bioinformática - biocristalografia.df.ibilce.unesp.br · 3. abasteça os reservatórios da fileira...
TRANSCRIPT

wfdaj.sites.uol.com.br
© 2006 Dr. Walter F. de Azevedo Jr.
Bioinformática Estrutural
Cristalografia de Proteínas
Prof. Dr. Walter F. de Azevedo Jr.

© 2006 Dr. Walter F. de Azevedo Jr.
Bioinformática Estrutural
ResumoDiagramas de fasesSalting-in e Salting-outMontagem de Gotas de CristalizaçãoMétodo da Matriz EsparsaKits da Hampton ResearchPlacas LinbroMatriz Esparsa (Passo a Passo)Caracterização de CristaisArtigos de cristalizaçãoLei de BraggProblema da faseReferências
wfdaj.sites.uol.com.br
wfdaj.sites.uol.com.br
Diagrama de Fases
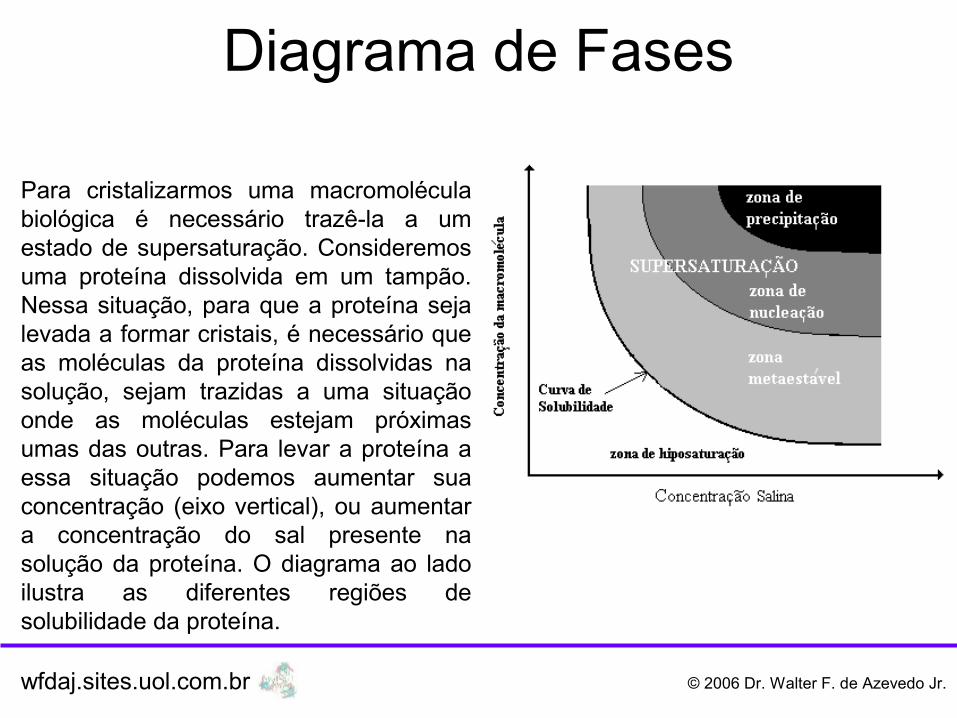
Para cristalizarmos uma macromolécula biológica é necessário trazê-la a um estado de supersaturação. Consideremos uma proteína dissolvida em um tampão. Nessa situação, para que a proteína seja levada a formar cristais, é necessário que as moléculas da proteína dissolvidas na solução, sejam trazidas a uma situação onde as moléculas estejam próximas umas das outras. Para levar a proteína a essa situação podemos aumentar sua concentração (eixo vertical), ou aumentar a concentração do sal presente na solução da proteína. O diagrama ao lado ilustra as diferentes regiões de solubilidade da proteína.
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
O processo de cristalização da proteína normalmente deve ser lento, ou seja, considerando-se a proteína inicialmente numa região abaixo da curva de solubilidade, devemos aumentar a concentração salina, ou da proteína, de modo a trazê-la na região de supersaturação, de forma lenta. Na região de supersaturação teremos as moléculas da proteína próximas umas das outras, o que, em casos favoráveis, promoverá o aparecimento dos primeiros núcleos cristalinos. Esse microcristais servirão de base para o crescimento de cristais maiores, adequados para experimentos de difração de raios X.
Diagrama de Fases
© 2006 Dr. Walter F. de Azevedo Jr.
wfdaj.sites.uol.com.br
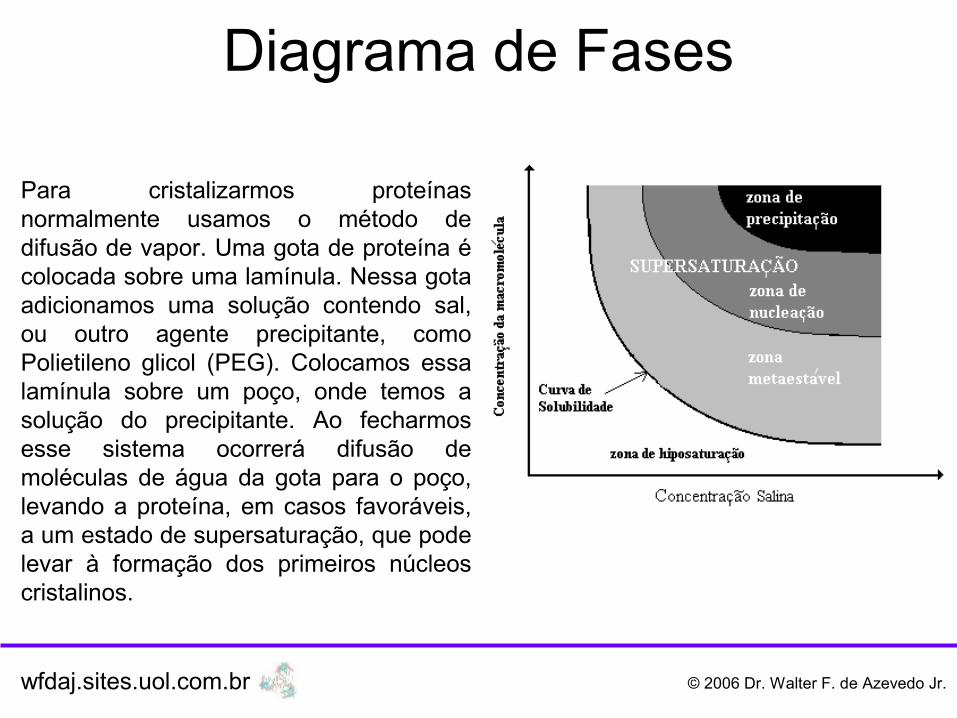
Para cristalizarmos proteínas normalmente usamos o método de difusão de vapor. Uma gota de proteína é colocada sobre uma lamínula. Nessa gota adicionamos uma solução contendo sal, ou outro agente precipitante, como Polietileno glicol (PEG). Colocamos essa lamínula sobre um poço, onde temos a solução do precipitante. Ao fecharmos esse sistema ocorrerá difusão de moléculas de água da gota para o poço, levando a proteína, em casos favoráveis, a um estado de supersaturação, que pode levar à formação dos primeiros núcleos cristalinos.
Diagrama de Fases
© 2006 Dr. Walter F. de Azevedo Jr.

Salting-in
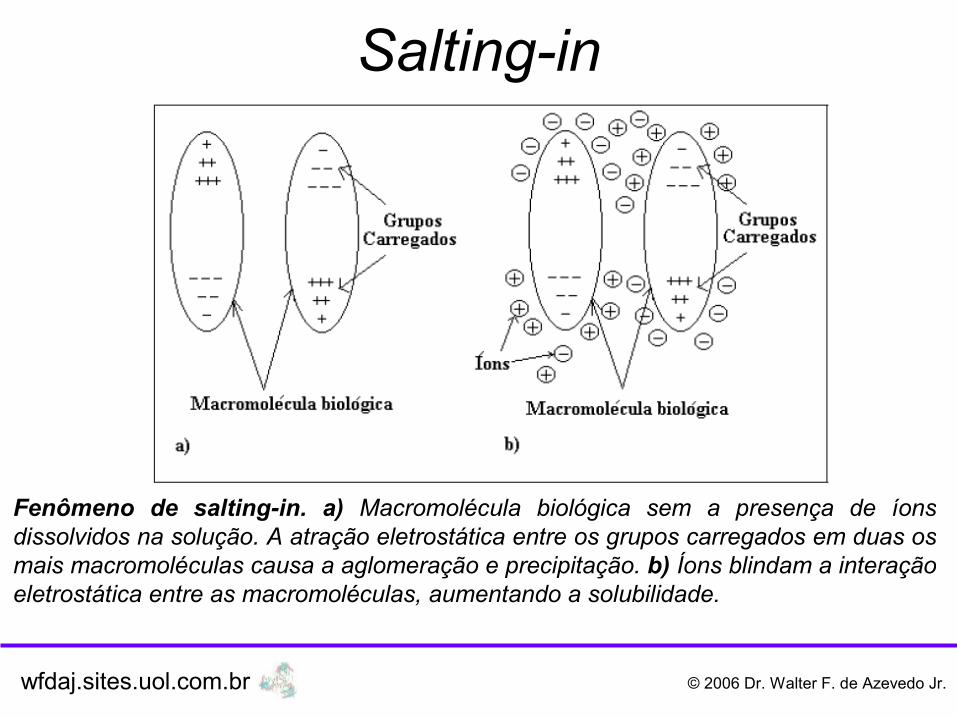
O aumento da solubilidade de uma macromolécula biológica a baixa concentração salina (<0,5M) é chamada salting-in. Este fenômeno é explicado pelas interações eletrostáticas não específicas entre a macromolécula carregada e as espécies iônicas do sal. Segundo a teoria de Debye-Hückel para soluções iônicas, um aumento na força iônica reduz a atividade dos íons em solução e aumenta a solubilidade do composto iônico. Uma forma alternativa de se tratar este fenômeno é considerar o salting-in como o resultado da competição entre grupos carregados na superfície da macromolécula e os íons em solução. Na ausência de íons de solvente a macromolécula precipita devido à atração de eletrostática entre cargas opostas em diferentes partes da macromolécula. Se os íons são adicionados à solução eles blindam os grupos carregados na macromolécula e aumentam a sua solubilidade.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Salting-in
wfdaj.sites.uol.com.br
Fenômeno de salting-in. a) Macromolécula biológica sem a presença de íons dissolvidos na solução. A atração eletrostática entre os grupos carregados em duas os mais macromoléculas causa a aglomeração e precipitação. b) Íons blindam a interação eletrostática entre as macromoléculas, aumentando a solubilidade.
© 2006 Dr. Walter F. de Azevedo Jr.

