conscientizaÇÃo sobre amiloidose · medula óssea, o tecido mole que preenche as cavidades de...
TRANSCRIPT

1Nome da seção aqui
CONSCIENTIZAÇÃO SOBRE AMILOIDOSE
Para pacientes e sua rede de apoio, incluindo médicos, enfermeiros e estudantes de medicina

íNDICE1 Visão geral de um minuto
2 O que é amiloidose?
3 Tipos de amiloidose
4 Diagnóstico
5 Tratamentos
6 Principais centros de amiloidose
7 Recursos online
1
2
7
17
26
37
39
Publicado em outubro de 2013.
Esta brochura foi feita com a orientação de Amyloidosis Support
Groups. Agradecimentos especiais aos doutores Morie Gertz, Angela
Dispenzieri, Martha Grogan, Shaji Kumar, Nelson Leung, Mathew
Maurer, Maria Picken, Janice Wiesman e Vaishali Sanchorawala.
Embora as informações aqui contidas tenham o propósito de ser
precisas, as ciências médicas estão sempre avançando. Assim,
o conteúdo desta publicação é apresentado apenas para fins
educacionais. Ele não se destina à recomendação médica. Todas as
decisões com relação ao tratamento médico devem ser discutidas com
um médico qualificado e no exercício da profissão.
Ilustrações © Fairman Studios, LLC.
Imagem da capa: A amiloidose ocorre frequentemente em indivíduos
de meia-idade ou mais velhos, mas também em pacientes com
30 ou 40 anos de idade e, ocasionalmente, até mais jovens.

21 O que é amiloidose?
1. VISÃO gERAL DE uM MINuTO
Todas as proteínas normais em nosso corpo são biodegradáveis e recicláveis. Amiloidose é uma doença em que proteínas anormais (amiloides) resistem à degradação. Como consequência, as proteínas amiloides se depositam e acumulam nos tecidos corporais. Caso o amiloide se acumule no rim, coração, fígado, trato gastrointestinal ou nervos, ele causará um funcionamento insuficiente desses órgãos. Portanto, os sintomas da amiloidose são associados ao funcionamento anormal dos órgãos envolvidos. Normalmente, os pacientes terão alguns dos seguintes sintomas: perda de peso inexplicável, fadiga, falta de ar, urina espumosa, inchaço nos tornozelos e pernas, assim como dormência e formigamento nas mãos e pés. Estas são as manifestações dos danos causados pela proteína amiloide insolúvel aos órgãos subjacentes. Os tratamentos são projetados para dissolver os depósitos amiloides ou interromper sua produção. Se não for tratada, a doença pode ser de risco à vida. Assim, o diagnóstico precoce e preciso é a chave para promover resultados positivos.
2. O QuE É AMILOIDOSE?
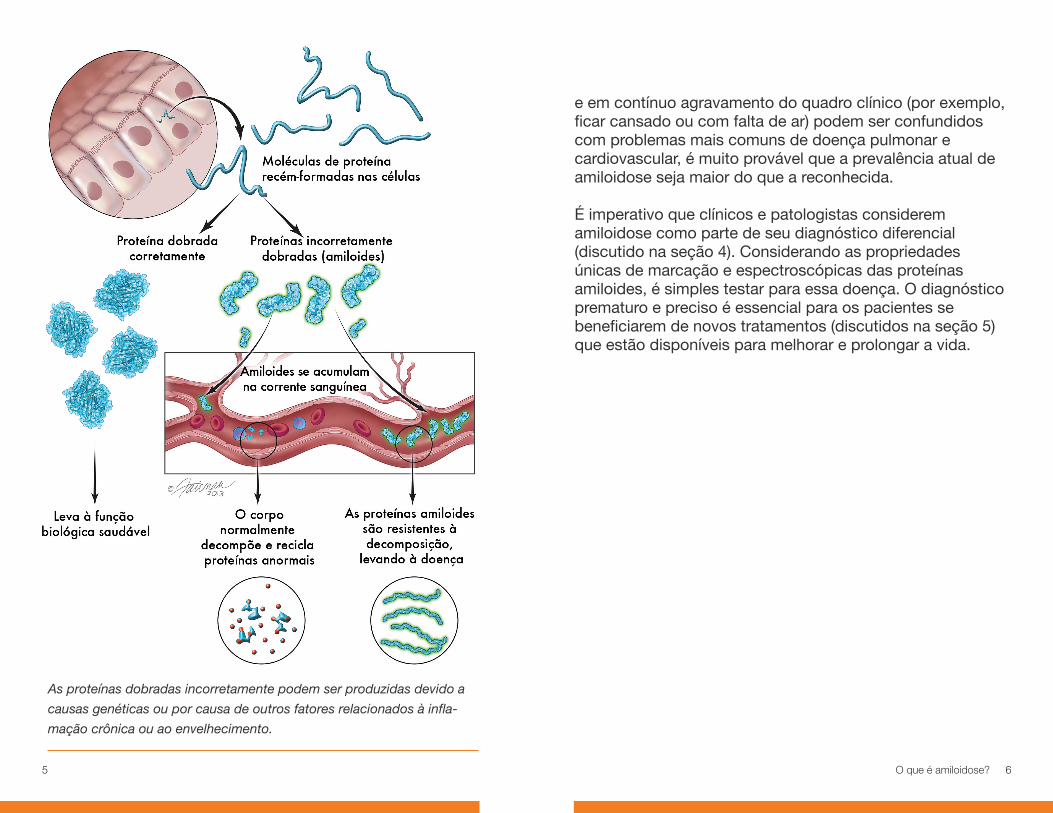
Durante a nossa vida, nosso DNA está codificando para a fabricação de pequenas moléculas chamadas proteínas. Essas proteínas fornecem a estrutura e função para quase todos os processos biológicos da vida. Enzimas que facilitam a química de nossas células, hormônios que afetam o crescimento de nosso corpo e anticorpos que formam nossa resposta imune são todos exemplos de proteínas em ação. Praticamente tudo em nossos corpos – da cor de nossos olhos, ao transporte de oxigênio no nosso sangue, ao fato de podermos ou não digerir leite – é determinado pelas proteínas que produzimos.
Uma vez produzidas no corpo, as proteínas irão se dobrar naturalmente em um formato particular. Essa forma natural de uma molécula de proteína é o que permite sua função especial. Colocado de maneira simples, quando as proteínas são dobradas de maneira correta, elas funcionam como deveriam e nós desfrutamos de saúde relativamente boa. Quando as proteínas são dobradas incorretamente, isso afeta a capacidade de nosso corpo funcionar e podem surgir problemas com o tempo.
As proteínas dobradas incorretamente podem ser produzidas devido a causas genéticas ou por causa de outros fatores relacionados à inflamação crônica ou ao envelhecimento. Independentemente, nossos corpos são normalmente capazes de identificar e remover essas proteínas anormais. Em alguns casos, no entanto, nós produzimos proteínas anormais em um número muito grande para que os nossos corpos possam lidar com elas ou não somos capazes de decompor e eliminar as proteínas. Tais defeitos na produção e no processamento de proteínas estão associados a muitas doenças.

