem sido
frequentemente utilizada como uma ferramenta no auxílio das otimizações em
química. Assim, muitas vezes, facilitam o entendimento das variáveis relevantes e
consequentemente, reduz o tempo e os custos das análises. Isso também favorece
a análise de dados de uma forma mais rápida, criteriosa e sistemática [76].

39
De forma geral, na aplicação da quimiometria, ressalta-se o potencial do
uso de planejamentos de experimentos como ferramenta auxiliar de otimização em
investigações envolvendo o preparo de amostras [66].
As técnicas de superfície de resposta são ferramentas matemáticas muito
úteis quando se está interessado na otimização de um processo em que se tem a
influência de vários fatores em uma variável resposta, ou seja, os modelos de
superfície de resposta podem ser explorados para determinar condições ótimas para
se trabalhar ou a sensibilidade da variável resposta a mudanças dos níveis dos
fatores de interesse. Para a fase de otimização, é importante ressaltar que a
visualização gráfica da superfície de resposta é possível quando se tem até 2
fatores.
A metodologia de superfícies de respostas é uma técnica de otimização
de sistemas que é baseada em planejamentos fatoriais. Possui duas etapas
distintas, a modelagem e o deslocamento. A primeira é feita ajustando-se modelos
simples às respostas obtidas com planejamentos fatoriais. A segunda trata-se do
deslocamento ao longo do caminho de máxima inclinação de um determinado
modelo, que é a trajetória na qual a resposta varia de forma mais pronunciada [76].
Os resultados obtidos a partir das superfícies de resposta contribuem
para encontrar as variáveis significativas, assim como as condições ótimas dos
fatores.
Alguns trabalhos são citados na literatura destacando a importância do
planejamento experimental para otimização dos procedimentos de extração.
Uma superfície de resposta foi obtida na otimização das condições de
extração em amostras de cevada: porcentagem de solventes orgânicos, temperatura
e tempo. A capacidade antioxidante aumentou ao utilizar 80,2% de metanol e 60,5°C
por 38,36 min, parâmetros obtidos através da superfície de resposta. Compostos
fenólicos de 6 amostras de cevada foram extraídos para obter a superfície de
resposta depois do desengorduramento com hexano e, subsequentemente, os
extratos foram avaliados quanto à sua atividade antioxidante. O teor de compostos
fenólicos totais medido de acordo com o método Folin-Ciocalteau, variou de 13,58 a
22,93 mg de ácido ferúlico por grama de amostra [77].
A determinação de compostos fenólicos foi otimizada por micro-ondas em
grãos de cevada torrada [51]. As melhores condições para obtenção da cevada
torrada com maior atividade antioxidante foi a potência de 600 W para micro-ondas,

40
tempo de aquecimento de 8,5 min e 65,5 g de grãos. A extração com acetona teve a
maior inibição da peroxidação lipídica. Foram avaliadas 3 variáveis independentes
no planejamento experimental tais como potência do micro-ondas, tempo e
quantidade de grãos de cevada com 3 níveis para cada variável, enquanto que as
variáveis dependentes foram atividade antioxidante com a % de inibição usando o
método DPPH e conteúdo de fenólicos totais. As superfícies de respostas
mostraram que mantendo o tempo e a quantidade de cevada constante, houve um
aumento significativo na atividade antioxidante com a elevação da potência das
micro-ondas. No entanto, as superfícies de respostas também mostraram uma
diminuição na concentração de compostos fenólicos devido à degradação, quando
elevadas potências do micro-ondas foram utilizadas. A atividade antioxidante
inicialmente aumentou e depois diminuiu com o tempo de aquecimento e quantidade
de grãos.
Com o objetivo de determinar compostos fenólicos em cascas de cevada,
após a auto-hidrólise com água líquida quente e a partir do fracionamento usando
coluna Sephadex LH-20, constatou-se que dentre os solventes testados, o acetato
de etila permitiu a maior extração de compostos fenólicos. As maiores
concentrações de compostos isolados foram para o ácido benzóico e o ácido
cinâmico. A partir da elaboração de um planejamento experimental de 23 com a
realização de 16 experimentos, considerando 3 variáveis independentes, tais como
pH, T°C e % de etanol. Foi possível concluir que a diminuição na concentração de
etanol diminuiu a concentração de compostos fenólicos para valores mais baixos de
pH. Já outra variável dependente evidenciou, a partir da superfície de resposta, o
aumento da atividade antioxidante com a maior concentração de etanol [78].
1.5.Técnicas de separação para determinação de compostos
fenólicos
As técnicas analíticas mais usadas para a determinação de compostos
fenólicos em cevada incluem a cromatografia líquida de alta eficiência (HPLC) [16,
48, 58] e a eletroforese capilar (CE) [53, 79]. Mais recentemente, a cromatografia
líquida de ultra alta eficiência, do inglês, Ultra High Performance Liquid
Cromatography (UHPLC) [80] têm sido usada para análise de compostos fenólicos.

41
1.5.1. Eletroforese Capilar
A eletroforese capilar (CE) é uma técnica analítica de separação baseada
na migração diferenciada de compostos iônicos ou ionizáveis em um campo elétrico.
Esta técnica que é aplicável na determinação de uma grande variedade
de analitos em amostras diversas possui uma série de vantagens como rapidez,
versatilidade, baixo custo das análises, alto poder de resolução e pouco consumo de
amostras, reagentes e solventes. Por outro lado, esta técnica oferece algumas
limitações como a sua baixa detectabilidade [81].
O equipamento é composto por uma fonte de alta tensão conectada por
dois eletrodos de platina a dois reservatórios contendo uma solução de eletrólitos,
além de um capilar com passagem por um centro óptico de um sistema de detecção,
conectado a um receptor de dados, e a um sistema de introdução da amostra que é
controlado por um computador [82]. A Figura 5 representa um esquema ilustrativo de
um equipamento de Eletroforese Capilar.
A utilização de capilares de sílica fundida na execução da técnica
introduziu a geração do chamado fluxo eletrosmótico (EOF). Este fluxo é a
consequência de uma interação entre a solução e as paredes do capilar. A sílica
fundida é quimicamente caracterizada pela presença de vários tipos de grupo silanol
(SiOH), os quais apresentam caráter ácido. Em contato com a solução aquosa,
alguns desses grupos são dissociados e por este motivo a superfície do capilar
torna-se negativamente carregada, gerando um saldo positivo de espécies
carregadas positivamente na solução. Quando um campo elétrico é aplicado à
superfície, forças elétricas causam um movimento unilateral de íons em direção ao
eletrodo de carga oposta [83].

42
Figura 5: Esquema ilustrativo de um equipamento de eletroforese capilar onde R1 e R2, e1 e e2 são os reservatórios e eletrodos, respectivamente. F representa a fonte de alta tensão, D é o detector, C é o computador para obtenção dos dados e EOF representa o fluxo eletrosmótico, que é gerado após ser aplicado o potencial. Adaptado de Pinheiro [82].
Assim como o EOF é influenciado pelo pH devido a dupla camada elétrica
que se desenvolve na interface entre a sílica e a solução, a migração eletroforética
também depende do pH do eletrólito de corrida, uma vez que a ionização das
moléculas depende do equilíbrio ácido-base. A completa ionização é alcançada
quando o pH do eletrólito estiver duas unidades de pH acima ou abaixo do valor de
pKa, para compostos ácidos e básicos, respectivamente. Assim, a escolha do pH é
uma ferramenta importante para a separação dos analitos [81].
Bonoli et al. [53] empregaram a técnica de CE e fizeram um estudo no
qual compostos fenólicos foram analisadosem 5,5 min, usando um tampão 20 mmol
L-1de tetraborato de sódio, 10 mmol L-1 de dodecil sulfato de sódio e 5 mmol L-1 de
KH2PO4 (pH 9), num capilar de 40 cm × 50 µm de diâmetro interno, 30 kV e 30 °C,
com detecção UV a 200 nm. A seletividade do método de extração para recuperação
dos compostos fenólicos foi avaliada tanto por CE como por espectrofotometria [53].
Os autores concluíram que as amostras de cevada continham principalmente
catequina e proantocianidinas como compostos fenólicos livres e ácidos
hidroxicinâmicos e seus derivados, como compostos fenólicos conjugados.
Verardo et al. [79], com o objetivo de prosseguirem os estudos de Bonoli
et al., compararam diferentes técnicas de separações para compostos fenólicos em
cevada usando UHPLC no modo fase reversa, cromatografia eletrocinética micelar,

43
do inglês, micellar electrokinetic chromatography (MECK) e espectrofotometria [79].
A quantificação por MEKC foi feita utilizando detecção UV, enquanto que no
cromatógrafo a líquido com ionização por eletrospray acoplado ao espectrometro de
massas (HPLC-ESI/MS), as análises foram realizadas apenas para coletar
informações estruturais. Os dois métodos determinaram elevadas concentrações de
flavono-3-ol. Em particular, a quantidade destes compostos encontrada por MECK e
HPLC estavam na razão de 126,9-163,7 mg 100 g-1 e 138,0-172,1 mg 100 g-1. Os
resultados encontrados na validação analítica tiveram semelhanças em termos de
precisão e exatidão para ambos os métodos.
Em 2008, Kavasnika et al. [59] fizeram a detecção de ácidos fenólicos em
cevada empregando a técnica de eletroforese capilar de zona, do inglês, Capillary
Zone Electrophoresis (CZE) com polaridade invertida. No entanto, apenas o ácido
ferúlico foi determinado em concentração máxima de 43,4 mg 100 g-1. Destaca-se o
menor tempo de análise por CE quando comparado com o método por HPLC, 15 e
25 min, respectivamente; melhor separação; melhor resolução; método
ambientalmente correto, com consumo de baixos volumes de eletrólitos. Por outro
lado, CZE tem menor detectabilidade do que HPLC e o tempo de migração é mais
variável do que o tempo de retenção por HPLC [59].
1.5.2. Cromatografia Líquida de Alta Eficiência
A cromatografia líquida de alta eficiência (HPLC) é uma das técnicas
cromatográficas mais empregadas na atualidade. Apresenta muitas vantagens para
as análises de combinações orgânicas. Amostras não voláteis e termolábeis são
preferencialmente analisadas por HPLC devido à sua rapidez na análise,
sensibilidade e precisão nas medições realizadas pelo equipamento, que apesar de
sofisticado e de alto custo, possui aplicação em diversos tipos de matrizes [84,85].
A técnica de HPLC é a mais usada e estabelecida para a determinação
de compostos fenólicos em cevada devido ao seu grande poder de separação e
possibilidade de acoplamento com diferentes detectores [84].
Zhao et al. [48] avaliaram o efeito de uma mistura de solventes na
atividade antioxidante, capacidade de extração e seletividade de compostos
fenólicos em cevada. A análise individual de compostos fenólicos foi realizada por

44
HPLC/UV. A separação dos compostos fenólicos foi alcançada usando
cromatografia no modo fase reversa com eluição por gradiente empregando fase
móvel A (água com 0,1% de ácido acético) e fase móvel B (metanol com 0,1% de
ácido acético) durante 60 min de análise [48]. Foram identificados e quantificados 9
compostos fenólicos tais como catequina, epicatequina, ácidos siringico, ferúlico,
protocatecóico, cafeico, vanílico, gálico e cumárico em diferentes solventes
extratores (etanol, metanol e água) e em 3 variedades de cevada. Os autores
determinaram 56,15µg g-1 para catequina; 12,45 µg g-1 para epicatequina; 10,29 µg
g-1 para o ácido siringico; 12,05 µg g-1 para o ácido ferúlico; 7,88µg g-1 para o ácido
cafeico; 3,62 µg g-1 para o ácido vanílico; 2,71 µg g-1 para o ácido gálico e 1,78 µg g-
1 para o ácido p-cumárico em grãos de cevada.
Os ácidos fenólicos que estão ligados às paredes celulares da cevada
germinada apresentam elevado potencial antioxidante na cerveja. Neste contexto, os
autores fizeram um estudo para determinar compostos fenólicos em cerveja. Depois
da hidrólise alcalina do extrato metanólico de cevada germinada (malte), 7
compostos fenólicos foram separados por HPLC/UV com coluna C18 e eluição por
gradiente usando fase móvel A (água com ácido ortofosfórico) e fase móvel B,
acetonitrila. Os 3 principais compostos, como ácidos trans-ferúlico, trans-cumárico e
cis-ferúlico (177,79; 78,50 e 47,15 µg g-1) respectivamente foram quantificados por
padronização externa. Os autores relataram que os ácidos fenólicos devem ser
protegidos antes do final do processo de malteação, que é um processo de
germinação controlada da cevada e que envolve a umidificação e a secagem, cujos
propósitos são amolecer os grãos para facilitar a moagem, além de introduzir cor e
aromas desejáveis à cerveja. Quando o processo de malteação não é controlado,
seus produtos de degradação, como por exemplo a descarboxilação do ácido
ferúlico podem ser responsáveis pela perda de sabor da cerveja [58].
Aleksenko et al. [86] identificaram vários compostos fenólicos por
HPLC/UV e ionização por eletrospray com espectrometria de massas sequencial
(ESI-MS/MS) em amostras de cevada. Os principais compostos encontrados foram
rutina, catequina, epicatequina, além de procianidinas B3 e C. A análise realizada
por HPLC/MS empregou analisador quadrupolo com ionização por ESI. A separação
foi alcançada a partir de uma coluna C18 com gradiente de eluição por gradiente
usando fase móvel contendo acetonitrila com 10 mmol L-1de ácido acético [86].

45
Um estudo realizado em 2010 por Li et al. [87] determinou 3 ácidos
fenólicos (ácido p-cumárico, ácido ferúlico e ácido sinápico) por HPLC/UV usando
coluna C18 e fase móvel com eluição por gradiente contendo água com 0,1% de
ácido acético no solvente A e acetronitrila com 0,1% de acetonitrila no solvente B em
5 amostras comerciais de cevada. O ácido ferúlico foi o composto fenólico
predominantemente encontrado na razão de 42 a 400 mg kg-1. Outros ácidos
fenólicos (cafeico, cumárico e sinápico) foram identificados por cromatografia líquida
de alta eficiência acoplada à espectrometria de massas sequencial (LC-MS/MS) [87].
Dvorakova et al. [47] constataram que catequina e prodelfinidina B3 foram
os principais compostos encontrados na avaliação de 10 variedades de amostras de
cevada usando HPLC/UV e HPLC-ESI-MS. O estudo qualitativo de compostos
fenólicos foi realizado por HPLC-ESI-MS com coluna C18, usando fase móvel A
contendo 100% de metanol e fase móvel B com 0,1 % de ácido fórmico, com eluição
por gradiente, vazão de 0,2 mL min-1 e volume de injeção de 20 µL, além de
ionização no modo negativo. Já a análise quantitativa dos extratos de acetona:água
(70:30, v/v) foi feita por HPLC/UV, com detecção em 280 nm, nas mesmas
condições cromatográficas empregadas em HPLC-ESI-MS.
Verardo et al. [79] fizeram um estudo comparando dois métodos de
separação de compostos fenólicos, HPLC/UV-MS e MECK. O objetivo do estudo foi
produzir farelo de cevada com compostos fenólicos para obter ingredientes
naturalmente enriquecidos para a indústria alimentícia. A análise por HPLC/UV foi
realizada no modo fase reversa com coluna C18 e detecção em 280 nm. Os autores
concluíram que as amostras de cevada analisadas continham principalmente
catequina e proantocianidinas como fenólicos livres, ácidos hidróxicinâmicos e seus
derivados na faixa de 130,6 a 166,5 mg 100 g-1 nas amostras de cevada [79]. Os
dados de quantificação mostram que tanto a MECK como HPLC apresentaram
resultados semelhantes em termos de precisão e quantidade de compostos
fenólicos (mesma ordem de grandeza).
Em 2011, Hao e Beta [80] realizaram a análise qualitativa e quantitativa
de compostos fenólicos em cascas de cevada no modo fase reversa por HPLC-
MS/MS e HPLC/UV, na qual determinaram ácido ferúlico, ácido p-cumárico, ácido
vanílico e vanilina nas concentrações de 2260,0; 1365,0; 58,6 e 157,2 mg kg-1
respectivamente. A análise por HPLC/UV com detecção em 280 nm foi realizada no
modo reverso com coluna C18, com fase móvel contendo água com 0,1% de ácido

46
acético no solvente A e acetronitrila com 0,1% de ácido acético no solvente B e
eluição por gradiente em 75 min. A identificação e caracterização dos compostos
fenólicos foi baseada na comparação do perfil dos espectros de UV e espectros de
MS/MS com os padrões. Os resultados quantitativos alcançados por HPLC/UV
mostraram que ácido ferúlico e ácido p-cumárico foram os compostos fenólicos
abundantes em cascas de cevada [80].
Um trabalho realizado para obtenção do perfil fitoquímico em cevada
negra constatou a predominância do ácido ferúlico e ácido p-cumárico por HPLC no
modo reverso com coluna C18 e eluição por gradiente usando 0,05% de ácido
trifluoracético em água (solvente A) e 0,05% ácido trifluoracético em acetonitrila
(solvente B). O comprimento de onda usado para detecção foi de 280 nm [88].
1.5.3. Cromatografia Líquida de Ultra Alta Eficiência
A cromatografia líquida de ultra alta eficiência (UHPLC) utiliza os mesmos
princípios da técnica de HPLC, porém com a vantagem de trabalhar com fases
estacionárias de partículas menores que 2 µm [89]. O uso destas partículas,
juntamente com as altas velocidades lineares da FM, proporciona um aumento na
resolução e na detectabilidade, assim como a diminuição no tempo das análises.
As colunas empregadas em UHPLC são de tamanhos reduzidos, 5-10 cm
de comprimento e diâmetros internos de 1,0-2,1 mm, permitindo que maiores
velocidades lineares de fase móvel possam ser alcançadas. Para tanto é necessário
o uso de equipamentos mais adequados capazes de operar a pressões mais
elevadas (9000-15000 psi) que HPLC, o qual opera na faixa de 4000-5000 psi [89,
90].
De modo geral, a UHPLC já está bem estabelecida, tanto em termos de
fases estacionárias, como de instrumentação. É uma técnica que apresenta
facilidade na transferência de um método desenvolvido por HPLC para UHPLC, uma
grande variedade de colunas, equipamentos e detectores disponíveis
comercialmente e isso indica que num futuro próximo esta técnica talvez consiga
superar a HPLC nas análises de rotina, além de usar menores volumes de solventes
orgânicos, gerando menor quantidade de resíduos, de acordo com os princípios da
Química Verde [90, 91].

47
A aplicação da técnica de UHPLC na determinação de compostos
fenólicos pode ser vista no trabalho de Gruz et al. [92]. Os autores determinaram
dezessete ácidos fenólicos em diferentes bebidas, usando UHPLC acoplada a um
espectrômetro de massas sequencial (MS/MS). O método apresentou resultados
satisfatórios quanto às figuras de mérito, como limites de detecção na faixa de 0,15
a 15 pmol e a precisão calculada com desvio padrão relativo de 4,4%. O método foi
aplicado para a análise de sucos de uva, amostras de vinho branco e chá verde. A
análise foi realizada num tempo muito menor (10 min) quando comparada as
análises de rotina comumente usadas por HPLC que variam na faixa de 40 min.
Recentemente, foi desenvolvido e validado um método por UHPLC-
MS/MS para determinação de 8 compostos derivados da catequina em várias
amostras de chá verde, branco e preto em um tempo de 4 min para cada análise. O
método foi validado a partir dos parâmetros de precisão intra e inter-dias, exatidão,
efeito matriz, linearidade, LD e LQ. A proposta do trabalho foi desenvolver um
rápido, eficiente e robusto método por UHPLC-MS/MS com analisador triplo
quadrupolo. Com isso foi possível minimizar o risco de degradação dos analitos,
uma vez que os compostos fenólicos são fotossensíveis, contribuindo para a análise
de um maior número de amostras e reduzindo o consumo de solventes orgânicos e
custo das análises. A coluna C18 usada no sistema UHPLC foi recheada com
partículas de 1,7 µm que permitiram maiores vazões de fase móvel e maior rapidez
nas análises. As amostras foram separadas usando eluição por gradiente composta
por 0,1% de ácido fórmico em água (solvente A) e 0,1 % de ácido fórmico em
metanol (solvente B). A média dos valores de catequinas em chás verdes, brancos e
pretos foi de 76,7 mg g-1; 66,1 mg g-1 e 22,7 mg g-1, respectivamente. Os resultados
indicaram que diferentes marcas de chá e custo não desempenharam um papel
significativo na quantidade total de catequinas [93].
1.6. Espectrometria de Massas
A espectrometria de massas pode ser compreendida como uma técnica
analítica que permite a identificação deuma amostra para um determinado composto
isolado, ou de diferentes substâncias em misturas complexas, através da
determinação de suas massas molares na forma iônica, (ou seja, com carga elétrica
líquida, positiva ou negativa), baseada na sua movimentação através de um campo

48
elétrico ou magnético. Esta movimentação é determinada pela razão entre a massa
de um determinado composto (analito) e sua carga líquida, representada por m/z
(razão massa/carga). Assim, conhecendo o valor de m/z de uma molécula é possível
deduzir sua composição química elementar, e com isso determinar sua estrutura
[94].
Dentre os principais componentes de um espectrômetro de massas estão
o sistema de introdução de amostras, uma fonte de ionização, onde são produzidos
íons na fase gasosa; um analisador de massas que identifica íons de acordo com
suas razões massa/carga (m/z); e um detector que geralmente é constituído por um
multiplicador de elétrons [95].
O desenvolvimento dos espectrômetros de massas de múltiplos estágios,
como os triplo e pentaquadrupolos, armadilha de íons (ion traps), setores elétricos e
magnéticos e equipamentos híbridos, do inglês, Time of Flight (QTOF, QTRAP)
permitiu o acesso a técnicas de espectrometria de massas sequencial (MS/MS ou
MSn) e, com isso, ocorreu um aumento significativo das potencialidades analíticas da
técnica.
O analisador de massas é responsável pela seleção e/ou separação de
acordo com a razão m/z dos íons.
O analisador triplo quadrupolo empregado em MS/MS é constituído por 3
quadruplos em série, sendo dois quadrupolos de transmissão (utilizados para
separar íons de mesma razão m/z) interligados por um quadrupolo que atua como
cela de colisão, no qual ocorre a fragmentação dos íons selecionados no primeiro
quadrupolo, geralmente por dissociação induzida por colisão (CID), e é empregado
como direcionador dos íons produzidos ao terceiro quadrupolo. A Figura 6 mostra
um analisador triplo quadrupolo empregado para análises de matrizes diversas [96].
Figura 6: Esquema de um analisador triplo quadrupolo operando no modo SRM onde Q1 e Q3 são quadrupolos de transmissão e Q2 um quadrupolo onde ocorre a fragmentação dos íons.

49
Na DIC, o íon precursor proveniente do primeiro quadrupolo é acelerado
por um potencial elétrico para uma região de alto vácuo no interior do segundo
quadrupolo, onde sofre repetidas colisões com um gás inerte de alta energia
(geralmente Ar ou He), induzindo um acréscimo na energia potencial do íon até
ocasionar sua fragmentação, com posterior formação de íons produto.
Assim, o fragmento de maior intensidade é empregado na quantificação
do analito, enquanto o segundo fragmento de maior intensidade é utilizado para
confirmação da autenticidade do analito.
As análises de íons pré-selecionados, formados através de processos de
dissociação ou reações íon/molécula, permitem a obtenção de informações mais
detalhadas quando comparadas àquelas obtidas através de espectros de massas
simples. Com técnicas de MSn obtém-se não somente a razão massa/carga (m/z)
dos íons, mas também, informações estruturais de cada um desses íons, permitindo,
portanto, a “reconstrução” da molécula precursora. A análise de misturas
diretamente por MSn também se tornou possível, já que os íons de cada um dos
seus constituintes podem ser individualmente separados e analisados. Desta forma,
é possível analisar misturas de compostos para as quais separações
cromatográficas não são indicadas [97].
A amostra a ser analisada é introduzida no espectrômetro de massas
através de um inlet (dispositivo para a introdução da amostra no espectrômetro) e
direcionada para a fonte de ionização. No início do desenvolvimento da técnica, a
amostra era introduzida pela vaporização direta; com o desenvolvimento das
técnicas cromatográficas, tornou-se bastante comum o uso de um cromatógrafo
como fonte de introdução da amostra no espectrômetro de massas [98]. Existe uma
grande variedade de técnicas de ionização, cuja escolha deve levar em conta as
propriedades físico-químicas do analito e a energia transferida durante o processo
de ionização.
Atualmente, para a análise de matrizes diversas, o modo mais empregado
é a cromatografia líquida acoplada à espectrometria de massas sequencial (LC-
MS/MS), com ionização por eletrospray e analisador triplo quadrupolo, devido à sua
elevada seletividade, que possibilita um aumento na detectabilidade e diminuição da
influência do efeito matriz [99].
A ionização por eletrospray (ESI) é uma das principais técnicas de
ionização em pressão atmosférica e também uma das mais importantes na

50
determinação estrutural de diversas moléculas, consistindo em mais uma das
técnicas no conceito de ionização suave, permitindo a formação de íons a partir de
macromoléculas, superando sua propensão em fragmentar-se quando ionizadas.
O processo de ionização por ESI ocorre quando um solvente volátil
contendo os analitos é bombeado através de um fino capilar de aço inoxidável. Uma
alta tensão é aplicada na ponta deste capilar, e, como consequência deste forte
campo elétrico, a amostra que sai pelo capilar estará dispersa em um aerosol
composto por gotículas de solvente e analito altamente carregadas [100].
Este processo é auxiliado por um gás inerte (geralmente nitrogênio e
chamado de gás de nebulização) que é introduzido na fonte ESI, fluindo
externamente ao capilar. Este fluxo de gás está em temperaturas elevadas (150-200
º C) e auxilia na evaporação do excesso solvente e na formação do aerosol.
O cone começa a se formar quando as microgotas de um solvente
eletricamente condutivo são expostas a fortes campos elétricos, causando uma
deformação em sua superfície, que então surge como um cone com os lados
convexos e ponta arredondada. Conforme o campo elétrico aumenta excedendo a
tensão do solvente, o cone se inverte e um jato é lançado, formando um spray.
Neste momento ocorre a formação de minúsculas gotas de solvente contendo o
analito e repleto de cargas elétricas positivas ou negativas (de acordo com o modo
de análise) [101]. Como as microgotas de solvente sofrem ação de um fluxo de N2
aquecido, elas passam a evaporar, tendo seu diâmetro reduzido rapidamente. Com
isso as cargas ali contidas passam a ter maior proximidade, e, consequentemente,
maior repulsão eletrostática. Quando esta repulsão atinge uma força maior do que a
tensão do solvente ocorre um colapso, que promove ruptura da microgota com
consequente formação de outras gotas de solvente e analito menores ainda. Várias
explosões se iniciam até que são produzidos íons do analito a partir dessas gotas,
os quais são transferidos para dentro do espectrômetro de massas por uma série de
dispositivos de focalização [102]. A Figura 7 apresenta a esquematização da
ionização por eletrospray, operando no modo positivo.