Salting-out
wfdaj.sites.uol.com.br
Outra forma de precipitar uma macromolécula é pela adição de sal (salting-out), este serve para imobilizar as moléculas de água, desta forma aumentando a concentração efetiva da macromolécula, ou seja, diminuindo a sua solubilidade. No fenômeno de salting-out a solubilidade da macromolécula biológica é governada principalmente por efeitos hidrofóbicos.
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
Montagem de Gotas de Cristalização
Seqüência de eventos para a montagem de uma gota de cristalização: a) Coloca-se 1-2µl da solução da macromolécula biológica sobre a lamínula de vidro. b) Adiciona-se 1-2µl da solução do reservatório à gota com a solução da macromolécula biológica. c) Ao final temos uma gota (2+2) com a solução de macromolécula biológica mais a solução do reservátorio.
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
Na cristalização de proteína podemos variar a forma de acomodar a gota de cristalização, como na figura acima. Tal variação permite modificações nas condições de difusão do vapor de água da gota para o poço, que pode favorecer o aparecimento de cristais de proteína.
Montagem de Gotas de Cristalização
© 2006 Dr. Walter F. de Azevedo Jr.

Com o aumento do número de macromoléculas biológicas cristalizadas com sucesso, tornou-se óbvio que muitas das condições de cristalização se assemelhavam, ou seja, havia uma concentração de resultados positivos de cristalização de macromoléculas biológicas usando-se número limitado de precipitantes, tampões e aditivos. Isto levou à proposição de diversos métodos de cristalização (Carter & Carter, 1979), onde um número limitado de condições de cristalização eram tentados, usando-se pequenas quantidades da macromolécula biológica, geralmente por volta de poucos miligramas. A partir da observação dos resultados preliminares destes experimentos era possível determinar que tampão, aditivo e agente precipitante seriam os mais favoráveis e a partir daí proceder-se a sucessivos melhoramentos até se conseguir cristais adequados, ou ainda, em casos favoráveis, obter-se cristais adequados já na primeira tentativa com as condições padrões. Dentro deste raciocínio a Dra. Jaru Jancarick da UC Berkeleley propôs o método da matriz esparsa ( Jancarick & Kim, 1991) onde diversas condições diferentes são tentadas para se cristalizar a macromolécula biológica.
Jancarik, J, & Kim, S. -H. (1991) J. Appl. Crystallogr. 24, 409-411.
Método da Matriz Esparsa
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Método da Matriz Esparsa
wfdaj.sites.uol.com.br
Parâmetros da matriz de cristalização (Jancarik & Kim, 1991)
Agentes precipitantesNão-Voláteis Sais Voláteis MisturaMPD Tartarato de Na,K 2-Propanol Sulfato de NH4 + PEGPEG 400 Fosfato de NH4 2-Propanol + PEGPEG 4000 Sulfato de NH4
PEG 8000 Acetato de NaSulfato de LiFormiato de NaFosfato de Na,KCitrato de NaFormiato de Mg
Faixa de pH: 4,6; 5,6; 6,5; 7,5 e 8,5
Aditivos: Cloreto de Ca, Citrato de Na, Cloreto de Mg, Acetato de NH4, Sulfato de NH4, Acetato de Mg, Acetato de Zn e Acetato de Ca.
© 2006 Dr. Walter F. de Azevedo Jr.

Método da Matriz Esparsa
wfdaj.sites.uol.com.br
Por tentativa e erro a matriz multimensional foi simplificada eliminando-se as condições que podem ser parcialmente representadas por resultados de outras condições, a proposta original apresenta 58 condições. Comercialmente a empresa Hampton Research, (USA) simplificou o método original, e disponibiliza um kit com 50 condições de cristalização. Comercialmente há outros kits usando-se como princípio a variação de pH, força iônica e agentes precipitantes.
Fonte: http://www.hamptonresearch.com/products/ProductDetails.aspx?cid=1&sid=17&pid=1
© 2006 Dr. Walter F. de Azevedo Jr.

Kits da Hampton Research
wfdaj.sites.uol.com.br
Fonte: http://www.hamptonresearch.com/products/ProductDetails.aspx?cid=1&sid=17&pid=1

Crystal Screen I (1-25)
wfdaj.sites.uol.com.br

wfdaj.sites.uol.com.br
Crystal Screen I (26-50)

Crystal Screen II (1-25)
wfdaj.sites.uol.com.br

Crystal Screen II (26-50)
wfdaj.sites.uol.com.br

wfdaj.sites.uol.com.br
Placas Linbro
Um dos sistema usados para cristalização de proteínas é a placa linbro, mostrada acima. Essa placa apresenta 24 poços, que permite testarmos diversas condições de cristalização. As lamínulas são colocadas sobre cada um dos poços, e vedadas com graxa de vácuo.Fonte: http://www.hamptonresearch.com

wfdaj.sites.uol.com.br
Matriz Esparsa (Passo a Passo)
Passo 1) Se a macromolécula biológica tiver de ser transportada mantenha-a em gelo (sem congelá-la) e adicione glicerol até 50% em volume e armazene-a em - 20oC. Use o necessário para o experimento de cristalização inicial, geralmente entre 1 e 2 mg é o suficiente, e coloque o restante de volta a -20oC. A fim de eliminar o glicerol da solução estoque, toma-se o volume desejado da macromolécula biológica em glicerol e adiciona-se um volume igual do tampão, então dialisa-se contra o tampão.Passo 2) concentre a proteína no mesmo tampão e acrescente DTT, ou EDTA se necessário. Se existe NaCl no tampão é melhor eliminá-lo, desde que o NaCl não seja necessário para a solubilidade da macromolécula biológica.Passo 3) Faça o fatorial à 4oC e à temperatura ambiente, se houver material suficiente.Passo 4) Examine a gotas imediatamente após montado o experimento de cristalização e anote: a) o número das gotas com precipitado.b) há presença de “sujeiras”.c) há presença de microcristais.
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
Matriz Esparsa (Passo a Passo)
Fonte: http://www.hamptonresearch.com
Passo 5) Confira as gotas diariamente por uma semana, fazendo as mesmas anotações do passo 4. E verifique também o surgimento de algum bom cristal. É importante verificar como a macromolécula biológica se comporta com o tempo em cada gota de cristalização.
Passo 6) Supondo que pequenos cristais se formaram em uma ou mais gotas do experimento inicial de cristalização, deve-se concentrar esforços nos melhores cristais e tentar a melhoria das condições de cristalização dos mesmos. Os procedimentos para a melhoria das condições de cristalização serão mostrados a seguir.
© 2006 Dr. Walter F. de Azevedo Jr.
wfdaj.sites.uol.com.br
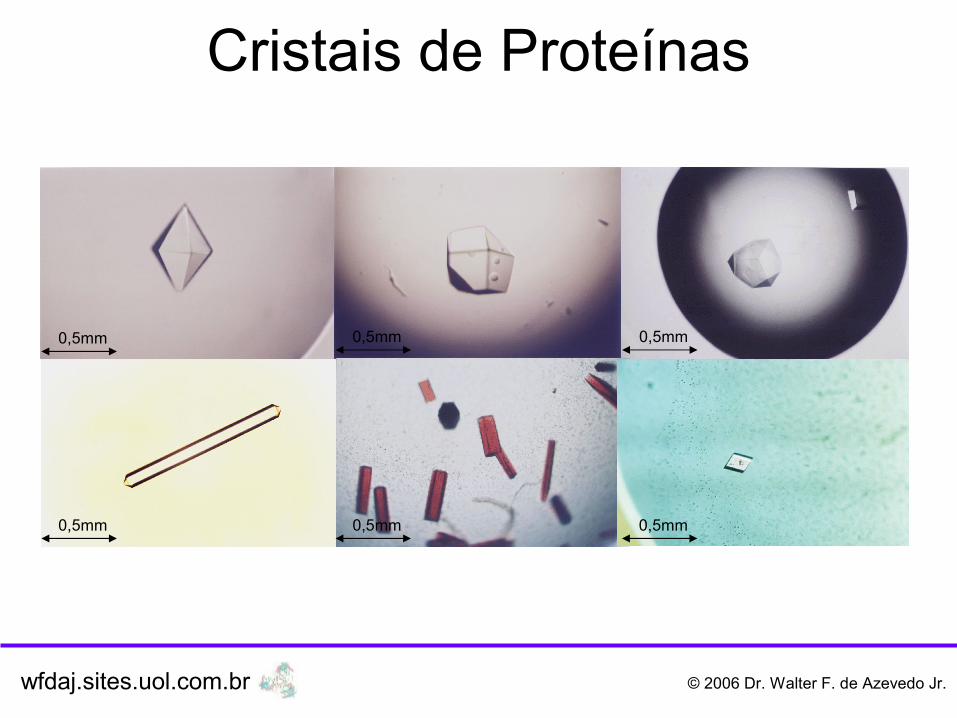
Cristais de Proteínas
0,5mm 0,5mm 0,5mm
0,5mm0,5mm0,5mm
© 2006 Dr. Walter F. de Azevedo Jr.
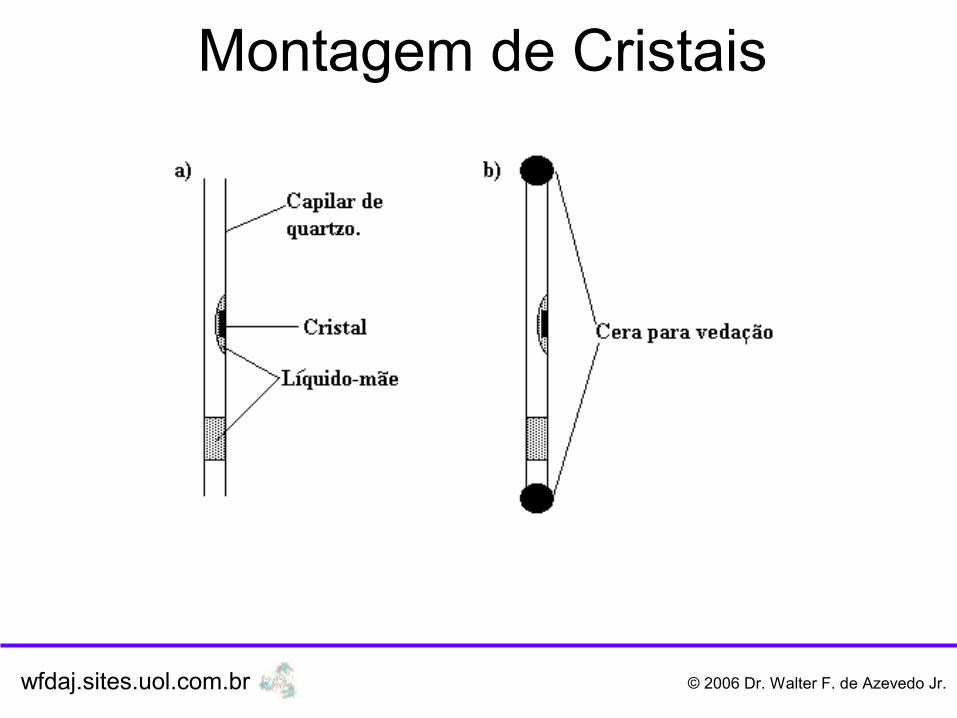
Montagem de Cristais
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Criocristalografia
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
Criocristalografia
Fonte: http://www.hamptonresearch.com

