universidade federal do cearÁ - repositorio.ufc.br · palavras-chave: ataque isquêmico...
TRANSCRIPT

1
UNIVERSIDADE FEDERAL DO CEARÁ
FACULDADE DE MEDICINA
DEPARTAMENTO DE MEDICINA CLÍNICA
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS MÉDICAS
CAROLINA MELO DE SOUZA
EFEITO NEUROPROTETOR DA CURCUMINA SOBRE O ESTRESSE
OXIDATIVO, INFLAMAÇÃO, MEMÓRIA E DANO NEURONAL DE RATOS
SUBMETIDOS À ISQUEMIA CEREBRAL TRANSITÓRIA
FORTALEZA
2012

2
CAROLINA MELO DE SOUZA
EFEITO NEUROPROTETOR DA CURCUMINA SOBRE O ESTRESSE OXIDATIVO,
INFLAMAÇÃO, MEMÓRIA E DANO NEURONAL DE RATOS SUBMETIDOS À
ISQUEMIA CEREBRAL TRANSITÓRIA
Tese de Doutorado apresentada ao Programa
de Pós-Graduação em Ciências Médicas, da
Faculdade de Medicina da Universidade
Federal do Ceará, como requisito parcial para
obtenção do Título de Doutor em Ciências
Médicas. Área de concentração: Biomedicina.
Orientadora: Profa. Dra. Geanne Matos de
Andrade.
FORTALEZA
2012

3
Dados Internacionais de Catalogação na Publicação Universidade Federal do Ceará
Biblioteca de Ciências da Saúde
S714e Souza, Carolina Melo de. Efeito neuroprotetor da curcumina sobre o estresse oxidativo, inflamação, memória e dano
neuronal de ratos submetidos à isquemia cerebral transitória/ Carolina Melo de Souza. – 2012.
99 f. : il.
Tese (Doutorado) - Universidade Federal do Ceará. Faculdade de Medicina. Programa de
Pós-Graduação em Ciências Médicas, Fortaleza, 2012.
Área de concentração: Biomedicina.
Orientação: Profa. Dra. Geanne Matos de Andrade.
1. Ataque Isquêmico Transitório. 2. Curcumina. 3. Memória. 4. Estresse Oxidativo. 5.
Inflamação I.Título.
CDD 616.81

4
CAROLINA MELO DE SOUZA
EFEITO NEUROPROTETOR DA CURCUMINA SOBRE O ESTRESSE OXIDATIVO,
INFLAMAÇÃO, MEMÓRIA E DANO NEURONAL DE RATOS SUBMETIDOS À
ISQUEMIA CEREBRAL TRANSITÓRIA
Tese de Doutorado apresentada ao Programa
de Pós-Graduação em Ciências Médicas, da
Faculdade de Medicina da Universidade
Federal do Ceará, como requisito parcial para
obtenção do Título de Doutor em Ciências
Médicas. Área de concentração: Biomedicina.
Data de aprovação 29/06/2012
BANCA EXAMINADORA
__________________________________________
Profa. Dra. Geanne Matos de Andrade (Orientadora)
Universidade Federal do Ceará - UFC
___________________________________________
Profa. Dra. Francisca Cléa Florenço de Sousa
Universidade Federal do Ceará - UFC
____________________________________________
Dr. Francisco Romero Cabral
Instituto do Cérebro –
Instituto Irsaelita de Ensino e Pesquisa Albert Einstein
______________________________________________
Profa. Dra. Tatiana Lima Ferreira
Universidade Federal do ABC - UFABC
_____________________________________________
Profa. Dra. Maria Erivalda Farias de Aragão
Universidade Estadual do Ceará - UECE

5
AGRADECIMENTOS
À minha família, por todo o amor e apoio incondicional.
À minha orientadora Geanne, por toda paciência e por me trazer de volta para o
mundo da ciência.
Agradeço aos meus amigos por trazerem leveza para minha vida.
Aos colegas do Laboratório de Neurociência e Comportamento pela convivência
harmoniosa.
Aos professores dos Programas de Pós-graduação em Ciências Médicas e de Pós-
graduação Farmacologia pelos ensinamentos.
Às secretárias e funcionários do Programas de Pós-graduação em Ciências Médicas
por toda a paciência e ajuda.
Às agências financiadoras CAPES, CNPq e FUNCAP por terem colaborado
financeiramente com todo o projeto de pesquisa.
À todos, minha humilde e sincera gratidão.

6
“A alegria não chega apenas no encontro do achado,
mas faz parte do processo da busca.
E ensinar e aprender não pode dar-se fora da procura,
fora da boniteza e da alegria.”
(Paulo Freire)

7
RESUMO
A fisiopatologia da isquemia cerebral envolve uma complexa cascata de eventos dentre eles
excitotoxicidade, inflamação, estresse oxidativo e apoptose. A curcumina é um polifenol
presente no rizoma da Curcuma longa e que apresenta propriedades antiinflamatória e
antioxidante. No presente estudo, foram investigados os efeitos da curcumina sobre os déficits
de memória, estresse oxidativo, inflamação e dano neuronal induzidos por isquemia cerebral
transitória (ICT). A ICT foi induzida por oclusão das artérias carótidas, por 30min, seguida de
reperfusão. Os animais receberam curcumina ou veículo (6% de DMSO em tampão fosfato)
por via oral 60 min antes e diariamente após a ICT. A avaliação dos déficits cognitivos foi
realizada através dos testes do labirinto em Y, da esquiva passiva e do labirinto aquático que
avaliam as memórias de trabalho, aversiva e espacial, respectivamente. A integridade
neuronal foi avaliada através de análise histológica do hipocampo utilizando as técnicas de
violeta de cresil e Fluoro Jade C. As dosagens de IL-1β e mieloperoxidase (MPO) em
homogenatos hipocampais foram utilizadas para a avaliação da inflamação. O estresse
oxidativo foi analisado através da quantificação de malondialdeído (MDA), nitrito/nitrato,
glutationa reduzida (GSH) e atividade da superóxido dismutase (SOD) hipocampais. Não
foram observadas alterações na atividade locomotora e nem na memória de trabalho. A
curcumina preveniu déficits nas memórias aversiva recente e tardia induzidas por ICT. A ICT
promoveu déficits no aprendizado e memória espacial. A curcumina não alterou a
aprendizagem, mas apresentou uma tendência estatística na prevenção do déficit de memória
espacial. A ICT promoveu o aumento na atividade de MPO e tendência a aumento nas
concentrações de IL-1β e esse efeito foi prevenido pelo tratamento com curcumina. A ICT
promoveu o aumento significativo nas concentrações de MDA. O tratamento com a
curcumina não alterou esse efeito 6h, mas preveniu o aumento 4 dias após a ICT. A
curcumina preveniu o aumento de nitrito e a depleção de GSH induzidos por ICT. A
curcumina protegeu os neurônios da morte 4 mas não 7 dias após a ICT. Os efeitos
neuroprotetores da curcumina parecem estar relacionados com suas propriedades anti-
inflamatórias e antioxidante.
Palavras-chave: Ataque Isquêmico Transitório. Curcumina. Memória. Estresse Oxidativo.
Inflamação.

8
ABSTRACT
The pathophysiology of cerebral ischemia involves a complex cascade of events which
includes excitotoxicity, inflammation, oxidative stress and apoptosis. Curcumin is a
polyphenol extracted from the rhizome of Curcuma longa. It has anti-inflammatory and
antioxidant properties. In this study, we investigated the effects of curcumin on memory
deficits, oxidative stress, inflammation and neuronal damage induced by transient cerebral
ischemia (TCI). The TCI was induced by bilateral common carotid artery occlusion for 30
min followed by reperfusion. Vehicle or curcumin (6% DMSO in phosphate buffer) were
orally administered 60 min before and daily after TCI. Cognitive evaluation was performed
by using Y-maze, passive avoidance and water maze tests to assess working, spatial and
aversive memories, respectively. To assess neuronal injury histological sections from
hippocampus were stained with cresyl violet or FluoroJade C. The interleukin-1β and
myeloperoxidase (MPO) contents from hippocampus were used for inflammatory evaluation.
Oxidative stress was assessed by quantification of malondialdehyde (MDA), nitrite/nitrate,
reduced glutathione (GSH) and superoxide dismutase (SOD) hippocampal. There were no
changes in locomotor activity, neither in working memory. Curcumin prevented the TCI-
induced early and late aversive memory deficits. The TCI impair spatial learning and
memory. Curcumin treatment did not affect spatial learning, but showed a tendency to prevent
the impairment on spatial memory. TCI promoted the increase in MPO activity and a
tendency to increased concentrations of IL-1β and this effect was prevented by curcumin
treatment. Curcumin treatment did not prevent the TCI-induced MDA increases 6h after
surgery. Four days after surgery TCI showed a tendency to increase MDA contents and
curcumin treatment lowered it. Curcumin prevented TCI- induced nitrite formation 6h after
surgery and GSH depletion 24 hours after surgery. Curcumin protected the neurons from
death 4d but not 7 days after TCI. Our results demonstrated that curcumin neuroprotection
effects may be due to its anti-inflammatory and antioxidant properties.
Key words: Ischemic Attack Transient. Curcumin. Memory. Oxidative Stress.
Inflammation.

9
LISTA DE ILUSTRAÇÕES
Figura 1 – Mecanismos potenciais de insulto neuronal após isquemia. ............................... 20
Figura 2 – Resumo dos principais tipos e fontes de EROs e o ponto de ação de antioxidantes.
..................................................................................................................................... 22
Figura 3 – Resumo dos principais eventos inflamatórios que acontecem durante a
isquemia/reperfusão ..................................................................................................... 24
Figura 4 – Via mitocondrial da apoptose. ............................................................................ 27
Figura 5 - Via da apoptose mediada por receptores de morte. .............................................. 28
Quadro 1 – Tipos de isquemia cerebral. .............................................................................. 29
Figura 6 – Esquema mostrando as artérias cerebrais anteriores e posteriores, arterias
comunicantes e posteriores e a carotida interna que formam o polígono Willis. ............ 30
Figura 7 – Taxonomia dos sistemas de memória de longa duração. ..................................... 33
Figura 8 – Estrutura química da curcumina. ........................................................................ 36
Figura 9 – Potencias doenças alvo da curcumina ................................................................. 38
Figura 10 - Protocolo experimental 1. ................................................................................ 43
Figura 11 – Protocolo experimental 2. ................................................................................. 44
Figura 12 – Campo Aberto. ................................................................................................ 45
Figura 13 - Labirinto em Y. ................................................................................................ 46
Figura 14 – Aparelho de Esquiva Passiva. .......................................................................... 48
Figura 15 - Labirinto Aquático. .......................................................................................... 49
Figura 16 - Efeito da curcumina (50 e 100mg/kg) sobre o número de crossings e de rearings
de ratos submetidos à isquemia cerebral transitória (ICT). ............................................ 54
Figura 17 - Efeito da curcumina (50 e 100mg/kg) sobre a memória de trabalho de ratos
submetidos à ICT. ........................................................................................................ 55
Figura 18 - Efeito da curcumina (50 e 100mg/kg) sobre a memória aversiva recente e tardia
de ratos submetidos à ICT. ........................................................................................... 56
Figura 19 - Efeito do tratamento com a curcumina (100 e 300mg/kg) sobre o aprendizado
espacial de ratos submetidos à ICT. .............................................................................. 57

10
Figura 20 – Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro latência para alcançar a plataforma, de ratos submetidos à ICT.... 58
Figura 21 – Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro número de cruzamentos, de ratos submetidos à ICT. ................... 59
Figura 22 – Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro tempo de permanência no quadrante, de ratos submetidos à ICT. 59
Figura 23 – Efeito da curcumina (100mg/kg) sobre a peroxidação lipídica em hipocampo de
ratos 6h após a ICT....................................................................................................... 60
Figura 24 – Efeito da curcumina (50 e 100mg/kg) sobre a peroxidação lipídica em
hipocampo de ratos 96h após a ICT. ............................................................................. 61
Figura 25 – Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em hipocampo
de ratos 24h após a ICT.. ............................................................................................. 62
Figura 26 – Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em hipocampo
de ratos 96h após a ICT. ............................................................................................... 63
Figura 27 – Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em estriado de
ratos 96h após a ICT. .................................................................................................... 63
Figura 28 – Efeito da curcumina (50 e 100mg/kg) sobre os níveis de GSH em hipocampo de
ratos 24h após a ICT.. .................................................................................................. 64
Figura 29 – Efeito da curcumina (50 e 100mg/kg) sobre os níveis de SOD em hipocampo de
ratos 96h após a ICT.. .................................................................................................. 65
Figura 30 – Efeito da curcumina (100mg/kg) sobre os níveis de MPO em hipocampo de ratos
24h após a ICT. ............................................................................................................ 66
Figura 31 – Efeito da curcumina (100mg/kg) sobre os níveis de IL-1β em hipocampo de ratos
24h após a ICT. ............................................................................................................ 67
Figura 32 – Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia. Coloração violeta de cresil. ... 69
Figura 33 – Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia. Coloração violeta de cresil. ... 70
Figura 34 – Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia. Fluoro Jade C ........................ 71
Figura 35 – Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia. Fluoro Jade C ........................ 72
Figura 36 – Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia. Coloração violeta de cresil. ... 73

11
Figura 37 – Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia. Coloração violeta de cresil. ... 74
Figura 38 – Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia. A e B: FO; C e D: ICT; E e F:
ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Fluoro Jade C ...... 75
Figura 39 – Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia. Fluoro Jade C ........................ 76

12
LISTA DE ABREVIATURAS E SIGLAS
2-VO Oclusão de dois vasos
4-VO Oclusão de quatro vasos
AA Ácido araquidônico
ACM Artéria cerebral média
AGL Ácidos graxos livres
AIT Ataque Isquêmico Transitório
AMPA Ácido amino-3-hidroxi-5-metil-isoxasole-propiônico
ANOVA Análise de variância
ATP Adenosina trifosfato
AVC Acidente Vascular Cerebral
CA Corno Amon
CAD DNAse ativada por caspase
CAMKII Cálcio-calmodulina quinase II
CAT Catalase
COX-2 Ciclooxigenase 2
CPF Córtex pré-frontal
DMSO Dimetilsulfóxido
DTNB Ácido 5,5'-ditiobis (ácido 2-nitrobenzóico)
EDTA Ácido Etilenodiamino Tetra-Acético
EPM Erro padrão da média
ERNs Espécies reativas de nitrogênio
EROs Espécies reativas de oxigênio
FADD Domínio de morte associado ao FAS
FO Falso-operado

13
GD Giro denteado
GPX Glutationa peroxidase
GSH Glutationa reduzida
GSSG Glutationa oxidada
H2O2 Peróxido de hidrogênio
HO2•
Radical peroxila
IAPs Proteínas inibidoras da apoptose
ICAD Inibidor de endonuclease
ICAM-1 Moléculas de adesão intercelular-1
ICE Enzima Conversora de interleucina-1 beta
ICT Isquemia cerebral transitória
IL-1β Interleucina-1 beta
iNOS Óxido nítrico sintase induzida
IRF-1 Fator regulador de interferon 1
LOX Lipooxigenase
LTP Potenciação de longa duração
MAP-2 Proteína associada à microtúbulos-2
MCP-1 Proteína quimiotática de monócito-1
MDA Malondialdeído
MIP-1α Proteína inflamatória de macrófagos 1
MPO Mieloperoxidase
NBT Azul de nitro-tetrazolio
NF-kb Fator nuclear kappa b
NMDA N-Metil-D-Aspartato
nNOS Óxido nítrico sintase neuronal

14
NO Óxido nítrico
NOS Óxido nítrico sintase
O˚ radical oxigênio
O2 −• Ânion superóxido
OH• Radical hidroxila
ONOO ̄ Peroxinitrito
PARP Proteínas reparadoras de DNA
PBS Tampão salina-fosfato
PLA2 Fosfolipase A
PLC Fosfolipase C
R • Radical lipídico alquila
RH- Lipídios
RLs Radicais livres
ROO • Radical lipídico peroxil
ROOH Lipídio hidroperóxido
SMAC Ativador de caspase derivado da mitocôndria
SNC Sistema Nervoso Central
SOD Superóxido dismutase
TBARS Substâncias reativas ao ácido tiobarbitúrico
TCA Ácido tricloro acético
TNF-α Fator de necrose tumoral-α
t-PA Ativador plasminogênio tecidual

15
SUMÁRIO
1 INTRODUÇÃO 15
1.1 Acidente Vascular Cerebral 15
1.1.1 Definição e classificação 15
1.1.2 Epidemiologia 15
1.2 Fisiopatologia da isquemia cerebral 16
1.3 Mecanismos de dano neuronal induzidos por isquemia 18
1.3.1 Depleção de ATP e despolarização da membrana 18
1.3.2 Excitotoxicidade e Ca2+
18
1.3.3 Estresse oxidativo 20
1.3.4 Inflamação 23
1.3.5 Apoptose 25
1.4 Modelos animais de isquemia cerebral 29
1.5 Memória 31
1.6 Curcumina 35
1.7 Relevância e justificativa 39
2 OBJETIVOS 41
2.1 Objetivo Geral 41
2.2 Objetivos Específicos 41
3. MATERIAIS E MÉTODOS 42
3.1 Animais 42
3.2 Drogas 42
3.3 Protocolo Experimental 42
3.4 Isquemia cerebral por oclusão bilateral da carótida 44
3.5 Avaliação da atividade locomotora – Teste do campo aberto 45
3.6 Avaliação da memória de trabalho – Teste do labirinto em Y 46
3.7 Avaliação da memória aversiva- Teste da esquiva passiva 47
3.8 Avaliação da memória espacial – Teste do labirinto aquático 48
3.9. Dissecção do hipocampo 49
3.10 Ensaio para mieloperoxidase (MPO) 49
3.11 Dosagem de substâncias reativas ao ácido tiobarbitúrico (TBARS) 50
3.12 Dosagem de superóxido dismutase (SOD) 50

16
3.13 Dosagem de nitrito 51
3.14 Determinação da concentração da glutationa reduzida (GSH) 51
3.15 Determinação da concentração de IL-1B 51
3.16 Análise histológica 52
3.16.1 Coloração de violeta de cresil 52
3.16.2 Fluoro Jade C 52
3.17 Análise estatística 52
4 RESULTADOS 54
4.1 Efeito da curcumina (50 e 100mg/kg) sobre a atividade locomotora de ratos
submetidos à isquemia cerebral transitória (ICT) 54
4.2 Efeito da curcumina (50 e 100mg/kg) sobre a memória de trabalho de ratos
submetidos à ICT. 55
4.3 Efeito da curcumina (50 e 100mg/kg) sobre a memória aversiva de ratos
submetidos à ICT. 56
4.4 Efeito da curcumina (50 e 100mg/kg) sobre o aprendizado e memória espacial
de ratos submetidos à ICT. 57
4.5 Efeito da curcumina (50 e 100mg/kg) sobre a peroxidação lipídica no
hipocampo e estriado de ratos submetidos à ICT. 60
4.6 Efeito da curcumina (50 e 100mg/kg) sobre a concentração de nitrito no
hipocampo e estriado de ratos submetidos à ICT. 62
4.7 Efeito da curcumina (50 e 100mg/kg) sobre a concentração de glutationa
reduzida (GSH) no hipocampo de ratos submetidos à ICT. 64
4.8 Efeito da curcumina (50 e 100mg/kg) sobre os níveis da superóxido dismutase
(SOD) no hipocampo de ratos submetidos à ICT. 65
4.9 Efeito da curcumina (100mg/kg) sobre a atividade da mieloparoxidase (MPO)
no hipocampo de ratos submetidos à ICT. 66
4.10 Efeito da curcumina (100mg/kg) sobre a concentração de IL-1β no hipocampo
e no estriado de ratos submetidos à ICT. 67
4.11 Efeito da curcumina (100 e 300mg/kg) sobre a integridade neuronal das
regiões CA1 e CA3 do hipocampo de ratos submetidos à ICT. 68
5 DISCUSSÃO 77
6 CONCLUSÃO 87
REFERÊNCIAS 88

15
1 INTRODUÇÃO
1.1 Acidente Vascular Cerebral
1.1.1 Definição e classificação
O Acidente Vascular Cerebral (AVC) é uma emergência médica caracterizada pelo
início agudo ocasionando déficit neurológico que persiste por pelo menos 24 horas. Difere do
Ataque Isquêmico Transitório (AIT) que é caracterizado por um déficit neurológico focal,
encefálico ou retiniano, súbito e reversível, com duração menor que 1 hora e sem evidência de
lesão isquêmica nos exames de imagem. No entanto, podemos citar que ambos são causados
pela redução do suprimento sanguíneo, resultando no decréscimo da tensão de oxigênio e dos
metabólitos de alta energia em uma área do cérebro (ATP e glicose, principalmente). O AIT e
AVC isquêmico são espectros de uma mesma doença vascular isquêmica. Cerca de 15 a 30%
dos pacientes que apresentam AVC isquêmico têm história prévia de ataque isquêmico
transitório, muitas vezes em um curto intervalo de tempo (MASSARO, 2006).
O AVC é comumente dividido em duas maiores categorias. A primeira e mais comum
é o AVC isquêmico ou tromboembólico (cerca de 80% dos casos), o qual se deve a uma
interrupção do suprimento sanguíneo como resultado da oclusão de uma artéria (BROUNS;
DE DEYN, 2009). A segunda categoria é o AVC hemorrágico, o qual envolve sangramento
no parênquima cerebral ou no espaço subaracnóideo.
1.1.2 Epidemiologia
Segundo dados da Organização Mundial de Saúde, o AVC representa a segunda causa
de morte em todo o mundo, sendo responsável por cerca de 6 milhões de mortes/ano. O AVC
já é a primeira ou segunda causa de morte em muitos países latino-americanos e caribenhos,
embora, na última década, as taxas estejam em queda (LAVADOS et al., 2007). O Brasil
apresenta nítidas diferenças, tanto regionais como entre classes, nas taxas de incidência e
mortalidade decorrentes do AVC sendo ainda a causa mais frequente de óbito em parte desta
população. A distribuição desigual de riqueza, os inadequados acessos aos avanços científico
e tecnológico e à assistência à saúde encontram-se entre os fatores responsáveis por essas
diferenças (FALCÃO et al., 2004; MASSARO, 2006).
Nas últimas quatro décadas, as taxas de incidência de AVC caíram 42% em países
ricos enquanto, em países de baixa e média renda houve uma duplicação dessas taxas. Entre