43
De um modo geral, a amiloidose é uma classe em uma lista crescente de disfunções de dobramento de proteínas. Embora haja muitos tipos distintos de amiloidose, em todos os casos, as proteínas dobradas incorretamente, chamadas amiloides (com significado de “semelhante a amido”), assumem um formato particular que torna difícil a decomposição pelo corpo. Devido a esse dobramento incorreto, as proteínas amiloides se ligam entre si para formar fibras rígidas, lineares (ou fibrilas) que se acumulam nos órgãos e tecidos do corpo. Dependendo de onde o amiloide se acumula, tal como no rim, coração e nervos, sintomas diferentes e quadros clínicos que representam um potencial risco à vida se tornam evidentes.
Embora a amiloidose seja conhecida desde o século 19, nosso entendimento sobre ela se desenvolveu apenas nas últimas décadas. Atualmente, existem mais de 25 proteínas diferentes que foram identificadas como contribuindo para amiloidose (das quais as formas mais importantes estão descritas na próxima seção). Tipos adicionais de proteínas precursoras que podem levar à formação de amiloide continuam a ser descobertos por meio de pesquisas contínuas.
Certamente, a amiloidose é um quadro clínico raro e frequentemente negligenciado. A cada ano, estima-se que 50.000 pessoas em todo o mundo são afetadas pela doença, com mais de 3.000 pessoas sendo diagnosticadas somente na América do Norte. Isto é cerca de 1/5 da incidência de mieloma múltiplo e uma incidência semelhante à da doença de Hodgkin ou da leucemia mieloide crônica. Devido à sua raridade, estudantes de medicina e médicos podem não esperar ver amiloidose em sua prática. Além disso, como os sintomas não específicos
O amiloide é uma substância semelhante ao amido causada pelo
dobramento incorreto de proteínas. O amiloide se liga entre si em fibras
rígidas e lineares (fibrilas) que se depositam em tecidos e órgãos.
O que é amiloidose?
65
e em contínuo agravamento do quadro clínico (por exemplo, ficar cansado ou com falta de ar) podem ser confundidos com problemas mais comuns de doença pulmonar e cardiovascular, é muito provável que a prevalência atual de amiloidose seja maior do que a reconhecida. É imperativo que clínicos e patologistas considerem amiloidose como parte de seu diagnóstico diferencial (discutido na seção 4). Considerando as propriedades únicas de marcação e espectroscópicas das proteínas amiloides, é simples testar para essa doença. O diagnóstico prematuro e preciso é essencial para os pacientes se beneficiarem de novos tratamentos (discutidos na seção 5) que estão disponíveis para melhorar e prolongar a vida.
As proteínas dobradas incorretamente podem ser produzidas devido a
causas genéticas ou por causa de outros fatores relacionados à infla-
mação crônica ou ao envelhecimento.
O que é amiloidose?

87
3. TIPOS DE AMILOIDOSE
Existem muitas proteínas diferentes em nossos corpos, que podem se dobrar incorretamente produzindo a amiloidose. A predisposição para formar proteínas anormais pode ser herdada dos pais ou até mesmo surgir de mutações no DNA adquiridas durante a nossa vida. Em alguns casos, a amiloidose resulta de doenças inflamatórias e infecciosas crônicas ou de diálise renal de longo prazo. A maioria dos casos diagnosticados, no entanto, é causada por um quadro clínico de medula óssea que tem semelhanças com mieloma múltiplo.
Conforme as proteínas amiloides se acumulam em nossa corrente sanguínea, elas vão se depositando em órgãos e tecidos. As fibrilas amiloides resultantes podem comprometer os sistemas de vários órgãos ou se localizar em uma área do corpo. O amiloide se deposita mais comumente no rim, coração e nervos, com fígado, baço, trato gastrointestinal e vias respiratórias também sendo afetados ocasionalmente. O amiloide é muitas vezes encontrado no pâncreas de pessoas que desenvolvem diabetes quando adultos.
Embora as proteínas precursoras que levam à amiloidose apareçam em diversos formatos e tamanhos, todas compartilham da mesma estrutura incorretamente dobrada em depósitos amiloides. Esta característica unificadora do amiloide, em que o formato de alfa-hélice de uma proteína normal se dobra incorretamente em uma folha-beta pregueada, permite o diagnóstico preciso e em tempo hábil da doença (discutido na próxima seção).
Embora os sintomas e tratamentos do quadro clínico dependam dos órgãos afetados, os diversos tipos de amiloidose podem ser classificados de acordo com a proteína precursora envolvida. Conforme observado na Tabela 1 (próxima página), um sistema de denominação conveniente é utilizado, de tal forma que o prefixo “A” se refere a amiloide, seguido por uma abreviação para a proteína associada. Por exemplo, AL designa amiloide derivado de anticorpos de cadeia leve (light-chain antibodies); AA designa proteína A amiloide sérica (serum amyloid A protein); e ATTR designa amiloide de transtiretina (amyloid from transthyretin).
À medida que novas proteínas amiloides são caracterizadas e o nosso entendimento médico se aprofunda, é possível discutir os tipos diferentes de amiloidose a partir de uma perspectiva mais ampla. Abaixo, encontra-se uma breve descrição de amiloidose AL, amiloidose AA, amiloidose familiar, amiloidose sistêmica senil, assim como amiloidose associada ao fator 2 quimiotático de leucócitos (Leukocyte Chemotactic Factor 2-associated Amyloidosis, ALECT2), relacionada à diálise e localizada.
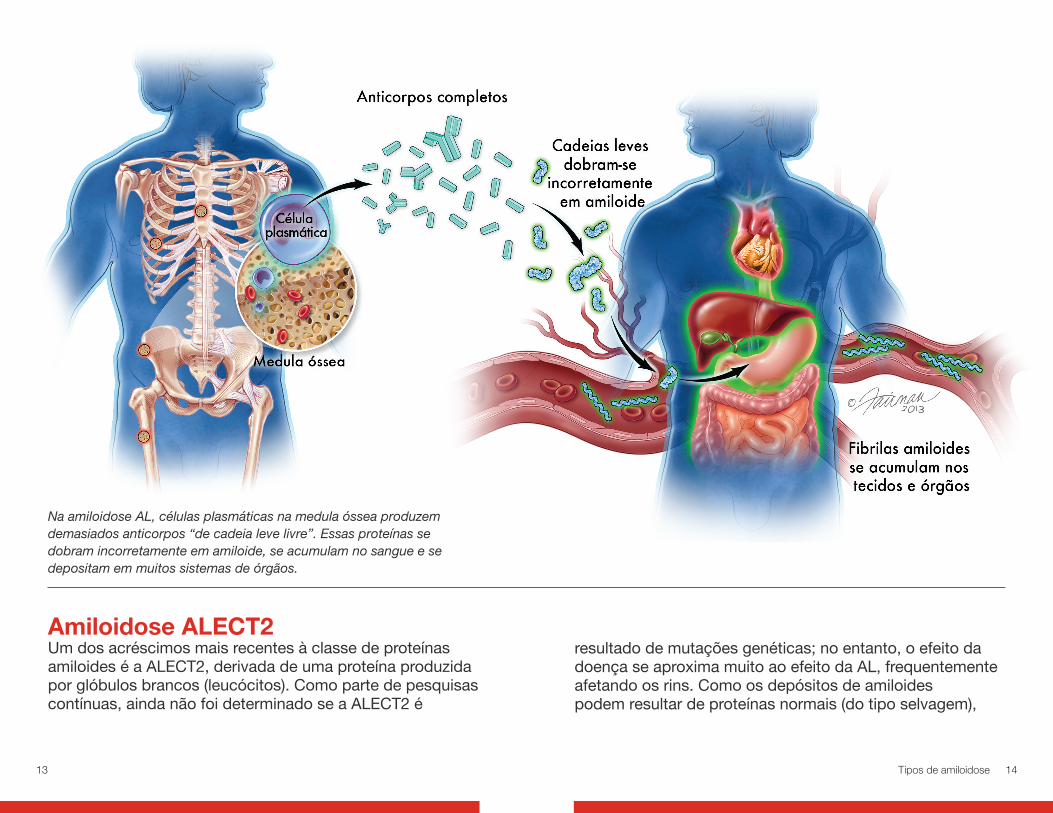
Amiloidose ALA amiloidose AL (ou primária) é a forma mais comumente diagnosticada da doença, representando 85% de todos os casos em países desenvolvidos. O distúrbio se inicia na medula óssea, o tecido mole que preenche as cavidades de nossos ossos, no qual os glóbulos vermelhos e brancos são formados. Um tipo de glóbulo branco, chamado célula plasmática, produz anticorpos que nos protegem de infecções. Essas proteínas de anticorpo (imunoglobulinas)
Tipos de amiloidose

