artigo linha de excipientes celulomax - embrafarma.com.br · a habilidade para medir com acurácia...
TRANSCRIPT

RELATÓRIO TÉCNICO
DESEMPENHO DA LINHA DE EXCIPIENTES CELULOMAX PARA
CÁPSULAS DURAS

1
1. Introdução e objetivo
A cápsula gelatinosa dura é a forma farmacêutica sólida oral de eleição na
farmácia magistral. A opção pela cápsula é devida principalmente à sua
versatilidade. As cápsulas permitem a veiculação de misturas de pós, líquidos
anidros, massas semissólidas e até mesmo de outras formas farmacêuticas de
menor volume. Além disso, é possível preparar cápsulas de liberação
modificada tais como, de liberação entérica e liberação lenta (Allen Jr., 2002). A
mistura de pós pode ser veiculada diretamente na cápsula sem envolver um
processo de granulação ou compressão prévio. Essa facilidade no preparo
aliada à possibilidade de se preparar um pequeno número de unidades por lote
faz com que a cápsula seja a forma farmacêutica de escolha para o preparo de
fórmulas individualizadas na farmácia e em estudos clínicos iniciais (Orelli &
Leuenberger, 2004).
Embora as cápsulas duras sejam relacionadas como uma forma
farmacêutica simples, o desenvolvimento de formulações para cápsulas pode
representar significantes desafios ao formulador (Guo et al., 2002). Por
exemplo, a seleção dos excipientes (agentes molhantes, lubrificantes,
desintegrantes etc) necessários para o preeenchimento da cápsula; problemas
tais como compatibilidade dos ingredientes e estabilidade; mistura dos pós e
homogeneidade; fluidez dos pós e lubrificação são frequentemente observados
e precisam ser levados em consideração durante o desenvolvimento das
formulações. A habilidade para medir com acurácia volumes precisos de um pó
ou granulado e a habilidade de transferir tais sólidos para os invólucros das
cápsulas são fatores determinantes na variação de peso e para o grau de
uniformidade de conteúdo (Guo et al., 2002). A complexa relação entre os
parâmetros de formulação (ex. tipos e quantidades de excipientes, tamanho de
cápsulas, etc), medidas de desempenho (ex. uniformidade de conteúdo e perfil
e taxa de dissolução), natureza do ingrediente ativo veiculado (ex. classificação
biofarmacêutica, higroscopia, compatibilidade física e química com outros
ingredientes da formulação etc) determina a necessidade da avaliação
laboratorial da formulação para fins de padronização.
Um ponto chave em comum na formulação de cápsulas e comprimidos é a
taxa de dissolução in vitro do fármaco veiculado. Antes de ser absorvido pela
mucosa do trato gastrintestinal, o fármaco deve ser primeiro liberado e
2
dissolvido nos fluidos gastrintestinais. O teste de dissolução é o primeiro
importante passo para se determinar a qualidade de certas preparações e para
orientar o desenvolvimento de novas formulações sólidas de uso oral (Orelli &
Leuenberger, 2004).
Em 1995, Amidon e colaboradores elaboraram um sistema de
classificação biofarmacêutica (SCB) para classificar os fármacos baseados nas
suas propriedades de solubilidade em meio aquoso e permeabilidade intestinal.
As substâncias farmacêuticas foram divididas em quatro classes,
conforme relacionado no Quadro 1. A via oral é a via de escolha para a
administração de medicamentos. Esta via, no entanto, apresenta limitações de
absorção e, por conseguinte, de biodisponibilidade. O fármaco presente na
forma farmacêutica é liberado no fluido gastrintestinal para formar uma solução.
Esse processo é limitado pela solubilidade. Uma vez que o fármaco se
encontra na forma de solução, atravessa as membranas das células que
revestem o trato gastrintestinal. Esse processo é limitado pela permeabilidade.
Posteriormente, o fármaco absorvido atinge a circulação sistêmica. Em resumo
a absorção oral e a biodisponibilidade são determinadas pela extensão e
permeabilidade do fármaco.
Quadro 1 : SISTEMA DE CLASSIFICAÇÃO BIOFARMACÊUTICA (SCB) Classe Solubilidade Permeabilidade Correlação
in Vivo / in Vitro
( IV/IV) I - Anfifílico Alta Alta Haverá correlação IV/IV se a
velocidade de dissolução for menor que a velocidade de esvaziamento gástrico; de outro modo não haverá correlação ou ela será limitada.
II - Lipofílico Baixa Alta Haverá correlação IV/IV se a velocidade de dissolução in vitro for similar à velocidade de dissolução in vivo; exceto se dose for muito alta.
III - Hidrofílico Alta Baixa Correlação IV/IV da absorção (permeabilidade) com a velocidade de dissolução limitada ou ausente.
IV - Hidrofóbico Baixa Baixa Correlação IV/IV limitada ou ausente.
Fonte: (Amidon et al., 1995; Lonenderg, Amidon, 2000)

