desenvolvimento de metodologia para … · vantagens é a alta sensibilidade, que pode chegar a...
TRANSCRIPT

Departamento de Química
DESENVOLVIMENTO DE METODOLOGIA PARA DETERMINAÇÃO
DE ELEMENTOS TERRAS RARAS (REE) EM AMOSTRA DE
ASFALTO.
Aluno: Rodrigo Manes de Souza Rocha
Orientador: Tatiana D. Saint'Pierre
Introdução
Elementos Terras Raras (do inglês, REE) vêm se tornando cada vez mais importantes
para as economias globais. Na tabela periódica, os REE são todos os lantanídeos e o ítrio e
escândio, que pertencem ao grupo 3B mas apresentam propriedades semelhantes aos
lantanídeos. Estes elementos são maleáveis e dúcteis, têm alta condutividade elétrica e
geralmente reativos a temperaturas elevadas. Têm raio iônico similar e o estado de oxidação é
geralmente 3+. Essas semelhanças em suas características e propriedades faz com que eles se
tornem difíceis de separar. [1]
Para algumas de suas aplicações na indústria, sua especificidade os torna
insubstituíveis, como na utilização do Er na fabricação de cabos de transmissão por fibra ótica
e do cério (o mais abundante e barato dos REE), cujo óxido atua como agente de polimento de
vidros. Catalisadores de elementos terras raras vêm sendo ambientalmente cruciais tanto no
craqueamento de fluidos de petróleo quanto na conversão de gases poluentes em automóveis
[2]. No entanto, esses elementos são considerados “raros” uma vez que dificilmente são
encontrados em concentrações que tornem a exploração economicamente viável. Assim, é
necessário o desenvolvimento de métodos analíticos para quantificá-los com exatidão em
amostras geológicas.
Os asfaltos compreendem a fração mais pesada do petróleo, composta de resinas e
asfaltenos. Logo, inevitavelmente o asfalto possui terras raras em sua composição. Pode
haver, portanto, uma contaminação dos solos por esses elementos desde que haja uma um
agente lixiviante que aja sobre o asfalto. Naturalmente, esse processo é feito pela chuva ácida,
cada vez mais comum nos dias atuais.
Existem dois obstáculos principais para a determinação de elementos terras raras: as
pequenas concentrações e o fato de eles serem refratários. Sendo assim, métodos analíticos
que visem esse objetivo devem ser, além de extremamente sensíveis, específicos, para evitar a
formação de óxidos ou carbetos. [3]
As duas técnicas mais eficazes para tal determinação são baseadas em análise
instrumental por ativação de nêutrons (INAA) e espectrometria de massa por diluição
isotópica (IDMS). Contudo, apesar de essas técnicas serem suficientemente sensíveis para a
quantificação dos elementos nas concentrações em que eles aparecem na natureza, elas são
muito caras e complexas, demandando muito tempo de análise. Ademais, a diluição isotópica
não pode ser aplicada a elementos mono-isotópicos, como é o caso do Y, Pr, Tb, Ho e Tm.
Assim, buscam-se alternativas mais simples e rentáveis, mas que também permitam a
exatidão desejada para a determinação desses elementos com tantas aplicações científicas e
industriais. [3]
ICP-MS
A espectrometria de massas com plasma acoplado indutivamente (ICP-MS) é uma
técnica analítica que se utiliza da medição da razão massa/carga para proporcionar uma
análise qualitativa ou quantitativa multielementar de uma amostra. Uma de suas principais

Departamento de Química
vantagens é a alta sensibilidade, que pode chegar a detectar espécies em concentrações até
pg/g.
Como é uma técnica bem estabelecida, apresenta-se o esquema abaixo (figura 1), que
representa bem as etapas de introdução da amostra, ionização no plasma e separação de cargas
até o detector.
