análises tipo f - universidade federal de são carlosevolucao/popgen/phylogeog2007.pdf · - pode...
TRANSCRIPT

1
1
2
43
5
11111
cba
xx
x
xxxx
xx
c
c
b
a
m1
m2
m3
m4
‘p1’= [ 1 0 0 0 ]‘p2’= [ 0 0 0 0 ]‘p3’= [ 0 0 1 0 ]‘p4’= [ 0 1 0 0 ]
‘p5’= [ 0 1 0 1 ]
m4m2 m3m1
Analysis of MOlecular VAriance
FST
PhiST
Vantagens
- Pode ser usado em vários tipos de marcadores que gerammatrizes de distância, como microssatélites, RAPD e isozimas;
- Simples de se implementar em programas rápidos;
Análises “tipo” FST
Desvantagens
- Índice único pode estar sendo influenciado por váriosfatores (fluxo gênico, sistemas de acasalamento, deriva);
- Requer conhecimento das populações para determinação daestrutura a ser estudada.
- Assume que todo processo de diferenciação esteja sendodeterminado por processos recorrentes;
Processos Recorrentes
•Sistemas de acasalamento
•Fluxo gênico
•Deriva genética
Eventos Históricos
•fragmentação (⇓ fluxo gênico, ⇑ deriva & endogamia)
•expansão na área (⇑ fluxo gênico, ⇓ deriva& endogamia)
•mudanças não recorrentes no tamanhopopulacional
- gargalos e efeito fundador- crescimento populacional
Tempo
↖ Análises do “tipo” Fst assumem processosrecorrentes determinando padrãoencontrado, mas muitas vezes não é assim.
Dificuldade está em se determinar quaiseventos, históricos e recorrentes, estãoassociados ao padrão de variação genéticaencontrado
FST ⇓
FST ⇑
Análises tipo Fst
Desequilíbrio de Ligação e Associações multiloci
Loci únicos alcançam equilíbrio de H.W. em uma geração
Já 2 ou mais loci…
Problemas:
1- LD deve-se a história comum ou subestruturapopulacional?
2- LD é uma medida invariavelmente entre dois loci
Investigar a história ao estudarpopulações ancestrais

2
PCA 1o PCA na Europa
Árvore evolutiva deve ser estimada apenas quandopopulações forem isoladas (apenas quando existir umaárvore de fato). Fluxo gênico neste caso faz papel da recombinação
NJ e árvores de populações
Como vimos, nem sempre uma distância genéticamaior do que 0 implica existência de isolamentohistórico, mas pode refletir o fluxo gênico.
Como distinguir os dois?
NJ e árvores de populações
Como distinguir fluxo gênico de vicariância
Se for vicariância:–Valores de distância genética devem seguir valores daárvore, e serem consistentes com outros loci.
Se for fluxo gênico:–Distância genética estará associada a distribuição geográfica, sendo maior em distâncias maiores.
Fluxo gênico restrito• Distância genética estará associada a distribuição
geográfica, sendo maior em distâncias maiores
0
1
2
500 600 700 800
Av er age clade dist ance (Km)

3
Correlação cofenética
Correlação entre as distâncias geradas pela matrizpopulacional e as esperadas por um modelo de árvoreestimado.
Permite quantificar quão bem uma matriz de distânciase adequa a modelos de árvores.
Ou seja, permite testar se existem árvores!
Correlação cofenética
Correlação cofenética
Se rejeitado, pode se dever a dois eventos:
•Fluxo gênico recorrente
•Fluxo gênico raro, de longa distância
Uma árvore não deve existir se fluxo gênico recorrentefor a força dominante na população ao invés de eventos históricos
Fluxo Gênico X Miscigenação
Amostragens mais detalhadas na região permitemdeterminar se efeito é global ou local.Amostragem de vários loci indica se padrão é geral ourestrito a alguns loci.Isolamento por distância não produz padrões globais, pois mutações ocorrem aleatoriamente e se espalhamgradualmente.Cria padrões que diferem entre loci.
Fluxo Gênico X Miscigenação
Fluxo gênico recorrente cria mais polimorfismos naspopulações com um fluxo gênico limitado pordistâncias geográficas.
Episódios de misturas genéticas raros criam padrãotemporal e espacial de mudanças de freqüênciasalélicas em vários loci ao mesmo tempo.
