universidade federal de santa catarina centro …livros01.livrosgratis.com.br/cp136398.pdf ·...
TRANSCRIPT

UNIVERSIDADE FEDERAL DE SANTA CATARINA
CENTRO TECNOLÓGICO DEPARTAMENTO DE ENGENHARIA QUÍMICA E
ENGENHARIA DE ALIMENTOS CURSO DE PÓS-GRADUAÇÃO EM ENGENHARIA QUÍMICA
VIVIAN STUMPF MADEIRA
APROVEITAMENTO DE RESÍDUOS DA MINERAÇÃO DE CARVÃO PARA FABRICAÇÃO DE PRODUTOS COM
ELEVADO VALOR AGREGADO
Florianópolis/SC Janeiro de 2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.


VIVIAN STUMPF MADEIRA
APROVEITAMENTO DE RESÍDUOS DA MINERAÇÃO DE CARVÃO PARA FABRICAÇÃO DE PRODUTOS COM
ELEVADO VALOR AGREGADO
Tese submetida ao Programa de Pós-Graduação em Engenharia Química da Universidade Federal de Santa Catarina, para a obtenção do título de Doutor em Engenharia Química.
Orientador: Profª. Drª. Regina de Fátima Peralta Muniz Moreira Co-orientadora: Prof. Dr. Humberto Jorge José
Florianópolis/SC Janeiro de 2010


AGRADECIMENTOS
À Profa Dra Regina F. P. M. Moreira pela especial amizade de todos estes anos, pelo companheirismo, confiança em mim depositada e valorosa orientação.
Ao Prof. Dro Humberto Jorge José pela contribuição em minha coorientação, pelo incentivo, confiança e pelas surpreendentes idéias inovadoras que foram fundamentais para esta pesquisa.
Ao meu marido, Eng. Ariovaldo Dezotti, pelo incentivo, orientação, carinho, amor e paciência nos momentos de crise quando nada dava certo!
Ao Dr. Alfredo Flávio Gazzolla, diretor industrial da Carbonífera Criciúma S/A e maior idealizador deste projeto. Agradeço especialmente pela confiança, pelo incentivo, compreensão e apoio mantidos durante todo o desenvolvimento deste trabalho.
Ao Sr. Antônio Rabelo Gaspar, pela amizade, paciência, orientação, ajuda e incentivo.
A todo pessoal do Laboratório de Pesquisa e Desenvolvimento da Carbonífera Criciúma S/A, especialmente as químicas Priscila dos Reis Martins e Gislaine Paganini dos Santos, que foram fundamentais em várias etapas deste trabalho.
A Prof.a Dra. Ticiane Sauer estimada amiga que teve uma participação importante no desenvolvimento deste trabalho e deixa saudades até hoje do período em que esteve na Carbonífera Criciúma S/A.
Ao geólogo Carlos Henrique Schneider que juntamente com a direção da Carbonífera Criciúma S/A foi um dos idealizados destes projetos de recuperação dos rejeitos da mineração de carvão.
A minha família, em especial o meu pai pelo incentivo e, sobretudo confiança em mim depositada.
E a todas as pessoas que de uma forma ou de outra, contribuíram à sua maneira na realização e conclusão deste trabalho.


RESUMO Os resíduos do processo de beneficiamento do carvão mineral contém minerais sulfetados, como a pirita, a pirolusita e a calcopirita. Quando estes minerais são expostos ao ar e em presença de umidade oxidam-se gerando a drenagem ácida de mina (DAM), e produzindo um efluente líquido com elevada concentração de acidez e de metais dissolvidos. O impacto ambiental decorrente desta carga poluidora ao longo dos anos, fez com que a região Sul catarinense fosse considerada como uma das 14 áreas críticas de poluição do país. A utilização dos resíduos líquidos gerados na indústria de mineração de carvão é exaustivamente explorada nesta tese de doutorado, que tem como objetivo principal recuperar e desenvolver materiais de elevado valor agregado, baseados no ferro e no sulfato, ambos presentes no rejeito líquido da mineração de carvão. O monitoramento da concentração de metais e de sulfato na DAM de uma indústria de mineração de carvão no município de Forquilhinha/SC foi realizado no período de 5 anos, indicando que este efluente líquido pode servir de matéria prima para a obtenção de coagulante químico férrico aluminoso, gesso para a construção civil e pigmentos amarelo e vermelho de óxido de ferro. Dois tipos de tratamento da DAM, denominados tratamento simples ou tratamento seqüencial, foram aplicados nesta tese. No tratamento simples, a DAM é neutralizada soda, causando a precipitação dos óxidos hidróxidos de ferro na concentração de 45% de Fe2O3 e 5% de Al2O3, quando a relação molar Fe/NaOH =0,45. A solubilização deste lodo com quantidade estequiométrica de ácido sulfúrico resulta na formação do coagulante férrico aluminoso contendo 24% de Fe2(SO4)3 e 8% de Al2(SO4)3. O lodo químico produzido no tratamento simples pode também ser utilizado na produção de pigmento de óxido de ferro de baixa cristalinidade. O tratamento seqüencial foi aplicado para a obtenção de produtos com maior valor agregado, especialmente os pigmentos amarelo e vermelho de óxido de ferro. Neste processo, a DAM é inicialmente neutralizada com cal, onde se formam os precipitados de sulfato de cálcio com impurezas de alumínio e ferro. Este gesso amarelo possui como fase cristalina somente o sulfato de cálcio dihidratado, (CaSO4.2H2O) e possui 80% em massa de CaSO4.2H2O e 20% de Al2O3+Fe2O3. As impurezas de alumínio e ferro do gesso Amarelo foram eficientemente removidas pelo tratamento ácido, através do qual se obtém o gesso com características adequadas para o seu uso na construção civil, e um subproduto líquido concentrado, o sulfato férrico aluminoso, que também pode ser utilizado como coagulante químico no tratamento de águas e efluentes líquidos. Dois diferentes processos de produção de pigmentos de óxidos de ferro foram aplicados ao sobrenadante do processo de neutralização com cal, denominados métodos do crescimento e método com formação de intermediário. No método do crescimento, o processo inclui uma etapa de neutralização da solução aquosa contendo sulfato ferroso (6 a 10 g/L) com tempo total de reação de 7 a 8 horas, com dosagem contínua de peróxido

de hidrogênio e soda cáustica, resultando na remoção de 83% do ferro contido na fase aquosa e 80% da acidez. O pigmento produzido através deste processo possui 74-80% de Fe2O3. Os ensaios de colorimetria indicam o valor do ∆Eab (amarelo)>10 e ∆Eab(vermelho)>34, e a difração de raios X indicou que as principais fases cristalinas são a hematita e goetita. No método com formação de intermediário, a DAM com concentração de Fe2+ 4 g/L é rapidamente neutralizada com adição de soda, na razão molar molFe+2/molOH-: 1,0 - 1,5, que resulta na formação de um lodo intermediário por decantação e espessamento. Em seguida, este lodo intermediário é oxidado lentamente para a formação do pigmento amarelo, que é então espessado, filtrado, lavado e seco, seguindo-se as operações de moagem e classificação granulométrica. O pigmento amarelo foi produzido em escala industrial, com características de fase cristalina de goetita (sem calcinação) ou hematita (após calcinação). O teor de Fe2O3 das amostras produzidas na planta obedeceu aos parâmetros especificados na NBR 5.801; ou seja, %Fe2O3 ≥ 83,0%, e o pico temperatura associado à mudança de fase gira entre 270 e 280 0C com uma perda de massa total em torno de 10%. Palavra-chave: Carvão mineral. Resíduos de Mineração. Rejeitos líquidos. Fabricação de produtos. Valor agregado.

ABSTRACT The wastes from the beneficiation of coal containing sulfide minerals such as pyrite, chalcopyrite and the pyrolusite. When these minerals are exposed to air and in the presence of moisture, they oxidize generating acid mine drainage (AMD), and producing an liquid effluent with high concentrations of acidity and dissolved metals. The environmental impact of this pollution load over the years, made the southern state of Santa Catarina was considered one of the 14 critical areas of pollution in the country. The use of liquid waste generated in the coal mining industry is thoroughly explored in this thesis, which has as main objective to recover and develop materials with high added value, based on iron and sulfate, both present in the waste liquid from the coal mining. The monitoring the concentration of metals and sulfate in a DAM coal mining industry in the municipality of Forquilhinha / SC was performed in 5 years, indicating that this effluent is useful as raw material to produce chemical coagulant ferric aluminous, gypsum and yellow and red iron oxides pigments.. Two types of treatment of AMD, called simple treatment or sequential treatment, were applied in the present thesis. In simple treatment, AMD is firstly neutralized with soda using the molar ratio Fe / NaOH = 0.45, causing the formation of a chemical sludge containing 45% Fe2O3 and 5% Al2O3,. The solubilization of this sludge using stoichiometric amount of sulfuric acid results in the formation of aluminium ferric coagulant, containing 24% Fe2 (SO4)3 and 8% Al2(SO4)3. The chemical sludge produced in the simple treatment can also be used in the production of iron oxide pigment of low crystallinity. The sequential treatment was applied to obtain products with higher added value, especially the yellow and red iron oxides pigment. In this case, AMD is firstly neutralized with lime, which forms the precipitates of calcium sulfate with impurities of iron and aluminum. This yellow gypsum presented only one crystalline phase of calcium sulfate dihydrate (CaSO4.2H2O) and has 80 wt% of CaSO4.2H2O and 20% Al2O3 + Fe2O3. Impurities in aluminum and cast iron from Yellow were efficiently removed by acid treatment, whereby you get the plaster with characteristics suitable for use in construction, and a byproduct liquid concentrate, alumina ferric sulphate, which can also be used as chemical coagulant in water treatment and wastewater. Two different processes for producing iron oxide pigments were applied to the supernatant from lime neutralization process, called growth method and method with formation of intermediate. In the method of growth, the process includes a step of neutralizing the aqueous solution containing ferrous sulfate (6-10 g / L) with total reaction time 7-8 hours with continuous dosing of hydrogen peroxide and caustic soda, resulting in removal of 83% of iron contained in the aqueous phase and 80% acidity. The pigment produced by this process has 74-80% of Fe2O3. Colorimetric tests indicate the value of ∆EAB (yellow)> 10 and ∆EAB (red)> 34 and X-ray diffraction indicated that the main crystalline phases are hematite

and goethite. In the method with the formation of intermediary, the AMD with Fe2 + concentration of 4 g/L is rapidly neutralized by adding soda in molar ratio molFe+2/molOH: 1.0 - 1.5, which results in the formation of a intermediate sludge after settling and thickening. Then, this intermediate sludge is slowly oxidized to the formation of yellow pigment, which is then thickened, filtered, washed and dried, followed by the operations of grinding and sieving. The yellow pigment was produced on an industrial scale, with characteristics of the crystalline phase of goethite (without calcination) or hematite (after calcination). The Fe2O3 content of the samples produced at the plant followed the parameters specified in the NBR 5801, ie, Fe2O3% ≥ 83.0%, and peak temperature associated with phase change is between 270 and 280oC with a total mass loss around 10%. Keywords: Coal. Mining waste. Liquid waste. Obtaining products. Added value.

LISTA DE ILUSTRAÇÕES Figura 1 – Constantes de velocidade das reações de oxidação da pirita
na ausência de bactérias em função do pH. ...................... 33 Figura 2 – Velocidade de oxidação do ferro +2 para ferro +3 em meio
ácido biótico e abiótico calculadas pelas equações 3 e 6. (pH< 2,0; p[O2]=1,0 atm; [bactérias]=0,02 NMP/L no meio biótico). ............................................................................. 36
Figura 3 - Sólido de cor do sistema L*a*b* - representação tridimensional e bidimensional. ........................................ 67
Figura 4 - Primeira rota experimental visando o aproveitamento da DAM como matéria-prima para obtenção de coagulante ou pigmento a base de ferro (realizada no período entre 2004 e 2005). ................................................................................ 81
Figura 5 - Segunda rota experimental para o aproveitamento da DAM como matéria-prima para obtenção de pigmento inorgânico, (pigmento 2 ou 3), bem como sub-produtos gerados – Gipsita branca e coagulante 2. ......................................... 82
Figura 6 – Esquema da planta de tratamento de efluentes implantada na Carbonífera Criciúma S.A. - Circuito 1. ........................... 88
Figura 7 - Esquema da planta de tratamento de efluentes implantada na Carbonífera Criciúma S.A. - Circuito 2. (a) fase de formação do intermediário; (b) fase de oxidação e formação do pigmento. ..................................................................... 89
Figura 8 – Concentração de ferro ferroso e ferro total presentes na DAM de Agosto de 2004 até Agosto de 2009. ................. 95
Figura 9 - Concentração de Alumínio presente na DAM de Agosto de 2004 até Agosto de 2009. ................................................. 95
Figura 10 - Concentração de sulfato e acidez presente na DAM de Agosto de 2004 a Agosto de 2009. ................................... 96
Figura 11 - Precipitação de ferro trivalente presente na DAM pela neutralização com NaOH (a) e precipitação de ferro divalente (b). ..................................................................... 99
Figura 12 - Equilíbrio da solubilidade iônica. Fonte: Sawyer e McCarty, 1978. ............................................................................... 100
Figura 13 - Difratograma de raios-X da amostra de Pigmento 1 - PG0. ................................................................................ 106
Figura 14 - Difratograma de raios-X da amostra de Pigmento 1 - PG1. ........................................................................................ 106

Figura 15 - Difratograma de raios-X da amostra de Pigmento 1 - PG2. ................................................................................ 107
Figura 16 - Análise termogravimétrica da amostra de Pigmento 1 - PG10.A (bruta). .............................................................. 110
Figura 17 - Análise termogravimétrica da amostra de Pigmento 1 - PG10.B (após lavagem alcalina). ................................... 110
Figura 18 - Difratogramas de raios-X para o Pigmento 1 - PG10A - seco a diferentes temperaturas. (a) 100°C; (b) 300°C; (c) 500°C; (d) 800°C (e) 1000°C. ........................................ 112
Figura 19 - Difratogramas de raios-X para o Pigmento 1 - PG10B (após lavagem alcalina). (a) 100°C; (b) 300°C; (c) 500°C; (d) 800°C (e) 1000°C. .......................................................... 113
Figura 20 - Difratograma de raios-X da amostra PG10.B (a) e dos pigmentos comerciais; (b) amarelo, (c) vermelho e (d) preto. ............................................................................... 114
Figura 21 - Concentração de FeT presente na DAM e no Sobrenadante 1 pela adição de cal (aplicação na planta industrial). ........ 121
Figura 22 – Concentração do Al+3 presente na DAM e no Sobrenadante 1 pela adição de cal (aplicação na planta industrial). ..... 122
Figura 23 - Acidez presente na DAM e no Sobrenadante 1 pela adição de cal (aplicação na planta industrial). ........................... 122
Figura 24 - Difratogramas de raios-X de duas amostras de Gesso Amarelo obtidas na planta industrial – B = Bassanita – CaSO4.0,5H2O. ............................................................... 126
Figura 25 - Difração de raios-X da amostra calcinada a 600C, 110 0C e 160 0C. B = Bassanita (CaSO4.1/2H2O) e G=Gipsita (CaSO4.2H2O). ............................................................... 127
Figura 26 - Difratograma de raios-X e fases identificadas do Gesso Amarelo (bruto) da Gipsita Branca (gesso limpo). ......... 130
Figura 27 – Análise térmica diferencial do Gesso Amarelo e da Gipsita Branca. ............................................................................ 131
Figura 28 - Difratograma de raios-x da amostra de Pigmento 2 - T104.bruta. ..................................................................... 137
Figura 29 - Difratograma de raios-x da amostra de Pigmento 2 - T104.lavada. ................................................................... 138
Figura 30 - Difratograma de raios-x da amostra de Pigmento 2 - T105.bruta. ..................................................................... 138
Figura 31 - Difratograma de raios-x da amostra de Pigmento 2 - T105.lavada. ................................................................... 139

Figura 32 - Difratograma de raios-x da amostra comercial de pigmento amarelo. .......................................................................... 139
Figura 33 - Difratogramas de raios-X da amostra T105.lavada - Pigmento2 (a) e PG10B - Pigmento1 (b). ....................... 140
Figura 34 - Análise termogravimétrica e térmica diferencial simultâneas - TG (a) e DTA (b). ......................................................... 142
Figura 35 - Difratograma de raios-X da amostra de Pigmento 2 produzida pelo método de crescimento, porém com concentração inicial de ferro no Sobrenadante 1 igual a 3,0 g/L (a). (b) Difratograma de raios-X do pigmento comercial amarelo. .......................................................................... 144
Figura 36 – Difratograma de raios-X da amostra de Pigmento 3 - T112 (R=0,62; CFe+2
inicial=5,9 g/L; alta aeração). .................... 148 Figura 37 - Difratograma de raios-X da amostra de Pigmento 3 - T114
(R=0,88; CFe+2inicial=5,4 g/L; alta aeração).................. 148 Figura 38 - Difratograma de raios-X da amostra de Pigmento 3 - T115
(R=0,9; CFe+2inicial=5,65 g/L; alta aeração).................. 149 Figura 39 - Difratograma de raios-X das amostras de Pigmento 3 -
T123.1 e T123.2 (método com formação de intermediário - alta velocidade de oxidação - R=0,9 – G=Goetita; L=Lepidocrocita). ........................................................... 149
Figura 40 - Difratograma de raios-X das amostras de Pigmento 3 - T123.1 e T124.1 (método com formação de intermediário - alta velocidade de oxidação - R=0,9 – G=Goetita; L=Lepidocrocita). ........................................................... 150
Figura 41 – Difratograma de raios-X da amostra de núcleo alcalino de Goetita - T116 (R=0,33; CFe+2
inicial=42 g/L; T=400C). ... 154 Figura 42 - Difratograma de raios-X da amostra de núcleo alcalino de
Goetita - T117 (R=0,33; CFe+2inicial=42 g/L; T=220C). .. 155
Figura 43 - Difratograma de raios-X da amostra de núcleo alcalino de Goetita - T118 (R=0,33; CFe+2
inicial=7 g/L; T=220C). .... 155 Figura 44 - Difratograma de raios-X da amostra de núcleo ácido de
Goetita - T119 (R=1,30; CFe+2inicial=24 g/L;T=400C). ... 156
Figura 45 - Difratograma de raios-X da amostra de núcleo ácido de Goetita - T120 (R=1,30; CFe+2
inicial=24 g/L;T=250C). ... 156 Figura 46 - Difratograma de raios-X da amostra de núcleo ácido de
Goetita - T121 (R=1,3; CFe+2inicial=9,5 g/L;
T=ambiente). ................................................................... 157 Figura 47 - Difratograma de raios-X da amostra de pigmento amarelo
comercial......................................................................... 157

Figura 48 - Difratogramas de raios-X da amostra de Pigmento 3 - T137 (Cinicial = 2,9g/L, Fe+2
sólido inicial= 4,8g/L) e T140 (Cinicial = 6,8 g/L, Fe+2
sólido inicial= 9,0g/L) ambas produzidas com R=0,33. M=Magnetita; H=Hematita. ............................. 164
Figura 49 - Difratograma de raios-X da amostra de Pigmento 3 - T136 (Cinicial = 6,0 g/L, Fe+2
sólido inicial =31g/L) produzida com R ≅ 0,33. ................................................................................ 164
Figura 50 - Difratogramas de raios-X de duas amostras de pigmento comercial de óxido de ferro. (a) Pigmento preto (magnetita) e (b) Pigmento vermelho (Hematita). ............................. 165
Figura 51 – Difratogramas de raios-X das amostras de Pigmento 3, T137 (Cinicial= 2,9g/L, Fe+2
sólido inicial =3,3g/L) e T140 (Cinicial= 6,8 g/L, Fe+2
sólido inicial =8,9g/L) produzidas com R=0,5. L=Lepidocrocita; G=Goetita. ............................. 166
Figura 52 – Difratogramas de raios-X das amostras de Pigmento 3 T142.am.1 (Cinicial=6,4g/L) e T142.am.3 (Cinicial=4,4 g/L), produzidas com R=0,5714. ............................................. 166
Figura 53 – Difratograma de raios-X da amostra de Pigmento 3 - T129.am.1 (Cinicial=2,9g/L). ............................................ 167
Figura 54 - Difratograma de raios-X de uma amostra de pigmento comercial amarelo (Goetita). .......................................... 167
Figura 55 - Acompanhamento da oxidação do ferro ferroso sólido (composto intermediário) no teste T140 e T137 (R≅0,5). .......................................................................... 169
Figura 56 - Acompanhamento da oxidação do ferro ferroso sólido nos testes T142 e T129 com R≅0,57(Pigmento 3). ............... 170
Figura 57 – Difratogramas de raios-X da amostra de Pigmento 3 - T135.am.1 e T135.am.3 (Cinicial=4g/L, diferente valor de R). ................................................................................... 173
Figura 58 - Difratogramas de raios-X da amostra de Pigmento 3 - T138.am.1, am.2 e am.3 (Cinicial=3,8g/L, diferentes valores de R). .................................................................. 173
Figura 59 - Difratogramas de raios-X das amostras de Pigmento 3 - T140.am.1, am.2, am.3 e am.4 (Cinicial=6,8g/L, diferentes valores de R). .................................................................. 174
Figura 60 - Difratogramas de raios-X das amostras de Pigmento 3 - T141.am.1, am.2 e am.3 (Cinicial=6,5g/L, diferentes valores de R). .................................................................. 174

Figura 61 - Difratogramas de raios-X de cinco amostras de Pigmento 3 produzidas na planta industrial da empresa Carbonífera Criciúma S.A .................................................................. 179
Figura 62 - Difratogramas de raios-X da amostra de Pigmento 3 produzida na planta industrial (Batelada 5), seca a 100 e 200oC. ............................................................................. 182
Figura 63 - Difratogramas de raios-X da amostra de Pigmento 3 produzida na planta industrial (Batelada 5), calcinada a 300 e 500oC............................................................................ 182
Figura 64 - Difratograma de raios-X de uma amostra de pigmento comercial vermelho......................................................... 183


LISTA DE TABELAS Tabela 1 - Métodos de controle e prevenção da DAM ......................... 43 Tabela 2 - Técnicas de remediação e tratamento da DAM. .................. 44 Tabela 3 - Padrões para o lançamento de efluentes da CONAMA No
357. ................................................................................... 47 Tabela 4 - Constituintes de três amostras de fosfogesso encontradas na
literatura. ........................................................................... 54 Tabela 5 - Composição química média do depósito de Gipsita do Polo
Gesseiro de Araripe/PE. .................................................... 57 Tabela 6 - Os óxidos de ferro. .............................................................. 63 Tabela 7 - Classificação com relação a diferença de cor utilizada na
indústria de tintas de impressão. ....................................... 67 Tabela 8 - Métodos para a obtenção do núcleo de Goetita (αFeO.OH)
utilizados industrialmente na produção de pigmento. ....... 70 Tabela 9 - Condições experimentais ótimas citadas na literatura para
obtenção da Goetita. ......................................................... 73 Tabela 10 - Valores ótimos para R citados na literatura para formação
da Goetita em função da temperatura - solução aquosa inicial de 0,1 mol/L de sulfato ferroso. ............................. 75
Tabela 11 - Histórico da caracterização da drenagem ácida de mina no período avaliado neste trabalho. ....................................... 92
Tabela 12 - Precipitação de ferro férrico a partir da DAM pela neutralização direta com NaOH. ....................................... 98
Tabela 13 - Precipitação de ferro ferroso a partir da DAM pela neutralização direta com NaOH. ....................................... 98
Tabela 14 - Obtenção do Coagulante 1 pela solubilização do Lodo químico 1 (ferro férrico) em ácido sulfúrico. .................. 101
Tabela 15 - Resultados do ensaio de coagulação, floculação, decantação utilizando-se um efluente têxtil e o Coagulante 1 versus um Sulfato férrico Comercial. ............................................... 102
Tabela 16 - Análise de custo para produção do Coagulante 1 (sulfato férrico aluminoso) a partir da neutralização direta da DAM com NaOH. ..................................................................... 103
Tabela 17 – Parâmetros utilizados nos testes para obtenção do Pigmento 1. ...................................................................... 105
Tabela 18 - Descrição de duas lavagens básicas feitas na amostra de Pigmento 1 - PG10 - para remoção de SO4
2-. ................. 108

Tabela 19 - Perda de massa e teor de Fe2O3 nos pigmentos PG10A e PG10B (Pigmento 1), após calcinação. .......................... 109
Tabela 20 - Caracterização dos pigmentos comerciais e do Pigmento 1, (PG10B) obtido nesta etapa do trabalho. ........................ 115
Tabela 21 - Tratamento parcial da DAM realizado em laboratório – Remoção do alumínio, ferro férrico, sulfato e acidez. .... 119
Tabela 22 - Caracterização do Gesso Amarelo produzido pelo circuito 1 em escala de laboratório (amostra úmida filtrada).......... 124
Tabela 23 - Caracterização do Gesso Amarelo produzido pelo circuito 1 na planta industrial (em base seca). ................................ 125
Tabela 24 - Condições experimentais e resultados obtidos na variação do volume de água para reação. ...................................... 129
Tabela 25 - Remoção do ferro ferroso presente no Sobrenadante 1 pela neutralização lenta com NaOH. Circuito 2 – Método de crescimento em escala de laboratório. ............................ 133
Tabela 26 - Porcentagens de remoção do Fe2+, Al3+, SO4-2 e acidez do
Sobrenadante 1 pela neutralização com NaOH. Circuito 2 – Método de crescimento em escala de laboratório. .......... 135
Tabela 27 - Colorimetria e caracterização química das amostras de Pigmento 2 produzidas pelo método de crescimento. ..... 140
Tabela 28 - Distribuição do tamanho de partículas por difração a laser. ............................................................................... 141
Tabela 29 - Condições experimentais dos testes para a obtenção do Pigmento 3 com alta velocidade de oxidação. ................ 147
Tabela 30 - Caracterização físico-química e colorimétrica das amostras preliminares de Pigmento 3 e de uma amostra comercial de pigmento amarelo. .......................................................... 152
Tabela 31 - Condições experimentais dos testes realizados para a produção do núcleo de Goetita. ...................................... 154
Tabela 32 - Caracterização físico-quimica das amostras de núcleo de Goetita produzidas pelo método alcalino e método ácido. .............................................................................. 160
Tabela 33 - Condições experimentais dos testes realizados para avaliação do valor de R versus a concentração inicial de ferro ferroso na região do alcalino ao estequiométrico. .. 163
Tabela 34 - Caracterização físico-quimica e colorimétrica das amostras de Pigmento 3 produzidas com R na região do alcalino ao estequiométrico. Caracterização também de amostras de pigmento comerciais. ...................................................... 171

Tabela 35 - Condições experimentais dos testes realizados para avaliação do valor de R versus a concentração inicial de ferro ferroso no método ácido. ........................................ 172
Tabela 36 - Caracterização físico-quimica e colorimétrica das amostras de Pigmento 3 produzidas com R ácido e de uma amostra de pigmento amarelo comercial. ..................................... 175
Tabela 37 - Produção de pigmento amarelo na planta industrial pelo método com formação de intermediário. ........................ 178
Tabela 38 - Caracterização das amostras de pigmento produzidas na planta industrial. ............................................................. 180
Tabela 39 - Diferença de cor das amostras de pigmento amarelo produzidas na planta industrial da CCSA para três amostras comerciais de pigmento amarelo. ................................... 181


SUMÁRIO 1 INTRODUÇÃO ................................................................................ 25 2 OBJETIVOS GERAIS ..................................................................... 29 2.1 OBJETIVOS ESPECÍFICOS .......................................................... 29 3 REVISÃO BIBLIOGRÁFICA ........................................................ 31 3.1 GERAÇÃO DE DRENAGEM ÁCIDA DE MINA DE CARVÃO 31 3.1.1 Oxidação da pirita ........................................................................ 31 3.1.2 Bio-oxidação da pirita .................................................................. 37 3.2 PREVENÇÃO DA DRENAGEM ÁCIDA DE MINA ................... 42 3.3 TRATAMENTO DA DRENAGEM ÁCIDA DE MINA ................ 44 3.3.1 Tratamento químico ativo da DAM ............................................. 46 3.2.2 Tratamento da DAM – Estudo de caso......................................... 49 3.4 APROVEITAMENTO DE RESÍDUOS DA MINERAÇÃO ......... 51 3.4.1 Aproveitamento de resíduos contendo ferro ................................. 51 3.4.2 Aproveitamento de resíduos contendo sulfato de cálcio .............. 53 3.5 GESSO PARA A CONSTRUÇÃO CIVIL ..................................... 56 3.6 COAGULANTES INORGÂNICOS À BASE DE FERRO ............ 60 3.7 OS ÓXIDOS DE FERRO ............................................................... 63 3.7.1 Pigmentos inorgânicos de óxidos de ferro .................................... 64 3.7.1 Colorimetria ................................................................................. 66 4 METODOLOGIA EXPERIMENTAL ........................................... 81 4.1 METODOLOGIA APLICADA A PRIMEIRA ROTA
EXPERIMENTAL ......................................................................... 83 4.1.1 Caracterização da drenagem ácida de mina .................................. 83 4.1.2 Neutralização da DAM com NaOH ............................................. 83 4.2 METODOLOGIA APLICADA À SEGUNDA ROTA
EXPERIMENTAL ..................................................................... 85 4.2.1 Neutralização parcial da DAM com cal ....................................... 85 4.2.2 Neutralização do Sobrenadante 1 com NaOH .............................. 86 4.3 ESTAÇÃO DE TRATAMENTO DE EFLUENTES DA
CARBONÍFERA CRICIÚMA S.A. ............................................... 88 5 RESULTADOS DA PRIMEIRA ROTA EXPERIMENTAL ...... 91 5.1 CARACTERIZAÇÃO DA DRENAGEM ÁCIDA DE MINA ....... 91 5.2 NEUTRALIZAÇÃO DA DAM COM NAOH ................................ 96 5.2.1 Obtenção do Lodo Químico 1 e do Coagulante 1 ........................ 96

5.2.2 Obtenção do Lodo Químico 1 e do Pigmento 1 ......................... 104 6 RESULTADOS DA SEGUNDA ROTA EXPERIMENTAL ..... 117 6.1 NEUTRALIZAÇÃO PARCIAL DA DAM COM CAL ............... 117 6.2 CARACTERIZAÇÃO E PURIFICAÇÃO DO GESSO
AMARELO .................................................................................. 123 6.2.1 Caracterização do Gesso Amarelo ............................................. 123 6.2.2 Purificação do Gesso Amarelo ................................................... 127 6.3 NEUTRALIZAÇÃO DO SOBRENADANTE 1 COM NAOH.... 131 6.3.1 Método de Crescimento - Obtenção do Pigmento 2 ................. 131 6.3.2 Método com formação de intermediário - Obtenção do Pigmento 3
................................................................................................. 144 7 PRODUÇÃO DO PIGMENTO AMARELO NA PLANTA
INDUSTRIAL ............................................................................. 177 8 CONCLUSÕES .............................................................................. 184 REFERENCIAS ................................................................................ 187 ANEXO A - PRODUÇÃO TÉCNICA E BIBLIOGRAFICA ....... 197

1 INTRODUÇÃO A elevação ininterrupta da demanda por carvão mineral após o
ano 2000 revela que os países voltaram a considerar este mineral como um importante recurso para o primeiro grande objetivo de políticas energéticas, ou seja, a segurança no fornecimento (Heck, R. F., 2008).
Segundo dados da British Petroleum, 2008, as reservas mundiais de carvão mineral foram estimadas ao final de 2007 em 847.488,0 milhões de toneladas. Existem reservas de carvão em todos os continentes, entretanto há uma concentração maior na Europa e Eurásia, (32% das reservas mundiais), América do Norte (com 30%), Ásia e Oceania (também com 30% das reservas).
As cinco maiores reservas de carvão mineral se encontram nos Estados Unidos (242.721 milhões de toneladas), na Rússia (157.010 milhões de toneladas), China (114.500 milhões de toneladas), Austrália (76.600 milhões de toneladas) e Índia (56.498 milhões de toneladas).
A qualidade desse recurso mineral é representada pela quantidade de carbono fixo contido em sua estrutura. Desta forma, o carvão mineral é dividido em classificações. A turfa é um dos primeiros estágios do carvão, com o menor teor de carbono fixo, é seguida pelo lignito, pelo carvão betuminoso, o mais utilizado como fonte combustível, e por fim, pelo carvão antracito o carvão mineral mais puro e de melhor qualidade com mais de 90% de carbono fixo em sua estrutura.
Grande parte dos problemas da utilização do carvão mineral como fonte de energia decorrem das externalidades negativas geradas por este mineral. Na geração de eletricidade, o carvão é um grande emissor de óxidos de nitrogênio (NOx), dióxido de carbono (CO2), material particulado e dióxido de enxofre (SOx), sendo este último o maior responsável pelo fenômeno conhecido como “chuva ácida”. Dessa forma, antecedendo a utilização do carvão mineral, deve ser realizado um processo de concentração ou beneficiamento do carvão o qual visa a remover o enxofre presente no mesmo evitando a geração de SOx.
Especificamente no Brasil, no estado de Santa Catarina, o carvão mineral caracteriza-se pela elevada presença de enxofre, em torno de 4 a 5% e elevado teor cinzas, em torno de 58 a 62%. Isto resulta em um carvão com baixo poder calorífico e baixo teor de carbono fixo, de 2.700 a 2.800 kcal/kg e de 21,4 a 26,5%, respectivamente (DNPM, 2000).
Essas características do carvão catarinense associadas ao modelo de aproveitamento atual conferem a este carvão grande capacidade poluidora devido ao volume de resíduos gerados tanto na mineração
25

quanto na geração termoelétrica. Os resíduos do processo de beneficiamento do carvão contém
minerais sulfetados, tais como a pirita, a pirolusita e a calcopirita. Estes minerais quando expostos ao ar e em presença de umidade oxidam-se gerando a drenagem ácida de mina (DAM). Ocorre a dissolução dos componentes contidos nas rochas, produzindo um efluente líquido com elevada concentração de acidez e de metais dissolvidos. O impacto ambiental decorrente desta carga poluidora ao longo dos anos, fez com que a região Sul catarinense fosse considerada como uma das 14 áreas críticas de poluição do país, conforme o Decreto Federal no. 85206 de 25/09/1980.
A crescente preocupação com a conservação do meio ambiente tem atingido todos os setores industriais, inclusive a indústria de mineração. É, portanto, necessário inovar no gerenciamento de resíduos e desenvolver novas tecnologias que visem o reaproveitamento destes materiais. A introdução do conceito “resíduo zero” na atividade industrial tem oportunizado não somente a minimização da geração de resíduos, sejam eles sólidos, líquidos ou gasosos, quanto a criação de novas oportunidades de geração de emprego e fonte de renda.
A utilização dos resíduos líquidos gerados na indústria de mineração de carvão é exaustivamente explorada nesta tese de doutorado, que tem como objetivo principal recuperar e desenvolver materiais de elevado valor agregado, baseados no ferro e no sulfato, ambos presentes no rejeito líquido da mineração de carvão. Esta tese apresentara no capítulo 2 os objetivos gerais e específicos e no capítulo 3 será apresentada a revisão bibliográfica e o estado da arte do aproveitamento e do tratamento dos resíduos da mineração de carvão.
A revisão bibliográfica iniciar-se-á com a geração da drenagem ácida de mina, seguida das tecnologias de tratamento desta drenagem e as experiências encontradas para o reaproveitamento dos resíduos da mineração. Após isto, será apresentada uma revisão sobre minerais sulfatados, mais especificamente sobre o gesso para a construção civil, e em seguida será apresentada uma revisão bibliográfica sobre os coagulantes inorgânicos a base de ferro, os óxidos de ferro e especificamente sobre pigmentos inorgânicos de óxidos de ferro.
No capítulo 4 serão apresentadas a duas abordagens metolodológicas utilizadas assim como a descrição experimental e as técnicas analíticas.
No capítulo 5 serão apresentados os resultados obtidos da aplicação da primeira abordagem metodológica e no capitulo 6 os
26

resultados da segunda abordagem. Os resultados obtidos neste trabalho confirmam a viabilidade de
obtenção de produtos de elevado valor agregado a partir do tratamento da drenagem ácida de mina de carvão. Com isso, foi projetada e implanta uma planta industrial de tratamento da drenagem ácida de mina que visa a produzir pigmentos inorgânicos de óxido de ferro além dos subprodutos associados. Os resultados da operação desta planta, bem como a caracterização do pigmento obtido serão apresentados no capítulo 7.
No Anexo A são apresentados resumidamente os pedidos de privilégio de invenção depositados até a presente data.
27


2 OBJETIVOS GERAIS O objetivo geral deste trabalho é recuperar e desenvolver
materiais de elevado valor agregado, baseados no ferro e no sulfato, partindo-se do rejeito líquido gerado na mineração de carvão, mais especificamente a partir da drenagem ácida de mina de carvão (DAM).
2.1 OBJETIVOS ESPECÍFICOS Os objetivos específicos da tese são: a) Desenvolver um método para produção de coagulante
inorgânico à base de ferro, a partir da drenagem ácida de mina. - Caracterizar o coagulante desenvolvido.
b) Desenvolver um método para produção de pigmento
inorgânico amarelo de óxido de ferro, a partir da drenagem ácida de mina. - Caracterizar o pigmento inorgânico obtido durante todo o
processo de desenvolvimento do método, de tal forma que se obtenha um produto final com a mesma qualidade dos pigmentos inorgânicos ja existentes no mercado.
c) Desenvolver um método para fabricação de gesso para
construção civil ou gesso agrícola a partir da drenagem ácida de mina. - Caracterizar o gesso obtido, de tal forma que se obtenha um
produto final com a mesma qualidade do gesso existente no mercado nacional.


3 REVISÃO BIBLIOGRÁFICA
3.1 GERAÇÃO DE DRENAGEM ÁCIDA DE MINA DE CARVÃO A poluição hídrica causada pelas drenagens ácidas é o impacto
ambiental mais significativo das operações de mineração, beneficiamento e re-beneficiamento do carvão mineral. Esta poluição é decorrente da percolação da água de chuva através dos rejeitos gerados nas atividades de lavra e beneficiamento, alcançando os corpos hídricos superficiais e/ou subterrâneos.
A drenagem ácida de mina é rica em íons metálicos solúveis, gerados pelo contato de água e ar com minerais sulfetados. O rejeito sólido oriundo da mineração de carvão é rico em minerais sulfetados, tais como a pirita, a pirolusita e a calcopirita. O contato desse rejeito com água e ar provoca a lixiviação das pilhas de rejeito, resultando em elevadas concentrações de sulfatos metálicos em solução aquosa, entre eles, o FeSO4, CuSO4, ZnSO4 e Al2(SO4)3. Como o rejeito é predominantemente pirita, FeS2, os componentes majoritários da solução aquosa são Fe+2 e SO4
-2 e a DAM é atribuída principalmente a oxidação da pirita (Johnson e Hallberg, 2005). O impacto da drenagem ácida de mina em ecossistemas de rios e córregos se da com o aumento da acidez, a precipitação de íons férricos, a diminuição do oxigênio dissolvido, e aumento da concentração de metais pesados.
3.1.1 Oxidação da pirita A oxidação da pirita ocorre quando o mineral sulfetado é exposto
ao contato com ar e água. O processo é complexo e participam diversos tipos de reações de oxi-redução, hidrólise e formação de íons complexos, através de mecanismos químicos e biológicos. A velocidade da reação de oxidação da pirita depende de vários fatores, entre eles, a concentração de oxigênio e ferro (ferroso e férrico), quantidade de CO2, pH, atividade bacteriana, temperatura, área superficial do mineral sulfetado e cristalinidade do mineral (Johnson e Hallberg, 2005). Em condições ótimas de temperatura e pH, as reações assistidas pelas bactérias são muito mais rápidas do que aquelas promovidas pelo mecanismo químico e consequentemente a velocidade de geração da drenagem ácida é muito maior.

Tem sido reportado que a oxidação inicial da pirita ocorre pelo oxigênio atmosférico produzindo H2SO4 e ferro ferroso em solução (Fe+2). O ferro ferroso pode ser oxidado a ferro férrico (Fe+3) pelo oxigênio dissolvido no meio com a posterior hidrólise e precipitação na forma de oxi-hidróxido-férrico - Fe(OH)3. Com isso, adicional quantidade de ácido é liberada para a solução e o pH do meio reacional torna-se mais baixo (Evangelou, V.P., 1995).
Durante este estágio inicial, a oxidação da pirita é um processo lento (Ivanov, 1962). Entretanto, na medida em que a oxidação continua e o pH se torna mais baixo, alcançando valores menores do que 3.5, a formação de hidróxido férrico é substancialmente reduzida e o ferro férrico obtido fica disponível no meio reacional. Nestas condições, a oxidação da pirita ocorre preferencialmente pelo ferro férrico, e este se torna o principal mecanismo de produção de ácido da drenagem ácida (Singer and Stumm, 1970).
Em baixos valores de pH, um tipo de bactéria acidofílica, quimioautotrófica, ferro oxidante, denominada Thiobacillus ferroxidans, catalisa e acelera a oxidação de Fe+2 para Fe+3 pelo oxigênio dissolvido. Com isso, o processo de oxidação da pirita pelo Fe+3 é consideravelmente acelerado, e desta forma fecha-se um ciclo contínuo de oxidação da pirita controlado pelo pH. Desta forma, as reações de oxidação da pirita quando exposta ao ar e água podem ser descritas como:
Oxidação da pirita pelo oxigênio molecular dissolvido no meio líquido:
)()(2
4)(2
)(2)(2)(2 222
7aqaqaqaqaqs
HSOFeOHOFeS +−+ ++→++ (1)
Oxidação do ferro ferroso em meio ácido a ferro férrico na
presença de oxigênio dissolvido:
OHFeHOFe 23
22
2
1
4
1+→++ +++ (2)
Hidrólise e precipitação de ferro férrico que ocorre em pH ≥ 3,5:
++ +→+ HOHFeOHFe 3)(3 323 (3)
Oxidação da pirita, ou qualquer outro mineral sulfetado, pelo
32

ferro férrico em pH < 3,5: +−++ ++→++ HSOFeOHFeFeS 16215814 2
42
23
2 (4)
oSFeMMSFe ++→+ +++ 223 22 (5)
Apesar de o oxigênio ser necessário para a liberação do ferro a
partir da pirita, (Reação 1), é consenso na literatura que a velocidade de oxidação da pirita, tanto em meio abiótico quanto biótico é muito mais rápida quando realizada com o íon férrico como oxidante. Na Figura 1, (Evangelou, V.P.), observa-se claramente a influência do pH nas constantes de velocidade das reações de oxidação da pirita na ausência das bactérias Thiobacillus ferroxidans. Ressalta-se novamente que a reação 1 é a oxidação da pirita pelo oxigênio dissolvido, a reação 2 é a oxidação do ferro ferroso a ferro férrico e a reação 4 é a oxidação da pirita pelo ferro férrico.
Reação 1
Reação 2
Reação 4
Figura 1 – Constantes de velocidade das reações de oxidação da pirita na
ausência de bactérias em função do pH. Fonte: Nordstrom, 1982.
Observa-se, na ausência de bactérias, que em pH < 4,5 a pirita é
oxidada pelo Fe+3, mais rapidamente do que pelo oxigênio, e é oxidada pelo oxigênio mais rapidamente do que o ferro +2 é oxidado a ferro +3. Por essa razão, na ausência de bactérias e em pH < 4,5 a oxidação de ferro ferroso é a etapa limitante da oxidação da pirita (Singer and Stumm, 1970). Neste caso, se a intenção for gerar a drenagem ácida para
33

a posterior recuperação de metais, a introdução das bactérias Thiobacillus ferroxidans pode acelerar em até 106 vezes a velocidade de oxidação do ferro ferroso, (reação 2), suprindo o insumo necessário para a oxidação da pirita.
Em pH próximo à neutralidade (pH > 4,5), a oxidação do ferro ferroso a férrico ocorre rapidamente, mas a concentração de ferro trivalente (Fe+3) diminui devido a hidrólise e precipitação (reação 3). Neste caso, há muito pouca participação das bactérias na oxidação da pirita e em tais condições, o oxigênio é mais importante oxidante do que o ferro trivalente (Goldhaber, 1983; Hood, 1991).
Williamson e Rimstidt (1994) propuseram uma equação cinética para descrever a velocidade da oxidação da pirita na presença de oxigênio molecular e ausência de bactérias (eq.1).
[ ][ ]
)./(10][ 2
40,02
93,0307,62 smmol
Fe
Fe
dt
FeSd
+
+−=− Equação 1
Na ausência do oxigênio e das bactérias, a velocidade de
oxidação da pirita pelo íon férrico (reação 4), é diferente daquela encontrada em ambiente oxigenado, tornando-se mais dependente do pH, conforme descrito abaixo (Williamson e Rimstidt, 1994).
[ ][ ] [ ]
)./(10][ 2
32,047,02
30,0358,82 smmol
HFe
Fe
dt
FeSd
++
+−=− Equação 1
As equações 1 e 2 mostram que a velocidade de oxidação da
pirita é fortemente dependente da relação Fe+3/Fe+2 em solução aquosa, ou seja, quanto maior a relação Fe+3/Fe+2, maior a velocidade da reação de oxidação.
Lowson (1982) reportou três equações cinéticas para descrever a velocidade de oxidação do ferro ferroso, reação 2, pelo oxigênio dissolvido em função do pH, temperatura e concentração de ferro e oxigênio (equações 3 a 5).
[ ] [ ] )0,2(2
222
<=− ++
pHpOCFekdt
dCFe Equação 2
34

[ ][ ][ ] )0,50,2(22
2
<<=− −++
pHOHpOCFekdt
dCFe Equação 3
[ ][ ] [ ] )0,5(2
22
2
>=− −++
pHOHpOCFekdt
dCFe Equação 4
Em pH < 2,0 e 25oC, a velocidade da reação é de segunda ordem
em relação a concentração de Fe2+, primeira ordem em relação à concentração de oxigênio, e é independente do pH. O valor da constante cinética encontra-se entre 6,5 e 40 x 10-7 M-1.atm-1.min-1. Com o aumento do pH, a velocidade da reação torna-se mais dependente do pH e menos dependente da concentração de ferro.
Para pH > 3,5 Singer e Stumm (1970) chegaram a mesma equação de velocidade do ferro ferroso que Lowson, 1982 para pH > 5,0, ou seja, a ordem da reação é 1 para o ferro ferroso e o oxigênio e 2 para o pH, sendo a constante cinética igual a 8x1013 M-2.atm-1.min-1 a 25 0C. A velocidade de oxidação do ferro ferroso a ferro férrico pelo oxigênio molecular, intermediada pelas bactérias Thiobacillus
Ferroxidans é descrita pela Equação 6:
[ ][ ][ ][ ]
[ ][ ][ ] )2,2(exp10.62,1
)2,2(exp10.62,1
77,58
2211
)2
77,58
2211
)2
<=−
>=−
−+
+
−++
+
pHpOCFeCdt
dCFe
pHHpOCFeCdt
dCFe
RTbact
RTbact Equação 5
Neste caso, em pH >2,2 a velocidade da reação é diretamente
proporcional à concentração de bactérias, Fe+2, O2 e H+ e o valor da constante de velocidade da reação é multiplicado pela exponencial 1011.
Se compararmos as equações 3 e 6, ou seja, as velocidades de oxidação de ferro ferroso em pH<2,0 em meio abiótico e biótico, observa-se que em pH<2,0 e sem a presença de bactérias a velocidade de oxidação do ferro ferroso é praticamente desprezível, ao passo que com um mínimo de 0,02 NMP/L de Thiobacillus ferroxidans, a velocidade torna-se significativamente maior (Figura 2).
35

-
20.000.000
40.000.000
60.000.000
80.000.000
100.000.000
120.000.000
- 0,05 0,10 0,15 0,20 0,25 0,30 0,35 0,40
[Fe+2], mol/L
- d[
Fe+
2]/d
t, (m
ol/L
) / m
in
meio biótico meio abiótico
Figura 2 – Velocidade de oxidação do ferro +2 para ferro +3 em meio ácido biótico e abiótico calculadas pelas equações 3 e 6. (pH< 2,0; p[O2]=1,0 atm;
[bactérias]=0,02 NMP/L no meio biótico). Mare et al. (2004) estudaram um processo integrado de oxidação
e neutralização do ferro ferroso presente na drenagem ácida de mina. Eles propuseram que a velocidade de oxidação abiótica de ferro ferroso, em pH 4,5 a 5,5 e alta concentração de sólidos suspensos, obtida através do reciclo do lodo decantado, não depende somente da concentração de ferro, pH e quantidade de oxigênio dissolvido na água. Nestas condições, a velocidade é influenciada, também, pela área superficial do reator, concentração de sólidos suspensos e intensidade da mistura, de acordo com a Eq. 7.
[ ] [ ] [ ] [ ] [ ] [ ] 5,05,05,05,15,02
5,022 ][
MSSRSAOHOFekdt
Fed −++
=− Equação 6
sendo [Fe+2] a concentração de Fe+2 (mol.L-1), [O2] a
concentração de oxigênio dissolvido (mol.L-1); [OH-] a concentração de
íons hidroxila (mol.L-1); RSA é a área superficial do reator em m2/m3, SS é a concentração de sólidos suspensos (g/L), e M é a intensidade da mistura em rpm. A dependência da constante cinética com a temperatura (entre 5 e 37oC) foi estabelecida de acordo com a equação de Arrhenius e resultou na energia de ativação de 21,55 kcal/mol (Mareet al., 2004).
36

3.1.2 Bio-oxidação da pirita O processo de bio-oxidação da pirita, ou biolixiviação da pirita, é
a lixiviação da pirita catalisada por bactérias. O mesmo consiste de uma série de reações bioquímicas e químicas que solubilizam o metal de uma forma direta, quando a própria bactéria ataca o sulfeto metálico, ou de uma forma indireta, quando os produtos do metabolismo bacteriano atuam sobre o sulfeto metálico. Dentre os agentes que podem participar da biolixiviação da pirita, destacam-se o oxigênio dissolvido, o sulfato férrico, o ácido sulfúrico e o Thiobacillus ferroxidans, sendo este último o responsável pela oxidação do ferro +2 para ferro +3 ou pela oxidação direta da pirita.
As bactérias autotróficas Thiobacillus ferrooxidans e Thiobacillus
thiooxidans, são as principais bactérias responsáveis pela ação microbiana na oxidação da pirita. A descoberta do T. ferrooxidans como a principal bactéria envolvida no processo de oxidação da pirita, determinou um crescente interesse de pesquisadores pelos aspectos tecnológicos e científicos do fenômeno. T. ferrooxidans é uma bactéria não patogênica, que se apresenta como bastonetes gram negativos, com dimensões médias de 0,50 a 0,80 µm de largura por 0,90 a 1,50 µm de comprimento; sua reprodução é por divisão binária e apresenta um flagelo polar e cílios. A faixa de temperatura de seu crescimento situa-se entre 5 e 40 oC, sendo que 30 oC é a temperatura ótima. A espécie é aeróbia estrita e o pH ótimo de crescimento é em torno de 2,0 ocorrendo, porém, crescimento na faixa de 1,2 a 4,0.
T. ferrooxidans é uma bactéria quimiolitotrófica, ou seja, a energia necessária para o seu crescimento é obtida pela oxidação de substratos inorgânicos a qual é utilizada para a fixação do CO2 atmosférico. Dentre os substratos inorgânicos oxidáveis utilizados como fonte de energia, se pode citar o íon Fe+2, na forma de sulfato ferroso, o enxofre elementar, o tiossulfato ou ainda um sulfeto metálico qualquer, como por exemplo, a pirita. Além dos substratos oxidáveis, esta bactéria necessita basicamente de suprimentos de nitrogênio, fósforo e magnésio. Uma outra característica fisiológica marcante dessa espécie é a sua generalizada resistência a altas concentrações de metais; dentre eles, 0,37 M de Alumínio, 0,15 M de Zinco, 0,17 M de Cobalto, 0,18 M de Manganês, 0,16 M de Cobre e 0,1 M de Cromo.
37