10
são compostas de moléculas de cadeias leves e pesadas. Normalmente, nossas células plasmáticas produzem anticorpos completos e o nosso corpo decompõe essas proteínas e as recicla após um curto período. Na AL, no entanto, muitas cadeias leves não montadas e incorretamente dobradas estão sendo produzidas. Essas “cadeias leves livres” (e, em casos raros, cadeias pesadas livres) não podem ser decompostas de forma eficiente. Elas se ligam entre si para formar fibrilas amiloides que se acumulam no espaço extracelular de órgãos e tecidos. Dessa maneira, o funcionamento normal do corpo fica comprometido. Os problemas surgem normalmente no rim, coração, fígado, baço, nervos, intestinos, pele, língua e vasos sanguíneos.
Amiloidose AAA amiloidose AA (ou secundária) resulta de níveis aumentados de proteína amiloide A sérica circulante. O amiloide A sérico se eleva em nosso sangue como resposta natural a infecções e inflamações. Em geral, se um paciente tem um quadro clínico de infecção ou inflamação por seis meses ou mais, ele tem o risco de desenvolver AA. A amiloidose surge de quadros clínicos inflamatórios crônicos e infecciosos, incluindo: doença reumática, doença inflamatória intestinal, tuberculose, osteomielite, lúpus e síndromes de febre hereditária, tal como a febre familiar do Mediterrâneo. A deposição de amiloide geralmente se inicia nos rins, mas o fígado, baço, linfonodos e intestino também são comumente afetados.
-
SÍNDROME
Forma primária de amiloidose, semelhante ao mieloma múltiplo, afetando os rins, coração, fígado, trato gastrointestinal e nervos.
Secundária a doenças inflamatórias crônicas e infecciosas, afetando os rins e fígado.
Clinicamente se aproxima da AL, afetando os rins e fígado.
Relacionada à diálise, afetando as articulações e tendões.
Hereditária com mais de 100 mutações, afetando o sistema nervoso, coração e rins. A mutação Val-122-Ile é comum em afro-americanos, causando doençacardíaca. Uma forma não heredi-tária, do tipo selvagem e senil, causa doença cardíaca nos idosos.
Hereditária, afetando os rins.
Hereditária, afetando o fígado, coração, rins e nervos.
Hereditária, afetando o trato gastro-intestinal e rins.
Hereditária, afetando a pele, nervos e rins.
Ocorre principalmente na bexiga, pele e vias respiratórias.
FONTE DO AMILOIDE
Células plasmáticas na medula óssea
(Cadeias leves ou pesadasde imunoglobulina,
ou ambas)
Proteína inflamatória circulante
(Amiloide A sérico)
Glóbulos brancos(Fator 2 quimiotático de
leucócitos)
Proteína sérica circulante(β2-microglobulina)
Proteína mutante e do tipo selvagem produzida
no fígado(Transtiretina)
Proteína mutante produzida no fígado (Cadeia de A-alfa de
fibrinogênio)
Proteína sérica circulante(Apolipoproteína AI)
Proteína sérica circulante(Lisozima)
Proteína sérica circulante(Gelsolina)
Células plasmáticas em tecidos locais
(Cadeias leves de imunoglobulina)
TIPO
AL, AH, ALH
AA
ALECT2
Aβ2M
ATTR
AFib
AapoAI
ALys
AGel
Localizada
(Proteína precursora)
Tipos de amiloidose
Tabela 1: Exemplos de amiloidose. O sistema de denominação combina um “A” de amiloide com uma abreviação da proteína do quadro clínico subjacente.

1211
Amiloidose familiarA amiloidose familiar (ou hereditária), como o nome sugere, é uma forma herdável da doença. Quer uma mutação tenha ocorrido no DNA da própria pessoa, quer tenha sido herdada de um dos pais, a forma mais comum de amiloidose familiar está associada à proteína transtiretina (TTR) mutante produzida no fígado. A TTR é uma proteína que ajuda a transportar tiroxina (um hormônio da tireoide) e retinol (vitamina A) pelo corpo. Existem mais de 100 mutações conhecidas da TTR que fazem com que a proteína se torne instável e se dobre incorretamente em amiloide. Vários órgãos são afetados, especialmente o sistema nervoso e o coração, com sintomas ocorrendo da meia-idade à idade avançada. Caso os nervos sejam afetados primeiramente, o quadro clínico é conhecido como polineuropatia amiloide familiar; se o coração for afetado primeiramente, ele será conhecido como cardiomiopatia amiloide familiar.
A mutação conhecida mais comum de TTR é chamada Val-30-Met e causa dano nervoso e problemas elétricos no coração. Outra mutação comum nos Estados Unidos é a Thr-60-Ala, que causa espessamento do músculo cardíaco. Enquanto a amiloidose mediada por transtiretina (ATTR) ocorre em famílias de quase todas as etnias, existe uma variante de TTR, Val-122-Ile, que parece ser comum na população afro-americana. Estima-se que 4% dos afro-americanos possuam esse gene mutante, compreendendo mais de 25% de pacientes afro-americanos com amiloidose. Essa mutação pode ser uma causa frequentemente não reconhecida de doença cardíaca em afro-americanos.
Além da ATTR, existem outras mutações no gene para diferentes proteínas que levam à amiloidose. Embora muito raras, algumas delas incluem: AFib (da cadeia A-alfa de fibrinogênio), AApoAI (da apolipoproteína AI), ALys (da lisozima) e AGel (da gelsolina).
Amiloidose sistêmica senilA amiloidose sistêmica senil (ou relacionada à idade) é uma doença de início tardio que é adquirida, não herdada. Os depósitos de amiloide se acumulam no corpo a partir de proteínas normais (do tipo selvagem). O melhor exemplo conhecido de amiloidose sistêmica senil surge do acúmulo de transtiretina (TTR) do tipo selvagem nos corações de idosos. Ao contrário da amiloidose familiar, não existem mutações no gene TTR, mas a doença cardíaca progressiva mais lenta tem sintomas semelhantes. Seja mutante ou do tipo selvagem, acredita-se que a amiloidose mediada por TTR seja mais comum que a amiloidose AL, embora ela frequentemente não seja diagnosticada. Por exemplo, a TTR do tipo selvagem é encontrada em até 30% dos pacientes que demonstram “insuficiência cardíaca com fração de ejeção preservada” clínica.
Outros exemplos de amiloidose senil incluem: APro (da prolactina), ACal (da calcitonina), AIAPP (da amilina) e AANF (do fator natriurético atrial). Todos são derivados do dobramento incorreto de proteínas do tipo selvagem. Apesar de seu nome, esse quadro clínico não tem nenhuma relação com a senilidade ou demência.
Tipos de amiloidose
1413
Na amiloidose AL, células plasmáticas na medula óssea produzem demasiados anticorpos “de cadeia leve livre”. Essas proteínas se dobram incorretamente em amiloide, se acumulam no sangue e se depositam em muitos sistemas de órgãos.
Tipos de amiloidose
Amiloidose ALECT2Um dos acréscimos mais recentes à classe de proteínas amiloides é a ALECT2, derivada de uma proteína produzida por glóbulos brancos (leucócitos). Como parte de pesquisas contínuas, ainda não foi determinado se a ALECT2 é
resultado de mutações genéticas; no entanto, o efeito da doença se aproxima muito ao efeito da AL, frequentemente afetando os rins. Como os depósitos de amiloides podem resultar de proteínas normais (do tipo selvagem),

