vÂnia maria teixeira carneiro - livros grátislivros01.livrosgratis.com.br/cp021557.pdf ·...
TRANSCRIPT

VÂNIA MARIA TEIXEIRA CARNEIRO
Síntese de piretróides e estudo de sua atividade inseticida Dissertação apresentada à Universidade
Federal de Viçosa, como parte das exigências do Programa de Pós-Graduação em Agroquímica, para obtenção do título de Magister Scientiae.
VIÇOSA MINAS GERAIS – BRASIL
2006

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

Ficha catalográfica preparada pela Seção de Catalogação e Classificação da Biblioteca Central da UFV
T Carneiro, Vânia Maria Teixeira, 1982- C289s Síntese de piretróides e estudos de sua atividade de inseticida 2006 / Vânia Maria Teixeira Carneiro. – Viçosa : UFV, 2006. xiii, 190f. : il. ; 29cm. Inclui anexos. Orientador: Elson Santiago Alvarenga. Dissertação (mestrado) - Universidade Federal de Viçosa. Referências bibliográficas: f. 101-104. 1. Piretróides - Síntese. 2. Inseticidas. 3. Química orgânica. 4. Fotoquímica. I. Universidade Federal de Viçosa. II.Título. CDD 22.ed. 668.651

VÂNIA MARIA TEIXEIRA CARNEIRO
Síntese de piretróides e estudo de sua atividade inseticida Dissertação apresentada à Universidade
Federal de Viçosa, como parte das exigências do Programa de Pós-Graduação em Agroquímica, para obtenção do título de Magister Scientiae.
APROVADA: 28 de julho de 2006
________________________________ Profa. Mayura M. Magalhães Rubinger (Co-Orientadora) ________________________________ Prof. Luciano Sindra Virtuoso
________________________________ Prof. Marcelo Coutinho Picanço (Co-Orientador) ________________________________ Profa. Vanderlúcia Fonseca de Paula
_______________________________ Prof. Elson Santiago de Alvarenga
(Orientador)

ii
AGRADECIMENTOS
Gostaria de agradecer aos meus pais Walter e Vitolina, pelo apoio em todos os
momentos da minha vida.
À Universidade Federal de Viçosa e ao Departamento de Química, pela
oportunidade de desenvolvimento desta dissertação.
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES),
pela concessão da minha bolsa de estudos.
Ao Prof. Elson Santiago de Alvarenga pela orientação e aos colegas de
Laboratório pela cooperação no decorrer deste trabalho.
À Profa. Mayura M. Magalhães Rubinger e ao Prof. Marcelo Coutinho Picanço
pelas sugestões e apoio durante o desenvolvimento deste trabalho.
À minha irmã Wilza por todo amor que ela tem por mim e, principalmente, por
ter me apoiado e ajudado durante o processo de escrita desta dissertação.

iii
BIOGRAFIA
Vânia Maria Teixeira Carneiro, filha caçula de Vitolina Maria Teixeira Carneiro
e Walter Cotta Carneiro, nasceu em 06 de fevereiro de 1982, na cidade de Alvinópolis
no estado de Minas Gerais.
Viveu seus primeiros 18 anos em Dom Silvério, cidade pequena pertencente à
Zona da Mata Mineira, onde estudou até concluir o ensino médio. Cursou ensino
fundamental na “Escola Estadual Nossa Senhora da Saúde” e o ensino médio na “Escola
Estadual Presidente Tancredo Neves”.
Ingressou em 2000 no curso de Química pela Universidade Federal de Viçosa,
graduando-se em julho de 2004.
Em agosto de 2004 foi admitida no programa de Pós-Graduação em
Agroquímica pela mesma universidade, na área de Síntese Orgânica, sob orientação do
professor Elson Santiago de Alvarenga. Em 28 de julho de 2006 teve sua dissertação
aprovada para a obtenção do título de Magister Scientiae.

iv
SUMÁRIO
AGRADECIMENTOS............................................................................................................................... ii
BIOGRAFIA.............................................................................................................................................. iii
LISTA DE ABREVIATURAS ................................................................................................................. vi
RESUMO .................................................................................................................................................viii
ABSTRACT............................................................................................................................................... xi
INTRODUÇÃO GERAL........................................................................................................................... 1
GENERALIDADES METODOLÓGICAS.............................................................................................. 7
TÉCNICAS EXPERIMENTAIS E INSTRUMENTOS........................................................................................... 7 Reator de UV de Alta Pressão............................................................................................................. 7 Reator de Luz UV de Baixa Pressão................................................................................................... 8 Temperaturas de Fusão....................................................................................................................... 8 Rotação Específica.............................................................................................................................. 8 Teste para Detecção de H2O2 .............................................................................................................. 8 Cromatografia em Camada Delgada (CCD)...................................................................................... 9 Separações Cromatográficas em Coluna............................................................................................ 9
TÉCNICAS ESPECTROSCÓPICAS................................................................................................................. 9 Espectroscopia no Infravermelho....................................................................................................... 9 Espectroscopia de Ressonância Magnética Nuclear.......................................................................... 9 Espectroscopia de Massas................................................................................................................. 10
TRATAMENTO DE SOLVENTES E REAGENTES.......................................................................................... 10 Secagem de Acetona.......................................................................................................................... 10 Secagem de Tetraidrofurano, Tolueno, Benzeno e Éter Dietílico ................................................... 10 Secagem de Diclorometano............................................................................................................... 11 Tratamento de Cloreto de Zinco....................................................................................................... 11 Tratamento de Furfural.................................................................................................................... 11
CAPÍTULO 1 - SÍNTESE DE PIRETRÓIDES..................................................................................... 12
1.1. INTRODUÇÃO ......................................................................................................................... 12 1.2. RESULTADOS E DISCUSSÃO................................................................................................ 17 1.3. CONCLUSÃO ........................................................................................................................... 30 1.4. EXPERIMENTAL ..................................................................................................................... 31
1.4.1. Síntese do metoxicarbonilmetileno(trifenil)fosforano...................................................... 31

v
1.4.2. Síntese dos Sais de Wittig S1-S5....................................................................................... 32 1.4.3. Síntese do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2)................................................... 35 1.4.4. Síntese do 2,3-O-isopropilideno-D-gliceraldeído (1.3)..................................................... 36 1.4.5. Síntese dos isômeros (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5)........................................................................... 36 1.4.6. Síntese do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropa- no-1-carboxilato de metila (1.6)................................................................................................................. 38 1.4.7. Síntese do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7)....... 39 1.4.8. Síntese do (1R,3R)-3-[(R)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopro-pano-1-carboxilato de metila (1.8)................................................................................................................. 40 1.4.9. Síntese do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9)....... 40 1.4.10. Síntese de piretróides......................................................................................................... 41 1.4.11. Isomerização Fotoquímica da mistura de E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14)......................................................................... 47
CAPÍTULO 2 - ADIÇÃO FOTOQUÍMICA DE ÁLCOOL ISOPROPÍL ICO COMO ETAPA INTERMEDIÁRIA NA SÍNTESE DE PIRETRÓIDES............ ........................................................... 48
2.1. INTRODUÇÃO ......................................................................................................................... 48 2.2. RESULTADOS E DISCUSSÃO................................................................................................ 55 2.3. CONCLUSÃO ........................................................................................................................... 65 2.4. EXPERIMENTAL ..................................................................................................................... 66
2.4.1. Síntese da 5H-furan-2-ona (2.2a)..................................................................................... 66 2.4.2. Síntese da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3)...................................... 67 2.4.3. Síntese da 4-isopropeniltetraidrofuran-2-ona (2.4a) e da 4-(1-metiletilideno)tetraidrofuran-2-ona (2.4b)....................................................................................... 67 2.4.4. Síntese da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) a partir da mistura de (2.4a) e (2.4b)..................................................................................................................................... 68 2.4.5. Síntese da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) a partir da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3).............................................................................................. 69 2.4.6. Síntese da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6)........................................... 70 2.4.7. Síntese da (S)-5-hidroximetil(5H)furan-2-ona (2.7)........................................................ 71 2.4.8. Síntese da (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8)............................. 71 2.4.9. Síntese da (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-metiletil) tetraidrofuran-2-ona (2.9)................................................................................................................. 72 2.4.10. Síntese da (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil) tetraidrofuran-2-ona (2.10). ............................................................................................................................................ 73 2.4.11. Síntese da (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)-tetraidrofuran-2-ona (2.11) ............................................................................................................................................ 74
CAPÍTULO 3 - OUTRAS REAÇÕES FOTOQUÍMICAS................................................................... 76
3.1. INTRODUÇÃO ......................................................................................................................... 76 3.2. RESULTADOS E DISCUSSÃO................................................................................................ 78 3.3. CONCLUSÃO ........................................................................................................................... 85 3.4. EXPERIMENTAL ..................................................................................................................... 86
3.4.1. Síntese do ácido (2E, 4E)-hexa-2,4-dienóico (3.2)........................................................... 86 3.4.2. Síntese do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3)........................................ 86 3.4.3. Síntese da 3-hidroximetil-2-metilciclobutan-1-ona (3.4)................................................. 87 3.4.4. Síntese do 5-hidroxi-4-oxopentanoato de metila (3.6)...................................................... 88
CAPÍTULO 4 – ATIVIDADE INSETICIDA DOS PIRETRÓIDES.. ................................................. 89
4.1. INTRODUÇÃO ......................................................................................................................... 89 4.2. RESULTADOS E DISCUSSÃO................................................................................................ 92 4.3. CONCLUSÃO ........................................................................................................................... 99 4.4. EXPERIMENTAL ................................................................................................................... 100
REFERÊNCIAS BIBLIOGRÁFICAS ................................................................................................. 101

vi
LISTA DE ABREVIATURAS
λ comprimento de onda
Bz benzila
BuLi butilítio
CCD cromatografia em camada delgada
CG cromatografia gasosa
COSY Correlated Spectroscopy
d dupleto
DCM diclorometano
dd dupleto duplo
DDT Dicloro difenil tricloroetano
δ deslocamento químico
DME dimetoxietano
DMS dimetilsulfóxido
ee excesso enatiomérico
EM espectro de massas
HETCOR Heteronuclear Chemical Shift Correlation
IV infravermelho
J constante de acoplamento
m multipleto
Me metil
Ph fenil
Rf fator de retenção
RMN de 13C Ressonância Magnética Nuclear de Carbono 13

vii
RMN de 1H Ressonância Magnética Nuclear de Hidrogênio
s simpleto
t tripleto
td tripleto duplo
THF tetraidrofurano
UV ultravioleta

viii
RESUMO
CARNEIRO, Vânia Maria Teixeira, M.Sc., Universidade Federal de Viçosa, julho de 2006. Síntese de piretróides e estudo de sua atividade inseticida. Orientador: Elson Santiago de Alvarenga. Co-Orientadores: Mayura Marques Magalhães Rubinger e Marcelo Coutinho Picanço.
O termo piretróide é usado para designar inseticidas sintéticos derivados
estruturalmente das piretrinas. Décadas de pesquisas feitas pela indústria agroquímica e
por laboratórios governamentais e acadêmicos têm resultado numa ampla mudança da
estrutura dos piretróides e numa multiplicidade de usos na agricultura, veterinária e
controle de pestes domésticas. Sabe-se que o uso prolongado de defensivos agrícolas
acarreta o desenvolvimento de mecanismos de resistência por várias espécies, além dos
efeitos tóxicos causados ao meio ambiente. Estas desvantagens justificam a necessidade
da busca por compostos mais específicos e com maior ação inseticida. Dessa forma, a
primeira parte deste trabalho teve como objetivo a síntese de piretróides contendo
variados grupos aromáticos em substituição ao grupo dimetilvinil presente na estrutura
de vários piretróides e piretrinas. A rota sintética mostrada no primeiro capítulo utilizou
como material de partida o D-manitol (1.1), que foi submetido a uma reação com
acetona em presença de cloreto de zinco, levando a formação do 1,2:5,6-di-O-
isopropilideno-D-manitol (1.2) em 87% de rendimento. O diacetal (1.2) foi clivado com
periodato de sódio para a formação do 2,3-O-isopropileno-D-gliceraldeído (1.3). O
aldeído (1.3) foi submetido à reação de Wittig utilizando-se
metoxicarbonilmetileno(trifenil)fosforano em metanol produzindo os ésteres (S)-(Z)-
4,5-O-isopropilidenopent-2-enoato de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-

ix
enoato de metila (1.5) (8 : 1) em 24% de rendimento a partir do diacetal (1.2). O éster
(1.4) foi utilizado na síntese do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.6) em 50% de rendimento, utilizando-se
sal de Wittig. O composto (1.6) foi hidrolisado com ácido perclórico em THF e clivado
com periodato de sódio originando o (1S,3S)-3-formil-2,2-dimetilciclopropano-1-
carboxilato de metila (1.7) em 80% de rendimento (e 40% de rendimento a partir de
1.4). Já o éster (1.5) foi submetido à mesma seqüência de reações do éster (1.4)
produzindo o (1R,3R)-3-[(R)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-
carboxilato de metila (1.8) e posteriormente o (1R,3R)-3-formil-2,2-
dimetilciclopropano-1-carboxilato de metila (1.9) em 10% de rendimento a partir de
(1.5). Finalmente, os piretróides (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.10), (1S,3S)-3-[2-(3-metoxifenil)eten-1-
il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.11), (1S,3S)-3-[2-(2-
clorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.12), (1S,3S)-3-
[2-(pentafluorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.13) e
(1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila
(1.14) foram produzidos por meio de reação de Wittig, a partir do aldeído (1.7) com
rendimentos de 46, 45, 64, 50 e 65%, respectivamente. Já o segundo capítulo trata de
uma rota alternativa para a síntese de piretróides tendo como etapa intermediária uma
reação fotoquímica de adição de álcool isopropílico. Esta rota utiliza o furfural (2.1)
como material de partida que foi inicialmente submetido a uma reação de oxidação
originando o 5H-furan-2-ona (2.2a) em 40% de rendimento. Este último foi irradiado
utilizando-se lâmpada de mercúrio de baixa pressão (λ = 254 nm) na presença de álcool
isopropílico, levando a formação da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3)
em 92% de rendimento. Este álcool foi desidratado utilizando-se pentacloreto de fósforo
em DCM originando os alquenos 4-isopropeniltetraidrofuran-2-ona (2.4a) e 4-(1-
metiletilideno)tetraidrofuran-2-ona (2.4b) em 90% de rendimento. Estes alquenos foram
submetidos à reação com tribrometo de fósforo em DCM na presença de óxido de
silício, produzindo o brometo 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) em
39% de rendimento. Este brometo foi tratado com tert-butóxido de potássio em THF
anidro, levando a formação da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6) em
40% de rendimento. A lactona (2.6) é um importante intermediário para síntese do ácido
trans-crisantêmico e vários piretróides. O éster (1.4) foi tratado com ácido sulfúrico em
metanol originando a (S)-5-hidroximetil(5H)furan-2-ona (2.7) em 73% de rendimento.

x
Posteriormente, o grupo hidroxila da lactona (2.7) foi protegido utilizando-se cloreto de
tert-butildimetilsilano em DCM na presença de imidazol, levando a formação do (S)-5-
O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8) em 60% de rendimento. As lactonas
(2.8) e (2.7) foram irradiadas na presença de álcool isopropílico produzindo os álcoois
(4S,5S)-5-O-tert-butildimetilsiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona
(2.9) e (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.10) em
66 e 90% de rendimento, respectivamente. O álcool (2.10) foi convertido na (4S,5S)-5-
benzoiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.11) em 90% de
rendimento, utilizando-se cloreto de benzoíla em piridina anidra. No Capítulo 3, são
mostrados os resultados da irradiação (λ = 254 nm) do éster (1.4) e do ácido (2E, 4E)-
hexa-2,4-dienóico (3.2) na presença de álcool isopropílico. Primeiramente o sorbato de
potássio (3.1) foi tratado com ácido clorídrico para formar o ácido (2E, 4E)-hexa-2,4-
dienóico (3.2) em 87% de rendimento. Este ácido foi dissolvido em álcool isopropílico e
irradiado por 96h, levando a formação do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico
(3.3) em 10% de rendimento. Este último foi reduzido com hidreto de lítio e alumínio
para formar o 3-hidroximetil-2-metilciclobutan-1-ona (3.4) em 74% de rendimento. A
irradiação do ácido (3.2) utilizando-se acetonitrila levou a formação do ácido (2Z,4E)-
hexa-2,4-dienóico (3.5). Após irradiação do éster (1.4), durante 1h, foi observada a
formação do 5-hidroxi-4-oxopentanoato de metila (3.6) em 85% de rendimento.
Finalmente, foram feitos ensaios biológicos com o objetivo de avaliar a ação inseticida
dos compostos (1.7), (1.9) a (1.14) e (2.6). Para realização destes testes foram utilizados
adultos de Acanthoscelides obtectus (Say) (Coleoptera: Bruchidae) e Sitophilus zeamais
Mots. (Coleoptera: Curculionidae), larvas de segundo instar de Ascia monuste orseis
(Godart) (Lepidoptera: Pieridae) e ninfas de segundo instar de Periplaneta americana
(L.) (Blattaria: Blattidae). Todos os piretróides testados apresentaram ação inseticida
significativa para as quatro espécies, 48h após sua aplicação.

xi
ABSTRACT
CARNEIRO, Vânia Maria Teixeira, M.Sc., Universidade Federal de Viçosa, july of 2006. Pyrethroids synthesis and study of its insecticide activity . Adviser: Elson Santiago de Alvarenga. Co-Advisers: Mayura Marques Magalhães Rubinger and Marcelo Coutinho Picanço.
The term pyrethroid is used to designate synthetic insecticides derived
structurally of pyrethrins. Decades of researches done by the industry agrochemistry and
for government and academic laboratories have been resulting in a wide change of
structure of pyrethroids and in a multiplicity of uses in the agriculture, veterinary and
control of domestic plagues. It is known that the prolonged use of agricultural
defensives takes the development of resistance mechanisms for several species, besides
the toxicant effects caused to the environment. These disadvantages justify the need of
search for agricultural chemicals more specific and with larger insecticide action. In that
way, the first part of this work had as objective the pyrethroids synthesis containing
several aromatic groups in substitution to the group dimethylvinyl presents in the
structure of several pyrethroids and pyrethrins. The synthetic route shown in the first
chapter used as first material D-mannitol (1.1), that was submitted to a reaction with
ketone in presence of zinc chloride, taking the formation of 1,2:5,6-di-O-
isopropylidene-D-mannitol (1.2) in 87% of yielding. The diacetal (1.2) was broken with
sodium periodate for the formation of 2,3-O-isopropylidene-D-glyceraldehyde (1.3).
The aldehyde (1.3) was submitted to the reaction of Wittig being used
methoxycarbonyl(triphenyl)phosphonium in methanol for the formation of esters methyl
(S)-(Z)-4,5-O-isopropylidenepent-2-enoate (1.4) and methyl (S)-(E)-4,5-O-

xii
isopropylidenepent-2-enoate (1.5) (8: 1) in 24% of yielding starting from the diacetal
(1.2). The ester (1.4) was used in the synthesis of methyl (1S,3S)-3-[(S)-2,2-dimethyl-
1,3-dioxolan-4-il]-2,2-dimethylcyclopropane-1-carboxylate (1.6) in 50% of yielding,
with Wittig salt. The product (1.6) was hydrolyzed with perchloric acid in THF and
broken with sodium periodate that resulted the methyl (1S,3S)-3-formil-2,2-
dimethylcyclopropane-1-carboxylate (1.7) in 80% of yielding (and 40% of yielding
starting from 1.4). The esters (1.5) was submitted to the same sequence of reactions of
esters (1.4) taking the formation of methyl (1R,3R)-3-[(R)-2,2-dimethyl-1,3-dioxolan-4-
il]-2,2-dimethylcyclopropane-1-carboxylate (1.8) and later on of methyl (1R,3R)-3-
formil-2,2-dimethylcyclopropane-1-carboxylate (1.9) in 10% of yielding starting from
(1.5). Finally, the pyrethroids methyl (1S,3S)-3-[2-(2-methoxyphenyl)eten-1-il]-2,2-
dimethylcyclopropane-1-carboxylate (1.10), methyl (1S,3S)-3-[2-(3-
methoxyphenyl)eten-1-il]-2,2-dimethylcyclopropane-1-carboxylate (1.11), methyl
(1S,3S)-3-[2-(2-chlorophenyl)eten-1-il]-2,2-dimethylcyclopropane-1-carboxylate (1.12),
methyl (1S,3S)-3-[2-(pentafluorophenyl)eten-1-il]-2,2-dimethylcyclopropane-1-
carboxylate (1.13) and methyl (1S,3S)-3-[2-(4-ethoxyphenyl)eten-1-il]-2,2-
dimethylcyclopropane-1-carboxylate (1.14) were produced through the reaction of
Wittig, starting from the aldehyde (1.7) with yielding of 46, 45, 64, 50 and 65%,
respectively. The second chapter is about an alternative route for pyrethroids synthesis
that has as intermediary stage a reaction photochemistry of photoadition of isopropylic
alcohol. This route uses the phurphural (2.1) as first material that was initially submitted
to an oxidation reaction to give the 5H-furan-2-one (2.2a) in 40% of yielding. This last
one was irradiated being used low-pressure mercury lamp (= 254 nm) in the presence of
isopropylic alcohol, resulting 4-(1'-hydroxy-1'-methylethyl)tetrahydrofuran-2-one (2.3)
in 92% of yielding. This alcohol was dehydrated being used phosphorus pentachloride
in DCM for formation of alkenes 4-isopropeniltetrahydrofuran-2-one (2.4a) and 4-(1-
methylethylideno)tetrahydrofuran-2-one (2.4b) in 90% of yielding. These alkenes were
submitted to the reaction with phosphorus tribromide in DCM in the presence of silicon
oxide for formation of bromide 4-(1'-bromide-1'-methylethyl)tetrahydrofuran-2-one
(2.5) in 39% of yielding. This bromide was treated with potassium tert-butoxide in THF
dry, taking the formation of 6,6-dimethyl-3-oxabicyclo[3.1.0]hexan-2-one (2.6) in 40%
of yielding. The lactone (2.6) is an important middleman for synthesis of acid trans-
chrysanthemic and several pyrethroids. The esters (1.4) was treated with sulfuric acid in
methanol for the formation of (S)-5-hydroxymethyl(5H)furan-2-one (2.7) in 73% of

xiii
yielding. Later on, the group hydroxyl of lactone (2.7) was protected being used tert-
butyldimethylsilyl chloride in DCM in the presence of the imidazole, resulting (S)-5-O-
tert-butyldimethylsiloxymethyl(5H)furan-2-one (2.8) in 60% of yielding. The lactones
(2.8) and (2.7) were irradiated in the presence of isopropylic alcohol taking the
formation of alcohols (4S,5S)-5-O-tert-butyldimethylsiloxymethyl-4-(1'-hydroxy-1'-
methylethyl)tetrahydrofuran-2-one (2.9) and (4S,5S)-5-hydroxymethyl-4-(1'-hydroxy-1'-
methylethyl)tetrahydrofuran-2-one (2.10) in 66 and 90% of yielding, respectively. The
alcohol (2.10) was transformed into the (4S,5S)-5-benzoiloxymethyl-4-(1'-hydroxy-1'-
methylethyl)tetrahydrofuran-2-one (2.11) in 90% of yielding, being used benzoil
chloride in pyridine dry. In the Chapter 3, the results of irradiation are shown (= 254
nm) of esters (1.4) and of (2E,4E)-hexa-2,4-dienoic acid (3.2) in the presence of
isopropylic alcohol. In the beginning, the potassium sorbate (3.1) was treated with
hydrochloric acid to form the (2E,4E)-hexa-2,4-dienoic acid (3.2) in 87% of yielding.
This acid was dissolved in isopropylic alcohol and irradiated by 96h, taking the
formation of 2-methyl-3-oxo-1-cyclobutanecarboxylic acid (3.3) in 10% of yielding.
This last one was reduced with lithium aluminum hydride to form the 3-hydroxymethyl-
2-methylcyclobutan-1-one (3.4) in 74% of yielding. The irradiation of acid (3.2) being
used acetonitrile took the formation of (2Z,4E)-hexa-2,4-dienoic acid (3.5). After
irradiation of esters (1.4), during 1h, was observed the formation of methyl 5-hydroxy-
4-oxopentanoate (3.6) in 85% of yielding. Finally, were made biological tests with the
objective of evaluating the insecticide action of compositions (1.7), (1.9) to (1.14) and
(2.6). Adults of Acanthoscelides obtectus (Say) (Coleoptera: Bruchidae) and Sitophilus
zeamais Mots. (Coleoptera: Curculionidae), larvas of Ascia monuste orseis (Godart)
(Lepidoptera: Pieridae) and nymphs of Periplaneta americana (L.) (Blattaria: Blattidae)
were used to carry out these tests. All pyrethroids, were tested, presented significant
insecticide action for the four species, 48h after its application.

-1-
INTRODUÇÃO GERAL
O mais antigo registro do uso de compostos químicos para o controle de pragas é
atribuído aos sumérios, em 2500 a.C., pela utilização de enxofre para combater insetos.
Mas foi a partir do século XIX que surgiram os primeiros estudos sistemáticos sobre o
uso de compostos químicos para o controle de pragas agrícolas, devido ao constante
desenvolvimento da agricultura. Os pesticidas utilizados nesta época eram, geralmente,
compostos inorgânicos ou extratos vegetais (BARBOSA, 2004).
Com o desenvolvimento da química orgânica sintética a partir de 1828, data da
primeira síntese de um composto orgânico (a uréia), várias metodologias têm sido
desenvolvidas para permitir a síntese de compostos orgânicos nos moldes dos
compostos naturais. Os pesticidas sintéticos foram amplamente usados a partir da
Segunda Guerra Mundial, quando, juntamente com o desenvolvimento de armas
químicas, ocorreu uma intensa busca por novos inseticidas que pudessem proteger as
tropas nas regiões de combate que eram sujeitas à ação de insetos causadores de
doenças. A descoberta da atividade inseticida do DDT em 1939, por Muller, contribuiu
bastante para a larga aplicação dos pesticidas sintéticos (BARBOSA, 2004).
O “piretro” é o nome dado ao inseticida natural obtido na forma de extrato ou
como pó de flores secas de plantas do gênero Chrysanthemum (recentemente
denominado Tanacetum). Acredita-se que o uso do piretro como inseticida teve origem
na Pérsia (atual Irã) e que a fonte mais antiga conhecida deste inseticida natural é o C.
coccineum. Posteriormente a espécie C. cinerariaefolium passou a ser mais
eficientemente usada como fonte deste inseticida. O C. cinerariaefolium cresce
naturalmente na costa da Dalmatia, atual Sérvia e Montenegro. Esta planta foi

-2-
introduzida no Japão em 1881, tendo este país se tornado o principal produtor de piretro
no período que vai da Primeira à Segunda Guerra Mundial. A partir de 1920, plantações
de C. cinerariaefolium foram introduzidas na França, Inglaterra e Quênia. Com o
aumento do consumo deste inseticida durante a Segunda Guerra, a produção de piretro
foi estendida para as terras-altas da Tanzânia e sudeste de Uganda. Plantações de C.
cinerariaefolium foram introduzidas em muitos outros países tais como Ruanda,
Equador, Índia, Zaire, Nepal, China, Brasil e Tasmânia na Austrália (WANDAHWA et
al., 1996).
Os compostos responsáveis pela ação inseticida do piretro são seis ésteres
conhecidos conjuntamente como piretrinas. Os quatro principais ingredientes ativos do
piretro são as piretrinas I e II e cinerinas I e II. Também estão presentes no piretro,
pequenas quantidades de jasmolinas I e II. Os compostos designados por I são derivados
do ácido crisantêmico enquanto os compostos designados por II são derivados do ácido
pirétrico. Entre estes compostos a piretrina I é o componente que apresenta maior ação
inseticida. As estruturas e proporções relativas destes seis ésteres são mostradas na
Tabela 1 (NAUMANN, 1990).
Tabela 1: Estruturas e proporções relativas das piretrinas presentes no piretro natural (NAUMANN, 1990).
Grupo alquila (R)
O
O
O
O
O
O
COR
R = OH → Ácido Crisantêmico
Piretrina I 35 %
Jasmolina I 5 %
Cinerina I 10 %
CORCH3OOC
R = OH → Ácido Pirétrico
Piretrina II 32 %
Jasmolina II 4 %
Cinerina II 14 %

-3-
As piretrinas são bastante tóxicas contra um grande número de espécies de insetos
sendo praticamente inofensivas a animais de sangue quente e ao homem. Outra importante
característica destes compostos é o efeito “Knock-down” (que pode ser traduzido como
efeito de “choque”), que significa que o inseto é derrubado quase instantaneamente ao ser
exposto ao inseticida. Como as piretrinas possuem baixa estabilidade na presença de luz e
oxigênio, elas não são utilizadas de maneira eficiente para controle de pragas em lavouras,
restringindo seu uso a ambientes fechados (HASSAL, 1990).
A partir de 1970, com o desenvolvimento de inseticidas sintéticos mais estáveis
estruturalmente relacionados às piretrinas, a produção e a utilização do piretro diminuíram
significativamente. Atualmente, o Quênia é o maior produtor de piretro, sendo responsável
por aproximadamente 70% da produção mundial. Outros países que ainda se destacam na
produção deste inseticida são Ruanda, Tanzânia e Tasmânia, sendo responsáveis por quase
30% da produção mundial. O piretro produzido nestes países é exportado para os Estados
Unidos, Europa e Ásia (PKF Consulting Ltd, 2005).
Os análogos sintéticos das piretrinas são conhecidos como piretróides. Décadas de
pesquisas feitas pela indústria agroquímica e por laboratórios governamentais e acadêmicos
têm resultado numa ampla mudança estrutural dos piretróides e numa multiplicidade de
usos na agricultura, veterinária e controle de pestes domésticas (SODERLUND et al.,
2002).
O primeiro piretróide a ser sintetizado foi a aletrina (Figura 1), produzida em 1949
por Schechter na forma de uma mistura de oito isômeros derivados dos ácidos cis e trans
crisantêmicos. Esta mistura de isômeros apresenta toxidade semelhante à das piretrinas
naturais, mas um baixo efeito “Knock-down”. Em 1965, Elliott e colaboradores
sintetizaram a bioresmetrina (Figura 1) que é aproximadamente 100 vezes mais tóxica para
moscas que os ésteres naturais. Um aspecto marcante deste trabalho foi que a síntese da
bioresmetrina teve como objetivo principal mimetizar a geometria (forma tridimencional)
da piretrina I (NAUMANN, 1990).
A partir desta data, muitos grupos de pesquisa tiveram como objetivo a síntese de
uma grande variedade de álcoois para serem ligados a derivados do ácido
ciclopropanocarboxílico presente nas piretrinas naturais. Em 1972, alguns trabalhos
mostraram que eram formados ésteres bastante estáveis em condições atmosféricas ao ligar
um álcool apropriado a análogos diclorovinil do ácido ciclopropanocarboxílico. Um
exemplo importante deste tipo de substância é a permetrina (Figura 1), cuja descoberta foi
um ponto de mudança na química de piretróides. A permetrina não possui efeito tão rápido

-4-
como os compostos naturais, mas é altamente tóxica a insetos e muito estável na presença
de luz, sendo o primeiro piretróide suficientemente fotoestável para uso na agricultura. Na
mesma época, japoneses prepararam piretróides sem o anel ciclopropano, cujo
representante mais bem sucedido desta nova classe foi o fenvalerato (Figura 1). Este último,
também possui um grupo α-ciano em sua estrutura da mesma forma que dois outros
importantes piretróides estáveis à luz: cipermetrina e deltametrina, cujas estruturas estão
também representadas na Figura 1 (HASSAL, 1990).
O
O
O
H
Aletrina
DeltametrinaCipermetrina
Fenvalerato
Bioresmetrina
O
O
O
Permetrina
O
OO
Cl
Cl
O
OO
CNCl
Cl
Cl
O
OO
CNBr
Br
O
O
H CNO
Figura 1: Estruturas de alguns piretróides importantes.
É interessante resaltar que a presença do grupo α-ciano altera de modo significante
algumas respostas tóxicas desenvolvidas por organismos vertebrados e invertebrados. Em
1981, Gammon e colaboradores dividiram os piretróides em duas classes: piretróides tipo I
– incluindo piretrinas e piretróides que não possuem grupo α-ciano em sua estrutura;
piretróides tipo II – piretróides que possuem grupo α-ciano (HASSAL, 1990).
De modo geral, como o desenvolvimento de piretróides envolve um interativo
processo de modificação estrutural e avaliação biológica, muitos inseticidas identificados
como piretróides são bastante diferentes estruturalmente dos inseticidas naturais
(SODERLUND et al., 2002).
Os piretróides, da mesma forma que o DDT e análogos, são compostos
neurotóxicos que se ligam aos canais de sódio, presentes nas membranas dos neurônios,
alterando a transmissão de impulsos elétricos ao longo do axônio (HASSAL, 1990).

-5-
Estudos da relação estrutura-atividade indicam que a efetiva ação inseticida dos piretróides
está altamente relacionada à forma tridimensional da molécula como um todo. Assim, ao
mesmo tempo em que quase todas as partes da molécula molde (geralmente piretrina I)
podem ser substituídas simultaneamente por unidades estruturais análogas sem perda da
atividade inseticida, outras mudanças aparentemente menos drásticas podem levar à
produção de compostos inativos. Sabe-se que algumas características estruturais estão
relacionadas com a atividade inseticida, como por exemplo: cadeia lateral insaturada; a
presença de um grupo gem-dimetil na parte ácida da molécula; a estereoquímica de C1 e C3
em moléculas contendo anel ciclopropano; presença de uma ligação éster central ou análoga
(ligação éter no caso do Etofenprox mostrado na Figura 2); ponte de metileno ou análogo
separando uma região planar de uma região terminal insaturada (ELLIOTT, 1978). A
Figura 2 resume a dependência da atividade inseticida com a estrutura dos piretróides
(SODERLUND et al., 2002).
O
O
O
H
Fenvalerato
Bioresmetrina
O
O
O
Piretrina I
O
OO
CNCl
EtofenproxO
O
C2H5O
cadeia lateral insaturada(ou equivalente) grupo dimetil
geminal
ligação éster central(ou equivalente)
região planar ponte metileno(ou equilavente)
centro de insaturação não coplanar
Figura 2: Características estruturais dos piretróides relacionadas com a atividade, ilustradas pela Piretrina I e três piretróides sintéticos (SODERLUND et al., 2002).