3.1.2.1 Bio-oxidação da pirita pelo mecanismo direto No caso da oxidação microbiológica da pirita pelo mecanismo
direto, a interferência das bactérias se da por adesão na superfície do mineral sulfetado, oxidação localizada e liberação dos produtos da oxidação. As bactérias aderem em um ponto da estrutura cristalina, preferencialmente às falhas e desuniformidades da estrutura, formando um ponto de corrosão e oxidação. Dentre as etapas da oxidação microbiológica direta da pirita, pode-se citar:
a) Adesão das bactérias na pirita com destruição da estrutura cristalina.
b) Oxidação da pirita diretamente pela bactéria com a liberação dos produtos de oxidação (reação 6).
424)(2)(2)(2 2
7SOHFeSO
BactériaOHOFeS aqaqs
+ →++ (6)
c) Oxidação do íon ferroso com a liberação de íon férrico
utilizado para nova oxidação da pirita, conforme as reações 7 e 8.
OHSOFeBactéria
SOHOFeSO 23424224 )(2
12 + →++ (7)
0
43422 23)( SFeSOQuímica
SOFeFeS + →+ (8)
A reação de oxidação química da pirita pelo ferro férrico, reação
8, ocorre somente em pH < 3,5. O enxofre elementar obtido na reação 8 pode ser oxidado pelo íon férrico ou pelo O2 sendo rapidamente convertido a sulfato (reações 9 e 10). Esta reação é acelerada na presença de bactérias oxidantes, tais como o grupo Thiobacillus
thiooxidans.
+−++ ++→++ HSOFeOHFeS o 162128122 24
22
3 (9)
+−+→++ HSOOHOS o 42232 2
422 (10) Como visto anteriormente, em pH < 4,5 a velocidade de oxidação
38

do íon ferroso somente na presença de oxigênio é muito baixa, sendo então importante o papel desempenhado pelas bactérias Thiobacillus
ferrooxidans conforme a reação 7. Em pH > 4,5 a precipitação de hidróxidos férricos toma espaço não havendo a oxidação química da pirita, (reação 8), e somente a oxidação pelo oxigênio dissolvido (reação 6). Em pH < 3,5 o Fe+3 oxida diretamente a pirita sendo rapidamente convertido a Fe+2. Este último é submetido a uma nova ação microbiana via Thiobacillus ferrooxidans, resultando em Fe3+, e fechando desta forma um ciclo envolvendo a ação microbiana controlada pelo pH. O ciclo permanece até que toda pirita acessível aos agentes de reação tenha sido consumida. Assim, a percolação de água ácida pela pirita resulta na diminuição do pH da água e aumento da concentração de metais pesados, notadamente o ferro, e secundariamente, alumínio, manganês, cobre, zinco, chumbo e cromo.
3.1.2.2 Bio-oxidação da pirita pelo mecanismo indireto No caso da oxidação microbiológica pelo mecanismo indireto, as
bactérias agem somente na obtenção do íon férrico, reação 7, acelerando a velocidade de oxidação do ferro ferroso. A energia necessária para os processos bioquímicos e fisiológicos do Thiobacillus ferrooxidans, vem da oxidação do íon Fe+2. Os elétrons transferidos na reação de oxidação do Fe+2, liberam energia para a fosforilação do ADP, sendo o O2 o aceptor terminal de elétrons, conforme descrito no Esquema 1.
A taxa de oxidação do íon ferroso pelas bactérias é bem rápida, mas a quantidade de energia liberada na oxidação é baixa (Lundgren et. Al, 1986).
Tuovinen & Kelly (1972) calcularam que nas ótimas condições de reação, cerca de 7 kcal/mol Fe+2 oxidado são produzidos. Como para a formação de ATP a partir de ADP+Pi são necessárias cerca de 14 Kcal/mol, é preciso então serem oxidados 2 moles de Fe+2 para a formação de 1 mol de ATP. Assim, grandes quantidades de Fe+2 têm que ser processadas para fornecer um suprimento adequado de energia (ATP) para a fixação do CO2 atmosférico e demais metabolismos celulares. Lundgren et. al.(1986) estimaram que cerca de 2,8 x 106 átomos de Fe+2 são oxidados por segundo por célula, admitindo-se igual atividade de todas as células de uma cultura.
39

OHeHO
ATPPiADP
eFeBactéria
Fe
22
Kcal 14
32
22
1
Kcal 14222
→++
→+
++ →
−+
−++
Esquema 1 Nengovhela et al (2004) concluíram que a velocidade de
oxidação do ferro +2 catalisada pelas bactérias aumenta através da adição de um meio suporte, para possibilitar a formação de um biofilme multicelular, adição de nutrientes, (nitrogênio e fósforo), adição de CO2 e adição de oxigênio molecular. Portanto, os principais fatores que influenciam no processo da biolixiviação da pirita são:
a) pH As bactérias T. ferroxidans necessitam de baixo pH para se
desenvolverem. A ação bacteriana nesse aspecto é ativa, pois ha produção de ácido sulfúrico durante a oxidação da pirita. Durante o processo, a precipitação do íon férrico na forma de hidróxidos ou óxidos também aporta mais radical ácido ao meio reacional. Na presença de grande quantidade de íon sulfato, o ferro precipita não como hidróxido, mas como um complexo de sulfato mineral denominado jarosita, de fórmula aproximada MFe3(SO4)2(OH)6, em que M pode ser K+, NH4
+ ou H+. Trata-se de um precipitado amarelado ou pardo, característico da presença de oxidação bacteriana.
b) Eh (potencial de óxido-redução) O potencial de equilíbrio para a reação de oxidação do ferro
ferroso, é de +750 mV. Durante o crescimento de T. ferrooxidans em meios contendo Fe2+ como fonte de energia e pH menor que 2,5 os valores de Eh atingem justamente pontos entre 700 e 750 mV, mostrando que o sistema encontra seu equilíbrio eletroquímico na mesma faixa do equilíbrio da reação de oxidação do Fe2+. Potenciais maiores que o de equilíbrio favorecem a formação de Fe3+, e potenciais menores favorecem a formação de Fe2+.
c) Temperatura As reações envolvidas no processo de lixiviação bacteriana são
exotérmicas. A velocidade das reações é baixa, mas se o processo se da numa grande massa mineral, como uma pilha de grandes proporções, o interior da massa mineral pode atingir temperaturas elevadas, na faixa
40

de 60 a 80 oC. A oxidação química nessas temperaturas é também mais rápida.
d) Disponibilidade de O2 e CO2 Para acelerar os processos oxidativos durante a lixiviação, é
necessário adicionar oxigênio suficiente para a oxidação de ferro ferroso. O oxigênio é necessário nas reações químicas como oxidante, e no metabolismo das bactérias como aceptor terminal de elétrons na cadeia respiratória. O gás carbônico é a fonte de carbono para as bactérias. Se não houver um sistema de aeração do interior das massas minerais em lixiviação, seja por convecção natural, seja por convecção forçada, pode-se gerar um ambiente redutor. O acesso desses gases podem se dar também por sua dissolução na solução lixiviante. Na lixiviação, a difusão de O2 e CO2, do ar para a película líquida percolante é o principal mecanismo de transporte de gases para o interior da massa rochosa fragmentada. Experimentos realizados com ar enriquecido artificialmente em CO2, mostraram que é possível obter-se maiores velocidades de oxidação, tanto química como, e principalmente, biológica, do que quando se emprega ar atmosférico.
e) Irrigação do minério com lixívia A recirculação da solução lixiviante na pilha de rejeito é
importante no processo de lixiviação bacteriana. Ela mantém sempre úmida a superfície mineral, criando um ambiente propício para a ação bacteriana transportar O2 e CO2 para as células e para as reações químicas na superfície mineral.
f) Concentração de íons metálicos e de nutrientes minerais na
lixívia Os ambientes minerais, como o solo, contém todos os nutrientes,
em quantidades adequadas para o crescimento das bactérias, à exceção talvez de nitrogênio e fósforo. Dessa maneira, em ambientes pobres em um ou mais macronutrientes, a adição deles à lixívia melhora as condições da ação bacteriana. As lixívias, por sua elevada acidez, solubilizam e carregam muitos metais durante a sua percolação pelas massas minerais. A concentração desses metais pode atingir níveis tóxicos para as próprias bactérias do processo. Esse nível de toxidez é variável de metal para metal e de microrganismo para microrganismo. As bactérias do processo, particularmente o T. ferrooxidans, desenvolvem resistência a esses íons metálicos.
41

g) Composição do mineral e tamanho da partícula Rochas porosas, permeáveis ou com fissuras facilitam a
penetração da lixívia, ao passo que rochas impermeáveis não expõem seu interior à ação lixiviante. A forma como o mineral de interesse se apresenta também influi, pois o processo demanda contato íntimo entre este e a lixívia. Nas rochas permeáveis, a penetração da lixívia pelo interior rochoso torna o processo relativamente independente da granulometria. Nas rochas impermeáveis, a ação se da apenas na superfície exposta, daí a importância da granulometria do material que esta sendo lixiviado. Quanto menor a granulometria, maior área exposta e, conseqüentemente, maior área do mineral de interesse é acessível à ação lixiviante. Em lixiviações in-situ lança-se mão de explosivos para fraturar convenientemente a rocha, permitindo a percolação da lixívia.
3.2 PREVENÇÃO DA DRENAGEM ÁCIDA DE MINA Embora o escopo desta tese seja direcionado ao aproveitamento
do ferro e do sulfato a partir da drenagem ácida de mina, serão apresentados brevemente os métodos de controle e prevenção da DAM.
Os métodos de controle da DAM podem ser classificados em três categorias (Kontopoulos, 1998): métodos preventivos, métodos de controle da migração e remediação (Tabela 1).
A característica comum dos métodos preventivos é a redução drástica da velocidade de acidificação da água, através da redução do contato dos sulfetos com água e/ou oxigênio ou eliminando as bactérias responsáveis pela catálise das reações (Kontopoulos, 1998)
Uma possibilidade para minas abandonadas é a inundação e o selamento das minas. O oxigênio dissolvido inicialmente presente na água de inundação, (8,0 a 9,0 mg/L), será rapidamente consumido pelos microorganismos do meio, (tanto para a respiração quanto para a oxidação mineral), e a difusão de mais oxigênio será impedida devido ao selamento da mina. Este método, entretanto, só será eficiente em casos onde todo acesso do ar ao redor da mina, (poços e galerias), for devidamente conhecido e selado, evitando-se com isso qualquer introdução do oxigênio presente na atmosfera.
42
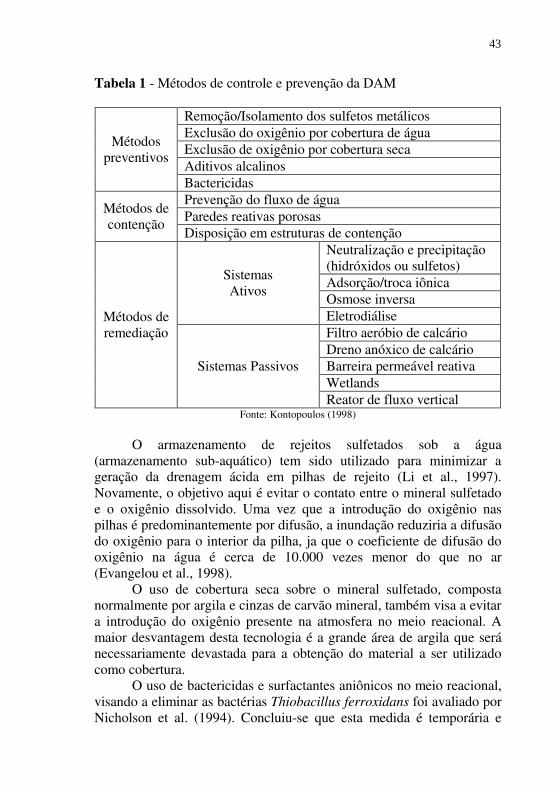
Tabela 1 - Métodos de controle e prevenção da DAM
Métodos preventivos
Remoção/Isolamento dos sulfetos metálicos Exclusão do oxigênio por cobertura de água Exclusão de oxigênio por cobertura seca Aditivos alcalinos Bactericidas
Métodos de contenção
Prevenção do fluxo de água Paredes reativas porosas Disposição em estruturas de contenção
Métodos de remediação
Sistemas Ativos
Neutralização e precipitação (hidróxidos ou sulfetos) Adsorção/troca iônica Osmose inversa Eletrodiálise
Sistemas Passivos
Filtro aeróbio de calcário Dreno anóxico de calcário Barreira permeável reativa Wetlands Reator de fluxo vertical
Fonte: Kontopoulos (1998)
O armazenamento de rejeitos sulfetados sob a água
(armazenamento sub-aquático) tem sido utilizado para minimizar a geração da drenagem ácida em pilhas de rejeito (Li et al., 1997). Novamente, o objetivo aqui é evitar o contato entre o mineral sulfetado e o oxigênio dissolvido. Uma vez que a introdução do oxigênio nas pilhas é predominantemente por difusão, a inundação reduziria a difusão do oxigênio para o interior da pilha, ja que o coeficiente de difusão do oxigênio na água é cerca de 10.000 vezes menor do que no ar (Evangelou et al., 1998).
O uso de cobertura seca sobre o mineral sulfetado, composta normalmente por argila e cinzas de carvão mineral, também visa a evitar a introdução do oxigênio presente na atmosfera no meio reacional. A maior desvantagem desta tecnologia é a grande área de argila que será necessariamente devastada para a obtenção do material a ser utilizado como cobertura.
O uso de bactericidas e surfactantes aniônicos no meio reacional, visando a eliminar as bactérias Thiobacillus ferroxidans foi avaliado por Nicholson et al. (1994). Concluiu-se que esta medida é temporária e
43

requer múltiplas aplicações para ser efetiva na redução da velocidade de oxidação da pirita, inviabilizando econômica e tecnicamente a aplicação desta tecnologia.
3.3 TRATAMENTO DA DRENAGEM ÁCIDA DE MINA Devido ás dificuldades práticas e econômicas de se inibir a
formação da drenagem ácida de mina, técnicas de remedição e tratamento podem ser utilizadas para que sejam evitados maiores danos ambientais às águas superficiais e/ou subterrâneas.
As técnicas de remediação podem ser divididas em tratamento ativo e passivo; biótico e abiótico, conforme descrito na Tabela 2.
Tabela 2 - Técnicas de remediação e tratamento da DAM.
RE
ME
DIA
ÇÃ
O D
A D
AM
Abiótica (ausência de
bactérias)
Sistema Ativo �Neutralização, oxidação e precipitação. Sistema Passivo � Canais abertos com calcário; Poços de desvio; Dreno anóxico de calcário.
Biótica (presença de
bactérias)
Sistema Ativo � Processo Thiopaq e processo BioSulphide por bactérias redutoras de sulfato. Sistema Passivo � Filtro aeróbio de rocha; Wetlands aeróbios e anaeróbios; Barreira Reativa Permeável; Reator de Fluxo Vertical
Os processos de tratamento ativo são usualmente implementados
durante a fase operacional do empreendimento mineiro, quando os recursos de manutenção e controle da planta de tratamento estão disponíveis. A grande vantagem do tratamento ativo é que as variações de demanda do efluente podem ser prontamente atendidas com uma dosagem proporcional dos reagentes químicos utilizados.
Os sistemas de tratamento passivo da DAM são os métodos mais indicados para áreas descomissionadas, com atividades paralisadas, e principalmente onde as vazões dos efluentes ácidos a serem tratados são relativamente baixas.
No caso de tratamento passivo não biológico, ou seja, sistema
44

abiótico passivo, realiza-se a neutralização da drenagem ácida de mina forçando o fluxo da água a passar por um canal de drenagem contendo calcário ou magnesita granular como material alcalino. Este canal pode ser aberto, onde o meio reacional está em equilíbrio com o oxigênio dissolvido na atmosfera; ou fechado, onde temos ausência total de oxigênio dissolvido. Porém, ambos os sistemas são apropriados somente para águas com baixos teores de metais uma vez que ocorre gradativamente a precipitação dos hidróxidos metálicos na superfície do calcário reduzindo com isso a sua eficiência de neutralização.
O tratamento passivo biológico da DAM é uma alternativa eficiente para a remoção conjunta de sulfato, íons metálicos e de matéria orgânica, sendo os primeiros oriundos da DAM e o segundo oriundo de um esgoto doméstico qualquer.
Nas reações de oxidação da matéria orgânica presente na água, sob condições aeróbias, o oxigênio dissolvido é utilizado como aceptor terminal de elétrons. Porém, quando o oxigênio dissolvido não esta presente, (condições anaeróbias), outras moléculas podem agir como aceptores de elétrons, como o sulfato e o nitrato. Na respiração anaeróbia, ha transferência de hidrogênio da matéria orgânica para o sulfato, utilizando oxigênio combinado como aceptor terminal de elétrons e formando produtos ainda oxidáveis, como o HCO3
-1 e o CH4 (Nunes A. J., 2001).
Neste contexto, a oxidação anaeróbia da matéria orgânica só ocorrera quando não estiver presente no meio o oxigênio dissolvido. Assim, as bactérias redutoras de sulfato, (SRB), são bactérias anaeróbias estritas. Estas bactérias reduzem o sulfato a sulfeto utilizando compostos orgânicos como doadores de elétrons conforme descrito na reação 11 itens (a) e (b), (Gazea et al, 1996).
5.622)(
5.622)(
3)(2
42
3)(22
42
>++→+
<+→+
+−−−
−−
pHHHCOHSSOOCHb
pHHCOSHSOOCHa
aqu
gás
(11) Uma parcela dos íons sulfeto formados pela Reação 11, combina-
se com os metais dissolvidos na água precipitando sulfetos metálicos, conforme a Reação 12. Também os íons de bicarbonato e carbonato obtidos pela oxidação da matéria orgânica se combinam com os íons metálicos dissolvidos na água aumentando a remoção destes por precipitação. (Gazea et al, 1996).
45

++−
++
+→+
+→+
HPbSPbHSb
HFeSFeSHa
s
s
2)(
2)(
)(2
)(2
2 (12)
As características principais das bactérias redutoras de sulfato
utilizadas no tratamento biótico da DAM são: - Aumentam o pH do meio, removendo sulfato e metais
pesados; - O volume de lodo produzido por SRB é muito menor do que
o volume de lodo produzido por precipitação química; - Bactérias extremamente anaeróbias; - Utilizam compostos orgânicos como doadores de elétrons; - Nutrientes essenciais: nitrogênio e fósforo. Os sistemas ativos e passivos de tratamento da drenagem ácida de
mina por bactérias redutoras de sulfato diferem entre si, principalmente pelas maiores velocidades de degradação da matéria orgânica e consequentemente redução do sulfato e metais, nos sistemas ativos. Estes sistemas operados normalmente suprindo ao meio as condições ótimas para que as reações anaeróbias ocorram, tais como; potencial redox entre -100 a -300 mV; temperatura entre 25 e 300C; pH entre 6,5-7,5; relações estequiométricas ótimas para DQO/SO4
2-; N2/DQO e P/N2; reciclo de lodo para aumentar a concentração de bactérias no meio e meio suporte para as bactérias.
3.3.1 Tratamento químico ativo da DAM O tratamento químico ativo da drenagem ácida de mina para o
descarte em um corpo hídrico qualquer deve ser realizado de tal forma que o efluente tratado atenda a todos os padrões estabelecidos pela Resolução do CONAMA n° 357 de 17 de março de 2005. Nesta resolução, estão descritas no capítulo 4, Tabela X, as condições e padrões a serem atendidos para o lançamento de efluentes em um corpo hídrico receptor. Na Tabela 3, apresentada a seguir, chama a atenção os baixos teores de metais, principalmente os metais pesados, tais como arsênio, cádmio e chumbo.
46
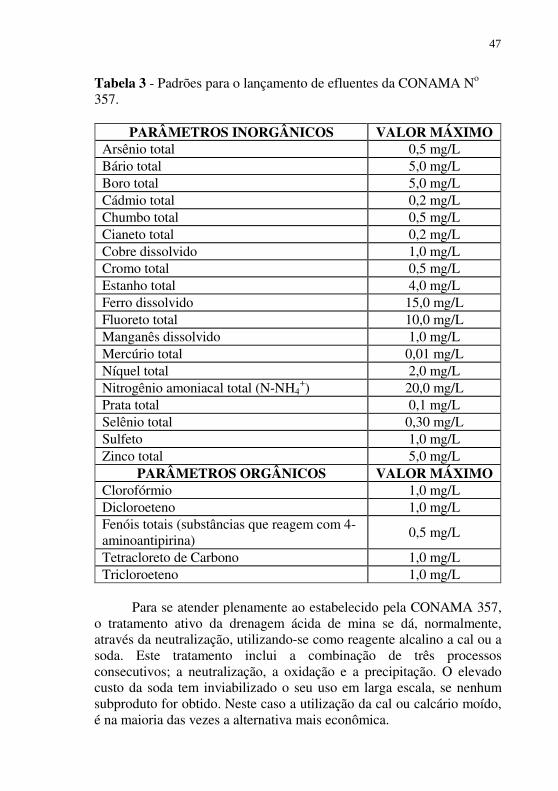
Tabela 3 - Padrões para o lançamento de efluentes da CONAMA No 357.
PARÂMETROS INORGÂNICOS VALOR MÁXIMO
Arsênio total 0,5 mg/L Bário total 5,0 mg/L Boro total 5,0 mg/L Cádmio total 0,2 mg/L Chumbo total 0,5 mg/L Cianeto total 0,2 mg/L Cobre dissolvido 1,0 mg/L Cromo total 0,5 mg/L Estanho total 4,0 mg/L Ferro dissolvido 15,0 mg/L Fluoreto total 10,0 mg/L Manganês dissolvido 1,0 mg/L Mercúrio total 0,01 mg/L Níquel total 2,0 mg/L Nitrogênio amoniacal total (N-NH4
+) 20,0 mg/L Prata total 0,1 mg/L Selênio total 0,30 mg/L Sulfeto 1,0 mg/L Zinco total 5,0 mg/L
PARÂMETROS ORGÂNICOS VALOR MÁXIMO Clorofórmio 1,0 mg/L Dicloroeteno 1,0 mg/L Fenóis totais (substâncias que reagem com 4-aminoantipirina)
0,5 mg/L
Tetracloreto de Carbono 1,0 mg/L Tricloroeteno 1,0 mg/L
Para se atender plenamente ao estabelecido pela CONAMA 357,
o tratamento ativo da drenagem ácida de mina se dá, normalmente, através da neutralização, utilizando-se como reagente alcalino a cal ou a soda. Este tratamento inclui a combinação de três processos consecutivos; a neutralização, a oxidação e a precipitação. O elevado custo da soda tem inviabilizado o seu uso em larga escala, se nenhum subproduto for obtido. Neste caso a utilização da cal ou calcário moído, é na maioria das vezes a alternativa mais econômica.
47

A adição do material alcalino visa aumentar o pH da água ácida, acelerar a velocidade das reações de oxidação dos metais, principalmente o Fe+2, e facilitar a remoção destes metais através da precipitação na forma de hidróxidos e/ou carbonatos metálicos. O líquido sobrenadante obtido após a decantação dos metais apresenta concentrações na faixa de 0,5 a 1,0 mg/L de metais. Por este método, alguma remoção de sulfato, como CaSO4.2H2O, também pode ser obtida quando se utilizam reagentes contendo cálcio para a neutralização e desde que os teores de sulfato no efluente bruto sejam altos. Neste caso, a menor concentração de sulfato obtida será de 1.500 mg/L, devido à solubilidade do sulfato de cálcio ser 3.000 mg/L a 25 0C.
O custo do tratamento químico da drenagem ácida de mina com a cal está diretamente relacionado com:
- A grande quantidade de cal necessária para elevar o pH da água de 3,5 a 4,0 para 6,5 a 7,0. Ressalta-se porém, que para se tratar drenagens ácidas contendo manganês além de ferro, alumínio, cobre e zinco, (metais mais comumente encontrados nas drenagens ácidas de mina de carvão), o pH da água deve ser elevado para 8,5 a 9,0, ao invés de 6,5 a 7,0, para a remoção efetiva do manganês. Neste caso o consumo de reagentes alcalinos e consequentemente o custo, aumentam consideravelmente.
- A necessidade de uso de cal com baixa granulometria, preferencialmente < 200 mesh Tyler (0,075mm), disponibilizando grande área superficial para a reação de neutralização.
- Longo tempo de residência necessário para a completa oxidação de Fe2+ a Fe3+.
- Grande volume de lodo gerado que deve ser disposto em aterro controlado.
Além do uso de Ca(OH)2 ou CaO, pode ser utilizado um processo
combinado de oxidação, tal como aeração e adição de peróxido de hidrogênio ou aeração e cloração, para se aumentar a velocidade de oxidação do ferro ferroso reduzindo com isso o tempo de residência para a reação e conseqüentemente as dimensões dos tanques de reação.
O tratamento ativo somente com calcário, não é aplicado em larga escala devido principalmente à baixa velocidade de solubilização do calcário e a baixa capacidade de neutralização deste reagente. Usualmente usa-se uma combinação de 30% de calcário e 70% de cal.
48

Grandes volumes de lodo, rico em ferro e outros hidróxidos metálicos, são gerados pelo tratamento químico da DAM. A sua disposição em aterros controlados eleva substancialmente o custo do tratamento, devido ao volume do lodo gerado com cerca de 7 a 10% de sólidos totais. Uma alternativa para reduzir o volume de lodo gerado e aumentar a eficiência da cal adicionada no meio é realizar-se um reciclo parcial do lodo, podendo-se desta forma alcançar valores de concentração de sólidos maiores do que 30 % no lodo (Kalin et. al., 2006).
O uso deste lodo com 30% de sólidos, rico em sulfato de cálcio e metais, quando da utilização da cal (Ca(OH)2 ou CaO); ou rico em carbonato de cálcio e metais, quando da utilização de carbonato de cálcio, como matéria-prima para a produção de algum produto comercial traria um retorno financeiro para a empresa de mineração, o qual em última instância poderia subsidiar os custos referentes ao tratamento da drenagem ácida de mina. Entretanto, poucos estudos são reportados na literatura sobre o reaproveitamento deste então denominado resíduo sólido.
3.2.2 Tratamento da DAM – Estudo de caso O sistema desenvolvido pela Carbonífera Criciúma S.A., empresa
mineradora de carvão localizada em Forquilhinha/SC, para tratamento da drenagem ácida de mina, é uma alternativa técnica e economicamente viável para solucionar os problemas relacionados com os elevados custos de tratamento, com reagentes químicos e energia, assim como para superar as dificuldades envolvendo a manipulação e o descarte dos resíduos sólidos gerados (Schneider, 2003).
Além de tratar o excedente hídrico oriundo do subsolo, esta indústria de mineração de carvão deve suprir a necessidade de água para a unidade de beneficiamento onde a separação sólido-sólido é feita com o uso intensivo de água em equipamentos como o jigue, espirais, mesas concentradoras e hidrociclones.
Neste sentido, o tratamento químico da DAM na CCSA é realizado ao mesmo tempo em que é suprida a necessidade de água para a usina de beneficiamento de carvão. A drenagem ácida de mina é direcionada do subsolo para uma bacia de adução onde é captada e realizada a adição dos reagentes químicos alcalinos, este último na sucção das bombas que direcionam a água para o beneficiamento. Por
49

este método se obtém uma grande interação entre o reagente químico alcalino e o efluente a ser tratado, (água ácida). Os elementos ingressam no processo de beneficiamento de carvão, permitindo uma maior aeração do efluente, levando a uma rápida oxidação dos metais e elevação do pH, num fluxo turbulento onde as partículas suspensas se chocam umas contra as outras, favorecendo, desta forma, a sua coagulação, floculação para posterior decantação. Esta última acontece após o lançamento do efluente nas bacias de decantação. Nestas bacias os sólidos suspensos, hidróxidos metálicos e finos de carvão e rejeito de carvão, passam a decantar, restando um sobrenadante de caráter químico neutro a básico, isento dos metais contaminantes (Schneider, 2003).
As vantagens deste sistema de tratamento envolvem o baixo custo inicial de implantação de uma ETE, (estação de tratamento de efluentes), normalmente equipada com bombas, aeradores, tanques de mistura, bacias de decantação, entre outros, uma vez que o sistema proposto aproveita as próprias instalações existentes na usina de beneficiamento de carvão. Outra vantagem associada é a resolução do problema associado à manipulação dos resíduos sólidos uma vez que os metais decantam juntamente às partículas e fragmentos finos carbonosos e alumino-silicatados suspensos no efluente liberado pelo beneficiamento, os quais podem ser dragados das bacias de decantação e transportados e acondicionados apropriadamente.
Da mesma forma, soluciona-se a questão da captação de água necessária ao beneficiamento, situação esta que é suprida pelo próprio efluente contaminado, que passa por tratamento capaz de enquadrá-lo aos parâmetros da CONAMA 357.
Outras vantagens indiretas deste sistema de tratamento são: - Permite a utilização de reagentes químicos alcalinos
residuários, tendo em vista que os metais contaminantes presentes nestes reagentes são eliminados juntamente com aqueles presentes na drenagem ácida;
- A redução dos custos de manutenção da usina de beneficiamento de carvão uma vez que as peças metálicas de desgaste não serão atacadas com água ácida;
- A menor reatividade posterior dos minerais sulfetados oriundos do beneficiamento, (rejeito), quando expostos às condições atmosféricas;
- Baixo custo de implantação; - Menor custo relativo de operação; - Simplicidade operacional.
50

3.4 APROVEITAMENTO DE RESÍDUOS DA MINERAÇÃO
3.4.1 Aproveitamento de resíduos contendo ferro Alguns trabalhos têm demonstrado as inúmeras alternativas de
aproveitamento de materiais que em alguma situação foram considerados resíduos.
No Brasil, por exemplo, uma produtora de pigmento de dióxido de titânio, até 1996, descartava uma grande quantidade de resíduo sólido contendo elevada concentração de ferro, que era então depositado em aterros industriais, caracterizando-se assim um grande passivo ambiental. Desde aquele ano, a partir do resíduo é produzido sulfato ferroso e sulfato férrico, que são produtos amplamente utilizados no tratamento de água potável e efluentes industriais (Millenium, 2009).
Santos et al. (2004) avaliaram a possibilidade da remoção seletiva do ferro da drenagem ácida de mina, por precipitação e biosorção, para posterior recuperação dos metais. O objetivo daquele trabalho era precipitar seletivamente o ferro aumentando a sua remoção a partir da DAM de forma a se obter uma corrente líquida com baixa concentração de ferro, mas elevada concentração de cobre e zinco, para posterior recuperação desses metais. Os resultados foram parcialmente atingidos, pois não era possível remover seletivamente o ferro.
A possibilidade da recuperação de ouro a partir da DAM foi reportada por Leppinen et al. (1997). Através de um processo de flotação, os íons metálicos eram removidos da fase líquida. Na fase concentrada, os produtos de maior valor comercial eram o enxofre e o ouro, embora as concentrações de cobre, zinco e prata tivessem sido também significativas. Uma estimativa do valor potencial do concentrado, considerando-se somente a recuperação de ouro e enxofre, seriam US$ 4 a 5 milhões por ano, para uma produção de 50.000 toneladas por ano. Se somente o ouro fosse recuperado, essa estimativa era de US$ 1,5 milhão/ano, se 100% do ouro fosse recuperado.
O rejeito piritoso é considerado o pior resíduo sólido da atividade de mineração de carvão, sendo disposto em pilhas denominadas módulos de rejeito. Entretanto, Zoulboulis et al (1995) demonstraram sua aplicabilidade no tratamento de efluentes líquidos, como alternativa econômica para a redução de Cr(VI). Após a redução do Cr(VI) para Cr(III), as partículas de pirita eram também capazes de reter as espécies reduzidas, mesmo em pH ácido (1-2) e concluíram que a pirita atua como adsorvente e coagulante para os complexos de Cr(III).
51

A utilização da DAM como coagulante foi pioneiramente descrita por Rao et al. (1992). A elevada concentração de ferro presente neste resíduo líquido, em geral maior do que 2,5 g/L sugeria que a DAM pudesse ser utilizada como coagulante no tratamento de esgoto doméstico. Os resultados de coagulação/decantação demonstraram que é possível alcançar boa eficiência na remoção de sólidos suspensos, turbidez e ferro. Os resultados apresentados por aqueles autores mostraram ainda que a DAM tinha eficiência, algumas vezes, superior àquela resultante do uso de cloreto férrico comercial, utilizado com base em quantidade equivalente de Fe3+. Este resultado foi associado à presença de Al3+ na DAM. Entretanto, devido à presença de outros metais pesados naquela drenagem, especialmente o zinco, seria necessário pré-tratar o efluente da DAM antes de seu uso como coagulante no tratamento de esgoto doméstico. Além disso, a viabilidade econômica desta aplicação está diretamente relacionada ao ponto de utilização da DAM como coagulante, uma vez que o transporte da drenagem ácida até áreas distantes da mina, inviabilizaria a sua aplicação devido ao custo elevado.
Sandström e Mattsson (2001) reportaram a recuperação de ferro, na forma de hidróxidos e óxidos, a partir da DAM. A proposta de recuperação incluía, inicialmente, a oxidação de Fe+2 com bactérias, para permitir à precipitação seletiva de Fe+3 em pH próximo de 3, sendo objetivada a produção de pigmentos inorgânicos de óxidos de ferro a partir do lodo precipitado. Entretanto, os pigmentos obtidos naquele caso não obtiveram qualidade similar aos produtos existentes no mercado, principalmente no que diz respeito a pureza e cristalinidade do produto.
Wang et al. 1996, obtiveram óxidos de ferro magnéticos, (ferrita) a partir de DAM. Foi realizado um processo capaz de remover a maior parte dos íons divalentes presentes na drenagem. O resíduo sólido produzido, contendo ferrita, teria uso potencial como mídia de gravação, marcadores magnéticos e matéria-prima na produção do aço. As mídias de gravação, por exemplo, são feitas de uma variedade de materiais magnéticos, incluindo óxidos de ferro, óxidos de ferro dopados com cobalto, bário e cromo. (Wang et al., 1996).
Nanopartículas magnéticas de óxidos de ferro também podem ser obtidas a partir da DAM (Wei e Viadero Jr, 2007). Para tal é realizada a co-precipitação a temperatura ambiente, sendo necessário para isso que a razão Fe2+/Fe3+ seja de 2:1 (como na estrutura da magnetita), um pH = 9,5 e ausência de oxigênio. Se oxigênio estiver presente, formariam os
52

oxihidróxidos e/ou hidróxidos de ferro. O lodo químico produzido no tratamento da DAM foi utilizado
como matéria prima para a produção de pigmentos por Marcello et al. (2008), após o tratamento térmico a 1250ºC. O sólido resultante da calcinação continha 26,5% de Fe2O3 e contaminantes SiO2 (11,4%), Al2O3 (25,6%), CaO (21,4%), MgO (11,2), MnO (3,1) e outros contaminantes presentes em menor quantidade. Essa composição indica que o pigmento não atende às normas ABNT (NBR 5801) para sua utilização como pigmento de óxido de ferro. A substituição do pigmento comercial (Cr-Fe-Zn) por este material não produziu bons resultados ao esmalte cerâmico (Marcello et al., 2008), entretanto a sua adição em pequenos percentuais (5 a 7%) produziu bons resultados de pigmentação do esmalte cerâmico.
3.4.2 Aproveitamento de resíduos contendo sulfato de cálcio A literatura tem reportado vários estudos que fazem a reutilização
de resíduos e/ou a obtenção de novos produtos a partir de resíduos industriais. Em geral, estes produtos podem ser utilizados nas mais diversas áreas, como na construção civil, na indústria cerâmica e na remediação de solos (Pappu et al., 2007; Singh e Garg, 2000; Valss e Varquez, 2001; Basegio et al., 2002; Fauziah et al., 1996; Degirmenci, 2007).
O uso de cal ou carbonato de cálcio para o tratamento de efluentes ácidos contendo H2SO4, tal como a drenagem ácida de mina, é consideravelmente mais barato do que o uso de reagentes alcalinos que não contenham cálcio, tal como a soda ou a barrilha. Porém isto leva a geração em grande quantidade de um lodo químico rico em sulfato de cálcio di-hidratado. Um produto similar a esse gerado em grande quantidade no Brasil e no mundo é o fosfogesso ou gesso químico como é chamado (Canut, C.M.M. et. al., 2007; Singh, M. 2002; Degirmenci, N. 2007).
O fosfogesso é um resíduo sólido gerado na indústria de ácido fosfórico, originado pelo ataque da rocha fosfática com ácido sulfúrico (reação 13). O problema é que para cada tonelada de ácido fosfórico produzido são produzidas de 4 a 5 toneladas de fosfogesso (reação 13).
HFPOHOHCaSOOHSOHFPOCa 262.102010)( 432424226410 ++→++ (13)
53
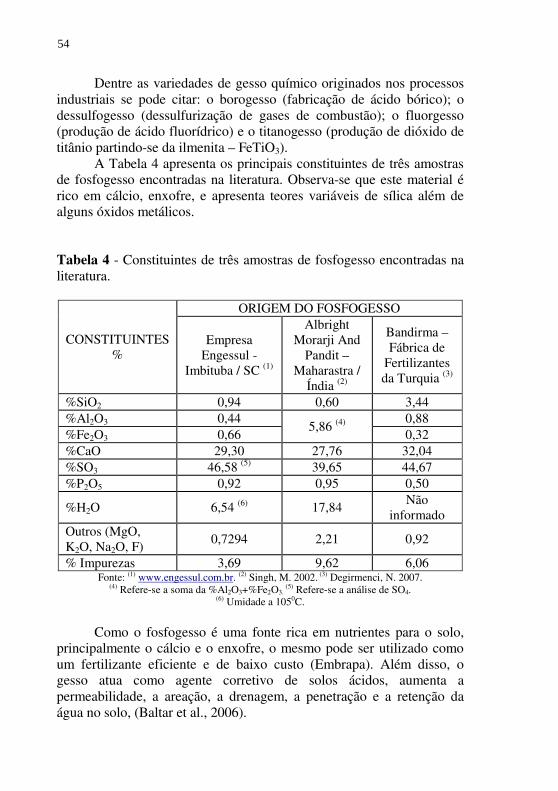
Dentre as variedades de gesso químico originados nos processos industriais se pode citar: o borogesso (fabricação de ácido bórico); o dessulfogesso (dessulfurização de gases de combustão); o fluorgesso (produção de ácido fluorídrico) e o titanogesso (produção de dióxido de titânio partindo-se da ilmenita – FeTiO3).
A Tabela 4 apresenta os principais constituintes de três amostras de fosfogesso encontradas na literatura. Observa-se que este material é rico em cálcio, enxofre, e apresenta teores variáveis de sílica além de alguns óxidos metálicos.
Tabela 4 - Constituintes de três amostras de fosfogesso encontradas na literatura.
CONSTITUINTES %
ORIGEM DO FOSFOGESSO
Empresa Engessul -
Imbituba / SC (1)
Albright Morarji And
Pandit – Maharastra /
Índia (2)
Bandirma –Fábrica de
Fertilizantes da Turquia (3)
%SiO2 0,94 0,60 3,44 %Al2O3 0,44
5,86 (4) 0,88
%Fe2O3 0,66 0,32 %CaO 29,30 27,76 32,04 %SO3 46,58 (5) 39,65 44,67 %P2O5 0,92 0,95 0,50
%H2O 6,54 (6) 17,84 Não
informado Outros (MgO, K2O, Na2O, F)
0,7294 2,21 0,92
% Impurezas 3,69 9,62 6,06 Fonte: (1) www.engessul.com.br. (2) Singh, M. 2002. (3) Degirmenci, N. 2007.
(4) Refere-se a soma da %Al2O3+%Fe2O3. (5) Refere-se a análise de SO4.
(6) Umidade a 1050C.
Como o fosfogesso é uma fonte rica em nutrientes para o solo,
principalmente o cálcio e o enxofre, o mesmo pode ser utilizado como um fertilizante eficiente e de baixo custo (Embrapa). Além disso, o gesso atua como agente corretivo de solos ácidos, aumenta a permeabilidade, a areação, a drenagem, a penetração e a retenção da água no solo, (Baltar et al., 2006).
54

Ao ser incorporado ao solo, o gesso sofre dissolução conforme a reação 14:
OHCaSOSOCaOHOHCaSO 242
42
224 32.2 +++→+−+ (14)
Temos no território nacional, uma ocorrência superior a 60% de
solos ácidos, com deficiência de cálcio ou excesso de alumínio tóxico, que constituem uma barreira química à penetração das raízes. A dissolução do gesso no solo libera o íon Ca2+ que pode reagir no complexo de troca do solo, deslocando Al+3, K+ e Mg2+ para a solução de solo. Estes últimos podem por sua vez, reagir com o SO4
2- formando AlSO4
+ (que é menos tóxico às plantas) e os pares iônicos neutros K2SO4, MgSO4 e CaSO4. Os pares iônicos apresentam grande mobilidade ao longo do perfil do solo, ocasionando uma descida de cátions para as camadas mais profundas (Dias, 1992).
Na fabricação de cimento, o gesso ou seus resíduos são adicionados ao clínquer durante a moagem, na proporção de 3 a 5%, com a finalidade de retardar o tempo de pega do cimento (Lyra, 2006).
No estudo da reciclagem e reutilização de resíduos inorgânicos apresentados por Pappu et al. (2007), um resíduo de Gipsita, tal como o fosfogesso ou gesso químico e um lodo de cal foram reciclados para a fabricação do cimento Portland, cimento de alvenaria e gesso Gipsita.
Valss e Varques (2001) utilizaram um lodo obtido de uma planta de tratamento de esgoto urbano, com gesso em sua composição, como um componente de argamassa e concreto.
Silva et al. (2006) avaliaram a incorporação de rejeitos de gesso da construção civil em formulações de massa para a cerâmica vermelha. Observou-se que as formulações que apresentaram os maiores valores de tensão de ruptura, e, portanto a maior resistência mecânica, foi de 2:1 de argila plástica para não plástica e 2:1 com 15% de incorporação de rejeitos de gesso.
Andrade et al. (2006) estudaram também a incorporação de resíduos de gesso da construção civil na fabricação da cerâmica vermelha (tijolos e telhas). Os autores observaram que a adição de 20% do resíduo de gesso na massa cerâmica resultou em uma absorção de água pelo corpo de prova de 14%, o que está dentro das especificações técnicas de tijolos e telhas.
O processo de beneficiamento do gesso, a partir de rejeitos industriais, pode compreender a lavagem com água, tratamentos térmicos e químicos. Assim, o fosfogesso foi adequado para ser usado
55

na fabricação de cimento e Gipsita através do tratamento com pequenas quantidades de solução aquosa de ácido cítrico e subseqüente lavagem com água (Singh, 2002).
Uma das maiores aplicações de gesso se encontra ainda na indústria da construção civil: no revestimento de paredes, placas, blocos e painéis. Para isso, entretanto, o gesso deve atender a uma série de normas, entre as quais as NBR’s 13.207, 12.127 e 12.128 além de apresentar um limite máximo de cerca de 6% de impurezas (Silva et al., 2006).
Neste contexto, uma alternativa bastante atraente, econômica e tecnicamente, é a utilização deste resíduo, ou seja, do gesso químico obtido no tratamento da drenagem ácida de mina, como matéria-prima rica em ferro, alumínio e sulfato de cálcio, para, após tratamento adequado, produzir um coagulante férrico e aluminoso, a ser utilizado no tratamento de água potável e esgoto doméstico, bem como um gesso branco para a indústria da construção civil.
3.5 GESSO PARA A CONSTRUÇÃO CIVIL Tradicionalmente, a obtenção do gesso para a construção civil se
da através da Gipsita. O termo Gipsita é utilizado para designar um mineral em estado natural abundante na natureza. Quimicamente a Gipsita é um sulfato de cálcio dihidratado cuja fórmula química é CaSO4.2H2O. Uma composição química média do minério de Gipsita encontrado no Pólo Gesseiro de Araripe no estado de Pernambuco (Brasil) é apresentada na Tabela 5.
Observa-se que este minério (Tabela 5) é de ótima qualidade, pois apresenta um teor máximo de 6,35% de impurezas e 93,65% de Gipsita.
Quando calcinada às temperaturas na faixa de 140 a 800 ºC, a Gipsita desidrata-se podendo perder ε moléculas de água, sendo que ε pode variar de 0 a 2. A temperatura e o tempo necessários para a desidratação da Gipsita dependem da qualidade do produto a que se deseja obter e da pressão parcial de vapor de água (Pereira, 1973). Conforme as condições experimentais de desidratação da Gipsita, as reações esperadas podem ser descritas nas reações 15 a 17.
56

Tabela 5 - Composição química média do depósito de Gipsita do Polo Gesseiro de Araripe/PE.
CONSTITUINTE VALOR Umidade a 60 0C 0,08%
Água combinada a 200 0C 19,58% Perda ao fogo a 1.000 0C 1,62%
Resíduos insolúveis 0,28% Sílica (SiO2) 0,32%
Ferro e alumínio (R2O3) 0,20% Cálcio (CaO) 32,43%
Magnésio (MgO) 0,31% Sulfatos (SO3) 45,04%
Cloretos (NaCl) 0,15% Teor de Gipsita 93,65%
Fonte: Peres et al. (2001).
H 1,5 + 5,0 2H 224)/81,23(
160 - 140
24
0
OOHCaSOOCaSOgcal
C
⋅ →⋅ (15)
OCaSOOCaSO C
224 190 >
24 2.H ).-(1+ OH. 2H 0
εε →⋅ (16)
OCaSOOCaSO C
24 350 >
24 H + H 0
εε →⋅ (17) É possível se admitir a existência de mais de um hemihidratado,
dependendo da temperatura e pressão do processo, com teor de água podendo variar de 0,06 a 0,66 moléculas de água. O hemihidratado com 0,50 moléculas de água é denominado gesso.
Os graus de hidratação e cristalinidade do produto estão associados ao processo de obtenção e as condições do tratamento térmico. Se a desidratação da Gipsita é realizada em autoclave, em pressões superiores a 100 kPa, a formação do semi-hidratado dá-se lentamente por um mecanismo de dissolução - cristalização em meio líquido. O produto bem cristalizado é denominado semi-hidratado alfa (α). Se, entretanto, a desidratação é realizada à pressão atmosférica, com pressão parcial de vapor d’água baixa, isso leva a uma má formação dos cristais devido à saída abrupta da água de cristalização, obtendo-se assim, um sólido mal cristalizado denominado de semi-hidratado beta -
57

β (Santos et al., 2004). Dentre os tipos de gesso beta, destacam-se os de fundição e os de
revestimento que são utilizados na indústria da construção civil, cerâmica e indústria de modelagem. O gesso alfa, um produto mais homogêneo, é utilizado em bandagens de alta resistência, matrizes para a indústria cerâmica, indústria de modelagem, ortopedia, odontologia e indústria automobilística (Baltar et al., 2006).
A anidrita solúvel ( OH. 24 εCaSO ) ou anidrita III, constitui a fase intermediária da anidrita insolúvel, (anidrita II), e pode conter de 0,11 a 0,06 moléculas de água (Angeler et al., 1983).
Esta anidrita é reativa transformando-se no semi-hidratado com a umidade do ar. Essa hidratação é conhecida como “estabilização” do gesso, tendo-se verificado que ela se da após doze horas de armazenamento do produto em atmosfera com umidade relativa de 80 % (Santos et al., 2004).
Ao se alcançar temperaturas maiores que 4000C, a anidrita III transforma-se em anidrita II ( 4CaSO ), conforme a reação 17. Sendo exotérmico, este processo de transformação é extremamente rápido e não reversível. Com isso, a hidratação da anidrita II torna-se bem mais lenta, e na medida em que a temperatura de calcinação aumenta, a velocidade de rehidratação da anidrita torna-se cada vez menor. A Gipsita calcinada a 600 0C, por exemplo, necessita de mais de dez dias para rehidratar metade da anidrida, e a 800 0C, o produto é considerado de difícil rehidratação. A anidrita II é também chamada de anidrita supercalcinada (Santos et al., 2004).
A produção de gesso no Brasil concentra-se na região nordeste, mais especificamente no extremo oeste do estado de Pernambuco, a cerca de 700 km de Recife. As reservas estimadas do Pólo Gesseiro do Araripe são da ordem de um bilhão de toneladas de minério bruto, totalizando uma produção atual de 2,6 milhões de toneladas/ano o que representa 95% de todo o gesso produzido no Brasil (Baltar et al., 2006).
A fabricação do gesso para a construção civil compreende as seguintes etapas (Peres, Benachour e Santos, 2001):
- Extração e pré-seleção manual da Gipsita. - Britagem, moagem e peneiramento. - Calcinação e estabilização. Uma faixa granulométrica ampla para a Gipsita a ser calcinada
dificulta o controle das condições de calcinação e causa
58

desuniformidades nos produtos obtidos. Além disso, pode causar sérios problemas ambientais pelo arraste excessivo de sólidos em fornos contínuos.
A calcinação da Gipsita à pressão atmosférica, por via seca, pode ser realizada utilizando-se diferentes tipos de fornos, os quais devem assegurar uma distribuição e um cozimento regular do material.
Dentre estes fornos pode-se citar (Monção Junior, 2008): - Fornos rotativos a contracorrente� os diâmetros variam
de 1 a 3 m e os comprimentos variam de 8 a 20 m. Dentro destes fornos, a Gipsita, com granulometria adequada, é introduzida numa extremidade (mais alta do forno) e segue até a outra extremidade, onde entram os gases quentes.
- Fornos rotativos concorrentes � a Gipsita é introduzida pela extremidade onde a temperatura é mais elevada, e é transportada para a outra extremidade por arraste com auxílio de gases quentes.
- Fornos marmita vertical ou horizontal � a Gipsita é depositada sobre uma superfície metálica e entra em contato indireto com gases quentes, normalmente a temperaturas entre 600 e 800 ºC.
- Fornos a leito fluidizado � as partículas de Gipsita são mantidas em suspensão durante a calcinação, pela ação de um fluxo de gás quente.
Para promover um contato gás-sólido mais efetivo em fornos
onde se estabelece esse fenômeno de forma direta, a maioria desses fornos é construída com chicanas (Monção Junior, 2008). Esses acessórios, paralelamente colocados ao longo do comprimento axial do equipamento, têm a função de transportar e distribuir os sólidos por toda a área da seção transversal do forno, como resultado do movimento de rotação do mesmo. Um bom projeto para as chicanas é essencial à obtenção de uma rápida e homogênea troca de calor entre os componentes da mistura em escoamento.
Entre os fornos de maior rendimento térmico no processo de produção de gesso, encontram-se o do tipo rotativo de contato direto e concorrente. O regime concorrente evita com que os cristais do semi-hidrato (gesso) entrem em contato com gases em temperaturas elevadas. Caso isso ocorresse, existiriam possibilidades da perda dessa meia molécula de água em função do elevado gradiente de temperatura existente, responsável pelo fenômeno de transferência de calor ao longo
59

do equipamento. Um forno rotativo de calcinação de gesso compreende um
cilindro metálico que gira em torno de sua maior dimensão, seu eixo, no interior do qual escoam minério de Gipsita e gases produzidos pela combustão. O calor dos gases é aproveitado para fornecer condições energéticas à ocorrência da desidratação térmica da Gipsita, dando origem à produção de seu semi-hidrato, comercialmente denominado de gesso. Para isso, o minério de Gipsita deve ser adequadamente preparado, entrando no forno em regime de escoamento concorrente com os gases quentes, a uma granulometria reduzida para facilitar a troca de calor durante o percurso no interior do forno. A rotação e a inclinação do forno são responsáveis pelo tempo de contato entre os componentes envolvidos. Dessa forma, são diretamente responsáveis pela conversão da reação: a granulometria, o tempo de contato entre sólido e gás, as dimensões do forno, sua rotação e as temperaturas dos gases. Esta última não deve exceder determinada faixa, sob pena de promover uma desidratação total do minério de Gipsita, dando origem a um sulfato de cálcio totalmente isento de água, ou seja, a anidrita II ou III (Monção Junior, 2008).
3.6 COAGULANTES INORGÂNICOS À BASE DE FERRO A coagulação química é um dos processos primários para a
remoção de sólidos dissolvidos e sólidos suspensos no tratamento de águas naturais e efluentes líquidos industriais. Em geral, uma pequena concentração de um cátion metálico polivalente, tal como o ferro ou o alumínio, é adicionado à água para desestabilizar e agregar partículas coloidais, bem como adsorver matéria orgânica e/ou inorgânica dissolvida, facilitando a sua remoção subseqüente por meio da sedimentação e filtração.
Os sais de alumínio e ferro (Al3+, Fe2+, Fe3+) são os principais coagulantes utilizados atualmente no tratamento de água potável e efluente industrial. Dentre os seus compostos principais podemos destacar o sulfato de alumínio (Al2(SO4)3), o cloreto férrico (FeCl3) e o sulfato férrico (Fe2(SO4)3).
Os coagulantes que têm mostrado melhores resultados do que o sulfato de alumínio, em relação a coagulação-floculação, e tem merecido maior interesse na substituição aos sais de alumínio devido aos efeitos nocivos do alumínio, como o desenvolvimento do mal de
60