Sistema Criogênico
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização de PNP Humana
Equipamentos e reagentes:
- Placa Linbro;- PNP (De Azevedo et al., 2003) a 12mg/mL em tampão fosfato de potássio a 10 mM, pH 7,1 (solução A);- 40 % de sulfato de amônio em tampão citrato de sódio a 0,05 M, pH 5,3 (solução B);- Microfiltro de 0,22 μm;- Graxa de vácuo;- Lamínulas.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização de PNP Humana
Método:1. Prepare a soluções estoques de 40 % de sulfato de amônio em tampão citrato de sódio a 0,05 M, pH 5,3 e PNP a 12mg/mL em tampão fosfato de potássio a 10 mM, pH 7,1. Filtre a solução com um microfiltro de 0,22 μm;2. Prepare uma placa Linbro;3. Abasteça os reservatórios da fileira A da placa Linbro com solução B variando de 15-40 % em passos de 5 %;4. Em uma lamínula, misture 1 μL da solução A com 1 μL do reservatório. Rápido e em série engraxe a borda dos reservatórios e cubra com a lamínula;5. Abasteça os reservatórios da fileira B com solução B variando de 19-29 % em passos de 2%;6. Abasteça os reservatórios da fileira C com solução B variando de 15-20 % em passo de 1 %;7. Use a fileira D para duplicar ou testar parâmetros particulares (por exemplo, volume da gota para observar se a influência nos efeitos cinéticos ou no crescimento);8. Mantenha os experimentos a 18 ºC;9. Observe os experimentos no dia seguinte;
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização de PNP Humana
wfdaj.sites.uol.com.br
Resultados.Os cristais da PNP humana apareceram após 24 horas e apresentam dimensões aproximadas de 0,5 x 0,5 x 0,6 mm.
© 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização da Lisozima
wfdaj.sites.uol.com.br
Equipamentos e reagentes:
- Placa Linbro;- lisozima 40mg/mL em acetato a 50 mM, pH 4,5;- NaCl 3 M ;- Microfiltro de 0,22 μm;- Graxa de vácuo;- Lamínula.
© 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização da Lisozima
Método:1. Prepare as soluções estoques de NaCl a 3 M e lisozima a 40 mg/mL (2,74 mM) em acetato a 50 mM, pH 4,5. Filtre todas as soluções com um microfiltro de 0,22 μm;2. Prepare uma placa Linbro;3. Abasteça os reservatórios da fileira A com soluções de NaCl variando de 0,5-1,5M em passos de 0,2 M;4. Em uma lamínula, misture 4 μL da solução estoque com 4 μL do reservatório. Rápido e em série, engraxe a borda dos reservatórios e cubra com a lamínula;5. Abasteça os reservatórios da fileira B com solução de NaCl variando de 0,8-1,8 M em passos de 0,2 M. Repita o experimento na fileira B depois de diluir a solução estoque de proteína à metade para obter uma nova concentração de 20 mg/mL (1,37 mM);6. Abasteça os reservatórios da fileira C com soluções de NaCl variando de 1,5-2,5 M em passo de 0,2 M. Repita o experimento depois de diluir a solução estoque de proteína pela metade;7. Use a fileira D para duplicar ou testar parâmetros particulares (por exemplo, volume da gota para observar se há influência nos efeitos cinéticos ou no crescimento);8. Mantenha os experimentos a 18 ºC;9. Observe os experimentos durante 2 dias; © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Cristalização da Lisozima
Resultados:Os cristais de lisozima apareceram após 24 horas e apresentam dimensões aproximadas de 0,5x0,5x0,5mm.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Otimização de Cristais
A grande maioria dos experimentos de cristalização de proteínas normalmente termina em insucesso. Os números variam, mas de uma forma geral aceita-se que para uma proteína qualquer a taxa de sucesso varia entre 10 e 20 %. Contudo, mas as que conseguimos cristalizar normalmente não é na primeira tentativa, assim um processo crucial na cristalização de uma proteína é a otimização. Normalmente ao usarmos os screens temos resultados como precipitado, ou gotas claras, ou ainda microcristais. Em cada uma dessas situações temos estratégias que podem levar à formação de cristais adequados para os experimentos de difração de raios X. Cristais adequados devem ter as seguintes características:1) Boas dimensões, normalmente maiores que 0,2 mm.2) Arestas bem definida.3) Relativa rigidez.4) Facilidade de manuseio.Mesmo satisfazendo às essas condições o cristal pode não difratar. O teste final é quando expomos o cristal ao feixe de raios X e vemos um padrão claro e com centenas de pontos de difração.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Otimização de Cristais
wfdaj.sites.uol.com.br
Consideremos o seguinte cenário onde temos pequenos cristais obtidos nos experimentos iniciais, com ou sem precipitado amorfo acompanhando-os. Para a melhoria dos cristais e eliminação do precipitado amorfo seguimos os seguintes passos:
Passo 1) Abaixe a concentração da macromolécula e tente as mesmas condições em que obteve os primeiros cristais. Tente varias concentrações, por exemplo, se os primeiros cristais foram conseguidos com a concentração de 10mg/ml tente 9, 8, 7, 6 e 5 mg/ml.
Espere por uma semana e se não houver melhora nos cristais,
Passo 2) Mantenha a concentração da macromolécula biológica constante e abaixea concentração do agente precipitante.
© 2006 Dr. Walter F. de Azevedo Jr.

Protocolo de Otimização de Cristais
wfdaj.sites.uol.com.br
Espere por uma semana e se não houver melhora nos cristais,
Passo 3) Adicione 0,2M NaCl ao reservatório. Se a gota de cristalização ficar completamente “clara” (sem precipitado ou cristais) após uma semana, aumente a concentração do precipitante em incrementos de 5% a cada 3 dias. Se a gota continuar sem cristais ou precipitado 0,2M de NaCl foi demasiado, tente novamente com 20mM NaCl o mesmo procedimento. A eliminação do precipitado, se presente, é importante visto que o mesmo age como sítio de nucleação e complica a manipulação futura do cristal. Se não houve melhoria nos cristais pode-se tentar microssemeadura (microseeding) caso tal tentativa não funcione pode-se tentar variar a temperatura, caso o experimento foi realizado a 4oC, deve-se tentar em temperatura ambiente e vice-versa.
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Uma etapa inicial importante na elucidação da estrutura tridimensional de proteínas, por meio de cristalografia por difração de raios X é determinação do conteúdo da cela unitária. Uma vez tenhamos determinado os parâmetros da cela unitária: a, b, c, alfa, beta e gama, temos que determinar quantas moléculas temos na unidade assimétrica. Para isto precisamos além dos parâmetros da cela unitária o peso molecular da proteína cristalizada. O método proposto por Matthews (1968) permite calcular o número de moléculas por unidade assimétrica. Matthews determinou que para cristais de proteínas a relação entre volume da cela unitária (Vcell) e o peso molecular da proteína cristalizada fica na faixa de 1,7 a 3,5 Å3/Da, com a maioria dos valores em torno de 2,15 Å3/Da, este valor é chamado volume de Matthews (VM)
VM = Vcell
Z.MW
Onde Z é o número de moléculas na cela unitária, MW o peso molecular da proteína cristalizada e Vcell o volume cela unitária, que para uma cela unitária qualquer é dada por:
Vcell = a.b.c [1 + 2 cos α cos β cos γ - cos2α - cos2β - cos2γ]1/2
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
A fração de volume ocupada por proteína (Vprotein) é dada por:
Vprotein = 1,23/VM
E a fração de volume de solvente no cristal é dada por:
Vsolvent = 1 – 1,23/VM
Vprotein pode ser determinado por:
Vprotein = (Volume específico da proteína em cm3/g) VM
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização
wfdaj.sites.uol.com.br
Principais seções de um artigo de cristalização de proteínas:
3) Clonagem, expressão e purificação da proteína4) Cristalização5) Coleta de dados de difração de raios X6) Outros (Teste de atividade, sequenciamento, solução da estrutura e refinamento cristalográfico parcial.
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(cristalização)
wfdaj.sites.uol.com.br
The purifed MtCS was concentrated and dialyzed against 50 mM Tris±HCl buffer pH 7.8 (Hampton Research, USA). The final protein concentration was about 10 mg.ml-1. Crystallization was performed by the hanging-drop vapour-diffusion and sparse-matrix methods (Jancarik & Kim, 1991) using tissue-culture multiwell plates with covers (Linbro, ICN Biomedicals, Inc, USA) at a temperature of 293 K. Each hanging drop was prepared by mixing 1 ml each of protein solution and reservoir solution and was placed over 700 µl reservoir solution. Initial conditions were screened using Crystal Screen I and II kits (Hampton Research, USA).
Hexagonal crystals of MtCS. Approximate dimensionsare 0.30 0.25 0.25 mm.
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
A data set was collected at a wavelength of 1.427 Åusing a synchrotron-radiation source (Station PCr, LNLS, Campinas- Brazil). The data set was collected from a single MtCS crystal using a MAR CCD image-plate system. The crystal was looped out from the drop and flash-cooled. The PEG 400 present in the crystallization conditions served as a cryoprotectant, X-ray diffraction data were collected at a temperature of 100 K under a cold nitrogen stream generated and maintained with an Oxford Cryosystem. The crystal was rotated through a total of 160o, with a 1o
oscillation range per frame, a crystal-to-detector distance of 130 mm and an exposure time of 60 s. Data were processed on a Silicon Graphics Octane2 computer using the programs MOSFLM (Leslie, 1990) and SCALA (CCP4, 1994).
Artigos de cristalização (dados de difração)
A typical diffraction pattern of the MtCS crystal with 1° oscillation range. The crystal diffracts to 2.8 Å resolution.
© 2006 Dr. Walter F. de Azevedo Jr.