3
Os fármacos encapsulados raramente são administrados isoladamente,
pois normalmente vêm acompanhados de excipientes. Estas substâncias são
produtos auxiliares diretamente envolvidas na composição das diversas
formulações farmacêuticas.
Embora historicamente os excipientes tenham sido considerados inertes,
já que não exercem ação terapêutica ou biológica, hoje são vistos como
capazes de influenciar a velocidade e/ou a extensão da absorção de um
fármaco (Ferreira, 2011).
As cápsulas de liberação lenta constituem um exemplo notório e ilustrativo
da influência exercida pelo excipiente na cinética de liberação do fármaco a
partir desta forma farmacêutica. Nessas cápsulas, a liberação lenta do fármaco
é devido a presença de um ou mais excipientes formadores de matriz hidrofílica
como, a hidroxipropilmetilcelulose(HPMC), carbômeros ou o alginato de sódio
(USP Pharmacists’ Pharmacopeia, 2008). Cápsulas e comprimidos com matriz
hidrofílica têm sido usados por vários anos e é eficiente na redução de
inconsistências devido uma rápida liberação e em grande quantidade de um
determinado fármaco. A redução da velocidade de liberação do fármaco
promovida pela matriz hidrofílica minimiza efeitos adversos associados aos
altos picos de concentração plasmática promovidos por alguns fármacos, bem
como estes passam a ser mais lentamente liberados e absorvidos (IJPC,
1999). Cápsulas de liberação lenta são relativamente de fácil preparação e
produzem resultados clínicos consistentes (IJPC, 1999). De um modo geral, a
velocidade de liberação é retardada conforme a proporção do polímero
matricial é aumentada em relação aos ingredientes hidrossolúveis que podem
estar presentes, como a lactose (USP Pharmacists’ Pharmacopeia, 2008).
Contudo, é difícil predizer o exato perfil de liberação para um fármaco à partir
destas cápsula de liberação modificada. Condição que torna ainda mais
importante padronizar cada passo envolvido no preparo destas cápsulas,
incluindo a realização de avaliações laboratoriais com a determinação do perfil
de dissolução da formulação e monitoramento contínuo da resposta clínica do
paciente (USP Pharmacists’ Pharmacopeia, 2008).
O estudo de dissolução é uma ferramenta indispensável nas várias
etapas dos processos de desenvolvimento do fármaco, identificação de
variáveis críticas na produção, formulação, controle de qualidade e no

4
estabelecimento de correlações in vitro/in vivo.Os ensaios de dissolução in vitro
constituem importante meio de caracterização da qualidade biofarmacêutica de
uma forma farmacêutica sólida oral, possibilitando o controle da qualidade da
formulação em estudo. (Ferreira, 2011).
A adesão dos pós no equipamento de encapsulação é um outro
problema sério e de difícil solução, podendo causar uma maior variabilidade de
peso entre as cápsulas. As principais razões para a adesão da formulação no
equipamento são as grandes forças de adesão ocasionadas por partículas
muito finas de pós, um baixo ponto de fusão do pó (< 100oC) e a tendência de
determinados pós em absorver umidade (ex. pós higroscópicos) (Podzeck &
Jones, 2004). A umidade representa um papel destacadamente negativo em
preparações farmacêuticas, particularmente em formas farmacêuticas sólidas e
excepcionalmente em cápsulas duras de gelatina, reconhecidamente sensíveis
à sua presença. Tanto a estabilidade química como a estabilidade física de
alguns ingredientes farmacêuticos ativos e do próprio invólucro da cápsula
podem ser afetados pela presença de umidade (Islam et al., 2008). A natureza
higroscópica de excipientes e ingredientes ativos deve ser considerada no
desenvolvimento da formulação. A utilização de uma quantidade suficiente de
excipiente apropriado na formulação pode prevenir problemas com substâncias
de baixo ponto de fusão (Gohil, 2002). Pós higroscópicos ou deliquescentes ou
que apresentem tendência a absorver umidade não devem ser misturados com
excipientes que contêm grande quantidade de água como amido. Excipientes
absorventes ou então, excipientes que apresentem baixa captação da umidade
tal como, a celulose microcristalina e o manitol devem ser preferidos nessa
situação (Podzeck & Jones, 2004; Islam et al., 2008). Os excipientes
absorventes diminuem a tendência dos pós em absorver a umidade
proveniente de susbtâncias higroscópicas e deliquescentes, minimizando o
contato de partículas de pós, reduzindo desta forma a tendência à liquefação
de misturas eutéticas (USP Pharmacist’s Pharmacopeia, 2008). Para uma
maior eficiência, é conveniente que a quantidade de excipiente empregada na
manipulação de cápsulas com misturas eutéticas, substâncias deliquescentes
ou higroscópicas corresponda a pelo menos 50% do total da formulação (Gohil,
2002).