Figura 1. Esquema simplificado de um espectrômetro de ICP-MS. [4]
Na câmera de nebulização, ocorre a introdução da amostra, que normalmente acontece
utilizando-se um nebulizador pneumático. Esse dispositivo é normalmente acoplado a
câmaras, onde as gotículas maiores são condensadas. Mais de 95% do volume é, então,
descartado e apenas as gotículas menores, formando uma nuvem mais homogênea de
aerossol, são levadas para a tocha. A ação pneumática funciona com base no efeito venturi,
pois o nebulizador promove uma área de baixa pressão e o líquido, então, é aspirado. [5]
Uma vez introduzida a amostra, precisa-se de uma fonte geradora de íons que, nesse
caso, é o plasma. O plasma é gerado em uma tocha de quartzo, cujo extremo é centralizado no
interior de uma bobina de indução eletromagnética alimentada por uma fonte de
radiofrequência. Essa tocha é constituída por três tubos concêntricos, por onde há a passagem
de gás, normalmente argônio. No tubo mais externo é introduzido, tangencialmente, o gás
com o qual o plasma é gerado. No intermediário, há um fluxo de gás auxiliar utilizado para a
estabilização do plasma gerado. Finalmente, no tubo mais interno é introduzido o gás que
permite o transporte da amostra na forma de aerossol. [6]
A fonte de rádio frequência (RF) induz um campo magnético que promove a aceleração
de elétrons por um efeito em cascata chegando a atingir uma densidade eletrônica da ordem
de 104 elétrons por cm3 a uma temperatura de 7500K. Nessas condições, atinge-se um grau de
ionização simples para a maioria dos elementos e dupla para os mais facilmente ionizáveis.
[7]
No quadrupolo os íons gerados são finalmente conduzidos até o detector, para que possa
ser obtido o espectro desejado. A condução desses íons acontece através da aplicação de um
campo eletromagnético entre placas com diferentes potenciais. As diferentes relações
massa/carga dos íons fazem com que o campo magnético os separe pelo desvio de suas
trajetórias. Os quadrupolos funcionam como filtros que conduzem os íons sucessivamente até
o detector. [6]

Departamento de Química
Apesar dos limites de quantificação adequados para a determinação de REE, a técnica
apresenta dois tipos de interferência que podem ser relevantes e devem ser superadas.
O primeiro são as interferências não-espectrais. Elas ocorrem em geral devido a fatores
que afetam o processo de transporte da amostra, a eficiência de produção de íons no plasma
ou que alteram a extração e condução dos íons para o analisador de massas. Essas
interferências são as mais facilmente resolvidas pelos métodos clássicos de adição padrão ou
padronização interna. [8]
Mais difíceis de contornar são as interferências espectrais. Estas podem ser divididas em
dois tipos: interferências isobáricas e interferências causadas por íons duplos ou íons contendo
mais de um átomo (geralmente óxidos). As isobáricas ocorrem quando isótopos de diferentes
elementos possuem a mesma massa e a mesma relação massa/carga, gerando a sobreposição
de sinais. O segundo tipo de interferência espectral ocorre na maioria das vezes devido à
formação de íons duplos de elementos com baixa energia de ionização ou a formação de
óxidos que possuem a mesma massa que um dos elementos a ser analisado. A formação dos
íons óxidos pode ocorrer no plasma ou na vizinhança da superfície do cone de amostragem.
Sua formação é favorecida pela introdução de alta taxa da amostra solvatada, devido ao
resfriamento provocado no canal central do plasma. [5]
Em nosso caso, uma interferência que sabidamente pode ser relevante é dos óxidos de
bário em európio, já que o BaO possui a mesma relação massa carga que o isótopo mais
abundante do európio, o que pode forçar a escolha de um isótopo alternativo menos abundante
e acarretar perda na sensibilidade.