-6-
Sabe-se que o uso prolongado de defensivos agrícolas acarreta o desenvolvimento
de mecanismos de resistência por várias espécies de insetos-praga, sem contar os efeitos
tóxicos causados ao meio ambiente quando resíduos destes defensivos contaminam rios e
lagoas (MARTIN et al., 2002). Estas desvantagens justificam a necessidade da constante
busca por compostos mais específicos e com maior ação inseticida. Dessa forma, a
primeira parte deste trabalho (Capítulo 1) tem como objetivo a síntese de piretróides
contendo variados grupos aromáticos em substituição ao grupo dimetilvinil ligado ao anel
ciclopropano presente nas piretrinas e em vários piretróides. Já o Capítulo 2 trata de uma
rota alternativa para síntese de piretróides, utilizando-se como etapa intermediária uma
reação de adição fotoquímica de álcool isopropílico à γ-lactonas α,β-insaturadas. O
Capítulo 3 mostra alguns resultados obtidos após tentativa de adição fotoquímica de
álcool isopropílico a ésteres α,β-insaturados. Finalmente, como discutido no Capítulo 4,
foram feitos ensaios biológicos com o objetivo de avaliar a ação inseticida de alguns
compostos sintetizados nos capítulos 1 e 2.

-7-
GENERALIDADES METODOLÓGICAS
Técnicas Experimentais e Instrumentos
Reator de UV de Alta Pressão
A reação de isomerização fotoquímica discutida no capítulo 1 foi feita
utilizando-se uma lâmpada de mercúrio de alta pressão de 125 W de potência (lâmpada
de mercúrio para iluminação pública da qual se retirou o bulbo externo de proteção) e
um reator de Pirex para acondicionar a amostra contendo sistema de resfriamento por
fluxo de água fria. Veja na Figura 3 as fotos da montagem:
(a)
(b)
Figura 3: (a) Foto do recipiente de Pirex utilizado para acondicionar a amostra durante a reação. (b) Foto da lâmpada de iluminação pública sem o bulbo externo.

-8-
Reator de Luz UV de Baixa Pressão
O reator de luz UV utilizado durante as reações fotoquímicas discutidas nos
capítulos 2 e 3 foi confeccionado utilizando-se uma caixa de madeira hexagonal de 60
cm de altura, contendo, em seu interior, quatro lâmpadas de mercúrio de baixa pressão
(15W, λ= 254nm) igualmente distanciadas. Esta caixa possui uma abertura na parte
superior para inserção de um tubo de quartzo contendo a mistura reacional, que deverá
ser suspenso por um suporte. As fotos do reator estão mostradas na Figura 4:
(a)
(b)
(c)
Figura 4: (a) Foto externa do reator. (b) Foto interna do reator. (c) Foto da abertura superior do reator.
Temperaturas de Fusão
As temperaturas de fusão dos compostos sólidos foram determinadas utilizando-
se o aparelho MQAPF-301.
Rotação Específica
As medidas de rotação específica foram feitas no polarímetro Bellingham +
Stanley Model D, do Departamento de Química da UFV. A concentração (c) das
amostras foi dada em g/100 mL.
Teste para Detecção de H2O2
Este teste foi realizado durante a primeira síntese mostrada no capítulo 2.
Utilizou-se dois tubos de ensaio (tubo 1 e tubo 2), onde foram adicionadas soluções de
sulfato ferroso amoniacal 1% (5 mL), H2SO4 0,5 mol.L-1 (0,5 mL) e tiocianato de
potássio 0,1 mol.L-1 (0,5 mL). Ao tubo 1 adicionou-se 5 mL de DCM puro e ao tubo 2
adicionou-se o mesmo volume da solução a ser testada quanto a presença de peróxido

-9-
de hidrogênio. Ambos os tubos foram agitados vigorosamente por 1 min. e os resultados
interpretados como descrito abaixo:
� Solução do tubo 2 apresentando cor amarela igual a solução do tubo 1 →
resultado negativo para presença de H2O2;
� Solução do tubo 2 apresentando cor vermelha → resultado positivo para
presença de H2O2.
Cromatografia em Camada Delgada (CCD)
Para cromatografia em camada delgada (CCD) analítica, foram utilizadas placas
de sílica poligrama / UV254 (MACHEREY – NAGEL).
As placas cromatográficas foram previamente observadas sob lâmpada de UV
(λ= 254 nm) antes de serem reveladas com solução de permanganato de potássio (3 g de
KMnO4, 20 g de K2CO3, 5 ml de NaOH 5% e 300 mL de H2O destilada) ou de ácido
fosfomolíbdico (12 g de 2H3PO4.20MoO3.48H2O em 250 mL de etanol).
Separações Cromatográficas em Coluna
As separações cromatográficas em coluna foram feitas utilizando-se sílica-gel
(70-230 Mesh, VETEC) e eluente apropriado. O material, quando sólido, foi
previamente incorporado à sílica em evaporador rotatório ou, quando líquido,
adicionado com auxílio de pipeta pelo topo da coluna previamente empacotada. As
frações coletadas foram analisadas por meio de CCD.
Técnicas Espectroscópicas
Espectroscopia no Infravermelho
Os espectros no Infravermelho foram obtidos em espectrofotômetro PERKIN
ELMER FT-IR 1000, pertencente ao Departamento de Química da UFV. As amostras
sólidas foram analisadas em pastilhas de KBr a 1% e as amostras oleosas como filme
sobre cristal de KBr, NaCl ou CsI.
Espectroscopia de Ressonância Magnética Nuclear
Os espectros de Ressonância Magnética Nuclear foram registrados em
espectrômetro VARIAN MERCURY (300 MHz), pertencente ao Departamento de
Química da UFV. Clorofórmio e metanol deuterados e tetracloreto de carbono foram

-10-
utilizados como solventes. TMS foi utilizado como padrão interno de referência (δ = 0).
As constantes de acoplamento (J) foram expressas em Hertz (Hz) e os deslocamentos
químicos em δ.
Espectroscopia de Massas
Os espectros de massas foram obtidos em espectrômetro GC-MS SHIMADZU
QP5050A, pertencente ao Departamento de Química da UFV. As amostras obtidas na
forma de misturas de isômeros geométricos (Capítulo 1) foram separadas com o uso de
coluna capilar (25 m x 0,25 mm) DB-5. A temperatura do injetor e da coluna foi fixada
em 250 °C. Hélio foi utilizado como gás carreador à pressão de 173,4 kPa e a vazão da
coluna foi de 2,5 mL/min.
Tratamento de Solventes e Reagentes
Secagem de Acetona
A acetona (600 mL) foi mantida sob refluxo na presença de carbonato de
potássio anidro (40 g, 7 % m/v) por 3 h. Em seguida, o solvente foi destilado e
armazenado em frascos contendo peneira molecular 4 Å tampados com septo de
borracha.
O carbonato de potássio anidro foi obtido através de secagem em mufla a 200 °C
durante 6 horas.
Secagem de Tetraidrofurano, Tolueno, Benzeno e Éter Dietílico
O tetraidrofurano (600 mL) foi mantido sob refluxo na presença de sódio
metálico (3 g, 0,5 % m/v) por 16 h. Adicionou-se benzofenona (100 mg) e manteve-se o
sistema sob refluxo até a mistura apresentar coloração azul escura. Em seguida, o THF
foi destilado e armazenado em frascos contendo peneira molecular 4 Å tampados com
septo de borracha.
Tolueno, benzeno e éter dietílico foram secados utilizando-se o mesmo
procedimento descrito para secagem de THF, sendo necessárias apenas 4 h de refluxo
na presença do sódio.

-11-
Secagem de Diclorometano
O diclorometano (600 mL) foi mantido sob refluxo na presença de hidreto de
cálcio (3 g, 0,5 % m/v) por 3 h. Em seguida, o DCM foi destilado e armazenado em
frascos contendo peneira molecular 4 Å tampados com septo de borracha.
Tratamento de Cloreto de Zinco
O cloreto de zinco comercial, muitas vezes contaminado com óxido de zinco, foi
dissolvido em ácido clorídrico concentrado (1 mL por g de ZnCl2) e a solução aquecida
até total eliminação de vapor de água e de HCl(g). Em seguida, o ZnCl2 foi transferido
para um frasco apropriado e mantido em dessecador.
Tratamento de Furfural
O furfural (100 g) foi destilado sob vácuo (30 mm) e agitação magnética em
banho de óleo (100º C) na presença de Na2CO3 (7 % m/m) obtendo-se um líquido
incolor.

-12-
CAPÍTULO 1 - SÍNTESE DE PIRETRÓIDES
1.1. INTRODUÇÃO
A elucidação da estrutura dos componentes do “piretro” e a observação da
superior ação inseticida da piretrina I inspiraram a síntese de compostos estruturalmente
relacionados a este produto natural, derivados do ácido trans-crisantêmico. Um
exemplo deste tipo de piretróide é a bioresmetrina, sintetizada em 1965 por Elliott e
colaboradores, cuja toxidade é superior à das piretrinas naturais (ELLIOTT et al., 1965).
As estruturas da bioresmetrina e piretrina I estão representadas abaixo:
O
O
O
HPiretrina I
BioresmetrinaO
O
O
Como o ácido trans-crisantêmico é um importante intermediário para a síntese
dos piretróides, várias metodologias foram desenvolvidas para a síntese deste composto.
Uma metodologia que se destacou foi o emprego de ilídeos de enxofre e fósforo em
reações com compostos contendo dupla ligação conjugada a um grupo carbonílico.

-13-
Em 1967, Corey observou que ao reagir o ilídeo de enxofre
isopropilidenodifenilsulfônio com compostos carbonílicos α,β-insaturados ocorria
adição do grupo isopropilideno á dupla ligação. Assim, ele utilizou esta metodologia
para a síntese do ácido (+)-trans-crisantêmico a partir do (E)-5-metilexa-2,4-dienoato de
metila como mostrada na Figura 1.1. Neste caso, o (E)-5-metilexa-2,4-dienoato de
metila reagiu com o isopropilidenodifenilsulfônio em dimetoxietano (-70°C a -20°C),
ocorrendo adição do grupo isopropil aos átomos de carbono da dupla ligação mais
próxima ao grupo éster. Esta reação ocorreu com rendimento de 72% e levou a
formação de uma mistura racêmica dos ésteres (+)-trans-crisantematos de metila. Em
seguida, a hidrólise alcalina dos ésteres levou a formação da mistura dos ácidos (+)-
trans-crisantêmicos (COREY, 1967).
CO2CH3
CO2CH3
-70 °C a -20 °C72 %
Ph2S C(CH3)2, DME
CO2H
ácido (+)-trans-crisatêmico
H2O / OH-
+ enantiômero
+ enantiômero
Figura 1.1: Síntese do ácido (+)-trans-crisantêmico utilizando ilídeo de enxofre.
Posteriormente, Krief estudou o emprego dos ilídeos de enxofre
(isopropilidenodifenilsulfônio) e de fósforo (isopropilidenotrifenilfosfônio) para
construção de anéis dimetilciclopropanos utilizando-se ésteres γ-alcóxi-α,β-insaturados.
Ele observou a estereoespecificidade da reação destes ésteres com os ilídeos de enxofre,
onde a olefina na configuração Z leva a formação do ciclopropano cis-substituído e a
olefina na configuração E leva a formação do ciclopropano trans-substituído. Estes
estudos mostraram também a estereosseletividade da reação com ilídeo de fósforo, onde
as olefinas tanto na configuração Z quanto na E levam a formação de ciclopropanos
trans-substituídos (KRIEF, 1989). A Figura 1.2 mostra um resumo dos resultados
obtidos para estas reações.

-14-
O
O CO2MeO
OCO2Me
O
O
CO2Me
O
O
CO2MeO
O
CO2Me
O
O
CO2Me
OHC CO2Me
S Ri) HClO4, THFii) NaIO4, MeOH
60%ee 96%
Ph3P=C(CH3)2, THF
61%0 °C a 20 °C
Ph2S=C(CH3)2, DME -78 °C a -50 °C
84%
i) HClO4, THFii) NaIO4, MeOH
63%ee 98%
OHC CO2Me
SS
Ph2S=C(CH3)2, DME -78 °C a -50 °C
92%
Ph3P=C(CH3)2, THF
55%0 °C a 20 °C
i) HClO4, THFii) NaIO4, MeOH
63%ee 97%
OHC CO2Me
R Ri) HClO4, THFii) NaIO4, MeOH
66%ee 71%
OHC CO2Me
SS
Figura 1.2: Formação de anel dimetilciclopropano pelo uso de ilídeos de enxofre e fósforo.
Os mecanismos mostrados na Figura 1.3, propostos por Krief, explicam a
estereoquímica dos produtos formados pela reação entre o ilídeo de enxofre e os ésteres
Z e E γ-alcóxi-α,β-insaturados. Neste caso, o ataque do ilídeo de enxofre ocorre pela
face Re do carbono β para ambos os ésteres E e Z. Consequentemente, forma-se um
intermediário contendo um anel de quatro átomos. Este intermediário se decompõe,
levando a formação do produto contendo um anel dimetilciclopropano pela eliminação
do Ph2S. Esta reação apresenta elevada estereoespecificidade para ambos isômeros Z e
E, produzindo anéis ciclopropano com configurações cis e trans, respectivamente
(KRIEF, 1989).

-15-
Re
Z
O
O CO2CH3
H
C SPh2
H3C
H3C
- Ph2S
O
O
CO2CH3
H
CS
Ph
CH3
H3C
Ph
O
O
CO2CH3
H
E
C SPh2
H3C
H3C
Re- Ph2S
O
O
H
CO2CH3
SS- 78 °C a - 50 °C
DME
- 78 °C a - 50 °CDME
O
O CO2CH3
H
CS
Ph
CH3
H3C
Ph
O
O
H
CO2CH3
SR
Figura 1.3: Mecanismo da reação de ciclopropanação utilizando-se ilídeo de enxofre.
Os mecanismos para as reações entre o ilídeo de fósforo e os ésteres Z e E γ-
alcóxi-α,β-insaturados são mostrados na Figura 1.4. O ataque do ilídeo de fósforo
ocorre pela face Re do carbono β para o éster contendo a dupla ligação conjugada na
configuração Z e pela face Si do carbono β para o éster contendo a dupla ligação
conjugada na configuração E. O intermediário formado após ataque do ilídeo de fósforo
ao carbono β possui cadeia aberta, permitindo rotação da ligação Cα-Cβ, ao contrário
do intermediário formado após ataque do ilídeo de enxofre. A natureza do intermediário
formado explica o fato de ambos isômeros E e Z levarem a formação de produtos
contendo anel ciclopropano com configuração trans (KRIEF, 1989).

-16-
Re
Z
O
O CO2CH3
H
C PPh3
H3C
H3C
O
OC
H
CCH3
CH3 P
Ph PhPh
OCH3
OO
O
C
H
CCH3
CH3P
Ph PhPh
OCH3
O
O
O
H
CO2CH3
SS
SiO
O
CO2CH3
H C
PPh3
CH3
H3CE
- PPh3
THF, 0 °C
O
O
C
H
O
OCH3
C
CH3
CH3
P
Ph
Ph
Ph
- PPh3
O
O
H
CO2CH3
RR
THF, 0 °C
Figura 1.4: Mecanismo da reação de ciclopropanação utilizando-se ilídeo de fósforo.
Neste capítulo será discutida a síntese de piretróides derivados do ácido trans-
crisantêmico contendo grupos aromáticos no lugar do grupo dimetilvinil (presente na
piretrina I e em muitos piretróides) como mostrado na estrutura abaixo. A etapa chave
desta rota sintética será a utilização da reação entre o ilídeo isopropildimetilfosforano e
um éster γ-alcóxi-α,β-insaturado obtido a partir do D-manitol.
CO2CH3R1
R2 Sendo R1 = H e R2 = Ar ou R1 = Ar e R2 = H.

-17-
1.2. RESULTADOS E DISCUSSÃO
Na rota sintética apresentada na Figura 1.5 utilizou-se D-manitol (1.1) como
material de partida para produção dos ésteres (S)-(Z)-4,5-O-isopropilidenopent-2-enoato
de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5).
CO2CH3
OO
OO
CHO
H
HO
OH
HO
HO
OH
OH
O
O
O
O
OH
HO
(1.1) (1.2)
(1.3)
(1.4)
CO2CH3
OO
(1.5)
+
acetona, ZnCl2
87%
NaIO4 Ph3PCHCO2Me
MeOH48%
50%
Figura 1.5: Síntese dos ésteres (1.4) e (1.5) a partir do D-manitol (1.1).
Utilizou-se reação entre D-manitol (1.1) e acetona anidra para síntese do 1,2:5,6-
di-O-isopropilideno-D-manitol (1.2) em 87% de rendimento. Esta reação foi catalisada
por cloreto de zinco, empregando procedimento modificado de Baer (BAER, 1945).
Primeiramente dissolveu-se o catalisador em acetona, formando uma solução
transparente. Em seguida adicionou-se D-manitol (1.1) formando uma suspensão, pois
este reagente é pouco solúvel em acetona. Como o produto (1.2) é bastante solúvel neste
solvente, o término da reação foi determinado pela total dissolução do soluto
inicialmente em suspensão. A reação foi encerrada por meio de neutralização com
solução aquosa de carbonato de potássio.
Durante a elaboração desta reação, observou-se hidrólise do produto (1.2) em
quantidades significativas ao evaporar a acetona e a água utilizando-se evaporador
rotatório a 40 °C. Para evitar hidrólise do diacetal utilizou-se fluxo de ar comprimido
para evaporação da acetona e parte da água. Em seguida, o resíduo foi extraído com éter
dietílico que foi evaporado sob pressão reduzida à temperatura ambiente, originando um
sólido branco contendo o diacetal (1.2) e resíduos de triacetais. O sólido branco foi
lavado com hexano, solvente no qual os triacetais foram altamente solúveis, originando
o produto (1.2) em 87% de rendimento, com pureza suficiente para ser utilizado na
próxima etapa da rota. A Tabela 1.1 mostra uma comparação dos resultados obtidos a
partir de diversos procedimentos encontrados na literartura para obtenção de (1.2).

-18-
Tabela 1.1: Resumo dos procedimentos (em ordem cronológica) para obtenção de 1,2:5,6-di-O-isopropilideno-D-mannitol (1.2) e seus respectivos rendimentos.
Reagentes Tempo de reação
Método de Purificação Rendimento Referências
D-manitol, ZnCl2, acetona (1:8:94)
18-19 h
Recristalização com éter de petróleo (60-800C).
54,8% BAER, 1939.
D-manitol, ZnCl2, acetona (1:2:25)
2 h
Recristalização com éter n-butílico (1350C).
42% BAER, 1945.
D-manitol, 2,2-dimetoxipropano, 1,2-dimetoxietano, SnCl2.
35 min (refluxo)
Recristalização com éter n-butílico (1350C).
54-58 % CHITTENDEN,
1980.
D-manitol, 2,2-dimetoxipropano, 1,2-
dimetoxietano.
20-24 h (refluxo)
Recristalização com éter n-butílico (1350C).
52 % CHITTENDEN,
1980.
D-manitol, 2-metoxipropeno, N,N-
dimetilformamida, ácido p-toluenosulfônico.
3-4 h
92%
36%
40%
DEBOST et al., 1983.
KUSZMANN et al., 1984.
CHITTENDEN, 1991.
D-manitol, ZnCl2, acetona (1:2:8)
18 h
Recristalização com tolueno (1100C).
60-61% MORPAIN et al.,
1990.
D-manitol, 2,2-dimetoxipropano, 1,2-dimetoxietano, SnCl2.
1h (refluxo)
Lavagem com diclorometano.
54 % SCHMID, 1993.
D-manitol, ZnCl2, acetona (1:3:39)
5 h Lavagem com Hexano. 87% *
* procedimento descrito neste trabalho.
Para testar a estabilidade do diacetal (1.2) foram mantidos em repouso e à
temperatura ambiente (25 °C) três soluções deste composto (50 mg/mL): uma em éter
dietílico, outra em acetona e a última em diclorometano. Não foram observados sinais
de decomposição do material em éter ou acetona durante 30 dias. Já a solução em
diclorometano apresentou-se turva após 3 dias, sendo um claro indício de
decomposição. A instabilidade da última solução pode ser explicada pela acidez do
diclorometano levando à hidrólise dos grupos acetal e conseqüente precipitação do D-
manitol (1.1) que é insolúvel neste solvente.
Observou-se que o aquecimento do produto, por alguns minutos, a 40 °C leva a
decomposição de quantidades consideráveis do diacetal (1.2). Desta forma, uma
provável explicação para os menores rendimentos (42-61%) descritos na literatura para
esta reação entre D-manitol e acetona na presença de ZnCl2 é devido a hidrólise do

-19-
produto durante a etapa de recristalização, que utiliza aquecimento do material em
solução, como indicado na Tabela 1.1.
O espectro no IV do diacetal (1.2), Anexo 1, mostra uma banda larga em ν
3436 cm-1 referente ao estiramento das duas ligações O-H. Os sinais no espectro de
RMN de 1H (Anexo 2) do produto foram completamente atribuídos com o auxílio da
técnica de dupla irradiação e de NOEDIF. Os simpletos em δ 1,36 e δ 1,42 integrados
para 6H cada e atribuídos aos grupos CH3’ e CH3, respectivamente, e o dupleto em δ
2,66 integrando para 2H, atribuídos aos grupos hidroxila são fortes indícios da formação
do produto esperado. As atribuições dos sinais no espectro de RMN de 13C mostrado no
Anexo 3 foram confirmadas por meio de HETCOR. Os sinais em δ 25,7 e δ 27,2
atribuídos respectivamente aos grupos CH3’ e CH3 e o sinal em δ 109,6 atribuído aos
átomos de carbono não hidrogenados C(CH3)2, confirmam a formação do diacetal (1.2).
O 2,3-O-isopropileno-D-gliceraldeído (1.3) foi preparado a partir da clivagem
oxidativa do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2) utilizando-se 2 equi. molares
de metaperiodato de sódio em DCM, na presença de solução saturada de bicarbonato de
sódio (SCHMID & BRYANT, 1993). Utilizou-se solução de bicarbonato de sódio para
minimizar a formação do hidrato a partir do aldeído (1.3), altamente solúvel em água.
Devido a sua baixa estabilidade, o aldeído (1.3) foi utilizado na próxima etapa da síntese
imediatamente após sua preparação, sem ser purificado.
Os ésteres (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4) e (S)-(E)-
4,5-O-isopropilidenopent-2-enoato de metila (1.5) foram preparados por meio de reação
de Wittig entre o aldeído 2,3-O-isopropiledeno-D-gliceraldeído (1.3) e
metoxicarbonilmetileno(trifenil)fosforano em metanol (MANN et al., 1987). Estes
isômeros (1.4) e (1.5) foram obtidos na proporção de 8 : 1, em 24% de rendimento a
partir do diacetonídeo (1.2). Esta reação foi conduzida em banho de gelo, sob controle
cinético, favorecendo a formação do produto cis menos estável. Os isômeros foram
separados por cromatografia em coluna, caracterizados e utilizados nas próximas
sínteses.
No espectro no IV do composto (1.4) dado no Anexo 4 pode-se notar a presença
de uma banda intensa em 1726 cm-1 referente ao estiramento da ligação C=O e outra
banda em 1646 cm-1 referente ao estiramento da ligação C=C. No espectro de RMN de 1H (Anexo 5) o simpleto em δ 3,70 integrado para 3H e atribuído ao grupo OCH3 é um
indício da formação do éster (1.4). Neste espectro observa-se um dupleto duplo em δ

-20-
5,84 atribuído a H2 e originado pelo acoplamento deste com H3 (J2-3 = 11,7 Hz) e H4
(J2-4 = 1-8 Hz) e um dupleto duplo em δ 6,35 atribuído a H3 e originado do
acoplamento deste com H2 (J3-2= 11,7 Hz) e H4 (J3-4= 6,6 Hz). As constantes de
acoplamento entre os átomos de hidrogênio olefínicos H2 e H3 estão de acordo com os
valores descritos na literatura para isômeros Z, confirmando a geometria do produto
(1.4) (SILVERSTEIN & WEBSTER, 2000). No espectro de RMN de 13C (Anexo 6)
observa-se o sinal em δ 51,7 atribuído ao carbono da metoxila e os sinais em δ 120,5
e δ 149,7 atribuídos a C2 e C3 respectivamente, que confirmam a formação deste éster
α,β-insaturado (1.4).
No espectro no IV do isômero (1.5) (Anexo 7) pode-se notar a presença de uma
banda intensa em 1730 cm-1 referente ao estiramento da ligação C=O e outra banda em
1666 cm-1 referente ao estiramento da ligação C=C. No espectro de RMN de 1H (Anexo
8) o simpleto em δ 3,75 integrado para 3H e atribuído ao grupo OCH3 é um indício da
formação do éster (1.5). Neste espectro observa-se um duplo dupleto em δ 6,10
atribuído a H2 e originado pelo acoplamento deste com H3 (J2-3 = 15,6 Hz) e H4 (J2-4 =
1,5 Hz) e um duplo dupleto em δ 6,89 atribuído a H3 e originado do acoplamento deste
com H2 (J3-2= 15,6 Hz) e H4 (J3-4= 5,4 Hz). As constantes de acoplamento entre os
átomos de hidrogênio olefínicos H2 e H3 estão de acordo com os valores descritos na
literatura para isômeros E, confirmando a geometria do produto (1.5) (SILVERSTEIN
& WEBSTER, 2000). No espectro de RMN de 13C (Anexo 9) observa-se o sinal em δ
51,9 atribuído ao carbono da metoxila e os sinais em δ 122,2 e δ 145,2 atribuídos a
C2 e C3 respectivamente, que confirmam a formação deste éster α,β-insaturado (1.5).
Após serem separados, os isômeros (S)-(Z)-4,5-O-isopropilidenopent-2-enoato
de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5) foram
submetidos à reação com o ilídeo isopropilidenotrifenilfosfônio em THF a 0 °C, sob
condições anidras (KRIEF, 1989). Os produtos formados contendo anel ciclopropano
(1.6) e (1.8) foram convertidos nos aldeídos (1.7) e (1.9), respectivamente, como
representado na Figura 1.6.

-21-
OO
CO2CH3
(1.8)
(1.7)
OHC CO2CH3
(1.9)
OHC CO2CH3
OO
CO2CH3
(1.6)
(1.4)
OO
CO2CH3
OO
CO2CH3
(1.5)
10% (a partir de 1.5)(impuro)
i) HClO4, THF
ii) NaIO4, MeOH80%
Ph3P=C(CH3)2
50% THF, 0°C
i) HClO4, THF
ii) NaIO4, MeOH
Ph3P=C(CH3)2
THF, 0°C
Figura 1.6: Formação dos aldeídos (1.7) e (1.9).
A reação com o isômero (1.4) levou a formação do (1S,3S)-3-[(S)-2,2-dimetil-
1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.6) em 50% de
rendimento. Não foi possível isolar outro estereoisômero desta reação. A
enantiosseletividade desta reação com ílideo de fósforo foi previamente estudada por
Krief e colaboradores (KRIEF, 1989). Eles propuseram um mecanismo capaz de
explicar a formação preferencial deste estereoisômero (1.6) discutido na introdução
deste capítulo e representado na Figura 1.4.
No espectro no IV mostrado no Anexo 10 observa-se uma banda intensa em
1734 cm-1 referente ao estiramento da ligação C=O e desaparecimento da banda em
1646 cm-1 referente ao estiramento da ligação C=C do reagente (1.4) (Anexo 4).
Analisando o espectro de RMN de 1H do composto (1.6) (Anexo 11) observa-se a
presença de dois simpletos em δ 1,25 e δ 1,26 atribuídos aos átomos de hidrogênio dos
grupos metila geminais ligados ao anel ciclopropano. Pode-se observar também a
presença de dois simpletos em δ 1,34 e δ 1,43 atribuídos aos grupos metila geminais
pertencentes ao anel de 5 membros. A presença destes quatro sinais juntamente com o
simpleto em δ 3,66 atribuído aos átomos de hidrogênio do grupo OCH3 e o
desaparecimento dos sinais dos átomos de hidrogênio olefínicos do reagente (1.4)
(Anexo 5) são fortes indicativos da formação do produto (1.6). A constante de
acoplamento entre H1 e H3 (J1-3 = 5,4 Hz) está dentro da faixa esperada para o isômero
ciclopropano contendo átomos de hidrogênio na configuração trans (SILVERSTEIN &

-22-
WEBSTER, 2000). Baseado no valor da constante de acoplamento entre H1 e H3 e nos
estudos feitos por Krief podemos definir a estereoquímica do produto (1.4) como 1S-
trans. No espectro de RMN de 13C (Anexo 12) os quatro sinais entre δ 21,1 e δ 27,5
atribuídos aos átomos de carbono metílicos e o desaparecimento dos sinais dos átomos
de carbonos olefínicos (Anexo 6) confirmam a adição do grupo isopropil ao éster (1.4)
formando o produto (1.6).
O grupo acetal do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.6) foi hidrolisado em solução de ácido
perclórico e THF sob agitação a temperatura ambiente durante 2 horas. O consumo do
reagente (1.6) foi acompanhado por CCD. Em seqüência, adicionou-se solução saturada
de bicarbonato de sódio à mistura reacional até atingir pH = 7,5 e 2 equi. molares de
metaperiodato de sódio. Após 1,5 horas de agitação observou-se o término da reação de
clivagem do diol por meio de CCD e fez-se extração do produto com éter dietílico. O
solvente foi concentrado sob pressão reduzida e o resíduo da evaporação foi purificado
por cromatografia em coluna, levando a formação do aldeído (1S,3S)-3-formil-2,2-
dimetilciclopropano-1-carboxilato de metila (1.7) em 80% de rendimento. Pode-se
observar que o produto (1.7) foi obtido a partir do reagente (1.6) com rendimento
superior ao descrito por Krief (63%). Provavelmente este aumento significativo de
rendimento tenha ocorrido devido a conversão de (1.6) em (1.7) em uma única etapa
sem isolamento do intermediário diol. Em seus trabalhos, Krief purificou o diol antes da
clivagem oxidativa que leva ao aldeído. Assim, o menor rendimento obtido por ele para
formação deste aldeído pode ser explicado pela dificuldade em purificar o intermediário
diol, devido a sua alta polaridade.
No espectro no IV (Anexo 13) do aldeído (1.7) pode-se observar a presença de
duas bandas em 1734 e 1704 cm-1 referentes ao estiramento das duas ligações C=O do
grupo éster e aldeído, respectivamente. No espectro de RMN de 1H (Anexo 14) pode-se
observar um dupleto em δ 9,59 atribuído ao hidrogênio do grupo aldeído. Nos espectros
de RMN de 1H e 13C (Anexos 14 e 15, respectivamente) as atribuições dos sinais
confirmam a formação do aldeído esperado.
De acordo com os estudos de Krief, a reação entre o ilídeo
isopropilidenotrifenilfosfônio e o isômero (1.5) levou a formação do (1R,3R)-3-[(S)-2,2-
dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.8). Não
foi possível purificar este produto para obtenção dos dados espectroscópicos. Dessa
forma, o composto (1.8) impuro foi submetido à reação de hidrólise do grupo acetal

-23-
formando o diol correspondente para posterior clivagem deste diol, utilizando-se o
mesmo procedimento descrito para o ciclopropano (1.6). Esta seqüência de reações
levou a formação do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila
(1.9), em 10% de rendimento a partir do éster (1.5).
Como os aldeídos (1.7) e (1.9) são enantiômeros, seus espectros no IV e RMN
de 1H e 13C são semelhantes. No espectro no IV (Anexo 16) do aldeído (1.9) pode-se
observar a presença de duas bandas em 1738 e 1714 cm-1 referentes ao estiramento das
duas ligações C=O dos grupos éster e aldeído, respectivamente. As atribuições dos
sinais nos espectros de RMN de 1H e 13C nos Anexos 17 e 18, respectivamente,
confirmam a formação do aldeído esperado. No espectro de RMN de 1H (Anexo 17)
pode-se observar um dupleto em δ 9,55 atribuído ao hidrogênio do grupo aldeído. Pode-
se observar no Anexo 17 que os sinais referentes a H1 e H3 têm deslocamentos
químicos semelhantes, impedindo a confirmação da estereoquímica do produto (1.9).
Dessa forma, considerou-se que o aldeído possui estereoquímica 1R-trans com base nos
estudos de Krief (KRIEF, 1989).
O aldeído (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila
(1.7) foi submetido a reações de Wittig em THF anidro a 0 °C, sob atmosfera de N2.
Estas reações levaram a formação de uma mistura de isômeros Z e E dos piretróides
(1.10), (1.11), (1.12), (1.13) e (1.14). A Tabela 1.2 apresenta os compostos formados,
rendimentos e proporções aproximadas de cada isômero.
Não foi possível separar os isômeros Z e E de cada piretróide por meio de
cromatografia em coluna, logo eles foram utilizados na forma de mistura para os testes
de atividade discutidos no Capítulo 4. As proporções dos isômeros foram estimadas
pelos valores das integrais dos sinais referentes a cada isômero nos espectros de RMN
de 1H. Estas proporções também foram confirmadas pelos cromatogramas obtidos pela
separação dos isômeros em cromatógrafo a gás acoplado a espectrômetro de massas.
Em cada cromatograma mostrado na Figura 1.7, podem-se ver dois picos principais
contendo aproximadamente a mesma área, com exceção do cromatograma da mistura de
piretróides (1.12), onde o pico com tempo de retenção menor possui aproximadamente
o dobro da área do pico com tempo de retenção maior. Através do espectro de RMN de 1H descobriu-se que o isômero Z está em maior quantidade que o isômero E na mistura
de piretróides (1.12). Desta forma, no cromatograma obtido para (1.12), mostrado na
Figura 1.7, o pico com tempo de retenção menor (e área maior) se refere ao isômero Z
do piretróide (1.12).

-24-
Tabela 1.2: Estruturas, rendimentos e proporções dos piretróides sintetizados a partir do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7).
CO2CH3
R1
R2
CO2CH3OHC sal de Wittig, BuLi
THF
Composto R1 R2 Proporção
a : b Rendimento
%
(1.10a) H- OCH3
(1.10b) OCH3
H-
1 : 1 46
(1.11a) H- OCH3
(1.11b)
OCH3
H-
1 : 1 45
(1.12a) H- Cl
(1.12b) Cl
H-
2 : 1 64
(1.13a) H- F
F
F
F
F
(1.13b) F
F
F
F
F
H-
1 : 1 50
(1.14a) H- EtO
(1.14b) EtO
H-
1 : 1 65

-25-
(1.10)
14.5 15.0 15.5 16.0 16.5 17.0 17.5
10e6
20e6
30e6 TIC
(1.11)
(1.12)
12 13 14 15 16 17 18 19
10e6
20e6
30e6TIC
(1.13)
8 9 10 11 12
10e6
20e6TIC
(1.14)
16 17 18 19 20 21
10e6
20e6
30e6TIC
Figura 1.7: Cromatogramas das misturas contendo os isômeros Z e E (1.10) a (1.14).