5
Excipientes farmacêuticos constituem elementos de elevado destaque
na formulação dos medicamentos, uma vez que exercem efetivo papel na
garantia de obtenção da forma farmacêutica estável, adequada ao uso e ao
efeito terapêutico desejado, regendo e influenciando de maneira significativa a
cedência do ingrediente ativo contido no medicamento (Aulton, 2005). Portanto,
o desenvolvimento de uma formulação eficiente na forma de cápsulas
necessita levar em consideração aspectos farmacotécnicos e biofarmacêuticos,
contemplando assim uma escolha criteriosa dos excipientes a serem utilizados.
A linha de excipientes Celulomax foi desenvolvida com o intuito de
viabilizar para a farmácia magistral, excipientes com funcionalidade e qualidade
avaliadas através de estudos laboratoriais, incluindo a adequação dos mesmos
em função das características físicas, químicas dos ingredientes farmacêuticos
veiculados. Foram considerados aspectos biofarmacêuticos, farmacotécnicos e
de estabilidade física, química e microbiológica adotados como requisitos de
qualidade de medicamentos na forma de cápsulas duras.
Os estudos relacionados a seguir tiveram como objetivo a avaliação de
desempenho do excipiente Celulomax® E
em cápsulas duras contendo sua
mistura com ingredientes farmacêuticos ativos (IFAs) pouco solúveis; do
Celulomax SL® em cápsulas duras contendo sua mistura com IFAs solúveis;
do Celulomax® HG em cápsulas duras contendo sua mistura com IFAs
higroscópicos.

6
2. Materiais e métodos
O desempenho do excipiente Celulomax E®
foi avaliado através do
controle de qualidade e perfil de dissolução, adotando o sistema de
classificação biofarmacêutica como critério de escolha dos IFAs de baixa
solubilidade (classe II e classe IV) avaliados.
As formulações foram preparadas exclusivamente com o IFA e o excipiente
Celulomax E®
utilizado em quantidade suficiente para o preenchimento total
do volume da cápsula. As formulações foram preparadas por um manipulador
previamente treinado no laboratório de Pesquisa e Desenvolvimento da
empresa Ortofarma, sob condições de temperatura e umidade controladas.
As formulações avaliadas são listadas a seguir com as suas respectivas
classificações biofarmacêuticas:
• Cápsulas com Acetato de medroxiprogesterona 5mg e Celulomax E
qsp cápsula dura de gelatina número 2 – classe biofarmacêutica II;
• Cápsulas com Carbamazepina 200mg e Celulomax E qsp cápsula
dura de gelatina número 0– classe biofarmacêutica II;
• Cápsulas com Furosemida 40mg e Celulomax E qsp cápsula dura
de gelatina número 1 – classe biofarmacêutica II e IV.
O desempenho do excipiente Celulomax®
SL foi avaliado através do
controle de qualidade e perfil de dissolução, adotando o sistema de
classificação biofarmacêutica como critério de escolha dos IFAs solúveis
(Classe I e III) avaliados.
As formulações foram preparadas exclusivamente com o IFA e o excipiente
Celulomax® SL utilizado em quantidade suficiente para o preenchimento total
do volume da cápsula.
As formulações avaliadas são listadas a seguir com as suas respectivas
classificações biofarmacêuticas:
• Cápsulas com Fluoxetina (como cloridrato) 20mg e Celulomax® SL
qsp cápsula dura de gelatina número 2 branca/branca– classe
biofarmacêutica I;