ICP-OES
A espectrometria de emissão ótica com plasma indutivamente acoplado (ICP-OES) é
uma técnica analítica também multielementar. Ela refere-se a fenômenos que envolvem
elétrons de valência, abrangendo a região do espectro visível e ultravioleta, com
comprimentos de onda entre 800 e 180 nm. Contudo, diferentemente de ICP-MS, que mede a
razão massa/carga, essa técnica detecta oticamente a energia emitida pelos átomos e íons
quando estes são excitados a estados eletrônicos mais elevados. [6]
As etapas de introdução de amostra e ionização no plasma são semelhantes às descritas
para ICP-MS. Contudo, uma vez ionizadas, as espécies não percorrem o caminho do
espectrômetro de massas. Nesse caso, a radiação emitida pela população de átomos é medida
na região do plasma denominada região normal, que possui temperaturas entre 6500 e 6800
K. Essa radiação é direcionada para o sistema ótico, constituído por uma fenda de entrada
capaz de selecionar uma fina banda com as mesmas dimensões da fenda e alinhada para
focalizar a região do plasma onde os analitos apresentem a máxima razão entre intensidade da
emissão e intensidade de fundo. [6]
As interferências não espectrais estão associadas as mesmas causas que as no ICP-MS e
são resolvidas das mesmas formas. Já a principal interferência espectral associada ao ICP-
OES é devido a superposição das linhas espectrais de diferentes elementos. Essa superposição
acontece devido aos espectros de linha serem muito complexos em decorrência da
temperatura atingida no plasma. Pode acontecer a coincidência (total ou parcial) de linhas, a
alteração do fundo em um dos extremos da linha de interesse em razão da proximidade de
outra linha e o aumento do fundo. A relevância das interferências está relacionada à razão
entre as concentrações do interferente e do analito. [9,10] No caso das interferências devido à
proximidade ou coincidência de linhas de emissão, a cautelosa escolha da linha do analito
utilizada pode resolver a questão. No caso de o interferente estar em concentrações baixas e
apenas afetar o fundo, o problema pode ser facilmente resolvido utilizando-se o corretor de
fundo, que faz medidas nas regiões adjacentes ao sinal do analito. Contudo, há uma
degradação significativa nos limites de detecção. [6]

Departamento de Química
Objetivos
O trabalho visa o desenvolvimento de um procedimento de preparação de amostra
adequada para a determinação de REE em amostra de pavimento asfáltico, a partir das
técnicas de ICP-OES e ICP-MS. Além disso, objetiva simular o possível efeito da chuva ácida
sobre o asfalto, através da lixiviação ácida usando soluções aquosas de ácido nítrico ou ácido
sulfúrico.
Metodologia
Neste trabalho, as técnicas utilizadas para a determinação dos REE foram a
espectrometria de massas com plasma acoplado indutivamente (ICP-MS) e a espectrometria
de emissão ótica com plasma indutivamente acoplado (ICP-OES). As duas técnicas utilizadas
são bastante sensíveis e apresentam limites de quantificação adequados para a determinação
desses elementos que, na maioria das vezes, se encontram em concentrações traço e ultra-
traço.
Instrumentação – ICP-MS:
As medições foram realizadas em um espectrômetro de massa com plasma
indutivamente acoplado, dotado de célula de reação dinâmica modelo ELAN DRC II (Perkin
Elmer - Sciex, Norwalk, Estados Unidos). A figura 2 apresenta uma foto do equipamento
utilizado neste trabalho, localizado no Labspectro, do Departamento de Química da PUC-Rio
e na tabela 1 são mostradas as condições operacionais empregadas.
Figura 2. Fotografia do ICP-MS, modelo ELAN DRC II (PerkinElmer), instalado no
Labspectro da PUC-Rio.
Tabela 1. Condições operacionais empregadas em ICP-MS
Parâmetro Valor
Potência do ICP 1100 L min-1
Vazão de Argônio (plasma) 15 L min-1
Vazão de Argônio (auxiliar) 1 L min-1
Taxa de aspiração 1,5 L min-1
Vazão de Argônio (nebulizador) 0,92 L min-1
Leituras por replicata 3

Departamento de Química
Curvas analíticas empregadas em ICP-MS:
As soluções de calibração foram preparadas a partir de dissolução multielementar para
os elementos: La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb e Lu, utilizando-se uma
solução mista de REEs a 10 mg L-1 (PerkinElmer, PE-17), contendo, além dos citados, os
elementos Sc, Y e Th. A faixa de concentração das soluções de calibração variou de 2,0 - 30
μg L-1 para as determinações feitas por ICP-MS. Em todas as determinações foi utilizada a
calibração externa. Os isótopos (m/z) monitorados usando esta técnica foram 139La, 140Ce, 141Pr, 144Nd, 152Sm, 153Eu, 157Gd, 159Tb, 163Dy, 163Ho, 166Er, 169Tm, 174Yb e 175Lu.