-26-
Os espectros no IV mostrados nos Anexos 19 a 23 foram obtidos a partir da
mistura de isômeros geométricos Z e E. Nestes espectros pode-se observar uma banda
intensa entre 1725 e 1734 cm-1 referente ao estiramento da ligação C=O do grupo éster
de cada piretróide. Pode-se observar também bandas de deformação angular fora do
plano das ligações =C-H do anel aromático. Na Tabela 1.3 temos um resumo destas
bandas.
Tabela 1.3: Bandas dos espectros de IV de cada mistura de isômeros Z e E dos piretróides (1.10) a (1.14).
Compostos IV Estiramento C=O (cm-1)
Deformação angular de C-H aromático (cm-1)
(1.10) Anexo 19 1724 752 (F)
(1.11) Anexo 20 1726 779 (m), 691 (m)
(1.12) Anexo 21 1728 751 (F)
(1.13) Anexo 22 1734 ------
(1.14) Anexo 23 1725 841 (F)
Intensidade: F = forte; m = média
Também não foi possível a atribuição de todos os sinais nos espectros de RMN
de 1H e 13C das misturas de isômeros geométricos. Nos espectros de RMN de 1H das
misturas de piretróides, os sinais de H4 de ambos isômeros puderam ser facilmente
observados nos deslocamentos listados na Tabela 1.4. De acordo com a constante de
acoplamento (J4-5 / Hz) foi possível determinar qual sinal correspondia ao isômero E ou
Z. Utilizou-se também espectro de COSY para atribuir os sinais de H3 e H1, onde se
observou correlações entre (H4 - H3) e (H3 - H1) de cada isômero E e Z. Os sinais
referentes a H5 dos isômeros Z e E puderam ser atribuídos utilizando-se os valores das
constantes de acoplamento e espectro de COSY para as mistura de isômeros (1.12),
como mostrado no Anexo 26. No caso das misturas (1.11) e (1.14) os sinais de H5cis e
H5trans apresentaram deslocamentos químicos coincidentes (Anexos 25 e 28,
respectivamente). Os sinais de C1 e C3 dos isômeros Z e E no RMN de 13C (Anexos 29
a 33) foram atribuídos por meio de HETCOR.

-27-
Tabela 1.6: Atribuições dos sinais de H4, H3 e H1 nos espectros de RMN de 1H.
Composto RMN de 1H
Config. C=C δδδδ H4 J5-4 * δδδδ H3 J4-3 * δδδδ H1 J3-1 *
(1.10a) Anexo 24 Z 5,45 (dd) 11,5 2,34
(dd) 9,0 1,57 (d) 5,4
(1.10b) Anexo 24 E 5,96 (dd) 15,9 2,26
(dd) 8,7 1,69 (d) 5,4
(1.11a) Anexo 25 Z 5,40 (dd) 11,7 2,45
(dd) 8,7 1,57 (d) 5,4
(1.11b) Anexo 25 E 5,95 (dd) 15,9 2,22
(dd) 8,4 1,67 (d) 5,7
(1.12a) Anexo 26 Z 5,55 (dd) 11,4 2,27
(m) 9,0 1,60
(d) 5,7
(1.12b) Anexo 26 E 5,97 (dd) 15,9 2,27
(m) 9,0 1,73
(d) 5,4
(1.13a) Anexo 27 Z 5,75 (dd) 11,1 2,16
(m) 9,6 1,65
(d) 5,4
(1.13b) Anexo 27 E 6,27 (dd) 16,2 2,33
(m) 9,0 1,75
(d) 5,4
(1.14a) Anexo 28 Z 5,30 (dd) 11,5 2,40
(m) 8,4 1,54
(d) 5,4
(1.14b) Anexo 28 E 5,82 (dd) 15,6 2,18
(m) 8,7 1,64
(d) 5,4
* J em Hz.
Os espectros de massas de cada isômero Z e E foram obtidos após separação
destes isômeros em CG-EM, mas por serem idênticos estão apresentados neste trabalho
apenas os espectros de massas dos isômeros que apresentaram menor tempo de retenção
durante a cromatografia gasosa (Anexos 34 a 38). Para todos os piretróides (1.10),
(1.11), (1.12), (1.13) e (1.14) foi possível a observação do pico do íon molecular no
EM.
Sabe-se que ao absorver luz UV num determinado comprimento de onda uma
molécula passa do estado fundamental para o estado excitado. Durante a excitação de
um composto contendo dupla ligação, um elétron pertencente ao orbital π ligante (que
contribui para formação da ligação π) é promovido para o orbital π antiligante (que
contribui para a quebra da ligação π). Esta transição eletrônica provoca a perda, no
estado excitado, do caráter de dupla ligação existente na molécula inicialmente no
estado fundamental. Logo, quando a molécula retorna ao seu estado fundamental ela

-28-
pode continuar na configuração original ou ter a configuração invertida (isomerização).
Dessa forma, a isomerização efetiva de uma mistura de isômeros Z ou E pode ocorrer
quando um dos isômeros absorve luz mais efetivamente que outro. Neste caso o
isômero que apresenta menor absorção terá tendência de permanecer em maior
quantidade durante a irradiação (COYLE, 1985).
Uma solução de 1000 ppm contendo a mistura de piretróides Z e E (1.14) em
benzeno foi irradiada com lâmpada de mercúrio de alta pressão com intuito de observar
uma possível isomerização das espécies (BROWN e WONG, 2004). Fez-se CG-EM das
alíquotas retiradas antes do início da irradiação e após 0,5 h, 1 h, 2 h e 5 h de irradiação.
De acordo com os cromatogramas obtidos para cada amostra (Figura 1.8) não se
observou mudança na proporção dos isômeros durante a irradiação. Dessa forma, não
ocorreu isomerização efetiva de nenhum dos dois compostos Z e E. Uma possível
explicação para estas observações experimentais pode estar no fato de ambos isômeros
apresentarem absorções semelhantes de luz UV na faixa de comprimento de onda
utilizada. Assim, não ocorre favorecimento da formação de nenhuma das duas espécies.

-29-
(a)
16 17 18 19 20 21
10e6
20e6
30e6TIC
(b)
16 17 18 19 20 21
10e6
20e6
30e6TIC
(c)
16 17 18 19 20 21
10e6
20e6
30e6TIC
(d)
16 17 18 19 20 21
10e6
20e6
30e6TIC
(e)
16 17 18 19 20 21
10e6
20e6
30e6TIC
Figura 1.8: Cromatogramas da mistura dos isômeros Z e E (1.14). (a) antes da irradiação com luz UV; (b) após 0,5 h de irradiação; (c) após 1 h de irradiação; (d) após 2 h de irradiação; (e) após 5 h de irradiação.

-30-
1.3. CONCLUSÃO
Sintetizou-se o (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila
(1.7) em 8,3% de rendimento a partir do D-manitol (1.1). Esta síntese teve como etapa
intermediária uma reação de ciclopropanação do éster γ-alcóxi-α,β-insaturado (1.4)
utilizando-se o ilídeo isopropilidenotrifenilfosfônio, de acordo com metodologia
descrita por Krief e colaboradores.
O aldeído (1.7) foi convertido nos piretróides (1.10), (1.11), (1.12), (1.13) e
(1.14) por meio de reação de Wittig. Estes piretróides foram obtidos como uma mistura
de isômeros Z e E, cuja separação não foi possível por cromatografia em coluna. A
mistura de isômeros geométricos (1.14) foi irradiada com lâmpada de mercúrio de alta
pressão durante 5 horas, mas não se observou mudança na proporção dos isômeros.
O (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9) foi
sintetizado a partir do D-manitol (1.1) em baixo rendimento, seguindo metodologia
semelhante a utilizada na síntese de seu enantiômero (1.7).

-31-
1.4. EXPERIMENTAL
1.4.1. Síntese do metoxicarbonilmetileno(trifenil)fosforano
Br CO
O
Tolueno
PPh3Br Ph3P
O
O
Ph3PO
OOH- / H2O
ilídeosal de Wittigbromoacetato de metila
Adicionou-se trifenilfosfina (37 g, 141 mmol) a uma solução de bromoacetato de
metila (20 g, 131 mmol) em tolueno anidro (300 mL). O balão contendo a mistura foi
tampado com um tubo de cloreto de cálcio e o sistema mantido sob agitação magnética
por 48 h. O sólido branco formado foi filtrado sob vácuo, lavado com éter dietílico (100
mL) e mantido em dessecador sob vácuo por 4 h. Obteve-se o brometo de
metoxicarbonilmetileno(trifenil)fosforano (54 g, 130 mmol) em rendimento
quantitativo.
Adicionou-se água destilada (400 mL) ao sal de Wittig observando a dissolução
quase completa do material. Adicionou-se lentamente solução de NaOH 2 mol.L-1 (400
mL) e observou-se a formação de um precipitado amarelado que foi separado por
filtração à vácuo em funil de vidro sinterizado. O sólido foi dissolvido em DCM (150
mL), a solução foi secada com MgSO4 e filtrada. O DCM foi evaporado sob pressão
reduzida obtendo-se o metoxicarbonilmetileno(trifenil)fosforano (39 g, 117 mmol) em
93% de rendimento global na forma de um sólido amarelado.
Características do sal de Wittig:
Tf = 164,5-166,5 ºC.
IV (KBr, cm -1) ν : 3057, 1942, 1619, 1482, 1441, 1435, 1435, 1349, 1181, 1122, 1111,
1103, 885, 748, 721, 712, 692, 527.
RMN de 1H (300 MHz, CDCl3) δδδδ: 3,57 (s, 3H, OCH3), 5,57 (d, 2H, JH-P = 13,5, CH2),
7,75 (m, 15H, PPh3).
RMN de 13C (75 MHz; CDCl3) δδδδ: 32,9 (d, JCH2-P = 57,2, CH2), 53,4 (CH3), 118,0 (d,
JC-P = 88,7, =C-P), 130,1 (d, JCo-P = 13,1, Co), 133,8 (d, JCm-P = 10,3, Cm), 135,1 (d, JCp-
P = 3,5, Cp), 165,1 (C=O).

-32-
1.4.2. Síntese dos Sais de Wittig S1-S5
Em um balão de 50 ml equipado com condensador, barra magnética e tubo de
cloreto de cálcio, adicionou-se o haleto (10 mmol), tolueno seco (25 mL) e
trifenilfosfina (2,9 g, 11 mmol). A mistura foi refluxada sob agitação magnética (veja o
tempo de reação na Tabela 1), observando-se a formação de um sólido branco. O sólido
foi filtrado sob vácuo em funil de vidro sinterizado, lavado com éter dietílico (15 mL) e
secado em dessecador sob vácuo por 2 h. Os rendimentos de cada reação são dados na
Tabela 1.7:
Tabela 1.7: Condições para formação dos sais de Wittig S1 – S5.
Sal Haleto Massa haleto Tempo de
reação (h)
Rendimento
(%)
S1 (H3C)2CHI 1,70 48 42
S2 o-MeOPhCH2Cl 1,57 48 88
S3 m-MeOPhCH2Cl 1,57 48 72
S4 o-ClPhCH2Br 2,10 21 99
S5 F5PhCH2Br 2,60 21 98
Iodeto de isopropiltrifenilfosfônio (S1):
I -CH P
H3C
H3C
IV (KBr, cm -1) ν : 2878, 1629, 1439, 1112, 755, 723, 693, 531.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,33 (dd, 6H, JCH3-CH = 6,9 e JCH3-P = 18,9, 2x CH3),
5,33 (m, 1H, CH), 7,65 (m, 15 H, PPh3).

-33-
Cloreto de 2-metoxibenziltrifenilfosfônio (S2):
1
23
4
5 6
CH2 P
OCH3
Cl-
Tf = 251,2 - 251,5 °C
IV (KBr, cm -1) ν : 3044, 2844, 1598, 1495, 1437, 1251, 1113, 765, 747, 692, 508.
RMN de 1H (300 MHz, CDCl3) δδδδ: 3,15 (s, 3H, OCH3), 5,17 (d, 2H, J H-P = 13,5, CH2),
6,55 (d, 1H, , J3-4 = 8,1, H3), 6,76 (t, 1H, J5-4 = 7,8 e J5-6 = 7,8, H5), 7,19 (t, 1H, J4-3 =
8,1 e J4-5 = 7,8, H4), 7,32 (d, 1H, J5-6 = 7,8, H6), 7,65 (m, 15H, PPh3).
Cloreto de 3-metoxibenziltrifenilfosfônio (S3):
1
23
4
5 6
CH2 P
CH3O
Cl-
Tf = 270,2 – 270,9 °C
IV (KBr, cm -1) ν : 2990, 2859, 2782, 1596, 1483, 1437, 1250, 1111, 1041, 746, 716,
699, 502.
RMN de 1H (300 MHz, CDCl3) δδδδ: 3,45 (s, 3H, OCH3), 5,34 (d, 2H, J H-P = 14,4, CH2),
6,66 (m, 3H, H2, H4 e H6), 6,96 (t, 1H, J5-4 = 7,8 e J5-6 = 7,8, H5), 7,65 (m, 15H, PPh3).

-34-
Brometo de 2-clorobenziltrifenilfosfônio (S4):
1
23
4
5 6
CH2 P
Cl
Br-
IV (KBr, cm -1) ν : 3047, 2841, 2773, 1436, 1110, 782, 759, 752, 691, 592.
RMN de 1H (300 MHz, CDCl3) δδδδ: 5,59 (d, 2H, JCH2-P = 14,1, CH2), 7,15 (m, 4H, H3-
H6), 7,70 (m, 15 H, PPh3).
Brometo de pentafluorobenziltrifenilfosfônio (S5):
CH2 P
F
FF
F
F
Br-
Tf = 240,0 – 240,5 °C
IV (KBr, cm -1) ν : 2859, 1524, 1505, 1439, 1111, 981, 751, 726, 690.
RMN de 1H (300 MHz, CDCl3) δδδδ: 5,61 (d, 2H, JCH2-P = 13,8, CH2), 7,8 (m, 15 H,
PPh3).

-35-
1.4.3. Síntese do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2)
O
OHO H
OHH
O
O
H
H'
H
H
'H
H
CH3
CH3'
'H3C
H3C
1
2
3
(1.2)
A um balão de fundo redondo (500 mL) equipado com barra magnética foram
adicionados acetona anidra (250 mL) e cloreto de zinco (40 g, 294 mmol). O balão foi
tampado e a mistura mantida sob agitação magnética até total dissolução do sólido (30
min). Adicionou-se D-manitol (1.1) (20 g, 110 mmol) à mistura reacional, formando
uma suspensão. A mistura foi mantida sob agitação magnética por 5 horas, observando-
se formação de uma solução incolor. A mistura reacional foi neutralizada com solução
de carbonato de potássio (45 g; 326 mmol) em água (45 mL). O sólido obtido (KCl e
ZnCO3) foi separado por filtração simples e lavado com 50 mL de acetona anidra. O
filtrado foi concentrado sob ar comprimido até início da cristalização. Adicionou-se éter
dietílico (100 mL) ao resíduo aquoso, separou-se a fase orgânica e, a fase aquosa foi
extraída com éter dietílico (3 x 50 mL). As fases orgânicas foram reunidas, secadas com
MgSO4 e concentradas sob pressão reduzida, observando-se a formação de um sólido
branco. Adicionou-se hexano (100 mL) a este resíduo, a mistura foi mantida em
geladeira por 2 horas e filtrada sob vácuo sendo o sólido lavado com hexano (100 mL)
previamente resfriado. O sólido foi seco sob vácuo em dessecador por 2 horas, obtendo-
se o 1,2:5,6-di-O-isopropilideno-D-manitol (1.2) (25 g, 95 mmol) em 87% de
rendimento, com pureza suficiente para ser utilizado na próxima etapa da síntese.
T f: 117-119 oC.
CCD: Rf = 0,20 (hexano : éter dietílico 1:1).
Rotação específica: 20][ Dα = +10,7 ° (c = 1,74, acetona).
IV (KBr, cm -1) ν : 3403, 3280, 1384, 1372, 1259, 1213, 1159, 1069, 859, 846, 654.

-36-
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,36 (s, 6H, CH3’), 1,42 (s, 6H, CH3), 2,66 (d, 2H,
J3-OH = 6,0, OH), 3,74 (dd, 2H, J3-OH = 6,0, J3-2 = 5,7, H3), 3,98 (dd, 2H, J1-2 = 5,4, J1-1’ =
8,4, H1), 4,09-4,22 (m, 4H, H1’ e H2).
RMN de 13C (75 MHz; CDCl3) δδδδ: 25,4 (CH3’), 26,9 (CH3), 67,0 (C1), 71,3 (C3), 76,3
(C2) e 109,6 [C(CH3)2].
1.4.4. Síntese do 2,3-O-isopropilideno-D-gliceraldeído (1.3)
OO
CHOH
(1.3)
A um balão de 250 mL adicionou-se 1,2:5,6-di-O-isopropilideno-D-manitol
(1.2) (23 g; 87,8 mmol), DCM (125 mL) e solução saturada de NaHCO3 (9,5 mL, 0,4
mL por g de diacetal). A mistura foi agitada por 5 min e colocada em banho de gelo.
Adicionou-se periodato de sódio (37,4 g, 175 mmol) e manteve-se a mistura sob
agitação em banho de gelo por 2,5 h. Adicionou-se MgSO4 (11,5 g, 0,5 g por g de
diacetal) mantendo a agitação por mais 20 min. A mistura foi filtrada e o sólido lavado
com DCM (3 x 50 mL). O filtrado foi concentrado sob pressão reduzida obtendo-se o
2,3-O-isopropileno-D-gliceraldeído (1.3) na forma de um óleo castanho impuro (15 g),
que foi usado na etapa seguinte da rota sintética.
CCD: Rf = 0,25 (hexano : acetato de etila 1:1).
1.4.5. Síntese dos isômeros (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5)
OO
CO2CH3
(1.4)
23
45
OO
CO2CH3(1.5)
23
45
A um balão de 250 mL foram adicionados 15 g do óleo castanho escuro obtido
na etapa anterior e metanol (75 mL). Manteve-se a mistura sob banho de gelo e
adicionou-se lentamente metoxicarbonilmetileno(trifenil)fosforano (29 g, 86,8 mmol),
sob constante agitação. Para calcular a quantidade de ilídeo considerou-se 50 % de

-37-
rendimento para a reação de clivagem do diacetal. A mistura foi mantida sob agitação
em banho de gelo por 6 horas e o término da reação foi detectado por CCD (solução
reveladora de KMnO4). O metanol foi evaporado e o resíduo extraído a quente com
hexano : éter dietílico 7 : 3 (5 x 50 mL). Os extratos foram reunidos e concentrados sob
pressão reduzida. O resíduo da evaporação foi purificado por cromatografia em coluna
(hexano : éter 8 : 2), levando à obtenção dos isômeros (S)-(Z)-4,5-O-isopropilidenopent-
2-enoato de metila (1.4) e (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5)
(7,8 g e 1,0 g, respectivamente) em 24% de rendimento à partir do diacetal (1.2). Os
ésteres Z e E foram obtidos na forma de óleo amarelo claro.
(S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4)
CCD: Rf = 0,73 (hexano : éter dietílico 8:2).
Rotação específica: 20][ Dα = + 125,2 ° (c = 1,02, CHCl3).
IV (KBr, cm -1) ν : 2988, 2944, 2876, 1722, 1642, 1439, 1402, 1381, 1209, 1155, 1060,
1000, 859, 821.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,37 (s, 3H, CH3), 1,43 (s, 3H, CH3), 3,58 (dd, 1H,
J5-5’ = 6,9 e J5-4= 8,1, H5), 3,70 (s, 3H, OCH3), 4,35 (dd, 1H, J5’-5 = 6,9 e J5’-4 = 8,4,
H5’), 5,47 (m, 1H, H4), 5,84 (dd, 1H, J2-3 = 11,7 e J2-4 = 1,8, H2), 6,35 (dd, 1H, J3-2 =
11,7 e J3-4 = 6,6, H3).
RMN de 13C (75 MHz; CDCl3) δδδδ: 25,6 (CH3), 26,7 (CH3), 51,7 (OCH3), 69,5 (C5),
73,7 (C4), 109,9 [C(CH3)2)], 120,5 (C2), 149,7 (C3) e 166,3 (C=O).
(S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5)
CCD: Rf = 0,48 (Hexano : éter dietílico 8:2).
Rotação específica: 20][ Dα = + 43,3 ° (c = 1,02, CHCl3).
IV (KBr, cm -1) ν : 2988, 2948, 2878, 1727, 1664, 1437, 1360, 1307, 1263, 1212, 1164,
1063, 978, 852.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,40 (s, 3H, CH3), 1,44 (s, 3H, CH3), 3,67 (dd, 1H,
J5-5’ = 8,4 e J5-4 = 7,1, H5), 3,75 (s, 3H, OCH3), 4,18 (dd, 1H, J5’-5 = 8,4 e J5’-4 = 6,6,

-38-
H5’), 4,66 (m, 1H, H4), 6,10 (dd, 1H, J2-3 = 15,6 e J2-4 = 1,5, H2), 6,89 (dd, 1H, J3-2=
15,6 e J3-4= 5,4, H3).
RMN de 13C (75 MHz; CDCl3) δδδδ: 25,9 (CH3), 26,6 (CH3), 51,9 (OCH3), 69,0 (C5),
75,1 (C4), 110,4 [C(CH3)2], 122,2 (C2), 145,2 (C3) e 166,4 (C=O).
1.4.6. Síntese do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropa- no-1-carboxilato de metila (1.6)
OOCO2CH3
(1.6)
23
45 1
Adicionou-se solução comercial de butillítio (3,5 mL, BuLi 2 mol.L-1 em
hexano) a uma suspensão de iodeto de isopropiltrifenilfosfônio (S1) (3,0 g, 6,94 mmol)
em THF seco (15 mL), sob atmosfera de N2, em banho de gelo. Observou-se durante a
adição do BuLi a dissolução do sal e formação de uma solução avermelhada. O sistema
foi mantido sob agitação magnética por 10 min e adicionou-se solução do éster (1.4)
(1,2 g, 6,45 mmol) em THF seco (5 mL), observando-se mudança da cor vermelha para
amarela. A mistura foi mantida sob agitação em banho de gelo por três horas e o final da
reação foi detectado por CCD. O THF foi evaporado sob pressão reduzida. Adicionou-
se água destilada (10 mL) ao resíduo da evaporação e fez-se extração do material com
éter dietílico (5 x 20 mL). As fases orgânicas foram reunidas, o material secado com
MgSO4 e filtrado. O filtrado foi concentrado sob pressão reduzida e o resíduo da
evaporação submetido à cromatografia em coluna (hexano : acetato de etila 9:1) levando
à obtenção do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-
carboxilato de metila (1.6) (0,9 g, 3,95 mmol) em 50% de rendimento como um óleo
amarelo viscoso.
CCD: Rf = 0,66 (hexano : acetato de etila 4:1).
Rotação específica: 20][ Dα = + 30 ° (c = 1,02, CHCl3).
IV (KBr, cm -1) ν : 2985, 2874, 1729, 1447, 1374, 1234, 1213, 1175, 1116, 1065, 853.
RMN de 1H (300 MHz, CDCl3) δδδδ:1,25 (s, 3H, CH3), 1,26 (s, 3H, CH3), 1,28 (d, 1H, J1-
3 = 5,4, H1), 1,34 (s, 3H, CH3), 1,43 (s, 3H, CH3), 1,55 (dd, 1H, J3-1 = 5,4 e J3-4 = 6,6,
H3), 3,66 (s, 3H, OCH3), 3,70-3,72 (m, 2H, H4 e H5) e 3,97 (m, 1H, H5’).

-39-
RMN de 13C (75 MHz; CDCl3) δδδδ: 21,1 (CH3), 22 (CH3), 26,4 (CH3), 27,4 (C2), 27,5
(CH3), 30,9 (C1), 35,2 (C3), 52,1 (OCH3), 69,6 (C5), 76,1 (C4), 109,6 [C(CH3)2],
172,0 (C=O).
1.4.7. Síntese do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7)
(1.7)
23
1OHC CO2CH3
Adicionou-se solução de HClO4 2,5 mol.L-1 (12,5 mL) a uma balão de 100 mL
contendo solução do (1.6) (2,1 g, 9,21 mmol) em THF (25 mL). A mistura foi mantida
sob agitação magnética por 2 h em temperatura ambiente e o término da reação de
clivagem acompanhado por CCD. Adicionou-se solução saturada de NaHCO3 até cessar
a liberação de gás (pH = 7,5) e manteve-se a mistura sob agitação por 10 min.
Adicionou-se NaIO4 (4,0 g, 18,7 mmol), observando-se a formação de um precipitado
branco gelatinoso e desprendimento de gás. A mistura foi mantida sob agitação por 1,5
h e, em seguida, extraída com éter dietílico (5 x 30 mL). As fases etéreas foram
reunidas, o material foi secado com MgSO4 e filtrado. O filtrado foi concentrado sob
pressão reduzida e o resíduo da evaporação submetido à cromatografia em coluna
(hexano : acetato de etila 4:1) levando à obtenção do (1S,3S)-3-formil-2,2-
dimetilciclopropano-1-carboxilato de metila (1.7) (0,9 g, 3,95 mmol) em 80 % de
rendimento como um óleo incolor.
CCD: Rf = 0,50 (hexano : acetato de etila 4:1).
Rotação específica: 20][ Dα = - 18,6 ° (c = 1,3, acetona).
IV (KBr, cm -1) ν : 2955, 2932, 2743, 1734, 1442, 1339, 1242, 1171, 1106, 976, 910.
RMN de 1H (300 MHz, CDCl3) δδδδ:1,30 (s, 3H, CH3), 1,34 (s, 3H, CH3), 2,46 (m, 2H,
H1 e H3), 3,70 (s, 3H, OCH3), 9,59 (d, 1H, JOHC-H3 = 3,3, HC=O).
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,8 (CH3), 21,1 (CH3), 33,4 (C2), 34,4 (C1), 42,3
(C3), 52,0 (OCH3), 170,5 (COOCH3), 198,8 (HC=O).

-40-
1.4.8. Síntese do (1R,3R)-3-[(R)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopro-pano-1-carboxilato de metila (1.8)
OO
CO2CH3
(1.8)
23
45 1
Adicionou-se solução comercial de butillítio (3,5 mL, BuLi 2 mol.L-1 em
hexano) a uma suspensão do iodeto de isopropiltrifenilfosfônio (S1) (3,0 g, 6,94 mmol)
em THF seco (15 mL), sob atmosfera de N2, em banho de gelo. Observou-se durante a
adição do BuLi a dissolução do sal e formação de uma solução avermelhada. O sistema
foi mantido sob agitação magnética por 10 min e adicionou-se solução do éster (1.5)
(1,2 g, 6,45 mmol) em THF seco (5 mL), observando-se mudança da cor vermelha para
amarela. A mistura foi mantida sob agitação em banho de gelo por três horas e o final da
reação foi detectado por CCD. O THF foi evaporado sob pressão reduzida. Adicionou-
se água destilada (10 mL) ao resíduo da evaporação e fez-se extração do material com
éter dietílico (5 x 20 mL). As fases orgânicas foram reunidas, o material secado com
MgSO4 e filtrado. O filtrado foi concentrado sob pressão reduzida e o resíduo da
evaporação submetido à cromatografia em coluna (hexano : acetato de etila 4:1) levando
à obtenção de 0,9 g de um óleo amarelo viscoso contendo o (1R,3R)-3-[(R)-2,2-dimetil-
1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.8). Devido a
dificuldade de purificação do produto ele foi utilizado de forma impura para a próxima
etapa da síntese.
CCD: Rf = 0,66 (hexano : acetato de etila 4:1).
1.4.9. Síntese do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9)
(1.9)
23
1OHC CO2CH3
Adicionou-se solução de HClO4 2,5 mol.L-1 (8,5 mL) a uma balão de 50 mL
contendo solução do (1.8) (0,9 g de material impuro) em THF (15 mL). A mistura foi

-41-
mantida sob agitação magnética por 2 h em temperatura ambiente e o término da reação
de clivagem acompanhado por CCD. Adicionou-se solução saturada de NaHCO3 até
cessar a liberação de gás (pH = 7,5) e manteve-se a mistura sob agitação por 10 min.
Adicionou-se NaIO4 (1,7 g, 7,95 mmol), observando-se a formação de um precipitado
branco gelatinoso e desprendimento de gás. A mistura foi mantida sob agitação por 1,5
h e, em seguida, extraída com éter dietílico (5 x 15 mL). As fases etéreas foram
reunidas, o material foi secado com MgSO4 e filtrado. O filtrado foi concentrado sob
pressão reduzida e o resíduo da evaporação submetido à cromatografia em coluna
(hexano : acetato de etila 4:1) levando à obtenção do (1R,3R)-3-formil-2,2-
dimetilciclopropano-1-carboxilato de metila (1.9) (0,10 g, 0,64 mmol) em 10 % de
rendimento à partir do éster (1.5) e na forma de um óleo incolor.
CCD: Rf = 0,56 (hexano : acetato de etila 4:1).
Rotação específica: 20][ Dα = + 14,0 ° (c = 1,3, acetona).
IV (KBr, cm -1) ν : 2956, 2752, 1738, 1714, 1434, 1237, 1173, 1112, 975.
RMN de 1H (300 MHz, CDCl3) δδδδ:1,27 (s, 3H, CH3), 1,31 (s, 3H, CH3), 2,44 (m, 2H,
H1 e H3), 3,67 (s, 3H, OCH3), 9,55 (d, 1H, JHOC-H3 = 3,3, HC=O).
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,8 (CH3), 21,1 (CH3), 33,4 (C2), 34,3 (C1), 42,2
(C3), 52,2 (OCH3), 170,4 (COOCH3), 199,0 (HC=O).
1.4.10. Síntese de piretróides
Adicionou-se solução de butillítio (1,8 mL, BuLi 2 mol.L-1 em hexano) a uma
suspensão do sal de Wittig (S2 - S6) (3,53 mmol) em THF seco (15 mL), sob atmosfera
de N2, em banho de gelo. O sistema foi mantido sob agitação magnética por 10 min e
adicionou-se solução do aldeído (1.7) (0,50 g, 3,20 mmol) em THF seco (5 mL). A
mistura foi mantida sob agitação em banho de gelo por três horas e o final da reação foi
detectado por CCD. O THF foi evaporado sob pressão reduzida. Adicionou-se água
destilada (10 mL) ao resíduo da evaporação e fez-se extração do material com éter
dietílico (5 x 20 mL). As fases orgânicas foram reunidas, o material secado com MgSO4
e filtrado. O filtrado foi concentrado sob pressão reduzida e o resíduo da evaporação
submetido à cromatografia em coluna (hexano : éter dietílico 9:1) levando à obtenção de
uma mistura de isômeros geométricos. A proporção de cada isômero e o rendimento das
reações são dados na Tabela 1.8:

-42-
Tabela 1.8: Condições para formação dos piretróides (1.10) a (1.14).
Piretróides Sal de Wittig (g) Massa do sal Proporção
Z : E
Rendimento
(%)
(1.10) S2 1,47 1 : 1 46
(1.11) S3 1,47 1 : 1 45
(1.12) S4 1,64 2 : 1 64
(1.13) S5 1,85 1 : 1 50
(1.14) p-EtOPhCH2PPh3Br S6
1,69 1 : 1 65
Dados para a mistura dos isômeros E e Z (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.10):
(1.10a) (1.10b)
CO2CH3
CH3O1
2
3
45
4'5'
CO2CH3CH3O
1'2'
3'6'
1'2'
3'
4'5'
6'1
2
3
4
5
CCD: Rf = 0,45 (hexano : éter dietílico 9 : 1).
Rotação específica: 20][ Dα = - 84,4 ° (c = 1,60, acetona).
IV (KBr, cm -1) ν : 2950, 1724, 1598, 1438, 1244, 1167, 1029, 752.
RMN de 1H (300 MHz, CDCl3) δδδδ:1,20 (s, 6H, 2 x CH3), 1,25 (s, 3H, CH3), 1,30 (s, 3H,
CH3), 1,57 (d, 1H, J1-3cis = 5,4, H1cis), 1,69 (d, 1H, J1-3trans = 5,4, H1trans), 2,26 (dd, 1H,
J1-3trans = 5,4 e J3-4trans = 8,7, H3trans), 2,34 (dd, 1H, J1-3 cis = 5,4 e J3-4 cis = 9,0, H3cis),
3,65 (s, 3H, OCH3), 3,70 (s, 3H, OCH3), 3,83 (s, 3H, CH3O-Ar), 3,83 (s, 6H, CH3O-Ar),
5,45 (dd, 1H, J3-4cis = 9,0 e J4-5cis = 11,5, H4cis), 5,96 (dd, 1H, J3-4trans = 8,7 e J4-5trans =
15,9, H4trans), 6,68 (d, 1H, J4-5cis = 11,5, H5cis), 6,92 ( m, 5H, H5trans, H3’ e H5’), 7,30
(m, 4H, H4’ e H6’).