7
• Cápsulas com Doxazosina (como mesilato) 4mg e Celulomax® SL
qsp cápsula dura de gelatina número 2 branca/branca– classe
biofarmacêutica I;
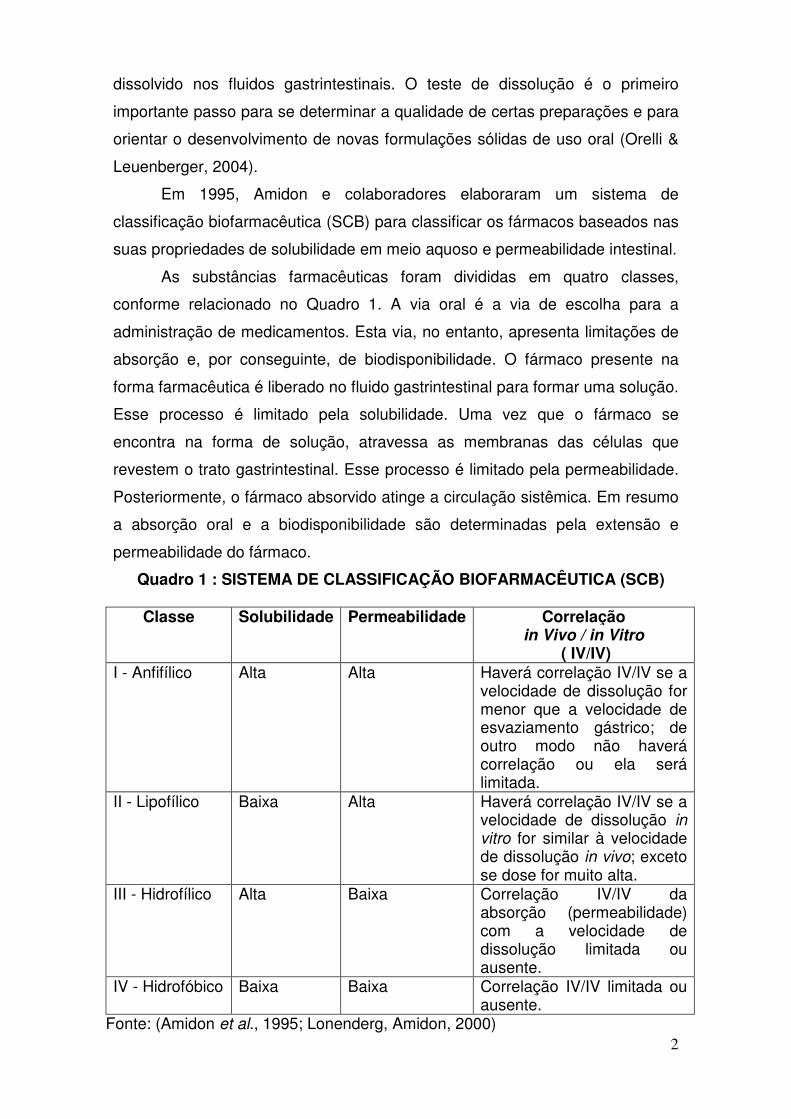
• Cápsulas com Anlodipino (como besilato) 5mg e Celulomax® SL
qsp cápsula dura de gelatina número 2 branca/branca – classe
biofarmacêutica I e III.
Métodos analíticos empregados:
Inicialmente os ingredientes farmacêuticos ativos foram submetidos ao
controle de qualidade físico-químico sendo realizados os seguintes ensaios:
- Descrição;
- Solubilidade;
- Identificação;
- Perda por dessecação;
- Teor médio.
Em seguida, foi realizado o controle de qualidade da forma farmacêutica de
acordo com os seguintes ensaios:
-Cápsulas com Furosemida: Farmacopeia Brasileira IV Edição
Identificação(por espectrofotômetro UV);Doseamento(por espectrofotômetro
UV); Uniformidade de conteúdo (por espectrofotômetro UV);Perfil de
dissolução (por espectrofotômetro UV).
-Cápsulas com Carbamazepina: Farmacopeia Brasileira IV Edição
Identificação (Infravermelho); Doseamento (por espectrofotômetro UV) ;
Água por Karl Fischer ;Uniformidade de conteúdo; Perfil de dissolução (por
espectrofotômetro UV);
-Cápsulas com Acetato de Medroxiprogesterona: Protocolo de Validação
RVCM FQ-23, Farmacopeia Brasileira IV Edição, USP 34
Identificação (Infravermelho);Doseamento (por HPLC) ; Uniformidade de
conteúdo (por HPLC) ; Perfil de dissolução (por HPLC);
-Cápsulas com Anlodipino (como besilato): Relatório de Validação de
Metodologia Analítica - RVMA-FQ-02 – Por espectrofotometria de absorção
no ultravioleta.

8
Identificação (por espectrofotômetro UV), Uniformidade de Doses Unitárias
(Uniformidade de conteúdo), Doseamento (por espectrofotômetro UV);
Teste de dissolução (por espectrofotômetro UV).
-Cápsulas com Doxazosina (como mesilato): Relatório de Validação de
Metodologia Analítica - RVMA-FQ-10 – Por espectrofotometria de absorção
no ultravioleta.
Identificação (por espectrofotômetro UV), Uniformidade de Doses Unitárias
(Uniformidade de conteúdo), Doseamento (por espectrofotômetro UV);
Teste de dissolução (por espectrofotômetro UV).
- Cápsulas com Fluoxetina (como cloridrato): Farmacopeia Brasileira 5ª
edição. – Por espectrofotometria de absorção no ultravioleta.
Identificação (por espectrofotômetro UV), Uniformidade de Doses Unitárias
(Uniformidade de conteúdo), Doseamento (por espectrofotômetro UV);
Teste de dissolução (por espectrofotômetro UV).
No estudo dos perfis de dissolução das formulações com Celulomax® E e
Celulomax® SL foram avaliadas as taxas de dissolução nos seguintes nos
tempos de 8 minutos, 16 minutos, 24 minutos, 30 minutos e 60 minutos.
O desempenho do excipiente Celulomax® HG
foi avaliado através de
estudo comparativo da captação de umidade pelo cloridrato de ranitidina e
misturas 1:1 do cloridrato de ranitidina com Celulomax® HG acondicionados na
forma de pó em uma placa de Petri e submetida a um ambiente de umidade e
temperatura controladas. O ambiente de umidade controlada a 75% de UR
(umidade relativa), foi produzido pelo uso de dessecador contendo solução
saturada de cloreto de sódio mantido a 25º C e controlado com o uso de
termohigrômetro, conforme método descrito por Islan e colaboradores. O
percentual de umidade captado em intervalos de tempo de 10, 20, 30, 60, 90,
120, 150 e 180 minutos foram calculados à partir do aumento do peso devido a
absorção da umidade determinados em balança eletrônica de precisão
analítica (ISLAM et al., 2008). Para fim exclusivo de uma observação visual da
estabilidade física, cápsulas duras de gelatina contendo Levocarnitina e
misturas 1:1 de Levocarnitina com lactose monoidratada, amido de milho,
celulose microcristalina e cápsulas contendo a mistura 1:1 de Levocarnitina e