16
os anos de 2000 e 2008, as taxas de incidência em países de baixa e média renda superaram,
pela primeira vez, as taxas daqueles de alta renda (FEIGIN et al., 2009). Avaliando as taxas
de mortalidade por AVC no Brasil, Garritano et al. (2012), observaram que entre os anos de
2000 e 2009, houve uma tendência de queda em todas as faixas etárias estudadas (a partir de
30 anos). Entretanto, esses valores continuam elevados, sendo superiores aos encontradas em
países desenvolvidos e ainda sendo considerada a quarta maior taxa entre todos os países da
América Latina.
O AVC representa a principal causa de incapacidade em todo o mundo. O seu pico de
incidência está entre a sétima e oitava década de vida, mas também pode ocorrer mais
precocemente, sendo que 10% dos pacientes têm idade inferior a 55 anos (ZÉTOLA et al.,
2001). Nos Estados Unidos aproximadamente 730.000 pessoas sofrem um AVC a cada ano,
sendo que 25% morrem no momento do evento ou logo depois e metade delas tornam-se
permanentemente incapacitadas. Os custos diretos e indiretos com AVC são de
aproximadamente 50 bilhões de dólares anuais (SICARD; FICHER, 2009). O fato do AVC
levar à incapacidade indivíduos em idade produtiva tem um forte impacto econômico. Dados
do Instituto Nacional de Seguridade Social demonstram que o AVC é responsável por 40%
das aposentadorias precoces no Brasil (FALCÃO et al., 2004).
Uma parte significativa do risco de AVC pode ser atribuída aos clássicos e evitáveis
fatores de risco cardiovasculares como hipertensão arterial, hiperlipidemias, diabetes,
tabagismo, sedentarismo e obesidade. Evitar esses modificáveis fatores de risco pode ter um
efeito substancial na expectativa e qualidade de vida. Embora fatores de risco
cardiovasculares possam ser mais prevalentes em países de alta renda do que em países de
baixa e média renda, esses fatores são geralmente mais bem geridos nos primeiros o que pode
explicar, em parte, as diferenças nas taxas de mortalidade por AVC. Em países de baixa renda
a pressão arterial pode chegar a grandes extremos, e mesmo assim continuar sem tratamento
(JOHNSTON; MENDIS; MATHERS, 2009).
1.2 Fisiopatologia da isquemia cerebral
A isquemia cerebral pode ser desencadeada através da oclusão de vasos cervico-
cranianos ou por hipoperfusão para o cérebro, causada por variados processos, tais como,
arterotrombose, embolia ou anormalidades hemodinâmicas (KAPOSZTA et al., 1999). A
arterotrombose ocorre nas artérias cervico-cranianas e nas pequenas artérias penetrantes

17
intracranianas. Nesta condição, um trombo é formado in situ em um estreitamento arterial
aterosclerótico que impede o fluxo sanguíneo distal e causa isquemia e o consequente infarto
do tecido cerebral suprido pelo vaso ocluído. Na embolia cerebral, a artéria cerebral é
subitamente bloqueada pelo material embólico que é geralmente um trombo originado do
coração e grandes vasos (aorta, carótidas e artérias cerebrais) (KAPOSZTA et al., 1999). O
infarto embólico é responsável por cerca de 30% dos casos de AVC isquêmico, a perda súbita
de perfusão arterial para uma determinada área do cérebro gera sinais clínicos abruptos. A
embolia, geralmente, ocorre devido a alterações cardíacas (sendo as mais comuns as
valvulopatias cardíacas, o aneurisma ventricular e as miocardiopatias), apesar de também
ocorrerem por problemas cirúrgicos em pulmões ou por fraturas ósseas que desenvolvem
embolia gordurosa (ROWLAND; MERRI, 2002).
Essas patologias apresentam conseqüências que variam dependendo da localização e
do tamanho da área cerebral que foi atingida e do tempo que o paciente levou para ser
atendido (melhor o prognóstico quanto mais rápido for iniciada a recuperação). As seqüelas
mais comuns são alterações na função motora, sensitiva, perceptiva, de linguagem e de
memória (KRIZ; LALANCETTE-HÉBERT, 2009).
Outra anormalidade associada à isquemia cerebral é a hipoperfusão sistêmica que é
caracterizada por fluxo sanguíneo cerebral criticamente diminuído causado por falência
cardíaca ou hipovolemia que leva a uma redução global no fluxo sanguíneo. A seqüela mais
comum está relacionada a déficits de memória (BLOCK, 1999). Dessa forma o SNC pode
sofrer lesões isquêmicas globais e focais, e para ambas as situações existem modelos
experimentais.
Os danos causados ao cérebro por isquemia podem provocar perda de sua função, mas
o cérebro pode superar essa situação pela neuroplasticidade (LIEPERT et al., 2000). A
neuroplasticidade se refere a todos os mecanismos implicados na capacidade do cérebro de
mudar sua estrutura e função durante processos de maturação, aprendizado e memória, assim
como em resposta a lesões traumáticas. As alterações relacionadas à neuroplasticidade
incluem modificações em sinapses, dendritos e síntese de fatores neurotróficos essenciais para
a sobrevivência de células nervosas (LLEDO; ALONSO; GRUBB, 2006).

18
1.3 Mecanismos de dano neuronal induzidos por isquemia
1.3.1 Depleção de ATP e despolarização da membrana
A fisiopatologia da isquemia envolve uma complexa cascata de eventos que se inicia
com a interrupção do suprimento de substratos metabólicos, particularmente de oxigênio e
glicose. O tecido cerebral apresenta uma alta taxa metabólica e depende quase exclusivamente
da fosforilação oxidativa para a produção de energia. Durante a isquemia o metabolismo
anaeróbico mantém os níveis de ATP por um curto período de tempo. Quando o tecido se
torna incapaz de sintetizar ATP ocorre à falência de proteínas transportadoras dependentes de
energia. A interrupção do transporte através da bomba de sódio-potássio (Na+/K
+) leva ao
acúmulo de sódio dentro da célula e a perda de potássio com consequente despolarização de
membrana em neurônios e células gliais (MARTIN; LLOYD; COWAN, 1994). A ordem dos
eventos a partir daí é constantemente debatida, mas o que se sabe é que as consequências
desses eventos levam à morte celular por múltiplos processos: excitotoxicidade,
acidotoxicidade, estresse oxidativo, inflamação e apoptose.
1.3.2 Excitotoxicidade e Ca2+
A excitotoxicidade representa um processo extensamente estudado de morte neuronal
e está envolvida em diversas patologias do sistema nervoso central como AVC, doenças
neurodegenerativas e epilepsia (SATTLER; TYMIANSKI, 2001; DOBREK; THOR, 2011).
Durante o período isquêmico e o início da reperfusão a intensa liberação de glutamato, o
acúmulo de Ca2+
citoplasmático e o aumento dos radicais livres são características da fase de
excitotoxicidade. O glutamato é o principal neurotransmissor excitatório no SNC de
mamíferos e desempenha importante papel no desenvolvimento neural e em processos de
aprendizagem e memória. Fisiologicamente, os níveis de glutamato nas sinapses são
regulados através de transportadores dependente de sódio localizados em membrana
plasmática de neurônios e células gliais. A concentração sináptica de glutamato se encontra na
faixa micromolar, enquanto a concentração citosólica é aproximadamente 10 mM (LEE et al.,
2000).
Devido a despolarização da membrana neuronal os canais de Ca2+
dependende de
voltagem tornam-se ativados, levando à entrada desse íon e à liberação de glutamato para o
meio extracelular. Outra consequência da falência energética decorrente da isquemia é o

19
acúmulo citoplasmático de sódio em astrócitos, que promove a reversão dos transportadores
de glutamato e que contribui para acúmulo desse neurotransmissor no meio extracelular. A
excitotoxicidade é decorrência do acúmulo sináptico do glutamato que causa a
superestimulação de seus receptores ionotrópicos, como NMDA e AMPA/KA e de
receptores metabotrópicos. Os receptores AMPA/KA regulam a entrada de Na+ e
despolarização da membrana. Os receptores NMDA são canais iônicos operados por ligantes,
que medeiam o influxo de Ca2+
e que contém um sítio de ligação a agonistas, um sítio
modulatório de glicina, assim como um sítio de ligação dentro do canal iônico, onde o
magnésio exerce um bloqueio voltagem dependente. A despolarização da membrana
promovida por canais AMPA/KA promove a saída do Mg2+
do receptor NMDA e
consequente influxo de Ca2+
.
A falência energética resultante da diminuição da oferta de oxigênio e glicose durante
a isquemia também impede a manutenção da baixa concentração de Ca2+
intracelular pela
Ca2+
ATPase (DOYLE; SIMON; STENZEL-POORE, 2008). O acúmulo de Ca2+
, que pode
ser demonstrado in vitro após exposição ao glutamato e in vivo em neurônios hipocampais
após isquemia global, é prejudicial aos neurônios por diversos mecanismos (CHOI, 1988;
HARTLEY et al., 1993; MELDRUM et al., 1985; ROTHMAN; OLNEY, 1986;
SAKAMOTO et al., 1986). Esses eventos incluem a ativação de fosfolipases, proteases,
endonucleases, calmodulina e óxido nítrico sintase (NOS) que promovem o estresse
oxidativo, processos inflamatórios e até mesmo morte por necrose ou apoptose (Figura 1).

20
Figura 1 – Mecanismos potenciais de insulto neuronal após a isquemia.
Fonte: Traystman, 2003.
1.3.3 Estresse oxidativo
Na estrutura dos átomos e das moléculas, os elétrons associam-se normalmente em
pares. Os radicais livres (RLs) são espécies altamente reativas que contêm um ou mais
elétrons não pareados. Em sistemas biológicos os RLs gerados a partir de oxigênio e
nitrogênio são classificados como espécies reativas de oxigênio (EROs) e espécies reativas de
nitrogênio (ERNs) . Esses radicais são produzidos fisiologicamente devido ao metabolismo
celular e são altamente controlados por sistemas de antioxidantes enzimáticos e não
enzimáticos. Também participam da resposta fisiológica à estímulos nocivos como, por
exemplo, na reposta à agentes infecciosos. Em concentrações baixas e moderadas participam
de cascatas de sinalização celular e, dessa maneira, modulam genes envolvidos no reparo de
DNA, no controle do ciclo celular, da resposta inflamatória e da apoptose (LU et al., 2011;
VALKO, 2007).
ISQUEMIA
DIMINUIÇÃO DA PRODUÇÃO DE ENERGIA
FALHA DAS
BOMBAS IÔNICAS
AUMENTO INTRACELULAR DE
Na+, Cl
-, Ca
++
LIBERAÇÃO DE EXCITOTOXINAS
LESÃO MITOCONDRIAL
ATIVAÇÃO DOS LEUCÓCITOS
MEDIADORES DA
INFLAMAÇÃO
FORMAÇÃO DE RADICAIS DE
OXIGÊNIO
ESTIMULAÇÃO DA FOSFOLIPASE
AUMENTO DOS ÁCIDOS GRAXOS LIVRES
ÁCIDO ARACDÔNICO
ENDOPEROXIDASES ÁCIDO
HIPERPERÓXIDO PROSTAGLANDINAS
LEUCOTRIENOS
LESÃO DA MEMBRANA CELULAR
MORTE NEURONAL
ESTIMULAÇÃO DE PROTEASES
CALMOMODULINA, NOS, NO, ONOO-
QUEBRA DO CITOESQUELETO E
DO DNA

21
O estresse oxidativo tem sido definido com um distúrbio no balanço pró-oxidante e
antioxidante em favor do primeiro, levando a reações potencialmente deletérias em lipídeos,
proteínas, RNA e DNA. Essas reações podem incluir: peroxidação dos ácidos graxos das
membranas celulares; oxidação dos grupos sulfidrila e inativação enzimática; inibição da
síntese de ATP e consumo das reservas de dinucleotídeos adenínicos da nicotinamida;
inibição da bomba de Na+/K
+; alterações do DNA que levam à mutagênese ou apoptose;
inativação direta do NO.
A produção do ânion superóxido (O2 −•) ocorre principalmente durante a fosforilação
oxidativa mitocondrial como bioproduto do metabolismo aeróbico celular. Esse ânion é
considerado uma espécie reativa primária e com fraco poder oxidante e sua ação tóxica
ocorre, na verdade, em função dos produtos de sua redução. Ele pode formar EROs
secundárias, como o peróxido de hidrogênio (H2O2), o radical peroxila (HO2•) e o radical
hidroxil (OH•) ou reagir com o NO originando o peroxinitrito. O peróxido de hidrogênio
(H2O2), apesar de não ser um radical livre, pois não tem elétrons desemparelhados na última
camada, é importante porque participa das reações que produzem o radical hidroxil (OH•), via
reação de Fenton ou de Haber-Weiss. O radical hidroxil é altamente reativo, podendo atacar o
DNA levando a alterações na expressão genética que podem causar apoptose ou mutação.
Também pode reagir com proteína e lipídios causando carbonilação e peroxidação lipídica
(LU et al., 2010; RIBEIRO et al., 2005) (Figura 2).
O NO está envolvido em diversos processos fisiológicos incluindo neurotransmissão,
regulação da pressão arterial, mecanismos de defesa, relaxamento da musculatura lisa e
regulação da imunidade (BERGENDI et al., 1999). A formação de NO é tanto constitutiva,
produzido pela enzima óxido nítrico sintase constitutiva (nNOS ou tipo 1), quanto induzida,
produzido pela NOS induzida (iNOS ou tipo 2) encontrada na microglia. Durante eventos
isquêmicos o aumento de concentração de Ca2+
citoplasmático mediada pelo glutamato leva
ao aumento da síntese do NO. Este atua dilatando vasos e, dessa maneira, aumentando a
oferta de glicose e oxigênio, mas também é capaz de reagir com o radical superóxido (O2 −•)
levando a danos nitrosativos (ESPEY et al., 2000). O estresse nitrosativo promovido pelo
peroxinitrito danifica proteínas, lipídeos de membrana e DNA (Figura 2).

22
Figura 2 - Resumo dos principais tipos e fontes de EROs e o ponto de ação de antioxidantes.
O2−•: ânion superóxido; HO2 •: radical perihidroxil; • OH: o radical hidroxil; H2O2: peróxido de hidrogênio; NO:
óxido nítrico: HOCl: ácido hipocloroso; ONOO-: peroxinitrito; RH: lipídios; R •: radical lipídico alquila; ROO
•: radical lipídico peroxil; ROOH: lipídio hidroperóxido; SOD: superóxido dismutase; CAT: catalase; GPX:
glutationa peroxidase.
Fonte: Lu et al., 2010.
Uma das consequências de aumento de EROs é a peroxidação lipídica, que se refere a
deteriorização oxidativa de lipídios que contenham carbono insaturados, tais como ácidos
graxos insaturados, glicolipídeos, ésteres de colesterol e colesterol. As EROs atacam os ácidos
graxos poliinsaturados e grupamentos metil com átomos de hidrogênio reativos, ocorrendo
reações em cadeia e como resultado ocorre a produção de malonaldeído (MDA). A
peroxidação lipídica altera estrutura e permeabilidade de membranas celulares e pode
culminar com a morte celular (LÜ et al., 1998).
Através da ação de mecanismos antioxidantes enzimáticos e não enzimáticos sistemas
biológicos mantêm as concentrações de RLs dentro de limites fisiológicos (REMACLE et al.,
1992). Os sistemas não enzimáticos são compostos por moléculas capazes de neutralizar os
RLs através da doação ou captação de elétrons. Essas moléculas podem reagir diretamente
com espécies reativas, neutralizando-as e tornando-se radicais menos reativos. Ácido
ascórbico (vitamina C), α-tocoferol, GSH, carotenóides e polifenois fazem parte dessas
moléculas.

23
O sistema enzimático é composto pelas enzimas superóxido dismutase (SOD), catalase
(CAT) e glutationa peroxidase (GPX). A SOD atua metabolizando o ânion superóxido e assim
formando o peróxido de hidrogênio (H2O2) (MCCORD; FRIDOVICH, 1969). O peróxido de
hidrogênio pode ser metabolizado através de duas vias antioxidantes: catalase e o sistema
glutationa. A catalase tem como principal função eliminar peróxido formado no peroxissoma,
diminuindo o risco da formação de radical hidroxil (NORDEBERG; ARNER, 2001). O
sistema glutationa peroxidase atua no citoplasma e na mitocôndria. É composto por glutationa
reduzida (GSH), glutationa oxidada (GSSG) e glutationa peroxidase (GPX). A glutationa
reduzida é um tripeptídeo formado por glicina, ácido glutâmico e cisteína. A glutationa
constitui o tiol redutor mais abundante no meio intracelular (REMACLE et al., 1992).
O estresse oxidativo representa uma dos principais responsáveis pelo dano tecidual
promovido por isquemia. O cérebro apresenta elevado consumo de oxigênio, auto-oxidação
de alguns neurotransmissores (ex. norepinefrina e dopamina), altos níveis de ácidos graxos
poliinsaturados, capacidade antioxidante relativamente baixa e um limitado sistema de reparo
celular que o torna particularmente suscetível ao estresse oxidativo (LOH et al., 2010; SUN et
al., 2009). A reperfusão restaura a oferta de nutrientes, porém, por fornecer oxigênio a região
em desequilíbrio metabólico, resulta na potencialização do estresse oxidativo.
1.3.4 Inflamação
Além dos eventos excitotóxicos que ocorrem durante a isquemia, a reação inflamatória
que se inicia horas e persiste por dias após a isquemia também representa um importante
papel na fisiopatologia da isquemia/reperfusão. A fase inflamatória é caracterizada por
migração de neutrófilos, ativação de células gliais e secreção de mediadores inflamatórios
potencialmente citotóxicos sintetizados por essas células, eventos que podem culminar com
prejuízo funcional ou morte das células afetadas (BLOCK, 1999; HUANG; UPADHYAY;
TAMARGO, 2006).
A ativação de segundos mensageiros por Ca2+
, o acúmulo de RLs, assim como a
própria hipóxia, promovem alteração na expressão de diversos genes por induzir a ação de
fatores de transcrição, incluindo o NF-kb, HIF-1 (hipoxia inducible factor-1) e o IRF-1
(interferon inducible factor-1) (DIRNAGL; IADECOLA; MOSKOWITZ, 1999). Dentre as
proteínas que passam a ser expressas encontram-se citocinas como IL-1β e TNF- α e

24
quimiocinas como a proteína quimiotática de monócito-1 (MCP-1) e a proteína inflamatória
de macrófagos (MIP-1α) (BROUNS; DE DEYN, 2009). Essas moléculas induzem a
expressão de moléculas de adesão intercelular (ICAM-1) pelas células endoteliais nos
capilares que promovem a adesão e migração transendotelial de neutrófilos e macrófagos (YI
et al., 2007). Poucas horas após o início da isquemia leucócitos circulantes aderem às paredes
dos vasos e migram para as regiões afetadas liberando mais mediadores inflamatórios e
intensificando o dano. Neutrófilos representam o primeiro subtipo de leucócito a migrar para
as regiões afetadas (BROUNS; DE DEYN, 2009).
O aumento da expressão de IL-1β e ICAM-1 assim como da migração de neutrófilos
para o hipocampo foram documentados em modelo de isquemia global (GALVÃO et al.,
2005; ZHUANG et al., 2009; MINAMI et al., 1992).
Figura 3 – Mecanismos inflamatórios induzidos por isquemia/reperfusão.
Fonte: Brouns e De Deyn, 2009.

25
A cascata inflamatória também inclui o aumento da expressão de diversas enzimas. O
aumento de Ca2+
intracelular inicia a cascata do ácido aracdônico. O acúmulo desse cátion
ativa a fosfolipase C (PLC) e a fosfolipase A2 (PLA2) resultando em hidrólise dos
fosfolipídios da membrana celular e liberando grande quantidade de ácidos graxos livres
(AGL), principalmente o ácido araquidônico (AA). A quebra desses fosfolipídeos, por sua
vez, potencializa a formação de RLs e inflamação. O AA pode ser metabolizado através da
ciclooxigenase (COX) ou lipooxigenase formando respectivamente protraglandina e
leucotrieno. A COX-2 está aumentada após eventos isquêmicos (BROUNS; DE DEYN,
2009). A redução do insulto isquêmica pode ser obtida através da ablação genética de PLA2,
assim como o uso de inibidor de PLC ou de calpaínas (BONVENTRE, 1997; MARKGRAF et
al., 1998; UMEMURA; MABE; NAGAI, 1992).
1.3.5 Apoptose
Em 1964, foi proposto o termo "morte celular programada" para designar um tipo de
morte celular que ocorre de forma não acidental. Em 1972, Kerr, Wyllie e Currie sugeriram o
termo apoptose para indicar esse tipo de morte.
A apoptose é um processo ativo de autodestruição celular que envolve mecanismos
codificados no genoma das células eucarióticas, sendo dependente de energia. Tem papel
importante em diversos processos fisiológicos - como o desenvolvimento embrionário - e
patológicos - como nas doenças neurodegenerativas e isquemia/reperfusão (BREDESEN,
1996).
Diversas cascatas apoptóticas têm sido descritas, como vias intrínsecas e extrínsecas,
caspases-dependentes e independentes, em associação com fases de início, comprometimento
e execução. Tem se tornado aparente, portanto, que a apoptose não é uma série de vias
claramente definidas, mas uma multiplicidade de vias interconectadas, altamente reguladas e
convergentes, resultando nas alterações morfológicas e bioquímicas características da
apoptose (ASHE; BERRY, 2003).
A apoptose é caracterizada por uma série de alterações morfológicas distintas como,
células em picnose, condensação da cromatina, formação de protuberâncias citoplasmáticas
na superfície da célula e de corpos apoptóticos caracterizados por pequenos corpúsculos

26
ligados à membrana contendo organelas intactas e/ou aglomerados densos de cromatina
condensada (WYLLIE et al., 1981).
A fragmentação do DNA e a condensação e fragmentação de cromossomos, são
alterações características da apoptose (DI IORIO et al., 1996). Outra característica única da
apoptose é a exposição da fosfatidilserina na superfície da célula como parte do processo pelo
qual a célula se destina para a fagocitose. Isto pode ser monitorizado por ligação da anexina
V, que preferencialmente se liga a este fosfolipídio na presença de Ca2+
(LIPTON,1999).
Durante a isquemia cerebral a incapacidade de recaptação de glutamato do meio
extracelular leva à ativação excessiva de receptores glutamatérgicos, ao acúmulo de Ca2+
citoplasmático, à produção excessiva de RLs, como também ao dano mitocôndrial e no DNA
que podem levar à morte celular por necrose ou apoptose. O tipo de morte depende da
natureza e intensidade do estímulo, do tipo de célula e de sua fase do ciclo celular ou
desenvolvimento (DIRNAGL; IADECOLA; MOSKOWITS, 1999).
As caspases constituem uma família de proteases que incluem no seu sítio catalítico
um pentapeptídeo contendo cisteína e apresentam a exigência de um resíduo aspartato na
porção N-terminal no sítio de clivagem do seu substrato. Elas participam da morte por
apoptose através de duas principais vias de ativação: a via mitocondrial ou intrínseca e a via
de morte mediada por receptor ou extrínseca. As caspases são sintetizadas como pró-enzimas
inativas (pró-caspase) e, quando ativadas, clivam diversos substratos proteicos que podem ser
outras pró-caspases, proteínas do citoesqueleto como a actina e a gelsolina, proteínas
reparadoras de DNA como a PARP, proteínas antiapoptóticas como Bcl-2 e/ou diversas
quinases que participam da regulação da transcrição gênica (LOVE, 2003).
A caspases podem ser divididas em dois grandes grupos: as da família ICE
(interleukin-1 converting enzyme) que estão envolvidas na maturação de citocinas e na
indução de inflamação (caspases 1, 4, 5, 11, 12 e 14) e as que participam diretamente da
apoptose (caspases 2, 3, 6, 7, 8, 9 e 10).
Na via intrínseca da apoptose, o citocromo c que é liberado da mitocôndria para
o citosol liga-se ao Apaf-1 formando um complexo (apoptossomo) e ativando caspase-9.
Dessa maneira se inicia a cascata da caspase citocromo c dependente. A caspase 9 ativa a

27
caspases 3, a qual causa a ativação em cascata de outras caspases (CHEN et al., 1998; NIWA
et al., 2001; ZHAN et al., 2001) (Figura 3).
Além de promover a ativação da cascata de caspases, a caspase 3 afeta o DNA através
de dois mecanismos, clivando o inibidor de endonuclease ICAD promovendo, dessa maneira,
a liberação da CAD (DNAse ativada por caspase) com consequente quebra do DNA ou
inativando a PARP, uma enzima reparadora de DNA. Dessa forma a caspase-3 garante a
quebra do DNA e, consequentemente, impede a sobrevivência celular (SUGAWARA et al.,
2004) (Figura 4).
Outra molécula que também é liberada por estímulos apoptóticos é o ativador de
caspase derivado da mitocôndria (SMAC) que, no citosol, impede a ação das IAPs. A família
de proteínas inibidoras da apoptose (IAPs) atuam através de dois mecanismos: prevenindo a
ativação de pró-caspase e inibindo a atividade das que se encontram ativadas (SUGAWARA
et al., 2004) (Figura 4).
As proteínas da família Bcl-2 exercem um papel crucial na transdução do sinal
apoptótico por regular a permeabilidade da membrana mitocondrial ao citocromo c. Elas
podem ser divididas em dois grupos: proteínas pró-apoptóticas como Bax, Bid, Bad e Bcl-xs e
as antiapoptóticas como Bcl-2 e Bcl-xl (SUGAWARA et al., 2004) (Figura 4).
Figura 4 - Via mitocondrial da apoptose.
Fonte: Sugawara et al., 2004.