-43-
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,4 (CH3), 20,7 (CH3), 22,4 (CH3), 22,5 (CH3),
29,7 [C(CH3)2], 28,8 [C(CH3)2], 33,7 (C3cis), 34,6 (C1trans), 35,7 (C1cis), 37,6 (C3trans),
51,7 (OCH3), 51,8 (OCH3), 55,7 (CH3OAr), 55,8 (CH3OAr), 110,5 – 130,2 (C4, C5 e
Ar), 156,5 (C2’), 157,1 (C2’), 172,5 (C=O), 172,7 (C=O).
EM, m/z (I): [ +•M ] 260 (35,02), 201 (100,00), 185 (46,55), 121 (56,32), 91 (95,16), 77
(57,62), 41 (73,00).
Dados para a mistura dos isômeros E e Z (1S,3S)-3-[2-(3-metoxifenil)eten-1-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.11):
(1.11a) (1.11b)
CO2CH3
CH3O
1
2
3
45
CO2CH3
OCH3
1'1'
2'
2'
3'
3'
4'
4'
5'
5'
6'6'1
2
3
4
5
CCD: Rf = 0,45 (hexano : éter dietílico 9:1).
IV (KBr, cm -1) ν : 2948, 1728, 1438, 1219, 767, 751.
Rotação específica: 20][ Dα = - 80,9 ° (c = 1,10, acetona).
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,24 (s, 6H, CH3 cis), 1,26 (s, 3H, CH3 trans), 1,28 (s,
6H, CH3 cis), 1,34 (s, 3H, CH3 trans), 1,60 (d, 2H, J1-3cis = 5,7, H1cis), 1,73 (d, 1H, J1-3trans
= 5,7, H1trans), 2,27 (m, 3H, H3cis e H3trans), 3,67 (s, 6H, OCH3 cis), 3,71 (s, 3H, OCH3
trans), 5,55 (dd, 2H, J3-4cis = 9,0 e J4-5cis = 11,4, H4cis), 5,97 (dd, 1H, J3-4trans = 9,0 e J4-
5trans = 15,9, H4trans), 6,67 (d, 2H, J4-5cis = 11,4, H5cis), 6,95 (d, 1H, J4-5trans = 15,9,
H5trans), 7,25 (m, 12H, Arcis e Artrans)*.
* Os isômeros Z e E estão na proporção de 2 : 1, aproximadamente.
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,4 (2 x CH3), 20,6 (CH3), 22,4 (CH3), 29,7 [2 x
C(CH3)2], 33,2 (C3cis), 34,8 (C1trans), 35,7 (C1cis), 37,0 (C3trans), 51,8 (2 x OCH3), 126,5
– 135,3 (C4, C5 e Ar), 172,4 (2 x C=O).

-44-
EM, m/z (I): [ +•M ] 260 (26,10), 201 (61,42), 185 (58,24), 159 (60,84), 115 (77,52),
102 (100,00), 91 (67,98), 77 (68,16), 41 (72,87).
Dados para a mistura dos isômeros E e Z (1S,3S)-3-[2-(2-clorofenil)eten-1-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.12):
(1.12a) (1.12b)
CO2CH3
Cl1
2
3
45
4
5CO2CH3
Cl1
2
3
1'2'
3'
4'5'
6'
6'
1'2'
3'
4'5'
CCD: Rf = 0,55 (hexano : éter dietílico 9:1).
Rotação específica: 20][ Dα = - 80,6 ° (c = 1,32, acetona).
IV (KBr, cm -1) ν : 2956, 2752, 1738, 1714, 1434, 1237, 1173, 1112, 975.
RMN de 1H (300 MHz, CDCl3) 1,20 (s, 6H, 2 x CH3), 1,25 (s, 6H, 2 x CH3) 1,57 (d,
1H, J1-3cis = 5,4, H1cis), 1,67 (d, 1H, J1-3trans = 5,7, H1trans), 2,22 (dd, 1H, J1-3trans = 5,7 e
J3-4trans = 8,4, H3trans), 2,45 (dd, 1H, J1-3cis = 5,4 e J3-4cis = 8,7, H3cis), 3,67 (s, 3H,
OCH3), 3,69 (s, 3H, OCH3), 3,80 (s, 6H, 2 x CH3O-Ar), 5,40 (dd, 1H, J3-4cis = 8,7 e J4-
5cis = 11,7, H4cis), 5,95 (dd, 1H, J3-4trans = 8,4 e J4-5trans = 15,9, H4trans), 6,54 (m, 2H,
H5trans e H5cis), 6,90 ( m, 6H, H2’, H4’ e H6’), 7,23 (m, 2H, H5’).
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,8 (CH3), 21,1 (CH3), 33,4 (C2), 34,3 (C1), 42,2
(C3), 52,2 (OCH3), 170,4 (COOCH3), 199,0 (HC=O).
EM, m/z (I): [ +•M +2] 266 (6,29), [ +
•M ] 264 (19,09), 207 (28,14), 205 (100,00), 139
(43,93), 125 (84,02), 115 (31,50), 77 (25,86).

-45-
Dados para a mistura dos isômeros E e Z (1S,3S)-3-[2-(pentafluorofenil)eten-1-il]-
2,2-dimetilciclopropano-1-carboxilato de metila (1.13):
CO2CH3
F
F
FF
F
1
2
3
45
4
5
CO2CH3F
F
F
FF
1
2
3
(1.13a) (1.13b)
CCD: Rf = 0,63 (hexano : éter dietílico 9:1).
Rotação específica: 20][ Dα = - 74,6 ° (c = 2,60, acetona).
IV (KBr, cm -1) ν : 2952, 1734, 1651, 1521, 1495, 1219, 1171, 1002, 869.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,23 (s, 6H, 2 x CH3), 1,34 (s, 6H, 2 x CH3) 1,65 (d,
1H, J1-3cis = 5,4, H1cis), 1,75 (d, 1H, J1-3trans = 5,4, H1trans), 2,16 (m, 1H, H3cis), 2,23 (m,
1H, H3trans), 3,65 (s, 3H, OCH3), 3,67 (s, 3H, OCH3), 5,75 (dd, 1H, J3-4cis = 9,6 e J4-5cis =
11,1, H4cis), 6,14 (d, 1H, J4-5cis = 11,1, H5cis), 6,27 (dd, 1H, J3-4trans = 9,0 e J4-5trans =
16,2, H4trans), 6,46 (d, 1H, J4-5trans = 16,2, H5trans).
RMN de 13C (75 MHz; CDCl3) δδδδ: 20,5 (2 x CH3), 22,3 (CH3), 22,5 (CH3), 29,8
[C(CH3)2], 30,0 [C(CH3)2], 34,1 (C3cis), 34,2 (C1trans), 35,7 (C1cis), 37,7 (C3trans), 51,9
(OCH3), 52,0 (OCH3), 114,7 (C5cis), 115,8 (C5trans), 137,4 (C4cis e C4 trans), 172,0 (2 x
C=O).
EM, m/z (I): [ +•M ] 320 (8,37), 261 (100,00), 195 (24,67), 181 (98,29), 139 (38,03), 59
(38,77), 41 (57,36).

-46-
Dados para a mistura dos isômeros E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-
dimetilciclopropano-1-carboxilato de metila (1.14):
(1.14a) (1.14b)
CO2CH3
OEt
1
2
3
45
4
5CO2CH3
EtO
1
2
3
1'2'
3'
4'
1'
2'
3'4'
CCD: Rf = 0,52 (hexano : éter dietílico 9:1).
Rotação específica: 20][ Dα = - 71,4 ° (c = 2,80, acetona).
IV (KBr, cm -1) ν : 2979, 2949, 1725, 1607, 1510, 1245, 1167, 1048, 841, 732.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,20 (s, 3H, CH3), 1,21 (s, 3H, CH3), 1,30 (s, 3H,
CH3), 1,32 (s, 3H, CH3), 1,54 (d, 1H, J1-3cis = 5,4, H1cis), 1,64 (d, 1H, J1-3trans = 5,4,
H1trans), 2,18 (m, 1H, H3trans), 2,40 (m, 1H, H3cis), 3,67 (s, 3H, OCH3), 3,69 (s, 3H,
OCH3), 4,03 (m, 4H, 2 x OCH2), 5,30 (dd, 1H, J3-4cis = 8,4 e J4-5cis = 11,5, H4cis), 5,82
(dd, 1H, J3-4trans = 8,7 e J4-5trans = 15,6, H4trans), 6,46 (m, 2H, H5trans e H5cis), 6,85 ( m,
4H, H3’), 7,25 (m, 4H, H2’).
RMN de 13C (75 MHz; CDCl3) δδδδ: 15,1 (2 x OCH2CH3), 20,4 (2 x CH3), 22,4 (2 x
CH3), 29,5 [C(CH3)2], 39,8 [C(CH3)2], 33,4 (C3cis), 34,5 (C1trans), 35,7 (C1cis), 37,1
(C3trans), 51,8 (2 x OCH3), 63,36 (2 x OCH2), 114,4 (C3’), 114,7 (C3’), 125,0 (C4trans),
126,7 (C4cis), 127,2 (2 x C1’), 129,8 (C2’), 130,2 (C2’), 131,5 (C5), 131,6 (C5), 158,1
(C4’), 158,5 (C4’), 172,7 (2 x C=O).
EM, m/z (I): [ +•M ] 274 (51,44), 215 (100,00), 199 (35,28), 107 (50,20), 77 (44,56), 29
(62,83).

-47-
1.4.11. Isomerização Fotoquímica da mistura de E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14)
Fez-se uma solução na concentração de 1000 ppm da mistura dos isômeros E e Z
(1.14) (30 mg) em benzeno anidro (30 mL). Reservou-se uma alíquota desta solução (1
mL) e transferiu-se o restante da solução para o reator de vidro mostrado na Figura 3(a),
ligado a um fluxo de água gelada. Borbulhou-se N2 durante 1 h tampando-se o reator. A
solução foi irradiada por 5 h utilizando-se lâmpada de mercúrio de alta pressão. Retirou-
se uma alíquota (1 mL) da mistura reacional 0,5 h, 1,0 h, 2,0 h e 5,0 h após irradiação.
As 5 alíquotas retiradas antes e durante a irradiação foram submetidas a CG-EM para
observação da possível mudança na proporção dos isômeros.

-48-
CAPÍTULO 2 - ADIÇÃO FOTOQUÍMICA DE ÁLCOOL ISOPROPÍL ICO COMO ETAPA INTERMEDIÁRIA NA SÍNTESE DE PIRETRÓIDES
2.1. INTRODUÇÃO
Sabe-se que alguns piretróides derivados do ácido cis-crisantêmico possuem
atividade superior quando comparados com os piretróides derivados do ácido trans-
crisantêmico. Um exemplo importante é o caso da deltametrina, sintetizada por Elliott e
colaboradores, derivada do ácido (1R,3S)-cis-crisantêmico. Ela possui elevada atividade
inseticida quando comparada com vários outros piretróides sintéticos e com a piretrina I
(ELLIOTT et al., 1974). Abaixo estão apresentadas as estruturas da piretrina I
(componente mais ativo do piretro natural) e da deltametrina (inseticida comercial para
proteção de grãos armazenados, componente ativo em loções e shampoos contra
piolhos, entre outras aplicações).
O
O
O
HPiretrina I
DeltametrinaBr
Br
O
O
H CNO
Com a descoberta de piretróides altamente ativos derivados do ácido cis-
crisantêmico, vários grupos de pesquisa se dedicaram à produção deste ácido. Uma rota

-49-
sintética bastante interessante foi desenvolvida por R. L. Funk e J. D. Munger em 1984,
tendo como material de partida o ácido 3,3-dimetilpent-4-inóico facilmente disponível
que foi convertido no ácido (±) cis-crisantêmico em cinco etapas como mostrado na
Figura 2.1 (FUNK & MUNGER, 1984). Na primeira reação utilizou-se butillítio para
retirar o hidrogênio do alquino terminal formando um carbânion que, em seguida, se
adicionou à carbonila da acetona. O produto da reação anterior foi submetido a uma
reação de hidrogenação catalítica parcial, ocorrendo adição sin de hidrogênio à tripla
ligação e subseqüente lactonização do cis-alqueno. A lactona foi submetida à reação de
sililação originando um acetal que sofreu rearranjo ao ser dissolvido em clorofórmio a
65 °C. Este rearranjo, conhecido como rearranjo de Claisen, ocorreu com a formação de
dois possíveis estados de transição mostrados na Figura 2.2. Um dos estados de
transição levou ao éster (1R, 3S)-cis-crisantêmico e o outro levou ao éster (1S, 3R)-cis-
crisantêmico. Finalmente, os ésteres foram hidrolisados com ácido fluorídrico em
acetonitrila para formar uma mistura dos ácidos cis-crisantêmicos.
OH
O
H
OH
O
OH
O
O
O
OR
H2/Pd-BaSO4
quinolina90%
i) (i-Pr)2NLi, THFii) t-BuMe2SiCl
CHCl3, 65 °C
97%
100%94%
R = t-BuMe2Si
HF, CH3CN
O
ORH
H
O
OHH
H
65%
i) BuLi, THF
ii) (CH3)2C=O (- 41°C)iii) ácido
+ enantiômero+ enantiômero
Figura 2.1: Síntese do ácido (±) cis-crisantêmico a partir do ácido 3,3-dimetilpent-4-inóico.

-50-
OR
H
HO
O
H
HOR
O
ORH
H O
ORH
H
O
OR(a)
(b)
Figura 2.2: Estados de transição formados durante o rearranjo de Claisen. (a) Formação do éster (1R, 3S)-cis-crisantêmico. (b) Formação do éster (1S, 3R)-cis-crisantêmico..
Em 1988, Krief e colaboradores estudaram a síntese do ácido (±) cis-
crisantêmico tendo como etapa chave a ciclopropanação da 2,2,5,5-tetrametilciclohexa-
1,3-diona em meio básico na presença de Br2, como mostrado na primeira etapa da
Figura 2.3. A redução do produto da ciclopropanação para produção do álcool exo foi
feita testando-se vários agentes redutores e condições reacionais, concluindo-se que o
melhor resultado foi obtido pela redução com 1 equi. de NaBH4 e 1 equi. de CeCl3 em
éter/metanol a -78 °C. Esta redução leva a formação de dois enantiômeros, um pela
redução de uma carbonila e outro pela redução da outra carbonila (apenas um
enantiômero foi representado na Figura 2.3). A mistura racêmica dos álcoois exo foi
convertida na mistura dos β-ceto mesilatos correspondentes por reação com cloreto de
metanossulfonila. Finalmente, ao reagir a mistura de mesilatos com hidróxido de
potássio ocorre a abertura do anel de cinco átomos de carbono, produzindo o ácido cis-
crisantêmico como uma mistura de enantiômeros (KRIEF, 1988).

-51-
O
O
O
O
O
OH
i) t-BuOK, THF
ii)Br2, +40 °C
67%
NaBH4, CeCl3
-78 °C
88%
MsCl, NEt3DCM, 20 °C
85%
KOH, DMS/H2O
70 °C69%
ácido (+) cis-crisantêmico
O
H
HO
O
H
MsO
Figura 2.3: Síntese do ácido (±) cis-crisantêmico a partir do 2,2,5,5-tetrametilciclohexa-1,3-diona.
Posteriormente, esta rota foi modificada para produção do ácido (1R, 3S)-cis-
crisantêmico a partir da síntese quimio e enantiosseletiva do β-ceto mesilato como
mostrado resumidamente na Figura 2.4. O diol foi obtido pela redução do reagente
dicarbonílico utilizando-se 2 equi. de NaBH4 e 2 equi. de CeCl3 em éter/metanol a -78
°C. Em seguida, o diol foi acetilado pela reação com anidrido acético. O produto
diacetilado foi hidrolisado por uma enzima estearase encontrada em fígado de porco
(PLE), quimio e enantioseletiva, levando a produção do álcool enantiomericamente puro
mostrado na Figura 2.4. O álcool foi oxidado com o uso de um complexo de óxido de
crômio (VI) e piridina, conhecido como reagente de Collins. O grupo acetila foi
hidrolisado e substituído pelo grupo mesila. Finalmente, o β-ceto mesilato formado na
etapa anterior foi convertido no ácido (1R, 3S)-cis-crisantêmico por meio de reação com
hidróxido de potássio em dimetilsulfóxido e água (KRIEF, 1993).
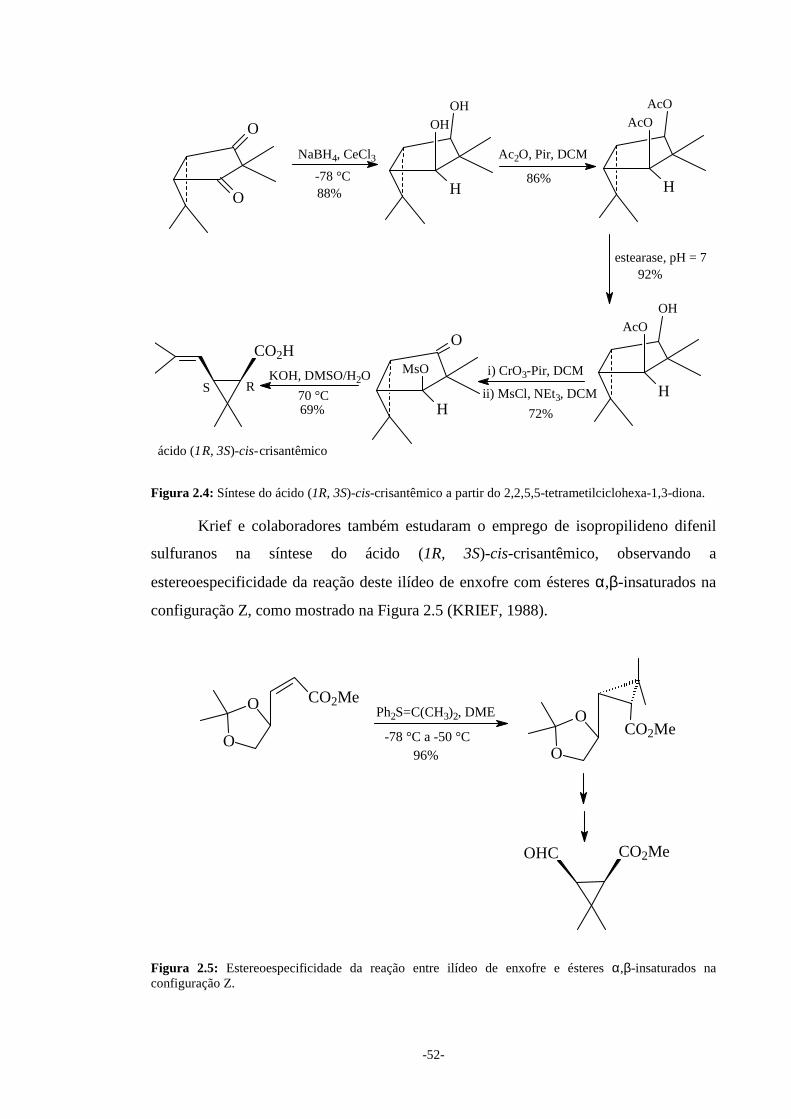
-52-
O
O
OH
H
OH
O
H
MsO
NaBH4, CeCl3
-78 °C
72%
ii) MsCl, NEt3, DCM
88%
ácido (1R, 3S)-cis-crisantêmico
AcO
H
AcO
OH
H
AcO
CO2H
KOH, DMSO/H2O
70 °C69%
i) CrO3-Pir, DCM
Ac2O, Pir, DCM
86%
RS
estearase, pH = 792%
Figura 2.4: Síntese do ácido (1R, 3S)-cis-crisantêmico a partir do 2,2,5,5-tetrametilciclohexa-1,3-diona.
Krief e colaboradores também estudaram o emprego de isopropilideno difenil
sulfuranos na síntese do ácido (1R, 3S)-cis-crisantêmico, observando a
estereoespecificidade da reação deste ilídeo de enxofre com ésteres α,β-insaturados na
configuração Z, como mostrado na Figura 2.5 (KRIEF, 1988).
O
O CO2MePh2S=C(CH3)2, DME
-78 °C a -50 °C96% O
OCO2Me
OHC CO2Me
Figura 2.5: Estereoespecificidade da reação entre ilídeo de enxofre e ésteres α,β-insaturados na configuração Z.

-53-
John Mann e Alexander Weymouth-Wilson desenvolveram uma nova rota para a
síntese de um importante precursor do ácido (1R, 3S)-cis-crisantêmico. Nesta rota
sintética, utilizaram-se como etapa chave uma reação de adição fotoquímica de álcool
isopropílico a γ-lactonas α,β-insaturadas, representada na Figura 2.6. O álcool terciário
produzido pela reação fotoquímica foi desidratado por meio de reação com pentacloreto
de fósforo em DCM. O alqueno foi submetido à reação de adição de brometo de
hidrogênio em ácido acético. O brometo formado foi ciclisado por reação com t-
butóxido de potássio em THF produzindo um composto contendo um anel ciclopropano
que pode ser facilmente convertido no ácido (1R, 3S)-cis-crisantêmico (MANN &
WEYMOUTH-WILSON, 1991).
O OHO
O OHO
OH
O OBzO
O OBzO
i-PrOH254 nm
O OBzO
Br
HBr, AcOH
94%
98%
ii) PCl5, DCM, 0 °C95%
t-BuOK, THF
51%
0 °C
i) BzCl, pir, 97%
Figura 2.6: Síntese de um precursor do ácido (1R, 3S)-cis-crisantêmico utilizando reação de fotoadição de álcool isopropílico a γ-lactonas α,β-insaturadas.
As reações apresentadas neste capítulo se baseiam no trabalho de John Mann
mostrado acima. Primeiramente, utilizou-se como material de partida para produção de
um intermediário para síntese do ácido (+) cis-crisantêmico o furfural que é um
reagente barato de fácil disponibilidade.
Posteriormente, utilizou-se a reação de adição fotoquímica de álcool isopropílico
a γ-lactonas α,β-insaturadas com substituinte na posição γ, para obtenção de alguns
compostos enantiomericamente puros. Estes compostos foram produzidos para serem
submetidos a testes de atividade inseticida, levando em consideração que alguns
piretróides com comprovada ação inseticida não possuem anel de três átomos de

-54-
carbono com metilas geminais. Abaixo estão mostradas as estruturas de dois
representantes importantes deste tipo de piretróides: o fenvalerato e o etofenprox.
Fenvalerato
O
OO
CNCl
EtofenproxO
O
C2H5O

-55-
2.2. RESULTADOS E DISCUSSÃO
A reação de adição fotoquímica de álcool isopropílico a γ-lactonas α,β-insaturadas
pode ser usada como etapa chave na síntese do ácido cis-crisantêmico (e também vários
piretróides), pois o produto da adição pode ser facilmente convertido a um composto
contendo um anel ciclopropano com duas metilas geminais (MANN & WEYMOUTH-
WILSON, 1991).
Na Figura 2.7 estão representadas as reações utilizadas na síntese da 6,6-dimetil-3-
oxabiciclo[3.1.0]hexan-2-ona (2.6), importante intermediário para a síntese do ácido cis-
crisantêmico.
(2.4a)
O
Br
O
(2.5)
O OO O
(2.6)
PCl5
PBr3/SiO2
DCM
DCM 90%
39%
50%
t-BuOK / THF
40%
i) PBr3/ DCM /pir.ii) SiO2
O O
+
(2.4b)
O CHO O OO
OH
O
(2.1) (2.2)(2.3)
i-PrOHH2O2/HCO2H
hν40%92%
Figura 2.7: Síntese da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6).
O furfural (2.1) foi oxidado pelo ácido perfórmico formado a partir da mistura de
ácido fórmico e peróxido de hidrogênio. De acordo com o mecanismo de oxidação do
furfural dado na Figura 2.8, além da formação da 5H-furan-2-ona (2.2a), forma-se também
a 3H-furan-2-ona (2.2b). Nesta reação utilizou-se N,N-dietiletanolamina para promover a
isomerização da 3H-furan-2-ona (2.2b) para 5H-furan-2-ona (2.2a) ao invés de N,N-
dimetiletanolamina, como descrito na literatura, e observou-se resultados semelhantes (40%
de rendimento). De acordo com a literatura, uma vantagem da utilização deste amino álcool
é que ele forma também um complexo com o ácido fórmico fazendo com que este não
passe eficientemente para a fase orgânica, o que torna o produto mais fácil para ser isolado.

-56-
Acredita-se que a oxidação ocorra na fase aquosa, pois reações testadas com furfurais
substituídos, menos solúveis em água, não foram bem sucedidas (NÄSMAN, 1990).
O OC
O
H
O O
H
H+
H+O O
O O
(2.2a) (2.2b)
(2.2a)
1
122
HCO2H
N,N-dietiletanolamina
OC H
O O C
O
H
O H
O CO
H
H OO
H
O
HCO2H
H2O
O O
Figura 2.8: Mecanismo da reação de oxidação do furfural (2.1).
Após o término da reação separou-se a fase orgânica, lavando-a com solução
saturada de bissulfito de sódio. Os íons bissulfito reagem com peróxidos eliminando os
resíduos presentes na fase orgânica. Em seguida, fez-se teste para detecção da presença de
resíduos de peróxido adicionando a fase orgânica a um tubo contendo solução ácida de
sulfato ferroso amoniacal (como fonte de Fe2+) e tiocianato de potássio. O peróxido oxida
os íons Fe2+ a Fe3+ que formam um complexo vermelho ao se coordenar com íons tiocianato
e água. Assim, a presença de peróxido é detectada pela mudança de coloração da solução de
amarelo claro para vermelho. Quando não ocorre mudança de coloração da solução tem-se
resultado negativo para presença de peróxido e pode-se continuar a elaboração da reação.
Abaixo estão representadas as equações balanceadas dessas reações:
Reação de eliminação de peróxido: +−− ++→+ 2H OH2SOOSO2H 22
42
5222
Reação de oxidação do Fe(II): O2H2Fe2HOH2Fe 23
222 +→++ +++
Reação de complexação: +−+ →++ 2522
3 ]O)[Fe(SCN)(HO5HSCNFe
vermelho

-57-
O espectro no IV do produto apresentado no Anexo 39 apresenta duas bandas
intensas em ν 1775 e 1741 cm-1 uma referente ao estiramento da ligação C=O e outra
devido à ressonância de Fermi. Os espectros de RMN de 1H e 13C (Anexos 40 e 41)
confirmam a formação da 5H-furan-2-ona (2.2a).
A 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) foi preparada pela adição
fotoquímica de álcool isopropílico ao carbono β da 5H-furan-2-ona (2.2a) (OHGA e
MATSUO, 1974). Esta reação se processou com rendimento quantitativo. Um possível
mecanismo para esta reação proposto por John Mann e colaboradores está representado na
Figura 2.9. Primeiramente, a lactona absorve um fóton de luz UV passando do estado
fundamental para o estado excitado. Em seguida, um elétron é transferido de uma molécula
de álcool isopropílico para a lactona no estado excitado. Ocorre então a transferência de um
próton do álcool para a lactona seguida da adição do radical do álcool para formar o produto
em sua forma enólica. Finalmente, ocorre tautomerização da forma enólica para a forma
ceto mais estável (MANN & WEYMOUTH-WILSON, 1994).
HO H
O O
HO H
O OH
OH
O Ohν
O O
OH
O
OH
OH
(2.3)
(2.2a)
O O *
Figura 2.9: Mecanismo da adição fotoquímica de álcool isopropílico à 5H-furan-2-ona (2.2a)
A banda larga observada no espectro no IV (Anexo 42) ν 3428 cm-1 refere-se ao
estiramento da ligação O-H. No espectro de RMN de 1H (Anexo 43) o aparecimento do
multipleto em δ 1,28 integrado para 6H e o desaparecimento dos sinais referentes aos
átomos de hidrogênios olefínicos evidenciam a formação do produto esperado (2.3). No
espectro de RMN de 13C (Anexo 44) os sinais em δ 27,9 e δ 28,3 atribuídos aos grupos CH3
e o sinal em δ 70,0 atribuído ao carbono não hidrogenado do álcool (2.3) confirmam sua
formação.

-58-
Os alquenos 4-isopropeniltetraidrofuran-2-ona (2.4a) e 4-(1-
metiletilideno)tetraidrofuran-2-ona (2.4b) foram preparados a partir da mistura racêmica da
4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) por eliminação de água, utilizando-se
PCl5 em DCM anidro, em 90% de rendimento total. O mecanismo da reação de eliminação
mostrando a formação do produto principal (2.4a) está representado na Figura 1.10. Os
produtos (2.4a) e (2.4b) não foram totalmente separados por cromatografia em coluna,
dessa forma, as frações obtidas puras foram usadas para identificação por meio de
espectroscopia de IV, RMN de 1H e RMN de 13C, e a mistura contendo quantidade
predominante do isômero (2.4a) foi utilizada na próxima etapa da rota.
O O
CH3
CH2
POCl3
HCl+
O O
CH3
H2CO P
Cl
Cl
Cl
ClH
O O
CH3
CH3O
H
P
Cl
Cl
Cl
ClCl
(2.3) (2.4a)
HCl
Figura 2.10: Mecanismo da reação de eliminação de água.
No espectro no IV do isômero (2.4a) (Anexo 45) pode-se observar uma banda em
ν 1650 cm-1 referente ao estiramento da ligação C=C e desaparecimento da banda larga em
ν 3428 cm-1, indicando o sucesso da reação de eliminação. No espectro de RMN de 1H do
Anexo 46 o simpleto em δ 1,73 integrado para 3H atribuído ao CH3 e o multipleto em δ
4,85 integrado para 2H atribuído ao =CH2 são os principais sinais que indicam a formação
do alqueno (2.4a). No espectro de RMN de 13C (Anexo 47) as atribuições dos sinais em δ
33,2 e δ 42,5 aos C3 e C4, respectivamente, foram confirmadas por meio do espectro de
DEPT.
No espectro no IV do isômero (2.4b) (Anexo 48) pode-se observar uma banda em
ν 1648 cm-1 referente ao estiramento da ligação C=C e desaparecimento da banda larga em
ν 3428 cm-1. As atribuições dos sinais em δ 1,60 e 1,68 aos grupos CH3 e CH3’,
respectivamente, no espectro de RMN de 1H (Anexo 49) foram feitas com o auxílio de
COSY. No espectro de COSY o sinal em δ 1,60 apresentou maior correlação com o
multipleto em δ 3,13 atribuído ao CH2 α a carbonila, logo corresponde ao CH3. Da mesma
forma, o sinal em δ 1,68 apresentou maior correlação com o multipleto em δ 4,83 atribuído

-59-
ao O-CH2, logo corresponde ao CH3’. Os sinais em δ 19,6 e δ 21,6 no espectro de RMN de 13C (Anexo 50) foram atribuídos aos grupos CH3 e CH3’, respectivamente, com auxílio de
HETCOR.
A reação da mistura de alquenos (2.4a) e (2.4b) com PBr3/SiO2 em DCM levou ao
produto da adição de HBr seguindo a regra de Markovnikov. A mistura de enantiômeros 4-
(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) foi obtida em 39% de rendimento. Esta
reação tem como vantagem não necessitar de condições anidras e não levar a formação de
produtos da adição anti-Markovnikov, comuns devido à presença de luz ou O2 no meio
reacional (SANSEVERINO e MATTOS, 2001). Uma proposta para o mecanismo desta
reação está representada na Figura 2.11.
Si
OH
OO
O H O H
SiO
OO
H
Br
P
Br
Br
Si
OH
OO
O H O H
SiO
OO
H
P
Br
Br
Si
OH
OO
O H O H
SiO
OO P
Br
Br
Br-
+
O
C
CH3
H
H
O
O O
CH3
CH3Br
(2.4a)
(2.5)
Figura 2.11: Mecanismo da adição de HBr ao alqueno 4-isopropeniltetraidrofuran-2-ona (2.4a).
Foi desenvolvida uma metodologia para conversão dos álcoois (2.3) no brometo
(2.5) sem a necessidade de isolar os alquenos (2.4a) e (2.4b). Nesta metodologia adicionou-
se lentamente um excesso de PBr3 a uma solução dos álcoois (2.3) e piridina (1 eq. molar)
em DCM e, após alguns minutos, adicionou-se SiO2. A mistura reacional juntamente com
sílica foi agitada por meia hora e filtrada em seguida. O filtrado foi lavado com solução de
NaHCO3 10% e solução de NaCl, secado com MgSO4 e concentrado sob pressão reduzida.
O resíduo foi purificado por meio de cromatografia em coluna, originando o brometo 4-(1’-

-60-
bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) em 50% de rendimento. Desta forma, há a
economia de uma etapa na rota sintética e aumento significativo no rendimento da
conversão de (2.3) em (2.5) de 35% (no caso da desidratação seguida por adição de HBr)
para 50% (no caso da conversão do álcool no brometo). Como se trata de um álcool
terciário não é provável que ocorra a substituição direta do grupo hidroxila pelo brometo
como esperado para álcoois primários e secundários. Assim, suspeita-se que ocorra
inicialmente a eliminação de água catalisada por PBr3 seguida pela adição de HBr aos
alquenos formados.
As características físicas e dados espectroscópicos de ambos os produtos das
reações acima coincidiram perfeitamente. No espectro no IV (Anexo 51) pode-se observar
uma banda intensa em ν 1779 cm-1 referente ao estiramento C=O. O espectro de RMN de 1H é mostrado no Anexo 52 e as atribuições dos sinais do RMN de 13C (Anexo 53) foram
feitas com auxílio de espectro de DEPT.
A 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) foi tratada com tert-butóxido
de potássio em THF anidro para formar a 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6),
em 40% de rendimento. A base forte retirou um hidrogênio α à carbonila e o carbânion
formado sofreu ciclização por substituição nucleofílica no carbono ligado ao brometo. No
espectro no IV da lactona (2.6) (Anexo 54) pode-se observar uma banda intensa em ν 1774
cm-1 referente ao estiramento da ligação C=O. No espectro de RMN de 1H (Anexo 55), de
acordo com os deslocamentos químicos supõe-se que os dois sinais em δ 1,88 e δ 2,01
correspondem a H3 e H4 e os sinais em δ 4,10 e δ 4,31 correspondem aos átomos de
hidrogênio do CH2. Para completar as atribuições destes quatro sinais foram feitos
experimentos de dupla irradiação. Primeiramente irradiou-se o sinal em δ 2,01 observando a
simplificação dos sinais em δ 4,10 e δ 4,31 e confirmando a existência de um maior
acoplamento (J = 5,2) com o sinal de deslocamento químico maior, assim, atribuiu-se
inicialmente o sinal em δ 2,01 ao H4, o sinal em δ 4,31 ao H5 (cis ao H4) e o sinal em δ
4,10 ao H5’ (trans ao H4). Ao irradiar o sinal em δ 1,88 observou-se o desacoplamento do
sinal em δ 4,10 e nenhuma mudança no sinal em δ 4,31, logo ao atribuir o sinal em δ 1,88
ao H3 observa-se um acoplamento a longa distância entre o H3 e o H5’ (J4 = 0,9). Para
confirmar estas atribuições irradiou-se o sinal em δ 4,31 e observou-se a simplificação do
sinal em δ 2,01 e nenhuma mudança no desdobramento do sinal em δ 1,88. Ao irradiar o
sinal em δ 4,10 observou-se a simplificação dos sinais em δ 2,01 e δ 1,88. Na Figura 2.12
está representado um esquema indicando as constantes de acoplamento do composto (2.6).