51
Figura 7: Mecanismo da evaporação do íon e da explosão coulômbica [103].
Durante a ionização por eletrospray, três tipos de íons podem ser
gerados: íons moleculares (M+˙ ou M-˙), moléculas protonadas ou desprotonadas
([M+H]+ou [M-H]-e moléculas catiônicas ou adutos (por exemplo, ([M+Na]+ ou [M+Cl]-
), sendo M a massa do analito em questão. O modo de operação positivo ou
negativo é estabelecido pelos modificadores adicionados à FM, como exemplo de
um ácido (modo positivo), geralmente ácido fórmico, ou uma base (modo negativo).
Uma das vantagens do eletrospray é que a ionização ocorre em solução,
de forma branda, favorecendo que os compostos termolábeis possam ser ionizados
sem que ocorra degradação [104].
1.6.1. Efeito matriz em espectrometria de massas
O efeito matriz é atribuído à variação da eficiência de ionização que pode
causar alterações negativa ou positiva no sinal do detector, devido à supressão ou
acréscimo de ionização e, como consequência, a quantificação dos analitos deixa de

52
ser confiável, uma vez que ocorreram alterações causadas na precisão, exatidão e
detectabilidade do método. O mecanismo exato do efeito matriz ainda não é
conhecido, mais é comum assumir que os compostos coeluídos competem com os
analitos e a ionização será favorecida para as moléculas que são mais ionizáveis e
estão em maiores quantidades na amostra [105].
O efeito matriz pode causar um aumento ou a diminuição da resposta do
espectrômetro para um analito presente no extrato da amostra comparado ao
mesmo analito em solvente orgânico. O estudo do efeito matriz é relevante quando
se deseja trabalhar com uma curva analítica em solvente, ou seja, sem a presença
da matriz [106].
As dificuldades encontradas nas análises em decorrência do efeito matriz
não se limitam a métodos analíticos baseados em UHPLC-MS/MS. Eles também
podem estar nos outros métodos mais convencionais baseados, por exemplo, em
HPLC-UV e cromatografia líquida de alta eficiência com detector de fluorescência
(HPLC-FLU). Geralmente os métodos de preparo de amostras quando se utiliza
UHPLC ou HPLC-MS/MS são normalmente mais simplificados e muitas vezes, não
há uma preocupação com a separação cromatográfica devido à alta seletividade da
detecção por MS/MS. Nesse sentido, o efeito matriz tornou-se mais evidente e mais
preocupante nos métodos UHPLC ou LC-MS/MS em comparação com os métodos
convencionais, devido a simplificação do método de preparo de amostra [105].
Mesmo que os mecanismos de supressão ou ganho de sinal não estejam
bem estabelecidos, o efeito matriz é altamente dependente da natureza do analito e
da matriz de análise. Observa-se que a eficiência de ionização de compostos
polares é mais pronunciadamente afetada por componentes da matriz coeluídos do
que quando se trabalha com compostos menos polares [107].
A etapa de preparo de amostras é considerada muito relevante para
minimizar o efeito da matriz, provocado pelo aumento ou diminuição do sinal
analítico. Portanto, a etapa de clean-up é fundamental para eliminar compostos
provenientes da matriz presentes no extrato, os quais podem interferir no método
analítico.
Algumas equações são empregadas para o cálculo do efeito matriz tais
como:

53
onde a % do efeito matriz é referente ao aumento ou supressão de sinal, pela razão
entre o coeficiente angular da curva analítica construída na matriz e o coeficiente
angular da curva analítica construída em solvente [108].
O efeito matriz (Matrix Effect – ME) também pode ser avaliado pela razão
da resposta analítica do extrato final da amostra, que foi fortificado com a solução
dos padrões, pela solução dos padrões em solvente, na mesma concentração [107].
Os valores de efeito matriz negativos sugerem a ocorrência de supressão
do sinal analítico, enquanto que os valores positivos sugerem ganho no sinal,
decorrentes da interação do analito com a matriz da amostra em análise.
Quando as variações nas respostas do detector entre o extrato fortificado
e a solução dos padrões em solventes, ambos na mesma concentração forem < ±
20%, considera-se que há um leve efeito matriz. Valores superiores a ± 20%
apontam que o efeito matriz é significativo e ele pode ser causado por supressão de
ionização, conduzindo à perda de sinal, ou acréscimo de ionização, que resulta em
um ganho de sinal analítico [109].
Um fator que pode reduzir o efeito matriz é a diluição da amostra. Em
2009, Kruve et al. concluíram que a diluição da amostra pode minimizar o aumento
dosinal analítico ou a supressão iônica [108]. Entretanto, essa prática da diluição
pode ser comprometida pelo aumento do limite de detecção, dependendo da
natureza da amostra.
1.7. Validação de métodos analíticos
A validação é uma etapa que deve ser realizada após o desenvolvimento
do método analítico e antes das análises das amostras disponíveis comercialmente,
com o objetivo de gerar resultados com credibilidade, precisão e exatidão

54
adequados. Assim, a validação pode ser compreendida como um processo de
verificação para garantir que o método desenvolvido é capaz de assegurar
confiabilidade às medidas experimentais do método [110, 111].
De maneira geral, o processo de validação de um método tem como
objetivo fornecer evidências de que o método é adequado para realizar o que se
propõe com exatidão, reprodutibilidade e credibilidade [112].
O desenvolvimento de um novo método já validado, quando bem definido
e documentado, deve sempre fornecer evidências organizadas, claras e objetivas de
que o método e o sistema são adequados para o uso desejado.
Não há um consenso comum a respeito de quais parâmetros devem ser
validados, o que varia de uma área para outra. No entanto alguns órgãos e
instituições criaram guias de validação que são facilmente aceitos [110]. Dentre os
órgãos podem-se citar: USA-FDA, ANVISA E MAPA entre outros.
Os parâmetros para validação de métodos têm sido definidos em
diferentes grupos de trabalho de organizações nacionais e internacionais. Assim,
órgãos como ICH, IUPAC, ISO, ANVISA, INMETRO exigem o item validação de
métodos analíticos como um requisito fundamental no credenciamento para
qualidade assegurada e demonstração de competência técnica [111]. Alguns
parâmetros como especificidade/seletividade, faixa de trabalho e faixa linear de
trabalho, linearidade, sensibilidade, limite de detecção, limite de quantificação,
exatidão e precisão são comumente avaliados no processo de validação.
Vale ressaltar que estes órgãos são responsáveis por acompanhar e
credenciar a competência de laboratórios de ensaios, porém as diferentes
terminologias e até algumas características de desempenho do método podem ter
alterações de acordo com o órgão competente.
1.7.1. Seletividade
Um método analítico é considerado seletivo quando é capaz de produzir
respostas para vários analitos, mas também é capaz de distinguir a resposta de um
analito dos outros [113]. É a capacidade de um método em quantificar um analito
sem equívocos na presença da matriz que podem estar presentes as impurezas,
produtos de degradação, entre outros.

55
Na cromatografia líquida, a seletividade pode ser alcançada através da
otimização das etapas de extração e de separação dos analitos. Em relação à
extração, a seletividade do método depende do tipo de técnica de preparo de
amostra escolhida e de todas as variáveis envolvidas no seu procedimento, como
tipo de solventes, tempo de extração e temperatura. Já a seletividade na etapa de
separação cromatográfica pode variar de acordo com o tipo de fase estacionária da
coluna cromatográfica e composição da fase móvel. A seletividade de um método
analítico pode ser avaliada utilizando-se detectores como arranjo de diodos e
espectrômetro de massas que permitem comparar o espectro do pico obtido na
amostra com o espectro do padrão puro [111].
1.7.2. Linearidade
A linearidade corresponde à capacidade do método em fornecer sinais
que sejam diretamente proporcionais à quantidade do analito em um determinado
intervalo de concentração. A correlação entre o sinal medido (área ou altura do pico)
e a concentração da espécie, na maioria dos casos é definida por uma equação
matemática, que pode ser expressa como uma equação de reta chamada de curva
analítica.
A estimativa dos coeficientes de uma curva analítica usando um conjunto
de medições experimentais pode ser realizada através do método matemático
conhecido como regressão linear, determinado pelo método dos mínimos
quadrados, e é definida pela equação da reta [114].
y = a.x + b, onde:
y = resposta medida ou sinal analítico
x = concentração do analito, variável independente
a = inclinação da curva analítica
b = intersecção da curva com a ordenada, quando x=0 (coeficiente linear).
A escolha da regressão pelo modelo linear e a verificação do ajuste em
função dos valores experimentais da curva podem ser avaliados pela análise da
variância. Calcula-se a porcentagem máxima de variação explicável para os dados
experimentais e a porcentagem de variação explicada (r2) [115].

56
1.7.3. Limite de detecção
É definido como a menor quantidade de um analito que pode ser
detectada, porém não necessariamente quantificada como um valor exato.
O limite de detecção (LD) pode ser expresso como:
Onde;
s = estimativa do desvio padrão da resposta
S = inclinação (coeficiente angular) da curva analítica
O “s” é a estimativa do desvio padrão da resposta, que pode ser a
estimativa do desvio padrão do branco, da equação da linha de regressão ou do
coeficiente linear da equação e S é a inclinação (tangente) ou coeficiente angular da
curva analítica [114].
1.7.4. Limite de quantificação
Limite de quantificação (LQ) é considerado como a menor concentração
do analito que pode ser medida, utilizando um determinado procedimento
experimental. O LQ segue o mesmo princípio do LD, porém com a relação
sinal/ruído de 10:1, ou seja:
Onde;
s = estimativa do desvio padrão da resposta
S = inclinação (coeficiente angular) da curva analítica
Na maioria das vezes, o LQ é determinado como sendo o ponto de menor
concentração da curva analítica, excluindo-se o branco [114].

57
Para as técnicas cromatográficas, a melhor condição para determinação
de LD e LQ é o baseado nos parâmetros da curva analítica, pois este é
estatisticamente mais confiável que os demais, uma vez que os valores destes
limites podem variar dependendo-se do tipo e tempo de uso da coluna
cromatográfica, das condições cromatográficas e até da sensibilidade dos analitos. A
curva analítica deve conter a concentração correspondente ao LQ [111].
1.7.5. Precisão
A precisão representa a dispersão de resultados entre ensaios
independentes, repetidos de uma mesma amostra, amostras semelhantes ou
padrões, sob condições definidas. Normalmente, a precisão é avaliada em termos
de desvio-padrão (DP) e desvio padrão relativo (DPR), conhecida como coeficiente
de variação (CV) e expressa pela equação 5:
= média aritmética das medições;
s = desvio padrão das medições;
A taxa de aceitação de DPR nos processos de validação de métodos
analíticos é de até 20%, dependendo da complexidade da amostra. Assim, a
precisão em validação de métodos analíticos é considerada em três níveis
diferentes: repetitividade, precisão intermediária e reprodutibilidade [111].
A repetitividade representa a concordância entre os resultados de
medições sucessivas de um mesmo método, sob as mesmas condições de medição:
mesmo procedimento, mesmo analista, mesmo local, mesmo instrumento usado sob
as mesmas condições e repetições em curto espaço de tempo.
A precisão intermediária representa a precisão sobre a mesma amostra
quando um método é aplicado várias vezes pelo mesmo laboratório, porém variando
condições como analistas, equipamentos e tempos diferentes [113].
A reprodutibilidade é o grau de concordância entre os resultados de
medições de uma mesma amostra, efetuadas sob condições variadas (mudança de
operador, local, equipamentos, etc...[113].

58
1.7.6. Exatidão
Consiste no grau de aproximação entre o resultado de um experimento e
o valor de referência aceito como verdadeiro [113].
A recuperação é definida como a proporção da quantidade da substância
de interesse, presente ou adicionada na proporção analítica do material teste que é
extraída e passível de ser quantificada. Segundo a ICH e a ANVISA é necessário um
mínimo de 9 determinações, envolvendo um mínimo de 3 níveis de concentração
para a determinação da exatidão.
Os processos mais utilizados para avaliar a exatidão de um método são:
materiais de referência; comparação de métodos; ensaios de recuperação e adição
padrão.
Os materiais de referência são acompanhados de um certificado que
possui o valor de concentração de uma dada substância, ou outra grandeza para
cada parâmetro e uma incerteza associada [111]. Já na comparação de métodos,
emprega-se o método em desenvolvimento e os resultados conseguidos através de
um método de referência, avaliando o grau de proximidade entre os resultados
obtidos pelos dois métodos, ou seja, o grau de exatidão do método. Nos ensaios de
recuperação, tem-se a proporção da quantidade da substância de interesse,
presente ou adicionada na porção analítica do material teste, que é extraída e
passível de ser quantificada. Na avaliação da exatidão usando o método adição
padrão, cabe o uso quando for difícil ou impossível preparar um branco da matriz
sem a substância de interesse [111]. No método de adição padrão, quantidades
conhecidas da substância são adicionadas em diferentes níveis numa matriz da
amostra, antes do procedimento de preparo da amostra, que já contenha
quantidades (desconhecidas) da substância a fim de recuperá-las no término da
análise.

59
2.OBJETIVOS
2.1. Objetivos gerais
O objetivo deste trabalho consistiu na avaliação de diferentes métodos de
preparo de amostras buscando a investigação (qualitativa e quantitativa) dos
compostos fenólicos presentes na cevada.
Após o desenvolvimento dos métodos de extração, fez-se a validação
usando a técnica de cromatografia líquida de ultra alta eficiência acoplada à
espectrometria de massas sequencial (UHPLC-MS/MS) e a aplicação de um dos
métodos em diferentes amostras de cevada de diferentes locais.
2.2. Objetivos específicos
� Avaliação de técnicas clássicas de extração e caracterização dos extratos por
ESI(-)-MS/MS e UHPLC-MS/MS, de modo a se detectarem os possíveis
compostos fenólicos ou derivados presentes na cevada.
� Otimização das variáveis de extração das técnicas clássicas por meio de um
planejamento experimental.
� Avaliação do método QuEChERS para extração de compostos fenólicos em
cevada.
� Validação dos métodos por UHPLC-MS/MS para a quantificação dos
compostos fenólicos nos extratos de cevada.
� Identificação e quantificação dos compostos fenólicos em diferentes amostras
de cevada de diferentes locais.

60
3. PARTE EXPERIMENTAL
3.1. Reagentes
Os padrões catequina, epicatequina, rutina, miricetina, ácido sinápico,
ácido vanílico, ácido cafeico, ácido ferúlico, ácido p-cumárico, ácido clorogênico e
ácido protocatecóico foram adquiridos da Sigma-Aldrich (Diadema, Brasil). Todos os
padrões foram de grau analítico (≥ 98%) e usados sem purificação adicional.
Água ultrapura foi obtida a partir de um sistema Milli-Q UV Synergy da
Millipore (Molsheim, França). Também foram usados papel de filtro qualitativo
Whatman com espessura de 55 mm, filtro Millex de 0,22 µm de porosidade da
Millipore e tubos de polipropileno para centrífuga tipo Falcon Nest (Xangai, China)
com volume de 50 mL e vials com capacidade de 2 mL da Agilent.
Foram empregados os reagentes etanol e dimetilsulfóxido (DMSO) grau
HPLC, Panreac (Barcelona, Espanha); hidróxido de amônio e ácido clorídrico, ácido
acético glacial e ácido fórmico, todos Synth (Diadema, Brasil); hexano grau HPLC e
acetonitrila grau HPLC, Tedia (Fairfield, EUA) e metanol grau HPLC, Merck (Madri,
Espanha). O óleo de silicone usado para o controle da temperatura na extração em
sistema de refluxo foi obtido da Synth (Diadema, Brasil).
Também foram utilizados os reagentes sólidos sulfato de magnésio, P.A,
Vetec (Rio de Janeiro, Brasil), acetato de sódio P.A, Sigma-Aldrich (Missouri, EUA),
sorvente de extração de sílica recoberta com grupos C-18, com tamanho de
partícula de 40µm, Agilent Technologies (Santa Clara, EUA) e amina primária-
secundária (PSA), Varian (São Francisco, EUA).
As soluções estoque dos padrões de concentração 1000 mg L-1 foram
preparadas dissolvendo-se a quantidade adequada dos padrõesapós pesagem
cuidadosa da respectiva massa, em metanol grau cromatográfico, filtrado em
membrana porosa e desgaseificado em ultrassom), e subsequente homogeneização
das soluções, as quais foram armazenadas em frascos âmbar fechados em
geladeira a 5 °C. As soluções de trabalho dos compostos fenólicos foram
preparadas pela diluição, em metanol, das soluções estoque.
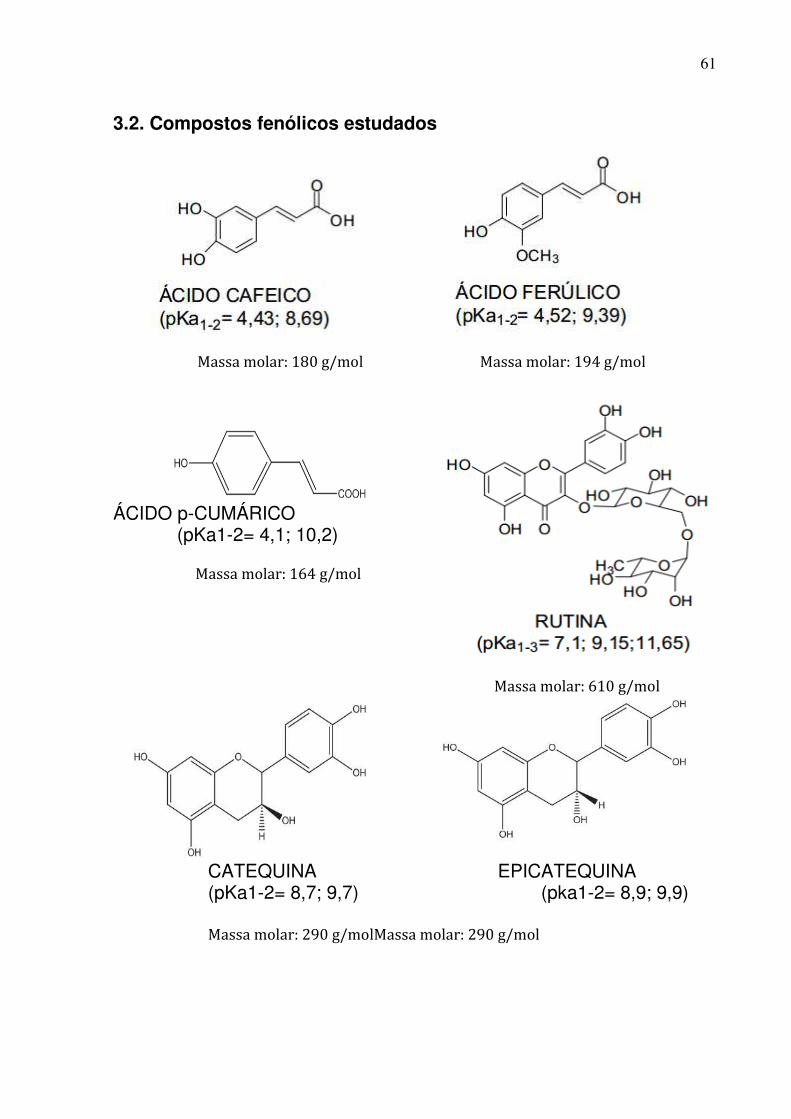
61
3.2. Compostos fenólicos estudados
Massa molar: 180 g/mol Massa molar: 194 g/mol
ÁCIDO p-CUMÁRICO (pKa1-2= 4,1; 10,2)
Massa molar: 164 g/mol
Massa molar: 610 g/mol
CATEQUINA EPICATEQUINA (pKa1-2= 8,7; 9,7) (pka1-2= 8,9; 9,9) Massa molar: 290 g/molMassa molar: 290 g/mol

62
ÁCIDO PROTOCATECÓICO ÁCIDO CLOROGÊNICO
(Ácido 5-cafeoilquínico) Massa molar: 182 g/mol Massa molar: 354 g/mol
MIRICETINA ÁCIDO VANÍLICO
Massa molar: 318 g/mol Massa molar: 168 g/mol
ÁCIDO SINÁPICO
Massa molar: 224 g/mol
Figura 8: Estruturas químicas dos compostos fenólicos em estudo.

63
3.3. Equipamentos
Na análise cromatográfica foi utilizado o UHPLC-MS/MS Acquity,
acoplado ao espectrômetro de massas Waters, modelo Micromass Quatro Micro TMAPI, com o analisador de massas do tipo triplo quadrupolo (QqQ) e fonte de
ionização por electrospray (ESI) no modo negativo, da Waters (Milford, EUA). O
sistema de aquisição de dados foi controlado pelo software Mass Lynx versão 4.1. A
coluna era Acquity UHPLC BEH C-18 1,7 µm (2,1 × 50 mm d.i.) da Waters.
As análises de infusão direta também foram realizadas por espectrometria
de massas sequencial (MS/MS), modelo Micromass Quatro Micro TMAPI, da Waters
(Milford, EUA) com uma fonte de ESI. Argônio ultra puro foi utilizado como gás de
colisão e nitrogênio (N2) de elevada pureza como gás de nebulização.
Outros equipamentos utilizados foram: agitador magnético, modelo 752,
Fisatom; agitador Vórtex, modelo AP 56, Phoenix; balança analítica com precisão de
0,1 mg, modelo A-250, Fischer Scientific; balança analítica com precisão de 0,01
mg, modelo CP225D, Sartorius ; banho de ultrassom, modelos T14 e T50, Thornton;
Centrífuga de alimentos modelo CF-02, Juicer Mondial; rotaevaporador modelo
LS541, Sinc; micropipetas de 0,5-10; 10-100; 100-1000; 500-5000 µL, Eppendorf
Research; pHmetro, 827 pH Lab da Metrohm e moinho Quadrumat Senior da
Brabender (Duisburg, Alemanha).
3.4. Amostras de cevada
Os grãos de cevada foram fornecidos pelo Centro de Pesquisa da
Embrapa Trigo de Passo Fundo e cultivados no município de Ibiaçá, Rio Grande do
Sul. Foram utilizadas as cultivares BRS Lagoa 225 e BRS Lagoa 26612 do ano de
2010. Os grãos foram previamente secos em estufa a 55°C e moídos em micro-
moinho, a fim de se obter tamanho de partículas <1 mm.

64
3.5. Métodos de Extração
3.5.1. Método A – com hidrólise ácida
Foi pesado 1,0 g de grãos moídos de cevada em um frasco com tampa de
rosca. Adicionaram-se 10 mL de acetonitrila e agitaram-se os frascos
vigorosamente. Adicionaram-se 6 mL de água deionizada e as amostras foram
colocadas em banho de ultrassom por um período de 2 h, aquecidas à temperatura
de 50°C. Após esta etapa, as amostras foram resfriadas até temperatura ambiente e
foram adicionados 4 mL de água deionizada para obter um volume total de líquido
de 20 mL. Esta amostra líquida foi tampada e guardada em geladeira a 4°C por 15 h
antes da hidrólise ácida.
Para a hidrólise, 4 mL do extrato líquido foram colocados em um tubo de
ensaio com tampa rosqueável com 1 mL de HCl 37% (v/v) e o tubo de ensaio foi
aquecido em um banho termostático a 80°C por um período de 2 h.
Após a hidrólise, o tubo foi resfriado e 2 mL de uma solução de NH4OH
30% v/v: ácido acético glacial: DMSO 50:5:45 (v/v/v) foram adicionados. O pH da
solução final foi ajustado para se obter um meio neutro com solução de NH4OH 30%
v/v. Esta solução final foi filtrada em membrana de 0,22 µm e colocada num vial.
Este método foi adaptado de Ribani [117].
3.5.2. Métodos B1, B2 e B3–com hidrólise básica
Foi pesado 1 g dos grãos moídos de cevada que foram pesados em um
béquer e adicionados 5 mL de hexano sob agitação por 60 min para remoção da
gordura. A solução contendo hexano e gordura foi descartada com o uso da pipeta
de Pasteur e à porção restante foram adicionados10 mL de água (método B1) ou 10
mL de etanol 50 % (v/v) (método B2) ou 10 mL de etanol P.A (método B3) e
deixados sob agitação com barra magnética por 20 min, à temperatura ambiente e
na presença de gás nitrogênio. Cada extrato foi centrifugado por 10 minutos e
filtrado em papel de filtro qualitativo e em membrana de 0,22 µm.
Os resíduos sólidos das extrações foram hidrolisados com 20 mL de
NaOH 2 mol/L por 1 h, em agitação com barra magnética, à temperatura ambiente e

65
na presença de gás nitrogênio. Os extratos alcalinos foram acidificados até pH 2
com 20 mL de HCl 2 mol/L e centrifugados a 5148 G por 10 minutos. Após a filtração
em papel de filtro qualitativo e em membrana de 0,22 µm os filtrados foram
colocados em vials para serem analisados por ESI(-)-MS/MS. Este método foi
adaptado de Inglett [41].
3.5.3. Método C1, C2, C3 e C4 - Extração hidroetanólica
Foram pesados 3 g de grãos moídos de cevada em um béquer e
adicionados 15 mL de hexano sob agitação por 60 min para remoção da gordura. A
solução contendo hexano e gordura foi descartada com o uso da pipeta de Pasteur e
na porção restante foram adicionados 45 mL de solução hidroetanólica 50% (v/v) no
método C1 ou 80% (v/v) etanol/água no método C2 com agitação magnética a 80 ºC
por 90 min, em chapa de aquecimento, seguido de filtração em papel de filtro
qualitativo. Já para o método C3, foram adicionados 45 mL de solução
hidroetanólica 80% (v/v) e deixada sob agitação magnética em manta de
aquecimento a 80 ºC por 90 min, em um balão de fundo redondo sob atmosfera
inerte (gás nitrogênio), seguido de filtração em filtro qualitativo. Para o método C4
adicionaram-se 45 mL de solução etanol/água (80% v/v), deixando em sistema de
refluxo por 90 min a 80 ºC, seguido de filtração em papel de filtro qualitativo.
Em seguida, o filtrado foi evaporado em rotaevaporador até a secagem e
ressuspendido em 2 mL de solução hidroetanólica 50% (v/v) ou etanol/água 80%
(v/v). Esta solução final foi filtrada em membrana de 0,22 µm e colocada num vial.
Este método foi adaptado de Brindzová [42].
3.5.4. Métodos D1, D2 e D3- Extração hidroetanólica com variação
da temperatura
Amostras de 100 mg de cevada foram extraídas com 10 mL de
água:etanol 50 (% v/v) em agitação por 30 s em um vórtex, seguido do controle de
temperatura a 25°C (método D1), aquecimento a 50°C (método D2) ou a 100°C

66
(método D3) por 5 min. Os extratos foram centrifugados por 10 minutos a 5148 G.
Após a filtração em papel de filtro qualitativo e em membrana de 0,22 µm, os
filtrados foram colocados em vials e analisados por ESI(-)-MS/MS. Este método foi
adaptado de Inglett [41].
3.5.5. Método QuEChERS adaptado (versão acetato)
Preparou-se um slurry a partir de 10 g de cevada e 15 mL de água.
Adicionou-se gelo seco finamente moído em 10 g de slurry e 10 mL de acetonitrila
contendo ácido acético 1% (v/v). Homogeneizou-se em vórtex por 1 min.
Adicionaram-se, em seguida, 4 g de sulfato de magnésio com 1,7 g de acetato de
sódio. Centrifugou-se a 5148 G, por 8 min.
Retirou-se do sobrenadante (fase superior) uma alíquota de 8 mL e
transferiu-se para outro frasco. Colocou-se o frasco durante 5 min no gelo seco.
Adicionaram-se 500 mg de C18, 100 mg de PSA e 600 mg de sulfato de magnésio.
Agitou-se, por 1 min, em vórtex.
Centrifugou-se a 5148 G, por 8 min. Retirou-se do sobrenadante uma
alíquota de 4 mL, filtrou-se em filtro tipo seringa com 0,22 µm de espessura e
transferiu-se diretamente para um vial. Este método foi adaptado de Lehotay et al.;
Prestes, Bandeira e Núñez [67, 71, 73, 118]. Na Figura 9 encontram-se as etapas do
procedimento correspondentes ao método QuEChERS.