Summary of data-collection statistics for CRL.X-ray wavelength (A ° ) 1.427Space group P6422 or P6222Unit-cell parameters (Å ) a = 129.74, b = 129.74,
c = 156.77Highest resolution shell (Å) 2.94–2.8Asymmetric unit content 2 moleculesTotal reflections measured 92610Number of independent reflection 19341Completeness (%) 97.9 (97.9)Rmerge (%) 5.6 (16.5)
Artigos de cristalização (dados de difração)
Values in parentheses are for the highest resolution shell (2.94–2.8 Å).
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Aplicações.
3) Determinação do conteúdo da unidade assimétrica da corismato sintase de Mycobacterium tuberculosis (Dias et al., 2004). A partir dos dados de difração de raios X foi determinado que os parâmetros da cela unitária.
a = 129,74 Åb = 129,74 Åc = 156,77 ÅGrupo espacial: P6422 ou P6222
MW = 41.800 kDa
Para cela hexagonal: Vcell = a.b.c.sen (γ)
Vcell = 129,74. 129,74 . 156,77. sen(120o) = 2.285.290,31 Å3
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Para determinarmos o conteúdo da unidade assimétrica calcularemos o volumede Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual valor fica mais próximo da faixa de 1,7 a 3,5 Å, como segue:
VM = Vcell
Z.MW
Z VM(Å3/Da)2 54,676 9,114 4,5618 3,0424 2,2830 1,82
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Os valores dentro das caixas estão dentro da faixa prevista por Matthews, contudosabemos que o grupo espacial é hexagonal primitivo, o que significa quetemos 6 unidades assimétricas, assim teremos um número de moléculas múltiplode 6, ou seja, 12, 18, 24, 30.., sendo o valor mais provável 24. Assim temos:
Vprotein = 1,23/VM = 0,5395 e Vsolvent = 0,4605
Z VM(Å3/Da)2 54,676 9,114 4,5618 3,0424 2,2830 1,82
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização (purificação)
wfdaj.sites.uol.com.br
C. roseum seeds were ground to a fine powder in a coffee mill. The powder was stirred with 0.15 M NaCl [1:10(w:v)] at room temperature for 4 h and then centrifuged at 10 000g for 20 min at 278 K. The resultant supernatant was applied onto a Sepharose-4B-mannose column (0.5 10 cm) equilibrated with 0.15 M NaCl containing 5 mMCaCl2 and 5 mMMnCl2. After removing unbound material, the lectin was eluted with 0.1 M glycine, 0.15 M NaCl pH 2.6. Purified CRL was monitored by SDS–PAGE as described by Laemmli (1970) and was used to perform further characterization. N-terminal sequence analysis was performed using an Applied Biosystems pulsed-liquid phase 477A protein sequencer with a 120A PTH aminoacid analyzer, following the method described by the manufacturer.
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,
Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 1(62), 235-237, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização (purificação)
wfdaj.sites.uol.com.br
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,
Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 1(62), 235-237, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(cristalização)
wfdaj.sites.uol.com.br
The lyophilized purified CRL was dissolved to a concentration of 12 mg ml-1in 20 mM Tris–HCl pH 8.0 containing 0.5 mM CaCl2 and MnCl2 and used for crystallization trials. Crystallization screening by the hanging-drop vapour-diffusion method was performed in Linbro plates at 293 K using Hampton Research Crystal Screens I and II, SaltRx, Index and PEG/Ion Screens (Hampton Research, Aliso Viejo, CA, USA). The drops were composed of equal volumes (2 ml) of protein solution and reservoir solution and were equilibrated against 500 ml reservoir solution. An example of a crystal of CRL isshown in Fig. 1(a).
© 2006 Dr. Walter F. de Azevedo Jr.

wfdaj.sites.uol.com.br
A crystal was transferred to a cryoprotectant solution consisting of 30% glycerol in the crystallization reservoir solution. Data were collected at 1.42 A ° wavelength at a synchrotron-radiation source (beamline MX1, CPr station, Laborato´ rio Nacional de LuzSíncrotron–LNLS, Campinas, Brazil) using a MAR Research CCD imaging plate at a crystal-to-detector distance of 70 mm. A set of 100 1° oscillation images was recorded (an image is shown in Fig. 1b). Diffraction data were indexed, integrated and scaled using MOSFLM and SCALA (Collaborative Computational Project, Number 4, 1994).
Artigos de cristalização (dados de difração)
© 2006 Dr. Walter F. de Azevedo Jr.

Summary of data-collection statistics for CRL.X-ray wavelength (A ° ) 1.427Space group P212121
Unit-cell parameters (A° ) a = 67.82, b = 103.14, c = 122.09Resolution limits (A ° ) 34.92–1.77Asymmetric unit content 4 moleculesTotal reflections measured 286361Unique reflections measured 80568Completeness (%) 97.00 (97.0)Rmerge (%) 5.4 (32.5)
Artigos de cristalização (dados de difração)
Values in parentheses are for the highest resolution shell (1.87–1.77 A ° ).
wfdaj.sites.uol.com.br
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,
Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 1(62), 235-237, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Aplicações.
3) Determinação do conteúdo da unidade assimétrica da Lectina de Cymbosema roseum seeds (Cavada et al., 2006). A partir dos dados de difração de raios X foi determinado que os parâmetros da cela unitária.
a = 67,82 Åb = 103,14 Åc = 122,09 ÅGrupo espacial: P212121
MW = 25kDa
Vcell = 67,82 . 103,14 . 122,09 = 854.014,03 Å3
Referência:Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,
Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 1(62), 235-237, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Para determinarmos o conteúdo da unidade assimétrica calcularemos o volumede Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual valor fica mais próximo da faixa de 1,7 a 3,5 Å, como segue:
VM = Vcell
Z.MWZ VM(Å3/Da)2 34,143 17,074 11,385 8,536 6,837 5,698 4,889 4,27
Z VM(Å3/Da)9 3,7910 3,4111 3,1012 2,8513 2,6314 2,4415 2,2816 2,14
Z VM(Å3/Da)17 2,0118 1,9019 1,8020 1,7121 1,6322 1,5523 1,4924 1,42
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Os valores dentro das caixas estão dentro da faixa prevista por Matthews, contudosabemos que o grupo espacial é ortorrômbico primitivo, o que significa quetemos 4 unidades assimétricas, assim teremos um número de moléculas múltiplode 4, ou seja, 12, 16 ou 20, sendo o valor mais provável 16. Assim temos:Vprotein = 1,23/VM = 0,575 e Vsolvent = 0,425
Z VM(Å3/Da)2 34,143 17,074 11,385 8,536 6,837 5,698 4,889 4,27
Z VM(Å3/Da)9 3,7910 3,4111 3,1012 2,8513 2,6314 2,4415 2,2816 2,14
Z VM(Å3/Da)17 2,0118 1,9019 1,8020 1,7121 1,6322 1,5523 1,4924 1,42
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(clonagem, expressão)
wfdaj.sites.uol.com.br
The pheA gene (Rv3838c) encoding prephenate dehydratase from M. tuberculosis was amplified by the polymerase chain reaction (PCR) from genomic DNA. The forward (50-TGCATATGGTGCGTATCGCTTACCTCGGTCC- 30) and reverse (50-ACAAGCTTTCATGCTTGCGCCCCCTGGTCG- 30) synthetic oligonucleotide primers were based on the amino-terminal coding and carboxyterminal non-coding strands of the pheA gene (Cole et al., 1998) containing 50 NdeI and 30 HindIII restriction sites, respectively. The PCR product was cloned into pET-23a(+) expression vector (Novagen) and the recombinant plasmid was sequenced to confirm the identity of the cloned DNA fragment and to ensure that no mutations had been introduced by the PCR amplification step. ...
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(purificação)
wfdaj.sites.uol.com.br
The supernatant was loaded onto a Q-Sepharose Fast Flow (2.6 8.2 cm) anion-exchange column (GE Healthcare) and fractionated using a 0.0–0.5 M NaCl linear gradient. The fractions were pooled and ammonium sulfate was added to a final concentration of 0.6 M; the mixture was then loaded onto a HiLoad 16/10 Phenyl Sepharose HP hydrophobic interaction column (GE Healthcare). The active fractions were loaded onto a Mono Q HR 16/10 anion-exchange column (GE Healthcare) and eluted using a 0.0–0.5 M NaCl linear gradient.Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006 © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(clonagem, expressão)
wfdaj.sites.uol.com.br
Crystallization trials were initially performed by the hanging-drop vapour-diffusion method at 292 K. Hampton Crystal Screen and Crystal Screen 2 kits (Hampton Research) were used to determine the initial crystallization conditions. Hanging drops were prepared by mixing 1 ml of a solution containing 10 mg ml1 recombinant protein in 50 mM Tris–HCl pH 7.8 and 1 ml reservoir solution. Crystals were obtained with a reservoir solution containing 0.1 M HEPES pH 7.5, 28%(v/v) PEG 400, 0.2 M calcium chloride.Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006 © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(dados de difração)
wfdaj.sites.uol.com.br
The data set for recombinant M. tuberculosis prephenate dehydratase was collected at a wavelength of 1.438 Å using a synchrotronradiation source (Station MX1, LNLS, Campinas) and a MAR CCD detector. The crystal was flash-frozen at 100 K in liquid nitrogen. The oscillation range used was 0.8, the crystal-to-detector distance was 150 mm and the exposure time was 90 s. The crystal diffracted to 3.2 Å resolution. All data were processed and scaled using the programs MOSFLM and SCALA from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994).
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006 © 2006 Dr. Walter F. de Azevedo Jr.