Alzheimer, são o sulfato e o cloreto férricos (Butler et al., 2004). Não existem evidências de que compostos de ferro apresentem
riscos a saúde humana tendo sido sugerido portanto, que água potável tratada com os sais férricos é segura para consumo humano (Hendrich et al., 2001).
O cloreto férrico é um agente coagulante inorgânico, à base de ferro trivalente, bastante eficiente nas operações de coagulação e muito utilizado no tratamento de água potável, esgotos domésticos, efluentes líquidos industriais e condicionamento de lodos de estações de tratamento de efluentes. Apresenta como principal problema, a excessiva corrosividade, que acaba acarretando danificações nas partes metálicas dos sistemas de tratamento. O cloreto férrico é produzido pela reação do ácido clorídrico com minério de ferro de alta pureza, o que resulta na formação de cloreto férrico com baixa concentração de contaminantes. Hoje existe uma tendência de se usar sucata como fonte de ferro, um procedimento que pode resultar em problemas de contaminação por metais pesados,uma vez que não ha um controle rígido da qualidade da sucata a ser utilizada.
O sulfato férrico também é um excelente coagulante inorgânico à base de ferro trivalente. É usado em sistemas de tratamento de água potável, esgotos domésticos, efluentes líquidos industriais e condicionamento de lodos de estações de tratamento. Os coágulos formados com sulfato férrico sedimentam mais rápido que os flocos de sulfato de alumínio devido ao seu maior peso, além de possuírem alta eficácia na remoção de algas. É um coagulante que se equivale ao cloreto férrico, quanto à efetividade, mas com a vantagem de apresentar um menor custo, menor acidez e índices de corrosão mais baixos. O sulfato férrico é obtido pela reação do minério de ferro ou sucata de ferro com ácido sulfúrico.
Coagulantes férricos ou de alumínio apresentam vantagens e desvantagens. Fan et al. (2003) reportaram que um coagulante misto, composto tanto de ferro quanto de alumínio, seria um coagulante ideal, por reunir as vantagens dos dois sistemas e minimizar as desvantagens.
A produção de sulfato férrico a partir de diferentes resíduos industriais tem sido proposta (Kirk e Othmer, 1995).
Cici e Cuci (1997) propuseram a produção de coagulantes férricos a partir de resíduos da indústria de galvanoplastia, que continham 16,2% de ferro e 31% de sulfato. Nesta proposta, o resíduo de galvanoplastia contendo sulfato ferroso reagia com ácido sulfúrico conforme a reação (18).
61

2 FeSO4. x H2O + 2 H2SO4
Fe2(SO
4)3 + SO
2 + (2 x + 2) H
2O
(18) As cinzas leves oriundas da combustão do carvão mineral foram
utilizadas para a produção de coagulantes complexos por Fan et al (2003). As concentrações de silício, ferro e alumínio nas cinzas utilizadas por esses autores encontravam-se no intervalo de 20 a 60% para o silício; de 5 a 35% para o ferro e 10 a 35% para o alumínio. Após o pré-tratamento das cinzas através da lavagem com água a 95oC, o coagulante era produzido a partir da reação dos óxidos de ferro e de alumínio com ácido sulfúrico às temperaturas na faixa de 80 a 120oC.
Butler et al. (2004) propuseram um processo para produção de sulfato (poli) férrico no qual SO2 é oxidado pelo clorato de sódio para produzir o ácido necessário para a oxidação de Fe2+. A absorção de SO2 e a conversão de Fe2+ ocorrem através das reações descritas abaixo.
3 SO
2 + ClO
3- + 3 H
2O 3 SO
42- + 6 H+ + Cl-
(19)
6 Fe2+ + ClO3- + 6 H+ 6 Fe3+ + 3 H2O + Cl-
(20) Onde clorato de sódio é utilizado para oxidar S(IV) para S(VI), e
Fe2+ para Fe3+. A oxidação de Fe+2 depende do ácido produzido na reação 19. Água é produzida na reação 20 e é incorporada no sulfato (poli) férrico através de subseqüentes reações de hidrólise, inclusão de sulfato e polimerização (reações 21 a 23).
x Fe3+ + y H2O Fex[(OH)y]
(3x-y)+ + y H+ (21)
Fex[(OH)y]
(3x-y)+ + (3x-y) SO42- Fe2x(OH)2y(SO4)(3x-y) (22)
m [Fe2x(OH)2y(SO4)(3x-y)] [ Fe2x(OH)2y(SO4)(3x-y)]m (23)
A estrutura do sulfato (poli) férrico também pode ser expressa na
forma [Fe2(OH)n(SO4)(3-n/2)]m, onde m é uma função de n, e n=2y/x, com n≤ 2 (Butler et al., 2004).
62

3.7 OS ÓXIDOS DE FERRO Os óxidos de ferro, nome genérico dado aos óxidos, hidróxidos,
oxi-hidróxidos e outros compostos de ferro, são conhecidos desde os primórdios da humanidade quando eram utilizados em inscrições rupestres. Há indícios da manipulação de várias tonalidades de óxidos de ferro, tais como o preto, o vermelho, o amarelo, o marrom e o verde, capazes de revelar o grau de conhecimento tecnológico dos nossos ancestrais (Lopes, 2005).
Os óxidos de ferro naturais e sintéticos podem, dependendo da sua estrutura cristalina e estado de oxidação, apresentar-se como várias fases. Entre elas podemos citar a Goetita (α-FeOOH), a Lepidocrocita (γ-FeOOH), a Hematita (α-Fe2O3), a Maghemita (γ-Fe2O3) e a Magnetita (Fe3O4). Cada uma destas fases apresenta propriedades bastante distintas entre si.
Totalizam 16 os óxidos de ferro conhecidos, conforme apresentado na Tabela 6. Estes materiais são compostos de ferro com oxigênio e/ou íons hidroxila. Observa-se pela tabela que na maioria dos compostos o ferro apresenta-se no estado de oxidação trivalente, porém são três os compostos onde o ferro encontra-se na forma divalente: FeO, Fe(OH)2 e Fe3O4 (Cornell e Schwertmann, 2003).
A maioria dos óxidos, hidróxidos e oxi-hidróxidos de ferro possuem estrutura cristalina, porém o grau de ordenamento estrutural e o tamanho do cristal variam de acordo com as condições sob as quais os cristais foram obtidos (Cornell e Schwertmann, 2003).
Os óxidos de ferro são de grande importância em uma variedade de processos, entre os quais, podemos citar o uso como pigmentos, como agentes anti-corrosivos, como dispositivos eletromagnéticos, adsorventes, catalisadores, matéria-prima para produção de coagulantes férricos, entre outros.
O aumento contínuo da importância dos óxidos de ferro como pigmentos coloridos está baseado na sua baixa toxicidade, estabilidade química, ampla variedade de cores, boa razão performance/custo, entre outros (Buxbaum, et. al, 2005).
63

Tabela 6 - Os óxidos de ferro.
Óxi-hidróxidos e hidróxidos de ferro Óxidos
Goetita � α-FeOOH Hematita � α-Fe2O3
Lepidocrocita � γ-FeOOH Magnetita � Fe3O4
(Fe+2Fe+32º4)
Akaganéita �β-FeOOH Maghemita � γ-Fe2O3
Schwertmannita � Fe16O16(OH)y(S04)z. n
H20 β-Fe2O3
δ-FeOOH ε - Fe2O3
Ferroxyhyta � δ’-FeOOH Wustita � FeO
High pressure FeOOH
Ferrihydrita � Fe5HO8 .4H20
Bernalita � Fe(OH)3
Fe(OH)2
Green Rusts � Fe+3xFe+2
y(OH)3x+2y-z(A-)z;
onde A-= Cl- ou ½ SO4-2
Fonte: Cornell e Schwertmann, 2003.
3.7.1 Pigmentos inorgânicos de óxidos de ferro Sob o ponto de vista dos pigmentos, os primeiros a serem
utilizados pelo homem foram os ocres que vem do grego e significa amarelo. A espécie química responsável pela cor do ocre é o óxido de ferro férrico monohidratado, ou seja, a Goetita, α-FeO.OH (Lopes, 2005).
A importância dos pigmentos para a civilização humana é evidente e bem documentada. Embora estes materiais tenham sido descobertos há tantos anos, as pesquisas continuam até hoje, pois as indústrias exigem frequentemente novos tons e cores cada vez mais reprodutíveis e estáveis, além de processos que sejam cada vez mais
64

sustentáveis e que estejam em plena concordância com as novas práticas ambientais.
A maior parte da produção mundial de pigmentos inorgânicos de óxido de ferro está na Alemanha, Estados Unidos, Inglaterra, Itália, Brasil e Japão, com mais de 50% do total sendo produzido na Alemanha. As duas companhias que mais produzem óxido de ferro sintético são a Bayer (Alemanha), cujo nome do produto comercial é Bayferrox, e a Harcross (U.K./U.S.A.) (Cornell e Schwertmann, 2003).
Os principais pigmentos sintéticos de óxido de ferro comercializados no mercado mundial são os amarelos (Goetita), os laranjas (Lepidocrocita), os vermelhos (Hematita), os marrons (Maghemita), os pretos (Magnetita), e a mistura destes (Cornell e Schwertmann, 2003). Estes pigmentos demandam uma porção de volume maior do que o mercado de todos os outros pigmentos coloridos combinados. O pigmento de óxido de ferro mais importante em termos de volume é o amarelo, e o baixo custo do pigmento (em relação aos orgânicos), combinado com algumas vantagens, como a opacidade elevada, o alto poder de cobertura, a boa resistência química e a estabilidade ao calor, mantém este pigmento em um patamar elevado de vendas.
O processo responsável pela cor dos óxidos de ferro é a absorção e o espalhamento da luz. As cores, vermelha e amarela, dos óxidos de ferro, por exemplo, resultam da absorção da parte azul e verde do espectro visível. Quando a absorção é muito maior que o espalhamento, em toda a região visível do espectro, os óxidos de ferro são pretos (Cornell e Schwertmann, 2003).
A cor dos pigmentos inorgânicos de óxido de ferro esta diretamente relacionada à área superficial especifica do pigmento, a composição química, estrutura cristalina e defeitos estruturais do pigmento e a opacidade do pigmento. Em geral, quanto maior a área superficial específica, ou seja, quanto menor o tamanho de partículas do pigmento, maior o espalhamento da luz e maior a intensidade da cor. A opacidade do pigmento, ou seja, a capacidade do pigmento em impedir a transmissão de luz, esta relacionada com as dimensões das partículas e a diferença entre o índice de refração do pigmento e o índice de refração da matriz na qual o pigmento se encontra disperso.
65

3.7.1 Colorimetria Para o estudo de pigmentos, sejam eles orgânicos ou inorgânicos
naturais ou sintéticos, é fundamental o uso de ferramentas quantitativas que possam avaliar e expressar as cores. Este suporte é fornecido pela colorimetria. Através dela a cor pode ser caracterizada pela tonalidade, luminosidade e saturação (Casqueira e Santos, 2008).
A tonalidade corresponde ao comprimento de onda predominante, o que é usualmente chamado de cor, é na realidade, o tom (vermelho, verde, entre outras).
A luminosidade descreve o quanto de luz é refletida ou absorvida por um objeto e a saturação depende das proporções ocupadas por cada comprimento de onda na radiação eletromagnética, ou seja, ela descreve a pureza do tom (Casqueira e Santos, 2008).
A transformação das cores em números foi realizada por meio da representação gráfica das variáveis cromáticas (luminosidade, tonalidade e saturação) em diagramas, de tal modo que cada ponto no plano ou no espaço representasse uma cor. O método mais utilizado que possibilita a realização deste tipo de medida colorimétrica é o método CIELab - Commission Internationale de L’Eclairage (Lewis, A. P., 1973). Este método permite medir a intensidade de absorção na região visível para obtenção dos parâmetros L*, referente à luminosidade que varia do negro (0) ao branco (100), a* referente a intensidade de cor vermelho(+)/verde(-) e b* a intensidade de cor amarelo(+)/azul(-) conforme mostrado na Figura 3 (Lewis, A. P., 1973).
Minuciosas diferenças de cor entre duas amostras podem ser facilmente identificadas por um observador treinado, entretanto, quando se necessita transcrever estas diferenças de cor de maneira compreensível, a colorimetria diferencial é uma ferramenta utilizada para resolver este problema.
A colorimetria diferencial transforma as diferenças de cor em números facilmente interpretáveis, utilizando-se a equação 8.
( ))();();(
;
212121
222
bbbaaaLLL
ondebaLEab
−=∆−=∆−=∆
∆+∆+∆=∆ Equação 7
Na Tabela 7 se observa uma classificação utilizada na indústria
de tintas de impressão para o controle de qualidade. Tintas que tenham um valor de ∆Eab acima do limite esperado são classificadas como cores diferentes.
66

Figura 3 - Sólido de cor do sistema L*a*b* - representação tridimensional e
bidimensional.
Tabela 7 - Classificação com relação a diferença de cor utilizada na indústria de tintas de impressão.
Diferenças (∆∆∆∆Eab) Classificação com relação à diferença de cor de uma amostra em relação à outra.
0,0< ∆Eab < 0,2 Imperceptível 0,2< ∆Eab < 0,5 Muito pequena 0,5< ∆Eab < 1,5 Pequena 1,5< ∆Eab < 3,0 Distinguível 3,0< ∆Eab < 6,0 Facilmente distinguível > 6,0 Muito grande
Fonte: Norma DIN 6174.
3.7.1.2 Obtenção de pigmento inorgânico de óxido de ferro Industrialmente, os pigmentos inorgânicos amarelos de óxido de
ferro são os pigmentos mais produzidos em relação às outras cores. Isto se justifica, porque além do maior uso das tonalidades amarelas ou ocres, a partir destes, ou seja, a partir da Goetita, é possível obter-se várias cores de pigmentos inorgânicos. Desta forma, esta revisão bibliográfica, assim como esta tese, enfatizara a obtenção da Goetita.
A Goetita é uma das formas de óxido de ferro mais estáveis termodinamicamente em temperatura ambiente, sendo, portanto, o
67

primeiro óxido de ferro a se formar em ambientes naturais (Cornell e Schwertmann, 2003).
O processo para produção de pigmento inorgânico de óxido de ferro amarelo pode ser realizado pelo método de precipitação através da mistura de FeCl2 ou FeSO4 com CaCO3 (Meisen et. al., 2005).
Neste processo, realiza-se a mistura de uma solução aquosa de FeCl2 com um núcleo alcalino que contenha α-FeO.OH. A mistura é aquecida a 750C e oxida-se o ferro ferroso a ferro férrico em uma única etapa cuja velocidade de oxidação é de 0,4 a 4,0 (mol Fe+2
oxidado/h)/mol Fe+2 inicial. Durante a oxidação do ferro ferroso é adicionado Ca(OH)2 ou
CaCO3 para manter o pH na faixa de 3,2 a 4,2. Ainda segundo Meisen et al., (2005), pigmentos mais claros, ou seja, com L* > 61, podem ser obtidos com o uso de núcleos mais finos, (BET > 60m2/g, dp 0,1 a 2 µm), pH após a oxidação entre 2,4 e 3,8 e baixas velocidades de oxidação, na faixa de 0,5 a 1,0 mol%Fe+3formado/h. Ao contrário, pigmentos mais escuros, ou seja, com L* < 61, são obtidos com núcleos mais grosseiros, (BET<60m2/g), pH final maior (3,2 a 5,2) e velocidade de oxidação mais rápida (>3 mol%Fe+3/h).
Rosenhahn (2007) utilizou uma reação entre o FeSO4 e a soda cáustica para produção de óxido de ferro amarelo. De maneira geral, em uma primeira etapa produz-se o gerador, (ou seja, o núcleo), a partir do qual em um próximo passo se permite o aumento da camada de α-FeO.OH de maneira relativamente lenta. A produção do gerador se da conforme a reação (24).
OHSONaOHFeOONaOHOHFeSO 242224 152.22/147.2 ++→++ (24)
Observa-se que o gerador de óxido de ferro amarelo é produzido
a partir de uma solução de sulfato ferroso adicionando-se NaOH, sob oxidação do ar. Assim se obtém α-FeO.OH na forma de microcristais (gerador). Para se conseguir um amarelo claro, a temperatura média da reação deve ser mantida abaixo de 35ºC.
A conseguinte produção do pigmento amarelo se da adicionando-se sucata de ferro em oxidação juntamente com o oxigênio ao gerador. Obtém-se desta forma cristais de α-FeO.OH em meio ácido, contendo sempre um excesso de FeSO4 oriundo da oxidação da sucata de ferro, conforme a reação (25).
68

242)(224
2442)(
42224
3.463
4.464
HSOHOHFeOFeOHOFeSO
HFeSOSOHFe
SOHOHFeOOHOFeSO
ferrodesucata
ferrodesucata
++→+++
+→+
+
+→++ (25)
O sólido assim obtido é lavado até que se retire todo sal, seco e
moído até a granulometria desejada. Meisen (2004) relata outro processo para produção de pigmento
amarelo de óxido de ferro pelo método de precipitação com solução de cloreto ferroso e soda cáustica. Neste caso, se utilizam uma solução de FeCl2 após prévia remoção de contaminantes, sendo estes o cromo e o alumínio. O processo compreende as seguintes etapas:
a) Adição rápida do núcleo alcalino a solução de FeCl2 obtida após a remoção dos contaminantes.
b) Conseqüente formação de ferro ferroso sólido, resultando em uma suspensão aquosa com pH entre 4,0 e 6,0.
c) Aquecimento da suspensão obtida a uma temperatura entre 30 e 40ºC.
d) Oxidação da suspensão em duas etapas: d.1 Etapa rápida: Oxidação da suspensão em uma primeira etapa,
até que todo ferro ferroso sólido formado da mistura do núcleo alcalino com o sobrenadante, seja oxidado. A velocidade de oxidação encontra-se no intervalo entre 4,0 e 7,0 mol%Fe+3/h, a uma temperatura entre 30 a 40ºC, obtendo-se pH final na faixa 2,40 - 2,80.
d.2 Etapa lenta: Oxidação do ferro ferroso presente em fase líquida a uma velocidade de oxidação entre 0,4 e 4,0 mol%Fe+3/h, à temperatura entre 60 e 75ºC e com um aumento lento do pH de 0,1 a 0,8 unidades de pH/h. Nesta etapa, mantém-se o pH da mistura entre 2,8 a 4,2 e a mesma é denominada etapa de crescimento do pigmento.
Neste trabalho Meisen (2004) cita ainda que o FeCl2 pode conter constituintes orgânicos que podem influenciar no processo de cristalização do pigmento.
São três os métodos de obtenção dos núcleos, ou geradores, utilizados industrialmente para a produção de pigmento amarelo:
- o método alcalino; - o método alcalino contendo alumínio; - o método ácido. Estes métodos estão descritos, respectivamente, por Meisen,
(2004; 2003) e Meisen et.al. (2005). A Tabela 8 apresenta os parâmetros utilizados nestes métodos para a produção do núcleo.
69

Tab
ela
8 -
Mét
odos
par
a a
obte
nção
do
núcl
eo d
e G
oetit
a (α
FeO
.OH
) ut
iliza
dos
indu
stri
alm
ente
na
prod
ução
de
pigm
ento
. P
AR
ÂM
ET
RO
S
Núc
leo
alca
lino(1
) :Pro
cess
o al
calin
o di
fere
do
proc
esso
áci
do
na q
uant
idad
e de
com
post
o al
calin
o ad
icio
nada
, na
tem
pera
tura
e n
o m
aior
risc
o de
fo
rmaç
ão d
a m
agne
tita.
Núc
leo
ácid
o(2) : F
eSO
4 é m
istu
rado
so
b fo
rte
agita
ção
ao N
aOH
. Apó
s is
to, i
nici
a-se
a o
xida
ção
com
ar a
te
mpe
ratu
ra e
ntre
20
e 50
ºC, a
té
com
plet
a es
tabi
lizaç
ão d
o pH
ou
pote
ncia
l red
ox.
Núc
leo
ácid
o co
nten
do a
lum
ínio
(3) :
FeC
l 2 é
mis
tura
do s
ob fo
rte
agita
ção
a A
lCl 3
e aq
ueci
do. N
aOH
é a
dici
onad
o,
após
isto
se
inic
ia a
oxi
daçã
o co
m a
r at
é a
com
plet
a es
tabi
lizaç
ão d
o pH
ou
pote
ncia
l red
ox.
Con
c. F
e+2
inic
ial
44 a
132
g/L
44
a 1
32 g
/L
40 a
65
g/L
R
elaç
ão F
e+2 /F
e+3 n
o nú
cleo
N
ão in
form
ado
0,74
g F
e+2 /g
Fe+
3 N
ão in
form
ado
Qua
ntid
ade
de A
l3+ n
o nú
cleo
N
ão in
form
ado
Não
info
rmad
o 0,
06 g
Al+
3 /g F
e+2
- 12
a 1
3 m
ol
%A
l/mol
Fe
Con
cent
raçã
o de
rea
gent
e al
calin
o 3
a 15
Eq
gr/L
N
ão in
form
ado
4 a
8 E
q-g/
L
Qua
ntid
ade
de r
eage
nte
alca
lino
adic
iona
da
0,05
4 a
0,09
0 E
q gr
/g F
e+2
De
150
a 25
0% d
a qu
antid
ade
este
quio
mét
rica
p/ f
orm
ar
Fe(O
H) 2
.
0,01
4 a
0,02
1 E
q g/
g Fe
+2
De
15 a
70%
da
quan
tidad
e es
tequ
iom
étri
ca p
/ for
mar
Fe
(OH
) 2.
30 a
60%
da
quan
tidad
e es
tequ
iom
étri
ca p
ara
(Fe+
2 +A
l+3 ).
pH f
inal
13
a 1
4 2,
8 a
3,0
Não
info
rmad
o
Tem
po d
e ox
idaç
ão
Até
com
plet
a ox
idaç
ão d
e Fe
+2 .
De
3,5
h ou
até
a e
stab
iliza
ção
do
pH e
m to
rno
de 2
,8.
Tem
po d
e ad
ição
NaO
H
30 a
40
min
(rá
pida
) T
empe
ratu
ra d
e ob
tenç
ão d
o pr
ecip
itado
e o
xida
ção
34 a
47°
C
Am
bien
te o
u de
34
a 47
°C
Am
bien
te o
u de
35
a 50
°C
Vel
ocid
ade
de o
xida
ção,
(m
olFe
+2 ox
id./h
)/m
olFe
+2 in
icia
l 15
a 3
0 10
a 3
0 10
a 3
5
Aer
ação
0,
18 a
0,6
3 (L
/h)/
g Fe
+2
0,11
(L
/h)/
g Fe
+2
Não
info
rmad
o Fo
nte:
(1) M
eise
n (2
004)
. (2) M
eise
n (2
005)
.(3) M
eise
n (2
003)
.
70

A precipitação/oxidação de ferro a partir de uma solução aquosa de FeSO4 pode ser utilizada para a síntese de vários compostos de ferro, tais como a α-FeO.OH (Goetita), a γ-FeO.OH (Lepidocrocita), a δ-FeO.OH (ferroxita), α-Fe2O3 (hematita) e Fe3O4 (magnetita). Dependendo das condições experimentais, pode ocorrer a alternância entre uma fase, outra ou ambas (Cornell R. M. and Schwertmann, 2003).
Segundo Olowe et al. (1991.a), os mecanismo de oxidação de hidróxido ferroso em meio aquoso sulfatado, tal como ocorre após a mistura de FeSO4 com NaOH, são extremamente complexos e dependem de uma série de fatores. Os principais fatores que influenciam na reação são:
- A concentração inicial de reagentes; - A relação Fe+2:OH; - A temperatura e a velocidade da reação de oxidação. Em função destes parâmetros é possível obter-se um tipo de
óxido-hidróxido de ferro ou outro. A reação estequiométrica que ocorre entre o ferro ferroso e a soda
cáustica está representada na reação 26.
)(42)(24 )(2líquidosólido
SONaOHFeNaOHFeSO +→+ (26)
Se definirmos R como sendo a relação molar entre
[FeSO4]:[NaOH] ou, seja, R=[Fe+2]/[OH-], tem-se: - Para R = 0,50 = 1/2 – método estequiométrico (reação 26). - Para R < 0,50, ou seja, quando a quantidade de base é maior
do que 2 moles para cada mol de ferro ferroso, tem-se o método básico. Neste caso, temos excesso de NaOH na mistura e todo Fe+2 será consumido para formar Fe(OH)2 como único produto sólido inicial e algum OH- e Na+ permanecerão em solução aquosa, conforme a reação 27.
)(50,0
;)2()2()( 4224
alcalinonúcleodoinicialcomposiçãobásicométodoy
xR
OHxyNaxySOxNaOHxFeyNaOHxFeSO
−<=
−+−++→+ −+
(27)
- Para R > 0,50, tem-se um excesso de íons Fe+2 em relação
ao OH- adicionado; método ácido. Neste caso, todo OH- adicionado será consumido para formar, inicialmente,
71

Fe(OH)2 deixando um excesso de íons Fe+2 e íons SO4-2 em solução aquosa, conforme a reação 28.
)(50,0
;)2/()2/(2
)(2
24
24224
ácidonúcleodoinicialcomposiçãoácidométodoy
xR
SOyxFeyxSONay
OHFey
yNaOHxFeSO
−>=
−+−++→+−+ (28)
Olowe etal. (1991a), observaram que quando se trabalha na
condição básica, ou seja, quando R < 0,50 somente um estágio de mudança de fase ocorre durante a oxidação do ferro ferroso sólido a ferro férrico e nenhum composto intermediário é formado neste estágio. Desta forma, a transformação global pode ser apresentada como: finalodutoOOHFe
EstágioPr)(
122 →+ ; e o processo de oxidação
finda em meio básico. Entretanto, para R > 0,50, ou seja, no método ácido, dois
diferentes estágios foram observados durante a precipitação e oxidação do ferro ferroso. Isto indica a presença de um composto intermediário de transição denominado Green Rust 2 - GR2 (Olowe et al. 1991.a).
Neste caso, a transformação global pode ser apresentada como segue: finalodutoermediárioGRsólidoferrosoFerro
EstágioEstágio Pr)(int2 21 → → ; e
o processo de oxidação finda em meio ácido. Embora a caracterização do GR2 tenha sido apresentada na literatura, sua composição pode variar desde Fe(OH)2 até formas mais cristalinas próximas a Goetita, Lepidocrocita e Magnetita, indicando que são necessários mais estudos para sua identificação.
Para se obter predominantemente a Goetita partindo-se de FeSO4 0,1 mol/L e NaOH 0,2 mol/L a 250C, foi demonstrado que a melhor faixa de R, em condições ácidas, situa-se entre 0,565 e 0,580 (Tabela 9).
As análises de difração de raios X mostraram ainda a predominância da Magnetita, para R=0,50 ou seja para a quantidade estequiométrica; predominância da Goetita, para R=0,571 (ácido) e Lepidocrocita para R=0,583. Ainda para meios extremamente básicos (R=0,2) ou extremamente ácidos (R=3), a Goetita aparece como única e bem definida fase cristalina (Olowe et. al., 1991a).
72
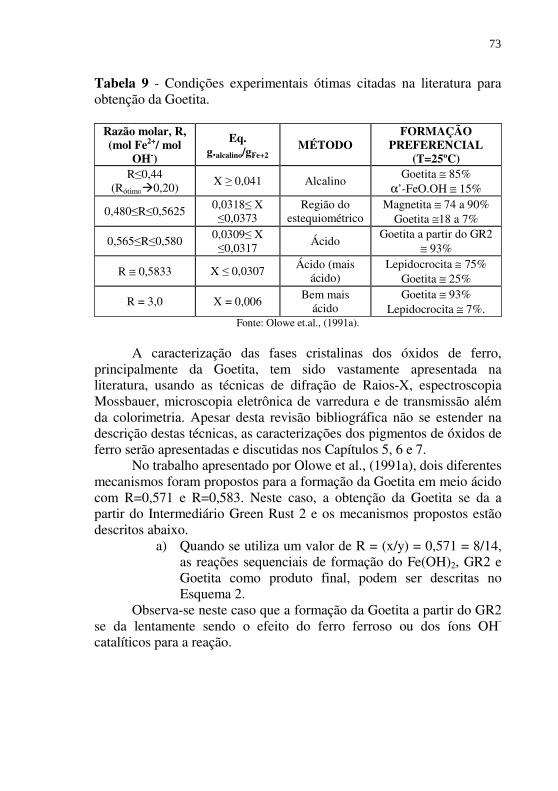
Tabela 9 - Condições experimentais ótimas citadas na literatura para obtenção da Goetita.
Razão molar, R, (mol Fe2+/ mol
OH-)
Eq. g.alcalino/gFe+2
MÉTODO FORMAÇÃO
PREFERENCIAL (T=25ºC)
R≤0,44 (Rótimo�0,20)
X ≥ 0,041 Alcalino Goetita ≅ 85%
α’-FeO.OH ≅ 15%
0,480≤R≤0,5625 0,0318≤ X ≤0,0373
Região do estequiométrico
Magnetita ≅ 74 a 90% Goetita ≅18 a 7%
0,565≤R≤0,580 0,0309≤ X ≤0,0317
Ácido Goetita a partir do GR2
≅ 93%
R ≅ 0,5833 X ≤ 0,0307 Ácido (mais
ácido) Lepidocrocita ≅ 75%
Goetita ≅ 25%
R = 3,0 X = 0,006 Bem mais
ácido Goetita ≅ 93%
Lepidocrocita ≅ 7%. Fonte: Olowe et.al., (1991a).
A caracterização das fases cristalinas dos óxidos de ferro,
principalmente da Goetita, tem sido vastamente apresentada na literatura, usando as técnicas de difração de Raios-X, espectroscopia Mossbauer, microscopia eletrônica de varredura e de transmissão além da colorimetria. Apesar desta revisão bibliográfica não se estender na descrição destas técnicas, as caracterizações dos pigmentos de óxidos de ferro serão apresentadas e discutidas nos Capítulos 5, 6 e 7.
No trabalho apresentado por Olowe et al., (1991a), dois diferentes mecanismos foram propostos para a formação da Goetita em meio ácido com R=0,571 e R=0,583. Neste caso, a obtenção da Goetita se da a partir do Intermediário Green Rust 2 e os mecanismos propostos estão descritos abaixo.
a) Quando se utiliza um valor de R = (x/y) = 0,571 = 8/14, as reações sequenciais de formação do Fe(OH)2, GR2 e Goetita como produto final, podem ser descritas no Esquema 2.
Observa-se neste caso que a formação da Goetita a partir do GR2 se da lentamente sendo o efeito do ferro ferroso ou dos íons OH- catalíticos para a reação.
73

4
.)(
2
2)12()(2/1)(7
2.2
222/1
1.2
22)()(7
1
117)(7148
571,014
8
22
441427
22
2
44142722
42
2
22
22
2
4414272
42
2
24
24224
etapa
OHFeOSOOHOFe
globaletapa
OHFeOHmSOOHOFeOSOFeOHFe
etapa
OHeOHO
etapa
eFeOmHSOOHOFeSOFeOHFe
etapa
SOFeSONaOHFeNaOHFeSO
ácidométodoy
xR
OHFeGR
mm
GR
mm
GR
mm
− →
−
++−+
→+++
→++
+++
→++
+++→+
−===
−+
−
−+
−
−+
−−
−+−
−+
−+
α
Esquema 2 - Formação da Goetita em fase líquida no meio ácido com R=0,571.
b) Quando se utiliza um valor de R=0,583 (7/12), ou seja,
meio um pouco mais ácido do que o anterior, neste caso, as reações sequenciais de formação do Fe(OH)2, GR2 e Goetita como produto final, podem ser descritas no Esquema 3.
4
.)(
2
)12()(2/1)(6
2.2
222/1
1.2
22)22()()(6
1
116)(6127
583,012
7
2
441427
2
2
44142722
42
2
22
2
2
4414272
42
2
24
24224
etapa
OHFeOSOOHOFe
globaletapa
OHmSOOHOFeOSOFeOHFe
etapa
OHeOHO
etapa
eHOHmSOOHOFeSOFeOHFe
etapa
SOFeSONaOHFeNaOHFeSO
ácidométodoy
xR
GR
mm
GR
mm
GR
mm
−→
−
−+
→+++
→++
++−+
→++
+++→+
−===
−
−
−+
−−
−+−
−+
−+
α
Esquema 3 - Formação da Goetita em fase líquida. Meio ácido com R=0,583.
Neste caso, com o meio um pouco mais ácido, não temos Fe+2 ou
74

íons OH- após a formação do intermediário Green Rust 2 e a Lepidocrocita pode ser um dos subprodutos da oxidação do GR2, sendo obtida diretamente pela oxidação deste último com O2, através da reação 29.
422441427 )24(.6)( FeSOOHmOHFeOOSOOHOFe mm +−+−→+− γ (29)
Ressalta-se, portanto que a adição extra de FeSO4 na solução
aquosa favorece a formação da Goetita, frente a Lepidocrocita. A Goetita então necessita de um excesso de íons Fe+2 ou excesso
de íons OH- para a sua formação frente a Lepidocrocita quando se origina do Intermediário Green Rust.
A influência da temperatura na oxidação do hidróxido ferroso em meio aquoso sulfatado foi investigada por Olowe et al. 1991(b). Os valores ótimos encontrados para R em função da temperatura, ou seja, os valores nos quais se obtiveram as maiores percentuais de Goetita como produto final, estão descritos na Tabela 10.
Tabela 10 - Valores ótimos para R citados na literatura para formação da Goetita em função da temperatura - solução aquosa inicial de 0,1 mol/L de sulfato ferroso.
R,
molFe+2/molOH Eq.gr.OH-
./gr.Fe+2 T, 0C
% Goetita
% Lepido-crocita
% Magnetita
0,40 0,045
29 40 50 60
66,1% 26,5% 8,0% 2,2%
0,0% 0,0% 0,0% 0,0%
11,8% 61,4% 86,6% 96,1%
0,5714 0,0313 25 92,6% 0,0% 0,0%
0,5833
0,0307
25 40 50 60
25,1% 81,9% 54,7% 11,5%
74,9% 18,1% 0,0% 0,0%
0,0% 0,0%
45,3% 88,5%
0,70
0,0256 30 40 50
66,7% 89,9% 90,6%
33,3% 10,1% 0,0%
0,0% 0,0% 9,4%
1,0 0,0180
9 26 29 40
5,9% 68,4% 77,4% 98,1%
94,1% 31,6% 22,6% 1,9%
0,0% 0,0% 0,0% 0,0%
Fonte: Olowe et.al., (1991.b).
75

Os autores observaram que, quanto mais ácido é o método utilizado para a obtenção da Goetita, maior deve ser a temperatura de oxidação. Isto ocorre porque em temperaturas baixas, no método ácido, há uma grande tendência de formação da Lepidocrocita (Tabela 10). Observou-se ainda que para a região de R entre 0,40 e 0,58, quanto maior a temperatura maior a porcentagem de magnetita formada chegando-se a obter 96% de magnetita para R=0,40 e 600C (Tabela 10).
Krehula et. al. (2002) estudaram a síntese de partículas aciculares de α-FeO.OH, partindo-se de ferro férrico em altos valores de pH. Segundo estes autores, uma rota para a produção de α-FeO.OH acicular é a hidrólise por um longo tempo em baixas temperaturas de uma solução diluída de Fe(NO3)3. Tal hidrólise pode ser acelerada pelo aumento da temperatura.
α-FeO.OH pode também ser obtida pela precipitação (através da adição de um meio alcalino), partindo-se de soluções aquosas de Fe2(SO4)3 ou NH4Fe(SO4)2. No caso de altas concentrações destes sais entretanto, a jarosita – H3OFe3(OH)6(SO4)2 ou NH4Fe3(OH)6(SO4)2 – também pode precipitar tornando a amostra menos cristalina. O rendimento nestes casos é pequeno e a Goetita produzida pode facilmente sofrer mudança de fase (Krehula et al., 2002).
A produção de α-FeO.OH é significativamente maior no processo de precipitação partindo-se de solução aquosa de FeSO4, tal como visto anteriormente. Entretanto, a formação da Goetita por este método é extremante sensível ás condições experimentais. Além disso, α-FeO.OH pode também ser produzida a partir da cristalização da ferridrita em altos valores de pH e temperatura (Music et. al, 1990).
Betancur et al. (2004) estudaram o efeito da quantidade de água nas propriedades magnéticas e estruturais da Goetita. As amostras de Goetita foram preparadas partindo-se de ferro ferroso e férrico como precursores. Observou-se neste trabalho que a Goetita preparada a partir de ferro ferroso como precursor, (método alcalino, R=0,45), contém mais água estrutural e adsorvida do que aquela produzida a partir de ferro férrico, menor grau de cristalinidade e mais larga distribuição no tamanho de partículas (granulometria mais dispersa). Ressalta-se ainda que o conteúdo de água que deixa a amostra em temperaturas menores do que 700C esteja associado à água adsorvida na superfície.
Para a utilização da Goetita como pigmento, a forma, o tamanho dos cristais e a coexistência de mais de uma fase cristalina ou até mesmo amorfa, são de fundamental importância (Meisen 2003 e 2004).
Kurokawa et al. (2006) estudaram a influência de diferentes
76

fontes de ferro ferroso no tamanho e morfologia dos cristais de Goetita produzida. No caso da obtenção da Goetita a partir de precursores de ferro ferroso, estes parâmetros, (ou seja, o tamanho e a morfologia dos cristais) são extremamente sensíveis aos tipos de precursores utilizados. No trabalho de Kurokawa, utilizaram-se como precursores:
- Fe(OH)2 – obtido pelo método alcalino de precipitação onde R=0,14 (1molFe+2 : 7moles base) e R=0,20 (1molFe+2 : 5moles base);
- Intermediário Green Rust II – obtido pelo método ácido de precipitação onde R=5,0 (1molFe+2 : 0,2moles base) e R=0,70 (1molFe+2 : 1,4moles base);
- FeCO3+Fe(OH)2 – obtido pelo método alcalino ou pelo método ácido de precipitação onde R=0,40 (1:2,5) e R=0,67 (1:1,5), respectivamente.
Os autores observaram que os melhores precursores para
obtenção de Goetita anisotrópica são, respectivamente, FeCO3+Fe(OH)2 método alcalino > Fe(OH)2 > Intermediário Green Rust II. A baixa viscosidade da mistura FeCO3+Fe(OH)2, ocasiona um aumento na velocidade de oxidação, devido à maior saturação com O2, isto também aumenta a finura (extremidade) dos cristalitos obtidos favorecendo o crescimento em uma única forma (anisotrópico). Durante a reação de precipitação e oxidação, também no caso da mistura de FeCO3+Fe(OH)2, a mudança no pH da solução é extremamente menor do que quando se parte do Intermediário Green Rust II como precursor (ou até mesmo do Fe(OH)2). Isto ocorre devido ao efeito tampão da coexistência de CO3
-2 e HCO3- na solução. O pH inicial da mistura
inicia-se em 9,4 e o pH final após a oxidação permanece alcalino em torno de 8,7, por exemplo.
Music et al (2007), também relataram que a composição da fase de óxido de ferro obtida pelo método de precipitação, partindo-se de FeSO4, é extremamente dependente da relação [Fe+2/OH-] no início do processo de precipitação, ou seja, do valor de R, da velocidade de oxidação, do tempo de precipitação, da temperatura, e do tipo de reagente químico alcalino adicionado (NaOH versus NH4OH, por exemplo). Também o tamanho das partículas obtidas para a Goetita a partir de Fe(OH)2 é fortemente dependente do tempo de oxidação.
Lepidocrocita foi sintetizada a partir de FeSO4 a 200C em pH 7,0. Ferroxita também pode ser sintetizada neste tipo de sistema, desde que as seguintes condições experimentais sejam satisfeitas:
77

- Rápida adição de O2 através da suspensão de Fe(OH)2; - Rápida oxidação de Fe(OH)2 pelo H2O2; - Secagem direta de Fe(OH)2 pelo O2; - pH do meio fortemente alcalino. Sabe-se que a precipitação de ferro ferroso com soda a partir de
FeSO4 ou FeCl2 pode levar à formação de vários óxidos de ferro, a natureza do produto final, bem como morfologia e cristalinidade, dependem da temperatura, concentração inicial de ferro ferroso, relação [Fe+2]/[NaOH], velocidade de oxidação e tipo de sal ferroso utilizado. Gilbert et al. (2008) avaliaram a influência da concentração inicial destes reagentes, bem como da agitação da mistura, nas propriedades da Goetita produzida a partir da suspensão aquosa de ferro ferroso, Fe(OH)2. Em relação ao tipo de sal ferroso utilizado, é sabido que íons cloreto favorecem a formação da Lepidocrocita (Detournay e.al., 1976; Refait et.al. 2003). Portanto, FeSO4 tem sido preferido para obtenção da Goetita. Com relação ao valor de R, (moles Fe+2/mol OH-), sabe-se que a Goetita forma-se, preferencialmente, com um excesso de íons OH-, ou seja, R=0,20, ou excesso de íons Fe+2, R=2,5 (Olowe et al., 1991a).
Com relação à cinética de oxidação do Fe(OH)2, sabe-se que a mesma é bastante influenciada pelo teor de oxigênio dissolvido no meio, o que é função da temperatura. Para uma temperatura maior do que 60 0C, por exemplo, têm-se baixas solubilidades do O2 e, portanto baixas velocidades de oxidação. Neste caso, forma-se como produto da oxidação, preferencialmente, a magnetita – Fe3O4 (Olowe etal., 1991b). Para uma temperatura menor do que 20 0C, têm-se maiores solubilidades do O2, com conseqüente maior velocidade de oxidação, e possibilidade de formação da Lepidocrocita. Isto explica que, para a formação da Goetita, a temperatura deve ser mantida entre 25 e 450C. Entretanto, o valor ótimo da temperatura deve ser avaliado juntamente com o valor de R, ou seja, meio ácido ou básico (Gilbert, F. et. al, 2008). Com isso, conclui-se que a formação da Goetita por oxidação de Fe(OH)2 precipitado em meio aquoso sulfatado é principalmente controlada pela razão R e pela temperatura. As condições experimentais ótimas que levaram à formação da Goetita foram definidas como:
- R=0,20 (método alcalino); 0,1mol/L Fe(OH)2; Tótima=250C. Rota de formação da Goetita: Fe(OH)2 � α-FeO.OH.
- R=2,50 (método ácido); 0,1mol/L Fe(OH)2; Tótima=450C. Rota de formação Goetita: Fe7O2m(OH)14-4mSO4 (catalisado pelo Fe+2) � α-FeO.OH.
78

No trabalho de Gilbert et al. (2008), variando-se a velocidade de agitação e a concentração inicial de FeSO4 observou-se que o tamanho dos cristais é alterado pela mudança na cinética de formação da Goetita, sendo tal reação controlada pela redução do O2 dissolvido no meio. Maior velocidade de oxidação, seja pela redução da relação [Fe+2]/[O2], seja pelo aumento da velocidade de agitação, (quando a velocidade é controlada pela transferência de massa e não pela reação química em si), causou um menor tamanho dos cristais formados (Gilbert et al, 2008).
Foi demonstrado, no trabalho de Gilbert et al. (2008), que quanto maior a velocidade da reação de oxidação do intermediário GR2 para Goetita, menor o tamanho dos cristais formados. Observou-se, também, que a cristalinidade do óxido de ferro era maior quando a oxidação ocorria lentamente, que ocorreu quando a concentração de ferro aumentou de 5,6 para 56 gFe/L. Adicionalmente, também houve a formação de Lepidocrocita para menores concentrações de ferro (5,6 g/L) e maiores velocidades de oxidação, sendo que os picos da Lepidocrocita no difratograma de raios X das amostras, desapareceram a partir de concentrações maiores do que 9,5 g/L.
Com relação à morfologia dos cristais; foram obtidos cristais aciculares, tipo agulha, com razão comprimento:raio 0,70:0,08, entretanto, aparecem dispersos no meio pequenos cristais com morfologia totalmente diferente (Gilbert et al, 2008).
Vale ainda ressaltar com relação a este trabalho que o aumento da velocidade da reação de oxidação, o que ocorreu com baixas concentrações de ferro, favoreceu a formação e crescimento da Lepidocrocita e impediu a formação dos cristais de Goetita. A Lepidocrocita foi formada para velocidades de oxidação mais alta, o que levou a formação de cristais δ-FeO.OH ao invés da Goetita. Entretanto, no método alcalino, não houve a formação da Lepidocrocita, somente no método ácido, pois no método alcalino forma-se diretamente o Fe(OH)2 que oxida-se para Goetita. No caso do método ácido forma-se o complexo Intermediário Green Rust 2 (Fe+2/Fe+3/SO4
2-/OH-), que é posteriormente oxidado para formação da Goetita (Gilbert et al., 2008).
79


4 METODOLOGIA EXPERIMENTAL Os trabalhos experimentais desta tese foram realizados no
Laboratório da Divisão Química da Carbonífera Criciúma S.A, em Criciúma – SC, no Laboratório de Energia e Meio Ambiente, LEMA, do Departamento de Engenharia Química e Engenharia de Alimentos da UFSC e na nova Estação de Tratamento de Efluentes da Carbonífera Criciúma S.A. implantada durante o desenvolvimento da tese.
A metodologia compreendeu duas rotas, mostradas esquematicamente nas Figuras 4 e 5.
Em uma primeira rota, Figura 4 utilizou-se diretamente a DAM como matéria-prima para produção de Pigmento e Coagulante.
Na segunda rota, Figura 5 verificou-se a necessidade de pré-tratar a DAM para a produção do pigmento amarelo; daí houve a obtenção do gesso, coagulante e finalmente do pigmento.
Nas Figuras 4 e 5 estão descritas as atividades realizadas em cada etapa do trabalho, bem como os produtos obtidos para cada atividade.
DAM
NEUTRALIZAÇÃO COM NAOH
OBTENÇÃO DO LODO QUÍMICO 1
TRATAMENTO COM H2SO4 OU HCL
COAGULANTE 1
CARACTERIZAÇÃO
LAVAGEM / SECAGEM / MOAGEM / CALCINAÇÃO
PIGMENTO 1
CARACTERIZAÇÃO
Opção 1 Opção 2
Figura 4 - Primeira rota experimental visando o aproveitamento da DAM como
matéria-prima para obtenção de coagulante ou pigmento a base de ferro (realizada no período entre 2004 e 2005).
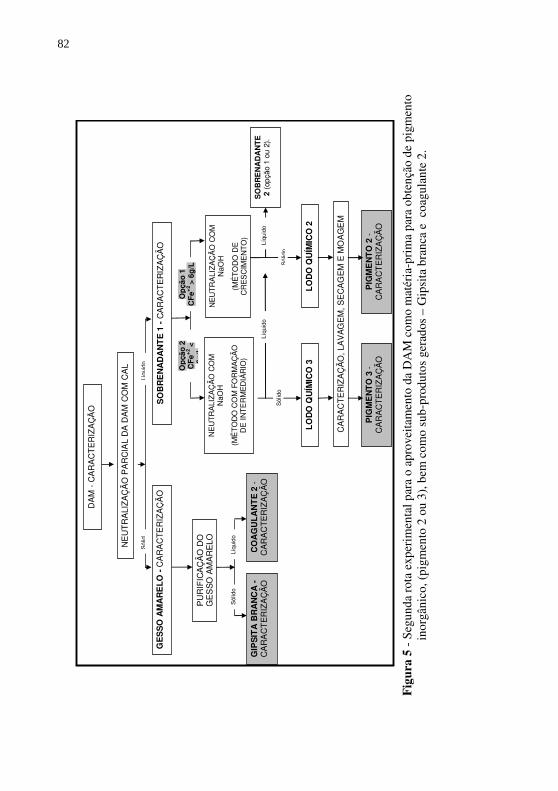
DA
M -
CA
RA
CT
ER
IZA
ÇÃ
O
NE
UT
RA
LIZ
AÇ
ÃO
PA
RC
IAL
DA
DA
M C
OM
CA
L
GE
SS
O A
MA
RE
LO
- C
AR
AC
TE
RIZ
AÇ
ÃO
S
OB
RE
NA
DA
NT
E 1
- C
AR
AC
TE
RIZ
AÇ
ÃO
PU
RIF
ICA
ÇÃ
O D
O
GE
SS
O A
MA
RE
LO
GIP
SIT
A B
RA
NC
A -
C
AR
AC
TE
RIZ
AÇ
ÃO
C
OA
GU
LA
NT
E 2
-
CA
RA
CT
ER
IZA
ÇÃ
O
NE
UT
RA
LIZ
AÇ
ÃO
CO
M
NaO
H
(MÉ
TO
DO
CO
M F
OR
MA
ÇÃ
O
DE
IN
TE
RM
ED
IÁR
IO)
LO
DO
QU
ÍMIC
O 3
NE
UT
RA
LIZ
AÇ
ÃO
CO
M
NaO
H
(MÉ
TO
DO
DE
C
RE
SC
IME
NT
O)
CA
RA
CT
ER
IZA
ÇÃ
O, L
AV
AG
EM
, SE
CA
GE
M E
MO
AG
EM
PIG
ME
NT
O 2
-
CA
RA
CT
ER
IZA
ÇÃ
O
PIG
ME
NT
O 3
-
CA
RA
CT
ER
IZA
ÇÃ
O
Op
ção
2 C
Fe+
2 <
6g/L
Op
ção
1 C
Fe+
2 >
6g
/L
Sól
idLí
quid
o
SO
BR
EN
AD
AN
TE
2
(opç
ão 1
ou
2).
Líqu
ido
Líqu
ido
LO
DO
QU
ÍMIC
O 2
Sól
ido
Sól
ido
Sól
ido
Líqu
ido
F
igur
a 5
- S
egun
da r
ota
expe
rim
enta
l par
a o
apro
veit
amen
to d
a D
AM
com
o m
atér
ia-p
rim
a pa
ra o
bten
ção
de p
igm
ento
in
orgâ
nico
, (pi
gmen
to 2
ou
3), b
em c
omo
sub-
prod
utos
ger
ados
– G
ipsi
ta b
ranc
a e
coa
gula
nte
2.
82