67
Figura 9: Fluxograma referente ao procedimento do método QuEChERS.
3.6. Caracterização dos extratos
3.6.1. Espectrometria de Massas - Infusão direta
Procedeu-se a análise dos diferentes extratos por meio da infusão direta
de soluções contendo 10 µL da amostra filtrada em 1 mL de ACN:H2O:ácido fórmico
80:19,9:0,1 (v/v/v) no espectrômetro de massas Waters, modelo Quattro Micro-API,
em uma vazão de 100 µL min-1. A caracterização foi realizada com ionização por
eletrospray no modo negativo (ESI(-)-MS), com voltagens de capilar e cone de 4,5
kV e 30 V respectivamente, utilizando N2 como gás de dessolvatação, a 400 °C, em
uma vazão de 800 L h-1 e no modo full scan, com varredura do espectro entre 150 e
1000 unidades de massa. Os picos de razão massa/ carga (m/z) de interesse foram
fragmentados a uma energia de colisão de 20 eV, utilizando-se argônio a 120 L h-1

68
como gás de colisão. Os dados foram analisados usando o software MassLynx
versão 4.1.
3.6.2. Cromatografia Líquida de Ultra Alta Eficiência acoplada à
Espectrometria de Massas Sequencial (UHPLC-MS/MS)
A otimização da análise cromatográfica empregando UHPLC-MS/MS foi
iniciada utilizando o monitoramento de íons no modofull scan, para localizar as
massasmolares dos compostos de interesse. Para a identificação e construção das
curvas analíticas foi empregado o modo SRM (do inglês, “selected reaction
monitoring”) e para a confirmação da presença dos compostos fenólicos na amostra,
foram monitoradas as transições m/z dos íons precursores e íons produtos.
A coluna cromatográfica empregada foi a Acquity UHPLC BEH C18 com
1,7 µm (50 × 2,1 mm d.i.) da Waters. As condições de análise otimizadas foram:
volume de injeção: 2 µL; vazão da fase móvel: 0,35 mL min-1; temperatura do forno
de 40 °C, eluição por gradiente: fase móvel A= H2O 0,1 % (v/v) ácido fórmico e B=
ACN 0,1% (v/v) ácido fórmico. Gradiente: 3% B por 1 min; 15% B em 4 min; 40% B
em 3 min; 3% B em 1 min. Adaptado de André [43]. A fase móvel foi utilizada em
uma vazão de 100 µL min-1. As análises foram realizadas com ionização por
eletrospray no modo negativo (ESI(-)-MS), com voltagens de capilar e cone de 4,5
kV e 30 V, respectivamente, utilizando N2 como gás de dessolvatação, a 400 °C, em
uma vazão de 800 L h-1 e varrendo-se (full scan) o espectro entre 150 e 1000
unidades de massa. Os picos de razão massa/ carga (m/z) de interesse foram
fragmentados a uma energia de colisão de 20 eV, utilizando-se argônio a 120 L h-1
como gás de colisão. Modo de Ionização: ESI(-) (ionização por eletrospray no modo
negativo, 30 kV).
3.7. Planejamento Experimental
Após verificar quais os métodos de extração clássicos (entre A e D) que
resultaram em maior quantidade de compostos fenólicos extraídos, foi feito um

69
planejamento experimental, a fim de otimizar as condições de extração. Para isso,
foi aplicado um planejamento fatorial completo com 3 fatores (Tabela 2) para os
métodos C3 (agitação magnética sob nitrogênio) e C4 (refluxo). Os fatores avaliados
foram modo de extração (agitação ou refluxo), temperatura (25 °C e 80 °C) e tempo
(10 e 90 min). Os valores de mínimo e máximo para os fatores temperatura e tempo
de extração foram escolhidos de acordo com a literatura sobre extração de
compostos fenólicos [17, 28]. O tratamento dos dados foi realizado no programa
Statistica 7.0.
Tabela 2: Ensaios para um planejamento fatorial 23 para otimização da extração de compostos fenólicos em cevada.
Ensaio* Temperatura (°C)
Tempo (min) Método
1 25 (-) 10 (-) agitação sob N2 2 80 (+) 10 (-) agitação sob N2 3 25 (-) 90 (+) agitação sob N2 4 80 (+) 90 (+) agitação sob N2 5 25 (-) 10 (-) refluxo 6 80 (+) 10 (-) refluxo 7 25 (-) 90 (+) refluxo 8 80 (+) 90 (+) refluxo
*Ensaios realizados em duplicata.
3.8. Validação dos métodos
A validação dos métodos foi feita de acordo com as recomendações da
Agência Brasileira de Vigilância Sanitária (ANVISA) [119] por meio das figuras de
mérito: seletividade, faixa linear, limite de detecção (LD), limite de quantificação
(LQ), exatidão (recuperação), precisão (inter-dias; intra-dia).
A seletividade foi avaliada pela comparação dos tempos de retenção,
massa molar e transições m/z do íon precursor e íons produtos dos analitos nos
extratos com relação aos padrões, pelo perfil dos espectros de massa dos analitos
nos extratos com relação aos padrões e pela fortificação das amostras para
construção da curva analítica.
A faixa de trabalho foi avaliada empregando-se o método de adição
padrão na matriz, ou seja, diferentes concentrações dos padrões foram adicionados

70
no extrato e também pelo método do padrão externo dissolvendo os padrões dos
compostos fenólicos em solvente (metanol). Injetaram-se, no sistema cromatográfico
2 µL das soluções, com concentrações variando entre 3-100 µg mL-1 e, 2 µL dos
extratos sem adição dos padrões. As injeções foram feitas em sete níveis de
concentrações e em triplicatas. A partir das áreas obtidas, foram construídas as
curvas analíticas. As linearidades dos métodos foram determinadas por meio da
regressão linear e obtiveram-se os coeficientes de regressão (a e b) e os
coeficientes de correlação. Também foram construídos os gráficos de resíduos para
verificar a qualidade da linearidade.
Os valores de limite de detecção (LDi) e limite de quantificação (LQi) do
instrumento foram alcançados a partir da transição dos íons de confirmação para
estimar a relação sinal/ruído (S/N), considerando 3 e 10 vezes, respectivamente, a
partir da adição dos padrões em solvente (metanol) O limite de quantificação do
método (LQm) foi determinado experimentalmente pela menor concentração do
extrato de cevada fortificado que alcançou recuperações entre 70 e 120% e com
RSD≤ 20%. A precisão foi avaliada em termos de repetitividade (precisão intra-dia) e
precisão intermediária (precisão inter-dia). A repetitividade foi expressa pelo
coeficiente de variação (CV) para 9 replicatas (3 níveis de fortificação, 3 repetições
cada um) realizados no mesmo dia. A precisão intermediária foi expressa pelo CV
para 9 replicatas (3 níveis de fortificação, 3 repetições cada um), realizados em dias
diferentes.
A avaliação da exatidão foi realizada pela obtenção da recuperação que
consiste na fortificação (spiking) da amostra antes da extração com concentração
conhecida do analito de interesse em três níveis distintos. A recuperação foi
calculada conforme a equação 6:
Cexperimental: é a concentração encontrada empregando a curva analítica
preparada com o extrato
Cteórica: é a concentração adicionada

71
Avaliou-se a eficiência de extração (EE) de acordo com Siqueira et al.
[120], realizando-se, em uma primeira etapa, a fortificação das amostras antes do
processo de extração e, em uma segunda etapa, a fortificação dos mesmos após o
processo de extração. A eficiência de extração foi calculada a partir da seguinte
equação:
O efeito matriz foi avaliado a partir do cálculo baseado na razão da
resposta analítica obtida no extrato final da amostra fortificada com solução
contendo os analitos de interesse, pela solução dos padrões sintéticos preparados
em solvente, na mesma concentração.
Avaliou-se o efeito matriz sobre o analito de acordo com a Equação 1.
3.9. Análise de amostras comerciais de cevada
Foram obtidas amostras de cevada de diferentes locais, comercializadas
nas seguintes cidades: A1: São Paulo, A2: Campinas, A3: Belo Horizonte, A4:
Florianópolis, A5: Curitiba, A6: Guarapuava e A7: Manaus.
Estas amostras de marcas diferentes foram analisadas empregando o
método QuEChERS.

72
4. RESULTADOS E DISCUSSÃO
4.1. Avaliação das diferentes técnicas de extração
4.1.1.Identificação dos compostos fenólicos por ESI(-)-MS/MS
A avaliação dos diferentes métodos de extração para a identificação dos
compostos fenólicos em cevada foi realizada por meio da caracterização dos
extratos pela técnica de infusão direta no espectrômetro de massas, a qual teve
como objetivo a visualização dos perfis espectrométricos de composição de cada
extrato, buscando identificar possíveis flavonóides e ácidos fenólicos desprotonados
presentes nos mesmos, através das razões m/z [M-H]-, onde M representa a massa
da molécula avaliada. Mesmo na ausência da separação cromatográfica (como na
LC-MS/MS), esta análise permite a confirmação dos compostos fenólicos baseada
no perfil da fragmentação obtido para as moléculas a partir de seus padrões
comerciais. Inicialmente foram avaliados os modos de ionização positivo (ESI(+)-
MS/MS) e negativo (ESI(-)-MS/MS) utilizando a solução dos padrões dos compostos
fenólicos por meio da infusão direta. A ionização dos compostos fenólicos, em modo
negativo, apresentou melhor detectabilidade e, portanto, os espectros de massas
foram obtidos neste modo, produzindo uma molécula desprotonada [M-H]- [121]. Os
espectros de massasdos padrões de compostos fenólicos no modo negativo pode
ser visto nas Figuras de 10-16.

73
Figura 10: Espectro de massas de ESI(-)-MS/MS do íon m/z193 (ácido ferúlico) no padrão.
Figura 11: Espectro de massas de ESI(-)-MS/MS do íon m/z 163 (ácido p-cumárico) no padrão.
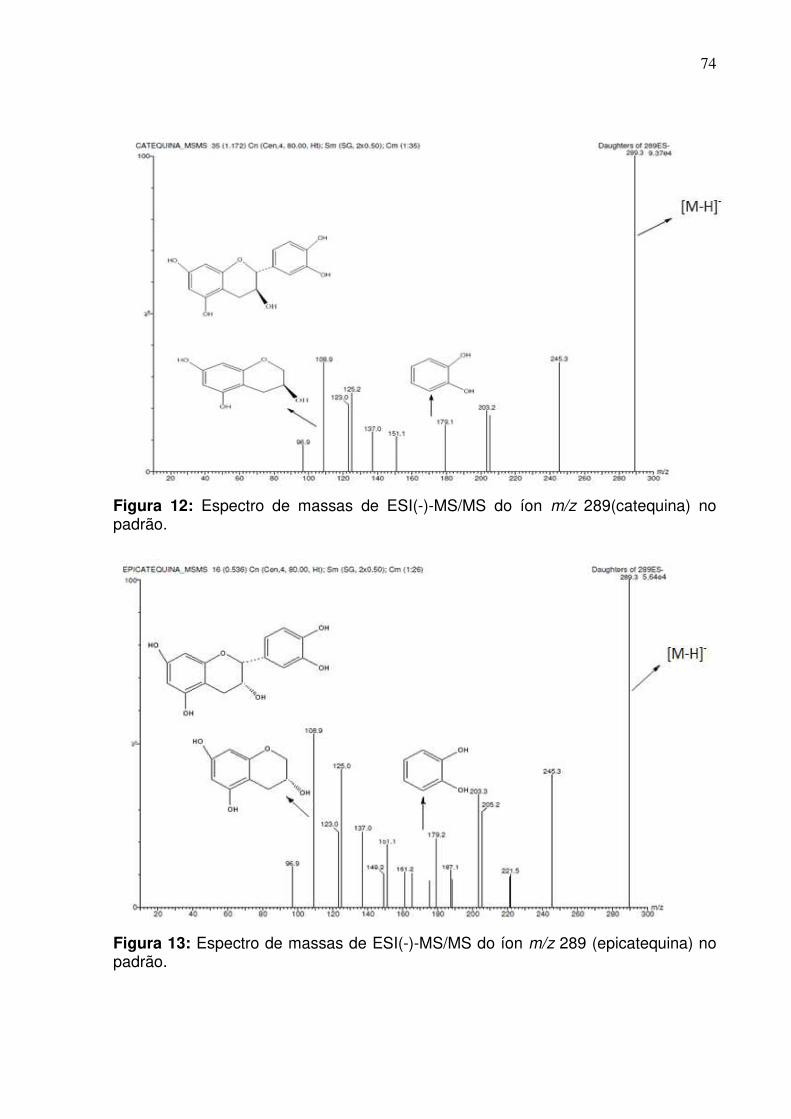
74
Figura 12: Espectro de massas de ESI(-)-MS/MS do íon m/z 289(catequina) no padrão.
Figura 13: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (epicatequina) no padrão.

75
Figura 14: Espectro de massas de ESI(-)-MS/MS do íon m/z 179 (ácido cafeico) no padrão.
Figura 15: Espectro de massas de ESI(-)-MS/MS do íon m/z 609 (rutina) no padrão.
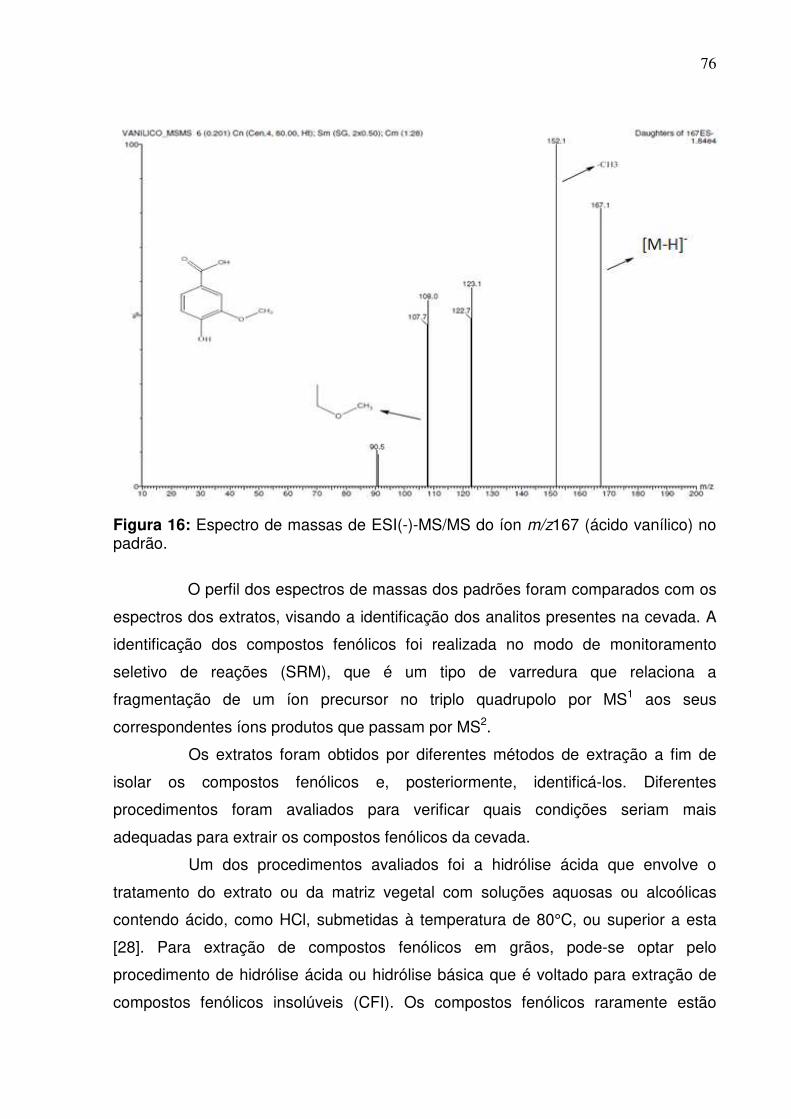
76
Figura 16: Espectro de massas de ESI(-)-MS/MS do íon m/z167 (ácido vanílico) no padrão.
O perfil dos espectros de massas dos padrões foram comparados com os
espectros dos extratos, visando a identificação dos analitos presentes na cevada. A
identificação dos compostos fenólicos foi realizada no modo de monitoramento
seletivo de reações (SRM), que é um tipo de varredura que relaciona a
fragmentação de um íon precursor no triplo quadrupolo por MS1 aos seus
correspondentes íons produtos que passam por MS2.
Os extratos foram obtidos por diferentes métodos de extração a fim de
isolar os compostos fenólicos e, posteriormente, identificá-los. Diferentes
procedimentos foram avaliados para verificar quais condições seriam mais
adequadas para extrair os compostos fenólicos da cevada.
Um dos procedimentos avaliados foi a hidrólise ácida que envolve o
tratamento do extrato ou da matriz vegetal com soluções aquosas ou alcoólicas
contendo ácido, como HCl, submetidas à temperatura de 80°C, ou superior a esta
[28]. Para extração de compostos fenólicos em grãos, pode-se optar pelo
procedimento de hidrólise ácida ou hidrólise básica que é voltado para extração de
compostos fenólicos insolúveis (CFI). Os compostos fenólicos raramente estão

77
presentes nos vegetais em sua forma livre, mas comumente, apresentam-se em sua
forma glicosilada [46].
O método A utilizou hidrólise ácida no extrato de acetonitrila com adição
de ácido clorídrico 37% (v/v) e aquecimento a 80°C. A hidrólise ácida tem o objetivo
de quebrar a ligação entre o composto fenólico e a parte glicosídica transformando
todos os compostos fenólicos nas formas agliconas. Por outro lado, a hidrólise
básica pode quebrar as ligações do éster, removendo grupos ácidos que estão
ligados à parte glicosídica dos compostos fenólicos.
Em seguida o pH do extrato foi ajustado com NH4OH para que os
compostos fenólicos ficassem na forma desprotonada, favorável para análise dos
mesmos no modo negativo por meio da infusão direta. A Figura 18 mostra a
fragmentação obtida para o extrato A, indicando os fragmentos do ácido ferúlico.
Figura 17: Espectro de massas de ESI(-)-MS/MS do íon m/z 193 (ácido ferúlico) no extrato A.
A molécula de ácido ferúlico (m/z 193) eliminou o dióxido de carbono (m/z
44) para formar íons de m/z 149. O fragmento m/z 178 corresponde à perda de um
grupo metila. Um dos picos mais abundante deste ácido (m/z 134) foi formado por
eliminação de dióxido de carbono (CO2) e um grupo metila.
Também foi avaliada a hidrólise básica no método B, consistindo no
tratamento dos resíduos da extração com solução de NaOH 2 mol L-1.

78
No entanto, não foram identificados compostos fenólicos nos resíduos
sólidos obtidos após extração com os métodos B1, B2 e B3. Provavelmente, os
compostos fenólicos foram extraídos antes da hidrólise, indicando que neste caso, a
extração com etanol, água ou a mistura etanólica são eficientes para a extração dos
compostos fenólicos. Outra possibilidade é que as condições de hidrólise
empregadas foram brandas.
Compostos fenólicos com alta polaridade, como alguns ácidos fenólicos
(ácido cafeico, ferúlico e clorogênico), podem não ser adequadamente extraídos
com solventes orgânicos puros, sendo que, normalmente, as soluções extratoras
consistem de solventes em diferentes frações aquosas [28, 122]. O solvente extrator
empregado no método C, etanol, já foi testado e avaliado como um dos mais
eficientes para a extração de compostos fenólicos em cevada [39, 42], sendo,
portanto, empregado neste trabalho, a mistura hidroetanólica 80%(v/v). Neste
procedimento foi avaliada a influência da atmosfera inerte durante o aquecimento,
uma vez que os compostos fenólicos podem ser degradados quando expostos ao
oxigênio do ambiente. Além da extração sob agitação, foi avaliada a extração
contínua pelo sistema de refluxo. Este tipo de extração foi testada por ser um
método eficiente e bem estabelecido na literatura para extração de compostos
fenólicos [117, 123]. Vale ressaltar que tanto nos métodos B quanto nos métodos C
foi adicionado hexano nos extratos, sob agitação magnética, para a remoção da
gordura para posterior etapa de extração com adição da mistura hidroetanólica. Esta
etapa de clean-up foi usada para minimizar os interferentes na análise
cromatográfica.
Para a identificação dos compostos fenólicos nos extratos, foram
avaliados os espectros de massas dos íons produto. Nos métodos C1 e C2 que
empregaram agitação magnética em chapa de aquecimento, foram identificados o
ácido ferúlico, ácido p-cumárico, catequina e epicatequina (Figuras 18- 24).
A molécula de ácido ferúlico (m/z 193) nos extratos C2 também eliminou o
dióxido de carbono (m/z 44) para formar íons de m/z 149. O fragmento m/z 178
corresponde à perda de um grupo metila. O pico mais abundante deste ácido (m/z
134) foi formado por eliminação de dióxido de carbono (CO2) e um grupo metila.
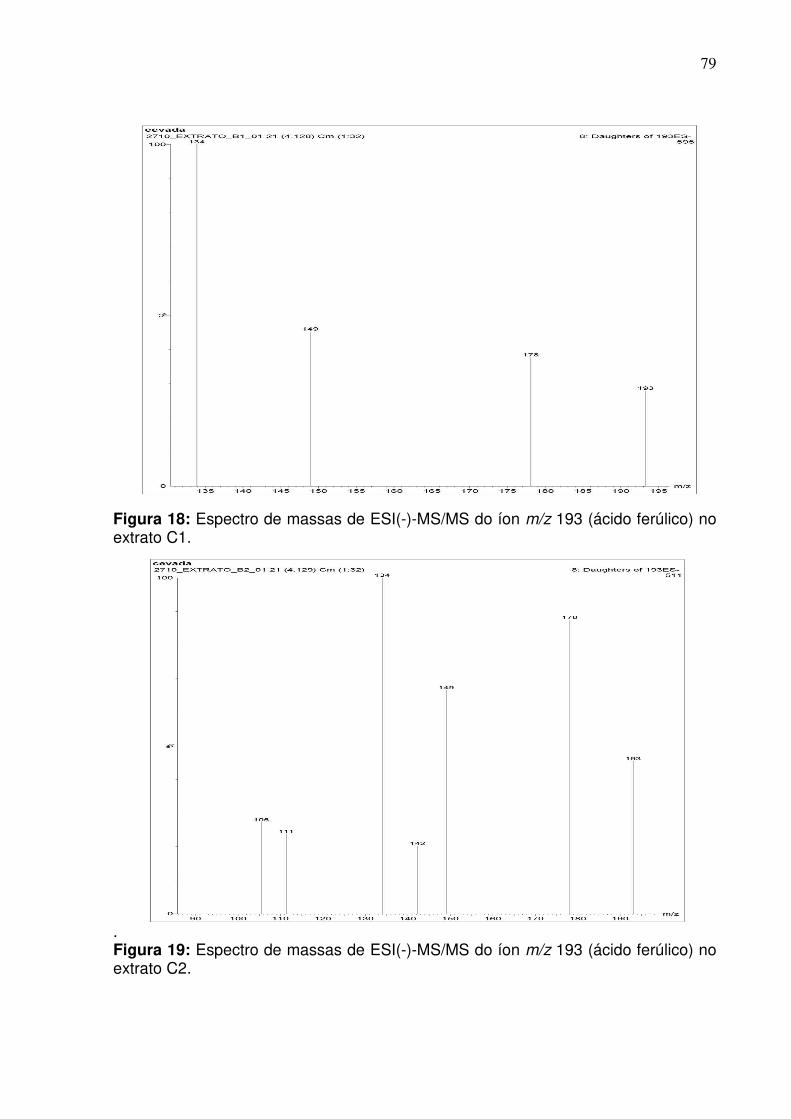
79
Figura 18: Espectro de massas de ESI(-)-MS/MS do íon m/z 193 (ácido ferúlico) no extrato C1.
. Figura 19: Espectro de massas de ESI(-)-MS/MS do íon m/z 193 (ácido ferúlico) no extrato C2.
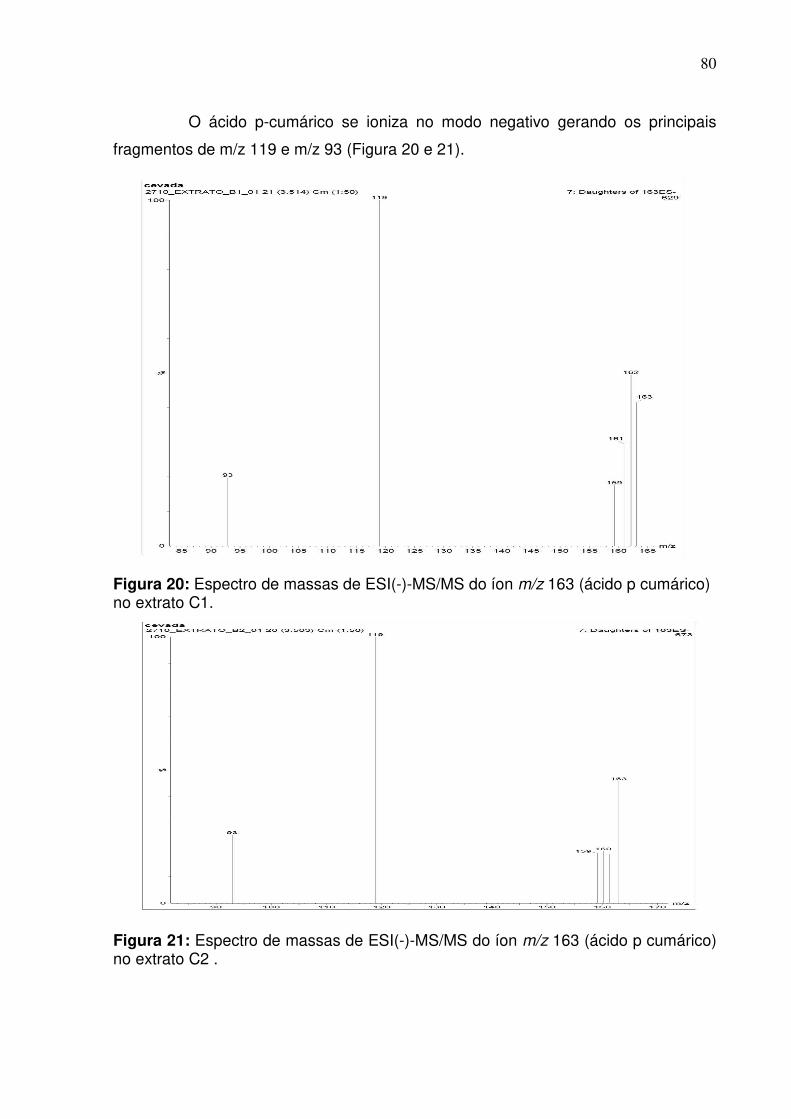
80
O ácido p-cumárico se ioniza no modo negativo gerando os principais
fragmentos de m/z 119 e m/z 93 (Figura 20 e 21).
Figura 20: Espectro de massas de ESI(-)-MS/MS do íon m/z 163 (ácido p cumárico) no extrato C1.
Figura 21: Espectro de massas de ESI(-)-MS/MS do íon m/z 163 (ácido p cumárico) no extrato C2 .
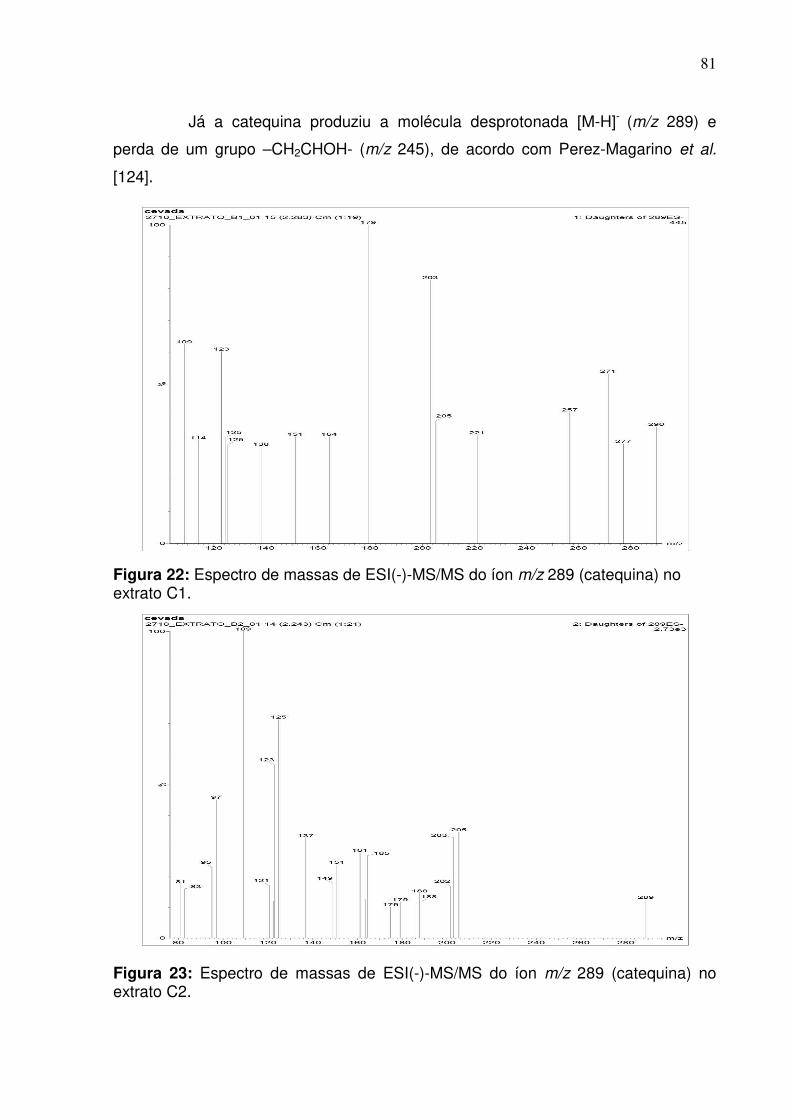
81
Já a catequina produziu a molécula desprotonada [M-H]- (m/z 289) e
perda de um grupo –CH2CHOH- (m/z 245), de acordo com Perez-Magarino et al.
[124].
Figura 22: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (catequina) no extrato C1.
Figura 23: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (catequina) no extrato C2.