-61-
As atribuições dos sinais no espectro de RMN de 13C (Anexo 56) foram confirmadas por
espectro de DEPT.
O O
HH
H
H
34
5
5'0,9
1,29,9
6,3
5,2
Figura 2.12: Esquema indicando as constantes de acoplamento do composto (2.6)
A Figura 2.13 apresenta a rota utilizada na síntese dos álcoois enantiomericamente
puros (2.9), (2.10) e (2.11), de acordo com a estereosseletividade da reação de adição
fotoquímica de álcool isopropílico às γ-lactonas α,β-insaturadas (2.7) e (2.8) (MANN &
WEYMOUTH-WILSON, 1994).
O
OH
OHO
OO
CO2CH3
O OHO O OSiO
O OSiO
OH
(2.9)
O
OH
OO
O
(2.11)
(1.4)
(2.7) (2.8)
(2.10)
H2SO4 cat.
MeOH
TBDMSCl
imidazol / DCM
i-PrOHhν
i-PrOHhν
BzClpir.
73% 60%
66%90%
90%
S
S
S
S
S
S
S
S
Figura 2.13: Síntese das lactonas (2.9) e (2.11).
Bz = benzoíla

-62-
A γ-lactona α,β-insaturada (S)-5-hidroximetil(5H)furan-2-ona (2.7) foi obtida em
73% de rendimento pela hidrólise do grupo acetal do (S)-(Z)-4,5-O-isopropilidenopent-2-
enoato de metila (1.4), seguida por ciclização do diol formado (MANN et al., 1987). Na
Figura 2.14 está representada uma proposta para o mecanismo desta reação. A síntese do
éster (1.4) foi discutida no Capítulo 1.
OO
C
O
OCH3H
OHO
H
OCH3
OH
H
OOC
O
OCH3
HH
H
+ H+
OHO O H
H
OHO O
H
OOC
O
OCH3
HHH
H
OHO
OCH3
OH
H
H
HOCH3
H2O/H+
H+
(1.4)
(2.7)
Figura 2.14: Mecanismo da reação de formação da (S)-5-hidroximetil(5H)furan-2-ona (2.7).
No espectro no IV (Anexo 57) podemos observar uma banda larga em ν 3414 cm-1
referente ao estiramento da ligação O-H, uma banda intensa em ν 1746 cm-1 referente ao
estiramento da ligação C=O e uma banda em ν 1602 cm-1 referente ao estiramento da
ligação C=C, que são fortes indicativos da formação do composto (2.7). No espectro de
RMN de 1H (Anexo 58) podemos notar o desaparecimento dos sinais das metilas geminais
do grupo acetal e da metila do grupo éster presentes no espectro de RMN de 1H do reagente
(1.4). A análise e atribuição dos sinais observados no espectro de RMN de 1H (Anexo 58) e
RMN de 13C (Anexo 59) confirmam a formação do produto esperado.
Formou-se a lactona (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8) pela
proteção do grupo hidroxila da (S)-5-hidroximetil(5H)furan-2-ona (2.7) por reação com
cloreto de tert-butildimetilsilano em DCM anidro na presença de imidazol em 60% de
rendimento. No espectro no IV (Anexo 60) observou-se o desaparecimento da banda larga
de estiramento da ligação O-H acima de ν 3000 cm-1, sendo um forte indício da formação
do produto esperado (2.8). No espectro de RMN de 1H (Anexo 61) nota-se a presença de

-63-
dois simpletos em δ 0,06 e δ 0,07 integrados para 6H atribuídos às metilas ligadas ao Si e de
um simpleto em δ 0,87 integrado para 9H atribuídos às metilas do grupo tert-butil. No
espectro de 13C (Anexo 62) observam-se sinais em δ -5,0, δ 18,7 e δ 26,3 atribuídos a
Si(CH3)2], [C(CH3)3] e [C(CH3)3], respectivamente, confirmando a formação do produto.
A reação de adição fotoquímica de álcool isopropílico à 5H-furan-2-ona (2.2a)
ocorre com a produção de uma mistura racêmica do ácool terciário (2.3). Já no caso da
fotoadição aos compostos (S)-5-hidroximetil(5H)furan-2-ona (2.7) e (S)-5-O-tert-
butildimetilsiloximetil(5H)furan-2-ona (2.8) esta reação, além de ser regioseletiva, é
também estereosseletiva. No caso do substrato (2.8), esta estereosseletividade é facilmente
explicada pela presença do grupo protetor que causa impedimento espacial em uma das
faces da molécula provocando a adição preferencial na face oposta, para formar a (4S,5S)-5-
O-tert-butildimetilsiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.9) em 66%
de rendimento. Já no caso do substrato (2.7), a estereosseletividade pode ser a conseqüência
da formação da ligação de hidrogênio entre o grupo hidroxila do substrato com o álcool
isopropílico, favorecendo a adição na fase oposta ao grupo hidroximetil, formando a
(4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil)tetraidro- furan-2-ona (2.10) em 90% de
rendimento (MANN & WEYMOUTH-WILSON, 1994).
No Anexo 63 tem-se o espectro no IV do produto da fotoadição do álcool
isopropílico à lactona (2.8), mostrando o aparecimento de uma banda larga em ν 3459 cm-1
de estiramento da ligação O-H. No espectro de RMN de 1H (Anexo 64) observa-se o
aparecimento de dois simpletos em δ 1,20 e δ 1,23 atribuídos as duas metilas geminais
ligadas ao C-OH e de um sinal largo em δ 1,91 atribuído ao hidrogênio do grupo OH, além
do desaparecimento dos sinais dos átomos de hidrogênio olefínicos do substrato (2.8).
Observa-se também o aparecimento de um multipleto em δ 2,43 atribuído aos H3 e H4 e
um dupleto duplo em 2,68 atribuído ao H3’. A análise do espectro de RMN de 1H
juntamente com o RMN de 13C (Anexo 65) confirma a formação da lactona (2.9).
No espectro no IV (Anexo 66) do produto da fotoadição de álcool isopropílico a (S)-
5-hidroximetil(5H)furan-2-ona (2.7) observa-se duas bandas largas parcialmente
sobrepostas em ν 3349 cm-1 e ν 3412 cm-1 referentes ao estiramentos das ligações O-H de
duas hidroxilas e a ausência da banda de estiramento C=C em ν 1602 cm-1 presente no
espectro no IV do substrato (Anexo 57). No espectro de RMN de 1H (Anexo 67) observa-se
a presença de dois simpletos em δ 1,08 e δ 1,10 integrados para 6H e atribuídos às metilas
geminais e ausência dos sinais dos átomos de hidrogênio olefínicos do substrato (2.7)

-64-
indicando a formação do produto (2.10), confirmada pela análise e atribuição dos sinais do
espectro de RMN de 13C (Anexo 68).
O (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.11)
foi preparado a partir da benzoilação do álcool primário da lactona (2.10). No espectro no
IV (Anexo 69) pode-se observar duas bandas em ν 1708 cm-1 e ν 1744 cm-1 referentes ao
estiramento da ligação C=O de duas carbonilas e uma banda em ν 3441 cm-1 referente ao
estiramento da ligação O-H do álcool terciário presentes no composto (2.11). No espectro
de RMN de 1H (Anexo 70) o principal indicativo da formação do produto benzoilado (2.11)
é o aparecimento de sinais entre δ 7-8 referentes aos átomos de hidrogênio aromáticos do
grupo benzoíla. No espectro de RMN de 13C (Anexo 71) observamos também a presença de
quatro sinais (entre δ 128,7 e δ 133,6) referentes aos átomos de C aromáticos e o
aparecimento de um sinal em δ 166,5 referente ao carbono da carbonila do grupo benzoíla,
confirmando a formação do composto (2.11).

-65-
2.3. CONCLUSÃO
A reação de adição fotoquímica de álcool isopropílico foi uma maneira eficiente
para produção dos compostos (2.3), (2.9) e (2.10) a partir de γ-lactonas α,β-insaturadas.
O álcool 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) formado em 100%
de rendimento pela reação de fotoadição de álcool isopropílico a 5H-furan-2-ona (2.2a)
pôde ser facilmente convertido na 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6), que
é um intermediário chave para a síntese do ácido (+) cis-crisantêmico.
Os álcoois (4S,5S)-5-O-tert-butildimetilsiloximetil-4-(1’-hidroxi-1’-
metiletil)tetraidrofuran-2-ona (2.9) e (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-
metiletil)tetraidrofuran-2-ona (2.10) foram obtidos em bons rendimentos (66% e 90%,
respectivamente) pela adição de álcool isopropílico às lactonas correspondentes. Estes
compostos poderão ser utilizados para a formação de produtos contendo anel gem-
dimetilciclopropano assim como o álcool (2.3).

-66-
2.4. EXPERIMENTAL
2.4.1. Síntese da 5H-furan-2-ona (2.2a)
O O
(2.2a)
A um balão tritubulado de 500 mL equipado com dois condensadores ligados a
um refrigerador de água, barra magnética e um funil de adição de líquido, adicionou-se
furfural (2.1) (21 mL), DCM (100 mL), Na2SO4 (10,7 g) e N,N-dietiletanolamina (8,5
mL). A mistura foi mantida sob agitação vigorosa durante a adição gota-a-gota de ácido
fórmico (19 mL) seguida da adição rápida de H2O2 30% (5 mL). A mistura reacional foi
mantida sob refluxo por 10 min. O sistema foi resfriado à temperatura ambiente e H2O2
30% (50 mL) foi adicionado gota-a-gota durante 3 h sob agitação vigorosa. A mistura
reacional foi mantida sob refluxo durante 18 h. O sistema foi resfriado a temperatura
ambiente, a fase orgânica foi separada e a fase aquosa extraída com DCM (3 x 20 mL).
As fases orgânicas foram reunidas e lavadas com solução saturada de Na2S2O5 (3 x 10
mL). Fez-se teste para detecção de peróxido (resultado negativo) e o DCM foi
evaporado sob pressão reduzida. O produto foi purificado por cromatografia em coluna
(acetato de etila : hexano 2:1) obtendo-se o 5H-furan-2-ona (2.2a) (12,23 g, 145,6
mmol) em 40% de rendimento bruto como um líquido amarelado com pureza suficiente
para utilização na próxima etapa da síntese.
CCD: Rf = 0,60 (acetato de etila : hexano 2:1).
IV (KBr, cm -1) ν : 3108, 2942, 2872, 1775, 1742, 1599, 1160, 1095, 1035, 811, 692.
RMN de 1H (300 MHz, CDCl3) δδδδ: 5,03 (m, 2H, H5 e H5’), 6,25 (m, 1H, H3), 7,77 (m,
1H, H4).
RMN de 13C (75 MHz, CDCl3) δδδδ: 71,8 (C5), 122,0 (C3), 152,5 (C4), 172,2 (C=O).

-67-
2.4.2. Síntese da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3)
O
OH
O
(2.3)
Adicionou-se a um tubo de quartzo 5H-furan-2-ona (2.2a) (4,5 g; 53,57 mmol) e
álcool isopropílico (100 mL). Borbulhou-se N2 durante 1 h tampando-se o tubo com
rolha esmerilhada. A solução foi irradiada por 12 h (λ = 254 nm). O término da reação
foi determinado por CCD. O álcool isopropílico foi evaporado sob pressão reduzida a
35 °C e o resíduo da evaporação submetido à cromatografia em coluna (acetato de etila :
hexano 2:1) obtendo-se o 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) (7,0 g;
48,61 mmol) em 100% de rendimento como um óleo incolor.
CCD: Rf = 0,52 (acetato de etila : hexano 2:1).
IV (KBr, cm -1) ν : 3426, 2978, 2928, 1774, 1378, 1186, 1016, 956, 850.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,28 (m, 6H, 2 x CH3), 2,61 (m, 3H, H3, H3’ e H4),
2,8 (s, 1H, OH), 4,4 (m, 2H, H5 e H5’).
RMN de 13C (75 MHz, CDCl3) δδδδ: 27,9 (CH3), 28,3 (CH3), 30,1 (C3), 46,1 (C4), 69,7
(C5), 70,0 [C(CH3)2], 178,0 (C=O).
EM, m/z (I): [ +•M ] 144 (1,43), 126 (3,40), 111 (3,60), 85 (56,39), 59 (100,00).
2.4.3. Síntese da 4-isopropeniltetraidrofuran-2-ona (2.4a) e da 4-(1-metiletilideno)tetraidrofuran-2-ona (2.4b)
(2.4a)
O O O O
(2.4b)
Adicionou-se uma suspensão de PCl5 (20 g; 95,92 mmol) em DCM anidro (25
mL) a uma solução de 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) (7g; 48,61
mmol) em DCM anidro (40 mL) em banho-de-gelo. O sistema foi deixado sob agitação
por 5 min, adicionou-se H2O (150 mL) e fez extração com éter dietílico (2 x 150 mL). A

-68-
fase orgânica foi lavada com solução saturada de NaCl (3 x 25 mL), secada com MgSO4
e concentrada sob pressão reduzida. Não foi possível a separação completa dos
compostos (2.4a) e (2.4b) por cromatografia em coluna (hexano : acetato de etila 1:1).
Assim, foram obtidas pequenas quantidades de cada isômero puro para identificação
espectroscópica. Obteve-se também uma mistura dos dois isômeros (5,5 g) em 90% de
rendimento global, contendo o composto (2.4a) em maior quantidade. A mistura foi
utilizada na próxima etapa da síntese.
(2.4a)
CCD: Rf = 0,65 (hexano : acetato de etila 1:1).
IV (KBr, cm -1) ν : 3083, 2975, 2916, 1794, 1650, 1179, 1021, 900.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,73 (s, 3H, CH3), 2,43 (dd, 1H, Jgem= 17,1, J3-4=
8,5, H3), 2,62 (dd, 1H, Jgem= 17,1 e J3’-4= 8,5, H3), 3,15 (m, 1H, H4), 4,07 (dd, 1H,
Jgem= 8,8 e J5-4= 7,8, H5), 4,42 (dd, 1H, Jgem= 8,8 e J5’-4= 7,8, H5’), 4,85 (m, 2H, =CH2).
RMN de 13C (75 MHz, CDCl3) δδδδ: 20,5 (CH3), 33,2 (C3), 42,5 (C4), 71,9 (C5), 112,4
(=CH2), 176,9 (C=O).
EM, m/z (I): [ +•M ] 126 (12,36), 125 (28,86), 95 (22,98), 68 (88,19), 67 (100,00), 59
(34,65).
(2.4b)
CCD: Rf = 0,68 (hexano : acetato de etila 1:1).
IV (KBr, cm -1) ν : 2974, 2912, 1784, 1650, 1362, 1180, 1025, 847, 558.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,60 (m, 3H, CH3), 1,68 (m, 3H, CH3’), 3,13 (m,
2H, H3 e H3’), 4,83 (m, 2H, H5 e H5’).
RMN de 13C (75 MHz, CDCl3) δδδδ: 19,6 (CH3), 21,6 (CH3’), 32,4 (C3), 71,6 (C5), 121,8
(C4), 126,8 (C6), 176,7 (C=O).
2.4.4. Síntese da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) a partir da mistura de (2.4a) e (2.4b)
O
Br
O
(2.5)

-69-
Adicionou-se gota-a-gota solução de PBr3 (1,6 mL; 17,02 mmol) em DCM (10
mL) a uma suspensão contendo os isômeros (2.4a) e (2.4b) (5,5 g; 43,65 mmol), SiO2
(15 g) e DCM (50 mL). A mistura reacional foi mantida sob agitação por 1 h, filtrada
sob vácuo e o SiO2 lavado com DCM (50 mL). O filtrado foi lavado com NaHCO3 10%
(2 x 20 mL) e solução saturada de NaCl (2 x 20 mL), secado com MgSO4 e concentrado
sob pressão reduzida. O resíduo foi submetido à cromatografia em coluna (hexano :
acetato de etila 1:1) obtendo-se o 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5)
(3,5 g; 17,02 mmol) em 39% de rendimento como um sólido branco.
CCD: Rf = 0,52 (acetato de etila : hexano 1:1).
IV (KBr, cm -1) ν : 2973, 2917, 1779, 1373, 1175, 1115, 1027, 629.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,75 (m, 6H, 2xCH3), 2,64 (m, 3H, H3, H3’ e H4),
4,29 (m, 1H, H5), 4,45 (m, 1H, H5’).
RMN de 13C (75 MHz, CDCl3) δδδδ: 31,8 (C3), 32,0 (CH3), 32,3 (CH3), 48,2 (C4), 66,5
(C-Br), 70,5 (C5), 175,7 (C=O).
EM, m/z (I): [ +•M + 2] 209 (1,63), [ +
•M ] 207 (1,67), 126 (17,59), 111 (60,31), 83
(100,00), 68 (54,27).
2.4.5. Síntese da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5) a partir da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3)
O
Br
O
(2.5)
Adicionou-se gota-a-gota solução de PBr3 (2,0 mL; 21,28 mmol) em DCM (10
mmL) a uma solução de 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3) (7,00 g;
48,61 mmol) e piridina (4,0 mmL; 49,56 mmol) em DCM (25 mL) mantida sob agitação
vigorosa. Após 1h, adicionou-se novamente gota-a-gota solução de PBr3 (2,0 mL; 21,28
mmol) em DCM (10 mmL), após 20 min, adicionou-se SiO2 (15 g) e, após 40 min, fez-
se filtração a vácuo lavando-se o SiO2 com DCM (50 mL). O filtrado foi lavado com
NaHCO3 10% (2 x 25 mL) e solução saturada de NaCl (25 mL), secado com MgSO4 e
concentrado sob pressão reduzida. O resíduo foi submetido à cromatografia em coluna

-70-
(hexano : acetato de etila 1:1) obtendo-se a 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-
ona (2.5) (5,03 g; 24,30 mmol) em 50% de rendimento como um sólido branco.
CCD: Rf = 0,52 (acetato de etila: hexano 1:1).
IV (KBr, cm -1) ν : 2973, 2917, 1779, 1373, 1175, 1115, 1027, 629.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,75 (m, 6H, 2xCH3), 2,64 (m, 3H, H3, H3’ e H4),
4,29 (m, 1H, H5), 4,45 (m, 1H, H5’).
RMN de 13C (75 MHz, CDCl3) δδδδ: 31,8 (C3), 32,0 (CH3), 32,3 (CH3), 48,2 (C4), 66,5
(C-Br), 70,5 (C5), 175,7 (C=O).
EM, m/z (I): [ +•M + 2] 209 (1,63), [ +
•M ] 207 (1,67), 126 (17,59), 111 (60,31), 83
(100,00), 68 (54,27).
2.4.6. Síntese da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6)
(2.6)
O O
Adicionou-se gota-a-gota uma solução da 4-(1’-bromo-1’-
metiletil)tetraidrofuran-2-ona (2.5) (2,00 g; 9,66 mmol) em THF anidro (20 mL) a uma
suspensão de tert-butóxido de potássio (2,20 g; 19,64 mmol) em THF anidro (30 mL)
em banho-de-gelo. A mistura resultante foi mantida sob agitação por 5 min. Adicionou-
se solução saturada de NH4Cl (30 mL) e fez-se extração com DCM (3 x 50 mL). A fase
orgânica foi secada com MgSO4 e concentrada sob pressão reduzida. O resíduo foi
submetido à cromatografia em coluna (hexano: acetato de etila 1:1) obtendo-se a 6,6-
dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6) (0,4870 g; 3,87 mmol) em 40% de
rendimento como um óleo incolor.
CCD: Rf = 0,52 (acetato de etila: hexano 1:1).
IV (KBr, cm -1) ν : 3068, 2961, 2909, 1774, 1368, 1186, 1046, 975, 892.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,10 (s, 6H, 2 x CH3), 1,88 (dd, 1H, J3-4 = 6,3 e J3-5’
= 0,9, H3), 2,01 (ddd, 1H, J3-4 = 6,3, J4-5 = 5,2 e J4-5’ = 1,2, H4), 4,10 (dt, 1H, Jgem = 9,9,
J3-5’ = 0,9 e J4-5’ = 1,2, H5’), 4,31 (dd, 1H, Jgem = 9,9 e J4-5 = 5,2, H5).
RMN de 13C (75 MHz, CDCl3) δδδδ: 14,6 (CH3), 23,3 [C(CH3)2], 25,4 (CH3), 30,3 (C4),
30,7 (C3), 66,8 (C5), 175,3 (C=O).

-71-
EM, m/z (I): [ +•M ] 126 (3,67), 125 (9,74), 111 (18,33), 97 (25,60), 85 (28,50), 83
(31,98), 71 (53,30), 69 (47,64), 57 (100,00), 55 (67,45).
2.4.7. Síntese da (S)-5-hidroximetil(5H)furan-2-ona (2.7)
O OHO
(2.7)
Adicionou-se 6 gotas de H2SO4 20% a uma solução do (S)-(Z)-4,5-O-
isopropilidenopent-2-enoato de metila (1.4) (6,0g, 32,26 mmol) em metanol (40mL) em
banho-de-gelo. A mistura foi mantida sob agitação magnética por 1 h, adicionou-se
mais 6 gotas de H2SO4 20% e manteve-se a agitação por mais 3 h sempre em banho-de-
gelo. O término da reação foi determinado por CCD. Adicionou-se KOH 2 mol.L-1 (12
gotas) e concentrou-se a mistura sob pressão reduzida. Em seguida, adicionou-se éter
dietílico (70 mL), secou-se com MgSO4 e evaporou-se o éter. O resíduo foi submetido à
cromatografia em coluna (acetato de etila) obtendo-se a (S)-5-hidroximetil(5H)furan-2-
ona (2.7) (2,68g, 23,51 mmol) em 73% de rendimento como um sólido branco.
Tf = 40-41°C
CCD: Rf = 0,48 (acetato de etila).
Rotação específica: 20][ Dα = - 148 ° (c = 1,08, água) (lit., - 151,3 °, MANN et al., 1987)
IV (KBr, cm -1) ν : 3400, 3111, 2932, 2879, 1746, 1602, 1332, 1171, 1112, 1053, 829.
RMN de 1H (300 MHz, CDCl3) δδδδ: 2,65 (s, 1H, OH), 3,73 (dd, 1H, Jgem = 12,3 e J5-6 =
4,8, H6), 3,97 (dd, 1H, Jgem = 12,3 e J5-6’ = 3,9, H6’), 5,12 (m, 1H, H5), 6,16 (dd, J3-4 =
5,8 e J3-5 = 2,2, H3), 7,45 (dd, 1H, J3-4 = 5,8 e J3-5 = 1,8, H4).
RMN de 13C (75 MHz, CDCl3) δδδδ: 62,4 (CH2), 84,4 (C5), 123,1 (C3), 154,0 (C4), 173,6
(C=O).
2.4.8. Síntese da (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8)
O OSiO
(2.8)

-72-
Adicionou-se uma solução de cloreto de tert-butildimetilsilano (2,64 g; 17,54
mmol) em DCM anidro (20 mL) a uma solução de (S)-5-hidroximetil(5H)furan-2-ona
(2.7) (1,00 g, 8,77 mmol), imidazol (1,19 g, 17,54 mmol) em DCM anidro (40 mL)
previamente resfriada em banho-de-gelo. A mistura foi deixada sob agitação magnética
por 20 min em banho de gelo e por mais uma hora à temperatura ambiente. O término
da reação foi determinado por CCD. A mistura foi filtrada a vácuo em funil contendo
sílica, lavou-se com DCM (40 mL) e o filtrado foi concentrado sob pressão reduzida. O
resíduo foi submetido à cromatografia em coluna (hexano : éter dietílico 2:1) obtendo-se
a (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8) (1,20 g, 5,26 mmol) em 60%
de rendimento como um sólido branco.
Tf = 30-31°C
CCD: Rf = 0,60 (acetato de etila).
Rotação específica: 20][ Dα = - 147,7 ° (c = 1,08, água) (lit., - 151,3 °, MANN et al.,
1987)
IV (KBr, cm -1) ν : 3095, 2955, 2930, 2858, 1755, 1603, 1257, 1138, 1100, 837, 779.
RMN de 1H (300 MHz, CCl4) δδδδ: 0,06 (s, 3H, Si-CH3), 0,07 (s, 3H, Si-CH3), 0,87 (s,
9H, [C(CH3)3]), 3,65 (dd, 1H, Jgem = 10,7 e J5-6 = 5,4, H6), 3,85 (dd, 1H, Jgem = 10,7 e
J5-6’ = 4,4, H6’), 4,85 (m, 1H, H5), 6,00 (dd, 1H, J3-4 = 5,9 e J3-5 = 2,0, H3), 7,35 (dd,
1H, J3-4 = 5,9 e J4-5 = 1,5, H4).
RMN de 13C (75 MHz, CCl4) δδδδ: -5,0 [Si(CH3)2], 18,7 [C(CH3)3], 26,3 [C(CH3)3], 63,7
(C6), 82,6 (C5), 123,2 (C3), 153,2 (C4), 170,8 (C=O).
2.4.9. Síntese da (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-metiletil) tetraidrofuran-2-ona (2.9)
O OSiO
OH
(2.9)
Adicionou-se a um tubo de quartzo a (S)-5-O-tert-
butildimetilsiloximetil(5H)furan-2-ona (2.8) (0,6062 g; 2,66 mmol) e álcool isopropílico

-73-
(75 mL). Borbulhou-se N2 durante 1 h tampando-se o tubo com rolha esmerilhada. A
solução foi irradiada (λ = 254 nm). O término da reação foi determinado por CCD. O
álcool isopropílico foi evaporado sob pressão reduzida a 35 ºC e o resíduo da
evaporação submetido à cromatografia em coluna (hexano : acetato de etila 1:1)
obtendo-se a (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-
metiletil)tetraidrofuran-2-ona (2.9) (0,4929 g; 1,75 mmol) em 66% de rendimento como
um líquido incolor.
CCD: Rf = 0,59 (hexano : acetato de etila 1:1).
Rotação específica: 20][ Dα = - 147,7 ° (c = 1,08, água) (lit., - 151,3 °, MANN et al.,
1987)
IV (KBr, cm -1) ν : 3459, 2954, 2858, 1766, 1472, 1255, 1183, 1125, 837, 779.
RMN de 1H (300 MHz, CDCl3) δδδδ: -0,06 (s, 3H, Si-CH3), -0,07 (s, 3H, Si-CH3), 0,87 (s,
9H, [C(CH3)3]), 1,20 (s, 3H, OC-CH3), 1,23 (s, 3H, OC-CH3), 1,91 (s 1H, OH), 2,43 (m,
2H, H3 e H4), 2,68 ( dd, 1H, Jgem = 16,8 e J3’-4 = 9,6, H3’), 3,69 (dd, 1H, Jgem = 11,1 e
J5-6 = 2,5, H6), 3,86 (dd, 1H, Jgem = 11,1 e J5-6’ = 3,5, H6’), 4,57 (m, 1H, H5).
RMN de 13C (75 MHz, CDCl3) δδδδ: -5,5 (Si-CH3), -5,6 (Si-CH3), 18,2 [C(CH3)3], 25,7
[C(CH3)3], 26,1 (CH3), 27,8 (CH3), 31,4 (C4), 47,1 (C3), 65,2 (C6), 71,0 (C-OH), 81,2
(C5), 177,1 (C=O).
2.4.10. Síntese da (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil) tetraidrofuran-2-ona (2.10)
O OHO
OH
(2.10)
Adicionou-se a um tubo de quartzo a (S)-5-hidroximetil(5H)furan-2-ona (2.7)
(1,56 g; 13,68 mmol) e álcool isopropílico (100 mL). Borbulhou-se N2 durante 1 h
tampando-se o tubo com rolha esmerilhada. A solução foi irradiada por 1 h (λ = 254
nm). O término da reação foi determinado por CCD. O álcool isopropílico foi
evaporado sob pressão reduzida a 35 °C e o resíduo da evaporação submetido à
cromatografia em coluna (acetato de etila) obtendo-se a (4S,5S)-5-hidroximetil-4-(1’-

-74-
hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.10) (2,14 g; 12,32 mmol) em 90% de
rendimento como um sólido branco.
Tf = 103-104°C
CCD: Rf = 0,40 (acetato de etila).
Rotação específica: 20][ Dα = + 25 ° (c = 1,08, água) (lit., + 25 °, MANN e
WEYMOUTH-WILSON, 1991)
IV (KBr, cm -1) ν : 3412, 3349, 2972, 2936, 2878, 1729, 1366, 1197, 1068, 696.
RMN de 1H (300 MHz, CD3COD) δδδδ: 1,08 (s, 3H, CH3), 1,10 (s, 3H, CH3), 2,37 (m,
2H, H3 e H4), 2,60 ( dd, 1H, Jgem = 18,2 e J3’-4 = 10,2, H3’), 3,49 (dd, 1H, Jgem = 12,3 e
J5-6 = 4,5, H6), 3,72 (dd, 1H, Jgem = 12,3 e J5-6’ = 2,8, H6’), 4,52 (m, 1H, H5).
RMN de 13C (75 MHz, CD3COD) δδδδ: 25,4 (CH3), 26,2 (CH3), 30,7 (C4), 46,4 (C3),
64,0 (C6), 70,1 [C(CH3)2], 82,7 (C5), 178,7 (C=O).
EM, m/z (I): [ +•M - 15] 159 (1,46), 97 (20,37), 69 (33,45), 59 (100,00).
2.4.11. Síntese da (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)-tetraidrofuran-2-ona (2.11)
O
OH
OO
O
H'H
(2.11)
3
Adicionou-se cloreto de benzoíla (1 mL, ρ = 1,20 g/cm3) a uma solução da
(4S,5S)-5-hidroximetil-4-(1-hidroxi-1-metiletil)tetraidrofuran-2-ona (2.10) (1,00 g, 5,75
mmol) em piridina seca (5 mL) sob atmosfera de N2 e em banho-de-gelo. Manteve-se a
mistura sob agitação por 3 h ainda em banho-de-gelo, adicionou-se água (10 mL) e fez-
se extração com DCM (3 x 20 mL). A fase orgânica foi lavada com água (25mL),
solução diluída de NaHCO3 (25 mL) e água (25 mL), secada com MgSO4 e concentrada
sob pressão reduzida. Adicionou-se tolueno (4 x 10 mL) ao resíduo e concentrou-se o
material sob pressão reduzida. O resíduo foi submetido à cromatografia em coluna
(hexano : acetato de etila 1:2) obtendo-se a (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-
metiletil)tetraidrofuran-2-ona (2.11) (1,43, 5,17 mmol) em 90% de rendimento como
um sólido branco.

-75-
Tf = 120,1-121,1°C
CCD: Rf = 0,48 (hexano : acetato de etila 1:2).
Rotação específica: 20][ Dα = + 45 ° (c = 1,12, água).
IV (KBr, cm -1) ν : 3444, 2978, 2882, 1744, 1708, 1601, 1452, 1278, 720, 713.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,23 (s, 3H, CH3), 1,27 (s, 3H, CH3), 2,20 (s, 1H,
OH), 2,43 (m, 1H, H4), 2,53 (dd, 1H, Jgem = 18,3 e J3-4 = 6,0, H3), 2,71 (dd, 1H, Jgem =
18,3 e J3’-4 = 10,5, H3’), 4,42 (dd, 1H, Jgem = 12,3 e J5-6 = 5,0, H6), 4,56 (dd, 1H, Jgem =
12,3 e J5-6’ = 2,7, H6’), 4,89 (m, 1H, H5), 7,43 (t, 2H, Jo = 7,6, Hm), 7,56 (m, 1H, Hp),
7,98 (dd, 2H, Jo = 7,6 e Jm = 1,5, Ho).
RMN de 13C (75 MHz, CDCl3) δδδδ: 26,6 (CH3), 28,4 (CH3), 30,9 (C4), 47,0 (C3), 66,8
(C6), 70,9 [C(CH3)2], 89,0 (C5), 128,7 (Cm), 129,4 (Co), 129,8 (=C-C=O), 133,6 (Cp),
166,5 (Ar-C=O), 176,5 (C=O).
EM, m/z (I): [ +•M - 1] 277 (0,03), 105 (100,00), 97 (26,40), 77 (45,10), 59 (73,51).

-76-
CAPÍTULO 3 - OUTRAS REAÇÕES FOTOQUÍMICAS
3.1. INTRODUÇÃO
A fotoquímica abrange todos os aspectos da química e física dos estados
eletronicamente excitados da matéria, desde a sua criação até a sua eventual desativação
ao estado fundamental (NEUMANN & QUINA, 2002).
As reações fotoquímicas englobam todas as reações que ocorrem via estado
excitado da matéria. O estudo destas reações fotoquímicas é conhecido como
fotoquímica orgânica. A molécula inicialmente no estado fundamental pode absorver
um fóton de energia quando se irradia luz em um determinado comprimento de onda.
Ao absorver energia um elétron na molécula é promovido de um orbital molecular de
mais baixa energia para outro de mais alta energia e diz-se que a molécula encontra-se
excitada. Se durante a promoção do elétron para o orbital de mais alta energia não
ocorre inversão do spin eletrônico, forma-se um estado simpleto excitado (Figura 3.1).
Mas se ocorre inversão do spin do elétron forma-se um estado tripleto excitado cujo
diagrama de níveis de energia está representado na Figura 3.1. Muitas moléculas não
atingem diretamente o estado tripleto excitado, necessitando da ação de outra molécula
denominada sensitizador. O sensitizador aborve energia formando o estado tripleto
excitado e em seguida transfere esta energia para outra molécula inicialmente no estado
fundamental, fazendo com que esta atinja o estado tripleto excitado. Como os estados
simpleto e tripleto possuem energias diferentes, reações que passam pela formação de
um estado simpleto podem levar a produtos diferentes das reações que passam pela
formação do estado tripleto (COYLE, 1986).

-77-
Estado Fundamental Estado Simpleto ExcitadoEstado Tripleto Excitado
EnergiaEnergiaEnergia
2 S + 1 = 1 2 S + 1 = 3
HOMO
LUMO
Figura 3.1: Representação dos diagramas de níveis de energia de orbitais moleculares no estado fundamental, simpleto excitado e tripleto excitado.
Ao irradiar dois compostos carbonílicos α,β-insaturados com lâmpada de
mercúrio de baixa pressão na presença de álcool isopropílico não ocorreu reação de
adição do álcool como observado para os compostos mostrados no Capítulo 2. Neste
capítulo, serão discutidas estas duas reações fotoquímicas que levaram a resultados
diferentes do esperado.