Instrumentação – ICP-OES:
As medições foram realizadas em um espectrômetro de emissão óptica com plasma
indutivamente acoplado modelo Optima 4300 DV PerkinElmer (Perkin Elmer-Sciex, USA), a
figura 3 apresenta uma foto do equipamento em questão e as condições operacionais são
mostradas na tabela 2.
Figura 3. Fotografia do ICP-OES, modelo Optima 4300 DV (PerkinElmer), instalado no
Laboratório de Espectrometria de Emissão da PUC-Rio.
Tabela 2. Condições operacionais empregadas.
Parâmetro Valor
Potência do ICP 1400 W
Vazão de Argônio (plasma) 15 L min-1
Vazão de Argônio (auxiliar) 1 L min-1
Vazão de Argônio (nebulizador) 0,55 L min-1
Taxa de aspiração: 1,5 L min-1
Aquisição de dados Área de Pico
Leituras por replicata 3
As linhas de emissão empregadas para a determinação de REE nesse trabalho foram,
em nm: La (II) 408,672; Ce (II) 418,660; Pr (II) 422,293; Nd (II) 406,109; Sm (II) 359,260;
Eu (II) 381,967; Gd (II) 335,047; Tb (II) 350,917; Dy (II) 353,170; Ho (II) 345,600; Er (II)
349,910; Tm (II) 346,220; Yb (II) 328,937 e Lu (II) 291,139. Empregou-se a visão axial do
plasma para todas as medidas.
Curvas analíticas empregadas em ICP-OES
A preparação das soluções para calibração foi feita a partir de padrões
multielementares de 100 mg L-1 (Multielementar Terras Raras MICPG21V; Qhemis High
Purity®, São Paulo, Brasil). Foram preparadas curvas analíticas com as concentrações entre

Departamento de Química
10 e 1000 μg L-1 para os elementos: La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb e
Lu.
Foram preparadas duas curvas de calibração, usando ácido nítrico 10% ou ácido
sulfúrico 10%, dependendo da matriz da amostra.
Preparo de amostra para análise por ICP-MS e ICP-OES
Quando se analisa asfalto, a dissolução total da amostra, de modo a liberar todos os
metais em solução, é muito difícil. Sendo assim, é mais interessante a utilização de ácidos
num processo de lixiviação semelhante ao que a chuva ácida faz naturalmente nos solos e no
próprio asfalto. Verificou-se experimentalmente que tanto o ácido nítrico quanto o sulfúrico
são eficientes para essa etapa.
A grande vantagem do uso do ácido sulfúrico é que os íons sulfato reagem com o Ba2+
formando a precipitação do BaSO4 (reação 1) e, assim, reduzindo drasticamente o número de
íons bário em solução. Assim, a lixiviação ácida das amostras foi feita utilizando os dois
ácidos para avaliar esse efeito.
Ba2+ (aq) + SO4
2-(aq) → BaSO4(s) (Equação 1)
A amostra de asfalto foi obtida no bairro do Jardim Botânico, no município do Rio de
Janeiro. Cerca de 50 g de asfalto foram coletados e moídos em almofariz de porcelana até
obter uma mistura homogênea. A amostra moída foi peneirada em peneiras da marca
GRANUTEST, de abertura 0,180 mm. O almofariz e a peneira foram devidamente lavados e
descontaminados com acetona, água acidificada (HNO3, 10 %) e água, e posteriormente secos
em estufa.
Na lixiviação (procedimentos 1 e 2) empregou-se uma chapa aquecida a 80 °C por 1 h e,
posteriormente, a amostra foi levada a um banho de ultrassom modelo T50 (Thorton, Brasil)
por um tempo de 15 min. Este procedimento foi realizado três vezes. Após isso, foi separado
o sobrenadante do resíduo sólido e filtrado para garantir a ausência total de material
particulado proveniente do asfalto. O mesmo procedimento foi também realizado utilizando
os dois ácidos.
As amostras foram centrifugadas e filtradas previamente à análise. Os filtros utilizados
foram filtros de membrana provenientes da Minisart® (Surfactant-free celulose acetate
(SFCA) 24mm, Syringe filter 0,45μm pore size).