82
A catequina e epicatequina são considerados isômeros, portanto
possuem a mesma massa molar. O que difere uma molécula de outra é o desvio da
luz polarizada na posição da hidroxila no sentido S, levógero (anti-horário) e no
sentido R, destrógiro (horário). Portanto, estes isômeros apresentam os mesmos
fragmentos.
A epicatequina também produziu a molécula desprotonada [M-H]- (m/z
289) e a perda de um grupo –CH2CHOH- (m/z 245) [124].
Figura 24: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (epicatequina) no extrato C2.
Pelo método C3, o qual diferiu dos métodos C1 e C2 devido a utilização
de uma manta de aquecimento sob atmosfera inerte (gás nitrogênio), foram
identificados os mesmos compostos: ácido ferúlico, ácido p-cumárico, catequina e
epicatequina, e mais o ácido cafeico. Já no método C4 que utilizou sistema de
refluxo, foram identificados os ácidos cafeico, ácido p-cumárico, catequina,
epicatequina, além da rutina e ácido vanílico (Figuras 25-35).
Nas Figuras 25 a 35 são apresentados os espectros de identificação dos
extratos C3 e C4.

83
Figura 25: Espectro de massas de ESI(-)-MS/MS do íon m/z 193 (ácido ferúlico) no extrato C3.
O espectro de massas do ácido cafeico (Figuras 26 e 27) desprotonado
(m/z 179) produziu um pico com m/z 135, devido à perda de CO2, mas sem outros
fragmentos abundantes.
Nas Figuras 28 e 29, a catequina produziu a molécula desprotonada [M-
H]- (m/z 289) e perda de um grupo –CH2CHOH- (m/z 245), de acordo com Perez-
Margarino et al. [124].
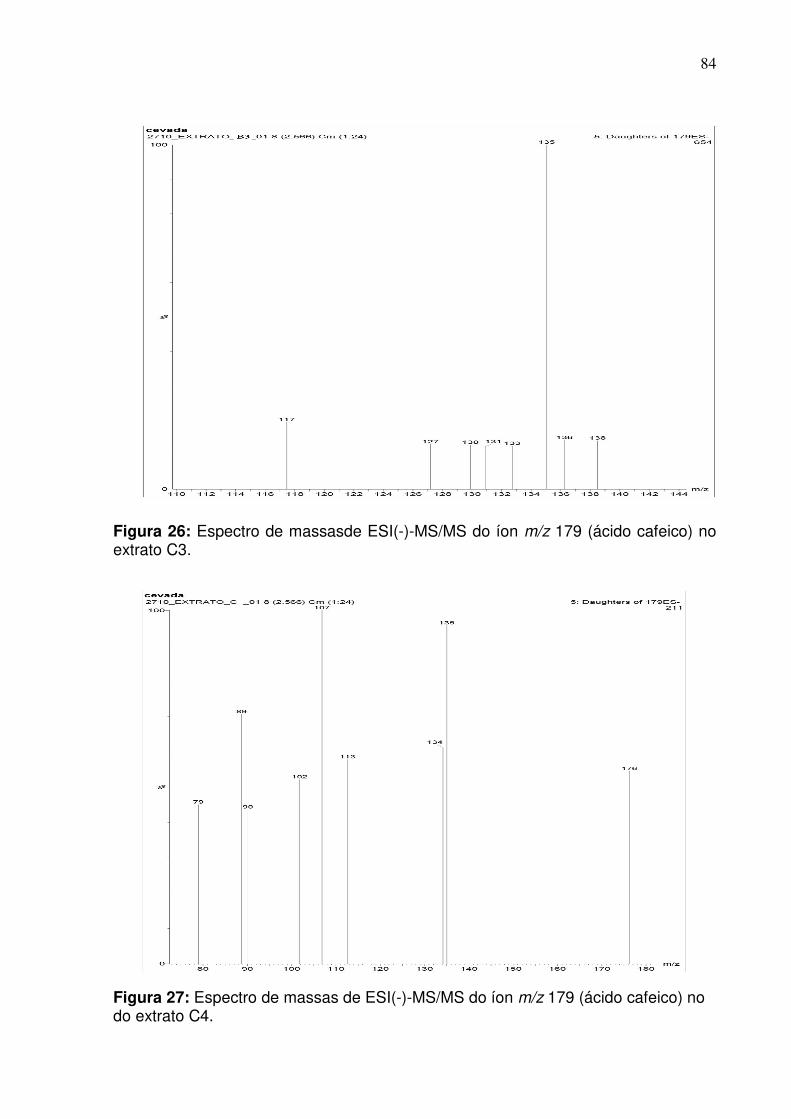
84
Figura 26: Espectro de massasde ESI(-)-MS/MS do íon m/z 179 (ácido cafeico) no extrato C3.
Figura 27: Espectro de massas de ESI(-)-MS/MS do íon m/z 179 (ácido cafeico) no do extrato C4.
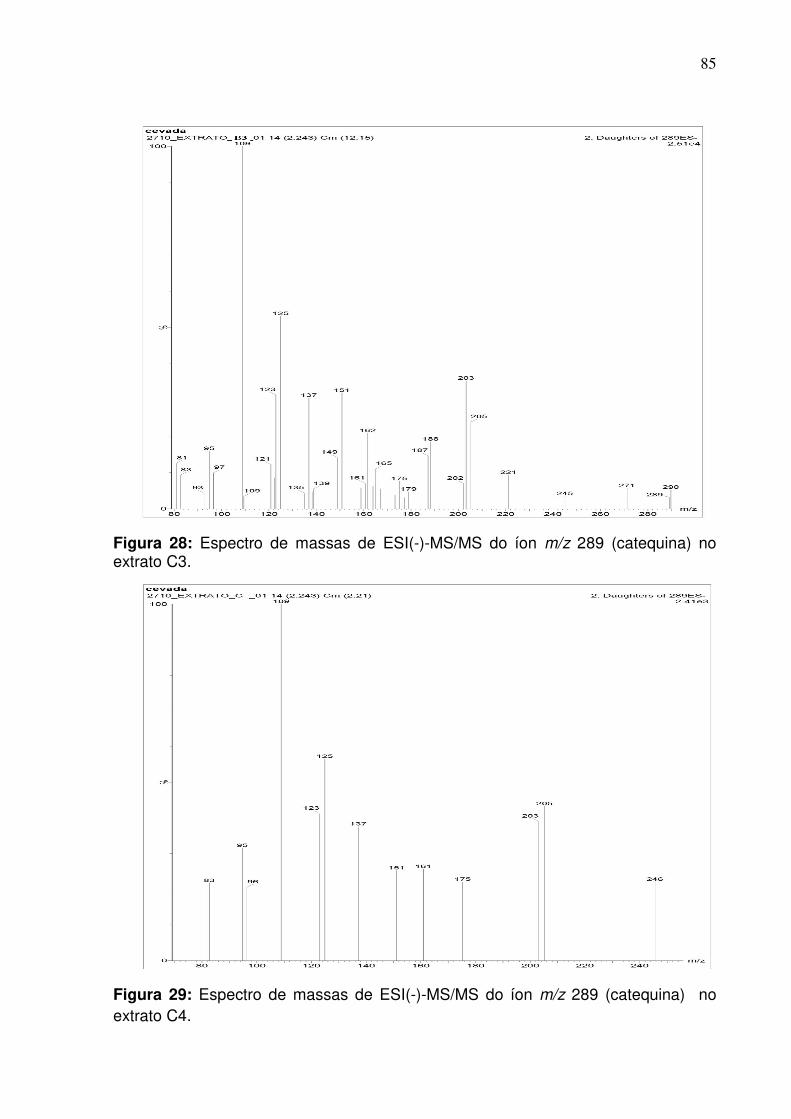
85
Figura 28: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (catequina) no extrato C3.
Figura 29: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (catequina) no extrato C4.

86
Figura 30: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (epicatequina) no extrato C3.
Figura 31: Espectro de massas de ESI(-)-MS/MS do íon m/z 289 (epicatequina) no extrato C4.
A epicatequina também produziu a molécula desprotonada [M-H]- (m/z
289) e a perda de um grupo –CH2CHOH- (m/z 245) (Figuras 30 e 31). O ácido p-

87
cumárico se ioniza no modo negativo gerando o principal fragmento de m/z 119. Os
espectros para os métodos C3 e C4 podem ser vistos nas Figuras 32 e 33.
Figura 32: Espectro de massas de ESI(-)-MS/MS do íon m/z 163 (ácido p-cumárico) no extrato C3.
Figura 33: Espectro de massas de ESI(-)-MS/MS do íon m/z 163 (ácido p-cumárico) no extrato C4.
Para flavonóis O-glicosilados, tais como rutina, o espectro gerado mostrou
a molécula desprotonada [M-H-] do glicosídeo e o íon correspondente à aglicona
desprotonada (a-H)-. Assim, para a rutina (Figura 34) foi observada a perda da
unidade de ramnose-glucose (m/z 301) [125].
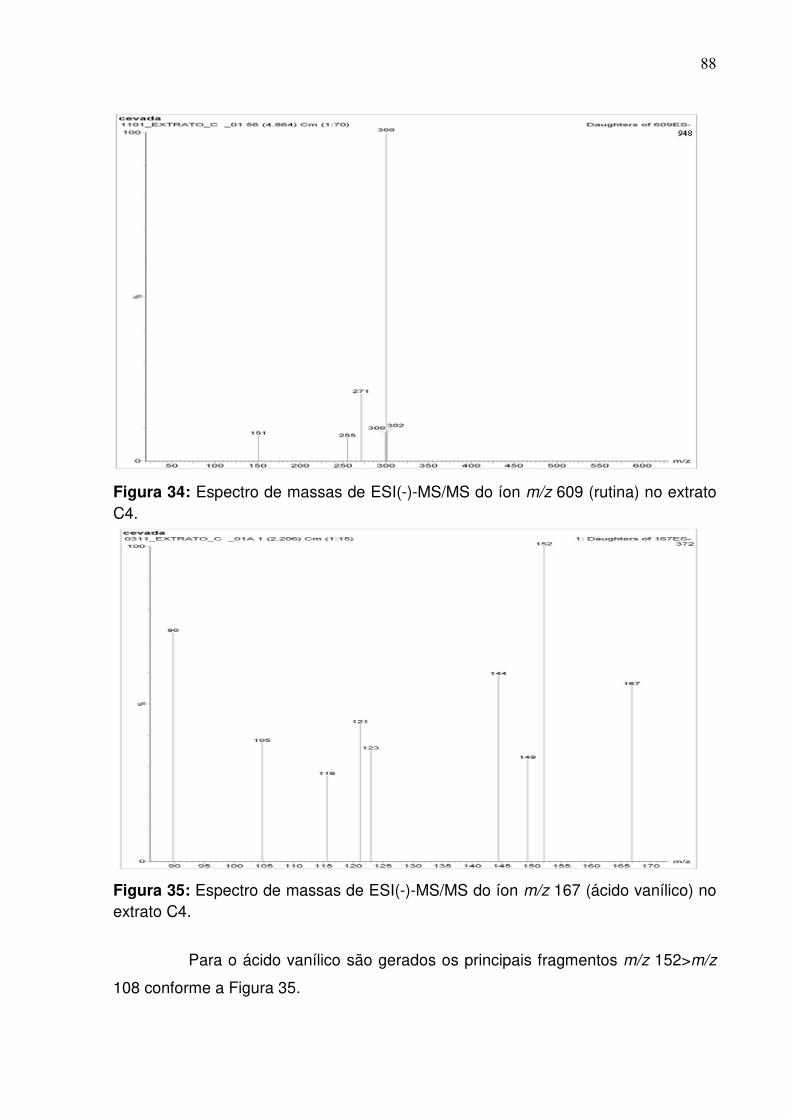
88
Figura 34: Espectro de massas de ESI(-)-MS/MS do íon m/z 609 (rutina) no extrato C4.
Figura 35: Espectro de massas de ESI(-)-MS/MS do íon m/z 167 (ácido vanílico) no extrato C4.
Para o ácido vanílico são gerados os principais fragmentos m/z 152>m/z
108 conforme a Figura 35.

89
Na Tabela 3 são apresentados os compostos fenólicos identificados nos
diferentes extratos.
Tabela 3: Presença de compostos fenólicos em diferentes extratos.
A partir da quantidade de compostos fenólicos identificados nos extratos,
selecionaram-se os métodos C3 e C4 para otimização dos parâmetros de extração
uma vez que estes métodos indicaram um maior número de compostos
identificados.
4.1.2. Determinação das condições cromatográficas e
espectrométricas por UHPLC-MS/MS
Após a identificação dos compostos fenólicos pela técnica de ESI-MS/MS,
optou-se em fazer também a identificação e quantificação pela técnica de UHPLC-
MS/MS, devido à possibilidade de separação prévia dos compostos antes da
ionização no espectrômetro de massas.
Para isto, primeiramente fez-se a otimização das condições
cromatográficas e espectrométricas para os analitos ácido cafeico, ácido
clorogênico, ácido ferúlico, ácido p-cumárico, ácido protocatecóico, ácido sinápico,
ácido vanílico, catequina, epicatequina, miricetina e rutina.

90
Inicialmente, foram otimizados os parâmetros da fonte de ionização do
espectrômetro de massas do tipo triplo quadrupolo a fim de se conseguir o melhor
sinal (maior detectabilidade) do íon molecular e de dois íons produtos mais
abundantes de cada composto fenólico, através da infusão dos mesmos em solução,
diretamente no espectrômetro de massas. Neste tipo de analisador os íons
moleculares são fragmentados, obtendo-se as transições de quantificação e
confirmação, recomendadas para uma determinação analítica empregando
espectrometria de massas.
A infusão consistiu no bombeamento de soluções de compostos fenólicos
a 5 mg L-1 em metanol, na vazão de 50 µL min-1, combinada com os solventes
auxiliares acetonitrila com ácido fórmico 0,1% (v/v) : água com ácido fórmico 0,1%
(v/v) (50:50) na vazão de 0,1 mL min-1.
Para o modo de ionização, foram avaliados o modo positivo e o modo
negativo. Os testes em ambos os modos de ionização consistiram em aplicar
diferentes voltagens do cone com o objetivo de obter o espectro com melhor
ionização e maior intensidade dos analitos. Foi realizada uma varredura da voltagem
do cone de 10 V a 60 V.
Com a utilização de ESI no modo positivo, apenas 4 analitos foram
capazes de sofrer ionização, obtendo as melhores intensidades de sinal com as
respectivas voltagens: ácido protocatecóico, 10 V; catequina ou epicatequina, 60 V;
miricetina e rutina, 60 V, conforme mostra a Figura 36.

91
Figura 36: ESI no modo positivo dos íons precursores da mistura contendo 11 padrões de compostos fenólicos com voltagens do cone avaliadas em (A) 60, (B) 45, (C) 30, (D) 20, (E) 10 V.
Os analitos investigados no modo de ionização positivo com suas
respectivas razões m/z estão apresentados na Tabela 4. Para tal investigação,
foram identificados apenas o ácido protocatecóico, catequina ou epicatequina,
miricetina e rutina.
A Figura 37 representa os espectros obtidos por ESI no modo negativo da
mistura de compostos fenólicos com voltagem do cone avaliada em 30 V. Foram
avaliadas todas as voltagens de 10 a 60 V, sendo que a melhor voltagem para
avaliação dos compostos fenólicos estudados foi fixada em 30 V.

92
Tabela 4: Compostos fenólicos analisados com ionização no modo positivo.
Substância [M+H]+
(m/z) Ácido cafeico 181
Ácido clorogênico 355 Ácido ferúlico 195
Ácido p-cumárico 165 Ácido protocatecóico 183
Ácido sinápico 225 Ácido vanílico 169
Catequina 291 Epicatequina 291
Miricetina 319 Rutina 611
Como os compostos fenólicos apresentam os grupos ácidos carboxílicos
em sua estrutura química, estes podem se dissociar de acordo com a variação nos
valores de pH da fase móvel [39, 43].
Figura 37: Espectro de massas obtido por ESI no modo negativo da mistura contendo 11 padrões de compostos fenólicos com voltagem do cone avaliada em 30 V.
A escolha pelo modo negativo de análise ocorreu pois os 11 analitos
investigados foram ionizados neste modo (Figura 37) enquanto que no modo

93
positivo apenas 4 analitos foram capazes de sofrer ionização (Figura 36).As
condições otimizadas da fonte do espectrômetro de massas podem ser verificadas
na Tabela 5.
Tabela 5: Melhores condições de operação do espectrômetro de massas para a análise dos compostos fenólicos.
Parâmetros Condição
Modo de ionização negativo
Capilar (kV) 4,5
Temperatura da Fonte (°C) 120
Temperatura de Dessolvatação (°C) 400
Vazão do gás no Cone (L h-1) 900
Vazão do gás de dessolvatação (L h-1) 30
Multiplier 650
Pressão na cela de Colisão (mbar) 2,43 × 10-3
Na Tabela 6 estão apresentados os íons precursores [M-H]– selecionados
para obtenção das transições SRM, os íons produto, a voltagem do cone e energia
de colisão (CE) para as análises dos compostos fenólicos utilizados neste trabalho.
Tabela 6: Parâmetros do espectrômetro de massas para a análise dos compostos fenólicos.
Substância Íon precursor (m/z)
Íons produto (m/z)
Voltagem do cone (V)
Energia de Colisão (eV)
Ácido cafeico 179 117 135
35 35 15
Ácido clorogênico 353 85 191
30 45 15
Ácido ferúlico 193 134 178
30 15 10
Ácido p-cumárico 163 52 119
30 35 15
Ácido protocatecóico
181 108 153
40 20 15
Ácido sinápico 223 149 164
30 20 15
Ácido vanílico 167 123 152
30 10 15
Catequina 289 123 245
35 30 15
Epicatequina 289 109 245
35 15 25
Miricetina 317 151 179
45 25 20
Rutina 609 271 300
60 50 40
* As transições em negrito referem-se aos íons de quantificação
Na etapa seguinte, após o estabelecimento das condições
espectrométricas, realizou-se a otimização da separação cromatográfica. No UHPLC

94
foram testados os tipos de eluição isocrático e gradiente, diferentes tipos de
solventes e proporções de fase móvel, como solventes orgânicos (metanol e
acetonitrila, tanto acidificados com 0,1 % (v/v) de ácido fórmico, como não
acidificados) e solventes aquosos (ácido fórmico 0,1% (v/v) e tampão formiato de
amônio ou NH4OH em 0,025% (v/v).
Foi avaliada também a fase móvel acetonitrila com ácido fórmico 0,1%
(v/v): água com hidróxido de amônio 0,1% (v/v) [50:50, v/v] na vazão de 0,1 mL min-1
onde foi observada uma pronunciada diminuição da intensidade do sinal dos
analitos, mostrando a inviabilidade do uso desse tipo de fase móvel comparado à
anterior que utilizou os solventes auxiliares acetonitrila com ácido fórmico 0,1% (v/v)
: água com ácido fórmico 0,1% (v/v) (50:50) na vazão de 0,1 mL min-1.
Os testes de diferentes condições cromatográficas, utilizando diferentes
combinações de fase móvel, estão apresentados nas Figuras 38 e 39. Na Figura 38,
os cromatogramas foram obtidos com a ionização no modo negativo e os solventes
auxiliares acetonitrila com ácido fórmico 0,1% (v/v): água com ácido fórmico 0,1%
(v/v); [50:50, v/v]. Já na Figura 39 foram utilizados os solventes aquosos com ácido
fórmico 0,1% (v/v) e tampão formiato de amônio ou NH4OH em 0,025% (v/v) como
fase móvel.
A separação dos analitos foi alcançada em fase reversa. Este tipo de
separação se deve principalmente a interações entre a parte apolar do soluto e a
fase estacionária, isto é, à repulsão desta parte do soluto pela fase móvel aquosa.
Assim, os compostos fenólicos são bem separados com fase móvel contendo
acetonitrila com ácido fórmico 0,1% (v/v): água com ácido fórmico 0,1% (v/v); [50:50,
(v/v)]. Com o uso do meio ácido na fase móvel, os compostos fenólicos encontram-
se na sua forma protonada (molecular), que favorece a retenção dos mesmos em
fase reversa. Por outro lado, quando foi utilizada a fase móvel contendo solventes
aquosos com ácido fórmico 0,1% (v/v) e tampão formiato de amônio ou NH4OH em
0,025% (v/v), a retenção dos compostos foi praticamente nula, pois os mesmos
ficaram ionizados (desprotonados) em meio básico, minimizando a interação com a
fase estacionária do tipo C18.
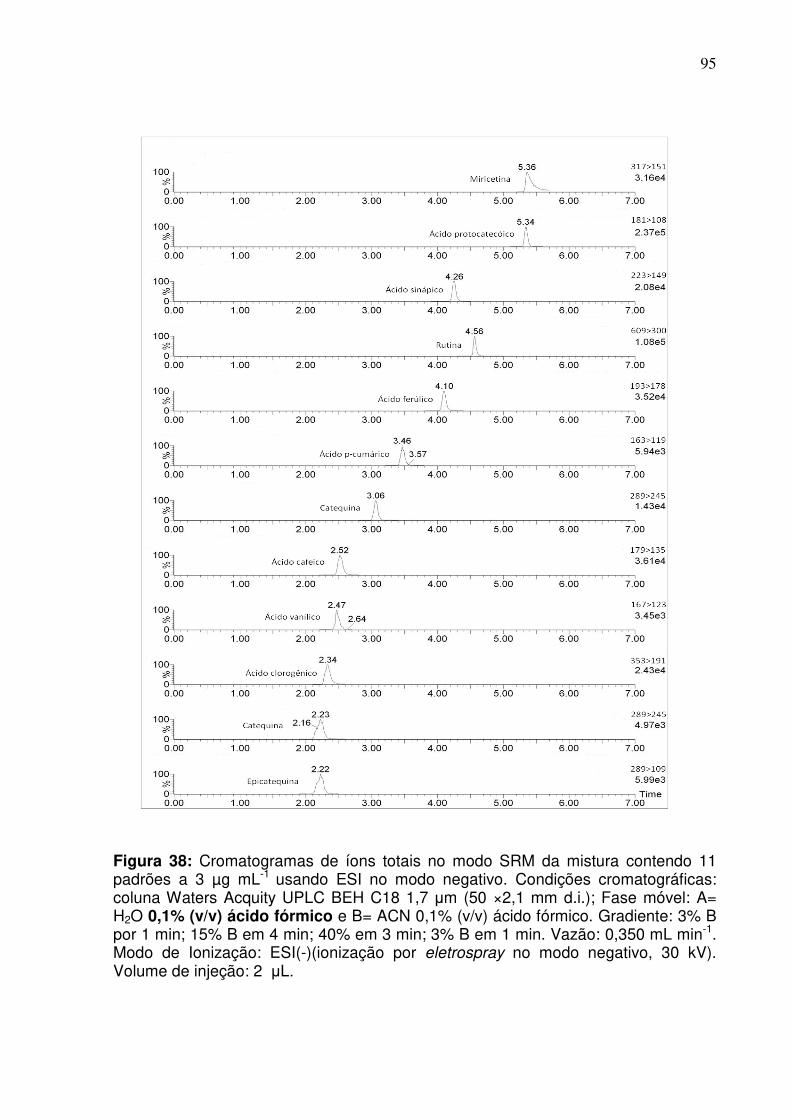
95
Figura 38: Cromatogramas de íons totais no modo SRM da mistura contendo 11 padrões a 3 µg mL-1 usando ESI no modo negativo. Condições cromatográficas: coluna Waters Acquity UPLC BEH C18 1,7 μm (50 ×2,1 mm d.i.); Fase móvel: A= H2O 0,1% (v/v) ácido fórmico e B= ACN 0,1% (v/v) ácido fórmico. Gradiente: 3% B por 1 min; 15% B em 4 min; 40% em 3 min; 3% B em 1 min. Vazão: 0,350 mL min-1. Modo de Ionização: ESI(-)(ionização por eletrospray no modo negativo, 30 kV). Volume de injeção: 2 μL.

96
Figura 39: Cromatogramas de íons totais no modo SRM da mistura contendo 11 padrões a 3 µg mL-1 com ESI no modo negativo com adição de NH4OH 0,025%(v/v).Condições cromatográficas: coluna Waters Acquity UPLC BEH C18 1,7 μm (50 ×2,1 mm d.i.); Fase móvel: A= H2O (0,025%NH4OH) e B= ACN (0,1% ácido fórmico). Gradiente: 3% B por 1 min; 15% B em 4 min; 40% B em 3 min; 3% B em 1 min. Vazão: 0,350 mL min-1. Modo de Ionização: ESI-(ionização por eletronebulização no modo negativo, 30 kV). Volume de injeção: 2 μL.

97
Pela observação dos cromatogramas, os gradientes que proporcionaram
melhor linha de base dos picos cromatográficos e melhor separação foram aqueles
onde se utilizou como fase móvel água acidificada combinada com acetonitrila
acidificada.
Foram avaliados diferentes programas de gradiente e o mais promissor foi
o gradiente adaptado de Andre [43]. O gradiente de eluição foi composto por 3% B
por 1 min; 15% B em 4 min; 40% B em 3 min; 3% B em 1 min. Neste programa, foi
possível alcançar a separação da maioria dos compostos fenólicos estudados e a
identificação de todos analitos num tempo de retenção menor que 6 minutos.
Pela avaliação e comparação das Figuras 38 e 39, os cromatogramas
comprovam que a maior resolução para a maioria dos analitos foi alcançada com o
uso da fase móvel A contendo água com ácido fórmico 0,1% (v/v): fase móvel B
contendo acetonitrila com ácido fórmico 0,1% 50:50 (v/v), no modo de ionização
negativo.
Já a adição de NH4OH 0,025% (v/v) na fase móvel diminui a intensidade e
resolução dos analitos, além de prejudicar a linha de base dos mesmos e causar
coeluição de picos.
É importante destacar que devido à alta seletividade do espectrômetro do
tipo triplo quadrupolo, quando o mesmo é utilizado no modo de aquisição SRM, é
possível identificar os compostos fenólicos sem a necessidade de separá-los
completamente; os analitos podem coeluir sem causar problemas de interferência
pois os mesmos são identificados individualmente, em canais isolados.
Na Figura 40 pode ser visualizado o cromatograma de íons totais,
adquiridos em canais distintos de aquisição, no modo SRM, nas quais foram
analisadas as transições de quantificação e confirmação, de uma mistura contendo
todos os compostos fenólicos.
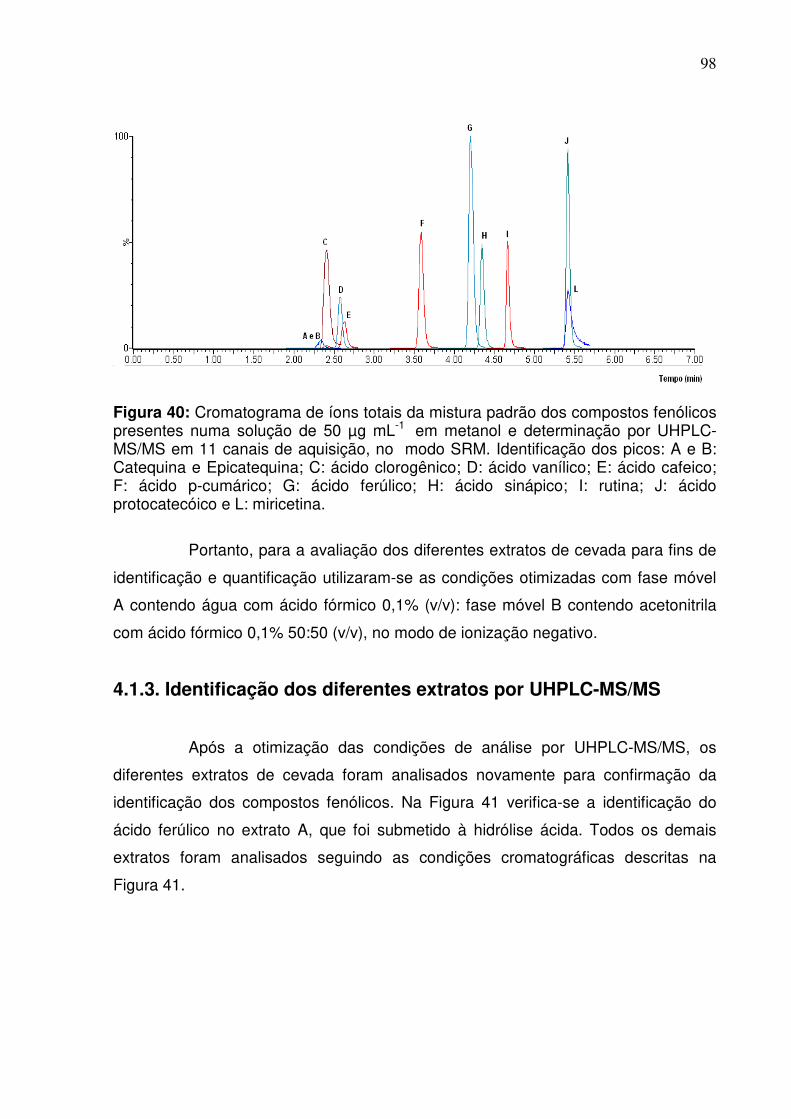
98
Figura 40: Cromatograma de íons totais da mistura padrão dos compostos fenólicos presentes numa solução de 50 µg mL-1 em metanol e determinação por UHPLC-MS/MS em 11 canais de aquisição, no modo SRM. Identificação dos picos: A e B: Catequina e Epicatequina; C: ácido clorogênico; D: ácido vanílico; E: ácido cafeico; F: ácido p-cumárico; G: ácido ferúlico; H: ácido sinápico; I: rutina; J: ácido protocatecóico e L: miricetina.
Portanto, para a avaliação dos diferentes extratos de cevada para fins de
identificação e quantificação utilizaram-se as condições otimizadas com fase móvel
A contendo água com ácido fórmico 0,1% (v/v): fase móvel B contendo acetonitrila
com ácido fórmico 0,1% 50:50 (v/v), no modo de ionização negativo.
4.1.3. Identificação dos diferentes extratos por UHPLC-MS/MS
Após a otimização das condições de análise por UHPLC-MS/MS, os
diferentes extratos de cevada foram analisados novamente para confirmação da
identificação dos compostos fenólicos. Na Figura 41 verifica-se a identificação do
ácido ferúlico no extrato A, que foi submetido à hidrólise ácida. Todos os demais
extratos foram analisados seguindo as condições cromatográficas descritas na
Figura 41.