28
Outra via apoptótica é iniciada por sinais externos através de receptores de morte
celular, como receptores de Fas e de TNF. Por exemplo, a ligação da molécula de Fas ligante
presente no meio extracelular ao seu receptor promove, no citoplasma, a clivagem da pró-
caspase 8. A caspase 8 ativa a caspase 3 que por sua vez age em diversos alvos. Outra ação da
caspase 8 é a ativação da proteína pró-apoptótica Bid que inicia a via da mitocôndria
(SUGAWARA et al., 2004) (Figura 5).
Em episódios isquêmicos em humanos, os achados diferem consideravelmente se o
infarto é devido à oclusão arterial arterotrombótica ou a um período de parada cardíaca. Em
indivíduos que sofreram parada cardíaca seguida de ressuscitação foram observados caspase 3
ativada e degradação de PARP em neurônios corticais (LOVE, 2003).
Figura 5 - Via da apoptose mediada por receptor de morte.
Fonte: Sugawara et al., 2004.
Em modelos de isquemia global transitória neurônios piramidais da região CA1
hipocampal são especialmente suscetíveis à morte por apoptose devido a alta densidade de
receptores glutamatérgicos. Nessa população celular a saída do citocromo c e do SMAC da
mitocôndria precedem a ativação da caspase 3, sugerindo a participação da via intrínseca da
apoptose (SUGAWARA et al., 2002). Jin et al. (2001) observaram o aumento de Faz, FasL,
caspase 10 e FADD além de colocalizar caspase 10 e FADD após o insulto isquêmico,
indicando que a via extrínseca também participa desse porcesso.

29
1.4 Modelos animais de isquemia cerebral
A fisiopatologia do AVC isquêmico é extremamente complexa e o desenvolvimento
de modelos animais para o estudo dessa doença representa uma ferramenta indispensável na
investigação de sua fisiopatologia e no desenvolvimento de novos fármacos para seu
tratamento. O pesquisador Seymour Levine (1960) foi o primeiro a propor um modelo de
isquemia cerebral através da combinação de hipóxia e oclusão unilateral da artéria carótida e,
em meados de 1960, seu trabalho foi adaptado por Brown, Brierly e colaboradores que assim
estabeleceram as bases para o estudo do dano isquêmico cerebral em roedores (LIPTON,
1999).
De acordo com a localização os modelos de isquemia cerebral podem ser classificados
como global ou focal (Quadro 1). A isquemia global ocorre quando há falência circulatória e a
isquemia focal ocorre quando o fluxo sanguíneo cerebral é limitado em uma região específica,
geralmente resultado da oclusão de uma artéria cerebral (BRAEUNINGER;
KLEINSCHNITZ, 2009).
Quadro 1 - Tipos de isquemia Cerebral
Fonte: Adaptado de Braeuninger e Kleinschnitz, 2009.
Os modelos de isquemia estão relacionados ao tipo de isquemia (temporária ou
permanente), ao seu tempo de duração, ao tempo de reperfusão, ás artérias ocluídas no
Isquemia
Cerebral
Etiologia Permanente ou
com reperfusão
Modelos
Global Intravascular Transitória ou
permanente
Parada cardíaca com ou sem
ressucitação cardiopulmonar
Extravascular Transitória ou
permanente
Compressão cervical pelo pescoço;
ligação das principais artérias
irrigadoras do cérebro
Focal Intravascular Transitória ou
permanente
Modelo de oclusão da artéria
cerebral média com fio
intraluminal
Extravascular Transitória ou
permanente
Oclusão cirúrgica da artéria
cerebral média: eletrocoagulação,
clampeadura; indução da olusão da
artéria cerebral média da
endotelina-1
Multifocal Intravascular Transitória ou
permanente
Modelo de embolização com
coágulos de sangue; microesferas
ou outro material embolizante

30
experimento (carótidas ou vertebrais) (TARDINI et al., 2003). A presença ou ausência de
reperfusão caracteriza o modelo como temporário/transitório ou permanente
(BRAEUNINGER; KLEINSCHNITZ, 2009).
Os principais vasos de irrigação do cérebro são as artérias carótidas e as artérias
vertebrais, que se comunicam através do polígono de Willis. Este é uma anastomose arterial
que fornece o suprimento sangüíneo para os hemisférios cerebrais, sendo formado pelas
artérias cerebrais anteriores e posteriores, artérias comunicantes anteriores e posteriores e pela
carótida interna (Figura 6).
Figura 6 - Esquema mostrando as artérias cerebrais anteriores e posteriores, artérias
comunicantes anteriores e posteriores e a carótida interna que formam o polígono de Willis.
Fonte: WIKIMIDIA COMMONS, 2008.
Os danos da isquemia global são mais comumente produzidos pela oclusão das
artérias cervicais do que pela interrupção total da circulação sanguínea. Os modelos de
oclusão de artérias mais utilizados são: oclusão de 4 vasos, onde se faz a coagulação
permanente das artérias vertebrais e posterior clampeadura das carótidas, mais utilizado em
ratos (PULSINELLI; BRIERLY; PLUM, 1982); oclusão de 2 vasos, também realizado em
ratos, onde apenas as duas carótidas são clampeadas aliadas a redução da pressão arterial
50mmHg (SMITH; AUER; SIESJO, 1984); e oclusão das duas carótidas em gerbils sem
redução da pressão arterial, pois nestes animais não há o polígono de Willis (KIRINO, 1982).

31
O modelo utilizado no presente trabalho foi o da oclusão bilateral das carótidas com
posterior reperfusão e sem redução da pressão arterial. Esse modelo pode causar desde
prejuízos na LTP até morte de neurônios hipocampais com conseqüente prejuízo em testes de
memória explícita (LIPTON, 1999; MAROSI et al., 2008; WALKER; ROSENBERG, 2009).
Atualmente os modelos mais utilizados de isquemia cerebral focal são os de oclusão
da artéria cerebral média (ACM). A oclusão da ACM pode ser feita através do uso de um fio
intraluminal ou através de técnicas cirúrgicas como eletrocauterização, clampeamento e
ligadura da artéria. A eletrocauterização da ACM leva a interrupção permanente do fluxo
sanguíneo, enquanto que a ligadura e o campleamento permitem a reperfusão. Existe ainda o
modelo de fototrombose, onde uma lesão cortical do cérebro é induzida por injeção sistêmica
de um corante fotossensível, como o rosa de Bengala, e posterior irradiação focal do crânio
(BACIGALUPPI; COMI; HERMANN, 2010).
Na isquemia focal a zona central (core) contém neurônios necróticos resultantes da
falência da membrana associada a perda da homeostase do Ca2+
e glutamato. Nessa região o
fluxo sanguíneo é reduzido para menos de 15%. A penumbra isquêmica é definida como a
zona de tecido adjacente a zona central de infarto isquêmico. Geralmente contém neurônios
eletricamente silencioso, com seus gradientes iônicos intactos e com neurônios que podem ter
suas membranas despolarizadas se o fluxo não for restaurado rapidamente (GILGUN-
SHERKI et al., 2002; WEINSTEIN; HONG; SHARP, 2004). Na penumbra o nível de ATP é
mantido em torno de 50 a 70% do normal, não caindo o suficiente para permitir
despolarizações anóxicas. O fluxo sanguíneo nessa região reduz de 20 a 40% do normal
(HOSSMANN, 1996). Os neurônios perifocais na zona de penumbra são os de mais alto risco
e com o tempo a zona de infarto vai crescer em tamanho, com mais células da penumbra
sendo recrutadas para a zona central.
1.5 Memória
Aprendizagem significa o processo de aquisição de novas informações que serão
armazenadas na memória. Ela representa o modo pelo qual as experiências modificam o
sistema nervoso, e desta forma, também alteram o comportamento de homens e animais.
Difere da memória, pois essa é a habilidade de armazenar seletivamente informações
aprendidas para que possam ser recuperadas e utilizadas posteriormente consciente ou

32
inconscientemente. Esta fornece a base para nossos conhecimentos, habilidades, sonhos,
planos e anseios.
As formas mais usuais de classificação dos tipos de memórias se referem ao tempo de
duração e à sua natureza. Quanto à sua natureza a memória é dividida em: (1) declarativa ou
explícita, pode ser descrita por meio de palavras; (2) implícita, não declarativa ou de
procedimento não pode ser descrita por meio de palavras; (3) de trabalho ou operacional,
permite o raciocínio e planejamento do comportamento (DEW; CABEZA, 2011).
A memória operacional representa o armazenamento temporário de informação
utilizado para planejar uma ação futura e é processada principalmente no córtex pré-frontal. A
memória explícita envolve a recuperação consciente das informações e pode ser dividida em
episódica e semântica. A memória episódica está relacionada a fatos datados e é individual –
cada pessoa possui suas próprias lembranças. A memória semântica está relacionada com a
cultura e envolve conhecimentos gerais de um povo. A memória declarativa pode ser de curto
ou longo prazo e é processada por estruturas do córtex temporal medial como hipocampo e
estruturas adjacentes como os córtices entorrinal, perirrinal e para-hipocampal. A memória
implícita é inconsciente, requer mais tempo e treinamento para se formar, mas persiste por
períodos maiores. Engloba diversos tipos e inclui: a representação perceptual, que representa
imagens sem significado conhecido; a memória procedural (ou de procedimento), que é
demonstrada através de um procedimento motor, por exemplo, andar de bicicleta ou dirigir
um carro. Esse tipo de memória envolve habilidades, hábitos e associações do tipo estímulo-
resposta. Acredita-se que a memória implícita seja processada pelo núcleo estriado e/ou
diencéfalo. Estas categorias de memória não são excludentes, quando um organismo aprende
alguma coisa importante, vários destes sistemas de memória podem ser empregados (DEW;
CABEZA, 2011; SQUIRE; ZOLA, 1996) (Figura 7).
Apesar de estarmos expostos a diversos estímulos ambientais os sistemas de memória
filtram as informações e, apenas parte delas, passa pelo processo de aquisição A aquisição
representa a etapa na qual determinados estímulos alteram circuito neurais por um curto
período de tempo. Após essa fase as informações adquiridas podem ser armazenadas por
períodos mais longos. Essa é a fase de retenção, período no qual as informações permanecem
disponíveis para serem lembradas. Após essa etapa, as informações são consolidadas ou
esquecidas e, dentre os fatores determinantes para seu destino está o tipo de utilização de cada

33
evento memorizado. A consolidação corresponde ao armazenamento das informações por
períodos ainda maiores e é dependente de síntese protéica. Informações consolidadas podem
ser evocadas, ou seja, lembradas ou acessadas quando necessário (NADEL et al., 2012).
Figura 7 - Taxonomia dos sistemas de memória de longa duração.
Fonte: Squire e Zola, 1996.
Outro tipo de classificação da memória envolve o tempo de sua duração, podendo ser
(1) ultra-rápida ou imediata, cuja retenção não dura mais que alguns segundos; (2) memória
de curta duração, que dura minutos ou horas; (3) memória de longa duração, que dura de dias
a anos. A conversão da memória de curto prazo em memória de longo prazo é chamada de
consolidação (IZQUIERDO et al., 2006).
A primeira evidência de que o córtex temporal medial é essencial para os processos de
aquisição e consolidação da memória surgiu com o trabalho de Willian Scoville e Brenda
Milner. Em 1957 a dupla relatou a história do paciente H. M. que aos 27 anos de idade teve a
superfície interna de seu lobo temporal medial retirado cirurgicamente como tratamento para
suas severas crises epilético. A cirurgia foi eficaz ao resolver as crises epiléticas, mas
promoveu um devastador déficit de memória. H. M. passou a apresentar severa amnésia
anterógrada tornando-se incapaz de converter uma memória de curta duração em memória
permanente de longa duração, além de amnésia retrógada referente a fatos acontecido pouco
tempo antes da cirurgia. Rempel-Clower et al. (1996) descreveram casos de pacientes que

34
haviam sofrido lesão bilateral limitada à região CA1 hipocampal devido a episódio isquêmico
e que apresentavam amnésia anterógrada e retrógada relacionada a fatos que aconteceram até
15 anos antes do insulto.
O fato de indivíduos que sofreram lesões hipocampais se lembrarem de fatos ocorrido
muito antes do insulto indica que apesar de essencial para os processos de armazenamento o
hipocampo não é o local de armazenamento dessas memórias. Atualmente, acredita-se que
embora não haja um único sítio de armazenamento para a memória, ela não se encontra
homogeneamente dispersa ao longo do sistema nervoso. Diversas regiões do encéfalo estão
envolvidas na representação até mesmo de um simples evento, mas cada região contribui de
forma diferente para a representação como um todo. O somatório total das alterações no
encéfalo que codificam inicialmente uma experiência e que então constituem o registro
daquela experiência é denominada engrama (SQUIRE; ZOLA, 1996; SQUIRE; KANDEL,
2003) (Figura 7).
Em 1973, os fisiologistas Timothy Bliss e Terje Lφmo demonstraram em neurônios
localizados no hipocampo que a estimulação elétrica de alta freqüência num axônio pré-
sináptico durante alguns segundos produz um aumento na magnitude da resposta pós-
sináptica. A duração da resposta pode variar entre alguns minutos a vários meses, ou até
mesmo uma vida inteira. Este fenômeno foi denominado pelos pesquisadores de
potencialização de longa duração (long-term potentiation- LTP). Dentre os mecanismos
envolvidos nesse processo temos: (1) a liberação do neurotransmissor glutamato pelo
neurônio pré-sináptico; (2) a ativação pós-sináptica de receptores glutamatérgicos dos tipos
AMPA, NMDA e metabotrópico; (3) a despolarização da membrana e aumento do Ca++
citoplasmático no neurônio pós-sináptico, com conseqüente ativação de enzimas como a Ca++
-
calmodulina-cinase; (4) a síntese e difusão de NO pelo neurônio pós-sináptico com
conseqüente aumento da liberação de glutamato pelo neurônio pré-sináptico. Do ponto de
vista funcional, a LTP corresponde a um processo de facilitação do sistema nervoso, cujo
estabelecimento depende da duração e da freqüência do estímulo repetitivo ou numa analogia,
depende do “treinamento” e, portanto, de um processo de aprendizagem. Apesar de ter sido
mais exaustivamente estudada no hipocampo os processos de LTP também são encontrados
em outras regiões envolvidas nos mecanismos de memória como a amígdala, o córtex pré-
frontal e o cerebelo (IZQUIERDO; MEDINA, 1997; MORRIS et al., 1986; MORRIS et al.,
2003).

35
Uma questão que foi durante muito tempo pesquisada buscava esclarecer se a memória
de curta duração é uma etapa da consolidação da memória de longa duração, ou se esses dois
processos são independentes. O pesquisador Izquierdo et al. (2002), utilizando a tarefa da
esquiva inibitória, observaram que tratamentos farmacológicos que interferem com sistemas
de neurotransmissores no hipocampo, ou nos córtices entorrinal ou parietal, afetam
diferentemente esses dois tipos de memória: podem bloquear a memória de curta duração sem
afetar a memória de longa duração; ou podem alterar ambas de forma distinta (melhorando
uma e dificultando a outra). Tais resultados sugerem, claramente, que esses dois processos
envolvem mecanismos diferentes e, em certa medida, independentes.
A memória não é igual para todos os eventos. Lembramos mais facilmente quando de
um evento envolvendo raiva ou felicidade. Isso significa que a memória é modulada pelas
emoções e o sistema límbico do qual fazem parte, o hipocampo, amígdala, septo medial,
bulbo olfatório e áreas talâmicas anteriores estão envolvidos.
A isquemia causa déficits cognitivos que podem estar ou não correlacionados com a
morte neuronal. Em roedores, a isquemia global promove déficits na memória declarativa
acessada através dos testes da esquiva passiva e do labirinto aquático que pode estar ou não
relacionado com a morte de neurônios hipocampais (KIM et al., 2008). Os mecanismos
responsáveis por esses efeitos envolvem alterações em receptores glutamatérgicos,
diminuição da expressão de proteínas citosólicas envolvidas nos processos de aprendizagem e
memória morte neuronal por apoptose (KIM et al., 2007; MAIA et al., 2004; MAROSI et al.,
2009; WALKER; ROSENBERG, 2008; YAMAMOTO et al., 2009; YOSHIOKA et al.,
1999).
1.6 Curcumina
Cúrcuma é um tempero indiano derivado do rizoma da planta Curcuma longa que tem
sido utilizada na medicina Ayurvédica para tratar infecções oculares feridas, picadas de cobra,
queimadura e acne. A Cúrcuma longa pertence à família Zingiberaceae e é amplamente
cultivada na Índia e em outras partes do sudeste asiático. O principal constituinte ativo da
cúrcuma e um dos responsáveis por sua cor amarelo vibrante é a curcumina (HATCHER et
al., 2008).

36
A curcumina foi isolada pela primeira vez em 1815 por Vogel e Pelleitier, obtida na
forma cristalina e identificada como 1,7-bis(4-hidroxi-3-metoxifenil)-1,6-heptadieno-3,5-
diona ou diferuloilmetano em 1870 e, finalmente, teve sua estrutura confirmada por Lampe
em 1910 (AGGARWAL; SUNG, 2008). Embora a curcumina seja o diferuloimetano, a sua
formulação comercial também contém em menor proporção demetoxicurcumina e
bisdemetoxicurcumina.
A curcumina tem peso molecular de 368,4 g/mol e fórmula molecular C21H20O6
(Figura 8). Ela é um polifenol que existe sob condições fisiológicas nas formas enólica e
cetônica. Em condições ácidas e neutras e em fase sólida, a forma cetônica predomina e a
curcumina atua como um dador potente de átomos de hidrogênio. Em contraste, sob
condições alcalinas (pH >7), a forma enólica predomina, e a região fenólica da molécula
desempenha o papel principal como doador de elétrons. A curcumina é solúvel em etanol,
acetona e dimetilsulfóxido (DMSO) e pouco solúvel em água (TONNESEN; KARLSEN,
1985).
Figura 8 - Estrutura química da curcumina.
Fonte: Hatcher et al., 2008.
Quando administrada oralmente a curcumina pode sofrer reações de glucuronidação,
sulfatação e redução no intestino formando produtos como: curcumina glicuronido, curcumina
sulfato, tetrahidrocurcumina, hexadihidrocurcumina e hexahidrocurcumino (IRESON et al.,
2002). A farmacocinética da curcumina tem sido estudada desde os anos 70, sua excreção
pela urina é insignificante sendo aproximadamente 75% da dose excretada pelas fezes
(WAHSTROMAND; BLENNOW,1978).

37
Estudos clínicos demosntraram a eficácia do tratamento com a curcumina em doença
inflamatória intestinal, artrite reumatóide, e diversos tipos de câncer (ALI; SHAKIR;
MORTON, 2012; CHANDRAN; GOEL, 2012; CHENG et al., 2001; SHEHZAD; WAHID;
LEE, 2010). De acordo com autoridades em saúde como o FDA (Food and Drug
Administration) e a Organização Mundial de Saúde a curcumina é considerada segura.
Diversos estudos pré-clínicos in vitro e in vivo tanto com a curcumina quanto com a cúrcuma
têm sido realizados. Além disso, para avaliar a eficácia terapêutica da curcumina existem mais
de 40 ensaios clínicos finalizados e cerca de 30 em andamento nas fases I e II. Em muitos
destes estudos além da avaliação da eficácia também foram incluídas observações
clínicas sobre possíveis efeitos adversos, fornecendo assim oportunidade para acessar com
segurança a tolerabilidade da curcumina (GOEL et al., 2008). Segundo Cheng et al. (2001) a
curcumina em doses elevadas como 12g/dia por via oral por 3 (três) meses não
causou quaisquer efeitos adversos e foi bem tolerada em humanos. Em tais
investigações, nenhuma toxicidade limitante da dose da curcumina foi obsevada. Além
disso, a segurança da curcumina também é evidenciada por uma variedade de estudos
experimentais em animais e in vitro. Apesar da sua eficácia e segurança, a curcumina ainda
não foi aprovada como agente terapêutico sendo a sua biodisponibilidade o principal
problema. Dentre as razões para a biodisponibilidade reduzida estão a baixa taxa de absorção
e seu metabolismo de primeira passagem.
A cucurmina apresenta efeito neuroprotetor em modelos de isquemia cerebral tanto
global quanto focal. Seus efeitos são mediados por suas propriedades antioxidante
(GHONEIM et al., 2002; THIYAGARAJAN; SHARMA, 2004), anti-inflamatória (DOHARE
et al., 2008) e anti-apoptótica (WANG et al., 2005). Outros estudos demonstraram o efeito
neuroprotetor da curcumina em modelo de doença de Parkinson (JAGATHA et al., 2008;
MANSOURI et al., 2010; RAJESWARI; SABESAN, 2008), de Alzheimer (AHMED;
ENAM; GILANI, 2010; BEGUM et al., 2008; GARCIA-ALLOZA et al., 2007; LIM et al.,
2001), de epilepsia (NOOR et al., 2012), de depressão (ARORA et al., 2011) e de estresse
crônico (XU et al., 2009).

38
Figura 9 – Potencias doenças alvo da curcumina.
Essa ampla variedade de efeitos deve-se ao seu grande número de alvos moleculares
que podem ser divididos em duas categorias: aqueles que a curcumina liga-se diretamente
modulando sua atividade e outra categoria que envolve modulação indireta ou secundária.
Dentre os alvos diretos da curcumina encontram-se enzimas como a ciclooxigenase-2 (COX-
2) e a lipooxigenase (LOX) (GAFENR et al., 2004; HONG et al., 2004). Os alvos indiretos
podem ser regulados positiva ou negativamente dependendo do alvo e da célula. Esse tipo de
ação ocorre, por exemplo, através de sua ação sobre fatores de transcrição como o NFkB.
A ação antioxidante da curcumina ocorre de maneira direta agindo como sequestrador
de radicais livres, tais como peróxido de hidrogênio e radical hidroxil, e ligando-se à cátions
divalentes como ferro e cobre (BARZEGAR; MOOSAVI-MOVAHEDI, 2011; BAUN; NG,
2004). De maneira indireta a curcumina altera os níveis de compostos do sistema de limpeza,
por exemplo, ativando o fator de transcrição Nrf-2 que, por sua vez, vai induzir o aumento da
expressão de diversos genes como o da enzima glutationa-S-transferase (BALOGUN et al.,
2003).
Devido a sua atividade antioxidante, a curcumina protege os animais dos danos
cognitivos, melhorando a memória de ratos. Em modelo de excitotoxicidade cerebral induzido
pela hemocisteína ela apresentou proteção na memória espacial e na memória aversiva
(AWASTHI et al., 2010).