Artigos de cristalização(dados de difração)
wfdaj.sites.uol.com.br
Summary of data-collection statistics for M. tuberculosis prephenate dehydratase.
X-ray wavelength (A ° ) 1.438Temperature (K) 100Resolution range (A° ) 65.94–3.20 (3.37–3.20)Total/unique reflections 71611/23215Space group I222 or I212121Matthews coefficient (Å3 Da-1) 2.7Unit-cell parameters
a (Å) 98.26b (Å ) 133.22c (Å ) 225.01
Mosaicity () 0.43Data completeness (%) 94.4 (97.2)Average I/(I) 5.7 (1.5)Multiplicity 3.1 (3.0)Rmerge(%) 0.120 (0.434)
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Aplicações.
3) Determinação do conteúdo da unidade assimétrica da MtPD. A partir dos dados de difração de raios X foi determinado que os parâmetros da cela unitária.
a = 98,26 Åb = 133,22 Åc = 225,01 ÅGrupo espacial: I222 ou I212121
MW = 33,6 kDa
Vcell = 98,26 . 133,22 . 225,01 = 2.945.425,3 Å3
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Para determinarmos o conteúdo da unidade assimétrica calcularemos o volumede Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual valor fica mais próximo da faixa de 1,7 a 3,5 Å, como segue:
VM = Vcell
Z.MW
Z VM(Å3/Da)2 87,668 10,9616 5,4824 3,6532 2,7440 2,1948 1,83
© 2006 Dr. Walter F. de Azevedo Jr.

Caracterização de Cristais
wfdaj.sites.uol.com.br
Os valores dentro das caixas estão dentro da faixa prevista por Matthews, contudosabemos que o grupo espacial é ortorrômbico com centragem I, o que significa quetemos 16 unidades assimétricas, assim teremos um número de moléculas múltiplode 8, ou seja, 8, 16, 24, 32,…, sendo o valor mais provável 32. Assim temos:
Vprotein = 1,23/VM = 0,4489 e Vsolvent = 0,5511
Z VM(Å3/Da)2 87,668 10,9616 5,4824 3,6532 2,7440 2,1948 1,83
© 2006 Dr. Walter F. de Azevedo Jr.
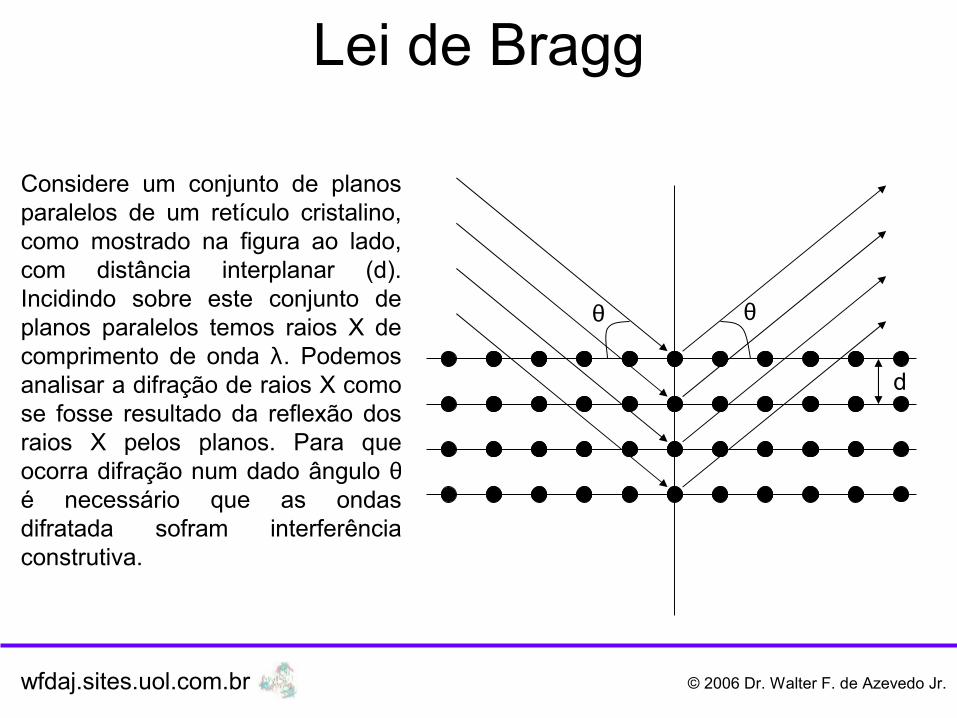
Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
θ θ
Considere um conjunto de planos paralelos de um retículo cristalino, como mostrado na figura ao lado, com distância interplanar (d). Incidindo sobre este conjunto de planos paralelos temos raios X de comprimento de onda λ. Podemos analisar a difração de raios X como se fosse resultado da reflexão dos raios X pelos planos. Para que ocorra difração num dado ângulo θé necessário que as ondas difratada sofram interferência construtiva.
d

Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Analisemos a diferença de caminho ótico dos feixes 1 e 2, indicados na figura. O feixe 2 percorre a distância A + B a mais que o feixe 1. Assim, para que as ondas dos feixes 1 e 2 sofram interferência construtiva, a diferença de caminho ótico entre elas deve ser um número inteiro de comprimentos de onda.
θ θ
θθθ θ
A B
A + B = 2.A = 2 d.sen θ
dd
θd
d.sen θ
1
2
1
2

Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
A diferença de caminho ótico (2 d.sen θ ) tem que ser um número inteiro de comprimento de onda (n.λ), onde n é inteiro, assim temos:
2 d.sen θ = n.λ (Lei de Bragg)
θ θ
θθθ θ
A Bd
d
θd
d.sen θ
1
2
1
2

Aplicação da Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Num experimento típico de difração de raios X, temos a fonte de radiação o cristal e detector, como mostrado no diagrama esquemático abaixo. Normalmente os ângulos de difração são expressos em relação ao feixe incidente, ou seja, 2 θ.
2θ
θ
θFonte de raios X
Cristal
Detector
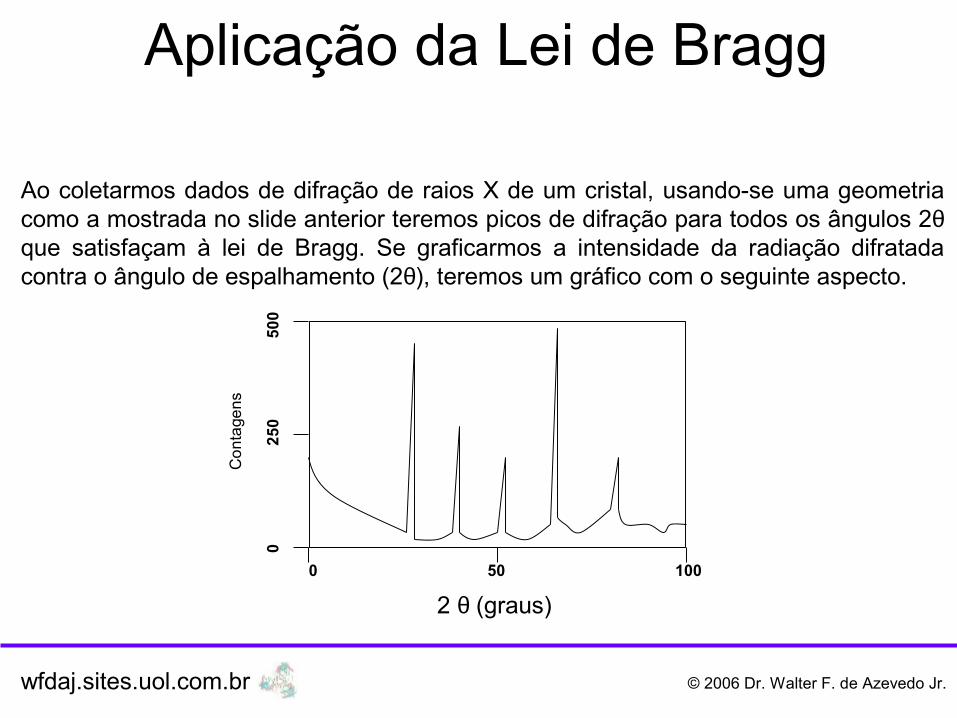
Aplicação da Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Ao coletarmos dados de difração de raios X de um cristal, usando-se uma geometria como a mostrada no slide anterior teremos picos de difração para todos os ângulos 2θque satisfaçam à lei de Bragg. Se graficarmos a intensidade da radiação difratada contra o ângulo de espalhamento (2θ), teremos um gráfico com o seguinte aspecto.
2 θ (graus)0 50 100
0
250
5
00
Con
tage
ns

Aplicação da Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
No caso de colocarmos um filme fotográfico para registrar a imagem de difração de raios X, como mostrado no diagrama abaixo, teremos um padrão de difração de raios X bidimensional, quanto mais distante o ponto de difração de raios X do ponto central da figura (feixe direto) maior o ângulo de espalhamento (2θ).
2θ
θ
θ
Cristal
Filme fotográfico ou placa de imagem
Feixe de raios X
Feixedireto

Aplicação da Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Consideremos um cristal cúbico primitivo com parâmetro de cela unitária a = 4 Å. Determine a posição angular, das 4 primeiras linhas de difração de raios X desse cristal, sabendo-se que o comprimento de onda da radiação incidente é 1,54 Å.
a = 4 Å
Pela lei de Bragg temos: 2 d.sen θ = n.λ , determinaremos para n=1,2,3 e 4. Isolando-seo ângulo θ na lei de Bragg temos:
sen θ = n.λ/2.d θ = arcsen (n.λ/2.d )
Sabemos o comprimento de onda (λ) e a distância interplanar (a = 4 Å), variando-seo n de 1 a 4 teremos os ângulos de difração.