99
Figura 41: Cromatogramadeidentificação do ácido ferúlico no extrato A. Condições cromatográficas: coluna Waters Acquity UPLC BEH C-18 1,7 μm (50 × 2,1 mm d.i.); Fase móvel: A= H2O (0,1% (v/v) ácido fórmico) e B= ACN (0,1% (v/v) ácido fórmico). Gradiente: 3% B por 1 min; 15% B em 4 min; 40% B em 3 min; 3% B em 1 min adiquiridos em canais de aquisição no modo SRM. Vazão: 0,350 mL min-1. Modo de Ionização: ESI-(ionização por eletrospray no modo negativo, 30 kV). Volume de injeção: 2 μL.
Nos resíduos dos extratos do método B que passaram pela hidrólise
básica também não foram encontrados compostos fenólicos pela técnica de UHPLC-
MS/MS, assim como por ESI(-)-MS/MS.
Na Figura 42 estão os cromatogramas de identificação dos ácidos
ferúlico, p-cumárico e cafeico, além de catequina e epicatequina obtidos do extrato
C1, que empregou agitação magnética em chapa de aquecimento. Ao empregar
ESI(-)-MS/MS foram identificados apenas os ácidos ferúlico, p-cumárico e catequina.
Figura 42: Cromatogramas dosácidos ferúlico, p-cumárico e cafeico; catequina e epicatequina obtidos para o extrato C1. Condições cromatográficas idem asda Figura 41.

100
Nas Figuras 43 e 44 estão os cromatogramas de identificação dos extratos C2
e C3, respectivamente, sendo este último realizado em atmosfera de nitrogênio.
Foram identificados os ácidos ferúlico, p-cumárico e cafeico, catequina e
epicatequina. Todos os analitos dos extratos C2 e C3 também foram identificados
por ESI(-)-MS/MS, exceto o ácido cafeico no extrato C2.
Figura 43: Cromatogramas dos ácidos ferúlico, p-cumárico e cafeico; catequina e epicatequina no extrato C2. Condições cromatográficas idem asda Figura 41.

101
Figura 44: Cromatogramas dosácidos ferúlico, p-cumárico e cafeico; catequina e epicatequina no extrato C3. Condições cromatográficas idem asda Figura 41.
Já os compostos identificados por UHPLC-MS/MS do extrato C4 sob
refluxo, são mostrados na Figura 45.
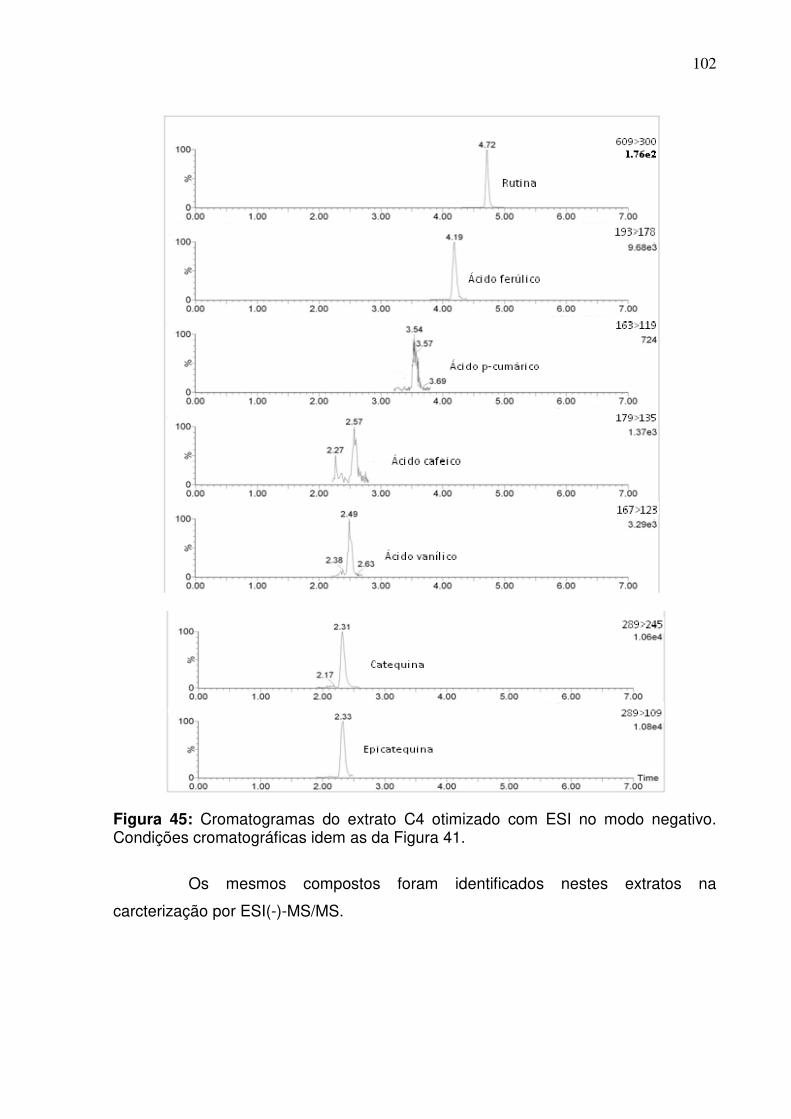
102
Figura 45: Cromatogramas do extrato C4 otimizado com ESI no modo negativo. Condições cromatográficas idem as da Figura 41.
Os mesmos compostos foram identificados nestes extratos na
carcterização por ESI(-)-MS/MS.
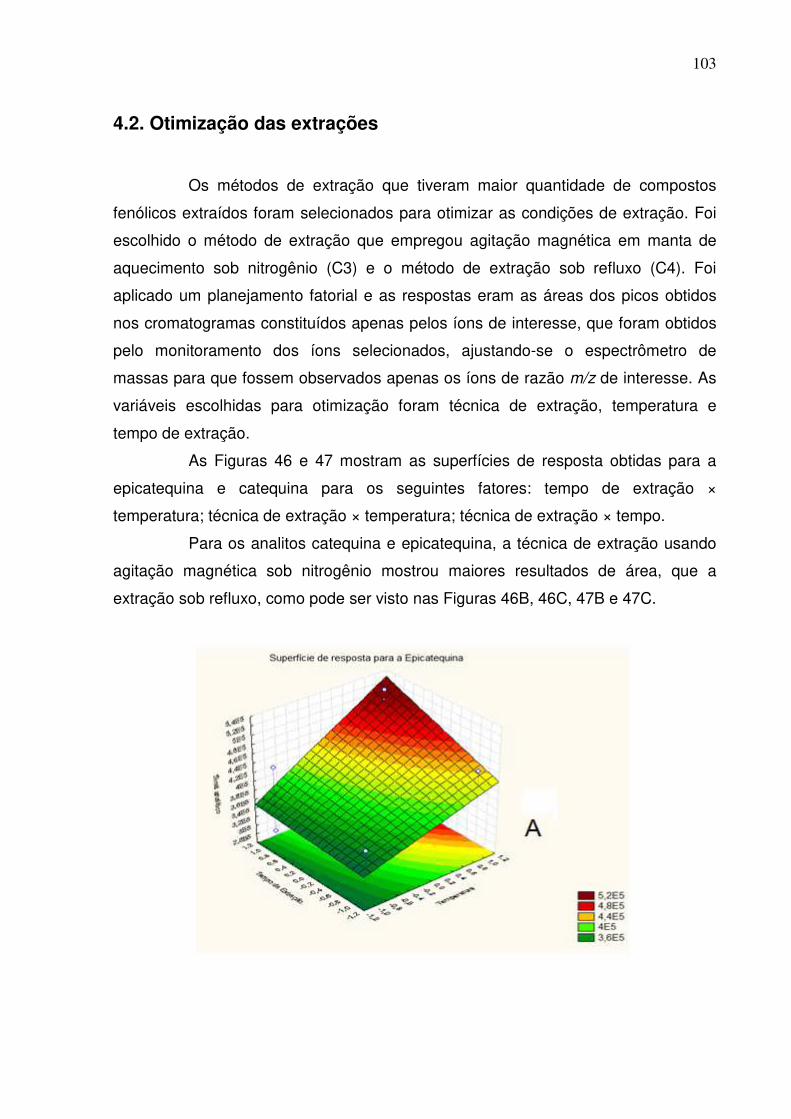
103
4.2. Otimização das extrações
Os métodos de extração que tiveram maior quantidade de compostos
fenólicos extraídos foram selecionados para otimizar as condições de extração. Foi
escolhido o método de extração que empregou agitação magnética em manta de
aquecimento sob nitrogênio (C3) e o método de extração sob refluxo (C4). Foi
aplicado um planejamento fatorial e as respostas eram as áreas dos picos obtidos
nos cromatogramas constituídos apenas pelos íons de interesse, que foram obtidos
pelo monitoramento dos íons selecionados, ajustando-se o espectrômetro de
massas para que fossem observados apenas os íons de razão m/z de interesse. As
variáveis escolhidas para otimização foram técnica de extração, temperatura e
tempo de extração.
As Figuras 46 e 47 mostram as superfícies de resposta obtidas para a
epicatequina e catequina para os seguintes fatores: tempo de extração ×
temperatura; técnica de extração × temperatura; técnica de extração × tempo.
Para os analitos catequina e epicatequina, a técnica de extração usando
agitação magnética sob nitrogênio mostrou maiores resultados de área, que a
extração sob refluxo, como pode ser visto nas Figuras 46B, 46C, 47B e 47C.
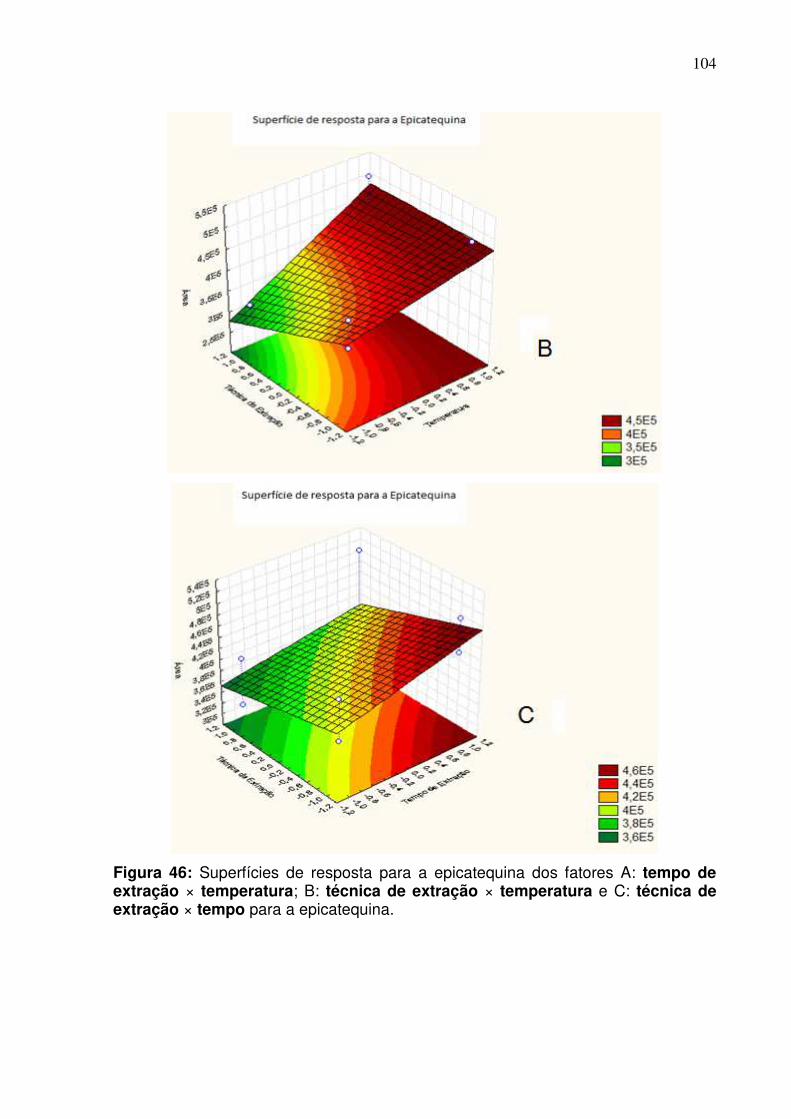
104
Figura 46: Superfícies de resposta para a epicatequina dos fatores A: tempo de extração × temperatura; B: técnica de extração × temperatura e C: técnica de extração × tempo para a epicatequina.

105

106
Figura 47: Superfícies de resposta para a catequina dos fatores A: tempo de extração × temperatura; B: técnica de extração × temperatura e C: técnica de extração × tempo para a catequina.
Para os analitos ácido cafeico e ácido ferúlico, a extração em sistema de
refluxo (método C4) mostrou maiores valores de área dos picos. As superfícies de
resposta indicaram que a variável 1 (temperatura de extração) é a mais significativa
quando comparada com as outras variáveis de acordo com as Figuras48 A e 48 B;
49 A e 49 B.
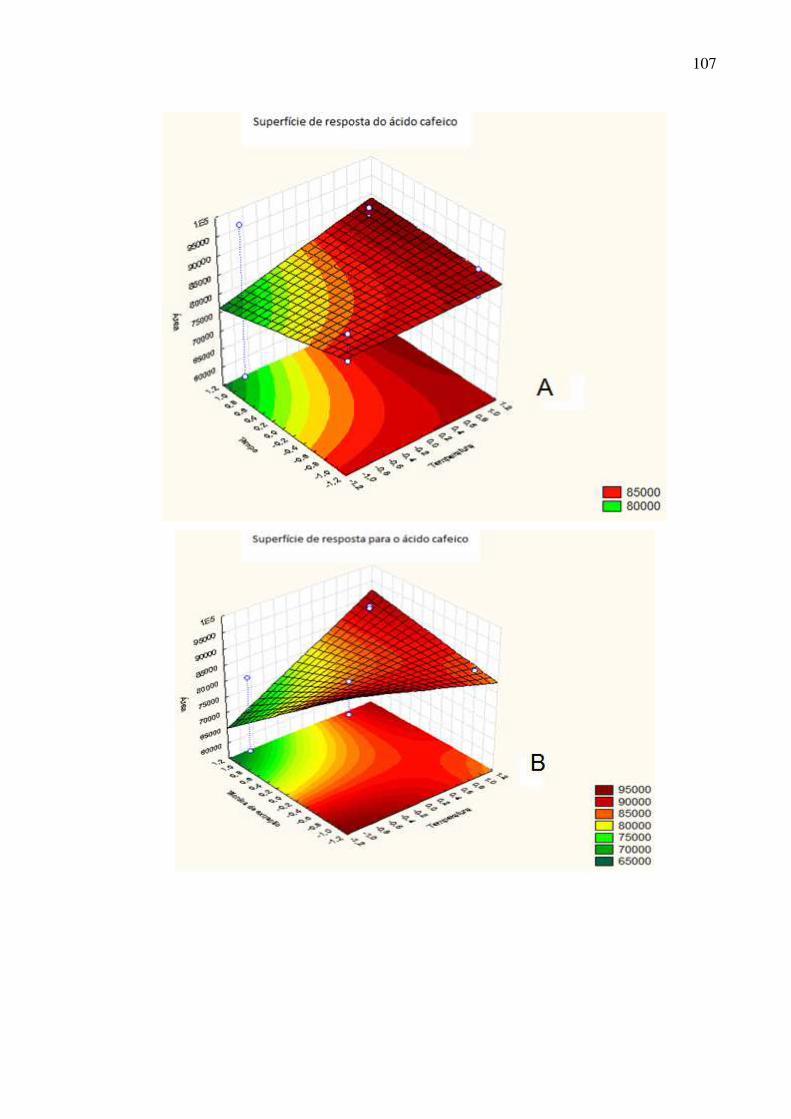
107
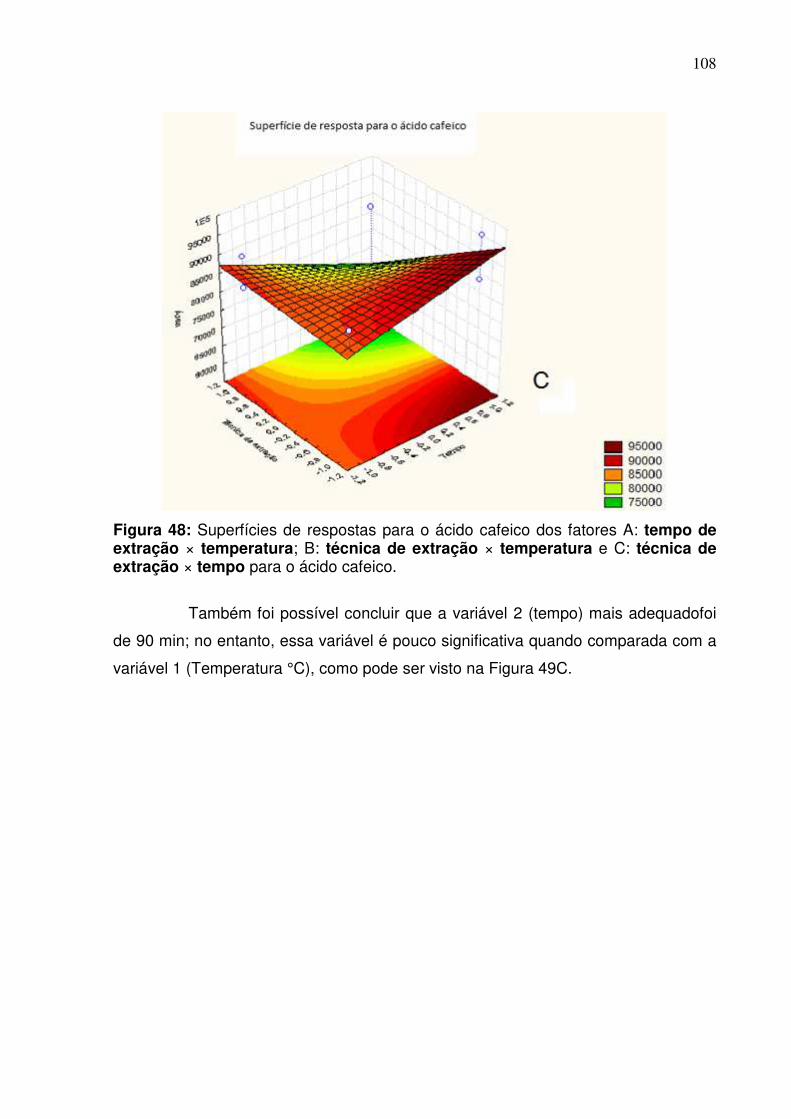
108
Figura 48: Superfícies de respostas para o ácido cafeico dos fatores A: tempo de extração × temperatura; B: técnica de extração × temperatura e C: técnica de extração × tempo para o ácido cafeico.
Também foi possível concluir que a variável 2 (tempo) mais adequadofoi
de 90 min; no entanto, essa variável é pouco significativa quando comparada com a
variável 1 (Temperatura °C), como pode ser visto na Figura 49C.
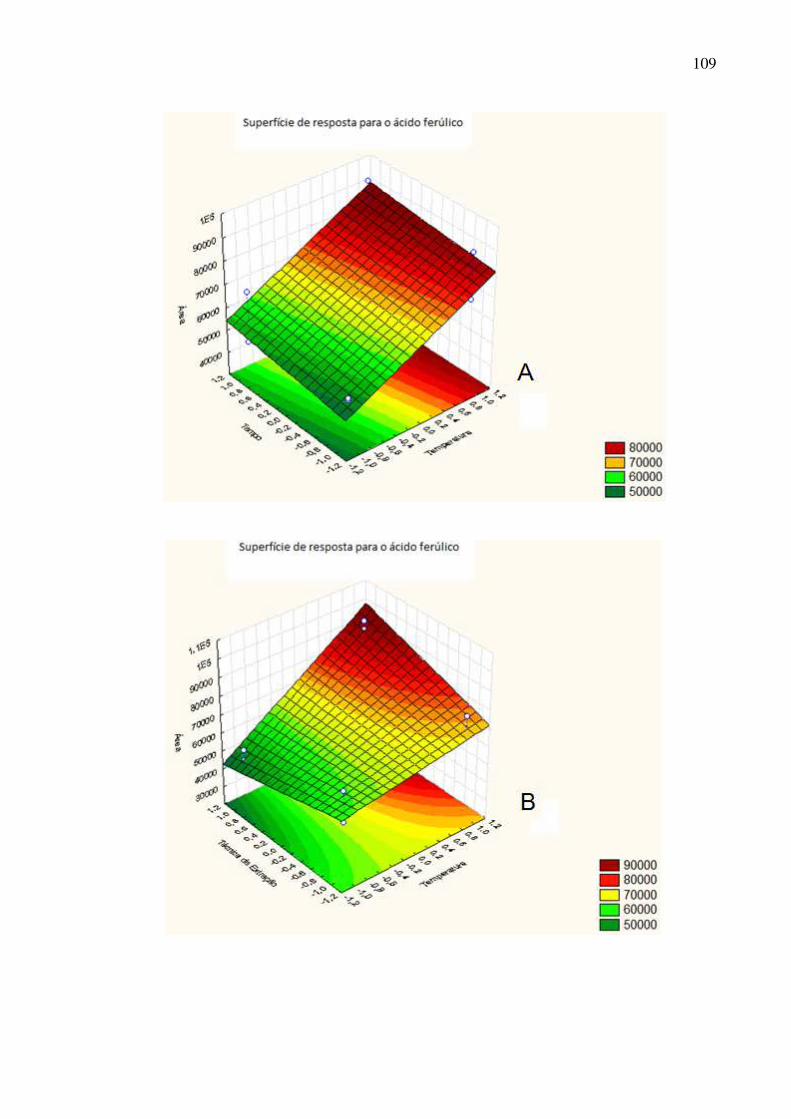
109

110
Figura 49: Superfícies de respostas para o ácido ferúlico dos fatores A: tempo de extração × temperatura; B: técnica de extração × temperatura e C: técnica de extração × tempo para o ácido ferúlico.
Para analitos como o ácido p-cumárico, a extração realizada em manta de
aquecimento ou por refluxo não sofreu alteração em termos de aumento nas áreas
dos picos assim como o tempo de extração, como pode ser observado na Figura
50B e Figura 50C, sendo considerada significativa apenas a variável 1 (Temperatura
°C), conforme a Figura 50A e Figura 50B.
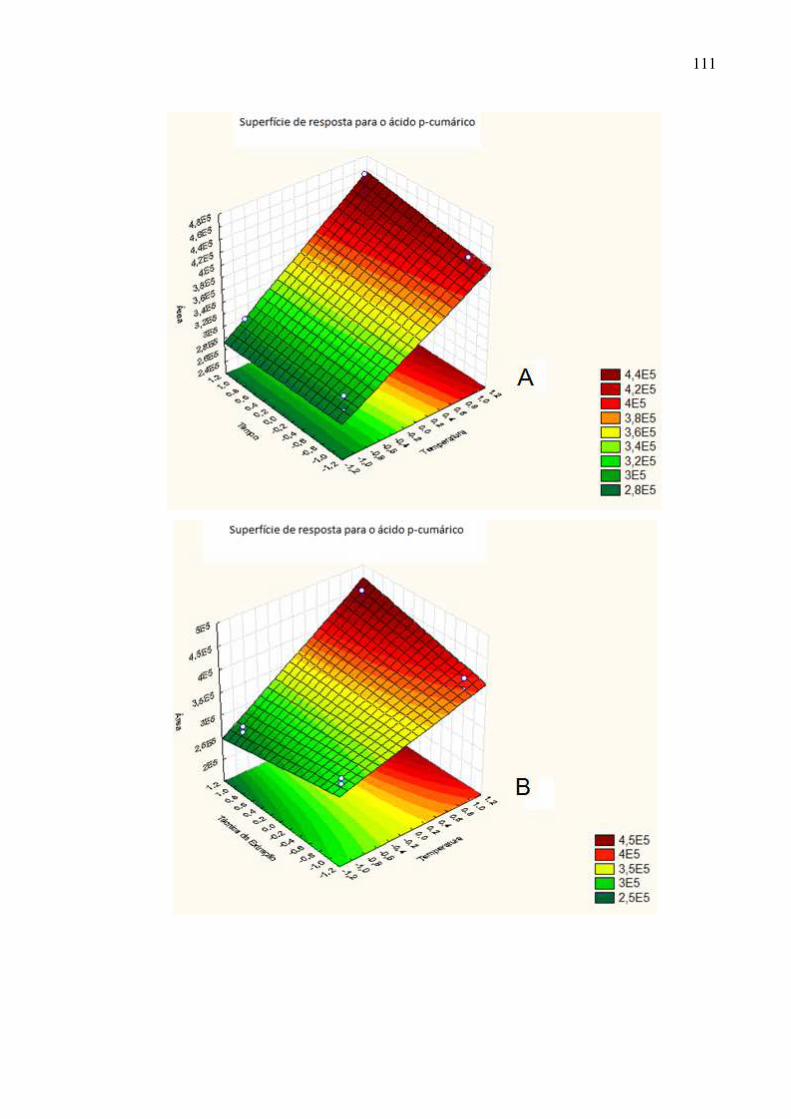
111

112
Figura 50: Superfícies de respostas para o ácido p-cumárico dos fatores A: tempo de extração × temperatura; B: técnica de extração × temperatura e C: técnica de extração × tempo para o ácido p-cumárico.
Para os analitos ácido protocatecóico, ácido clorogênico, ácido vanílico,
ácido sinápico, miricetina e rutina, não foram obtidas as superfícies de resposta, pois
os valores de área foram muito baixos.
Dentre as superfícies de respostas apresentadas, notou-se que a variável
1 (Temperatura) é a mais significativa quando comparada com as outras variáveis e
a temperatura ótima para extração dos compostos fenólicos foi de 80°C. A variável 2
(tempo) conforme visto nas Figuras 46, 48, 49 e 50 também foi significativa e a
condição mais favorável foi um período de extração de 90 min.
A partir da avaliação da superfície de resposta dos analitos foi possível
estabelecer as condições otimizadas de extração para determinação de compostos
fenólicos em cevada. Como a técnica de extração não foi significativa para a maioria
dos analitos avaliados, escolheu-se a extração em sistema de refluxo, por ser um
método comumente usado para extração deste tipo de compostos [9, 43, 39,123].
Assim, a temperatura escolhida foi de 80 °C e o tempo de extração foi de 90 min.