39
A curcumina reduz a inflamação e a neurodegeneração diminuindo a interleucina 1β
(IL-1β) e o marcador astrocitário GFAP (BEGUM et al., 2008). Assim como outros anti-
inflamatórios não-esteroidais, a curcumina mostrou-se capaz de suprimir a transcrição do
fator NF-kb, que controla a expressão de genes como a ciclooxigenase-2 e ciclina D1, levando
a inibição da proliferação de células tumorais (TAKADA et al., 2004). Outro estudo mostrou
que os níveis neuronais de espécies reativas de oxigênio (EROs), de peroxinitrito e de NO
foram significativamente inibidas após o tratamento com a curcumina (DOHARE et al.,
2008).
Apesar da biodisponibilidade da curcumina ser limitada e sua meia-vida muito curta,
já foi demonstrado em estudos clínicos sua presença nas porções mais distais do trato
gastrointestinal (HATCHER et al., 2008) e, em estudos pré-clínicos, quando administrada por
via oral foi evidenciada sua presença no cérebro, demosntrando sua capacidade de atravessar
a barreira hematoencefálica (BEGUM et al., 2008).
1.7 Relevância e justificativa
Segundo dados da Organização Mundial de Saúde, o AVC representa a segunda causa
de morte em todo o mundo, sendo responsável por cerca de 6 milhões de mortes/ano. No
Brasil apesar de haver uma tendência de queda esses valores continuam elevados, sendo
superiores aos encontradas em países desenvolvidos e ainda sendo considerada a quarta maior
taxa entre todos os países da América Latina.
O AVC representa a principal causa de incapacidade em todo o mundo. O seu pico de
incidência está entre a sétima e oitava década de vida, mas também pode ocorrer mais
precocemente, sendo que 10% dos pacientes têm idade inferior a 55 anos (ZÉTOLA et al.,
2001). Nos Estados Unidos os custos diretos e indiretos com AVC são de aproximadamente
50 bilhões de dólares anuais (SICARD; FICHER, 2009). O fato do AVC levar à incapacidade
indivíduos em idade produtiva tem um forte impacto econômico. Dados do Instituto Nacional
de Seguridade Social demonstram que o AVC é responsável por 40% das aposentadorias
precoces no Brasil (FALCÃO et al., 2004). Dentre as seqüelas encontram-se apraxia, afasia,
alterações visuais e de memória (KRIZ; LALANCETTE-HÉBERT, 2009).
Atualmente o tratamento mais utilizado é o ativador plasminogênio tecidual (rt-PA)
que possui baixa efetividade principalmente devido a curta janela para a utilização com

40
eficácia, devendo ser administrado até 3 horas após o aparecimento dos sintomas, e também
pelo temor de complicar eventos hemorrágicos. Devemos procurar drogas neuroprotetoras
capazes de impedir a lesão por reperfusão e bloquear cascatas de sinalização responsáveis
pelo dano neuronal. Algumas terapias foram testadas experimentalmente sem, no entanto
obter sucesso clínico que justifica seu uso nesta patologia. A lista de fracassos inclui
antagonistas de canais glutamatérgico e de cálcio, fármacos que inibem a liberação de
glutamato, fármacos que potencializam os efeitos do GABA e removedores de radicais livres
(GINSBERG, 2008; TARAWNEH; GALVIN, 2010).
Os benefícios do consumo de polifénois têm sido observados em estudos
epidemiológicos e experimentais (SCAPAGNINI et al., 2011). A curcumina apresentou
atividade neuroprotetora em diversos modelos de isquemia cerebral, entretanto seu efeito
sobre os déficits de memória induzidos por esse tipo de insulto ainda não foi avaliado
(RATHORE et al., 2008; DOHARE et al., 2008).

41
2 OBJETIVOS
2.1 Objetivo Geral
Estudar os efeitos da curcumina sobre o déficit cognitivo, estresse oxidativo e
processo inflamatório de ratos submetidos à ICT.
2.2 Objetivos Específicos
Produzir dano cognitivo usando modelo de ICT por oclusão bilateral das artérias
carótidas em ratos;
Investigar se o tratamento com a curcumina previne o déficit cognitivo promovido
pela ICT;
Verificar o efeito da curcumina sobre a resposta inflamatória produzida pela ICT
através da determinação da atividade da enzima mieloperoxidase e concentrações de
IL-1β no hipocampo de ratos;
Avaliar o efeito do tratamento com a curcumina sobre o estresse oxidativo induzidos
pela ICT através das dosagens de nitrito, peróxidos lipídicos, GSH e SOD no
hipocampo de ratos;
Determinar o efeito da curcumina sobre a lesão neuronal produzido pela ICT através
das técnicas histoquímicas de Fluoro-jade C e violeta de cresil.

42
3 MATERIAL E MÉTODOS
3.1 Animais
Foram utilizados ratos (Rattus novergicus albinus, Wistar), com massa corpórea entre
230-280 gramas, provenientes do biotério central do Campus do Pici, da Universidade Federal
do Ceará (UFC) e transferidos para o biotério setorial do Departamento de Fisiologia e
Farmacologia, Faculdade de Medicina, UFC. Os animais foram mantidos em gaiolas plásticas
apropriadas, forradas com raspas de madeira, com ciclo de claro/escuro de 12h/12h e
alimentados com ração padrão e água à vontade.
No que se refere aos cuidados com os animais, este estudo seguiu os princípios éticos
da experimentação animal, estabelecidos pelo Colégio Brasileiro de Experimentação Animal
(COBEA). O estudo foi submetido e aprovado pelo comitê de ética em pesquisa animal da
Universidade Federal do Ceará sob o número de registro 052/07.
3.2 Drogas
Curcumina (C1368 - SIGMA); Cloridrato de Xilazina 2% (Kensol® Laboratórios König S.A,
Argentina); Cloridrato de Ketamina 5% (Vetanarcol®, Laboratórios König S.A, Argentina).
Todos os reagentes utilizados eram de grau analítico. O veículo utilizado para diluição da
droga foi fluído DMSO 6% em tampão fosfato 50mM.
3.3 Protocolo Experimental
Para a avaliação da atividade locomotora e memórias de trabalho e aversiva diferentes
doses de curcumina (50 ou 100mg/Kg) ou veículo foram administrados por via oral 1h antes,
24 e 48h após a cirurgia. A avaliação do estresse oxidativo e análise histológica 4d após a
cirurgia foram realizadas nos animais utilizados nos testes comportamentais já descritos. Para
a avaliação do estresse oxidativo, 6h e 24h após a cirurgia, e a avaliação dos parâmetros
inflamatórios, 24h após a cirurgia, a curcumina ou veículo foram administrados apenas 1h
antes da cirurgia. Ver protocolo experimental 1 (Figura 10).
Para a avaliação da memória espacial os animais foram tratados com curcumina (100
ou 300mg/Kg) ou veículo 1h antes e, diariamente, por mais 6 dias. A avaliação histológica 7d
após a Cirurgia foi realizada com esses animais. Ver protocolo experimental 2 (Figura 11).

43
Figura 10 - Protocolo experimental 1.

44
Figura 11 - Protocolo experimental 2.
3.4 Isquemia cerebral por oclusão bilateral da carótida (ULRICH et al., 1998)
Os animais foram anestesiados com xilazina (10mg/kg) e ketamina (90mg/kg)
administrados por via intraperitoneal para o procedimento cirúrgico.
Inicialmente foi feito uma incisão vertical na altura da traquéia para a exposição das
carótidas do animal. Após a separação do nervo vago as artérias carótidas foram pinçadas com
pinça buldogue e submetidas à isquemia durante 30 min. A temperatura foi monitorada e
mantida em torno de 37C. Ao fim deste período as pinças foram removidas para que

45
ocorresse a reperfusão. Após a isquemia o local da incisão foi suturado e os animais foram
colocados em gaiolas para recuperação da cirurgia com livre acesso a água e comida. Os
animais falso-operados (FO) foram submetidos aos procedimentos descritos para a cirurgia
com exceção do clampeamento das artérias carótidas.
3.5 Avaliação da atividade locomotora - Teste do Campo Aberto (Broadhurst, 1957).
O teste do campo aberto foi originalmente descrito por Hall (1934), para analisar o
estado emocional em ratos. O teste utilizado nesse trabalho foi baseado no modelo de
Broadhurst (1957) e foi utilizado com o intuito de aferir a capacidade locomotora dos animais.
O campo aberto consiste de uma arena quadrada (50 x 50 cm) com piso dividido em 4
quadrados iguais. No teste o animal foi colocado na arena e deixado para explorar o ambiente
por 5 minutos, durante este período foi registrado o número de quadrantes atravessados pelo
animal (crossing). Também foi avaliado o número de vezes que o animal se levantou para
explorar o ambiente, mantendo-se suspenso apenas pelas patas traseiras, caracterizando o
comportamento exploratório do tipo rearing. A arena foi limpa com álcool a 20% após cada
animal ser retirado, para evitar interferência do cheiro de urina e fezes no teste.
Figura 12 - Campo Aberto.
Fonte: arquivo pessoal.

46
3.6 Avaliação da memória de trabalho - Teste do labirinto em Y (SARTER et al., 1988).
A memória operacional ou de trabalho foi avaliada através do teste do labirinto em Y.
Nesse teste o animal é colocado em um labirinto em forma de Y com os três braços iguais
(SARTER et al., 1988). Os animais apresentam forte tendência de alternar a entrada nos
diferentes ambientes.
O labirinto em Y é composto por 3 braços de acrílico com 35 cm de altura, 12 cm de
largura e 40 cm de comprimento. Para a avaliação da memória os braços foram numerados.
Figura 13 - Labirinto em Y.
Fonte: arquivo pessoal.
O animal foi colocado no aparelho e durante 8 minutos o número de cada braço que o
animal entrou foi anotado. Foi considerado acerto cada vez que o animal entrou em 3
diferentes braços sem repetição. O resultado foi expresso em porcentagem e obtido através da
seguinte fórmula matemática:

47
O sucesso do teste é indicado pela alta taxa de alternância nos grupos controle,
indicando que os animais podem se lembrar em qual braço eles entraram por último (STONE
et al., 1991). Entre cada sessão, o labirinto foi higienizado com uma solução de álcool a 20%
e secado com toalhas de papel.
3.7 Avaliação da memória aversiva – Teste da Esquiva Passiva (De Noble, 1986)
Uma forma de avaliar o condicionamento pavloviano ao medo e a reposta instrumental
(operante) é através do teste da esquiva passiva. Durante o teste, a inibição da tendência
natural do animal de entrar no compartimento escuro, devido à presença de um componente
aversivo, constitui uma resposta condicionada.
O aparelho consiste de uma caixa de acrílico (48 x 22 x 22), dividida em dois
compartimentos separados por uma porta, um branco (iluminado) e um preto (escuro), este
tem o piso eletrificado. A porta permaneceu aberta durante todo o protocolo. O animal foi
colocado no compartimento iluminado e deixado para ambientação no aparelho durante um 1
min., quando então foi retirado. Após 30 segundos, o animal foi colocado novamente no
compartimento iluminado. O animal ao entrar no compartimento escuro, recebeu um choque
de 1,0 mA, durante 1 seg., com o tempo de latência para entrar sendo registrado, até um
máximo de 300 seg. (treino). Retirou-se o animal (alguns animais saíram espontaneamente
após o choque) e após 15 min. este foi colocado novamente no compartimento iluminado e
registrou-se a latência de entrada (avaliação da memória recente). A retenção do aprendizado
(avaliação da memória tardia) foi testada após 24 h, quando o animal era colocado no
compartimento iluminado e o tempo de latência para a entrada no compartimento escuro foi
registrado, nessa etapa o animal não recebe o choque.

48
Figura 14. Aparelho de Esquiva Passiva.
Fonte: Ugo Basili.
3.8 Avaliação da memória espacial - Teste do Labirinto Aquático (MORRIS et al. 1982)
Nesta versão do labirinto aquático os roedores aprendem a nadar a uma distância mais
curta possível das bordas de um tanque de água para uma plataforma escondida abaixo da
superfície. Eles aprendem guiados por pistas nas paredes ou outros estímulos visuais externos
ao tanque. Esta tarefa espacial é dependente do hipocampo.
O animal foi colocado de forma aleatória em uma piscina circular (180 cm de
diâmetro e 60 cm de profundidade) contendo água turva (3 cm de altura) com tinta branca
não tóxica 27 C, dividida espacialmente em quatro quadrantes, devendo encontrar uma
plataforma (7cm de diâmetro) submersa 2cm. O animal teve 60s para achar a plataforma (que
permaneceu no mesmo local em todos os treinos) e lá permaneceu por 10s. Quando o animal
não encontrava a plataforma sozinho ele era colocado sobre a plataforma. Este treino foi
realizado seis vezes por dia com intervalos de 30s entre os treinos, durante dois dias
(aprendizagem) e 48h após o último treino, os animais foram submetidos ao teste, agora sem a
plataforma, onde foi avaliada a retenção da memória. Na ocasião, os mesmos permaneciam na
piscina por 60s e foram registrados o tempo em que os animais permaneciam no quadrante em
que a plataforma deveria estar, o tempo de latência para encontrar o local da plataforma e o
número de vezes em que eles cruzaram o local exato da plataforma.

49
Figura 15 - Labirinto aquático.
Fonte: arquivo pessoal.
3.9 Dissecção do hipocampo
Os animais foram eutanasiados, seus cérebros rapidamente removidos e os
hipocampos foram retirados, pesados e em seguida congelados a -80˚C.
3.10 Ensaio para Mieloperoxidase (MPO) (BRADLEY; CHRISTENSEN; ROTHSTEIN,
1982)
Mieloperoxidase (MPO) é uma enzima presente nos grânulos de neutrófilos. Essa
enzima é utilizada como indicador de processo inflamatório, mais especificamente, como
marcador de migração de neutrófilos dos tecidos. Neste ensaio, a H2O2 é clivada por meio da
MPO presente nas amostras de tecido. O radical oxigênio (O˚) resultante se combina com
diidrocloreto de O-dianisidina que é convertido a um composto colorido. O aparecimento
deste composto, ao longo do tempo, é medido por espectrofotômetro para determinar o
conteúdo de MPO do ensaio.
Amostras do hipocampo foram homogeneizadas (50mg/mL) em uma solução de
brometo de hexadeciltrimetilamônio 0,5 % (HTAB) em tampão fosfato 50 mM, pH 6,0. Em
seguida, as amostras foram centrifugadas a 14000 rpm a 4˚C por 2 minutos. Adicionou-se

50
30μL do sobrenadante da amostra e 200μL da solução contendo 0,167 mg/ml de hidrocloreto
de -dianisidina e 0,0005 % de peróxido de hidrogênio. A absorbância foi medida nos
tempos 0,1 e 3 minutos no comprimento de onda 460nm. Os resultados foram dados como
unidades de MPO por mg de tecido. E uma unidade de MPO equivale à quantidade que
degrada 1μmol/min de peróxido de hidrogênio.
3.11 Dosagem de substâncias reativas ao àcido tiobarbitúrico (TBARS) (DRAPER;
HADELY, 1998).
O estresse oxidativo foi avaliado através do ensaio de TBARS (Substâncias Reativas
ao Ácido Tiobarbitúrico), um indicador de peroxidação lipídica em homogenatos
hipocampais. No dia do ensaio 250 µL do homogenato (10% em tampão fosfato) foi
introduzido em tubo de ensaio e incubado no banho de água a temperatura de 37ºC. Após a
incubação, 400 µL de ácido perclórico a 35% foram adicionados para parar a peroxidação,
após agitação e centrifugação a 14000 rpm por 10 minutos, o sobrenadante foi retirado e a
este 400µl de ácido tiobarbitúrico 0,6%. A mistura foi levada ao banho de água por 30
minutos a uma temperatura variável de 95 - 100ºC. A solução então foi retirada e colocada
para esfriar, após isso foi feita à leitura da absorbância no comprimento de 532nm. A curva
padrão foi obtida mediante leitura de várias concentrações de malondialdeído (MDA) padrão
(1,56; 3,12; 6,25; 12,5; 25; 50; 100) e os resultados foram expressos em concentração (µM).
3.12 Dosagem da atividade da Superóxido Dismutase (SOD) (BEAUCHAMP;
FRIDOVICH, 1971) .
Para avaliar a atividade dos sistemas antioxidantes foi realizado o ensaio da SOD. A
quantidade da enzima superóxido dismutase foi avaliada medindo-se sua capacidade de inibir
a redução fotoquímica do azul de nitro-tetrazolio (NBT). Nesse método a riboflavina reduzida
fotoquimicamente gera O2- o qual reduz o NBT produzindo formazan que absorve no
comprimento de onda de 560nm. Na presença de SOD a redução do NBT é inibida. Os
resultados são expressos em unidades da enzima, que é a quantidade de SOD necessária para
inibir a taxa de redução do NBT em 50%. O homogenato de hipocampo (10% em tampão
fosfato) foi centrifugado (10 minutos 3600rpm a 4°C). O sobrenadante foi retirado e
centrifugado novamente (20 min 12000rpm 4°C). Para o ensaio foi utilizado o sobrenadante.
Numa câmara escura foram misturados 1mL do meio de reação (tampão fosfato 50mM,
EDTA 100nM e L-metionina 13mM pH 7,8) 30 μL do sobrenadante ou do tampão fosfato,

51
1 0μL do NBT 7 0μM e 300 μL riboflavina 10μM. Os tubos contendo a solução obtida
foram expostos à luz fluorescente (15W) por 15minutos. Ao término do tempo, o material foi
lido em espectofotômetro a 560nm (BEAUCHAMP; FRIDOVICH, 1971).
3.13 Dosagem de Nitrito/nitrato (GREEN et al., 1981)
Nesse ensaio o reativo de Griess (N-1-naftiletilenodiamina a 0,025% e sulfanilamida a
0,25% em ácido fosfórico 2,5%) revela a presença de nitrito em uma amostra por uma reação
de diazotização que forma um cromóforo de cor róseo, com um pico de absorbância em 560
nm. Para realização do ensaio homogenatos de hipocampo (10% em tampão) foram
centrifugados (12000 rpm por 10 min) e 100 μL dos sobrenadantes foram adicionados a 100
μL do reagente de Griess. Para o branco utilizamos 100 L do tampão fosfato e 100 L
reagente de Griess. A curva padrão foi obtida mediante leitura de várias concentrações de
nitrito padrão (1,56; 3,12; 6,25; 12,5; 25; 50; 100) e os resultados foram expressos em
concentração (µM).
3.14 Determinação da concentração da glutationa reduzida (GSH) (SEDLAK;
LINDSAY, 1968)
O método utilizado baseia-se na reação do reagente de Ellman (ácido 5,5'-ditiobis
(ácido 2-nitrobenzóico) - DTNB) com o tiol livre originando um dissulfeto misto mais ácido
2-nitro-5-tiobenzóico com pico de absorbância em 412 nm. O preparo das amostras foi feito
da seguinte forma: 40 µL de cada amostra (homogenato do hipocampo a 10% em tampão
fosfato) foi adicionada a um eppendorf seguido da adição de 50 µL de água destilada e 10 µL
de TCA (ácido tricloro acético) 50%. O material foi centrifugado a 3000 rpm por 10 min e
retirado 60 µL do sobrenadante que foi adicionado a solução de DTNB em tampão Tris. A
curva padrão foi obtida mediante leitura de várias concentrações de GSH padrão (1,56; 3,12;
6,25; 12,5; 25; 50; 100) e os resultados foram expressos em µg.
3.15 Determinação da concentração Il-1β
Para a dosagem de IL-1β foi utilizado o kit de ELISA Ready-SET-Go. As amostras de
homogenatos de hipocampo (5% em tampão fosfato) foram centrifugadas (1600 rpm, 15 min)
e seus sobrenadantes utilizados para a quantificação. O protocolo seguiu as especificações do
fabricante.

52
3.16 Análise histológica
Os animais foram perfundidos através do coração, pelo ventrículo esquerdo com salina
gelada, seguido de paraformaldeído a 4% em PBS. Os cérebros foram removidos e pós-
fixados com formol tamponado por 24 horas, após esse período foram armazenados em
solução crioprotetora de sacarose a 30%. O tecido foi cortado no criostato e montado em
lâminas gelatinizadas.
3.16.1 Coloração de violeta de cresil (BITTENCOURT, 2007).
A coloração de Nissl é uma das técnicas mais utilizadas para investigação do sistema
nervoso. Ela permite a evidenciação da substância de Nissl, material granular constituído por
DNA ribossomal, o qual se concentra principalmente no nucléolo, possibilitando a
discriminação desta estrutura celular específica (BITTENCOURT, 2007). As lâminas foram
mergulhadas em água destilada durante 1 minuto para posterior incubação na solução de
violeta de cresil 0,5%, por um período de 3 minutos. Em seguida, as lâminas foram
desidratadas em álcool (50, 70 e 100%), diafanizadas e montadas em meio à base de xilol
(Entelan). A análise dos resultados foi realizada de maneira qualitativa.
3.16.2 Avaliação da morte celular através do Fluoro-jade C (SCHMUED et al., 2005).
Foi utilizada a técnica de marcação fluorescente com Fluoro-jade C, este liga-se
especificamente à neurônios em processo de degeneração (SCHMUED et al., 2005). As
lâminas foram hidratadas em água destilada. Depois, colocados em solução de permanganato
de potássio 0,06% por 6 minutos. Foram então lavadas em banho de água destilada durante 1
minuto. Em seguida, foram incubadas em solução de Fluoro-jade C 0,001%, em ácido acético
0,1%, durante 15 minutos. Após esse período, as lâminas foram lavadas 3 vezes com água
destilada por 3 minutos. Para a montagem das lâminas fez-se a desidratação com álcool em
álcool (50, 70 e 100%), diafanizadas e montadas em meio à base de xilol (Entelan).
3.17 Análise estatística
A avaliação dos testes comportamentais foi realizada utilizando o teste de Kruskal-
Wallis e, para comparação entre os grupos, foi utilizado o teste de Mann-Whitney. Para os
testes da esquiva passiva e treino do labirinto aquático também foram realizados o teste
ANOVA de medidas repetidas e, para comparações dentro dos grupos, o teste de Bonferronni.

53
Para avaliação dos demais resultados foi utilizada a análise de variância (ANOVA), seguido
do teste de Tukey. Em todos os testes o critério de significância de p < 0,05. Os resultados
foram expressos como média ± erro padrão da média. O programa de computador usado foi
Graph Pad Instat® 5.0.

54
4 RESULTADOS
4.1 Efeito da curcumina (50 e 100mg/kg) sobre a atividade locomotora de ratos
submetidos à isquemia cerebral transitória (ICT)
Na avaliação da atividade locomotora não foram observadas alterações significativas
que pudessem prejudicar os animais para a execução dos testes de memória. A figura 16
representa o número de crossings (FO: 20,0±1,5; FO + C100: 21,5±2,9; ICT: 18,0±1,3; ICT +
C50: 18,0±1,2; ICT + C100: 22,0±1,6) e o número de rearings (FO: 6,0±1,2; FO + C100:
5,0±1,3; ICT: 5,5±1,2; ICT + C50: 7,0±0,91; ICT + C100: 5,0±0,56) (Figura 16).
Figura 16 - Efeito da curcumina (50 e 100mg/kg) sobre o número de crossings e de rearings
de ratos submetidos à isquemia cerebral transitória (ICT).
FO
FO +
C10
0IC
T
ICT +
C50
ICT +
C10
0
0
10
20
30Crossings
Rearings
Nú
mero d
e e
ven
tos
Os animais foram tratados 1h antes, 24 e 48h após a cirurgia. N=8-10/grupo. Os valores
representam a média EPM.

55
4.2 Efeito da curcumina (50 e 100mg/kg) sobre a memória de trabalho de ratos
submetidos à ICT
A ICT ou o tratamento com curcumina não afetou a memória de trabalho dos animais,
como observado pela porcentagem de alternações espontâneas no labirinto em Y (FO:
70,5±1,9; FO + C100: 67,0±3,3; ICT: 67,0±1,8; ICT + C50: 71,5±2,5; ICT + C100: 70,5±3,9)
(Figura 17).
Figura 17 - Efeito da curcumina (50 e 100mg/kg) sobre a memória de trabalho de ratos
submetidos à ICT.
FO
FO +
C10
0IC
T
ICT +
C50
ICT +
C10
00
20
40
60
80
Alt
ern
açõ
es e
sp
on
tân
eas (
%)
Os animais foram tratados 1h antes, 24 e 48h após a cirurgia. N=8-10/grupo. Os valores representam a média
EPM.

56
4.3 Efeito da curcumina (50 e 100mg/kg) sobre a memória aversiva de ratos
submetidos à ICT
Animais submetidos à ICT apresentaram déficits significativos de memória tanto
recente quanto tardia quando comparado com o controle (Memória recente: FO: 117,8±36,2;
ICT: 35,3±7,9) (Memória tardia: FO: 221,1±39,8; ICT: 96,7±44,4). O tratamento com
curcumina (100 mg/Kg) foi capaz de reverter os déficits observados na memória recente (ICT
+ C50: 166,8±50,6; ICT + C100: 160,2±38,4), e na memória tardia (ICT + C50: 219,1±52,2;
ICT + C100: 234,7±42,5).
Através da uma análise dentro de cada grupo foi observado que animais FO
aprenderam a tarefa mais rapidamente, pois apresntaram latência significativamente maior
tanto na avaliação da memória recente quanto da tardia quando comparado com o treino.
Animais submetidos à ICT e que receberam veículo ou foram tratados com curcumina na dose
de 50 mg/kg apresentaram latência significativamente maior que a do treino apenas na
avaliação da memória tardia. Animais isquemiados e tratados com curcumina na dose de 100
mg/kg apresentaram desempenho semelhante aos animais FO.
Figura 18 - Efeito da curcumina (50 e 100mg/kg) sobre a memória aversiva recente e tardia
de ratos submetidos à ICT.
FO
FO +
C10
0IC
T
ICT +
C50
ICT +
C10
00
100
200
300Treino
Memória Recente
Memória Tardia
*
*
#
#
La
tên
cia
(se
g)
Os animais foram tratados 1h antes, 24 e 48h após a cirurgia. N=8-10/grupo. Os valores representam a média EPM. * vs FO, # vs ICT, p<0,05, testes de Kruskall-Wallis e Mann-Whitney.
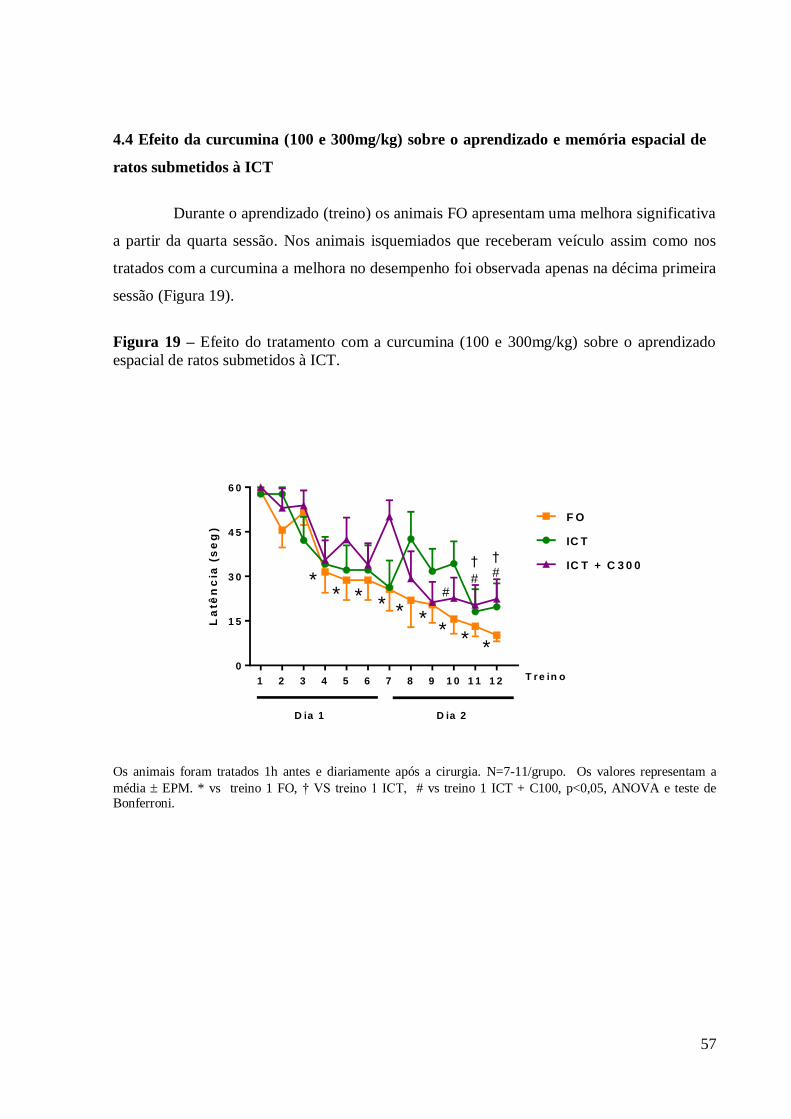
57
4.4 Efeito da curcumina (100 e 300mg/kg) sobre o aprendizado e memória espacial de
ratos submetidos à ICT
Durante o aprendizado (treino) os animais FO apresentam uma melhora significativa
a partir da quarta sessão. Nos animais isquemiados que receberam veículo assim como nos
tratados com a curcumina a melhora no desempenho foi observada apenas na décima primeira
sessão (Figura 19).
Figura 19 – Efeito do tratamento com a curcumina (100 e 300mg/kg) sobre o aprendizado
espacial de ratos submetidos à ICT.
1 2 3 4 5 6 7 8 9 1 0 1 1 1 2
0
1 5
3 0
4 5
6 0
F O
IC T
IC T + C 3 0 0
T r e in o
D ia 1 D ia 2
La
tên
cia
(s
eg
)
** *
******
##
#† †
Os animais foram tratados 1h antes e diariamente após a cirurgia. N=7-11/grupo. Os valores representam a
média EPM. * vs treino 1 FO, † VS treino 1 ICT, # vs treino 1 ICT + C100, p<0,05, ANOVA e teste de Bonferroni.

58
Na avaliação da retenção da memória espacial a isquemia promoveu um déficit
significativo nos parâmetros latência para alcançar a região onde estava a plataforma e
números de cruzamentos, e uma tendência a piorar no tempo de permanência no quadrante
(Latência: FO: 13,0±5,5; FO + C300: 26,0 ± 8,7; ICT: 45,0±7,9; Número de cruzamentos:
FO: 2,0±0,5; FO + C100: 1,0± 0,4; ICT: 1,0±0,3; Tempo de permanência no quadrante: FO:
16,0±2,2; FO + C100: 12,0± 3,7; ICT: 13,0±2,1). O tratamento com a curcumina (300mg/kg)
preveniu, embora não significativamente, o aumento da latência e promoveu tendência a
proteção nos parâmetros número de cruzamentos e tempo no quadrante (Latência: ICT +
C100: 60,0±8,3; ICT + C300: 12,0±8,7; Número de cruzamentos: ICT + C100: 0,0±0,42; ICT
+ C300: 2,0±0,78; Tempo de permanência no quadrante: ICT + C100: 10,0±2,7; ICT + C300:
16,0±2,7) (Figuras 20, 21 e 22).
Figura 20 - Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro latência para alcançar a plataforma, de ratos submetidos à ICT.
FO
FO +
C30
0IC
T
ICT +
C10
0
ICT +
C30
00
20
40
60
*
Latê
ncia
(seg
)
Os animais foram tratados 1h antes e diariamente após a cirurgia. N=7-11/grupo. Os valores representam a
média EPM. * vs FO, p<0.05, testes de Kruskall-Wallis e Mann-Whitney.

59
Figura 21 - Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro número de cruzamentos, de ratos submetidos à ICT.
FO
FO +
C30
0IC
T
ICT +
C10
0
ICT +
C30
00
1
2
3
4
*
Nú
mero
de c
ruzam
en
tos
Os animais foram tratados 1h antes e diariamente após a cirurgia. N=7-11/grupo. Os valores representam a
média EPM. * vs FO, p<0.05, testes de Kruskall-Wallis e Mann-Whitney.
Figura 22 - Efeito da curcumina (100 e 300mg/kg) na retenção de memória espacial, avaliada
através do parâmetro tempo de permanência no quadrante, de ratos submetidos à ICT.
FO
FO +
C30
0IC
T
ICT +
C10
0
ICT +
C30
00
5
10
15
20
25
Tem
po
no
qu
ad
ran
te (
seg
)
Os animais foram tratados 1h antes e diariamente após a cirurgia. N=7-11/grupo. Os valores representam a
média EPM.

60
4.5 Efeito da curcumina (50 e 100mg/kg) sobre a peroxidação lipídica no hipocampo de
ratos submetidos à ICT
As figuras 23 e 24 mostram as concentrações de MDA 6 e 96 horas após a ICT no
hipocampo. Após 6 horas da cirurgia a isquemia promove o aumento dos níveis de MDA (FO:
12,1±3; ICT: 19,7±1,8). O tratamento com a curcumina não preveniu esse aumento (ICT +
C100: 17,8±1,9) (Figura 23). Após 96 horas da cirurgia a isquemia promove o aumento dos
níveis de MDA, embora não estatisticamente significativo. O tratamento com a curcumina
preveniu esse aumento (FO: 2,3±0,2; FO + C100: 2,7±0,3; ICT: 3,0±0,5; ICT + C50: 2,6±0,3;
ICT + C100: 1,9±0,2) (Figura 24).
Figura 23 - Efeito da curcumina (100mg/kg) sobre a peroxidação lipídica em hipocampo de
ratos 6h após a ICT.
FO
FO +
C10
0IC
T
ICT +
C10
00
5
10
15
20
25
*
MD
A (
M)
Os animais foram tratados 1h antes da cirurgia. N=5-6/grupo. Os valores representam a média EPM. Os
valores representam a média EPM. . * vs FO, p<0.05,ANOVA e teste de Tukey.

61
Figura 24 - Efeito da curcumina (50 e 100mg/kg) sobre a peroxidação lipídica em hipocampo
de ratos 96h após a ICT.
FO
FO
+ C
100
ICT
ICT
+ C
50
ICT
+ C
100
0
1
2
3
4
#
MD
A (
M)
Os animais foram tratados 1h antes, 24 e 48h após a cirurgia. N=6-10/grupo. Os valores representam a média
EPM. Os valores representam a média EPM. . # vs ICT, p<0.05,ANOVA e teste de Tukey.

62
4.6 Efeito da curcumina (50 e 100mg/kg) sobre a concentração de nitrito no hipocampo
de ratos submetidos à ICT
A partir a dosagem de nitrito podemos avaliar a presença de NO no tecido, visto que
este é um de seus metabólitos. As figuras 25, 26 e 27 mostram as concentrações de nitrito no
hipocampo 6, 24 e 96 horas após a cirurgia, respectivamente.
A ICT promoveu um aumento significativo de nitrito 6 horas após a cirurgia no
hipocampo e a curcumina foi capaz de prevenir este aumento (FO: 6,4±0,5; FO + C100:
6,5±0,3; ICT: 12,4±1,7; ICT + C100: 7,3±0,7) (Figura 25).
No hipocampo 24 (figura 26) e 96 horas (figura 27) após a cirurgia não foram
observadas alterações entre os grupos (24h: FO: 3,9±1,9; FO + C100: 3,2±1,6; ICT: 2,2±1,1;
ICT + C50: 1,2±0,7; ICT + C100: 2,0±0,9; 96h: FO: 3,5±0,6; ICT: 2,8±3,4; ICT + C50:
3,8±0,7; ICT + C100: 3,2±0,4).
Figura 25 - Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em hipocampo
de ratos 6h após a ICT.
FO
FO +
C10
0IC
T
ICT +
C10
00
5
10
15 *
#
Nit
rito
(
M)
Os animais foram tratados 1h antes da cirurgia. N=5-6/grupo. Os valores representam a média EPM. *vs FO, # vs ICT, p<0,05, ANOVA e teste de Tukey.
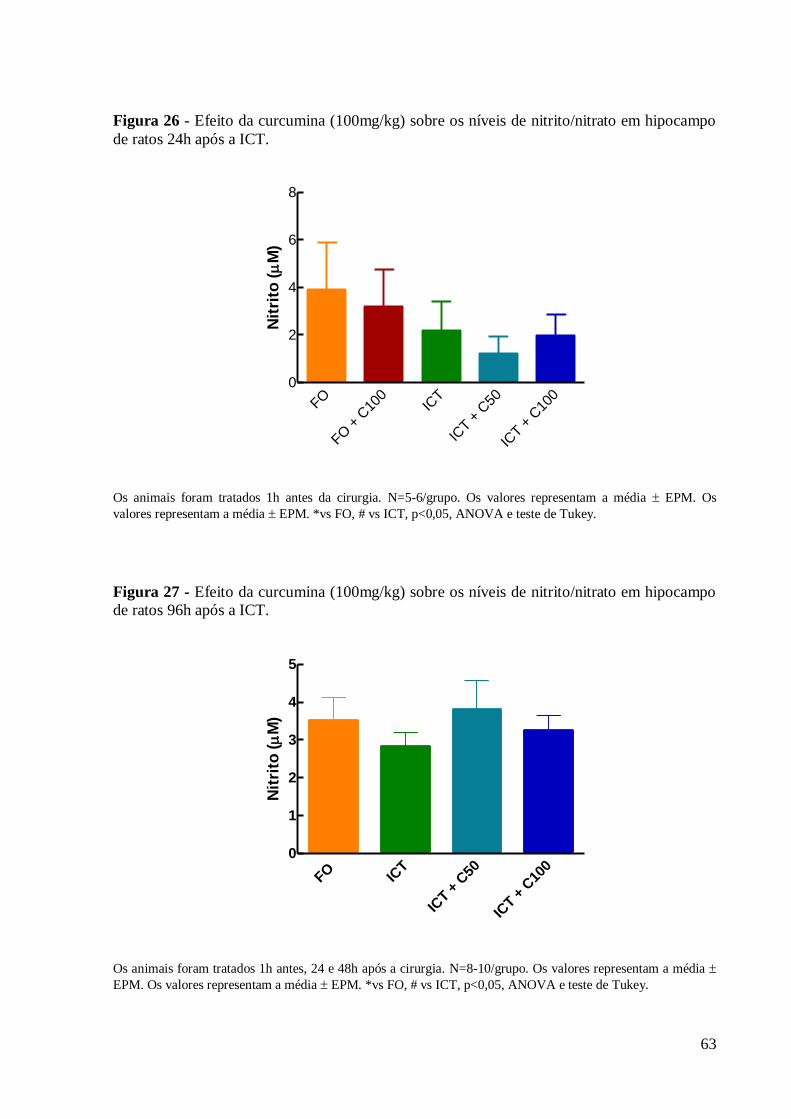
63
Figura 26 - Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em hipocampo
de ratos 24h após a ICT.
FO
FO +
C10
0IC
T
ICT +
C50
ICT +
C10
00
2
4
6
8
Nit
rito
(
M)
Os animais foram tratados 1h antes da cirurgia. N=5-6/grupo. Os valores representam a média EPM. Os
valores representam a média EPM. *vs FO, # vs ICT, p<0,05, ANOVA e teste de Tukey.
Figura 27 - Efeito da curcumina (100mg/kg) sobre os níveis de nitrito/nitrato em hipocampo
de ratos 96h após a ICT.
FO
ICT
ICT +
C50
ICT +
C10
00
1
2
3
4
5
Nit
rito
(
M)
Os animais foram tratados 1h antes, 24 e 48h após a cirurgia. N=8-10/grupo. Os valores representam a média
EPM. Os valores representam a média EPM. *vs FO, # vs ICT, p<0,05, ANOVA e teste de Tukey.

64
4.7 Efeito da curcumina (50 e 100mg/kg) sobre a concentração de glutationa reduzida
(GSH) no hipocampo de ratos submetidos à ICT
A figura 28 mostra a concentração de GSH 24 horas após indução da ICT no
hipocampo dos ratos. Podemos observar que a ICT não foi capaz de diminuir
significativamente os níveis deste peptídeo, mas a curcumina eleva esses níveis em ratos
isquemiados (FO: 11,8±8,0; FO + C100: 1,8±1,1; ICT: 4,9±2,5; ICT + C50: 17,8±5,2; ICT +
C100: 14,8±5,3).
Figura 28 - Efeito da curcumina (50 e 100mg/kg) sobre os níveis de GSH em hipocampo de
ratos 24h após a ICT.
FO
FO +
C10
0IC
T
ICT +
C50
ICT +
C10
00
5
10
15
20
25 #
GS
H (
g)
Os animais foram tratados 1h antes da cirurgia. N=5-6/grupo. Os valores representam a média EPM. Os
valores representam a média EPM. # vs ICT, p<0,05, ANOVA e teste de Tukey.

65
4.8 Efeito da curcumina (50 e 100mg/kg) sobre os níveis da superóxido dismutase (SOD)
no hipocampo de ratos submetidos à ICT
96 horas após a cirurgia a ICT promoveu uma diminuição significativa nos níveis de
SOD. A curcumina aumentou esses níveis embora não significativamente. (FO: 22,9±1,0; FO
+ C100: 21,7±2,6; ICT: 19,5±0,6; ICT + C50: 21,9±1,1; ICT + C100: 21,8±1,3) (Figura 29).
Figura 29 - Efeito da curcumina (50 e 100mg/kg) sobre os níveis de SOD em hipocampo de
ratos 96h após a ICT.
FO
FO+C
100
ICT
ICT +
C50
ICT +
C10
00
10
20
30
*
SO
D (
U/m
g p
rote
ína)
Os animais foram tratados 1h antes, 24 e 48 h após a cirurgia. N=6/grupo. Os valores representam a média
EPM. Os valores representam a média EPM. *vs FO, p<0,05, ANOVA e teste de Tukey.

66
4.9 Efeito da curcumina (100mg/kg) sobre a atividade da mieloparoxidase (MPO) no
hipocampo de ratos submetidos à ICT
24h após a indução da ICT os animais isquemiados apresentaram aumento
significativo da MPO (FO: 2,29±0,3; ICT: 3,51±0,3). O tratamento com a curcumina foi
capaz de diminuir esses valores, porém não significativamente (ICT + C100: 2,32±0,5)
(Figura 30).
Figura 30 - Efeito da curcumina (100mg/kg) sobre os níveis de MPO em hipocampo de ratos
24h após a ICT.
FO
ICT
ICT
+ C
100
0
1
2
3
4
5
*
MP
O (
U/m
g d
e t
ec
ido
)
Os animais foram tratados 1h antes da cirurgia. N=5-8/grupo. Os valores representam a média EPM. Os
valores representam a média EPM. *vs FO, p<0,05, ANOVA e teste de Tukey.

67
4.10 Efeito da curcumina (100mg/kg) sobre a concentração de IL-1β no hipocampo de
ratos submetidos à ICT
A reação inflamatória foi avaliada também através da dosagem de IL-1β no
hipocampo dos animais 24 horas após indução da ICT. A figura 27 mostra que houve uma
tendência de aumento na quantidade de IL-1β no grupo ICT em relação ao controle no
hipocampo (FO: 17,7±3,1; ICT: 23,6±2,4) e o tratamento com a curcumina foi capaz de
diminuir esse aumento significativamente (ICT + C100: 9,7±1,8) (Figura 31).
Figura 31 - Efeito da curcumina (100mg/kg) sobre as concentrações de IL-1β em hipocampo
de ratos 24h após a ICT.
FO
ICT
ICT
+ C
100
0
1 0
2 0
3 0
#
IL-1
b (
pg
)
Os animais foram tratados 1h antes da cirurgia. N=4-5/grupo. Os valores representam a média EPM. Os
valores representam a média EPM. # vs ICT, p<0,05, ANOVA e teste de Tukey.

68
4.11 Efeito da curcumina (100 e 300mg/kg) sobre a integridade neuronal das regiões
CA1 e CA3 do hipocampo de ratos submetidos à ICT
A avaliação do dano neuronal foi realizada 4 e 7 dias após a cirurgia através das
técnicas histológicas Violeta de Cresil e Fluoro-jade C. Através da técnica de coloração com
Violeta de Cresil podemos observar a morfologia de neurônios. Quatro dias após a cirurgia as
regiões CA1 (Figura 32) e CA3 (Figura 33) hipocampais de animais submetidos à ICT
apresentaram neurônios com citoarquitetura celular anormal, com células vacuolizadas e
picnóticas, sem visualização de nucléolos (Figuras 32D e 33D). A curcumina (100 mg/kg)
(Figuras 32F e 33F) mostrou uma ação citoprotetora, com células apresentando citoarquitetura
normal, com núcleos e nucléolos bem visualizados, semelhantes ao FO (Figuras 32B e 33B).
A técnica do Fluoro-jade C é utilizada como marcador de morte neuronal. No
hipocampo dos animais submetidos à ICT, particularmente na região CA1 (Figura 34C e 34D)
observamos uma aumento da fluorescência, com maior número de células Fluoro-jade C
positivas quando comparados ao animal FO (Figuras 34A e 34B). O tratamento com
curcumina (100 mg/kg) (Figuras 34E e 34F) preveniu esse aumento. Na região CA3 (Figura
35) não observamos alterações na marcação de células Fluoro-jade C positivas entre os
grupos.
Sete dias após a cirurgia neurônios das regiões CA1 (Figura 36) e CA3 (Figura 37)
hipocampais de animais FO apresentam citoarquitetura preservada (Figuras 36B e 37B) e do
grupo ICT apresentaram neurônios com citoarquitetura celular anormal, com células
vacuolizadas (Figuras 36D e 37D). O tratamento com curcumina (300 mg/kg) não preveniu
essas alterações, apresentando neurônios com citoarquitetura anormal, alguns apresentando
picnose (Figuras 36F e 37F).
Sete dias após a cirurgia os animais submetidos à ICT apresentaram um aumento da
fluorescência em CA1 e CA3 (Figura 38D e 39D), com maior número de células Fluoro-jade
C positivas quando comparados ao animal FO (Figuras 38B e 39B). O tratamento com
curcumina (300 mg/kg) preveniu esse aumento apenas na região CA1 (38F) e não na CA3
(39F).

69
Figura 32 - Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Coloração
violeta de cresil.

70
Figura 33 - Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Coloração violeta de cresil.

71
Figura 34 - Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Fluoro-jade C.

72
Figura 35 - Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (100mg/kg) 4d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Fluoro-jade C.

73
Figura 36 - Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Coloração
violeta de cresil.
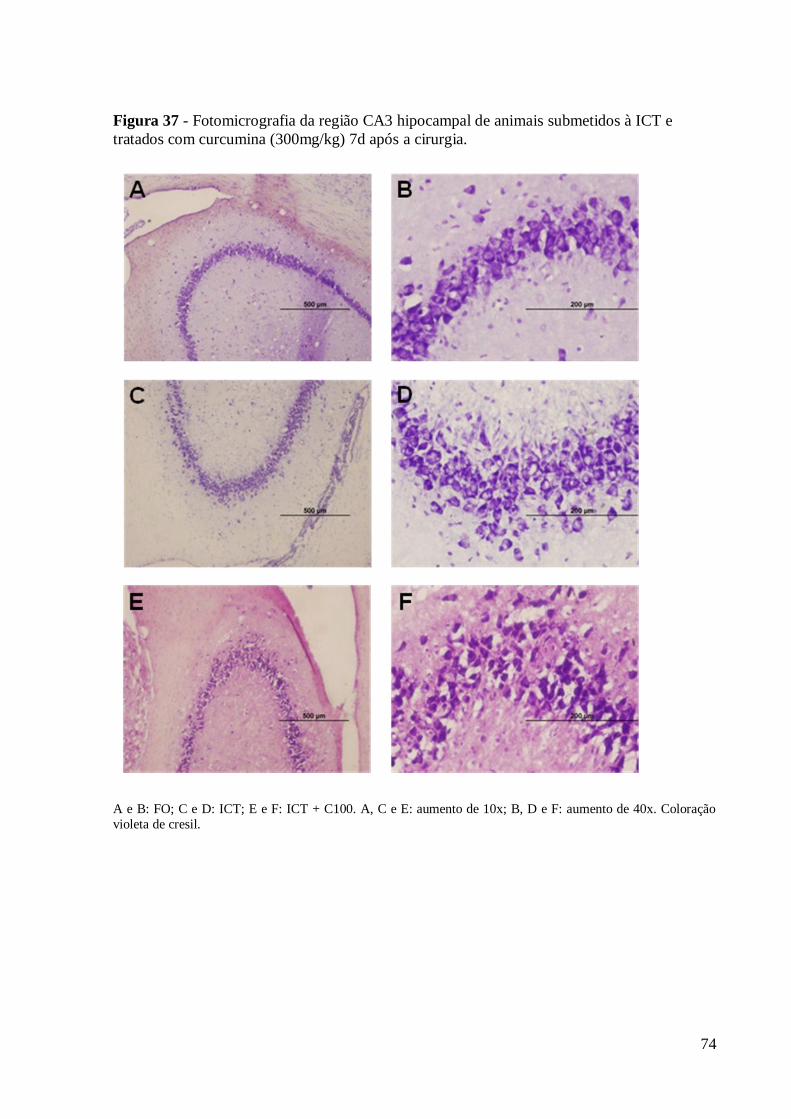
74
Figura 37 - Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Coloração
violeta de cresil.

75
Figura 38 - Fotomicrografia da região CA1 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Fluoro-jade C.

76
Figura 39 - Fotomicrografia da região CA3 hipocampal de animais submetidos à ICT e
tratados com curcumina (300mg/kg) 7d após a cirurgia.
A e B: FO; C e D: ICT; E e F: ICT + C100. A, C e E: aumento de 10x; B, D e F: aumento de 40x. Fluoro-jade C.

77
5 DISCUSSÃO
O Acidente Vascular Cerebral (AVC) representa a segunda causa de morte em todo o
mundo e a primeira causa de incapacitação em adultos (DONNAN et al., 2008). Cerca de
80% dos casos de AVC são do tipo isquêmico. As sequelas decorrentes do AVC variam
dependendo da localização e do tamanho da área cerebral que foi atingida, assim como do
tempo que o paciente levou para ser atendido. Alterações motoras e sensoriais, afasia,
prejuízo na orientação espacial e déficits de memória têm sido descritas (BOKURA;
ROBINSON, 1997; BRADVIK; SONESSON; HOLTAS, 1989; GODEFROY et al., 1994).
As sequelas causadas por isquemia cerebral apresentam graves consequências para os
indivíduos afetados como também para a sociedade. Além da isquemia cerebral há diversas
situações que podem levar ao AVC como os embólicos ou hipotensão severa durante
operações cardiopulmonares e hipoperfusão sistêmica causada por falência cardíaca ou
hipovolemia. Dos indivíduos que sofrem um ataque cardíaco e são reanimados a sequela mais
comum está relacionada a déficits neurológicos principalmente a memória semântica
(ALEXANDER, 1997; REMPEL-CLOWER et al., 1996). Portanto, é de grande importância a
compreensão dos processos fisiopatológicos que levam ao dano isquêmico assim como a
descoberta de novas estratégias terapêuticas.
A fisiopatologia da isquemia cerebral envolve a liberação de glutamato e ativação
excessiva de seus receptores com aumento do Ca2+
citoplasmático, estresse oxidativo,
inflamação e apoptose. Atualmente, inúmeros ensaios clínicos têm testado diversos tipos de
agentes farmacológicos e dentre eles encontram-se antagonistas de receptores
glutamatérgicos, antioxidantes e antiinflamatórios, mas nenhum deles se mostrando eficaz,
ficando ainda como primeira escolha o rtPA (ativador de plasmonigênio tecidual).
O consumo de vegetais, frutas, raízes e folhas ricos em polifenois têm apresentado
efeitos benéficos na proteção contra doenças ligadas ao envelhecimento como câncer e
doenças cardiovasculares e neurodegenerativas. Esses efeitos envolvem suas propriedades
antiinflamatória e antioxidante (SUN et al., 2011). A curcumina é um polifenol com atividade
antioxidante e antiinflamatória.
No presente trabalho, 3 dias após a ICT não foi observada alteração motora
significativa, o que tornou possível a utilização destes animais nos testes de memória. Nossos
resultados corroboram com os resultados de Maia et al. (2004) que, utilizando protocolo

78
semelhante, também não observaram alterações nesse parâmetro. Diversos autores têm
demonstrado que, em gerbil, a isquemia global por oclusão das artérias carótidas promove um
aumento transitório na atividade locomotora (DUSZCZYK et al., 2006; KARASAWA et al.,
1994; LU et al., 2012). Block e Schwarz (1996) também observaram esse efeito utilizando o
modelo de oclusão dos 4-VO em ratos. O mecanismo responsável por essa alteração não foi
esclarecido, mas pode estar relacionado ao aumento da liberação de dopamina no estriado ou
a uma falha na habituação relacionada com morte de neurônios hipocampais (LIPTON, 1999).
No presente trabalho, a curcumina não alterou a atividade locomotora. Porém, Wang et
al. (2005) demonstraram que a curcumina preveniu o aumento na atividade locomotora
induzida por isquemia global em gerbil. No entanto, esses autores não avaliaram as possíveis
alterações no estriado e associaram a ação comportamental à neuroproteção observada no
hipocampo. Em modelo de Parkinson induzido por hemocisteína a curcumina preveniu a
diminuição na atividade locomotora e essa ação foi correlacionada com a neuroproteção na
substância negra (MANSOURI et al., 2012). Dessa maneira, a ação da curcumina sobre a
atividade locomotora estaria relacionada à neuroproteção em situações patológicas.
Atualmente, acredita-se que embora não haja um único sítio de armazenamento para a
memória, ela não se encontra homogeneamente dispersa ao longo do sistema nervoso. A
memória parece ser armazenada, transitória ou permanentemente, no mesmo conjunto de
estruturas encefálicas que participam da percepção inicial e do processamento do que será
lembrado. Assim, ela é armazenada em diversas áreas corticais, de acordo com sua função:
memórias auditivas no córtex auditivo, memórias visuais no córtex visual, e assim por diante.
O somatório das alterações no encéfalo que codificam inicialmente uma experiência e que
então constituem o seu registro é denominado engrama (SQUIRE; ZOLA, 1996; KANDEL,
2006).
A memória de trabalho ou operacional pode ser definida como uma memória ultra-
rápida que representa o armazenamento temporário de informação utilizado para planejar
uma ação futura. É utilizada em diversas habilidades cognitivas como raciocínio,
compreensão da linguagem e planejamento espacial (BADDELEY, 1986; DESPOSITO,
2007). A memória de trabalho não está localizada em uma única região cerebral. Ela envolve
interações funcionais entre o córtex pré-frontal (CPF) e diversas regiões cerebrais
(BADDELEY, 1986; DESPOSITO, 2007). O labirinto em Y, no qual a alternação entre seus

79
braços é avaliada, têm sido proposto para avaliar esse tipo de memória. Além do CPF, outras
regiões como hipocampo, estriado, diencéfalo e cerebelo influenciam o desempenho dos
animais nesse teste (LALONDE, 2002). A participação do hipocampo está relacionada com
a identificação do ambiente através de células piramidais denominadas “células de lugar”
que disparam seletivamente quando o animal se move circulando num determinado
ambiente.
No presente trabalho, 3 dias após a cirurgia, nem a ICT nem a curcumina promoveram
alterações na memória de trabalho. Nossos achados estão de acordo com Fontenele Filho
(2009) que utilizando o protocolo semelhante ao nosso não encontrou alteração nesse tipo de
memória. Entretanto, outros estudos utilizando o modelo obstrução transitória de 2 vasos em
camundongo demonstraram que a lesão isquêmica promove prejuízo na memória de trabalho
(KIM et al., 2006; KIM et al., 2007; YAMAMOTO et al., 2009). Kim et al. (2006)
associaram os déficits nas memórias de trabalho, aversiva e espacial promovidos por ICT com
a morte neuronal e a ativação da microglia nas regiões CA1 e GD hipocampais. Além disso,
esses autores observaram que o uso do flavonóide oroxilina A promoveu a prevenção da
morte neuronal e da ativação da microglia além da melhora cognitiva.
Apesar de não ser o local onde novos registros ficam guardados, o hipocampo atua
processando e intermediando o armazenamento nas áreas corticais. Em humanos, lesões
hipocampais têm sido associadas a déficits de memória declarativa. Esses indivíduos
apresentam amnésias anterógrada e retrógrada relacionada a fatos que aconteceram pouco
tempo antes da lesão (REMPEL-CLOWER et al., 1996). Em diversos mamíferos como rato,
camundongo e macaco foram demonstrados que lesões no hipocampo ou em suas estruturas
anatomicamente relacionadas, promovem déficits de memória declarativa (MORRIS, 1984;
KANDEL, 2009). Os mecanismos moleculares relacionados aos processos de aprendizagem e
memória envolvem, inicialmente, a ativação de receptores glutamatérgicos, entrada de Ca2+
,
ativação de proteínas como CaMKII, PKC e a proteína quinase Fyn. Os processos de
consolidação envolvem alteração gênica, síntese de novas proteínas e alterações morfológicas
(KANDEL, 2009).
A memória declarativa é elaborada para representar objetos e eventos no mundo
externo e as relações entre eles. Uma característica chave da memória declarativa é que a
resultante é flexível. Animais podem aprender relações entre itens armazenados e, então,

80
expressar esse conhecimento de relações em situações novas. Em roedores como rato e
camundongo esse tipo a memória declarativa pode ser acessada através de testes como o da
esquiva passiva e do labirinto aquático (CLARK; SQUIRE, 2010; DEW; CABEZA, 2011).
No teste da esquiva passiva após um único treino o animal deve lembrar-se do ambiente no
qual levou um choque e evitá-lo. Esse teste possui um componente emocional sendo
considerada uma memória aversiva. O sistema límbico do qual fazem parte o hipocampo,
amígdala, septo medial, bulbo olfatório e áreas talâmicas anteriores participam modulando
esse tipo de memória (IZQUIERDO et al., 2006). O teste do labirinto aquático é amplamente
utilizado no estudo da memória espacial. A versão utilizada no presente trabalho, no qual o
animal é colocado em diferentes pontos de uma piscina e deve encontrar uma plataforma
submersa, é dependente da integridade do hipocampo (MORRIS, 1984).
Na avaliação da memória aversiva, os resultados desse estudo mostram que a ICT
promoveu déficits nas memórias recente e tardia. Estudos utilizando o modelo de ICT em
camundongo e ratos demonstraram que o insulto isquêmico promove prejuízo na memória
aversiva avaliada através do teste da esquiva passiva (KIM et al., 2006; KIM et al., 2007;
YAMAMOTO et al., 2009). O estudo anterior realizado em nosso grupo (MAIA et al., 2004)
mostrou correlação entre o déficit cognitivo com aumento nas concentrações de MDA e
nitrito no hipocampo.
O tratamento com curcumina na dose de 100mg/kg foi capaz de reverter esses efeitos.
Corroborando com nossos resultados, outros autores demonstraram que a curcumina previne o
déficit na memória aversiva em modelos de neurotoxicidade induzida alumínio (PAN et al.,
2008) e de disfunção cognitiva induzida por antiepiléticos (REETA; MEHLA; GUPTA,
2010). Além disso, Ataie et al. (2010) utilizando modelo de demência induzida por
hemocisteína, demonstraram que o tratamento com curcumina promoveu a diminuição do
estresse oxidativo e a neuroproteção no GD, resultando na prevenção do déficit mnemônico.
No presente trabalho a ICT promoveu déficits no aprendizado e memória espacial.
Estudos utilizando o modelo de ICT em camundongo e ratos demonstraram que o insulto
isquêmico promove prejuízo na memória espacial avaliada através do teste do labirinto
aquático (KIM et al., 2006; KIM et al., 2007; MAIA et al., 2004). Hartman et al. (2005)
utilizaram o modelo de oclusão de 2 vasos com hipotensão em rato correlacionaram o déficit
de memória com a morte de neurônios da região CA1.

81
A curcumina não reverteu o déficit no aprendizado. No entanto, quando avaliamos a
memória, observamos que o tratamento com curcumina (300mg/kg) preveniu, embora não
significativamente, o aumento da latência para alcançar a plataforma e promoveu tendência a
proteção nos parâmetros número de cruzamentos e tempo de permanência no quadrante.
Trabalhos demonstram que a curcumina previne o déficit na memória espacial induzido por
estresse crônico (XU et al., 2009) e demência induzidas por alumínio (KUMAR; DOGRA;
PRAKASH, 2009) e por β- amilóide (AHMED; ENAM; GILANI, 2010).
A indução da isquemia global em ratos e camundongos por oclusão das artérias
carótidas promove a diminuição do fluxo sanguíneo cerebral causando lesão hipóxica em
áreas fracamente perfundidas e vulneráveis. O tecido cerebral é extremamente sensível à
isquemia/reperfusão. Um breve evento isquêmico pode desencadear uma complexa
sequência de eventos podendo ou não culminar com a perda da funcionalidade sináptica e
morte neuronal. Neurônios piramidais da camada CA1 hipocampal são particularmente
sensíveis a esses eventos devido à alta densidade de receptores glutamatérgicos do tipo
NMDA e AMPA que deixa a região mais exposta a excitotoxicidade. No entanto, a
depender da intensidade do estímulo, neurônios de outras regiões hipocampais, como as
zonas CA2 e CA3 e o GD, assim como de outras áreas cerebrais como estriado e córtex
cerebral também podem ser afetados (KIM et al., 2006; LIPTON, 1999; WALKER;
ROSENBERG, 2009).
Déficits cognitivos promovidos por isquemia não ocorrem apenas quando há morte
neuronal. Yamamoto et al. (2009) demonstraram que 20 minutos de isquemia por oclusão das
artérias carótidas promoveu morte neuronal em CA1 e déficit de memória e, que ao diminuir
o tempo de isquemia para 5 minutos, a morte neuronal não ocorria, mas o déficit de memória
persistia. Esse déficit foi correlacionado com a diminuição da expressão de CaMKII, MAP-2
e o receptor GluR1. Marosi et al. (2009) demonstraram que 3 dias após a indução de ICT
ocorria o prejuízo na LTP com ausência de degeneração em neurônios hipocampais.
No presente trabalho a avaliação do dano neuronal foi realizada 4 e 7 dias após a
cirurgia. Quatro dias após a cirurgia os animais submetidos à ICT apresentaram neurônios da
região CA1 e CA3 com citoarquitetura celular anormal, com células vacuolizadas e
picnóticas, sem visualização de nucléolos. Na região CA1 também foi observado o aumento
de células Fluoro-jade C positivas, indicando morte celular. Sete dias após a cirurgia a ICT

82
promoveu alterações morfológicas como neurônios vacuolizados, assim como aumento de
células Fluoro-jade C positivas nas regiões CA1 e CA3 hipocampais, indicando neurônios em
degeneração. Nossos resultados corroboram com dados da literatura que demonstram que, em
modelo de ICT por oclusão 2-VO, os neurônios hipocampais entram em apoptose de 3 a 7
dias após o insulto isquêmico (CHEN et al., 1998; MAGNONI et al., 2004; OGURO et al.,
2001; WALKER; ROSENBERG, 2009). Diversos mecanismos têm sido propostos para essa
morte neuronal incluindo ativação excessiva de receptores glutamatérgicos, excesso de Ca2+
citoplasmático, estresse oxidativo e inflamação.
Quatro dias após a indução de ICT animais tratados com curcumina (100 mg/kg)
apresentaram neurônios das regiões CA1 e CA3 hipocampais com citoarquitetura normal,
com núcleos e nucléolos bem visualizados além de apresentar uma diminuição de neurônios
em degeneração (Fluoro-jade C positivas) na região CA1. Nossos achados demonstraram que
a curcumina preveniu o déficit na memória aversiva e a morte neuronal em CA1 quatro dias
após a isquemia. Esses dados corroboram com o trabalho de Wang et al. (2005) que
demonstraram que o tratamento com curcumina previne a morte neuronal na região CA1
hipocampal de animais submetido à isquemia/reperfusão 96h após cirurgia. Os autores
demonstraram que ação neuroprotetora da curcumina foi mediada pela diminuição da
expressão de NK-kb e ICAM-1. Sete dias após a isquemia a curcumina também impediu a
morte em CA1. Entretanto, o tratamento não preveniu as alterações morfológicas em CA1 e
CA3 e nem a morte em CA3. Portanto, a curcumina promoveu um atraso na lesão neuronal
que poderia ser responsável pela tendência a proteção do déficit de memória espacial.
Em humanos, o consumo de curcumina tem sido associado a uma menor incidência de
doenças de Alzheimer e de Parkinson além de melhora cognitiva (NG et al., 2006). Em
modelos animais os mecanismos responsáveis pela prevenção dos déficits cognitivos parecem
ser mediados por suas propriedades antiapoptóticas (PAN et al. 2008) e sua capacidade de
alterar bioquímica e morfologicamente as sinapses (AHMED; ENAM; GILANI, 2010; XU et
al. 2009). In vitro a curcumina preveniu a apoptose através da inibição da ativação de JNK em
modelos de toxicidade induzida por glutamato (SUH; KANG; KWON, 2007) e MPTP (YU et
al., 2010). Em modelo animal de excitotoxicidade por glutamato sua ação ocorreu por
ativação da via BDNF/TrkB (WANG et al., 2008) e inibição das vias JNK/c-Jun e p38/p53
(CHEN et al., 2003). Rathore et al. (2008) utilizando modelo de isquemia focal transitória

83
demonstraram que a curcumina preveniu a ativação da caspase-3, apoptose, estresse
oxidativo, déficit neurológico e alteração motora avaliados 24 h após a isquemia.
EROs e ERNs são produzidas fisiologicamente através do metabolismo celular e, em
concentrações moderadas, desempenham importantes papéis em processos de sinalização
celular, apoptose, expressão gênica e transporte iônico. Essas moléculas são rigidamente
controladas por antioxidantes que podem agir de maneira direta, atuando como “scavenger”
(seqüestradores), ou de maneira indireta através do aumento da expressão de enzimas
antioxidantes (LU et al., 2011). Entretanto, o excesso desses radicais causa o estresse
oxidativo, um processo deletério que pode ser um importante mediador dos danos a estruturas
celulares, como lipídeos, proteínas e DNA (VALKO et al., 2007). O dano celular associado à
isquemia envolve a formação de RLs e ocorre através de diversos mecanismos como o
desarranjo mitocondrial e acúmulo de Ca2+
citoplasmático. Durante a fase isquêmica ocorre o
desacoplamento da cadeia transportadora de elétrons na mitocôndria e, quando ocorre a
reperfusão com consequente retorno da oferta de oxigênio, o estresse oxidativo é intensificado
(LIPTON, 1999).
Um dos principais componentes celulares afetados pelo estresse oxidativo é a
membrana celular que sofre um processo de peroxidação. O MDA é um aldeído de cadeia
curta, sendo o principal produto da peroxidação lipídica. No presente trabalho a ICT
promoveu o aumento das concentrações de MDA no hipocampo 6h e 4d após a cirurgia.
Nossos dados estão de acordo com a literatura. O tratamento com curcumina não preveniu
essa alteração 6h após a cirurgia. Quando avaliado 4 dias após a cirurgia a curcumina
promoveu a diminuição de MDA em animais submetidos à ICT. A inibição da peroxidação
lipídica pela curcumina foi demonstrada em modelos de isquemia/reperfusão tanto global
(GHONEIM et al., 2002) quanto focal (RATHORE et al., 2008). A falha na diminuição da
peroxidação lipídica pela curcumina presente no nosso trabalho pode estar relacionada à
diferente via de administração, pois os outros autores utilizaram a via intraperitoneal.
Durante a isquemia/reperfusão o NO apresenta efeitos protetores, dilatando vasos e
dessa maneira aumentando a oferta de glicose e oxigênio, e deletérios reagindo com o radical
superóxido formando então o íon peroxinitrito. Essa molécula é extremamente tóxica e
danifica proteínas, lipídeos de membrana e DNA. De maneira indireta podemos quantificar a
produção do NO através da dosagem de seu metabólito nitrito. No hipocampo a ICT

84
promoveu o aumento de nitrito 6h, mas não 24h e nem 4d após a cirurgia. Galvão et al. (2004)
observaram o aumento nos níveis de nitrito 24h após a ICT, porém o período do insulto
isquêmico utilizado nesse trabalho foi maior (45min). O tratamento com curcumina preveniu
o aumento dos níveis de nitrito em modelo de isquemia tromboembólico (DOHARE;
VARMA; RAY, 2008) e também preveniu o aumento de nitrito e da expressão de iNOS em
modelo de isquemia focal transitória (JIANG et al., 2007).
A enzima superóxido dismutase é uma das responsáveis pelo “seqüestro” de radicais
livres, mais especificamente retirando o anion superóxido com conseqüente formação de
peróxido de hidrogênio (LU et al., 2011). No presente trabalho, a ICT promoveu uma
depleção significativa da SOD 4d após a cirurgia. O aumento da expressão da SOD
mitocondrial torna animais menos susceptíveis ao insulto isquêmico (SUGAWARA et al.,
2002) e a sua depleção foi demonstrada em diversos modelos de isquemia cerebral. No
presente trabalho, o tratamento com a curcumina atenuou a depleção dessa enzima. Rathore et
al. (2008) demonstraram que a administração de curcumina reverteu a depleção de SOD,
aumento de EROs e ativação de caspase-3 induzidos por isquemia focal transitória. Shukla et
al. (2008) demonstraram que 5d de pré-tratamento com a curcumina, por via oral, preveniu a
depleção de SOD causada por isquemia focal transitória.
O sistema glutationa é capaz de degradar o peróxido de hidrogênio e assim atuar como
scavenger em situações de estresse oxidativo. Para que isso ocorra, a glutationa reduzida
(GSH) é utilizada, levando a sua depleção (LU et al., 2011). A ICT promoveu a depleção de
GSH 24h após a cirurgia. A curcumina reverteu a depleção de GSH em modelo de isquemia
tromboembólico (DOHARE et al., 2008) e em modelo de isquemia cerebral transitória
(GHOENEIM et al., 2002), corroborando com nossos resultados.
A ativação de segundos mensageiros por Ca2+
, o acúmulo de RLs, assim como a
própria hipóxia, promovem alteração na expressão de diversos genes através da ação de
fatores de transcrição como o NF-kb (DIRNAGL; IADECOLA; MOSKOWITZ, 1999).
Dentre as proteínas que passam a ser expressas encontram-se citocinas como IL-1β, TNF- α e
quimiocinas como MCP-1 e MIP-1α (BROUNS; DE DEYN, 2009). Esses mediadores
induzem a expressão de moléculas de adesão intracelular (ICAM-1) pelas células endoteliais
nos capilares que promovem a adesão e migração transendotelial de neutrófilos e macrófagos
(YI et al., 2007). Poucas horas após o início da isquemia leucócitos circulantes aderem às

85
paredes dos vasos e migram para as regiões afetadas liberando mais mediadores inflamatórios
e intensificando a lesão, os neutrófilos representam o primeiro subtipo de leucócito a migrar
para as regiões afetadas (BROUNS; DE DEYN, 2009).
No presente trabalho 24h após a ICT ocorreu um aumento, embora não significativo,
dos níveis de IL-1β no hipocampo. Sairanen et al. (1997) demonstraram que ratos submetidos
à oclusão bilateral das artérias carótidas (20 minutos de isquemia) juntamente com hipotensão
seguido de reperfusão apresentavam aumento da expressão de mRNA da IL-1β ao longo de
24horas em diversas regiões hipocampais. O aumento dos níveis de IL-1β cerebral foi
associado com prejuízo na LTP hipocampal em modelo de isquemia global (YOSHIKA et al.,
1999; TOGASHI et al., 2001) e de inflamação por LPS (TANAKA et al., 2011). No presente
trabalho, o tratamento com a curcumina diminuiu os níveis dessa citocina. Esse resultado
corrobora com outros autores que demonstraram que a curcumina previne o aumento de IL-1β
em modelo de doença de Alzheimer (BEGUM et al., 2008; LIM et al., 2001). Em modelo de
hipóxia esse diminuição foi mediado por inibição da expressão de NF-kb (HIMADRI et al.,
2010). No nosso trabalho, a prevenção do aumento da IL-1β pode ser um dos fatores
associados a proteção cognitiva descrita anteriormente.
Os nossos achados demonstram que 24h após a ICT houve um aumento da atividade
de MPO no hipocampo, indicando migração de neutrófilos. Galvão et al. (2005) observaram
que a ICT promoveu o aumento de MPO e alterações morfológicas no hipocampo e que o uso
do antiinflamatório tenoxicam preveniu esses efeitos. No presente trabalho o tratamento com
curcumina reverteu, embora não significativamente, esse aumento. Outros autores
demonstraram que a curcumnina previne o aumento de ICAM-1 via inibição da expressão de
NF-kb em modelo de ICT (ZHUANG et al., 2009) e de hipóxia (HIMADRI et al., 2010).
Dohare et al. (2008) demonstraram que a administração de curcumina por via intraperitoneal
preveniu a migração de neutrófilos em modelo de isquemia cerebral tromboembólica.
No presente trabalho a administração oral de curcumina atenuou os efeitos deletérios
promovidos por isquemia cerebral transitória. A curcumina reduz a inflamação, o estresse
oxidativo e a morte neuronal em diversos modelos de doenças neurodegernerativas. Ela
também apresenta potente efeito neuroprotetor em diversos modelos de isquemia cerebral
quando administrada por via intravenosa ou intraperitoneal (DOHARE et al., 2008; JIANG et
al., 2008; LIM et al., 2001; PAN et al., 2008; THIYAGARAJAN; SHARMA, 2004; WANG

86
et al., 2004). Portanto, as limitações de sua disponibilidade poderiam justificar a ausência
proteção em alguns parâmetros avaliados. Atualmente, há a procura por novas formas de
melhoramento de sua molécula para que seus limitações farmacocinética e de
biodisponibilidade sejam superadas. Em humanos, a revascularização dos vasos cerebrais
durante o AVC isquêmico agudo através do uso do tPA apesar de ser a primeira escolha
apresnta séria limitações. A curcumina, devido ao seu amplo espectro de ação, é uma forte
candidata ao papel de droga neuroprotetora capaz de impedir a progressão do dano tecidual
durante a reperfusão cerebral.

87
6 CONCLUSÃO
A isquemia cerebral transitória produziu morte neuronal nas regiões CA1 e CA3
hipocampais, bem como produziu déficits nas memórias aversiva e espacial. Estes efeitos
foram atenuados pela curcumina.
Em relação à inflamação a isquemia cerebral transitória promoveu o aumento atividade
da Mieloperoxidase bem como tendência ao aumento dos níveis de IL-1β no hipocampo. A
curcumina apresentou tendência na prevenção do efeito na mieloperoxidase e impediu o
aumento de IL-1β.
Em relação ou estresse oxidativo a isquemia cerebral transitória promoveu o aumento
dos níveis de MDA e nitrito. A curcumina preveniu o aumento de nitrito e de MDA 4d após a
cirurgia de MDA. Entretanto, a curcumina não foi capaz de bloquear o aumento de MDA 6h
após a cirurgia.
Em relação aos sistemas antioxidantes a isquemia cerebral transitória promoveu a
depleção de SOD e tendência à diminuição de GSH. A curcumina aumentou os níveis de GSH
e promoveu tendência à reversão da SOD em animais submetidos à ICT
Os resultados do presente estudo sugerem que a curcumina apresenta um importante
potencial neuroprotetor. Entretanto, há a necessidade de que novas formas farmacêuticas
sejam desenvolvidas com intuito de que a curcumina possa efetivamente exercer ação
neuroprotetoras em pacientes com AVC.

88
REFERÊNCIAS
AGGARWAL, B. B.; SUNG, B. Pharmacological basis for the role of curcumin in chronic
diseases: an age-old spice with modern targets. Trends Pharmacol. Sci., v. 30, n. 2, p. 85-94,
2008.
AHMED, T.; ENAM, S. A.; GILANI, H. Curcuminoids enhance memory in an amyloid-
infused rat model of Alzheimer’s disease. Neuroscience, v. 169, p. 1296-1306, 2010.
ALEXANDER, M. P. Specific semantic memory loss after hypoxic-ischemic injury.
Neurology, v. 48, p. 165-173,1997.
ALI, T.; SHAKIR, F.; MORTON, J. Curcumin and Inflammatory Bowel Disease: Biological
Mechanisms and Clinical Implication. Digestion, v. 85, p. 249-255, 2012.
ARORA, V.; KUHAD, A.; TIWARI, V.; CHOPRA, K. Curcumin ameliorates reserpine-
induced pain-depression dyad: behavioural, biochemical, neurochemical and molecular
evidences. Psychoneuroendocrinology, v. 36, n. 10, p. 1570-1581, 2011.
ASHE, P. C.; BERRY, M. D. Apoptotic signaling cascades. Prog Neuropsychopharmacol
Biol. Psychiatry, v. 27, n. 2, p. 199-214, 2003.
ATAIE, A.; SABETKASAEI, M.; HAGHPARAST, A.; MOGHADDAM, A. H.;
KAZEMINEJAD, B. Neuroprotective effects of the polyphenolic antioxidant agent,
Curcumin, against homocysteine-induced cognitive impairment and oxidative stress in the rat.
Pharmacol. Biochem. Behav., v. 96, p. 378- 385, 2010.
AWASTHI, H.; TOTA, S.; HANIF, K.; NATH,C.; SHUKLA,R. Protective effect of curcumin
against intracerebral streptozotocin induced impairment in memory and cerebral blood flow.
Life Sci., v. 86, p. 87-94, 2010.
BACIGALUPPI, M.; COMI, G.; HERMANN, D. M. Animal Models of Ischemic Stroke. Part
One: Modeling Risk Factors. Open Neurol. J., v. 4, p. 26-33, 2010.
BADDELEY, A. Working memory. Oxford: Oxford University Press, 1986.
BALOGUN, E.; HOQUE, M.; GONG, P.; KILEEN, E.; GREEN, C. J.; FORESTI, R.;
ALAM, J.; MOTTERLINI, R. Curcumin activates the haem oxygenase-1 gene via regulation
of Nrf2 and the antioxidant-responsive element. Biochem. J., v. 371, p. 887-895, 2003.
BARZEGAR, A.; MOOSAVI-MOVAHEDI, A. A. Intracellular ROS Protection Efficiency
and Free Radical-Scavenging Activity of Curcumin. PLoS ONE, v. 6, n. 10, p. e26012, 2011.
BAUM, L.; NG, A. Curcumin interaction with copper and iron suggests one possible
mechanism of action in Alzheimer’s disease animal models. J. Alzheimers Dis., v. 6, p. 367-
377, 2004.
BEAUCHAMP, C.; FRIDOVICH, I. Superoxide dismutase: improved assays and an assay
applicable to acrylamide gels. Anal, Biochem., v.44, n.1, p.276-287, Nov.1971.

89
BEGUM, A. N.; JONES, M. R.; LIM, G. P.; MORIHARA, T.; KIM, P.; HEATH, D. D.;
ROCK, C. L.; PRUITT, M. A.; YANG, F.; HUDSPETH, B.; HU, S.; FAULL, K. F.; TETER,
B.; COLE, G. M.; FRAUTSCH, S. A. Curcumin Structure-Function, Bioavailability, and
Efficacy in Models of Neuroinflammation and Alzheimer’s Disease. J. Pharmacol. Exp.
Therap., v. 326, n. 1, p.196-208, 2008.
BITTENCOURT, J. C. Métodos em neurociência. São Paulo: Roca, 2007.
BLOCK, F. Global ischemia and behavioural deficits. Prog. Neurobiol., v. 58, p. 279-295,
1999.
BLOCK, F.; SCHWARZ, M. Dextromethorphan reduces functional deficits and neuronal
damage after global ischemia in rats. Brain Res., v.741, p. 153-159, 1996.
BOKURA, H.; ROBINSON, R. G. Long-term cognitive impairment associated with caudate
stroke. Stroke, v. 28, p. 970-975, 1997.
BONVENTRE, J. V. Roles of phospholipases A2 in brain cell and tissue injury associated
with ischemia and excitotoxicity. J. Lipid Mediat. Cell Signal., v. 16, n. 3, p. 199-208, 1997.
BRADLEY, P. P.; CHRISTENSEN, R. D.; ROTHSTEIN, G. Cellular and extracellular
myeloperoxidase in pyogenic inflammation. Blood. v. 60, p. 618–622, 1982
BRADVIK, B.; SONESSON, B.; HOLTAS, S. Spatial impairment following right
hemisphere transient ischaemic attacks in patients without carotid artery stenosis. Acta
Neurol. Scand., v. 80, p. 411-418, 1989.
BRAEUNINGER, S.; KLEINSCHNITZ, C. Rodent models of focal cerebral ischemia:
procedural pitfalls and translational problems. Exp. Translat. Stroke Med., v. 1, p. 1-11,
2009.
BREDESEN, D. E. Keeping neurons alive: the molecular comtrol of apoptosis.
Neuroscientistis, v. 2, p. 211-216, 1996.
BROADHURST, P. L. Determinants of emotionality in the rat: I situational factors. Br. J.
Psychol., v. 48, p. 1-12, 1957.
BROUNS, R.; DE DEYN, P. P. The complexity of neurobiological processes in acute
ischemic stroke. Clin. Neurol. Neurosurg., v. 111, p. 483–495, 2009.
CHANDRAN, B.; GOEL, A. A Randomized, Pilot Study to Assess the Efficacy and Safety of
Curcumin in Patients with Active Rheumatoid Arthritis. Phytother. Res., 2012
CHEN, J.; NAGAYAMA, T.; JIN, K.; STETLER, R.A.; ZHU, R.L.; GRAHAM, S.H.;
SIMON, R.P. Induction of caspase-3-like protease may mediate delayed neuronal death in the
hippocampus after transient cerebral ischemia. J. Neurosci., v. 18, p. 4914-4928, 1998.

90
CHEN, R. W.; QIN, Z. H.; REN, M.; KANAI, H.; CHALECKA-FRANASZEK, E.; LEEDS,
P.; CHUANG, D. M. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA
binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium
neuroprotection. J. Neurochem., v. 84, n. 3, p. 566-575, 2003.
CHENG, A. L.; HSU, C. H.; LIN, J. K.; HSU, M. M.; HO, Y. F.; SHEN, T. S.; ET, A. L.;
KO, J. Y.; LIN, J. T.; LIN, B. R.; MING-SHIANG, W.; YU, H. S.; JEE, S. H.; CHEN, G. S.;
CHEN, T. M.; CHEN, C. A.; LAI, M. K.; PU, Y. S.; PAN, M. H.; WANG, Y. J.; TSAI, C. C.;
HSIEH, C. Y. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with
high-risk or pre-malignant lesions. Anticancer Res., v. 21, p. 2895-900, 2001.
CHOI, D. W. Calcium-mediated neurotoxicity: relationship to specific channel types and role
in ischemic damage. Trends Neurosci., v. 11, p. 465-469, 1988.
CLARK, R. E.; SQUIRE, L. R. An animal model of recognition memory and medial temporal
lobe amnesia: History and current issues. Neuropsychologia, v. 48, n. 8, p. 2234-2244, 2010.
DeNOBLE, V. J.; REPETTI, S. J.; GELPKE, L.W.; WOOD, L. M.; KEIM, K. L.
Vinpocetine: nootropic effects on scopolamine-induced and hypoxia-induced retrieval
deficits of a step-through passive avoidance response in rats. Pharmacol. Biochem.
Behav., v. 24, n. 4, p.1123-1128, 1986.
DEW, I.T. Z.; CABEZA, R. The porous boundaries between explicit and implicit memory:
behavioral and neural evidence. Ann. NY Acad. Sci., v.1224, p. 174-190, 2011.
DIRNAGL, U.; ISADECOLA, C.; MOSKOWITZ M. A. Pathobiology of ischaemic stroke:
an integrated view. Trends Neurosci., v. 22, p. 391–397, 1999.
DOBREK, L.; THOR, P. Glutamate NMDA receptors in pathophysiology and
pharmacotherapy of selected nervous system diseases. Postepy. Hig. Med. Dosw., v. 65, p.
338-346, 2011.
DOHARE, P.; GARG, P.; JAIN, V.; NATH, C.; RAY, M. Dose dependence and therapeutic
window for the neuroprotective effects of curcumin in thromboembolic model of rat. Behav.
Brain Res., v. 193, p. 289-297, 2008.
DOHARE, P.; VARMA, S.; RAY, M. Curcuma oil modulates the nitric oxide system
response to cerebral ischemia/reperfusion injury. Nitric Oxide, v. 19, p. 1-11, 2008.
DONNAN, G. A; FISHER, M., MACLEOD, M.; DAVIS, S. M. Stroke. Lancet, v. 371, p.
1612-1623, 2008.
DOYLE, K. P.; SIMON, R. P.; STENZEL-POORE, M. P. Mechanisms of Ischemic Brain
Damage. Neuropharmacology, v. 55, n. 3, p. 310-318, 2008.
DUSZCZYK M, ZIEMBOWICZ A, GADAMSKI R, LAZAREWICZ JW. Behavioral
evaluation of ischemic damage to CA1 hippocampal neurons: effects of preconditioning. Acta
Neurobiol. Exp., v. 66, n. 4, p. 311-319, 2006.

91
ESPEY, M. G.; MIRANDA, K. M.; THOMAS, D. D.; XAVIER, S.; CITRIN, D.; VITEK, M.
P.; WINK, D. A. Mechanism of cell death governed by the balance between nitrosative and
oxidative stress. Ann. NY Acad. Sci., v. 899, p. 209-221, 2000.
FALCÃO, I. V.; CARVALHO, E. M. F.; BARRETO, K. M. L.; LESSA, F. J. D.; LEITE, V.
M. V. Acidente vascular cerebral precoce: implicações para adultos em idade produtiva
atendidos elo Sistema Único de Saúde. Rev. Bras. Saúde Matern. Infant., v. 4, p. 95-101,
2004.
FEIGIN, V. L.; LAWES, C. M. M.; BENNETT, D. A.; BARKER-COLLO, S. L.; PARAG,
V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies:
a systematic review. Lancet Neurol., v. 8, p. 355-369, 2009.
FONTENELE FILHO, A. T. Efeito neuroprotetor do pré-condicionamento por estresse de
contensão sobre lesão induzida por breve mudança subcrítica isquêmica: papel dos
receptores A1 da adenosina. Dissertação (Mestrado) - Programa de Pós-graduação em
Farmacologia, Universidade Federal do Ceará, Fortaleza, 2009.
GAFNER, S.; LEE, S. K.; CUENDET, M.; BARTHÉLÉMY, S.; VERGNES, L.;
LABIDALLE, S.; MEHTA, R. G.; BOONE, C. W.; PEZZUTO, J. M. Biologic evaluation of
curcumin and structural derivatives in cancer chemoprevention model systems.
Phytochemistry, v. 65, p. 2849-2859, 2004.
GALVÃO, R. I. M.; DIÓGENES, J. P. L.; MAIA, G. C. L.; FILHO, E. A. S.;
VASCONCELOS, S. M. M.; MENEZES,D. B.; CUNHA, G. M. A.; VIANA, G. S. B.
Tenoxicam Exerts a Neuroprotective Action after Cerebral Ischemia in Rats. Neurochem.
Res., v. 30, n. 1, p. 39-46, 2005.
GARCIA-ALLOZA, M.; BORRELLI, L. A.; ROZKALNE, A.; HYMAN, B. T.; BACSKAI,
B. J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially
restores distorted neurites in an Alzheimer mouse model. J. Neurochem., v. 102, n. 4, p.
1095-1104, 2007.
GARRITANO, C. R.; LUZ, P. M.; PIRES, M. L. E.; BARBOSA, M. T. S.; BATISTA, K. M.
Analysis of the Mortality Trend due to Cerebrovascular Accident in Brazil in the XXI
Century. Arq. Bras. Cardiol., v. 98, n. 6, p. 519-527, 2012.
GHONEIM, A. I.; ABDEL-NAIM, A. B.; KHALIFA, A. E.; EL-DENSHARY, E. S.
Protective effects of curcumin against ischaemia/reperfusion insult in rat forebrain.
Pharmacol. Res., v. 46, n. 3, p. 273-279, 2002.
GILGUN-SHERKI, Y.; ROSENBAUM, Z.; MELAMED, E.; OFFEN D. Antioxidant therapy
in acute central nervous system injury: current state. Pharmacol. Rev., v. 54, n. 2, p. 271-284,
2002.
GINSBERG, M. D. Neuroprotection for ischemic stroke: Past, present and future
Neuropharmacology, v. 55, p. 363-389, 2008.

92
GODEFROY, O.; ROUSSEAUX, M.; PRUVO, J. P.; CABARET, M.; LEYS, D.
Neuropsychological changes related to unilateral lenticostriate infarcts. J. Neurol.
Neurosurg. Psyc., v. 57, p. 480-485, 1994.
GREEN, L.C.; WAGNER, D.A.; GLOGOWSKI, J.; SKIPPER, P.L.; WISHNOK, J.S.;
TANNENBAUM, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids.
Anal. Biochem., v.126, n.1, p.131-138, 1982.
HARTLEY, D. M.; KURTH, M. C.; BJERKNESS, L.; WEISS, J. H.; CHOI, D. W.
Glutamate receptor-induced Ca2+
accumulation in cortical cell culture correlates with
subsequent neuronal degeneration. J. Neurosc., v. 13, n. 1993-2000, 1993.
HARTMAN, R. E.; LEE, J. M.; ZIPFEL, G. J.; WOZNIAK, D. F. Characterizing learning
deficits and hippocampal neuron loss following transient global cerebral ischemia in rats.
Brain Res., v. 1043, p. 48-56, 2005.
HATCHER, H.; PLANALP, R.; CHO, J.; TORTI, F. M.; TORTI, S. V. Curcumin: From
ancient medicine to current clinical trials. Cell Mol. Life Sci., v. 65, p. 1631-1652, 2008.
HIMADRI, P.; KUMARI, S. S.; CHITHARANJAN, M.; DHANANJAY, S. Role of oxidative
stress and inflammation in hypoxia-induced cerebral edema: a molecular approach. High Alt.
Med. Biol., v. 11, n. 3, p. 231-244, 2010.
HONG, J.; BOSE, M.; J. U, J.; RYU, J. H.; CHEN, X.; SANG, S.; LEE, M. J.; YANG, C. S.
Modulation of arachidonic acid metabolism by curcumin and related b-diketone derivatives:
effects on cytosolic phospholipase A(2), cyclooxygenases and 5-lipoxygenase.
Carcinogenesis, v. 25, p. 1671-1679, 2004.
HOSSMANN, K. A. Animal models of cerebral ischemia. Cerebrovasc. Dis., v. 1, p. 2-15,
1991.
HUANG, J.; UPADHYAY, U. M.; TAMARGO, R. J. Inflammation in stroke and focal
cerebral ischemia. Surg. Neurol., v. 66, p. 232-245, 2006.
IRESON, C.; ORR, S.; JONES, D. J. L.; VERSCHOYLE, R.; LIM, C. K.; LUO, J. L.;
HOWELLS, L.; PLUMMER, S.; JUKES, R.; WILLIAMS, M.; STEWARD, W. P.;
GESCHER, A. Characterization of Metabolites of the Chemopreventive Agent Curcumin in
Human and Rat Hepatocytes and in the Rat in Vivo, and Evaluation of Their Ability to Inhibit
Phorbol Ester-induced Prostaglandin E2 Production1. Cancer Res., v. 61, p. 1058–1064,
2001.
IZQUIERDO, I.; BEVILAQUA, L. R. M.; ROSSATO, J. I.; BONINI, J. S.; MEDINA, J. H.;
CAMMAROTA, M. Different molecular cascades in different sites of the brain control
memory consolidation. Trends Neurosc., v. 29, n .9, p. 496-505, 2006.
IZQUIERDO, I.; MEDINA, H. Memory Formation: The Sequence of Biochemical Events in
the Hippocampus and Its Connection to Activity in Other Brain Structures. Neurobiol. Learn
Mem., v. 68, p. 285-316, 1997.

93
JAGATHA, B.; MYTHRI, R. B.; VALI, S.; BHARATH, M. S. S. Curcumin treatment
alleviates the effects of glutathione depletion in vitro and in vivo: Therapeutic implications for
Parkinson's disease explained via in silico studies. Free Rad. Biol. Med., v. 44, p. 907-917,
2008.
JIANG, J.; WANG, W.; SUN, Y. J.; HU, M.; LI, F.; ZHU, D. Y. Neuroprotective effect of
curcumin on focal cerebral ischemic rats by preventing blood–brain barrier damage. Eur. J.
Pharmacol., v. 561, p. 54-62, 2007
JIN, K.; GRAHAM, S. H.; MAO, X.; NAGAYAMA, T.; SIMON, R. P.; GREENBERG, D.
A. Fas (CD95) may mediate delayed cell death in hippocampal CA1 sector after global
cerebral ischemia. J. Cereb. Blood Flow Metab., v. 21, p. 1411-1421, 2001.
JOHNSTON, S. C.; MENDIS, S.; MATHERS, C. D. Global variation in stroke burden and
mortality: estimates from monitoring, surveillance, and modelling. Lancet Neurol., v. 8, p.
345-354, 2009.
KANDEL, E. R. The Biology of Memory: A Forty-Year Perspective. J Neurosc., v. 29, n. 41,
p. 12748 -12756, 2009.
KAPOSZTA, Z.; TOUNG, E.; BATH, P. M. W.; MARKUS, H. S.Clinical application of
asymptomatic embolic signal detection in acute stroke : a prospective study. Stroke, v. 30,
p.1814–1818, Sept. 1999.
KARASAWA, Y.; ARAKI, H.; OTOMO, S. Changes in locomotor activity and passive
avoidance task performance induced by cerebral ischemia in mongolian gerbils. Stroke, v.
25, p. 645-650, 1994.
KIM, D. H.; JEON, S. J.; SON, K. H.; JUNG, J. W.; LEE, S.; YOON, B. H.; CHOI, J. W.;
CHEONG, J. H.; KO, K. H.;, RYU, J. H. Effect of the flavonoid, oroxylin A, on transient
cerebral hypoperfusion induced memory impairment in mice. Pharmacol. Biochem. Behav.,
v. 85, p. 658–668, 2006.
KIM, D.H.; YOON, B. H.; KIM, YW.; LEE, S.; SHIN, B. Y.; JUNG, J. W.; KIM, H. J., LEE,
Y. S.; CHOI, J. S.; KIM, S. Y.; LEE, K.T.; RYU, J. H. The Seed Extract of Cassia obtusifolia
Ameliorates Learning and Memory Impairments Induced by Scopolamine or Transient
Cerebral Hypoperfusion in Mice. J. Pharmacol. Sci., v. 105, p. 82- 93, 2007.
KIRINO, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain
Res., v. 239,p. 57–69, 1982.
KRIZ, J.; LALANCETTE-HÉBERT, M. Inflammation, plasticity and real-time imaging after
cerebral ischemia. Acta Neuropathol, v. 117, p. 497-509, 2009.
KUMAR, A.; DOGRA, S.; PRAKASH, A. Protective effect of curcumin (Curcuma longa),
against aluminium toxicity:Possible behavioral and biochemical alterations in rats. Behav.
Brain Res., v. 205, p. 384-390, 2009.
LALONDE, R. The neurobiological basis on spontaneous alternation. Neurosc. Biobehav.
Rev., v.26, p.91-104, 2002.