1615 Tipos de amiloidose
assim como na amiloidose sistêmica senil, essa forma da doença pode ser diagnosticada incorretamente ou subdiagnosticada. Um estudo, na realidade, sugeriu que a ALECT2 era o tipo de amiloide mais comum e não diagnosticado, especialmente entre os pacientes de origem mexicana. Em uma análise de amostras de rim contendo amiloide durante os últimos oito anos, ALECT2 foi o terceiro tipo mais comum (2,5%), quando comparado ao AL (86%), AA (7%) e ATTR (1,4%).
Amiloidose Aβ2MAβ2M (ou relacionada à diálise) ocorre frequentemente em pacientes que sofrem de insuficiência renal e estiveram em diálise por muitos anos. Uma proteína sérica circulante, a beta-2 microglobulina (β2M), se acumula no sangue, pois é incapaz de atravessar o filtro de diálise. Como a β2M não pode ser excretada do corpo, o amiloide resultante se acumula nos tecidos, particularmente nas articulações e nos tendões. Isto causa dor, rigidez e fluido nas articulações, assim como síndrome do túnel carpal.
Amiloidose localizadaEmbora as formas principais de amiloidose estejam descritas acima, é importante reconhecer que os depósitos de amiloide podem ocasionalmente ocorrer em áreas isoladas, sem evidência de doença sistêmica. Estes depósitos localizados, semelhantes a um tumor, ocorrem mais frequentemente na bexiga e nas vias respiratórias (por exemplo, traqueia ou pulmões). Depósitos também foram diagnosticados no olho, trato gastrointestinal, pele e seio. Semelhante à amiloidose AL, os depósitos de amiloide localizados são compostos de proteínas de cadeia leve. No entanto, na amiloidose localizada, as células plasmáticas
anormais que produzem as cadeias leves de amiloides estão nos tecidos afetados, não na medula óssea.
Outros tipos de amiloidose não sistêmica estão associados a proteínas hormonais, envelhecimento ou áreas específicas do corpo. Um caso especial de amiloidose localizada é a angiopatia amiloide cerebral (cerebral amyloid angiopathy, CAA). Embora a causa ainda seja desconhecida, em alguns indivíduos a CAA pode ser hereditária. A proteína amiloide se deposita nas paredes das artérias cerebrais, aumentando o risco de acidente vascular cerebral e demência. Este quadro clínico neurológico é observado na maioria dos casos em pacientes mais idosos e não está relacionado à doença de Alzheimer.

1817
4. DIAgNÓSTICO
De certa maneira, a amiloidose não é fácil de reconhecer. Seus sintomas são vagos e não específicos, frequentemente imitando aqueles de outros quadros clínicos mais comuns. Por exemplo, falta de ar pode ser um indicador de doença cardíaca, que é normalmente causada por aterosclerose e doença arterial coronariana. Não se pensaria inicialmente em amiloidose. Além disso, proteína na urina é um sinal precoce de doença renal, como em pacientes com diabetes, mas, novamente, não se pensaria de imediato em amiloidose.
A amiloidose normalmente aparece em indivíduos de meia-idade ou mais velhos, mas também pode ocorrer durante os 30 ou 40 anos de idade e, ocasionalmente, até mesmo em idade mais jovem. Os depósitos de amiloide podem causar perda de peso, fadiga, falta de ar, tontura ao estar em pé, inchaço nos tornozelos e nas pernas, dormência ou formigamento nas mãos e nos pés, urina espumosa, episódios alternados de constipação e diarreia, assim como sentir-se saciado rapidamente após comer. Além disso, se hematomas se formam facilmente em um paciente, especialmente ao redor dos olhos (púrpura periorbital), ou se ele tem uma língua aumentada (macroglossia), a amiloidose muito provavelmente é a causa subjacente.
À medida que uma constelação de sintomas persiste e piora, muitos médicos não consideram (ou não se lembram de procurar por) uma doença tão rara e insidiosa. Não é incomum um indivíduo visitar diversos médicos antes de uma biópsia (amostra de tecido) ser feita ou de que se desenvolva insuficiência em um órgão antes de
Os sintomas são frequentemente vagos, mimetizando os de outros quadros clínicos comuns. Assim, uma abordagem multidisciplinar entre especialistas médicos é essencial para o diagnóstico. Em alguns casos, os sinais indicadores de amiloidose são língua aumentada (macroglossia) ou formação de hematomas ao redor dos olhos (púrpura periorbital).
um diagnóstico apropriado ser feito. Na verdade, se os patologistas forem aguardar por informações clínicas para buscar por amiloidose, eles não o farão na maioria dos casos.
Embora a amiloidose possa afetar apenas um único órgão, ela frequentemente causa problemas sistêmicos (ou seja, afeta mais de um sistema de órgãos). Os órgãos mais frequentemente envolvidos são os rins (aproximadamente
Diagnóstico

2019
70% dos pacientes), coração (50%), sistema nervoso (30%) e trato gastrointestinal. Portanto, em pacientes com combinações de doença renal, cardíaca, nervosa, gastrointestinal ou hepática – sem nenhuma causa óbvia – os médicos deveriam testar para amiloidose.
Como parte do diagnóstico diferencial (ou seja, o processo de distinguir uma doença de outra), os quatro contextos clínicos mais comuns nos quais a amiloidose deveria ser considerada são:
1. Perda de quantidades massivas de proteína na urina (proteinúria)
2. Coração enrijecido ou espessado (cardiomiopatia restritiva), como observado no ecocardiograma; baixa voltagem observada no eletrocardiograma; batimento cardíaco irregular (arritmia) que seja resistente ao tratamento convencional, frequentemente associado à pressão sanguínea normal ou baixa; ou insuficiência cardíaca inexplicável
3. Fígado aumentado (hepatomegalia) sem consumo de álcool ou outra explicação, frequentemente com testes sanguíneos hepáticos anormais
4. Dormência ou dor nos dedos das mãos ou pés (neuropatia periférica), tal como a síndrome do túnel carpal ou episódios alternados de constipação e diarreia (neuropatia autonômica), enquanto também sente tontura (baixa pressão sanguínea) ao estar em pé.
Testes para amiloidoseUma vez que se suspeite de amiloidose, ela pode geralmente ser identificada, se presente, com um procedimento ambulatorial muito simples. A detecção precoce e avaliação precisa são essenciais para que os pacientes se beneficiem das muitas terapias agora disponíveis (discutidas na próxima seção).
Testes de sangue e de urina podem revelar uma proteína anormal no corpo, mas o padrão ouro para detecção de depósitos de amiloide é aplicar coloração vermelho Congo (Congo red stain) em uma amostra de tecido. Biópsias podem ser retiradas de gengivas, nervos, rim, fígado ou reto. No entanto, a maneira mais fácil de se conseguir uma amostra de tecido é aspirar gordura do abdômen. Neste procedimento não invasivo, a pele da barriga é anestesiada com um anestésico local e uma agulha é utilizada para realizar uma minilipoaspiração em células de gordura abaixo da pele. Devido à estrutura incorretamente dobrada comum a todos os amiloides, eles têm uma cor rosa quando marcados com vermelho Congo no laboratório e uma birrefringência verde-maçã característica quando visualizados com um microscópio de polarização. Esta técnica distintiva é capaz de diagnosticar amiloidose em 70-80% dos pacientes.
Caso o aspirado do depósito de gordura seja negativo para amiloidose, mas a suspeita para a doença seja alta, uma biópsia direta do órgão envolvido (por exemplo, coração, rim ou fígado) deve ser realizada. Caso o amiloide esteja presente, a utilização da coloração vermelho Congo fornecerá um diagnóstico definitivo em quase 100% dos casos.
A imuno-histoquímica deve ser realizada e interpretada com cautela. É importante evitar a supercoloração da amostra de tecido com o vermelho Congo, pois isso pode dar resultados falsos. A visualização do tecido com um microscópio eletrônico mostrará a estrutura clássica das fibrilas amiloides, confirmando assim a sua presença.
Diagnóstico