4.1 METODOLOGIA APLICADA A PRIMEIRA ROTA
EXPERIMENTAL
4.1.1 Caracterização da drenagem ácida de mina Em todas as fases experimentais desta tese procurou-se obter
produtos de elevado valor agregado a partir da drenagem ácida de mina de carvão. Neste sentido, foram realizados testes para se determinar as condições ótimas de recuperação do ferro e do sulfato presentes na DAM.
A composição química da drenagem ácida de mina foi acompanhada por um período total de 5 anos, englobando os testes tanto da primeira rota experimental quanto da segunda. A concentração dos metais ferro, alumínio, manganês, cobre e zinco assim como as análises de acidez, pH e sulfato foram realizadas utilizando-se os métodos colorimétricos de acordo com o Standard Methods for Examination for Water and Wastewater, 1995.
4.1.2 Neutralização da DAM com NaOH
4.1.2.1 Obtenção do Lodo Químico 1 A precipitação de ferro foi realizada utilizando-se NaOH, em
diferentes valores de pH’s, em escala de laboratório. Além do pH, foi também medido o potencial de oxidação do meio uma vez que para se alcançar elevada recuperação do ferro dissolvido na DAM, é necessário que o meio tenha elevado potencial redox.
Para se aumentar o potencial de oxidação foi utilizado o peróxido de hidrogênio, como oxidante adicional, além do oxigênio dissolvido. Amostras de 5 a 50 litros da DAM foram tratadas com NaOH com ou sem suprimento adicional de peróxido de hidrogênio, em diferentes valores de pH’s. O rendimento da recuperação do ferro foi calculado pelo balanço de massa entre a quantidade de ferro total dissolvido antes e após a neutralização. A caracterização do lodo químico 1 foi realizada pela dissolução completa do material sólido em HCl concentrado conforme os métodos padrões de análises químicas (AWWA,1995)
83

4.1.2.2 Obtenção do Coagulante 1 O lodo químico 1, produzido pela neutralização direta da DAM
com soda, foi tratado com quantidades estequiométricas, com base nas quantidades de ferro e alumínio, de ácido sulfúrico ou ácido clorídrico, em frascos com sistema de agitação constante. Após a solubilização total do lodo químico 1 no meio ácido as concentrações de ferro e alumínio presentes no coagulante produzido (Coagulante 1), foram medidas através de métodos colorimétricos (AWWA, 1995).
4.1.2.3 Obtenção do Pigmento 1 Diferentes amostras do lodo químico 1, obtidas pela variação no
pH e no método de precipitação, foram secas a 110ºC e posteriormente calcinadas a diferentes temperaturas, visando à produção de pigmento inorgânico de óxido de ferro (Pigmento 1).
Foi verificada a necessidade de eliminação dos íons sulfato adsorvidos no lodo químico 1. Para tal, foi utilizado o método de lavagens sucessivas com água destilada submetida a diferentes valores de pH, até a completa eliminação do sulfato (determinada por análise colorimétrica). Após a secagem, foi realizada a calcinação em diferentes temperaturas em forno mufla.
A caracterização do Pigmento 1 foi realizada através da obtenção dos espectros de difração de raios-X, análise termogravimétrica e análise colorimétrica.
A determinação das coordenadas colorimétricas (Cielab) foi realizada no Centro de Tecnologia em Materiais do Senai em Criciúma (Senai SC/CTCmat) utilizando-se um espectrofotômetro HunterLab modelo ColorQuest 45/0. A análise de difratometria de raios-X em pó foi realizada no Laboratório de Caracterização Microestrutural do Departamento de Engenharia Mecânica da UFSC, utilizando-se um difratômetro Philips X’PERT. As análises termograviétricas foram realizadas no Senai-SC/CTCmat, utilizando-se o Método PR-CC-128, taxa de aquecimento constante 10ºC/min, atmosfera de ar sintético, fluxo 70 cm3/min.
84

4.2 METODOLOGIA APLICADA À SEGUNDA ROTA EXPERIMENTAL
A segunda rota experimental envolveu o tratamento seqüencial da
drenagem ácida de mina cuja necessidade foi verificada após a realização dos primeiros testes.
O tratamento seqüencial da drenagem ácida tem por objetivo a remoção seletiva dos componentes majoritários presentes na água, ou seja, do ferro, do alumínio e do sulfato, obtendo-se com isso um teor mais elevado de ferro em um lodo precipitado, bem como de sulfato e alumínio no outro.
4.2.1 Neutralização parcial da DAM com cal
4.2.1.1 Obtenção do Gesso Amarelo e do Sobrenadante 1 A precipitação seletiva do alumínio foi realizada na faixa de pH
entre 4,0 e 4,5, utilizando-se solução de hidróxido de cálcio a 15% como agente precipitante. O volume de DAM para os testes variou entre 50 e 70 litros. Com isso, obteve-se um precipitado contendo sulfato de cálcio e hidróxido de alumínio e uma fase líquida sobrenadante.
O precipitado, designado como Gesso Amarelo, foi caracterizado de acordo com sua composição química e mineralógica. O líquido sobrenadante foi designado como Sobrenadante 1 e o mesmo, após caracterização, foi direcionado para uma próxima etapa de tratamento. O pH de precipitação foi ajustado de forma a se alcançar a máxima remoção do alumínio e mínima remoção do ferro.
4.2.1.2 Caracterização e purificação do Gesso Amarelo A purificação do Gesso Amarelo foi realizada por meio de uma
reação controlada com a adição de solução ácida ao lodo filtrado. A quantidade de ácido utilizada foi àquela necessária para reagir com todas as impurezas de ferro e alumínio presentes no gesso. A mistura, lodo úmido filtrado + solução ácida, foi agitada e filtrada obtendo-se com isso duas fases distintas; uma fase líquida, extremamente ácida rica em ferro e alumínio; e uma fase sólida, rica em Gipsita Branca. A Gipsita foi seca a 110oC por 3 horas, moída e caracterizada. A amostra líquida,
85

denominada Coagulante 2, foi caracterizada para se determinar as concentrações de alumínio e ferro contidas na mesma.
4.2.2 Neutralização do Sobrenadante 1 com NaOH
4.2.2.1 Método de Crescimento – Obtenção do Pigmento 2 Completada a primeira etapa de tratamento da DAM, ou seja, a
remoção do alumínio, o Sobrenadante 1 (um sulfato ferroso com concentração de ferro ≥ 6g/L) foi submetido à segunda etapa, que envolvia a remoção seletiva do ferro com obtenção do pigmento pelo método de crescimento.
Nesta etapa, o pH da água ácida foi elevado lentamente com a adição de soda cáustica diluída, enquanto Fe2+ foi oxidado a Fe3+ sob aeração.
Obtinham-se, então, duas fases distintas: uma fase sólida, rica em Fe3+ e uma fase líquida ainda ácida, porém com baixas concentrações de íons metálicos. Os testes para obtenção do pigmento pelo método de crescimento foram realizados variando-se a dosagem de soda cáustica, o tempo da reação de oxidação, a dosagem de peróxido de hidrogênio e o reciclo de lodo decantado.
4.2.2.2 Método com Formação de Intermediário – Obtenção do
Pigmento 3 Durante o desenvolvimento deste trabalho a concentração de
ferro ferroso na drenagem ácida de mina reduziu drasticamente devido às alterações nas pilhas de rejeito piritoso da indústria. Em função disto, a qualidade do Pigmento 2, obtido pelo método de crescimento, ficou bastante prejudicada, principalmente no que diz respeito a sua cristalinidade.
Por este motivo foi necessário o desenvolvimento de outro método de obtenção de pigmento, partindo-se agora de uma drenagem ácida contendo ferro ferroso com concentrações menores do que 6 g/L.
Neste método, após a remoção do alumínio o Sobrenadante 1 foi misturado, sem aeração, com diferentes quantidades de NaOH obtendo-se um lodo verde intermediário como sólido precipitado. O lodo verde, rico em Fe2+, foi separado por decantação e espessamento e então
86

submetido a diferentes velocidades de oxidação com ou sem aquecimento. Após a oxidação, o lodo de Fe3+ foi lavado sucessivas vezes, até a completa remoção dos sais incorporados, co-precipitados e adsorvidos no lodo. Após a lavagem, o sólido foi moído, seco a 1100C por 12 horas e caracterizado. Nestes testes, variou-se a dosagem de soda, (ou seja, o valor de R), a concentração inicial de ferro, a temperatura da reação de oxidação, a velocidade da oxidação, e a adição (ou não) de núcleo inicial de Goetita. A produção do núcleo de Goetita foi também avaliada pelo método alcalino e método ácido conforme descrito na Tabela 8.
Os testes de produção do Pigmento 3 foram realizados também com o objetivo de se obter um produto mais cristalino e, portanto, de melhor qualidade do que a amostra de pigmento obtida no item anterior (Pigmento 2). Além disso, houve a necessidade de se adequar o processo de produção do pigmento à qualidade da matéria-prima disponível na indústria.
4.2.2.3 Caracterização dos Pigmentos 2 e 3 A caracterização dos pigmentos 2 e 3 foi realizada em diferentes
laboratórios, selecionados pela maior confiabilidade em suas análises. As análises mineralógicas qualitativas, visando a identificação das fases cristalinas presentes, foram realizadas por difratometria de raios-X. Estas análises foram feitas no Laboratório de Análises de Minerais e Rochas (LAMIR), da Universidade Federal do Paraná. Foi utilizado um Difratômetro Philips PW 1830 e as fases cristalinas foram identificadas por comparação com os padrões do Joint Committee on Power Diffraction Standards (International Centre for Diffraction Data).
A determinação das coordenadas colorimétricas (Cielab) foi realizada no Centro de Tecnologia em Materiais do Senai em Criciúma (Senai-SC - Criciúma/CTCmat) utilizando-se um espectrofotômetro HunterLab modelo ColorQuest 45/0.
As análises de distribuição do tamanho de partículas foram realizadas no Analisador de Partículas a Laser (CILAS 1064 líquido) pelo método de difratometria a laser e como agente dispersante foi utilizada a água. Este método baseia-se no princípio de que o ângulo de difração é inversamente proporcional à dimensão da partícula. Por este método foram determinados os valores em micrometros de D10 (10% do material é menor do que aquele tamanho determinado), D50, D90 e o
87

valor de Dmédio. Estas análises também foram realizadas no Senai-SC - Criciúma/CTCmat.
4.3 ESTAÇÃO DE TRATAMENTO DE EFLUENTES DA
CARBONÍFERA CRICIÚMA S.A. Uma nova planta em escala industrial de tratamento de efluentes
com produção de pigmentos e subprodutos associados foi projetada, adaptada e implantada de acordo com o desenvolvimento deste trabalho. O esquema da planta é mostrado nas Figuras 6 e 7.
Na Figura 6, esta apresentado o Circuito 1 onde ocorre a produção do Gesso Amarelo e do Sobrenadante 1.
Na Figura 7 esta apresentado o Circuito 2, onde ocorre a produção do pigmento pelo método com formação de intermediário.
Na Figura 7 (a), é apresentada a primeira fase do Circuito 2, onde é obtido, a partir da mistura do Sobrenadante 1 com a soda, o lodo verde intermediário. Na Figura 7 (b) esta apresentada a etapa de oxidação do composto intermediário com a consequente formação do pigmento amarelo.
Figura 6 – Esquema da planta de tratamento de efluentes implantada na
Carbonífera Criciúma S.A. - Circuito 1.
88
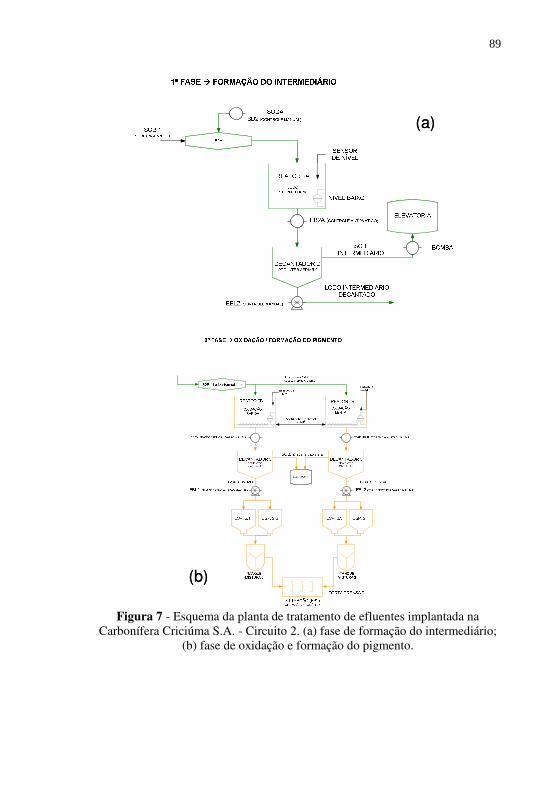
(a)(a)
(b)(b)
Figura 7 - Esquema da planta de tratamento de efluentes implantada na
Carbonífera Criciúma S.A. - Circuito 2. (a) fase de formação do intermediário; (b) fase de oxidação e formação do pigmento.
89


5 RESULTADOS DA PRIMEIRA ROTA EXPERIMENTAL
5.1 CARACTERIZAÇÃO DA DRENAGEM ÁCIDA DE MINA A caracterização da drenagem ácida de mina de carvão utilizada
neste trabalho foi realizada no período de agosto de 2004 a agosto de 2009. A Tabela 11 mostra alguns dos valores encontrados para os metais assim como para o sulfato presentes nesta drenagem.
Observa-se (Tabela 11) que, no período entre agosto e dezembro de 2004, a água apresentava uma concentração média de Fe2+ de 12g/L; 1,4g/L de alumínio; 0,3g/L de manganês e 27g/L de sulfato. Ressalta-se ainda que em quase todas as amostras, 100% do ferro estava na forma de Fe+2.
No período entre maio e agosto de 2005, a concentração de Fe+2
variou entre 5 e 13g/L (com 89% do ferro na forma divalente) e a concentração média de alumínio ficou em 1,4g/L. Exceto para o alumínio, os outros metais encontravam-se em concentrações inferiores a 200 mg/L. Estes valores de concentração, a princípio, não seriam problema para a obtenção de um lodo puro em ferro a partir do tratamento desta drenagem ácida.
No período entre janeiro de 2006 e agosto de 2007, as concentrações dos metais presentes na DAM reduziram significativamente devido a alterações realizadas nos módulos de rejeito piritoso da indústria. Neste período, obtiveram-se valores médios de Fe+2 de 6 a 8g/L, Al3+ de 1,0 a 1,5g/L, sulfato de 21 a 26g/L e acidez entre 29 e 33gCaCO3/L.
No período entre 2008 e 2009, as concentrações médias de ferro, alumínio e sulfato ficaram entre 3,0 e 3,5g/L; 0,3 e 0,5g/L e 9 e 12g/L, respectivamente.
As Figuras 8, 9 e 10 resumem a variação na composição da drenagem ácida de mina durante o desenvolvimento deste trabalho. Atualmente as concentrações médias dos metais são de 4g/L para o Fe+2
e 0,45g/L para o alumínio. Os valores da acidez e sulfato situam-se entre 10 e 12 g/L, respectivamente.

Tab
ela
11 -
His
tóri
co d
a ca
ract
eriz
ação
da
dren
agem
áci
da d
e m
ina
no p
erío
do a
valia
do n
este
trab
alho
.
Dat
a pH
F
e+2 , g
/L
Fer
ro T
otal
, g/
L
%F
e+2
Al3+
,mg/
L
Mn,
m
g/L
SO
42-,
mg/
L
Cu2+
, m
g/L
Z
n2+,
mg/
L
Aci
dez,
m
g/L
04
/08/
04
3,0
15,6
15
,6
100%
1.
319,
0 29
5,0
21.4
72
- -
- 09
/08/
04
3,8
14,1
14
,1
100%
-
- 28
.400
-
- -
10/0
8/04
3,
4 15
,2
15,2
10
0%
- -
28.3
70
- -
- 11
/08/
04
3,0
16,0
16
,0
100%
-
- 28
.370
-
- -
12/0
8/04
3,
1 16
,0
16,0
10
0%
- -
28.3
70
- -
- 19
/08/
04
3,0
12,0
12
,0
100%
-
- -
- -
- 26
/10/
04
2,8
9,1
10,4
88
%
1.97
8,0
298,
3 21
.472
-
- -
10/1
1/04
2,
7 6,
8 7,
0 97
%
1.38
3,2
225,
4 27
.900
-
- -
22/1
1/04
2,
9 11
,5
11,5
10
0%
1.09
1,8
468,
8 33
.890
-
- -
23/1
1/04
3,
5 9,
2 9,
2 10
0%
673,
5 31
8,4
23.9
00
- -
- 24
/11/
04
3,8
11,9
11
,9
100%
1.
800,
0 25
1,7
18.3
50
- -
- 02
/12/
04
2,9
17,4
17
,4
100%
1.
255,
0 27
3,5
31.3
20
- -
- 03
/12/
04
2,9
10,5
10
,5
100%
1.
814,
0 17
1,0
34.5
30
- -
- 20
04
3,1
12,7
12
,8
99%
1.
414,
3 28
7,8
27.1
95
- -
- 31
/05/
05
3,1
9,3
9,3
100%
1.
152,
0 16
7,5
25.6
60
13,6
35
,6
24.8
30
13/0
6/05
3,
2 8,
1 8,
3 98
%
1.56
8,0
153,
0 24
.140
41
,0
59,1
28
.740
21
/06/
05
3,1
13,4
13
,4
100%
1.
128,
0 16
6,1
34.1
30
9,9
76,0
32
.160
22
/06/
05
3,1
6,0
8,0
75%
-
- -
- -
- 21
/06/
05
2,9
8,6
8,9
97%
1.
442,
5 16
6,1
- -
- 32
.160
25
/06/
05
3,1
8,9
8,9
100%
1.
330,
9 16
3,2
25.0
00
4,6
65,9
22
.720
30
/06/
05
3,1
4,7
9,0
53%
1.
490,
0 -
- -
- 32
.000
C
onti
nua
92

Dat
a pH
F
e+2 , g
/L
Fer
ro T
otal
, g/
L
%F
e+2
Al3+
,mg/
L
Mn,
m
g/L
SO
42-,
mg/
L
Cu2+
, m
g/L
Z
n2+,
mg/
L
Aci
dez,
m
g/L
C
onti
nuaç
ão
03/0
7/05
4,
0 6,
9 7,
6 91
%
460,
0 -
- -
- -
12/0
7/05
2,
9 7,
2 8,
2 89
%
1.50
6,4
- 22
.400
-
- -
25/0
7/05
3,
1 8,
2 8,
3 99
%
1.39
4,5
156,
0 30
.220
7,
9 2,
7 20
.480
19
/08/
05
3,1
6,6
8,8
75%
1.
535,
0 16
0,3
23.4
80
9,1
- 32
.670
20
05
3,1
8,0
8,9
89%
1.
394,
2 16
1,7
26.4
33
14,3
47
,9
28.2
20
01/0
8/06
3,
1 6,
9 8,
4 82
%
1.50
3,2
125,
3 25
.441
6,
9 -
31.7
35
15/0
8/06
3,
3 5,
0 9,
6 52
%
1.37
8,5
166,
1 26
.608
9,
5 -
34.7
60
01/0
9/06
2,
7 7,
1 9,
0 79
%
1.64
0,7
148,
8 32
.613
14
,0
0,0
25.2
00
15/0
9/06
2,
7 4,
3 8,
4 51
%
1.53
8,4
150,
2 28
.918
9,
1 0,
0 29
.920
01
/11/
06
2,6
7,2
8,0
91%
1.
496,
8 10
7,0
14.7
91
5,8
0,0
30.3
05
15/1
1/06
2,
8 4,
1 8,
0 52
%
1.25
7,0
148,
8 27
.179
5,
4 0,
0 26
.950
20
06
2,9
5,8
8,6
68%
1.
469,
1 14
1,0
25.9
25
8,5
0,0
29.8
12
01/0
2/07
2,
8 9,
2 9,
2 10
0%
1.08
6,6
223,
4 29
.360
0,
1 0,
1 38
.074
02
/03/
07
3,0
9,2
9,2
100%
1.
128,
2 16
2,8
21.4
47
- -
32.8
55
12/0
3/07
3,
0 8,
1 8,
1 10
0%
1.05
9,1
- 19
.469
-
- 30
.353
22
/03/
07
2,8
9,1
9,1
100%
1.
066,
4 -
20.0
62
- -
33.9
74
02/0
6/07
2,
8 6,
2 6,
6 94
%
1.01
1,8
- 22
.238
-
- 36
.032
12
/06/
07
3,1
8,9
9,6
92%
1.
102,
8 -
23.6
23
- -
30.6
18
01/0
7/07
2,
7 5,
6 6,
8 83
%
1.05
5,5
- 21
.051
-
- 22
.938
10
/07/
07
3,0
8,7
8,7
100%
1.
157,
3 -
24.8
10
- -
- 19
/07/
07
2,9
9,5
9,5
100%
1.
175,
5 -
25.0
08
- -
30.8
51
28/0
7/07
3,
2 8,
3 8,
3 10
0%
891,
8 -
20.8
54
- -
23.2
31
Con
tinu
a
93

Dat
a pH
F
e+2 , g
/L
Fer
ro T
otal
, g/
L
%F
e+2
Al3+
,mg/
L
Mn,
m
g/L
SO
42-,
mg/
L
Cu2+
, m
g/L
Z
n2+,
mg/
L
Aci
dez,
m
g/L
C
onti
nuaç
ão
06/0
8/07
3,
0 8,
7 14
,0
62%
99
7,3
- 17
.317
-
- 41
.067
15
/08/
07
3,1
9,3
14,4
65
%
1.01
9,1
- 17
.836
-
- 43
.177
24
/08/
07
2,9
7,4
7,4
100%
96
8,2
- 16
.540
-
- 32
.420
20
07
3,0
8,3
9,3
92%
1.
055,
4 19
3,1
21.5
09
0,1
0,1
32.9
66
15/8
/08
- 1,
8 2,
0 88
%
246,
4 -
9.68
9 -
- 5.
513
15/9
/08
- 2,
8 3,
4 83
%
421,
6 -
13.6
71
- -
9.77
7 15
/10/
08
- 2,
8 3,
0 93
%
414,
8 -
11.0
40
- -
8.68
9 15
/11/
08
- 2,
7 2,
7 97
%
342,
8 -
10.6
93
- -
8.08
9 15
/12/
08
- 3,
0 3,
2 94
%
517,
5 -
11.8
55
- -
9.20
0 20
08
- 2,
6 2,
9 91
%
388,
6 -
11.3
90
- -
8.25
4 15
/1/0
9 3,
0 3,
6 3,
7 99
%
618,
4 -
12.2
13
- -
10.6
57
15/2
/09
2,9
2,6
2,7
95%
34
3,1
- 10
.947
-
- 7.
627
15/3
/09
2,8
2,4
2,6
94%
36
9,9
- 9.
381
- -
6.95
8 15
/4/0
9 3,
1 3,
2 3,
3 96
%
412,
0 -
10.4
25
- -
8.38
4 15
/5/0
9 3,
4 4,
0 4,
0 10
0%
541,
9 -
13.5
89
- -
10.6
68
15/6
/09
3,1
3,6
3,6
100%
47
7,4
- 13
.125
-
- 10
.499
15
/7/0
9 3,
2 4,
0 4,
0 10
0%
483,
1 -
13.9
44
- -
10.4
88
15/8
/09
3,1
3,4
3,5
100%
43
1,8
- 12
.617
-
- 9.
528
2009
3,
1 3,
4 3,
4 98
%
459,
7 -
12.0
30
- -
9.35
1 Fo
nte
(1)
Sobr
enad
ante
obt
ido
após
a r
emoç
ão d
o al
umín
io.
94

A Figura 8 mostra os três períodos de concentração alta, média e baixa do ferro ferroso presente na DAM. Além disso, esta figura aponta os produtos recuperados no tratamento ativo da DAM para a produção do Lodo Químico 1 e Pigmento 1 (alta concentração de ferro), Pigmento 2 (média concentração de ferro) e o Pigmento 3 (baixa concentração de ferro na DAM).
-
2,0
4,0
6,0
8,0
10,0
12,0
14,0
16,0
18,0
23/4/04 9/11/04 28/5/05 14/12/05 2/7/06 18/1/07 6/8/07 22/2/08 9/9/08 28/3/09 14/10/09
data
Co
nce
ntr
ação
, g/L
Fe+2 FeT
Lodo Químico 1 - Pigmento 1
Pigmento 2 - Método de crescimento
Pigmento 3 - Método com formação de intermediário e
adição de núcleo.
Figura 8 – Concentração de ferro ferroso e ferro total presentes na DAM de
Agosto de 2004 até Agosto de 2009.
-
200
400
600
800
1.000
1.200
1.400
1.600
1.800
2.000
23/04/04 09/11/04 28/05/05 14/12/05 02/07/06 18/01/07 06/08/07 22/02/08 09/09/08 28/03/09 14/10/09
Con
cent
raçã
o, m
g/L
Alumínio Figura 9 - Concentração de Alumínio presente na DAM de Agosto de 2004 até
Agosto de 2009.
95

-
5.000
10.000
15.000
20.000
25.000
30.000
35.000
40.000
45.000
23/04/04 09/11/04 28/05/05 14/12/05 02/07/06 18/01/07 06/08/07 22/02/08 09/09/08 28/03/09 14/10/09
Con
cent
raçã
o, m
g/L
Acidez Sulfato Figura 10 - Concentração de sulfato e acidez presente na DAM de Agosto de
2004 a Agosto de 2009.
5.2 NEUTRALIZAÇÃO DA DAM COM NAOH
5.2.1 Obtenção do Lodo Químico 1 e do Coagulante 1 A utilização da drenagem ácida de mina diretamente como
coagulante pode ser justificada devido à elevada concentração de ferro presente na DAM (3 a 12 g/L, Tabela 11). Porém a viabilidade econômica desta aplicação é limitada ao ponto de aplicação do coagulante, uma vez que o custo elevado do transporte pode inviabilizar a sua comercialização.
Considerando-se que os coagulantes férricos comerciais têm concentração total de ferro na faixa de 100 a 200 g/L, torna-se necessário avaliar os métodos para se aumentar a concentração de ferro na DAM, podendo-se citar as seguintes alternativas:
Biolixiviação da pirita catalisada pelas bactérias Thiobacillus
ferroxidans; Evaporação da água presente na drenagem ácida, com
conseqüente aumento da concentração de ferro; Precipitação seletiva do ferro, obtendo-se um lodo úmido filtrado
com alta concentração de ferro, para posterior solubilização deste lodo em ácido sulfúrico ou ácido clorídrico concentrado.
96

Dos três métodos citados acima, optou-se pela produção de lodo férrico ou ferroso filtrado a qual pode ser obtida através das reações 30 (ferroso) ou 31 (férrico).
)(42)(22
42 )(2
líquidosólidoSONaOHFeNaOHSOFe +→++
−+
(30)
)(42)(32
43 3)(2632
líquidosólidoSONaOHFeNaOHSOFe +→++
−+
(31) Após a adição da soda, se obtém um lodo decantado rico em
ferro. Este lodo pode ser filtrado e solubilizado em ácido sulfúrico e/ou ácido clorídrico, obtendo-se com isso o sulfato férrico ou ferroso ou o cloreto férrico ou ferroso. As reações de obtenção dos coagulantes estão descritas abaixo sendo a reação 32 a obtenção de coagulantes férricos e a reação 33 a obtenção de coagulantes ferrosos.
OHFeClHClOHFe
OHSOFeSOHOHFe
líquido
líquidosólido
2)(33
2)(34242)(3
33)(
6)(3)(2
+→+
+→+ (32)
OHFeClHCLOHFe
OHFeSOSOHOHFe
líquidosólido
líquidosólido
2)(2)(2
2)(442)(2
22)(
2)(
+→+
+→+ (33)
A adição de soda na DAM, porém, provoca não somente a
precipitação do ferro, (Fe2+ e/ou Fe3+), como também a precipitação dos outros metais presentes na água (alumínio e manganês, principalmente) conforme as Reações 34 e 35.
4232
43 3)(2632 SONaOHAlNaOHSOAl +→++
−+ (34)
42242 )(2 SONaOHMnNaOHSOMn +→+++ (35)
Para se avaliar a precipitação seletiva do ferro a partir da DAM,
assim como avaliar a eficiência obtida no tratamento da drenagem ácida de mina pela neutralização com NaOH, foram realizados vários testes variando-se a dosagem de soda e a forma de precipitação do ferro (Fe+2 ou Fe+3).
Nesta etapa do trabalho, foi definido o parâmetro “R1” como
97

sendo a relação molar entre o ferro total presente na água e o OH- adicionado; ou seja, R1 = FeT/OH-.
A Tabela 12 apresenta os resultados da adição de soda cáustica na drenagem ácida contendo ferro férrico, a qual foi obtida após a oxidação com peróxido de hidrogênio. A Tabela 13 apresenta os resultados da adição de soda na drenagem ácida contendo Fe2+. Também a Figuras 11 (a) e (b) apresenta os resultados da neutralização da DAM com NaOH.
Tabela 12 - Precipitação de ferro férrico a partir da DAM pela neutralização direta com NaOH.
[Fe+3]inicial
na DAM., g/L
R1, molFeT/molOH-
Hdec.
% lodo
decant, 2h
% remoção do Fe da
DAM
Pureza do Lodo químico 1 seco, g/g
%Fe2O3 %
Mn %Al2O3
Fe2O3/ Al2O3
7,20 1,49 3,83 31% 29,0% 71,36% 0,08% 3,02% 23,6 14,70 0,87 3,90 69% 51,0% 59,80% 0,18% 14,97% 4,0 5,99 0,45 3,75 33% 79,0% 45,20% 0,07% 4,78% 9,5 8,27 0,35 3,77 68% 69,0% 36,04% 0,14% 12,57% 2,9
Tabela 13 - Precipitação de ferro ferroso a partir da DAM pela neutralização direta com NaOH.
[Fe+2]inicial
na DAM., g/L
R1, molFeT/molOH-
pHdec.
% lodo
decant, 2h
% remoção do Fe da
DAM
Pureza Lodo químico 1 seco, g/g
%Fe2O3 %Mn %Al3O3 Fe2O3
/Al2O3 11,5 0,41 8,90 88% 100,0% 34,07% 0,65% 9,62% 3,5 12,8 0,71 6,30 64% 61,6% 47,90% 1,63% 9,22% 5,2 12,8 - 5,65 47% 39,0% 40,62% 0,12% 5,48% 7,4 12,8 1,12 5,10 46% 22,2% 34,33% 0,14% 8,16% 4,2
Durante os testes, é possível observar que a adição de soda na
DAM previamente oxidada produz um lodo de coloração vermelha intensa ao passo que a adição de soda na DAM reduzida produz um lodo verde azulado.
No caso da precipitação de FeIII, observa-se (Tabela 12 e Figura 11 (a)) que a porcentagem de Fe2O3 no lodo precipitado aumenta à medida que a razão molar R1 aumenta, indicando que quanto mais ácido é o método de precipitação mais puro será o lodo obtido.
98

0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6
R, mol Fe / molOH-
%remoção Fe+3 DAM %Fe2O3 lodo qmco 1 %Al2O3 lodo qmco 1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
- 0,2 0,4 0,6 0,8 1,0 1,2
R, mol Fe / mol OH-
% remoção Fe+2 DAM %Fe2O3 lodo qmco 1 %Al2O3 lodo qmco 1
(a)
(b)
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6
R, mol Fe / molOH-
%remoção Fe+3 DAM %Fe2O3 lodo qmco 1 %Al2O3 lodo qmco 1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
- 0,2 0,4 0,6 0,8 1,0 1,2
R, mol Fe / mol OH-
% remoção Fe+2 DAM %Fe2O3 lodo qmco 1 %Al2O3 lodo qmco 1
(a)
(b)
Figura 11 - Precipitação de ferro trivalente presente na DAM pela neutralização
com NaOH (a) e precipitação de ferro divalente (b). Neste caso, a maior porcentagem de Fe2O3 obtida para o lodo
ocorre com R1=1,5. A caracterização deste lodo foi de 71% de Fe2O3; 3,1% de Al2O3 e 0,08% de manganês. Entretanto, com este valor de R1, a porcentagem de lodo férrico obtido, assim como a porcentagem de remoção do ferro presente na DAM, foi muito baixa (≅30%). Por este método, obtem-se um lodo puro, entretanto com um rendimento muito baixo tanto em produto formado quanto no tratamento da DAM. No caso da utilização de R1=0,45, ou seja, com um excesso de NaOH, se
99

obtém uma porcentagem maior de remoção do ferro a partir da drenagem ácida (79%) porém um lodo com 45% de Fe2O3 e 4,8% de Al2O3.
A precipitação direta do Fe2+ presente na drenagem ácida com hidróxido de sódio mostrou que a porcentagem de Fe2O3 no precipitado, assim como a %Al2O3, praticamente não é afetada pelo valor de R1 (Tabela 12 e Figura 11 (b)). A caracterização média do lodo obtido pela precipitação de Fe+2 foi de 39% de Fe2O3, 8% de Al2O3 e 0,6% de manganês. Neste caso, a qualidade do lodo não é influenciada pelos valores de R1. Portanto, com valores de R1 abaixo do estequiométrico, ou seja, com um excesso de base, obtêm-se 100% de remoção do ferro presente na DAM, e aproximadamente 90% de lodo decantado. Com isso, o rendimento é muito maior, tanto em relação ao produto formado quanto no tratamento da DAM.
Estes resultados estão de acordo com as solubilidades dos hidróxidos de Fe+2, Fe+3 e Al+3 em função do pH conforme apresentado na Figura 12.
Fe3+
0
-2
-4
-6
-80 2 4 6 8
pH
Mn3+
Al3+
Cr3+ Cu2+
Zn2+ Cd2+
Mg2+ Ca2+Mn2+
Fe2+
Pb2+
Figura 12 - Equilíbrio da solubilidade iônica. Fonte: Sawyer e McCarty, 1978.
As amostras do Lodo químico 1, produzidas pela precipitação de
ferro férrico com NaOH a partir da DAM foram filtradas, caracterizadas e solubilizadas em ácido sulfúrico para obtenção de sulfato férrico aluminoso, conforme mostra a reação 36.
OHSOAlSOFeSOHOHAlOHFe 23423424233 12)()(6)(2)(2 ++→++ (36)
100
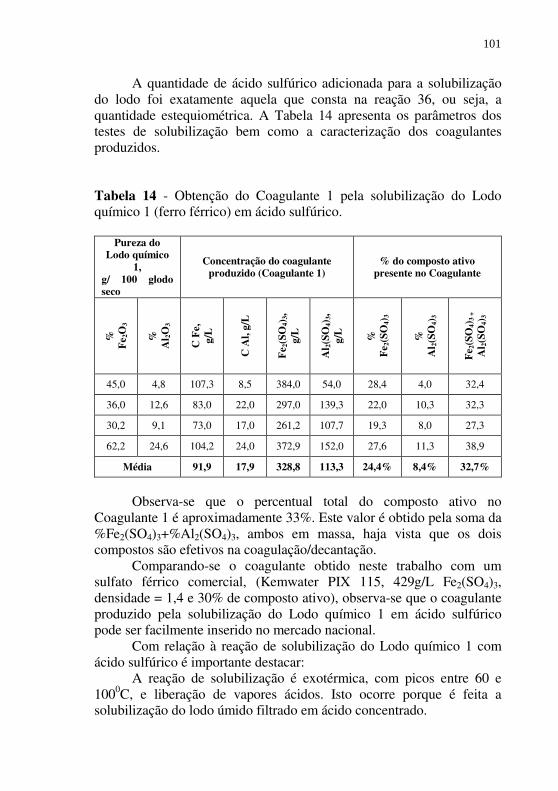
A quantidade de ácido sulfúrico adicionada para a solubilização do lodo foi exatamente aquela que consta na reação 36, ou seja, a quantidade estequiométrica. A Tabela 14 apresenta os parâmetros dos testes de solubilização bem como a caracterização dos coagulantes produzidos.
Tabela 14 - Obtenção do Coagulante 1 pela solubilização do Lodo químico 1 (ferro férrico) em ácido sulfúrico.
Pureza do
Lodo químico 1,
g/ 100 glodo seco
Concentração do coagulante produzido (Coagulante 1)
% do composto ativo presente no Coagulante
%
Fe 2
O3
%
Al 2
O3
C F
e,
g/L
C A
l, g/
L
Fe 2
(SO
4)3,
g/L
Al 2
(SO
4)3,
g/L
%
Fe 2
(SO
4)3
%
Al 2
(SO
4)3
Fe 2
(SO
4)3
+
Al 2
(SO
4)3
45,0 4,8 107,3 8,5 384,0 54,0 28,4 4,0 32,4
36,0 12,6 83,0 22,0 297,0 139,3 22,0 10,3 32,3
30,2 9,1 73,0 17,0 261,2 107,7 19,3 8,0 27,3
62,2 24,6 104,2 24,0 372,9 152,0 27,6 11,3 38,9
Média 91,9 17,9 328,8 113,3 24,4% 8,4% 32,7%
Observa-se que o percentual total do composto ativo no
Coagulante 1 é aproximadamente 33%. Este valor é obtido pela soma da %Fe2(SO4)3+%Al2(SO4)3, ambos em massa, haja vista que os dois compostos são efetivos na coagulação/decantação.
Comparando-se o coagulante obtido neste trabalho com um sulfato férrico comercial, (Kemwater PIX 115, 429g/L Fe2(SO4)3, densidade = 1,4 e 30% de composto ativo), observa-se que o coagulante produzido pela solubilização do Lodo químico 1 em ácido sulfúrico pode ser facilmente inserido no mercado nacional.
Com relação à reação de solubilização do Lodo químico 1 com ácido sulfúrico é importante destacar:
A reação de solubilização é exotérmica, com picos entre 60 e 1000C, e liberação de vapores ácidos. Isto ocorre porque é feita a solubilização do lodo úmido filtrado em ácido concentrado.
101

A densidade do sulfato férrico aluminoso produzido é aproximadamente 1,40 g/cm3.
A velocidade de solubilização é muito alta, sendo que em 30 minutos de reação sob agitação, todo lodo úmido filtrado é completamente solubilizado, sem a produção de qualquer resíduo sólido para descarte.
Foi avaliada a aplicação do Coagulante 1, (com 92g/L de FeT e 18g/L de Al), no tratamento de um efluente têxtil. Comparou-se a sua eficiência com um coagulante comercial (Sulfato férrico Kemwater PIX 115 com 120g/L de FeT). Para tal foi utilizado um efluente proveniente de uma indústria têxtil situada no vale do Rio Itajaí (SC), que foi coletado imediatamente após o tanque de equalização (pH≅9,0).
O teste de coagulação, floculação, decantação foi realizado com metodologia padrão de teste de jarros, utilizando-se diferentes dosagens de coagulante, tempo de coagulação (velocidade de mistura rápida 90 rpm, durante 7 minutos; tempo de floculação: 30 minutos em 15 rpm; tempo de decantação: 50 minutos). O sobrenadante, obtido após a decantação, foi centrifugado e os parâmetros DQO e Cor predominante (λmáximo 620 nm) foram medidos de acordo com métodos analíticos padrões. Os resultados estão apresentados na Tabela 15.
Observa-se que o Sulfato férrico aluminoso produzido neste trabalho, ou seja, o Coagulante 1 mostrou-se mais eficiente do que o Sulfato férrico comercial, principalmente em relação a remoção de cor (para 74 mg/L de ferro, 90% de remoção contra 50%). Isto se justifica pela presença do alumínio no Coagulante 1, que também atua como íon coagulante na formação dos flocos.
Tabela 15 - Resultados do ensaio de coagulação, floculação, decantação utilizando-se um efluente têxtil e o Coagulante 1 versus um Sulfato férrico Comercial.
Dosagem de
ferro para ambos os
coagulante, mg/L
% Remoção de DQO % Remoção cor
Coagulante 1
Coagulante férrico
comercialmente disponível
Coagulante 1
Coagulante férrico
comercialmente disponível
37,85 31,00 17,00 47,00 17,00 74,80 55,00 40,00 90,00 50,00
145,35 65,00 61,00 95,00 92,00
102
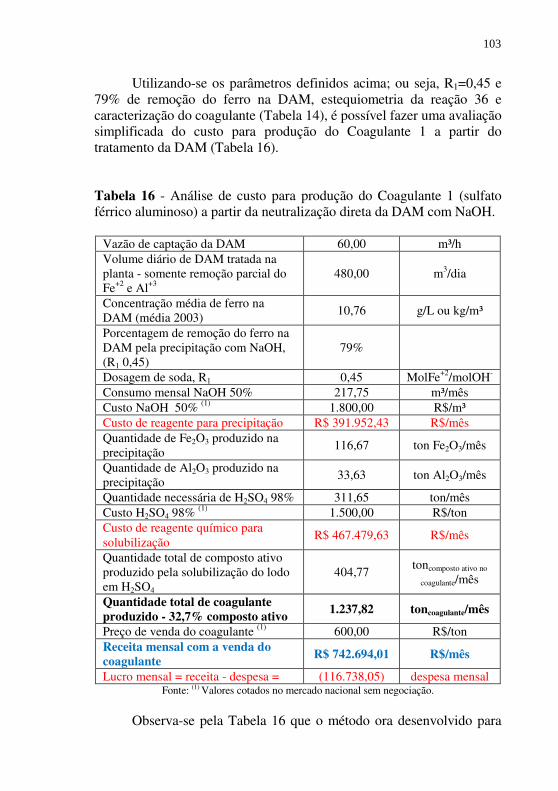
Utilizando-se os parâmetros definidos acima; ou seja, R1=0,45 e 79% de remoção do ferro na DAM, estequiometria da reação 36 e caracterização do coagulante (Tabela 14), é possível fazer uma avaliação simplificada do custo para produção do Coagulante 1 a partir do tratamento da DAM (Tabela 16).
Tabela 16 - Análise de custo para produção do Coagulante 1 (sulfato férrico aluminoso) a partir da neutralização direta da DAM com NaOH.
Vazão de captação da DAM 60,00 m³/h Volume diário de DAM tratada na planta - somente remoção parcial do Fe+2 e Al+3
480,00 m3/dia
Concentração média de ferro na DAM (média 2003)
10,76 g/L ou kg/m³
Porcentagem de remoção do ferro na DAM pela precipitação com NaOH, (R1 0,45)
79%
Dosagem de soda, R1 0,45 MolFe+2/molOH- Consumo mensal NaOH 50% 217,75 m³/mês Custo NaOH 50% (1) 1.800,00 R$/m³ Custo de reagente para precipitação R$ 391.952,43 R$/mês Quantidade de Fe2O3 produzido na precipitação
116,67 ton Fe2O3/mês
Quantidade de Al2O3 produzido na precipitação
33,63 ton Al2O3/mês
Quantidade necessária de H2SO4 98% 311,65 ton/mês Custo H2SO4 98% (1) 1.500,00 R$/ton Custo de reagente químico para solubilização
R$ 467.479,63 R$/mês
Quantidade total de composto ativo produzido pela solubilização do lodo em H2SO4
404,77 toncomposto ativo no
coagulante/mês
Quantidade total de coagulante produzido - 32,7% composto ativo
1.237,82 toncoagulante/mês
Preço de venda do coagulante (1) 600,00 R$/ton Receita mensal com a venda do coagulante R$ 742.694,01 R$/mês
Lucro mensal = receita - despesa = (116.738,05) despesa mensal Fonte: (1) Valores cotados no mercado nacional sem negociação.
Observa-se pela Tabela 16 que o método ora desenvolvido para
103

produção do Coagulante 1 a partir do tratamento da DAM não tem pré-viabilidade econômica.
Isto ocorre porque o preço de venda do coagulante (≅ 600 R$/ton) não justifica a utilização de insumos tão caros, tal como a soda, para a sua produção. Ressalta-se ainda que a soda contribui com praticamente metade dos custos de produção do coagulante, considerando-se apenas os insumos utilizados.
Porém, ainda que a neutralização da DAM com NaOH para produção de coagulante não suporte economicamente o custo do tratamento da drenagem ácida, é importante lembrar que este tratamento é ambientalmente exigido para o seu descarte no meio ambiente. Neste sentido, ressalta-se que o tratamento proposto neste trabalho utilizando-se um valor de R1 igual a 0,45 moles FeT/mol NaOH, é capaz de produzir um efluente tratado com acidez consideravelmente mais baixa do que a DAM. Este efluente será submetido então para uma próxima etapa de tratamento, adequando-o para o descarte final (CONAMA 357).
5.2.2 Obtenção do Lodo Químico 1 e do Pigmento 1 Os primeiros pigmentos produzidos neste trabalho a partir da
neutralização direta da DAM com NaOH, denominados de PG0, PG1, PG2 e PG10, foram obtidos através da adição de soda cáustica 50%, aeração, até a completa oxidação de ferro ferroso a férrico, (alternativamente adição de peróxido de hidrogênio para completar a oxidação), decantação do lodo férrico obtido em diferentes valores de pH’s, secagem, moagem e caracterização.
A justificativa para a busca do pigmento adveio da análise de custo a qual mostrou que somente com a obtenção de produtos de elevado valor agregado seria possível viabilizar o tratamento da DAM pela neutralização com NaOH. A Tabela 17 descreve as condições experimentais dos testes realizados assim como a porcentagem de Fe2O3 nas amostras de pigmento PG0, PG1, PG2 e PG10, cabendo destacar aqui que a concentração de Fe2+ na DAM nesta etapa do trabalho situava-se entre 7,6 e 10,0 g/L.
A diferença principal entre as amostras PG0, PG1 e PG2, é o pH de neutralização e o processo de oxidação. As análises de difração de raios-X dos pigmentos PG0, PG1 e PG2 foram realizadas a partir das amostras secas a 110ºC, moídas e peneiradas em 325 mesh.
104
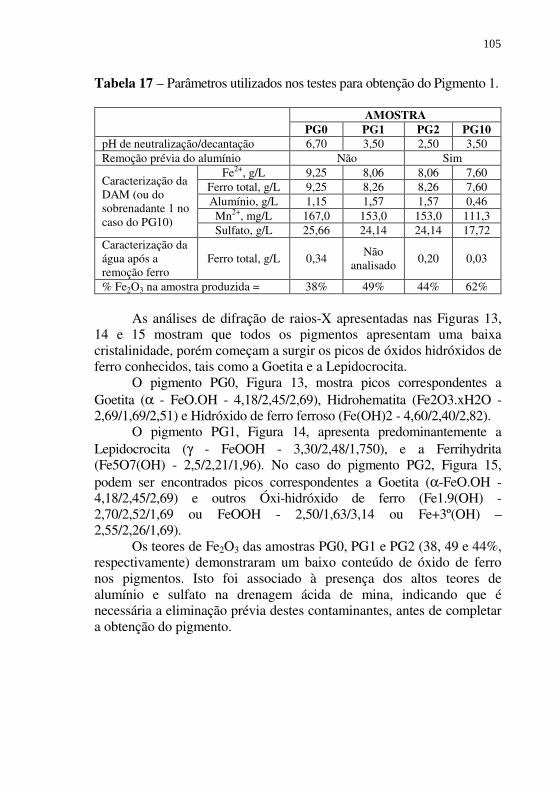
Tabela 17 – Parâmetros utilizados nos testes para obtenção do Pigmento 1.
AMOSTRA
PG0 PG1 PG2 PG10 pH de neutralização/decantação 6,70 3,50 2,50 3,50 Remoção prévia do alumínio Não Sim
Caracterização da DAM (ou do sobrenadante 1 no caso do PG10)
Fe2+, g/L 9,25 8,06 8,06 7,60 Ferro total, g/L 9,25 8,26 8,26 7,60 Alumínio, g/L 1,15 1,57 1,57 0,46 Mn2+, mg/L 167,0 153,0 153,0 111,3 Sulfato, g/L 25,66 24,14 24,14 17,72
Caracterização da água após a remoção ferro
Ferro total, g/L 0,34 Não
analisado 0,20 0,03
% Fe2O3 na amostra produzida = 38% 49% 44% 62% As análises de difração de raios-X apresentadas nas Figuras 13,
14 e 15 mostram que todos os pigmentos apresentam uma baixa cristalinidade, porém começam a surgir os picos de óxidos hidróxidos de ferro conhecidos, tais como a Goetita e a Lepidocrocita.
O pigmento PG0, Figura 13, mostra picos correspondentes a Goetita (α - FeO.OH - 4,18/2,45/2,69), Hidrohematita (Fe2O3.xH2O - 2,69/1,69/2,51) e Hidróxido de ferro ferroso (Fe(OH)2 - 4,60/2,40/2,82).
O pigmento PG1, Figura 14, apresenta predominantemente a Lepidocrocita (γ - FeOOH - 3,30/2,48/1,750), e a Ferrihydrita (Fe5O7(OH) - 2,5/2,21/1,96). No caso do pigmento PG2, Figura 15, podem ser encontrados picos correspondentes a Goetita (α-FeO.OH - 4,18/2,45/2,69) e outros Óxi-hidróxido de ferro (Fe1.9(OH) - 2,70/2,52/1,69 ou FeOOH - 2,50/1,63/3,14 ou Fe+3º(OH) – 2,55/2,26/1,69).
Os teores de Fe2O3 das amostras PG0, PG1 e PG2 (38, 49 e 44%, respectivamente) demonstraram um baixo conteúdo de óxido de ferro nos pigmentos. Isto foi associado à presença dos altos teores de alumínio e sulfato na drenagem ácida de mina, indicando que é necessária a eliminação prévia destes contaminantes, antes de completar a obtenção do pigmento.
105

20 40 60 80 1000
25
50
75
100
125
150
Inte
nsi
dad
e
2θθθθ
4,94
A
4,16
A 2,77
A
2,63
A
2,46
A
1,63
A
Figura 13 - Difratograma de raios-X da amostra de Pigmento 1 - PG0.
20 40 60 80 1000
50
100
150
Inte
nsi
dad
e
2θθθθ
5,2
A
3,1
A
2,5
A
2,2
A
1,97
A
1,83
A
Figura 14 - Difratograma de raios-X da amostra de Pigmento 1 - PG1.
106
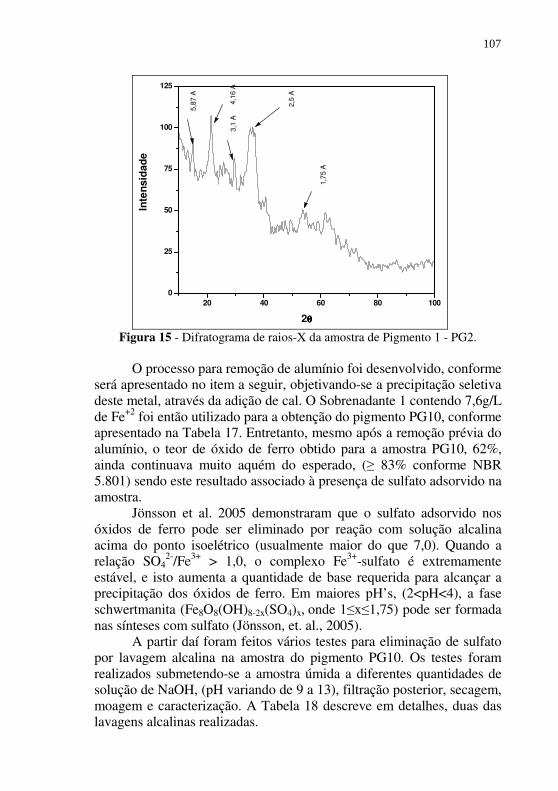
20 40 60 80 1000
25
50
75
100
125
Inte
nsi
dad
e
2θθθθ
5,87
A
4,16
A3,
1 A
2,5
A
1,75
A
Figura 15 - Difratograma de raios-X da amostra de Pigmento 1 - PG2. O processo para remoção de alumínio foi desenvolvido, conforme
será apresentado no item a seguir, objetivando-se a precipitação seletiva deste metal, através da adição de cal. O Sobrenadante 1 contendo 7,6g/L de Fe+2 foi então utilizado para a obtenção do pigmento PG10, conforme apresentado na Tabela 17. Entretanto, mesmo após a remoção prévia do alumínio, o teor de óxido de ferro obtido para a amostra PG10, 62%, ainda continuava muito aquém do esperado, (≥ 83% conforme NBR 5.801) sendo este resultado associado à presença de sulfato adsorvido na amostra.
Jönsson et al. 2005 demonstraram que o sulfato adsorvido nos óxidos de ferro pode ser eliminado por reação com solução alcalina acima do ponto isoelétrico (usualmente maior do que 7,0). Quando a relação SO4
2-/Fe3+ > 1,0, o complexo Fe3+-sulfato é extremamente estável, e isto aumenta a quantidade de base requerida para alcançar a precipitação dos óxidos de ferro. Em maiores pH’s, (2<pH<4), a fase schwertmanita (Fe8O8(OH)8-2x(SO4)x, onde 1≤x≤1,75) pode ser formada nas sínteses com sulfato (Jönsson, et. al., 2005).
A partir daí foram feitos vários testes para eliminação de sulfato por lavagem alcalina na amostra do pigmento PG10. Os testes foram realizados submetendo-se a amostra úmida a diferentes quantidades de solução de NaOH, (pH variando de 9 a 13), filtração posterior, secagem, moagem e caracterização. A Tabela 18 descreve em detalhes, duas das lavagens alcalinas realizadas.
107