94
LAVADOS, P. M.; HENNIS, A. J. M.; FERNANDES, J. G.; MEDINA, M. T.; LEGETIC,
B.; HOPPE, A.; SACKS, C.; JADUE, L.; SALINAS, R. LEE, J,; GRABB, M. C.; ZIPFEL G.
J.; CHOI, D. W. Stroke epidemiology, prevention, and management strategies at a regional
level : Latin America and the Caribbean. Lancet Neurol., v. 6, p. 362-372, 2007.
LEE, J. M.; GRABB, M. C.; ZIPFEL, G. J.; CHOI, D. W. Brain tissue responses to ischemia.
J. Clin. Invest., v. 106, n. 6, p. 723-731, 2000.
LIEPERT, J.; BAUDER, H.; MILTNER, W. H. R.; TAUB, E.; WEILER, C. Treatment-
Induced Cortical Reorganization After Stroke in Humans. Stroke, v. 31, p. 1210-1216, 2000.
LIM, G. P.; CHU, T.; YANG, F.; BEECH, W.; FRAUTSCHY, S. A.; COLE, G. M. The curry
spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic
mouse. J. Neurosci., v. 21, n. 21, p. 8370-8377, 2001.
LIPTON, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev., v. 79, p. 1431-1568, 1999.
LLEDO, P. M.; ALONSO, M.; GRUBB, M.S. Adult neurogenesis and functional plasticity in
neuronal circuits. Nat. Rev. Neurosc., v. 7, p. 179-193, 2006.
LOH, K. P.; QI, J.; TAN, B. K.; LIU, X. H.; WEI, B. G.; ZHU, Y. Z. Leonurine protects
middle cerebral artery occluded rats through antioxidant effect and regulation of
mitochondrial function. Stroke, v. 41, n. 11, p. 2661-2668, 2010.
LOVE, S. Apoptosis and brain ischemia. Prog. Neuropsychopharmacol. Biol. Psychiatry,
v. 27, p. 267-282, 2003.
LÜ, J. M.; LIN, P. H.; YAO, Q.; CHEN, C. Chemical and molecular mechanisms of
antioxidants: Experimental approaches and model systems. J. Cell Mol. Med., v. 14, n. 4), p.
840-860, 2010.
MAGNONI, S.; BAKER, A.; GEORGE, S. J.; DUNCAN, W. C.; KERR, L. E.;
MCCULLOCH, J.; HORSBURGH, K. Differential alterations in the expression and activity
of matrix metalloproteinases 2 and 9 after transient cerebral ischemia in mice. Neurobiol.
Dis., v. 17, p. 188-197, 2004.
MAIA, F. D.; PITOMBEIRA, B. S. S.; ARAÚJO, D. T.; CUNHA, G. M. A.; VIANA, G. S.
B. l-Deprenyl Prevents Lipid Peroxidation and Memory Deficits Produced by Cerebral
Ischemia in Rats. Cell Mol. Neurobiol., v. 24, n. 1, 2004.
MANSOURI,, Z.; SABETKASAEI, M.;MORADI, F.; MASOUDNIA, F.; ATAIE, A.
Curcumin has Neuroprotection Effect on Homocysteine Rat Model of Parkinson. J. Mol.
Neurosci., v. 47, n.2, p. 234-242, 2012.
MARKGRAF, C.G.; VELAYO, N. L.; JOHNSON, M. P.; MCCARTY, D. R.; MEDHI, S.;
KOEHL, J. R.; CHMIELEWSKI, P. A.; LINNIK, M. D. Six-hour window of opportunity for
calpain inhibition in focal cerebral ischemia in rats. Stroke, v. 29, n. 1, p. 152-158, 1998.

95
MAROSI, M.; FUZIK, J.; NAGY, D.; RÁKOS, G.; KIS, Z.; VÉCSEI, L.; TOLDI, J.;
RUBAN-MATUZANI, A.; TEICHBERG, V. I.; FARKAS, T. Oxaloacetate restores the long-
term potentiation impaired in rat hippocampus CA1 region by 2-vessel occlusion. Eur. J.
Pharmacol., v. 604, p. 51-57, 2009.
MARTIN, R. L.; LLOYD H. G.; COWAN, A. I. The early events of oxygen and glucose
deprivation: setting the scene for neuronal death? Trends Neurosci., v. 17, p. 251–257, 1994.
MASSARO, A. R. Triagem do AVC isquêmico agudo. Rev. Soc. Cardiol. Rio Grande do
Sul, v. 7, 2006.
McCORD, J. M.; FRIDOVICH, I. Superoxide dismutase an enzymic function for
erythrocuprein Hemocuprein. J. Biol. Chem., v. 244, p. 6049-6055, 1969.
MELDRUM, B.; EVANS, M.; GRIFFITHS, T.; SIMON, R. Ischaemic brain damage: the role
of excitatory activity and of calcium entry. Br. J. Anaesth., v. 57, p. 44-46, 1985.
MINAMI, M.; KURAISHI, Y.; YABUUCHI, K.; YAMAZAKI, A.; SATOH, M. Induction of
interleukin-1 beta mRNA in rat brain after transient forebrain ischemia. J. Neurochem., v. 58,
n. 1, p. 390-392, 1992.
MORRIS, R. Developments of a water-maze procedure for stugying spatial learning in the rat.
J, Neurosci.Methods,v.11, p. 47-60, 1984.
MORRIS, R. G. M.; MOSER, E. I.; RIEDEL, G.; MARTIN, S. J.; SANDIN, J.; DAY, M.;
OCARROL, C. Elements of a neurobiological theory of the hippocampus: the role of activity-
dependent synaptic plasticity in memory. Phil. Trans. R. Soc., v. 358, p. 773-786, 2003.
MORRIS, R.G.; ANDERSON, E.; LYNCH, G.S.; BAUDRY, M.. Selective impairment of
learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor
antagonist, AP5. Nature, v. 319, p. 774-776, 1986.
MORRIS, R.G.M.; GARRUD, P.; RAWLIN, J.N.P.; O’KEEF, J. Place Navigation Impaired
in Rats with Hippocampal Lesions. Nature, v.297, p. 681-683, 1982.
NADEL, L.; HUPBACH, A.; GOMEZ, R.; NEWMAN-SMITH, K. Memory formation,
consolidation and transformation. Neurosci. Biobehav. Rev., v. 36, n. 7, p. 1640-1645, 2012.
NG, T.; CHIAM, P.; LEE, T.; CHUA, H.; LIM, L.; KUA, E. Curry Consumption and
Cognitive Function in the Elderly. Am. J. Epidemiol., v. 164, p. 1-9, 2006.
NIWA, M.; HARA, A.; IWAI, T.; WANG, S.; HOTTA, K.; MORI, H.; UEMATSU, T.
Caspase activation as an apoptotic evidence in the gerbil hippocampal CA1 pyramidal cells
following transient forebrain ischemia. Neurosci. Lett., v. 300, p. 103-106, 2001
NOOR , N. A.; ABOUL-EZZ, H. S.; FARAAG, A. R.; KHADRAWY, Y. A. Evaluation of
the antiepileptic effect of curcumin and Nigella sativa oil in the pilocarpine model of epilepsy
in comparison with valproate. Epilepsy Behav., v. 24, n. 2,p. 199-206, 2012.

96
NORDBERG, J.; ARNÉR, E. S. Reactive oxygen species, antioxidants, and the mammalian
thioredoxin system. Free Radic. Biol. Med., v. 31, n. 11, p. 1287-312, 2001.
OGURO, K.; JOVER, T.; TANAKA, H.; LIN, Y.; KOJIMA, T.; OGURO, N.; ROOMS, S.Y.;
BENNETT, M.V.; ZUKIN, R.S. Global ischemia-induced increases in the gap junctional
proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32
knock-out mice. J. Neurosci., v. 21, p. 7534-7542, 2001.
PAN, R.; QIU, S.; LU D; DONG J. Curcumin improves learning and memory ability and its
neuroprotective mechanism in mice. Chin. Med. J., v. 121, n. 9, p. 832-839, 2008.
PULSINELLI, W. A.; BRIERLY, J. B.; PLUM, F. Temporal profile of neuronal damage in a
model of transient forebrain ischemia. Ann. Neurol., v. 11, p. 491-498, 1982.
RAJESWARI ,A; SABESAN, M. Inhibition of monoamine oxidase-B by the polyphenolic
compound, curcumin and its metabolite tetrahydrocurcumin, in a model of Parkinson’s
disease induced by MPTP neurodegeneration in mice. Inflammopharmacology, v. 16, p. 96-
99, 2008.
RATHORE, P.; DOHARE, P.; VARMA, S.; RAY, A.; SHARMA, U.; JAGANATHANAN,
N. R.; RAY, M. Curcuma Oil: Reduces Early Accumulation of Oxidative Product and is Anti-
apoptogenic in Transient Focal Ischemia in Rat Brain. Neurochem. Res., v. 33, p. 1672-1682,
2008.
REETA, R. H.; MEHLA, J.; GUPTA, Y. K. Curcumin ameliorates cognitive dysfunction and
oxidative damage in phenobarbitone and carbamazepine administered rats. Eur. J.
Pharmacol., v. 644, p. 106-112, 2010.
REMACLE, J.; LAMBET, D.; RAES, M.; PIGEOLET, E.; MICHELS, C.; TOUISSANT, O.
Importance of various antioxidant enzymes for cell stability. Confrontation between
theoretical and experimental data. Biochem. J., v. 286, p. 41-46. 1992.
REMPEL-CLOWER, N. L.; ZOLA, S. M.; SQUIRE, L. R.; AMARAL, D. G. Three cases of
enduring memory impairment after bilateral damage limited to the hippocampal formation. J.
Neurosci., v. 16, p. 5233-5255, 1996.
RIBEIRO, S. M. R.; QUEIROZ, J. H.; PELUZO, M. C. G.; COSTA, N. M. B.; MATTA, S.
L. P.; QUEIROZ, M. E. R. L. A formação e os efeitos das espécies reativas de oxigênio no
meio biológico. Biosc. J., v. 21, p. 133-149, 2005.
ROTHMAN, S. M.; OLNEY, J. W. Glutamate and the pathophysiology of hypoxic-ischemic
brain damage. Ann. Neurol., v. 19, p.105-111, 1986.
ROWLAND, L. P.; MERRI, T. T. Tratado de Neurologia. 10.ed. Rio de Janeiro: Guanabara
Koogan, 2002.
SAIRANEN, T. R.; LINDSBERG, P. J.; BRENNER, M.; SIREN, A. Global Forebrain
Ischemia Results in Differential Cellular Expression of Inter1eukin-lβ (IL-lβ) and its Receptor
at mRNA and Protein Level. J. Cer. Blood Flow Met., v. 17, p. 1107-1120, 1997.

97
SAKAMOTO, N.; KOGURE, K., KATO, H.; OHTOMO, H. Disturbed Ca2+
homeostasis in
the gerbil hippocampus following brief transient ischemia. Brain Res., v. 364, p. 372-376,
1986.
SAKAMOTO, N.; KOGURE, K.; KATO, H.; OHTOMO, H. Disturbed Ca2+ homeostasis in
the gerbil hippocampus following brief transient ischemia. Brain Res., v. 364, p. 372-376,
1986.
SARTER, M.; BODEWITZ, G; STEPHENS, D. N. Attenuation of scopolamine-induced
impairment of spontaneous alteration behaviour by antagonist but not inverse agonist and
agonist beta-carbolines. Psychopharmacology, v. 94, n. 4, p. 491-495, 1988.
SATTLER, R.; TYMIANSKI, M. Molecular Mechanisms of Glutamate Receptor-Mediated
Excitotoxic Neuronal Cell Death. Mol. Neurobiol., v. 4, p. 107-129, 2001.
SCAPAGNINI, G.; SONYA, V.; NADER, A. G.; CALOGERO, C.; ZELLA, D.; FABIO, G.
Modulation of Nrf2/ARE Pathway by Food Polyphenols: A Nutritional Neuroprotective
Strategy for Cognitive and Neurodegenerative Disorders. Mol. Neurobiol., v. 44, p. 192-201,
2001.
SCHMUED, L. C.; STOWERS, C. C.; SCALLET, A. C.; XU. L. Fluoro-Jade C results in
ultra high resolution and contrast labeling of degenerating neurons. Brain Res., v. 1035, p.
24-31, 2005.
SEDLAK J., LINDSAY R. H. Estimation of total, protein-bound, and nonprotein sulfhydryl
groups in tissue with Ellman's reagent. Anal. Biochem., v. 25, p. 192-195, 1968.
SHEHZAD, A.; WAHID, F.; LEE, Y. S.Curcumin in Cancer Chemoprevention: Molecular
Targets, Pharmacokinetics, Bioavailability, and Clinical Trials. Arch. Pharm. Chem. Life
Sci., v. 9, p. 489-499, 2010.
SHUKLA, P. K.; KHANNA, V. K.; ALI, M. M.; KHAN, M. Y.; SRIMAL, R. C. Anti-
ischemic Effect of Curcumin in Rat Brain. Neurochem. Res., v. 33, p. 1036-1043, 2008.
SICARD, K. M.; FICHER, M. Animal models of focal brain ischemia. Exp. Trans. Stroke
Med., v. 1, p.7, 2009.
SMITH, M.-L.; AUER, R. N.; SIESJO, B. K.. The density and distrubution of ischemic brain
injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol., v. 64, p. 319-
332, 1984.
SQUIRE, L.R.; ZOLA, S.M. Structure and function of declarative and nondeclarative
memory systems. Proc. Natl. Acad. Sci., v. 26, n. 93, p. 13515-13522, 1996.
STONE, W. S.; WALSER, B.; GOLD, S. D.; GOLD, P. E. Scopolamine- and morphine-
induced impairments of spontaneous alternation performance in mice: reversal with glucose
and with cholinergic and adrenergic agonists. Behav. Neurosci., v. 105, n. 2, p. 264-271,
1991.

98
SUGAWARA, T.; FUJIMURA, M.; NOSHITA, N.; KIM, G. W.; SAITO, A.; HAYASHI,T.;
NARASIMHAN, P.; MAIER, C. M.; CHAN, P. H. Neuronal Death/Survival Signaling
Pathways in Cerebral Ischemia. NeuroRX, v. 1, p. 17-25, 2004.
SUGAWARA, T.; NOSHITA, N.; LEWE, A.; GASCHE, Y.; FERRAND-DRAKE, M.;
FUJIMURA, M.; MORITA-FUJIMURA, Y.; CHAN, P. H Overexpression of Copper/Zinc
Superoxide Dismutase in Transgenic Rats Protects Vulnerable Neurons against Ischemic
Damage by Blocking the Mitochondrial Pathway of Caspase Activation. J. Neurosc., v. 22, n.
1, p. 209-217, 2002.
SUH, H.; HANG, S.; KWON, K. Curcumin attenuates glutamate-induced HT22 cell death by
suppressing MAP kinase signaling. Mol. Cell Biochem., v. 298, p. 187-194, 2007.
SUN, M.; ZHAO, Y.; GU, Y.; XU, C. Inhibition of nNOS reduces ischemic cell death
through down-regulating calpain and caspase-3 after experimental stroke. Neurochem. Int.,
v. 54, p. 339-346, 2009.
TAKADA, Y.; BHARDWAJ, A.; POTDAR, P.; AGGARWAL, BB. Nonsteroidal anti-
inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of
expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation.
Oncogene, v. 23, n. 57, p. 9247-9258, 2004.
TANAKA, S.; KONDO, H.; KANDA, K.; ASHINO, T.; NAKAMACHI, T.; SEKIKAWA,
K.; IWAKURA, Y.; SHIODA, S.; NUMAZAWA, S.; YOSHIDA, T. Involvement of
interleukin-1 in lipopolysaccaride-induced microglial activation and learning and memory
deficits. J. Neurosci. Res., v. 89, n. 4, p. 506-514, 2011.
TARAWNEH, R.; GALVIN, J. E. Potential future neuroprotective therapies for
neurodegenerative disorders and stroke. Clin. Geriatr. Med., v. 26, n. 1, p. 125-147, 2010.
TARDINI, D. M.S.; YOSHIDA, W. B.; NOVELLI, E. L.B.; SEQUEIRA, J. L. Avaliação de
dois modelos experimentais de isquemia e reperfusão cerebral em ratos com oclusão
temporária carotídea associada ou não à oclusão vertebral. Acta Cir. Bras., v. 18, n. 6, p.
502-508, 2003.
THIYAGARAJAN, M.; SHARMA, S. S. Neuroprotective effect of curcumin in middle
cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sci., v. 74, p. 969-985,
2004.
TOGASHI, H.; MORI, K.; ITOH, Y.; MATSUMOTO, M.; UENO, K.; OHASHI, S.; OTANI,
H.; YOSHIOKA, M. Involvement of interleukin-1beta/nitric oxide pathway in the
postischemic impairment of long-term potentiation of the rat hippocampus. Neurosci. Lett.,
v. 313, n. 3, p. 133-136, 2001.
TONNESEN, H. H.; KARLSEN, J. Studies on curcumin and curcuminoids. VI. Kinetics of
curcumin degradation in aqueous solution. Z. Lebensm. Unters Forsch., v. 180, p. 402-404,
1985.
TRAYSTMAN, A. Acute and chronic estradiol treatments reduce memory deficits induced by
transient global ischemia in female rats. Horm Behav. Stroke, v. 31, p. 1939-1944, 2000.

99
ULRICH, P.T.; KROPPENSTEDT, S.; HEIMANN, A.; KEMPSKI, O. Laser-Doppler
scanning of local cerebral blood flow and reserve capacity and testing of motor and memory
functions in a chronic 2-vessel occlusion model in rats. Stroke, v. 29, n. 11, p. 2412-2020,
1998.
UMEMURA, A.; MABE, H.; NAGAI, H. A phospholipase C inhibitor ameliorates
postischemic neuronal damage in rats. Stroke , v.23, n. 8, p. 1163-1166, 1992.
VALKO, M.; LEIBFRITZ, D.; MONCOL, J.; CRONIN, M. T. D.; MAZUR, M.; TELSER, J.
Free radicals and antioxidants in normal physiological functions and human disease. Int. J.
Biochem. Cell Biol., v. 39, p. 44-84, 2007.
WAHLSTRÖM, B.; BLENNOW, G. A study on the fate of curcumin in the rat. Acta
Pharmacol. Toxicol., v. 43, n. 2, p. 86-92, 1978.
WALKER, E. J.; ROSENBERG, G. A. TIMP-3 and MMP-3 contribute to delayed
inflammation and hippocampal neuronal death following global ischemia. Exp. Neurol., v.
216, p. 122–131, 2009.
WANG, Q.; SUN, A. Y.; SIMONYI, A.; JENSEN, M. D.; SHELAT, P. B.;
ROTTINGHAUS, G. E.; MACDONALD, R. S.; MILLER, D. K.; LUBAHN, D. E.;
WEISMAN, G. A.; SUN, G. Y. Neuroprotective Mechanisms of Curcumin Against Cerebral
Ischemia-Induced Neuronal Apoptosis and Behavioral Deficits. J. Neurosci. Res., v. 82, p.
138-148, 2005.
WANG, R.; LI, Y. B.; LI, Y. H.; XU, Y.; WU, H.; LI, X. J. Curcumin protects against
glutamate excitotoxicity in rat cerebral cortical neurons by increasing brain-derived
neurotrophic factor level and activating TrkB. Brain Res., v. 1210, p. 84-91, 2008.
WEINSTEIN, P. R.; HONG, S.; SHARP, F. R. Molecular identification of the ischemic
penumbra. Stroke, v.35, p. 2666-2670, 2004.
WYLLIE, A. H.; BEATTIE, G. J.; HARGREAVES, A. D. Chromatin changes in apoptosis.
Histochem. J., v. 13, p. 681-692, 1981.
XU, Y.; LIN, D.; LI, S.; LI, G.; SHYAMALA, S. G.; BARISH, P. A.; VERNON, M. M.;
PAN, J.; OGLE, W. O. Curcumin reverses impaired cognition and neuronal plasticity induced
by chronic stress. Neuropharmacology, v. 57, n. 4, p. 463-471, 2009.
YAMAMOTO, A.Y.; SHIODA, N. F.; HANB, F.; MORIGUCHIA, S.; NAKAJIMAC, A.;
YOKOSUKAD, A.; MIMAKID, Y.; SASHIDAD, Y.; YAMAKUNIC, T.; OHIZUMIE, Y.;
FUKUNAGA, K. Nobiletin improves brain ischemia-induced learning and memory deficits
through stimulation of CaMKII and CREB phosphorylation. Brain Res., v. 1295, p. 218-219,
2009.
YI, J.; PARK, S.; KAPADIA, R.; VEMUGANTI, R. Role of transcription factors in
mediating post-ischemic cerebral inflammation and brain damage. Neurochem. Int., v. 50, n.
7, p. 1014-1027, 2007.

100
YOSHIOKA, M.; ITOH, Y.; MORI, K.; UENO, K.; MATSUMOTO, M.; TOGASHI, H.
Effects of an interleukin-1beta analogue [Lys-D-Pro-Thr], on incomplete cerebral ischemia-
induced inhibition of long-term potentiation in rat hippocampal neurons in vivo. Neurosci.
Lett., v. 261, n. 3, p. 171-174, 1999.
YU, S., ZHENG, W., XIN, N.; CHI, Z.; WANG, N.; NIE, Y.; FENG, W.; WANG, Z.
Curcumin Prevents Dopaminergic Neuronal Death Through Inhibition of the c-Jun N-
Terminal Kinase Pathway. Rejuv. Res., v. 13, n. 1, p. 55-64, 2010.
ZÉTOLA, V. H. F.; NÓVAK, E. M.; CAMARGO, C. H. F.; JÚNIOR, H. C.; CORAL, P.;
MUZZIO, J. A.; IWAMOTO, F. M.; COLETA, M. V. D.; WERNECK, L. N. Acidente
Vascular Cerebral em pacientes jovens. Arq. Neuro-Psiquiatr., v. 59, n. 3B, p. 740-
745, 2001.
ZHAN, R. Z.; WU, C.; FUJIHARA, H.; TAGA, K.; NAITO, M.; SHIMOJI, K. Both caspase-
dependent and caspase-independent pathways may be involved in hippocampal CA1 neuronal
death because of loss of cytochrome c from mitochondria in a rat forebrain ischemia model. J.
Cereb. Blood Flow Metab., v. 21, p. 529-540, 2001.
ZHUANG, R.; LIN, M. X.; SONG, Q. Y.; LI, J. Effects of curcumin on the expression of
nuclear factor-kappaB and intercellular adhesion molecular 1 in rats with cerebral ischemia-
reperfusion injury. Nan Fang Yi Ke Da Xue Xue Bao, v. 29, n. 6, p. 1153-1135, 2009.