2221
O padrão ouro para detectar amiloidose utiliza a coloração vermelho Congo em uma amostra de tecido, a qual aparece verde-maçã quando visualizada com um microscópio de polarização. A microdissecção a laser seguida por espectrometria de massa pode determinar o tipo de amiloide em praticamente 100% dos casos.
Tipagem da amiloidoseSe a amiloidose for encontrada na biópsia do tecido ou os resultados forem inconclusivos, é recomendado entrar em contato com centros de amiloidose especializados (ver seção 6) que utilizam métodos mais sofisticados. Provar que
o amiloide está presente em um órgão é apenas o início do processo. Depois, deve-se determinar que tipo de amiloide está causando a doença para planejar um tratamento apropriado e individualizado.
Em todos os casos, a identificação do tipo de amiloide deve se basear na avaliação de proteínas anormais depositadas nos tecidos afetados. Um teste de sangue simples para medir a produção anormal de cadeias leves livres séricas mostrará níveis desproporcionalmente elevados em aproximadamente 98% dos pacientes com amiloidose AL (ou primária). Subsequentemente, uma biópsia da medula óssea, com análise de coloração imuno-histoquímica apropriada ou de citometria de fluxo, demonstrará uma população clonal de células plasmáticas na maioria dos pacientes, que estão produzindo as cadeias leves defeituosas do anticorpo. Caso esses testes sejam negativos, uma forma hereditária da doença deve ser investigada. Testes moleculares e genéticos podem ser realizados em amostras de sangue para observar se o paciente tem algum dos tipos de amiloide familiar (por exemplo, TTR, fibrinogênio, lisozima, apolipoproteínas AI e AII e gelsolina). Caso tenha tal mutação, então seus filhos terão uma chance de 50% de herdá-la.
Deve-se enfatizar que a presença de uma mutação genética nem sempre se correlaciona com o tipo de amiloidose. Por exemplo, um paciente pode ter amiloidose AL ao mesmo tempo em que também possui uma variante genética que não é a causa de sua doença. Por esse motivo, a identificação inequívoca da proteína amiloide deve ser feita juntamente com outras técnicas de diagnóstico. (Lembre-se: nos Estados Unidos, a Lei de não discriminação por informação genética (Genetic Information Nondiscrimination Act) legisla que os pacientes que têm uma disposição
Diagnóstico
23
genética para doenças, tal como a amiloidose, não podem ser discriminados com respeito ao emprego ou seguro de saúde.)
Para pacientes com quadros clínicos inflamatórios crônicos ou infecciosos ou diálise renal de longo prazo, os testes de sangue sugerirão a presença de amiloidose AA ou Aβ2M. Caso o paciente tenha mais de 50 anos e apresente insuficiência cardíaca congestiva, ou sinais de demência ou acidente vascular cerebral, o médico deve considerar tipos clinicamente isolados da doença, tal como amiloidose sistêmica senil (SSA) ou angiopatia amiloide cerebral (CAA), conforme evidenciado em ecocardiogramas e imagem por ressonância magnética.
Entretanto, avanços recentes no domínio de proteômica prometem revolucionar o diagnóstico preciso de amiloidose. A proteômica envolve o estudo de todo o complemento de proteínas em um organismo ou ambiente. Ao contrário de técnicas padrão de imunoquímica, que podem ser limitadas em sua disponibilidade, especificidade e sensibilidade, a proteômica pode identificar qualquer proteína – com ou sem mutações genéticas – em um único teste. Isto proporciona imensa economia de tempo e de custos na identificação precisa de proteínas amiloides a partir de amostras de tecido disponíveis.
Portanto, a microdissecção a laser seguida por espectrometria de massa (LMD-MS) é a técnica principal para tipagem de amiloidose. Para realizar esse teste, amostras positivas de vermelho Congo são dissecadas e decompostas em componentes menores de moléculas de proteínas (chamadas peptídeos). Os peptídeos são
TIPA
GEM
Coloração vermelho Congo Microscopia eletrônica
Birrefringência positiva Fibrilas de 8-10 nm
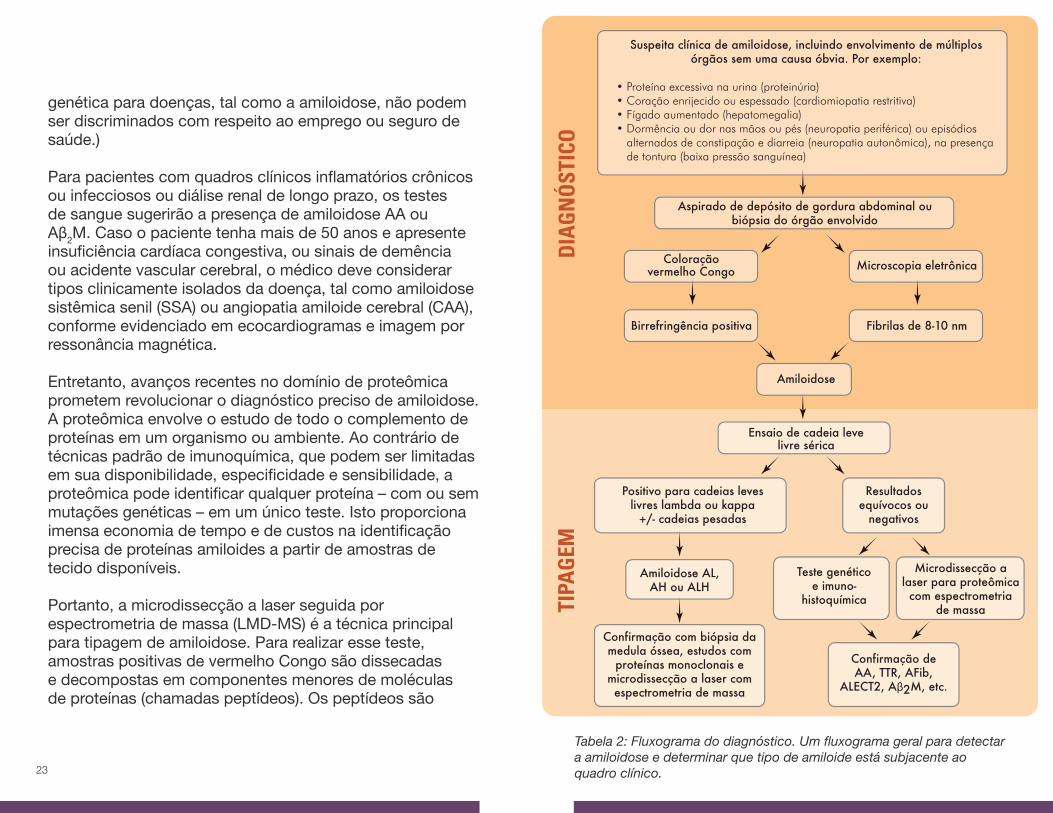
Suspeita clínica de amiloidose, incluindo envolvimento de múltiplos órgãos sem uma causa óbvia. Por exemplo:
• Proteína excessiva na urina (proteinúria)• Coração enrijecido ou espessado (cardiomiopatia restritiva)• Fígado aumentado (hepatomegalia)• Dormência ou dor nas mãos ou pés (neuropatia periférica) ou episódios alternados de constipação e diarreia (neuropatia autonômica), na presença de tontura (baixa pressão sanguínea)
Aspirado de depósito de gordura abdominal ou biópsia do órgão envolvido
Amiloidose
Ensaio de cadeia leve livre sérica
Positivo para cadeias leves livres lambda ou kappa
+/- cadeias pesadas
Resultados equívocos ou
negativos
Confirmação com biópsia da medula óssea, estudos com
proteínas monoclonais e microdissecção a laser com espectrometria de massa
Confirmação de AA, TTR, AFib,
ALECT2, Aβ2M, etc.
Amiloidose AL, AH ou ALH
Teste genético e imuno-
histoquímica
Microdissecção a laser para proteômica com espectrometria
de massa
DIAGNÓST
ICO
Tabela 2: Fluxograma do diagnóstico. Um fluxograma geral para detectar a amiloidose e determinar que tipo de amiloide está subjacente ao quadro clínico.