9
Celulomax® HG foram acondicionadas diretamente em uma placa de Petri e
submetidas a um ambiente de umidade controlada de 75%UR a 25º C, durante
um período de 24 horas. Adicionalmente, foi observado a estabilidade física de
uma formulação de cápsulas preparada com a mistura 1:1 de Levocarnitina e
Celulomax® HG mantida em condição ambiental média de 50 +/- 5% UR e
21ºC +/- 2ºC de temperatura após o período de 6 (seis) meses. As amostras
após exposição às condições anteriormente relacionadas foram fotografadas
para geração das evidências.
Equipamentos:
- Dissolutor da marca Nova Ética, modelo 299 com aparato de dissolução 2
(pás) e uso de sinker para imersão das cápsulas no meio de dissolução.
- Cromatógrafo Líquido de Alta Eficiência Young Lin.
- Espectrofotômetro UV/Vis da marca Varian modelo Care 50 Probe.
- Balança analítica de precisão marca GEHAKA, mod. AG 200.
- Termohigrômetro da marca Instrutherm, modelo HT200.
- Dessecadores de vidro.
3. Resultados Controle da qualidade dos ingredientes farmacêuticos ativos:
Os relatórios de ensaio correspondentes ao controle de qualidade dos
ingredientes farmacêuticos ativos demonstram que estes atendem ás
especificações dos ensaios aos quais foram submetidos, garantindo desta
forma o atendimento a requisitos qualitativos e quantitativos da qualidade para
as formulações estudadas.
Controle da qualidade das formulações:
Os resultados do controle de qualidade das formulações avaliadas para
o Celulomax® E e o Celulomax® SL apresentaram resultados satisfatórios e de
acordo com as especificações preconizadas, como pode ser evidenciado nos
relatórios de ensaios nº. 8994/11 – Cápsulas com Furosemida ; nº.8992/11 –

10
Cápsulas com Carbamazepina ; nº. 8996/11 – Cápsulas com Acetato de
medroxiprogesterona; nº.13717/12Cápsulas com Fluoxetina;
nº.13718/12Cápsulas com Doxazosina; nº 13904/12 e Cápsulas com anlodipino
e nº10954/12.
Estudo do perfil de dissolução das formulações utilizando o excipiente
Celulomax E®
Os resultados do estudo de perfil de dissolução das formulações
testadas atendem as especificações preconizadas como pode ser
evidenciado nos relatórios de ensaio nº. 8994/11 – Cápsulas com Furosemida;
8992/11 – Cápsulas com Carbamazepina; 8996/11 – Cápsulas com Acetato de
medroxiprogesterona
Os perfis de dissolução destas podem ser vistos abaixo:
Perfil de dissoluçãoCápsulas de Furosemida 40 mg
0
20
40
60
80
100
0 10 20 30 40 50 60
Tempo (min)
Lib
eraç
ão (
%)
Especificação: Não menos que 80% (T) da quantidade declarada de furosemida se dissolvem em 60 minutos

11
Perfil de dissoluçãoCápsulas de Carbamazepina 200 mg
0
20
40
60
80
100
120
0 10 20 30 40 50 60 70
Tempo (min)
Lib
eraç
ão (
%)
Especificação: Não menos que 75% da quantidade declarada de carbamazepina se dissolvem em 60 minutos
Perfil de dissoluçãoCápsulas de Medroxiprogesterona (acetato) 5 mg
0
20
40
60
80
100
0 10 20 30 40 50
Tempo (min)
Lib
eraç
ão (
%)
Especificação: Não menos que 50% (Q) do valor rotulado de acetato de medroxiprogesterona se dissolvem em 45 minutos