113
4.3. Quantificação dos compostos fenólicos pelo método otimizado
4.3.1. Validação do método otimizado
Após a otimização das condições de extração, o método foi validado
seguindo as figuras de mérito de acordo com os critérios descritos pela ANVISA [93].
A amostras de cevada utilizada nesta etapa foi da cultivar BRS Lagoa 225.
A seletividade do método foi avaliada para algumas amostras de cevada
(extratos de cevada com fortificação) que foram injetadas para monitorar, através
das janelas de aquisição, os cromatogramas dos analitos individualmente sem a
necessidade de separá-los completamente, pois os analitos podem coeluírem sem
causar problemas de interferência no modo de aquisição SRM, conforme visto na
Figura 40. Esse monitoramento foi realizado para verificar se nos tempos de
retenção correspondentes aos compostos fenólicos estudados havia a coeluição de
interferentes da matriz. Os cromatogramas do extrato fortificado a 3 µg mL-1 são
observados na Figura 51, que ilustra a seletividade do método, embora haja
coeluição de alguns picos no extrato fortificado.
Os resultados mostrados na Figura 51 mostram que mesmo na presença
da matriz, não foram encontrados sinal de falso positivo, ou seja, o sinal obtido é
gerado apenas pelos compostos fenólicos, e não por interferentes provenientes das
matrizes. Dessa forma, o método é considerado seletivo.
De acordo com a Tabela 6, foram selecionadas as transições entre o íon
precursor e o respectivo íon produto de maior intensidade dos compostos fenólicos
estudados, para que pudesse ser realizada a identificação e a quantificação destes
compostos fenólicos com base nestas transições. Todos os compostos fenólicos
estudados neste trabalho apresentaram íons precursores, assim como íons produtos
distintos uns dos outros, o que assegura seletividade ao método.

114
Figura 51: Cromatogramas obtidos por UHPLC-MS/MS para separação da mistura de compostos fenólicos do extrato C4 fortificado (3 µg mL-1).Condições cromatográficas idem as da Figura 41.

115
Outra forma de verificar se o método de análise com detecção por MS/MS
é seletivo, é analisando se os espectros de massas obtidos pela janela de retenção
de cada um dos compostos fenólicos estudados apresentam intensidade de 100%
para os íons produto monitorados, o que também comprovou a seletividade do
método [111].
O coeficiente de determinação (r2) e a faixa linear foram determinados
pela obtenção das curvas analíticas (0,3 até 50 µg mL-1) preparadas pelas soluções
dos compostos (padrões) em solvente (metanol) e também na matriz.
A curva analítica preparada com os compostos na matriz foi comparada
com as curvas preparadas em solvente. Avaliou-se a detectabilidade ou inclinação
da curva analítica obtida para os dois tipos de curvas.
Pela Figura 52, pode-se analisar os comportamentos dos compostos
fenólicos nos extratos e em solvente. Verificou-se três comportamentos distintos dos
compostos fenólicos encontrados em cevada. Alguns analitos, catequina e ácido
cafeico, apresentaram grande efeito matriz, uma vez que a curva preparada em
solvente puro mostrou sensibilidade bem menor em relação às curvas preparadas
no extrato (cevada). Outros compostos apresentaram pequeno efeito matriz, como o
ácido p-cumárico e ácido protocatecóico, uma vez que a curva preparada em
solvente puro e a preparada na matriz com fortificação tiveram comportamentos
muito semelhantes. O composto fenólico que praticamente não apresentou efeito
matriz foi a miricetina, pois as curvas analíticas tiveram comportamentos
semelhantes.
A avaliação do efeito matriz foi realizada comparando as áreas obtidas
em duas curvas analíticas nos mesmos pontos, uma no extrato de cevada e outra no
solvente, empregando a Equação 1.
A obtenção de valores negativos indicam a supressão de sinais da matriz,
enquanto que resultados positivos mostram o aumento do sinal devido à matriz [108,
126].
Quando as variações nas respostas do detector entre o extrato fortificado
e a solução dos padrões em solventes, ambos na mesma concentração, for < ±20%
considera-se que há um leve efeito matriz. As concentraçõesusadas nas
fortificações foram de 1 µg g-1, 3 µg g-1 e 6 µg g-1. Valores superiores a ± 20%
apontam que o efeito matriz é significativo e ele pode ser causado por supressão de
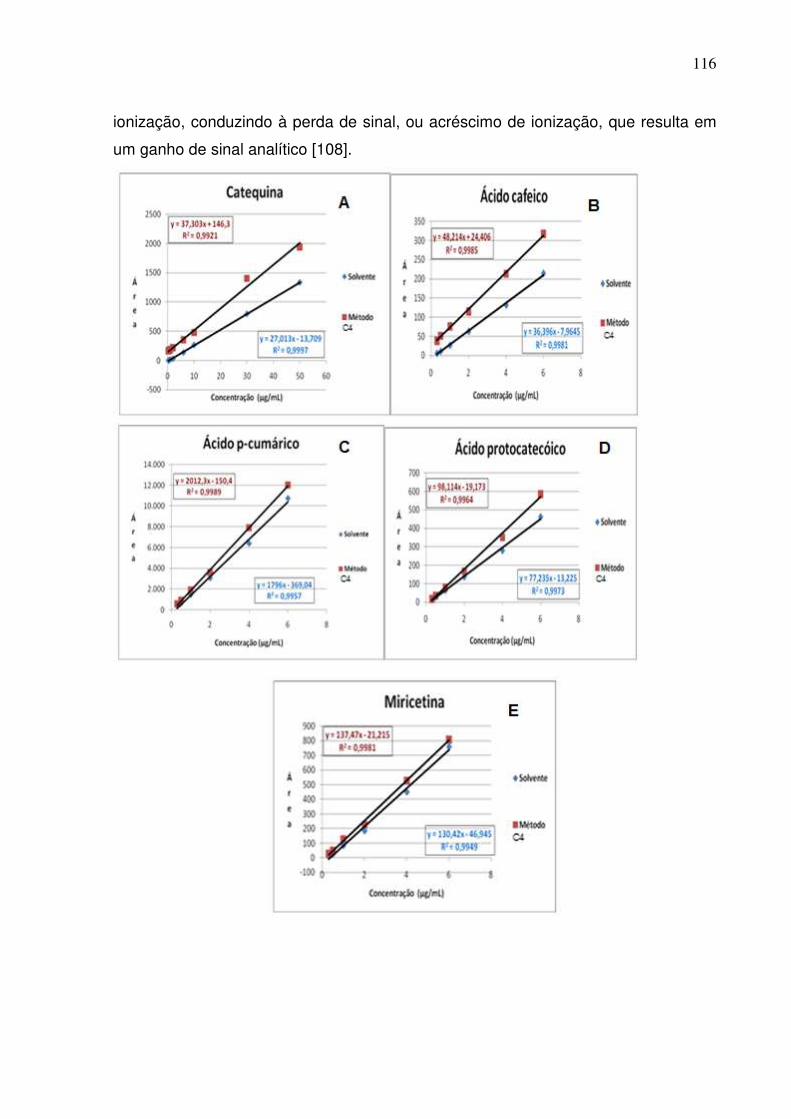
116
ionização, conduzindo à perda de sinal, ou acréscimo de ionização, que resulta em
um ganho de sinal analítico [108].

117
Figura 52: Curvas analíticas preparadas em solvente e na matriz pelo método de extração em refluxo. A: catequina, B: ácido cafeico, C: ácido p-cumárico, D: ácido protocatecóico, E: miricetina, F: ácido clorogênico, G: rutina, H: ácido sinápico, I: epicatequina, J: ácido ferúlico e K: ácido vanílico.

118
Pelo efeito matriz concluiu-se que a curva analítica não pode ser
construída somente no solvente, pois apresentou respostas do detector entre o
extrato fortificado e a solução dos padrões em solventes, ambos na mesma
concentração, com variação > ± 20%, considerando, portanto, que há um efeito
matriz relevante [109].Os valores do efeito matriz (%) para cada composto fenólico
estão representados na Figura 53.
Figura 53: Efeito matriz (%) para os compostos fenólicos em cevada usando UHPLC-MS/MS. (1) catequina; (2) epicatequina; (3) ácido clorogênico; (4) ácido vanílico; (5) ácido cafeico; (6) ácido p-cumárico; (7) ácido ferúlico; (8) ácido sinápico; (9) rutina; (10) ácido protocatecóico; (11) miricetina.
Portanto, para garantir um comportamento semelhante entre a curva e as
amostras analisadas, optou-se pela construção da curva com os compostos
fenólicos fortificados no extrato. Destaca-se que para os analitos catequina e
epicatequinafoi feita uma diluição de 5 vezes nos extratos de cevada, após a etapa
da extração, para a construção das curvas analíticas com o objetivo de alcançar
respostas dentro de uma faixa linear.
Tão importante como avaliar o percentual do efeito matriz é examinar
cuidadosamente os resíduos deixados pelo modelo. Num modelo bem ajustado,
esses resíduos devem dar a impressão de terem sido produzidos por uma
distribuição normal, ou melhor, não devem apresentar nenhum indício de
anormalidade.

119
Osresíduos da regressão linear obtida para os analitos correspondentes à
curva analítica construída na matriz podem ser vistos na Figura 54.
Os resultados enquadram-se em três possibilidades. O primeiro caso
mostra a situação ideal, em que o modelo está bem ajustado: os resíduos estão
distribuídos aleatoriamente, e não há nada que indique que sua variância não é
constante. Esta situação ocorreu para os ácidos ferúlico, vanílico e sinápico.
Já nos ácidos p-cumárico, protocatecóico, clorogênico e para a miricetina
embora a distribuição continue aleatória, pode-se perceber que os resíduos crescem
com o aumento do valor da concentração. Para a rutina e epicatequina houve a
maior evidência do terceiro caso com o aparecimento de uma estrutura não
aleatória, de aspecto curvilíneo. Os resíduos são menores nas extremidades da
faixa de calibração, e maiores na região intermediária. A catequina foi uma exceção
aos modelos de resíduos apresentados com indício de anormalidade em
concentrações baixas.
Ácido p-cumárico
Ácido protocatecóico
Ácido clorogênico

120
Ácido cafeico
Ácido ferúlico
Miricetina
Rutina
Ácido vanílico

121
Ácido sinápico
Catequina
Epicatequina
Figura 54: Resíduos da regressão linear obtida para os compostos fenólicos analisados por UHPLC-MS/MS para a curva analítica construída na matriz.
A exatidão foi avaliada experimentalmente pela recuperação através da
fortificação dos extratos em 1, 3 e 6 µg g-1 em quintuplicata, resultando num total de
15 determinações. A recuperação R (%) foi avaliada por meio da comparação do
sinal analítico dos compostos com o sinal analítico das soluções dos compostos
fenólicos antes da extração e todos os valores estiveram entre 70 e 120%. Esse
parâmetro foi obtido pelas injeções de cinco amostras, em três níveis de fortificação,
e os resultados estão na Tabela 6.
A precisão foi avaliada em termos de repetitividade e da precisão
intermediária. A repetitividade foi obtida pela média de três injeções feitas em três
níveis de fortificações e em quintuplicatas. A precisão intermediária foi avaliada
realizando-se ensaios em 3 dias consecutivos e variou entre 2,3 e 19,2 %, que estão
dentro dos valores estabelecidos pela ANVISA, que são abaixo de 20% [119].
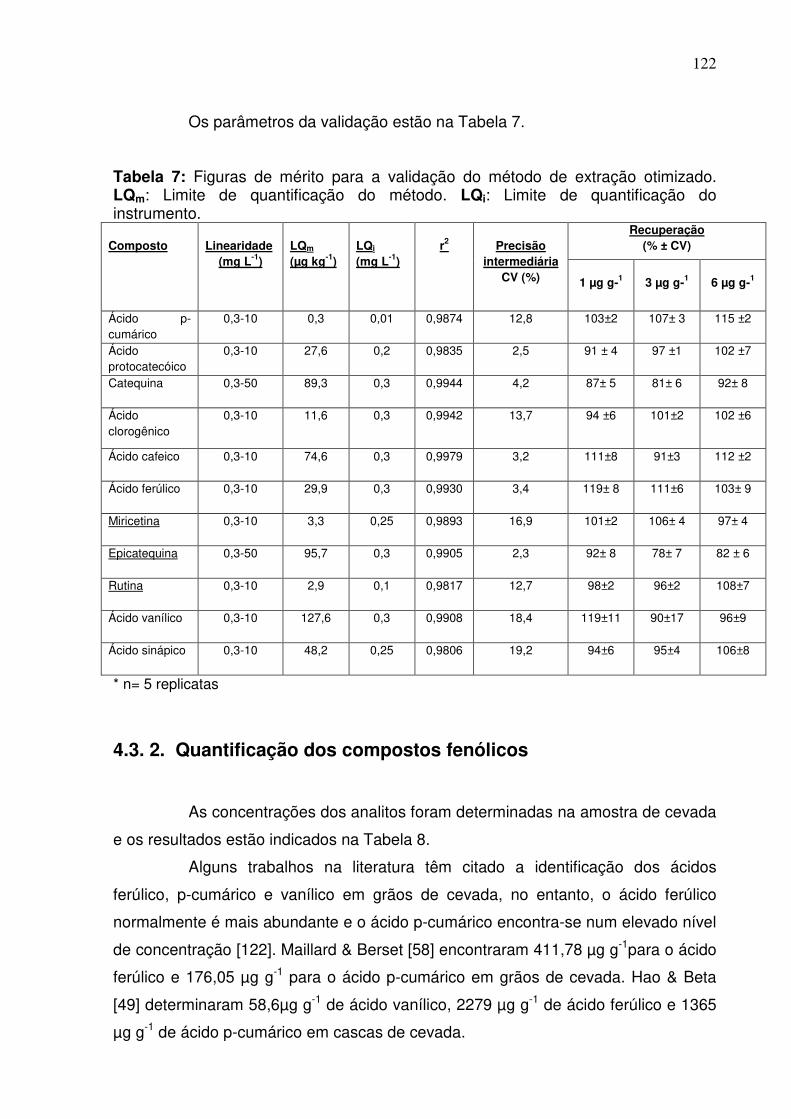
122
Os parâmetros da validação estão na Tabela 7.
Tabela 7: Figuras de mérito para a validação do método de extração otimizado. LQm: Limite de quantificação do método. LQi: Limite de quantificação do instrumento.
Composto
Linearidade
(mg L-1)
LQm
(µg kg-1)
LQi (mg L-1)
r2
Precisão
intermediária CV (%)
Recuperação (% ± CV)
1 µg g-1
3 µg g-1
6 µg g-1
Ácido p-cumárico
0,3-10 0,3 0,01 0,9874 12,8 103±2 107± 3 115 ±2
Ácido protocatecóico
0,3-10 27,6 0,2 0,9835 2,5 91 ± 4 97 ±1 102 ±7
Catequina
0,3-50 89,3 0,3 0,9944 4,2 87± 5 81± 6 92± 8
Ácido clorogênico
0,3-10 11,6 0,3 0,9942 13,7 94 ±6 101±2 102 ±6
Ácido cafeico
0,3-10 74,6 0,3 0,9979 3,2 111±8 91±3 112 ±2
Ácido ferúlico
0,3-10 29,9 0,3 0,9930 3,4 119± 8 111±6 103± 9
Miricetina
0,3-10 3,3 0,25 0,9893 16,9 101±2 106± 4 97± 4
Epicatequina
0,3-50 95,7 0,3 0,9905 2,3 92± 8 78± 7 82 ± 6
Rutina
0,3-10 2,9 0,1 0,9817 12,7 98±2 96±2 108±7
Ácido vanílico
0,3-10 127,6 0,3 0,9908 18,4 119±11 90±17 96±9
Ácido sinápico
0,3-10 48,2 0,25 0,9806 19,2 94±6 95±4 106±8
* n= 5 replicatas
4.3. 2. Quantificação dos compostos fenólicos
As concentrações dos analitos foram determinadas na amostra de cevada
e os resultados estão indicados na Tabela 8.
Alguns trabalhos na literatura têm citado a identificação dos ácidos
ferúlico, p-cumárico e vanílico em grãos de cevada, no entanto, o ácido ferúlico
normalmente é mais abundante e o ácido p-cumárico encontra-se num elevado nível
de concentração [122]. Maillard & Berset [58] encontraram 411,78 µg g-1para o ácido
ferúlico e 176,05 µg g-1 para o ácido p-cumárico em grãos de cevada. Hao & Beta
[49] determinaram 58,6µg g-1 de ácido vanílico, 2279 µg g-1 de ácido ferúlico e 1365
µg g-1 de ácido p-cumárico em cascas de cevada.

123
Tabela 8: Concentração dos compostos fenólicos na amostra de cevada BRS lagoa 225.
Substância Concentração
(µg g-1) ± CV(%)
Ácido p- cumárico 116,7 ± 5
Ácido protocatecóico n.d ±
Catequina 9170 ± 8
Ácido clorogênico n.d
Ácido cafeico 716,7 ± 13
Ácido ferúlico 649 ± 6
Miricetina n.d
Epicatequina 8163,3 ± 12
Rutina 143,3 ± 9
Ácido vanílico 650 ± 7
Ácido sinápico n.d
* n.d = não detectado
Zhao et al. [48] determinaram 56,15 µg g-1 para catequina, 12,45 µg g-1
para epicatequina, 12,05 µg g-1 para o ácido ferúlico e 1,78 µg g-1 para o ácido p-
cumárico em grãos de cevada.Bezerra [9] avaliou diferentes cultivares e destacou-se
a determinação de 334,5 µg g-1 para a rutina e 50,5 µg g-1 para o ácido cafeico.
Bezerra [39] também avaliou a quantificação de compostos fenólicos em diferentes
cultivares entre os anos 2008 até 2010. Os valores variaram de 1320,0 µg g-1 para
484,6 µg g-1 correspondentes ao ácido p-cumárico e de 260,8 µg g-1 para 316,5 µg g-
1 correspondentes ao ácido ferúlico nos respectivos anos. Com base na Tabela 7,
notou-se que as concentrações da catequina e epicatequina foram as mais elevadas
nas amostras avaliadas neste trabalho, dentre todos os analitos. Já os ácidos
protocatecóico e clorogênico tiveram as concentrações mais baixas. Os resultados
foram apresentados considerando a matéria seca para a determinação das
concentrações.
4.4. Eficiência de extração
Na Tabela 9 os valores de eficiência de extração variaram de 84,1 a 99,8%.

124
Tabela 9:Eficiência de extraçãodos compostos fenólicos no método por refluxo.
4.5. Método QuEChERS
O método QuEChERS foi empregado neste trabalho na tentativa de se
obterem extratos mais limpos, com boa recuperação para os compostos fenólicos e
para se avaliar a aplicabilidade do método para estes compostos, uma vez que
normalmente o método QuEChERS é empregado para extração de agrotóxicos.
Os métodos QuEChERS original e modificado (versão acetato) foram
avaliados para determinação de compostos fenólicos em cevada. Os resultados
mostraram melhores recuperações dos analitos para o método QuEChERS
modificado (versão acetato), sendo este o escolhido para o processo de validação
analítica. O método QuEChERS versão acetato tem sido adequado para a extração
de agrotóxicos em matrizes gordurosas tais como o abacate, leite e carne bovina
[73]. Como a cevada trata-se de um cereal com um considerável teor de gordura
(6,4%), o método QuEChERS versão acetato foi escolhido na tentativa de alcançar
melhores resultados nas figuras de mérito do que os métodos de extração
convencionais.
A amostra de cevada utilizada nesta etapa foi da cultivar BRS lagoa
26612.
Composto
Eficiência de extração (%)
Ácido p-cumárico
86,7
Ácido protocatecóico
99,8
Catequina 98,5 Ácido clorogênico
99,7
Ácido cafeico 98,8 Ácido ferúlico 84,1 Miricetina 96,5 Epicatequina 99,2 Rutina 93,1 Ácido vanílico 98,0 Ácido sinápico 84,1

125
4.5.1. Validação do Método QuEChERS
A seletividade do método foi avaliada para algumas amostras de cevada
(extratos de cevada com fortificação) que foram injetadas para monitorar, através
das janelas de aquisição, os cromatogramas dos analitos individualmente sem a
necessidade de separá-los completamente. Na Figura 55 estão os cromatogramas
do extrato obtido utilizando-se o método QuEChERS para a amostra fortificada. O
cromatograma de cada composto fenólico foi obtido no canal correspondente à sua
transição.

126
Figura 55: Cromatogramas do extrato fortificado com 3 µg mL-1 obtidos pelo método QuEChERS.
Neste método foi feita uma diluição de 5 vezes nos extratos de cevada
após a etapa de extração a fim de melhorar a resolução dos picos e diminuir o efeito
matriz. Pode-se notar que o fator de diluição nos extratos de cevada melhorou a
resolução dos picos cromatográficos quando comparados com os cromatogramas do
método de extração sob refluxo apresentado anteriormente (Figura 51).
O coeficiente de determinação (r2) e a faixa linear foram determinados
pela obtenção das curvas analíticas (0,3 até 100 µg mL-1) preparadas pelas soluções
dos compostos em solvente e na matriz. A Figura 56 mostra os comportamentos
distintos das curvas analíticas.
Foi possível observar que existe uma diferença significativa nos
coeficientes angulares para alguns compostos, indicando a presença do efeito matriz
pronunciado quando o espectrômetro de massas sequencial foi empregado.
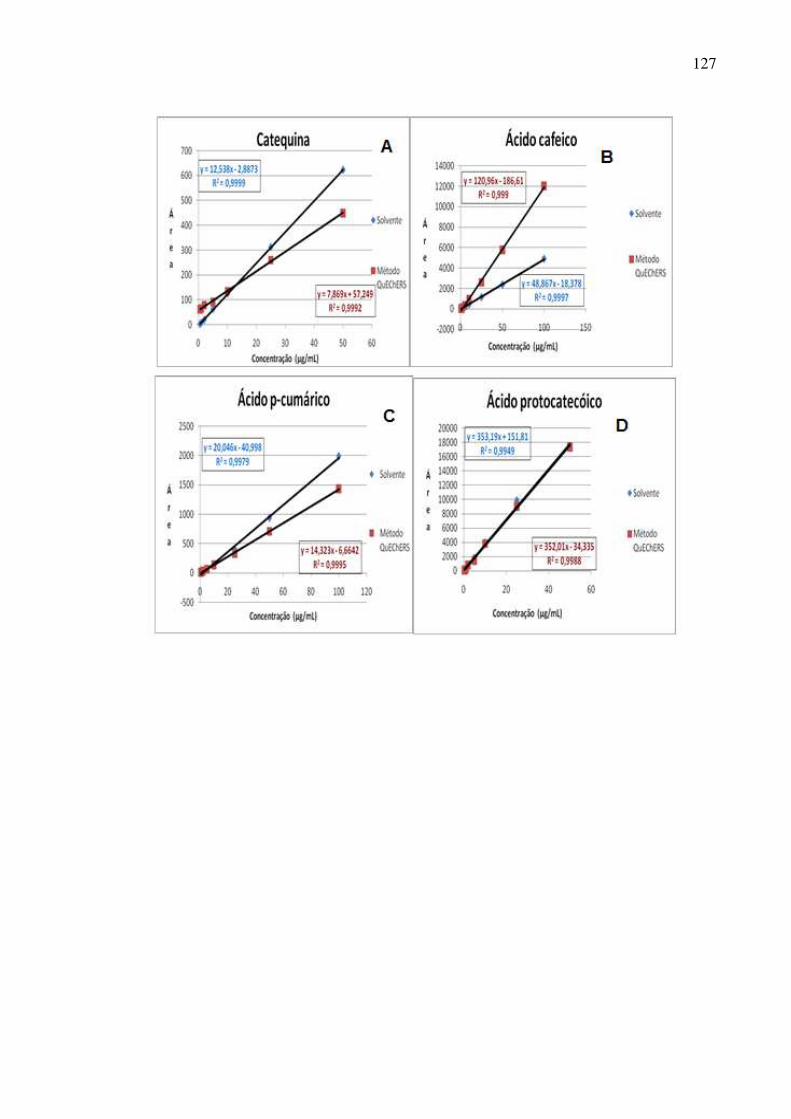
127

128

129
Figura 56: Curvas analíticas preparadas em solvente e na matriz pelo método QuEChERS.A: catequina, B: ácido cafeico, C: ácido p-cumárico, D: ácido protocatecóico, E: ácido clorogênico, F: rutina, G: ácido sinápico, H: epicatequina, I: ácido ferúlico, J: ácido vanílico e K: miricetina.
Os analitos que apresentaram maiores efeito matriz foram a miricetina e o
ácido cafeico, sendo que para a miricetina, a curva preparada em solvente mostrou
sensibilidade bem maior em relação à curva preparada na matriz, enquanto que para
o ácido cafeico a curva preparada em solvente mostrou sensibilidade bem menor em
relação à curva preparada na matriz. Os compostos fenólicos que apresentaram um
médio efeito matriz foram o ácido p-cumárico e a catequina. O composto fenólico
que praticamente não apresentou efeito matriz foi o ácido protocatecóico, pois as
curvas analíticas tiveram comportamentos semelhantes.
Outros analitos que apresentaram grande influência do efeito matriz foram
os ácidos sinápico, ferúlico, vanílico e epicatequina. A epicatequina e o ácido ferúlico
apresentaram respostas cromatográficas (área dos picos) maiores quando estes se
encontravam na presença da matriz do que em solvente. Os ácidos sinápico e
vanílico apresentaram respostas cromatográficas (área dos picos) menores quando
estes se encontravam na presença da matriz do que em solvente (supressão iônica).
Já os compostos ácido clorogênico e rutina apresentaram um leve efeito matriz.
O efeito matriz também foi avaliado neste método. Os valores do efeito
matriz (%) para cada composto fenólico estão mostrados na Figura 57.