A Figura 4 apresenta um fluxograma para melhor visualização dos procedimentos da
lixiviação ácida usados no preparo das amostras.

Departamento de Química
Figura 4. Fluxograma dos procedimentos estudados para preparo da amostra via lixiviação
ácida.
A fim de determinar em qual fase do material (inorgânica ou orgânica) os terras raras se
encontram, utilizou-se uma outra porção da amostra para realizar uma extração utilizando
solvente orgânico (diclorometano e tolueno) assistida por ultrassom, previamente à lixiviação.
Nesse procedimento, 1,0 g de asfalto com granulometria de 0,180 mm foi extraído
utilizando 3 mL de tolueno ou diclorometano como solvente extrator por 45 min (dividido em
três ciclos de 15 minutos). A amostra passou pelo banho de ultrassom para assistir na
solubilização da fase orgânica no solvente. A solução obtida foi centrifugada e a separação
dos extratos foi realizada por filtração simples após cada 15 minutos usando unidades
filtrantes. O sólido remanescente após a extração foi seco usando diclorometano que foi,
posteriormente, evaporado. Após isso, foi feita a lixiviação ácida do sólido utilizando tanto
ácido nítrico quanto sulfúrico. Após esse procedimento, pode-se assumir que todo o sólido
remanescente faz parte da matriz inorgânica do material.
A filtragem das amostras foi feita com unidades filtrantes, tipo Millex em polietileno
com membrana PTFE com 13 mm de diâmetro e poros de 0,22 µm (Merck Millipore). O
extrato filtrado (líquido escuro) foi, posteriormente, concentrado na capela, em atmosfera de
argônio, até peso constante e conservado em geladeira.

Departamento de Química
Resultados e discussões:
Parâmetros de validação: linearidade, limites de detecção e quantificação:
As curvas de calibração foram feitas com soluções contendo entre 10 e 1000 µg L-1,
para o caso das análises no ICP-OES. Essa faixa de concentração usada para a curva de
calibração foi escolhida observando a faixa de concentração dos analitos nos materiais
certificados e nas amostras. Os coeficientes de correlação das curvas de calibração foram
melhores que 0,99 para todos os analitos estudados tanto na curva usando ácido sulfúrico
quanto ácido nítrico (tabela 3). Os limites de detecção instrumental (expressos para a solução
de leitura, em µg L-1) e limites de quantificação do método (expressos para a amostra sólida,
em µg kg-1) também estão apresentados na tabela 3, para as soluções em ácido nítrico ou em
ácido sulfúrico.
Tabela 3. Coeficiente de determinação (R2) e LODs e LOQs obtidos por ICP-OES, nas
soluções em ácido nítrico ou sulfúrico.
Ácido nítrico Ácido sulfúrico
Analito R2 LOD
µg L-1
LOQ
µg kg-1 R2
LOD
µg L-1
LOQ
µg kg-1
La 0,9997 0,27 35,7 0,9999 0,32 43,2
Ce 0,9998 0,75 100 0,9999 0,93 124
Pr 0,9999 2,46 329 0,9999 3,07 410
Nd 0,9999 0,51 67,6 0,9999 0,61 81,7
Sm 0,9999 1,46 194 0,9999 1,70 226
Eu 0.9997 0,14 18,3 0,9998 0,16 21,2
Gd 0,9999 1,92 256 0,9999 2,25 300
Tb 0,9998 1,62 216 0,9998 1,90 254
Dy 0,9999 0,76 101 0,9999 0,90 121
Ho 0,9998 0,39 52,5 0,9999 0,46 61,6
Er 0,9999 1,06 142 0,9999 1,27 169
Tm 0,9999 0,67 89,9 0,9999 0,80 106
Yb 0,9999 0,05 6,55 0,9999 0,06 7,69
Lu 0,9998 0,05 6,63 0,9998 0,06 7,82

Departamento de Química
Da mesma forma, os parâmetros de mérito obtidos para as análises por ICP-MS estão
apresentados na tabela 4. As concentrações empregadas para construção das curvas analíticas
foram entre 2 e 30 µg L-1 de cada analito. Com base em trabalhos anteriores, adicionou-se
uma concentração de 80 µg L-1 de 103Rh a cada solução, como padrão interno.