2625
então analisados utilizando um processo conhecido como “cromatografia líquida acoplada à espectrometria de massas em tandem com ionização por electrospray” (“liquid chromatography electrospray tandem mass spectrometry”). A LMD-MS pode ser realizada em quaisquer amostras de tecido, incluindo aspirado de depósito de gordura, se o amiloide estiver presente. Estudos mostraram que a LMD-MS tem a capacidade de identificar todas as proteínas amiloides conhecidas com praticamente 100% de precisão, assim como a capacidade de caracterizar as novas. Independentemente dos sintomas de um paciente, nenhum conhecimento clínico prévio é necessário na tipagem da amiloidose com LMD-MS.
Enquanto certas formas de amiloidose são frequentemente subdiagnosticadas – tal como a variante de TTR, Val-122-Ile, que causa doença cardíaca em afro-americanos, e a proteína do tipo selvagem ALECT2, que causa doença renal em pacientes de origem mexicana – com a LMD-MS este não deve mais ser o caso.
Em resumo, a amiloidose afeta indivíduos de várias idades e etnias e o risco de desenvolver a doença é maior em pessoas que:
• Têm 50 anos ou mais• Têm uma infecção ou doença inflamatória crônica• Têm histórico familiar de amiloidose• Têm mieloma múltiplo (cerca de 10% dos pacientes com
mieloma múltiplo também desenvolvem amiloidose)• Têm doença renal que exige diálise por muitos anos.
5. TRATAMENTOS
Alguns médicos costumavam presumir que nada poderia ser feito para um paciente com amiloidose. Isto simplesmente não é verdade, principalmente porque os tratamentos se tornaram mais efetivos. Eventualmente, à medida que as terapias continuam a se desenvolver e melhorar, a amiloidose será um pouco mais que um incômodo tratável.
Trabalhar com uma equipe de médicos – incluindo hematologistas, cardiologistas, nefrologistas e neurologistas, entre outros – é importante para conseguir um diagnóstico conclusivo e preciso da doença o mais rápido possível. Os tratamentos disponíveis dependem do tipo de amiloidose e dos órgãos afetados, assim como do quadro clínico, idade e preferência pessoal do paciente. Se não tratados em tempo hábil, os depósitos de amiloide continuarão a danificar os tecidos até a insuficiência do órgão e, possivelmente, óbito. O tratamento de amiloidose é um processo em duas partes: (1) gerenciar os sintomas e promover o bem-estar, a qualidade de vida e a sobrevivência do paciente; e (2) eliminar o suprimento de proteína amiloide para melhorar a função do órgão.
Existem três abordagens gerais para interromper a formação e deposição de proteína amiloide, as quais variam de acordo com o tipo de amiloidose. O tratamento mais comum interfere com a produção da proteína precursora que leva à doença. Um segundo método utiliza a terapia com medicamento para estabilizar a estrutura normal da
Tratamentos

2827
proteína precursora, evitando assim o seu dobramento incorreto em amiloide. Uma terceira estratégia deve ter como alvo os depósitos de amiloide diretamente, desestabilizando as fibrilas amiloides para que elas não possam mais permanecer incorretamente dobradas.
Todas as terapias têm efeitos colaterais e o médico se certificará de recomendar as melhores opções. Em muitos casos, se a fonte de proteína anormal for removida, os depósitos de amiloide existentes podem ser reabsorvidos com o tempo e a função do órgão pode ser restaurada.
Amiloidose ALPara amiloidose AL (ou primária), a forma mais comumente diagnosticada da doença, o envolvimento extensivo de órgãos é usual. Sem tratamento, a taxa de sobrevivência média é de cerca de 12-18 meses e apenas cerca de 6 meses para pacientes com função cardíaca gravemente comprometida.
Quimioterapia, oral ou intravenosa, é fundamental para o tratamento para amiloidose AL. O objetivo é interromper o crescimento de células plasmáticas que produzem proteínas de anticorpos de cadeia leve anormais. Por muitos anos, terapias que utilizam melfalan (também conhecido como Alkeran) ou ciclofosfamida (Cytoxan) foram o tratamento de escolha. Medicamentos mais novos que são utilizados no tratamento de mieloma múltiplo, tais como bortezomibe (Velcade), lenalidomida (Revlimid) ou carfilzomibe (Kyprolis), também provaram ser eficazes. Essas terapias são frequentemente utilizadas em combinação com dexametasona, um esteroide para ajudar na resposta imune. Submeter-se à quimioterapia pode ter efeitos colaterais, tais como náusea, vômitos, perda de cabelo, infecção e fadiga extrema. Caso os
efeitos colaterais interfiram na qualidade de vida da pessoa, diferentes tratamentos podem estar disponíveis.Em pacientes cuidadosamente selecionados, a quimioterapia é combinada com transplante de célula-tronco. As células-tronco são encontradas na medula óssea e se desenvolvem em diversos tipos de células sanguíneas, incluindo nossas células plasmáticas. Uma vez que as células plasmáticas sejam destruídas utilizando altas doses de quimioterapia, a medula óssea é reabastecida com células-tronco frescas do corpo do próprio paciente (transplante autólogo). Com a erradicação das células plasmáticas defeituosas, a produção de amiloide é retardada ou interrompida e a medula óssea pode se tornar saudável novamente.
A quimioterapia seguida por transplante de célula-tronco frequentemente obtém uma resposta excelente, com melhora significativa ou estabilização da função do órgão. No entanto, nem todos os pacientes conseguem tolerar esse regime agressivo, particularmente aqueles com problemas cardíacos avançados. Considerando a complexidade da doença, recomenda-se que o tratamento seja realizado em um centro médico que tenha experiência com amiloidose (ver a próxima seção). Alternativamente, os pacientes podem ter uma avaliação inicial em tal centro, com comunicação contínua durante o tratamento em sua comunidade local.
Outro medicamento sendo desenvolvido almeja atacar diretamente os depósitos de amiloide de cadeia leve que se acumularam no corpo. Atualmente em estudos clínicos, esse tratamento utiliza pequenas moléculas chamadas anticorpos monoclonais para encontrar e se ligar especificamente às proteínas incorretamente dobradas das fibrilas amiloides. Os anticorpos monoclonais
Tratamentos