Tabela 18 - Descrição de duas lavagens básicas feitas na amostra de Pigmento 1 - PG10 - para remoção de SO4
2-.
PARÂMETROS DA LAVAGEM
LAVAGEM 1 LAVAGEM 2
Concentração de SO42- na
amostra seca, g SO4/g lodo seco
0,14 0,14
Concentração máxima de SO4
2- a ser obtida na fase líquida (mg/L)
5.009,83 4.870,64
Relação estequiométrica entre OH-/SO4
2- para lavagem (molOH-/molSO4
-2)
2,00 2,00
pH inicial da solução básica 12,67 12,36 ANÁLISE DA SOLUÇÃO BÁSICA APÓS A LAVAGEM
Concentração SO4-2, mg/L
Lavagem 1 Lavagem 2 5.000,0 3.426,0
Concentração máxima teórica SO4
-2, mg/L 5.009,8 4.870,6
% Remoção de SO4-2 com
base na solução 100% 70%
ANÁLISE DO PIGMENTO ANTES E APÓS A LAVAGEM ALCALINA
Parâmetro Lavagem 1 Lavagem 2
antes depois %
rem. antes depois
% rem.
% Fe2O3 (b.s.) 62,5 84,5 - 62,5 88,9 - % SO4
-2 (b.s.) 13,8 1,3 90,5 13,8 4,3 68,7 Observa-se pela Tabela 18 que após a lavagem alcalina, a
amostra apresentou concentração de sulfato menor do que 5%, e teor de Fe2O3 maior do que 83%, isto indica que a eliminação do sulfato é bastante eficiente na lavagem.
A Tabela 19 mostra os resultados das análises de perda ao fogo nas temperaturas de 300, 500, 800 e 1000°C e o teor de Fe2O3 nos pigmentos PG10A e PG10B, sendo respectivamente a amostra bruta e a amostra lavada.
Observa-se que, aumentando a temperatura de calcinação, aumenta-se o teor de Fe2O3 presente na amostra. Isto ocorre devida a perda de água absorvida e estrutural e perda do sulfato adsorvido. Em paralelo, foi também observado mudança de coloração do pigmento durante o aquecimento, do vermelho amarelado ao vermelho escuro.
108
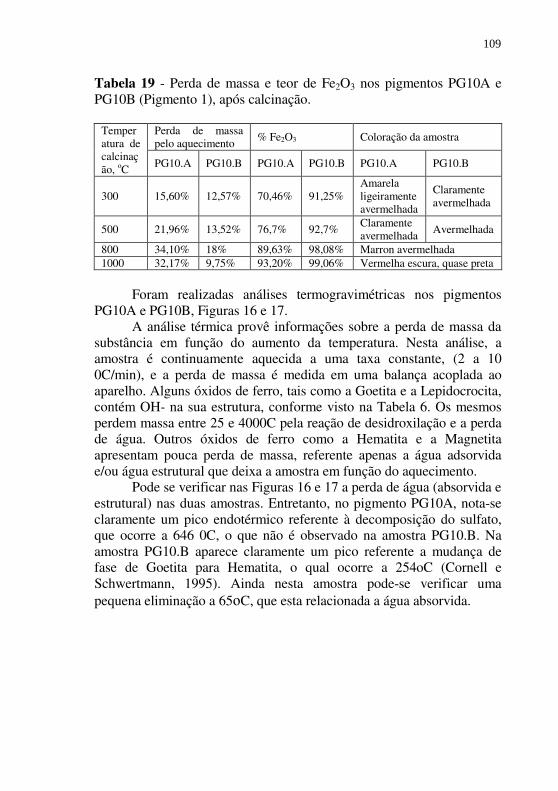
Tabela 19 - Perda de massa e teor de Fe2O3 nos pigmentos PG10A e PG10B (Pigmento 1), após calcinação.
Temperatura de calcinação, oC
Perda de massa pelo aquecimento
% Fe2O3 Coloração da amostra
PG10.A PG10.B PG10.A PG10.B PG10.A PG10.B
300 15,60% 12,57% 70,46% 91,25% Amarela ligeiramente avermelhada
Claramente avermelhada
500 21,96% 13,52% 76,7% 92,7% Claramente avermelhada
Avermelhada
800 34,10% 18% 89,63% 98,08% Marron avermelhada 1000 32,17% 9,75% 93,20% 99,06% Vermelha escura, quase preta
Foram realizadas análises termogravimétricas nos pigmentos
PG10A e PG10B, Figuras 16 e 17. A análise térmica provê informações sobre a perda de massa da
substância em função do aumento da temperatura. Nesta análise, a amostra é continuamente aquecida a uma taxa constante, (2 a 10 0C/min), e a perda de massa é medida em uma balança acoplada ao aparelho. Alguns óxidos de ferro, tais como a Goetita e a Lepidocrocita, contém OH- na sua estrutura, conforme visto na Tabela 6. Os mesmos perdem massa entre 25 e 4000C pela reação de desidroxilação e a perda de água. Outros óxidos de ferro como a Hematita e a Magnetita apresentam pouca perda de massa, referente apenas a água adsorvida e/ou água estrutural que deixa a amostra em função do aquecimento.
Pode se verificar nas Figuras 16 e 17 a perda de água (absorvida e estrutural) nas duas amostras. Entretanto, no pigmento PG10A, nota-se claramente um pico endotérmico referente à decomposição do sulfato, que ocorre a 646 0C, o que não é observado na amostra PG10.B. Na amostra PG10.B aparece claramente um pico referente a mudança de fase de Goetita para Hematita, o qual ocorre a 254oC (Cornell e Schwertmann, 1995). Ainda nesta amostra pode-se verificar uma pequena eliminação a 65οC, que esta relacionada a água absorvida.
109

Figura 16 - Análise termogravimétrica da amostra de Pigmento 1 - PG10.A
(bruta).
Figura 17 - Análise termogravimétrica da amostra de Pigmento 1 - PG10.B
(após lavagem alcalina).
110

As Figuras 18 e 19 de (a) até (e), mostram, respectivamente os resultados das análises de difração de raios-X nas amostras de pigmento PG10A e PG10B, calcinadas a diferentes temperaturas.
Observa-se pela Figura 18 que o pigmento PG10A é praticamente amorfo a 100°C, com apenas um pico em 2θ= 35,73º, que é atribuído à Goetita, porém com baixa definição e intensidade.
Um aumento na cristalinidade do pigmento PG10A é obtido somente após a calcinação da amostra a 500°C, associado ao aparecimento dos picos referentes da Hematita, com traços da Goetita (Figura 18).
Após a calcinação a 800 e 1000°C, são verificados no pigmento PG10A apenas picos referentes à Hematita, com aumento da resolução dos sinais, o que implica em aumento da cristalinidade.
Na amostra PG10B seca a 100οC, (amostra lavada - Figura 19) temos uma amostra pouco cristalina, entretanto, não tão amorfa quanto a amostra PG10A, o que demonstra que a lavagem para a remoção do sulfato deixa a amostra mais cristalina.
Observa-se claramente a formação de Goetita, mesmo que com baixa cristalinidade, pelos picos em 2θ aproximadamente iguais a 21; 33; 34; 36; 40; 53; 59 e 61. Picos referentes a Maghemita, com presença de Hematita e Goetita (traços) assim como uma melhor cristalinidade são encontrados na amostra a 300οC (Figura 19).
A 500οC observa-se picos referentes à Hematita e presença da Maghemita, com um aumento da resolução dos sinais, conseqüência de uma boa cristalinidade da amostra. Uma cristalinidade bastante acentuada é verificada nas amostras calcinadas a temperaturas mais altas e observa-se uma fase praticamente pura de Hematita, sem traços de Magnetita. Estes resultados mostram que as eliminações das águas adsorvidas e do sulfato, permitem uma melhora acentuada na cristalinidade das amostras.
111
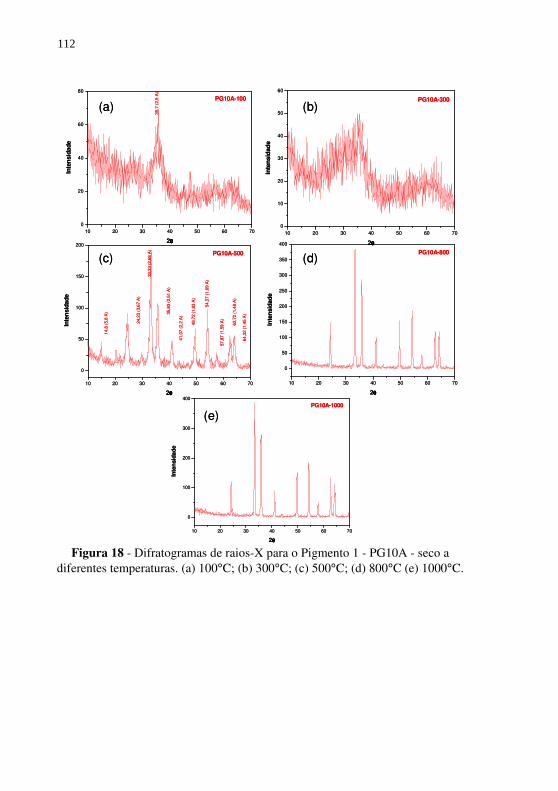
10 20 30 40 50 60 700
20
40
60
80
Inte
nsi
dade
2θθθθ
35,7
(2,
5 A
)
PG10A-100
10 20 30 40 50 60 700
10
20
30
40
50
60
PG10A-300
Inte
nsi
dad
e
2θθθθ
10 20 30 40 50 60 70
0
50
100
150
200
Inte
nsi
dade
2θθθθ
14,8
(5,
8 A
)
24,2
3 (3
,67
A)
33,3
3 (2
,68
A)
35,8
3 (2
,51
A)
41,0
7 (2
,2 A
)
49,7
2 (1
,83
A)
54,3
7 (1
,69
A)
57,8
7 (1
,59
A)
62,7
2 (1
,48
A)
64,3
2 (1
,45
A)
PG10A-500
10 20 30 40 50 60 70
0
50
100
150
200
250
300
350
400
PG10A-800
Inte
nsi
dade
2θθθθ
10 20 30 40 50 60 70
0
100
200
300
400
PG10A-1000
Inte
nsi
dade
2θθθθ
(b)(a)
(c) (d)
(e)
10 20 30 40 50 60 700
20
40
60
80
Inte
nsi
dade
2θθθθ
35,7
(2,
5 A
)
PG10A-100
10 20 30 40 50 60 700
10
20
30
40
50
60
PG10A-300
Inte
nsi
dad
e
2θθθθ
10 20 30 40 50 60 70
0
50
100
150
200
Inte
nsi
dade
2θθθθ
14,8
(5,
8 A
)
24,2
3 (3
,67
A)
33,3
3 (2
,68
A)
35,8
3 (2
,51
A)
41,0
7 (2
,2 A
)
49,7
2 (1
,83
A)
54,3
7 (1
,69
A)
57,8
7 (1
,59
A)
62,7
2 (1
,48
A)
64,3
2 (1
,45
A)
PG10A-500
10 20 30 40 50 60 70
0
50
100
150
200
250
300
350
400
PG10A-800
Inte
nsi
dade
2θθθθ
10 20 30 40 50 60 70
0
100
200
300
400
PG10A-1000
Inte
nsi
dade
2θθθθ
(b)(a)
(c) (d)
(e)
Figura 18 - Difratogramas de raios-X para o Pigmento 1 - PG10A - seco a
diferentes temperaturas. (a) 100°C; (b) 300°C; (c) 500°C; (d) 800°C (e) 1000°C.
112

10 20 30 40 50 60 70 80 90 100
0
50
100
150
200
Inte
nsi
dad
e
2θθθθ
24,1
8 (3
,6 A
)
33,1
8 (2
,6 A
)
35,7
8 (2
,5 A
)41
,02
(2,1
A)
49,5
(1,8
A)
54,3
(1,7
A)
57,7
(1,6
A)
62,5
(1,5
A)
64,1
(1,4
5 A
)
PG10B 300
20 40 60 80 100-25
0
25
50
75
100
125
Inte
nsi
dad
e
2θθθθ
21,2
8 (4
,2 A
)
33,4
3 (2
,68
A)
34,8
(2,
6 A
)
40,0
2 (2
,25
A)
36,6
8 (2
,44
A)
41,4
2 (2
,18
A)
53,4
7 (1
,71
A)
59,2
7 (1
,56
A)
61,5
2 (1
,51
A)
PG10B 100
(b)(a)
(c) (d)
(e)
20 40 60 80 100
0
50
100
150
200
250
Inte
nsi
dad
e
2θθθθ
24,1
8 (3
,67
A)
33,2
0 (2
,68
A)
35,8
(2,
51 A
)41
,02
(2,1
A)
49,5
2 (1
,82
A)
54,3
7 (1
,7 A
)
57,6
7 (1
,59
A)
62,5
2 (1
,47
A)
64,1
2 (1
,45
A)
PG10B 500
20 40 60 80 100
0
100
200
300
400
500
Inte
nsi
dad
e
2θθθθ
PG10B 800
20 40 60 80 100
0
50
100
150
200
250
300
350
Inte
nsi
dad
e
2θθθθ
PG10B 1000
10 20 30 40 50 60 70 80 90 100
0
50
100
150
200
Inte
nsi
dad
e
2θθθθ
24,1
8 (3
,6 A
)
33,1
8 (2
,6 A
)
35,7
8 (2
,5 A
)41
,02
(2,1
A)
49,5
(1,8
A)
54,3
(1,7
A)
57,7
(1,6
A)
62,5
(1,5
A)
64,1
(1,4
5 A
)
PG10B 300
20 40 60 80 100-25
0
25
50
75
100
125
Inte
nsi
dad
e
2θθθθ
21,2
8 (4
,2 A
)
33,4
3 (2
,68
A)
34,8
(2,
6 A
)
40,0
2 (2
,25
A)
36,6
8 (2
,44
A)
41,4
2 (2
,18
A)
53,4
7 (1
,71
A)
59,2
7 (1
,56
A)
61,5
2 (1
,51
A)
PG10B 100
(b)(a)
(c) (d)
(e)
20 40 60 80 100
0
50
100
150
200
250
Inte
nsi
dad
e
2θθθθ
24,1
8 (3
,67
A)
33,2
0 (2
,68
A)
35,8
(2,
51 A
)41
,02
(2,1
A)
49,5
2 (1
,82
A)
54,3
7 (1
,7 A
)
57,6
7 (1
,59
A)
62,5
2 (1
,47
A)
64,1
2 (1
,45
A)
PG10B 500
20 40 60 80 100
0
100
200
300
400
500
Inte
nsi
dad
e
2θθθθ
PG10B 800
20 40 60 80 100
0
50
100
150
200
250
300
350
Inte
nsi
dad
e
2θθθθ
PG10B 1000
Figura 19 - Difratogramas de raios-X para o Pigmento 1 - PG10B (após
lavagem alcalina). (a) 100°C; (b) 300°C; (c) 500°C; (d) 800°C (e) 1000°C. Os difratogramas de raios-X de três amostras de pigmentos
comerciais, (amarelo, vermelho e preto), assim como o difratograma de raios-X da amostra PG10B seca a 1000C, são mostrados lado a lado na Figura 20. Verificam-se nos pigmentos comerciais fases puras com alto grau de cristalinidade da Goetita (para o pigmento amarelo, Figura 20b), Hematita (para o pigmento vermelho, Figura 20c) e Magnetita (pigmento preto, Figura 20d).
113
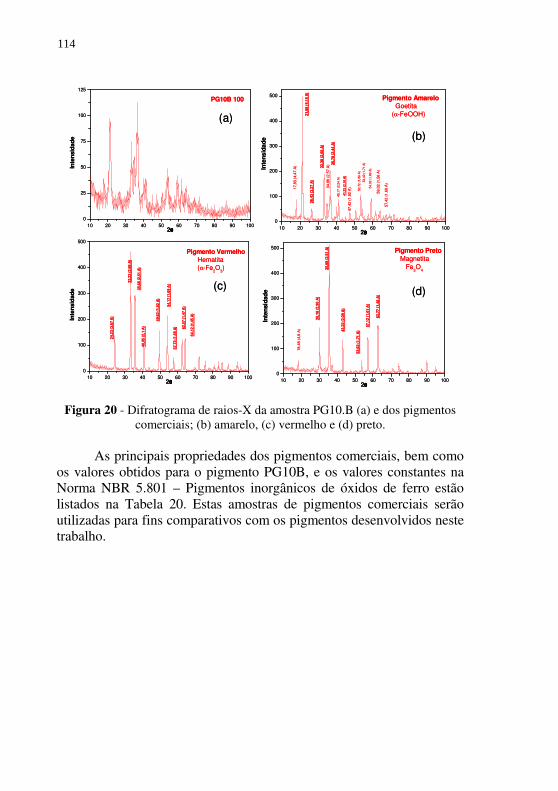
10 20 30 40 50 60 70 80 90 1000
25
50
75
100
125
Inte
nsi
dad
e
2θθθθ
PG10B 100
(b)
(a)
(c) (d)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsid
ade
2θθθθ
17,8
3 (4
,47
A)
21,8
8 (4
,18
A)
26,4
3 (3
,37
A)
33,3
8 (2
,68
A)
34,8
8 (2
,57
A)
40,1
7 (2
,24
A)
36,7
8 (2
,44
A)
41,3
2 (2
,18
A)
47,4
2 (1
,92
A)
50,7
2 (1
,80
A)
53,4
2 (1
,71
A)
54,3
2 (1
,69
A)
59,0
2 (1
,56
A)
57,4
2 (1
,60
A)
Pigmento Amarelo Goetita (α-FeOOH)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsi
dad
e
2θθθθ
24,2
3 (3
,67
A)
33,2
3 (2
,68
A)
35,6
8 (2
,51
A)
40,9
5 (2
,1 A
)
49,6
2 (1
,82
A)
54,1
7 (1
,69
A)
57,7
2 (1
,59
A) 62
,57
(1,4
7 A
)
64,1
2 (1
,45
A)
Pigmento Vermelho Hematita (α-Fe
2O
3)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsid
ade
2θθθθ
18,4
8 (4
,8 A
)
30,1
8 (2
,96
A)
35,6
8 (2
,51
A)
43,2
2 (2
,09
A)
53,6
2 (1
,71
A)
57,1
2 (1
,61
A)
62,7
7 (1
,48
A)
Pigmento Preto Magnetita Fe
3O
4
10 20 30 40 50 60 70 80 90 1000
25
50
75
100
125
Inte
nsi
dad
e
2θθθθ
PG10B 100
(b)
(a)
(c) (d)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsid
ade
2θθθθ
17,8
3 (4
,47
A)
21,8
8 (4
,18
A)
26,4
3 (3
,37
A)
33,3
8 (2
,68
A)
34,8
8 (2
,57
A)
40,1
7 (2
,24
A)
36,7
8 (2
,44
A)
41,3
2 (2
,18
A)
47,4
2 (1
,92
A)
50,7
2 (1
,80
A)
53,4
2 (1
,71
A)
54,3
2 (1
,69
A)
59,0
2 (1
,56
A)
57,4
2 (1
,60
A)
Pigmento Amarelo Goetita (α-FeOOH)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsi
dad
e
2θθθθ
24,2
3 (3
,67
A)
33,2
3 (2
,68
A)
35,6
8 (2
,51
A)
40,9
5 (2
,1 A
)
49,6
2 (1
,82
A)
54,1
7 (1
,69
A)
57,7
2 (1
,59
A) 62
,57
(1,4
7 A
)
64,1
2 (1
,45
A)
Pigmento Vermelho Hematita (α-Fe
2O
3)
10 20 30 40 50 60 70 80 90 1000
100
200
300
400
500
Inte
nsid
ade
2θθθθ
18,4
8 (4
,8 A
)
30,1
8 (2
,96
A)
35,6
8 (2
,51
A)
43,2
2 (2
,09
A)
53,6
2 (1
,71
A)
57,1
2 (1
,61
A)
62,7
7 (1
,48
A)
Pigmento Preto Magnetita Fe
3O
4
Figura 20 - Difratograma de raios-X da amostra PG10.B (a) e dos pigmentos
comerciais; (b) amarelo, (c) vermelho e (d) preto. As principais propriedades dos pigmentos comerciais, bem como
os valores obtidos para o pigmento PG10B, e os valores constantes na Norma NBR 5.801 – Pigmentos inorgânicos de óxidos de ferro estão listados na Tabela 20. Estas amostras de pigmentos comerciais serão utilizadas para fins comparativos com os pigmentos desenvolvidos neste trabalho.
114

Tab
ela
20 -
Car
acte
riza
ção
dos
pigm
ento
s co
mer
ciai
s e
do P
igm
ento
1, (
PG10
B)
obtid
o ne
sta
etap
a do
trab
alho
.
Par
âmet
ros
da n
orm
a N
BR
5.8
01 –
Óxi
do
de f
erro
com
o pi
gmen
to.
Coo
rden
adas
col
orim
étri
cas
CIE
LA
B
Teo
r de
Fe
2O3
Um
idad
e (1
10 °
C)
Inso
lúve
is
HC
l R
esíd
uos
(100
0°C
) L
* (b
ranc
o)
A*
(ver
mel
ho)
B*
(am
arel
o)
∆E
ab(3
) ∆
Eab
(4)
Nor
ma
(1) ≥
83%
≤
1,0
0%
≤ 0
,5%
N
ão
cont
empl
ado
Ton
alid
ade
de a
cord
o co
m o
pad
rão
apro
vado
.
Am
arel
o 83
%
0,79
%
< 0
,1%
<
1%
51,7
1 11
,44
41,6
5 0,
00
37,8
7 V
erm
elho
98
%
0,74
%
< 0
,1%
<
1%
25,0
4 25
,39
18,6
6 37
,87
0,00
P
reto
>
99%
0,
65%
<
0,1
%
<1%
-
- -
- -
PG10
.B
87%
3,
61%
(2)
Não
ana
lisad
o 36
,78
19,8
5 31
,87
19,7
3 18
,52
Con
diçõ
es e
spec
ífic
as p
ara
pigm
ento
s si
ntét
icos
de
óxid
o de
fer
ro a
mar
elo.
Pe
rda
de á
gua
a 14
00C
. D
ifer
ença
de
cor
em r
elaç
ão a
o pi
gmen
to a
mar
elo.
D
ifer
ença
de
cor
em r
elaç
ão a
o pi
gmen
to v
erm
elho
.
115

O pigmento PG10B, pigmento preliminar produzido pela neutralização do Sobrenadante 1 (isento de alumínio e contendo 7,6g/L de Fe+2, com NaOH 50%, lavagem alcalina e secagem a 1000C), encontra-se exatamente em um patamar intermediário entre o pigmento vermelho e o pigmento amarelo, tendo uma ligeira tendência para o pigmento vermelho com ∆Eab em relação ao vermelho é menor (Tabela 20).
Observa-se também que este pigmento tem um teor muito elevado de água absorvida e/ou água estrutural, 3,61%, parâmetro este que diverge dos pigmentos comerciais.
Com relação ao grau de cristalinidade, este pigmento apresenta-se pouco cristalino, quando seco a 100oC, apesar de aparecerem os picos da Goetita e da Hematita.
Os resultados destes testes preliminares para produção de pigmento inorgânico a partir da DAM indicaram a formação predominante do pigmento vermelho α-Fe2O3 (ou seja, da Hematita) como uma consequência direta de fatores como pH de precipitação, eliminação de sulfato, temperaturas de secagem e calcinação. Isto indica que outros pigmentos com colorações distintas assim como com maiores valores de Fe2O3 e melhor cristalinidade, poderão ser obtidos, desde que sejam alteradas e melhor estudadas as condições acima mencionadas.
Embora esta tese não objetive a realização de análise econômica, vale ressaltar que o valor agregado aos pigmentos sintéticos de óxido de ferro é cerca de 6 a 10 vezes maior do que o valor agregado aos coagulantes férricos. Assim, mais atenção será dada para a obtenção de pigmentos sintéticos de valor agregado superior, tal como o pigmento amarelo, ou seja, a Goetita, haja vista que a partir dela é possível produzir-se o pigmento vermelho, (a Hematita) além da maior demanda existente no mercado nacional pelos pigmentos amarelos.
116

6 RESULTADOS DA SEGUNDA ROTA EXPERIMENTAL A necessidade de se realizar o tratamento da drenagem ácida de
mina em etapas foi verificada após a realização da primeira rota experimental utilizada nesta tese. Naqueles testes, ficou demonstrado que para a obtenção do pigmento com elevado grau de pureza é necessário que se proceda à remoção prévia do alumínio e parcialmente do sulfato antes da remoção do ferro. Portanto o tratamento da drenagem ácida de mina com produção de pigmento será realizado em duas etapas distintas. Em uma primeira etapa será removido o alumínio e parcialmente o sulfato e acidez presentes na água e em uma próxima etapa será removido o ferro.
6.1 NEUTRALIZAÇÃO PARCIAL DA DAM COM CAL O tratamento da drenagem ácida de mina com cal virgem ou
hidratada é economicamente mais viável, porém, produz um lodo rico em hidróxidos metálicos e sulfato de cálcio hidratado conforme o Esquema 4. Ao mesmo tempo, o tratamento da DAM com soda ou qualquer outro meio alcalino isento de cálcio, produz um lodo rico somente em óxidos e hidróxidos metálicos conforme visto anteriormente.
50,8.......................;4)()(
50,6..........................;4)()(
00,4...................;3)(2)(332
50,3....................;3)(2)(332
)()(222
42
)()(222
42
)(4322
43
4322
43
)(
)()(
≥+→++
≥+→++
≥+→++
≥+→++
−+
−+
−+
−+
pHCaSOOHMnOHCaSOMn
pHCaSOOHFeOHCaSOFe
pHCaSOOHAlOHCaSOAl
pHCaSOOHFeOHCaSOFe
ss
ss
ss
ss
Esquema 4 – Precipitação seletiva de metais em função do pH pela adição de
cal. Considerando estas alternativas, o tratamento da DAM
desenvolvido neste trabalho foi realizado em etapas consecutivas de precipitação, onde em cada etapa do processo são removidos contaminantes específicos.
Em uma primeira etapa do tratamento sequencial, através de uma pequena elevação no pH da água pela adição de cal, é possível remover o alumínio, o ferro férrico e, parcialmente, o sulfato e a acidez. Através

disto, obtêm-se duas fases distintas; uma fase sólida, rica em ferro, alumínio e sulfato, e uma fase líquida, ainda ácida, rica em ferro ferroso.
A sequência de fotos abaixo ilustra como se dá a remoção do alumínio através da adição de cal na DAM e a Tabela 21 mostra as análises na DAM antes e após o tratamento com diferentes dosagens de cal.
Foto 1 – pH = 3,11tempo = 0:00
Foto 2 – pH = 3,30tempo adição cal = 0:02 min
Foto 3 – pH 4,02tempo = 0:05
Foto 4 – pH=4,12tempo = 1:35
Foto 6 – pH=4,20; tempo = 4:10.
Foto 5 – pH=4,12; tempo = 3:25.
Foto 4 – pH=4,12tempo = 1:35
Foto 6 – pH=4,20; tempo = 4:10.
Foto 5 – pH=4,12; tempo = 3:25.
Foto 7 – lodo de alumínio decantado.
118
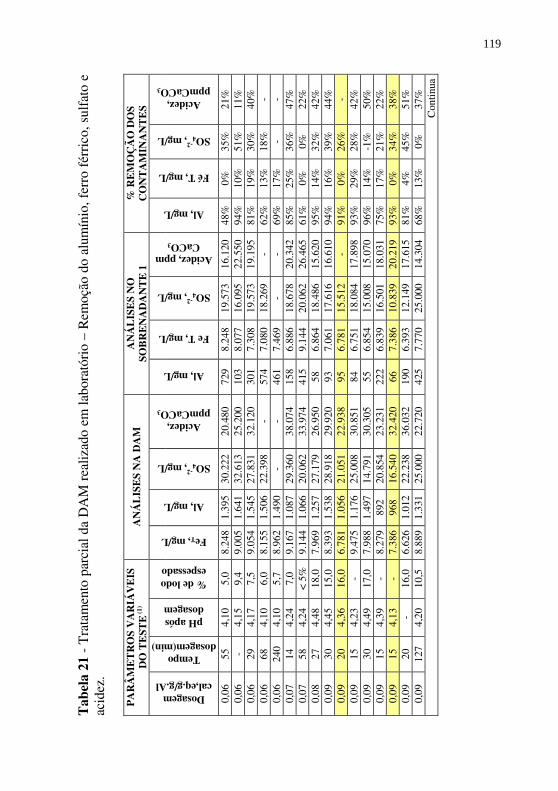
Tab
ela
21 -
Tra
tam
ento
par
cial
da
DA
M r
ealiz
ado
em la
bora
tóri
o –
Rem
oção
do
alum
ínio
, fer
ro f
érri
co, s
ulfa
to e
ac
idez
. P
AR
ÂM
ET
RO
S V
AR
IÁV
EIS
D
O T
EST
E (
1)
AN
ÁL
ISE
S N
A D
AM
A
NÁ
LIS
ES
NO
SO
BR
EN
AD
AN
TE
1
% R
EM
OÇ
ÃO
DO
S C
ON
TA
MIN
AN
TE
S
Dosagem cal,eq.g/g.Al
Tempo dosagem(min)
pH após dosagem
% de lodo espessado
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
Al, mg/L
Fe T, mg/L
SO4-2
, mg/L
Acidez, ppm CaCO3
Al, mg/L
Fé T, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
0,06
55
4,
10
5,0
8.24
8 1.
395
30.2
22
20.4
80
729
8.24
8 19
.573
16
.120
48
%
0%
35%
21
%
0,06
-
4,15
9,
4 9.
005
1.64
1 32
.613
25
.200
10
3 8.
077
16.0
95
22.5
50
94%
10
%
51%
11
%
0,06
29
4,
17
7,5
9.05
4 1.
545
27.8
31
32.1
20
301
7.30
8 19
.573
19
.195
81
%
19%
30
%
40%
0,
06
68
4,10
6,
0 8.
155
1.50
6 22
.398
-
574
7.08
0 18
.269
-
62%
13
%
18%
-
0,06
24
0 4,
10
5,7
8.96
2 1.
490
- -
461
7.46
9 -
- 69
%
17%
-
- 0,
07
14
4,24
7,
0 9.
167
1.08
7 29
.360
38
.074
15
8 6.
886
18.6
78
20.3
42
85%
25
%
36%
47
%
0,07
58
4,
24
< 5
%
9.14
4 1.
066
20.0
62
33.9
74
415
9.14
4 20
.062
26
.465
61
%
0%
0%
22%
0,
08
27
4,48
18
,0
7.96
9 1.
257
27.1
79
26.9
50
58
6.86
4 18
.486
15
.620
95
%
14%
32
%
42%
0,
09
30
4,45
15
,0
8.39
3 1.
538
28.9
18
29.9
20
93
7.06
1 17
.616
16
.610
94
%
16%
39
%
44%
0,
09
20
4,36
16
,0
6.78
1 1.
056
21.0
51
22.9
38
95
6.78
1 15
.512
-
91%
0%
26
%
- 0,
09
15
4,23
-
9.47
5 1.
176
25.0
08
30.8
51
84
6.75
1 18
.084
17
.898
93
%
29%
28
%
42%
0,
09
30
4,49
17
,0
7.98
8 1.
497
14.7
91
30.3
05
55
6.85
4 15
.008
15
.070
96
%
14%
-1
%
50%
0,
09
15
4,39
-
8.27
9 89
2 20
.854
23
.231
22
2 6.
839
16.5
01
18.0
31
75%
17
%
21%
22
%
0,09
15
4,
13
- 7.
386
968
16.5
40
32.4
20
66
7.38
6 10
.839
20
.219
93
%
0%
34%
38
%
0,09
20
-
16,0
6.
626
1.01
2 22
.238
36
.032
19
0 6.
393
12.1
49
17.6
15
81%
4%
45
%
51%
0,
09
127
4,20
10
,5
8.88
9 1.
331
25.0
00
22.7
20
425
7.77
0 25
.000
14
.304
68
%
13%
0%
37
%
Con
tinua
119

PA
RÂ
ME
TR
OS
VA
RIÁ
VE
IS
DO
TE
STE
(1)
A
NÁ
LIS
ES
NA
DA
M
AN
ÁL
ISE
S N
O
SOB
RE
NA
DA
NT
E 1
%
RE
MO
ÇÃ
O D
OS
CO
NT
AM
INA
NT
ES
Dosagem cal,eq.g/g.Al
Tempo dosagem(min)
pH após dosagem
% de lodo espessado
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
Al, mg/L
Fe T, mg/L
SO4-2
, mg/L
Acidez, ppm CaCO3
Al, mg/L
Fé T, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
Con
tinua
ção
0,09
23
4,
50
9,0
8.83
0 1.
535
23.4
85
32.6
70
1,7
8.81
2 14
.139
17
.215
10
0%
0%
40%
47
%
0,09
21
4,
17
16,0
9.
187
1.12
8 21
.447
32
.855
95
6.
781
15.5
12
32.8
55
92%
26
%
28%
0%
0,
11
138
4,20
10
,0
8.62
4 1.
442
- -
288
7.18
7 -
- 80
%
17%
-
- 0,
11
20
4,33
16
,0
8.69
7 1.
157
24.8
10
- 95
6.
781
15.5
12
- 92
%
22%
37
%
- 0,
13
23
4,35
8,
0 9.
570
1.37
9 26
.608
34
.760
14
3 8.
482
19.3
55
23.3
20
90%
11
%
27%
33
%
0,12
43
4,
40
12,0
8.
399
1.50
3 25
.441
31
.735
32
7.
451
14.7
91
16.9
40
98%
11
%
42%
47
%
Méd
ia
4,30
11
,41
8.48
6 1.
293
24.2
93
29.8
46
209
7.39
1 17
.038
19
.433
84
%
12%
28
%
35%
Fo
nte:
(1)
Par
âmet
ros
fixo
s do
s te
stes
: Vol
ume
méd
io d
e dr
enag
em c
apta
da p
ara
cada
test
e =
75L
; Agi
taçã
o pr
opor
cion
ada
por
aera
dor
de b
olha
gr
ossa
inst
alad
o na
bas
e do
rea
tor
com
pre
ssão
tota
l de
ar d
e 10
psi;
Con
cent
raçã
o m
édia
da
cal a
dici
onad
a =
197
gC
a(O
H)2
/L; T
empo
tota
l de
deca
ntaç
ão =
1,0
hora
.
Obs
erva
-se
pela
Tab
ela
21 q
ue o
tra
tam
ento
pro
post
o é
efet
ivo
e se
letiv
o pa
ra a
rem
oção
do
alum
ínio
, re
mov
endo
em
tor
no d
e 84
% d
o m
esm
o e
efet
ivo
para
a n
ão r
emoç
ão d
e fe
rro,
um
a ve
z qu
e re
mov
e em
tor
no d
e 12
% d
o fe
rro
tota
l pre
sent
e na
DA
M.
Com
re
laçã
o à
rem
oção
do
su
lfat
o e
da
acid
ez,
obse
rvam
-se
valo
res
em
torn
o de
28
%
e 35
%
resp
ectiv
amen
te. A
con
diçã
o ót
ima
de t
rata
men
to, o
u se
ja, a
quel
a co
ndiç
ão e
m q
ue s
e ob
tém
a m
áxim
a re
moç
ão
do a
lum
ínio
e a
mín
ima
rem
oção
do
ferr
o é
alca
nçad
a co
m a
dos
agem
na
faix
a de
0,0
9 a
0,13
equ
ival
ente
- g
ram
a al
calin
o /g
ram
a A
l+3 , c
om u
m te
mpo
de
dosa
gem
/rea
ção
entr
e 10
a 2
0 m
inut
os e
um
pH
de
neut
raliz
ação
em
torn
o de
4,3
5.
120
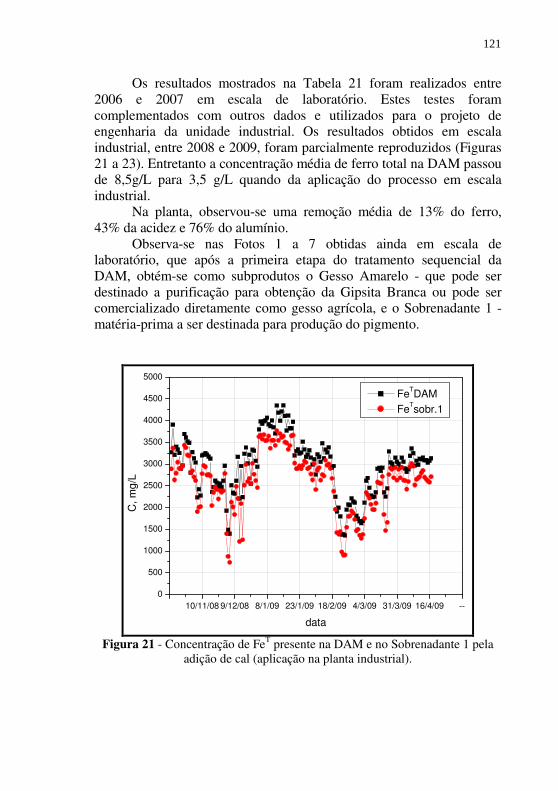
Os resultados mostrados na Tabela 21 foram realizados entre 2006 e 2007 em escala de laboratório. Estes testes foram complementados com outros dados e utilizados para o projeto de engenharia da unidade industrial. Os resultados obtidos em escala industrial, entre 2008 e 2009, foram parcialmente reproduzidos (Figuras 21 a 23). Entretanto a concentração média de ferro total na DAM passou de 8,5g/L para 3,5 g/L quando da aplicação do processo em escala industrial.
Na planta, observou-se uma remoção média de 13% do ferro, 43% da acidez e 76% do alumínio.
Observa-se nas Fotos 1 a 7 obtidas ainda em escala de laboratório, que após a primeira etapa do tratamento sequencial da DAM, obtém-se como subprodutos o Gesso Amarelo - que pode ser destinado a purificação para obtenção da Gipsita Branca ou pode ser comercializado diretamente como gesso agrícola, e o Sobrenadante 1 - matéria-prima a ser destinada para produção do pigmento.
10/11/08 9/12/08 8/1/09 23/1/09 18/2/09 4/3/09 31/3/09 16/4/09 --0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
C, m
g/L
data
FeTDAM FeTsobr.1
Figura 21 - Concentração de FeT presente na DAM e no Sobrenadante 1 pela
adição de cal (aplicação na planta industrial).
121
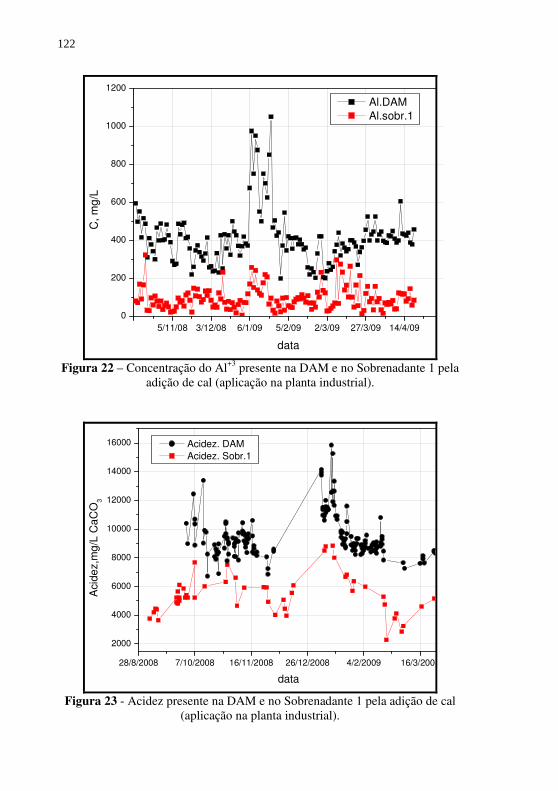
5/11/08 3/12/08 6/1/09 5/2/09 2/3/09 27/3/09 14/4/090
200
400
600
800
1000
1200C
, mg/
L
data
Al.DAM Al.sobr.1
Figura 22 – Concentração do Al+3 presente na DAM e no Sobrenadante 1 pela
adição de cal (aplicação na planta industrial).
28/8/2008 7/10/2008 16/11/2008 26/12/2008 4/2/2009 16/3/2009
2000
4000
6000
8000
10000
12000
14000
16000
Aci
dez,
mg/
L C
aCO
3
data
Acidez. DAM Acidez. Sobr.1
Figura 23 - Acidez presente na DAM e no Sobrenadante 1 pela adição de cal
(aplicação na planta industrial).
122

As reações sequenciais que ocorrem para a remoção do alumínio da DAM e formação do Gesso Amarelo, podem ser descritas como:
2;3)(23)(32
1;3)(3)(333
)(432
4)(23
)(4)(222
42
)( ls
ss
FeSOOHAlSOOHFeAl
CaSOOHFeOHCaSOFe
s+→++
+→++
−+
−+
(37)
Ou seja, primeiramente a cal adicionada reage com o Fe2+
presente na DAM formando um sólido de Fe+2 (Fe(OH)x). Após isto, o hidróxido ferroso reage com o alumínio presente na fase líquida, formando um hidróxido de alumínio e/ou hidróxi-sulfato de alumínio retornando o ferro ferroso para a solução. Além disso, o ferro trivalente presente na DAM reage com a cal adicionada e precipita juntamente com o alumínio. Isto é inevitável uma vez que o Fe+3 inicia a precipitação em pH mais baixo do que o alumínio (3,5 versus 4,0). Desta forma na reação global o ferro ferroso não participa e somente o alumínio, o ferro trivalente e o sulfato participam da formação dos sub-produtos, conforme Esquema 5.
)()(3)(322
433
)()(322
43
)(432
4)(23
)()(222
42
46)(2)(2)(6622
3;43)(2)(332
2;3)(23)(32
1;43)(3)(333
)(
sss
ss
ls
ss
CaSOOHFeOHAlOHCaSOFeAl
CaSOOHFeOHCaSOFe
FeSOOHAlSOOHFeAl
CaSOOHFeOHCaSOFe
s
++→+++
+→++
+→+
+→++
−++
−+
−+
−+
Esquema 5
Os resultados da caracterização completa do Gesso Amarelo
assim como o estudo para obtenção da Gipsita Branca para construção civil e do Coagulante 2 serão apresentados no próximo item.
6.2 CARACTERIZAÇÃO E PURIFICAÇÃO DO GESSO AMARELO
6.2.1 Caracterização do Gesso Amarelo A caracterização do Gesso Amarelo, obtido na primeira etapa do
tratamento sequencial da DAM esta apresentada nas Tabelas 22 e 23. Esta caracterização foi realizada primeiramente nos testes em
escala de laboratório (2006 e 2007) e posteriormente na planta industrial (2008 e 2009). Ressalta-se aqui que na planta industrial este processo
123

corresponde ao Circuito 1, conforme descrito na Figura 6. Foram produzidos diferentes lotes do Gesso Amarelo,
principalmente na planta industrial. Isto se deveu à variabilidade das características da DAM.
Os parâmetros utilizados para o tratamento parcial da DAM pela adição de cal, com consequente formação do Gesso Amarelo e do Sobrenadante 1, foram àqueles definidos nos testes de laboratório, ou seja, dosagem cal de 0,09 a 0,13 eq.g/g Al+3, tempo de reação entre 10 e 30 minutos obtendo-se um pH de neutralização entre 4,30 e 4,50.
Observa-se pelas Tabelas 22 e 23 que o Gesso Amarelo, é um material sólido rico em sulfato, alumínio e ferro. Se todo sulfato presente no Gesso Amarelo estiver como CaSO4.2H2O, observa-se que o teor de Gipsita neste material varia entre 79 a 81%, tanto nos testes de laboratório quanto na planta industrial. Além disso, o teor de alumínio e ferro fica entre 19 e 21% (como R2O3).
Tabela 22 - Caracterização do Gesso Amarelo produzido pelo circuito 1 em escala de laboratório (amostra úmida filtrada).
Nomenclatura do lodo úmido
filtrado
Base úmida
Base seca
Base úmida Base seca
Base úmida
Base seca
Um
idad
e%
% Al
% Al2O3
% Al2O3
% Fe
% Fe2O3
% Fe2O3
% SO4
% SO4
T63 5,0 9,4 14,4 3,9 5,5 8,4 38,3 58,4 34,4 T67 3,0 5,7 12,6 1,4 2,0 4,3 29,0 63,7 54,5 T71 4,2 7,9 19,4 1,5 2,1 5,2 18,5 45,3 59,2 T74 3,7 7,0 18,5 1,4 2,1 5,5 11,8 31,5 62,5 T80 2,7 5,1 8,5 0,9 1,3 2,1 11,5 19,0 39,3 T98 2,9 5,4 14,6 1,0 1,4 3,9 17,2 46,4 63,0 T96 2,2 4,1 11,1 1,1 1,6 4,3 17,6 47,6 63,0
Média 3,4 6,4 14,2 1,6 2,3 4,8 20,6 44,6 53,7
Duas amostras do Gesso Amarelo foram destinadas à análise de
difração de raios-X. Para tal, as mesmas foram secas a 1100C graus por 4horas, moída e passada na peneira 325 mesh. Os difratogramas de raios-X das amostras, (produzidas na planta industrial) estão apresentados na Figura 24.
124

Tabela 23 - Caracterização do Gesso Amarelo produzido pelo circuito 1 na planta industrial (em base seca).
Data %Al2O3 %Fe2O3 %R2O3
(1) %SO42-
4/9/08 6,5 15,7 22,2 47,7 5/9/08 7,4 13,7 21,1 45,9 8/9/08 7,4 14,5 21,9 44,1
15/10/08 7,4 9,4 16,7 40,6 28/10/08 12,9 5,3 18,1 41,9 29/10/08 7,0 7,2 14,2 47,4 31/10/08 6,7 4,4 11,2 43,0 31/10/08 8,2 3,9 12,1 49,9 7/11/08 11,1 8,8 19,9 42,2 11/11/08 10,6 6,2 16,8 41,5 12/11/08 11,0 6,2 17,1 48,5 13/11/08 8,0 6,7 14,7 48,8 13/11/08 13,4 7,7 21,1 48,6 13/11/08 12,6 7,7 20,3 46,3 18/11/08 10,2 6,8 16,9 38,7 27/11/08 9,0 9,0 18,0 45,2 01/12/08 8,3 5,7 14,0 46,0 05/12/08 13,4 6,7 20,1 44,9 05/12/08 11,1 7,2 18,3 45,2 8/1/09 13,41 5,70 19,1 37,0 9/1/09 10,84 5,22 16,1 41,33 12/1/09 11,73 5,40 17,1 39,22 15/1/09 14,23 7,02 21,3 48,51 16/1/09 10,94 6,33 17,3 32,00 29/1/09 11,51 7,12 18,6 50,56 3/4/09 13,16 8,40 21,6 46,84 9/4/09 10,25 4,41 14,7 35,35 23/4/09 8,43 11,56 20,0 41,45 23/4/09 9,26 12,44 21,7 44,06 Média 10,2 7,8 18,0 43,9
Fonte: (1) %R2O3 = %Fe2O3+%Al2O3
125

0
200
400
600
800
1000
1200
1400
4 7 10 13 16 19 22 25 28 31 34 37 40 43 46 49 52 55 58
Posição (’2theta)
Inte
nsi
dad
e
gesso amarelo.30/04/09 gesso amarelo.31/10/08
B
B
B
BB
B
B
B
Figura 24 - Difratogramas de raios-X de duas amostras de Gesso Amarelo
obtidas na planta industrial – B = Bassanita – CaSO4.0,5H2O. Observa-se que esta amostra possui como fase cristalina o Sulfato
de cálcio hemi-hidratado, (CaSO4.0,5H2O) e é detectada a possível presença do Sulfato de alumínio hidróxi-hidratado, (3Al2O3.4SO3.xH2O – picos em 11,5; 19,5; 25,6; 29; 36,47,6) e da Boehmita (AlO.OH – picos em 14,7; 28,5; 38,6; 46,1; 49,1).
A calcinação da Gipsita é um processo de aquecimento no qual ocorre a desidratação do cristal conforme o Esquema 6 (Pereira, 1973).
140ºC – 160ºC - OHOHCaSOOHCaSO 22424 5,15,0.2. +→ - Bassanita
160ºC – 250ºC - OHOHCaSOOHCaSO 22424 2.2. +→ ε - Anidrita III
250ºC – 800ºC - OHCaSOOHCaSO 2424 22. +→ - Anidrita II Esquema 6 – Desidratação da Gispsita indo para Bassanita e Anidrita.
A desidratação da Gipsita depende da temperatura, do tempo de
exposição àquela temperatura e do tamanho de partículas utilizado para calcinação.
O Gesso Amarelo obtido na planta industrial foi calcinado em três temperaturas diferentes, 60, 100 e 160 0C. Esta calcinação sucedeu-se pelo mesmo tempo de contato, 3 horas e com um mesmo tamanho de partículas, uniforme de 3,0 a 5,0 mm. O objetivo deste teste foi a definição da temperatura e tempo de contato necessários para obtenção somente da Bassanita (CaSO4.0,5H2O) a partir da Gipsita.
126