-78-
3.2. RESULTADOS E DISCUSSÃO
Na tentativa de adicionar álcool isopropílico ao carbono β de compostos
carbonílicos α,β-insaturados de cadeia aberta utilizou-se a mesma metodologia descrita
no Capítulo 2 para as γ-lactonas α,β-insaturadas. Os substratos foram dissolvidos em
excesso do álcool em um recipiente de quartzo, borbulhou-se N2 na solução por 1 hora
para eliminar o excesso de oxigênio dissolvido e irradiou-se a solução com luz UV (λ =
254 nm). O consumo dos reagentes foi acompanhado por meio de CCD. O álcool
isopropílico foi evaporado sob pressão reduzida e os produtos purificados por meio de
cromatografia em coluna. Em todas as reações não se observou formação do produto da
fotoadição de álcool isopropílico.
A Figura 3.2 mostra um esquema das reações discutidas neste capítulo, partindo
do sorbato de potássio (3.1), um reagente barato e encontrado comercialmente para uso
como conservante de alimentos.
OH
O
(3.2)
O
O
HO
(3.3)
O
HO (3.4)
OK
O
(3.1)
HCl , DCM
87%
LiAlH 4, Et2O74%
i-PrOH
254 nm, 96 h10%
CH3CN
254 nm, 96 h
HO O
(3.5)
Figura 3.2: Síntese da 3-hidroximetil-2-metilciclobutan-1-ona (3.4) a partir do sorbato de potássio (3.1).
O ácido (2E, 4E)-hexa-2,4-dienóico (3.2) foi obtido a partir do sorbato de
potássio (3.1) pela reação com ácido clorídrico 1 mol.L-1. O produto (3.2) foi separado
da fase aquosa por meio de extração com DCM. A evaporação do solvente levou a
obtenção do ácido (3.2) em 87% de rendimento.
O espectro no IV (Anexo 72) mostra uma banda larga entre 3600 e 2500 cm-1
referente ao estiramento da ligação O-H dos dímeros do ácido carboxílico. Pode-se
observar uma banda em 1693 cm-1, referente ao estiramento da ligação C=O conjugada

-79-
e duas bandas em 1638 cm-1 e 1613 cm-1 referentes ao estiramento das duas ligações
C=C. Os espectros de RMN de 1H e de 13C do ácido (3.2) encontram-se nos Anexos 73 e
74, respectivamente.
Com o objetivo de adicionar fotoquimicamente álcool isopropílico ao ácido (2E,
4E)-hexa-2,4-dienóico (3.2) dissolveu-se este substrato no álcool e irradiou-se a solução
com luz UV (λ = 254 nm) na ausência de oxigênio. Esta reação não levou a formação
do produto esperado, mas produziu o ácido 2-metil-3-oxo-1-ciclobutanocarboxílico
(3.3) em 10% de rendimento, como mostrado na Figura 3.2.
Para estudar o efeito da presença de oxigênio no meio reacional, esta reação foi
conduzida primeiramente sem borbulhar N2 na mistura reacional, depois borbulhando
N2 e por último borbulhando-se O2. Para os dois primeiros casos não se observou
diferenças nos tempos de reação e nos rendimentos do produto (3.3). Já no último caso a
reação não se completou mesmo após 120 h de irradiação e não foi possível isolar o
produto (3.3). Estes resultados sugerem que o átomo de oxigênio do grupo cetona do
produto (3.3) não tenha sido proveniente do O2 presente no meio reacional.
Para avaliar a importância da utilização do álcool isopropílico nesta reação
substituiu-se o álcool por acetonitrila e não se observou formação do produto (3.3) após
96 h de reação. Dessa forma, suspeita-se que o átomo de oxigênio do grupo cetona do
produto (3.3) seja proveniente do álcool isopropílico. É interessante ressaltar que
durante a irradiação do ácido (2E, 4E)-hexa-2,4-dienóico (3.2) em acetonitrila
observou-se a formação do ácido (2Z, 4E)-hexa-2,4-dienóico (3.5) proveniente da
isomerização fotoquímica do reagente.
Nas primeiras 48h de irradiação do ácido (2E, 4E)-hexa-2,4-dienóico (3.2) em
álcool isopropílico observou-se também a formação do seu isômero (3.5), cujos
espectros de RMN de 1H e 13C encontram-se nos Anexos 75 e 76. Com o decorrer da
reação ambos isômeros geométricos foram totalmente consumidos, sendo possível isolar
apenas o produto (3.3).
O Anexo 77 mostra o espectro no IV do composto (3.3) onde pode-se observar
uma banda larga entre 3500 e 2500 cm-1 referente ao estiramento da ligação O-H dos
dímeros do ácido carboxílico. Podem-se observar também duas bandas em 1971 cm-1 e
1713 cm-1, referentes ao estiramento das ligações C=O da cetona e do ácido carboxílico,
respectivamente. Os sinais nos espectros de RMN de 1H e RMN de 13C (Anexos 78 e
79) foram atribuídos com auxílio de RMN de COSY e HETCOR. No espectro de RMN
de 1H mostrado no Anexo 78 pode-se observar um multipleto em δ 1,65 integrado para

-80-
3H e atribuído ao CH3. Podem-se observar outros dois multipletos em δ 3,05 e δ 5,15
integrados para 2H cada, sendo o primeiro atribuído ao grupo CH2 e o último aos dois
átomos de hidrogênio dos grupos CH. No Anexo 79 encontra-se o espectro de RMN de 13C do ácido (3.3). Como os sinais referentes aos átomos de hidrogênio dos dois grupos
CH apresentaram deslocamento químico coincidentes no espectro de RMN de 1H e o
composto foi obtido na forma de um óleo não foi possível elucidar a estereoquímica do
produto.
Acredita-se que a formação do produto (3.3) a partir do ácido (3.2) tenha como
etapa intermediária uma reação de ciclização eletrocíclica fotoquímica representada na
Figura 3.3. Durante a ciclização eletrocíclica os dienos devem estar na conformação s-
cis. Esta ciclização ocorre através do mecanismo disrotatório, como descrito na
literatura (COYLE, 1986). Isso significa que durante a ciclização os grupos CH3 e
COOH giram de acordo com a direção das setas mostradas na Figura 3.3, dando origem
a ciclobutenos contendo substituintes com estereoquímica definida. A suspeita em
relação a formação do produto (3.3) como uma mistura de enantiômeros e as
observações a respeito da ocorrência de isomerização fotoquímica do ácido (3.2) levam
a supor que a ciclização eletrocíclica ocorra com apenas um dos isômeros (3.2) ou (3.5).
Assim o intermediário ciclobuteno pode ter sido formado por um dos caminhos (a) ou
(b) representados na Figura 3.3. No caminho (a) o substrato (3.2) passa para a
conformação s-cis sofrendo, em seguida, uma ciclização eletrocíclica disrotatória e
originado uma mistura de enantiômeros com ambos substituintes na configuração cis.
Neste caso, o composto (3.5) formado pela isomerização fotoquímica é novamente
convertido ao ácido (3.2). No caminho (b) o substrato (3.2) é isomerizado ao ácido
(3.5), este passa para a conformação s-cis e sofre ciclização eletrocíclica disrotatória
para dar origem a uma mistura de enantiômeros ciclobuteno contendo substituintes na
configuração trans.

-81-
HO2CCO2H
s-transHO2C
νh
CO2H
s-cis(3.2)
s-trans
νh
HO2C
H s-cis
νh
+
HO2C
HO2C
+
HO2C
ciclização eletrocíclica disrotatória
(a)
(b)(3.5)
Figura 3.3: Mecanismo da ciclização eletrocíclica disrotatória.
A partir da observação da dependência da presença de álcool isopropílico e do
efeito da presença ou ausência de oxigênio para o sucesso da formação do produto (3.3),
acredita-se que o intermediário ciclobuteno sofra rapidamente reação de adição de
álcool isopropílico formando outro intermediário contendo grupo éter. Assim, este
último se decompõe levando ao ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3),
como mostrado na Figura 3.4.
O
HOCO2H
s-transHO2C
νh
O
HO
O
HH
i-PrOH
O
HO
O
H2 ++
νh
s-cis(3.2)
(3.3)
Figura 3.4: Mecanismo para formação do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3).

-82-
Foram feitas tentativas para redução da carbonila do grupo cetona do ácido (3.3)
com NaBH4 em metanol, com o objetivo de elucidar a estereoquímica relativa dos
substituintes do anel. Em todos os casos, o material de partida foi recuperado e nenhum
produto de redução foi isolado. Posteriormente a redução do composto (3.3) foi feita
com LiAlH4 em éter dietílico sob refluxo, de acordo com o procedimento descrito na
literatura para redução da ciclobutanona (ROBERTS & SAUER, 1949).
Surpreendentemente, observou-se a redução da carbonila do grupo ácido carboxílico e
nenhuma alteração na carbonila do grupo cetona, formando a 3-hidroximetil-2-
metilciclobutan-1-ona (3.4) em 74% de rendimento.
O espectro no IV do composto (3.4) encontra-se no Anexo 80 onde se pode
observar uma banda larga em 3358 cm-1 referente ao estiramento da ligação O-H.
Podem-se observar também bandas de estiramento das ligações C-H entre 2930 e 2870
cm-1 e uma banda em 1966 cm-1 referente ao estiramento da ligação C=O da cetona. Os
sinais no espectro de RMN de 1H (Anexo 81) foram atribuídos com auxílio de
experimento de dupla irradiação. No espectro de RMN de 1H pode-se observar um
dupleto duplo em δ 1,63 atribuído ao CH3 e um simpleto em δ 2,04 atribuído ao OH.
Observa-se também um multipleto em δ 2,24 atribuído aos H4 e H4’, um tripleto em δ
3,67 atribuído aos átomos de hidrogênio do CH2 vizinho ao OH e um multipleto em δ
5,06 atribuídos aos H2 e H3. No espectro de RMN de 13C (Anexo 82) observa-se o
desaparecimento do sinal referente ao carbono do grupo COOH e a permanência do
sinal da carbonila do grupo cetona. Os demais sinais neste espectro de RMN de 13C
foram atribuídos com auxílio de HETCOR.
Este resultado é bastante incomum, pois em geral o grupo cetona é reduzido sob
condições mais brandas que o grupo ácido carboxílico. Dessa forma, não foi possível
elucidar a estereoquímica dos C1 e C2 do ácido (3.3), pois o espectro de RMN de 1H da
3-hidroximetil-2-metilciclobutan-1-ona (3.4) apresentou sinais referentes aos grupos CH
sobrepostos.
A tentativa de fazer reação de adição fotoquímica de álcool isopropílico ao
carbono β do éster (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4) levou a
formação do 5-hidroxi-4-oxopentanoato de metila (3.6) em 85% de rendimento ao invés
do produto da fotoadição. Na Figura 3.5 está representada uma proposta para o
mecanismo desta reação. Ao absorver um fóton de luz o éster (1.4) vai para seu estado
eletronicamente excitado, que por sua vez recebe um elétron de uma molécula de álcool

-83-
isopropílico. A espécie contendo elétron desemparelhado sofre rearranjo dando origem
ao produto 5-hidroxi-4-oxopentanoato de metila (3.6).
OO
C
O
OCH3
OOC
O
OCH3
OH
OO
C
O
OCH3
OH
H
OHO
C
O
OCH3
νh
H
OOC
O
OCH3
H
H
OO
C
O
OCH3
H
H
OH2
O
OO
C
O
OCH3
O
H
(1.4) (3.6)
Figura 3.5: Mecanismo proposto para a formação do 5-hidroxi-4-oxopentanoato de metila (3.6) a partir do (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4).
No espectro no IV (Anexo 83) observa-se a presença de uma banda larga em ν
3437 cm-1 referente ao estiramento da ligação O-H e uma banda intensa em ν 1734 cm-1
referente ao estiramento da ligação C=O. No espectro de RMN de 1H (Anexo 84) pode-
se observar um multipleto em δ 2,60 integrado para 4H e atribuído aos átomos de
hidrogênio dos dois grupos CH2 internos da molécula. O simpleto em δ 3,60 integrado
para 3H foi atribuído ao CH3 e o simpleto em δ 4,21 integrado para 2H foi atribuído aos
átomos de hidrogênio do grupo CH2 vizinho ao OH.
No espectro de RMN de 13C (Anexo 85) observa-se a presença de dois sinais em
δ 173,4 e δ 208,9 atribuídos a carbonila do grupo éster e do grupo cetona,
respectivamente. As atribuições dos demais sinais no espectro de RMN de 13C foram
feitas com o auxílio de espectro de RMN de DEPT. Observou-se que os sinais em δ
27,6 e δ 33,0 correspondem aos grupos CH2 internos e o sinal em δ 52,2 corresponde a
CH3. O sinal em δ 68,2 foi atribuído ao CH2 vizinho do grupo OH. Os dados

-84-
espectroscópicos obtidos para este composto (3.6) coincidem com os dados encontrados
na literatura (LUÕND et al., 1992).

-85-
3.3. CONCLUSÃO
A tentativa de adicionar fotoquimicamente álcool isopropílico ao carbono β do
ácido (2E, 4E)-hexa-2,4-dienóico (3.2) levou a formação do ácido 2-metil-3-oxo-1-
ciclobutanocarboxílico (3.3) ao invés do produto da adição. Não foi encontrado na
literatura nenhum relato de síntese do produto (3.3). Assim, apesar do rendimento desta
reação ter sido baixo (10%) ela tem grande importância sintética por ser a única
maneira, até então estudada, para produção deste composto (3.3).
No caso da irradiação do éster (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de
metila (1.4) em álcool isopropílico observou-se a formação do 5-hidroxi-4-
oxopentanoato de metila (3.6) ao invés do produto da adição do álcool ao éster. Este
éster (3.6) pode ser utilizado como intermediário na síntese de vários outros compostos.

-86-
3.4. EXPERIMENTAL
3.4.1. Síntese do ácido (2E, 4E)-hexa-2,4-dienóico (3.2)
OH
O
(3.2)
A um funil de separação de 250 mL, adicionou-se sorbato de potássio (3.1) (20
g, 133 mmol), DCM (100 mL) e solução de ácido clorídrico 1 mol.L-1 (100 mL). A
mistura foi agitada por 10 min, as fases orgânica e aquosa separadas e a fase aquosa
extraída com DCM (3 x 70 mL). As fases orgânicas foram reunidas, secadas com
MgSO4 e concentradas sob pressão reduzida, obtendo-se o ácido (2E, 4E)-hexa-2,4-
dienóico (3.2) (13 g, 116 mmol) em 87% de rendimento na forma de um sólido branco
cristalino.
CCD: Rf = 0,58 (hexano : acetato de etila 2:1).
IV (KBr, cm -1) ν : 3600 – 2500, 1693, 1939, 1613, 1415, 1330, 1266, 1155, 998, 921.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,87 (dd, 3H, J6-5 = 3,3 e J6-4 = 2,1, CH3), 5,74 (d,
1H, J2-3 = 15,6, H2), 6,20 (m, 2H, H4 e H5), 7,32 (m, 2H, H3).
RMN de 13C (75 MHz, CDCl3) δδδδ: 19,0 (CH3), 118,0 (C2), 129,9 (C4), 141,1 (C5),
147,6 (C3), 172,8 (C=O).
3.4.2. Síntese do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3)
O
O
HO
(3.3)
1
23
4
Adicionou-se a um tubo de quartzo o ácido (2E, 4E)-hexa-2,4-dienóico (3.2) (2,0
g; 17,86 mmol) e álcool isopropílico (150 mL). Borbulhou-se N2 durante 1 h tampando-
se o tubo com rolha esmerilhada. A solução foi irradiada por 96 h (λ = 254 nm). O
término da reação foi determinado por CCD. O álcool isopropílico foi evaporado sob
pressão reduzida a 35 °C e o resíduo da evaporação submetido à cromatografia em

-87-
coluna (hexano : acetato de etila 2:1) obtendo-se o ácido 2-metil-3-oxo-1-
ciclobutanocarboxílico (3.3) (0,228 g; 1,78 mmol) em 10% de rendimento como um
óleo incolor.
CCD: Rf = 0,58 (hexano : acetato de etila 2:1).
IV (filme, cm -1) ν : 3500-2500, 1971, 1713, 1410, 1284, 1220, 945, 872.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,65 (m, 3H, CH3), 3,05 (m, 2H, H4 e H4’), 5,15
(m, 2H, H1 e H2).
RMN de 13C (75 MHz, CDCl3) δδδδ: 14,3 (CH3), 34,9 (C4), 83,0 (CH), 87,2 (CH), 172,8
(C=O ácido), 206,2 (C=O cetona).
3.4.3. Síntese da 3-hidroximetil-2-metilciclobutan-1-ona (3.4)
O
HO(3.4)
1
2
34
Adicionou-se uma solução do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico
(3.3) (0,100 g; 0,78 mmol) em éter dietílico anidro (5 mL) a uma suspensão do LiAlH4
(0,080 g; 2,10 mmol) em éter dietílico anidro (10 mL), sob atmosfera de N2. O sistema
foi mantido sob agitação vigorosa e refluxo por 1,5 h. Em seguida, o sistema foi
resfriado a temperatura ambiente. Adicionou-se solução saturada de NaHCO3 (15 mL) e
éter dietílico (50 mL). A mistura foi transferida para um funil de separação onde as
fases orgânica e aquosa foram separadas. A fase aquosa foi extraída com éter dietílico
(3 x 25 mL). As fases orgânicas foram reunidas, secadas com MgSO4 e concentradas em
evaporador rotatório. O resíduo da evaporação foi submetido à cromatografia em coluna
(hexano: acetato de etila 2:1) obtendo-se a 3-hidroximetil-2-metilciclobutan-1-ona (3.4)
(0,066 g; 0,58 mmol) em 74% de rendimento como um óleo incolor.
CCD: Rf = 0,58 (hexano : acetato de etila 2:1).
IV (filme, cm -1) ν : 3358, 2927, 2875, 1966, 1442, 1371, 1050, 873, 703, 561.
RMN de 1H (300 MHz, CDCl3) δδδδ: 1,63 (dd, 3H, JCH3-H2 = 6,9 e JCH3-H3 = 3,3, CH3),
2,04 (s, 1H, OH), 2,24 (m, 2H, H4 e H4’), 3,67 (t, 2H, JCH2-H4 = 6,3 CH2OH), 5,06 (m,
2H, H2 e H3).

-88-
RMN de 13C (75 MHz, CDCl3) δδδδ: 14,7 (CH3), 32,4 (C4), 62,2 (CH2OH), 86,4 (CH),
86,8 (CH), 205,7 (C=O).
3.4.4. Síntese do 5-hidroxi-4-oxopentanoato de metila (3.6)
HO O
O
O(3.6)
Adicionou-se a um tubo de quartzo o (S)-(Z)-4,5-O-isopropilidenopent-2-enoato
de metila (1.4) (1,0 g; 5,38 mmol) e álcool isopropílico (100 mL). Borbulhou-se N2
durante 1 h tampando-se o tubo com rolha esmerilhada. A solução foi irradiada por 1 h
(λ = 254 nm). O término da reação foi determinado por CCD. O álcool isopropílico foi
evaporado sob pressão reduzida a 35 °C e o resíduo da evaporação submetido à
cromatografia em coluna (éter dietílico) obtendo-se o 5-hidroxi-4-oxopentanoato de
metila (3.6) (0,667 g; 4,57 mmol) em 85% de rendimento como óleo incolor.
CCD: Rf = 0,40 (éter dietílico).
IV (KBr, cm -1) ν : 3437, 2955, 1728, 1634, 1442, 1439, 1212, 1079.
RMN de 1H (300 MHz, CDCl3 e CD3COD) δδδδ: 2,60 (m, 4H, CH2 e CH2’), 3,60 (s, 3H,
CH3), 4,21 (s, 2H, CH2-O).
RMN de 13C (75 MHz, CDCl3 e CD3COD) δδδδ: 27,6 (CH2), 33,0 (CH2), 52,2 (CH3),
68,2 (CH2-O), 173,4 (C=O éster), 208,9 (C=O cetona).

-89-
CAPÍTULO 4 – ATIVIDADE INSETICIDA DOS PIRETRÓIDES
4.1. INTRODUÇÃO
Os piretróides são inseticidas sintéticos neurotóxicos que possuem baixa
toxidade a mamíferos e animais de sangue quente. Muitos piretróides são conhecidos
por apresentarem efeito de choque “Knock-down”, o que significa que os insetos são
derrubados imediatamente após exposição ao inseticida (NAUMANN, 1990). Estes
inseticidas, da mesma forma que o DDT e análogos, ligam-se aos canais de sódio
presentes na membrana dos neurônios aumentando o fluxo de Na+ para o interior da
membrana. Este desequilíbrio causa hiperexcitação ao inseto, levando-o a morte
(HASSALL, 1990).
Acanthoscelides obtectus (Say) (Coleoptera: Bruchidae), popularmente
conhecido como caruncho-do-feijão é uma praga de grãos armazenados, adaptada para
viver em condições de baixa umidade (ROMANO et al., 2006). As larvas de A. obtectus
se alimentam dos cotilédones e podem provocar a destruição completa dos grãos devido
ao seu rápido desenvolvimento (LOECK, 2002). Os ovos são colocados nas vagens
ainda no campo ou diretamente nos grãos armazenados. Os adultos (Figura 4.1) são
bons voadores e são responsáveis pela infestação dos campos e armazéns, porém não se
alimentam e têm vida curta (ROMANO et al., 2006). O controle desta praga geralmente
é feito pelo uso de inseticidas inorgânicos tais como fosfeto de alumínio e fosfeto de
magnésio. Também podem ser usados inseticidas organofosforados e deltametrina
(piretróide) para o controle desta praga (AGROFIT, 2006).

-90-
Uma das pragas chave do milho armazenado é o Sitophilus zeamais Mots.
(Coleoptera: Curculionidae) popularmente conhecido como gorgulho (Figura 4.1). Os
prejuízos causados por esta praga ocorrem devido à alimentação de larvas e adultos no
interior dos grãos, os quais podem ser totalmente destruídos (TOSCANO et al., 1999).
A principal forma de controle da infestação por S. zeamais é feita pelo emprego de
inseticidas (OLIVEIRA et al., 2005). Piretróides como deltametrina, permetrina e
bifentrina são utilizados comercialmente no Brasil para controle de S. zeamais
(AGROFIT, 2006).
(a)
(b)
Figura 4.1: (a) Acanthoscelides obtectus. (b) Sitophilus zeamais.
O curuquerê-da-couve, Ascia monuste orseis (Godart) (Lepidoptera: Pieridae) é
uma das pragas chaves de culturas de hortaliças, tais como repolho (Brassica Oleraceae
var. capitata), couve-flor (B. Oleraceae var. bortrytis) e couve-comum (B. Oleraceae
var. acephala) (PICANÇO & MARQUINI, 1999; CRESPO et al., 2002). Os prejuízos
causados por esta praga estão relacionados com perdas pela desfolha (Figura 4.2) e seu
controle é feito pelo uso de inseticidas (PICANÇO & MARQUINI, 1999). No Brasil,
vários inseticidas piretróides encontram-se registrados para uso no controle de A.
monuste orseis, tais como deltametrina, permetrina, bifentrina, lambda-cialotrina e beta-
ciflutrina (AGROFIT, 2006).

-91-
(a)
(b)
Figura 4.2: (a) Foto de uma folha de couve-comum infestada por Ascia monuste orseis. (b) Foto de uma
plantação de couve-comum infestada por A. monuste orseis.
Periplaneta americana (L.) (Blattaria: Blattidae) é uma espécie de barata. Este
inseto é uma praga responsável pela contaminação de diversos ambientes com os quais
entra em contato. Isto ocorre devido à alternância de habitat deste inseto durante o dia e
a noite. Sendo que, durante o dia repousam em ambientes escuros, úmidos e quentes
como tubulações de esgotos, fossas sépticas e latrinas. À noite invadem habitações,
como armazéns, restaurantes, cozinhas e hospitais. Dessa forma, as baratas atuam como
vetores mecânicos de agentes patogênicos, tais como diversos vírus, fungos e
protozoários (VIANNA, 2001). Encontram-se comercialmente vários produtos para
controle de P. americana contendo piretróides como princípio ativo, entre eles os mais
comuns são deltametrina, permetrina, cipermetrina, etc.

-92-
4.2. RESULTADOS E DISCUSSÃO
Os piretróides (1.7), (1.9) e (1.10) a (1.14) cuja síntese foi discutida no Capítulo
1 e o composto (2.6) cuja síntese foi discutida no Capítulo 2 foram submetidos a testes
de atividade inseticida. Nos bioensaios de toxicidade foram usados adultos de
Acanthoscelides obtectus (Say) (Coleoptera: Bruchidae) e Sitophilus zeamais Mots.
(Coleoptera: Curculionidae), larvas de segundo instar de Ascia monuste orseis (Godart)
(Lepidoptera: Pieridae) e ninfas de segundo instar de Periplaneta americana (L.)
(Blattaria: Blattidae). Estes insetos foram escolhidos devido a sua importância como
insetos-pragas. A. obtectus, S. zeamais, A. monuste orseis e P. americana constituem
importantes pragas do feijão e milho em armazenamento, brássicas e domissanitária,
respectivamente (ROMANO et al., 2006; TOSCANO et al., 1999; CRESPO et al.,
2002; VIANNA, 2001).
Verificou-se que os oito piretróides exibiram ação inseticida sobre as quatro
espécies de insetos a partir de 12 horas após a aplicação tópica, com exceção do
piretróide (1.7) que só apresentou ação inseticida sobre A. monuste orseis a partir de 24
horas após a aplicação (Tabelas 4.1 a 4.4).
Os oito piretróides foram igualmente tóxicos a A. obtectus a partir de 12 horas
após aplicação tópica. Para S. zeamais os piretróides (1.7) e (1.9) apresentaram toxidade
inferior aos demais piretróides. Tanto para A. obtectus como para S. zeamais não se
observou aumento da mortalidade a partir de 12 horas após a aplicação (Tabelas 4.1 e
4.2).
Para A. monuste orseis o piretróide (1.7) apresentou toxidade inferior aos demais
piretróides 48 horas após a aplicação. Observou-se aumento da mortalidade de A.
monuste orseis de 12 para 24 horas após a aplicação dos piretróides (1.7), (1.10), (1.11)
e (1.14). Verificou-se que a mortalidade de A. monuste orseis causada pelo piretróide
(1.9), ocorrida 12 horas após a aplicação foi menor que 48 horas após a aplicação, sendo
que a mortalidade ocorrida 24 horas após a aplicação foi intermediária nestes dois
tempos. Para os piretróides (2.6), (1.13) e (1.12) não houve efeito do tempo em relação
ao aumento da mortalidade, observando-se mortalidade de 100% completadas 12 horas
após a aplicação tópica (Tabela 4.3).
Os oito piretróides foram igualmente tóxicos a P. americana completadas 48
horas após a aplicação tópica. Não se observou aumento da mortalidade com o aumento
do tempo a partir de 12 horas após a aplicação dos piretróides (2.6), (1.10), (1.11) e
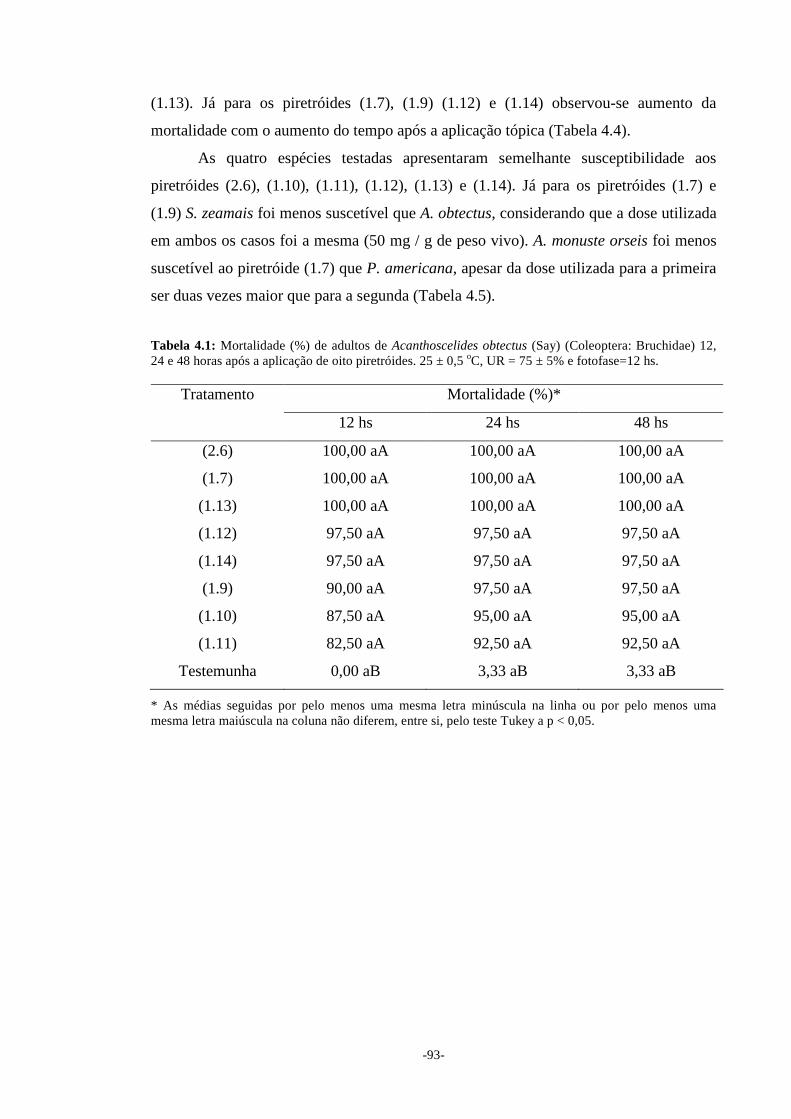
-93-
(1.13). Já para os piretróides (1.7), (1.9) (1.12) e (1.14) observou-se aumento da
mortalidade com o aumento do tempo após a aplicação tópica (Tabela 4.4).
As quatro espécies testadas apresentaram semelhante susceptibilidade aos
piretróides (2.6), (1.10), (1.11), (1.12), (1.13) e (1.14). Já para os piretróides (1.7) e
(1.9) S. zeamais foi menos suscetível que A. obtectus, considerando que a dose utilizada
em ambos os casos foi a mesma (50 mg / g de peso vivo). A. monuste orseis foi menos
suscetível ao piretróide (1.7) que P. americana, apesar da dose utilizada para a primeira
ser duas vezes maior que para a segunda (Tabela 4.5).
Tabela 4.1: Mortalidade (%) de adultos de Acanthoscelides obtectus (Say) (Coleoptera: Bruchidae) 12, 24 e 48 horas após a aplicação de oito piretróides. 25 ± 0,5 oC, UR = 75 ± 5% e fotofase=12 hs.
Tratamento Mortalidade (%)*
12 hs 24 hs 48 hs
(2.6) 100,00 aA 100,00 aA 100,00 aA
(1.7) 100,00 aA 100,00 aA 100,00 aA
(1.13) 100,00 aA 100,00 aA 100,00 aA
(1.12) 97,50 aA 97,50 aA 97,50 aA
(1.14) 97,50 aA 97,50 aA 97,50 aA
(1.9) 90,00 aA 97,50 aA 97,50 aA
(1.10) 87,50 aA 95,00 aA 95,00 aA
(1.11) 82,50 aA 92,50 aA 92,50 aA
Testemunha 0,00 aB 3,33 aB 3,33 aB
* As médias seguidas por pelo menos uma mesma letra minúscula na linha ou por pelo menos uma mesma letra maiúscula na coluna não diferem, entre si, pelo teste Tukey a p < 0,05.

-94-
Tabela 4.2: Mortalidade (%) de adultos de Sitophilus zeamais Mots. (Coleoptera: Curculionidae) 12, 24 e 48 horas após a aplicação de oito piretróides. 25 ± 0,5 oC, UR = 75 ± 5% e fotofase=12 hs.
Tratamento Mortalidade (%)*
12 hs 24 hs 48 hs
(1.13) 97,50 aA 100,00 aA 100,00 aA
(1.14) 97,50 aA 100,00 aA 100,00 aA
(1.11) 92,50 aA 97,50 aA 97,50 aA
(1.10) 95,00 aA 95,00 aA 95,00 aA
(2.6) 85,00 aA 90,00 aA 95,00 aA
(1.12) 82,50 aA 90,00 aA 95,00 aA
(1.9) 42,50 aB 45,00 aB 52,50 aB
(1.7) 45,00 aB 47,50 aB 50,00 aB
Testemunha 0,00 aC 3,33 aC 6,67 aC
* As médias seguidas por pelo menos uma mesma letra minúscula na linha ou por pelo menos uma mesma letra maiúscula na coluna não diferem, entre si, pelo teste Tukey a p < 0,05.
Tabela 4.3: Mortalidade (%) de larvas de Ascia monuste orseis (Godart) (Lepidoptera: Pieridae) 12, 24 e 48 horas após a aplicação de oito piretróides. 25 ± 0,5oC, UR = 75 ± 5%.
Tratamento Mortalidade (%)*
12 hs 24 hs 48 hs
(2.6) 100,00 aA 100,00 aA 100,00 aA
(1.13) 100,00 aA 100,00 aA 100,00 aA
(1.12) 100,00 aA 100,00 aA 100,00 aA
(1.11) 75,00 bB 95,00 aA 100,00 aA
(1.14) 75,00 bB 92,50 aA 100,00 aA
(1.10) 62,50 bBC 95,00 aA 100,00 aA
(1.9) 50,00 cC 67,50 bB 85,00 aA
(1.7) 27,50 bD 47,50 aC 57,50 aB
Testemunha 11,67 aD 13,33 aD 13,33 aC
* As médias seguidas por pelo menos uma mesma letra minúscula na linha ou por pelo menos uma mesma letra maiúscula na coluna não diferem, entre si, pelo teste Tukey a p < 0,05.