3029
imitam os anticorpos que o nosso sistema imune produz naturalmente para nos proteger de doenças. Ao ter como alvo e desestabilizar os depósitos de amiloide dessa maneira, o corpo da pessoa se torna potencialmente capaz de identificá-los e removê-los mais eficientemente. Nos próximos anos, esse medicamento promissor pode contribuir com a peça que faltava no quebra-cabeça para ajudar na restauração da função do órgão e da saúde em geral.
Amiloidose AAA amiloidose AA (ou secundária) é a segunda forma mais comum da doença em todo o mundo. Com as suas doenças inflamatórias crônicas associadas (por exemplo, artrite reumatoide, doença de Crohn e febre familiar do Mediterrâneo), a deposição de amiloide é muito gradual. A taxa de sobrevivência é frequentemente maior do que 10 anos, particularmente com tratamento para doença renal. Em contraste, infecções não tratadas, tais como osteomielite ou tuberculose, podem causar acumulação mais rápida de amiloide.
Em todos os casos, o alicerce da terapia é tratar o quadro clínico de infecção ou inflamatório subjacente. Isto pode retardar ou interromper o acúmulo progressivo de amiloide reduzindo a proteína precursora circulante, o amiloide A sérico.
Além disso, descobriu-se que um medicamento oral chamado eprodisate (Kiacta) inibe a formação de fibrilas amiloides. O Kiacta evita que o amiloide A sérico interaja com outras moléculas que facilitam o seu dobramento incorreto em amiloide. Estudos clínicos demonstraram
que, como o amiloide não pode mais ser formado e depositado, esse tratamento pode retardar ou interromper a deterioração da função renal de forma eficaz. Antecipa-se que o Kiacta seja aprovado para utilização ampla nos próximos anos, após a confirmação de sua eficácia por meio de estudos internacionais.
Para aqueles pacientes com insuficiência renal, diálise e transplante de rim são tratamentos possíveis. No entanto, assim como com transplantes de rim para amiloidose AL, se a fonte de proteína anormal não for tratada, o amiloide pode eventualmente aparecer no rim do doador.
Amiloidose ATTRNa amiloidose familiar (ou hereditária) mediada por TTR, o coração e o sistema nervoso são os mais comumente afetados. Sem intervenção, a taxa de sobrevivência varia entre 5-15 anos a partir do início da doença.
Como a maioria das proteínas anormais é produzida no fígado, transplantar o fígado pode permitir a produção de TTR normal. Os períodos de espera para doação de órgão são frequentemente longos, mas o transplante é uma opção viável para alguns pacientes cuja doença não está tão avançada.
Entretanto, novos medicamentos estão sendo desenvolvidos para evitar, em primeiro lugar, a formação de depósitos de amiloide familiar. Assim como com o transplante de fígado, evidências mostram que ao reduzir a proteína anormal disponível para se tornar amiloide, a função do órgão pode melhorar. Dois medicamentos desse tipo, que podem evitar que a TTR mutante se dobre
Tratamentos
3231
incorretamente em amiloide, são diflunisal e tafamidis. Essas pequenas moléculas se ligam às proteínas precursoras e estabilizam a sua estrutura, para que elas não formem fibrilas amiloides e se acumulem no corpo.
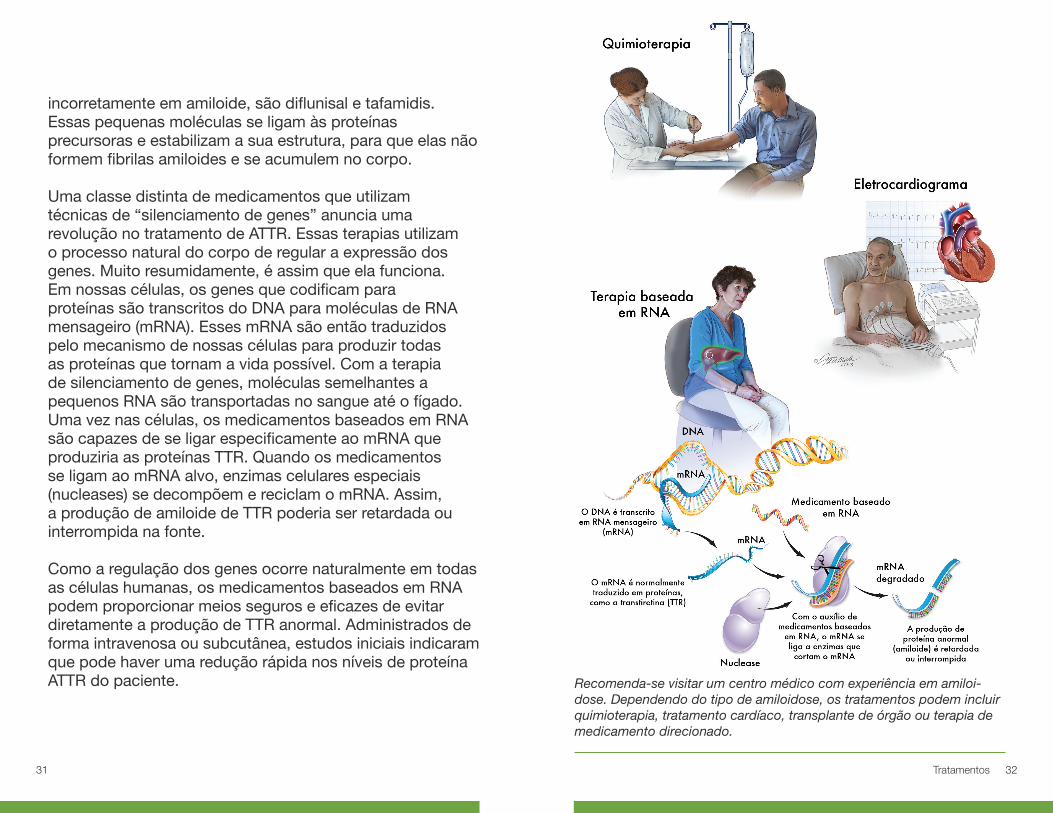
Uma classe distinta de medicamentos que utilizam técnicas de “silenciamento de genes” anuncia uma revolução no tratamento de ATTR. Essas terapias utilizam o processo natural do corpo de regular a expressão dos genes. Muito resumidamente, é assim que ela funciona. Em nossas células, os genes que codificam para proteínas são transcritos do DNA para moléculas de RNA mensageiro (mRNA). Esses mRNA são então traduzidos pelo mecanismo de nossas células para produzir todas as proteínas que tornam a vida possível. Com a terapia de silenciamento de genes, moléculas semelhantes a pequenos RNA são transportadas no sangue até o fígado. Uma vez nas células, os medicamentos baseados em RNA são capazes de se ligar especificamente ao mRNA que produziria as proteínas TTR. Quando os medicamentos se ligam ao mRNA alvo, enzimas celulares especiais (nucleases) se decompõem e reciclam o mRNA. Assim, a produção de amiloide de TTR poderia ser retardada ou interrompida na fonte.
Como a regulação dos genes ocorre naturalmente em todas as células humanas, os medicamentos baseados em RNA podem proporcionar meios seguros e eficazes de evitar diretamente a produção de TTR anormal. Administrados de forma intravenosa ou subcutânea, estudos iniciais indicaram que pode haver uma redução rápida nos níveis de proteína ATTR do paciente. Recomenda-se visitar um centro médico com experiência em amiloi-
dose. Dependendo do tipo de amiloidose, os tratamentos podem incluir quimioterapia, tratamento cardíaco, transplante de órgão ou terapia de medicamento direcionado.
Tratamentos