As análises de difração de raios-X das amostras calcinadas estão apresentadas na Figura 25.
0
500
1000
1500
2000
2500
3000
3500
4000
4 8 12 16 20 24 28 32 36 40 44 48 52 56 60
Posição (’2theta)
Inte
nsi
dad
e
60 graus 110 graus 160 graus
G G
G
G
G
G
B
B
B
B
B
Figura 25 - Difração de raios-X da amostra calcinada a 600C, 110 0C e 160 0C.
B = Bassanita (CaSO4.1/2H2O) e G=Gipsita (CaSO4.2H2O). Observa-se claramente a mudança de fase da Gipsita para a
Bassanita quando a temperatura aumenta de 600C para 1600C, mantendo-se o mesmo tempo de secagem.
A amostra do Gesso Amarelo seca a 60 graus apresenta apenas os picos referentes à Gipsita (CaSO4.2H2O); enquanto que a amostra seca a 1100C apresenta os picos da Gipsita e começam a surgir os picos da Bassanita (CaSO4.1/2H2O).
Aumentando a temperatura de 60 para 160ºC, toda Gipsita se transforma em Bassanita e não aparecem os picos da Anidrita. Conclui-se, portanto que a calcinação da Gipsita com o objetivo de transforma – lá somente em Bassanita, ou seja, no gesso utilizado pela indústria da construção civil, pode ser realizada utilizando-se um diâmetro de partículas de 3,0-5,0 mm, temperatura de secagem de 1600C e tempo de residência no secador de 3 horas.
6.2.2 Purificação do Gesso Amarelo Embora o objetivo deste trabalho seja o de se obter produtos de
elevado valor agregado, o que exige a purificação do Gesso Amarelo, ressalta-se que este material possa ser utilizado diretamente um como
127

insumo agrícola, após a etapa de secagem, como fonte de cálcio e enxofre para o solo.
Além de insumo agrícola, outros usos possíveis para o Gesso Amarelo são a adição, a úmido, na composição da massa para a cerâmica vermelha bem como a substituição deste pelo minério de Gipsita na produção de cimento.
A purificação do Gesso Amarelo foi realizada por meio de uma reação controlada com a adição de solução ácida, a qual pode ser preparada com ácido sulfúrico e/ou ácido clorídrico e água destilada, para a solubilização do ferro e alumínio. A mistura, lodo úmido filtrado + solução ácida, foi mantida sob agitação mecânica por um período de tempo variável. Esta suspensão, com um teor de sólido extremamente alto e também variável, foi filtrada obtendo-se com isso duas fases distintas; uma fase líquida, extremamente ácida rica em ferro e alumínio; e uma fase sólida, rica em Gipsita. Após a filtração, a Gipsita obtida foi lavada com água destilada para remoção da umidade ácida residual presente na torta, seca a 110oC por 3 horas, moída e caracterizada. A escolha deste processo de purificação do gesso foi baseada na formação de Sulfato de alumínio e ferro na fase liquida, Coagulante 2, um sub-produto adicional deste tratamento.
Os testes para a purificação do Gesso Amarelo foram realizados adicionando-se o volume de ácido estequiométrico para reação com o Al2O3+Fe2O3 presentes na amostra. Variou-se o volume de água adicionada para a mistura e o tempo da reação de solubilização (para avaliação da completa remoção dos contaminantes). O objetivo destes testes foi o de determinar os parâmetros de operação para a obtenção de Gipsita Branca, bem como o de se obter uma solução ácida oriunda da reação o mais concentrada possível para ser comercializada como um coagulante férrico aluminoso, conforme a reação 38.
)()()()( 42243232 62.
lsss SOHOHCaSOOAlOFe +++
OHOlHCaSOSOAlSOFe sll 2)(24)342342 62.)()( )( +++ (38)
A Tabela 24 apresenta as condições experimentais utilizadas em
duas baterias de testes, bem como os resultados de caracterização da Gipsita Branca, do Coagulante 2 e a porcentagem de remoção dos contaminantes que foi alcançada.
128

Tab
ela
24 -
Con
diçõ
es e
xper
imen
tais
e r
esul
tado
s ob
tidos
na
vari
ação
do
volu
me
de á
gua
para
rea
ção.
Par
âmet
ros
vari
ávei
s do
tes
te
Coa
gula
nte
2 G
ipsi
ta
bran
ca, (
bu)
Ges
so
Am
arel
o, (
bu)
%
Rem
oção
Tes
te
m3 so
l.ác./
to
n L.u
.f
tem
po
reaç
ão,
min
rela
ção,
m
in/g
Fe
T, g
/L
Al+
3 , g/L
R
endi
men
to,
%
%Fe
%
Al
%Fe
%
Al
Fe
Al
4 0,
41
60
0,60
11
,6
44,0
64
,0
0,1
0,3
1,4
3,0
91
90
5 0,
11
0,60
28
,8
86,8
45
,0
0,3
1,0
1,4
3,0
78
67
6 0,
30
0,60
31
,2
120,
7 61
,0
0,3
0,6
1,5
4,2
80
86
20
0,39
120
0,30
19
,0
66,8
-
0,1
0,1
0,9
2,7
93
98
21
0,49
0,
30
8,5
31,6
-
0,1
0,1
0,9
2,7
91
97
22
0,31
0,
30
10,4
38
,8
- 0,
1 0,
1 0,
9 2,
7 94
98
23
0,
59
0,30
8,
6 28
,4
- 0,
1 0,
1 0,
9 2,
7 93
98
Fo
nte:
L.U
.F =
lodo
úm
ido
filtr
ado.
Com
o m
ostr
ado
na T
abel
a 24
, as
im
pure
zas
de a
lum
ínio
e f
erro
con
tidas
na
amos
tra
ante
s do
tra
tam
ento
fo
ram
efi
cien
tem
ente
rem
ovid
as.
Obs
erva
-se
tam
bém
que
par
a as
men
ores
rel
açõe
s de
sol
ução
áci
da p
ara
lodo
úm
ido
filt
rado
, ou
sej
a, p
ara
0,11
e 0
,30
mL
/g,
mai
ores
for
am a
s co
ncen
traç
ões
obtid
as p
ara
o C
oagu
lant
e 2.
E
ntre
tant
o, a
con
sist
ênci
a da
sus
pens
ão f
icou
mui
to p
asto
sa, e
torn
a-se
dif
ícil
tran
spor
tá-l
a po
r bo
mbe
amen
to, p
or
exem
plo.
Def
iniu
-se
em f
unçã
o di
sto
o us
o da
rel
ação
vol
ume/
mas
sa,
mL
/g i
gual
a 0
,32.
A p
artir
do
espe
ctro
de
difr
ação
de
ra
ios-
X
da
amos
tra
ante
s e
após
o
trat
amen
to
ácid
o,
Figu
ra
26,
veri
fica
-se
um
aum
ento
na
cr
ista
linid
ade
do g
esso
lim
po p
ela
mai
or in
tens
idad
e do
s pi
cos.
129

0
200
400
600
800
1000
1200
1400
1600
1800
2000
4 7 10 13 16 19 22 25 28 31 34 37 40 43 46 49 52 55 58
Posição (2theta)
Inte
nsi
dad
e
gesso limpo.31/10/08 gesso amarelo.31/10/08
B
G
B
B
B
B
B
B
G
G
Figura 26 - Difratograma de raios-X e fases identificadas do Gesso Amarelo
(bruto) da Gipsita Branca (gesso limpo). A análise térmica diferencial da Gipsita Branca bem como do
Gesso Amarelo está apresentada na Figura 27 (a) e (b). Observa-se que, para o Gesso Amarelo, ocorrem dois picos
endotérmicos: o primeiro, a 1490C, com perda de massa de 8,98% é referente à mudança de fase da Gipsita para a Bassanita; o outro pico ocorre em temperatura igual a 7880C, com perda de massa de 5,3%, e se refere à mudança de fase da Bassanita para a Anidrita.
A análise térmica da Gipsita Branca mostrou três picos endotérmicos. O primeiro, a 1520C, com perda de massa de 13,3%, é atribuído à mudança de fase da Gipsita para a Bassanita. O segundo, entre 583 e 913 0C, com perda de massa de 9,19%; e um terceiro pico endotérmico entre 913 e 1102 0C, com perda de massa de 2,5%. Ou seja, três mudanças de fase podem ser observadas para a Gipsita branca; da transformação da Gipsita para a Bassanita; da Bassanita para a Anidrita III e desta última para a Anidrita II.
130
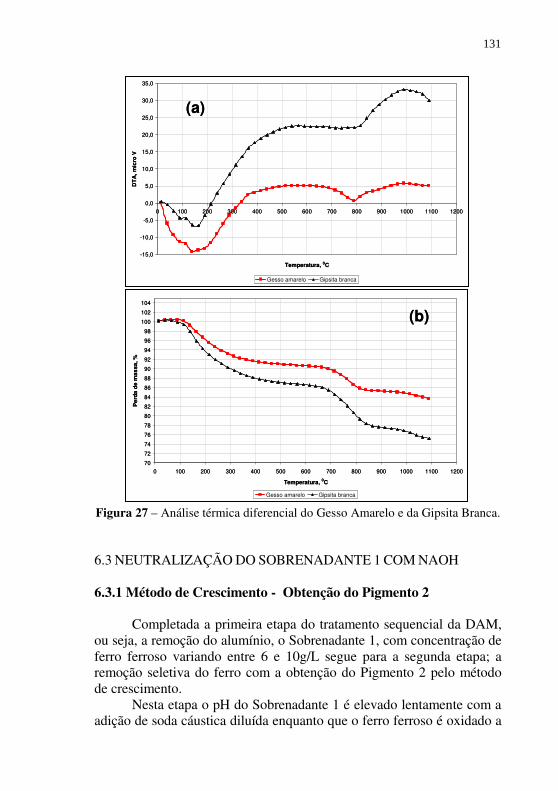
-15,0
-10,0
-5,0
0,0
5,0
10,0
15,0
20,0
25,0
30,0
35,0
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
DT
A, m
icro
V
Gesso amarelo Gipsita branca
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
102
104
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
Pe
rda
de
mas
sa, %
Gesso amarelo Gipsita branca
(a)
(b)
-15,0
-10,0
-5,0
0,0
5,0
10,0
15,0
20,0
25,0
30,0
35,0
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
DT
A, m
icro
V
Gesso amarelo Gipsita branca
70
72
74
76
78
80
82
84
86
88
90
92
94
96
98
100
102
104
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
Pe
rda
de
mas
sa, %
Gesso amarelo Gipsita branca
(a)
(b)
Figura 27 – Análise térmica diferencial do Gesso Amarelo e da Gipsita Branca.
6.3 NEUTRALIZAÇÃO DO SOBRENADANTE 1 COM NAOH
6.3.1 Método de Crescimento - Obtenção do Pigmento 2 Completada a primeira etapa do tratamento sequencial da DAM,
ou seja, a remoção do alumínio, o Sobrenadante 1, com concentração de ferro ferroso variando entre 6 e 10g/L segue para a segunda etapa; a remoção seletiva do ferro com a obtenção do Pigmento 2 pelo método de crescimento.
Nesta etapa o pH do Sobrenadante 1 é elevado lentamente com a adição de soda cáustica diluída enquanto que o ferro ferroso é oxidado a
131

ferro férrico e precipitado. Daí obtém-se novamente duas fases distintas; uma fase sólida rica em ferro férrico e uma fase líquida ainda ácida, porém com baixas concentrações dos sulfatos metálicos.
As condições experimentais foram definidas para se evitar a co-precipitação dos outros metais presentes no Sobrenadante 1, principalmente o manganês, assim como para se obter um lodo férrico filtrado com o máximo teor de Fe2O3 possível, o mínimo teor de contaminantes e a máxima cristalinidade. Neste sentido, os testes foram realizados variando-se a dosagem de soda cáustica adicionada, o tempo total de adição da soda e consequentemente o tempo total da reação de oxidação/formação do pigmento e a dosagem de peróxido de hidrogênio.
As Tabelas 25 e 26 mostram os resultados alcançados com relação a qualidade do Sobrenadante 1 e Sobrenadante 2 (Tabela 25) assim como as porcentagens de remoção alcançadas e a análise química do lodo férrico obtido (Tabela 26). Todos estes testes foram realizados em escala de laboratório objetivando-se desenvolver o método para a produção do pigmento inorgânico, bem como levantar os dados de engenharia para o projeto da unidade industrial.
132

Tab
ela
25 -
Rem
oção
do
ferr
o fe
rros
o pr
esen
te n
o S
obre
nada
nte
1 pe
la n
eutr
aliz
ação
lent
a co
m N
aOH
. Cir
cuito
2
– M
étod
o de
cre
scim
ento
em
esc
ala
de la
bora
tóri
o.
Teste
PA
RÂ
ME
TR
OS
VA
RIÁ
VE
IS D
O T
EST
E
AN
ÁL
ISE
S N
O S
OB
RE
NA
DA
NT
E 1
A
NÁ
LIS
ES
NO
SO
BR
EN
AD
AN
TE
2
Relação eq.g/g Fe+2
Tempo de dosagem NaOH, h
pH após a dosagem / pH decantação
Tempo de dosagem H2O2, h
Relação gr.H2O2/g Fe
+2
% de lodo férrico obtido
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
1 0,
018
4,5
3,77
3,
0 0,
2 19
%
6.83
9 22
2 16
.501
18
.031
2.
431
91
- 10
.789
2
0,01
8 7,
5 3,
53
7,5
0,2
19%
6.
751
84
18.0
84
17.8
98
3.22
4 59
13
.930
8.
478
3 0,
018
5,0
2,48
3,
0 0,
4 19
%
6.78
0 99
14
.919
23
.668
2.
506
99
14.9
19
7.53
3 4
0,01
8 7,
5 3,
42
7,5
0,2
19%
7.
098
55
16.6
99
23.8
80
2.37
4 22
13
.534
3.
499
5 0,
018
7,5
3,75
7,
5 0,
1 -
9.76
3 11
3 12
.653
22
.842
1.
872
31
9.80
2 4.
731
6 0,
019
2,0
5,57
2,
4 0,
1 80
%
6.57
7 21
26
.195
19
.170
55
1
23.6
81
320
8 0,
021
7,5
3,80
7,
5 0,
2 -
9.44
3 12
5 14
.422
-
280
25
12.6
87
9
0,02
2 7,
0 3,
75
7,0
0,2
- 6.
853
139
13.4
30
23.1
57
2.29
7 18
8.
786
7.37
0 10
0,
022
7,5
3,75
7,
5 0,
2 -
5.83
1 -
13.6
89
13.2
47
751
- 11
.098
3.
192
11
0,02
2 7,
0 3,
71
7,0
0,2
- 7.
674
66
11.0
98
20.2
19
1.22
2 -
11.0
98
5.08
0 13
0,
031
- 3,
50 /
3,40
-
0,6
6,2%
7.
569
461
- -
31
402
17.7
19
7.64
0 14
0,
031
3,0
5,75
3,
1 0,
1 60
%
6.78
1 95
17
.153
-
176
1 17
.153
21
3 15
0,
037
3,5
5,44
3,
3 0,
2 68
%
6.57
7 16
4 21
.963
-
0 0
22.0
09
300
16
0,04
0 3,
0 4,
65
2,9
0,2
66%
4.
322
156
17.1
73
11.3
75
914
0 17
.173
32
5 C
ontin
ua
Con
tinua
ção
17
0,04
1 3,
2 5,
60
3,2
0,2
45%
6.
886
158
18.6
78
NA
17
6 1
17.1
53
213
133

Teste P
AR
ÂM
ET
RO
S V
AR
IÁV
EIS
DO
TE
STE
A
NÁ
LIS
ES
NO
SO
BR
EN
AD
AN
TE
1
AN
ÁL
ISE
S N
O S
OB
RE
NA
DA
NT
E 2
Relação eq.g/g Fe+2
Tempo de dosagem NaOH, h
pH após a dosagem / pH decantação
Tempo de dosagem H2O2, h
Relação gr.H2O2/g Fe
+2
% de lodo férrico obtido
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
FeT, mg/L
Al, mg/L
SO4-2
, mg/L
Acidez, ppmCaCO3
18
0,04
2 3,
0 4,
16
3,0
0,4
78%
5.
506
282
19.5
09
11.8
81
49
26
19.5
09
405
19
0,04
3 4,
13
4,80
/ 3,
50
- -
42%
8.
422
205
- -
834
50
20.4
36
1.50
4 20
0,
045
9,7
5,10
/ 4,
85
- -
62%
8.
482
143
19.3
55
23.3
20
55
0,07
17
.662
44
0 22
0,
054
1,6
5,30
/ 4,
40
- -
77%
7.
308
301
19.5
73
19.1
95
2.84
9 33
19
.500
6.
463
24
0,06
2 1,
5 2,
84
3,6
0,2
47%
6.
864
58
18.9
66
15.6
20
1.01
5 16
18
.966
2.
640
25
0,07
2 8,
4 4,
10 /
3,63
0,
0 0,
0 38
%
7.45
1 32
14
.791
16
.940
2.
487
2,1
10.9
79
3.90
5 26
0,
075
6,4
4,20
/ 4,
00
6,4
0,9
47%
8.
812
1,7
16.8
25
17.2
15
40
0,04
16
.825
50
9
0,04
5,
07
4,23
4,
84
0,28
46
%
7.14
2 14
7 17
.723
17
.956
1.
162
38,7
1 16
.254
3.
500
Font
e (1
) Pa
râm
etro
s fi
xos
dos
test
es: V
olum
e de
Sob
rena
dant
e 1
= 6
0 a
65L
; Agi
taçã
o pr
opor
cion
ada
por
aera
dor
de b
olha
fin
a in
stal
ado
na b
ase
do
reat
or c
om p
ress
ão to
tal d
e ar
de
10 p
si; C
once
ntra
ção
méd
ia d
a so
da a
dici
onad
a =
350
gN
aOH
/L; C
once
ntra
ção
méd
ia d
o pe
róxi
do =
290
gH
2O2/
L;
Tem
po m
édio
de
deca
ntaç
ão =
6,0
hora
s.
Obs
erva
-se
pela
Tab
ela
25 q
ue o
Sob
rena
dant
e 1,
mat
éria
-pri
ma
para
a o
bten
ção
do p
igm
ento
pel
o m
étod
o de
cre
scim
ento
, te
m c
once
ntra
ção
de f
erro
fer
roso
que
var
ia e
ntre
6 e
10g
/L e
de
alum
ínio
ent
re 2
e 2
00m
g/L
. O
tr
atam
ento
des
ta á
gua
com
sod
a e
peró
xido
de
hidr
ogên
io v
isan
do a
rem
oção
sel
etiv
a de
fer
ro f
oi b
asta
nte
efic
ient
e, u
ma
vez
que
rem
oveu
em
torn
o de
83%
do
ferr
o, 1
0% d
o su
lfat
o e
80%
da
acid
ez.
134

Tab
ela
26 -
Por
cent
agen
s de
rem
oção
do
Fe2+
, A
l3+,
SO4-2
e a
cide
z do
Sob
rena
dant
e 1
pela
neu
tral
izaç
ão c
om
NaO
H. C
ircu
ito 2
– M
étod
o de
cre
scim
ento
em
esc
ala
de la
bora
tóri
o.
Tes
te
PA
RÂ
ME
TR
OS
VA
RIÁ
VE
IS D
O T
EST
E
AN
ÁL
ISE
S N
O L
OD
O S
EC
O
(SE
M L
AV
AG
EM
) %
RE
MO
ÇÃ
O P
EL
O T
RA
TA
ME
NT
O
Rel
ação
eq.
gr/g
r Fe
+2
%Fe
2O3
%A
l2O
3 %
SO4
FeT
Al+
3 SO
4-2
Aci
dez
1 0,
018
- -
- 64
%
59%
-
40%
2
0,01
8 -
- -
52%
30
%
23%
53
%
3 0,
018
- -
- 63
%
0%
0%
68%
4
0,01
8 -
- -
67%
59
%
19%
85
%
5 0,
018
- -
- 81
%
73%
23
%
79%
6
0,01
9 50
,1
0,0
10,8
99
%
95%
10
%
98%
8
0,02
1 69
,0
0,5
10,0
97
%
80%
12
%
- 9
0,02
2 -
- -
66%
87
%
35%
68
%
10
0,02
2 68
,0
0,0
13,0
87
%
- 19
%
76%
11
0,
022
66,0
0,
4 12
,0
84%
10
0%
0%
75%
13
0,
031
64,0
1,
9 13
,8
100%
13
%
- -
14
0,03
1 -
- -
97%
99
%
0%
- 15
0,
037
- -
- 10
0%
100%
0%
-
16
0,04
0 -
- -
79%
10
0%
0%
97%
17
0,
041
50,4
0,
2 17
,9
97%
10
0%
8%
- 18
0,
042
99
%
91%
0%
97
%
19
0,04
3 57
,5
2,1
20,5
90
%
76%
-
- 20
0,
045
76,1
2,
4 15
,7
99%
10
0%
9%
98%
22
0,
054
49,1
10
,7
22,1
61
%
89%
0%
66
%
24
0,06
2 52
,2
0,0
13,5
85
%
73%
0%
83
%
Con
tinu
a
135

Tes
te
PA
RÂ
ME
TR
OS
VA
RIÁ
VE
IS D
O T
EST
E
AN
ÁL
ISE
S N
O L
OD
O S
EC
O
(SE
M L
AV
AG
EM
) %
RE
MO
ÇÃ
O P
EL
O T
RA
TA
ME
NT
O
Rel
ação
eq.
gr/g
r Fe
+2
%Fe
2O3
%A
l2O
3 %
SO4
FeT
Al+
3 SO
4-2
Aci
dez
Con
tinu
ação
25
0,
072
64,9
0,
04
5,43
67
%
93%
26
%
77%
26
0,
075
63,4
0,
12
13,2
10
0%
100%
0%
97
%
27
0,10
3 56
,5
5,4
- -
- -
-
0,04
58
,42
2,29
14
,77
83%
75
%
10%
80
%
Ent
reta
nto,
res
salta
-se
que
se a
inda
tiv
er a
lum
ínio
no
Sob
rena
dant
e 1,
o m
esm
o se
rá r
emov
ido
junt
amen
te
com
o f
erro
tra
balh
ando
-se
com
dos
agen
s de
sod
a m
aior
es d
o qu
e 0,
018
eq.g
.alc
alin
o/g.
Fe+
2 e p
H m
aior
do
que
3,80
(T
abel
as 2
5 e
26).
A
inda
com
rel
ação
ao
trat
amen
to s
eque
ncia
l da
DA
M,
a re
moç
ão d
o fe
rro
no S
obre
nada
nte
1 re
duz
a ac
idez
da
água
de
uma
méd
ia d
e 17
.956
par
a 3.
500
mgC
aCO
3/L
(T
abel
a 25
). E
sta
redu
ção
da a
cide
z é
uma,
ent
re
as v
anta
gens
que
dev
em s
er r
essa
ltada
s pe
lo m
étod
o or
a pr
opos
to u
ma
vez
que
em u
ma
próx
ima
etap
a de
tr
atam
ento
o S
obre
nada
nte
2, o
btid
o ap
ós a
rem
oção
do
ferr
o fe
rros
o e
prod
ução
do
Pig
men
to,
terá
que
ser
ad
equa
do a
todo
s os
par
âmet
ros
da C
ON
AM
A 3
57 e
isso
impl
icar
a em
rem
over
a a
cide
z ai
nda
rem
anes
cent
e.
Por
tant
o,
o m
étod
o de
ob
tenç
ão
do
Pig
men
to
2,
deno
min
ado
mét
odo
de
cres
cim
ento
, co
nsis
te
na
neut
raliz
ação
len
ta d
e um
a so
luçã
o aq
uosa
de
sulf
ato
ferr
oso
prod
uzid
a a
part
ir d
a dr
enag
em á
cida
de
min
a,
cont
endo
ent
re 6
e 1
0g/L
de
Fe+
2 , com
um
tem
po to
tal d
e re
ação
de
7 a
8h, u
ma
dosa
gem
con
tínua
de
peró
xido
de
hidr
ogên
io j
unta
men
te c
om a
sod
a cá
ustic
a du
rant
e to
da r
eaçã
o (n
as q
uant
idad
es d
e 0,
20 g
H2O
2/gF
e+2 e
0,0
18
eq.g
r. a
lcal
ino
/grF
e+2 ),
um
con
trol
e no
pH
da
reaç
ão m
ante
ndo-
o en
tre
3,70
e 3
,90
e a
real
izaç
ão d
e re
cicl
o de
lo
do ú
mid
o de
cant
ado
e es
pess
ado
para
aum
enta
r a
velo
cida
de d
e de
cant
ação
bem
com
o se
rvir
com
o nú
cleo
par
a in
icia
r a
reaç
ão d
e fo
rmaç
ão d
o pi
gmen
to a
mar
elo.
136

Os parâmetros assim definidos foram utilizados para o projeto de engenharia da unidade industrial implantada na empresa Carbonífera Criciúma S/A.
6.3.1.1 Caracterização do Pigmento 2 As análises de difração de raios-X de quatro amostras de
pigmento produzidas pelo método de crescimento, (Pigmento 2), estão apresentadas nas Figuras 28 a 31. Todas as análises foram realizadas nas amostras secas a 1100C por 3horas, cominuídas em moinho de bolas e passadas pela peneira de 325 mesh (45µm).
Quatro amostras foram caracterizadas; a amostra T104; a amostra T104 após lavagem alcalina (T104.lavada); a amostra T105, a qual foi obtida também pelo método de crescimento porém com reciclo da amostra lavada e a amostra T105.lavada.
Para fins comparativos a Figura 32 apresenta o difratograma de raios-X de uma amostra comercial de pigmento amarelo (Goetita).
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
03308B.CAF
Figura 28 - Difratograma de raios-x da amostra de Pigmento 2 - T104.bruta.
137
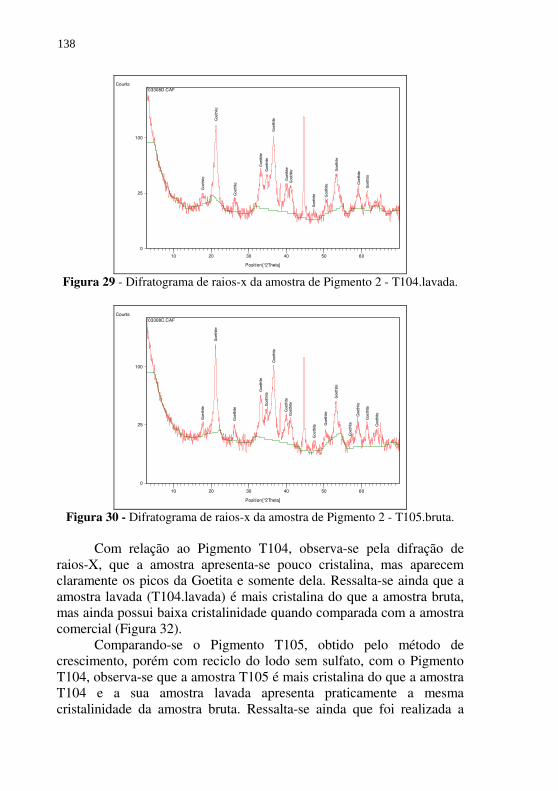
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
03308D.CAF
Figura 29 - Difratograma de raios-x da amostra de Pigmento 2 - T104.lavada.
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
03308C.CAF
Figura 30 - Difratograma de raios-x da amostra de Pigmento 2 - T105.bruta.
Com relação ao Pigmento T104, observa-se pela difração de
raios-X, que a amostra apresenta-se pouco cristalina, mas aparecem claramente os picos da Goetita e somente dela. Ressalta-se ainda que a amostra lavada (T104.lavada) é mais cristalina do que a amostra bruta, mas ainda possui baixa cristalinidade quando comparada com a amostra comercial (Figura 32).
Comparando-se o Pigmento T105, obtido pelo método de crescimento, porém com reciclo do lodo sem sulfato, com o Pigmento T104, observa-se que a amostra T105 é mais cristalina do que a amostra T104 e a sua amostra lavada apresenta praticamente a mesma cristalinidade da amostra bruta. Ressalta-se ainda que foi realizada a
138
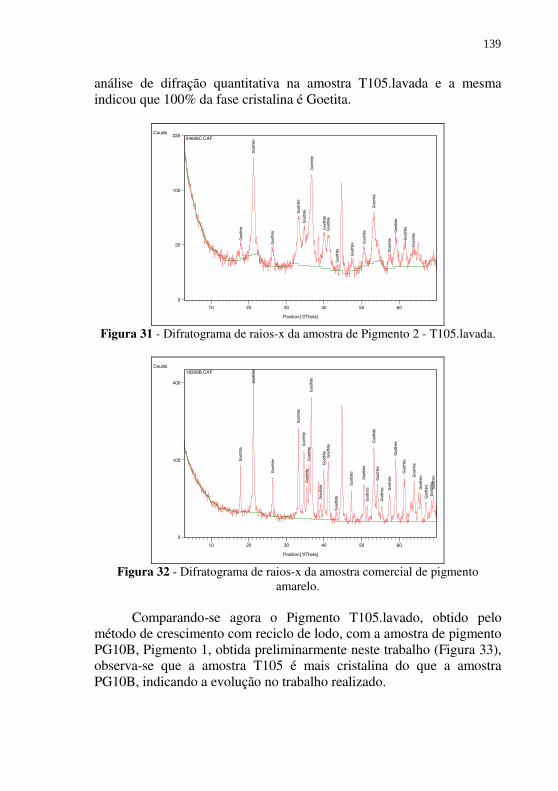
análise de difração quantitativa na amostra T105.lavada e a mesma indicou que 100% da fase cristalina é Goetita.
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
04608C.CAF
Figura 31 - Difratograma de raios-x da amostra de Pigmento 2 - T105.lavada.
Position [°2Theta]
10 20 30 40 50 60
Counts
0
100
400
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
18308B.CAF
Figura 32 - Difratograma de raios-x da amostra comercial de pigmento
amarelo. Comparando-se agora o Pigmento T105.lavado, obtido pelo
método de crescimento com reciclo de lodo, com a amostra de pigmento PG10B, Pigmento 1, obtida preliminarmente neste trabalho (Figura 33), observa-se que a amostra T105 é mais cristalina do que a amostra PG10B, indicando a evolução no trabalho realizado.
139

10 20 30 40 50 60
0
25
100
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
10 20 30 40 50 60 70 80 90 1000
25
50
75
100
Inte
nsi
dad
e
2θθθθ
(b)(a)
10 20 30 40 50 60
0
25
100
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
10 20 30 40 50 60 70 80 90 1000
25
50
75
100
Inte
nsi
dad
e
2θθθθ
(b)(a)
Figura 33 - Difratogramas de raios-X da amostra T105.lavada - Pigmento2 (a)
e PG10B - Pigmento1 (b). Ao confrontarmos a cristalinidade da amostra de Pigmento 2,
(T105.lavada), com a cristalinidade da amostra comercial, (Figura 31 e 32) observa-se que a amostra de Pigmento 2 esta pouca coisa menos cristalina do que a amostra comercial. Apesar disto, somente os picos da Goetita são encontrados naquele pigmento e vale ressaltar que esta análise (difração de raios-X) foi realizada na amostra tal qual é produzida; ou seja, com uma granulometria bastante desuniforme e maior do que a amostra comercial, como será visto mais adiante.
A Tabela 27 abaixo apresenta as análises colorimétricas das amostras secas a 1100C, bem como a composição química das mesmas.
Tabela 27 - Colorimetria e caracterização química das amostras de Pigmento 2 produzidas pelo método de crescimento.
Pigmento 2
Colorimetria Composição, %base seca
L* a* b* ∆Eab(1) ∆Eab(2) Fe2O3 SO4 Al2O3 T104 52,64 19,84 41,32 11,30 36,14 66,0 - 69,0 11,2 0,37 T104
lavada 56,19 19,48 47,71 13,81 43,00 77,0 - 78,0 2,1 0,73
T105 56,13 18,78 44,42 10,91 40,91 71,0 12,0 1,14 T105
lavada 55,44 18,61 45,24 11,57 40,95 74,0 – 78,0 0,2 1,00
Comercial Amarelo
58,72 11,49 36,73 0,00 40,67 84,2 - 86,0 0,4 <0,01
Comercial Vermelho
25,04 25,39 18,66 40,67 0,00 - - -
Diferença de cor em relação ao pigmento amarelo comercial. Diferença de cor em relação ao pigmento vermelho comercial.
De maneira geral as amostras lavadas apresentam uma % de
Fe2O3 em torno de 74 a 80% e um teor de sulfato entre 2 e 8%. Com
140

relação a colorimetria observa-se que as amostras têm um ∆Eab > 10 quando comparadas com a amostra amarela e ∆Eab > 34 quando comparadas com a amostra vermelha. Estes valores determinam uma diferença de cor muito grande das amostras produzidas pelo método de crescimento com relação as amostras comerciais conforme definição da Tabela 7. Entretanto os valores de ∆Eab indicam também que a tonalidade das amostras tende para o amarelo e não mais para o vermelho como era o caso da amostra de Pigmento 1 (PG10B).
Tem-se que considerar ainda que o tamanho de partícula dos pigmentos influência enormemente na cor (Lewis, A. P., 1973).
Para a amostra T105.lavada e a amostra de pigmento comercial, foram analisadas a distribuição granulométrica das partículas e as mesmas, apresentadas abaixo, indicam que a amostra comercial tem uma granulometria bem menor e mais uniforme do que a amostra produzida neste trabalho. Isto pode ser um dos motivos pelo qual esta amostra é mais clara e mais cristalina do que o pigmento produzido.
Tabela 28 - Distribuição do tamanho de partículas por difração a laser.
PARÂMETROS PIGMENTO AMARELO
COMERCIAL PIGMENTO
T105.LAVADO Diâmetro à 10% 0,40 µm 0,60 µm Diâmetro à 50% 0,55 µm 5,11 µm Diâmetro à 90% 0,85 µm 16,23 µm Área específica 11,0 m2/g 4,7 m2/g
A Figura 35 (a) e (b) apresenta a análise térmica diferencial das
amostras de Pigmento 2 (T105.bruto e T105.lavado) e do pigmento amarelo comercial.
Observa-se que nas três amostras, ocorrem picos endotérmicos com perda de massa entre 80 e 1800C. Esta perda de massa nos óxidos, hidróxidos de ferro esta associada a água adsorvida e/ou água estrutural presente nas amostras. O valor em si depende do tamanho de partículas da amostra e do método de obtenção da mesma (Schwetmann, 1984).
Para as amostra produzidas neste trabalho, a saída da água inicia-se em temperaturas relativamente menores do que no caso da amostra comercial (Figura 34a).
A análise da amostra T105.bruta mostrou nitidamente dois picos endotérmicos com perda de massa após a saída da água adsorvida. Um
141
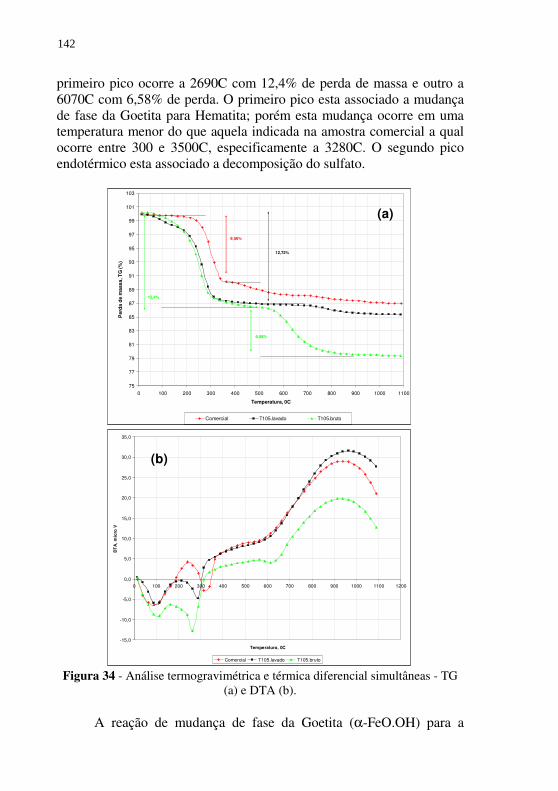
primeiro pico ocorre a 2690C com 12,4% de perda de massa e outro a 6070C com 6,58% de perda. O primeiro pico esta associado a mudança de fase da Goetita para Hematita; porém esta mudança ocorre em uma temperatura menor do que aquela indicada na amostra comercial a qual ocorre entre 300 e 3500C, especificamente a 3280C. O segundo pico endotérmico esta associado a decomposição do sulfato.
75
77
79
81
83
85
87
89
91
93
95
97
99
101
103
0 100 200 300 400 500 600 700 800 900 1000 1100
Temperatura, 0C
Per
da
de
mas
sa, T
G (%
)
Comercial T105.lavado T105.bruto
12,4%
12,72%
9,56%
6,58%
(a)
-15,0
-10,0
-5,0
0,0
5,0
10,0
15,0
20,0
25,0
30,0
35,0
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
DT
A,
mic
ro V
Comercial T105.lavado T105.bruto
(b)
75
77
79
81
83
85
87
89
91
93
95
97
99
101
103
0 100 200 300 400 500 600 700 800 900 1000 1100
Temperatura, 0C
Per
da
de
mas
sa, T
G (%
)
Comercial T105.lavado T105.bruto
12,4%
12,72%
9,56%
6,58%
(a)
-15,0
-10,0
-5,0
0,0
5,0
10,0
15,0
20,0
25,0
30,0
35,0
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Temperatura, 0C
DT
A,
mic
ro V
Comercial T105.lavado T105.bruto
(b)
Figura 34 - Análise termogravimétrica e térmica diferencial simultâneas - TG
(a) e DTA (b). A reação de mudança de fase da Goetita (α-FeO.OH) para a
142

Hematita (α-Fe2O3); ou seja, a reação de dehidroxilação da Goetita, se processa diretamente sem a formação de uma fase intermediária, conforme a reação 39. A perda de massa associada somente a esta reação é de aproximadamente 10%.
OHOFeOHFeO 232.2 +→ (39)
A temperatura da mudança de fase da Goetita para Hematita, ou
seja, a temperatura do pico endotérmico depende da cristalinidade da Goetita, do tamanho dos cristais e da porcentagem de substituição do ferro pelo alumínio (presente em algumas amostras de Goetita) (Schwetmann, 1984).
Em geral, a temperatura de mudança de fase aumenta com o aumento do tamanho do cristal, o aumento da cristalinidade da Goetita e o aumento do teor de alumínio na amostra (Schwetmann, 1984). Quando a cristalinidade da Goetita é baixa, por exemplo, o pico endotérmico aparece em 2600C, entretanto quando uma amostra bem cristalina é submetida ao aquecimento o pico aparece em 3200C (Schwetmann, 1984).
Para a amostra T105.lavada o pico endotérmico a 6070C associado ao sulfato desaparece; e aparece apenas o pico endotérmico da mudança de fase da Goetita para Hematita o qual ocorre a 2820C. A perda de massa associada a este pico foi de 12,72%.
Ressalta-se neste ponto que tanto o resultado colorimétrico, quanto os difratogramas de raios-X e a análise termogravimétrica indicam a melhora na qualidade do Pigmento 2, obtido pelo método de crescimento após a remoção prévia do alumínio, quando comparado com o Pigmento 1 (PG10.B).
Com relação a colorimetria, a amostra de Pigmento 2 apresenta-se mais clara, ou seja, com L* maior e com ∆Eab mais próximo do amarelo. Para o ajuste da cor na amostra de Pigmento 2 (T105.lavada) conforme os parâmetros de cor da amostra comercial de pigmento amarelo (L*a*b*), é necessário que seja realizada a moagem e classificação granulométrica da amostra obtendo um tamanho de partículas < 1µm.
Também a análise termogravimétrica e a difração de raios-X indicam uma melhora na cristalinidade da amostra de Pigmento 2 quando comparada com o Pigmento 1. O pico de mudança de fase da Goetita para a Hematita subiu de 250 para 2900C e houve uma melhora na cristalinidade observada pela difração de raios-X.
143
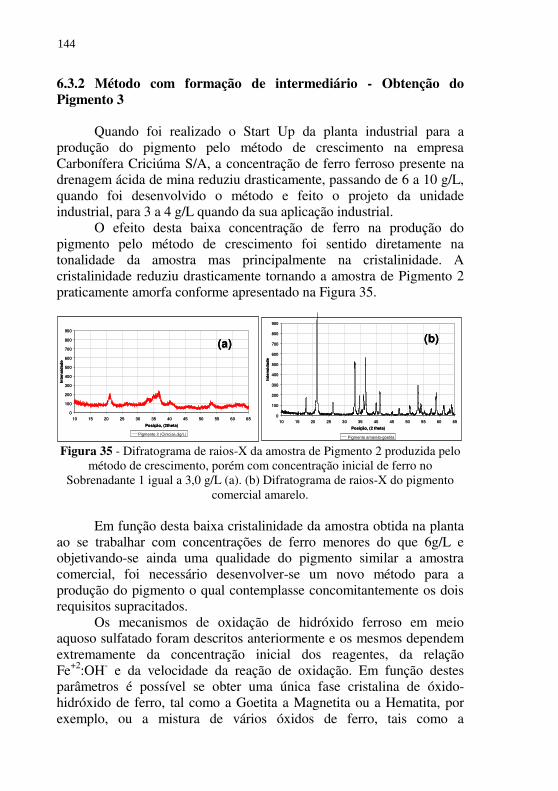
6.3.2 Método com formação de intermediário - Obtenção do Pigmento 3
Quando foi realizado o Start Up da planta industrial para a
produção do pigmento pelo método de crescimento na empresa Carbonífera Criciúma S/A, a concentração de ferro ferroso presente na drenagem ácida de mina reduziu drasticamente, passando de 6 a 10 g/L, quando foi desenvolvido o método e feito o projeto da unidade industrial, para 3 a 4 g/L quando da sua aplicação industrial.
O efeito desta baixa concentração de ferro na produção do pigmento pelo método de crescimento foi sentido diretamente na tonalidade da amostra mas principalmente na cristalinidade. A cristalinidade reduziu drasticamente tornando a amostra de Pigmento 2 praticamente amorfa conforme apresentado na Figura 35.
0
100
200
300
400
500
600
700
800
900
10 15 20 25 30 35 40 45 50 55 60 65
Posição, (2theta)
Inte
nsi
dad
e
Pigmento 2 (Cinicial=3g/L)
0
100
200
300
400
500
600
700
800
900
10 15 20 25 30 35 40 45 50 55 60 65
Posição, (2 theta)
Inte
nsi
dad
e
Pigmento amarelo-goetita
(a) (b)
0
100
200
300
400
500
600
700
800
900
10 15 20 25 30 35 40 45 50 55 60 65
Posição, (2theta)
Inte
nsi
dad
e
Pigmento 2 (Cinicial=3g/L)
0
100
200
300
400
500
600
700
800
900
10 15 20 25 30 35 40 45 50 55 60 65
Posição, (2 theta)
Inte
nsi
dad
e
Pigmento amarelo-goetita
(a) (b)
Figura 35 - Difratograma de raios-X da amostra de Pigmento 2 produzida pelo
método de crescimento, porém com concentração inicial de ferro no Sobrenadante 1 igual a 3,0 g/L (a). (b) Difratograma de raios-X do pigmento
comercial amarelo. Em função desta baixa cristalinidade da amostra obtida na planta
ao se trabalhar com concentrações de ferro menores do que 6g/L e objetivando-se ainda uma qualidade do pigmento similar a amostra comercial, foi necessário desenvolver-se um novo método para a produção do pigmento o qual contemplasse concomitantemente os dois requisitos supracitados.
Os mecanismos de oxidação de hidróxido ferroso em meio aquoso sulfatado foram descritos anteriormente e os mesmos dependem extremamente da concentração inicial dos reagentes, da relação Fe+2:OH- e da velocidade da reação de oxidação. Em função destes parâmetros é possível se obter uma única fase cristalina de óxido-hidróxido de ferro, tal como a Goetita a Magnetita ou a Hematita, por exemplo, ou a mistura de vários óxidos de ferro, tais como a
144

Lepidocrocita e a Goetita ou a Magnetita e a Goetita. É possível até mesmo obter-se uma fase amorfa conforme observado na Figura 35.a.
Com o objetivo de se obter uma amostra de pigmento inorgânico amarelo, ou seja, uma única fase bem cristalina de Goetita, partindo-se agora de uma drenagem ácida de mina pré-tratada, com 3,0 a 4,0 g/L de Fe+2, foram estudados e otimizados os parâmetros descritos acima.
Foi avaliada a influência da concentração inicial de ferro ferroso, da velocidade de oxidação e da relação Fe+2:OH- (valor de R) na qualidade do pigmento obtido. Além disso, foi produzido um núcleo de Goetita pelos métodos descritos na literatura visando a utilização do mesmo como gerador inicial dos cristalitos de Goetita.
Após a conclusão dos testes em laboratório e o desenvolvimento do novo método para a produção do pigmento, a partir de uma drenagem ácida de mina mais diluída, a planta industrial foi modificada e adaptada para a aplicação deste método operando conforme descrito na Figura 7.
A operação da planta para a produção do pigmento, (Circuito 2 – método com formação de intermediário), deu inicio em meados de Julho/2009 e finalmente foi produzido o pigmento inorgânico de óxido de ferro a partir da drenagem ácida de mina. Os resultados de 5 bateladas produzidas serão apresentados no capítulo 7.
6.3.2.1 Método com formação de intermediário - Alta velocidade de
oxidação A Tabela 29 resume as condições experimentais utilizadas nos
primeiros testes para a obtenção do pigmento amarelo pelo método com formação de intermediário.
Nestes testes a adição do reagente alcalino ao Sobrenadante 1 foi realizada de uma só vez, ou seja, adição rápida, e houve a consequente formação de um lodo verde intermediário. Sem que houvesse a decantação, o espessamento e estabilização deste lodo o mesmo foi imediatamente oxidado através da introdução de ar na mistura.
Os testes T112, 114 e 115 foram realizados em um reator piloto com 5,0 metros de altura e 300mm de diâmetro. Na base do reator foi fixado um difusor para distribuição de ar ligado ao compressor. O fornecimento de ar foi mantido constante durante todo o experimento a uma pressão de 10psi e o volume total de Sobrenadante 1 utilizado em cada teste foi de 260L. Quando necessário foi adicionado FeSO4 comercial para se aumentar a concentração inicial de ferro no
145

Sobrenadante 1 até se atingir o valor desejado objetivando-se com isto estudar a formação do pigmento em diferentes valores de concentração inicial de ferro ferroso.
Os testes T123.1, 123.2 e 124.1 foram realizados utilizando-se a mesma metodologia descrita acima, porém em escala de laboratório. Neste caso, o volume total de Sobrenadante 1 foi de 2L por teste. O ar foi fornecido por meio de compressores de laboratório com sistema de distribuição via placa porosa.
Conforme definido anteriormente, R é a relação molar entre o ferro ferroso e os íons hidroxila adicionados na mistura. Para R=0,50=1/2, tem-se a quantidade estequiométrica da reação e a mesma se processa conforme descrito abaixo.
)(42)(24 )(2líquidosólido
SONaOHFeNaOHFeSO +→+ (40)
Para R<0,50, tem-se um excesso de base na mistura e todo NaOH
será consumido para formar Fe(OH)2 deixando um excesso de base na solução, conforme a reação 41.
−+ −+−++→+⇒<= OHxyNaxySOxNaOHxFeyNaOHxFeSO
y
xR )2()2()(50,0 4224
(41) Já para R>0,50, tem-se, ao contrário, um excesso de FeSO4 em
relação ao NaOH adicionado e neste caso, todo OH- adicionado será consumido para formar, inicialmente, Fe(OH)2 deixando um excesso de íons Fe+2 e SO4
-2 em solução aquosa, conforme a reação 42.
24
24224 )2/()2/(
2)(
250,0 −+ −+−++→+⇒>= SOyxFeyxSONa
yOHFe
yyNaOHxFeSO
y
xR
(42) Portanto, dependendo do valor de R, o lodo verde intermediário
pode ser composto por Fe(OH)2 em equilíbrio com NaOH; Fe(OH)2 ou Fe(OH)2 em equilíbrio com FeSO4. No último caso, dá origem a um composto intermediário denominado GR2 (Green rust 2) (Olowe, et.al., 1991).
Nos testes T114, 115, 123.1, 123.2 e 124.1, se trabalharam no método ácido com R ≅ 0,9. No teste T112, o valor de R aproximou-se mais do valor estequiométrico (Tabela 29).
146

Tabela 29 - Condições experimentais dos testes para a obtenção do Pigmento 3 com alta velocidade de oxidação.
TESTE Cinicial Fe+2,
mg/L(1)
R (molFe+2
/ molOH-)
T adição soda (min)
pH
soda
(2)
Cinicial Fe+2
sólido mg/L(3)
pH após
oxidação
Aeração(L/min)/gFe+2
sól (4)
T112 5.900,0 0,62 35 5,90 5.390,0 3,8 10psi T114 5.400,0 0,88 16 7,24 2.842,0 3,6
T115 5.650,0 0,93 15 6,80 3.400,0 3,5 T123.1 7.045,8 0,90 - 7,95 4.893,5 3,6 0,204 T123.2 7.045,8 0,90 - 7,95 4.862,3 3,7 0,743 T124.1 7.560,4 0,90 - 7,56 8.317,3 3,6 0,367
(1) Solução preparada a partir do Sobrenadante 1 mais suprimento de sulfato ferroso até se alcançar a concentração desejada para o estudo. (2) pH obtido na suspensão aquosa após adição do volume de soda calculado para dar o valor de R desejado. (3) Concentração de ferro ferroso no estado sólido calculada pela diferença entre o ferro ferroso total na suspensão aquosa obtida após adição de soda menos o ferro ferroso total na fase líquida. (4) Aeração adicionada para a oxidação - calculada conforme equação:
=
+
+
)/(*)(
min)/(/min)/(,
2
2
LgCFeLsuspensãoVolume
LcompressorarVazãogFeLAeração
sólido
Observa-se pela Tabela 29 que a principal diferença entre os
testes T112, 114 e 115 esta no valor de R; ou seja, na quantidade de reagente alcalino adicionado para a formação do lodo verde intermediário. O teste T112 foi realizado com um valor de R mais próximo ao estequiométrico e os outros testes com um método mais ácido (R≅0,90).
Já entre as amostras T123.1 e T124.1 as diferenças estão na concentração inicial de ferro ferroso como sólido ao iniciar a oxidação e na velocidade de oxidação.
As Figuras 36 a 38 apresentam os difratogramas de raios-X das três amostras produzidas em escala piloto e as Figuras 39 e 40 apresentam os difratogramas das amostras produzidas em escala de laboratório.
147
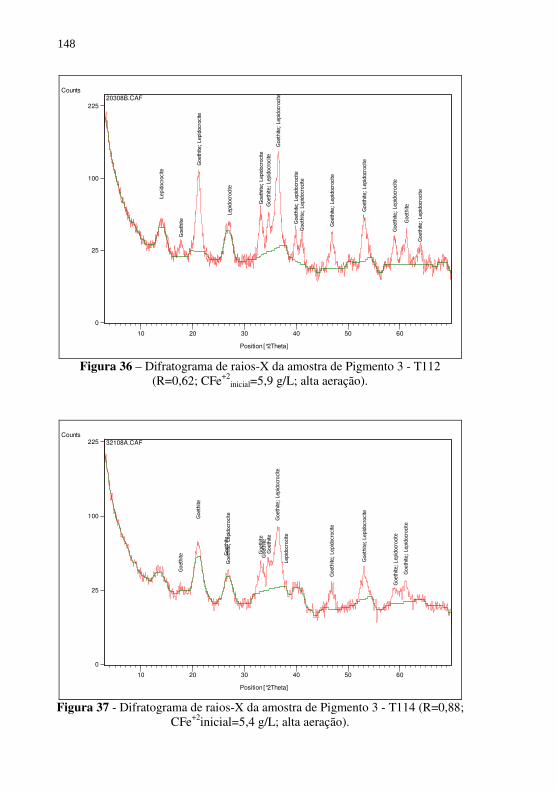
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Lepidocrocite
Goethite
Goethite; Lepidocrocite
Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite
Goethite; Lepidocrocite
20308B.CAF
Figura 36 – Difratograma de raios-X da amostra de Pigmento 3 - T112
(R=0,62; CFe+2inicial=5,9 g/L; alta aeração).
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Goethite
Goethite
Goethite
Goethite; Lepidocrocite
Goethite
Goethite
Goethite
Goethite; Lepidocrocite
Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
32108A.CAF
Figura 37 - Difratograma de raios-X da amostra de Pigmento 3 - T114 (R=0,88;
CFe+2inicial=5,4 g/L; alta aeração).
148
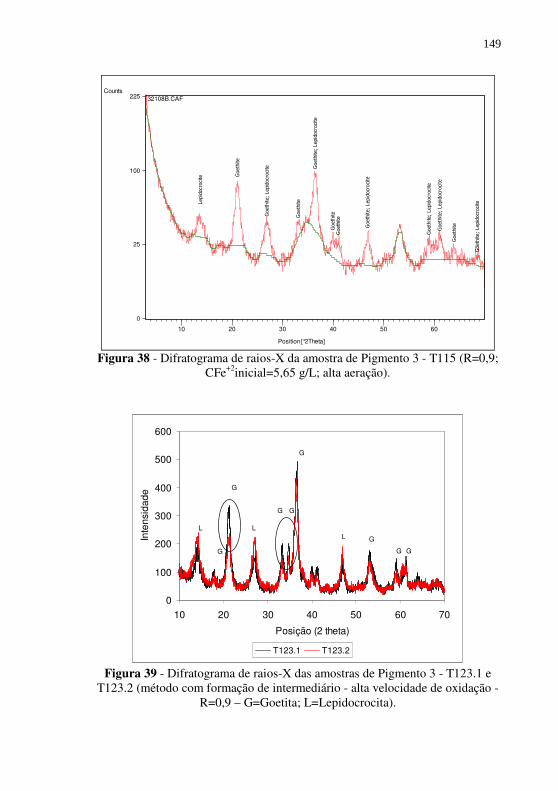
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Lepidocrocite Goethite
Goethite; Lepidocrocite
Goethite
Goethite; Lepidocrocite
Goethite
Goethite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite
Goethite; Lepidocrocite
32108B.CAF
Figura 38 - Difratograma de raios-X da amostra de Pigmento 3 - T115 (R=0,9;
CFe+2inicial=5,65 g/L; alta aeração).
0
100
200
300
400
500
600
10 20 30 40 50 60 70
Posição (2 theta)
Inte
nsid
ade
T123.1 T123.2
L LL
G
G
G
G G
G
G
G
Figura 39 - Difratograma de raios-X das amostras de Pigmento 3 - T123.1 e
T123.2 (método com formação de intermediário - alta velocidade de oxidação - R=0,9 – G=Goetita; L=Lepidocrocita).
149