Tabela 4. Coeficiente de determinação (R2) e LODs e LOQs obtidos por ICP-MS, nas
soluções em ácido nítrico ou sulfúrico.
Ácido nítrico Ácido sulfúrico
Analito R2 LOD
µg L-1
LOQ
µg kg-1 R2
LOD
µg L-1
LOQ
µg kg-1
La 0,9997 0,005 12,4 0,9999 0,006 17,1
Ce 0,9998 0,005 13,4 0,9998 0,005 12,5
Pr 0,9996 0,004 4,77 0,9998 0,003 3,97
Nd 0,9992 0,010 27,2 0,9997 0,014 36,6
Sm 0,9976 0,006 8,62 0,9995 0,008 10,4
Eu 0,9993 0,004 0,48 0,9990 0,003 0,33
Gd 0,9985 0,010 13,7 0,9998 0,011 15,3
Tb 0,9986 0,002 2,69 0,9999 0,002 2,91
Dy 0,9981 0,007 9,15 0,9994 0,009 11,4
Ho 0,9982 0,002 1,30 0,9997 0,001 0,61
Er 0,9991 0,005 3,23 0,9996 0,002 1,63
Tm 0,9995 0,003 0,79 0,9991 0,003 0,67
Yb 0,9989 0,006 4,28 0,9996 0,004 2,36
Lu 0,9986 0,003 1,81 0,9996 0,002 1,60
Avaliação da exatidão do método:
Para avaliar se o método desenvolvido é adequado para a determinação de REE,
determinou-se os REE por ICP-MS e ICP-OES no material certificado GSP-1. Os percentuais
de recuperação estão representados na tabela 5.
Departamento de Química
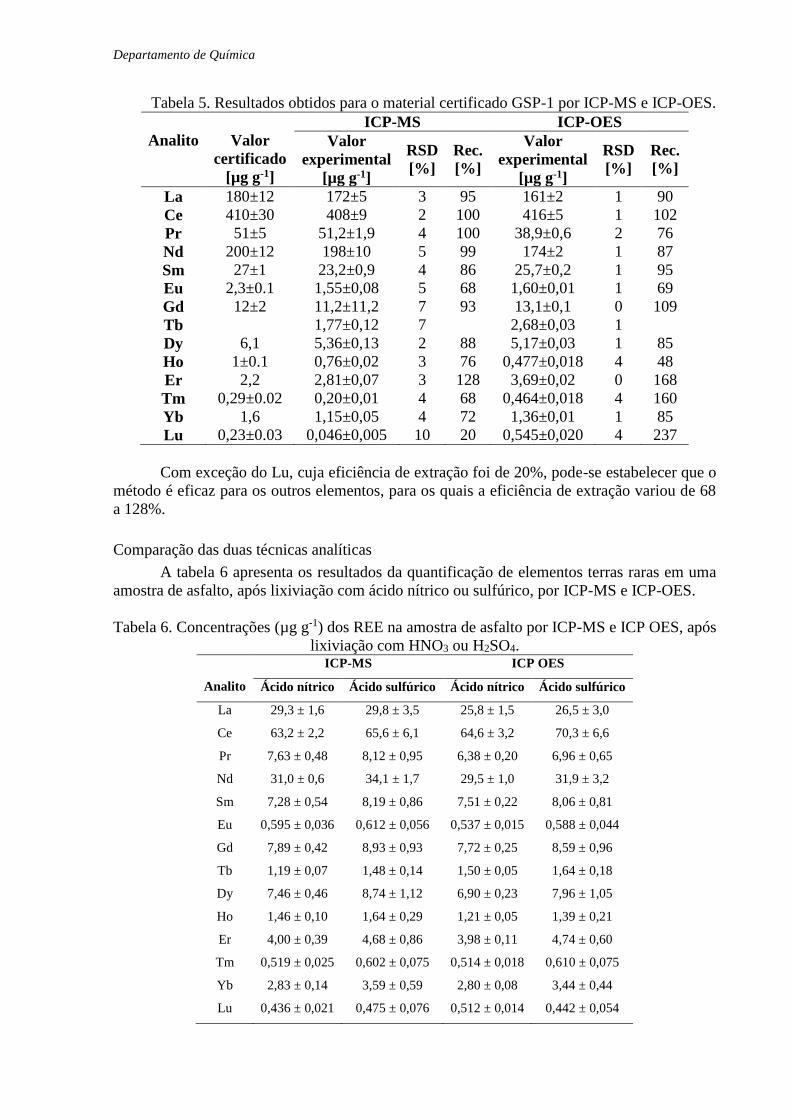
Tabela 5. Resultados obtidos para o material certificado GSP-1 por ICP-MS e ICP-OES.