130
Figura 57: Percentual do efeito matriz (%) empregando QuEChERS acetato para extração dos compostos fenólicos em cevada usando UHPLC-MS/MS. (1) catequina; (2) epicatequina; (3) ácido clorogênico; (4) ácido vanílico; (5) ácido cafeico; (6) ácido p-cumárico; (7) ácido ferúlico; (8) ácido sinápico; (9) rutina; (10) ácido protocatecóico; (11) miricetina.
Pode-se observar que alguns compostos como miricetina, ácido p-
cumárico, ácido sinápico e ácido vanílico apresentaram respostas cromatográficas
(área dos picos) menores quando estes se encontravam na presença da matriz do
que em solvente (supressão do sinal).
Concluiu-se que o efeito matriz é significativo para grande parte dos
compostos fenólicos estudados, sendo necessário o emprego na presença da matriz
para a quantificação dos compostos fenólicos, a fim de corrigir os efeitos da matriz
provocados por interferentes coextraídos.
Osresíduos da regressão linear obtida para os analitos correspondentes à
curva analítica construída na matriz podem ser vistos na Figura 58.
Os resíduos das curvas analíticas preparadas na matriz mostram a
situação ideal, em que o modelo está bem ajustado: os resíduos estão distribuídos
aleatoriamente, e não há nada que indique que sua variância não é constante.
Não houve nenhum analito no qual os resíduos cresceram com o
aumento do valor da concentração. Para o ácido p-cumárico, há maior evidência do
terceiro caso, com o aparecimento de uma estrutura não aleatória, de aspecto
curvilíneo. Os resíduos são menores nas extremidades da faixa de calibração e
maiores na região intermediária.
131
Ácido p-cumárico
Ácido protocatecóico
Ácido clorogênico
Ácido cafeico
Ácido Ferúlico
Miricetina
132
Rutina
Ácido vanílico
Catequina
Sinápico
Epicatequina
Figura 58: Resíduos da regressão linear obtida para os compostos fenólicos analisados por UHPLC-MS/MS para a curva analítica construída na matriz pelo método QuEChERS.
Os valores das figuras de mérito referentes à validação analítica do
método QuEChERS são encontrados na Tabela 8.
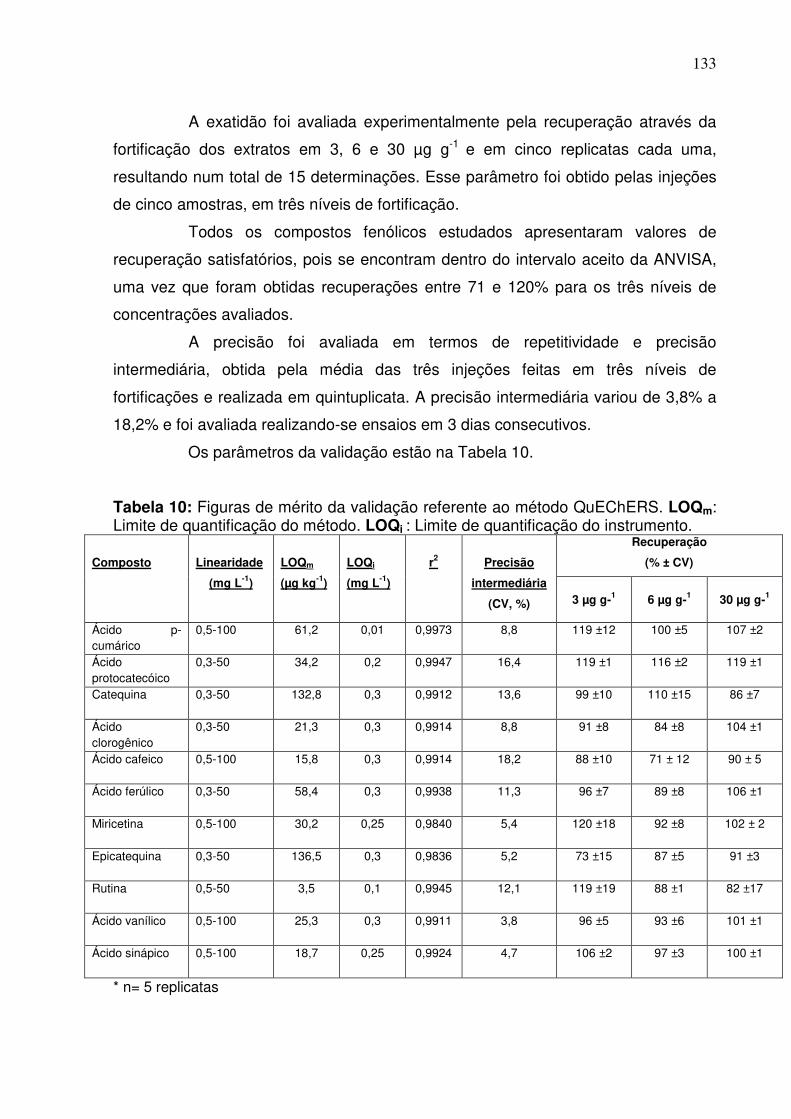
133
A exatidão foi avaliada experimentalmente pela recuperação através da
fortificação dos extratos em 3, 6 e 30 µg g-1 e em cinco replicatas cada uma,
resultando num total de 15 determinações. Esse parâmetro foi obtido pelas injeções
de cinco amostras, em três níveis de fortificação.
Todos os compostos fenólicos estudados apresentaram valores de
recuperação satisfatórios, pois se encontram dentro do intervalo aceito da ANVISA,
uma vez que foram obtidas recuperações entre 71 e 120% para os três níveis de
concentrações avaliados.
A precisão foi avaliada em termos de repetitividade e precisão
intermediária, obtida pela média das três injeções feitas em três níveis de
fortificações e realizada em quintuplicata. A precisão intermediária variou de 3,8% a
18,2% e foi avaliada realizando-se ensaios em 3 dias consecutivos.
Os parâmetros da validação estão na Tabela 10.
Tabela 10: Figuras de mérito da validação referente ao método QuEChERS. LOQm: Limite de quantificação do método. LOQi : Limite de quantificação do instrumento.
Composto
Linearidade
(mg L-1)
LOQm
(µg kg-1)
LOQi
(mg L-1)
r2
Precisão
intermediária
(CV, %)
Recuperação
(% ± CV)
3 µg g-1
6 µg g-1
30 µg g-1
Ácido p-cumárico
0,5-100 61,2 0,01 0,9973 8,8 119 ±12 100 ±5 107 ±2
Ácido protocatecóico
0,3-50 34,2 0,2 0,9947 16,4 119 ±1 116 ±2 119 ±1
Catequina
0,3-50 132,8 0,3 0,9912 13,6 99 ±10 110 ±15 86 ±7
Ácido clorogênico
0,3-50 21,3 0,3 0,9914 8,8 91 ±8 84 ±8 104 ±1
Ácido cafeico
0,5-100 15,8 0,3 0,9914 18,2 88 ±10 71 ± 12 90 ± 5
Ácido ferúlico
0,3-50 58,4 0,3 0,9938 11,3 96 ±7 89 ±8 106 ±1
Miricetina
0,5-100 30,2 0,25 0,9840 5,4 120 ±18 92 ±8 102 ± 2
Epicatequina
0,3-50 136,5 0,3 0,9836 5,2 73 ±15 87 ±5 91 ±3
Rutina
0,5-50 3,5 0,1 0,9945 12,1 119 ±19 88 ±1 82 ±17
Ácido vanílico
0,5-100 25,3 0,3 0,9911 3,8 96 ±5 93 ±6 101 ±1
Ácido sinápico
0,5-100 18,7 0,25 0,9924 4,7 106 ±2 97 ±3 100 ±1
* n= 5 replicatas

134
4.5.2. Quantificação dos compostos fenólicos pelo método
QuEChERS
Após a validação do método QuEChERS, as concentrações dos analitos
foram determinadas na amostra de cevada. É importante destacar que esta amostra
é diferente daquela usada no método de extração por refluxo e, os resultados são
mostrados na Tabela 11.
Notou-se que as concentrações da catequina e epicatequina também
foram as mais elevadas, dentre todos os analitos. Em seguida, ácido ferúlico foi o
analito mais abundante. Já os ácidos protocatecóico e clorogênico também tiveram
as concentrações baixas quando comparados ao método de extração por refluxo. No
entanto, o ácido p-cumárico teve a menor concentração dentre todos os analitos.
As diferentes concentrações de alguns analitos avaliados no método
QuEChERS quando comparados com o método de extração por refluxo podem ser
atribuídos à influência da sazonalidade dos diferentes lotes de amostragem.
Tabela 11: Concentração dos compostos fenólicos na amostra de cevada.
Substância Concentração
(µg g-1) ± CV(%)
Ácido p- cumárico 162,5 ± 7
Ácido protocatecóico 330 ± 9±
Catequina 15750 ± 5
Ácido clorogênico 262,5 ± 14
Ácido cafeico 5375 ± 8
Ácido ferúlico 12750 ± 9
Miricetina 412,5 ± 13
Epicatequina 16750 ±5
Rutina 487,5 ±6
Ácido vanílico 987,5 ±8
Ácido sinápico 512,5 ±12
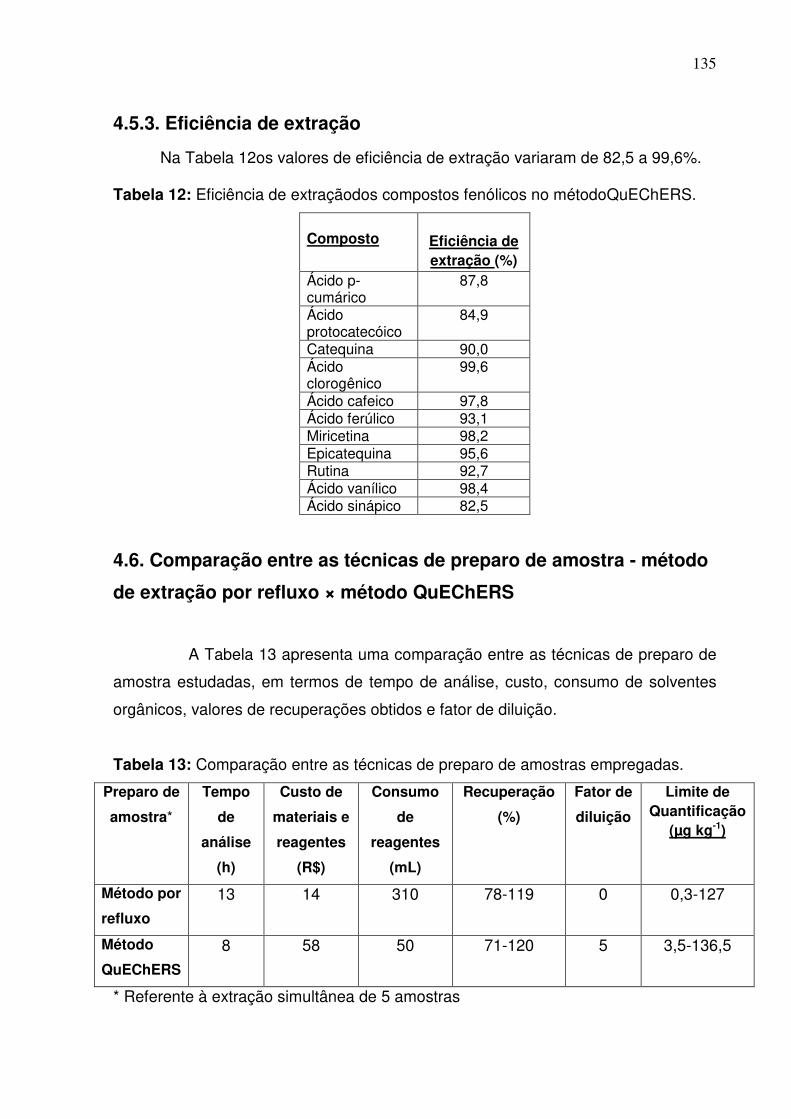
135
4.5.3. Eficiência de extração
Na Tabela 12os valores de eficiência de extração variaram de 82,5 a 99,6%. Tabela 12: Eficiência de extraçãodos compostos fenólicos no métodoQuEChERS.
Composto
Eficiência de extração (%)
Ácido p-cumárico
87,8
Ácido protocatecóico
84,9
Catequina 90,0 Ácido clorogênico
99,6
Ácido cafeico 97,8 Ácido ferúlico 93,1 Miricetina 98,2 Epicatequina 95,6 Rutina 92,7 Ácido vanílico 98,4 Ácido sinápico 82,5
4.6. Comparação entre as técnicas de preparo de amostra - método
de extração por refluxo × método QuEChERS
A Tabela 13 apresenta uma comparação entre as técnicas de preparo de
amostra estudadas, em termos de tempo de análise, custo, consumo de solventes
orgânicos, valores de recuperações obtidos e fator de diluição.
Tabela 13: Comparação entre as técnicas de preparo de amostras empregadas.
Preparo de
amostra*
Tempo
de
análise
(h)
Custo de
materiais e
reagentes
(R$)
Consumo
de
reagentes
(mL)
Recuperação
(%)
Fator de
diluição
Limite de Quantificação
(µg kg-1)
Método por
refluxo
13 14 310 78-119 0 0,3-127
Método
QuEChERS
8 58 50 71-120 5 3,5-136,5
* Referente à extração simultânea de 5 amostras

136
A técnica de preparo de amostra hidroetanólica sob refluxo mostrou-se
mais demorada e com várias etapas. O método por refluxo apresentou um tempo de
análise maior do que o método QuEChERS. Vale ressaltar que o consumo de
reagentes e a produção de resíduos foi consideravelmente maior na extração por
refluxo que o método QuEChERS.
O método QuEChERS foi realizado em um menor tempo de análise,
possivelmente por envolver um menor número de etapas, consumir menor volume
de solvente e, consequentemente, a produção de resíduos (solventes orgânicos) é
reduzida quando comparada com o método por refluxo. Obtiveram-se recuperações
satisfatórias para todos os analitos estudados e conseguiu-se atingir coeficientes de
variação menores. É importante destacar que não existem trabalhos citados na
literatura que utilizam o método QuEChERS para determinação de compostos
fenólicos em amostras de cevada, uma vez que esta técnica é muito bem
estabelecida para a análise de multirresíduos de agrotóxicos em alimentos.
Portanto, pode-se concluir que o método QuEChERS foi mais adequado
em termos de rapidez, consumo de solventes, resíduos gerados, atende os
princípios da Química Verde, eficiência e repetibilidade de extração.
Por outro lado, nota-se que para a realização das réplicas das 5 amostras
de cevada foi gasto um valor de aproximadamente R$ 58,00. Enquanto que para a
extração pelo método por refluxo foi gasto um valor de aproximadamente R$14,00.
Para avaliação dos diferentes métodos de preparo de amostras, o mesmo
lote de amostragem BRS 225, 026607 foi analisado tanto pelo método por refluxo
como pelo método QuEChERS.
Na Tabela 14 estão apresentados os valores obtidos para os compostos
fenólicos ácido cafeico, ácido ferúlico, ácido p-cumárico, rutina, catequina e
epicatequina analisadas por UHPLC-MS/MS pelas técnicas por refluxo e pelo
método QuEChERS.
Dentre as amostras analisadas, para o método por refluxo, foram
encontrados os analitos ácido cafeico, ácido ferúlico, ácido vanílico, ácido p-
cumárico, rutina, catequina e epicatequina em concentrações entre 208,8 e 11248,6
µg g-1.
Empregando o método QuEChERS, dentre as amostras analisadas,
foram detectados os ácidos cafeico, ácido ferúlico, ácido p-cumárico, além de
catequina e epicatequina em quantidades entre 172,5 e 16750 µg g-1. Isto significa

137
que o método QuEChERS permite maior detectabilidade quando comparado com o
método por refluxo em que as concentrações dos analitos variaram entre 208,8 e
11248,6 µg g-1.
As pequenas diferenças entre os 2 métodos de extração pode ser
justificada com base no uso de diferentes solventes extratores, nas proporções dos
volumes de solventes etanol/água e acetonitrila (45 e 10 mL). Embora as
quantidades de amostra entre os métodos C e QuEChERS sejam bem próximas, 3 e
4 g respectivamente, vale ressaltar que o método C foi concentrado previamente a
partir da secagem dos extratos em rotaevaporador e ressuspensão do mesmos em 2
mL de metanol antes das análises por UHPLC-MS/MS, enquanto que no método
QuEChERS foram realizadas as diluições com o objetivo de minimizar o efeito
matriz.
Tabela 14: Concentração de compostos fenólicos em grãos de cevada avaliada pelo método por refluxo e pelo método QuEChERS. Composto Método por Refluxo
Concentração (µg g-1)
Método QuEChERS
Concentração (µg g-1)
Ácido p-cumárico 208 ± 4 172 ± 8
Catequina 11248,6± 6 15750± 13
Ácido cafeico 7706,6± 9 5375± 5
Ácido ferúlico 9490± 11 12750± 9
Epicatequina 10155 ± 10 16750± 12
Rutina 454 ± 7 487 ± 7
Ácido Vanílico 1011 ± 8 987 ± 11
Assim, o maior volume da mistura extratora empregado no método C,
como a influência da temperatura podem ter colaborado para a pequena variação de
valores obtidos das concentrações dos analitos no método C quando comparados
com o método QuEChERS. Neste contexto, ambos os métodos apresentam
diferentes condições de extração, no entanto os analitos apresentaram
concentrações aproximadamente similares. Um teste estatístico foi realizado para
fins de comparação entre os dois métodos. O teste t para diferença nas médias foi
calculado conforme a Equação 9 e seus valores podem ser visualizados na Tabela
15.

138
O teste estatístico t foi determinado por:
Equação 9
Tabela 15: Comparação entre a diferença nas médias do método por refluxo e método QuEChERS.
Composto t calculado H°
Ácido p-cumárico 2,65 Aceita
Catequina 1,22 Aceita
Ácido cafeico 6,12 Rejeitada
Ácido ferúlico 7,23 Rejeitada
Epicatequina 1,83 Aceita
Rutina 1,85 Aceita
Ácido Vanílico 0,46 Aceita
* 95 % de confiança
A comparação foi feita com o t tabelado com N1+ N2 – 2 para alguns
graus de liberdade [84]. Considerando-se que t tabelado é 2,78 a 95 % de confiança
para 4 graus de liberdade foram rejeitadas as hipóteses nulas para o ácido cafeico e
ácido ferúlico, cujos valores encontrados não foram aceitáveis.
4.7. Avaliação de amostras comerciais de cevada
A fim de comparar a concentração de compostos fenólicos em amostras
de diferentes locais, algumas amostras comerciais foram adquiridas e avaliadas:
A1= São Paulo-SP, A2= Campinas-SP, A3= Belo Horizonte-MG, A4= Florianópolis-
SC, A5= Curitiba-PR, A6= Guarapuava-PR e A7= Manaus-AM.
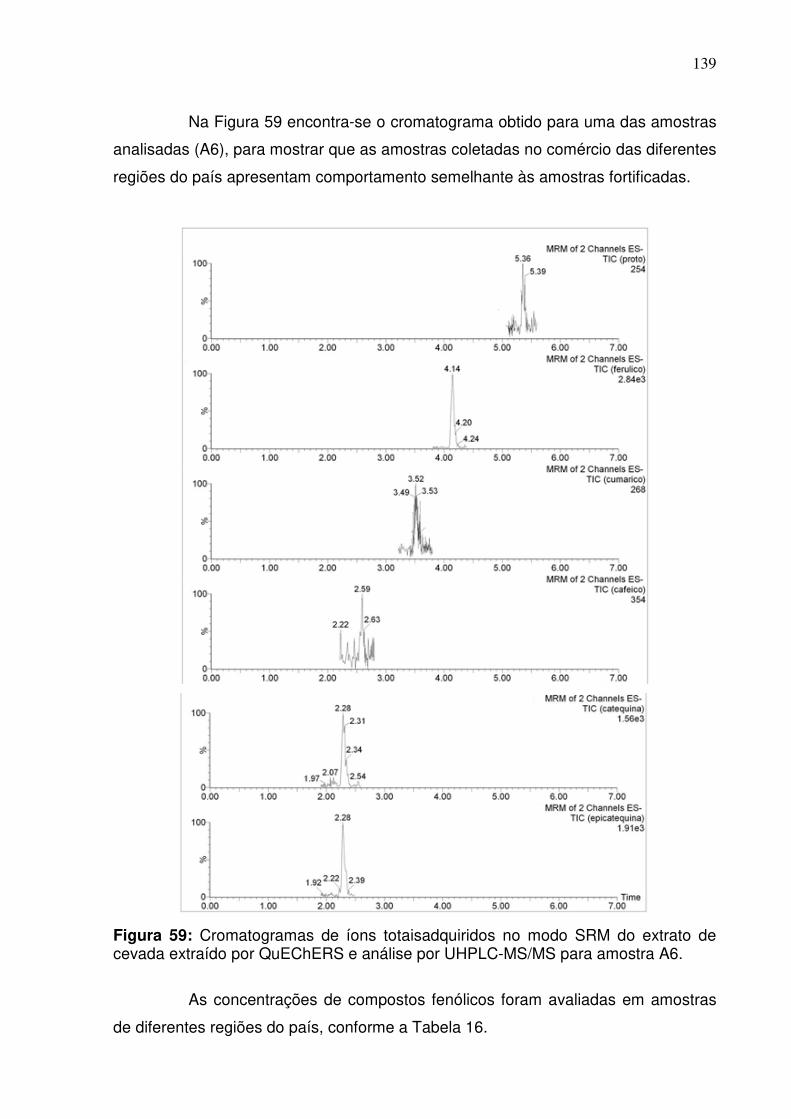
139
Na Figura 59 encontra-se o cromatograma obtido para uma das amostras
analisadas (A6), para mostrar que as amostras coletadas no comércio das diferentes
regiões do país apresentam comportamento semelhante às amostras fortificadas.
Figura 59: Cromatogramas de íons totaisadquiridos no modo SRM do extrato de cevada extraído por QuEChERS e análise por UHPLC-MS/MS para amostra A6.
As concentrações de compostos fenólicos foram avaliadas em amostras
de diferentes regiões do país, conforme a Tabela 16.
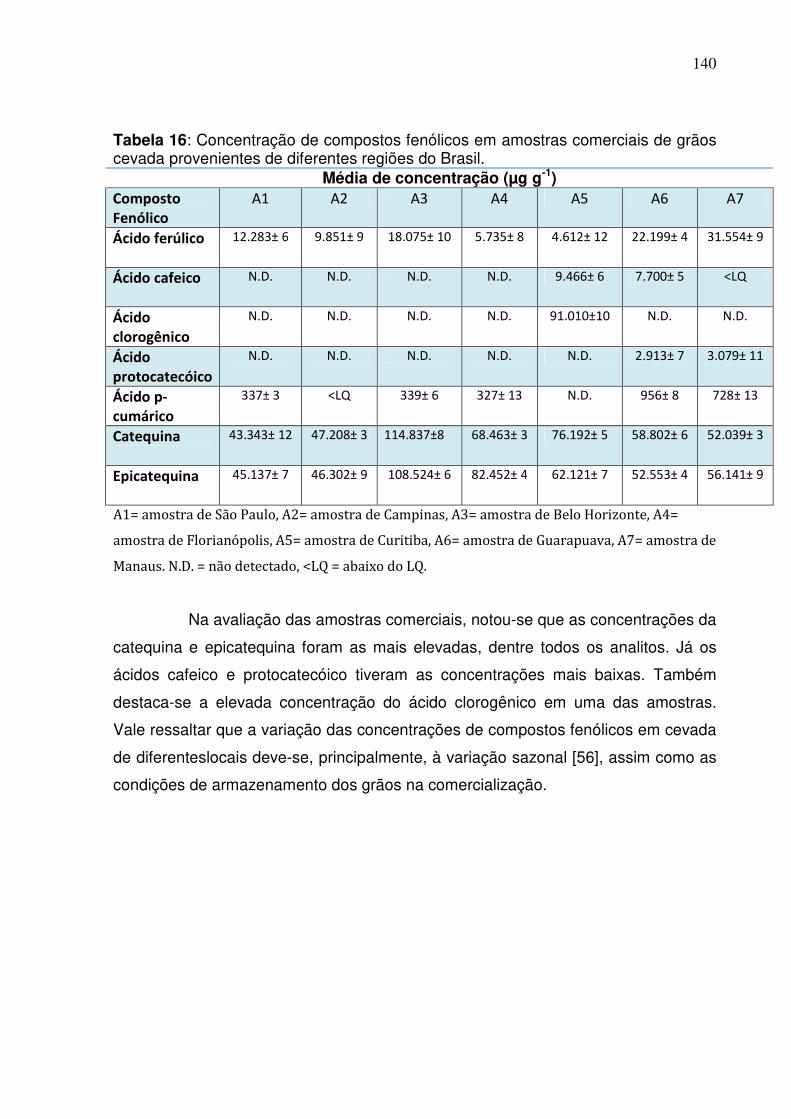
140
Tabela 16: Concentração de compostos fenólicos em amostras comerciais de grãos cevada provenientes de diferentes regiões do Brasil.
Média de concentração (µg g-1) Composto Fenólico
A1 A2 A3 A4 A5 A6 A7
Ácido ferúlico
12.283± 6 9.851± 9 18.075± 10 5.735± 8 4.612± 12 22.199± 4 31.554± 9
Ácido cafeico
N.D. N.D. N.D. N.D. 9.466± 6 7.700± 5 <LQ
Ácido clorogênico
N.D. N.D. N.D. N.D. 91.010±10 N.D. N.D.
Ácido protocatecóico
N.D. N.D. N.D. N.D. N.D. 2.913± 7 3.079± 11
Ácido p-cumárico
337± 3 <LQ 339± 6 327± 13 N.D. 956± 8 728± 13
Catequina
43.343± 12 47.208± 3 114.837±8 68.463± 3 76.192± 5 58.802± 6 52.039± 3
Epicatequina
45.137± 7 46.302± 9 108.524± 6 82.452± 4 62.121± 7 52.553± 4 56.141± 9
A1= amostra de São Paulo, A2= amostra de Campinas, A3= amostra de Belo Horizonte, A4=
amostra de Florianópolis, A5= amostra de Curitiba, A6= amostra de Guarapuava, A7= amostra de
Manaus. N.D. = não detectado, <LQ = abaixo do LQ.
Na avaliação das amostras comerciais, notou-se que as concentrações da
catequina e epicatequina foram as mais elevadas, dentre todos os analitos. Já os
ácidos cafeico e protocatecóico tiveram as concentrações mais baixas. Também
destaca-se a elevada concentração do ácido clorogênico em uma das amostras.
Vale ressaltar que a variação das concentrações de compostos fenólicos em cevada
de diferenteslocais deve-se, principalmente, à variação sazonal [56], assim como as
condições de armazenamento dos grãos na comercialização.

141
5. CONCLUSÕES
Para identificar e quantificar compostos fenólicos em cevada, foram
testadas diferentes técnicas de extração tais como refluxo, aquecimento com
agitação magnética, extração em atmosfera inerte e hidrólise, que foram avaliadas
utilizando-se ESI(-)-MS/MS e UHPLC-MS/MS.
Com a realização de testes de ESI no modo positivo, apenas 4 analitos foram
capazes de sofrer ionização. Portanto, fez-se a escolha pelo modo negativo de
ionização, sendo que este foi favorável para 11 analitos investigados. Para a maioria
dos compostos fenólicos, a melhor condição cromatográfica foi obtida com a mistura
binária de acetonitrila acidificada: água com ácido fórmico 0,1 % (v/v) e por gradiente
de eluição.
O desenvolvimento do método por UHPLC/MS-MS foi eficaz para
monitorar 11 analitos simultaneamente num tempo de 7 min de análise e 10 min
entre as replicatas das injeções. Destaca-se que o tempo de análise é menor
comparado com outros métodos de análise de compostos fenólicos em cevada
citados na literatura, como o HPLC.
A elaboração de um planejamento fatorial (23) foi importante para fornecer
dados para otimização do método por refluxo. Notou-se que a variável temperatura
foi a mais significativa quando comparada com as outras variáveis e a temperatura
ótima para extração dos compostos fenólicos foi de 80°C. A variável tempo também
foi significativa para obtenção de uma extração eficiente e a condição mais favorável
foi um período de extração de 90 min.
Os métodos de preparo de amostras validados foram capazes de
determinar compostos fenólicos solúveis em cevada com a mistura hidoetanólica no
método por refluxo e com acetonitrila pelo QuEChERS.
O método QuEChERS foi empregado neste trabalho na tentativa de se
obterem extratos mais limpos, com boa recuperação para os compostos fenólicos e
para avaliar a aplicabilidade do método para este tipo de composto, uma vez que
normalmente o método QuEChERS é empregado para extração de agrotóxicos.
A validação analítica mostrou que ambos os métodos respondem
adequadamente para analisar os compostos fenólicos em cevada.
Concluiu-se que a melhor técnica de preparo de amostra para
determinação de compostos fenólicos em cevada em termos de rapidez, consumo

142
de solventes, resíduos gerados, que atende os princípios da Química Verde,
eficiência e repetibilidade de extração é o QuEChERS.
Para avaliação dos diferentes métodos de preparo de amostras, o mesmo
lote de amostragem dos grãos de cevada foi analisado tanto pelo método por refluxo
como pelo método QuEChERS. Vale ressaltar que ambos os métodos podem ser
utilizados na análise de compostos fenólicos em cevada.
Conclui-se que os métodos de preparo de amostras validados podem ser
utilizados para controle de qualidade de cevada em cervejarias, uma vez que onze
compostos fenólicos foram estudados e as figuras de mérito comprovaram a
confiabilidade dos resultados em diferentes amostras.
A fim de comparar a concentração de compostos fenólicos em amostras
de diferentes locais, algumas amostras comerciais foram adquiridas e avaliadas,
indicando diferenças nas concentrações que podem ser decorrentes da variação
sazonal como diferentes climas, solos, temperaturas, durante cada estação do ano e
quantidade de chuva.