Haplótipos e história evolutiva
Haplótipos são geralmente definidos para pequenasregiões em que há pouca ou nenhuma recombinação.Desta forma, mantém história evolutiva original porvárias gerações ou mesmo toda sua existência.Isso é de grande valia para estudos populacionais.
Particularmente importante para estudos de fluxogênico são os haplótipos raros.

4
Haplótipos e história evolutiva
Particularmente importante para estudos de fluxogênico são os haplótipos raros.
–Geralmente são geograficamente restritos pois leva tempo para que aumentem de freqüência.–Servem de marcadores para uma melhor identificação de fluxo gênico.–Com o aumento de dados de seqüência aumentou o númerode haplótipos raros.
Templeton e Georgiadis (1996)
1. Seronera
2. Lobo
3. Loliondo
4 Colina de Naabi
5. Cratera de Ngorongoro
6. Tarangire
7. Nairobi
8. Chobe
12 34 56
7
8
Impala (Aepyceros melampus)
Búfalo (Syncerus caffeer)
Como separar história evolutiva de estrutura populacional
São encontrados em savanas com árvores, de que se alimentam, junto com grama. Ecologicamente mais especializados. Fêmeas se agregam em grupos o ano todo, com cerca de 2% de dispersão entregrupos. Machos se dispersam para acasalar.
Impalas são simpátricos em grande parte da savana Africana, sãopolíginos, vivem em manadas e tem dispersão preferencial de machos
Como separar história evolutiva de estrutura populacional
Búfalos preferem savanas com alta biomassa de gramíneas, mastambém são encontrados em áreas de floresta, formam grandesagregações que permanecem relativamente estáveis. São maisfilopátricos do que os impalas
Como separar história evolutiva de estrutura populacional
Impala (Aepyceros melampus) FST = 0.10
Búfalo (Syncerus caffeer) FST = 0.08
12 34 56
7
8
Ao usar árvore de haplótipos estamos inferindo padrões temporais e espaciais de variação genética, enquanto Fsts lidam apenas com padrões espaciais

5
12 34 56
7
8
Como separar história evolutiva de estrutura populacional Filogeografia intraespecífica - Avise 1987
Problema: definir probabilidades de eventos
12 34 56
7
8
Isolamento por distância
Análise de Clados Aninhados
Quantifica a associação entre geografia, tempo e árvorede haplótipos.Como funciona?Usa informação histórica fornecida pela árvore de haplótipos para definir certas temporalidades e eventos.Começar pela árvore de haplótipos.
Uso de métodos não apropriados para estudos intraspecíficos
Porque não apropriados:• baixa divergência• ancestral não está extinto• Politomia• reticulação• tamanho amostral grande

6
Árvore de haplótipos
Em estudos intraspecíficos:•NÃO esperamos que o ancestral esteja extinto;• Politomias são esperadas. Na verdade, politomias são PROVÁVEIS;• Como alelo ancestral não está extinto, esperamos que alelos mais antigos tenham maior freqüência. Por outro lado, novos alelos devem ter baixa freqüência;• É mais provável que um alelo raro seja derivado de um alelo comum do que de outro raro;
Podemos usar estas esperanças para resolver“loops” ou homoplasias nos dados
Máxima parcimônia Parcimônia estatística
Grupos externos nem sempre são eficientes para estudos intraspecíficos
Hsa6
Hsa5
Hsa1,3,4Hsa2
Ppa1
Ppa2
P pa3
Ptr1
Ptr2,4
Ptr3
Homo sapiens P an troglodytes P an paniscus
Substituições de a.a.
Substituições silentes
1 Mutação
Ptr5
Ppa4 Ggo1
Ggo3
Ggo4
Ggo2
Ggo6
Ggo5
Gorilla gorilla
PontasInteriores
Ambígüos
Fixados Interespecíficos
]PolimorfismosIntraspecíficos
Templeton 1995
Árvores mais parcimoniosas são estimadas pelo comprimento geral da árvore
Hsa6
Hsa5
Hsa1,3,4Hsa2
Ppa1Ppa2
Ppa3
Ptr1
Ptr2,4
Ptr3Homo sapiens Pan troglodytes Pan paniscus
Substituições de a.a.