Aplicação da Lei de Bragg
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
n θ (° ) 2. θ (° )1 11,10 22,202 22,64 45,284 35,28 70,564 50,35 100,70
Para n = 1 temos:θ = arcsen (n.λ/2.d ) = arcsen (1.1,54/2.4) = arcsen (0,1925 ) = 11,10 °
Para n = 2 temos:θ= arcsen (n.λ/2.d ) = arcsen (2.1,54/2.4) = arcsen (0,385 ) = 22,64 °
Para n = 3 temos:θ= arcsen (n.λ/2.d ) = arcsen (3.1,54/2.4) = arcsen (0,5775 ) = 35,28 °
Para n = 4 temos:θ= arcsen (n.λ/2.d ) = arcsen (4.1,54/2.4) = arcsen (0,77 ) = 50,35 °

Densidade Eletrônica
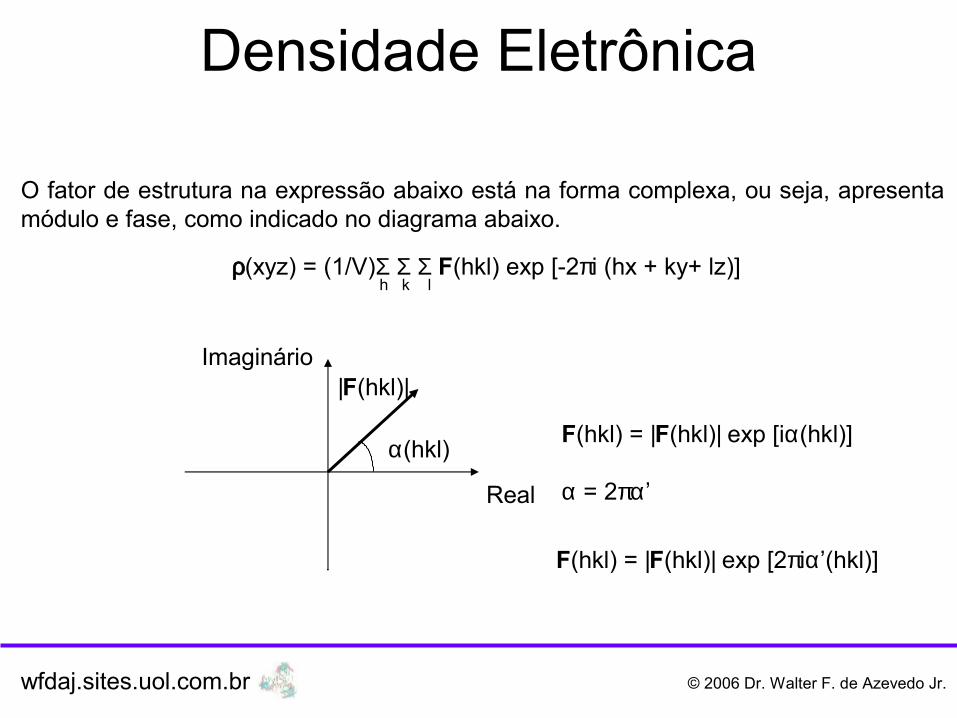
Quando os raios X incidem sobre um cristal o mesmo gera feixes difratados, esta radiação difratada é resultado da interação dos campos elétricos dos raios X com a nuvem eletrônica dos átomos no cristais. Assim podemos afirmar que a interpretação do padrão de difração de raios X fornece informação sobre a parte eletrônica do cristal, mas especificamente sobre a densidade eletrônica do cristal. Tal grandeza física, indicada por ρ(xyz) e cujas as unidades são e/Å3, pode ser usada para localizar a posição dos átomos na cela unitária. Seu cálculo é expresso pela equação abaixo:
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ F(hkl) exp [-2πi (hx + ky+ lz)] h k l
V é o volume da cela unitária
Densidade Eletrônica
O fator de estrutura na expressão abaixo está na forma complexa, ou seja, apresenta módulo e fase, como indicado no diagrama abaixo.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ F(hkl) exp [-2πi (hx + ky+ lz)] h k l
Real
Imaginário
α(hkl)
|F(hkl)|
F(hkl) = |F(hkl)| exp [iα(hkl)]
α = 2πα’
F(hkl) = |F(hkl)| exp [2πiα’(hkl)]
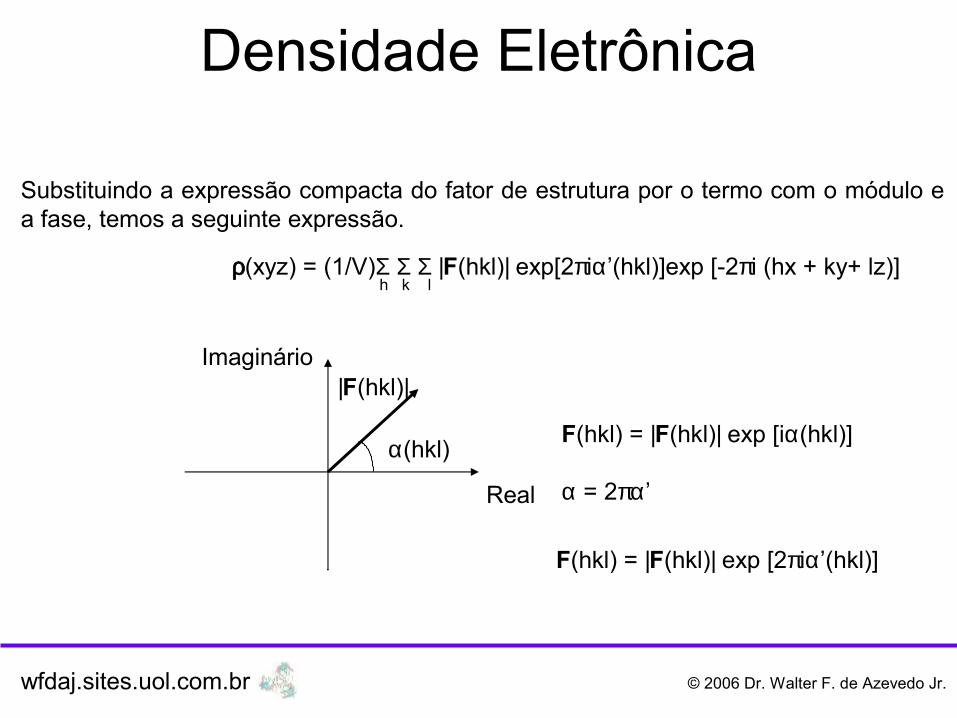
Densidade Eletrônica
Substituindo a expressão compacta do fator de estrutura por o termo com o módulo e a fase, temos a seguinte expressão.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ |F(hkl)| exp[2πiα’(hkl)]exp [-2πi (hx + ky+ lz)] h k l
Real
Imaginário
α(hkl)
|F(hkl)|
F(hkl) = |F(hkl)| exp [iα(hkl)]
α = 2πα’
F(hkl) = |F(hkl)| exp [2πiα’(hkl)]

Densidade Eletrônica
Manipulando-se a expressão temos:
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ |F(hkl)| exp {-2πi [(hx + ky+ lz) - α’(hkl)]}h k l
Real
Imaginário
α(hkl)
|F(hkl)|
F(hkl) = |F(hkl)| exp [iα(hkl)]
α = 2πα’
F(hkl) = |F(hkl)| exp [2πiα’(hkl)]

Densidade Eletrônica
Para determinarmos a densidade eletrônica para cada posição xyz temos que sabem os módulos do fator de estrutura e a fase.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ |F(hkl)| exp {-2πi [(hx + ky+ lz) - α’(hkl)]} h k l
Real
Imaginário
α(hkl)
|F(hkl)|
F(hkl) = |F(hkl)| exp [iα(hkl)]
α = 2πα’
F(hkl) = |F(hkl)| exp [2πiα’(hkl)]

Problema da Fase
Num experimento típico de difração de raios X temos o registro das intensidades para cada reflexão hkl, sabemos que a intensidade de cada reflexão I(hkl) é proporcional ao módulo do fator de estrutura, como segue:
I(hkl) α |F(hkl)|2
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Na figura ao lado a intensidade é representada pelo grau de enegrecimento do ponto de difração, quanto mais escuro maior a intensidade do feixe difratado.

Problema da Fase
Contudo nenhuma informação sobre a fase pode ser obtida diretamente do experimento de difração de raios X. Para o cálculo da função densidade eletrônica é necessário do termo de fase. A solução da fase é chamada solução do problema da fase.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ |F(hkl)| exp {-2πi [(hx + ky+ lz) - α’(hkl)]} h k l
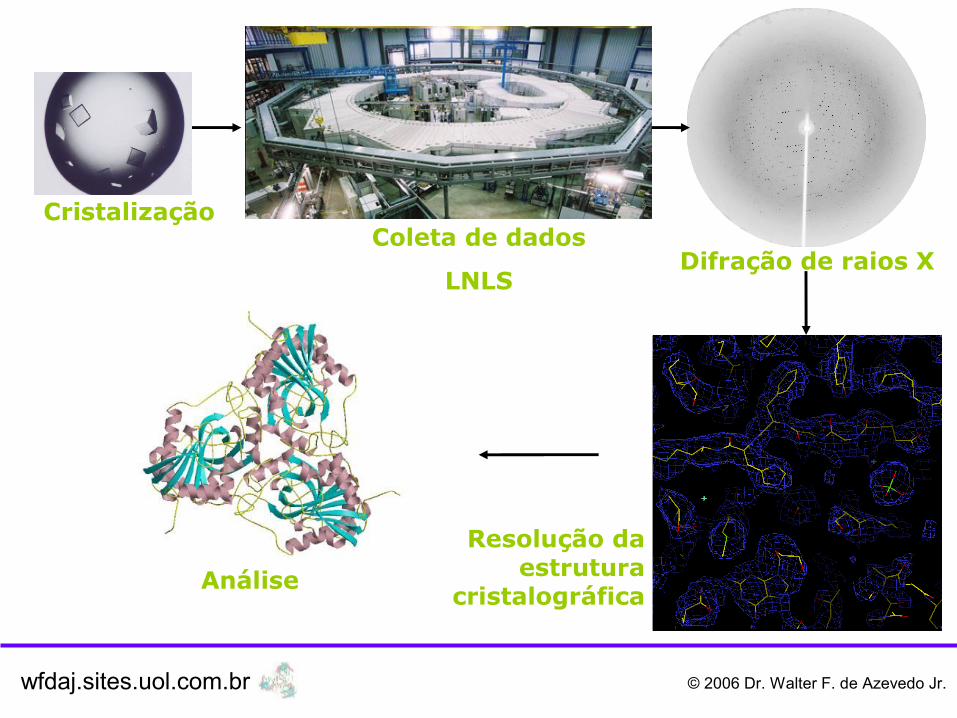
CristalizaçãoColeta de dados
LNLSDifração de raios X
Resolução da estrutura
cristalográficaAnálise
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Problema da Fase
Vimos que ao realizarmos a coleta de dados difração de raios X, obtemos informações sobre as intensidades difratadas para cada reflexão I(hkl), sendo que a intensidade é proporcional ao módulo do fator de estrutura, como segue:
I(hkl) α |F(hkl)|2
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.