3433
Amiloidose sistêmica senilNa amiloidose sistêmica senil (ou relacionada à idade), a terapia é principalmente de apoio à doença cardíaca do paciente. Em alguns casos, pode ser feito um transplante cardíaco.
Para formas de amiloidose senil mediadas por TTR, os mesmos tratamentos com medicamentos utilizados em amiloidose familiar (por exemplo, diflunisal, tafamidis ou medicamentos baseados em RNA) podem inibir a produção de amiloide de TTR. Isto poderia ajudar a prolongar a vida ao evitar a formação posterior de amiloide e a deposição no músculo cardíaco.
Amiloidose Aβ2MNa amiloidose Aβ2M (ou relacionada à diálise), o transplante renal é considerado a melhor opção terapêutica. Membranas de diálise com pouco cobre podem evitar ou adiar o início da doença.
Amiloidose localizadaPara depósitos de amiloide que ocorrem em áreas isoladas, tais como bexiga ou vias respiratórias, a terapia de radiação pode retardar a progressão da doença. A remoção cirúrgica do depósito de amiloide pode ser apropriada, uma vez que o quadro clínico sistêmico esteja descartado. Assim como com todas as formas de amiloidose, os pacientes são encorajados a fazer exames médicos periódicos para monitorarem o seu quadro clínico.
Para a angiopatia amiloide cerebral (CAA), que afeta o cérebro, não existe tratamento efetivo conhecido. O objetivo é aliviar os sintomas. Isto pode incluir medicamentos que ajudem a melhorar a memória, tais
como os utilizados para tratar a doença de Alzheimer. Convulsões, algumas vezes chamadas de “episódios amiloides” (“amyloid spells”), podem ser tratadas com anticonvulsivantes, tais como fenitoína (Dilantin) ou carbamazepina (Tegretol). Em alguns casos, terapias de fala e físicas são necessárias.
Tratando os sintomas da amiloidoseÉ muito importante tratar não somente as causas subjacentes da amiloidose, mas também os sintomas da doença. Isto assegurará qualidade de vida e longevidade ao paciente.
Atividades normais, cotidianas podem ser realizadas como sempre. No entanto, caso ocorra fadiga ou falta de ar, é necessário descansar. Não se deve fazer esforço além do que é recomendado por seu médico.
Para tratamentos de apoio dos rins e coração, os pacientes podem precisar tomar um medicamento diurético para urinar, conforme prescrito por seus médicos; limitar a quantidade de sal em sua dieta; ou vestir meias elásticas e elevar as pernas para diminuir o inchaço. Para o trato gastrointestinal, certos medicamentos podem ajudar na diarreia e constipação. Em geral, pode ser útil fazer algumas alterações dietéticas para ajudar a aliviar os sintomas ou manter o peso corpóreo.
Conforme o tratamento progride, o dano aos nervos (neuropatia) pode melhorar. Pode levar até 12-24 meses para que os nervos se recuperem, mas a dormência e a fraqueza podem diminuir. Medicamentos para aliviar a dor podem ser tomados por via oral ou aplicados na pele. Para
Tratamentos

3635
o desconforto, formigamento ou sensação de ardor, o uso de um massageador para os pés com água-quente, durante 15 minutos antes de deitar-se, pode ajudar a dormir. A água quente e as vibrações estimulam nervos que não transmitem a dor e bloqueiam os nervos de transmissão de dor.
Participação em uma pesquisa clínicaEstudos clínicos são estudos de pesquisa que testam novas maneiras de diagnosticar e tratar doenças. Tal pesquisa é essencial para melhorar nosso entendimento da amiloidose e desenvolver terapias mais eficazes. Os tratamentos que estão disponíveis hoje foram todos desenvolvidos e refinados por meio desta pesquisa clínica em andamento. Agora os pacientes podem alcançar remissão durável, de longo prazo de sua doença, juntamente com melhora do sistema de órgãos principal.
Para pacientes que se qualifiquem, existe uma oportunidade de participar de estudos clínicos. Sempre que novos tratamentos são testados, espera-se que eles sejam tão bons quanto ou melhores do que os tratamentos padrão. Todos os estudos clínicos propostos devem ser aprovados e supervisionados por um Conselho de Revisão Institucional (Institutional Review Board, IRB). O IRB é composto por médicos, cientistas, clero e leigos. Eles estão lá para garantir a segurança dos estudos e a precisão dos resultados.
Os novos tratamentos são avaliados em grupos de pessoas que atendem a certas exigências do estudo. A participação em estudos clínicos é completamente voluntária e os pacientes participantes assinam um termo de consentimento livre e esclarecido. Também é possível se retirar do estudo a qualquer momento. Em muitos casos, os custos do tratamento podem ser cobertos como parte do estudo.
Estar envolvido em pesquisa clínica permite aos pacientes beneficiar-se de tratamentos novos e experimentais antes de eles estarem amplamente disponíveis. Em longo prazo, isto leva a medicamentos e terapias melhorados para todos. Para saber que estudos clínicos estão recrutando atualmente, pode-se consultar os centros de amiloidose ou visitar ‘www.ClinicalTrials.gov’. Os pacientes também podem pesquisar em ‘www.PubMed.gov’ para encontrar artigos científicos, revisados por especialistas.
O diagnóstico precoce e preciso, juntamente com um plano de trata-
mento individualizado, são a chave para alcançar resultados positivos
para pacientes e famílias. Com uma ampla comunidade de apoio de
profissionais de saúde e especialistas, vocês não estão sozinhos.
Tratamentos

3837
6. PRINCIPAIS CENTROS DE AMILOIDOSE
Existem muitos médicos qualificados para ajudar no diag-nóstico e tratamento de amiloidose. Como pacientes, vocês não estão sozinhos. Nos Estados Unidos, entre em contato com os grupos de apoio à amiloidose (Amyloidosis Support Groups) para obter auxílio e orientação 24 horas. O número de ligação gratuita é +1 (866) 404-7539 ou e-mail ‘[email protected]’.
A seguir, há uma lista dos principais centros de pesquisa e tratamento nos Estados Unidos e internacionais. Como a amiloidose varia a cada caso, a experiência inestimável desses centros ajudará a promover resultados positivos para pacientes e famílias.
Centros de amiloidose nos EUA• Mayo Clinic (Rochester, MN) –
www.mayoclinic.org/amyloidosis
• Boston University Amyloidosis Center (Boston, MA) – www.bu.edu/amyloid
• Brigham and Women’s Hospital Cardiac Amyloidosis Program (Boston, MA) – www.brighamandwomens.org/cvcenter/amyloidosis
• Columbia Multidisciplinary Amyloidosis Program (New York City, NY) – www.nyp.org/services/amyloidosis-program-overview.html
• Memorial Sloan-Kettering Cancer Center (New York City, NY) – www.mskcc.org
• Mount Sinai Hospital (New York City, NY) – www.mountsinai.org/patient-care/health-library/diseases-and-conditions/amyloidosis
• Cedars-Sinai Multiple Myeloma & Amyloidosis Program (Los Angeles, CA) – www.cedars-sinai.edu/Patients/Programs-and-Services/Multiple-Myeloma-and-Amyloidosis-Program
• Stanford Amyloid Center (Stanford, CA) – www.stanfordhospital.org/cardiovascularhealth/amyloid
• Indiana University School of Medicine Amyloid Research Group (Indianapolis, IN) – www.iupui.edu/~amyloid/team.htm
Centros de amiloidose internacionais• Centro de Estudos em Paramiloidose Antônio Rodrigues
de Mello (Rio de Janeiro, Brasil) – www.ceparm.com
• Center for the Study & Cure of Systemic Amyloidosis (Pavia, Itália) – www.amiloidosi.it
• Groningen Unit for Amyloidosis Research & Development (Países Baixos) – www.amyloid.nl
• National Centre for Amyloidosis (Londres, Reino Unido) – www.ucl.ac.uk/medicine/amyloidosis
• Princess Alexandra Hospital (Brisbane, Austrália) – www.health.qld.gov.au/pahospital
• Westmead Hospital (Sydney, Austrália) – www.wslhd.health.nsw.gov.au/Westmead-Hospital
• Kumamoto University Hospital (Kumamoto, Japão) – www.kuh.kumamoto-u.ac.jp
• Princess Margaret Cancer Centre (Toronto, Canadá) – www.theprincessmargaret.ca
Principais centros de amiloidose

39
7. RECuRSOS ONLINE
Para mais informações, incluindo reuniões de apoio locais e uma lista detalhada de médicos regionais, visite:
Amyloidosis Support Groupswww.AmyloidosisSupport.com
Outros recursos úteis incluem:
• Amyloidosis Foundation www.amyloidosis.org
• Amyloid Support Group U.K. www.amyloidsupportgroup.co.uk
• Canadian Amyloidosis Support Network www.thecasn.org
• Leukemia & Lymphoma Society www.lls.org
• National Organization for Rare Disorders www.rarediseases.org
• RareConnect www.rareconnect.org
O conteúdo desta publicação não foi influenciado por nossos patrocinadores.
Viabilizado com o apoio destas empresas líderes de diagnósticos e terapêuticas:
www.auventx.comwww.pfizer.com www.millennium.com
www.alnylam.com www.isisph.com
www.prothena.com www.thebindingsite.com