Substituições silentes
1 Mutação
Ptr5
Ppa4 Ggo1Ggo3
Ggo4
Ggo2Ggo6
Ggo5
Gorilla gorilla
Ptr2,4
5Ptr1 Ptr3
Pan troglodytesPtr
Homo sapiens
Hsa6
Hsa5
Hsa1,3,4Hsa2
Ppa1Ppa2
Ppa3
Pan paniscus
Ppa4 Ggo1Ggo3
Ggo4
Ggo2Ggo6
Ggo5
Gorilla gorilla
Substituições de a.a.
Substituições silentes
1 Mutação
Ptr2,4
5Ptr1 Ptr3
Pan troglodytesPtr
Homo sapiens
Hsa6
Hsa5
Hsa1,3,4Hsa2
Ppa1Ppa2
Ppa3
Pan paniscus
Ppa4 Ggo1Ggo3
Ggo4
Ggo2Ggo6
Ggo5
Gorilla gorilla
Para se estimar estas conexões que estão mais distantes, temos que estimar os limites da coalescência.
• Determina até que ponto parcimônia é modelo apropriado para nossos dados (> 95% probabilidade de NÃO haver ocorrido uma homoplasia)• Usamos estimativa de theta para calcular adequação a IAM, segundo Hudson:
Árvores são estimadas considerando os alelos filogeneticamente mais próximos
Geralmente neste ponto checamos porrecombinação, mas aqui estamos assumindoque isto não ocorre.
Na prática usamos o programa TCS, mascuidado:
• Dados ausentes podem alterar o resultado
• Erros nas seqüências também, principalmente por estarmos lidandocom pequenas diferenças
D. serido tipo B
D. serido tipo A e D
7
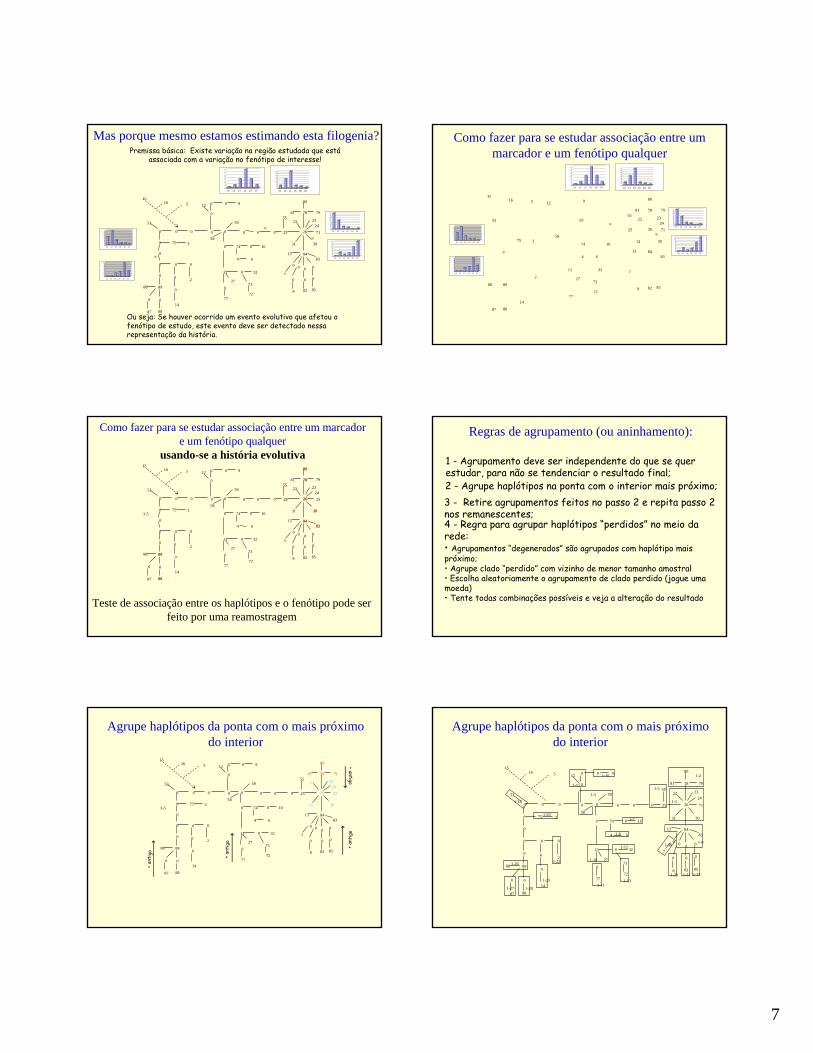
Mas porque mesmo estamos estimando esta filogenia?Premissa básica: Existe variação na região estudada que está
associada com a variação no fenótipo de interesse!