0
100
200
300
400
500
600
10 20 30 40 50 60
Posição (2 theta)
Inte
nsid
ade
T123.1 T124.1
L
L
LG
G
G
G
G
G
G
Figura 40 - Difratograma de raios-X das amostras de Pigmento 3 - T123.1 e
T124.1 (método com formação de intermediário - alta velocidade de oxidação - R=0,9 – G=Goetita; L=Lepidocrocita).
Pelos difratogramas de raios-X, vê-se claramente a formação da
Goetita em todas as amostras, porém, para estas velocidades de introdução de ar na mistura, os picos da Lepidocrocita também apareceram.
Fica claro ao observarmos os difratogramas das amostras T112, 114 e 115 que a obtenção do pigmento com um valor de R mais próximo ao estequiométrico deixou a amostra pouca coisa mais cristalina. Ou seja, a amostra T112 esta mais cristalina do que as amostras T114 e T115. Porém, isto só pode ser afirmado para esta concentração inicial, esta temperatura e esta velocidade de introdução de ar na mistura.
Comparando-se as amostras T123.1 e T123.2, obtidas com diferentes velocidades de oxidação, (Figura 39) observa-se que aparecem claramente os picos da Goetita e da Lepidocrocita para ambas as amostras. Porém, a amostra obtida com menor velocidade de oxidação (T123.1) apresenta-se pouca coisa mais cristalina e com os picos da Goetita mais intensos. A melhora é pequena porque a diferença de velocidade entre as duas amostras é baixa. Ressalta-se ainda que os picos da Lepicocrocita sejam ressaltados nas amostras obtidas com maior velocidade de oxidação. Comportamento similar também pode ser
150

observado ao comparamos as amostras T123.1 e T124.1 (Figura 40). Ao se realizar estes testes, observou-se que analisando o ferro
ferroso no sobrenadante obtido após a mistura de FeSO4+NaOH não se consegue acompanhar a oxidação do Fe+2 haja vista que o mesmo mantém-se aproximadamente constante na fase líquida. Concluiu-se, portanto que deveria ser analisado o Fe+2 como sólido, conforme descrito na Tabela 29, e que este composto é quem estaria sendo oxidado para a formação dos produtos finais, ou seja, este seria o composto intermediário que após a oxidação iria se transformar nos produtos finais, sejam eles a Goetita, a Lepidocrocita ou até mesmo a Magnetita.
A Tabela 30 apresenta a caracterização das amostras obtidas nesta etapa do trabalho, bem como a caracterização de uma amostra comercial de pigmento amarelo.
Ao correlacionarmos a cristalinidade das amostras T112, 114 e 115 com os valores da perda ao fogo, (Tabela 30), observa-se que as amostras mais amorfas, (T114 e T115) apresentaram também uma perda ao fogo maior o que é outro indicativo de maior grau de amorficidade.
Com relação aos teores de Fe2O3, observa-se que os mesmos não estão suficientemente altos, (≥ 83%). Isto ocorre devido ao alto teor de sulfato ainda presente nas amostras. O teor de sulfato na amostra T112 esta extremamente alto, (9,7%). Ressalta-se que esta amostra não foi lavada previamente a caracterização. Também as amostras T114 e T115 não foram eficientemente lavadas, haja vista que os teores de sulfato encontrados nas mesmas, 3,3% e 3,7% ainda estão muito aquém da amostra comercial (0,4%).
A Tabela 30 apresenta ainda as análises colorimétricas das amostras preliminares de Pigmento 3. A diferença de cor de todas as amostras com relação a amostra comercial esta muito grande, (∆Eab >
10), mas ressalta-se que a amostra obtida com um valor de R mais próximo ao estequiométrico (T112) apresentou-se nitidamente mais escura. Isto pode ser explicado em partes pelo maior teor de manganês o qual esta associado a colorações mais escuras de amostras de óxidos de ferro.
151
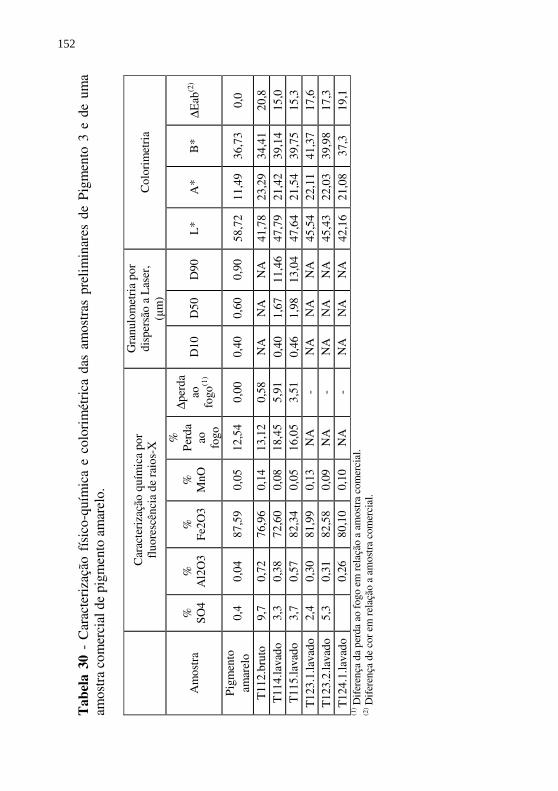
Tab
ela
30 -
Car
acte
riza
ção
físi
co-q
uím
ica
e co
lori
mét
rica
das
am
ostr
as p
reli
min
ares
de
Pig
men
to 3
e d
e um
a am
ostr
a co
mer
cial
de
pigm
ento
am
arel
o.
C
arac
teri
zaçã
o qu
ímic
a po
r fl
uore
scên
cia
de r
aios
-X
Gra
nulo
met
ria
por
disp
ersã
o a
Las
er,
(µm
) C
olor
imet
ria
Am
ostr
a %
S
O4
%
Al2
O3
%
Fe2
O3
%
MnO
%
Per
da
ao
fogo
∆pe
rda
ao
fogo
(1)
D10
D
50
D90
L
* A
* B
* ∆
Eab
(2)
Pig
men
to
amar
elo
0,4
0,04
87
,59
0,05
12
,54
0,00
0,
40
0,60
0,
90
58,7
2 11
,49
36,7
3 0,
0
T11
2.br
uto
9,7
0,72
76
,96
0,14
13
,12
0,58
N
A
NA
N
A
41,7
8 23
,29
34,4
1 20
,8
T11
4.la
vado
3,
3 0,
38
72,6
0 0,
08
18,4
5 5,
91
0,40
1,
67
11,4
6 47
,79
21,4
2 39
,14
15,0
T
115.
lava
do
3,7
0,57
82
,34
0,05
16
,05
3,51
0,
46
1,98
13
,04
47,6
4 21
,54
39,7
5 15
,3
T12
3.1.
lava
do
2,4
0,30
81
,99
0,13
N
A
- N
A
NA
N
A
45,5
4 22
,11
41,3
7 17
,6
T12
3.2.
lava
do
5,3
0,31
82
,58
0,09
N
A
- N
A
NA
N
A
45,4
3 22
,03
39,9
8 17
,3
T12
4.1.
lava
do
0,
26
80,1
0 0,
10
NA
-
NA
N
A
NA
42
,16
21,0
8 37
,3
19,1
(1
) Dif
eren
ça d
a pe
rda
ao f
ogo
em r
elaç
ão a
am
ostr
a co
mer
cial
. (2
) Dif
eren
ça d
e co
r em
rel
ação
a a
mos
tra
com
erci
al.
152

Com relação a granulometria, observa-se que as amostras de Pigmento 3, assim como as amostras de Pigmento 2, tem uma granulometria muito pouco uniforme e muito maior do que a amostra comercial. Também como no caso do Pigmento 2, esta distribuição granulométrica diferente é um dos principais fatores responsáveis pela diferença de cor entre as amostras. Para a obtenção de uma granulometria mais uniforme e menor (d90 < 1µm), é necessário aprimorar a etapa de moagem antes da caracterização. Isto será realizado no final desta etapa do trabalho após a definição das condições operacionais ótimas, para obtenção da Goetita partindo-se de uma drenagem ácida de mina com 4g/L de Fe+2.
6.3.2.2 Método com formação de intermediário – Obtenção do núcleo
de Goetita Foram realizados 6 testes em escala de laboratório reproduzindo
as mesmas condições experimentais descritas por Meisen, 2004 para a obtenção do núcleo de Goetita. A partir deste núcleo e do Sobrenadante 1, realizar-se-ia a conseguinte produção do pigmento amarelo conforme descrito na literatura.
Nos testes T116 ao T118, foi utilizado o método alcalino para a obtenção do núcleo. Neste caso, utilizou-se o mesmo valor de R para as três amostras, ou seja, R=0,33 e variou-se a concentração inicial de Fe+2 na solução e a temperatura da reação de oxidação (Tabela 31).
Nos testes T119 ou T121, foi utilizado o método ácido para a produção do núcleo. Neste caso, R=1,30 para todos os testes e variou-se a concentração inicial de ferro ferroso e a temperatura da reação (Tabela 31).
As condições experimentais dos testes para a produção do núcleo de Goetita estão descritas na Tabela 31 e os difratogramas de raios-X das amostras produzidas pelo método alcalino e pelo método ácido estão apresentados nas Figuras 41 a 46. Na Figura 47 é apresentado o difratograma de raios-X da amostra comercial de pigmento amarelo.
153

Tabela 31 - Condições experimentais dos testes realizados para a produção do núcleo de Goetita.
Método de
produção do núcleo
Teste Valor de
R cinicial Fe+2
solução, mg/l (1)
Temperatura reação
oxidação, 0c
Método alcalino
T116 0,33
42,2 400C T117 42,2 220C T118 7,00 220C
Método ácido
T119 1,30
24,0 400C T120 24,0 220C T121 9,50 220C
(1) Solução inicial de sulfato ferroso. (2) Aeração de 0,4(L/h)/gFe+2; Tempo adição soda = 30 minutos; tempo reação ≅8,5 horas.
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
Goethite
Goethite
Goethite
Magnetite
Goethite
Goethite
Goethite; Magnetite Goethite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
Magnetite
Goethite
Goethite
Goethite
Goethite
32108C.CAF
Figura 41 – Difratograma de raios-X da amostra de núcleo alcalino de Goetita -
T116 (R=0,33; CFe+2inicial=42 g/L; T=400C).
154

Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
GoethiteGoethite
Goethite
Goethite Goethite
30208A.CAF
Figura 42 - Difratograma de raios-X da amostra de núcleo alcalino de Goetita -
T117 (R=0,33; CFe+2inicial=42 g/L; T=220C).
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
30208B.CAF
Figura 43 - Difratograma de raios-X da amostra de núcleo alcalino de Goetita -
T118 (R=0,33; CFe+2inicial=7 g/L; T=220C).
155

Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Goethite
Goethite
Goethite
Magnetite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
MagnetiteGoethite
Goethite
Goethite Goethite
32108D.CAF
Figura 44 - Difratograma de raios-X da amostra de núcleo ácido de Goetita -
T119 (R=1,30; CFe+2inicial=24 g/L;T=400C).
Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
Goethite
Goethite
Goethite
Magnetite
Goethite
Goethite
Goethite; MagnetiteGoethite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite; Magnetite
Goethite
Goethite
Magnetite
Goethite
Goethite
Goethite
32108E.CAF
Figura 45 - Difratograma de raios-X da amostra de núcleo ácido de Goetita -
T120 (R=1,30; CFe+2inicial=24 g/L;T=250C).
156

Position [°2Theta]
10 20 30 40 50 60
Counts
0
25
100
225
Lepidocrocite
Goethite
Goethite
Lepidocrocite
Goethite
Goethite
Goethite; Lepidocrocite
Goethite
Goethite
Goethite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
Goethite
Goethite; Lepidocrocite
Goethite; Lepidocrocite
30208C.CAF
Figura 46 - Difratograma de raios-X da amostra de núcleo ácido de Goetita -
T121 (R=1,3; CFe+2inicial=9,5 g/L; T=ambiente).
Position [°2Theta]
10 20 30 40 50 60
Counts
0
100
400
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
Goethite
GoethiteGoethite
Goethite
Goethite
Goethite
18308C.CAF
Figura 47 - Difratograma de raios-X da amostra de pigmento amarelo
comercial.
157

Avaliando-se o método alcalino utilizado para a produção do núcleo de Goetita, observa-se, para a mesma concentração inicial de ferro ferroso, (42g/L) e diferente temperatura de reação, (400C versus 220C), que o pigmento obtido pelo método alcalino em temperaturas maiores apresenta traços da Magnetita, o que não ocorre com aquele obtido em temperatura ambiente (Figuras 41 e 42). Isto esta plenamente de acordo com a literatura que descreve que em valores de R alcalinos, quanto maior a temperatura da reação, maior a probabilidade de formação da Magnetita frente a Goetita (Olowe, et. al, 1991.b).
Comparando-se agora a cristalinidade da amostra obtida pelo método alcalino de produção do núcleo com a cristalinidade do pigmento comercial, (T117 versus amostra comercial), observa-se que esta amostra é pouca coisa menos cristalina do que a amostra comercial. Ressalta-se ainda que para R=0,33, Ci=42g/L e temperatura ambiente somente aparecem os picos da Goetita.
Ao avaliarmos o efeito da redução da concentração inicial de ferro na solução no método alcalino, (T117 x T118), observa-se que a mesma praticamente não influenciou na cristalinidade da amostra obtida. Ou seja, a aplicação do método alcalino com uma concentração inicial baixa, (≈7g/L) não deixou a amostra amorfa e não apareceram os picos da Lepidocrocita.
Avaliando-se agora o método ácido de produção do núcleo, para as amostras T119 x T120, (mesma concentração inicial e diferente temperatura), observa-se que apareceram os picos da Goetita+Magnetita nas duas amostras, porém, ao contrário do que ocorre no método alcalino, no método ácido o pico da magnetita é mais sentido na amostra produzida em temperatura ambiente. Ou seja, para a produção da Goetita frente a Magnetita no método ácido, é preferível trabalhar-se com temperaturas elevadas.
Ressalta-se ainda que se realizarmos a oxidação no tempo aproximadamente igual ao citado na literatura, ou seja, entre 3 e 4 horas, há pouca oxidação da amostra e sobra muito Fe+2 ainda na mistura. Ou seja, o núcleo ácido de Goetita não necessariamente é composto apenas por Fe+3 como era o esperado.
Já a redução da concentração inicial de ferro ferroso no método ácido ocasionou uma drástica redução na cristalinidade da amostra de Goetita produzida (T121 x T120) e, além disso, no método ácido com baixa concentração inicial de ferro ferroso, ressalta-se 9,5g/L, apareceram os picos da Lepidocrocita o que não ocorre no método alcalino.
158

Vale lembrar que ao reduzirmos a concentração inicial de ferro ferroso e mantendo-se a mesma velocidade de introdução de ar na mistura, estaremos consequentemente aumentando a relação [(L/min)ar/gFe+2
sólido] e, tendo mais oxigênio disponível na mistura, a oxidação do composto intermediário para a formação dos produtos finais ocorrerá mais rapidamente. Foi visto no item anterior que maiores velocidades de oxidação aumentam a formação da Lepidocrocita frente a Goetita. Fica claro, portanto, que no método ácido a temperatura ambiente e concentração inicial baixa correm-se os riscos de termos uma amostra mais amorfa e/ou “contaminada” com cristais de Lepidocrocita.
A Tabela 32 apresenta a caracterização das amostras de núcleo obtidas assim como a caracterização da amostra comercial de pigmento amarelo.
Pela Tabela 32 observa-se que tanto no método ácido quanto no método alcalino de produção do núcleo, as amostras produzidas com maior velocidade de oxidação, (T117 e T120 x as amostras T116 e T119, respectivamente), apresentam um menor tamanho de partículas. Para a mesma velocidade de introdução de ar na mistura e a mesma concentração inicial de ferro ferroso, as amostras produzidas em temperaturas menores terão maior eficiência na transferência do oxigênio da fase gasosa para a fase líquida, devido a maior solubilidade do oxigênio, consequentemente maior velocidade de oxidação.
159

Tab
ela
32 -
Car
acte
riza
ção
físi
co-q
uim
ica
das
amos
tras
de
núcl
eo d
e G
oetit
a pr
oduz
idas
pel
o m
étod
o al
calin
o e
mét
odo
ácid
o.
Mét
odo
de
prod
ução
do
núcl
eo
Am
ostr
a %
SO
4 %
Fe
2O3
Col
orim
etri
a D
ispe
rsão
a L
aser
G
ranu
lom
etri
a (µ
m)
L*
A*
B*
∆E
ab
D10
D
50
D90
P
igm
ento
am
arel
o 0,
40
87,6
59
,2
15,4
6 47
,88
0,0
0,40
0,
60
0,90
Mét
odo
alca
lino
T11
6 0,
17
87,0
31
,34
4,33
16
,71
43,2
6 0,
41
1,81
8,
81
T11
7 0,
20
88,0
44
,36
7,55
26
,74
27,0
1 0,
31
0,94
3,
45
T11
8 0,
10
82,0
46
,63
14,2
8 39
,38
15,2
2 0,
70
2,23
8,
87
Mét
odo
ácid
o
T11
9 0,
50
77,0
40
,49
15,8
8 30
,73
25,3
8 0,
67
1,99
12
,62
T12
0 0,
50
80,0
33
,72
9,80
22
,42
36,4
6 0,
52
1,92
9,
24
T12
1 0,
40
72,0
47
,94
22,7
6 42
,16
14,5
9 0,
6 2,
65
13,2
4 C
om r
elaç
ão a
cor
não
é c
orre
to c
orre
laci
onar
mos
os
valo
res
de L
*a*b
* da
s am
ostr
as d
e nú
cleo
com
a
amos
tra
com
erci
al, h
aja
vist
a qu
e no
tem
po t
otal
de
oxid
ação
do
mét
odo
para
a p
rodu
ção
do n
úcle
o, (
tant
o ác
ido
quan
to a
lcal
ino)
, as
amos
tras
pro
duzi
das
não
esta
vam
com
plet
amen
te o
xida
das
e o
inte
rmed
iári
o da
pro
duçã
o de
G
oetit
a, s
eja
ele;
o F
e(O
H) 2
ou
GR
2, a
inda
est
ava
pres
ente
na
amos
tra.
160

Resumindo em tópicos as conclusões principais deste item e do item anterior de produção do pigmento com formação de intermediário, pode-se dizer que:
- No método ácido, alta velocidade de introdução de oxigênio na mistura e, consequentemente alta velocidade de oxidação do composto intermediário para obtenção dos produtos finais, pode levar a formação da Lepidocrocita juntamente com a Goetita.
- Para que a reação de oxidação seja direcionada para a formação preferencial da Goetita, no método ácido, é recomendado que sejam aumentadas a temperatura da reação ou a concentração inicial de ferro ferroso na solução ou ainda que seja reduzida a velocidade de introdução de ar na mistura. Com uma, ou outra destas ações estar-se-á reduzindo a relação (L/min)ar/gFe+2
sólido e consequentemente reduzir-se-á a velocidade da reação de oxidação.
- Para uma concentração inicial entre 6,0 e 7,0, g/L de ferro ferroso, trabalhando-se com R mais próximo ao estequiométrico, a amostra obtida torna-se mais cristalina do que quando se trabalha com R ácido. Porém, partindo-se da amostra de drenagem ácida de mina pré-tratada, ou seja, do Sobrenadante 1, o pigmento produzido nesta condição poderá tornar-se mais escuro em função dos contaminantes existentes da DAM, principalmente do manganês.
- No método alcalino com alta concentração de ferro ferroso, a formação da Goetita pode ser acompanhada da formação indesejada da Magnetita. Para que a reação de oxidação do composto intermediário seja direcionada para os cristais de Goetita, é preferível trabalhar-se com temperaturas de oxidação menores.
A partir dessas condições gerais, é ainda necessário avaliar o
efeito da concentração inicial de ferro e a influência do valor de R para cada concentração inicial, uma vez que a concentração de ferro no Sobrenadante 1 pode variar em torno de 3 a 4 g/L e este efeito deve ser avaliado antes da aplicação do processo em escala industrial, como será descrito a seguir.”
Método com formação de intermediário – Avaliação do valor de R versus a concentração inicial de Fe+2
Foram realizados vários testes a partir da mistura do
161

Sobrenadante 1 (obtido na planta industrial) com NaOH e, em alguns casos, suprimento adicional de sulfato ferroso. O objetivo destes testes era determinar quais as condições ótimas para a obtenção da Goetita a partir do Sobrenadante 1 bem como avaliar a influência do valor de R e da concentração inicial de ferro ferroso na produção da Goetita.
Trabalhou-se com R na faixa do estequiométrico, ou seja, R ≅ 0,5 a 0,6 mol Fe+2/mol OH-, e valores de R ácidos, ou seja, R ≥ 0,9. A aeração foi mantida baixa haja vista que alta relação [(L/min)ar/gFe+2sólido] aumenta a probabilidade de formação da Lepidocrocita. Abaixo estão apresentados os testes, separados em método alcalino a estequiométrico e método ácido.
Avaliação do valor de R na faixa do alcalino ao estequiométrico
Conforme a literatura, para a obtenção de Goetita a partir da
mistura FeSO4+NaOH em temperatura ambiente, os valores de R ótimos situam-se entre 0,20 e 0,40, para o método alcalino e entre 0,565 e 0,580 para a região do estequiométrico. O valor ótimo encontrado para a obtenção da Goetita foi exatamente igual a 0,5714 com concentração inicial de ferro ferroso de 5,6 g/L entretanto, com R=0,5833 ocorre preferencialmente a formação da Lepidocrocita (Olowe, et. al, 1991).
Em função disto foram realizados vários testes utilizando-se estas faixas de valores para R e variando-se a concentração inicial de ferro ferroso presente na solução aquosa.
Partiu-se sempre de um teste com concentração inicial igual a do Sobrenadante 1, ou seja entre 3,0 e 4,0 g/L e suprimento de FeSO4 adicional para valores maiores do que isto.
A Tabela 33 apresenta os parâmetros dos testes realizados e as Figuras 48 a 53 apresentam os difratogramas de raios-X das amostras obtidas.
162

Tab
ela
33 -
Con
diçõ
es e
xper
imen
tais
dos
tes
tes
real
izad
os p
ara
aval
iaçã
o do
val
or d
e R
ver
sus
a co
ncen
traç
ão
inic
ial d
e fe
rro
ferr
oso
na r
egiã
o do
alc
alin
o ao
est
equi
omét
rico
.
TE
STE
F
e+2 in
icia
l, m
g/L
(s
oluç
ão)
R
pH
soda
(1
)
Fe+
2 in
icia
l,
mg/
L (
sólid
o)
Ar,
(L
/min
)/gF
e+2 só
l. pH
após
oxi
daçã
o R
elaç
ão
Al+
3 /gF
e+2
(2)
137
am.1
2.
960,
1 0,
33>
0,3
3 12
,04
4.83
7,2
0,03
9,
68
0,03
14
0 am
.1
6.76
8,7
12,5
0 9.
006,
2 0,
03
11,9
2 0,
04
136
am.1
6.
031,
0 11
,31
30.8
57,7
0,
01
4,40
0,
01
140
am.2
6.
768,
7 ≅
0,5
0 8,
02
8.92
5,7
0,03
4,
19
0,04
13
7 am
.2
2.96
0,1
7,73
3.
322,
0 0,
03
4,84
0,
03
142
am.1
6.
445,
1 0,
5714
> 0
,571
4 8,
50
11.5
72,2
0,
03
3,73
0,
01
142
am.3
4.
456,
8 8,
34
8.16
7,5
0,03
3,
66
0,02
12
9.am
.1
2.95
6,3
7,60
3.
735,
8 0,
01
3,70
0,
05
(1)
O v
olum
e de
sod
a a
ser
adic
iona
do f
oi c
alcu
lado
em
fun
ção
da c
once
ntra
ção
inic
ial d
e fe
rro
ferr
oso
e da
con
cent
raçã
o in
icia
l de
soda
. O
bjet
ivan
do-s
e o
valo
r de
R e
stip
ulad
o. E
ste
valo
r de
pH
é o
pH
obt
ido
na m
istu
ra a
pós
a ad
ição
do
volu
me
calc
ulad
o.
(2)
Rel
ação
exi
sten
te e
ntre
o a
lum
ínio
e o
fer
ro f
erro
so n
a so
luçã
o in
icia
l (So
bren
adan
te 1
e/o
u So
bren
adan
te 1
+ F
eSO
4) u
tiliz
ada
para
a o
bten
ção
do
lodo
ver
de in
term
ediá
rio.
163

R alcalino = 0,33
0
20
40
60
80
100
120
140
160
180
200
10 15 20 25 30 35 40 45 50 55 60 65 70
Posição (2 theta)
Inte
nsid
ade
T137 - Ci=3g/L T140 - Ci=6,8g/L
M, H
M, HM
H
Figura 48 - Difratogramas de raios-X da amostra de Pigmento 3 - T137 (Cinicial = 2,9g/L, Fe+2
sólido inicial= 4,8g/L) e T140 (Cinicial = 6,8 g/L, Fe+2sólido inicial= 9,0g/L)
ambas produzidas com R=0,33. M=Magnetita; H=Hematita.
R alcalino = 0,33
1100
1600
2100
2600
3100
3600
4100
4600
10 14 18 22 26 30 34 38 42 46 50 54 58 62 66 70
Posição (2 theta)
Inte
nsid
ade
T136 - Ci=6g/L
M
M
M
M
M
M
M
M
Figura 49 - Difratograma de raios-X da amostra de Pigmento 3 - T136 (Cinicial =
6,0 g/L, Fe+2sólido inicial =31g/L) produzida com R ≅ 0,33.
164
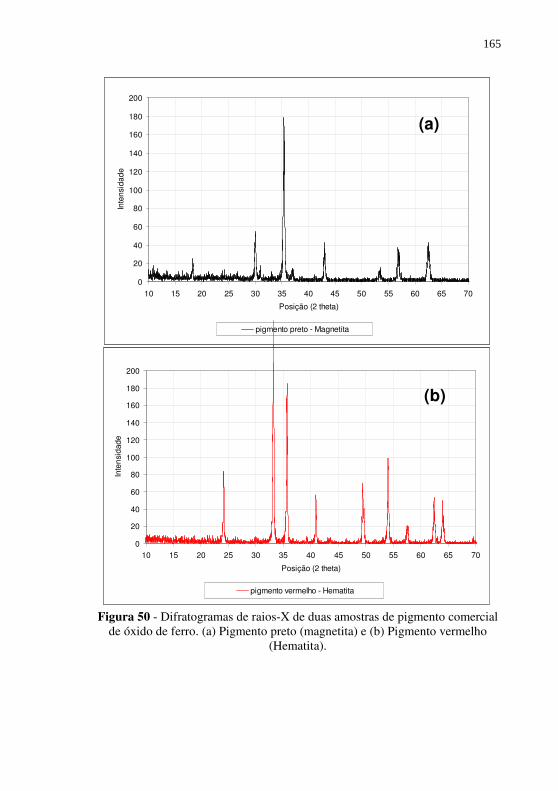
0
20
40
60
80
100
120
140
160
180
200
10 15 20 25 30 35 40 45 50 55 60 65 70
Posição (2 theta)
Inte
nsid
ade
pigmento preto - Magnetita
0
20
40
60
80
100
120
140
160
180
200
10 15 20 25 30 35 40 45 50 55 60 65 70
Posição (2 theta)
Inte
nsi
dade
pigmento vermelho - Hematita
(a)
(b)
0
20
40
60
80
100
120
140
160
180
200
10 15 20 25 30 35 40 45 50 55 60 65 70
Posição (2 theta)
Inte
nsid
ade
pigmento preto - Magnetita
0
20
40
60
80
100
120
140
160
180
200
10 15 20 25 30 35 40 45 50 55 60 65 70
Posição (2 theta)
Inte
nsi
dade
pigmento vermelho - Hematita
(a)
(b)
Figura 50 - Difratogramas de raios-X de duas amostras de pigmento comercial
de óxido de ferro. (a) Pigmento preto (magnetita) e (b) Pigmento vermelho (Hematita).
165
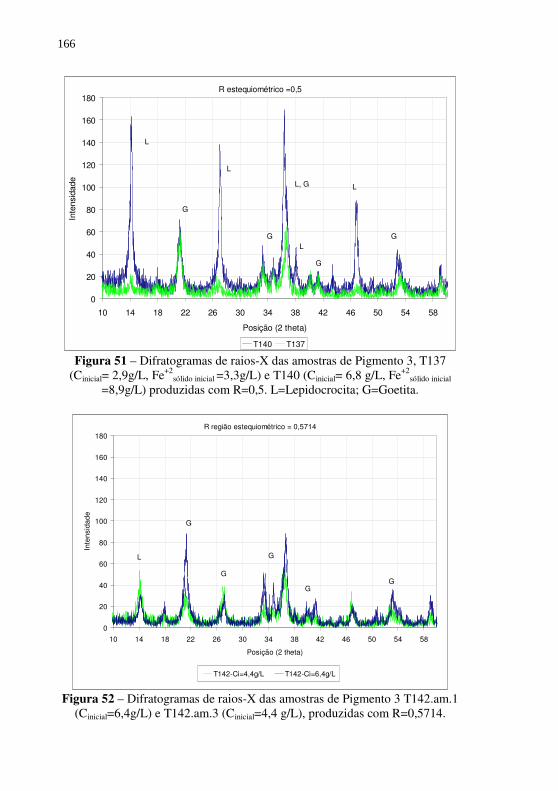
R estequiométrico =0,5
0
20
40
60
80
100
120
140
160
180
10 14 18 22 26 30 34 38 42 46 50 54 58
Posição (2 theta)
Inte
nsid
ade
T140 T137
G
L
L
L, G
G
L
G
L
G
Figura 51 – Difratogramas de raios-X das amostras de Pigmento 3, T137
(Cinicial= 2,9g/L, Fe+2sólido inicial =3,3g/L) e T140 (Cinicial= 6,8 g/L, Fe+2
sólido inicial
=8,9g/L) produzidas com R=0,5. L=Lepidocrocita; G=Goetita.
R região estequiométrico = 0,5714
0
20
40
60
80
100
120
140
160
180
10 14 18 22 26 30 34 38 42 46 50 54 58
Posição (2 theta)
Inte
nsid
ade
T142-Ci=4,4g/L T142-Ci=6,4g/L
GG
G
G
G
L
Figura 52 – Difratogramas de raios-X das amostras de Pigmento 3 T142.am.1
(Cinicial=6,4g/L) e T142.am.3 (Cinicial=4,4 g/L), produzidas com R=0,5714.
166
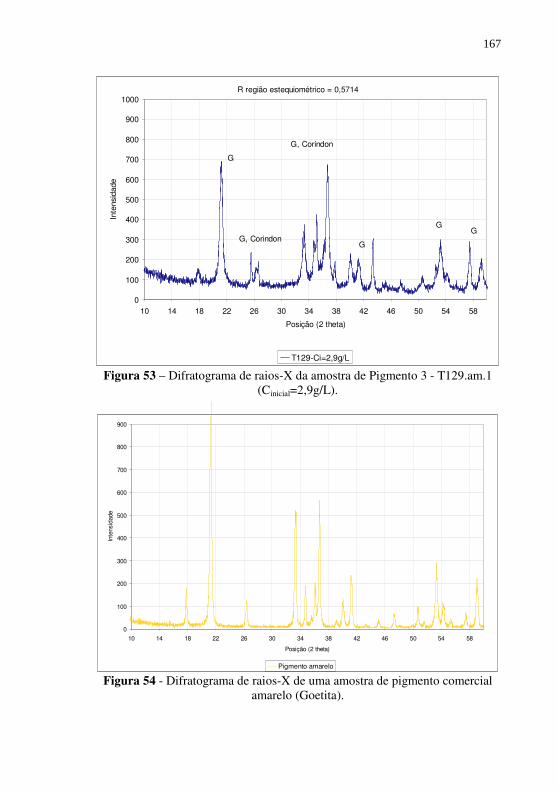
R região estequiométrico = 0,5714
0
100
200
300
400
500
600
700
800
900
1000
10 14 18 22 26 30 34 38 42 46 50 54 58
Posição (2 theta)
Inte
nsid
ade
T129-Ci=2,9g/L
GG
G
G, Corindon
G, Corindon
G
Figura 53 – Difratograma de raios-X da amostra de Pigmento 3 - T129.am.1
(Cinicial=2,9g/L).
0
100
200
300
400
500
600
700
800
900
10 14 18 22 26 30 34 38 42 46 50 54 58
Posição (2 theta)
Inte
nsid
ade
Pigmento amarelo Figura 54 - Difratograma de raios-X de uma amostra de pigmento comercial
amarelo (Goetita).
167

No método alcalino, a temperatura ambiente com R=0,33 e concentrações iniciais de 2,9 e 6,8 g/L de Fe+2, (Figura 48) obtiveram-se amostras muito pouco cristalinas e apareceram apenas os picos da Hematita e da Magnetita. Além disso, a amostra produzida com concentração inicial maior, (T140) apresentou-se pouca coisa mais cristalina do que aquela cuja concentração inicial era menor (Figura 48 e Tabela 33).
Já no teste T136, também no método alcalino e concentração inicial ≅ 6g/L e R≅0,33, obteve-se, surpreendentemente, uma única fase bem cristalina de Magnetita (Figura 49). A principal diferença entre as amostras T136 e T140 esta no valor da concentração inicial de ferro ferroso como sólido ao inicial a reação de oxidação. No caso da amostra T136, após a adição de soda no Sobrenadante1+FeSO4, o lodo verde intermediário produzido foi mantido em repouso por aproximadamente 24horas para que houvesse o espessamento do mesmo. Daí, após ser alcançada a concentração inicial de 31 g/L de ferro ferroso como sólido a oxidação para a obtenção dos produtos finais foi iniciada.
Mantendo-se as mesmas concentrações iniciais de ferro ferroso na solução aquosa e aumentando-se o valor de R dos testes T137 e T140, até a faixa do estequiométrico, ou seja, com R≅0,5, as amostras produzidas tornaram-se bem mais cristalinas do que aquelas obtidas com R=0,33 e neste caso, apareceram somente os picos da Goetita e da Lepidocrocita (Figura 51 versus Figura 48).
Ao confrontarmos as amostras T140 e T137, (Figura 51) produzidas com R=0,5 e concentração inicial diferente, observa-se que a amostra T140 teve uma grande formação da Lepidocrocita. Isto pode ser justificado pelo maior valor do pH após a adição da soda calculada e principalmente pela maior velocidade de oxidação do composto intermediário (Figura 55, abaixo).
O valor do pH da amostra T140 com R≅0,5 (Tabela 33) esta mais alto do que o pH da amostra T137 (pH=8,0 versus pH=7,7). Este valor diferente de pH demonstra que na realidade o valor de R das duas amostras não ficou exatamente igual e no caso da amostra T140, o valor do R aproximou-se mais do alcalino.
Com relação a velocidade de oxidação, a Figura 55 demonstra que mesmo que a vazão de ar adicionada tenha sido a mesma, ou seja, 0,03 (L/min)ar/gFe+2
sólido, a velocidade de oxidação da amostra T140 foi substancialmente maior do que aquela obtida para a amostra T137, justificando daí a formação preferencial da Lepidocrocita.
168
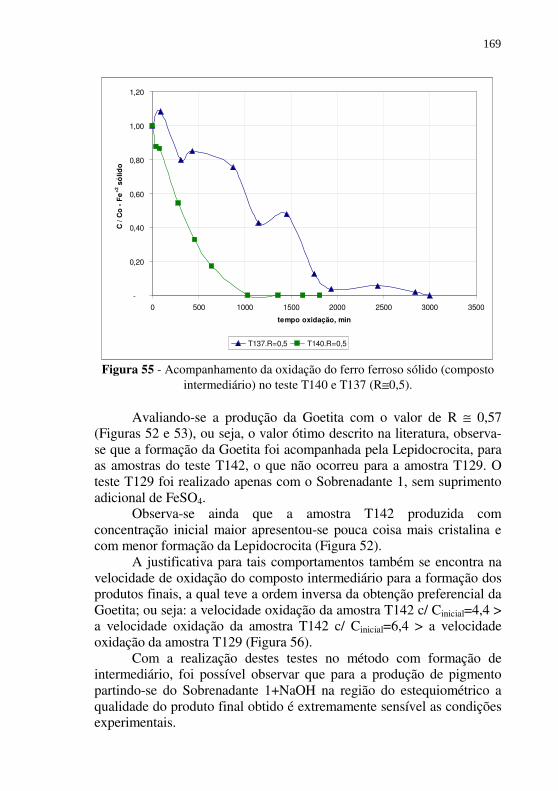
-
0,20
0,40
0,60
0,80
1,00
1,20
0 500 1000 1500 2000 2500 3000 3500
tempo oxidação, min
C /
Co
- F
e+2
só
lido
T137.R=0,5 T140.R=0,5
Figura 55 - Acompanhamento da oxidação do ferro ferroso sólido (composto
intermediário) no teste T140 e T137 (R≅0,5). Avaliando-se a produção da Goetita com o valor de R ≅ 0,57
(Figuras 52 e 53), ou seja, o valor ótimo descrito na literatura, observa-se que a formação da Goetita foi acompanhada pela Lepidocrocita, para as amostras do teste T142, o que não ocorreu para a amostra T129. O teste T129 foi realizado apenas com o Sobrenadante 1, sem suprimento adicional de FeSO4.
Observa-se ainda que a amostra T142 produzida com concentração inicial maior apresentou-se pouca coisa mais cristalina e com menor formação da Lepidocrocita (Figura 52).
A justificativa para tais comportamentos também se encontra na velocidade de oxidação do composto intermediário para a formação dos produtos finais, a qual teve a ordem inversa da obtenção preferencial da Goetita; ou seja: a velocidade oxidação da amostra T142 c/ Cinicial=4,4 > a velocidade oxidação da amostra T142 c/ Cinicial=6,4 > a velocidade oxidação da amostra T129 (Figura 56).
Com a realização destes testes no método com formação de intermediário, foi possível observar que para a produção de pigmento partindo-se do Sobrenadante 1+NaOH na região do estequiométrico a qualidade do produto final obtido é extremamente sensível as condições experimentais.
169
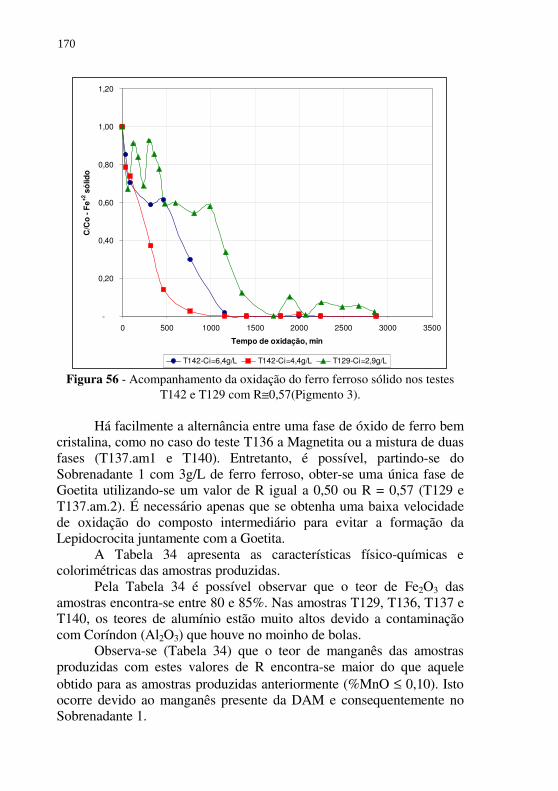
-
0,20
0,40
0,60
0,80
1,00
1,20
0 500 1000 1500 2000 2500 3000 3500
Tempo de oxidação, min
C/C
o -
Fe+2
só
lido
T142-Ci=6,4g/L T142-Ci=4,4g/L T129-Ci=2,9g/L
Figura 56 - Acompanhamento da oxidação do ferro ferroso sólido nos testes T142 e T129 com R≅0,57(Pigmento 3).
Há facilmente a alternância entre uma fase de óxido de ferro bem
cristalina, como no caso do teste T136 a Magnetita ou a mistura de duas fases (T137.am1 e T140). Entretanto, é possível, partindo-se do Sobrenadante 1 com 3g/L de ferro ferroso, obter-se uma única fase de Goetita utilizando-se um valor de R igual a 0,50 ou R = 0,57 (T129 e T137.am.2). É necessário apenas que se obtenha uma baixa velocidade de oxidação do composto intermediário para evitar a formação da Lepidocrocita juntamente com a Goetita.
A Tabela 34 apresenta as características físico-químicas e colorimétricas das amostras produzidas.
Pela Tabela 34 é possível observar que o teor de Fe2O3 das amostras encontra-se entre 80 e 85%. Nas amostras T129, T136, T137 e T140, os teores de alumínio estão muito altos devido a contaminação com Coríndon (Al2O3) que houve no moinho de bolas.
Observa-se (Tabela 34) que o teor de manganês das amostras produzidas com estes valores de R encontra-se maior do que aquele obtido para as amostras produzidas anteriormente (%MnO ≤ 0,10). Isto ocorre devido ao manganês presente da DAM e consequentemente no Sobrenadante 1.
170

Tab
ela
34 -
Car
acte
riza
ção
físi
co-q
uim
ica
e co
lori
mét
rica
das
am
ostr
as d
e P
igm
ento
3 p
rodu
zida
s co
m R
na
regi
ão d
o al
calin
o ao
est
equi
omét
rico
. Car
acte
riza
ção
tam
bém
de
amos
tras
de
pigm
ento
com
erci
ais.
Am
ostr
a
Car
acte
riza
ção
quím
ica
por
Flu
ores
cênc
ia d
e ra
ios-
X
Col
orim
etri
a G
ranu
lom
etri
a po
r di
sper
são
a L
aser
, (µ
m)
%
Al 2
O3
%
Fe 2
O3
%
MnO
%
per
da
ao f
ogo
L*
A*
B*
∆ ∆∆∆E
ab (1
) d
10%
d
50%
d
90%
Pig
men
to a
mar
elo
0,04
87
,59
0,05
12
,15
59,2
15
,46
47,8
8 -
0,4
- 0,
85
Pig
men
to p
reto
E
lem
ento
maj
orit
ário
: Fe
12,9
7 -1
,25
2,58
66
,85
NA
N
A
NA
P
igm
ento
ver
mel
ho
0,09
97
,9
0,35
1,
23
27,2
3 24
,87
17,3
1 45
,22
0,13
0,
44
1,97
T
137
am.1
(R
0,3
3)
2,48
(3)
71,8
3 1,
24
12,3
5 28
,52
15,7
5 20
,37
41,2
1 0,
49
1,9
8,13
T
140
am.1
(R 0
,33)
1,
27 (3
) 87
,57
0,84
5,
14
19,1
8 13
,4
12,3
6 53
,55
NA
N
A
NA
T
136
am.1
(R 0
,57)
19
,24
(3)
69,2
8 0,
44
2,42
17
,45
2,08
4,
77
6,00
(2)
0,64
2,
95
13,0
5 T
140
am.2
(R 0
,50)
1,
87 (3
) 85
,38
0,36
11
,38
30,2
6 18
,06
24,5
1 37
,29
NA
N
A
NA
T
137
am.2
(R 0
,50)
2,
37 (3
) 79
,85
0,45
15
,14
37,3
6 19
,54
31,0
5 27
,87
1,26
3,
7 6,
83
T14
2 am
.1(R
0,5
7)
0,07
85
,26
0,38
13
,47
43,4
7 19
,72
35,6
2 20
,39
NA
N
A
NA
T
142
am.3
(R 0
,57)
0,
09
84,9
5 0,
39
13,4
5 37
,91
21,0
5 31
,35
27,5
3 N
A
NA
N
A
T12
9.am
.1(R
0,5
7)
22,9
4 (3
) 55
,19
0,23
11
,04
44,7
5 19
,90
40,0
3 17
,03
0,68
5,
02
24,4
3 D
ifer
ença
de
cor
em r
elaç
ão a
am
ostr
a co
mer
cial
am
arel
a. (
2) D
ifer
ença
de
cor
em r
elaç
ão a
am
ostr
a co
mer
cial
pre
ta. (
3) C
onta
min
ação
da
amos
tra
com
Cor
índo
n du
rant
e a
moa
gem
.
Tam
bém
a c
or d
as a
mos
tras
est
a m
uito
aqu
ém d
aque
la d
esej
ada
para
a a
mos
tra
com
erci
al d
e pi
gmen
to
amar
elo;
ent
reta
nto,
a a
mos
tra
T13
6 te
m v
alor
es d
e L
*a*b
* m
uito
pró
xim
os d
a am
ostr
a co
mer
cial
de
pigm
ento
pr
eto
(Mag
netit
a). R
essa
lta-s
e po
rém
que
a g
ranu
lom
etri
a nã
o fo
i aju
stad
a de
aco
rdo
com
a a
mos
tra
com
erci
al.
171

Avaliação do valor de R na faixa de R ácido Foi avaliada a obtenção da Goetita a partir da mistura de
Sobrenadante 1+ NaOH a temperatura ambiente no método ácido. Os valores de R estudados foram R ≅ 1,0; 2,0; 2,5 e 3,0 molFe+2/mol NaOH.
Variou-se novamente a concentração inicial de ferro ferroso presente na solução aquosa partindo-se de um teste com concentração inicial igual ao Sobrenadante 1 para valores maiores do que isto. A velocidade de oxidação foi mantida baixa (≅ 0,03 (L/min)/gFe+2
sól.) a fim de evitar a formação da Lepidocrocita.
Tabela 35 - Condições experimentais dos testes realizados para avaliação do valor de R versus a concentração inicial de ferro ferroso no método ácido.
TESTE Solução inicial
Fe+2inicial,
mg/L (solução)
R pH
NaOH
Fe+2inicial,
mg/L (sólido)
Ar (L/min) /
g Fe+2
pH final
135 am. 1 Sob.1 4.003,0
2,00 6,39 4.414,5 0,034 4,00 135 am. 3 1,00 7,65 5.220,7 0,027 3,85 138 am. 1
Sob.1 3.797,8 1,50 6,71 2.832,7 0,062 3,90
138 am. 2 2,50 6,53 2.689,6 0,058 3,84 138 am.3 3,00 5,03 1.968,0 0,101 3,83 140 am. 1
Sob.1+ FeSO4
6.768,7
0,33 12,5 9.006,2 0,030 11,92 140 am. 2 0,50 8,02 8.925,7 0,030 4,19 140 am.3 1,00 6,38 6.148,8 0,031 3,74 140 am.4 1,50 5,31 4.963,5 0,033 3,89 141 am. 1 Sob.1+
FeSO4
6.482,7 3,00 3,73 3.134,1 0,037 3,18
141 am. 2 2,50 5,39 4.609,2 0,030 2,92 141 am. 3 2,00 6,45 4.342,1 0,031 2,91
A Tabela 35 apresenta os parâmetros dos testes realizados, as
Figuras 57 a 60 apresentam os difratogramas de raios-X das amostras obtidas pelo método ácido (com baixa concentração de ferro), e a Tabela 36 resume as principais análises dos pigmentos obtidos.
172
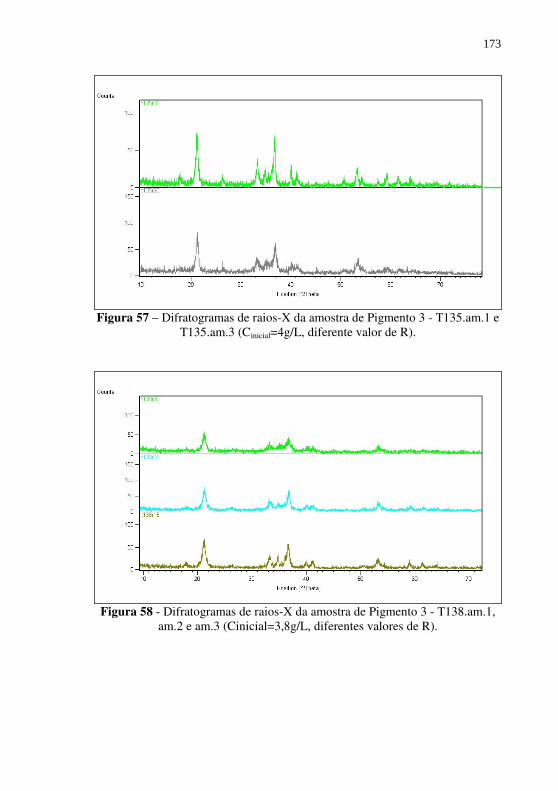
Figura 57 – Difratogramas de raios-X da amostra de Pigmento 3 - T135.am.1 e
T135.am.3 (Cinicial=4g/L, diferente valor de R).
Figura 58 - Difratogramas de raios-X da amostra de Pigmento 3 - T138.am.1,
am.2 e am.3 (Cinicial=3,8g/L, diferentes valores de R).
173
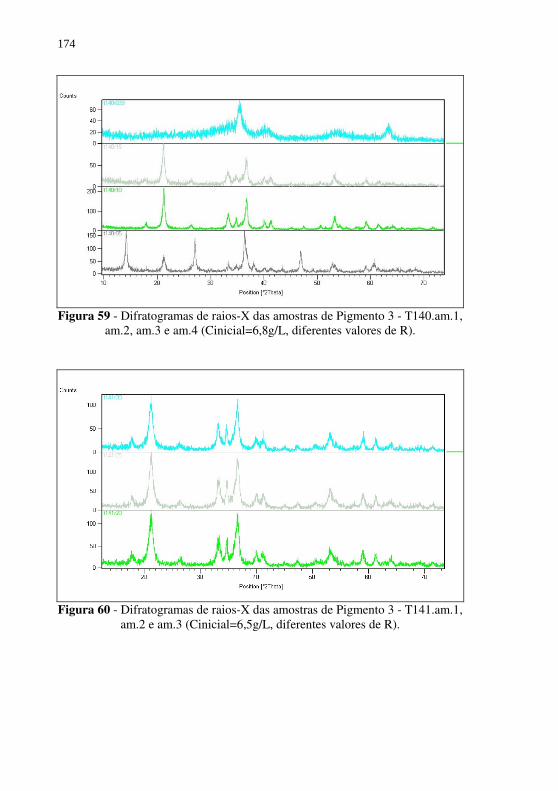
Figura 59 - Difratogramas de raios-X das amostras de Pigmento 3 - T140.am.1,
am.2, am.3 e am.4 (Cinicial=6,8g/L, diferentes valores de R).
Figura 60 - Difratogramas de raios-X das amostras de Pigmento 3 - T141.am.1,
am.2 e am.3 (Cinicial=6,5g/L, diferentes valores de R).
174