Analito
Valor
certificado
[µg g-1]
ICP-MS ICP-OES
Valor
experimental
[µg g-1]
RSD
[%]
Rec.
[%]
Valor
experimental
[µg g-1]
RSD
[%]
Rec.
[%]
La 180±12 172±5 3 95 161±2 1 90
Ce 410±30 408±9 2 100 416±5 1 102
Pr 51±5 51,2±1,9 4 100 38,9±0,6 2 76
Nd 200±12 198±10 5 99 174±2 1 87
Sm 27±1 23,2±0,9 4 86 25,7±0,2 1 95
Eu 2,3±0.1 1,55±0,08 5 68 1,60±0,01 1 69
Gd 12±2 11,2±11,2 7 93 13,1±0,1 0 109
Tb 1,77±0,12 7 2,68±0,03 1
Dy 6,1 5,36±0,13 2 88 5,17±0,03 1 85
Ho 1±0.1 0,76±0,02 3 76 0,477±0,018 4 48
Er 2,2 2,81±0,07 3 128 3,69±0,02 0 168
Tm 0,29±0.02 0,20±0,01 4 68 0,464±0,018 4 160
Yb 1,6 1,15±0,05 4 72 1,36±0,01 1 85
Lu 0,23±0.03 0,046±0,005 10 20 0,545±0,020 4 237
Com exceção do Lu, cuja eficiência de extração foi de 20%, pode-se estabelecer que o
método é eficaz para os outros elementos, para os quais a eficiência de extração variou de 68
a 128%.
Comparação das duas técnicas analíticas
A tabela 6 apresenta os resultados da quantificação de elementos terras raras em uma
amostra de asfalto, após lixiviação com ácido nítrico ou sulfúrico, por ICP-MS e ICP-OES.
Tabela 6. Concentrações (µg g-1) dos REE na amostra de asfalto por ICP-MS e ICP OES, após
lixiviação com HNO3 ou H2SO4.
Analito
ICP-MS ICP OES
Ácido nítrico Ácido sulfúrico Ácido nítrico Ácido sulfúrico
La 29,3 ± 1,6 29,8 ± 3,5 25,8 ± 1,5 26,5 ± 3,0
Ce 63,2 ± 2,2 65,6 ± 6,1 64,6 ± 3,2 70,3 ± 6,6
Pr 7,63 ± 0,48 8,12 ± 0,95 6,38 ± 0,20 6,96 ± 0,65
Nd 31,0 ± 0,6 34,1 ± 1,7 29,5 ± 1,0 31,9 ± 3,2
Sm 7,28 ± 0,54 8,19 ± 0,86 7,51 ± 0,22 8,06 ± 0,81
Eu 0,595 ± 0,036 0,612 ± 0,056 0,537 ± 0,015 0,588 ± 0,044
Gd 7,89 ± 0,42 8,93 ± 0,93 7,72 ± 0,25 8,59 ± 0,96
Tb 1,19 ± 0,07 1,48 ± 0,14 1,50 ± 0,05 1,64 ± 0,18
Dy 7,46 ± 0,46 8,74 ± 1,12 6,90 ± 0,23 7,96 ± 1,05
Ho 1,46 ± 0,10 1,64 ± 0,29 1,21 ± 0,05 1,39 ± 0,21
Er 4,00 ± 0,39 4,68 ± 0,86 3,98 ± 0,11 4,74 ± 0,60
Tm 0,519 ± 0,025 0,602 ± 0,075 0,514 ± 0,018 0,610 ± 0,075
Yb 2,83 ± 0,14 3,59 ± 0,59 2,80 ± 0,08 3,44 ± 0,44
Lu 0,436 ± 0,021 0,475 ± 0,076 0,512 ± 0,014 0,442 ± 0,054

Departamento de Química
Comparando-se as concentrações dos analitos nas soluções dos lixiviados da amostra de
asfalto, com ácido nítrico ou com ácido sulfúrico, empregando o teste t pareado, obteve-se o
valor de tcaldulado=3,51, para os resultados de ICP-MS, e tcaldulado=3,45, para os resultados de
ICP OES, ambos maiores do que o tcrítico=2,160. Esses valores de t calculados para os
resultados de concentração indicam que houve diferença significativa entre os dois
procedimentos de extração. Observa-se também que os valores obtidos com a lixiviação com
ácido sulfúrico foram um pouco maiores do que os valores obtidos com ácido nítrico, como
esperado, devido à maior força ácida e maior poder oxidante do ácido sulfúrico.
Conclusões
A partir da análise do material certificado GSP-1 pelas duas técnicas utilizando-se o
procedimento de preparação de amostra desenvolvido e o padrão interno 103Rh, foram
determinadas as concentrações de REE nos lixiviados e a eficiência de extração foi de 68 a
128% para todos os elementos analisados, com exceção do Lu, cujo percentual de recuperação
foi de apenas 20%. Assim, podemos afirmar que a metodologia desenvolvida é adequada para
a determinação desses elementos.
Com a comparação estatística dos resultados obtidos pelas duas técnicas, pudemos
comprovar que ambas são adequadas para determinação de REE para a maioria dos elementos
e, assim, a escolha de cada técnica deve ser função da exatidão desejada e das concentrações
de analito presentes, já que ICP-MS é mais sensível.
As concentrações de REE determinadas nos lixiviados da amostra de asfalto indicam
que a chuva ácida deve promover a extração dos elementos para o ambiente, representando
uma fonte de poluição do solo.
Referências
1. Alam, M. A., Zuga, L., Pecht, M. G., Economics of rare earth elements in ceramic
capacitors, Disponível em: < http://www.sciencedirect.com> Acesso em: 24 Jul.
2016.
2. HAXEL, B. G., HENDRICK, B. J., ORRIS, J. G., Rare Earth Elements—Critical
Resources for High Technology, USGS Fact Sheet 087-02, 2002
3. Ardini, F., Soggia, F., Rugi, F., Udisti, R., Grotti, M., Comparison of inductively
coupled plasma spectrometry techniques for the direct determination of rare
earth elements in digests from geological samples, Analytica Chimica Acta 678,
2010
4. Jarvis, K. E., Gray, A. L., Houk, R. S., Handbook of Inductively Coupled Plasma
Mass Spectrometry, 1992
5. Giné-Rosias, M. F., Espectrometria De Massas Com Fonte De Plasma (ICP-MS),
Piracicaba: CENA, 1999.
6. Giné-Rosias, M. F., Espectrometria De Emissão Atômica Com Plasma Acoplado
Indutivamente. (ICP-AES), Piracicaba: CENA, 1998.
7. Greenfield, S., Jones, I. L. I., Berry, T. C., High Pressure plasmas as spectroscopy
emission sources. Analyst, 1964. v. 89, 713-720p

Departamento de Química
8. EVANS, E. H., GIGLIO, J. J. Interferences in inductively coupled plasma mass
spectrometry. A review. Journal of Analytical Atomic Spectrometry, 1993, v. 8, n. 2,
1-18p.
9. Boumans, P.W.J.M.; Vrakking, J.J.A.M. Spectral interferences in Inductively
Coupled Plasma - Atomic Emission Spectrometry. I. A theoretical and
experimental study of the effect of spectral bandwidth on selectivity, limits of
determination, limits of detection and detection power. Spectrochimica Acta,
1985. , v. 40B, p. 1085-1125,
10. Boumans, P.W.J.M. Line selection and spectral interferences, Inductively
Coupled Plasma Emission Spectrometry. Part I. Methodology, Instrumentation
and Performance. New York: Wiley -Interscience, 1987. p 358-465.