-95-
Tabela 4.4. Mortalidade (%) de ninfas de Periplaneta americana (L.) (Blattaria: Blattidae) 12, 24 e 48 horas após a aplicação de oito piretróides. 25 ± 0,5oC, UR = 75 ± 5%.
Tratamento Mortalidade (%)*
12 hs 24 hs 48 hs
(2.6) 100,00 aA 100,00 aA 100,00 aA
(1.10) 100,00 aA 100,00 aA 100,00 aA
(1.13) 100,00 aA 100,00 aA 100,00 aA
(1.11) 92,00 aAB 92,00 aABC 100,00 aA
(1.7) 79,33 bABC 96,00 abAB 100,00 aA
(1.14) 75,00 bBC 75,00 bBCD 100,00 aA
(1.9) 67,00 bC 71,00 bCD 96,00 aA
(1.12) 67,00 bC 67,00 bD 100,00 aA
Testemunha 4,00 aD 6,00 aE 12,17 aB
* As médias seguidas por pelo menos uma mesma letra minúscula na linha ou por pelo menos uma mesma letra maiúscula na coluna não diferem, entre si, pelo teste Tukey a p < 0,05.
Tabela 4.5. Mortalidade (%) de quatro espécies de insetos 48 horas após aplicação de oito piretróides. 25 ± 0,5oC, UR = 75 ± 5%.
Tratamento Mortalidade (%)*
A. obtectus S. zeamais A. monuste orseis P. americana
(2.6) 100,00 a 95,00 a 100,00 a 100,00 a
(1.7) 100,00 a 50,00 b 57,00 b 100,00 a
(1.9) 97,50 a 52,50 b 85,00 a 96,00 a
(1.11) 92,50 a 97,50 a 100,00 a 100,00 a
(1.10) 95,00 a 95,00 a 100,00 a 100,00 a
(1.13) 100,00 a 100,00 a 100,00 a 100,00 a
(1.12) 97,50 a 95,00 a 100,00 a 100,00 a
(1.14) 97,50 a 100,00 a 100,00 a 100,00 a
* As médias seguidas por pelo menos uma mesma letra minúscula na linha ou por pelo menos uma mesma letra maiúscula na coluna não diferem, entre si, pelo teste Tukey a p < 0,05.
Para todos os piretróides testados observou-se efeito de choque (Knock-down)
imediatamente após aplicação nas quatro espécies. O efeito Knock-down é uma
importante característica da maioria dos inseticidas piretróides (HASSALL, 1990;
SODERLUND et al., 2002). Para os piretróides (1.7) e (1.9)-(1.14) observou-se que os

-96-
insetos apresentaram perda da coordenação motora e excitabilidade imediatamente após
aplicação do produto. Para o piretróide (2.6) além das alterações comportamentais
observadas para os outros compostos observou-se 5 min após aplicação 70% de
mortalidade para A. obtectus e S. zeamais, 60% de mortalidade para A. monuste orseis e
80% de mortalidade para P. americana. O composto (2.6) possui estereoquímica cis para
os átomos de hidrogênio ligados ao anel ciclopropano da mesma forma que a
deltametrina, que é um piretróide muito utilizado para controle de insetos-praga.
Fernandes et al. observou o efeito “Knock-down” da deltametrina para larvas de
Rhipicephalus sanguineus, carrapatos de cães (FERNANDES et al., 2001).
Para A. obtectus e S. zeamais os piretróides foram aplicados na dose de 50 mg/g
de peso vivo observando-se efetiva atividade inseticida destes compostos nas doses
utilizadas. Os oito piretróides foram igualmente tóxicos à A. obtectus a partir de 12 horas
após aplicação tópica. Para S. zeamais os piretróides (1.7) e (1.9) apresentaram toxidade
inferior aos demais piretróides. Tanto para A. obtectus como para S. zeamais observou-se
que todos os piretróides apresentaram toxidade máxima até 12 horas após a aplicação
tópica (Tabelas 4.1 e 4.2). Estes resultados estão de acordo com a alta velocidade de ação
desta classe de compostos (HASSALL, 1990; SODERLUND et al., 2002).
Várias características estruturais dos piretróides podem influenciar na atividade
inseticida destes compostos. Entre elas podemos citar a estereoquímica dos C1 e C3 em
moléculas contendo anel ciclopropano (ELLIOTT, 1978). Para A. monuste orseis o
piretróide (1.7) apresentou toxidade inferior aos demais piretróides 48 horas após a
aplicação. Este composto (1.7) possui a mesma estereoquímica que os piretróides (1.10)-
(1.14). Uma diferença estrutural marcante entre o composto (1.7) e os demais piretróides
(1.10)-(1.11) é a presença, nestes últimos, de um grupo aromático ligado à parte ácida da
molécula. Comparando a toxidade apresentada para estes piretróides, observa-se que a
presença do grupo aromático pode aumentar significativamente a atividade tóxica do
piretróide para esta espécie.
Para A. monuste orseis os oito piretróides foram aplicados na concentração de 10
mg/g de peso vivo. Nesta dose todos os piretróides apresentaram ação inseticida (Tabela
4.3). Doses muito baixas de piretróides são encontradas na literatura para esta espécie de
inseto. De acordo com Crespo et al. (2002), a concentração letal para 90% da população
(CL90) para soluções de deltametrina e permetrina utilizadas para mergulhar folhas de
couve usadas na alimentação das larvas de A. monuste orseis são de 0,0052 mg/mL e
0,0183 mg/mL, respectivamente (CRESPO et al., 2002). Os piretróides (2.6), (1.12) e

-97-
(1.13) apresentaram toxidade máxima para A. monuste orseis completadas 12 horas após
aplicação tópica. Observou-se aumento da mortalidade de A. monuste orseis de 12 para 24
horas após a aplicação dos piretróides (1.7), (1.10), (1.11) e (1.14) (Tabela 4.3). O
piretróide (2.6) possui estereoquímica cis ao passo que os piretróides (1.7), (1.10), (1.11)
e (1.14) possuem estereoquímica 1S-trans. Desta forma, os resultados obtidos levam à
observação da influência da estereoquímica do composto na velocidade de ação. Já os
piretróides (1.12) e (1.13) possuem estereoquímica 1S-trans, mas contém átomos de
halogênios em sua estrutura. Como os piretróides (1.12) e (1.13) possuem ação mais
rápida que os piretróides (1.10), (1.11) e (1.14), supõe-se que a presença dos átomos de
halogênio pode aumentar a velocidade de ação destes compostos para A. monuste orseis.
Verificou-se que a mortalidade para A. monuste orseis causada pelo piretróide (1.9),
ocorrida 48 horas após a aplicação, foi maior que a mortalidade ocorrida 12 horas após a
aplicação. A mortalidade ocorrida 24 horas após a aplicação foi intermediária nestes dois
tempos. Logo o composto (1.9) foi o que apresentou ação mais lenta a A. monuste orseis
e, esta diferença na velocidade de ação pode estar relacionada à estereoquímica da
molécula que é 1R-trans, diferente dos demais piretróides.
Os oito piretróides foram igualmente tóxicos a P. americana completadas 48
horas após a aplicação tópica. Como todos os piretróides foram altamente tóxicos na dose
utilizada não foi possível detectar possíveis relações entre a estrutura dos compostos e a
atividade tóxica. Observou-se, porém, diferenças na velocidade de ação destes compostos.
Os piretróides (2.6), (1.10), (1.11) e (1.13) apresentaram toxidade máxima até 12 horas
após a aplicação, o piretróide (1.7) apresentou toxidade máxima depois de completadas 24
horas da aplicação e os piretróides (1.9), (1.12) e (1.14) apresentaram toxidade máxima
somente 48 horas após a aplicação (Tabela 4.4). Estas diferenças de velocidade de ação
podem estar relacionadas com características estruturais destes compostos que
influenciam na velocidade de penetração do inseticida através do corpo do inseto.
Sabe-se que a estereoquímica dos piretróides pode influenciar a ação tóxica destes
compostos (SODERLUND et al., 2002; ELLIOTT, 1978). Os piretróides (1.7) e (1.9) são
enantiômeros, mas não apresentaram diferenças de atividade para A. obtectus e S.
zeamais. Já para A. monuste orseis o piretróide (1.7) foi menos tóxico, ao passo que o
piretróide (1.9) mostrou ação mais lenta. A toxidade máxima para o piretróide (1.9) e para
o piretróide (1.7) foi observada 48 horas e 24 horas após a aplicação, respectivamente.
Para P. americana os dois enantiômeros foram igualmente tóxicos 48 horas após

-98-
aplicação, mas o piretróide (1.9) apresentou ação inseticida mais lenta a esta espécie que o
piretróide (1.7).
As quatro espécies testadas apresentaram semelhante susceptibilidade aos
piretróides (2.6) e (1.10)-(1.14). Já para os piretróides (1.7) e (1.9) S. zeamais foi menos
suscetível que A. obtectus, considerando que a dose utilizada em ambos as espécies foi a
mesma (50 mg/g de peso vivo). A. monuste orseis foi menos suscetível ao piretróide (1.7)
que P. americana, apesar da dose utilizada para a primeira ser duas vezes maior que
utilizada para a segunda (Tabela 4.5).
Possíveis razões para a variação da toxidade são as taxas de penetração das
substâncias através da cutícula dos insetos, as doses usadas, os mecanismos de
decomposição e excreção das substâncias dentro do corpo do inseto e diferenças nos
canais de sódio destas espécies que são o sítio de ação dos piretróides.

-99-
4.3. CONCLUSÃO
Verificou-se que todos os oito piretróides exibem ação inseticida sobre A.
obtectus, S. zeamais, A. monuste orseis e P. americana a partir de 12 horas após a
aplicação tópica, com exceção do piretróide (1.7) que só apresenta ação inseticida sobre
A. monuste orseis a partir de 24 horas após a aplicação.
As características estruturais dos piretróides estão intimamente relacionadas à
atividade destes compostos. Dessa forma, os enantiômeros (1.7) e (1.9) apresentam ação
inseticida semelhante para A. obtectus e S. zeamais, ao passo que para A. monuste orseis
e P. americana sua atividade inseticida apresentam diferenças significativas.

-100-
4.4. EXPERIMENTAL
Os testes de atividade dos piretróides foram realizados no Laboratório de
Manejo Integrado de Pragas da Universidade Federal de Viçosa (UFV), Viçosa, MG,
em 2006.
Nos bioensaios de toxicidade foram usados adultos de Acanthoscelides obtectus
(Say) (Coleoptera: Bruchidae) e Sitophilus zeamais Mots. (Coleoptera: Curculionidae),
larvas de segundo ínstar de Ascia monuste orseis (Godart) (Lepidoptera: Pieridae) e
ninfas de segundo instar de Periplaneta americana (L.) (Blattaria: Blattidae). A. obtectus
e S. zeamais foram criados em laboratório em feijão e milho, respectivamente, a
temperatura de 25 ± 0,5°C, U.R. = 75 ± 5%. A. monuste orseis foi criada em folhas de
couve e P. americana em ração de gato.
O delineamento experimental foi inteiramente casualizado com quatro
repetições. Cada parcela experimental foi constituída de placa de Petri (9 cm de
diâmetro por 2 cm de altura) contendo 10 insetos.
As doses usadas foram: 5 (para P. americana), 10 (para A. monuste orseis) e 50
mg/ g de peso vivo (para A. obtectus e S. zeamais). Para obtenção das doses pesou-se 10
insetos de cada espécie para obtenção da massa média destes. Os piretróides foram
dissolvidos em acetona e aplicou-se topicamente 0,5 µL da solução em cada inseto. Na
testemunha os insetos foram tratados topicamente com igual volume da acetona pura.
Os insetos foram confinados em placas de Petri sem alimentação (para A. obtectus e S.
zeamais) e com alimento (para A. monuste orseis e P. americana).
As placas de Petri foram acondicionadas em estufa incubadora a 25 ± 0,5°C,
U.R.= 75 ± 5% e fotofase de 12 horas. Os insetos vivos e mortos foram contados 12, 24
e 48 horas após o tratamento. Foram considerados mortos os insetos que deixaram de
apresentar capacidade de locomoção. Os dados de mortalidade dos insetos foram
submetidos à análise de variância e as médias comparadas pelo teste de Tukey a p <
0,05.

-101-
REFERÊNCIAS BIBLIOGRÁFICAS
AGROFIT, Relatório de pragas e doenças. Disponível em: <http://extranet.agricultura.gov.br/agrofit_cons/principal_agrofit_cons>. Acessado em: 17 jul. 2006.
BAER, E. 1,2,5,6-diacetone d-mannitol and 1,2,5,6-diactonide d-mannitol. J. Am. Chem. Soc. v. 67, 1945, p. 338-339.
BAER, E.; FISCHER, H. O. L. Studies on acetone-glyceraldehyde. VI. Preparation of d(+)-acetone glycerol. J. Biol Chem. v. 128, 1939, p. 463 – 473.
BARBOSA, L. C. A. Os Pesticidas o Homem e o Meio Ambiente. Viçosa: Editora UFV, 2004, p. 215.
BROWN, G. D.; WONG, H. Total synthesis of (+) maculalactone A, maculalactone B and Maculalactone C and the determination of the absolute configuration of natural (+) maculalactone A by asymmetric synthesis. Tetrahedron. v. 60, 2004, p. 5439-5451.
CHITTENDEN, G. J. F. Isopropylidenation of D-mannitol under neutral conditions. Carbohydr. Res. v. 87, 1980, p. 219-226.
COREY, E. J. Constrution of ring systems containing the gem-dimethylcyclopropane unit using diphenylsulfonium isopropylide. J. Am. Chem. Soc. v. 89, n. 15, 1967, p. 3912-3914.
COYLE, J. D. Introduction to Organic Photochemistry. Nova York: Wiley, 1986, p. 176.
CRESPO, A. L. B. et al. Seletividade fisiológica de inseticidas a Vespidae predadores de Ascia monuste orseis. Pesq. Agropec. bras. v. 37, n. 3, 2002, p. 237-242.
DEBOST, J. L.; GELAS, J.;HORTON, D. Selective preparation of mono- and diacetals of D-mannitol. J. Org. Chem. v. 48, 1983, p. 1381-1382.

-102-
ELLIOTT, M. et al. New pyrethrin-like esters with high insetcticidal activity. Nature. v. 207, 1965, p. 938-939.
ELLIOTT, M. et al. Synthetic insecticide with a new order of activity. Nature. v. 248, 1978, p. 710-711.
ELLIOTT, M.; JANES, N. F. Synthetic Pyrethroids – A new class of Insecticide. 1978, p. 474 – 505.
FERNANDES, F. F. et al. Efeitos toxicológicos e ineficiência in vitro de deltametrina sobre larvas de Rhipicephalus sanguineus, de Goiânia, Goiás, Brasil. Rev. Soc. Bras. Med. Trop. v. 34, n. 2, 2001, p. 159-165.
FUNK, R. L.; MUNGER, J. D. The stereospecific total synthesis of (+)-cis-chrysanthemic acid via the alicyclic Claisen Rearrangement. J. Org. Chem. v. 50, 1985, p. 707-709.
HASSALL, K. A. Cap. 7 - Natural e synthetic pyrethroids. In: The Biochemistry ad Uses of Pesticides. 2a ed. New York: Macmillan. 1990, p. 185-207.
KRIEF, A. et al. Stereoselective synthesis of methyl (1R) trans- and cis- hemicaronaldehydes from natural tartaric acid: application to the synthesis of S-bioallethrin and deltametrin insecticides. Tetrahedron. v. 45, n. 10, 1989, p. 3039-3052.
KRIEF, A.; DUMONT, W. From tartaric acid to the most biologically active insecticides: straightforward enantioselective synhtesis of deltamethrin. Tetrahedron Lett.. v. 29, n. 9, 1988, p. 1083-1084.
KRIEF, A.; SURLERAUX, D.; FRAUENRATH, H. Novel stereoselective route to cis-chrysanthemic acid. Tetrahedron Lett.. v. 29, n. 47, 1988, p. 6157-6160.
KRIEF, A.; SURLERAUX, D.; ROPSON, N. Novel enantioselective syntheses of optically active (1R)-cis- e (1R)-trans- chrysanthemic acids. Tetrahedron: Asymmetry. v. 4, n. 3, 1993, p. 289-292.
KUSZMANN, J.; TOMORI, E.; MEERWALD, I. The synthesis of 1,2:5,6-di-O-isoporpylidene-D-mannitol: a comparative study. Carbohydr. Res. v. 128, 1984, p. 87-99.
LOECK, A. E. Pragas de Produtos Armazenados. Pelotas: EGUFPEL, 2002. 113 p.
LÜÕND, R. M.; WALKER, J.; NEIER, R. W. Assessment or the active-site requirements of 5-aminolaevulinic acid dehydratase: evaluation of substrate and product analogues as competitive inhibitors. J. Org. Chem. v. 57, 1992, p. 5005 - 5013.
MANN, J.; PARTLETT, N. K.; THOMAS, A. A pratical synthesis of (S)-5-hydroxymethylfuran-2(5H)-one from 1,2:5,6-Di-O-isopropyllidene-D-mannose. J. Chem. Res. (S), 1987, p. 369.
MANN, J.; WEYMOUTH-WILSON, A. A new approach to cis-chrysanthemic acid. Carb. Res. v. 201, 1991, p. 511-515.

-103-
MANN, J.; WEYMOUTH-WILSON, A. Photocatalysed addition of alcohols to 5-substituted 2,5-dihydrofuran-2-ones: novel synthesis of (3’R)-2’,3’-dideoxy-3’-hydroxymethyl nucleosides. J. Chem. Soc. Perkin Trans. 1. 1994, p. 3141-3148.
MARTIN, T. et al. Pyrethroid resistance mechanisms in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) from West Africa. Pesticide Biochemistry and Physiology. v. 74, 2002, p. 17–26.
MORPAIN, C. et al. Improved preparation of Di-O-isopropylidene-1,2;5,6-D-Mannitol. Organic Preparation Procedures International. v. 22, n. 4, 1990, p. 540-543.
NÄSMAN, J. H. 3-Methyl-2(5H)-furanone. Org. Syntheses. v. 68, 1990, p. 162-174.
NAUMANN, K. Synthetic Pyrethroid Insecticides: Structures and Properties. Berlin: Springer-Verlag, 1990, p. 3-15.
NEUMANN, M. G.; QUINA, F. H. A fotoquímica no Brasil. Quim. Nova. v. 25, 2002, p. 34 – 38.
OHGA, K.; MATSUO, T. Photoinduced addition of isopropil alcohol to α,β-unsaturated lactones. J. Org. Chem., v. 39, n. 1, 1974, p. 106-108.
OLIVEIRA, E. E. et al. Resistência vs Susceptibilidade a Piretróides em Sitophilus zeamais Motschulsky (Coleoptera: Curculionidae): há vencedor? Crop protection. 2005, p. 981-990.
PICANÇO, M. C.; MARQUINI, F. Manejo integrado de pragas de hortaliças em ambiente protegido. Informe Agropecuário. Belo Horizonte, v. 20, n. 200/201, 1999, p. 126-133.
PKF CONSULTING LTD. Kenya’s pyrethrum industry 2005. Disponível em: <http://www.epzakenya.com/userfiles/file/kenyapyrethrum.pdf>. Acessado em: 01 jul. 2006.
ROBERTS, J. D.; SAUER, C. W. Small-ring compounds. III. Synthesis of Cyclobutane, Cyclobutanol, Cyclobutene and Cyclobutane. J. Org. Chem. 1949, p. 3925 – 3929.
ROMANO, C. M. et al. Aplicação de pó inerte no controle do Acanthoscelides obtectus em grãos de feijão armazenado. Disponível em: <http://www.cnpaf.embrapa.br/conafe/pdf/conafe2005-0318.pdf>. Acessado em: 17 jul. 2006.
SANSEVERINO, A. M.; MATTOS, M. C. S. Hydrobromination of alkenes with PBr3/SiO2: a simple and efficient regiospecific preparation of alkyl bromides. J. Braz. Chem. Soc. v. 12, n. 5, 2001, p. 685-687.
SCHMID, C. R. et al. Synthesis of 2,3-O-isopropilidene-D-glyceraldehyde in high chemical and optical purity: observations on the development of a pratical bulk process. J. Org. Chem. v. 56, 1991, p. 4056-4058.
SCHMID, C. R.; BRYANT, J. D. D-(R)-Glyceraldehyde Acetonide [1,3-Dioxolane-4-carboxaldehyde, 2,2-dimethyl-, (R)-]. Organic Syntheses. v. 72, 1993, p. 6-10.

-104-
SILVERSTEIN, R. M.; WEBSTER, F. X. Identificação espectroscópica de compostos orgânicos. 6a. ed. Rio de Janeiro: LTC, 2000, p. 460.
SODERLUND, D. M. et al. Mechanismis of pyretroid neurotoxicity: implications for cumulative risk assessment. Toxicology. v. 171, 2002, p. 3-59.
TOSCANO, L.C. et al. Resistência e mecanismos envolvidos em genótipos de milho em relação ao ataque do gorgulho, Sitophilus zeamais Mots (Coleoptera, Curculionidae). In: Soc. Entomol. Bras. v. 28, n. 1, 1999, p.141-147.
VIANNA, E. E. S.; BERNE, M. E. A.; RIBEIRO, P. B. Desenvolvimento e longevidade de Periplaneta americana Linneu, 1758 (Blattodea: Blattidae). In: Rev. Bras. de Agrociência. v. 7, n. 2, 2001, p. 111-115.
WANDAHWA, P.; RANST, E. V.; DAMME, P. V. Pyrethrum (Chrysanthemum cinerariaefolium Vis.) cultivation in West Kenya: origin, ecological conditions and management. Industrial Crops and Products. v. 5, 1996, p. 307-322.

-105-
Anexos

-106-
Anexo 1: Espectro no IV do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2).

-107-
Anexo 2: Espectro de RMN de 1H do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2).

-108-
Anexo 3: Espectro de RMN de 13C do 1,2:5,6-di-O-isopropilideno-D-manitol (1.2).

-109-
Anexo 4: Espectro no IV do (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4).

-110-
Anexo 5: Espectro de RMN de 1H do (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4).
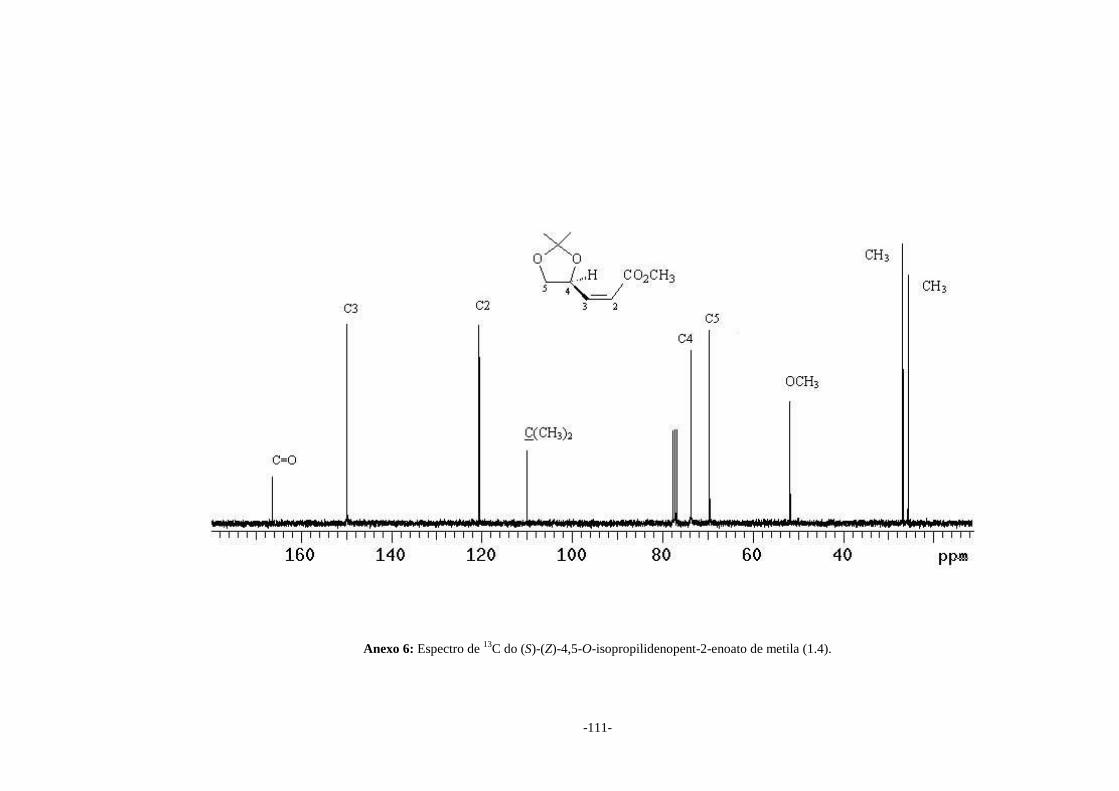
-111-
Anexo 6: Espectro de 13C do (S)-(Z)-4,5-O-isopropilidenopent-2-enoato de metila (1.4).

-112-
Anexo 7: Espectro no IV do (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5).

-113-
Anexo 8: Espectro de RMN de 1H do (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5).
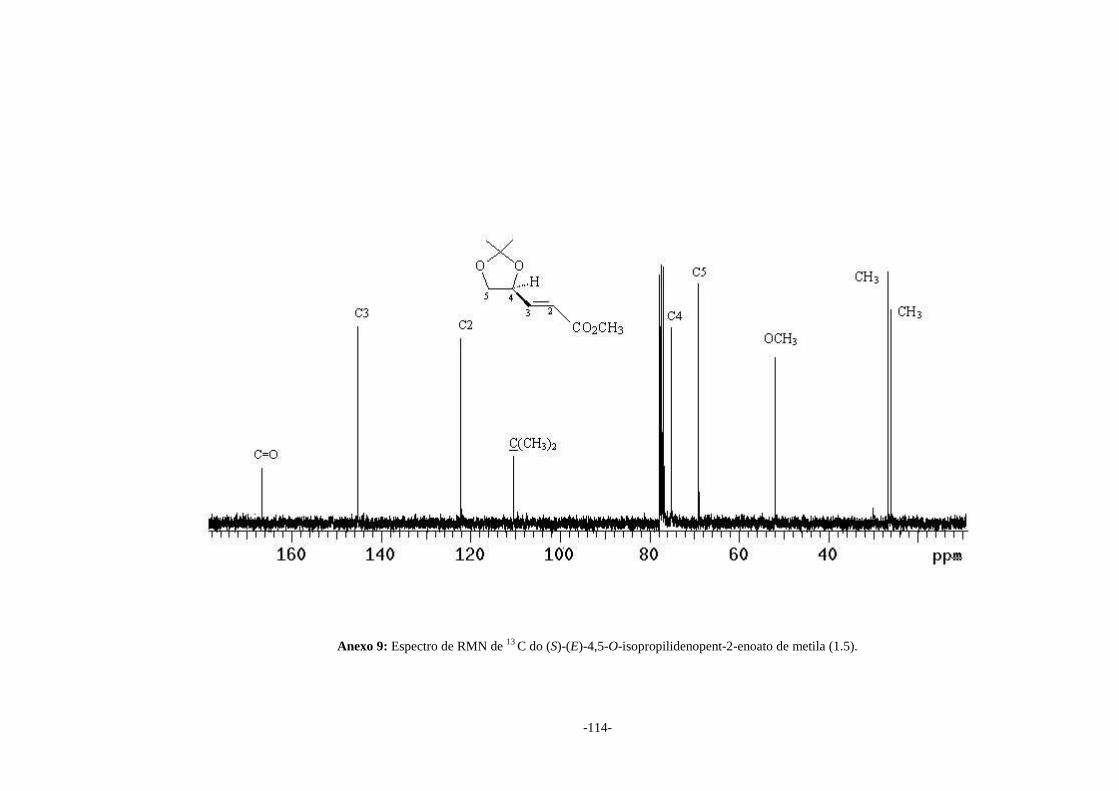
-114-
Anexo 9: Espectro de RMN de 13 C do (S)-(E)-4,5-O-isopropilidenopent-2-enoato de metila (1.5).

-115-
Anexo 10: Espectro no IV do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.6).
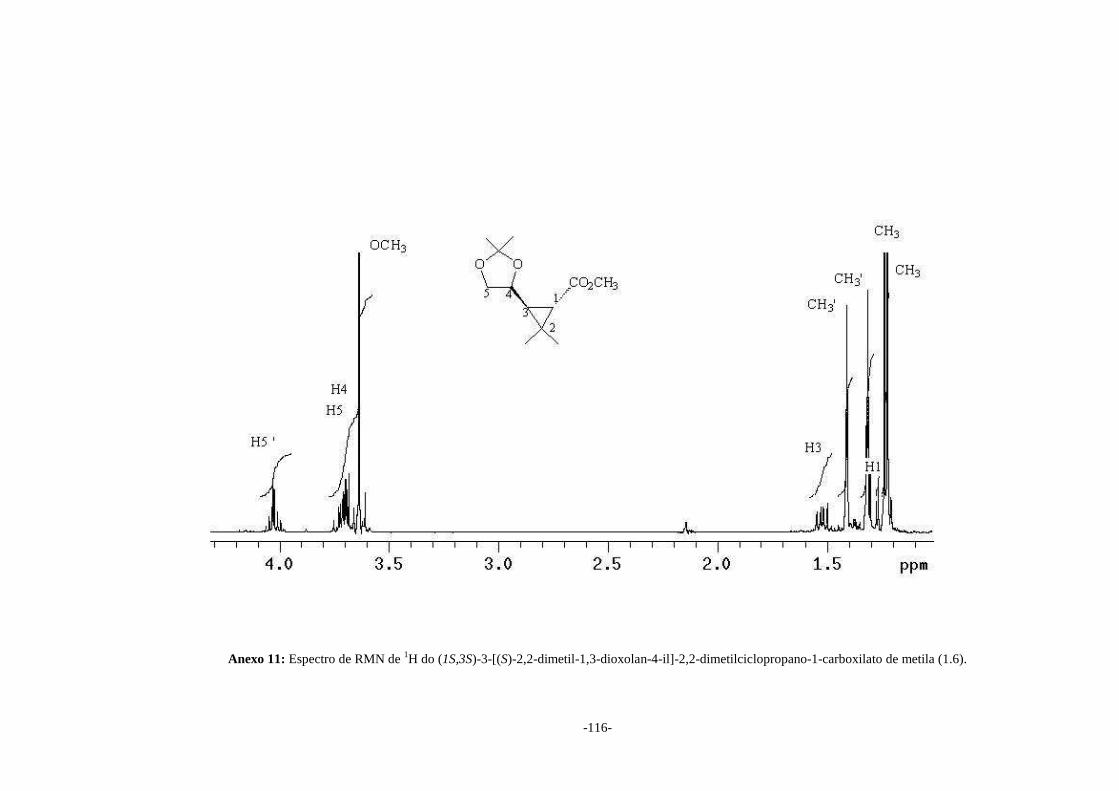
-116-
Anexo 11: Espectro de RMN de 1H do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.6).