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981 78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
0
912 0
0 0
55
0
0
0
0
Ou seja: Se houver ocorrido um evento evolutivo que afetou o fenótipo de estudo, este evento deve ser detectado nessa representação da história.
**
*
02468
1012141618
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 2002468
1012141618
10 12 14 16 18 20
0
2
4
6
8
10
12
14
16
18
10 12 14 16 18 20
02468
101214161820
10 12 14 16 18 20
Como fazer para se estudar associação entre um marcador e um fenótipo qualquer
82
32
4
58
74
6
86
8887
89
14
2
75
53
515
3
16
26
22
71
30
7981 78
84
25
83
858
7
13
31
2423
80
11
59
27
77
10
73
72
912
55
**
*
0
2468
10121416
18
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 2002468
1012141618
10 12 14 16 18 20
0
24
68
1012
1416
18
10 12 14 16 18 20
02468
101214161820
10 12 14 16 18 20
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
25
30
7981 78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
3-5
0
912 0
0 0
55
0
0
0
0
Como fazer para se estudar associação entre um marcador e um fenótipo qualquer
usando-se a história evolutiva
88
89
26
30
8483
25
5578
80
Teste de associação entre os haplótipos e o fenótipo pode ser feito por uma reamostragem
Regras de agrupamento (ou aninhamento):
1 - Agrupamento deve ser independente do que se quer estudar, para não se tendenciar o resultado final;2 - Agrupe haplótipos na ponta com o interior mais próximo;3 - Retire agrupamentos feitos no passo 2 e repita passo 2 nos remanescentes;4 - Regra para agrupar haplótipos “perdidos” no meio darede:• Agrupamentos “degenerados” são agrupados com haplótipo mais próximo;• Agrupe clado “perdido” com vizinho de menor tamanho amostral• Escolha aleatoriamente o agrupamento de clado perdido (jogue uma moeda)• Tente todas combinações possíveis e veja a alteração do resultado
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
25
30
7981 78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
3-5
0
912 0
0 0
55
0
0
0
0
+ an
tigo + an
tigo + an
tigo
+ ant igo
Agrupe haplótipos da ponta com o mais próximo do interior
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981
1-2
78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
1-30
1-321-311-29
1-12
1-131-11
1-14
1-10
1-20
0
912 0
1-15
1-16
1-5
1-7
1-8
1-27 1-28
1-22
1-25
0 0
55
0
0
1-26
0
0
1-1
1-3
1-9
Agrupe haplótipos da ponta com o mais próximo do interior
8
00
8232
4
0
0
58
74
60
0
0
0
0
0
86
8887
89
0
14
2
75
53
0
0
0
515
3
16
26
22
71
30
7981
1-2
78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
1-30
1-321-311-291-12
1-131-11
1-14
1-10
1-20
0
12 0
1-15
1-16
1-5
1-7
1-8
1-27 1-28
1-221-25
0
055
0
0
1-26
0
0
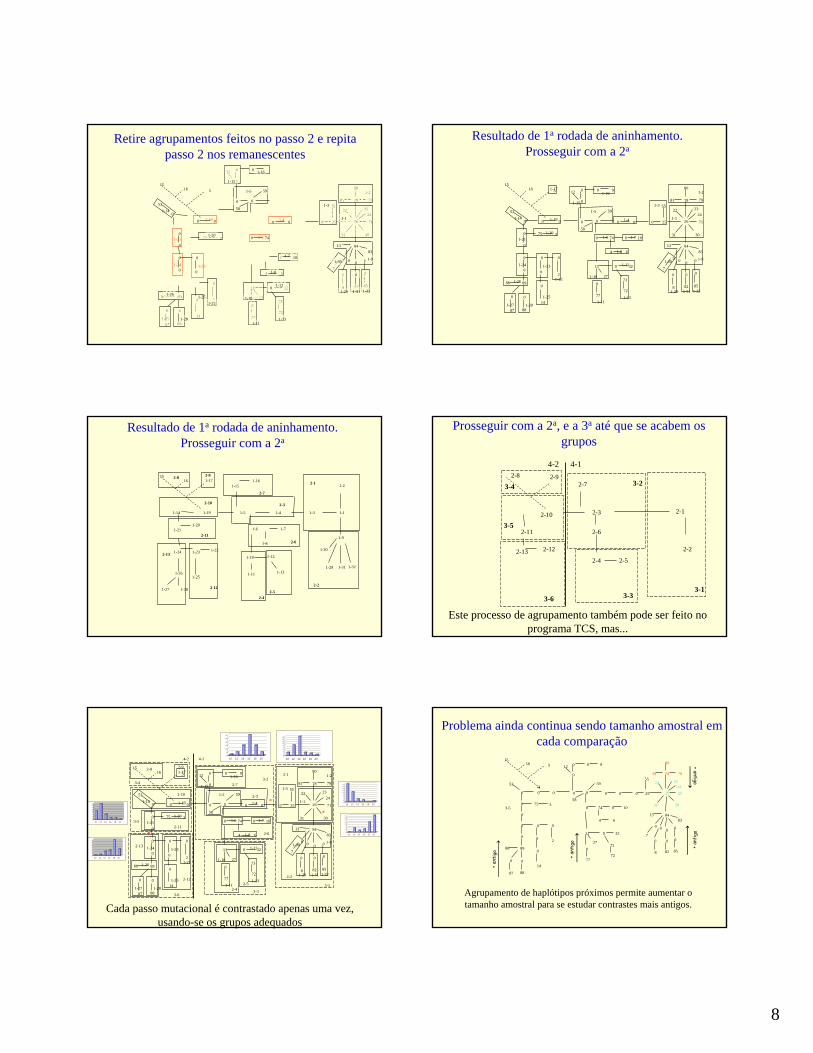
Retire agrupamentos feitos no passo 2 e repitapasso 2 nos remanescentes
1-1
1-3
1-9
1-6
1-4
1-21
1-19
1-24 1-23
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981
1-2
78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
1-11-4
1-91-30
1-321-311-29
1-12
1-131-11
1-14
1-10
1-201-21
0
912 0
1-15
1-16
1-5
1-7
1-19
1-8
1-27
1-24
1-28
1-17
1-23
1-22
1-25
0 0
551-3
1-6
0
0
1-26
0
0
Resultado de 1a rodada de aninhamento. Prosseguir com a 2a
1-27 1-28
1516
1-2
1-11-4
1-9
1-30
1-321-311-29
1-12
1-131-11
1-14
1-10
1-201-21
1-151-16
1-5
1-7
1-19
1-8
1-24
1-17
1-23 1-22
1-25
1-3
1-6
1-26
2-1
2-2
2-42-5
2-12
2-13
2-7
2-11
2-6
2-3
2-8 2-9
2-10
Resultado de 1a rodada de aninhamento. Prosseguir com a 2a
Prosseguir com a 2a, e a 3a até que se acabem os grupos
2-1
2-2
2-4 2-5
2-122-13
2-7
2-11 2-6
2-3
2-8 2-9
2-10
3-4
3-5
3-2
3-13-33-6
4-14-2
Este processo de agrupamento também pode ser feito no programa TCS, mas...
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981
1-2
78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
4-14-2
3-4
3-5
3-2
3-1
2-1
1-11-4
2-2
1-91-30
1-321-311-29
1-12
1-132-5
2-41-11
1-14
1-10
3-3
2-12
3-6
2-13
1-201-21
0
912 0
2-71-15
1-16
1-5
1-7
1-19
1-8
1-27
1-24
1-28
1-17
1-23
1-22
1-25
2-11
2-6
2-3
0 0
551-3
1-6
0
0
2-8 2-9
1-26
0
0
2-10
*
*
*
0
2468
10121416
18
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 20
0
5
10
15
20
25
30
10 12 14 16 18 2002468
1012141618
10 12 14 16 18 20
0
2
4
68
10
1214
16
18
10 12 14 16 18 20
02468
101214161820
10 12 14 16 18 20
Cada passo mutacional é contrastado apenas uma vez, usando-se os grupos adequados
Problema ainda continua sendo tamanho amostral em cada comparação
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
25
30
7981 78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
3-5
0
912 0
0 0
55
0
0
0
0
+ an
tigo + an
tigo + an
tigo
+ ant igo
Agrupamento de haplótipos próximos permite aumentar o tamanho amostral para se estudar contrastes mais antigos.

9
Este método descrito anteriormente pode até funcionar para se estudar um fenótipo qualquer, mas… será que podemos tratar a localização geográfica como um fenótipo?
O que é um fenótipo?
- Qualquer característica física individual que possa ser quantificada de forma precisa.
- Ergo...
Análise de Clados Aninhados Teste inicial de associação com haplótipos
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981
1-2
78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
4-14-2
3-4
3-5
3-2
3-1
2-1
1-11-4
2-2
1-91-30
1-321-311-29
1-12
1-132-5
2-41-11
1-14
1-10
3-3
2-12
3-6
2-13
1-201-21
0
912 0
2-71-15
1-16
1-5
1-7
1-19
1-8
1-27
1-24
1-28
1-17
1-23
1-22
1-25
2-11
2-6
2-3
0 0
551-3
1-6
0
0
2-8 2-9
1-26
0
0
2-10
Associação geográfica entre haplótipos e distribuição geográfica
82
32
4
58
74
6
86
8887
89
14
2
75
53
515
3
16
26
22
71
30
7981 78
84
25
83
858
7
13
31
2423
80
11
59
27
77
10
73
72
912
55
**
*
Dc mede o quanto um haplótipo está disperso no ambiente
Dn mede o quanto um haplótipo está distante da médiade outros haplótipos que são seus parentes
Para se incorporar a localização geográfica ao teste anterior dois índices são usados:
Teste inicial de associação com haplótipos
a b c1 2 0 02 3 6 03 4 4 4
1 12 2 2
3 3 3 3
3 3 3 3
3 3 3 3
2 2 2 2 2 2
a
b
c
Testa-se associação de haplótipos com localidades sem considerar distâncias. Signif. por randomização.
Para se incorporar a localização geográfica aoteste anterior dois índices são usados:
1
3
2
Resto docladograma
1 12 2 2
3 3 3 3
3 3 3 3
3 3 3 3
2 2 2 2 2 2
1
2
3
N
Dc Distribuição do clado
Dn Dispersão aninhadodo clado
N Centro Geográfico do clado1
2
3
1
2
3
N

10
Haplótipos antigos e recentes.
00
82
32
4
0 0
58
74
6
0
0
0
0
0
0
86
8887
89
0
14
2
75
53
0 0
0
515
3
16
26
22
71
30
7981 78
84
00
250
83
0
858
0
00
07
13
31
2423
80
11
59
27
0
0
77
10
73
72
0
912 0
0 0
55
0
0
0
0
Este é a única parte da análise para a qual um bom outgroup seria benvindo para definir polaridade, mas...
Simulações indicam que podemos usar a expectativa da coalescência para definir probabilidades de serem mais antigos
Como em geral haplótipos localizados no interior da rede são maisantigos do que os das pontas, comparamos os valores médios de Dc e Dn entre eles:Índices Dc(I), Dn(I), Dc(T) e Dn(T) medem a dispersão média de haplótipos de ponta (tips) daquele clado em relação a de haplótiposno interior.
1 12 2 2
3 3 3 3
3 3 3 3
3 3 3 3
2 2 2 2 2 2 Dc Distribuição do clado
Dn Dispersão aninhadodo clado
N Centro Geográfico do clado1
2
3
1
2
3
N
Resto docladograma
1
3
2 PontasInterior
Dc(I) = 3
Dn(I) = 3
Dc(T) = 1 2+
Dn(T) = 21 +
Análise conjunta de DC, Dn e I-T permite diferenciaçãode migrações curtas ou de longa distância, mas apenasquando sabemos o que esperar das forças evolutivas.
Como as forças evolutivas determinam os padrõesgeográficos de distribuição de haplótipos?
Para se incorporar a localização geográfica aoteste anterior dois índices são usados:
Dc mede o quanto um haplótipo está disperso no ambienteDn mede o quanto um haplótipo está distante da média de
outros haplótipos que são seus parentesI-T Medida de Dc e Dn que indica uma relação entre haplótipos
mais novos (tips) e mais antigos (interiores).
Processos Recorrentes
• Sistemas de acasalamento
• Fluxo gênico
• Deriva genética
Eventos Históricos
• fragmentação (⇓ fluxo gênico, ⇑ deriva & endogamia)
• expansão da área (⇑ fluxo gênico, ⇓deriva e endogamia)
• mudanças não recorrentes no tamanho populacional
- gargalos e efeito fundador- crescimento populacional
Forças Evolutivas
Hipótese nula: não associação entre marcador e distribuição geográfica
Se Ho é rejeitada, testamos para isolamento pordistância.
O que se espera em padrão de isolamento por distância?
Separando forças evolutivas Fluxo gênico restrito por isolamento por distância
1 - Pontas serão menos dispersas do que interiores a elas conectados
2 - Média de Dc(I) - Dc(T) será signif. grande3 - Média de Dc aumenta com o aumento donível do clado
0
1
2
500 600 700 800
Av er age clade di st ance (Km)
Dc signif pequenos para pontas
11
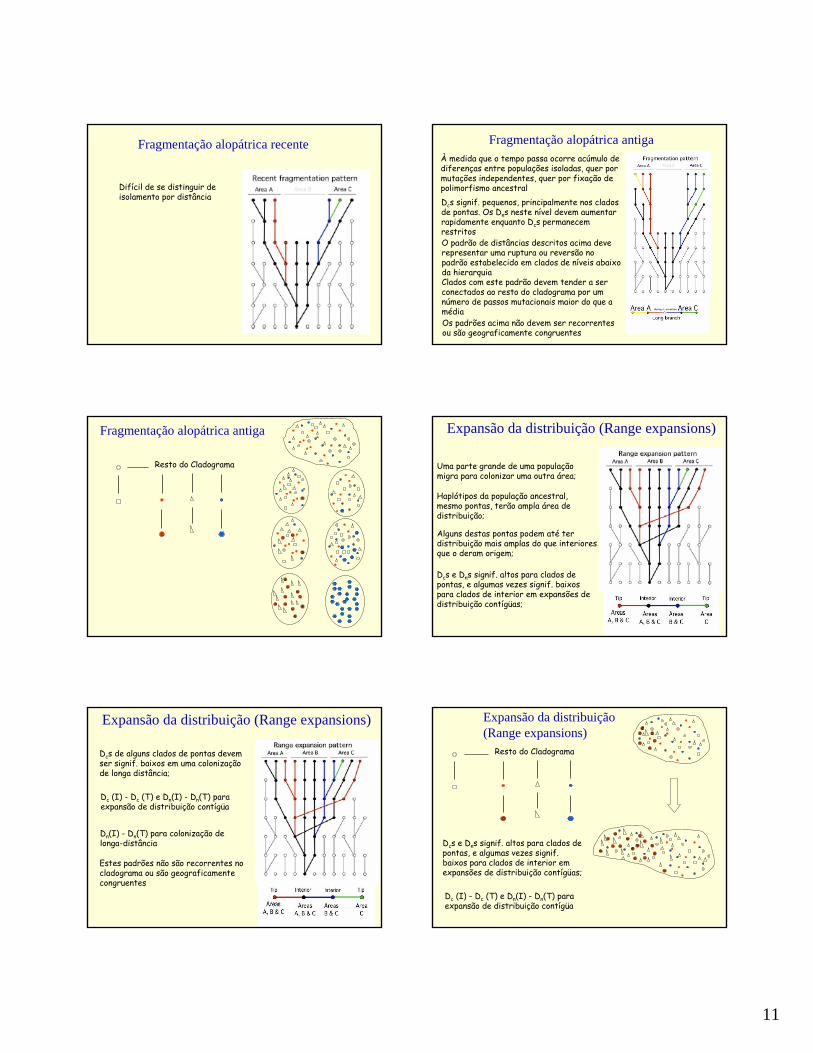
Fragmentação alopátrica recente
Difícil de se distinguir de isolamento por distância
Fragmentação alopátrica antigaÀ medida que o tempo passa ocorre acúmulo de diferenças entre populações isoladas, quer por mutações independentes, quer por fixação de polimorfismo ancestralDcs signif. pequenos, principalmente nos cladosde pontas. Os Dns neste nível devem aumentar rapidamente enquanto Dcs permanecem restritosO padrão de distâncias descritos acima deve representar uma ruptura ou reversão no padrão estabelecido em clados de níveis abaixo da hierarquiaClados com este padrão devem tender a ser conectados ao resto do cladograma por um número de passos mutacionais maior do que a médiaOs padrões acima não devem ser recorrentes ou são geograficamente congruentes
Fragmentação alopátrica antiga
Resto do Cladograma
Expansão da distribuição (Range expansions)
Uma parte grande de uma população migra para colonizar uma outra área;
Haplótipos da população ancestral, mesmo pontas, terão ampla área de distribuição;
Alguns destas pontas podem até ter distribuição mais amplas do que interiores que o deram origem;
Dcs e Dns signif. altos para clados de pontas, e algumas vezes signif. baixos para clados de interior em expansões de distribuição contígüas;
Expansão da distribuição (Range expansions)
Dcs de alguns clados de pontas devemser signif. baixos em uma colonizaçãode longa distância;
Dc (I) - Dc (T) e Dn(I) - Dn(T) paraexpansão de distribuição contígüa
Dn(I) - Dn(T) para colonização de longa-distância
Estes padrões não são recorrentes no cladograma ou são geograficamentecongruentes
Resto do Cladograma
Dcs e Dns signif. altos para clados de pontas, e algumas vezes signif. baixos para clados de interior em expansões de distribuição contígüas;
Dc (I) - Dc (T) e Dn(I) - Dn(T) para expansão de distribuição contígüa