Problema da Fase
Mas a informação sobre a fase de cada reflexão é perdida no processo de registro da informação. Para o cálculo da função densidade eletrônica é necessário o termo de fase. A obtenção da fase para os fatores de estrutura é chamada solução do problema da fase.
Os dois principais métodos para a solução do problema da fase para cristais de macromoléculas biológicas serão discutidos na presente aula.
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
ρ(xyz) = (1/V)Σ Σ Σ |F(hkl)| exp {-2πi [(hx + ky+ lz) - α’(hkl)]} h k l

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Função de Patterson
A função de Patterson é dada pela somatória de Fourier dos módulos dos fatores de estrutura, como indicado na equação abaixo:
A função de Patterson é calculada para cada coordenada fracionária uvw. Fisicamente esta função é um mapa de vetores entre átomos, há um pico na função de Patterson para cada vetor interatômico. A grande vantagem da função de Patterson, é que a mesma não necessita de informação sobre a fase dos fatores de estrutura, e fornece alguma informação estrutural, visto que a partir da interpretação do mapa de Patterson (a representação gráfica da função de Patterson) podemos obter informação sobre posição dos átomos.
P(uvw) = Σ Σ Σ |F(hkl)|2 cos {2π[(hu + kv+ lw)} h k l

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Função de Patterson
Consideremos uma cela unitária bidimensional, com apenas 2 átomos, como representado na figura da esquerda. A função de Patterson é uma representação dos vetores interatômicos, ou seja, apresentará valores diferentes de zero somente para valores de uvw que indiquem distâncias interatômicas, assim a representação gráfica da função de Patterson, da cela unitária bidimensional é representada à direita.
u
uu
uu
uu
uu

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Função de Patterson
Vamos considerar agora um exemplo numérico. Seja uma cela unitária tridimensional com dois átomos, com as seguintes coordenadas fracionárias: Átomo 1: 0,20, 0,31, 0,33 e Átomo 2: 0,15, 0,18, 0,22. Haverá um máximo da função de Patterson para o vetor interatômico, como segue:
Átomo 1: 0,20, 0,31, 0,33 Átomo 2: 0,15, 0,18, 0,22
Pico do mapa de Patterson:
u = x1 – x2 = 0,20 – 0,15 = 0,05
v = y1 – y2 = 0,31 – 0,18 = 0,13
w = z1 – z2 = 0,33 – 0,22 = 0,11

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
A função de Patterson permite a localização de átomos pesados em estrutura de cristais. Consideremos como exemplo um cristal de organometálico, onde temos um átomo de platina, como na estrutura do composto trans-dicloro[(trietilfosfina)(2-metilsulfinil)piridina)] platina (II), onde o átomo de platina está ligado a dois átomos de cloro e dois enxofres. O pico mais alto do mapa de Patterson dará indicação do vetor interatômico Pt-Pt, como só temos um átomo de platina, a distância será entre duas platinas relacionadas por simetria. Esta estrutura foi relatada por De Azevedo et al. 1995, o cristal é do grupo espacial P21/c, onde temos átomos nas seguinte posições:
1) x, y, z2) -x, -y, -z3) -x, y+1/2, -z+1/24) x, -y+1/2, z+1/2
Pt
ClCl
S S
CH 2 CH 2
OO
CH 2 CH 2
CH 2
CH 3
CH 2
H 3 C

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
As distâncias entre os átomos, relacionados por simetria será dada por:
Relação Diferença Coordenadas interatômicasA 1-2 ou 3-4 ±2x ±2y ±2zB 1-4 ou 2-3 0 ±2y± ½ ± ½C 1-3 ou 2-4 ±2x ½ ±2z± ½
Pt
ClCl
S S
CH 2 CH 2
OO
CH 2 CH 2
CH 2
CH 3
CH 2
H 3 C
Fonte: De Azevedo, W. F., Mascarenhas, Y. P., Sousa, G. F. and Filgueiras, C. A. L.
Acta Crystallogr. Sect.C - Cryst. Struct. Commun., 51, 619-621, 1995.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
A partir dos dados de difração de raios X calculamos os picos do mapa de Patterson, que estão listados a seguir:
Número do pico x y z1 0,000 0,100 0,5002 0,238 0,500 0,7063 0,239 0,400 0,206...
Pt
ClCl
S S
CH 2 CH 2
OO
CH 2 CH 2
CH 2
CH 3
CH 2
H 3 C
Fonte: De Azevedo, W. F., Mascarenhas, Y. P., Sousa, G. F. and Filgueiras, C. A. L.
Acta Crystallogr. Sect.C - Cryst. Struct. Commun., 51, 619-621, 1995.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
Considerando-se o pico 2, localizamos as coordenadas fracionárias x e z a partir da relação C, como segue:2 x = 0,238 → x = 0,119-2 z + 0,500 = 0,706 → z = 0,603
Número do pico x y z1 0,000 0,100 0,5002 0,238 0,500 0,7063 0,239 0,400 0,206...
Pt
ClCl
S S
CH 2 CH 2
OO
CH 2 CH 2
CH 2
CH 3
CH 2
H 3 C
Fonte: De Azevedo, W. F., Mascarenhas, Y. P., Sousa, G. F. and Filgueiras, C. A. L.
Acta Crystallogr. Sect.C - Cryst. Struct. Commun., 51, 619-621, 1995.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
Considerando-se agora o pico 1, localizamos a coordenada fracionária y partir da relação B, como segue:-2 y + 0,500 = 0,100 → y = 0,200
Número do pico x y z1 0,000 0,100 0,5002 0,238 0,500 0,7063 0,239 0,400 0,206...
Pt
ClCl
S S
CH 2 CH 2
OO
CH 2 CH 2
CH 2
CH 3
CH 2
H 3 C
Fonte: De Azevedo, W. F., Mascarenhas, Y. P., Sousa, G. F. and Filgueiras, C. A. L.
Acta Crystallogr. Sect.C - Cryst. Struct. Commun., 51, 619-621, 1995.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Aplicação da Função de Patterson
A função de Patterson permite a localização de átomos pesados em estrutura de cristais de pequenas moléculas. Para macromoléculas biológicas sua aplicação restringe-se à determinação da posição de átomos pesados inseridos em cristais de proteínas, como será discutido no método de substituição isomórfica.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
A primeira estrutura de proteína resolvida foi a mioglobina em 1959. Essa estrutura foi resolvida por Kendrew e Perutz. Para a resolução da estrutura cristalográfica da mioglobina foi usado o método de substituição isomórfica múltipla. Este método usa uma abordagem reducionista para a solução do problema da fase. Podemos pensar no cristal de proteína como um “mar”, quase homogêneo, de densidade eletrônica. Num mapa como este, a inclusão de densidades eletrônicas maiores destacariam-se, no mar de densidade eletrônica. Tal efeito é obtido com a inclusão de metais pesados no cristal. Tal inclusão, permite a obtenção de informações iniciais sobre a fase, que permitem a solução do problema da fase. Num experimento típico de substituição isomórfica, coletamos dados de difração de raios X de um cristal nativo, sem metal pesado adicionado ao retículo cristalino, e outros conjuntos de dados com metais pesados adicionados ao retículo cristalino, chamados derivados isomorfos. Assim, temos pelo menos dois conjuntos de dados de difração de raios X: um nativo e outro com metal pesado.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
Chamaremos de FP o fator de estrutura do cristal nativo, sendo que este fator de estrutura apresenta fase αP. O fator de estrutura FPH é o fator de estrutura do derivado com metal pesado, que apresenta fase αPH. Vetorialmente temos que o fator de estrutura FPH é dado por:
FPH = FP + FH
Como ilustrado no diagrama.
Eixo real
FPH
FH
FP
Eixo imaginário
AB
O

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
Analisando-se o triângulo OAB temos: αP - αPH + [180o – ( αP - αH )] + γ = 180o
γ = αPH - αH . Pela lei dos cossenos temos:
|FPH|2 = |FP|2 + |FH|2 - 2|FP| |FH|cos[180o – (αP - αH)]
|FPH|2 = |FP|2 + |FH|2 + 2|FP| |FH|cos[(αP - αH)]
(|FPH|2 - |FP|2 - |FH|2 )
2|FP| |FH|
(|FPH|2 - |FP|2 - |FH|2 )
2|FP| |FH| Eixo real
FPH
FH
FP
AB
O
αPHαP
γαP - αH
αH
αPH - αH
[ ]αP - αH = arcos
cos[(αP - αH)] =