12
Estudo do perfil de dissolução das formulações utilizando o excipiente
Celulomax®
SL
Os resultados do estudo de perfil de dissolução das formulações
testadas atendem as especificações preconizadas como pode ser
evidenciado nos relatórios de ensaio nº13717/12 – Cápsulas com Fluoxetina;
nº13718/12 – Cápsulas com Doxazosina ; nº13904/12– Cápsulas com
Anlodipino.
Os perfis de dissolução destas podem ser vistos abaixo:
Especificação (Farmacopeia Brasileira, 5ª Edição, p. 831, 2010): Não menos que 70% (+5%) da quantidade declarada de fluoxetina (C12H18F3NO) se dissolvem em 45 minutos.
Especificação (USP 35, p. 2977, 2012) : Não menos que 70% (+5%) da quantidade declarada de mesilato de doxazosina (C23H25N5O5.CH4SO3) se dissolvem em 30 minutos.
13
Especificação(USP 35, p. 2187, 2012): Não menos que 75% (+5%) da quantidade declarada de anlodipina (C20H25N2O5Cl) se dissolvem em 30 minutos.
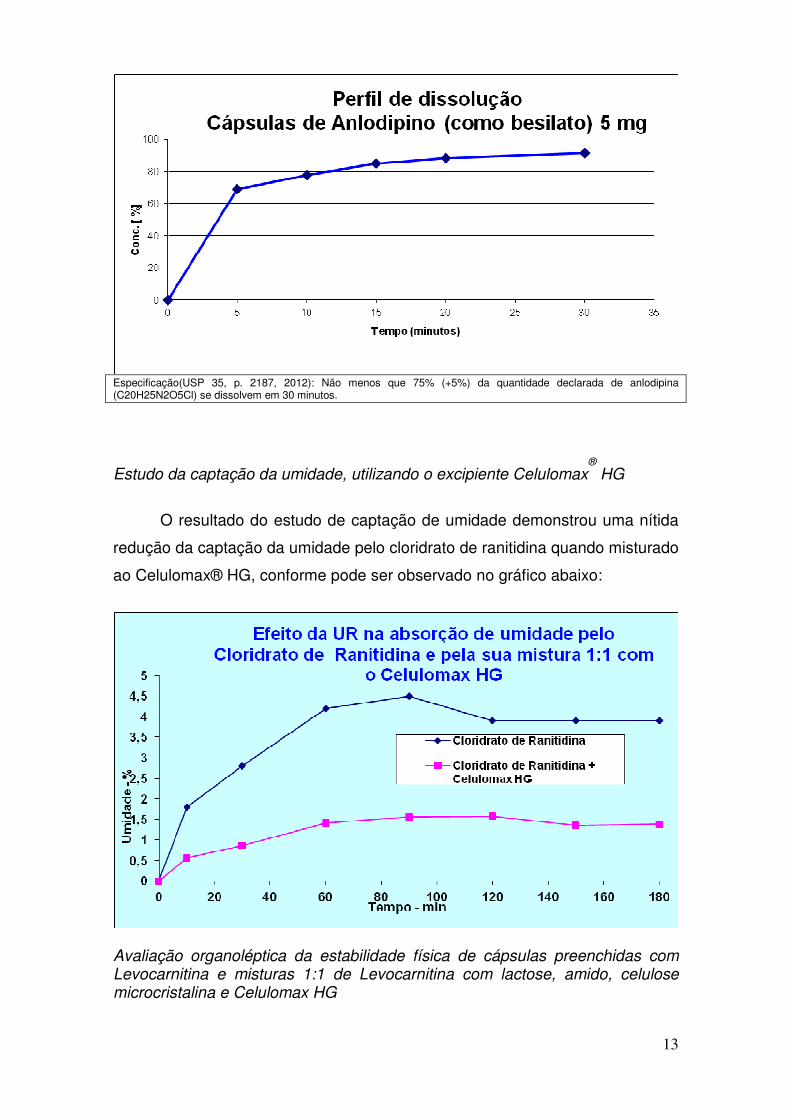
Estudo da captação da umidade, utilizando o excipiente Celulomax®
HG
O resultado do estudo de captação de umidade demonstrou uma nítida
redução da captação da umidade pelo cloridrato de ranitidina quando misturado
ao Celulomax® HG, conforme pode ser observado no gráfico abaixo:
Avaliação organoléptica da estabilidade física de cápsulas preenchidas com Levocarnitina e misturas 1:1 de Levocarnitina com lactose, amido, celulose microcristalina e Celulomax HG

14
As fotos a seguir mostram as características das cápsulas preenchidas
com a Levocarnitina e com misturas 1:1 da Levocarnitina com os excipientes
avaliados após a exposição direta a uma condição de umidade relativa de 75%
a 25º C:
Foto 1: Cápsula preenchida exclusivamente com Levocarnitina após 24h de exposição direta a ambiente com 75% UR a 25º C.
Foto 2: Cápsula preenchida com mistura 1:1 de Levocarnitina + lactose monoidratada após 24h de exposição direta a ambiente com 75% UR a 25º C.

15
Foto 3: Cápsula preenchida com mistura 1:1 de Levocarnitina + amido de milho após 24h de exposição direta a ambiente com 75% UR a 25º C.
Foto 4: Cápsula preenchida com mistura 1:1 de Levocarnitina + celulose microcristalina (Avicel® PH 101) após 24h de exposição direta a ambiente com 75% UR a 25º C.
Foto 5: Cápsula preenchida com mistura 1:1 de Levocarnitina + Celulomax® HG após 24h de exposição direta a ambiente com 75% UR a 25º C.
As cápsulas contendo a mistura 1:1 de levocarnitina e Celulomax® HG
mantiveram-se íntegras mesmo nesta condição ambiental extrema, enquanto

16
as demais cápsulas preparadas com levocarnitina e com a mistura de
levocarnitina e os outros excipientes investigados apresentaram deformação,
rachaduras ou sinais de deliquescência.
Avaliação organoléptica da estabilidade física de cápsulas preenchidas com mistura 1:1 de Levocarnitina e Celulomax® HG em condições ambientais de temperatura e umidade após 6 meses de armazenamento As cápsulas preenchidas com a mistura 1:1 de Levocarnitina e
Celulomax® HG, embaladas em frasco plástico de polietileno de alta densidade
com o uso de algodão para o preenchimento do espaço vazio; apresentaram
mínimo sinal de alteração física. Os invólucros das tampas e corpos das
cápsulas mantiveram-se íntegros e sem sinais de deformação ou rachadura. O
conteúdo de pó encapsulado apresentou sinais mínimos de captação de
umidade, constatada pela presença de alguns grumos. Considerando que a
Levocarnitina é um ingrediente ativo muito higroscópico e deliquescente, as
propriedades absorventes e a baixa captação de umidade conferidas pelo
Celulomax® HG foram evidentes na manutenção da estabilidade física desta
preparação durante este período de armazenamento.
Foto 5: Cápsula preenchida com mistura 1:1 de Levocarnitina + Celulomax® HG embalada em pote de polietileno de alta densidade, após 6 meses em ambiente com média de 50% +/- 5% de UR e temperatura média de 21ºC +/- 2ºC.