143
6. REFERÊNCIAS
1.Kendall, N. T. Barley and Malt. Handbook of Brewing. Food Science and Tecnology.
Ed. By William A. Hardwick. Marcel Dekker, Inc. p. 109-132, 1994.
2. Stephen, R. M.; Eisendrath, B. Understanding cereal crops I wheat, oats, barley
and rye, 1986.
3. Borowski, D. Z., Efeito do genótipo, ambiente e suas interações em características
agronômicas e de qualidade em cevada cervejeira no sul do Brasil.Dissertação de
Mestrado.Instituto de Agronomia. 2012, Universidade de Passo Fundo.
4. FAO, Food and Agriculture Organization of the United Nations. Word crop
production. 2012.
5. Baik, B. K.; Ullrich, S.E. Barley for food: Characteristics, improvement, and renewed
interest.Journal of Cereal Science, 2008. 48(2): p. 233-242.
6. Mori, C.; Minella, E.; Aspectos econômicos e conjunturais da cultura da cevada
Passo Fundo: Embrapa Trigo, 2012. 28 p. html. (Embrapa Trigo. Documentos Online,
139). Disponível em: <http://www.cnpt.embrapa.br/biblio/do/p_do139.htm>.
7. Vicenzo, R. Tópicos avançados em alimentos II- Bebidas. Universidade do
Noroeste do Estado do RS (UNIJUÍ).
8. Yalçn, E. et al. Effects of genotype and environment on β-glucan and dietary fiber
contents of hull-less barley grown in Turkey. Food Chemistry, 2007. 101 (1) p. 171-
176.
9. Bezerra, A.S. Caracterização de compostos antioxidantes em grãos de diferentes
cultivares de cevada (Hordeum vulgare L.). Dissertação de Mestrado. Centro de
Ciências Rurais. 2009, Universidade Federal de Santa Maria.
10. Anderson, A.A.M., R. Anderson, and P. Aman, Air classification of barley flours.
2000. Cereal Chemistry, 2000. 77: p. 463-467.
11. Slavin, J.L., Dietary fiber and body weight. Nutrition, 2005. 21(3): p. 411-418.
12. Fischer, H. and P. Griminger, Cholesterol lowering effects of certain grains and of
oat fractions in chickens. Proceedings of the Society for Experimental. Biology and.
Medicine, 1967. 126: p. 108-111.

144
13. Bengtsson, E.A., Chemical studies on mixed-linked B-Glucans in hull-less barley
cultivars giving different hypercholesterolaemic responses in chickens. Journal of the
Science ofFood and Agriculture, 1990. 52: p. 435-445.
14. Trowell, H., Coronary heart disease and dietary fiber. The American Journal
Clinical Nutrition, 1975. 28: p. 798-800.
15. Rimm, E.B., Vegetable, fruit, and cereal fiber intake and risk of coronary heart
disease among men. The Journal of the American Medical Association, 1996. 275(6):
p. 447-451.
16. Ferreres, F.; Krsková, Z.; Gonçalves, R. F.; Valentão, P.; Pereira, J. A.; Dusek, J.;
Martin, J.; Andrade, P. B, Free water-soluble phenolics profiling in barley (Hordeum
vulgare L.). Journal of Agricultural and Food Chemistry, 2009. 57(6): p. 2405-9.
17. Acosta-Estrada, B.A.; Gutierrez-Uribe, J. A.; Serna-Saldivar, S.O.Bound phenolics in
foods, a review. Food Chemistry, 2014. 152: p. 46-55.
18. Boroski, M.; Visentainer, J. V.; Cottica, S. M.; Morais, D. R. Antioxidantes.
Princípios e métodos analíticos.Vol. 1. 2015, Curitiba-PR. Editora Appris. p. 139.
19. Lima, F.O., Estudo comparativo da Bioatividade de compostos fenólicos em
plantas medicinais, Química Analítica. Tese de Doutorado. 2013, Universidade
Federal de Santa Maria (UFSM): Santa Maria, RS.
20. Soares, S.E., Ácidos fenólicos como antioxidantes. Revista de Nutrição, 2002.
15(1). p. 71-81.
21. Berger, M.M., Can oxidative damage be treated nutritionally. American Journal
Clinical Nutrition, 2005. 24: p. 172-183.
22. Mathew, S.; Abraham T.E, Studies on the antioxidant activities of cinnamon
(Cinnamomum verum) bark extracts, through various in vitro models. Food
Chemistry, 2006. 94(4): p. 520-528.
23. Freitas, G. L. Potencial antioxidante e compostos fenólicos na cerveja, choop,
cevada (Hordeum vulgare L.) e no bagaço de bressagem. Dissertação de Mestrado.
Ciência e Tecnologia de Alimentos. 2006, Universidade Federal de Santa Catarina .
Florianópolis-SC.
24. Bianchi, M.L.P.; Antunes, L. M. G, Radicais livres e os principais antioxidantes da
dieta. Revista Nutrição, 1999. 12(2): p. 123-130.

145
25. Sousa, C. M. M., Rocha, H.; Vieira-Jr, G. M.; Ayres, M. C. C.; Costa, C. L. S.; Araújo,
D. S.; Cavalcante, L.C.D.; Barros, E.D.S.; Araújo, P.B.M.; Brandão, M. S.; Chaves, M.H.
Fenóis totais e atividade antioxidante de cinco plantas medicinais.Química Nova,
2007. 30(2): p. 351-355.
26. Angelo, P. M.; Jorge, N., Compostos fenólicos em alimentos – Uma breve revisão.
Revista Instituto Adolfo Lutz, 2007. 66(1): p. 1-9.
27. Naczk, M.; Shahidi, F., Extraction and analysis of phenolics in food. Journal of
Chromatography A, 2004. 1054(1-2): p. 95-111.
28. Stalikas, C.D., Extraction, separation, and detection methods for phenolic acids
and flavonoids. Journal Separation Science, 2007. 30(18): p. 3268-3295.
29. Maillard, M.N.; Soum, M. H.; Boivin, P.; Berset, C, Antioxidant Activity of Barley
and Malt: Relationship with Phenolic Content.LWT - Food Science and Technology,
1996. 29(3): p. 238-244.
30. Yu, J., Vasanthan, T.; Temelli, F., Analysis of Phenolic Acids in Barley by High-
Performance Liquid Chromatography. Journal of Agricultural and Food Chemistry,
2001. 49(9): p. 4352-4358.
31. Adom, K.K.; Liu R.H, Antioxidant Activity of Grains. Journal of Agricultural and
Food Chemistry, 2002. 50(21): p. 6182-6187.
32. Beckman, C. H. Phenolic-storing cells: keys to programmed cell death and
periderm formation in wilt disease resistance and in general defence responses in
plants? Physiological and Molecular Plant Pathology, 2000. 57, (3), p. 101-110.
33. Faulds, C. B.; Williamson, G. The role of hydroxycinnamates in the plant cell wall.
Journal of the Science of Food and Agriculture, 1999.79, (3), p. 393-395.
34. Tian, S.; Nakamura, K.; Kayahara, H. Analysis of phenolic compounds in white
rice, brown rice and germinated brown rice. Journal of Agricultural and Food
Chemistry, 2004. 52(15):p. 4808-4813.
35. Karakaya, S. Bioavailability of phenolic compounds. Critical Reviews in Food
Science and Nutrition, 2004. 44 (6), p. 453-464.
36. Andreasen, M.F., Kroon, P. A.; Williamson, G.; Garcia Conesa, M. T, Esterase
Activity Able To Hydrolyze Dietary Antioxidant Hydroxycinnamates Is Distributed
along the Intestine of Mammals. Journal of Agricultural and Food Chemistry, 2001.
49(11): p. 5679-5684.

146
37. Roginsky, V.; Eduardo, A.L., Review of methods to determine chain-breaking
antioxidant activity in food. Food Chemistry, 2005. 92(2): p. 235-254.
38. Abe, L.T.E. A., Compostos fenólicos e capacidade antioxidante de cultivares de
uvas Vitis labrusca L. e Vitis vinifera L. Ciência e Tecnologia de alimentos, 2007.
27(2): p. 394-400. 53
39. Bezerra, A.S., Caracterização de compostos antioxidantes em grãos de
diferentes cultivares de cevada (Hordeum vulgare L.), Tese de Doutorado. Centro de
Ciências Rurais. 2012, Universidade Federal de Santa Maria: Santa Maria, RS, Brasil.
40. Wang, J., Chow, W; Leung, D., Applications of LC/ESI-MS/MS and UHPLC QqTOF
MS for the determination of 148 pesticides in fruits and vegetables. Analytical and
Bioanalytical Chemistry, 2010. 396(4): p. 1513-1538.
41. Inglett, G.E., Chen, D.; Berhow, M.; Lee, S., Antioxidant activity of commercial
buckwheat flours and their free and bound phenolic compositions. Food Chemistry,
2011. 125(3): p. 923-929.
42. Brindzová, L., Zalibera, M.; Simon, P.; Certík, M.; Takácsová, M.; Mikulajová, A.;
Mikusová, L.; Rapta, P., Screening of cereal varieties for antioxidant and radical
scavenging properties applying various spectroscopic and thermoanalytical methods.
International Journal of Food Science & Technology, 2009. 44(4): p. 784-791.
43. André, M.F., Determinação de compostos fenólicos em extrato de carqueja
empregando técnicas cromatográficas e eletroforéticas, Tese de Doutorado.
Química Analítica. Instituto de Química. 2014, Unicamp.
44. Medeiros, C. R., Determinação de compostos fenólicos em extratos aquosos de
resíduos sólidos por micro-extração em fase sólida e cromatografia a gás acoplada
à espectrometria de massas, Dissertação de Mestrado. Centro de Ciências Físicas e
Matemáticas. 2013, Universidade Federal de Santa Catarina.
45. Vidal, J. L. E. A., Application of gas cromatography-tandem mass spectrometry to
the analysis of pesticides in fruits and vegetables. Journal of Cromatography A, 2002.
959: p. 203-213.
46. Annegowda, H. V.; Nee, C. W.; Mordi, M. M.; RAmanathan, S.; Mansor, S. M.
Evaluation of phenolic content and antioxidant property of hydrolysed extracts of
Terminalia catappa L. leaf. Asian Journal of Plant Sciences, 2010, 9(8) p. 479–485.

147
47. Dvorakova, M., Moreira, M. M.; Dostalek, P.; Skulivova, Z.; Guido, L. F.; Barros,
A.A., Characterization of monomeric and oligomeric flavan-3-ols from barley and malt
by liquid chromatography-ultraviolet detection-electrospray ionization mass
spectrometry. Journal of Cromatography A, 2008. 1189(1-2): p. 398-405.
48. Zhao, H.,Dong, J.; Lu, J.; Chen, J.; Li, Y.; Shan, L.; Lin, Y.; Fan, W.; Gu, G., Effects of
extraction solvent mixtures on antioxidant activity evaluation and their extraction
capacity and selectivity for free phenolic compounds in barley (Hordeum vulgare L.).
Journal of Agricultural and Food Chemistry, 2006. 54(19): p. 7277-86.
49. Hao, M.; Beta, T., Qualitative and quantitative analysis of the major phenolic
compounds as antioxidants in barley and flaxseed hulls using HPLC/MS/MS. Journal
of the Science of Food and Agriculture, 2012. 92(10): p. 2062-8.
50. Lehotay, S.J., et al., Comparison of QuEChERS sample preparation methods for the
analysis of pesticide residues in fruits and vegetables. Journal of Cromatography A,
2010. 1217(16): p. 2548-60.
51. Omwamba, M.; Hu, Q., Antioxidant activity in barley (Hordeum Vulgare L.) grains
roasted in a microwave oven under conditions optimized using response surface
methodology. Journal of Food Science, 2010. 75(1): p. C66-73.
52. Gong, L., Huang, L.; Zhang, L., Effect of steam explosion treatment on barley bran
phenolic compounds and antioxidant capacity. Journal of Agricultural and Food
Chemistry, 2012. 60(29): p. 7177-84.
53. Bonoli, M., Marconi, E.; Caboni, M.F., Free and bound phenolic compounds in
barley (Hordeum vulgare L.) flours. Evaluation of the extraction capability of different
solvent mixtures and pressurized liquid methods by micellar electrokinetic
chromatography and spectrophotometry. Journal of Chromatography A, 2004.
1057(1-2): p. 1-12.
54. Gallegos-Infante, J.A.,Rocha-Guzman, N.E.; Gonzalez-Laredo, R.F.; Pulido-Alonso,
J., Effect of processing on the antioxidant properties of extracts from Mexican barley
(Hordeum vulgare) cultivar. Food Chemistry, 2010. 119: p. 903-906.
55. Siddhuraju, P., & Becker, K. Effect of various domestic processing methods on
antinutrients and in vitro protein and starch digestibility of two indigenous varieties of
Indian tribal pulse, Mucuna pruriens var. utilis. Journal of Agriculture and Food
Chemistry, 2001. 49(6), 3058–3067.

148
56. Bezerra, A.S.; Nornberg, J. L.; Lima, F.O.; Rosa, M.B.; Carvalho, L.M., Parâmetros
climáticos e variação de compostos fenólicos em cevada. Ciência Rural, 2013. 43(9):
p. 1546-1552.
57. Sarkar, S., Alvarez, V.H.; Saldaña, M.D.A., Relevance of ions in pressurized fluid
extraction of carbohydrates andphenolics from barley hull. The Journal of
Supercritical Fluids, 2014. 93: p. 27-37.
58. Maillard, M.N.; Berset, C., Evolution of Antioxidant Activity during Kilning: Role of
Insoluble Bound Phenolic Acids of Barley and Malt. Journal of Agricultural and Food
Chemistry, 1995. 43(7): p. 1789-1793.
59. Kvasnička, F., et al., Determination of phenolic acids by capillary zone
electrophoresis and HPLC. Central European Journal of Chemistry, 2008. 6(3): p. 410-
418.
60. Pereira de Abreu, D.A., K.V. Rodriguez, and J.M. Cruz, Extraction, purification and
characterization of an antioxidant extract from barley husks and development of an
antioxidant active film for food package. Innovative Food Science & Emerging
Technologies, 2012. 13: p. 134-141.
61. Freitas, G.L., Potencial antioxidante e compostos fenólicos na cerveja, chopp,
cevada (Hordeum Vulgare L.) e no bagaço de brassagem, Dissertação de Mestrado.
Ciência dos Alimentos. 2006, Universidade Federal de Santa Catarina: Florianópolis.
62. Prestes, O.D.; Adaime, M.B, Zanella, R., QuEChERS: possibilidades e tendências no
preparo de amostra para determinação multirresíduo de pesticidas em alimentos.
Scientia Chromatographica, 2011. 3(1): p. 51-64.
63. Vigna, C.R.M., Aplicação de Polissiloxanos imobilizados sobre sílica como fase
estacionária e como sorvente na determinação de agrotóxicos em água e caldo de
cana, Tese de Doutorado. Química Analítica. 2010, Unicamp: Instituto de Química.
64. Anastassiades, M.; Lehotay, S.J., Fast and Easy Multiresidue Method Employing
Acetonitrile Extraction/Partitioning and “Dispersive Solid-Phase Extraction”for the
Determination of Pesticide Residues in Produce. Journal of AOAC INTERNATIONAL,
2003. 86(2). p. 412-431.
65. Chiaradia, M.C., Desenvolvimento, validação e aplicação de métodos para
análise multirresidual de agrotóxicos em suco de laranja e tangerina utilizando

149
CLAE-DAD, CL-EM-EM e CLUE-DAD, Tese de Doutorado. Química Analítica. 2009,
Unicamp: Campinas.
66. Borges, K. B.; Figueiredo, E. C.; Queiroz, M.E.C., Preparo de amostras para análise
de compostos orgânicos. 2015. 263.
67. Lehotay, S.J., Mastovska, K.; Lightfield, A.R., Use of Buffering and Other Means to
Improve Results of Problematic Pesticides in a Fast and Easy Method for Residue
Analysis of Fruits and Vegetables. Journal of AOAC International, 2005. 88(2).p. 615-
629.
68. INTERNATIONAL., A., Official method 2007.01: Pesticide residues in foods by
acetonitrile extraction and partioning with magnesium sulphate., A. International,
Editor. 2007.
69. Anastassiades, M.; Scherbaum, E.; Tasdelen, B.; Stajnbaher, D. Chapter 46. Recent
developments in QuEChERS methodology for pesticide multiresidue analysis.
Pesticide Chemistry: Crop Protection, Public Health, Environmental Safety, 2007.
p.439-458.
70. CEN (COMITÉ EUROPEEN DE NORMALISATION), C.T.W.N., Food of plant origin,
Determination of pesticide residues using GC-MS and/or LC-MS (/MS) following
acetonitrile extraction/partitioning- QuEChERS Method, 2006.
71. Prestes, O.D., Desenvolvimento e validação de método multirresíduo para
determinação de pesticidas em arroz polido utilizando método QuEChERS
modificado, clean-up dispersivo e GC-MS (NCI-SIM). Dissertação de Mestrado.
Centro de Ciências Naturais e exatas. 2007, Universidade Federal de Santa Maria.
72. Hackbart, H.C.S.; Souza, M. M.; Scaglioni, P. T.; Primel, E. G, Garda-Buffon, J.;
Badiale-Furlong, E., Método QuEChERS para determinação de ocratoxina e citrinina
em arroz e farelo de arroz. Química Nova, 2012. 35(9). p. 1733-1737.
73. Bandeira, D.D., Determinação de resíduos de agrotóxicos em leite bovino
empregando método QuEChERS modificado e GC-MS/MS. Dissertação de Mestrado.
Centro de Ciências Naturais e Exatas. 2012, Universidade Federal de Santa Maria.
74. Prestes, O.D.E.A., QuEChERS – Um método moderno de preparo de amostra para
determinação multirresíduo de pesticidas em alimentos por métodoscromatográficos
acoplados à espectrometria de massas. Química Nova, 2009. 32(6): p. 1620-1634.

150
75. Fontana, A.R.; R. Bottini, High-throughput method based on quick, easy, cheap,
effective, rugged and safe followed by liquid chromatography-multi-wavelength
detection for the quantification of multiclass polyphenols in wines. Journal of
Chromatography A, 2014. 1342: p. 44-53.
76. Neto, B. B.; Scarminio, I. S.; Bruns, R. E. Como fazer experimentos, Campinas-SP:
Editora Unicamp. 2007.
77. Madhujith, T.; Shahidi, F., Optimization of the extraction of antioxidative
constituents of six barley cultivars and their antioxidant properties. Journal of
Agricultural and Food Chemistry, 2006. 54(21): p. 8048-57.
78. Conde, E.;Moure, A.;Domínguez, H.;Parajó, J. C.Fractionation of Antioxidants from
Autohydrolysis of Barley Husks. Journal of Agricultural and Food Chemistry, 2008. 56
(22) p.10651–10659.
79. Verardo, V.; Gomez-Caravaca, A. M.; Marconi, E.; Fiorenza Caboni, M., Air
classification of barley flours to produce phenolic enriched ingredients: Comparative
study among MEKC-UV, RP-HPLC-DAD-MS and spectrophotometric determinations.
LWT - Food Science and Technology, 2011. 44(7): p. 1555-1561.
80. Hao, M.; Beta, T, Development of Chinese steamed bread enriched in bioactive
compounds from barley hull and flaxseed hull extracts. Food Chemistry, 2012. 133(4):
p. 1320-1325.
81. Mamani, M. C. V., Desenvolvimento e validação de métodos para a
determinação de antimicrobianos em leite e fármacos usando a cromatografia
líquida de alta eficiência e eletroforese capilar. Tese de Doutorado. Química
Analítica. 2007, Unicamp. Instituto de Química.
82. Pinheiro, A. D. N., Avaliação da eletroforese capilar para quantificação do
nebivolol em forma farmacêutica sólida e análise enantiosseletivada da ligação do
nebivolol às proteínas plasmáticas. Dissertação de Mestrado. Medicamentos e
Cosméticos. 2015, Universidade de São Paulo. Faculdade de Ciências Farmacêuticas
de Ribeirão Preto.
83. Spudeit, D. A; Dolzan, M. D.; Micke, G. A, Conceitos básicos em Eletroforese
Capilar. Scientia Chromatographica, 2012. 4 (4): p.287-297.
84. Cass, Q. B.; Cassiano, N. Cromatografia Líquida. Novas tendências e aplicações. 1.
ed. Rio de Janeiro: Elsevier Editora Ltda, 2015. 199 p.

151
85. Lanças, F. M. Scientia Chromatographica, 2010. 2 (1): p. 49-57.
86. Aleksenko, S. S. Antioxidant activity and phenolic compounds of buckwheat and
barley by the data of spectrophotometry and HPLC. Journal of Analytical Chemistry,
2013. 68(5): p. 458-465.
87. Li, W.; Friel, J.; Beta, T. An evaluation of the antioxidant properties and aroma
quality of infant cereals. Food chemistry, 2010. 121: p. 1095-1102.
88. Siebenhandl, S.; Grausgruber, H.; Pellegrini, N.; Rio, D. D; Fogliano, V; Pernice, R;
Berghofer, E. Phytochemical profile of main antioxidants in different fractions of
purple and blue wheat, and black barley. Journal of agricultural and food chemistry,
2007, 55 (21): p. 8541-8547.
89. Maldaner, L.; Jardim, I.C.S.F., O estado da arte da cromatografia líquida de ultra
eficiência. Quimica Nova, 2009. 32(1): p. 214-222.
90. Maldaner, L.; Jardim, I.C.S.F., UHPLC Uma abordagem atual: desenvolvimentos e
desafios recentes. Scientia Chromatographica, 2012. 4(3): p. 197-207.
91. Nunez, O.; Gallart- Ayala, H.; Martins, C.P.; Lucci, P., New trends in fast liquid
chromatography for food and environmental analysis. Journal of Cromatography A,
2012. 1228: p. 298-323.
92. Gruz, J.; Novák, O.; Strnad, M. Rapid analysis of phenolic acids in beverages by
UPLC–MS/MS. Food Chemistry, 2008. 111(3): p.789–794.
93. Svoboda, P.; Vlcková, H.; Nováková, L. Development and validation of UHPLC-
MS/MS method for determination of eight naturally occurring catechin derivatives in
various tea samples and the role of matrix effects. Journal of Pharmaceutical and
Biomedical Analysis, 20015. 114: p. 62-70.
94. Van Bramer, S.E., An introduction to mass spectrometry. 1998, Widener: Chester
PA.
95. Konato, F.R.B., Desenvolvimento e validação de métodos analíticos para
determinação de multirresíduos de agrotóxicos em cultura de alface, por
cromatografia líquida acoplada à espectrometria de massas sequencial, Tese de
Doutorado. Química Analítica. 2014, Unicamp: Instituto de Química.
96. Waters. Micromass Quattro micro API Mass Spectrometer: Operator´s Guide, p.
1-230, 2002.

152
97. Diniz, M.E. R. Uso da técnica de espectrometria de massas com ionização por
electrospray (ESI-MS) para o estudo do mecanismo de reações orgânicas e
avaliação do perfil de fragmentação de bis-hidroxiiminas aromáticas, Dissertação
de Mestrado. Departamento de Química. 2011, Universidade Federal de Minas
Gerais: Instituto de Ciências Exatas.
98. Lanças, F.M., A cromatografia líquida moderna e a espectrometria de massas:
Finalmente "compatíveis"? II. A escolha do analisador de massas. Scientia
Chromatographica, 2013. 5(1): p. 27-46.
99. Di Stefano, V.; Avellone, G.; Bongiorno D.; Cunsolo, V.; Muccilli, V.; Sforza, S.;
Dossena, A.; Drahos, L.; Vékey, K. Applications of liquid chromatography-mass
spectrometry for food analysis. Journal of Chromatography A, 2012. 1259: p. 74-85.
100. Yamashita, M.; Fenn, J.B., Electrospray ion source. Another variation on the free-
jet theme. The Journal of Physical Chemistry, 1984. 88(20): p. 4451-4459.
101. Souza, L. M. Aplicações da espectrometria de massas e da cromatografia
líquida na caracterização estrutural de biomoléculas de baixa massa molecular.
Ciências Biológicas. 2008, Universidade Federal do Paraná. Departamento de
Bioquímica e Biologia Molecular.
102. Chiaradia, M.C., C.H. Collins, and I.C.S.F. Jardim, O estado da arte da
cromatografia associada à espectrometria de massas acoplada à espectrometria de
massas na análise de compostos tóxicos em alimentos. Química Nova, 2008. 31(3): p.
623-636.
103. Lanças, F.M., A Cromatografia Líquida Moderna e a Espectrometria de Massas:
finalmente “compatíveis”? Scientia Chromatographica, 2009. 1(2): p. 35-61.
104. Ignat, I.; Volf, I.; Popa, V.I. A critical review of methods for characterisation of
polyphenolic compounds in fruits and vegetables. Food Chemistry, 2011. 126(4): p.
1821-35.
105. Trufelli, H.; Palma, P; Famiglini, G; Cappiello, A. Mass Spectrom, 2011. Rev. 30.
p. 491.
106. Lourenço, M. R. Avaliação de métodos multirresíduos de preparo de amostra
para determinação de antimicrobianos em alimentos: QueChERS e MEPS. Tese de
Doutorado. Química Analítica e Inorgânica. 2013, Usp: Instituto de Química de São
Carlos.

153
107. Kruve, A., Matrix effects in liquid cromatograph electrospray mass-
spectrometry, Instituto de Química.2011, Universidade de Tartu.
108. Economou, A., Botitsi, H.; Antoniou, S.; Tsipi, D., Determination of multi-class
pesticides in wines by solid-phase extraction and liquid chromatography-tandem
mass spectrometry. Journal of Chromatography A, 2009. 1216(31): p. 5856-5867.
109. Kmellar, B., Pareja, L.; Ferrer, C.; Fodor, P.; Fernadéz-Alba, A. R., Study of the
effects of operational parameters on multiresidue pesticide analysis by LC-MS/MS.
Talanta, 2011. 84(2): p. 262-73.
110. Lanças, F. M., Validação de métodos cromatográficos de análise - São Carlos,
SP, Editora Rima, 2004.
111. Ribani, M.; Botolli, C.B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.
Validação em métodos cromatográficos e eletroforéticos. Química Nova, 2004. 25
(5), p.771-780.
112. Huber, L., A primer good laboratory practice and current good manufacturing
practice - Agilent Technologies Deutschland GmbH, Germany, 2000.
113. Instituto Nacional de Metrologia, Normalização e Qualidade Industrial
(INMETRO); Vocabulário Internacional de Termos Fundamentais e Gerais de
Metrologia, 2 a. ed., 2000.
114. International conference on Harmonisation (ICH); Validation of Analytical
Procedures: Definitions and Terminology. Q2A (CPMP/ICH/381/95), 1995.
115. Barros Neto, B.; Scarminio, I. S.; Bruns, R.E.; Planejamento e otimização de
experimentos. Editora da Unicamp, Campinas, 1996.
116. Kruve, A.; Herodes, K.; Leito, Leito, I. Matrix effects in pesticide multi-residue
analysis by liquid chromatography-mass spectrometry. Journal of Cromatography A,
2008. 1187: 58-66.
117. Ribani, M., Obtenção e aplicação de padrões de isoflavonas de soja, Tese de
Doutorado. Química Analítica. Instituto de Química. 2008, UNICAMP.
118. Núñez, M.; Borrull, F.; Fontanals, N.; Pocurull, E., Determination of
pharmaceuticals in bivalves using QuEChERS extraction and liquid chromatography-
tandem mass spectrometry. Analytical and Bioanalytical Chemistry, 2015. 407(13):
p. 3841-9. 118.

154
119. ANVISA. Resolução - RE n. 27 de 17 de Maio de 2012: Guia para validação de
métodos analíticos e bioanalíticos.
120. Siqueira, S. R. R.; Donato, J. L.; Nucci, G.; Reyes, F. G. R. A high troughput method
for determining chloramphenicol residues in poultry, egg, shrimp, fish, swine and
bovine using LC-ESI-MS/MS. Journal Separation Science. 2009.32: 4012-4019.
121. Guo, H.; Liu, A.H.; Ye, M.; Yang, M.; Guo, D.A., Characterization of phenolic
compounds in the fruits of Forsythia suspensa by high-performance liquid
chromatography coupled with electrospray ionization tandem mass spectrometry.
Rapid Communications in Mass Spectrometry, 2007. 21(5): p.715–729.
122. Naczk, M.; Shahidi, F., Phenolics in cereals, fruits and vegetables: occurrence,
extraction and analysis. Journal of Pharmaceutical and Biomedical Analysis, 2006.
41(5): p. 1523-42.
123. Dadakova, E.; Kalinova, J., Determination of quercetin glycosides and free
quercetin in buckwheat by capillary micellar electrokinetic chromatography. Journal
Separation Science, 2010. 33(11): p. 1633-8.
124. Pérez-Magariño, S.; Revilla, I.; González-SanJosé, M. L, Beltrán, S. Various
applications of liquid chromatography–mass spectrometry to the analysis of
phenolic compounds. Journal of Chromatography A, 1999. 847 (1–2), p.75–81.
125. Michodjehoun-Mestres, L. et al. Monomeric phenols of cashew apple
(Anacardium occidentale L.). Food Chemistry, 2009. 112 (4), p. 851-857.
126. Taylor, P.J., Matrix effects: the Achilles heel of quantitative high-performance
liquid chromatography-electrospray-tandem mass spectrometry. Clinical
Biochemistry, 2005.38(4): p. 328-34.