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
Assumindo-se que medimos FPH e FP e que sabemos a posição dos átomos pesados na cela unitária do cristal, ou seja, podemos determinar FH e αH, temos que as fases dos fatores de estrutura da proteína αP são dados por:
(|FPH|2 - |FP|2 - |FH|2 ) 2|FP| |FH|
Ou seja:
αP = αH ± α’
Há dois valores possíveispara αP
Eixo real
FPH
FH
FP
AB
O
αPHαP
γαP - αH
αH
αPH - αH[ ]αP = αH + arcos
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
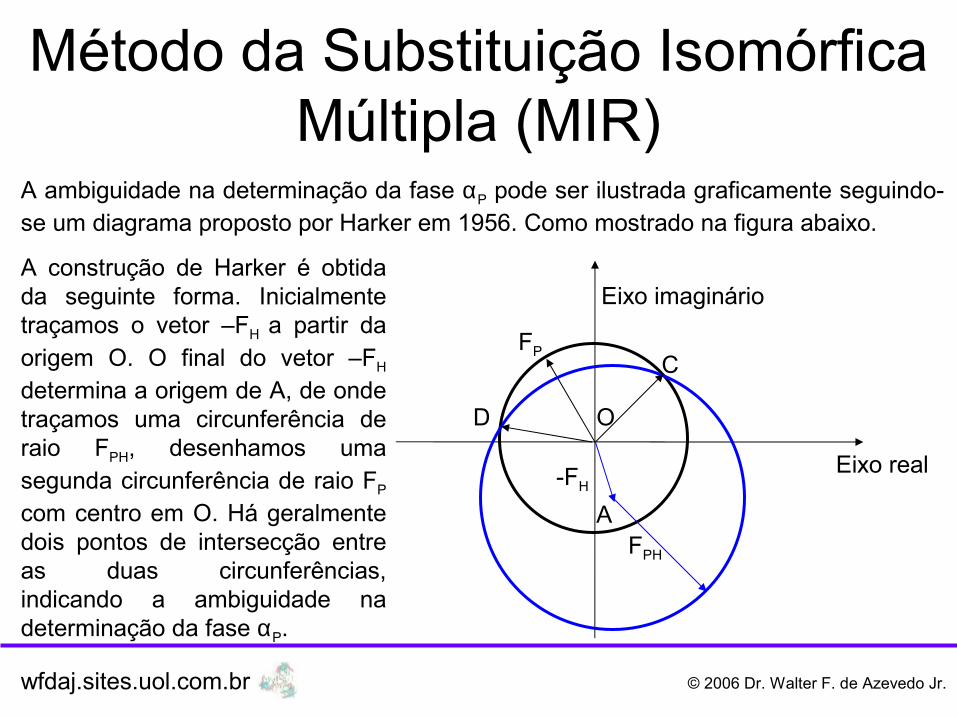
A ambiguidade na determinação da fase αP pode ser ilustrada graficamente seguindo-se um diagrama proposto por Harker em 1956. Como mostrado na figura abaixo.
A construção de Harker é obtida da seguinte forma. Inicialmente traçamos o vetor –FH a partir da origem O. O final do vetor –FH
determina a origem de A, de onde traçamos uma circunferência de raio FPH, desenhamos uma segunda circunferência de raio FP
com centro em O. Há geralmente dois pontos de intersecção entre as duas circunferências, indicando a ambiguidade na determinação da fase αP.
FPH
-FH
FP
Eixo real
A
D
Eixo imaginário
C
O

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
Os vetores OB e OC indicam dois valores possíveis para o fator de estrutura da proteína nativa (FP). Destacamos, que inicialmente só sabemos os módulos FP, FPH e a fase (αH) e o módulo do fator de estrutura para o átomo pesado. Vimos anteriormente que: FPH = FP
+ FH (soma vetorial), como o objetivo é a determinação do vetor FP, assim isolando-se FP, temos: FP = FPH – FH, esta é a razão de representarmos graficamente o –FH na construção de Harker.
FPH
-FH
FP
Eixo real
A
D
Eixo imaginário
C
O

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
A informação do átomo pesado é obtida pelo método de Patterson. Para acabarmos a ambiguidade necessitamos de informação adicional, que pode ser obtida com a coleta de dados de raios X de um terceiro conjunto, com átomos pesados inseridos no retículo cristalino.
FP = FPH – FH
FPH
-FH
FP
Eixo real
A
D
Eixo imaginário
C
O

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Método da Substituição Isomórfica Múltipla (MIR)
A inclusão da informação, de um segundo derivado de átomo pesado, acaba com a ambiguidade na determinação do termo de fase. A circunferência referente ao FPH2
cruza a circunferência do FP em dois pontos (E e F), sendo que E coincide com C. Contudo, vale destacar, que o diagrama de Harker ilustra uma situação idealizada. Conjuntos de dados de difração de raios X reais apresentam erros, e tais erros podem ser interpretados como um circunferência com o traço espesso, que gera uma certa indeterminação no valor exato da fase, muitas vezes faz-se necessário a coleta de vários derivados isomorfos, para a determinação da fase.
-FH2
FPH2
FPH
-FH
FP
Eixo real
O
A
D
Eixo imaginário
C≡E
F

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
O crescimento do número de estruturas de proteínas resolvidas gerou uma vasta base de dados estruturais, armazenadas no PDB (protein data bank) (www.rcsb.org/pdb). Partindo-se do pressuposto que proteínas que compartilham alta identidade na estrutura primária, apresentam o mesmo enovelamento, Rossmann propôs em 1962 um método onde as informações sobre a fase de uma proteína, com estrutura 3D desconhecida, mas que apresentasse estrutura primária similar com outra resolvida, poderia usar as informações estruturais da proteína resolvida como informação inicial. Tal método é chamado de substituição molecular.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
O fluxograma a seguir ilustra o principais passos na resolução de uma estrutura de proteína a partir do método de substituição molecular. Para o uso do método da substituição molecular, recomenda-se realizar uma procura no PDB usando-se a sequência de aminoácidos da estrutura a ser determinada e selecionar, entras as estruturas resolvidas, a que apresentar mais alta identidade. Há outros fatores a serem considerados tais como: Resolução dos dados de difração do modelo, presença ou não de ligantes e estado oligomérico.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
Arquivo PDB de proteína similar. Dados de difração de raios Xda nova proteína (Fobs)
Cálculo do fator de Estrutura (Fcalc)
Rotação e translação domodelo
Cálculo do Coeficientede Correlção
Listagem da rotação, translação e CC

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
A procura de rotação e translação é uma busca com 6 graus de liberdades, se realizarmos as duas buscas concomitantemente teremos um alto custo computacional. Consideremos que temos 10 posições para cada ângulo e 10 posições para cada eixo de translação. Teremos neste caso 106 cálculos, dividindo-se em busca de rotação primeiro e em seguida a busca de translação teremos 103 cálculos para rotação e 103
cálculos para translação, ou seja, 2000 cálculos, uma redução considerável. Suponha que cada cálculo demore 1 s de CPU, para o cálculo dividindo-se a busca em rotação e translação teremos 2000 s, pouco mais de meia hora, para a procura concomitante teremos 1.000.000 de segundos 277,77 horas (mais de 11 dias!!!).

REMARK Written by O version 5.10.2REMARK Sun Feb 25 14:05:51 1996REMARK Walter F. de Azevedo Jr.CRYST1 72.307 73.069 54.284 90.00 90.00 90.00ORIGX1 1.000000 0.000000 0.000000 0.00000ORIGX2 0.000000 1.000000 0.000000 0.00000ORIGX3 0.000000 0.000000 1.000000 0.00000SCALE1 0.013830 0.000000 0.000000 0.00000SCALE2 0.000000 0.013686 0.000000 0.00000SCALE3 0.000000 0.000000 0.018422 0.00000ATOM 1 CB MET 1 103.933 112.272 94.785 1.00 50.37 6ATOM 2 CG MET 1 104.548 112.540 96.126 1.00 55.72 6ATOM 3 SD MET 1 106.336 112.671 95.934 1.00 62.79 16ATOM 4 CE MET 1 106.542 114.250 95.159 1.00 54.71 6ATOM 5 C MET 1 103.199 114.420 93.762 1.00 47.20 6ATOM 6 O MET 1 102.995 114.577 92.561 1.00 51.55 8ATOM 7 HT1 MET 1 102.092 112.026 92.841 1.00 0.00 1ATOM 8 HT2 MET 1 100.857 112.905 93.606 1.00 0.00 1ATOM 9 N MET 1 101.710 112.330 93.759 1.00 48.54 7ATOM 10 HT3 MET 1 101.467 111.494 94.328 1.00 0.00 1ATOM 11 CA MET 1 102.732 113.140 94.479 1.00 47.79 6ATOM 12 N GLU 2 103.906 115.275 94.503 1.00 44.44 7ATOM 13 H GLU 2 104.333 114.933 95.316 1.00 0.00 1ATOM 14 CA GLU 2 104.085 116.695 94.178 1.00 40.49 6ATOM 15 CB GLU 2 104.531 117.459 95.428 1.00 43.49 6ATOM 16 CG GLU 2 103.464 117.597 96.515 1.00 52.62 6
wfdaj.sites.uol.com.br
|Fcalc (hkl)| = { [Σ fj cos (2π (hxj + kyj + lzj) ]2
+ [Σ fj sen (2π (hxj + kyj + lzj) ]2 }1/2
j=1
N
j=1
N
{Xj + Yj + Zj
© 2006 Dr. Walter F. de Azevedo Jr.
A partir das coordenadas atômicas podemos determinar os fatores de estrutura, usando-se a equação ao lado.
Substituição Molecular

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
Modelo alfa(o) beta(o) gama(o) Tx Ty Tz CC1 0 0 0 0 0 0 0,122 10 0 0 0 0 0 0,05.......
O coeficiente de correlação é um indicador estatístico, que assume valores próximos a 1 quando há correlação, e valores próximos a 0 quando não há correlação. Para seu usoem cristalografia, este é calculado usando-se os fatores de estrutura calculados (Fcalc) e os fatores de estrutura observados (Fobs). Os Fcalc são obtidos diretamente das coordenadas atômicas do modelo de busca, e os Fobs do experimento de difração de raios X. As coordenadas atômicas do modelo de busca são submetidas a rotações e translações. Para cada nova posição é calculado o Fcalc e um novo coeficiente de correlação. Esta informação é armazenada, assim no final temos uma listagem com as seguintes informações.

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
O coeficiente de correlação é dado pela seguinte equação:
CC = Σ ab – (Σ a Σ b)/N
{ [ Σ a2 – (Σ a)2/N ]1/2 } { [ Σ b2 – (Σ b)2/N ]1/2 }
Onde a = |Fobs| e b = |Fcalc|

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Substituição Molecular
Para solução da estrutura, usando-se o método da substituição molecular, faz-se necessário somente um conjunto de dados de difração de raios X. Tal vantagem possibilita que a estrutura seja obtida de forma mais rápida que quando aplicamos o método de substituição isomórfica, contudo, nem sempre é possível aplicar-se o método de substituição molecular, mesmo quando há alta identidade entre a estrutura e o modelo de busca, as principais causas de insucesso do método de subsituição molecular são as seguintes:
3) Diferenças devido ao empacotamento cristalino4) Diferenças devido ao estado oligomérico5) Baixa qualidade estrutural do modelo de busca6) Baixa qualidade dos dados de difração de raios X

wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.
Outros Métodos
Há ainda dois outros método usados na solução do problema da fase:
3) Métodos diretos4) Dispersão anômala

Drenth, J. (1994). Principles of Protein X-ray Crystallography. New York: Springer-Verlag.
Rhodes, G. (2000). Crystallography Made Crystal Clear. 2nd ed.San Diego: Academic Press.
Stout, G. H. & Jensen, L. H. (1989). X-Ray Structure Determination. A Practical Guide. 2nd ed. New York: John Wiley & Sons.
Referências
wfdaj.sites.uol.com.br © 2006 Dr. Walter F. de Azevedo Jr.