17
4. Discussão e Conclusão Na prática farmacêutica magistral a cápsula parece ser a escolha mais
adequada para o atendimento de formulações individualizadas. Contudo, a
diversidade da natureza dos ingredientes ativos veiculados nesta forma
farmacêutica implica em uma série de variáveis que devem ser consideradas
na escolha de excipientes para cápsulas. A necessidade prática de
padronização neste processo de escolha, envolve uma conciliação complexa
de considerações técnicas e por conseguinte, abre oportunidade para o
desenvolvimento de excipientes funcionalmente avaliados.
O Celulomax® E
é um excipiente composto por celulose microcristalina
modificada com a presença croscarmelose sódica como superdesintegrante,
um lubrificante e um agente deslizante e dessecante. É alegado pelo fabricante
que esta composição confere propriedades de fluxo, compactação e
estabilidade apropriadas e com baixo potencial de incompatibilidade. A
presença da croscarmelose sódica poderia melhorar potencialmente as
características de desintegração e consequente dissolução de fármacos pouco
solúveis, podendo resultar em uma maior biodisponibilidade. Neste estudo o
excipiente Celulomax E® foi avaliado na manipulação de cápsulas com
fármacos pouco solúveis, classificados como Classe II e Classe IV de acordo
com o Sistema de classificação biofarmacêutica (SCB). Os resultados obtidos
permitiram concluir o potencial de aplicação do Celulomax E® como excipiente
para cápsulas contendo fármacos pouco solúveis. Os ensaios de dissolução
resultaram em taxas de dissolução que atenderam as especificações
farmacopeicas para os medicamentos avaliados. Os medicamentos avaliados
continham fármacos de baixa solubilidade e alta permeabilidade (Classe II) e
de baixa solubilidade e baixa permeabilidade (Classe IV) e apresentaram
resultados de controle de qualidade em conformidade para as especificações
de cápsulas de Furosemida, Carbamazepina e Acetato de
Medroxiprogesterona. Adicionalmente, os parâmetros de peso médio e
uniformidade de doses unitárias conformes podem ter sido favorecidos pela

18
boa propriedade de fluxo do Celulomax E®que influencia no preenchimento
uniforme de cápsulas.
O Celulomax® SL é um excipiente composto por celulose microcristalina
de tamanho médio de partícula na ordem de 180 Microns, amido pré-
gelatinizado e dióxido de silício coloidal como agente deslizante e dessecante.
Esta composição melhora propriedades de fluxo, compactação, reduzindo
problemas relacionados à variação de peso. Apresenta estabilidade apropriada,
baixo potencial de incompatibilidade, com pouca interferência na taxa de
dissolução. É recomendado pelo fabricante para fármacos solúveis e/ou com
pobres características de fluxo. Neste estudo o excipiente Celulomax® SL foi
avaliado na manipulação de cápsulas com fármacos solúveis, classificados
como Classe I e Classe III de acordo com o Sistema de classificação
biofarmacêutica (SCB). Os resultados obtidos permitiram concluir o potencial
de aplicação do Celulomax® SL como excipiente para cápsulas contendo
ingredientes farmacêuticos ativos solúveis. Os ensaios de dissolução
resultaram em taxas de dissolução que atenderam as especificações
farmacopeicas para os medicamentos avaliados. Os medicamentos avaliados
continham fármacos solúveis e de alta permeabilidade (Classe I) e solúveis de
baixa permeabilidade (Classe III). Os medicamentos apresentaram resultados
de controle de qualidade em conformidade com as especificações de cápsulas
com Fluoxetina, Doxazosina e anlodipino. Adicionalmente, os parâmetros de
peso médio e uniformidade de doses unitárias conformes podem ter sido
favorecidos pela notável propriedade de fluxo do Celulomax SL®que influencia
no preenchimento uniforme de cápsulas.
O Celulomax® HG
é um excipiente composto por celulose
microcristalina de tamanho médio de partícula na ordem de 100 Microns,
estearato de magnésio como lubrificante, silicato de magnésio como
absorvente e dióxido de silício coloidal como agente deslizante e absorvente.
Esta composição confere, segundo o fabricante, uma baixa captação de
umidade aliada a uma ação absorvente, boas características de fluxo e