-117-
Anexo 12: Espectro de RMN de 13C do (1S,3S)-3-[(S)-2,2-dimetil-1,3-dioxolan-4-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.6).
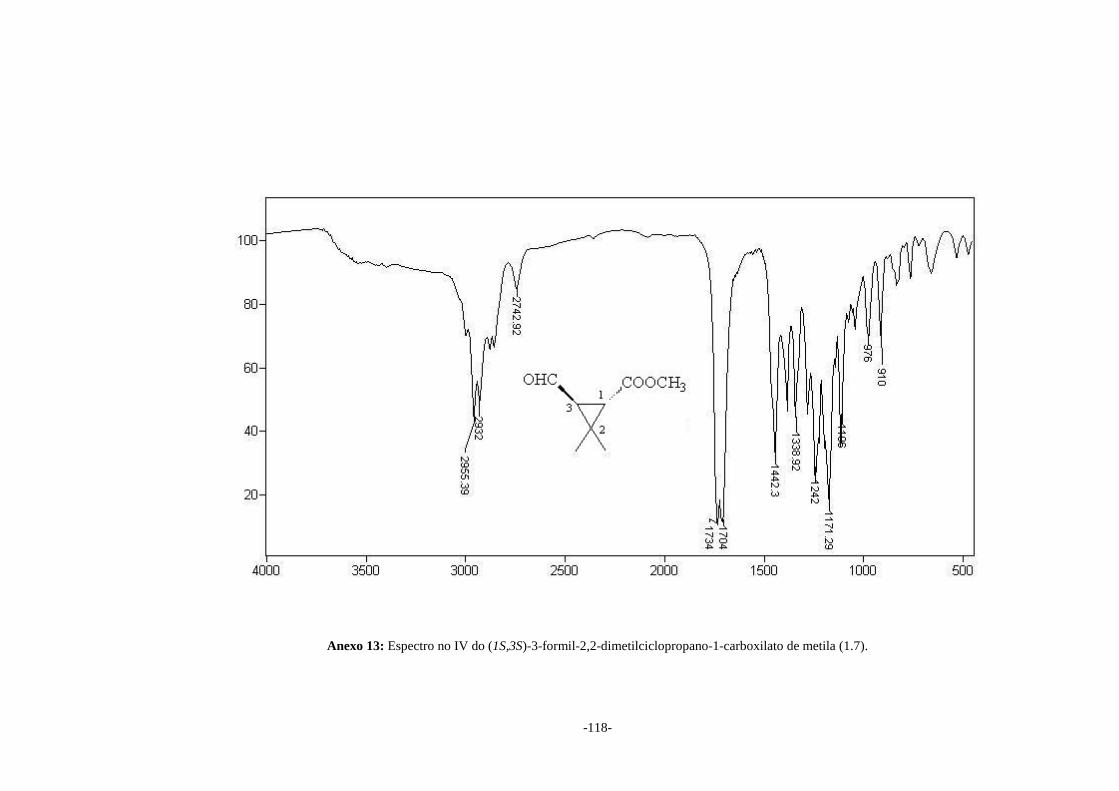
-118-
Anexo 13: Espectro no IV do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7).
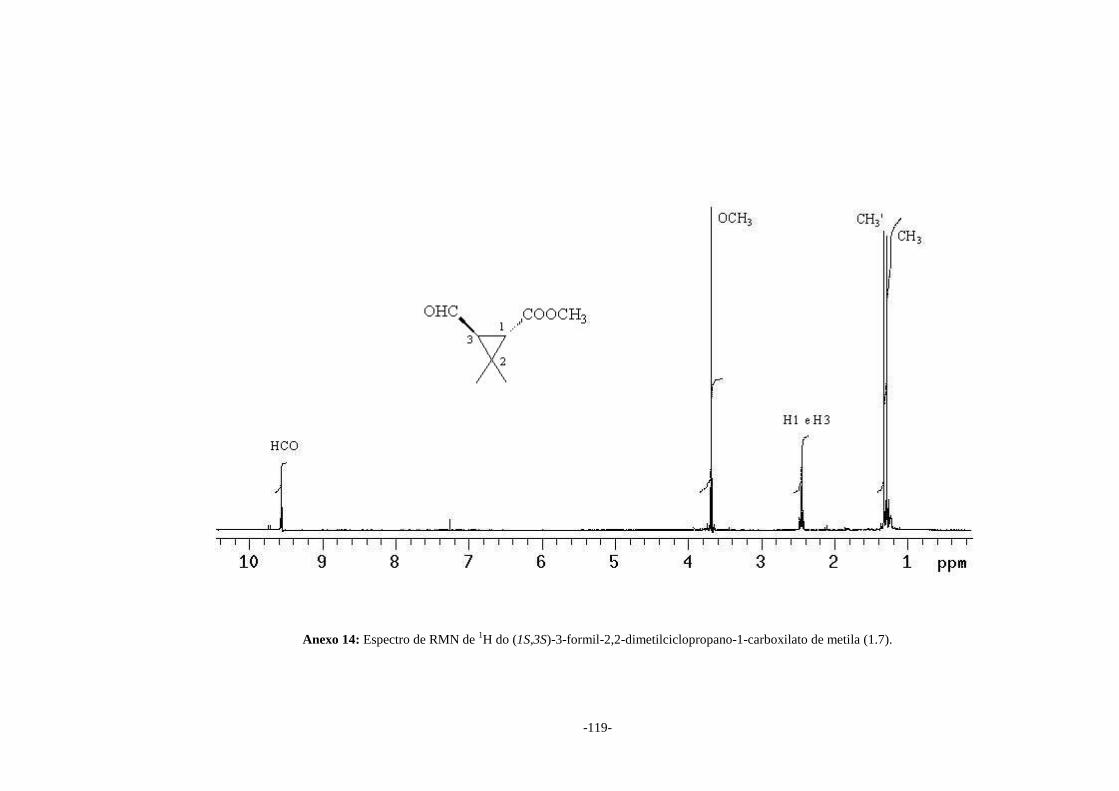
-119-
Anexo 14: Espectro de RMN de 1H do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7).
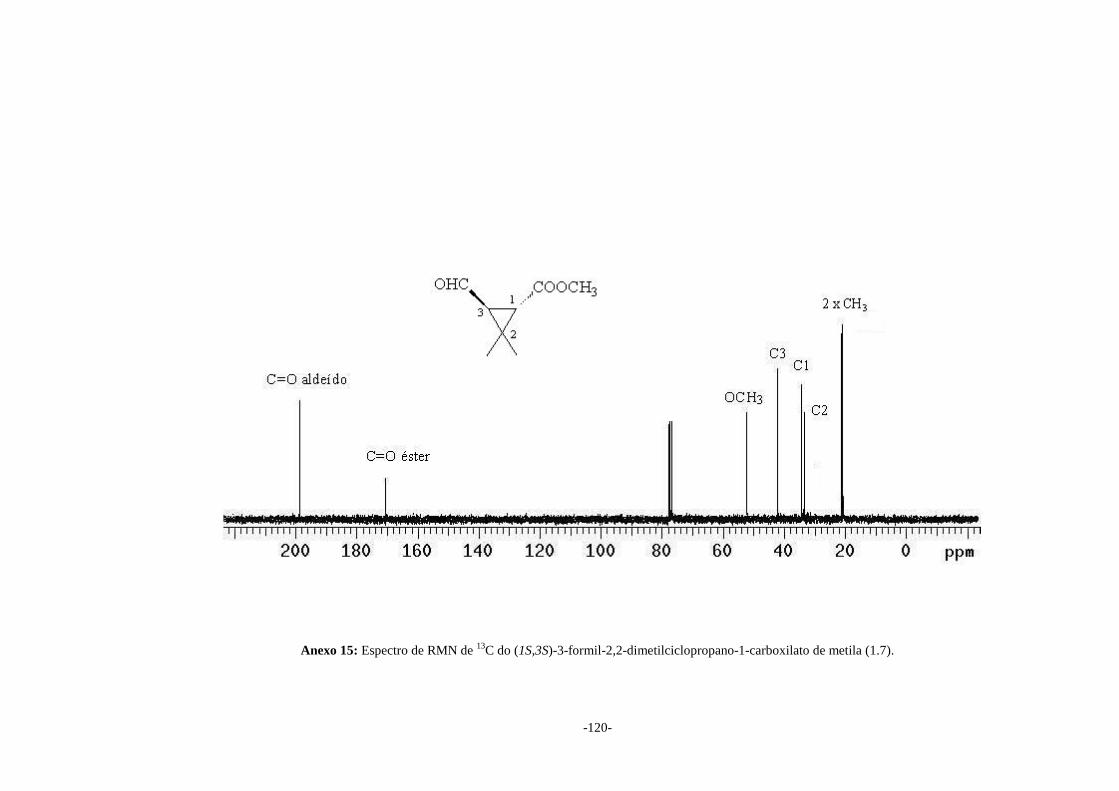
-120-
Anexo 15: Espectro de RMN de 13C do (1S,3S)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.7).

-121-
Anexo 16: Espectro no IV do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9).

-122-
Anexo 17: Espectro de RMN de 1H do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9).

-123-
Anexo 18: Espectro de RMN de 13C do (1R,3R)-3-formil-2,2-dimetilciclopropano-1-carboxilato de metila (1.9).

-124-
Anexo 19: Espectro no IV dos isômeros E e Z (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.10).

-125-
Anexo 20: Espectro no IV dos isômeros E e Z (1S,3S)-3-[2-(3-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.11).

-126-
Anexo 21: Espectro no IV dos isômeros E e Z (1S,3S)-3-[2-(2-clorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.12).
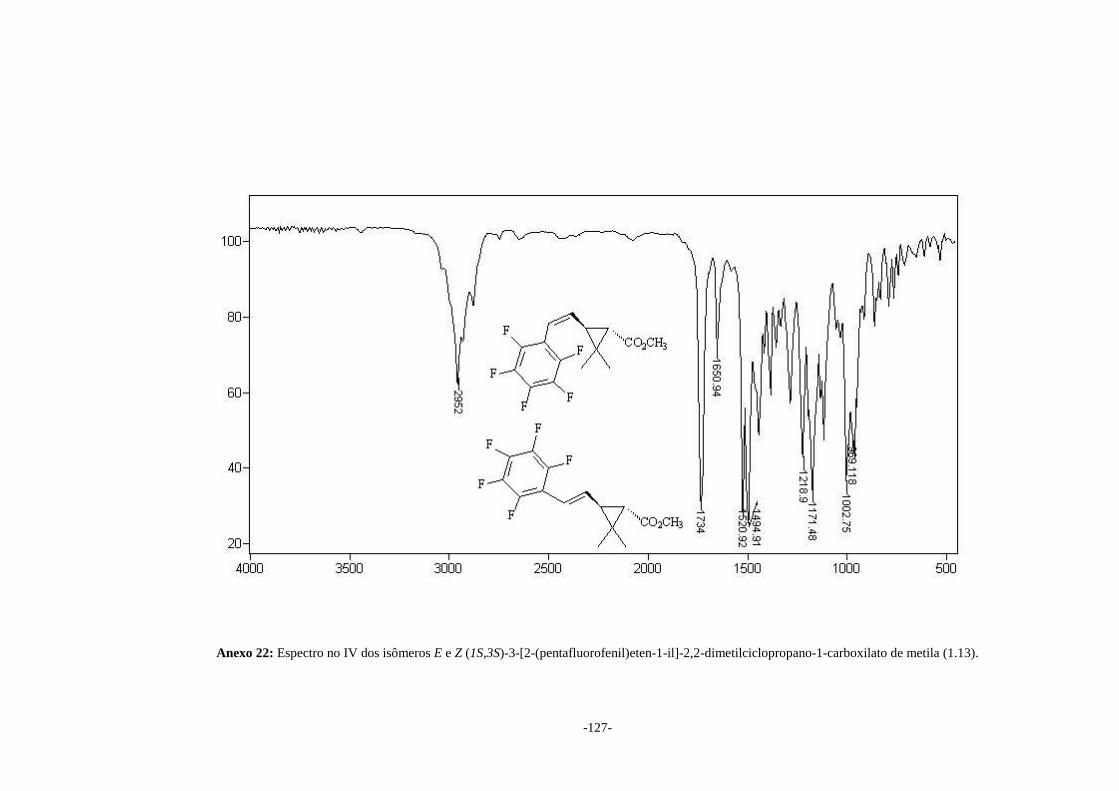
-127-
Anexo 22: Espectro no IV dos isômeros E e Z (1S,3S)-3-[2-(pentafluorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.13).

-128-
Anexo 23: Espectro no IV dos isômeros E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14).

-129-
Anexo 24: Espectro de RMN de 1H dos isômeros E e Z (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.10).

-130-
Anexo 25: Espectro de RMN de 1H dos isômeros E e Z (1S,3S)-3-[2-(3-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.11).
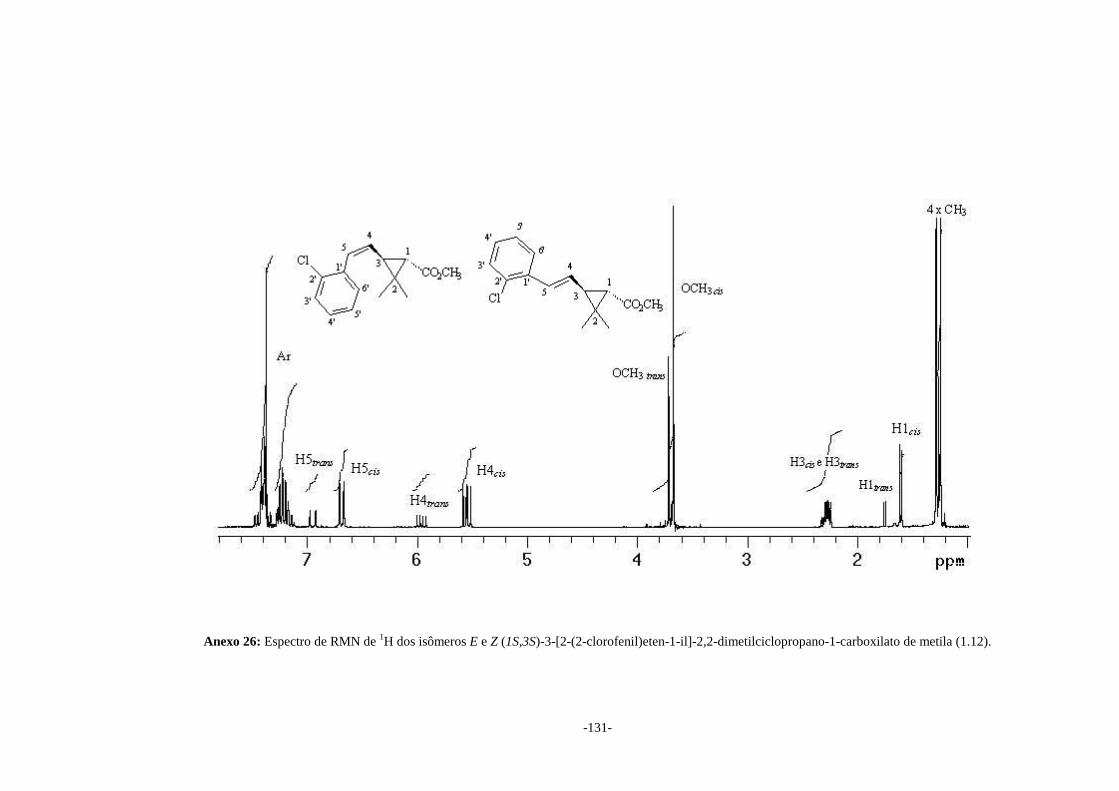
-131-
Anexo 26: Espectro de RMN de 1H dos isômeros E e Z (1S,3S)-3-[2-(2-clorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.12).

-132-
Anexo 27: Espectro de RMN de 1H dos isômeros E e Z (1S,3S)-3-[2-(pentafluorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.13).

-133-
Anexo 28: Espectro de RMN de 1H dos isômeros E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14).

-134-
Anexo 29: Espectro de RMN de 13C dos isômeros E e Z (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.10).

-135-
Anexo 30: Espectro de RMN de 13C dos isômeros E e Z (1S,3S)-3-[2-(3-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.11).

-136-
Anexo 31: Espectro de RMN de 13C dos isômeros E e Z (1S,3S)-3-[2-(2-clorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.12).

-137-
Anexo 32: Espectro de RMN de 13 C dos isômeros E e Z (1S,3S)-3-[2-(pentafluorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.13).

-138-
Anexo 33: Espectro de RMN de 13C dos isômeros E e Z (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14).

-139-
Anexo 34: Espectro de Massas do (1S,3S)-3-[2-(2-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.10).
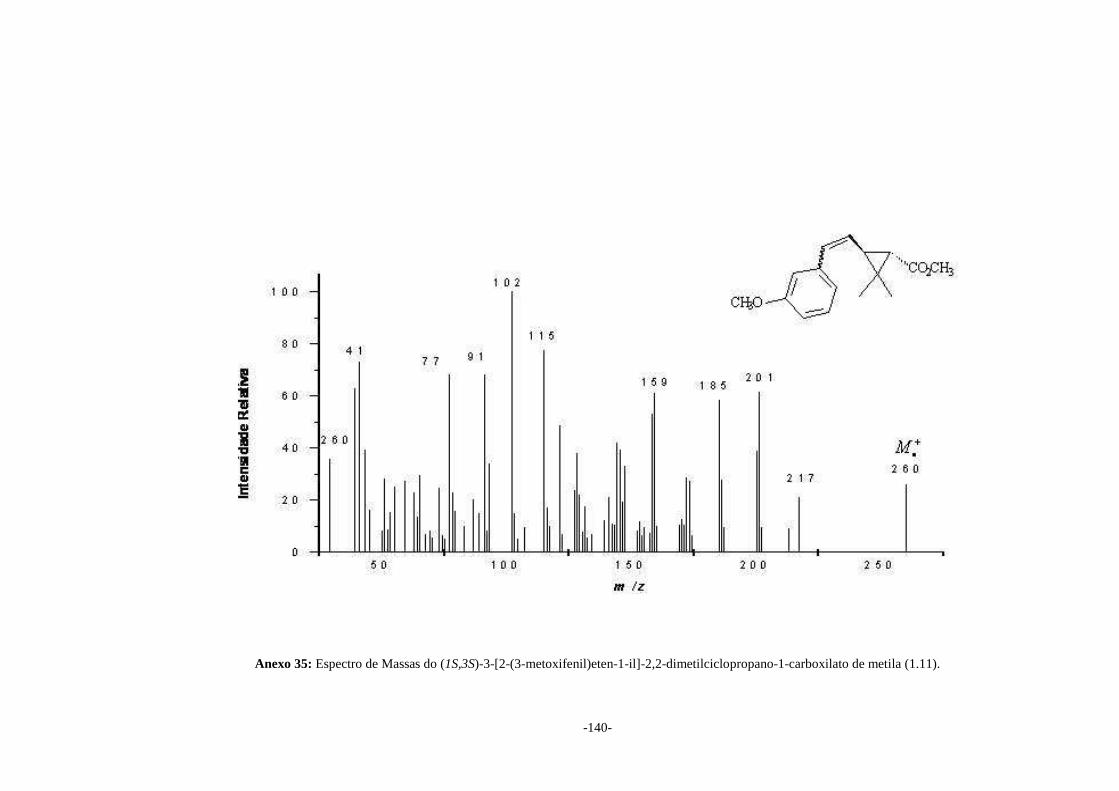
-140-
Anexo 35: Espectro de Massas do (1S,3S)-3-[2-(3-metoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.11).

-141-
Anexo 36: Espectro de Massas do (1S,3S)-3-[2-(2-clorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.12).

-142-
Anexo 37: Espectro de Massas do (1S,3S)-3-[2-(pentafluorofenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.13).

-143-
Anexo 38: Espectro de Massas do (1S,3S)-3-[2-(4-etoxifenil)eten-1-il]-2,2-dimetilciclopropano-1-carboxilato de metila (1.14).

-144-
Anexo 39: Espectro no IV da 5H-furan-2-ona (2.2a).

-145-
Anexo 40: Espectro de RMN de 1H da 5H-furan-2-ona (2.2a).

-146-
Anexo 41: Espectro de RMN de 13C da 5H-furan-2-ona (2.2a).

-147-
Anexo 42: Espectro no IV da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3).
-148-
Anexo 43: Espectro de RMN de 1H da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3).

-149-
Anexo 44: Espectro de RMN de 13C da 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.3).

-150-
Anexo 45: Espectro no IV da 4-isopropeniltetraidrofuran-2-ona (2.4a).

-151-
Anexo 46: Espectro de RMN de 1H da 4-isopropeniltetrahidrofuran-2-ona (2.4a).

-152-
Anexo 47: Espectro de RMN de 13C da 4-isopropeniltetrahidrofuran-2-ona (2.4a).

-153-
Anexo 48: Espectro no IV da 4-(1-metiletilideno)tetraidrofuran-2-ona (2.4b).

-154-
Anexo 49: Espectro de RMN de 1H da 4-(1-metiletilideno)tetraidrofuran-2-ona (2.4b).

-155-
Anexo 50: Espectro de RMN de 13C da 4-(1-metiletilideno)tetraidrofuran-2-ona (2.4b).

-156-
Anexo 51: Espectro no IV da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5).

-157-
Anexo 52: Espectro de RMN de 1H da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5).

-158-
Anexo 53: Espectro de RMN de 13C da 4-(1’-bromo-1’-metiletil)tetraidrofuran-2-ona (2.5).

-159-
Anexo 54: Espectro no IV da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6).

-160-
Anexo 55: Espectro de RMN de 1H da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6).

-161-
Anexo 56: Espectro de RMN de 13C da 6,6-dimetil-3-oxabiciclo[3.1.0]hexan-2-ona (2.6).

-162-
Anexo 57: Espectro no IV da (S)-5-hidroximetil(5H)furan-2-ona (2.7).

-163-
Anexo 58: Espectro de RMN de 1H da (S)-5-hidroximetil(5H)furan-2-ona (2.7).

-164-
Anexo 59: Espectro de RMN de 13C da (S)-5-hidroximetil(5H)furan-2-ona (2.7).

-165-
Anexo 60: Espectro no IV da (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8).

-166-
Anexo 61: Espectro de RMN de 1H da (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8).
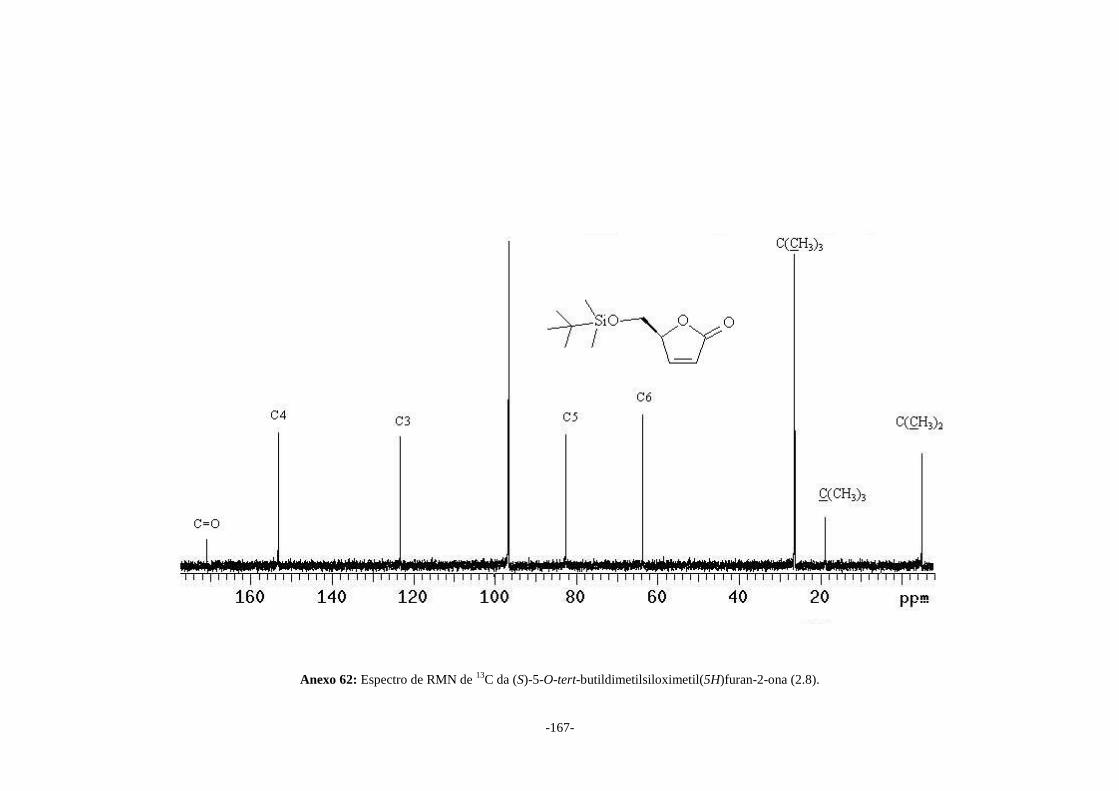
-167-
Anexo 62: Espectro de RMN de 13C da (S)-5-O-tert-butildimetilsiloximetil(5H)furan-2-ona (2.8).
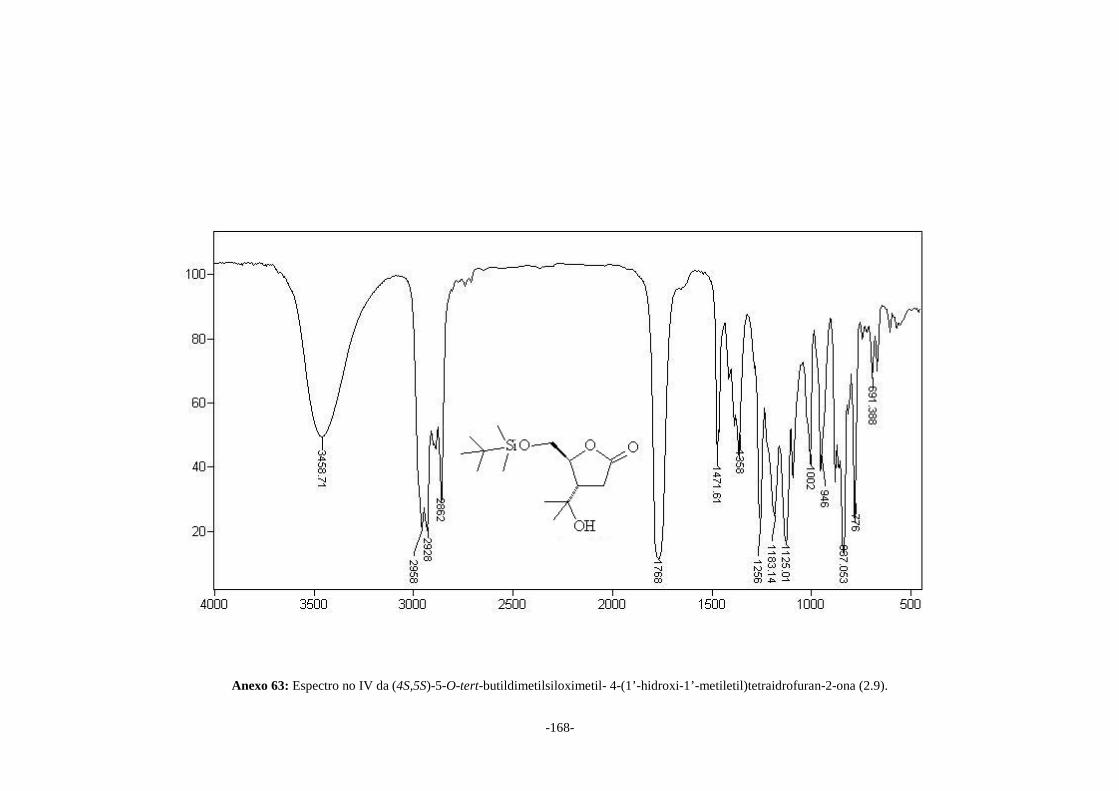
-168-
Anexo 63: Espectro no IV da (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.9).

-169-
Anexo 64: Espectro de RMN de 1H da (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.9).
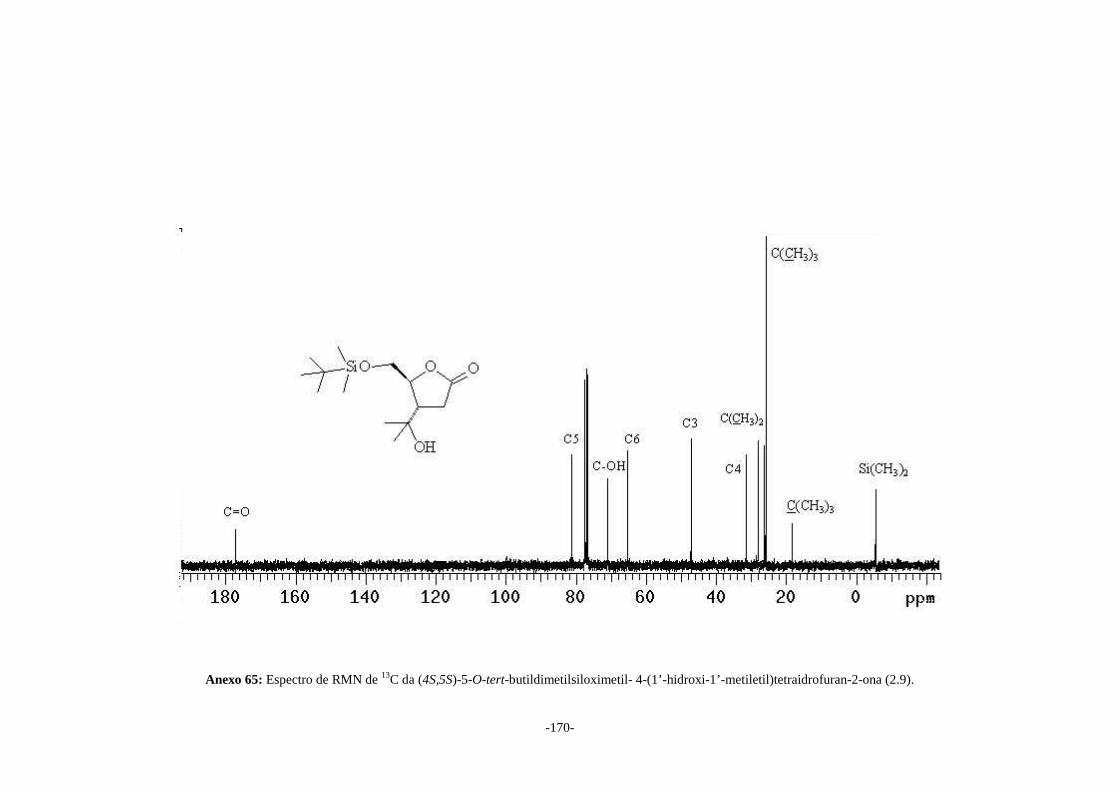
-170-
Anexo 65: Espectro de RMN de 13C da (4S,5S)-5-O-tert-butildimetilsiloximetil- 4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.9).

-171-
Anexo 66: Espectro no IV da (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.10).

-172-
Anexo 67: Espectro de RMN de 1H da (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.10).

-173-
Anexo 68: Espectro de RMN de 13C da (4S,5S)-5-hidroximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.10).

-174-
Anexo 69: Espectro no IV da (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.11).

-175-
Anexo 70: Espectro de RMN de 1H (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.11).

-176-
Anexo 71: Espectro de RMN de 13C da (4S,5S)-5-benzoiloximetil-4-(1’-hidroxi-1’-metiletil)tetraidrofuran-2-ona (2.11).
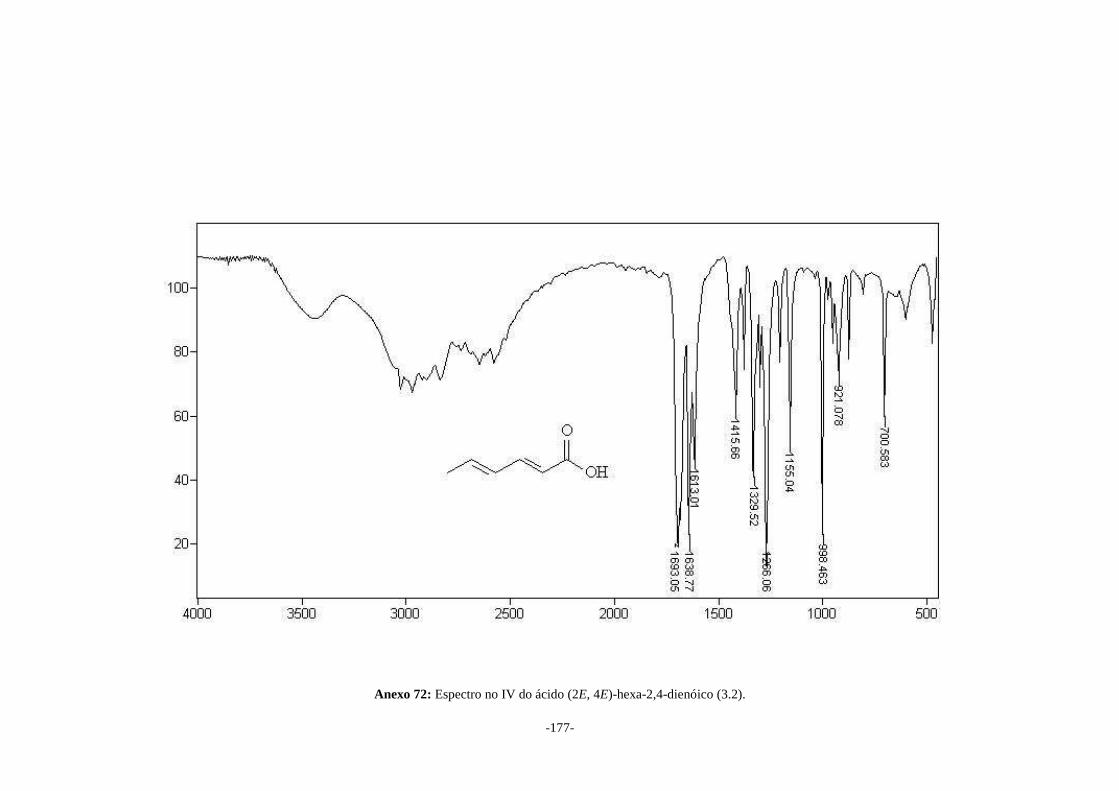
-177-
Anexo 72: Espectro no IV do ácido (2E, 4E)-hexa-2,4-dienóico (3.2).

-178-
Anexo 73: Espectro de RMN de 1H do ácido (2E, 4E)-hexa-2,4-dienóico (3.2).

-179-
Anexo 74: Espectro de RMN de 13C do ácido (2E, 4E)-hexa-2,4-dienóico (3.2).

-180-
Anexo 75: Espectro de RMN de 1H do ácido (2Z,4E)-hexa-2,4-dienóico (3.5).

-181-
Anexo 76: Espectro de RMN de 13C do ácido (2Z,4E)-hexa-2,4-dienóico (3.5).

-182-
Anexo 77: Espectro no IV do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3).

-183-
Anexo 78: Espectro de RMN de 1H do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3).
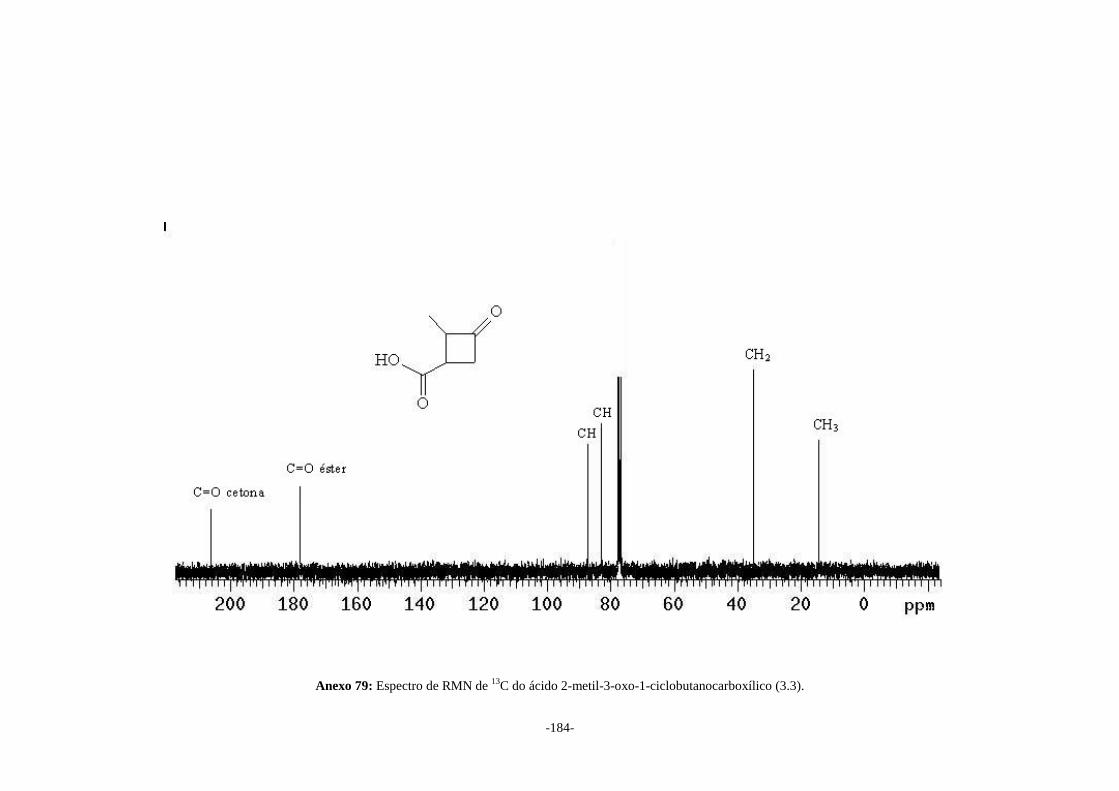
-184-
Anexo 79: Espectro de RMN de 13C do ácido 2-metil-3-oxo-1-ciclobutanocarboxílico (3.3).

-185-
Anexo 80: Espectro no IV da 3-hidroximetil-2-metilciclobutan-1-ona (3.4).

-186-
Anexo 81: Espectro de RMN de 1H da 3-hidroximetil-2-metilciclobutan-1-ona (3.4).

-187-
Anexo 82: Espectro de RMN de 13C da 3-hidroximetil-2-metilciclobutan-1-ona (3.4).

-188-
Anexo 83: Espectro no IV do 5-hidroxi-4-oxopentanoato de metila (3.6).

-189-
Anexo 84: Espectro de RMN de 1H do 5-hidroxi-4-oxopentanoato de metila (3.6).

-190-
Anexo 85: Espectro de RMN de 13C do 5-hidroxi-4-oxopentanoato de metila (3.6).

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo