universidade federal de sÃo joÃo del - ufsj.edu.br · anderson fernandes de melo mecanismos...
TRANSCRIPT

UNIVERSIDADE FEDERAL DE SÃO JOÃO DEL - REI - UFSJ
CAMPUS CENTRO-OESTE DONA LINDU - CCO
PROGRAMA MULTICÊNTRICO EM BIOQUÍMICA E BIOLOGIA MOLECULAR
ANDERSON FERNANDES DE MELO
MECANISMOS BIOQUÍMICOS ENVOLVIDOS NA DOENÇA
HEPÁTICA GORDUROSA NÃO ALCÓOLICA EM RATOS
ALIMENTADOS COM A DIETA CAFETERIA
DIVINÓPOLIS - MG
ABRIL – 2016

ANDERSON FERNANDES DE MELO
MECANISMOS BIOQUÍMICOS ENVOLVIDOS NA DOENÇA
HEPÁTICA GORDUROSA NÃO ALCÓOLICA EM RATOS
ALIMENTADOS COM A DIETA CAFETERIA
Dissertação apresentada ao Programa
Multicêntrico em Bioquímica e Biologia
Molecular da Universidade Federal de São
João Del Rei, como requisito parcial para a
obtenção do grau de Mestre.
Orientador: Valéria Ernestânia Chaves
Co-orientador: Helena Coutinho Franco de Oliveira
DIVINÓPOLIS – MG
ABRIL – 2016

Catalogação na fonte
Universidade Federal de São João del-Rei
Biblioteca Campus Centro Oeste – Dona Lindu

ii
Aos meus pais e irmãos que me garantiram a realização
dessa etapa importante em minha vida.
A minha esposa e companheira Danielle pela
fundamental presença em minha vida, por todos os dias
dedicados a mim, pela paciência, pela compreensão e companheirismo.
E a todos que contribuíram de alguma forma para esta conquista!

iii
AGRADECIMENTOS
Agradeço primeiramente a Deus que se faz sempre presente em minha vida. Por ter
guiado meu caminho, sempre me iluminando e me tranquilizando para que eu
conseguisse chegar até aqui.
Agradeço por toda a vida, a minha orientadora Dra. Valéria Ernestânia Chaves por
me aceitar como seu aluno, por todos os conselhos, aprendizados, preocupações,
discussões e por não ter desistido de mim. Por sempre me escutar, sempre disposta
a me ensinar. Serei eternamente grato à você.
Agradeço a minha co-orientadora Dra.Helena Coutinho Franco de Oliveira por me
aceitar como seu aluno e me acolher em seu laboratório permitindo assim o meu
crescimento como pessoa e pesquisador. A todo pessoal da Unicamp, em especial o
Thiago Hentz e Helena Raposo pela grande oportunidade de aprendizado e
amizade, e por estarem sempre dispostos a me ensinarem.
Agradeço a todos os meus amigos do Laboratório de Fisiologia, em especial
Gleuber, Willian, Bárbara, Reysla e Thais por sempre me apoiarem e socorrerem na
hora do aperto e pela troca de conhecimento.
Agradeço em especial à professora Dra.Cristiane Queixa Tilelli pelas palavras sábias
e confortantes e por permitir o convívio e o aprendizado.
Agradeço a toda UFSJ por ter me proporcionado a realização desse sonho, em
especial agradeço ao professor Dra. Hélio Batista dos Santos e sua aluna Camila
Sales pela ajuda na histologia. Agradeço também a todos do Laboratório de
Bioquimica Celular, em especial a professora Dra.Hérica de Lima Santos e o
professor Dr.Leandro Augusto de Oliveira Barbosa, que além de ceder
equipamentos, me concederam um pouco de seus conhecimentos.
A minha amada família por entender as minhas faltas e pelo amor incondicional,
sempre apoiando minhas escolhas. Ao pai e amigo Antônio (Jiló) e a minha amada
mãe, quero agradecer por tudo, sem vocês não poderia ter chegado até aqui. Aos
meus amados irmãos Marco Antônio, Marcelo e André por me compreenderem e me
amarem do jeito que sou.
Agradeço ao grande amor da minha vida, minha companheira, amiga, cúmplice e
esposa Danielle. Deus colocou você em minha vida e ela mudou completamente.
Agradeço pela confiança e pela paciência. Somente pessoas especiais como você

iv
poderiam suportar todos os problemas que passamos juntos. Te amo pra todo o
sempre.

v
“A ignorância gera mais frequentemente confiança do que o conhecimento: são os
que sabem pouco, e não aqueles que sabem muito, que afirmam de uma forma tão
categórica que este ou aquele problema nunca será resolvido pela ciência.”.
(Charles Darwin)

vi
RESUMO
A obesidade é uma condição em que ocorre aumento na gordura corporal, podendo
prejudicar a saúde e o bem-estar. A obesidade está correlacionada com hipertensão,
resistência à insulina, dislipidemia e doença hepática gordurosa não alcóolica
(DHGNA). Embora considerada benigna por muitos estudos, a DHGNA pode evoluir
para processos inflamatórios (esteatohepatite), cirrose e carcinoma hepático. O
acúmulo de lipídios intracelulares na DHGNA é resultado de um desequilíbrio entre,
1) lipogênese de novo, 2) lipólise hepática 3) oxidação lipídica 4) captação de ácidos
graxos hepáticos e 5) exportação de lipídios via partículas de VLDL. Este estudo
teve como objetivo investigar os mecanismos bioquímicos envolvidos no
desenvolvimento da DHGNA em ratos alimentados com a dieta cafeteria. Ratos
Wistar foram alimentados com uma dieta comercial suplementada com 12 itens
hipercalóricos e água contendo 20% de sacarose (grupo cafeteria) ou com uma dieta
comercial e água ad libitum (grupo controle) por 24 dias. O peso corporal foi
monitorado a cada dois dias. Após eutanásia os órgãos e o sangue foram coletados.
A concentração de triacilglicerol (TAG), colesterol, glicose e ácido úrico séricos
foram avaliados utilizando kits comerciais. A concentração de lipídios totais, TAG e
colesterol no fígado foram avaliados pelo método Folch (1957) ou por método
colorimétrico (kit comercial). A análise histológica do tecido hepático foi realizada
através da coloração com Hematoxilina e Eosina e Oil Red. A expressão dos gênica
foi avaliada por RT-PCR. A secreção de VLDL foi calculada pela diferença de TAG
no soro antes e depois da administração de Triton WR 1339 (20%) em ratos em
jejum. A dieta cafeteria induziu um aumento no peso dos tecidos adiposo branco
epididimal e retroperitoneal sem alterar o peso corporal final, assim como um
aumento na concentração de TAG sérico. Também houve um aumento na
concentração dos lipídios totais, de TAG e o colesterol no tecido hepático. A dieta
cafeteria induziu um aumento na expressão do gene da carnitina palmitoil
transferase I (CPT-1), da proteína de transferência microssomal de triacilglicerol
(MTP), mas reduziu a expressão de ApoB apesar do aumento na secreção de VLDL.
Nossos dados demonstram que a dieta cafeteria induziu esteatose hepática e
hipertrigliceridemia, acompanhado de um aumento na secreção de VLDL, contudo
nossos estudos prévios indicam uma sensibilidade insulínica preservada neste
tecido. Desta forma, sugerimos que, apesar da dieta cafeteria induzir um acúmulo de

vii
lipídios no tecido hepático, mecanismos compensatórios parecem estar estimulados,
provavelmente minimizando o efeito desta dieta neste acúmulo lipídico.
Palavras chave: dieta cafeteria, doença hepática gordurosa não alcóolica, secreção
de VLDL, triacilglicerol.

viii
ABSTRACT
The obesity, a condition characterized by an increase in body fat, is correlated with
hypertension, insulin resistance, dyslipidemia and non-alcoholic fatty liver disease
(NAFLD). Although considered benign by many studies, NAFLD may progress to
inflammation (NASH), cirrhosis and hepatic carcinoma. The intracellular
accumulation of lipids in NAFLD results of an imbalance between, 1) lipogenesis de
novo, 2) hepatic lipolysis, 3) lipid oxidation, 4) uptake of fatty acids by liver and 5)
lipid export via VLDL particles. This study aimed to investigate the biochemical
mechanisms involved in the development of NAFLD in rats fed the cafeteria diet.
Wistar rats were fed a commercial diet supplemented with 12 hypercaloric items and
water containing 20% sucrose (cafeteria group) and a commercial diet and water ad
labitum (control group) for 24 days. Body weight was monitored every two days. After
euthanasia, organs and blood were collected. Serum concentration of triacylglycerol
(TAG), cholesterol, uric acid, and glucose were evaluated using commercial kits.
Concentration of total lipids, TAG and cholesterol in the liver were assessed by the
Folch method or by colorimetric method (commercial kit). Histological analysis of liver
tissue was performed by staining with hematoxylin & eosin and oil red. Genes
expression was assessed by RT -PCR. VLDL secretion was calculated by difference
of TAG in the serum before and after the administration of Triton WR 1339 (20%) in
fasted rats. The cafeteria diet induced an increase in the weight of epididymal and
retroperitoneal adipose tissue without to change the final body weight, as well as an
increase in serum concentrations of TAG. There was also an increase in the
concentration of total lipids, TAG and cholesterol in the liver tissue. The cafeteria diet
induced an increase in the gene expression of carnitine palmitoyl transferase (CPT-
1), microsomal triacylglycerol transfer protein (MTTP), but reduced apolipoprotein B
(ApoB) expression, despite the increased secretion of VLDL. Our data showed that
cafeteria diet induced fatty liver disease and hypertriglyceridaemia, followed by an
increase in the VLDL secretion, in addition our previous studies indicate an insulin
sensitivity preserved in this tissue. Thus, we suggest that despite the cafeteria diet
induce an accumulation of lipids in the liver tissue, compensatory mechanisms
appear to be stimulated, probably minimizing the effect of this diet in this lipid

ix
accumulation. Keywords: cafeteria diet, non-alcoholic fatty liver disease, secretion of
VLDL, triacylglycerol.

x
SUMÁRIO
LISTA DE ABREVIATURAS ..................................................................................... xii
1. INTRODUÇÃO ........................................................................................................ 1
1.1 Geração de TAG no tecido hepático ...............................................................5
1.2 Mobilização de TAG hepático ..........................................................................7
1.3 Oxidação hepática de ácidos graxos ...............................................................7
1.4 Secreção de VLDL ..........................................................................................7
1.5 Resistência à insulina e DHGNA ....................................................................9
1.6 Dieta cafeteria................................................................................................10
2. OBJETIVOS ................................................................................................ ..........12
2.1 Objetivo geral................................................................................................12
2.2 Objetivos específicos .................................................................................... 12
3. MATERIAL E MÉTODOS ..................................................................................... 13
3.1 Animais e seu tratamento ............................................................................. 13
3.2 Histologia ...................................................................................................... 14
3.2.1 Coloração com Hematoxilina e Eosina...................................................14
3.2.2 Coloração com Oil Red...................................................................................15
3.3 Determinação do conteúdo lípidico do fígado ............................................... 15
3.4 Avaliação da secreção de VLDL-TAG pelo fígado ........................................ 16
3.5 Reação em cadeia da polimerase – transcrição reversa em tempo real (Real
Time-PCR) .............................................................................................................. 16
3.6 Determinação dos níveis séricos de glicose, triacilglicerol, colesterol, ácido
úrico, AST e ALT. ...................................................................................................... 18
3.7 Análise estatística..........................................................................................18
4. RESULTADOS ...................................................................................................... 19
4.1 Parâmetros séricos ....................................................................................... 19
4.1.1 Ganho de peso corporal dos ratos.........................................................19

xi
4.1.2 Peso dos tecidos....................................................................................20
4.1.3 Parâmetros séricos.................................................................................22
4.2 Parâmetros hepáticos...................................................................................24
4.2.1 Histologia...............................................................................................24
4.2.2 Concentração de lipídios hepáticos.......................................................29
4.2.3 Prova de função hepática.......................................................................32
4.2.4 Expressão gênica no fígado...................................................................34
4.2.4.1 Expressão gênica de fatores de transcrição relacionados com a
regulação da expressão gênica de enzimas envolvidas no metabolismo
de lipídios..............................................................................................34
4.2.4.2 Expressão dos genes relacionados com a oxidação lipídica...36
4.2.4.3 Expressão dos genes relacionados com a mobilização de
lipídios no fígado...................................................................................37
4.2.4.4 Expressão dos genes relacionados com a secreção de
VLDL.....................................................................................................38
4.2.5 Velocidade de secreção de VLDL-TAG..................................................39
5. DISCUSSÃO..........................................................................................................41
5.1 Lipogênese hepática.................................................................................46
5.2 Lipólise hepática.......................................................................................49
5.3 Oxidação de Lipídios Hepáticos................................................................53
5.4 Captação de Ácidos Graxos Hepático......................................................55
5.5 Secreção de VLDL....................................................................................55
6. CONSIDERAÇÕES FINAIS .................................................................................. 62
7. REFERÊNCIAS BIBLIOGRÁFICAS ..................................................................... 63
8. ANEXO...................................................................................................................81

xii
LISTA DE ABREVIATURAS
ACC Acetil CoA carboxilase
ACO Acil-CoA oxidade
AGL Ácido graxo livre
AICAR 5-aminoimidazole-4-carboxamida-ribonucleotídeo
AKT Serina/treonina quinase
ALT Alanina aminotransferase
AMP Adenosina monofosfato
AMPK 5' Adenosina monofosfato quinase
ApoB Apolipoproteina B
AST Aspartato aminotransferase
ATGL Lipase do triacilglicerol do tecido adiposo
ATP Adenosina trifosfato
CGI-58 Comparative gene identification-58
ChREBP Proteína de ligação do elemento de resposta aos carboidratos
CPT-I Carnitina palmitoil transferase I
Crtc2 CREB regulated transcription coactivator 2
DAG Diacilglicerol
DGAT Diacilglicerol aciltransferase
DHGNA Doença hepática gordurosa não alcoólica
FAS Ácido graxo sintase
FATP5 Transportador de ácido graxo 5
Foxo1 Forkhead box O1
G0S2 G0/G1 switch gene 2
G3P Glicerol 3 fosfato
HMG-CoA
redutase 3-hidroxi-3-metil-glutaril-CoA redutase
HNF-4α Fator nuclear de hepatócito
IDL Lipoproteína de densidade intermediária
IL10 Interleucina 10
KLF6 Kruppel-like factor 6
LDL Lipoproteína de baixa densidade
LHS Lipase hormônio sensível

xiii
L-PK Piruvato quinase do fígado
LPL Lipase lipoproteica
LXR Receptor X do fígado
MAG Monoacilglicerol
MCP1 Macrophage chemotatic protein 1
mTOR Complexo alvo da rampamicina em mamíferos 1
MTP Proteína microssomal de transferência de TAG
NAFLD Non-alcoholic fatty liver disease
NASH Non-alcoholic steatohepatitis
PCR Reação em cadeia de polimerase
PEPCK Fosfoenolpiruvato carboxiquinase
PGC-1α Coativador-1α do PPAR-γ
PI3K Fosfatidilinositoil-3-quinase
PKA Proteína quinase dependente de cAMP
PP2A Proteína fosfatase 2A delta
PPARα Peroxisome proliferator-activated receptors alpha
PPARγ Peroxisome proliferator-activated receptors gamma
RNA Ácido ribonucléico
SCD1 Stearol-CoA desaturase 1
SOD2 Superóxido desmutase 2
SREBP-1c Proteína de ligação ao elemento regulador de esterol 1c
TAB Tecido adiposo branco
TAG Triacilglicerol
TGH Hidrolase de triacilglicerol
TNFα Fator de necrose tumoral alfa
UCP2 Proteína desacopladora 2
VLDL Lipoproteína de muito baixa densidade
Xu-5-P Xilulose-5-fosfato

1
1. INTRODUÇÃO
Segundo a Organização Mundial da Saúde, a obesidade é uma condição em
que ocorre o aumento na gordura corporal, podendo prejudicar a saúde e o bem-
estar (OMS, 2016). A obesidade é hoje uma pandemia e se a tendência mundial for
mantida é esperado que a obesidade atinga 10% da população mundial até 2030
(ASRIH; JORNAYVAZ, 2015). Esse quadro vem aumentando demasiadamente na
população, principalmente entre as crianças e os adolescentes de países
desenvolvidos e em desenvolvimento (PRENTICE, 2006). Mudanças nos hábitos
alimentares e atividades físicas reduzidas parecem estar relacionados com o quadro
epidêmico de obesidade (BASCIANO et al., 2005).
A maioria dos tecidos não adiposos apresenta uma pequena reserva lipídica,
funcionando como fonte imediata de energia e substrato para funções essenciais,
como manutenção estrutural, fluidez da membrana e sinalização intracelular
(RASOULI et al., 2007). Obesidade e síndrome metabólica ocasionam alterações
nesse acúmulo natural de lipídios, propiciando deposição lipídica em órgãos não
específicos, como fígado, músculo esquelético e cardíaco, pâncreas e rins
(MEDINA-GOMEZ et al., 2007). Essa deposição ectópica de lipídios leva à disfunção
destes tecidos por lipotoxicidade, e morte celular programada induzida por lipídios
(lipoapoptose) (UNGER, 2003). O acúmulo ectópico de lipídios pode ser relacionado
à limitação da capacidade de expansão do tecido adiposo para armazenar os
nutrientes em excesso. Tem sido sugerido que a expansibilidade do tecido adiposo é
limitada em cada indivíduo, e que, ao atingir seu máximo, desencadeia um aumento
do fluxo lipídico para órgãos não adiposos. Nestas condições, as células adiposas
secretam quimiocinas, por exemplo, MCP1 (macrophage chemotatic protein 1), que
atraem macrófagos, os quais por sua vez são ativados e secretam citocinas, que
prejudicam a sensibilidade à insulina na célula adiposa, aumentando a lipólise e
bloqueando o recrutamento de pré-adipócitos (Figura 1) (VIRTUE; VIDAL PUIG,
2010).

2
Figura 1 – Limitação na expansibilidade do tecido adiposo.
Fonte: VIRTUE e VIDAL PUIG (2010)
Em indivíduos obesos, o maior acúmulo de lipídios na região abdominal em
relação ao acúmulo subcutâneo está associado ao desenvolvimento de deposição
de lipídios em tecidos não adiposos e resistência à insulina (DESPRÉS, LEMIEUX,
2006; CALI et al., 2009). Tem sido sugerido que o acúmulo lipídico abdominal,
independentemente da adiposidade total do indivíduo, está relacionado à deposição
ectópica de lipídios, especialmente no fígado (ADAMS et al., 2007). Este fato tem
sido atribuído a um comportamento diferencial (região-específico) do adipócito
visceral frente aos hormônios lipolíticos e lipogênicos (FLIER; FOSTER, 1998). Tais
adipócitos são mais responsivos às catecolaminas e menos responsivos à insulina
que aqueles do tecido subcutâneo (LAFONTAN et al., 1979; BOLINDER et al.,
1983), aumentando rapidamente e localmente a oferta de ácidos graxos e
adipocinas na circulação portal hepática, resultando em supressão da ação da
insulina no fígado (LARA-CASTRO et al., 2008).