19
compatibilidade; reduzindo problemas relacionados à higroscopia e
deliquescência, sem impactação negativa na desintegração da forma
farmacêutica e na posterior dissolução do ingrediente ativo. É recomendado
pelo fabricante para o preparo de cápsulas duras contendo ingredientes
farmacêuticos ativos de moderada a alta higroscopicidade, deliquescentes;
incluindo também extratos secos fitoterápicos e aminoácidos. Neste estudo, o
excipiente Celulomax® HG foi avaliado em relação a sua influência na
captação da umidade ambiental quando misturado a um fármaco
reconhecidamente higroscópico como o cloridrato de ranitidina. Os resultados
obtidos permitiram observar uma significativa redução da captação da umidade
pela mistura do Celulomax® HG com a ranitidina quando comparada à
captação da umidade pela ranitidina somente. O potencial de aplicação do
Celulomax® HG como excipiente para cápsulas contendo ingredientes
farmacêuticos ativos higroscópicos e deliquescentes também foi evidenciada
com a observação da estabilidade física de cápsulas duras preparadas com
levocarnitina, ingrediente ativo notoriamente reconhecido como muito
higroscópico, expostas a condições desafiadoras de um ambiente de alta
umidade relativa. As cápsulas contendo a mistura levocarnitina com
Celulomax® HG obtiveram um melhor desempenho quando comparadas às
cápsulas preenchidas somente com levocarnitina ou com as misturas de
levocarnitina e os demais excipientes avaliados. O bom desempenho do
Celulomax® HG também foi confirmado no teste de prateleira onde as
propriedades absorventes e a baixa captação de umidade conferidas pelo
Celulomax® HG foram evidentes na manutenção da estabilidade física desta
preparação durante este período de armazenamento.
A preparação da forma farmacêutica cápsula na farmácia representa um
desafio ao profissional farmacêutico, devendo o mesmo estabelecer critérios
definidos para escolha de excipientes apropriados que atendam aos requisitos
regulatórios e aos requisitos técnicos de qualidade e desempenho, incluindo
aspectos biofarmacêuticos, farmacotécnicos e de conveniência terapêutica. A
linha de excipientes Celulomax® é abrangente no atendimento dos diversos
aspectos envolvidos no desenvolvimento desta forma farmacêutica e pode

20
representar uma importante ferramenta para a padronização de excipientes na
farmácia magistral.
Referências: 1. Allen Jr., L. V. The Art, Science and Technology of Pharmaceutical
Compounding. 2nd ed. Washington:AphA, 2002. p.133-159.
2. Allen Jr., L..V.; Popovich, N.G.; Ansel, H.C. Anse’ls Pharmaceutical Dosage
Forms and Drug Delivery Systems. 8th edition. Baltimore: Lippincott Williams &
Wilkins, 2005. Chapter 7, p.204- 226.
3. Amidon, G.L., H. Lennernäs, V.P. Shah, and J. R. Crison, "A theoretical basis
for a biopharmaceutics Drug Classification: The correlation of in vitro drug
product dissolution and in vivo bioavailability." Pharmaceutical Research 12 (3,
March), 413-420, 1995.
4.Aulton, M.E. Delineamento de Formas Farmacêuticas. 2a edição. Porto
Alegre: Artmed Editora. P.453-465.
5.Guo, M.; Kaira,G.; Wilson, W.; Peng, Y.; Augsburger, L.L. A Prototype
Intelilligent Hybrid System for Hard Gelatin Capsule Formulation Development.
Pharmaceutical Technology, 2002. p.44-60.
6.Lobenberg, R.L. & Amidon, G.L. Modern bioavailability, bioequivalence and
biopharmaceutics classification system. New scientific approaches to
international regulatory standards.European Journal of Pharmaceutics and
Biopharmaceutics 50 (2000) 3-12.
7. Orelli, J.V. & Leuenberger, H. Search for Technological reasons to develop a
capsule or a tablet formulation with respect to wettability and dissolution.
International Journal of Pharmaceutics 287 (2004) 135-145.

21
8. Farmacopeia Brasileira, 5ª edição, 2010.
9. USP34-NF29-United States Pharmacopeia, 34TH ed., 2011.
10. Ferreira, A.O. Guia Prático da Farmácia Magistral. 4ª Ed. Vol.1. São Paulo:
Pharmabooks, 2011.
11. USP Pharmacists’ Pharmacopeia. 2nd ed. Washington: The United States
Pharmacopeial Convention, 2008.
12. IJPC–International Journal of Pharmaceutical Compounding,
September/October1999 pg 401.
13. Podzeck, F.; Jones, B.E. Pharmaceutical Capsules. 2nd ed. London:
Pharmaceutical Press, 2004. p.101-117.
14.Islam, S.M.A. et al. Study of Moisture Absorption by Ranitidine
Hydrochloride: Effect of % RH, Excipients, Dosage Forms and Packing
Materials. J. Pharm. Sci. 7(1): 59-64, 2008.
15. Gohil, U.C. Investigations into the Filling Properties of Powder Mixtures into
Hard Shell Capsules. PhD Thesis, University of London, 2002. 184:202-212.
16. USP35-NF30-United States Pharmacopeia, 35TH ed., 2012.