Expansão da distribuição(Range expansions)

12
Dcs de alguns clados de pontas devem ser signif.baixos em uma colonização de longa distância;
Dn(I) - Dn(T) para colonização de longa-distância
Resto do Cladograma
Expansão da distribuição(Range expansions) Exceção na detecção de Range Expansions
D. buzzatii na Europa
Integrando resultados de vários loci
• Nenhum locus pode capturar todos as forças queafetaram a distribuição de uma espécie
• Mesmo que não esteja enviesado por seleção, como mutação e deriva são processos aleatórios, determinação será afetada pelo acaso.
Podemos replicar o experimento, ou…
Estudar vários loci diferentes
Integrando resultados de vários loci
Estudar vários loci diferentes:preferencialmente nas mesmas regiões, com mesmo tamanho amostral, uma vez que o resultado final é afetado pelo:
número de localidadestamanho amostral
Tipo de região amostrada influencia o resultado, uma vez que mutações devemocorrer na “hora e lugar certo”
Integrando resultados de vários loci
Influencia determinação de forças históricas e forçasrecorrentes:
Recorrência indica que força ocorreu diversasvezes na escala da coalescência
A dispersão em torno da média é alta em virtude davariância do tempo de coalescência ser alta. Issoafeta estimativa de loci diferentes.
Como combinar dados de marcadores diferentes?
Integrando resultados de vários loci
Como saber se o último evento de coalescência de loci diferentes mede a mesma escala temporal?
Takahata et al. sugerem que pode-se calibrar o relógio molecular ao dividir o número médio de diferenças em uma espécie A pela metade dadiferença entre esta espécie e um grupo externo B. Este valor é multiplicado pelo tempo de divergência entre as espécies A e B e estima o tempo médio de coalescência dos polimorfismosna espécie A.
Requer também uma estimativa da variância

13
Integrando resultados de vários loci
Como saber se o último evento de coalescência de loci diferentes mede a mesma escala temporal?
Estimativa de Takahata et al. e variância permitemfazer um teste de verossimilhança para estimar se um ou mais eventos estão envolvidos no padrãoencontrado em loci diferentes.
mtDNA
MADAGASCAR
ICELAND
by Isolation by Distance
Restricted
Gene Flow
Restricted Gene Flow AfterRange Expansion Mostly ThroughIsolation by Distance But With
Some Recurrent LongDistance Interchange
Hb
MADAGASCAR
ICELAND
by Isolation by Distance
Restricted
Gene Flow
The OldestEvent Is APopulation
Range ExpansionOut of Africa
After Expansion Out ofAfrica, Gene Flow Was
Established Among All OldWorld Populations, AndA Secondary PopulationExpansion Occurred OutOf SE Asia/Indonesia,
But Without Replacementof Earlier Populations
História Evolutiva Humana