3
A partir da década de 1980, alterações morfológicas hepáticas relacionadas à
esteatose com etiologias diversas, excetuando-se o álcool, passaram a ser
denominadas Doença Hepática Gordurosa Não Alcoólica (DHGNA) ou non-alcoholic
fatty liver disease (NAFLD) (SILVA; ESCANHOELA, 2009). Estudos relatam duas
categorias de DHGNA, a esteatose simples, a qual raramente progride para cirrose,
e a esteatohepatite, ou non-alcoholic steatohepatitis (NASH), processo que pode
levar ao desenvolvimento de cirrose e carcinoma hepatocelular (HCC) (FARRELL et
al., 2006; CASTRO et al., 2012).
A esteatose simples, embora considerada benigna por muitos autores, não
está isenta de consequências estruturais, podendo progredir para cirrose em 4% dos
casos e levar à morte em 2% dos pacientes (MATTEONI et al., 1999). Cerca de 30%
dos pacientes com DHGNA têm esteatohepatite (TRAUNER et al., 2010), a qual é
caracterizada pela presença de esteatose macrovesicular, inflamação lobular,
degeneração dos hepatócitos e fibrose pericelular, tratando-se de uma doença
progressiva que leva à disfunção hepática grave, como a cirrose, em 20-25% dos
casos (VUPPALANCHI et al., 2009; TRAUNER et al., 2010) (Figura 2).
Figura 2 – Evolução da doença hepática gordurosa não alcóolica (DHGNA).
Fonte: TRAUNER et al. (2010)
Embora os mecanismos responsáveis pela esteatose hepática na DHGNA
não estejam inteiramente elucidados, a característica principal da esteatose hepática
é a acumulação de triacilglicerol no citoplasma dos hepatócitos. Isto resulta de um

4
desequilíbrio entre a aquisição de lipídios (isto é, a captação de ácidos graxos e a
lipogênese de novo) e de remoção (isto é, a oxidação de ácido graxo mitocondrial e
exportação como componente de partículas de VLDL) como resumido na Figura 3.
O consumo de uma dieta hipercalórica associado a um estilo de vida
sedentário são possíveis fatores de risco para o desenvolvimento de esteatose
hepática (CASTRO et al., 2012). A esteatose hepática está associada a diversos
estados patológicos, tais como, obesidade, diabetes tipo 2, síndrome metabólica,
desordens nutricionais, lipodistrofias, hiperhomocisteinemia, uso de terapia
antiretroviral, etc. (BROWNING et al., 2004; FARREL et al., 2006; GUILLÉN et al.,
2009), portanto, também é considerada um grande problema de saúde para a
sociedade ocidental (LARTER et al., 2009).
Figura 3 – Mecanismo do metabolismo hepatocelular e sua desregulação na doença
hepática gordurosa não alcóolica
Fonte: KAWANO e COHEN (2013)
A teoria atualmente mais aceita para explicar a gênese da DHGNA sugere
que dois eventos devem ocorrer (DAY et al., 1998): 1) mudanças no metabolismo
lipídico favorecendo depósito de triacilglicerol nos hepatócitos, ocasionado, por

5
exemplo, por resistência à insulina ou por defeito genético, e 2) produção excessiva
de espécies reativas de oxigênio pela mitocôndria e/ou pelo sistema do citocromo
P450 2E1 no fígado. O sistema citocromo P450 2E1 (CYP2E1) está relacionado à
peroxidação lipídica e dano hepático em modelos de esteatose hepática alcoólica, e
também tem sido relacionado com a progressão de DHGNA (DAY et al., 1998;
DUVNJAK et al., 2009, FISHER et al., 2009).
Vários processos medeiam o balanço lipídico intra-hepático, como captação
de lipoproteínas plasmáticas mediada por receptores, captação de AGL mediada por
proteínas transportadoras, velocidade de oxidação lipídica, velocidade de síntese de
novo, esterificação e secreção de lipídios e lipoproteínas. A disponibilidade
aumentada de ácidos graxos no fígado colabora para o desenvolvimento de
esteatose em pacientes com diabetes tipo 2. Como os ácidos graxos são
prontamente levados aos hepatócitos, a resistência à insulina aumenta
extremamente a quantidade de ácidos graxos disponíveis ao fígado (BEGRICH et
al., 2006).
1.1 Geração de TAG no tecido hepático
Os ácidos graxos sintetizados mais ativamente no fígado também contribuem
para o acúmulo de lipídios hepático (BEGRICH et al., 2006). Certamente, os níveis
aumentados de insulina e glicose estimulam a síntese do ácido graxo e triacilglicerol
com ativação da expressão de enzimas-chaves envolvidas na lipogênese,
promovendo a formação e deposição de triacilglicerol no fígado (MATSUSUE et al.,
2003). Na DHGNA, a lipogênese de novo contribui com o acúmulo de TAG no tecido
hepático (DONNELLY et al., 2005). As concentrações de glicose e insulina
plasmática são necessárias para a ativação da lipogênese. Em situação de excesso
de energia, a glicose é convertida em ácidos graxos através da conversão de glicose
em piruvato, que é translocado para o interior da mitocôndria, em seguida convertido
a acetil-CoA que se condensa com oxaloacetato para a formação de citrato, que
será transportado para o citosol (Figura 4). Estes ácidos graxos sintetizados de
novo são utilizados para sintetizar triglicerídeos, a principal fonte de armazenamento
e de transporte de energia (BROWNING; HORTON, 2004).
A glicose e a insulina promovem a lipogênese por ativação do ChREBP e a
SREBP1c, respectivamente. Estes são fatores de transcrição mestre, que por sua
vez, promovem a transcrição de genes lipogênicos tais como, ácido graxo sintase

6
(FAS), acetil CoA carboxilase (ACC), estearil CoA desaturase 1 (SCD1), lipin-1 e
piruvato quinase do fígado (L-PK) (KAWANO; COHEN, 2013). SREBP-1c é
considerada como o principal regulador da transcrição de genes relacionado a
síntese de ácido graxo e TAG em resposta à estimulação da insulina (XU et al.,
2013). A insulina estimula o SREBP-1c através de uma cascata de sinalização que
envolve a proteína serina/treonina quinase (AKT), o receptor X do fígado (LXR) e
mTOR (complexo alvo da rampamicina em mamíferos 1) (COHEN et al.,
2011). SREBP-1c é expresso a um nível baixo no fígado de animais em jejum, mas
sua expressão é dramaticamente induzida mediante a alimentação, situação em que
as concentrações de insulina encontram-se aumentadas. A insulina também regula
SREBP-1c a nível pós-traducional, através de um processamento proteolítico
permitindo o seu deslocamento para o núcleo (XU et al., 2013). Modificações pós-
traducionais de ChREBP são necessárias para a sua ativação. A fosforilação /
desfosforilação de ChREBP tem sido proposta ser importante para a translocação de
ChREBP e sua ativação nuclear (XU et al., 2013). No tecido hepático, a glicose é
convertida em xilulose-5-fosfato (Xu-5-P) a partir da derivação da hexose
monofosfato, e Xu-5-P ativa a proteína fosfatase 2A delta (PP2A delta) que irá
desfosforilar ChREBP permitindo assim sua migração para o núcleo seguido da sua
ativação (IIZUKA; HORIKAWA, 2008).
Figura 4 – Via metabólica da produção de TAG e a relação sobre a resistência à
insulina.
Fonte: BROWNING e HORTON (2004)

7
1.2 Mobilização de TAG hepático
A lipólise no tecido hepático é um mecanismo capaz de garantir a homeostasia
do metabolismo lipídico (REID et al., 2008). Mutações genéticas de enzimas
envolvidas na mobilização de TAG no tecido hepático podem levar a
esteatose. Mutações na ATGL ou no seu co-ativador, comparative gene
identification-58 (CGI-58), impedem a mobilização de ácidos graxos livres a partir de
gotículas lipídicas nos hepatócitos (ZIMMERMANN et al. 2004). Estudos tem
demonstrado que o tecido hepático possui uma baixa expressão das hidrolases LHS
e a ATGL, em relação aos demais tecidos (ZIMMERMANN et al. 2004). No entanto,
são as principais hidrolases capazes de hidrolisar o TAG hepático, direcionando os
ácidos graxos para oxidação ou secreção (REID et al., 2008; ONG et al., 2011). A
regulação dessas duas enzimas é bem distinta. A ativação da LHS ocorre por
modificação covalente, exigindo a presença de hormônios (glucagon, catecolaminas)
e sua translocação do citosol para a gota lipídica (ZIMMERMANN et al., 2004). No
estado basal, CGI-58, ativador da ATGL, esta associado à perilipina, e a partir do
estímulo pela PKA (proteína quinase dependente de cAMP), o co-ativador CGI-58 se
transloca para ATGL permitindo sua ativação (GRANNEMAN et al., 2007). A
deficiência no funcionamento dessas hidrolases pode levar a um aumento da massa
de gordura em diversos tecidos, incluindo o tecido hepático (REID et al., 2008; ONG
et al., 2011). Alguns trabalhos sugerem a possibilidade de haver um aumento na
expressão das hidrolases hepática, dentre elas a ATGL, com o intuito de proteger o
fígado do acúmulo de TAG (WANG et al., 2013; ZANG et al., 2014). Nesses
trabalhos, o estado nutricional interfere na expressão do G0/G1 switch gene (G0S2),
um inibidor da ATGL. Em jejum, G0S2 aumenta sua expressão no tecido hepático
inibindo a atividade de ATGL protegendo TAG da degradação. Além dessa questão,
a atividade das lipases no tecido hepático é necessária para o fornecimento de
ácidos graxos para β-oxidação ou para a geração de TAG e sua secreção via
molécula de VLDL (DOLINSKY et al., 2004; LO et al., 2010).
1.3 Oxidação hepática de ácidos graxos
Os peroxisome proliferator-activated receptors (PPAR) são receptores
hormonais nucleares ativados por ácidos graxos e por proliferadores de
peroxissomos, que desempenham um papel importante na homeostasia do
metabolismo de lipídios (KELLER et al., 1992; LEMBERGER et al., 1996). Existem

8
três isoformas do PPAR, sendo a isoforma α a mais comum no fígado
(LEMBERGER et al., 1996). O PPAR- α é um fator de transcrição que regula a β-
oxidação peroxissomal e mitocondrial através da ativação da transcrição de genes
importantes para esse processo, dentre eles acil-CoA oxidade (ACO) e a carnitina
palmitoil transferase I (CPT-I) no tecido hepático (KELLER et al., 1993;
LEMBERGER et al., 1996; LOUET et al., 2001; MINNICH et al., 2001). No estado
pós-prandial, a β-oxidação no fígado é suprimida. Isto ocorre, pelo menos em parte
devido ao efeito antilipolíticos da insulina. No entanto, estudos realizados em
pacientes com DHGNA tiveram resultados contraditórios no que diz respeito a
alterações nas taxas de oxidação de ácidos graxos (KAWANO; COHEN, 2013). A
redução na oxidação dos ácidos graxos, associada com a diminuição da expressão
ou atividade de CPT-1a, tem sido relatada em seres humanos e em roedores com
DHGNA (BONFLEUR et al., 2015). Mutações nas enzimas necessárias para a
oxidação de ácidos graxos livres nas mitocôndrias (as transferases de hidróxiacil-
CoA) também causam esteatose hepática (COHEN et al,. 2011). Outros estudos
relataram evidências de aumento nas taxas de oxidação de ácidos graxos em
DHGNA. Atualmente tem sido sugerido que o aumento na atividade mitocondrial em
consumir ácidos graxos pode promover o stress oxidativo no fígado e contribuir para
a evolução da esteatose para esteatohepatite (KAWANO; COHEN, 2013).
1.4 Secreção de VLDL
A redução na secreção de VLDL-TAG pode estar relacionada ao acúmulo de
TAG na DHGNA (KOO, 2013). No entanto, mesmo com DHGNA, pacientes podem
ter um aumento na secreção de VLDL, muito provavelmente por causa da taxa de
secreção não acompanhar a taxa de biossíntese de TAG (KOO, 2013). Muitos
estudos sugerem que esse aumento na secreção de VLDL-TAG é em decorrência
da resistência à insulina no tecido hepático (BARTELS et al., 2002; KAMAGATE et
al., 2008). A montagem e a secreção hepática de VLDL é um processo complexo
que envolve muitos genes e proteínas, incluindo ApoB (apolipoproteina B), MTP
(proteína microssomal de transferência de triacilglicerol) e a mobilização de lipídios
(SPARKS; DONG, 2009). A proteína MTP é responsável pela transferência de TAG
para ApoB, proteína estrutural do quilomicron, bem com das lipoproteínas VLDL, IDL
(lipoproteína de densidade intermediaria) e LDL (lipoproteína de densidade baixa).
No retículo endoplasmático dos hepatócitos é formada a molécula de pré-VLDL.

9
Posteriormente esse pré-VLDL é direcionado para o complexo de golgi onde será
completada sua lipidação e consequentemente a secreção de VLDL-TAG (HAAS et
al., 2013).
Alguns estudos sugerem que a insulina desempenha um papel fundamental
na regulação da secreção de VLDL. A insulina inibe a transcrição de MTP via
FOXO1 (forkhead box O1) (KAMAGATE et al.,2008). A fosforilação de FOXO1 por
AKT leva a exclusão de FOXO1 do núcleo impedindo sua ação transcricional
(BRUNET et al., 1999). A insulina também é capaz de diminuir a secreção de VLDL
regulando ApoB a nível traducional e pós traducional (HAAS et al., 2013). Por um
processo ainda não muito compreendido, a insulina estimula a degradação de ApoB
pelo sistema proteossomal e lisossomal (FISHER; GINSBERG, 2002). A sugestão é
que a insulina através de sua cascata de sinalização ative a proteína Akt que, por
sua vez, ativa mTORC1, suprimindo a tradução e a degradação de ApoB. No
entanto, a insulina pode suprimir sortilin, uma proteína envolvida no direcionamento
da ApoB para a degradação pelo lisossoma (HAAS et al.,2013). Assim, a insulina
pode promover secreção ApoB, protegendo-a da degradação via sortilin, embora
também iniba a tradução e a lipidação de ApoB. Corroborando com essa ideia,
vários trabalhos mostram que na DHGNA pode ocorrer um aumento na secreção de
VLDL. Possivelmente, uma excessiva produção de TAG, aumento da lipólise e uma
alteração na sinalização insulínica hepática podem contribuir para o aumento na
secreção de VLDL na DHGNA (KAWANO; COHEN, 2013; FABRINNI et al., 2008,
CHOI; GINSBERG, 2011). Recentemente alguns trabalhos sugerem que a insulina é
capaz de diminuir a secreção de VLDL somente de forma aguda e que a
disponibilidade de TAG nos hepatócitos é o regulador dominante para a secreção de
VLDL (SPARKS et al., 2011; MOON et al., 2012). Ao que parece, a taxa de
produção de lipídios hepáticos não é acompanhada pela oxidação dos ácidos graxos
intra ou extra- hepáticos, e pela secreção de VLDL.
1.5 Resistência à insulina e DHGNA
Alguns estudos relacionam a DHGNA com um estado de resistência à insulina
associado a obesidade resumido na Figura 4 (BROWNING, HORTON, 2004;
KAWANO, COHEN, 2013). Uma série de alterações moleculares e fisiológicas
ocorrem na configuração da resistência à insulina que resulta na acumulação de
triglicerídeos no fígado. A resistência à insulina manifesta-se por hiperinsulinemia,

10
aumento da produção de glicose hepática e diminuição da utilização de glicose pelo
tecido muscular e adiposo. Em adipócitos, a resistência à insulina aumenta a
atividade da lipase hormônio sensível (LHS), resultando em elevadas taxas de
lipólise de triglicérides e maior fluxo de AGL para o fígado. Os AGL podem ser
oxidados nas mitocôndrias para formar ATP ou esterificados para produzir
triglicerídeos de armazenagem ou incorporação em partículas de VLDL
(BROWNING; HORTON, 2004). Desta forma, apesar de diversos estudos
relacionarem a resistência à insulina no fígado, no tecido adiposo e no tecido
muscular esquelética com a porcentagem de gordura no fígado, hoje em dia, não se
sabe se a DHGNA é causa, ou é uma consequência da resistência à insulina, ou
possivelmente ambas (FABBRINI et al., 2010). A complexidade da relação entre a
esteatose hepática e resistência à insulina é reforçada pela observação de que a
esteatose não está sempre associada com a resistência à insulina. Uma dissociação
entre esteatose e a resistência à insulina foi relatada em modelos animais
selecionados geneticamente manipulados farmacologicamente e também em seres
humanos. A superexpressão de diacilglicerol aciltransferase (DGAT) hepática,
o bloqueio da secreção de VLDL hepática e o bloqueio farmacológico da β-
oxidação em ratos provocam esteatose hepática, mas não causam resistência
insulínica no tecido hepático ou no tecido muscular esquelético. Dessa mesma forma
a inibição hepática da síntese de TAG em camundongos obesos diminui a esteatose
hepática, mas não faz melhorar a sensibilidade à insulina. Além disso, a esteatose
hepática causada por deficiência genética da síntese de apoB e a diminuição da
secreção de VLDL hepática em pacientes com hipobetalipoproteinemia familiar não
é acompanhada pela resistência à insulina hepática ou periférica (FABBRINI et al.,
2010). Contudo, a maioria dos estudos demonstra uma relação direta entre a
esteatose hepática com diabetes e resistência à insulina causada por diabetes do
tipo 2.
1.6 Dieta cafeteria
O primeiro trabalho, disponível no PUBMED, a usar alimentos da dieta
humana, palatáveis, de alto valor energético na experimentação com animais,
permitindo aos ratos o livre acesso aos alimentos ocorreu por Sclafani e Springer
(1976). No entanto, Rothwell e Stock (1979) foram os primeiros a usar o termo
cafeteria para a dieta sugerida acima. Esse modelo de obesidade induzido por dieta

11
do tipo cafeteria é um modelo de obesidade simples e possivelmente o que tem
maior proximidade com a realidade dos humanos (SCLAFANI; SPRINGER, 1976). A
utilização deste modelo rápido e potente para indução de obesidade pode ser útil
para identificar novos caminhos para intervenção preventiva ou terapêutica da
obesidade humana (SAMPEY et al., 2011). A dieta de cafeteria induz um aumento
na síntese de ácidos graxos de novo no tecido adiposo branco, com acúmulo de
gordura corporal e hiperinsulinemia, sem alteração nas concentrações séricas de
ácidos graxos e glicose (CHAVES et al., 2006). No tecido hepático, a dieta cafeteria
induz um aumento de 120% na síntese de ácidos graxos de novo (CHAVES et al.,
2012), acompanhado de um aumento da glicólise e diminuição da gliconeogênese
(MARTINS-SANTOS, 2008), e aumento na atividade das lipases citosólicas
hepáticas (MOREIRA & BOTION, dados não publicados).
No presente projeto, os mecanismos bioquímicos envolvidos no
desenvolvimento da DHGNA serão analisados nos ratos alimentados com a dieta
hipercalórica e hiperlipídica, do tipo cafeteria diet.

12
2. OBJETIVOS
2.1 – Objetivo Geral
Avaliar os mecanismos bioquímicos envolvidos na doença hepática gordurosa
não alcóolica de ratos alimentados com uma dieta cafeteria.
2.2 – Objetivos Específicos
Investigar o efeito da dieta cafeteria nos seguintes parâmetros
a) Séricos:
1. triacilglicerol, colesterol, glicose e ácido úrico.
b) Hepáticos:
1. conteúdo de lipídios totais, triacilglicerol e colesterol;
2. morfologia através de análise histologia por Hematoxilina & Eosina e Oil
Red;
3. expressão dos genes acil-CoA oxidade (ACO), apolipoproteina B (ApoB),
lipase do triacilglicerol do tecido adiposo (ATGL), proteína de ligação do elemento de
resposta aos carboidratos (ChREBP), carnitina palmitoil transferase I (CPT-1), lipase
hormônio sensível (LHS), proteína microssomal de transferência de triacilglicerol
(MTP), peroxisome proliferator-activated receptors alpha (PPAR-alfa), proteína de
ligação ao elemento regulador de esterol 1c (SREBP-1c);
4. velocidade de secreção de VLDL.

13
3. MATERIAL E MÉTODOS
3.1 Animais e seu Tratamento
Foram utilizados ratos machos Wistar com idade inicial de aproximadamente
30 dias de vida e peso inicial de aproximadamente 70g, proveniente do Biotério
Central da Universidade Federal de São João Del Rei – UFSJ. Os ratos foram
mantidos no Laboratório de Fisiologia do Campus Centro-Oeste Dona Lindu em
ambiente com ciclo claro-escuro de 12 horas. Ao grupo experimental foi oferecida
por 24 dias uma dieta hipercalórica do tipo cafeteria, obtida pela suplementação da
dieta balanceada comercial com diversos ingredientes palatáveis. A cada dia, quatro
ingredientes diferentes, entre doze itens previamente selecionados (Tabela I), foram
oferecidos aos animais, sendo que todos os itens apresentaram densidade calórica
superior à da dieta comercial (ROTHWELL et al., 1982). A água dos animais do
grupo experimental foi acrescida de sacarose 20%. Os ratos controles receberam a
dieta balanceada comercial (Nuvilab®) e água ad libitum.
A distribuição dos macronutrientes e a densidade calórica de cada um dos
itens alimentares oferecidos aos animais cafeteria foram obtidas da tabela de
informações nutricionais presente no rótulo dos alimentos (Tabela I). A distribuição
dos macronutrientes e a densidade calórica da ração comercial Nuvilab® foram
fornecidos pelo produtor (Tabela I).
Todos os experimentos foram realizados em ratos alimentados, pesando
aproximadamente de 230g, entre 8:00 e 10:00 da manhã ao final dos 24 dias de
tratamento.
Todos os procedimentos foram aprovados pelo Comitê de Ética para o Uso de
Animais da UFSJ – CEUA/UFSJ de acordo com o protocolo n° 016/2014.

14
Tabela I – Distribuição dos macronutrientes e densidade calórica dos
alimentos oferecidos aos animais alimentados com a dieta cafeteria
Carboidrato Lipídios Proteínas Densidade
Calórica
(g/100g) (g/100g) (g/100g) (kcal/g)
Bala de Caramelo 72,70 9,09 18,18 4,45
Bacon 00,00 58,0 9,00 5,69
Batata Frita 45,00 35,00 5,00 5,15
Bis® 63,13 23,3 8,10 4,95
Bolacha de Maisena 62,5 12,5 7,5 4,02
Castanha-do-Pará 7,00 67,00 17,00 6,99
Chocolate 50,00 33,30 10,00 5,40
Chocooky® 63,33 23,33 3,33 5,00
Pé de Moleque 58,82 26,47 11,76 5,29
Snack® 65,00 17,50 7,50 4,50
Torrada 73,33 6,67 13,33 4,00
Torrone 80,00 7,50 10,00 3,80
Ração Nuvilab® 55,00 4,50 22,00 2,85
Os ratos do grupo cafeteria receberam água ad libitum acrescida de sacarose 20%
3.2 Histologia
Os ratos foram eutanasiados por guilhotina e foram retiradas amostras com
aproximadamente 3mm de espessura da porção medial do maior lóbulo do fígado.
3.2.1 Coloração com Hematoxilina e Eosina
Os fragmentos do tecido hepático foram fixados no líquido Bouin por 24h, e
passaram pelas etapas de desidratação em série crescente de álcoois (70º, 80º, 90º
e 100º), diafanizada com xilóis e incluídos em parafina, seccionados com 5 μm de

15
espessura e corados com hematoxilina e eosina. A área dos hepatócitos (μm2) foram
obtidos utilizando microscópio de luz acoplado ao software Motic Imagens Plus 2.0.
Foram utilizados 6 ratos para cada grupo e 10 fotos foram retiradas em campos
aleatórios para cada animal. Em cada foto foram mensuradas as áreas de 10
hepatócitos, totalizando 600 células por grupo. Apenas hepatócitos que tiveram o
núcleo seccionado foram mensurados.
3.2.2 Coloração com Oil Red
A visualização e a quantificação de lipídios neutros foram realizadas através
da coloração com Oil Red O. As amostras foram congeladas em nitrogênio líquido e
posteriormente embebidas em Tissue-Tek® OCT e fatias com 20 μm de espessura
foram obtidas com auxílio do criostato. Foram confeccionadas 3 lâminas do tecido
hepático de cada rato de diferentes profundidades do tecido, sendo 6 ratos por
grupo. Os cortes foram lavados com paraformaldeído 10%, isopropanol 60% e
coradas durante 12mim na solução de Oil Red O (0,4% em isopropanol, seguida de
uma diluição para 0,24% em água destilada e uma filtração). As lâminas foram
lavadas mais duas vezes, uma em isopropanol 60% e a outra em água destilada.
Por último, as lâminas foram mergulhadas na hematoxilina de Mayer e novamente
lavadas em água destilada. A quantificação do acúmulo de lipídios foi realizada
através da mensuração da intensidade das áreas em vermelho através do software
Image J (MEHLEM et al., 2013).
3.3 Determinação do conteúdo lipídico do fígado
A extração dos lipídios foi realizada pelo método de FOLCH et al. (1957). O
fígado (1g) foi homogeneizado em 20 mL de clorofórmio:metanol (2:1). Em seguida,
o material foi filtrado utilizando papel Whatman nº1 e ao filtrado foi adicionado água
deionizada. Após separação da fase clorofórmica e aquosa, uma alíquota da fase
clorofórmica foi transferida para tubos previamente pesados para evaporação à
50ºC. A quantidade da gordura foi obtida pelo método gravimétrico. Os lipídios de
cada tubo foram solubilizados com 1mL de álcool isopropílico e as concentrações de
triacilglicerol e colesterol foram determinados utilizando kits comerciais (Bioclin®,
Belo Horizonte, MG).

16
3.4 Avaliação da secreção de VLDL-TAG pelo fígado
Para avaliar a secreção de VLDL-TAG, os animais foram deixados em jejum
por 12 horas e após esse período, a concentração plasmática de TAG foi
determinada antes e 2 horas após administração por via intravenosa (veia jugular
sob anestesia) de uma solução a 20% (v/v) de tyloxapol (Triton WR – 1339, Sigma-
Aldrich, USA) em salina na dose de 400 mg/kg de peso. A velocidade de secreção
de VLDL-TAG foi estimada pela diferença entre as concentrações plasmática de
TAG antes e após administração de tyloxapol, um inibidor da ação da lipase
lipoprotéica (BORENSZTAJN et al., 1976). As concentrações de TAG foram
determinadas por kit enzimático (Bioclin®, Belo Horizonte, MG).
3.5 Reação em cadeia da polimerase – transcrição reversa em tempo real (Real
Time-PCR)
Para determinação da expressão gênica das proteínas envolvidas no
metabolismo de lipídios, foi feita a extração de RNA e análise por RT-PCR em
Tempo Real. RNA total foi extraído de aproximadamente 50 mg de tecido congelado
em nitrogênio líquido com o reagente Trizol (Gibco-Life Technology). A integridade
do RNA foi confirmada por eletroforese em gel de agarose a 1,2% com tampão Tris-
borato 89 mM, EDTA 2 mM, e revelado com brometo de etídio (Imagem 1).
Imagem 1 – Eletroforese em gel de agarose corado com brometo de etídeo.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
28S rRNA18S rRNA
Nota: O RNA total do tecido hepático dos ratos controles estão apresentados nas
canaletas ímpares e dos ratos alimentados com a dieta cafeteria nas canaletas
pares.
A pureza e quantificação foram determinadas em espectrofotômetro
NanoVue™ Plus Spectrophotometers, (GE Healthcare, Little Chalfont,
Buckinghamshire UK). Para verificação de contaminação com DNA genômico, foi
realizado PCR simples com 1 μg de RNA total, o que não resultou em nenhum

17
produto de PCR. O cDNA foi obtido a partir de 2 μg de RNA total por transcrição
reversa usando kit da Applied biosystems (High-Capacity cDNA reverse transcription
kit) com amplímetros aleatórios (10X RT Random Primers), dNTPs 100 mM e a
transcriptase reversa (MultiScribeTM Reverse Transcriptase) em volume final de 20
μl, sendo a incubação realizada a 25°C por 10 min., seguidos de 120 min. a 37°C e 5
min. a 85ºC, conforme descrito pelo fabricante. As expressões gênicas no tecido
hepático foram determinadas por Real time PCR, sendo utilizados SYBR Green PCR
Master Mix (Applied Biosystems, Foster City, CA), sistema de detecção 7500 Fast
Real time PCR System (Applied Biosystems, Foster City, CA) e pares específicos de
primers (senso e antisenso) mostrados a baixo (Tabela II). A quantificação foi feita
pela medida do ciclo threshold normalizado pelo controle interno β-actina e expressa
em relação aos ratos controles.
Tabela II – Primers para PCR em tempo real.
GENE PRIMER
Beta actina Senso 5’-CATGAAGATCAAGATCATTGCTCCT-3’
Antisenso 5’-CTGCTTGCTGATCCACATCTG-3’
ACO Senso 5’-GGCCGCTATGATGGAAATGTG-3’
Antisenso 5’-GGGCTTCAAGTGCTTGTGGTAA-3’
Apo B 100 Senso 5’-CATGCTCCATTCTCACATGTTTA-3’
Antisenso 5’-AGAGAACAAAGCAGAGATTGTGG-3’
ATGL Senso 5’- CTGTGGCCTCATTCCTCCTACC-3’
Antisenso 5’- GTGGCAAGTTGTCTGAAATGCCG-3’
ChREBP Senso 5’-AAAGGCCTCAAGTTGCTATG-3’
Antisenso 5’-AGACAACAGCCTCAGGTTTC-3’
CPT 1 Senso 5’-ACGTGAGTGACTGGTGGGAAGAAT-3’
Antisenso 5’-TCTCCATGGCGTAGTAGTTGCTGT-3’
LHS Senso 5’-CAGGAGTGCTCTTCTTTGAG-3’
Antisenso 5’-CAGCCTTTATGTAGCGTGAC-3’
MTP Senso 5’-AACCAGAAATGTCAATGGCTAGA-3’
Antisenso 5’-AGTGATTTGATGTCCAAAATGCT-3´
PPAR alfa Senso 5’-TACCACTATGGAGTCCACGCATGT-3’
Antisenso 5’-TTGCAGCTTCGATCACACTTGTCG-3’
SREBP 1c Senso 5’-GCCCACAATGCCATTGAGA-3’
Antisenso 5’-GCAGATTTATTCAGCTTTGCCTCA-3’

18
3.6 Determinação dos níveis séricos de glicose, triacilglicerol, colesterol, ácido
úrico, AST e ALT.
As concentrações de glicose, triacilglicerol, colesterol, ácido úrico, AST e ALT
séricos foram determinados utilizando kits comerciais (Bioclin®, Belo Horizonte, MG).
3.7 Análise estatística
Para os dados paramétricos, os resultados foram expressos em média ± erro
padrão e as diferenças entre grupos foram analisadas utilizando o test t de Student.
Para os dados não-paramétricos, os resultados foram expressos em percentis e as
diferenças entre os grupos analisados utilizando o teste Mann-Whitney Rank Sum.
Em todos os casos foi adotado P < 0,05 para nível de significância.

19
0 5 10 15 20 25
0
30
60
90
120
150
180
210
240
Pe
so (
g)
Tempo (dias)
Controle Cafeteria
4. RESULTADOS
4.1 Parâmetros Gerais
4.1.1 Ganho de peso corporal dos ratos
O peso corporal dos ratos foi avaliado a cada dois dias (Gráfico 1). O ganho
de peso dos ratos alimentados com a dieta cafeteria foi similar ao dos ratos controle
durante todo o período experimental.
Gráfico 1 – Efeito da dieta cafeteria no peso corporal (g) de ratos Wistar. Os valores
representam a média ± erro padrão de 8 animais. Este experimento foi repetido 3
vezes.

20
4.1.2 Peso dos tecidos
Os ratos alimentados com a dieta cafeteria apresentaram um aumento de
aproximadamente 68% e 225% no peso do TAB epididimal e retroperitoneal,
respectivamente. A dieta cafeteria não afetou o peso do fígado e coração (Tabela III
e Gráfico 2).
Tabela III – Peso relativo dos tecidos adiposos brancos (TAB) epididimal e
retroperitoneal, fígado e coração de ratos alimentados com as dietas controle e
cafeteria.
Controle Cafeteria
TAB epididimal (g/100g) 0,60 ± 0,04 (8) 1,01 ± 0,09* (8)
TAB retroperitoneal (g/100g) 0,27 ± 0,03 (8) 0,88 ± 0,10* (7)
Fígado (g/100g) 4,75 ± 0,12 (8) 4,50 ± 0,08 (8)
Coração (g/100g) 0,47 ± 0,01 (8) 0,46 ± 0,01 (8)
Os valores representam a média ± erro padrão. O valor entre parênteses representa
o número de ratos utilizados. *P < 0,05 vs. controle.

21
0,0
0,5
1,0
5
6
0,0
0,5
1,0
5
6
**
Coração FígadoRim DRim ERetroEpi
Peso
(g
/10
0g
)
Controle Cafeteria
**
Coração FígadoRetroEpi
Peso (
g/1
00g)
Controle Cafeteria
Gráfico 2 – Efeito da dieta cafeteria no peso dos tecidos adiposos branco epididimal
(Epi) e retroperitoneal (Retro), coração e fígado (g/100g) de ratos Wistar. As colunas
e barras representam a média ± erro padrão de 7-8 ratos. *P < 0,05 vs. controle.

22
4.1.3 Parâmetros séricos
A dieta cafeteria não alterou as concentrações séricas de glicose, ácido úrico
e colesterol, mas induziu um aumento significativo (171%) na concentração sérica
de triacilglicerol (Tabela IV e Gráfico 3).
Tabela IV – Concentração sérica de glicose, triacilglicerol, colesterol total e ácido
úrico de ratos alimentados com as dietas controle e cafeteria.
Controle Cafeteria
Glicose (mg/dL) 116 ± 5 (8) 113 ± 8 (8)
Triacilglicerol (mg/dL) 42 ± 4 (8) 114 ± 10* (7)
Colesterol (mg/dL) 68 ± 10 (8) 81 ± 14 (7)
Ácido Úrico (mg/dL) 1,74 ± 0,06 (8) 1,76 ± 0,08 (8)
Os valores representam a média ± erro padrão. O valor entre parênteses representa
o número de ratos utilizados. *P < 0,05 vs. controle.

23
0,0
0,5
1,0
1,5
40
60
80
100
120
Ácido úricoColesterolTriacilglicerolGlicose
*
Concentr
ações s
éricas (
mg/d
L)
Controle Cafeteria
Gráfico 3 – Efeito da dieta cafeteria nas concentrações séricas de glicose,
triacilglicerol, colesterol e ácido úrico de ratos Wistar. As colunas e barras
representam a média ± erro padrão de 7-8 ratos. *P < 0,05 vs. controle.

24
4.2 Parâmetros hepáticos
4.2.1 Histologia
A análise histológica do tecido hepático revelou que o grupo alimentado com
a dieta cafeteria apresentou um aumento na mediana de aproximadamente 36% na
área dos hepatócitos em relação ao grupo alimentado com a dieta controle (Tabela
V, Gráfico 4). O citoplasma dos hepatócitos dos ratos do grupo cafeteria apresentou
um aspecto vacuolar com o núcleo centralizado, sugerindo um acúmulo lipídico
microvesicular (Prancha 1).
A técnica de coloração com Oil Red O (ORO) permitiu verificar que os ratos
alimentados com a dieta cafeteria apresentaram um aumento na concentração de
lipídios neutros no tecido hepático na mediana de aproximadamente 260 vezes em
relação aos ratos alimentados com dieta controle (Tabela VI; Gráfico 5; Prancha 1).
Tabela V – Área dos hepatócitos (µm2) de ratos alimentados com as dietas controle
e cafeteria.
Controle Cafeteria
Mínimo 5,6 9,7
Percentil 25 12,3 17,6
Percentil 50 14,7 20,1*
Percentil 75 17,4 24,1
Máximo 35,8 44,7
Os valores representam a distribuição em percentis (P) da área de 600 hepatócitos.
*P<0,05 vs. controle.

25
0
10
20
30
40
50
60
CafeteriaControle
Áre
a d
os h
ep
ató
cito
s (m
2)
Gráfico 4 - Efeito da dieta cafeteria na área dos hepatócitos. As colunas
representam os percentis 25, 50 e 75 e as barras representam os valores mínimos e
máximos de 600 hepatócitos. O símbolo representa a média.

26
Tabela VI – Lipídios neutros (unidades arbitrárias) no tecido hepático de ratos
alimentados com as dietas controle e cafeteria.
Controle Cafeteria
Mínimo 0,049 0,023
Percentil 25 0,108 0,209
Percentil 50 0,190 49,156*
Percentil 75 0,427 95,270
Máximo 0,906 239,425
Os valores representam a distribuição em percentis dos lipídios neutros de 18
lâminas do tecido hepático provenientes de 6 ratos. *P<0,05 vs. controle.

27
0,0
0,2
0,4
0,6
0,8
50
100
150
200
250
300
CafeteriaControle
Lip
ídio
s n
eu
tro
s (
un
ida
des a
rbitrá
ria
s)
Gráfico 5 - Efeito da dieta cafeteria na concentração de lipídios neutros no tecido
hepático. As colunas representam os percentis 25, 50 e 75 e as barras representam
os valores mínimos e máximos de 18 lâminas proveniente de 6 ratos. O símbolo
representa a média.

28
Prancha 1 - Efeito da dieta cafeteria no parênquima do tecido hepático. A coloração
com hematoxilina e eosina do tecido hepático dos ratos alimentados com a dieta
controle (A, C) e cafeteria (B, D) estão apresentadas no aumento de 10x (A, B) e
40x (C, D). A coloração com Oil Red O do tecido hepático dos ratos alimentados
com a dieta controle (E) e cafeteria (F) está apresentada no aumento de 10x. As
barras representam 100 µm (A,B,E,F) e 25 µm (C,D).

29
4.2.2 Concentração de lipídios hepáticos
Os ratos alimentados com a dieta cafeteria apresentaram um aumento de
aproximadamente 25% na concentração hepática de lipídios totais, caracterizando
um quadro de esteatose (Gráfico 6). No grupo controle as concentrações de
triacilglicerol e colesterol hepático representaram, aproximadamente, 10% e 4% da
concentração de lipídios totais. A dieta cafeteria induziu um aumento de
aproximadamente 120% e 30% na concentração hepática de triacilglicerol e
colesterol, respectivamente (Tabela VII; Gráfico 7).
Tabela VII - Concentração hepática de lipídios totais, triacilglicerol e colesterol total
de ratos alimentados com as dietas controle e cafeteria.
Controle Cafeteria
Lipídios totais (mg/g) 44,00 ± 1,30 (8) 55,00 ± 3,50* (8)
Triacilglicerol (mg/g) 4,50 ± 0,27 (8) 9,90 ± 0,94* (7)
Colesterol (mg/g) 1,90 ± 0,04 (8) 2,45 ± 0,20* (8)
Os valores representam a média ± erro padrão. O valor entre parênteses representa
o número de ratos utilizados. *P < 0,05 vs. controle.

30
0
20
40
60
*
Lip
ídio
s h
epáticos t
ota
is (
mg/g
)
Controle Cafeteria
Gráfico 6 – Efeito da dieta cafeteria na concentração hepática dos lipídios totais de
ratos Wistar. As colunas e barras representam a média ± erro padrão de 8 ratos. *P
< 0,05 vs. controle.

31
0
2
4
6
8
10
12 Controle Cafeteria
*
*
ColesterolTriacilglicerol
Lip
ídio
s h
ep
áticos (
mg
/g)
Gráfico 7 – Efeito da dieta cafeteria na concentração hepática de triacilglicerol e
colesterol de ratos Wistar. As colunas e barras representam a média ± erro padrão
de 8 ratos. *P < 0,05 vs. controle.

32
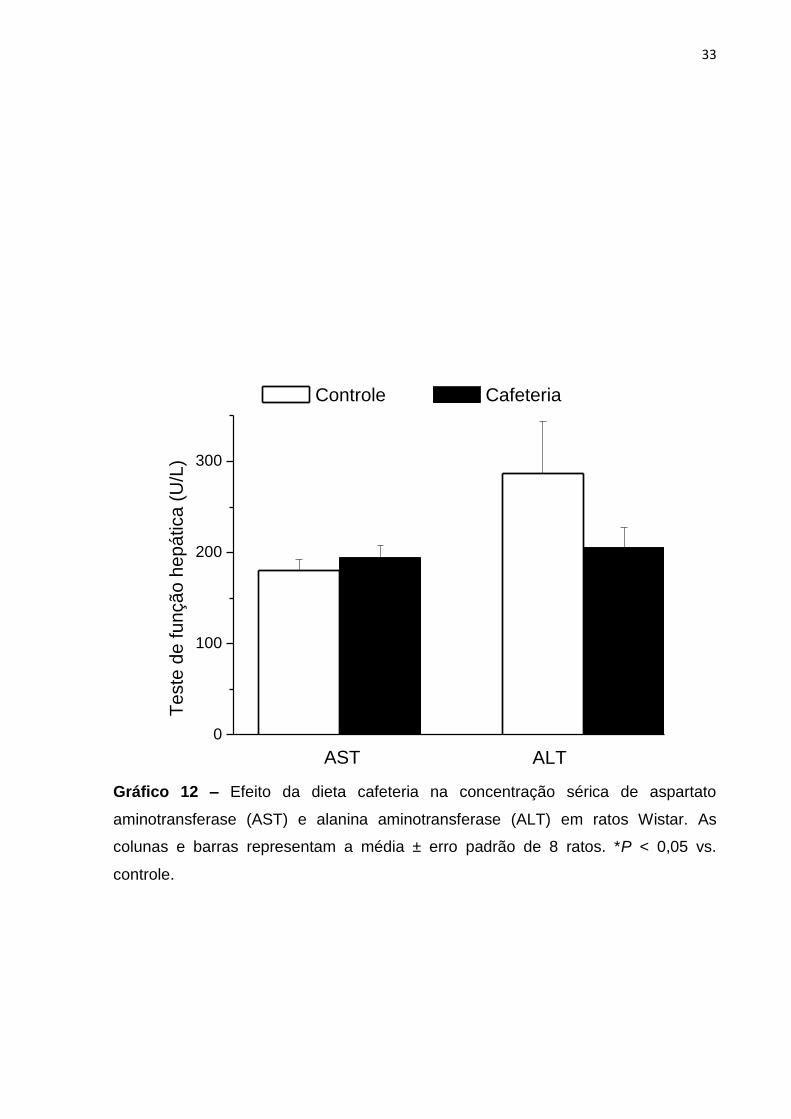
4.2.3 Prova de função hepática
A função hepática foi determinada através da análise sérica das enzimas
aspartato aminotransferase (AST) e alanina aminotransferase (ALT). Como já
evidenciado, os ratos alimentados com a dieta cafeteria apresentaram um percentual
lípidico maior do que o grupo alimentado com a dieta controle. Contudo, esse
acúmulo de lipídios foi insuficiente para causar alterações séricas das enzimas
analisadas, não evidenciando um quadro de lesão hepática (esteatohepatite)
(Tabela VIII; Gráfico 8).
Tabela VIII. Concentrações séricas de aspartato aminotransferase (AST) e alanina
aminotransferase (ALT) nos ratos alimentados com a dieta controle e cafeteria.
Controle Cafeteria
AST (U/L) 179,90 ± 12,23 (8) 194,10 ± 13,89 (8)
ALT (U/L) 286,50 ± 53,81 (7) 205,40 ± 22,05 (7)
Os valores representam a média ± erro padrão. O valor entre parênteses representa
o número de ratos utilizados. *P < 0,05 vs. controle.
33
0
100
200
300
Controle Cafeteria
AST ALT
Teste
de f
unção h
epática (
U/L
)
Gráfico 12 – Efeito da dieta cafeteria na concentração sérica de aspartato
aminotransferase (AST) e alanina aminotransferase (ALT) em ratos Wistar. As
colunas e barras representam a média ± erro padrão de 8 ratos. *P < 0,05 vs.
controle.

34
4.2.4 Expressão gênica no fígado
4.2.4.1 Expressão gênica de fatores de transcrição relacionados com a
regulação da expressão gênica de enzimas envolvidas no metabolismo de
lipídios
Como observado no Gráfico 9 e na Tabela IX a dieta cafeteria não foi capaz
de alterar a expressão do RNAm dos fatores de transcrição ChREBP e SREBP1c no
tecido hepático, quando comparado aos controle.
Tabela IX – Expressão de genes envolvidos no metabolismo de lipídios no tecido
hepático de ratos alimentados com as dietas controle e cafeteria. Valores arbitrários
de RNAm.
Gene Controle Cafeteria
ACO 1,02 ± 0,09 0,89 ± 0,06
ApoB 1,01 ± 0,06 0,74 ± 0,06*
ATGL 1,02 ± 0,09 1,24 ± 0,09
ChREBP 1,02 ± 0,07 0,86 ± 0,08
CPT1 1,01 ± 0,07 2,32 ± 0,33*
LHS 1,01 ± 0,07 0,83 ± 0,10
MTP 1,03 ± 0,10 1,34 ± 0,10*
PPAR-alfa 1,03 ± 0,10 1,36 ± 0,19
SREBP-1c 1,00 ± 0,16 1,09 ± 0,12
Todos os valores foram normalizados pela expressão do gene da β-actina e estão
expressos em unidades arbitrárias. Os valores representam a média ± erro padrão
de 8 ratos. *P < 0,05 vs. controle.

35
0,0
0,3
0,6
0,9
1,2
1,5
SREBP-1cChREBP
Ge
ne
s/
-actin
a (
un
idad
es a
rbitrá
ria
s)
Controle Cafeteria
Gráfico 9 - Efeito da dieta cafeteria na expressão do RNAm da proteína de ligação
ao elemento de resposta ao carboidrato (ChREBP) e da proteína de ligação ao
elemento regulatório de esterol 1c (SREBP-1c) no tecido hepático. As colunas e
barras representam a média ± erro padrão de 8 ratos. *P < 0,05 vs. controle.

36
0,0
0,5
1,0
1,5
2,0
2,5
3,0
*
Genes/
-actina (
Unid
ades a
rbitrá
rias)
PPAR-alfaCPT1ACO
Controle Cafeteria
4.2.4.2 Expressão dos genes relacionados com a oxidação lipídica
Em relação aos genes envolvidos na oxidação lipídica, não houve alterações
no RNAm da ACO e PPAR-α no tecido hepático dos ratos alimentados com a dieta
cafeteria quando comparado ao controle. Contudo, apesar da expressão de RNAm
do PPAR-α não estar alterada, a expressão de CPT1, seu gene alvo, está
aumentada em aproximadamente 122% no tecido hepático de ratos alimentados
com a dieta cafeteria quando comparado ao controle, sem alterar a expressão de
ACO, outro gene alvo do PPAR-α (Gráfico 10, Tabela IX).
Gráfico 10 - Efeito da dieta cafeteria na expressão de RNAm da acil CoA oxidase
(ACO), carnitina palmitoiltransferase 1 (CPT1) e peroxisome proliferator-activated
receptors alpha (PPAR-α) no tecido hepático. As colunas e barras representam a
média ± erro padrão de 8 ratos. *P < 0,05 vs. controle.

37
0,0
0,3
0,6
0,9
1,2
1,5
LHSATGL
Ge
ne
s/
-actin
a (
un
idad
es a
rbitrá
ria
s)
Controle Cafeteria
4.2.4.3 Expressão dos genes relacionados com a mobilização de lipídios no
fígado
O Gráfico 11 e a Tabela IX mostram que tanto os ratos alimentados com a
dieta cafeteria quanto a dieta controle não apresentaram alterações na expressão do
RNAm da ATGL e LHS, duas enzimas responsáveis pela hidrólise do triacilglicerol,
no tecido hepático quando comparado ao controle.
Gráfico 11 - Efeito da dieta cafeteria na expressão do RNAm da lipase do
triacilglicerol do adipócito (ATGL) e lipase hormônio sensível (LHS) no tecido
hepático. As colunas e barras representam a média ± erro padrão de 8 ratos. *P <
0,05 vs. controle.

38
0,0
0,3
0,6
0,9
1,2
1,5
1,8
MTPApoB
*
*
Genes/
-actina (
unid
ades a
rbitrá
rias)
Controle Cafeteria
4.2.4.4 Expressão dos genes relacionados com a secreção de VLDL
Os ratos alimentados com a dieta cafeteria apresentaram uma redução de
aproximadamente 36% na expressão de ApoB no tecido hepático quando
comparado ao controle. Para o RNAm da MTP, proteína responsável pela lipidação
de ApoB, houve um aumento de 31% no tecido hepático dos ratos alimentados com
a dieta cafeteria quando comparados com os ratos da dieta controle (Gráfico 12,
Tabela IX).
Gráfico 12 - Efeito da dieta cafeteria na expressão de RNAm da apolipoproteína B
(ApoB) e proteína de transferência microssomal de triacilglicerol (MTP) no tecido
hepático. As colunas e barras representam a média ± erro padrão de 8 ratos. *P <
0,05 vs. controle.

39
4.2.5 Velocidade de secreção de VLDL-TAG
A secreção hepática de VLDL-TAG foi verificada pela determinação da
concentração sérica de TAG após administração de tyloxapol (Triton WR – 1339). A
secreção de VLDL-TAG nos ratos alimentados com a dieta cafeteria foi 22% maior
quando comparado com os animais alimentados com a dieta controle (Tabela X,
Gráfico 13).
Tabela X – Velocidade de secreção de VLDL-TAG hepática de ratos alimentados
com as dietas controles e cafeteria.
Controle Cafeteria
VLDL-TAG (mg.dL-1.h-1) 278,8 ± 21,72 (8) 360,1 ±14,8 (7)*
Os valores representam a média ± erro padrão. O valor entre parênteses representa
o número de ratos utilizados. *P < 0,05 vs. controle.

40
0
70
140
210
280
350
420
Controle Cafeteria
*
Se
cre
çã
o d
e V
LD
L-T
AG
(m
g.d
L-1.h
-1)
Gráfico 13 – Efeito da dieta cafeteria na velocidade de secreção de VLDL-TAG
(mg.dL-1.h-1) pelo tecido hepático. As colunas e barras representam a média ± erro
padrão de 7-8 ratos. *P < 0,05 vs. controle.

41
5. DISCUSSÃO
O presente estudo avaliou os efeitos da dieta cafeteria no metabolismo lipídico
sérico e hepático de ratos Wistar machos após 24 dias de tratamento. Parâmetros
metabólicos decorrentes dessa dieta hipercalórica foram analisados, sendo
demostrantrado alterações na adiposidade, nos lipídios séricos e hepáticos, na
expressão dos genes CPT-1, MTP e ApoB 100 e na secreção de VLDL.
De fato, como observado por outros autores, a oferta de ração comercial
suplementada com itens de alta densidade calórica e sacarose na água de beber
(dieta cafeteria) a ratos recém-desmamados durante 24 dias promove alteração na
composição corporal sem afetar o peso corporal final (CHAVES et al., 2006). No
presente estudo, os ratos alimentados com a dieta cafeteria apresentaram um
aumento no peso dos tecidos adiposos epididimal e retroperitoneal de 68% e 225%,
respectivamente, em comparação ao grupo alimentado com a dieta controle,
estando de acordo outros estudos. A oferta de uma dieta hipercalórica na forma de
pellet durante 12 semanas a camundongos com idade inicial de 10 semanas não
altera o peso corporal final apesar de induzir um aumento no peso do tecido adiposo
retroperitoneal (126%) e epididimal (56%) (HIGA et al., 2014). Os ratos alimentados
com a dieta cafeteria apresentam um quadro de hiperinsulinemia, um aumento na
síntese de ácidos graxos de novo e um aumento (24%) na atividade da LPL no
tecido adiposo (CHAVES et al., 2006). O aumento da insulina plasmática pós-
prandial é um importante estimulador da lipogênese de novo e a atividade da LPL no
tecido adiposo. A LPL é uma lipase encontrada no endotélio do tecido adiposo,
capaz de hidrolisar o TAG circulante, permitindo a captação dos ácidos graxos pelos
adipócitos, que por sua vez serão reesterificados a TAG formando as gotas lipídicas
(PICARD et al., 1999). O outro fator observado nos ratos alimentados com a dieta
cafeteria é um aumento na geração de G3P através da via glicolítica, uma vez que
esses animais apresentam um aumento de 50% na captação na glicose sérica por
esses tecidos em comparação aos ratos alimentados com a dieta controle (CHAVES
et al., 2006). Desta forma, possivelmente, o acúmulo de lipídios nos tecido adiposo
retroperitoneal e epididimal nos ratos alimentados com a dieta cafeteria seja devido
a uma maior biossíntese de ácidos graxos e uma maior captação de ácidos graxos
das lipoproteínas circulantes pela LPL, ambos estimulados pela insulina.

42
Apesar da dieta cafeteria apresentar um acréscimo de 20% de sacarose na
água de beber, os ratos alimentados com essa dieta apresentaram um quadro de
normoglicemia. Este dado esta de acordo com nosso estudo prévio (CHAVES et al.,
2006). Os ratos alimentados com a dieta cafeteria além da maior captação de
glicose plasmática pelo tecido adiposo branco e marrom (CHAVES et al., 2006;
CHAVES et al., 2008), apresentam um aumento no fluxo glicolítico acompanhado de
uma redução na gliconeogênese, avaliada pela incorporação de piruvato em glicose
e pela atividade da PEPCK, em fatias de fígado (MARTINS-SANTOS, 2008). Todos
esses dados indicam que a normoglicemia pode ser devido ao fato da sensibilidade
tecidual à insulina estar preservada nos ratos alimentados com a dieta cafeteria.
A frutose contida nos alimentos e na água de beber (20% de sacarose) dos
ratos alimentados com a dieta cafeteria não alterou o ácido úrico sérico, apesar de
alguns estudos sugerirem que o consumo de frutose aumenta os níveis séricos e
citológicos de ácido úrico devido a maior fosforilação da frutose e consequentemente
maior formação de AMP, um subproduto para síntese de ácido úrico
(PERHEENTUPA et al., 1967; van dem BERGHE et al., 1977). No entanto, outros
estudos contradizem o papel da frutose dietética impactando na concentração de
ácido úrico. Um estudo realizado com 366 homens de 20 a 60 anos divididos em 4
grupos submetidos a dietas com diferentes fontes de açúcar no leite (grupo I- 9%
das calorias proveniente da frutose, grupo II- 18% de xarope de milho, grupo III- 18%
de sacarose e grupo IV- 9% de glicose), não apresentaram diferenças na
concentração de ácido úrico depois de 10 semanas de dieta (ANGELOPOULOS et
al., 2015). Em outro estudo, ratos Sprague-Dawley machos inicialmente com 6
semanas de vida alimentados com ração comercial e água acrescida com 10% de
frutose durante 3 meses, não apresentam aumento no ácido úrico sérico em
comparação aos ratos controle (CITIL et al., 2014). Estudos mostram que doses
elevadas de frutose na dieta contribuem para um aumento de ácido úrico na urina,
contudo, não se sabe ao certo a relação entre a frutose dietética e a concentração
de ácido úrico sérico (MOORE et al., 2014). No entanto, um trabalho de meta-
análise com 425 indivíduos diabéticos e não diabéticos mostrou que a substituição
isocalórica de amido, glicose ou sacarose por frutose, não induziu alteração no ácido
úrico sérico em ambos os indivíduos. Em contrapartida, em condições de dieta
hipercalórica, a dieta controle acrescida com excesso de energia (>30% de energia
em excesso ou >200g/dia de frutose) a partir da frutose, aumenta de forma

43
significante a concentração sérica de ácido úrico em comparação a dieta controle
nos indivíduos não diabéticos (WANG et al., 2012). A medida que a frutose fornece
substrato para a lipogênese de novo, ela fornece substrato também para a produção
de ácido úrico. (MOORE et al., 2014). Desta forma, a frutose da dieta cafeteria está
fornecendo substrato para a produção de TAG sem causar alterações nos níveis
plasmáticos do ácido úrico.
No presente estudo, a dieta cafeteria não induziu alteração no colesterol total
sérico em comparação ao grupo que recebeu a dieta controle. Ratos Wistar machos,
com idade inicial de 10 semanas, alimentados com ração comercial suplementada
com patê de fígado de galinha, bolacha, batata chips, leite achocolatado e bacon
durante 56 dias, caracterizando uma dieta cafeteria, não apresentam diferença no
colesterol total sérico quando comparado ao grupo alimentado com a ração
comercial (ANDRADE et al., 2013). No entanto, alguns trabalhos mostram que ratos
Wistar com idade inicial de 4 a 6 semanas alimentados com uma dieta do tipo
cafeteria por 6 semanas apresentam um aumento no colesterol total sérico de
aproximadamente 25% em comparação ao grupo alimentado com a dieta controle
(KUMAR et al., 2011; SHIVAPRASAD et al., 2014). Essa diferença nos resultados
pode estar relacionado ao tipo e ao tempo de tratamento, pois, apesar de todos
esses artigos estudarem os efeitos da dieta cafeteria, a composição, a ingestão
calórica e a contribuição dos grupos alimentares para esse perfil energético é
diferente da dieta cafeteria do presente estudo, além do tempo de experimentação
também ser diferente. Por outro lado, o consumo de uma dieta com elevados teores
de colesterol não resulta obrigatoriamente em elevados níveis de colesterol sérico,
devido a diversos mecanismos adaptativos como o aumento da degradação ou
transformação desse colesterol dietético em sais biliais (BOONE et al., 2011). Outro
ponto importante para a não co-existência da relação citada acima é a premissa de
que diversos tipos de animais apresentam sensibilidade ou resistência ao colesterol
dietético (BOONE et al., 2011). Coelho, hamster e camundongos C57BL/6 não são
resistentes ao colesterol dietético, podendo apresentar alterações séricas quando
submetidos a uma dieta rica em colesterol, por outro lado, ratos Sprague-Dawley,
Wistar ou Fischer apresentam pequena ou nenhuma alteração plasmática no
colesterol total se submetidos a dietas rica em colesterol, devido ao efeito protetor
que HMG-CoA redutase hepática garante (NESS; GERTZ, 2004).

44
Em nosso estudo, a dieta cafeteria induziu um aumento de aproximadamente
170% na concentração sérica de TAG em relação ao grupo controle. Considerando
que a dieta cafeteria aumentou (24%) a atividade da LPL no tecido adiposo
retroperitoneal (CHAVES et al., 2006) e a secreção de VLDL-TAG na mesma
proporção, talvez o alto conteúdo de triacilglicerol sérico nos ratos alimentados com
a dieta cafeteria seja decorrente do maior consumo de lipídios dietéticos e uma
menor excreção desses lipídios nas fezes. Ratas Wistar, inicialmente com 45 dias de
vida, submetidas à dieta cafeteria, por 42 dias, contendo itens de alta densidade
calórica (leite condensado, pão, chocolate, coco seco, biscoito, queijo, batatas)
durante 6 semanas apresentam um aumento de 322% na concentração de
triacilglicerol sérico em relação ao grupo alimentado com a dieta controle
(SHIVAPRASAD et al., 2014). Ainda nesse estudo, analisando as fezes desses
animais durante 24h, as ratas alimentadas com a dieta cafeteria apresentam uma
redução drástica de TAG em suas fezes em comparação as ratas alimentadas com a
dieta controle. Desta forma, é possível que a dieta cafeteria tenha aumentado a
absorção de gordura intestinal.
A dieta cafeteria induziu um aumento de 25% no conteúdo de lipídios totais no
tecido hepático avaliado pelo método gravimétrico. Desses lipídios hepáticos, a dieta
cafeteria induziu um aumento de 120% e 30% na concentração de TAG e colesterol
respectivamente, em comparação aos ratos da dieta controle, que apresentaram um
percentual de 10% na concentração de TAG e 4% na concentração de colesterol
dos lipídios totais. Corroborando tais resultados, a coloração com óleo vermelho
(ORO), que é capaz de estimar o teor e a distribuição tecidual de lipídios neutros
(MEHLEN et al., 2013), detectou um aumento na mediana de 260 vezes no teor de
lipídios neutros nos ratos alimentados com a dieta cafeteria em comparação aos
ratos alimentados com a dieta controle. O menor limiar sugerido para esteatose tem
sido 5% de hepatócitos contendo gotas lipídicas (PASCHOS; PALETAS, 2009),
determinado após coloração com hematoxilina-eosina (KLEINER et al., 2005;
HÜBSCHER, 2006). Em humanos através de ressonância magnética nuclear o limiar
de 5% de conteúdo lipídico intra-hepático também tem sido usado como parâmetro
para a classificação da DHGNA (PERSEGHIN et al., 2006). Desta forma, os ratos
alimentados com a dieta cafeteria apresentaram um quadro de DHGNA. Através da
coloração com hematoxilina e eosina, foi possível visualizar que os ratos
alimentados com a dieta cafeteria apresentaram esteatose simples microvesicular,

45
vacuolização e um inchaço nos hepatócitos provavelmente causado pela presença
de TAG em comparação aos ratos alimentados com a dieta controle. Nos ratos
alimentados com a dieta cafeteria apesar de caracterizado um perfil de esteatose
hepática, esse acúmulo de lipídios provavelmente não foi suficiente para causar uma
lesão hepática, pois esse grupo não apresentou diferença na concentração sérica
para as enzimas AST e ALT. Ratos Sprague Dawley com idade inicial de 5-6
semanas, quando submetidos a uma alimentação contento 10% de frutose na água
de beber por 28 dias, apresentam um quadro de esteatose hepática microvesicular
sem apresentar alterações nas enzimas séricas AST e ALT (LI et al., 2015). Os
valores séricos de AST e ALT tem sido usado como um marcador indireto de lesão
hepática (KARMEN et al., 1954), contudo, muitas vezes, os valores dessas enzimas
não correlacionam com a gravidade das lesões (MOFRAD et al., 2002).
A dieta cafeteria induziu um aumento de aproximadamente 30% no colesterol
hepático comparado à dieta controle. O fígado de ratos Sprague Dawley perfundidos
com uma solução contendo somente albumina ou com diferentes ácidos graxos
(palmítico, oleico e linoleico) ligados à albumina apresentam um aumento na
atividade da enzima HMG-CoA redutase, enzima chave para a biossíntese de
colesterol (GOH; HEIMBERG, 1976). A dieta cafeteria induziu um aumento de 30%
na atividade das hidrolases de TAG citosólicas no tecido hepático (MOREIRA;
BOTION, dados não publicados). Desta forma, a ação das hidrolases de TAG pode
estar fornecendo os ácidos graxos necessários para ativação da HMG-CoA redutase
e consequentemente contribuindo para o aumento do colesterol hepático nos ratos
alimentados com a dieta cafeteria em comparação aos ratos alimentados com a
dieta controle.
Estes animais também apresentaram um aumento de 120% na concentração
de TAG no tecido hepático em comparação aos animais alimentados com a dieta
controle. O acúmulo de lipídios intracelulares na DHGNA é resultado de um
desequilíbrio entre: 1) lipogênese de novo, 2) lipólise hepática, 3) oxidação lipídica,
4) a captação de ácidos graxos hepáticos, e 5) exportação de TAG via partículas de
VLDL (REID et al., 2008, NESS; KASER, 2016). Cada um destes processos serão
discutidos separadamente nos itens abaixo.

46
5.1 Lipogênese hepática
A dieta cafeteria induz um aumento (120%) na síntese de ácidos graxos de
novo no tecido hepático (CHAVES et al., 2012). No presente trabalho, a dieta
cafeteria não induziu alterações na expressão hepática do gene dos fatores de
transcrição SREBP-1c e ChREBP. Entretanto, vários trabalhos têm sugerido que a
insulina induz um aumento na expressão de SREBP-1c (SHIMOMURA et al. 1999,
HORTON et al., 1998, LETEXIER et al., 2005). Ratos Sprague–Dawley diabéticos
por estreptozotocina apresentam uma redução de 80% na expressão do RNAm de
SREBP-1c em comparação ao controle, enquanto a administração de insulina aos
ratos diabéticos restaura quase completamente a expressão do RNAm deste fator
de transcrição no tecido hepático (SHIMOMURA et al., 1999). Em camundongos, o
jejum (1, 3, 6, 9, 12 e 24h) induz uma redução de 60% na expressão hepática do
SREBP-1c quando comparado ao grupo não jejuado alimentado com uma dieta
balanceada (HORTON et al., 1998). O jejum de 48 horas também induz uma
redução na concentração plasmática de insulina e na expressão SREBP-1c no
tecido hepático de ratos Wistar previamente alimentados com uma dieta rica em
carboidratos (70%) ou rica em lipídios saturados (60%) (LETEXIER et al., 2005). A
realimentação com uma dieta hiperglicídica e hipolipídica, por 12 horas, aos
camundongos previamente jejuados por 24 horas, induz um aumento de
aproximadamente 4 vezes na expressão de SREBP-1c no tecido hepático
(HORTON et al., 1998). Mais recentemente foi demonstrado em hepatócitos isolados
que a adição de 10 nM de insulina no meio de incubação por 6 horas aumenta
acentuadamente (25 vezes) a expressão do RNAm deste fator de transcrição (LI et
al., 2010). Este efeito é bloqueado pela presença de wortimanina (inibidor de PI3K)
ou de Akti-1/2 (inibidor de AKT) ou de rapamicina (inibidor de mTORC1) no meio de
incubação (LI et al., 2010), identificando os intermediários da cascata de sinalização
de insulina responsáveis pelo aumento da expressão do gene de SREBP-1c. Por
outro lado, camundongos com atividade constitutiva de mTOR (LTsc1KO) ao serem
alimentados com uma dieta hiperlipídica apresentam, no tecido hepático, uma
redução na expressão do RNAm de SREBP-1c e a jusante da expressão dos genes
da ácido graxo sintase e estearoil-CoA desaturase 1, dois genes envolvidos na
lipogênese, quando comparado ao grupo controle (YECIES et al., 2011). Nos
camundongos com atividade constitutiva de AKT (Pten -/-) não ocorre alteração na
expressão gênica de SREBP-1c (KENERSON et al., 2015). Em camundongos com

47
fenótipo de ambas, mTORC e AKT, constitutivas (Tsc1 -/-; Pten -/-) ocorre um
aumento na expressão de SREBP-1c (KENERSON et al., 2015). Desta forma, são
claros os efeitos da redução plasmática de insulina e da reposição aguda deste
hormônio na regulação da expressão deste fator de transcrição. Por outro lado,
apesar da similaridade na concentração de insulina, foi demonstrado que ratos
Wistar alimentados, durante seis semanas, com uma dieta rica em carboidratos
apresentam aumento na expressão do RNAm de SREBP-1C no tecido hepático
quando comparado aos ratos alimentados com uma dieta rica em lipídios,
(LETEXIER et al., 2005). No presente estudo, os ratos foram tratados com a dieta
cafeteria por 24 dias e apresentam aumento na concentração plasmática de insulina
(CHAVES et al., 2006), sugerindo que a ausência de alteração na expressão de
SREBP-1c pode ser decorrente do tempo de exposição à hiperinsulinemia,
provavelmente devido a contínua ativação de AKT. Corroborando nossa hipótese, foi
recentemente demonstrado que a oferta de uma dieta hiperlipídica (60% das calorias
provenientes de lipídios), durante 18 semanas, a camundongos deficientes em
Crtc2-/- (coativador transcricional 2 regulado por CREB) induz um aumento
significativo na concentração plasmática de insulina e no conteúdo de SREBP-1c
nuclear, sem afetar a transcrição hepática do RNAm de SREBP-1c (HAN et al.,
2015).
Em hepatócitos HepG2, foi demonstrado que a adição de 10 ou 100 nM de
insulina durante 4 horas aumenta a expressão de ChREBP (SIREK et al., 2009).
Contudo, os pesquisadores não encontraram alteração no RNAm de ChREBP
quando estes hepatócitos foram incubados na presença de 1 nM de insulina. Nossos
dados prévios demonstram que em ratos Wistar tratados com a dieta cafeteria a
concentração plasmática de insulina é inferior a 1 nM (CHAVES et al., 2006). Ratos
Wistar alimentados, durante seis semanas, com uma dieta rica em carboidratos ou
rica em lipídios não apresentam alteração no RNAm de ChREBP no tecido hepático
e na concentração de insulina quando ambas são comparadas (LETEXIER et al.,
2005). Contudo, ao serem submetidos ao jejum de 48 horas, apesar de reduzir a
concentração de insulina para valores inferiores a 0,35 nM, também não ocorre
alteração na expressão deste fator de transcrição quando comparado aos ratos
alimentados com cada uma das dietas (LETEXIER et al., 2005). Posteriormente, foi
demonstrado que a realimentação de camundongos ob/ob por 18 horas com uma
dieta rica em carboidratos induziu um aumento de duas vezes na expressão de

48
ChREBP acompanhado de um aumento de duas vezes na concentração plasmática
de insulina, atingindo uma concentração de 2,5 nM (DENTIN et al., 2006). Além do
efeito da insulina, a glicose também pode explicar o aumento da expressão de
ChREBP nestes animais, sendo demonstrado um aumento do RNAm deste fator de
transcrição em hepatócitos isolados incubados na presença de 25 mM de glicose, no
entanto, nenhum efeito é observado na presença de 2,5 ou 5 mM de glicose (IIZUKA
et al., 2009). A realimentação dos camundongos ob/ob por 18 horas com uma dieta
rica em carboidratos também induz um aumento de aproximadamente duas vezes
na concentração plasmática de glicose, atingindo uma concentração de 25 mM
(DENTIN et al., 2006). No entanto, o jejum de 48 horas de ratos Wistar previamente
alimentados com uma dieta rica em carboidratos (70%) ou rica em lipídios saturados
(60%) apesar de reduzir a concentração plasmática de glicose de 8 mM para
aproximadamente 4 mM não altera a expressão de ChREBP no tecido hepático
(LETEXIER et al., 2005). Desta forma, estes estudos sugerem que, alterações na
concentração de insulina e glicose decorrentes do estado nutricional não controlam
a expressão de ChREBP no tecido hepático. No presente estudo, os ratos
alimentados com a dieta cafeteria apresentam 6,4 mM de glicose plasmática e 351
pmol/L de insulina plasmática (CHAVES et al., 2006), explicando a ausência de
efeito da dieta regulando ChREBP hepático.
Vários trabalhos demonstram uma correlação positiva entre a expressão de
SREBP-1c ou ChREBP e a expressão das enzimas lipogênicas, como acetil-CoA
carboxilase e ácido graxo sintase (SHIMOMURA et al., 1999; LETEXIER et al. 2005;
DENTIN et al., 2006; BENHAMED et al., 2012; KENERSON et al., 2015). Contudo,
camundongos deficientes no coativator transcricional 2 regulado pela proteína de
ligação ao elemento de resposta ao AMPc (CREB) apresentam um aumento no
RNAm das enzimas lipogênicas, sem alteração no RNAm de SREBP-1C e ChREBP
(HAN et al., 2015). A deficiência do coativator transcricional 2 regulado por CREB,
no entanto, aumentou o conteúdo da proteína SREBP1 nuclear (HAN et al., 2015),
demonstrando a importância da regulação pós-traducional de SREBP no controle da
lipogênese. Previamente, foi demonstrado que camundongos C57B1/6 nocaute para
ChREBP não apresentam modificação na expressão das enzimas acetil-CoA
carboxilase, acetil-CoA sintetase, estearoil-CoA dessaturase e glicerol-3-fosfato
aciltransferase (CHA; RAPA, 2007). Este fator de transcrição também sofre
modificação pós-traducional (ILZUKA et al., 2009; XU et al., 2013). Apesar de a dieta

49
cafeteria induzir um aumento na síntese de novo de ácidos graxos no tecido
hepático in vivo (CHAVES et al., 2012), no presente trabalho foi demonstrado que
esta dieta não altera a expressão do RNAm de SREBP-1c e ChREBP, sendo
possível um maior conteúdo nuclear destes fatores de transcrição no tecido hepático
dos ratos alimentados com a dieta cafeteria.
5.2 Lipólise hepática
A lipólise fornece ácidos graxos, di e monoglicerídeos; estes substratos são
importantes na produção de energia, assim como na síntese de outros lipídios e/ou
na sinalização celular (ZIMMERMANN et al., 2009). A mobilização de ácidos graxos
de TAG acontece principalmente nos tecidos adiposos e requer enzimas lipolíticas,
sendo que a lipase hormônio sensível (LHS) e a lipase do triacilglicerol do tecido
adiposo (ATGL) correspondem a 90% dessas enzimas nesse tecido (ZIMMERMANN
et al., 2004). Essas enzimas estão também presentes em tecidos não adiposos,
porém parece haver uma diferença na expressão dessas enzimas entre os tecidos.
A análise do RNAm da ATGL por Northen Blot em diferentes tecidos de
camundongos jejuados sugere uma expressão quase imperceptível dessa enzima
no tecido hepático quando comparado ao tecido adiposo, porém o mesmo foi
observado para a β-actina (ZIMMERMANN et al., 2004). No tecido hepático de
camundongos machos C57BL/6J com idade final de 10 semanas, a expressão da
ATGL é 70% menor quando comparado com essa enzima no tecido adiposo
mesentérico (KERSHAW et al., 2006). Se comparado ao tecido adiposo perigonodal
a expressão da ATGL é 90% menor (KERSHAW et al., 2006), demonstrando uma
baixa expressão dessa enzima no tecido hepático. O conteúdo de LHS e ATGL
quando corrigido pelo gene constitutivo GAPDH é 48 e 24 vezes respectivamente,
maior no tecido adiposo quando comparado ao tecido hepático em camundongos B6
(REID et al., 2008). Apesar dessa baixa expressão e conteúdo, autores têm sugerido
um envolvimento da ATGL no desenvolvimento do quadro de esteatose hepática. A
redução em 70% da expressão de ATGL hepática, utilizando um adenovírus, resulta
em um aumento de 30% no peso do fígado, acompanhado de um aumento de
aproximadamente 2 vezes no conteúdo de TAG no tecido hepático em
camundongos alimentados durante 7 dias com uma dieta comercial ou hiperlipídica
(ONG et al., 2011). Por outra lado, a superexpressão de ATGL no tecido hepático de
camundongos fêmeas ob/ob reduz a concentração de TAG hepático (REID et al.,

50
2008). A superexpressão da LHS em camundongos fêmeas ob/ob também reduz a
concentração de TAG hepático, sugerindo que ambas, ATGL e LHS podem
participar do desenvolvimento de esteatose hepática (REID et al., 2008). Contudo, a
redução no conteúdo de TAG hepático é maior em camundongos ob/ob
superexpressando ATGL (25 vezes) comparados com camundongos
supreexpressando LHS (100 vezes) (REID et al., 2008). Este efeito pode ser
decorrente da especificidade de cada uma destas enzimas por seus substratos. A
ATGL tem uma maior afinidade por TAG enquanto LHS tem maior por DAG e MAG
(VILLENA et al., 2004; ZIMMERMANN et al., 2004). No presente estudo, a
expressão de ATGL e LHS nos ratos alimentados com a dieta cafeteria foi similar ao
grupo alimentado com a dieta controle apesar de uma maior concentração
plasmática de insulina no grupo alimentado com a dieta cafeteria (CHAVES et al.,
2006). Em camundongos FVB machos com 8 semanas de vida quando submetidos
a um jejum de 6 à 24h, no tecido adiposo houve um aumento na expressão de ATGL
e a realimentação durante 12h ou 24h após o jejum de 24h foi seguida por uma
diminuição na expressão da mesma hidrolase, contudo, a expressão de LHS não
sofreu alteração por tais estados nutricionais (KERSHAW et al., 2006). Ainda nesse
mesmo estudo, adipócitos incubados com diferentes concentrações de insulina (10-4
a 104 ng/mL) apresentaram uma redução na expressão de ATGL num padrão dose
dependente (KERSHAW et al., 2006). Posteriormente, foi demonstrado que a
inibição da expressão de ATGL em adipócitos pela insulina se dá através da via de
sinalização mediada pelo fator de transcrição FOXO1 (CHAKRABARTI; KANDROR,
2009). No fígado de ratos Sprague-Dawley diabéticos por estreptozocina, a
concentração nuclear de FOXO1 é maior que no grupo controle não diabético,
demonstrando que essa via de sinalização esta ativa no tecido hepático (XIA et al.,
2011). Recentemente foi demonstrado que FOXO1 é capaz de estimular a
expressão de ATGL em hepatócitos primários de camundongos e inibir a expressão
do seu inativador G0S2 nessas mesmas células (ZHANG et al., 2016). Nesse
mesmo estudo, quando esses hepatócitos foram incubados na presença de 100 nM
de insulina durante 4h os resultados foram contrários aos encontrados
primeiramente, corroborando a idéia de que insulina pode regular a mobilização de
TAG via FOXO1. Os nossos dados demonstram que os ratos alimentados com a
dieta cafeteria, apesar do quadro de hiperinsulinemia plasmática, não apresentam
redução na expressão de ATGL no tecido hepático.

51
Tem sido demonstrado que o aumento na atividade de AMPK em células
HepG2 incubadas com seu ativador AICAR (5-aminoimidazole-4-carboxamida-
ribósido) durante 24h é seguido de um aumento na expressão de ATGL (KUO et al.,
2012). AMPK é um sensor de energia que pode ser ativado por diversos fatores tais
como jejum, estado redox (AMP/ATP) e a privação de glicose (SUCHANKOVA et al.,
2009). A adição de glicose ou piruvato regula a atividade de AMPK independente da
relação AMP/ATP (SUCHANKOVA et al., 2009). Desta forma, no presente estudo,
devido a um quadro de normoglicemia entre os grupos, podemos sugerir níveis
normais de AMPK seguido também de níveis normais do RNAm para ATGL.
Ácidos graxos livres também podem regular da expressão de ATGL e a
atividade das lipases totais do fígado (JAERGER et al., 2015). Camundongos
deficientes em CGI-58, um ativador de ATGL, apresentam uma diminuição na
concentração de ácidos graxos livres acompanhado da diminuição do RNAm de
ATGL no tecido hepático (JAERGER et al., 2015). Nesse mesmo estudo, a
administração de azeite de oliva (200 mL/camundongo) por sonda gástrica seguido
de uma injeção de heparina (10 IU/camundongo) induz aumentos de ambos analitos
(ATGL e CGI-58). Em ratos alimentados com a dieta cafeteria, a concentração de
ácidos graxos livres plasmático é similar ao dos animais alimentados com a dieta
controle (CHAVES et al., 2006), no entanto tem sido demostrado um aumento de
30% na atividade das lipases citosólicas do tecido hepático nesse mesmos animais
(MOREIRA; BOTION, dados não publicados), sugerindo uma maior mobilização de
ácidos graxos do TAG endógeno. Desta forma é possível que o efeito inibitório da
insulina reduzindo a expressão de ATGL possa ser contra-balanceado pelo efeito
estimulatório dos ácidos graxos endógenos. Apesar de ter sido demonstrado que o
estado nutricional não modifica a expressão de LHS (KERSHAW et al., 2006), tem
sido demonstrado em células HepG2 que a presença de 100 µM ATP (3µCi γ-32P-
ATP/sample) e 10 IU PKA (proteína quinase dependente de AMPc) induz um
aumento na fosforilação dessa enzima, aumentando a sua atividade (ZIMMERMANN
et al., 2004). Nesse mesmo estudo foi demonstrado que PKA não regula a atividade
de ATGL diretamente. Ainda que exista similaridade na expressão no RNAm da LHS
e ATGL no tecido hepático dos ratos alimentados com a dieta cafeteria quando
comparado aos animais da dieta controle, essas enzimas sofrem regulação pós
traducionais. É esperado que a maior concentração de insulina plasmática dos ratos
alimentados com a dieta cafeteria estimule a enzima fosfodiesterase reduzindo a

52
concentração intracelular de AMPc e atividade de PKA resultando diretamente na
inibição de LHS e indiretamente de ATGL via CGI-58 (GRANNEMAN et al., 2007;
GAIDHU et al., 2009). Contudo, a realimentação durante 6 horas induz uma redução
de G0S2, um inibidor de ATGL, no tecido hepático (ZHANG et al., 2014),
possibilitando uma maior atividade de ATGL. Camundongos fêmeas nocaute para
G0S2 (G0S2-/-) apresentam resistência ao desenvolvimento de esteatose hepática
induzida pela dieta hiperlipídica devido a um aumento na atividade da enzima ATGL
(ZHANG et al., 2014). Alguns estudos sugerem um importante papel das hidrolases
ATGL e LHS na homeostasia de lipídios hepáticos (REID et al., 2008; ONG et al.,
2011). Camundongos com alterações no funcionamento de ATGL ou seu co-ativador
CGI-58 hepático, ou na LHS, apresentam um desequilíbrio na homeostasia dos
lipídios e um quadro de esteatose hepática (REID et al., 2008; ONG et al., 2011).
Além disso, enquanto no tecido adiposo as enzimas LHS e ATGL correspondem a
90% das hidrolases totais (ZIMMERMANN et al., 2004), no tecido hepático, tem sido
estimado, que tais enzimas contribuem apenas com aproximadamente 40% desta
atividade (REID et al., 2008; ONG et al., 2011). Através da utilização de adenovírus
para ATGL (shRNAm ATGL) hepatócitos apresentam uma redução de 70% no
RNAm e 80% na concentração proteica desta enzima, porém esses hepatócitos
mantem aproximadamente 60% da atividade hidrolítica de suas lipases totais (ONG
et al., 2011). Considerando os valores normais da expressão de ATGL e LHS entre
nossos grupos analisados, o aumento de 30% na atividade das lipases citosólicas
hepáticas nos ratos alimentados com a dieta cafeteria (MOREIRA; BOTION, dados
não publicados) e a regulação pós traducional destas enzimas pela insulina,
podemos sugerir que além da redução na inibição de ATGL exercida por G0S2
(ZANG et al., 2014), outras hidrolases podem estar ativas no tecido hepático.
Em 1997, utilizando hepatócitos de porco, foi purificada uma enzima
associada a microssomas com a capacidade de hidrolisar TAG, essa enzima
recebeu o nome de hidrolase de triacilglicerol (TGH) (LEHNER; VERGER, 1997).
Esta enzima parece participar da hidrólise do pool de TAG armazenado no tecido
hepático e formação da VLDL (DOLINSKY et al., 2004; LEHNER et al., 1999; LO et
al., 2010). Tem sido demostrado que a expressão da arilacetamida deacetilase em
células RH7777 reduz a concentração de TAG e secreção de ApoB, mas aumenta a
oxidação de ácidos graxos (LO et al., 2010). Esta enzima está associada ao retículo
endoplasmático (LO et al., 2010) e compartilha homologia com a LHS (PROBST et

53
al., 1994). O aumento na atividade das lipases totais do tecido hepático dos ratos
alimentados com a dieta cafeteria pode, portanto, ser decorrente de uma maior
atividade da ATGL assim como de outras hidrolases, como TGH, uma vez que
mesmo após a centrifugação de 100.000g tem sido demostrado a presença desta
enzima na fração citosólica no adiposo branco (SONI et al. 2004; WEI et al., 2007a).
Entretanto, poucos estudos investigam a regulação da expressão e atividade de
TGH no tecido hepático. O efeito direto da insulina regulando TGH ainda não foi
demonstrado, mas a expressão de TGH está diminuída no fígado de camundongos
com resistência insulínica induzida por TNF-alfa (RUAN et al., 2002). Apesar da
regulação dessas outras hidrolases ainda requisitar de mais investigações, tem sido
sugerido que o destino do ácido graxo proveniente da lipólise depende da lipase
responsável pela hidrólise do TAG (WEI et al., 2007). Os ácidos graxos provenientes
da ação da LHS são usados preferencialmente na β-oxidação (PEASE et al., 1999).
Estudos recentes fornecem evidencias que a ATGL consegue distribuir os ácidos
graxos hidrolisados entre as vias oxidativas e a síntese de lipoproteína de muito
baixa densidade (VLDL) (ONG et al., 2011). E como já mostrado acima, TGH é
capaz de mobilizar TAG e fornecer substrato melhorando a secreção de VLDL (LO
et al., 2010).
5.3 Oxidação de lipídios hepáticos
Tem sido amplamente demonstrado que o PPAR-α regula a β-oxidação
peroxissomal e mitocondrial através da ativação da transcrição de genes
importantes para esse processo, dentre eles acil-CoA oxidase (ACO) e a carnitina
palmitoiltransferase I (CPT-I) no tecido hepático (KELLER et al., 1993; LEMBERGER
et al., 1996; MINNICH et al., 2001; EVANS et al., 2004). No presente trabalho, a
dieta cafeteria não promoveu alteração na expressão de PPAR-α e de ACO no
tecido hepático. Alguns trabalhos correlacionam o conteúdo dos RNAm desses dois
genes (ZOU et al., 2008; BONFLEUR et al., 2015). No entanto, outros estudos
sugerem uma inexistência dessa relação entre o conteúdo do RNAm PPAR-α e de
ACO (NOH et al., 2013; GARCIA-CARABALLO et al., 2013). Ratos Fisher tratados
com dexametasona (40 µg/Kg-1) apresentam um aumento na expressão de PPAR-α
sem alterar a expressão de ACO (LEMBERGER et al., 1996). No entanto, a
administração de WY-14643, um ativador de PPAR-α, e dexametasona induz um
aumento de 6,5 vezes na expressão de ACO em comparação aos animais que só

54
receberam WY-14643 (LEMBERGER et al., 1996). Portanto, a ativação do receptor
é um pré-requisito para o efeito dos glicocorticóides nos genes alvos de PPAR-α.
Alguns estudos têm demonstrado que a insulina pode controlar a β-oxidação
através da regulação da expressão da CPT-I (CHATELAIN et al., 1995; PARK et al.,
1995; AKKAOUI et al., 2009). O jejum de 24h induz uma diminuição na concentração
sérica de insulina acompanhada de um aumento na expressão de CPT-I em
camundongos quando comparado àqueles alimentados com a dieta comercial
(ESTALL et al., 2009). Este mesmo efeito é observado em camundongos
heterozigotos para PGC-1α (ESTALL et al., 2009). De fato, previamente foi
demonstrado que a adição de insulina (100 nM) em células de hepatoma de ratos
reduz a expressão desta proteína (PARK et al., 1995). Por outro lado, uma dieta rica
em proteínas (35%) induz um aumento na expressão de CPT-I sem afetar a
concentração sérica de insulina no tecido hepático de camundongos C57BL/6J
(GARCIA-CARABALLO et al., 2013), demonstrando que a regulação desse gene
pode ocorrer de forma independente da insulina. De fato, a adição de ácido linoléico
(0,5 mM) no meio de incubação de hepatócitos estimula a transcrição de CPT-I
(CHANTELAIN et al., 1995). No presente estudo, no tecido hepático dos ratos
alimentados com a dieta cafeteria foi observado um aumento de CPT-I, mesmo na
presença de altas concentrações séricas de insulina, sem alteração na expressão de
PPAR-α. Em hepatomas de ratos foi demonstrado que o ácido linoléico (18:2, ω-6)
estimula a transcrição do gene da CPT-I independente de PPAR-α (LE MAY et al.,
2005). Por outro lado, a adição de insulina (1, 10 e 100 nM) no meio de incubação
inibe o efeito estimulatório do ácido linoléico (0,5 mM) na expressão de CPT-I em
hepatócito de ratos (CHANTELAIN et al., 1995). Entretanto, este efeito não é
observado na concentração de 0,1 nM de insulina (CHANTELAIN et al., 1995).
Considerando que a dieta cafeteria induz um aumento de 62% na concentração
sérica de insulina atingindo 351 pmol/L (CHAVES et al., 2006), acompanhado de
30% de aumento na atividade das lipases citosólicas no tecido hepático (MOREIRA;
BOTION, dados não publicados), podemos sugerir que o aumento na mobilização de
ácidos graxos estimulando a expressão de CPT-I prevalece ao efeito inibitório da
insulina.

55
5.4 Captação de ácidos graxos hepáticos
A captação hepática de ácidos graxos contribui para o equilíbrio constante de
triacilglicerol nesses hepatócitos, bem como para a DHGNA (KAWANO; COHEN,
2013). A taxa de absorção de ácidos graxos plasmático pelo fígado depende da
concentração de ácidos graxos no sangue e proteínas transportadoras como, FATP2
e CD36 (BRADBURY, 2006). Camundongos C57BL/6 submetidos a uma dieta rica
em lipídios durante 6 semanas, apresentam um quadro de esteatose hepática em
comparação aos camundongos alimentados com a dieta controle (FALCON et al.,
2006). Estes camundongos transfectados com adenovírus shRNA FATP2 (nocaute)
e tratados com a dieta rica em lipídios por mais 6 semanas apresentam uma
redução de TAG hepático quando comparado aos camundongos que receberam
somente o veículo. Os ácidos graxos captados pelo fígado podem ser provenientes
da dieta ou da lipólise do tecido adiposo (BRADBURY, 2006). Os ratos alimentados
com a dieta cafeteria não apresentam alterações nos ácidos graxos livres,
comparado aos ratos alimentados com a dieta controle (CHAVES et al.,2006). No
entanto, os adipócitos dos ratos alimentados com a dieta cafeteria apresentam um
aumento de 29% na lipólise basal, avaliada pela liberação de glicerol in situ,
utilizando a técnica de microdiálise (CARVALHO, 2010). A administração de ácidos
graxos marcados com isótopos estáveis a indivíduos obesos por via oral durante 4
dias demonstrou que aproximadamente 60% dos ácidos graxos incorporado à
molécula de TAG no tecido hepático foram provenientes da captação plasmática
(DONNELLY et al., 2005). Portanto, nos ratos alimentados com a dieta cafeteria,
possivelmente os ácidos graxos provenientes da lipólise do TAB pode esta sendo
esterificado no fígado, contribuindo para o aumento de TAG nesse tecido.
5.5 Secreção de VLDL
A montagem e a secreção de VLDL pelo fígado é um processo complexo que
necessita da proteína ApoB (apolipoproteina B) e da sua lipidação pela proteína
microssomal de transferência de triacilglicerol (MTP) (HUSSAIN et al., 2003). Vários
estudos têm demostrado que a insulina é capaz de regular de forma negativa a
expressão de MTP (HAGAN et al., 1994; LIN et al., 1995; AU et al., 2003). Nestes
trabalhos hepatócitos incubados com diferentes doses de insulina (1nM à 100nM)
por até 6h apresentam uma redução de 20 à 80% no RNAm de MTP de uma forma
dose-dependente. Tem sido sugerido que FOXO1 regula a expressão de MTP

56
(KAMAGATE et al., 2008). Neste estudo, células HepG2 foram transfectadas com
um vetor para FOXO1 e incubadas por 24h na ausência ou na presença de insulina
(100nM), sendo observado que a presença da insulina reduz a expressão de MTP.
Ainda nesse mesmo estudo, camundongos expressando de forma constitutiva
FOXO1 no tecido hepático apresentam um aumento na expressão de MTP seguido
de um aumento na produção de VLDL quando comparados ao controle.
Previamente, foi observado que camundongos ob/ob resistentes à insulina
apresentam um aumento na expressão de MTP (BARTELS et al., 2002), e o jejum
de 24h em camundongos machos C57BL/6J aumentam a presença nuclear (ativa)
de FOXO1 no tecido hepático (QU et al., 2006). No entanto, o presente estudo
mostrou que os ratos alimentados com a dieta cafeteria apresentaram uma maior
expressão de MTP em relação aos ratos alimentados com a dieta controle, apesar
da hiperinsulinemia. Nesses animais, a sensibilidade hepática a insulina é
evidenciada pelo aumento no fluxo glicolítico e pela diminuição da gliconeogênese
em fatias de fígado (MARTINS-SANTOS, 2008). Além disso, os ratos alimentados
com a dieta cafeteria por 5, 6, 8 e 12 semanas não apresentam alteração na curva
de tolerância à glicose (CARVALHO, 2010). Tais achados sugerem que os altos
níveis plasmáticos da insulina foram suficientes para superar alguma possível
resistência de sua ação tecidual. Vários estudos veem sugerindo que a insulina não
é o principal regulador capaz de alterar a expressão de MTP (BARTELS et al., 2002;
SPARKS et al., 2011; SHI et al., 2013). Em camundongos C57BL/6 diabéticos
induzidos por estreptozotocina não houve alteração na expressão da proteína MTP
em comparação aos camundongos não diabéticos (BARTELS et al., 2002). Em
camundongos nocaute para apoB (apobec-1-/-), a administração de insulina
(10µL/kg) após um jejum de 12h não induz alteração no RNAm da MTP em
comparação ao grupo salina (SPARKS et al., 2011). Em outro estudo, ratos Wistar
resistentes à insulina alimentados com uma dieta rica em frutose por 8 semanas
apresentaram um redução no RNAm de MTP quando comparados aos ratos
alimentados com a dieta controle (SHI et al., 2013). Sendo assim, a insulina é capaz
de regular a expressão de MTP negativamente talvez em situação mais intensa e de
pouca duração. Na região promotora do gene MTP existem regiões de
reconhecimento para o fator nuclear de hepatócito (HNF) 1 e 4α (HAGAN et al.,
1994). O fator de transcrição HNF-4α demostrou ser um regulador positivo para a
expressão de MTP. Camundongos nocaute para HNF-4α apresentam uma redução

57
quase que total na expressão de MTP seguido de uma diminuição na secreção de
VLDL (HAYHURST et al., 2001). A atividade transcricional de HNF-4α pode ser
regulada por diferentes tipos de ácidos graxos, com variações no tamanho da cadeia
de carbono e também no seu nível de saturação (HERTZ et al., 1998). Neste
trabalho, células COS-7 co-transfectadas com um vetor de expressão HNF4-α CAT
(cloranfenicol-acetiltransferase) e incubadas com diferentes tipos de ácidos graxos,
por uma hora, apresentam um aumento na atividade de HNF4-α na presença de
ácidos graxos de cadeia com 14 e 16 carbonos saturados. Nos ratos alimentados
com a dieta cafeteria o aumento na atividade das hidrolases citosólicas (MOREIRA;
BOTION, dados não publicados) e um aumento na síntese de ácidos graxos de novo
no tecido hepático (CHAVES et al., 2012) podem estar fornecendo substrato para
maior ativação de HNF4-α e consequentemente aumentando a expressão de MTP
nesse grupo, superando um possível efeito inibitório exercido pela insulina. Outro
trabalho sugere que o colesterol pode regular a expressão de MTP. Na região
promotora de MTP foi identificado um elemento de resposta ao esterol
possibilitando assim que o colesterol pudesse regular a expressão de MTP. Células
HepG2 transfectadas com o gene para MTP humano e incubados por 48h na
presença de 1,0µg/mL de 25-OH colesterol e 10µg/mL colesterol apresentam um
aumento na expressão de MTP comparado as células incubadas com etanol
(controle) (HAGAN et al., 1994). Os ratos alimentados com a dieta cafeteria
apresentaram um aumento de 30% no colesterol hepático quando comparado aos
ratos alimentados com a dieta controle. Portanto, a diferença na expressão de MTP
entre os grupos analisados também pode ter sido devido a maior concentração de
colesterol no tecido hepático nos ratos alimentados com a dieta cafeteria.
No presente estudo, os ratos alimentados com a dieta cafeteria apresentaram
um redução na expressão da ApoB 100 (apolipoproteina B) em relação aos ratos
alimentados com a dieta controle. A regulação na expressão de ApoB ainda não é
totalmente compreendida, no entanto, muitos estudos sugerem que o gene ApoB é
expresso constitutivamente (DASHTI et al., 1989; PULLINGER et al., 1989; FISHER
et al., 2001; SPARKS et al., 2006). Células HepG2 submetidas a diversos fatores
regulatórios para a secreção de ApoB tais como a insulina (1, 10 e 100nM), ácido
oleico (0,8mM) não apresentam alteração no RNAm de ApoB quando comparado a
β-actina (DASHTI et al., 1989; PULLINGER et al., 1989). No entanto, outros estudos
sugerem que em determinadas circunstâncias, tais como: animais diabéticos por

58
estreptozotocina, ações farmacológicas e alguns tipos de dieta (rica em colesterol ou
alguns aminoácidos), a expressão de ApoB pode sofrer regulação ainda não
totalmente compreendida (SPARKS et al., 1992; DASHTI 1992; CIANFLONE et al.,
1996; SINGH et al., 2002; SPARKS et al., 2006; ASHUR-FABIAN et al., 2010). Ratos
Sprague Dawley diabéticos por estreptozotocina não apresentam alteração na
expressão de ApoB comparado aos ratos não diabéticos, no entanto, os ratos
diabéticos quando tratados com insulina durante 7 dias (dose única e diária
200µL/Kg) apresentam um aumento na expressão da proteína ApoB no tecido
hepático comparado aos ratos diabéticos que não receberam insulina (SPARKS et
al., 1992). Células HepG2 incubadas com colesterol (10-40µg/mL) de 16 a 20h
apresentam um aumento na expressão de ApoB (55%) comparado ao grupo
controle (DASHTI 1992). Alguns estudos demonstram que tanto in vitro quanto in
vivo, alguns aminoácidos como, por exemplo, asparagina, glutamina e a metionina,
podem interferir na expressão de ApoB (CIANFLONE et al., 1996; SPARKS et al.,
2006). Mais recentemente, foi identificado na região promotora do gene da ApoB um
elemento de resposta a p53 (p53RE), um gene supressor de tumor capaz de ativar a
transcrição de vários genes (ASHUR-FABIAN et al., 2010). Nesse trabalho,
camundongos C57BL/6 tratados durante 5 dias com adriamicina, um potente
ativador de p53, apresentam um aumento na expressão de ApoB no tecido hepático
em comparação ao controle. Camundongos C57BL/6J com idade inicial de 5
semanas, tratados com um silenciador do gene ApoB durante 6 semanas ( 2 vezes
por semana) apresentam um redução de aproximadamente 90% no RNAm de ApoB,
sem causar alteração no conteúdo da proteína (CROOKE et al., 2005). De fato os
estudos prévios tem sugerido que a expressão de ApoB pode interferir na secreção
de VLDL, no entanto, esses mesmos trabalhos demostram que as alterações na
transcrição de ApoB não é acompanhada na mesma proporção das alterações na
produção dessa proteína, sugerindo a existência de mecanismos regulatórios co e
pós transcricionais/traducionais (DASHTI, 1992; SPARKS et al., 1992; FISHER et al.,
2001; SINGH et al., 2002; CROOKE et al., 2005). Embora as alterações nos níveis
de transcrição e tradução de ApoB possam desempenhar um papel na produção da
proteína, a secreção de ApoB é regulada principalmente a nível de degradação
proteica (FISHER et al., 2014). A falta de co-relação entre a concentração de RNAm
e da proteína ApoB demonstra uma necessidade de mais estudos, no entanto, tem
sido fortemente sugerido que os fatores envolvidos na regulação da taxa de

59
secreção de VLDL são: a) a quantidade de proteina ApoB que escapa das diversas
formas de degradação, b) a disponibilidade de lipídios a serem secretados e c) a
expressão de MTP ( DAVIS, 1999; FISHER et al., 2001; SPARKS et al., 2006).
Os ratos alimentados com a dieta cafeteria apresentaram um aumento de
aproximadamente 20% na taxa de secreção de VLDL-TAG quando comparados aos
ratos alimentados com a dieta controle. Apesar de um quadro de hiperinsulinemia e
normoglicemia (CHAVES et al., 2006), a sinalização da insulina no tecido hepático,
avaliada pelo fluxo da glicólise e da gliconeogênese esta preservada nos ratos
alimentados com a dieta cafeteria (citado acima) sugerindo que a secreção de VLDL
deveria estar reduzida. Tem sido sugerido que a insulina reduz a expressão de MTP
através de FOXO1, altera a tradução do RNAm e estimula de várias formas a
degradação da proteína ApoB (HAAS et al., 2013). Desta forma, ratos ob/ob
resistentes à insulina apresentam maior secreção de VLDL-TAG (BARTELS et al.,
2002). Contudo, células HepG2 incubadas a uma concentração de 100nM de
insulina apresentam uma redução na tradução do RNAm da proteína ApoB no
período entre 4 e 16h, mas entre 15 mim e 1h de incubação, a insulina aumenta a
tradução de ApoB (KARIMIAN POUR; ADELI, 2011). Em outro trabalho, células
HepG2 transfectadas com plasmídeos contendo as regiões 5’ UTR ou 3’UTR no
gene para ApoB e incubadas na ausência ou na presença de 1µg/mL de insulina
durante 16h demostram que células contendo a região 5’UTR incubadas na
presença de insulina apresentam uma redução na tradução desta proteína
(SIDIROPOULOS et al., 2005). Este estudo sugere que a insulina diminui a ligação
da proteína a região 5’ UTR que conduz o RNAm para a tradução. A insulina diminui
a ligação da proteína a região 5’UTR através da sua cascata de sinalização
envolvendo PI3K e mTOR (SIDIROPOULOS et al., 2007). Grande parte da ApoB
produzida é degradada por diversos mecanismos, sendo as que escapam desse
processo utilizadas para a secreção de VLDL (FISHER et al., 2001). A degradação
de apoB pode acontecer pela via proteassômica ou não proteassômica, como por
exemplo estimulada por ácido graxo omega 3 ou insulina (LIANG et al., 2001;
FISHER; GINSBERG, 2002). A insulina é capaz de diminuir a secreção de VLDL
induzindo a proteína ApoB para uma degradação (SPARKS et al., 1990). Em
hepatócitos de ratos incubados com 10nM de insulina durante 12 à 14h, foi
demonstrado uma redução na secreção de ApoB quando comparados aos controles
(0,1nM de insulina) devido a uma menor concentração de ApoB (SPARKS et al.,

60
1990). Por outro lado, humanos alimentados com uma dieta rica em carboidratos
apresentam aumento na velocidade de secreção da fração TAG de VLDL, sem
alterar a velocidade de secreção da fração apoB de VLDL (MELISH et al., 1980),
indicando que alterações na produção de VLDL-TAG não são acompanhadas pela
produção de VLDL-apoB. Em acordo com essa ideia, hepatócitos cultivados na
presença de 1mM de ácido oleico, um potente estimulador da síntese de TAG,
apresentam um aumento na secreção de VLDL-TAG sem alterar a síntese proteica
de ApoB (DAVIS; BOOGAERTS, 1982). Ainda nesse estudo, a inibição da síntese
proteica de ApoB por cicloheximida induz uma redução na secreção de VLDL
mesmo na presença do ácido oleico, sugerindo que a ApoB desempenha um papel
importante na determinação da capacidade do hepatócito em aumentar a secreção
de TAG. A disponibilidade de lipídios e o nível de expressão e atividade de MTP
protegem ApoB da degradação e consequentemente pode melhorar a secreção de
VLDL (FISHER et al., 2001). Células HepG2 incubadas na presença de RTV, um
inibidor de proteassoma, apresentam um acúmulo de ApoB intracelular, no entanto,
essas células apresentam uma redução na secreção de ApoB (LIANG et al., 2001).
Entretanto, ao serem incubadas na presença do RTV e 0,4 mM de ácido oleico
ocorre um aumento de 65% na taxa de secreção de ApoB em comparação as
células que receberam somente o acido oleico, demonstrando que o fornecimento
de lipídios melhora secreção de ApoB protegendo da degradação proteassômica,
assim esses mecanismos são capazes de controlar a secreção de lipídios hepáticos.
Em outro trabalho, hepatócitos de ratos incubados com diferentes ácidos graxos
(concentração de 1mM) apresentaram um aumento na secreção de VLDL- [³H]TAG
visualizado através da incorporação de [³H]glicerol (DAVIS; BOOGAERTS, 1982).
Nesse trabalho, o ácido oleico estimulou um aumento de 2 a 3 vezes na secreção de
VLDL-TAG. Outro passo limitante para a secreção de VLDL é a transferência de
lipídios para ApoB pela proteína MTP, sendo que alterações tanto na expressão
quanto na atividade de MTP são capazes de prejudicar a secreção de VLDL
(WETTERAUL et al., 1992). Em camundongos machos nocaute para ApoB
(apobec-1-/-), a insulina (10µ/Kg) não é capaz de alterar a expressão e a atividade de
MTP no tecido hepático quando comparado aos camundongos salina (SPARKS et
al., 2011). Ainda nesse trabalho, hepatócitos que receberam um adenovírus para
superexpressarem MTP (HMTP) apresentam maior secreção de ApoB em relação
ao controle. No entanto, os hepatócitos HMTP na presença de 100nM de insulina

61
tiveram uma redução na secreção de ApoB quando comparado aos hepatócitos
HMTP que receberam doses basais de insulina (1nM). Desta forma, mesmo em
situações em que a secreção de ApoB está aumentada pela maior atividade de
MTP, parte da ApoB produzida ainda esta sujeita a degradação pela insulina.
Portanto, os ratos alimentados com a dieta cafeteria apresentam um aumento na
secreção de VLDL-TAG possivelmente devido a um maior fornecimento de lipídios
para a secreção proveniente do aumento da lipogênese de novo (CHAVES et al.,
2012), do aumento na atividade das hidrolases citosólicas hepáticas (MOREIRA;
BOTION, dados não publicados), e de um possível aumento na captação pelo tecido
hepático de ácidos graxos livres provenientes da maior lipólise nos TAB
(CARVALHO, 2010), conciliado com uma maior expressão de MTP quando
comparado aos ratos alimentados com a dieta controle, superando um efeito da
insulina na tentativa de diminuir a secreção de VLDL. Desta forma, mesmo com um
aumento na secreção de VLDL-TAG, a dieta cafeteria foi capaz de induzir um
quadro de esteatose hepática.

62
6. CONSIDERAÇÕES FINAIS
Nossos dados demonstram que a dieta cafeteria induziu um aumento na
adiposidade, na concentração hepática de lipídios e na concentração sérica de
triacilglicerol. O aumento acentuado na síntese de ácidos graxos de novo hepática
(CHAVES et al., 2012), não compensada pelo aumento na secreção de VLDL e na
degradação de lipídios contribuem para o desenvolvimento da esteatose hepática. A
dieta cafeteria foi capaz de causar um desequilíbrio na concentração e na secreção
de triacilglicerol revelando uma incapacidade do tecido hepático em reestabelecer
esse equilíbrio lipídico. Um efeito oposto na expressão dos genes envolvidos na
montagem da VLDL, que são, aumento na expressão do RNAm de MTP e
diminuição do RNAm da apoB, podem representar uma alteração na capacidade
secretora de triacilglicerol via VLDL nos ratos alimentados com a dieta cafeteria,
provavelmente sobre ação da insulina, hormônio esse, importante na homeostasia
dos lipídios hepáticos. O aumento da atividade das lipases hepáticas (MOREIRA,
BOTION, dados não publicados) e um aumento acentuado do RNAm de CPT1
sugerem a ativação de processos de degradação, podendo representar uma
resposta adaptativa ao excesso de lípidos intracelulares. No entanto, mesmo com
um pequeno aumento da secreção de VLDL, a ativação do catabolismo de lipídios
intracelulares e a sensibilidade insulínica preservada no tecido hepático, esse tecido
permanece com teor de lipídios em excesso nos animais alimentados com a dieta
cafeteria. Portanto, o aumento acentuado na síntese dos ácidos graxos parece ser
determinante em induzir esteatose hepática nos ratos alimentados com a dieta
cafeteria ainda que mecanismos compensatórios possam estar estimulados,
provavelmente minimizando o efeito desta dieta neste acúmulo lipídico.

63
7. REFERÊNCIAS BIBLIOGRÁFICAS
Abdelmalek MF, Liu C, Shuster J, Nelson DR, Asal NR. Familial aggregation of
insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver
disease. Clin Gastroenterol Hepatol. 4(9):1162-9, 2006.
Adams LA, Lindor KD.Nonalcoholic fatty liver disease. Ann Epidemiol. 17(11):863-9,
2007.
Akkaoui M, Cohen I, Esnous C, Lenoir V, Sournac M, Girard J, Prip-Buus C.
Modulation of the hepatic malonyl-CoA–carnitine palmitoyltransferase 1A partnership
creates a metabolic switch allowing oxidation of de novo fatty acids. J. Biochem.
420(3):429–438, 2009.
Andrade GF, de Almeida Cd, Espeschit AC, Dantas MI, Benjamin Ldos A, Ribeiro
SM, Martino HS. The addition of whole soy flour to cafeteria diet reduces metabolic
risk markers in wistar rats. Lipids Health Dis. 11;12:145, 2013.
Angelopoulos TJ1, Lowndes J, Sinnett S, Rippe JM. Fructose containing sugars do
not raise blood pressure or uric acid at normal levels of human consumption. J Clin
Hypertens (Greenwich). 17(2):87-94, 2015.
Ashur-Fabian O, Har-Zahav A, Shaish A, Wiener Amram H, Margalit O, Weizer-Stern
O, Dominissini D, Harats D, Amariglio N, Rechavi G. apoB and apobec1, two genes
key to lipid metabolism, are transcriptionally regulated by p53. Cell Cycle.
9(18):3761-70, 2010.
Asrih M, Jornayvaz FR. Metabolic syndrome and nonalcoholic fatty liver disease: Is
insulin resistance the link? Mol Cell Endocrinol. 418 (1):55-65, 2015.
Au WS, Kung HF, Lin MC. Regulation of microsomal triglyceride transfer protein gene
by insulin in HepG2 cells: roles of MAPKerk and MAPKp38. Diabetes. 52(5):1073-
80, 2003.
Bartels ED, Lauritsen M, Nielsen LB. Hepatic expression of microsomal triglyceride
transfer protein and in vivo secretion of triglyceride-rich lipoproteins are increased in
obese diabetic mice. Diabetes. 51(4):1233-9, 2002.
Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic
dyslipidemia. Nutr Metab. 2(1):5, 2005 .

64
Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH:
Causes, consequences and possible means to prevent it. Mitochondrion. 6(1):1-28,
2006.
Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel
J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, Postic C. The
lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin
resistance in mice andhumans. J Clin Invest. 122(6):2176-94, 2012.
Bolinder J, Kager L, Ostman J, Arner P. “Differences at the receptor and postreceptor
levels between human omental and subcutaneous adipose tissue in the action of
insulin on lipolysis”. Diabetes. 32(2):117-23, 1983.
Bonfleur ML, Borck PC, Ribeiro RA, Caetano LC, Soares GM, Carneiro EM, Balbo
SL. Improvement in the expression of hepatic genes involved in fatty acid metabolism
in obese rats supplemented with taurine. Life Sci. 135:15-21, 2015.
Boone LR1, Brooks PA, Niesen MI, Ness GC. Mechanism of resistance to dietary
cholesterol. J Lipids. 2011:101242, 2011.
Borensztajn J, Rone MS, Kotlar TJ. The inhibition in vivo lipoprotein lipase (clearing-
fator lipase) activity by triton WR-1339. Biochemical J. 156(3):539-543, 1976.
Brain, liver, and serum salusin-alpha and -beta alterations in Sprague-Dawley rats
with or without metabolic syndrome. Med Sci Monit. 20:1326-33, 2014.
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J
Clin Invest. 114(2):147–152, 2004.
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC,
Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting
a Forkhead transcription factor. Cell. 96(6):857-68, 1999.
Cali AMG, Caprio S. Ectopic fat deposition and the metabolic syndrome in obese
children and adolescents. Horm Res. 71(suppl 1):2–7, 2009.
Carvalho L. Efeitos da dieta hipercalórica no metabolismo de proteínas em
músculo esquelético e na atividade lipolítica do tecido adiposo branco de
ratos. Ribeirão Preto. Dissertação (Mestrado – Programa de Pós-Graduação em
Bioquímica). Faculdade de Medicina de Ribeirão Preto da Universidade de São
Paulo. 2010.

65
Castro GSF, Almeida BB, LeonardI DS, Ovídio PP, Jordão AA. Association between
hepatic cholesterol and oleic acid in the liver of rats treated with partially
hydrogenated vegetable oil. Rev. Nutr. 1(25), 2012.
Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The
carbohydrate-response element-binding protein is a target gene of LXR. J Biol
Chem. 282(1):743-51, 2007.
Chakrabarti P, Kandror KV. FoxO1 controls insulin-dependent adipose triglyceride
lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem. 284(20):13296-
300, 2009.
Chatelain F, Kohl C, Esser V, Mcgarry JD, Girard J, Pegoriek JP. Cyclic AMP and
fatty acids increase carnitine palmitoyltransferase I gene transcription in cultured fetal
rat hepatocytes. J. Biochem. 235:789-798, 1996.
Chaves VE, Frasson D, Garófalo MA, Navegantes LC, Migliorini RH, Kettelhut IC.
Increased glyceride-glycerol synthesis in liver and brown adipose tissue of rat: in-vivo
contribution of glycolysis and glyceroneogenesis. Lipids. 47(8):773-80, 2012.
Chaves VE, Frasson D, Martins-Santos ME, Boschini RP, Garófalo MA, Festuccia
WT, Kettelhut IC, Migliorini RH. Glyceroneogenesis is reduced and glucose uptake is
increased in adipose tissue from cafeteria diet-fed ratsindependently of tissue
sympathetic innervation. J Nutr. 136(10):2475-80, 2006.
Chaves VE, Frasson D, Martins-Santos ME, Navegantes LC, Galban VD, Garófalo
MA, Kettelhut IC, Migliorini RH. Fatty acid synthesis and generation of glycerol-3-
phosphate in brown adipose tissue from rats fed a cafeteria diet. Can J Physiol
Pharmacol. 86(7):416-23, 2008.
Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion,
hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 22(9):353-63,
2011.
Cianflone K, Zhang Z, Vu H, Kohen-Avramoglu R, Kalant D, Sniderman AD. The
effect of individual amino acids on ApoB 100 and Lp(a) secretion by HepG2 cells. J
Biol Chem. 271(46):29136-45, 1996.

66
Citil C, Konar V, Aydin S, Yilmaz M, Albayrak S, Ozercan IH, Ozkan Y. Cohen JC,
Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights.
Science. 332(6037):1519-23, 2011.
Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S, Perera RJ. An
apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic
mice without causing hepatic steatosis. J Lipid Res. 46(5):872-84, 2005.
Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new
insights. Science. 332(6037):1519-23, 2011.
Dashti N, Williams DL, Alaupovic P. Effects of oleate and insulin on the production
rates and cellular mRNA concentrations of apolipoproteins in HepG2 cells. J Lipid
Res. 30(9):1365-73,1989.
Dashti N. The effect of low density lipoproteins, cholesterol, and 25-
hydroxycholesterol on apolipoprotein B gene expression in HepG2 cells. J Biol
Chem. 267(10):7160-9, 1992.
Davis RA, Boogaerts JR. Intrahepatic assembly of very low density lipoproteins.
Effect of fatty acids on triacylglycerol and apolipoprotein synthesis. J Biol Chem.
257(18):10908-13, 1982.
Davis RA. Cell and molecular biology of the assembly and secretion of apolipoprotein
B-containing lipoproteins by the liver. Biochim Biophys Acta. 1440(1):1-31, 1999.
Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology.
114(4):842-5, 1998.
Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic
C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin
resistance in ob/ob mice. Diabetes. 55(8):2159-70, 2006.
Després JP, Lemieux I. Abdominal obesity and metabolic syndrome.
Nature.444(7121):881-7, 2006.
Dolinsky VW, Douglas DN, Lehner R, Vance DE. Regulation of the enzymes of
hepatic microsomal triacylglycerol lipolysis and re-esterification by theglucocorticoid
dexamethasone. J. Biochem. 378(3):967-74, 2004.

67
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources
of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic
fatty liver disease. J Clin Invest. 115(5):1343-51, 2005.
Duvnjak M, Tomasic V, Gomercic M, Duvnjak S, Barsic N, Lerotic I. Therapy of
nonalcoholic fatty liver disease: current status. Journal of Physiology and
Pharmacology. 60(7):57-66, 2009.
Estall JL, Kahn M, Cooper MP, Fisher FM, Wu MK, Laznik D, Qu L, Cohen DE,
Shulman GI, Spiegelman BM. Sensitivity of Lipid Metabolism and Insulin Signaling to
Genetic Alterations in Hepatic Peroxisome Proliferator–Activated Receptor-γ
Coactivator-1α Expression. Diabetes. 58(7):1499–1508, 2009.
Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat.
Med. 10(4):355-61, 2004.
Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S.
Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with
nonalcoholic fatty liver disease. Gastroenterology. 134(2):424-31, 2008.
Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease:
biochemical, metabolic, and clinical implications. Hepatology. 51(2):679-89, 2010.
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis.
Hepatology. 43(2 Suppl 1):S99-S112, 2006.
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis.
Hepatology. 43(2 Suppl 1): S99–S112, 2006.
Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS,
Cherrington NJ. Hepatic cytochrome P450 enzyme alterations in humans with
progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos.
37(10):2087-94, 2009.
Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and
secretion of apolipoprotein B-containing lipoproteins. J Biol Chem. 277(20):17377-
80, 2002.
Fisher E, Lake E, McLeod RS. Apolipoprotein B100 quality control and the regulation
of hepatic very low density lipoprotein secretion. J Biomed Res. 28(3):178-93, 2014.

68
Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The
triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative
pathways. J Biol Chem. 276(30):27855-63, 2001.
Flier JS, Foster DW. Eating disorders: obesity, anorexia nervosa, and bulimia
nervosa. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams
textbook of Endocrinology. 9th edition. Philadelphia: WB Saunders; p.1061-97,
1998.
Folch J, Lees M, Stanley GA. A simple method for the isolation and purification of
total lipids from animal tissues. Journal of Biological Chemistry; 226(1):497-509,
1957.
Gaidhu MP, Fediuc S, Anthony NM, So M, Mirpourian M, Perry RL, Ceddia RBJ.
Prolonged AICAR induced AMP-kinase activation promotes energy dissipation in
white adipocytes: novelmechanisms integrating HSL and ATGL. Lipid Res.
50(4):704-15, 2009.
Garcia-Caraballo SC, Comhaira TM, Verheyen F, Gaemers I, Schaap FG, Houten
SM, Hakvoort TBM, Dejong CHC, Lamers WH, Koehler SE. Prevention and reversal
of hepatic steatosis with a high-protein diet in mice. Biochim Biophys Acta.
1832(5):685-95, 2013.
Goh EH, Heimberg M. Effects of free fatty acids on activity of hepatic microsomal 3-
hydroxy-3-methylglutaryl coenzyme A reductase and on secretion of triglyceride and
cholesterol by liver. J Biol Chem. 252(9):2822-6, 1977.
Goldberg IJ, Ginsberg HN. Ins and outs modulating hepatic triglyceride and
development of nonalcoholic fatty liver disease. Gastroenterology.130(4):1343–
1346, 2006.
Granneman JG, Moore HP, Granneman RL, Greenberg AS, Obin MS, Zhu Z.
Analysis of lipolytic protein trafficking and interactions in adipocytes. J Biol Chem.
282(8):5726-35, 2007.
Guillén N, Navarro MA, Arnal C, Noone E, Arbonés-Mainar JM, Acin S, Surra JC,
Muniesa P, Roche HM, Osada J. Microarray analysis of hepatic gene expression
identifies new genes involved in steatotic liver. Physiol Genomics. 37(3):187–198,
2009.

69
Haas ME, Attie AD, Biddinger SB. The regulation of ApoB metabolism by insulin.
Trends Endocrinol Metab. 24(8):391-7, 2013.
Hagan DL, Kienzle B, Jamil H, Hariharan N. Transcriptional regulation of human and
hamster microsomal triglyceride transfer protein genes. Cell type-specific expression
and response to metabolic regulators. J Biol Chem. 269(46):28737-44, 1994.
Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, Wang Y. The CREB coactivator
CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature.
524(7564):243-6, 2015.
Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor
4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression
and lipid homeostasis. Mol Cell Biol. 21(4):1393-403, 2001.
Hertz R, Magenheim J, Berman I, Bar-Tana J. Fatty acyl-CoA thioesters are ligands
of hepatic nuclear factor-4alpha. Nature. 392(6675):512-6, 1998.
Higa TS, Spinola AV, Fonseca-Alaniz MH, Evangelista FS. Comparison between
cafeteria and high-fat diets in the induction of metabolic dysfunction in mice. Int J
Physiol Pathophysiol Pharmacol. 6(1):47-54,2014.
Hoekstra M, Li Z, Kruijt JK, Eck MV, Berkel TJCV, Kuiper J. The expression level of
non-alcoholic fatty liver disease-related gene PNPLA3 in hepatocytes is highly
influenced by hepatic lipid status. Journal of Hepatology. 52(2):244–251. 2010.
Horton JD, Bashmakov Y, Shimomura I, Shimano H. Regulation of sterol regulatory
element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci.
95(11):5987-92, 1998.
Hübscher SG. Histological assessment of non-alcoholic fatty liver disease.
Histopathology. 49(5):450-65, 2006.
Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in
apoB-lipoprotein assembly. J Lipid Res. 44(1):22-32, 2003.
Iizuka K, Horikawa Y. ChREBP: a glucose-activated transcription factor involved in
the development of metabolic syndrome. J. Endocr. 55(4):617-24, 2008.

70
Iizuka K, Takeda J, Horikawa Y. Hepatic overexpression of dominant negative Mlx
improves metabolic profile in diabetes-prone C57BL/6J mice. Biochem Biophys Res
Commun. 379(2):499-504, 2009.
Jaeger D, Schoiswohl G, Hofer P, Schreiber R, Schweiger M, Eichmann TO, Pollak
NM, Poecher N, Grabner GF, Zierler KA, Eder S, Kolb D, Radner FP, Preiss-Landl K,
Lass A, Zechner R, Kershaw EE, Haemmerle G. Fasting-induced G0/G1 switch gene
2 and FGF21 expression in the liver are under regulation of adipose tissuederived
fatty acids. J. Hepatol. 63(2):437-45, 2015.
Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, Meseck M, Dong HH.
FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J
Clin Invest. 118(6):2347-64, 2008.
Karimian Pour N, Adeli K. Insulin silences apolipoprotein B mRNA translation by
inducing intracellular traffic into cytoplasmic RNA granules. Biochemistry.
50(32):6942-50, 2011.
Karmen A, Wroblewski F, Ladue Js. Transaminase activity in human blood. J Clin
Invest. 34(1):126-31, 1955.
Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-
alcoholic fatty liver disease. J Gastroenterol. 48(4):434-41, 2013.
Keller H, Dreyer C, Medin J, Mahfoudl A, Ozato K, Wahli W. Fatty acids and retinoids
control lipid metabolism through activation of peroxisome proliferator-activated
receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. 90:2160-2164,
1993.
Kenerson HL, Subramanian S, McIntyre R, Kazami M, Yeung RS. Livers with
constitutive mTORC1 activity resist steatosis independent of feedback suppression of
Akt. PLoS One. 10(2), 2015.
Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Adipose
triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin.
Diabetes. 55(1):148-57, 2006.
Kleiner DE, Brunt EM, Natta MV, Behling C, Contos MJ, Cummings OW, Ferrell LD,
Liu YC, Torbenson MS, Arida AU,Yeh M, McCullough AJ, Sanyal AJ. Design and

71
Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease.
Hepatology. 41 (6):1313-21, 2005.
Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic
steatosis. Clin Mol Hepatol. 19(3):210-5, 2013.
Kumar S, Alagawadi KR, Rao MR. Effect of Argyreia speciosa root extract on
cafeteria diet-induced obesity in rats. Indian J Pharmacol. 43(2):163-7, 2011.
Kuo YT, Lin TH, Chen WL, Lee HM. Alpha-lipoic acid induces adipose triglyceride
lipase expression and decreases intracellular lipid accumulation inHepG2 cells. Eur J
Pharmacol. 692(1-3):10-8, 2012.
Lafontan, M, Dang-Tran, L, Berlan, M. “Alpha-adrenergic antilipolytic effect of
adrenaline in human fat cells of the thigh: comparison with adrenaline
responsiveness of different fat deposits”. Eur J Clin Invest. 9(4): 261-6, 1979.
Lara-Castro C, Garvey WT. Intracellular lipid accumulation in liver and muscle and
the insulin resistance syndrome. Endocrinol Metab Clin N Am. 37(4):841–856.
2008.
Larter CZ, Yeh MM, Rooven DMV, Teoh NC, Brooling J, Hou JY, Williams J, Clyne
M, Nolan CJ, Farrell GC. Roles of adipose restriction and metabolic factors in
progression of steatosis to steatohepatitis in obese, diabetic mice. Journal of
Gastroenterology and Hepatology. 24(10):1658–1668, 2009.
Le May C, Cauzac M, Diradourian C, Perdereau D, Girard J, Burnol AF, Pegorier JP.
Fatty Acids Induce L-CPT I Gene Expression through a PPARα-Independent
Mechanism in Rat Hepatoma Cells. J. Nutr. 135:2313-2319, 2005.
Lehner R, Cui Z, Vance DE. Subcellullar localization, developmental expression and
characterization of a liver triacylglycerol hydrolase. J. Biochem 338 (3):761-8, 1999.
Lehner R, Verger R. Purification and characterization of a porcine liver microsomal
triacylglycerol hydrolase. Biochemistry. 36(7):1861-8. 1997.
Lemberger T, Saladin R, Vázquez M, Assimacopoulos F, Staels B, Desvergne B,
Wahli W, Auwerx J. Expression of the Peroxisome Proliferator-activated Receptor α
Gene Is Stimulated by Stress and Follows a Diurnal Rhythm. J. Biol. Chem.
271(3):1764–1769, 1996.

72
Letexier D, Peroni O, Pinteur C, Beylot M. In vivo expression of carbohydrate
responsive element binding protein in lean and obese rats. Diabetes Metab.
31(6):558-66, 2005.
Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver:
mTORC1 required for stimulation of lipogenesis, but notinhibition of
gluconeogenesis. Proc Natl Acad Sci U S A. 107(8):3441-6, 2010.
Li X, Xu Z, Wang S, Guo H, Dong S, Wang T, Zhang L, Jiang Z. Emodin ameliorates
hepatic steatosis through endoplasmic reticulum-stress sterol regulatory element-
binding protein 1c pathway in liquid fructose-feeding rats. Hepatol Res. 12538.
[Epub ahead of print] 2015.
Liang JS, Distler O, Cooper DA, Jamil H, Deckelbaum RJ, Ginsberg HN, Sturley SL.
HIV protease inhibitors protect apolipoprotein B from degradation by the proteasome:
a potential mechanism for protease inhibitor-induced hyperlipidemia. Nat Med.
7(12):1327-31, 2001.
Lin MC, Gordon D, Wetterau JR. Microsomal triglyceride transfer protein (MTP)
regulation in HepG2 cells: insulin negatively regulates MTP gene expression. J Lipid
Res. 36(5):1073-81, 1995.
Lo V, Erickson B, Thomason-Hughes M, Ko KW, Dolinsky VW, Nelson R, Lehner R.
Arylacetamide deacetylase attenuates fatty-acid-induced triacylglycerol accumulation
in rat hepatoma cells. J Lipid Res. 51(2):368-77, 2010.
Louet JF, Chatelain F, Decaux JF, Park EA, Kohl C, Pineau T, Girard J, Pegorier JP.
Long-chain fatty acids regulate liver carnitine palmitoyltransferase I gene (L-CPT I)
expression through a peroxisome-proliferator-activated receptor alpha (PPARalpha)-
independent pathway. Biochem J. 354(Pt 1):189-97, 2001.
Martins-Santos ME, Gliceroneogênese e fontes de glicerol-3-fosfato para a
síntese de glicerídeo-glicerol no fígado de ratos em diversas situações
experimentais. Ribeirão Preto. Tese (Doutorado – Programa de Pós-Graduação em
Bioquímica). Faculdade de Medicina de Ribeirão Preto. Universidade de São Paulo.
2008.
Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr,
Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARγ in leptin-deficient mice

73
improves fatty liver but aggravates diabetic phenotypes. J. Clin Invest. 111(5):737-
47, 2003.
Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, Mccullough AJ.
Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity.
Gastroenterology.116(6):1413-1419, 1999.
Medina-Gomez G, Gray S, Vidal-Puig A. Adipogenesis and lipotoxicity: role of
peroxisome proliferator-activated receptor gamma (PPARgamma) and
PPARgammacoactivator-1 (PGC1). Public Health Nutrition. 10(10A):1132–1137,
2007.
Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by
oil red O for analyzing the metabolic status in health and disease. Nat Protoc.
8(6):1149-54, 2013.
Melish J, Le NA, Ginsberg H, Steinberg D, Brown WV. Dissociation of apoprotein B
and triglyceride production in very-low-density lipoproteins. Am J Physiol.
239(5):E354-62, 1980.
Miele L, Beale G, Patman G, Nobili V, Leathart J. The Kruppel-like factor 6 genotype
is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology.
135(1):282–91, 2008.
Minnich A, Tian N, Byan L, Bilder G. A potent PPARα agonist stimulates
mitochondrial fatty acid b-oxidation in liver and skeletal muscle. J Physiol
Endocrinol Metab 280:270–279, 2001.
Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, Sterling RK,
Shiffman ML, Stravitz RT, Sanyal AJ. Clinical and histologic spectrum of nonalcoholic
fatty liver disease associated with normal ALT values. Hepatology. 37(6):1286-92,
2003.
Moon BC, Hernandez-Ono A, Stiles B, Wu H, Ginsberg HN. Apolipoprotein B
secretion is regulated by hepatic triglyceride, and not insulin, in a model of increased
hepatic insulin signaling. Arterioscler Thromb Vasc Biol. 32(2):236-46, 2012.
Moore JB, Gunn PJ, Fielding BA. The role of dietary sugars and de novo lipogenesis
in non-alcoholic fatty liver disease. Nutrients. 6(12):5679-703, 2014.

74
Ness GC, Gertz KR. Hepatic HMG-CoA reductase expression and resistance to
dietary cholesterol. Exp Biol Med (Maywood). 229(5):412-6, 2004.
Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin N Am.
39(1):1–7, 2010.
Noh H, Lee H, Kim E, Mu L, Rhee Y K, Cho CW, Chung J. Inhibitory Effect of a
Cirsium setidens Extract on Hepatic Fat Accumulation in Mice Fed a High-Fat Diet
via the induction of Fatty Acid β –Oxidation. Biosci. Biotechnol. Biochem.
77(7):1424-1429, 2013.
Olofsson SO, Borén J. Apolipoprotein B secretory regulation by degradation.
Arterioscler Thromb Vasc Biol. 32(6):1334-8, 2012.
Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase
is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid
signaling and partitioning. Hepatology. 53(1):116-26, 2011.
Organização Mundial de Saúde, disponível em
http://www.who.int/features/factfiles/obesity/facts/en/. Acesso: 01/06/2016
Park EA, Mynatt RL, Cook GA, Kashfi K. Insulin regulates enzyme activity, malonyl-
CoA sensitivity and mRNA abundance of hepatic carnitine palmitoyltransferase-I. J.
Biochem. 310:853-858, 1995.
Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome.
Hippokratia. 13(1):9-19, 2009.
Pease RJ, Wiggins D, Saggerson ED, Tree J, Gibbons GF. Metabolic characteristics
of a human hepatoma cell line stably transfected with hormone-sensitive lipase. J.
Biochem. 341(2):453-60, 1999.
Perheentupa J, Raivio K. Fructose-induced hyperuricaemia. Lancet. 2(7515):528-
31,1967.
Perseghin G, Bonfanti R, Magni S, Lattuada G, De Cobelli F, Canu T, Esposito A,
Scifo P, Ntali G, Costantino F, Bosio L, Ragogna F, Del Maschio A, Chiumello G,
Luzi L. Insulin resistance and whole body energy homeostasis in obese adolescents
with fatty liver disease. Am J Physiol Endocrinol Metab. 291(4):E697-703, 2006.

75
Picard F, Naími N, Richard D, Deshaies Y. Response of adipose tissue lipoprotein
lipase to the cephalic phase of insulin secretion. Diabetes. 48(3):452-9, 1999.
Prentice AM. The emerging epidemic of obesity in developing countries. Int J
Epidemiol. 35(1):93-9, 2006.
Probst MR, Beer M, Beer D, Jenö P, Meyer UA, Gasser R. Human liver
arylacetamide deacetylase. Molecular cloning of a novel esterase involved in the
metabolicactivation of arylamine carcinogens with high sequence similarity to
hormone-sensitive lipase. J Biol Chem. 269(34):21650-6, 1994.
Pullinger CR, North JD, Teng BB, Rifici VA, Ronhild de Brito AE, Scott J. The
apolipoprotein B gene is constitutively expressed in HepG2 cells: regulation of
secretion by oleic acid, albumin, and insulin, and measurement of the mRNA half-life.
J Lipid Res. 30(7):1065-77, 1989.
Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH.
Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism.
Endocrinology. 147(12):5641-52, 2006.
Rasouli N, Molavi B, Elbein SC, Kern PA. Ectopic fat accumulation and metabolic
syndrome. Diabetes Obes Metab. 9(1):1-10, 2007.
Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS, Goldberg
IJ, Schwabe RF, Chua SC Jr, Huang LS. Hepatic overexpression of hormone-
sensitive lipase and adipose triglyceride lipase promotes fatty acidoxidation,
stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem.
283(19):13087-99, 2008.
Rothwell NJ, Saville, ME, Stock M. Effects of feeding a “cafeteria” diet on energy
balance and diet-induced thermogenesis in four strains of rats. J Nutr 112(8): 1515-
1524, 1982.
Rothwell NJ, Stock MJ. Regulation of energy balance in two models of reversible
obesity in the rat. J Comp Physiol Psychol. 93(6):1024-34, 1979.
Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF. Profiling
gene transcription in vivo reveals adipose tissue as an immediate target of tumor
necrosis factor-alpha: implications for insulin resistance. Diabetes. 51(11):3176-88,
2002.

76
Sampey BP, Vanhoose AM, Winfield HM, Freemerman AJ, Muehlbauer MJ, Fueger
PT, Newgard CB, Makowski L. Cafeteria diet is a robust model of human metabolic
syndrome with liver and adipose inflammation: comparison to high-fat diet. Obesity
(Silver Spring). 19(6):1109-17, 2011.
Sclafani A, Springer D. Dietary obesity in adult rats: similarities to hypothalamic and
human obesity syndromes. Physiol Behav. 17(3):461-71,1976.
Shi L, Shi L, Zhang H, Hu Z, Wang C, Zhang D, Song G. Oxymatrine ameliorates
non-alcoholic fatty liver disease in rats through peroxisome proliferator-activated
receptor-α activation. Mol Med Rep. 8(2):439-45, 2013.
Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin
selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-
induced diabetes. Proc Natl Acad Sci U S A. 96(24):13656-61,1999.
Shivaprasad HN, Gopalakrishna S, Mariyanna B, Thekkoot M, Reddy R, Tippeswamy
BS. Effect of Coleus forskohlii extract on cafeteria diet-induced obesity in rats.
Pharmacognosy Res. 6(1):42-5, 2014.
Sidiropoulos KG, Meshkani R, Avramoglu-Kohen R, Adeli K. Insulin inhibition of
apolipoprotein B mRNA translation is mediated via the PI-3 kinase/mTOR signaling
cascade but does not involve internal ribosomal entry site (IRES) initiation. Arch
Biochem Biophys. 465(2):380-8, 2007.
Sidiropoulos KG, Pontrelli L, Adeli K. Insulin-mediated suppression of apolipoprotein
B mRNA translation requires the 5' UTR and is characterized by decreased binding
of an insulin-sensitive 110-kDa 5' UTR RNA-binding protein. Biochemistry.
44(37):12572-81, 2005.
Silva GH, Escanhoela CAF. Nonalcoholic fatty liver disease: pathogenesis and
histological findings, with emphasis on mitochondrial alterations. Rev. Ciênc. Méd.
18(5/6):269-279, 2009.
Singh K, Batuman OA, Akman HO, Kedees MH, Vakil V, Hussain MM. Differential,
tissue-specific, transcriptional regulation of apolipoprotein B secretion by
transforming growth factor beta. J Biol Chem. 277(42):39515-24, 2002.
Sirek AS, Liu L, Naples M, Adeli K, Ng DS, Jin T. Insulin stimulates the expression of
carbohydrate response element binding protein (ChREBP) by attenuating

77
therepressive effect of Pit-1, Oct-1/Oct-2, and Unc-86 homeodomain protein octamer
transcription factor-1. Endocrinology. 150(8):3483-92. 2009.
Soni KG, Lehner R, Metalnikov P, O'Donnell P, Semache M, Gao W, Ashman K,
Pshezhetsky AV, Mitchell GA. Carboxylesterase 3 (EC 3.1.1.1) is a major adipocyte
lipase. J Biol Chem. 279(39):40683-9, 2004.
Sparks JD, Chamberlain JM, O'Dell C, Khatun I, Hussain MM, Sparks CE. Acute
suppression of apo B secretion by insulin occurs independently of MTP. Biochem
Biophys Res Commun. 406(2):252-6, 2011.
Sparks JD, Collins HL, Chirieac DV, Cianci J, Jokinen J, Sowden MP, Galloway CA,
Sparks CE. Hepatic very-low-density lipoprotein and apolipoprotein B production are
increased following in vivo induction of betaine-homocysteine S-methyltransferase.
Biochem J. 395(2):363-71, 2006.
Sparks JD, Sparks CE. Insulin modulation of hepatic synthesis and secretion of
apolipoprotein B by rat hepatocytes. J Biol Chem. 265(15):8854-62, 1990.
Sparks JD, Zolfaghari R, Sparks CE, Smith HC, Fisher EA. Impaired hepatic
apolipoprotein B and E translation in streptozotocin diabetic rats. J Clin Invest.
89(5):1418-30, 1992.
Sparks JD1, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol.
20(3):217-226, 2009.
Suchankova G, Nelson LE, Gerhart-Hines Z, Kelly M, Gauthier MS, Saha AK, Ido Y,
Puigserver P, Ruderman NB. Concurrent regulation of AMP-activated protein kinase
and SIRT1 in mammalian cells. Biochem Biophys Res Commun. 378(4):836-41,
2009.
Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta.
1801(3):299-310, 2010.
Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic
lipids in the metabolic syndrome. Endocrinology. 144(12): 5159–5165, 2003.
van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of
adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study
of liver adenylate deaminase. Biochem J. 162(3):601-9, 1977.

78
Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene
encoding a novel patatin domain-containing protein, is induced by fasting and
glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J
Biol Chem. 279(45):47066-75, 2004.
Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic
Syndrome--an allostatic perspective. Biochim Biophys Acta. 1801(3):338-49, 2010.
Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic
steatohepatitis: selected practical issues in their evaluation and management.
Hepatology. 49(1):306-17, 2009.
Wang DD, Sievenpiper JL, de Souza RJ, Chiavaroli L, Ha V, Cozma AI, Mirrahimi A,
Yu ME, Carleton AJ, Di Buono M, Jenkins AL, Leiter LA, Wolever TM, Beyene J,
Kendall CW, Jenkins DJ. The effects of fructose intake on serum uric acid vary
among controlled dietary trials. J Nutr. 142(5):916-23, 2012.
Wang MY, Grayburn P, Chen S, Ravazzola M, Orci L, Unger RH. Adipogenic
capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc Natl
Acad Sci U S A. 105(16):6139-44, 2008.
Wang Y, Zhang Y, Qian H, Lu J, Zhang Z, Min X, Lang M, Yang H, Wang N, Zhang
P. The g0/g1 switch gene 2 is an important regulator of hepatic triglyceride
metabolism. PLoS One. 8(8):e72315, 2013.
Wei E, Alam M, Sun F, Agellon LB, Vance DE, Lehner R. Apolipoprotein B and
triacylglycerol secretion in human triacylglycerol hydrolase transgenic mice. J Lipid
Res. 48(12):2597-606, 2007.
Wei E, Gao W, Lehner R. Attenuation of adipocyte triacylglycerol hydrolase activity
decreases basal fatty acid efflux. J Biol Chem. 282(11):8027-35, 2007 (a).
Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, Schmitz
J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein
in individuals with abetalipoproteinemia. Science. 258(5084):999-1001, 1992.
Xia X, Yan J, Shen Y, Tang K, Yin J, Zhang Y, Yang D, Liang H, Ye J, Weng J.
Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic
gluconeogenesis. PLoS One. 6(2):e16556, 2011.

79
Xu X, So JS, Park JG, Lee AH. Transcriptional control of hepatic lipid metabolism by
SREBP and ChREBP. Semin Liver Dis. 33(4):301-311, 2013.
Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C,
Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD. Akt stimulates hepatic
SREBP1c and lipogenesis through parallel mTORC1-dependent and independente
pathways. Cell Metab. 14(1):21-32, 2011.
Younossi ZM, Baranova A, Ziegler K, Del Giacco L, Schlauch K, Elariny H, Gorreta
F, VanMeter A, Younoszai A, Ong JP, Goodman Z, Chandhoke V. A genomic and
proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology.
42(3):665–74, 2005.
Zhang W, Bu SY, Mashek MT, O-Sullivan I, Sibai Z, Khan SA, Ilkayeva O, Newgard
CB, Mashek DG, Unterman TG. Integrated Regulation of Hepatic Lipid and Glucose
Metabolism by Adipose Triacylglycerol Lipase and FoxO Proteins. Cell Rep.
15(2):349-59, 2016.
Zhang X, Xie X, Heckmann BL, Saarinen AM, Czyzyk TA, Liu J. Targeted disruption
of G0/G1 switch gene 2 enhances adipose lipolysis, alters hepatic energy balance,
and alleviates high-fat diet-induced liver steatosis. Diabetes. 63(3):934-46, 2014.
Zimmermann R, Lass A, Haemmerle G, Zechner R. Fate of fat: the role of adipose
triglyceride lipase in lipolysis. Biochim Biophys Acta. 1791(6):494-500, 2009.
Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R,
Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat
mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science.
306(5700):1383-6, 2004.
Zou Y, Du H, Yin M, Zhang L, Mao L, Xiao N, Ren G, Zhang C, Pan J. Effects of high
dietary fat and cholesterol on expression of PPARα, LXRα, and their responsive
genes in the liver of apoE and LDLR double deficient mice. Mol Cell Biochem.
323:195–205, 2009.

80
8. ANEXO
Comissão de Ética no Uso de Animais da UFSJ – CEUA/UFSJ
CERTIFICADO
Certificamos que o protocolo para uso de animais em experimentação 016/2014 “Mecanismos
bioquímicos envolvidos na esteatose hepática nos ratos alimentados com dieta cafeteria”,
coordenado pela Prof.ª Valéria Ernestânia Chaves, está de acordo com os Princípios Éticos na
Experimentação Animal, dispostos na Lei Federal no 11.794, de 08.10.2008 e foi aprovado pela
Comissão de Ética no Uso de Animais – CEUA/UFSJ em reunião do dia 12 de dezembro de 2014.
São João del-Rei, 12 de dezembro de 2014.
Prof.ª Leila de Genova Gaya
Coordenadora da Comissão de Ética no Uso de Animais
CEUA-UFSJ