Tab
ela
36 -
Car
acte
riza
ção
físi
co-q
uim
ica
e co
lori
mét
rica
das
am
ostr
as d
e P
igm
ento
3 p
rodu
zida
s co
m R
áci
do e
de
um
a am
ostr
a de
pig
men
to a
mar
elo
com
erci
al.
AM
OST
RA
F
luor
escê
ncia
de
raio
s-X
A
nális
e T
erm
ogra
vim
étri
ca
Col
orím
etri
a G
ranu
lom
etri
a po
r di
sper
são
a L
aser
, (µ
m)
R
%,
Al 2O
3 %
, Fe
2O3
%,
MnO
Perd
a ao
fo
go
Pico
1,
0 C
Perd
a de
m
assa
Pico
2,
0 C
Perd
a de
m
assa
L
* A
* B
* ∆
Eab
(1)
d 10
%
d 50
%
d 90
%
d m
édio
Pigm
ento
am
arel
o -
0,04
87
,59
0,05
12
,1%
-
- 31
8 10
,4%
59
,2
15,4
6 47
,88
0,0
0,40
0,
60
0,90
-
T13
5.am
.1
2,0
7,06
71
,80
0,05
16
,7%
44
,0
5,80
%
269
10,6
%
43,6
3 22
,09
37,6
1 19
,8
1,69
5,
25
9,56
5,
50
T13
5.am
.3
1,0
3,31
79
,57
0,25
14
,0%
63
,0
3,50
%
267
10,0
%
38,5
1 19
,42
32,7
8 25
,9
1,36
4,
05
7,82
4,
41
T13
8.am
.1
3,0
5,97
72
,8
0,04
15
,5%
61
,5
- 27
7 18
,6%
42
,15
20,6
0 33
,93
22,6
1,
65
5,97
10
,3 9 6,
07
T13
8.am
.2
2,5
4,5
76,1
0,
05
15,2
%
38,0
5,
30%
26
9 11
,7%
41
,37
21,8
8 36
,26
22,2
2,
11
5,92
10
,1 8 6,
06
T13
8.am
.3
1,5
2,43
79
,76
0,11
15
,0%
-
- 26
0 14
,0%
43
,01
22,0
0 39
,13
19,5
1,
35
4,06
7,
4 4,
23
T
140.
am.1
0,
33
1,27
87
,57
0,84
5,
1%
75,7
-
191
12,0
%
19,1
8 13
,40
12,3
6 53
,5
- -
- -
T14
0.am
.2
0,50
1,
87
85,3
8 0,
36
11,4
%
65,9
2,
80%
27
1 10
,6%
30
,26
18,0
6 24
,51
37,3
-
- -
- T
140.
am.3
1,
0 3,
27
82,4
2 0,
12
12,0
%
66,9
-
269
15,0
%
36,9
5 19
,06
34,0
7 26
,4
- -
- -
T14
0.am
.4
1,5
4,41
80
,14
0,06
13
,2%
82
,9
- 26
0 13
,4%
37
,32
20,0
6 35
,12
25,7
-
- -
- T
141.
am.1
3,
0 0,
33
82,6
9 0,
10
14,5
%
14
,90%
25
1 5,
0%
46,2
8 21
,22
41,3
4 15
,6
- -
- -
T14
1.am
.2
2,5
0,3
82,0
6 0,
11
14,6
%
17
,20%
24
9 4,
6%
45,7
6 20
,82
39,8
6 16
,5
- -
- -
T14
1.am
.3
2,0
0,23
82
,93
0,11
14
,5%
19,5
0%
247
4,0%
45
,13
20,7
9 39
,3
17,3
-
- -
- Pi
co 1
� p
ico
endo
térm
ico
refe
rent
e a
perd
a de
águ
a ad
sorv
ida
e/ou
águ
a es
trut
ural
pre
sent
e na
am
ostr
a de
óxi
do d
e fe
rro.
Pi
co 2
� p
ico
endo
térm
ico
refe
rent
e a
mud
ança
de
fase
da
Goe
tita
par
a a
Hem
atita
.
175

No teste T135 com concentração inicial de 4g/L e R ácido, (R = 1,0 ou 2,0), observam-se pelos difratogramas de raios-X que foi obtida somente a Goetita como fase cristalina e praticamente não houve diferença na cristalinidade das amostras para os dois valores de R (Figura 57). Houve apenas uma pequena melhora da cristalinidade da amostra com R=1,0.
Comportamento similar foi observado no teste T138, (Figura 58), onde a concentração inicial de ferro ferroso no Sobrenadante 1 também era baixa (≅3,7g/L), e os valores de R variaram entre 1,5; 2,5 e 3,0. Ou seja, o melhor valor de R encontrado foi de 1,5 molFe+2/mol OH-.
No teste T140 com Cinicial = 6,8g/L, o melhor valor de R encontrado também foi R=1,0, haja vista que com R=0,5 houve a formação da Lepidocrocita. Já no teste T141, (Cinicial = 6,5g/L), praticamente não houve diferença entre R=2,0; 2,5 e 3,0 (Figuras 59 e 60, respectivamente).
Com relação ao teor de Fe2O3 das amostras, Tabela 36, observa-se que o mesmo ficou entre 72 e 85%, sendo os valores baixos justificados pela contaminação com Al2O3 no moinho de bolas.
Com a realização destes testes foi possível definir o novo método para a produção do pigmento amarelo a ser utilizado na planta industrial.
Este método consiste na adição de soda ao Sobrenadante 1, obtendo-se um valor de R entre 1,0 e 1,5 molFe+2/molOH-, formação de um lodo verde intermediário por decantação e espessamento até obter-se uma concentração de ferro ferroso como sólido no lodo ≥ 4,5g/L; oxidação deste composto intermediário para a formação do pigmento amarelo com uma baixa velocidade de oxidação; espessamento, filtração, lavagem, secagem, moagem e classificação granulométrica do pigmento.
176

7 PRODUÇÃO DO PIGMENTO AMARELO NA PLANTA INDUSTRIAL
Após a definição do novo método industrial para a produção do
pigmento amarelo a partir da drenagem ácida de mina, a planta, ora projetada para trabalhar no método de crescimento, foi adaptada para a operação dentro dos novos conceitos implantados; ou seja, a formação de um composto intermediário antecedendo a obtenção do pigmento.
A operação da planta para a produção do pigmento amarelo deu inicio em meados de Julho de 2009 e a mesma seguiu conforme descrito nas Figuras 6 e 7; ou seja:
Circuito 1 – remoção do alumínio presente na DAM com produção do Gesso Amarelo e Sobrenadante 1.
Circuito 2 – produção do pigmento amarelo a partir do Sobrenadante 1 via formação de um composto intermediário.
As Tabelas 37 e 38 apresentam os resultados experimentais de cinco bateladas de pigmento amarelo produzidas. Ressalta-se que o Circuito 1 e a primeira fase do Circuito 2 operam em sistema contínuo, e os resultados de caracterização do Sobrenadante 1 e do Sobrenadante intermediário referem-se a uma média de no mínimo 15 dias de operação. Já a segunda etapa do Circuito 2, ou seja, a fase de oxidação do composto intermediário com formação do pigmento em si, opera em bateladas.
A Figura 61 apresenta os difratogramas de raios-X das amostras produzidas sobrepostos ao difratograma de raios-X da amostra comercial de pigmento amarelo. A Tabela 39 apresenta a caracterização das amostras.
177
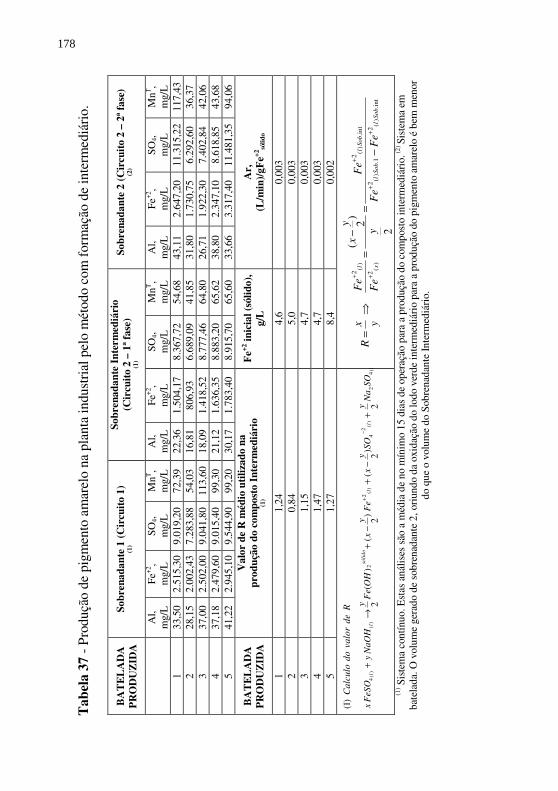
Tab
ela
37 -
Pro
duçã
o de
pig
men
to a
mar
elo
na p
lant
a in
dust
rial
pel
o m
étod
o co
m f
orm
ação
de
inte
rmed
iári
o.
BA
TE
LA
DA
P
RO
DU
ZID
A
Sobr
enad
ante
1 (
Cir
cuit
o 1)
(1
)
Sobr
enad
ante
Int
erm
ediá
rio
(C
ircu
ito
2 –
1ª fa
se)
(1)
Sobr
enad
ante
2 (
Cir
cuit
o 2
– 2ª
fase
) (2
)
A
l, m
g/L
Fe
+2 ,
mg/
L
SO4,
m
g/L
M
nT,
mg/
L
Al,
mg/
L
Fe+
2 , m
g/L
SO
4,
mg/
L
MnT
, m
g/L
A
l, m
g/L
Fe
+2 ,
mg/
L
SO4,
m
g/L
M
nT,
mg/
L
1 33
,50
2.51
5,30
9.
019,
20
72,3
9 22
,36
1.50
4,17
8.
367,
72
54,6
8 43
,11
2.64
7,20
11
.315
,22
117,
43
2 28
,15
2.00
2,43
7.
283,
88
54,0
3 16
,81
806,
93
6.68
9,09
41
,85
31,8
0 1.
730,
75
6.29
2,60
36
,37
3 37
,00
2.50
2,00
9.
041,
80
113,
60
18,0
9 1.
418,
52
8.77
7,46
64
,80
26,7
1 1.
922,
30
7.40
2,84
42
,06
4 37
,18
2.47
9,60
9.
015,
40
99,3
0 21
,12
1.63
6,35
8.
883,
20
65,6
2 38
,80
2.34
7,10
8.
618,
85
43,6
8 5
41,2
2 2.
945,
10
9.54
4,90
99
,20
30,1
7 1.
783,
40
8.91
5,70
65
,60
33,6
6 3.
317,
40
11.4
81,3
5 94
,06
BA
TE
LA
DA
P
RO
DU
ZID
A
Val
or d
e R
méd
io u
tiliz
ado
na
prod
ução
do
com
post
o In
term
ediá
rio
(1)
Fe+
2 inic
ial (
sólid
o),
g/L
A
r,
(L/m
in)/
gFe+
2 sóli
do
1 1,
24
4,6
0,00
3 2
0,84
5,
0 0,
003
3 1,
15
4,7
0,00
3 4
1,47
4,
7 0,
003
5 1,
27
8,4
0,00
2
(4
2)
(2
4)
(2
2)
()
(4
2)
2(
)2
()
(2
)1(
ll
lsó
lid
o
ll
SO
Na
yS
Oy
xF
ey
xO
HF
ey
Na
OH
yF
eS
Ox
Rd
eva
lor
do
Ca
lcu
lo
+−
+−
+→
+−
+
in
t.
)(
21.
)(
2
int
.)
(2
)(
2
)(
2
2
)2
(
So
bl
So
bl
So
bl
sl
Fe
Fe
Fe
y
yx
Fe
Fe
yxR
++
+
++
−=
−=
⇒=
(1) S
iste
ma
cont
ínuo
. Est
as a
nális
es s
ão a
méd
ia d
e no
mín
imo
15 d
ias
de o
pera
ção
para
a p
rodu
ção
do c
ompo
sto
inte
rmed
iári
o. (
2) Si
stem
a em
ba
tela
da. O
vol
ume
gera
do d
e so
bren
adan
te 2
, ori
undo
da
oxid
ação
do
lodo
ver
de in
term
ediá
rio
para
a p
rodu
ção
do p
igm
ento
am
arel
o é
bem
men
or
do q
ue o
vol
ume
do S
obre
nada
nte
Inte
rmed
iári
o.
178
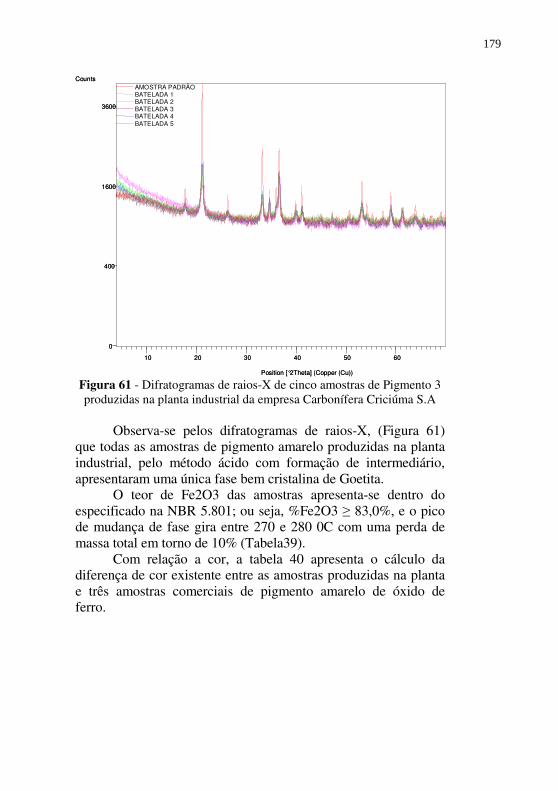
0
400
1600
3600
Counts
Position [°2Theta] (Copper (Cu))
10 20 30 40 50 60
BATELADA 5BATELADA 4BATELADA 3BATELADA 2BATELADA 1AMOSTRA PADRÃO
0
400
1600
3600
Counts
Position [°2Theta] (Copper (Cu))
10 20 30 40 50 60
BATELADA 5BATELADA 4BATELADA 3BATELADA 2BATELADA 1AMOSTRA PADRÃO
Figura 61 - Difratogramas de raios-X de cinco amostras de Pigmento 3 produzidas na planta industrial da empresa Carbonífera Criciúma S.A
Observa-se pelos difratogramas de raios-X, (Figura 61)
que todas as amostras de pigmento amarelo produzidas na planta industrial, pelo método ácido com formação de intermediário, apresentaram uma única fase bem cristalina de Goetita.
O teor de Fe2O3 das amostras apresenta-se dentro do especificado na NBR 5.801; ou seja, %Fe2O3 ≥ 83,0%, e o pico de mudança de fase gira entre 270 e 280 0C com uma perda de massa total em torno de 10% (Tabela39).
Com relação a cor, a tabela 40 apresenta o cálculo da diferença de cor existente entre as amostras produzidas na planta e três amostras comerciais de pigmento amarelo de óxido de ferro.
179

Tab
ela
38 -
Car
acte
riza
ção
das
amos
tras
de
pigm
ento
pro
duzi
das
na p
lant
a in
dust
rial
.
BA
TE
LA
DA
%
SO
4
Car
acte
riza
ção
quím
ica
por
Flu
ores
cênc
ia d
e ra
ios-
X
TG
A
Dis
pers
ão a
Las
er
Col
orím
etri
a
Fe2
O3,
%
A
l2O
3,
%
MnO
, %
P
.Fog
o,
%
Pic
o m
udan
ça
de f
ase
Per
da
de
mas
sa
d10
d50
d90
L
a b
Pig
men
to
Am
arel
o co
mer
cial
1 0,
10
a 0,
40
83,2
3 a
86,0
0 ≅
0,2
2 ≅
0,0
3 -
326
9,4%
0,
30
a 0,
40
0,50
a
0,60
0,75
a
0,85
56,3
5 15
,49
46,0
5 2
- 32
7 9,
6%
58,7
2 11
,49
36,7
3 3
- 32
5 9,
0%
56,4
2 17
,58
45,5
4 1
0,00
83
,20
0,40
0,
14
13,8
4 26
9 10
,6%
3,
2 12
,8
32,0
42
,20
18,4
0 37
,80
2 0,
00
84,1
3 0,
15
0,18
13
,56
269
10,8
%
2,3
13,8
46
,1
45,2
2 17
,39
36,3
9 3
0,00
84
,01
0,32
0,
16
13,4
4 27
6 9,
9%
0,9
8,6
29,3
46
,83
17,8
1 37
,4
4 0,
00
83,9
8 0,
35
0,15
13
,37
274
11,1
%
0,5
7,3
28,4
46
,72
17,8
6 38
,05
5
83,2
9 0,
49
0,12
13
,94
276
10,5
%
0,6
12,6
33
,2
47,0
7 18
,00
39,0
4 5
(1)
- -
- -
- -
- -
- <
1,5
49
,5
15,9
0 37
,33
(1)
Am
ostr
a m
oída
.
Obs
erva
-se
pela
Tab
ela
40 q
ue a
dif
eren
ça d
e co
r en
tre
as a
mos
tras
de
pigm
ento
am
arel
o pr
oduz
idas
na
plan
ta d
a C
CSA
e a
s am
ostr
as d
e pi
gmen
to a
mar
elo
com
erci
al g
irou
ent
re 1
0 a
15 (
∆E
ab 1
, 2 o
u 3)
. Por
ém p
ode
ser
obse
rvad
o pe
la t
abel
a qu
e os
men
ores
val
ores
de
dife
renç
a de
cor
for
am o
btid
os p
ara
a am
ostr
a 5
moí
da
(∆E
ab1
= 1
1; ∆
Eab
2 =
10,
2; ∆
Eab
3 =
10,
9). O
bser
va-s
e ai
nda
que
a di
fere
nça
de c
or e
ntre
as
amos
tras
com
erci
ais
de p
igm
ento
am
arel
o ta
mbé
m c
hega
a 1
0,4
e 11
,0 (
Tab
ela
40).
Ou
seja
, de
uma
amos
tra
com
erci
al p
ara
outr
a há
a
mes
ma
dife
renç
a de
cor
que
exi
ste
entr
e a
amos
tra
de p
igm
ento
pro
duzi
da n
a pl
anta
da
CC
SA e
a a
mos
tra
com
erci
al.
180
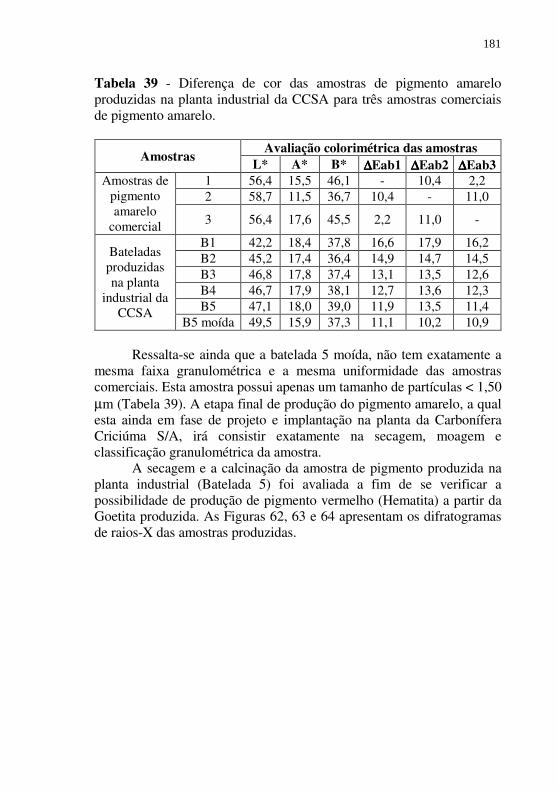
Tabela 39 - Diferença de cor das amostras de pigmento amarelo produzidas na planta industrial da CCSA para três amostras comerciais de pigmento amarelo.
Amostras Avaliação colorimétrica das amostras
L* A* B* ∆∆∆∆Eab1 ∆∆∆∆Eab2 ∆∆∆∆Eab3 Amostras de
pigmento amarelo
comercial
1 56,4 15,5 46,1 - 10,4 2,2 2 58,7 11,5 36,7 10,4 - 11,0
3 56,4 17,6 45,5 2,2 11,0 -
Bateladas produzidas na planta
industrial da CCSA
B1 42,2 18,4 37,8 16,6 17,9 16,2 B2 45,2 17,4 36,4 14,9 14,7 14,5 B3 46,8 17,8 37,4 13,1 13,5 12,6 B4 46,7 17,9 38,1 12,7 13,6 12,3 B5 47,1 18,0 39,0 11,9 13,5 11,4
B5 moída 49,5 15,9 37,3 11,1 10,2 10,9 Ressalta-se ainda que a batelada 5 moída, não tem exatamente a
mesma faixa granulométrica e a mesma uniformidade das amostras comerciais. Esta amostra possui apenas um tamanho de partículas < 1,50 µm (Tabela 39). A etapa final de produção do pigmento amarelo, a qual esta ainda em fase de projeto e implantação na planta da Carbonífera Criciúma S/A, irá consistir exatamente na secagem, moagem e classificação granulométrica da amostra.
A secagem e a calcinação da amostra de pigmento produzida na planta industrial (Batelada 5) foi avaliada a fim de se verificar a possibilidade de produção de pigmento vermelho (Hematita) a partir da Goetita produzida. As Figuras 62, 63 e 64 apresentam os difratogramas de raios-X das amostras produzidas.
181

500
1000
1500
2000
2500
3000
3500
4000
4500
10 20 30 40 50 60 70
Posição (2 theta)
Inte
nsi
dad
e
bat.5 - 200 oC bat.5 - 100 oC pigmento amarelo
Figura 62 - Difratogramas de raios-X da amostra de Pigmento 3 produzida na planta industrial (Batelada 5), seca a 100 e 200oC.
1500
2000
2500
3000
3500
4000
20 30 40 50 60 70
Posição (2 theta)
Inte
nsi
dad
e
bat.5 - 300 oC bat.5 - 500 oC
Figura 63 - Difratogramas de raios-X da amostra de Pigmento 3 produzida na planta industrial (Batelada 5), calcinada a 300 e 500oC.
182

0
50
100
150
200
250
300
20 30 40 50 60 70
Posição (2 theta)
Inte
nsi
dad
e
pigmento vermelho (Hematita)
Figura 64 - Difratograma de raios-X de uma amostra de pigmento comercial vermelho.
Observa-se que para a obtenção do pigmento vermelho, partindo-
se do pigmento amarelo produzido na planta industrial da CCSA é necessário apenas que seja realizada a calcinação da amostra a temperaturas entre 300 e 500 0C.
Com a realização destes testes, foi possível produzir o pigmento amarelo de óxido de ferro e o pigmento vermelho a partir da drenagem ácida de mina.
183

8 CONCLUSÕES A principal conclusão deste trabalho é a de que é possível, a
partir de uma drenagem ácida de mina contendo principalmente ferro ferroso como componente majoritário, produzir pigmentos inorgânicos de óxido de ferro, amarelo, vermelho ou preto, com qualidade similar aos pigmentos comerciais. Abaixo, estão descritos em itens as conclusões obtidas de cada etapa.
O Lodo Químico 1, obtido através da adição de soda cáustica diretamente na drenagem ácida de mina, possui em torno de 45% de Fe2O3 e 5% de Al2O3.
Este lodo foi obtido utilizando-se um valor de R1=0,45 molFeT/molNaOH, ou seja, um excesso de NaOH em relação ao FeSO4 presente na drenagem.
Pela solubilização do Lodo Químico 1 em ácido sulfúrico, utilizando-se a quantidade estequiométrica de ácido para a reação com o ferro e alumínio presentes no lodo, foi possível produzir um coagulante férrico aluminoso, o Coagulante 1, com 24% de Fe2(SO4)3 e 8% de Al2(SO4)3.
A utilização deste coagulante no tratamento físico-químico de um efluente têxtil industrial apresentou bons resultados com relação a remoção de cor, turbidez e DQO presentes no efluente.
A caracterização das amostras preliminares de Pigmento 1, obtidas a partir do Lodo Químico 1, mostrou que estes pigmentos apresentam uma baixa cristalinidade, porém que começam a surgir os picos da Goetita e da Lepidocrocita.
Os teores de Fe2O3 nas amostras de Pigmento 1, em torno de 35 a 50%, ficaram muito baixos. Isto foi associado á presença dos altos teores de alumínio e sulfato nas amostras.
A eliminação do alumínio foi realizada pelo tratamento prévio da DAM com cal e a eliminação do sulfato foi realizada por lavagem alcalina no lodo de óxido-hidróxido de ferro obtido.
Aumentando-se a temperatura secagem e calcinação do Pigmento 1, de 100 para 300, 500, 800 e 1000°C, observa-se um aumento no teor de Fe2O3 das amostras devido a perda da água, perda do sulfato e mudança de fase do óxido hidróxido de ferro.
Na amostra final de Pigmento 1, PG10.B o pico endotérmico referente á mudança de fase da Goetita para Hematita ocorreu em 254oC e a difração de raios-X ilustra uma amostra pouco cristalina mas claramente aparecem os picos da Goetita.
Com relação a cor, o Pigmento 1, PG10B, encontra-se

exatamente em um patamar intermediário entre o pigmento vermelho e o pigmento amarelo, tendo uma ligeira tendência para o pigmento vermelho.
Para os pigmentos comerciais de óxido de ferro, verificaram-se fases puras com alto grau de cristalinidade da Goetita para o pigmento amarelo, Hematita para o pigmento vermelho e Magnetita para o pigmento preto.
Com relação ao tratamento sequencial da DAM, observa-se que o primeiro circuito (Circuito 1) é bastante efetivo para a remoção do alumínio, removendo em torno de 85 a 90% do mesmo, é efetivo para a não remoção do ferro, uma vez que remove em torno de 8 á 10% do ferro e remove em torno de 30 a 40% do sulfato e da acidez presentes na drenagem.
A condição ótima de operação do Circuito 1, ou seja, aquela condição em que se obtém a máxima remoção do alumínio e a mínima remoção do ferro, é alcançada com dosagens na faixa de 0,09 a 0,13 equivalente - grama alcalino /grama Al+3.
Através da aplicação do Circuito 1, é possível obter-se o Sobrenadante 1, um sulfato ferroso líquido isento de alumínio que será utilizado para a produção do pigmento no Circuito 2 e o Gesso Amarelo.
Pelos difratogramas de raios-X e pela caracterização química das amostras de Gesso Amarelo observa-se que esta amostra possui como fase cristalina somente o sulfato de cálcio dihidratado, (CaSO4.2H2O) e possui 80% em massa de CaSO4.2H2O e 20% de Al2O3+Fe2O3.
As impurezas de alumínio e ferro do Gesso Amarelo podem ser eficientemente removidas pelo tratamento ácido através do qual é possível obter uma amostra sólida bem cristalina de Gipsita Branca e uma amostra líquida bem concentrada de Coagulante férrico aluminoso (Coagulante 2).
A mudança de fase da Gipsita para a Bassanita, (gesso utilizado na construção civil), pode ser processada pela calcinação da amostra a 1600C por 3,0 horas.
A aplicação do Circuito 2 para a produção do pigmento pelo método de crescimento (Pigmento 2), produz uma fase sólida pura rica em ferro férrico e uma fase líquida ainda ácida, porém com baixas concentrações de íons metálicos (Sobrenadante 2).
O método de crescimento, consiste na neutralização lenta de uma solução aquosa de sulfato ferroso, contendo entre 6 e 10g/L de Fe+2, com um tempo total de reação de 7 a 8h, e uma dosagem contínua de peróxido de hidrogênio e soda cáustica.
O tratamento do Sobrenadante 1 no Circuito 2 pelo método de
185

crescimento é bastante eficiente para a remoção do ferro e da acidez removendo em torno de 83% do ferro e 80% da acidez.
O Pigmento 2, produzido pelo método de crescimento, apresenta uma % de Fe2O3 em torno de 74 a 80% e um teor de sulfato entre 2 e 8%.
Com relação a colorimetria este pigmento têm um ∆Eab(amarelo)>10 e ∆Eab(vermelho)>34 indicando que a tonalidade tende para o amarelo e não mais para o vermelho como era o caso do Pigmento 1.
Para a amostra de Pigmento 2, o pico da mudança de fase da Goetita para Hematita ocorre a 2820C com uma perda de massa total de 12,72%.
A produção de pigmento pelo método de crescimento a partir de uma drenagem ácida de mina com baixa concentração de ferro ferroso (≅ 4g/L) não é possível. A amostra produzida neste caso é praticamente amorfa e não cristalina.
O método para a produção de pigmento amarelo a partir da drenagem ácida de mina com 4g/L de ferro ferroso consiste na adição rápida de soda ao sobrenadante 1, obtendo-se um valor de R entre 1,0 e 1,5 molFe+2/molOH-; formação de um lodo intermediário por decantação e espessamento; oxidação deste composto intermediário para a formação do pigmento amarelo; espessamento; filtração; lavagem; secagem; moagem e classificação granulométrica.
Foi possível produzir na planta industrial da Carbonífera Criciúma S/A várias amostras de pigmento amarelo de óxido de ferro. Estas amostras apresentaram uma única fase bem cristalina de Goetita e após calcinação, Hematita. O teor de Fe2O3 das amostras produzidas na planta ficou dentro do especificado na NBR 5.801; ou seja, %Fe2O3 ≥ 83,0%, e o pico de mudança de fase gira entre 270 e 280 0C com uma perda de massa total em torno de 10%.
186

REFERENCIAS
A Malik, M. G. Dastidar, P. K. Roychoudhury. Factors limiting bacterial iron oxidation in biodesulphurization system, Int. J. Miner. Process, 2003.
Albertino Frello, Avaliação da biodessulfurização do carvão mineral de Santa Catarina por intemperismo natural, lixiviação com água ácida de mina e inoculação microbiana, Dissertação de Mestrado, UFSC, 1998.
Andrade A.F.C.; Trindade Filho M.O.; Borja E.V.; Souza Junior C.F. e Silva, G.G., Estudo de compósitos cerâmicos com resíduos de gesso da indústria da construção civil. I Congresso de Pesquisa e Inovação da Rede Norte Nordeste de Educação Tecnológica, 2006.
Baltar C.A.M.; Bastos F.F. e Luz A.B., Diagnóstico do pólo gesseiro de Pernambuco (Brasil) com ênfase na produção de Gipsita para fabricação de cimento. Disponível em http://www.minas.upm.es/catedra-anefa/Consultas.htm . Acesso em 24 maio 2006.
Basegio T.; Berutti F.; Bernardes A. and Bergmann, C.P., Environmental and technical aspects of the utilization of tannery sludge as a raw material for clay products. Journal of the European Ceramic Society, v. 22, p. 2251-2259, 2002.
Betancur, J.D.; Barreto C.A.; Greneche, J.M., Goya, G.F. The effect of water content on the magnetic and structural properties of goethite. J. of Alloys and Compounds, 369, 247-251, 2004.
Bioreactor systems for treatment of acid mine drainage, Canmet Mining and Mineral Sciences Laboratories, Canada, 2002.
Butler A.D.; Fan M.; Brown R.C.; Leeuwen J.; Sung S. and Duff B., Pilot scale tests of poly ferric sulfate synthetized using SO2 at Des Moines Water Works, Chemical Engineering Processing, 44, 413-419, 2004.
3 BP Statistical Review of World Energy June 2008
Buxbaum, G.; Pfaff G. Industrial inorganic pigments. WILEY-VCH

GmbH & Co.: Weinheim, Germany, 2005.
Cabral T., Ignatiadis I., Mechanistic study of the pyrite – solution interface during the oxidative bacterial dissolution of pyrite by using electrochemical thecnical. Int. J. Miner. Process., 62, 41-64, 2001.
Canut M.M.C., Estudo da viabilidade do uso de resíduo fosfogesso como material de construção. Dissertação de Mestrado. Universidade Federal de Minas Gerais, 2006.
Casqueira, R.G. e Santos, S.F. Pigmentos inorgânicos: Propriedades, Métodos de síntese e Aplicações. Série Rochas e Minerais Industriais, 12. Rio de Janeiro. CETEM/MCT, 2008.
Cici M, Cuci, Production of some coagulant materials from galvanizing workshop waste, Waste Management, 17, 407-410, 1998.
CIE, International Commsion on Illumination. Colorimetry. 2ºed, CIE Publications nº 15,2, 1986.
Colmer, A.R.; Temple, K.L.; Hinckle, M.E. (1950). An iron-oxidizing bacterium from the acid drainage of some bituminous coal mines. Journal of Bacteriology, 59, 317.
CONAMA, Resolução CONAMA Nº 357, de 17 de Março de 2005.
Cornell R. M.; Schwertmann U. The Iron Oxides – Structure, Properties, Reactions, Occurrences and Uses. 2nd Edition, 2003.
Crundwell F., The formation of biofilms of iron-oxidising bacteria on pyrite. Minerals Engineering, Vol.9, No 10, pp 1081-1089, 1996.
Degirmenci N., The using of waste phosphogypsum and natural gypsum in adobe stabilization. Construction and Building Materials, article in press, doi:10.1016/j.conbuildmat.2007.01.027, 2007.
Detournay J.; Derie R.; Ghodsi M. Etude de l’oxydation par aération de Fe(OH)2. Z. Anorg. Allg. Chem. 427, 265-273, 1976.
Dias L.E., Uso de gesso como insumo agrícola. Comunicado Técnico, CNPBS Nº7, pp. 1-6, 1992.
188

DIN 6174. Farbmetrische Bestimmung von Farbabständen bei Körperfarben nach der CIELAB Formel, Beuth Verlag, Berlim und Köln 1979.
Evangelou, V.P., Pyrite Oxidation and Its Control: solution chemistry, surface chemistry, acid mine drainage, molecular oxidation mechanisms, microbial role, kinetics, control, ameliorates and limitations microencapsulation, 1a Ed., Florida, 1995.
Fan M, Brown RC, Leewen JV, Nomura M, Zhuang Y, The kinetics of producing sulfate-based complex coagulant from fly ash, Chemical Engineering and Processing, 42 (2003) 1019-1025.
Fan M, Sung S, Brown RC, Wheelock TD, Laabs FC, Synthesis, characterization and coagulation performance of polymeric ferric sulfate, J. Environ. Eng. 128(6) (2002) 483-490.
Fauziah I.; Zauyah S. e Jamal T., Characterization and land application of red gypsum: a waste product from the titanium dioxide industry. The Science of the Total Environmental, v. 188, p. 243-251, 1996.
Fowler T. A., Holmes P.R., Crundwell F. K., On the kinetics and mechanism of the dissolution of pyrite in the presence of Thiobacillus ferroxidans, Hydrometalurgy. 59, 257-270, 2001.
Gazea B., Adam K, Kontopoulos A, A review of passive systems for the treatment of acid mine drainage, Minerals Engineering, 9,23 -42, 1996.
Gilbert, F.; Refait P.; Lévêque F.; Remazeilles C. e Conforto E. Synthesis of goethite from Fe(OH)2 precipitates: Influence of Fe(II) concentration and stirring speed. J. Physics and Chemistry of Solids 69, 2124-2130, 2008.
Hendrich S.; Fan M.; Sung S.; Brown R.C.; Mazur M.L.; Myers R. e Osweiler G.S., Toxic evaluation of polymeric ferric sulfate, Int. J. Environ. Technol. Manag. 1, 464-471, 2001.
Herbert Jr. R. B., Sulfide oxidation in mine waste deposits - A review with emphasis on dysoxic weathering. — 45 p., Luleå (MiMi Print),1999.
189

Herbert Jr., R. B. MiMi — Sulfide oxidation in mine waste deposits —Hood, T.A., The kinetics of Pyrite Oxidation in Marine Systems, Ph.D. Thesis, University of Miami, 1999.
Hutchins, S.R.; Davidson, M.S.; Brierley, J.A.; Brierly,C.L. (1986). Microorganisms in reclamation of metals. Ann. Rev. Microbiol., 40, p. 311-336.
Ivanov, V. I., Effect of some factors on iron oxidation by cultures of Thiobacillus Ferroxidans, Microbiology, 31, p.645, 1962.
Jae-Young Yu, Terence J. McGenity, Max L. Coleman. Solution chemistry during the lag phase and exponential phase of pyrite oxidation by Thiobacillus ferrooxidans, Chemical Geology, 175, 307-317, 2001.
Johnson, D.B. e Hallberg, K.B. Acid mine drainage options: a review. Science of the Total Environment, v.338, p.3-14, 2005.
Kalin, M; Fyson, A; Wheeler, W N, The chemistry of conventional and alternative treatment systems for the neutralization of acid mine drainage, Science of The Total Environment, 366, 395-408, 2006.
Karavaiko, G.I.; Groudev, S.N. (1985). Biogeotechnology of metals, its history, tasks and trends of development. In International Seminar on Modern Aspects of Microbiological Hydrometallurgy, Moscou; International Training Course on Microbiological Leaching of Metals from Ores, Sophia, 1982. Collected papers. Moscou, Centre of International Projects GKNT, p. 6-24.
Kargi, F. (1982). Coal dessulfurization by leaching involving acidophilic and thermophilic microorganisms. Biotechnology and Bioengineering, 24, p. 743-8.
Kirk- Othmer, Encyclopedia of Chemical Technology, John Wiley & Sons, Inc.,1995.
Kontopoulos, A. Acid mine drainage control. In: Effluent treatment in the mining industry. Castro SH, Vergara F, Sánchez MA (editors). University of Concepciòn, 57-118 (1998).
Krehula, S.; Popovic, S. e Music, S. Synthesis of acicular α-FeO.OH
190

particles at a very high pH. Materials Letters, 54, 108-113, 2002.
Kurokawa, H. e Senna, M. Bench scale control of the crystallite size and morphology of goethite particles by different ferrous source. Materials Science and Engineering B, 135, 55-59, 2006.
L. Falco, C. Pogliani, G. Curutchet, E. Donati. A comparison of bioleaching of covellite using pure cultures of Acidithiobacillus ferrooxidans and Acidithiobacillus thiooxidans or a mixed culture of Leptospirillum ferrooxidans and Acidithiobacillus thiooxidans, Hydrometallurgy, 2003.
Leppinen J.O.; Salonsaari P. and Palosaari V., Flotation in acid mine drainage control: Beneficiation of concentrate, Canadian Metallurgical Quaterly 36, 225-230, 1997.
Lewis, A. P. Pigment Handbook: Characterization and Physical Relationships, 1a Ed., New York, Published by Wiley, 1973.
Lopes, N.F. Espectroscopia Raman aplicada ao estudo de pigmentos em bens culturais: I – Pinturas Rupestres. Dissertação de Mestrado. Universidade de São Paulo. Instituto de Química. São Paulo, 2005.
Lowson, R.T., Aqueous oxidation of pyrite by molecular oxygen. Chem. Rev. 82, 461–499, 1982.
Lyra A.C., O Mercado de Gipsita no Brasil. Disponível em www.prossiga.br/gesso. Acesso em 24 maio 2006.
Marcello RR, Galato S, Peterson M, Riella HG, Bernardin AM, Inorganic pigments made from the recycling of coal mine drainage treatment sludge, Journal of Environmental Management, 88, 1280-1284, 2008.
Maree J.P.; Nengovhela N.R.; Strydom C.A. e Greben H.A., Chemical and Biological Oxidation of Iron in Acid Mine Water, Mine Water and the Environment, 45, 76-80, 2004.
Meisen, U. Method of preparing an aluminum-containing iron oxide nucleus. United States. Patent Aplication Publication, Jan. 21, 2003.
191

Meisen, U. Process for production yellow iron oxide pigments. United States. Patent Aplication Publication, Feb. 10, 2004.
Meisen, U.; Brunn, H.; Van Bonn, K. H. e Friedrich H. Process for preparing yellow iron oxide pigments with CaCO3 precipitant. United States. Patent Aplication Publication, Mar. 3, 2005.
Millenium, http://www.millenniumchem.com Acessado em 26 de novembro de 2009.
Monção Junior, A.R., Otimização das condições experimentais na desidratação da Gipsita para obtenção de um gesso beta reciclável. Dissertação de Mestrado, Universidade Católica de Pernambuco, Recife/PE, Janeiro de 2008.
Mota M.K.F; Soares F.P.P; Junior C.F.S e Borja E.V., Reaproveitamento de resíduos de gesso na argila para fabricação de tijolos. Congresso Brasileiro de Engenharia e Ciência de Materiais, 2008.
Music, S. e Gotic, M. Mossbauer, FT-IR and FE SEM investigation of iron oxides precipitated from FeSO4 solutions. J. of Molecular Structure, 834-836, 445-453, 2007.
Music, S.; Popovic, S.; Gotic, M. Mössbauer spectroscopy and X-ray diffraction of oxide precipitates formed from FeSO4 solution, Journal of Materials Science. 25, 3186-3190, 1990.
NBR 5801 - Óxido de ferro, Associação Brasileira de Normas Técnicas - ABNT.
Nicholson RV, Iron-sulfide oxidatin mechanisms: Laboratory studies. Em: J L Jambor e D W Blowes (eds.), The Environmental Geochemistry of Sulfide Mine-Wastes, Short Course Handbook, Mineralogical Association of Canada, Waterloo, Ontario, Canada, 22, 163-183 (1994).
Nordstrom, D.K.; Alpers, C.N.; Ptacek, C.J. e Blowes, D.W., Negative ph and extremely acidic mine waters from Iron Mountain, Califórnia. Environmental Science and Technology, v.34, p.254-258, 2000.
Nunes, J.A., Tratamento físico-químico de água residuárias
192

industriais.3nd ed., Brasil, 2001.
Olowe, A. A. e Génin M.R. The mechanism of oxidation of ferrous hydroxide in sulphated aqueous media: Importance of the initial ratio of the reactants. Corrosion Science, Vol. 32, N0 9, pp.965-984, 1991(a).
Olowe, A. A.; Pauron, B. e Génin M.R. The influence of temperature on the oxidation of ferrous hydroxide in sulphated aqueous media: Activation energies of formation of the products and hyperfine structure of magnetite. Corrosion Science, Vol. 32, N0 9, pp.985-1001, 1991(b).
Pappu A.; Saxena M. e Asolekar S.R., Solid wastes generation in Índia and their recycling potential in building materials. Building and Environmental, vol. 42, pp. 2311-2320, 2007.
Pereira E.B., Perfil analítico da Gipsita, Boletim 15, DNPM - MME, Rio janeiro, 1973.
Peres L.S.; Benachour M. e Santos V.A., O Gesso: Produção e Utilização na Construção Civil. Recife: Bagaço, 2001. 166p.
Rao S.R.; Gehr R.; Riendeau M.; Lu D. e Finch J.A., Acid mine drainage as a coagulant, Minerals Engineering 5, 1011-1020, 1992.
Refait Ph.; Memet J.B.; Bon C.; Sabot R. and Génin J.-M.R., Formation of the Fe(II)-Fe(III) hydroxysulphate green rust during marine corrosion of steel. Corros. Sci., 45, 833-845, 2003.
Rimstidt D. e Williamson M.A., The kinetics and electrochemical rate determining step of aqueous pyrite oxidation, Geochimica et Cosmochimica Acta, 58, 5.443-5.454, 1994.
Rosenhahn, C. Yellow iron oxide pigment. United States. Patent Aplication Publication, Nov. 15, 2007.
Sand W., Gehrke T., Jozsa P. G., Schippers A., Biochemistry of bacterial leaching – direct vs indirect bioleaching. Hydrometallurgy, 59, 159-175, 2001.
Sandström A. e Mattson E., Bacterial ferrous iron oxidation of acid mine drainage as pre-treatment for subsequent metal recovery, Int. J.
193

Miner. Process. 62, 309-320, 2001.
Santos S.; Machado R.; Correia M.J.N. e Carvalho J.R., Treatment of acid mining waters, Minerals Engineering 17, 225-232, 2004.
Schneider, C.H., Sistema para tratamento de drenagem ácida de mina, Instituto Nacional da Propriedade Industrial, PI0301571-8 A2, Maio/2003.
Sharma, P.K. Das A., K. Rao H, Forssberg. K.S. E. Surface characterization of Acidithiobacillus ferrooxidans cells grown under different conditions, Hydrometallurgy, 2003. 71, 285-292, 2003.
Silva J.B.; Dutra R.P.S.; Nascimento R.M.; Martinelli A.E. e Gomes, U.U., Avaliação da incorporação de rejeitos de gesso de construção em formulações de massa cerâmica. Anais do Congresso Brasileiro de Engenharia e Ciência dos Materiais, pg.2149-2160, 2006.
Silva J.B.; Silva M.P.F.; Cândido T.A.V.; Souza J.; Nascimento R.M. e Gomes, U.U., Caracterização e estudo de formulações da massas para a cerâmica vermelha de argilas provenientes do município de Goianinha-RN. Parte II: Influência do tempo de isoterma. Anais do 510 Congresso Brasileiro de Cerâmica, 2007.
Silverman, M.P.; Lundgren, D.G. (1959). Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans. I. An improved medium and a harvesting procedure for securing high cell yields. J. Bacteriol., 77, p. 642-647.
Sing M. e Garg M. Making of anhydrite cement from waste gypsum. Cement and Concret Research, v. 30, p. 571-577, 2000.
Singer, P.C. e Stumm, W., 1970. The solubility of ferrous iron in carbonate bearing waters. J. Am. Water. Works Ass. 62, 198–202.
Singer, P.C. e Stumm, W., Acidic mine drainage: the reate-determining step, Science, 167, p.1121, 1970.
Singh M., Treating waste phosphogypsum for cement and plaster manufactured. Cement and Concrete Research, vol. 32, pp. 1033-1038, 2002.
194

Site da Embrapa www.embrapa.br, acessado em Março de 2009.
Site da empresa Engessul, www.engessul.com.br., acessado em Março de 2009.
Standard Methods for examination of water and wastewater. 19th edition. Publication office American Public Health Association, Washington. APHA, AWWA, WEF., 1995.
Stumm W. e Lee G.F., Oxygenation of Ferrous iron. Industrial and Engineering Chemistry, 53 (2), 143 – 146, 1961.
Stumm, W., Morgan, J.J., 1970. Aquatic Chemistry: An Introduction Emphasizing Chemical Equilibria in Natural Waters. Wiley-Interscience, New York.
Temple, K.L.; Colmer, A.R. (1951). The autotrophic oxidation of iron by a new bacterium: Thiobacillus ferrooxidans. Journal of Bacteriology, 62, p. 605-611.
Tuovinen, O.H. e Kelly, D.P., Biology of Thiobacillus ferroxidans in relation to the microbiological leaching of sulphide ore. Z. Allg. Microbiologie, 12 (4): 311-346, 1972.
Valls S.E. e Vàzquez E., Accelerated carbonation of sewage sludge-cement-sand mortars and its environmental impact. Cement and concrete research, v. 31, p. 1271-1276, 2001.
Wang W, Xu Z, Finch J, Fundamental study of an ambient temperature ferrite process in the treatment of acid mine drainage, Environmental Science and Technology, 30, 2604-2608, 1996.
Wei X, Viadero Jr RC, Synthesis of magnetite nanoparticles with ferric iron recovered from acid mine drainage: Implications for environmental engineering, Colloids and Surfaces A: Physicochem. Eng. Aspects 294, 280-286, 2007.
Zahed A.H; Zhu J.X. e Grace J.R., Modeling and Simulation of Batch and Continuous Fluidized Bed dryers. Drying Thecnology, 13(1&2), 1-28, 1995.
195

Zouboulis A.I.; Kydros K.A. e Matis K.A., Removal of hexavalent chromium anions from solutions by pyrite fines, Water Research, 29, 1755-1760, 1995.
196

ANEXO A - PRODUÇÃO TÉCNICA E BIBLIOGRAFICA ARTIGOS CIENTÍFICOS MADEIRA, V. S., MOREIRA, R. F. P. M., ANJOS, A., PERUCH, M. G. B. Recuperação de produtos de alto valor agregado a partir da drenagem ácida de mina de carvão mineral. In: XVI Seminário de Iniciação Científica da UFSC, 2006, Florianópolis. Anais do XVI Seminário de Iniciação Científica da UFSC. Florianópolis: Editora da UFSC, 2006. v.01. p.01 – 01. PRODUTOS E PROCESSOS TECNOLÓGICOS COM REGISTROS OU PATENTES MADEIRA, V. S., MOREIRA, R. F. P. M., SCHNEIDER, C. H. Processo para a fabricação de coagulante férrico a partir da drenagem acida de mina de carvão e respectivo coagulante., 2004. Patente: Privilégio de Inovação n.PI0400047-1 A2, PROCESSO PARA A FABRICAÇÃO DE COAGULANTE. 15 de Janeiro de 2004 (Depósito); 18 de Maio de 2004 (Exame); 13 de Setembro de 2005 (Concessão). MADEIRA, V. S., MOREIRA, R. F. P. M., JOSÉ, H. J., CASARIL, L., Dantas, T.L.P. Compósito granular para a remoção de contaminantes em águas e processo para a produção do mesmo., 2004. Patente: Privilégio de Inovação n.PI0405916-6, Compósito granular para a remoção de con. 21 de Dezembro de 2004 (Depósito); 09 de Fevereiro de 2005 (Exame); 03 de Outubro de 2006 (Concessão). MADEIRA, V. S., MOREIRA, R. F. P. M., JOSÉ, H. J., Dantas, T.L.P. Processo catalítico para a destruição de contaminantes em água utilizando peróxido de hidrogênio e catalisador heterogêneo., 2004. Patente: Privilégio de Inovação n.PI0405915-8 A2, PROCESSO CATALÍTICO PARA A DESTRUIÇÃO DE. 21 de Dezembro de 2004 (Depósito); 09 de Fevereiro de 2005 (Exame); 03 de Outubro de 2006 (Concessão). MADEIRA, V. S., MOREIRA, R. F. P. M., JOSÉ, H. J. Processo de
197

obtenção de pigmento inorgânico de oxido de ferro, pigmento amarelo e vermelho, a partir da drenagem acida de mina de carvão e pigmentos obtidos., 2010. Patente: Privilégio de Inovação n.020100025667 (Protocolo INPI), Processo de obtenção de pigmento inorgân. 24 de Março de 2010 (Depósito); MADEIRA, V. S. Processo para a fabricação de cloreto férrico puro e impuro a partir de óxidos de ferro oriundos da ustulacao da pirita., 2010. Patente: Privilégio de Inovação n.020100007046 (Protocolo INPI), Processo para fabricação de cloreto férr. 26 de Janeiro de 2010 (Depósito); MADEIRA, V. S., MOREIRA, R. F. P. M., SCHNEIDER, C. H. Processo para a produção de sulfato ferroso liquido concentrado e respectivo sulfato ferroso obtido., 2010. Patente: Privilégio de Inovação n.020100023571 (Protocolo INPI), Processo para a produção de sulfato ferr. 18 de Março de 2010 (Depósito); MADEIRA, V. S. Processo para obtenção de bassanita a partir de um fosfogesso ou gesso quimico., 2010. Patente: Privilégio de Inovação n.020100007047 (Protocolo INPI), Processos para a obtenção de bassanita a. 26 de Janeiro de 2010 (Depósito);
198

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo